-
Technisches Gebiet
-
Die
vorliegende Erfindung bezieht sich auf eine neue kondensierte heterocyclische
Verbindung mit Affinität
zum Dopamin-D4- (hierin nachstehend als
D4 bezeichnet, wobei dieselbe Abkürzung bezüglich der
folgenden Dopaminrezeptorsubtypen eingesetzt wird) und Serotonin-2-Rezeptor
(hierin nachstehend als 5-HT2 bezeichnet), die die NMDA-Rezeptorunterfunktion
blockiert und die auf medizinischem Gebiet als Zentralnervenmittel,
insbesondere als Antipsychotikum verwendet wird.
-
Stand der Technik
-
Die
folgenden Patentveröffentlichungen
sind bezüglich
Zentralnervenmittel mit Affinität
zum D4-Rezeptor veröffentlicht worden. Die WO94/10162,
WO94/21630 und WO94/21626 offenbaren heterotricyclische aromatische
Verbindungen mit Affinität
zum D4-Rezeptor, WO94/21627, WO94/21628
und WO94/24105 offenbaren Indolderivate mit Affinität zum D4-Rezeptor, WO94/20459 und WO94/20497 offenbaren
Pyrropyridinderivate mit Affinität
zum D4-Rezeptor und WO94/21615 und WO94/22839
offenbaren Benzimidazolderivate mit Affinität zum D4-Rezeptor.
Die WO94/10145 und WO94/20471 offenbaren Pyrazolderivate und Chinolonderivate,
die jeweils Affinität
zum D4-Rezeptor aufweisen.
-
Die
japanische ungeprüfte
Patentveröffentlichung
Nr. 157576/1979 offenbart, daß durch
Dapiprazol, [3-(2-(4-(2-Tolyl)-1-piperazinyl)ethyl)-5,6,7,8-tetrahydro-s-triazol[4,3-a]pyridin],
repräsentierte
Cycloalkyltriazole zur Therapie von Glaukom, Psychose und dergleichen
verwendet werden können.
-
Nahezu
alle auf Schizophrenie anwendbaren Antipsychotika zeigen eine gemeinsame
pharmakologische Wirkung des Blockierens des Dopaminrezeptors, der
einer der zerebralen Neurotransmitter ist, und zeigen eine besonders
starke D2-Rezeptor Blockadewirkung. Diese Arzneimittel
(typischerweise antipsychotische Mittel) sind gegen positive Symptome
wirksam, die sich auf Halluzinieren und Wahn konzentrieren, die
für den akuten
Zustand von Schizophrenie kennzeichnend sind, aber kaum wirksam
gegen negative Symptome einer Apathie, Abulie und Autismus. Außerdem sind
sie mit ernsten Nebenwirkungsproblemen wie extrapyramidale Symptome
(z. B. verzögerte
Dyskenie, akute Dystonie, Akathisie usw.) und endokriner Störung (z.
B. Hyperprolaktinämie)
verbunden, die bei akuter Verabreichung und fortlaufender Langzeitverabreichung
beobachtet werden.
-
Der
Dopaminrezeptor wird herkömmlicherweise
durch pharmakologische Verfahren gemäß dem Typ der Ligandenbindung
und Assoziationsweise mit Adenylatcyclase [Nature, Bd. 227, S. 93
(1979)] in zwei Rezeptorsubtypen eingeteilt. Das bedeutet ein D1-Rezeptortyp, der Adenylatcyclase über ein
akzelerierendes G-Protein
unter Produzieren von cyclischem AMP fördert, und ein D2-Rezeptortyp,
der Adenylatcyclase über ein
supprimierendes G-Protein unter Unterdrücken der Produktion von cyclischem
AMP unterdrückt.
Aufgrund der revolutionären
Entwicklung der Molekularbiologie in den letzten Jahren wurden fünf verschiedene
Gene von Dopaminrezeptoren kloniert und die Dopaminrezeptoren werden
nun in der D1-Familie angehörende D1- und D5-Rezeptoren
und der D2-Familie angehörende D2-,
D3 und D4-Rezeptoren
eingeteilt [Trends in Pharmacol. Sci., Bd. 15, S. 264 (1994)].
-
Es
wurde dokumentiert, daß Haloperidol,
das ein typisches Antipsychotikum ist, eine höhere Affinität zum D2-Rezeptor als zum D4-Rezeptor
aufweist und Clozapin, das mit weniger extrapyramidalen Nebenwirkungen
verbunden ist und auch gegen negative Symptome wirksam ist, eine
10 mal höhere
Affinität
zum D4-Rezeptor
als zum D2-Rezeptor aufweist [Nature, Bd.
350, S. 610 (1991)], [Trends in Pharmacol. Sci., Bd. 15, S. 264
(1994)]. Es wurde auch berichtet, daß die wirksame therapeutische
Plasmakonzentration von Clozapin mit der Affinitätskonstanten für den D4-Rezeptor korreliert [Trends in Pharmacol.
Sci., Bd. 15, S. 264 (1994)]. Ein Bericht wurde über einen Bindungstest unter
Verwenden des Gehirns eines Schizophreniepatienten postmortem dokumentiert,
wobei der D4-Rezeptor einen 6 mal größeren Spiegel
als bei einer gesunden Person zeigte [Nature, Bd. 365, S. 441 (1993)].
Es scheint daher, daß der
D4-Rezeptor sehr wahrscheinlich Schizophrenie
verursacht oder an der Wirkungsstelle eines therapeuti schen Mittels
vorhanden ist. Es sind Veränderungen
bei der Verteilung des Dopaminrezeptors in dem Gehirn gefunden worden,
die auf Subtypen zurückzuführen sind,
wobei der D2-Rezeptor am häufigsten
im Corpus striatum gefunden wird und der D4-Rezeptor
am häufigsten
im Lobus frontalis des Cortex cerebralis gefunden wird, der für die emotionellen
Funktionen verantwortlich ist.
-
Aus
klinischen Anwendungen wurde deutlich, daß die gleichzeitige Verwendung
von Ritanserin (das ein 5-HT2-Rezeptorblocker
ist) mit einem typischen Antipsychotikum negative Symptome und emotionale
Störungen
wie etwa Angst [Current Therapeutics Research, Bd. 10, S. 492 (1986)]
verbessert. Von den durch Antipsychotika hervorgerufenen Nebenwirkungen
werden maligne Symptome, die am ernstesten und tödlich sind, wenngleich in geringer
Häufigkeit,
gemäß einer
hypothetischen Betrachtungsweise durch das Ungleichgewicht von Dopamin/Serotonin-Nervenfunktionen
im Körpertemperatur-Kontrollzentrum
verursacht [Japanese Journal of Psychopharmacology, Bd. 11, S. 17
(1989)] und es wird eine Unterdrückung
des Auftretens der Krankheit durch die Anwendung der 5-HT2-Rezeptorblockadewirkung erwartet.
-
Es
gibt auch eine hypothetische Betrachtungsweise zur Ursache von Schizophrenie,
die lautet, daß die
Verschlechterung der Funktion des NMDA-Nervensystems (N-Methyl-D-asparaginsäure), die
vom Cortex cerebralis zum Subcortex zielt, die suppressive Rückmeldefunktion
des Informationskontrollkreises verschlechtert, was wiederum den
Schizophreniezustand wie die übermäßige Förderung
der Aktivität
des subcorticalen Dopaminnervensystems verschlimmert und von Clopazin
wurde berichtet, daß es
neben der Wirkung auf das Dopamin- und Serotoninnervensystem eine
Blockade der NMDA-Rezeptorunterfunktion zeigt [Trends Neurosci.
Bd. 13, S. 272 (1990)].
-
Aus
dem Vorangehenden wird von einer Verbindung, die eine D4-Rezeptor-
und 5-HT2-Rezeptorblockadewirkung aufweist
und die die NMDA-Rezeptorunterfunktion blockiert, erwartet, daß sie ein
Antipsychotikum darstellt, das mit weniger extrapyramidalen Nebenwirkungen
verbunden ist und sowohl gegen positive als auch negative Symptome
wirksam ist. Ein Gegenstand der vorliegenden Erfindung ist daher
das Bereitstellen einer Verbindung, die sowohl eine starke Blockadewirkung
auf den D4-Rezeptor und 5-HT2-Rezeptor
als auch eine Blockade der NMDA-Rezeptorunterfunktion zeigt, die
verglichen mit herkömmlichen
Verbindungen sowohl gegen negative Symptome als auch positive Symptome
wirksam ist und die mit weniger Nebenwirkungen verbunden ist.
-
Offenbarung der Erfindung
-
Die
Erfinder haben eingehende Untersuchungen ausgeführt und fanden, daß eine neue
kondensierte heterocyclische Verbindung der folgenden Formel (I),
ein optisches Isomer davon und ein pharmazeutisch annehmbares Salz
davon eine stärkere
Blockadewirkung auf den D4-Rezeptor und
5-HT2-Rezeptor als auf den D2-Rezeptor aufweisen
und daß sie
eine Blockade der NMDA-Rezeptorunterfunktion zeigen. Die Erfinder
haben weiter gefunden, daß diese
Verbindungen brauchbare, nicht nur gegen positive Symptome, die
sich auf Halluzinieren und Wahn konzentrieren, die für den akuten
Zustand von Schizophrenie kennzeichnend sind, sondern auch gegen
negative Symptome einer Apathie, Abulie und Autismus wirksame Antipsychotika
darstellen können,
die weniger Nebenwirkungen wie etwa extrapyramidale Symptome und
eine endokrine Störung verursachen,
die beobachtet werden, wenn herkömmliche
Antipsychotika mit D2-Rezeptor-Blockadewirkung verabreicht
werden, was zum Abschluß der
vorliegenden Erfindung führte.
-
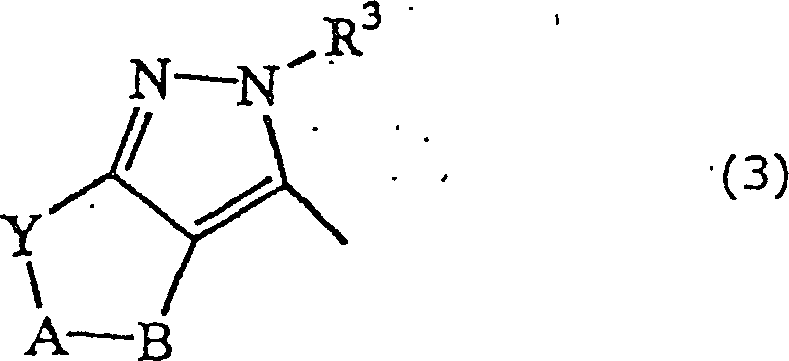
Demgemäß bezieht
sich die vorliegende Erfindung auf eine Verbindung der Formel (I)
wobei
R eine Gruppe
mit der folgenden Formel (3)
ist, wobei
Y ein lineares
oder verzweigtes C
1-C
4-Alkylen
ist, das gegebenenfalls einen Substituenten R
1a an
einer optionalen Position hat, wobei R
1a ein
aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl,
Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl,
Hydroxy, ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy, Amino
oder ein aus Methylamino, Dimethylamino, Ethylamino, Diethylamino,
Propylamino und Dipropylamino ausgewähltes Alkylamino ist, A fehlt
oder ein Sauerstoffatom, ein Schwefelatom, SO, SO
2 oder
N-R
7 ist, wobei R
7 Wasserstoff,
ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl,
Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Benzyl,
2-Phenylethyl, 3-Phenylpropyl, 2-Methyl-2-phenylethyl, 3-Methyl-3-phenylpropyl,
4-Chlorbenzyl, 2-(4-Chlorphenyl)ethyl,
3-(4-Chlorphenyl)propyl, 2-Methyl-2-(4-chlorphenyl)ethyl und 3-Methyl-3-(4-chlorphenyl)propyl
ausgewähltes
Aralkyl oder ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl
ausgewähltes
Acyl ist,
B ein lineares oder verzweigtes C
1-C
4-Alkylen ist, das gegebenenfalls einen Substituenten
R
1b an einer optionalen Position hat, wobei
R
1b ein aus Methyl, Ethyl, Propyl, Isopropyl,
Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl
und Octadecyl ausgewähltes
Alkyl, Hydroxy, ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy,
Isobutoxy, tert-Butoxy,
Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy, Amino,
ein aus Methylamino, Dimethylamino, Ethylamino, Diethylamino, Propylamino
und Dipropylamino ausgewähltes
Alkylamino ist, und
R
3 Wasserstoff,
ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl,
Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Formyl,
Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl
oder ein aus Phenyl, Naphthyl und 2-Indenyl ausgewähltes Aryl
ist,
D fehlt oder ein lineares oder verzweigtes Alkylen mit
1 bis 8 Kohlenstoffatomen ist,
Q-T eine CH-, CH
2-N-,
(CH
2)
2-N-, CH
2-C(R
4)-Bindung ist,
wobei R
4 Wasserstoff, Hydroxy, ein aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl,
Hexadecyl und Octadecyl ausgewähltes Alkyl
oder ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy
oder CH=C ist,
G fehlt oder ein lineares oder verzweigtes Alkylen
mit 1 bis 8 Kohlenstoffatomen oder ein Carbonyl ist,
R
5 Wasserstoff oder ein aus Methyl, Ethyl,
Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl,
Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl ist und
R
6 ein Aryl, das aus Phenyl, Naphthyl und
Indenyl ausgewählt
ist, das gegebenenfalls durch (einen oder mehrere) Substituenten
substituiert ist, der (die) aus einem Halogen, Trifluormethyl, einem
aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl
ausgewählten
Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), ein Heteroaryl, das aus Pyridyl, Furyl, Thienyl und Pyrimidinyl
ausgewählt
ist, das gegebenenfalls durch (einen oder mehrere) Substituenten
substituiert ist, der (die) aus einem Halogen, Trifluormethyl, einem
aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl
ausgewählten
Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy,
Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), oder ein kondensiertes Heteroaryl, das aus 1,2-Benzoisoxazol-3-yl,
1,2-Benzoisothiazol-3-yl, Indol-3-yl, Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl
ausgewählt
ist, das gegebenenfalls durch (einen oder mehrere) Substituenten
substituiert ist (sind), der (die) aus einem Halogen, Trifluormethyl,
einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und
tert-Butyl ausgewählten
Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind),
ist,
ein optisches Isomer davon oder ein pharmazeutisch annehmbares
Salz davon.
-
Die
Verbindung der Formel (I) schließt die folgende Verbindung
ein
-
Bezüglich der
vorstehenden Formel (I) ist Alkyl als R1a zum
Beispiel Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl,
Pentyl, Hexyl, Heptyl, Octyl, Decyl, Hexadecyl und Octadecyl, wobei
Alkyl mit 1 bis 4 Kohlenstoffatomen der Vorzug gegeben wird. Alkoxy
ist zum Beispiel Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy, wobei
Alkoxy mit 1 bis 4 Kohlenstoffatomen der Vorzug gegeben wird. Alkylamino
ist zum Beispiel Methylamino, Dimethylamino, Ethylamino, Diethylamino,
Propylamino und Dipropylamino.
-
Alkyl,
Alkoxy und Alkylamino als R1b sind die bezüglich Alkyl,
Alkoxy und Alkylamino als R1a angeführten.
-
Alkyl
als R3 wird durch die bezüglich Alkyl
als R1a angeführten veranschaulicht. Acyl
ist zum Beispiel Formel, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl,
wobei Acetyl der Vorzug gegeben wird. Aryl ist zum Beispiel Phenyl,
Naphthyl und 2-Indanyl, wobei Phenyl mit 1 oder 2 Substituenten
wie etwa Halogen, Methyl, Trifluormethyl und Methoxy der Vorzug
gegeben wird.
-
Alkyl
und Alkoxy als R4 werden durch die bezüglich Alkyl
und Alkoxy als R1a angeführten veranschaulicht.
-
Alkyl
als R5 wird durch die bezüglich Alkyl
als R1a angeführten veranschaulicht.
-
Aryl
als R6 ist zum Beispiel Phenyl, Naphthyl
und 2-Indanyl. Heteroaryl ist zum Beispiel Pyridyl, Furyl, Thienyl
und Pyrimidinyl. Kondensiertes Heteroaryl ist zum Beispiel 1,2-Benzisoxazol-3-yl,
1,2-Benzoisothiazol-3-yl, Indol-3-yl, Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl.
Der Substituent dafür
kann Halogen (z. B. Fluor, Chlor und Brom), Halogenalkyl (z. B.
Trifluormethyl), Alkyl (z. B. Methyl, Ethyl, Propyl, Isopropyl,
Butyl, Isobutyl und tert-Butyl), Alkoxy (z. B. Me thoxy, Ethoxy,
Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy,
Hexyloxy, Heptyloxy und Octyloxy), Hydroxy, Nitro, Amino, Methylamino
und Dimethylamino sein.
-
Alkyl
als R7 wird durch die bezüglich Alkyl
als R1a veranschaulichten veranschaulicht.
Aralkyl ist zum Beispiel Benzyl, 2-Phenylethyl, 3-Phenylpropyl,
2-Methyl-2-phenylethyl,
3-Methyl-3-phenylpropyl, 4-Chlorbenzyl, 2-(4-Chlorphenyl)ethyl,
3-(4-Chlorphenyl)propyl,
2-Methyl-2-(4-chlorphenyl)ethyl und 3-Methyl-3-(4-chlorphenyl)propyl,
wobei Benzyl der Vorzug gegeben wird. Acyl ist zum Beispiel Formyl,
Acetyl, Propionyl, Benzoyl und Benzylcarbonyl, wobei Acetyl der
Vorzug gegeben wird.
-
Alkyl
als R8 wird durch die bezüglich Alkyl
als R1a angeführten veranschaulicht. Acyl
wird durch die bezüglich
Acyl als R3 angeführten veranschaulicht.
-
Gerades
oder verzweigtes Alkylen mit 1 bis 4 Kohlenstoffatomen als Y wird
durch Methylen, Ethylen, Trimethylen, Tetramethylen, Methylmethylen,
Dimethylmethylen, 1-Methylethylen, 2-Methylethylen, 1,1-Dimethylethylen,
2,2-Dimethylethylen,
Ethylmethylen, Diethylmethylen, 1-Ethylethylen, 2-Ethylethylen, 1-Methyltrimethylen,
1,1-Dimethyltrimethylen, 2-Methyltrimethylen,
2,2-Dimethyltrimethylen, 3-Methyltrimethylen, 3,3-Dimethyltrimethylen,
1-Ethyltrimethylen, 2-Ethyltrimethylen und 3-Ethyltrimethylen veranschaulicht, wobei
Ethylen und Trimethylen der Vorzug gegeben wird.
-
Gerades
oder verzweigtes Alkylen mit 1 bis 4 Kohlenstoffatomen als B wird
durch die bezüglich
des linearen oder verzweigten Alkylens mit 1 bis 4 Kohlenstoffatomen
als Y veranschaulicht.
-
Gerades
oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen als D wird
durch Methylen, Ethylen, Trimethylen, Tetramethylen, Pentamethylen,
Hexamethylen, Octamethylen, Methylmethylen, Dimethylmethylen, 1-Methylethylen,
2-Methylethylen,
1,1-Dimethylethylen, 2,2-Dimethylethylen, Ethylmethylen, Diethylmethylen,
1-Ethylethylen, 2-Ethylethylen, 1-Methyltrimethylen, 1,1-Dimethyltrimethylen,
2-Methyltrimethylen, 2,2-Dimethyltrimethylen, 3- Methyltrimethylen, 3,3-Dimethyltrimethylen,
1-Ethyltrimethylen, 2-Ethyltrimethylen und
3-Ethyltrimethylen veranschaulicht, wobei Ethylen, Trimethylen und
Tetramethylen der Vorzug gegeben wird.
-
Gerades
oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen als G wird
durch die bezüglich
des geraden oder verzweigten Alkylens mit 1 bis 8 Kohlenstoffatomen
als D angeführten
veranschaulicht.
-
Die
Gruppe der Formel
wird speziell durch die
folgenden Gruppen veranschaulicht.
-
-
Bezüglich der
Formel (I) ist eine bevorzugte Verbindung die Verbindung, bei der
R
eine Gruppe mit der Formel (3) ist, wobei
Y ein lineares oder
verzweigtes C1-C4-Alkylen
ist, das gegebenenfalls einen Substituenten R1a an
einer optionalen Position hat, wobei R1a ein
Alkyl ist, das aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl,
tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl
ausgewählt
ist,
A fehlt oder ein Sauerstoffatom, ein Schwefelatom oder
N-R7 ist, wobei R7 Wasserstoff,
ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl,
Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Benzyl,
2-Phenylethyl, 3-Phenylpropyl, 2-Methyl-2-phenylethyl, 3-Methyl-3-phenylpropyl, 4-Chlorbenzyl,
2-(4-Chlorphenyl)ethyl,
3-(4-Chlorphenyl)propyl, 2-Methyl-2-(4-chlorphenyl)ethyl und 3-Methyl-3-(4-chlorphenyl)propyl
ausgewähltes
Aralkyl oder ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl
ausgewähltes
Acyl ist,
B ein lineares oder verzweigtes C1-C4-Alkylen ist, das gegebenenfalls einen Substituenten
R1b an einer optionalen Position hat, wobei
R1b ein aus Methyl, Ethyl, Propyl, Isopropyl,
Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl
und Octadecyl ausgewähltes
Alkyl ist, und
R3 Wasserstoff, ein
aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl,
Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl,
ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl
oder ein aus Phenyl, Naphthyl und 2-Indenyl ausgewähltes Aryl
ist,
D fehlt oder ein lineares oder verzweigtes Alkylen mit
1 bis 8 Kohlenstoffatomen ist,
Q-T eine CH-, CH2-N-,
(CH2)2-N-, CH2-C(R4)-Bindung ist,
wobei R4 Wasserstoff, Hydroxy, ein aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl,
Hexadecyl und Octadecyl ausgewähltes
Alkyl oder ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy,
Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy
ausgewähltes
Alkoxy oder CH=C ist,
G fehlt,
R5 Wasserstoff
oder ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl,
tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl
ausgewähltes
Alkyl ist und
R6 ein Aryl, das aus
Phenyl, Naphthyl und 2-Indenyl ausgewählt ist, das gegebenenfalls
durch (einen oder mehrere) Substituenten substituiert ist, der (die)
aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl,
Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy,
Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy,
Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro,
Amino, Methylamino und Dimethylamino ausgewählt ist (sind), ein Heteroaryl,
das aus Pyridyl, Furyl, Thienyl und Pyrimidinyl aus gewählt ist,
das gegebenenfalls durch (einen oder mehrere) Substituenten substituiert
ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl,
einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), oder ein kondensiertes Heteroaryl, das aus 1,2-Benzoisoxazol-3-yl,
1,2-Benzoisothiazol-3-yl, Indol-3-yl, Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl
ausgewählt
ist, das gegebenenfalls durch (einen oder mehrere) Substituenten
substituiert ist (sind), der (die) aus einem Halogen, Trifluormethyl,
einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und
tert-Butyl ausgewählten
Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind).
-
Bezüglich der
Formel (I) ist eine bevorzugtere Verbindung die Verbindung, bei
der R eine Gruppe mit der Formel (3) ist,
wobei
Y ein
lineares Alkylen mit 1 bis 4 Kohlenstoffatomen ist,
A fehlt
oder ein Sauerstoffatom oder N-R7 ist, wobei
R7 Wasserstoff oder ein aus Formyl, Acetyl,
Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl ist,
B ein
lineares oder verzweigtes Alkylen mit 1 bis 4 Kohlenstoffatomen
ist,
R3 Wasserstoff, ein Alkyl mit
1 bis 4 Kohlenstoffatomen, ein aus Formyl, Acetyl, Propionyl, Benzoyl
und Benzylcarbonyl ausgewähltes
Acyl ist,
D fehlt oder ein lineares oder verzweigtes Alkylen
mit 1 bis 8 Kohlenstoffatomen ist,
Q-T eine CH2-N-,
CH2-C(R4)-Bindung
ist, wobei R4 Wasserstoff, Hydroxy, ein
aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl,
Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl
oder ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy,
Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy oder CH=C ist,
G
fehlt,
R5 Wasserstoff oder ein Alkyl
mit 1 bis 4 Kohlenstoffatomen ist und
R6 ein
Aryl, das aus Phenyl, Naphthyl und 2-Indenyl ausgewählt ist,
das gegebenenfalls durch (einen oder mehrere) Substituenten substituiert
ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl,
einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy,
Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), ein Heteroaryl, das aus Pyridyl, Furyl, Thienyl und Pyrimidinyl
ausgewählt
ist, das gegebenenfalls durch (einen oder mehrere) Substituenten substituiert
ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl,
einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), oder ein kondensiertes Heteroaryl, das aus 1,2-Benzoisoxazol-3-yl,
1,2-Benzoisothiazol-3-yl, Indol-3-yl, Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl
ausgewählt
ist, das gegebenenfalls durch (einen oder mehrere) Substituenten
substituiert ist (sind), der (die) aus einem Halogen, Trifluormethyl,
einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und
tert-Butyl ausgewählten
Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind).
-
Bezüglich Formel
(I) ist eine noch bevorzugtere Verbindung die Verbindung, bei der
R
eine Gruppe mit der Formel (3) ist,
wobei
Y Ethylen ist,
A
fehlt oder ein Sauerstoffatom oder N-R7 ist,
wobei R7 Wasserstoff oder ein aus Formyl,
Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl
ist,
B ein lineares oder verzweigtes Alkylen mit 1 bis 3 Kohlenstoffatomen
ist und
R3 Wasserstoff oder ein Alkyl
mit 1 bis 4 Kohlenstoffatomen ist,
D fehlt oder ein lineares
oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen ist,
Q-T
eine CH2-N-, CH2-C(R4)-Bindung ist, wobei R4 Wasserstoff,
Hydroxy, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl,
tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl
ausgewähltes
Alkyl oder ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy,
Isobutoxy, tert-Butoxy,
Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy
oder CH=C ist,
G fehlt,
R5 Wasserstoff
oder ein Alkyl mit 1 bis 4 Kohlenstoffatomen ist und
R6 ein Aryl, das aus Phenyl, Naphthyl und
2-Indenyl ausgewählt
ist, das gegebenenfalls durch (einen) Substituenten substituiert
ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl,
einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), ein Heteroaryl, das aus Pyridyl, Furyl, Thienyl und Pyrimidinyl
ausgewählt
ist, das gegebenenfalls durch (einen) Substituenten substituiert
ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl,
einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind), oder ein kondensiertes Heteroaryl, das aus 1,2-Benzoisoxazol-3-yl,
1,2-Benzoisothiazol-3-yl, Indol-3-yl,
Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl ausgewählt ist,
das gegebenenfalls durch (einen) Substituenten substituiert ist
(sind), der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl,
Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl,
einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy,
Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist
(sind).
-
Bezüglich der
Formel (I) ist eine besonders bevorzugte Verbindung die Verbindung,
bei der R eine Gruppe mit der Formel (3) ist,
wobei
Y
Ethylen ist,
A fehlt oder ein Sauerstoffatom oder N-R7 ist, wobei R7 Wasserstoff
oder ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl
ausgewähltes
Acyl ist,
B ein lineares Alkylen mit 1 bis 3 Kohlenstoffatomen
ist und
R3 Wasserstoff oder Methyl
ist,
D Trimethylen ist,
Q-T eine CH2-N-,
CH2-CH- oder CH=C-Bindung ist,
G fehlt,
R5 Wasserstoff ist und
R6 ein
Aryl ist, das aus Phenyl, Naphthyl und 2-Indenyl ausgewählt ist,
das gegebenenfalls ein Halogen oder ein Alkyl mit 1 bis 4 Kohlenstoffatomen
hat.
-
Bezüglich der
Formel (I) sind die bevorzugtesten Verbindungen die aus der folgenden
Gruppe von Verbindungen ausgewählten,
wobei die Zahl in Klammern der Beispielnummer entspricht.
- (104) 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol,
- (116) 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol,
- (117) 3-(3-(4-(4-Methylphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol,
- (118) 3-(3-(4-Phenylpiperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol,
- (137) 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydropyrano[4,3-c]pyrazol,
- (141) 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
und
- (142) 5-Acetyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin.
-
Das
pharmazeutisch annehmbare Salz der Verbindung der Formel (I) ist
zum Beispiel ein Säureadditionssalz
mit einer anorganischen Säure
(z. B. Salzsäure,
Bromwasserstoffsäure,
Schwefelsäure,
Phosphorsäure
und Salpetersäure)
oder organischen Säure
(z. B. Essigsäure,
Propionsäure,
Bernsteinsäure,
Glykolsäure,
Milchsäure, Äpfelsäure, Weinsäure, Citronensäure, Maleinsäure, Fumarsäure, Methansulfonsäure, Benzolsulfonsäure, p-Toluolsulfonsäure, Camphersulfonsäure und
Ascorbinsäure).
-
Die
Verbindung der Formel (I) und pharmazeutisch annehmbare Salze davon
können
in Form eines Hydrats oder eines Solvats vorliegen und ein derartiges
Hydrat und Solvat ist ebenfalls in der vorliegenden Erfindung eingeschlossen.
Beispiele davon schließen
ein 1/10-Hydrat, ¼-Hydrat, ½-Hydrat,
Monohydrat, Dihydrochlorid-1/2-hydrat, Dihydrochlorid-dihydrat,
Dihydrochlorid-3/2-hydrat und dergleichen ein. Wenn die Verbindung
der Formel (I) ein asymmetrisches Kohlenstoffatom aufweist, liegen
wenigstens zwei Arten optische Isomeren vor. Diese optischen Isomeren
und Racemate davon sind im Umfang der vorliegenden Erfindung eingeschlossen.
-
Die
Verbindung der Formel (I) und die in der Formel (I) eingeschlossenen
erfinderischen Verbindungen können
durch die folgenden Verfahren synthetisiert werden. Jedes Symbol
in den folgenden Reaktionsformeln ist solange nicht besonders angegeben
wie vorstehend definiert.
-
3.
Verbindung, bei der R eine Gruppe der Formel (3) ist:
-
-
Eine
Verbindung der Formel (36) wird mit einer Verbindung der Formel
(37), worin J Chlor, Imidazol, Cyan und dergleichen ist, in einem
geeigneten Lösungsmittel,
das die Reaktion nicht stört
(z. B. Tetrahydrofuran, Diethylether, Methylenchlorid, Chloroform,
Dimethylformamid und Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart eines geeigneten Dehydrierungsmittels
(z. B. Lithiumdiisopropylamid, Lithiumbistrimethylsilylamid, Kalium-t-butoxid
und Triethylamin) 1–24
Stunden von –78°C bis unter
Eiskühlen
unter Ergeben einer Verbindung der Formel (38) umgesetzt. Diese
Verbindung wird in einem geeigneten Lösungsmittel (z. B. Methanol,
Ethanol, Butanol, Ethylenglykol, Methylenchlorid, Chloroform und
Lösungsmittelgemische wahlfreier
Mitglieder davon) in Gegenwart einer Verbindung der Formel (39)
1–24 Stunden
bei Raumtemperatur bis zur Rückflußtemperatur
des Lösungsmittels
unter Ergeben einer Verbindung der Formel (I-3) umgesetzt.
-
-
Gemäß dem in
Org. Syntheses Coll., Bd. 5, S. 808 (1973), beschriebenen Verfahren
