DE69822909T2 - Verfahren zur Herstellung von Deoxypyridinolin - Google Patents
Verfahren zur Herstellung von Deoxypyridinolin Download PDFInfo
- Publication number
- DE69822909T2 DE69822909T2 DE69822909T DE69822909T DE69822909T2 DE 69822909 T2 DE69822909 T2 DE 69822909T2 DE 69822909 T DE69822909 T DE 69822909T DE 69822909 T DE69822909 T DE 69822909T DE 69822909 T2 DE69822909 T2 DE 69822909T2
- Authority
- DE
- Germany
- Prior art keywords
- give
- protected
- deoxypyridinoline
- dmso
- oxidizing agent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- ZAHDXEIQWWLQQL-IHRRRGAJSA-N Deoxypyridinoline Chemical compound OC(=O)[C@@H](N)CCCC[N+]1=CC(O)=C(C[C@H](N)C([O-])=O)C(CC[C@H](N)C(O)=O)=C1 ZAHDXEIQWWLQQL-IHRRRGAJSA-N 0.000 title claims description 33
- 238000000034 method Methods 0.000 title claims description 20
- 238000002360 preparation method Methods 0.000 title claims description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 47
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 34
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 30
- 239000002904 solvent Substances 0.000 claims description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 16
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 12
- 239000007800 oxidant agent Substances 0.000 claims description 11
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 10
- 238000006243 chemical reaction Methods 0.000 claims description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 9
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 claims description 9
- 150000001875 compounds Chemical class 0.000 claims description 9
- 239000004472 Lysine Substances 0.000 claims description 8
- -1 5-benzyloxycarbonylamino-5-t-butoxycarbonylpentyl Chemical group 0.000 claims description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 7
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 6
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 5
- XSEHBPXXAIDEFE-UHFFFAOYSA-N 1,2-dihydropyridin-3-ol Chemical compound [O-]C1=CC=C[NH2+]C1 XSEHBPXXAIDEFE-UHFFFAOYSA-N 0.000 claims description 4
- 150000002009 diols Chemical class 0.000 claims description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 claims description 4
- LRQKBLKVPFOOQJ-YFKPBYRVSA-M L-2-aminohexanoate Chemical compound CCCC[C@H](N)C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-M 0.000 claims description 3
- BDGDWWGTAFXEEW-UHFFFAOYSA-N methylsulfinylmethane;oxalyl dichloride Chemical compound CS(C)=O.ClC(=O)C(Cl)=O BDGDWWGTAFXEEW-UHFFFAOYSA-N 0.000 claims description 3
- 125000006239 protecting group Chemical group 0.000 claims description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 3
- GRFNBEZIAWKNCO-UHFFFAOYSA-N 3-pyridinol Chemical group OC1=CC=CN=C1 GRFNBEZIAWKNCO-UHFFFAOYSA-N 0.000 claims description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical class C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 2
- MHCFAGZWMAWTNR-UHFFFAOYSA-M lithium perchlorate Chemical compound [Li+].[O-]Cl(=O)(=O)=O MHCFAGZWMAWTNR-UHFFFAOYSA-M 0.000 claims description 2
- 229910001486 lithium perchlorate Inorganic materials 0.000 claims description 2
- 239000000543 intermediate Substances 0.000 claims 3
- 239000003054 catalyst Substances 0.000 claims 1
- 238000002955 isolation Methods 0.000 claims 1
- AYGNOFSNYMGKMQ-UHFFFAOYSA-O tert-butyl 6-[3-[4-butoxy-4-oxo-3-(phenylmethoxycarbonylamino)butyl]-5-hydroxy-4-[3-[(2-methylpropan-2-yl)oxy]-3-oxo-2-(phenylmethoxycarbonylamino)propyl]pyridin-1-ium-1-yl]-2-(phenylmethoxycarbonylamino)hexanoate Chemical class C=1C=CC=CC=1COC(=O)NC(C(=O)OCCCC)CCC(C(=C(O)C=1)CC(NC(=O)OCC=2C=CC=CC=2)C(=O)OC(C)(C)C)=C[N+]=1CCCCC(C(=O)OC(C)(C)C)NC(=O)OCC1=CC=CC=C1 AYGNOFSNYMGKMQ-UHFFFAOYSA-O 0.000 claims 1
- SZHOJFHSIKHZHA-UHFFFAOYSA-N tridecanoic acid Chemical compound CCCCCCCCCCCCC(O)=O SZHOJFHSIKHZHA-UHFFFAOYSA-N 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 26
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 14
- 239000000203 mixture Substances 0.000 description 13
- 235000019439 ethyl acetate Nutrition 0.000 description 10
- 239000000047 product Substances 0.000 description 8
- LCYXYLLJXMAEMT-SAXRGWBVSA-N Pyridinoline Chemical compound OC(=O)[C@@H](N)CCC1=C[N+](C[C@H](O)CC[C@H](N)C([O-])=O)=CC(O)=C1C[C@H](N)C(O)=O LCYXYLLJXMAEMT-SAXRGWBVSA-N 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 5
- XQFGVGNRDPFKFJ-UHFFFAOYSA-N 1,2,3,5,6,7-hexahydropyrrolo[1,2-b]pyridazine Chemical compound N1CCC=C2CCCN21 XQFGVGNRDPFKFJ-UHFFFAOYSA-N 0.000 description 4
- 102000008186 Collagen Human genes 0.000 description 4
