EP0681565B1 - Verfahren zum herstellen alpha-verzweigter aliphatischer monocarbonsäuren - Google Patents
Verfahren zum herstellen alpha-verzweigter aliphatischer monocarbonsäuren Download PDFInfo
- Publication number
- EP0681565B1 EP0681565B1 EP94905102A EP94905102A EP0681565B1 EP 0681565 B1 EP0681565 B1 EP 0681565B1 EP 94905102 A EP94905102 A EP 94905102A EP 94905102 A EP94905102 A EP 94905102A EP 0681565 B1 EP0681565 B1 EP 0681565B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- reaction
- branched aliphatic
- monocarboxylic acids
- reaction mixture
- aliphatic monocarboxylic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- -1 aliphatic monocarboxylic acids Chemical class 0.000 title claims abstract description 15
- 238000004519 manufacturing process Methods 0.000 title claims description 6
- 238000000034 method Methods 0.000 claims abstract description 40
- 239000000344 soap Substances 0.000 claims abstract description 18
- 150000001298 alcohols Chemical class 0.000 claims abstract description 15
- 239000003085 diluting agent Substances 0.000 claims abstract description 10
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 4
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 3
- 238000006243 chemical reaction Methods 0.000 claims description 37
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 23
- 239000011541 reaction mixture Substances 0.000 claims description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 12
- 239000007788 liquid Substances 0.000 claims description 12
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 229910052783 alkali metal Inorganic materials 0.000 claims description 5
- 239000003701 inert diluent Substances 0.000 claims description 5
- 239000007787 solid Substances 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 239000002826 coolant Substances 0.000 claims description 3
- 238000001704 evaporation Methods 0.000 claims description 3
- 239000007789 gas Substances 0.000 claims description 2
- 239000000126 substance Substances 0.000 claims description 2
- 238000000354 decomposition reaction Methods 0.000 claims 1
- 239000003513 alkali Substances 0.000 abstract description 9
- 150000001447 alkali salts Chemical class 0.000 abstract description 3
- 239000003518 caustics Substances 0.000 abstract 1
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 7
- 150000002763 monocarboxylic acids Chemical class 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- ZONJATNKKGGVSU-UHFFFAOYSA-N 14-methylpentadecanoic acid Chemical compound CC(C)CCCCCCCCCCCCC(O)=O ZONJATNKKGGVSU-UHFFFAOYSA-N 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- 238000005187 foaming Methods 0.000 description 4
- XULHFMYCBKQGEE-UHFFFAOYSA-N 2-hexyl-1-Decanol Chemical compound CCCCCCCCC(CO)CCCCCC XULHFMYCBKQGEE-UHFFFAOYSA-N 0.000 description 3
- VGANCIUXOAKSHS-UHFFFAOYSA-N 24-methylpentacosanoic acid Chemical compound CC(C)CCCCCCCCCCCCCCCCCCCCCCC(O)=O VGANCIUXOAKSHS-UHFFFAOYSA-N 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 238000007664 blowing Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- 239000008149 soap solution Substances 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- XUJLWPFSUCHPQL-UHFFFAOYSA-N 11-methyldodecan-1-ol Chemical compound CC(C)CCCCCCCCCCO XUJLWPFSUCHPQL-UHFFFAOYSA-N 0.000 description 1
- KGHVQLDYCDULEN-UHFFFAOYSA-N 22-methyltricosanoic acid Chemical compound CC(C)CCCCCCCCCCCCCCCCCCCCC(O)=O KGHVQLDYCDULEN-UHFFFAOYSA-N 0.000 description 1
- GCUBDUZUXAQLEY-UHFFFAOYSA-N 26-methylheptacosanoic acid Chemical compound CC(C)CCCCCCCCCCCCCCCCCCCCCCCCC(O)=O GCUBDUZUXAQLEY-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 238000007869 Guerbet synthesis reaction Methods 0.000 description 1
- 241000589614 Pseudomonas stutzeri Species 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000003990 capacitor Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 150000001734 carboxylic acid salts Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000002737 fuel gas Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 229940043348 myristyl alcohol Drugs 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- HGASFNYMVGEKTF-UHFFFAOYSA-N octan-1-ol;hydrate Chemical compound O.CCCCCCCCO HGASFNYMVGEKTF-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000007711 solidification Methods 0.000 description 1
- 230000008023 solidification Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000004753 textile Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/295—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with inorganic bases, e.g. by alkali fusion
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/41—Preparation of salts of carboxylic acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the invention relates to a method for producing ⁇ -branched aliphatic monocarboxylic acids having 12 to 48 carbon atoms.
