-
Hintergrund der Erfindung
-
Derzeit
sind viele Arzneimittel zur Behandlung von Störungen des zentralen Nervensystems
verfügbar. Unter
diesen Arzneimitteln befindet sich eine Kategorie, die als Antipsychotika
bekannt sind, welche zur Behandlung von ernsten mentalen Zuständen verwendet
werden, wie Schizophrenie und Erkrankungen mit schizophrener Form.
Derzeit verfügbare
Behandlungen für
solche Zustände
sind oft mit unerwünschten
schädlichen
Ereignissen assoziiert. Daher bleibt ein Bedarf für neue Verbindungen,
die die Symptome von solchen mentalen Zuständen mit verbesserten Nebenwirkungsprofilen
kontrollieren oder eliminieren.
-
Patienten,
die an Schizophrenie leiden, einem Zustand unbekannter Ätiologie,
zeigen eine Gruppe an sowohl positiven als auch negativen Symptomen.
Positive Symptome umfassen Wahnvorstellungen, Halluzinationen, gestörtes Denken
und unorganisierte Sprache, während
negative Symptome wenig Einflussnahme, Anhedonie, sozialen Rückzug, emotionalen
Rückzug,
kognitive Defizite und arme Sprache umfassen. Schizophrenie verursacht
nicht nur ein persönliches
Leiden des Patienten, sie beeinflusst stark die beruflichen und sozialen
Funktionen des Patienten, so dass der Patient oft einer Heimaufbewahrung
unterzogen werden muss, was zu hohen Kosten für die Gesellschaft führt.
-
Eine
führende
Hypothese legt nahe, dass die positiven Symptome der Schizophrenie
wirksam durch Verbindungen behandelt werden können, die als Antagonisten
an bestimmten Dopaminrezeptoren wirken. Derzeit wurden 5 prinzipielle
Dopaminrezeptoren (D1-D5)
identifiziert. Antipsychotische Wirksamkeit ist am engsten mit der
Blockade der D2 Klasse der Dopaminrezeptoren
assoziiert. Eine Klasse an antipsychotischen Mitteln, die als "typische" antipsychotische
Mittel bekannt sind (beispielsweise Haloperidol) sind bei der Kontrolle
der positiven Symptome der Schizophrenie wirksam. Jedoch behandeln
sie nicht adäquat
die negativen Symptome und sind mit signifikanten Nebenwirkungen
assoziiert, vor allem Hyperprolaktinämie, tardiver Dyskenesie und
extrapyramidalen Nebenwirkungen (EPS).
-
Ein
Ansatz zur Entwicklung besserer antipsychotischer Mittel umfasst
die Identifizierung von Verbindungen, die eine D2 Rezeptorblockade
mit Wirkungen an anderen Rezeptoren kombinieren. Ein solches Mittel ist
Clozapin.
-
Clozapin
war das erste Arzneimittel, das als "atypisches" Antipsychotikum identifiziert wurde,
das heißt ein
Arzneimittel, das bei der Behandlung sowohl der positiven als auch
negativen Symptome der Schizophrenie wirksam ist. Zusätzlich hat
es eine verringerte Neigung zur Induktion von EPS, Hyperprolaktinämie und
tardiver Dyskinesie, die bei klassischen, "typischen" Antipsychotika beobachtet wird. Obwohl
Clozapin ein wirksames Arzneimittel ist, ist dessen Brauchbarkeit
aufgrund der klinischen Beobachtung bei der Behandlung der Schizophrenie
beschränkt,
dass 1 bis 2% der behandelten Patienten eine potentiell fatale Blutstörung entwickelten,
nämlich
Agranulocytose. Kürzlich
wurde Olanzapin breit als Antipsychotikum mit relativ wenigen Nebenwirkungen
akzeptiert.
-
Jedoch
wurde eine Gewichtszunahme während
der Behandlung mit vielen der atypischen antipsychotischen Verbindungen
beobachtet (Wetterling, "Body
Weight Gain with Atypical Antipsychotics, A Comparative Review", Drug Safety 24,
59-73 (2001), Wirshing et al, "Novel
Antipsychotics: Comparison of Weight Gain Liabilities", J. Clin. Psychiatry
60, 358-363 (1999), Allison et al., "Antipsychotic-Induced Weight Gain: A
Comprehensive Research Synthesis",
Am. J. Psychiatry 156, 1686-1696 (1999), R. Ganguli, Weight gain
associated with antipsychotic drugs, J. Clin. Psychiatry 60 (Suppl.
2), 20-24, (1999). Arzneimittel mit dem klinischen Wirksamkeits-
und Sicherheitsprofil der atypischen Antipsychotika, aber mit verringerter
Neigung zur Induktion einer Gewichtszunahme repräsentieren verbesserte Mittel
zur Behandlung der Schizophrenie, bipolaren Störungen und verwandten Störungen.
-
Atypische
Antipsychotika, wie Clozapin und Olanzapin sind D2 Rezeptorantagonisten,
aber Wechselwirken auch mit anderen Neurotransmitterrezeptoren,
einschließlich
anderen Subtypen für
Dopamin und bestimmten Rezeptorsubklassen für Serotonin, Norepinephrin,
Histamin und Acetylcholin. Man nimmt an, dass einige dieser Rezeptoraktivitäten für die verbesserte
Wirksamkeit der atypischen Antipsychotika verantwortlich sind und
die schädlichen
Wirkungen dieser Mittel können
durch Wechselwirkungen mit anderen vermittelt werden. Insbesondere
wurde vorgeschlagen, dass die Gewichtszunahmeeffekte der atypischen
Antipsychotika aufgrund der Blockade des Histamin H1 Rezeptors auftreten
(Wetterling, "Body
Weight Gain with typical Antipsychotics, A Comparative Review", Drug Safety 24,
59-73 (2001), Wirshing et al., "Novel
Antipsychotics: Comparison of Weight Gain Liabilities", J. Clin. Psychiatry
60, 358-363 (1999),
Kroeze et al., "H1
Histamin Receptor Affinity Predicts Short-Term Weight Gain for Typical
and Atypical Antipsychotics Drugs", Neuropsychopharmacology 28, 519-526
(2003), N. Orthen-Gambill, Antihistaminic drugs increase feeding,
while Histidin suppresses feeding in rats. Pharmacol. Biochem. Behau.
31, 81-86 (1988). Daher repräsentiert
die Entwicklung von typischen Antipsychotika mit verringerter Affinität für den Histamin
H1 Rezeptor einen Mechanismus zur Identifizierung von Antipsychotika
mit verbessertem Nebenwirkungsprofil.
-
Die
US 5 824 676 A beschreibt
Verbindungen, die selektiv an den Serotonin Rezeptor 5-HT2 binden und
die Herstellung solcher Verbindungen und die Verwendung solcher
Verbindungen zu therapeutischen Zwecken und Arzneimittelscreeningzwecken.
-
Das
Journal of Medicinal Chemistry (1982), 25 (10), 1133-1140 beschreibt
die Wirkungen der konformationsmäßig beschränkten 4-Piperazinyl-10H-thienobenzodiazepinneuroleptika
auf die zentralen dopaminergen und cholinergen Systeme.
-
Die
EP 0 354 781 A beschreibt
Thiazolo-[1,5]benzodiazepinverbindungen und ihre Verwendung als Pharmazeutika.
-
Die
US 5 602 121 A beschreibt
alkylsubstituierte Verbindungen mit einer Dopaminrezeptoraffinität.
-
Die
EP 1 016 664 A beschreibt
kondensierte Thiophenverbindungen, die zur Behandlung von Schizophrenie,
Alzheimerscher Erkrankung, manisch-depressiver Erkrankung und dergleichen
brauchbar sind, die pharmazeutische Verwendung hiervon und ein synthetisches
Zwischenprodukt hiervon.
-
Die
vorliegende Erfindung liefert antipsychotische Verbindungen und
Verfahren zur Verwendung dieser Verbindungen zur Behandlung von
psychotischen Störungen,
insbesondere Schizophrenie und Stimmungsstörungen, wie bipolaren Störungen.
Diese Verbindungen bieten bestimmte Verbesserungen und Vorteile
gegenüber
den derzeit erhältlichen
antipsychotischen Mitteln, beispielsweise unter anderem verbesserte Nebenwirkungsprofile.
Insbesondere haben viele Verbindungen der Erfindung aufgrund ihrer
verringerten Affinität
für den
H1 Rezeptor eine verringerte Neigung, eine
Gewichtszunahme zu verursachen.
-
Kurze Zusammenfassung der Erfindung
-
Ein
Aspekt der vorliegenden Erfindung liefert Verbindungen der Formel
(I)
worin
für einen otional benzofusionierten
fünf- oder
sechsgliedrigen aromatischen Ring mit 0 bis 3 Heteroatomen steht,
die unabhängig
ausgewählt
sind aus N, S und O,
Alk für
(C
1-C
4)-Alkylen
oder hydroxysubstituiertes (C
1-C
4)-Alkylen steht,
X für Sauerstoff
oder Schwefel steht,
R
1 für Wasserstoff,
(C
1-C
6)-Fluoralkyl,
(C
3-C
6)-Cycloalkyl
oder (C
1-C
4)-Alkyl
steht, worin das (C
1-C
4)-Alkyl
unsubstituiert oder substituiert ist durch Hydroxy, Methoxy, Ethoxy,
OCH
2CH
2OH oder -CN,
R
2 für
H, Halogen, (C
1-C
6)-Fluoralkyl,
(C
3-C
6)-Cycloalkyl,
OR
4, SR
4, NO
2, CN, COR
4, C(O)OR
4, CONR
5R
6, NR
5R
6,
SO
2NR
5R
6,
NR
5COR
4, NR
5SO
2R
4,
das optional durch eine aromatische Gruppe substituiert ist, oder
für (C
1-C
6)-Alkyl steht,
worin das (C
1-C
6)-Alkyl
unsubstituiert oder durch Hydroxy substituiert ist,
R
3 steht für
Wasserstoff, (C
1-C
4)-Fluoralkyl,
(C
2-C
6)-Alkenyl,
Ar oder (C
1-C
4)-Alkyl,
worin C
1-C
4 Alkyl
unsubstituiert oder mit einer Ar Gruppe substituiert ist
R
4 für
Wasserstoff, (C
1-C
6)-Alkyl,
(C
1-C
6)-Fluoralkyl
oder eine optional substituierte aromatische Gruppe steht,
R
5 und R
6 unabhängig für Wasserstoff,
(C
1-C
6)-Alkyl oder
eine optional substituierte aromatische Gruppe stehen,
R
7 für
Wasserstoff, (C
1-C
6)-Alkyl,
(C
1-C
6)-Fluoralkyl
oder eine optional substituierte aromatische Gruppe steht,
R
8 und R
9 unabhängig für Wasserstoff,
(C
1-C
6)-Alkyl oder
eine optional substituierte aromatische Gruppe stehen,
Ar für optional
substituiertes Phenyl, Naphthyl, eine monocyclische heteroaromatische
Gruppe oder eine bicyclische heteroaromatische Gruppe steht,
Z
1 und Z
2 unabhängig ausgewählt sind
aus Wasserstoff, Halogen, (C
1-C
6)-Alkyl,
(C
1-C
6)-Fluoralkyl,
OR
7, SR
7, NO
2, CN, COR
7, CONR
8R
9, NR
8R
9 und einer optional substituierten aromatischen
Gruppe,
und alle Salze, Solvate, optischen und geometrischen
Isomeren und kristallinen Formen hiervon.
-
Bevorzugt
unter den Verbindungen der Formel (I) sind jene, worin
R1 für
Wasserstoff oder (C1-C4)-Alkyl
steht, das unsubstituiert oder substituiert ist durch Hydroxy, Methoxy, Ethoxy,
-OCH2CH2OH oder
-CN,
R2 steht für H, (C1-C6)-Alkyl, Halogen, (C1-C6)-Fluoralkyl, -OR4,
-SR4, -NO2, -CN,
-COR4, -C(O)OR4,
-CONR5R6, -NR5R6, -SO2NR5R6, -NR5COR4 oder -NR5SO2R4, oder eine optional
substituierte aromatische Gruppe, und
R3 steht
für Wasserstoff,
(C1-C4)-Alkyl, Ar
oder (C1-C4)-Alkyl-Ar.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene der Formel
(Ia)
worin
Alk für (C
1-C
4)-Alkylen steht,
R
1 für
Wasserstoff, (C
1-C
6)
Fluoralkyl, (C
3-C
6)
Cycloalkyl oder (C
1-C
4 Alkyl)
steht, worin (C
1-C
4)
Alkyl unsubstituiert oder mit Hydroxy, Methoxy, Ethoxy, -OCH
2CH2OH oder -CN substituiert ist,
R
2 für
H, Halogen, (C
1-C
6)-Fluoralkyl,
(C
3-C
6)-Cycloalkyl,
-OR
4, -SR
4, -NO
2, -CN, -COR
4, -C(O)OR
4, -CONR
5R
6, Phenyl oder (C
1-C
6)-Alkyl steht, worin das (C
1-C
6)-Alkyl unsubstituiert oder durch eine Hydroxylgruppe
substituiert ist,
R
3 für Wasserstoff,
(C
1-C
6)-Fluoralkyl,
(C
2-C
6)-Alkenyl,
Phenyl oder (C
1-C
4)-Alkyl
steht, worin (C
1-C
4)-Alkyl
unsubstituiert oder durch eine Phenylgruppe substituiert ist,
R
4 für
Wasserstoff, (C
1-C
6)-Alkyl
oder (C
1-C
6)-Fluoralkyl
steht,
R
5 und R
6 unabhängig für Wasserstoff
oder (C
1-C
6)-Alkyl
stehen,
R
7 für Wasserstoff, (C
1-C
6)-Alkyl oder (C
1-C
6)-Fluoralkyl steht,
Z
1 und
Z
2 unabhängig
ausgewählt
sind aus Wasserstoff, Halogen, (C
1-C
6)-Alkyl, (C
1-C
6)-Fluoralkyl, -OR
7,
-SR
7, -NO
2, -CN
und -COR
7, und
Phenyl unsubstituiert
oder substituiert ist durch ein bis drei Substituenten, die unabhängig ausgewählt sind
aus Wasserstoff, Halogen, (C
1-C
6)-Alkyl,
(C
1-C
6)-Fluoralkyl,
-OH, (C
1-C
6)-Alkoxy,
(C
1-C
6)-Fluoralkoxy, (C
1-C
6)-Alkylthio,
(C
1-C
6)-Acyl, (C
1-C
4)-Alkylsulfonyl,
-OCF
3, -NO
2, -CN
oder Carboxamido, welche am Stickstoff substituiert sind durch ein
oder zwei (C
1-C
4)-Alkylgruppen
und -NH
2, worin eines der Wasserstoffatome
durch eine (C
1-C
4)-Alkylgruppe
ersetzt sein kann und das andere Wasserstoffatom durch eine (C
1-C
4)-Alkylgruppe,
eine (C
1-C
4)-Acylgruppe
oder eine (C
1-C
4)-Alkylsulfonylgruppe
substituiert sein kann.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
die Stereokonfiguration des Kohlenstoffs der an Alk gebundenen Piperazingruppe "S" ist. Bevorzugte sind jene "S"-Konfigurationsverbindungen,
worin Alk für
C2-C4 Alkylen steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
die Stereokonfiguration des an Alk gebundenen Kohlenstoffs "R" ist. Bevorzugter sind die "R"-Konfigurationsverbindungen, worin Alk für Methylen
steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
Alk für
-CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH(CH3)- oder -CH2C(CH3)2-
steht. Bevorzugter sind Verbindungen, worin Alk für -CH2CH2CH2-
oder -CH2CH2- steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
X für O
steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
R1 für
C1-C4 Alkyl steht. Bevorzugt
sind Verbindungen, worin R1 für Methyl
steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
R2 für
C1-C5 Alkyl oder C1-C6 Fluoralkyl steht.
Bevorzugter sind Verbindungen, worin R2 für -CF3, Methyl oder Isopropyl steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
R3 für
C1-C4 Alkyl steht. Bevorzugter
sind Verbindungen, worin R3 für Methyl
oder Ethyl steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
Z1 und Z2 unabhängig für Wasserstoff
oder Halogen stehen. Bevorzugter sind Verbindungen, worin zumindest
eines von Z1 und Z2 für Halogen
steht. Noch bevorzugter sind die Verbindungen, worin das Halogen
für Fluor
steht.
-
Ebenfalls
bevorzugt unter den Verbindungen der Formel (I) sind jene, worin
steht für
-
Noch
bevorzugter sind Verbindungen, worin
steht für
-
Ein
weiterer Aspekt der Erfindung liefert eine pharmazeutische Zusammensetzung,
die eine wirksame Menge einer Verbindung der Formel (I) zusammen
mit einem pharmazeutisch annehmbaren Träger, Verdünnungsmittel oder Hilfsstoff
umfasst.
-
Ein
weiterer Aspekt der Erfindung liefert eine pharmazeutische Zusammensetzung,
die eine Verbindung der Formel (I) in einer Menge umfasst, die zur
Antagonisierung der D2 Rezeptorstimulierung
wirksam ist und einen pharmazeutisch annehmbaren Träger, Verdünnungsmittel
oder Hilfsstoff.
-
Ein
weiterer Aspekt der Erfindung liefert eine pharmazeutische Zusammensetzung,
die eine Verbindung der Formel (I) in einer Menge umfasst, die zur
Antagonisierung der 5-HT2A Rezeptorstimulierung
wirksam ist und einen pharmazeutisch annehmbaren Träger, Verdünnungsmittel
oder Hilfsstoff umfasst.
-
Ein
weiterer Aspekt der Erfindung liefert eine pharmazeutische Zusammensetzung,
die eine Verbindung der Formel (I) in einer Menge umfasst, die zur
Antagonisierung der 5-HT5 Rezeptorstimulierung
wirksam ist und einen pharmazeutisch annehmbaren Träger, Verdünnungsmittel
oder Hilfsstoff umfasst.
-
Ein
weiterer Aspekt der Erfindung liefert eine Verbindung der Formel
(I) zur Verwendung bei der Behandlung einer psychotischen Störung. In
einer bevorzugten Ausführungsform
ist die psychotische Störung Schizophrenie,
eine Störung
schizophrener Form oder eine schizoaffektive Störung.
-
Ein
weiterer Aspekt der Erfindung liefert die Verwendung einer Verbindung
der Formel (I) zur Herstellung eines Arzneimittels zur Behandlung
einer psychotischen Störung.
In einer bevorzugten Ausführungsform ist
die psychotische Störung
Schizophrenie, eine Störung
schizophrener Form oder eine schizoaffektive Störung.
-
Ein
weiterer Aspekt der Erfindung liefert eine Verbindung der Formel
(I) zur Verwendung bei der Behandlung einer Stimmungsstörung. In
einer bevorzugten Ausführungsform
ist die psychotische Störung
eine bipolare Störung.
In einer bevorzugteren Ausführungsform
ist die bipolare Störung
akute Manie oder bipolare Depression.
-
Ein
weiterer Aspekt der Erfindung liefert die Verwendung einer Verbindung
der Formel (I) zur Herstellung eines Arzneimittels zur Behandlung
einer Stimmungsstörung.
In einer bevorzugten Ausführungsform
ist die Stimmungsstörung
eine bipolare Störung.
In einer bevorzugteren Ausführungsform
ist die bipolare Störung akute
Manie oder bipolare Depression.
-
Ein
weiterer Aspekt der Erfindung liefert Verbindungen der Formel (VIz)
worin
R
2 für H, C
1-C
6 Alkyl, Halogen,
C
1-C
6 Fluoralkyl,
OR
4, SR
4, NO
2, CN, COOR
4, C(O)OR
4, CONR
5R
6, SO
2NR
5R
6, NR
5R
6,
NR
5COR
4, NR
5SO
2R
4 oder
optional substitiertes Phenyl steht,
R
4 für Wasserstoff,
C
1-C
6 Alkyl, C
1-C
6 Fluoralkyl oder
optional substituiertes Phenyl steht,
R
5 und
R
6 unabhängig
für Wasserstoff,
C
1-C
6Alkyl oder
optional substituiertes Phenyl stehen,
Z
1 und
Z
2 unabhängig
aus Wasserstoff, Halogen, C
1-C
6 Alkyl,
C
1-C
6 Fluoralkyl,
OR
7, SR
7, NO
2, CN, COR
7, CONR
8R9, SO
2NR
8R
9, NR
8SO
2R
7, NR
8R
9 oder optional substituiertem Phenyl ausgewählt sind,
R
7 für
Wasserstoff, C
1-C
6 Alkyl,
C
1-C
6 Fluoralkyl
oder optional substituiertes Phenyl steht,
R
8 und
R
9 unabhängig
für Wasserstoff,
C
1-C
6 Alkyl oder
optional substituiertes Phenyl stehen,
W für Sauerstoff oder Schwefel
steht
und Tautomere und Säureadditionssalze
hiervon, die als Zwischenprodukte bei der Herstellung der erfindungsgemäßen Verbindungen
brauchbar sind.
-
Ein
weiterer Aspekt der Erfindung liefert Verbindungen der Formel (Vz)
worin
X für Sauerstoff
oder Schwefel steht,
Alk für
-CH
2CH
2- steht,
das optional mit einer oder zwei Methylgruppen oder einer Ethylgruppe
substituiert ist,
R
3 für C
1-C
4 Alkyl, Ar oder
C
1-C
4 Alkyl-Ar steht,
worin Ar für
optional substituiertes Phenyl, Naphthyl, monocyclische Heteroaromaten
oder bicyclische Heteroaromaten steht,
und Säureadditionssalze
hiervon, die als Zwischenprodukte zur Herstellung der erfindungsgemäßen Verbindungen
brauchbar sind.
-
Ein
weiterer Aspekt der Erfindung umfasst verbesserte Nebenwirkungsprofile
(beispielsweise verringerte Gewichtszunahme) gegenüber derzeit
verfügbaren
antipsychotischen Mitteln und/oder bessere Dopamin D2 Bindung.
-
Detaillierte Beschreibung der Erfindung
-
Die
Ausdrücke
und Symbole, die hierin verwendet werden, stimmen mit der Verwendung
in der allgemeinen chemischen Literatur überein falls nichts anderes
angegeben ist.
-
Beispielsweise
umfasst der Ausdruck "C1-C6 Alkyl" gesättigte Alkylgruppen,
die verzweigt oder unverzweigt sein können, wie Methyl, Ethyl, n-Propyl,
Isopropyl, n-Butyl, Isobutyl, sek-Butyl, tert-Butyl, Pentyl, 2-Pentyl,
3-Pentyl, Neopentyl, n-Hexyl und dergleichen.
-
Der
Ausdruck "C1-C4 Alkyl" umfasst gesättigte Reste
und diese können
verzweigt oder unverzweigt sein, wie Methyl, Ethyl, n-Propyl, Isopropyl,
n-Butyl, Isobutyl, sek-Butyl, tert-Butyl und dergleichen.
-
Der
Ausdruck "C2-C6 Alkenyl" umfasst ungesättigte Alkylgruppen,
die verzweigt oder unverzweigt sein können, mit 2 bis 6 Kohlenstoffatomen,
wie Vinyl, Alkyl, 1-Buten-4-yl, 2-Buten-4-yl, -CHC(=CH2)CH3, -CH=CH2CH2CH3, -CH=C(CH3)2, -CH=CH-CH2CH2CH3 ,
-CH=CHCH2CH2CH2CH3 und dergleichen.
-
Der
Ausdruck "C1-C4 Alkylen" bezieht sich auf
geardkettige Alkylengruppen, wie -CH2-,
-CH2CH2-, -CH2CH2CH2-,
-CH2CH2CH2CH2- oder verzweigte
Alkylengruppen, wie -CH2C(CH3)2- oder -CH2CH(CH3)-, -CH2CH2CH(CH3)-, -CH2CH(CH3)CH2- und derglichen.
-
Der
Ausdruck "C3-C6 Cycloalkyl" bezieht sich auf
Cyclopropyl, Cyclobutyl, Cyclopentyl und Cyclohexyl.
-
Der
Ausdruck "Halogen" umfasst Fluor, Chlor,
Brom und Iod.
-
Der
Ausdruck "C1-C6 Fluoralkyl" bezieht sich auf
eine C1-C6 Alkylgruppe,
die mit 1 bis 6 Fluoratomen substituiert ist, wie Fluormethyl, Difluormethyl,
Trifluormethyl, 2-Fluorethyl, 2,2,2-Trifluorethyl, 1,1,2,2,2-Pentafluorethyl,
3-Fluorpropyl, 3,3,3-Trifluorpropyl, 1,1,1,3,3,3-Hexafluorprop-2-yl
und 6-Fluorhexyl
und dergleichen.
-
Der
Ausdruck "C1-C6 Alkoxy" umfasst solche Gruppen,
wie Methoxy, Ethoxy, Isopropoxy, sek-Butoxy, tert-Butoxy, 2-Pentoxy, 3-Hexyloxy
und dergleichen.
-
Der
Ausdruck "C1-C6 Fluoralkoxy" bezieht sich auf
eine C1-C6 Fluoralkylgruppe,
die an einen Sauerstoff gebunden ist.
-
Der
Ausdruck "C1-C6 Alkylthio" umfasst solche Gruppen,
wie Methylthio, Ethylthio, Isopropylthio, sek-Butylthio, tert-Butylthio,
1-Hexylthio und dergleichen.
-
Der
Ausdruck "Acyl" umfasst beispielsweise
Formyl, Acetyl, Propanoyl, Butanoyl, 2-Methylpropanoyl, Hexanoyl
und dergleichen.
-
Der
Ausdruck "C1-C4 Alkylsulfonyl" umfasst Methansulfonyl,
Ethansulfonyl, Propansulfonyl, Isopropansulfonyl, 1-Butansulfonyl
und dergleichen.
-
Der
Ausdruck "optional
substituierter Aromat" bezieht
sich auf eine Phenyl-, Napthyl-, monocyclische heteroaromatische
oder bicyclische heteroaromatische Gruppe, die mit 1 bis 3 Substituenten
substituiert ist, die unabhängig
ausgewählt
sind aus Wasserstoff, Halogen, C1-C6 Alkyl, C1-C6 Fluoralkyl, -OH, C1-C6 Alkoxy, C1-C6 Fluoralkoxy, C1-C6 Alkylthio, Acyl, C1-C4 Alkylsulfonyl, -NO2,
-CN, Carboxamido, das am Stickstoff durch 1 oder 2 C1-C4 Alkylgruppen substituiert sein kann und
NH2, worin einer der Wasser stoffe durch
eine C1-C4 Alkylgruppe
substituiert sein kann und der andere Wasserstoff entweder durch
eine C1-C4 Alkylgruppe,
eine Acylgruppe oder eine C1-C4 Alkylsulfonylgruppe
ersetzt sein kann.
-
Der
Ausdruck "monocyclischer
Heteroaromat" bezieht
sich auf einen fünf-
oder sechsgliedrigen aromatischen Ring, der 1 bis 3 Heteroatome
enthält,
die aus N, O und S ausgewählt
sind. Es sollte erkannt werden, dass falls eines der Heteroatome
O oder S ist, der heteroaromatische Ring ein fünfgliedriger Ring sein muss
und dass alle anderen Heteroatome, die hierin enthalten sind, für N stehen
müssen.
Beispiele für
solche monocyclischen heteroaromatischen Systeme umfassen Furan,
Thiophen, Pyridin, Pyrimdidin, Thiazol, 1,2,3-Triazol und dergleichen.
-
Der
Ausdruck "bicyclischer
Heteroaromat" bezieht
sich auf bicyclische aromatische Systeme, die 1 bis 3 Heteroatome
enthalten, welche aus N, O oder S ausgewählt sind. Beispiele umfassen
Indol, Benzofuran, Benzothiophen, Chinolin, Isochinolin, Indazol,
Benzothiazol und dergleichen.
-
Der
Ausdruck "optional
substituiertes Phenyl" bezieht
sich auf Phenyl, das mit 1 bis 3 Substituenten substituiert sein
kann, die unabhängig
ausgewählt
sind aus Wasserstoff, Halogen, C1-C6 Alkyl, C1-C6 Fluoralkyl, -OH, C1-C6 Alkoxy, C1-C6 Fluoralkoxy, C1-C6 Thioalkyl, Acyl, C1-C4 Alkylsulfonyl, -NO2,
-CN, Carboxamido, das am Stickstoff durch 1 oder 2 C1-C4 Alkylgruppen substituiert sein kann und
NH2, worin einer der Wasserstoffe durch
eine C1-C4 Alkylgruppe
ersetzt sein kann und der andere Wasserstoff entweder durch eine
C1-C4 Alkylgruppe,
eine Acylgruppe oder eine C1-C4 Alkylsulfonylgruppe
ersetzt sein kann.
-
Der
Ausdruck "optional
substituierte Phenyl-, Naphthyl-, monocyclische heteroaromatische
oder bicyclische heteroaromatische Gruppe" bezieht sich auf eine Phenyl-, Naphthyl-,
monocyclische heteroaromatische oder bicyclische heteroaromatische
Gruppe, die mit 1 bis 3 Substituenten substituiert sein kann, die
unabhängig
ausgewählt
sind aus Wasserstoff, Halogen, C1-C6 Alkyl, C1-C6 Fluoralkyl, -OH, C1-C6 Alkoxy,
C1-C6 Fluoralkoxy,
C1-C6 Thioalkyl,
Acyl, C1-C4 Alkylsulfonyl,
-NO2, -CN, Carboxamido, das am Stickstoff
mit 1 oder 2 C1-C4 Alkylgruppen
substituiert ist, und NH2, worin die Wasserstoffe
durch eine C1-C4 Alkylgruppe
ersetzt werden können
und der andere Wasserstoff entweder durch eine C1-C4 Alkylgruppe, eine Acylgruppe oder eine C1-C4 Alkylsulfonylgruppe
ersetzt sein kann.
-
Im
Fall eines optional benzofusionierten fünf- oder sechsgliedrigen aromatischen
Rings mit 0 bis 3 Heteroatomen, die unabhängig aus N, O und S ausgewählt sind,
sind die zwei Atome des aromatischen Rings, die an den angefügten siebengliedrigen
Ring fusioniert sind, zwangsweise beide Kohlenstoff. Falls der aromatische
Ring zwei zusätzliche
benachbarte Kohlenstoffatome enthält, kann ein Benzolring an
den aromatischen Ring an die zwei benachbarten Kohlenstoffatome
fusioniert sein. Beispiele für
optional benzofusionierte fünf- oder
sechsgliedrige aromatische Ringe mit 0 bis 3 Heteroatomen die unabhängig aus
N, S und O ausgewählt sind,
umfassen Benzol, Pyridin, Furan, Pyrrol, Thiophen, Thiazol, Oxazol,
Pyrazol, Imidazol, 1,2,3-Triazol, Naphthalin, Chinolin, Isochinolin,
Indol, Benzofuran, Benzothiophen und dergleichen.
-
Die
Verbindungen der vorliegenden Erfindung können in Abhängigkeit ihrer Struktur und
der Art der Synthese und Isolierung als pharmazeutisch annehmbares
Solvat vorkommen. Diese Solvate umfassen Wasser, Methanol und Ethanol.
Solvatisierte Formen der Verbindungen der vorliegenden Erfindung
stellen eine weitere Ausführungsform
der vorliegenden Erfindung dar.
-
Die
Verbindungen der vorliegenden Erfindung können in Abhängigkeit ihrer Struktur und
Art der Synthese und Isolierung als pharmazeutisch annehmbare Hydrate
vorkommen. Hydratisierte Formen der erfindungsgemäßen Verbindungen
stellen eine weitere Ausführungsform
der vorliegenden Erfindung dar.
-
Die
vorliegende Erfindung liefert auch neue kristalline Formen der Verbindungen
der Formel (I). Neue kristalline Formen können durch Kristallisation
unter kontrollierten Bedingungen hergestellt werden. Die Kristallisation
aus einer Lösung
und Aufschlämmtechniken
sind im Umfang des vorliegenden Verfahrens enthalten. In der Praxis
können
mehrere Faktoren die erhaltene Form beeinflussen, einschließlich Temperatur,
Lösemittelzusammensetzung
und auch optimales Beimpfen. Impfkristalle können aus vorherigen Synthesen
der Verbindungen erhalten werden, bei denen Kristalle isoliert wurden.
Die neuen kristallinen Formen der vorliegenden Erfindung können auch
durch Lösen
der Verbindungen der Formel (I) in einem Lösemittel und anschließende Bildung
des Hydrochloridsalzes durch die Zugabe einer Lösung, die Chlorwasserstoffsäure enthält, und einer
Ermöglichung
der Kristallisation unter Kontrolle der Temperatur hergestellt werden.
Ebenfalls können
die neuen Formen der vorliegenden Erfindung auch durch Lösen der
Anhydratsalzform der Verbindungen der Formel (I) in Wasser und einer
Beimpfung mit einer kristallinen Form hergestellt werden.
-
Es
sind mehrere Verfahren zur Charakterisierung der kristallinen Formen
der organischen Verbindungen verfügbar. Beispielsweise umfassen
Verfahren Differentialscanningkalorimetrie, Festphasen NMR Spektrometrie,
Infrarotspektrometrie und Röntgenbeugung
am Pulver. Unter diesen sind die Röntgenbeugung am Pulver und
die Festphasen NMR Spektroskopie zur Identifizierung und Unterscheidung
zwischen den Kristallformen sehr brauchbar.
-
Differentielle
thermische/themographische Analysen DTA/TGA) werden auf einer TA
simultanen DTA/TGA Einheit (Modell SDT2960) ausgeführt. Die
Proben werden in offenen Aluminiumpfannen von 25 bis 295°C mit 10°C/min mit
einer Stickstoffspülung
von 150 ml/min erhitzt. Die Temperatur wird mit Indium kalibriert.
Die Gewichtskalibrierung wird mit Standards, die vom Hersteller
geliefert werden, ausgeführt
und gegen Natriumtartratdesolvatisierung verifiziert.
-
Die
Röntgenbeugungsanalysen
am Pulver werden durch eine Vielzahl an Methoden ausgeführt, die dem
Fachmann bekannt sind. Diese Methoden können zur Erhöhung der
Empfindlichkeit variiert werden durch die Probenpräparationstechniken
und durch die Verwendung intensiverer Strahlung, kleineren Scanstufen
und langsameren Scangeschwindigkeiten. Ein Verfahren ist folgendes.
Entweder mit oder ohne leichtem Vermahlen der Probe mit einem Achatmörser und
Stößel wird
die Probe in einen Probenhalter für die Röntgenstreuungsmessung am Pulver
geladen. Es werden Mikroröntgenbeugungsmuster
am Pulver (μ-xrpd)
auf einem Bruker AXS Röntgenpulverdiffraktometer
erhalten, das mit einer CuKα Quelle
(λ = 1,54056
Angström)
und einem Hi Star Flächendetektor
ausgestattet ist, der bei 40 kV und 50 mA arbeitet. Das Instrument
wird mit einem einzelnen Göebel
Spiegel am eintreffenden Strahl, einem 0,05 mm Collimator und einem
Abstand zwischen Probe und Detektor von 15 cm konfiguriert. Die
Daten werden über
den Bereich von 5,5 bis 35° 2θ integriert.
-
Es
ist in der Kristallographietechnik gut bekannt, dass für jede gegebene
Kristallform die relativen Intensitäten und die Peakbreiten der
Streuungspeaks aufgrund mehrerer Faktoren variieren können, einschließlich der
Effekte der bevorzugten Orientierung und/oder Partikelgröße. Wenn
die Effekte der bevorzugten Orientierung und/oder Partikelgröße vorhanden
sind, können
die Peakintensitäten
verändert
sein, aber die charakteristischen Peakpositionen oder das Polymoph
sind unverändert.
Siehe beispielsweise The United States Pharmacopoeia Nr. 24, National
Formulary Nr. 19, Seiten 1843-1844, 2000.
-
Das
Vermahlen kann verwendet werden, um die Peakintensität zu minimieren.
Falls jedoch das Vermahlen signifikant das Diffraktogramm verändert oder
den kristallinen Zustand der Probe ändert, dann sollte das Diffraktogramm
der ungemahlenen Probe verwendet werden. Das Vermahlen wird in einem
kleinen Achatmörser
und Stößel ausgeführt. Der
Mörser
wird während
dem Vermahlen gehalten und es wird ein leichter Druck auf den Stößel ausgeübt.
-
So
kann eine gut präparierte
Probe der kristallinen Verbindung der Formel I durch einen oder
mehrere 2 θ Werte
in einem Röntgenbeugungsmuster
charakterisiert werden, das wie oben erhalten wird.
-
Kristalline
Verbindungen der Formel I können
auch durch Festphasen NMR Spektroskopie charakterisiert werden.
Chemische Festphasen 13C Verschiebungen
reflektieren nicht nur die molekulare Struktur, sondern auch die
elektronische Umgebung des Moleküls
im Kristall.
-
Die
Verbindungen der Formel (I) können
in optisch isomeren Formen existieren, das heißt stereoisomeren Formen. Das
heißt
diese Verbindungen haben zumindest ein chirales das heißt asymmetrisches
Zentrum am Kohlenstoffatom des Piperazinrings, an den "Alk" gebunden ist. Eine
solche Asymmetrie führt
zu mindestens einem Enantiomerenpaar. Ein gleiches Gemisch an Enantiomeren
ist auch als "razemisches" Gemisch oder als "Razemat" bekannt. Die Darstellung
der Formel (I) soll jedes der Stereoisomere darstellen und Gemische
hiervon.
-
Die
Ausdrücke "R" und "S" werden
hierin verwendet, wie sie herkömmlich
in der organischen Chemie verwendet werden, um eine spezifische
Konfiguration eines chiralen Zentrums zu bezeichnen. Es ist verständlich,
dass die erfindungsgemäßen Verbindungen
als Stereoisomere vorkommen können.
Alle solchen Enantiomere, Diastereomere und Gemische hiervon sind
im Umfang der Erfindung enthalten. Wenn spezifische Stereochemien
in dieser Beschreibung spezifiziert sind, werden die Cahn-Prelog-Ingold Bezeichnungen
(R)- und (S)- und die cis und trans Bezeichnung der relativen Stereochemie
verwendet, um die spezifischen Isomere und die relative Stereochemie
zu benennen. Einige der Verbindungen der Formel (I) haben zwei oder
mehr chirale Zentren.
-
Einige
der erfindungsgemäßen Verbindungen
können
auch in Bezug auf eine oder mehrere Doppelbindungen isomer sein,
was geometrische Isomere, das heißt cis und trans einführt. Eine
Diskussion der optischen und geometrischen Isomere kann in organischen
Standardchemielehrbüchern
gefunden werden, wie March's
Advanced Organic Chemistry, 5. Ausgabe, Kapitel 4, Wiley-Interscience,
John Wiley & Sons,
Inc. New York (2001), hierin später "March" genannt. Wenn hierin
eine Verbindung der vorliegenden Erfindung erwähnt wird oder die Struktur
gezeigt wird, ohne dass eine Angabe der asymmetrischen Form gemacht
wird, sind alle möglichen
asymmetrischen Formen gemeint. Die Erfindung ist nicht auf ein bestimmtes
isomer beschränkt, umfasst
aber alle möglichen
individuellen Isomere und Razemate.
-
Die
Verbindungen der Formel (Ie), die in Tabelle 1 angegeben sind, sind
von besonderem Interesse:
-
Die
absolute Konfiguration ist "S" am Kohlenstoff der
Piperazingruppe, die an Alk gebunden ist, falls nichts anderes angegeben
ist. Tabelle 1
| Bsp.
Nr. | Alk-X-R3 | R1 | R2 | Z1 | Z2 |
| 198 | CH2CH2OCH3 | H | CH3 | H | H |
| 217 | CH2CH2OCH3 | CH3 | CH3 | H | H |
und alle Salze, Solvate und geometrischen Isomere
und kristallinen Formen hiervon.
- Bsp. Nr. entspricht der
Beispielnummer im Beispielteil
-
Die
Verbindungen der Formel (If), die in Tabelle 2 angegeben sind, sind
von besonderem Interesse:
-
Die
absolute Konfiguration ist "S" am Kohlenstoff der
Piperazingruppe, die an Alk gebunden ist, falls nichts anderes angegeben
ist. Tabelle 2
| BSP.
NR. | Alk-X-R3 | R1 | R2 | Z3 | Z2 |
| 199 | CH2CH2OCH3 | H | CH3 | H | H |
| 200 | CH2CH2OCH3 | H | CH(CH3)2 | H | H |
| 201 | CH2CH2CH2OCH3 | H | CH(CH3)2 | H | H |
| 202 | CH2CH2OCH3 | H | CF3 | H | H |
| 203 | CH2CH2CH2OCH3 | H | CF3 | H | H |
| 204 | CH2CH2OCH3 | H | CH3 | Cl | H |
| 205 | CH2CH2OCH3 | H | CH(CH3)2 | Cl | H |
| 206 | CH2CH2OCH3 | H | CH3 | F | H |
| 207 | CH2CH2OCH3 | H | CF3 | F | H |
| 208 | CH2CH2OCH3 | H | CH(CH3)2 | F | H |
| 209 | CH2CH2OCH3 | H | CH(CH3)2 | H | F |
| 210 | CH2CH2OCH3 | H | CF3 | H | F |
| 211 | CH2CH2CH2OCH3 | H | CF3 | H | F |
| 212 | CH2CH2CH2OCH3 | H | CF3 | F | H |
| 213 | CH2CH2OCH3 | H | Cyclopropyl | H | F |
| 214 | CH2CH2OCH3 | H | CF3 | F | F |
| 215 | CH2CH2OCH3 | H | CF3 | Cl | H |
| 215a | CH2CH2OCH3 | H | Cl | H | H |
| 216 | CH2CH2OCH3 | CH3 | CH3 | H | H |
| 218 | CH3CH2OCH3 | CH3 | CH(CH3)2 | H | H |
| 219 | CH2CH2CH2OCH3 | CH3 | CH(CH3)2 | H | H |
| 221 | CH2CH2OCH3 | CH3 | CF3 | H | H |
| 222 | CH2CH2CH2OCH3 | CH3 | CF3 | H | H |
| 224 | CH2CH2OCH3 | CH3 | CH3 | Cl | H |
| 225 | CH2CH2OCH3 | CH3 | CH(CH3)2 | Cl | H |
| 227 | CH2CH2OCH3 | CH3 | CH3 | F | H |
| 228 | CH2CH2OCH3 | CH3 | CF3 | F | H |
| 229 | CH2CH2OCH3 | CH3 | CH(CH3)2 | F | H |
| 230 | CH2CH2OCH3 | CH | CH(CH3)2 | H | F |
| 231 | CH2CH2OCH3 | CH3 | CF3 | H | F |
| 232 | CH2CH2CH2OCH3 | CH3 | CF3 | H | F |
| 233 | CH2CH2CH2OCH3 | CH3 | CF3 | F | H |
| 234 | CH2CH2OCH3 | CH3 | Cyclopropyl | H | F |
| 235 | CH2CH2OCH3 | CH3 | CF3 | F | F |
| 236 | CH2CH2OCH3 | CH3 | CF3 | Cl | H |
| 236a | CH2CH2OCH3 | CH3 | Cl | H | H |
und alle Salze, Solvate, optischen und geometrischen
Isomere und kristallinen Formen hiervon.
- Bsp. Nr. entspricht
der Beispielnummer im Beispielteil
-
Die
Verbindungen der Formel (Ig), die in Tabelle 3 angegeben sind, sind
von besonderem Interesse:
worin
die absolute Konfiguration "S" am Kohlenstoff der Piperazingruppe
ist, die an Alk gebunden ist, falls nichts anderes angegeben ist,
und
E
2 für C steht und E
3 für S steht. Tabelle 3
| Bsp.
Nr. | E1 | Alk-X-R3 | R1 | R2 | Z1 | Z2 |
| 237 | CH | (R)CH2OH | H | CH3 | H | H |
| 238 | CH | (R)CH2OCH3 | H | CH3 | H | H |
| 239 | CH | (R)CH2OCHC(=CH2)(CH3) | H | CH3 | H | H |
| 240 | CH | CH2CH2OH | H | CH3 | H | H |
| 241 | CH | (R)CH2CH2OH | H | CH3 | H | H |
| 241a | CH | CH2CH2OCH3 | H | H | H | H |
| 242 | CH | CH2CH2OCH3 | H | CH3 | H | H |
| 242a | CH | CH2CH2Ophenyl | H | CH3 | H | H |
| 243 | CH | CH2CH2OCH2CH3 | H | CH3 | H | H |
| 244 | CH | (R)CH2OPh | H | CH3 | H | H |
| 245 | CH | CH2CH2OCH3 | H | CH(CH3)2 | H | H |
| 246a | CH | CH2CH2CH2OCH3 | H | H | H | H |
| 246b | CH | CH2CH2CH2OCH3 | H | CH3 | H | H |
| 247 | CH | CH2CH2CH2CH2OCH3 | H | CH3 | H | H |
| 247a | CH | CH2CH2OCH3 | H | H | H | F |
| 247b | CH | CH2CH2CH2OCH3 | H | H | H | F |
| 248 | CH | CH2CH2OCH3 | H | CH3 | H | F |
| 248c | CH | CH2CH2CH2OCH3 | H | CH3 | H | F |
| 249 | CM | CH2CH2OCH3 | H | CH3 | F | H |
| 249a | CH | CH2CH2CH2OCH3 | H | CH3 | F | H |
| 249b | CH | CH2CH2OH | H | CH3 | H | F |
| 250 | CH | CH2CH2OCH2CH3 | H | CH3 | H | F |
| 251 | CH | CH2CH2OCH2CH3 | H | CH3 | F | H |
| 252 | CH | CH2CH2OCH3 | H | CH2CH3 | H | F |
| 253 | CH | CH2CH2OCH3 | H | CH(CH3)2 | H | F |
| 253a | CH | CH2CH2OCH3 | H | H | F | F |
| 254 | CH | CH2CH2OCH3 | H | CH3 | F | F |
| 254d | CH | CH2CH2OH | H | CH3 | F | F |
| 255 | CH | CH2CH2OCH2CH3 | H | CH3 | F | F |
| 256 | CH | CH2CH2OCH3 | H | CH2CH3 | F | F |
| 257 | CH | CH2CH2OCH3 | H | CH3 | H | Cl |
| 257a | CH | CH2CH2OCH3 | H | H | Cl | H |
| 257b | CH | CH2CH2CH2OCH3 | H | H | Cl | H |
| 258 | CH | CH2CH2OCH3 | H | CH3 | Cl | H |
| 258b | CH | CH2CH2OH | H | CH3 | Cl | H |
| 259 | CH | (R)CH2OH | CH3 | CH3 | H | H |
| 260 | CH | (R)CH2OCH3 | CH3 | CH3 | H | H |
| 261 | CH | (R)CH2OCH2CH3 | CH3 | CH3 | H | H |
| 262 | CH | (R)CH2OCH2CH=CH2 | CH3 | CH3 | H | H |
| 263 | CH | (R)CH2OCH2CH2CH3 | CH3 | CH3 | H | H |
| 264 | CH | (R)CH2OCH2C(=CH2)(CH3) | CH3 | CH3 | H | H |
| 265 | CH | (R)CH2Ophenyl | CH3 | CH3 | H | H |
| 266 | CH | CH2CH2OH | CH3 | CH3 | H | H |
| 266a | CH | CH2CH2OCH3 | CH3 | H | H | H |
| 266b | CH | CH2CH2CH2OCH3 | CH3 | H | H | H |
| 267 | CH | CH2CH2OCH3 | CH3 | CH3 | H | H |
| 268 | CH | CH2CH2OCH2CH3 | CH3 | CH3 | H | H |
| 269 | CH | CH2CH2Ophenyl | CH3 | CH3 | H | H |
| 270 | CH | (R)CH2OCH2phenyl | CH3 | CH3 | H | H |
| 271 | CH | (R)CH2Ophenyl | CH3 | CH3 | H | H |
| 272 | CH | CH2CH2OCH3 | CH3 | CH(CH3)2 | H | H |
| 273a | CH | CH2CH2CH2OCH3 | CH3 | CH3 | H | H |
| 274 | CH | CH2CH2CH2CH2OCH3 | CH3 | CH3 | H | H |
| 276 | CH | CH2CH2OCH3 | CH3 | CH3 | H | H |
| 277 | CH | CH2CH2CH2OCH3 | CH3 | CF3 | H | H |
| 278a | CH | CH2CH2OCH3 | CH3 | H | H | F |
| 278b | CH | CH2CH2CH2OCH3 | CH3 | H | H | F |
| 278c | CH | CH2CH2OH | CH3 | C H3 | H | F |
| 279 | CH | CH2CH2OCH3 | CH3 | CH3 | H | F |
| 280 | CH | CH2CH2OCH3 | CH3 | CH2CH3 | H | F |
| 281 | CH | CH2CH2OCH2CH3 | CH3 | CH3 | H | F |
| 282 | CH | CH2CH2OCH3 | CH3 | CH(CH3)2 | H | F |
| 283 | CH | CH2CH2CH2OCH3 | CH3 | CH3 | H | F |
| 283a | CH | CH2CH2CH2OCH3 | CH3 | CH3 | F | H |
| 284 | CH | CH2CH2OCH3 | CH3 | CF3 | H | F |
| 285 | CH | CH2CH2CH2OCH3 | CH3 | CF3 | H | F |
| 286 | CH | CH2CH2OCH3 | CH3 | CH3 | F | H |
| 287 | CH | CH2CH2OCH2CH3 | CH3 | CH3 | F | H |
| 287a | CH | CH2CH2OH | CH3 | CH3 | F | F |
| 288 | CH | CH2CH2OCH3 | CH3 | CH3 | F | F |
| 289 | CH | CH2CH2OCH2CH3 | CH3 | CH3 | F | F |
| 290 | CH | CH2CH2OCH3 | CH3 | CH2CH3 | F | F |
| 290a | CH | CH2CH2OCH3 | CH3 | H | F | F |
| 290b | CH | CH2CH2OCH3 | CH3 | CF3 | F | F |
| 291 | CH | CH2CH2OCH3 | CH3 | CH3 | H | Cl |
| 291a | CH | CH2CH2OCH3 | CH3 | H | Cl | H |
| 291b | CH | CH2CH2CH2OCH3 | CH3 | H | Cl | H |
| 291c | CH | CH2CH2OH | CH3 | CH3 | Cl | H |
| 292 | CH | CH2CH2OCH3 | CH3 | CH3 | Cl | H |
| 293 | CH | CH2CH2OCH3 | CH3 | CF3 | Cl | H |
| 294 | CH | CH2CH2CH2OCH3 | CH3 | CF3 | Cl | H |
| 295 | CH | CH2CH2SPhenyl | H | CH3 | H | H |
| 296 | CH | CH2CH2SPhenyl | CH3 | CH3 | H | H |
| 297 | CH | CH2CH2SCH3 | CH3 | CH3 | H | F |
| 300 | N | CH2CH2OH | H | CH3 | H | H |
| 301 | N | CH2CH2OCH3 | H | CH3 | H | H |
| 302 | N | CH2CH2OCH2OCH3 | H | CH3 | H | H |
| 303 | N | CH2CH2OCH3 | H | CH2CH3 | H | H |
| 304 | N | CH2CH2OCH3 | H | CH2CH2CH3 | H | H |
| 305 | N | CH2CH2OCH3 | H | CH2CH2CH2CH3 | H | H |
| 307 | N | CH2CH2OH | H | CH(CH3)2 | H | H |
| 308 | N | CH2CH2OCH3 | H | CH(CH3)2 | H | H |
| 309 | N | CH2CH2OCH2CH3 | H | CH(CH3)2 | H | H |
| 310 | N | CH2CH2CH2OCH3 | H | CH(CH3)2 | H | H |
| 311 | N | CH2CH2OCH3 | H | Cyclopentyl | H | H |
| 312 | N | CH2CH2OCH3 | H | CH2OH | N | H |
| 313 | N | CH2CH2OCH3 | H | C(O)OCH2CH3 | H | H |
| 314 | N | CH2CH2OCH3 | H | CF3 | H | H |
| 316 | N | CH2CH2OCH3 | H | CF2H | H | H |
| 318 | N | CH2CH2OCH3 | H | CH2CH2CF3 | H | H |
| 319 | N | CH2CH2OH | CH3 | CH3 | H | H |
| 320 | N | CH2CH2OCH3 | CH3 | CH3 | H | H |
| 321 | N | CH2CH2OCH2CH3 | CH3 | CH3 | H | H |
| 322 | N | CH2CH2OCH3 | CH3 | CH2CH3 | H | H |
| 323 | N | CH2CH2OCH3 | CH3 | CH2CH2CH3 | H | H |
| 324 | N | CH2CH2OCH3 | CH3 | (CH2)3CH3 | H | H |
| 326 | N | CH2CH2OH | CH3 | CH(CH3)2 | H | H |
| 327 | N | CH2CH2OCH3 | CH3 | CH(CH3)2 | H | H |
| 328 | N | CH2CH2OCH2CH3 | CH3 | CH(CH3)2 | H | H |
| 329 | N | CH2CH2CH2OCH3 | CH3 | CH(CH3)2 | H | H |
| 331 | N | CH2CH2OCH3 | CH3 | Cyclopentyl | H | H |
| 332 | N | CH2CH2OCH3 | CH3 | CH2OH | H | H |
| 333 | N | CH2CH2OCH3 | CH3 | C(O)OCH2CH3 | H | H |
| 334 | N | CH2CH2OCH3 | CH3 | CF3 | H | H |
| 336 | N | CH2CH2OCH3 | CH3 | CF2H | H | H |
| 338 | N | CH2CH2OCH3 | CH3 | CH2CH2CF3 | H | H |
| 339 | N | CH2CH2OCH3 | cyclopropyl | CH(CH3)2 | H | H |
| 340 | N | CH2CH2OCH3 | CH2CH3 | CH(CH3)2 | H | H |
| 341 | N | CH2CH2OCH3 | CH2CH2CH3 | CH(CH3)2 | H | H |
| 342 | N | CH2CH2OCH3 | CH2CH2OH | CH(CH3)2 | H | H |
| 343 | N | CH2CH2OCH3 | CH2CH2OCH3 | CH(CH3)2 | H | H |
| 344 | N | CH2CH2OCH3 | CH2CH3 | CF3 | H | H |
| 346 | N | CH2CH2OCH3 | (CH2)2CH3 | CF3 | H | H |
| 348 | N | CH2CH2OCH3 | C(O)CH2F | CF3 | H | H |
| 349 | N | CH2CH2OCH3 | CH2CH2F | CF3 | H | H |
| 351 | N | CH2CH2OCH3 | CH2CH2CH2F | CF3 | H | H |
| 353 | N | CH2CH2OCH3 | CH2CH2OH | CF3 | H | H |
| 355 | N | CH2CH2OCH3 | CH2CH2CH2OH | CF3 | H | H |
| 357 | N | CH2CH2OCH3 | CH2CH2OCH2CH2OH | CF3 | H | H |
| 359 | CH | *CH2CH(CH3)OCH3 | H | CH3 | H | H |
| 360 | CH | *CH2CH(CH3)OCH3 | CH3 | CH3 | H | H |
| 361 | N | *CH2CH(CH3)OCH3 | H | CH(CH3)2 | H | H |
| 362 | N | (S,S)CH2CH(CH3)OCH3 | H | CH(CH3)2 | H | H |
| 363 | N | (S,R)CH2CH(CH3)OCH3 | H | CH(CH3)2 | H | H |
| 364 | N | *CH2CH(CH3)OCH3 | CH3 | CH(CH3)2 | H | H |
| 365 | N | (S,S)CH2CH(CH3)OCH3 | CH3 | CH(CH3)2 | H | H |
| 366 | N | (S,R)CH2CH(CH3)OCH3 | CH3 | CH(CH3)2 | H | H |
| 367 | N | CH2C(CH3)2OH | H | CH(CH3)2 | H | H |
| 368 | N | CH2C(CH3)2OH | CH3 | CH(CH3)2 | H | H |
| 369 | CH | CH2C(CH3)2OH | H | CH3 | H | H |
| 370 | CH | CH2C(CH3)2OH | CH3 | CH3 | H | H |
| 371 | CH | *CH2CH(CH3)OH | H | CH3 | H | H |
| 372 | CH | (S,S)CH2CH(CH3)OH | H | CH3 | H | H |
| 373 | CH | (S,R)CH2CH(CH3)OH | H | CH3 | H | H |
| 374 | CH | Isomer
1 | CH3 | CH3 | H | H |
| 375 | CH | Isomer
2 | CH3 | CH3 | H | H |
und alle Salze, Solvate, optischen und geometrischen
Isomere und kristallinen Formen hiervon.
- Bsp. Nr. entspricht
der Beispielnummer im Beispielteil
- * Diastereoisomerengemisch
-
Die
Verbindungen der Formel (Ih), die in Tabelle 4 angegeben sind, sind
von besonderem Interesse:
-
Die
absolute Konfiguration ist "S" am Kohlenstoffatom
der Piperazingruppe, die an Alk gebunden ist, falls nichts anderes
angegeben ist. Tabelle 4
| Bsp.
Nr. | Alk-X-R3 | R1 | R2 | Z1 | Z2 |
| 380 | CH2CH2OCH3 | H | H | CF3 | H |
| 382 | CH2CH2OCH3 | CH3 | H | CF3 | H |
| 384 | CH2CH2OCH3 | H | H | H | H |
| 385 | CH2CH2OCH3 | CH3 | H | H | H |
| 387 | CH2CH2OCH3 | H | H | F | H |
| 388 | CH2CH2OCH3 | CH3 | H | F | H |
| 390 | CH2CH2OCH3 | H | H | H | F |
| 391 | CH2CH2CH2OCH3 | H | H | H | F |
| 392 | CH2CH2OCH3 | CH3 | H | H | F |
| 393 | CH2CH2CH2OCH3 | CH3 | H | H | F |
| 395 | CH2CH2OCH3 | H | H | F | F |
| 396 | CH2CH2OCH3 | CH3 | H | F | F |
und alle Salze, Solvate, optischen und geometrischen
Isomere und kristallinen Formen hiervon.
- Bsp. Nr. entspricht
der Beispielnummer im Beispielteil.
-
Da
die erfindungsgemäßen Verbindungen
von Natur aus basisch sind, reagieren sie mit mehreren anorganischen
und organischen Säuren
unter Bildung von Säureadditionssalzen.
Für die
therapeutische Brauchbarkeit, die hierin beschrieben ist, müssen die
Salze der beanspruchten Verbindungen pharmazeutisch annehmbar sein.
Säuren,
die herkömmlich
zur Bildung von pharmazeutisch annehmbaren Salzen verwendet werden,
sind anorganische Säuren,
wie Chlorwasserstoffsäure,
Bromwasserstoffsäure,
Iodwasserstoffsäure, Schwefelsäure, Phosphorsäure und
organische Säuren,
wie p-Toluolsulfonsäure,
Methansulfonsäure,
Oxalsäure,
p-Bromphenylsulfonsäure,
Kohlensäure,
Bernsteinsäure,
Citronensäure,
Benzoesäure,
Essigsäure, Milchsäure, Maleinsäure, Weinsäure und
dergleichen. Für
weitere Details bezüglich
pharmazeutisch annehmbarer Salze siehe Journal of Pharmaceutical
Science, 66, 1 (1977). Salze, die nicht pharmazeutisch annehmbar
sind, können
als Zwischenprodukte unter Bildung von anderen Verbindungen der
Formel (I) oder eines pharmazeutisch annehmbaren Salzes der Verbindungen
der Formel (I) verwendet werden und liegen innerhalb des Umfangs
der vorliegenden Erfindung. Besondere phamazeutisch annehmbare Salze
sind jene, die mit Chlorwasserstoffsäure, Schwefelsäure, Fumarsäure oder
Phosphorsäure
gebildet werden.
-
Salze
von Verbindungen der Formel (I) existieren bekanntermaßen als
Anhydratformen und verschiedene hydratisierte Formen.
-
Die
Zwischenprodukte und Endprodukte, die hierin beschrieben sind, können durch
die herkömmlichen
Techniken isoliert und gereinigt werden, die dem Fachmann der organischen
Chemie bekannt sind. Beispielsweise können die gut bekannten Techniken
der Chromatographie, Umkristallisation, Destillation und Sublimation
einzeln und sequenziell verwendet werden.
-
Allgemeine Syntheseverfahren
-
Verbindungen
der Formel (I) der Erfindung können
durch mehrere Verfahren hergestellt werden, die allgemein in der
Technik der organischen Chemie bekannt sind. Die Ausgangsmaterialien,
deren Herstellung nicht beschrieben ist, sind im Handel erhältlich oder
können
leicht durch bekannte Techniken aus im Handel erhältlichen
Ausgangsmaterialien hergestellt werden.
-
Wie
in Schema 1 gezeigt können
die Verbindungen der Formel (I) bequem aus Verbindungen der Formel
(IIa) hergestellt werden, indem die Schutzgruppe "ProG" vom Aminstickstoff
des siebengliedrigen Rings des tricyclischen Ringsystems entfernt
wird. Die Verfahren zur Einführung
und Entfernung dieser Schutzgruppen sind in der Technik bekannt.
Siehe T.W. Green, Protective Groups in Organic Synthesis, John Wiley
and Sons, Inc. (1981). Beispiele solcher ProG Gruppen umfassen Benzyl,
Acetyl, t-Butoxycarbonyl,
Methansulfonyl und dergleichen.
-
-
Wie
hierin verwendet steht "Pg" entweder für Wasserstoff
oder eine Aminschutzgruppe ProG. Für die Beispiele, worin Pg für eine Aminschutzgruppe
steht, kann das vorletzte Zwischenprodukt in die Verbindung der
Formel (I) durch die Entfernung der Schutzgruppe umgewandelt werden.
Im folgenden Text kann für
die Zwischenprodukte, die eine Gruppe Pg enthalten, worin Pg für eine Aminschutzgruppe
steht, die Schutzgruppe unter Bildung des ungeschützten Amins
entfernt werden. Ähnlich
kann für
die Zwischenprodukte, worin Pg für Wasserstoff
steht, eine Aminschutzgruppe in das Zwischenprodukt eingebaut werden.
-
Die
Verbindungen der Formel (IIb), worin R
1 für Wasserstoff
steht, können
zu Verbindungen der Formel (IIc) umgewandelt werden, worin R
1 für
C
1-C
4Alkyl steht,
das wahlweise mit einem Substituenten substituiert ist, der aus
der Gruppe ausgewählt
ist, die besteht aus Hydroxy, Methoxy, Ethoxy oder OCH
2CH
2OH oder -CN. Die Umwandlung kann, wie dies
in Schema 1a gezeigt ist, durch die Behandlung der Formel (IIb)
mit einem Alkylierungsmittel erreicht werden. Alkylierungsmittel
umfassen Alkylhalogenide und Alkylsulfonatester. Beispiele umfassen
Methyliodid, 1-Brombutan, 2-Propylmethansulfonat und Bromethylmethylether.
Diese Umsetzung wird im allgemeinen in Gegenwart einer Base und
eines Lösemittels
ausgeführt.
Die Base kann entweder eine organische Base sein, wie Pyridin oder
Diisopropylethylamin oder eine anorganische Base, wie Kaliumcarbonat.
Lösemittel
umfassen Methanol, Ethanol, THF und DMF. Die Transformation kann
auch durch die reduktive Alkylierung des Piperazins durch Behandlung
mit einem Aldehyd oder Keton unter reduzierenden Bedingungen erreicht
werden. Beispiele für
geeignete Aldehyde umfassen Formaldehyd, Acetaldehyd, Propionaldehyd,
Butyraldehyd, Isobutyraldehyd und dergleichen. Geeignete Ketone
umfassen Aceton, Methylethylketon und dergleichen. Reduktive Alkylierungen
werden oft unter katalytischen Hydrierungsbedingungen ausgeführt. Andere
Reduktionsmittel umfassen Ameisensäure, Natriumborhydrid, Natriumcyanoborhydrid
und Natriumtriacetoxyborhydrid. Die Umwandlung kann auch durch Acylierung
des Piperazinstickstoffs unter Bildung eines Amids und der Reduktion
des Amids unter Bildung des alkylierten Piperazins erreicht werden.
Beispiele für
Acylierungsmittel umfassen Acylhalogenide, wie Acetylchlorid, Propionylchlorid,
Pivaloylchlorid und Cyclopropylcarbonylchlorid, Carbonsäureanhydride,
wie Formylessigsäureanhydrid
und Essigsäureanhydrid und
Carbonsauren in Gegenwart eines Aktivierungsmittels, wie Dicyclohexylcarbodiimid
oder Carbonyldiimidazol. Die entstehenden Amide können zu
den tertiären
Aminen mit Reduktionsmitteln reduziert werden, wie Lithiumaluminiumhydrid
oder Boran. Schema
1a

-
Wie
in Schema 2 gezeigt, können
die Verbindungen der Formel (II) durch Umsetzung eines geeignet substituierten
Piperazins der Formel (V) mit einem tricyclischen Zwischenprodukt
der Formel (IV) hergestellt werden. "LG" steht
für eine
Abgangsgruppe, wobei Beispiele NH
2, Halogen,
OY
1 oder SY
1 umfassen,
worin Y
1 für Niederalkyl steht, wie Methyl,
Ethyl oder Propyl oder optional substituiertes Phenyl oder OP(=O)R
10. R
10 kann für Morpholin
stehen. Die Umsetzung kann bequemerweise unter Erhitzen in einem
Lösemittel,
wie DMSO, Toluol, IPA, DMF und N-Methylpyrrolidon oder einem Gemisch
aus Lösemitteln
ausgeführt
werden, wie DMSO und Toluol in Verhältnissen von (1:2, 1:3 oder
1:4). Für
Verbindungen der Formel (II), worin LG für SY
1 steht,
kann die Äquivalenz
des Piperazins auf 1 oder 2 verringert werden, wenn es in IPA erhitzt
wird. Schema
2
-
Alternativ
dazu können,
wie es in Schema 3 gezeigt ist, tricyclische Amid- und Thioamidzwischenprodukte
der Formel (VI), worin X
1 jeweils für O oder
S steht, mit substituierten Piperazinen der Formel (V) unter Bildung
der entsprechenden Verbindungen der Formel (II) reagieren. Diese
Umsetzung wird bequemerweise in einem polaren Lösemittel ausgeführt, wie
Pyridin und Methylenchlorid und kann in Gegenwart oder Abwesenheit
einer Lewissäure
ausgeführt
werden, wie TiCl
4. Schema
3
-
In
Schema 4 können
Verbindungen der Formel (VIb), worin X
1 für S steht,
aus Verbindungen der Formel (VIa), worin X
1 für O steht,
durch Behandlung mit einem dehyratisierenden, thiolisierenden Mittel
in Gegenwart eines inerten Lösemittels
hergestellt werden. Beispiele solcher dehydratisierenden, thiolisierenden Mittel
umfassen P
2S
5 und
Lawesson's Reagenz
(2,4-Bis(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetan-2,4-disulfid). Für eine Beschreibung
des Lawesson's Reagenz
und dessen Verwendung siehe M.P. Cava und M.I. Levinson, Tetrahedron,
41, 5061 (1985). Schema
4
-
Wie
in Schema 5 gezeigt, können
tricyclische Zwischenprodukte der Formel (IV) aus den entsprechenden
tricyclischen Amid- und Thioamidzwischenprodukten der Formel (VI)
hergestellt werden. Die O-Alkylierung eines Amids der Formel (Via)
(X1 = O) liefert einen Iminoether der Formel
(IV) (LG = OY1). Geeignete Alkylierungsmittel
umfassen Meerwein Reagenz und Methylfluorsulfonat. Iminothioether
der Formel (IV), worin LG für SY1 steht, können durch S-Alkylierung von
Thioamiden der Formel (VIb) (X1 = S) hergestellt
werden. Geeignete Alkylierungsmittel umfassen Alkylhalogenide, Alkylsulfonate,
wie Methyltrifluormethansulfonat, Meerweinreagenz und Methylfluorsulfonat.
Die Umsetzung eines Amids der Formel (IVa) (X1 =
O) mit einem dehydratisierenden, halogenierenden Mittel liefert
ein Iminohalogenid der Formel (IV), worin LG für eine Halogengruppe steht.
Geeignete dehydratisierende, halogenierende Mittel umfassen POCl3, SOCl2, PCl3, PCl5, PBr3, PPh3/B2, P(OPh)3/I2 und PPh3/Mel.
-
-
Die
Verbindungen der Formel (IV), worin LG für NH2,
OY1 oder SY1 steht,
können
aus Verbindungen der Formel (VI), worin LG für Halogen steht, durch Umsetzung
mit einem geeigneten Nukeophil, wie Ammoniak, einem Alkohol oder
einem Thiol unter Bildung von Verbindungen der Formel (IV) hergestellt
werden, worin LG jeweils für
NH2, OY1 oder SY1 steht. Die Umsetzung kann bequemerweise
in einem Lösemittel,
wie DMF und unter basischen Bedingungen hergestellt werden, wie
K2CO3.
-
Wie
in Schema 6 gezeigt, können
Verbindungen der Formel (II) auch durch den Ringschluss eines Zwischenprodukts
der Formel (XIIIa) hergestellt werden. Diese Umsetzung kann durch
die Behandlung eines Amids der Formel (XIIIa) mit einem Aktivierungsmittel
in Gegenwart eines inerten Lösemittels
bewirkt werden. Beispiele für
solche Aktivierungsmittel umfassen TiCl4,
POCl3, P2S5 und Lawesson's Reagenz.
-
-
Gemäß Schema
7 können
die Verbindungen der Formel (VIa) durch Cyclisierung einer Aminverbindung
der Formel (XIIIb) hergestellt werden, worin Y2 für OY7 oder NY8Y9 steht, worin Y7,
Y8 und Y9 unabhängig für Wasserstoff
oder Niederalkyl stehen, wie Methyl, Ethyl oder Propyl.
-
-
Wie
es in Schema 8 ersichtlich ist, können die Amine der Formel (XIIIb)
aus Verbindungen der Formel (XIIIc) hergestellt werden. Das Symbol
Y3 steht für eine Gruppe, die in eine
Aminogruppe umgewandelt werden kann, wie NO2,
COOH und NHCOOY4, worin Y4 ein
wahlweise substituiertes Alkyl sein kann, wie unter anderem Methyl,
Ethyl, 2-Phenylethyl, t-Butyl, 2-(Trimethylsilyl)ethyl, 2,2,2-Trichlorethyl,
Vinyl, Allyl oder eine optional substituierte Benzylgruppe, wie
unter anderem Benzyl, p-Methoxybenzyl, p-Nitrobenzyl oder Diphenylmethyl.