kann eine Enaminverbindung der Formel (40) aus einer Verbindung
der Formel (36) und einem sekundären
Amin (z. B. Morpholin, Pyrrolidin und Piperidin) erhalten werden.
Gemäß dem in
Synthesis, S. 510 (1970), beschriebenen Verfahren kann eine Verbindung
der Formel (38) aus dieser Verbindung und einer Verbindung der Formel
(37) erhalten werden. Diese Verbindung wird in einem geeigneten
Lösungsmittel
(z. B. Methanol, Ethanol, Butanol, Ethylenglykol, Methylenchlorid,
Chloroform und Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart einer Verbindung der Formel
(39) 1–24
Stunden bei Raumtemperatur bis zur Rückflußtemperatur des Lösungsmittels
unter Ergeben einer Verbindung der Formel (I-3) umgesetzt.
-
-
-
Eine
durch das in Chemische Berichte, Bd. 92, S. 652 (1959), beschriebene
Verfahren erhaltene Verbindung der Formel (41) wird mit einer Verbindung
der Formel (26) unter Verwenden eines Kondensationsmittels wie etwa
1,3-Dicyclohexylcarbodiimid, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid
und Cyanphosphonsäurediester
in einem geeigneten Lösungsmittel,
das die Reaktion nicht stört
(z. B. Tetrahydrofuran, Dichlormethan, Dimethylformamid und Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart eines tertiären Amins
wie etwa Triethylamin 1–24
Stunden von unter Eiskühlen
bis Raumtemperatur unter Ergeben einer Verbindung der Formel (42)
umgesetzt. Wenn ein reaktionsfähiges
Derivat (Säurechlorid
und Acylimidazol) der Verbindung der Formel (41) verwendet wird,
läuft die
Reaktion in einem geeigneten Lösungsmittel,
das die Reaktion nicht stört
(z. B. Tetrahydrofuran, Dichlormethan, Chloroform, Benzol und Lösungsmittelgemische
wahlfreier Mitglieder davon), in Gegenwart eines tertiären Amins
wie etwa Triethylamin oder Pyridin 1–24 Stunden unter Eiskühlen bis
Raumtemperatur ab.
-
Diese
Verbindung wird in einem geeigneten Lösungsmittel (z. B. Methanol,
Ethanol, Butanol, Ethylenglykol, Methylenchlorid, Chloroform und
Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart einer Verbindung der Formel
(39) 1–24
Stunden bei Raumtemperatur bis zur Rückflußtemperatur des Lösungsmittels
unter Ergeben einer Verbindung der Formel (43) oder eines Gemisches
da von umgesetzt. Im Falle eines Gemisches kann es durch ein Reinigungsverfahren
wie etwa Kieselgel-Säulenchromatographie
und Umkristallisation getrennt werden. Die so erhaltene Verbindung
der Formel (43) oder (44) wird in einem geeigneten Lösungsmittel,
das die Reaktion nicht stört
(z. B. Tetrahydrofuran, Diethylether, Toluol und Lösungsmittelgemische
wahlfreier Mitglieder davon) unter Verwenden eines Reduktionsmittels
wie etwa Lithiumaluminiumhydrid und Boran 1–24 Stunden bei –78°C bis zur
Rückflußtemperatur
des Lösungsmittels
unter Ergeben einer Verbindung der Formel (45) reduziert.
-
-
Eine
Verbindung der Formel (41) wird in einem geeigneten Lösungsmittel
(z. B. Methanol, Ethanol, Butanol, Ethylenglykol, Methylenchlorid,
Chloroform und Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart einer Verbindung der Formel
(39) 1–24
Stunden bei Raumtemperatur bis zur Rückflußtemperatur des Lösungsmittel
unter Ergeben einer Verbindung der Formel (47) oder (48) oder eines
Gemisches davon umgesetzt. Diese Verbindung wird mit einer Verbindung
der Formel (26) unter Verwenden eines Kondensationsmittels wie etwa
1,3-Dicyclohexylcarbodiimid,
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid und Cy anphosphonsäurediester
in einem geeigneten Lösungsmittel,
das die Reaktion nicht stört
(z. B. Tetrahydrofuran, Dichlormethan, Dimethylformamid und Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart eines tertiären Amins
wie etwa Triethylamin 1–24
Stunden von unter Eiskühlung
bis Raumtemperatur unter Ergeben einer Verbindung der Formel (43)
oder (44) oder eines Gemisches davon umgesetzt. Im Falle eines Gemisches
kann es durch ein Reinigungsverfahren wie etwa Kieselgel-Säulenchromatographie
und Umkristallisation getrennt werden. Wenn ein reaktionsfähiges Derivat
(Säurechlorid
und Acylimidazol) der Verbindung der Formel (47) verwendet wird,
verläuft
die Reaktion in einem geeigneten Lösungsmittel, das die Reaktion nicht
stört (z.
B. Tetrahydrofuran, Dichlormethan, Chloroform, Benzol und Lösungsmittelgemische
wahlfreier Mitglieder davon) in Gegenwart eines tertiären Amins
wie etwa Triethylamin oder Pyridin 1–24 Stunden unter Eiskühlung bis
Raumtemperatur.
-
Die
so erhaltene Verbindung der Formel (43) wird in einem geeigneten
Lösungsmittel,
das die Reaktion nicht stört
(z. B. Tetrahydrofuran, Diethylether, Toluol und Lösungsmittelgemische
wahlfreier Mitglieder davon) unter Verwenden eines Reduktionsmittels
wie etwa Lithiumaluminiumhydrid und Boran 1–24 Stunden bei –78°C bis zur
Rückflußtemperatur
des Lösungsmittels
unter Ergeben einer Verbindung der Formel (45) reduziert.
-
Das
pharmazeutisch annehmbare Salz der Verbindung der Formel (I) wird
durch ein Säureadditionssalz
mit einer anorganischen Säure
oder organischen Säure
veranschaulicht, das durch Behandeln der Verbindung der Formel (I)
mit einer anorganischen Säure
(z. B. Salzsäure,
Bromwasserstoffsäure,
Schwefelsäure, Phosphorsäure und
Salpetersäure)
oder organischen Säure
(z. B. Essigsäure,
Propionsäure,
Bernsteinsäure, Glykolsäure, Milchsäure, Äpfelsäure, Weinsäure, Citronensäure, Maleinsäure, Fumarsäure, Methansulfonsäure, Benzolsulfonsäure, p-Toluolsulfonsäure, Camphersulfonsäure und
Ascorbinsäure)
durch ein herkömmliches
Verfahren veranschaulicht. Bei der Kristallisation der Verbindung
kann Oxalsäure
unter Ergeben eines Oxalats zugesetzt werden.
-
Die
auf diese Weise erhaltene erfinderische Verbindung kann durch ein
herkömmliches
Verfahren wie etwa Umkristallisation und Säulenchromatographie getrennt
und gereinigt werden. Wenn das erhaltene Produkt ein Racemat ist, können die
gewünschten
optisch aktiven Verbindungen durch präparative Umkristallisation
aus einem Salz mit einer optisch aktiven Säure oder durch Fließenlassen
durch eine mit einem optisch aktiven Träger gepackte Säule erhalten
werden. Jedes Diastereomer kann durch präparative Umkristallisation, Chromatographie
und dergleichen getrennt werden. Diese können auch durch die Verwendung
eines optisch aktiven Ausgangsmaterials erhalten werden. Außerdem kann
ein Stereoisomer durch Umkristallisation, Säulenchromatographie und dergleichen
getrennt werden.
-
Insofern,
als die kondensierte heterocyclische Verbindung der vorliegenden
Erfindung, ein optisches Isomer davon und ein pharmazeutisch annehmbares
Salz davon sowohl eine starke Blockadewirkung auf den D4-Rezeptor
und 5-HT2-Rezeptor als auch eine Blockade der
NMDA-Rezeptorunterfunktion zeigen, können sie brauchbare Antipsychotika
sein, die nicht nur bei positiven Symptomen, die sich auf Halluzinieren
und Wahn konzentrieren und für
den Akutzustand von Schizophrenie kennzeichnend sind, sondern auch
bei negativen Symptome einer Apathie, Abulie und Autismus wirksam
sind. Von ihnen wird auch erwartet, daß sie Antipsychotika darstellen,
die mit weniger Nebenwirkungen wie etwa extrapyramidale Symptome
und endokrine Störung
verbunden sind, die beobachtet werden, wenn ein herkömmliches
Antipsychotikum mit einer D2-Rezeptorblockadewirkung
verabreicht wird. Daher kann die erfinderische Verbindung als Therapeutikum
für Krankheiten
wie etwa Schizophrenie verwendet werden.
-
Wenn
die erfinderische Verbindung als Pharmazeutikum verwendet wird,
wird die erfinderische Verbindung mit pharmazeutisch annehmbaren
Trägern
(z. B. Arzneimittelträger,
Bindemittel, Zerfallhilfsmittel, Korrektiva, Korrigentien, Emulgatoren,
Verdünnungsmittel
und Löslichmacher)
unter Ergeben einer pharmazeutischen Zusammensetzung vermischt,
die anschließend
durch ein herkömmliches
Verfahren unter Ergeben von Tabletten, Pillen, Kapseln, Granulaten,
Pulvern, Sirupen, Emulsionen, Elixieren, Suspensionen, Lösungen,
Injektionen, Transfusionen und Suppositorien formuliert wird, die
oral oder parenteral verabreicht werden können.
-
In
der vorliegenden Beschreibung wird unter parenteral die subkutane
Injektion, intravenöse
Injektion, intramuskuläre
Injektion, intraperitoneale Injektion, Transfusion und dergleichen
verstanden. Eine Zubereitung zur Injektion wie etwa sterile wäßrige und ölige Suspensionen
zur Injektion können
durch ein auf diesem Gebiet bekanntes Verfahren unter Verwenden
eines geeigneten Dispergiermittels, Feuchtemittels und Suspendiermittels
hergestellt werden. Die sterile Zubereitung zur Injektion kann eine
sterile injizierbare Lösung
(z. B. wäßrige Lösung) oder
Suspension in einem Verdünnungsmittel
oder Lösungsmittel
sein, das nichttoxisch und parenteral verabreichbar ist. Beispiele
eines verwendbaren Trägers
und Lösungsmittels
schließen
Wasser, Ringer-Lösung,
isotone Kochsalzlösung
und dergleichen ein. Außerdem
kann ein steriles nichtflüchtiges Öl allgemein
als Lösungsmittel
oder als Suspendiermittel verwendet werden. Jedes nichtflüchtige Öl oder Fettsäure kann
zu diesem Zweck verwendet werden und Beispiele davon schließen natürliche,
synthetische oder halbsynthetische Lipidöle oder Fettsäure und
natürliche,
synthetische oder halbsynthetische Mono-, Dioder Triglyceride ein.
Ein Suppositorium zur rektalen Verabreichung kann beim Mischen eines
Wirkstoffs mit einem geeigneten, nichtreizenden Träger wie
etwa Kakaobutter und Polyethylenglykole, die bei Normaltemperatur
fest und flüssig bei
der Temperatur des Darms sind und die im Rektum unter Freisetzen
des Wirkstoffs schmelzen, hergestellt werden.
-
Die
feste Dosierungsform zur oralen Verabreichung kann zum Beispiel
ein vorstehend angeführtes Pulver,
Granulat, Tablette, Pille oder Kapsel sein. Bei diesen Dosierungsformen
kann die aktive Verbindung mit wenigstens einem Additiv wie etwa
Sucrose, Lactose, Cellulosezucker, Mannit, Maltit, Dextran, Stärken, Agar, Arginate,
Chitine, Chitosane, Pektine, Tragacanthgummi, Gummiarabicum, Gelatinen,
Kollagene, Casein, Albumin, synthetische oder halbsynthetische Polymere
und Glycerine vermischt werden. Ein Produkt mit einer derartigen
Dosierungsform kann weiter verschiedene Additive wie üblich wie
etwa inerte Verdünnungsmittel, Schmiermittel
wie etwa Magnesiumstearat, Konservierungsmittel wie etwa Natrium-p-hydroxybenzoat
und Sorbinsäuren,
Antioxidantien wie etwa Ascorbinsäure, α-Tocopherol und Cystein, Zerfallhilfsmittel,
Bindemittel, Verdickungsmittel, Puffer, Süßstoffe, Aromen, Duftstoffe
und dergleichen enthalten. Tabletten und Pillen können magensaftresistent
beschichtet sein. Die flüssige
Zubereitung zur oralen Verabreichung wird durch eine pharmazeutisch
annehmbare Emulsion, Sirup, Elixier, Suspension, Lösung und
dergleichen veranschaulicht, die ein normalerweise auf diesem Gebiet
verwendetes inertes Verdün nungsmittel
wie etwa Wasser enthalten kann.
-
Die
Dosis wird unter Berücksichtigung
des Alters, Körpergewichts,
allgemeinen Gesundheitszustands, Geschlechts, Ernährung, Verabreichungszeit,
Verabreichungsweg, Ausscheidungsrate, Kombination von Wirkstoffen,
des zu behandelnden Krankheitszustands des Patienten zu diesem Zeitpunkt
und anderen Faktoren bestimmt. Die erfinderische Verbindung, ein
optisches Isomer davon und ein pharmazeutisch annehmbares Salz davon
sind wenig toxisch und können
sicher verwendet werden. Die Tagesdosis, die einer Änderung
gemäß dem Zustand
und Körpergewicht
eines Patienten, der Art der Verbindung, Verabreichungsweg und dergleichen
unterliegt, ist etwa 0,01–50
mg/Patient/Tag, vorzugsweise 0,01–20 mg/Patient/Tag bei subkutaner,
intravenöser,
intramuskulärer
oder intraperitonealer Verabreichung und etwa 0,01–150 mg/Patient/Tag, vorzugsweise
0,1–100
mg/Patient/Tag bei oraler Verabreichung.
-
Die
vorliegende Erfindung wird unter Bezug auf die Ausgangsmaterial-Synthesebeispiele,
Beispiele und Formulierungsbeispiele, auf die die vorliegende Erfindung
nicht beschränkt
ist, genauer beschrieben.
-
Ausgangsmaterial-Synthesebeispiel
1
-
Hydrazinhydrat
(1,1 g) wurde einer Lösung
(40 ml) von 4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure (4,1 g)
in Ethanol zugesetzt und das Gemisch wurde 1 Stunde unter Rückfluß erhitzt.
Nach Abschluß der
Reaktion wurde das Reaktionsgemisch abgekühlt und die ausgefallenen Kristalle
wurden unter Ergeben von 3,9 g 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure durch
Filtration gesammelt; Schmp. 135–136°C.
-
Ausgangsmaterial-Synthesebeispiel
2
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(2-oxocyclohexyl)propionsäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wurde, wurde 2-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)essigsäure erhalten;
Schmp. 155–156°C.
-
Ausgangsmaterial-Synthesebeispiel
3
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(2-oxocyclohexyl)valeriansäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wurde, wurde 4-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)-n-buttersäure erhalten;
Schmp. 94–96°C.
-
Ausgangsmaterial-Synthesebeispiel
4
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(2-oxocyclopentyl)-n-buttersäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wurde, wurde 3-(2,4,5,6-Tetrahydrocyclopentapyrazol-3-yl)propionsäure erhalten;
Schmp. 157–159°C.
-
Ausgangsmaterial-Synthesebeispiel
5
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(2-oxocycloheptyl)-n-buttersäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wurde, wurde 3-(2,4,5,6,7,8-Hexahydrocycloheptapyrazol-3-yl)propionsäure erhalten;
Schmp. 102–105°C.
-
Ausgangsmaterial-Synthesebeispiel
6
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(4-oxo-2,3,5,6-tetrahydro-4H-pyran-3-yl)-n-buttersäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wird, wird 3-(2,4,6,7-Tetrahydropyrano[4,3-c]pyrazol-3-yl)propionsäure erhalten.
-
Ausgangsmaterial-Synthesebeispiel
7
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(4-oxo-2,3,5,6-tetrahydro-4H-thiopyran-3-yl)-n-buttersäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wird, wird 3-(2,4,6,7-Tetrahydrothiopyrano[4,3-c]pyrazol-3-yl)propionsäure erhalten.
-
Ausgangsmaterial-Synthesebeispiel
8
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Oxo-4-(1-benzoyl-4-oxopiperidin-3-yl)-n-buttersäure anstatt
4-Oxo-4-(2-oxocyclohexyl)-n-buttersäure als
Ausgangsmaterial verwendet wurde, wurde 3-(5-Benzoyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin-3-yl)propionsäure erhalten;
Schmp. 143–145°C.
-
Ausgangsmaterial-Synthesebeispiel
9
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß Methylhydrazin
anstatt Hydrazinhydrat als Ausgangsmaterial verwendet wird, wird
3-(4,5,6,7-Tetrahydro-2-methyl-2H-indazol-3-yl)propionsäure erhalten.
-
Ausgangsmaterial-Synthesebeispiel
10
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 2-Pyridylhydrazin anstatt
Hydrazinhydrat als Ausgangsmaterial verwendet wird, wird 3-(4,5,6,7-Tetrahydro-2-(2-pyridyl)-2H-indazol-3-yl)propionsäure erhalten.
-
Ausgangsmaterial-Synthesebeispiel
11
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß Phenylhydrazin
anstatt Hydrazinhydrat als Ausgangsmaterial verwendet wird, wird
3-(4,5,6,7-Tetrahydro-2-phenyl-2H-indazol-3-yl)propionsäure erhalten.
-
Ausgangsmaterial-Synthesebeispiel
12
-
Auf
dieselbe Weise wie bei Ausgangsmaterial-Synthesebeispiel 1, außer daß 4-Chlorphenylhydrazin anstatt
Hydrazinhydrat als Ausgangsmaterial verwendet wird, wird 3-(2-(4-Chlorphenyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)propionsäure erhalten.
-
-
Einer
Lösung
von 1-Morpholino-1-cyclohepten in Chloroform werden Triethylamin
und 4-(4-(4-Chlorphenyl)piperidin-1-yl)butanoylchlorid unter Rühren zugesetzt
und das Gemisch wird bei Raumtemperatur gerührt. Nach Abschluß der Reaktion
wird das Reaktionsgemisch in Wasser gegossen und mit Chloroform
extrahiert und der Extrakt wird mit Wasser gewaschen und über Magnesiumsulfat
getrocknet. Die Lösung
wird unter verringertem Druck eingeengt. Einer Mischlösung des
erhaltenen 2-(4-(4-(4-Chlorphenyl)piperidin-1-yl)butanoyl)cycloheptanons
in Chloroform und Methanol wird Hydrazinmonohydrat unter Rühren zugesetzt
und das Gemisch wird 6 Stunden bei Raumtemperatur gerührt. Nach
Abschluß der
Reaktion wird das Reaktionsgemisch in Wasser gegossen und das Gemisch
wird mit Chloroform extrahiert, mit Wasser gewaschen und über Magnesiumsulfat
getrocknet. Die Lösung
wird unter verringertem Druck eingeengt und der erhaltene Rückstand
wird der Kieselgel-Säulenchromatographie
unter Ergeben von 3-(3-(4-(4-Chlorphenyl)piperidin-1-yl)propyl)-2,4,5,6,7,8-hexahydrocyeloheptapyrazol
unterzogen.
-
-
Durch
dieselbe Reaktion und Behandlung wie in Beispiel 94 unter Verwenden
von 1-Morpholino-1-cyclohepten und 4-(4-(4-Chlorphenyl)-1,2,3,6-tetrahydropyridin-1-yl)butanoylchlorid
wird 3-(3-(4-(4-Chlorphenyl)-1,2,3,6-tetrahydropyridin-1-yl)propyl)-2,4,5,6,7,8-hexahydrocycloheptapyrazol
erhalten.
-
-
Durch
dieselbe Reaktion und Behandlung wie in Beispiel 94 unter Verwenden
von 1-Morpholino-1-cyclohepten und 4-(4-(4-Chlorphenyl)piperazin-1-yl)butanoylchlorid
wird 3-(3-(4-(4-Chlorphenylpiperazin-1-yl)propyl)-2,4,5,6,7,8-hexahydrocycloheptapyrazol
erhalten.
-
-
Einer
Lösung
von in Ausgangsmaterial-Synthesebeispiel 1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure (0,7
g), 4-(4-Chlorphenyl)piperidin (0,7 g) und Triethylamin (1,7 ml)
in Dimethylformamid (10 ml) wurde tropfenweise Diethylcyanphosphat
(0,7 ml) unter Rühren
zugesetzt und das Gemisch wurde 3 Stunden gerührt. Nach Abschluß der Reaktion
wurde das Reaktionsgemisch eingeengt, mit Chloroform extrahiert,
mit Wasser gewaschen und über
Magnesiumsulfat getrocknet. Die Lösung wurde unter verringertem Druck
eingeengt und Lithiumaluminiumhydrid wurde einer Lösung des
erhaltenen 4-(4-Chlorphenyl)-1-(3-(4,5,6,7-tetrahydro-2H-indazol-3-yl)propionyl)piperidins
in Tetrahydrofuran unter Eiskühlung
und Rühren
zugesetzt, woran sich 1 Stunde Rühren
bei Raumtemperatur anschloss. Nach Abschluß der Reaktion wird das Reaktionsgemisch
mit einem Lösungsmittelgemisch
aus Wasser-Tetrahydrofuran behandelt und durch Celite filtriert.
Das Filtrat wird unter verringertem Druck eingeengt, der erhaltene
Rückstand
wird der Kieselgel-Säulenchromatographie
unter Ergeben von 3-(3-(4-(4-Chlorphenyl)piperidin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
unterzogen.
-
-
Auf
dieselbe Weise wie bei Beispiel 102 wurde unter Verwenden von 4-(4-Chlorphenyl)-1,2,3,6-tetrahydropyridin
anstatt 4-(4-Chlorphenyl)piperidin 3-(3-(4-(4-Chlorphenyl)-1,2,3,6-tetrahydropyridin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol erhalten.
1H-MMR (CDCl3) δ: 1.67–1.85 (4H,
m) 1.89 (2H, tt, J = 6,7 Hz), 2.35–247 (2H, m) 2.52 (2H, t, J
= 7 Hz), 2.53–2.71
(6H, m), 2.71 (2H, t, J = 6 Hz), 3.12–3.22 (2H, m), 7.21–7.36 (4H,
m).
-
-
Auf
dieselbe Weise wie bei Beispiel 102 wurde unter Verwenden von 1-(4-Chlorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol-1/10-hydrat
erhalten; Schmp. 109–111°C.
-
-
Auf
dieselbe Weise wie bei Beispiel 102, außer daß 1-(4-Fluorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol
erhalten; Schmp. 106–108°C. Die Verbindung
wurde in ein Säureadditionssalz
von Maleinsäure
unter Ergeben von 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol-maleat
umgewandelt; Schmp. 141–142°C. Die Verbindung
wurde in ein Säureadditionssalz
von Salzsäure
unter Ergeben von 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol-hydrochlorid
umgewandelt; Schmp. 210–211°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, außer daß 1-(4-Methylphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde und anschließend durch
eine herkömmliche
Behandlung mittels Maleinsäure wurde
3-(3-(4-(4-Methylphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol-maleat
erhalten; Schmp. 144–146°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, außer daß 1-Phenylpiperazin anstatt
4-(4-Chiorphenyl)piperidin
verwendet wurde, wurde 3-(3-(4-Phenylpiperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol
erhalten; Schmp. 92–94°C. Die Verbindung
wurde in ein Säureadditionssalz
von Maleinsäure
unter Ergeben von 3-(3-(4-Phenylpiperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol-maleat
umgewandelt; Schmp. 123–125°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(4-Bromphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(4-Bromphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol-1/4-hydrat
er halten; Schmp. 145–147°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(3-Trifluormethylphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(3-Trifluormethylphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol erhalten.
1H-NMR (CDCl3) δ: 1.68–1.94 (6H,
m), 2.36–2.53
(4H, m), 2.58–2.74
(8H, m), 3.27 (4 H, t, J = 5 Hz), 7.01–7.17 (3H, m), 7.33 (1H, t,
J = 8 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(2-Chlorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(2-Chlorphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.66–1.92 (6H,
m), 2.32–2.45
(4H, m), 2.55–2.65
(8H, m) 3.12–3.20
(4H, m), 6.98 (1H, dt, J = 1,8 Hz), 7.07 (1H, dd, J = 2.8 Hz), 7.21
(1H, dt, J = 1,8 Hz), 7.34 (1H, dd, J = 1,8 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(2,5-Dichlorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(2,5-Dichlorphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.65–1.90 (6H,
m), 2.32–2.44
(4H, m), 2.52–2.73
(8H, m), 3.08–3.22
(4H, m), 6.95 (1H, dd, J = 2.9 Hz), 7.02 (1H, d, t, J = 3 Hz), 7.25
(1H, d, J = 9 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(3-Chlorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(3-Chlorphenyl)piperazin-1-yl)propyl-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.68–1.94 (6H,
m), 2.38–2.55
(4H, m), 2.58–2.65
(8H, m), 3.23–3.27
(4H, m), 6.79 (1H, dt, J = 2.8 Hz), 6.87 (1H, d, J = 2 Hz), 7.15
(1H, t, J = 8 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(5,6,7,8-Tetrahydronaphthalin-1-yl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(5,6,7,8-Tetrahydronaphthalin-1-yl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.75–1.92 (10H,
m), 2.41–2.52
(4H, m), 2.62–2.78
(12H, m), 2.94–2.97
(4H, m), 6.83 (1H, d, J = 7 Hz), 6.89 (1H, d, J = 7 Hz), 7.08 (1H,
t, J = 8 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(3,4-Dichlorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(3,4-Dichlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.68–1.94 (6H,
m), 2.33–2.51
(4H, m), 2.53–2.70
(8H, m), 3.19 (4H, t, J = 5 Hz), 6.72 (1H, dd, J = 3.9 Hz), 6.94
(1H, d, J = 3 Hz), 7.25 (1H, d, J = 9 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(2-Methoxyphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.68–1.90 (6H,
m), 2.38–2.50
(4H, m), 2.55–2.68
(8H, m), 3.19–3.25
(4H, m), 3.86 (3H, s), 6.84–7.04
(4H, m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
2 erhaltenes 1-(2-Naphthyl)piperazin anstatt 4-(4-Chlor phenyl)piperidin
verwendet wurde, wurde 3-(3-(4-(2-Naphthyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.73–2.10 (6H,
m), 2.41–2.52
(4H, m), 2.55–2.70
(8H, m), 3.10–3.25
(4H, m), 7.13 (1H, d, J = 2 Hz), 7.24–7.31 (2H, m), 7.37–7.42 (1H,
m), 7.73 (3H, t, J = 8 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(3-Methoxyphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(3-Methoxyphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.70–1.91 (6H,
m), 2.40–2.48
(4H, m), 2.59–2.66
(8H, m), 3.21–3.26
(4H, m), 3.78 (3H, s), 6.41 (1H, dd, J = 2 Hz, 8 Hz), 6.46 (1H,
t, J = 2 Hz), 6.53 (1H, dd, J = 2 Hz, 8 Hz), 7.16 (1H, t, J = 8
Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 4-(6-Fluor-1,2-benzoisoxazol-3-yl)piperidin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(6-Fluor-1,2-benzoisoxazol-3-yl)piperidin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol-monohydrat
erhalten; Schmp. 124–126°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(1-Naphthyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(1-Naphthyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.65–1.91 (6H,
m), 2.35–2.52
(4H, m), 2.55–2.70
(8H, m), 3.00–3.12
(4H, m), 7.14 (1H, d, J = 7 Hz), 7.40 (1H, t, J = 8 Hz), 7.45–7.50 (2H,
m), 7.58 (1H, d, J = 8 Hz), 7.81–7.85 (1H, m), 8.12–8.15 (1H,
m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(5-Methylbenzo[b]furan-3-yl)piperidin anstatt
4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(5-Methylbenzo[b]furan-3-yl)piperidin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.70–1.98 (6H,
m), 2.04–2.39
(6H, m), 2.36–2.55
(4H, m), 2.45 (3 H, s), 2.58–2.72
(4H, m), 3.01–3.22
(3H, m), 7.12 (1H, d, J = 9 Hz), 7.42 (1H, d, J = 9 Hz), 7.54 (1H,
s), 7.78 (1H, s).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
2 erhaltene 2-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)essigsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 3-(2-(4-(4-Fluorphenyl)piperazin-1-yl)ethyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
3 erhaltene 4-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)-n-buttersäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 3-(4-(4-(4-Fluorphenyl)piperazin-1-yl)butyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.52–1.83 (8H,
m), 2.35–2.44
(4H, m), 2.57–2.65
(8H, m), 3.11–3.15
(4H, m), 6.83–6.99 (4H,
m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
4 erhaltene 3-(2,4,5,6-Tetrahydrocyclopentapyrazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, und anschließend
durch eine herkömmliche
Behandlung mittels Salzsäure
wurde 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,5,6-tetrahydrocyclopentapyrazol-dihydrochlorid-dihydrat
erhalten; Schmp. 228–230°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
4 erhaltene 3-(2,4,5,6-Tetrahydrocyclopentapyrazol-3-yl)propionsäure und
1-(4-Methylphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 3-(3-(4-(4-Methylphenynpiperazin-1-yl)propyl)-2,4,5,6-tetrahydrocyclopentapyrazol
erhalten; Schmp. 101–102°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
5 erhaltene 3-(2,4,5,6,7,8-Hexahydrocycloheptapyrazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet wurden, wurde 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,5,6,7,8-hexahydrocycloheptapyrazol
erhalten.
1H-NMR (CDCl3) δ: 1.57–1.70 (4H,
m), 1.77–1.87
(4H, m), 2.43–2.48
(4H, m), 2.61– 2.66
(6H, m), 2.70–2.75 (2H,
m), 3.14–3.18
(4H, m), 6.48–7.00
(4H, m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
6 erhaltene 3-(2,4,6,7-Tetrahydropyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydropyrano[4,3-c]pyrazol
erhalten.
1H-NMR (CDCl3) δ: 1.85 (2H,
tt, J = 6.6 Hz), 2.48 (2H, t, J = 7 Hz), 2.57–2.71 (6H, m), 2.78 (2H, t,
J = 6 Hz), 3.18 (4H, t, J = 5 Hz), 3.93 (2H, t, J = 6 Hz), 4.65
(2H, s), 6.82–7.01
(4H, m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
7 erhaltene 3-(2,4,6,7-Tetrahydrothiopyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3- yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydrothiopyrano[4,3-c]pyrazol erhalten;
Schmp. 110–111°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
8 erhaltene 3-(5-Benzoyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 5-Benzyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
erhalten.
1H-NMR (CDCl3) δ: 1.82 (2H,
