EP0000392B1 - Cephalosporin und Penicillin-derivate, Verfahren zu deren Herstellung, und deren pharmazeutische Präparate - Google Patents
Cephalosporin und Penicillin-derivate, Verfahren zu deren Herstellung, und deren pharmazeutische Präparate Download PDFInfo
- Publication number
- EP0000392B1 EP0000392B1 EP78100352A EP78100352A EP0000392B1 EP 0000392 B1 EP0000392 B1 EP 0000392B1 EP 78100352 A EP78100352 A EP 78100352A EP 78100352 A EP78100352 A EP 78100352A EP 0000392 B1 EP0000392 B1 EP 0000392B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- compounds
- formula
- salts
- acid
- parts
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- 238000000034 method Methods 0.000 title claims description 17
- 238000002360 preparation method Methods 0.000 title claims description 10
- 229930182555 Penicillin Natural products 0.000 title description 6
- 229930186147 Cephalosporin Natural products 0.000 title description 5
- 229940124587 cephalosporin Drugs 0.000 title description 5
- 150000001780 cephalosporins Chemical class 0.000 title description 5
- 150000002960 penicillins Chemical class 0.000 title description 5
- 239000008194 pharmaceutical composition Substances 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 63
- -1 1,3,5-trimethyl pyrazolyl Chemical group 0.000 claims description 42
- 239000000203 mixture Substances 0.000 claims description 38
- 150000003839 salts Chemical class 0.000 claims description 24
- 239000002253 acid Substances 0.000 claims description 20
- 239000002904 solvent Substances 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 239000011230 binding agent Substances 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 7
- 150000007513 acids Chemical class 0.000 claims description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 6
- 150000002148 esters Chemical class 0.000 claims description 6
- 239000003782 beta lactam antibiotic agent Substances 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- 150000001540 azides Chemical class 0.000 claims description 4
- 229910052799 carbon Inorganic materials 0.000 claims description 4
- 125000002541 furyl group Chemical group 0.000 claims description 3
- 125000001544 thienyl group Chemical group 0.000 claims description 3
- 125000003460 beta-lactamyl group Chemical group 0.000 claims description 2
- 125000001841 imino group Chemical group [H]N=* 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 60
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 41
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 38
- 239000000243 solution Substances 0.000 description 38
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 29
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 26
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 239000000126 substance Substances 0.000 description 22
- 238000006243 chemical reaction Methods 0.000 description 19
- 239000002585 base Substances 0.000 description 17
- 239000004480 active ingredient Substances 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- 239000000460 chlorine Substances 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 235000019441 ethanol Nutrition 0.000 description 12
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 0 *C(c1ccc[o]1)N Chemical compound *C(c1ccc[o]1)N 0.000 description 8
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 229910052801 chlorine Inorganic materials 0.000 description 8
- 239000000825 pharmaceutical preparation Substances 0.000 description 8
- 241000588769 Proteus <enterobacteria> Species 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 239000000376 reactant Substances 0.000 description 7
- 235000015424 sodium Nutrition 0.000 description 7
- 241000894006 Bacteria Species 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 239000013543 active substance Substances 0.000 description 6
- 230000000844 anti-bacterial effect Effects 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 239000000969 carrier Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- 244000052769 pathogen Species 0.000 description 6
- HSHGZXNAXBPPDL-HZGVNTEJSA-N 7beta-aminocephalosporanic acid Chemical compound S1CC(COC(=O)C)=C(C([O-])=O)N2C(=O)[C@@H]([NH3+])[C@@H]12 HSHGZXNAXBPPDL-HZGVNTEJSA-N 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- 241000588653 Neisseria Species 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 239000003513 alkali Substances 0.000 description 5
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 229910052794 bromium Inorganic materials 0.000 description 5
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 150000007529 inorganic bases Chemical class 0.000 description 5
- 150000007530 organic bases Chemical class 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 5
- 159000000000 sodium salts Chemical class 0.000 description 5
- 241000607528 Aeromonas hydrophila Species 0.000 description 4
- 241000588779 Bordetella bronchiseptica Species 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 241000588724 Escherichia coli Species 0.000 description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- 241000606768 Haemophilus influenzae Species 0.000 description 4
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 4
- 241000588915 Klebsiella aerogenes Species 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical group CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- 241000588768 Providencia Species 0.000 description 4
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 4
- 241000607715 Serratia marcescens Species 0.000 description 4
- 241000191967 Staphylococcus aureus Species 0.000 description 4
- 241000193998 Streptococcus pneumoniae Species 0.000 description 4
- 241000193996 Streptococcus pyogenes Species 0.000 description 4
- 241000186064 Trueperella pyogenes Species 0.000 description 4
- 208000025087 Yersinia pseudotuberculosis infectious disease Diseases 0.000 description 4
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 238000000354 decomposition reaction Methods 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 229940047650 haemophilus influenzae Drugs 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 239000006187 pill Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 4
- 239000002132 β-lactam antibiotic Substances 0.000 description 4
- 229940124586 β-lactam antibiotics Drugs 0.000 description 4
- 150000003952 β-lactams Chemical class 0.000 description 4
- ODIGIKRIUKFKHP-UHFFFAOYSA-N (n-propan-2-yloxycarbonylanilino) acetate Chemical compound CC(C)OC(=O)N(OC(C)=O)C1=CC=CC=C1 ODIGIKRIUKFKHP-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- KGGVRDQTCVGNFX-UHFFFAOYSA-N 3-(furan-2-ylmethylideneamino)-2-oxoimidazole-1-carbonyl chloride Chemical compound O=C1N(C(=O)Cl)C=CN1N=CC1=CC=CO1 KGGVRDQTCVGNFX-UHFFFAOYSA-N 0.000 description 3
- BNVQSFCIMDJHJB-UHFFFAOYSA-N 3-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-3h-furan-2-yl]propanoic acid Chemical compound CC(C)(C)OC(=O)NC1(CCC(O)=O)CC=CO1 BNVQSFCIMDJHJB-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 241000607534 Aeromonas Species 0.000 description 3
- 241000606125 Bacteroides Species 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 241000588807 Bordetella Species 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- 241000588914 Enterobacter Species 0.000 description 3
- 241000606790 Haemophilus Species 0.000 description 3
- 241000588748 Klebsiella Species 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 241000606860 Pasteurella Species 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 241000589516 Pseudomonas Species 0.000 description 3
- 241000607142 Salmonella Species 0.000 description 3
- 241000607768 Shigella Species 0.000 description 3
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 150000003973 alkyl amines Chemical class 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 235000012216 bentonite Nutrition 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000008298 dragée Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 150000008282 halocarbons Chemical class 0.000 description 3
- 238000002329 infrared spectrum Methods 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 235000011118 potassium hydroxide Nutrition 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- VHRXWLDSKMPYRE-ZEPSKSRBSA-N (6R)-3-(acetyloxymethyl)-7-[[2-amino-3-(furan-2-yl)propanoyl]amino]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound NC(C(=O)NC1[C@@H]2N(C(=C(CS2)COC(C)=O)C(=O)O)C1=O)CC1=CC=CO1 VHRXWLDSKMPYRE-ZEPSKSRBSA-N 0.000 description 2
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HJJWHNKVHFBCER-UHFFFAOYSA-N 3-(furan-2-ylmethylideneamino)-2-oxoimidazolidine-1-carbonyl azide Chemical compound N(=[N+]=[N-])C(=O)N1C(N(CC1)N=CC1=CC=CO1)=O HJJWHNKVHFBCER-UHFFFAOYSA-N 0.000 description 2
- ZWXXVWSSKFZMOJ-UHFFFAOYSA-N 3-(furan-2-ylmethylideneamino)-2-oxoimidazolidine-1-carbonyl chloride Chemical compound O=C1N(C(=O)Cl)CCN1N=CC1=CC=CO1 ZWXXVWSSKFZMOJ-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241001112741 Bacillaceae Species 0.000 description 2
- 241000193830 Bacillus <bacterium> Species 0.000 description 2
- 241000193738 Bacillus anthracis Species 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- 241000606126 Bacteroidaceae Species 0.000 description 2
- 241000508772 Brucella sp. Species 0.000 description 2
- 101100352919 Caenorhabditis elegans ppm-2 gene Proteins 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 240000001817 Cereus hexagonus Species 0.000 description 2
- 241000193403 Clostridium Species 0.000 description 2
- 241000193468 Clostridium perfringens Species 0.000 description 2
- 241000193449 Clostridium tetani Species 0.000 description 2
- 241000186031 Corynebacteriaceae Species 0.000 description 2
- 241000186216 Corynebacterium Species 0.000 description 2
- 241001508000 Corynebacterium bovis Species 0.000 description 2
- 241000186227 Corynebacterium diphtheriae Species 0.000 description 2
- 241000186225 Corynebacterium pseudotuberculosis Species 0.000 description 2
- 241000186246 Corynebacterium renale Species 0.000 description 2
- 241000223936 Cryptosporidium parvum Species 0.000 description 2
- 241000186427 Cutibacterium acnes Species 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 241000588697 Enterobacter cloacae Species 0.000 description 2
- 241000588921 Enterobacteriaceae Species 0.000 description 2
- 241001495410 Enterococcus sp. Species 0.000 description 2
- 241000588698 Erwinia Species 0.000 description 2
- 241000192125 Firmicutes Species 0.000 description 2
- 241000605986 Fusobacterium nucleatum Species 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 241000606826 Haemophilus haemoglobinophilus Species 0.000 description 2
- 241001490623 Helicobacter suis Species 0.000 description 2
- 241000588747 Klebsiella pneumoniae Species 0.000 description 2
- 241000588744 Klebsiella pneumoniae subsp. ozaenae Species 0.000 description 2
- 241000186610 Lactobacillus sp. Species 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241000192017 Micrococcaceae Species 0.000 description 2
- 241000219470 Mirabilis Species 0.000 description 2
- 241000186360 Mycobacteriaceae Species 0.000 description 2
- 241000186359 Mycobacterium Species 0.000 description 2
- 241000186367 Mycobacterium avium Species 0.000 description 2
- 241000186366 Mycobacterium bovis Species 0.000 description 2
- 241000186362 Mycobacterium leprae Species 0.000 description 2
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 241000588652 Neisseria gonorrhoeae Species 0.000 description 2
- 241000588650 Neisseria meningitidis Species 0.000 description 2
- 240000006691 Nymphoides peltata Species 0.000 description 2
- 208000005141 Otitis Diseases 0.000 description 2
- 241000606856 Pasteurella multocida Species 0.000 description 2
- 201000007100 Pharyngitis Diseases 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- 241000588774 Providencia sp. Species 0.000 description 2
- 241000947836 Pseudomonadaceae Species 0.000 description 2
- 206010037596 Pyelonephritis Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 241001138501 Salmonella enterica Species 0.000 description 2
- 241001354013 Salmonella enterica subsp. enterica serovar Enteritidis Species 0.000 description 2
- 241000531795 Salmonella enterica subsp. enterica serovar Paratyphi A Species 0.000 description 2
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 2
- 241000607149 Salmonella sp. Species 0.000 description 2
- 241000607720 Serratia Species 0.000 description 2
- 241000607764 Shigella dysenteriae Species 0.000 description 2
- 241000607758 Shigella sp. Species 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- 241000123534 Sphaerophorus Species 0.000 description 2
- 241000191940 Staphylococcus Species 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 241000194017 Streptococcus Species 0.000 description 2
- 208000003217 Tetany Diseases 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 241000607734 Yersinia <bacteria> Species 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- 241000606834 [Haemophilus] ducreyi Species 0.000 description 2
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 2
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 2
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 230000000845 anti-microbial effect Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 229940065181 bacillus anthracis Drugs 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 206010006451 bronchitis Diseases 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 150000004292 cyclic ethers Chemical class 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 201000003146 cystitis Diseases 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 208000019258 ear infection Diseases 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 206010014665 endocarditis Diseases 0.000 description 2
- 229940092559 enterobacter aerogenes Drugs 0.000 description 2
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000003925 fat Substances 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 235000001727 glucose Nutrition 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 2
- 229910000103 lithium hydride Inorganic materials 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000011368 organic material Substances 0.000 description 2
- 238000001139 pH measurement Methods 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 229940051027 pasteurella multocida Drugs 0.000 description 2
- 206010034674 peritonitis Diseases 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000001376 precipitating effect Effects 0.000 description 2
- 229940007042 proteus vulgaris Drugs 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 229940007046 shigella dysenteriae Drugs 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- IXZDIALLLMRYOU-UHFFFAOYSA-N tert-butyl hypochlorite Chemical compound CC(C)(C)OCl IXZDIALLLMRYOU-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- CBDKQYKMCICBOF-UHFFFAOYSA-N thiazoline Chemical compound C1CN=CS1 CBDKQYKMCICBOF-UHFFFAOYSA-N 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- JWCKWKGDWUVGON-JFIBYHEFSA-N (6R)-3-(acetyloxymethyl)-7-[[3-(furan-2-yl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoyl]amino]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound C(C)(C)(C)OC(=O)NC(C(=O)NC1[C@@H]2N(C(=C(CS2)COC(C)=O)C(=O)O)C1=O)CC1=CC=CO1 JWCKWKGDWUVGON-JFIBYHEFSA-N 0.000 description 1
- HNOQAFMOBRWDKQ-UHFFFAOYSA-N 1,3,5-trimethylpyrazole Chemical compound CC=1C=C(C)N(C)N=1 HNOQAFMOBRWDKQ-UHFFFAOYSA-N 0.000 description 1
- HMTUBXVXHHITGO-UHFFFAOYSA-N 1,3,5-trimethylpyrazole-4-carbaldehyde Chemical compound CC1=NN(C)C(C)=C1C=O HMTUBXVXHHITGO-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- NDVMCQUOSYOQMZ-UHFFFAOYSA-N 2,2-bis(trimethylsilyl)acetamide Chemical compound C[Si](C)(C)C(C(N)=O)[Si](C)(C)C NDVMCQUOSYOQMZ-UHFFFAOYSA-N 0.000 description 1
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 1
- HFIXMOCDYMVWEV-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonylamino]-2-(1,3,5-trimethylpyrazol-4-yl)acetic acid Chemical compound CC1=NN(C)C(C)=C1C(NC(=O)OC(C)(C)C)C(O)=O HFIXMOCDYMVWEV-UHFFFAOYSA-N 0.000 description 1
- LMKWQHJJTTWIEJ-UHFFFAOYSA-N 2-amino-2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]acetic acid Chemical compound CC(C)(C)OC(=O)NC1=NC(C(N)C(O)=O)=CS1 LMKWQHJJTTWIEJ-UHFFFAOYSA-N 0.000 description 1
- STHZFZDWFBBWOW-UHFFFAOYSA-N 2-azaniumyl-2-(1,3,5-trimethylpyrazol-4-yl)acetate Chemical compound CC1=NN(C)C(C)=C1C(N)C(O)=O STHZFZDWFBBWOW-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- CCGVLYVPYIMSRC-UHFFFAOYSA-N 3-(2-amino-3h-furan-2-yl)propanoic acid Chemical compound OC(=O)CCC1(N)CC=CO1 CCGVLYVPYIMSRC-UHFFFAOYSA-N 0.000 description 1
- PYSGFFTXMUWEOT-UHFFFAOYSA-N 3-(dimethylamino)propan-1-ol Chemical compound CN(C)CCCO PYSGFFTXMUWEOT-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N Azide Chemical compound [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 241000606124 Bacteroides fragilis Species 0.000 description 1
- 239000004604 Blowing Agent Substances 0.000 description 1
- 241001622982 Bombus soroeensis proteus Species 0.000 description 1
- 241001261624 Brevundimonas bacteroides Species 0.000 description 1
- UBJTVPVKYYBAAK-IQTBQJLQSA-N C/C(/N=C(\C)/SC)=C/CS Chemical compound C/C(/N=C(\C)/SC)=C/CS UBJTVPVKYYBAAK-IQTBQJLQSA-N 0.000 description 1