-
-
Falls
Y3 für
NO2 steht, führt die Behandlung der Verbindungen
der Formel (XIIIc) unter reduzierenden Bedingungen zu den entsprechenden
Verbindungen der Formel (XIIIb). Beispiele für solche reduzierenden Bedingungen
umfassen katalytische Hydrierungsbedingungen oder SnCl2.
Die Verbindungen der Formel (XIIIc), worin Y3 für NHCOOY4 steht, können zu den entsprechenden
Verbindungen der Formel (XIIIb) unter Bedingungen umgewandelt werden,
die eine Entfernung der COOY4 Gruppe erlauben.
Falls Y4 für optional substituiertes Alkyl
steht, können
solche Bedingungen eine Hydrolyse unter sauren oder basischen Bedingungen umfassen.
Falls Y4 für optional substituiertes Benzyl
steht, liefert die Behandlung unter reduzierenden Bedingungen, vorzugsweise
die katalytischen Hydrierungsbedingungen, die entstehende Verbindung
der Formel (XIIIb). Falls Y4 für t-Butyl
steht, liefert die Behandlung mit Säure eine Verbindung der Formel
(XIIIb). Falls Y4 für 2,2,2-Trichlorethyl steht,
ergeben reduzierende Bedingungen, vorzugsweise Zinkmetall in einem
sauren Medium, eine Verbindung der Formel (XIIIb). Falls Y4 für
2-(Trimethylsilyl)ethyl steht, ergibt die Behandlung mit Fluoridionen
eine Verbindung der Formel (XIIIb).
-
Die
Verbindungen der Formel (XIIIb) können auch durch eine Curtius
Umlagerung der entsprechenden Verbindung der Formel (XIIIc) hergestellt
werden, worin Y3 für COOH steht. Die Curtius Umlagerung
tritt durch thermische Umlagerung des Acylazids der Formel (XIIIc)
auf, worin Y3 für CON3 steht,
um ein Isocyanat der Formel (XIIIc) zu ergeben, worin Y3 für NCO steht.
Dieses Isocyanat kann entweder direkt oder über das Urethan, worin Y3 für
NHCO2Y4 steht, unter
Bildung der entsprechenden Verbindung der Formel (XIIIb) hydrolysiert werden.
-
Gemäß Schema
9 können
Verbindungen der Formel (IVa), worin LG für NH2 steht,
durch Cyclisierung von Aminonitrilverbindungen der Formel (XIIId)
hergestellt werden.
-
-
Gemäß Schema
10 können
Aminonitrilverbindungen der Formel (XIIId) aus den entsprechenden
Verbindungen der Formel (XIIIe) auf die Weise hergestellt werden,
die für
Schema 8 beschrieben ist. Alternativ dazu können die Verbindungen der Formel
(XIIId) durch eine Curtius Umlagerung unter Bedingungen hergestellt
werden, die auch für
Schema 8 beschrieben sind.
-
-
Wie
in Schema 11 gezeigt, können
die Verbindungen der Formel (XIIIc), worin alle Gruppen wie oben definiert
sind, aus den entsprechenden Verbindungen der Formel (XIIIf) hergestellt
werden, worin Y3 für eine Gruppe steht, die in
eine Aminogruppe umgewandelt werden kann.
-
-
Gemäß Schema
12 können
Verbindungen der Formel (XIIIf), worin Y3 für eine Gruppe
steht, die zu einer wie oben definierten Aminogruppe umgewandelt
werden kann und alle anderen Gruppen wie oben definiert sind, durch
die Kupplung einer Verbindung der Formel (V) mit einer Verbindung
der Formel (XIIIg) hergestellt werden. Solche Kupplungsreaktionen
können
unter Bedingungen ausgeführt
werden, die herkömmlich zur
Bildung von Amidbindungen verwendet werden können. Kupplungsreagenzien umfassen
Dicyclohexylcarbodiimid (DCC), Diphenylphosphorylazid (DPPA) und
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(EDC).
-
-
Wie
in Schema 13 gezeigt, können
Verbindungen der Formel (XIII), worin Y
3 für NH
2 oder eine Gruppe stehen kann, die wie oben
beschrieben in eine Aminogruppe umgewandelt werden kann, Y
10 für
Wasserstoff, CN, COOY
7 oder CONY
8Y
9 stehen kann,
worin Y
7, Y
8 und
Y
9 unabhängig
für Wasserstoff
oder Niederalkyl stehen können
oder NY
8Y
9 für die Gruppe
(XVI) stehen, durch Umsetzung von Verbindungen der Formel (XIV), worin
Y
11 für
eine Halogengruppe oder OSO
2CF
3 stehen
kann, mit Verbindungen der Formel (XVa) hergestellt werden. Diese
Umsetzung kann unter basischen Bedingungen in einem polaren, aprotischen
Lösemittel
ausgeführt
werden. Geeignete Basen umfassen NaH, KH, Kalium-tert-butoxid, Lithiumhydroxid
und Cäsiumcarbonat.
Geeignete Lösemittel
umfassen DMF, N-Methylpyrrolidinon, DMSO und THF. Die Kupplung der
Verbindungen der Formel (XIV) mit Verbindungen der Formel (XVa)
unter Bildung einer Verbindung der Formel (XIII) kann auch in Gegenwart
eines Metallkatalysators ausgeführt
werden. Bedingungen für
diese Transformation können
gefunden werden in Hartwig, Angew. Chem. Int. Ed. 37, 2046-2067
(1998), Wolff et al., Acc. Chem. Res. 31, 805-818 (1998), Yang und
Buchwald, J. Organomet. Chem. 576, 125-146 (1999),
US 6 271 225 A ,
US 6 455 542 A und die hierin
zitierten Literaturangaben.
-
-
Die
Verbindungen der Formel (XIV) können
durch in der Technik bekannte Verfahren hergestellt werden.
-
Alternativ
dazu können,
wie dies in Schema 14 gezeigt ist, die Verbindungen der Formel (XIII),
worin Y3 für NH2 oder
eine Gruppe stehen kann, die wie oben beschrieben in eine Aminogruppe
umgewandelt werden kann, Y10 für Wasserstoff,
CN, COOY7 oder CONY8Y9 stehen kann, worin Y7,
Y8 und Y9 unabhängig für Wasserstoff
oder Niederalkyl stehen können
oder NY8Y9 für die Gruppe
(XVI) steht und die anderen Gruppen wie oben definiert sind, auch
durch Umsetzung der Verbindungen der Formel (XIVa) mit Verbindungen
der Formel (XV) hergestellt werden, worin Y12 für eine Halogengruppe
oder O-SO2CF3 stehen kann.
Diese Umsetzung kann unter basischen Bedingungen in einem polaren,
aprotischen Lösemittel
ausgeführt
werden. Geeignete Basen umfassen NaH, KH, Kalium-tert-butoxid, Lithiumhydroxid
und Cäsiumcarbonat.
Geeignete Lösemittel umfassen
DMF, N-Methylpyrrolidinon, DMSO und THF. Die Kupplung der Verbindungen
der Formel (XIVa) mit Verbindungen der Formel (XV) unter Bildung
einer Verbindung der Formel (XIII) kann auch in Gegenwart eines Metallkatalysators
ausgeführt
werden. Die Bedingungen für
diese Umwandlung könne
in Hartwig, Angew. Chem. Int. Ed. 37, 2046-2067 (1998), Wolff et
al., Acc. Chem. Res. 31, 805-818 (1998) und Yang und Buchwald, J.
Organomet. Chem. 576, 125-146 (1999) und den hierin zitierten Literaturangaben
gefunden werden.
-
-
Die
Verbindungen der Formel (XIVa) können
durch in der Technik bekannte Verfahren hergestellt werden.
-
Gemäß Schema
15 kann eine Verbindung der Formel (VIa) auch durch Cyclisierung
von Isocyanat (XIIIh) unter sauren Bedingungen hergestellt werden.
Isocyanat (XIIIh) kann aus Verbindungen der Formel (XIII), worin
Y10 für
Wasserstoff steht und Y3 für eine Aminogruppe
steht, durch Umsetzung mit Ameisensäureanhydrid und einer Dehydratisierung
des entstehenden Formamids mit einem Dehydratisierungsmittel, wie
POCl3 oder P2O5 hergestellt werden. Isocyanat (XIIIh) kann
auch aus Verbindungen der Formel (XIII), worin Y10 für Wasserstoff
steht und Y3 für COOH steht, durch Curtious
Umlagerung hergestellt werden, wie dies vorher beschrieben ist.
Alternativ dazu kann eine Verbindung der Formel (IIb) auch durch
Umsetzung des Harnstoffs (XIIIi) in Gegenart einer Lewissäure hergestellt
werden. Der Harnstoff (XIIIi) kann durch Umsetzung von Isocyanat (XIIIh)
mit einem Amin der Formel (V) hergestellt werden.
-
-
In
Schema 16 können
die Verbindungen, worin der aromatische Ring
für Thiazol steht, durch Cyclisierung
des Zwischenprodukts der Formel (XVIII) mit einem dehydratisierenden, thiolisierenden
Mittel hergestellt werden, wie P
2S
5 oder Lawesson's Reagenz. Die Verbindungen der Formel (VId),
worin der aromatische Ring
für einen Oxazolring steht, können durch
Cyclisierung des Zwischenprodukts der Formel (XVIII) mit einem Dehydratisierungsmittel
hergestellt werden, wie P
2O
5 oder
PPh
3/Tf
2O.
-
-
Gemäß Schema
17 werden die Verbindungen der Formel (XVIII) durch Acylierung eines
Amins der Formel (XIX) hergestellt. Diese Umsetzung wird gewöhnlich durch
die Behandlung der Formel (XIX) mit einem Säurechlorid oder einem Säureanhydrid
in Gegenwart einer Base in einem inerten Lösemittel ausgeführt. Verfahren
zur Synthese von Verbindungen der Formel (XIX) sind in der Technik
bekannt, siehe beispielsweise Hagishita et al., Bioorg. Med. Chem.,
5 (7), 1433-1446, (1997).
-
-
Wie
in Schema 18 gezeigt können
die Verbindungen der Formel (VIe), worin der A Ring
für Pyrazol steht oder (VIf),
worin der A Ring
für Pyrimidin steht, auch durch
Umsetzung von Verbindungen der Formel (XX) jeweils mit einem substituierten Hydrazin
oder einem Amidin hergestellt werden.
-
Die
Verbindungen der Formel (XX) werden hergestellt, wie dies in Roma
et al., Farmaco, Ed. Sci., 38, 546-558 (1983) beschrieben ist.
-
-
Die
Verfahren zur Herstellung der Verbindung der Formel (XVa) und Formel
(XVc) sind in der Technik bekannt und variieren in Abhängigkeit
der Art des aromatischen Rings A
-
Der
Fachmann erkennt, dass die Substituenten R2 und
Z1 und Z2 in den
Verbindungen der Formel (I) in den Vorläufermolekülen der Formeln (XIV), (XIVa),
(XVb) und (XVc) vorkommen können.
Alternativ dazu können
diese Substituenten an jeder bequemen Stelle während der Synthese entweder
durch den Ersatz eines Wasserstoffs (beispielsweise durch eine elektrophile,
aromatische Substitutionsreaktion) oder durch Umwandlung eines existierenden
Substituenten in die in den Verbindungen der Formel (I) vorkommenden
Substituenten eingeführt
werden. Beispiele für
elektrophile aromatische Substitutionsreaktionen umfassen Halogenierung,
Nitrierung, Friedel-Crafts Acylierung und elektrophile Trifluormethylierung
unter Bedingungen, die in der Literatur beschrieben sind. Beispiele
für die
Umwandlung eines existierenden Substituenten in einen, der in der
schließlichen
Verbindung vorkommt, umfassen die Umwandlung eines Br Substituenten
in einem Substituenten, wie SR11 oder COR11 durch Metallierung mit einem Organolithiumreagenz
und die Umsetzung mit einem Elektophil, wie R11SSR11 oder R11COOMe.
R11 kann für C1-C6 Alkyl, C1-C6 Fluoralkyl, Benzyl oder optional substituiertes
Phenyl stehen. Zusätzlich
kann ein Br Substituent in einen optional substituierten aromatischen
Ring durch Umsetzung mit einer optional substituierten Phenylborsäure in Gegenwart
eines Palladiumkatalysators umgewandelt werden. Viele andere solche
Umwandlungen funktioneller Gruppen sind in der Literatur angegeben.
-
Allgemeine
Verfahren und spezifische Beispiele der Synthese dieser Verbindungen
können
in den folgenden Literaturangaben gefunden werden:
Chakrabarti
et al., J. Med. Chem., 23, 878-884 (1980),
Chakrabarti et al.,
J. Med. Chem., 23, 884-889 (1980),
Chakrabarti et al., J. Med.
Chem., 25, 1133-1140 (1982),
Chakrabarti et al., J. Med. Chem.,
32, 2573-2582 (1989),
Liegeois et al., J. Med. Chem., 36, 2107-2114,
(1993),
Liegeois und Delarge,
US 5 393 752 A (1995),
Chakrabarti und
Hotten,
EP 0 354 781
A (1990),
Bolton et al.,
WO 97 00 252 A (1997),
Chakrabarti
et al.
EP 0 027 390
B1 (1981),
Tehim et al.,
US 5 602 124 A (1998),
Tehim
et al.,
US 5 824 676
A (1998),
Elingsfeld und Swybold,
DE 27 13 573 (1978),
Gallemaers
et al., Tetrahedron Lett., 693-694 (1976),
Durnow und Abele,
Chem. Ber., 97, 3349-3353 (1964),
Klempier et al., J. Heterocyclic
Chem., 29, 93-95 (1992).
-
In
Schema 19 können
die Verbindungen der Formel (XVd) durch regioselektive Nitrierung
von 3-Brombenzothiophenverbindungen
unter Bildung der 2-Nitro-3-brombenzothiophenverbindungen der Formel
(XVe) hergestellt werden. Geeignete Nitrierungsbedingungen umfassen
Salpetersäure
(optional in Gegenwart einer anderen Säure, wie Trifluoressigsäure, Schwefelsäure oder
Essigsäure
oder in Gegen wart eines inerten Lösemittels, wie Dichlormethan
oder Wasser), rauchende Salpetersäure oder Natriumnitrit in Gegenwart
einer Säure.
Die Verdrängung
der 3-Bromgruppe mit Cyanid kann unter Verwendung von CuCN in Gegenwart
eines polaren Lösemittels
erreicht werden, wie DMF oder N-Methylpyrrolidinon unter Bildung
von Verbindungen der Formel (XVf). Die Reduktion der Nitrogruppe
zum Amin kann durch Reduktionsmittel, wie SnCl2/HCl,
Zn/HOAc und Pd-C/H2 unter Bildung von Verbindungen
der Formel (XVd) erreicht werden, worin Pg für Wasserstoff steht. Eine Schutzgruppe
kann anschließend
eingeführt
werden
-
-
Die
Verbindungen der Formel (V) der Erfindung können aus den Verbindungen der
Formel (XXIVb), wie dies in Schema 20 gezeigt ist, worin einer der
Stickstoffe des Piperazinrings durch eine Aminschutzgruppe geschützt sein
kann, durch Entfernung dieser Schutzgruppe hergestellt werden. In
dieser Gleichung steht ProG2 für eine Aminschutzgruppe.
Beispiele für
solche ProG2 Gruppen umfassen Benzyl, Acetyl,
t-Butoxycarbonyl, Methansulfonyl und dergleichen. Beispiele für zusätzliche
ProG2 Gruppen und Verfahren zur Einführung und
Entfernung solcher Gruppen können
in T.W. Green, Protective Groups in Organic Synthesis, John Wiley and
Sons, Inc. (1981) gefunden werden. Im folgenden Text steht Pg2 entweder für Wasserstoff oder eine Aminschutzgrupe
ProG2. Im folgenden Text kann für jene Zwischenprodukte,
die eine Gruppe Pg2 enthalten, worin Pg2 für
eine Aminschutzgruppe steht, die Schutzgruppe unter Bildung des
ungeschützten
Amins entfernt werden. Ähnlich
kann für
jene Zwischenprodukte, worin Pg2 für Wasserstoff
steht, eine Aminschutzgruppe in das Zwischenprodukt eingeführt werden.
Die Verfahren zur Einführung
und Entfernung dieser Schutzgruppen sind in der Technik bekannt.
-
-
Gemäß Schema
21 können
die Verbindungen der Formel (XXIVa) der Erfindung aus Verbindungen
der Formel (XXVa) durch Entfernung der Aminschutzgruppe ProG
1 hergestellt werden. Beispiele für solche
ProG
1 Aminschutzgruppen umfassen Benzyl,
Acetyl, t-Butoxycarbonyl, Methansulfonyl und dergleichen. Beispiele
für zusätzliche
ProG
1 Gruppen und Verfahren zur Einführung und
Entfernung solcher Gruppen können
in T.W. Green, Protective Groups in Organic Synthesis, John Wiley
and Sons, Inc. 1981 gefunden werden. Es ist ersichtlich, dass in
einigen Fällen
in den Verbindungen der Formel (XXVa) so wohl Pg
2 als
auch ProG
1 für Schutzgruppen stehen, die
unter denselben Reaktionsbedingungen entfernt werden können. In
diesen Fällen
führt die
Schutzgruppenabspaltung an der Verbindung zu Verbindungen der Formel
(V), worin R
1 für Wasserstoff steht. In Verbindungen
der Formel (XXIVa) führt,
falls Pg
2 für eine Aminschutzgruppe ProG
2 steht, die Alkylierung der Formel (XXIVa)
zu Verbindungen der Formel (XXIV), worin R
1 für C
1-C
4 Alkyl steht,
das optional mit einem Substituenten substituiert ist, der aus der
Gruppe ausgewählt
ist, die besteht aus Hydroxy, Methoxy, Ethoxy, -OCH
2CH
2OH oder -CN. Schema
21
-
In
Schema 22 können
die Verbindungen der Formel (XXV), worin alle Gruppen wie oben definiert
sind, durch die Reduktion von entweder Ketopiperazin der Formel
(XXVI) oder einem Diketopiperazin der Formel (XXVII) hergestellt
werden. Pg1 steht entweder für Wasserstoff,
für C1-C4 Alkyl, das optional
mit einem Substituenten substituiert ist, der aus der Gruppe ausgewählt ist,
die besteht aus Hydroxy, Methoxy, Ethoxy, -OCH2CH2OH, -CN oder eine Aminschutzgruppe ProG1. Geeignete Reduktionsmittel für diese
Umwandlung umfassen Aluminiumhydrid und Boran. Verfahren zur Synthese
von Ketopiperazinen und Diketopiperazinen sind in der Technik bekannt.
-
-
Wie
in Schema 23 gezeigt, können
die Verbindungen (XXVI) und (XXVII) durch die Alkylierung des entsprechenden
Ketopiperazins (XXVIII) und Diketopiperazins (XXIX) mit einem Alkylierungsmittel
der Formel Lg-Alk-X-R3 hergestellt werden,
worin Lg für
eine Abgangsgruppe steht, wie eine Halogen-, Alkylsulfonyloxy- oder
Arylsulfonyloxygruppe. Beispiele für Alkylsulfonyloxygruppen umfassen
Methansulfonyloxy und Ethansulfonyloxy und Beispiele von Arylsulfonyloxygruppen
umfassen Toluolsulfonyloxy- und
Benzolsulfonyloxygruppen. Die Alkylierungsreaktion wird in Gegenwart
einer Base ausgeführt.
Geeignete Basen umfassen Lithiumdiisopropoxid, Lithiumhexamethyldisilazid,
Natriumhydrid, Kalium-t-butoxid
und dergleichen.
-
-
Ferner
können,
wie dies in Schema 24 gezeigt ist, die Verbindungen der Formel (XXVb),
worin Alk für -CH2CH2-, -CH2CH2CH2-
und -CH2CH2CH2CH2- steht, aus
einem geeignet geschützten
2-substituierten
Piperazin der Formel (XXX) durch Verwendung einer Hydroborierung/Oxidationssequenz
hergestellt werden.
-
-
Daher
liefert die Umsetzung einer Verbindung der Formel (XXX) mit einem
Boran HBZZ', worin
Z und Z unabhängig
stehen für
H, Alkyl, wie Methyl, Ethyl, Propyl oder Alkoxy, wie Methoxy, Ethoxy
oder Propoxy, ein Organoboran der Formel (XXXI). Geeignete Borane
HBZZ' umfassen Boran,
Trisiamylboran, Catecholboran und 9-Borabicyclo[3.3.0]nonan (9-BBN)
Das entstehende Organoboran wird dann zum Alkohol (XXVb) unter Verwendung
eines Oxidationsmittels oxidiert, wie Wasserstoffperoxid oder t-Butylhydroperoxid.
-
Die
Verbindungen der Formel (XXVc) können
aus den Verbindungen (XXX) durch Hydrierung des Olefins gebildet
werden. Diese Hydrierung wird typischerweise unter sauren Bedingungen
ausgeführt
oder kann auch durch eine Oxymercurisierungs/Reduktionssequenz ausgeführt werden.
Die Oxymercurisierung wird typischerweise durch eine Behandlung
des Olefins mit einem Quecksilber-(II)-salz ausgeführt, wie
Hg(OAc)2. Das Quecksilberatom wird aus der
Zwischenproduktverbindung durch die Reduktion mit NaBH4 entfernt.
-
Es
sollte erwähnt
werden, dass die Verbindungen (XXVb) und (XXVc) Regioisomere voneinander
sind. Gemische dieser Verbindungen können entweder aus der Hydroborierung/Oxidation,
Säurekatalysierten
Hydrierung oder Oxymercurisieurng/Reduktion Sequenz hergestellt
werden, falls die regiochemische Kontrolle dieser Verfahren limitiert
ist.
-
Die
Verbindungen der Formel (XXX) (m = 0) können durch das in Tsuda et
al., J. Org. Chem., 55, 3388-3390 (1990) und Uozumi et al., J. Org.
Chem., 58, 6826-6832 (1993) beschriebene Verfahren hergestellt werden.
-
In
Schema 25 können
die Verbindungen der Formel (XXX) (m = 1,2) durch eine Alkylierung
der Formel (XXVIII) mit einem Allylhalogenid oder einem Homoallylhalogenid
und unter Bildung der Verbindungen der Formel (XXXII) und Reduktion
mit Lithiumaluminiumhydrid unter Bildung der Verbindungen der Formel
(XXX) (m = 1,2) hergestellt werden.
-
-
Wie
in Schema 26 gezeigt, können
die Verbindungen der Formel (XXVb) (m = 0,1) und (XXVc) (m = 0,1)
jeweils zum entsprechenden Aldehyd (XXXIII) (m = 0,1) und Keton
(XXXIV) (m = 0,1) oxidiert werden. Geeignete Oxidationsreagenzien
umfassen Pyridiniumchlorchromat, DMSO/Oxalylchlorid (Swernoxidation)
und Dimethylsulfid/N-Chlorsuccinimid (Corey-Kim Oxidation). Die
Verbindungen (XXXIII) und (XXXIV) können jeweils mit einem Organoalkylreagenz,
MR12 unter Bildung der Alkohole (XVc) (m
= 0,1) und (XXVe) (m = 0,1) behandelt werden. Geeignete Organoalkylreagenzien
umfassen Organolithiumreagenzien, wie Methyllithium und Ethyllithium,
Grignard Reagenzien, wie Methylmagnesiumbromid und Ethylmagnesiumchlorid
und dergleichen.
-
-
Wie
in Schema 27 gezeigt kann ein Alkohol, der Formel (XXV f) zu den
entsprechenden Ethern und Thioethern (XXVh) (X = O, S) und (XXV
g) durch mehrere Verfahren umgewandelt werden. Der Sauerstoff des Alkohols
(XXV f) kann dann mit einem Alkylierungsmittel unter Bildung des
Ethers (XXV g) behandelt werden, worin R3 für eine Alkylgruppe
steht. Geeignete Alkylierungsmittel umfassen Dimethylsulfat, Alkylhalogenide, wie
Methyliodid, Ethylbromid und Benzylchlorid und Sulfonatester, wie
Methyltosylat, Ethylmethansulfonat und Methyltrifluormethansulfonat.
Die Alkylierung wird gewöhnlich
unter basischen Bedingungen ausgeführt.
-
Alternativ
dazu kann, wie dies in Schema 27 gezeigt ist, die Verbindung (XXV
f) in eine Verbindung der Struktur (XXV) umgewandelt werden, worin
Lg für
eine Abgangsgruppe steht. Beispiele für Abgangsgruppen umfassen Halogen,
die Alkylsulfonyloxygruppe und die Arylsulfonyloxygruppe. Beispiele
für Alkylsulfonyloxygruppen
umfassen Methansulfonyloxy und Ethansulfonyloxy und Beispiele für Arylsulfonyloxygruppen
umfassen Toluolsulfonyloxy- und Benzolsulfonyloxygruppen. Verbindungen,
worin Lg für
Halogen steht, wie Chlor oder Brom, können aus der Verbindung (XXV
f) durch Umsetzung mit einem anorganischen Halogenid hergestellt
werden, wie Thionylchlorid, Phosphorpentachlorid oder Phosphortribromid.
Verbindungen, worin Lg für eine
Alkylsulfonyloxygruppe oder Arylsulfonyloxygruppe steht, können durch
Umsetzung der Verbindung (XXV f) mit dem entsprechenden Alkylsulfonylhalogenid,
Arylsulfonylhalogenid, Alkylsulfonsäureanhydrid oder Arylsulfonsäureanhydrid
in Gegenwart einer Base hergestellt werden. Die Umsetzung der Verbindung
(XXXV) mit einem Alkohol R3OH oder einem
Thiol R3SH liefert die entsprechenden Ether
und Thioether (XXV h) (X = O, S). Diese Umsetzung wird typischerweise
unter basischen Bedingungen in einem inerten Lösemittel ausgeführt. Geeignete
Basen umfassen Natriumhydrid, Natriumhydroxid und Kaliumhydrid.
-
-
Alternativ
dazu kann, wie dies auch in Schema 27 gezeigt ist, die Verbindung
(XXV f) direkt zur Verbindung (XXV h) (X = O, S). durch Behandlung
mit einem Alkohol R3OH oder einem Thiol
R3SH unter Mitsunobubedingungen umgewandelt
werden. Klassische Mitsunobubedingungen verwenden Triphenylphosphin und
Diethylazodicarboxylat. Die Mitsunobureaktion wurde in den folgenden
Literaturstellen zusammengefasst:
David L. Hughes, Organic
Reactions, 42, 335-656 (1992),
David L. Hughes, Organic Preparations
and Procedures International, 28, 127-164 (1996).
-
Wie
in Schema 28 gezeigt können
die Thioether (XXV i) in die entsprechenden Sulfoxide (XXV j) m
= 1 und Sulfone (XXV j) m = 2 durch Umsetzung mit einem geeigneten
Oxidationsmittel umgewandelt. Die Oxidationsmittel umfassen molekularen
Sauerstoff, Wasserstoffperoxid, t-Butylhydroperoxid, Peroxyessigsaure, meta-Chlorperoxybenzoesäure, Ozon
und Oxon (Kaliumperoxymonosulfat).
-
-
Der
Fachmann erkennt, dass viele der vorher erwähnten Reaktionen in jeder bequemen
Reihenfolge ausgeführt
werden können. Ähnlich erkennt
der Fachmann für
die Verbindungen, die ein asymmetrisches Zentrum enthalten, dass
die vorher erwähnten
Reaktionen entweder mit reinen Isomeren oder einem Isomerengemisch
ausgeführt
werden. Die Isomere können
an jeder bequemen Stufe während
der Synthese getrennt werden.
-
Pharmazeutische Aktivität
-
Die
Verbindungen der Formel (I) haben eine moderate bis hohe Bindeaktivität für mehrere
Neurotransmitterrezeptoren und insbesondere die Dopaminrezeptoren.
Daher hat der Fachmann der Neuropharmakologie und verwandten Fachgebieten
die Dopaminrezeptorbindungsaktivität als indikativ für antipsychotische,
insbesondere antischizophrene Eigenschaften erkannt. Siehe P. Seeman
et al., Nature, 261, 717-718 (1976), P. Seeman, Synapse, 1, 133
(1987), H. Howard et al., 28, 39 (1993) und J. Schaus et al., Annual
Reports in Medicinal Chemistry, 33, 1 (1998). Die Klonierungsstudien
haben derzeit 5 hautsächliche
Dopaminrezeptorsubtypen erzeugt, die in zwei Hauptklassen fallen,
D
1-ähnlich
und D
2-ähnlich.
Die D
1-ähnliche
Klasse umfasst die D
1 und D
5 Subtypen
und die D
2-ähnliche Klasse umfasst die
D
2, D
3 und D
4 Subtypen. Die Tabelle 5 zeigt die relative
Bindeaktivität
von ausgewählten
Verbindungen der Formel (I) für
den D
2 Rezeptor. Das experimentelle Protokoll
für den
Test, der diese Daten generiert, ist der folgende Beispielabschnitt. Tabelle 5 Relative D
2 Rezeptorbindungsaktivität für Verbindungen
der Formel (I)
| Verb. Nr. | Affinität Ki* | | Verb. Nr. | Affinität Ki | Verb. Nr. | Affinität Ki | | Verb. Nr. | Affinität Ki |
| 198 | + | | 324 | +++ | | 269 | +++ | | 340 | +++ |
| 199 | ++ | | 314 | ++++ | | 242 | +++ | | 341 | +++ |
| 216 | ++ | | 334 | ++++ | | 267 | +++ | | 343 | ++ |
| 303 | ++++ | | 346 | ++++ | | 319 | +++ | | 342 | +++ |
| 304 | ++++ | | 344 | ++++ | | 327 | ++++ | | 359 | +++ |
| 313 | ++ | | 351 | +++ | | 320 | +++ | | 360 | ++ |
| 318 | ++++ | | 316 | ++++ | | 326 | ++++ | | 361 | ++++ |
| 322 | +++ | | 336 | ++++ | | 331 | +++ | | 367 | ++++ |
| 323 | +++ | | 355 | +++ | | 309 | +++ | | 368 | ++++ |
| 333 | ++ | | 353 | +++ | | 328 | +++ | | 369 | ++ |
| 338 | +++ | | 357 | +++ | | 302 | +++ | | 370 | ++ |
| 312 | ++ | | 240 | +++ | | 268 | +++ | | | |
| 327 | +++ | | 241 | ++ | | 321 | ++ | | | |
| 305 | +++ | | | | | 339 | +++ | | | |
- *Ki ist im allgemeinen
als Bindungsaffinitätskonstante
(das heißt
Dissoziationskonstante) eines unmarkierten Liganden und einem Bindungstest
mit radioaktiven Liganden definiert. Siehe beispielsweise Neurotransmitter Receptor
Binding, Second Edition, Herausgeber H.I. Yamamura, S.J. Enna und
M.J. Kuhar, Rauen Press (1985).
- *++++ = < 10
nM, +++ = 10-100 nM, ++ = 100-1000 nM, + ≥ 1000 nM
-
Unter
Verwendung des relativen Ki Maßstabs
der Tabelle 5 weist Clozapin eine ++ Affinität auf und Olanzapin eine +++
Affinität.
Daher weisen viele der Verbindungen der Formel (I) eine D2 Rezeptoraffinität auf, die größer als
die von sowohl Clozapin als auch Olanzapin ist. Die Verbindungen
der Formel (I) haben eine erwünschte
D2 Bindeaffinität von vorzugsweise kleiner
oder gleich 1000 nM, vorzugsweise kleiner oder gleich 200 nM und
noch bevorzugter kleiner oder gleich 50 nM.
-
Ähnlich wie
Clozapin und Olanzapin zeigen die Verbindungen der Formel (I) auch
eine Affinität
für den 5-HT6 Rezeptor. Da Clozapin und Olanzapin eine
größere Wirksamkeit
bei der Behandlung von kognitiven Störungen von Schizophrenie aufweisen
als typische Antipsychotika (Purdon et al., Arch. Gen. Psych., 57,
249 (2000)) und selektive 5-HT6 Antagonisten
bei den Modellen der kognitiven Verstärkung aktiv sind, ist diese
Aktivität
in einem antipsychotischen Arzneimittel erwünscht.
-
Viele
atypischen Antipsychotika haben eine hohe Affinität für den 5-HT2A Rezeptor. Forscher nehmen an, dass eine
hohe Affinität
für den
5-HT2A Rezeptor bei der Behandlung der negativen
Symptome von Schizophrenie und der Verhinderung von einigen der
motorischen Nebenwirkungen hilft (H. Meltzer et al., J. Pharm. Exp.
Ther. 25, 238 (1989)). Jedoch sind selektive 5-HT2A Antagonisten
keine effektiven Antipsychotika als Monotherapie. Daher ist ein
5-HT2A Antagonismus wahrscheinlich unter
den anderen Neurorezeptoraffinitäten
einer überlegenen
antipsychotischen Verbindung. Die Verbindungen der Formel (I) zeigen
ein gewünschtes
Niveau an 5-HT2A Affinität.
-
Antipsychotika
dürften
zumindest einen Teil ihrer therapeutischen Effekte durch die Blockierung
des Dopamin D2 Rezeptors ausüben. Die
Fähigkeit
einer Verbindung zur Blockade von Dopamin D2 Rezeptoren
in der Ratte wurde in in vivo durch Messen des Effekts der Verbindung
auf den Spiegel von DOPAC (3,4-Dihydroxyphenylessigsäure) bestimmt,
einem Dopaminmetaboliten im Nukleus accumbens der Ratte. Dopamin
D2 Rezeptorantagonisten erhöhen die
Freisetzung von Dopamin in der Synapse durch die Blockade des Dopamin D2 Autorezeptors. Diese erhöhte Freisetzung
von Dopamin kann nicht direkt gemessen werden, da die Wirksamkeit
des Dopaminwiederaufnahmesystems die Erhöhung der synaptischen Dopaminkonzentrationen
verhindert. Anstelle dessen spiegeln erhöhte Mengen der Dopaminmetaboluten
DOPAC (3,4-Dihydroxyphenylessigsäure)
und HVA (Homovanillinsäure)
eine erhöhte
neuronale dopaminerge Aktivität
in vivo wider. Beispielsweise erhöhen Olanzapin und andere Dopamin
D2 Rezeptorantagonisten die Konzentrationen
an DOPAC und HVA im Striatum und Nucleus accumbens ohne beträchtliche
Veränderung
der Dopaminkonzentrationen. Die Potenz einer Verbindung zur Blockade
der Dopamin D2 Rezeptoren wird durch die Dosis bestimmt, die zur Erhöhung der
DOPAC Spiegel auf 200% der Kontrolle erforderlich sind. Dieser Wert
wird ED200 genannt.
-
Antipsychotika
dürften
zumindest einen Teil ihrer Gewichtszunahmeeffekte durch die Blockade
der Histamin H1 Rezeptoren im Hypothalamus
induzieren.
-
Ihre
Fähigkeit
zur Blockade der Histamin H1 Rezeptoren
kann in vitro durch Messen der in vitro Histamin H1 Rezeptoraffinität gemessen
werden. Verbindungen mit verringerter Affinität für Histamin H1 Rezeptoren neigen
weniger zur Induktion einer Gewichtszunahme. Das Verhältnis der
in vitro Histamin H1 Rezeptoraffinität dividiert
durch die in vitro Dopamin D2 Rezeptoraffinität, die beide
als Ki Werte ausgedrückt
werden, ist eine Abschätzung
der Wahrscheinlichkeit einer Verbindung, bei therapeutischen Mengen
eine Gewichtszunahme zu verursachen. Je größer das Verhältnis, desto
weniger wahrscheinlich wird eine Verbindung eine Gewichtszunahme
verursachen. Die Verhältnisse
von Clozapin und Olanzapin betragen jeweils 0,01 und 0,3. Erfindunsgemäße Verbindungen
haben vorzugsweise H1/D2 Verhältnisse,
die größer oder
gleich 1 sind und vorzugsweise H1/D2 Verhältnisse,
die größer oder
gleich 3 sind.
-
Die
in vitro Potenz einer Verbindung zur Belegung der Hypothalamus Histamin
H1 Rezeptoren in der Ratte wird mittels
eines Histamin H1 ex vivo Bindungstest bestimmt.
Die ED50 ist die Dosis, die zur Belegung von
50% der Histamin H1 Rezeptoren der Ratte
erforderlich ist. Je größer die
ED50 desto weniger wahrscheinlich ist, dass
die Verbindung erneut eine Gewichtszunahme verursachen wird. Die
erfindungsgemäßen Verbindungen
weisen vorzugsweise eine Histamin H1 ex
vivo Bindungs-ED50 von größer oder
gleich 10 mg/kg p.o. und bevorzugter ED50 Werte
von mehr als 30 mg/kg p.o. auf.
-
Histamin H1 ex
vivo Bindung und DOPAC Konzentrationen
-
Verfahren
-
Männliche
Sprague Dawley Ratten (Harlan Sprague Dawley, Inc. Indianapolis,
IN), die 110 g wiegen, lässt
man über
Nacht fasten. Den Tieren wird Clozapin (RBI, Inc.) oder die Verbindung
von Interesse verabreicht und sie werden 90 Minuten später getötet. Clozapin
wird mit 5 mg/kg in 5% Akaziengummisuspension verabreicht. Alle
anderen Verbindungen werden mit 5 ml/kg in verdünnter Milchsäure verabreicht.
Die Gewebe werden entnommen, in Trockeneis eingefroren und bei –70°C vor der
Analyse gelagert.
-
Histamin H1 ex
vivo Bindung
-
Es
wird die ex vivo Bindung des Histamin H1 Antagonsiten
[3H]-Pyrilamin (NEN Life Science Products) an
Rattenhypothalamushomogenaten bestimmt. Die Gewebe werden in 600 μl Inkubationspuffer
(monobasisches 50 mM Natriumphosphat, pH 7,4) homogenisiert und
10 Minuten bei 37°C
unter Entfernung des endogenen Histamins präinkubiert. Dreifache Röhrchen,
die jeweils 100 μl
Homogenat enthalten, werden mit 1 ml Puffer inkubiert, der 3 nM
[3H]-Pyrilamin enthält und 30 Minuten bei 25°C inkubiert.
Die unspezifische Gewebebindung wird auch zweifach in Röhrchen gemessen,
die 10 μM
Clozain enthalten. Die [3H]-Pyrilaminbindung wird
nach der Separationsfiltration mittels eines Brandell Zellernetegeräts mit GF/C
Filtern gemessen, die mit 0,1% Polyethylenimin getränkt sind.
Die ED50 Werte werden mittels des statistischen
Allfitprogramms für
die Verdrängungsbildung
bestimmt.
-
DOPAC Konzentrationen
-
Die
DOPAC (3,4-Dihydroxyphenyslessigsäure) Konzentrationen im Nukleus
accumbens der Ratte werden mittels Hochdruckflüssigchromatographie mit einer
elektrochemischen Detektion (HPLC-EC) gemessen. Die Gewebe werden
in 1 ml an 0,1 N TCA ultrabeschallt. Nach der Zentrifugation wird
ein 25 μl
Aliquot des Überstands
auf eine BDS Hypersyl C18 Säule
(150 × 4,6
mm, Keystone Scientific) injiziert.
-
Der
Elutionspuffer enthält
monobasisches 75 mM Natriumphosphat, 0,5 mM EDTA, 350 mg/l 1-Octansulfonat-Natrium,
7% Acetonitril (V/V) und 0,7% Tetrahydrofuran (V/V), pH 3,0. Die
Flussrate beträgt
1,2 ml/min bei 40°C.
Die Peakhöhen
werden bei 750 mV bei einer Empfindlichkeit von 10 nA gemessen und
mit Proben verglichen, die bekannte Mengen an DOPAC Standards enthalten.
Die Dosen, die die DOPAC Mengen auf 200% der Kontrollwerte erhöhen (ED
200), werden mittels einer linearen Best-Fit Regressionsanalyse
berechnet. Tabelle
6
| Test | Clozapin | Beispiel
279 | Beispiel
292 | Beispiel
288 | Beispiel
327 |
| Rezeptoraffinität (Ki, nM) | | | | | |
| D2 | 194 | 6 | 13 | 11 | 9 |
| H1 | 2,9 | 24 | 48 | 80 | 91 |
| In
vivo Aktivität (mg/kg,
p.o.) | | | | | |
| D2 1 | 45,6 | 6 | 7,5 | 6,4 | 4 |
| H1 2 | 10 | > 30 | > 30 | > 30 | > 30 |
- 1 DOPAC Eröhung (ED200)
- 2 H1 ex vivo
Bindung (ED50)
-
Die
Verbindungen der Formel (I) sind zur Behandlung von pathologischen
psychologischen Zuständen,
speziell Psychose, mit minimalen Nebenwirkungen brauchbar. Pathologische
psychologische Zustände, die
Psychose sind oder mit psychotischen Merkmalen assoziiert sein können, sind
unter anderem die psychotischen Störungen, die charakterisiert
wurden in DSM-IV-TR, Diagnostic and Statistical Manual of Mental
Disorders, überarbeitete
4. Ausgabe, Textrevision (2000). Siehe auch DSM-IV, Diagnostic and
Statistical Manual of Metal Disorders, 4. Ausgabe (1994). Das DSM-IV
und das DSM-IV-TR wird durch die Arbeitsgruppe über Nomenklatur und Statistik
der American Psychiatric Association erstellt und liefert Beschreibungen
der diagnostischen Kategorien. Der Fachmann erkennt, dass es alternative
Nomenklaturen, Nosologien und Klassifikationssysteme für pathologische
psychologische Bedingungen gibt und dass diese Systeme sich mit
dem medizinischen Fortschritt entwickeln. Beispiele für pathologische
Zustände,
die mit Psychose assoziiert sind, welche mit den erfindungsgemäßen Verbindungen
behandelt werden kann, umfassen unter anderem Schizophrenie, Störung schizophrener
Form, schizoaffektive Störung,
Wahnstörung,
kurze psychotische Störung,
verteilte psychotische Störung,
psychotische Störung
aufgrund des allgemeinen medinzinischen Zustands, Substanz-induzierte
psychotische Störung,
schizotypische, schizoide, paranoide Persönlichleitsstörung und
nicht weiter spezifizierte psychotische Störung, siehe DSM-IV, Abschnitt:
Schizophrenia and Other Psychotic Disorders, Seiten 273 bis 316.
-
Die
Verbindungen der vorliegenden Erfindung sind zur Behandlung von
Depression und Stimmungsstörungen
brauchbar, die in DSM-IV, Diagnostic and Statistical Manual of Mental
Disorders, 4. Ausgabe (1994) Abschnitt: Mood Disorders, Seiten 317
bis 392 gefunden werden. Die Störungen
umfassen unter anderem Stimmungsstörungen, wie größere Depressionsepisoden,
manische Episode, gemischte Episode, hypomanische Episode, Depressionsstörungen,
wie größere Depressionsstörung, dysthyme
Störung,
nicht weiter spezifzierte Depressionsstörung, bipolare Störung, wie
bipolare Störung
I, bipolare Störung
II, cyclothme Störung, nicht
weiter spezifizierte bipolare Störung,
andere Stimmungsstörungen,
wie Stimmungsstörung
aufgrund allgemeiner medizinischer Zustände, Substanz-induzierte Stimmungsstörung, nicht
weiter spezifizierte Stimmungsstörung
und Stimmungsstörung,
wie mild, moderat und schwer ohne psychotische Merkmale, schwer mit
psychotischen Merkmalen, in teilweiser Remission, in voller Remission,
mit katatonischen Merkmalen, mit melancholischen Merkmalen, mit
atypischen Merkmalen und mit Postpartumbeginn.
-
Der
Fachmann erkennt, dass die erfindungsgemäßen Verbindungen bei der Behandlung
von depressiven Episoden sehr brauchbar sind, die mit bipolaren
Störungen
assoziiert sind, der Behandlung von manischen Episoden, die mit
bipolaren Störungen
assoziiert sind, wie unter anderem der Behandlung von akuten manischen
Episoden, die mit bipolarer Störung
I assoziiert sind.
-
Die
erfindungsgemäßen Verbindungen
sind brauchbar zur Behandlung von kognitiven Störungen, altersbedingter kognitiver
Störung,
milder kognitiver Beeinträchtigung,
postconcussionaler Störung,
milder neurokognitiver Störung,
Angst (insbesondere einschließlich
generalisierter Angststörung,
Panikstörung
und obsessiv-kompulsiver Störung)
und Migräne
(einschließlich
Migränekopfschmerz).
Diese Verbindungen sind auch brauchbar bei der Behandlung von Substanzentzug
(einschließlich
Substanzen, wie Opiate, Nikotin, Tabakprodukte, Alkohol, Benzodiazepine,
Kokain, Sedativa, Hypnotika, Koffein usw.). Andere Zustände, die
mit den erfindungsgemäßen Verbindungen
behandelt werden könne,
umfassen unter anderem Demenz, Demenz mit Vehaltensstörungen,
Bewegungsstörungen,
Persönlichkeitsstörungen,
Borderlinepersönlichkeitsstörungen,
pervasive Entwicklungsstörungen,
Essstörungen,
prämenstruelle
Dysphoriestörung,
Tickstörungen,
Sexualstörungen,
Delirium, Übelkeit,
Substanz-bezogene Störungen,
Impulskontrollstörungen,
psotpsychotische Depressionsstörung
der Schizophrenie, einfache verschlechternde Störung (einfache Schizophrenie),
leichte Depressionsstörung,
wiederholte, kurze Depressionstörung
und gemischte Angst-Depressionsstörung.
-
Die
Verbindungen der vorliegenden Erfindung sind bei der Behandlung
der kognitiven Defizite brauchbar, die mit der obigen Liste assoziiert
sind, wie unter anderem psychologische Zustände, wie Schizophrenie, Stimmungsstörungen und
andere psychotische Störungen.
-
Eine
wirksame Menge kann leicht durch den betreuenden Diganostiker als
Fachmann durch die Verwendung bekannter Techniken und durch Beobachtung
von Ergebnissen bestimmt werden, die unter analogen Umständen erhalten
werden. Bei der Bestimmung der wirksamen Dosis werden mehrere Faktoren
durch den betreuenden Diagnostiker in Betracht gezogen, wie unter
anderem: Die Art des Säugers,
dessen Größe Säugers, dessen
Größe, Alter
und allgemeiner Gesundheitszustand, die spezifische Erkrankung oder
Störung,
das Ausmaß oder
die Schwere der involvierten Migräne, die Reaktion des einzelnen
Patienten, die im einzelnen verabreichte Verbindung, die Verabreichungsart,
die Bioverfügbarkeitscharakteristiken
der verabreichten Präparation,
der ausgewählte
Dosierungsplan und die Verwendung von begleitender Medikation und
andere relevante Umstände.
-
Die
erfindungsgemäßen Verbindungen
sind über
einen weiten Dosierungsbereich wirksam, aber die tatsächlich verabreichte
Dosis hängt
von dem zu behandelnden Zustand ab. Während die Exakte Dosis durch die
Beurteilung des behandelnden Gesundheitsspezialisten verabreicht
wird, können
typischer bei der Behandlung von erwachsenen Menschen Dosierungen
von 0,1 bis 500 mg, vorzugsweise 0,25 mg bis 100 mg pro Tag verwendet
werden. Normalerweise ist eine Dosierung pro Tag ausreichend, obwohl
verteilte Dosen verabreicht werden können. Beispielsweise ist zur
Behandlung von psychotischen Störungen
ein Dosisbereich von 0,1 mg bis 500 mg, vorzugsweise 0,25 mg bis
100 mg pro Tag geeignet.
-
Bei
der Auswahl des geeigneten Dosierungsplans für Patienten, die an psychotischen
Zuständen
leiden, können
Zusammensetzungen, die Verbindungen der Formel (I) als Wirkstoff
enthalten, formuliert werden, um eine schnelle, anhaltende oder
verzögerte
Freisetzung des Wirkstoffs nach einer Verabreichung an den Patienten
zu erreichen. In Abhängigkeit
des Verabreichungsverfahrens können
die Zusammensetzungen als Tabletten, Kapseln, Suspensionen oder
Elixiere zur oralen Verwendung oder Injektionslösungen oder Zäpfchen zur
parenteralen Verwendung formuliert werden. Vorzugsweise werden die
Formulierungen in einer Einheitsdosierungsform formuliert, wobei
jede Dosis 0,1 mg bis 500 mg, gewöhnlicher 0,25 mg bis 100 mg
des Wirkstoffs enthält.
-
Eine
bevorzugte Formulierung der Erfindung ist eine Kapsel oder eine
Tablette, die 0,1 bis 500 mg Wirkstoff zusammen mit einem pharmazeutisch
annehmbaren Träger
umfasst. Eine weitere bevorzugte Formulierung ist eine Injektion,
in der die Einheitsdosierungsform 0,1 mg bis 500 mg Wirkstoff zusammen
mit einem pharmazeutisch annehmbaren Verdünnungsmittel umfasst. Eine
anhaltend freisetzende Formulierung ist auch eine bevorzugte Formulierung.
-
Pharmazeutische Formulierungen
-
Während es
möglich
ist, eine Verbdinu8ng der Formel (I) ohne zusätzliche Inhaltsstoffe einem
behandlungsbedürftigen
Patienten zu verabreichen, ist es wesentlich erwünschter, eine solche Verbindung
in Form einer pharmazeutischen Zusammensetzung zu verabreichen.
Pharmazeutische Zusammensetzungen, die eine Verbindung der Formel
(I) als Wirkstoff enthalten, liefern eine Kontrolle der Dosierung
und der Absorbtionsgeschwindigkeit in den Körper und die Stabilität des Produkts
beim Transport und der Lagerung. Ferner sind pharmazeutische Formulierungen
sind für
den zu behandelnden Patienten annehmbarer und erhöhen so die
Kompliance mit einem Behandlungsprogramm. Solche Zusammensetzungen
umfassen zumindest einen pharmazeutisch annehmbaren Träger und
sind aufgrund des Vorkommens der Verbindungen der Formel (I) hierin
wertvoll und neu. Die Formulierung von pharmazeutischen Zusammensetzungen
ist eine Wissenschaft für
sich, über
die viel veröffentlicht
wurde. Die erfin dungsgemäßen Verbindungen
können
in pharmazeutische Zusammensetzungen durch im wesentlichen jedes
Verfahren der Technik formuliert werden, einschließlich unter
anderem den unten beschriebenen Verfahren.
-
Die
gewöhnlichen
Formulierungsverfahren, die in der pharmazeutischen Wissenschaft
verwendet werden, und die gewöhnlichen
Zusammensetzungstypen können
verwendet werden, einschließlich
Tabletten, Kautabletten, Kapseln, Lösungen, parenterale Lösungen,
intranasale Sprays oder Pulver, Pastillen, Zäpfchen, Transdermalpflaster
und Suspensionen. Im allgemeinen enthalten die Zusammensetzungen
insgesamt etwa 0,5% bis etwa 50% der Verbindung der Formel I in
Abhängigkeit
der gewünschten
Dosen und dem Typ der zu verwendenden Zusammensetzung. Die Menge
der Verbindung der Formel (I) wird jedoch am besten als die wirksame
Menge definiert, die die Menge jeder Verbindung ist, welche einem
behandlungsbedürftigen
Patienten die erforderliche Dosis liefert. Die Aktivität der Verbindungen
hängt nicht
von der Art der Zusammensetzung ab, und so werden die Zusammensetzungen
alleine nach Bequemlichkeit und Ökonomie
ausgewählt
und formuliert. Jede Verbindung kann in jeder gewünschten
Form der Zusammensetzung formuliert werden. Eine Diskussion der
unterschiedlichen Zusammensetzungen wird bereitgestellt, wonach
einige typische Formulierungen folgen.
-
Kapseln
werden durch Mischen der Verbindung mit einem geeigneten Verdünnungsmittel
und Einfüllen
der richtigen Menge des Gemisches in Kapseln hergestellt. Die gewöhnlichen
Verdünnungsmittel
umfassen inerte pulverisierte Substanzen, wie verschiedene Stärkearten,
pulverisierte Cellulose, speziell kristalline und mikrokristalline
Cellulose, Zucker, wie Fructose, Mannit und Saccharose, Getreidemehle
und ähnliche
essbare Pulver.
-
Tabletten
werden durch direktes Verpressen, Nassgranulierung oder Trockengranulierung
hergestellt. Ihre Formulierungen umfassen gewöhnlich Verdünnungsmittel, Bindemittel,
Gleitmittel und Zerfallshilfsmittel, wie auch die Verbindung. Typische
Verdünnungsmittel
sind unter anderem Stärkearten,
Lactose, Mannit, Kaolin, Calciumphosphat oder -sulfat, anorganische
Salze, wie Natriumchlorid, und pulverisierter Zucker. Pulverisierte
Cellulosederivate sind auch brauchbar. Typische Tablettenbindemittel
sind Substanzen, wie Stärke, Gelatine
und Zucker, wie Lactose, Fructose, Glucose und dergleichen. Natürliche und
synthetische Gummis sind auch verwendbar, einschließlich Akaziengummi,
Alginate, Methylcellulose, Polyvinylpyrrolidon und dergleichen.
Polyethylenglycol, Ethylcellulose und Wachse können auch als Bindemittel dienen.
-
Ein
Gleitmittel ist in einer Tablettenformulierung erforderlich, um
die Tablette und die Körner
vor dem Festkleben in der Speiseröhre zu bewahren. Das Gleitmittel
wird aus schmierigen Feststoffen ausgewählt, wie Talkum, Magnesium-
und Calciumstearat, Stearinsäure
und hydrierten Pflanzenölen.
-
Tablettenzerfallshilfsstoffe
sind Substanzen, die quellen, wenn sie nass werden, um die Tablette
aufzubrechen und den Wirkstoff freizusetzen. Sie umfassen Stärkearten,
Tonerden, Cellulosen, Algine und Gummis. Insbesondere können Mais-
und Kartoffelstärken,
Methylcellulose, Agar, Bentonit, Holzcellulose, pulverisierter natürlicher
Schwamm, Kationenaustauscherharze, Alginsäure, Guargummi, Citruspulpe
und Carboxymethylcellulose beispielsweise verwendet werden, wie
auch Natriumlaurylsulfat.
-
Enterische
Formulierungen werden oft verwendet, um einen Wirkstoff vor den
starken Säuren
des Magens zu schützen.
Solche Formulierungen werden durch Überziehen einer festen Dosierungsform
mit einem Polymerfilm erzeugt, der in sauren Umgebungen unlöslich und
in basischen Umgebungen lös lich
ist. Beispielsgemäße Filme
sind Celluloseacetatphthalat, Polyvinylacetatphthalat, Hydroxypropylmethylcellulosephtalat und
Hydroxypropylmethylcelluloseacetatsuccinat.
-
Tabletten
werden oft mit Zucker als Geschmack und Versiegelung oder mit filmbildenden
Schutzmitteln überzogen,
um die Auflösungseigenschaften
der Tablette zu modifizieren. Die Verbindungen können auch als Kautabletten
formuliert werden, wobei man große Mengen an angenehm schmeckenden
Substanzen, wie Mannit, in der Formulierung verwendet, wie es derzeit
gut etablierte Praxis ist. Sofort auflösende tablettenähnliche
Formulierungen werden jetzt auch häufig verwendet, um sicherzustellen,
dass der Patient die Dosierungsform konsumiert und um die Schwierigkeit
beim Schlucken fester Objekte zu vermeiden, die manche Patienten
belästigt.
-
Wenn
es gewünscht
ist, die Kombinationen als Zäpfchen
zu verabreichen, können
die gewöhnlichen Grundlagen
verwendet werden. Cacaobutter ist eine übliche Zäpfchengrundlage, die durch
die Zugabe von Wachsen modifiziert werden kann, um deren Schmelzpunkt
leicht anzuheben. Wassermischbare Zäpfchengrundlagen, die insbesondere
Polyethylenglycole mit verschiedenen Molekulargewichten umfassen,
werden ebenfalls häufig
verwendet.
-
Transdermalpflaster
wurden kürzlich
populär.
Typischerweise weisen sie eine harzartige Zusammensetzung auf, worin
die Arzneimittel sich lösen
oder teilweise lösen,
die mit der Haut durch einen Film in Kontakt gehalten wird, der
die Zusammensetzung schützt.
Es tauchten in diesem Feld kürzlich
viele Patente auf. Andere kompliziertere Pflasterzusammensetzungen
werden auch verwendet, insbesondere die mit einer mit Poren gelochten
Membran, durch die die Arzneimittel durch osmotische Wirkung gepumpt
werden.
-
Beispiele
-
Beispiel 1
-
2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-on
-
Es
werden 2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-ylaminhydrochlorid
(
US Patent 5 229 382 ) (50
g, 0,19 mol) und festes Kaliumcarbonat (182 g, 132 mol) in einem
Gemisch aus Wasser (1 l) und Ethanol (450 ml) vereinigt und bei
85°C für 48 h erhitzt.
Es wird auf Raumtemperatur gekühlt
und in ein Eisbad für
2 h gegeben. Das gewünschte
Material fällt
als gelber Feststoff aus. Der Feststoff wird durch Filtration gesammelt, mit
kaltem Wasser gewaschen, für
wenige Minuten luftgetrocknet und in einen Vakuumofen bei 45°C für 48 h gegeben,
um 41,9 g der Titelverbindung zu erhalten, die im nächsten Schritt
direkt ohne weitere Reinigung verwendet wird. Massenspektrum (APCI):
m/z = 231,0 (M+1).
-
Beispiel 2
-
2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
-
Es
werden 2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-on
(21 g, 0,09 mol) und Phosphorsäurepentasulfid
(53 g, 0,12 mol) in Pyridin (350 ml) vereinigt und bei Raumtemperatur über Nacht
gerührt
und am Rückfluss
für 5 h
erhitzt. Es wird auf Raumtemperatur gekühlt und der Niederschlag setzt
sich ab, der Feststoff wird durch Filtration gesammelt, der Kuchen
wird mit kaltem Ethanol/Wasser gewaschen und für weinige Minuten unter Bildung
von 29 g des rohen Materials luftgetrocknet. Der Feststoff wird
in einer kleinem Menge Pyridin aufgenommen, ein kleines Volumen
Ethanol/Wasser wird zugegeben, für
wenige Minuten ultrabeschallt und wieder durch Filtration gesammelt.
Der Kuchen wird luftgetrocknet und dann in einen Vakuumofen bei
40°C für 48 h unter
Bildung von 15,9 g der Titelverbindung als oranger Feststoff gegeben,
der direkt im nächsten
Schritt ohne weitere Reinigung verwendet wird: Schmelzpunkt: 259,0-259,5°C. Massenspektrum (APCI):
m/z = 247 (M+1).
-
Beispiel 3
-
2-Methyl-4,9-dihydro-3-thia-6-fluor-4,9-diazabenzo[f]azulen-10-thion
-
2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-on
(1,2 g, 4,8 mmol) wird in trockenem Toluol mit Lawesson's Reagenz (1,1 g,
2,7 mmol) unter Stickstoff suspendiert. Das Reaktionsgemisch wird
unter Rückfluss
für eine
Stunde erhitzt und kann dann über
Nacht abkühlen.
Das gewünschte
Material fällt
aus und wird durch Filtration gesammelt, für mehrere Minuten unter Bildung
von 529 mg eines gelben Feststoffs luftgetrocknet, der im nächsten Schritt
ohne weitere Reinigung verwendet werden kann. Massenspektrum (FIA)
265 (M+1).
NMR (1H, 300 MHz, CDCl3) δ 6,98
(s, 1H), 6,75 (t, 1H), 6,6 (t, 1H), 6,43 (d, 1H), 3,29 (s, 1H),
2,21 (s, 3H).
-
Beispiel 4
-
2-(5-Fluor-2-nitro-phenylamino)-5-ethyl-thiophen-3-carbonitril
-
2-Amino-5-ethyl-thiophen-3-carbonitril
(8,2 g, 54 mmol) und 2,4-Difluornitrobenzol (8,6 g, 54 mmol) werden
in Tetrahydrufuran (20 ml) gelöst
und zu einer gerührten
Lösung
aus Natriumhydrid (50% Dispersion in Mineralöl) (4,1 g, 1,4 Äquivalente)
in Tetrahydrofuran (50 ml) unter einer Stickstoffatmosphäre gegeben.
Die Zugabegeschwindigkeit wird so aufrechterhalten, dass die Temperatur
unter 45°C
bleibt und die Gasentwicklung unter Kontrolle ist. Es wird für 18 h gerührt. Das
Gemisch wird in ein Gemisch aus Eis und 2 N HCl gegossen, in Ethylacetat
extrahiert, getrocknet (MgSO4) und unter
verringertem Druck konzentriert. Es wird in Ethanol (100 ml) gelöst und 2-(5-Fluor-2-nitro-phenylamino)-5-ethyl-thiophen-3-carbonitril
wird durch Filtration als gelber Feststoff (7,9 g, 50%) gesammelt.
Massenspektrum (LCMS) 292 (M+1), 314 (M+Na).
NMR (1H,
300 MHz, CDCl3): δ 9,7 (1H, breit), 8,25 (1H,
m), 6,80 (1H, s), 6,75 (1H, dd), 6,65 (1H, s), 2,80 (2H, q), 1,35
(3H, t).
-
Beispiel 5
-
6-Fluor-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(5-Fluor-2-nitrophenylamino)methyl-thiophen-3-carbonitril
(0,79 g, 27 mmol) wird in Ethanol (70 ml) suspendiert, wasserfreies
Zinn(II)chlorid (15 g, 79 mmol) in konzentrierter Chlorwasserstoffsäure (60
ml) wird zugegeben und das Gemisch wird unter Rückfluss für 6 Stunden erhitzt. Das Gemisch
wird mit Wasser verdünnt
und kann abkühlen.
Der gebildete Niederschlag wird durch Filtration gesammelt und unter
Hochvakuum unter Bildung von 6-Fluor-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
als gelber Feststoff (6,5 g, 82%) getrocknet. Massenspektrum (FIA)
262 (M+1).
-
Beispiel 6
-
2-(5-Chlor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
-
2-Amino-5-methyl-thiophen-3-carbonitril
(0,79 g, 5,7 mmol) und 2-Fluor-4-chlornitrobenzol (1 g, 5,7 mmol)
werden in Dimethylsulfoxid (15 ml) gelöst und unter Stickstoff gerührt. Lithiumhyroxidmonohydrat
(250 mg, 6 mmol) wird zugegeben und das Gemisch wird in einem Ölbad bei
60°C über Nacht
erhitzt. Das Gemisch wird in gesättigtes
NH4Cl(wässrig) gegossen,
in Ethylacetat extrahiert, getrocknet (MgSO4)
und unter verringertem Druck konzentriert. Eine Chromatographie
auf Silicagel (Eluent Cyclohexan/Ethylacetat) ergibt 2-(5-Chlor-2-nitrophenylamino)-5-methyl-thiophen-3-carbonitril
als gelben Feststoff (0,58 g, 35%).
NMR (1H,
300 MHz, CDCl3): δ 9,6 (1H, breit), 8,21 (1H,
d), 7,09 (1H, s), 6,92 (1H, d), 6,80 81H, s), 2,49 (3H, s).
-
Beispiel 7
-
6-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(5-Chlor-2-nitro-phenylamino)-5-methyl-thiophen-3-carbonitril
(0,58 g, 1m,98 mmol) wird in Ethanol (20 ml) suspendiert, wasserfreies
Zinn(II)chlorid (1,5 g, 6,66 mmol) in konzentrierter Chlorwasserstoffsäure (6 ml)
wird zugegeben und das Gemisch wird unter Rückfluss für 75 Minuten erhitzt. Das Gemisch
wird mit Wasser verdünnt
und kann sich abkühlen.
Der Niederschlag wird durch Filtration gesammelt und unter Hochvakuum
unter Bildung von 6-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
als gelber Feststoff (0,537 g, 90%) getrocknet. Massenspektrum (FIA)
300/302 (M+1).
-
Beispiel 8
-
2-(4-Chlor-2-nitrophenylamino)-5-methyl-thiophen-3-carbonitril
-
2-Amino-5-methyl-thiophen-3-carbonitril
(4,14 g, 30 mmol) und 2,4-Dichlornitrobenzol (5,76 g, 30 mmol) werden
in Dimethylsulfoxid (70 ml) gelöst
und unter Stickstoff gerührt.
Lithiumhydroxidmonohydrat (2,0 g, 47,7 mmol) wird zugegeben und
das Gemisch wird in einem Ölbad
bei Raumtemperatur für
18 Stunden erhitzt. Das Gemisch wird in Eiswasser gegossen, mit
2 N HCl angesäuert,
in Ethylacetat extrahiert, getrocknet (MgSO4)
und unter verringertem Druck konzentriert. Es wird aus Ethanol unter
Bildung von 2-(4-Chlor-2-nitrophenylamino)-5-methyl-thiophen-3-carbonitril
als gelber Feststoff (2,92 g, 33%) kristallisiert.
NMR (1H, 300 MHz, CDCl3): δ 9,5 (1H,
breit), 8,25 (1H, s), 7,45 (1H, d), 7,1 (1H, d), 6,25 (1H, s), 2,5
(3H, s).
-
Beispiel 9
-
7-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(4-Chlor-2-nitrophenylamino)-5-methyl-thiophen-3-carbonitril
(2,92 g) wird in Ethanol (40 ml) suspendiert, wasserfreies Zinn(II)chlorid
(5 g) in 1:1 konzentrierter Chlorwasserstoffsäure/Wasser (32 ml) wird zugegeben
und das Gemisch wird unter Rückfluss
für 2 Stunden
erhitzt. Das Gemisch wird mit Wasser verdünnt und kann sich abkühlen. Der
gebildete Niederschlag wird durch Filtration gesammelt und unter
Hochvakuum unter Bildung von 7-Chlor-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-ylaminhydrochlorid
als gelber Feststoff (3,3 g, 90%) gesammelt: Massenspektrum (FIA)
264/266 (M+1).
-
Beispiel 10
-
4-Methyl-2-(2-nitrophenylamino)benzonitril
-
Es
werden 1-Fluor-2-nitrobenzol (5,34 g, 37,83 mmol), 2-Amino-4-methyl-benzonitril
(5,00 g, 37,83 mmol), Lithiumhydroxidmonohydrat (3,17 g, 75,66 mmol)
und DMSO (70,0 ml) vereinigt und bei 55°C gerührt. Nach 16 Stunden wird auf
Umgebungstemperatur gekühlt,
das Gemisch wird auf Eischips gegossen und gerührt. Nach 1 Stunde wird der
entstehende gelbe Niederschlag durch Vakuumfiltration entfernt.
Der Niederschlag wird unter Vakuum getrocknet und in Ethanol unter
Bildung von 5,15 g (54%) an feinen, hellbraun gefärbten Nadeln
umkristallisiert. Smp. 162-164°C.
Massenspektrum (Ionenspray): m/z = 254,0 (M+1).
-
Beispiel 11
-
5-Methyl-2-(2-nitrophenylamino)benzonitril
-
1-Fluor-2-nitrobenzol
(4,34 g, 30,79 mmol), 2-Amino-5-methyl-benzonitril (4,07 g, 30,79
mmol), Lithiumhydroxidmonohydrat (2,58 g, 61,58 mmol) und DMSO (50,0
ml) werden vereinigt. Das Gemisch wird bei 55°C für 22 Stunden gerührt, auf
Umgebungstemperatur gekühlt
und das Gemisch wird auf Eischips gegossen und gerührt. Nach
1 Stunde wird der entstehende Niederschlag durch Vakuumfiltration
entfernt. Der Niederschlag wird unter Vakuum getrocknet und auf
Silicagel mittels Dichlormethan/Hexan (75:25) unter Bildung von 4,45
g (57%) eines orangen Feststoffs gereinigt. Smp. 135-139°C. Massenspektrum
(Ionenspray): m/z = 254,0 (M+1).
-
Beispiel 12
-
2-Amino-5-methylbenzonitril
-
2-Brom-4-methylphenylamin
(8,00 g, 43,00 mmol), CuCN (4,62 g, 51,6 mmol) und NMP (30,0 ml)
werden vereinigt und am Rückfluss
gerührt.
Nach 75 Minuten wird auf Umgebungstemperatur gekühlt, das Gemisch wird auf Eischips
gegossen und für
1 Stunde gerührt.
Der entstehende Niederschlag wird durch Vakuumfiltration entfernt.
Der Niederschlag wird in NH4OH gelöst und mit
Dichlormethan extrahiert. Es wird vereinigt, gewaschen (Kochsalzlösung), getrocknet
(Natriumsulfat) und die Extrakte werden zu einem Rückstand reduziert.
Der Rückstand
wird auf Silicagel mittels Dichlormethan/Hexan (75:25) unter Bildung
von 3,39 g (60%) als oranger Feststoff gereinigt. Massenspektrum
(Ionenspray): m/z = 133,1 (M+1).
-
Beispiel 13
-
2-Methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
5-Methyl-2-(2-nitrophenylamino)benzonitril
(4,03 g, 15,91 mmol), Zinn(II)chloriddihydrat (10,77 g, 47,74 mmol),
5 N HCl (65 ml) und Ethanol (40,0 ml) werden vereinigt und das Gemisch
wird am Rückfluss
gerührt.
Nach 7 Stunden wird auf Umgebungstemperatur gekühlt und in einem Kühlschrank über Nacht
gekühlt. Der
entstehende Niederschlag wird durch Vakuumfiltration entfernt und
der Niederschlag wird in Ethanol (100,0 ml) und 5 N HCl (20,0 ml)
gegeben und am Rückfluss
für 19
Stunden erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur
gekühlt
und in einem Kühlschrank
gekühlt.
Der entstehende Niederschlag wird durch Vakuumfiltration abfiltriert
und in einem Vakuumofen unter Bildung von 2,59 g (63%) eines orangen
Feststoffs getrocknet: Massenspektrum (Ionenspray): m/z 224,0 (M+1).
-
Beispiel 14
-
3-Methyl-5H-dibenzo[b,e]diazepin-11-ylaminhydrochlorid
-
4-Methyl-2-(2-nitrophenylamino)benzonitril
(2,46 g, 9,71 mmol), Zinn(II)chloriddihydrat (6,57 g, 29,71 mmol),
5 N HCl (40 ml) und Ethanol (40,0 ml) werden vereinigt und das Gemisch
wird am Rückfluss
gerührt. Nach
8 Stunden wird auf Umgebungstemperatur gekühlt. Das Gemisch kann bei Umgebungstemperatur über Nacht
stehen und wird für
weitere 3 Stunden in einem Kühlschrank
gekühlt.