tt, J = 7.7 Hz), 2.46 (2H, t, J = 7 Hz), 2.55–2.67 (6H, m), 2.77 (4H, s),
3.12–3.20 (4H,
m), 3.44 (2H, s), 3.72 (2H, s), 6.82–6.98 (4H, m), 7.21–7.40 (5
H, m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
8 erhaltene 3-(5-Benzoyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 5- Benzyl-3-(3-(4-(4-chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
erhalten.
1H-NMR (CDCl3) δ: 1.82 (2H,
tt, J = 7.7 Hz), 2.45 (2H, t, J = 7 Hz), 2.52–2.66 (6H, m), 2.76 (4H, s),
3.14–3.24 (4H,
m), 3.43 (2H, s), 3.72 (2H, s), 6.84 (2H, t, J = 9 Hz), 7.19 (2H,
t, J = 9 Hz), 7.27–7.40
(5H, m).
-
-
In
Beispiel 139 erhaltenes 5-Benzyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
(0,5 g) wurde in Ethanol (10 ml) gelöst und Raneynickel (0,2 g)
wurde zum Ausführen
einer katalytischen Reduktion zugesetzt. Nach Abschluß der Reaktion
wurde das Reaktionsgemisch durch Celite filtriert und das Filtrat
wurde unter verringertem Druck eingeengt. Der erhaltene Rückstand
wurde der Kieselgel-Säulenchromatographie
unter Ergeben von 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
unterzogen.
1H-NMR (CDCl3) δ: 1.86 (2H,
tt, J = 7.6 Hz), 2.45 (2H, t, J = 7 Hz), 2.55–2.66 (4H, m), 2.89 (2H, t,
J = 6 Hz), 3.00–3.14
(6H, m), 3.16–3.23
(2H, s), 6.81–6.98
(4H, m).
-
-
In
Beispiel 141 erhaltenes 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
(0,3 g) wurde in einer Mischlösung
von Chloroform (5 ml) und gesättigter,
wäßriger Natriumhydrogencarbonatlösung (5
ml) gelöst
und Acetylchlorid (0,3 ml) wurde tropfenweise unter Eiskühlung zuge setzt. Nach
der tropfenweisen Zugabe wurde die Chloroformschicht verteilt und über Magnesiumsulfat
getrocknet. Das Lösungsmittel
wurde unter verringertem Druck verdampft und der erhaltene Rückstand
wurde der Kieselgel-Säulenchromatographie
unter Ergeben von 5-Acetyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin
unterzogen.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
9 erhaltene 3-(4,5,6,7-Tetrahydro-2-methyl-2H-indazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2-methyl-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.69–1.82 (6H,
m), 2.37–2.45
(4H, m), 2.57–2.65
(8H, m), 3.10–3.14
(4H, m), 3.75 (3H, s), 6.83–6.99
(4H, m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
9 erhaltene 3-(4,5,6,7-Tetrahydro-2-methyl-2H-indazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet wurden, wurde 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2-methyl-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.76 (6H,
m), 2.42 (4H, m), 2.56–2.65
(8H, m), 3.18 (4H, m), 3.76 (3 H, s), 6.83 (2H, d, J = 7 Hz), 7.18
(2H, d, J = 7 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
9 erhaltene 3-(4,5,6,7-Tetrahydro-2-methyl-2H-indazol-3-yl)propionsäure und
1-(4-Methylphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet werden, wird 4,5,6,7-Tetrahydro-2-methyl-3-(3-(4-(4-methylphenyl)piperazin-1-yl)propyl)-2H-indazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
9 erhaltene 3-(4,5,6,7-Tetrahydro-2-methyl-2H-indazol-3-yl)propionsäure und
1-Phenylpiperazin anstatt in Ausgangsmaterial-Synthesebeispiel 1
erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 4,5,6,7-Tetrahydro-2-methyl-3-(3-(4-phenylpiperazin-1-yl)propyl)-2H-indazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
10 erhaltene 3-(4,5,6,7-Tetrahydro-2-(2-pyridyl)-2H-indazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet wurden, wurde 3-(3-(4-(4-Chlorphenylpiperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2-(2-pyridyl)-2H-indazol
erhalten; Schmp. 101–103°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
10 erhaltene 3-(4,5,6,7-Tetrahydro-2-(2-pyridyl)-2H-indazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet werden, wird 3-(3-(4-(4-Fluorphenylpiperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2-(2-pyridyl)-2H-indazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
11 erhaltene 3-(4,5,6,7-Tetrahydro-2-phenyl-2H-indazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet wurden, wurde 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2-phenyl-2H-indazol
erhalten; Schmp. 127–129°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
11 erhaltene 3-(4,5,6,7-Tetrahydro-2-phenyl-2H-indazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2-phenyl-2H-indazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
12 erhaltene 3-(2-(4-Chlorphenyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, wurde 2-(4-Chlorphenyl)-3-(3-(4-(4-chlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol erhalten;
Schmp. 129–131°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
12 erhaltene 3-(2-(4-Chlorphenyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)propionsäure und
1-(4-Fluorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 2-(4-Chlorphenyl)-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
4 erhaltene 3-(2,4,5,6-Tetrahydrocyclopentapyrazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet wurden, und anschließend
durch eine herkömmliche
Behandlung mittels Salzsäure
wurde 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-2,4,5,6-tetrahydrocyclopentapyrazol-hydrochlorid
erhalten; Schmp. 228–230°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
4 erhaltene 3-(2,4,5,6-Tetrahydrocyclopentapyrazol-3-yl)propionsäure und
1-Phenylpiperazin anstatt in Ausgangsmaterial-Synthesebeispiel 1
erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 2,4,5,6-Tetrahydro-3-(3-(4-phenylpiperazin-1-yl)propyl)cyclopentapyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial- Synthesebeispiel
5 erhaltene 3-(2,4,5,6,7,8-Hexahydrocycloheptapyrazol-3-yl)propionsäure und
1-(4-Methylphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 2,4,5,6,7,8-Hexahydro-3-(3-(4-(4-methylphenyl)piperazin-1-yl)propyl)cycloheptapyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
5 erhaltene 3-(2,4,5,6,7,8-Hexahydrocyclopentapyrazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet werden, wird 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-2,4,5,6,7,8-hexahydrocycloheptapyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
5 erhaltene 3-(2,4,5,6,7,8-Hexahydrocycloheptapyrazol-3-yl)propionsäure und
1-Phenylpiperazin anstatt in Ausgangsmaterial-Synthesebeispiel 1
erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 2,4,5,6,7,8-Hexahydro-3-(3-(4-phenylpiperazin-1-yl)propyl)cycloheptapyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
6 erhaltene 3-(2,4,6,7-Tetrahydropyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-(4-Methylphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet werden, wird 2,4,6,7-Tetrahydro-3-(3-(4-(4-methylphenyl)piperazin-1-yl)propyl)pyrano[4,3-c]pyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
6 erhaltene 3-(2,4,6,7-Tetrahydropyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 3-(3-(4-(4-Chlorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydropyrano[4,3-c]pyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
6 erhaltene 3-(2,4,6,7-Tetrahydropyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-Phenylpiperazin anstatt in Ausgangsmaterial-Synthesebei spiel 1
erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 2,4,6,7-Tetrahydro-3-(3-(4-phenylpiperazin-1-yl)propyl)pyrano[4,3-c]pyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
7 erhaltene 3-(2,4,6,7-Tetrahydrothiopyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-(4-Methylphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet werden, wird 2,4,6,7-Tetrahydro-3-(3-(4-(4-methylphenyl)piperazin-1-yl)propyl)thiopyrano[4,3-c]pyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
7 erhaltene 3-(2,4,6,7-Tetrahydrothiopyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-(4-Chlorphenyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet werden, wird 3-(3-(4-(4-chlorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydrothiopyrano[4,3-c]pyrazol
erhalten.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
7 erhaltene 3-(2,4,6,7-Tetrahydrothiopyrano[4,3-c]pyrazol-3-yl)propionsäure und
1-Phenylpiperazin anstatt in Ausgangsmaterial-Synthesebeispiel 1
erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und 4-(4-Chlorphenyl)piperidin
verwendet werden, wird 2,4,6,7-Tetrahydro-3-(3-(4-phenylpiperazin-1-yl)propyl)thiopyrano[4,3-c]pyrazol
erhalten.
-
-
Einem
Gemisch aus 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
(3,1 g), Dimethylformamid (30 ml) und Triethylamin (2,5 ml) wurde
tropfenweise Acetylchlorid (0,72 ml) unter Eiskühlung zugesetzt. Das Gemisch
wurde so wie es war 30 Minuten gerührt. Das Reaktionsgemisch wurde
in Eiswasser (200 ml) gegossen. Das Gemisch wurde mit Ethylacetat
extrahiert, mit Wasser gewaschen und über wasserfreiem Magnesiumsulfat
getrocknet. Das Filtrat wurde unter verringertem Druck unter Ergeben
von 3,4 g öliger
Substanz eingeengt. Das Öl
wurde der Kieselgel-Säulenchromatographie
(Elutionsmittel: Hexan:Ethylacetat = 1:2) unter Ergeben von 0,60
g 1-Acetyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-1H-indazol
(später
eluierte Komponente (Verbindung A)) und 0,30 g 2-Acetyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
(zuerst eluierte Komponente (Verbindung B)) unterzogen. 1-Acetyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-1H-indazol
(Verbindung A: Schmp. 60–61°C); 2-Acetyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
(Verbindung B; Schmp. 104–106°C).
-
-
Auf
dieselbe Weise wie in Beispiel 194, ausgenommen, daß Benzoylchlorid
(1,2 ml) anstatt Acetylchlorid verwendet wurde, wurden 1,0 g 1-Benzoyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-1H-indazol
(später
eluierte Komponente (Verbindung A)) und 0,45 g 2-Benzoyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
(zuerst eluierte Komponente (Verbindung B)) erhalten. 1-Benzoyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-1H-indazol
(Verbindung A: Schmp. 71–72°C); 2-Benzoyl-3-(3-(4-(4-fluorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol (Verbindung B:
Schmp. als Maleat 147–148°C).
-
-
Aus
Diisopropylamin (2,8 ml) und 1 M n-Butyllithium (11 ml) in Tetrahydrofuran
(40 ml) hergestellte Lithiumdiisopropylamidlösung wurde auf –78°C gekühlt und
es wurde Cyclohexanon (2,0 ml) zugesetzt. Das Gemisch wurde 1 Stunde
bei –78°C gerührt und
aus N-tert-Butoxycarbonylisonipecotinsäure hergestelltes 1-tert-Butoxycarbonyl-4-imidazocarbonylpiperidin
(4,8 g) wurde zugesetzt. Das Gemisch wurde 1 Stunde bei Raumtemperatur
gerührt.
Das Reaktionsgemisch wurde eingeengt und dem erhaltenen Rückstand
wurde Wasser zugesetzt. Das Gemisch wurde mit Ethylacetat extrahiert
und der Extrakt wurde über
wasserfreiem Magnesiumsulfat getrocknet. Das Filtrat wurde unter
Ergeben von 3,8 g einer öligen
Substanz unter verringertem Druck eingeengt. Das Öl wurde
der Kieselgel-Säulenchromatographie
(Elutionsmittel: Chloroform:Methanol = 20:1) unter Ergeben von 2-((1-tert-Butoxycarbonylpiperidin-4-yl)carbonyl)cyclohexanon
unterzogen; Schmp. 71–72°C.
-
2-((1-tert-Butoxycarbonylpiperidin-4-yl)carbonyl)cyclohexanon
(1,0 g) wurde in Trifluoressigsäure
(10 ml) gelöst
und die Lösung
wurde 30 Minuten bei Raumtemperatur gerührt. Das Lösungsmittel wurde verdampft und
der Rückstand
wurde mit wäßriger Kaliumcarbonatlösung alkalisch
gemacht und mit Ethylacetat extrahiert. Nach dem Trocknen über Magnesiumsulfat
wurde das Lösungsmittel
unter verringertem Druck verdampft und dem erhaltenen Rückstand
wurde Dimethylformamid (10 ml), 4-Fluorphenylessigsäure (0,5
g) und Triethylamin (0,9 ml) zugesetzt. Diethylcyanphosphat (0,5
ml) wurde tropfenweise in einem Eisbad unter Rühren des Gemischs zugesetzt.
Das Gemisch wurde 3 Stunden weitergerührt. Nach Abschluß der Reaktion
wurde das Lösungsmittel
unter verringertem Druck verdampft und dem erhaltenen Rückstand
wurde Wasser zugesetzt. Das Gemisch wurde mit Chloroform extrahiert
und der Extrakt wurde über
Magnesiumsulfat getrocknet. Das Lösungsmittel wurde unter verringertem
Druck ver dampft. Einer Lösung
(50 ml) des erhaltenen 2-((1-(2-(4-Fluorphenyl)acetyl)piperidin-4-yl)carbonyl)cyclohexanons
in Ethanol wurde Hydrazinhydrat (0,2 g) zugesetzt und das Gemisch
wurde 1 Stunde unter Rückfluß erhitzt.
Das Lösungsmittel
wurde unter verringertem Druck verdampft und der erhaltene Rückstand
wurde der Kieselgel-Säulenchromatographie
unter Ergeben von 3-(1-(2-(4-Fluorphenyl)acetyl)piperidin-4-yl)-4,5,6,7-tetrahydro-2H-indazol
unterzogen.
1H-NMR (CDCl3) δ: 1.45–1.98 (8H,
m), 2.39 (2H, t, J = 6 Hz), 2.59 (2H, t, J = 6 Hz), 2.63–2.91 (2H,
m), 3.14 (2H, d, t, J = 3.13 Hz), 3.72 (2H, s), 3.94 (1H, d, J =
14 Hz), 4.78 (1H, d, J = 14 Hz) 6.93–7.06 (2H, m), 7.18–7.30 (2H,
m).
-
Einer
Lösung
des erhaltenen 3-(1-(2-(4-Fluorphenyl)acetyl)piperidin-4-yl)-4,5,6,7-tetrahydro-2H-indazols
in Tetrahydrofuran wurde Lithiumaluminiumhydrid unter Eiskühlen und
Rühren
zugesetzt und das Gemisch wurde 1 Stunde bei Raumtemperatur gerührt. Nach
Abschluß der
Reaktion wurde das Reaktionsgemisch mit einem Lösungsmittelgemisch aus Wasser – Tetrahydrofuran
behandelt und durch Celite filtriert. Das Filtrat wurde unter verringertem
Druck eingeengt und der erhaltene Rückstand wurde der Kieselgel-Säulenchromatographie
unter Ergeben von 3-(1-(2-(4-Fluorphenyl)ethyl)piperidin-4-yl)-4,5,6,7-tetrahydro-2H-indazol-dihydrochlorid-monohydrat
unterzogen; Schmp. 143–145°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß in Ausgangsmaterial-Synthesebeispiel
3 erhaltene 4-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)-n-buttersäure und
1-(2-Pyridyl)piperazin anstatt in Ausgangsmaterial-Synthesebeispiel
1 erhaltener 3-(4,5,6,7-Tetrahydro-2H-indazol-3-yl)propionsäure und
4-(4-Chlorphenyl)piperidin verwendet wurden, wurde 3-(4-(4-(2-pyridyl)piperazin-1-yl)butyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.55–1.80 (8H,
m), 2.34–2.44
(4H, m), 2.52–2.64
(8H, m), 3.53–3.57
(4H, m), 6.58–6.65 (2H,
m), 7.43–7.49
(1H, m), 8.18 (1H, dd, J = 2.6 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(2-Pyridyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(2-Pyridyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.66–1.92 (6H,
m), 2.40–2.48
(4H, m), 2.56–2.72
(8H,m ), 3.55–3.59
(4H, m), 6.59–6.65 (2H,
m), 7.43–7.50
(1H, m), 8.18 (1H, dd, J = 2.4 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(4-Chlorphenyl)-3-methylpiperazin anstatt
4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(4-Chlorphenyl)-2-methylpiperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol erhalten.
1H-NMR (CDCl3) δ: 1.01 (3H,
d, J = 6 Hz), 1.68–1.94
(6H, m), 2.30–2.56
(6H, m), 2.58–2.73
(5H, m), 2.79–2.89 (1H,
m), 3.10–3.23
(2H, m), 3.78–3.88
(1H, m), 6.83 (2H, d, J = 9 Hz), 7.19 (2H, d, J = 9 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 4-Phenylpiperidin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-Phenyl piperidin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.69–1.92 (10H,
m), 1.98–2.13
(2H, m), 2.40–2.52
(5H, m), 2.61–2.71
(4H, m), 3.02–3.24 (2H,
m), 7.14–7.34
(5H, m).
-
-
In
Beispiel 138 erhaltenes 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydrothiopyrano[4,3-c]pyrazol
(0,5 g) wurde in Ameisensäure
(5 ml) gelöst
und 30%iges Wasserstoffperoxid (0,27 ml) wurde tropfenweise unter
Eiskühlen
zugesetzt und das Gemisch wurde 30 Minuten bei höchstens 5°C gerührt. Das Reaktionsgemisch wurde
in Eiswasser gegossen, mit Kaliumcarbonat alkalisch gemacht und
mit Ethylacetat extrahiert. Nach dem Trocknen über Magnesiumsulfat wurde die
Lösung
eingeengt und unter Ergeben von 3-(3-(4-(4-Fluorphenyl)piperazin-1-yl)propyl)-2,4,6,7-tetrahydrothiopyrano[4,3-c]pyrazol-5-oxid
aus Isopropylalkohol-Isopropylether (1:2) umkristallisiert; Schmp.
140–142°C.
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(2,3-Dichlorphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 3-(3-(4-(2,3-Dichlorphenyl)piperazin-1-yl)propyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.68–1.95 (6H,
m), 2.43 (2H, t, J = 6 Hz), 2.51 (2H, t, J = 7 Hz), 2.56–2.77 (8H,
m), 3.02–3.16
(4H, m), 6.92–6.99
(1H, m), 7.08–7.17
(2H, m).
-
-
Auf
dieselbe Weise wie in Beispiel 102, ausgenommen, daß 1-(2,3-Dimethylphenyl)piperazin
anstatt 4-(4-Chlorphenyl)piperidin verwendet wurde, wurde 4,5,6,7-Tetrahydro-3-(3-(4-(2,3-dimethylphenyl)piperazin-1-yl)propyl)-2H-indazol erhalten.
1H-NMR (CDCl3) δ: 1.68–1.92 (6H,
m), 2.21 (3H, m), 2.26 (3H, s), 2.43 (2H, t, J = 6 Hz), 2.49 (2H,
t, J = 7 Hz), 2.55–2.76
(8H, m), 2.89–3.02
(4H, m), 6.99 (1H, d, J = 7 Hz), 6.93 (1H, d).
-
-
Auf
dieselbe Weise wie in Beispiel 133, ausgenommen, daß 1-(2,3-Dimethylphenyl)piperazin
anstatt 1-(4-Fluorphenyl)piperazin verwendet wurde, wurde 3-(4-(4-(2,3-Dimethylphenyl)piperazin-1-yl)butyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.56–1.83 (8H,
m), 2.26 (3H, s), 2.34 (3H, s), 2.41–2.46 (4H, m), 2.57–2.64 (8H,
m), 2.90–2.94
(4H, m), 6.90 (2H, t, J = Hz), 7.07 (1H, t, J = 8 Hz).
-
-
Auf
dieselbe Weise wie in Beispiel 133, ausgenommen, daß 1-(2,3-Dichlorphe nyl)piperazin
anstatt 1-(4-Fluorphenyl)piperazin verwendet wurde, wurde 3-(4-(4-(2,3-Dichlorphenyl)piperazin-1-yl)butyl)-4,5,6,7-tetrahydro-2H-indazol
erhalten.
1H-NMR (CDCl3) δ: 1.53–1.88 (8H,
m), 2.41–2.47
(4H, m), 2.57–2.65
(8H, m), 3.07–3.10
(4H, m), 6.96 (1H, dd, J = 3.6 Hz), 7.13–7.15 (2H, m).
-
Formulierungsbeispiel
1
-
Die
erfinderische Verbindung (0,5 Teile), Lactose (25 Teile), kristalline
Cellulose (35 Teile) und Maisstärke
(3 Teile) wurden gründlich
vermischt und mit einem aus Maisstärke (2 Teile) hergestellten
Bindemittel gut verknetet. Das verknetete Produkt wurde durch ein
16-Mesh-Sieb geführt,
im Ofen bei 50°C
getrocknet und durch ein 24-Mesh-Sieb geführt. Das so erhaltene verknete
Produkt, Maisstärke
(8 Teile), kristalline Cellulose (11 Teile) und Talk (9 Teile) wurden
gründlich
gemischt und unter Ergeben 0,5 mg aktiven Bestandteil je Tablette
enthaltender Tabletten verpreßt.
-
Formulierungsbeispiel
2
-
Die
erfinderische Verbindung (1,0 mg) und Natriumchlorid (9,0 mg) werden
in injizierbarem Wasser gelöst
und die Lösung
wird zur Pyrogenentfernung filtriert. Das Filtrat wird aseptisch
in Ampullen gefüllt
und sterilisiert, was vom Zuschmelzen der Ampullen unter Ergeben
1,0 mg aktiven Bestandteil enthaltender Injektionen gefolgt wird.
-
Die überlegene
pharmakologische Aktivität
der Verbindung der Formel (I) wurde durch die folgende Reihe von
Rezeptorbindungstests, die Anti-Methamphetaminwirkung,
Katalepsieinduktion und supprimierende Wirkung auf die Neurotoxizität durch
MK-801 ermittelt.
-
Versuchsbeispiel 1: Affinität zum D4-Rezeptor, 3H-Spiperonbindung
-
Die
D4-Rezeptorexpressionszellmembranprobe und 3H-Spiperon wurden 2 Stunden bei 27°C in Gegenwart
einer Testverbindung inkubiert. Unmittelbar nach Abschluß der Reaktion
wurde das Reaktionsgemisch durch ein Whatman GF/B-Filter (Warenzeichen)
saugfiltriert und die Radioaktivität auf dem Filter wurde auf
einem Flüssigszintillationszähler bestimmt.
Das Ausmaß des
nicht-spezifischen Bindens wurde in Gegenwart von 10 μM Haloperidol
bestimmt. Die zum Hemmen um 50% notwendige Konzentration der Testverbindung
(IC50) wurde aus der nichtlinearen Regressionskurve
berechnet, auf deren Grundlage die Hemmkonstante (Ki-Wert) bestimmt
wurde.
-
Versuchsbeispiel 2: Affinität zum D2-Rezeptor, 3H-Spiperonbindung
-
Die
Präparation
einer rohen synaptischen Membran und der Bindungstest folgte dem
Verfahren von I. Creese et al., European Journal of Pharmacology,
Bd. 46, S. 377 (1977). Die rohe synaptische Membran wurde aus dem
kältekonservierten
Corpus striatum von Ratten präpariert
und die Membranprobe und 3H-Spiperon wurden
20 Minuten bei 37°C
in Gegenwart einer Testverbindung umgesetzt. Unmittelbar nach dem
Abschluß der
Reaktion wurde das Reaktionsgemisch durch ein Whatman GF/B-Filter
(Warenzeichen) saugfiltriert und die Radioaktivität auf dem
Filter wurde auf einem Flüssigszintillationszähler bestimmt.
Das Ausmaß des nicht-spezifischen
Bindens wurde in Gegenwart von 100 μM (+)-Sulpirid bestimmt. Die
zum Hemmen um 50% notwendige Konzentration der Testverbindung (IC50) wurde aus der nichtlinearen Regressionskurve
berechnet, auf deren Grundlage die Hemmkonstante (Ki-Wert) bestimmt
wurde.
-
Als
Ergebnis der Versuchsbeispiele 1 und 2 zeigte die erfinderische
Verbindung einen Ki-Wert für
den D4-Rezeptor von 0,01–10 nM, wogegen er für den D2-Rezeptor
mindestens 10 nM war. Daher wurde von der erfinderischen Verbindung
bestätigt,
daß sie
eine stärkere
Affinität
zum D4-Rezeptor als zum D2-Rezeptor
aufwies. Im Gegensatz dazu zeigte Dapiprazol für den D4-Rezeptor
oder D2-Rezeptor
kaum eine Affinität.
-
Versuchsbeispiel 3: Affinität für den 5-HT2-Rezeptor
-
Ein
spezieller Serotonin-2-(5-HT2)-Rezeptorbindungstest
folgte dem Verfahren von Mol. Pharmacol., Bd. 21, S. 301 (1981).
-
Eine
rohe Synaptosomenfraktion wurde aus dem Hippocampus 9–10 Wochen
alter Wistar-Ratten abgetrennt und zu dem Versuch in 50 mM Tris-HCl-Puffer
(pH 7,7) suspendiert. Die Testverbindung in mehreren Konzentrationen
und tritiiertes Ketanserin (Endkonzentration 0,2 nM) wurden der
Synaptosomensuspension zugesetzt und jedes Gemisch wurde 20 Minuten
bei 37°C
inkubiert. Nach der Inkubation wurde das Gemisch durch ein Whatman
GF/B-Glasfilter (Warenzeichen) saugfiltriert. Das Filter wurde mit
50 nM Tris-HCl-Puffer (pH 7,7) gewaschen und die Radioaktivität auf dem
Filter wurde durch einen Flüssigszintillationszähler gemessen.
Das nicht-spezifische Binden wurde in Gegenwart von 105 Mianserin
bestimmt. Die zum Hemmen um 50% notwendige Konzentration (IC50) wurde aus einer graphischen Darstellung
berechnet und die Hemmkonstante (Ki-Wert) wurde berechnet.
-
Als
Ergebnis zeigte die erfinderische Verbindung eine starke Affinität von 0,01–50 nM für den 5-HT2-Rezeptor.
-
Versuchsbeispiel 4: Bestimmung
der Maus-anti-Methamphetaminwirkung (Hauptwirkung)
-
Männliche
ddY-Mäuse
(20–30
g, 4 Wochen alt, 15 Mäuse
je Gruppe) wurden zu dem Versuch verwendet. Eine Testverbindung
wurde den Mäusen
oral verabfolgt und eine Stunde später wurde eine wäßrige Lösung von
Methamphetamin (DAINIPPON PHARMACEUTICAL CO., LTD., 1 mg/kg) in
physiologischer Kochsalzlösung
subkutan verabfolgt. Unmittelbar danach wurden die Mäuse in ein
Meßgerät gesetzt,
das mit einem Paar Infrarotstrahlen ausgerüstet war und ein Innenmaß von 25 × 15 × 14 (Höhe) cm aufwies.
Die Anzahl der Kreuzungen der Infrarotstrahlen in 30 Minuten von
10 Minuten bis 40 Minuten nach Beobachtungsbeginn wurde als Index
zum Aufzeigen einer durch Methamphetamin geförderten Bewegung verwendet
und die supprimierende Wirkung des ED50-Werts
der Testverbindung wurde berechnet.
-
Im
Ergebnis zeigte die erfinderische Verbindung eine durch einen ED50-Wert von höchstens 1,0 mg/kg (p. o.) ausgedrückte starke
Aktivität.
-
Versuchsbeispiel 5: Bestimmung
der Katalepsieinduktion (Nebenwirkungen)
-
Männliche
ddY-Mäuse
(20–30
g, 4 Wochen alt, 8 Mäuse
je Gruppe) wurden zu dem Versuch verwendet. Eine Testverbindung
wurde den Mäusen
verabfolgt und 1, 3, 5 und 7 Stunden später wurde die Zeit (Katalepsiezeit)
bis zu einem Höchstwert
von 30 Sekunden gemessen, währenddessen
die Vordergliedmaßen
auf einen Stab gestellt waren, der in einer Höhe von 4 cm in horizontaler
Richtung befestigt war und die Körperposition
wurde in einem Winkel von etwa 45 Grad gehalten. Die Stärke der
Katalepsieinduktion der Testverbindung wurde durch Addieren der
Katalepsiezeiten an 4 Bestimmungspunkten bei jeder Dosis (wobei
der Gesamtwert ein Summenwert ist) und Berechnen der ED10s durch
Regression zu der Dosis, die die Durchschnittszeit 10 Sekunden werden
ließ,
ermittelt.
-
Im
Ergebnis war die ED10s der erfinderischen
Verbindung mindestens 20 mg/kg, was eine schwache Katalepsieinduktion
anzeigt.
-
Versuchsbeispiel 6: Unterdrückende Wirkung
auf die Neurotoxizität
durch MK-801
-
Weibliche
SD-Ratten (200–300
g, 9–12
Wochen alt) wurden zu dem Test verwendet. Eine wäßrige Lösung der Testverbindung (0,1
ml/kg) wurde intraperitoneal verabfolgt und eine wäßrige Lösung von
(+)-MK-801 (0,5 mg/kg, 0,1 ml/kg, Research Biochemicals International,
Natick, MA, USA) wurde 15 Minuten später subkutan verabfolgt. 4
Stunden nach der Verabfolgung von MK-801 wurden die Ratten mit Pentobarbital
betäubt. Eine
Injektionsnadel wurde in die linke Ventrikel der Ratten eingeführt und
die linke Auricula wurde eröffnet. Nach
dem Ausbluten durch Perfusion mit physiologischer Kochsalzlösung (ca.
100 ml) wurden die Ratten unter Perfusion mit 4% Paraformaldehyd – 1,5% Glutaraldehyd – 0,1 M
Phosphatpuffer (ca. 400–500
ml) fixiert. Der Kopf wurde eröffnet
und das Gehirn wurde entnommen. Ein Teil einschließlich des
hinteren Gyrus cinguli wurde herausgeschnitten und in das Fixiermittel
zur Nachfixierung gelegt. Nach der Paraffineinbettung wurde ein dünner Schnitt
(3 μm) hergestellt
und mit Hämatoxylin
und Eosin angefärbt.
Nach dem Entwässern
und Abdecken wurde der Objektträger
mit einem Lichtmikroskop betrachtet.
-
Als
Testergebnis wurde von der erfinderischen Verbindung gefunden, daß sie die
Neurotoxizität
(Zellvakuolosierung) des hinteren Gyrus cinguli, die bei der Verabfolgung
des NMDA-Rezeptor-antagonistischen Wirkstoffs MK-801 beobachtet
wird, unterdrückt.
Die Testergebnisse machen deutlich, daß die erfinderische Verbindung
die Verschlechterung der NMDA-Rezeptorfunktion verbessert.
-
Versuchsbeispiel 7: Akute
Toxizität
-
Die
erfinderische Verbindung (100 mg/kg) wurde oral an 4 weibliche SD-Ratten
(5 Wochen alt) verabfolgt, aber es wurde kein Tod beobachtet.
-
Die
erfinderische kondensierte heterocyclische Verbindung, ein optisches
Isomer davon und ein pharmazeutisch annehmbares Salz davon zeigen
sowohl eine starke Blockadewirkung auf den D4-Rezeptor
und 5-HT2-Rezeptor als auch eine Blockade
der NMDA-Rezeptorunterfunktion. Außerdem besitzt die erfinderische Verbindung
eine pharmakologische Wirkung wie etwa Anti-Methamphetaminwirkung,
die als Antipsychotikum notwendig ist, sowie eine starke unterdrückende Wirkung
auf die durch MK-801 ausgelöste
Neurotoxizität. Zum
anderen wurde gefunden, daß das
Auslösen
einer Katalepsie bei Mäusen,
das ein Index für
extrapyramidale Nebenwirkungen ist, extrem schwach ist. Die Ergebnisse
zeigen eine stärke
Abtrennung der Hauptwirkung von den Nebenwirkungen der erfinderischen
Verbindung.
-
Aus
den vorangehenden Ergebnissen ist deutlich, daß die erfinderischen Verbindungen
brauchbare Antipsychotika darstellen können, die nicht nur gegen positive
Symptome, die sich auf Halluzinieren und Wahn konzentrieren, die
für den
Akutzustand von Schizophrenie kennzeichnend sind, sondern auch gegen
negative Symptome einer Apathie, Abulie und Autismus wirksam sind.
Von ihnen wird erwartet, daß sie
hochsichere Antipsychotika sind, die mit weniger Nebenwirkungen
wie etwa extrapyramidalen Symptomen und endokriner Störung verbunden
sind, die beobachtet werden, wenn herkömmliche Antipsychotika mit
D2-Rezeptorblockadewirkung
verabreicht werden. Somit können
die erfinderischen Verbindungen als Therapeutika für Krankheiten
wie etwa Schizophrenie verwendet werden.
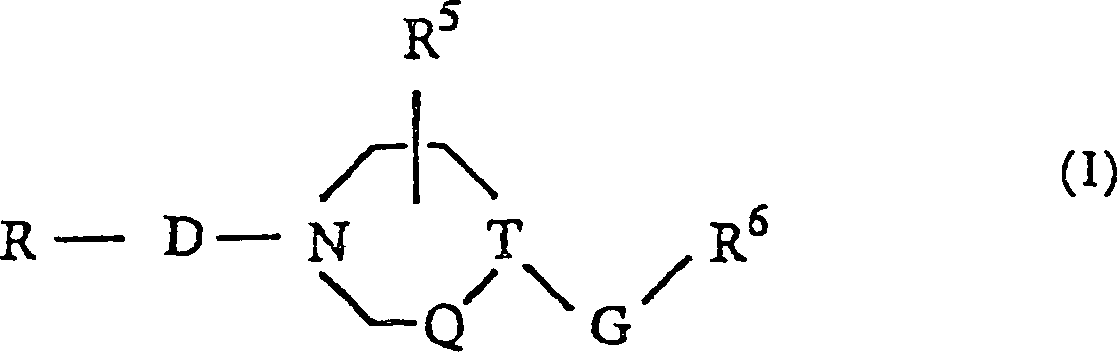
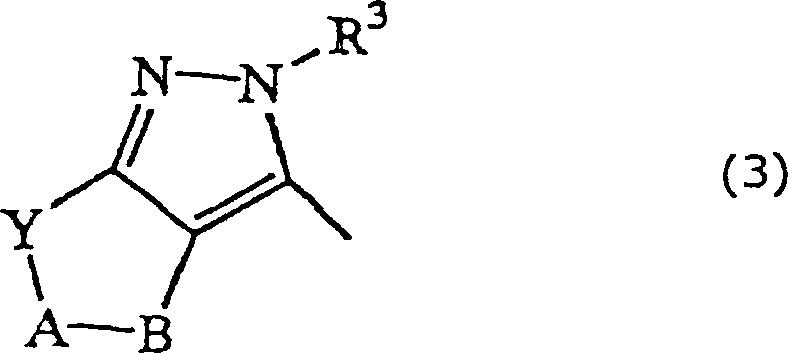