- RAMUMXGBSGIOJN-KWRQCDRVSA-N C/C=C\NSC(SC)=N Chemical compound C/C=C\NSC(SC)=N RAMUMXGBSGIOJN-KWRQCDRVSA-N 0.000 description 1
- YPSFTKUZIONOFD-UHFFFAOYSA-N C/N=N\N(C(SC)=N)[U]C Chemical compound C/N=N\N(C(SC)=N)[U]C YPSFTKUZIONOFD-UHFFFAOYSA-N 0.000 description 1
- LZNXKARYDGKIDU-UHFFFAOYSA-N CC(C(C)SC(C)(C)C(C)C(O)=O)C(N)=O Chemical compound CC(C(C)SC(C)(C)C(C)C(O)=O)C(N)=O LZNXKARYDGKIDU-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- 206010013786 Dry skin Diseases 0.000 description 1
- 241000588722 Escherichia Species 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- UIOFUWFRIANQPC-JKIFEVAISA-N Floxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(F)C=CC=C1Cl UIOFUWFRIANQPC-JKIFEVAISA-N 0.000 description 1
- 241000605952 Fusobacterium necrophorum Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- 241000579664 Grateloupia proteus Species 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 241000580463 Nicrophorus Species 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 108010087702 Penicillinase Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- URWAJWIAIPFPJE-UHFFFAOYSA-N Rickamicin Natural products O1CC(O)(C)C(NC)C(O)C1OC1C(O)C(OC2C(CC=C(CN)O2)N)C(N)CC1N URWAJWIAIPFPJE-UHFFFAOYSA-N 0.000 description 1
- 241000293871 Salmonella enterica subsp. enterica serovar Typhi Species 0.000 description 1
- LUSZGTFNYDARNI-UHFFFAOYSA-N Sesamol Natural products OC1=CC=C2OCOC2=C1 LUSZGTFNYDARNI-UHFFFAOYSA-N 0.000 description 1
- 229930192786 Sisomicin Natural products 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 241000295644 Staphylococcaceae Species 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 235000009470 Theobroma cacao Nutrition 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000016383 Zea mays subsp huehuetenangensis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- JVVXZOOGOGPDRZ-SLFFLAALSA-N [(1R,4aS,10aR)-1,4a-dimethyl-7-propan-2-yl-2,3,4,9,10,10a-hexahydrophenanthren-1-yl]methanamine Chemical compound NC[C@]1(C)CCC[C@]2(C)C3=CC=C(C(C)C)C=C3CC[C@H]21 JVVXZOOGOGPDRZ-SLFFLAALSA-N 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910000095 alkaline earth hydride Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 125000005233 alkylalcohol group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000002647 aminoglycoside antibiotic agent Substances 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000012501 ammonium carbonate Nutrition 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000003674 animal food additive Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 150000003974 aralkylamines Chemical class 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 102000006635 beta-lactamase Human genes 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- VXIVSQZSERGHQP-UHFFFAOYSA-N chloroacetamide Chemical compound NC(=O)CCl VXIVSQZSERGHQP-UHFFFAOYSA-N 0.000 description 1
- 125000004775 chlorodifluoromethyl group Chemical group FC(F)(Cl)* 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- POBMBNPEUPDXRS-UHFFFAOYSA-N ethyl 2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetate Chemical compound CCOC(=O)C(=NOC)C1=CSC(N)=N1 POBMBNPEUPDXRS-UHFFFAOYSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 239000010642 eucalyptus oil Substances 0.000 description 1
- 229940044949 eucalyptus oil Drugs 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- SZMNOLSLNRNAJC-UHFFFAOYSA-N formaldehyde;propane-1,2,3-triol Chemical compound O=C.OCC(O)CO SZMNOLSLNRNAJC-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 230000002949 hemolytic effect Effects 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Substances C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000010985 leather Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 229910001623 magnesium bromide Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 235000012254 magnesium hydroxide Nutrition 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 235000009973 maize Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 208000027531 mycobacterial infectious disease Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 150000002902 organometallic compounds Chemical class 0.000 description 1
- UWYHMGVUTGAWSP-JKIFEVAISA-N oxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1 UWYHMGVUTGAWSP-JKIFEVAISA-N 0.000 description 1
- 229960001019 oxacillin Drugs 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- RBKMMJSQKNKNEV-RITPCOANSA-N penicillanic acid Chemical compound OC(=O)[C@H]1C(C)(C)S[C@@H]2CC(=O)N21 RBKMMJSQKNKNEV-RITPCOANSA-N 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229950009506 penicillinase Drugs 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- NHKJPPKXDNZFBJ-UHFFFAOYSA-N phenyllithium Chemical compound [Li]C1=CC=CC=C1 NHKJPPKXDNZFBJ-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 1
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 229910052573 porcelain Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 150000003139 primary aliphatic amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000009419 refurbishment Methods 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 150000005619 secondary aliphatic amines Chemical class 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 229960005456 sisomicin Drugs 0.000 description 1
- URWAJWIAIPFPJE-YFMIWBNJSA-N sisomycin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC=C(CN)O2)N)[C@@H](N)C[C@H]1N URWAJWIAIPFPJE-YFMIWBNJSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000004328 sodium tetraborate Substances 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- VYPDUQYOLCLEGS-UHFFFAOYSA-M sodium;2-ethylhexanoate Chemical compound [Na+].CCCCC(CC)C([O-])=O VYPDUQYOLCLEGS-UHFFFAOYSA-M 0.000 description 1
- 229940075554 sorbate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 229960004793 sucrose Drugs 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- QQWYQAQQADNEIC-RVDMUPIBSA-N tert-butyl [(z)-[cyano(phenyl)methylidene]amino] carbonate Chemical compound CC(C)(C)OC(=O)O\N=C(/C#N)C1=CC=CC=C1 QQWYQAQQADNEIC-RVDMUPIBSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003510 tertiary aliphatic amines Chemical class 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 229960000707 tobramycin Drugs 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/48—Acylated amino or imino radicals by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof, e.g. carbonylguanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/587—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with aliphatic hydrocarbon radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms, said aliphatic radicals being substituted in the alpha-position to the ring by a hetero atom, e.g. with m >= 0, Z being a singly or a doubly bound hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D499/00—Heterocyclic compounds containing 4-thia-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. penicillins, penems; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
Definitions
- the present invention relates to new ⁇ -lactam compounds, processes for their preparation and their use as medicaments, in particular as antimicrobial agents and as agents for promoting the growth and improving the use of feed in animals.
- novel ⁇ -lactam antibiotics according to the invention differ chemically from the known compounds of the prior art, above all by the a-position heterocyclic ring in the acyl side chain, which surprisingly shows an improved activity.
- the compounds according to the invention have a broad antibacterial action, i.e. Effect against several bacterial families in the gram-negative area and against ⁇ -lactamase formers. Because of their strong antibacterial properties and because of their. Ability to improve the growth and feed conversion in animals, the compounds of the invention thus represent an enrichment of the technology.
- lower alkyl means everywhere, also in connection with 1 other atoms or groups (for example lower alkoxy, HCON- (lower alkyl), etc.) straight-chain or branched alkyl with preferably 1 to 6, in particular 1 to 4 Carbon atoms. Examples of optionally substituted methyl, ethyl, n- and i-propyl, n-, i- and t-butyl may be mentioned.
- “Lower alkyl” can be substituted by 1 to 5, in particular 1 to 3, identical or different halogen atoms, the halogen atoms preferably being fluorine, chlorine and bromine, in particular fluorine and chlorine. Examples include trifluoromethyl, chlorodifluoromethyl, bromomethyl, 2,2,2-trifluoroethyl and pentafluoroethyl.
- alkyl in alkyl-CO-O- preferably denotes alkyl having 1 to 4, in particular 1 or 2, carbon atoms. Examples include methyl and ethyl, with methyl being particularly preferred.
- the heterocyclic ring Het in ⁇ S ⁇ Het (definition of T) consists of 5 or 6 ring members and contains 1 to 4, preferably 1 to 3 identical or different heteroatoms, oxygen, sulfur and nitrogen being heteroatoms.
- the heterocyclic ring is preferably unsaturated and particularly preferably contains 2 double bonds.
- the heterocyclic ring can contain one or more, preferably 1 or 2, in particular one, substituent.
- substituents are: halogen, such as fluorine, chlorine and bromine, preferably chlorine and bromine, amino, Niederalkylamino, Diniederalkylamino, Niederalkyl, Cycloalkyl (with 3 to 7, preferably 5 or 6 carbon atoms in the cycloalkyl part), Niederalkyloxy (meaning of "Niederalkyl” see above), Trifluormethyl, Phenyl, Benzyl and Acylaminomit preferably 2 to 5, in particular 2 or 3 carbon atoms .
- the -S-phenyl radical in the definition of T can carry one or more, preferably 1 to 3, in particular 1 or 2 identical or different substituents, preference being given to those which are listed above as possible substituents for the -S-Het radical .
- R in the meaning of lower alkoxy preferably denotes an alkoxy group having 1 to 6, in particular 1 to 4, carbon atoms, but in particular methoxy or ethoxy.
- Halogen W represents fluorine, chlorine and bromine, preferably bromine or chlorine, in particular chlorine.
- Nucleofugic leaving groups in the definition of W are to be understood as meaning all nucleofugic groups usually used in organic chemistry, and above all those which are described in Angewandte Chemie, 81 (1969), page 543.
- the compounds of the formula (I) are often obtained in the form of salts during manufacture or can be easily converted into them.
- the pharmaceutically usable salts of the compounds of the formula (I) are particularly important for use as medicaments.
- Pharmaceutically usable salts of the compounds of formula (I) are salts of these compounds with inorganic and organic bases on the acidic carboxyl group or the acidic carboxyl and sulfonic acid groups. All bases normally used in pharmaceutical chemistry, in particular in the chemistry of antibiotics, can be used as bases for this purpose.
- inorganic bases are: alkali and alkaline earth metal hydroxides, alkali and alkaline earth metal carbonates and alkali metal hydrogen carbonates, such as sodium and potassium hydroxide, calcium and magnesium hydroxide, sodium and potassium carbonate, calcium carbonate, sodium and potassium hydrogen carbonate; Aluminum hydroxide and ammonium hydroxide.
- Primary, secondary and tertiary aliphatic amines and heterocyclic amines can be used as organic amines.
- organic amines examples include: di- and tri-lower alkylamines, e.g. Diethylamine, triethylamine, tri-J3-hydroxyethylamine, procain, dibenzylamine, N, N'-dibenzylethylenediamine, N-benzyl-ß-phenyl-ethylamine, N-methyl- and N-ethylmorpholine, 1-ephenamine, dehydroabietylamine, N, N ' -Bis-dehydroabietylethylenediamine, N-lower alkylpiperidine.
- So-called basic amino acids such as lysine or arginine can also advantageously be used as bases.
- Particularly preferred salts are the sodium salts.
- Salts of the compounds of the formula II which can be used are preferably salts with bases which are listed as being suitable for salt formation with compounds of the formula.
- the sodium salts are particularly preferred.
- the compounds of general formula III used as starting materials can be obtained by known methods. They can be obtained, for example, in the following way (see also JACS 78 (1956) 5349):
- Phosgenation is also possible without prior silylation directly in an inert organic solvent in the presence of a base.
- the compounds of formula 11 used as starting materials are known or can be prepared by methods known per se (cf. DOS 2 555 159).
- Suitable diluents in the process according to the invention are water and all inert organic solvents, preferably those which are miscible with water. These primarily include lower dialkyl ketones, e.g. Acetone, methyl ethyl ketone, cyclic ether, e.g. Tetrahydrofuran and dioxane; Nitriles, e.g. Acetonitrile; lower dialkylformamides, e.g. Dimethylformamide; lower alkyl alcohols, e.g. Ethanol and isopropanol as well as dimethyl sulfoxide. These solvents can also be used in mixtures with one another and in any mixtures of one or more of these solvents with water.
- solvents can also be used in mixtures with one another and in any mixtures of one or more of these solvents with water.
- the process according to the invention can therefore be carried out in the presence of: (a) exclusively water, (b) exclusively one or more organic solvents or (c) water and one or more organic solvents. If a pH measurement is possible during the reaction according to the invention due to the presence of water, the pH of the reaction mixture is preferably kept between 6.5 to 7.5 by adding bases or by using buffer mixtures. However, the process according to the invention can also be carried out very well in another pH range, for example between 4.5 and 9.0 or at pH 2.0 to 4.5. It is also possible to carry out the reaction in water-immiscible solvents, e.g.
- halogenated hydrocarbons such as chloroform or methylene chloride
- organic bases preferably lower alkylamines, e.g. Triethylamine, diethylamine or cyclic bases, e.g. Perform N-ethyl piperidine.
- the reaction can be carried out in a mixture of water and a water-immiscible solvent, e.g.
- Lower alkyl ethers such as diethyl ether, halogenated hydrocarbons such as chloroform and methylene chloride; Carbon disulfide; Isobutyl methyl ketone; Esters such as ethyl acetate; aromatic hydrocarbons such as benzene, whereby it is advisable to stir vigorously and to adjust the pH by adding a base or using conventional buffer solutions, e.g. Phosphate, acetate or citrate buffers, between 4.5 and 9.0 or e.g. Keep 2.0 and 4.5.
- the reaction can also be carried out in water alone in the absence of organic solvents in the presence of an organic or inorganic base or with the addition of customary buffer substances.
- Acid binders customarily used in the chemistry of antibiotics can be used as acid binders. These include inorganic bases and organic bases, which, for. B. are difficult to acylate due to steric hindrance. Sodium and potassium hydroxide may be mentioned as examples of inorganic bases. Practically all open-chain or cyclic amines which are difficult or difficult to acylate and also heteroaromatic bases are suitable as organic bases. Examples of bases which may be mentioned are tertiary amines, preferably lower alkylamines, for example triethylamine and / or cyclic bases, for example pyridine, and dicyclohexylamine as secondary amine which is difficult to acylate.
- the addition of a base is only necessary if acidic compounds are formed during the reaction, e.g. in the case where W is halogen or azide.
- reaction temperatures can be varied within a wide range. Generally one works between about -20 ° C and about + 50 ° C, preferably between 0 and + 20 ° C. As with most chemical reactions, higher or lower temperatures can in principle be used.
- the reaction can be carried out under normal pressure, but also under reduced or elevated pressure. Generally one works at normal pressure.
- the proportions of the reactants of the formulas (11) and (111) can be varied within wide limits without the result being adversely affected.
- the starting materials can e.g. are reacted with one another in equimolecular amounts.
- the reactants of the general formula 11 can be used in an excess of 0.1 to 0.3 mol equivalents, and thereby a lower decomposition of the reactants of the general formula III in a water-containing solvent mixture can be achieved.
- the excess of the reactants of the general formula 11 can be easily removed because of the good solubility in aqueous mineral acids when working up the reaction mixture.
- the reactants of the general formula III with an excess of, for example, 0.1 to 1.0 molar equivalents. This will make the Reaction partners of the general formula II are better utilized and the decomposition of the reactants of the general formula III taking place as a side reaction in water-containing solvents is compensated. Since the compounds of the general formula III added in excess quickly convert into neutral nitrogen-containing heterocycles in water which can be easily removed, the purity of the antibiotics is hardly impaired thereby.
- the amount of the bases used is e.g. determined by the desired compliance with a certain pH value. Where there is no pH measurement and adjustment, or because of the lack of sufficient amounts of water in the diluent it is not possible or is not sensible, 2 molar equivalents of base are preferably added.