- ⁇ -branched aliphatic monohydric alcohols (Guerbet alcohols) are converted into the alkali metal salts of the corresponding ⁇ -branched aliphatic monocarboxylic acids in the presence of alkali metal hydroxide, and the ⁇ -branched aliphatic monocarboxylic acids are released from the alkali metal salts by soap splitting in a second process stage .
- the Guerbet reaction turns unbranched, saturated primary alcohols of the form R-CH 2 -CH 2 OH into ⁇ -branched alcohols of the form prepared, wherein R represents a hydrocarbon radical having 4 to 20 carbon atoms.
- esters with multiple alcohols such as pentaerythritol or trimethylolpropane are advantageously characterized by low vapor pressures and low pour points.
- the object of the invention is to develop a process which can be carried out on an industrial scale in which the problem of foaming is solved without having to accept the disadvantage of a reduction in the space-time yield.
- process step a namely the conversion of the Guerbet alcohols into the alkali metal salts of the corresponding monocarboxylic acids, is carried out as a reaction between solid alkali metal hydroxide and liquid alcohol and an inert diluent is added to the reaction mixture after the end of this reaction to lower the viscosity.
- the process is carried out continuously.
- the choice of the viscosity depressant and diluent is preferably based on the derivatization following the preparation of the salts of the branched monocarboxylic acids.
- the starting materials are fed together and completely to the reactor when cold.
- the alkali preferably NaOH in solid form, can be precisely metered in a simple manner compared to the melt process mentioned above.
- the process according to the invention is therefore a solid-liquid reaction in which the temperatures for an alkali melt are not reached. It is proposed in particular that in process step a, namely the conversion of the Guerbet alcohols into the alkali salts of the corresponding monocarboxylic acids, the alkali metal hydroxide is suspended in the liquid alcohol.
- the reaction is preferably carried out at temperatures of at most 350 ° C.
- the temperatures can be between 250 and 350 ° C.
- a substance which can be vaporized at the reaction temperature in particular water, is proposed for use as a coolant and diluent.
- Water is not only very suitable for keeping the reaction mixture liquid, it also enables the reaction mixture to be cooled very quickly to temperatures below 200 ° C. In a further embodiment of the invention it is therefore proposed that the reaction mixture be cooled to temperatures below 200 ° C.
- the water can be added in different ways. Blowing in water vapor and injecting liquid water have proven particularly useful.
- the greater part of the water evaporates in accordance with the phase equilibrium between soap and water.
- the evaporated water is condensed and returned to the reaction mixture.
- the reaction is carried out under superatmospheric pressure, in particular under a pressure of up to 10 bar, in order to reduce the gas velocity of the hydrogen formed and thus the foaming.
- the reaction is preferably carried out under an inert gas, e.g. B. nitrogen gas.
- the economy of the process is also increased if the hydrogen reaction formed in the first process step, the conversion of the Guerbet alcohols into the corresponding monocarboxylic acids, is collected and utilized. It can be used in particular as fuel gas.
- the soaps formed are split. Since only stoichiometric amounts of alkali are used in the process according to the invention, the soap need not be separated from the alkali in the reactor, but can remain in the reactor for soap splitting.
- Soap splitting can be carried out as described in the literature. This can be done under normal pressure, for. B. sulfuric acid or hydrochloric acid. Soap splitting under pressure is also possible and is within the scope of the method according to the invention.
- the periphery consists of a dephlegmator, a capacitor and a phase separator.
- Example 1 The feed materials were introduced as described in Example 1, but the reactor was pressurized with 4 bar of nitrogen before heating. The further procedure was carried out as in Example 1 and 8 150 kg of product (SZ 201, OHZ ⁇ 3) and about 50 kg of distillate were obtained.
- Example 1 180 kg of isotridecyl alcohol and 33.6 kg of NaOH were used in the 350 l reactor and heated to 340 ° C. under an N 2 pressure of 6 bar. Otherwise, the procedure was as in Example 1. A product with an SZ of 240 and an OHZ of 6.3 was obtained.
- Example 2 The process was carried out as described in Example 1 until the soap solution.
- the product was then diluted in the ratio of 2 parts water to 1 part soap and placed in an autoclave.