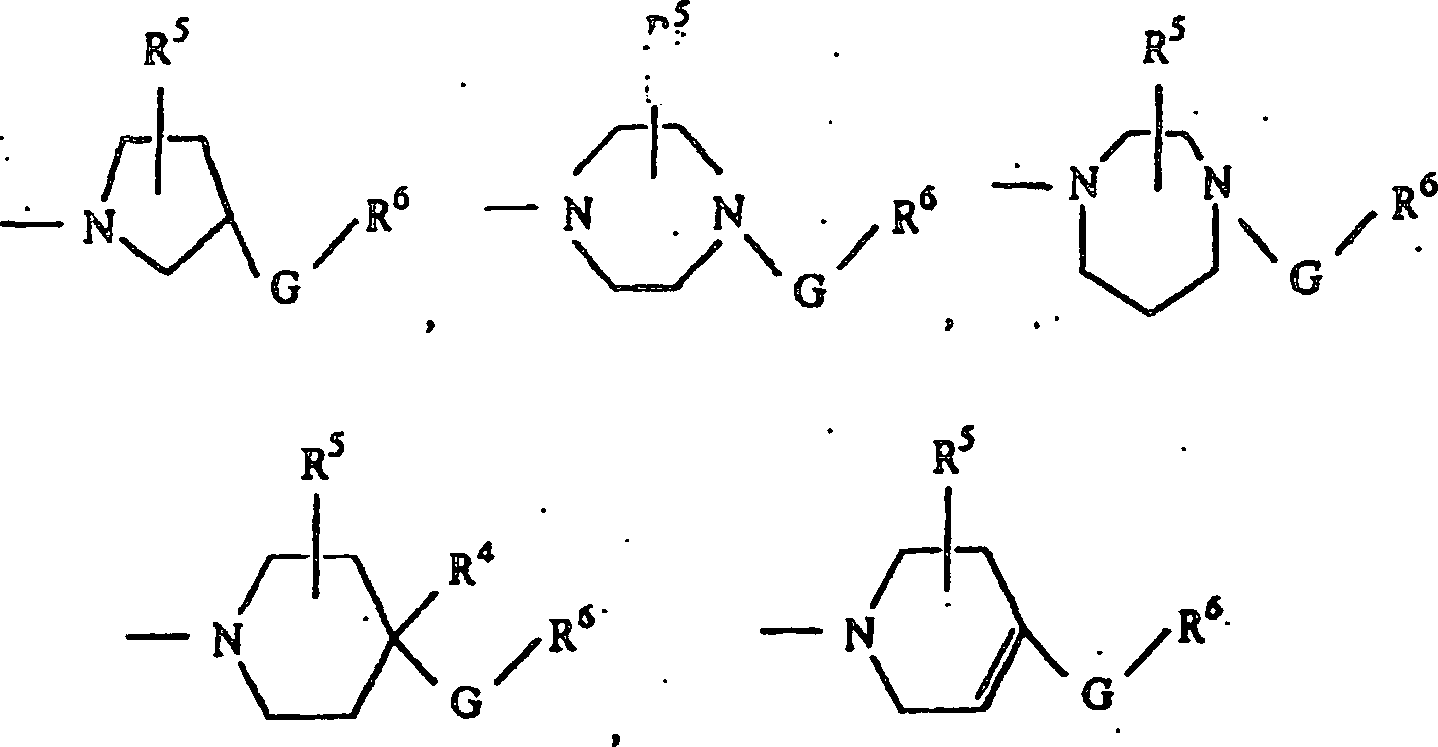
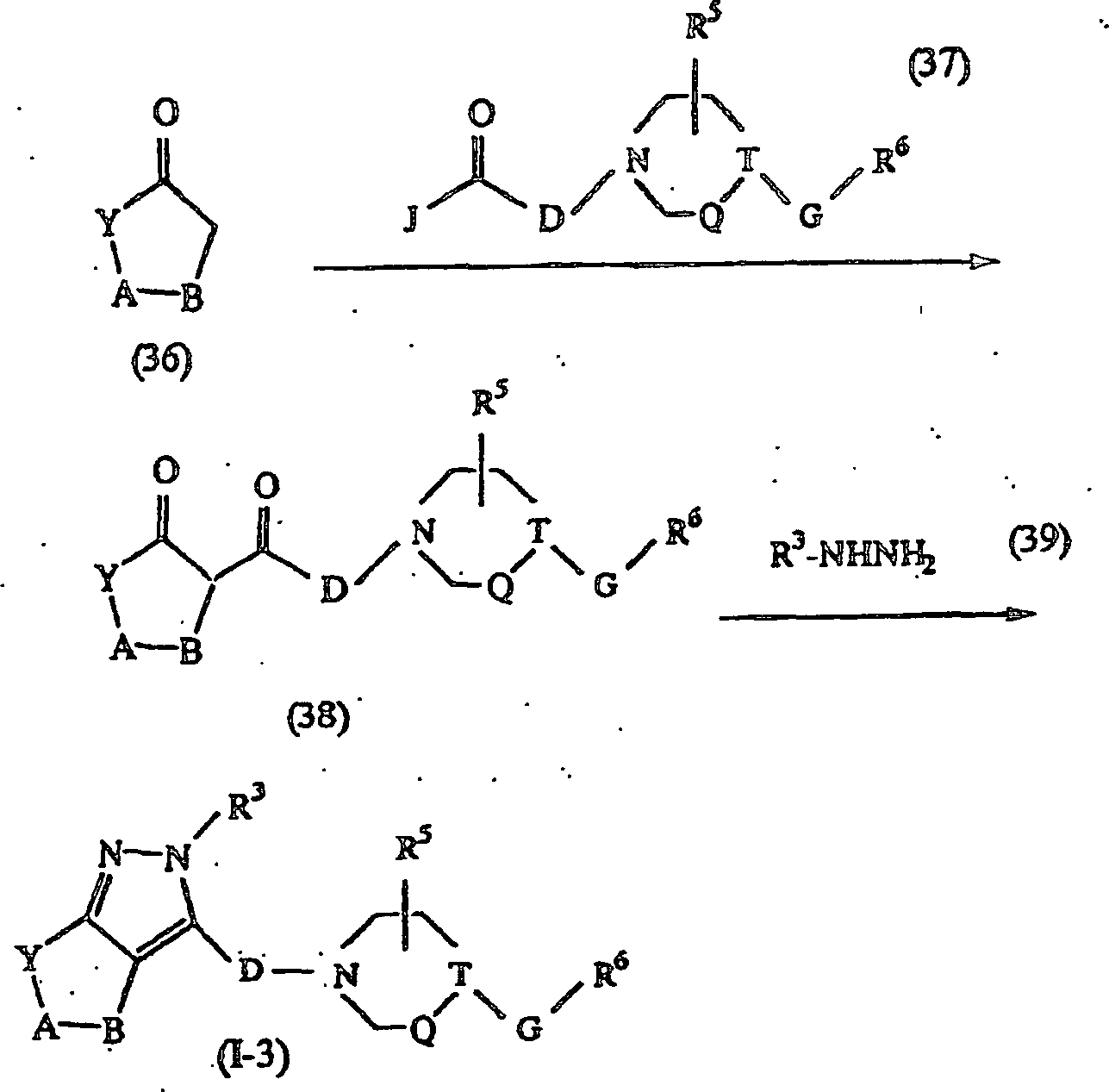
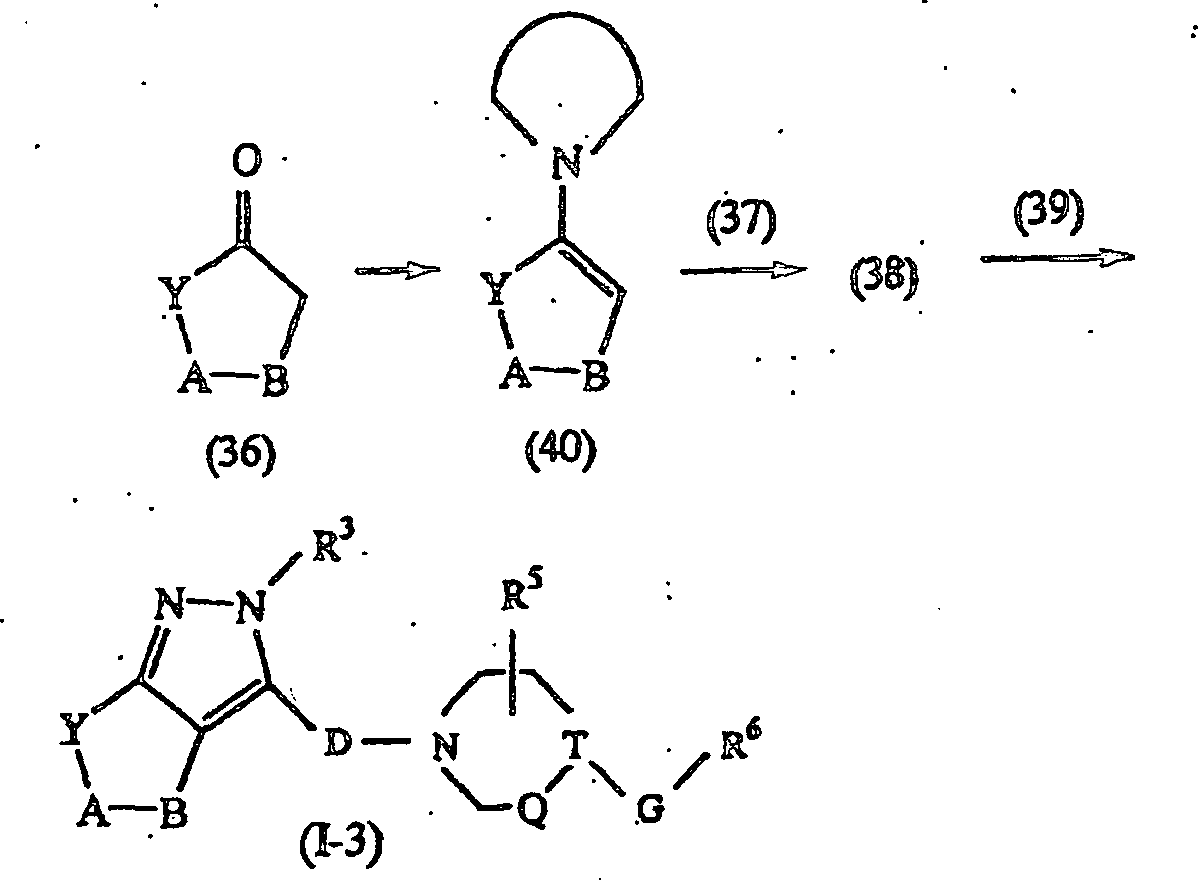