- reaction batches for the preparation of the compounds according to the invention and their salts are worked up in the manner generally known for these bodies.
- the isolation and purification of the compounds according to the invention and the liberation of the free acids from salts or the conversion of the free acids into salts are also carried out by generally known methods of organic chemistry, which are familiar to any person skilled in the art.
- a ⁇ -lactam compound of the formula (I) in which R is hydrogen is put in with 2-10 equivalents per equivalent of a ⁇ -lactam compound of a base in the presence of an excess of an alcohol of the formula R'OH the R 'denotes lower alkyl in an inert organic solvent, adds between about 1 to about 8 equivalents of an N-halogenating agent and isolates the compound of the formula.
- R denotes lower alkoxy, optionally after prior removal of the acid protecting group, conversion into isolated a salt or a pharmaceutically acceptable ester.
- compounds which transmit positive chlorine such as t-butyl hypochlorite or chloroacetamide, are preferably used as the N-halogenating agent.
- Suitable bases are complex and simple, but preferably simple, alkali and alkaline earth hydrides, organometallic compounds and Grignard compounds.
- Examples include: lithium hydride, sodium hydride, butyl lithium, phenyl lithium, alkyl magnesium bromide, for example methyl magnesium bromide, or other known acid binders such as alkali and alkaline earth metal alcoholates or carbonates, alkali metal and alkaline earth metal bicarbonates or oxides such as sodium carbonate or other acid binders such as borax or open-chain or cyclic organic bases such as trialkyl or aralkylamines or cyclic amidines such as 2,3,4,6,7,8-hexahydro-pyrrolo [1,2-a] pyrimidine (DBN) or 2,3,4,6,7,8,9,10-octahydro-pyrimido [1,2-a] azepine (DBU).
- DBN 2,3,4,6,7,8,9,10-octahydro-
- Suitable solvents are, for example, open-chain or cyclic ethers, aliphatic and aromatic hydrocarbons or halogenated hydrocarbons or the alcohols R'OH. Tetrahydrofuran is particularly suitable.
- reaction temperatures should be kept below 0 ° C, preferably between -70 ° C and -45 ° C.
- the compounds of the general formula I are both crystalline and amorphous in the form of the free acid and are both antibacterially active in the same way both anhydrous and in various forms of hydrate.
- the compounds of general formula I are also in the form of their salts, e.g. Sodium salts, both crystalline and amorphous and both water-free and water-containing, for example as a hydrate, antibacterial in the same way.
- the active compounds according to the invention have a strong and broad antimicrobial activity. These properties enable their use as chemotherapeutic agents in medicine and as substances for the preservation of inorganic and organic materials, in particular of all kinds of organic materials, e.g. Polymers, lubricants, paints, fibers, leather, paper and wood, food and water.
- organic materials e.g. Polymers, lubricants, paints, fibers, leather, paper and wood, food and water.
- the active compounds according to the invention are active against a very broad spectrum of microorganisms. With their help, gram-negative and gram-positive bacteria and bacterial micro-organisms can be fought, and the diseases caused by these pathogens can be prevented, improved and / or cured.
- the active compounds according to the invention are particularly effective against bacteria and bacteria-like microorganisms. They are therefore particularly suitable for the prophylaxis and chemotherapy of local and systemic infections in human and veterinary medicine, which are caused by these pathogens.
- the present invention includes pharmaceutical preparations which, in addition to non-toxic, inert pharmaceutically suitable excipients, contain one or more active compounds according to the invention or which consist of one or more active compounds according to the invention, and processes for the preparation of these preparations.
- the present invention also includes pharmaceutical preparations in dosage units.
- the preparations in the form of individual parts e.g. Tablets, coated tablets, capsules, pills, suppositories and ampoules are present, the active ingredient content of which corresponds to a fraction or a multiple of a single dose.
- the dosage units can e.g. 1, 2, 3 or 4 single doses or 1/2, 1/3 or 1/4 of a single dose.
- a single dose preferably contains the amount of active ingredient which is administered in one application and which usually corresponds to a whole, a half or a third or a quarter of a daily dose.
- Non-toxic, inert pharmaceutically suitable carriers are to be understood as solid, semi-solid or liquid diluents, fillers and formulation auxiliaries of all kinds.
- Tablets, dragees, capsules, pills, granules, suppositories, solutions, suspensions and emulsions, pastes, ointments, gels, creams, lotions, powders and sprays may be mentioned as preferred pharmaceutical preparations.
- Tablets, dragees, capsules, pills and granules can contain the active ingredient (s) in addition to the usual carriers, such as (a) fillers and extenders, for example starches, milk sugar, cane sugar, Glucose, mannitol and silica, (b) binders, e.g. carboxymethyl cellulose, alginates, gelatin, polyvinylpyrrolidone, (c) humectants, e.g. glycerin, (d) disintegrants, e.g. agar-agar, calcium carbonate and sodium carbonate, (e) dissolving agents, e.g. paraffin and (f) absorption accelerators, e.g.
- fillers and extenders for example starches, milk sugar, cane sugar, Glucose, mannitol and silica
- binders e.g. carboxymethyl cellulose, alginates, gelatin, polyvinylpyrrolidone
- quaternary ammonium compounds (g) wetting agents, e.g. cetyl alcohol, glycerol monostearate, (h) adsorbents, e.g. kaolin and bentonite and (i) lubricants, e.g. talc, calcium and magnesium stearate and solid polyethylene glycols or mixtures of the (a ) to (i) listed substances.
- wetting agents e.g. cetyl alcohol, glycerol monostearate
- adsorbents e.g. kaolin and bentonite
- lubricants e.g. talc, calcium and magnesium stearate and solid polyethylene glycols or mixtures of the (a ) to (i) listed substances.
- the tablets, dragees, capsules, pills and granules can be provided with the customary coatings and casings optionally containing opacifying agents and can also be composed in such a way that they release the active ingredient (s) only or preferably in a certain part of the intestinal tract, if necessary with a delay, using as a preventative e.g. Polymer substances and waxes can be used.
- the active ingredient (s) can optionally also be in microencapsulated form with one or more of the above-mentioned excipients.
- suppositories can contain the usual water-soluble or water-insoluble carriers, for example polyethylene glycols, fats, for example cocoa fat and higher esters (for example C 4 alcohol with C 6 fatty acid) or mixtures of these substances.
- water-soluble or water-insoluble carriers for example polyethylene glycols, fats, for example cocoa fat and higher esters (for example C 4 alcohol with C 6 fatty acid) or mixtures of these substances.
- ointments, pastes, creams and gels can contain the usual carriers, e.g. animal and vegetable fats, waxes, paraffins, starches, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide or mixtures of these substances.
- carriers e.g. animal and vegetable fats, waxes, paraffins, starches, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide or mixtures of these substances.
- Powders and sprays can contain the usual excipients in addition to the active ingredient (s), e.g. Milk sugar, talc, silica, aluminum hydroxide, calcium silicate and polyamide powder or mixtures of these substances. Sprays can also use the usual blowing agents e.g. Contain chlorofluorocarbons.
- active ingredient e.g. Milk sugar, talc, silica, aluminum hydroxide, calcium silicate and polyamide powder or mixtures of these substances.
- Sprays can also use the usual blowing agents e.g. Contain chlorofluorocarbons.
- solutions and emulsions can contain the usual carriers such as solvents, solubilizers and emulsifiers, e.g. Water, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils, especially cottonseedol, peanut oil, maize germol, olive oil, castor oil and sesamol, glycerol, glycerol formaldehyde, fatty alcohol sorbate, tetrahydrofuran glycolate, tetrahydrofuran desorbate or mixtures of these substances.
- solvents e.g. Water, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol
- solutions and emulsions can also be in sterile and blood isotonic form.
- suspensions can contain the usual carriers such as liquid diluents, e.g. Water, ethyl alcohol, propylene glycol, suspending agents e.g. Contain athoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar and tragacanth or mixtures of these substances.
- liquid diluents e.g. Water, ethyl alcohol, propylene glycol
- suspending agents e.g. Contain athoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar and tragacanth or mixtures of these substances.
- the formulation forms mentioned can also contain colorants, preservatives and odor and taste-improving additives, e.g. Peppermint oil and eucalyptus oil and sweeteners, e.g. Saccharin.
- the therapeutically active compounds should be present in the pharmaceutical preparations listed above preferably in a concentration of approximately 0.1 to 99.5, preferably approximately 0.5 to 95 percent by weight of the total mixture.
- the pharmaceutical preparations listed above can also contain further pharmaceutical active substances.
- the pharmaceutical preparations listed above are prepared in a conventional manner by known methods, e.g. by mixing the active substance or substances with the carrier substance or substances.
- the present invention also includes the use of the active compounds according to the invention and of pharmaceutical preparations which contain one or more active compounds according to the invention in human and veterinary medicine for preventing, ameliorating and / or curing the diseases mentioned above.
- the active substances or the pharmaceutical preparations can be administered locally, orally, parenterally, intraperitoneally and / or rectally, preferably orally or parenterally, such as intravenously or intramuscularly.
- the active compound (s) according to the invention in total amounts of about 5 to about 1000, preferably 20 to 200 mg / kg of body weight per 24 hours, optionally in the form of several individual doses to deliver the results you want.
- a single dose contains the active ingredient (s) according to the invention, preferably in amounts of about 1 to about 250, in particular 10 to 100 mg / kg of body weight.
- the new compounds When used as a feed additive, the new compounds can be given in the usual concentrations and preparations together with the feed or with feed preparations or with the drinking water. This can prevent, ameliorate and / or cure an infection by gram-negative or gram-positive bacteria and also promote growth and improve the utilization of the feed.
- the new penicillins and cephalosporins are characterized by strong antibacterial effects that have been tested in vivo and in vitro.
- penicillins and cephalosporins according to the invention can be used with other antimicrobial active ingredients, e.g. can be combined with penicillins that are particularly resistant to penicillinase. Such a combination would be e.g. those with oxacillin or dicycloxacillin.
- penicillins and cephalosporins according to the invention can also be used with aminoglycoside antibiotics, for example for the purpose of widening the activity spectrum and to achieve an increase in activity.
- Gentamicin, Kanamicin, Sisomicin, Amikacin or Tobramicin can be combined.
- THF prepared from 5.0 parts by weight of 7-ACS and 5.6 parts by weight of bistrimethylsilylacetamide
- the mixture was stirred at ⁇ 20 ° for 10 minutes and allowed to come to RT.
- Ice water added, adjusted to pH 7.3 and the THF removed.
- the aqueous phase is extracted with ethyl acetate, acidified at 0-5 ° and extracted again with ethyl acetate.
- the latter extracts of ethyl acetate are dried over magnesium sulfate and concentrated.
- the product (7.5 parts by weight) is obtained as a yellow foam which is processed directly.
- the aqueous solution is extracted with ethyl acetate, cooled to 5 °, with 150 parts by volume. Layered ethyl acetate and acidified with 1 N hydrochloric acid, the product acid precipitating (3.5 parts by weight). It will be in 35 VoLTln. Suspended water, dissolved with 0.5 N sodium hydroxide solution and lyophilized this solution. 3.0 parts by weight are obtained. Sodium salt from decomposition. p. 170-180 °.
- ⁇ -lactam content 85% (substance contains approx. 6% ⁇ -lactam open product)
- the substance contains about 0.08 molar equivalents of sodium 2-ethylhexanoate and about 3.2 molar equivalents of water.
- reaction solution is poured into 80 parts by volume. ice-cold concentrated hydrochloric acid, rinsed with water and passes 50-60 g of HCl gas at 0-5 ° C into the solution, which is then left overnight (deduction). After dilution with 100 parts by volume. Water, 2.5 hours of refluxing and concentration, the residue is smoked twice with water in a porcelain bowl (to remove HCl).
- the loaded ion exchanger is suctioned off, washed well with water and finally eluted with ammonium carbonate solution. This solution is evaporated to dryness. The residue is washed with ethanol and 7.4 parts by weight from the mother liquors. (40%) of the desired amino acid.
- 7-Aminocephalosporanic acid 5.44 parts by weight 7-Aminocephalosporanic acid are in 100 vol. 80% aqueous tetrahydrofuran and 2% by weight tin. Triethylamine dissolved and cooled to -10 ° C. To do this, drop A at -10 ° C. The mixture is stirred for a further 1.5 hours at -10 ° C. and for a further 2 hours, allowing to come to room temperature.
- reaction solution is in 100 parts by volume. Given water, washed with ethyl acetate, acidified (pH 1.8), extracted with ethyl acetate and this ethyl acetate solution washed with saturated NaCl solution. After drying over MgS0 4 , the mixture is concentrated and the residue is dried under high vacuum.
- the trifluoroacetic acid is then largely distilled off (initial charge cooled to -70 ° C., high vacuum) and the residue in 10 parts by volume. Dissolved methylene chloride and stirred for 20 min. Then 100 vol. Parts of ether / ligroin are added 1: 1 and the mixture is stirred for a further 30 minutes.
- the pH is adjusted to 7 with amberlite (liquid ion exchanger, chloride as counterion), the phases are separated and the aqueous phase is washed very thoroughly with ether. The aqueous phase is freeze-dried.
- Thin layer chromatogram (eluent see example 5.1) a substance stain at the starting point.
- reaction solution is then placed in a 10% ammonium chloride solution and maintains the pH at 7 with further addition of dilute hydrochloric acid.
- the tetrahydrofuran is distilled off in vacuo and the aqueous phase is washed with ethyl acetate. Then you give 10 Vol.Tle. Acetone and, while stirring, slowly adjust the pH to 4 with dilute hydrochloric acid.
- the penicillanic acid slowly crystallizes out.
- the precipitate is filtered off and dissolved in water with the addition of 1 N sodium hydroxide solution at pH 7.
- the solution is finally lyophilized.
- Example 7.3 5.0 g of a substance prepared according to Example 7.3 (contains 2.0 g of NaCl) are suspended or dissolved in 70 ml of 60% aqueous tetrahydrofuran and the mixture is brought to pH 7.5 using 2N sodium hydroxide solution. Then 2.72 g of 1-chlorocarbonyl-2-oxo-3-furylideneaminoimidazolidine are introduced and the pH is kept at 7.5 using sodium hydroxide solution. When the pH no longer changes, the tetrahydrofuran is removed in vacuo, acidified to pH 2.3 with 2N hydrochloric acid, the precipitated low whipped and washed with water. The precipitate is suspended in hot aqueous ethanol, cooled and suction filtered.
- Example 9.5 0.7 g of the substance shown in Example 9.5 are suspended in 2.8 ml of anisole, cooled in ice / water, mixed with 14 ml of trifluoroacetic acid and the mixture is left to stand at 4 ° C. overnight. The solution is then evaporated in vacuo at -25 ° C., the residue is triturated with ether, suction filtered and dried in a desiccator over P 4 O 10 .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Cephalosporin Compounds (AREA)
Description
- Die vorliegende Erfindung betrifft neue β-Lactam-Verbindungen, Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel, insbesondere als antimikrobielle Mittel und als Mittel zur Förderung des Wachstums und zur Verbesserung der Futterverwendung bei Tieren.
- Es ist bereits bekannt geworden, daß bestimmte α-(Imidazolidin-2-oxo-1-yl-carbonylamino)-benzylpenicilline antibakteriell wirksam sind (vgl. BE-PS 767 647 und 767 648, DT-OS 2 152 968 und FR-PS 2292471).
- Die erfindungsgemäßen neuen ß-Lactam-Antibiotica unterscheiden sich chemisch von den bekannten Verbindungen des Standes der Technik vor allem durch den a-ständigen heterocyclischen Ring in der Acylseitenkette, wodurch sie überraschenderweise eine verbesserte Wirksamkeit zeigen.
- Die Erfindung betrifft neue β-Lactam-Verbindungen der Formel (I)