- the mixture was stirred at 30 ° C. under 60 bar CO 2 for 2 hours. After phase separation, an SZ of 189.2 was measured in the fat phase.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Inorganic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
- Die Erfindung betrifft ein Verfahren zum Herstellen α-verzweigter aliphatischer Monocarbonsäuren mit 12 bis 48 Kohlenstoffatomen. Dabei überführt man in einer ersten Verfahrensstufe a α-verzweigte aliphatische einwertige Alkohole (Guerbetalkohole) in Gegenwart von Alkalihydroxid in die Alkalisalze der entsprechenden α-verzweigten aliphatischen Monocarbonsäuren und setzt die α-verzweigten aliphatischen Monocarbonsäuren in einer zweiten Verfahrensstufe b aus den Alkalisalzen durch Seifenspaltung frei.
- In der fettchemischen Industrie werden mit der Guerbet-Reaktion aus unverzweigten, gesättigten primären Alkoholen der Form R-CH2-CH2OH α-verzweigte Alkohole der Form
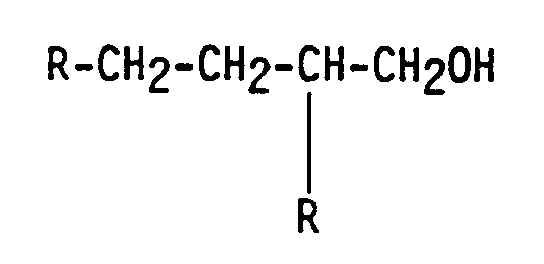
- Diese Alkohole und ihre Derivate werden in einer Vielzahl von Rezepturen der Schmierstoffindustrie, der Kosmetik sowie der Textilpflege eingesetzt.
- Bei den Fettsäuren sind ähnlich verzweigte Produkte bisher großtechnisch nicht herstellbar gewesen, obwohl für diese Fettsäuren und ihre Derivate entsprechende Anwendungsmöglichkeiten gegeben sind. Besonders die Ester mit Mehrfach-Alkoholen wie Pentaerythrit oder Trimethylolpropan zeichnen sich vorteilhaft durch niedrige Dampfdrücke und niedrige Stockpunkte aus.
- Es hat daher nicht an Versuchen gefehlt, Herstellverfahren für die verzweigten Fettsäuren zu finden.
- In einem in der DE-A-2 320 461 beschriebenen Verfahren der eingangs genannten Art wird zuerst eine Umsetzung von Guerbetalkoholen mit Alkali zu den carbonsauren Salzen vorgeschlagen. Die Darstellung der entsprechenden Monocarbonsäure gelingt anschließend über eine Seifenspaltung mit Mineralsäuren. Die erste Stufe dieses Verfahrens wird in einer oxidativen Alkalischmelze bei entsprechend hohen Temperaturen durchgeführt. Aufgrund der Schmelzpunkte von NaOH bzw. KOH, die aus wirtschaflichen Gründen eingesetzt werden, sind daher Betriebstemperaturen von mehr als 370°C erforderlich.
- Auch die Stockpunkte der Seifen, die nach diesem Stand der Technik in flüssiger Form vorliegen müssen, um aus dem Reaktionsgemisch abgetrennt zu werden, liegen oberhalb von 340 °C. Der bei der Reaktion entstehende Wasserstoff führt bei der erhöhten Temperatur zu Sicherheitsproblemen. Neben der Bildung von Verblockungen in den Rohrleitungen stellte die hohe Korrosivität der Alkalischmelze gegenüber nahezu allen bekannten Werkstoffen ein Haupthindernis auf dem Weg zur großtechnischen Durchführung dieses Verfahrens dar.
- In einem aus EP 31 694 B1 bekannten Verfahren zur Herstellung von Salzen von Carbonsäuren aus den entsprechenden Alkoholen und einer Base wird die Entstehung von festen Reaktionsprodukten und das Aufschäumen der Reaktionsmischung bei der alkalischen Oxidation der Alkohole durch Einsatz von inerten Viskositätserniedrigern verhindert. Vorgegeben wird hier vor Beginn der Reaktion eine Mischung der Base und eines Katalysator in einer inerten Verdünnungsflüssigkeit. Erst danach wird der umzusetzende Alkohol dieser Mischung unter Reaktionsbedingungen zudosiert. Die Zugabe des Verdünnungsmittels schon vor der Reaktion führt aber zur Absenkung der Raumzeitausbeute auf Werte zwischen 20 bis 80 %.
- Die in den Vergleichsbeispielen in EP 31 694 B1 genannte Wasserzugabe dient ausschließlich dazu, die Verseifungsreaktion und damit die Wasserstoffbildung zu verlangsamen und so ein Aufschäumen zu verhindern.