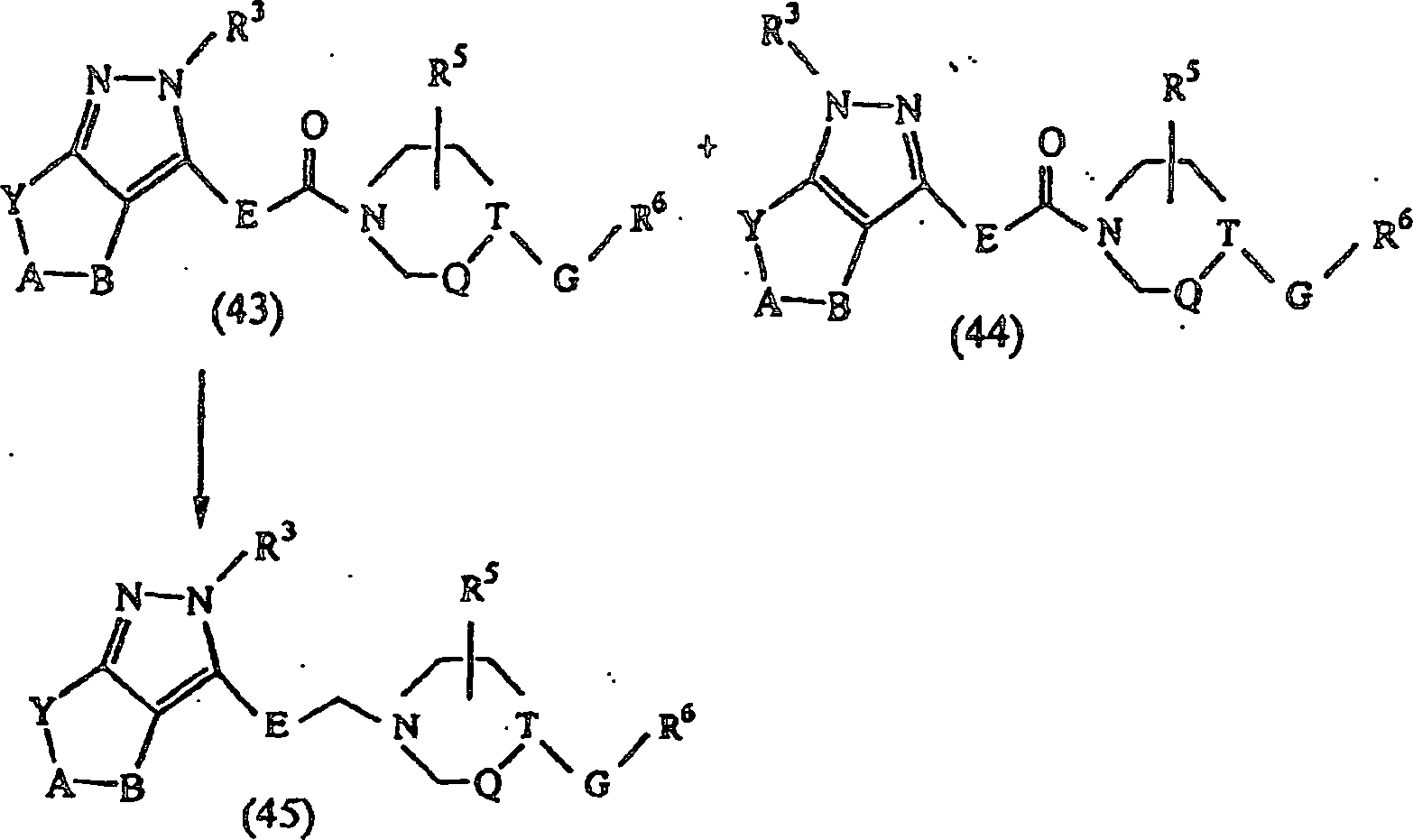
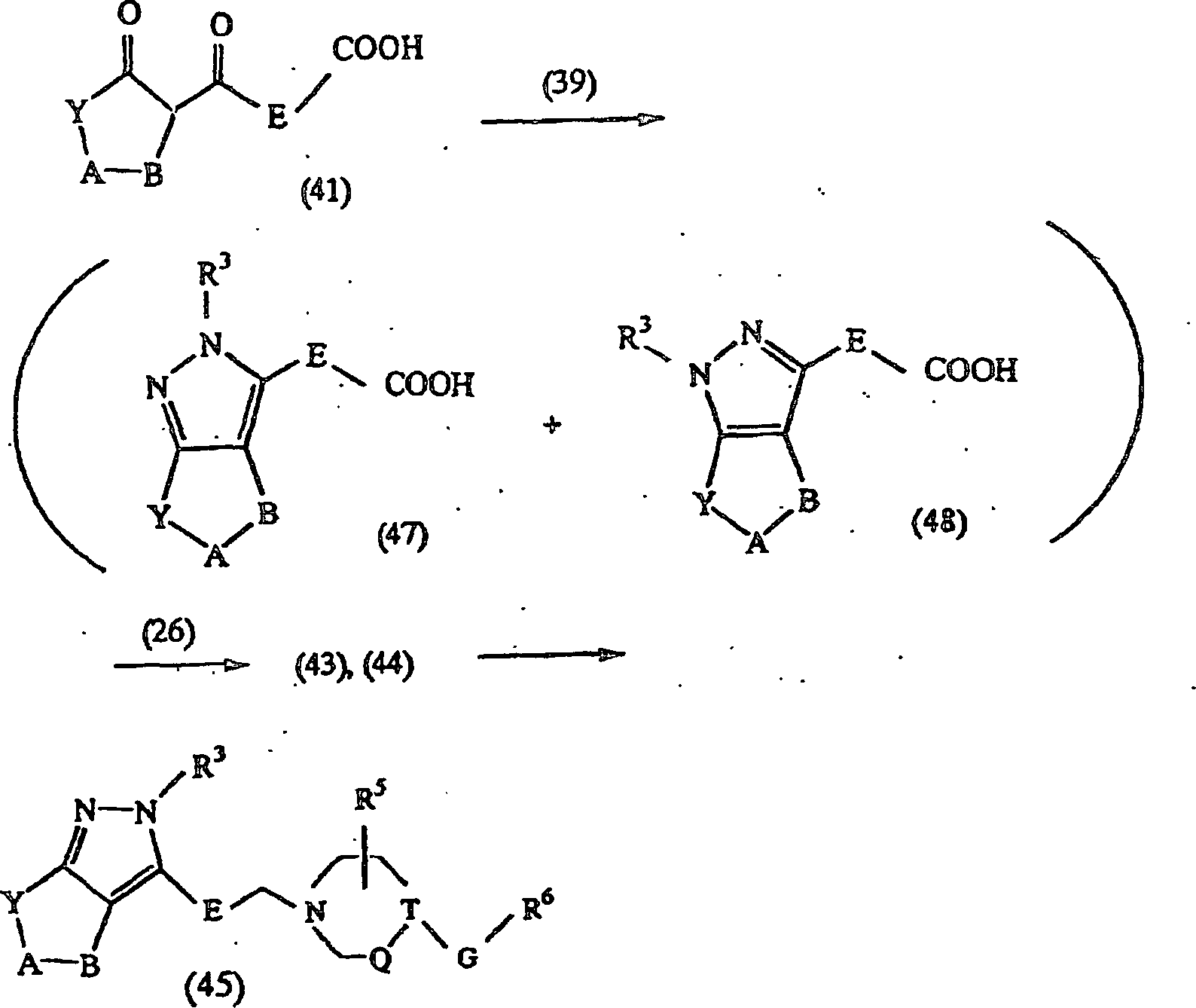