- Z R4

- R für Wasserstoff oder Niederalkoxy steht,
- A für -CH2-CH2-, ―CH2―CH2―CH2― oder

- B für einen Furyl-, Thienyl-, 1,3,5-Trimethyl-pyrazolyl- oder 2-Aminothiazolylrest steht, und
- Y für die Gruppen

- T für Wasserstoff, -O-CH-CH3, Hydroxyl oder für gegebenenfalls durch Alkyl mit 1 bis 4 Kohlenstoffatomen oder CF3 substituiertes Thiadiazolylthio oder Tetrazolylthio steht, wobei diese Verbindungen der Formel 1 bezüglich des Chiralitätszentrums C in den beiden möglichen R- und S-Konfigurationen sowie als Gemische der daraus resultierenden Diastereomeren vorliegen können, und wobei die Verbindungen der Formel I bezüglich der Iminogruppe im Rest Z sowohl in der syn-Form als auch in der anti-Form vorliegen können und wobei diese Verbindungen der Formel I auch in den verschiedenen Hydratformen sowie in Form ihrer pharmazeutisch verwendbaren Salze vorliegen können.
- Weiterhin wurde gefunden, daß man die neuen ß-Lactam-Antibiotica der Formel 1 erhält, wenn man Verbindungen der Formel II

- R, B, C, X und Y die oben angegebene Bedeutung haben oder deren Salze, mit Verbindungen der Formel III

- Z und A die oben angegebene Bedeutung haben und
- W für Halogen, Azid oder eine andere nukleofuge Abgangsgruppe steht,
- Die erfindungsgemäßen Verbindungen der Formel (I), in welcher R für Niederalkoxy steht, sind ebenfalls herstellbar durch Alkoxylierung der entsprechenden Wasserstoffderivate (R=H), wobei es vorteilhaft ist, bei der Alkoxylierung solche Verbindungen zu verwenden, in denen E eine geeignete Schutzgruppe bedeutet.
- Die erfindungsgemäßen Verbindungen zeigen neben guter Verträglichkeit und Löslichkeit eine breite antibakterielle Wirkung, d.h. Wirkung gegenüber mehreren Bakterienfamilien im gram-negativen Bereich und gegenüber β-Lactamasebildnern. Aufgrund ihrer starken antibakteriellen Eigenschaften und aufgrund ihrer. Fähigkeit, das Wachstum und die Futterverwertung bei Tieren zu verbessern, stellen die erfindungsgemäßen Verbindungen somit eine Bereicherung der Technik dar.
- Verwendet man beispielsweise α-Amino-furfurylacetylpenicillin und 1-Chlor arbonyl-3-furfuryl- idenamino-imidazolidin-2-on als Ausgangsstoffe, so kann der Reaktionsablauf durch das folgende Formelschema wiedergegeben werden:

- In der Definition von T bedeutet Alkyl in Alkyl-CO-O- vorzugsweise Alkyl mit 1 bis 4, insbesondere 1 oder 2 Kohlenstoffatome. Beispielhaft seien Methyl und Athyl genannt, wobei Methyl besonders bevorzugt ist.
- Der heterocyclische Ring Het in ―S―Het (Definition von T) besteht aus 5 oder 6 Ringgliedern und enthält 1 bis 4, vorzugsweise 1 bis 3 gleiche oder verschiedene Heteroatome, wobei als Heteroatome Sauerstoff, Schwefel und Stickstoff stehen. Bevorzugt ist der heterocyclische Ring ungesättigt und enthält besonders bevorzugt 2 Doppelbindungen. Der heterocyclische Ring kann einen oder mehrere, vorzugsweise 1 oder 2, insbesondere einen Substituenten enthalten. Als Substituenten seien beispielhaft aufgeführt: Halogen, wie Fluor, Chlor und Brom, vorzugsweise Chlor und Brom, Amino, Niederalkylamino, Diniederalkylamino, Niederalkyl, Cycloalkyl (mit 3 bis 7, vorzugsweise 5 oder 6 Kohlenstoffatomen im Cycloalkylteil), Niederalkyloxy (Bedeutung von "Niederalkyl" siehe oben), Trifluormethyl, Phenyl, Benzyl und Acylaminomit vorzugsweise 2 bis 5, insbesondere 2 oder 3 Kohlenstoffatomen. Als -S-Het seien als besonders bevorzugt aufgeführt:



- R in der Bedeutung von Niederalkoxy bezeichnet bevorzugt eine Alkoxygruppe mit 1 bis 6, insbesondere 1 bis 4 Kohlenstoffatomen, insbesondere aber Methoxy oder Äthoxy.
- Ganz besonders bevorzugt sind erfindungsgemäße Verbindungen, in welchen C in der D- = R-Konfiguration vorliegt.
- Alle Kristallformen und Hydratformen der erfindungsgemäßen Verbindungen der allgemeinen Formel (I) und ihrer Salze sind in gleicher Weise antibakteriell wirksam.
- Halogen W steht für Fluor, Chlor und Brom, vorzugsweise für Brom oder Chlor, insbesondere für Chlor.
- Unter nukleofugen Abgangsgruppen in der Definition von W sind alle üblicherweise in der organischen Chemie verwendeten nukleofugen Gruppen und vor allem solche zu verstehen, welche in Angewandte Chemie, 81 (1969), Seite 543 beschrieben sind.
- Die Verbindungen der Formel (I) fallen bei der Herstellung vielfach in Form von Salzen an oder können leicht in diese überführt werden. Besonders wichtig für den Gebrauch als Arzneimittel sind die pharmazeutisch verwendbaren Salze der Verbindungen gemäß Formel (I).
- Pharmazeutisch verwendbare Salze der Verbindungen der Formel (I) sind Salze dieser Verbindungen mit anorganischen und organischen Basen an der sauren Carboxylgruppe bzw. den sauren Carboxyl- und Sulfonsäuregruppen. Als Basen können hierzu alle in der pharmazeutischen Chemie, insbesondere in der Chemie der Antibiotika, üblicherweise verwendeten Basen eingesetzt werden. Als anorganische Basen seien beispielhaft genannt: Alkali- und Erdalkalihydroxide, Alkali- und Erdalkalicarbonate und Alkalihydrogen-carbonate, wie Natrium- und Kaliumhydroxid, Calcium- und Magnesiumhydroxid, Natrium- und Kaliumcarbonat, Calciumcarbonat, Natrium- und Kaliumhydrogencarbonat; Aluminiumhydroxid und Ammoniumhydroxid. Als organische Amine können primäre, sekundäre und tertiäre aliphatische Amine sowie heterocyclische Amine eingesetzt werden. Beispielhaft seien genannt: Di- und Triniedrigalkylamine, z.B. Diäthylamin, Triäthylamin, Tri-J3-hydroxyäthylamin, Procain, Dibenzylamin, N,N'-Dibenzyläthylendiamin, N-Benzyl-ß-phenyl-äthylamin, N-Methyl- und N-Äthylmorpholin, 1-Ephenamin, Dehydroabietylamin, N,N'-Bis-dehydroabietyläthylendiamin, N-Niedrigalkylpiperidin. Auch sogenannte basische Aminosäuren wie Lysin oder Arginin können vorteilhaft als Basen Verwendung finden. Besonders bevorzugte Salze sind die Natriumsalze.
- Ganz besonders bevorzugte Verbindungen der Formel 1 sind solche, in welchen
- R für Wasserstoff oder Methoxy und
- A für -CH2-CH2-
- Y für die Gruppen

- T für Wasserstoff, -O-CO-CH3, Hydroxyl oder für gegebenenfalls durch Alkyl mit 1 bis 4 Kohlenstoffatomen oder CF3 substituertes Thiadiazolylthio oder Tetrazolylthio steht; und
- C liegt in der D- = R-Konfiguration vor;
- Die als Ausgangsstoffe verwendeten Verbindungen der allgemeinen Formel II sind bereits bekannt oder nach bekannten Methoden erhältlich.
- Alle Kristallformen, Hydratformen und Salze der Verbindungen der allgemeinen Formel II sind als Ausgangsmaterialien für das erfindungsgemäße Verfahren geeignet.
- Als Beispiele seien genannt:
- α-Amino-furfurylacetyl-penicillin, 7-(α-Amino-furfuryl-acetamido-3-acetoxymethyl-ceph-3-em-4-carbonsäure.
- Als Salze der Verbindungen der Formel II können vorzugsweise Salze mit Basen eingesetzt werden, welche als für die Salzbildung mit Verbindungen der Formel geeignet aufgeführt werden. Besonders bevorzugt sind die Natriumsalze.
- Die als Ausgangstoffe verwendeten Verbindungen der allgemeinen Formel III sind nach bekannten Methoden erhältlich. Sie können z.B. auf folgendem Wege erhalten werden (vgl. auch J.A.C.S. 78 (1956) 5349):