Das in der EP 31 694 B1 genannte Kriterium zur Auswahl des Verdünnungsmittels ist die bei Reaktionsbedingungen nur geringe Flüchtigkeit, so daß das Verdünnungsmittel in der flüssigen Phase verbleibt. - Der Erfindung liegt die Aufgabe zugrunde, ein großtechnisch durchführbares Verfahren zu entwickeln, bei dem das genannte Problem des Aufschäumens gelöst ist, ohne daß der Nachteil einer Verringerung der Raumzeitausbeute in Kauf genommen werden muß. Dazu schlägt die Erfindung vor, daß man die Verfahrensstufe a, nämlich die Überführung der Guerbetalkohole in die Alkalisalze der entsprechenden Monocarbonsäuren, als Reaktion zwischen festem Alkalihydroxid und flüssigem Alkohol durchführt und dem Reaktionsgemisch nach Abschluß dieser Reaktion ein inertes Verdünnungsmittel zum Absenken der Viskosität zugibt. Insbesondere führt man das Verfahren kontinuierlich durch.
- Die Wahl des Viskositätserniedrigers und Verdünnungsmittels richtet sich bevorzugt nach der an die Herstellung der Salze der verzweigten Monocarbonsäuren anschließenden Derivatisierung.
- Im erfindungsgemäßen Verfahren werden die Einsatzstoffe dem Reaktor im kalten Zustand gemeinsam und vollständig zugeführt. Der Alkali, vorzugsweise NaOH in fester Form, läßt sich gegenüber dem oben genannten Schmelze-Verfahren in einfacher Weise genau dosieren.
- Bereits ab einer Reaktortemperatur von 200 bis 250 °C beginnt während des Aufheizens die Reaktion zwischen flüssigem Alkohol und dem festen Alkalihydroxid. Bei dieser Reaktion wird Wasserstoff frei, der den Reaktor gasförmig verläßt. Auch gegen Reaktionsende werden die Temperaturen für eine Alkalischmelze unterschritten, um die bekannte Korrosivität zu vermeiden. Die Reaktoren können dadurch in preiswerten Werkstoffen ausgeführt werden. Die Temperaturen nach Beendigung der Reaktionsphase liegen zwischen 250 bis 350 °C. Die Stockpunkte der reinen Seifen werden damit teilweise nicht überschritten. Ein Festwerden der Seife vor dem Reaktionsende wird durch den noch vorhandenen Alkohol vermieden. Mit Erreichen einer Ausbeute von 98 % und mehr, bezogen auf den eingesetzten Alkohol, steigt schließlich die Viskosität stark an. Erst dann wird dem Reaktionsgemisch ein inertes Verdünnungsmittel zugegeben, um es hantierbar zu halten und den Stockpunkt der Reaktionsprodukte zu erniedrigen. Dabei reichen schon geringe gelöste Wassermengen, etwa 1 bis 5 Gew.-%, bei Temperaturen oberhalb von 200 °C aus.
- Der zur Viskositätserniedrigung nach Reaktionsende bei 250 bis 350 °C eingeblasene Wasserdampf verläßt zum größten Teil den Reaktionsraum dampfförmig und führt erst nach einer Kondensation und erneuten Verdampfung zur Abkühlung und Erhöhung der Wasserlöslichkeit.
- Bei dem erfindungsgemäßen Verfahren handelt es sich also im Gegensatz zum Stand der Technik um eine Fest-Flüssig-Reaktion, bei der die Temperaturen für eine Alkalischmelze unterschritten werden. Es wird insbesondere vorgeschlagen, daß in der Verfahrensstufe a, nämlich der Überführung der Guerbetalkohole in die Alkalisalze der entsprechenden Monocarbonsäuren, das Alkalihydroxid im flüssigem Alkohol suspendiert ist.
- Bei der erfindungsgemäßen Durchführung der Fest-Flüssig-Reaktion unter Zugabe eines inerten Verdünnungsmittels zur Erniedrigung der Viskosität geht man vorzugsweise von in kaltem Zustand in den Reaktor vorgelegtem Alkalihydroxid und Guerbetalkohol aus, heizt das Reaktionsgemisch allmählich von etwa Raumtemperatur auf Reaktionstemperatur auf und kühlt nach Abschluß der Reaktion durch Zugabe des Verdünnungsmittels ab. Dabei führt man die Reaktion vorzugsweise bei Temperaturen von höchstens 350 °C durch. Die Temperaturen können dabei etwa zwischen 250 und 350 °C liegen.