Der entstehende Niederschlag wird durch Vakuumfiltration entfernt
und unter Vakuum unter Bildung von 1,24 g (49%) der gewünschten
Verbindung als gelber Feststoff getrocknet. Massenspektrum (Ionenspray):
m/z = 224,0 (M+1).
-
Beispiel 15
-
2-Amino-5-isopropylbenzonitril
-
2-Brom-4-isopropylanilin
(7,5 g, 35 mmol) und Kupfer(I)cyanid (3,76 g, 42 mmol) werden in
NMP (30,0 ml) vereinigt und bei 200°C für 2 Stunden erhitzt. Es wird
auf Umgebungstemperatur gekühlt
und mit Wasser (300 ml) verdünnt.
Es wird mit Ethylacetat unter Bildung von 4,58 g des rohen Produkts
extrahiert. Eine Silicagelchromatographie unter Elution mit Methylenchlorid
ergibt 3,20 g der Titelverbindung als rotes Öl: Massenspektrum (Ionenspray):
m/z = 161 (M+1).
1H (300 MHz, DMSO-dδ): δ 7,21 (m,
2H), 6,73 (d, 1H), 5,79 (s, 2H), 2,73 (Quintet, 1H), 1,12 (d, 6H).
-
Beispiel 16
-
5-Isopropyl-2-(2-nitrophenylamino)benzonitril
-
2-Amino-5-isopropylbenzonitril
(3,19 g, 20 mmol), 1-Fluor-2-nitrobenzol (2,1 ml, 20 mmol) und Lithiumhydroxid
(1,68 g, 40 mmol) werden in DMSO (40,0 ml) vereinigt und bei 55°C für 19 Stunden
erhitzt. Es wird auf Umgebungstemperatur gekühlt und mit Wasser (200 ml)
verdünnt.
Die Titelverbindung fällt
mit 4,56 g als oranger Feststoff aus. Smp. 91-96°C. Massenspektrum (Ionenspray):
m/z = 280 (M+1).
-
Beispiel 17
-
2-Isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
5-Isopropyl-2-(2-nitrophenylamino)benzonitril
(4,54 g, 16,1 mmol) und Zinn(II)chlorid (10,92 g, 48,4 mmol) werden
in 65,0 ml einer 5 N HCl Lösung
und 65,0 ml Ethanol vereinigt. Das Gemisch wird bei 86°C für 18 Stunden
erhitzt. Durch Kühlen
des Gemisches fällt
die Titelverbindung mit 4,22 g als gelber Feststoff aus. Smp > 250°C Massenspektrum
(Ionenspray): m/z = 252 (M+1).
-
Beispiel 18
-
2-(2-Nitrophenylamino)-5-trifluormethylbenzonitril
-
Cäsiumcarbonat
(1,3 g, 4 mmol) wird zu einer Lösung
aus 2-Nitroanilin (276 mg, 2 mmol) und 2-Fluor-5-trifluormethylbenzonitril (378
mg, 2 mmol) in DMF (10 ml) bei Raumtemperatur gegeben und dann wird die
entstehende dunkelrote Lösung
bei Raumtemperatur für
16 Stunden und für
2 Stunden bei 50°C
gerührt. Es
wird abgekühlt
und in ein Gemisch aus Eis und konzentrierter Chlorwasserstoffsäure (50
ml, V/V) gegossen. Die wässrige
Phase wird mit Dichlormethan (3 × 300 ml) extrahiert, mit Wasser
und Kochsalzlösung
gewaschen und über
MgSO4 unter Bildung der Titelverbindung
als gelber Feststoff (480 mg, 80%) getrocknet. Smp. 160-161°C.
1H NMR (CDCl3): δ 7,14 (ddd,
1H), 7,48 (dd, 1H), 7,58 (dd, 1H), 7,60 (d, 1H), 7,76 (dd, 1H),
7,92 (d, 1H), 8,27 (dd, 1H), 9,63 (bs, 1H). MS (ESI/neg) m/z (relative
Intensität)
306,1 (100).
-
Beispiel 19
-
2-Trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus Zinn(II)chlorid (567 mg, 3 mmol) in 12 N Chlorwasserstoffsäure (1,8
ml) wird zu einer Lösung
aus 2-(2-Nitrophenylamino)-5-trifluormethylbenzonitril (307 mg,
1 mmol) in Ethanol (10 ml) gegeben. Es wird für 24 Stunden am Rückfluss
erhitzt, dann unter Vakuum konzentriert und Wasser wird zugegeben
und es wird filtriert. Der entstehende Feststoff wird mit Wasser
und Dichlormethan gewaschen und dann unter Vakuum unter Bildung
der Titelverbindung als gelber Feststoff (282 mg, 90%) getrocknet.
Smp. 334-336°C.
1H (DMSO-d6) δ 7,05-7,19
(m, 4H), 7,34 (d, 1H), 7,86 (dd, 1H), 7,87 (s, 1H), 8,79 (s, 1H),
9,26 (s, 1H), 9,75 (s, 1H), 12,40 (s, 1H). MS (ESI/neg) m/z (relative
Intensität)
276,1 (100).
-
Beispiel 20
-
2-(4-Fluor-2-nitrophenylamino)-5-methylbenzonitril
-
4-Fluor-2-nitrophenylamin
(2,9 g, 18,50 mmol), 2-Fluor-5-methyl-benzonitril (2,5 g, 18,50
mmol) und Lithiumhydroxidmonohydrat (2,4 g, 57,20 mmol) werden in
Methylsulfoxid (DMSO, 40 ml) vereinigt. Das entstehende Gemisch
wird auf 55°C
für 40
Stunden erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur
gekühlt,
in etwa 250 ml Eiswasser gegossen und für eine Stunde gerührt. Das
entstehende Gemisch wird filtriert und der Niederschlag wird gesammelt.
Der Feststoff wird mittels Blitzchromatographie chromatographiert
und mit folgender mobilen Phase: 90% Hexan, 5% Ethylacetat und 5%
Dichlormethan unter Bildung von 2,267 g der Titelverbindung (8,36
mmol, 45% Ausbeute) als oranger amorpher Feststoff eluiert. Massenspektrum
(m/e): 272 (M+1).
-
Beispiel 21
-
8-Fluor-2-methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 2-(4-Fluor-2-nitrophenylamino)-5-methylbenzonitril (1,747 g,
6,44 mmol) in Ethanol (35 ml) wird auf 60°C erhitzt. Eine Lösung aus
Zinn(II)chlorid (6,06 g, 31,96 mmol) in 5,0 N Chlorwasserstoffsaure
(35 ml) wird zugegeben und das entstehende Gemisch wird am Rückfluss
für 40
Stunden erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt und
in einen Gefrierschrank für
16 Stunden gegeben. Nachdem das Produkt durch Filtration gesammelt
wurde, fällt
es aus der Lösung
unter Bildung von 1,3 g der Titelverbindung (4,68 mmol, 73% Ausbeute)
als gelb-grüner
amorpher Feststoff aus: Massenspektrum (m/e): 241 (M+1).
-
Beispiel 22
-
2-Amino-5-isopropylbenzonitril
-
Ein
Gemisch aus Kupfer(I)cyanid (2,5 g, 28,02 mmol) und 2-Brom-4-isopropylphenylamin
(5,0 g, 23,35 mmol) in 1-Methyl-2-pyrrolidinon (20 ml) wird auf
195°C für vier Stunden
erhitzt. Das Reaktionsgemisch wird mit 100 ml Ethylacetat verdünnt und
die dunkle Lösung
wird zweimal mit 28% wässrigem
Ammoniumhydroxid, zweimal mit gesättigtem wässrigem Natriumchlorid (Kochsalzlösung) und
zweimal mit Wasser gewaschen. Die organische Phase wird gesammelt, über Natriumsulfat
getrocknet und das Lösemittel
wird unter verringertem Druck entfernt. Dann wird mittels Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Hexan und
ansteigend bis 80% Hexan mit 20% Ethylacetat unter Bildung von 3,31
g (20,66 mmol, 88% Ausbeute) der Titelverbindung als oranges Öl gereinigt.
Massenspektrum (m/e): 161 (M+1).
-
Beispiel 23
-
2-(4-Fluor-2-nitrophenylamino)-5-isopropylbenzonitril
-
Eine
Lösung
aus 2-Amino-5-isopropylbenzonitril (1,482 g, 9,25 mmol) wird mit
1,4-Difluor-2-nitrobenzol
(1,47 g, 9,25 mmol) und Lithiumhydroxidmonohydrat (0,78 g, 18,50
mmol) in DMSO (20 ml) auf 70°C
für 38
Stunden erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
in etwa 200 ml Eiswasser gegossen und für eine Stunde gerührt. Die
Titelverbindung wird durch Filtration gesammelt und fällt aus.
Es ist keine weitere Reinigung nötig,
um 2,236 g (7,47 mmol, 81% Ausbeute) der Titelverbindung als oranger
amorpher Feststoff zu erhalten. Massenspektrum (m/e): 300 (M+1).
-
Beispiel 24
-
8-Fluor-2-isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Mittels
eines Verfahrens, das für
8-Fluor-2-methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
gefunden wird und mittels 2-(4-Fluor-2-nitrophenylamino)-5-isopropylbenzonitril
(0,559 g, 1,87 mmol), Zinn(II)chlorid (1,06 g, 5,60 mmol) wird die
Titelverbindung (0,422 g, 1,38 mmol, 74% Ausbeute) als gelber amorpher
Feststoff erhalten. Massenspektrum (m/e): 270 (M+1).
-
Beispiel 25
-
2-(4-Fluor-2-nitrophenylamino)-5-trifluormethylbenzonitril
-
4-Fluor-2-nitrophenylamin
(5,0 g, 32,03 mmol), 2-Fluor-5-trifluormethylbenzonitril (6,07 g,
32,03 mmol) und Lithiumhydroxidmonohydrat (4,03 g, 96,08 mmol) in
Methylsulfoxid (DMSO, 60 ml) werden vereinigt. Das entstehende Gemisch
wird für
16 Stunden auf 70°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt und
dann in etwa 400 ml Eiswasser gegossen und für eine Stunde gerührt. Das
entstehende Gemisch wird filtriert und der Niederschlag wird unter
Bildung von 9,995 g der Titelverbindung (30,73 mmol, 96% Ausbeute)
als oranger amorpher Feststoff erhalten. Das Produkt wird ohne weitere
Reinigung verwendet: Massenspektrum (m/e): 326 (M+1).
-
Beispiel 26
-
8-Fluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 2-(4-Fluor-2-nitrophenylamino)-5-trifluormethylbenzonitril (9,995
g, 30,73 mmol) in Ethanol (170 ml) wird auf 60°C erhitzt. Zu dieser Lösung wird
Zinn(II)chlorid (29,1 g, 153,67 mmol) in 5,0 N Chlorwasserstoffsäure (170
ml) gegeben. Das entstehende Gemisch wird am Rückfluss für 18 Stunden erhitzt. Die Reaktion
wird auf Raumtemperatur gekühlt
und in einen Gefrierschrank für
24 Stunden gegeben. Das Produkt fällt aus der Lösung aus
und wird durch Filtration unter Bildung von 2,253 g der Titelverbindung
(6,79 mmol, 22% Ausbeute) als gelber amorpher Feststoff gesammelt:
Massenspektrum (m/e): 296 (M+1).
-
Beispiel 27
-
2-Amino-5-trifluormethylbenzonitril
-
Ein
Gemisch aus Kupfer(I)cyanid (2,24 g, 25,00 mmol) und 2-Brom-4-trifluormethylphenylamin
(5,0 g, 20,83 mmol von Avocado) in 1-Methyl-2-pyrrolidinon (20 ml)
wird für
vier Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird mit 100 ml Ethylacetat verdünnt und
die dunkle Lösung
wird zweimal mit 28% wässrigem Ammoniumhydroxid,
zweimal mit gesättigtem
wässrigem
Natriumchlorid (Kochsalzlösung)
und zweimal mit Wasser gewaschen. Die organische Phase wird gesammelt, über Natriumsulfat
getrocknet und das Lösemittel wird
unter verringertem Druck entfernt. Der Rückstand wird durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Hexan und
weiter bis 70% Hexan mit 30% Ethylacetat unter Bildung von 1,821
g (9,78 mmol, 47% Ausbeute) der Titelverbindung als grüner amorpher
Feststoff gereinigt. Massenspektrum (m/e): 187 (M+1).
-
Beispiel 28
-
2-(5-Fluor-2-nitrophenylamino)-5-trifluormethylbenzonitril
-
2,4-Difluor-1-nitrobenzol
(5,36 g, 33,69 mmol), 2-Amino-5-trifluormethylbenzonitril (5,701
g, 30,63 mmol) und Lithiumhydroxidmonohydrat (2,57 g, 61,25 mmol)
in Methylsulfoxid (DMSO, 70 ml) werden vereinigt. Das Reaktionsgemisch
wird für
16 Stunden auf 55°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt, dann
in etwa 250 ml Eiswasser gegossen und für eine Stunde gerührt. Das
entstehende Gemisch wird filtriert, mit einer großen Menge
Wasser gewaschen und der Niederschlag wird unter Bildung von 9,53
g der Titelverbindung (29,30 mmol, 96% Ausbeute) als gelber amorpher
Feststoff gesammelt. Massenspektrum (m/e): 324 (M-1).
-
Beispiel 29
-
7-Fluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhyrochlorid
-
Eine
Lösung
aus 2-(5-Fluor-2-nitrophenylamino)-5-trifluormethylbenzonitril (1,62
g, 4,98 mmol) in Ethanol (24 ml) wird auf 60°C erhitzt. Zu dieser Lösung wird
Zinn(II)chlorid (2,83 g, 14,94 mmol) in 5,0 N Chlorwasserstoffsäure (24
ml) gegeben. Das entstehende Gemisch wird für 20 Stunden am Rückfluss
erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt, 30
ml an 5,0 N Chlorwasserstoffsäure
werden zugegeben und das Gemisch wird für 16 Stunden in einen Gefrierschrank
gegeben. Das Produkt fällt
aus der Lösung
aus und wird durch Filtration gesammelt. Der Feststoff wird mit
5,0 N Chlorwasserstoffsäure
unter Bildung von 1,44 g der Titelverbindung (4,34 mmol, 87% Ausbeute)
als gelber amorpher Feststoff erhalten. Massenspektrum (m/e): 295
(M+1).
-
Beispiel 30
-
2-(5-Fluor-2-nitrophenylamino)-5-isopropylbenzonitril
-
Eine
Lösung
aus 2-Amino-5-isopropylbenzonitril (1,21 g, 7,55 mmol) wird mit
2,4-Difluor-1-nitrobenzol (1,20
g, 7,55 mmol) und Lithiumhydroxid (0,54 g, 22,66 mmol) in DMSO (15
ml) für
18 Stunden auf 70°C
erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
dann in etwa 200 ml Eiswasser gegossen und für eine Stunde gerührt. Die
Titelverbindung fällt
aus und wird durch Filtration gesammelt. Der Feststoff wird mittels
Blitzchromatographie unter Elution mit 95% Hexan : 5% Ethylacetat
als mobiler Phase unter Bildung von 0,372 g (1,24 mmol, 16% Ausbeute)
der Titelverbindung als oranger amorpher Feststoff gereinigt: Massenspektrum
(m/e): 298 (M-1).
-
Beispiel 31
-
7-Fluor-2-isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 2-(5-Fluor-2-nitro-phenylamino)-5-isopropylbenzonitril (0,372
g, 1,24 mmol) in Ethanol (6 ml) wird auf 60°C erhitzt. Eine Lösung aus
Zinn(II)chlorid (0,707 g, 3,73 mmol) in 5,0 N Chlorwasserstoffsaure
(6 ml) wird zugegeben und das entstehende Gemisch wird am Rückfluss
für 16
Stunden erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt, 30
ml an 5,0 N Chlorwasserstoffsäure
werden zugegeben und das Gemisch wird in einen Gefrierschrank für 16 Stunden
gegeben. Das Produkt fällt
aus der Lösung
aus und wird durch Filtration gesammelt. Der Feststoff wird mit
5,0 N Chlorwasserstoffsäure
unter Bildung von 0,365 g der Titelverbindung (1,19 mmol, 96% Ausbeute)
als gelber amorpher Feststoff gewaschen. Massenspektrum (m/e): 270
(M+1).
-
Beispiel 32
-
2-Amino-5-cyclopropylbenzonitril
-
Es
werden 2-Amino-5-brombenzonitril (1,76 g, 8,95 mmol), Cyclopropylborsäure (1,0
g, 11,64 mmol), Tricyclohexylphosphin (0,251 g, 0,89 mmol) und Kaliumphosphat
(6,65 g, 31,26 mmol) in einem Gemisch aus Toluol (40 ml) und Wasser
(2 ml) vereinigt und das Gemisch wird für 10 Minuten gerührt. Palladiumacetat (0,101
g, 0,45 mmol) wird zugegeben und das Gemisch wird auf 100°C erhitzt.
Nach 3 Stunden werden weiteres Tricyclohexylphosphin (0,251 g, 0,89
mmol) und Palladiumacetat (0,101 g, 0,45 mmol) zugegeben. Nach weiteren
3 Stunden bei 100°C
werden 20 ml Wasser zugegeben und das Gemisch wird dreimal mit 50
ml Ethylacetat extrahiert. Die organischen Phasen werden vereinigt, über Natriumsulfat
getrocknet und konzentriert. Der Rückstand wird mittels Blitzchromatographie
und Elution mit einem linearen Gradienten ausgehend von 100% Hexan
und bis 70% Hexan : 30% Ethylacetat unter Bildung von 0,988 g der
Titelverbindung (6,24 mmol, 70% Ausbeute) als klares gelbes Öl gereinigt.
Massenspektrum (m/e): 159 (M+1).
-
Beispiel 33
-
5-Cyclopropyl-2-(5-fluor-2-nitrophenylamino)benzonitril
-
Eine
Lösung
aus 2-Amino-5-cyclopropylbenzonitril (0,980 g, 6,16 mmol) wird mit
2,4-Difluor-1-nitrobenzol
(0,676 g, 6,16 mmol) und Lithiumhydroxidmonohydrat (0,517 g, 12,33
mmol) in DMSO (12 ml) für
18 Stunden auf 55°C
erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
dann werden etwa 150 ml Eiswasser zugegeben und das Gemisch wird
für eine
Stunde gerührt.
Die Titelverbindung fällt
aus und wird durch Filtration gesammelt. Der Feststoff wird mittels
einer Blitzchromatographie unter Elution mit einem linearen Gradienten
ausgehend von 100% Hexan und bis 70% Hexan : 30% Ethylacetat unter
Bildung von 0,452 g (1,51 mmol, 25% Ausbeute) der Titelverbindung
als gelber amorpher Feststoff gereinigt. Massenspektrum (m/e): 298
(M+1).
-
Beispiel 34
-
2-Cyclopropyl-7-fluor-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 5-Cyclopropyl-2-(5-fluor-2-nitrophenylamino)benzonitril (0,447
g, 1,50 mmol) in Ethanol (7 ml) wird auf 60°C erhitzt. Eine Lösung aus
Zinn(II)chlorid (0,855 g, 4,51 mmol) in 5,0 N Chlorwasserstoffsaure
(7 ml) wird zugegeben und das entstehende Gemisch wird für 16 Stunden
am Rückfluss
erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt, 25
ml an 5,0 N Chlorwasserstoffsäure
werden zugegeben und das Gemisch wird für 16 Stunden in einen Gefrierschrank
gegeben. Das ausgefallene Produkt wird durch Filtration aus der
Lösung
gesammelt. Der Feststoff wird mit 5,0 N Chlorwasserstoffsaure unter
Bildung von 0,277 g der Titelverbindung (0,91 mmol, 61% Ausbeute)
als gelber amorpher Feststoff gewaschen. Massenspektrum (m/e): 268
(M+1).
-
Beispiel 35
-
2-(4,5-Difluor-2-nitrophenylamino)-5-trilfuormethylbenzonitril
-
Eine
Lösung
aus 2-Fluor-5-trifluormethylbenzonitril (1,0 g, 5,74 mmol) wird
mit 4,5-Difluor-2-nitrophenylamin
(1,09 g, 5,74 mmol) und Lithiumhydroxidmonohydrat (0,48 g, 11,49
mmol) in DMSO (12 ml) für
16 Stunden auf 55°C
erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
dann werden etwa 150 ml Eiswasser zugegeben und das Gemisch wird
für eine
Stunde gerührt.
Es wird mit drei 200 ml Portionen an Dichlormethan extrahiert. Die
organischen Phasen werden vereinigt, über Natriumsulfat getrocknet
und das Lösemittel
wird eingedampft. Der Rückstand
wird mittels Blitzchromatographie unter Elution mit einem linearen Gradienten
ausgehend von 100% Hexan und bis 70% Hexan : 30% Ethylacetat unter
Bildung von 0,995 g (2,90 mmol, 50% Ausbeute) der Titelverbindung
als gelber amorpher Feststoff gereinigt. Massenspektrum (m/e): 344 (M+1).
-
Beispiel 36
-
7,8-Difluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 2-(4,5-Difluor-2-nitrophenylamino)-5-trifluormethylbenzonitril
(0,995 g, 2,90 mmol) in Ethanol (15 ml) wird auf 60°C erhitzt.
Hierzu wird eine Lösung
aus Zinn(II)chlorid (1,65 g, 8,70 mmol) in 5,0 N Chlorwasserstoffsaure
(15 ml) gegeben. Das entstehende Gemisch wird für 18 Stunden am Rückfluss
erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt, 30
ml an 5,0 N Chlorwasserstoffsäure
werden zugegeben und das Gemisch wird in einen Gefrierschrank für 16 Stunden
gegeben. Das ausgefallene Produkt wird aus der Lösung durch Filtration gesammelt.
Der Feststoff wird mit 5,0 N Chlorwasserstoffsäure unter Bildung von 0,648 g
der Titelverbindung (1,85 mmol, 64% Ausbeute) als gelber amorpher
Feststoff gewaschen. Massenspektrum (m/e): 314 (M+1).
-
Beispiel 37
-
2-(4-Chlor-2-nitrophenylamino)-5-methylbenzonitril
-
Eine
Lösung
aus 2-Fluor-5-methylbenzonitril (20 g, 14,80 mmol) wird mit 4-Chlor-2-nitrophenylamin (2,5
g, 14,80 mmol) und Lithiumhydroxidmonohydrat (1,24 g, 29,60 mmol)
in DMSO (30 ml) für
14 Stunden auf 55°C
erhitzt. Weitere 0,61 g (14,80 mmol) an Lithiumhydroxidmonohydrat
werden zugegeben und das Erhitzen wird auf 70°C gesteigert, und für weitere
14 Stunden fortgesetzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
in ein Gemisch aus 100 ml Wasser und 200 ml Ethylacetat gegossen.
Es wird mit drei 200 ml Portionen an Ethylacetat extrahiert. Die
organischen Phasen werden vereinigt, über Natriumsulfat getrocknet
und das Lösemittel
wird eingedampft. Der Rückstand
wird mittels Blitzchromatographie unter Elution mit einem Stufengradienten
ausgehend von 90% Hexan : 10% Ethylacetat bis zu 40% Hexan : 60%
Ethylacetat unter Bildung von 1,63 g (5,67 mmol, 38% Ausbeute) der
Titelverbindung als rot-oranger amorpher Feststoff gereinigt: Massenspektrum
(m/e): 288 (M+1).
-
Beispiel 38
-
8-Chlor-2-methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 2-(4-Chlor-2-nitrophenylamino)-5-methylbenzonitril (1,63 g,
6,44 mmol) in Ethanol (25 ml) wird auf 60°C erhitzt. Hierzu wird eine
Lösung
aus Zinn(II)chlorid (3,22 g, 17,00 mmol) in 5,0 N Chlorwasserstoffsaure
(25 ml) gegeben. Das entstehende Gemisch wird für 16 Stunden am Rückfluss
erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt und
in einen Gefrierschrank für
16 Stunden gegeben. Das ausgefallene Produkt wird aus der Lösung durch
Filtration unter Bildung von 1,325 g der Titelverbindung (4,50 mmol, 80%
Ausbeute) als gelber amorpher Feststoff gewaschen. Massenspektrum
(m/e): 258 (M+1).
-
Beispiel 39
-
2-(4-Chlor-2-nitrophenylamino)-5-isopropylbenzonitril
-
1,4-Dichlor-2-nitrobenzol
(0,513 g, 2,67 mmol), 2-Amino-5-isopropylbenzonitril (0,514 g, 3,21
mmol), Kaliumcarbonat (0,754 g, 5,45 mmol) und feines granuläres Kupfer
(0,031 g, 0,48 mmol) in Dimethylformamid (5 ml) werden vereinigt.
Das Reaktionsgemisch wird für
16 Stunden auf 160°C
erhitzt und auf Umgebungstemperatur gekühlt. Das Reaktionsgemisch wird
mit 50 ml Ethylacetat verdünnt
und dreimal mit 50 ml Wasser und einmal mit 50 ml Kochsalzlösung gewaschen.
Die organische Phase wird gesammelt, über Natriumsulfat getrocknet
und das Lösemittel
wird eingedampft. Der Rückstand
wird durch Blitzchromatographie unter Elution mit einem Stufengradienten
ausgehend von 90% Hexan : 10% Ethylacetat und bis 85% Hexan : 15%
Ethylacetat unter Bildung von 0,316 g (1,00 mmol, 37% Ausbeute)
der Titelverbindung als oranger amorpher Feststoff gereinigt: Massenspektrum
(m/e): 316 (M+1).
-
Beispiel 40
-
8-Chlor-2-isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Eine
Lösung
aus 2-(4-Chlor-2-nitrophenylamino)-5-isopropylbenzonitril (0,313
g, 0,99 mmol) in Ethanol (6 ml) wird auf 60°C erhitzt. Hierzu wird eine
Lösung
aus Zinn(II)chlorid (0,752 g, 3,96 mmol) in 5,0 N Chlorwasserstoffsäure (6 ml)
gegeben und das entstehende Gemisch wird für 16 Stunden am Rückfluss
erhitzt. Die Reaktion wird auf Raumtemperatur gekühlt und
in einen Gefrierschrank für
16 Stunden gegeben. Das ausgefallene Produkt wird aus der Lösung durch
Filtration unter Bildung von 0,316 g der Titelverbindung (0,98 mmol, 99%
Ausbeute) als gelber amorpher Feststoff gewaschen. Massenspektrum
(m/e): 286 (M+1).
-
Beispiel 41
-
2-(4-Chlor-2-nitrophenylamino)-5-trifluormethylbenzonitril
-
Mittels
eines Verfahrens, das zu Beispiel 39 ähnlich ist, aber unter Verwendung
von 4-Chlor-2-nitrophenylamin
(6,4 g, 37,3 mmol), 2-Fluor-5-trifluormethylbenzonitril (7,05 g,
37,3 mol) werden 12,03 g der Titelverbindung (95% Ausbeute) als
amorpher gelber Feststoff erhalten. Das Produkt kann wie es ist
ohne weitere Reinigung verwendet werden: Massenspektrum (m/e): 342
(M+1).
-
Beispiel 42
-
8-Chlor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
-
Mittels
eines Verfahrens, das zu Beispiel 40 ähnlich ist, aber unter Verwendung
von 2-(4-Chlor-2-nitrophenylamino)-5-trifluormethylbenzonitril
(12,03 g, 35,2 mmol) werden 8,4 g der Titelverbindung (68% Ausbeute)
als amorpher gelber Feststoff erhalten. Massenspektrum (m/e): 313
(M+1).
-
Beispiel 43
-
2-(5-Fluor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
-
2-Amino-5-methylthiophen-3-carbonitril
(10,0 g, 72,4 mmol) und 2,4-Difluor-1-nitrobenzol (8,00 ml, 73,0
mmol) werden zu DMSO (130 ml) gegeben und unter Stickstoff bei Umgebungstemperatur
gerührt.
Lithiumhydroxidmonohydrat (6,10 g, 145 mmol) wird in einer Portion
zugegeben und bei Umgebungstemperatur gerührt. Nach 18 Stunden wird entionisiertes
Wasser (390 ml) tropfenweise bei 10-20°C zugegeben. Der pH wird mit
konzentrierter HCl (~6 ml) auf pH 7-8 eingestellt und für 4 Stunden
gerührt.
Das rohe Produkt wird filtriert und mit 3:1/Wasser : DMSO und dann
Wasser gewaschen. Es wird bei 50°C
zu einem konstanten Gewicht getrocknet. Es wird durch Blitzchromatographie
unter Elution mit Methylenchlorid unter Bildung von 10,3 g (62%)
der Titelverbindung gereinigt.
1H NMR
(400 MHz, DMSO-d6): δ 9,83 (bs, 1H), 8,28 (m, 1H),
7,12 (s, 1H), 6,91 (m, 1H), 6,73 (m, 1H), 2,53 (s, 3H). HRMS (ES)
exakte Masse M+H berechnet für
C12H8FN3O2S 300,0219, gefunden 300,0219.
-
Beispiel 44
-
6-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(5-Fluor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
(113 g, 0,408 mol) wird als eine Suspension in EtOH (1,1 l bei Umgebungstemperatur
gerührt.
Wässriges
6 N HCl (1,1 l) und Zinn(II)chlorid (232 g, 1,22 mol) werden unter
Rühren
zugegeben. Es wird am milden Rückfluss
(85°C) für 3-4 Stunden
erhitzt und kann dann sich dann auf Umgebungstemperatur abkühlen. Das
rohe Produkt wird filtriert und mit 1:1EtOH : 6 N HCl, dann entionisiertem
Wasser gewaschen und in einem Vakuumofen bei 50°C zu einem konstanten Gewicht
getrocknet. Das rohe Produkt (139 g) wird als eine Suspension in
wässrigem
1 N HCl (6 l) am milden Rückfluss
(95°C) für 2 Stunden
gerührt
und kann sich auf Umgebungstemperatur abkühlen. Das Produkt wird filtriert
und mit 1 N HCl und Wasser gewaschen und bei 50°C unter Bildung von 101 g (87%)
der Titelverbindung getrocknet.
1H
(400 MHz, DMSO-d6) δ 11,33 (bs, 1H), 9,80 (s, 1H),
9,16 (bs, 1H), 8,88 (bs, 1H), 6,99 (m, 1H), 6,89 (m, 1H), 6,84 (s,
1H), 2,28 (s, 3H).
HRMS (ES) exakte Masse M+H berechnet für C12H11ClFN3S 248,0658, gefunden 248,0657.
-
Beispiel 45
-
6-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-on
-
6-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(82,5 g, 0,291 mol) wird als eine Suspension in entionisiertem Wasser
(2475 ml) bei Umgebungstemperatur gerührt. EtOH (830 ml) wird zugegeben
und für
15 Minuten gerührt.
Kaliumcarbonat (301 g, 2,18 mol) wird unter Rühren zugegeben und am milden
Rückfluss
(80-85°C)
für 3-4
Tage erhitzt. Es kann sich auf Umgebungstemperatur abkühlen. Das
Produkt wird filtriert und mit 3:1/Wasser : EtOH und dann Wasser
gewaschen. Der Feststoff wird bei 50-60°C unter Bildung von 55,2 g (76%)
der Titelverbindung getrocknet.
1H
NMR (400 MHz, DMSO-d6) δ 9,18 (bs, 1H), 9,00 (bs, 1H),
6,90 (m, 1H), 6,73 (m, 1H), 6,60 (m, 2H), 2,24 (s, 3H).
HRMS
(ES) exakte Masse M+H berechnet für C12H9FN2OS 249,0498,
gefunden 249,0488.
-
Beispiel 46
-
6-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-thion
-
6-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-on
(65,2 g, 0,263 mol) und 2,4-Bis-(4-methoxyphenyl)-[1,3,2,4]dithiadiphosphetan-2,4-disulfid
(Lawesson's Reagenz,
63,7 g, 0,157 mol) werden zu 1,2-Dichlorethan (DCE, 3500 ml) unter
Rühren
gegeben. Es wird am milden Rückfluss
(80-83°C) für 30 Minuten
erhitzt und kann dann auf Umgebungstemperatur abkühlen. Das
Produkt wird filtriert und mit DCE gewaschen. Es wird bei 50-60°C unter Bildung
von 61,9 g (89%) der Titelverbindung gewaschen.
1H
NMR (400 mHz, DMSO-d6) δ 10,98 (bs, 1H), 8,87 (bs, 1H),
7,00 (m, 1H), 6,88 (s, 1H), 6,78 (m, 1H), 6,56 (m, 1H), 2,22 (s,
3H).
HRMS (ES) exakte Masse M+H berechnet für C123H9FN2S2 265,0269,
gefunden 265,0276.
-
Beispiel 47
-
6-Fluor-2-methyl-10-methylsulfanyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-thion
(61,5 g, 0,233 mol) und DMF (310 ml) werden unter Stickstoff für 15 Minuten
bei Umgebungstemperatur gerührt.
Pulverisiertes Kaliumcarbonat (67,7 g, 0,490 mol) wird zugegeben
und für
15 Minuten bei Umgebungstemperatur gerührt. Jodmethan (22 ml, 0,35
mol) wird zugegeben und für
4 Stunden bei Umgebungstemperatur gerührt. Es wird auf 0-5°C gekühlt und
entionisiertes Wasser (150 ml) wird tropfenweise zugegeben, wobei
die Temperatur unter 15°C
gehalten wird. Das Produkt wird mit Ethylacetat extrahiert. Die
organische Lösung
wird mit Kochsalzlösung
viermal gewaschen, dann über
Magnesiumsulfat getrocknet, filtriert und unter verringertem Druck
unter Bildung von 74,0 g des rohen Produkts konzentriert. Eine Reinigung
durch Blitzchromatographie unter Elution mit 2:1 Methylenchlorid
: Heptan, dann Methylenchlorid ergibt 55,0 g (85%) der Titelverbindung.
1H NMR (400 MHz, DMSO-d6) δ 8,11 (bs,
1H), 6,87 (m, 1H), 6,73 (m, 1H), 6,50 (s, 1H), 6,44 (m, 1H), 2,41
(s, 3H), 2,25 (s, 3H).
HRMS (ES) exakte Masse M+H berechnet
für C13H11FN2S2 279,0426, gefunden 279,0416.
-
Beispiel 48
-
5-Amino-2-methyl-thiazol-4-carbonsäureethylester
-
Acetoamidocyanoacetat
(1000 g, 5,88 mol) wird zu einem 22 l Dreihalsrundbodenkolben gegeben,
der mit einem Rückflusskühler, einem
Thermometer, einem mechanischen Rührer ausgestattet ist, und
Toluol (12 l) wird zugegeben. Zu dieser Suspension wird bei RT Lawesson's Reagenz (11,8 g,
2,93 mol) gegeben. Die entstehende gelbe Aufschlämmung wird bei 70°C für 16 h gerührt und
auf RT gekühlt.
Die obere gelbe Lösung wird
von dem gummiartigen Material auf dem Boden des Kolbens in einen Trenntrichter
abgegossen. 1 N HCl Lösung
(2,5 l) und TBEA (2,5 l) werden zugegeben und das Gemisch wird gerührt. Nach
15 min wird die biphasische Lösung
in die Toluollösung
in dem Trichter vereinigt. Das gummiartige Material kann in dem
Kolben zurückbleiben.
Das obige Verfahren wird wiederholt. Die wässrige Phase wird abgetrennt
und die organische Lösung
wird mit 1 N HCl (2 × 2,5
l) gewaschen. Die organische Phase wird abgetrennt und die wässrige Phase wird
vereinigt und mit 2 N KOH Lösung
basisch gemacht. Ethylacetat (3 × 4 l) wird zugegeben und das
Produkt wird extrahiert. Die organische Phase wird vereinigt, über wasserfreiem
Natriumsulfat getrocknet und unter Bildung von 552 g als blassgelber
Feststoff eingedampft. Der restliche Gummi wird in Methanol (1 l)
gelöst
und zur Trockne eingedampft. MTBE (2,5 l) und 1 N HCl (4 l) werden
zugegeben und das Gemisch wird gerührt. Nach 15 min wird die organische
Phase abgetrennt und die wässrige
wird mit 2 N KOH Lösung
basisch gemacht. Das Produkt wird mit Ethylacetat (2 × 2 l) extrahiert.
Die organischen Phasen werden vereinigt und über wasserfreiem Natriumsulfat
getrocknet und unter Bildung von 165 g als blassgelber Feststoff
eingedampft. (Gesamt: 717g, 65%).
Massenspektrum (m/e): 187
(M+1)
1H NMR (300 MHz, DMSO-d6, ppm): δ 1,21
(t, 3H), 2,38 (s, 3H), 4,21 (q, 2H), 7,21 (bs, 2H).
13C NMR 75 MHz, DMSO-ppm): δ 15,1, 19,2,
59,8, 119,3, 145,6, 161,7, 164,1. Formel: C7H10N2O2S.
-
Beispiel 49
-
2-Methyl-5-(2-nitrophenylamino)thiazol-4-carbonsäure-ethylester
-
Eine
Lösung
aus Ethyl-5-amino-2-methylthiazol-4-carboxylat (120 g, 645 mmol)
und 2-Fluornitrobenzol (68 ml, 645 mmol) in Dimethylsulfoxid (1
l) wird zu einem 2 l Dreihalsrundbodenkolben, der mit einem Rückflusskühler, einem
Thermometer und einem mechanischen Rührer ausgestattet ist, gegeben.
Lithiumhydroxidmonohydrat (54 g, 1290 mmol) wird zu der Lösung gegeben
und bei 50°C
für 3 Stunden
unter Stickstoff erhitzt. Die purpurfarbene Lösung wird gekühlt und
in Eiswasser gegossen, wo sie für
eine Stunde rühren
kann, wird filtriert und mit Wasser gewaschen und bei 50°C unter verringertem
Druck unter Bildung von 190 g (96%) als oranger Feststoff getrocknet.
Massenspektrum
(m/e): 308 (M+1).
1H NMR (300 MHz,
DMSO-d6, ppm) δ 1,25 (tr, 3H), 2,56 (s, 3H),
4,25 (q, 2H), 7,20 (m, 1H), 7,78 (m, 2H), 8,20 (d, 1H), 11,42 (s,
1H, NH).
13C NMR (75 MHz, DMSO, ppm): δ 24,4, 29,2,
71,2, 127,8, 132,5, 132,8, 137,8, 146,5, 147,0, 147,5, 160,2, 161,5,
173,7. Formel: C13H3N3O4S.
-
Beispiel 50
-
2-Methyl-5-(2-nitrophenylamino)thiazol-4-carbonsäureamid
-
Ethyl-2-methyl-5-(2-nitroanilino)thiazol-4-carboxylat
(80 g, 260 mmol) und Formamid (52 ml, 1,3 mol) werden in DMF (200
ml) vereinigt und bei 105°C
erhitzt, wobei zu dieser Zeit die gelbe Aufschlämmung zu einer dunklen Lösung wird.
Zu diesem Reaktionsgemisch wird bei 105°C tropfenweise 25% Natriummethoxid
in Methanol (40 ml, 182,4 mmol) für einen Zeitraum von 45 min
gegeben und es wird auf 115°C
erhitzt und das Rühren
wird für
60 h fortgesetzt. Die Reaktion wird auf RT gekühlt und in eine kalte gesättigte NaHCO3 Lösung gegossen.
Die entstehende Aufschlämmung
wird für
1 h gerührt,
filtriert und der Feststoff wird mit DMF/H2O (2:1)
gewaschen. Es wird in einem Vakuumofen unter Bildung eines dunkelbraunen
Feststoffs (62 g, 86%) getrocknet. Eine weiterer Ansatz der von
100 g Ethyl-2-methyl-5-(2-nitroanilino)thiazol-4-carboxylat
ausgeht, ergibt 82 g (90%) des rohen Produkts.
Massenspektrum
(m/e): 279 (M+1)
1H NMR (300 MHz, DMSO-d6, ppm): 2,5 (s, 3H), 7,05 (m, 1H), 7,51
(d, 1H), 7,65 (m, 2H), 8,10 (d, 1H), 12,18 (s, 1H).
13C NMR (75 MHz, DMSO, ppm): δ 19,4, 116,8,
121,7, 127,3, 129,9, 136,3, 137,0, 137,8, 145,6, 151,2, 166,1.
Formel:
C11H10N4O3S.
-
Beispiel 51
-
2-Methyl-5-(2-nitrophenylamino)thiazol-4-carbonitril
-
Es
werden 2-Methyl-5-(2-nitroanilino)thiazol-4-carbonsäureamid
(60 g, 215 mmol) und Toluol vereinigt und POCl3 (40
ml, 430 mmol) wird zugegeben und das Reaktionsgemisch wird am Rückfluss
erhitzt. Nach 2,5 Stunden wird auf 0°C gekühlt. Gesättigte NaHCO3 Lösung wird
zum Abstoppen des extra POCl3 (Vorsicht
!) zugegeben, bis die wässrige
Phase einen pH um 8 erreicht. Ethylacetat (2 × 2 l) wird zur Extraktion
des Produkts zugegeben. Die organische Phase wird vereinigt und
mit Kochsalzlösung
(2 × 1
l) gewaschen, über MgSO4 getrocknet und unter Bildung eines rötlichen
Feststoffs eingedampft, der mit 25% Ethylacetat in Hexan unter Bildung
eines rötlichen
Feststoffs (36 g) behandelt wird. Das Filtrat wird auf die Hälfte des
Volumens unter Bildung eines zweiten Ansatzes der Verbindung (3,2
g) eingedampft. Gesamtausbeute (39,2 g, 70%): Massenspektrum (m/e):
261 (M+1).
1H NMR (300 MHz, DMSO-d6, ppm): δ 2,70
(s, 1H), 7,02 (t, 1H), 7,22 (d, 1H), 7,58 (t, 1H), 8,25 (d, 1H),
9,78 (s, 1H).
13C NMR (75 MHz, DMSO-d6, ppm): δ 20,4,
113,1, 116,2, 118,7, 121,2, 127,0, 134,9, 136,6, 140,0, 148,6, 161,5.
Formel: C11H8N4O2S
-
Beispiel 52
-
2-Methyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-ylaminhydrochlorid
-
Eine
Suspension aus 2-Methyl-5-(2-nitroanilino)thiazol-4-nitril (36 g,
138,5 mmol) in Isopropanol (400 ml) wird in einem 2,0 Liter Dreihalsrundbodenkolben,
der mit einem Rückflusskühler, einem
Thermometer und einem magnetischen Rührstab ausgestattet ist vereinigt
und unter Rühren
auf 65°C
(orange Lösung)
erhitzt. Zinn(II)chloridhydrat (78,7 g, 415,4 mmol) in Chlorwasserstoffsäure (400
ml, 5 M) wird zugegeben und die entstehende Lösung wird am Rückfluss
erhitzt. Nach 2,5 h wird die Reaktion auf 15°C gekühlt, die Suspension wird filtriert,
mit IsopropanolWasser (2:1) gewaschen und bei 50°C unter verringertem Druck unter
Bildung eines gelben Feststoffs (36,7 g) getrocknet. Das Filtrat
wird zu etwa 200 ml unter Bildung einer gelben Aufschlämmung eingedampft.
Die Aufschlämmung
wird wieder filtriert und bei 50°C
unter verringertem Druck unter Bildung eines gelben Feststoffs (10
g) getrocknet. Der Feststoff wird vereinigt und in 1 N HCl (700
ml) suspendiert und am Rückfluss
für 20
min erhitzt und auf 15°C
gekühlt.
Die entstehende gelbe Aufschlämmung wird
filtriert und bei 50°C
unter verringertem Druck unter Bildung eines gelben Feststoffs (32,4
g, 88%) getrocknet. Massenspektrum (m/e): 231 (M+1).
1H NMR (300 MHz, DMSO-d6,
ppm): δ 2,5
(s, 3H), 6,78 (dd, 1H), 6,85 (dd, 1H), 6,98 (t, 1H), 7,02 (t, 1H),
8,80 (s, 1H), 9,10 (s, 1H), 9,98 (s, 1H), 10,78 (s, 1H).
13C NMR (75 MHz, DMSO, ppm): δ 19,6, 120,1,
120,8, 123,6, 125,8, 127,8, 129,2, 137,6, 154,4, 159,3, 160,4. Formel
C11H11N4S.
-
Beispiel 53
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)propionamid
-
2-Amino-1H-1,5-benzodiazepin-2,4-(3H,5H)-dion
(5,7 g, 30,0 mmol) und Triethylamin (3,33 g, 33,0 mmol) werden in
120 ml DMF vereinigt und Propionylchlorid wird tropfenweise bei
RT zugegeben. Nach dem Rühren über Nacht
wird DMF unter verringertem Druck entfernt, der Rückstand
wird in einem gemischten Lösemittel
(CHCl3/i-PrOH = 3/1, 400 ml) suspendiert.
Es wird ein nicht ganz weißer
Feststoff mittels Unterdruckfiltration unter Bildung der Titelverbindung
gesammelt. Das Filtrat wird mit NaHCO3 (gesättigt 2 × 100 ml)
gewaschen und mit Na2SO4 getrocknet.
Das organische Lösemittel
wird zu einem Rückstand
runterkonzentriert, mit Ether behandelt und der Feststoff wird gesammelt:
Massenspektrum (APCI) (m/e): 248,1 (M+1).
1H
NMR (400 MHz, DMSO-d6): δ 10,70 (s, 2H), 8,19 (d, 1H,
J = 7,2 Hz), 7,23-7,15 (m, 4H), 4,73 (d, 1H, J = 8,0 Hz), 2,27 (q,
2 , J = 8,0 Hz), 0,94 (t, 3H, J = 8,0 Hz).
-
Beispiel 54
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)butyramid
-
Mittels
eines Verfahrens, das zu Beispiel 53 ähnlich ist, aber unter Verwendung
von 3-Amino-1H-1,5-benzodiazepin-2,4-(3H,5H)dion
(4,0 g, 20,9 mmol), Butyrylchlorid (2,45 g, 23,0 mmol) und einem Stehen
bei RT über
Nacht wird die Titelverbindung (4,16 g, Ausbeute 76%) erhalten.
1H NMR (300 MHz, DMSO-d6): δ 10,70 (s,
2H), 8,21 (d, 1H, J = 7,8 Hz), 7,24-7,15 (m, 4H), 4,74 (d, 1H, J
= 7,5 Hz), 2,24 (t, 2H, J = 7,2 Hz), 1,51-1,43 (m, 2H), 0,83 (t,
3H, J = 7,5 Hz).
-
Beispiel 55
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)oxalaminsäureethylester
-
Mittels
eines Verfahrens, das zu Beispiel 53 ähnlich ist, aber unter Verwendung
von 3-Amino-1H-1,5-benzodiazepin-2,4-(3H,5H)dion
(1,91 g, 10,0 mmol) und Ethyloxalylchlorid (1,50 g, 11,0 mmol) und einem
Stehen bei RT über
Nacht wird die Titelverbindung (1,54 g, 76%) als nicht ganz weißer Feststoff
erhalten.
Massenspektrum (APCI) (m/e): 292,11 (M+1).
1H NMR (300 MHz, DMSO-d6): δ 10,95 (s,
2H), 8,32 (d, 1H, J = 6,6 Hz), 7,29-7,17 (m, 4H), 4,74 (d, 1H, J
= 6,6 Hz), 4,27 (q, 2H, J = 6,9 Hz), 1,27 (t, 3H, J = 7,2 Hz).
-
Beispiel 56
-
2-Ethyl-4,9-dihydro-3-thia-1,4,9-triazabenzo[f]azulen-10-thion
-
Es
werden N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)propionamid
(3,7 g, 15,0 mmol) und Lawesson's
Reagenz (9,09 g, 22,5 mmol) in 225 ml an 1,2-Dichlorethan vereinigt
und am Rückfluss unter
N2 erhitzt. Nach dem Erhitzen am Rückfluss über Nacht
wird die Reaktion auf RT gekühlt,
der orange Feststoff wird mittels Unterdruckfiltration gesammelt
und unter Vakuum unter Bildung von 3,3 g des rohen Materials getrocknet.
Das rohe Material (1,0 g) wird genommen, mit Lawesson's Reagenz (0,75 g)
in 1,2-Dichlorethan (30 ml) gemischt, am Rückfluss über Nacht erhitzt, auf RT gekühlt und
der orange-rote Feststoff wird mittels Unterdruckfiltration unter
Bildung der Titelverbindung gesammelt. Das verbleibende Zwischenprodukt wird ähnlich unter
Bildung der zusätzlichen
Titelverbindung (23 g) behandelt:
Massenspektrum (Elektrospray)
(m/e): 261,8 (M+1), 260,0 (M-1).
1H
NMR (300 MHz, DMSO-d6): δ 10,97 (s, 1H), 9,17 (s, 1H),
7,00-6,91 (m, 3H), 6,79-6,70 (m, 1H), 2,73 (q, 2H, J = 7,5 Hz),
1,16 (t, 3H, J = 7,5 Hz).
-
Beispiel 57
-
2-Propyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
-
Es
werden N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)butyramid
(2,3 g, 8,80 mmol) und Lawesson's
Reagenz (4,45 g, 11,0 mmol) in 70 ml an 1,2-Dichlorethan vereinigt
und unter N2 am Rückfluss erhitzt. Nach 1,5 Stunden
wird die Reaktion auf RT gekühlt,
der rötliche
Feststoff wird durch Unterdruckfiltration gesammelt:
1H NMR (300 MHz, DMSO-d6): δ 10,97 (s,
1H), 9,06 (s, 1H), 7,00-6,89 (m, 3H), 6,78-6,75 (m, 1H), 2,68 (t,
2H, J = 7,2 Hz), 1,64-1,56 (m, 2H), 0,91 (t, 3H, J = 7,2 Hz).
-
Beispiel 58
-
10-Thioxo-9,10-dihydro-4H-3-thia-1,4,9-triaza-benzo[f]azulen-2-carbonsäureethylester
-
Es
werden N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)oxalaminsäureethylester (0,73
g, 2,5 mmol) und Lawesson's
Reagenz (1,52 g, 3,75 mmol) in 25 ml Toluol vereinigt und unter
N2 am Rückfluss
erhitzt. Nach dem Erhitzen über
Nacht wird die Reaktion auf RT gekühlt, der orange Feststoff wird durch
Unterdruckfiltration unter Bildung des rohen Produkts gesammelt
und eine Reinigung durch Blitzchromatographie ergibt die Titelverbindung:
Massenspektrum
(APCI) (m/e): 306,0 (M+1).
1H NMR (300
MHz, DMSO-d6): δ 11,24 (s, 1H), 9,60 (s, 1H),
7,05-6,96 (m, 3H), 6,78-6,76 (m, 1H), 4,32 (q, 2H, J = 6,8 Hz),
1,30 (t, 3H, J = 6,8 Hz).
-
Beispiel 59
-
Pentansäure-(2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)amid
-
Valerylchlorid
(3,92 ml, 33,0 mmol) wird tropfenweise zu einer Lösung aus
3-Amino-1,5-dihydro-benzo[b][1,4]diazepin-2,4-dion
(5,74 g, 30,0 mmol) und Triethylamin (4,60 ml, 33,0 mmol) in wasserfreiem
Dimethylformamid (123 ml) gegeben und gerührt. Nach 6 Stunden wird unter
verringertem Druck zu einem Rückstand
konzentriert und der Rückstand
wird in einer Lösung
aus Isopropanol : Chloroform (1:3, 500 ml) rekonstituiert. Es wird über Nacht
unter Bildung eines Feststoffs gerührt und der Feststoff wird
durch Unterdruckfiltration isoliert und der Feststoff wird mit Dichlormethan
gewaschen. Der Feststoff wird bei Umgebungstemperatur für 2 Stunden
unter Bildung der Titelverbindung vakuumgetrocknet. Das Filtrat
wird mit einer gesättigten Lösung aus
Natriumbicarbonat (2 × 200
ml) gewaschen und das Extraktionsgemisch wird unter Entfernung des Salzes
in den Waschschritten entfernt. Die organische Phase wird getrennt
und mit gesättigtem
wässrigem
Natriumchlorid (150 ml) gewaschen. Die wässrige Bicarbonatphase wird
mit Dichlormethan rückextrahiert.
Alle organischen Phasen werden vereinigt, getrocknet (Natriumsulfat),
filtriert und unter verringertem Druck unter Bildung eines Rückstands
konzentriert. Der Rückstand
wird in Diethylether behandelt, der entstehende Feststoff wird filtriert
und mit Diethylether gewaschen, was 2 × wiederholt wird. Der Feststoff
wird bei Umgebungstemperatur unter Vakuum unter Bildung der Titelverbindung
getrocknet. Massenspektrum (APCI, m/e): 276 (M+1).
NMR (1H , 300 MHz, DMSO-d6): δ 10,68 (s,
2H), 8,23 (d, 1H, J = 7,5 Hz), 7,20 (m, 4H), 4,71 (d, 1H, J = 7,5 Hz),
2,25 (t, 2H, J = 7,5 Hz), 1,43 (m, 2H), 1,25 (m, 2H), 0,83 (t, 3H,
J = 7,5 Hz).
-
Beispiel 60
-
2-Butyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
-
Pentansäure-(2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)amid
(4,13 g, 15,0 mmol) und Lawesson's
Reagenz (9,10 g, 22,5 mmol) werden in wasserfreiem Dichlorethan
(250 ml) vereinigt, auf 85°C
erhitzt und gerührt.
Nach 16 h wird auf Umgebungstemperatur gekühlt, der Reaktionsfeststoff
wird durch Unterdruckfiltration gesammelt und der Feststoff wird
bei Umgebungstemperatur unter Vakuum unter Bildung der Titelverbindung
getrocknet:
Massenspektrum (APCI, m/e): 290 (M+1).
NMR
(1H, 300 MHz, DMSO-d6): δ 10,95 (s,
1H), 9,01 (s, 1H), 6,93 (m, 3H), 6,71 (d, 1H, J = 7,5 Hz), 2,68
(t, 2H, J = 7,5 Hz), 1,53 (m, 2H), 1,30 (m, 2H), 0,85 (t, 3H, J
= 7,5 Hz).
-
Beispiel 62
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)-2,2,2-trifluoracetamid
-
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(7,67 g, 40,0 mmol) und 4-(Dimethylamino)pyridin (0,244 g, 2,00
mmol) werden zu einer Lösung
aus 3-Amino-1,5-dihydro-benzo[b][1,4]diazepin-2,4-dion (7,65 g,
40,0 mmol) in wasserfreiem N,N-Dimethylformamid (50 ml) gegeben.
Die Feststoffe werden in der Reaktion mit wasserfreiem N,N-Dimethylformamid
(50 ml) gewaschen und die Reaktion wird auf 0°C in einem Eis/Wasserbad gekühlt. Mittels
einer Spritze wird Trifluoressigsäure (3,08 ml, 40,0 mmol) zugegeben. Nach
10 Minuten wird das Kühlen
entfernt und nach 5,5 Stunden bei Umgebungstemperatur werden zusätzliche
0,2 Äquivalente
an 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid (1,53
g) und Trifluoressigsäure
(0,62 ml) zugegeben und bei Umgebungstemperatur gerührt. Nach
einer Übernachtperiode
wird unter verringertem Druck unter Bildung eines Rückstands
konzentriert. Der Rückstand
wird in Isopropanol : Chloroform (1:3, 20 ml) rekonstituiert und
5 Minuten gehalten. Der durch Unterdruck gebildete Feststoff wird
gesammelt, mit Isopropanol : Chloroform (3:1) gewaschen und bei
Umgebungstemperatur unter Vakuum unter Bildung der Titelverbindung
getrocknet. Das Filtrat wird filtriert, das den ausgefallenen Feststoff
enthält
und dieser Feststoff wird bei Umgebungstemperatur unter Vakuum unter
Bildung eines zweiten Kristallisats der Titelverbindung getrocknet:
Massenspektrum
(ES neg., m/e): 286,0 (M-1)
NMR (1H,
300 MHz, DMSO-d6) δ 10,93 (s, 2H), 9,42 (d, 1H,
J = 6,9 Hz), 7,29-7,15 (m, 4H), 4,91 (d, 1H, J = 7,2 Hz).
-
Beispiel 63
-
2-Trifluormethyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)-2,2,2-trifluoracetamid
(3,02 g, 10,5 mmol) wird mit Lawesson's Reagenz, [2,4-Bis-(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetan-2,4-disulfid], (6,38
g, 15,8 mmol) in wasserfreiem Toluol (60 ml) am Rückfluss
erhitzt und gerührt.
Nach 16 Stunden wird gekühlt
und für
wenige Stunden gerührt.
Der Reaktionsfeststoff wird durch Unterdruckfiltration gesammelt,
mit einer kleinen Menge an Toluol gewaschen und bei 40°C für wenige
Stunden unter Bildung des rohen Produkts (3,6 g) getrocknet. Das
Material wird auf Silicagel 60 adsorbiert und durch Blitzchromatographie
unter Elution mit einer Lösung
aus 35% Ethylacetat in Hexan gereinigt. Die das Produkt enthaltenden
Fraktionen werden vereinigt und unter verringertem Druck konzentriert
und das Produkt wird bei 54°C
unter Vakuum für
4,5 Stunden unter Bildung der Titelverbindung konzentriert.
Massenspektrum
(APCI, m/e): 302 (M+1).
NMR (1H, 300
MHz, DMSO-d6): δ 11,39 (s, 1H), 9,57 (s, 1H),
7,03 (m, 3H), 6,77 (m, 1H).
-
Beispiel 64
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)-2,2-difluoracetamid
-
Mittels
des Verfahrens von Beispiel 62 und unter Verwendung von 3-Amino-1,5-dihydro-benzo[b][1,4]diazepin-2,4-dion
und Difluoressigsäure
und unter Rühren über Nacht
bei Umgebungstemperatur nach der Zugabe der Säure und Waschen des Reaktionsfeststoffs
mit Dichlormethan an Stelle von Isopropanol : Chloroform (3:1) wird
die Titelverbindung erhalten.
Massenspektrum (APCI, m/e): 270
(M+1).
NMR (1H, 300 MHz, DMSO-d6): δ 10,88
(s, 2H), 9,13 (d, 1H, J = 6,9 Hz), 7,29-7,13 (m, 4H), 6,49 (t, 1H, 2J(H,F) = 53,7 Hz),
4,79 (d, 1H, J = 6,9 Hz).
-
Beispiel 65
-
2-Difluormethyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)-2,2-difluoracetamid
(3,0 g, 11,1 mmol) wird mit Lawesson's Reagenz, (2,4-Bis(4-methoxyphenyl)-1,3-dithia-2,4-diphosphetan-2,4-disulfid],
(6,8 g, 16,7 mmol) in wasserfreiem Toluol (60 ml) vereinigt, auf
100°C erhitzt
und gerührt.
Nach 20 Stunden wird gekühlt,
und der Reaktionsfeststoff wird durch Unterdruckfiltration gesammelt
und mit einer kleinen Menge Dichlormethan gewaschen. Der Feststoff
wird mit dem Filtrat, das das Produkt enthält, vereinigt und Methanol und
Acetonitril werden zugegeben. Das unlösliche Material wird abfiltriert,
das Filtrat wird auf Silicagel 60 adsorbiert und eine Vorsäule wird
gemacht. Das Material wird durch Blitzchromatographie unter Elution
mit Hexan gereinigt und dann mit einem Gradienten aus Hexan : Ethylacetat
(0-35% über
55 Minuten). Die das Produkt enthaltenden Fraktionen werden zu einem
Feststoff vereinigt, in Hexan behandelt, filtriert und der filtrierte Feststoff
wird wieder durch Blitzchromatographie unter Elution mit Isopropylacetat:
Chloroform filtriert. Der ausgefällte
Feststoff wird aus einer gemischten Fraktion unter Bildung der Titelverbindung
filtriert. Die reinen Fraktionen werden unter verringertem Druck
vereinigt und unter Bildung einer zusätzlichen Titelverbindung konzentriert.
Das gemischte Material wird durch Blitzchromatographie unter Elution
mit einem Gradienten aus Isopropylacetat : Chloroform (2%-5% Isopropylacetat
in Chloroform über
61 Minuten) gereinigt und kombiniert und die das Produkt enthaltenden
Fraktionen werden unter verringertem Druck zu einem Feststoff konzentriert. Der
Feststoff wird in Hexan ultrabeschallt und filtriert. Der filtrierte
Feststoff wird in Chloroform: Hexan (1:1) ultrabeschallt und unter
Bildung der Titelverbindung filtriert:
Massenspektrum (APCI,
m/e): 284 (M+1)
NMR (1H, 300 MHz, DMSO-d6): δ 11,28
(s, 1H), 9,42 (s, 1H), 7,13 (t, 1H, 2J(H,F) = 54,3 Hz), 7,07-6,96 (m, 3H), 6,76
(m, 1H).
-
Beispiel 66
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)-4,4,4-trifluorbutyramid
-
3-Amino-1H-1,5-benzodiazepin-2,4-(3H,5H)-dion
(5,60 g, 29,3 mmol), 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimidhydrochlorid
(6,75 g, 35,2 mmol) und 4-(Dimethylamino)pyridin (0,183 g, 1,5 mmol)
werden in 100 ml DMF vereinigt und 4,4,4-Trifluorbuttersäure wird
tropfenweise bei 0-5°C
zugegeben. Nach der Zugabe wird das Reaktionsgemisch in Eiswasser
gerührt.
Nach einer halben Stunde wird auf RT erwärmt. Nach dem Rühren über Nacht
wird das gelöste
DMF unter verringertem Druck entfernt und der Rückstand wird in 200 ml gemischtem
Lösemittel
(CHCl3 : i-PrOH = 3:1) suspendiert. Der
Feststoff wird mittels Vakuumfiltration unter Bildung der Titelverbindung
(6,87 g, Ausbeute 74%) gesammelt.
Massenspektrum (APCI) (m/e):
316,1 (M+1)
1H NMR (400 MHz, DMSO-d6): δ 10,72
(s, 2H), 8,60 (d, 1H, J = 7,2 Hz), 7,24-7,15 (m, 4H), 4,74 (d, 1H,
J = 7,2 Hz), 2,58 (t, 2H, J = 7,2 Hz), 2,39-2,35 (m, 2H).
-
Beispiel 67
-
2-(3,3,3-Trifluorpropyl)-4,9-dihydro-3-thia-1,4,9-triazabenzo[f]azulen-10-thion
-
Mittels
des Verfahrens von Beispiel 57 und unter Verwendung von N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)-4,4,4-trifluorbutyramid
(1,58 g, 5,0 mmol) und Lawesson's
Reagenz (3,04 g, 7,5 mmol) wird die Titelverbindung erhalten:
Massenspektrum
(APCI) (m/e): 330,0 (M+1).
NMR (300 MHz, DMSO-d6): δ 11,01 (s,
1H), 9,06 (s, 1H), 7,01-6,92 (m, 3H), 6,77-6,74 (m, 1H), 3,00 (t,
2H, J = 7,8 Hz), 2,71-2,61 (m, 2H).
-
Beispiel 68
-
Cyclopentancarbonsäure-(2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)amid
-
Mittels
des Verfahrens von Beispiel 53 und mittels 3-Amino-1H-1,5-benzodiazepin-2,4-(3H,
5H)dion (7,0 g, 36,6 mmol), Cyclopentancarbonylchlorid (5,34 g,
40,3 mmol) und Triethylamin (4,07 g, 40,3 mmol) wird über Nacht
bei RT die Titelverbindung (9,13 g, Ausbeute 87%) erhalten.
1H NMR (300 MHz, DMSO-d6): δ 10,70 (s,
2H), 8,11 (d, 1H, J = 7,8 Hz), 7,24-7,15 (m, 4H), 4,73 (d, 1H, J
= 7,5 Hz), 2,96-2,87 (m, 1H), 1,74-1,45 (m, 8H).
-
Beispiel 69
-
2-Cyclopentyl-4,9-dihydro-3-thia-1,4,9-triazabenzo[f]azulen-10-thion
-
Mittels
des Verfahrens von Beispiel 57 werden Cyclopentancarbonsäure-(2,4-dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazapin-3-yl)amid
(1,90 g, 6,6 mmol) und Lawesson's
Reagenz (4,01 g, 9,9 mmol) in 120 ml an 1,2-Dichloridethan vereinigt
und am Rückfluss
unter N2 erhitzt. Die Reaktion wird auf
RT gekühlt
wird, nach 4 Stunden werden 1,56 g oranger Feststoff mittels Unterdruckfiltration
gesammelt, Ausbeute 78%.
1H NMR (400
MHz, DMSO-d6): δ 10,96 (s, 1H), 9,05 (s, 1H),
7,00-6,89 (m, 3H), 6,78-6,76 (m, 1H), 3,21-3,10 (m, 1H), 2,00-1,90
(m, 2H), 1,7-1,55 (m, 6H).
-
Beispiel 70
-
Cyanoisobutyrylaminoessigsäureethylester
-
Wässriges
gesättigtes
Natriumbicarbonat (560 ml) wird mit entionisiertem Wasser (700 ml)
verdünnt und
die Lösung
wird unter Zugabe von portionsweise Ethylcyanoglyoxylat-2-oxim (70,0
g, 493 mmol) gerührt (Anmerkung:
Es wird etwas Entgasen und eine leichte Endothermie beobachtet).
Natriumdithionit (238 g, 1,37 mol, 2,8 Äquivalente) wird portionsweise
zugegeben und bei RT gerührt.
Nach 2,5-3 Stunden wird die Lösung mit
Natriumclorid (400 g) gesättigt,
während
dieser Zeit wird die Reaktion durch TLC (EtOAc, I2 Färbung) verfolgt
und das Produkt wird mit CH2Cl2 (1 × 500 ml,
3 × 250
ml) extrahiert, wobei sichergestellt wird, dass festes NaCl während den
Extraktionen sichtbar ist (weiteres wird wenn nötig zugegeben). Die organischen
Phasen werden vereinigt, über
(MgSO4) getrocknet, filtriert und das Filtrat
wird im Vakuum auf einem Rotationsverdampfer bei geringer Badtemperatur
(30-35°C)
unter Bildung von 19,6 g (31%) des rohen Aminocyanoessigsäureethylesters
konzentriert, der sofort in der nächsten Reaktion verwendet wird.
-
Eine
Lösung
aus Aminocyanoessigsäueethylester
(19,0 g, 148 mmol) in CH2Cl2 (300
ml) wird unter N2 auf 0-5°C gekühlt. Pyridin
(12,0 ml, 148 mmol) wird zugegeben, gefolgt von Isobuttersäureanhydrid
(24,6 ml, 148 mmol). Die Reaktionslösung kann sich auf RT über Nacht
erwärmen,
bis sie gemäß TLC (ETOAc)
vollständig
ist. Die Lösung
wird mit wässriger
1 N HCl, Wasser, wässrigem
gesättigtem
NaHCO3 und dann Kochsazlösung (150 ml jeweils) gewaschen.
Die organische Phase wird über
MgSO4 getrocknet, filtriert und das Filtrat
wird im Vakuum auf einem Rotationsverdampfer zu einem Feststoff
zur Trockne konzentriert. Der Feststoff wird mit Et2O
(500 ml) behandelt, filtriert und unter Bildung von 22,0 g (75%)
der Titelverbindung getrocknet (50°C Vakuumofen).
1H NMR (300 MHz, DMSO-d6) δ 9,05 (d,
1H, J = 7,32 Hz), 5,67 (d, 1H, J = 7,32 Hz), 4,25-4,13 (m, 2H),
2,46 (dq, 1H, J = 6,95 Hz), 1,21 (t, 3H, J = 6,95 Hz), 1,03 (d,
6H, J = 6,95 Hz). 13C NMR (75 MHz, DMSO-d6) δ 176,51,
164,13, 115,60, 62,33, 44,07, 33,29, 18,93, 18,86, 13,74.
IR
(CHCl3) 3425, 3028, 2975, 2933, 2905, 2874,
1757, 1687, 1492, 1370, 1284, 1189 cm–1.
HRMS (FAB+) m/z berechnet für
C9H15N2O3 (M+H) 199,1083, gefunden 199,1075.
-
Beispiel 71
-
Cyanoisobutyrylaminoessigsäureethylester
-
Ethylcyanoglyoxylat-2-oxim
(20,0 g, 141 mmol) und 5% Pt/C (2,0 g, 10 Gewichtsprozentbeladung)
in Essigsäure
(120 ml) und EtOAc (60 ml) werden vereinigt und unter 40 psi H2 über
Nacht hydriert, bis die Reaktion gemäß TLC (5:1/Heptan : EtOAc,
I2 Färbung)
vollständig
ist. Der verbrauchte Katalysator wird mittels partiellem Vakuum
durch ein Glasfiberpapier filtriert und mit HOAc/EtOAc gewaschen,
während
der Kuchen austrocknen kann. Das Filtrat wird im Vakuum auf einem
Rotationsverdampfer zu einem Öl
konzentriert, wobei 25,5 g (96%) des rohen Aminocyanoessigsäureethylesters
als HOAc Salz zurückbleiben.
Eine Portion (13,0 g) des HOAc Salzes wird zwischen EtOAc (70 ml)
und Wasser (35 ml) aufgeteilt. Die biphasische Lösung wird gerührt und
es wird tropfenweise wässriges
5 N NaOH (16,5 ml) zum Einstellen des pH auf 8,0-8,2 zugegeben. Die
Phasen werden getrennt und die wässrige
Phase wird mit weiterem EtOAc (3 × 25 ml) extrahiert. Die organische
Phase wird vereinigt, getrocknet (MgSO4),
filtriert und das Filtrat wird im Vakuum auf einem Rotationsverdampfer
bei niedriger Radtemperatur (30-35°C) unter
Bildung von 5,68 g (65%) des rohen Aminocyanoessigsäureethylesters
konzentriert, der sofort in der nächsten Reaktion verwendet wird.
-
Eine
Lösung
des rohen Aminocyanoessigsäureethylesters
(5,68 g, 44,3 mmol) in CH2Cl2 (60
ml) wird unter N2 auf 0-5°C gekühlt. Pyridin
(3,60 ml, 44,5 mmol) gefolgt von Isobuttersäureanhydrid (7,40 ml, 44,6 mmol)
wird zugegeben. Die Reaktionslösung
kann sich auf RT über
Nacht (18 h) erwärmen
bis sie gemäß TLC (3:1EtOAc:
Heptan, I2 Färbung, ein weiterer Fleck ist
nötig,
um zwischen SM und Unreinheit zu unterscheiden) vollständig ist.