- 108010035532 Collagen Proteins 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229920001436 collagen Polymers 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 150000002978 peroxides Chemical class 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 229910004373 HOAc Inorganic materials 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 150000002118 epoxides Chemical class 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 206010065687 Bone loss Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 229960004132 diethyl ether Drugs 0.000 description 2
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical group NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 2
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- QWPPLZZDMBQLCG-UHFFFAOYSA-O tert-butyl 6-[3-hydroxy-5-[4-[(2-methylpropan-2-yl)oxy]-4-oxo-3-(phenylmethoxycarbonylamino)butyl]-4-[3-[(2-methylpropan-2-yl)oxy]-3-oxo-2-(phenylmethoxycarbonylamino)propyl]pyridin-1-ium-1-yl]-2-(phenylmethoxycarbonylamino)hexanoate Chemical class C=1C=CC=CC=1COC(=O)NC(C(=O)OC(C)(C)C)CCCC[N+](C=C1CCC(NC(=O)OCC=2C=CC=CC=2)C(=O)OC(C)(C)C)=CC(O)=C1CC(C(=O)OC(C)(C)C)NC(=O)OCC1=CC=CC=C1 QWPPLZZDMBQLCG-UHFFFAOYSA-O 0.000 description 2
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 0 *CC(*)Cc(c(CCC([*+])*1CC1)c[n+](CCCCC(*)**)c1)c1O Chemical compound *CC(*)Cc(c(CCC([*+])*1CC1)c[n+](CCCCC(*)**)c1)c1O 0.000 description 1
- KPZGRMZPZLOPBS-UHFFFAOYSA-N 1,3-dichloro-2,2-bis(chloromethyl)propane Chemical class ClCC(CCl)(CCl)CCl KPZGRMZPZLOPBS-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- LCWXJXMHJVIJFK-UHFFFAOYSA-N Hydroxylysine Natural products NCC(O)CC(N)CC(O)=O LCWXJXMHJVIJFK-UHFFFAOYSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 229910013684 LiClO 4 Inorganic materials 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- LSTJLLHJASXKIV-UHFFFAOYSA-N amino hexanoate Chemical compound CCCCCC(=O)ON LSTJLLHJASXKIV-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000005868 electrolysis reaction Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- QJHBJHUKURJDLG-UHFFFAOYSA-N hydroxy-L-lysine Natural products NCCCCC(NO)C(O)=O QJHBJHUKURJDLG-UHFFFAOYSA-N 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- GAPYKZAARZMMGP-UHFFFAOYSA-N pyridin-1-ium;acetate Chemical compound CC(O)=O.C1=CC=NC=C1 GAPYKZAARZMMGP-UHFFFAOYSA-N 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 239000003351 stiffener Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000011345 viscous material Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/65—One oxygen atom attached in position 3 or 5
- C07D213/66—One oxygen atom attached in position 3 or 5 having in position 3 an oxygen atom and in each of the positions 4 and 5 a carbon atom bound to an oxygen, sulphur, or nitrogen atom, e.g. pyridoxal
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
- HINTERGRUND DER ERFINDUNG
- Kollagen ist in verschiedenen Formen in allen Geweben vorhanden. Es ist allgemein anerkannt, dass Kollagen die Form von Aminosäure-Ketten aufweist, die durch Pyridinium-Vernetzungen vernetzt sind. Die Pyridinium-Vernetzungen werden aus drei Hydroxylysin-Resten gebildet, von denen zwei aus den endständigen (nicht-helikalen) Peptiden des Kollagen-Moleküls, welche enzymatisch in Aldehyde vor der Reaktion überführt werden, und aus einem dritten Hydroxylysin stammen, das im helikalen Teil eines benachbarten Kollagen-Moleküls vorliegt. Zwei Pyridinium-Vernetzungen, nämlich Pyridinolin (PYD) und Deoxypyridinolin (DPD), sind identifiziert worden. Es sind in der Literatur Verfahrensweisen zur Messung von Pyridinolin in Urin durch Enzym-markiertes anti-PYD zur Bildung eines Pyridinolin-Enzym-markierten Komplexes beschrieben worden, der mit einem Enzym-gebundenen Imunosorbens-Assay nachgewiesen werden kann. Während sich die PYD-Analyse als Mittel zur Untersuchung von Osteoporose und rheumatoider Arthritis eignet, kann dessen Vorliegen und Vorhandensein in verbindendem Gewebe sowie im Knochen verfälschte Ergebnisse für die Diagnose von Osteoporose oder von Knochenabbau verursachen. Demzufolge haben Immunoassayverfahren für Deoxypyridinolin (DPD), das nur im Knochen vorgefunden wird, Vorrang gegenüber denjenigen für PYD zum frühen Nachweis eines Knochenabbaus erhalten.
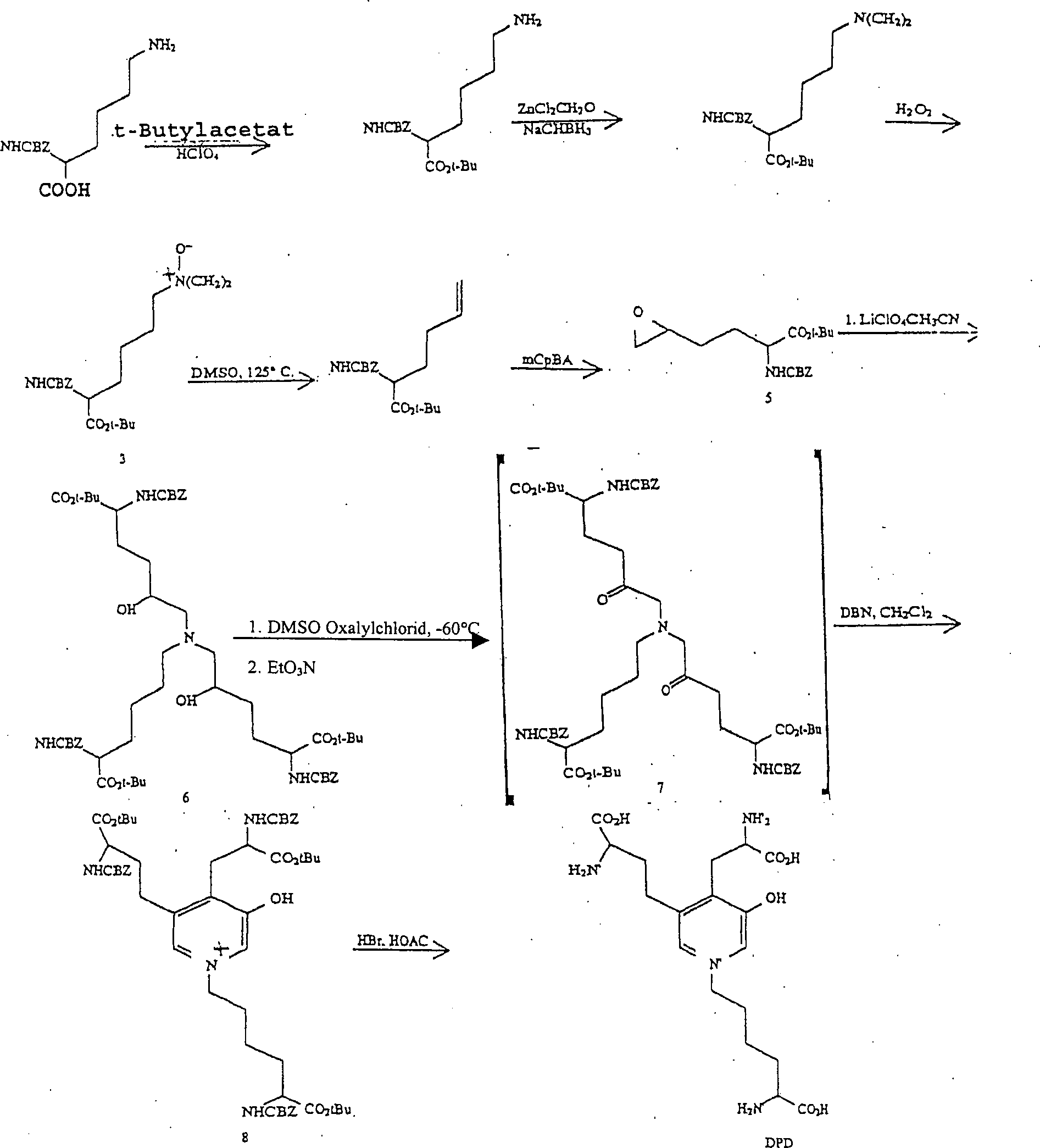
- Ein Testverfahren für DPD kann durchgeführt werden, wobei man eine fluide Testprobe, z. B. Urin, in Kontakt mit einem für DPD spezifischen markierten Antikörper bringt. Ein besonders bequemes Verfahren zur DPD-Analyse beinhaltet die Anwendung eines Teststreifens des in