- Zum Einsatz als Kühl- und Verdünnungsmittel wird in einer Ausgestaltung der Erfindung ein bei Reaktionstemperatur verdampfbarer Stoff vorgeschlagen, insbesondere Wasser. Wasser ist nämlich nicht nur sehr gut geeignet, den Reaktionsansatz flüssig zu halten, sondern es ermöglicht auch eine sehr schnelle Kühlung des Reaktionsgemisches auf Temperaturen unter 200 °C. In einer weiteren Ausführungsform der Erfindung wird daher vorgeschlagen, daß man das Reaktionsgemisch auf Temperaturen unter 200 °C abkühlt. Die Zugabe des Wassers kann dabei auf unterschiedliche Weise erfolgen. Besonders bewährt hat sich das Einblasen von Wasserdampf sowie das Einspritzen von flüssigem Wasser.
- Die Zugabe von 5 bis 30 Gew.-% Wasser zum Reaktionsgemisch reicht aus, um die gebildeten Seifen auch bei Raumtemperatur flüssig zu halten. Nach der Spaltung der Seifen kann das eingebrachte Wasser durch eine einfache Phasentrennung vollständig entfernt werden.
- Bei der Abkühlung des Reaktionsgemisches durch Zugabe von Wasser verdampft der größere Teil des Wassers entsprechend dem Phasengleichgewicht zwischen Seife und Wasser. Das verdampfte Wasser wird in einer anderen vorteilhaften Ausführungsform kondensiert und in das Reaktionsgemisch zurückgeführt.
- In einer anderen Ausführung des erfindungsgemäßen Verfahrens wird die Reaktion unter Überdruck durchgeführt, insbesondere unter einem Druck bis zu 10 bar, um die Gasgeschwindigkeit des entstehenden Wasserstoffs und damit das Aufschäumen zu verringern. Die Reaktion wird vorzugsweise unter Inertgas, z. B. Stickstoffgas, durchgeführt.
- Die Wirtschaftlichkeit des Verfahrens wird außerdem erhöht, wenn man den in der ersten Verfahrensstufe, der Überführung der Guerbetalkohole in die entsprechenden Monocarbonsäuren, enstehenden Reaktionswasserstoff auffängt und verwertet. Er kann dabei insbesondere als Brenngas verwendet werden.
- Nach der Umsetzung des Reaktionsgemisches zu den Salzen der verzweigten Monocarbonsäuren werden die entstandenen Seifen gespalten. Da im erfindungsgemäßen Verfahren lediglich stöchiometrische Mengen an Alkali eingesetzt werden, braucht die Seife im Reaktor nicht vom Alkali getrennt zu werden, sondern kann zur Seifenspaltung im Reaktor verbleiben.
- Die Seifenspaltung kann wie in der Literatur beschrieben durchgeführt werden. Dazu kann unter Normaldruck z. B. Schwefelsäure oder Salzsäure eingesetzt werden. Aber auch eine Seifenspaltung unter Druck ist möglich und liegt im Rahmen des erfindungsgemäßen Verfahrens.
- Die nachfolgenden Beispiele sollen den Gegenstand der Erfindung näher erläutern, ohne ihn darauf zu beschränken.
- Zur Herstellung wurde ein Rührreaktor verwendet. Die Peripherie besteht aus einem Dephlegmator, einem Kondensator und einem Phasenabscheider.
- In den Reaktor wurden im kalten Zustand
- 8 000 kg
- 2-Hexyldecanol
- 1 280 kg
- NaOH
- Es entstanden ca. 7 800 kg Reaktionsprodukt mit einer Säurezahl SZ von 200 und einer Hydroxylzahl OHZ < 3. Während der Reaktionsphase wurden ca. 400 kg Destillat aufgefangen, das in weiteren Ansätzen als Einsatzstoff verwendet werden kann.
- Die Einsatzstoffe wurden wie in Beispiel 1 beschrieben vorgelegt, jedoch wurde der Reaktor vor dem Aufheizen mit einem Druck von 4 bar Stickstoff beaufschlagt. Das weitere Verfahren wurde wie in Beispiel 1 durchgeführt und 8 150 kg Produkt (SZ 201, OHZ < 3) und ca. 50 kg Destillat wurden erhalten.
- In einem 350 l Reaktor wurde 180 kg Octanol, 4,5 kg KOH und 80 g ZnO vorgelegt. Der Reaktor wurde auf 200 °C aufgeheizt. Nach Reaktionsbeginn wurde nach dem Kondensator ein Octanol-Wasser-Gemisch abgetrennt und die Alkoholphase in den Reaktor zurückgeführt. Die Reaktortemperatur stieg im Verlauf der Reaktion auf bis zu 250 °C an. In dem Reaktionsgemisch befand sich nach der Beendigung der Wasserausschleusung laut GC-Analyse ca. 8 % Octanol, 78 % 2-Hexyldecanol und ca. 8 % 2-4 Dihexyldodecanol. Durch Destillation bei 240 °C und 100 mbar wurde der Octanolgehalt auf unter 1 % gesenkt. Nach Entspannen auf Normaldruck wurden 24 kg NaOH in den Reaktor dosiert und der Ansatz wie in Beispiel 1 weitergeführt.
