DE69735339T2 - Verfahren zur Herstellung von Benzothiophenen - Google Patents
Verfahren zur Herstellung von Benzothiophenen Download PDFInfo
- Publication number
- DE69735339T2 DE69735339T2 DE69735339T DE69735339T DE69735339T2 DE 69735339 T2 DE69735339 T2 DE 69735339T2 DE 69735339 T DE69735339 T DE 69735339T DE 69735339 T DE69735339 T DE 69735339T DE 69735339 T2 DE69735339 T2 DE 69735339T2
- Authority
- DE
- Germany
- Prior art keywords
- reaction
- compound
- formula
- cation exchange
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000000034 method Methods 0.000 title claims description 33
- 230000008569 process Effects 0.000 title claims description 13
- 238000002360 preparation method Methods 0.000 title claims description 9
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical class C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 title description 23
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 66
- 150000001875 compounds Chemical class 0.000 claims description 25
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 24
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 22
- -1 methylpyrrolidinyl Chemical group 0.000 claims description 22
- 239000003729 cation exchange resin Substances 0.000 claims description 19
- 239000011347 resin Substances 0.000 claims description 18
- 229920005989 resin Polymers 0.000 claims description 18
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 claims description 13
- 230000008707 rearrangement Effects 0.000 claims description 12
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 10
- 239000011541 reaction mixture Substances 0.000 claims description 10
- 150000003839 salts Chemical class 0.000 claims description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 4
- 239000004793 Polystyrene Substances 0.000 claims description 4
- 229920002223 polystyrene Polymers 0.000 claims description 4
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 4
- 239000012453 solvate Substances 0.000 claims description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 2
- 125000003386 piperidinyl group Chemical group 0.000 claims description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 2
- 238000006243 chemical reaction Methods 0.000 description 45
- 239000002904 solvent Substances 0.000 description 26
- 238000007363 ring formation reaction Methods 0.000 description 20
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 18
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical compound CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- 239000002253 acid Substances 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 239000000203 mixture Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- 239000003377 acid catalyst Substances 0.000 description 6
- 229940023913 cation exchange resins Drugs 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 239000006227 byproduct Substances 0.000 description 5
- 238000006462 rearrangement reaction Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- 229920005654 Sephadex Polymers 0.000 description 4
- 239000012507 Sephadex™ Substances 0.000 description 4
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 150000001768 cations Chemical class 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 229920001429 chelating resin Polymers 0.000 description 4
- 229920000137 polyphosphoric acid Polymers 0.000 description 4
- HRWAGCVMOGWQJF-UHFFFAOYSA-N 6-methoxy-2-(4-methoxyphenyl)-1-benzothiophene Chemical compound C1=CC(OC)=CC=C1C1=CC2=CC=C(OC)C=C2S1 HRWAGCVMOGWQJF-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 238000010520 demethylation reaction Methods 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 239000002841 Lewis acid Substances 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 239000003849 aromatic solvent Substances 0.000 description 2
- 238000010533 azeotropic distillation Methods 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 150000007517 lewis acids Chemical class 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 2
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 125000000542 sulfonic acid group Chemical group 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- UAYWVJHJZHQCIE-UHFFFAOYSA-L zinc iodide Chemical compound I[Zn]I UAYWVJHJZHQCIE-UHFFFAOYSA-L 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- HMGCGUWFPZVPEK-UHFFFAOYSA-N 2-naphthalen-2-ylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C1=CC=C(C=CC=C2)C2=C1 HMGCGUWFPZVPEK-UHFFFAOYSA-N 0.000 description 1
- WHBMMWSBFZVSSR-UHFFFAOYSA-N 3-hydroxybutyric acid Chemical compound CC(O)CC(O)=O WHBMMWSBFZVSSR-UHFFFAOYSA-N 0.000 description 1
- BTHAMNJTBJFIQX-UHFFFAOYSA-N 4-(1-benzothiophen-2-yl)phenol Chemical compound C1=CC(O)=CC=C1C1=CC2=CC=CC=C2S1 BTHAMNJTBJFIQX-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- KSPMZOIHTPOBNW-UHFFFAOYSA-N 4-methoxy-2-(4-methoxyphenyl)-1-benzothiophene Chemical compound C1=CC(OC)=CC=C1C1=CC2=C(OC)C=CC=C2S1 KSPMZOIHTPOBNW-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- KHNUUOBBTJPQOV-UHFFFAOYSA-N 6-methoxy-3-(4-methoxyphenyl)-1-benzothiophene Chemical compound C1=CC(OC)=CC=C1C1=CSC2=CC(OC)=CC=C12 KHNUUOBBTJPQOV-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 239000007848 Bronsted acid Substances 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- GRMDZNADEAHPLE-UHFFFAOYSA-N COc1cccc2c1cc(-c(cc1)ccc1O)[s]2 Chemical compound COc1cccc2c1cc(-c(cc1)ccc1O)[s]2 GRMDZNADEAHPLE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- CYTYCFOTNPOANT-UHFFFAOYSA-N Perchloroethylene Chemical group ClC(Cl)=C(Cl)Cl CYTYCFOTNPOANT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 1
- MKLKHSYFDZTITR-UHFFFAOYSA-N [2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]-[4-(2-morpholin-4-ylethoxy)phenyl]methanone Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCOCC3)=CC=2)C2=CC=CC=C2S1 MKLKHSYFDZTITR-UHFFFAOYSA-N 0.000 description 1
- UAUVDTRIPPZZCC-UHFFFAOYSA-N [2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]-[4-(2-piperidin-1-ylethoxy)phenyl]methanone Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=CC=C2S1 UAUVDTRIPPZZCC-UHFFFAOYSA-N 0.000 description 1
- SEZLKRUKAYSYCE-UHFFFAOYSA-N [2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]-[4-(2-pyrrolidin-1-ylethoxy)phenyl]methanone Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCC3)=CC=2)C2=CC=CC=C2S1 SEZLKRUKAYSYCE-UHFFFAOYSA-N 0.000 description 1
- YLFHUDANHUPRPK-UHFFFAOYSA-N [4-(2-aminoethoxy)phenyl]-(1-benzothiophen-3-yl)methanone Chemical class C1=CC(OCCN)=CC=C1C(=O)C1=CSC2=CC=CC=C12 YLFHUDANHUPRPK-UHFFFAOYSA-N 0.000 description 1
- FUYHPEFCMUFNLQ-UHFFFAOYSA-N [4-(2-aminoethoxy)phenyl]-[6-hydroxy-2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]methanone Chemical compound C1=CC(OCCN)=CC=C1C(=O)C1=C(C=2C=CC(O)=CC=2)SC2=CC(O)=CC=C12 FUYHPEFCMUFNLQ-UHFFFAOYSA-N 0.000 description 1
- DTRYPUDAGJUMRK-UHFFFAOYSA-N [4-[2-(azepan-1-yl)ethoxy]phenyl]-[6-hydroxy-2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]methanone Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCCC3)=CC=2)C2=CC=C(O)C=C2S1 DTRYPUDAGJUMRK-UHFFFAOYSA-N 0.000 description 1
- LZRVYDUXPBPSJL-UHFFFAOYSA-N [4-[2-(dimethylamino)ethoxy]phenyl]-[2-(4-hydroxyphenyl)-1-benzothiophen-3-yl]methanone Chemical compound C1=CC(OCCN(C)C)=CC=C1C(=O)C1=C(C=2C=CC(O)=CC=2)SC2=CC=CC=C12 LZRVYDUXPBPSJL-UHFFFAOYSA-N 0.000 description 1
- WAIPAZQMEIHHTJ-UHFFFAOYSA-N [Cr].[Co] Chemical compound [Cr].[Co] WAIPAZQMEIHHTJ-UHFFFAOYSA-N 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 159000000032 aromatic acids Chemical class 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- JXLHNMVSKXFWAO-UHFFFAOYSA-N azane;7-fluoro-2,1,3-benzoxadiazole-4-sulfonic acid Chemical compound N.OS(=O)(=O)C1=CC=C(F)C2=NON=C12 JXLHNMVSKXFWAO-UHFFFAOYSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- YMEKEHSRPZAOGO-UHFFFAOYSA-N boron triiodide Chemical compound IB(I)I YMEKEHSRPZAOGO-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- JOYKCMAPFCSKNO-UHFFFAOYSA-N chloro benzenesulfonate Chemical compound ClOS(=O)(=O)C1=CC=CC=C1 JOYKCMAPFCSKNO-UHFFFAOYSA-N 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229940114081 cinnamate Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- 230000001335 demethylating effect Effects 0.000 description 1
- 230000017858 demethylation Effects 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethanethiol Chemical compound CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 229910000856 hastalloy Inorganic materials 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 230000009878 intermolecular interaction Effects 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- QALQIBQJEQZNTE-UHFFFAOYSA-N n-(hydroxymethyl)-n,2-dimethylprop-2-enamide Chemical compound OCN(C)C(=O)C(C)=C QALQIBQJEQZNTE-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 125000005489 p-toluenesulfonic acid group Chemical class 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 1
- 229920000867 polyelectrolyte Polymers 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000011175 product filtration Methods 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000011973 solid acid Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000003774 sulfhydryl reagent Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical compound [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- KKEYFWRCBNTPAC-UHFFFAOYSA-L terephthalate(2-) Chemical compound [O-]C(=O)C1=CC=C(C([O-])=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-L 0.000 description 1
- PUGUQINMNYINPK-UHFFFAOYSA-N tert-butyl 4-(2-chloroacetyl)piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCN(C(=O)CCl)CC1 PUGUQINMNYINPK-UHFFFAOYSA-N 0.000 description 1
- 229950011008 tetrachloroethylene Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M trans-cinnamate Chemical compound [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- 229940071104 xylenesulfonate Drugs 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D333/56—Radicals substituted by oxygen atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Catalysts (AREA)
Description
- Diese Erfindung betrifft das Gebiet pharmazeutischer Chemie und stellt ein vorteilhaftes Verfahren bereit zur Herstellung einer Gruppe von Benzothiophenen aus Dialkoxyacetophenonen. Das Verfahren stellt die gewünschten Verbindungen in hervorragender Ausbeute in einem großen Maßstab bereit.
- Die Herstellung von Benzothiophenen durch ein Dialkoxybenzothiophenzwischeprodukt wurde zuvor beschrieben in
US 4 380 635 , dessen Offenbarung hierin durch Bezugnahme einverleibt wird, das die intramolekulare Cyclisierung von α-(3-Methoxyphenylthio)-4-methoxyacetophenon in Gegenwart von Polyphosphorsäure (PPA) lehrt. Ein Erwärmen des Acetphenonausgangsmaterials in PPA bei etwa 85°C über einen Zeitraum von etwa 1 Stunde stellt ein Gemisch von etwa 3:1 der zwei Isomeren 6-Methoxy-2-(4-methoxyphenyl)benzo[b]thiophen und 4-Methoxy-2-(4-methoxyphenyl)benzo[b]thiophen bereit. Wenn diese Umsetzung jedoch in einem Herstellungsmaßstab durchgeführt wird, kristallisieren die isomeren Benzothiophene und erzeugen eine dicke Paste, die nicht hinreichend in einer herkömmlichen Herstellungsanlage gerührt werden kann. - Die Verwendung eines Lösemittels, um das Problem, das durch eine Paste in einem anderen Reaktionsschema hervorgerufen wird zu beseitigen, wurde vorgeschlagen von Guy et al., Synthesis, 222 (1980). Wenn dieser Vorschlag jedoch auf das vorliegende Schema angewendet wird, führt die Zugabe eines Lösemittels zu unvollständiger Cyclisierung des Ausgangsacetophenons, unvollständiger Umlagerung von 6-Methoxy-3-(4-methoxyphenyl)benzo[b]thiophen und dramatisch erhöhten Reaktionszeiten.
- Daher gibt es einen Bedarf an einem verbesserten Verfahren, das wechselnde oder alternierende Katalysatoren einsetzt für die Umsetzung von Dialkoxyacetophenonderivaten zu Benzothiophenen mit geeigneten Ausbeuten und annehmbaren Reaktionszeiten.
- Die europäische Patentanmeldung 95 305 085 offenbart ein Verfahren zur Herstellung von 3-(4-Aminoethoxybenzoyl)benzo[b]thiophenen und Dialkoxy/benzo[b]thiophenverbindungen.
- Die vorliegende Erfindung stellt ein Verfahren bereit zur Herstellung von Benzothiophenen unter Verwendung eines Kationenaustauschharzes. Diese Herstellung beruht auf einer intermolekularen Cyclisierung eines Dialkoxyacetophenonderivats, um ein Benzothiophen zu ergeben.
- Daher stellt die Erfindung ein Verfahren bereit zur Herstellung einer Verbindung der Formel 1wobei die Gruppen R gleich oder verschieden sind und für C1-C6-Alkyl stehen, wobei das Verfahren einschließt ein Cyclisieren einer Verbindung der Formel II
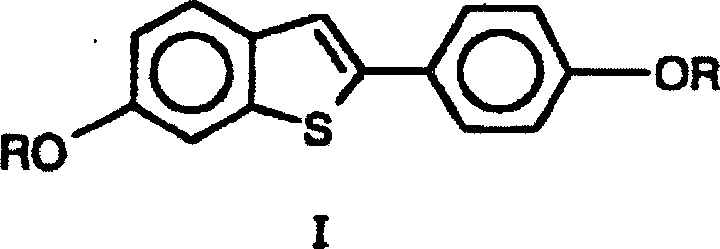 wobei die Gruppen R wie oben definiert sind, in Gegenwart eines Kationenaustauschharzes.
wobei die Gruppen R wie oben definiert sind, in Gegenwart eines Kationenaustauschharzes.
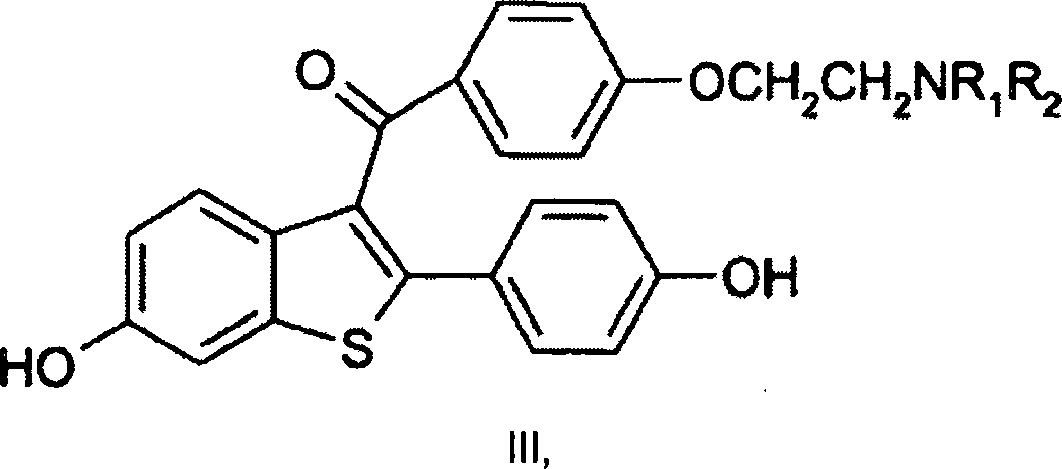
- Benzothiophen ist ein Schlüsselzwischenprodukt bei der Synthese von Raloxifen, einem selektiven Östrogenrezeptormodulator, oder SERM. Außer der Bereitstellung eines verbesserten Verfahrens zur Herstellung dieses Zwischenprodukts stellt die vorliegende Erfindung außerdem ein verbessertes Verfahren bereit zur Herstellung einer Verbindung der Formel IIIwobei:
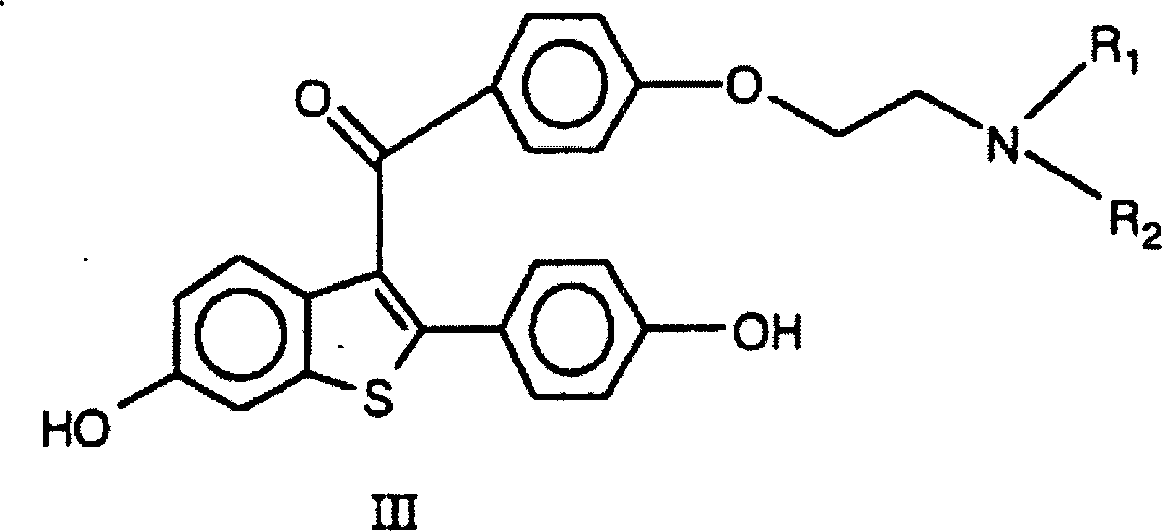
R1 und R2 unabhängig für C1-C6-Alkyl stehen oder verbunden sind, um mit dem Stickstoff, an den sie gebunden sind, Piperidinyl, Pyrrolidinyl, Methylpyrrolidinyl, Dimethylpyrrolidinyl oder Hexamethylenimino zu bilden, oder der pharmazeutisch annehmbaren Salze oder Solvate davon; wobei das Verfahren umfasst:
ein Cyclisieren einer Verbindung der Formel IIwobei die R-Gruppen gleich oder verschieden sind und für C1-C6-Alkyl stehen in Gegenwart eines Kationenaustauschharzes.
- Viele der Ausgangsmaterialien und Verbindungen, die hergestellt werden durch das Verfahren dieser Erfindung werden weiter bereitgestellt in
US 4 133 814 undUS 4 380 635 , deren Offenbarungen hierin durch Bezugnahme einverleibt sind. - In dieser Beschreibung werden alle Temperaturen in Grad Celsius angegeben. Alle Mengen, Verhältnisse, Konzentrationen, Anteile und dergleichen werden in Gewichtseinheiten angegeben, sofern es nicht anders angegeben ist mit Ausnahme der Verhältnisse von Lösemitteln, die in Volumeneinheiten angegeben sind.
- Der Begriff "Säurekatalysator", der hierin verwendet wird, bedeutet eine Lewis-Säure oder eine Brønsted-Säure. Stellvertretende Lewis-Säuren sind Zinkchlorid, Zinkiodid, Aluminiumchlorid und Aluminiumbromid. Stellvertretende Brønsted-Säuren schließen anorganische Säuren ein, wie Schwefel- und Phosphorsäuren, Carbonsäuren, wie Essig- und Trifluoressigsäuren, Sulfonsäuren, wie Methansulfon-, Benzolsulfon-, 1-Naphthalinsulfon-, 1-Butansulfon-, Ethansulfon-, 4-Ethylbenzolsulfon-, 1-Hexansulfon-, 1,5-Naphthalindisulfon-, 1-Octansulfon-, Camphersulfon-, Trifluormethansulfon-, p-Toluolsulfonsäuren. Außerdem schließt der Begriff „Säurekatalysator" Kationenaustauschharze ein, auf die auch als auf Harz basierende Säurekatalysatoren Bezug genommen wird. Diese Kationenaustauschharze sind definitionsgemäß unlösliche, saure Harze. Die Kationenaustauschharze schließen, ohne darauf beschränkt zu sein, Kationenaustauscher an/auf Dextran ein, wie z.B. CM Sephadex (Carboxymethyl Sephadex), SP Sephadex (Sulfopropyl Sephadex) und dergleichen; Kationenaustauscher an/auf Agarose, wie z.B. CM Sepharose, S Sepharose und dergleichen; Kationenaustauscher an/auf Cellulose, wie z.B. CM Cellulose, Cellulosephosphat, Sulfoxyethylcellulose und dergleichen; Kationenaustauscher an/auf Polystyrol, wie z.B. sulfonierte Polystyrolharze (die typischerweise in der Gesamtzahl von Sulfonsäuregruppen an dem Harz unterschiedlich sind), die Amberlyst® XN-1010, Amberlyst®15, Amberlite®, XE586® und dergleichen einschließen; sulfonierte Polyfluorkohlenstoffharze, die Nafion-H® Harz; Oxycellulose; SP Trisacryl® Harze, wie z.B. SP Trisacryl Plus M® und SP Trisacryl Plus LS®; Poly(N-tris[hydroxymethyl]methylmethacrylamidharz und dergleichen einschließen.
- Der Begriff „Halo" oder „Halogen" bezieht sich auf Fluor-, Chlor-, Brom- oder Iodgruppen.
- Der Begriff „C1-C6-Alkyl" steht für eine gerade oder verzweigte Alkylkette, die von 1 bis 6 Kohlenstoffatome aufweist. Typische C1-C6-Alkylgruppen schließen Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, sek.-Butyl, tert.-Butyl, n-Pentyl, Isopentyl, n-Hexyl, 2-Methylpentyl und dergleichen ein. Der Begriff C1-C4-Alkyl" steht für eine gerade oder verzweigte Alkylkette, die von 1 bis 4 Kohlenstoffatome aufweist und Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, sek.-Butyl, i-Butyl und tert.-Butyl einschließt.
- Geeignete aktivierende Estergruppen sind in der Technik bekannt. Zahlreiche Reaktionen für die Bildung und Entfernung von Schutzgruppen sind beschrieben in einer Vielzahl von Standardwerken, die z.B. Protective Groups in Organic Chemistry, Plenum Press (London und New York, 1973); T.W. Green, Protective Groups in Organic Synthesis, Wiley (New York, 1981); und The Peptides, Band 1, Schrooder und Lubke, Academic Press (London und New York, 1965) einschließen. Verfahren für eine nicht regioselektive Entfernung von Hydroxyschutzgruppen, insbesondere Methyl, sind in der Technik bekannt. Verbindungen der Formel III, die zuvor an der 6- und 4'-Position mit Methoxy geschützt worden sind, können selektiv gespalten werden, um Verbindungen der Formel III zu erzeugen mit einer 4'-Methoxygruppe. Im Allgemeinen schließt das Verfahren zur Spaltung einer Methoxygruppe an der 4'-Position die Kombination eines 6-, 4'-Dimethoxysubstrats mit einem Demethylierungsreagenz ein, das ausgewählt wird aus der Gruppe von Bortribromid, Bortrichlorid oder Bortriiodid, oder mit AlCl3 und verschiedenen Thiolreagenzien, wie EtSH. Die Umsetzung wird unter einer inerten Atmosphäre durchgeführt, wie Stickstoff, mit ein oder mehreren Molen des Reagenzes pro Mol an Methoxygruppe, die gespalten wird.
- Geeignete Lösemittel für die Entschützungsreaktion oder Reaktion zur Entfernung der Schutzgruppe sind solche Lösemittel oder Gemische von Lösemitteln, die inert bleiben während der Demethylierungsreaktion. Halogenierte Lösemittel, wie Dichlormethan, 1,2-Dichlorethan und Chloroform, oder aromatische Lösemittel, wie Benzol oder Toluol, sind bevorzugt. Die bei dieser Reaktion oder Umsetzugn eingesetzte Temperatur sollte ausreichend sein, um die Vervollständigung der Demethylierungsreaktion zu bewirken. Es ist jedoch vorteilhaft die Temperatur unterhalb von 0°C zu halten, um die Selektivität zur Spaltung der 4'-Methoxygruppe zu maximieren und die Bildung von ungewünschten Nebenprodukten, insbesondere dem Produkt 6,4'-Dihydroxyanalogons, entstehend durch überschüssige Demethylierung, zu vermeiden. Unter den bevorzugten Reaktionsbedingungen wird ein selektiv dealkyliertes Produkt gebildet nach einem Rühren der Umsetzung oder Reaktion für etwa 1 bis 24 h. Eine bevorzugte Variation schließt die Verwendung von Bortribromid in der Menge von etwa 1,5 Mol mit 1 Mol des 6-, 4'-Dimethoxysubstrats in Dichlormethan ein unter einer Stickstoffatmosphäre bei einer Temperatur von –20°C über einen Zeitraum von 1 bis 4 Stunden.
- Die Ausgangsmaterialien für die Verfahren der vorliegenden Erfindung können erhalten werden durch eine Vielzahl von Reaktionswegen, die solche einschließen, die offenbart sind in
US 4 133 814 undUS 4 380 635 . Das Verfahren zur Herstellung von Verbindungen der Formel I, wie bereitgestellt durch die vorliegende Erfindung, ist unten gezeigt in Schema I: Schema I
- Das gesamte Reaktionsverfahren umfasst eine erste Cyclisierungsstufe und eine nachfolgende Umlagerungsstufe. Eine Verbindung der Formel I ist das gewünschte Produkt. Die Cyclisierungsstufe in der ersten Stufe findet statt mit einer Vielzahl von Säurekatalysatoren, und findet im Allgemeinen 50- bis 100 mal schneller statt als die nachfolgende Umlagerungsreaktion. Die vorliegende Erfindung setzt ein Kationenaustauschharz als Säurekatalysator ein.
- Die Cyclisierungsreaktionsrate kann erhöht werden durch Erhöhung der Menge an eingesetztem Harz in dem Reaktionsgemisch. Die Wirkung der Katalysatorbeladung auf die Reaktionsausbeute wurde untersucht, wenn das A15 Harz als Kationenaustauschharz eingesetzt wurde. Über den Bereich von 5 bis 33 ml Katalysator/Gramm Reaktant gab es keine Wirkung auf die Ausbeute oder auf den Grad der Desmethylbildung. Die Reaktionsrate ist jedoch direkt proportional zu der Katalysatorbeladung.
- Ein beliebiges Kationenaustauschharz oder eine Kombination von Kationenaustauschharzen kann in der Cyclisierungsstufe eingesetzt werden. Bevorzugt für die Praxis der vorliegenden Erfindung sind Kationenaustauschharze auf Polystyrol-Basis. Besonders bevorzugt sind Sulfonsäurekatalysatoren auf Polystyrol-Basis.
- Die Kationenaustauschharze können leicht getrennt werden von dem Gesamtreaktionsgemisch durch beliebige Mittel, die, ohne darauf beschränkt zu sein, Filtration einschließen, und alle wiedergewonnenen Harze können wiederverwendet werden. Ein Filtration kann erreicht werden durch beliebige Mittel, die die Verwendung von Whatman-Papier, 100 mesh Sieb, 5–20 μm Filterkartuschen und dergleichen einschließen.
- Die Reaktion oder Umsetzung lässt man typischerweise unter Rückfluss laufen mit der azeotropen Entfernung von Wasser. Die Wirkung von Wasser auf die Aktivität oder Wirksamkeit von Sulfonsäureharzen in einigen Reaktionen oder Umsetzungen wurde zuvor diskutiert von A.R. Pitochelli, Ion Exchange Catalysis and Matrix Effects, Broschüre veröffentlicht von Rohm und Haas, Inc., 1975. Siehe auch G. Zundel, Hydration and Intermolecular Interaction Infrared Investigations with Polyelectrolyte Membranes, Academic Press, New York, 1969 und G. Zundel et al., Physik. Chem., 59, 225, 1968.
- Verschiedene Desmethylnebenprodukte können entstehen während der Reaktion. Die Strukturen von vier verschiedenen Desmethylnebenprodukten sind bereitgestellt in dem Schema II unten: Schema II
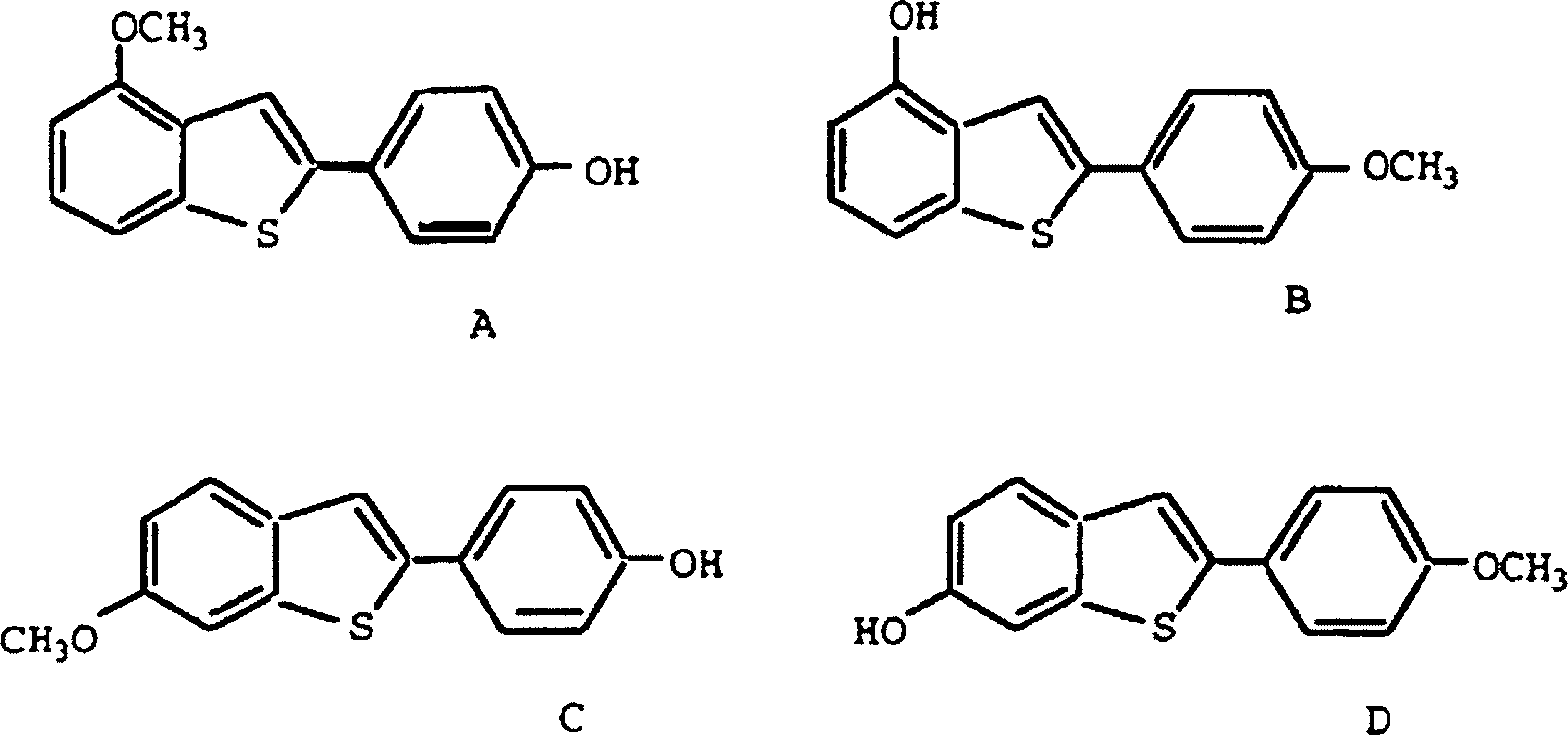
- Die Isomere A und B stammen von einer Verbindung der Formel Ia', während die Isomere C und D von einer Verbindung der Formel I stammen. Das Verhältnis der Isomere A:B:C:D in einem typischen Reaktionsgemisch war ungefähr 1:1:9:9. Die Isomeridentität wurde im Allgemeinen bestätigt durch HPLC. Das Isomerenverhältnis und daher die letzte Ausbeute wird bestimmt durch die kinetisch gesteuerte Cyclisierungsreaktion. Unter Verwendung eines Kationenaustauschharzes in Toluol wurde ein bevorzugtes Isomerverhältnis von 88:12 (I/Ia') in der Cyclisierungsstufe erhalten im Vergleich zu einem Isomerenverhältnis von 75:25, das erhalten wird, wenn Polyphosphorsäure in der Cyclisierungsstufe verwendet wurde. Eine weitere Äquilibrierung oder Gleichgewichtseinstellung zwischen ortho- und para-Isomeren während dieses Verfahrens wurde nicht beobachtet.
- Die Umlagerungsreaktion ist eine thermodynamisch gesteuerte oder kontrollierte Reaktion. Die Gleichgewichtskonstanten für die Reaktion sind wie folgt: K1 ist > 100, während K2 etwa 7–9 beträgt. Unter Verwendung eines Kationenaustauschharzes und Toluol/Heptan als Lösemittelsystem kristallisiert eine Verbindung der Formel Ia, wenn sie sich im Reaktionsgemisch bildet, um dadurch die Reaktion oder Umsetzung zur Vervollständigung zu treiben. Die Umlagerung des ungewünschten Isomers, einer Verbindung der Formel Ib', war 3–5 mal schneller als die Umlagerung des gewünschten Isomers, einer Verbindung der Formel Ib.
- Lösemittel, die Lösemittelgemische und Co-Lösemittel einschließen, die in der Praxis der vorliegenden Erfindung eingesetzt werden, können die Gesamtreaktion beeinflussen, einschließlich der Reaktionsprodukte und Gesamtausbeute. Typischerweise ist das Lösemittel der Wahl eine sehr schwache Base. Außerdem sollte das Lösemittel das Sulfonsäureproton des Harzes nicht solvatisieren. Das bevorzugte Lösemittel für die Praxis der vorliegenden Erfindung ist ein aromatisches Lösemittel, wobei angemessene oder vernünftige Ergebnisse erzielt werden sowohl in aliphatischen als auch chlorierten Lösemitteln. Beispielhafte Lösemittel schließen Toluol, Heptan, Xylol, Chlorbenzol, Dimethoxyethan und Tetrachlorethylen ein. Bevorzugt für die Praxis der vorliegenden Erfindung ist Toluol. Besonders bevorzugt ist Toluol mit zugesetzter Methansulfonsäure. Die zugegebene Methansulfonsäure erleichtert die nachfolgende Umlagerungsreaktion. Ausreichend Methansulfonsäure muss zugegeben werden zu dem Toluol, so dass sich eine getrennte MSA Phase bildet.
- Heptan ist ein ebenfalls bevorzugtes Lösemittel, das die Kristallisation des Benzothiophenprodukts beeinflusst oder bewirkt. Diese Kristallisation erzeugt eine dramatische Reduktion in der Löslichkeit desselben, was dadurch das Gleichgewicht der Reaktion treibt. Heptan wird am besten zu dem Reaktionsgemisch vor dem Gleichgewicht gegeben.
- Gemäß der Erfindung wird die Cyclisierungsreaktion bei Temperaturen von etwa 50°C bis etwa 110°C, vorzugsweise von etwa 75°C bis 110°C und am meisten bevorzugt von etwa 80 bis 110°C, durchgeführt. Die Ausbeute der Cyclisierungsreaktion ist die gleiche ob die Reaktion bei Rückfluss oder bei 70°C durchgeführt wird. Rückfluss wird jedoch sehr bevorzugt, da die Reaktionsrate oder Reaktionsgeschwindigkeit 10 bis 20 mal schneller ist. Die Verwendung höherer Temperaturen während der Reaktion ist nicht erwünscht, da dies zu erhöhten Gehalten an Desmethylnebenprodukten führt.
- Das Acetophenonausgangsmaterial wird in Gegenwart eines Kationenaustauschharzes und Toluol während eines Zeitraums von mindestens 30 min erwärmt, und vorzugsweise von etwa 60 bis 180 min. Wie gegenwärtig in der Praxis ausgeführt, wird das Acetophenon bei etwa 100°C etwa 3 bis 5 h lang cyclisiert. Nachfolgend zu dieser Anfangserwärmungsperiode wird die Reaktion auf eine Temperatur von etwa 50 bis 90°C gekühlt, und das Kationenaustauschharz wird durch Filtration entfernt. Wenn die Reaktion auf unter 50°C gekühlt wird, kann eine leichte Kristallisation von Benzothiophen auftreten, was eine Funktion ist der Menge der Umlagerung, die auftrat während der Cyclisierungsstufe. Typischerweise tritt 1 bis 7% Umlagerung während der Cyclisierungsreaktion auf.
- Die Umlagerungsreaktion findet typischerweise in Gegenwart der Methansulfonsäure und des Toluolreaktionsgemischs statt. Die weitere Zugabe von Heptan zu diesem Zeitpunkt ist gegebenenfalls möglich, kann aber eine gesteigerte Reaktionsausbeute bereitstellen. Nach der Zugabe von Heptan während der Reaktion ist eine Erhöhung der Temperatur von 90° auf 106° unerwünscht aufgrund der negativen Wirkung auf die Ausbeute.
- Ein geeignetes Lösemittel oder Lösemittelgemisch kann ferner zugegeben werden zu dem Reaktionsgemisch am Ende der Umlagerungsreaktion, um die Reaktion zu stoppen oder zu quenchen. Ein Beispiel eines geeigneten Lösemittels ist, ohne darauf beschränkt zu sein, Isopropanol (IPA) und dergleichen. Diese Lösemittelzugabe verringert die Löslichkeit des Produkts und verbessert die Reinheit desselben.
- Das Gesamtverfahren kann als eine „Ein-Topf"-Synthese, chargenweise, halbkontinuierlich, kontinuierlich und dergleichen betrieben werden. Ein Fachmann wird die Unterschiede zwischen diesen Betriebsarten abschätzen, einschließlich welche Reaktion eingesetzt wird für einen gegebenen Zweck. Zum Beispiel werden im halbkontinuierlichen oder kontinuierlichen Betrieb das Ausgangsmaterial und Lösemittel einer gepackten Säule des festen Säureharzes zugeführt. Die Gewinnung und Isolierung von überschüssigem Lösemittel und Produkt kann bewerkstelligt werden durch Destillation. Ferner wird die Reaktion gegebenenfalls in Gegenwart eines organischen Lösemittels durchgeführt, das ein Azeotrop mit Wasser bildet, und erleichtert auf diese Weise die Entfernung von Nebenprodukt durch azeotrope Destillation während des Reaktionsverfahrens. Beispiele von solchen Lösemitteln, die eingesetzt werden können, schließen aromatische Kohlenwasserstoffe ein, wie Benzol, Toluol, Xylol und dergleichen.
- Das Benzothiophenprodukt kann mit einer Standardextraktionsaufarbeitung isoliert werden durch Zugabe von Wasser, Trennung der Phasen, gegebenenfalls Extraktion der wässrigen Phase wieder mit dem organischen Lösemittel, Kombination oder Vereinigung der organischen Phasen und Konzentration der vereinigten organischen Phasen. Wenn das Ausgangsmaterial das Methoxyderivat ist, kristallisiert die gewünschte 6-Alkoxyverbindung in dem konzentrierten Lösemittel, während das 4-Alkoxyisomer in der Lösung verbleibt. Die gewünschte 6-Alkoxyverbindung kann durch Filtration gesammelt werden.
- Bei einem bevorzugten Cyclisierungsverfahren gemäß der Erfindung ist das Ausgangsmaterial α-(3-Methoxyphenylthio)-4-methoxyacetophenon, das nach Aufarbeitung nach Cyclisierung 6-Methoxy-2-(4-methoxyphenyl)benzo[b]thiophen ergibt. Dieses Material kann danach in eine Verbindung der Formel III überführt werden, wie z.B. 6-Hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-aminoethoxy)benzoyl]benzo[b]thiophen. Eine Umsetzung von 6-Alkoxy-2-(4-alkoxyphenyl)benzo[b]thiophen zu Verbindungen der Formel III kann bewerkstelligt werden gemäß den Umsetzungen, wie sie bereitgestellt werden in
US 4 380 635 . - Die Verbindungen der Formel III werden sehr oft in der Form von Säureadditionssalzen verabreicht. Die Salze werden geeigneterweise gebildet wie es üblich ist in der organischen Chemie durch Umsetzung der hergestellten Verbindung gemäß dieser Erfindung mit einer geeigneten Säure. Die Salze werden schnell gebildet in hohen Ausbeuten bei moderaten oder mäßigen oder mittleren Temperaturen und werden oft her gestellt durch bloßes Isolieren der Verbindung aus einer geeigneten Säurewäsche als letzter Stufe der Synthese. Zum Beispiel können Salze gebildet werden mit anorganischen oder organischen Säuren.
- Typische anorganische Säuren, die verwendet, um solche Salze zu bilden, schließen Chlorwasserstoffsäure, Bromwasserstoffsäure, Iodwasserstoffsäure, Salpetersäure, Schwefelsäure, Phosphorsäure, Hypophosphorsäure und dergleichen ein. Salze, die abgeleitet sind aus organischen Säuren, wie aliphatischen Mono- und Dicarbonsäuren, phenylsubstituierten Alkansäuren, Hydroxyalkan- und Hydroxyalkandisäuren, aromatischen Säuren, aliphatischen und aromatischen Sulfonsäuren, können ebenso verwendet werden. Solche pharmazeutisch annehmbaren Salze schließen daher Acetat, Phenylacetat, Trifluoracetat, Acrylat, Ascorbat, Benzoat, Chlorbenzoat, Dinitrobenzoat, Hydroxybenzoat, Methoxybenzoat, Methylbenzoat, o-Acetoxybenzoat, Naphthalin-2-benzoat, Bromid, Isobutyrat, Phenylbutyrat, β-Hydroxybutyrat, Butin-1,4-dioat, Hexin-1,4-dioat, Caprat, Caprylat, Chlorid, Cinnamat, Citrat, Formiat, Fumarat, Glykolat, Heptanoat, Hippurat, Lactat, Malat, Maleat, Hydroxymaleat, Malonat, Mandelat, Mesylat, Nicotinat, Isonicotinat, Nitrat, Oxalat, Phthalat, Terephthalat, Phosphat, Monohydrogenphosphat, Dihydrogenphosphat, Metaphosphat, Pyrophosphat, Propiolat, Propionat, Phenylpropionat, Salicylat, Sebacat, Succinat, Suberat, Sulfat, Bisulfat, Pyrosulfat, Sulfit, Bisulfit, Sulfonat, Benzolsulfonat, p-Bromphenylsulfonat, Chlorbenzolsulfonat, Ethansulfonat, 2-Hydroxyethansulfonat, Methansulfonat, Naphthalin-1-sulfonat, Naphthalin-2-sulfonat, p-Toluolsulfonat, Xylolsulfonat, Tartrat und dergleichen ein. Ein bevorzugtes Salz ist das Hydrochloridsalz.
- Die folgende Gruppe von Verbindungen wird bereitgestellt als weitere Veranschaulichung des Gesamtverfahrens, das hierin offenbart ist:
6-Hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-dimethylaminoethoxy)benzoyl]benzo[b]thiophen;
3-[4-(2-Ethoxymethylaminoethoxy)benzoyl]-6-hydroxy-2-(4-hydroxylphenyl)benzo[b]thiophen;
3-[4-(2-Ethoxylisopropylaminoethoxy)benzoyl]-6-hydroxy-2-(4-hydroxyphenyl))benzo[b]thiophen;
3-(4-(2-Dibutylaminoethoxy)benzoyl]-5-hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophen;
3-[4-(2-(1-Methylpropyl)methylaminoethoxy]benzoyl]-6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophen;
6-Hydroyx-2-(4-hydroxyphenyl)-3-[4-[2-di(2-methylpropyl)aminoethoxy]benzoyl]benzo[b]thiophen;
6-Hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-pyrrolidinoethoxy)benzoyl]benzo[b]thiophen;
6-Hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-piperidinoethoxy)benzoyl]benzo[b]thiophen;
6-Hydroxy-2-(4-hydroxyphenyl)-3-[4-(2-morpholinoethoxy)benzoyl]benzo[b]thiophen;
3-[4-(2-Hexamethyleniminoethoxy)benzoyl]-6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophen. - Die folgenden Beispiele werden bereitgestellt, um die Praxis der vorliegenden Erfindung besser zu erläutern und sollten nicht so ausgelegt werden, um den Schutzbereich derselben in irgendeiner Weise zu beschränken. Leute vom Fach werden erkennen, dass verschiedene Modifizierungen oder Veränderungen gemacht werden können, ohne von dem Geist und dem Schutzbereich der Erfindung abzurücken. Alle genannten Veröffentlichungen und Patentanmeldungen in der Beschreibung geben das Niveau der Fachleute an, an die sich diese Erfindung richtet.
- Beispiele
- Alle Experimente wurden unter Überdruck von trockenem Stickstoff durchgeführt. Alle Lösemittel und Reagenzien wurden verwendet, wie sie erhalten wurden. Die Prozentangaben sind im Allgemeinen berech net auf der Basis von Gewicht (w/w), mit Ausnahme der Lösemittel für die Hochleistungs-Flüssigchromatographie (HPLC), die berechnet sind auf einer Volumenbasis (v/v). Die protonenkernmagnetischen Resonanz (1H NMR)-Spektren und 13C kernmagnetischen Resonanzspektren (13C NMR) wurden auf einem Bruker AC-300 FTNMR Spektrometer erhalten bei 300,135 MHz oder einem GE QE-300 Spektrometer bei 300,15 MHz. Kieselgel-Flashchromatographie wurde durchgeführt wie beschrieben von Sill et al. unter Verwendung von Kieselgel 60 (230–400 mesh, E. Merck). Still et al., J. Org. Chem., 43, 2923 (1978). Die Elementaranalysen für Kohlenstoff, Wasserstoff und Stickstoff wurden bestimmt auf einem Control Equipment Corporation 440 Elemental Analyzer. Elementaranalysen für Schwefel wurden bestimmt auf einem Brinkman Colorimetric Elemental Analyzer. Die Schmelzpunkte wurden bestimmt in offenen Glaskapillaren auf einer Gallenkamp Heißluftbadschmelzpunktapparatur oder einem Mettler FP62 Automatic Instrument und sind unkorrigiert. Felddesorptionsmassenspektren (FDMS) wurden erhalten unter Verwendung eines Varian Instruments VG 70-SE oder VG ZAB-3F Massenspektrometer. Hochauflösungs-Free Atom Bombardment Massenspektren (FABMS) wurden erhalten unter Verwendung eines Varian Instruments VG ZAB-2SE Massenspektrometer.
- Die Ausbeuten von 6-Methoxy-2-(4-methoxyphenyl)benzo[b]thiophen können bestimmt werden durch Hochleistungs-Flüssigchromatographie (HPLC) im Vergleich zu einer authentischen Probe dieser Verbindung hergestellt durch veröffentlichte Syntheserouten. Siehe z.B. das US Patent mit der Nummer
US 4 133 814 . - Beispiel 1
- Cyclisierung:
- 40 g α-(3-Methoxyphenylthio)-4-methoxyacetophenon, 4 g trockenes Amberlyst® 15 (A15) Harz (erhältlich von Rohm & Haas) und 120 ml Toluol (Drum Stock) wurden zugegeben zu einem 1 l Dreihalsrundbodenkolben, ausgestattet mit einem Rückflusskühler und einer Dean-Stark-Falle. Die Falle ist entweder vorgefüllt mit Toluol oder extra Lösemittel wird zu dem Reaktor gegeben. Das Gemisch wird bis zum Rückfluss erwärmt und während eines Zeitraums von 3 bis 5 h gerührt während einer azeotropen Entfernung des Wassers. Dieses Gemisch wurde dann gekühlt auf 50 bis 70°C. Das Harz wurde abgefiltert unter Verwendung eines 4,25 cm Büchner Trichters und mit 20 ml Toluol gewaschen. Das Gesamtgewicht des Filtrats wurde aufgezeichnet, und das Volumen des Toluols, das benötigt wird zur Verwendung als Waschflüssigkeit für den Transfer des Filtrats in einen 500 ml Umlagerungskolben, wurde berechnet. [Waschvolumen = (Gewicht-161,5)/0,866.] [Beachte: Dieses Volumen an Waschflüssigkeit berücksichtigt den Verdampfungsverlust, der auftritt während der heißen Toluolfiltration.]
- Umlagerung:
- Das Filtrat wurde in einen 500 ml Rundbodenkolben überführt, der ausgestattet war mit einem Rückflusskühler. 14 g Methansulfonsäure (MSA) wurden über einen Zeitraum von 2 bis 5 min mit einem Tropftrichter zugegeben. Das Gemisch wurde mit 3 ml Toluol gewaschen und bei 90°C 3 bis 5 h lang gerührt. 56 ml Heptan (Drum Stock) wurde während eines Zeitraums von 5 bis 20 min zugegeben. Das Gemisch wurde dann bei 90°C 1 h lang gerührt und bei 80°C 3 bis 4 h lang. 98 ml Isopropanol (IPA) (Drum Stock) wurden über einen Zeitraum von 5 bis 20 min zugegeben, und dann über einen Zeitraum von 30 min bei etwa 83°C refluxiert. Das Gemisch wurde dann auf 0°C gekühlt mit einer Rate von nicht schneller als 50°C pro Stunde. Dies wurde dann 1 h lang bei 0°C gerührt, filtriert, zweimal mit 75 ml 70/30 (Toluol/IPA) gewaschen und bei 60°C unter Vollvakuum getrocknet. Ausbeute = 77 bis 80,4%; 100% Wirkstoffgehalt; 0,1% Desmethyl; 0,1% Verbindung D; 0,3% TRS.
- Beispiel 2
- Die folgende Umsetzung wurde in der Pilotanlage durchgeführt. Die Cyclisierung und Umlagerung wurden beide in 50 Gallonen Hastelloy C Reaktoren durchgeführt. Sofern es nicht anders angegeben ist, sind die Reaktionsbedingungen wie in Beispiel 1 angegeben. Cyclisierung:
α-(3-Methoxyphenylthio)-4-methoxyacetophenon: 14 kg A15 Harz: 1,4 kg Toluol: 42 l Cyclisierungsrückflusszeit: 2,5 h A15 Toluol Waschflüssigkeit: 6 l A15 Harz Filtrationstemperatur: 60°C MSA: 4,9 kg MSA/Toluol Waschflüssigkeit: 1 l Rührzeit bei 90°C vor Heptan: 3 h Rührzeit bei 90°C nach Heptan: 1 h Rührzeit bei 80°C nach Heptan: 3 h Heptan: 20 l Heptanzugabezeit: 20 min IPA Stoppmenge: 34 l IPA Zugabezeit: 17 min Rückflusszeit nach IPA Zugabe: 30 min Kühlungsrate: 50°C pro h Endtemperatur vor Produktfiltration: 0°C Waschen des Filterkuchens: 2 × 26 l 70/30 Toluol/IPA - Die erhaltenen Ergebnisse waren:
77,5% Ausbeute 100,1% Wirkstoffgehalt 0,21% rel Ersätze (rel subs) 0,08% Desmethyl - Beispiel 3
- Die Reaktionsbedingungen, die verwendet wurden für dieses Beispiel waren identisch zu solchen in Beispiel 2 außer für die folgenden Variablen: 50 Gallonen mit Glas ausgekleidete Reaktoren wurden verwendet anstelle der 50 Gallonen Hastelloy Reaktoren; die Rührzeit bei 90°C vor der Heptanzugabe wurde erhöht auf 4 h; und die Rührzeit bei 80°C nach der Heptanzugabe wurde erhöht auf 4 h. Die erhaltenen Ergebnisse waren wie folgt:
55% Ausbeute 99,5% Wirkstoffgehalt 0,30% rel Ersätze (rel subs) 0,09% Desmethyl - Beispiel 4
- Die folgende Umsetzung wurde durchgeführt in der Pilotanlage:
Die Cyclisierung und Umlagerung wurden beide durchgeführt in einem 50 Gallonen mit Glas ausgekleideten Reaktor. Sofern es nicht anders angegeben ist, sind die Reaktionsbedingungen wie angegeben in Beispiel 1. Die Menge an α-(3-Methoxyphenylthio)-4-methoxyacetophenon wurde erhöht auf 16,5 kg, und alle anderen Mengen wurden dementsprechend skaliert oder angepasst. Das Gesamtvolumen im Reaktionsgefäß wurde daher erhöht. Das Rühren des Reaktionsgemischs wurde erhöht von 95 auf 115 U/min. Die erhaltenen Ergebnisse waren:79,6% Ausbeute 100,6% Wirkstoffgehalt 0,25% rel Ersätze (rel subs) 0,08% Desmethyl
Claims (11)
- Verfahren zur Herstellung einer Verbindung der Formel Ibwobei die R-Gruppen gleich oder verschieden sind und für C1-C6-Alkyl stehen, wobei das Verfahren umfasst: Cyclisieren einer Dialkoxyverbindung der Formel II
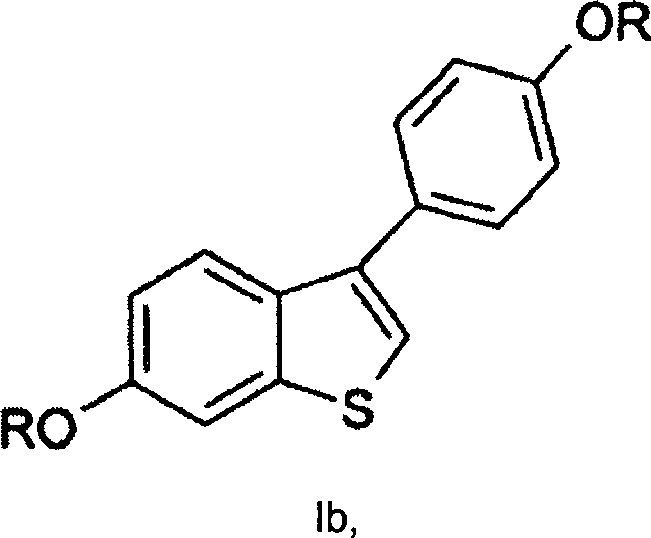 in Gegenwart eines Kationenaustauschharzes.
in Gegenwart eines Kationenaustauschharzes.
- Verfahren gemäß Anspruch 1, wobei R für Methyl steht.
- Verfahren gemäß Anspruch 1 oder 2, wobei das Kationenaustauschharz für ein Sulfonsäureharz auf Polystyrolbasis steht.
- Verfahren zur Herstellung einer Verbindung der Formel Iwobei die R-Gruppen gleich oder verschieden sind und für C1-C6-Alkyl stehen, wobei das Verfahren umfasst ein Herstellen einer Verbindung der Formel Ib gemäß dem Verfahren nach einem der Ansprüche 1 bis 3 und Bewirken einer nachfolgenden Umlagerung.

- Verfahren gemäß Anspruch 4, das ein Kontaktieren des Reaktionsgemischs mit Methansulfonsäure in Toluol umfasst.
- Verfahren gemäß Anspruch 5, das ein Kontaktieren des Reaktionsgemischs mit Heptan umfasst.
- Verfahren gemäß einem der Ansprüche 1 bis 7, wobei das Cyclisieren bei einer Temperatur von etwa 70°C bis etwa 90°C ausgeführt wird.
- Verfahren gemäß Anspruch 1, wobei das Verfahren als Zeitstaffelbetrieb ausgeführt wird.
- Verfahren gemäß Anspruch 1, wobei das Verfahren als Dauerbetrieb ausgeführt wird.
- Verfahren zur Herstellung einer Verbindung der Formel IIIwobei: R1 und R2 unabhängig für C1-C6-Alkyl stehen oder verbunden sind, um mit dem Stickstoff, an den sie gebunden sind, Piperidinyl, Pyrrolidinyl, Methylpyrrolidinyl, Dimethylpyrrolidinyl oder Hexamethylenimino zu bilden, oder die pharmazeutisch annehmbaren Salze oder Solvate davon; wobei das Verfahren umfasst: ein Cyclisieren einer Verbindung der Formel II
 wobei die R-Gruppen gleich oder verschieden sind und für C1-C6-Alkyl stehen in Gegenwart eines Kationenaustauschharzes.
wobei die R-Gruppen gleich oder verschieden sind und für C1-C6-Alkyl stehen in Gegenwart eines Kationenaustauschharzes.
- Verfahren gemäß Anspruch 11, wobei R für Methyl steht.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2669596P | 1996-09-25 | 1996-09-25 | |
| US26695P | 1996-09-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69735339D1 DE69735339D1 (de) | 2006-04-27 |
| DE69735339T2 true DE69735339T2 (de) | 2006-10-12 |
Family
ID=21833302
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69735339T Expired - Lifetime DE69735339T2 (de) | 1996-09-25 | 1997-09-22 | Verfahren zur Herstellung von Benzothiophenen |
Country Status (31)
| Country | Link |
|---|---|
| US (1) | US5977383A (de) |
| EP (1) | EP0832889B1 (de) |
| JP (1) | JP2001501208A (de) |
| KR (1) | KR20000048539A (de) |
| CN (1) | CN1088704C (de) |
| AR (1) | AR013321A1 (de) |
| AT (1) | ATE318805T1 (de) |
| AU (1) | AU718919B2 (de) |
| BR (1) | BR9712844A (de) |
| CA (1) | CA2266617A1 (de) |
| CO (1) | CO4900043A1 (de) |
| CZ (1) | CZ92899A3 (de) |
| DE (1) | DE69735339T2 (de) |
| EA (1) | EA001914B1 (de) |
| EG (1) | EG21037A (de) |
| ES (1) | ES2257761T3 (de) |
| HU (1) | HUP9904228A3 (de) |
| ID (1) | ID21349A (de) |
| IL (1) | IL129001A (de) |
| IN (1) | IN183239B (de) |
| MY (1) | MY118009A (de) |
| NO (1) | NO991193L (de) |
| NZ (1) | NZ334591A (de) |
| PE (1) | PE2699A1 (de) |
| PL (1) | PL332208A1 (de) |
| TR (1) | TR199900672T2 (de) |
| TW (1) | TW472053B (de) |
| UA (1) | UA62938C2 (de) |
| WO (1) | WO1998013363A1 (de) |
| YU (1) | YU15399A (de) |
| ZA (1) | ZA978372B (de) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2002230409A1 (en) * | 2000-11-27 | 2002-06-03 | Eli Lilly And Company | Process for preparing 3-aryl-benzo(b)thiophenes |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1138163A (en) * | 1965-05-21 | 1968-12-27 | Bristol Myers Co | Benzothiophene derivatives having anti-fertility properties |
| US3394125A (en) * | 1965-10-23 | 1968-07-23 | Bristol Myers Co | 2-phenyl-3-tertiary-aminoalkoxy phenyl-and corresponding tertiaryaminoalkyl thio benzfurans substituted in the benzo nucleus with an alkoxy or tertiaryamino alkoxy or alkylthio group |
| US4133814A (en) * | 1975-10-28 | 1979-01-09 | Eli Lilly And Company | 2-Phenyl-3-aroylbenzothiophenes useful as antifertility agents |
| US4230862A (en) * | 1975-10-28 | 1980-10-28 | Eli Lilly And Company | Antifertility compounds |
| AT364852B (de) * | 1977-11-05 | 1981-11-25 | Thomae Gmbh Dr K | Verfahren zur herstellung von kondensierten isothiazol-3(2h)-on-1,1-dioxiden und von deren salzen |
| US4436748A (en) * | 1980-10-20 | 1984-03-13 | Hoechst-Roussel Pharmaceuticals Inc. | Benzo[b]thiophenes |
| US4358593A (en) * | 1981-04-03 | 1982-11-09 | Eli Lilly And Company | Process for preparing 3-(4-aminoethoxybenzoyl)benzo[b]thiophenes |
| IL65379A0 (en) * | 1981-04-03 | 1982-05-31 | Lilly Co Eli | Process for preparing acylated benzothiophenes |
| US4418068A (en) * | 1981-04-03 | 1983-11-29 | Eli Lilly And Company | Antiestrogenic and antiandrugenic benzothiophenes |
| NZ200204A (en) * | 1981-04-03 | 1985-05-31 | Lilly Co Eli | Benzothiophene derivatives and process for preparation |
| IL65378A (en) * | 1981-04-03 | 1986-02-28 | Lilly Co Eli | Process for preparing 3-(4-aminoethoxybenzoyl)benzo-(b)thiophenes |
| US4380635A (en) * | 1981-04-03 | 1983-04-19 | Eli Lilly And Company | Synthesis of acylated benzothiophenes |
| US4544746A (en) * | 1982-05-17 | 1985-10-01 | Ciba-Geigy Corporation | Process for preparing 2-anilinoacridone |
| JPH02501301A (ja) * | 1987-09-25 | 1990-05-10 | チバ‐ガイギー アクチェンゲゼルシャフト | 4‐(トリアルキルベンジル)‐ピペラジニル化合物のジアシル誘導体 |
| US5395842A (en) * | 1988-10-31 | 1995-03-07 | Endorecherche Inc. | Anti-estrogenic compounds and compositions |
| DE4117512A1 (de) * | 1991-05-25 | 1992-11-26 | Schering Ag | 2-phenylbenzo(b)furane und -thiophene, verfahren zu deren herstellung und diese enthaltende pharmazeutische praeparate |
| JP3157882B2 (ja) * | 1991-11-15 | 2001-04-16 | 帝国臓器製薬株式会社 | 新規なベンゾチオフエン誘導体 |
| DE4204969A1 (de) * | 1992-02-19 | 1993-08-26 | Basf Ag | Verfahren zur herstellung von benzo(b)thiophenen |
| US6756388B1 (en) * | 1993-10-12 | 2004-06-29 | Pfizer Inc. | Benzothiophenes and related compounds as estrogen agonists |
| US5523416A (en) * | 1994-07-22 | 1996-06-04 | Eli Lilly And Company | Process for preparing 3-(4-aminoethoxy-benzoyl) benzo (B)-thiophenes |
| US5606075A (en) * | 1995-06-07 | 1997-02-25 | Eli Lilly And Company | Process for the synthesis of benzo[b]thiophenes |
| US5606076A (en) * | 1995-06-07 | 1997-02-25 | Eli Lilly And Company | Process for the synthesis of benzo[b]thiophenes |
-
1997
- 1997-09-17 IN IN1714CA1997 patent/IN183239B/en unknown
- 1997-09-17 PE PE1997000827A patent/PE2699A1/es not_active Application Discontinuation
- 1997-09-17 MY MYPI97004310A patent/MY118009A/en unknown
- 1997-09-17 ZA ZA978372A patent/ZA978372B/xx unknown
- 1997-09-17 AR ARP970104264A patent/AR013321A1/es unknown
- 1997-09-18 EG EG96197A patent/EG21037A/xx active
- 1997-09-19 YU YU15399A patent/YU15399A/sh unknown
- 1997-09-19 IL IL12900197A patent/IL129001A/xx not_active IP Right Cessation
- 1997-09-19 PL PL97332208A patent/PL332208A1/xx unknown
- 1997-09-19 BR BR9712844-9A patent/BR9712844A/pt not_active IP Right Cessation
- 1997-09-19 JP JP10515741A patent/JP2001501208A/ja active Pending
- 1997-09-19 CN CN97198100A patent/CN1088704C/zh not_active Expired - Fee Related
- 1997-09-19 EA EA199900330A patent/EA001914B1/ru not_active IP Right Cessation
- 1997-09-19 KR KR1019990702454A patent/KR20000048539A/ko not_active Ceased
- 1997-09-19 UA UA99031657A patent/UA62938C2/uk unknown
- 1997-09-19 ID IDW990074A patent/ID21349A/id unknown
- 1997-09-19 HU HU9904228A patent/HUP9904228A3/hu unknown
- 1997-09-19 CA CA002266617A patent/CA2266617A1/en not_active Abandoned
- 1997-09-19 AU AU43561/97A patent/AU718919B2/en not_active Ceased
- 1997-09-19 CZ CZ99928A patent/CZ92899A3/cs unknown
- 1997-09-19 NZ NZ334591A patent/NZ334591A/xx unknown
- 1997-09-19 TR TR1999/00672T patent/TR199900672T2/xx unknown
- 1997-09-19 WO PCT/US1997/016683 patent/WO1998013363A1/en not_active Ceased
- 1997-09-22 US US08/934,999 patent/US5977383A/en not_active Expired - Lifetime
- 1997-09-22 ES ES97307377T patent/ES2257761T3/es not_active Expired - Lifetime
- 1997-09-22 CO CO97055060A patent/CO4900043A1/es unknown
- 1997-09-22 EP EP97307377A patent/EP0832889B1/de not_active Expired - Lifetime
- 1997-09-22 AT AT97307377T patent/ATE318805T1/de not_active IP Right Cessation
- 1997-09-22 DE DE69735339T patent/DE69735339T2/de not_active Expired - Lifetime
- 1997-12-26 TW TW086113989A patent/TW472053B/zh not_active IP Right Cessation
-
1999
- 1999-03-11 NO NO991193A patent/NO991193L/no not_active Application Discontinuation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE19549755B4 (de) | Verfahren zur Herstellung eines nicht solvatisierten kristallinen Raloxifenhydrochlorids | |
| DE69520738T2 (de) | 2-amino-3-aroyl-benzo beta thiophene und verfahren zu deren herstellung | |
| DE69712590T2 (de) | Verfahren zur Herstellung von Benzothiophenen | |
| DE69731043T2 (de) | Benzo[B]thiophen-Verbindungen, Zwischenprodukte, Verfahren und Zusammensetzungen | |
| DE69735339T2 (de) | Verfahren zur Herstellung von Benzothiophenen | |
| DE69708418T2 (de) | Naphthylverbindungen und Zusammensetzungen | |
| DE69726513T2 (de) | Benzo[B]thiophenverbindungen,Zwischenprodukte,Verfahren,Zusammensetzungen und Methode | |
| DE69724062T2 (de) | Benzothiophenderivate, Zwischenprodukte, Verfahren, Zubereitungen und Methoden | |
| DE69522762T2 (de) | Verfahren zur Herstellung von 3-(4-Aminoethoxy-benzoyl)-benzo[beta]-thiophenen | |
| DE69724548T2 (de) | Substituierte 2,3-Aryl-Benzothiophene und ihre östrogene Aktivität | |
| DE69727529T2 (de) | Benzothiophenverbindungen, Zwischenprodukte, Zusammensetzungen und Methode | |
| DE69513022T2 (de) | Verfahren zur herstellung von hydroxythioacetamiden und deren verwendung zur synthese von benzothiophenen | |
| HK1009812A (en) | Process for the synthesis of benzothiophenes | |
| DE69735340T2 (de) | Benzo(b)thiophenderivate, Zwischenprodukte, Zubereitungen und Verfahren | |
| IL143559A (en) | Process for the synthesis of benzothiophenes | |
| DE69711528T2 (de) | Naphthofluorenverbindungen, Zwischenverbindungen, Zusammensetzungen und Verfahren | |
| HK1010883A (en) | Process for the synthesis of benzothiophenes | |
| MXPA99004323A (en) | Process for the synthesis of benzothiophenes | |
| DD201794A5 (de) | Verfahren zur herstellung von 6-hydroxy-2-(4-hydroxyphenyl) 3-(4-(2-aminoethoxy)benzoyl)benzo(b)thiophenen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8328 | Change in the person/name/address of the agent |
Representative=s name: KROHER, STROBEL RECHTS- UND PATENTANWAELTE, 80336 |
|
| 8328 | Change in the person/name/address of the agent |
Representative=s name: DR. SCHOEN & PARTNER, 80336 MUENCHEN |