1 dargestellten Typs. Was1 betrifft, bindet der Steifen10 , der einen markierten anti-DPD-Antikörper-Komplex aufweist (in typischer Weise mit Gold-Sol als das Markierungsmaterial, um einen Gold-Sol-DPD-Antikörper-Komplex zu ergeben), mit DPD in der fluiden Testprobe, die auf die Aufbringzone12 des Streifens10 aufgetragen wurde, worauf der Komplex durch die erste Einfangzone14 und die zweite Einfangzone16 wandert. In der ersten Einfangzone14 liegt immobilisiertes DPD vor, das ungebundenes, markiertes anti-DPD einfängt. Der markierte Antikörper, der nicht in der ersten Einfangzone eingefangen wurde, weil er mit DPD in der fluiden Testprobe verbunden wurde, wird in der zweiten Einfangzone16 durch anti-DPD-Antikörper eingefangen, die in dieser Zone immobilisiert sind. Die DPD-Konzentration in der Testprobe kann durch spektrofotometrische Messung der Menge an markiertem DPD, die in der ersten Einfangzone14 eingefangen wurde, oder noch genauer durch Anwendung einer algorithmischen Behandlung von Reflexionsmessungen aus beiden Zonen14 und16 ermittelt und bestimmt werden. - In der ersten Einfangzone
12 wird immobilisiertes DPD benötigt, mit welchem das markierte anti-DPD, das nicht mit DPD in der fluiden Probe reagiert hat, verbunden werden kann, um in dieser Einfangzone immobilisiert zu werden. Diese Sorte eines verfügbaren Testsystems bedarf der Anwendung beachtlicher Mengen an DPD, das, bei Erhalt aus tierischem Knochen, ziemlich teuer ist. Der mit der Bereitstellung von natürlichem DPD verbundene Aufwand hat zu Versuchen Anlass gegeben, dieses Material zu synthetisieren und dadurch die Kosten diagnostischer Teststreifen zu senken, in denen DPD in DPD-Nachweissystemen zur Anwendung gelangt. - Ein Verfahren zur Synthese von DPD sowie von PYD und Derivaten davon ist in der veröffentlichten
EP 0 556 152 A1 offenbart. In diesem Verfahren wird eine Verbindung der Formel:mit einer R-Gruppe zur Reaktion gebracht, die als X5-CH2-CHX1-Z-R3 definiert ist, worin die Gruppen R, X, Y und Z ausgewählt sind, um DPD, PYD oder verschiedene Derivate davon zu ergeben. In jedem Fall wird der Pyridin-Ring vor der Zufügung der Seitenkette zum Stickstoffatom gebildet. Im Verfahren gemäß der vorliegenden Erfindung zur Herstellung des DPD sind weniger Stufen enthalten, weil es nicht notwendig ist, X5-CH2-CHX1-Z-R3 getrennt herzustellen, wodurch ein weniger Labor-intensives Verfahren angegeben und zur Verfügung gestellt wird.
- BESCHREIBUNG DER ERFINDUNG
- Die Synthese von DPD, welche durch die vorliegende Erfindung in Betracht gezogen und durchgeführt wird, beinhaltet die Stufen, in denen man:
- a) ein Nα-geschütztes-O-geschütztes Lysin (Verbindung 1, Schema 1) mit mindestens 2 Äquivalenten des 5,6-Epoxy-2-N-geschützten-O-geschützten(2S)-2-aminohexanoats (Verbindung 5, Schema 1) in einem geeigneten Lösungsmittel zur Reaktion bringt, um das entsprechende Aminodiol zu erzeugen (Verbindung 6, Schema 1). Geeignete Lösungsmittel schließen Acetonitril, Methanol und Wasser/Alkohol ein. Im Schema 1 ist Lithiumperchlorat als ein Zugabestoff angegeben, welcher zur Katalyse der Reaktion vorgesehen ist. 2 Äquivalente des Epoxids pro Äquivalent des Aminohexanoats werden benötigt, weil eine Dialkylierung im Verfahren durchgeführt werden muss.
- b) Das Aminodiol 6 wird dann mit einem geeigneten Oxidiermittel, wie Oxalylchlorid-DMSO, in einem geeigneten Lösungsmittel wie Methylenchlorid oxidiert, um das entsprechende Aminodiketon zu ergeben (Verbindung 7, Schema 1). Geeignete oxidierende Mittel, die sich von Oxalylchlordi-DMSO unterscheiden, schließen Thionylchlorid-DMSO, Trifluoressigsäureanhydrid-DMSO und Essigsäureanhydrid-DMSO ein. Geeignete Lösungsmittel, die sich von Methylenchlorid unterscheiden, schließen Diethylether, Tetrahydrofuran und Chloroform ein.
- c) Das Aminodiketon von Stufe (b) wird mit einer Base wie Natriumhydroxid in Alkohol oder einem tertiären Amin, z. B. mit Triethylamin, weiter umgesetzt, um das 3-Hydroxydihydropyridin zu erzeugen, das mit einem oxidierenden Mittel wie der Base (DBN) in der Gegenwart von O2 in einem geeigneten Lösungsmittel wie Methylenchlorid zusammengebracht wird, um den 3-Hydroxypyridin-Ring zu bilden, um das Pyridinium-Produkt 8 in Schema 1 zu ergeben. Weitere geeignete oxidierende Mittel schließen Dicyanodichlorchinon (DDQ), Brom oder MnO2, ein, wobei die Reaktion auch in weiteren Lösungsmitteln, wie Methylenchlorid, Essigsäure oder Methanol, durchgeführt werden kann. Aus dem Produkt 8 werden die Schutzgruppen abgespalten, um Deoxypyridinolin zu ergeben:
- Schema 1
- Die vorliegende Erfindung wird nun durch das folgende Beispiel noch weiter erläutert.