- Es ergaben sich aus diesem Versuch 135 kg Produkt mit einer SZ von 205 und einer OHZ < 3. In der Destillation wurden 25,2 kg abgetrennt, die zu gleichen Teilen aus Octanol und Guerbetalkohol bestanden. Das Destillat kann als Einsatzstoff für weitere Ansätze verwendet werden.
- In den 350 l Reaktor wurden 180 kg Isotridecylalkohol und 33,6 kg NaOH eingesetzt und unter einem N2-Druck von 6 bar auf 340 °C aufgeheizt. Ansonsten wurde das Verfahren wie unter Beispiel 1 durchgeführt. Ein Produkt mit einer SZ von 240 und einer OHZ von 6,3 wurde erhalten.
- In den Reaktor wurden 180 kg Lauryl- und Myristylalkohol zu gleichen Teilen zusammen mit 1,34 kg KOH und 80 g ZnO vorgelegt. Mit einem Verfahren entsprechend Beispiel 3 ergaben sich in der Destillation ca. 25 kg Gemischalkohol, der aus Monomeren und Dimeren zu gleichen Teilen bestand. Das nach der Spaltung erhaltene Produkt (145 kg) hatte eine SZ = 148 und eine OHZ < 2. Nach GC-Anlayse bestand dieses Gemisch zu 19 % aus Isotetracosansäure, zu 51 % aus Iso-Hexacosansäure (Isocerotinsäure) und zu 25 % aus Iso-Octacosansäure.
- Das Verfahren wurde, wie in Beispiel 1 beschrieben, bis zur Seifenlösung durchgeführt. Anschließend wurde das Produkt im Verhältnis von 2 Teilen Wasser zu 1 Teil Seife verdünnt und in einen Autoklaven eingesetzt. Der Ansatz wurde bei 30 °C unter 60 bar CO2 2 Stunden gerührt. Nach einer Phasentrennung wurde in der Fettphase eine SZ von 189,2 gemessen.
Claims (13)
- Verfahren zum Herstellen α-verzweigter aliphatischer Monocarbonsäuren mit 12 bis 48 Kohlenstoffatomen, wobei mana) α-verzweigte aliphatische einwertige Alkohole (Guerbetalkohole) in Gegenwart von Alkalihydroxid in die Alkalisalze der entsprechenden α-verzweigten aliphatischen Monocarbonsäuren überführt undb) aus den Alkalisalzen durch Seifenspaltung die α-verzweigten aliphatischen Monocarbonsäuren freisetzt,dadurch gekennzeichnet,
daß man die Verfahrensstufe a als Reaktion zwischen festem Alkalihydroxid und flüssigem Alkohol durchführt und dem Reaktionsgemisch nach Abschluß dieser Reaktion ein inertes Verdünnungsmittel zum Absenken der Viskosität zugibt, wobei man das Verfahren insbesondere kontinuierlich durchführt. - Verfahren nach Anspruch 1,
dadurch gekennzeichnet,
daß in der Verfahrensstufe a das Alkalihydroxid im flüssigen Alkohol suspendiert ist. - Verfahren nach Anspruch 1 oder 2,
dadurch gekennzeichnet,
daß man in der Verfahrensstufe a das Reaktionsgemisch allmählich von etwa Raumtemperatur auf Reaktionstemperatur aufheizt und nach Abschluß der Reaktion durch Zugabe des Verdünnungsmittels abkühlt. - Verfahren nach Anspruch 3,
dadurch gekennzeichnet,
daß man die Reaktion bei Temperaturen von höchstens 350 °C durchführt. - Verfahren nach einem der Ansprüche 1 bis 4,
dadurch gekennzeichnet,
daß man als Kühl- und Verdünnungsmittel einen bei Reaktionstemperatur verdampfbaren Stoff einsetzt. - Verfahren nach Anspruch 5,
dadurch gekennzeichnet,
daß man als Kühl- und Verdünnungsmittel Wasser einsetzt. - Verfahren nach Anspruch 6,
dadurch gekennzeichnet,
daß man das Reaktionsgemisch auf Temperaturen unter 200 °C abkühlt. - Verfahren nach Anspruch 6 oder 7,
dadurch gekennzeichnet,
daß man Wasserdampf einbläst. - Verfahren nach Anspruch 6 oder 7,
dadurch gekennzeichnet,
daß man flüssiges Wasser einspritzt. - Verfahren nach einem der Ansprüche 6 bis 9,
dadurch gekennzeichnet,
daß man 5 bis 30 Gew.-% Wasser dem Reaktionsgemisch zugibt. - Verfahren nach einem der Ansprüche 6 bis 10,
dadurch gekennzeichnet,
daß man das verdampfte Wasser kondensiert und in das Reaktionsgemisch zurückführt. - Verfahren nach einem der Ansprüche 1 bis 11,
dadurch gekennzeichnet,
daß man die Verfahrensstufe a unter Überdruck durchführt. - Verfahren nach einem der Ansprüche 1 bis 12,
dadurch gekennzeichnet,
daß man den in der Verfahrensstufe a entstehenden Wasserstoff auffängt und verwertet und insbesondere als Brenngas verwendet.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4302463A DE4302463A1 (de) | 1993-01-29 | 1993-01-29 | Verfahren zum Herstellen alpha-verzweigter aliphatischer Monocarbonsäuren |