- Die Phosgenierung ist auch ohne vorherige Silylierung direkt in einem inerten organischen Lösungsmittel in Gegenwart einer Base möglich.
- Als Beispiele für erfindungsgemäße Ausgangsverbindungen der allgemeinen Formel (11) seien genannt:
- 1-Chlorcarbonyl-2-oxo-3-furfurylidenamino-imidazolidin
- 1 -Azidocarbonyl-2-oxo-3-furfurylidenamino-imidazolidin
- 1-Chlorcarbonyl-2-oxo-3-crotonylidenamino-imidazolidin
- Diejenigen Verbindungen der allgemeinen Formel (111), in denen W Azid ist, werden in üblicher Weise z.B. aus den entsprechenden Verbindungen (111), in denen W Halogen ist, durch Umsetzung beispielsweise mit Alkaliaziden erhalten.
- Die als Ausgangsstoffe eingesetzten Verbindungen der Formel 11 sind bekannt oder können nach an sich bekannten Methoden hergestellt werden (vgl. DOS 2 555 159).
- Als Verdünnungsmittel kommen beim erfindungsgemäßen Verfahren Wasser sowie alle inerten organischen Lösungsmittel, vorzugsweise solche, welche mit Wasser mischbar sind, in Frage. Hierzu gehören vor allem niedere Dialkylketone, z.B. Aceton, Methylathylketon, cyclische Aether, z.B. Tetrahydrofuran und Dioxan; Nitrile, z.B. Acetonitril; niedere Dialkylformamide, z.B. Dimethylformamid; niedere Alkylalkohole, z.B. Aethanol und Isopropanol sowie Dimethylsulfoxid. Diese Losungsmittel können auch in Mischungen untereinander sowie in beliebigen Mischungen einzelner oder mehrerer dieser Lösungsmittel mit Wasser verwendet werden. Das erfindungsgemäße Verfahren kann also durchgeführt werden in Gegenwart von: (a) ausschließlich Wasser, (b) ausschließlich einem oder mehreren organischen Lösungsmitteln oder (c) Wasser und einem oder mehreren organischen Lösungsmitteln. Ist wegen des Vorhandenseins von Wasser eine pH-Messung wahrend der erfindungsgemaßen Reaktion möglich, wird der pH der Reaktionsmischung durch Zusatz von Basen oder durch Verwendung von Puffergemischen vorzugsweise zwischen 6,5 bis 7,5 gehalten. Das erfindungsgemäße Verfahren läßt sich aber auch sehr gut in einem anderen pH-Bereich, beispielsweise zwischen 4,5 und 9,0 oder bei pH 2,0 bis 4,5, durchführen. Ferner ist es möglich, die Reaktion in mit Wasser nicht mischbaren Lösungsmitteln, z.B. halogenierten Kohlenwasserstoffen, wie Chloroform oder Methylenchlorid, unter Zusatz von organischen Basen, vorzugsweise Niederalkylaminen, z.B. Triäthylamin, Diäthylamin oder cyclischen Basen, z.B. N-Äthylpiperidin durchzuführen. Weiterhin läßt sich die Reaktion in einer Mischung aus Wasser und einem mit Wasser nicht mischbaren Lösungsmittel, wie z.B. Niederalkyläthern, wie Diäthyläther, halogenierten Kohlenwasserstoffen, wie Chloroform und Methylenchlorid; Schwefelkohlenstoff; Isobutylmethylketon; Estern wie Essigsäureäthylester; aromatischen Kohlenwasserstoffen wie Benzol, ausführen, wobei es zweckmäßig ist, kräftig zu rühren und den pH-Wert durch Basenzusatz oder Verwendung von üblichen Pufferlösungen, z.B. Phosphat-, Acetat- oder Citratpuffer, zwischen 4,5 und 9,0 oder z.B. 2,0 und 4,5 zu halten. Man kann die Reaktion aber auch in Wasser allein in Abwesenheit von organischen Lösungsmitteln in Gegenwart einer organischen oder anorganischen Base oder unter Zusatz von üblichen Pufferstoffen durchführen.
- Als Säurebindemittel können alle in der Chemie der Antibiotica üblicherweise verwendeten Säurebinder verwendet werden. Hierzu gehören anorganische Basen und organische Basen, welche z.B. durch sterische Hinderung schwer acylierbar sind. Als Beispiele für anorganische Basen seien Natrium-und Kaliumhydroxid genannt. Als organische Basen kommen praktisch alle nicht oder schwer acylierbaren offenkettigen oder cyclischen Amine und auch heteroaromatische Basen infrage. Als Basen seien beispielhaft tertiäre Amine, vorzugsweise Niederalkylamine, z.B. Triäthylamin und/oder cyclische Basen, z.B. Pyridin sowie als schwer acylierbares sekundäres Amin Dicyclohexylamin genannt.
- Beim erfindungsgemäßen Verfahren ist der Zusatz einer Base nur dann erforderlich, wenn während der Reaktion saure Verbindungen entstehen, z.B. im Falle, daß W für Halogen oder Azid steht.
- Die Reaktionstemperaturen können in einem größeren Bereich variiert werden. Im allgemeinen arbeitet man zwischen etwa -20°C und etwa +50°C, vorzugsweise zwischen 0 und +20°C. Wie bei den meisten chemischen Reaktionen können jedoch prinzipiell auch höhere oder niedrigere Temperaturen verwendet werden.
- Die Umsetzung kann bei Normaldruck, aber auch bei vermindertem oder erhöhtem Druck ausgeführt werden. Im allgemeinen arbeitet man bei Normaldruck.
- Bei der Durchführung der erfindungsgemäßen Verfahren können die Anteile der Reaktionspartner der Formel (11) und (111) in weiten Grenzen variiert werden, ohne daß das Ergebnis nachteilig beeinflußt wird. Die Ausgangsstoffe können z.B. in äquimolekularen Mengen miteinander zur Reaktion gebracht werden. Es kann jedoch zweckmäßig sein, einen der beiden Reaktionspartner im Überschuß zu verwenden, um sie die Reinigung oder Reindarstellung des gewünschter Penicillins zu erleichtern und die Ausbeute zu erhöhen.
- Beispielsweise kann man die Reaktionspartner der allgemeinen Formel 11 mit einem Ueberschuß von 0,1 bis 0,3 Molaquivalenten einsetzen und dadurch einen geringere Zersetzung der Reaktionspartner der allgemeinen Formel III in einem wasserhaltigen Lösungsmittelgemisch erreichen. Der Ueberschuß der Reaktionspartner der allgemeinen Formel 11 laßt sich wegen der guten Löslichkeit in wäßrigen Mineralsauren beim Aufarbeiten des Reaktionsgemisches leicht entfernen.
- Andererseits kann man aber auch mit Vorteil die Reaktionspartner der allgemeinen Formel III mit einem Ueberschuß von beispielsweise 0,1 bis 1,0 Molaquivalenten einsetzen. Dadurch werden die Reaktionspartner der allgemeinen Formel II besser ausgenutzt und die als Nebenreaktion in wasserhaltigen Losungsmitteln ablaufende Zersetzung der Reaktionsteilnehmer der allgemeinen Formel 111 kompensiert. Da die im Ueberschuß zugesetzt a Verbindungen der allgemeinen Formel III sich in Wasser rasch in neutrale stickstoffhaltige Heterocyclen umwandeln, die sich leicht entfernen lassen, wird die Reinheit der antibiotica hierdurch kaum beeintrachtigt.
- Die Menge der gegebenenfalls verwendeten Basen ist z.B. durch die gewünschte Einhaltung eines bestimmten pH-Wertes festgelegt. Wo eine pH-Messung und Einstellung nicht erfolgt oder wegen des Fehlens von ausreichenden Mengen Wasser im Verdünnungsmittel nicht möglich ist oder nicht sinnvoll ist, werden vorzugsweise 2 Moläquivalente Base zugesetzt.
- Die Aufarbeitung der Reaktionsansätze zur Herstellung der erfindungsgemäßen Verbindungen und ihrer Salze erfolgt durchweg in der bei diesen Körpern allgemein bekannten Art und Weise. Auch die Isolierung und Reinigung der erfindungsgemäßen Verbindungen sowie die Freisetzung der freien Säuren aus Salzen oder die Umwandlung der freien Säuren in Salze werden nach allgemein üblichen Methoden der organischen Chemie, welche jedem Fachmann geläufig sind, vorgenommen.
- Alternativ sind die Verbindungen der Formel (1), bei denen R Niederalkoxy bedeutet auch durch Alkoxylierung der entsprechenden Wasserstoffderivate (R=H) erhältlich, wobei E eine geeignete Schutzgruppe bedeutet wie eine leicht entfernbare esterbildende Gruppe, eine Acetoxymethylgruppe, den kationischen Rest einer Base, vorzugsweise eines Alkali- oder Erdalkalimetallhydroxid oder Wasserstoff bezeichnet.
- Bei diesem Verfahren setzt man eine ß-Lactam-Verbindung der Formel (I), in der R Wasserstoff bedeutet, mit 2-10 Äquivalenten pro Äquivalent β-Lactam-Verbindung einer Base in Anwesenheit eines Überschusses eines Alkohols der Formel R'OH, in der R' Niederalkyl bezeichnet in einem inerten organischen Lösungsmittel um, fügt zwischen etwa 1 bis etwa 8 Äquivalente eines N-Halogenierungsmittels zu und isoliert die Verbindung der Formel.(I) in der R Niederalkoxy bedeutet, gegebenenfalls nach vorheriger Abspaltung der Säureschutzgruppe, Überführung in ein Salz oder einen pharmazeutisch verwendbare Ester isoliert.
- Bei diesem erfindungsgemäßen Verfahren werden als N-Halogenierungsmittel, vorzugsweise positives Chlor übertragende Verbindungen, wie t-Butylhypochlorit oder Chloracetamid verwendet.
- Als Basen eignen sich komplexe und einfache, vorzugsweise jedoch einfache, Alkali- und Erdalkalihydride, metallorganische Verbindungen sowie Grignard-Verbindungen. Als Beispiele seien genannt: Lithiumhydrid, Natriumhydrid, Butyl-Lithium, Phenyl-Lithium, Alkyl-Magnesiumbromid, beispielsweise Methyl-Magnesiumbromid, oder andere bekannte Säurebindemittel wie Alkali- und Erdalkali-alkoholate oder -carbonate, Alkali- und Erdalkali-bicarbonate oder -oxide wie beispielsweise Natriumcarbonat oder andere Säurebindemittel wie Borax oder offenkettige oder cyclische organische Basen wie Trialkyl- oder Aralkylamine oder cyclische Amidine wie 2,3,4,6,7,8 - Hexahydro - pyrrolo[1,2 - a]pyrimidin (DBN) oder 2,3,4,6,7,8,9,10 - Octahydro - pyrimido[1,2 - a]azepin (DBU).
- Als Lösungsmittel eignen sich beispielsweise offenkettige oder cyclische Äther, aliphatische und aromatische Kohlenwasserstoffe oder Halogenkohlenwasserstoffe oder die Alkohole R'OH. Besonders geeignet ist Tetrahydrofuran.
- Die Reaktionstemperaturen sind möglichst unter 0°C zu halten, vorzugsweise zwischen -70°C und -45°C.
- Die Verbindungen der allgemeinen Formel I sind in Form der freien Säure sowohl kristallin wie amorph und sowohl wasserfrei wie in verschiedenen Hydratformen in gleicher Weise antibakteriell wirksam. Ebenfalls sind die Verbindungen der allgemeinen Formel I in Form ihrer Salze, z.B. Natriumsalze, sowohl kristallin wie amorph und sowohl wasserfrei wie wasserhaltig, beispielsweise als Hydrat, in gleicher Wiese antibakteriell wirksam.
- Als neue Wirkstoff seien beispielhaft genannt (Formeln (IV) und (V)