- Beispiel
- A. Herstellung von Nα-CNBZ-O-t-butyl-L-lysin (1)
- Eine Mischung aus 25 g Nα-CBZ-L-lysin von Sigma, worin CBZ Benzyloxycarbonyl bedeutet, aus 250 mL t-Butylacetat und 14 mL 70%-iger Perchlorsäure wurde gründlich durchmischt, bis alle Feststoffe gelöst waren. Nach Rühren über Nacht wurden 375 mL Ethylacetat und dann 250 mL Wasser zugegeben. Der pH-Wert der wässrigen Schicht wurde auf 5,5 mit 20%-iger NaOH angehoben, worauf die organische Phase abgetrennt wurde. Die wässrige Phase wurde mit der Ethylacetat-Lösung extrahiert, und der pH-Wert wurde auf 10,5 erneut angehoben, worauf das Ganze mit 200 mL frischem Ethylacetat erneut extrahiert wurde. Die organischen Extrakte wurden vereinigt, mit gesättigter Kochsalzlösung gewaschen, über Na2SO4 getrocknet und eingeengt. Der Rückstand wurde über Nacht unter Hoch-Vakuum getrocknet, um dann 23,2 g eines farblosen, viskosen Materials zu ergeben.
1H-NMR (60 MHz, CDCl3) δ: 7,35 (s, 5H), 5,1 (s, 2H), 4,2 (m, 1H), 3,1 (m, 2H), 1,4 (s, 9H), 2,1–1,4 (m, 4H) - B. Nε-Dimethylamino-Nα-CBZ-O-t-butyl-L-lysin (2)
- Eine Mischung aus 3,7 g (27,2 mMol) ZnCl2, 3,2 g (53 mMol) NaCNBH3 und aus 60 mL Methanol wurde hergestellt, und es wurde eine Lösung von 18 g (51,7 mMol) des Amins (1) in 180 mL Methanol zugegeben. Nach Kühlung auf 10 bis 15°C wurden 12,4 mL 37%-iger Formaldehyd (165 mMol) über eine Dauer von 2 bis 3 min zugetropft. Die Reaktion wurde 1 h lang gerührt und durch Silika-Gel-TLC unter Elution mit einer 60 : 10 : 1 (V/V/V)-Mischung aus Chloroform/Methanol/konzentriertem Ammoniak (Lösungsmittel A) verfolgt. 100 mL Wasser wurden zugegeben, und die Mischung wurde auf ein Volumen von 200 mL eingeengt. Ethylacetat (200 mL) wurde zugegeben, und der pH-Wert wurde auf 10,5 mit 10%-iger NaOH angehoben. Das Ethylacetat wurde abgetrennt, und der pH-Wert der wässrigen Phase wurde auf 10,5 erneut angehoben, worauf diese mit 200 mL frischem Ethylacetat extrahiert wurde. Die Ethylacetat-Extrakte wurden vereinigt, mit NaCl gewaschen, über Na2SO4 getrocknet, filtriert und eingeengt, um nach Trocknung über Nacht unter Hochvakuum 14,86 g viskoses Öl zu ergeben.
1H-NMR (300 MHz, d6-DMSO), δ: 7,56 (d, 1H), 7,35 (s, 5H), 5,05 (s, 2H), 3,85 (m, 1H), 2,15 (t, 2H), 2,08 (s, 6H), 1,60 (d von t, 2H), 1,40 (s, 9H), 1,35 (m, 4H) - C. Nε-Dimethylamino-Nα-CBZ-O-t-butyl-L-lysin-N-oxid (3)
- Eine Lösung aus 25,3 g des Dimethylamins (2), 7,9 mL 30%-igem Wasserstoffperoxid und aus 150 mL Methanol wurde 5 h lang gerührt, worauf weitere 7,9 g des 30%-igen Wasserstoffperoxids zugegeben wurden. Die Reaktion wurde 48 h lang gerührt und mit Silika-Gel-TLC unter Eluieren mit dem Lösungsmittel A verfolgt. Eine 1 mL wässrige Aufschlämmung von ca. 5 mg Platin-Schwarz wurde zugegeben, worauf die Reaktion 7 h lang gerührt und eine weitere Aufschlämmung von 5 bis 10 mg Platin-Schwarz zugegeben wurden. Die Mischung wurde über Nacht gerührt und bezüglich Peroxid mit Peroxid-Testpapier unter Erwärmung auf 60°C zur gegebenenfalls notwendigen Entfernung von Peroxiden verfolgt. Sobald die Reaktion negativ bezüglich der Peroxide getestet war, wurde die Mischung filtriert und eingeengt. Der Rückstand wurde in 300 mL EtOAc gelöst, worauf das Ganze über Na2SO4 getrocknet, filtriert, eingeengt und an 400 g Silika-Gel unter Eluieren mit dem Lösungsmittel A chromatografiert wurde, um 13 g Produkt als farbloses Öl zu erzeugen.
1H-NMR (300 MHz, d6-DMSO) δ: 7,78 (d, 1H), 7,37 (s, 5H), 5,05 (dd, 2H), 2,88 (m, 1H), 3,05 (t, 2H), 2,95 (s, 6H), 1,70 (m, 4H), 1,40 (s, 9H), 1,32 (m, 2H) - Die Synthese des Nα-geschützten-O-geschützten Lysins ist in der Literatur bekannt (Scott et al, Commun. 1981, 11(4) 303–314).
- D. N-CBZ-2-Amino-5-hexenoat-5-t-butylester (4)
- Eine Lösung von 14,8 g des N-Oxids 3 in 250 mL DMSO wurde auf 125°C 2 h lang unter Spülen mit Argon erhitzt. Das DMSO wurde bei 70°C unter Hochvakuum abdestilliert. Unter Chromatografie des Rückstands an 450 mL Silika-Gel und Eluieren mit 20 : 80 (V/V) EtOAc/Hexan wurde das Produkt in den Fraktionen 70 bis 120 (18 mL Fraktionen) gewonnen. Diese wurden vereinigt und eingeengt, um 3,15 g des Produkts als farbloses Öl zu ergeben.
1H-NMR (d6-DMSO) δ: 7,62 (d, 1H), 7,35 (s, 5H), 5,78 (m, 1H), 5,08 (dd, 2H), 4,98 (m, 2H), 3,86 (m, 1H), 2,08 (dt, 2H), 1,68 (m, 2H), 1,38 (s, 9H), 1,35 (m, 2H) - E. N-CBZ-2-Amino-5,6-epoxyhexanoat-5-t-butylester (5), ausgehend von Verbindung 4 (Stufe D)
- Die 85%-ige m-Chlorperbenzoesäure (mCpBA) von Aldrich Chemical Co. wurde durch Auflösen in Methylenchlorid und waschen mit einem Puffer von pH = 7,5 gereinigt. Die organische Schicht wurde abgetrennt, über Na2SO4 getrocknet, filtriert und eingeengt. Der Feststoff wurde über Nacht unter Hochvakuum getrocknet und bei 3°C aufbewahrt.