| DE4302463 | 1993-01-29 | ||
| PCT/EP1994/000155 WO1994017026A1 (de) | 1993-01-29 | 1994-01-21 | VERFAHREN ZUM HERSTELLEN α-VERZWEIGTER ALIPHATISCHER MONOCARBONSÄUREN |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0681565A1 EP0681565A1 (de) | 1995-11-15 |
| EP0681565B1 true EP0681565B1 (de) | 1997-06-18 |
Family
ID=6479185
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP94905102A Expired - Lifetime EP0681565B1 (de) | 1993-01-29 | 1994-01-21 | Verfahren zum herstellen alpha-verzweigter aliphatischer monocarbonsäuren |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US5654453A (de) |
| EP (1) | EP0681565B1 (de) |
| JP (1) | JPH08506809A (de) |
| AT (1) | ATE154584T1 (de) |
| DE (2) | DE4302463A1 (de) |
| DK (1) | DK0681565T3 (de) |
| ES (1) | ES2102823T3 (de) |
| GR (1) | GR3024140T3 (de) |
| WO (1) | WO1994017026A1 (de) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19521900A1 (de) * | 1995-06-16 | 1996-12-19 | Rwe Dea Ag | Verfahren zur kontinuierlichen Herstellung von Monocarbonsäuren |
| GB2385589A (en) * | 2000-08-07 | 2003-08-27 | Sofitech Nv | Surfactant |
| CN103012105B (zh) * | 2011-09-20 | 2015-03-04 | 沈阳张明化工有限公司 | 一种2-丙基庚酸的制备方法 |
| WO2018214089A1 (zh) * | 2017-05-25 | 2018-11-29 | 沈阳张明化工有限公司 | 2-丙基庚酸的制备方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2293649A (en) * | 1941-07-15 | 1942-08-18 | Du Pont | Preparation of high molecular weight branched chain acids |
| NL6702490A (de) * | 1966-02-28 | 1967-08-29 | ||
| GB1450897A (en) * | 1972-11-20 | 1976-09-29 | Ici Ltd | Production of salts of carboxylic acids |
| DE2320461A1 (de) * | 1973-04-21 | 1974-11-07 | Henkel & Cie Gmbh | Neue alpha-verzweigte aliphatische monocarbonsaeuren sowie deren herstellung |
| AU541663B2 (en) * | 1979-12-26 | 1985-01-17 | Stauffer Chemical Company | Process for producing carboxylic acid salts |
-
1993
- 1993-01-29 DE DE4302463A patent/DE4302463A1/de not_active Withdrawn
-
1994
- 1994-01-21 AT AT94905102T patent/ATE154584T1/de not_active IP Right Cessation
- 1994-01-21 WO PCT/EP1994/000155 patent/WO1994017026A1/de active IP Right Grant
- 1994-01-21 JP JP6516658A patent/JPH08506809A/ja active Pending
- 1994-01-21 ES ES94905102T patent/ES2102823T3/es not_active Expired - Lifetime
- 1994-01-21 EP EP94905102A patent/EP0681565B1/de not_active Expired - Lifetime
- 1994-01-21 DK DK94905102.3T patent/DK0681565T3/da active
- 1994-01-21 DE DE59403185T patent/DE59403185D1/de not_active Expired - Fee Related
- 1994-01-21 US US08/500,905 patent/US5654453A/en not_active Expired - Fee Related
-
1997
- 1997-07-16 GR GR970401790T patent/GR3024140T3/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ES2102823T3 (es) | 1997-08-01 |
| DK0681565T3 (da) | 1997-12-29 |
| WO1994017026A1 (de) | 1994-08-04 |
| DE4302463A1 (de) | 1994-08-04 |
| GR3024140T3 (en) | 1997-10-31 |
| DE59403185D1 (de) | 1997-07-24 |
| ATE154584T1 (de) | 1997-07-15 |
| JPH08506809A (ja) | 1996-07-23 |
| EP0681565A1 (de) | 1995-11-15 |
| US5654453A (en) | 1997-08-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE19949319A1 (de) | Verfahren zur Herstellung von Arylalkylethern | |
| WO2007012097A1 (de) | Verfahren zur herstellung von carbonsäurealkylestern | |
| DE69302557T2 (de) | Verfahren zur Herstellung von Fettsäureestern und -amiden von Sulfonsäuresalzen | |
| DE2447551B2 (de) | Verfahren zur Herstellung von Methylchlorid | |
| DE2438432A1 (de) | Verfahren zur herstellung von pisopropenylphenol | |
| EP0681565B1 (de) | Verfahren zum herstellen alpha-verzweigter aliphatischer monocarbonsäuren | |
| DE2131753A1 (de) | Verfahren zum Umwandeln der Carboxylgruppen von Carbonsaeuren in Hydroxymethylgruppen durch Hydrogenolyse | |
| DE69001980T2 (de) | Kontinuierte Synthese von Estern aus Merkaptocarbonsäuren. | |