Die Lösung
wird mit wässriger
1 N HCl, Wasser, wässrigem
gesättigtem
NaHCO3 und dann Kochsalzlösung (50
ml jeweils) gewaschen. Die organische Phase (MgSO4)
wird getrocknet, filtriert und das Filtrat wird im Vakuum auf einem
Rotationsverdampfer zu einem Feststoff konzentriert. Der Feststoff
wird mit Et2O (150 ml) behandelt, filtriert
und unter Bildung von 4,33 g (49%) der Titelverbindung getrocknet
(50°C Vakuumofen).
-
Beispiel 80
-
5-Amino-2-isopropylthiazol-4-carbonsäurethylester
-
Cyanoisobutyrylaminoessigsäureethylester
(139 g, 701 mmol) wird mechanisch als eine Aufschlämmung in
Toluol (1,4 l) bei RT unter N2 gerührt. Lawesson's Reagenz (170 g,
420 mmol, 0,6 Äquivalente)
wird portionsweise zugegeben und die dicke Aufschlämmung wird
auf 70°C
erhitzt und für
12 Stunden gerührt,
bis sie gemäß TLC (2:1/Heptan:
THF) vollständig
ist. Das Gemisch wird gekühlt
und im Vakuum auf einem Rotationsverdampfer unter Bildung von 353
g eines dicken gelben Öls
zur Trockne konzentriert, das teilweise durch Silicagelkissen (1
kg Silicagel 60, 1,5 Volumina warmes 2:1/THF: Heptan als Verdünnungsmittel,
2:1/Heptan : THF als Eluent) gereinigt wird. Die das Produkt enthaltenden
Filtrate werden vereinigt und im Vakuum auf einem Rotationsverdampfer
unter Bildung von 194 g des rohen Feststoffs konzentriert. Der Feststoff
wird in EtOAc (400 ml) bei 50-60°C
unter Rühren
gelöst
und kann sich dann schrittweise auf RT abkühlen. Das Produkt fällt aus
und wird unter Rühren
für 30
Minuten auf 0-5°C
gekühlt,
durch Unterdruckfiltration isoliert, mit kaltem EtOAc (2 × 50 ml)
gewaschen und dann in einem Vakuumofen bei 50°C unter Bildung einer ersten
Kristallisats von 76,3 g (51%) der Titelverbindung getrocknet. Ein
zweites Kristallisat von 17,6 g (12%) aus dem Filtrat nach der Konzentration
im Vakuum und Silicagelchromatographie (1 kg Silicagel 60, 2:1 Heptan
: THF) wird erhalten.
1H NMR (300 MHz,
DMSO-d6): δ 7,23 (bs, 2H), 4,21 (q, 2H,
J = 6,95 Hz), 3,02 (dq, 1H, J = 6,95 Hz), 1,27 (t, 3H, J = 6,95
Hz), 1,22 (d, 6H, J = 6,95 Hz).
13C
NMR (75 MHz, DMSO-d6) δ 163,72, 160,08, 156,20, 118,08,
58,99, 32,27, 22,35 (2) 14,46.
IR (CHCl3)
3483, 3347, 2975, 2933, 2868, 1668, 1582, 1530, 1494, 1464, 1409,
1382 cm–1.
HRMS
(ES) m/z berechnet für
C9H14N2O2S 215,0854, gefunden 215,0842.
-
Beispiel 81
-
2-Isopropyl-5-(2-nitrophenylamino)thiazol-4-carbonsäureethylester
-
Eine
Lösung
aus 5-Amino-2-isopropylthiazol-4-carbonsäureethylester (8,71 g, 40,6
mmol) und 2-Fluornitrobenzol
(4,28 ml, 40,6 mmol) in DMSO (105 ml) wird vereinigt und bei RT
unter N2 als LiOH (1,95 g, 81,4 mmol, 2,0 Äquivalente)
oder LiOH Monohydrat (2 Äquivalente)
in einer Portion zugegeben. Die Reaktion wird dunkel. Das Reaktionsgemisch
wird für
3 h auf 55°C
erhitzt, bis sie gemäß HPLC (Zorbax
SB C18 25 cm, 60:40/ACN : 0,1% TFA in Wasser, 233 nm, 1,0 ml/min)
vollständig
ist. Es wird über
Nacht auf RT gekühlt,
die Reaktion wird unter Rühren
auf 0-5°C
gekühlt
während
entionisiertes Wasser (315 ml) bei einer derartigen Geschwindigkeit
zugegeben wird, dass die Temperatur unter 20°C beträgt. Das Produkt fällt aus
und die Reaktionsfarbe verändert
sich von braun bis rostorange. Die Aufschlämmung wird für 3-4 h
bei RT gerührt,
durch Vakuum filtriert und mit minimal 3:1/H2O
: DMSO ge waschen, in einem Vakuumofen bei 60°C unter Bildung von 12,4 g (91%)
der Titelverbindung als oranger Feststoff gewaschen.
1H NMR (300 MHz, DMSO-d6) δ 11,52 (bs,
1H), 8,23 (d, 1H, J = 8,05 Hz), 7,80 (m, 2H), 7,21 (m, 1H), 4,36
(q, 2H, J = 7,32 Hz; 6,95 Hz), 3,23 (dq, 1H, J = 6,95 Hz), 1,34
(m, 9H).
13C NMR (75 MHz, DMSO-d6) δ 163,22,
161,85, 149,08, 136,99, 136,37, 136,10, 126,53, 126,48, 121,94, 117,05,
60,45, 32,47, 22,34(2), 14,24.
IR (CHCl3)
2976, 2932, 2867, 1709, 1677, 1611, 1580, 1550, 1512, 1415, 1340
cm–1.
HRMS
(ES) M/z berechnet für
C15H17N3O4S 336,1018, gefunden 336,1009.
-
Beispiel 82
-
2-Isopropyl-5-(2-nitrophenylamino)thiazol-4-carbonsäureamid
-
Eine
Lösung
aus 2-Isopropyl-5-(2-nitrophenylamino)thiazol-4-carbonsäureethylester
(68,6 g, 204 mmol) wird unter N2 als eine
Aufschlämmung
in DMF (205 ml) gerührt.
Formamid (32,4 ml, 816 mmol, 4,0 Äquivalente) wird in einer Portion
zugegeben und die dicke rote Aufschlämmung wird auf 100°C erhitzt.
Es bildet sich eine rote/purpurfarbene Lösung. Über 20-30 min wird 25% NaOMe
in MeOH (32,6 ml, 143 mmol, 0,7 Äquivalente)
tropfenweise zugegeben. Die Temperatur wird auf 120°C erhöht und die
dunkle Lösung
wird bei 120°C über Nacht
gerührt,
bis sie gemäß HPLC (Zorbax
SB C18 25 cm, 60:40/ACN : 0,1% TFA in Wasser, 233 nm, 1,0 ml/min)
vollständig
ist (< 2% Me Ester
+ SM). Nach dem Kühlen
der Reaktion auf RT wird wässriges
5% NH4Cl (410 ml) derartig zugegeben, dass
die Temperatur unter 35°C
bleibt, wobei nicht extern gekühlt
wird. Das Produkt fällt
aus, die Aufschlämmung
wird auf 0-5°C
gekühlt,
durch Vakuumfiltration filtriert und in einem Vakuumofen bei 60°C unter Bildung
von 52,7 g (84% Ausbeute) der rohen Titelverbindung als purpurfarbener Feststoff
gekühlt,
der ohne weitere Reinigung verwendet wird. Eine wässrige Aufarbeitung
kann schlechte Emulsionenlangsame Trennungen ergeben:
1H NMR (300 MHz, DMSO-d6) δ 12,22 (bs,
1H), 8,21 (d, 1H, J = 7,69 Hz), 7,78 (m, 2H), 7,59 (bs, 1H), 7,53
(bs, 1H), 7,15 (m, 1H), 3,23 (dq, 1H, J = 6,95 Hz), 1,35 (d, 6H,
J = 6,95 Hz).
13C NMR (75 MHz, DMSO-d6): δ 165,53,
161,44, 144,58, 137,12, 136,31, 135,72, 128,90, 126,58, 121,08, 116,28,
32,32, 22,35(2):
IR (CHCl3) 3520, 3400,
3004, 2967, 2925, 2866, 1658, 1611, 1578, 1513, 1427, 1342 cm–1:
HRMS
(ES) M/z berechnet für
C13H14N4O3S1 329,0684, gefunden
329,0667.
-
Beispiel 83
-
2-Isopropyl-5-(2-nitrophenylamino)thiazol-4-carbonitril
-
Es
werden 2-Isopropyl-5-(2-nitrophenylamino)thiazol-4-carbonsäureamid
(47,5 g, 155 mmol) und 2-Dichlorethan (475 ml) vereinigt und bei
RT unter N2 als dunkle Lösung gerührt. POCl3 (14,5
ml, 155 mmol) wird in die Lösung
gegossen und die Reaktion wird am Rückfluss (80-83°C) für 2-3 h
erhitzt, bis sie gemäß HPLC (Zorbax
SB C18 25 cm, 60:40/ACN : 0,1% TFA in Wasser, 233 nm, 1,0 ml/min)
vollständig
ist. Die Reaktion wird auf RT gekühlt und dann weiter auf 0-5°C gekühlt. Der
pH wird durch die Zugabe von wässrigem
2 N NaOH (275 ml) bei einer derartigen Geschwindigkeit auf pH 8-9
eingestellt, dass die Temperatur unter 20°C beliebt. Die Phasen werden
getrennt, die wässrige
Phase wird mit CH2Cl2 (2 × 100 ml)
extrahiert. Die organische Phase wird vereinigt, mit Kochsalzlösung (2 × 100 ml)
gewaschen, getrocknet (MgSO4), filtriert
und das Filtrat wird im Vakuum zu einem dunklen Öl/festen Rückstand (40 g) konzentriert.
Das rohe Produkt wird durch Silicagelchromatographie (1200 g Silicagel
60, CH2Cl2) unter
Bildung von 29,4 g (66%) der Titelverbindung als roter Feststoff
gereinigt:
1H NMR (300 MHz, DMSO-d6) δ 9,78
(bs, 1H), 8,15 (dd, 1H, J = 6,95 Hz), 7,67 (dt, 1H, J = 7,32 Hz,
1,46 Hz), 7,26 (dd, 1H, J = 7,32, 1,10 Hz), 7,15 (dt, 1H, J = 6,95
Hz, 1,10 Hz), 3,26 (dq, 1H, J = 6,95 Hz), 1,33 (d, 6H, J = 6,95
Hz).
13C NMR (75 MHz, DMSO-d6) δ 171,85,
150,83, 139,10, 136,22, 136,00, 126,05, 121,41, 118,64, 116,09, 113,73,
33,01, 22,07(2).
IR (CHCl3) 3311, 3021,
2970, 2928, 2868, 2223, 1613, 1583, 1518, 1492, 1448, 1403, 1341
cm–1.
HRMS
(ES) M/z berechnet für
C13H12N4O2S 289,0759, gefunden 289,0744.
-
Beispiel 84
-
2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-ylaminhydrochlorid
-
Es
werden 2-Isopropyl-5-(2-nitrophenylamino)thiazol-4-carbonitril (35,1
g, 122 mmol) und IPA (525 ml) unter N2 vereinigt
und bei 60°C
zum Lösen
erhitzt. Eine Lösung
aus SnCl2 (70,0 g, 369 mmol, 3,0 Äquivalente) in
wässrigem
5 M HCl (525 ml) wird tropfenweise über 30 min zugegeben. Das Reaktionsgemisch
wird am Rückfluss
für 1 h
erhitzt (80-85°C),
bis sie gemäß HPLC (Zorbax
SB C18 25 cm, 60:40/ACN : 0,1% TFA in Wasser, 233 nm, 1,0 ml/min)
vollständig
ist. Die Reaktion wird auf 50°C
gekühlt.
Der Großteil
des Lösemittels wird
im Vakuum entfernt. Der wässrige
feste Rückstand
(188 g) wird mit IPA (500 ml) behandelt und für wenige Minuten unter Bildung
einer homogenen Aufschlämmung
auf 60-70°C
erhitzt. Die Aufschlämmung
wird auf RT gekühlt
und dann für
1-2 h auf 0-5°C.
Das Produkt wird durch Vakuumfiltration isoliert und in einem Vakuumofen
bei 60°C
unter Bildung von 45,9 g (128%) des rohen Produkts getrocknet, das
mit restlichen Zinn stark kontaminiert ist. Das rohe Produkt wird
in wässrigem
1 N HCl (2,25 l) suspendiert und am Rückfluss (95°C) für 1 h erhitzt, während der
Zeit sind die meisten Feststoffe gelöst. Es wird auf RT gekühlt, das
Produkt wird durch Vakuumfiltration isoliert, mit wässrigem
1 N HCl gewaschen und in einem Vakuumofen bei 70°C unter Bildung von 34,5 g (97%)
der Titelverbindung als gelboranger Feststoff getrocknet. Analytische
Analyse: Sn (9,0%), H2O (1,2%).
1H NMR (300 MHz, DMSO-d6) δ 10,96 (bs,
1H), 10,15 (bs, 1H), 8,94 (bs, 2H), 7,10-6,95 (m, 2H), 6,93-6,82 (m, 2H), 3,10
(dq, 1H, J = 6,95 Hz), 1,28 (s, 3H), 1,26 (s, 3H).
13C NMR (75 MHz, DMSO-d6) δ 164,42,
159,24, 158,94, 137,01, 128,58, 127,111, 125,09, 122,87, 120,14, 119,23,
32,41, 21,98(2).
IR (KBr) 3301, 3249, 2964, 1653, 1614, 1553,
1509 cm–1.
HRMS
(ES) M/z berechnet für
C13H15N4S
259,1017 (M+-Cl), gefunden 259,1010.
-
Beispiel 85
-
-
Diethylketomalonat
(50 g, 0,287 mol) in 3 A Ethanol (250 ml) wird vereinigt und N-Methylhydroxylamin-HCl
(23,9 g, 0,287 mol) wird zugegeben. Pyridin (22,7 g, 0,287 mol)
wird zugegeben und am Rückfluss (78°C) wird die
farblose homogene Lösung
unter Rühren
unter Stickstoff für
etwa 2,5 Stunden erhitzt, ehe auf Umgebungstemperatur gekühlt wird.
Es wird bei Umgebungstemperatur für etwa 48 Stunden gerührt, wobei durch
HPLC Analyse (Zorbax RX C18, 55% ACN/45% 0,1% TFA, 1 ml/min, 233
nm) aufgezeichnet wird. Das Reaktionsgemisch wird im Vakuum zu einem
nassen, wachsartigen weißen
Feststoff konzentriert und zwischen 100 ml EtOAc und 100 ml H2O aufgeteilt. Die wässrige Phase wird mit 3 × 100 ml
Portionen EtOAc extrahiert. Die EtOAc Phasen werden vereinigt, mit
100 ml Kochsazlösung
gewaschen, über
MgSO4 getrocknet, filtriert und zu einem
gelben Öl
eingedampft. Das rohe Produkt wird auf Silicagel 60 (Merck, 230-400
mesh) absorbiert und mit 4 l einer 9:1 Hexan : EtOAc Lösung als
Eluent eluiert. Der Eluent wird im Vakuum unter Bildung von 56,3
g (96,6% Ausbeute) der Titelverbindung als blaßgelbes homogenes Öl eingedampft.
1H NMR (300 MHz, CDCl3) δ 4,29 (m,
4H), 4,05 (s, 3H), 1,259 (t, 6H, J = 7,32 Hz).
13C
NMR (75 MHz, CDCl3) δ 159,9, 158,9, 143,3, 64,2,
62,3, 62,1, 13,7.
IR (CHCl3) 3029,
2986, 2944, 1744, 1605, 1329, 1301, 1264, 1237, 1103, 1043 cm–1.
UV
(ETOH ) λmax
231 nm (ε 8582)
HRMS
(ES) exakte Masse berechnet für
C8H13NO5 203,0794,
gefunden 203,0797.
Analyse berechnet für C8H13NO5: C 47,29, H
6,45, N 6,89. Gefunden: C 46,28, H 6,36, N 4,86.
-
Beispiel 86
-
1,5-Dihydro-benzo[b][1,4]diazepin-2,3,4-trion-3-(O-methyloxim)
-
Es
wird 21 Gewichtsprozent NaOEt in EtOH Lösung (164 g, 0,507 mol) in
2-B-3-Ethanol (331 ml) gelöst
und 1,2-Phenylendiamin (27,4 g, 0,253 mol) wird zugegeben. Das entstehende
Gemisch kann bei Umgebungstemperatur für etwa 30 Minuten rühren, wobei
sich während
der gesamten Zeit das gesamte feste Material unter Bildung einer
dunklen bernsteinfarbenen Lösung
löst. Es
wird 2-Methoxyiminomalonsäurediethylether
(51,5 g, 0,253 mol) zugegeben. Das entstehende Gemisch wird auf
70°C erhitzt
und unter Stickstoff für etwa
4 Stunden gerührt,
während
durch HPLC Analyse/Zorbax RX C18, 55% ACN/45% 0,1% TFA, 1 ml/min, 233
nm) aufgezeichnet wird. Das Reaktionsgemisch kann sich auf Umgebungstemperatur
abkühlen.
Der pH des Reaktionsgemisches wird von 11,6 auf 1,2 durch die Zugabe
von 700 ml wässriger
1 N HCl Lösung
eingestellt. Das Reaktionsgemisch wird im Vakuum zur Entfernung
des Ethanols konzentriert. Die entstehende bernsteinfarbene Aufschlämmung wird
durch eine Glasfritte filtriert. Der Feststoff wird gesammelt und
mit wässriger
1 N HCl gewaschen und unter Vakuum bei 50°C unter Bildung von 42 g (76%
Ausbeute) der Titelverbindung als gelber Feststoff getrocknet.
1H NMR (300 MHz, CDCl3) δ 11,1 (s,
1H), 10,9 (s, 1H), 7,22 (s, 4H), 3,86 (s, 3H).
13C
NMR (75 MHz, CDCl3) δ 161,7, 159,4, 148,1, 128,5,
127,8, 125,8, 125,7, 122,8, 122,7, 63,1.
IR (CHCl3)
3062, 2970, 2902, 1704, 1656, 1606, 1505, 1414, 1058, 1043, 754
cm–1.
UV
(ETOH) λmax
278 nm (ε 4623),
276 nm (ε 4618),
216 nm (ε 41495).
HRMS
(ES+) exakte Masse berechnet für
C10H9N3O3 242,0542, gefunden 242,0553.
Analyse
berechnet für
C10H9N3O3: C 54,79, H 4,14, N 19,17, gefunden: C
54,60, H 4,15, N 18,94.
-
Beispiel 87
-
3-Amino-1,5-dihydro-benzo[b][1,4]diazepin-2,4-dion
-
Zu
nassem 10% Pd/C (37,5 g) mit Eisessig (HOAc, 3,75 l) wird 1,5-Dihydro-benzo[b][1,4]diazepin-2,3,4-trion-3-(O-methyloxim)
(150 g, 0,684 mol) gegeben und das entstehende Gemisch wird einer
katalytischen Hydrierung unter 50 psi H2 bei
Umgebungstemperatur für
~18 Stunden unterzogen, bis sie gemäß HPLC vollständig ist.
Der Katalysator wird durch Filtration über Glasfiberpapier/Hyflo entfernt
und mit HOAc (2 l) gewaschen. Das Filtrat wird im Vakuum unter Bildung
des rohen Produkts als bernsteinfarbener wachartiger Feststoff konzentriert.
Der Feststoff wird mit Ethylacetat (EtOAc, 3 l) behandelt und unter
Rühren
für 1 Stunde auf
70°C erhitzt.
Die Aufschlämmung
kann sich auf Umgebungstemperatur abkühlen und wird dann verstärkt auf
0°C gekühlt. Das
Produkt wird durch Filtration isoliert und unter Bildung von 109,9
g (84%) der Titelverbindung getrocknet.
1H
NMR (300 MHz, DMSO-d6) δ 10,48 (bs, 2H), 7,18 (m, 4H),
3,75 (s, 1H), ~3,5 (bs, 2H).
13C NMR
(75 MHz, DMSO-d6) δ 167,9, 164,5, 129,2, 125,1,
122,2, 55,8, 55,7.
IR (KBr) 3375, 3306, 3282, 3047, 2750, 1697,
1670, 1559, 1500, 1433, 1411, 1337, 1307, 1258, 1183 cm–1.
UV
(EtOH) λmax
217 nm (ε 33772),
285 nm (ε 3210).
HRMS (ES+) exakte Masse berechnet für C9H9N3O2 192,0773,
gefunden 192,0768.
-
Beispiel 88
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)isobutyramid
-
3-Amino-1,5-dihydro-benzo[b][1,4]diazepin-2,4-dion
(10,0 g, 52,3 mmol) wird als hellbraune gefärbte Aufschlämmung in
THF (300 ml) bei 0-3°C
unter Stickstoff gerührt.
Triethylamin (8,00 ml, 57,0 mmol) wird in einer Portion zugegeben.
Isobutyrylchlorid (6,00 ml, 57,0 mmol) wird tropfenweise zugegeben,
wobei die Temperatur unter 3°C
bleibt. Die entstehende hellgelb gefärbte Aufschlämmung wird
bei 0-3°C
für 3,5
Stunden gerührt,
bis die Reaktion gemäß HPLC vollständig ist.
Es wird entionisiertes Wasser (450 ml) über 15 bis 20 Minuten zugegeben,
wobei die Gefäßtemperatur
unter 10°C
gehalten wird. Nach dem Rühren
bei 0-10°C
für 1,5 Stunden
wird das Produkt durch Filtration isoliert, mit kaltem (0-3°C) 2:1/Wasser
: THF (50 ml) dann Wasser (50 ml) gewaschen. Der Feststoff wird
in einem Vakuumofen bei 50°C
unter Bildung von 10,5 g (77%) der Titelverbindung getrocknet.
1H NMR (500 MHz, DMSO-d6) δ 10,7 (s,
2H), 8,15 (d, 1H), 7,2 (m, 4H), 4,7 (d, 1H), 2,75 (m, 1H), 1,0 (s,
6H).
-
Beispiel 89
-
2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
-
N-(2,4-Dioxo-2,3,4,5-tetrahydro-1H-benzo[b][1,4]diazepin-3-yl)isobutyramid
(10,0 g, 38,3 mmol) und Lawesson's
Reagenz (23,2 g, 57,4 mmol) werden vereinigt und als eine Aufschlämmung in
1,2-Dichlorethan (DCE,
600 ml) unter Stickstoff bei 80°C
für 5 Stunden
gerührt,
bis die Reaktion gemäß HPLC vollständig ist. Das
Reaktionsgemisch kann sich schrittweise über Nacht auf Umgebungstemperatur
abkühlen.
Das Produkt wird durch Filtration isoliert, mit DCE (4 × 40 ml)
gewaschen und dann in einem Vakuumofen bei 50-60°C über Nacht unter Bildung von
7,96 g (76%) der Titelverbindung getrocknet (Gewicht und Ausbeute
um 7,5 Gewichtsprozent der übrigen
DCE mittels Protonen NMR korrigiert).
1H
NMR (300 MHz, DMSO-d6) δ 10,97 (bs, 1H), 8,93 (bs, 1H),
7,06-6,90 (m, 3H), 6,81-6,74 (m, 1H), 3,11-2,97 (m, 1H), 1,24 (s,
3H), 1,22 (s, 3H).
13C NMR (75 MHz,
DMSO-d6) δ 192,3,
163,0, 150,1, 141,6, 135,0, 131,1, 126,0, 124,1, 123,0, 119,2, 32,6, 22,3.
IR
(KBr) 3198, 3147, 2965, 1600, 1534, 1505, 1480, 1397, 1306, 1095,
1049 cm–1.
HRMS
(ES+) exakte Masse berechnet für
C13H13N3S2 276,0629, gefunden 276,0613.
-
Beispiel 90
-
2-(4-Fluor-2-nitrophenylamino)benzo[b]thiophen-3-carbonitril
-
2-Aminobenzo[b]thiophen-3-carbonitril
(3,75 g, 21,5 mmol), 1,4-Difluor-2-nitrobenzol (2,33 ml, 21,5 mmol)
und Lithiumhydroxid (1,03 g, 43,0 mmol) werden in wasserfreiem Dimethylsulfoxid
(50 ml) vereinigt und bei 55°C
für etwa
5 Stunden erhitzt. Es wird auf Umgebungstemperatur gekühlt, in
einen Becher mit Eis/entionisiertem Wasser (200 ml) gefüllt und
für 30
Minuten gerührt.
Das ausgefallene Material wird durch Unterdruckfiltration isoliert,
der Feststoff wird mit Dichlormethan gewaschen und unter verringertem
Druck unter Bildung der Titelverbindung (0,618 g) getrocknet. Entionisiertes
Wasser und Ethylacetat werden zu dem Filtrat gegeben und dann wird
die organische Phase abgetrennt. Die wässrige Phase wird mit Ethylacetat
mehrmals und dann mit Dichlormethan mehrmals extrahiert. Die organischen
Phasen werden dann mit entionisiertem Wasser (3 ×) gewaschen. Die Waschschritte
werden vereinigt und dann mit Ethylacetat extrahiert. Die organischen
Phasen werden mit einer gesättigten
Lösung
aus Natriumchlorid gewaschen und dann getrocknet (Natriumsulfat),
filtriert und die organischen Bestandteile werden unter verringertem
Druck zu einem dunklen Feststoff konzentriert. Nach der Behandlung
mit Methanol wird der entstehende Feststoff durch Unterdruckfiltration isoliert,
mit Methanol gewaschen und bei Umgebungstemperatur unter verringertem
Druck unter Bildung der Titelverbindung (4,422 g) getrocknet. Gesamter
Feststoff: 5,04 g (74,8%). Massenspektrum (ES+, m/e): 314 (M+1)
NMR
(1H 300 MHz, CDCl3) δ (ppm): 9,96
(s, 1H), 8,06-7,98 (m, 1H), 7,85-7,77 (m, 1H), 7,77-7,69 (m, 1H), 7,68-7,59
(m, 1H), 7,56-7,47 (m, 1H), 7,45-7,34 (m, 2H).
-
Beispiel 91
-
8-Fluor-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrochlorid
-
2-(4-Fluor-2-nitrophenylamino)benzo[b]thiophen-3-carbonitril
(5,04 g, 16,1 mmol) und Zinn(II)chlorid (9,15 g, 48,3 mmol) in absolutem
Ethanol und 5 N HCl (60 ml jeweils) werden vereinigt und die Suspension wird
für 4 Stunden
am Rückfluss
erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
der Reaktionsfeststoff wird durch Unterdruckfiltration gesammelt,
mit kaltem Ethanol gewaschen und bei Umgebungstemperatur unter verringertem
Druck unter Bildung eines Feststoff (6,18 g) getrocknet. Der Feststoff
wird in Methanol und dann entionisiertem Wasser aufgenommen und
der Feststoff wird durch Unterdruckfiltration isoliert. Der Feststoff
wird über
Nacht unter verringertem Druck unter Bildung der Titelverbindung
(5,00 g, 97,3%) getrocknet.
Massenspektrum (APCI+, m/e): 284
(M+1-HCl).
NMR (1H, 300 MHz, DMSO-d6) δ (ppm):
11,65 (s, 1H), 9,98 (s, 1H), 9,19-9,00 (m, 2H), 7,90-7,82 (m, 1H), 7,72-7,63
(m, 1H), 7,46-7,36 (m, 1H), 7,33-7,24 (m, 1H), 7,11-6,97 (m, 2H),
6,96-6,88 (m, 1H).
-
Beispiel 92
-
2-(2-Nitro-4-trifluormethylphenylaminobenzo[b]thiophen-3-carbonitril
-
2-Amino-benzo[b]thiophen-3-carbonitril
(3,48 g, 20,0 mmol), 1-Fluor-2-nitro-4-trifluormethylbenzol (4,31
g, 20,0 mmol) und Lithiumhydroxid (0,958 g, 40,0 mmol) in wasserfreiem
Dimethylsulfoxid (50 ml) werden vereinigt und bei 55°C für 2 Stunden
erhitzt. Es wird auf Umgebungstemperatur gekühlt in einen Becher mit Eisentionisiertem
Wasser (50 ml) gefüllt
und für
30 Minuten gerührt.
Entionisiertes Wasser und Dichlormethan werden zugegeben und dann
wird die organische Phase abgetrennt. Die wässrige Phase wird mit Dichlormethan
mehrmals extrahiert. Dann werden die organischen Phasen mit entionisiertem
Wasser (3 ×)
gewaschen. Die Waschschritte werden vereinigt und dann mit Dichlormethan
rückextrahiert.
Die organischen Waschschritte werden mit einer gesättigten
Lösung
aus Natriumchlorid gewaschen und die vereinigten Waschschritte werden
mit Dichlormethan rückextrahiert.
Es wird getrocknet (Natriumsulfat), filtriert und die organischen
Bestandteile werden unter verringertem Druck zu einem Rückstand
konzentriert. Nach der Behandlung des Rückstands mit Methanol wird
der entstehende Feststoff durch Unterdruckfiltration isoliert, mit
Methanol gewaschen und bei Umgebungstemperatur unter verringertem
Druck unter Bildung der Titelverbindung (4,61 g) getrocknet. Das
Filtrat wird unter verringertem Druck konzentriert und durch Blitzchromatographie
unter Elution mit einem Gradienten aus Ethylacetat : Dichlormethan
: Hexan (0,25: 5 : 5, 0-100% in Hexan) unter Bildung der Titelverbindung
(0,53 g) gereinigt.
Gesamtausbeute: 5,14 g (70,8%)
NMR
(1H, 300 MHz, CDCl3) δ (ppm): 10,26
(s, 1H), 8,63-8,56 (m, 1H), 7,93-7,85 (m, 1H), 7,84-7,75 (m, 2H), 7,64-7,45
(m, 3H).
-
Beispiel 93
-
8-Trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrochlorid
-
2-(2-Nitro-4-trifluormethylphenylamino)benzo[b]thiophen-3-carbonitril
(5,14 g, 14,1 mmol) und Zinn(II)chlorid (8,05 g, 42,4 mmol) in absolutem
Ethanol und 5 N HCl (60 ml jeweils) werden vereinigt und die Suspension
wird für
1,5 Stunden am Rückfluss
erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt. Der
Reaktionsfeststoff wird durch Unterdruckfiltration gesammelt, mit
kaltem Ethanol gewaschen und bei 40°C unter verringertem Druck unter
Bildung der Titelverbindung (5,083 g) getrocknet. Ein zweites Kristallisat
des Materials aus dem anfänglichen
Filtrat wird isoliert und bei 40°C
unter verringertem Druck unter Bildung der Titelverbindung (0,482
g) getrocknet.
Gesamtausbeute: 5,20 g (94,6%).
Massenspektrum
(APCI+, m/e) 334 (M+1-HCl).
NMR (1H,
300 MHz, DMSO-d6) δ (ppm): 11,71, (s, 1H), 10,32
(s, 1H), 9,32-9,10 (m, 2H), 7,94-7,85 (m, 1H), 7,74-7,65 (m, 1H),
7,60-7,51 (m, 1H), 7,49-7,40 (m, 1H), 7,39-7,27 (m, 2H), 7,23-7,14
(m, 1H).
-
Beispiel 94
-
3-Brom-2-nitro-benzo[b]thiophen
-
Es
wird tropfenweise rauchende Salpetersäure (90%, 8,6 ml, 183 mmol)
zu einem Gemisch aus 3-Brombenzo[b]thiophen (39 g, 183 mmol) in
TFA (100 ml) und Dichlormethan (400 ml) bei 0°C gegeben. Die Reaktion wird
grünlich
und dann fällt
ein gelber Niederschlag aus. Zu diesem Reaktionsgemisch wird Dichlormethan
(200 ml) gegeben und die Reaktion wird bei 0°C für 30 min gerührt. Dann
wird die Reaktion in Eiswasser (2 l) gegossen. Sie wird mit Dichlormethan
(3 × 500
ml) extrahiert und die organische Phase wird über MgSO4 getrocknet.
Eine Eindampfung ergibt einen gelben Feststoff. Der entstehende
gelbe Feststoff wird mit Diethylether unter Bildung eines gelben
Feststoffs behandelt. (Gesamt: 34,8 g, 73%).
Massenspektrum
(m/e): 259 (M+1)
1H NMR (300 MHz, DMSO-d6) δ ppm:
7,70 (tt, 2H), 8,04 (d, 1H), 8,17 (d, 1H).
13C
NMR (75 MHz, DMSO-d6) δ ppm: 112,5, 124,8, 126,9, 127,9,
131,3, 137,0, 137,2, 166,1.
-
Beispiel 95
-
2-Nitrobenzo[b]thiophen-3-carbonitril
-
3-Brom-2-nitrobenzo[b]thiophen
(33,0 g, 127,4 mmol) und Kupfercyanid (17,1 g, 191,1 mmol) in DMF (150
ml) werden vereinigt und für
drei Stunden auf 120°C
erhitzt. Die Reaktion wird auf RT gekühlt, auf Eis gegossen und dann
filtriert. Der Filterkuchen wird mit Dichlormethan gewaschen. Die
organische Phase wird abgetrennt und über MgSO4 getrocknet,
und unter Bildung einer DMF Lösung
eingedampft. Wasser (400 ml) wird zugegeben und der gelbe Feststoff
fällt aus.
Nach der Filtration wird ein bräunlicher
Feststoff (23,5 g, 90%) erhalten.
Massenspektrum (m/e): 205
(M+1).
1H NMR (300 MHz, DMSO-d6) δ 7,78
(m, 2H), 8,04 (d, 1H), 8,29 (d, 1H).
13C
NMR (75 MHz, DMSO-d6) δ ppm: 105,9, 112,1, 125,0, 125,2,
128,8, 131,2, 135,9, 137,8, 158,0.
-
Beispiel 96
-
2-Aminobenzo[b]thiophen-3-carbonitril
-
In
einem 500 ml Schlenk-Kolben werden 2-Nitrobenzo[b]thiophen-3-carbonitril
(5,8 g, 28,4 mmol) und Pd/C (3,0 g, 10 G/G, 2,84 mmol) in 1,2-Dichlorethan
(120 ml) vereinigt und das Reaktionsgemisch wird mit einem Wasserstoffballon
befüllt.
Nach dem Rühren über Nacht,
wird der Wasserstoff abgelassen, der Katalysator wird durch Filtration
entfernt und der Katalysator wird durch 1,2-Dichlorethan mehrmals
gewaschen. Dann wird zu einem Rückstand
runter konzentriert, der durch Blitzchromatographie auf Silicagel,
Gradient (100% Hexan bis 100% Hexan : CH2Cl2 : EtOAc = 50:50:2,5) unter Bildung eines
bräunlichen
Feststoffs mit 3,6 g der Titelverbindung gereinigt wird. (Ausbeute:
73%).
Massenspektrum: ES(+) (m/e): 175 (M+1).
1H NMR (300 MHz, DMSO-d6,
ppm: δ 7,81
(br, 2H), 7,65-7,62 (m, 1H), 7,28-7,24 (m, 2H), 7,11-7,01 (m, 1H).
-
Beispiel 97
-
2-(5-Fluor-2-nitrophenylamino)benzo[b]thiophen-3-carbonitril
-
2-Aminobenzo[b]thiophen-3-carbonitril
(2,25 g, 12,5 mmol), 2,4-Difluornitrobenzol (1,99 g, 12,5 mmol) und
Lithiumhydroxid (0,58 g, 25 mmol) werden in 30 ml DMSO vereinigt
und auf 50°C
erhitzt. Nach 4 Stunden wird die Reaktion auf RT gekühlt und
auf Eis gegossen, für
30 min gerührt,
mit CH2Cl2 extrahiert,
das vereinigte Lösemittel
wird mit Wasser und Kochsalzlösung
gewaschen und über
Na2SO4 getrocknet.
Dann wird zu einem Rückstand
runterkonzentriert, mit MeOH behandelt und der orange Niederschlag
wird durch Unterdruckfiltration unter Bildung der Titelverbindung
2,15 g gesammelt. Das Filtrat wird konzentriert und durch Blitzchromatographie
unter Bildung von 0,22 g eines orangen Feststoffs gereinigt. Gesamtausbeute:
2,35 g, Ausbeute 61%.
Massenspektrum: ES(+) (m/e): 314 (M+1)
1H NMR (300 MHz, DMSO-d6) δ: 10,35 (br,
1H), 8,31-8,25 (m, 1H), 8,00-7,96 (m, 1H), 7,68-7,65 (m, 1H), 7,53-7,37
(m, 3H), 7,13-7,07 (m, 1H).
13C NMR
(75 MHz, DMSO-d6) δ ppm: 165,5 (d, J = 254,5 Hz),
156,2, 139,8, (d, J = 12,5 Hz), 135,9, 134,8, 132,2, 129,3 (d, J
= 11,7 Hz), 126,3 125,2, 123,0, 120,4, 113,5, 110,4 (d, J = 24,0
Hz), 107,6 (d, J = 27,4 Hz), 92,9.
-
Beispiel 98
-
9-Fluor-11H-12-thia-6,11-diazadibenzo[a,f]azulen-5-ylaminhydrochlorid
-
2-(5-Fluor-2-nitrophenylamino)benzo[b]thiophen-3-carbonitril
(2,15 g, 6,87 mmol) und Zinn(II)chloriddihydrat (4,65 g, 20,6 mmol)
werden in einem gemischten Lösemittel
aus EtOH (25 ml) und 5,0 N HCl (25 ml) vereinigt, die Suspension
wird für
3 Stunden am Rückfluss
erhitzt und auf RT gekühlt.
Eine Unterdruckfiltration ergibt die Titelverbindung mit 1,73 g
(Ausbeute 78%) als gelber Feststoff. Massenspektrum: ACPI (m/e):
284 (M+1-HCl)
1H NMR (300 MHz, DMSO-d6) δ 11,46
(br, 1H), 10,02 (br, 1H), 9,02 (br, 2H), 7,90-7,87 (m, 1H), 7,71-7,68 (m, 1H), 7,46-7,40
(m, 1H), 7,33-7,28 (m, 1H), 7,12-6,94 (m, 2H), 6,85-6,81 (m, 1H).
-
Beispiel 99
-
2-Amino-5-tert-butylthiophen-3-carbonitril
-
Zu
einer Lösung
aus 3,3-Dimethylbutyraldehyd (20 g, 200 mmol) in EtOH (40 ml) wird
tropfenweise ein Gemisch aus Schwefel (6,4 g, 200 mmol), Malononitril
(13,2 g, 200 mmol) und Triethylamin (14,3 ml, 100 mmol) in EtOH
(400 ml) bei 0°C
gegeben. Das Gemisch wird bei Raumtemperatur für 20 Minuten nachdem die Zugabe
vollständig
ist, gerührt
und dann am Rückfluss
für 2 Stunden
erhitzt. Es wird gekühlt
und zu einer Paste konzentriert. Diethylether (200 ml) und 2 N HCl
(200 ml) werden zugegeben. Die organische Phase wird wieder mit
2 N HCl gewaschen, getrocknet (Na2SO4) und konzentriert. Der Rückstand
wird mittels Säulenchromatographie
unter Elution mit Methylenchlorid unter Bildung der Titelverbindung
als hellbraune Kristalle (16,9 g, 47%) gereinigt.
1H
NMR (CDCl3): δ 1,27 (s, 9H), 4,60 (bs, 2H),
6,36 (s, 1H).
-
Beispiel 100
-
2-Amino-5-isopropyl-thiophen-3-carbonitril
-
Isovaleraldehyd
wird an Stelle von 3,3-Dimethylbutyraldehyd eingesetzt und das Verfahren
von Beispiel 99 wird unter Bildung der Titelverbindung als brauner
Feststoff verwendet.
1H NMR (CDCl3) δ 1,24
(d, 6H), 2,93 (Septet, 1H), 6,37 (s, 1H).
-
Beispiel 101
-
2-Amino-5-cyclopentylthiophen-3-carbonitril
-
2-Cyclopentylacetaldehyd
wird an Stelle von 3,3-Dimethylbutyraldehyd eingesetzt und DMF wird
an Stelle von EtOH eingesetzt und das Verfahren von Beispiel 99
wird unter Bildung der Titelverbindung (16,1 g, 57%) als gelber
Feststoff verwendet.
1H NMR (CDCl3) δ 1,48-1,58
(m, 2H), 1,61-1,69 (m, 2H), 1,72-1,78 (m, 2H), 1,98-2,07 (m, 2H),
3,01 (m, 1H), 4,58 (bs, 2H), 6,38 (s, 1H).
-
Beispiel 102
-
5-tert-Butyl-2-(2-nitrophenylamino)thiophen-3-carbonitril
-
Eine
Lösung
aus 2-Amino-5-tert-butyl-thiophen-3-carbonitril (16,9 g, 94 mmol)
in THF (50 ml) wird zu einem Gemisch aus gewaschenem NaH (aus 6,76
g an 60% Mineralöldispersion)
in THF (200 ml) in einem Wasserbad bei Raumtemperatur gegeben. Es
wird für
15 Minuten gerührt
und dann wird eine Lösung
aus 2-Fluornitrobenzol (13,2 g, 94 mmol) in THF (50 ml) tropfenweise
zugegeben. Es wird über
Nacht gerührt.
Das purpurfarbene Reaktionsgemisch wird auf 6 N HCl (400 ml) gegossen.
Das Gemisch wird mit Diethylether (400 ml) extrahiert. Die Etherphase
wird mit 2 N HCl (400 ml), Kochsalzlösung (250 ml) gewaschen, getrocknet (Na2SO4) und unter Bildung
eines Gemisches aus Kristallen in einem dunklen öligen Rückstand konzentriert. Die Kristalle
werden mit Hexan behandelt und unter Bildung der Titelverbindung
als rotes Pulver (21,2 g, 75%) filtriert. Smp 85-90°C.
1H NMR (CDCl3) δ 1,39 (s,
9H), 6,81 (s, 1H), 6,97 (t, 1H), 7,23 (d, 1H), 7,53 (t, 1H), 8,25
(d, 1H), 9,66 (bs, 1H).
-
Gemäß dem Verfahren
von Beispiel 102 werden die folgenden Verbindungen hergestellt und
als freie Base isoliert.
| Nr | R2 | Daten |
| 103 | i-Pr | 1H NMR (CDCl3) δ 1,35 (d,
6H), 3,13 (Septett, 1H), 6,80 (s, 1H), 6,96 (t, 1H), 7,22 (d, 1H),
7,54 (t, 1H), 8,24 (d, 1H), 9,65 (s, 1H). |
| 104 | c-Pentyl | 1H NMR (CDCl3) δ 1,56-1,84
(m, 6H), 2,11-2,19 (m, 2H), 3,18 (Pentett, 1H), 6,80 (s, 1H), 6,96
(t, 1H), 7,21 (d, 1H), 7,53 (t, 1H), 8,25 (d, 1H), 9,64 (s, 1H) |
-
Beispiel 105
-
2-tert-Butyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
5-tert-Butyl-2-(2-nitrophenylamino)thiophen-3-carbonitril
(21,2 g, 70 mmol) wird zu einer Lösung aus Zinn(II)chloriddihydrat
(46,1 g, 209 mmol) in konz. HCl (200 ml) und Ethanol (600 ml) gegeben.
Das Gemisch wird für
2 Stunden am Rückfluss
erhitzt. Die Lösung
wird auf 200 ml konzentriert und zu Wasser (1 l) gegeben. Es wird
filtriert und mit Wasser und dann Hexan unter Bildung der Titelverbindung
als oranges Pulver (19,4 g) gewaschen.
1H
NMR (DMSO-d6) δ 1,27 (s, 9H), 6,86 (d, 1H),
6,89 (s, 1H), 6,95 (d, 1H), 7,03 (t, 1H), 7,11 (t, 1H), 8,69 (s, 1H),
9,11 (s, 1H), 9,52 (s, 1H), 10,88 (s, 1H).
MS (APCI) m/z (relative
Intensität)
272 (100).
-
Gemäß dem Verfahren
von Beispiel 105 werden die folgenden Verbindungen hergestellt und
als freie Base isoliert:
| Nr | R | Daten |
| 106 | i-Pr | 1H NMR (DMSO-d6) δ 1,16 8d, 6H),
2,88 (Septet, 1H), 6,82 (s, 1H), 6,83 (d, 1H), 6,91 (d, 1H), 6,99
(t, 1H), 7,07 (t, 1H), 8,71 (s, 1H), 9,09 (s, 1H), 9,54 (s, 1H), 10,94
(s, 1H). |
| 107 | Cyclopentyl | 1H NMR (DMSO-d6) δ 1,42-1,70 (m,
6H), 1,92-2,00 (m, 2H), 2,99 (Pentet, 1H), 6,81 (s, 1H), 6,82 (d, 1H),
6,91 (d, 1H), 6,99 (t, 1H), 7,07 (t, 1H), 8,63 (bs, 1H), 9,05 (bs,
1H), 9,50 (bs, 1H), 10,79 (bs, 1H). |
-
Beispiel 108
-
2-(4,5-Difluor-2-nitrophenylamino)benzo[b]thiophen-3-carbonitril
-
2-Amino-benzo[b]thiophen-3-carbonitril
(3,93 g, 22,6 mmol) und 2,4,5-Trifluornitrobenzol (4,0 g, 22,6 mmol)
werden in 30 ml THF vereinigt, Natriumhydrid (3,54 g, 88,5 mmol)
wird portionsweise bei 0-5°C zugegeben
und nach der Zugabe wird die Reaktion über Nacht bei RT gerührt. Die
Reaktion wird auf Eiswasser (200 ml) gegossen, mit Dichlormethan
(3 × 50
ml) extrahiert, die vereinigte organische Phase wird mit Wasser und
Kochsalzlösung
gewaschen und über
Na2SO4 getrocknet.
Es wird zu einem Rückstand
einkonzentriert, durch Chromatographie gereinigt, Gradient Hexan
bis Hexan : CH2Cl2 :
EtOAc = 5:5:0,5, und es wird ein rot-oranger Feststoff mit 1,71
g als Titelverbindung erhalten.
1H
NMR (300 MHz, DMSO-d6) δ 10,34 (br, 1H), 8,41-8,35 (m,
1H), 7,96-7,94 (m, 1H), 7,85-7,79 (m, 1H), 7,64-7,61 (m, 1H), 7,52-7,49
(m, 1H), 7,42-7,36 (m, 1H).
-
Beispiel 109
-
8,9-Difluor-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrochlorid
-
2-(4,5-Difluor-2-nitrophenylamino)benzo[b]thiophen-3-carbonitril
(2,03 g, 6,13 mmol) und Zinn(II)chlorid (3,49 g, 18,4 mmol) werden
in einem gemischten Lösemittel
aus EtOH (20 ml) und 5,0 N HCl (20 ml) vereinigt, die Suspension
wird am Rückfluss über Nacht
erhitzt, und auf RT gekühlt.
Die Titelverbindung wird mit 2,05 g als gelber Feststoff durch Unterdruckfiltration
erhalten.
1H NMR (DMSO-d6) δ 11,56 (br,
1H), 10,06 (br, 1H), 9,14 (br, 2H), 7,96-7,85 (m, 1H), 7,71-7,64
(m, 1H), 7,49-7,42 (m, 1H), 7,34-7,29 (m, 1H), 7,22-7,16 (m, 1H),
7,11-7,04 (m, 2H).
-
Beispiel 110
-
2-(4-Chlor-2-nitrophenylamino)thiophen-3-carbonitril
-
1,4-Dichlor-2-nitrobenzol
(3,87 g, 20,13 mmol), 2-Aminothiophen-3-carbonitril (2,50 g, 20,13
mmol) und DMSO (25,0 ml) werden vereinigt. Lithiumhydroxidmonohydrat
(1,69 g, 40,27 mmol) wird auf einmal zugegeben und dann wird das
Gemisch bei Umgebungstemperatur für 24 Stunden gerührt. Das
Gemisch wird auf Eischips gegossen und für 1,5 Stunden gerührt. Der
entstehende orange Niederschlag wird durch Vakuumfiltration entfernt
und dann unter Vakuum unter Bildung von 4,85 g (86%) der Titelverbindung
getrocknet: Massenspektrum (Ionenspray): m/z = 279,1 (M+1).
-
Beispiel 111
-
7-Chlor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(4-Chlor-2-nitrophenylamino)thiophen-3-carbonitril
(4,85 g, 17,35 mmol) wird in Ethanol (40,0 ml) suspendiert. Zinn(II)chloriddihydrat
(11,75 g, 52,06 mmol) wird in 5 N HCl (40,0 ml) gelöst und dann
wird es zu der Suspension gegeben. Die Reaktion wird am Rückfluss
für 48
Stunden erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
dann in einem Kühlschrank
für 2 Stunden
gekühlt.
Der entstehende Niederschlag wird durch Vakuumfiltration gesammelt
und dann unter Vakuum unter Bildung von 3,53 g (71%) der Titelverbindung
getrocknet: Massenspektrum (Ionenspray): m/z = 250,0 (M+1).
-
Beispiel 112
-
2-(5-Fluor-2-nitrophenylamino)thiophen-3-carbonitril
-
2,4-Difluor-1-nitrobenzol
(5,24 g, 32,94 mmol), 2-Aminothiophen-3-carbonitril (4,09 g, 32,94
mmol) und DMSO (30,0 ml) werden vereinigt. Lithiumhydroxidmonohydrat
(2,76 g, 65,88 mmol) wird auf einmal zugegeben und dann wird das
Gemisch bei 55°C
für 22
Stunden gerührt.
Das Gemisch wird auf Umgebungstemperatur gekühlt und dann auf Eischips gegossen.
Es wird mit Ethylacetat extrahiert und dann gewaschen (Kochsalzlösung), getrocknet
(Natriumsulfat) und die organische Phase wird zum Rückstand
eingedampft. Der Rückstand
wird auf Silicagel mittels Hexan/Dichlormethan (35:65) unter Bildung
von 3,89 g (45%) der Titelverbindung als roter Feststoff gereinigt:
Massenspektrum (Ionenspray): m/z = 262,0 (M-1).
-
Beispiel 113
-
6-Fluor-4H-3-thia-4,9-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(5-Fluor-2-nitrophenylamino)thiophen-3-carbonitril
(3,89 g, 14,78 mmol) wird in Ethanol (40,0 ml) suspendiert. Zinn(II)chloriddihydrat
(10,00 g, 44,33 mmol) wird in 5 N HCl (40,0 ml) gelöst und dann
zu der Suspension gegeben. Die Reaktion wird am Rückfluss
für 2,5
Stunden erhitzt und dann auf Umgebungstemperatur gekühlt. Der
entstehende Niederschlag wird durch Vakuumfiltration gesammelt und dann
unter Vakuum unter Bildung von 3,32 g (83%) der Titelverbindung
getrocknet: Massenspektrum (Ionenspray): m/z = 234,1 (M+1).
-
Beispiel 114
-
2-(2-Nitrophenylaminho)benzo[b]thiophen-3-carbonitril
-
2-Aminobenzo[b]thiophen-3-carbonitril
(3,56 g, 20,5 mmol), 2-Fluornitrobenzol (2,88 g, 20,5 mmol) und
Lithiumhydroxid (0,96 g, 41,0 mmol) werden in 50 ml DMSO vereinigt
und auf 50°C
erhitzt. Nach dem Erhitzen über
Nacht wird die Reaktion auf RT gekühlt und auf Eis gegossen, für 30 min
gerührt,
mit CH2Cl2 extrahiert,
das vereinigte Lösemittel
wird mit Wasser und Kochsalzlösung
gewaschen und über
Na2SO4 getrocknet.
Dann wird auf einen Rückstand
einkonzentriert, mit MeOH behandelt und der Niederschlag wird durch
Unterdruckfiltration gesammelt. Das Filtrat wird konzentriert und
durch Blitzchromatographie unter Bildung von 5,0 g, 83% Ausbeute,
gereinigt. Massenspektrum: ES(+) (m/e): 296,0 (M+1).
1H NMR (400 MHz, DMSO-d6): δ 10,27 (s,
1H), 8,13 (dd, 1H, J = 1,7 Hz, J = 8,3 Hz), 7,91-7,89 (m, 1H), 7,76-7,72
(m, 1H), 7,64-7,57 (m, 2H), 7,48-7,44 (m, 1H), 7,36-7,32 (m, 2H).
-
Beispiel 115
-
11H-12-Thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrochlorid
-
2-(2-Nitrophenylamino)benzo[b]thiophen-3-carbonitril
(5,0 g, 17,0 mmol) und Zinn(II)chlorid (9,65 g, 51,0 mmol) werden
in einem gemischten Lösemittel
aus EtOH (50 ml) und 5,0 N HCl (50 ml) vereinigt und die Suspension
wird für
3 Stunden am Rückfluss
erhitzt und auf RT gekühlt.
Eine Unterdruckfiltration ergibt die Titelverbindung mit 4,65 g
(Ausbeute 91%) als gelben Feststoff durch Massenspektrum: ACPI (m/e):
266,0 (M+1-HCl)
1H NMR (300 MHz, DMSO-d6): δ 11,7
(br, 1H), 10,00 (br, 1H), 9,10 (br, 2H), 7,90-7,85 (m, 1H), 7,72-7,65 (m, 1H), 7,48-7,38
(m, 1H), 7,35-7,28 (m, 1H), 7,22-6,98 (m, 4H).
-
Beispiel 116
-
2-(4,5-Difluor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
-
Natriumhydrid,
60% Dispersion in Mineralöl
(55,0 g × 60%
= 33,0 g, 1,38 mol) wird zu THF (950 ml) unter Rühren bei 0-5°C unter Stickstoff
gegeben. Eine Lösung
aus 2-Amino-5-methyl-thiophen-3-carbonitril (95,0
g, 0,687 mol) und 1,2,4-Trifluor-5-nitrobenzol (121,7 g, 0,687 mol)
in THF (1325 ml) wird tropfenweise über ~1 Stunde zugegeben, wobei
die Temperatur unter 10°C
gehalten wird. Die Reaktion kann sich auf Umgebungstemperatur erwärmen und
wird für
8-20 Stunden gerührt.
Dann wird auf 10-15°C gekühlt und
wässrige 1
N HCl (550 ml) wird tropfenweise über 10-15 Minuten zugegeben,
um den pH auf 7,0-7,2 einzustellen, wobei die Temperatur unter 15°C gehalten
wird. Der Großteil
der organischen Portion wird unter verringertem Druck entfernt und
dann filtriert und mit Wasser gewaschen. Es wird bei 50-60°C unter Bildung
des rohen Produkts getrocknet. Es wird durch Blitzchromatographie
unter Elution mit 1:1Methylenchlorid : Heptan unter Bildung von
89,1 g (44%) der Titelverbindung gereinigt.
1H
NMR (400 MHz, DMSO-d6) δ 9,77 (bs, 1H), 8,33 (m, 1H),
7,09 (s, 1H), 7,07 (m, 1H), 2,47 (s, 3H).
HRMS (ES) exakte
Masse M+H berechnet für
C12H7F2N3O2S 318,0125, gefunden
318,0133.
-
Beispiel 117
-
6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(4,5-Difluor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
(94,0 g, 318 mmol) und Zinn(II)chloriddihydrat (225 g, 997 mmol)
werden zu Ethanol (1400 ml) und wässrigem 6 N HCl (1400 ml) unter
Rühren unter
Stickstoff gegeben. Es wird für
3 Stunden auf mildem Rückfluss
(86-87°C)
erhitzt und kann dann auf Umgebungstemperatur abkühlen. Es
wird filtriert, mit Wasser (3 × 250
ml) gewaschen und bei 50-60°C
unter Bildung von 95,7 g (100%) der Titelverbindung getrocknet.
1H NMR (400 MHz, DMSO-d6) δ 11,41 (bs,
1H), 9,84 (bs, 1H), 9,33 (bs, 1H), 8,93 (bs, 1H), 7,00 (m, 2H),
6,85 (s, 1H), 2,28 (s, 3H).
HRMS (ES) exakte Masse M+H berechnet
für C12H9F2N3S (freie Base) 266,0563, gefunden 266,0555.
-
Beispiel 118
-
2-(4-Chlor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
-
In
einem Dreihalsrundbodenkolben der 500 ml fasst, werden 2-Amino-5-methyl-thiophen-3-carbonitril (124,11
g, 0,898 mol, 1 Äquivalent),
1,4-Dichlor-2-nitrobenzol (174,15 g, 0,907 mol, 1,01 Äquivalente)
und DMSO (1,6 l, 13 Volumina) für
15 min gerührt
(dunkle Lösung).
In einer Portion wird LiOH × H2O (75,37 g, 1,796 mol, 2 Äquivalente)
zugegeben. Es beginnt sich eine Exothermie von 20,4°C bis 25,5°C über 10 min
zu entwickeln. Nach 30 Minuten erreicht die Temperatur 28,1°C und nach
1 Stunde 30,4°C.
Leitungswasser wird in das Bad um den Kolben zur Kontrolle der Exothermie
gegeben. Nach 23 h wird durch HPLC überprüft, die 81,2% des gewünschten
Produkts anzeigt. Es wird bei Umgebungs temperatur für eine weitere
Reaktionsumwandlung weitergerührt.
Nach 39 Stunden wird durch HPLC überprüft, die
83,5% des einen Produkts anzeigt. Es wird insgesamt für 40 Stunden
gerührt.
Es wird mit einem Eiswasserbad auf 17°C gekühlt. Die pH Einstellung wird durch
langsame Zugabe von 1 N HCl (1020 ml) zur Einstellung von einem
pH = 7,1 begonnen. Ein Halten der Exothermie wird bei 15-22°C während der
Zugabe erreicht. Eine Aufarbeitung schließt mit ein: Gießen des
Reaktionsgemisches in einen 22 l Bodenauslasskolben, Waschen mit
CH2Cl2 (5,6 l) und
5% LiCl Lösung
(3,6 l), Rühren
für etwa
10 min und ein Zulassen der Phasentrennung. Die untere Phase kann
dunkler als die obere Phase sein. Die wässrige Phase wird mit CH2Cl2 (1,5 l) rückextrahiert.
Die organischen Phasen werden vereinigt und mit einer 5% LiCl Lösung (3 × 1,3 l)
gewaschen. Die organische Phase wird mit Na2SO4 (800 g) getrocknet, wobei für 1 Stunde
gerührt
wird, dann wird filtriert und zur Trockne eingedampft. Gewicht:
376 g. Die vereinigten wässrigen
Phasen werden mit CH2Cl2 (0,5
l) rückextrahiert.
Die wässrige
Phase wird durch HPLC mit ~2 ml einer wässrigen Phase in 15 ml eines
Eluenten als Produkt in der wässrigen
Phase überprüft. Die organische
Phase wird mit Kochsalzlösung
(2 × 500
ml) und schließlich
mit 5% LiCl (500 ml) gewaschen. Über das
Wochenende wird mit NaSO4 (80 g) getrocknet.
Es wird filtriert und zu einer anderen organischen Phase gegeben.
Zur Chromatographie wird das rohe Material in Methylenchlorid (950
g) gelöst
und auf einem Silicagel 60 Bett (2,8 kg mit Heptan vorher naß gemacht)
auf einem Glastrichter gegeben. Dasselbe Produkt kann oben auf dem
Silicakuchen ausfallen. Der Kolben wird gewaschen und auf den Kuchen
oben mit CH2Cl2 (0,3 l)
gegeben bis der gesamte Niederschlag gelöst und auf der Säule ist.
Es wird mit Heptan (~16 l), dann 25% CH2Cl2 in Heptan (~8 l), dann 37,5% CH2Cl2 in Heptan (~8
l) gefolgt von 50% CH2Cl2 in
Heptan und 100% CH2Cl2 eluiert.
Die ersten Fraktionen können
kein Produkt enthalten. Die gemischten Fraktionen die das Produkt
enthalten werden identifiziert und getrennt einrotiert, bis im wesentlichen
das gesamte Methylenchlorid entfernt ist, wobei das Produkt in Heptan
zurückbleibt
und auf 0°C
gekühlt
und filtriert wird. Eine Filtratanalyse zeigt 5% Produkt durch prozentuale
Flächenintegration.
Das Produkt wird nur fraktionsweise vereinigt und zur Trockne eingedampft.
Der Kuchen wird aus gemischten Fraktionen zugegeben. Vereinigtes
Trockengewicht = 197,4 g. HPLC Flächenprozent: 97,0%. NMR stimmt
mit dem gewünschten
Produkt überein:
Ausbeute: 74,8%.
-
Beispiel 119
-
7-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
In
einem 12 l fassenden Dreihalsrundbodenkolben 2-(4-Chlor-2-nitrophenylamino)-5-methylthiophen-3-carbonitril
(204,9 g, 0,6976 mol, 1 Äquivalent),
3 A EtOH (2 l) und 6 N HCl (2 l) für 10 Minuten gerührt. Zur
Aufschlämmung
wird Zinn-(II)-chlorid (404,1 g, 2,131 mol, 3,06 Äqu.) in
einer Portion gegeben und bis zum leichten Rückfluss bei 85°C für 3 Stunden
erhitzt. Die HPLC nach 3 Stunden zeigt < 0,4% Ausgangsmaterial. Das Gemisch
kann abkühlen
und über
Nacht rühren.
Während
der Aufarbeitung besteht die Möglichkeit,
dass Zinnsalze im Produkt ausfallen und es wird nicht wie im Laborversuch
gekühlt,
wobei die Aufarbeitung umfasst: Filtration des Reaktionsgemisches,
Waschen mit 1:1 3A EtOH/6 N HCl (2 × 500 ml) bei Umgebungstemperatur, Waschen
mit DI Wasser (2 × 50
ml) und Trocknen im Va kuumofen bei 60°C für 2 Tage. Gewicht = 231,5 g
und Analyse. Es wird eine sehr kleine Menge Niederschlag verworfen,
die im Filtrat auftritt. Die Zinnentfernung in heißer 1 N
HCl umfasst das Plazieren von 231,5 g des gelben Rohprodukts in
einem 22 l fassenden Kolben, Zugabe von 1 N HCl (11,6 l), Erhitzen
auf 95°C
und Halten der Temperatur für
1 Stunde, Abstellen der Heizung und Abkühlung mit einem Kühlmantel über Nacht.
Am nächsten
Morgen beträgt
die Temperatur 29°C.
Der Mantel wird entfernt und durch ein Wasserbad ersetzt und auf
22°C abgekühlt und
filtriert. Der Kolben wird gewaschen und mit 1 N HCl (4 l) gefolgt
von DI Wasser (4 l) zum Kuchen gegeben. Es wird Luft durch den Kuchen für 10 Minuten
gesaugt und er wird bei 60°C
für 40
Stunden in den Vakuumofen gestellt (Anmerkung: Nur 1 Gramm Gewicht
wird über
die letzten 16 Stunden Trocknungszeit verloren). Die trockene Titelverbindung
beträgt
200,4 g (95,7%). Es wird eine Zinnanalyse ausgeführt. Die NMR stimmt mit dem
Produkt überein.
Die HPLC zeigt 98,5% an.
-
Beispiel 120
-
7-Chlor-2-methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
-
In
einem 22 l fassenden Dreihalsrundbodenkolben werden 7-Chlor-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-ylaminhydrochlorid
(199 g, 0,6629 mol, 1 Äquivalent),
DI Wasser (6 l) und 3 A EtOH (2 l) für 15 min gerührt. Zu
der Aufschlämmung
wird in einer Portion pulverisiertes Kaliumcarbonat (690,6 g, 7,54 Äquivalente)
gegeben, für
10 min gerührt
und dann am milden Rückfluss
bei 84-85°C
erhitzt. Nach 3 Tagen zeigt die HPLC 10,7% Ausgangsmaterial. Die
Reaktion wird am Rückfluss
für einen
weiteren Tag aufrechterhalten. Nach 4 Tagen zeigt die HPLC, dass
10,1% des Ausgangsmaterials verbleiben. Wenn die Reaktion nicht
fortschreitet, kann sich das Gemisch auf 20-25°C abkühlen und wird filtriert und
mit 3:1 3 A EtOH/DI Wasser (1,2 l) gefolgt von DI Wasser (1,2 l)
gewaschen. Der Feststoff wird in einem Vakuumofen für 48 Stunden
bei 50°C getrocknet.
Nach 24 Stunden verliert der Feststoff immer noch an Gewicht. Trockengewicht
= 157,6 g. HPLC 85,2% des gewünschten
Amids, 9,8% Amidinausgangsmaterial. Die NMR zeigt das Produkt und
andere Resonanzen. Die Ausbeute der Titelverbindung beträgt 157,6
g (89,8%). Im Filtrat zeigt sich eine kleine Menge des Niederschlags.
Filtration, Waschen mit DI Wasser und Trocknen ergibt die Titelverbindung
als trockenes Gewicht = 1,45 g. Eine HPLC der Mutterlauge zeigt,
dass 10,9% des gewünschten
Produkts zurückbleiben.
-
Beispiel 120a
-
7-Chlor-2-methyl-10-methylsulfanyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
7-Chlor-2-methyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-thion
(59,1 g, 0,210 mol) und DMF (236,4 ml) werden unter Stickstoff für 15 Minuten
bei Umgebungstemperatur gerührt.
Pulverisiertes Kaliumcarbonat (61,1 g, 0,442 mol) wird zugegeben
und für
15 Minuten bei Umgebungstemperatur gerührt. Jodmethan (26,2 ml, 0,421
mol) wird zugegeben und für
2,5 Stunden bei Umgebungstemperatur gerührt. MTBE (591 ml) wird zugegeben
und für
15 Minuten bei Umgebungstemperatur gerührt. Die Feststoffe werden
mit MTBE (59,1 ml) filtriert und gewaschen. Das organische Filtrat
wird mit Wasser (3 × 591
ml) gewaschen, dann über
Natriumsulfat getrocknet, filtriert und unter verringertem Druck
unter Bildung von 60,0 g (96,7%) des rohen Produkts konzentriert.
1H NMR (500 MHz, DMSO-d6) δ 2,22 (s,
3H), 2,39 (s, 3H), 6,47 (d, 1H), 6,57 (d, 1H), 6,86 (d, 1H), 6,97
(dd, 1H), 8,09 (bs, 1H).
13C NMR (100
MHz, DMSO-d6) δ 13,24, 14,70, 120,58, 120,62,
121,67, 125,64, 127,51, 127,52, 127,77, 141,65, 142,16, 153,99,
165,48.
-
Beispiel 121
-
2-Isopropyl-10-methylsulfanyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Ein
750 ml fassender Dreihals Eurokolben wird mit einem magnetischen
Rührstab,
einer Trennwand mit Thermometerführung,
einem Zugabetrichter, einem Stickstoffeinlass und einem Kühlbad ausgestattet.
Der Kolben wird mit der Verbindung 2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion (49,8 g, 0,181
mol, enthält
9,7% (Gewichtsprozent) an 1,2-Dichlorethan, wobei diese Tatsache
während
der Berechnungen der Anzahl an Äquivalenten
der anderen in der Reaktion verwendeten Reagenzien nicht berücksichtigt wird)
und DMF (249 ml, 5 Volumina basierend auf dem Ausgangsmaterial)
befüllt.
Das entstehende Gemisch kann bei Umgebungstemperatur für 5 Minuten
rühren,
bis die gesamten Feststoffe gelöst
sind. Pulverisiertes K2CO3 (325
Mesh, 138,2 g, ~0,381 mol, ~2,1 Äquivalente)
wird zu dem Reaktionsgefäß gegeben
und für
5 Minuten kontinuierlich gerührt.
Methyliodid (22,5 ml, ~0,362 mol, ~2,0 Äquivalente) wird zu dem Zugabetrichter und
tropfenweise zu dem obigen Gemisch für 20 Minuten gegeben. Die Gefäßtemperatur
wird unter 28,0°C mittels
der Zugabe von kaltem Leitungswasser zum Kühlbad gehalten. Das entstehende
Gemisch kann bei Umgebungstemperatur für eine weitere Stunde rühren. Eine
Probe wird aus dem Reaktionsgemisch entfernt und durch TLC (60/40
V/V Hexan/Aceton, UV) analysiert. Es wird festgestellt, dass das
Reaktionsgemisch kein Ausgangsmaterial. mehr enthält. MTBE
(500 ml, 10 Volumina) wird zu dem Reaktionsgemisch gegeben und für 20 Minuten
kontinuierlich gerührt.
Anschließend
wird das Gemisch filtriert und die Feststoffe werden mit MTBE (250
ml, 5 Volumina) gewaschen. Das Filtrat wird in einen Trenntrichter überführt und
mit entionisiertem Wasser (500 ml, 10 Volumina) verdünnt. Nach
dem Schütteln
werden die Phasen getrennt und die wässrige Phase wird mit MTBE
(250 ml, 5 Volumina) reextrahiert. Die organischen Portionen werden
vereinigt und mit entionisiertem Wasser (3 × 500 ml) gewaschen, über Na2SO4 getrocknet,
filtriert und im Vakuum konzentriert. Das entstehende dicke orange Öl kann unter
Vakuum über
Nacht bei Umgebungstemperatur zie hen. Eine Kristallisation des Materials
ergibt 45,17 g der gelb-orangen Feststoffe: Das rohe Material wird
durch HPLC mittels des folgenden Systems analysiert: Säule = Zorbax
C-8, Fluß =
1 ml/min, A = ACN, B = 0,1% wässriges
TFA, Gradient = 95% A/5% B bis 5% A/95% B über 10 Minuten. Es wird bei
5/95 A/B für
3 Minuten gehalten und zurück
auf 95/5 A/B für
2 Minuten. Säulentemperatur
= 30°C,
Wellenlänge
= 250 nm. Mittels des obigen HPLC Systems wird das rohe Material
untersucht und es wird gefunden, dass es zu 97,1% rein ist.
1H NMR (500 MHz, DMSO-d6): δ 1,19 (d,
6H), 2,32 (s, 3H), 2,99-3,02 (m, 1H), 6,50 (d, 1H), 6,80 (d, 1H), 6,82-6,90
(m, 2H), 8,10 (bs, 1H).
13C NMR (100
MHz, DMSO-d6): δ 13,29, 22,88, 32,92, 119,85,
124,88, 127,19, 129,92, 132,94, 140,40, 142,17, 151,59, 164,42,
164,63.
MS (80:20 MeOH : H2O Gewicht/6,5
mM NH4OAc). Berechnet: 289,07, gefunden:
ES+ 290,0. ES– 288,0.