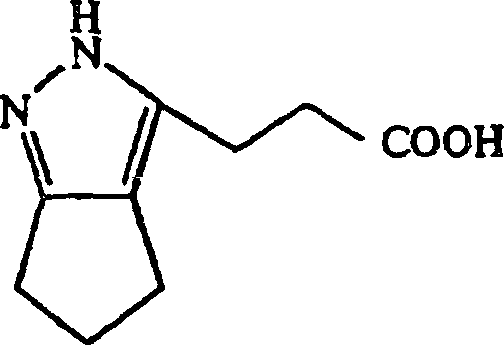

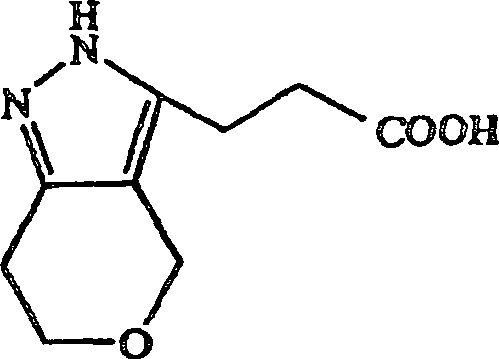
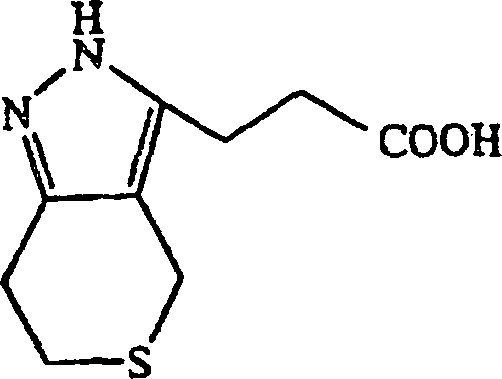

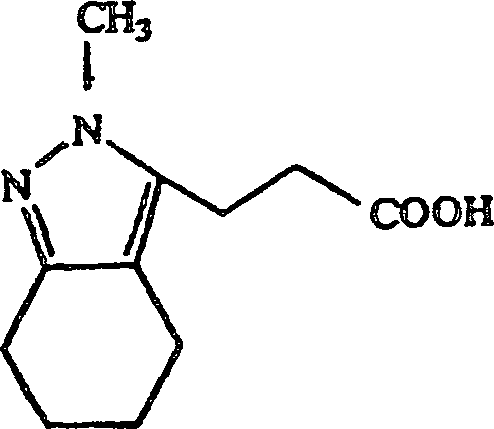

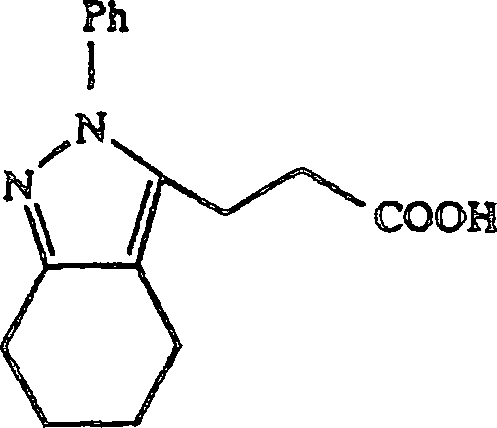

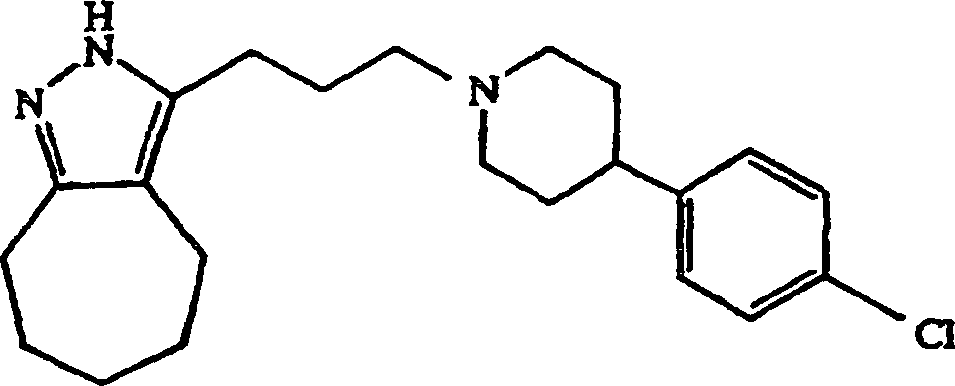






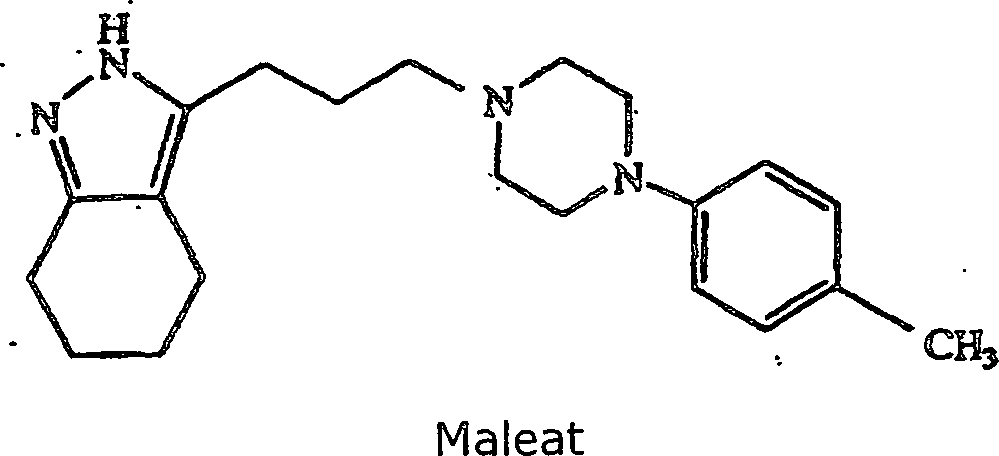
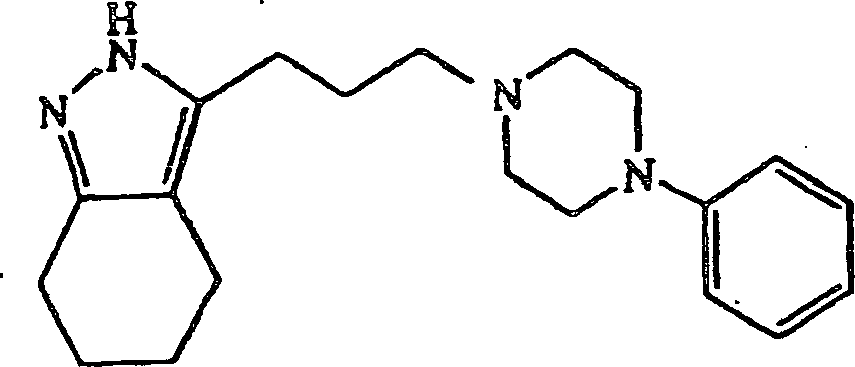
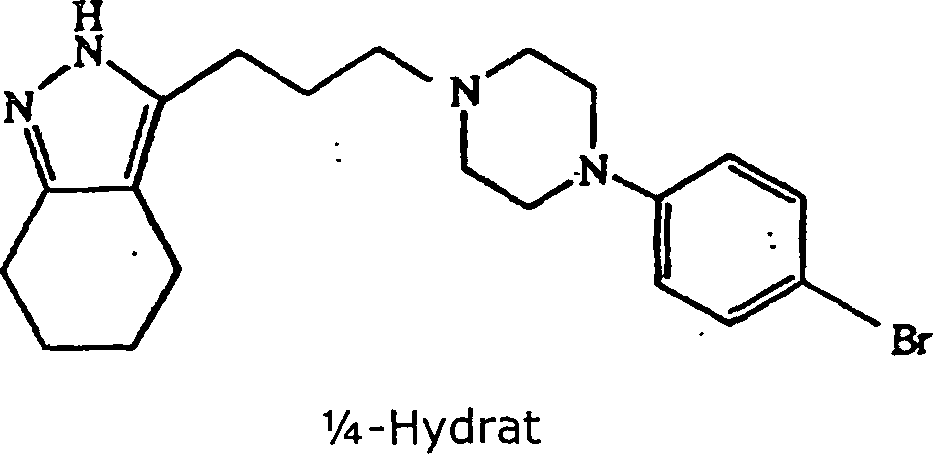
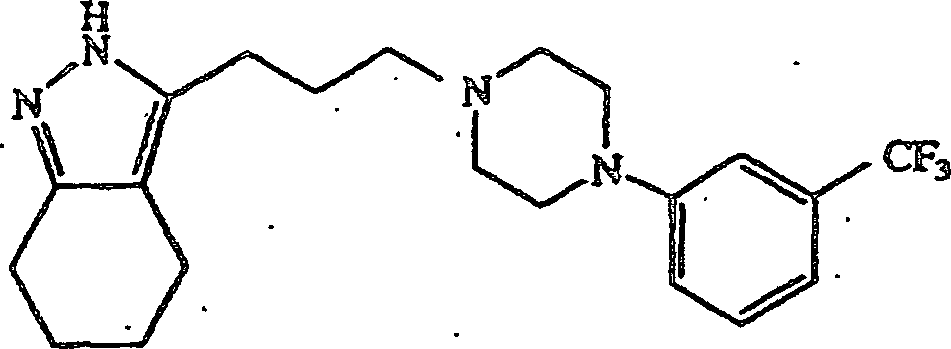


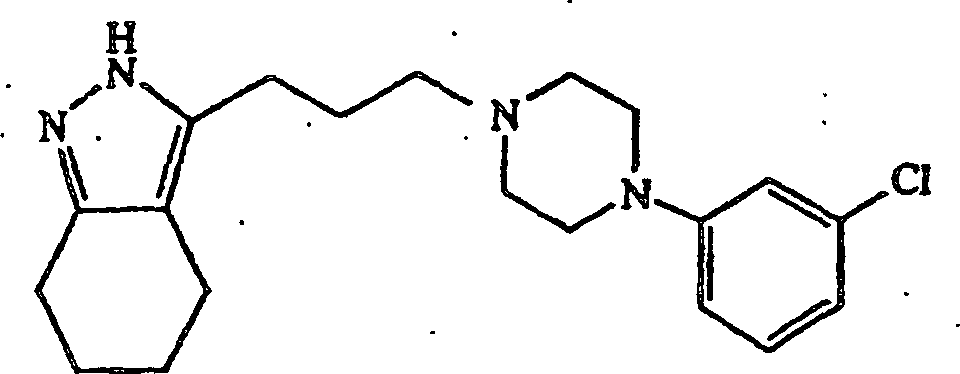


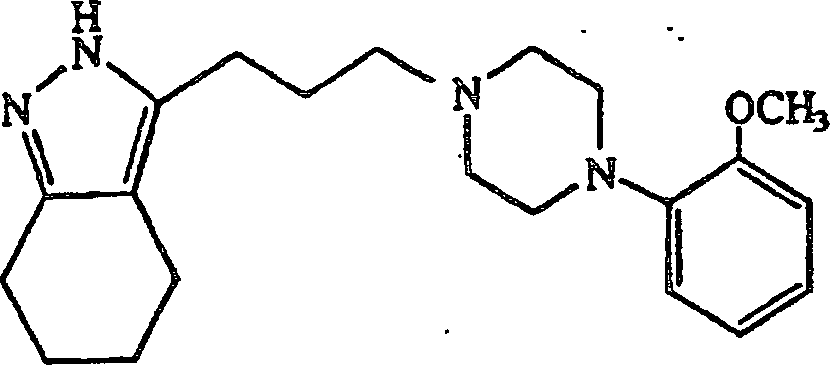
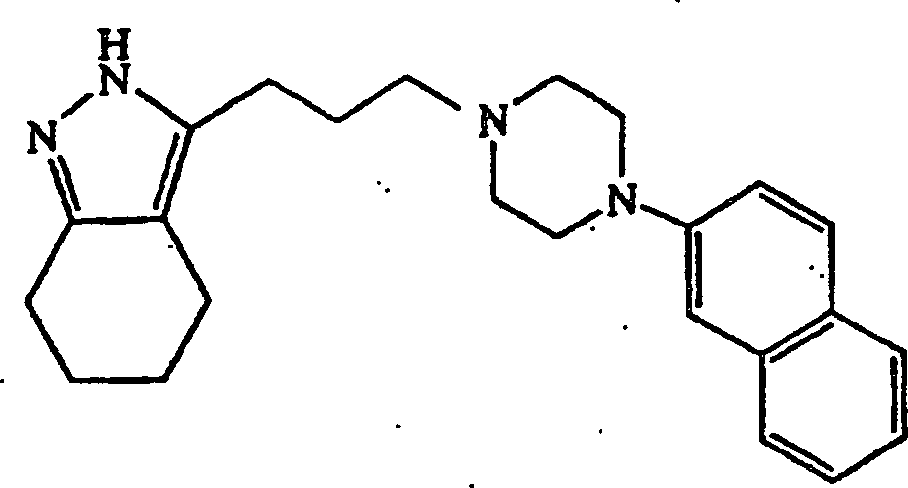

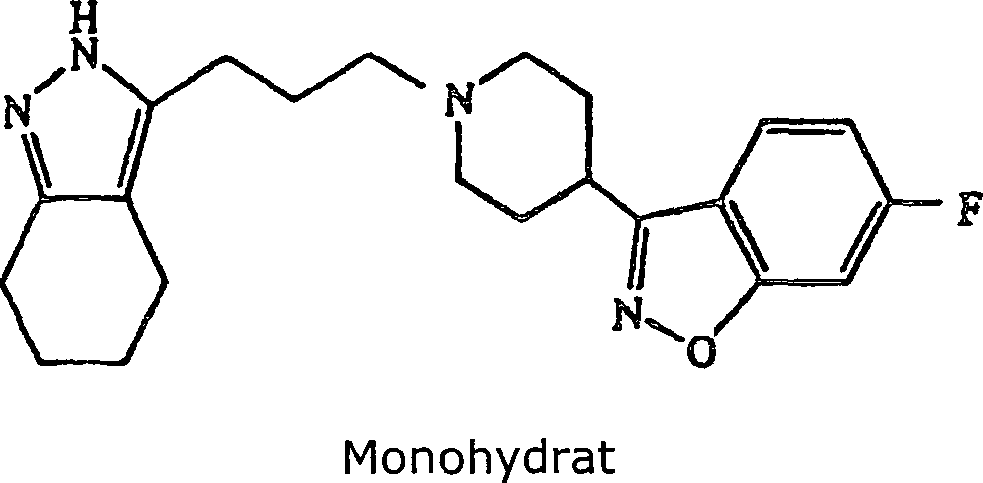
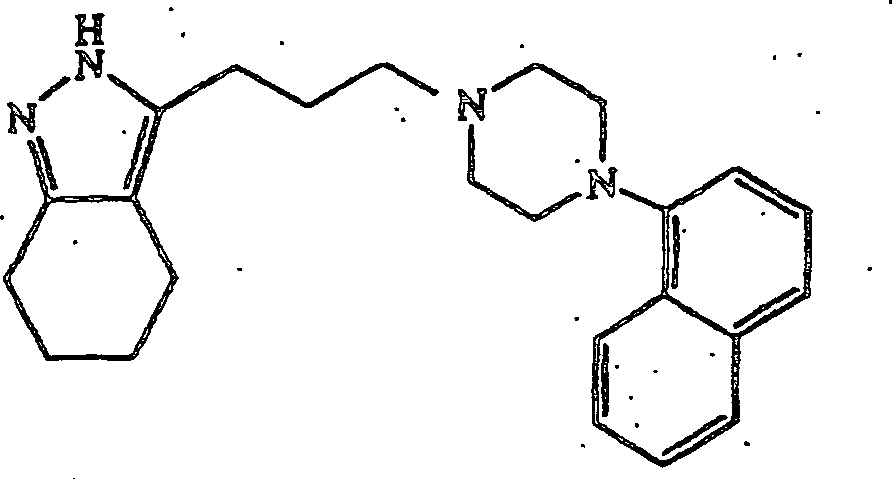
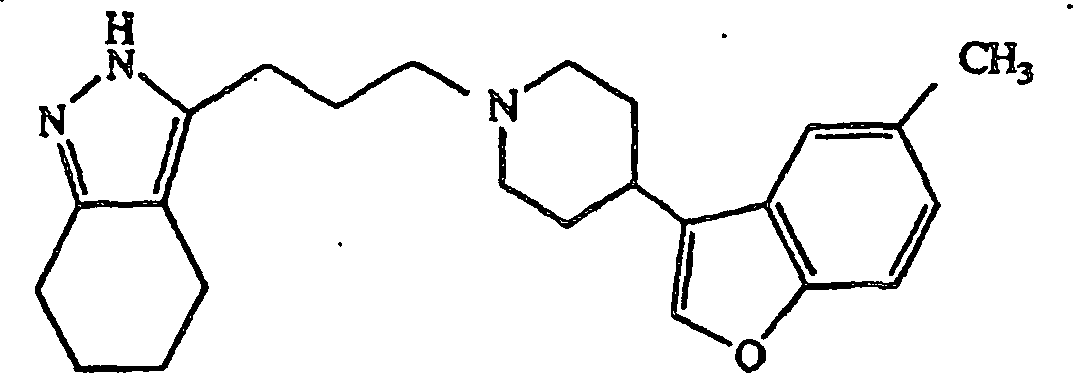
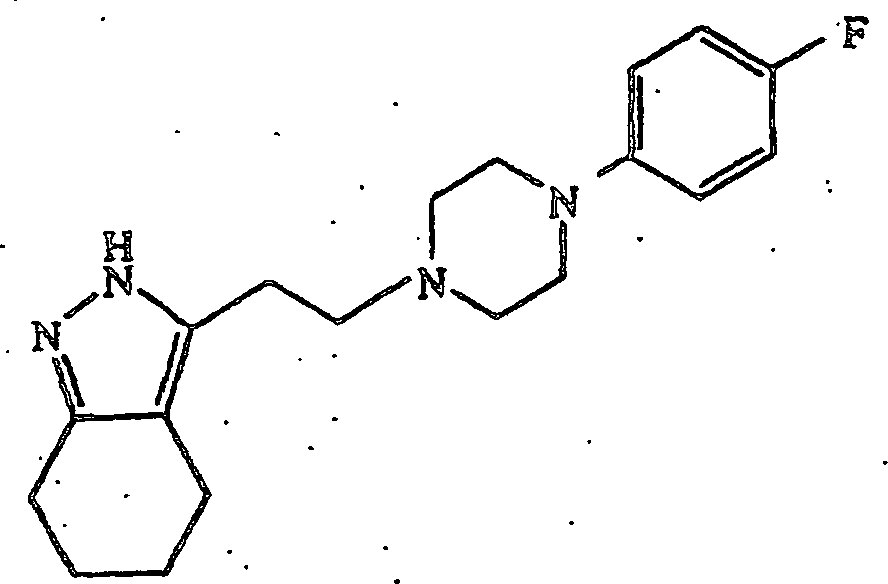
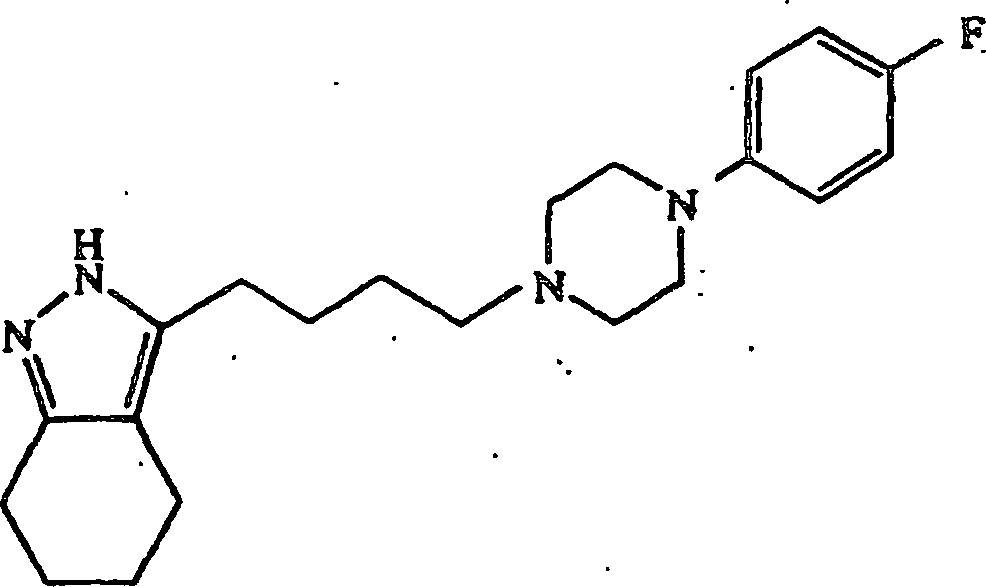