- Eine Mischung aus 3,15 g des Hexanoats 4 (9,75 mMol) und 1,76 g mCpBA (10,2 mMol) in 25 mL Methylenchlorid wurde 48 h lang bei Raumtemperatur gerührt. Die Reaktion wurde mit 125 mL EtOAc verdünnt und 2 Mal mit 5%-iger NaOH und dann mit Kochsalzlösung gewaschen. Nach Trocknung über Na2SO4 wurde die Reaktionsmischung filtriert und eingeengt, um 2,9 g des Produkts als farbloses Öl zu erzeugen.
1H-NMR (d6-DMSO) δ: 7,35 (s, 5H), 5,05 (s, 2H), 3,90 (m, 1H), 2,65 (m, 1H), 2,50 (m, 1H), 2,42 (m, 1H), 1,8–1,5 (m, 2H), 1,40 (s, 9H), 1,35 (m, 2H)
MS (FAB): 358 (m+ + Na+) - F. 2,12-Benzyloxycarbonylamino-7-(5-benzyloxycarbonylamino-5-t-butoxycarbonylpentyl-)aza-1,13-di-t-butyl-5,9-dihydroxytridecanodiat (6)
- Das Epoxid 5 (1,68 g, 5,01 mMol) wurde in 1,5 mL trockenem Acetonitril gelöst, worauf 0,53 g (5,01 mMol) wasserfreies LiClO4 zugegeben wurden. Als der Feststoff aufgelöst war, wurden 0,8 g (23 mMol) des Amins (1) in 1,5 mL Acetonitril zugegeben. Die Mischung wurde bei 50°C über Nacht gerührt und dann unter vermindertem Druck eingeengt. Der Rückstand wurde an 150 g Silika-Gel unter Eluieren mit 7 : 1 (V/V) Chloroform/Lösungsmittel A chromatografiert, um 1 g Aminodiol 6 und 0,5 g rückgewonnenes Epoxid zu ergeben. Das Diol wurde in einer bekannten Menge Methylenchlorid gelöst und über 3 Å Molekularsieben zum späteren Gebrauch aufbewahrt.
MS (FAB): 1007 (M+) - G. 3-Hydroxy-1-(5-benzyloxycarbonylamino-5-t-butoxycarbonylpentyl)-4-(2-benzyloxycarbonylamino-2-t-butoxycarbonylethyl)-5-(3-benzyloxycarbonylamino-3-t-butoxycarbonylpropyl)pyridinium-Salz (8)
- Zu 1 mL Methylenchlorid wurden 0,6 mL 2 M (1,2 mMol) Oxalylchlorid in Methylenchlorid gegeben und die Mischung auf –55°C gekühlt. Dimethylsulfoxid (DMSO) (78 μL, 1,1 mMol) in 0,25 mL Methylenchlorid wurde dann zugetropft. Nach 15 min wurden 400 mg des Diols 6 in 1 mL Methylenchlorid zugetropft. Die Mischung wurde auf –30°C unter Rühren über 20 min erwärmt, und die Reaktionsmischung wurde dann zurück auf –55°C gekühlt, worauf 0,26 g (2,6 mMol) Triethylamin zugegeben wurden. Die Reaktionsmischung wurde auf Raumtemperatur erwärmt und dann mit 25 mL EtOAc verdünnt, mit 5%-iger NaOH und dann mit Kochsalzlösung gewaschen und über Na2SO4 getrocknet. Nach Filtrieren wurde die Lösung eingeengt, um 0,39 g 2,12-Benzyloxycarbonylamino-7-(5-benzyoxycarbonylamino-5-t-butoxycarbonylpentyl)aza-1,13-di-t-butyl-5,9-dioxotridecanodiat 7 zu ergeben, das wegen seiner Instabilität nicht charakterisiert wurde. Der Rückstand wurde in 4 mL Methylenchlorid gelöst, worauf 0,25 g Diazabicyclo[4.3.0]non-5-en (DBN) zugegeben wurden. Die Mischung wurde über Nacht unter Schutz eines Trocknungsrohrs gerührt. Essigsäure (100 μL) wurde zugefügt, und das Lösungsmittel wurde eingedampft. Der Rückstand wurde an 100 g Silika-Gel unter Eluieren mit 20 : 1 : 0,5 Chloroform/Methanol/Essigsäure chromatografiert. Die Fraktionen 11 bis 14 (18 mL Fraktionen) enthielten 0,26 g Pyridinium-Produkt 8.
- H. Deoxypyridinolin (DPD)
- Eine Mischung aus 160 mg des Pyridiniumacetats 8 und aus 1 mL HBr in HOAc wurde 30 min lang gerührt, worauf noch einmal 1 mL HBr/HOAc zugegeben wurde. Nach 1 h wurde das Lösungsmittel entfernt, und der Rückstand wurde in 50 mL einer 4 : 1 : 1 (V/V/V)-Mischung aus n-Butanol/Wasser/HOAc (Lösungsmittel B) gelöst, worauf 5 g Faser-Cellulose zugegeben wurden. Die Suspension wurde 30 min lang stehen gelassen und filtriert, worauf weitere 3 g Cellulose zum Filtrat gegeben und nach 2 h erneut filtriert wurde. Die zwei Cellulose-Feststoffe wurden vereinigt und in 50 mL Wasser aufgeschlämmt und nach 30 min filtriert. Ein Mengenanteil wurde entnommen, dessen Absorption in einem Puffer von pH = 8 bei 326 nm unter Zugrundelegen eines molaren Extinktionskoeffizienten von 5290 und eines Molekulargewichts von 413 28 mg DPD anzeigte. Die Lösung wurde lyophilisiert, und der Rückstand wurde in 10 mMol HCl und 8 g gewaschenem BioRad AG-1X10 (Cl–-Form) aufgelöst. Nach 1 h wurden das entstandene Produkt filtriert und das Filtrat zur Trockene in einem Savant Speed-Vac®-Konzentrator eingeengt, um 56 mg des sehr hygroskopischen Tetrachlorid-Salzes zu ergeben.
1H-NMR (300 MHz, d6-DMSO) δ: 8,65 (s, 1H), 8,50 (s, 1H), 4,51 (t, 2H, J = 7,5), 4,28 (t, 1H, J = 7), 4,10 (t, 1H, J = 6), 3,99 (t, 1H, J = 7), 3,4 (m, 2H), 3,2–2,9 (m, 2H), 2,2 (m, 2H), 2,1 (m, 2H), 2,0 (m, 2H), 1,5–1,3 (m, 2H) - Die O-t-Butylester- und CBZ-Schutzgruppen können mit einer Vielzahl von Behandlungsverfahren entfernt werden, mit denen der Fachmann vertraut ist, wie beschrieben von Greene et al in Protective Groups in Organic Synthesis; John Wiley & Sons: New York, 1991: S. 246 und 335–7. Somit kann der t-Butylester mit weiteren Säuren wie mit Ameisen-, Salz-, p-Toluolsulfon-, Trifluormethansulfon-, Methansulfon- und mit Trifluoressigsäure entfernt werden. Die CBZ-Gruppe kann mit einer Vielzahl von Verfahren wie z. B. durch Hydrogenolyse, mit Lewis-Säuren wie Aluminiumchlorid, Trimethylsilyljodid und Bortribromid, durch Fotolyse, Elektrolyse, mit Bariumhydroxid und mit weiteren Säuren wie mit Trifluor- und Methansulfonsäure entfernt werden.