-
Beispiel 122
-
2-(4,5-Difluor-2-nitrophenylamino)thiophen-3-carbonitril
-
2,4,5-Trifluornitrobenzol
(5,00 g, 24,24 mmol), 2-Aminothiophen-3-carbonitril (3,51 g, 28,24
mmol) und wasserfreies THF (30,0 ml) werden vereinigt. Das Gemisch
wird auf –10°C gekühlt und
dann wird langsam NaH (2,26 g, 56,47 mmol, 60% Dispersion in Mineralöl) zugegeben,
während
die Temperatur unter 10°C
gehalten wird. Das Gemisch wird auf Umgebungstemperatur erwärmt, nachdem
die NaH Zugabe vollständig
ist. Das Gemisch wird bei Umgebungstemperatur für 24 Stunden gerührt und
dann auf Eischips gegossen. Die wässrige Phase wird mit Ethylacetat
extrahiert und dann gewaschen (Kochsalzlösung), getrocknet (Natriumsulfat)
und die organische Phase wird zum Rückstand reduziert. Der Rückstand
wird auf Silicagel mittels Hexan/Dichlormethan (35:65) unter Bildung
von 4,06 g (51%) der Titelverbindung als roter Feststoff gereinigt: Massenspektrum
(Ionenspray): m/z = 280,0 (M-1).
-
Beispiel 123
-
6,7-Difluor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
-
2-(4,5-Difluor-2-nitrophenylamino)thiophen-3-carbonitril
(4,02 g, 14,30 mmol) wird in Ethanol (40,0 ml) suspendiert. Zinn(II)chlorid
(8,14 g, 42,91 mmol) wird in 5 N HCl (40,0 ml) gelöst und dann
zu der Suspension gegeben. Die Reaktion wird am Rückfluss
für 5,5
Stunden erhitzt und dann auf Umgebungstemperatur gekühlt. Das
Gemisch wird für
16 Stunden in einem Gefrierschrank gekühlt. Der entstehende Niederschlag
wird durch Vakuumfiltration gesammelt und dann durch Unterziehen
eines Vakuums unter Bildung von 3,53 g (86%) der Titelverbindung
als gelber Feststoff getrocknet: Massenspektrum (Ionenspray): m/e
= 252,1 (M+1).
-
Beispiel 153
-
(S)-4-Benzyl-1-methyl-2-(2-methylsulfanylethyl)piperazin
-
(S)-1-Benzyl-3-(2-methylsulfanylethyl)piperazin
(0,900g, 3,59 mmol) und 37% Formaldehydlösung (0,4 ml, 5,39 mmol) werden
in Methylenchlorid (15 ml) vereinigt. Es wird für 10 Minuten gerührt und
Natriumtriacetoxyborhydrid (3,05 g, 14,4 mmol) wird zugegeben. Es
wird weitere 90 Minuten gerührt
und dann wird die Lösung
auf 1 N Natriumhydroxidlösung
gegossen. Es wird mit Methylenchlorid unter Bildung von 0,947 g
des rohen Produkts extrahiert. Eine Silicagelchromatographie unter
Elution mit Methylenchlorid : 2 N Ammoniak/Methanol (100:2) ergibt
0,906 g der Titelverbindung als farbloses Öl. Massenspektrum (Ionenspray):
m/z = 265 (M+1).
-
Beispiel 154
-
(S)-1-Methyl-2-(2-methylsulfanylethyl)piperazin
-
Zu
einer kalten Lösung
aus (S)-4-Benzyl-1-methyl-2-(2-methylsulfanylethyl)piperazin (0,450
g, 1,7 mmol) in 1,2-Dichlorethan (5,0 ml) wird tropfenweise 1-Chlorethylchlorformiat
(0,24 ml, 2,2 mmol) gegeben. Nach dem Rühren bei Umgebungstemperatur
für 18
Stunden wird das 1,2-Dichlorethan verdampft und 40 ml Methanol werden
zu dem Rückstand
gegeben. Diese Lösung
wird für
2 Stunden am Rückfluss
erhitzt. Ein Eindampfen des Methanols ergibt das rohe Produkt. Eine
Silicagelchromatographie unter Elution mit Methylenchlorid : 2 N
AmmoniakMethanol (90:10) ergibt 0,175 g der Titelverbindung als
hellgelbes Öl.
Massenspektrum (Ionenspray): m/z = 175 (M+1).
-
Beispiel 155
-
(S)-1,4-Dibenzyl-2-vinylpiperazin
-
Beispiel 156
-
(R)-1,4-Dibenzyl-2-vinylpiperazin
-
Wasserfreies
Tetrahydrofuran (4,5 l) wird zu einem 10 l Flanschhalskolben, der
mit einem Rührstab und
einem Rührblatt,
einem Thermometer und einem Stickstoffeinlass und Auslassröhrchen ausgestattet
ist, gegeben. Der Flüssigkeitskörper wird
mit trockenem Stickstoffgas (Eislassröhrchen weist ein gesintertes
Ende zur maximalen Gasverteilung auf) für 1 h gespült und Tris(dibenzylidenaceton)dipalladium(0)chloroformaddukt (36,0
g, 34,8 mmol) wird zugegeben. Isopropylphosphit (67,8 ml, 0,275
mol) wird in einer Portion zu dem Gemisch immer noch unter Stickstoff
gegeben und gerührt.
Nach 5 Minuten erhält
sich die Farbe von Purpur nach Bernstein. Dibenzylethylendiamin
(322,0 g, 1,34 mol) wird in einer Portion zugegeben, gefolgt von
der tropfenweisen Zugabe von cis-1,4-Diacetoxy-2-buten (214 ml,
1,34 mol) über
15 Minuten und es wird unter Stickstoff für 18 Stunden gerührt. Das
Lösemittel
wird im Vakuum bei 40°C
entfernt und das restliche Öl
wird in Diethylether (2,5 l) gelöst
und mit 1 N wässrigem
Natriumhydroxid (2 × 2
l) extrahiert. Die vereinigten wässrigen
Extrakte werden mit Diethylether (2 ×) gewaschen und mittels 5
N wässrigem
Natriumhydroxid auf pH 14 basisch gemacht und mit Diethylether (3 ×) extrahiert.
Die vereinigten Etherextrakte werden über Magnesiumsulfat getrocknet,
filtriert und im Vakuum bei 40°C
zur Trockne eingedampft. Eine Reinigung durch Chromatographie auf
Silica (1,17 kg) mittels 1% MethanolEther (Dichlormethan kann ebenfalls
verwendet werden) ergibt ein blassgelbes Öl (377,35 g, 96%).
1H NMR und Massenspektrum stimmen mit dem
Produkt überein.
-
Das
Gemisch der Isomere wird in Ethylacetat (3670 ml) gelöst und portionsweise
zu einer heißen
Lösung
aus (S)-(+)Mandelsäure
(385 g, 2 Äquivalente)
in Ethylacetat (3850 ml) ausgehend von 72°C gegeben. Das Gemisch wird
auf 0°C
gekühlt
und mit Kristallen (erhalten aus einer früheren Auftrennung) angeimpft.
Das Gemisch wird in einen Gefrierschrank (–20°C) über Nacht gegeben. Der kristalline
Feststoff wird von den Kolbenwänden
weggekratzt und das Gemisch kann sich auf 0°C erwärmen. Der Feststoff wird trocken
isoliert. Dann wird das Material im Vakuum bei Raumtemperatur weiter
getrocknet. Ausbeute: 252,6 g, weißer, kristalliner Feststoff
des S-Mandelsäuresalzes
des (R)-1,4-Dibenzyl-2-vinylpiperazins.
-
Das
Filtrat wird im Vakuum bei 40°C
zur Trockne eingedampft um ein bernsteinfarbenes Öl zu erhalten. Das
Filtrat wird in Dichlormethan (2 l) gelöst und die Lösung wird
mit 1 N wässrigem
Natriumhydroxid (2 l + 1 l), Kochsalzlösung (1 l) gewaschen und über Magnesiumsulfat
getrocknet. Es wird filtriert und im Vakuum bei 45°C unter Bildung
der erhaltenen freien Base zur Trockne konzentriert. Dann wird weiter
durch Vakuum getrocknet. Die wässrigen
Flüssigkeiten
werden mit Dichlormethan zur weiteren Gewinnung jeder verbleibenden freien
Base (207,6 g) extrahiert. Eine Chiral HPLC zeigt, dass das Material
in einem 85:15 Verhältnis
der Isomere zugunsten des gewünschten
Isomers übereinstimmt.
-
(R)-(–)Mandelsäure (216
g, 1,42 mol) und Ethylacetat (2,5 l) werden zu einem 10 Liter fassenden Flanschhalskolben
gegeben, der mit einem Rührstab
und einem Rührblatt,
einem Thermometer und einem Wasserkühler ausgestattet ist, und
die Suspension wird auf 60°C
erwärmt.
Eine Lösung
der freien Base (207,6 g, 0,71 mol) in Ethylacetat (500 ml) wird
zugegeben und kann auf Raumtemperatur abkühlen und wird über Nacht
in einen Gefrierschrank gegeben (bei 35°C beginnt der Feststoff auszufallen).
Der kristalline Feststoff wird durch Filtration isoliert und bis
zur Trockne abgezogen. Weiter wird im Vakuum bei Raumtemperatur (290,34
g) getrocknet.
-
Es
wird aus heißem
Ethylacetat (2,3 l) bei 70°C
umkristallisiert. Diese Lösung
kann sich nach dem Animpfen über
Nacht auf Raumtemperatur abkühlen.
Eine Filtration und Trocknen im Vakuum bei Raumtemperatur ergibt
das (R) Mandelsäuresalz
des (S)-1,4-Dibenzyl-2-vinylpiperazins, aus dem die freie Base hergestellt
werden kann (225,44 g). Eine Chiral HPLC zeigt: 98,74% + 1,26%.
1H NMR (DMSO-d6); δ 7,20-7,35
(m, 10H), 5,75-5,90 (m, 1H), 5,15-5,30 (q, 2H), 3,85-3,95 (d, 1H),
3,40-3,45 (s, 2H), 3,00-3,10 (d, 1H), 2,80-2,90 (t, 1H), 2,55-2,60
(d, 3H), 1,95-2,10 (m, 3H).
-
Beispiel 157
-
(S)-1,4-Dibenzyl-2-vinylpiperazin
-
(S)-1,4-Dibenzyl-2-vinylpiperazinmandelsäure (200,0
g, 0,450 mol), Wasser (1 l) und 5 N Natriumhydroxid (112,5 ml, 0,562
mol) werden vereinigt und bei Umgebungstemperatur für 15 Minuten
gerührt.
MTBE (1 l) wird zugegeben und bei Umgebungstemperatur für 1 Stunde
gerührt.
Die Phasen werden getrennt und die organischen Phasen werden mit
Wasser (2 × 1
l) gewaschen. Die organischen Phasen werden über Natriumsulfat getrocknet,
filtriert und im Vakuum unter Bildung von 131,2 g der Titelverbindung
als Öl
(99,7%) konzentriert.
1H NMR (500 MHz,
DMSO-d6) δ 2,00
(bt, 1H), 2,07 (d, 2H), 2,56 (m, 1H), 2,58 (d, 2H), 2,84 (t, 1H),
3,02 (d, 1H), 3,49 (s, 2H), 3,91 (d, 1H), 5,15 (d, 1H), 5,25 (d,
1H), 5,81 (m, 1H), 7,27 (m, 10H). MS (ES+) M+H berechnet für C20H24N2 292,43,
gefunden: 293,10.
-
Beispiel 158
-
(S)-2-(1,4-Dibenzylpiperazin-2-yl)ethanol
-
(S)-1,4-Dibenzyl-2-vinylpiperazin
(131,0 g, 0,448 mol) und THF (500 ml) werden bei Umgebungstemperatur
vereinigt. Eine 0,5 M THF Lösung
aus 9-Borabicyclo[3,3,1]nonan (9-BBN) (985,5 ml, 0,493 mol) wird über 20 Minuten
zugegeben, wobei die Gefäßtemperatur
unter 25°C
gehalten wird. Das entstehende Gemisch wird bei Umgebungstemperatur
für 16-18
Stunden gerührt.
Die Lösung
wird auf 0-5°C
gekühlt
und mit 3 N NaOH (164,3 ml, 0,493 mol) über 5-10 Minuten gestoppt.
Es wird bei 0-5°C
für 10-15 Minuten
gerührt
und das Kühlbad
wird entfernt. Eine 30% wässrige
Lösung
aus Wasserstoffperoxid (160,2 ml, 1,57 mol) wird über 1 Stunde
zugegeben, wobei die Gefäßtemperatur
bei 30-35°C
gehalten wird. Das entstehende Gemisch wird für 1 Stunde gerührt und
mit Wasser (1,31 l) und MTBE (1,31 l) verdünnt. Die Phasen werden getrennt
und die wässrige
Phase wird mit MTBE (655 ml) rückextrahiert.
Die organischen Phasen werden vereinigt und mit Wasser (2 × 655 ml)
und Kochsalzlösung
(2 × 655
ml) gewaschen. Die MTBE Lösung
wird über
Natriumsulfat getrocknet, filtriert und im Vakuum unter Bildung
von 169,1 g des rohen Öls
(Theorie = 139,1 g) konzentriert. Das rohe Material wird mit 5 N
HCl (358 ml, 1,79 mol) behandelt und für 5 Minuten gerührt. Wasser
(250 ml) wird zugegeben und für
15-20 Minuten gerührt.
Heptan (1,31 l) wird zugegeben und für 5 Minuten gerührt. Die Phasen
werden getrennt und die wässrige
Phase wird mit MTBE (655 ml) rückextrahiert.
Diese organischen Extrakte werden verworfen und 5 N NaOH (403 ml,
2,02 mol) wird zu der wässrigen
Portion gegeben. Es wird für
5 Minuten gerührt,
dann mit MTBE (1,31 l) verdünnt
und die Phasen werden getrennt. Die wässrige Phase wird mit MTBE
(655 ml) rückextrahiert.
Die MTBE Extrakte werden vereinigt und mit Wasser (655 ml) gewaschen,
dann über
Natriumsulfat getrocknet, filtriert und im Vakuum unter Bildung
von 137,8 g (99,0%) der Titelverbindung konzentriert.
1H NMR (500 MHz, DMSO-d6) δ 1,87 (m,
1H), 2,03 (m, 1H), 2,33 (m, 2H), 2,43 (m, 1H), 2,50 (d, 1H), 2,66
(d, 1H), 2,83 (m, 1H), 2,93 (m, 1H), 3,39 (d, 1H), 3,50 (m, 1H),
3,74 (m, 1H), 3,88 (m, 1H), 4,18 (d, 1H), 4,80 (bs, 1H), 7,26 (m,
2H), 7,32 (m, 8H).
-
Beispiel 159
-
(S)-1,4-Dibenzyl-2-(2-methoxyethyl)piperazin
-
Natriumhydrid
(52,9 g, 1,32 mol) und THF (500 ml) werden vereinigt und die entstehende
Aufschlämmung
wird auf 0-5°C
gekühlt.
(S)-2-(1,4-Dibenzylpiperazin-2-yl)ethanol (137,0 g, 0,441 mol) wird
in THF (500 ml) gekühlt
und zu dem Natriumhydrid/THF Gemisch über 30 Minuten gegeben, wobei
eine Gefäßtemperatur von
0-10°C aufrechterhalten
wird. Das Gemisch wird bei 0-10°C
für 15-20
Minuten gerührt,
dann wird Dimethylsulfat (55,6 g, 0,441 mol) über 1 Stunde zugegeben, wobei
eine Gefäßtemperatur
von 0-10°C
aufrechterhalten wird. Das Kühlbad
wird entfernt und das Reaktionsgemisch kann sich über 1 Stunde
auf Umgebungstemperatur erwärmen.
Es wird für
eine weitere Stunde bei Umgebungstemperatur gerührt, auf 0-10°C gekühlt und
dann mit 1 N Ammoniumchlorid (863 ml) gestoppt. Das Gemisch wird
mit Ethylacetat (2 × 700
ml) extrahiert. Die Ethylacetatextrakte werden vereinigt und mit
gesättigtem
wässrigem
Natriumbicarbonat (2 × 2
l) gewaschen, über
Magnesiumsulfat getrocknet und im Vakuum unter Bildung von 162,1
g eines Öls
konzentriert. Das Öl
wird in Heptan (900 ml) gelöst
und Wasser (450 ml) wird zugegeben. Während dem Rühren wird 3 N HCl zu einem
pH von 1-2 zugegeben. Die Phasen werden getrennt und die wässrige Phase
wird mit 50% Kalilauge auf einen pH von 13-14 behandelt. Diese wässrige Lösung wird
mit Methylenchlorid (2 × 1
l) extrahiert, die organischen Extrakte werden vereinigt und über Magnesiumsulfat
getrocknet. Es wird im Vakuum unter Bildung von 136,3 g (93,3%)
der Titelverbindung konzentriert.
1H
NMR (400 MHz, DMSO-d6) δ 1,80 (m, 2H), 2,20 (bm, 2H,
1H), 2,41 (m, 1H), 2,55 (m, 2H), 2,60 (d, 1H), 3,18 (s, 3H), 3,35
(bm, 2H, 1H), 2,40 (d, 1H), 2,52 (d, 1H), 3,90 (d, 1H). MS (ES+)
berechnet für
C21 H28N 324,47, gefunden
325,2.
-
Beispiel 160
-
(S)-2-(2-Methoxyethyl)piperazin
-
(S)-1,4-Dibenzyl-2-(2-methoxyethyl)piperazin
(2,0 g, 6,2 mmol) in Ethanol (15 ml) wird in einem geeigneten Hydrierungsgefäß gelöst. Palladiumhydroxid
wird zugegeben (Pearlman's
Katalysator, 400 mg), das Gefäß wird mit
Stickstoff gespült
und dann auf 60 psi unter Druck gesetzt. Es wird auf 50°C erhitzt
und für
18-24 Stunden kräftig
gerührt.
Das Gemisch kann sich auf Umgebungstemperatur abkühlen. Der
Katalysator wird abfiltriert und mit Ethanol (5 ml) gewaschen. Es
wird im Vakuum mittels eines 35°C
Bades unter Bildung der Titelverbindung (0,81 g, 91,1%) konzentriert.
1H NMR (500 MHz, DMSO-d6) δ 1,41 (m,
2H), 2,12 (t, 1H), 2,43 (td, 1H), 2,53 (m, 2H, 1H), 2,63 (d, 1H),
2,72 (m, 2H), 3,19 (s, 3H), 3,34 (m, 2H).
-
Beispiel 161
-
(S)-(4-Benzyl-3,6-dioxopiperazin-2-yl)essigsäuremethylester
-
Kommerziell
erhältlicher
N-t-Boc-L-Asparaginsäure-β-methylester
(40 g, 0,16 mol) wird in Dichlormethan (800 ml) gelöst, auf
0°C gekühlt und
N-Benzylglycinmethylester (28 g, 0,15 mol wird als eine Lösung in 100
ml Dichlormethan) gefolgt nacheinander von N,N-Diisopropylethylamin
(28 ml, 0,16 mol), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimidhydrochlorid
(EDAC, 31 g, 0,16 mol) und 1-Hydroxybenzotriazol (22 g, 0,16 mol)
zugegeben. Es wird bei Raumtemperatur über das Wochenende gerührt und
im Vakuum zu einem orangen Öl
konzentriert. Das Öl
wird zwischen 2 N Chlorwasserstoffsäure und Ethylacetat aufgetrennt,
die wässrige
Phase wird abgetrennt und mit einer zweiten Portion Ethylacetat
extrahiert. Die organischen Extrakte werden vereinigt, im Vakuum
konzentriert und mit 10% wässrigem
Kaliumcarbonat gewaschen. Die organische Phase wird über Magnesiumsulfat
getrocknet, filtriert und im Vakuum unter Bildung von 64 g (95%)
des gewünschten
Dipeptids als öliger
Rückstand
konzentriert.
-
Das
rohe Dipeptid wird in 150 ml Trifluoressigsäure gelöst, bei Raumtemperatur für 1 h gerührt und dann
wird das Lösemittel
im Vakuum entfernt. Der entstehende Rückstand wird auf 800 ml kommerziell
erhältlichem
2 N Ammoniak in einer Methanollösung
aufgenommen und bei Raumtemperatur über Nacht gerührt. Das
Gemisch wird bei 70°C
für mehrere
Stunden erhitzt, dann auf Raumtemperatur gekühlt und das Lösemittel wird
im Vakuum entfernt. Der Rückstand
wird in Dichlormethan rückgelöst, der
entstehende Niederschlag wird abfiltriert und das Filtrat wird im
Vakuum konzentriert. Der Rückstand
wird auf eine Silicagelsäule
aufgetragen. Es wird mit einem 2% Gemisch aus 2 N Ammoniak – Methanol
in Dichlormethan unter Bildung von 31,9 g (72%) der Titelverbindung
als gelbes Öl
eluiert: Massenspektrum (APCI): m/e 277,1 (M+1).
-
Beispiel 162
-
(S)-(–)-2-(4-Benzylpiperazin-2-yl)ethanol
-
Zu
einer 0°C
Lösung
aus (S)-(4-Benzyl-3,6-dioxopiperazin-2-yl)essigsäuremethylester (31,9 g, 0,12 mol)
in Tetrahydrofuran (1 l) wird Lithiumaluminiumhydrid mittels einer
langsamen Kanülierung
(350 ml einer kommerziell erhältlichen
1,0 M Lösung
in Tetrahydrofuran) gegeben. Es wird bei Raumtemperatur über Nacht gerührt, durch
die folgende vorsichtige Zugabe von 13,3 ml Wasser, 13,3 ml einer
15% wässrigen
Natriumhydroxidlösung
und 39,9 ml Wasser unter dauerndem kräftigem Rühren gestoppt um die Bildung
eines feinen Niederschlags sicherzustellen. Es wird durch eine Glasnutsche
filtriert und die Feststoffe werden mit Tetrahydrofuran und Dichlormethan
gut gewaschen. Es wird im Vakuum unter Bildung von 26,5 g eines öligen Rückstands
konzentriert und direkt auf eine Silicagelsäule gegeben. Es wird mit einem
5% Gemisch aus 7 N Ammoniak-Methanol in Dichlormethan unter Bildung
des gewünschten
Produkts als oranges Öl
eluiert, das sich unter Vakuum verfestigt. Der Feststoff wird in
Acetonitril aufgenommen und für
wenige Minuten ultrabeschallt. Der entstehende Niederschlag wird
unter Bildung von 7,5 g (25%) der Titelverbindung als nicht ganz
weißer kristalliner
Feststoff filtriert. Smp 78,9-80,4°C. Die Mutterlauge wird unter
Bildung von 6,8 g (23%) eines leicht weniger reinen Materials als
amorpher Feststoff konzentriert. Massenspektrum (ES): m/e 221,3
(M+1). Spezifische Rotation: –7,84.
-
Beispiel 163
-
(S)-4-Benzyl-2-(2-hydroxyethyl)piperazin-1-carbonsäure-tert-butylester
-
Zu
einer Lösung
aus (S)-(–)-2-(4-Benzylpiperazin-2-yl)ethanol
(14,9 g, 67,6 mmol) in Dichlormethan (200 ml) wird Di-tert-butyldicarbonat
(15,5 g, 71 mmol) als eine Lösung
in Dichlormethan (30 ml) gegeben. Es wird bei Raumtemperatur für 4 h gerührt, zwischen
gesättigtem
wässrigem
Bicarbonat und Dichlormethan aufgeteilt und die wässrige Phase
wird mit zusätzlichem
Dichlormethan extrahiert. Die organischen Extrakte werden vereinigt, über Natriumsulfat
getrocknet, filtriert und im Vakuum zu einem Rückstand konzentriert. Der Rückstand
wird auf eine Silicagelsäule
unter Elution mit 5% an 2 N Ammoniak-Methanol in Dichlormethan unter
Bildung der Titelverbindung als gelbes Öl gegeben. Massenspektrum (APCI):
m/e 321,2 (M+1).
-
Beispiel 164
-
(R)-2-Piperazin-2-yl-ethanol
-
Unter
Verwendung einer Sequenz, die zu der in den Beispielen 161 und 162
beschriebenen ähnlich ist,
wird N-t-Boc-D-Asparaginsäure-β-benzylester
mit Glycinmethylester unter Bildung der Titelverbindung als gelbes Öl vereinigt.
Massenspektrum (APCI): m/e 131,1 (M+1).
-
Beispiel 165
-
(S)-2-(1,4-Dibenzylpiperazin-2-yl)ethanol
-
(S)-1,4-Dibenzyl-2-vinylpiperazin
(16,2 g, 55,5 mmol) wird in Tetrahydrofuran (370 ml) gelöst, 9-BBN (0,56 l einer
0,5 M Lösung
in Tetrahydrofuran) wird mittels eines Zugabetrichters zugegeben
und bei Raumtemperatur über
Nacht gerührt.
Es wird auf 0°C
gekühlt
und mit 30% wässrigem
Wasserstoffperoxid (195 ml) gefolgt von 3 N wässrigem Natriumhydroxid (195
ml) behandelt. Es kann Raumtemperatur erreichen und wird für 24 h gerührt. Es
wird in einen Trenntrichter gegossen, die organische Phase wird
abgetrennt und das Lösemittel
wird im Vakuum entfernt. Der Rückstand
wird in Dichlormethan aufgenommen, Wasser wird zugegeben und mit
der wässrigen
Phase von oben vereinigt. Es wird in einen Trenntrichter gegossen,
die organische Phase wird abgetrennt und die wässrige Phase wird mit mehreren
Portionen an Dichlormethan extrahiert. Alle organischen Phasen werden
vereinigt und das Lösemittel
wird im Vakuum entfernt. Der Rückstand
wird in 1 l Methanol aufgenommen, 185 g SCX Harz werden zugegeben.
Die Aufschlämmung
wird durch einen Büchner Trichter
filtriert und mit Methanol gut gewaschen. Um das Produkt zu eluieren,
wird der Kuchen vorsichtig mit 50% an 7 N Ammoniak-Methanol in Dichlormethan
gewaschen. Das Filtrat wird im Vakuum unter Bildung der Titelverbindung
(16,6 g, 96%) als dickes bräunliches Öl konzentriert.
Massenspektrum (APCI): m/z = 311,2 (M+1).
-
Beispiel 166
-
(S)-(1,4-Dibenzylpiperazin-2-yl)acetaldehyd
-
Eine
Lösung
aus Oxalylchlorid (0,183 ml, 21,0 mmol) in Dichlormethan (20 ml)
wird mit einer Lösung aus
Dimethylsulfoxid (2,34 ml, 33,0 mmol) in Dichlormethan (10 ml) bei –78°C vereinigt
und für
15 Minuten gerührt.
Eine Lösung
aus (S)-2-(1,4-Dibenzylpiperazin-2-yl)ethanol (0,60 g, 1,82 mmol)
in Dichlormethan (10 ml) wird bei –78°C mittels einer Kanüle zugegeben.
Es wird bei –78°C für eine Stunde
gerührt,
Triethylamin (10,5 ml, 75,0 mmol) wird zugegeben und es wird über Nacht
auf Raumtemperatur erwärmt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Es wird durch Blitzchromatographie unter Elution mit einem Stufengradienten
ausgehend von Dichlormethan bis hin zu 6% an 2 N Ammoniak-Methanol
in Dichlormethan unter Bildung der Titelverbindung (3,66 g, 11,9
mmol, 79%) als braunes Öl
gereinigt. Massenspektrum (APCI): m/z = 309,4 (M+1).
-
Beispiel 167
-
(S)-2-Piperazin-2-yl-ethanol
-
(S)-2-(1,4-Dibenzylpiperazin-2-yl)ethanol
(11,7 g, 0,038 mol) wird in 380 ml Ethanol gelöst, 10% Palladium auf Kohle
(3,7 g eines nassen Reagenzes, 50 Gewichtsprozent) wird als eine
Suspension in wenigen ml Ethanol gelöst. Überschüssiges Ammoniumformiat (16,8
g, 0,27 mol) wird auf einmal zugegeben. Es wird am Rückfluss
für 5 h
erhitzt, auf Raumtemperatur gekühlt
und durch ein Celitekissen filtriert und mit Ethanol gut gewaschen.
Das Filtrat wird im Vakuum unter Bildung der Titelverbindung (5
g, quantitativ) als trüber
Rückstand
konzentriert. Es wird im nächsten
Schritt ohne weitere Reinigung verwendet: Massenspektrum (APCI): m/z
= 131,1 (M+1).
-
Beispiel 168
-
(S)-2-(2-Hydroxyethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
(S)-2-Piperazin-2-yl-ethanol
(4 g, 30,7 mmol) wird in 150 ml Dichlormethan gelöst und einige
wenige Milliliter Ethanol werden zum Lösen des Materials zugegeben.
Di-tert-butyldicarbonat (28 g, 0,13 mol) wird in zwei Portionen
zu Beginn zugegeben und nach dem Rühren bei Raumtemperatur für 4 h wieder.
Das Gemisch wird auf gesättigtes
wässriges
Natriumbicarbonat gegossen und mit Dichlormethan extrahiert. Die
organischen Extrakte werden vereinigt, über Natriumsulfat getrocknet,
filtriert und das Filtrat wird im Vakuum zu einem Rückstand
konzentriert. Eine Reinigung mittels Silicagelchromatographie unter
Elution mit einem Stufengradienten von 2% an 2 N Ammoniak-Methanol
in Dichlormethan, 4% und 6% ergibt die Titelverbindung als gelbes Öl, das sich
während
dem Stehen verfestigt. Massenspektrum (FAB): m/z = 331,20.
-
Beispiel 169
-
(S)-2-(2-Oxoethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
Eine
Lösung
aus Oxalylchlorid (0,238 ml, 2,72 mmol) in Dichlormethan (25 ml)
wird mit Dimethylsulfoxid (0,322 ml, 4,54 mmol) bei –78°C vereinigt
und gerührt.
Nach 15 Minuten wird eine Lösung
aus (S)-2-(2-Hydroxyethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(0,60 g, 1,82 mmol) in Dichlormethan bei –78°C mittels einer Kanüle zugegeben
und bei –78°C gerührt. Nach
einer Stunde wird Triethylamin (1,27 ml, 9,08 mmol) zugegeben und über Nacht
auf Raumtemperatur erwärmt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
der Titelverbindung konzentriert. Massenspektrum (APCI): m/z = 129,1
(M+1-2BOC).
-
Beispiel 170
-
(S)-2-(2-Methoxyethyl)piperazin
-
Natriumhydrid
(0,675 g, 16,9 mmol) wird portionsweise zu einer 0°C Lösung aus
(S)-2-(2-Hydroxyethyl)lpiperazin-1,4-dicarbonsäure-di-tert-butylester
(3,72 g, 11,3 mmol) in Tetrahydrofuran (50 ml) gegeben und gerührt. Nach
20 Minuten wird Methyliodid (1,4 ml, 22,5 mmol) tropfenweise zugegeben.
Das Gemisch kann über
Nacht Raumtemperatur erreichen, wird mit gesättigtem Ammoniumchlorid verdünnt und
dreimal mit Ethylacetat extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Es wird durch Blitzchromatographie unter Elution mit einem Stufengradienten
ausgehend von Dichlormethan bis zu 7% an 2 N Ammoniak-Methanol in Dichlormethan
unter Bildung von 2-(2-Methoxyethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester gereinigt.
-
Zu
dem obigen Material in Dichlormethan (50 ml) wird Trifluoressigsäure (15
ml) bei 0°C
gegeben und gerührt.
Nach 30 Minuten kann sich das Gemisch auf Raumtemperatur erwärmen und
wird gerührt.
Nach 1 h wird das Lösemittel
im Vakuum unter Bildung eines goldenen gelben Öls entfernt. Der Rückstand
wird mit Methanol verdünnt
und auf eine 30 g SCX Säule
aufgetragen. Die Säule
wird mit Methanol gewaschen, mit 2 N Ammoniak in Methanol unter
Bildung der Titelverbindung als dickes farbloses Öl (1,13
g, 7,84 mmol, 89%) eluiert. Massenspektrum (APCI): m/z = 145,2 (M+1)
-
Beispiel 172
-
(S)-4-Benzyl-2-(2-methansulfonyloxyethyl)piperazin-1-carbonsäure-tert-butylester
-
Zu
einer Lösung
aus (S)-4-Benzyl-2-(2-hydroxyethyll)piperazin-1-carbonsäure-tert-butylester
(4,87 g, 15,2 mmol) in Dichlormethan (200 ml) wird Pyridin (1,84
ml, 22,8 mmol) gefolgt von Methansulfonylchlorid (1,29 ml, 16,7
mmol) gegeben. Es wird bei Raumtemperatur über Nacht gerührt und
dann mit gesättigtem wässrigem
Natriumbicarbonat verdünnt.
Es wird dreimal mit Dichlormethan extrahiert, die vereinigten organischen
Extrakte werden über
Natriumsulfat getrocknet, filtriert und im Vakuum unter Bildung
eines braunen Rückstands
konzentriert. Der Rückstand
wird in Dichlormethan rückgelöst und auf
ein Silicagelkissen aufgetragen. Das Kissen wird mit 5% an 2 N Ammoniak-Methanol
in Dichlormethan unter Bildung von (S)-4-Benzyl-2-(2-methansulfonyloxyethyl)piperazin-1-carbonsäure-tert-butylester
(3,74 g, 62%) als gelb-braunes dickes Öl gewaschen.
-
Beispiel 173
-
(S)-2-(2-Phenoxyethyl)piperazin
-
(S)-4-Benzyl-2-(2-methansulfonyloxyethyl)piperazin-1-carbonsäure-tert-butylester
(2,82 g, 7,08 mmol), Natriumiodid (0,106 g, 0,708 mmol) und Phenol
(3,33 g, 35,4 mmol) werden in Dimethylformamid (40 ml) vereinigt
und bei 100°C
für 20
Stunden gerührt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über Natriumsulfat
getrocknet und unter verringertem Druck konzentriert. Es wird durch
Blitzchromatographie unter Elution mit einem Stufengradienten (100%
Dichlormethan bis 5% an 2 N Ammoniak-Methanol in Dichlormethan)
unter Bildung von (S)-4-Benzyl-2-(2-phenoxyethyl)piperazin-1-carbonsäure-tert-butylester
(0,703 g, 25%) gereinigt. Massenspektrum (APCI): m/z = 397,2 (M+1).
-
Das
Material von oben (0,890 g, 2,24 mmol), Palladium auf Kohle (0,090
g, 10%) und Ammoniumformiat (0,707 g, 11,22 mmol) in Ethanol (20
ml) werden vereinigt und am Rückfluss
für 8 Stunden
erhitzt. Es wird abgekühlt,
filtriert und das Gemisch wird unter Bildung von (S)-2-(2-Phenoxyethyl)piperazin-1-carbonsäure-tert-butylester
eingedampft (0,639 g, 93%). Massenspektrum (APCI): m/z = 307,2 (M+1).
-
Trifluoressigsäure (3 ml)
wird zu dem Material von oben (0,626 g, 2,04 mmol) in Dichlormethan
(10 ml) bei 0°C
gegeben und gerührt.
Nach 1 Stunde wird das Gemisch auf Raumtemperatur erwärmt und
für zwei Stunden
gerührt
und dann eingedampft. Das Gemisch wird mit 1 N Natriumhydroxid verdünnt und
dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
der Titelverbindung (0,334 g, 79%) konzentriert: Massenspektrum
(APCI): m/z = 207,1 (M+1).
-
Beispiel 174
-
(S)-2-(2-Ethoxyethyl)piperazin
-
Natriumhydrid
(0,558 g, 14,0 mmol) wird portionsweise zu (S)-2-(2-Hydroxyethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(3,075 g, 9,31 mmol) in Dimethylformamid (60 ml) gegeben und für 15 Minuten
gerührt.
Es wird auf 0°C
gekühlt
und Ethyliodid (1,5 ml, 18,6 mmol) wird tropfenweise zugegeben und
bei 0°C gerührt. Nach
30 Minuten wird das Gemisch auf Raumtemperatur erwärmt und über Nacht
gerührt.
Das Gemisch wird mit Ethylacetat verdünnt und die organischen Phasen
werden sechsmal mit Kochsalzlösung
gewaschen. Es wird über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von (S)-2-(2-Ethoxyethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester (3,17
g, 95%) als gelbes Öl
konzentriert.
-
Trifluoressigsäure (2 ml)
wird zu dem Material von oben (0,350 g, 0,976 mmol) in Dichlormethan
(6 ml) bei 0°C
gegeben und für
30 Minuten gerührt.
Das Gemisch wird auf Raumtemperatur erwärmt und für zwei Stunden gerührt und
dann zu einem gelben Öl
eingedampft. Das Gemisch wird mit Methanol verdünnt und auf eine 5 g SCX Säule aufgetragen.
Es wird mit Methanol und dann 2 N Ammoniak in Methanol unter Bildung
der Titelverbindung (144 mg, 94%) als Öl eluiert. Massenspektrum (APCI):
m/z = 159,2 (M+1).
-
Beispiel 175
-
2(S)-(2(S)-Methoxypropyl)piperazin 2(S)-(2(R)-Methoxypropyl)piperazin
-
Methylmagnesiumbromid
(3,61 ml, 10,8 mmol, 3 M Lösung
in Diethylether) wird zu einer Lösung
aus 2-(2-Oxoethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester (3,036
g, 0,84 mmol) in Tetrahydrofuran (30 ml) bei 0°C gegeben. Das Gemisch wird
auf Raumtemperatur erwärmt
und für
5 Stunden gerührt.
Das Gemisch wird mit gesättigtem
Ammoniumchlorid verdünnt
und dreimal mit Ethylacetat extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von 2(S)-(2(S)-Hydroxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
und 2(S)-(2(R)-Hydroxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(3,37 g, 99%) als gelber öliger
Feststoff konzentriert. Massenspektrum (APCI: m/z = 245,2 (M+1-BOC).
-
Zu
dem Material von oben (3,35 g, 9,73 mmol) in Dimethylformamid (70
ml) wird Natriumhydrid (0,583 g, 14,6 mmol) portionsweise gegeben, über 15 Minuten
gerührt
und auf 0°C
gekühlt.
Methyliodid (1,21 ml, 19,5 mmol) wird tropfenweise zugegeben und
bei 0°C
gerührt.
Nach 30 Minuten wird das Gemisch auf Raumtemperatur erwärmt und über Nacht
gerührt.
Das Gemisch wird mit Ethylacetat verdünnt und die organischen Bestandteile
werden sechsmal mit Kochsalzlösung
gewaschen. Es wird über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Es wird durch Blitzchromatographie unter Elution mit einem Stufengradienten (100%
Dichlormethan bis 3% an 2 N Ammoniak-Methanol in Dichlormethan)
unter Bildung von 2(S)-(2(S)-Methoxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
und 2(S)-(2(R)-Methoxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(Gemisch des Diastereoisomere) (2,67 g, 77%) als gelbes Öl gereinigt.
-
Trifluoressigsäure (15
ml) wird zu dem Material von oben (2,65 g, 7,39 mmol) in Dichlormethan
(45 ml) bei 0°C
gegeben und für
30 Minuten gerührt.
Das Gemisch wird auf Raumtemperatur erwärmt und für 6 Stunden gerührt und
dann zu einem gelben Öl
eingedampft. Das Gemisch wird mit Methanol verdünnt und auf eine 30 g SCX Säule aufgetragen.
Es wird mit Methanol gewaschen und mit 2 N Ammoniak in Methanol
unter Bildung der Titelverbindungen (Gemisch des Diastereoisomere)
(1,06 g, 91%) als dickes braunes Öl eluiert. Massenspektrum (APCI):
m/z = 159,2 (M+1).
-
Beispiel 176
-
2(S)-(2(S)-Hydroxypropyl)piperazin 2(S)-(2(R-Hydroxypropyl)piperazin
-
Zu
einer Lösung
aus (S)-(2(S)-Hydroxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
und (S)-(2(R-Hydroxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(5,11 g, 14,8 mmol) in Dichlormethan (100 ml) unter Rühren bei
0°C wird
Trifluoressigsäure
(25 ml) gegeben. Das Gemisch wird auf Raumtemperatur erwärmt und
gerührt.
Nach 18 Stunden wird es zu einem gelben Öl eingedampft. Das Gemisch
wird mit Methanol verdünnt
und auf eine 60 g SCX Säule
gegeben. Es wird mit Methanol gewaschen und mit 2 N Ammoniak in
Methanol unter Bildung der Titelverbindung (Gemisch der Diastereoisomere)
als Öl
(1,932 g, 90%) eluiert: Massenspektrum (APCI): m/z = 145,1 (M+1).
-
Beispiel 177
-
(S)-2-Methyl-1-piperazin-2-yl-propan-2-ol
-
Oxalylchlorid
(2,52 ml, 28,9 mmol) wird zu einer Lösung aus Dimethylsulfoxid (3,42
ml, 48,2 mmol) in Dichlormethan (130 ml) bei –78°C gegeben. Nach 15 Minuten bei –78°C wird (S)-2-(2-Hydroxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(6,64 g, 19,3 mmol) in Dichlormethan (20 ml) zugegeben. Das Gemisch
wird für
1 Stunde bei –78°C gerührt, Triethylamin
(13,4 ml, 96,4 mmol) wird zugegeben und über Nacht auf Raumtemperatur
erwärmt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Es wird durch Blitzchromatographie unter Elution mit einem Stufengradienten
(100% Dichlormethan bis 5% an 2 N Ammoniak-Methanol in Dichlormethan)
unter Bildung von (S)-2-(2-Oxo-propyl)piperazin-1,4-dicarbonsäure-di-tert-butylester (5,77
g, 87%) gereinigt. Massenspektrum (APCI): m/z = 143,1 (M+1-2BOC).
-
Zu
einer Lösung
des Aldehyds von oben (0,263 g, 0,768 mmol) in Tetrahydrofuran (10
ml) bei 0°C
wird Methylmagnesiumbromid (0,28 ml, 0,845 mmol, 3 M Lösung in
Diethylether) gegeben. Das Gemisch wird am Rückfluss erhitzt und über Nacht
gerührt.
Das Gemisch wird mit gesättigtem
Ammoniumchlorid verdünnt
und dreimal mit Ethylacetat extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
von (S)-2-(2-Hydroxy-2-methylpropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(0,258 g, 0,720 mmol, 94%) konzentriert.
Massenspektrum (APCI):
m/z = 359,3 (M+1).
-
Zu
dem Material von oben (4,3 g, 12,0 mmol) in Dichlormethan (100 ml)
bei 0°C
wird Trifluoressigsäure (25
ml) gegeben und für
1 Stunde gerührt.
Das Gemisch wird auf Raumtemperatur erwärmt, über Nacht gerührt und
zu einem braunen Öl
eingedampft. Das Gemisch wird mit Methanol verdünnt und auf eine 60 g SCX Säule aufgetragen.
Es wird mit Methanol gewaschen und mit 2 N Ammoniak in Methanol
unter Bildung der Titelverbindung (1,9 g, 100%) eluiert. Massenspektrum
(APCI): m/z = 159,2 (M+1).
-
Beispiel 178
-
(S)-2-(2-Phenylsulfanylethyl)piperazin
-
Zu
einer Lösung
aus (S)-4-Benzyl-2-(2-hydroxyethyl)lpiperazin-1-carbonsäure-tert-butylester
(4,87 g, 15,2 mmol) in Dichlormethan (200 ml) wird Pyridin (1,84
ml, 22,8 mmol) gefolgt von Methansulfonylchlorid (1,29 ml, 16,7
mmol) gegeben. Es wird bei Raumtemperatur über Nacht gerührt und
mit gesättigtem
wässrigem Natriumbicarbonat
verdünnt.
Es wird dreimal mit Dichlormethan extrahiert, die vereinigten organischen
Extrakte werden über
Natriumsulfat getrocknet, filtriert und im Vakuum unter Bildung
eines braunen Rückstands
konzentriert. Der Rückstand
wird in Dichlormethan rückgelöst und auf
ein Kissen aus Silicagel aufgetragen. Das Kissen wird mit 5% an
2 N Ammoniak-Methanol in Dichlormethan unter Bildung von (S)-4-Benzyl-2-(2-methansulfonyloxyethyl)piperazin-1-carbonsäure-tert-butylester
(3,74 g, 62%) als gelb-braunes dickes Öl gewaschen.
-
Kaliumhydrid
(9,6 g, 84 mmol, 35%) und Thiophenol (9,6 ml, 93,3 mmol) werden
in Tetrahydrofuran (250 ml) bei 0°C
vereinigt und auf Raumtemperatur über 25 Minuten erwärmt. Das
Gemisch wird wieder auf 0°C
gekühlt
und das Mesylat von oben (3,72 g, 9,33 mmol) wird als eine Lösung in
Tetrahydrofuran (50 ml) zugegeben. Das Gemisch wird über Nacht
bei Raumtemperatur gerührt.
Das Gemisch wird mit gesättigtem wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Es wird durch Blitzchromatographie unter Elution mit einem Stufengradienten
ausgehend von Dichlormethan bis zu 5% an 2 N Ammoniak Methanol in
Dichlormethan unter Bildung von (S)-4-Benzyl-2-(2-phenylsulfanylethyl)piperazin-1-carbonsäure-tert-butylester
(2,46 g, 64%) als gelbes Öl
gereinigt. Massenspektrum (APCI): m/z = 413,2 (M+1).
-
Chlorethylchlorformiat
(1,16 ml, 10,7 mmol) und das Material von oben (2,01 g, 4,87 mmol)
werden in Dichlorethan (30 ml) bei 0°C vereinigt und am Rückfluss über Nacht
erhitzt. Das Gemisch wird eingedampft, mit Methanol verdünnt, für 2 Stunden
am Rückfluss
erhitzt und eingedampft. Das Gemisch wird mit Methanol verdünnt und
auf eine 10 g SCX Säule
aufgetragen. Die Säule
wird mit Methanol gewaschen, mit 2%, 5% und 100% an 2 N Ammoniak-Methanol
in Dichlormethan unter Bildung der Titelverbindung (0,470 g, 43%)
als Öl eluiert.
Massenspektrum (APCI): m/z = 223,1 (M+1).
-
Beispiel 180
-
(S)-(1,4-Dibenzylpiperazin-2-yl)acetaldehyd
-
Wasserfreies
DMSO (3,57 ml, 50,25 mmol) wird in wasserfreies Dichlormethan (68,0
ml) gegeben. Es wird auf –78°C gekühlt und
gerührt.
Oxalylchlorid (2 M in Dichlormethan, 12,06 ml, 24,125 mmol) wird
tropfenweise zugegeben. Es wird bei –78°C für 30 Minuten gerührt. (1,4-Dibenzylpiperazin-2-yl)ethanol (6,241
g, 20,1 mmol) in Dichlormethan (12,0 ml) wird zugegeben und gerührt. Nach
1 Stunde wird Triethylamin (14,01 ml, 100,5 mmol) zugegeben und
für eine
weitere Stunde gerührt.
Das Reaktionsgemisch kann sich schrittweise auf Umgebungstemperatur
erwärmen.
Gesättigtes
Ammoniumchlorid (200 ml) wird zugegeben. Die wässrige Portion wird mit Ethylacetat
dreimal extrahiert. Die organische Lösung wird vereinigt, über Natriumsulfat
getrocknet, filtriert und unter verringertem Druck unter Bildung
eines rohen Rückstands
konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Elution mit 2 M Ammoniak in
Methanol : Dichlormethan (5:95) unter Bildung der Titelverbindung
gereinigt. Massenspektrum (m/e): 309,03 (M+1).
-
Beispiel 181
-
(S)-1,4-Dibenzyl-2-(3-methoxyallyl)piperazin
-
LiHMDS
(1 M in THF, 21,38 mmol) wird zu (Methoxymethyl)triphenylphosphoniumchlorid
(7,329 g, 21,38 mmol) in THF (51,0 ml) mittels einer Spritze bei
0°C gegeben
und gerührt.
Nach 30 Minuten wird (S)-(1,4-Dibenzylpiperazin-2-yl)acetaldehyd
(4,71 g, 15,27 mmol) in THF (51,0 ml) mittels einer Spritze zugegeben.
Das Eisbad wird entfernt und bei Umgebungstemperatur gerührt. Nach
4 Stunden wird Wasser zugegeben und mit Ethylacetat zweimal extrahiert.
Die organische Lösung
wird vereinigt, über
Natriumsulfat getrocknet, filtriert und unter verringertem Druck
unter Bildung des rohen Rückstands
konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Elution mit Ethylacetat: Hexan
(40:60) unter Bildung der Titelverbindung gereinigt. Massenspektrum
(m/e): 337,18 (M+1).
-
Beispiel 182
-
(S)-2-(3-Methoxypropyl)piperazin
-
Es
wird 20% Pd(OH)2/C (2,15 g) zu (S)-1,4-Dibenzyl-2-(3-methoxyallyl)piperazin
(3,382 g, 10,05 mmol) in Ethanol (100 ml) unter einer Stickstoffatmosphäre bei 40°C über Nacht
gegeben. Es wird unter Bildung der Titelverbindung filtriert: Massenspektrum
(m/e): 159,1 (M+1).
-
Beispiel 183
-
(S)-2-(3-Ethoxycarbonylallyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
Ein
Gemisch aus (S)-2-(2-Oxoethyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(1,38 g, 4,21 mmol) und (Carbethoxymethylen)triphenylphosphoran
(1,87 g, 5,38 mmol) in THF (17 ml) wird am Rückfluss für 4 h erhitzt. Das Gemisch
wird unter verringertem Druck konzentriert und durch Silicagelchromatographie
unter Elution mit 20% bis 50% EtOAc in Hexan gereinigt. Die gereinigten
Fraktionen werden vereinigt, unter verringertem Druck konzentriert
und mit CH2Cl2/Hexan
(1:2) azeotrop gemacht und unter Vakuum unter Bildung der Titelverbindung
gesetzt. Gelber Teer (0,961 g), Massenspektrum (m/e): 416,09 (M+NH4).
-
Beispiel 184
-
(S)-2-(3-Ethoxycarbonylpropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
Alternativ
dazu wird die Aufschlämmung
aus (S)-2-(3-Ethoxycarbonylallyl)piperazin-1,4-dicarbonsäure-di-tert-butylester (0,757
g, 1,89 mmol) und 10% Pd/C (0,198 g) in Dichlorethan für 2 Tage
evakuiert und mit H2 (3 ×) versetzt. Das Gemisch wird
durch ein Bett aus Celite mit CH2Cl2 (50 ml), EtOH (50 ml), CH2Cl2 (50 ml) und EtOH (50 ml) gewaschen. Der Überstand
wird unter verringertem Druck konzentriert und unter Vakuum unter
Bildung der Titelverbindung gegeben: Gelber Feststoff (0,747 g),
Massenspektrum (m/e): 401,10 (M+H).
-
Beispiel 185
-
(S)-2-(3-Carboxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
Eine
Lösung
aus (S)-2-(3-Ethoxycarbonylpropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(0,739 g, 1,84 mmol) wird in MeOH gerührt und 1,0 N NaOH (2,13 ml)
wird zugegeben und das Rühren
wird über Nacht
fortgesetzt. Das Gemisch wird unter verringertem Druck konzentriert,
mit H2O (40 ml) verdünnt und mit Et2O
(3 × 30
ml) gewaschen. Die wässrige
Phase wird mit 10% NaHSO4 (20 ml) angesäuert und
mit EtOAc (3 × 50
ml) extrahiert. Die organische Phase wird mit Na2SO4 getrocknet, filtriert und unter verringertem
Druck konzentriert und der Rückstand
wird unter Vakuum gegeben, um die Titelverbindung zu erhalten: Weißer Feststoff
(0,618 g), Massenspektrum (m/e): 373,07 (M+H).
-
Beispiel 186
-
(S)-2-(4-Hydroxybutyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
In
eine gerührte
Lösung
aus (S)-2-(3-Carboxypropyl)piperazin-1,4-dicarbonsäure-di-tert-butylester (0,596
g, 1,60 mmol) in THF wird 1,0 M BH3 in THF
(3,2 ml, 3,20 mmol) gegeben und das Rühren wird über Nacht fortgesetzt. Das
Gemisch wird unter verringertem Druck konzentriert, mit H2O (30 ml) verdünnt und mit EtOAc (3 × 50 ml)
extrahiert. Die organischen Phasen werden mit Na2SO4 getrocknet, filtriert, unter verringertem
Druck konzentriert, der Rückstand
wird mit CH2Cl2/Hexan
(1:2) azeotrop gemacht und der Rückstand
wird unter Vakuum gegeben, um die Titelverbindung zu erhalten: Farbloser
Feststoff (0,604 g), Massenspektrum (m/e): 359,12 (M+H).
-
Beispiel 187
-
(S)-2-(4-Methoxybutyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
-
In
einer bei Raumtemperatur gerührten
Lösung
aus (S)-2-(4-Hydroxybutyl)piperazin-1,4-dicarbonsäure-di-tert-butylester
(0,344 g, 0,95 mmol) und Mel (0,179 ml, 2,87 mmol) in DMF (5 ml)
wird NaH (0,042 g, 1,05 mmol, 20% in Mineralöl) gegeben. Es wird über Nacht
gerührt.
Das Gemisch wird mit 75% Kochsalzlösung (50 ml) verdünnt und
mit EtOAc (3 × 100
ml) extrahiert. Die organischen Phasen werden unter verringertem
Druck konzentriert, mit EtOAc (40 ml) verdünnt und mit 75% Kochsalzlösung (5 × 25 ml)
gewaschen. Die organische Phase wird über Na2SO4 getrocknet und filtriert. Das Gemisch wird
unter verringertem Druck konzentriert und durch Silicagelchromatographie
unter Elution mit 20% bis 98% EtOAc in Hexan gereinigt. Die gereinigten
Fraktionen werden vereinigt, unter verringertem Druck konzentriert
und unter Bildung der Titelverbindung unter Vakuum gegeben: Gelber
Teer (0,209 g), Massenspektrum (m/e): 373,13 (M+H).
-
Beispiel 188
-
(S)-2-(4-Methoxybutyl)piperazin
-
In
eine bei Raumtemperatur gerührte
Lösung
aus (S)-2-(4-Methoxybutyl)piperazin-1,4-dicarbonsaure-di-tert-butylester
(0,198 g, 0,53 mmol) in CH2Cl2 (2,6
ml) wird TFA (2,6 ml) gegeben und für 2 h gerührt. Das Gemisch wird unter
verringertem Druck konzentriert, mit H2O
verdünnt
und auf eine SCX Säule
(10 g) geladen. Das Harz wird mit H2O (3 × 100 ml)
und MeOH (3 × 100
ml) gewaschen und das Produkt wird mit 2 M NH3 in MeOH
(3 × 100
ml) eluiert. Die gereinigten Fraktionen werden unter verringertem
Druck konzentriert und der Rückstand
wird mit CH2Cl2/Hexan
(1:2) azeotrop gemacht. Der Rückstand
wird unter Bildung der Titelverbindung unter Vakuum gegeben: Orange
Kristalle (0,093 g), Massenspektrum (m/e): 173,4 (M+H).
-
Beispiel 190
-
2-(R)-tert-Butoxycarbonylamino-3-(2-methylallyloxy)propionsäure
-
Eine
60% Dispersion aus Natriumhydrid in Mineralöl (4,4 g, 115 mmol) wird mit
Hexan gewaschen. DMF (30 ml) wird zugegeben und es wird in einem
Eisbad gekühlt.
Eine Lösung
aus N-(tert-Butoxycarbonyl)-L-serin
(10,0 g, 49 mmol) in DMF (160 ml) wird tropfenweise zugegeben. Es
wird auf Raumtemperatur erwärmt
und gerührt
bis die Wasserstoffentwicklung nach einer Stunde aufhört. Es wird
in einem Eisbad gekühlt und
3-Brom-2-methylpropen (7,40 g, 55 mmol) wird zugegeben. Es wird
für 2 Stunden
gerührt.
Das Gemisch wird in 5% wässriges
Natriumbicarbonat (800 ml) gegossen. Es wird zweimal mit Ethylacetat
gewaschen. Ethylacetat (200 ml) wird zugegeben und die wässrige Phase
wird in Anwesenheit eines pH 3 mit konzentrierter Chlorwasserstoffsäure angesäuert. Die
organische Phase wird mit Wasser gewaschen, getrocknet (Na2SO4) und unter Bildung
der Titelverbindung als klares Öl
(12,7 g) konzentriert.
1H NMR (CDCl3) δ 1,45
(s, 9H), 1,70 (s, 3H), 3,65 (m, 1H), 3,85 (m, 1H), 3,86-3,94 (m,
2H), 4,44 (m, 1H), 4,89 (s, 1H), 4,93 (s, 1H), 5,43 (d, 1H).
-
Beispiel 190a
-
3-(R)-(2-Methylallyloxymethyll)piperazin-2,5-dion
-
Durch
das Ringschlussverfahren in Beispiel 161 mittels 2-(R)-tert-Butoxycarbonylamino-3-(2-methylallyloxy)propionsäure (12,7
g) wird die Titelverbindung als weißer Feststoff (8,95 g) erhalten.
1H NMR (DMSO-d6): δ 1,60 (s,
3H), 3,42 (dd, 1H), 3,57 (d, 1H), 3,66 (d, 1H), 3,75 (d, 1H), 3,80
(m, 1H), 3,81 (s, 2H), 4,82 (s, 1H), 4,86 (s, 1H), 8,03 (s, 1H),
8,11 (s, 1H).
-
Beispiel 190b
-
2-(R)-(2-Methylallyloxymethyl)piperazin
-
Durch
das Reduktionsverfahren in Beispiel 162 und mittels 3-(R)-(2-Methylallyloxymethyl)piperazin-2,5-dion
(8,9 g) wird die Titelverbindung als gelber Feststoff (4,6 g) erhalten.
1H NMR (CDCl3): δ 1,73 (s,
3H), 2,48 (t, 1H), 2,72-3,02 (m, 6H), 3,25 (m, 1H), 3,36 (dd, 1H),
3,88 (s, 1H), 4,89 (s, 1H), 4,94 (s, 1H).
-
Beispiel 191
-
2-(R)-tert-Butoxycarbonylamino-3-(2-methylallyloxy)propionsäure
-
Durch
das Allylierungsverfahren in Beispiel 190 und mittels 2-(R)-tert-Butoxycarbonylamino-3-hydroxy-propionsäure (25,0
g, 122 mmol) wird die Titelverbindung als ein weißer Feststoff
(27,0 g, 90%) erhalten.
1H NMR (CDCl3) δ 1,46
(s, 9H), 2,94 (d, 2H), 3,67 (dd, 1H), 4,00 (d, 2H), 5,23 (dd, 2H),
5,87 (m, 1H), 10,50 (bs, 1H).
-
Beispiel 191a
-
3-(R)-(2-Allyloxymethyl)piperazin-2,5-dion
-
Durch
das Cyclisierungsverfahren in Beispiel 161 und mittels 2-(R)-tert-Butoxycarbonylamino-3-(2-allyloxy)propionsäure (27,0
g) wird die Titelverbindung als ein weißer Feststoff (8,5 g) erhalten.
1H NMR (DMSO-d6) δ 3,49 (dd,
1H), 3,60 (dd, 1H), 3,73-3,85 (m, 3H), 3,96 (dt, 2H), 5,14 (dq,
1H), 5,23 (dq, 1H), 5,84 (m, 1H), 8,05 (bs, 1H), 8,13 (bs, 1H).
-
Beispiel 191b
-
2-(R)-(2-Allyloxymethyl)piperazin
-
Durch
das Reduktionsverfahren in Beispiel 162 und mittels 3-(R)-(2-Allyloxymethyl)piperazin-2,5-dion (8,5 g, 46,1
mmol) wird die Titelverbindung als ein weißer Feststoff (5,0 g, 69%)
erhalten.
1H NMR (CDCl3) δ 2,02 (bs,
2H), 2,71-3,69 (m, 9H), 3,98 (dt, 2H), 5,19 (dq, 1H), 5,27 (dq,
1H), 5,90 (m, 1H).
-
Beispiel 193
-
2-(R)-tert-Butoxycarbonylamino-3-methoxypropionsäure
-
Durch
das Alkylierungsverfahren in Beispiel 190 und mittels 2-(R)-tert-Butoxycarbonylamino-3-hydroxypropionsäure wird
die Titelverbindung erhalten.
-
Beispiel 194
-
(R)-3-Methoxymethylpiperazin-2,5-dion
-
Durch
das Cyclisierungsverfahren das für
Beispiel 161 beschrieben ist, und mittels 2-(R)-tert-Butoxycarbonylamino-3-methoxy-propionsäure (40
g) wird die Titelverbindung als weißes Pulver (20,0 g) erhalten.
1H NMR (DMSO-d6): δ 3,22 (s,
3H), 3,38 (dd, 1H), 3,56 (d, 1H), 3,64 (dd, 1H), 3,72 (d, 1H), 3,79
(m, 1H), 8,00 (bs, 1H), 8,09 (bs, 1H).
-
Beispiel 194a
-
(R)-2-(Methoxymethyl)piperazin
-
Durch
das Reduktionsverfahren das für
Beispiel 162 beschrieben ist und mittels (R)-3-(Methoxymethylpiperazin-2,5-dion
(4,8 g) wird die Titelverbindung als ein gelbes Öl (2,6 g, 67%) erhalten.
1H NMR (CDCl3) δ 2,48 (dd,
1H), 2,72-3,00 (m, 6H), 3,23 (dd, 1H), 3,34 (dd, 1H), 3,35 (s, 3H).
MS (APCI) m/z (relative Intensität)
131 (100).
-
Beispiel 195
-
(R)-2-Phenoxymethylpiperazin
-
Zu
einer Lösung
aus Diethylazodicarboxylat (3,46 ml, 22 mmol) in THF (6 ml) wird
eine Lösung
aus (R)-1,4-Dibenzylpiperazin-2-yl)methanol (5,92 g, 20 mmol) (J.
Med. Chem., 1993, Vol. 36, 2075-2083 und J. Med. Chem., 1993, Vol
36, 990-1000), Phenol (2,07 g, 22 mmol) und Triphenylphosphin (5,78
g, 22 mmol) in THF (100 ml) bei 0°C
gegeben. Es wird bei Raumtemperatur über Nacht gerührt, zwischen
Wasser und Ether aufgeteilt und konz. HCl wird zugegeben, bis die
wässrige
Phase unter pH = 2 beträgt.
Die wässrige
Phase wird abgetrennt und mit Diethylether gewaschen. Kaliumcarbonat
wird zu der wässrigen
Phase gegeben, bis der pH 10 beträgt und mit Ethylacetat extrahiert,
getrocknet und unter Bildung eines gelben Öls (7,4 g) konzentriert. Ethanol
(200 ml) und konz. HCl (2 ml) werden zu dem rohen Öl gegeben
und die Lösung
wird in eine Parrflasche gegeben. Die Atmosphäre wird zweimal mit Stickstoff ersetzt
und 10% Pd/C (200 mg) wird zugegeben. Die Parrflasche wird gewaschen
und mit Wasserstoff zweimal ausgetauscht. Sie wird auf 50°C erhitzt und über Nacht
geschüttelt.
Es wird gekühlt,
durch Celite filtriert und das Celitekissen wird dreimal mit Wasser gewaschen.
Der Ethanol wird aus den vereinigten Filtraten mittels Eindampfen
im Vakuum entfernt. Kaliumcarbonat wird zu dem Gemisch gegeben,
bis der pH 10 beträgt.
Das Gemisch wird dreimal mit 25% Isopropanol in CH2Cl2 extrahiert. Es wird getrocknet und die
vereinigten organischen Phasen werden unter Bildung des Titelpiperazins
als weißer
Feststoff (3,1 g, 81%) konzentriert.
1H
NMR (CDCl3) δ 2,57 (dd, 1H), 2,75-3,02 (m,
5H), 3,08 (m, 1H), 3,80 (dd, 1H), 3,86 (dd, 1H), 6,86 (d, 2H), 6,92
(t, 1H), 7,24 (t, 2H). MS (APCI) m/z (relative Intensität) 193 (100)
-
Beispiel 196
-
2-(R)-tert-Butoxycarbonylamino-3-(2-ethoxy)propionsäure
-
Durch
das Alkylierungsverfahren in Beispiel 190 und mittels 2-(R)-tert-Butoxycarbonylamino-3-hydroxypropionsäure (50,0
g) wird die Titelverbindung als klares Öl (41 g) erhalten.
1H NMR (CDCl3) δ 1,17 (t,
3H), 1,45 (s, 9H), 3,53 (q, 2H), 3,67 (dd, 1H), 3,90 (dd, 1H), 4,43
(m, 1H), 5,44 (d, 1H).
-
Beispiel 196a
-
(R)-3-Ethoxymethylpiperazin-2,5-dion
-
Durch
das Cyclisierungsverfahren das für
Beispiel 161 beschrieben ist und mittels 2-(R)-tert-Butoxycarbonylamino-3-methoxypropionsäure (24
g) wird die Titelverbindung als weißes Pulver (6,34 g) erhalten.
1H NMR (DMSO-d6) δ 1,03 (t,
3H), 3,40 (q, 2H), 3,41 (dd, 1H), 3,56 (d, 1H), 3,70-3,80 (m, 3H),
7,99 (bs, 1H), 8,13 (bs, 1H).
-
Beispiel 196b
-
(R)-2-Ethoxymethylpiperazin
-
Durch
das Reduktionsverfahren das für
Beispiel 162 beschrieben ist und mittels (R)-3-Ethoxymethylpiperazin-2,5-dion (6,0
g) wird die Titelverbindung als gelbes Öl (4,61 g) erhalten.
1H NMR (CDCl3) δ 1,20 (t,
3H), 2,48 (dd, 1H), 2,70-3,40 (m, 8H), 3,42 (q, 2H). MS (APCI) m/z
(relative Intensität)
145 (100).
-
Beispiel 197
-
(R)-3-Benzyloxymethylpiperazin-2,5-dion
-
Durch
das Cyclisierungsverfahren das für
Beispiel 161 beschrieben ist und mittels 3-(R)-Benzyloxy-2-tert-butoxycarbonylaminopropionsäure (50,0
g) wird die Titelverbindung als weißer Feststoff (26,4 g, 66%)
erhalten.
1H NMR (DMSO-d6) δ 3,52 (dd,
1H), 3,57 (d, 1H), 3,73 (d, 1H), 3,76 (dd, 1H), 3,84 (m, 1H), 4,47
(s, 2H), 7,23-7,35 (m, 5H), 8,02 (s, 1H), 8,12 (s, 1H). MS (APCI)
m/z (relative Intensität)
235 (100).
-
Beispiel 197a
-
(R)-2-Benzyloxymethylpiperazin
-
Durch
das Reduktionsverfahren in Beispiel 162 und mittel (R)-3-Benzyloxymethylpiperazin-2,5-dion (16 g) wird
die Titelverbindung als oranges Öl
(9,0 g, 64%) erhalten.
1H NMR (CDCl3) δ 2,16
(dd, 1H), 2,45 (ddd, 1H), 2,53 (ddd, 1H), 2,64 (d, 1H), 2,69 (m,
1H), 2,72 (d, 1H), 3,22 (m, 2H), 3,36 (m, 1H), 4,44 (m, 2H), 7,24-7,34
(m, 5H). MS (APCI) m/z (relative Intensität) 207 (100).
-
Beispiel 198
-
(S)-3-Methyl-11-[3-(2-methoxyethyll)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
3-Methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(400,0 mg, 1,54 mmol), (S)-2-(2-Methoxyethyl)piperazin
(444,2 mg, 3,08 mmol), N,N-Diisopropylethylamin (199,0 mg, 1,54
mmol), DMSO (0,7 ml) und Toluol (2,8 ml) werden vereinigt und gerührt und
das Gemisch wird bei 110°C
erhitzt. Nach 48 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt
und mit Ethylacetat verdünnt.
Die or ganische Phase wird mit 0,1 N NaOH und Kochsalzlösung gewaschen.
Es wird getrocknet (Natriumsulfat) und die organische Phase wird
zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels Dichlormethan/Methanol (90:10) unter
Bildung von 257,1 mg (48%) eines gelben Schaums gereinigt. Smp.
64°C Zers.
Massenspektrum (Ionenspray): m/z = 433,1 (M+1).
-
Beispiel 199
-
(S)-2-Methyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
2-Methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(600,0 mg, 2,31 mg), (S)-2-(2-Methoxyethyl)piperazin
(666,3 mg, 4,62 mmol), N,N-Diisopropylethylamin (298,6 mg, 2,31
mmol), DMSO (1,0 ml) und Toluol (4,0 ml) werden vereinigt und gerührt und
das Gemisch wird bei 110°C
erhitzt. Nach 46 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt
und mit Ethylacetat verdünnt.
Die organische Phase wird mit 0,1 N NaOH und Kochsalzlösung gewaschen,
getrocknet (Natriumsulfat) und die organische Phase wird zu einem
Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels Dichlormethan/Methanol (90:10) unter Bildung
von 425,5 mg (53%) eines gelben Schaums gereinigt. Smp 63°C, Zers.
Massenspektrum (Ionenspray): m/z = 351,2 (M+1).
-
Beispiel 200
-
(S)-2-Isopropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
2-Isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylamin
(0,969 g, 3,85 mmol) und (S)-2-(2-Methoxyethyl)piperazin (0,556 g, 3,85
mmol) werden in NMP (7,0 ml) vereinigt und bei 200°C für 4 Stunden
erhitzt. Es wird auf Umgebungstemperatur gekühlt und mit Wasser verdünnt. Es
wird mit Ethylacetat unter Bildung von 1,51 g des rohen Produkts
extrahiert. Eine Silicagelchromatographie und Elution mit Methylenchlorid
: 2 N NH3/Methanol (100:4) ergibt 0,560
g der Titelverbindung als hellbraunen Feststoff: Smp. 158-160°C Massenspektrum
(Ionenspray): m/z = 379 (M+1). Analyse für C23H30N4O (0,2 H2O): berechnet: C 72,29, H 8,02, N 14,66,
gefunden: C 72,20, H 7,70, N 14,60.
-
Beispiel 201
-
(S)-2-Isopropyl-11-[3-(3-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
Mittels
eines ähnlichen
Verfahrens zu Beispiel 203 und unter Verwendung von 2-Isopropyl-5H-dibenzo[b,e][1,4]diazepin-1l-ylamin
wird die Titelverbindung erhalten. Massenspektrum (m/e): 393,12
(M+1).