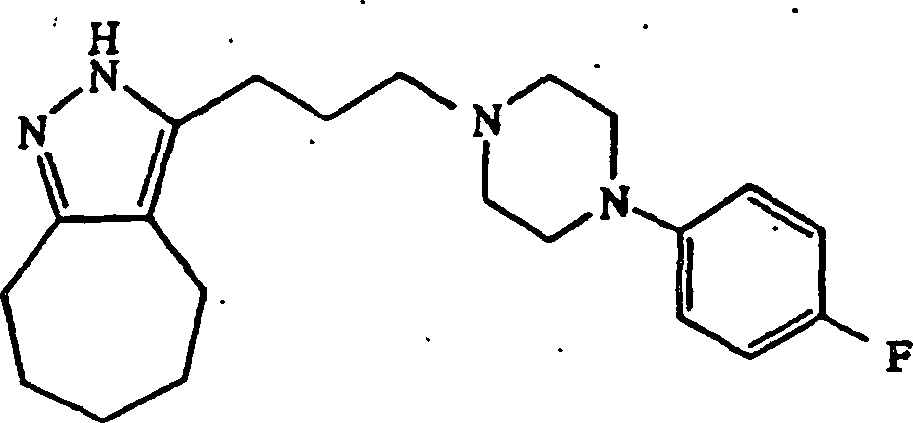
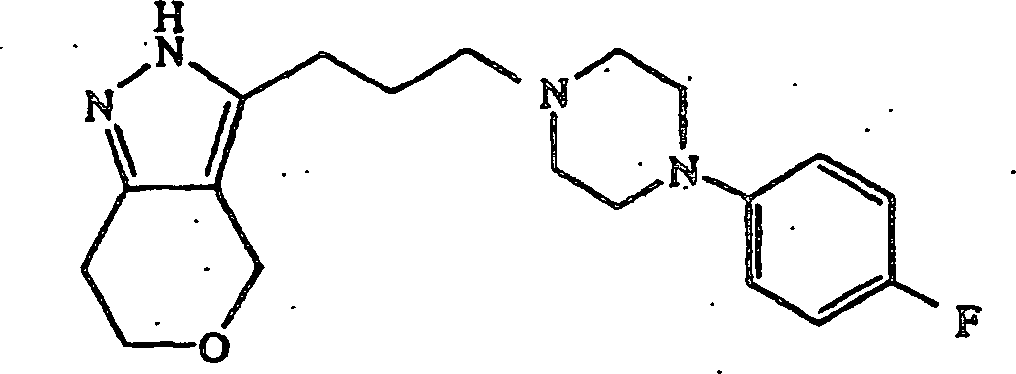
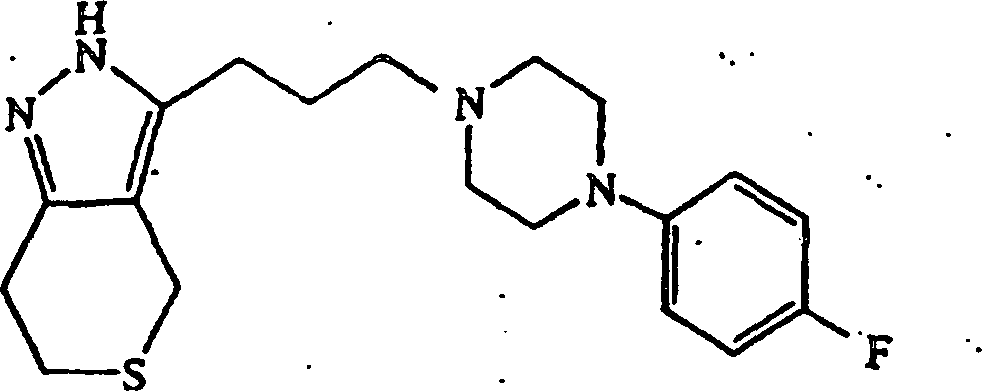


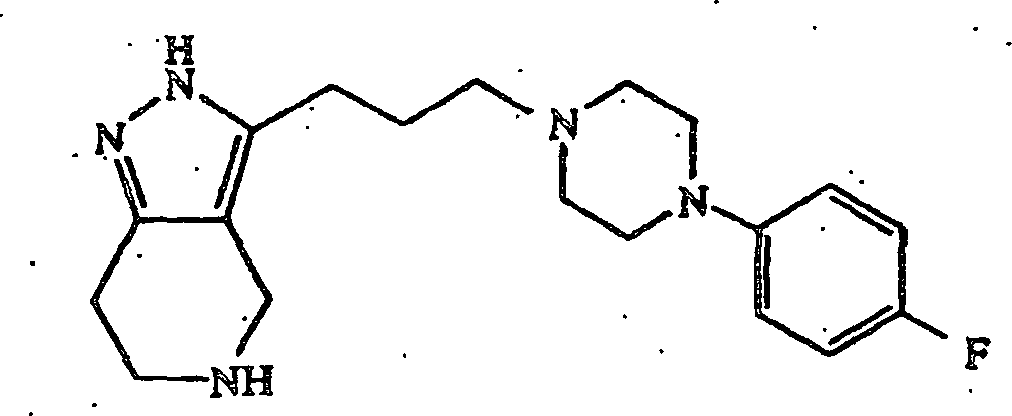
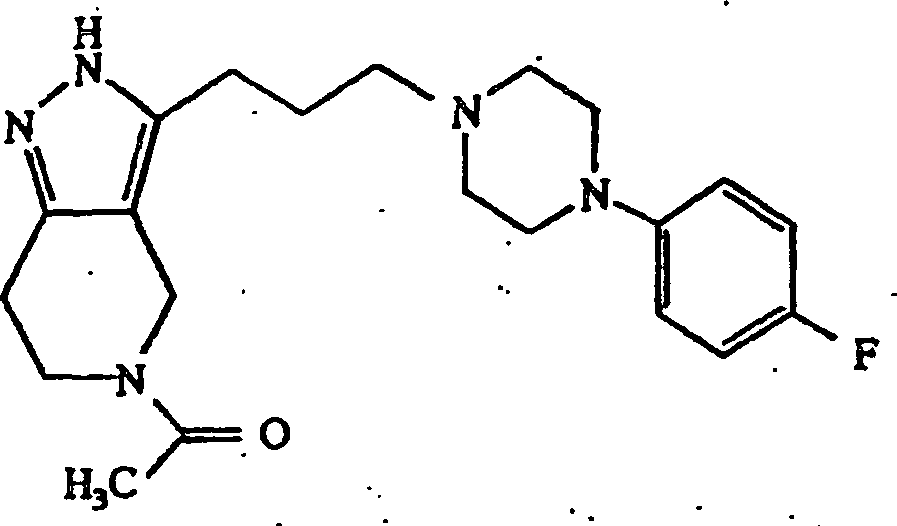
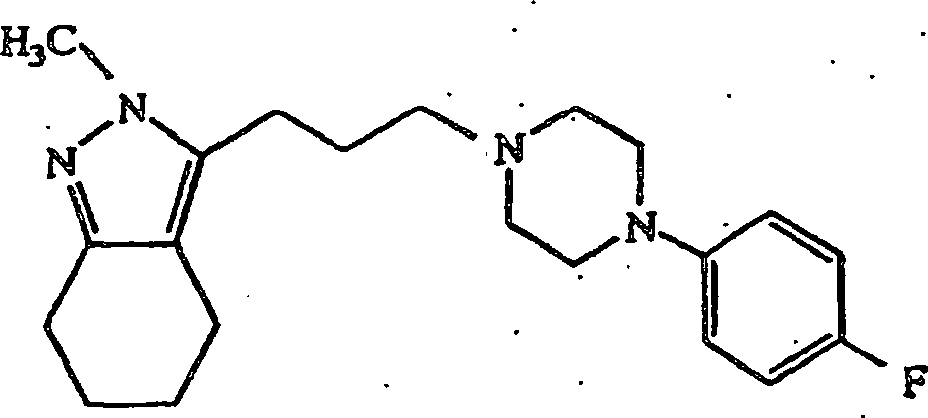
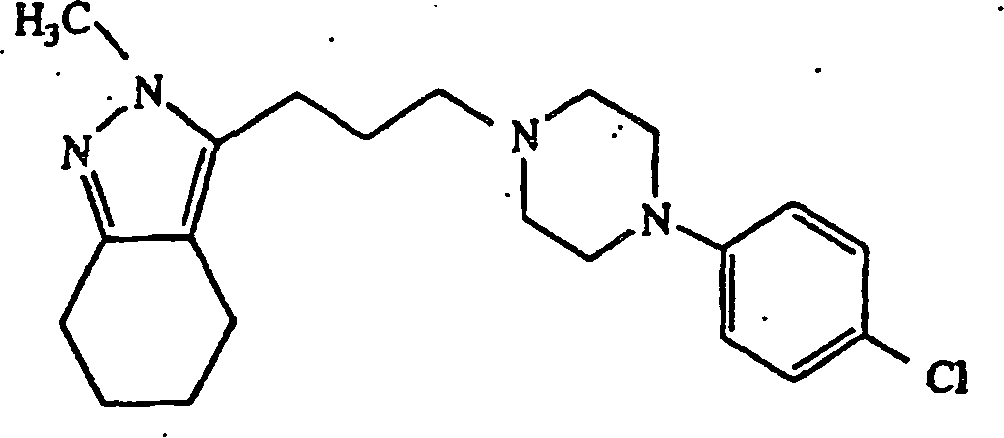
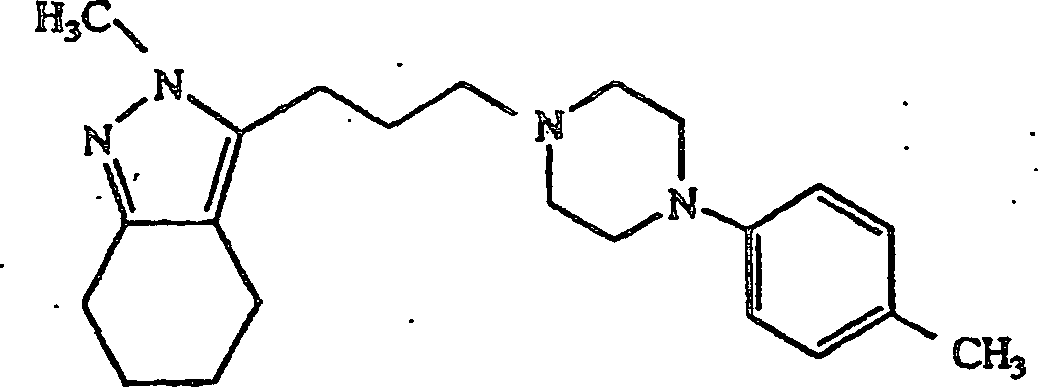
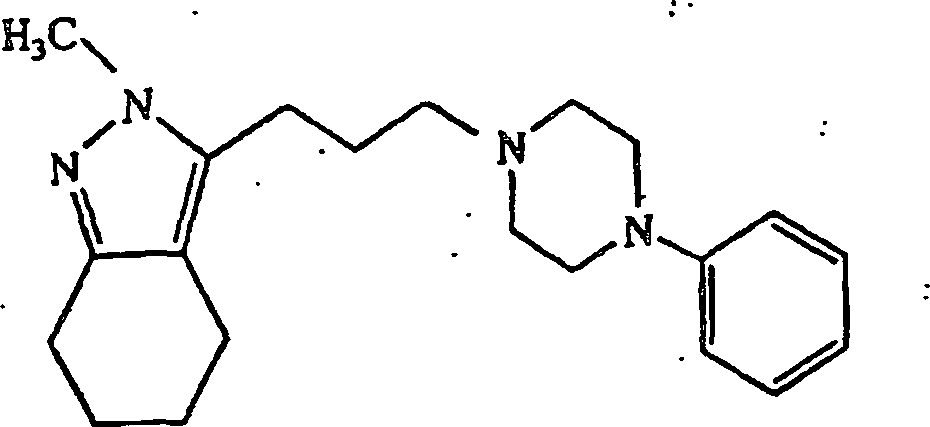
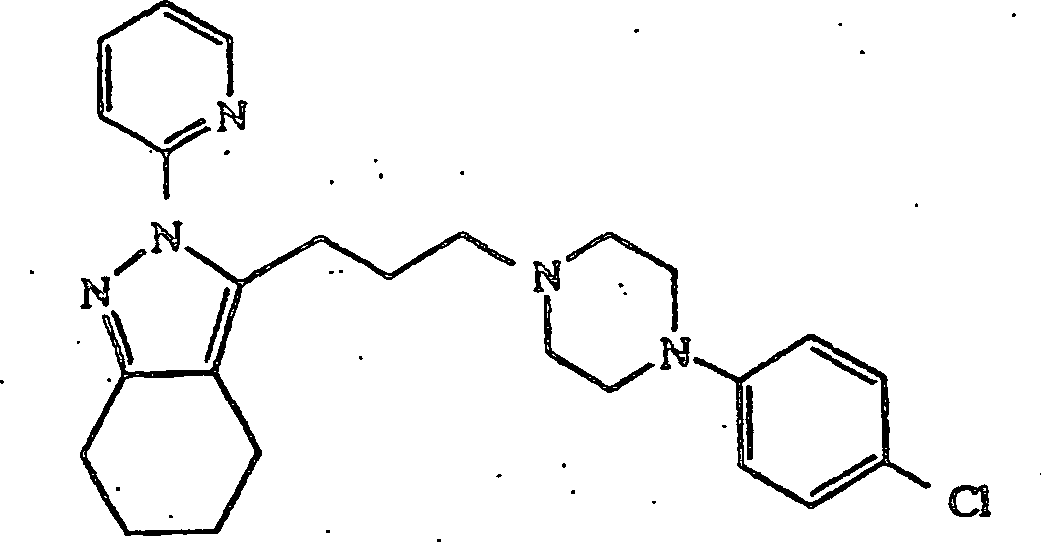
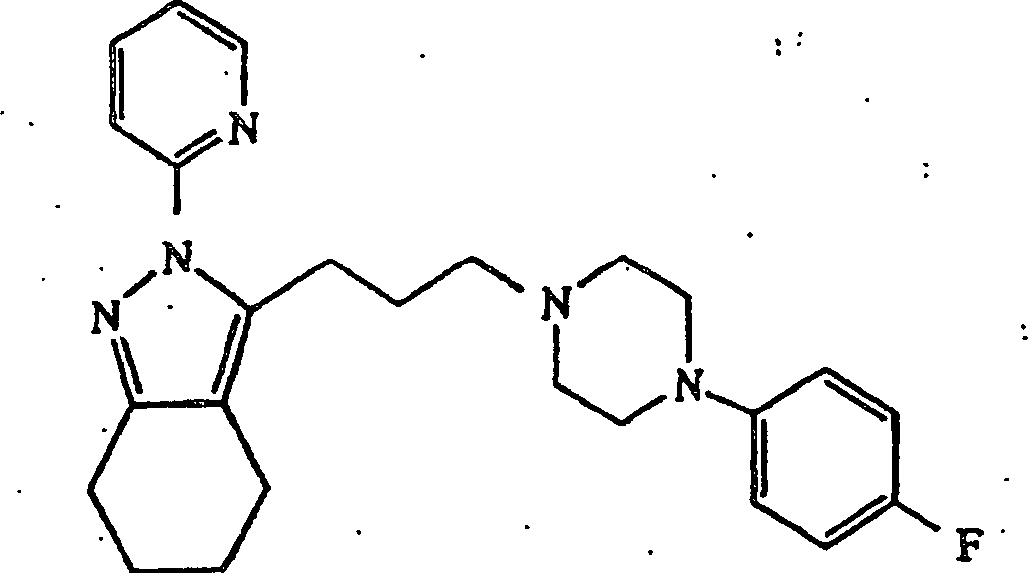
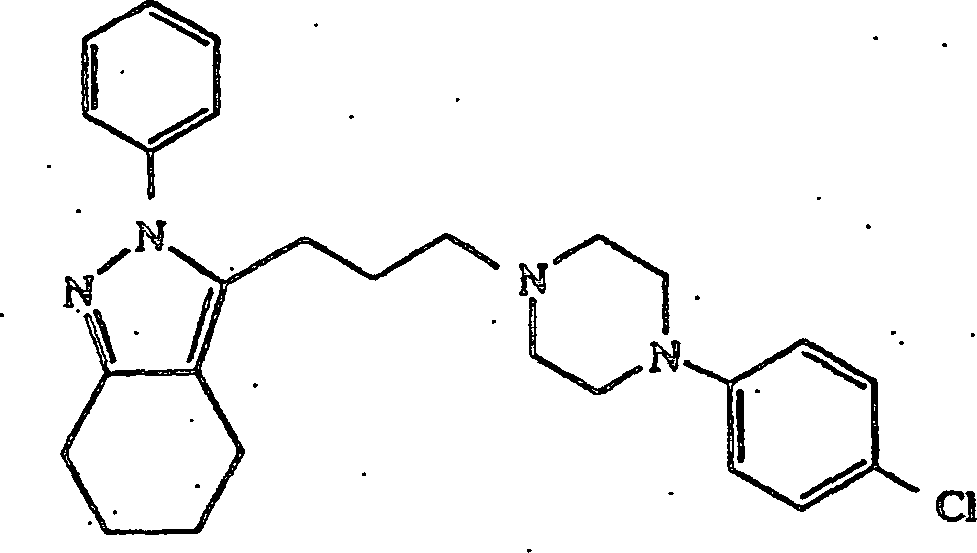
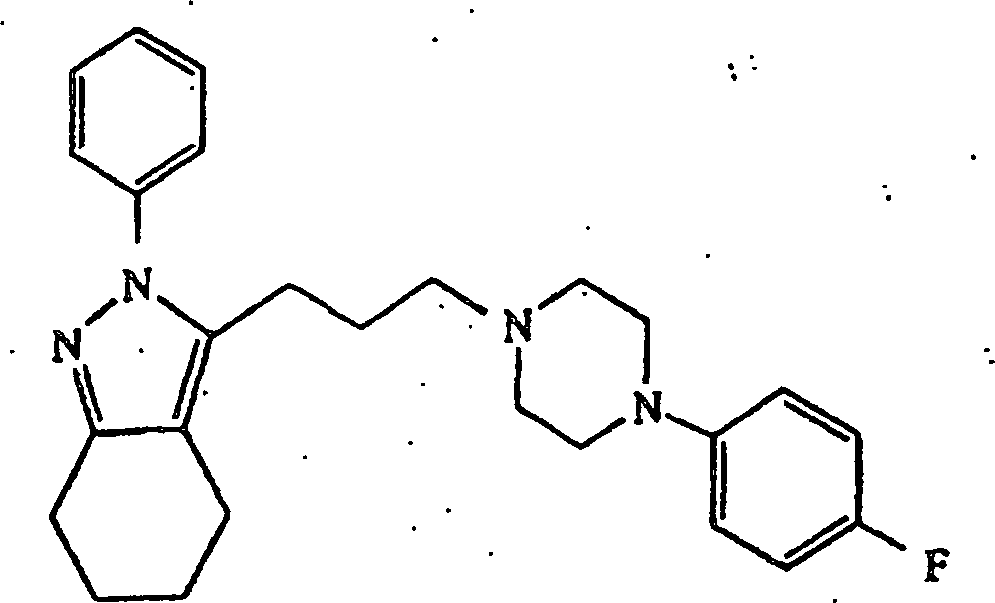

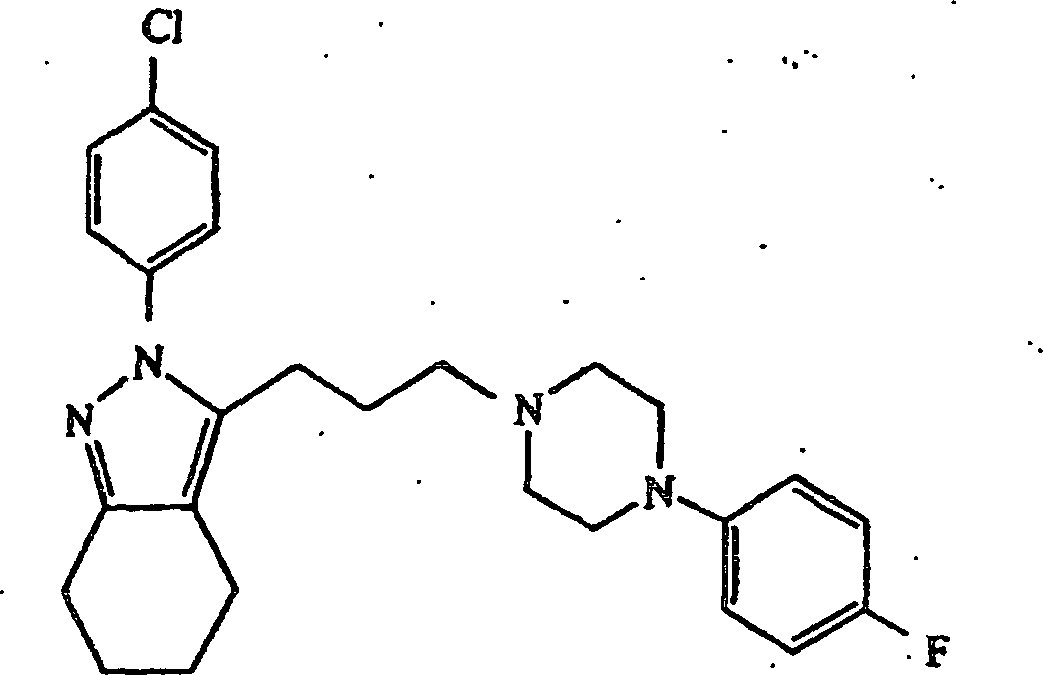
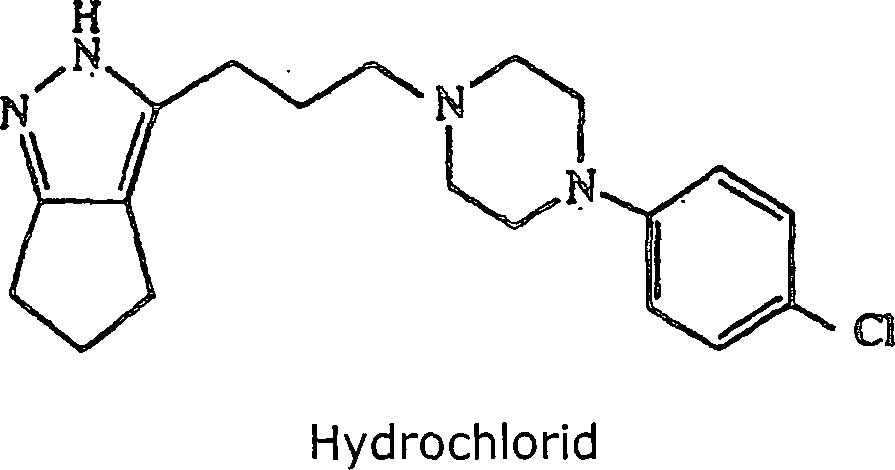