Claims (10)
- Verfahren zur Herstellung von Deoxypyridinolin, welches die Stufen umfasst, in denen man: a) ein Nα-geschütztes-O-geschütztes Lysin der Formel:mit mindestens 2 Äquivalenten des 5,6-Epoxy-2-N-geschützten-O-geschützten-(2S)-2-aminohexanoats der Formel zur Reaktion bringt:
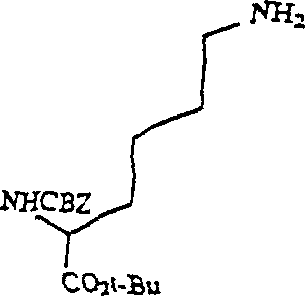 um das entsprechende Aminodiol der Formel zu erzeugen:
um das entsprechende Aminodiol der Formel zu erzeugen: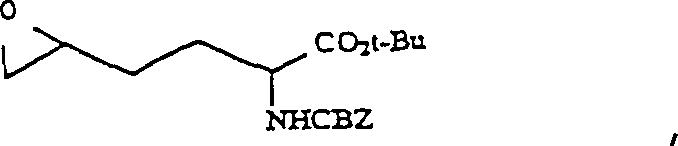 b) das Aminodiol mit einem oxidierenden Mittel zur Reaktion bringt, um das entsprechende Aminodiketon der Formel zu ergeben:
b) das Aminodiol mit einem oxidierenden Mittel zur Reaktion bringt, um das entsprechende Aminodiketon der Formel zu ergeben: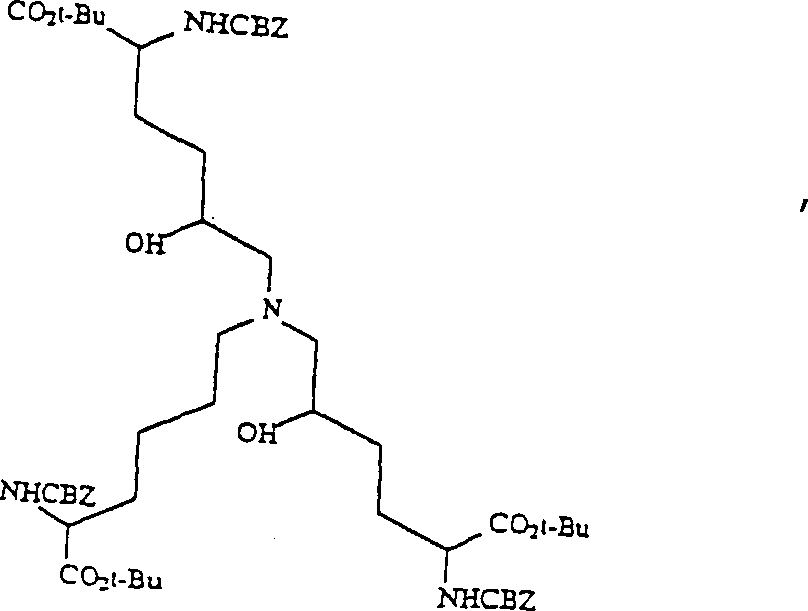 c) das Aminodiketon mit einer Base zur Reaktion bringt, um das entsprechende 3-Hydroxydihydropyridin zu erzeugen, und man dann das 3-Hydroxydihydropyridin mit einem oxidierenden Mittel zur Reaktion bringt, um den 3-Hydroxypyridinium-Ring zu bilden, um dadurch eine Verbindung der Formel zu ergeben:
c) das Aminodiketon mit einer Base zur Reaktion bringt, um das entsprechende 3-Hydroxydihydropyridin zu erzeugen, und man dann das 3-Hydroxydihydropyridin mit einem oxidierenden Mittel zur Reaktion bringt, um den 3-Hydroxypyridinium-Ring zu bilden, um dadurch eine Verbindung der Formel zu ergeben: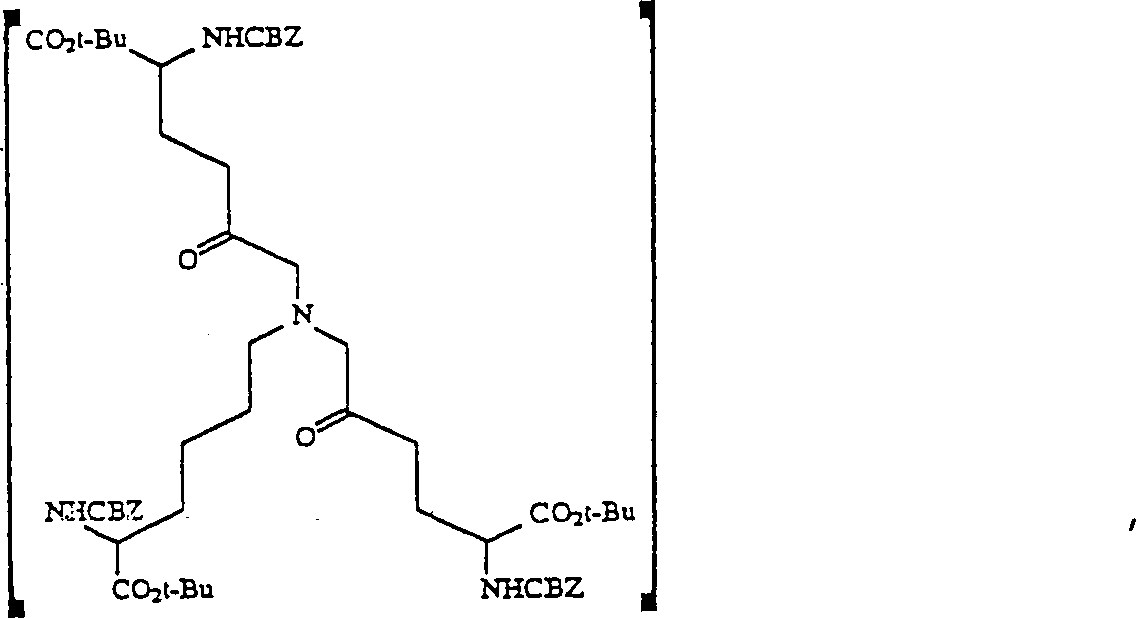 und man d) die Schutzgruppe aus der Verbindung der Stufe c) abspaltet, um Deoxypyridinolin der Formel zu ergeben:
und man d) die Schutzgruppe aus der Verbindung der Stufe c) abspaltet, um Deoxypyridinolin der Formel zu ergeben: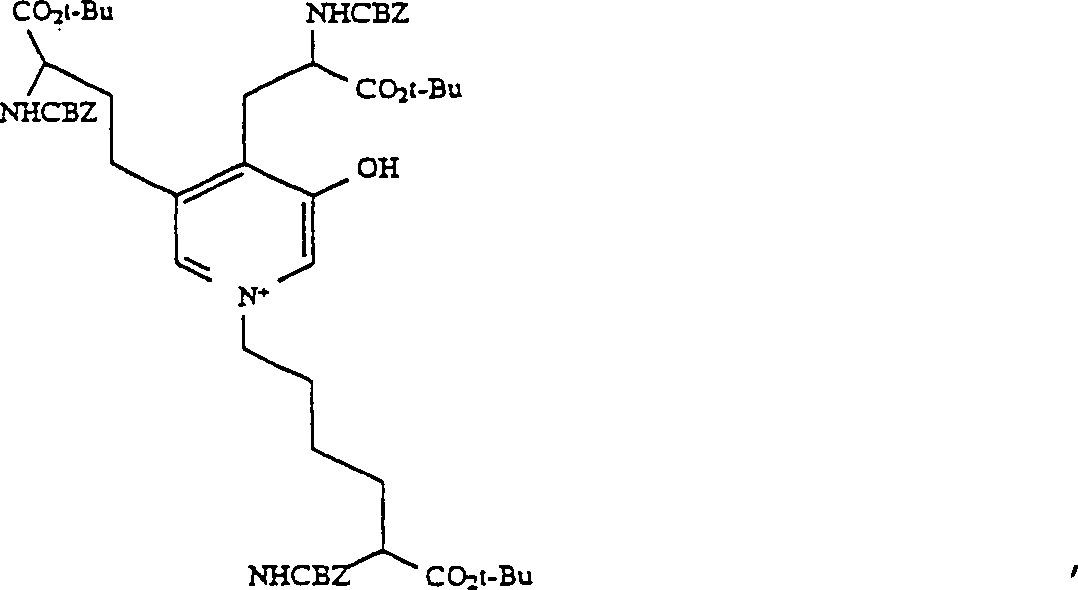

- Verfahren nach Anspruch 1, worin alle Zwischenprodukte mit Ausnahme des Aminodiketons gewonnen und gereinigt werden.