-
Beispiel 202
-
(S)-2-Trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
Eine
Suspension aus 2-Trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochloridsalz
(626 mg) und (S)-2-(2-Methoxyethyl)piperazin (863 mg) in DMSO (1,42
ml, DMSO pro mmol Amin) und Toluol (5,68 ml pro mmol Amin) wird
am Rückfluss
für 48
Stunden erhitzt. Das Toluol wird unter Vakuum eingedampft und die
entstehende Lösung
wird in Wasser (5,71 ml pro mmol Amin) gegossen. Der entstehende
braune Feststoff wird durch Blitzchromatographie (Methylenchlorid/Methanol
(95:5) unter Bildung der Titelverbindung als gelber Feststoff (499
mg, 60%) gereinigt. Smp. 71-82°C.
1H NMR (CDCl3) δ 1,75-1,60
(m, 2H), 2,64 (dd, 1H), 3,07-2,94 (m, 4H), 3,31 (s, 3H), 3,48 (ddd,
2H), 3,78 (bs, 2H), 5,09 (s, 1H), 6,69 (dd, 1H), 6,93-6,88 (m, 2H),
7,00 (dt, 1H), 7,10 (dd, 1H), 7,57-7,50 (m, 2H). MS (ESI/ pos) m/z
(relative Intensität)
405,3 (100).
-
Beispiel 203
-
(S)-2-Trifluormethyl-11-[3-(3-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
2-Trifluormethyl-5H-dibenzo[b,e]1,4]diazepin-11-ylamin
(0,438 g, 1,58 mmol), (S)-2-(3-Methoxypropyl)piperazin (0,25 g,
1,58 mmol) wird zu NMP (2,8 ml) gegeben. Es wird bei 200°C unter Rühren erhitzt.
Nach 2 Stunden wird das Erhitzen gestoppt und das Reaktionsgemisch
kann sich auf Umgebungstemperatur abkühlt. Kochsalzlösung wird
zugegeben und es wird mit Ethylacetat extrahiert. Die organische
Lösung
wird mit Kochsalzlösung
dreimal gewaschen, über
Natriumsulfat getrocknet, filtriert und unter verringertem Druck
unter Bildung des rohen Rückstands
konzentriert. Eine Reinigung des Rückstands durch Blitzchromatographie
unter Elution mit 2 M Ammoniak in Methanol : Dichlormethan (5:95)
ergibt die Titelverbindung. Massenspektrum (m/e) 419,05 (M+1).
-
Beispiel 204
-
(S)-8-Chlor-2-methyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 8-Chlor-2-methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,2 g, 0,68 mmol) und Diisopropylethylamin (0,264 g, 2,04 mmol)
wird in 1-Methyl-2-pyrrolidinon (5 ml) für 30 Minuten gerührt. (S)-2-(2-Methoxyxethyl)piperazin
(0,294 g, 2,04 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und zweimal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet. Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Gemisch aus 50% Hexan : 50% Dichlormethan
plus 2% Isopropylamingesamtvolumen ergibt die freie Base der Titelverbindung
(0,097 g, 0,25 mmol, 37% Ausbeute) als gelben amorphen Feststoff.
Das Produkt wird durch Lösen
des Produkts in Methanol und Zugabe von einem Äquivalent Bernsteinsäure in das
Produkt umgewandelt, das Gemisch wird verwirbelt oder ultrabeschallt,
bis keine feste Bernsteinsäure
verbleibt und das Lösemittel
wird unter verringertem Druck unter Bildung der Titelverbindung
entfernt. Massenspektrum (m/e): 385 (M+1).
-
Beispiel 205
-
(S)-8-Chlor-2-isopropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 8-Chlor-2-isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,285 g, 0,88 mol) und Diisopropylethylamin (0,137 g, 1,06 mmol)
wird in 1-Methyl-2-pyrtrolidinon (7 ml) für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,383 g, 2,65 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und zweimal mit Kochsalzlösung,
zweimal mit Wasser und einmal wieder mit Kochsalzlösung gewaschen.
Die organische Phase wird gesammelt und über Natriumsulfat getrocknet.
Das Lösemittel
wird unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von 99% Dichlormethan
: 1% an 2 M Ammoniak in Methanol bis zu 97% : 3% ergibt die freie
Base der Titelverbindung (0,132 g, 0,32 mmol, 36% Ausbeute) als
hellbraunen amorphen Feststoff. Das Produkt wird wie vorher beschrieben
unter Bildung der Titelverbindung in das Succinatsalz umgewandelt.
Massenspektrum (m/e): 413 (M+1). Exakte Massenberechnung: Berechnet
413,2108, gefunden 413,2104.
-
Beispiel 206
-
(S)-8-Fluor-2-methyl-11-[3-(2-methoxyethyllpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 8-Fluor-2-methyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,2 g, 0,72 mmol) und Diisopropylethylamin (0,19 g, 1,44 mmol)
in 1-Methyl-2-pyrrolidinon (5 ml) wird für 30 Minuten gerührt. (S)-2-(2-Methoxyethyll)piperazin
(0,31 g, 2,16 mmol) wird zugegeben und das Reaktionsgemisch wird für 16 Stunden
auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 100 ml Ethylacetat verdünnt
und zweimal mit Kochsalzlösung,
zweimal mit Wasser und einmal mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet. Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Gemisch aus 50% Hexan : 50% Dichlormethan
plus 2% Gesamtvolumen an Isopropylamin ergibt die freie Base der
Titelverbindung (0,073 g, 0,20 mmol, 28% Ausbeute) als gelben amorphen
Feststoff. Das Produkt wird wie vorher beschrieben unter Bildung
der Titelverbindung in das Succinatsalz umgewandelt: Massenspektrum (m/e):
369 (M+1): Exakte Massenspezifität:L
Berechnet 369,2091, gefunden: 369,2109.
-
Beispiel 207
-
(S)-8-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 8-Fluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhzydrochlorid
(0,2 g, 0,60 mmol) und Diisopropylethylamin (0,078 g, 0,60 mmol)
wird in 1-Methyl-2-pyrrolidinon (5 ml) für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,261 g, 1,81 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und einmal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet, Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von 99% Dichlormethan:
1% an 2 M Ammoniak in Methanol und bis zu 95% : 5% ergibt die freie
Base der Titelverbindung (0,061 g 0,14 mmol, 24% Ausbeute) als gelb-braunen
amorphen Feststoff. Das Produkt wird wie vorher beschrieben unter
Bildung der Titelverbindung in das Succinatsalz umgewandelt. Massenspektrum
(m/e): 423 (M+1): Exakte Massenspezifität: Berechnet: 423,1808. gefunden: 423,1799.
-
Beispiel 208
-
(S)-8-Fluor-2-isorpropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 8-Fluor-2-isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,3 g, 0,98 mmol) und Diisopropylethylamin (0,139 g, 2,94 mmol)
in 1-Methyl-2-pyrrolidinon (6 ml) wird für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,139 g, 1,08 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und einmal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet. Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von 100% Dichlormethan
und bis zu 90% Dichlormethan : 10% an 2% Ammoniak in Methanol ergibt
die freie Base der Titelverbin dung (0,094 g 0,24 mmol, 24% Ausbeute)
als gelb-braunen amorphen Feststoff. Das Produkt wird wie vorher
beschrieben unter Bildung der Titelverbindung in das Succinatsalz
umgewandelt. Massenspektrum (m/e): 397 (M+1).
-
Beispiel 209
-
(S)-7-Fluor-2-isorpopyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 7-Fluor-2-isopropyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,2 g, 0,65 mmol) und Diisopropylethylamin (0,093 g, 1,96 mmol)
in 1-Methyl-2-pyrrolidinon (4 ml) wird für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,28 g, 1,96 mmol) wird zugegeben und das entstehende Gemisch wird für 16 Stunden
auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und einmal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet. Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem linearen Gradienten ausgehend von 100% Dichlormethan
und bis zu 90% Dichlormethan : 10% an 2% Ammoniak in Methanol ergibt
die freie Base der Titelverbindung (0,085 g 0,21 mmol, 33% Ausbeute)
als gelb-braunen amorphen Feststoff. Das Produkt wird wie vorher
beschrieben unter Bildung der Titelverbindung in das Succinatsalz
umgewandelt. Massenspektrum (m/e): 397 (M+1).
-
Beispiel 210
-
(S)-7-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 7-Fluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,4 g, 1,21 mmol) und Diisopropylethylamin (0,171 g, 1,33 mmol)
in 1-Methyl-2-pyrrolidinon (8 ml) wird für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,52 g, 3,62 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und einmal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über Natriumsulfat
getrocknet. Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem linearen Gradienten ausgehend von 99% Dichlormethan
: 1% an 2 M Ammoniak in Methanol und bis zu 90% Dichlormethan :
10% an 2 M Ammoniak in Methanol ergibt die freie Base der Titelverbindung
(0,153 g 0,36 mmol, 30% Ausbeute) als gelb-orangen amorphen Feststoff.
Das Produkt wird wie vorher beschrieben unter Bildung der Titelverbindung
in das Succinatsalz umgewandelt. Massenspektrum (m/e): 423 (M+1).
Exakte Massenspezifität:
Berechnet: 423,1808, gefunden: 423,1790.
-
Beispiel 211
-
(S)-7-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
7-Fluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,399 g, 1,20 mmol), (S)-2-(3-Methoxypropyl)piperazin (0,381 g,
2,41 mmol) und Diisopropylethylamin (0,21 ml, 1,20 mmol) werden in
einem Gemisch aus Toluol (4,5 ml) und Dimethylsulfoxid (1,5 l) vereinigt
und bei 110°C
für 24
Stunden gerührt.
Das Gemisch wird eingedampft und dann durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 7% an 2 N Ammoniak-Methanol in Dichlormethan unter Bildung von
(S)-7-Fluor-11-[3-(3-methoxypropyl)piperazin-1-yl]-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin
(0,268 g, 0,614 mmol, 51%) als gelbes Öl gereinigt. Massenspektrum
(APCI): m/z = 437,2 (M+1).
-
Beispiel 212
-
(S)-8-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
8-Fluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,201 g, 0,606 mmol), (S)-2-(3-Methoxypropyl)piperazin (0,192 g,
1,21 mmol) und Diisopropylethylamin (0,106 ml, 0,606 mmol) werden
in einem Gemisch aus Toluol (3 ml) und Dimethylsulfoxid (1 ml) vereinigt
und bei 110°C
für 24
Stunden gerührt.
Das Gemisch wird eingedampft und dann durch Blitzchromatographie
unter Flution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 7% an 2 N Ammoniak-Methanol in Dichlormethan unter Bildung
von 8-Fluor-11-[3-(3-methoxypropyl)piperazin-1-yl]-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin
(0,151 g, 0,346 mmol, 57%) als gelbes Öl gereinigt. Massenspektrum
(APCI): m/z = 437,2 (M+1).
-
Beispiel 213
-
(S)-2-Cyclopropyl-7-fluor-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 2-Cyclopropyl-7-fluor-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,120 g, 0,40 mmol) und Diisopropylethylamin (0,058 g, 1,22 mmol)
in 1-Methyl-2-pyrrolidinon (4 ml) wird für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,176 g, 1,22 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 50 ml Ethylacetat verdünnt
und einmal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet. Das Lösemittel wird
unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem linearen Gradienten ausgehend von 100% Dichlormethan
bis zu 90% Dichlormethan : 10% an 2 M Ammoniak in Methanol ergibt
die freie Base der Titelverbindung (0,067 g 0,17 mmol, 43% Ausbeute)
als roten amorphen Feststoff. Das Produkt wird wie vorher beschrieben
unter Bildung der Titelverbindung in das Succinatsalz umgewandelt.
Massenspektrum (m/e): 395 (M+1). Exakte Massenspezifität: Berechnet:
395,2247, gefunden: 395,2244
-
Beispiel 214
-
(S)-7,8-Difluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Eine
Lösung
aus 7,8-Difluor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,2 g, 0,57 mmol) und Diisopropylethylamin (0,081 g, 0,63 mmol)
in 1-Methyl-2-pyrrolidinon (4 ml) wird für 30 Minuten gerührt. (S)-2-(2-Methoxyethyl)piperazin
(0,25 g, 1,72 mmol) wird zugegeben und das entstehende Gemisch wird
für 16
Stunden auf 195°C
erhitzt. Das Reaktionsgemisch wird auf Umgebungstemperatur gekühlt. Es
wird mit 40 ml Ethylacetat verdünnt
und einmal mit Kochsalzlösung,
zweimal mit Wasser und erneut mit Kochsalzlösung gewaschen. Die organische
Phase wird gesammelt und über
Natriumsulfat getrocknet. Das Lösemittel
wird unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem linearen Gradienten ausgehend von 100% Dichlormethan
bis zu 85% Dichlormethan : 15% an 2 M Ammoniak in Methanol ergibt
die freie Base der Titelverbindung (0,103 g 0,23 mmol, 41% Ausbeute)
als hellbraunen amorphen Feststoff. Das Produkt wird wie vorher
beschrieben unter Bildung der Titelverbindung in das Succinatsalz
umgewandelt. Massenspektrum (m/e): 441 (M+1). Exakte Massenspezifität: Berechnet: 441,1714,
gefunden: 441,1718.
-
Beispiel 215
-
(S)-8-Chlor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Mittels
eines Verfahrens, das zu dem Verfahren von (S)-8-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat ähnlich ist
und unter Verwendung von 8-Chlor-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,706 g, 2,02 mmol) werden 0,081 g (9% Ausbeute) der freien Base
als gelber Feststoff erhalten. Das Produkt wird wie vorher beschrieben unter
Bildung der Titelverbindung in das Succinatsalz umgewandelt. Massenspektrum
(m/e): 440 (M+1).
-
Beispiel 215a
-
(S)-2-Chlor-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
2-Chlor-5H-dibenzo[b,e][1,4]diazepin-11-ylaminhydrochlorid
(0,840 g, 3,0 mmol), (S)-2-(2-Methoxyethyl)piperazin
(0,865 g, 6,0 mmol), N,N-Diisopropylethylamin (0,53 ml, 3,0 mmol),
DMSO (1,25 ml) und Toluol (5,0 ml) werden vereinigt und gerührt und
das Gemisch wird auf 110°C
erhitzt. Nach 46 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt
und mit Ethylacetat verdünnt.
Die organische Phase wird mit 0,1 N NaOH und Kochsalzlösung gewaschen
getrocknet (Natriumsulfat) und die organische Phase wird zu 0,985
g des rohen Produkts konzentriert. Eine Silicagelchromatographie
unter Elution mit Dichlormethan/Methanol (90:10) ergibt 0,501 g
der Titelverbindung als gelben Schaum: Massenspektrum (Ionenspray):
m/z = 371 (M+1).
-
Beispiel 216
-
(S)-2-Methyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
(S)-2-Methyl-11-[3-(2-methoxyethyl)piperazin-2-yl]-5H-dibenzo[b,e][1,4]diazepin
(300,0 mg, 0,86 mmol), Formaldehyd (76,4 μl, 0,94 mmol, 37% in Wasser)
und 1,2-Dichlorethan (28,0 ml) werden vereinigt und das Gemisch
wird bei Umgebungstemperatur für
5 Minuten gerührt
und Natriumtriacetoxyborhydrid (272,1 mg, 1,28 mmol) wird zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt,
die wässrige
Phase wird mit Dichlormethan extrahiert und vereinigt, gewaschen
(Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand
wird auf Silicagel mittels Dichlormethan/Methanol (90:10) unter
Bildung von 260,4 mg (89%) eines gelben Schaums gereinigt. Smp.
60°C. Zers.
Massenspektrum (Ionenspray): m/z = 365,2 (M+1).
-
Beispiel 217
-
(S)-3-Methyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
(S)-3-Methyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(215,4 mg, 0,61 mmol), Formaldehyd (54,9 μl, 0,68 mmol, 37% in Wasser)
und 1,2-Dichlorethan (20,0 ml) werden vereinigt. Das Gemisch wird
bei Umgebungstemperatur für
5 Minuten gerührt
und Natriumtriacetoxyborhydrid (195,4 mg, 0,92 mmol) wird dann zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt,
die wässrige
Phase wird mit Dichlormethan extrahiert und vereinigt, gewaschen
(Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand
wird auf Silicagel mittels Dichlormethan/Methanol (90:10) unter
Bildung von 171,1 mg (76%) eines gelben Schaums gereinigt. Smp.
100-108°C. Massenspektrum
(Ionenspray): m/z = 365,2 (M+1).
-
Beispiel 218
-
(S)-3-Isopropyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinydrochlorid
-
(S)-2-Isopropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,405 g, 1,07 mmol) und 37% Formaldehydlösung (0,1 ml, 1,12 mmol) in
1,2-Dichlorethan (25 ml) werden vereinigt. Es wird für 10 Minuten
gerührt
und Natriumtriacetoxyborhydrid (0,343 g, 1,60 mmol) wird zugegeben.
Es wird für
weitere 30 Minuten gerührt
und dann wird die Lösung
auf gesättigte
Natriumbicarbonatlösung
gegossen. Es wird mit Methylenchlorid unter Bildung von 0,489 g
des rohen Produkts extrahiert. Eine Silicagelchromatographie unter
Elution mit Methylenchlorid : Methanol (100:10) ergibt 0,354 g der
Titelverbindung als freie Base. Das Dihydrochloridsalz fällt in Ethylacetat
als Feststoff aus. Smp. 210°C.
Massenspektrum (Ionenspray): m/z = 393 (M+1). Analyse für C24H34Cl2N4O: Berechnet: C 61,93, H 7,36, N 12,04.
Gefunden: C 61,74, H 7,47, N 11,86.
-
Beispiel 219
-
(S)-2-Isopropyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
Mittels
eines Verfahrens, das zu dem von Beispiel 222 ähnlich ist, wird die Titelverbindung
erhalten. Massenspektrum (m/e): 407,15 (M+1).
-
Beispiel 220
-
(S)-2-Isopropyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepindihydrochlorid
-
Mittels
eines Verfahrens, das zu dem von Beispiel 223 ähnlich ist, wird die Titelverbindung
erhalten. Massenspektrum (m/e): 407,15 (M+1).
-
Beispiel 221
-
(S)-2-Trifluormethyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
Wässriges
37% Formaldehyd (1,1 Äquivalente)
wird zu einer Lösung
aus (S)-2-Trifluormethyl-11-[3-(3-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(470 mg) in Dichlorethan (0,2 M) gegeben. Das Gemisch wird für 2 Minuten
gerührt
und Natriumtriacetoxyborhydrid (1,5 Äquivalente) wird zugegeben.
Die Suspension wird für
30 Minuten gerührt
und mit einer gesättigten
wässrigen
Lösung
aus Natriumbicarbonat gestoppt. Die wässrige Phase wird dreimal mit
Dichlormethan extrahiert und die organischen Phasen werden vereinigt, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird mittels Chromaotgraphie
auf Silicagel (Methylenchlorid/Methanol (90:10) unter Bildung der
Titelverbindung als gelber Feststoff (394 mg, 81%) gereinigt. Smp.
48-56°C.
1H NMR (CDCl3): δ 1,70-1,56
(m, 1H), 2,04-1,92 (m, 1H), 2,31 (m, 1H), 2,37 (s, 3H), 2,43 (dt,
1H), 2,81 (t, 1H), 2,89 (d, 1H), 3,13 8t, 1H), 3,25 (s, 3H), 3,38
(t, 2H), 3,73 (bs, 2H), 5,10 (s, 1H), 6,69 (dd, 1H), 6,93-6,88 (m, 2H),
7,00 (dt, 1H), 7,10 (dd, 1H), 7,56-7,50 (m, 2H): MS (ESI/ neg.)
m/z (relative Intensität)
417,4 (100).
-
Beispiel 222
-
(S)-2-Trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
-
(S)-2-Trifluormethyl-11-[3-(3-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,167 g, 0,399 mmol), Formaldehyd (40,8 g, 0,499 mmol, 37 5) und
Natriumtriacetoxyborhydrid (0,127 g, 0,598 mmol) werden in Dichlormethan
(3 ml) gegeben. Es wird bei Umgebungstemperatur über Nacht gerührt. Das
Gemisch wird mit Wasser verdünnt
und mit Dichlormethan extrahiert. Die organische Phase wird zweimal
mit Kochsalzlösung
gewaschen, über
Natriumsulfat getrocknet, filtriert und unter verringertem Druck
unter Bildung des rohen Rückstands
konzentriert. Der Rückstand
wird mittels Blitzchromatographie unter Elution mit 2 M Ammoniak
in Methanol : Dichlormethan (5:95) unter Bildung der Titelverbindung
gereinigt: Massenspektrum (m/e) 433,08 (M+1).
-
Beispiel 223
-
(S)-2-Trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepindihydrochlorid
-
2
M HCl wird in Diethylether (2,0 ml) zu (S)-2-Trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,112 g, 0,259 mmol) in Ethylether (1,0 ml) gegeben und dann unter verringertem
Druck konzentriert. Hexan wird zugegeben und der Feststoff aus der
Vakuumfiltration wird unter Bildung der Titelverbindung transferiert.
Massenspektrum (m/e): 433,08 (M+1).
-
Beispiel 224
-
(S)-8-Chlor-2-methyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-8-Chlor-2-methyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,070 g, 0,18 mmol) wird in Dichlormethan (4 ml) gelöst. Natriumtriacetoxyborhydrid
(0,116 g, 0,55 mmol) und Formaldehyd (0,011 g, 0,36 mmol, 0,030
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für zwei Stunden bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 15 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringertem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem Gemisch aus 75% Hexan und 25% Chloroform mit 5%
Gesamtvolumen an 2 M Ammoniak in Methanol ergibt die freie Base
der Titelverbindung (0,039 g, 0,10 mmol, 54% Ausbeute) als gelben
amorphen Feststoff. Es wird dann in das Succinatsalz, wie vorher
beschrieben, umgewandelt. Massenspektrum (m/e): 399 (M+1).
-
Beispiel 225
-
(S)-8-Chlor-2-isopropyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]
diazepinsuccinat
-
(S)-8-Chlor-2-isopropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat (0,089
g, 0,22 mmol) wird in Dichlormethan (12 ml) gelöst. Natriumtriacetoxyborhydrid
(0,137 g, 0,65 mmol) und Formaldehyd (0,013 g, 0,43 mmol, 0,035
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für zwei Stunden bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 50 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden vereinigt, über Natriumsulfat
getrocknet und das Lösemittel
wird unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Gemisch aus 75% Hexan und 25% Chloroform
mit 1% Gesamtvolumen an Isopropylamin ergibt die freie Base der
Titelverbindung (0,027 g, 0,06 mmol, 29% Ausbeute) als gelben amorphen
Feststoff. Es wird dann in das Succinatsalz, wie vorher beschrieben,
umgewandelt. Massenspektrum (m/e): 427 (M+1). Exaktes Massenspektrum:
Berechnet: 427,2265, gefunden: 427,2279.
-
Beispiel 227
-
(S)-8-Fluor-2-methyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-8-Fluor-2-methyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat (0,043
g, 0,12 mmol) wird in Dichlormethan (4 ml) gelöst. Natriumtriacetoxyborhydrid
(0,074 g, 0,35 mmol) und Formaldehyd (0,007 g, 0,23 mmol, 0,019
g einer 37% wässrigen
Lösung)
werden zugegeben. und das Gemisch wird für zwei Stunden bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 50 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden vereinigt, über Natriumsulfat
getrocknet und das Lösemittel
wird unter verringertem Druck entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Gemisch aus 75% Hexan und 25% Dichlormethan mit
3% Gesamtvolumen an 2 M Ammoniak in Methanol ergibt die freie Base
der Titelverbindung (0,032 g, 0,08 mmol, 72% Ausbeute) als gelben
amorphen Feststoff. Es wird dann in das Succinatsalz, wie vorher
beschrieben, umgewandelt. Massenspektrum (m/e): 383 (M+1). Exaktes
Massenspektrum: Berechnet: 383,2247, gefunden: 383,2251.
-
Beispiel 228
-
(S)-8-Fluor-2-triffluormethyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-8-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,031 g, 0,07 mmol) wird in Dichlormethan (4 ml) gelöst. Natriumtriacetoxyborhydrid
(0,047 g, 0,21 mmol) und Formaldehyd (0,004 g, 0,14 mmol, 0,012
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für zwei Stunden bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 50 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringertem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem Gemisch aus 75% Hexan und 25% Chloroform mit 5%
Gesamtvolumen an 2 M Ammoniak in Methanol ergibt die freie Base
der Titelverbindung (0,021 g, 0,05 mmol, 66% Ausbeute) als gelben
amorphen Feststoff. Es wird dann in das Succinatsalz, wie vorher
beschrieben, umgewandelt. Massenspektrum (m/e): 437 (M+1).
-
Beispiel 229
-
(S)-8-Fluor-2-isopropyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-8-Fluor-2-isopropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat (0,046
g, 0,12 mmol) wird in Dichlormethan (6 ml) gelöst. Natriumtriacetoxyborhydrid
(0,049 g, 0,23 mmol) und Formaldehyd (0,004 g, 0,14 mmol, 0,011
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für zwei Stunden bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 50 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringer tem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem Gemisch aus 50% Hexan und 50% Dichlormethan mit
1% Gesamtvolumen an Isopropylamin ergibt die freie Base der Titelverbindung
(0,030 g, 0,07 mmol, 63% Ausbeute) als gelben amorphen Feststoff.
Es wird dann in das Succinatsalz, wie vorher beschrieben, umgewandelt.
Massenspektrum (m/e): 411 (M+1).
-
Beispiel 230
-
(S)-7-Fluor-2-isopropyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-7-Fluor-2-isopropyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat (0,063
g, 0,16 mmol) wird in Dichlormethan (8 ml) gelöst. Natriumtriacetoxyborhydrid
(0,067 g, 0,32 mmol) und Formaldehyd (0,005 g, 0,16 mmol, 0,013
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für zwei Stunden bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 30 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit 50 ml Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringertem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem linearen Gradienten ausgehend von 99% Dichlormethan:
1% an 2 M Ammoniak in Methanol bis zu 90% Dichlormethan : 10% 2
M Ammoniak in Methanol ergibt die freie Base der Titelverbindung
(0,041 g, 0,10 mmol, 63% Ausbeute) als gelben amorphen Feststoff.
Es wird dann in das Succinatsalz, wie vorher beschrieben, umgewandelt.
Massenspektrum (m/e): 411 (M+1). Exaktes Massenspektrum: Berechnet:
411,2560, gefunden: 411,2557.
-
Beispiel 231
-
(S)-7-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-7-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
(0,048 g, 0,11 mmol) wird in Dichlormethan (6 ml) gelöst. Natriumtriacetoxyborhydrid
(0,048 g, 0,23 mmol) und Formaldehyd (0,003 g, 0,11 mmol, 0,009
g einer 37% wässrigen
Lösung)
werden zuge geben und das Gemisch wird für eine Stunde bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 10 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit 20 ml Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringertem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem linearen Gradienten ausgehend von 99% Dichlormethan
: 1% an 2 M Ammoniak in Methanol und bis zu 90% Dichlormethan :
10% an 2 M Ammoniak in Methanol ergibt die freie Base der Titelverbindung
(0,049 g, 0,11 mmol, 98% Ausbeute) als gelben amorphen Feststoff.
Es wird dann in das Succinatsalz, wie vorher beschrieben, umgewandelt.
Massenspektrum (m/e): 437 (M+1). Exaktes Massenspektrum: Berechnet:
437,1965, gefunden: 437,1948.
-
Beispiel 232
-
(S)-7-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepindihydrochlorid
-
(S)-7-Fluor-2-trifluormethyl-11-[3-(2-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
(0,259 g, 0,593 mmol), Formaldehyd (67 μl, 0,831 mmol, 37%) und Natriumtriacetoxyborhydrid
(0,189 g, 0,890 mmol) werden in Dichlorethan (10 ml) vereinigt und
bei Raumtemperatur über
Nacht gerührt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumcarbonat verdünnt
und dreimal mit Methylenchlorid extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
des rohen Produkts entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt (S)-7-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin (0,220
g, 0,488 mmol, 82%) als gelbes I. Massenspektrum (APCI) m/z 451,3
(M+1). Das reine Produkt wird als entsprechendes Dihydrochlorid
auf folgende Weise isoliert: Der gelbe Schaum wird in Ethanol (5
ml) gelöst
und eine Lösung
aus etwa 5 Äquivalenten
HCl in Ethanol (5 ml) wird zugegeben. Das Gemisch wird unter Bildung von
(S)-7-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepindihydrochlorid
eingedampft.
-
Beispiel 233
-
(S)-8-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepindihydrochlorid
-
(S)-8-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
(0,144 g, 0,330 mmol), Formaldehyd (37 μl, 0,461 mmol, 37%) und Natriumtriacetoxyborhydrid
(0,105 g, 0,495 mmol) werden in Dichlorethan (5 ml) vereinigt und
bei Raumtemperatur über
Nacht gerührt.
Das Gemisch wird mit gesättigtem
Natriumbicarbonat verdünnt
und dreimal mit Methylenchlorid extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung des
rohen Produkts entfernt. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt (S)-8-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,115 g, 0,255 mmol, 77%) als gelbes Öl. Massenspektrum (APCI) m/z
451,3 (M+1). Das reine Produkt wird als entsprechendes Dihydrochlorid
auf folgende Weise isoliert: Der gelbe Schaum wird in Ethanol (5
ml) gelöst
und eine Lösung
aus etwa 5 Äquivalenten
HCl in Ethanol (5 ml) wird zugegeben. Das Gemisch wird unter Bildung von
(S)-8-Fluor-2-trifluormethyl-11-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepindihydrochlorid
eingedampft.
-
Beispiel 234
-
(S)-2-Cyclopropyl-7-fluor-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-2-Cyclopropyl-7-fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
(0,039 g, 0,10 mmol) wird in Dichlormethan (5 ml) gelöst. Natriumtriacetoxyborhydrid
(0,042 g, 0,20 mmol) und Formaldehyd (0,003 g, 0,10 mmol, 0,008
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für eine Stunde bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 25 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit 20 ml Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringertem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem linearen Gradienten ausgehend von 99% Dichlormethan
: 1% an 2 M Ammoniak in Methanol und bis zu 90% Dichlormethan: 10%
an 2 M Ammoniak in Methanol ergibt die freie Base der Titelverbindung
(0,034 g, 0,08 mmol, 84% Ausbeute) als oranger amorpher Feststoff.
Es wird dann in das Succinatsalz, wie vorher beschrieben, umgewandelt.
Massenspektrum (m/e): 409 (M+1). Exaktes Massenspektrum: Berechnet:
409,2404, gefunden: 409,2417.
-
Beispiel 235
-
(S)-7,8-Difluor-2-trifluormethyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
(S)-7,8-Difluor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
(0,067 g, 0,15 mmol) wird in Dichlormethan (7 ml) gelöst. Natriumtriacetoxyborhydrid
(0,064 g, 0,30 mmol) und Formaldehyd (0,005 g, 0,15 mmol, 0,012
g einer 37% wässrigen
Lösung)
werden zugegeben und das Gemisch wird für eine Stunde bei Umgebungstemperatur
gerührt.
Das Gemisch wird mit 30 ml an gesättigtem wässrigem Natriumchlorid verdünnt und
dreimal mit 20 ml Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und das Lösemittel wird unter verringertem
Druck entfernt. Eine Reinigung durch Blitzchromatographie unter
Elution mit einem linearen Gradienten ausgehend von 100% Dichlormethan
bis zu 85% Dichlormethan : 15% an 2 M Ammoniak in Methanol ergibt
die freie Base der Titelverbindung (0,055 g, 0,12 mmol, 80% Ausbeute)
als gelben amorphen Feststoff. Es wird dann in das Succinatsalz,
wie vorher beschrieben, umgewandelt. Massenspektrum (m/e): 455 (M+1).
Exaktes Massenspektrum: Berechnet: 455,1870, gefunden: 455,1874.
-
Beispiel 236
-
(S)-8-Chlopr-2-trifluormethyl-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat
-
Mittels
eines Verfahrens, das zu Beispiel (S)-8-Fluor-2-trifluormethyl-11-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinsuccinat ähnlich ist
und unter Verwendung von (S)-8-Chlor-2-trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,051 g, 0,12 mmol) werden 0,054 g (100% Ausbeute) der freien Base
als gelbes Öl
erhalten, das wie vorher beschrieben in das Succinatsalz umgewandelt
wird. Massenspektrum (m/e): 454 (M+1).
-
Beispiel 236a
-
(S)-2-Chlor-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepinhydrochlorid
-
(S)-2-Chlor-11-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
(0,474 g, 1,27 mmol) und 37% Formaldehydlösung (0,1 ml, 1,34 mmol) werden
in 1,2-Dichlorethan (20 ml) vereinigt. Es wird für 10 Minuten gerührt und
Natriumtriacetoxyborhydrid (0,542 g, 2,55 mmol) wird zugegeben.
Es wird für weitere
30 Minuten gerührt
und dann wird die Lösung
auf eine gesättigte
Natriumbicarbonatlösung
gegossen. Es wird mit Methylenchlorid unter Bildung von 0,555 g
des rohen Produkts extrahiert. Eine Silicagelchromatographie unter
Elution mit Methylenchlorid : Methanol (100:5) ergibt 0,490 g der
Titelverbindung als freie Base. Das Dihydrochloridsalz fällt in Ethylacetat
als ein Feststoff aus. Smp. 205°C
Massenspektrum (Ionenspray): m/z = 385 (M+1).
-
Beispiel 237
-
(R)-[4-(2-Methyl-4H-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol
-
Mittels
eines Kupplungsverfahrens, das zu (S)-2-Trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin ähnlich ist
und unter Verwendung von 2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-ylaminhydrochlorid
ergibt die Titelverbindung als hellbraunes Pulver: Smp. 90°C, 1H NMR (CDCl3) δ 2,31 (s,
3H), 2,89 (ddd, 1H), 3,00-3,25 (m, 4H), 3,55 (dd, 1H), 3,72 (dd,
1H), 3,72-3,87 (m, 2H), 5,02 (s, 1H), 6,29 (s, 1H), 6,60 (d, 1H),
6,87 (t, 1H), 6,96 (t, 1H), 7,01 (d, 1H), MS (APCI) m/z (rel. Intensität) 329 (100).
-
Beispiel 238
-
(R)-10-(3-Methoxymethylpiperazin-1-yl)-2-melhyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Unter
Verwendung eines ähnlichen
Kupplungsverfahrens zu (S)-2-Trifluormethyl-11[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
unter Verwendung von 2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10ylaminhydrochlorid
(1,9 g) und (R)-2-Methoxymethylpiperazin (2,6 g) wird die Titelverbindung als
gelber Feststoff (974 mg, 41%) erhalten. Smp 87-95°C.
1H NMR (CDCl3) δ 2,32 (d,
3H), 2,69 (dd, 1H), 3,10-2,87 (m, 4H), 3,33 (dd, 1H), 3,38 (s, 3H),
3,49-3,43 (m, 1H), 3,94
(d, 1H), 4,02 (d, 1H), 4,98 (s, 1H), 6,30 (s, 1H), 6,61 (dd, H),
6,88 (dt, 1H), 6,97 (dt, 1H), 7,03 (dd, 1H). MS (APCI) m/z (relative
Intemsität)
343,3 (100).
-
Beispiel 239
-
(R)-10-[3-(2-Methylallyloxymethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Mittels
eines Kupplungsverfahrens, das zu (S)-2-Trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin ähnlich ist
wird die Titelverbindung als gelber Feststoff erhalten.
1H NMR (CDCl3) δ 1,74 (s,
3H), 2,31 (s, 3H), 2,68 (dd, 1H), 2,92-2,96 (m, 2H), 3,03-3,10 (m,
2H), 3,35 (dd, 1H), 3,47 (dd, 1H), 3,90 (s, 2H), 3,95-4,04 (m, 2H),
4,90 (s, 1H), 4,96 (s, 2H), 6,30 (s, 1H), 6,60 (d, 1H), 6,87 (t,
1H), 6,97 (t, 1H), 7,02 (d, 1H). MS (ES) m/z (relative Intensität) 383 (100).
-
Beispiel 240
-
(S)-2-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2-yl]ethanoldihydrochlorid
-
(S)-2-Piperazin-2-yl-ethanol
(1,48 g, 11,35 mmol) und 2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-ylamin
als freie Base (1,3 g, 5,67 mmol) werden in 9 ml Dimethylsulfoxid
und 27 ml Toluol vereinigt. Der Reaktionskolben wird mit einem Rückflusskühler ausgestattet,
und das Gemisch wird bei 110°C
unter mildem positivem Stickstoffdruck für 3 Tage erhitzt. Es wird mit
Methanol verdünnt
und direkt auf eine 46 g SCX Säule aufgetragen.
Die Säule
wird mit Methanol gewaschen, mit 2 N Ammonaik-Methanol eluiert.
Der Eluent wird im Vakuum unter Bildung eines dunkelbraunen Rückstands
konzentriert und auf Silicagel gereinigt. Es wird mit einem Gradienten
aus 2,5% an 2 N Ammoniak-Methanol in Dichlormethan und dann 5% und
dann 10% unter Bildung der rohen freien Base der Titelverbindung
(0,48 g, 25%) als braunes Öl
eluiert. Massenspektrum (APCI): m/z = 343,1 (M+1).
-
Das
reine Produkt wird als das entsprechende Dihydrochlorid auf folgende
Weise isoliert. Es werden 220 mg des braunen Öls der freien Base zwischen
Wasser und Dichlormethan aufgeteilt. Die organische Phase wird abgetrennt
und das Lösemittel
wird im Vakuum entfernt, der Rückstand
wird in Methanol aufgenommen und auf eine SCX Säule aufgetragen. Die Säule wird
mit Methanol gewaschen, mit Chlorwasserstoffsäure in Ethanol (hergestellt
durch die Zugabe von 20 ml Acetylchlorid zu 500 ml Ethanol) eluiert
und das entstehende gelbe Band das langsam die Säule herunterwandert wird gesammelt.
Die gelben Fraktionen werden vereinigt und im Vakuum konzentriert.
Der entstehende schaumige Rückstand
wird in einem 1:1 Gemisch aus Wasser und Acetonitril rückgelöst und über Nacht
unter Bildung von 197 mg (74%) des Dihydrochlorids der Titel verbindung
als flockiger oranger Feststoff lyophilisiert: Schmelzpunkt: >250°C (Zers) Massenspektrum (APCI)
m/z = 343,1 (M+1 der freien Base).
-
Beispiel 241
-
(R)-2-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-19-yl)piperazin-2-yl]ethanoldihydrochlorid
-
(R)-2-Piperazin-2-yl-ethanol
(0,92 g, 7 mmol) und 2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(1,7 g, 7 mmol) werden in Pyridin (14 ml) vereinigt. Es wird bei
120°C über Nacht
erhitzt und dann wird das Lösemittel
im Vakuum entfernt und in Methanol rückgelöst. Es wird auf 20 g SCX Harz gereinigt,
mit Methanol gewaschen und zuerst mit 5% an 2 N Ammoniak-Methanol
in Dichlormethan eluiert und dann mit starker 2 N Ammoniak-Methanol.
Die Produktfraktionen werden vereinigt und dann weiter auf Silicagel
mittels eines Stufengradienten aus 1,5% bis 5% an 2 N Ammoniak-Methanol
in Dichlormethan gereinigt. Es wird als das Dihydrochloridsalz (243
mg, 8%) durch die Behandlung einer ehtanolischen Lösung der
freien Base mit einer Lösung
aus 5 Äquivalenten
Chlorwasserstoffsäure
in Ethanol isoliert und dann eingedampft. Schmelzpunkt >250°C (Zers): Massenspektrum (APCI)
m/z =) 343,1 (M+1 der freien Base).
-
Beispiel 241a
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-4H-j3-thia-4,9-diaza-benzo[f]azulen
-
4H-3-Thia-4,9-diaza-benzo[f]azulen-10-ylamin
(1,26 g, 5,85 mmol), (S)-2-(2-Methoxyethyl)piperazin (2,53 g, 17,56
mmol), DMSO (4,0 ml) und toluol (16,0 ml) werden vereinigt. Es wird
gerührt
und das Gemisch wird auf 105°C
erhitzt. Nach 24 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt.
Das Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 0,1 N NaOH und Kochsalzlösung gewaschen. Es wird getrocknet
(Natriumsulfat und die organische Phase wird zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan zu
Dichlormethan/Methanol (85:15) unter Bildung von 395,8 mg (20%)
als brauner Schaum gereinigt. Massenspektrum (Ionenspray): m/z =
343,1 (M+1).
-
Beispiel 242
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulenhydrochlorid
-
Auf
eine Weise die zu der in Beispiel 242a beschriebenen ähnlich ist,
wird (S)-2-(2-Methoxyethyll)piperazin (161 mg, 1 mmol) in die Titelverbindung
umgewandelt: Massenspektrum (APCI) m/z = 357,2 (M+1).
-
Beispiel 242a
-
(S)-10-[3-(2-Phenoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(0,382 g, 1,55 mmol), (S)-2-(2-Phenoxyethyllpiperazin
(0,320 g, 1,55 mmol) und Pyridin (5 ml) werden vereinigt und für 36 Stunden
am Rückfluss
erhitzt. Das Gemisch wird eingedampft und das Material wird auf
10 g SCX gegeben und dann mit Methanol gefolgt von 5% an 2 N Ammoniak-Methanol
in Dichlormethan und dann 2 N Ammoniak-Methanol eluiert. Eine Reinigung durch
Blitzchromatographie unter Elution mit einem Stufengradienten ausgehend
von Dichlormethan bis 7% an 2 N Ammoniak-Methanol in Dichlormethan
ergibt die Titelverbindung: Massenspektrum (APCI): m/z = 419,1 (M+1)
-
Beispiel 243
-
(S)-10-[3-(2-Ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Auf
eine Weise die zu der in Beispiel 311 beschriebenen ähnlich ist,
werden 2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(0,487 g, 1,98 mmol) und (S)-2-(2-Ethoxyethyl)piperazin (0,313 g, 1,98
mmol) unter Bildung der Titelverbindung vereinigt: Massenspektrum
(APCI) m/z = 371,2 (M+1).
-
Beispiel 244
-
(R)-10-(3-Phenoxymethylpiperazin-1-yl)-2-methy-4H-3-thia-4,9-diazabenzo[f]azulen
-
Durch
ein Verfahren, das zu dem Kupplungsverfahren von (S)-2-Trifluormethyl-11-[3-(2-methoxyethyl)piperazin-1-yl]-5H-dibenzo[b,e][1,4]diazepin
ergeben 2-Methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(1,2 g) und (R)-2-Phenoxymethylpiperazin (2,6 g) die Titelverbindung
als gelben Feststoff (728 mg): Smp = 64-81°C.
1H
NMR (CDCl3) δ 2,32 (s, 3H), 2,86 (dd, 1H),
3,14-2,94 (m, 3H), 3,32-3,24 (m, 1H), 3,98-3,78 (m, 2H), 4,02 (dd,
1H), 4,11 (bd, 1H), 4,96 (bs, 1H), 6,32-6,30 (m, 1H), 6,61 (dd,
1H), 7,00-6,86 (m, 5H), 7,04 (dd, 1H), 7,34-7,25 (m, 2H). MS (APCI)
m/z (relative Intensität)
405,4 (100).
-
Beispiel 245
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
2-Isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,32 g, 1,07 mmol) wird zu einer Lösung aus (S)-2-(2-Methoxyethyl)piperazin
(0,31 g, 2,15 mmol) in Dimethylsulfoxid gegeben: Toluol (1:3, 4
ml). Diisopropylethylamin (0,19 ml, 1,09 mmol) wird zugegeben, auf
110°C erhitzt
und gerührt.
Nach einem Zeitraum über
Nacht wird unter Bildung der rohen Titelverbindung auf Umgebungstemperatur
gekühlt.
-
Beispiel 246
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
2-Isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,31 g, 1,04 mmol) und (S)-2-(2-Methoxyethyl)piperazin (0,30 g,
2,08 mmol) wird mit wasserfreiem Pyridin (4 ml) vereinigt und auf 110°C erhitzt
und über
Nacht gerührt.
Es wird unter Bildung des rohen Materials auf Umgebungstemperatur gekühlt. Eine
Reinigung durch Kationenaustauschchromatographie unter Elution mit
Lösungen
aus 2 M Ammoniak in Methanol in Dichlormethan (2%, 5%, 10%) und
2 M Ammoniak in Methanol gefolgt von einer Blitzchromatographie
unter Elution mit einem Gradienten der Lösungen aus 2 M Ammoniak in
Methanol, in Dichlormethan (2-4%) wird die Titelverbindung erhalten.
Massenspektrum (APCI, m/e): 385 (M+1)
NMR (1H,
300 MHz, DMSO-d6) δ (ppm) 7,60 (s, 1H), 6,86-6,73
(m, 3H), 6,67 (m, 1H), 6,30 (s, 1H), 3,87 (m, 2H), 3,38-3,33 (m,
2H), 3,19 (s, 3H), 3,01-2,60 (m, 6H), 2,39 (m, 1H), 1,51 (m, 2H),
1,17 (d, 6H, J = 7,5 Hz).
-
Beispiel 246a
-
(S)-10-[3-(3-Methoxypropyl)piperazin-1-yl]-4H-3-thia-4,9-diazabenzo[f]azulen
-
4H-3-Thia-4,9-diaza-benzo[f]azulen-10-ylamin
(1,10 g, 5,11 mmol), (S)-2-(3-Methoxypropyl)piperazin (1,62 g, 10,22
mmol), DMSO (4,0 ml) und Toluol (16,0 ml) werden vereinigt. Es wird
gerührt
und das Gemisch wird auf 105°C
erhitzt. nach 45 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt.
Das Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 0,1 N NaOH und Kochsazlösung
gewaschen. Es wird getrocknet (Natriumsulfat) und die organische
Phase wird zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (85:15) unter Bildung ovn 296,0 mg (16%)
der Titelverbindung gereinigt. Massenspektrum (Ionenspray): m/z
= 357,2 (M+1).
-
Beispiel 246b
-
(S)-10-[3-(3-Methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Mittels
eines Verfahrens, das zu dem Beispiel 203 ähnlich ist und unter Verwendung
von 2-Methyl-4H-3-hia-4,9-diaza-benzo[f]azulen-10-ylamin
wird die Titelverbindung erhalten: Massenspektrum (m/e): 371,08
(M+1).
-
Beispiel 247
-
(S)-10-[3-(4-Methoxybutyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Eine
gerührte
Lösung
aus (S)-2-(4-Methoxybutyl)piperazin (0,086 g, 0,49 mnmol) und 2-Methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylamin
(0,114 g, 0,49 mmol) wird in NMP (1 ml) in einem 211°C Ölbad für 4 h erhitzt.
Es wird auf Raumtemperatur gekühlt,
die Lösung
wird mit Kochsalzlösung
(15 ml) verdünnt und
mit EtOAc (3 × 30
ml) extrahiert. Die organischen Phasen werden unter verringertem
Druck auf ein Volumen von 20 ml kombiniert und mit 75% Kochsalzlösung (4 × 20 ml)
gewaschen. Die organische Phase wird über Na2SO4 getrocknet und filtriert. Das Gemisch wird
unter verringertem Druck konzentriert und durch Silicagelchromatographie
unter Elution mit 10% (33% an 2 M NH3 in
MeOH/64% EtOH)/CH2Cl2 gereinigt.
Die gereinigten Fraktionen werden vereinigt, unter verringertem
Druck konzentriert, mit CH2Cl2/Hexan
azeotrop gemacht und unter Bildung der Titelverbindung unter Vakuum
gegeben: brauner Feststoff (0,036 g), Massenspektrum (m/e): 385,08
(M+1H).
-
Beispiel 247a
-
(S)-6-Fluor-10-[3-(2-methoxyelhyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(1,20 g, 4,45 mmol), (S)-2-(2-Methoxyethyl)piperazin
(1,60 g, 11,12 mmol), DMSO (4,0 ml) und Toluol (16,0 ml) werden
vereinigt. Es wird gerührt
und das Gemisch wird auf 105°C
erhitzt. Nach 40 Stunden wird das Gemisch auf Umgebungstemperatur gekühlt. Das
Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 0,1 N NaOH und Kochsalzlösung gewaschen. Es wird getrocknet
(Natriumsulfat) und die organische Phase wird zu einem Rückstand konzentriert.
Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 400,4 mg (25%)
als hellbrauner Schaum gereinigt. Massenspektrum (Ionenspray): m/z
= 361,1 (M+1).
-
Beispiel 247b
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(742,0 mg, 2,75 mmol), (S)-2-(3-Methoxypropyl)piperazin
(870,7 mg, 5,50 mmol), DMSO (2,0 ml) und Toluol (8,0 ml) werden
vereinigt. Es wird gerührt
und das Gemisch wird auf 105°C
erhitzt. Nach 48 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt.
Das Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 0,1 N NaOH und Kochsalzlösung gewaschen. Es wird getrocknet
(Natriumsulfat) und die organische Phase wird zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (85:15) unter Bildung von 220,1 mg (21%)
als Titelverbindung gereinigt. Massenspektrum (Ionenspray): m/z
= 375,1 (M+1).
-
Beispiel 248
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(500 mg, 1,9 mmol) wird in Dichlormethan (30 ml) suspendiert, unter
Stickstoff gerührt
und in einem EisWasserbad gekühlt.
Methyltrifluormethansulfonat (500 μl) wird zugegeben und über Nacht
gerührt.
Das Reaktionsgemisch wird unter verringertem Druck konzentriert,
in Pyridin (10 ml) aufgenommen und (S)-2-(2-Methoxyethyl)piperazin
(400 mg, 2,78 mmol) wird zugegeben und das Reaktionsgemisch wird
unter Stickstoff gerührt
und bei 90°C
für drei
Tage erhitzt. Das Reaktionsgemisch wird unter verringertem Druck
konzentriert und durch Blitzchromatographie auf Silicagel (Eluent
Dichlormethan/Methanol) unter Bildung des gewünschten gelben Feststoffs mit
727 mg gereinigt. Massenspektrum (FIA) 375 (M+1).
-
Beispiel 248a
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-2-methyl-10-methylsulfanyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(3,40 g, 12,2 mmol) und (S)-2-(2-Methoxyethyl)pipierazin (2,20 g,
15,3 mmol) werden in Isopropylalkohol (20 ml) bei Umgebungstemperatur
unter Stickstoff für
20-30 Minuten zum Lösen
gerührt.
Es wird am Rückfluss
(80-83°C)
für 3-4
Tage erhitzt und kann dann auf Umgebungstemperatur abkühlen. Das
Gemisch wird unter verringertem Druck unter Bildung eines Rückstands
konzentriert. Eine Reinigung des Rückstands durch Blitzchromatographie
unter Elution mit 90:10:1→80:20:1/EtOAc
: MeOH : konz. NH4OH ergibt 4,06 g (89%)
der Titelverbindung.
1H NMR (400 MHz,
DMSO-d6): δ 7,75 (bs, 1H), 6,78 (m, 1H),
6,68 (, 1H), 6,53 (m, 1H), 6,35 (s, 1H), 3,80 (m, 2H), 3,40 (m,
2H), 3,23 (s, 3H), 2,88 (m, 1H), 2,71 (m, 3H), 2,41 (m, 1H), 2,30
(bs, 1H), (s, 3H), 1,53 (m, 2H). HRMS (ES) exakte Masse M+H berechnet
für C19H23FN4OS
375,1655, gefunden 375,1663.
-
Beispiel 248b
-
(S)-6-fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
6-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylamin
(2,16 g, 8,7 mmol) und (S)-2-(2-Methoxyethyl)piperazin
(1,39 g, 9,6 mmol) werden in NMP (16,0 ml) vereinigt und bei 190°C für 1 Stunde
erhitzt. Es wird auf Umgebungstemperatur gekühlt und mit Wasser verdünnt. Es
wird mit Ethylacetat unter Bildung von 2,25 g des rohen Produkts
extrahiert. Eine Silicagelchromatographie unter Elution mit Methylenchlorid:
2 N NH3/Methanol (100:7) ergibt 0,616 g
der Titelverbindung als Öl
Das Dihydrochloridsalz fällt
in Ethylacetat als hellgrüner
Feststoff aus: Smp 90°C
Massenspektrum (Ionenspray): m/z = 375 (M+1). Analyse für C19H25FN4OS (0,8
H2O): berechnet C 49,41, H 5,81, N 12,13.
Gefunden: C 49,75, H 5,74, N 11,75.
-
Beispiel 248c
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
6-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,304 g, 1,07 mmol), (S)-2-(3-Methoxypropyl)piperazin (0,339 g,
2,14 mmol) und Diisopropylethylamin (0,19 ml, 1,07 mmol) werden in
einem Gemisch aus Toluol (3 ml) und Dimethylsulfoxid (1 ml) werden
vereinigt und bei 110°C
für 24
Stunden gerührt.
Das Gemisch wird dann durch Blitzchromatographie unter Elution mit
einem Stufengradienten ausgehend von Dichlormethan bis zu 5% an
2 N Ammoniak-Methnaol in Dichlormethan unter Bildung von (S)-6-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (0,110
g, 0,283 mmol, 26%) als gelbes Öl
eingedampft. Massenspektrum (APCI): m/z = 398,1 (M+1).
-
Beispiel 249
-
(S)-7-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Auf ähnliche
Weise zu Beispiel 248 und unter Verwendung von 7-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-19-thion
(1,25 g, 4,73 mmol) und (S)-2-(3-Methoxyethyl)piperazin (800 mg, 5,5
mmol) wird die Titelverbindung mit 1,73 g (4,62 mmol) erhalten.
Massenspektrum (FIA): 375 (M+1).
NMR 1H,
300 MHz, CDCl3): δ 6,5-6,73 (m, 3H), 6,27 (s,
1H), 5,12 (s, 1H), 4,20 (d, 1H), 4,04 (d, 1H), 3,4-3,65 (m, 5H),
3,34 (s, 3H), 3,13-3,3 (m, 2H), 2,32 (s, 3H), 2,07-2,18 (m, 1H),
1,75 (m, 1H).
-
Beispiel 249a
-
(S)-7-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
7-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(270 mg, 0,95 mmol) und (S)-2-(3-Methoxypropyl)piperazin (330 g,
2 mmol) werden in einem Gemisch aus Toluol und Dimethylsulfoxid (1:2)
vereinigt und bei 110°C
für 24
Stunden gerührt.
Das Gemisch wird dann durch Blitzchromatographie unter Elution mit
einem Stufengradienten mit Dichlormethan bis zu 5% an 2 N Ammoniak-Methanol
in Dichlormethan unter Bildung von (S)-7-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(320 mg, 0,82 mmol) eingedampft. (APCI): m/z = 389,1 (M+1).
-
Beispiel 249b
-
(S)-6-Fluor-10-[3-(2-hydroxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(2,94 mg, 10,35 mmol), (S)-2-Piperazin-2-yl-ethanol (2,7 g, 20,7
mmol), Diisopropylethylamin (3,6 ml, 20,7 mmol), Dimethylsulfoxid (4,25
ml) und Toluol (17 ml) werden vereinigt und bei 110°C für 42 Stunden
erhitzt. Es wird auf Umgebungstemperatur gekühlt und mit Ethylacetat (200
ml) verdünnt.
Es wird mit gesättigter
Natriumbicarbonatlösung
und Kochsalzlösung
gewaschen und unter Bildung von 2,64 g des rohen Produkts eingedampft.
Eine Silicagelchromatographie unter Elution mit Methylenchlorid
: Acetonitril : 7 N NH3/Methanol (85:10:7)
ergibt 0,523 g der Titelverbindung als braunen festen Schaum. Massenspektrum
(Ionenspray): m/z = 361 (M+1).
-
Beispiel 250
-
(S)-6-Fluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Mittels
eines Verfahrens, das zu dem Verfahren von (S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen ähnlich ist
und unter Verwendung von 2-Methyl-4,9-dihydro-3-thia-6-fluor-4,9-diaza-benzo[f]azulen-10-thion
und (S)-2-(2'-Ethoxy)ethylpiperazin
wird die Titelverbindung erhalten: Massenspektrum (FIA): 389 (M+1).
NMR
(1H, 300 MHz, CDCl3) δ 6,93 (m,
1H), 6,66 (m, 1H), 6,41 (m, 1H), 6,26 (s, 1H), 4,92 (s, 1H), 4,14-4,27 (breit,
2H), 3,9-4,1 (m, 2H), 3,58 (m, 2H), 3,47 (q, 2H), 3,0-3,2 (m, 3H),
2,80 (m, 1H), 2,30 (s, 3H), 1,75 (m, 1H), 1,17 (t, 3H).
-
Beispiel 251
-
(S)-7-Fluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Mittels
eines Verfahrens, das zu dem Verfahren von Beispiel 248 ähnlich ist
und unter Verwendung von 7-Fluor-2-methyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-thion
(750 mg, 2,84 mmol) und (S)-2-(2-Ethoxy)ethylpiperazin (510 mg,
3,3 mmol) wird die Titelverbindung (955 mg, 2,46 mmol) erhalten: Massenspektrum
(FIA): 389 (M+1).
-
Beispiel 252
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Fluor-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,6 g, 2,0 mmol) und (S)-2-(2-Methoxy)ethylpiperazin (0,864 g,
6,0 mmol) werden in Dimethylsulfoxid/Toluol (1:2) (15 ml) gelöst, unter
Stickstoff gerührt
und in einem Ölbad
am Rückfluss
für 18
Stunden erhitzt. Das Gemisch wird in Wasser gegossen und mit Ethylacetat
extrahiert. Die organische Phase (MgSO4)
wird getrocknet, filtriert und unter verringertem Druck konzentriert.
Eine Chromatographie auf Silicagel (Eluent Dichlormethan/Methanol)
ergibt (S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-2-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen (558
mg, 82%).
Massenspektrum (LCMS) 389 (M+1).
-
Beispiel 253
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Auf ähnliche
Weise zu dem Beispiel 248 und unter Verwendung von 6-Fluor-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-thion
(402 mg, 1,34 mmol) und (S)-2-(2-Methoxy)ethylpiperazin (350 mg,
2,43 mmol) wird die Titelverbindung mit 410 mg, (102 mmol) erhalten:
FIA M+1 403.
-
Beispiel 253a
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6,7-Difluor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(876,9 mg, 3,05 mmol), (S)-2-(2-Methoxyethyl)piperazin
(1,32 g, 9,16 mmol), DMSO (3,0 ml) und Toluol (12,0 ml) werden vereinigt.
Es wird gerührt
und das Gemisch wird auf 105°C
erhitzt. Nach 63 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt.
Das Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 1 N NaOH und Kochsalzlösung
gewaschen. Es wird getrocknet (Natriumsulfat) und die organische
Phase wird zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (85:15) unter Bildung von 561,6 mg (49%)
der Titelverbindung gereinigt: Massenspektrum (Ionenspray): m/z
= 379,1 (M+1).
-
Beispiel 254
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(65,0 g, 215 mmol) und 2-(2-Methoxyethyll)piperazin (96,3 g, 668
mmol) wird zu einer Lösung
aus DMSO (650 ml) und Toluol (1300 ml) unter Rühren unter Stickstoff gegeben.
Es wird am milden Rückfluss
(115°C)
für 2 Tage
erhitzt und kann dann auf Umgebungstemperatur abkühlen. Der
Großteil
der organischen Portion wird unter verringertem Druck entfernt und
dann wird der Rückstand
zwischen Ethylacetat (450 ml) und wässriger gesättigter Ammoniumchloridlösung (450
ml) aufgeteilt. Die Phasen werden getrennt und dann wird die wässrige Phase
mit Ethylacetat (3 × 200
ml) extrahiert. Die vereinigten organischen Phasen werden mit Kochsalzlösung (3 × 200 ml)
extrahiert. Die organische Phase wird unter verringertem Druck konzentriert.
Das rohe Produkt wird durch Blitzchromatographie unter Elution mit
90:10/Methylenchlorid : MeOH unter Bildung von 56 g (66%) der Titelverbindung
gereinigt.
1H NMR (400 MHz, DMSO-d6) δ 7,70
(s, 1H), 6,74 (m, 2H), 6,36 (s, 1H), 3,86 (bm, 2H), 3,39 (7m, 2H),
3,23 (s, 3H), 2,88 (bd, 1H), 2,72 (bm, 3H), 2,44 (bt, 1H), 2,31
(s, 3H), 2,31 (bm, 1H), 1,53 (m, 2H). HRMS (ES) exakte Masse M+H
berechnet für
C19H22F2N4OS 393,1561, gefunden 393,1554.
-
Beispiel 254a
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,90 g, 3 mmol) und 2-(2-Methoxyethyl)piperazin (1,34 g, 9,3 mmol)
werden in Dimethylsulfoxid/Toluol (1:2) (30 ml) gelöst und unter Stickstoff
gerührt
und in einem Ölbad
bei 140°C
für 48
Stunden erhitzt. Das Gemisch wird in Wasser gegossen und mit Ethylacetat
extrahiert. Die organische Phase wird getrocknet (MgSO4),
filtriert und unter verringertem Druck unter Bildung des gewünschten
Produkts konzentriert. Die wässrige
Phase wird mit einem losen SCX-2 Harz behandelt. Das Harz wird unter
Bildung von weiterem gewünschtem
Produkt durch Elution mit 2 N NH3 in MeOH
abfiltriert. Eine Reinigung durch Chromatographie auf Florisil (Eluent
Ethylacetat) des vereinigten Materials ergibt (S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(0,67 g, 57%). Massenspektrum (LCMS) 393 (M+1)
1H
NMR (400 MHz, DMSO-d6) δ 6,80 (m, 1H), 6,45 (m, 1H),
6,30 (s, 1H), 4,82 (s, 1H), 4,00 (bt, 2H), 3,50 (t, 2H), 3,30 (s,
3H), 3,05 (m, 1H), 2,90 (m, 3H), 2,60 (m, 1H), 2,31 (s, 3H), 1,68
(q, 2H).
-
Beispiel 254b
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6,7-Difluor-2-methyl-4H-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(1,7 g, 5,66 mmol) und (S)-2-(2-Methoxyethyl)piperazin (2,45 g,
17,0 mmol) werden in Dimethylsulfoxid/Toluol (1:2) (21 ml) gelöst, unter
Stickstoff gerührt
und in einem Ölbad
bei 130°C
für 43
Stunden erhitzt. Es wird auf Umgebungstemperatur gekühlt. Das
Gemisch wird mit einem Volumen Wasser und zwei Volumen Ethylacetat
verdünnt,
die Phasen werden getrennt und die wässrige Phase wird mit Ethylacetat
extrahiert. Die wässrige
Phase wird mit Ammoniumhydroxid basisch gemacht und mit Ethylacetat
extrahiert. Alle Ethylacetatextrakte werden vereinigt und mit Wasser
und Kochsalzlösung
gewaschen. Die organische Phase wird getrocknet (Na2SO4), filtriert und unter verringertem Druck
konzentriert. Eine Reinigung durch Silicagelchromatographie (Eluent:
0-10% an 2 N Ammoniak in Methanol/Dichlormethan) unter Bildung der
Titelverbindung (840 mg, 38%) gereinigt. Massenspektrum (ESMS) 393
(M+1), 391 (M-1).
-
Beispiel 254c
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulenhydrochlorid
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(730 mg, 1,86 mmol) wird in Ethylacetat gelöst. Es werden 2,5 Äquivalente
Chlorwasserstoff in Ethanol zugegeben. Nach 3 Stunden wird der Niederschlag
filtriert, mit Ethylacetat gewaschen und unter Vakuum unter Bildung
der Titelverbindung (856 mg, 99%) getrocknet. Massenspektrum (ESMS)
393 8M+1), 391 8M-1) Smp. 198-202°C (Zers.).
-
Beispiel 254d
-
(S)-2-[4-(6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]ethanol
-
6,7-Difluor-2-methyl-4H-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(1,51 g, 4,99 mmol), N,N-Diisopropylethylamin (0,91 ml, 5,24 mmol)
und (S)-2-Piperazin-2-yl-ethanol (1,3 g, 9,99 mmol) werden in Dimethylsulfoxid/Toluol
(1:2) (18 ml) gelöst,
unter Stickstoff gerührt
und in einem Ölbad
bei 120°C
für 72
Stunden gerührt.
Das Gemisch wird mit Wasser (20 ml) und Ethylacetat (40 ml) verdünnt, die
Phasen werden getrennt und die wässrige
Phase wird mit Ethylacetat extrahiert. Alle Ethylacetatextrakte
werden vereinigt und mit Wasser und Kochsalzlösung gewaschen. Die organische
Phase (Na2SO4) wird
getrokcnet, filtriert und unter verringertem Druck konzentriert.
Es wird durch Silicagelchromatographie (Eluent: 0-13% an 2 N Ammoniak
in Methanol/Dichlormethan) unter Bildung von 780 mg des unreinen
Produkts gereinigt. Eine Reinigung durch Silicagelchromatographie
(Eluent: 85% Dichlormethan/10% Acetonitril/0% an 2 N Ammoniak in
Methanol bis 80% Dichlormethan/10% Acetonitril/10% an 2 N Amoniak
in Methanol) ergibt die Titelverbindung (516 mg, 27%). Massenspektrum
(ESMS) 379 (M+1), 377 (M-1).
-
Beispiel 255
-
(S)-6,7-Difluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Auf ähnliche
Weise zu Beispiel 249a und mittels 6,7-Difluor-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-ylaminhydrochlorid
(500 mg, 1,66 mmol) und (S)-2-(2-Ethoxy)ethylpiperazin (780 mg,
4,93 mmol) wird die Titelverbindung mit 353 mg (0,87 mmol) erhalten.
FIA M+1 407.
-
Beispiel 256
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Auf ähnliche
Weise zu Beispiel 248 und mittels 6,7-Difluor-2-ethyl-4,9-dihydro-3-thia-4,9-diaza-benzo[f]azulen-10-thion
(1,13 g, 3,83 mmol) und (S)-2-(2-Methoxy)ethylpiperazin (623 mg,
4,33 mmol) wird die Titelverbindung mit 440 mg (1,08 mmol) erhalten.
FIA M+1 407.
-
Beispiel 257
-
(S)-6-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
6-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,537 g, 1,8 mmol) und (S)-2-(2-Methoxy)ethylpiperazin (0.8 g,
5,5 mmol) werden in Dimethylsulfoxid/Toluol (1:2) (15 ml) gelöst, unter Stickstoff
gerührt
und in einem Ölbad
bei 120°C
für 6 Tage
erhitzt. Das Gemisch wird in Wasser gegossen und mit Ethylacetat
extrahiert. Die organische Phase wird getrocknet (MgSO4),
filtriert und unter verringertem Druck konzentriert. Eine Chromatographie
auf Silicagel (Eluent Dichlormethan/ Methanol) ergibt (S)-6-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(195 mg, 27%). Massenspektrum (FIA) 391/393 (M+1).
-
Beispiel 257a
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
7-Chlor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(1,20 g, 4,19 mmol), (S)-2-(2-Methoxyethyl)piperazin
(1,51 g, 10,48 mmol), DMSO (4,0 ml) und Toluol (16,0 ml) werden
vereinigt. Es wird gerührt
und das Gemisch wird auf 105°C
erhitzt. Nach 24 Stunden wird das Gemisch auf Umgebungstemperatur gekühlt und
für 16
Stunden gerührt.
Das Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 0,1 N NaOH und Kochsalzlösung gewaschen. Es wird getrocknet
(Natriumsulfat) und die organische Phase wird zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 571,5 mg (36%)
eines braunen Feststoffs gereinigt. Massenspektrum (Ionenspray)
m/z = 377,1 (M+1).
-
Beispiel 257b
-
(S)-7-Chlor-10-[3-(3-methoxypropyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
7-Chlor-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(786,7 mg, 2,75 mmol), (S)-2-(3-Methoxypropyl)piperazin
(870 mg, 5,50 mmol), DMSO (2,0 ml) und Toluol (8,0 ml) werden vereinigt. Es
wird gerührt
und das Gemisch wird auf 105°C
erhitzt. Nach 48 Stunden wird das Gemisch auf Umgebungstemperatur
gekühlt.
Das Gemisch wird mit Ethylacetat verdünnt und die organische Phase
wird mit 0,1 N NaOH und Kochsalzlösung gewaschen. Es wird getrocknet
(Natriumsulfat) und die organische Phase wird zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 280,3 mg (26%)
der Titelverbindung gereinigt. Massenspektrum (Ionenspray) m/z =
391,1 (M+1).
-
Beispiel 258
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-7-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,60 g, ~2,0 mmol) und (S)-2-(2-Methoxy)ethylpiperazin (0,87 g,
5,5 mmol) werden in Dimethylsulfoxid/Toluol (1:2) (15 ml) vereinigt,
unter Stickstoff gerührt
und in einem Ölbad
bei 120°C
für 4 Stunden
erhitzt. Das Gemisch wird in Wasser gegossen und mit Ethylacetat
extrahiert. Die organische Phase (MgSO4)
wird getrocknet, filtriert und unter verringertem Druck konzentriert.
Eine Chromatographie auf Silicagel (Eluent Dichlormethan/Methanol)
ergibt die Titelverbindung: Massenspektrum (ESMS) 391/393 (M+1).
Smp. 179-183°C
Zers.
NMR (1H, 300 MHz, CDCl3) δ 7,0
(d, 1H), 6,8 (1H, dd), 6,5 (d, 1H), 6,28 (s, 1H), 4,6 (s, 1H), 4,02
(m, 2H), 3,5 (m, 2H), 3,35 (s, 3H), 3,05 (m, 1H), 2,4 (m, 3H), 2,6
(m, 1H), 2,3 (s, 3H), 1,7 (m, 2H).
-
Beispiel 258a
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
7-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
(1,46 g, 4,86 mmol) und (S)-2-(2-Methoxyethyl)piperazin (2,1 g,
14,6 mmol) werden in Dimethylsulfoxid/Toluol (1:2) (18 ml) gelöst, unter Stickstoff
gerührt
und in einem Ölbad
bei 130°C
für 28,5
Stunden erhitzt. Es wird für
16 Stunden bei Umgebungstemperatur gerührt. Das Gemisch wird mit Wasser
und Ethylacetat verdünnt
und mit Ethylacetat extrahiert. Die Ethylacetatextrakte werden vereinigt,
mit Wasser und Kochsalzlösung
gewaschen, die organische Phase (Na2SO4) wird getrocknet, filtriert und unter verringertem
Druck konzentriert. Eine Chromatographie auf Silicagel (Eluent:
0-10% an 2 N Ammoniak in Methanolo/Dichlormethan) ergibt die Titelverbindung
(1,14 g, 60%). Massenspektrum (ESMS) 391393 (M+1), Smp. 179-183°C (Zers.).