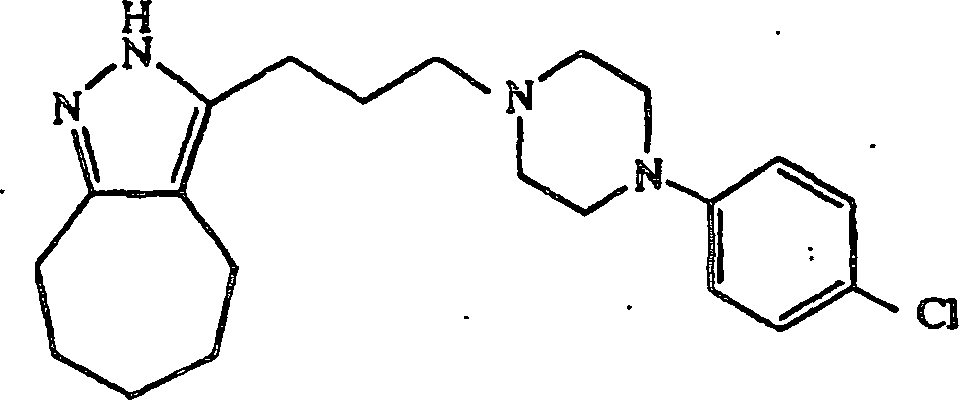

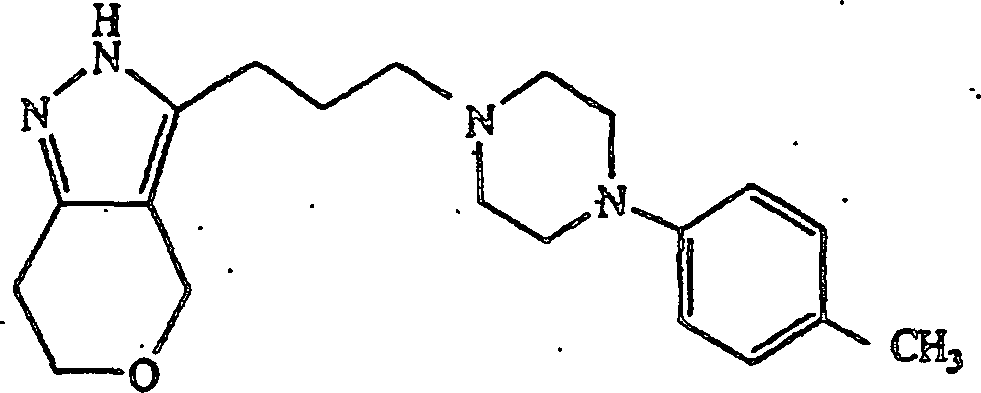
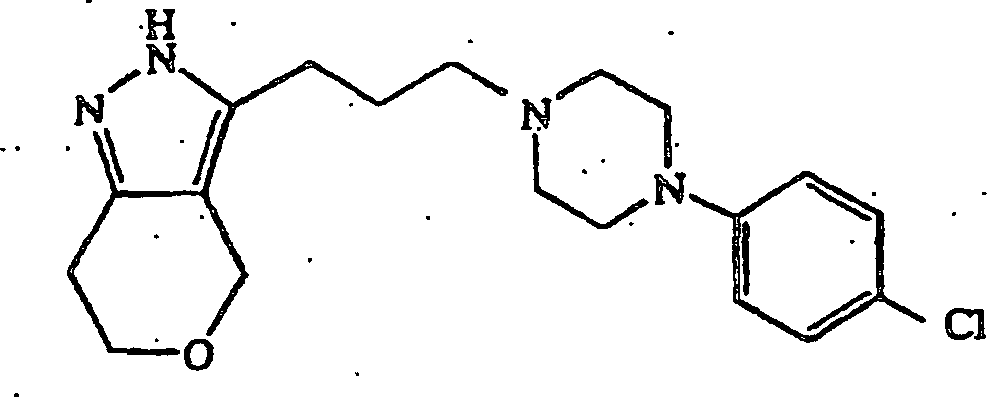

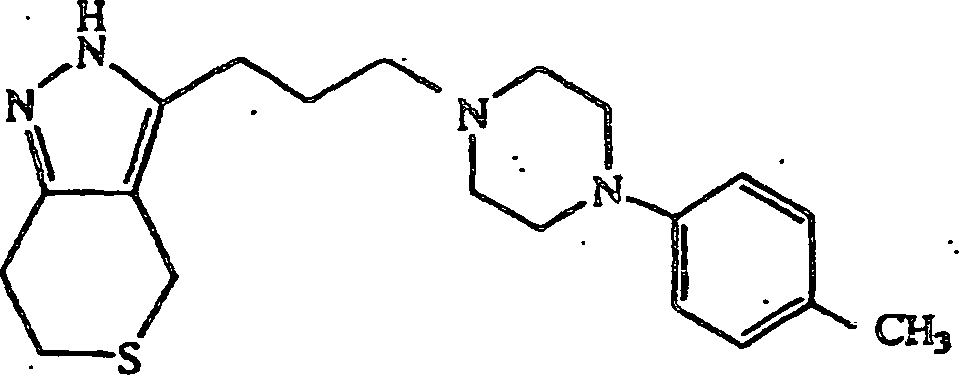
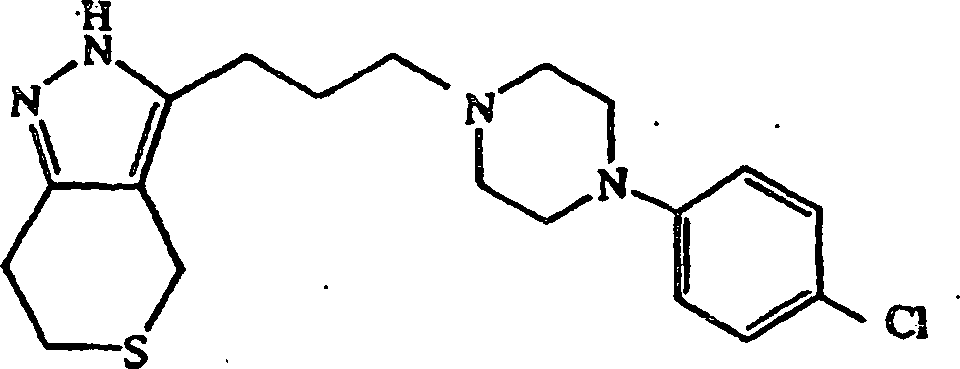
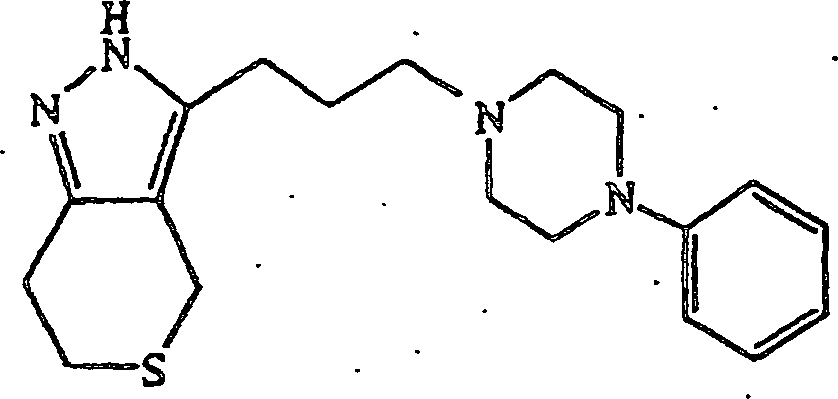
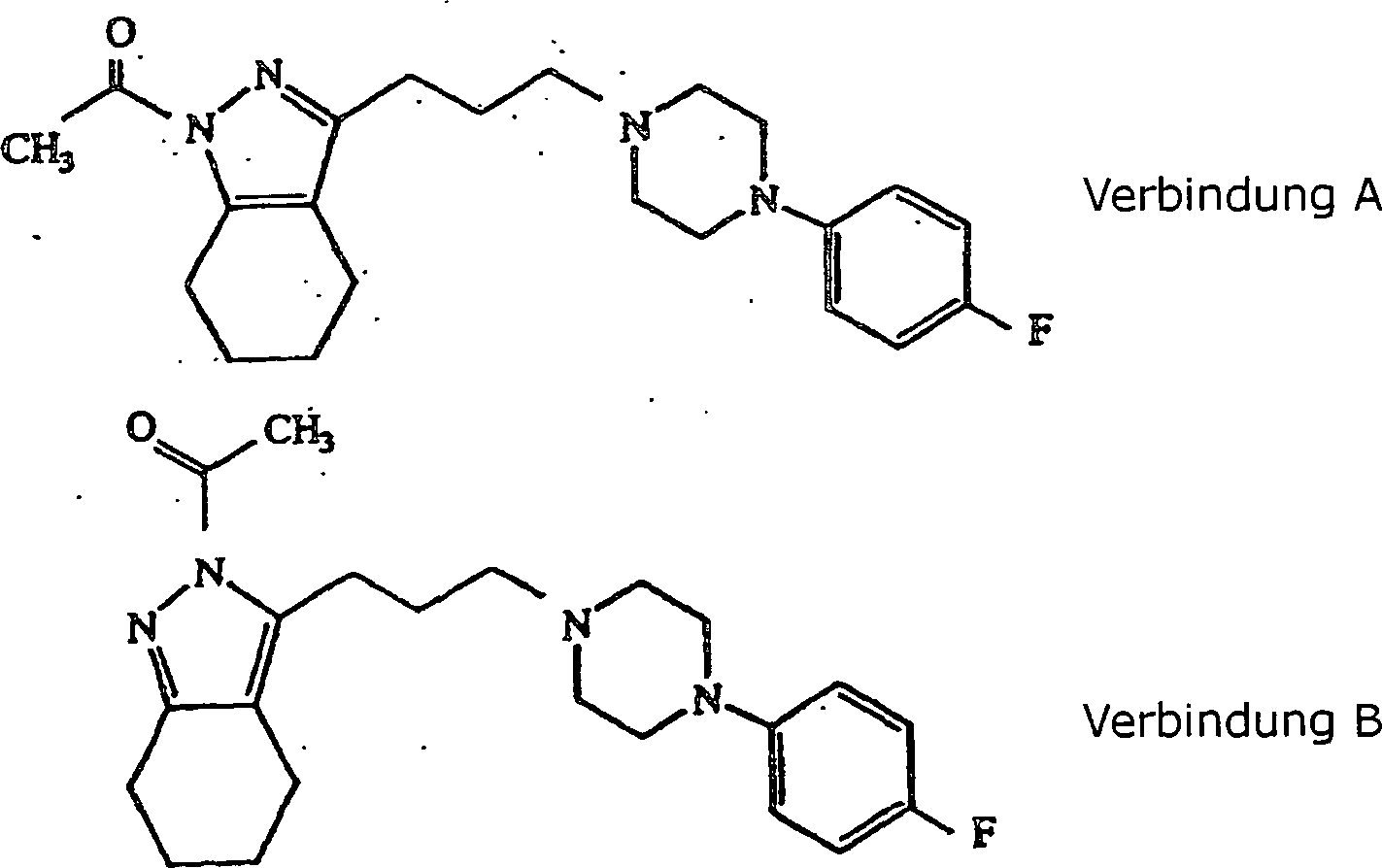

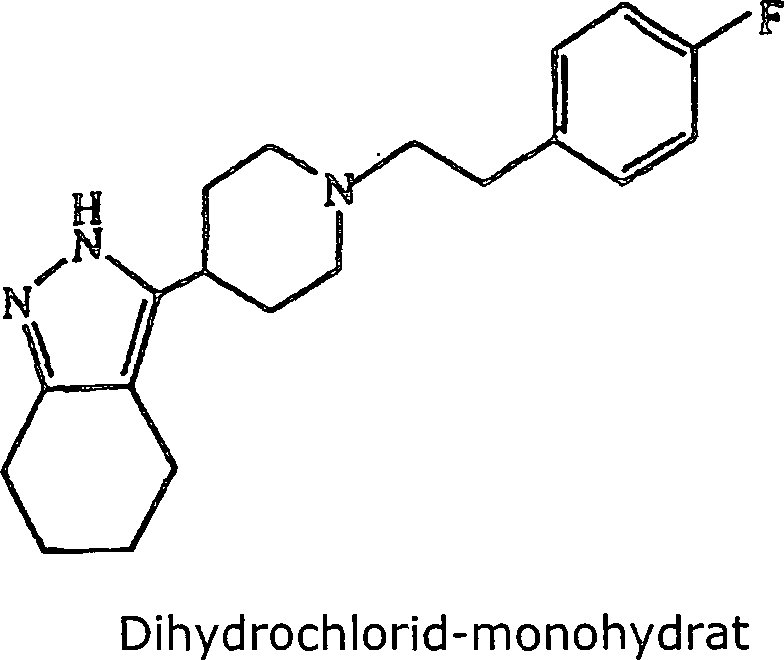


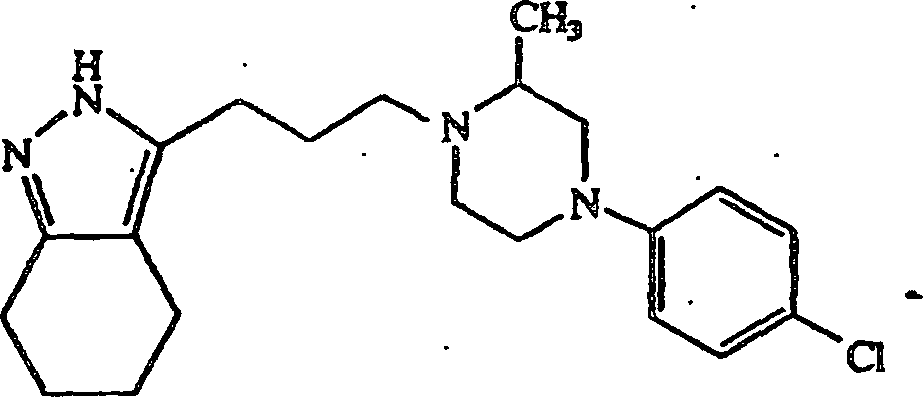


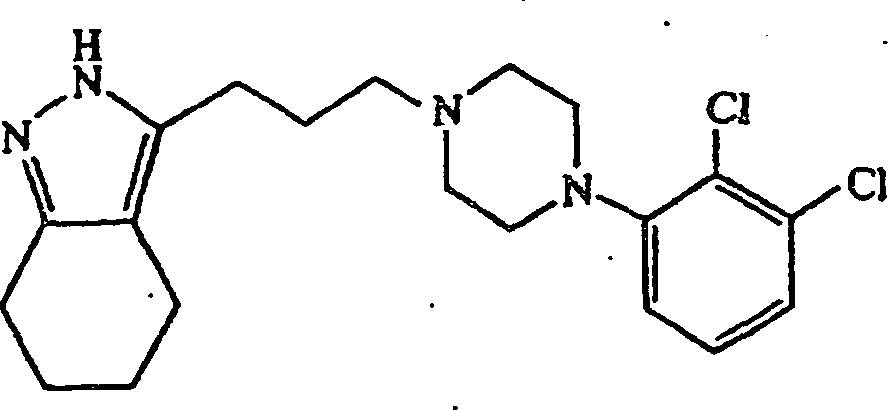
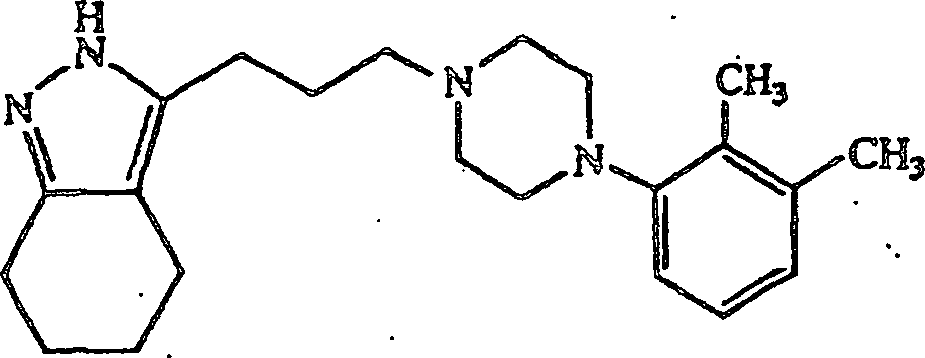


 ist, wobei Y ein lineares oder verzweigtes C1-C4-Alkylen ist, das gegebenenfalls einen Substituenten R1a an einer optionalen Position hat, wobei R1a ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octa decyl ausgewähltes Alkyl, Hydroxy, ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy, Amino oder ein aus Methylamino, Dimethylamino, Ethylamino, Diethylamino, Propylamino und Dipropylamino ausgewähltes Alkylamino ist, A fehlt oder ein Sauerstoffatom, ein Schwefelatom, SO, SO2 oder N-R7 ist, wobei R7 Wasserstoff, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Benzyl, 2-Phenylethyl, 3-Phenylpropyl, 2-Methyl-2-phenylethyl, 3-Methyl-3-phenylpropyl, 4-Chlorbenzyl, 2-(4-Chlorphenyl)ethyl, 3-(4-Chlorphenyl)propyl, 2-Methyl-2-(4-chlorphenyl)ethyl und 3-Methyl-3-(4-chlorphenyl)propyl ausgewähltes Aralkyl oder ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl ist, B ein lineares oder verzweigtes C1-C4-Alkylen ist, das gegebenenfalls einen Substituenten R1b an einer optionalen Position hat, wobei R1b ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, Hydroxy, ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy, Amino, ein aus Methylamino, Dimethylamino, Ethylamino, Diethylamino, Propylamino und Dipropylamino ausgewähltes Alkylamino ist, und R3 Wasserstoff, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl oder ein aus Phenyl, Naphthyl und 2-Indenyl ausgewähltes Aryl ist, D fehlt oder ein lineares oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen ist, Q-T eine CH-, CH2-N-, (CH2)2-N-, CH2-C(R4)-Bindung ist, wobei R4 Wasserstoff, Hydroxy, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl oder ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy oder CH=C ist, G fehlt oder ein lineares oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen oder ein Carbonyl ist, R5 Wasserstoff oder ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl ist und R6 ein Aryl, das aus Phenyl, Naphthyl und 2-Indenyl ausgewählt ist, das gegebenenfalls durch (einen) Substituenten substituiert ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind), ein Heteroaryl, das aus Pyridyl, Furyl, Thienyl und Pyrimidinyl ausgewählt ist, das gegebenenfalls durch (einen) Substituenten substituiert ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind), oder ein kondensiertes Heteroaryl, das aus 1,2-Benzoisoxazol-3-yl, 1,2-Benzoisothiazol-3-yl, Indol-3-yl, Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl ausgewählt ist, das gegebenenfalls durch (einen) Substituenten substituiert ist (sind), der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind), ist, ein optisches Isomer davon oder ein pharmazeutisch annehmbares Salz davon.
ist, wobei Y ein lineares oder verzweigtes C1-C4-Alkylen ist, das gegebenenfalls einen Substituenten R1a an einer optionalen Position hat, wobei R1a ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octa decyl ausgewähltes Alkyl, Hydroxy, ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy, Amino oder ein aus Methylamino, Dimethylamino, Ethylamino, Diethylamino, Propylamino und Dipropylamino ausgewähltes Alkylamino ist, A fehlt oder ein Sauerstoffatom, ein Schwefelatom, SO, SO2 oder N-R7 ist, wobei R7 Wasserstoff, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Benzyl, 2-Phenylethyl, 3-Phenylpropyl, 2-Methyl-2-phenylethyl, 3-Methyl-3-phenylpropyl, 4-Chlorbenzyl, 2-(4-Chlorphenyl)ethyl, 3-(4-Chlorphenyl)propyl, 2-Methyl-2-(4-chlorphenyl)ethyl und 3-Methyl-3-(4-chlorphenyl)propyl ausgewähltes Aralkyl oder ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl ist, B ein lineares oder verzweigtes C1-C4-Alkylen ist, das gegebenenfalls einen Substituenten R1b an einer optionalen Position hat, wobei R1b ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, Hydroxy, ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy, Amino, ein aus Methylamino, Dimethylamino, Ethylamino, Diethylamino, Propylamino und Dipropylamino ausgewähltes Alkylamino ist, und R3 Wasserstoff, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl, ein aus Formyl, Acetyl, Propionyl, Benzoyl und Benzylcarbonyl ausgewähltes Acyl oder ein aus Phenyl, Naphthyl und 2-Indenyl ausgewähltes Aryl ist, D fehlt oder ein lineares oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen ist, Q-T eine CH-, CH2-N-, (CH2)2-N-, CH2-C(R4)-Bindung ist, wobei R4 Wasserstoff, Hydroxy, ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl oder ein aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewähltes Alkoxy oder CH=C ist, G fehlt oder ein lineares oder verzweigtes Alkylen mit 1 bis 8 Kohlenstoffatomen oder ein Carbonyl ist, R5 Wasserstoff oder ein aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, tert-Butyl, Pentyl, Hexyl, Octyl, Decyl, Hexadecyl und Octadecyl ausgewähltes Alkyl ist und R6 ein Aryl, das aus Phenyl, Naphthyl und 2-Indenyl ausgewählt ist, das gegebenenfalls durch (einen) Substituenten substituiert ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind), ein Heteroaryl, das aus Pyridyl, Furyl, Thienyl und Pyrimidinyl ausgewählt ist, das gegebenenfalls durch (einen) Substituenten substituiert ist, der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind), oder ein kondensiertes Heteroaryl, das aus 1,2-Benzoisoxazol-3-yl, 1,2-Benzoisothiazol-3-yl, Indol-3-yl, Benzo[b]furan-3-yl und Benzo[b]thiophen-3-yl ausgewählt ist, das gegebenenfalls durch (einen) Substituenten substituiert ist (sind), der (die) aus einem Halogen, Trifluormethyl, einem aus Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl und tert-Butyl ausgewählten Alkyl, einem aus Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, tert-Butoxy, Pentyloxy, Hexyloxy, Heptyloxy und Octyloxy ausgewählten Alkoxy, Hydroxy, Nitro, Amino, Methylamino und Dimethylamino ausgewählt ist (sind), ist, ein optisches Isomer davon oder ein pharmazeutisch annehmbares Salz davon.