- Die erfindungsgemäßen Wirkstoffe weisen bei geringer Toxizität eine starke und breite antimikrobielle Wirksambeit auf. Diese Eigenschaften ermöglichen ihre Verwendung als chemotherapeutische Wirkstoffe in der Medizin sowie als Stoffe zur Konservierung von anorganischen und organischen Materialien, insbesondere von organischen Materialien aller Art, z.B. Polymeren, Schmiermitteln, Farben, Fasern, Leder, Papier und Holz, von Lebensmitteln und von Wasser.
- Die erfindungsgemaßen Wirkstoffe sind gegen ein sehr breites Spektrum von Mikroorganismen wirksam. Mit ihrere Hilfe können gramnegative und grampositive Bakterien und bakterienahnliche Mikrooganismen bekampft sowie die durch diese Erreger hervorgerufenen Erkrankungen verhindert, gebessert und/oder geheilt werden.
- Besonders wirksam sind die erfindungsgemaßen Wirkstoffe gegen Bakterien und bakterienähnliche Mikroorganismen. Sie und daher besonders gut zur Prophylaxe und Chemotherapie von lokalen und systemischen Infektionen in der Human- und Tiermedizin geeignet, die durch diese Erreger hervorgerufen werden.
- Beispielsweise können lokale und/oder systemische Erkrankungen behandelt und/oder verhindert werden, die durch die folgenden Erreger oder durch Mischungen der folgenden Erreger verursacht werden:
- Micrococcaceae, wie Staphylokokken, z.B. Staphylococcus aureus, Staph. epidermidis, Staph. aerogenes und Gaffkya tetragena (Staph. = Staphylococcus);
- Lactobacteriaceae, wie Streptokokken, z.B. Streptococcus pyogenes, a-bzw. ß-hämolysierende Streptokokken, nicht (y-)-hämolysierende Streptokokken, Str. viridans, Str. faecalis (Enterokokken), Str. agalactiae, Str. lactis, Str. equi, Str. anaerobis und Diplococcus pneumoniae (Pneumokokken) (Str. = Streptococcus);
- Neissericeae, wie Neisserien, z.B. Neisseria gonorrhoeae (Gonokokken), N.meningitidis (Meningokokken), N.catarrhalis und N.flava (N. = Neisseria);
- Corynebacteriaceae, wie Corynebakterien, z.B. Corynebacterium diphtheriae, C.pyogenes, C.- diphtheroides, C.acnes, C.parvum, C.bovis, C.renale, C.ovis, C.murisepticum.
- Mycobacteriaceae, wie Erreger von Mykobakteriosen, z.B. Mycobacterium tuberculosis, M.bovis, M.avium, sogenannte atypische Mykobakterien der Runyon-Gruppen, I, 11, III und IV, M.leprae (M. = Mycobacterium);
- Enterobacteriaceae, wie Escherichiae-Bakterien der Coli-Gruppe: Escherichia-Bakterien, z.B. Escherichia coli, Enterobacter-Bakterien, z.B. E.aerogenes, E.cloacae, Klebsiella-Bakterien, z.B. K.pneumoniae, K.pneumoniae, K.ozaenae, Erwiniae, z.B. Erwinia spec., Serratia, z.B. Serratia marcescens (E. = Enterobacter) (K. = Klebsiella), Proteae-Bakterien der Proteus-Gruppe: Proteus, z.B. Proteus vulgaris, Pr.morganii, Pr.rettgeri, Pr.mirabilis, Providencia, z.B. Providencia sp. (Pr. = Proteus), Salmonelleae: Salmonella-Bakterien, z.B. Salmonella paratyphi A und B. S.typhi, S.enteritidis, S.- cholerae suis, S.typhimurium (S. = Salmonella, Shigella-Bakterien, z.B. Shigella dysenteriae, Sh.ambi- gua, Sh.flexneri, Sh.boydii, Sh.sonnei (Sh. = Shigella);
- Pseudomonadaceae, wie Pseudomonas-Bakterien, z.B. Pseudomonas aeruginosa, Ps.pseudo- mallei (Ps. = Pseudomonas), Aeromonas-Bakterien, z.B. Aeromonas liquefaciens, A.hydrophila (A. = Aeromonas);
- Parvobacteriaceae, wie Pasteurella-Bakterien, z.B. Pasteurella multocida, Past.pestis (Yersinia), Past.pseudotuberculosis,Haemophilus-Bakterien, z.B. Haemophilus influenzae, H.ducreyi, H.suis, H.- canis, H.aegypitcus (H = Haemophilus), Bordetella-Bakterien, z.B. B.bronchiseptica (B. = Bordetella).
- Bacteroidaceae, wie Bacteroides-Bakterien, z.B. Bacteroides fragilis, B.serpens (B. = Bacteroides), Fusiforme-Bakterien, z.B. Fusobacterium fusiforme, Sphaerophorus-Bakterien, z.B.Sphaerophorus necrophorus, Sph-necroticus, Sph.pyrogenes (Sph. = Sphaerophorus);
- Bacillaceae, wie aerobe Sporenbildner, z.B. Bacillus anthracis (B.subtilis, B.cereus) B. = Bacillus), Anaerobe Sporenbildner-Chlostridien, z.B. Clostridium perfringens, CI.septicium, CI.oedematien, Cl.hi- stolyticum, Cl.tetani, CI.botulinum (Cl. = Clostridium).
- Die obige Aufzahlung von Erregern ist lediglich beispielhaft und keineswegs beschrankend aufzufassen.
- Als Krankheiten, die durch die erfindungsgemaßen Wirkstoffe verhindert, gebessert und/oder geheilt werden können, seien beispielsweise genannt:
- Erkrankungen der Atmungswege und des Rachenraumes;
- Otitis; Pharyngitis; Pneumonie; Peritonitis; Pyelonephritis; Cystitis; Endocarditis; Systeminfektionen; Bronchitis; Arthritis; lokale Infektionen.
- Zur vorliegenden Erfindung gehoren pharmazeutische Zubereitungen, die neben nichttoxischen, inerten pharmazeutisch geeigneten Tragerstoffen einen oder mehrere erfindungsgemäße Wirkstoffe enthalten oder die aus einem oder mehreren erfindungsgemäßen Wirkstoffen bestehen sowie Verfahren zur Herstellung dieser Zubereitungen.
- Zur vorliegenden Erfindung gehören auch pharmazeutische Zubereitungen in Dosierungseinheiten. Dies bedeutet, daß die Zubereitungen in Form einzelner Teile, z.B. Tabletten, Dragees, Kapseln, Pillen, Suppositorien und Ampullen vorliegen, deren Wirkstoffgehalt einem Bruchteil oder einem Vielfachen einer Einzeldosis entsprechen. Die Dosierungseinheiten können z.B. 1, 2, 3 oder 4 Einzeldosen oder 1/2, 1/3 oder 1/4 einer Einzeldosis enthalten. Eine Einzeldosis enthält vorzugsweise die Menge Wirkstoff, die bei einer Applikation verabreicht wird und die gewöhnlich einer ganzen, einer halben oder einem Drittel oder einem Viertel einer Tagesdosis entspricht.
- Unter nichttoxischen, inerten pharmazeutisch geeigneten Tragerstoffen sind feste, halbfeste oder flüssige Verdünnungsmittel, Füllstoffe und Formulierungshilfsmittel jeder Art zu verstehen.
- Als bevorzugte pharmazeutische Zubereitungen seien Tabletten, Dragees, Kapseln, Pillen, Granulate, Suppositorien, Lösungen, Suspensionen und Emulsionen, Pasten, Salben, Gele, Cremes, Lotions, Puder und Sprays genannt.
- Tabletten, Dragees, Kapseln, Pillen und Granulate können den oder die Wirkstoffe neben den üblichen Trägerstoffen enthalten, wie (a) Füll- und Streckmittel, z.B. Stärken, Milchzucker, Rohrzucker, Glukose, Mannit und Kieselsäure, (b) Bindemittel, z.B. Carboxymethylcellulose, Alginate, Gelatine, Polyvinylpyrrolidon, (c) Feuchthaltemittel, z.B. Glycerin, (d) Sprengmittel, z.B. Agar-Agar, Calciumcarbonat und Natriumcarbonat, (e) Lösungsverzögerer, z.B. Paraffin und (f) Resorptionsbeschleuniger, z.B. quarternäre Ammoniumverbindungen, (g) Netzmittel, z.B. Cetylalkohol, Glycerinmonostearat, (h) Adsorptionsmittel, z.B. Kaolin und Bentonit und (i) Gleitmittel, z.B. Talkum, Calcium- und Magnesiumstearat und feste Polyäthylenglykole oder Gemische der unter (a) bis (i) aufgeführten Stoffe.
- Die Tabletten, Dragees, Kapseln, Pillen und Granulate können mit den üblichen gegebenenfalls Opakisierungsmittel enthaltenden Ueberzügen und Hüllen versehen sein und auch so zusammengesetzt sein, daß sie den oder die Wirkstoffe nur oder bevorzugt in einem bestimmten Teil des Intestinaltraktes gegebenenfalls verzögert abgeben, wobei als Hintettungsmassen z.B. Polymersubstanzen und Wachse verwendet werden können.
- Der oder die Wirkstoffe können gegebenenfalls mit einem oder mehreren der oben angegebenen Trägerstoffen auch in mikroverkapselter Form vorliegen.
- Suppositorien konnen neben dem oder den Wirkstoffen die üblichen wasserloslichen oder wasserunloslichen Trägerstoffe enthalten, z.B. Polyathylenglykole, Fette, z.B. Kakaofett und hohere Ester (z.B. C'4-Alkohol mit C,6-Fettsaure) oder Gemische dieser Stoffe.
- Salben, Pasten, Cremes und Gele können neben dem oder den Wirkstoffen die üblichen Trägerstoffe enthalten, z.B. tierische und pflanzliche Fette, Wachse, Paraffine, Starke, Tragant, Cellulosederivate, Polyäthylenglykole, Silicone, Bentonite, Kieselsäure, Talkum und Zinkoxid oder Gemische dieser Stoffe.
- Puder und Sprays können neben dem oder den Wirkstoffen die üblichen Trägerstoffe enthalten, z.B. Milchzucker, Talkum, Kieselsäure, Aluminiumhydroxid, Calciumsilikat und Polyamidpulver oder Gemische dieser Stoffe. Sprays können zusätzlich die üblichen Treibmittel z.B. Chlorfluorkohlenwasserstoffe enthalten.
- Lösungen und Emulsionen konnen neben dem oder den Wirkstoffen die üblichen Trägerstoffe wie Lösungsmittel, Losungsvermittler und Emulgatoren, z.B. Wasser, Aethylalkohol, Isopropylalkohol, Aethylcarbonat, Aethylacetat, Benzylalkohol, Benzylbenzoat, Propylenglykol, 1,3-Butylenglykol, Dimethylformamid, Oele, insbesondere Baumwollsaatol, Erdnußol, Maiskeimol, Olivenöl, Ricinusöl und Sesamol, Glycerin, Glycerinformal, Tetrahydrofurfurylalkohol, Polyathylenglykole und Fettsaureester des Sorbitans oder Gemische dieser Stoffe enthalten.
- Zur parenteralen applikation konnen die Lösungen und Emulsionen auch in steriler und blutisotonischer Form vorliegen.
- Suspensionen konnen neben dem oder den Wirkstoffen die üblichen Trägerstoffe wie flüssige Verdünnungsmittel, z.B. Wasser, Aethylalkohol, Propylenglykol, Suspendiermittel, z.B. athoxylierte Isostearylalkohole, Polyoxyäthylensorbit- und Sorbitanester, mikrokristalline Cellulose, Aluminiummetahydroxid, Bentonit, Agar-Agar und Tragant oder Gemische dieser Stoffe enthalten.
- Die genannten Formulierungsformen können auch Färbemittel, Konservierungsstoffe sowie geruchs- und geschmacksverbessernde Zusätze,.z.B. Pfefferminzöl und Eukalyptusöl und Süßmittel, z.B. Saccharin, enthalten.
- Die therapeutisch wirksamen Verbindungen sollen in den oben aufgeführten pharmazeutischen Zubereitungen vorzugsweise in einer Konzentration von etwa 0,1 bis 99,5, vorzugsweise von etwa 0,5 bis 95 Gewichtsprozent der Gesamtmischung vorhanden sein.
- Die oben aufgeführten pharmazeutischen Zubereitungen können außer den erfindungsgemäßen Wirkstoffen auch weitere pharmazeutische Wirkstoffe enthalten.
- Die Herstellung der oben aufgeführten pharmazeutischen Zubereitungen erfolgt in üblicher Weise nach bekannten Methoden, z.B. durch Mischen des oder der Wirkstoffe mit dem oder den Tragerstoffen.
- Zur vorliegenden Erfindung gehört auch die Verwendung der erfindungsgemaßen Wirkstoffe sowie von pharmazeutischen Zubereitungen, die einen oder mehrere erfindungsgemäße Wirkstoffe enthalten, in der Human- und Veterinarmedizin zur Verhutung, Besserung und/oder Heilung der oben angeführten Erkrankungen.
- Die Wirkstoffe oder die pharmazeutischen Zubereitungen können lokal, oral, parenteral, intraperitoneal und/oder rectal, vorzugsweise oral oder parenteral wie intravenös oder intramuskular appliziert werden.
- Im allgemeinen hat es sich sowohl in der Human- als auch in der Veterinärmedizin als vorteilhaft erwiesen, den oder die erfindungsgemäßen Wirkstoffe in Gesamtmengen von etwa 5 bis etwa 1000, vorzugsweise 20 bis 200 mg/kg Körpergewicht je 24 Stunden, gegebenenfalls in Form mehrerer Einzelgaben, zur Erzielung der gewünschten Ergebnisse zu verabreichen. Eine Einzelgabe enthält den oder die erfindungsgemäßen Wirkstoffe, vorzugsweise in Mengen von etwa 1 bis etwa 250, insbesondere 10 bis 100 mg/kg Körpergewicht. Es kann jedoch erforderlich sein, von den genannten Dosierungen abzuweichen, und zwar in Abhängigkeit von der Art und dem Körpergewicht des zu behandelnden Objekts, der Art und der Schwere der Erkrankung, der Art der Zubereitung und der Applikation des Arzneimittels sowie dem Zeitraum bzw. Intervall, innerhalb welchem die Verabreichung erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der oben genannten Menge Wirkstoff auszukommen, während in anderen Fallen die oben angeführte Wirkstoffmenge überschritten werden muß. Die Festlegung der jeweils erforderlichen optimalen Dosierung und Applikationsart der Wirkstoff kann durch jeden Fachmann aufgrund seines Fachwissens leicht erfolgen.
- Im Falle der Anwendung als Futterzusatzmittel können die neuen Verbindungen in den üblichen Konzentrationen und Zubereitungen zusammen mit dem Futter bzw. mit Futterzubereitungen oder mit dem Trinkwasser gegeben werden. Dadurch kann eine Infektion durch gramnegative oder grampositive Bakterien verhindert, gebessert und/oder geheilt werden und ebenso eine Förderung des Wachstums und eine Verbesserung der Verwertung des Futters erreicht werden.
- Die neuen Penicilline und Cephalosporine zeichnen sich durch starke antibakterielle Wirkungen, die in vivo und in vitro geprüft wurden, aus.
- Die erfindungsgemäßen Penicilline und Cephalosporine können zum Zwecke der Erweiterung des Wirkungsspektrums und um eine Wirkungssteigerung speziell bei β-lactamasebildenden Bakterien zu erzielen mit anderen antimikrobiellen Wirkstoffen z.B. mit Penicillinen, die besonders penicillinasefest sind, kombiniert werden. Eine solche Kombination wäre z.B. die mit Oxacillin oder Dicycloxacillin.
- Die erfindungsgemäßen Penicilline und Cephalosporine können zum Zweck der Erweiterung des Wirkungsspektrums und um eine Wirkungssteigerung zu erreichen auch mit Aminoglykosidantibiotica, wie z.B. Gentamicin, Kanamicin, Sisomicin, Amikacin oder Tobramicin, kombiniert werden.
- Die Wirksamkeit der erfindungsgemäßen ß-Lactamantibiotika kann durch die folgenden in vitro- und in vivo-Versuche beispielhaft demonstriert werden:
- Die Beispiele 2.3 und 3.1, welche als typische Vertreter der erfindungsgemäßen Verbindungen betrachtet werden können, wurden mit Müller-Hinton-Nährbrühe unter Zusatz von 0,1% Glucose auf einen Gehalt von 100 µg/ml verdünnt. In der Nährlösung befanden sich jeweils 1 × 105 bis 2 × 105 Bakterien pro Milliliter. Die Röhrchen mit diesem Ansatz wurden jeweils 24 Stunden bebrütet und anschließend wurde der Trübungsgrad bestimmt. Trübungsfreiheit gibt Wirkung an. Bei der Dosierung von 100 µg/ml waren die folgenden Bakterienkulturen trübungsfrei (sp. = species):
- Klebsiella pneumoniae; Enterobacter aerogenes sp.; Providencia; Serratia marcescens; E.coli BE; Salmonella sp.; Shigella sp.; Proteus, indolnegativ und indolpositiv; Pasteurella. pseudotuberculosis; Brucella sp.; Haemophilus influenzae; Bordetella bronchiseptica; Staphylococcus aureus 133; Neisseria catarrhalis sp.; Diplococcus pneumoniae sp.; Streptococcus pyogenes W.; Enterococcus sp.; Lactobacillus sp.; Corynebacterium diphteriae gravis; Corynebacterium pyogenes M; Clostridium tetani; Pseudomonas aeruginosa sp.
- Bei den NMR-Spektren der erfindungsgemäßen Verbindungen bedeuten die Bezeichnungen:


- Die Ausbeuteangaben in % bedeuten Ausbeuten in % der Theorie.
-

- IR(KBr): 3357, 1720, 1686, 1513, 1154, 747 cm-1.
- NMR(CD3OD): seudo-s 7.42(1 H), pseudo-s (6.35(2H), s 5.30(1 H), s 1.45(9H) ppm (8).
-

-

-

- IR (KBr): 3420, 1765, 1725, 1675, 1605, 1415, 1235 cm-1.
- NMR(CD30D):
- s 7.75(1 H), pseudo-s 7.64(1 H), pseudo-s 7.50(1 H), d 6.89(1 H), m um 6.5(3H), m um 5.65(2H), m um 5.0(teilweise verdeckt vom Signal der austauschbaren Protonen), s(breit) 3.93(4H), AB bei 3.4 und 3.6 (verdeckt vom Lösungsmittelsignal), s 2.02(3H) ppm (8).