-
Beispiel 258b
-
(S)-7-Chlor-10-[3-(2-hydroxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines Verfahrens, das zu Beispiel 258 ähnlich ist, ergeben 7-Chlor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
und (S)-2-(2-Hydroxyethyl)piperazin die Titelverbindung.
-
Beispiel 258c
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
7-Chlor-2-methyl-10-methylsulfanyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(42,74 g, 145 mmol) und (S)-2-(2-Methoxyethyl)piperazin (26,1 g,
181 mmol) werden in Isopropylalkohol (190 ml) vereinigt. Es wird
auf 78-81°C
für 3-4
Tage erhitzt und kann dann auf Umgebungstemperatur kühlen. Die
Feststoffe werden filtriert und mit 1:1 MTBE/Ligroin (2 × 300 ml)
gewaschen. Ein Trocknen bei 30°C
ergibt 39,1 g (69,5%) der Titelverbindung.
1H
NMR (500 MHz, DMSO-d6): δ 1,52 (bm, 2H), 2,27 (s, 3H),
2,45 (bt, 1H), 2,65 (bm, 2H), 2,75 (bt, 1H), 2,86 (bd, 1H), 3,20
(s, 3H), 3,38 (m, 2H), 3,80 (m, 1H), 3,90 (m, 1H), 6,33 (s, 1H),
6,67 (d, 1H), 6,75 (d, 1H), 6,82 (dd, 1H), 7,70 (s, 1H).
13C NMR (100 MHz, DMSO-d6): δ 15,07, 25,49,
33,28, 45,111, 47,36, 52,55, 57,92, 69,18, 118,38, 119,81, 122,29,
122,56, 126,41, 126,99, 128,43, 142,54, 142,90, 152,84, 157,82.
MS
(ES+) M+H berechnet für
C19H23ClN4OS 390,94, gefunden 391,96.
-
Beispiel 259
-
(R)-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol
-
Wässrige 37%
Formaldehyd (1,1 Äquivalente)
werden zu einer Lösung
aus [(R)-4-(2-Methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol
(1 mmol) in Dichlorethan (60 ml pro 1 mmol nicht alkyliertes Ausgangsmaterial)
gegeben. Das Gemisch wird für
2 Minuten gerührt
und Natriumtriacetoxyborhydrid (1,5 mmol pro 1 mmol nicht alkyliertes
Ausgangsmaterial) wird zugegeben. Die Suspension wird für 30 Minuten
gerührt
und mit einer gesättigten
wässrigen
Lösung
aus Natriumbicarbonat gestoppt. Die wässrige Phase wird dreimal mit
Dichlormethan extrahiert und die organischen Phasen werden vereinigt, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Eine Reinigung des Rückstands
mittels Chromatographie auf Silicagel (Methylenchlorid/Methanol
(90:10) ergibt die Titelverbindung als gelbes Pulver. Smp. 118-124°C.
1H NMR (CDCl3): δ 2,31 (s,
3H), 2,43 (s, 3H), 2,45 (m, 1H), 2,59 (m, 1H), 2,82 (m, 1H), 3,31
(m, 1H), 3,58 (dd, 1H), 3,67-3,79 (m, 3H), 3,86 (dd, 1H), 4,96 (s,
1H), 6,30 (s, 1H), 6,60 (d, 1H), 6,87 (t, 1H), 6,96 (t, 1H), 7,01
(d, 1H). MS (APCI) m/z (relative Intensität) 343 (100).
-
Beispiel 260
-
(R)-10-(3-Mewthoxymethyl-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines zu (R)-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol ähnlichen
Verfahrens und mittels (R)-10-(3-Methoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(800 mg) wird die Titelverbindung als gelber Feststoff (758 mg)
erhalten. Smp. 76-80°C.
1H NMR (CDCl3) δ 2,31 (s,
3H), 2,42-2,27 (m, 2H), 2,39 (s, 3H), 2,84 (dt, 1H), 2,99 (dd, 1H),
3,14 (ddd, 1H), 3,36 (s, 3H), 3,46 (dd, 1H), 3,54 (dd, 1H), 3,91
(d, 1H), 4,02 (d, 1H), 4,96 (s, 1H), 6,30 (d, 1H), 6,60 (dd, 1H), 6,87
(dt, 1H), 6,96 (dt, 1H), 7,04 (dd, 1H). MS (APCI) m/z (relative
Intensität)
357,2 (100).
-
Beispiel 261
-
(R)-10-(3-Ethoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines zu (R)-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol ähnlichen
Verfahrens und mittels (R)-10-(3-Methoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(375 mg, 1 mmol) wird die Titelverbindung als gelber Feststoff (77
mg, 21%) erhalten.
1H NMR (CDCl3) δ 1,08
(t, 3H), 2,30 (s, 3H), 2,31-2,38 (m, 2H), 2,38 (s, 3H), 2,85 (m,
1H), 2,95 (m, 1H), 3,15 (m, 1H), 3,45-3,58 (m, 4H), 3,93 (m, 1H),
4,04 (m, 1H), 5,08 (s, 1H), 6,31 (s, 1H), 6,59 (d, 1H), 6,86 (t,
1H), 6,96 (t, 1H), 7,03 (d, 1H). MS (ES) m/z (relative Intensität) 371,2
(100).
-
Beispiel 262
-
(R)-10-(3-Allyloxymethyl-4-methyl-piperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines zu (R)-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol ähnlichen
Verfahrens wird die Titelverbindung als gelber Feststoff (28 mg)
erhalten.
1H NMR (CDCl3) δ 2,30 (s,
3H), 2,35 (m, 2H), 2,39 (s, 3H), 2,85 (d, 1H), 2,97 (t, 1H), 3,15
(t, 1H), 3,53 (m, 2H), 3,92-4,06 (m, 4H), 4,95 (s, 1H), 5,18 (d,
1H), 5,26 8d, 1H), 5,90 (m, 1H), 6,31 (s, 1H), 6,60 (d, 1H), 6,87
(t, 1H), 6,96 (t, 1H), 7,02 (d, 1H). MS (ESI) m/z (relative Intensität) 383 (100).
-
Beispiel 263
-
(R)-10-(4-Methyl-3-propoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(R)-10-(3-Allyloxymethyl-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(270 mg), 20% Palladiumhydroxid (Degussa Typ, 15 mg) wird in Ethanol
(0,014 M) unter einer Wasserstoffatmosphäre (1 atm) für 4 Stunden
bei Raumtemperatur gerührt.
Es wird filtriert und das Lösemittel
wird unter Bildung der Titelverbindung als gelber Feststoff (268
mg) eingedampft.
1H NMR (CDCl3): δ 0,90
(t, 3H), 1,58 (m, 2H), 2,30 (s, 3H), 2,33 (m, 1H), 2,39 (s, 3H),
2,84 (m, 2H), 3,14 ((, 1H), 3,38 (t, 2H), 3,49 (d, 1H), 3,58 (d,
1H), 3,95 (d, 1H), 4,07 (d, 1H), 4,94 (s, 1H), 6,31 (s, 1H), 6,60
(d, 1H), 6,85 (t, 1H), 6,95 (t, 1H), 7,02 (d, 1H). MS (ESI) m/z
(relative Intensität)
385 (100).
-
Beispiel 264
-
(R)-10-[4-Methyl-3-(2-melhylallyloxymethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaZa-benzo[f]azulen
-
Mittels
eines zu (R)-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]methanol ähnlichen
Verfahrens wird die Titelverbindung als oranges Pulver erhalten.
1H NMR (CDCl3) δ 1,73 (s,
3H), 2,30 (s, 3H), 2,31-2,39 (m, 2H), 2,39 (s, 3H), 2,85 (ddd, 1H),
2,93 (dd, 1H), 3,14 (ddd, 1H), 3,45 (dd, 1H), 3,56 (dd, 1H), 3,88
(s, 2H), 3,97 (m, 1H), 4,07 (m, 1H), 4,88 (s, 1H), 4,94 (s, 1H), 5,04
(s, 1H), 6,31 (s, 1H), 6,60 (d, 1H), 6,87 (t, 1H), 6,96 (t, 1H),
7,02 (d, 1H). MS (ES) m/z (relative Intensität) 397 (100).
-
Beispiel 265
-
(R)-10-(4-Methyl-3-phenoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Wässriges
37% Formaldehyd (1,1 Äquivalente)
wird zu einer Lösung
aus (R)-10-3-Phenoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(520 mg) in Dichlorethan (60 ml pro 1 mmol nicht alkyliertes Ausgangsmaterial)
gegeben. Das Gemisch wird für
2 Minuten gerührt
und Natriumtriacetoxyborhydrid (1,5 mmol pro 1 mmol nicht alkyliertes
Ausgangsmaterial) wird zugegeben. Die Suspension wird für 30 Minuten gerührt und
mit einer gesättigten
wässrigen
Lösung
aus Natriumbicarbonat gestoppt. Die wässrige Phase wird dreimal mit
Dichlormethan extrahiert und die organischen Phasen werden vereinigt, über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wird mittels Chromatographie
auf Silicagel (Methylenchlorid/Methanol (90:10) unter Bildung der
Titelverbindung als gelber Feststoff (265 mg, 49%) gereinigt. Smp. 74-88°C.
1H NMR (CDCl3): δ 2,28 (s,
3H), 2,47 (s, 3H), 2,50-2,39 (m, 1H), 2,66-2,58 (m, 1H), 2,91 (dt,
1H), 3,11 (dd, 1H), 3,26 (dt, 1H), 3,93 (bd, 1H), 4,14-4,03 (m,
3H), 4,91 (bs, 1H), 6,31-6,29 (m, 1H), 6,59 (dt, 1H), 7,00-6,86
(m, 5H), 7,03 (dd, 1H), 7,32-7,27 (m, 2H). MS (APCI) m/z (relative
Intensität)
419,3 (100).
-
Beispiel 266
-
(S)-2-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2-yl]ethanoldihydrochlorid
-
(S)-2-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2-yl]ethanoldihydrochlorid
(110 mg, 0,26 mmol) werden in Dichlormethan (7 ml) vereinigt und
Diisopropylethylamin (226 μl,
1,30 mmol) werden zugegeben. Nach wenigen Minuten wird 37% wässriges
Formaldehyd (23 μl,
0,31 mmol) gefolgt von Natriumtriacetoxyborhydrid auf einmal zugegeben
(83 mg, 0,39 mmol). Es wird bei Raumtemperatur für 30 min gerührt. Es
wird mit Methanol verdünnt
und im Vakuum konzentriert, in Methanol rückgelöst und auf eine SCX Säule gegeben.
Es wird mit Methanol gewaschen und mit 2,5% an 7 N Ammoniak-Methanol
in Dichlormethan unter Bildung der freien Base der Titelverbindung
als gelbes Öl
eluiert. Es wird als reines Dihydrochlorid (39 mg, 35%) derartig
isoliert, wie es in Beispiel 319 beschrieben ist. Massenspektrum
(APCI): m/z = 357,1 8M+1 der freien Base).
-
Beispiel 266a
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
(395,8 mg, 1,16 mmol), Formaldehyd (103,2 μl, 1,27 mmol, 37% in Wasser)
und 1,2-Dichlorethan (25,0 ml) werden vereinigt. Das Gemisch wird
bei Umgebungstemperatur für
5 Minuten gerührt
und dann wird Natriumtriacetoxyborhydrid (367,4 mg, 1,73 mmol) zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt
und gewaschen (Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand wird
auf Silicagel mittels eines Gradienten aus Dichlormethan bis Dichlormethan/Methanol
(80:20) unter Bildung von 306,9 mg (75%) der Titelverbindung als
brauner Schaum gereinigt. Massenspektrum (Ionenspray): m/z = 357,2
(M+1).
-
Beispiel 266b
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-10-[3-(3-Methoxypropyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
(296,0 mg, 0,83 mmol), Formaldehyd (74,1 μl, 0,91 mmol, 37% in Wasser)
und 1,2-Dichlorethan (20,0 ml) werden vereinigt. Das Gemisch wird
bei Umgebungstemperatur für
5 Minuten gerührt
und dann wird Natriumtriacetoxyborhydrid (264,0 mg, 1,25 mmol) zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt
und gewaschen (Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 269,6 mg (88%)
der Titelverbindung gereinigt. Massenspektrum (Ionenspray): m/z
= 371,2 (M+1).
-
Beispiel 267
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
-
Auf
eine Weise, die zu der in Beispiel 269 beschriebenen ähnlich ist
und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
wird das Dihydrochloridsalz der Titelverbindung (102 mg, 22%) erhalten.
Massenspektrum (ES): m/z 371,2 (M+1 der freien Base).
-
Beispiel 268
-
(S)-10-[3-(2-Ethoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
-
Auf
eine Weise, die zu der in Beispiel 331 beschriebenen ähnlich ist
und unter Verwendung von (S)-10-[3-(2-Ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
wird die freie Base der Titelverbindung (0,105 g, 0,273 mmol, 15%)
als gelbes Öl
erhalten. Massenspektrum (APCI): m/z 385,2 (M+1). Das reine Produkt
wird als das entsprechende Dihydrochlorid auf die gleiche Weise,
wie in Beispiel 331 beschrieben, isoliert. Exakte Masse, berechnet:
385,2062, gefunden 385,2070.
-
Beispiel 269
-
(S)-10-[4-Methyl-3-(2-phenoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
-
(S)-10-[3-(2-Phenoxyethyl)piperazin-2-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen,
Formaldehyd (1,1 Äquivalente,
37% wässrige
Lösung)
und Natriumtriacetoxyborhydrid (1,5 Äquivalente) in Dichlormethan (10
ml) werden vereinigt und bei Raumtemperatur für 2 Stunden gerührt. Das
Gemisch wird mit gesättigtem wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Eine Reinigung durch Blitzchromatographie unter Elution mit einem
Stufengradienten ausgehend von Dichlormethan bis zu 6% an 2 N Ammoniak-Methanol
in Dichlormethan ergibt die freie Base der Titelverbindung. Es wird
als Dihydrochloridsalz durch Lösen
der freien Base in Ethanol und Zugabe einer Lösung aus 5 Äquivalenten Chlorwasserstoffsäure in Ethanol
isoliert. Eine Eindampfen der Lösung
und Trocknen des Salzes ergibt die Titelverbindung (0,059 g, 0,129
mmol, 8%) als orangen Feststoff. Massenspektrum (APCI): m/z = 433,2
(M+1 der freien Base).
-
Beispiel 270
-
(R)-10-(3-Benzyloxymethyl-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines Verfahrens, das zu Beispiel 269 ähnlich ist, aber unter Verwendung
von (R)-10-(3-Benzyloxymethyl)piperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
wird die Titelverbindung als gelber Feststoff erhalten: Smp. 55-65°C.
1H NMR (DMSO-d6): δ 2,17 (s,
3H), 2,19 (m, 1H), 2,20 (s, 3H), 2,27 (m, 1H), 2,69 (m, 2H), 2,91
(t, 1H), 3,33 (t, 1H), 3,62 (dd, 1H), 3,68 (d, 1H), 3,84 (d, 1H),
4,43 (q, 2H), 6,28 (s, 1H), 6,63 (dd, 1H), 6,77 (m, 3H), 7,27 (m, 5H),
7,55 (s, 1H). MS (ESI) m/z (relative Intensität) 433 (100)
-
Beispiel 271
-
(R)-10-(4-Methyl-3-phenoxymethylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines Verfahrens, das zu Beispiel 269 ähnlich ist, aber unter Verwendung
von (R)-10-(3-Phenoxymethyl)piperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
wird die Titelverbindung als gelber Feststoff erhalten: Smp. 74-88°C.
1H NMR (DMSO-d6): δ 2,28 (s,
3H), 2,47 (s, 3H), 2,50-2,39 (m, 1H), 2,66-2,58 (m, 1H), 2,91 (dt,
1H), 3,11 (dd, 1H), 3,26 (dt, 1H), 3,93 (bd, 1H), 4,14-4,03 (m,
3H), 4,91 (bs, 1H), 6,31-6,29 (m, 1H), 6,59 (t, 1H), 7,00-6,86 (m,
5H), 7,03 (dd, 1H), 7,32-7,27 (m, 2H). MS (APCI) m/z (relative Intensität) 419,3
(100). 49%, 520 mg ergibt 265 mg.
-
Beispiel 272
-
(R)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl)-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Natriumtriacetoxyborhydrid
(0,15 g, 0,71 mmol) und wässriges
Fomraldehyd (37% G/G, 0,05 ml, 0,62 mmol) werden zu einer Lösung aus
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(0,18 g, 0,47 mmol) in Dichlorethan (12 ml) gegeben und gerührt. Nach
5 Stunden wird mit einer gesättigten
wässrigen
Lösung
aus Natriumbicarbonat verdünnt
und die Phasen werden getrennt. Die wässrige Phase wird mit Dichlormethan
(3 ×)
extrahiert, die organischen Phasen werden vereinigt und getrocknet
(Natriumsulfat), filtriert und unter verringertem Druck zu einem Öl (0,24
g) konzentriert. Das Öl
wird durch Blitzchromatographie unter Elution mit einem Gradienten
einer 5% Lösung
aus 2 M Ammoniak in Methanol in Dichlormethan (0-100% über 30 Minuten)
und dann mit einer 5 Lösung
aus 2 M Ammoniak in Methanol in Dichlormethan unter Bildung der
Titelverbindung (0,14 g, 74 %) gereinigt. Massenspektrum (APCI,
m/e): 399 (M+1).
NMR (1H, 300 MHz,
DMSO-d6): δ 7,63 (s, 1H), 6,80 (m, 3H),
6,66 (m, 1H), 6,30 (s, 1H), 3,70 (m, 2H), 3,35-3,28 (m, 2H), 3,20
(s, 3H), 2,99-2,88 (m, 2H), 2,80-2,60 (m, 2H), 2,29-2,05 (m, 5H),
1,66 (m, 2H), 1,17 (d, 6H, J = 7,5 Hz).
-
Beispiel 273
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl)-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
Eine
Lösung
aus Acetylchlorid (0,13 ml, 1,76 mmol) wird in absolutem Ethanol
zu einer Lösung
aus (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl)-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(0,14 g, 0,35 mmol) in absolutem Ethanol gegeben und gerührt. Es
wird unter verringertem Druck unter Bildung der Titelverbindung
(0,16 g, 94%) konzentriert. Massenspektrum (APCI, m/e): 399 (M+1).
Exakte Massenspektrum (ES+, m/e, C22H30N4OS × 2 HCl):
berechnet 399,2218 (M+1-1HCl), gefunden 399,2226.
-
Beispiel 273a
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
Mittels
eines Verfahrens, das zu Beispiel 222 ähnlich ist, wird die Titelverbindung
erhalten. Massenspektrum (m/e): 385,12 (M+1).
-
Beispiel 273b
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulenhydrochlorid
-
Mittels
eines Verfahrens, das zu Beispiel 223 ähnlich ist, wird die Titelverbindung
erhalten. Massenspektrum (m/e): 385,12 (M+1).
-
Beispiel 274
-
(S)-10-[3-(4-Methoxybutyl)-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
In
eine bei Raumtemperatur gerührte
Lösung
aus (S)-10-[3-(4-Methoxybutyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(0,031 g, 0,08 mmol) und Formaldehydlösung (0,082 ml, 0,10 mmol, 37%
in H2O) in Dichlorethan (0,5 ml) wird NaHB(OAc)3 (0,026 g, 0,12 mmol) gegeben und über Nacht
gerührt. Die
Lösung
wird mit H2O (20 ml), 1,0 N NaOH (1 ml)
verdünnt
und mit CH2Cl2 (3 × 40 ml)
extrahiert. Die vereinigten organischen Phasen werden über Na2SO4 getrocknet und
filtriert. Das Gemisch wird unter verringertem Druck konzentriert
und durch Silicagelchromatographie unter Elution mit 10 (33% an
2 M NH3 in MeOH64% EtOH)CH2cl2 gereinigt. Die gereinigten Fraktionen werden
vereinigt, unter verringertem Druck konzentriert und unter Vakuum
unter Bildung der Titelverbindung gegeben: Oranger Feststoff (0,014
g), Massenspektrum (m/e): 399,3 (M+H).
-
Beispiel 275
-
(S)-10-[3-(4-Methoxybutyl)-4-methylpiperazin-1-yl)-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
In
eine bei Raumtemperatur gerührte
Lösung
aus (S)-10-[3-(4-Methoxybutyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(0,015 g, 0,038 mmol) in CH2Cl2 wird
2,0 N HCl in Et2O (0,038 ml, 0,07 mmol)
gegeben und für
1 Stunde gerührt.
Das Gemisch wird unter verringertem Druck konzentriert und in einem
Vakuumofen bei 69°C über Nacht
unter Bildung der Titelverbindung konzentriert.: Brauner Feststoff (0,018
g), Massenspektrum (m/e): 399,0 (M+1).
-
Beispiel 276
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl)-2-trifluormethyl-4H-3-thia-4,9-diaza-benzo[f]azulenmaleatsalz
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl)-4H-3-thia-4,9-diaza-benzo[f]azulen
(250,0 mg, 0,70 mmol), S-(Trifluormethyl)-3,7-dinitrodibenzothiophentrifluormethansulfonat
(345,2 mg, 0,70 mmol) und DMSO (5 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
2 Stunden gerührt.
Das Gemisch wird zwischen 1 N NaOH und Dichlormethan aufgeteilt,
die organischen Bestandteile werden entfernt, mit Kochsalzlösung gewaschen, über Natriumsulfat
getrocknet und dann zu einem Rückstand
konzentriert. Der Rückstand
wird auf Silicagel mittels Dichlormethan/2 N Ammoniak in Methanol
(95:5) gereinigt und die das gewünschte
Produkt enthaltenden Fraktionen werden reduziert. Der Rückstand
wird wieder mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3)
unter Bildung von 53,6 mg (18%) des gewünschten Produkts als Öl gereinigt.
Das Maleatsalz des Produkts wird in Diethylether hergestellt: Massenspektrum
(Ionenspray): m/z = 425,1 (M+1).
-
Beispiel 277
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl)-2-trifluormethyl-4H-3-thia-4,9-diaza-benzo[f]azulenmaleatsalz
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl)-4H-3-thia-4,9-diaza-benzo[f]azulen
(235,1 mg, 0,63 mmol), S-(Trifluormethyl)-3,7.-dinitrodibenzothiophentrifluormethansulfonat
(343,6 mg, 0,70 mmol) und DMSO (5 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
4 Stunden gerührt.
Das Gemisch wird zwischen DI H2O und Ethylacetat
aufgeteilt, die organische Phase wird entfernt, mit Kochsalzlösung gewaschen,
die organische Phase wird über
Natriumsulfat getrocknet und dann zu einem Rückstand reduziert. Der Rückstand
wird mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3)
unter Bildung 33,8 mg (12%) des gewünschten Produkts als braunes Öl gereinigt.
Das Maleatsalz wird aus dem Produkt in Diethylether hergestellt:
Massenspektrum (Ionenspray): m/z = 439,0 (M+1).
-
Beispiel 278a
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl)-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl)-4H-3-thia-4,9-diaza-benzo[f]azulen
(379,6 mg, 1,05 mmol), Formaldehyd (97,7 μl, 1,20 mmol, 37% in Wasser)
und Dichlormethan (15,0 ml) vereinigt. Das Gemisch wird bei Umgebungstemperatur
für 5 Minuten
gerührt
und dann wird Natriumtriacetoxyborhydrid (334,8 mg, 1,58 mmol) zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt
und gewaschen (Kochsalzlösung), getrocknet
(Natriumsulfat) und die Extrakte werden zu einem Rückstand
verringert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 339,8 mg (86%)
der Titelverbindung als hellbrauner Schaum gereinigt. Massenspektrum
(Ionenspray): m/z = 375,1 (M+1).
-
Beispiel 278b
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl)-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl)-4H-3-thia-4,9-diaza-benzo[f]azulen
(220,1 mg, 0,59 mmol), Formaldehyd (52,5 μl, 0,64 mmol, 37% in Wasser)
und Dichlormethan (8,0 ml) vereinigt. Das Gemisch wird bei Umgebungstemperatur
für 5 Minuten
gerührt
und dann wird Natriumtriacetoxyborhydrid (186,8 mg, 0,88 mmol) zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Der organische Teil wird entfernt und
gewaschen (Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
eingeengt. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 195,1 mg (86%)
der Titelverbindung gereinigt. Massenspektrum (Ionenspray): m/z
= 389,1 (M+1).
-
Beispiel 278c
-
(S)-6-Fluor-10-[3-(2-hydroxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-2-Methyl-10-[3-(2-hydroxyethyl)piperazin-1-yl]piperazin-1-yl]-4H-3-thia-6-fluor-4,9-diaza-benzo[f]azulen (0,508
g, 1,40 mmol) und 37% Formaldehydlösung (0,11 ml, 1,48 mmol) in
1,2-Dichlorethan
(20 ml) vereinigt. Es wird für
15 Minuten gerührt
und Natriumtriacetoxyborhydrid (0,597 g, 2,81 mmol) wird zugegeben.
Es wird für
weitere 60 Minuten gerührt
und dann wird die Lösung
auf gesättigte
Natriumbicarbonatlösung
gegossen. Es wird mit Methylenchlorid unter Bildung von 0,521 g
des rohen Produkts extrahiert. Eine Silicagelchromatographie unter
Elution mit Methylenchlorid : 2 N NH3/Methanol (100:7,5) ergibt
0,271 g der Titelverbindung als freie Base. Das Dihydrochloridsalz
fällt in
Ethylacetat als gelber Feststoff aus: Smp. = 230°C. Massenspektrum (Ionenspray):
m/z = 375 (M+1)
-
Beispiel 279
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulenhydrochlorid
-
(S)-6-Fluor-10-[3-(2-methoxyethyl]piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(5,00 g, 13,4 mmol) wird unter Stickstoff gerührt, um in 1,2-Dichlorethan
(DCE, 55 ml) gelöst
zu werden. Es wird auf 15-20°C
gekühlt
und Formaldehyd (37% G/G in Wasser, 1,03 g Lösung, 0,38 g, 12,7 mmol) in
einer Portion gegeben. Natriumtriacetoxyborhydrid (3,41 g, 16,1
mmo0l) wird portionsweise über
5-10 Minuten zugegen, wobei die Gefäßtemperatur unter 20°C gehalten
wird. Es wird bei 5-20°C
für 15
Minuten gerührt
und kann sich dann auf Umgebungstemperatur erwärmen. Es wird auf 2 Stunden
gerührt
und dann wird wässriges
gesättigtes
Natriumbicarbonat (50 ml) zugegeben. Es wird für 15 Minuten gerührt und
dann werden die Phasen getrennt. Das Produkt wird mit Methylenchlorid
extrahiert. Die vereinigten organischen Phasen werden mit Kochsalzlösung extrahiert, über Magnesiumsulfat
getrocknet und dann unter verringertem Druck zu einem Rückstand
konzentriert. Methylenchlorid wird zu dem Rückstand gegeben und unter verringertem
Druck unter Bildung von 4,94 g (100%) der freien Base der Titelverbindung
konzentriert.
1H NMR (400 MHz, DMSO-d6) δ 7,78
(bs, 1H), 6,79 (m, 1H), 6,68 (m, 1H), 6,53 (m, 1H), 6,34 (s, 1H),
3,74 (m, 2H), 3,36 (m, 3H), 3,22 (s, 3H), 2,95 (m, 1H), 2,77 (m,
1H), 2,69 (m, 1H), 2,30 (s, 3H), 2,23 (s, 3H), 2,17 (bs, 1H), 1,84
(m, 1H), 1,52 (m, 1H). HRMS (ES) exakte Masse M+H berechnet für C20H25FN4OS
389,1811, gefunden 389,1802.
-
Herstellung der kristallinen Form I (Dihydrochlorid
hydratisierter Zustand)
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen freie
Base (100 mg) wird in ein Szintillationsgefäß gegeben. IPA (5 ml) wird
zugegeben. Es wird auf etwa 60°C
auf einer Rührplatte
erhitzt. 2 M Äquivalente
aus HCl (1 N HCl) wird zugegeben. Es wird zur Trockne eingedampft.
IPA (5 ml) wird zu der Aufschlämmung/Suspension
zurückgegeben.
Der Feststoff wird durch Vakuumfiltration isoliert. Es kann über Nacht
unter Bildung der kristallinen Form I luftgetrocknet werden. Nach 194,08°C zersetzt
es sich (TGA-DTA).
-
Herstellung der kristallinen Form II (Dihydrochlorid
hydratisierter Zustand)
-
Gesättigte 400
Mikroliter aus Wasser mit (S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochloridanhydrat.
Es wird mit Form I angeimpft und bei Raumtemperatur über Nacht
gemischt. Der Feststoff wird filtriert und als kristalline Form
II charakterisiert: Schmelzpunkt (Beginn): 195,92°C (DSC).
-
Bestätigung
der kristallinen Form II
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochloridanhydrat
(60 mg) wird zu Wasser (1 ml) gegeben. Es wird mit dem Material
angeimpft, das aus der anfänglichen
Herstellung der kristallinen Form II (Dihydrochlorid hydratisierter
Zustand) erhalten wird. Es kann über
Nacht rühren.
Der Feststoff wird filtriert und charakterisiert.
-
Herstellung der kristallinen Form III
(Dihydrochloridanhydrat)
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (38,2
g, 98,3 mmol) wird unter Stickstoff gerührt um in 2-Propanol (IPA,
380 ml) bei Umgebungstemperatur zu lösen. Es wird auf 70-75°C erhitzt.
Eine Lösung
aus konzentrierter HCl (19,0 ml, 228 mmol) in IPA (190 ml) wird
derartig zugegeben, dass die Temperatur über 70°C gehalten wird. Das Produkt
sollte innerhalb von wenigen Minuten kristallisieren. Die Produktaufschlämmung wird
für 10-15
Minuten bei 70-75°C
erhitzt und kann dann auf Umgebungstemperatur kühlen. Es wird auf 0-5°C gekühlt und
für 1-2
Stunden gerührt.
Es wird filtriert und mit kaltem IPA gewaschen. Es wird bei 50°C unter Bildung
von 41,9 g (92%) der kristallinen Form III (Dihydrochloridanhydrat)
getrocknet. Analyse für
C20H26Cl2FN4OS: berechnet
C 52,06, H 5,90, N 12,14. Gefunden C 51,75, H 5,82, N 11,95. HRMS
(ES) exakte Masse M+H berechnet für C20H25FN4OS (freie Base)
389,1811, gefunden 389,1805.
-
Beispiel 279a
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(1,08 g, 2,88 mmol) wird in 1,2-Dichlorethan (30 ml) gelöst und bei
Raumtemperatur gerührt.
Es wird eine 37% wässrige
Formaldehydlösung
(2 ml) gefolgt von Natriumtriacetoxyborhydrid (0,65 g, 3,06 mmol)
gegeben. Das Reaktionsgemisch wird bei Raumtemperatur über Nacht
gerührt.
Gesättigtes
wässriges
Natrium wird zugegeben und die organische Phase wird gesammelt,
getrocknet und zu 1,2 g eines dunklen Öls konzentriert. Es wird in Methanol
(20 ml) aufgenommen, 2 N Chlorwasserstoffsäure (5 ml) wird zugegeben und
das Gemisch wird bei Raumtemperatur für 2 Stunden gerührt. Das
Reaktionsgemisch wird zwischen Dichlormethan und 2 N Natriumhyroxidlösung konzentriert.
Die organische Phase wird gesammelt, getrocknet und zu 1 g eines
dunklen Öls konzentriert.
Dieses Material wird durch Blitzsäulenchromatographie auf Florisil
(Eluent Dichlormethan/Methanol) unter Bildung von 0,6 g gelbes Öl gereinigt.
Dieses Material wird in Ethanol (20 ml) gelöst, 2 N Chlorwasserstoffsäure (2 ml)
wird zugegeben und das Gemisch wird konzentriert und unter Hochvakuum
unter Bildung des gewünschten
orangen Feststoffs (610 mg) konzentriert. Smp. 190-192°C. Massenspektrum
(FIA) 389 (M+1).
NMR (1H, 300 MHz,
CDCl3) (auf freier Base) δ 6,93 (m,
1H), 6,68 (m, 1H), 6,37 (d, 1H), 4,90 (s, 1H), 3,88 (m, 2H), 3,43
(m, 2H), 3,32 (s, 3H), 3,10 (m, 1H), 2,72 (m, 2H), 2,32 (s, 3H),
2,30 (s, 3H), 2,2-2,3 (m, 2H), 1,98 (m, 1H), 1,65 (m, 1H).
-
Beispiel 280
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(558 mg, 1,44 mmol) in 1,2-Dichorethan (8 ml) gelöst und bei
Raumtemperatur gerührt.
Es wird 37% wässrige
Formaldehydlösung
(200 μl),
gefolgt von Natriumtriacetoxyborhydrid (370 mg, 1,73 mmol) wird
zugegeben. Das Reaktionsgemisch wird für 2 Stunden gerührt. Gesättigtes
wässriges
Natriumcarbonat wird zugegeben und die organische Phase wird gesammelt,
getrocknet und zu einem dunklen Öl
konzentriert. Dieses Material wird durch Blitzsäulenchormatographie auf Florisil
(Eluent Dichlormethan/Methanol) unter Bildung von 340 mg des gelben Öls gereinigt.
Dieses Material wird in Ethylacetat gelöst und 2 N Chlorwasserstoffsäure wird
in Ether gelöst
und das Gemisch wird konzentriert und unter Hochvakuum unter Bildung
des gewünschten
Produkts als oranger Feststoff mit 350 mg getrocknet. Massenspektrum
(FIA) 403 (M+1) Smp. 186-188°C.
-
Beispiel 281
-
(S)-6-Fluor-10-[3-(2-ethoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulenhydrochlorid
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-6-Fluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(285 mg, 0,73 mmol) wird die Titelverbindung mit 142 mg (0,32 mmol) erhalten.
Massenspektrum (FIA) 403 (M+1) Smp. 180-182°C.
-
Beispiel 282
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpipierazin-1-yl]-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-6-Fluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(384 mg, 0,96 mmol) wird die Titelverbindung mit 274 mg (0,56 mmol)
erhalten.
NMR (1H) (DMSO-d6) δ 11,6961
(2H, bs), 9,6123 (1H, bs), 7,3012 (1H, bs), 7,0050 (1H, bt), 6,8960
(1H, bd), 6,6748 (1H, s), 4,4881 (1H, bs), 4,0587 (1H, bs), 3,8868
(2H, bs), 3,6106 (2H-H2O, bs), 3,4060 (3H,
bs) 3,1656 (3H, bs), 3,0294 (1H, m), 2,8423 (3H, bs), 2,2724 (1H,
bs), 1,8456 (1H, bs), 1,2292 (6H, d). Massenspektrum M+H = 417,1.
-
Beispiel 283
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (0,105
g, 0,270 mmol), Formaldehyd (31 μl,
0,378 mmol, 37%) und Natriumtriacetoxyborhydrid (0,086 g, 0,405
mmol) werden in Dichlorethan (6 ml) vereinigt und bei Raumtemperatur über Nacht
gerührt.
Das Gemisch wird mit gesättigtem
Natriumbicarbonat verdünnt
und dreimal mit Methylenchlorid extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung des
rohen Produkts konzentriert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt (S)-6-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (0,097
g, 0,240 mmol), 89%) als gelbes I. Massenspektrum (APCI): m/z =
403,1 (M+1). Das reine Produkt wird das das entsprechende Dichydrochlorid
auf folgende Weise isoliert: Der gelbe Schaum wird in Ethanol (5
ml) gelöst
und eine Lösung
von etwa 5 Äquivalenten
an HCl in Ethanol (5 ml) wird zugegeben. Das Gemisch wird unter
Bildung von (S)-6-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
erhalten.
-
Beispiel 283a
-
(S)-7-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-7-Fluor-10-[3-(3-methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(320 mg, 0,825 mmol) wird die Titelverbindung mit 210 mg (0,48 mmol)
erhalten.
Massenspektrum (APCI): m/z = 403,1 (M+1).
-
Beispiel 284
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
(309,0 mg, 0,83 mmol), (S)-(Trifluormethyl)-3,7-dinitrodibenzothiophentrifluormethansulfonat
(406,2 mg, 0,83 mmol) und DMSO (7,0 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
24 Stunden gerührt. Das
Gemisch wird mit DI H2O verdünnt und
der Niederschlag wird durch Vakuumfiltration entfernt. Das Filtrat wird
durch die Zugabe von 0,1 N NaOH basisch gemacht. Die wässrige Phase
wird mit Ethylacetat extrahiert, mit Kochsalzlösung gewaschen, die organische
Phase wird über
Natriumsulfat getrocknet und dann zu einem Rückstand reduziert. Der Rückstand
wird mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3) unter
Bildung von 60,2 mg (17%) des gewünschten Produkts gereinigt.
Das Dihydrochloridsalz des Produkts wird in Diethylether hergestellt:
Massenspektrum (Ionenspray): m/z = 443,1 (M+1).
-
Beispiel 285
-
(S)-6-Fluor-10-[3-(2-methoxypropyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
-
(S)-6-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen (170,6
mg, 0,44 mmol), (S)-(Trifluormethyl)-3,7-dinitrodibenzothiophentrifluormethansulfonat
(216,2 mg, 0,44 mmol) und DMSO (5 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
24 Stunden gerührt.
Das Gemisch wird mit DI H2O verdünnt und
der Niederschlag wird durch Vakuumfiltration entfernt. Das Filtrat
wird durch die Zugabe von 0,1 N NaOH basisch gemacht. Die wässrige Phase
wird mit Ethylacetat extrahiert, mit Kochsalzlösung gewaschen, die organische
Phase wird über
Natriumsulfat getrocknet und dann zu einem Rückstand reduziert. Der Rückstand
wird mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3)
unter Bildung von 21,8 mg (11%) des gewünschten Produkts gereinigt.
Das Dihydrochloridsalz des Produkts wird in Diethylether hergestellt:
Massenspektrum (Ionenspray): m/z = 457,1 (M+1).
-
Beispiel 286
-
(S)-7-Fluor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-7-Fluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(1,73 g, 4,62 mmol) wird die Titelverbindung mit 965 mg (2,09 mmol) erhalten.
Massenspektrum
(FIA) 389 (M+1). Smp. 192-194°C.
-
Beispiel 287
-
(S)-7-Fluor-10-[3-(2ethoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-7-Fluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(734 mg, 1,89 mmol) wird die Titelverbindung mit 312 mg (0,66 mmol) erhalten.
Massenspektrum
(FIA) 403 (M+1). Smp. 193-195°C.
-
Beispiel 287a
-
(S)-2-[4-(6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]ethanol
-
(S)-2-[4-(6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]ethanol
(436 mg, 1,15 mmol) wird in 1,2-Dichlorethan (5 ml) gelöst, Formaldehyd
(91 μl,
1,15 mmol, 37% in Wasser) wird zugegeben und unter Stickstoff für 10 min
gerührt.
Natriumtriacetoxyborhydrid (452 mg, 2,13 mmol) wird zugegeben und
für 1 Stunde
bei Umgebungstemperatur gerührt.
Das Gemisch wird mit gesättigter
Natriumbicarbonatlösung
verdünnt
und mit Dichlormethan extrahiert. Die Extrakte werden mit Kochsazlösung gewaschen und
die organische Phase (Na2SO4)
wird getrocknet, filtriert und unter verringertem Druck konzentriert.
Eine Reinigung durch Silicagelchromatographie (Eluent: 0-5% an 2
N Ammoniak in Methanol/Dichlormethan) ergibt 255 mg des unreinen
Produkts. Eine Reinigung durch Silicagelchromatographie (Eluent:
0-10% an 2 N Ammoniak in MethanolDichlormethan) ergibt 120 mg des
unreinen Produkts und 113 mg des reinen Produkts. Eine Reinigung
durch Radialsilicagelchromatographie mittels einer 2 mm Platte (Eluent:
1-2% an 2 N Ammoniak in Methanol/Dichlormethan) ergibt 76 mg des
reinen Produkts. Beide Lots des reinen Produkts werden unter Bildung
von 189 mg (42%) der Titelverbindung vereinigt. Massenspektrum (ESMS)
393 (M+1), 391 (M-1).
-
Beispiel 287b
-
(S)-2-[4-(6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]ethanoldihydrochlorid
-
(S)-2-[4-(6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]ethanol (171 mg,
0,44 mmol) wird in Ethylacetat gelöst. 3 Äquivalente Chlorwasserstoff
werden in Ethanol zugegeben. Nach 18 Stunden wird der Niederschlag
unter einer Stickstoffatmosphäre
filtriert, mit Ethylacetat gewaschen und unter Vakuum unter Bildung
der Titelverbindung (198 mg, 98%) getrocknet. Massenspektrum (LCMS)
393 (M+1), 391 (M-1).
-
Beispiel 288
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulenhydrochlorid
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (26,1
g, 66,5 mmol) wird in 1,2-Dichlorethan (DCE, 260 ml) unter Rühren bei
Umgebungstemperatur unter Stickstoff gelöst und auf 10-15°C gekühlt. Wässriges
37% G/G Formaldehyd (5,40 g × 37%
= 2,00 g, 66,6 mmol) wird in einer Portion zugegeben. Pulverisiertes
Natriumtriacetoxyborhydrid (19,8 g, 93,4 mmol) wird in wenigen Portionen über 10-15
Minuten zugegeben und die Temperatur wird unter 20°C gehalten.
Der Rückstand
wird mit DCE (26 ml) gewaschen. Die Reaktionslösung wird bei Umgebungstemperatur
für 1-2
Stunden gerührt.
Wässriges
gesättigtes
Natriumbicarbonat (260 ml) wird tropfenweise zugegeben und dann
bei Umgebungstemperatur für
20-30 Minuten gerührt.
Die Phasen werden getrennt und dann wird die wässrige Phase mit Methylenchlorid
(50 ml) extrahiert. Die vereinigten organischen Phasen werden mit
Wasser (100 ml) und dann Kochsalzlösung (100 ml) extrahiert. Die
organische Lösung
wird über
Magnesiumsulfat unter Rühren
getrocknet, filtriert und dann unter verringertem Druck zu einem öligen schaumigen
Rückstand
konzentriert. Es wird in Methylenchlorid gelöst und unter verringertem Druck
unter Bildung von 26,0 (96%) der freien Base der Titelverbindung
als Schaum konzentriert.
1H NMR (400
MHz, DMSO-d6): δ 7,70 (s, 1H), 6,74 (m, 2H),
6,36 (s, 1H), 3,75 (bm, 2H), 3,35 (m, 2H), 3,20 (s, 3H), 2,95 (bt,
1H), 2,75 (m, 2H), 2,30 (s, 3H), 2,22 (s, 3H), 2,15 (bm, 2H), 1,82
(m, 1H), 1,50 (m, 1H). HRMS (ES) exakte Masse M+H berechnet für C20H24F2N4OS 407,1717, gefunden 407,1716.
-
Herstellung der kristallinen Form I (Dihydrochlorid
hydratisierter Zustand)
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (123
mg) wird in ein Szintillationsgefäß gewogen. Aceton (4 ml) wird
zugegeben. Es wird auf etwa 60°C
erhitzt. 2 M Äquivalente
an HCl (1 N HCl stock) werden zugegeben. Es wird bei dieser Temperatur
für wenige
Minuten gerührt.
Es zeigt sich eine Suspension. Es wird auf Raumtemperatur über Nacht
gekühlt.
Es wird durch Vakuumfiltration zur Isolierung des Feststoffs filtriert.
Es kann lufttrocknen und wird als kristalline Form I charakterisiert.
-
Herstellung der kristallinen Form II (Dihydrochlorid
hydratisierter Zustand)
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen (3,00
g, 7,38 mmol) werden in Aceton (98 ml) unter Rühren bei Umgebungstemperatur
unter Stickstoff gelöst
und auf 50-55°C
erhitzt. Wässriges
1 N HCl (15,5 ml, 15,5 mmol), die das suspendierte Produkt enthält, wird
zugegeben und die Kristalle (30 mg) werden in einer Portion angeimpft.
Es wird auf 50-55°C
für 20-30
Minuten erhitzt, damit das Produkt ausfallen kann und dann kann
es auf Umgebungstemperatur abkühlen.
Es wird auf 0-5°C
gekühlt
und für
1-2 Stunden gerührt.
Es wird filtriert und mit kaltem Aceton (2 × 20 ml) gewaschen und bei
45-50°C
getrocknet. Es kann bei Umgebungstemperatur stehen, bis das konstante
Gewicht unter Bildung von 3,40 g (96%) der kristallinen Form II
(Dihydrochlorid hydratisierter Zustand) erreicht ist. HRMS (ES)
exakte Masse M+H berechnet für
C20H24F2N4OS (als freie Base) 407,1717, gefunden 407,1721.
-
Beispiel 288a
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-2-methyl-4H-3-thia-4,9-dibenzo[f]azulendihydrochlorid
-
Mittels
eines Verfahrens, das zu dem Beispiel (S)-6-Fluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid ähnlich ist
und unter Verwendung von 6,7-Difluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhydrochlorid
und (S)-2-(2-Methoxy)ethylpiperazin wird die Titelverbindung hergestellt.
1H NMR (DMSO-d6) δ 11,7110
(2H, bs), 9,3793 (1H, bs), 7,3795 (1H, bs), 7,0701 (1H, bt), 6,6326
(1H, bs), 4,1000 (3H, bm), 3,6041 (3H, bs), 3,4347 (3H, bs), 3,2081
(3H, bs), 2,8371 (3H, bs), 2,3319 (3H, bs), 2,2842 (1H, bm), 1,8410
(1H, bs). Massenspektrum M+H = 407,1.
-
Beispiel 289
-
(S)-6,7-Difluor-10-[3-(2-ethoxyethyl)-4-methyl-piperazin-1-yl]-2-methyl-4H-3-thia-4,9-dibenzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-7,8-Difluor-10-[3-(2-ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(353 mg, 0,868 mmol) wird die Titelverbindung mit 172 mg (0,35 mmol)
erhalten.
1H NMR (DMSO-d6): δ 11,6927
(2H, bs), 9,3610 (1H, bs), 7,3801 (1H, bs), 7,0656 (1H, bt), 6,6658
(1H, bs), 4,2374 (2H, bs), 4,0610 (1H, bs), 3,6059 (2H, bs). 3,3831
(3H, bs), 2,8383 (3H, bs), 2,3242 (3H, bs), 2,2500 (1H, bd), 1,8171
(1H, bs), 1,0375 (3H, bs). Massenspektrum M+H = 421,1.
-
Beispiel 290
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diazabenzo[f]azulensuccinat
-
Auf ähnliche
Weise zu Beispiel 280 und mittels (S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-4,9-diaza-benzo[f]azulensuccinat
(440 mg, 1,08 mmol) wird die freie Base der Titelverbindung (236
mg, 1,78 mmol) erhalten. Diese wird in DCM gelöst und mit einer Lösung aus
Bernsteinsäure (66,4
mg, 0,57 mmol) in Ethanol unter Bildung der Titelverbindung mit
300 mg (0,56 mmol) nach dem Eindampfen behandelt.
1H
NMR (DMSO-d6): δ 12,14 (2H, bs), 7,73 (1H, s),
6,72 (2H, m), 6,35 (1H, s), 3,76 (2H, bm), 3,33 (4H, m), 2,99 (1H,
m), 2,77 (2H, m)=, 2,65 (2H, m), 2,41 (5H, m), 2,41 (5H, m), 2,25
(5H, m), 1,85 (1H, m), 1,51 (1H, m), 1,16 (3H, m). Massenspektrum
M+H = 421,1.
-
Beispiel 290a
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
(207,2 mg, 0,55 mmol), Formaldehyd (48,9 μl, 0,60 mmol, 37% in Wasser)
und Dichlormethan (8,0 ml) werden vereinigt. Das Gemisch wird bei
Umgebungstemperatur für
5 Minuten gerührt
und dann wird Natriumtriacetoxyborhydrid (174,0 mg, 0,82 mmol) zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt
und gewaschen (Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 176,2 mg (82%)
der Titelverbindung gereinigt. Massenspektrum (Ionenspray) : m/z
= 393,2 (M+1).
-
Beispiel 290b
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-6,7-Difluor-10-[3-(2-methoxyethyl)-4-methyl-piperazin-1-yl]-4H-3-thia-4,9-diazabenzo[f]azulen (381,0
mg, 0,97 mmol), S-(Trifluormethyl)-3,7-dinitrodibenzothiophentrifluormethansulfonat
(479,0 mg, 0,97 mmol) und DMSO (10,0 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
24 Stunden gerührt.
Das Gemisch wird mit DI H2O verdünnt und
der Niederschlag wird durch Vakuumfiltration entfernt. Das Filtrat
wird durch die Zugabe von 0,1 N NaOH basisch gemacht. Die wässrige Phase
wird Ethylacetat extrahiert, mit Kochsalzlösung gewaschen, die organische
Phase wird über
Natriumsulfat getrocknet und dann zu einem Rückstand reduziert. Der Rückstand
wird mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3)
unter Bildung von 58,7 mg (13%) des gewünschten Produkts gereinigt.
Das Dihydrochloridsalz des Produkts wird in Diethylether hergestellt:
Massenspektrum (Ionenspray): m/z = 461,2 (M+1).
-
Beispiel 291
-
(S)-6-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-6-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(195 mg, 0,5 mmol) wird in 1,2-Dichlorethan (3 ml) gelöst und bei
Raumtemperatur gerührt.
37% wässrige
Formaldehydlösung
(50 μl)
gefolgt von Natriumtriacetoxyborhydrid (140 mg) wird zugegeben.
Das Reaktionsgemisch wird bei Raumtemperatur über Nacht gerührt. Gesättigtes
wässriges
Natriumcarbonat wird zugegeben und die organische Phase wird gesammelt,
getrocknet und zu einem dunklen Öl
konzentriert. Dieses Material wird durch Blitzsäulenchromatographie auf Florisil
(Eluent Dichlormethan/Methanol) unter Bildung von 71 mg eines gelben Öls gereinigt.
Dieses Material wird in Ethylacetat gelöst und 2 N Chlorwasserstoffsäure in Ether
wird zugegeben und das Gemisch wird konzentriert und unter Hochvakuum
unter Bildung von (S)-6-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
als das gewünschte
Produkt als oranger Feststoff mit 76 mg getrocknet. Massenspektrum
(FIA) 405/407 (M+1) Smp. = 175-176°C.
-
Beispiel 291a
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(571,5 mg, 1,52 mmol), Formaldehyd (135,4 μl, 1,67 mmol, 37% in Wasser)
und Dichlormethan (20,0 ml) werden vereinigt. Das Gemisch wird bei
Umgebungstemperatur für
5 Minuten gerührt
und dann wird Natriumtriacetoxyborhydrid (482,0 mg, 2,27 mmol) zugegeben.
Nach dem Rühren
für 30
min bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt
und gewaschen (Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 418,3 mg (71%)
der Titelverbindung als brauner Schaum gereinigt. Massenspektrum
(Ionenspray) m/z = 391,1 (M+1).
-
Beispiel 291b
-
(S)-7-Chlor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-7-Chlor-10-[3-(3-methoxypropyl)piperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
(280,3 mg, 0,72 mmol), Formaldehyd (64,0 μl, 0,79 mmol, 37% in Wasser)
und Dichlormethan (10,0 ml) werden vereinigt. Das Gemisch wird bei
Umgebungstemperatur für
5 Minuten gerührt
und dann wird Natriumtriacetoxyborhydrid (227,9 mg, 1,08 mmol) zugegeben.
Nach dem Rühren
für 30
Minuten bei Umgebungstemperatur wird die Reaktion mit gesättigtem
Natriumbicarbonat gestoppt. Die organische Portion wird entfernt
und gewaschen (Kochsalzlösung),
getrocknet (Natriumsulfat) und die Extrakte werden zu einem Rückstand
reduziert. Der Rückstand
wird auf Silicagel mittels eines Gradienten aus Dichlormethan bis
Dichlormethan/Methanol (90:10) unter Bildung von 223,0 mg (77%)
der Titelverbindung gereinigt. Massenspektrum (Ionenspray): m/z
= 405,1 (M+1).
-
Beispiel 291c
-
(S)-7-Chlor-10-[3-(2-hydroxyethyl)-4-methyl-piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
-
Mittels
eines Verfahrens, das zu dem Beispiel 287 a ähnlich ist und unter Verwendung
von 7-Chlor-10-[3-(2-hydroxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
wird die Titelverbindung hergestellt.
-
Beispiel 292
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
(1,15 g) wird in 1,2-Dichlorethan (150 ml) gelöst und bei Raumtemperatur gerührt. 37%
wässrige
Formaldehydlösung
(0,18 ml) gefolgt von Natriumtriacetoxyborhydrid (940 mg) wird zugegeben.
Das Reaktionsgemisch wird bei Raumtemperatur über Nacht gerührt. Gesättigtes
wässriges
Natriumcarbonat wird zugegeben und die organische Phase wird gesammelt,
getrocknet und zu einem dunklen Öl
konzentriert. Dieses Material wird durch Blitzsäulenchromatographie auf Florisil
(Eluent Dichlormethan/Methanol) unter Bildung von 400 mg eines gelben Öls gereinigt.
Dieses Material wird in Ethylacetat gelöst und 2 N Chlorwasserstoffsäure in Ether wird
zugegeben und das Gemisch wird konzentriert und unter Hochvakuum
unter Bildung des gewünschten Produkts
als oranger Feststoff mit 224 mg getrocknet. Massenspektrum (LCMS)
405/407 (M+1) Smp. = 200-202°C.
-
Beispiel 292a
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
(33,3 g, 85,2 mmol) und 1,2-Dichlorethan (DCE, 500 ml) werden vereinigt.
Formaldehyd (37% G/G in Wasser, 6,91 g Lösung, 2,56 g, 85,2 mmol) wird
in einer Portion zugegeben und bei Umgebungstemperatur für 5 Minuten
gerührt.
Natriumtriacetoxyborhydrid (3,41 g, 16,1 mmol) wird portionsweise über 5-10
Minuten zugegeben, wobei die Gefäßtemperatur
unter 25°C
bleibt. Es wird bei Umgebungstemperatur über Nacht gerührt. Gesättigtes
wässriges
Natriumcarbonat (333 ml) wird zugegeben. Es wird für 15 Minuten
gerührt
und dann werden die Phasen getrennt. Die wässrige Phase wird mit Methylenchlorid
(250 ml) extrahiert. Die vereinigten organischen Phasen werden mit
Wasser (3 × 333
ml) gewaschen, über
Natriumsulfat getrocknet und dann im Vakuum zu einem Rückstand
(>100% CH2Cl2 bis 93/7 CH2Cl2/MeOH) gereinigt, wobei 91,2% der Titelverbindung
erhalten werden.
1H NMR (500 MHz, DMSO-d6) δ 1,49
(m, 1H), 1,82 (m, 1H), 2,13 (m, 2H), 2,20 (s, 3H), 2,27 (s, 3H),
2,74 (m, 2H), 2,98 (bt, 1H), 3,19 (s, 3H), 3,32 (m, 2H), 3,77 (bm,
2H).
-
Beispiel 292b
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulendihydrochlorid
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(39,7 g, 97,6 mmol) und 2-Propanol (IPA, 397 ml) werden bei Umgebungstemperatur
vereinigt und auf 80-81°C
erhitzt. Eine Lösung
aus konzentriertem HCl (17,1 ml, 206 mmol) in IPA (79 ml) wird über 20-30
Minuten zugegeben. Die Temperatur des entstehenden Gemisches wird
bei 79-80°C
für 1,5
Stunden gehalten. Die Heizquelle wird geschlossen und das Gemisch
kann sich langsam auf 30°C über 3 Stunden
erhitzen. Es wird auf 0-5°C
gekühlt
und für
eine weitere Stunde gerührt.
Die Feststoffe werden filtriert und mit eiskaltem IPA (3 × 50 ml)
gewaschen. Es wird bei 50°C
unter Bildung von 46,5 g (99,3%) der Titelverbindung getrocknet.
KF Analyse ergibt 5,47% H2O, die Chloridanalyse
ergibt einen Wert von 12,8%:
-
Beispiel 293
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-4,9-diaza-benzo[f]dihydrochlorid
-
(S)-7-Chlor-10-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen
(381,2 mg, 0,98 mmol), S-(Trifluormethyl)-3,7-dinitrodibenzothiophentrifluormethansulfonat
(480,0 mg, 0,98 mmol) und DMSO (8,0 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
24 Stunden gerührt. Das
Gemisch wird mit DI H2O verdünnt und
der Niederschlag wird durch Vakuumfiltration entfernt. Das Filtrat wird
durch die Zugabe von 0,1 N NaOH basisch gemacht. Die wässrige Phase
wird mit Ethylacetat extrahiert, mit Kochsalzlösung gewaschen, die organische
Phase wird über
Natrium sulfat getrocknet und dann zu einem Rückstand reduziert. Der Rückstand
wird mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3) unter
Bildung von 78,3 mg (18%) des gewünschten Produkts gereinigt.
Das Dihydrochloridsalz des Produkts wird in Ethylacetat hergestellt:
Massenspektrum (Ionenspray): m/z = 459,1 (M+1).
-
Beispiel 294
-
(S)-7-Chlor-10-[3-(2-methoxypropyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-4,9-diaza-benzo[f]dihydrochlorid
-
(S)-7-Chlor-10-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-4H-3-thia-4,9-diaza-benzo[f]azulen (196,8
mg, 0,49 mmol), S-(Trifluormethyl)-3,7-dinitrodibenzothiophentrifluormethansulfonat
(239,2 mg, 0,49 mmol) und DMSO (5 ml) werden vereinigt. Das Gemisch
wird bei Umgebungstemperatur für
24 Stunden gerührt.
Das Gemisch wird mit DI H2O verdünnt und
der Niederschlag wird durch Vakuumfiltration entfernt. Das Filtrat
wird durch die Zugabe von 0,1 N NaOH basisch gemacht. Die wässrige Phase
wird mit Ethylacetat extrahiert, mit Kochsalzlösung gewaschen, die organische
Phase wird über
Natriumsulfat getrocknet und dann zu einem Rückstand reduziert. Der Rückstand
wird mittels Hexan/THF/Ethanol/2 N Ammoniak in Methanol (65:30:5:3)
unter Bildung von 34,5 mg (15%) des gewünschten Produkts gereinigt.
Das Dihydrochloridsalz des Produkts wird in Diethylether hergestellt:
Massenspektrum (Ionenspray): m/z = 473,0 (M+1).
-
Beispiel 295
-
(S)-10-[3-(2-Phenylsulfanylethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
-
2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(0,455 g, 1,85 mmol), (S)-2-(2-ÜPhenylsulfanylethyl)piperazin
(0,411 g, 1,85 mmol) und Pyridin (5 ml) werden vereinigt und für 36 Stunden
am Rückfluss
erhitzt. Das Gemisch wird eingedampft und das Material wird auf
10 g SCX aufgetra gen, dann mit Methanol gefolgt von 5% an 2 N Ammoniak-Methanol
in Dichlormethan und dann 2 N Ammoniak-Methanol eluiert. Eine Reinigung
mittels Blitzchromatographie unter Elution mit einem Stufengradienten
ausgehend von Dichlormethan bis 7 5 an 2 N Ammoniak-Methanol in
Dichlormethan ergibt (S)-10-[3-(2-Phenylsulfanylethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
(0,126 g, 0,290 mmol, 16%). Massenspektrum (APCI): m/z = 435,1 (M+1).
-
Beispiel 296
-
(S)-10-[4-Methyl-3-(2-phenylsulfanylethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]dihydrochlorid
-
(S)-10-[3-(2-Phenylsulfanylethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen
(0,118 g, 0,271 mmol), Formaldehyd (24 μl, 0,299 mmol, 37% wässrige Lösung) und
Natriumtriacetoxyborhydrid (86 mg, 0,407 mmol) werden in Dichlorethan
(10 ml) vereinigt und bei Raumtemperatur für 2 Stunden gerührt. Das
Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Eine Reinigung durch Blitzchromatographie unter Elution mit einem
Stufengradienten ausgehend von Dichlormethan bis zu 6% an 2 N Ammoniak-Methanol
in Dichlormethan ergibt die freie Base der Titelverbindung. Das
reine Produkt wird als das entsprechende Dihydrochlorid durch Lösen der
freien Base in Ethanol isoliert und eine Lösung aus 5 Äquivalenten Chlorwasserstoffsaure
in Ethanol werden zugegeben. Ein Eindampfen der Lösung und
ein Trocken des Salzes ergibt die Titelverbindung (0,089 g, 0,189
mmol, 70%) als braunen Feststoff. Massenspektrum (APCI) m/z = 449,1
(M+1 der freien Base).
-
Beispiel 297
-
(S)-6-Fluor-10-[4-methyl-3-(2-methylsulfanylethyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diaza-benzo[f]dihydrochlorid
-
Ein
Gemisch aus 6-Fluor-2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-ylaminhyrochloridsalz (0,135
g, 0,476 mmol), (S)-1-Methyl-2-(2-methylsulfanylethyl)piperazin
(0,166 g, 0,95 mmol) und Diisopropylethylamin (0,083 ml, 0,476 mmol)
in DMSO (0,2 ml) und Toluol (0,8 ml) wird bei 110°C für 46 Stunden
erhitzt. Das Reaktionsgemisch wird mit Ethylacetat und 0,1 N Natriumhydroxidlösung verdünnt. Der
Ethylacetatextrakt ergibt 0,196 g des rohen Produkts. Eine Silicagelchromatographie
unter Elution mit Methylenchlorid : Methanol (100:5) ergibt 0,081
g der Titelverbindung als oranges Öl. Das Dihydrochloridsalz fällt in Ethylacetat
als oranger Feststoff aus: Smp. 194°C. Massenspektrum (Ionenspray):
m/z = 405 (M+1).
-
Beispiel 300
-
(S)-2-[4-(2-Methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-2-yl]ethanol
-
2-Methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-ylaminhydrochlorid
(0,458 g, 1,72 mmol), (S)-2-Piperazin-2-yl-ethanol
(0,518 g, 3,98 mmol), Diisopropylethylamin (0,346 ml, 1,99 mmmol),
Dimethylsulfoxid (1,5 ml) und Toluol (4,5 ml) werden vereinigt und
für 48
Stunden auf 110°C
erhitzt. Das Gemisch wird eingedampft und das Material wird auf
20 g SCX aufgetragen, mit Methanol gefolgt von 5% an 2 N Ammoniak-Methanol
in Dichlormethan und schließlich
100% an 2 N Ammoniak-Methanol eluiert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 11)% an 2 N Ammoniak-Methanol in Dichlormethan ergibt (S)-2-[4-(2-Methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-2-yl]ethanol
(0,082 g, 0,239 mmol, 14%): Massenspektrum (APCI): m/z = 344,4 (M+1).
-
Beispiel 301
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Auf
eine Weise, die zu der in Beispiel 300 beschriebenen ähnlich ist
werden 2-Methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-ylaminhydrochlorid
(0,437 g, 1,64 mmol) und (S)-2-(2-Methoxyethyl)piperazin (0,474 g, 3,29
mmol) unter Bildung der Titelverbindung vereinigt. Massenspektrum
(APCI): m/z = 358,0 (M+1).
-
Beispiel 302
-
(S)-10-[3-(2-Ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Auf
eine Weise, die zu der in Beispiel 300 beschriebenen ähnlich ist
werden 2-Methyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-ylamin
(0,534 g, 2,01 mmol) und (S)-2-(2-Ethoxyethyl)piperazin (0,635 g,
4,01 mmol) unter Bildung der Titelverbindung vereinigt. Exakte Masse
berechnet: 372,1858, gefunden 372,1867.
-
Beispiel 303
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
2-Ethyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(1,05 g, 4,0 mmol) wird in 8 ml CH2Cl2 vereinigt und Methyltrifluormethansulfonat
(0,98 g, 0,68 ml, 6,0 mmol) wird zugegeben und über Nacht gerührt. Wenn
die Reaktion nicht vollständig
ist, wird zusätzliches
Methyltrifluormethansulfonat zugegeben. Das Reaktionsgemisch wird
unter verringertem Druck unter Bildung eines rot-braunen Feststoffs
(1,62 g) konzentriert. Die Hälfte
des Materials (0,81 g, ~1,9 mmol) wird genommen, mit 2(S)-(2-Methoxyethyl)piperazin
(0,27 g, 1,9 mmol) in 5 ml Pyridin gemischt und die Reaktion wird
auf 100°C
erhitzt. Die Reaktion wird nach dem Erhitzen über Nacht gekühlt und
zu einem Rückstand
konzentriert. Eine Reinigung durch Blitzchromatographie auf Silicagel
(35 g) (Gradient 100% CH2Cl2 bis
100% gemischtes Lösemittel
aus (CH2Cl2 : 2
N NH3/MeOH = 15:1) über 55 min ergibt 265 mg der
Titelverbindung. Massenspektrum (Elektrospray) (m/e): C19H25N5OS × HCl, berechnete
Masse (M+1-2 HCl): 372,1658, gefunden: 372,1876.
1H
NMR (300 MHz, CDCl3) δ 7,05-6,96 (m, 2H), 6,89-6,84
(m, 1H), 6,64-6,64 (m, 1H), 4,99 (s, 1H), 4,20 (br, 1H), 3,51 (t,
2H, J = 6,3 Hz), 3,32 (s, 3H), 3,01-2,97 (m, 4H), 2,85 (q, 2H, J
= 6,9 Hz), 2,71-2,63
(m, 1H), 1,87-1,63 (m, 4H), 1,30 (t, 3H, J = 7,2 Hz).
-
Beispiel 304
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-propyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 303 wird die Titelverbindung (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-propyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
als hellgelber Feststoff erhalten.
1H
NMR (400 MHz, CDCl3): δ 7,05-6,98 (m, 2H), 6,92-6,89
(m, 1H), 6,67-6,50 (m, 1H), 5,40 (br, 1H), 4,20 (br, 1H), 3,54 (m,
2H), 3,34 (s, 3H), 3,25-2,95 (m, 7H), 2,78 (t, 2H, J = 7,6 Hz),
1,84-1,80 (m, 2H), 1,78-1,63 (m, 2H), 0,98 (t, 3H, J = 7,6 Hz).
-
Beispiel 305
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-butyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Methyltrifluormethansulfonat
(0,51 ml, 4,5 mmol) wird zu einer 0°C Lösung aus 2-Butyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(0,87 g, 3,0 mmol) in wasserfreiem Dichlormethan (4 ml) gegeben.
Die Feststoffe werden in die Reaktion mit Dichlormethan (3 ml) gewaschen
und gerührt
und die Reaktion kann langsam Umgebungstemperatur erreichen. Nach
einer Übernachtperiode
wird unter verringertem Druck unter Bildung des rohen methylierten
Zwischenprodukts (1,30 g) konzentriert. Das Zwischenprodukt (1,30
g, 2,87 mmol) und wasserfreies Pyridin (10 ml) werden vereinigt,
auf 100°C
erhitzt und gerührt.
Nach einer Übernachtperiode
wird auf Umgebungstemperatur gekühlt
und unter verringertem Druck zu einem Öl (2,16 g) konzentriert. Das Öl wird durch
Blitzchromatographie unter Elution mit einem Gradienten aus einer
7% Lösung
an 2 M Ammoniak in Methanol in Dichlormethan (0-100% über 55 Minuten)
unter Bildung der Titelverbindung (0,37 g, 32%) gereinigt. Massenspektrum
(APCI, m/e): 400 (M+1).
NMR (1H, 300
MHz, DMSO-d6) δ 7,88 (s, 1H), 6,82 (m, 3H),
6,67 (m, 1H), 4,07 (m, 2H), 3,45-3,27 (m, 3H), 3,20 (s, 3H), 3,15-2,82
(m, 4H), 2,82-2,68 (m, 3H), 1,76-1,53 (m, 4H), 1,30 (m, 2H), 0,86
(t, 3H, J = 7,2 Hz).
-
Beispiel 306
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-butyl-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-2-Butyl-10-[3-(2-methoxyethyl)piperazin-1-yl]-4H-3-thia-1,4,9-triaza-benzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten: Massenspektrum (APCI, m/e): 400 (M+1).
Exaktes Massenspektrum (ES+, m/e, C21H29N5OS × 2 HCl):
berechnet : 400,2171 8M+1-2HCl),
gefunden 400,2193.
-
Beispiel 307
-
(S)-2-[4-(2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)piperazin-2-yl]ethanol
-
Auf
eine Weise, die zu der in Beispiel 300 beschriebenen ähnlich ist,
werden (2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,558 g, 1,89 mmol) und (S)-2-Piperazin-2-yl-ethanol (0,493 g, 3,79 mmol) der Titelverbindung
erhalten Massenspektrum (APCI = m/z) = 372,3 (M+1).
-
Beispiel 308
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,481 g, 1,63 mmol), k (S)-2-(2-Methoxyethyl)piperazin (0,471 g,
3,27 mmol) und Diisopropylethylamin (0,284 ml, 1,63 mmol) werden in
einem Gemisch auf Toluol (4,5 ml) und Dimethylsulfoxid (1,5 ml)
vereinigt und bei 110°C
für 24
Stunden gerührt.
Das Gemisch wird eingedampft und dann mit Methanol verdünnt und
auf zwei 10 g SCX Säulen
aufgetragen. Es wird mit Methanol eluiert und dann ergibt ein Stufengradient
ausgehend von Dichlormethan bis zu 10% an 2 N Ammoniak-Methanol
in Dichlormethan gefolgt von 2 N Ammoniak-Methanol das gewünschte Material in einem rohen
Status. Eine Reinigung durch Blitzchromatographie unter Elution
mit einem Stufengradienten ausgehend von Dichlormethan bis zu 9%
an 2 N Ammoniak-Methanol
in Dichlormethan ergibt die Titelverbindung (0,186 g, 0,482 mmol,
30%) als gelbes Öl:
Massenspektrum (APCI): m/z = 386,2 (M+1).
-
Beispiel 308a
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(2,61 g, 9,48 mmol) wird mit Trifluormethansulfonsäuremethylester
(1,61 ml, 14,2 mmol) in Dichlormethan (20 ml) vereinigt. Es wird
bei Raumtemperatur für
2 h gerührt
und dann wird das Lösemittel
im Vakuum entfernt. Der Rückstand
wird in Pyridin (20 ml) suspendiert und (S)-2-(2-Methoxyethyl)piperazin
(1,37 g, 9,48 mmol) wird zugegeben. Es wird bei 115°C für 8 erhitzt
und dann wird das Lösemittel
im Vakuum entfernt und der Rückstand
wird auf eine Silicagelsäule gegeben.