| DE69509156T2 (de) | Verfahren zur Herstellen einer Vinylverbindung mit einer Hydroxygruppe | |
| EP0285899B1 (de) | Verfahren zur Herstellung von Maleinsäuredimethylester | |
| EP0080023B1 (de) | Verfahren zum kontinuierlichen Verestern von Methacrylsäure | |
| DE1203760B (de) | Verfahren zur Herstellung von Sorbinsaeure-alkylestern | |
| DE69806957T2 (de) | Vinylether derivate | |
| DE19944874C1 (de) | Verfahren zur Herstellung von Cyclooctanol | |
| DE1568783C3 (de) | ||
| DE1568777A1 (de) | Verfahren zur kontinuierlichen Herstellung von Anlagerungsprodukten des AEthylenoxids an Carbonsaeuren | |
| DE3332436T1 (de) | Bildung von Isobuttersäure oder Methylisobutyrat | |
| DE888736C (de) | Verfahren zur Herstellung hochwirksamer Trockenstoffkombinationen | |
| WO2024099859A1 (de) | Verfahren zur herstellung von c4- bis c12- (meth)acrylaten | |
| EP1501788B1 (de) | Verfahren zur herstellung von 3-trifluormethylphenyl-4-cyanobenzylketon | |
| DE2221277B2 (de) | Verfahren zur Herstellung von Fettsäure-Milchsäure-Kondensationsprodukten | |
| DE19848548A1 (de) | Aufarbeitung verunreinigter Fettalkohol-Gemische | |
| DE4438581A1 (de) | Verfahren zur Herstellung von Dialkylethern | |
| AT265232B (de) | Verfahren zur Herstellung substituierter und unsubstituierter Adipinsäuren | |
| DE3033655C2 (de) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 19950720 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI NL PT SE |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| 17Q | First examination report despatched |
Effective date: 19960807 |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LI NL PT SE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 19970618 Ref country code: FR Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 19970618 |
|
| REF | Corresponds to: |
Ref document number: 154584 Country of ref document: AT Date of ref document: 19970715 Kind code of ref document: T |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| ET | Fr: translation filed | ||
| REF | Corresponds to: |
Ref document number: 59403185 Country of ref document: DE Date of ref document: 19970724 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2102823 Country of ref document: ES Kind code of ref document: T3 |
|
| ITF | It: translation for a ep patent filed | ||
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 19970901 |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: FG4A Free format text: 3024140 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: SC4A Free format text: AVAILABILITY OF NATIONAL TRANSLATION Effective date: 19970917 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980121 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980121 Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980121 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980122 Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980122 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980131 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980131 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980131 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980202 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| BERE | Be: lapsed |
Owner name: HENKEL K.G.A.A. Effective date: 19980131 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980801 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19980121 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 19980801 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19981001 |
|
| EUG | Se: european patent has lapsed |
Ref document number: 94905102.3 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19990731 |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: MM4A Free format text: LAPSE DUE TO NON-PAYMENT OF FEES Effective date: 19990731 |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 19990211 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20050121 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19980121 |