-
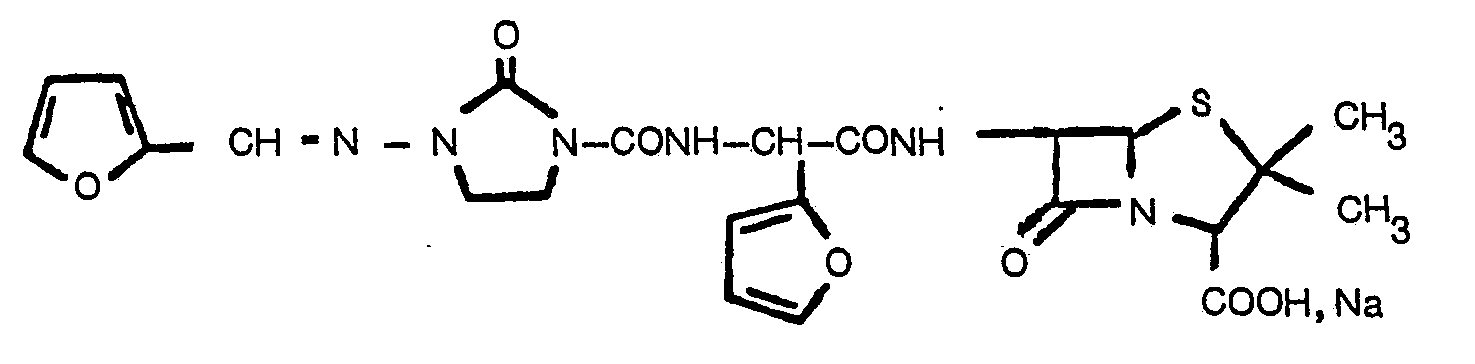
- IR(KBr): 1760, 1715, 1660, 1605 cm-1.
- NMR(CD30D):
- s 7.72(1 H), d 7.70(1 H), pseudo-s 7.50(1 H), d 6.85(1 H), m um 6.60(3H), s 5.55(1 H), AB 5.38 und 5.42(2H), s 4.12(1 H), s(breit) 3.80(4H), s 1.53(3H), s 1.48(3H) ppm (8).
-

- Ausbeute: 0,7 Gew.-Tle.
- β-Lactamgehalt: 85% (Substanz enthält ca. 6% β-Lactamoffenes Produkt)
- IR (Nujol; Carbonylbereich):
- 1760 (Schulter), 1750, 1715, 1655, 1595, 1520-10 cm-1. NMR (CD30D/D20):
- 7,7-6,9 m (5H. Thiophen, Furan, -CH=N=), 6,8 d (1 H, Furan), 6,5 m (1 H, Furan), 5,8 s (1 H, >N―CH―CO―), 5,5 pseudo-s (2H, 5.6-H), 4,2 s (1 H, 3-H), 3,9-3,6 s, breit (4H, CH2CH2), 1,55 s (3H) und 1,45 s (3H) ppm (δ).
- Nach dem NMR-Spektrum enthält die Substanz etwa 0,08 Moläquivalente Natrium-2- äthylhexanoat und etwa 3,2 Moläquivalente Wasser.
-
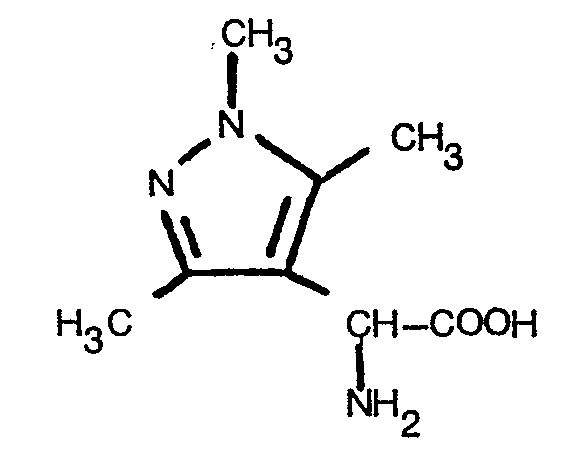
- Dann gibt man die Reaktionslösung in 80 Vol.Tle. eiskalte konzentrierte Salzsäure, spült mit Wasser nach und leitet 50-60 g HCI-Gas bei 0-5°C in die Lösung ein, die man anschließend über Nacht stehenlaßt (Abzug). Nach Verdünnung mit 100 Vol.Tln. Wasser, 2,5 Stdn. Rückflußkochen und Einengen wird der Rückstand zweimal mit Wasser in einer Porzellanschale (zur Entfernung von HCI) abgeraucht.
- Schließlich nimmt man in Aethanol auf, filtriert vom Ungelösten ab, engt zur Trockne ein, nimmt wieder in Wasser auf und verrührt mit ca. 50 g saurem Ionenaustauscher (Dowex der Fa. Serva, Heidelberg) (5 Stunden).
- Dann saugt man den beladenen Ionenaustauscher ab, wäscht gut mit Wasser nach und eluiert schließlich mit Ammonium-carbonatlösung. Diese Lösung wird zur Trockne eingeengt. Der Rückstand wird mit Aethanol gewaschen und aus den Mutterlaugen 7,4 Gew.Tle. (40%) der gewünschten Aminosäure erhalten.
- 60 MHz-NMR-Spektrum (D2O):
- *)Aufgrund des NMR-Spektrums und der guten Wirksamkeit muß man annehmen, daß die D-Form vorliegt. Da vom Racemat der a-Amino-thienylessigsäure ausgegangen wurde, muß auf der Stufe des a-Amino-thienylmethylpenicillins eine Diastereomerentrennung eingetreten sein.
- 2,05 ppm s (3H), 2,15 s (3H), 3,6 s (3H), 4,8 (Wasser), 4,9 s (1 H).
- Dünnschichtchromatographisch einheitlich Rf = 0.
- (Laufmittel: 200 ml n-Butylacetat, 36 ml n-Butanol, 100 ml Essigsäure (behandelt mit 150 ml Phosphatpuffer pH 6)).
-

- 3.52 Gew.-Tln. Natriumhydroxid
- 18 Vol.-Tln. t-Butanol
- 9 Vol.-Tln. Wasser
- 60 MH2-NMR-Spektrum (CDCl3):
- 1,4 ppm s (9H), 2,4 s (3H), 2,45 s (3H), 3,7 s (3H), 5,1 d (1H), 5,8 d (1H), 12,6 s (1H).
- Ausbeute: 15 Gew.-Tle. (63%) 2-(1,3,5-Trimethylpyrazol-4-yl)-2-tert.-butoxycarbonylaminoessigsäure.
-

- 5,44 Gew.-Tle. 7-Aminocephalosporansäure werden in 100 Vol.-Tln. 80%igem wäßrigen Tetrahydrofuran und 2 Gew.-Tin. Triäthylamin gelöst und auf -10°C gekühlt. Hierzu tropft man Lösung A bei -10°C. Es wird 1,5 Std. bei -10°C und noch weitere 2 Stdn., wobei man auf Raumtemperatur kommen läßt, nachgerührt.
- Die Reaktionslösung wird in 100 Vol.-Tle. Wasser gegeben, mit Essigester gewaschen, angesäuert (pH 1,8), extrahiert mit Essigester und diese Essigesterlösung mit gesättigter NaCI-Lösung gewaschen. Nach dem Trocknen über MgS04 wird eingeengt und der Rückstand im Hochvakuum getrocknet.
- Ausbeute: 7,5 Gew.-Tle. (70%) der oben bezeichneten Verbindung.
- Dünnschichtchromatogram einheitlich Rf -Wert 0,3 (Laufmittelgemisch wie in Beispiel 7.1)
- 100 MHz-NMR-Spektrum (CDCl3):
- 1,4 ppm s (9H), 2,1 s (3H) 2,3 m (6H), 3,4 a,b(2H), 3,7 m(3H), 4,95 a,b (2H), 5,05 d (1 H) (I=3Hz), 5,3 d (1 H) (I=3Hz), 5,8 d (1 H), 7,3 d (1H), 10,8 s (1H).
-

- Anschließend wird die Trifluoressigsäure weitgehend abdestilliert (Vorlage auf -70°C gekühlt, Hochvakuum) und der Rückstand in 10 Vol.-Tln. Methylenchlorid gelöst und 20 Min. gerührt. Dann gibt man 100 Vol.-Teile Äther/Ligroin 1:1 zu und rührt weitere 30 Min. nach, saugt den Neiderschlag ab und nimmt ihn in Wasser auf.
- Nach Überschichten mit Essigester/Äther stellt man mit Amberlite (flüssiger lonenaustauscher, Chlorid als Gegenion) auf pH 7, trennt die Phasen und wäscht die wäßrige Phase sehr gründlich mit Äther. Die wäßrige Phase wird gefriergetrocknet.
- Ausbeute: 3 Gew.-Tle. 7-[2-(1,3,5-Trimethylpyrazol-4-yl)-2-amino]-acetamidocephalosporan- säure.
- Dünnschichtchromatogram (Laufmittel siehe Beispiel 5.1) ein Substanzfleck am Startpunkt.
- IR(KBr) 3422, 2923, 1763, 1689, 1599, 1383, 1235, 1064, 1026, 739.
- 100 MHz-NMR-Spektrum (D2O):
- 2,1 ppm 2 s (Abstand 0,5 Hz) 3H (Verhältnis 1:1), 2,2 m (3H) 2,3 m (3H), 3,4u 3,6 a,b (2H), 3,7s (3H), 4,7 s (4 ausgetauschte H + 1 x H20), 5,15 m (3H), 5,7 m (1H).
-

- Aufarbeitung wie in Beispiel 2.3. beschrieben.
- Ausbeute: 1,2 Gew.Tie. (78%) der oben bezeichneten Verbindung.
- IR-Spektrum (Nujol) ß-Lactambande bei 1765.
- Dünnschichtchromatogramm (Laufmittel wie Beispiel 5.1.) Rf-Wert: 0,2 (einheitlich).
- 100 MHz-NMR-Spektrum (CD3OD+D2O):
- 2,15 ppm s (3H), 2,3 m (3H), 2,45 m (3H), 3,4 a,b (verdeckt durch Lösungsmittelsignal und folgende Signale) (2H), 3,8s (3H), 4,0 s (breit) (4H), 4,9-5,2 m (teilweise verdeckt durch ausgetauschte Wasserstoffe) (4H), 5,5 d (J=0,5Hz) (1 H), 6,65 q (1 H), 7,0 d (1 H), 7,8 d (1 H), 7,85 s (1 H).
-

- Die Reaktionslösung wird dann in eine 10%-ige Ammoniumchlorid-Lösung gegeben und hält den pH-Wert unter weiterer Zugabe von verdünnter Salzsäure bei 7. Das Tetrahydrofuran wird im Vakuum abdestilliert, und die wäßrige Phase mit Essigester gewaschen. Dann gibt man 10 Vol.Tle. Aceton zu und stellt den pH-Wert unter Rühren langsam mit verdünnter Salzsäure auf 4. Die Penicillansäure kristallisiert langsam aus. Den Niederschlag saugt man ab und löst ihn in Wasser unter Zugabe von 1 N Natronlauge bei pH-Wert 7. Die Lösung wird schließlich lyophilisiert.
- Man erhält 1,8 Gew.Tle. (86%) der oben bezeichneten Verbindung.
- Dünnschichtchromatogramm: Rf-Wert 0,6 einheitlich.
- IR-Spektrum (KBr):
- 3436, 1762, 1726, 1674, 1604, 1529, 1476, 1413, 1270, 1235, 1136, 740. 100 MHz-NMR-Spektrum (CD30D):
- 1,3 ppm s (3H), 1,5 s (3H), 3,6 s (3H), 4,0 s (breit) (4H), 4,3 s (1 H), 5,65 s (1 H), 6,0 d (1 H), 6,65 q (1 H), 7,0 d (1 H), 7,1-7,5 m (5H), 7,75 d (1 H), 7,8 s (1 H).
-

- NMR (60 MHz; 8 in CCI,) 7,05 (s; Thiazolin 5-H)
-

- Ausbeute 4,15 g
- IR(Nujol); Carboxylbereich 1730; 1710, 1540 cm-1.
- Schmelzpunkt: ab etwa 130°C Zers.
-

- IR(Nujol; Carbonylbereich) 1715; 1600; 1545 cm-'.
-

- Ausbeute 3,8 g
- Schmelzpunkt 226°C
- NMR (60 MHz; in d6-DMSO-CD3OD) 7,55-7,8 (s breit; Furan 5-H; -N=CH-) 7,05 (s; Thiazolin 5-H) 6,75 (d; Furan 3-H) 6,5 (m; Furan 4-H) 5,4 (s; CH-N) 3,75 (s breit; ―CH2―CH2) 1,45 (s; -C(CH3)3) ppm.
-