- Verfahren gemäß Anspruch 1 zur Herstellung von Deoxypyridinolin, welches die Stufen umfasst, in denen man: (a) mindestens 2 Äquivalente N-CBZ-2-Amino-5,6-epoxyhexanoat-5-t-butylester in einem geeigneten Lösungsmittel mit Nα-CBZ-O-t-Butyl-L-lysin zur Reaktion bringt, um 2,12-Benzyloxycarbonylamino-7-(5-benzyloxycarbonylamino-5-t-butoxycarbonylpentyl)aza-1,13-di-t-butyl-5,9-dihydroxytridecanoat zu ergeben, und dieses Diol gewinnt, (b) das in Stufe (a) hergestellte Diol mit einem geeigneten Oxidiermittel in einem geeigneten Lösungsmittel zur Reaktion bringt, um 2,12-Benzyloxycarbonylamino-7-(5-benzylcarbonylamino-5-t-butoxycarbonylpentyl)aza-1,13-di-t-butyl-5,9-dioxotridecanodiat zu ergeben, und man (c) ohne dessen Isolierung das Tridecanoat mit einer Base zur Reaktion bringt, um 3-Hydroxy-1-(5-benzoxycarbonylamino-5-t-butoxycarbonylpentyl)-4-(2-benzyloxycarbonylamino-2-t-butoxycarbonylethyl)-5-(3- benzyloxycarbonylamino-3-butoxycarbonylpropyl)-pyridinium-Salz zu ergeben, und man (d) das in Stufe (c) hergestellte Pyridinium-Salz mit HBr in einem geeigneten Lösungsmittel zur Reaktion bringt, um Deoxypyridinolin zu ergeben.
- Verfahren gemäß Anspruch 3, worin die Reaktion der Stufe (a) in Acetonitril, Methanol oder in einer Kombination aus Wasser und Alkohol als Lösungsmittel durchgeführt wird.
- Verfahren gemäß Anspruch 3, worin Lithiumperchlorat in die Stufe (a) als Katalysator und Acetonitril als das Lösungsmittel eingebracht werden.
- Verfahren gemäß Anspruch 3, worin das oxidierende Mittel in Stufe (b) Oxalylchlorid-DMSO, Thionylchlorid-DMSO, Trifluoressigsäureanhydrid-DMSO oder Essigsäureanhydrid-DMSO ist.
- Verfahren gemäß Anspruch 6, worin das oxidierende Mittel Oxalylchlorid-DMSO ist.
- Verfahren gemäß Anspruch 3, worin das Lösungsmittel in Stufe (b) Methylenchlorid, Diethylether, Tetrahydrofuran oder Chloroform ist.
- Verfahren gemäß Anspruch 3, worin die Base in Stufe (c) Triethylamin ist.
- Verfahren gemäß Anspruch 1 zur Herstellung von Deoxypyridinolin, wobei man (a) ein Nα-geschütztes-O-geschütztes Lysin mit mindestens 2 Äquivalenten des 5,6-Epoxy-2-N-geschützten-O-geschützten-(2S)-2-aminohexanoats in einem geeigneten Lösungsmittel zur Reaktion bringt, um das entsprechende Aminodiol zu erzeugen, (b) das Aminodiol mit einem oxidierenden Mittel zur Reaktion bringt, um das entsprechende Aminodiketon zu ergeben, (c) das Aminodiketon mit einer Base zur Reaktion bringt, um die entsprechende 3-Hydroxydihydropyridinolin-Verbindung als eine Zwischenproduktverbindung zu erzeugen, und man dann diese Zwischenproduktverbindung mit einem oxidierenden Mittel zur Reaktion bringt, um die entsprechende Pyridinium-Verbindung zu bilden, und man (d) aus der Pyridinium-Verbindung die Schutzgruppe abspaltet, um Deoxypyridinolin zu ergeben.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US782214 | 1997-01-13 | ||
| US08/782,214 US5723619A (en) | 1997-01-13 | 1997-01-13 | Method for the synthesis of deoxypyridinoline |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69822909D1 DE69822909D1 (de) | 2004-05-13 |
| DE69822909T2 true DE69822909T2 (de) | 2005-04-07 |
Family
ID=25125363
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69822909T Expired - Fee Related DE69822909T2 (de) | 1997-01-13 | 1998-01-03 | Verfahren zur Herstellung von Deoxypyridinolin |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5723619A (de) |
| EP (1) | EP0857720B1 (de) |
| JP (1) | JPH10195052A (de) |
| AU (1) | AU725687B2 (de) |
| CA (1) | CA2225198C (de) |
| DE (1) | DE69822909T2 (de) |
| DK (1) | DK0857720T3 (de) |
| IL (1) | IL122213A (de) |
| NO (1) | NO309601B1 (de) |
| NZ (1) | NZ329034A (de) |
| ZA (1) | ZA9710340B (de) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5834610A (en) * | 1997-05-06 | 1998-11-10 | Johnson; Gary M. | Conversion of pyridinoline to deoxypyridinoline |
| US6824985B1 (en) * | 1997-09-09 | 2004-11-30 | Bayer Corporation | Formulation for reducing urea effect for immunochromatography assays using urine samples |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US527715A (en) * | 1894-10-16 | Car-fender | ||
| GB9202139D0 (en) * | 1992-01-31 | 1992-03-18 | Sandoz Ltd | Improvements in or relating to organic compounds |
| US5527715A (en) * | 1992-07-31 | 1996-06-18 | Metra Biosystems, Inc. | Method and kit for pyridinoline assay |
| US5350855A (en) * | 1992-09-30 | 1994-09-27 | Metra Biosystems, Inc. | Derivatized D-acyl pyridinium reagent |
-
1997
- 1997-01-13 US US08/782,214 patent/US5723619A/en not_active Expired - Lifetime
- 1997-10-24 NZ NZ329034A patent/NZ329034A/en unknown
- 1997-11-17 ZA ZA9710340A patent/ZA9710340B/xx unknown
- 1997-11-17 IL IL12221397A patent/IL122213A/xx not_active IP Right Cessation
- 1997-11-24 NO NO975384A patent/NO309601B1/no unknown
- 1997-12-18 CA CA002225198A patent/CA2225198C/en not_active Expired - Fee Related
- 1997-12-19 JP JP9350499A patent/JPH10195052A/ja active Pending
-
1998
- 1998-01-03 DK DK98100050T patent/DK0857720T3/da active
- 1998-01-03 DE DE69822909T patent/DE69822909T2/de not_active Expired - Fee Related
- 1998-01-03 EP EP98100050A patent/EP0857720B1/de not_active Expired - Lifetime
- 1998-01-08 AU AU50389/98A patent/AU725687B2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| DK0857720T3 (da) | 2004-07-26 |
| EP0857720A1 (de) | 1998-08-12 |
| NO975384L (no) | 1998-07-14 |
| AU5038998A (en) | 1998-07-16 |
| ZA9710340B (en) | 1998-06-10 |
| CA2225198C (en) | 2003-10-07 |
| NO309601B1 (no) | 2001-02-26 |
| NO975384D0 (no) | 1997-11-24 |
| CA2225198A1 (en) | 1998-07-13 |
| AU725687B2 (en) | 2000-10-19 |
| NZ329034A (en) | 1998-07-28 |
| EP0857720B1 (de) | 2004-04-07 |
| US5723619A (en) | 1998-03-03 |
| DE69822909D1 (de) | 2004-05-13 |
| JPH10195052A (ja) | 1998-07-28 |
| IL122213A (en) | 2000-07-26 |
| IL122213A0 (en) | 1998-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69332988T2 (de) | Reagenzien zum Nachweis und zur Quantifizierung von Thyroxin in flüssigen Proben | |
| DE3587793T2 (de) | Lumineszierende metallchelatmarker und mittel zu deren nachweis. | |
| DE3856358T3 (de) | Für Tests verwendbare chemilumineszierende Ester, Thioester und Amide | |
| CH636625A5 (de) | Guajaconsaeure a, verfahren zu ihrer herstellung und diese substanz enthaltende diagnostische mittel. | |
| DE2920292A1 (de) | L- gamma -glutamyl-3-carboxy-4-hydroxyanilid und dessen salze, verfahren zu dessen herstellung und dessen verwendung zur bestimmung der aktivitaet von gamma -glutamyltranspeptidase | |
| DE1620411A1 (de) | Elektronenspinresonanzmarkierung von Biomolekuelen | |
| EP1576059A2 (de) | Carboxamid-substituierte farbstoffe für analytische anwendungen | |
| DE19521231A1 (de) | Neue Oxazinfarbstoffe und ihre Verwendung als Fluoreszenzmarker | |
| DE69223851T2 (de) | Nitro- oder Nitroso-substituierte polyhalogenierte Phenolsulfonphtaleine | |
| DE2305775A1 (de) | Neue analytische verbindungen und deren verwendung | |
| DE3000879C2 (de) | ||
| DE2919545C3 (de) | L-Leucin-4-hydroxyanilid-derivate, Verfahren zu deren Herstellung sowie deren Verwendung zur Bestimmung der Aktivität von Leucinaminopeptidase | |
| DE2808111A1 (de) | Neue dipeptidderivate und deren verwendung zur messung der enzymatischen aktivitaet | |
| DE2921782A1 (de) | Chemilumineszente naphthalin-1,2- dicarbonsaeure-hydrazid-markierte konjugate und verfahren zu deren herstellung | |
| EP0747699B1 (de) | Entstörungsreagenz für die Bestimmung eines Analyten mit einem lumineszenzfähigen Metallkomplex | |
| DE69822909T2 (de) | Verfahren zur Herstellung von Deoxypyridinolin | |
| EP0442372B1 (de) | Verbesserte markierte Haptene, Verfahren zu deren Herstellung sowie Verwendung dieser markierten Haptene in Immunoassays | |
| DE69431396T2 (de) | Verfahren zur herstellung von n-tert-butyl-2-pyrazincarboxamid und n-tert-butyl-2-piperazincarboxamid | |
| DE2735904A1 (de) | 1,5-disubstituierte-2-pyrrolidone und verfahren zu ihrer herstellung | |
| EP0465998B1 (de) | Neue Beta-Galactosidase-Substrate für den CEDIA | |
| DE3034618C2 (de) | Verfahren zur Bestimmung der Aktivität des Angiotensin-konvertierenden Enzyms | |
| DE1961947A1 (de) | 6,7-Diacyloxy-tetrahydroisochinolinverbindungen und Verfahren zu ihrer Herstellung | |
| CH640870A5 (de) | Verfahren zur herstellung von 9-desacetyl-9-formyl-n-trifluoracetyldaunorubicin. | |
| EP0342157B1 (de) | Aromatische Säuren | |
| DE2661113C2 (de) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8332 | No legal effect for de | ||
| 8370 | Indication related to discontinuation of the patent is to be deleted | ||
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |