-
TECHNISCHES
GEBIET
-
Diese
Erfindung betrifft drei chimäre
Gene, wobei das erste Dihydrodipicolinsäuresynthase (DHDPS) kodiert,
die gegen Inhibition durch Lysin unempfindlich ist und auf funktionsfähige Weise
mit einer pflanzlichen Chloroplasttransitsequenz verknüpft ist,
ein zweites, das ein lysinreiches Protein kodiert und ein drittes,
das eine pflanzliche Lysinketoglutaratreduktase kodiert, die alle
auf funktionsfähige
Weise mit pflanzensamenspezifischen Regulationssequenzen verknüpft sind.
Es werden Methoden zu ihrer Anwendung zum Herstellen erhöhter Niveaus
von Lysin in den Samen transformierter Pflanzen bereitgestellt.
Außerdem
werden transformierte Mais-, Raps- und Sojabohnenpflanzen bereitgestellt,
wobei die Samen Lysin in höheren
Niveaus aufspeichern als untransformierte Pflanzen.
-
HINTERGRUND
DER ERFINDUNG
-
Der
menschlichen Nahrung und Tierfutter, die von vielen Getreidearten
deriviert sind, mangelt es an einigen der zehn essentiellen Aminosäuren, die
in der tierischen Nahrung erforderlich sind. Beim Mais (Zea mays
L.) ist Lysin die am stärksten
einschränkende
Aminosäure
bezüglich
der Ernährungserfordernisse
vieler Tiere. Von anderen Nutzpflanzen, z.B. der der Sojabohne (Glycine
max L.) oder Raps (Brassica napus) deriviertes Mehl wird als Zusatzmittel
zu Tierfutter auf Maisbasis verwendet, um den Lysinmangel auszugleichen. Auch
wird zusätzliches
Lysin, das durch fermentieren von Mikroben hergestellt wird, als
Ergänzung
in Tierfutter verwendet. Eine Erhöhung des Lysingehalts von aus
pflanzlichen Quellen deriviertem Mehl würde die Notwendigkeit des Ergänzens Getreidemischfutter
mit mikrobiell hergestelltem Lysin reduzieren oder eliminieren.
-
Der
Aminosäuregehalt
von Samen wird hauptsächlich
(90-99%) durch die Aminosäureszusammensetzung
der Proteine im Samen und in geringerem Maße (1-10%) durch die Pools
freier Aminosäure
bestimmt. Die Menge an gesamtem Protein in Samen variiert zwischen
ca. 10%, auf das Trockengewicht bezogen, bei Cerealien Getreiden
und 20-40%, auf das Trockengewicht bezogen, bei Hülsenfrüchten. Ein
Großteil
der proteingebundenen Aminosäuren
ist in den Samenspeicherproteinen enthalten, die während der
Samenentwicklung synthetisiert werden und als Hauptnahrungsreserve
auf das Keimen hin dienen. In vielen Samenkörnern machen die Speicherproteine
50% oder mehr des gesamten Proteins aus.
-
Um
die Aminosäureszusammensetzung
von Samen zu verbessern, wird die genetische Engineeringtechnologie
zum Isolieren und Exprimieren von Genen für Speicherproteine in transgenen
Pflanzen angewendet. Beispielsweise ist ein Gen aus der Paranuss
für ein
Samen-2S-Albumin, das aus 26% schwefelhaltigen Aminosäuren besteht,
isoliert (Altenbach et al. (1987) Plant Mol. Biol. 8:239-250) und
in den Samen von transformiertem Tabak unter der Kontrolle der Regulationssequenzen
aus einem Bohnenphaseolin-Speicherprotein-Gen exprimiert worden.
Die Speicherung von schwefelreichem Protein in den Tabaksamen führte zu
einer bis zu 30%igen Erhöhung
des Niveaus an Methionin in den Samen (Altenbach et al. (1989) Plant
Mol. Biol. 13:513-522). Jedoch sind keine pflanzlichen Samenspeicherproteine,
die ähnlich
mit Lysin im Vergleich mit dem durchschnittlichen Lysingehalt von
Pflanzenproteinen angereichert sind, bisher identifiziert worden,
was diesen Ansatz daran hindert, zum Erhöhen von Lysin verwendet zu
werden.
-
Ein
alternativer Ansatz besteht darin, die Erzeugung und Speicherung
von Lysin durch genetische Engineeringtechnologie zu erhöhen. Zusammen
mit Threonin, Methionin und Isoleucin ist Lysin eine Aminosäure, die
von Aspartat deriviert ist und die Regulierung der Biosynthese jedes
Mitglied dieser Familie ist kompliziert, gegenseitig in Bezug stehend
und, besonders bei Pflanzen, nicht klar verständlich. Die Regulierung des
Stoffwechselflusses auf dem Stoffwechselweg scheint hauptsächlich durch
Endprodukte in Pflanzen zu erfolgen. Der Aspartatfamilienweg wird
ebenfalls an den Verzweigungspunktreaktionen reguliert. Bei Lysin
ist das die Kondensation von Aspartyl-β-semialdehyd mit Pyruvat, die
durch Dihydrodipicolinsäuresynthase
(DHDPS) katalysiert ist.
-
Das
E. coli dapA-Gen kodiert ein DHDPS-Enzym, das gegen Inhibition durch
Lysin etwa 20 mal weniger empfindlich ist als ein typisches pflanzliches
DHDPS-Enzym, z.B. Weizenkeim-DHDPS. Das E. coli dapA-Gen ist mit
dem 35S-Promotor des Blumenkohlmosaikvirus und einer pflanzlichen
Chloroplasttransitsequenz in Verbindung gebracht worden. Das chimäre Gen wurde
durch Transformation in Tabakzellen eingeführt und es wurde gezeigt, dass
es eine wesentliche Erhöhung
der Niveaus an freiem Lysin in den Blättern hervorruft [Glassman
et al. (1989) PCT-Patentanmeldung PCT/US89/01309, Shaul et al. (1992)
Plant Jour. 2:203-209, Galili et al. (1992), EPA-Patentanmeldung
91119328.2, Falco, PCT/US93/02480 (internationale Veröffentlichung
Nr. WO 93/19190)]. Jedoch wurde der Lysingehalt der Samen bei keiner
der in diesen Studien beschriebenen transformierten Pflanzen erhöht. Das
gleiche chimäre
Gen wurde auch in Kartoffelzellen eingeführt und führt zu einer geringen Erhöhung des
freien Lysins in den Blättern,
Wurzeln und Knollen regenerierter Pflanzen [Galili et al. (1992)
EPA-Patentanmeldung 91119328.2, Perl et al. (1992) Plant Mol. Biol 19:815-823].
-
Falco,
PCT/US93/02480 (internationale Veröffentlichung Nr. WO 93/19190)
hat das E. coli dapA-Gen mit
dem Bohnenphaseolinpromotor und einer pflanzlichen Chloroplasmansitsequenz
verknüpft,
um die Expression in den Samen zu erhöhen, beobachteten jedoch immer
noch keine Erhöhung
des Lysinniveaus in Samen. Wie oben bemerkt, wird der erste Schritt
des Lysinbiosynthesewegs durch Aspartochinase (AK) katalysiert und
dieses Enzym hat sich als ein wichtiges Target für die Regulierung bei vielen
Organismen erwiesen. Falco hat einen Mutanten des E. coli lysC-Gens
isoliert, das eine für
das Lysin-Feedback unempfindliche AK kodiert und sie mit dem Bohnenphaseolinpromotor
und einer pflanzlichen Chloroplasttransitsequenz verknüpft. Die
Expression dieses chimären
Gens in den Samen von transformiertem Tabak führte zu einer wesentlichen Erhöhung des
Niveaus an Threonin, jedoch nicht an Lysin. Galili et al. (1992),
EPA-Patentanmeldung 91119328.2 schlagen vor, dass das Transformieren
von Pflanzen mit chimären
Genen durch Verknüpfen
samenspezifischer Promotoren mit einem pflanzlichen Chloroplasttransitsequenz/E.
coli dapA-Gen und pflanzlichen Chloroplasttransitsequenz/mutantem
E. coli lysC-Gen zu erhöhten
Lysinniveaus in Samen führt.
Falco, PCT/US93/02480 (internationale Veröffentlichung Nr. WO 93/19190)
führte
diesen Versuch durch Transformieren von Tabak mit einem Konstrukt
aus, das beide chimäre
Gene, das Bohnenphaseolinpromotor/pflanzliche Chloroplasmansitsequenz/E.
Coli dapA-Gen und das Bohnenphaseolinpromotor/pflanzliche Chloroplasmansitsequenz/mutante
E. Coli lysC-Gen enthält.
Die gleichzeitige Exprimierung beider Gene hatte keine signifikante
Wirkung auf den Lysingehalt der Samen. Jedoch ist beobachtet worden,
dass ein Abbauprodukt von Lysin, nämlich α-Aminoadipinsäure, sich
in den Samen ansammelt. Dies weist darauf hin, dass die Ansammlung von
freiem Lysin in Samen aufgrund von Lysinkatabolismus verhindert
wurde. Bei einem Versuch, die Biosyntheserate von Lysin zu erhöhen, isolierte
Falco, PCT/US93/02480 (internationale Veröffentlichung Nr. WO/93/19190)
das Corynebacterium glutamicum dapA-Gen, das ein gegen Lysin vollständig unempfindliches DHDPS-Enzym
kodiert. Falco transformiert Tabak mit einem Konstrukt, das das
chimäre
Gen Bohnenphaseolinpromotor/pflanzliche Chloroplasttransitsequenz/Corynebacterium
glutamicum dapA-Gen mit Bohnenphaseolinpromotor/pflanzlicher Chloroplasttransitsequenz/mutantem
E. Coli lysC-Gen verknüpft
enthält.
Die gleichzeitige Exprimierung dieser beiden gegen Lysin unempfindlichen
Enzyme hat immer noch keine signifikante Wirkung auf den Lysingehalt
der Samen.
-
So
ist es klar, dass das begrenzte Verständnis der Einzelheiten der
Regulation des Lysinbiosynthesewegs in Pflanzen, insbesondere in
Samen, die Anwendung genetischer Engineeringtechnologie zum Erhöhen des
Lysingehalts ungewiss macht. Es ist bei den meisten Pflanzen nicht
bekannt, ob Lysin in Samen synthetisiert oder zu den Samen von den
Blättern
transportiert wird. Außerdem
ist wenig bekannt über
die Speicherung oder den Katabolismus von Lysin in Samen. Da freie
Aminosäuren
nur einen geringen Bruchteil des gesamten Aminosäuregehalts von Samen ausmachen,
muss die übermäßige Speicherung
vielfach sein, um die gesamte Aminosäureszusammensetzung der Samen
wesentlich zu beeinflussen. Außerdem
sind die Wirkungen der übermäßigen Speicherung
einer freien Aminosäure
wie Lysin auf die Samenentwicklung und Lebensfähigkeit nicht bekannt.
-
Vor
der hier beschriebenen Erfindung war keine Methode zum Erhöhen des
Lysingehalts von Samen durch genetisches Engineering bekannt und
es waren keine Beispiele von Samen mit erhöhten Lysinniveaus, die durch
genetisches Engineering erhalten worden waren, bekannt.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Diese
Erfindung betrifft ein neuartiges chimäres Gen und Pflanzen, die durch
Anwendung des neuartigen Gens transformiert worden sind, wobei ein
Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die gegen Inhibition durch Lysin unempfindlich ist, auf
funktionsfähige
Weise mit einer pflanzlichen Monokotchloroplasttransitsequenz und
einer monokoten pflanzlichen samenspezifischen Regulationssequenz verknüpft ist.
Bei einer bevorzugten Ausführungsform
umfasst das Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die Nukleotidsequenz, die in SEQ ID NO:3 gezeigt ist, die
Dihydrodipicolinsäuresynthase
aus Corynebacterium glutamicum kodiert. Bei besonders bevorzugten
Ausführungsformen
ist die pflanzliche Chloroplasttransitsequenz von einem Gen deriviert,
das die kleine Untereinheit von Ribulose-1,5-bisphosphatcarboxylase
kodiert, und die samenspezifische Regulationssequenz stammt aus
dem Gen, das die β-Untereinheit
des Samenspeicherproteins Phaseolin aus dem Bohnen-Phaseolus vulgaris,
dem Kunitz-Trypsin-Inhibitor-3-Gen
von Glycine max. oder einem monokoten embryospezifischen Promotor,
bevorzugt aus dem Globulin 1-Gen von Zea-Mais kodiert.
-
Die
beschriebene Gene können
beispielsweise zum Transformieren von Pflanzen, bevorzugt Maispflanzen,
verwendet werden. Ebenfalls beansprucht sind Samen, die von den
transformierten Pflanzen erhalten werden. Die Erfindung kann zur
Erzeugung transformierter Pflanzen führen, wobei die Samen der Pflanzen Lysin
bis zu einem Niveau aufspeichern, das mindestens zehn Prozent höher ist
als bei Samen untransformierter Pflanzen, bevorzugt zehn bis vier
hundert Prozent höher
ist als bei untransformierten Pflanzen.
-
Die
Erfindung betrifft des Weiteren eine Methode zum Erhalten einer
Pflanze, bevorzugt einer Mais-, Raps- oder Sojabohnenpflanze, wobei
die Samen der Pflanzen Lysin in einem 10 Prozent bis 400 Prozent
höheren
Niveau aufspeichern als die Samen von untransformierten Pflanzen,
umfassend
- (a) das Transformieren von Pflanzenzellen,
bevorzugt Mais-, Raps- oder Sojabohnenzeilen, mit einem chimären Gen
umfassend ein Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die gegen Inhibition durch Lysin unempfindlich ist und
funktionsfähig
mit einer Pflanzenchloroplasmansitsequenz und einer pflanzensamenspezifischen
Regulationssequenz verknüpft
ist;
- (b) das Regenerieren fruchtbarer reifer Pflanzen aus den aus
Schritt (a) erhaltenen transformierten Pflanzenzellen unter Bedingungen,
die zum Erhalten von Samen geeignet sind;
- (c) das Screenen des Abkömmlingsamens
aus Schritt (b) auf den Lysingehalt hin; und
- (d) das Auswählen
derjenigen Linien, deren Samen erhöhte Niveaus an Lysin enthalten.
Transformierte Pflanzen, die durch diese Methode erhalten werden,
sind ebenfalls beansprucht.
-
Die
Erfindung betrifft zusätzlich
ein Nukleinsäurefragment
umfassend
- (a) ein erstes chimäres Gen,
wobei ein Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die gegen Inhibition durch Lysin unempfindlich ist, auf
funktionsfähige
Weise mit einer Pflanzenchloroplasmansitsequenz und einer pflanzensamenspezifischen
Regulationssequenz verknüpft
ist und
- (b) ein zweites chimäres
Gen, wobei ein Nukleinsäurefragment,
das ein lysinreiches Protein kodiert, wobei der Gewichtsprozentsatz
von Lysin mindestens 15% beträgt,
auf funktionsfähige
Weise mit einer pflanzensamenspezifischen Regulationssequenz verknüpft ist
und
-
Ebenfalls
beschrieben ist ein Nukleinsäurefragment
umfassend
- (a) ein erstes chimäres Gen,
wobei ein Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die gegen Inhibition durch Lysin unempfindlich ist, auf
funktionsfähige
Weise mit einer Pflanzenchloroplasttransitsequenz und einer pflanzensamenspezifischen
Regulationssequenz verknüpft
ist; und
- (b) ein zweites chimäres
Gen, wobei ein Nukleinsäurefragment,
das ein lysinreiches Protein kodiert, eine Nukleinsäuresequenz
umfasst, die ein Protein kodiert, das n Heptadeinheiten (defgabc)
umfasst, wobei jedes Heptad entweder gleich oder verschieden ist,
wobei:
n mindestens 4 beträgt;
a
und d unabhängig
aus der Gruppe ausgewählt
werden bestehend aus Met, Leu, Val, Ile und Thr;
e und g unabhängig aus
der Gruppe ausgewählt
werden, bestehend aus den Säure/Basenpaaren
Glu/Lys, Lys/Glu, Arg/Glu, Arg/Asp, Lys/Asp, Glu/Arg, Asp/Arg und
Asp/Lys; und
b, c und f unabhängig irgendwelche Aminosäuren, mit
Ausnahme von Gly oder Pro, sind und mindestens zwei Aminosäuren von
b, c und f in jedem Heptad aus der Gruppe ausgewählt sind, bestehend aus Glu, Lys,
Asp, Arg, His, Thr, Ser, Asn, Ala, Gln und Cys,
wobei das Nukleinsäurefragment
funktionsfähig
mit einer pflanzensamenspezifischen Regulationssequenz verknüpft ist.
-
Des
Weiteren ist hier ein Nukleinsäurefragment
beschrieben umfassend:
- (a) ein erstes chimäres Gen,
wobei ein Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die gegen Inhibition durch Lysin unempfindlich ist, auf
funktionsfähige
Weise mit einer Pflanzenchloroplasttransitsequenz und einer pflanzensamenspezifischen
Regulationssequenz verknüpft
ist und
- (b) ein zweites chimäres
Gen, wobei ein Nukleinsäurefragment,
das ein lysinreiches Protein kodiert, eine Nukleinsäuresequenz
umfasst, die ein Protein kodiert, das die Aminosäuresequenz (MEEKLKA)6(MEEKMKA)2 aufweist,
funktionsfähig
mit einer pflanzensamenspezifischen Regulationssequenz verknüpft ist.
-
Ebenfalls
hier beansprucht sind Pflanzen, die verschiedene Ausführungsformen
der beschriebenen ersten chimären
Gene und zweiten chimären
Gene enthalten und die beschriebenen Nukleinsäurefragmente und aus derartigen
Pflanzen erhaltenen Samen.
-
Die
Erfindung betrifft des Weiteren ein Nukleinsäurefragment umfassend
- (a) ein erstes chimäres Gen, wobei ein Nukleinsäurefragment,
das Dihydrodipicolinsäuresynthase
kodiert, die gegen Inhibition durch Lysin unempfindlich ist, auf
funktionsfähige
Weise mit einer Pflanzenchloroplasttransitsequenz und einer pflanzensamenspezifischen
Regulationssequenz verknüpft
ist; und
- (b) ein zweites chimäres
Gen, wobei ein Nukleinsäurefragment,
das eine Lysinketoglutaratreduktase kodiert, funktionsfähig in der
Sense- oder Antisenseorientierung mit einer pflanzensamenspezifischen
Regulationssequenz verknüpft
ist. Ebenfalls beansprucht ist eine Pflanze, die in ihrem Genom
dieses Nukleinsäurefragment
umfasst und ein von einer derartigen Pflanze erhaltener Samen.
-
KURZE BESCHREIBUNG
DER ZEICHNUNGEN UND BESCHREIBUNGEN DER SEQUENZEN
-
Die
Erfindung kann aus der folgenden genauen Beschreibung und den beiliegenden
Zeichnungen und den Sequenzbeschreibungen, die einen Teil dieser
Anmeldung bilden, noch vollständiger
verstanden werden.
-
1 zeigt
eine Alpha-Helix als Seiten- und Draufsichten.
-
2 zeigt die End- (2a)
und Seiten- (2b) Ansichten einer spiralenförmigen Alpha-Helixspiralstruktur.
-
3 zeigt die chemische Struktur von Leucin
und Methionin, wobei ihre ähnlichen
Gestalten hervorgehoben werden.
-
4 zeigt
eine schematische Darstellung einer samenspezifischen Genexpressionskassette.
-
5A zeigt eine Karte des binären Plasmidvektors pZSl99; 5B zeigt eine Karte des binären Plasmidvektors pFS926.
-
6A zeigt eine Karte des Plasmidvektors pBT603; 6B zeigt eine Karte des Plasmidvektors pBT614.
-
7 zeigt
die Strategie für
das Bilden eines Vektors (pSKS) zur Verwendung bei der Konstruktion und
Expression der SSP-Gensequenzen.
-
8 zeigt
die Strategie für
das Insertieren von Oligonukleotidsequenzen in die einzige Ear I-Stelle der Basengensequenz.
-
9 zeigt
die Insertion der Basengenoligonukleotide in die Nco I/EcoR I-Stellen
von pSKS zur Bildung des Plasmids pSK6. Diese Basengensequenz wurde
wie in 8 zum Insertieren der verschiedenen SSP-Kodierregionen
an der einzigen Ear I-Stelle zur Bildung der aufgelisteten geklonten
Segmente verwendet.
-
10 zeigt die Insertion der 63 bp-„Segment"-Oligonukleotide,
die zum Bilden sich nicht wiederholender Gelsequenzen zur Verwendung
im Duplikationsschema in 11 verwendet
werden.
-
11 (A und B) zeigt die Strategie für das Multiplizieren
sich nicht wiederholender Gen-„Segmente", bei denen in-frame-Fusionen
verwendet werden.
-
12 zeigt die Vektoren, die samenspezifische Promotor-
und 3'-Sequenzkassetten
enthalten. SSP-Sequenzen wurden in diese Vektoren unter Anwendung
der Nco I und Asp718-Stellen insertiert.
-
13 zeigt eine Karte des binären Plasmidvektors pZS97.
-
14 zeigt eine Karte des Plasmidvektos pML63.
-
15 zeigt eine Karte des Plasmidvektors pML102,
der ein chimäres
Gen trägt,
wobei samenspezifische Regulationsseguenzen (aus dem Sojabohnen-Kunitz-Trypsininhibitor
3-Gen) mit einer Chloroplasttransitsequenz (aus der kleinen Untereinheit
von Sojabohnenribulose-bisphosphatcarboxylase) und der Kodiersequenz
für gegen
Lysin unempfindliche Dihydrodipicolinsäuresynthase (das dapA-Gen von
Corynebacterium glutamicum) verknüpft sind.
- SEQ ID No:
1 und 2 wurden in Beispiel 1 als PKR-Primer für das Isolieren des Corynebacterium
DapA-Gens verwendet.
- SEQ ID No: 3 zeigt die Nukleotid- und Aminosäuresequenz der kodierenden
Region des Corynebacterium dapA-Gens vom Wildtyp, die gegen Lysin
unempfindliche DHDPS-Säure,
wie in Beispiel 1 beschrieben, kodiert.
- SEQ ID No: 4 zeigt in Oligonukleotid, das in Beispiel 2, zum
Bilden einer Nco I-Stelle am Translationsstartcodon des E. Coli
dapA-Gens verwendet wird.
- SEQ ID No: 5 zeigt die Nukleotid- und Aminosäuresequenz der kodierenden
Region des E. Coli lysC-Gens vom Wildtyp, die AKIII, wie in Beispiel
3 beschrieben kodiert.
- SEQ ID No: 6 und 7 wurden in Beispiel 3 zum Bilden einer Nco
I-Stelle am Translationsstartcodon des E. Coli lysC-Gens verwendet.
- SEQ ID No: 8, 9, 10 und 11 wurden in Beispiel 4 zum Bilden einer
Chloroplasttransitsequenz und der Verknüpfung der Sequenz mit den E.
coli lysC-M4-, E. Coli dapA- und Corynebacterium dapA-Genen verwendet.
- SEQ ID No: 12 und 13 wurden in Beispiel 4 zum Bilden einer Kpn
I-Stelle, sofort auf den vom Translationsstopcodon des E. coli dapA-Gens
hin verwendet.
- SEQ ID No: 14 und 15 wurden in Beispiel 4 als PKR-Primer zum
bilden einer Sojabohnenchloroplasttransitsequenz und Verknüpfung der
Sequenz mit dem Corynebacterium dapA-Gen verwendet.
- SEQ ID No: 16-92 stellen die Nukleinsäurefragmente und die Polypeptide
dar, die sie kodieren, die zum Bilden chimärer Gene für lysinreiche synthetische
Samenspeicherproteine verwendet werden, die für die Expression in den Samen
von Pflanzen geeignet sind.
- SEQ ID No: 93-98 wurden in Beispiel 12 zum Bilden einer Maischloroplasttransitsequenz
verwendet.
- SEQ ID No: 99 und 100 wurden in Beispiel 12 als PKR-Primer zum
Bilden einer Maischloroplasttransitsequenz und einer Verknüpfung der
Sequenz zum E. coli dapA-Gen verwendet.
-
Die
Sequenzbeschreibungen enthalten die Einbuchstabenkode für Nukleotidsequenzschriftzeichen und
die Dreibuchstabenkode für
Aminosäuren,
wie den IUPAC-IYUB-Standards entsprechend definiert, die in Nucleic
Acids Research 13:3021-303 (1985) und im Biochemical Journal 219
(Nr. 2): 345-373 (1984) beschrieben sind, die hier summarisch eingefügt werden.
-
GENAUE BESCHREIBUNG
DER ERFINDUNG
-
Die
unten aufgeführten
Lehren beschreiben Nukleinsäurefragmente
und Verfahrensweisen, die für
das Erhöhen
der Aufspeicherung von Lysin in den Samen tansformierter Pflanzen
im Vergleich mit Lysinniveaus in untransformierten Pflanzen nützlich sind.
Um die Aufspeicherung von freiem Lysin in den Samen von Pflanzen durch
genetisches Engineering zu erhöhen,
wurde eine Bestimmung durchgeführt,
welche Enzyme auf diesem Weg den Weg in den Samen von Pflanzen regulieren.
Um dies zu erreichen, wurden Gene, die Enzyme in dem Weg kodieren,
von Bakterien isoliert. Intrazelluläre Lokalisationssequenzen und
geeignete Regulationssequenzen für
die Expression in den Samen von Pflanzen wurden zur Bildung chimärer Gene
verknüpft.
Die chimären
Gene wurden dann durch Transformation in Pflanzen eingeführt und
auf ihre Fähigkeit
hin beurteilt, die Aufspeicherung von Lysin in Samen herbeizuführen. Die
Expression von gegen Lysin unempfindlicher Dihydrodipicolinsäuresynthase
(DHDPS) unter der Steuerung durch eine starken samenspezifischen
Promotor hat sich als dahingehend erwiesen, die Niveaus von freiem
Lysin in Mais-, Raps- und Sojabohnensamen 10- bis 100-fach zu erhöhen.
-
Es
ist entdeckt worden, dass das volle Potential zum Aufspeichern von überschüssigem freiem
Lysin in Samen durch Lysinkatabolismus reduziert wird. Es werden
hier zwei alternative Möglichkeiten
zum Verhindern des Verlusts von überschüssigem Lysin
aufgrund von Katabolismus geboten. Beim ersten Ansatz wird der Lysinkatabolismus
durch Reduktion der Aktivität
des Enzyms Lysinketoglutaratreduktase (LKR) verhindert, die den
ersten Schritt beim Lysinabbau katalysiert. Es wird eine Vorgehensweise
zum Isolieren von pflanzlichen LKR-Genen bereitgestellt. Es werden
chimäre
Gene für
die Expression von Antisense-LKR-RNA oder zum gleichzeitigen Unterdrücken von
LKR in den Samen von Pflanzen gebildet. Das chimäre Gen wird dann mit dem chimären DHDPS-Gen
verknüpft
und beide gleichzeitig durch Transformation in Pflanzen eingeführt, oder
die Gene werden durch Kreuzen von unabhängig transformierten Pflanzen
mit jedem der chimären
Gene zusammengebracht.
-
Beim
zweiten Ansatz wird überschüssiges freies
Lysin in einer Form eingearbeitet, die gegen Abbau unempfindlich
ist, z.B. durch Einarbeiten desselben in ein Di-, Tri- oder Oligopeptid,
oder ein lysinreiches Speicherprotein. Es wird das Design von Polypeptiden
bereitgestellt, die in vivo exprimiert werden können, um als lysinreiche Samenspeicherproteine
zu dienen. Gene, die lysinreiche synthetische Speicherprotein (SSP)
kodieren, werden synthetisiert und es werden chimäre Gene,
in denen die SSP-Gene mit geeigneten Regulationssequenzen zur Expression
in den Samen von Pflanzen verknüpft
sind, gebildet. Das chimäre
SSP-Gen wird dann mit dem chimäre
DHDPS-Gen verknüpft
und beide werden durch gleichzeitige Transformation in Pflanzen
eingeführt,
oder die Gene werden durch Kreuzen von unabhängig transformierten Pflanzen
mit jedem der chimären
Gene zusammengebracht.
-
Eine
Methode für
das Transformieren von Pflanzen, bevorzugt Mais, Raps- und Sojabohnenpflanzen, wird
hier gelehrt, wobei die dabei gebildeten Samen der Pflanzen mindestens
zehn Prozent, bevorzugt zehn Prozent bis 400 Prozent mehr Lysin
als die Samen von untransformierten Pflanzen enthalten. Es werden
hier als Beispiel transformierte Rapspflanzen mit im Vergleich mit
untransformierten Pflanzen um 100% erhöhten Samenlysinniveaus, Sojapflanzen
mit im Vergleich mit untransformierten Pflanzen um 400% erhöhten Samenlysinniveaus
und transformierte Maispflanzen mit im Vergleich mit untransformierten
Pflanzen um 130% erhöhten
Samenlysinniveaus bereitgestellt.
-
Im
Zusammenhang mit dieser Offenbarung werden eine Reihe von Begriffen
verwendet. Der Begriff „Nukleinsäure", wie der hier verwendet
wird, bezieht sich auf ein großes
Molekül,
das einstrangig oder doppelstrangig sein und aus Monomeren (Nukleotiden)
bestehen kann, die einen Zucker, Phosphat oder ein Purin oder Pyrimidin
enthalten. Ein „Nukleinsäurefragment" ist eine Fraktion
eines vorgegebenen Nukleinsäuremoleküls. In Pflanzen
höherer
Ordnung ist die Desoxyribonukleinsäure (DNA) das genetische Material,
während die
Ribonukleinsäure
(RNA) bei der Übertragung
der Information in den DNA in Proteine involviert. Ein „Genom" ist der gesamte
Körper
von genetischem Material, das in jeder Zelle eines Organismus enthalten
ist. Der Begriff „Nukleotidsequenz", bezieht sich auf
ein Polymer von DNA oder RNA, das ein- oder doppelstrangig sein kann
und gegebenenfalls synthetische, nichtnatürliche oder geänderte Nukleotidbasen
enthält,
die in der Lage sind, in DNA- oder RNA-Polymere eingearbeitet zu
werden.
-
„Gen" bezieht sind auf
ein Nukleinsäurefragment,
das ein spezifisches Protein exprimiert, das Regulationssequenzen
enthält,
die der Kodierregion vorhergehen, (5'-nichtkodierend) und auf diese folgen
(3'-nichtkodierend). „Natives" Gen bezieht sich
auf das Gen, wie es in der Natur mit seinen eigenen Regulationssequenzen
aufgefunden wird. „Chimäres" Gen bezieht sich
auf ein Gen, das heterogene Regulations- und Kodiersequenzen umfasst. „Endogenes" Gen bezieht sich
auf das native Gen, das normalerweise an seiner natürlichen
Position im Genom vorzufinden ist. Ein „fremdes" Gen bezieht sich auf ein Gen, das normalerweise im
Wirtsorganismus nicht aufzufinden ist, das jedoch durch Genübertragung
eingeführt
wird.
-
„Kodiersequenz" bezieht sich auf
eine DNA-Sequenz, die für
ein spezifisches Protein kodiert und die nichtkodierenden Sequenzen
ausschließt.
-
„Initiationscodon" und Terminationscodon" beziehen sich auf
eine Einheit von drei nebeneinanderliegenden Nukleotiden in einer
Kodiersequenz, die jeweils die Initialion und den Kettenabbruch
bei der Proteinsynthese (mRNA-Translation) spezifiziert. „Offenes
Leseraster" bezieht
sich auf die Aminosäuresequenz,
die zwischen Translationsinitiations- und Abbruchcodons einer Kodiersequenz
kodiert ist.
-
Geeignete „Regulationssequenzen", wie hier verwendet,
bezieht sich auf Nukleotidsequenzen, die stromaufwärts (5'), innerhalb und/oder
stromabwärts
(3') von einer Kodiersequenz
positioniert sind, die die Transkription und/oder Expression der
Kodiersequenzen reguliert, potentiell in Verbindung mit dem Proteinbiosyntheseapparat
der Zelle. Diese Regulationssequenzen umfassen Promotoren, Translations-Leader-Sequenzen,
Transkriptionsabbruchsequenzen und Polyadenylationssequenzen.
-
„Promotor" bezieht sich eine
auf DNA-Sequenz in einem Gen, die sich gewöhnlich stromaufwärts (5') ihrer Kodiersequenz
befindet, die die Expression der Kodiersequenz durch Liefern der
Erkennung bezüglich RNA-Polymerase
und anderer Faktoren, die für
eine richtige Transkription erforderlich sind, reguliert. Ein Promotor
kann auch DNA-Sequenzen enthalten, die beim Binden von Proteinfaktoren
involviert sind, die die Wirksamkeit der Transkriptionsinitiation
als Reaktion auf physiologische oder Entwicklungsbedingungen regulieren. Er
kann auch Enhancerelemente enthalten.
-
Ein „Enhancer" ist eine DNA-Sequenz,
die die Promotoraktivität
stimulieren kann. Es kann sich um ein inhärentes Element des Promotors
oder ein heterologes Element handeln, das zum Verbessern des Niveaus und/oder
der Gewebespezifizität
eines Promotors eingeführt
worden ist. „Konstitutive" Promotoren bezieht
sich auf diejenigen, die die Genexpression in allen Geweben und
zu allen Zeitpunkten dirigieren. „Organspezifische" oder „entwicklungsspezifische" Promotoren, wie
hier auf diese Bezug genommen wird, sind diejenigen, die die Genexpression
fast ausschließlich
in spezifischen Organen wie beispielsweise Blättern oder Samen, oder in spezifischen
Entwicklungsstufen in einem Organ, wie beispielsweise jeweils in
der frühen
oder späten Embryogenese,
dirigieren.
-
Der
Begriff „funktionsfähig verknüpft" bezieht sich auf
Nukleinsäuresequenzen
an einem einzigen Nukleinsäuremolekül, die so
assoziiert sind, dass die Funktion einer derselben durch die andere
beeinflusst wird. Beispielsweise wird ein Promotor funktionsfähig mit
einem Strukturgen verknüpft,
wenn er in der Lage ist, die Expression dieses Strukturgens zu beeinflussen
(d.h., dass das Strukturgen unter der transkriptionellen Kontrolle
des Promotors steht).
-
Der
Begriff „Expression", wie er hier verwendet
wird, soll die Erzeugung des Proteinprodukts, das durch ein Gen
kodiert ist, bedeuten. Noch spezifischer bezieht sich „Expression" auf die Transkription
und beständige
Ansammlung der Sense-(mRNA) oder der Antisense-RNA, das von dem
erfindungsgemäßen Nukleinsäurefragment
bzw. den erfindungsgemäßen Nukleinsäurefragmenten
deriviert ist, das bzw. die in Verbindung mit dem Proteinapparat
der Zelle zu geänderten
Niveaus des Proteinprodukts führt
bzw. führen. „Antisense-Inhibition" bezieht sich auf
die Erzeugung von Antisense-RNA-Transkripten, die in der Lage sind,
die Expression des Zielproteins zu verhindern. „Überexpression" bezieht sich auf
die Erzeugung eines Genprodukts in transgenen Organismen, die die
Niveaus der Erzeugung in normalen oder nichttransformierten Organismen übersteigt. „Kosuppression" bezieht sich auf
die Expression eines fremden Gens, das im Wesentlichen zum einem
endogenen Gen homolog ist, was zur Unterdrückung der Expression sowohl
des fremden als auch des endogenen Gens führt. „Geänderte Niveaus" bezieht sich auf
die Erzeugung von Genprodukt(en) in transgenen Organismen in Mengen
oder Anteilen, die von demjenigen normaler oder transformierter
Organismen verschieden sind.
-
Die „3'-nichtkodierenden
Sequenzen" bezieht
sich auf den Anteil der DNA-Sequenz eines Gens, der ein Polyadenylationssignal
und irgendein anderes Regulationssignal enthält, das in der Lage ist, die mRNA-Verarbeitung
oder Genexpression zu beeinflussen.
-
Das
Polyadenylationssignal ist gewöhnlich
dadurch gekennzeichnet, dass es die Addition von Polyadenylsäuretrakten
an das 3'-Ende des
mRNA-Vorläufers
beeinflusst.
-
„Translationsleadersequenz" bezieht sich auf
denjenigen Anteil einer DNA-Sequenz eines Gens, der zwischen dem
Promotor und der kodierenden Sequenz liegt, die in die RNA transkribiert
ist und im vollständig verarbeiteten
mRNA stromaufwärts
(5') des Translationsstartcodons
vorliegt. die Translationsleadersequenz kann das Verarbeiten des
primären
Transkripts zu mRNA, die mRNA-Stabilität oder -translationseffizienz
beeinflussen.
-
„Reifes" Protein bezieht
sich auf ein posttranslationell verarbeitetes Polypeptid ohne dessen
Targetingsignal. „Vorläufer"-Protein bezieht
sich auf das primäre
Produkt der Translation von mRNA. Ein „Chloroplasttargetingsignal" ist eine Aminosäuresequenz,
die in Verbindung mit einem Protein translatiert wird und dieses dem
Chloroplast zuleitet. „Chloroplasttransitsequenz" bezieht sich auf
eine Nukleotidsequenz, die ein Chloroplasttargetingsignal kodiert.
-
„Transformation" bezieht sich hier
auf die Übertragung
eines fremden Gens in das Genom eines Wirtsorganismus und dessen
genetisch beständiges
Erbtum. Beispiele von Pflanzentransformationsmethoden umfassen durch
Agrobacterium vermittelte Transformation und teilchenbeschleunigte
oder „Genpistolen"-Transformationstechnologie.
-
„Aminosäuren" bezieht sich hier
auf die natürlich
vorkommenden L-Aminosäuren
(Alanin, Arginin, Aspartinsäure,
Asparagin, Cystin, Glutamsäure,
Glutamin, Glycin, Histidin, Isoleucin, Leucin, Lysin, Methionin, Prolin,
Phenylalanin, Serin, Threonin, Tryptophan und Valin). „Essentielle
Aminosäuren" sind diejenigen
Aminosäuren,
die durch Tiere nicht synthetisiert werden können. Ein „Polypeptid" oder „Protein", wie es hier verwendet
wird, bezieht sich auf ein Molekül,
das aus Monomeren (Aminosäuren)
besteht, die linear durch Amidbindungen (auch als Peptidbindungen
bekannt) verknüpft
sind.
-
„Synthetisches
Protein" bezieht
sich hier auf ein Protein, das aus Aminosäurensequenzen besteht, von denen
nicht bekannt ist, dass sie in der Natur vorkommen. Die Aminosäuresequenz
kann von einem Konsensus natürlich
vorkommender Proteine deriviert sein oder sie kann vollständig neuartig
sein.
-
„Primäre Sequenz" bezieht sich auf
die Konnektivitätsordnung
von Aminosäuren
in einer Polypeptidkette ohne Bezugnahme auf die Konformation des
Moleküls.
Primäre
Sequenzen werden üblicherweise
vom Aminoende zum Carboxyende der Polypeptidkette hin geschrieben.
-
„Sekundäre Struktur" bezieht sich hier
auf physikalisch-chemisch bevorzugte regelmäßige Rückgradkettenanordnungen einer
Polypeptidkette ohne Bezugnahme auf Variationen der Seitenkettenidentitäten oder -konformationen. „Alpha-Helices", wie es hier verwendet
wird, bezieht sich auf rechtsgedrehte mit ca. 3,6 Resten pro Helixdrehung.
Eine „amphiphatische
Helix" bezieht sich
hier auf ein Polypeptid in einer Helixkonformation, wobei eine Seite
der Helix überwiegend
hydrophob und die andere Seite überwiegend
hydrophil ist.
-
„Spiralenförmige Spirale" bezieht sich hier
auf eine Ansammlung von zwei parallelen rechts gerichteten Alpha-Helices,
die umeinander herumgewunden sind unter Bildung einer linksgerichteten
Superhelix.
-
„Salzbrücken" wie hier besprochen,
beziehen sich auf Säure-Basepaare
von beladenen Aminosäureseitenketten,
die räumlich
so angeordnet sind, dass eine elektrostatische Anziehungswechselwirkung
zwischen zwei Teilen einer Polypeptidkette oder zwischen einer Kette
und anderen aufrechterhalten wird.
-
„Wirtszelle" bedeutet, dass die
Zelle mit dem eingeführten
genetischen Material transformiert wird.
-
ISOLIERUNG
VON DHDPS-GENEN
-
Das
E. coli dapA-Gen (ecodapA) wurde als bacteriophager Lambda-Klon
aus einer geordneten Bibliothek von 3400 sich überlappenden Segmenten von
E. coli-DNA erhalten, die durch Kohara, Akiyame und Isono [Kohara
et al. (1987) Cell 50:495-508] konstruiert wurde. Einzelheiten der
Isolierung und Modifizierung von eoodapA sind in Beispiel 1 aufgeführt. Das
ecodapA-Gen kodiert ein DHDPS-Enzym das der Inhibition durch Lysin
gegenüber
mindestens 20 mal weniger empfindlich ist als ein typisches Pflanzenenzym
z.B. Weizen-DHDPS. Zum Zweck der vorliegenden Erfindung wird der
Ausdruck „Inhibition
durch Lysin 20 mal weniger empfindlich" als gegen Lysin unempfindlich bezeichnet.
-
Das
Corynebacterium dapA-Gen (cordapA) wurde von genomer DNA aus dem
ATCC-Stamm 13032 unter Anwendung der Polymerasekettenreaktion (PKR)
isoliert. Die Nukleotidsequenz des Corynebacterium dapA-Gens ist
veröffentlicht
worden [Bonnassie et al. (1990) Nucleic Acids Res. 18:6421]. Aus
dieser Sequenz war es möglich,
Oligonukleotidprimer für
die Polymerasekettenreaktion (PKR) zu konstruieren, die die Amplifikation
eines das Gen enthaltenden DNA-Fragments erlauben und gleichzeitig
einzige Restriktionsendonukleasestellen am Startcodon und gerade
am Startcodon des Gens vorbei zufügen würden, um weitere, das Gen involvierenden
Konstruktionen zu erleichtern. Die Einzellheiten des Isolierens
des Corynebacterium dapA (cordapA) Gens sind in Beispiel 1 aufgeführt. Das
cordapA-Gen kodiert ein bevorzugtes gegen Lysin unempfindliches
DHDPS-Enzym, das durch das Vorliegen von 70 mM Lysin in der Enzymreaktionsmischung
nicht beeinflusst wird.
-
Die
Isolierung anderer DHDPS kodierender Gene ist in der Literatur beschrieben
worden. Eine DHDPS aus Weizen kodierende cDNA [Kaneko et al. (1990)
J. Biol. Chem. 265:17451-17455] und eine DHDPS aus Mais kodierende
cDNA [Frisch et al. (1991) Mol. Gen. Genet. 228:287-293] sind Beispiele
pflanzlicher DHDPS-Gene, die isoliert und sequenziert worden sind.
Die Pflanzengene kodieren gegen Lysin unempfindliche DHDPS-Enzyme
vom Wildtyp. Jedoch erhielten Negrutui et al. [(1984) Theor. Appl.
Genet. 68:11-20] zwei AEC-resistente Tabakmutanten, bei denen die
DHDPS-Aktivität
gegen Inhibition durch Lysin weniger empfindlich war als das Enzym
vom Wildtyp. Das zeigt, dass diese Tabakmutanten DHDPS-Gene enthalten, die
gegen Lysin widerstandsfähiges
Enzym kodierten. Diese Gene könnten
ohne Weiteres aus Tabakmutanten unter Anwendung der für das Isolieren
der Weizen- oder Maisgene beschriebenen Methode entsprechend oder alternativ
durch Anwendung der Weizen- oder Maisgene als heterologe Hybridisierungssonden
isoliert werden.
-
Noch
andere Gene, die DHDPS kodieren, können durch Anwendung entweder
des E. coli dapA-Gens des
cordapA-Gens oder eines der pflanzlichen DHDPS-Gene als DNA-Hybridisierungssonden
isoliert werden. Als Alternative könnten andere Gene, die DHDPS
kodieren, durch funktionelles Ergänzen eines E. coli dapA-Mutanten
isoliert werden, wie zum Isolieren des cordapA-Gens [Yeh et al.
(1988) Mol. Gen. Genet 212:105-111] und des Mais-DHDPS-Gens durchgeführt wurde.
-
KONSTRUKTION
CHIMÄRER
GENE FÜR
DIE EXPRESSION EINER dapA-KODIERREGION IN PFLANZEN
-
Die
Expression fremder Gene in Pflanzen ist gut eingeführt [De
Blaere et al. (1987) Meth. Enzymol. 143:277-291]. Das richtige Expressionsniveau
von dapA mRNA kann unter Umständen
die Verwendung verschiedener chimärer Gene, bei denen verschiedene
Promotoren verwendet werden, erforderlich machen. Derartige chimäre Gene
können
in Wirtspflanzen entweder zusammen in einem einzigen Expressionsvektor
oder sequentiell unter Anwendung von mehr als einem Vektors übertragen
werden. Eine bevorzugte Klasse heterologer Wirte für die Expression
der Kodiersequenz der dapA-Gene sind eukaryontische Wirte, insbesondere die
Zellen von Pflanzen höherer
Ordnung. Besonders bevorzugt unter den Pflanzen höherer Ordnung
und den von diesen derivierten Samen sind Rapssamen (Brassica napus,
B. campestris) und Sojabohne (Glycine max).
-
Der
Ursprung des Promotors, der zum Antreiben der Expression der Kodiersequenz
ausgewählt
wird, ist nicht kritisch, so lange er eine ausreichende Transkriptionsaktivität zur Erreichung
der Erfindung durch Exprimieren translatierbarer mRNA für dapA-Gene
in dem erwünschten
Wirtsgewebe aufweist. Bevorzugte Promotoren sind diejenigen, die
die Expression des Proteins spezifisch in Samen erlauben. Dies kann
besonders nützlich
sein, das Samen die Hauptquelle von pflanzlichen Aminosäuren sind
und auch da die samenspezifische Expression irgendwelche unerwünschte Auswirkung
in Nichtsamenorganen vermeidet. Beispiele samenspezifischer Promotoren
umfassen, sind jedoch nicht darauf beschränkt, die Promotoren von Samenspeicherproteinen.
Die Samenspeicherproteine werden streng reguliert, indem sie fast
ausschließlich
in Samen auf hoch organspezifische und stufenspezifische Weise exprimiert
werden [Higgins et al. (1984) Ann. Rev. Plant Physiol. 35:191-221;
Goldberg et al. (1989) Cell 56:149-160; Thompson et al. (1989).
Bio Essays 10:108-113]. Außerdem
können
verschiedene Samenspeicherproteine in verschiedenen Stufen der Samenentwicklung
exprimiert werden.
-
Es
gibt im Augenblick zahlreiche Beispiele der samenspezifischen Expression
von Samenspeicherproteingenen in transgenen zweikeimblättrigen
Pflanzen. Diese umfassen Gene aus zweikeimblättrigen Pflanzen für Bohnen-β-Phaseolin
[Sengupta-Gopalan et al. (1985) proc. Natl. Acad. Sci. USA 82:3320-3324;
Hoffman et al. (1988) Plant Mol. Biol 11:717-729], Bohnenlectin
[Voellcer et al. (1987), EMBO J. 6:3571-3577], Sojabohnenlectin
[Okamwo et al. (1986) Proc. Natl. Acad. Sci. USA 83:8240-8244],
Sojabohnen-Kunitz-Trypsininhibitor [Perez-Grau et al. (1989) Plant
Cell 1:095-1109], Sojabohnen-β-Conglycinin
[Beachy et al. (1985) EMBO J. 4:3047-3053; Barker et al. (1988)
Proc. Natl. Acad. Sci. USA 85:458-462; Chen et al. (1988) EMBO J.
7:297-302; Chen et al. (1989) Dev. Genet. 10:112-122; Naito et al
(1988) Plant Mol. Biol. 11:109-123], Erbsenvicilin [Higgins et al.
(1988) Plant Mol. Biol. 11:683-695], Erbsenconvicilin [Newbigin
et al. (1990) Planta 180:461] Erbsenlegumin [Shirsat et al. (1989)
Mol. Gen. Genetics 215:326], Raspssamennapin [Radke et al. (1988)
Theor. Appl. Genet. 75:685-694] sowie Gene aus einkeimblättrigen
Pflanzen wie beispielsweise bei Maiszein von 15 kD [Hoffman at al.
(1987) EMBO J. 6:3213-3221; Schernthaner et al. (1988) EMBO J. 7:1249-1253;
Williamson et al. (1988) Plant Physiol. 88:1002-1007, Gersten-β-Hordein
[Marris et al. (1988) Plant Mol. Biol 10:359-366] und Weizenglutenin
[Colot et al. (1987) EMBO J. 6:3559-3564]. Außerdem behalten Promotoren
samenspezifischer Gene, die funktionsfähig mit heterologen Kodiersequenzen
in chimären Genkonstrukten
verknüpft
sind, ebenfalls in ihr zeitliches und räumliches Expressionsmuster
in transgenen Pflanzen bei. Derartige Beispiele umfassen Arabidopsis
thaliana 2S-Samenspeicherproteingenpromotor zum Exprimieren von
Enkephalinpeptiden in Arabidopsis- und B. napus-Samen [Vandekerckhove
et al. (1989) Bio/Technology 7:929-932], Bohnenlectin- und Bohnen-β-Phaseolinpromotoren
zum Exprimieren von Luciferase [Riggs et al (1989) Plant Sci. 63:47-57]
und Weizengluteninpromotoren zum Exprimieren von Chloramphenicolacetyltransferase
[Colot et al. (1987) EMBO J.6:3559-3564].
-
Von
besonderem Nutzen beim Exprimieren des erfindungsgemäßen Nukleinsäurefragments
sind die Promotoren verschiedener umfassend gekennzeichneter Samenspeicherproteingene,
wie beispielsweise derjenigen für
Bohnen-β-Phaseolin
[Sengupta-Goplalan et al. (1985) Proc. Natl. Acad. Sci. USA 82:3320-3324; Hoffman et
al. (1988) Plant Mol. Biol. 11:717-729], Sojabohnen-Kunitz-Trypsininhibitor
[Jofuku et al. (1989) Plant Cell 1:1079-1093; Perez-Grau et al.
(1989) Plant Cell 1:1095-1109], Sojabohnen-β-Conglycinin [Harada et al. (1989) Plant
Cell 1:415-425] und Rapssamennapin [Radke et al. (1988) Theor. Appl.
Genet. 75:685-694]. Promotoren von Genen für β-Phaseolin- und Sojabohnen-β-Conglycininspeicherprotein
werden beim Exprimieren der dapA-mRNA in den Keimblättern in
den mittleren bis späten
Stufen der Samenentwicklung besonders nützlich sein.
-
Ebenfalls
von besonderem Nutzen beim Exprimieren der erfindungsgemäßen Nukleinsäurefragmente werden
die heterologen Promotoren aus verschiedenen umfassend gekennzeichneten
Maissamenspeicherproteingenen, wie beispielsweise endospermspezifische
Promotoren aus dem 10 kD-Zein [Kirihara et al. (1988) Gene 71:359-370],
dem 27 kD-Zein [Prat et al. (1987) Gene 52:51-49; Gallardo et al.
(1988) Plant Sci. 54:211-281; Reina et al. (1990) Nukleic Acids
Res. 18:6426-6426]
und dem 19 kD-Zein [Marks et al. (1985) J. Biol. Chem. 260:16451-16459]
sein. Von den relativen transkriptionellen Aktivitäten dieser
Promotoren in Mais ist schon berichtet worden [Kodrzyek et al. (1989)
Plant Cell 1:105-114], die die Basis für das Auswählen eines Promotors zur Verwendung
in chimären
Genkonstrukten für
Mais bieten. Zum Exprimieren in Maisembryos kann der stark embryospezifische
Promotor aus dem Globulin 1-(GLB1) Gen [Kriz (1989) Biochemical
Genetics 27:239-251, Wallace et al. (1991) Plant Physiol. 95:973-975]
verwendet werden.
-
Es
ist geplant, dass das Einführen
Enhancern oder enhancerähnlichen
Elementen in andere Promotorenkonstrukte auch erhöhte Niveaus
an primärer
Transkription für
dapA-Gene zum Erreichen der Erfindung bereitstellen wird. Diese
würden
Virusenhancer, wie beispielsweise derjenige, der in dem 355-Promotor aufzufinden
ist [Odell et al. (1988) Plant Mol. Biol. 10:263-272], Enhancer
aus den opinen Genen [Fromm et al (1989) Plant Cell 1:977-984] oder
Enhancer aus irgendeiner anderen Quelle, die zu einer erhöhten Transkription
führen,
wenn sie in einen Promotor positioniert werden, der funktionsfähig mit
dem erfindungsgemäßen Nukleinsäurefragment
verknüpft
ist.
-
Von
besonderer Wichtigkeit ist das DNA-Sequenzelement, das aus dem Gen
für die α'-Untereinheit von β-Conglycinin isoliert worden
ist, das eine 40-fache samenspezifische Verbesserung bei einem konstitutiven
Promotor hervorrufen kann [Chen et al. (1988) EMBO J. 7:297-302;
Chen et al (1989) Dev. Genet. 10:112-122]. Ein Fachmann kann dieses
Element ohne weiteres isolieren und es in die Promotorregion irgendeines
Gens insertieren, um eine samenspezifisch verbesserte Expression
mit dem Promotor in transgenen Pflanzen zu erhalten. Die Insertion
eines derartigen Elements in irgendein samenspezifisches Gen, das zu
verschiedenen Zeiten als das β-Conglyciningen
exprimiert wird, führt
zum Exprimieren in transgenen Pflanzen für eine längere Zeitspanne während der
Samenentwicklung.
-
Irgendeine
nichtkodierende 3'-Region,
die in der Lage ist, ein Polyadenylationssignal bereitzustellen, und
andere Regulationssequenzen, die eventuell für die richtige Expression der
dapA-Kodierregionen
erforderlich sind, können
zum Erzielen der Erfindung verwendet werden. Dies würde das
3'-Ende von irgendeinem Speicherprotein,
wie beispielsweise das 3'-Ende
des Bohnenphaseolingens, das 3'-Ende
des Sojabohnen-β-Conglyciningens,
das 3'-Ende viraler
Gene, wie beispielsweise das 3'-Ende 35S-
oder des 19S-Blumenkohlmosaikvirustranskripts, das 3'-Ende der opinen
Synthesegene, die 3'-Enden
von Ribulose-1,5-bisphosphatcarboxylase oder Chlorophyll a-/b-Bindungsprotein
oder 3'-Endesequenzen
von irgendeiner Quelle, derart einschließen, dass die angewendete Sequenz
die nötige
Regulationsinformation innerhalb ihrer Nukleinsäuresequenz liefert, um zur
richtigen Expression der Kombination von Promotor/Kodierregion zu
führen,
mit der sie funktionsfähig
verknüpft
ist. Es gibt zahlreiche Beispiele im Stand der Technik, die die
Nützlichkeit
verschiedener nichtkodierender 3'-Regionen lehren [man
vergleiche beispielsweise Ingelbrecht et al. (1989) Plant Cell 1:671-680].
-
DNA-Sequenzen,
die für
intrazelluläre
Lokalisationsequenzen kodieren, können der dapA-Kodiersequenz zugegeben
werden, falls dies für
die richtige Exprimierung der Proteine zum Erreichen der Erfindung notwendig
ist. Pflanzliche, biosynthetische Aminosäureenzyme sind dafür bekannt,
dass sie in Chloroplasten lokalisiert sind und deshalb mit einem
Chloroplasttargetingsignal synthetisiert werden. Bakterienproteine
wie beispielsweise Corynebactrium DHDPS besitzen kein derartiges
Signal. Eine Chloroplasttansitsequenz könnte aus diesem Grund an die
dapA-Kodiersequenz fusioniert werden. Bevorzugte Chloroplasttransitsequenzen sind
diejenigen der kleinen Untereinheit von Ribulose-1,5-bisphosphatcarboxylat,
z.B. aus Sojabohnen [Berry-Lowe et al. (1982) J. Mol. Appl. Genet.
1:483-498] zur Verwendung in zweikeimblättrigen Pflanzen und aus Mais
[Lebrun et al (1987) Nukleic Acids. Res. 15:4360] zur Verwendung
in einkeimblättrigen
Pflanzen.
-
EINFÜHRUNG CHIMÄRER dapA-GENE
IN PFLANZEN
-
Verschiedene
Methoden zum Einführen
einer DNA-Sequenz (d.h. zum Transformieren) in eukaryontischene
Zellen von Pflanzen höherer
Ordnung stehen zur Verfügung
(man vergleiche die EPA-Veröffentlichungen
0 295 959 A2 und 0 138 341 A1. Derartige Methoden umfassen diejenigen
auf der Basis von Transformationsvektoren auf der Basis von Ti-
und Ri-Plasmiden von Agrobacterium spp. Es wird besonders bevorzugt, den
binären
Typ dieser Vektoren zu verwenden. Von Ti derivierte Vektoren transformieren
eine umfangreiche Reihe verschiedener Pflanzen höherer Ordnung, einschließlich einkeimblättrige und
zweikeimblättrige
Pflanzen, wie beispielsweise Sojabohnen, Baumwolle und Raps [Pacciotti
et al. (1985) Bio/Technology 3:241; Byrne et al. (1987) Plant Cell,
Tissue and Organ Culture 8:3; Sukhapinda et al. (1987) Plant Mol.
Biol. 8:209-216; Lorz et al. (1985) Mol. Gen. Genet. 199:178; Potrykus
(1985) Mol. Gen. Genet. 199:183]
-
Zum
Einführen
in Pflanzen können
die erfindungsgemäßen chimären Gene
in binäre
Vektoren, wie sie in den Beispielen 6-12 beschrieben sind, insertiert
werden. Die Vektoren stellen einen Teil eines binären Ti-Plasmidvektorsystems
[Bevan, (1984) Nucl. Acids. Res. 12:8711-8720] von Agrobacterium
tumefaciens dar.
-
Andere
Transformationsmethoden stehen den mit dem Stand der Technik vertrauten
Fachleuten zur Verfügung,
wie beispielsweise Direkt-Uptake von fremden DNA-Konstrukten [man
vergleiche EPA-Veröffentlichung
0 295 959 A2] Elektroporationstechniken [man vergleiche Fromm et
al. (1986) Nature (London) 319:791] oder ballistisches Hochgeschwindigkeitsbombardieren
mit Metallteilchen, die mit den Nukleinsäurekonstrukten beschichtet
sind [man vergleiche Kline et al. (1987) Nature (London) 327:70
und vergleiche die US-Patentschrift Nr. 4.945.050]. Nach dem Transformieren
können
die Zellen durch mit dem Stand der Technik vertraute Fachleute regeneriert
werden.
-
Von
besonderer Relevanz sind vor kurzem beschriebene Methoden zum Transformieren
fremder Gene in handelsmäßig wichtige
Anbaupflanzen wie Rapssamen [man vergleiche De Block et al. (1989)
Plant Physiol. 91:694-701], Sonnenblumen [Everett et al. (1987)
Bio/Technology 5:1201], Sojabohnen [McCabe et al. (1988) Bio/Technology
6:923; Hinchee et al. (1988) Bio/Technology 6:915; Chee et al. (1989)
Plant Physiol. 91:1212-1218; Christou et al. (1989) Proc. Natl.
Acad. Sci USA 86:7500-7504; EPA-Veröffentlichung
0 301 749 A2] und Mais [Gordon-Kamm et al. (1990) Plant Cell 2:603-618;
Fromm et al. (1990) Biotechnology 8:833-839].
-
Zum
Einführen
in Pflanzen durch ballistisches Hochgeschwindigkeitsbombardieren
können
die erfindungsgemäßen chimären Gene
in geeignete Vektoren, wie in Beispiel 6 beschrieben, insertiert
werden.
-
EXPRIMIERUNG VON CHIMÄREN dapA-GENEN
IN RAPS-, SOJABOHNEN UND MAISPFLANZEN
-
Zum
Analysieren bezüglich
der Expression des chimären
dapA-Gens in Samen und bezüglich
der Folgen der Expression des Aminosäuregehalts in den Samen kann
ein Samenmehl wie in den Beispielen 5 oder 6 beschrieben oder durch
irgendeine andere Methode zubereitet werden. Das Samenmehl kann,
falls erwünscht,
beispielsweise durch Hexanextraktion teilweise oder vollständig entfettet
werden. Proteinextrakte können
aus dem Mehl zubereitet und bezüglich
ihrer DHDPS-Enzymaktivität
analysiert werden. Als Alternative ist es möglich, immunologisch auf das
Vorliegen des DHDPS-Proteins hin durch den mit dem Stand der Technik
vertrauten Fachleuten allgemein bekannte Methoden geprüft werden.
Fast alle Transformanten exprimierten das fremde DHDPS-Protein (man
vergleiche Beispiele 5, 6 und 13). Zum Messen der Zusammensetzung der
freien Aminosäure
der Samen, können
freie Aminosäuren
von dem Mehl extrahiert und durch den mit dem Stand der Technik
vertrauten Fachleuten bekannte Methoden analysiert werden (man vergleiche
Beispiele 5 und 6 bezüglich
geeigneter Vorgehensweisen).
-
Rapssamentransformanten,
die DHDPS-Protein exprimieren, wiesen eine mehr als 100-fache Erhöhung des
Niveaus an freiem Lysin in ihren Samen auf. Es bestand eine gute
Korrelation zwischen Transformanten, die höhere Niveaus an DHDPS-Protein
exprimieren, und denjenigen, die höhere Niveaus an freiem Lysin
aufweisen. Unter den Transformanten fand keine größere Ansammlung
von freiem Lysin aufgrund der Expression eines gegen Lysin unempfindlichen
AK-Enzyms zusammen mit einer gegen Lysin unempfindlichen DHDPS,
im Vergleich mit der Expression einer gegen Lysin unempfindlichen
DHDPS als solcher statt. So ist die Exprimierung einer gegen Lysin
unempfindlichen DHDPS in Samen im Falle von Rapssamen notwendig und
ausreichend, um eine starke Erhöhung
des freien Lysins zu verursachen. Ein hohes Niveau an α-Aminoadipinsäure, das
einen Lysinkatabolismus anzeigt, wurde in allen den transformierten
Linien bei erhöhten
Niveaus an freiem Lysin beobachtet.
-
Um
die gesamte Aminosäurezusammensetzung
von reifem Rapssamensaatgut zu messen, wurde entfettetes Mehl wie
in Beispiel 5 beschrieben analysiert. Relative Aminosäureniveaus
in den Samen wurden als Prozentsätze
von Lysin auf die gesamten Aminosäuren bezogen, verglichen. Die
höchsten
Expressionslinien zeigten eine fast zweifache Erhöhung des
Lysinniveaus in den Samen, so dass Lysin ca. 12% der gesamten in
den Samen enthaltenen Aminosäuren
ausmacht.
-
Einundzwanzig
von dreiundzwanzig Sojabohnentransformanten exprimierten das DHDPS-Protein. Die
Analyse einzelner Samen dieser Transformanten zeigten eine ausgezeichnete
Korrelation zwischen der Expression des GUS-Transformationsmarkergens
und DHDPS in einzelnen Samen. Aus diesem Grund sind die GUS- und
DHDPS-Gene an der gleichen Stelle im Sojabohnengenom integriert.
-
Es
bestand eine ausgezeichnete Korrelation zwischen Transformanten,
die das Corynebacteria DHDPS-Protein exprimieren und denjenigen,
die höhere
Niveaus an freiem Lysin aufweisen. 20-fache bis 120-fache Erhöhungen des
freien Lysinniveaus wurden bei Samen beobachtet, die Corynebacteria
DHDPS exprimieren.
-
Die
Analysen der Niveaus an freiem Lysin in einzelnen Samen von Transformanten,
in denen die Transgene sich an einer einzigen Stelle absonderten,
zeigten, dass die Erhöhung
des Niveaus an freiem Lysin in etwa einem Viertel der Samen signifikant
stärker
war. Da zu erwarten ist, dass ein Viertel der Samen für das Transgen
homozygot sind, ist es wahrscheinlich, dass die Samen mit höherem Lysingehalt
die Homozygoten sind. Des Weiteren weist dies darauf hin, dass das
Erhöhungsniveau
an freiem Lysin von der Kopiezahl des DHDPS-Gens abhängt. Aus
diesem Grund könnten
Lysinniveaus durch Herstellen von Hybriden von zwei verschiedenen
Transformanten noch weiter erhöht
werden und es könnten
Abkömmlinge
erhalten werden, die an beiden Transgen-Loci homozygot sind, wodurch
die Kopiezahl des DHDPS-Gens von zwei auf vier erhöht wird.
-
Ein
hohes Niveaus an Saccharopin, das auf den Lysinkatabolismus hindeutet,
wurde in Samen beobachtet, die hohe Niveaus an Lysin enthielten.
So sollte die Verhinderung des Lysinkatabolismus durch die Inaktivierung
von Lysinketoglutaratreduktase die Ansammlung von freiem Lysin den
Samen noch weiter erhöhen.
Als Alternative würde
das Einarbeiten von Lysin in ein Peptid oder lysinreiches Protein
den Katabolismus verhindern und zu einer Erhöhung der Ansammlung von Lysin
in den Samen führen.
-
Die
gesamten Lysinniveaus waren in Samen, die das Corynebacteria DHDPS-Protein
exprimieren, signifikant erhöht.
Es wurden Samen mit einer 10-260%-igen Erhöhung des Lysinniveaus im Vergleich
mit untransformierter Kontrolle beobachtet. Die Expression von DHDPS
zusammen mit einem gegen Lysin unempfindlichem Aspartokinaseenzym
führte
zu einer Erhöhung
von Lysin von mehr als 400%. So enthalten diese Samen viel mehr
Lysin als irgendein früherer
Sojabohnensamen.
-
Die
Expression des Corynebacterium DHDPS-Proteins, die entweder durch
den Maisglobulin-1-Promotor
zur Expression im Embryo oder den Maisglutelin 2-Promotor zur Expression
in Endosperm angetrieben wird, wurde in den Maissamen beobachtet.
Die Niveaus an freiem Lysin in den Samen stiegen von ca. 1,4% freier
Aminosäure
in Kontrollsamen auf 15-27% freier Aminosäure in Samen, die Corynebacterium
DHDPS aus dem Globulin 1-Promotor exprimieren. Eine geringere Erhöhung des
freien Lysins wurde in Samen beobachtet, die Corynebacterium DHDPS
aus dem Glutelin 2-Promotor exprimieren. So ist es zum Erhöhen von Lysin
eventuell besser, dieses Enzym im Embryo anstatt dem Endosperm zu
exprimieren. Ein hohes Niveau an Saccharopin, das auf den Lysinkatabolismus
hindeutet, wurde in Samen beobachtet, die hohe Niveaus an Lysin
enthielten. Die erhöhte
Ansammlung von freiem Lysin in Samen, die Corynebacterium DHDPS
aus dem Globulin 1-Promotor exprimieren, reichte aus, um zu einer
wesentlichen Erhöhung
(35%-130%) des gesamten Lysingehalts der Samen zu führen.
-
ISOLIERUNG
EINES PFLANZENLYSINKETOGLUTARAT-REDUKTASEGENS
-
Zum
Akkumulieren höherer
Niveaus an freiem Lysin, kann es wünschenswert sein, dem Lysinkatabolismus
zu verhindern. Beweise zeigen an, dass Lysin in Pflanzen durch den
Saccharopinweg katabolisiert wird. Der erste enzymatische Beweis
des Vorliegens dieses Wegs bestand aus der Erfassung der Aktivität der Lysinketoglutaratreduktase
(LKR) im unreifen Endosperm sich entwickelnder Maissamen [Aruda
et al. (1982) Plant Physiol. 69:988-989]. LKR katalysiert den ersten
Schritt des Lysinkatabolismus, nämlich
die Kondensation von L-Lysin mit einem α-Ketoglutarat zu Saccharopin
unter Anwendung von NADPH als Co-Faktor. Die LKR-Aktivität erhöht sich
stark vom Einsetzen der Endospermentwicklung in Mais an, erreicht
einen Höhepunkt
ca. 2 Tage nach dem Bestäuben
und nimmt dann ab [Arruda et al. (1983) Phytochemicals 22:2687-2689].
Um den Katabolismus von Lysin zu verhindern, wäre es wünschenswert, die LKR-Expression oder
-aktivität
zu reduzieren oder eliminieren. Dies ließe sich durch Klonieren der
LKR, Zubereiten eines chimären
Gens zum gleichzeitigen Unterdrücken
von LKR oder Zubereiten eines chimären Gens zum Exprimieren von
Antisense-RNA für
LKR und Einführen
des chimären
Gens in Pflanzen durch Transformation erreichen.
-
Mehrere
Methoden zum Klonieren eines pflanzlichen LKR-Gens stehen den mit
dem Stand der Technik vertrauten Fachleuten zur Verfügung. Das
Protein kann aus Maisendosperm, wie bei Brochetto-Braga et al. [(1992)
Plant Physiol. 98:1139-1147 beschrieben] gereinigt und zum Züchten von
Antikörpern
verwendet werden. Die Antikörper
können
dann zum Screenen einer cDNA-Expressionsbibliothek für LKR-Klone
verwendet werden. Als Alternative kann das gereinigt Protein zum
Bestimmen der Aminosäuresequenz
am Aminoende des Proteins oder aus Protease derivierten internen
Peptidfragmenten verwendet werden. Es können entartete Oligonukleotidsonden
auf der Basis der Aminosäuresequenz
zubereitet und zum Screenen einer pflanzlichen cDNA- oder genomischen
DNA-Bibliothek durch Hybridisierung verwendet werden. Bei einer
anderen Methode wird ein E. coli-Stamm verwendet, der nicht in der
Lage ist, in einem synthetischen Medium, das 20 μg/ml L-Lysin enthält, zu wachsen.
Die Expression von LKR-cDCNA voller Länge in diesem Stamm macht die Wachstumsinhibition
durch Reduzieren der Lysinkonzentration rückgängig. Die Konstruktion eines
geeigneten E. coli-Stamms und dessen Verwendung zum Auswählen von
Klonen aus einer eine pflanzliche cDNA-Bibliothek, die zu gegen
Lysin widerstandsfähigem
Wachstum führt,
ist in Beispiel 7 beschrieben.
-
Um
die Expression des LKR-Gens in transformierten Pflanzen zu blockieren,
kann ein chimäres
Gen, das für
die gleichzeitige Unterdrückung
von LKR konstruiert ist, durch Verknüpfen des LKR-Gens oder -Genfragments
mit irgendeiner der oben beschriebenen Pflanzenpromotorsequenzen
konstruiert werden (US-Patent Nr. 5231020). Als Alternative kann
ein chimäres
Gen, das zu Exprimieren von Antisense-RNA für das gesamte oder einen Teil
des LKR-Gens konstruiert ist, durch Verknüpfen des LKR-Gens oder -Genfragments
in Umkehrorientierung mit irgendeiner der oben beschriebenen pflanzlichen
Promotorsequenzen konstruiert werden (Europäische Patentanmeldung Nr. 84112647.7).
Entweder das Kosuppresions- oder chimäre Antisense-Gen könnte durch
Transformation in Pflanzen eingeführt werden. Transformanten,
bei denen die Expression des endogenen LKR-Gens reduziert oder eliminiert
ist, werden ausgewählt.
-
Bevorzugte
Promotoren für
die chimären
Gene wären
samenspezifische Promotoren. Für
Sojabohnen-, Raps- und andere zweikeimblättrige Pflanzen wären stark
samenspezifische Promotoren aus einem Bohnenphaseolin-Gen, einem
Sojabohnen-β-Conglycinin-Gen,
Glycinin-Gen, Kunitz-Trypsin-Inhibitor-Gen oder
Rapssamennapin-Gen bevorzugt. Für
Mais und andere einkeimblättrige
Pflanzen würde
eine stark endospermspezifischer Promotor, d.h. der 10 kD- oder
27 kD Zein-Promotor bevorzugt.
-
Transformierte
Pflanzen, die irgendeines der chimären LKR-Gene enthalten, können durch
die oben beschriebenen Methoden erhalten werden. Um transformierte
Pflanzen zu erhalten, die ein chimäres Gen für die Kosuppression von LKR
oder Antisense-LKR, sowie ein chimäres Gen exprimieren, das gegen
Lysin unempfindliche DHDPS kodiert, könnte das Kosuppressions- oder
Antisense-LKR-Gens mit dem chimären
Gen verknüpft
werden, das das gegen Lysin unempfindliche DHDPS kodiert, und die
beiden Gene könnten
durch Transformation in Pflanzen eingeführt werden. Als Alternative
könnte
das chimäre
Gen für
die Kosuppression von LKR oder Antisense-LKR in vorher transformierte
Pflanzen eingeführt
werden, die gegen Lysin unempfindliches DHDPS exprimieren, oder
das Kosuppressions- oder Antisens-LKR-Gen könnte in normale Pflanzen eingeführt und
die dabei erhaltenen Transformanten könnten mit Pflanzen gekreuzt
werden, die gegen Lysin unempfindliche DHDPS exprimieren.
-
KONSTRUKTION
VON LYSINREICHEN POLYPEPTIDEN
-
Es
kann wünschenswert
sein, die hohen Niveaus an gebildetem Lysin in eine Form umzuwandeln,
die gegen Abbau unempfindlich ist, d.h. durch Einbauen desselben
in ein Bi- Tri- oder Oligopeptid oder ein lysinreiches Speicherprotein.
Es sind keine natürlichen
lysinreichen Proteine bekannt.
-
Eine
Ausgestaltung dieser Erfindung besteht aus der Entwicklung von Polypeptiden,
die in vivo exprimiert werden können,
um als lysinreiche Samenspeicherproteine zu dienen. Polypeptide
sind lineare Polymere von Aminosäuren,
wobei die α-Carboxylgruppe
einer Aminosäure
kovalent an die α-Aminogruppe der nächsten Aminosäure in der
Kette gebunden ist. Nichtkovalente Wechselwirkungen zwischen den
Resten der Kette und mit dem umgebenden Lösungsmittel bestimmen die endgültige Konformation
des Moleküls.
Diejenigen Fachleute, die mit dem Stand der Technik vertraut sind,
müssen
elektrostatische Kräfte,
Wasserstoffbindungen, Van-der-Waals-Kräfte, hydrophobe Wechselwirkungen
und konformationelle Vorlieben für
einzelne Aminosäurereste
im Design einer beständigen
gefalzten Peptidkette in Betracht ziehen [vergleiche beispielsweise: Creighton
(1984) Proteins, Structures and Molecular Properties (Proteine,
Strukturen und molekulare Eigenschaften), W. H. Freeman and Company,
New York, Seite 133-197 oder Schulz et al. (1979) Principles of
Protein Structure (Prinzipien der Proteinstruktur), Springer Verlag,
New York, Seite 27-45]. Die Anzahl von Wechselwirkungen und ihre
Komplexität
weist daraufhin, dass der Designvorgang durch Verwendung natürlicher Proteinmodelle,
soweit möglich,
unterstützt
werden kann.
-
Die
synthetischen Speicherproteine (SSP), die in dieser Erfindung verkörpert sind,
werden so gewählt, dass
es sich um Polypeptide mit dem Potential handelt, mit Bezug auf
die durchschnittlichen Proteinniveaus in Pflanzensamen lysinangereichert
zu sein. Lysin ist bei physiologischem pH-Wert eine geladene Aminosäure und
aus diesem Grund am häufigsten
auf der Oberfläche
von Proteinmolekülen
aufzufinden [Chotia (1976) Journal of Molecular Biology 105:1-14].
Um den Lysingehalt zu maximieren, haben die Anmelder für die in
dieser Erfindung verkörperten
synthetischen Speicherproteine eine Molekulargestalt mit einem hohen
Oberflächen-zu-Volumenverhältnis gewählt. Die
Alternativen bestanden darin, entweder die allgemeine kugelförmige Gestalt
der meisten Proteine unter Bildung einer stabähnlichen verlängerten
Struktur zu strecken oder die kugelförmige Gestalt zu einer scheibenähnlichen
Struktur abzuflachen. Die Anmelder haben die erstere Konfiguration
gewählt,
da es mehrere natürliche
Modelle für
lange stabförmige
Proteine in der Klasse fibröser
Proteine gibt [Creighton (1984) Proteins, Structures and Molecular
Properties (Proteine, Strukturen und Molekulareigenschaften), W.H.
Freeman and Company, New York, Seite 191].
-
Gewundene
Spiralen stellen eine eingehend studierte Untergruppe der Klasse
von fibrösen
Proteinen dar [man vergleiche Cohen et al. (1986) Trends Biochem.
Sci. 11:235-248]. Natürliche
Beispiele sind in Form von α-Keratinen,
Paramyosin, leichtem Meromyosin und Tropomyosin zu finden. Die Proteinmoleküle bestehen
aus zwei parallelen α-Helices,
die in Form einer linksdrehenden Superspirale umeinander herumgewickelt sind.
Der Wiederholungsabstand dieser Superspirale beträgt 140 Å (im Vergleich
mit einem Widerholungsabstand von 4,5 Å für eine Windung der einzelnen
Helices. Die Superhelix verursacht eine geringe Verzerrung (10 °) zwischen
den Achsen der beiden einzelnen α-Helices.
-
Bei
einer gewundenen Spirale liegen 3,5 Reste pro Umdrehung der einzelnen
Helices vor, was zu genau 7 Restperiodizitäten bezüglich der Superhelixachse führt (vergleiche 1).
Aus diesem Grund besetzt jede siebte Aminosäure in der Polypeptidkette
eine äquivalente
Position bezüglich
der Helixachse. Die Anmelder bezeichnen die sieben Positionen in
dieser Heptat-Einheit der Erfindung als (defgabc), wie in den 1 und 2a gezeigt.
Dieses entspricht der in der Literatur der gewundenen Spiralen üblichen
Gepflogenheit.
-
Die
a- und d-Aminosäuren
des Heptads folgen einem 3,4-Wiederholungsmuster in der primären Sequenz
und fallen auf eine Seite einer einzelnen Alpha-Helix (vergleiche 1).
Wenn die Aminosäuren
auf einer Seite einer Alpha-Helix alle nicht polar sind, so ist
diese Seite der Helix hydrophob und wird sich mit anderen hydrophoben
Flächen,
beispielsweise der nichtpolaren Seite anderer ähnlicher Helices assoziieren.
Eine gewundene Spiralenstruktur entsteht, wenn zwei Helices sich
derart dimerisieren, dass ihre hydrophoben Seiten aufeinander aufgerichtet
sind (man vergleiche 2a).
-
Die
Aminosäuren
auf den Außenseiten
der Komponente Alpha-Helices (b, c, e, f, g) sind in natürlichen gewundenen
Spiralen dem erwarteten Muster ausgesetzter oder verborgener Resttypen
entsprechend in kugelförmigen
Proteinen gewöhnlich
polar [Schulz et al. (1979) Principles of Protein Structure (Prinzipien
der Proteinstruktur). Springer Verlag, New York, Seite 12; Talbot
et al. (1982) Acc. Chem. Res. 15:224-230; Hodges et al. (1981) Journal
of Biological Chemistry 256:1214-1224]. Es können manchmal geladene Aminosäure aufgefunden
werden, die Salzbrücken
zwischen den Positionen e und g' oder
den Positionen g und e' auf
der gegenüberliegenden
Kette bilden (vergleiche 2a).
-
So
werden zwei amphiphatische Helices, wie diejenige, die in 1 gezeigt
ist, durch eine Kombination von hydrophoben Wechselwirkungen zwischen
den a, a', d und
d'-Resten und durch
Salzbrücken
zwischen den e und g'-
und/oder g und e'-Resten
zusammengehalten. Durch Zusammenpacken der hydrophoben Reste in
der Superspirale werden die Ketten „im Register" gehalten. Für kurze
Polypeptidketten, die nur einige wenige Windungen der Komponente
Alpha-Helixketten umfassen, kann die 10 °-Verzerrung zwischen den Helixachsen
ignoriert und die beiden Ketten als parallel behandelt werden (wie
in 2a gezeigt).
-
In
der Literatur ist von einer Anzahl synthetischer gewundener Spiralen
berichtet worden (Lau et al (1984) Journal of Biological Chemistry
259:13253-13261; Hodges et al. (1988) Peptide Research 1:19-30;
DeGrado et al. (1989) Science 243:622-628; O'Neil et al. (1990) Science 250:646-651].
Obwohl diese Polypeptide größenmäßig unterschiedlich
sind, haben Lau et al. gefunden, dass 29 Aminosäuren für die Dimerisierung unter Bildung
der gewundenen Spiralenstruktur ausreichten [Lau et al. (1984) Journal
of Biological Chemistry 259:13253-13261]. Die Anmelder haben bei
dieser Erfindung Polypeptide als 28-Rest- und größere Ketten aus Gründen der
Konformationsstabilität
konstruiert.
-
Die
erfindungsgemäßen Polypeptide
sind so konstruiert, dass sie sich in wäßrigen Umgebungen mit einem
gewundenen Spiralenmotif dimerisieren. Die Anmelder haben eine Kombination
von hydrophoben Wechselwirkungen und elektrostatischen Wechselwirkungen
zum Stabilisieren der gewundenen Spiralenkonformation verwendet.
Die meisten nichtpolaren Reste sind auf die a- und d-Position beschränkt, was
einen hydrophoben Streifen bildet, der zur Achse der Helix parallel
liegt. Das ist die Dimerisierungsseite. Die Anmelder haben große voluminöse Aminosäuren dieser
Seite entlang vermieden, um die sterische Störung bei der Dimerisierung
zu minimieren und die Bildung der beständigen gewundenen Spiralenstruktur
zu erleichtern.
-
Trotz
kürzlicher
Berichte in der Literatur, die darauf hindeuteten, das Methionin
an den Positionen a und d durch gewundene Spiralen in der Leucin-Zipper-Untergruppe
destabilisiert wird [Landschulz et al. (1989) Science 243:1681-1688
und Hu et al. (1990) Science 250:1400-1403] haben die Anmelder es
gewählt,
Leucin auf der hydrophoben Seite der SSP-Polypeptide durch Methioninreste
zu ersetzen. Methionin und Leucin weisen eine ähnliche Molekulargestalt auf
(3). Die Anmelder haben gezeigt, dass
irgendeine Destabilisierung der gewundenen Spirale, die durch Methionin
in dem hydrophoben Kern hervorgerufen werden kann, in Sequenzen
ausgeglichen zu sein scheint, wo die Bildung von Salzbrücken (e-g' und g-e') an allen möglichen
Positionen innerhalb der Helix erfolgt (d.h. zweimal pro Heptad).
-
Bis
zu dem Grad, in dem es mit dem Ziel des Schaffens eines mit Lysin
angereicherten Polypeptids vereinbar ist, haben die Anmelder die
unausgeglichenen Ladungen im Polypeptid minimiert. Dies kann dazu beitragen,
unerwünschte
Wechselwirkungen zwischen den synthetischen Speicherproteinen und
anderen pflanzlichen Proteinen zu verhindern, wenn die Polypeptide
in vivo exprimiert werden.
-
Die
erfindungsgemäßen Polypeptide
sind so konstruiert, dass sie sich spontan zu einer definierten, konformationell
beständigen
Struktur, der gewundenen Alpha-Helixspirale, mit minimalen Restriktionen
auf der primären
Sequenz falzen. Das erlaubt es, synthetische Speicherproteine spezielle
auf die spezifischen Anforderungen der Endanwender zuzuschneiden.
Irgendeine Aminosäure
kann mit einer Frequenz von bis zu einem je sieben Resten unter
Anwendung der b-, c- und f-Positionen in der Heptadwiederholungseinheit
eingearbeitet werden. Die Anmelder weisen darauf hin, dass bis zu
43% einer essentiellen Aminosäure
aus der Gruppe von Isoleucin, Leucin, Lysin, Methionin, Threonin
und Valin eingearbeitet werden können
und dass bis zu 14% der essentiellen Aminosäuren aus der Gruppe Phenylalanin,
Tryptophan und Tyrosin in die erfindungsgemäßen synthetischen Speicherproteine
eingearbeitet werden können.
-
In
den SSP sind nur Met, Leu, Ile, Val oder Thr im hydrophoben Kern
positioniert. Des Weiteren sind die e-, g-, e'- und g'-Positionen in den SSP derart eingeschränkt unterworfen,
dass eine elektrostatische Anziehungswechselwirkung immer an diesen
Positionen zwischen den beiden Polypeptidketten in einem SSP-Dmer
erfolgt. Dadurch werden die SSP-Polypeptide als Dimere stabiler.
-
So
stellen die neuartigen synthetischen Speicherproteine, die in dieser
Erfindung beschrieben sind, eine spezifische Untergruppe möglicher
gewundener Spiralenpolypeptide dar. Nicht alle Polypeptide, die
in einer wäßrigen Lösung eine
amphipatische Alpha-Helixkonformation annehmen, sind für die oben
beschriebenen Anwendungen geeignet.
-
Folgende
Regeln, die von den Arbeiten der Anmelder abgeleitet sind, definieren
die SSP-Polypeptide, die
die Anmelder bei ihrer Erfindung verwenden:
Das synthetische
Polypeptid umfasst n Heptadeinheiten (defgabc), wobei jedes Heptad
entweder gleich oder verschieden ist, wobei:
n mindestens 4
beträgt;
a
und d unabhängig
aus der Gruppe ausgewählt
werden bestehend aus Met, Leu, Val, Ile und Thr;
e und g unabhängig aus
der Gruppe ausgewählt
werden, bestehend aus den Säure/Basenpaaren
Glu/Lys, Lys/Glu, Arg/Glu, Arg/Asp, Lys/Asp, Glu/Arg, Asp/Arg und
Asp/Lys; und
b, c und f unabhängig irgendwelche Aminosäuren, mit
Ausnahme von Gly oder Pro, sind und mindestens zwei Aminosäuren von
b, c und f in jedem Heptad aus der Gruppe ausgewählt sind, bestehend aus Glu,
Lys, Asp, Arg, His, Thr, Ser, Asn, Gln, Cys und Ala.
-
CHIMÄRE GENE, DIE LYSINREICHE POLYPEPTIDE
KODIEREN
-
DNA-Sequenzen,
die die oben beschriebenen Polypeptide kodieren, können auf
der Basis der genetischen Code konstruiert werden. Wo multiple Codons
für spezifische
Aminosäuren
existieren, sollten die Codons aus denjenigen ausgewählt werden,
die für
die Translation in Pflanzen vorzuziehen sind. Oligonukleotide, die
diesen DNA-Sequenzen entsprechen, können unter Anwendung eines
ABI-DNA-Synthetisierers
synthetisiert, mit Oligonukleotiden einem Annealing unterworfen
werden, die dem komplementären
Strang entsprechen und in einen Plasmidvektor durch Methoden, die
den mit dem Stand der Technik vertrauten Fachleuten bekannt sind,
insertiert werden. Die kodierten Polypeptidsequenzen können durch
Insertieren zusätzlicher
Oligonukleotide, die einem Annealing unterworfen worden sind, an
Restriktionsendonukleasestellen verlängert werden, die in die synthetischen
Gene einengineered worden sind. Einige repräsentative Strategien für das Konstruieren
von Genen, die lysinreiche erfindungsgemäße Polypeptide kodieren, sowie
DNA- und Aminosäuresequenzen
bevorzugter Ausführungsformen
sind in Beispiel 8 bereitgestellt.
-
Ein
chimäres
Gen, das zum Exprimieren von RNA für ein synthetisches Speicherprotein-Gen
konstruiert ist, das ein lysinreiches Polypeptid kodiert, kann durch
Verknüpfen
des Gens mit irgendeiner der oben beschriebenen pflanzlichen Promotorsequenzen
konstruiert werden. Bevorzugte Promotoren wären samenspezifischen Promotoren.
Für Sojabohnen-,
Raps- und andere zweikeimblättrige
Pflanzen würden
stark samenspezifische Promotoren aus einem Bohnenphaseolingen,
einem Sojabohnen-β-conglyciningen,
Glyciningen, Kunitz-Trypsininhibitorgen oder Rapssamennapingen bevorzugt
werden. Für
Mais- und andere einkeimblättrige
Pflanzen würde
ein stark endospermspezifischer Promotor, z.B. der 10 kD- oder 27
kD-Zeinpromotor oder ein stark embryospezifischer Promotor, z.B.
der Maisglobulin 1-Promotor, bevorzugt werden.
-
Um
Pflanzen zu erhalten, die ein chimäres Gen für ein synthetisches Speicherproteingen
exprimieren, das ein lysinreiches Polypeptid kodiert, können Pflanzen
durch irgendeine der oben beschriebenen Methoden transformiert werden.
Um Pflanzen zu erhalten, die sowohl ein chimäres SSP-Gen als auch ein chimäres Gen exprimieren,
das gegen Lysin unempfindliche DHDPS kodiert, könnte das SSP-Gen mit dem chimären Gen verknüpft werden,
das die gegen Lys unempfindliche DHDPS kodiert, und die beiden Gene
könnten
durch Transformation in Pflanzen eingeführt werden. Als Alternative
könnte
das chimäre
SSP-Gen in vorher transformierte Pflanzen eingeführt werden, die die gegen Lysin
unempfindliche DHDPS exprimieren, oder das SSP-Gen könnte in
normale Pflanzen eingeführt
werden und die erhaltenen Transformanten könnten mit Pflanzen gekreuzt
werden, die gegen Lysin unempfindliche DHDPS exprimieren.
-
Die
Ergebnisse genetischer Kreuzungen transformierter Pflanzen, die
Lysinbiosynthesegene enthalten, mit transformierten Pflanzen, die
lysinreiche Proteingene enthalten (man vergleiche Beispiel 10) zeigen, dass
die gesamten Lysinniveaus in Samen durch koordinierte Expression
dieser Gene erhöht
werden können. Dieses
Ergebnis war besonders auffallend, weil die Genkopiezahl aller Transgene
im Hybrid reduziert war. Es ist zu erwarten, dass das Lysinniveau
noch weiter erhöht
würde,
wenn die Biosynthesegene und die lysinreichen Proteingene alle homozygot
wären.
-
BEISPIELE
-
Die
vorliegende Erfindung wird des Weiteren in den folgenden Beispielen
definiert, in denen alle Teile und Prozentsätze auf das Gewicht bezogen
und Grade in Celsius ausgedrückt
sind, es sei denn, es wird etwas anderes angegeben.
-
BEISPIEL 1
-
ISOLATION DER E. COLI-
und CORYNEBACTERIUM GLUTAMICUM-dapA-GENE
-
Das
E. coli-dapA-Gen (EcodapA) ist vorher kloniert, durch Restriktionsendonuklease
kartiert und sequenziert worden [Richaud et al (1986) J. Bacteriol.
166:297-300]. Für
die vorliegende Erfindung wurde das dapA-Gen auf einem bakteriophagen
Lambda-Klon aus einer geordneten Bibliothek von 3400 überlappenden Segmenten
klonierter E. coli-DNA erhalten, die durch Kohara, Akiyama und Isono
[Kohara et al. (1987) Cell 50:595-508] konstruiert wurde. Aus der
Kenntnis der Kartenposition von dapA bei 53 min auf der genetischen E.
coli-Karte [Bachman (1983) Mikrobiol. Rev. 47:180-230], der Restriktionsendonukleasekarte
des klonierten Gens [Richaud et al. (1986) J. Bacteriol. 166:297-300]
und der Restriktionsendonukleosekarte der klonierten DNA-Fragmente
in der E. coli-Bibliothek [Kohara et al. (1987) Cell 50:595-508]
war es möglich
Lambda-Phagen 4C11 und 5A8 [Kohara et al. (1987) Cell 50:595-508] als wahrscheinliche
Kandidaten für
das Tragen des dapA-Gens zu wählen.
Die Phagen wurden in flüssiger
Kultur aus einzelnen Plaques wie beschriebe [vergleiche Current
Protocols in molecular Biology (Gegenwärtige Protokolle in der molekularen
Biologie) Ausubel et al. Verlag., John Wiley & Sons New York] unter Anwendung von
LE392 als Wirt gezüchtet
[man vergleiche Sambrook et al. (1989) Molecular Cloning: a Laboratory
Manual (Molekulares Klonieren: ein Laborhandbuch), Cold Spring Harbor
Laboratory Press]. Phagen-DNA wurde durch Phenolextraktion, wie
beschrieben [man vergleiche Current Protocols in Molecular Biology
(gegenwärtige
Protokolle in der molekularen Biologie (1987) Ausubel et al. Verlag
John Wiley & Sons,
New York] hergestellt. Beide Phagen enthielten ungefähr 2,8 kb
Pst I-DNA-Fragment,
wie für
das dapA-Gen zu erwarten war [Richaud et al. (1986) J. Bacteriol.
166:297-300]. Das Fragment wurde vom Digest der Phage 5A8 isoliert
und in Plasmid pBT427, das digerierten Pst-I-Vektor pBR322 ergibt,
insertiert.
-
Das
Corynebacterium dapA-Gen (cordapA) wurde genomischer DNA vom ATCC-Stamm
13032 unter Anwendung der Polymerasekettenreaktion (PKR) isoliert.
Die Nukleotidsequenz des Corynebacterium dapA-Gens ist veröffentlicht
worden [Bonnassie et al. (1990) Nucleic Acids Res. 18:6421]. Aus
dieser Sequenz war es möglich,
Oligonukleotidprimer für
die PKR zu konstruieren, die die Amplifikation eines DNA-Fragments erlauben
würden,
das das Gen enthält,
und gleichzeitig einzige Restriktionsendonukleasestellen am Startcodon
(Nco I) und gerade nach dem Stopcodon (EcoR I) des Gens zufügen würde. Die
verwendeten Oligonukleotidprimer waren:
SEQ ID NO: 1:
CCCGGGCCAT
GGCTACAGGT TTAACAGCTA AGACCGGAGT AGAGCACT
SEQ ID NO: 2:
GATATCGAAT
TCTCATTATA GAACTCCAGC TTTTTC
-
Die
PKR wurde unter Anwendung eines Perkin-Elmer Cetus-Kits den Anleitungen
der Verkäuferfirma an
einem durch die gleiche Firma hergestellten Wärmezykler durchgeführt. Das
Reaktionsprodukt, wenn es einem Arbeitsgang auf Agarosegel unterworfen
und mit Ethidiumbromid gefärbt
wird, zeigte eine starke DNA-Bande der Größe, die für das Corynebacterium dapA-Gen
zu erwarten ist, nämlich
ca. 900 bp. Das durch PKR gebildete Fragment wurde mit den Restriktionsendonukleasen
Nco I und EcoR I digeriert und in den Expressionsvektor pBT430 (vergleiche
Beispiel 2), der mit den gleichen Enzymen digeriert worden war,
insertiert. Zusätzlich
zum Einführen
einer Nco-Stelle am Translationsstartcodon führten die PKR-Primer auch zu
einer Änderung
des zweiten Codons von der AGC-Kodierung
von Serin zur GCT-Kodierung für
Alanin. Mehrere Klone, die aktive, gegen Lysin unempfindliche DHDPS
(vergleiche Beispiel 2) exprimieren, wurden isoliert, was anzeigt,
dass die zweite Codonaminosäuresubstitution
die Aktivität
nicht beeinflusste; ein Klon wurde mit FS766 bezeichnet.
-
Das
Nco I bis EcoR I-Fragment, das das durch PKR gebildete Corynebacterium
dapA-Gen trägt,
wurde in den Phagemidvektor pGEM-9Zf(–) von Promega subkloniert,
eine einstrangige DNA wurde zubereitet und sequenziert. Dieses Sequenz
ist in SEQ ID NO:3 gezeigt.
-
Von
den schon erwähnten
Unterschieden im zweiten Codon abgesehen, entsprach die Sequenz
der veröffentlichten
Sequenz bis auf zwei Positionen, die Nukleotide 798 und 799. In
der veröffentlichten
Sequenz sind diese TC, während
sie bei dem in SEQ ID NO:3 gezeigten Gen C7 sind. Diese Änderung
führt zu
einer Aminosäuresubstitution
von Leucin für
Serin. Der Grund für
diesen Unterschied ist nicht bekannt. Er kann einem Fehlers in der
veröffentlichten
Sequenz, dem Unterschied in zum Isolieren des Gens verwendeten Stämmen oder
einem durch PKR hervorgerufenen Fehler beruhen. Letzteres scheint
unwahrscheinlich, da die gleiche Änderung in mindestens 3 unabhängig isolierten
durch PKR gebildeten dapA-Genen beobachtet wurde. Der Unterschied
hat keine Auswirkung auf die DHDPS-Enzymaktivität (vergleiche Beispiel 2).
-
BEISPIEL 2
-
HOCHNIVEAU-EXPRESSION
DER E. COLI UND CORYNEBACTERIUM GLUTAMICUM dapA-GENE IN E. COLI
-
Eine
Nco I (CCATGG)-Stelle wurde am Translationinitiationscodon des E.
coli dapA-Gens unter Anwendung der durch Oligonukleotid dirigierten
Mutagenese insertiert. Das 2,8 kb Pst I DNA-Fragment, das das dapA-Gen
im Plasmid pBT427 trägt
(vergleiche Beispiel 1) wurde in die Pst I-Stelle des Phagemidvektors pTZ18R
(Pharmacia) unter Erzeugung von pBT431 insertiert. Die Orientierung
des dapA-Gens war
derart, dass der Kodierstrang auf der einstrangingen Phagemid-DNA
vorhanden wäre.
Die durch Oligonukleotid dirigierte Mutagenese wurde unter Anwendung
eines Muta-Gen-Kits von Bio-Rad, dem Protokoll des Herstellers gemäß mit dem
unten gezeigten mutagenen Primer durchgeführt:
SEQ ID NO:4:
CTTCCCGTGA
CCATGGGCCA TC
-
Mutmaßliche Mutanten
wurden auf das Vorliegen einer Nco I-Stelle hin gescreened und es
wurde aufgezeigt, dass ein Plasmid, das pBT437 genannt wird, die
richtige Sequenz in der Nähe
der Mutation durch das DNA-Sequenzieren aufweist. Die Addition einer
Nco I-Stelle am Translationsstartcodon führte auch zu einer Änderung
des zweiten Codons von der TTC-Kodierung für Phenylalanin zur GTC-Kodierung
für Valin.
-
Um
eine Hochniveauexpression der dapA-Gene in E. coli zu erreichen,
wurde der Bakterienexpressionsvektor pBT430 verwendet. Dieser Expressionsvektor
ist ein Derivat von pET-3a [Rosenberg et al. (1987) (Gene 56:125-135],
bei dem das bacteriophage T7 RNA-Polymerase/T7-Promotorsystem verwendet wird. Der Plasmid
pBT430 wurde konstruiert, indem die Ecor I und Hind III-Stellen in pET-3a
zuerst an ihren Orginalpositionen zerstört wurden. Ein Oligonukleotidadaptor,
der Eeor I- und
Hind-Stellen enthält,
wurde an der BamH I-Stelle von pET-3a insertiert. Dieses bildete
pET-3aM mit zusätzlichen
einzigen Klonierstellen für
die Insertion von Genen in den Expressionsvektor. Daraufhin wurde
die Nde I-Stelle an der Position der Translationsinitiation zu einer
Nco I-Stelle unter Anwendung von durch Oligonukleotid dirigierter
Mutagenese umgewandelt. Die DNA-Sequenz von pET-3aM in dieser Region,
nämlich
5'-CATATGG, wurde zu 5'-CCCATGG in
pBT430 umgewandelt.
-
Das
E. coli dapA-Gen wurde aus dem Plasmid pBT437 als 1150 bp Nco I-Hind
III-Fragment ausgeschnitten und in den Expressionvektor pBT430,
der mit dem gleichen Enzymen digeriert worden war, unter Erzeugung
des Plasmids pBT422 insertiert. Zum Exprimieren des Corynebacterium
dapA-Gens wurde das 917 bp Nco I bis EcoR I-Fragment von SEQ ID
NO:3 in pBT430 insertiert (pFS766, vergleiche Beispiel 1) verwendet.
-
Für die Hochniveauexpression
wurde jedes der Plasmide in den E. coli-Stamm BL21(DE3) transformiert
[Studier et al. (1986) J. Mol. Biol. 189:113-130]. Es wurden Kulturen
in Ampicillin (100 mg/l) enthaltendem LB-Medium bei 25 °C gezüchtet. Bei
einer optischen Dichte bei 600 nM von ca. 1 wurde IPTG (Isopropylthio-β-Galactosid,
der Induzierer) einer Endkonzentration von 0,4 nM hinzugegeben und
die Inkubation 3 Stunden bei 25 °C
fortgeführt.
Die Zellen wurden durch Zentrifugieren aufgefangen und erneut in
1/20tel oder (1/100tel) des Originalkulturvolumens in 50 nM NaCl;
50 mM Tris-Cl, pH-Wert
7,5; 1 mM EDTA suspendiert und bei –20 °C eingefroren. Gefrorene Aliquote
von 1 ml wurden bei 37 °C
aufgetaut und in einem Eiswasserbad zum Lysieren der Zellen einer
Schallbehandlung unterworfen. Das Lysat wurde bei 4 °C 5 Minuten
lang bei 15.000 UpM zentrifugiert. Der Überstand wurde entfernt und
das Granulat erneut in 1 ml des obigen Puffers suspendiert.
-
Der Überstand
und die Granulatfraktionen uninduzierter und IPTG-induzierter Kulturen
von BL21 (DE3)/pBT442 oder BL21(D3)/pFS766 wurden durch SDS-Polyacrylamidgelelektrophorese
analysiert. Der Hauptteil des durch Coomassie-Blau-Färben im Überstand
sichtbaren Proteins und der Granulatfraktionen beider induzierter
Kulturen wiesen eine Molmasse von 32-34 kd, der für DHDPS
zu erwartenden Größe, auf. Selbst
in uninduzierten Kulturen war dieses Protein das auffälligste
gebildete Protein.
-
In
den IPTG-induzierten BL21(DE3)/pBT442-Kulturen befanden sich etwa
80% des DHDPS-Proteins im Überstand
und die DHDPS stellte 10-20% des gesamten Proteins im Extrakt dar.
In den IPTG-induzierten BL21(DE3)/pFS766-Kulturen
befanden sich mehr als 50% des DHDPS-Proteins in der Granulatfraktion.
Die Granulatfraktionen bestanden in beiden Fällen aus 90-95% reiner DHDPS,
wobei kein anderes einzelnes Protein in signifikanten Mengen vorlag.
So waren diese Fraktionen rein genug zur Verwendung bei der Bildung von
Antikörpern.
Die Granulatfraktionen, die 2-4 Milligramm entweder von E. coli
DHDPS oder Corynebacterium DHDPS enthielten, wurden in 50 mM NaCl;
50 mM Tris-Cl, pH-Wert
7,5; 1 mM EDTA, 0,2 mM Dithiothreitol, 0,2% SDS löslich gemacht
und an die Hazelton Research Facility (310 Swampridge Road, Denver,
PA 17517) geschickt, um Kaninchenantikörper gegen diese Protein züchten zu
lassen.
-
Die
DHDPS-Enzymaktivität
wurde wie folgt durch Assay bestimmt: Assay-Mischung (für Assayröhren von
10 × 1,0
ml oder 40 × 0,25
ml für
Mikrotiterplatten) vor der Verwendung frisch hergestellt:
2,5
ml H
2O
0,5 ml 1,0 m Tris-HCl, pH-Wert
8,0
0,5 ml 0,1 M Na Pyruvat
0,5 ml o-Aminobenzaldehyd
(10 mg/ml in Ethanol)
25 μl
1,0 M DL-Aspartin-β-Semialdehyd
(ASA) in 1,0 N HCl
-
Bei
30 °C für die erwünschte Zeitspanne
inkubieren. Durch Zusatz von:
abbrechen.
-
Man
lässt die
Farbe sich 30-60 Minuten lang entwickeln. Die Ausfällung wird
in einer Eppendorf-Zentrifuge
geschleudert. OD540 gegen 0 min als Blindwert
ablesen. Für
den MikroAssay wird ein Aliquot 1 von 0,2 ml in eine Mikrotitervertiefung
eingegeben und bei OD530 abgelesen.
-
Die
spezifische Aktivität
der E. coli DHDPS in der überstehenden
Fraktion von induzierten Extrakten betrug ca. 50 OD540 Einheiten
pro Minute pro Milligramm Protein in einem Assay von 1,0 ml. Die
E. coli DHDPS war für
die Anwesenheit von L-Lysin im Assay empfindlich. Bei einer Konzentration
von ca. 0,5 mM wurde eine Inhibition von fünfzig Prozent aufgefunden.
Für Corynebacterium
DHDPS wurde die Aktivität
in der überstehenden
Fraktion uninduzierter Extrakte anstatt induzierter Extrakte gemessen.
Die Enzymaktivität
betrug ca. 4 OD530-Einheiten pro Minute
pro Milligramm Protein in einem Assay von 0,25 ml. Im Gegensatz
zur E. coli DHDPS wurde Corynebacterium DHDPS durch L-Lysin selbst
in einer Konzentration von 70 mM nicht inhibiert.
-
BEISPIEL 3
-
ISOLIERUNG DES E. COLI
lysC-GENS UND MUTATIONEN IN lysC, DIE ZU GEGEN LYSIN UNEMPFINDLICHER
AKIII FÜHREN
-
Das
E. coli lysC-Gen ist schon vorher kloniert, die Restriktionsendonuklease
kartiert und sequenziert worden [Cassan et al. (1986) J. Biol. Chem.
261:1052-1057]. Für
die vorliegende Erfindung wurde das lysC-Gen auf einem bakteriophagen
Lambda-Klon von einer geordneten Bibliothek von 3400 sich überlappenden
Segmenten von klonierter E. coli DNA erhalten, die durch Kohara,
Akiyama und Isono [Kohara et al. (1987) Cell 50:595-508] konstruiert
wurde. Diese Bibliothek bietet eine physikalische Karte des gesamten
E. coli-Chromosoms und stellt die Verbindung zwischen der physikalischen
Karte und der genetischen Karte her. Aufgrund der Kenntnis der Kartenposition
von lysC bei 90 min auf der genetischen Karte von E. coli [Theze
et al. (1974) J. Bacteriol. 117:133-143], der Restriktionsendonukleasekarte
des klonierten Gens [Cassan et al. (1986) J. Biol. Chem. 261:1052-1057]
und der Restriktionsendonukleasekarte der klonierten DNA-Fragmente in
der E. coli-Bibliothek [Kohara et al. (1987) Cell 50:595-508], war
es möglich
die Lambda-Phagen 4E5 und 7A4 [Kohara et al. (1987) Cell 50:595-508]
als wahrscheinliche Kandidaten für
das Tragen des lysC Gens zu wählen.
Die Phagen wurden in flüssiger
Kultur aus einzelnen Plaques wie beschrieben [vergleiche Current
Protocols in Molecular Biology (Gegenwärtige Protokolle in der Molekularbiologie)
(1987) Ausubel et al. John Wiley & Sons,
New York] unter Anwendung von LE392 als Wirt gezüchtet [vergleiche Sambrook
et al. (1989) Molecular Cloning: a Laboratory Manual, Cold Spring
Harbor Laboratory Press]. Phagen-DNA wurde, wie beschrieben, durch
Phenolextraktion zubereitet [vergleiche Current Protocols in Molecular
Biology (gegenwärtige Protokolle
in der Molekularbiologie) (1987) Ausubel et al., John Wiley & Sons, New York].
-
Aus
der Sequenz des Gens wurden mehrere Restriktionsendonukleasefragemente,
die für
das lysC-Gen diagnostisch
sind, vorausgesagt, einschließlich
eines 1860 bp EcoR I-Nhe I-Fragments, eines 2140 bp EcoR I-Xmn I-Fragments
und eines 1600 bp EcoR I-BamH I-Fragments. Jedes dieser Fragmente
wurde in beiden Phagen-DNA erfasst, was bestätigt, dass diese das lysC-Gen
trugen. Das EcoR I-Nhe I-Fragment wurde isoliert und im Plasmid
pBR322, das mit den gleichen Enzymen digeriert worden war, unter
Erzeugung eines gegen Ampicillin resistenten, gegen Tetracyclin
empfindlichen E. coli-Transformanten subkloniert. Das Plasmid wurde
mit pBT436 bezeichnet.
-
Um
festzustellen, dass das klonierte lysC-Gen funktionsfähig war,
wurde pBT436 in den E. coli- Stamm Gif106M1
(E. coli-Stamm aus dem Genetic Stock Center-Stamm CGSC-5074) transformiert,
das in jedem der drei E. coli AK-Gene Mutationen aufweist [Theze
et al. (1974) J. Bacteriol. 117:133-143]. Diesem Stamm fehlt jegliche
AK-Aktivität
und er erfordert deshalb Diaminopimelat (einen Vorläufer zu
Lysin, der auch für
die Zellwandbiosynthese wesentlich ist), Threonin und Methionin.
Bei dem transformierten Stamm sind alle diese Ernährungserfordernisse
nicht erforderlich, was zeigt, dass das klonierte lysC-Gen funktionsfähige AKIII
kodiert.
-
Der
Zusatz von Lysin (oder Diaminopimelat, das ohne Weiteres in vivo
zu Lysin umgewandelt wird) in einer Konzentration von ca. 0,2 mM
zum Wachstumsmedium hemmt das Wachstum von Gif106M1, das mit pBT436
transformiert worden ist. M9-Medium [vergleiche Sambrook et al.
(1989) Molecular Cloning; a Laboratory Manual (Molekulares Klonieren:
ein Laborhandbuch), Cold Spring Harbor Laboratory Press], dem zum Ergänzen Arginin
und Isoleucin, die für
das Wachstum von Gifl06M1 erforderlich sind, und Ampicillin zum
Aufrechterhalten der Selektion zum pBT436 Plasmid zugesetzt worden
wind, wurde angewendet. Diese Inhibition wird durch Zugabe von Threonin
plus Methionin zum Wachsumsmedium rückgängig gemacht. Diese Ergebnisse
zeigten, dass AKIII durch exogenes Zugeben von Lysin gehemmt werden
konnte, was zu einem vollständigen
Mangel anderer Aminosäuren,
die von Aspartat deriviert sind, führt. Diese Eigenschaft von
durch pBT436 transformiertem Gif106M1 wurde benutzt, um Mutationen
in lysC_auszuwählen,
das gegen Lysin unempfindlichen AKIII kodiert.
-
Einzelne
Kolonien von Gif106M1, die mit pBT436 transformiert sind, wurden
ausgesucht und erneut in 200 μl
einer Mischung von 100 μl
1% Lysin plus 100 μl
M9-Medium suspendiert. Die gesamte Zellsuspension, die 107-108 Zellen enthält, wurde
in einer Petrischale ausgebreitet, die M9-Medium enthielt, dem zusätzlich Arginin,
Isoleucin und Ampicillin zugegeben worden war. Sechzehn Petrischalen
wurden auf diese Weise zubereitet. 1 bis 20 Kolonien erschienen
auf 11 von 16 Petrischalen. Eine oder zwei (falls vorhanden) Kolonien
wurden ausgesucht und auf Lysinresistenz hin geprüft und daraus
wurden neun gegen Lysin widerstandsfähige Klone erhalten. Plasmid-DNA
wurde aus acht derselben zubereitet und wieder in Gif106M1 transformiert,
um zu bestimmen, ob der Lysinwiderstandsdeterminant von Plasmid
getragen wurde. Sechs der acht Plasmid-DNA ergaben gegen Lysin widerstandsfähige Kolonien.
Drei dieser sechs trugen lysC-Gene, die AKIII kodieren, die durch
15 mM Lysin nicht inhibiert wurde, während AKIII vom Wildtyp durch
0,3-0,4 mM Lysin um 50% gehemmt und durch 1 mM Lysin > 90% gehemmt wird (man
vergleiche Beispiel 2 bezüglich
der Einzelheiten).
-
Um
die molekulare Basis für
die Widerstandsfähigkeit
gegen Lysin zu bestimmen, wurden die Sequenzen des lysC-Gens vom
Wildtyp und von drei mutanten Genen bestimmt. Eine Methode für das „Anwenden von
Miniprepplasmid-DNA für
das Sequenzieren doppelstrangiger Matrizen mit SequenaseTM [Kraft et al. (1988) Biotechniques 6:544-545]
wurde angewendet. Oligonukleotidprimer, die auf der veröffentlichten
lysC-Sequenz basieren und ungefähr
alle 200 bp voneinander entfernt sind, wurden synthetisiert, um
das Sequenzieren zu erleichtern. Die Sequenz des lysC-Gens vom Wildtyp,
die in pBT436 kloniert ist (SEQ ID NO:5), war von der veröffentlichten
lysC-Sequenz in der Kodierregion an den Positionen 5 verschieden.
Vier dieser Nukleotidunterschiede befanden sich an der dritten Position
in einem Codon und würden
nicht zu einer Änderung
der Aminosäuresequenz
des AKIII-Proteins führen.
Einer der Unterschiede würde
zu einer Cystein- zu Glycinsubstitution bei der Aminosäure 58 von
AKIII führen.
Diese Unterschiede sind wahrscheinlich den verschiedenen Stämmen zuzuschreiben,
von denen die lysC-Gene
kloniert wurden.
-
Die
Sequenzen der drei mutanten lysC-Gene, die gegen Lysin unempfindliche
AK kodierten, unterschieden sich jeweils von der Sequenz vom Wildtyp
durch ein einziges Nukleotid, was zu einer einzigen Aminosäuresubstitution
im Protein führte.
Beim mutaten M2 war ein G durch ein A am Nukleotid 954 von SEQ ID NO:
5 substituiert, was zu einer Substitution von Isoleucin für Methionin
an der Aminosäure
318 führte
und die mutanten M3 und M4 wiesen identische T für C-Substitutionen am Nukleotid
1055 von SEQ ID NO: 5 auf, was zu einer Substitution von Isoleucin
für Threonin
an der Aminosäure
352 führte.
So reicht eine dieser einfachen Aminosäuresubstitutionen aus, um das
AKIII-Enzym gegen
Lysininhibition unempfindlich zu machen.
-
Eine
Nco I-(CCATGG)-Stelle wurde am Translationsinitiationscodon des
lysC-Gens unter Anwendung folgender Oligonukleotide insertiert:
SEQ
ID NO: 6
GATCCATGGC TGAAATTGTT GTCTCCAAAT TTGGCG
SEQ ID
NO: 7
GTACCGCCAA ATTTGGAGAC AACAATTTCA GCCATG
-
Beim
Annealing weisen dies Oligonukleotide BamH I und Asp 718 „klebrige" Enden auf. Das Plasmid pBT436
wurde mit BamH I, das stromaufwärts
von der lysC-Kodiersequenz, und Asp 718, das 31 Nukleotide stromabwärts vom
Initiationscodon überschneidet,
digeriert. Die Oligonukleotide, die ein Annealing durchgemacht haben,
wurden an den Plasmidvektor ligiert und es wurden E. coli-Transformanten erhalten.
Plasmid-DNA wurde zubereitet und auf die Insertion der Oligonukleotide
hin aufgrund des Vorliegens einer Nco I-Stelle einem Screenen unterzogen.
Ein die Stelle enthaltendes Plasmid wurde sequenziert, um sicherzustellen,
dass die Insertion richtig war, und wurde mit pBT457 bezeichnet.
Zusätzlich
zum Bilden einer Nco 1-Stelle am Initiationscodon von lysC, änderte diese
Oligonukleotidinsertion den zweiten Codon von TCT, der Serin kodiert,
zu GCT, das Alanin kodiert. Diese Aminosäuresubstitution hat keine offensichtliche
Wirkung auf die AKIII-Enzymaktivitität.
-
Das
lysC-Gen wurde aus dem Plamid pBT457 als 1560 bp Nco I-EcoH I-Fragment
ausgeschnitten und in den Expressionsvektor pBT430 insertiert, der
mit den gleichen Enzymen unter Erzeugung des Plamids pBT461 digeriert
wird. Für
das Exprimieren des mutanten lysC-M4-Gens wurde pBT461 mit Kpn I-EcoR
I digeriert, wodurch das lysC-Gen vom Wiltyp von ca. 30 Nukleotiden
stromabwärts
vom Translationsstartcodon entfernt wird, und durch Insertieren
der analogen Kpn I-EcoR I-Fragmente aus den mutanten Genen unter
Erzeugung des Plasmids pBT492.
-
BEISPIEL 4
-
KONSTRUKTION
VON CHIMÄREN
dapA-GENEN ZUM EXPRIMIEREN IN DEN SAMEN VON PFLANZEN
-
Die
samenspezifische Expressionskassette (4) besteht
aus dem Promotor und dem Transkriptionsterminator aus dem Gen, das
die β-Untereinheit
des Samenspeicherproteins Phaseolin aus der Bohne Phaseolus vulgaris
kodiert [Doyle et al. (1986) J. Biol. Chem. 261:9228-9238]. Die
Phaseolinkassette enthält ca.
500 Nukleotide stromaufwärts
(5') vom Translationsinitiationscodon
und ca. 1650 Nukleotide stromabwärts (3') vom Translationsstopcodon
von Phaseolin. Zwischen den 5'-
und 3'-Regionen befinden
sich die einzigen Restriktionsendonukleasestellen Nco I (die den
ATG- Translationsinitiationscodon
umfassen, Sma I, Kpn I und Xba I. Die gesamte Kassette ist von Hind
III-Stellen flankiert.
-
Pflanzliche
biosynthetische Aminosäureenzyme
sind dafür
bekannt, dass sie in den Chloroplasten lokalisiert sind und aus
diesem Grund werden sie mit einem Chloroplasttargetingsignal synthetisiert.
Bakterienproteine, wie beispielsweise DHDPS und AKIII, besitzen
kein derartiges Signal. Eine Chloroplasttransitsequenz (cts) wurde
deshalb an die dapA- und lysC-M4-Kodiersequenz in den chimären Genen
fusioniert. Die verwendete cts basierte auf der cts der kleinen
Untereinheit von Ribulose-1,5-bisphosphatcarboxylase
aus Sojabohnen [Berry-Lowe et al. (1982) J. Mol. Appl. Genet. 1:483-498].
Die Oligonukleotide SEQ ID NOS:8-11 wurden synthetisiert und wie
unten beschrieben verwendet.
-
Es
wurden drei chimäre
Gene geschaffen:
- Nr. 1) Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3'-Region
- Nr. 2) Phaseolin 5'-Region/cts/ecodapA/Phaseolin
3'-Region
- Nr. 3) Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region
-
Die
Oligonukleotide SEQ ID NO:8 und SEQ ID NO:9, die einen endständigen Carboxylteil
des Chloroplasttargetingsignals kodieren, wurden einem Annealing
unterzogen, was zu mit Nco I kompatiblen Enden führt, durch Polyacrylgelelektrophorese
gereinigt und in mit Nco I digeriertes pBT461 insertiert. Die Insertion der
richtigen Sequenz in der richtigen Orientierung wurde durch DNA-Sequenzieren
nachgeprüft,
was zu pBT496 führte.
Die Oligonukleotide SEQ ID NO:10 und SEQ ID NO:11, die den Aminoendteil
des Chloroplasttargetingsignals kodieren, wurden einem Annealing
unterzogen, was zu mit Nco I verträglichen Enden führt, durch
Polyacrylamidgelelektrophorese gereinigt und in durch Nco I digeriertes
pBT496 insertiert. Die Insertion der richtigen Sequenz in der richtigen
Orientierung wurde durch DNA-Sequenzieren
nachgeprüft,
was pBT521 ergab. So wurden die cts an das lysC-Gen fusioniert.
-
Zum
Fusionieren der cts an das lysC-M4-Gen wurde pBT521 mit Sal I digeriert,
und ein DNA-Fragment von
ca. 900 bp das die cts und die Aminoendkodierregion von lysC enthielt,
wurde isoliert. Dieses Fragment wurde in Sal I, das mit pBT492 digeriert
ist, insertiert, wodurch die endständige Aminokodierregion von
lysC-M4 effektiv durch die fusionierte cts und die endständige Aminokodierregion
von lysC ersetzt wurde. Da die Mutation, die zur Lysinunempfindlichkeit
führte,
sich nicht in dem ersetzten Fragment befand, trug das neue Plasmid,
pBT523, die an lysC-M4 fusionierten cts.
-
Das
Nco I-Hpa I-Fragment von 1600 bp, das die an lysC-M4 fusionierte
cts plus ca. 90 bp der 3'- nichtkodierenden
Sequenz enthielt, wurde isoliert und in die samenspezifische Expressionskassette
insertiert, die mit Nco I und Sma I (chimäres Gen Nr. 1) digeriert war,
unter Bildung des Plasmids pBT544 insertiert.
-
Vor
der Insertion in die Expressionskassette wurde das ecodapA-Gen modifiziert,
um eine Restriktionsendonukleasestelle, Kpn I, gerade nach dem Translationsstopcodon
zu insertieren. Die Oligonukleotide SEQ ID NO:12-13 wurden zu diesem
Zweck synthetisiert:
SEQ ID NO:12:
CCGGTTTGCT GTAATAGGTA
CCA
SEQ ID NO:13:
AGCTTGGTAC CTATTACAGC AAACCGGCAT G
-
Die
Oligonukleotide SEQ ID NO:12 und SEQ ID NO:13 wurden einem Annealing
unterworfen, was zu einem mit Sph I kompatiblen Ende an einem Ende
und einem mit Hind III kompatiblen Ende am anderen führte, und
in Sph I plus Hind III insertiert, das mit pBT437 digeriert worden
war. Die Insertion der richtigen Sequenz wurde durch DNA-Sequenzierung
unter Bildung von pBT443 nachgeprüft.
-
Ein
Nco I-Kpn I-Fragment von 880 bp aus pBT443, das die gesamte ecodapA-Kodierregion
enthielt, wurde aus einem Agarosegel auf die Elektrophorese hin
isoliert und in die samenspezifische Expressionskassette, die mit
Nco I und Kpn I digeriert wurde, unter Bildung des Plasmids pBT494
insertiert. Die Oligonukleotide SEQ ID NO:8-11 wurden wie oben beschrieben
verwendet, um eine cts der ecodapA-Kodierregion in der samenspezifischen
Expressionskassette hinzuzufügen,
unter Bildung des chimären
Gens Nr. 2 in pBT520.
-
Ein
Nco I-EcoR I-Fragment von 870 bp aus pFS766, das die gesamte cordapA-Kodierregion
enthielt, wurde aus einem Agarosegel auf die Elektrophorese hin
isoliert und in die Blattexpressionskassette insertiert, die mit
Nco I und EcoR I digeriert worden war, unter Bildung des Plasmids
pFS789. Um das cts an das cordapA-Gen anzuknüpfen, wurde ein DNA-Fragment,
das die gesamte cts enthielt, unter Anwendung der PKR zubereitet.
Die Matrizen-DNA war pBT544 und die verwendeten Oligonukleotidprimer
waren:
SEQ ID NR:14:
GCTTCCTCAA TGATCTCCTC CCCAGCT
SEQ
ID NO:15:
CATTGTACTC TTCCACCGTT GCTAGCAA
-
Die
PKR wurde unter Anwendung eines Perkin-Elmer Cetus-Kits den Anweisungen
der Verkäuferfirma entsprechend
auf einem Thermozykler, der von der gleichen Firma hergestellt worden
war, durchgeführt.
Das durch PKR gebildete 160 bp-Fragment wurde mit T4 DNA-Polymerase
in Gegenwart der4 Desoxyribonukleotidtriphosphate unter Erzielung
eines stumpfkantigen Fragments behandelt. Das cts-Fragment wurde in
das Nco I insertiert, das den Startcodon des cordapA-Gens enthielt,
das digeriert und mit dem Klenow-Fragment von DNA-Polymerase behandelt
worden war, um die 5'-Überhänge zu füllen. Das
insertierte Fragment und die Vektor/Insertionsverbindungen wurden
durch DNA-Sequenzieren als richtig bestimmt.
-
Ein
Nco I-Kpn I-Fragment von 1030 bp, das das an die cordapA-Kodierregion
angeknüpfte
cts enthielt, wurde aus einem Agarosegel auf die Elektrophorese
hin isoliert und in die Phaseolinsamenexpressionskassette insertiert,
die mit Nco I und Kpn I digeriert worden war, unter Erzielung des
Plasmids pFS889, die das chimäre
Gen Nr. 3 enthielt.
-
BEISPIEL 5
-
TRANSFORMATION VON RAPSSAMEN
MIT DEN CHIMÄREN
PHASEOLINPROMOTOR/CTS/CORDAPA- UND PHASEOLINPROMOTOR/CTS/LYSC-M4-GENEN
-
Die
chimären
Genkassette Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region, Phaseolin
5'-Region/cts/lysC-M4/Phaseolin
3'- und Phaseolin
5'-Region/cts/cordapA/Phaseolin
3'-Region plus Phaseolin
5'-Region/cts/lysC-M4/Phaseolin
3' (Beispiel 4)
wurden in den binären
Vektor pZS199 insertiert (5A).
In pZS199 treibt der 35S-Promotor aus Blumenkohlmosaikvirus die
Expression des NPT II.
-
Die
chimäre
Genkassette der Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region wurde
unter Anwendung von Oligonukleotidadaptoren zum Umwandeln der Hind
III-Stellen an jedem Ende in BamH I-Stellen modifiziert. Die Genkassette
wurde dann als BamH I-Fragment von 2,7 kb isoliert und in mit BamH
I digeriertem pZS199 unter Bildung des Plasmids pFS926 (5B) insertiert. Bei diesem binären Vektor ist das chimäre Gen Phaseolin
5'-Region/cts/cordapA/Phaseolin
3'-Region in der
gleichen Orientierung wie das Markergen 35S/NPT II/nos 3' insertiert.
-
Um
die Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3'-Region zu insertieren,
wurde die Genkassette als 3,3 kb EcoR I bis Spe I-Fragment isoliert
und in EcoR I plus Xba I, das mit pZS199 digeriert worden war, unter
Bildung des Plasmids pBT593 insertiert. Bei diesem binären Vektor
ist das chimäre
Gen Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3'-Region in der
gleichen Orientierung wie das Markergen 35S/NPT II/nos 3' insertiert.
-
Um
die beiden Kassette zu kombinieren, wurde die EcoR I-Stelle von
pBT593 in eine BamH I-Stelle unter
Anwendung von Oligonukleotidadaptoren konvertiert, der dabei entstehende
Vektor wurde mit BamH I geschnitten, und die Genkassette 5'-Region/cts/cordapA/Phaseolin
3'-Region wurde
als BamH I-Fragment
von 2,7 kb isoliert und unter Bildung von pBT597 insertiert. Bei
diesem binären
Vektor sind beide chimären
Gene Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region und Phaseolin
5'-Region/cts/lysC-M4/Phasoelin
3'-Region in der
gleichen Orientierung wie das Markergen 35S/NPT II/nos 3' insertiert.
-
Der
Brassica napus-Kultivar „Westar" wurde durch gleichzeitiges
kultivieren von Stücken
von Keimlingen mit disarmiertem Agrobacterium tumefaciens-Stamm
LBA4404, der den geeigneten binären
Vektor trägt, transformiert.
-
Samen
von B. napus wurden durch Rühren
in 10% Chlorox, 0,1% SDS für
dreißig
min. sterilisiert und dann gründlich
mit sterilem destilliertem Wasser gespült. Die Samen wurden auf einem
sterilen Medium, das 30 mM CaC12 und 1,5% Agar enthielt, gekeimt
und sechs Tage lang im Dunkeln bei 24 °C gezüchtet.
-
Flüssige Kulturen
von Agrobacterium für
die Pflanzentransformation wurden über Nacht bei 28 °C in Minimal
A-Medium gezüchtet,
das 100 mg/l Kanamycin enthielt. Die Bakterienzellen wurden durch
Zentrifugieren pelletiert und erneut in einer Konzentration von
108 Zellen/ml in flüssigem Murashige- und organischem Skoo-Minimalmedium
suspendiert das 100 μm
Acetosyringon enthielt.
-
Keimlinghypokotyle
von B. napus wurden in Segmente von 5 mm geschnitten, die sofort
in die Bakteriensuspension eingegeben wurden. Nach 30 min wurden
die Hypokotylstücke
aus der Bakteriensuspension entfernt und auf BC-35-Callusmedium
aufgegeben, das 100 μm
Acetosyringon enthielt. Das Pflanzengewebe und Agrobakterien wurden
gleichzeitig drei Tage lang bei 24 °C in abgeblendetem Licht kultiviert.
-
Die
gleichzeitige Kultivierung wurde durch Übertragen der Hypokotylstücke auf
BC-35-Callusmedium, das
200 mg/l Carbenicillin, um die Agrobakterien abzutöten, und
25 mg/l Kanamycin, um transformiertes Pflanzenzellwachstum zu wählen, enthielt,
beendet. Die Keimlingstücke
wurden auf diesem Medium drei Wochen lang bei 24 °C unter kontinuierlichem
Licht inkubiert.
-
Nach
drei Wochen wurden die Segmente auf BS-48-Regenerierungsmedium übertragen,
das 200 mg/l Carbenicillin und 25 mg/l Kanamycin enthielt. Das Pflanzengewebe
wurde all zwei Wochen auf frisches selektives Regenerationsmedium
unter den gleichen Kultivierungsbedingungen, wie für das Callusmedium
beschrieben, subkultiviert. Mutmaßlich transformierte Calli
wuchsen schnell auf Regenerierungsmedium; als die Calli einen Durchmesser
von ca. 2 mm erreichten, wurden sie von den Hypocotylstücken entfernt
und auf das gleiche Medium, das kein Kanamycin aufwies, aufgegeben.
-
Es
begannen innerhalb mehrere Wochen nach der Übertragung auf das BS-48-Regenerierungsmedium
Schößlinge zu
erscheinen. Sobald die Schößlinge wahrnehmbare
Stiele gebildet hatten, wurden sie von den Calli abgeschnitten,
auf MSV-1A Verlängerungsmedium übertragen
und zu einer Photoperiode von 16:8-h bei 24 °C überführt.
-
Sobald
Schößlinge mehrere
Stengelglieder gewachsen waren, wurden sie über der Agaroberfläche abgeschnitten
und die Schnittenden wurden in Rootone getaucht. Die behandelten
Schößlinge wurden
direkt in bodenfreies Topferdemedium Metro-Mix 350 eingepflanzt.
Die Töpfe
wurden mit Kunststoffbeuteln bedeckt, die entfernt wurden, als die
Pflanzen klar ersichtlich wuchsen, und zwar nach ca. zehn Tagen.
Die Ergebnisse der Transformation sind in Tabelle 1 gezeigt. Transformierte
Pflanzen wurden mit jedem der binären Vektoren erhalten.
-
Bakterienwachstumsmedium
Miminal A
-
Man
löst Folgendes
in destilliertem Wasser:
| 10,5
g | zweibasisches
Kaliumphosphat |
| 4,5
g | einbasisches
Kaliumphosphat |
| 1,0
g | Ammoniumsulfat |
| 0,5
g | Natriumcitratdihydrat |
-
Man
füllt mit
destilliertem Wasser auf 979 ml auf.
-
Autoklavieren
-
Man
setzt 20 ml filtersterilisierte 10%-ige Saccharose hinzu.
-
Man
setzt 1 ml filtersterilisiertes 1 M MgSO4 hinzu.
-
Brassica Callus-Medium
BC-35
-
Pro Liter:
-
- Organisches Murashige- und Skoog-Minimalmedium
- (MS-Salze, 100 mg/l i-Inositol, 0,4 mg/l Thiamin; GIBCO #510-3118)
- 30 g Saccharose
- 15 g Mannit
- 0,5 mg/l 2,4-D
- 0,3 mg/l Kinetin
- 0,6% Agarose
- pH-Wert 5,8
-
Brassica-Regenerierungsmedium
BS-48
-
- Organisches Murashige- und Skoog-Minimalmedium
- Gamborg B5 Vitamine (SIGMA #1019)
- 10 g Glukose
- 250 mg Xylose
- 600 mg MES
- 0,4% Agarose
- pH-Wert 5,7
-
Man
filtersterilisiert und setzt nach dem Autoklavieren folgendes hinzu:
2,0
mg/l Zeatin
0,1 mg/l IAA
-
Verlängerungsmedium MSV-1A für Brassica-Schößlinge
-
- Organisches Murashige- und Skoog-Minimalmedium
- Gamborg B5 Vitamine
- 10 g Saccharose
- 0,6% Agarose
- pH-Wert 5,8
-
TABELLE
1: RAPS-TRANSFORMANTEN
-
Die
Pflanzen wurden unter einer Photoperiode von 16:8-h bei einer Tagestemperatur
von 23 °C
und einer Nachtemperatur von 17 °C
gezüchtet.
Als der primäre
Blütenstengel
sich zu verlängern
begann, wurde er mit einem aus Maschengewebe bestehenden Beutel
zum Zusammenhalten von Pollen zum Verhindern des Auskreuzens bedeckt.
Die Selbstbestäubung
wurde durch mehrmaliges Schütteln
der Pflanzen am Tag erleichtert. Reife Samen, die aus der Selbstbestäubung erhalten
wurden, wurden ca. drei Monate nach dem Auspflanzen geerntet.
-
Ein
teilweise entfettetes Samenmehl wurde wie folgt zubereitet: 40 Milligramm
reife trockene Samen wurden mit einer Reibeschale und Stößel unter
flüssigem
Stickstoff zu einem feinen Pulver zerrieben. 1 Milliliter Hexan
wurde zugegeben und die Mischung wurde 15 min lang bei Raumtemperatur
geschüttelt.
Das Mehl wurde in einer Eppendorfzentrifuge pelletiert, das Hexan
entfernt und die Hexanextraktion wiederholt. Das Mehl wurde daraufhin
10 min lang bei 65 °C
getrocknet, bis das Hexan unter Zurücklassen eines trockenen Pulvers
vollständig
verdampft war. Die gesamten Proteine wurden wie folgt aus reifen
Samen extrahiert. Ca. 30-40 mg Samen wurden in eine wegwerfbare
Mikrofugenröhre
von 1,5 ml eingegeben und in 0,25 ml 50 mM Tris-HCl, pH-Wert 6,8,
2 mM EDTA, 1% SDS, 1% β-Mercaptoethanol gemahlen.
Das Mahlen erfolgte unter Anwendung einer motorisierten Mahlvorrichtung
mit wegwerfbaren Kunststoffwellen, die so konstruiert waren, dass
sie in die Mikrofugenröhre
passten. Die dabei gebildeten Suspensionen wurden 5 min lang bei
Raumtemperatur in einer Mikrofuge zum Entfernen von teilchenförmigen Substanzen
zentrifugiert. Drei Volumen Extrakt wurden mit 1 Volumen Probepuffer
von 4 × SDS-Gel
(0,17 m Tris-HCl, pH-Wert 6,8, 6,7% SDS, 16,7% β-Mercaptoethanol, 33% Glycerin)
gemischt und von jedem Extrakt wurden 5 μl pro Bahn auf einem SDS-Polyacrylamidgel
einem Arbeitsgang unterworfen, wobei bakteriell hergestellte DHDPS
oder AKIII als Größenstandard diente,
und Protein, das aus untransformierten Tabaksamen extrahiert worden
waren, als negative Kontrolle diente. Die Proteine wurden dann elektrophoretisch
auf eine Nitrocellulosemembran geblottet. Die Membranen wurden den
DHDPS- oder AKIII-Antikörpern
in einer Verdünnung
von 1:5000 des Kaninchenserums unter Anwendung eines Standardprotokolls,
das von BioRad mit ihrem Immun-Blotkit bereitgestellt wurde, ausgesetzt. Auf
das Spülen
zum Entfernen von ungebundendem primärem Antikörper hin, wurden die Membranen
dem sekundären
Antikörper,
nämlich
Esel-Anti-Kaninchen-Ig, das an Meenettichperoxidase (Amersham) konjugiert worden
war, in einer Verdünnung
von 1:3000 ausgesetzt. Auf das Spülen zum Entfernen von ungebundenem sekundärem Antikörper hin,
wurden die Membranen einem Chemilumineszenzreagenz von Amersham
und Röntgenfilm
ausgesetzt.
-
Acht
von acht FS926-Transformanten und sieben von sieben BT597-Transformanten
exprimierten das DHDPS-Protein. Der einzige BP593-Transformant und
fünf von
sieben BT597-Transformanten exprimierten das AKIII-M4-Protein (Tabelle
2).
-
Um
die freie Aminosäurezusammensetzung
der Samen zu messen, wurden freie Aminosäuren aus 40 Milligramm des
entfetteten Mehls in 0,6 ml Methanol/Chloroform/Wasser, das im Verhältnis von
12 Vol./5 Vol./Vol. (MCW) bei Raumtemperatur gemischt worden war,
extrahiert. Die Mischung wurde in der Wirbelvorrichtung behandelt
und dann in einer Eppendorf-Mikrozentrifuge ca. 3 min lang zentrifugiert.
Ca 0,6 ml der überstehenden
Flüssigkeit
wurden dekantiert und zusätzliche
0,2 ml MCW dem Granulat hinzugegeben, das dann in der Wirbelvorrichtung
behandelt und wie oben zentrifugiert wurde. Die zweite überstehende
Flüssigkeit,
ca. 0,2 ml, wurde der ersten hinzugegeben. Diesem wurden 0,2 ml
Chloroform gefolgt von 0,3 ml Wasser hinzugegeben. Die Mischung
wurde in der Wirbelvorrichtung behandelt und dann in einer Eppendorf-Mikrozentrifuge
ca. 3 min lang zentrifugiert, die obere wässrige Phase, ca. 1,0 ml wurde
entfernt und in einem Savant Speed Vac-Konzentrator getrocknet.
Die Proben wurden in 6N Salzsäure,
0,4% β-Mercaptoethanol
unter Stickstoff 24 h bei 110-120 °C hydrolysiert; 1/4 der Probe
wurde auf einem Beckman Aminosäureanalysator, Modell
6300, unter Anwendung der Nachsäulen-Ninhydrinerfassung
einem Arbeitsgang unterzogen. Die relativen Niveaus freier Aminosäure in den
Samen wurden als Verhältnisse
von Lysin oder Threonin zu Leucin verglichen, wobei Leucin so als
interner Standard verwendet wurde.
-
Es
bestand eine gute Korrelation zwischen Transformanten, die höhere Niveaus
an DI-IDPS-Protein exprimierten
und denjenigen, die höhere
Niveaus an freiem Lysin aufweisen. Die höchsten Expressionslinien zeigten
eine mehr als 100 fache Erhöhung
des freien Lysinniveaus in den Samen auf. Es erfolgte keine stärkere Ansammlung
von freiem Lysin aufgrund der Expression von AKIII-M4 zusammen mit
Corynebacteria DHDPS im Vergleich mit der Expression von Corynebacteria
DHDPS als solcher. Der Transformant, der AKIII-M4 in Abwesenheit
der Corynebacteria DHDPS exprimiert, zeigte eine 5-fache Erhöhung des
Niveaus von freiem Threonin in den Samen. Ein höheres Niveau an α-Aminoadipinsäure, das
auf einen Lysinkatabolismus hindeutet, wurde bei vielen der transformierten
Linien beobachtet. So sollte die Verhinderung von Lysinkatabolismus
durch Inaktivierung von Lysinketoglutaratreduktase die Erhöhung der
Ansammlung von freiem Lysin in den Samen noch weiter erhöhen. Als
Alternative würde
das Einarbeiten von Lysin in ein Peptid oder lysinreiches Protein
den Katabolismus verhindern und zu einer Erhöhung der Ansammlung von Lysin
in den Samen führen.
-
Um
die gesamte Aminosäurezusammensetzung
reifer Samen zu messen, wurden 2 Milligramm des entfetteten Mehls
in 6N Salzsäure,
0,4% β-Mercaptoethanol
unter Stickstoff 24 h bei 110-120 °C hydrolysiert; 1/100 der Probe
wurde auf einem Beckman Aminosäureanalysator,
Modell 6300, unter Anwendung der Nachsäulen-Ninhydrinerfassung einem
Arbeitsgang unterzogen. Die relativen Niveaus von Aminosäure in den
Samen wurden als Prozentsätze
von Lysin, Threonin oder α-Aminoadipinsäure mit
den gesamten Aminosäuren verglichen.
Es bestand eine gute Korrelation zwischen Transformanten, die das DHDPS-Protein
exprimieren, und denjenigen, die hohe Niveaus an Lysin aufweisen.
Samen mit einer Erhöhung
des Lysinniveaus von 5-100% im Vergleiche mit den untransformierten
Kontrollen, wurden beobachtet. In den Samen mit den höchsten Niveaus
bildet Lysin 11-13% der gesamten Samenaminosäuren, was beträchtlich
höher ist
als bei irgendeinem vorher bekannten Rapssamen. TABELLE
2 FS926
Transformanten: Phaseolin 5'-Region/cts/cordapA/Phaseolin
3' BT593
Transformanten: Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3' BT597
Transformanten: Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3' Phaseolin
5'-Region/cts/cordapA/Phaseolin
3'
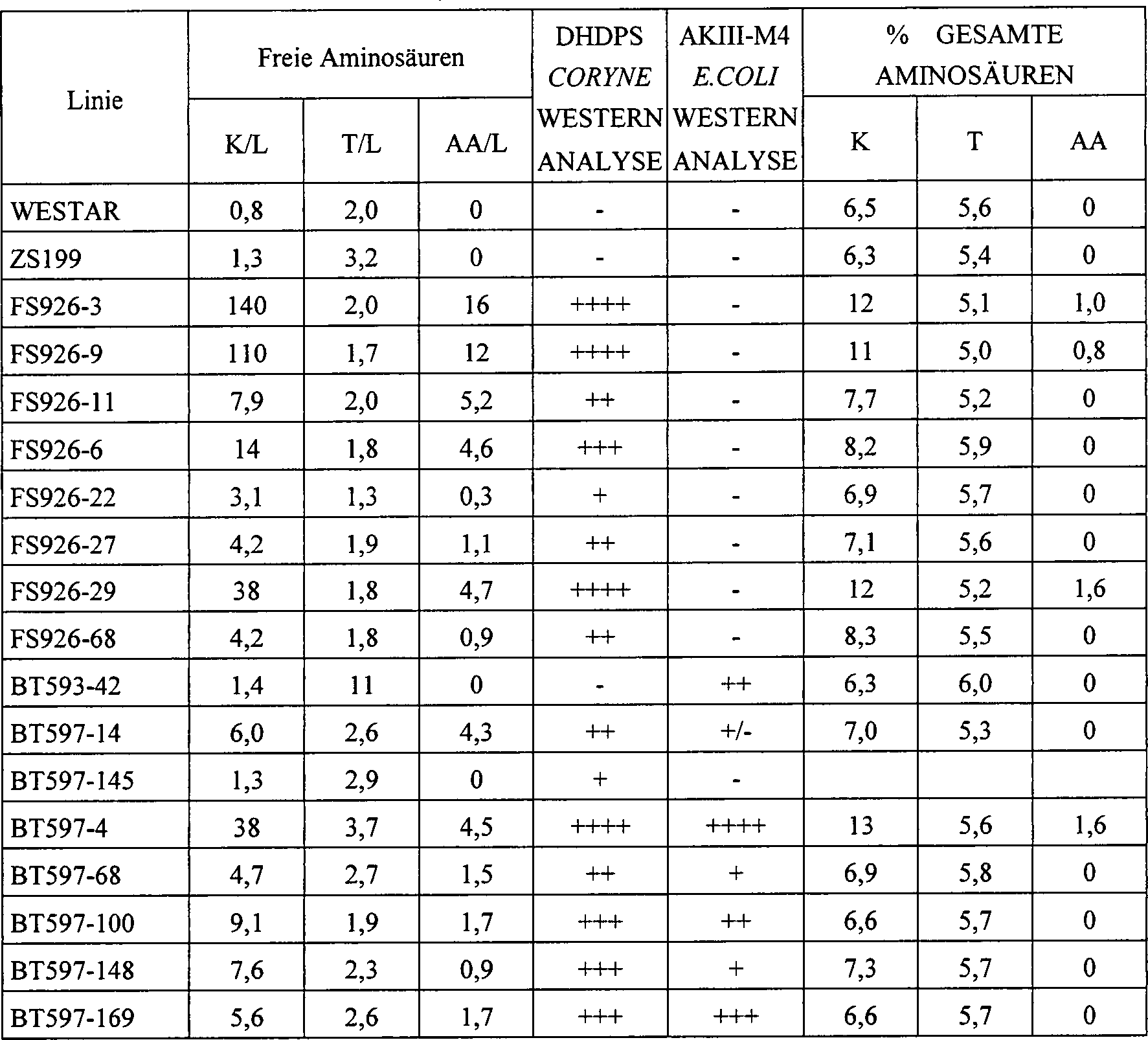
- AA ist α-Aminoadipinsäure
-
BEISPIEL 6
-
TRANSFORMATION VON SOJABOHNEN
MIT DEN CHIMÄREN
GENEN PHASEOLINPROMOTOR/CTS/CORDAPA UND PHASEOLINPROMOTOR/CTS/LYSC-M$
-
Die
chimären
Genkassetten Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region plus Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3', (Beispiel 4)
wurden in den Sojabohnentransformationsvektor pBT603 insertiert
(6A). Dieser Vektor weist ein Sojabohnentransformations-Markergen
auf, das aus dem 35S-Promotor
aus Blumenkohlmosaikvirus besteht, das die Expression des E. coli-β-Glucornidase-(GUS)- Gens treibt [Jefferson
et al. (1986) Proc. Natl. Acad. Sci. USA 83:8447-8451], wobei die
Region Nr. 3' sich
in einem modifizierten pGEM9Z-Plasmid befindet.
-
Zum
Insertieren der Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3'-Region wurde
die Genkassette als Hind III-Fragment von 3,3 kb isoliert und in
Hind III insertiert, das mit pBT603 digeriert worden war, unter
Bildung des Plasmids pBT609. Bei diesem Vektor ist das chimäre Gen Phaseolin
5'-Region/cts/lysC-M4/Phaseolin 3'-Region in der entgegengesetzten
Orientierung vom 35S/GUS/Nr. 3'-Markergen
insertiert.
-
Die
chimäre
Genkassette Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region wurde
unter Anwendung von Oligonukleotidadaptoren zum Umwandeln der Hind
III-Stellen an jedem Ende zu BamH T-Stellen modifiziert. Die Genkassette
wurde dann als BamH I-Fragment von 2,7 kb isoliert und in BamH I
insertiert, das mit pBT609 digeriert worden war, unter Bildung des
Plasmids pBT614 (6B). Bei diesem Vektor sind
beide chimären
Gene, Phaseolin 5'-Region/cts/cordapA/Phaseolin
3'-Region plus Phaseolin
5'-Region/cts/lysC-M4/Phaseolin
3' in der gleichen
Orientierung insertiert und beide befinden sich in entgegengesetzter
Orientierung zum 35S/GUS/Nr. 3'-Markergen.
-
Das
Plasmid pBT614 wurde in Sojabohnen durch Transformation durch Agractus
Company (Middleton, WI) dem im Patent der Vereinigten Staaten Nr.
5015580 beschriebenen Verfahren gemäß eingeführt. Samen von fünf transformierten
Linien wurden erhalten und analysiert.
-
Es
war zu erwarten, dass die Transgene in den RI-Samen der transformierten
Pflanzen getrennt vorliegen würden.
Um Samen, die die Transformationsmarkergene trugen, zu identifizieren,
wurde ein kleines Stückchen
des Samens mit einer Rasierklinge abgeschnitten und in eine Vertiefung
einer wegwerfbaren Mikrotiterplatte aus Kunststoff hineingegeben.
Eine GUS-Assaymischung, die aus 100 mM NaH2PO4, 10 mM EDTA, 0,5 mM K4Fe
(CN)6, 0,1% Triton X-100, 0,5 mg/ml 5-Brom-4-chlor-3-indolyl-β-D-glucuonsäure bestand, wurde
zubereitet und 0,15 ml wurden in jede Mikrotitervertiefung eingegeben.
Die Mikrotiterplatte wurde 45 min lang bei 37 °C inkubiert. Die Entwicklung
von blauer Farbe zeigte die Expression von GUS in dem Samen an.
-
Vier
von fünf
transformierte Linien zeigten eine Segregation von ca. 3:1 für die GUS-Expression
(Tabelle 3). Dies zeigt, dass das GUS-Gen an einer einzigen Stelle
im Sojabohnengenom insertiert war. Der andere Transformant zeigte
eine Segregation von 9:1, was darauf hinweist, dass das GUS-Gen
an zwei Stellen insertiert worden war.
-
Ein
Mehl wurde aus einem Fragment einzelner Samen durch Mahlen zu einem
feinen Pulver zubereitet. Die gesamten Proteine wurden aus dem Mehl
durch Zusetzen von 1 mg zu 0,1 ml 43 mM Tris-HCl, pH-Wert 6,8, 1,7% SDS, 4,2% β-Mercaptoethanol,
8% Glycerin, Behandeln der Suspension in der Wirbelvorrichtung, Kochen
für 2-3
Minuten und nochmaligem Behandeln in der Wirbelvorrichtung extrahiert.
Die dabei gebildeten Suspensionen wurden 5 min lang bei Raumtemperatur
in einer Mikrofuge zentrifugiert, um teilchenförmige Substanzen zu entfernen,
und von jedem Extrakt wurden 10 μl
pro Bahn auf einem SDS-Polyacrylamidgel einem Arbeitsgang unterworfen,
wobei bakteriell hergestellte DHDPS oder AKIII als Größenstandard
diente. Die Proteine wurden dann elektrophoretisch auf eine Nitrocellulosemembran
geblottet. Die Membranen wurden den DHDPS- oder AKIII-Antikörpern in
einer Verdünnung
von 1:5000 bzw. 1:1000 des Kaninchenserums unter Anwendung des Standardprotokolls,
das von BioRad mit ihrem Immun-Blotkit bereitgestellt wurde, ausgesetzt. Auf
das Spülen
zum Entfernen von ungebundendem primärem Antikörper hin, wurden die Membranen
dem sekundären
Antikörper,
nämlich Esel-Anti-Kaninchen
Ig, das an Meerrettichperoxidase (Amersham) konjugiert worden war,
in einer Verdünnung
von 1:3000 ausgesetzt. Auf das Spülen zum Entfernen von ungebundenem sekundärem Antikörper hin,
wurden die Membranen einem Chemilumineszenzreagenz von Amersham
und Röntgenfilm
ausgesetzt.
-
Vier
von fünf
Transformanten exprimierten das DHDPS-Protein. Bei den vier Transformanten,
die DHDPS exprimieren, bestand eine ausgezeichnete Korrelation zwischen
der Expression von GUS und DHDPS in einzelnen Samen (Tabelle 3).
Aus diesem Grund sind die GUS- und DHDPS-Gene an der gleichen Stelle
im Sojabohnengenom integriert. Zwei von fünf Transformanten exprimierten
das AKIII-Protein
und es bestand wiederum eine ausgezeichnete Korrelation zwischen
der Expression von AKIII, GUS und DHDPS in einzelnen Samen (Tabelle
3). So sind bei diesen beiden Transformanten die GUS-, AKIII- und
DHDPS-Gene an der gleichen Stelle in das Sojabohnengenom integriert.
Ein Transformant exprimierte nur GUS in seinen Samen.
-
Um
die freie Aminosäurezusammensetzung
der Samen zu messen, wurden freie Aminosäure aus 8-10 Milligramm des
Mehls in 1,0 ml Methanol/Chloroform/Wasser extrahiert, die in einem
Verhältnis
von 12 Vol./5 Vol./3 Vol. (MCW) bei Raumtemperatur gemischt waren.
Die Mischung wurde in einer Wirbelvorrichtung behandelt und daraufhin
in einer Eppendorfzentrifuge ca. 3 min lang zentrifugiert; ca. 0,8
ml der überstehenden Flüssigkeit
wurden dekantiert. Dieser überstehenden
Flüssigkeit
wurden 0,2 ml Chloroform gefolgt von 0,3 ml Wasser zugegeben. Die
Mischung wurde in der Wirbelvorrichtung behandelt und dann in einer
Eppendorf-Mikrozentrifuge ca. 3 min lang zentrifugiert, die obere
wässrige
Phase, ca. 1,0 ml, wurde entfernt und in einem Savant Speed Vac-Konzentrator
getrocknet. Die Proben wurden in 6N Salzsäure, 0,4% β-Mercaptoethanol unter Stickstoff
24 h bei 110-120 °C
hydrolysiert; 1/4 der Probe wurde auf einem Beckman Aminosäureanalysator, Modell
6300, unter Anwendung der Nachsäulen-Ninhydrinerfassung
einem Arbeitsgang unterzogen. Die relativen Niveaus freier Aminosäure in den
Samen wurden als Verhältnisse
von Lysin oder Threonin zu Leucin verglichen, wobei Leucin so als
interner Standard verwendet wurde.
-
Es
bestand eine ausgezeichnete Korrelation zwischen Transformanten,
die Corynebacteria DHDPS-Protein exprimieren und denjenigen, die
höhere
Niveaus an freiem Lysin aufweisen. Es wurden 20-fache bis 120-fache
Erhöhungen
des Niveaus an freiem Lysin in Samen beobachtet, die Corynebacteria
DHDPS exprimieren. Ein hohes Niveau an Saccharopin, das auf einen
Lysinkatabolismus hinweist, wurde in Samen beobachtet, die hohe
Niveaus an Lysin enthielten.
-
Um
die gesamte Aminosäurezusammensetzung
von reifen Samen zu messen, wurden 1-1,4 Milligramm des Samenmehls
in 6 N Salzsäure,
0,4% β-Mercaptoethanol
unter Stickstoff 24 h lang bei 110-120 °C hydrolysiert;
1/50 der Probe wurde auf einem Aminosäureanalysator von Beckman,
Modell 6300 unter Anwendung der Postsäulenninhydrierungserfassung
einem Arbeitslauf unterzogen. Die Lysin- (und andere Aminosäure-) Niveaus
in den Samen wurden als Prozentsätze
der gesamten Aminosäuren
verglichen.
-
Es
bestand eine ausgezeichnete Korrelation zwischen Samen, die Corynebacteria
DHDPS-Protein exprimieren und denjenigen, die hohe Niveaus an Lysin
aufweisen. Es wurden Samen mit einer 5-35%-igen Erhöhung des
Lysinniveaus im Vergleich der untransformierten Kontrolle beobachtet.
In diesen Samen stellt Lysin 7,5-7,7% der gesamten Samenaminosäuren dar,
was wesentlich höher
ist, als bei irgendeinem vorher bekannten Sojabohnensamen.
-
-
-
Es
wurden achtzehn zusätzliche
transformierte Sojabohnenlinien erhalten. Einzelne Samen aus den Linien
wurden auf GUS-Aktivität
hin, wie oben beschrieben, analysiert und alle Linien wiesen GUS
positive Samen auf. Es wurde Mehl aus einzelnen Samen oder in manchen
Fällen
einer Ansammlung von mehreren Samen zubereitet und auf die Expression
von DHDPS- und AKIII-Proteinen durch Western-Blot hin untersucht. Siebzehn
der achtzehn Linien exprimierten DHDPS und fünfzehn der achtzehn exprimierten
AKIII. Wiederum bestand eine ausgezeichnete Korrelation zwischen
Samen, die GUS, DHDPS und AKIII exprimierten, was darauf hinweist,
dass die Gene in den transformierten Linien verknüpft sind.
-
Die
Aminosäurezusammensetzung
der Samen aus diesen Linien wurde wie oben beschrieben bestimmt.
Wiederum wiesen Samen, die Corynebacteria DHDPS-Protein exprimierten,
erhöhte
Niveaus an Lysin auf. Die Expression von DHDPS als solcher führte zu
Erhöhungen
des gesamten Samenlysins von 5% bis 40%. Die Expression von DHDPS
zusammen mit AKIII-M4 führt
zu Erhöhungen
des Lysin von mehr als 400%. Eine Zusammenfassung aller verschiedener
transformierten Linien ist in Tabelle 3A gezeigt.
-
-
-
P
zeigt an, dass die Samen vor der Mehlextraktion und dem Assay gepoolt
wurden
-
BEISPIEL 7
-
ISOLIERUNG EINES PFLANZENICHEN
LYSINKETOGLUTARATREDUKTASE-GENS
-
Die
Lysinketoglutaratreduktase-(LKR)Enzymaktivität ist in unreifem Endosperm
von sich entwickelnden Maissamen beobachtet worden [Arruda et al.
(1982) Plant Physiol. 69:988-989]. Die LKR-Aktivität steigt scharf mit dem Einsetzen
der Endospermentwicklung an, erreicht einen Höhepunkt ca. 20 Tage nach der
Bestäubung
und nimmt dann ab [Arruda et al. (1983) Phytochemistry 22:2687-2689].
-
Um
das Mais LKR-Gen zu klonieren, wurde RNA aus sich entwickelnden
Samen 19 Tage nach der Bestäubung
isoliert. Diese RNA wurde an Clontech Laboratories Ine., (Palo Alto,
CA) zur genau zugeschnittenen Synthese einer cDNA-Bibliothek im
Vektor Lambda Zap II geschickt. Die Umwandlung der Lambda Zap II-Bibliothek
in eine Phagemidbibliothek und daraufhin in eine Plasmidbibliothek
wurde dem vom Clontech bereitgestellten Protokoll gemäß durchgeführt. Nach
der Umwandlung in eine Plasmidbibliothek tragen die erhaltenen gegen
Ampicillin resistenten Klone die cDNA-Insertion im Vektor pBluescript
SK(–).
Die Expression der cDNA steht unter der Kontrolle des lacZ-Promotors
am Vektor.
-
Es
wurden zwei Phagemidbibliotheken unter Anwendung der Mischungen
von Lambda Zap II-Phage und
der filamentösen
Helferphage von 100 μl
zu 1 μl
gebildet. Es wurden zwei zusätzliche
Bibliotheken unter Anwendung von Mischungen von 100 μl Lambda
Zap II zu 10 μl
Helferphage und 20 μl
Lambda Zap II 10 μl Helferphage
gebildet. Die Titer der Phagemidzubereitungen waren ähnlich,
gleichgültig,
welche Mischung verwendet wurde, und es lagen ca. 2 × 103 gegen Ampicillin resistente Transfektanden
pro ml beim E. coli-Stamm XLI-Blau als Wirt und ca. 1 × 103 bei DE126 (vergleiche unten) als Wirt vor.
-
Um
Klone auszuwählen,
die das LKR-Gen trugen, wurde ein spezifisch entworfener E. coli-Wirt, DE126,
konstruiert. Die Konstruktion von DE126 erfolgte in mehreren Stufen.
- (1) Ein verallgemeinerter transduzierender
Stamm von Coliphage Plvir wurde durch Infizieren einer Kultur von
TST1 [F–,
araD139, delta(argF-lac)205, flb5301, ptsF25, relAl, rpsL150, malE52::Tn10,
decoCl, λ–]
(E. coli Genetic Stock Center #6137) unter Zuhilfenahme einer Standardmethode
(bezüglich
der Methoden vergleiche J. Miller, Experiments in Molecular Genetics
(Versuche in der molekularen Genetic)) hergestellt.
- (2) Dieser Phagenstamm wurde als Donator in einer Transduktionskreuzung
(bezüglich
der Methode vergleiche J. Miller, Experiments in Molecular Genetics
(Versuche in der molekularen Genetic)) mit dem Stamm GIF106M1 [F–,
arg-, ilvA296, lysC1001, thrA1101, metL1000, λ–,
rpsL9, malT1, xyl-7, mtl-2, thil(?), supE44(?)] E. coli Genetic
Stock Center #5074) als Empfänger
verwendet. Es wurden Rekombinante an reichem Medium [mit DAP ergänztes L],
das das Antibiotikum Tetracyclin enthielt, ausgewählt. Das
Transposon Tn10, das Resistenz gegen Tetracyclin verleiht, wird
in das malE-Gen des Stamms TST1 insertiert. Gegen Tetracyclin resistente
Transduktanten, die aus dieser Kreuzung deriviert sind, enthalten
wahrscheinlicherweise bis zu 2 min des E. coli-Chromosoms in der
Nähe von
malE. Die Gene malE und lysC werden weniger als 0,5 Minuten, also
absolut innerhalb des Transduktionsabstands, getrennt,
- a. 200 gegen Tetracyclin resistente Transduktanten, wurden gründlich phänotypiert;
es wurden geeignete Fermentations- und Ernährungscharakterzüge aufgezeichnet.
Dem Empfängerstamm
GIF106M1 mangeln Aspartokinaseisozyme aufgrund von Mutationen in
thrA, metL und lysC vollständig
und er erfordert deshalb die Anwesenheit von Threonin, Methionin,
Lysin und Mesodiaminopimelinsäure
(DAP) zum Wachstum. Es wäre
zu erwarten, dass die Transduktanten, die lysC+ mit
malE::Tn10 aus TST1 geerbt hatten auf einem minimalen Medium wachsen,
das Vitamin B1, L-Arginin, L-Isoleucin und L-Valin zusätzlich zur
Glukose enthält,
die als Kohlenstoff- und Energiequelle dient. Außerdem exprimieren Stämme, die
die genetische Konstitution von lysC+, metL-
und thrA- aufweisen, nur die für
Lysin empfindliche Aspartokinase. Daher sollte der Zusatz von Lysin
zum Minimalmedium das Wachstum des lysC+-Rekombinanten dadurch verhindert, dass
es zum Verhungern für
Threonin, Methionin und DAP führt.
Von den 200 untersuchten gegen Tetracyclin resistenten Transduktanten,
wuchsen 49 auf dem Minimalmedium, das von Threonin, Methionin und DAP
frei war. Außerdem
wurden alle 49 durch Zusetzen von L-Lysin zum Minimalmedium inhibiert.
Einer dieser Transduktanten wurde DE125 genannt. DE125 hat den Phänotyp von
Tetracyclinresistenz, Wachstumserfordernisse für Arginin, Isoleucin und Valin
und Empfindlichkeit gegen Lysin. Der Genotyp dieses Stamms ist F-
malE52::Tn10 arg- ilvA296 thrA1101 metL1000 lambda-rpsL9 ma1T1 xyl-7
mtl-2 thil(?) supE44(?).
- b. Dieser Schritt involviert die Herstellung eines männlichen
Derivats des Stamms DE125. Der Stamm DE125 wurde mit dem männlichen
AB1528 [F'16/delta
(gpt-proA)62, lacY1 oder lacZ4, glnV44, galK2 rac–(?),
hisG4, rflid1, mgl-51, kdgK51(?), ilvC7, argE3, thi-1] (E. coli
Genetic Stock Center #1528) durch die Konjugationsmethode gepaart.
F'16 trägt das ilvGMEDAYC-Gencluster.
Die beiden Stämmne
wurden auf reichem Medium, das das Wachstum jedes Stamms erlaubt,
quer ausgestrichen. Nach der Inkubation wurde die Platte mit Bezug
auf ein synthetisches Medium, das Tetracyclin, Arginin, Vitamin
B und Glukose enthielt, replikationsplattiert. DE125 kann auf diesem
Medium nicht wachsen, weil es Isoleucin nicht synthetisieren kann.
Das Wachstum von AB 1528 wird durch Einschluss des antibiotischen
Tetracyclins und das Weglassen von Prolin und Histidin aus dem synthetischen
Medium verhindert. Ein Fleck von Zellen wuchs auf diesem selektiven
Medium.
-
Diese
rekombinanten Zellen machten eine einzige Kolonieisolation auf dem
gleichen Medium durch. Der Phänotyp
eines Klons wurde dahingehend bestimmt, dass es sich um Ilv+, Arg–, TetR, gegen Lysin
empfindlichen männlichen
spezifischen (MS2)-empfindlichen Phagen handelt, was mit der einfachen Übertragung von
F'16 von AB1528
auf DE125 übereinstimmt.
Dieser Klon wurde DE126 genannt und weist den Genotyp F'16/malE52::Tn10,
arg–,
ilvA296, thrA1101, metL100, lysC+, λ–,
rpsL9, malT1, xyl-7, mtl-2, thi-1?, supE44?, auf. Er wird durch
20 μg L-Lysin
in einem synthetischen Medium inhibiert.
-
Um
Klone aus der Mais cDNA-Bibliothek auszuwählen, die das LKR-Gen tragen,
wurden 100 μl
der Phagemidbibliothek mit 100 μl
einer Übernachtkultur
von DE126, die in L-Bouillon gezüchtet
worden war, gemischt und die Zellen wurden auf synthetisches Medium,
das Vitamin B1, L-Arginin, Glukose als Kohlenstoff- und Energiequellen,
100 μg/ml
Ampicillin und L-Lysin in einer Menge von 20, 30 oder 40 μg/ml enthielt,
aufplattiert. Es wurden vier Platten mit jeder der drei verschiedenen
Lysinkonzentrationen zubereitet. Es wurde erwartet, dass die Menge
an Phagemid und DE126-Zellen ca. 1 × 105 gegen
Ampicillin resistente Transfektanten pro Platte ergäbe. Zehn
bis dreißig
gegen Lysin resistente Kolonien wuchsen pro Platte (ca. 1 gegen
Lysin resistente pro 5000 gegen Ampicillin resistente Kolonien).
-
Plasmid-DNA
wurde aus 10 unabhängigen
Klonen isoliert und erneut in DE126 transformiert. Sieben der zehn
DNA ergaben gegen Lysin resistente Klone, was beweist, dass der
gegen Lysin resistente Charakter auf dem Plasmid getragen wurde.
Mehrere der klonierten DNA wurden sequenziert und biochemisch charakterisiert.
Es wurde gefunden, dass die insertierten DNA-Fragmente vom E. coli-Genom
anstatt einer Mais-cDNA derivierten, was anzeigt, dass die cDNA-Bibliothek,
die von Clontech bereitgestellt worden war, kontaminiert war. Eine
neue cDNA-Bibliothek wird deshalb zubereitet und wie oben beschrieben
einem Screenen unterzogen.
-
BEISPIEL 8
-
KONSTRUKTION
VON SYNTHETISCHEN GENEN IM EXPRESSIONSVEKTOR pSK5
-
Um
die Konstruktion und Expression der unten beschriebenen synthetischen
Gene zu erleichtern, war es notwendig, einen Plasmidvektor mit folgenden
Attributen zu konstruieren:
- 1. Keine Ear I
Restriktionsendonukleasestellen derart, dass die Insertion von Sequenzen
eine einzige Stelle erzeugen würde.
- 2. Enthaltend ein Tetracyclinresistenzgen, um Verluste an Plasmid
während
des Wachstums und der Expression von toxischen Proteinen zu vermeiden.
- 3. Enthaltend ca. 290 bp vom Plasmid pBT430, einschließlich dem
T7-Promotor und Terminatorsegment zum Exprimieren insertierter Sequenzen
in E. coli.
- 4. Enthaltend einzige EcoR I- und Nco I-Restriktionsendonukleaseerkennungsstellen
in der richtigen Position hinter dem T7-Promotor, um die Insertion
von Oligonukleotidsequenzen zu erlauben.
-
Um
die Attribute 1 und 2 zu erhalten, haben die Anmelder das Plasmid
pSKI verwendet, das ein spontaner Mutant von pBR322 war, wobei das
Ampicillingen und die Ear I-Stelle in der Nähe des Gens deletiert worden
waren. Das Plasmid pSKI behielt das Tetracyclinresistenzgen, die
einzigen EcoR I Restriktionsstellen an Base 1 und eine einzige Ear
I-Stelle an Base 2353 bei. Zum Entfernen der Ear I-Stelle an Base
2353 von pSKI, wurde eine Polymerasekettenreaktion (PKR) unter Anwendung
von pSKI als Matrize durchgeführt.
Ca. 10 Femtomole pSK1 wurden mit 1 μg von jedem der Oligonukleotide
SM70 und SM71 gemischt, die auf einem ABI1306B-DNA-Synthetisierer
unter Anwendung der Vorgehensweisen des Herstellers synthetisiert
worden waren.
SM70 5'-CTGACTCGCTGCGCTCGGTC
3' SEQ ID NO:16
SM71
5'-TA7TTTCTCCTTACGCATCTGTGC-3' SEQ ID NO:17
-
Die
Primierstellen dieser Oligonukleotide an der pSKI-Matrize sind in 7 gezeigt.
Die PKR wurde unter Anwendung eines Perkin-Elmer Cetus-Kits (Emeryville,
CA) den Anleitungen der Verkäuferfirma
entsprechend auf einem Thermozykler, der von der gleichen Firma
hergestellt worden war, durchgeführt.
Die 25 Zyklen fanden für
1 min bei 95 °C,
2 min bei 42 °C
und 12 min bei 72 °C
statt. Die Oligonukleotide waren so entworfen, dass sie die Replikation
des gesamten pSKI-Plasmids ausschließlich eines Fragments von 30
b um die Ear I-Stelle herum (man vergleiche 7) primieren.
10 Mikroliter des Reaktionsprodukts von 100 μl wurden auf einem 1%-igen Agarosegel
einem Arbeitslauf unterworfen und mit Ethidiumbroimid gefärbt, um
eine Bande von ca. 3,0 kb zum Vorschein zu bringen, die der vorausgesagten
Größe des replizierten
Plasmids entsprach.
-
Der
Rest der PKR-Reaktionsmischung (90 μl) wurde mit 20 μl von 2,5
mm Desoxynukleotidtriphosphaten (dATP, dTTP, dGTP und dCTP) gemischt,
30 Einheiten Klenowenzym wurde zugegeben und die Mischung wurde
bei 37 °C
30 min lang, gefolgt von 65 °C
für 10
min inkubiert. Das Klenowenzym wurde verwendet, um die zerfetzten
Enden, die durch die PKR gebildet worden waren, zu füllen. Die
DNA wurde durch Ethanol ausgefällt,
mit 70% Ethanol gewaschen, unter Vakuum getrocknet und erneut in
Wasser suspendiert. Die DNA wurde dann mit T4 DNA-Kinase in Gegenwart
von 1 mM ATP in Kinasepuffer behandelt. Diese Mischung wurde dann
30 min lang bei 37 °C,
gefolgt von 10 min bei 65 °C,
inkubiert. 10 μl
der kinasierten Zusammensetzung wurden 2 μl 5X Ligationspuffer und 10
Einheiten der T4 DNA-Ligase zugesetzt. Die Ligation wurde 16 h lang bei
15 °C durchgeführt. Auf
die Ligation hin wurde die DNA halbiert und eine Hälfte mit
Ear I-Enzym digeriert. Die Klenow-, Kinase-, Ligations- und Restriktionsendonukleasereaktionen
wurden wie bei Sambrook et al., [Molecular Cloning, A Laboratory
Manual, 2. Ausgabe (1989) Cold Spring Harbor Laboratory Press] beschrieben,
durchgeführt.
Klenow-, Kinase-, Lipase- und die meisten Restriktionsendonukleasen
wurden von BRI erworben. Einige Restriktionsendonukleasen wurden
von NEN Biolabs (Beverley, MA) oder Boehringer Mannheim (Indianapolis,
IN) erworben. Beide ligierten DNA-Proben wurden getrennt in kompetente
JM 103-[supE thi del (lac-proAB) F' (traD36 porAB, lacIq lacZ del M15]
Restriktion minus Zellen] unter Anwendung der CaCl2-Methode,
wie bei Sambrook et al [Molecular Cloning, A Laboratory Manual,
2. Ausgabe (1989) Cold Spring Harbor Laboratory Press] beschrieben,
transformiert und auf Medien plattiert, die 12,5 μ/ml Tetracyclin enthielten.
Mit oder ohne Ear I-Digestion, wurde die gleiche Anzahl von Transformanten
gewonnen, was darauf hinweist, dass die Ear I-Stelle von diesen
Konstrukten entfernt worden war. Die Klone wurden durch Zubereiten von
DNA durch das alkalische Lyse-Miniprep-Verfahren, wie bei Sambrook
et al. [Molecular Cloning, A Laboratory Manual, 2. Ausgabe (1989)
Cold Spring Harbor Laboratory Press] beschrieben, gefolgt von einer
Restriktionsendonukleasedigestionsanalyse, gescreent. Ein einziger
Klon wurde gewählt,
der gegen Tetracyclin widerstandsfähig war und keine Ear I-Stellen
enthielt. Dieser Vektor wurde pSK2 genannt. Die verbleibende EcoR
I-Stelle von pSK2 wurde durch Digerieren des Plasmids mit EcoR I
bis zum Abschluss, Füllen
der Enden mit Klenow und Ligieren zerstört. Ein Klon, der keine EcoR
I-Stelle enthielt, wurde pSK3 genannt.
-
Um
die obigen Attribute 3 und 4 zu erhalten, wurde das bakteriophage
T7 RNA-Polymerasepromotor/Terminatorsegment
vom Plasmid pBT430 (vergleiche Beispiel 2) durch PKR amplifiziert.
Die Oligonukleotidprimer SM78 (SEQ ID NO:18) und SM79 (SEQ ID NO:19)
wurden so konstruiert, dass sie ein 300b-Fragment von pBT430, das
die 77 Promotor/Terminatorsequenzen überspannt, primiert (vergleiche 7).
SM78
5'-TTCATCGATAGGCGACCACACCCGTCC-3' SEQ ID NO:18
SM79
5'-AATATCGATGCCACGATGCGTCCGGCG-3' SEQ ID NO:19
-
Die
PKR-Reaktion wurde wie oben beschrieben unter Anwendung von pBT430
als Matrize durchgeführt
und es wurde ein Fragment von 300 bp gebildet. Die Enden des Fragments
wurden unter Anwendung von Klenow-Enzym gefüllt und wie oben beschrieben
kinasiert. DNA aus dem Plasmid pSK3 wurde vollständig mit PvuII-Enzym digeriert
und dann mit alkalischer Kalbdarmphosphatase (Boehringer Mannheim)
zum Entfernen des 5'-Phosphats
behandelt. Der Vorgang war wie bei Sambrook et al. [Molecular Cloning,
A Laboratory Manual, 2. Ausgabe (1989) Cold Spring Harbor Laboratory
Press] beschrieben. Die geschnittene und phosphatasierte pSK3-DNA
wurde durch Ausfällen
mit Ethanol gereinigt und ein Teil wurde in einer Ligationsreaktion
mit dem durch PKR gebildeten Fragment, das die T7-Promotorsequenz
enthält,
gereinigt. Die Ligationsmischung wurde zu JM103 [supE thi del (lac-proAB)
F'[traD36 porAB,
lacIq lacZ del M15] Restriktion minus] transformiert und gegen Tetracyclin
widerstandsfähige
Kolonien wurden gescreened. Plasmid-DNA wurde durch die alkalische
Lyse-Miniprepmethode
zubereitet und es wurde eine Restriktionsendonukleaseanalyse durchgeführt, um
die Insertion und Orientierung des PKR-Produkts zu erfassen. Zwei
Klone wurden zur Sequenzanalyse ausgewählt: beim Plasmid pSKS war
das Fragment in der in 7 gezeigten Orientierung vorhanden.
Die an alkalischer denaturierter doppelstrangiger DNA unter Anwendung
von Sequenase® T7-DNA-Polymerase (US Biochemical
Corp) und des vom Hersteller vorgeschlagenen Protokolls durchgeführte Sequenzanalyse
brachte zum Vorschein, dass pSKS keine PKR-Replikationsfehler innerhalb
der T7-Promotor/Terminatorsequenz
aufwies.
-
Die
Strategie zur Konstruktion wiederholter synthetischer Gensequenzen
auf der Basis der EAR I-Stelle
ist in 8 gezeigt. Der erste Schritt
bestand aus der Insertion einer Oligonukleotidsequenz, die ein Basengen
von 14 Aminosäuren
kodiert. Diese Oligonukleotidinsertion enthielt eine einzige Ear
I-Restriktionsstelle
für die
darauffolgende Insertion von Oligonukleotiden, die ein oder mehrere
Heptadwiederholungen kodieren und fügte eine einzige Asp 718-Restriktionsstelle
zur Verwendung beim Übertragen
von Gensequenzen auf Pflanzenvektoren hinzu. Die überhängenden
Enden des Oligonukleotidsatzes erlaubten die Insertion in die einzigen Nco
I und EcoR I-Stellen des Vektors pSKS.
-
-
DNA
aus dem Plasmid pSKS wurde vollständig mit Nco I und EcoR I-Restriktionsendonukleasen
digeriert und durch Agarosegelelektrophorese gereinigt. Gereinigte
DNA (0,1 μg)
wurde mit 1 μg
jeweils des Oligonukleotids SM80 (SEQ ID NO:14) und SM81 (SEQ ID
NO:13) gemischt und ligiert. Die Ligationsmischung wurde in den
E. coli-Stamm JM103 [supE thi del (lac-proAB) F' (traD36 porAB, IacIq lacZ del M15]
Restriktion minus] transformiert und gegen Tetracyclin widerstandsfähige Transformanten
wurden durch Hochgeschwindigkeitsplasmid-DNA-Zubereitungen, gefolgt
von der Restriktionsverdaungsanalyse gescreened. Ein Klon wurde
gewählt,
der jeweils eine von Ear I, Nco I, Asp 718 und EcoR I-Stellen aufwies,
was die richtige Insertion der Oligonukleotide anzeigt. Dieser Klon
wurde pSK6 genannt (9). Das Sequenzieren der Region
von DNA auf den T7-Protor hin bestätigte die Insertion von Oligonukleotiden
der erwarteten Sequenz.
-
Repetitive
Heptadkodiersequenzen wurden dem Basengenkonstrukt des oben Beschriebenen
durch Bilden von Oligonukleotidpaaren hinzugefügt, die direkt in die einzige
Ear I-Stelle des Basengens ligiert werden konnten. Die Oligonukleotide
SM84 (SEQ ID NO:23) und SM85 (SEQ ID NO: 24) kodieren Wiederholungen
des SSPS-Heptads. Die Oligonukleotide SM82 (SEQ ID NO:25) und SM83
(SEQ ID NO:26) kodieren Wiederholungen des SSP7-Heptads.
-
-
Es
wurden Oligunkleotidsätze
ligiert und gereinigt, um DNA-Fragmente, die multiple Heptadwiederholungen
kodieren, zur Insertion in den Expressionvektor zu erhalten. Oligonukleotide
aus jedem Satz von insgesamt ca. 2 μg wurden kinasiert und 2 h lang
bei Raumtemperatur ligiert. Die ligierten Multimere der Oligonukleotidsätze wurden
auf einem 18%-igen nichtdenaturierenden Polyacrylamidgel von 20 × 20 × 0,015
cm (Acrylamid: Bisacrylamid = 19:1) getrennt. Multimere Formen die
sich auf dem Gel als 168 bp (8n) oder größer abtrennen, wurden durch
Schneiden eines kleinen Stücks
Polyacrylamid, das die Bande enthält, in feine Stückchen,
Zugeben von 1,0 ml 0,5 M Ammoniumacetat, 1 mM EDTA (pH-Wert 7,5) und Rotieren
der Röhre
bei 37 ° über Nacht
gereinigt. Das Polyacrylamid wurde durch Zentrifugieren heruntergeschleudert,
1 μg tRNA
wurde der überstehenden
Substanz zugegeben, die DNA-Fragmente
wurden mit 2 Volumen Ethanol bei –70 ° ausgefällt, mit 70% Ethanol gewaschen,
getrocknet und erneut in 10 μl
Wasser suspendiert.
-
Zehn
Mikrogramm pSK6 DNA wurden vollständig mit Ear I-Enzym digeriert
und mit alkalischer Kalbsdarmphosphatase behandelt. Die geschnittene
und phosphatasierte Vektor-DNA wurde auf die Elektrophorese hin
in einem Agarosegel von niedrigem Schmelzpunkt durch Ausschneiden
der bandenförmigen
DNA, Verflüssigen
der Agarose bei 55 ° und
Reinigen über
NACS PREPACTM Säulen (BRL) den vom Hersteller
vorgeschlagenen Vorgehensweisen entsprechend, isoliert. Ca 0,1 μg gereinigte,
durch Ear I digerierte und mit Phosphatase behandelte pSK6-DNA wurde
mit 5 μl
der mit Gen gereinigten multimeren Oligonukleotidsätze gemischt und
ligiert. Die ligierte Mischung wurde in den E. coli-Stamm JM103
(supE thi del (lac-proAB) F' (traD36
porAB, lacIq lacZ del M15) Restriktion minus] transformiert und
es wurden gegen Tetracyclin widerstandsfähige Kolonien ausgewählt. Die
Klone wurden durch Restriktionsdigeste von Schnellplasmidprep-DNA
gescreent, um die Länge
der insertierten DNA zu bestimmen. Restriktionsendonukleaseanalysen
wurden gewöhnlich
durch Digerieren der Plasmid-DNA mit Asp 718 und Bgl II, gefolgt
vom Trennen von Fragmenten auf 18%-igen nichtdenaturierten Polyacrylamidgelen,
gereinigt. Beim Sichtbarmachen der Fragmente mit Ethidiumbromid
kam zum Vorschein, dass ein Fragment von 150 bp gebildet wurde,
wenn nur das Basengensegment vorlag. Insertionen der Oligonukleotidfragmente
erhöhten
diese Größe um Mehrfache
von 21 Basen. Aus diesem gescreenten Material wurden mehrere Klone
für die
DNA-Sequenzanalyse und Expression von kodierten Sequenzen in E.
coli ausgewählt.
Die ersten und letzten SSPS-Heptade, die die Sequenz jedes Konstrukts
flankieren, stammen aus dem oben beschriebenen Basengen. Die Insertionen
sind durch Unterstreichen (Tabelle 4) angegeben.
-
TABELLE
4 – SEQUENZ
DURCH HEPTAD
-
Da
die Gelreinigung der oligomeren Formen der Oligonukleotide nicht
die erwartete Anreicherung längerer
(d.h. > 8n) Insertionen
ergab, wandten die Anmelder eine andere Vorgehensweise für eine darauffolgende
Runde von Insertionskonstruktionen an. Für diese Serie von Konstrukten
wurden vier weitere Sätze
von Oligonukleotiden gebildet, die jeweils SSP 8, 9, 10 und 11 Aminosäuresequenzen
kodieren:
-
Das
folgende HPLC-Verfahren wurde zum Reinigen multimerer Formen der
Oligonukleotidsätze
nach dem Kinasieren und Ligieren der Oligonukleotide, wie oben beschrieben,
angewendet. Es wurde eine Chromatographie auf einem Hewlett Packard
Flüssigchromatographeninstrument,
Modell 1090M, durchgeführt.
Die Ausflussextinktion wurde bei 260 nm überwacht. Ligierte Oligonukleotide
wurden 5 min lang bei 12.000 xg zentrifugiert und in eine TSK DEAE-NPR-Ionenaustauschsäule von
2,5 μ (Innendurchmesser
35 cm × 4,6
mm), die mit einem In-line-Filter von 0,5 μ (Supelco) ausgestattet war,
zentrifugiert. Die Oligonukleotide wurden aufgrund der Länge unter
Anwendung einer Gradientenelution und einer mobilen Zwei-Pufferphase
[Puffer A: 25 mM Tris-C., pH-Wert 9,0 und Puffer B: Puffer A + 1M
NaCl] getrennt. Beide Puffer A und B wurden vor der Anwendung durch
Filter von 0,2 μ hindurchgeführt.
-
Es
wurde folgendes Gradientenprogramm mit einer Fließrate von
1 ml pro min bei 30 °C
angewendet.
-
-
Bruchteile
(500 μl)
wurden zwischen 3 min und 9 min aufgefangen. Die Fraktionen, die
der Länge
zwischen 120 bp und 2000 bp entsprachen, wie durch die Kontrolltrennungen
von Restriktionsdigesten von Plasmid-DNA bestimmt, werden gepoolt.
-
Die
4,5 ml gepolter Fraktionen für
jeden Oligonukleotidsatz wurden durch Zugeben von 10 μg tRNA und
9,0 ml Ethanol ausgefällt,
zweimal mit 70% Ethanol gespült
und erneut in 50 μl
Wasser suspendiert. Zehn Mikroliter der erneut suspendierten, durch
HPLC gereinigten Oligonukleotide wurden 0,1 μg der oben beschriebenen, von
Ear I geschnittenen, phosphatasierten pSK6-DNA zugegeben und über Nacht
bei 15 °C
ligiert. Alle sechs oben beschriebenen Oligonukleotidsätze, die
kinasiert und selbstligiert, jedoch nicht durch Gel oder HPLC gereinigt
worden waren, wurden ebenfalls in einzelnen Ligationsreaktionen
mit dem pSK6-Vektor verwendet. Die Ligationsmischungen wurden in
den E. coli-Stamm
DHSa [supE44 del lacU169 (phi 80 lacZ del M15) hsdR17 recA] endA1
end196 thil relA1] transformiert und es wurden gegen Tetracyclin
widerstandsfähige Kolonien
ausgewählt.
Die Anmelder entschlossen sich, den DH5α- [supE44 del lacU169 (phi 80
lacZ del M15) hsdR17 recA1 endA1 gyr196 thil relA1] Stamm für alle darauffolgenden
Arbeiten zu wählen,
weil dieser Stamm eine sehr hohe Transformationsrate aufweist und
recA- ist. Der recA-Phänotyp
eliminiert Bedenken, dass diese repetitiven DNA-Struktwen Substrate
für eine
homologe Rekombination sein könnten,
die zur Deletion multimerer Sequenzen führt.
-
Es
wurden Klone wie oben beschrieben gescreened. Mehrere Klone wurden
ausgewählt,
um Insertionen jedes der sechs Oligonukleotidsätze darzustellen. Die ersten
und letzten SSP-Heptade, die die Sequenz flankieren, stellen die
Basengensequenz dar. Die insertierten Sequenzen sind unterstrichen.
Die Klonzahlen, die den Buchstaben „H" einschließen, bezeichnen durch HPLC
gereinigte Oligonukleotide (Tabelle 5).
-
-
Der
Verlust der ersten Basengenwiederholung in Klon 82-4 kann aus der
homologen Rekombination zurischen den Basengenwiederholungen 5.5
herrühren,
bevor der Vektor pSK6 in den recA-Stamm überführt worden war. Das HPLC-Verfahren
verbesserte die Insertion langer multimerer Formen der Oligonukleotidsätze in das
Basengen nicht, sondern diente als effiziente Reinigung der ligierten
Oligonukleotide.
-
Es
wurden Oligonukleotide konstruiert, die Mischungen der SSP-Sequenzen
kodieren und die die Codonanwendung soweit wie möglich variierten. Dies erfolgte,
um die Möglichkeit
der Deletion repetitiver Insertionen durch Rekombination zu reduzieren,
sobald die synthetischen Gene zu Pflanzen transformiert wurden, und
um die Länge
der konstruierten Gensegmente zu vergrößern. Diese Oligonukleotide
kodieren vier Wiederholungen von Heptadkodiereinheiten (20 Aminosäurereste)
und können
an der einzigen Ear I- Stelle
in irgendeinem der vorher konstruierten Klone insertiert werden.
SM96 und SM97 kodieren SSP(5)4, SM98 und SM99
kodieren SSP(7)4 und SM100 plus SM101 kodieren
SSP8.9.8.9.
-
-
DNA
aus den Klonen 82-4 und 84-H3 wurden vollständig mit Ear I-Enzym digeriert,
mit Phosphatase behandelt und mit Gel gereinigt. Ca. 0,2 μg dieser
DNA wurden mit 1,0 μg
jedes der Oligonukleotidsätze
SM96 und SM97, SM98 und SM99 oder SM100 und SM101 gemischt, die
vorher kinasiert worden waren. Die DNA und Oligonukleotide wurden über Nacht
ligiert und dann wurden die Ligationsmischungen in den E. coli-Stamm DHSa
transformiert. Gegen Tetracyclin widerstandsfähige Kolonien wurden wie oben
auf das Vorliegen der Oligonukleotidinsertionen hin gescreened.
Es wurden Klone für
die Sequenzanalyse aufgrund ihrer Restriktionsendonukleasedigestionsbildern
(Tabelle 6) gewählt.
-
-
Insertierte
Oligonukleotidsegmente sind unterstrichen.
-
Der
Klon 2-9 wurde aus den Oligonukleotiden SM100 (SEQ ID NO:71) und
SM101 (SEQ ID NO:72) deriviert, die in die Ear I-Stelle des Klons
82-4 ligiert worden waren (vergleiche oben). Der Klon 3-5 (SEQ ID NO:78)
wurde aus der Insertion der ersten 22 Basen des Oligonukleotidsatzes
SM96 (SEQ ID NO:65) und SM97 (SEQ ID NO:66) in die Ear I-Stelle
des Klons 82-4 (SEQ ID NO:53) deriviert. Diese teilweise Insertion kann
ein unrichtiges Annealing dieser äußerst repetitiven Oligos widerspiegeln.
Der Klon 5-1 (SEQ ID NO:76) wurde aus den Oligonukleotiden SM98
(SEQ ID NO:68) und SM99(SEQ ID NO:69) deriviert, die in die Ear I-Stelle
des Klons 84-H3 (SEQ ID NO:55)ligiert worden waren (vergleiche Abschnitt).
-
STRATEGIE II
-
Eine
zureite Strategie für
die Konstruktion synthetischer Gensequenzen wurde durchgeführt, um
eine größere Flexibilität sowohl
bei der DNA als auch der Aminosäuresequenz
zu gestatten. Diese Strategie ist in 10 und 11 dargestellt. Der erste Schritt bestand
aus der Insertion einer Oligonukleotidsequenz, die ein Basengen
von 16 Aminosäuren
kodiert, in den Originalvektor pSKS. Diese Oligonukleotidinsertion
enthielt eine einzige Ear I-Stelle, wie im vorherigen Basengenkonstrukt,
zur Verwendung bei darauffolgenden Insertion von Oligonukleotiden,
die eine oder mehrere Heptadwiederholungen kodieren. Das Basengen
umfasst auch eine BspH I-Stelle am 3'-Terminus. Die überhängenden Enden dieser Spaltungsstelle
sind so konstruiert, dass sie „in
frame"-Proteinfusionen
unter Anwendung von Nco I-Überhängungsenden
gestatten. Aus diesem Grund können
Gensegmente durch Anwendung der in 11 beschriebenen
Duplikationsschemen multipliziert werden. Die überhängenden Enden des Oligonukleotidsatzes
erlauben die Insertion in die einzigen Nco 1 und EcoR I-Stellen
des Vektors pSKS.
-
-
Der
Oligonukleotidsatz wurde in den pSKS-Vektor, wie oben unter Strategie
I beschrieben, insertiert. Das dabei entstehende Plasmid wurde mit
PSK34 bezeichnet.
-
Oligonukleotidsätze, die
35 Aminosäure-„Segmente" kodieren, wurden
in die einzige Ear I-Stelle des pSK34-Basengens unter Anwendung
von Verfahren wie oben beschrieben, ligiert. In diesem Fall wurden
die Oligonukleotide nicht durch Gel oder HPLC gereinigt, sondern
einfach einem Annealing unterworfen und in den Ligationsreaktionen
verwendet. Die folgenden Oligonukleotidsätze wurden verwendet:
-
Es
wurden Klone auf das Vorliegen der insertierten Segmente hin durch
Restriktionsdigestion gefolgt vom Trennen von Fragmenten auf 6%
Acrylamidgelen gescreened. Die richtige Insertion von Oligonukleotiden wurde
durch DNA-Sequenzanalysen bestätigt.
Klone, die jeweils die Segmente 3, 4 und 5 enthielten, wurden pSKseg3,
pSKseg4 und pSKsegS genannt.
-
Diese „Segment"-Klone wurden bei
einem Duplikationsschema, wie in 11 gezeigt,
angewendet. 10 μg
des Plasmids pSKseg3 wurden vollständig mit Nhe I und BspH I digeriert
und das Fragment von 1503 bp wurde von einem Agarosegel unter Anwendung
der Whatmannpapiertechnik isoliert. Zehn μg des Plasmids pSKseg4 wurden
vollständig
mit Nhe I und Nco I digeriert und die Bande von 2109 bp durch Gel
isoliert. Gleiche Mengen dieser Fragmente wurden ligiert und Rekombinanten
auf Tetracyclin ausgewählt.
Es wurden Klone durch Restriktionsdigestionen gescreened und ihre
Sequenzen bestätigt.
Das dabei entstehende Plasmid wurde pSKseg34 genannt.
-
pSKseg34-
und pSKsegS-Plasmid-DNA wurden digeriert, Fragmente isoliert und
auf ähnliche
Weise wie oben unter Bildung eines Plasmids ligiert, das DNA-Sequenzen
enthielt, die das an die Segmente 3 und 4 fusionierte Segment 5
kodieren. Dieses Konstrukt wurde pSKseg534 genannt und kodiert die
folgende Aminosäuresequenz.
-
BEISPIEL 9
-
KONSTRUKTION VON CHIMÄREN SSP-GENEN
ZUR EXPRESSION IN DEN SAMEN VON PFLANZEN
-
Um
die in Beispiel 8 beschriebenen synthetischen Genprodukte in Pflanzensamen
zu exprimieren, wurden die Sequenzen in die Samenpromotorvektoren
CW108, CW109 oder ML113 (12) übertragen.
Die Vektoren CW108 und ML113 enthalten den Bohnenphaseolinpromotor
(von Base +1 bis Base –494)
und 1191 Basen der 3'-Sequenzen
aus Bohnenphaseolingen. CW109 enthält den Sojabohnen-β-Conglycininpromotor (von
Base +1 bis Base –619)
und die gleichen 1191 Basen von 3'-Sequenzen aus dem Bohnenphaseolingen. Diese
Vektoren wurden so konstruiert, dass sie das direkte Klonieren von
Kodiersequenzen in einzige Nco I- und Asp 718-Stellen gestatten.
Diese Vektoren bieten auch Stellen (Hind III oder Sal I) an den
5'- und 3'-Enden, um die Übertragung
der Promotor/Kodienegion/3'-Sequenzen direkt
auf geeignete binäre
Vektoren zu gestatten.
-
Um
die synthetischen Speicherproteingensequenzen zu insertieren, wurden
10 μg Vektor-DNA
vollständig
mit Asp 718- und Nco I-Restriktionsendonukleasen digeriert. Der
linearisierte Vektor wurde durch Elektrophorese auf einem 1,0%-igen
Agarosegel über
Nacht bei 15 Volt gereinigt. Das Fragment wurde durch Schneiden
der Agarose vor der Bande, Eingeben eines 10 × 5 mm großen Stücks Whatmann 3 MM-Papier in die
Agarose und Behandeln des Fragments durch Elektrophorese auf dem
Papier aufgefangen [Errington (1990) Nukleic Acids Research, 18:17].
Das Fragment und der Puffer wurden durch Zentrifugieren aus dem Papier
herausgeschleudert und die DNA in den –100 μl wurde durch Zugeben von 10
mg tRNA, 10 μl
3 M Natriumacetat und 200 μl
Ethanol ausgefällt.
Die ausgefällte
DNA wurde zweimal mit 70% Ethanol gewaschen und unter Vakuum getrocknet.
Das DNA-Fragment wurde erneut in 20 μl Wasser suspendiert und ein
Teil 10fach zur Verwendung bei Ligationsreaktionen verdünnt.
-
Plasmid-DNA
(10 mg) aus den Klonen 3-5 und pSK534 wurden vollständig mit
Asp 718- und Nco I-Restriktionsendonukleasen digeriert. Die Digestionsprodukte
wurden auf einem 18%-igen nichtdenaturierenden Polyacrylamidgel,
wie in Beispiel 8 beschrieben, getrennt. Gelstücke, die die erwünschten
Fragmente enthielten, wurden von dem Gel abgeschnitten und durch
Eingeben der Gelstücke
in ein 1%-iges Agarosegel und Behandeln durch Elektrophorese für 20 min
bei 100 Volt gereinigt. DNA-Fragmente
wurde auf 10 × 5
mm großen
Stücken
von Whatmann 3 MM-Papier aufgefangen, der Puffer und die Fragmente
wurden durch Zentrifugieren herausgeschleudert und die ausgefällte DNA
mit Ethanol ausgefällt.
Die Fragmente wurden erneut in 6 μl
Wasser suspendiert. Ein Mikroliter des oben beschriebenen verdünnten Vektorfragments,
2 μl 5 × Ligationspuffer
und 1 μl
T4 DNA-Ligase wurden zugegeben. Die Mischung wurde über Nacht
bei 15 °C
ligiert.
-
Die
Ligationmischungen wurden in den E. coli-Stamm DHSa [supE44 del
lacU169 (phi 80 lacZ del M15) hsdR17 recA1 endA1 gyr196 thil relA1]
transformiert und es wurden gegen Ampicillin widerstandsfähige Kolonien
ausgewählt.
Die Klone wurden durch Restriktionsendonukleasedigestionsanalysen
schneller Plasmid-DNA und durch DNA-Sequenzieren gescreent.
-
BEISPIEL 10
-
TABAKPFLANZEN, DIE DIE
CHIMÄREN
GENE PHASEOLINPROMOTOR/CTS/ECODAPA, PHASEOLIN PROMOTOR/CTS/LYSC-M4
UND β-CONGLYCIN-PROMOTOR7SSP3-5
ENTHALTEN
-
Der
binäre
Vektor pZS97 wurde zum Übertragen
des chimären
SSP3-5-Gens aus Beispiel 9 und der chimären E. coli dapA- und lysC-M4-Gene
aus Beispiel 4 auf Tabakpflanzen angewendet. Der binäre Vektor pZS97
(13) ist ein Teil des binären Ti-Plasmidvektorsystems
[Bevan, (1984) Nucl. Acids. Res. 12:8711-8720] von Agrobactrium
tumefaciens. Der Vektor enthält:
(1) die Norpalinsynthase des chimären Gens:: Neomycinphosphotransferase
(nos::NPTII) als auswählbaren
Marker für
transformierte Pflanzenzellen [Bevan et al., (1983) Nature 304:184-186],
(2) die linken und rechten Ränder
der T-DNA des Ti-Plasmids [Bevan, (1984) Nucl. Acids. Res. 12:8711-8720],
(3) das E. coli LacZ a-Komplementiersegment
[Viering et al., (1982) Gene 19:259-267] mit einer einzigen Sal
I-Stelle (pSK97K) oder einer einzigen Hind III-Stelle (pZS97) in
der Polylinkerregion, (4) den Bakterienreplikationsursprung aus
dem Pseudomonasplasmid pVS1 [Itoh et al., (1984) Plasmid 11:206-220]
und (5) das Bakterien-β-lactamasegen als
auswählbaren
Marker für
transformiertes A. tumefaciens.
-
Die
Plasmid pZS97-DNA wurde vollständig
mit Hind III-Enzym digeriert und das digerierte Plasmid wurde durch
Gel gereinigt. Die durch Hind III digerierte pZS97-DNA wurde mit
dem durch Hind III digerierten und gelisolierten chimären Genfragmenten
gemischt, ligiert, wie oben transformiert und es wurden Kolonien auf
Ampicillin ausgewählt.
-
Binäre Vektoren,
die die chimären
Gene enthielten, wurden durch triparentale Kopplungen [Ruvkin et al.,
(1981) Nature 289:85-88] auf den Agrobacterium-Stamm LBA4404/pAL4404
[Hockema et al., (1983), Nature 303:179-180] übertragen, der auf Carbenicillin
Widerstandsfähigkeit
hin ausgewählt
wurde.
-
Kulturen
von Agrobacterium, die den binären
Vektor enthielten, wurden zum Umwandeln von Tabakblattscheiben [Horsch
et al. (1985) Science 227:1229-1231] verwendet. Transgene Pflanzen
wurden in selektivem Medium, das Kanamycin enthielt, regeneriert.
-
Tranformierte
Tabakpflanzen, die das chimäre
Gen β-Conglycininpromotor/SSP3-5/Phaseolin
3'-Region enthalten,
wurden so erhalten. Zwei transformierte Linien, pSK44-3A und pSK44-9A,
die eine einzige Stelleninsertion des SSP3-5-Gens trugen, wurden
auf der Basis der 3:1-Segregation des Markergens für Kanamycinwiderstandsfähigkeit
identifiziert. Die Abkömmlinge
der primären
Transformanten, die für
das Transgen pSK44-3A-6 und pSK44-9A-5 homozygot waren, wurden auf
der Basis der 4:0-Segregation der Kanamycinwiderstandsfähigkeit
in Samen dieser Pflanzen identifiziert.
-
Auf ähnliche
Weise wurden transformierte Tabakpflanzen mit den chimären Genen
Phaseolin 5'-Region/cts/lysC-M4/Phaseolin
3'-Region und Phaseolin
5'-Region/cts/ecodapA/Phaseolin
3'-Region erhalten.
Eine transformierte Linie, BT570-45A, die eine einzige Stelleninsertion
der DHDPS- und AK-Gene
trug, wurde aufgrund der 3:1-Segregation des Markergens für Kanamycinwiderstandsfähigkeit
identifiziert. Abkömmlinge
des primären
Transformanten, die für
das Transgen BT570-45A-3 und Bt570-45A-4 homozygot waren, wurden dann auf
der Basis der 4:0-Segregation der Kanamycinwiderstandsfähigkeit
in Samen dieser Pflanzen identifiziert.
-
Um
Pflanzen, die alle drei chimären
Gene tragen, zu bilden, wurden genetische Kreuzungen unter Anwendung
der homozygoten Stammpflanzen durchgeführt. Die Pflanzen wurden bis
zur Reife unter Gewächshausbedingungen
gezüchtet.
Es wurden Blüten,
die als männlich
und weiblich verwendet werden sollten, einen Tag vor Öffnen ausgewählt und ältere Blüten am Blütenstand
entfernt. Für
die Kreuzung wurden weibliche Blüten
zum Zeitpunkt kurz vor dem Öffnen
gewählt,
als die Staubbeutel noch nicht dehiszent waren. Die Blumenkrone
wurde auf einer Seite geöffnet
und die Staubbeutel entfernt. Männliche
Blüten
wurden als Blüten
gewählt,
die sich am gleichen Tag geöffnet
hatten und dehiszente Staubbeutel aufwiesen, die reife Pollen abwarfen.
Die Staubbeutel wurden entfernt und zum Bestäuben der Stempel der der Staubbeutel
beraubten weiblichen Blüten
verwendet. Die Stempel wurden zum Verhindern weiterer Bestäubung mit
Kunststoffröhren
bedeckt. Man ließ die
Samenhülsen
sich entwickeln und 4-6 Wochen lang trocknen und sie wurden dann
geerntet. Zwei der drei einzelnen Hülsen wurden aus jeder Kreuzung
ie folgenden Kreuzungen wurden durchgeführt.
-
-
Getrocknete
Samenhülsen
wurden aufgebrochen und die Samen eingesammelt und von jeder Kreuzung
gepoolt. Dreißig
Samen wurden für
jede Kreuzung ausgezählt,
als Kontrollen wurden Samen aus selbstbefruchteten Blüten jeder
Stammpflanze verwendet. Duplikatsamenproben wurden hydrolysiert
und auf den gesamten Aminosäuregehalt
hin, wie in Beispiel 5 beschrieben, untersucht. Die mengenmäßige Erhöhung des Lysins
als Prozentsatz der gesamten Samenaminosäuren im Vergleich mit Samen
vom Wildtyp, die 2,56% Lysin enthalten, ist in Tabelle 7 zusammen
mit der Kopiezahl jedes Gens im Endosperm des Samens aufgeführt. TABELLE
7
- * Die Kopiezahl ist der Durchschnitt der
Samenpopulation
-
Die
Ergebnisse dieser Kreuzungen zeigen, dass die gesamten Lysinniveaus
in Samen durch die Koordinatexpression der Lysinbiosynthesegene
und des Proteins SSP3-5 von hohem Lysingehalt um 10-25 Prozent erhöht werden
können.
Bei Samen, die aus Hybridpflanzen deriviert sind, ist dieser Synergismus
am stärksten,
wenn die Biosynthesegene von der weiblichen Stammpflanze deriviert
sind, möglicherweise
aufgrund der Gendosierung im Endosperm. Es ist zu erwarten, dass
das Lysinniveau noch weiter steigen würde, wenn die Biosynthesegene
und lysinreichen Proteingene alle homozygot wären.
-
BEISPIEL 11
-
SOJABOHNENPFLANZEN, DIE
DIE CHIMÄREN
GENE PHASEOLINPROMTOR/CTS/CORDAPA UND PHASEOLINPROMTOR/SSP3-5 ENTHALTEN
-
Transformierte
Sojabohnenpflanzen, die das chimäre
Gen Phaseolinpromotor/cts/cordapA/Phaseolin 3'-Region exprimieren, sind in Beispiel
6 beschrieben worden. Transformierte Sojabohnenpflanzen, die das chimäre Gen Phaseolinpromotor/SSP3-5/Phaseolin
3'-Region exprimiert
wurden durch Insertieren des chimären Gens als isoliertes Hind
III-Fragment in ein äquivalentes
Sojabohnentransformationsvektorplasmid pML63 (14, Beispiel 6) und Durchführen der Transformation, wie
in Beispiel 6 beschrieben, erhalten.
-
Es
wurden aus Samen aus primären
Transformanten Proben genommen durch Schneiden kleiner Stückchen von
den Seiten der Samen und von der Embryonenachse hinweg. Die Stückchen wurden
auf GUS-Aktivität,
wie in Beispiel 6 beschrieben, untersucht, um zu bestimmen, welche
der segregierten Samen die Transgene trugen. Die Hälfte der
Samen wurde zu Mehl gemahlen und auf die Expression des SSP3-5-Proteins
hin durch enzymverbundenen Immunosorbensassay (ELISA) untersucht.
Der Elisa wurde wie folgt durchgeführt:
Es wurde in Fusionsprotein
von Glutathion-S-Transferase und dem SSP3-5-Genprodukt durch Verwendung des
PharmaciaTM pGEX GST-Genfusionssystems (Cunent
Protocols in Molecular Biology – Gegenwärtige Protokolle
in der molekularen Biologie) Band 2, Seite 16.7.1-8 (1989) John
Wiley and Sons) gebildet. Das Fusionsprotein wurde durch Affinitätschromatographie
auf Glutathionagarose-(Sigma) oder Glutathionsepharose (Pharmacia)
Perlen gereinigt, unter Anwendung von Centricon 10TM – (Amicon)
Filtern konzentriert und dann einer SDS-Polyacrylamidelektrophorese
(15% Acrylamid, 19:1 Acrylamid:Bisacrylamid) zur weiteren Reinigung
unterworfen. Das Gel wurde 30 min lang mit Coomassie-Blau gefärbt, in
50% Methanol, 10% Essigsäure entfärbt und
die Proteinbanden unter Anwendung eines AmiconWZ-Centiluter-Mikroelektroeluierers
(Paul T. Matsudaira, Verfasser, A Practical Guide to Protein and
Peptide Purification for Microsequencing (Ein praktischer Führer für die Protein-
und Peptidreinigung für
das Mikrosequenzieren) Acadmic Press Inc. New York, 1989) elektroeluiert.
Ein zweites Gel, das auf die gleiche Weise zubereitet und einem
Arbeitsgang unterworfen worden war, wurde in einer Nichtessigsäure gefärbt, die
Färbemittel
[9 Teile 0,1 % Coomassie-Blau G250 (Bio-Rad) in 50% Methanol und
1 Teil Serva Blue (Serva, Westbury, NY) in destilliertem Wasser
enthielt, 1-2 h lang gefärbt.
Das Gel wurde kurz in 20% Methanol, 3% Glycerin für 0,5-1
h entfärbt,
bis die GTS-SSP3-5-Bande gerade noch sichtbar war. Diese Bande wurde
aus dem Gel ausgeschnitten und mit dem elektroeluierten Material
an Hazelton Laboratories zur Verwendung als Antigen beim Immunisieren
eines Neuseelandkaninchens geschickt. Insgesamt 1 mg des Antigens
wurde verwendet (0,8 mg in Gel, 0,2 mg in Lösung). Alle drei Wochen wurden
von Hazelton Laboratories Testblutproben bereitgestellt. Der ungefähre Titer
wurde durch Western-Blotting von E. coli-Extrakten aus Zellen, die
das SSp3-5-Gen enthielten, unter der Kontrolle des T7-Promotors
in verschiedenen Verdünnungen
von Protein und Serum getestet.
-
IgG
wurde von dem Serum unter Anwendung einer Protein A-Sepharosesäule isoliert.
Das IgG wurde auf Mikrotiterplatten in einer Menge von 5 μg pro Vertiefung
schichtweise eingebracht. Eine getrennte Portion des IgG wurde biotinyliert.
-
Wässrige Extrakte
aus den transgenen Pflanzen wurden verdünnt und in die Vertiefungen
eingegeben, wobei gewöhnlich
mit der Probe, die 1 μg
des gesamten Proteins enthielt, begonnen wurde. Die Probe wurde mehrere
Male verdünnt,
um sicherzustellen, dass mindestens eine der Verdünnungen
ein Ergebnis erbrachte, das innerhalb der Reichweite einer Standardkurve
lag, die auf der gleichen Platte gebildet wurde. Die Standardkurve
wurde unter Anwendung von chemisch synthetisiertem SSP3-5-Protein
gebildet. Die Proben wurden eine Stunde lang bei 37 ° inkubiert
und die Platten wurden gewaschen. Das biotinylierte IgG wurde dann in
die Vertiefungen eingegeben. Die Platten wurden 1 h bei 37 ° inkubiert
und gewaschen. An Streptavidin konjugierte alkalische Phosphatase
wurde in die Vertiefungen eingegeben, 1 h bei 37 ° inkubiert
und gewaschen. Ein Substrat, das aus 1 mg/ml p-Nitrophenylphosphat
in 1M Diethanolamin bestand, wurde in die Vertiefungen hineingegeben
und die Platten wurden 1 h bei 37 °C inkubiert. Eine 5%-ige EDTA-Stopplösung wurde
in die Vertiefungen eingegeben und die Extinktion bei 405 nm minus
650 nm abgelesen. Die transgenen Sojabohnensamen enthielten 0,5
bis 2,0% durch Wasser extrahierbares Protein als SSP3-5.
-
Die
verbleibende Hälfte
Samen, die bezüglich
GUS und SSP3-5-Protein positiv waren. wurden ausgepflanzt und bis
zur Reife unter Gewächshausbedingungen
gezüchtet.
Um Homozygoten für
den GUS-Phänotyp zu
bestimmen, wurden Samen aus diesen R1-Pflanzen auf Segregierung
von GUS-Aktivität,
wie oben, gescreened. Pflanzen die für das Phaseolin/SSP3-5-Gen
homozygot waren, wurden mit homozygoten transgenen Sojabohnen gekreuzt,
die das Corynbacterium dapA-Genprodukt exprimieren.
-
Als
bevorzugte Alternative zum Zusammenbringen des chimären SSP-Gens
und von chimärem
cordapA-Gen A durch genetisches Kreuzen wurde ein einziger Sojabohnentransformationsvektor,
der beide Gene trägt,
konstruiert. Das Plasmid pML63, das das oben beschriebene chimäre Gen Phaseolinpromotor/SSP3-5/Phaseolin
3'-Region trägt, wurde
mit dem Restriktionsenzym BamH I gespalten und das BamH I-Fragment,
das das chimäre
Gen Phaseolinpromotor/cts/cordapA/Phaseolin 3'-Region (Beispiel 5) trägt, wurde
insertiert. Dieser Vektor kann in Sojabohnen, wie in Beispiel 6
beschrieben, transformiert werden.
-
BEISPIEL 12
-
KONSTRUKTION CHIMÄRER GENE
FÜR DAS
EXPRIMIEREN VON CORYNEBACTERIUM DHDPS UND SSP3-5 IM EMBRYO UND ENDOSPERM
VON TRANSFORMIERTEM MAIS
-
Die
folgenden chimären
Gene wurden zur Transformation in Mais
- Globulin 1 Promotor/mcts/cordapA/NOS
3'-Region
- Glutelin 2 Promotor/mcts/cordapA/NOS 3'-Region
- Globulin 1 Promotor/SSP3-5/Gluobulin 13'-Region
- Glutelin 2 Promotor/SSP3-5/10 kD 3'-Region
hergestellt.
-
Der
Glutelin 2-Promotor wurde aus genomischer Mais-DNA unter Anwendung
der PKR mit Primern auf der Basis der veröffentlichten Sequenz [Reina
et al. (1990) Nukleic Acids Res. 18:6426-6426] kloniert. Das Promotorfragment
enthält
1020 Nukleotide stromaufwärts
vom ATG-Translationsstartcodon. Durch PKR wurde eine Nco I-Stelle
an der ATG-Startstelle eingeführt,
um direkte Translationsfusionen zu gestatten. Eine BamH 1-Stelle
wurde am 5'-Ende
des Promotors eingeführt.
Das 1,02 kb BamH I bis Nco I-Promotorfragment
wurde in die BambH I bis Nco I-Stellen des Pflanzenexpressionsvektors
pML63 (vergleiche Beispiel 11) unter Ersetzen des 35S-Promotors
zur Bildung des Vektors pML90 kloniert. Dieser Vektor enthält den Glutelin
2-Promotor der mit der GUS-Kodierregion und NOS 3' verknüpft ist.
-
Die
10 kD Zein 3'-Region
wurde aus einem Zeingenklon von 10 kD deriviert, der durch PKR aus
genomischer DNA unter Anwendung von Oligonukleotidprimern auf der
Basis der veröffentlichten
Sequenz [Kirihara et al. (1988) Gene 71:359-370] gebildet wurde.
Die 3'-Region erstreckt
sich über
940 Nukleotide vom Stopcodon. Restriktionsendonukleasestellen für Kpn I-,
Sma I- und Xba I-Stellen wurden sofort auf den TAG-Stopcodon hin
durch Oligonukleotidinsertion zum Erleichtern des Klonierens zugegeben.
Ein Sma I- bis Hind III-Segment, das die 10 kD 3'-Region enthält, wurde isoliert und in Sma
I und Hind III, die durch pML90 digeriert worden waren, unter Ersetzen
der NOS 3'-Sequenz
durch die 10 kd 3'-Region
ligiert unter Bildung des Plasmids pML103. pML103 enthält den Glutelin
2-Promotor, eine Nco I-Stelle am ATG-Startcodon des GUS-Gens, Sma
I- und Xba I-Stellen nach dem Stopcodon und 940 Nukleotide der Zein
3'-Sequenz von 10
kD.
-
Die
Globulin 1-Promotor- und 3'-Sequenzen
wurden von einer genomischen Clontech Mais-DNA-Bibliothek unter Anwendung von Oligonukleotidsonden
auf der Basis der veröffentlichten
Sequenz des Globulin 1-Gens [Kriz et al. (1989) Plant Physiol. 91:636]
isoliert. Das klonierte Segment umfasst das Promotorfragment, das
sich über
1078 Nukleotide stromaufwärts
vom ATG Translationsstartcodon erstreckt, die gesamte Globulinkodiersequenz
einschließlich
Introne und die 3'-Sequenz,
die sich über
803 Basen vom Translationsstop erstreckt, umfasst. Um das Ersetzen
der Globulin I-Kodiersequenz durch andere Kodiersequenzen zu erlauben,
wurde eine Nco I-Stelle am ATG Startcodon eingeführt und kPN I- und Xba I-Stellen
wurden auf den Translationsstopcodon hin durch PKR zum Bilden des
Vektors pCC50 eingeführt.
Es besteht auch eine zureite Nco I-Stelle innerhalb des Globulin
1-Promotorfragments. Die Globulin 1-Genkasette ist durch Hind III-Stellen flankiert.
-
Die
biosynthetischen pflanzlichen Aminosäureenzyme sind dafür bekannt,
dass sie sich in den Chlorplasten befinden und aus diesem Grund
werden sie mit einem Chloroplasttargetingsignal synthetisiert. Bakterienproteine
wie DHDPS besitzen kein derartiges Signal. Eine Chloroplastübergangssequenz
(cts) wurde aus diesem Grund an die cordapA-Kodiersequenz in den
unten beschriebenen chimären
Genen fusioniert. Für Mais
basierte die verwendete cts auf der cts der kleinen Untereinheit
von Ribulose-1,5-bisphosphatcarboxylase
aus Mais [Lebrun et al. (1987) Nukleic Acids Res. 15:4360] und ist
so mit acts bezeichnet, um sie von der Sojabohnen-cts zu unterscheiden.
Die Oligonukleotide SEQ ID NO:93-98 wurden synthetisiert und im
Wesentlichen wie in Beispiel 4 beschrieben verwendet.
-
Die
Oligonukleotide SEQ ID NO:93 und SEQ ID NO:94, die den Carboxyterminalteil
des Maischloroplasttargetingsignals kodieren, wurden einem Annealing
unterzogen, was zu mit Xba I und Nco I verträglichen Enden führt, durch
Polyacrylamidgelelektrophorese gereinigt und in durch Xba I plus
Co I digerierte pBT492 insertiert (vergleiche Beispiel 3). Die Insertion
der richtigen Sequenz wurde durch DNA-Sequenzieren unter Bildung von pBT556
nachgeprüft.
Die Oligonukleotide SEQ ID NO:95 und SEQ ID NO:96, die den mittleren
Teil des Chloroplasttargetingsignals kodieren, wurden einem Annealing
unterzogen, was zu mit Bgl II und Xba I verträglichen Enden führt, durch
Polyacrylamidgelelektrophorese gereinigt und in durch Bgl II und
Xba I digerierte pBT556 insertiert. Die Insertion der richtigen
Sequenz wurde durch DNA-Sequezieren unter Bildung von pBT557 nachgeprüft. Die
Oligonukleotide SEQ ID NO:97 und SEQ ID NO:98, die den Aminoterminalteil
des Chloroplasttargetingsignals kodieren, wurden einem Annealing
unterzogen, was zu mit Nco I und Afl II verträglichen Enden führt, durch
Polyacrylamidgelelektrophorese gereinigt und in durch Nco I und
Afl II pBT557 insertiert. Die Insertion der richtigen Sequenz wurde
durch DNA-Sequenzieren unter Bildung von pBT558 nachgeprüft. So wurde
die mcts an das lysC-M4-Gen fusioniert.
-
Ein
DNA-Fragment, das die gesamte mcts enthielt, wurde unter Anwendung
von PKR zubereitet. Die Matrizen-DNA bestand aus pBT558 und die
verwendeten Oligonukleotidprimer waren:
SEQ ID NO:99:
GCGCCCACCG
TGATGA
SEQ ID NO:100:
CACCGGATTC TTCCGC
-
Das
mcts-Fragment wurde an den Aminoterminus des DHDPS-Proteins, das
durch das ecodapA-Gen kodiert
ist, durch Digerieren mit Nco I und Behandeln mit dem Klenow-Fragment
von DNA-Polymerase
zum Füllen
der 5'-Überhänge verknüpft. Das
insertierte Fragment und die Vektor-Insertionsverbindungen wurden durch
DNA-Sequenzieren, das pBT576 ergab, als richtig bestimmt.
-
Um
das chimäre
Gen: Globulin 1-Promotor/mcts/cordapA/NOS 3'-Region zu konstruieren, wurde ein Nco
I bis Kpn I-Fragment, das die mcts-ecodapA-Kodiersequenz enthielt,
vom Plasmid pBT576 (vergleiche Beispiel 6) isoliert und in durch
Nco I plus Kpn I digerierte pCC50 unter Bildung des Plasmids pBT662
insertiert. Dann wurde die ecodapA-Kodiersequenz durch die cordapA-Kodiersequenz
wie folgt ersetzt. Ein Afl II bis Kpn I-Fragment, das die distalen
zwei Drittel der an die cordapA-Kodiersequenz fusionierten mcts
enthielt, wurde in durch Afl II bis Kpn I digerierte pBT662 unter
Bildung des Plasmids pBT677 insertiert.
-
Um
das chimäre
Gen: Glutelin 2-Promotor/mcts/cordapA/NOS 3'-Region zu konstruieren, wurde ein Nco
I bis Kpn I-Fragment, das die mcts-cordapA-Kodiersequenz enthielt,
vom Plasmid pBT677 isoliert und in durch Nco I bis Kpn I digerierte
pML90 unter Bildung des Plasmids pBT679 insertiert.
-
Um
das chimäre
Gen Glutelin 2-Promotor/SSP3-5/10 kD 3'-Region zu konstruieren, wurde das Plasmid
pML 103 (oben), das den Glutelin 2-Promotor und die 10 kD Zein 3'-Region enthielt,
an den Nco I- und Sma
I-Stellen gespalten. Die SSP3-5-Kodienegion (Beispiel 9) wurde als
Fragment von Nco I zum stumpfen Ende durch Spalten mit Xba I gefolgt
vom Füllen
des klebrigen Endes unter Anwendung von Klenow-Fragment von DNA-Polymerase
und darauffolgendes Spalten mit Nco I isoliert. Das Fragment des
193 Basenpaars Nco I bis zum stumpfen Ende wurde in das durch Nco
I und Sma I geschnittene pML103 unter Bildung von pLH104 ligiert.
-
Um
das chimäre
Gen: Globulin 1-Protomor/SSP3-5/Globulin 1 3'-Region zu konstruieren, wurde das Fragment
des 193 Basenpaars Nco I und Xba I, das die SSP3-5-Kodierregion
(Beispiel 9) enthält,
in das Plasmid pCC50 (oben), das vollständig mit Xba I gespalten worden
war, insertiert und dann teilweise mit Nco I zum Öffnen des
Plasmids am ATG-Startcodon unter Bildung von pLH105 geschnitten.
-
BEISPIEL 13
-
MAISPFLANZEN, DIE CHIMÄRE GENE
ENTHALTEN, ZUM EXPRIMIEREN VON CORYNEBACTERIUM DHDPS IM EMBRYO UND
ENDOSPERM
-
Mais
wurde mit den chimären
Genen: Globulin 1-Promotor/mcts/cordapA/NOS 3'-Region oder Glutelin 2 Promotor/mcts/cordapA/NOS
3'-Region transformiert.
-
Entweder
ein oder beide Plasmidvektoren, die auswählbare Marker enthalten, wurden
bei den Transformationen verwendet. Ein Plasmid, pDETRIC, enthielt
das bar-Gen von Steptomyces hygroscopicus, das eine Resistenz gegen
das Herbizid Glufosinat [Thompson et al. (1987) The EMBO Journal
6:2519-2523] verleiht. In dem bakteriellen Gen wurde der Translationscodon
von GTG zu ATG zur richtigen Translationsinitiation in Pflanzen
geändert
[De Block et al. (1987) The EMBO Journal 6:2513-2518]. Das bar-Gen wurde durch den 35S-Promotor
aus Blumenkohlmosaikvirus getrieben und macht sich das Terminations-
und Polyadenylationssignal des Octopinsynthasegens aus Agrobacterium
tumefaciens zunutze. Als Alternative bestand der auswählbare Marker
aus 35S/Ac einem synthetischen Phosphinothricin-N-acetyltransferase-(pat)-Gen
unter der Kontrolle des 35S-Promotors und des 3-Terminator-Polyadenylationssignals aus
Blumenkohlmosaikvirus [Eckes et al., (1989) J. Cell Biochem. Suppl.
13 D].
-
Die
embryogenen Calluskulturen wurden aus unreifen Embryos (ca. 1,0
bis 1,5 mm) initiiert, die aus Körnern
einer Maislinie geschnitten worden waren, die zu Ergeben einer „Callus
Typ II"-Gewebenkulturreaktion gezüchtet worden
war. Die Embryos wurden 10 bis 12 Tage nach der Bestäubung zerschnitten
und mit der Achsenseite nach unten in Kontakt mit durch Agarose
verfestigtes N6-Medium [Chu et al. (1974) Sci Sin 18:659-668] eingegeben,
das mit 0,5 mg/l 2,4-D (N6-0,5) ergänzt worden war. Die Embryos
wurden bei 27 °C im
Dunkeln gehalten. Krümeliger
embryogener Callus, der aus undifferenzierten Massen von Zellen
bestand, wobei somatische Proembryos und somatische Embryos an Suspensorstrukturen
getragen wurden, proliferierten aus dem Scutellum der unreifen Embryos.
Klonale embryogene Calli, die aus einzelnen Embryos isoliert worden
waren, wurden identifiziert und auf N6-0,5-Medium alle 2 bis 3 Wochen subkultiviert.
-
Die
Teilchenbombadiermethode wurde zum Übertragen von Genen auf die
Calluskulturzeilen verwendet. Ein Biolistic PDS-1000/He (BioRAD
Laboratories, Hercules, CA) wurde für diese Versuche verwendet.
-
Zirkuläre Plasmid-DNA
oder DNA, die durch Restriktionsendonukleasedigestion linearisiert
worden war, wurde auf der Oberfläche
von Goldteilchen ausgefällt.
DNA aus zurei oder drei verschiedenen Plasmiden, wobei eines den
auswählbaren
Marker für
die Maistransformation und eines oder beide die chimären Gene zum
Verstärken
der Lysinansammlung in Samen enthielten, wurden gleichzeitig ausgefällt. Um
dies zu erreichen, wurden 1,5 μg
jeder DNA (in Wasser in einer Konzentration von ca. 1 mg/ml) 25
ml Goldteilchen (Durchschnittsdurchmesser 1,5 μm) zugegeben und in Wasser (60
mg Gold pro ml) suspendiert. Calciumchlorid (25 ml einer 2,5 M Lösung) und
Spermidin (10 ml einer 1,0 M Lösung)
wurden der Gold-DNA-Suspension zugegeben, während die Röhre in einer Wirbelvorrichtung
behandelt wurde. Die Goldteilchen wurden in einer Mikrofuge 10 sec
lang zentrifugiert und die überstehende Substanz
wurde entfernt. Die Goldteilchen wurden dann erneut in 200 ml absolutem
Ethanol suspendiert, nochmals zentrifugiert und die überstehende
Substanz entfernt. Schließlich
wurden die Goldteilchen nochmals in 25 ml absolutem Ethanol suspendiert
und zureimal eine sec lang beschallt. Fünf μl der mit DNA beschichteten
Goldteilchen wurden dann auf eine Makroträgerscheibe aufgeladen und man
ließ das
Ethanol verdampfen unter Zurücklassen
der mit DNA bedeckten Goldteilchen, die auf die Scheibe aufgetrocknet
waren.
-
Embryogener
Callus (von der #LH132.5.S genannten Calluslinie) wurde in einem
runden Bereich von ca. 6 cm Durchmesser im Mittelpunkt einer Petrischale
von 100 × 20
mm angeordnet, die N6-0,5-Medium
enthielt, das mit 0,25 M Sorbit und 0,25 M Mannit ergänzt worden
war. Das Gewebe wurde 2 h lang auf dieses Medium vor der Bombardierung
als Vorbehandlung aufgebracht und verblieb auf dem Medium während des Bombardierungsvorgangs.
Am Ende der Vorbehandlungsperiode von 2 h wurde die Petrischale,
die das Gewebe enthielt, in die Kammer des PDS-1000/He eingegeben.
Die Luft in der Kammer wurde dann bis zu einem Vakuum von 28 Zoll
Hg evakuiert. Der Makroträger
wurde mit einer Heliumschockwelle unter Anwendung einer Brechmembran,
die zerbricht, wenn der He-Druck in der Schockröhre 1100 psi erreicht, beschleunigt.
Das Gewebe wurde 8 cm vom Stoppschirm entfernt, positioniert. Vier
Plattengewebe wurden mit den mit DNA beschichteten Goldteilchen
bombardiert. Sofort auf das Bombardieren hin wurde das Callusgewebe
auf N6-0,5-Medium ohne ergänzendes
Sorbit oder Mannit übertragen.
-
Innerhalb
von 24 h nach dem Bombardieren wurde das Gewebe auf selektives Medium,
N6-0,5-Medium, übetragen,
das 2 mg/l Glufosinat enthielt und kein Casein oder Prolin aufwies.
Das Gewebe, das auf diesem Medium langsam weiterwuchs, wurde auf
frisches N6-0,5-Medium übetragen,
das alle zurei Wochen mit Glufosinat ergänzt wurde. Nach 6-12 Wochen
wurden Klone von aktiv wachsendem Callus identifiziert. Der Callus
wurde dann auf Medium übertragen,
das die Pflanzenregenerierung fördert.
-
Aus
transformiertem Callus regenerierte Pflanzen wurden auf das Vorliegen
der intakten Transgene durch Southern Blot oder PKR analysiert.
Die Pflanzen wurden selbstbefruchtet oder auf eine Elitlinie ausgekreuzt,
um R1- bzur. F1-Samen zu bilden. Einzelne R1-Samen oder sechs bis
acht F1-Samen wurden gepoolt und zur Expression des Corynebacterium
DHDPS Proteins durch Western Blot-Analyse untersucht. Die freie Aminosäurezusammensetzung
und die gesamte Aminosäureszusammensetzung
der Samen wurde, wie in früheren
Beispielen beschrieben, bestimmt.
-
Die
Expression des Corynebacterium DHDPS-Proteins, das entweder durch
den Globulin- oder den Glutelin-Promotor getrieben wird, wurde in
den Maissamen beobachtet (Tabelle 8). Die freien Lysinniveaus in den
Samen stiegen von ca. 1,4% freier Aminosäure in Kontrollsamen auf 15-27%
in Samen auf, die Corynebacterium DHDPS aus dem Globulin I-Promotor
exprimieren. Die höhere
DHDPS-Expression
und das höhere Lysinniveau
in den selbsbefruchteten Samen rührt
wahrscheinlich von der Tatsache her, dass zu erwarten ist, dass
der Hälfte
der gepoolten Samen in den ausgekreuzten Linien das Transgen aufgrund
von Segregation fehlt. Eine geringer Erhöhung des freien Lysins wurde
in Samen beobachtet, die Corynebacterium DHDPS aus dem Glutelin
2-Promotor exprimieren. So ist es zum Erhöhen des Lysins eventuell besser,
dieses Enzym im Embryo anstatt dem Endosperm zu exprimieren. Ein
hohes Niveaus an Saccharopin, das auf Lysinkatabolismus hinweist,
wurde in Samen beobachtet, die hohe Niveaus an Lysin enthielten.
-
Normalerweise
stellt Lysin ca. 2,3% des Aminosäuregehalts
der Samen dar. Es ist deshalb aus der Tabelle 8 offensichtlich,
dass wesentliche Erhöhungen
(35%-130%) an Lysin als Prozentsatz der gesamten Samenaminosäuren in
Samen aufgefunden worden sind, die Corynebacterium DHDPS aus dem
Globulin I-Promotor
exprimieren.
-
TABELLE
8 1088.1.3
Linie: Globulin 1 Promotor/mcts/cordapA/NOS 3-Region 1099.2.1
Linie: Globulin 1 Promotor/mcts/cordapA/NOS 3-Region 1090.2.1
Linie: Glutelin 2 Promotor/mcts/cordapA/NOS 3-Region
-
BEISPIEL 14
-
TRANSFORMATION VON SOJABOHNEN
MIT DEM CHIMÄREN
GEN KUNITYTRYPSININHIBITOR 3-PROMOTOR/CTS/CORDAPA
-
Eine
samenspezifische Expressionskassette, die aus dem Promotor und dem
Transkriptionsterminator aus dem Sojabohnenkunitztrypsininhibitor
3- (KT13-) Gen [Jofuku et al. (1989) Platn Cell 1:427-435] wurde gebildet.
Die KT13-Kasette umfasst ca. 2000 Nukleotide stromaufwärts (5') vom Translationsinitiationscodon und
ca. 200 Nukleotide stromabwärts
(3') vom Translationsstopcodon
des Kunitztrypsininhibitors 3. Zwischen den 5'- und 3'-Regionen wurden die Restriktionsendonukleasestellen
Nco I (die den ATG-Translationsinitiationscodon umfasst) und Kpn
I gebildet, um die Insertion des Corynebacterium dapA-Gens zu gestatten.
Die gesamte Kassette wurde durch BamH I- und Sal I-Stellen flankiert.
-
Wie
in Beispiel 4 beschrieben, wurde eine Chloroplasmansitsequenz (cts)
an die dapA-Kodiersequenz in
dem chimären
Gen fusioniert. Die verwendete cts basierte auf der cts der kleinen
Untereinheit von Ribulose-1,5-biphosphatcarboxylase aus Sojabohnen
[Berry-Lowe et al. (1982) J. Mol. Appl. Genet. 1:483-498]. Ein Nco
I-Kpn I-Fragment von 1030 bp, das die cts enthielt, die an der cordapA-Kodierregion angeknüpft war,
wurde aus einem Agarosegel auf die Elektrophorese hin isoliert und
in die KT13-Expressionskasette unter Bildung des Plasmids pML102
(15) insertiert.
-
Das
Plasmid pML102 wurde in Sojabohnen durch teilchenvermittelte Bombardierung
durch die Firma Agracetus (Middleton, WI) dem im Patent der Vereinigten
Staaten Nr. 5015580 vorgeschriebenen Verfahren entsprechend eingeführt. Zum
Screenen auf transformierte Zellen wurde das Plasmid pML102 gleichzeitig
mit einem anderen Plasmid bombardiert, das ein Sojabohnentransformationsmarkergen
trägt,
das aus dem 35S-Promotor aus Blumenkohlmosaikvirus besteht, das
die Expression des E. coli-β-Glucuronidase- (GUS-) Gens
[Jefferson et al., (1986) Proc. Natl. Acad. Sci. USA 83:8447-8451]
trägt,
mit der Nos 3'-Region
treibt.
-
Es
war zu erwarten, dass die Transgene sich in den R1-Samen der transformierten
Pflanzen absondern würden.
Um Samen zu identifizieren, die das Transformationsmarkergen trugen,
wurde ein kleines Stück des
Samens mit einer Rasierklinge abgeschnitten und in eine Vertiefung
in einer wegwerfbaren Mikrotiterplatte aus Kunststoff eingegeben.
Eine GUS-Assaymischung bestehend aus 100 mm NaH2PO4, 10 mM EDTA, 0,5 mM K4Fe(CN)6, 0,1% Triton X-100, 0,5 mg/ml 5-Brom-4-chlor-3-indolyl-β-D-glucuronsäure wurde
zubereitet und 0,15 ml wurden in jede Mikrotitervertiefung eingegeben.
Die Mikrotiterplatte wurde 45 Minuten bei 37 ° inkubiert. Die Entwicklung
einer blauen Farbe zeigt die Expression von GUS in dem Samen an.
-
Zum
Messen der gesamten Aminosäurezusammensetzung
reifer Samen wurden 1-1,4 Milligramm des Samenmehls in 6N-Salzsäure, 0,4% β-Mercaptoethanol
unter Stickstoff 24 h bei 110-120 °C hydrolysiert; 1/50 der Probe
wurde auf einem Aminosäureanalysator
von Beckman, Modell 6300, unter Anwendung der Postsäulenninhydrinerfassung
einem Arbeitslauf unterzogen. Die Lysin- (und anderen Aminosäure-) Niveaus
in den Samen wurden als Prozentsatz der gesamten Aminosäuren verglichen.
Sojabohnensamen vom Wiltyp enthalten 5,7-6,0% Lysin.
-
Einhundertundfünfzig einzelne
Samen aus sechzehn unabhängigen
transformierten Linien wurden analysiert (Tabelle 9). Zehn der sechzehn
Linien wiesen Samen mit einem Lysingehalt von 7% der gesamten Samenaminosäuren oder
mehr auf, was eine 16-22%-ige Erhöhung im Vergleich mit Samen
vom Wildtyp darstellt. So wurden bei mehr als 62% der Transformationsereignisse
das das cordapA-Gen tragende Plasmid zusammen mit dem das Plasmid
tragenden Marker-GUS-Gen integriert. Ca. 80% der Samen mit hohem
Lysingehalt waren GUS-positiv, was darauf hinweist, dass das Plasmid,
das das cordapA-Gen trägt,
gewöhnlich
an der gleichen Chromosomenstelle integriert wird, wie das Plasmid,
das das GUS-Gen trägt.
Jedoch bestand bei einigen transformierten Linien, z.B. 260-05,
kaum eine Korrelation zurischen den GUS-positiven Phänotypen und
den Phänotypen
von hohem Lysingehalt, was darauf hinweist, dass die beiden Plasmide
an unverknüpften Stellen
integriert wurden. Beide dieser Typen von Transformationsereignissen
waren aufgrund des für
die Transformation angewendeten Vorgangs zu erwarten.
-
Es
wurden Samen mit einem Lysingehalt von mehr als 20% der gesamten
Samenaminosäuren
erhalten. Dies stellt eine fast dreihundert prozentige Erhöhung des
Samenlysingehalts dar. TABELLE
9
SEQUENZAUFLISTUNG
























 SEQUENZAUFLISTUNG
SEQUENZAUFLISTUNG














