- 1,0 g der nach Beispiel 7.4 hergestellten Substanz wird in 30 ml Dichlormethan mit 0,29 ml Triäthylamin versetzt, die Lösung auf -40°C abgekühlt, mit 0,02 ml 3-Dimethylaminopropanol und 0,26 ml Chlorameisensäureäthylester versetzt, 60 Min. bei der gleichen Temperatur gerührt, weitere 0,13 ml Chlormeisensäureester und 0,15 ml Triäthylamin zugegeben und dieses nach 30 Min. wiederholt.
- Zur im Eisbad gerührten Suspension von 0,77 g 7-Amino-cephalosporansäure in 20 ml Dichlormethan gibt man 2,0 ml Triäthylamin und 20 ml auf -10°C vorgekühltes Aceton. Die Mischung wird dann auf -20°C gekühlt.
- Mischung A und B werden vereinigt und ohne Kältebad gerührt. Nach 2 Stunden wird mit Wasser verdünnt, derpH auf 7,5 gebracht, zweimal mit Essigester ausgeschüttelt und die wäßrige Phase auf pH 2 angesäuert. Es wird dreimal mit Essigester ausgeschüttelt, die vereinigten und getrockneten organischen Phasen im Vakuum eingedampft und der Rückstand im Exsiccator über P4011 getrocknet. Ausbeute 1,05 g
-

- Ausbeute 0,6 g
- IR-Spektrum(Carbonylbereich) 1740, 1715, 1660 und 1520-1500 cm-1 (Nujol).

in Gegenwart eines Lösungsmittels und gegebenenfalls eines Säurebindemittels bei Temperaturen von etwa -20°C bis etwa +50°C umsetzt und die erhaltenen ß-Lactam-Antibiotica gegebenenfalls in ihre pharmazeutisch verwendbaren Salze oder Ester überführt oder aus den erhaltenen Salzen gewünschtenfalls die freien Säuren hergestellt.
sowie die Natriumsalze dieser Verbindungen.
wird bei 0°C mit 20,3 Gew.-Tln. Di-t-butyl-dicarbonat langsam versetzt, noch weitere 18 Vol.-Tle. t-Butanol hinzugefügt und 14 Stunden bei 25°C, dann 12 Stunden bei 50°C nachgerührt. Man verdünnt mit 50 Vol.Tln. Wasser, wäscht je 3mal mit n-Pentan und Äther und säuert auf pH 2,4 an. Nach Extraktion mit Essigsäureäthylester (4 ml), Trocknen über Na2SO4 und Einengen wird im Hochvakuum getrocknet.
Claims (4)




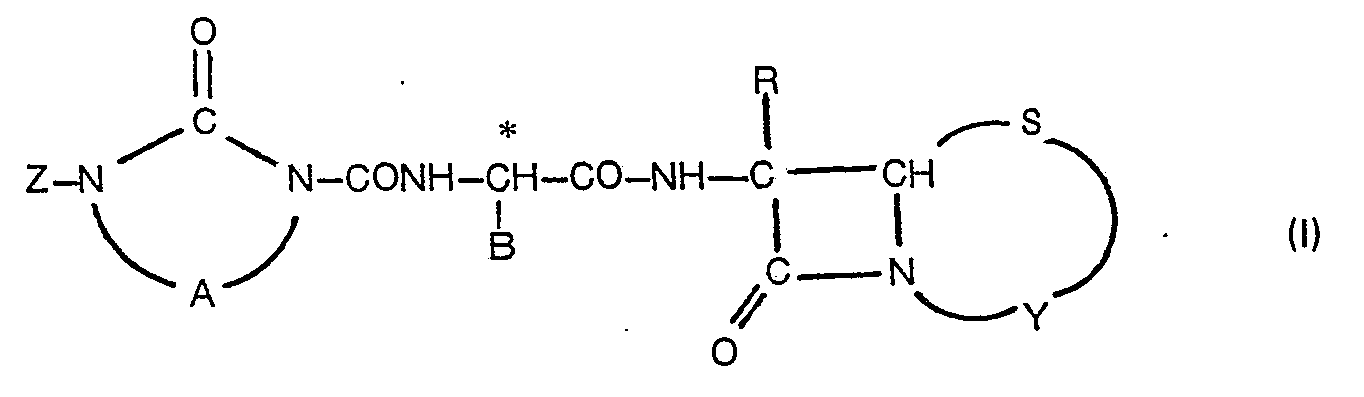


W für Halogen, Azid oder eine andere nukleofuge Abgangsgruppe steht, in Gegenwart eines Lösungsmittels und gegebenenfalls eines Säurebindemittels bei Temperaturen von etwa -20 bis etwa +50°C umsetzt und die erhaltenen β-Lactam-Antibiotika gegebenenfalls in ihre Salze oder pharmazeutisch verwendbaren Ester überführt oder aus den erhaltenen Salzen gewünschtenfalls die freien Säuren herstellt.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE2732283 | 1977-07-16 | ||
| DE19772732283 DE2732283A1 (de) | 1977-07-16 | 1977-07-16 | Beta-lactam-verbindungen |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0000392A2 EP0000392A2 (de) | 1979-01-24 |
| EP0000392A3 EP0000392A3 (en) | 1979-08-22 |
| EP0000392B1 true EP0000392B1 (de) | 1982-04-28 |
Family
ID=6014140
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78100352A Expired EP0000392B1 (de) | 1977-07-16 | 1978-07-11 | Cephalosporin und Penicillin-derivate, Verfahren zu deren Herstellung, und deren pharmazeutische Präparate |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US4215118A (de) |
| EP (1) | EP0000392B1 (de) |
| JP (1) | JPS5439091A (de) |
| AT (1) | AT365193B (de) |
| AU (1) | AU525027B2 (de) |
| CA (1) | CA1122593A (de) |
| DE (2) | DE2732283A1 (de) |
| DK (1) | DK318278A (de) |
| ES (1) | ES471770A1 (de) |
| IL (1) | IL55130A (de) |
| IT (1) | IT1097816B (de) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA813610B (en) * | 1980-06-06 | 1982-06-30 | Beecham Group Plc | Penicillin derivatives,processes for their preparation and compositions containing them |
| ZA813996B (en) * | 1980-06-26 | 1982-06-30 | Beecham Group Plc | Penicillin derivatives |
| US4436904A (en) * | 1981-02-14 | 1984-03-13 | Kanto Ishi Pharmaceutical Co., Ltd. | Cephalosporins |
| EP0066406A1 (de) * | 1981-05-28 | 1982-12-08 | Beecham Group Plc | Beta-Lactam-Antibiotika |
| DE3137038A1 (de) | 1981-09-17 | 1983-03-24 | Bayer Ag, 5090 Leverkusen | Ss-lactam antibiotika, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel |
| DE3215085A1 (de) * | 1982-04-22 | 1983-10-27 | Bayer Ag, 5090 Leverkusen | Ss-lactam-antibiotika, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel |
| DE3431273A1 (de) * | 1984-08-25 | 1986-03-06 | Bayer Ag, 5090 Leverkusen | Kristallines natriumsalz der d-6-((alpha)-(2-oxo-3-furfuryl-iden-amino-imidazolidin-1-yl)-carbonylamino)-thienyl-2-acetamido)-penicillansaeure, verfahren zur herstellung und seine verwendung in arzneimitteln |
| GB8519264D0 (en) * | 1985-07-31 | 1985-09-04 | Baldwin J E | Chemical compounds |
| GB8713061D0 (en) * | 1987-06-04 | 1987-07-08 | Beecham Group Plc | Compounds |
| AU782938B2 (en) * | 2001-08-28 | 2005-09-08 | Boss Rubber Technologies Pty Ltd | Resilient product |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687949A (en) * | 1969-01-21 | 1972-08-29 | Bristol Myers Co | Synthetic cephalosporins |
| US3959258A (en) * | 1970-05-25 | 1976-05-25 | Bayer Aktiengesellschaft | Ureidoacetamido-penicillins |
| US3974142A (en) * | 1971-10-23 | 1976-08-10 | Bayer Aktiengesellschaft | Penicillins |
| US4086340A (en) * | 1974-02-18 | 1978-04-25 | Bayer Aktiengesellschaft | Cephalosporins and their production |
| US4087424A (en) * | 1974-05-09 | 1978-05-02 | Toyama Chemical Co., Ltd. | Novel penicillins and cephalosporins and process for producing the same |
| GB1486349A (en) * | 1974-11-28 | 1977-09-21 | Bayer Ag | Beta-lactam antibiotics process for their preparation and their use as medicaments |
| JPS5852996B2 (ja) * | 1975-03-25 | 1983-11-26 | バイエル・アクチエンゲゼルシヤフト | β−ラクタム及び抗バクテリア剤 |
| DE2633317A1 (de) * | 1976-07-23 | 1978-01-26 | Bayer Ag | Beta-lactamantibiotika, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel |
-
1977
- 1977-07-16 DE DE19772732283 patent/DE2732283A1/de not_active Withdrawn
-
1978
- 1978-07-10 US US05/923,518 patent/US4215118A/en not_active Expired - Lifetime
- 1978-07-11 EP EP78100352A patent/EP0000392B1/de not_active Expired
- 1978-07-11 DE DE7878100352T patent/DE2861766D1/de not_active Expired
- 1978-07-13 IL IL55130A patent/IL55130A/xx unknown
- 1978-07-13 AU AU38018/78A patent/AU525027B2/en not_active Expired
- 1978-07-14 CA CA307,367A patent/CA1122593A/en not_active Expired
- 1978-07-14 DK DK783182A patent/DK318278A/da not_active Application Discontinuation
- 1978-07-14 ES ES471770A patent/ES471770A1/es not_active Expired
- 1978-07-14 AT AT0510878A patent/AT365193B/de not_active IP Right Cessation
- 1978-07-14 IT IT25714/78A patent/IT1097816B/it active
- 1978-07-14 JP JP8525378A patent/JPS5439091A/ja active Pending
-
1979
- 1979-06-04 US US06/045,474 patent/US4338434A/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| IT7825714A0 (it) | 1978-07-14 |
| ATA510878A (de) | 1981-05-15 |
| DE2732283A1 (de) | 1979-01-25 |
| AU525027B2 (en) | 1982-10-14 |
| AT365193B (de) | 1981-12-28 |
| DK318278A (da) | 1979-01-17 |
| EP0000392A3 (en) | 1979-08-22 |
| EP0000392A2 (de) | 1979-01-24 |
| CA1122593A (en) | 1982-04-27 |
| IT1097816B (it) | 1985-08-31 |
| US4338434A (en) | 1982-07-06 |
| DE2861766D1 (en) | 1982-06-09 |
| JPS5439091A (en) | 1979-03-24 |
| ES471770A1 (es) | 1979-02-01 |
| IL55130A0 (en) | 1978-09-29 |
| AU3801878A (en) | 1980-01-17 |
| IL55130A (en) | 1982-01-31 |
| US4215118A (en) | 1980-07-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0195947A2 (de) | Beta-Lactamantibiotika, Verfahren zur Herstellung und ihre Verwendung als und in Arzneimitteln | |
| EP0000392B1 (de) | Cephalosporin und Penicillin-derivate, Verfahren zu deren Herstellung, und deren pharmazeutische Präparate | |
| EP0004956B1 (de) | Beta-Lactamantibiotika, Verfahren zu ihrer Herstellung, Futterzusatzmittel, Arzneimittel sowie Verfahren zu deren Herstellung | |
| DE2407715C2 (de) | Cephalosporine, Verfahren zu ihrer Herstellung sowie Arzneimittel | |
| CH623326A5 (de) | ||
| EP0000380A1 (de) | Penicilline, Verfahren zu ihrer Herstellung und ihre Verwendung | |
| CH633802A5 (de) | Verfahren zur herstellung neuer beta-lactam-antibiotica. | |
| EP0197294A2 (de) | Beta-Lactam-Antibiotika, Verfahren zur Herstellung und ihre Verwendung als Arzneimittel | |
| EP0092722B1 (de) | Beta-Lactam-Antibiotika, Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel | |
| DE2354219A1 (de) | Beta-lactam-antibiotica, ein verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| EP0003992A2 (de) | Beta-lactam Verbindungen, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| EP0035161B1 (de) | Cephalosporine, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| DE2440268A1 (de) | Beta-lactamantibiotika, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| EP0075169A1 (de) | Beta-Lactam Antibiotica, Verfahren zu ihrer Herstellung sowie ihre Verwendung als Arzneimittel | |
| DE2720579A1 (de) | Beta-lactam-verbindungen, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| EP0098433B1 (de) | Beta-Lactamantibiotika, Verfahren zu deren Herstellung sowie sie enthaltende Mittel | |
| DE2456307A1 (de) | Beta-lactam-antibiotica, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| DE2528079A1 (de) | Penicilline, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| DE2720580A1 (de) | Beta-lactam-verbindungen, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| DE2658905A1 (de) | Beta-lactam-antibiotica, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| DE2727586A1 (de) | Methoxy-beta-lactam-antibiotica, verfahren zu ihrer herstellung sowie ihre verwendung | |
| DE2658718A1 (de) | Methoxy-beta-lactam-antibiotica, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| DE2548247A1 (de) | Cephalosporine, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| DE2658906A1 (de) | Beta-lactam-antibiotika, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel | |
| EP0000393A1 (de) | Beta-Lactam-Verbindungen, Verfahren zu ihrer Herstellung sowie ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB NL SE |
|
| 17P | Request for examination filed | ||
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB NL SE |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB NL SE |
|
| REF | Corresponds to: |
Ref document number: 2861766 Country of ref document: DE Date of ref document: 19820609 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19890617 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19890630 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19890717 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 19890719 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19890726 Year of fee payment: 12 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 19890731 Year of fee payment: 12 Ref country code: NL Payment date: 19890731 Year of fee payment: 12 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19900711 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19900712 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19900731 Ref country code: BE Effective date: 19900731 |
|
| BERE | Be: lapsed |
Owner name: BAYER A.G. Effective date: 19900731 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19910201 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19910329 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19910403 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| EUG | Se: european patent has lapsed |
Ref document number: 78100352.0 Effective date: 19910402 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |