-
Die
vorliegende Erfindung betrifft Verbindungen, bei denen es sich um
Antagonisten der Aktivität
des Gonadotropin-releasing-Hormons (GnRH) handelt. Die Erfindung
betrifft außerdem
pharmazeutische Formulierungen, die Verwendung einer Verbindung
der vorliegenden Erfindung bei der Herstellung eines Medikaments,
ein Verfahren zur therapeutischen Behandlung unter Einsatz einer
solchen Verbindung und Verfahren zur Herstellung der Verbindungen.
-
Bei
dem Gonadotropin-releasing-Hormon (GnRH) handelt es sich um ein
Decapeptid, das vom Hypothalamus als Reaktion auf neurale und/oder
chemische Stimuli in den hypophysären Portalkreislauf sezerniert wird,
was zur Biosynthese und Freisetzung von luteinisierendem Hormon
(LH) und follikelstimulierendem Hormon (FSH) durch die Hypophyse
führt.
GnRH ist auch unter anderen Namen bekannt, einschließlich Gonadoliberin,
LH-releasing-Hormon
(LHRH), FSH-releasing-Hormon (FSH RH) und LH/FSH-releasing-Faktor (LH/FSH
RF).
-
GnRH
spielt eine wichtige Rolle bei der Steuerung der Wirkung von LH
und FSH (indem es ihre Konzentrationen reguliert) und hat somit
eine Funktion bei der Steuerung der Konzentrationen an gonaden Steroiden
bei beiden Geschlechtern, einschließlich der Sexualhormone Progesteron, Östrogene
und Androgene. Eine ausführlichere
Diskussion von GnRH findet sich in
WO
98/5519 und
WO 97/14697 .
-
Man
nimmt an, daß bei
mehreren Krankheiten eine Steuerung der GnRH-Aktivität von Nutzen
wäre, insbesondere,
wenn man eine solche Aktivität
antagonisiert. Hierzu zählen
mit Sexualhormonen in Zusammenhang stehende Leiden, wie sexualhormonabhängiger Krebs,
benigne Prostatahypertrophie oder Gebärmuttermyom. Beispiele für sexualhormonabhängigen Krebs
sind Prostatakrebs, Gebärmutterkrebs,
Brustkrebs und Gonadotrophenadenom der Hypophyse.
-
In
den folgenden Dokumenten werden Verbindungen offenbart, die als
GnRH-Antagonisten wirken sollen:
WO
97/21435 ,
WO 97/21703 ,
WO 97/21704 ,
WO 97/21707 ,
WO 55116 ,
WO
98/55119 ,
WO 98/55123 ,
WO 98/55470 ,
WO 98/55479 ,
WO 99/21553 ,
WO 99/21557 ,
WO 99/41251 ,
WO 99/41252 ,
WO 00/04013 ,
WO 00/69433 ,
WO 99/51231 ,
WO 99/51232 ,
WO 99/51233 ,
WO 99/51234 ,
WO 99/51595 ,
WO 99/51596 ,
WO 00/53178 ,
WO 00/53180 ,
WO 00/53179 ,
WO 00/53181 ,
WO 00/53185 ,
WO 00/53602 ,
WO 02/066477 ,
WO 02/066478 ,
WO 02/06645 und
WO 02/092565 .
-
Es
wäre wünschenswert,
weitere als GnRH-Antagonisten wirkende Verbindungen bereitzustellen. Demgemäß stellt
die vorliegende Erfindung Verbindungen der Formel (I)
Formel
(I) wobei
A für
eine direkte Bindung oder gegebenenfalls substituiertes C
1-5-Alkylen steht;
B für eine Gruppe
der Formel (II):
Formel
(II) steht, wobei Formel (II) in Position (a) an das
Stickstoffatom gebunden ist und die Gruppe X an R
8 gebunden ist;
M
für -(CH
2)
0-2-O- steht;
R
1 für
Wasserstoff, gegebenenfalls substituiertes C
1-8-Alkyl oder (CH
2)
b-R
a steht,
wobei R
a für C
3-8-Cycloalkyl steht
und b für
Null oder eine ganze Zahl von 1 bis 6 steht;
R
2 für eine gegebenenfalls
substituierte mono- oder bicyclische aromatische Ringstruktur steht,
wobei die gegebenenfalls vorhandenen Substituenten ausgewählt sind
aus Cyano, NR
3R
3a,
gegebenenfalls substituiertem C
1-8-Alkyl, gegebenenfalls
substituiertem C
1-8-Alkoxy oder Halogen;
R
3 und R
3a unabhängig voneinander
ausgewählt
sind aus Wasserstoff, gegebenenfalls substituiertem C
1-8-Alkyl und
gegebenenfalls substituiertem Aryl;
R
5 ausgewählt ist
aus einem gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen
Ring mit 1 bis 4 unabhängig
voneinander aus O, N und S ausgewählten Heteroatomen, oder einer
Gruppe der Formel III-a, III-b, III-c, III-d, III-e, III-f, III-g,
III-h, III-i oder III-j;
wobei het für einen
gegebenenfalls substituierten 3- bis
8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander
aus O, N und S ausgewählten
Heteroatomen steht;
R
6 und R
6a unabhängig
voneinander ausgewählt
sind aus Wasserstoff und gegebenenfalls substituiertem C
1-8-Alkyl;
oder R
6 und R
6a zusammen
für Carbonyl
stehen;
R
7 für Wasserstoff oder gegebenenfalls
substituiertes C
1-8-Alkyl steht;
oder
zusammen einen gegebenenfalls
substituierten 3- bis 8gliedrigen heterocyclischen Ring 3 weiteren
unabhängig
voneinander aus mit 1 bis O, N und S ausgewählten Heteroatomen bildet und
R Wasserstoff und gegebenenfalls substituiertes C
1-8-Alkyl
steht;
X und R
8 ausgewählt sind
aus:
- (i) X steht für N und R8 ist
ausgewählt
aus:
Cyano, Wasserstoff, Hydroxy, -O-Rb,
-NRbRc -C(O)O-Rb, -CONRbRc oder NH-C(O)-Rb,
wobei Rb und Rc unabhängig voneinander
ausgewählt
sind aus Wasserstoff und C1-4-Alkyl, gegebenenfalls
substituiert durch Hydroxy, Amino, N-C1-4-Alkylamino, N,N-Di-C1-4-alkylamino, HO-C2-4-Alkyl-NH- oder HO-C2-4-Alkyl-N-(C1-4-alkyl)-;
- (ii) X steht für
CH und R8 steht für NO2;
und
- (iii) X-R8 steht für -O-;
R11 für eine Gruppe
der Formel: N(R9R10)
steht, wobei R9 für Wasserstoff, Aryl, einen
gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen
Ring oder gegebenenfalls substituiertes C1-8-Alkyl
steht und R10 für Wasserstoff oder gegebenenfalls
substituiertes C1-8-Alkyl steht; oder die Struktur N(R9R10) für einen
gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen
Ring steht, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander
aus O, N und S ausgewählte
Heteroatome enthält;
R12 und R12a unabhängig voneinander
ausgewählt
sind aus Wasserstoff oder gegebenenfalls substituiertem C1-8-Alkyl;
oder R12 und R12a zusammen
mit dem Kohlenstoff, an den sie gebunden sind, einen gegebenenfalls substituierten
3- bis 7gliedrigen Cycloalkylring bilden;
R13 und
R14 ausgewählt sind aus: - (i) R13 ist ausgewählt aus Wasserstoff, gegebenenfalls
substituiertem C1-8-Alkyl, gegebenenfalls
substituiertem Aryl, -Rd-Ar, wobei Rd für
C1-8-Alkylen steht und Ar für gegebenenfalls
substituiertes Aryl steht, und einem gegebenenfalls substituierten
3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis
3 weitere unabhängig
voneinander aus O, N und S ausgewählte Heteroatome enthält; und
R14 ist ausgewählt aus Wasserstoff, gegebenenfalls
substituiertem C1-8-Alkyl und gegebenenfalls
substituiertem Aryl;
- (ii) wenn R5 für eine Gruppe der Formel III-a,
III-b oder III-i steht, steht die Gruppe NR13(-R14) für
einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen
Ring, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und
S ausgewählte
Heteroatome enthält;
oder
- (iii) wenn R5 für Struktur III-e steht, steht
die Gruppefür einen gegebenenfalls substituierten
3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 4
unabhängig
voneinander aus O, N und S ausgewählte Heteroatome enthält; und
deren Salze, Prodrugs und Solvate bereit.
-
Gemäß einem
weiteren Merkmal des ersten Aspekts der Erfindung wird eine pharmazeutische
Formulierung bereitgestellt, die eine Verbindung der Formel (I)
oder ein Salz, eine Prodrug oder ein Solvat davon und ein pharmazeutisch
annehmbares Verdünnungsmittel
oder einen pharmazeutisch annehmbaren Träger enthält.
-
Gemäß einem
weiteren Merkmal des ersten Aspekts der Erfindung werden die folgenden
Verwendungen einer Verbindung der Formel (I) oder eines Salzes,
einer Prodrug oder eines Solvats davon bereitgestellt:
- (a) die Verwendung bei der Herstellung eines Medikaments zum
Antagonisieren der Aktivität
des gonadotropinfreisetzenden Hormons;
- (b) die Verwendung bei der Herstellung eines Medikaments zur
Verabreichung an einen Patienten, zum Absenken der Sezernierung
von luteinisierendem Hormon von der Hypophyse des Patienten; und
- (c) die Verwendung bei der Herstellung eines Medikaments zur
Verabreichung an einen Patienten, zur therapeutischen Behandlung
und/oder Prävention
eines mit Sexualhormonen im Zusammenhang stehenden Leidens des Patienten,
vorzugsweise eines mit Sexualhormonen im Zusammenhang stehenden
Leidens ausgewählt
aus Prostatakrebs und prämenopausalem
Brustkrebs.
-
Ebenfalls
beschrieben ist ein Verfahren zur Antagonisierung der Aktivität des gonadotropinfreisetzenden
Hormons in einem Patienten, bei dem man einem Patienten eine Verbindung
der Formel (I) oder ein Salz, eine Prodrug oder ein Solvat davon
verabreicht.
-
Pharmazeutisch
annehmbare Salze von erfindungsgemäßen Verbindungen sind bevorzugt,
jedoch können
auch andere, nicht pharmazeutisch annehmbare Salze von erfindungsgemäßen Verbindungen
von Nutzen sein, zum Beispiel bei der Herstellung von pharmazeutisch
annehmbaren Salzen von erfindungsgemäßen Verbindungen.
-
Die
Erfindung umfaßt
erfindungsgemäße Verbindungen
und deren Salze, Prodrugs und Solvate; in einer weiteren Ausführungsform
der Erfindung umfaßt
die Erfindung jedoch erfindungsgemäße Verbindungen und deren Salze.
-
In
der vorliegenden Beschreibung kann, wenn nicht anders angegeben,
eine Alkyl-, Alkylen- bzw. Alkenylgruppe jeweils geradkettig oder
verzweigt sein.
-
Der
Ausdruck "Alkylen" bezieht sich auf
-CH2-. C8-Alkylen ist somit
beispielsweise -(CH2)8-.
-
Der
Ausdruck "Aryl" bezieht sich auf
Phenyl oder Naphthyl.
-
Der
Ausdruck "Carbamoyl" bezieht sich auf
die Gruppe -CONH2.
-
Der
Ausdruck "Halogen" bezieht sich auf
Fluor, Chlor, Brom oder Iod.
-
Der
Ausdruck "Heterocyclyl" oder "heterocyclischer
Ring" bezieht sich
auf einen 5- bis 10gliedrigen aromatischen mono- oder bicyclischen
Ring oder einen 5- bis
10gliedrigen gesättigten
oder teilweise gesättigten
mono- oder bicyclischen Ring mit bis zu 5 Heteroatomen unabhängig voneinander
ausgewählt
aus Stickstoff, Sauerstoff oder Schwefel, der über Ringkohlenstoffatome oder
Ringstickstoffatome gebunden ist, wobei, wenn eine Bindung von einem
Stickstoff erlaubt ist, beispielsweise eine Bindung zu dem Stickstoff
eines Pyridinring nicht möglich
ist, aber eine Bindung über den
1-Stickstoff eines Pyrazolrings möglich ist. Beispiele für 5- oder
6gliedrige aromatische heterocyclische Ringe schließen Pyrrolyl,
Furanyl, Imidazolyl, Triazolyl, Pyrazinyl, Pyrimidinyl, Pyridazinyl,
Pyridinyl, Isoxazolyl, Oxazolyl, 1,2,4-Oxadiazolyl, Isothiazolyl, Thiazolyl
und Thienyl ein. Bei einem 9- oder 10gliedrigen bicyclischen aromatischen
heterocyclischen Ring handelt es sich um ein aromatisches bicyclisches
Ringsystem mit einem 6gliedrigen Ring, der entweder mit einem 5gliedrigen Ring
oder einem anderen 6gliedrigen Ring kondensiert ist. Beispiele für bicyclische
5/6- und 6/6-Ringsysteme schließen
Benzofuranyl, Benzimidazolyl, Benzothiophenyl, Benzothiazolyl, Benzisothiazolyl,
Benzoxazolyl, Benzisoxazolyl, Indolyl, Pyridoimidazolyl, Pyrimidoimidazolyl,
Chinolinyl, Isochinolinyl, Chinoxalinyl, Chinazolinyl, Phthalazinyl,
Cinnolinyl und Naphthyridinyl ein. Beispiele für gesättigte oder teilweise gesättigte heterocyclische
Ringe schließen
Pyrrolinyl, Pyrrolidinyl, Morpholinyl, Piperidinyl, Piperazinyl,
Dihidropyridinyl und Dihydropyrimidinyl ein. Diese Definition umfaßt weiterhin
schwefelhaltige Ringe, bei denen das Schwefelatom zu einer S(O)-
oder S(O2)-Gruppe oxidiert wurde.
-
Der
Ausdruck "aromatischer
Ring" bezieht sich
auf einen 5- bis 10gliedrigen aromatischen mono- oder bicyclischen
Ring, der gegebenenfalls bis zu 5 Heteroatome unabhängig voneinander
ausgewählt
aus Stickstoff, Sauerstoff oder Schwefel enthält. Beispiele für solche "aromatischen Ringe" schließen die
folgenden ein: Phenyl, Pyrrolyl, Furanyl, Imidazolyl, Triazolyl,
Pyrazinyl, Pyrimidinyl, Pyridazinyl, Pyridinyl, Isoxazolyl, Oxazolyl,
1,2,4-Oxadiazolyl, Isothiazolyl, Thiazolyl und Thienyl. Bevorzugte
aromatische Ringe schließen
Phenyl, Thienyl und Pyridyl ein.
-
Das
Symbol
zeigt an, wo die betreffende
Gruppe an den Rest des Moleküls
gebunden ist.
-
Um
Zweifel auszuschließen:
wenn
zusammen einen gegebenenfalls
substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis
3 weiteren Heteroatomen unabhängig
voneinander ausgewählt
aus O, N und S bilden, dann cyclisieren die gezeigten Gruppen unter
Bildung eines stickstoffhaltigen heterocylischen Rings, d.h.
der gegebenenfalls 1 bis
3 weitere Heteroatome unabhängig
voneinander ausgewählt
aus O, N und S enthält.
-
Beispiele
für C1-8-Alkyl schließen die folgenden ein: Methyl,
Ethyl, Phenyl, Propyl, Isopropyl, Butyl, Isobutyl, tert.-Butyl und
2-Methylpentyl ein; Beispiele für
C1-8-Alkylen schließen Methylen, Ethylen und 2-Methylpropylen ein;
Beispiele für
C1-8-Alkoxy schließen Methoxy, Ethoxy und Butyloxy
ein; Beispiele für
N-C1-4-Alkylamino
schließen N-Methylamino und N-Ethylamino ein; Beispiele für N,N-Di-C1-4-alkylamino, Beispiele für HO-C2-4-Alkyl-NH schließen Hydroxymethylamino, Hydroxyethylamino
und Hydroxypropylamino ein, Beispiele für HO-C2-4-Alkyl-N(C1-4-alkyl) schließen N-Methyl-hydroxymethylamino,
N-Ethyl-hydroxyethylamino und N-Propylhydroxypropylamino ein.
-
Es
versteht sich, daß insofern
als bestimmte erfindungsgemäße Verbindungen
auf Grund von einem oder mehreren asymmetrisch substituierten Kohlenstoffatomen
in optisch aktiven oder racemischen Formen vorliegen können, die
vorliegende Erfindung in ihrer Definition alle derartigen optisch
aktiven oder racemischen Formen einschließt, die dazu in der Lage sind,
die Aktivität
des Gonadotropin-releasing-Hormons (GnRH) zu antagonisieren. Die
Synthese von optisch aktiven Formen kann nach gut bekannten Standardmethoden
der organischen Chemie erfolgen, beispielsweise durch Synthese aus
optisch aktiven Ausgangsmaterialien oder durch Trennung einer racemischen
Form. Ganz ähnlich
kann die oben aufgeführte
Wirkung mit Hilfe der standardmäßigen Labortechniken,
auf die nachstehend Bezug genommen wird, beurteilt werden.
-
Die
Erfindung betrifft jegliche und alle tautomeren Formen der Verbindungen
der verschiedenen Merkmale der Erfindung, die dazu in der Lage sind,
die Aktivität
des Gonadotropin-releasing-Hormons (GnRH) zu antagonisieren.
-
Es
versteht sich auch, daß bestimmte
Verbindungen der vorliegenden Erfindung in solvatisierten, wie beispielsweise
hydratisierten Formen, sowie unsolvatisierten Formen existieren
können.
Es versteht sich, daß die
vorliegende Erfindung alle derartigen solvatisierten Formen einschließt, die
dazu in der Lage sind, die Aktivität des Gonadotropin-releasing-Hormons
(GnRH) zu antagonisieren.
-
Bevorzugte
Verbindungen der Formel (I) sind die, auf die eine der folgenden
Aussagen oder eine Kombination der folgenden Aussagen zutreffen.
-
A
steht vorzugsweise für
gegebenenfalls substituiertes C1-5-Alkylen,
besonders bevorzugt für
C1-4-Alkylen, ganz besonders bevorzugt für Methylen
oder Ethylen.
-
M
steht vorzugsweise für
-CH2-O-.
-
R1 steht vorzugsweise für Wasserstoff oder gegebenenfalls
substituiertes C1-6-Alkyl. Besonders bevorzugt
steht R1 für Wasserstoff, Methyl, Ethyl
oder tert.-Butyl.
Ganz besonders bevorzugt steht R1 für Wasserstoff.
-
R
2 steht vorzugsweise für eine gegebenenfalls substituierte
monocyclische aromatische Ringstruktur, bei der die gegebenenfalls
vorhandenen Substituenten ausgewählt
sind aus Cyano, NR
eR
f,
gegebenenfalls substituiertem C
1-8-Alkyl
(vorzugsweise C
1-4-Alkyl, zum Beispiel Methyl
oder Ethyl), gegebenenfalls substituiertem C
1-8-Alkoxy
(vorzugsweise C
1-6-Alkoxy, zum Beispiel
Methoxy, Ethoxy oder tert.-Butoxy) oder Halogen (vorzugsweise F,
Br oder Cl), wobei R
e und R
f unabhängig voneinander
ausgewählt
sind aus Wasserstoff, C
1-6-Alkyl oder Aryl.
Besonders bevorzugt steht R
2 für gegebenenfalls
substituiertes Phenyl, wobei die gegebenenfalls vorhandenen Substituenten
ausgewählt
sind aus Cyano, NR
eR
f,
gegebenenfalls substituiertem C
1-4-Alkyl,
gegebenenfalls substituiertem C
1-6-Alkoxy,
F, Br oder Cl, wobei R
e und R
f wie
oben definiert sind. Noch mehr bevorzugt steht R
2 für gegebenenfalls
substituiertes Phenyl, wobei die gegebenenfalls vorhandenen Substituenten
ausgewählt
sind aus Methyl, Ethyl, Methoxy, Ethoxy, tert.-Butoxy, F oder Cl.
Ganz besonders bevorzugt steht R
2 für
wobei Me für Methyl
steht. R
2 trägt vorzugsweise 1, 2 oder 3
Substituenten.
-
Vorzugsweise
sind R3 und R3a unabhängig voneinander
ausgewählt
aus Wasserstoff; gegebenenfalls substituiertem C1-6-Alkyl
und gegebenenfalls substituiertem Aryl. Besonders bevorzugt sind
R3 und R3a unabhängig voneinander
ausgewählt
aus Methyl, Ethyl, tert.-Butyl und Phenyl.
-
Vorzugsweise
ist R
5 ausgewählt aus einer Gruppe der Formel
III-a, III-g, III-h; III-i oder: III-i;
-
Weiter
bevorzugt ist R
5 ausgewählt aus einer der folgenden
Gruppen:
-
Noch
mehr bevorzugt ist R
5 ausgewählt aus
einer der folgenden Gruppen:
wobei
Me für
Methyl steht.
-
Ganz
besonders bevorzugt ist R
5 ausgewählt aus
einer der folgenden Gruppen:
-
Bei
einer Ausführungsform
stehen R6 und R6a jeweils
für Wasserstoff
und A steht für
C1-4-Alkylen (vorzugsweise Methylen).
-
Bei
einer weiteren Ausführungsform
steht R6 für Wasserstoff, R6a steht
für Methyl
und A steht für C1-4-Alkylen (vorzugsweise Methylen).
-
Vorzugsweise
ist R7 ausgewählt aus Wasserstoff oder gegebenenfalls
substituiertem C1-6-Alkyl. Weiter bevor zugt
steht R7 für Wasserstoff, Methyl, Ethyl
oder tert.-Butyl.
-
Vorzugsweise
stehen X und R8 jeweils für:
- (a) X steht für N und R8 steht
für Cyano
oder -C(O)O-Rb; oder
- (b) X steht für
N und R8 steht für Wasserstoff.
-
Besonders
bevorzugt steht X für
N und R8 steht für Cyano oder -C(O)O-Rb; wobei Rb für C1-6-Alkyl steht. Bei einer weiteren Ausführungsform
der Erfindung steht X für
N und R8 steht für -CONRbRc, wobei Rb und Rc wie oben definiert sind.
-
Vorzugsweise
ist R9 Teil einer Gruppe N(R9R10) oder steht für Wasserstoff, gegebenenfalls
substituiertes Aryl, einen gegebenenfalls substituierten 3- bis
10gliedrigen heterocyclischen Ring, oder gegebenenfalls substituiertes
C1-4-Alkyl, wobei die gegebenenfalls vorhandenen
Substituenten ausgewählt
sind aus: Hydroxy, Amino, Nitro, Cyano, gegebenenfalls substituiertem
Aryl, gegebenenfalls substituiertem 3- bis 8gliedrigem Heterocyclyl
mit 1 bis 4 Heteroatomen unabhängig
voneinander ausgewählt
aus O, N und S, -O-Rb, C(O)NRbRc, -NRbRc,
-NRcC(O)-Rb; -C(O)NRbRc, -NRcS(O0-2)Rb, -S(O0-2)Rb, wobei Rb und Rc wie oben
definiert sind.
-
Steht
R9 für
eine C1-6-Alkylgruppe substituiert durch
einen gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen
Ring mit 1 bis 4 Heteroatomen unabhängig voneinander ausgewählt aus
O, N und S, so ist der heterocyclische Ring vorzugsweise ausgewählt aus
Pyridyl, Thienyl, Piperidinyl, Imidazolyl, Triazolyl, Thiazolyl,
Pyrrolidinyl, Piperazinyl, Morpholinyl, Imidazolinyl, Benzotriazolyl,
Benzimidazolyl, Pyrimidinyl, Pyrazinyl, Pyridazinyl, Oxazolyl, Furanyl,
Pyrrolyl, 1,3-Dioxolanyl, 2-Azetinyl, die jeweils gegebenenfalls substituiert
sind. Besonders bevorzugt eine Gruppe der Formel VI-a, VI-b, VI-c,
VI-d, VI-e, VI-f, VI-g, VI-h, VI-i, VI-j oder VI-k; wobei jede Gruppe
gegebenenfalls substituiert ist durch eine oder mehrere Gruppen
ausgewählt aus
R16.
-
-
Ganz
besonders bevorzugt eine Gruppe der Formel VI-b, VI-i oder VI-j:
wobei
R
16 für Wasserstoff,
Aryl, einen 3- bis 10gliedrigen heterocyclischen Ring oder gegebenenfalls
substituiertes C
1-4-Alkyl steht, wobei die
gegebenenfalls vorhandenen Substituenten ausgewählt sind aus: Hydroxy, Amino, Nitro,
Cyano, gegebenenfalls substituiertem Phenyl, gegebenenfalls substituiertem
3- bis 8gliedrigem Heterocyclyl mit 1 bis 4 Heteroatomen unabhängig voneinander
ausgewählt
aus O, N und S, -O-R
b, C(O)NR
bR
c, -NR
bR
c,
-NR
cC(O)R
b; -C(O)NR
bR
c, -N(R
c)S(O
0-2)R
b, -S(O
0-2)R
b, wobei R
b und R
c wie oben definiert sind.
-
Vorzugsweise
ist R10 Teil einer Gruppe N(R9R10) oder steht für gegebenenfalls substituiertes
C1-6-Alkyl. Weiter bevorzugt ist R10 Teil einer Gruppe N(R9R10) oder ist ausgewählt aus: Methyl, Ethyl oder
tert.-Butyl.
-
Steht
N(R
9R
10) für einen
gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen
Ring, so ist N(R
9R
10)
vorzugsweise ausgewählt
aus einem 5- oder 6gliedrigen monocyclischen Ring mit zwischen 1
und 3 (vorzugsweise 1 oder 2) Heteroatomen unabhängig voneinander ausgewählt aus
O, N und S. besonders bevorzugt einem 5- oder 6gliedrigen monocyclischen
Ring mit zwischen 1 und 3 (vorzugsweise 1 oder 2) Heteroatomen unabhängig voneinander
ausgewählt
aus O, N und S, ausgewählt
aus Pyrrolidinyl, Thienyl, Pyrazolidinyl, Piperidinyl, Morpholinyl,
Thiomorpholinyl, Piperazinyl, Imidazol, Azetidinyl oder Azetinyl.
Weiter bevorzugt handelt es sich bei der Struktur N(R
9R
10) um einen heterocyclischen Ring ausgewählt aus
einer gegebenenfalls substituierten Gruppe der Formel IV-a, IV-b,
IV-c, IV-d und IV-e, wobei die gegebenenfalls vorhandenen Substituenten
vorzugsweise aus den unter für
R
15 aufgeführten Gruppen ausgewählt sind
-
Weiter
bevorzugt ist die Struktur N(R
9R
10) ausgewählt aus einer Gruppe der Formel
V-a, V-b oder V-c:
-
Ganz
besonders bevorzugt handelt es sich bei der Struktur N(R9R10) um eine Gruppe
der Formel V-c;
R15 steht für Wasserstoff,
gegebenenfalls substituiertes Aryl, einen gegebenenfalls substituierten
3- bis 10gliedrigen
heterocyclischen Ring oder gegebenenfalls substituiertes C1-4-Alkyl, wobei die gegebenenfalls vorhandenen
Substituenten an Aryl, einem heterocyclischen Ring oder C1-4-Alkyl ausgewählt sind aus: Hydroxy, Amino,
Nitro, Cyano, Halogen, gegebenenfalls substituiertem Aryl, gegebenenfalls
substituiertem 3- bis 8gliedrigem Heterocyclyl mit 1 bis 4 Heteroatomen
unabhängig
voneinander ausgewählt
aus O, N und S. -O-Rb, C(O)RbRc, -NRbRc,
-NRcC(O)-Rb; -C(O)NRbRc, -N(Rc)S(O0-2)Rb, -S(O0-2)Rb, wobei Rb und Rc wie oben definiert sind. Vorzugsweise steht
R15 für
Heterocyclyl. Weiter bevorzugt ist R15 ausgewählt aus:
Pyridyl, Pyrazinyl, Pyridazinyl, Pyrimidinyl oder Thiazolyl. Ganz
besonders bevorzugt steht R15 für Pyridyl.
-
Bei
einer weiteren Ausführungsform
der Erfindung steht N(R9R10)
für einen
gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen
Ring, wobei die gegebenenfalls vorhandenen Substituenten ausgewählt sind
aus R15, wie oben definiert.
-
Vorzugsweise
sind R12 und R12a unabhängig voneinander
ausgewählt
aus: Wasserstoff, gegebenenfalls substituiertem C1-6-Alkyl,
oder R12 und R12a bilden
zusammen mit dem Kohlenstoff, an den sie gebunden sind, einen gegebenenfalls
substituierten 3- bis 6gliedrigen Cycloalkylring. Weiter bevorzugt
sind R12 und R12a unabhängig voneinander
ausgewählt
aus: Wasserstoff, Methyl, Ethyl oder tert.-Butyl. Ganz besonders
bevorzugt stehen R12 und R12a beide
für Methyl.
-
Vorzugsweise
sind R13 und R14 unabhängig voneinander
ausgewählt
aus Wasserstoff, gegebenenfalls substituiertem C1-6-Alkyl,
gegebenenfalls substituiertem Phenyl und -Rd-Phenyl,
wobei Rd für C1-6-Alkylen
oder einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen
Ring (vorzugsweise einen 5- oder 6gliedrigen monocyclischen Ring)
mit 1 bis 3 (vorzugsweise 1 oder 2) weiteren Heteroatomen unabhängig voneinander
ausgewählt
aus O, N und S steht. Weiter bevorzugt sind R13 und
R14 unabhängig voneinander ausgewählt aus
Wasserstoff oder C1-6-Alkyl.
-
Dort,
wo verschiedentlich eine gegebenenfalls vorhandene Substitution
erwähnt
wird, bezieht sich dies auf einen, zwei, drei oder mehr gegebenenfalls
vorhandene Substituenten. Wenn oben nicht anders angegeben (d.h.
wenn eine Liste gegebenenfalls vorhandener Substituenten angegeben
ist), können
die Substituenten jeweils unabhängig
voneinander ausgewählt
sein aus C1-8-Alkyl (zum Beispiel C2-6-Alkyl und ganz besonders bevorzugt Methyl,
Ethyl oder tert.-Butyl); C3-8-Cycloalkoxy,
vorzugsweise Cyclopropoxy, Cyclobutoxy oder Cyclopentoxy; C1-6-Alkoxy, vorzugsweise Methoxy oder C2-4-Alkoxy; Halogen, vorzugsweise Cl oder
F; Hal3C-, Hal2CH-,
HalCH2-, Hal3CO-,
Hal2CHO oder HalCH2O,
wobei Hal für
Halogen (vorzugsweise F) steht; RgCH2O-, RbC(O)N(R)-,
RhSO2N(R)- oder
RgRhN-, wobei Rg und Rh unanbhängig für Wasserstoff
oder C1-8-Alkyl (vorzugsweise Methyl oder
C2-6-Alkyl oder C2-4-Alkyl)
stehen, oder Rg-RhN-
für einen
gegebenenfalls substituierten heterocyclischen C3-8-,
vorzugsweise C3-6-, Ring steht, der gegebenenfalls
1 bis 3 weitere Heteroatome unabhängig voneinander ausgewählt aus
O, N und S enthält;
Wasserstoff; oder RkC(O)O- oder RkC(O)-, wobei Rk für Wasserstoff,
gegebenenfalls substituiertes Phenyl oder C1-6-Alkyl
(vorzugsweise Methyl, Ethyl, Isopropyl oder tert.-Butyl) steht.
Bei der durch Rg-RhN-
wiedergegebenen gegebenenfalls vorhandenen Substitution des heterocyclischen
Rings kann wenigstens ein Substituent (zum Beispiel ein, zwei oder
drei Substituenten) unabhängig
ausgewählt
werden aus C1-6-Alkyl (zum Beispiel C2-4-Alkyl, mehr bevorzugt Methyl); Phenyl;
CF3O-; F2CHO-; C1-8-Alkoxy, vorzugsweise Methoxy, Ethoxy
oder C3-6-Alkoxy; C1-8-C(O)-Alkoxy,
vorzugsweise Methoxycarbonyl, Ethoxycarbonyl, tert.-Butoxycarbonyl oder
C3-6-C(O)-Alkoxy; Phenoxycarbonyl; Phenoxy;
C1-8-Alkanoyl, vorzugsweise Acetyl, Ethanoyl
oder C3-6-Alkanoyl; Carboxy; C1-8-S(O)nn-Alkyl, wobei nn für eine ganze Zahl zwischen
0 und 2 steht; vorzugsweise Methylthio, Ethylthio, C3-6-Alkylthio,
Methylsulfinyl, Ethylsulfinyl, C3-6-Alkylsulfinyl,
Methylsulfonyl, Ethylsulfonyl oder C3-6-Alkylsulfonyl;
Hydroxy; Halogen (zum Beispiel F, Cl oder Br); RmRnN-, wobei Rm und
Rn unabhängig
voneinander für
Wasserstoff oder C1-6-Alkyl (vorzugsweise C2-4-Alkyl, mehr bevorzugt Methyl, ganz besonders
bevorzugt Rm = Rn =
Methyl) stehen; und Nitro.
-
Dort,
wo verschiedentlich eine gegebenenfalls vorhandene Substitution
eines Rings erwähnt
wird, bezieht sich dies ganz besonders bevorzugt auf einen, zwei,
drei oder mehrere Substituenten ausgewählt aus C1-8-Alkyl (zum Beispiel
C2-6-Alkyl, und ganz besonders bevorzugt
Methyl); C1-8-Alkoxy, vorzugsweise Methoxy, Ethoxy
oder C3-6-Alkoxy; C1-8-S(O)-Alkyl,
wobei nn für
eine ganz Zahl zwischen 0 und 2 steht; vorzugsweise Methylthio,
Ethylthio, C3-5-Alkylthio, Methylsulfinyl,
Ethylsulfinyl, C3-6-Alkylsulfinyl, Methylsulfonyl,
Ethylsulfonyl oder C3-6-Alkylsulfonyl; Halogen
(zum Beispiel F, Cl oder Br); Cyano; und NO2.
-
Eine
bevorzugte Gruppe erfindungsgemäßer Verbindungen umfaßt Verbindungen
der Formel (I), in denen:
R11 für eine Gruppe
der Formel: N(R9R10)
steht; und
N(R9R10)
für einen
gegebenenfalls substituierten heterocyclischen 3- bis 8gliedrigen
Ring steht, der gegebenenfalls 1 bis 3 weitere Heteroatome unabhängig voneinander
ausgewählt
aus O, N und S enthält,
vorzugsweise substituiert durch Heterocyclyl;
und deren Salze,
Prodrugs und Solvate.
-
Eine
bevorzugte Gruppe erfindungsgemäßer Verbindungen
umfaßt
Verbindungen der Formel (I), in denen:
R11 für eine Gruppe
der Formel: N(R9R10)
steht;
R9 für eine C1-6-Alkylgruppe
substituiert durch einen gegebenenfalls
substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis
4 Heteroatomen unabhängig
voneinander ausgewählt
aus O, N und S steht; und
R10 für Wasserstoff
oder C1-6-Alkyl steht
und deren Salze,
Prodrugs und Solvate.
-
Eine
bevorzugte Gruppe erfindungsgemäßer Verbindungen
umfaßt
Verbindungen der Formel (Ia):
Formel
(Ia) in denen:
A, B, M, X, R
1,
R
5, R
6, R
6a, R
7, R
8, R
9, R
10,
R
11, R
12 und R
12a Wie oben definiert sind; und deren Salze,
Prodrugs und Solvate.
-
Eine
bevorzugte Gruppe erfindungsgemäßer Verbindungen
umfaßt
Verbindungen der Formel (Ib):
Formel
(Ib) in denen:
R
5 ausgewählt ist
aus: III-a, III-b, III-g, III-i oder III-i:
und A,
B, M, X, R
1, R
5,
R
6, R
6a, R
7, R
8, R
9,
R
10, R
11, R
12, R
12a, R
13 und R
14 wie oben
definiert sind;
und deren Salze, Prodrugs und Solvate.
-
Eine
weitere bevorzugte Gruppe erfindungsgemäßer Verbindungen umfaßt Verbindungen
der Formel (Ic):
Formel
(Ic) in denen:
R
5 ausgewählt ist
aus: III-a, III-b, III-g, III-i oder III-j:
und A,
B, M, X, R
1, R
6,
R
6a, R
7, R
8, R
9, R
10,
R
11, R
12, R
12a, R
13 und R
14 wie oben definiert sind;
und deren
Salze, Prodrugs und Solvate.
-
Eine
noch weiter bevorzugte Gruppe von erfindungsgemäßen Verbindungen umfaßt Verbindungen
der Formeln (Ia), (Ib) und (Ic), in denen:
R
5 für eine Gruppe
der Formel III-a:
steht;
NR
13(-R
14) für
einen gegebenenfalls substituierten 7- bis 8gliedrigen bicyclischen heterocyclischen
Ring steht und A, B, X, R
1, M, R
6, R
6a, R
7, R
8, R
9,
R
10, R
11, R
12 und R
12a wie oben
definiert sind;
und deren Salze, Prodrugs und Solvate.
-
Eine
bevorzugte Verbindung gemäß der vorliegenden
Erfindung ist:
3-[2,2-Dimethyl-3-oxo-3-(azabicyclo[2.2.1]heptan-7-yl)propyl]-4-[1S-methyl-2-(N'-isopropoxycarbonyl-3-pyrid-4-ylpyrrolidin-1-ylcarboximidamido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol;
und
deren Salze, Prodrugs und Solvate.
-
Bei
einer weiteren Ausführungsform
sind besonders bevorzugte Verbindungen gemäß der vorliegenden Erfindung
die, bei denen die Verbindung ausgewählt ist aus:
(12)-({2-[3-(2,2-Dimethyl-3-oxo-3-pyrrolidin-1-ylpropoxy)-5-(3,5-dimethylphenyl)-1H-pyrazol-4-yl]ethyl}amino)(3-pyridin-4-ylpyrrolidin-1-yl)methylidencarbaminsäureisopropylester;
(12)-({2-[3-(2,2-Dimethyl-3-oxo-3-(7-azabicyclo[2.2.1]hegt-7-yl)propoxy)-5-(3,5-dimethylphenyl)-1H-pyrazol-4-yl]ethyl}amino)(3-pyridin-4-ylpyrrolidin-1-yl)methylidencarbaminsäureisopropylester; (1Z)-({2-[3-[3-(Diethylamino)-2,2-dimethyl-3-oxopropoxy]-5-(3,5-dimethylphenyl)-1H-pyrazol-4-yl]ethyl}amino)(3-pyridin-4-ylpyrrolidin-1-yl)methylidencarbaminsäureisopropylester;
N-{2-[3-[3-(Diethylamino)-2,2-dimethyl-3-oxopropoxy]-5-(3,5-dimethylphenyl)-1H-pyrazol-4-yl]ethyl}-3-pyridin-4-ylpyrrolidin-1-carbonsäureamid;
und
deren Salzen, Prodrugs und Solvaten.
-
Gemäß einem
weiteren Merkmal des ersten Aspekts der Erfindung wird eine pharmazeutische
Formulierung enthaltend eine Verbindung der Formel (Ia), der Formel
(Ib) oder der Formel (Ic) oder bevorzugte erfindungsgemäße Verbindungen
oder ein Salz, eine Prodrug oder ein Solvat davon und ein pharmazeutisch
annehmbares Verdünnungsmittel
oder einen pharmazeutisch annehmbaren Träger bereitgestellt.
-
Gemäß einem
weiteren Merkmal des ersten Aspekts der Erfindung werden die folgenden
Verwendungen einer Verbindung der Formel (Ia), der Formel (Ib) oder
der Formel (Ic) oder bevorzugter erfindungsgemäßer Verbindungen oder eines
Salzes, einer Prodrug oder eines Solvates davon bereitgestellt:
- (a) die Verwendung bei der Herstellung eines
Medikaments zum Antagonisieren der Aktivität des gonadotropinfreisetzenden
Hormons;
- (b) die Verwendung bei der Herstellung eines Medikaments zur
Verabreichung an einen Patienten, zum Absenken der Sezernierung
von luteinisierendem Hormon von der Hypophyse des Patienten; und
- (c) die Verwendung bei der Herstellung eines Medikaments zur
Verabreichung an einen Patienten, zur therapeutischen Behandlung
und/oder Prävention
eines mit Sexualhormonen im Zusammenhang stehenden Leidens des Patienten,
vorzugsweise eines mit Sexualhormonen im Zusammenhang stehenden
Leidens ausgewählt
aus Prostatakrebs und prämenopausalem
Brustkrebs.
-
Die
Verbindungen der Formel (I) können
in Form einer Prodrug verabreicht werden, die im Körper des Menschen
bzw. Tieren zur Verbindung der Formel (I) abgebaut wird. Zu den
Prodrugs zählen
beispielsweise invivo hydrolysierbare Ester einer Verbindung der
Formel (I). Im Stand der Technik sind verschiedene Prodrugs bekannt.
Beispiele für
solche Prodrug-Derivate finden sich in:
- a) Design of Prodrugs,
herausgegeben von H. Bundgaard, (Elsevier, 1985) und Methods in
Enzymology, Band 42, S. 309-396, herausgegeben von K. Widder, et
al. (Academic Press, 1985);
- b) A Textbook of Drug Design and Development, herausgegeben
von Krogsgaard-Larsen und H. Bundgaard, Kapitel 5 "Design and Application
of Prodrugs", von
H. Bundgaard S.113-191 (1991);
- c) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
- d) H. Bundgaard, et al., Journal of Pharmaceutical Sciences,
77, 285 (1988); und
- e) N. Kakeya, et al., Chem Pharm Bull, 32, 692 (1984).
-
Ein
in vivo hydrolysierbarer Ester einer Verbindung der Formel (I) mit
einer Carboxyl- oder Hydroxylgruppe ist beispielsweise ein pharmazeutisch
annehmbarer Ester, der im Körper
des Menschen oder eines Tieres zur Säure- bzw. Alkoholstammverbindung hydrolysiert
wird. Zu den geeigneten pharmazeutisch annehmbaren Estern für Carboxy
gehören
C1-6-Alkoxymethylester, beispielsweise Methoxymethyl,
C1-6-Alkanoyloxymethylester, beispielsweise
Pivaloyloxymethyl, Phthalidylester, C3-8-Cycloalkoxycarbonyloxy-C1-6-alkylester, beispielsweise 1-Cyclohexylcarbonyloxyethyl;
1,3-Dioxolen-2-onylmethylester, beispielsweise 5-Methyl-1,3-dioxolen-2-onylmethyl; und C1-6-Alkoxycarbonyloxyethylester.
-
Zu
den in vivo hydrolysierbaren Estern einer Verbindung der Formel
(I) mit einer Hydroxylgruppe gehören
anorganische Ester wie Phosphatester (einschließlich cyclischer Phosphoramidester)
und α-Acyloxyalkylether
und verwandte Verbindungen, bei denen der Ester in vivo unter Freigabe
der Hydroxyl-Ausgangsgruppe hydrolysiert wird. α-Acyloxyalkylether schließen beispielsweise
Acetoxymethoxy und 2,2-Dimethylpropionyloxymethoxy ein. Eine Auswahl
von Gruppen, die mit Hydroxyl in vivo hydrolysierbare Ester bilden,
schließt Alkanoyl,
Benzoyl, Phenylacetyl und substituiertes Benzoyl, sowie Phenylacetyl,
Alkoxycarbonyl (was Alkylcarbonsäureester
ergibt), Dialkylcarbamoyl und N-(Dialkylaminoethyl)-N-alkylcarbamoyl (was
Carbamate ergibt), Dialkylaminoacetyl und Carboxyacetyl ein.
-
Ein
geeignetes pharmazeutisch annehmbares Salz einer erfindungsgemäßen Verbindung
ist beispielsweise ein Säureadditionssalz
einer erfindungsgemäßen Verbindung
mit ausreichender Basizität,
beispielsweise ein Säureadditionssalz
mit beispielsweise einer anorganischen oder organischen Säure, beispielsweise
Salzsäure,
Bromwasserstoffsäure,
Schwefelsäure,
Phosphorsäure,
Trifluoressigsäure,
Citronensäure oder
Maleinsäure.
Ein geeignetes pharmazeutisch annehmbares Salz einer erfindungsgemäßen Verbindung mit
ausreichender Acidität
ist außerdem
ein Alkalimetallsalz, beispielsweise ein Natrium- oder Kaliumsalz,
ein Erdalkalimetallsalz, beispielsweise ein Calcium- oder Magnesiumsalz,
ein Ammoniumsalz oder ein Salz mit einer organischen Base, die ein
physiologisch annehmbares Kation liefert, beispielsweise ein Salz
mit Methylamin, Dimethylamin, Trimethylamin, Piperidin, Morpholin
oder Tris(2-hydroxyethyl)amin.
-
Die
Verbindungen der Formel (I) lassen sich nach einem Verfahren darstellen,
welches einen Schritt ausgewählt
aus:
(a) bis (f) wie folgt umfaßt; diese Verfahren werden
als ein weiteres Merkmal der Erfindung bereitgestellt:-
- (a) bei Verbindungen, in denen X für N steht und R8 für CN steht,
die Umsetzung einer Verbindung der Formel XXXII wie folgt
- b) bei Verbindungen, in denen X für N steht und R8 für Wasserstoff
steht, die Spaltung der Cyanogruppe einer Verbindung der Formel
XXXIII in Gegenwart einer Säure
unter Bildung einer Verbindung der Formel XXXIV
- (c) bei Verbindungen, in denen X für CH steht und R8 für NO2 steht, die Umsetzung einer Verbindung der Formel
XXXV wie folgt
- (d) bei Verbindungen, in denen X-R8 für 0 steht,
die Umsetzung eine Verbindung der Formel XXXVII wie folgt
- (e) bei Verbindungen, in denen X-R8 für O steht, die
Umsetzung einer Verbindung der Formel XXXIX wie folgt
- (f) zur Bildung einer Verbindung, in welcher X für Stickstoff
steht, die Umsetzung einer Verbindung der Formel XXXXI wie folgt und anschließend, falls
erforderlich:
i) die Umwandlung einer Verbindung der Formel
(I) in eine andere Verbindung der Formel (I);
ii) die Abspaltung
gegebenenfalls vorhandener Schutzgruppen;
iii) die Bildung
eines Salzes, einer Prodrug oder eines Solvats.
-
Wie
für den
Fachmann leicht ersichtlich ist, müssen bei den erfindungsgemäßen Verfahren
bestimmte funktionelle Gruppen, wie Hydroxyl- oder Aminogruppen,
in den Ausgangsmaterialien oder Zwischenverbindungen möglicherweise
durch Schutzgruppen geschützt
werden. Somit kann die Herstellung der Verbindungen der Formel (I)
die Abspaltung einer oder mehrerer Schutzgruppen in einer geeigneten
Stufe umfassen.
-
Die
Schätzung
und Entschützung
funktioneller Gruppen ist in „Protective
Groups in Organic Chemistry",
herausgegeben von J.W.F. McOmie, Plenum Press (1973), und „Protective
Groups in Organic Synthesis", 2.
Auflage, T.W. Greene & P.G.M.
Wuts, Wiley-Interscience (1991) beschrieben.
-
Geeignete
Schutzgruppen für
eine Amino- oder Alkylaminogruppe sind beispielsweise eine Acylgruppe,
zum Beispiel eine Alkanoylgruppe wie Acetyl, eine Alkoxycarbonylgruppe,
beispielsweise eine Methoxycarbonyl-, Ethoxycarbonyl- oder tert.-Butoxycarbonylgruppe,
eine Arylmethoxycarbonylgruppe, beispielsweise Benzyloxycarbonyl,
oder eine Aroylgruppe, beispielsweise Benzoyl. Die Entschützungsbedingungen
für die oben
aufgeführten
Schutzgruppen hängen
natürlich
von der gewählten
Schutzgruppe ab. So kann man beispielsweise eine Acylgruppe wie
eine Alkanoyl- oder Alkoxycarbonylgruppe oder eine Aroylgruppe zum
Beispiel durch Hydrolyse mit einer geeigneten Base wie einem Alkalimetallhydroxid,
beispielsweise Lithium- oder Natriumhydroxid, abspalten. Alternativ
dazu kann man eine Acylgruppe wie eine tert.-Butoxycarbonylgruppe beispielsweise
durch Behandlung mit einer geeigneten Säure wie Salzsäure, Schwefelsäure oder
Phosphorsäure
oder Trifluoressigsäure
entfernen, und eine Arylmethoxycarbonylgruppe wie eine Benzyloxycarbonylgruppe
kann zum Beispiel durch Hydrierung an einem Katalysator wie Palladium
auf Aktivkohle oder durch Behandeln mit einer Lewissäure, beispielsweise
Bortris(trifluoracetat), abgespalten werden. Eine geeignete alternative
Schutzgruppe für
eine primäre
Aminogruppe ist beispielsweise eine Phthaloylgruppe, die sich durch
Behandeln mit einem Alkylamin, beispielsweise Dimethylaminopropylamin,
oder mit Hydrazin, entfernen läßt.
-
Eine
geeignete Schutzgruppe für
eine Hydroxygruppe ist beispielsweise eine Acylgruppe, zum Beispiel
eine Alkanoylgruppe wie Acetyl, eine Aroylgruppe wie zum Beispiel
Benzoyl, oder eine Arylmethylgruppe, zum Beispiel Benzyl. Die Entschützungsbedingungen
für die
oben aufgeführten
Schutzgruppen hängen
natürlich
von der gewählten
Schutzgruppe ab. So kann man beispielsweise eine Acylgruppe wie
eine Alkanoyl- oder eine Aroylgruppe zum Beispiel durch Hydrolyse
mit einer geeigneten Base wie einem Alkalimetallhydroxid, beispielsweise
Lithium- oder Natriumhydroxid, entfernen. Alternativ dazu kann eine
Arylmethylgruppe wie z.B. eine Benzylgruppe zum Beispiel durch Hydrierung
an einem Katalysator wie Palladium auf Aktivkohle abgespalten werden.
-
Eine
geeignete Schutzgruppe für
eine Carboxygruppe ist beispielsweise eine Veresterungsgruppe, beispielsweise
eine Methyl- oder eine Ethylgruppe, die zum Beispiel durch Hydrolyse
mit einer Base wie Natriumhydroxid entfernt werden kann, oder beispielsweise
eine tert.-Butylgruppe,
die zum Beispiel durch Behandlung mit einer Säure, beispielsweise einer organischen
Säure wie
Trifluoressigsäure,
entfernt werden kann, oder beispielsweise eine Benzylgruppe, die
zum Beispiel durch Hydrierung an einem Katalysator wie Palladium
auf Aktivkohle entfernt werden kann.
-
EXPERIMENTELLER TEIL
-
ALLGEMEINE REAKTIONSSCHEMATA
-
-
Pyrazole
wie 3 lassen sich in zwei Schritten synthetisieren (Schema a):
- (1) durch die Umsetzung eines Lactons mit dem
entsprechenden Ester unter Anwendung einer Claisen-Kondensation unter
Bildung einer Verbindung der Formel 2, unter einer inerten Atmosphäre wie Argon bei
einer Temperatur von etwa 0ºC
in einem geeigneten Lösungsmittel
wie THF.
- (2) und anschließende
Cyclisierung einer Verbindung der Formel 2 mit Hydrazin unter Bildung
des Pyrazols 3 bei Raumtemperatur in einem geeigneten Lösungsmittel
wie Ethanol.
-
-
Das
Pyrazol 3 kann mit einer Verbindung der Formel 4 in einer inerten
Atmosphäre
wie Argon in Gegenwart einer geeigneten Base wie Kaliumcarbonat
in einem geeigneten Lösungsmittel
wie DMA bei einer Temperatur von etwa 90ºC eine selektive Alkylierung
unter Bildung einer Verbindung der Formel 5 durchlaufen. Das Amin
6 läßt sich
dann aus einer Verbindung der Formel 5 und Phthalimid in einer Mitsunobu-Reaktion
mit einem Aktivierungsmittel wie Azodicarbonsäurediethylester (DEAD), Azodicarbonsäurediisopropylester
oder dergleichen mit Triphenylphosphin, Tributylphosphin und dergleichen
in einem inerten Lösungsmittel
wie Benzol, Toluol, Tetrahydrofuran oder Mischungen davon und anschließendem Entschützen mit
Hydrazin (Schema b) darstellen.
-
-
Ein
geeignetes Pyrazol 6 läßt sich
entweder durch direktes Behandeln mit einem Isocyanat in einem inerten
Lösungsmittel
wie Methylenchlorid, Chloroform oder THF und dergleichen oder nach
einer zweistufigen Reaktionsvorschrift mit Triphosgen (6 → 7) und
anschließender
Zugabe eines entsprechend substituierten Amins (7 → 8) in einen
Harnstoff umwandeln, wodurch man 8 erhält (Schema c).
-
Ein
geeignetes Pyrazol (6) läßt sich
durch Umsetzung mit einem geeigneten Isothiocyanat (9) unter Bildung
einer Verbindung der Formel 10 und anschließender Verdrängung durch
ein geeignetes Amin (11) in ein Guanidin oder Guanidinderivat (12)
umwandeln (Schema d).
-
Gemäß einem
weiteren Merkmal der Erfindung wird daher ein Verfahren zur Synthese
eines substituierten Pyrazols der Formel XXXXIV bereitgestellt,
bei dem man eine Verbindung der Formel XXXXIII mit Hydrazin umsetzt.
wobei:
R
2 wie oben definiert ist; und
R
X und R
Y unabhängig voneinander
ausgewählt
sind aus: gegebenenfalls substituiertem Alkyl, gegebenenfalls substituiertem
Aryl oder gegebenenfalls substituiertem Heterocyclyl.
-
Gemäß einem
weiteren Merkmal der Erfindung wird ein wie oben definiertes Zwischenprodukt
der Formel XXXXIII bereitgestellt.
-
BEISPIELE
-
Die
Erfindung wird nun in den folgenden nichteinschränkenden Beispielen erläutert, wobei,
sofern nicht anders vermerkt:
- (i) Eindampfungen
am Rotationsverdampfer im Vakuum durchgeführt wurden und die Aufarbeitung
nach dem Abfiltrieren von restlichen Feststoffen wie Trockenmitteln
erfolgte;
- (ii) alle Umsetzungen bei Raumtemperatur, das heißt im Bereich
von 18-25°C,
und unter einer Inertgasatmosphäre
wie einer Argonatmosphäre
oder einer Stickstoffatmosphäre
durchgeführt
wurden;
- (iii) Ausbeuten nur zur Erläuterung
angegeben sind und nicht unbedingt das erzielbare Maximum darstellen;
- (iv) die Strukturen der Endprodukte der Formel (I) im allgemeinen
durch kernmagnetische Resonanz (NMR) (im allgemeinen Protonen-NMR)
und massenspektrometrische Techniken bestätigt wurden; chemische Verschiebungen
bei der NMR auf der Delta-Skala gemessen wurden und die Peak-Multiplizitäten wie
folgt angeführt
sind: s, Singulett; d, Dublett; t, Triplett; m, Multiplett; br,
breit; q, Quartett; quin, Quintett;
- (v) Zwischenprodukte nicht generell vollständig durchcharakterisiert wurden
und die Reinheit durch Dünnschichtchromatographie
(DC), Hochleistungsflüssigchromatographie
(HPLC), Infrarot- (IR-) oder NMR-Analyse abgeschätzt wurde;
- (vi) Chromatographie an Kieselgel (Merck Kieselgel: Art. 9385)
durchgeführt
wurde.
Abkürzungen | DCC | 1,3-Dicyclohexylcarbodiimid |
| DEAD | Azodicarbonsäurediethylether |
| DMSO | Dimethylsulfoxid |
| DMAP | 4-Dimethylaminopyridin |
| DMF | Dimethylformamid |
| DNS | 2,4-Dinitrobenzolsulfonyl |
| EDC | 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-hydrochlorid |
| HOBt | 1-Hydroxybenzotriazol |
| LHMDS | Lithiumbis(trimethylsilyl)amid |
| THF | Tetrahydrofuran |
-
Ausgangsmaterialien
-
Die
Ausgangsmaterialien wurden wie folgt dargestellt:-
-
Eine
Lösung
von 3,5-Dimethylbenzoesäuremethylester
(25 g; 152 mmol) und Butyrolacton (40 ml, 520 mmol) in THF (300
ml) wurde auf 0ºC
abgekühlt
und tropfenweise mit LHMDS (200 ml; 200 mmol; 1 M in Hexan) versetzt.
Die Mischung wurde gerührt
und über
Nacht auf Raumtemperatur erwärmen
gelassen. Das THF wurde abgedampft. Der Rückstand wurde in Et2O aufgenommen, und die organische Phase
wurde mit gesättigter
wäßriger NaHCO3-Lösung
und Kochsalzlösung
gewaschen und über
MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/Hexan
(20 bis 40% EtOAc) als Laufmittel aufgereinigt, was ein Öl lieferte,
das langsam kristallisierte, wodurch man 2 als einen weißen Feststoff
(9,2 g) erhielt. Bei der Chromatographie wurde 3,5-Dimethylbenzoesäuremethylester-Ausgangsmaterial
(12,4 g) zurückgewonnen.
Ausbeute:
55% basierend auf zurückgewonnenem
3,5-Dimethylbenzoesäuremethylester.
1H-NMR-Spektrum (CDCl3):
2,39 (s, 6H); 2,5 (m, 1H); 2,82 (m, 1H); 4,41 (m, 1H); 4,51 (m,
2H); 7,25 (s, 1H); 7,65 (s, 2H)
MS-ESI: 219 [M + H]+
-
Die
Verbindung 2 (7,43 g; 34 mmol) wurde in EtOH (200 ml) gelöst und mit
Hydrazin-hydrat (17,2 ml; 354 mmol) versetzt. Die Mischung wurde
30 min gerührt.
Das Lösungsmittel
wurde abgedampft und der Rückstand
wurde mit Pentan verrieben, wodurch man 3 als einen weißen Feststoff
erhielt (7,05 g).
Ausbeute: 90%
1H-NMR-Spektrum
(DMSO d6): 2,32 (s, 6H); 2,58 (t, 2H); 3,50
(t, 2H); 4,8 (br s, 1H); 7,01 (s, 1H); 7,14 (s, 2H); 9,5 (br s,
1H).
MS-ESI: 233 [M + H]+
-
Eine
Mischung von 3 (4,26 g; 18,4 mmol) und 4 (4,51 g; 19,3 mmol) in
DMA (40 ml) wurde unter Argon mit K2CO3 (5,07 g; 36,7 mmol) versetzt. Die Mischung
wurde unter Rühren
2 h auf 90ºC
erhitzt. Die Mischung wurde in gesättigte wäßrige NaHCO3-Lösung gegossen
und mit EtOAc extrahiert, und die organische Phase wurde mit Wasser
und Kochsalzlösung
gewaschen und über
MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (0 bis 100%
EtOAc) als Laufmittel aufgereinigt, wodurch man den Alkohol 5 als
ein hellgelbes Öl
(6,56 g) erhielt.
Ausbeute: 93%
1H-NMR-Spektrum
(DMSO d6): 1,30 (s, 6H); 1,8 (m, 4H); 2,33
(s, 6H); 2,55 (m, 2H); 3,32 (m, 2H); 3,5 (m, 4H); 4,17 (s, 2H);
4,62 (t, 1H); 7,04 (s, 1H); 7,16 (s, 2H); 11,9 (br s, 1H).
MS-ESI:
386 [M + H]+
-
Eine
Mischung von 5 (3,85 g; 10 mmol), Phthalimid (1,62 g; 11 mmol) und
Triphenylphosphin (10,5 g; 40 mmol) in THF (100 ml) wurde bei 0ºC unter
Argon mit DEAD (6,33 ml; 40 mmol) versetzt. Die Mischung wurde eine
Stunde lang bei dieser Temperatur gerührt und dann in Wasser gegeben.
Die Mischung wurde mit Et2O extrahiert,
und die organische Phase wurde mit Wasser und Kochsalzlösung gewaschen
und über
MgSO4 getrocknet.
-
Durch
Eindampfen erhielt man einen rohen Feststoff, der ohne weitere Aufreinigung
sofort in EtOH (50 ml) aufgenommen und mit Hydrazin-hydrat (5 ml;
100 mmol) versetzt wurde. Die Mischung wurde 1,5 h gerührt, und
anschließend
wurde das EtOH teilweise abgedampft. Die Zugabe von CH2Cl2 führte
zur Ausfällung von
Phthalsäurehydrazid,
das abfiltriert und mit CH2Cl2 gewaschen
wurde. Das Filtrat wurde eingedampft und der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (0 bis 100%
EtOAc) und dann MeOH/CH2Cl2 (0
bis 8% MeOH) als Laufmittel aufgereinigt, wodurch man 6 als einen
beigefarbenen Feststoff (2,34 g) erhielt.
Ausbeute: 61%
1H-NMR-Spektrum (DMSO d6):
1,30 (s, 6H); 1,79 (m, 4H); 2,33 (s, 6H); 2,52 (m, 2H); 2,67 (t,
2H); 3,5 (m, 4H); 4,18 (s, 2H); 7,03 (s, 1H); 7,14 (s, 2H); 8,95
(br s, 1H). MS-ESI: 385 [M + H]+
-
Das
Ausgangsmaterial 4 wurde wie folgt dargestellt:-
-
Eine
Mischung von 8 (14,48 g; 80 mmol) und Oxalsäurebromid (43,2 g; 200 mmol),
die einen Tropfen DMF erhielt, wurde 2 h auf 50ºC erhitzt und dann abgekühlt. Überschüssiges Oxalsäurebromid
wurde abgedampft, und der Rückstand
wurde azeotrop mit Toluol destilliert, wodurch man das Rohprodukt
9 erhielt, das direkt in CH2Cl2 (25
ml) aufgenommen und auf 0ºC
abgekühlt
wurde. Es wurde mit Diisopropylethylamin (14 ml; 80 mmol) und anschließend mit
einer Lösung
von Pyrrolidin (3,3 ml; 40 mmol) in CH2Cl2 (30 ml) versetzt. Die Mischung wurde über Nacht
auf Raumtemperatur erwärmen
gelassen und mit CH2Cl2 verdünnt, mit
wäßriger HCl-Lösung (2
N), Natronlauge (1 N), Wasser und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie unter
Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (5 bis 10% EtOAc)
als Laufmittel aufgereinigt, wodurch man 4 als einen weißen Feststoff
(6,5 g) erhielt.
Ausbeute: 70%
1H-NMR-Spektrum
(DMSO d6): 1,39 (s, 6H); 1,9 (m, 4H); 3,57
(m, 4H); 3,62 (s, 2H). MS-ESI: 235 [M + H]+
-
Beispiel
1 3-(2,2-Dimethyl-3-oxo-3-(pyrrolidin-1-yl)propoxy]-4-[2-(3-pyridin-4-ylpyrrolidin-1-carboxamido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol
-
Eine
Lösung
von 6 (150 ml, 0,39 mmol) in CH2Cl2 (2 ml) wurde auf 0ºC abgekühlt. Diisopropylethylamin (136 μl; 0,78 mmol)
wurde zugesetzt, gefolgt von einer Lösung von Chlorameisensäure-4-nitrophenylester
(83 mg; 0,41 mmol) in CH2Cl2 (2
ml). Die Mischung wurde 3 h gerührt
und dann mit einer Lösung
von 4-(3-Pyrrolidyl)pyridin (70 mg; 0,47 mmol) in CH2Cl2 (2 ml) versetzt, und die Mischung wurde über Nacht
auf Raumtemperatur erwärmen
gelassen. Die Mischung wurde direkt durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von MeOH/EtOAc
(0 bis 10% MeOH) als Laufmittel aufgereinigt, wodurch man Beispiel
1 als einen beigefarbenen Feststoff (95 mg) erhielt.
Ausbeute:
44%
1H-NMR-Spektrum (DMSO d6): 1,29 (s, 6H); 1,75 (m, 4H); 1,95 (m,
1H); 2,2 (m, 1H); 2,31 (s, 6H); 2,56 (m, 2H); 3,1-3,4 (m, 6H); 3,5
(m, 4H); 3,64 (m, 1H); 4,18 (s, 2H); 6,22 (t, 1H); 6,99 (s, 1H);
7,21 (s, 2H); 7,27 (d, 2H); 8,48 (d, 2H). MS-ESI: 559 [M + H]+
-
Beispiel
2 3-[2,2-Dimethyl-3-oxo-3-(azabicyclo[2.2.1]heptan-7-yl)propyl]-4-(2-(N'-isopropoxycarbonyl-3-pyrid-4-ylpyrrolidin-1-ylcarboxindantido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol
-
Eine
Lösung
von 10 (260 mg; 0,5 mmol) in CH2Cl2 (5 ml) wurde auf 0ºC abgekühlt. EDCl (145 mg; 0,75 mmol)
und Diisopropylethylamin (130 μl;
0,75 mmol) wurden zugesetzt, gefolgt von 4-(3-Pyrrolidyl)pyridin
(111 mg; 0,75 mmol), und die Mischung wurde über Nacht auf Raumtemperatur
erwärmen
gelassen. Die Mischung wurde mit CH2Cl2 verdünnt,
und die organische Phase wurde mit gesättigter wäßriger NaHCO3-Lösung und Kochsalzlösung gewaschen
und über
MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie unter
Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (0 bis 100%
EtOAc) als Laufmittel aufgereinigt, wodurch man Beispiel 2 als einen
beigefarbenen Feststoff (277 mg) erhielt.
Ausbeute: 86%
1H-NMR-Spektrum (DMSO-d6):
1,07 (m, 6H); 1,29 (s, 6H); 1,8 (m, 4H); 1,95 (m, 1H); 2,2 (m, 1H);
2,30 (s, 6H); 2,65 (m, 2H); 3,2-3,4 (m, 6H); 3,5 (m, 4H); 3,65 (m,
1H); 4,18 (s, 2H); 4,6 (m, 1H); 6,95 (m, 1H); 7,00 (s, 1H); 7,12
(s, 2H); 7,27 (d, 2H); 8,49 (d, 2H). MS-ESI: 644 [M + H]+
-
Das
Ausgangsmaterial 10 wurde wie folgt dargestellt:
-
Eine
Lösung
von 6 (200 mg; 0,52 mmol) in CH2Cl2 (2 ml) wurde auf 0ºC abgekühlt. Eine Lösung von 11 (115 mg; 0,78 mmol)
wurde zugesetzt, und die Mischung wurde im Verlauf von 1 h auf Raumtemperatur erwärmen gelassen.
Die Mischung wurde mit Wasser versetzt und mit CH2Cl2 verdünnt,
und die organische Phase wurde mit Kochsalzlösung gewaschen und über MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von Et2O/Hexan (0 bis 100% Et2O)
als Laufmittel aufgereinigt, wodurch man 10 als einen beigefarbenen
Feststoff (260 mg) erhielt.
Ausbeute: 94%
1H-NMR-Spektrum
(DMSO-d6): 1,22 (m, 6H); 1,31 (s, 6H); 1,8
(m, 4H); 2,32 (s, 6H); 2,71 (m, 2H); 3,5 (m, 4H); 3,74 (m, 2H);
4,20 (s, 2H); 4,83 (m, 1H); 7,02 (s, 1H); 7,17 (s, 2H); 9,89 (t,
1H); 10,81 (s, 1H). MS-ESI: 530 [M + H]+
-
Beispiel
3 3-(2,2-Dimethyl-3-oxo-3-(N,N-diethylamino)propyl]-4-(2-(N'-isopropoxycarbonyl-3-pyrid-4-ylpyrrolidin-1-ylcarboximidamido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol
-
Eine
Lösung
von Bb4 (156 mg; 0,29 mmol) in CH2Cl2 (2 ml) wurde auf 0ºC abgekühlt. EDCl (85 mg; 0,44 mmol)
und Diisopropylethylamin (77 μl;
0,44 mmol) wurden zugegeben, gefolgt von 4-(3-Pyrrolidyl)pyridin (56
mg; 0,38 mmol), und die Mischung wurde über Nacht auf Raumtemperatur
erwärmen
gelassen. Die Mischung wurde mit CH2Cl2 verdünnt,
und die organische Phase wurde mit gesättigter wäßriger NaHCO3-Lösung und
Kochsalzlösung
gewaschen und über
MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von MeOH/CH2Cl2 (0 bis 100%
MeOH) als Laufmittel aufgereinigt, wodurch man Beispiel 3 als einen
beigefarbenen Feststoff (180 mg) erhielt.
Ausbeute: 96%
1H-NMR-Spektrum (DMSO-d6):
1,07 (m, 12H); 1,31 (s, 6H); 1,93 (m, 1H); 2,23 (m, 1H); 2,30 (s,
6H); 2,65 (m, 2H); 3,2-3,55 (m, 10H); 3,57 (m, 1H); 4,17 (s, 2H);
4,58 (m, 1H); 6,95 (m, 1H); 7,00 (s, 1H); 7,13 (s, 2H); 7,27 (d,
2H); 8,50 (d, 2H); 11,9 (br s, 1H). MS-ESI: 646 [M + H]+
-
Das
Ausgangsmaterial Bb4 wurde wie folgt dargestellt:-
-
Eine
Mischung von 3 (1,23 g; 5,3 mmol) und Bb1 (1,32 g; 5,5 mmol) in
DMA (20 ml) wurde unter Argon mit K2CO3 (1,46 g; 10,6 mmol) versetzt. Die Mischung
wurde unter Rühren
2 h auf 70ºC
erhitzt. Die Mischung wurde in gesättigte wäßrige NaHCO3-Lösung gegossen
und mit EtOAc extrahiert, und die organische Phase wurde mit Wasser
und Kochsalzlösung
gewaschen und über
MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (0 bis 100%
EtOAc) als Laufmittel aufgereinigt, wodurch man den Alkohol Bb2
als eine hellgelbes Öl
(1,92 g) erhielt.
Ausbeute: 94%
1H-NMR-Spektrum
(DMSO d6): 1,08 (t, 6H); 1,32 (s, 6H); 2,33
(s, 6H); 2,57 (m, 2H); 3,38 (m, 4H); 3,5 (m, 1H); 4,18 (s, 2H);
4,61 (t, 1H); 7,04 (s, 1H); 7,16 (s, 2H); 11,9 (br s, 1H).
MS-ESI:
388 [M + H]+
-
Eine
Mischung von Bb2 (1,92 g; 4,96 mmol), Phthalimid (0,8 g; 5,46 mmol)
und Triphenylphosphin (5,24 g; 20 mmol) in THF (50 ml) wurde bei
0ºC unter
Argon mit DEAD (3,2 ml; 20 mmol) versetzt. Die Mischung wurde 2
h bei dieser Temperatur gerührt
und anschließend
mit Wasser versetzt. Die Mischung wurde mit Et2O extrahiert,
und die organische Phase wurde mit Wasser und Kochsalzlösung gewaschen
und über
MgSO4 getrocknet.
-
Durch
Eindampfen erhielt man einen rohen Feststoff, der ohne weitere Aufreinigung
direkt in EtOH (50 ml) aufgenommen und mit Hydrazinhydrat (2,5 ml;
50 mmol) versetzt wurde. Die Mischung wurde 2 h gerührt, worauf
das EtOH teilweise eingedampft wurde. Durch Zugabe von CH2Cl2 kam es zur Ausfällung von
Phthalhydrazid, das abfiltriert und mit CH2Cl2 gewaschen wurde. Das Filtrat wurde eingedampft
und der Rückstand wurde
durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (0 bis 100%
EtOAc) als Laufmittel aufgereinigt, wodurch man Bb3 als einen beigefarbenen
Feststoff (0,865 g) erhielt.
Ausbeute: 45%
1H-NMR-Spektrum
(DMSO d6): 1,06 (t, 6H); 1,30 (s, 6H); 2,32
(s, 6H); 2,47 (m, 2H); 2,66 (t, 2H); 3,35 (m, 4H); 4,16 (s, 2H);
7,02 (s, 1H); 7,13 (s, 2H); 11,9 (br s, 1H).
MS-ESI: 387 [M
+ H]+
-
Eine
Lösung
von Bb3 (210 mg; 0,544 mmol) in CH2Cl2 (5 ml) wurde auf 0ºC abgekühlt. Eine Lösung von 11 (120 mg; 0,82 mmol)
wurde zugesetzt, und die Mischung wurde im Verlauf von 1 h auf Raumtemperatur erwärmen gelassen.
Die Mischung wurde mit Wasser versetzt und mit CH2Cl2 verdünnt,
und die organische Phase wurde mit Kochsalzlösung gewaschen und über MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von CH2Cl2 als
Laufmittel aufgereinigt, wodurch man Bb4 als einen beigefarbenen
Feststoff (235 mg) erhielt.
Ausbeute: 81%
1H-NMR-Spektrum
(CDCl3): 1,18 (t, 6H); 1,27 (d, 6H); 1,44
(s, 6H); 2,38 (s, 6H); 2,87 (m, 2H); 3,45 (m, 4H); 3,88 (m, 2H);
4,36 (s, 2H); 4,93 (m, 1H); 7,04 (s, 1H); 7,11 (s, 2H); 7,81 (s,
1H); 8,9 (s br, 1H); 9,7 (s, 1H).
MS-ESI: 532 [M + H]+
-
Das
Ausgangsmaterial Bb1 wurde wie folgt dargestellt:-
-
Eine
Mischung von 8 (14,48 g; 80 mmol) und Oxalsäurebromid (43,2 g; 200 mmol),
die einen Tropfen DMF enthielt, wurde 2 h auf 50ºC erhitzt und dann abgekühlt. Überschüssiges Oxalsäurebromid
wurde abgedampft und der Rückstand
wurde azeotrop mit Toluol destilliert, wodurch man das Rohprodukt
9 erhielt, das direkt in CH2Cl2 (25
ml) aufgenommen und auf 0ºC
abgekühlt
wurde. Diisopropylethylamin (14 ml; 80 mmol) wurde zugesetzt, gefolgt
von einer Lösung
von Pyrrolidin (3,3 ml; 40 mmol) in CH2Cl2 (30 ml). Die Mischung wurde über Nacht
auf Raumtemperatur erwärmen
gelassen und dann mit CH2Cl2 verdünnt, mit
wäßriger HCl (2
N), Natronlauge (1 N), Wasser und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/CH2Cl2 (5 bis 10% EtOAc)
als Laufmittel aufgereinigt, wodurch man Bb1 als einen weißen Feststoff
(6,5 g) erhielt.
Ausbeute: 70%
1H-NMR-Spektrum
(DMSO d6): 1,19 (m, 6H); 1,42 (s, 6H); 3,41
(m, 4H); 3,65 (s, 2H). MS-ESI: 237 [M + H]+
-
Beispiel
4 3-(2,2-Dimethyl-3-oxo-3-(N,N-diethylamino)propyl]-4-[2-(3-pyridin-4-ylpyrrolidin-1-carboxamido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol
-
-
Eine
Lösung
von Bb3 (150 mg; 0,39 mmol) in CH2Cl2 (2 ml) wurde auf 0ºC abgekühlt. Diisopropylethylamin (135 μl; 0,78 mmol)
wurde zugesetzt, gefolgt von einer Lösung von Chlorameisensäure-4-nitrophenylester
(83 mg; 0,41 mmol) in CH2Cl2 (2
ml). Die Mischung wurde 3 h gerührt
und anschließend
mit einer Lösung von
4-(3-Pyrrolidyl)pyridin (70 mg; 0,47 mmol) in CH2Cl2 (2 ml) versetzt, und die Mischung wurde über Nacht auf
Raumtemperatur erwärmen
gelassen. Die Mischung wurde direkt durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von MeOH/EtOAc
(0 bis 10% MeOH) als Laufmittel aufgereinigt, wodurch man Beispiel
4 als einen beigefarbenen Feststoff (138 mg) erhielt.
Ausbeute:
63%
1H-NMR-Spektrum (DMSO d6): 1,05 (m, 6H); 1,32 (s, 6H); 1,89 (m,
1H); 2,2 (m, 1H); 2,31 (s, 6H); 2,57 (m, 2H); 3,1-3,4 (m, 10H);
3,64 (m, 1H); 4,17 (s, 2H); 6,22 (t, 1H); 6,99 (s, 1H); 7,22 (s,
2H); 7,27 (d, 2H); 8,48 (d, 2H); 11,9 (br s, 1H).
MS-ESI: 561
[M + H]+
-
Beispiel
5 3-[2,2-Dimethyl-3-oxo-3-(azabicyclo[2.2.1]heptan-7-yl)propyl]-4-(1S-methyl-2-(N'-isopropoxycarbonyl-3-pyrid-4-ylpyrrolidin-1-ylcarboximidamido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol
-
Eine
Lösung
von CNR (141 mg; 0,25 mmol) in CH2Cl2 (5 ml) wurde auf 0ºC abgekühlt. EDCl (72 mg; 0,37 mmol)
und Diisopropylethylamin (65 μl;
0,37 mmol) wurden zugesetzt, gefolgt von 4-(3-Pyrrolidyl)pyridin (46
mg; 0,31 mmol), und die Mischung wurde über Nacht auf Raumtemperatur
abkühlen
gelassen. Die Mischung wurde mit CH2Cl2 verdünnt,
und die organische Phase wurde mit gesättigter wäßriger NaHCO3-Lösung und
Kochsalzlösung
gewaschen und über
MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von zunehmend polareren Mischungen von EtOAc/CH2CH (0 bis 100% EtOAc) als Laufmittel aufgereinigt,
wodurch man Beispiel 5 als einen beigefarbenen Feststoff (128 mg)
erhielt.
Ausbeute: 78%
1H-NMR-Spektrum
(DMSO d6): 1,05 (m, 6H); 1,12 (m, 3H); 1,28
(s, 6H); 1,42 (m, 4H); 1,62 (m, 4H); 1,91 (m, 1H); 2,2 (m, 1H);
2,30 (s, 6H); 2,95 (m, 1H); 3,2-3,7 (m, 7H); 4,17 (s, 2H); 4,56
(m, 3H); 7,01 (s, 1H); 7,04 (s, 1H); 7,06 (s, 1H); 7,2 (s br, 1H);
7,27 (dd, 2H); 8,49 (dd, 2H); 11,79 (s, 1H).
MS-ESI: 684 [M
+ H]+
-
Das
Ausgangsmaterial CNR wurde wie folgt dargestellt:
-
Eine
Lösung
von Ce (150 mg; 0,35 mmol) in CH2Cl2 (5 ml) wurde auf 0ºC abgekühlt. Eine Lösung von 11 (77 mg; 0,53 mmol)
in CH2Cl2 (1 ml)
wurde zugesetzt, und die Mischung wurde im Verlauf von 1 h auf Raumtemperatur
erwärmen
gelassen. Die Mischung wurde mit Wasser versetzt und mit CH2Cl2 verdünnt, und
die organische Phase wurde mit Kochsalzlösung gewaschen und über MgSO4 getrocknet. Der Rückstand wurde durch Flash-Chromatographie
unter Verwendung von EtOAc/CH2Cl2 (0-20% EtOAc) als Laufmittel aufgereinigt,
wodurch man CNR als ein Gummi (141 mg) erhielt.
Ausbeute: 70%
1H-NMR-Spektrum (DMSO d6):
1,22 (d, 3H); 1,8 (m, 6H); 1,27 (m, 6H); 1,41 (m, 4H); 1,61 (m,
4H); 2,29 (s, 6H); 3,06 (q, 1H); 3,65 (m, 1H); 3,84 (m, 1H); 4,19
(m, 2H); 4,58 (s, 2H); 4,79 (m, 1H); 7,02 (s, 1H); 7,04 (s, 2H); 9,84
(s, 1H); 11,8 (s br, 1H).
MS-ESI: 570 [M + H]+
-
Beispiele 5.1-5.2
-
Die
folgenden Beispiele wurden ähnlich
Beispiel 5 dargestellt:
-
Die
Tabelle zeigt die R in bezug auf die obige Struktur, die Reaktionsbedingungen
und die Charakteristika für
die jeweiligen Beispiele entsprechend der obigen Beschreibung der
Darstellung von Beispiel 5:- Beispiel
5.1
- Chromatographie:
EtOAc/CH2Cl2 (0
bis 100% EtOAc) und dann MeOH/CH2Cl2 (0 bis 5% MeOH)
- 1H-NMR-Spektrum (DMSO d6):
0,9-1,2 (m, 14H); 1,28 (m, 6H); 1,43 (m, 4H); 1,5 (m, 2H); 1,6 (m,
6H); 2,31 (s, 6H); 2,7 (m, 2H); 2,95 (m, 1H); 3,2-3,7 (m, 5H); 3,75
(m, 2H); 3,83 (m, 2H); 4,16 (s, 2H); 4,57 (m, 3H); 7,02 (s, 1H);
7,04 (s, 2H); 7,46 (s br, 1H); 11,80 (s, 1H).
Beispiel
5.2 - Chromatographie: EtOAc/CH2Cl2 (0 bis 100% EtOAc) und dann MeOH/CH2Cl2 (0 bis 5% MeOH)
- 1H-NMR-Spektrum (DMSO d6):
1,07 (m, 6H); 1,11 (m, 3H); 1,28 (m, 6H); 1,42 (m, 4H); 1,5 (m,
2H); 1,62 (m, 4H); 1,72 (m, 2H); 2,30 (s, 6H); 2,7 (m, 1H); 2,9
(m, 2H); 2,95 (m, 1H); 3,2-3,4 (m, 2H); 3,85 (m, 2H); 4,17 (m, 2H);
4,57 (m, 3H); 7,01 (s, 1H); 7,06 (s, 2H); 7,18 (d, 2H); 7,5 (s br,
1H); 8,45 (d, 2H); 11,81 (s, 1H).
-
Beispiel
6 3-[2,2-Dimethyl-3-oxo-3-(azabicyclo[2.2.1]heptan-7-yl)propoxy]-4-[1S-methyl-2-(3-pyridin-4-ylpyrrolidin-1-carboxamido)ethyl]-5-(3,5-dimethylphenyl)-1H-pyrazol
-
Eine
Lösung
von Ce (170 mg; 0,4 mmol) in CH2Cl2 (5 ml) wurde auf 0ºC abgekühlt und mit DIEA (140 μl, 0,8 mmol)
versetzt. Eine Lösung
von Chlorameisensäure-4-nitrophenylester
(85 mg; 0,42 mmol) in CH2Cl2 (1
ml) wurde zugegeben, und die Mischung wurde 30 min rühren gelassen.
4-(3-Pyrrolidyl)pyridin (71 mg; 0,48 mmol) wurde zugesetzt, und
die Mischung wurde im Verlauf von 1 h auf Raumtemperatur erwärmen gelassen. Die
Mischung wurde direkt durch Flash-Chromatographie unter Verwendung
von MeOH/CH2Cl2 (0-10%
MeOH) als Laufmittel aufgereinigt, wodurch man Beispiel 6 als ein
hellgelbes Pulver (212 mg) erhielt.
Ausbeute: 88%
1H-NMR-Spektrum (DMSO d6):
1,11 (m, 3H); 1,28 (m, 6H); 1,42 (m, 4H); 1,62 (m, 4H); 1,95 (m,
1H); 2,22 (m, 1H); 2,29 (s, 6H); 2,93 (m, 1H); 3,2-3,7 (m, 6H);
3,67 (m, 1H); 4,17 (s, 2H); 4,58 (s, 2H); 6,21 (m, 1H); 7,00 (s, 1H);
7,14 (s, 2H); 7,26 (m, 2H); 8,47 (m, 2H); 11,79 (s, 1H).
MS-ESI:
599 [M + H]+
-
Beispiele 6.1-6.2
-
Die
folgenden Beispiele wurden ähnlich
Beispiel 6 dargestellt,
-
Die
Tabelle zeigt die R in bezug auf die obige Struktur, die Reaktionsbedingungen
und die Charakteristika für
die jeweiligen Beispiele entsprechend der obigen Beschreibung der
Darstellung von Beispiel 6:- Beispiel
6.1
- Chromatographie: MeOH/EtOAc (0 bis 5%
MeOH)
- 1H-NMR-Spektrum (DMSO d6):
1,11 (m, 3H); 1,28 (m, 6H); 1,42 (m, 4H); 1,62 (m, 4H); 1,99 (m,
1H); 2,20 (m, 1H); 2,29 (s, 6H); 2,93 (m, 1H); 3,2-3,5 (m, 6H);
3,68 (m, 1H); 4,17 (s, 2H); 4,58 (s, 2H); 6,15 (m, 1H); 7,00 (s, 1H);
7,14 (m, 2H); 7,24 (m, 1H); 7,29 (m, 1H); 7,72 (m, 1H); 8,49 (m,
1H); 11,74 (s, 1H).
Beispiel
6.2 - Chromatographie:
EtOAc/CH2Cl2 (0
bis 100% EtOAc)
- 1H-NMR-Spektrum (DMSO d6):
0,9 (m, 2H); 1,07 (m, 3H); 1,14 (m, 4H); 1,28 (m, 6H); 1,4-1,7 (m,
12H); 2,30 (s, 6H); 2,9 (m, 1H); 3,2-3,4 (m, 6H); 3,82 (m, 2H);
3,96 (m, 2H); 4,17 (m, 2H); 4,58 (m, 2H); 6,45 (m, 1H); 6,99 (s,
1H); 7,14 (s, 2H); 11,81 (s, 1H).
-
THERAPEUTISCHE ANWENDUNGEN
-
Die
Verbindungen der Formel I werden als Medikamente zum Antagonisieren
der Aktivität
von Gonadotropinreleasing-Hormon (GnRH) in einem Patienten, beispielsweise
in Männern
und/oder Frauen, bereitgestellt. Zu diesem Zweck kann eine Verbindung
der Formel (I) als Teil einer pharmazeutischen Formulierung, die
außerdem
ein pharmazeutisch annehmbares Verdünnungsmittel bzw. einen pharmazeutisch
annehmbaren Träger
(z.B. Wasser) enthält,
bereitgestellt werden. Die Formulierung kann in Form von Tabletten,
Kapseln, Granulaten, Pulvern, Sirupen, Emulsionen (z.B. Lipidemulsionen),
Zäpfchen,
Salben, Cremen, Tropfen, Suspensionen (z.B. wäßrigen oder öligen Suspensionen)
oder Lösungen
(z.B. wäßrigen oder öligen Lösungen) vorliegen.
Falls gewünscht
kann die Formulierung eine oder mehrere zusätzliche, unabhängig voneinander aus
Stabilisierungsmitteln, Netzmitteln, Emulgatoren, Puffern, Lactose,
Sialylsäure,
Magnesiumstearat, Terra alba, Saccharose, Maisstärke, Talkum, Gelatine, Agar-Agar,
Pectin, Erdnußöl, Olivenöl, Kakaobutter
und Ethylenglykol ausgewählte
Substanzen enthalten.
-
Die
Verbindung wird einem Patienten vorzugsweise oral verabreicht, jedoch
sind auch andere Verabreichungswege wie z.B. die parenterale oder
rektale Verabreichung möglich.
Bei der intravenösen,
subkutanen oder intramuskulären
Verabreichung kann der Patient eine Tagesdosis von 0,1 mgkg–1 bis
30 mgkg–1 (vorzugsweise
5 mgkg–1 bis
20 mgkg–1)
der Verbindung erhalten, wobei die Verbindung 1- bis 4mal täglich verabreicht wird.
Die intravenöse,
subkutane bzw. intramuskuläre
Dosis kann als Bolusinjektion verabreicht werden. Alternativ dazu
kann die intravenöse
Dosis als kontinuierliche Infusion über eine Zeitspanne verabreicht
werden. Alternativ dazu kann der Patient eine tägliche orale Dosis erhalten,
die ungefähr
der täglichen
parenteralen Dosis entspricht, wobei die Zusammensetzung 1- bis
4mal täglich
verabreicht wird. Eine geeignete pharmazeutische Formulierung eignet
sich zur oralen Verabreichung in Einzeldosisform, zum Beispiel als
Tablette oder Kapsel, die zwischen 10 mg und 1 g (vorzugsweise zwischen
100 mg und 1 g) der erfindungsgemäßen Verbindung enthält.
-
Für eine bessere
Formulierung können
Puffer, pharmazeutisch annehmbare Kosolventien (z.B. Polyethylenglykol,
Propylenglykol, Glycerin oder EtOH) oder Komplexbildner wie Hydroxypropyl-β-Cyclodextrin verwendet
werden.
-
Ein
Aspekt der Erfindung betrifft die Verwendung von erfindungsgemäßen Verbindungen
zur Verminderung der Sezernierung von LH und/oder FSH durch die
Hypophyse eines Patienten. In dieser Hinsicht kann die Verminderung
durch eine reduzierte Biosynthese von LH und FSH und/oder durch
eine verminderte Freisetzung von LH und FSH durch die Hypophyse
erfolgen. Die erfindungsgemäßen Verbindungen
können
somit zur therapeutischen Behandlung und/oder Prävention eines mit Sexualhormonen
in Zusammenhang stehenden Leidens im Patienten angewendet werden. "Prävention" bedeutet hier die
Verminderung des Risikos des Patienten, sich das Leiden zuzuziehen. "Behandlung" ist als die Ausmerzung
des Leidens im Patienten bzw. eine Verminderung des Schweregrads
des Leidens zu verstehen. Beispiele für mit Sexualhormonen im Zusammenhang
stehende Leiden sind: sexualhormonabhängiger Krebs, benigne Prostatahypertrophie,
Gebärmuttermyom,
Endometriose, polyzystisches Ovarialsyndrom, Gebärmutterfibroide, Prostatavergrößerung,
Myoma uteri, Hirsutismus und Pubertas praecox. Beispiele für sexualhormonabhängigen Krebs
sind: Prostatakrebs, Gebärmutterkrebs,
Brustkrebs und Gonadotrophenadenom der Hypophyse.
-
Die
erfindungsgemäßen Verbindungen
lassen sich zusammen mit anderen Arzneimitteln und Therapien zur
Behandlung/Prävention
von mit Sexualhormonen in Zusammenhang stehenden Leiden anwenden.
-
Bei
einer Formulierung als feste Dosis werden in solchen Kombinationsprodukten
die Verbindungen der vorliegenden Erfindung in dem hier beschriebenen
Dosisbereich und das andere pharmazeutisch wirksame Mittel in seinem
zugelassenen Dosierungsbereich verwendet. Ist eine Kombinationsformulierung
ungeeignet, so wird eine aufeinanderfolgende Anwendung in Betracht
gezogen.
-
Auf
dem Gebiet der medizinischen Onkologie schließen solche Kombinationen Kombinationen
mit den folgenden Katagorien therapeutischer Mittel ein:
- (i) antiangiogene Mittel (beispielsweise Linomid,
Inhibitoren der Integrin-ανβ3-Funktion,
Angiostatin, Endostatin, Razoxin, Thalidomid), und einschließlich des
vaskulären
endothelialen Wachstumsfaktors (vascular endothelial growth factor,
VEGF) Rezeptortyrosinkinaseinhibitoren (RTKIS)
(zum Beispiel den in den internationalen Patentanmeldungen Nr. WO-97/22596 , WO-97/30035 , WO-97/32856 und WO-98/13354 beschriebenen);
- (ii) Zytostatika, wie Antiöstrogene
(beispielsweise Tamoxifen, Toremifen, Raloxifen, Droloxifen, Iodoxyfen), Progestogene
(beispielsweise Megestrol acetat), Aromatase-Inhibitoren (beispielsweise
Anastrozol, Letrozol, Vorazol, Exemestan), Antiprogestogene, Antiandrogene
(beispielsweise Flutamid, Nilutamid, Bicalutamid, Cyproteronacetat),
Inhibitoren der Testosteron-5α-dihydroreduktase
(beispielsweise Finasterid), Antiinvasionsmittel (beispielsweise
Metalloproteinase-Inhibitoren wie Marimastat und Inhibitoren der
Urokinase-Plasminogenaktivator-Rezeptorfunktion) und Inhibitoren
der Wachstumsfaktorfunktion (Beispiele für derartige Wachstumsfaktoren
sind EGF (epidermal growth factor), Plättchenwachstumsfaktor und Hepatozytenwachstumsfaktor;
Beispiele für
derartige Inhibitoren sind Wachstumsfaktorantikörper, Wachstumsfaktorrezeptorantikörper, Tyrosinkinase-Inhibitoren
und Serin/Threoninkinase-Inhibitoren);
- (iii) die biologische Reaktion modifizierende Mittel (beispielsweise
Interferon);
- (iv) Antikörper
(beispielsweise Edrecolomab) und
- (v) antiproliferative/antineoplastische Arzneistoffe und Kombinationen
davon, wie sie in der medizinischen Onkologie zum Einsatz kommen,
wie Antimetabolite (beispielsweise Antifolate wie Methotrexat, Fluorpyrimidine
wie 5-Fluoruracil, Purin- und Adenosin-Analoge, Cytosinarabinosid);
Antitumor-Antibiotika (beispielsweise Anthracycline wie Doxorubicin,
Daunomycin, Epirubicin und Idarubicin, Mitomycin-C, Dactinomycin,
Mithramycin); Platinderivate (beispielsweise Cisplatin, Carboplatin);
Alkylierungsmittel (beispielsweise N-Lost, Melphalan, Chlorambucil, Busulfan,
Cyclophosphamid, Ifosfamid, Nitrosoharnstoffe, Thiotepa); Antimitotika
(beispielsweise Vinca-Alkaloide wie Vincristin und Taxoide wie Taxol,
Taxotere); Enzyme (beispielsweise Asparaginase) und Thymidylatsynthaseinhibitoren
(beispielsweise Raltitrexed); Topoisomeraseinhibitoren (beispielsweise
Epipodophyllotoxine wie Etoposid und Teniposid, Amsacrin, Topotecan sowie
Irinotecan).
-
Die
erfindungsgemäßen Verbindungen
können
auch in Kombination mit chirurgischen Eingriffen oder Strahlentherapie
eingesetzt werden.
-
ASSAYS
-
Die
Fähigkeit
von erfindungsgemäßen Verbindungen,
als GnRH-Antagonisten zu wirken, läßt sich mit den folgenden In-vitro-Assays
bestimmen.
-
Bindungsassay mit dem GnRH-Rezeptor
der Rattenhypophyse
-
Der
Assay wird wie folgt durchgeführt:
- 1. Rohe, aus Rattenhypophysengewebe gewonnene
Plasmamembranen werden in Tris.HCl-Puffer (pH-Wert 7,5, 50 mM) mit Rinderserumalbumin
(0,1%), [I-125]D-t-Bu-Ser6-Pro9-Ethylamid-GnRH und der Testverbindung
inkubiert. Die Inkubation erfolgt bei 4ºC über 90 Minuten bis 2 Stunden.
- 2. Die Plasmamembranen werden schnell abfiltriert und wiederholt
auf einem Glasfaserfilter gewaschen.
- 3. Die Radioaktivität
der membrangebundenen Radioliganden wird mit einem Gammazähler bestimmt.
-
Aus
diesen Daten läßt sich
der IC50-Wert der Testverbindung als die
zur Inhibierung der Radioligandenbindung an GnRH-Rezeptoren um 50%
erforderliche Konzentration an Verbindung bestimmen. Die Verbindungen
gemäß der vorliegenden
Erfindung sind bei einer Konzentration von 1 nM bis 5 μM aktiv.
-
Bindungsassay mit dem humanen GnRH-Rezeptor
-
Als
Quellen für
den GnRH-Rezeptor dienen aus humane GnRH-Rezeptoren exprimierenden
CHO-Zellen gewonnene rohe Membranen. Die Bindungsaktivität erfindungsgemäßer Verbindungen
läßt sich
als IC50-Wert bestimmen, bei dem es sich
um die zur Inhibierung der spezifischen Bindung von [125I]-Buserelin
an GnRH-Rezeptoren um 50% erforderliche Verbindungskonzentration
handelt. [125I]-Buserelin (ein Peptid-GnRH-Analogon)
wird hier als ein radioaktiv markierter Ligand des Rezeptors verwendet.
-
Assay zur Bestimmung der Inhibierung
der LH-Freisetzung
-
Mit
dem LH-Freisetzungsassay läßt sich
die antagonistische Wirkung von Verbindungen zeigen, die sich in
einer Reduktion der durch GnRH induzierten Freisetzung von LH äußert.
-
Hypophysenpräparation
-
Von
Ratten erhaltene Hypophysen werden wie folgt zubereitet. Geeignete
Ratten sind männliche
Wistar-Ratten (150-200
g), die bei konstanter Temperatur (z.B. 25ºC) mit einem hell-dunkel-Zyklus
von 12 Stunden Licht/12 Stunden Dunkelheit gehalten wurden. Die
Ratten werden durch Enthaupten getötet, worauf die Hypophysen
aseptisch in Röhrchen
mit Hank's Balanced
Salt Solution (HBSS) entnommen werden. Die Hypophysen werden weiter
verarbeitet, indem man:
- 1. 5 Minuten lang bei
250 × g
zentrifugiert;
- 2. die HBSS-Lösung
absaugt;
- 3. die Hypophysen in eine Petrischale gibt und dann mit einem
Skalpell zerkleinert;
- 4. das zerkleinerte Gewebe in ein Zentrifugenröhrchen gibt,
indem man das Gewebe dreimal hintereinander in 10-ml-Aliquots von
HBSS mit 0,2% Collagenase und 0,2% Hyaluronidase suspendiert;
- 5. die Zellen durch leichtes Rühren der Gewebesuspension,
bei dem das Röhrchen
bei 37ºC in
einem Wasserbad gehalten wird, dispergiert;
- 6. 20- bis 30mal mit einer Pipette aspiriert, wobei sich nichtverdaute
Hypophysenfragmente 3-5 Minuten lang absetzen können;
- 7. die suspendierten Zellen absaugt und anschließend 5 Minuten
lang bei 1200 × g
zentrifugiert;
- 8. die Zellen in DMEM-Kulturmedium mit 0,37% NaHCO3,
10% Pferdeserum, 2,5% fetalem Rinderserum, 1% nichtessentiellen
Aminosäuren,
1% Glutamin und 0,1% Gentamycin resuspendiert;
- 9. die nichtverdauten Hypophysenfragmente 3mal mit 30-ml-Aliquots
der Collagenase und Hyaluronidase behandelt;
- 10. die Zellsuspensionen poolt und auf eine Konzentration von
3 × 105 Zellen/ml verdünnt;
- 11. jeweils 1,0 ml dieser Suspension in die Vertiefungen eines
Tabletts mit 24 Vertiefungen gibt, wobei die Zellen 3 bis 4 Tage
lang in einer angefeuchteten Atmosphäre mit 5% CO2/95%
Luft bei 37°C
gehalten werden.
-
Test der Verbindungen
-
Die
Testverbindung wird in DMSO auf eine Endkonzentration von 0,5% im
Inkubationsmedium gelöst.
-
1,5
Stunden for dem Assay werden die Zellen dreimal mit DMEM, enthaltend
0,37% NaHCO3, 10% Pferdeserum, 2,5% fetales
Rinderserum, 1% nichtessentielle Aminosäuren (100 X), 1% Glutamin (100
X), 1% Penizillin/Streptomycin (jeweils 10000 Einheiten pro ml)
und 25 mM HEPES bei einem pH-Wert von 7,4, gewaschen. Unmittelbar
vor dem Assay werden die Zellen noch zweimal mit diesem Medium gewaschen.
-
Im
Anschluß daran
werden 1 ml frisches Medium mit der Testverbindung und 2 nM GnRH
in zwei Vertiefungen gegeben. Bei anderen Testverbindungen (wenn
mehr als eine Verbindung getestet werden soll) werden diese jeweils
in zwei andere Vertiefungen gegeben. Die Inkubation erfolgt dann
bei 37°C über drei
Stunden.
-
Nach
der Inkubation werden die einzelnen Vertiefungen analysiert, indem
man das Medium aus den Vertiefungen entnimmt und zum Entfernen von
gegebenenfalls vorhandenem Zellmaterial 15 Minuten lang bei 2000 × g zentrifugiert.
Der Überstand
wird abgenommen und mit einem Doppelantikörper-Radioimmunassay auf den
LH-Gehalt untersucht.
Durch Vergleich mit einer geeigneten Kontrolle (keine Testverbindung)
wird bestimmt, ob die Testverbindung die LH-Freisetzung reduziert.
Verbindungen gemäß der vorliegenden
Erfindung sind bei einer Konzentration von 1 nM bis 5 μM aktiv.


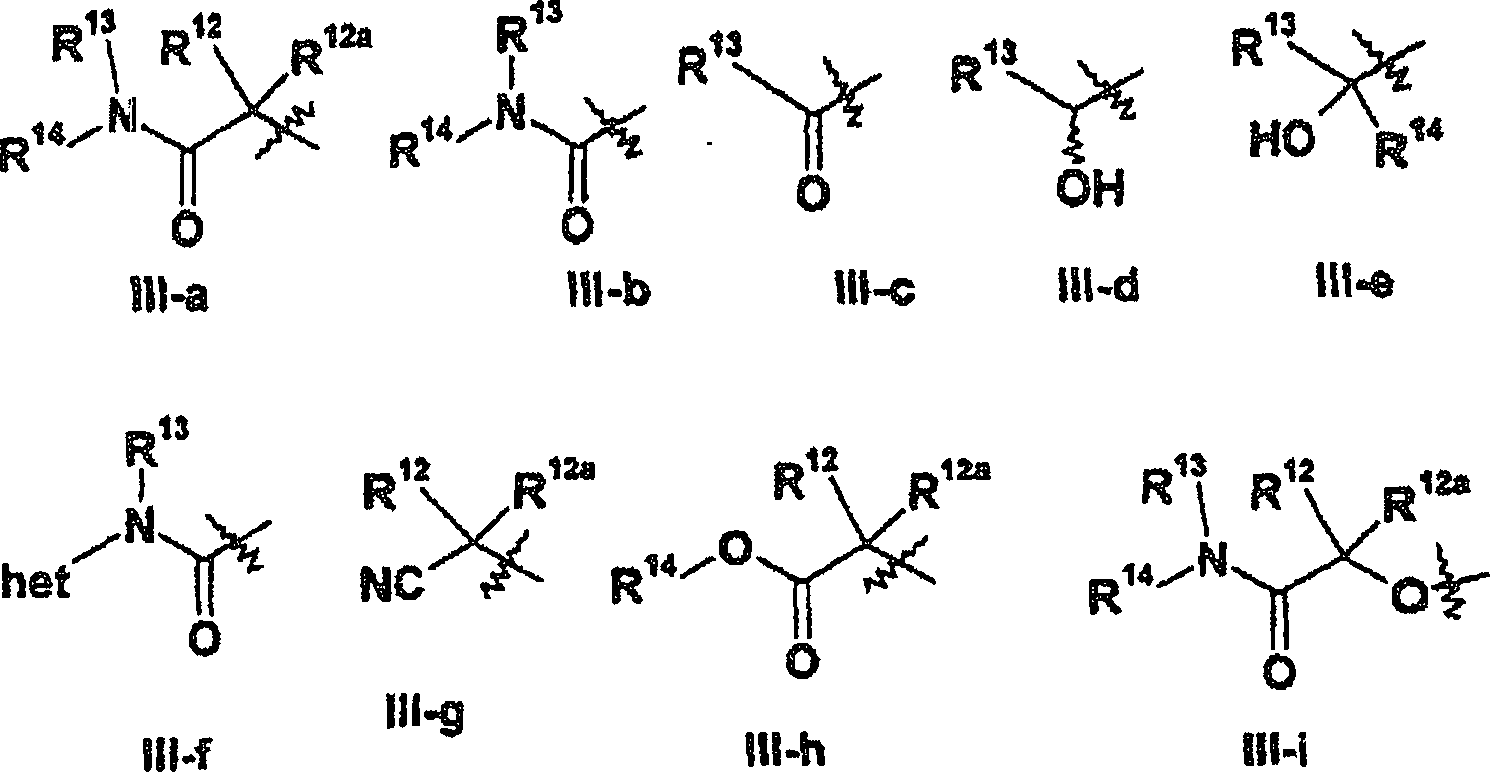 wobei het für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander aus O, N und S ausgewählten Heteroatomen steht;
wobei het für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander aus O, N und S ausgewählten Heteroatomen steht;



 der gegebenenfalls 1 bis 3 weitere Heteroatome unabhängig voneinander ausgewählt aus O, N und S enthält.
der gegebenenfalls 1 bis 3 weitere Heteroatome unabhängig voneinander ausgewählt aus O, N und S enthält.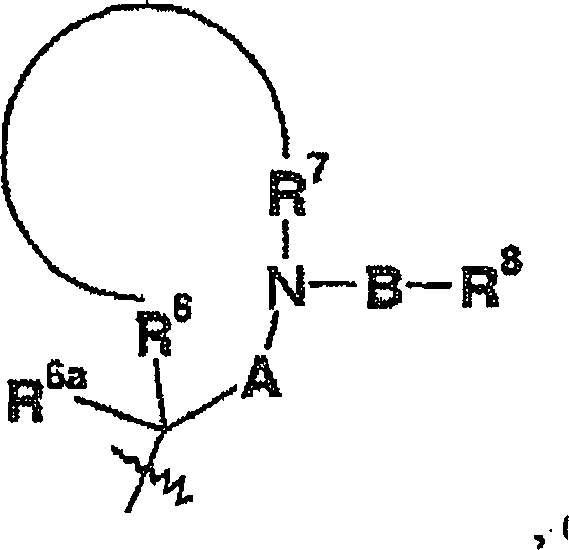

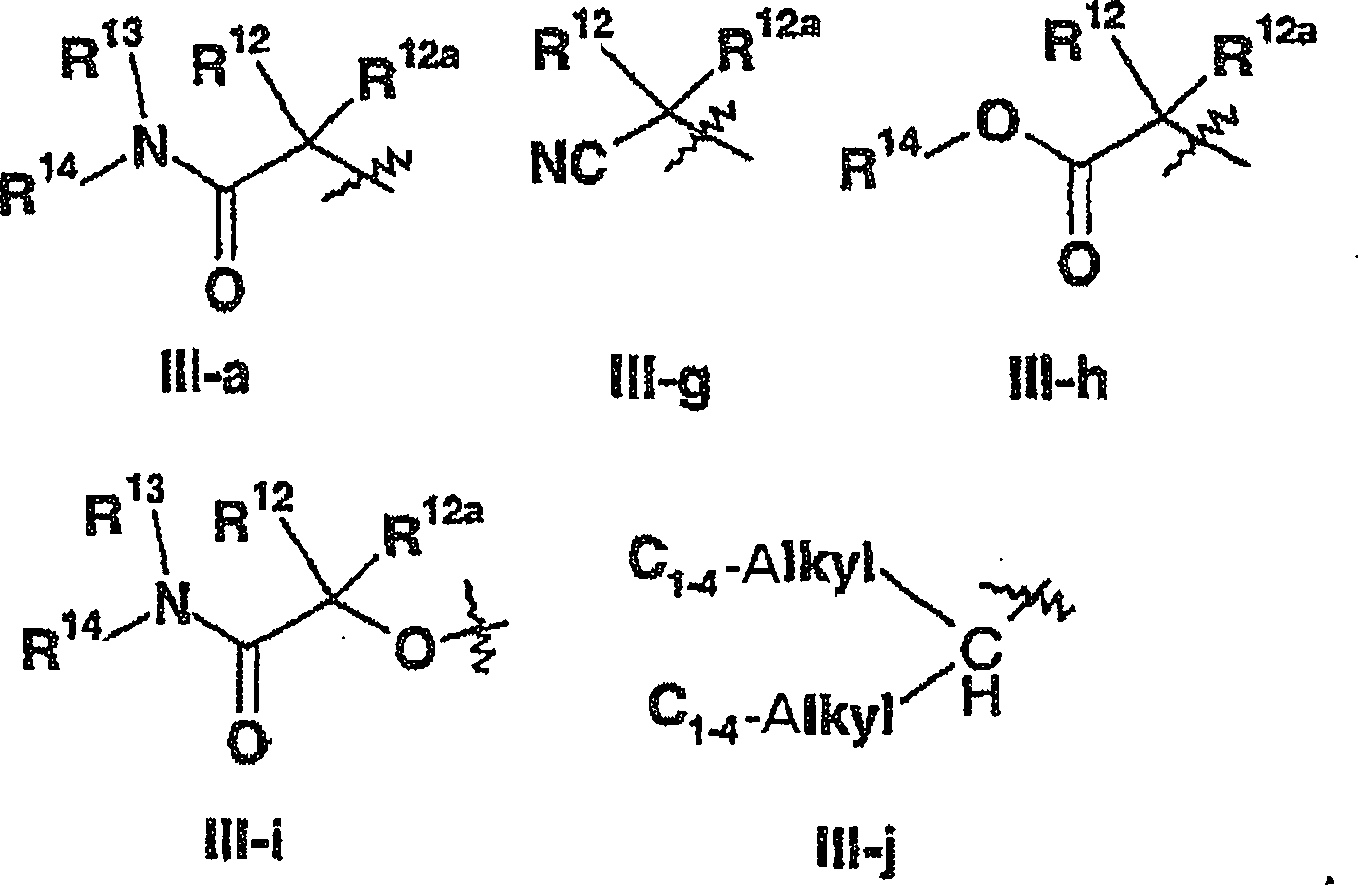



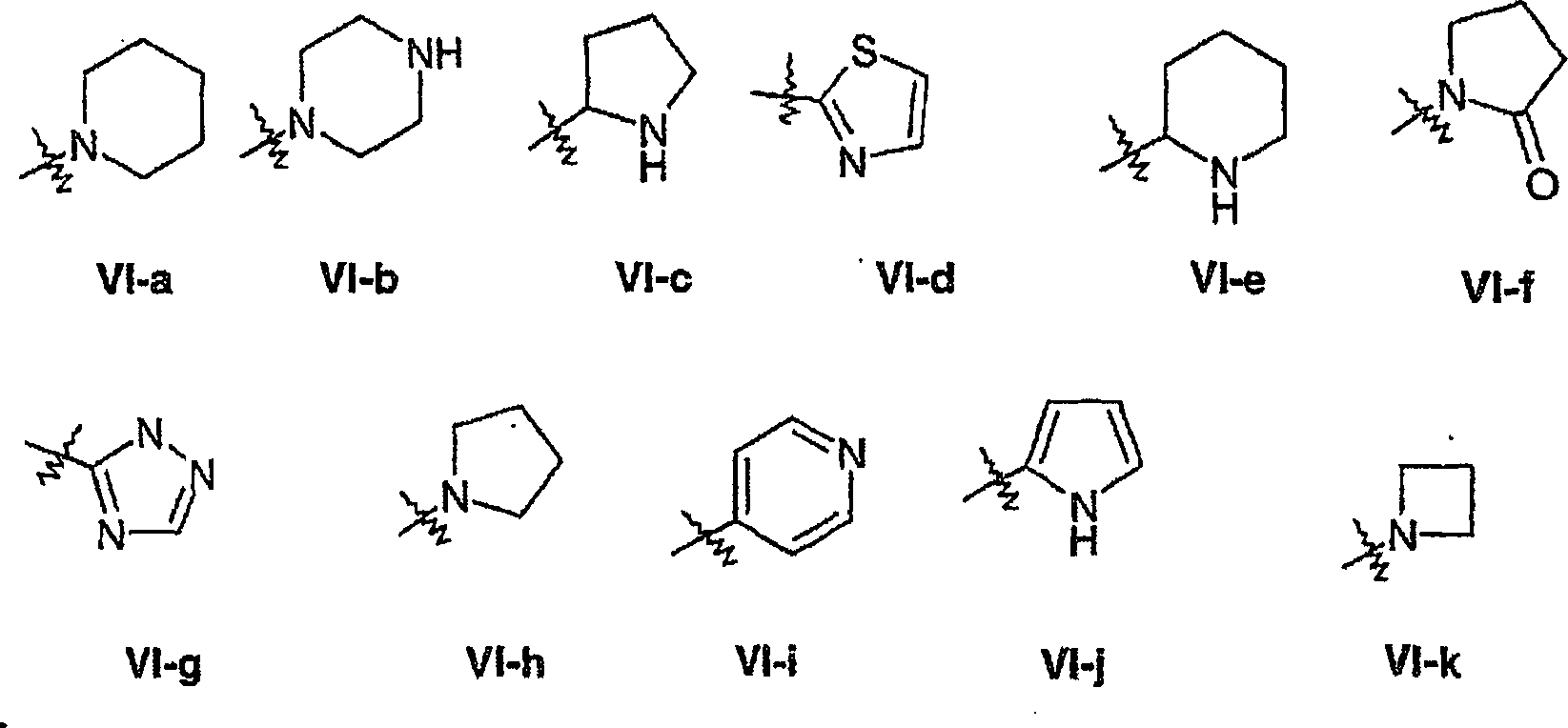



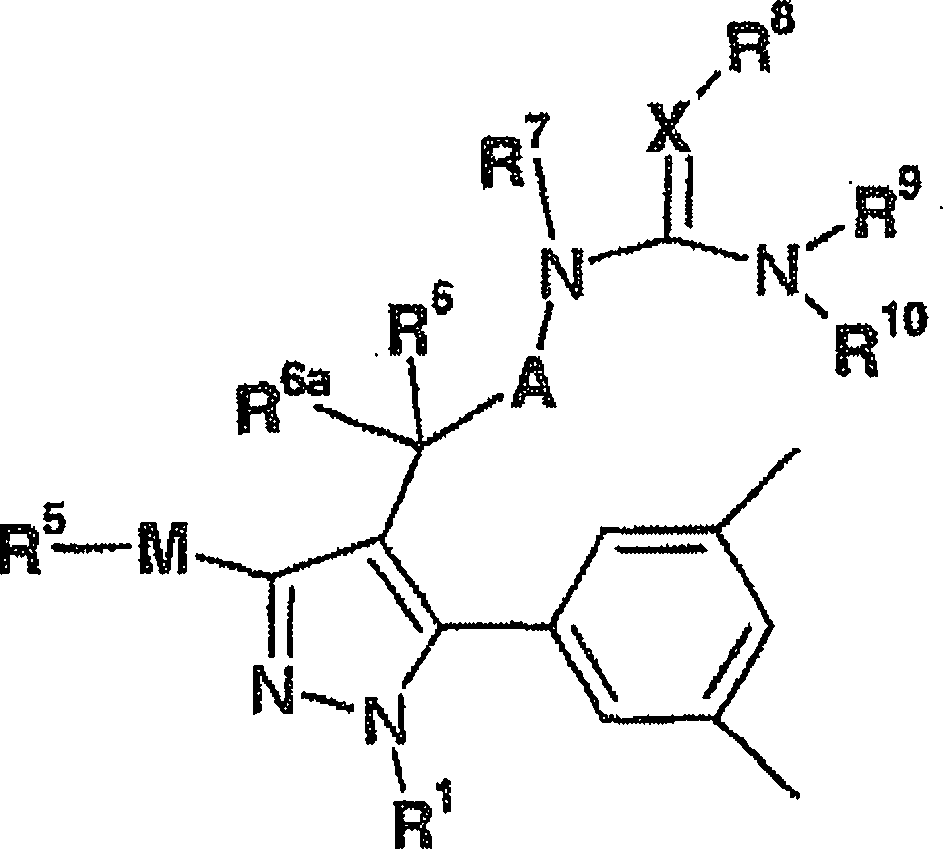
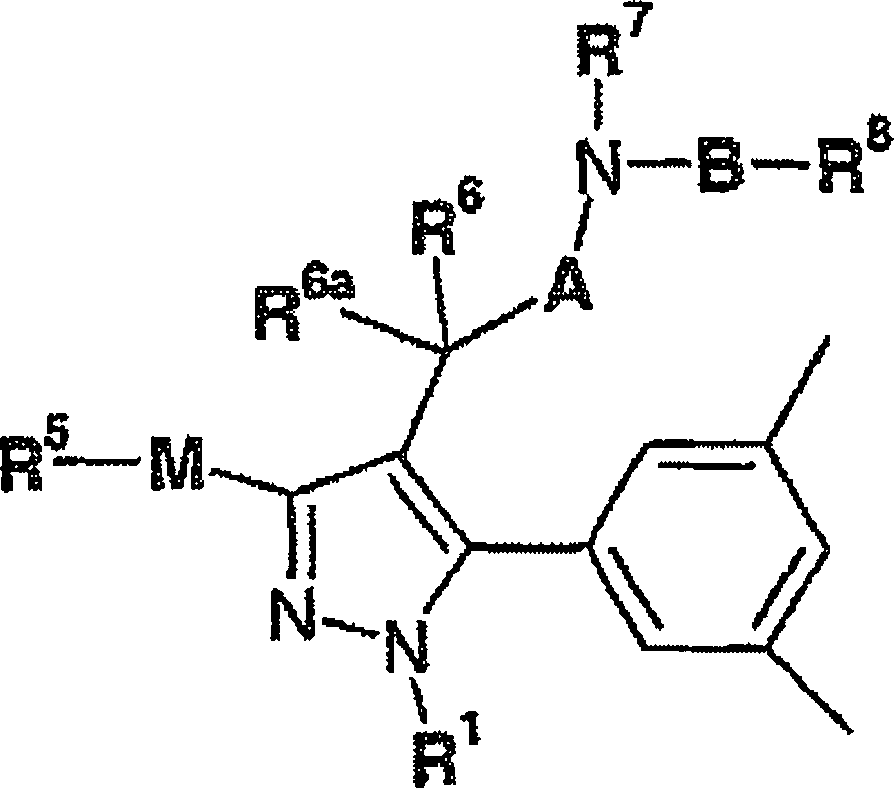








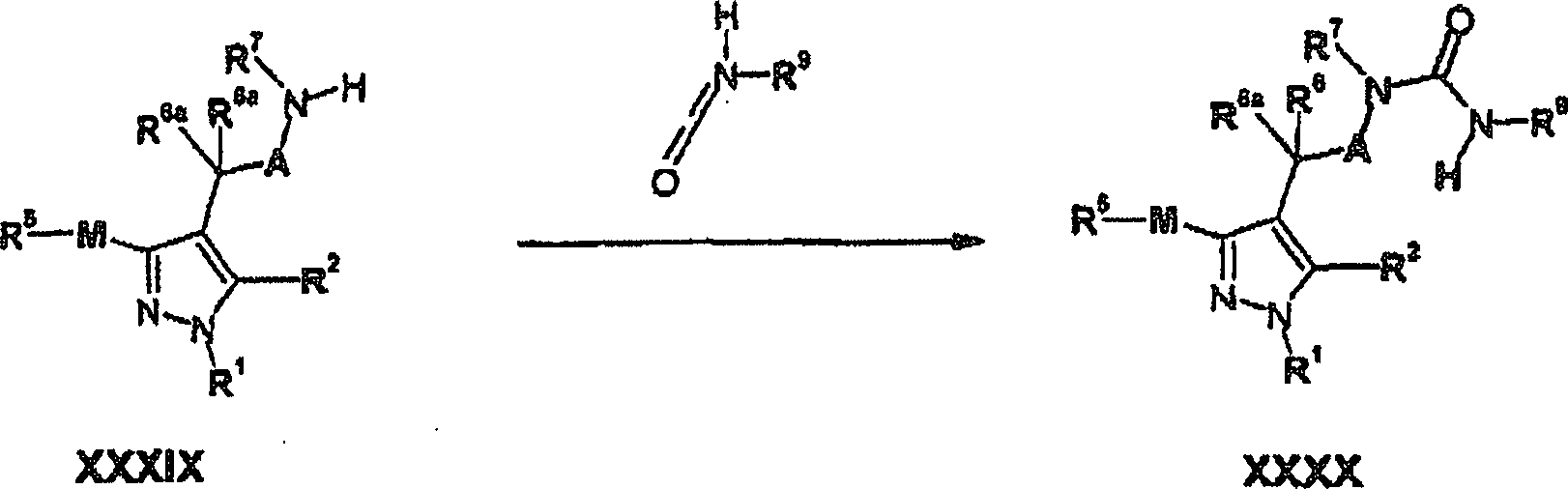




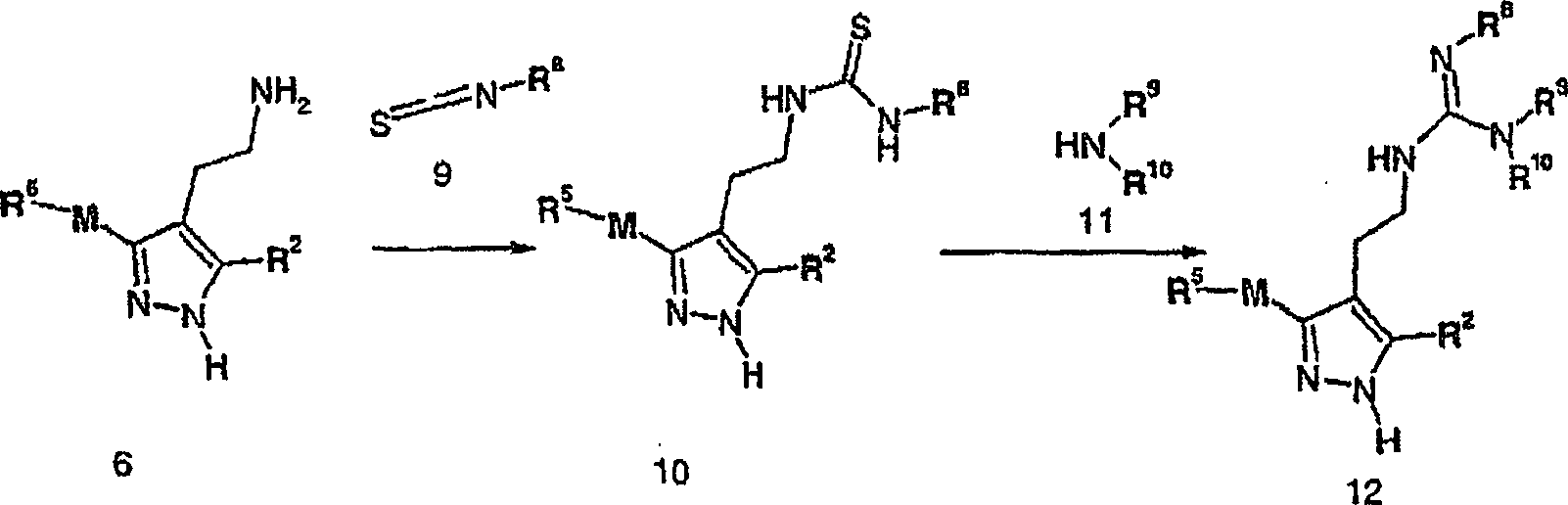







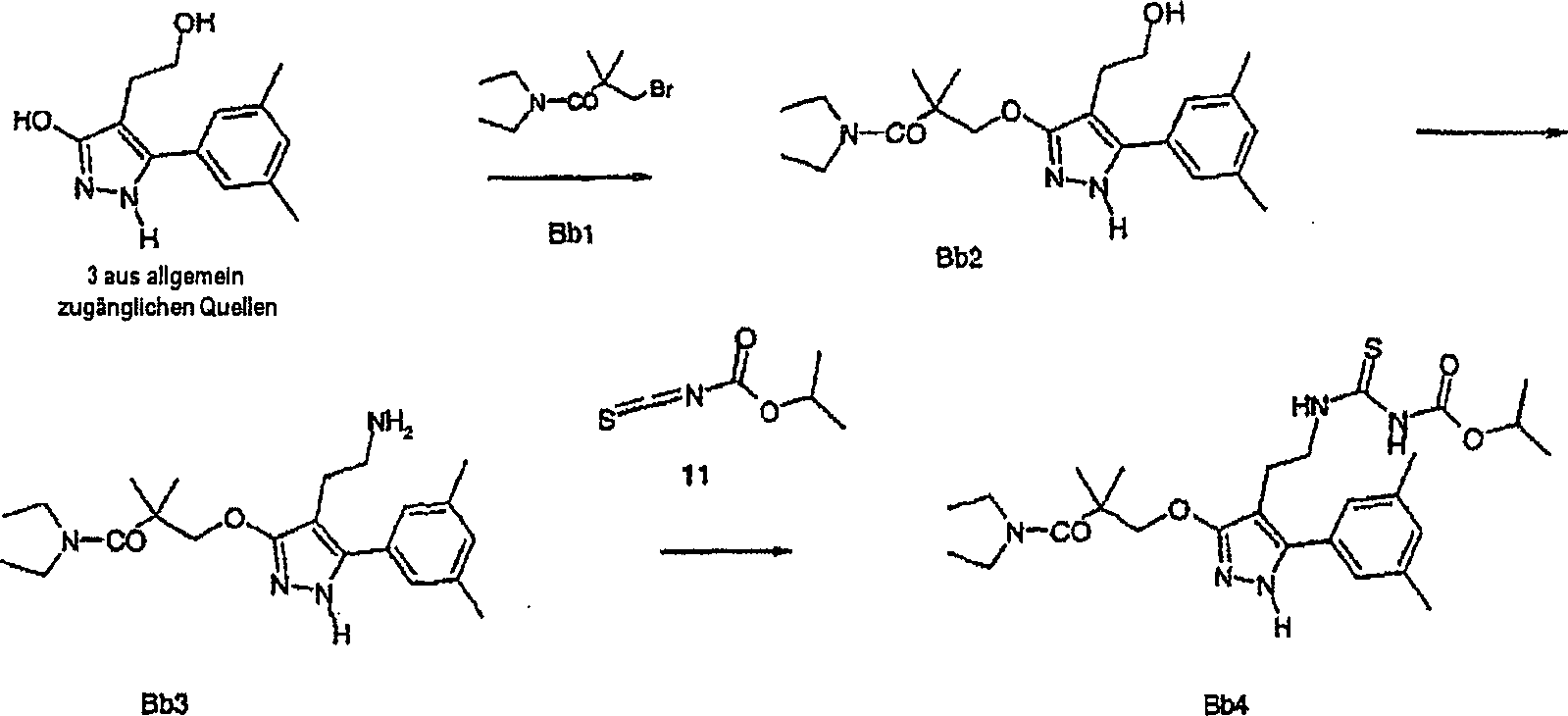



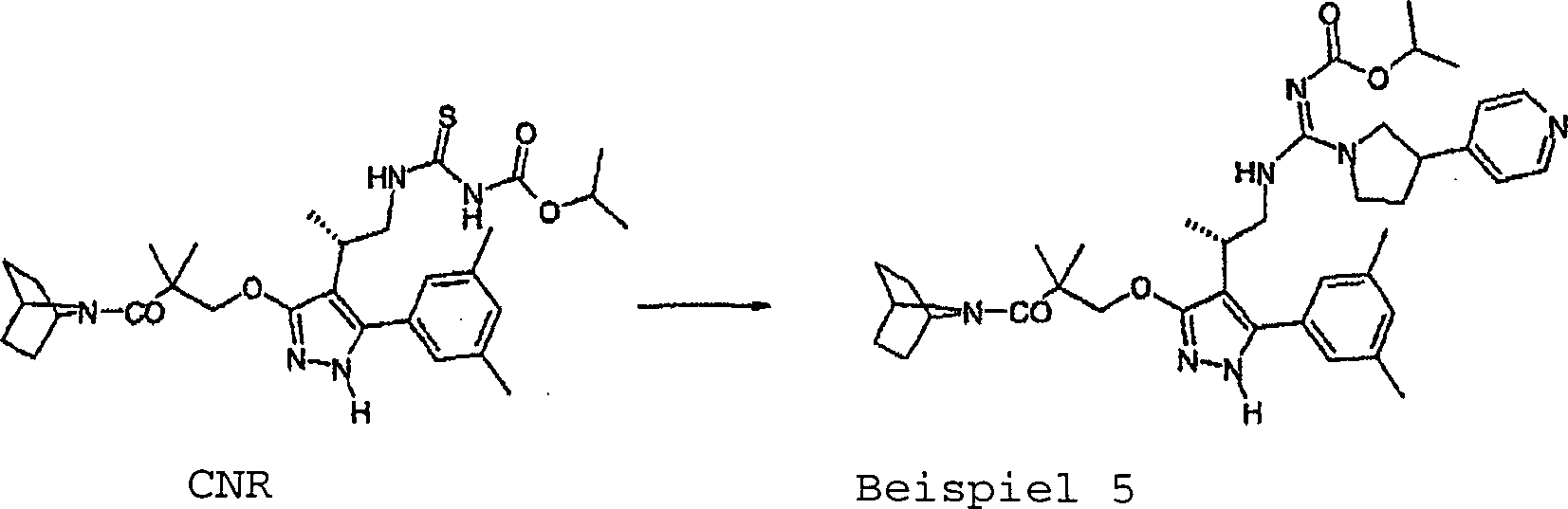



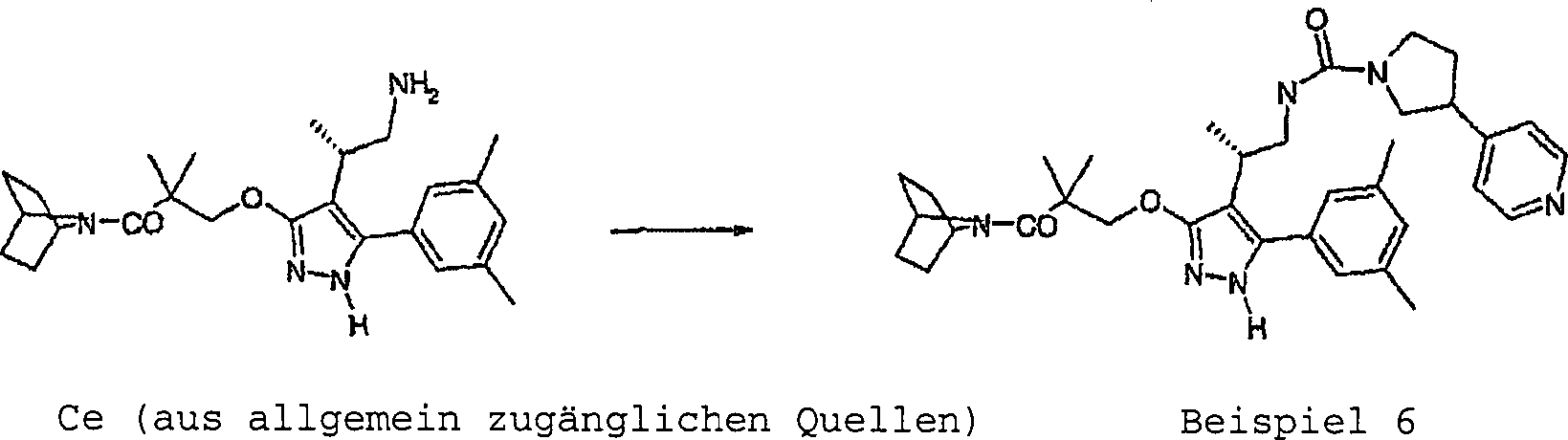




 Formel (II) steht, wobei Formel (II) in Position (a) an das Stickstoffatom gebunden ist und die Gruppe X an R8 gebunden ist; M für -(CH2)0-2-O- steht; R1 für Wasserstoff, gegebenenfalls substituiertes C1-8-Alkyl oder (CH2)b-Ra steht, wobei Ra für C3-8-Cycloalkyl steht und b für Null oder eine ganze Zahl von 1 bis 6 steht; R2 für eine gegebenenfalls substituierte mono- oder bicyclische aromatische Ringstruktur steht, wobei die gegebenenfalls vorhandenen Substituenten ausgewählt sind aus Cyano, NR3R3a, gegebenenfalls substituiertem C1-8-Alkyl, gegebenenfalls substituiertem C1-8-Alkoxy oder Halogen; R3 und R3a unabhängig voneinander ausgewählt sind aus Wasserstoff, gegebenenfalls substituiertem C1-8-Alkyl und gegebenenfalls substituiertem Aryl; R5 ausgewählt ist aus einem gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander aus O, N und S ausgewählten Heteroatomen, oder einer Gruppe der Formel III-a, III-b, III-c, III-d, III-e, III-f, III-g, III-h, III-i oder III-j;
Formel (II) steht, wobei Formel (II) in Position (a) an das Stickstoffatom gebunden ist und die Gruppe X an R8 gebunden ist; M für -(CH2)0-2-O- steht; R1 für Wasserstoff, gegebenenfalls substituiertes C1-8-Alkyl oder (CH2)b-Ra steht, wobei Ra für C3-8-Cycloalkyl steht und b für Null oder eine ganze Zahl von 1 bis 6 steht; R2 für eine gegebenenfalls substituierte mono- oder bicyclische aromatische Ringstruktur steht, wobei die gegebenenfalls vorhandenen Substituenten ausgewählt sind aus Cyano, NR3R3a, gegebenenfalls substituiertem C1-8-Alkyl, gegebenenfalls substituiertem C1-8-Alkoxy oder Halogen; R3 und R3a unabhängig voneinander ausgewählt sind aus Wasserstoff, gegebenenfalls substituiertem C1-8-Alkyl und gegebenenfalls substituiertem Aryl; R5 ausgewählt ist aus einem gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander aus O, N und S ausgewählten Heteroatomen, oder einer Gruppe der Formel III-a, III-b, III-c, III-d, III-e, III-f, III-g, III-h, III-i oder III-j;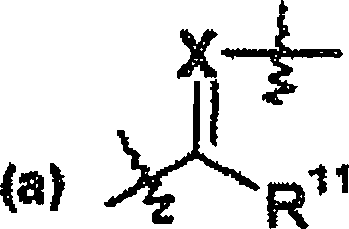 wobei het für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander aus O, N und S ausgewählten Heteroatomen steht; R6 und R6a unabhängig voneinander ausgewählt sind aus Wasserstoff und gegebenenfalls substituiertem C1-8-Alkyl; oder R6 und R6a zusammen für Carbonyl stehen; R7 für Wasserstoff oder gegebenenfalls substituiertes C1-8-Alkyl steht; oder
wobei het für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 4 unabhängig voneinander aus O, N und S ausgewählten Heteroatomen steht; R6 und R6a unabhängig voneinander ausgewählt sind aus Wasserstoff und gegebenenfalls substituiertem C1-8-Alkyl; oder R6 und R6a zusammen für Carbonyl stehen; R7 für Wasserstoff oder gegebenenfalls substituiertes C1-8-Alkyl steht; oder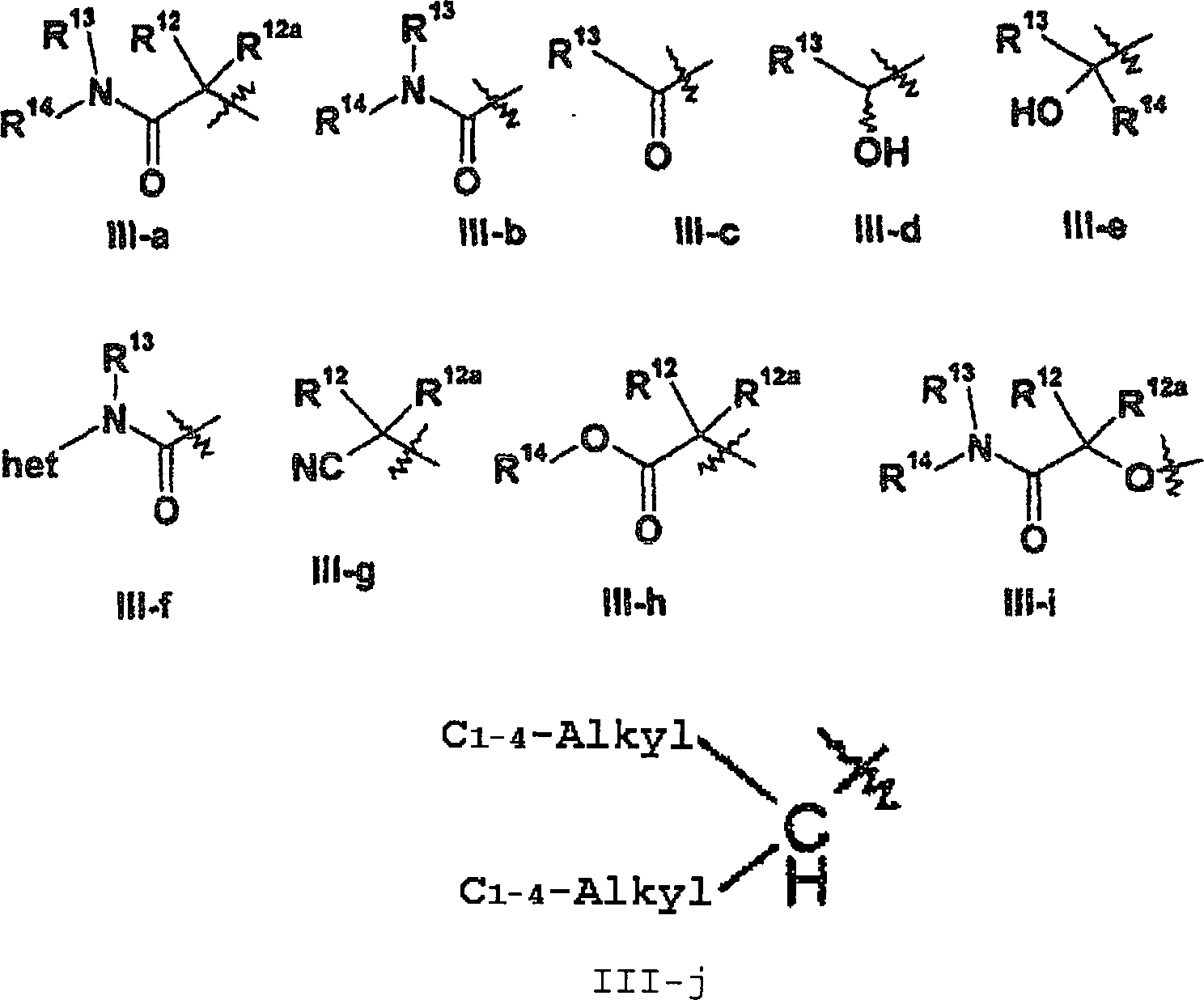 zusammen einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 3 weiteren unabhängig voneinander aus O, N und S ausgewählten Heteroatomen bildet und R6a für Wasserstoff und gegebenenfalls substituiertes C1-8-Alkyl steht; X und R8 ausgewählt sind aus: (i) X steht für N und R8 ist ausgewählt aus: Cyano, Wasserstoff, Hydroxy, -O-Rb, -NRbRc -C(O)O-R-b, -CONRbRc oder NH-C(O)-Rb, wobei Rb und Rc unabhängig voneinander ausgewählt sind aus Wasserstoff und C1-4-Alkyl, gegebenenfalls substituiert durch Hydroxy, Amino, N-C1-4-Alkylamino, N,N-Di-C1-4-alkylamino, HO-C2-4-Alkyl-NH- oder HO-C2-4-Alkyl-N-(C1-4-alkyl)-; (ii) X steht für CH und R8 steht für NO2; und (iii) X-R8 steht für -O-; R11 für eine Gruppe der Formel: N(R9R10) steht, wobei R9 für Wasserstoff, Aryl, einen gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen Ring oder gegebenenfalls substituiertes C1-8-Alkyl steht und R10 für Wasserstoff oder gegebenenfalls substituiertes C1-8-Alkyl steht; oder die Struktur N(R9R10) für einen gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen Ring steht, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; R12 und R12a unabhängig voneinander ausgewählt sind aus Wasserstoff oder gegebenenfalls substituiertem C1-8-Alkyl; oder R12 und R12a zusammen mit dem Kohlenstoff, an den sie gebunden sind, einen gegebenenfalls substituierten 3- bis 7gliedrigen Cycloalkylring bilden; R13 und R14 ausgewählt sind aus: (i) R13 ist ausgewählt aus Wasserstoff, gegebenenfalls substituiertem C1-8-Alkyl, gegebenenfalls substituiertem Aryl, -Rd-Ar, wobei Rd für C1-8-Alkylen steht und Ar für gegebenenfalls substituiertes Aryl steht, und einem gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; und R14 ist ausgewählt aus Wasserstoff, gegebenenfalls substituiertem C1-8-Alkyl und gegebenenfalls substituiertem Aryl; (ii) wenn R5 für eine Gruppe der Formel III-a, III-b oder III-i steht, steht die Gruppe NR13(-R14) für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; oder (iii) wenn R5 für Struktur III-e steht, steht die Gruppe
zusammen einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring mit 1 bis 3 weiteren unabhängig voneinander aus O, N und S ausgewählten Heteroatomen bildet und R6a für Wasserstoff und gegebenenfalls substituiertes C1-8-Alkyl steht; X und R8 ausgewählt sind aus: (i) X steht für N und R8 ist ausgewählt aus: Cyano, Wasserstoff, Hydroxy, -O-Rb, -NRbRc -C(O)O-R-b, -CONRbRc oder NH-C(O)-Rb, wobei Rb und Rc unabhängig voneinander ausgewählt sind aus Wasserstoff und C1-4-Alkyl, gegebenenfalls substituiert durch Hydroxy, Amino, N-C1-4-Alkylamino, N,N-Di-C1-4-alkylamino, HO-C2-4-Alkyl-NH- oder HO-C2-4-Alkyl-N-(C1-4-alkyl)-; (ii) X steht für CH und R8 steht für NO2; und (iii) X-R8 steht für -O-; R11 für eine Gruppe der Formel: N(R9R10) steht, wobei R9 für Wasserstoff, Aryl, einen gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen Ring oder gegebenenfalls substituiertes C1-8-Alkyl steht und R10 für Wasserstoff oder gegebenenfalls substituiertes C1-8-Alkyl steht; oder die Struktur N(R9R10) für einen gegebenenfalls substituierten 3- bis 10gliedrigen heterocyclischen Ring steht, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; R12 und R12a unabhängig voneinander ausgewählt sind aus Wasserstoff oder gegebenenfalls substituiertem C1-8-Alkyl; oder R12 und R12a zusammen mit dem Kohlenstoff, an den sie gebunden sind, einen gegebenenfalls substituierten 3- bis 7gliedrigen Cycloalkylring bilden; R13 und R14 ausgewählt sind aus: (i) R13 ist ausgewählt aus Wasserstoff, gegebenenfalls substituiertem C1-8-Alkyl, gegebenenfalls substituiertem Aryl, -Rd-Ar, wobei Rd für C1-8-Alkylen steht und Ar für gegebenenfalls substituiertes Aryl steht, und einem gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; und R14 ist ausgewählt aus Wasserstoff, gegebenenfalls substituiertem C1-8-Alkyl und gegebenenfalls substituiertem Aryl; (ii) wenn R5 für eine Gruppe der Formel III-a, III-b oder III-i steht, steht die Gruppe NR13(-R14) für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 3 weitere unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; oder (iii) wenn R5 für Struktur III-e steht, steht die Gruppe für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 4 unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; und deren Salze, Prodrugs und Solvate, wobei es sich bei einer Prodrug um einen in vivo hydrolysierbaren Ester einer Verbindung der Formel (I) mit einer Carboxyl- oder einer Hydroxygruppe ausgewählt aus (i) im Fall einer Carboxylgruppe C1-6-Alkoxymethylestern, C1-6-Alkanoyloxymethylestern, Phthalidylestern, C3-8-Cycloalkoxycarbonyloxy-C1-6-alkylestern, 1,3-Dioxolen-2-onylmethylestern und C1-6-Alkoxycarbonyloxyethylestern; und (ii) im Fall einer Hydroxylgruppe Phosphatestern, α-Acyloxyalkylethern und in vivo hydrolysierbaren, von Alkanoyl, Benzoyl, Phenylacetyl und substituiertem Benzoyl und Phenylacetyl, Alkoxycarbonyl, Dialkylcarbamoyl und N-(Dialkylaminoethyl)-N-alkylcarbamoyl, Dialkyl aminoacetyl und Carboxyacetyl gebildeten Estern handelt.
für einen gegebenenfalls substituierten 3- bis 8gliedrigen heterocyclischen Ring, der gegebenenfalls 1 bis 4 unabhängig voneinander aus O, N und S ausgewählte Heteroatome enthält; und deren Salze, Prodrugs und Solvate, wobei es sich bei einer Prodrug um einen in vivo hydrolysierbaren Ester einer Verbindung der Formel (I) mit einer Carboxyl- oder einer Hydroxygruppe ausgewählt aus (i) im Fall einer Carboxylgruppe C1-6-Alkoxymethylestern, C1-6-Alkanoyloxymethylestern, Phthalidylestern, C3-8-Cycloalkoxycarbonyloxy-C1-6-alkylestern, 1,3-Dioxolen-2-onylmethylestern und C1-6-Alkoxycarbonyloxyethylestern; und (ii) im Fall einer Hydroxylgruppe Phosphatestern, α-Acyloxyalkylethern und in vivo hydrolysierbaren, von Alkanoyl, Benzoyl, Phenylacetyl und substituiertem Benzoyl und Phenylacetyl, Alkoxycarbonyl, Dialkylcarbamoyl und N-(Dialkylaminoethyl)-N-alkylcarbamoyl, Dialkyl aminoacetyl und Carboxyacetyl gebildeten Estern handelt.

 (c) bei Verbindungen, in denen X für CH steht und R8 für NO2 steht, die Umsetzung einer Verbindung der Formel XXXV wie folgt
(c) bei Verbindungen, in denen X für CH steht und R8 für NO2 steht, die Umsetzung einer Verbindung der Formel XXXV wie folgt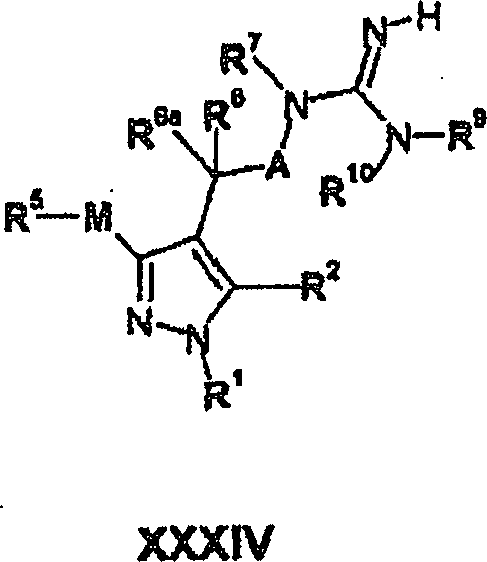 (d) bei Verbindungen, in denen X-R8 für O steht, die Umsetzung eine Verbindung der Formel XXXVII wie folgt
(d) bei Verbindungen, in denen X-R8 für O steht, die Umsetzung eine Verbindung der Formel XXXVII wie folgt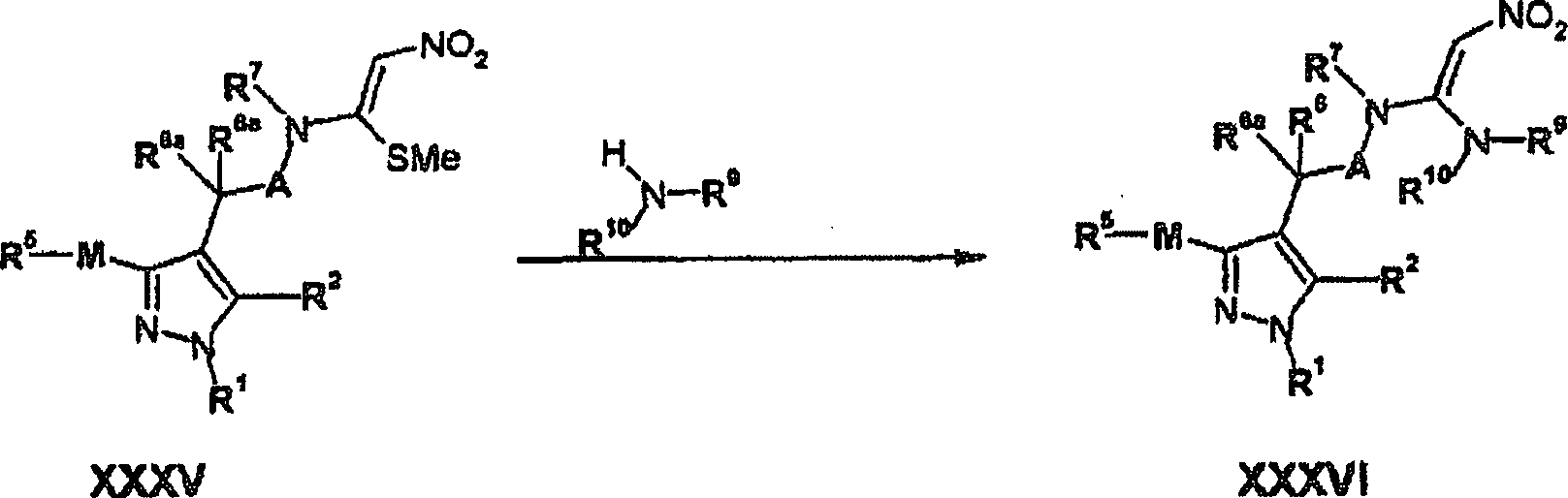 (e) bei Verbindungen, in denen X-R8 für O steht, die Umsetzung einer Verbindung der Formel XXXIX wie folgt
(e) bei Verbindungen, in denen X-R8 für O steht, die Umsetzung einer Verbindung der Formel XXXIX wie folgt (f) zur Bildung einer Verbindung, in welcher X für Stickstoff steht, die Umsetzung einer Verbindung der Formel XXXXI wie folgt
(f) zur Bildung einer Verbindung, in welcher X für Stickstoff steht, die Umsetzung einer Verbindung der Formel XXXXI wie folgt und anschließend, falls erforderlich: i) die Umwandlung einer Verbindung der Formel (I) in eine andere Verbindung der Formel (I); ii) die Abspaltung gegebenenfalls vorhandener Schutzgruppen; iii) die Bildung eines Salzes, einer Prodrug oder eines Solvats.
und anschließend, falls erforderlich: i) die Umwandlung einer Verbindung der Formel (I) in eine andere Verbindung der Formel (I); ii) die Abspaltung gegebenenfalls vorhandener Schutzgruppen; iii) die Bildung eines Salzes, einer Prodrug oder eines Solvats.