Die Säule
wird mit Dichlormethan gefolgt von 5% an 2 N Ammoniak-Methanol in
Dichlormethan unter Bildung der Titelverbindung (2,11 g, 58%) als
gelbes Öl
gegeben. Massenspektrum (APCI): m/z = 386,2 (M+1).
-
Beispiel 308b
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Ein
1 l Einzelhalsrundbodenkolben, der mit einem Kühler, einem magnetischen Rührstab,
einem Heizmantel und einem Stickstoffeinlass ausgestattet ist, wird
verwendet. Der Kolben wird mit 2-Isopropyl-10-methylsulfanyl-4H-3-thi-1,4,9-triaza-benzo[f]azulen
(44,9 g, 0,155 mol), (S)-2-(2-Methoxyethyl)piperazin (27,98 g, 0,194
mol, 1,25 Äquivalente)
und IPA (270 ml, 6 Volumina) befüllt.
Die entstehende Aufschlämmung
wird auf Rückfluss
erhitzt und durch HPLC aufgezeichnet. Säule = Zorbax C-8, Fluß = 1 ml/min,
A) ACN, B = 0,1% wässriges
TFA, Gradient = 95% A5% B bis 5% A95% B über 10 Minuten, bei 5/95 A/B
für 3 Minuten
gehalten und wieder zurück
auf 95/5 A/B über
2 Minuten. Säulentemperatur
= 30°C,
Wellenlänge
= 250 nm. Nach 3 Stunden am Rückfluss
zeigt die HPLC Analyse ein Produkt zu Ausgangsmaterialverhältnis (p/sm)
von 2,91,0. Das Reaktionsgemisch kann sich über Nacht unter einer Stickstoffspülung auf
Rückfluss
erhitzen. Nach 19,75 Stunden am Rückfluss zeigt die HPLC Analyse
ein p/sm Verhältnis
von 16,11,0. Das Gemisch kann am Rückfluss 2 Stunden länger erhitzen
und wird wieder durch HPLC untersucht. Die Indikationen dieser Analyse
zeigen keine Veränderung
im p/sm Verhältnis.
Zusätzliches
(S)-2-(2-Methoxyethyl)piperazin (2,24 g, 0,0155 mmol, 0,1 Äquivalente)
wird zu dem Reaktionsgefäß gegeben
und am Rückfluss
für 5 Stunden
weiter erhitzt. Die Indikationen der HPLC Analyse zeigen 3% verbleibendes
Ausgangsmaterial, 2-Iospropyl-10-methylsulfoanyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen. Während der
Reaktion entwickelt sich Mercaptan. In diesem Maßstab werden die Gerüche, die
mit dieser Chemie assoziiert sind, durch eine Stickstoffspülung kontrolliert,
die nach hinten in den Abzug geleitet wird.
-
Bei
zukünftigen
größeren Maßstäben sollte
Bleichlaugewäscher
in Betracht gezogen werden. Das Erhitzen wird gestoppt und das Reaktionsgemisch
kann sich auf 40-50°C
abkühlen.
Der Kolben wird in einen Rotationsverdampfer überführt und das IPA wird mittels
eines 40-50°C
Bads gestrippt. Das entstehende dicke, orange Öl wird in Ethylacetat (100
ml) gelöst,
in einen Trenntrichter überführt und
mit entionisiertem Wasser (2 × 250
ml) gewaschen. Nach einer Kochsalzwaschung (250 ml) wird die Lösung über Na2SO4 getrocknet,
filtriert und im Vakuum konzentriert. Das entstehende Öl kann unter
Vakuum über
Nacht bei Umgebungstemperatur abziehen. Eine Verfestigung des Materials über Nacht
in eine gelbe Masse (74,1 g, Theorie: 59,84 g). Die HPLC Analyse
zeigt 2,1% des verbleibenden 1-Isopropyl-10-methylsulfanyl-4H-3-thia-1,4,9-triaza-benzo[f]azulens.
Diese Feststoffe werden mit Ligroin (500 ml) und Ethylacetat (50
ml) behandelt. Ein magnetischer Rührstab wird in den Kolben gegeben
und das Gemisch wird bei Umgebungstemperatur kräftig gerührt, bis die gesamten Feststoffe
in kleine Partikel (6 Stunden) zerkleinert sind. Das Gemisch wird
filtriert und die Feststoffe werden mit 100/5 Ligroin/Ethylacetat
(105 ml) und dann Ligroin (2 × 200
ml) gewaschen. Die gelben Feststoffe werden auf eine auf Tara gesetzte
Wägeschale
gegeben und unter Vakuum bei Umgebungstemperatur getrocknet. Es
werden 53,67 g der Titelverbindung (89,7%) erhalten.
1H NMR (500 MHz, DMSO-d6): δ 1,23 (d,
6H), 1,49-1,53 (m, 2H), 2,20-2,39 (bm, 1H), 2,43-2,49 (m, 1H), 2,62-2,74
(m, 2H), 2,70-2,83 (m, 1H), 2,85-2,90 (m, 1H), 3,04-3,08 (m, 1H),
3,19 8s, 3H), 3,30-3,41
(m, 2H), 3,90-4,09 (bm, 2H), 6,67 (m, 1H), 6,69-6,79 (m, 2H), 6,79-6,86
(m, 1H).
13C NMR (100 MHz, DMSO-d6): δ 22,88,
22,91, 33,13, 33,85, 39,36, 40,61, 45,54, 52,88, 58,36, 69,67, 119,25, 123,43,
124,33, 127,96, 131,74, 141,41, 143,52, 151,04, 155,97, 165,59.
MS
(80:20 MeOH : H2O G/6,5 mM NH4OAc).
Berechnet: 385,53, gefunden: ES+ 386,2.
ES– 384,2.
-
Beispiel 309
-
(S)-10-[3-(2-Ethoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Auf
eine Weise, die zu der in Beispiel 300 beschriebenen ähnlich ist,
werden 2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-ylaminhydrochlorid
(0,523 g, 1,77 mmol) und (s)-2-(2-Ethoxyethyl)piperazin (0,561 g, 3,54
mmol) unter Bildung der Titelverbindung vereinigt. Massenspektrum
(APCI): m/z = 400,2 (M+1).
-
Beispiel 310
-
(S)-10-[3-(3-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
-
2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-19-thion
(0,5 g, 1,816 mmol) und Methyltriflat (0,306 ml, 2,723 mmol) werden
in Dichlormethan (4 ml) vereinigt und bei Umgebungstemperatur für 2 Stunden
gerührt.
Das Gemisch wird unter verringertem Druck eingedampft und dann wird
der Rückstand
mit (S)-2-(3-Methoxypropyl)piperazin (0,287 g, 1,816 mmol) und Pyridin
(4 ml) vereinigt und für
8 Stunden am Rückfluss
erhitzt. Das Reaktionsgemisch wird unter verringertem Druck konzentriert
und der Rückstand
wird mit Ethylacetat verdünnt.
Die organischen Phasen werden mit Kochsalzlösung zweimal gewaschen, über Natriumsulfat
getrocknet, filtriert und unter verringertem Druck unter Bildung
eines Rückstands
konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Elution mit 2 M Ammoniak in
Methanol : Dichlormethan (5:95) unter Bildung der Titelverbindung
gereinigt. Massenspektrum (m/e): 400,03 (M+1).
-
Beispiel 311
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-cyclopentyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(0,529 g, 1,75 mmol) und Methyltriflat (0,24 ml, 2,11 mmol) werden
in Dichlormethan (3 ml) vereinigt und bei 0°C über Nacht gerührt. Das
Gemisch wird bei 0°C
für 15
Minuten gerührt
und über
Nacht auf Raumtemperatur erwärmt.
Das Gemisch wird eingedampft und dann wird der Rückstand mit (S)-2-(2-Methoxyethyl)piperazin
(0,253 g, 1,75 mmol) und Pyridin (3 ml) vereinigt und am Rückfluss über Nacht
erhitzt. Das Gemisch wird eingedampft und dann mit Methanol verdünnt und
auf eine 10 g SCX Säule
aufgetragen. Es wird mit Methanol gewaschen und dann mit einem Stufengradienten
ausgehend von Dichlormethan bis 10% an 2 N Ammoniak-Methanol in
Dichlormethan gefolgt von 100% an 2 N Ammoniak-Methanol unter Bildung
des gewünschten
gekuppelten Produkts eluiert. Es wird durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan bis
zu 6% an 2 N Ammoniak-Methanol in Dichlormethan unter Bildung von
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-cyclopentyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,378 g, 0,918 mmol, 52%) als gelber amorpher Feststoff gereinigt.
Massenspektrum (APCI): m/z = 412,2 (M+1).
-
Beispiel 312
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-4H-3-thia-1,4,9-triaza-benzo[f]azulen-2-yl-methanol
-
(S)-10-[3-(2-Methoxyethyl)-4-methyl-piperazin-1-yl]-4H-3-thia-1,4,9-triaza-benzo[f]azulen-2-carbonsäureethylester
(0,49 g, 1,19 mmol) in 5,0 ml THF wird 2,95 ml an LiAlH4 (1,0
M THF) tropfenweise bei einem Eiswasserbad gegeben. Nach der Zugabe
wird das Eisbad entfernt und das Reaktionsgemisch wird bei RT gerührt. Nach
einer halben Stunde wird die Reaktion durch Zugabe von 1,0 N NaOH
vorsichtig gestoppt, bis keine Gasentwicklung auftritt. Die Suspension
wird durch ein Celitekissen gegeben, wiederholt mit Ether gewaschen
und die organische Lösung
wird zu einem Rückstand
konzentriert. Eine Reinigung durch Blitzchromatographie ergibt 0,22
g der Titelverbindung: Massenspektrum (Elektrospray) (m/e): 374,0
(M+1), 372,1 (M-1).
1H NMR (40 MHz,
DMSO-d6): 7,79 (s, 1H), 6,86-6,75 (m, 3H),
6,69-6,66 (m, 1H), 5,89 (br, 1H), 4,50 (d, 2H, J = 4,4 Hz), 4,04-3,93
(m, 2H), 3,38-3,333 (m, 2H), 2,84-2,60 (m, 4H), 2,46-2,41 (m, 1H),
1,49 (q, 2H, J = 6,6 Hz).
-
Beispiel 313
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-4H-3-thia-1,4,9-triaza-benzo[f]azulen-2-carbonsäureethylester
-
10-Thioxo-9,10-dihydro-4H-3-thia-1,4,9-triaza-benzo[f]azulen-2-carbonsäureethylester
(3,59 g, 11,76 mmol) in 25 ml CH2Cl2 wird vereinigt und Methyltriflouormethansulfonat
(2,41 g, 14,7 mmol) wird tropfenweise bei 0°C zugegeben, bei 0°C für eine halbe
Stunde gerührt
und auf RT erwärmt.
Nach 2 h wird das Reaktionsgemisch unter verringertem Druck konzentriert,
mit (S)-2-(2-Methoxyethyl)piperazin (0,147 g, 10,2 mmol) in 20 ml
Pyridin gemischt und auf 100°C
erhitzt. Nach 4 Stunden wird die Reaktion auf RT gekühlt und
zu einem Rückstand
konzentriert. Eine Reinigung durch Blitzchromatographie (zweimal)
auf Silicagel mittels eines Gradienten (100% CH2Cl2 bis 100% gemischtem Lösemittel aus (CH2Cl2: 2 N NH3/MeOH =
20:1) über
55 min ergibt die Titelverbindung: Massenspektrum (Elektrospray)
(m/e): C20H25N5O3S berechnet Masse
(M+1): 416,1756, gefunden: 416,1748.
1H
NMR (400 MHz, CDCl3) δ 7,05-6,98 (m, 2H), 6,91-6,85
(m, 1H), 6,65-6,62 (m, 1H), 5,51 (br, 1H), 4,40 (q, 2H, J = 7,5
Hz), 4,10 (br, 2H), 3,51 (t, 2H, J = 6,0 Hz), 3,32 (s, 3H), 3,30-3,00
(m, 4H), 2,79-2,72
(m, 1H), 1,79-1,66 (m, 2H), 1,38 (t, 3H, J = 7,5 Hz).
-
Beispiel 314
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 305 und mittels 2-Trifluormethyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
und Methyltrifluormethansulfonat und 2-(S)-(2-Methoxyethyl)piperazin,
gefolgt von einer Reinigung unter Elution mit einem Gradienten aus
einer 7% Lösung
aus 2 M Ammoniak in Methanol in Dichlormethan (0-100%) ergibt die
Titelverbindung: Massenspektrum (APCI, m/e): 412 (M+1)
NMR
(1H, 300 MHz, DMSO-d6) δ 8,71 (br.s,
1H), 8,60 (s, 1H9, 6,93 (m, 3H), 6,71 (m, 1H), 4,07 (br.m, 2H), 3,46-3,29
(m, 4H), 3,29-3,06 (m, 5H), 2,99 (m, 1H), 2,48 (m, 2H).
-
Beispiel 315
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und mittels (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur,
Rekonstitution des Produkts in entionisiertem Wasser : Aceton (1:1)
und Lyophilisierung wird die Titelverbindung erhalten. Massenspektrum
(APCI, m/e): 412 (M+1). Exakte Massenspektrum (ES+, m/e, C18H20F3N5OS × 2
HCl): berechnet 412,1419 (M+1-2HCl), gefunden 412,1429.
-
Beispiel 316
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 305 und mittels 2-Difluormethyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
und Methyltrifluormethansulfonat und 2-(S)-(2-Methoxyethyl)piperazin
und 20 Stunden bei 100°C
gefolgt von zwei Reinigungsschritten wird die Titelverbindung erhalten:
als erstes Elution mit einem Gradienten aus einer 4% Lösung aus
2 M Ammoniak in Methanol in Dichlormethan (0-100%) und als zweites Elution mit einem
Gradienten der Lösungen
aus 2 M Ammoniak in Methanol in Dichlormethan (2% bis 4%). Massenspektrum
(APCI, m/e): 394 (M+1)
NMR (1H, 300
MHz, DMSO-d6) δ 8,27 (s, 1H), 7,13 (t, 1H, 2J(H,F) = 54,3 Hz),
6,93-6,78 (m, 3H), 6,70 (m, 1H), 3,92 (br.m, 2H), 3,41-3,15 (m,
4H), 2,82 (m, 2H), 2,67 (m, 2H), 2,53-2,41 (m, 2H), 2,31 (br.s,
1H), 1,50 (m, 2H).
-
Beispiel 317
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triaza- benzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten: Massenspektrum (APCI, m/e): 394 (M+1).
Exakte Massenspektrum (ES+, m/e C17H21F2N5OS × 2 HCl):
berechnet 394,1513 (M+1-2HCl), gefunden 394,1490.
-
Beispiel 318
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-(3,3,3-trifluorpropyl)-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
2-(3,3,3-Trifluorpropyl)-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(1,11 g, 3,37 mmol) und 7 ml CH2Cl2 werden vereinigt, Methyltrifluormethansulfonat
(0,69 g, 4,22 mmol) wird bei 0°C
tropfenweise bei 0°C
zugegeben, bei 0°C
für eine
halbe Stunde gerührt
und für
4,5 h auf RT erwärmt.
Das Reaktionsgemisch wird unter verringertem Druck unter Bildung
von 1,58 g eines orangen Feststoffs konzentriert. Es werden 1,18 g
dieses Materials (1,18 g, 2,4 mmol) genommen, mit (S)-2-(2-Methoxyethylpiperazin
(0,346 g, 2,4 mmol) in 10 ml Pyridin gemischt und auf 100°C erhitzt.
Die Reaktion wird nach 3 Stunden auf RT gekühlt und zu einem Rückstand
konzentriert. Eine Reinigung durch Blitzchromatographie auf Silicagel
mittels eines Gradienten (100% CH2Cl2 bis 100% gemischtes Lösemittel aus (CH2Cl2 : 2 N NH3/MeOH
= 25:1) über
55 min ergibt 0,56 g an (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-(3,3,3-trifluorpropyl)-4H-3-thia-1,4,9-triaza-benzo[f]azulen
mit einer Ausbeute von 53%. Das Dihydrochloridsalz wird durch die
Zugabe von 5 Äquivalenten
an Acetylchlorid (156 mg, 1,99 mmol) zu der freien Base (75 mg,
0,55 mmol) in Ethanol (5 ml) gebildet. Das Lösemittel wird entfernt, in
12 ml gemischtem Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 186 mg des orangen Feststoffs als die Titelverbindung
lyophilisiert. Massenspektrum (Elektrospray) (m/e): C20H24F3N5OS × 2 HCl,
berechnet Masse (M+1-2 HCl): 440,1732, gefunden: 440,1716.
-
Beispiel 319
-
(S)-2-[1-Methyl-4-(2-methyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)piperazin-2-yl]ethanoldihydrochlorid
-
Das
Material von Beispiel 300 (0,080 g, 0,233 mmol), Formaldehyd (25 μl, 0,303
mmol, 37% wässrige Lösung) und
Natriumtriacetoxyborhydrid (74 mg, 0,349 mmol) werden in Dichlormethan
(8 ml) vereinigt und bei Raumtemperatur über Nacht gerührt. Das
Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck konzentriert.
Eine Reinigung durch Blitzchromatographie unter Elution mit einem
Stufengradienten ausgehend von Dichlormethan bis zu 6% an 2 N Ammoniak-Methanol
in Dichlormethan ergibt die freie Base der Titelverbindung (0,038
g, 0,106 mmol, 46%). Es wird als das Dihydrochloridsalz durch Lösen der
freien Base in Ethanol isoliert und eine Lösung aus 5 Äquivalenten Chlorwasserstoffsäure in Ethanol
wird zugegeben. Ein Eindampfen der Lösung und Trocknen des Salzes
ergibt die Titelverbindung als braunen Feststoff. Massenspektrum
(APCI): m/z = 358,3 8M+1 der freien Base).
-
Beispiel 320
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu der in Beispiel 319 beschriebenen und unter Verwendung
von (S)-10-[3-(2-Methoxyethylpiperazin-1-yl]-2-methyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
wird das Dihydrochloridsalz als gelber Schaum (0,125 g, 19%) erhalten.
Exakte Masse, berechnet 372,1858, gefunden 372,1866.
-
Beispiel 321
-
(S)-10-[3-(2-Ethoxyethyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
Auf ähnliche
Weise zu der in Beispiel 319 beschriebenen und unter Verwendung
von (S)-10-[3-(2-Ethoxyethyl)piperazin-1-yl]-2-methyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
wird die freie Base der Titelverbindung (0,207 g, 0,537 mmol, 33%)
als gelbes Öl
erhalten. Massenspektrum (APCI): m/z 414,2 (M+1). Das reine Produkt
wird als das entsprechende Dihydrochlorid auf die in Beispiel 319
beschriebenen Weise erhalten. Exakte Masse, berechnet 386,2015,
gefunden 386,2033.
-
Beispiel 322
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-ethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-ethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(180 mg, 0,48 mmol), Formaldehyd (37% G/G, wässrig) (49 mg, 0,6 mmol) und
Natriumtriacetoxyborhydrid (152,6 mg, 0,72 mmol) werden in 5 ml
1,2-Dichlorethan vereinigt und bei RT gerührt. Nach 18 h wird die Reaktion
durch die Zugabe von Wasser gestoppt, mit CH2Cl2 extrahiert und die vereinigten organischen
Lösemittel
werden über Na2SO4 getrocknet.
Eine Reinigung durch Blitzchromatographie auf Silicagel mittels
eines Gradienten (100% CH2Cl2 bis
100% gemischtes Lösemittel
aus (CH2Cl2 : 2
N NH3/MeOH = 15:1) über 55 min) ergibt 85 mg eines hellbraunen
Schaums der freien Base.
1H NMR (300
MHz, CDCl3): δ 6,98-6,89 (m, 2H), 6,82-6,77
(m, 1H), 6,56-6,53 (m, 1H), 4,99 (s, 1H), 4,00 (br, 2H), 3,38 (t,
2H, J = 6,9 Hz), 3,26 8s, 3H), 3,20-3,12 (m, 1H), 2,89-2,75 (m,
4H), 2,42-2,31 (m, 2H), 2,28 (s, 3H), 1,92-1,83 (m, 1H), 1,69-1,60
(m, 1H), 1,23 (t, 3H, J = 7,2 Hz).
-
Das
Dihydrochloridsalz wird durch die Zugabe von 4 Äquivalenten an Acetylchlorid
(69,3 mg, 0,88 mmol) der freien Base (85 mg, 0,22 mmol) in Ethanol
(5 ml) gebildet. Das Lösemittel
wird entfernt, der Rückstand
wird in 15 ml gemischtem Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 90 mg des orangen Feststoffs der Titelverbindung
lyophilisiert. Massenspektrum (Elektrospray) (m/z): C20H27N5OS × 2 HCl,
berechnet Masse (M+1-2HCl): 386,2028, gefunden: 386,2015.
-
Beispiel 323
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-propyl-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 322 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-propyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(390 mg, 1,01 mmol), Formaldehyd (37% G/G, wässrig) (102,2 mg, 1,26 mmol)
und Natriumtriacetoxyborhydrid (321,0 mg, 1,55 mmol) werden 200
mg eines gelben Schaums, (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-propyl-4H-3-thia-1,4,9-triaza-benzo[f)azulen
erhalten: Massenspektrum (Elektrospray) (m/e): 399,9 8M+1), 398,1
(M-1).
1H NMR (400 MHz, DMSO-d6):
7,04-6,97 (m, 2H), 6,89-6,85 (m, 1H), 6,63-6,61 (m, 1H), 5,29 8s,
1H), 4,10 (br, 1H), 3,45 (m, 2H), 3,31 (s, 3H), 3,26-3,23 (m, 1H),
3,01-2,77 (m, 4H), 2,49-2,43 (m, 1H), 2,36 (s, 3H), 2,02-1,93 (m,
1H), 1,77-1,60 (m, 5H), 0,99 8t, 3H, J = 6,8 Hz).
-
Das
Dihydrochloridsalz wird durch die Zugabe von 4 Äquivalenten aus Acetylchlorid
(157 mg, 2,0 mmol) zu der freien Base (200 mg, 0,50 mmol) in Ethanol
(8,0 ml) gebildet. Das Lösemittel
wird entfernt, der Rückstand
wird in 15 ml gemischte Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 213 mg der Titelverbindung lyophilisiert.
Massenspektrum (Elektrospray) (m/e): C21H29N5OS × 2 HCl
berechnete Masse (M+1-2HCl): 400,2171, gefunden: 400,2191.
-
Beispiel 324
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-butyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 272 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-butyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
und 2 Stunden erwärmen
auf Umgebungstemperatur und unter Verwendung eines gesättigten
wässrigen
Natriumchloridwaschschritts der organischen Bestandteile nach einem
gesättigten
wässrigen
Natriumbicarbonatwaschsschritt wird die Titelverbindung erhalten.
Massenspektrum (APCI, m/e): 414 (M+1).
NMR (1H,
300 MHz, CDCl3) δ 7,00 (m, 2H), 6,87 (dt, 1H,
J0 = 7,2 Hz, Jm =
1,5 Hz), 6,62 (dd, 1H, J0 = 7,8 Hz, Jm = 0,9 Hz), 5,00 (s, 1H), 4,05 (br.m, 2H),
3,45 (br.m, 2H), 3,32 (s, 3H), 3,23 (m, 1H), 2,94 (m, 1H), 2,82
(m, 3H), 2,45 (m, 1H), 2,35 (m, 4H), 1,95 (m, 1H), 1,69 (m, 3H),
1,39 (m, 2H), 0,93 (t, 3H, J = 7,5 Hz).
-
Beispiel 325
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-butyl-4H-3-thia-1,4,9-triaza-benzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-butyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
in absolutem Ethanol unter Zugabe von Methanoltropfen zum Lösen der
freien Base und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten. Massenspektrum (APCI, m/e) 414 (M+1)
Exakte Massenspektrum (ES+, m/e, C22H31N5OS × 2 HCl):
berechnet 414,2328 (M+1-2HCl), gefunden 414,2350.
-
Beispiel 324
-
(S)-2-[4-(2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]ethanol
-
2-[4-(2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)piperazin-2-yl]ethanol
(0,161 mg, 0,422 mmol), Formaldehyd (46 μl, 0,563 mmol, 37%) und Natriumtriacetoxyborhydrid
(0,138 g, 0,650 mmol) werden in Dichlorethan (15 ml) vereinigt und
bei Raumtemperatur über
Nacht gerührt.
Das Ge misch wird mit gesättigtem
Natriumbicarbonat verdünnt
und bei Raumtemperatur über
Nacht gerührt.
Das Gemisch wird mit gesättigtem
Natriumbicarbonat verdünnt
und dreimal mit Methylenchlorid extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
des rohen Produkts konzentriert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 8% an 2 N Ammoniak-Methanol in Dichlormethan ergibt (S)-2-[4-(2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]ethanol (0,059
g, 0,153 mmol, 35%) als gelbes Öl.
Massenspektrum (APCI): m/z = 386,4 (M+1). Das saubere Produkt wird
als das entsprechende Dihydrochlorid auf folgende Weise isoliert:
Das gelbe Öl
wird in Ethanol (5 ml) gelöst
und eine Lösung
von etwa 5 Äquivalenten
aus HCl in Ethanol (5 ml) wird zugegeben. Das Gemisch wird unter
Bildung von (S)-2-[4-(2-Isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]ethanoldihydrochlorid
eingedampft.
-
Beispiel 327
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(0,181 g, 0,469 mmol), Formaldehyd (50 μl, 0,610 mmol, 37%) und Natriumtriacetoxyborhydrid
(0,149 g, 0,704 mmol) werden in Dichlorethan (12 ml) vereinigt und
bei Raumtemperatur für
2 Stunden gerührt.
Das Gemisch wird mit gesättigtem
Natriumbicarbonat verdünnt
und dreimal mit Methylenchlorid extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
des rohen Produkts konzentriert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen (0,168
g, 0,420 mmol, 90%) als gelbes Öl:
Massenspektrum (APCI): m/z = 400,3 (M+1). Das klare Produkt wird
als das entsprechende Dihydrochlorid auf folgende Weise isoliert:
Der gelbe Schaum wird in Ethanol (5 ml) gelöst und eine Lösung von etwa
5 Äquivalenten
aus HCl in Ethanol (5 ml) wird zugegeben. Das Gemisch wird unter
Bildung der Titelverbindung aus brauner Schaum eingedampft. Exakte
Massen, berechnet 400,2171, gefunden 400,2171.
-
Beispiel 327a
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-triazabenzo[f]azulendihydrochlorid
-
Auf
dieselbe Weise wie für
Beispiel 327 beschrieben, wird (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(1,44 g, 3,73 mmol) in (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(1,22 g, 82%) umgewandelt. Gemäß den Standardverfahren
wird das Titelhydrochlorid (1,43 g, 100%) isoliert. Exakte Masse,
berechnet 400,2171, gefunden 400,2171.
-
Beispiel 327b
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
-
Ein
1 l fassender Dreihalsrundbodenkolben wird mit einer von oben kommenden
Rührapparatur,
einem Zugabetrichter, einer Trannwand, einer Thermometerführung und
einem Kühlbad
ausgestattet. Der Kolben wird mit (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(50,0 g, 0,129 mmol) und DCE (500 ml, 10 Volumina) ausgestattet.
Das entstehende Gemisch kann bei Umgebungstemperatur für 20 Minuten
rühren
und alle Feststoffe können
sich lösen.
Es wird 37 % wässriges
Formalin (9,7 ml, 0,129 mol, 1,0 Äquivalente) zu dem Zugabetrichter
gegeben und zu dem obigen Gemisch über 4 Minuten tropfenweise
gegeben. Natriumtriacetoxyborhydrid (31,6 g, 0,149 mol, 1,15 Äquivalente)
werden portionsweise über
10 Minuten zugegeben, wobei die Gefäßtemperatur unter 25°C bleibt.
Das Reaktionsgemisch wird über
Nacht bei Umgebungstemperatur gerührt. Nach 21 Stunden wird eine
kleine Probe (4 Tropfen) des Reaktionsgemisches in 0,4 ml gesättigtes
NaHCO3 gegossen. Die Probe wird mit 0,25
ml EtOAc verdünnt,
geschüttelt
und kann sich trennt. Eine Analyse einer Probe der organischen Phase
durch TLC (100% Aceton + 2,5% V/V an 2 M NH3 in
MeOH, UV) zeigt, dass kein Ausgangsmaterial verbleibt. Das Reaktionsgemisch
wird mit gesättigtem
NaHCO3 (500 ml) gestoppt und bei Umgebungstemperatur
für 20
Minuten gerührt.
Nach der Überführung in
einen Trenntrichter und Pha sentrennung wird die wässrige Phase
mit Methylenchlorid (500 ml) extrahiert. Die organischen Phasen
werden vereinigt und mit gesättigtem
NaHCO3 (2 × 500 ml) und entionisiertem
Wasser (500 ml) gewaschen. Die Lösung
wird über
Na2SO4 (50 g) in
Anwesenheit von Darco (1 T) getrocknet und durch ein Kissen aus
HiFlo, das mit einer 1/8'' Phase aus Kieselgel-60
Silica bedeckt ist, filtriert. Das Filtrat wird im Vakuum unter
Bildung von 58,6 g des orange-gelben Öls (Theorie 51,82 g) konzentriert.
Das rohe, übergewichtige Öl wird mit
Ligroin (500 ml) und EtOAc (50 ml) behandelt und kann bei Umgebungstemperatur
für 6 Stunden
rühren.
Die entstehenden Feststoffe werden filtriert, mit Ligroin (2 × 150 ml)
gewaschen und durch Vakuum unter Bildung von 43,6 g der freien Base
als blassgelber Feststoff (84,2%) getrocknet.
1H
NMR (500 MHz, DMSO-d6): δ 1,24 (d, 6H), 1,50-1,59 (m,
1H), 1,75-1,84 (m, 1H), 2,12-2,24 (m, 2H), 2,20 (s, 3H), 2,71-2,77
(m, 1H), 2,79-2,82 (m, 1H), 3,02-3,10 (m, 2H), 3,19 8s, 3H), 3,27-3,32
(m, 2H), 3,80-3,92 (bm, 2H), 6,67 (m, 1H), 6,77-6,81 (m, 2H), 6,84-6,87
(m, 1H), 7,82 (s, 1H).
-
Kristallisation
der freien Base: Ein 750 ml fassender Dreihals Euro-Kolben wird
mit einem magnetischen Rührstab,
einem Rückflusskühler, einem
Heizmantel und einer Trennwand mit Thermometerführung ausgestattet. Der Kolben
wird mit dem wiederaufgeschlämmten,
amorphen (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazinyl]-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(30,0 g, 0,075 mol), Heptan (300 ml, 10 Volumina) und Toluol (60
ml, 2 Volumina) befüllt.
Das entstehende Gemisch wird bei Umgebungstemperatur für 10 Minuten gerührt und
am Rückfluss
(99,8°C)
erhitzt. Alle Feststoffe werden gelöst, wobei die Gefäßtemperatur
97,1°C erreicht.
Die klare gelbe Lösung
wird für
5 Minuten am Rückfluss
erhitzt. Die Heizquelle wird abgeschalten, wobei der Mantel verbleibt,
so dass die Gefäßtemperatur
langsam absinken kann. Das Rühren
wird auf ein Minimum verlangsamt. Bei 70,1°C zeigen sich die ersten Zeichen
von Trübung
im Gemisch. Die Feststoffe bilden sich bei 69,3°C Gefäßtemperatur. Das Gemisch kann
sich auf eine Raumtemperatur von 25,7°C über 3,25 Stunden abkühlen. Der
Heizmantel wird entfernt und das Gemisch wird für weitere 50 Minuten gerührt, während die
schließliche
Temperatur 22,1°C
beträgt.
Das Material wird auf einen gesinterten Glastrichter Vakuum-filtriert
und die Feststoffe werden mit Ligroin (2 × 100 ml) gewaschen. Der Trichter
wird für
15 Minuten getrocknet, der Filterkuchen wird auf eine auf Tara gesetzte
Glasschale überführt und
weiter unter Vakuum bei 30°C
getrocknet. Es werden 27,9 g der gelben Feststoffe (92,9%) erhalten.
1H NMR (500 MHz, DMSO-d6): δ 1,24 (d,
6H), 1,50-1,59 (m, 1H), 1,75-1,84 (m, 1H), 2,12-2,24 (m, 2H), 2,20 (s,
3H), 2,71-2,77 (m, 1H), 2,79-2,82 (m, 1H), 3,02-3,10 (m, 2H), I
3,10 (m, 2H), 3,19 (s, 3H), 3,27-3,32
(m, 2H), 3,80-3,92 (bm, 2H), 6,67 (m, 1H), 6,77-6,81 (m, 2H), 6,84-6,87
(m, 1H), 7,82 (s, 1H).
-
Eine
kleine Probe des Materials wird unter einem Mikrokop beobachtet
und es wird gefunden, dass es eine Bröckligkeit zeigt.
-
Alternativ
dazu wird diffus amorphes (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazinyl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(65 ng) in n-Pentan (200 μl)
in amorphes (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazinyl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(65 mg) in Ethylacetat (210 μl)
gegossen. Die Kristalle werden gebildet und isoliert.
-
Beispiel 328
-
(S)-10-[3-(2-Ethoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Auf
eine Weise, die zu der in Beispiel 319 beschriebenen ähnlich ist,
und unter Verwendung von (S)-10-[3-(2-Ethoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
wird die Titelhyrochlorid (0,215 g, 0,491 mmol, 34%) als oranger
Feststoff erhalten. Exakte Masse berechnet: 414,2328, gefunden 414,2341.
-
Beispiel 329
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 222 wird die Titelverbindung erhalten.
Massenspektrum (m/e): 414,08 (M+1).
-
Beispiel 330
-
(S)-10-[3-(3-Methoxypropyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulenhydrochlorid
-
Mittels
des Verfahrens von Beispiel 223 wird die Titelverbindung erhalten.
Massenspektrum (m/e): 414,08 (M+1).
-
Beispiel 331
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-cyclopentyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-cyclopentyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,364 g, 0,884 mmol), Formaldehyd (93 μl, 1,15 mmol, 37%) und Natriumtriacetoxyborhydrid
(0,281 g, 1,33 mmol) werden in Dichlorethan (15 ml) vereinigt und
bei Raumtemperatur für
4 Stunden gerührt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
eines festen Rückstands
konzentriert. Eine Reinigung durch Blitzchromatographie unter Eution
mit einem Stufengradienten ausgehend von Dichlormethan bis 6% an
2 N Ammoniak-Methanol in Dichlormethan ergibt die freie Base der
Titelverbindung (0,342 g, 0,804 mmol, 91%) als gelbes Öl: Massenspektrum
(APCI): m/z) 386,4 (M+1) Das reine Produkt wird als das entsprechende
Dihydrochlorid auf folgende Weise isoliert: Der gelbe Schaum wird in
Ethanol (5 ml) gelöst
und eine Lösung
von etwa 5 Äquivalenten
Chlorwasserstoffsäure
in Ethanol (5 ml) wird zugegeben und dann wird das Lösemittel
unter Vakuum entfernt. Exakte Masse berechnet: 426,2328, gefunden:
426,2329.
-
Beispiel 332
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen-2-yl-methanoldihydrochlorid
-
Mittels
des Verfahrens von Beispiel 322 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen-2-yl-methanol
(210 mg, 0,564 mmol), Formaldehyd (37% wässrig, G/G) (57,2 mg, 0,706
mmol) und Natriumtriacetoxyborhydrid (179,2 mg, 0,846 mmol) werden
150 mg gelber Feststoff der freien Base erhalten: Massenspektrum
(Elektrospray) (m/e): 388,0 (M+1), 386,1 (M-1).
1H
NMR (400 MHz, DMSO-d6): 7,83 (s, 1H), 6,86-6,75
(m, 3H), 6,69-6,66 (m, 1H), 5,90 (t, 1H, J = 6,0 Hz), 4,51 (d, 2H,
J = 6,0 Hz), 3,83 (m, 2H), 3,33-3,31 (m, 2H), 3,29 (s, 3H), 3,06
(t, 1H, J = 10,4 Hz), 2,81-2,68 (m, 2H), 2,18 (s, 3H), 2,19-2,11
(m, 2H), 1,81-1,75 (m, 1H), 1,54-1,48 (m, 1H). Das Di hydrochloridsalz
wird durch die Zugabe von 5 Äquivalenten
Acetylchlorid (49,5 mg, 1,90 mmol) zu der freien Base (147 mg, 0,38
mmol) in Ethanol (5,0 ml) gebildet. Das Lösemittel wird entfernt, der
Rückstand
wird in 15 ml gemischtem Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 170 mg an (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen-2-yl-methanoldihydrochlorid
lyophilisiert. Massenspektrum (Elektrospray) (m/e): C19H25N5O2S × 2 HCl,
berechnete Masse (M+1-2 HCl) 388,1807, gefunden: 388,1794.
-
Beispiel 333
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen-2-carbonsäreethy-ester-dihydrochlorid
-
Mittels
des Verfahrens von Beispiel 322 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen-2-carbonsäureethylester
(300 mg, 0,72 mmol), Formaldehyd (37% wässrig, G/G) (72,9 mg, 0,90
mmol) und Natriumtriacetoxyborhydrid (228,9 mg, 1,08 mmol) werden
280 mg (Ausbeute 90%) an (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen-2-carbonsäure-ethy-ester
erhalten: Massenspektrum (Elektrospray) (m/e): 430,0 (M+1), 428,1 (M-1).
1H NMR (400 MHz, DMSO-d6):
8,50 (s, 1H), 6,91-6,80 (m, 3H), 6,69-6,67 (m, 1H), 4,30 (q, 2H,
J = 6,8 Hz), 3,83-3,81 (m, 2H), 3,32-3,30 (m, 2H), 3,13 (s, 3H),
3,12-3,06 (m, 1H), 2,81-2,70 (m, 2H), 2,22-2,13 (m, 5H), 1,83-1,75 (m, 1H), 1,53-1,46
(m, 1H), 1,27 (t, 3H, J = 7,2 Hz). Das Dihydrochlorid wird durch
die Zugabe von 5 Äquivalenten
an Actylchlorid (256,2 mg, 3,26 mmol) zu der freien Base (280 mg,
0,65 mmol) in Ethanol gebildet. Die Niederschläge werden durch Vakuumfiltration
unter Bildung von 300 mg eines gelben Feststoffs der Titelverbindung
gesammelt: Massenspektrum (Elektrospray) (m/e): C21H27N5O3S × 2 HCl,
berechnete Masse (M+1): 430,1913, gefunden: 430,1890.
-
Beispiel 334
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 272 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-4H-3-thia-1,4,9-triazabenzo[f]azulen
und nach 6,75 Stunden bei Umgebungstemperatur und unter Verwendung
eines gesättigten
wässrigen
Natriumchloridwaschschritts der organischen Bestandteile nach den gesättigten
wässrigen
Natriumbicarbonatwaschschritten wird die Titelverbindung erhalten.
Massenspektrum (APCI, m/e): 426 (M+1).
NMR (1H,
300 MHz, DMSO-d6): δ 8,47 (s, 1H), 6,k97-6,82 (m,
3H), 6,71 8m, 1H), 3,79 (br.m, 2H), 3,36-3,24 (m, 2H), 3,15 (s, 3H), 3,09 (m,
1H), 2,85-2,69 (m, 2H), 2,28-2,08 (m, 5H), 1,65 (m, 2H).
-
Beispiel 355
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur,
Rekonstitution des Produkts in entionisiertem Wasser : Aceton (1:1)
und Lyophilisierung wird die Titelverbindung erhalten: Massenspektrum
(APCI, m/e): 426 (M+1), exakte Massenspektrum (ES+, m/e, C19H22F3N5OS × 2
HCl): berechnet 426,1575 (M+1-2HCL),
gefunden 426,1565.
-
Beispiel 336
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 272 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und nach 24 Stunden bei Umgebungstemperatur und Reinigung durch
Blitzchromatographie unter Elution mit einem Gradienten aus Lösungen aus
2 M Ammoniak in Methanol in Dichlormethan (2%-6%) wird die Titelverbindung
erhalten. Massenspektrum (APCI, m/e): 408 (M+1).
NMR (1H, 300 MHz, DMSO-d6): δ 8,29 (s,
1H), 7,13 (t, 1H, 2J(H,F) =
54,3 Hz), 6,97-6,79 (m, 3H), 6,70 (m, 1H), 3,80 (br. m, 2H), 3,40-3,22
(m, 1H), 3,22-3,02 (m, 5H), 2,77 (m, 2H), 2,27-2,08 (m, 5H), 1,65
(m, 2H).
-
Beispiel 337
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)-4-methyl-piperazin-1-yl]-2-difluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten. Massenspektrum (APCI, m/e): 408 (M+1),
exaktes Massenspektrum (ES+, m/e, C19H23F2N5OS × 2 HCl):
berechnet 408,1670 (M+1-2HCl), gefunden 408,1652.
-
Beispiel 338
-
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-(3,3,3-trifluorpropyl)-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 322 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-(3,3,3-trifluorpropyl)-4H-3-thia-1,4,9-triazabenzo[f]azulen
(385 mg, 0,876 mmol), Formaldehyd (37% wässrige, G/G) 88,8 mg, 1,09
mmol) und Natriumtriacetoxyborhydrid (278,0 mg, 1,31 mmol) werden
203 g Schaum der freien Base erhalten.
NMR (300 MHz, CDCl3): δ 7,05-6,84
(m, 3H), 6,62-6,59 (m, 1H), 5,24 (s, 1H), 4,02 (br, m, 2H), 3,44
(t, 2H, J = 6,9 Hz), 3,31 (s, 3H), 3,26-3,03 (m, 1H), 2,97-2,81
(m, 4H), 2,65-2,37 (m, 4H), 2,34 (s, 3H), 1,97-1,92 (m, 1H), 1,73-1,66
(m, 1H). Das Dihydrochloridsalz wird durch die Zugabe von 5 Äquivalenten
Acetylchlorid (173 mg, 2,2 mmol) zu der freien Base (200 mg, 0,44
mmol) in Ethanol (10,0 ml) gebildet. Das Lösemittel wird entfernt, der
Rückstand
wird in 15 ml gemischtem Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 224 mg der Titelverbindung lyophilisiert.
Massenspektrum (Elektrospray) (m/e): C21H2F3N5OS × 2 HCl,
berechnet Masse (M+1-2HCl): 454,1888, gefunden: 454,1871.
-
Beispiel 339
-
(S)-10-[4-Cyclopropyl-3-(2-methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
(S)-10-[3-(2-Methoxyethyl)silanpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,100 g, 0,259 mmol) wird mit kommerziellem (1-Ethoxycyclopropoxy)trimethylsilan
(261 μl,
1,3 mmol), Natriumcyanoborhydrid (0,065 g, 1,04 mmol) und Essigsäure (0,148
ml, 2,59 mmol) in Methanol (3 ml) vereinigt und am Rückfluss
für 2 Stunden
erhitzt. Das Gemisch wird eingedampft und dann wird der Rückstand
mit gesättigtem wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
des rohen Produkts konzentriert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt die freie
Base der Titelverbindung (0,045 g, 41%) als gelbes Öl. Massenspektrum
(APCI): m/z = 426,2 (M+1). das reine Produkt wird als entsprechendes
Dihydrochlorid auf die in Beispiel 319 beschriebene Weise isoliert.
Exakte Masse, berechnet 426,2328, gefunden: 426,2346.
-
Beispiel 340
-
(S)-10-[4-Ethyl-3-(2-methoxyethyl)piperazin-2-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
(S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,103 g, 0,267 mmol), Acetaldehyd (30 μl, 0,534 mmol) und Natriumtriacetoxyborhydrid
(0,085 g, 0,401 mmoL) werden in Dichlorethan (7 ml) vereinigt und
bei Raumtemperatur für
6 Stunden gerührt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
des rohen Produkts konzentriert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 5% an 2 N Ammonaik-Methanol in Dichlormethan ergibt die freie
Base der Titelverbindung (0,79 g, 71%) als gelben Schaum. Massenspekt rum
(APCI): m/z = 414,2 (M+1). Das reine Produkt wird als das entsprechende
Dihydrochlorid auf die in Beispiel 319 beschriebene Weise isoliert.
Exakte Masse, berechnet: 414,2328, gefunden: 414,2326.
-
Beispiel 341
-
(S)-10-[3-(2-Methoxyethyl)-4-propylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Auf
eine ähnlich
Weise zu der in Beispiel 340 beschriebenen, aber unter Verwendung
von Propionaldehyd wird (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,295 g, 0,765 mmol) in die freie Base der Titelverbindung (0,208
g, 64%) umgewandelt. Massenspektrum (APCI): m/z = 428,2 (M+1). Das
reine Produkt wird als das entsprechende Dihydrochlorid auf die
in Beispiel 319 beschriebene Weise isoliert.
-
Beispiel 342
-
(S)-2-[4-(2-Isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)-2-(2-methoxyethyl)piperazin-1-yl]ethanoldihydrochlorid
-
Es
werden (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen (0,0972
g, 0,252 mmol), Kaliumcarbonat (0,348 g, 2,52 mmol), Kaliumiodid
(0,209 g, 1,26 mmol) und 2-Bromethanol (72 μl, 1,08 mmol) in Acetonitril
(5 ml) vereinigt und am Rückfluss
für 7 Stunden
erhitzt. Das Gemisch wird eingedampft. Eine Reinigung durch Silicagelchromatographie
unter Elution mit einem Gradienten ausgehend von Dichlormethan bis
zu 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt die freie
Base der Titelverbindung (0,042 g, 39%) als gelben Schaum. Massenspektrum
(APCI): m/z = 430,2 (M+1). Das reine Produkt wird als das entsprechende
Dihydrochlorid auf eine in Beispiel 319 beschriebene Weise isoliert.
Exakte Masse, berechnet: 430,2277, gefunden: 430,2277.
-
Beispiel 343
-
(S)-10-[3,4-Bis-(2-methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Auf
eine Weise die zu der in Beispiel 340 beschriebenen ähnlich ist
aber unter Verwendung von Methoxyacetyldehyd wird (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,331 g, 0,859 mmo) in die freie Base der Titelverbindung (0,266
g, 70%) umgewandelt. Massenspektrum (APCI): m/z = 444,2 (M+1). Das
reine Produkt wird als das entsprechende Dihydrochlorid auf eine
in Beispiel 319 beschriebenen Weise isoliert. Exakte Masse, berechnet:
444,2433, gefunden: 444,2414.
-
Beispiel 344
-
(S)-10-[4-Ethyl-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Mittels
des Verfahrens von Beispiel 346 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und Acetaldehyd bei Umgebungstemperatur und Reinigung durch Blitzchromatographie
und Elution mit einem Gradienten aus einer 5% Lösung aus 2 M Ammoniak in Methanol
in Dichlormethan (0-50% über
25 Minuten, 50% für
10 Minuten, 50-100% über
23 Minuten, 100% für
5 Minuten) wird die Titelverbindung erhalten. Massenspektrum (APCI,
m/e): 440 (M+1).
NMR (1H, 300 MHz,
DMSO-d6) δ (ppm): δ 8,47 (s,
1H), 6,96-6,81 (m, 3H), 6,71 (m, 1H), 3,50 (br.m, 3H), 3,35-3,21
(m, 2H), 3,21-3,07 (m, 4H), 2,76-2,51 (m, 3H), 2,37 (m, 2H), 1,67
(m, 2H), 0,96 (t, 3H, J = 6,9 Hz).
-
Beispiel 345
-
(S)-10-[4-Ethyl-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[4-Ethyl-3-(2-methoxyethylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten. Massenspektrum (APCI, m/e): 440 (M+1)
Exaktes Massenspektrum (ES+, m/e, C20H24F3N5OS × 2 HCl):
berechnet 440,1732 (M+1-2HCl), gefunden 440,1716.
-
Beispiel 346
-
(S)-10-[3-(2-Methoxyethyl)-4-propylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Natriumtriacetoxyborhydrid
(0,273 g, 1,29 mmol) und dann Propionaldehyd (0,083 ml, 1,15 mmol)
zu (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,353 g, 0,858 mmol) in wasserfreiem Dichlorethan (15 ml) gegeben
und bei Umgebungstemperatur über
Nacht gerührt.
Gesättigtes
wässriges
Natriumbicarbonat wird zugegeben und die wässrige Phase wird mit Dichlormethan
extrahiert. Die organischen Bestandteile werden mit gesättigtem
wässrigem
Natriumchlorid gewaschen und getrocknet (Natriumsulfat), filtriert
und unter verringertem Druck zu einem Rückstand (0,29 g) konzentriert.
Der Rückstand
wird durch Blitzchromatographie unter Elution mit einem Gradienten
aus einer 3% Lösung
aus 2 M Ammoniak in Methanol in Dichlormethan (0-100%) unter Bildung
der Titelverbindung (0,287 g, 74%) gereinigt. Massenspektrum (APCI,
m/e): 454 (M+1).
NMR (1H, 300 MHz,
DMSO-d5): δ 8,46 (s, 1H), 6,96-6,82 (m,
3H), 6,71 (m, 1H), 3,46 (br.m, 3H), 3,31-3,17 (m, 3H), 3,15 (s, 3H), 2,73 (m,
1H), 2,58-2,44 (m, 2H), 2,31 (m, 2H), 1,67 (m, 2H), 1,40 (m, 2H),
0,83 (t, 3H, J = 7,5 Hz).
-
Beispiel 347
-
(S)-10-[3-(2-Methoxyethyl)-4-propylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)-4-propylpiperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten. Massenspektrum (APCI, m/e): 454 (M+1)
Exaktes Massenspektrum (ES+, m/e, C21H26F3N5OS × 2 HCl):
berechnet 454,1888 (M+1-2HCl), gefunden 454,1893.
-
Beispiel 348
-
(S)-2-Fluor-1-[2-(2-methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-1-yl]ethanon
-
Eine
Lösung
aus Fluoracetylchlorid (0,133 g, 1,38 mmol) in wasserfreiem Dichlormethan
(1-2 ml) wird tropfenweise zu einer 0°C Lösung aus (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,283 g, 0,688 mmol) und Diisopropylethylamin (0,240 ml, 1,38 mmol)
in wasserfreiem Dichlormethan gegeben und bei 0°C für 3 Stunden gerührt. Die
Reaktion wird unter verringertem Druck unter Bildung eines Öls konzentriert.
Das Öl
wird durch Blitzchromatographie unter Flution mit einem Gradienten
aus einer Lösung
aus 2% an 2 M Ammoniak in Methanol in Dichlormethan (0-100°C in Dichlormethan über 30 Minuten,
dann 100% für
28 Minuten) unter Bildung der Titelverbindung konzentriert. 0,18 g
(56%). Massenspektrum (APCI+, m/e): 472 (M+1).
-
Beispiel 349
-
(S)-10-[4-(2-Fluorethyl)-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Eine
Lösung
aus Bortetrahydrofurankomplex (1,0 M in Tetrahydrofuran, 3,8 ml,
3,8 mmol) wird zu einer 0°C
Lösung
aus (S)-2-Fluor-1-[2-(2-methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-1-yl]ethanon
(0,18 g, 0,38 mmol) in wasserfreiem Tetrahydrofuran (3 ml) gegeben
und bei 0°C
für 3 Stunden
gerührt.
Die Reaktion wird in einem Eis/Wasserbad langsam mit Methanol gestoppt.
Die Reaktion wird unter verringertem Druck konzentriert und in wasserfreiem
Dichlorethan (6 ml) rekonstituiert. Ethylendiamin (0,23 ml, 3,4
mmol) wird zugegeben und am Rückfluss
für 45
Minuten erhitzt. Die Reaktion wird auf Umgebungstemperatur gekühlt und
mit entionisiertem Wasser und Dichlormethan (20 ml jeweils) verdünnt. Die organische
Phase wird abgetrennt und die wässrige
Phase wird mit Dichlormethan (2 ×) extrahiert. Die vereinigten
organischen Phasen werden mit entionisiertem Wasser gewaschen und
dann wird eine gesättigte
Lösung
aus Natriumchlorid zugegeben und dann getrocknet (Natriumsulfat),
filtriert und unter verringertem Druck zu einem orangen Öl (0,19
g) konzentriert. Das Öl
wird durch Blitzchromatographie unter Elution mit einem Gradienten
aus einer Lösung
aus Ethylacetat : Hexan (1:1, 0-50% in Hexan über 45 Minuten, dann 100% für 13 Minuten)
unter Bildung der Titelverbindung gereinigt. 0,050 g (29%). Massenspektrum
(APCI+, m/e) 458 (M+1).
-
Beispiel 350
-
(S)-10-[4-(2-Fluorethyl)-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[4-(2-Fluorethyl)-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,050 g, 0,11 mmol) und einer Lösung
aus Acetylchlorid (0,039 ml, 0,55 mmol) in absolutem Ethanol bei
Umgebungstemperatur wird die Titelverbindung erhalten. Massenspektrum
(APCI, m/e): 458 (M+1-2HCl) Exaktes Massenspektrum (ES+, m/e, C20H23F4N5OS × 2
HCl): berechnet 458,1638 (M+1-2HCl), gefunden 458,1627.
-
Beispiel 351
-
(S)-10-[4-(3-Fluorpropyl)-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
1-Brom-3-fluorpropan
(0,14 ml, 1,52 mmol) wird zu einer Aufschlämmung aus (S)-10-[4-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,52 g, 1,26 mmol), pulverisiertem Kaliumcarbonat (0,87 g, 6,32
mmol) und Natriumiodid (0,95 g, 6,32 mmol) in absolutem Ethanol
(7,8 ml) gegeben und am Rückfluss
erhitzt. Nach einer Übernachtperiode,
wird gekühlt,
gesättiges
wässriges
Natriumchlorid und Ethylacetat werden zugegeben und entionisiertes
Wasser wird zum Lösen
des ausgefallenen Salzes das sich während der Extraktion bildet,
zugegeben. Die wässrige
Phase wird mit Ethylacetat (2 ×)
extrahiert und getrocknet (Natriumsulfat), filtriert und die organischen
Bestandteile werden unter verringertem Druck zu eiem Öl (0,564
g) konzentriert. Das Öl
wird durch Blitzchromatographie unter Elution mit einem Gradienten aus
einer 2% Lösung
aus 2 M Ammoniak in Methanol in Dichlormethan (0-100°C) unter
Bildung der Titelverbindung konzentriert. Massenspektrum (APCI,
m/e): 472 (M+1).
NMR (1H, 300 MHz,
DMSO-d6): δ 8,47 (s, 1H), 6,96-6,82 (m,
3H), 6,71 (m, 1H), 4,55 (td, 2H, 2H(H,F) = 48 Hz, 3J( H ,H ) = 5,7 Hz), 3,47 (br.m, 3H), 3,31-3,18 (m,
3H), 3,15 (s, 3H), 2,70 (m, 2H), 2,55 (br.m, 1H), 2,38 (m, 2H), 1,86
(m, 2H), 1,86-1,67 (m, 3H), 1,61 (m, 1H).
-
Beispiel 352
-
(S)-10-[4-(3-Fluorpropyl)-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-10-[4-(3-Fluorpropyl)-3-(2-methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten.
Massenspektrum (APCI, m/e): 458
(M+1-2HCl) Exaktes Massenspektrum (APCI, m/e): 472 (M+1), exaktes Massenspektrum
(ES+, m/e, C21H25F4N5OS × 2 HCl):
berechnet 472,1794 (M+1-2HCl), gefunden 472,1771.
-
Beispiel 353
-
(S)-2-[2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-1-yl]ethanol
-
2-Iodethanol
(0,44 ml, 2,55 mmol) wird zu einer Aufschlämmung aus (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,75 g, 1,82 mmol) und pulverisiertem Kaliumcarbonat (1,26 g, 9,11
mmol) in absolutem Ethanol (7,5 ml) gegeben und am Rückfluss
erhitzt. Nach einer Übernachtperiode,
wird gekühlt,
gesättigtes
wässriges
Natriumchlorid und Ethylacetat werden zugegeben und entionisiertes
Wasser wird zum Lösen
des ausgefallenen Salzes das sich während der Extraktion bildet,
zugegeben. Die wässrige
Phase wird mit Ethylacetat (2 ×)
extrahiert und getrocknet (Natriumsulfat), filtriert und die organischen
Bestandteile werden unter verringertem Druck zu einem Öl (0,564
g) konzentriert. Absoluter Ethanol (8 ml), pulverisierte Kaliumcarbonat
(0,62 g, 4,50 mmol) gefolgt von 2-Iodethanol (0,098 ml, 1,26 mmol) werden
zugegeben und für
15,5 Stunden am Rückfluss
gerührt.
Zusätzliches
2-Iod-ethanol (0,010 ml, 0,13 mmol) wird zugegeben und am Rückfluss
gerührt.
Nach 4 Stunden wird auf Umgebungstemperatur gekühlt und entionisiertes Wasser
und Ethylacetat werden zugegeben. Die abgetrennte wässrige Phase
wird mit Ethylacetat extrahiert und vereinigt, getrocknet (Natriumsulfat),
filtriert und die organischen Bestandteile werden unter verringertem
Druck zu einem Rückstand
(0,86 g) konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Elution mit einem Gradienten
der Lösungen
von 2 M Ammoniak in Methanol: Ethylacetat (1%-5% an 2 M Ammoniak
in Methanol in Ethylacetat) unter Bildung der Titelverbindung (0,213
g, 26%) gereinigt. Massenspektrum (APCI, m/e): 456 (M+1).
NMR
(1H, 300 MHz, DMSO-d6): δ 8,45 (s,
1H), 6,96-6,81 (m, 3H), 6,71 (m, 1H), 4,38 (t, 1H, J = 5,4 Hz), 3,54-3,06
(m, 11H), 2,71 (m, 2H), 2,55 (m, 1H), 2,41 (m, 2H), 1,68 (m, 2H).
-
Beispiel 354
-
(S)-2-[2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl]piperazin-1-yl]ethanoldihydrochlorid
-
Eine
Lösung
aus Acetylchlorid (0,194 ml, 2,47 mmol) wird in absolutem Ethanol
wird zu einer Lösung aus
der freien Base (S)-2-[2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-1-yl]ethanol
(0,213 g, 0,468 mmol) in absolutem Ethanol gegeben und gerührt. Der
ausgefallene Feststoff wird durch Unterdruckfiltration isoliert,
mit Diethylether gewaschen und der Feststoff wird bei Umgebungstemperatur
unter verringertem Druck über
Nacht unter Bildung der Titelverbindung (0,197 g, 79,8%) getrocknet.
Massenspektrum (APCI, m/e): 456 (M+1) Exaktes Massenspektrum (ES+,
m/e, C20H24F3N5O2 × 2 HCl):
berechnet 456,1681 (M+1-2 HCl), gefunden 456,1668.
-
Beispiel 355
-
(S)-3-[2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-1-yl]propanol-1-ol
-
Mittels
des Verfahrens von Beispiel 351 und unter Verwendung von (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
und 3-Brompropan-1-ol unter 15 Stunden am Rückfluss und nach der Zugabe
von Alkohol und zwei Reinigungsschritten durch Blitzchromatographie
wird die Titelverbindung erhalten: Der Erste unter Elution mit einem
Gradienten einer 3% Lösung
aus Methanol in Ethylacetat (0-100%) und dann mit einem Gradienten
aus Methanol : Ethylacetat (3%-5% Methanol in Ethylacetat) und der
Zweite unter Elution mit einem Gradienten aus Lösungen aus 2 M Ammoniak in
Methanol in Ethylacetat (4%-5% über
40 Minuten). Massenspektrum (APCI, m/e): 470 (M+1).
NMR (1H, 300 MHz, DMSO-d6) δ 8,47 (s,
1H), 6,96-6,81 (m, 3H), 6,70 (m, 1H), 4,42 (m, 1H), 3,54-3,37 (m,
6H), 3,37-3,18 (m, 2H), 3,15 (s, 2H), 2,68 (m, 2H), 2,58-2,34 (m,
1H), 2,35 (m, 2H), 1,68 (m, 2H), 1,55 (m, 2H).
-
Beispiel 356
-
(S)-3-[2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl]piperazin-1-yl]propan-1-ol-dihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-3-[2-(2-Trifluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)piperazin-1-yl]propan-1-ol
und eine Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten. Massenspektrum (APCI, m/e): 470 (M+1).
Exaktes Massenspektrum (ES+, m/e C21H26F3N5O2S × 2
HCl) berechnet 470,1837 (M+1-2HCl), gefunden 470,1833.
-
Beispiel 357
-
(S)-(2-[2-(2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)ethoxy]ethanol
-
(S)-2-(2-Chlorethoxy)ethanol
(0,267 ml, 2,53 mmol) wird zu einer Aufschlämmung aus (S)-10-[3-(2-Methoxyethyl)piperazin-1-yl]-2-trifluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen
(0,52 g, 1,26 mmol) gegeben, pulverisiertes Kaliumcarbonat (0,87
g, 6,32 mmol) und Natriumiodid (0,95 g, 6,32 mol) in absolutem Ethanol
(5,2 ml) werden zugegeben und am Rückfluss erhitzt. Nach 22 Stunden
wird gekühlt
und pulverisiertes Kaliumcarbonat (0,09 g, 0,65 mmol), Natriumiodid
(0,09 g, 0,60 mmol) und (S)-2-(2-Chlorethoxy)ethanol
(0,100 ml, 0,938 mol) werden zugegeben. Die Feststoffe werden mit
absolutem Ethanol gewaschen und am Rückfluss erhitzt. Nach 4 Stunden
wird (S)-2-(2-Chlorethoxy)ethanol (0,307 ml, 2,88 mmol) zugegeben und
am Rückfluss
erhitzt. Nach 17,5 Stunden wird gekühlt und entionisiertes Wasser
und Ethylacetat werden zugegeben. Nach der Trennung der Phasen wird
die wässrige
Phase mit Ethylacetat (2 ×)
extrahiert und vereinigt, getrocknet (Natriumsuflat), filtriert
und die organischen Bestandteile werden unter verringertem Druck zu
einem Feststoff (1,54 g) konzentriert. Der Feststoff wird durch
Blitzchromatographie unter Elution mit einem Gradienten aus Lösungen aus
2 M Ammoniak in Methanol: Ethylacetat (1% bis 5% an 2 M Ammoniak
in Methanol in Ethylacetat über
61 Minuten) und dann mit einer 5% Lösung aus 2 M Ammoniak in Methanol
in Ethylacetat für
30 Minuten unter Bildung der Titelverbindung (0,146 g, 23%) gereinigt.
Massenspektrum (APCI, m/e) 500 (M+1)
NMR (1H,
300 MHz, DMSO-d6) δ 8,47 (s, 1H), 6,96-6,81 (m,
3H), 6,71 (m, 1H), 4,60 (t, 1H, J = 5,1 Hz,), 3,56-3,10 (m, 15H),
2,76 (m, 2H), 2,61-2,41 (m, 3H), 1,68 (m, 2H).
-
Beispiel 358
-
(S)-(2-[2-(2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-1-yl)ethoxy]ethanoldihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-(2-[2-(2-(2-Methoxyethyl)-4-(2-trifluormethyl-4H-3-thia-1,4,9-triaza-benzo[f]azulen-10-yl)piperazin-1-yl]ethoxy]ethanol
und einer Lösung
aus Acetylchlorid in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung erhalten. Massenspektrum (APCI, m/e): 500 (M+1).
Exaktes Massenspektrum (ES+, m/e C22H26F3N5O2S × 2
HCl) berechnet 500,1943 (M+1-2HCl), gefunden 500,1943.
-
Beispiel 359
-
10-[3(S)-(2(S)-Methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen 10-[3(S)-(2(R)-Methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
-
Auf
eine ähnliche
Weise zu der in Beispiel 311 beschriebenen werden 2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(0,711 g, 2,89 mmol) vereinigt und das Gemisch der Isomere von 2(S)-(2(S)-Methoxypropyl)piperazin
und 2(S)-(2(R)-Methoxypropyl)piperazin (0,456 g, 2,89 mmol) werden
unter Bildung des Gemisches der Isomere erhalten: 10-[3-(S)-(2-(S)-Methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
und 10-[3-(S)-(2-(R)-Methoxypropyl)piperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen
(0,54 g, 50%) werden als brauner amorpher Feststoff (Gemisch der
Isomere) erhalten. Massenspektrum (APCI): m/z = 371,2 (m+1).
-
Beispiel 360
-
10-[3-(S)-(2-(S)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid 10-[3-(S)-(2-(R)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9- diazabenzo[f]azulendihydrochlorid
-
Das
Material von Beispiel 359 (0,235 g, 0,634 mmol), Formaldehyd (67 μl, 0,825
mmol, 37%) und Natriumtriacetoxyborhydrid (0,202 g, 0,951 mmol)
in Dichloethan (15 ml) werden vereinigt und bei Raumtemperatur für 6 Stunden
gerührt.
Das Gemisch wird mit gesättigtem
wässrigem
Natriumbicarbonat verdünnt
und dreimal mit Dichlormethan extrahiert. Die organischen Phasen
werden vereinigt, über
Natriumsulfat getrocknet und unter verringertem Druck unter Bildung
des rohen Produkts konzentriert. Eine Reinigung durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend mit Dichlormethan
bis zu 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt die freie
Base der Titelverbindung. 10-[3-(S)-(2-(S)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
und 10-[3-(S)-(2-(R)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-methyl-4H-3-thia-4,9-diazabenzo[f]azulendihydrochlorid
(0,188 g, 77%) werden als gelber Schaum (Gemisch der Isomere) erhalten.
Massenspektrum (ASPCI): m/z = 385,2 8M+1) Das reine Produkt wird
als das entsprechende Dihydrochlorid auf die in Beispiel 319 beschriebene
Weise isoliert: exakte Masse, berechnet: 385,2062, gefunden: 385,2085.
-
Beispiel 361
-
10-[3-(S)-(2(S)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen 10-[3-(S)-(2(R)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
-
Aus
eine Weise, die zu der in Beispiel 359 beschriebenen ähnlich ist,
werden 2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(0,865 g, 3,14 mmol) und das Isomerengemisch aus 2-(S)-(2(S)-Methoxypropyl)piperazin
und 2-(S)-(2(R)-Methoxypropyl)piperazin (0,487 g, 3,14 mmol) unter
Bildung der Titelverbindung als ein Gemisch der Isomere hergestellt.
Massenspektrum (APCI): m/z = 400,2 (M+1).
-
Beispiel 362
-
10-[3-(S)-(2(S)-Methoxypropyl)piperazin-l-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
-
Beispiel 363
-
10-[3-(S)-(2(R)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Zu
einer Probe aus 225 mg an Diastereomerisch gemischtem 10-[3-(S)-(2(S)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulenhydrochlorid
und 10-[3-(S)-(2(R)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
werden chirale Chromatographievefahren Chiralpak AD 4,6 × 150 mm
Eluent: 0,2% DMEA, 15% MeOH, 15% an 3 A Alkohol bei Heptanfluss: 0,6
ml/min UV: 280 nm gegeben, um das Isomer 1 (66 mg) und das Isomer
2 (54 mg) als gelben Schaum zu erhalten. Beide Isomere (unbekannte
Stereochemie) werden als entsprechende Dihydrochlorid isoliert:
Isomer 1 RT 7,75 min. Exakte Masse berechnet: 400,2171, gefunden:
400,2159. Isomer 2 RT 10,20 min, exakte Masse berechnet: 400,2171,
gefunden: 400,2159.
-
Beispiel 364
-
10-[3-(S)-(2(S)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid 10-[3-(S)-(2(R)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Auf
eine Weise, die zu Beispiel 360 ähnlich
ist und unter Verwendung des Gemisches der Diastereoisomere aus
Beispiel 361 aus 10-[3-(S)-(2(S)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
und 10-[3-(S)-(2(R)-Methoxypropyl)piperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
wird die freie Base aus 10-[3-(S)-(2(S)-Methoxypropyl)-4-methylpropyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
(0,211 g, 0,510 mmol, 35%) als gelber Schaum (Gemisch des Diastereomere)
erhalten. Massenspektrum (APCI): m/z = 414,2 (M+1). Das reine Produkt
wird als das entsprechende Dihydrochlorid auf die in Beispiel 319
beschriebene Weise isoliert.
-
Beispiel 365
-
10-[3-(S)-(2(S)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
-
Beispiel 366
-
10-[3-(S)-(2(R)-Methoxypropyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
-
Die
individuellen Isomere aus dem Diastereomerengemisch von oben (203
mg) werden mittels einer Chiralpak AD 8 × 250 mm Säule getrennt. Es wird mit einem
85:15 Gemisch aus Heptan : Ethanol das 0,2% Dimethylethylamin enhält, eluiert.
Eine weitere Reinigung jedes Isomers durch Blitzchromatographie
unter Elution mit einem Stufengradienten ausgehend von Dichlormethan
bis zu 5% an 2 N Ammoniak-Methanol in Dichlormethan ergibt das Isomer
1 (0,058 g) und das Isomer 2 (0,052 g). Die reinen Produkte werden
als entsprechende Dihydrochloride auf die in Beispiel 319 beschriebene
Weise isoliert. Isomer 1: Exakte Masse, berechnet: 414,2328, gefunden:
414,2321. Isomer 2: Exakte Masse, berechnet: 414,2328, gefunden:
414,2314.
-
Beispiel 367
-
(S)-1-[4-(2-Isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)piperazin-2-yl]-2-methylpropan-2-ol-dihydrochlorid
-
Auf ähnliche
Weise zu der in Beispiel 359 beschriebenen werden 2-Isopropyl-4,9-dihydro-3-thia-1,4,9-triaza-benzo[f]azulen-10-thion
(0,686 g, 2,49 mmol) und (S)-2-Methyl-1-piperazin-2-yl-propan-2-ol
(0,394 g, 2,49 mmol) unter Bildung der Titelverbindung vereinigt:
Massenspektrum (APCI): m/z = 400,2 (M+1).
-
Beispiel 368
-
(S)-1-[4-(2-Isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)-1-methylpiperazin-2-yl]-2-methylpropan-2-oldihydrochlorid
-
Auf ähnliche
Weise zu der in Beispiel 360 beschriebenen und unter Verwendung
von (S)-1-[4-(2-Isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen-10-yl)-piperazin-2-yl]-2-methylpropan-2-ol
wird die freie Base der Titelverbindung (0,122 g, 0,295 mmol, 16%)
als gelbes Öl
erhalten. Massenspektrum (APCI): m/z = 414,2 (M+1). Das reine Produkt
wird als das entsprechende Dihydrochlorid auf die in Beispiel 319
beschriebene Weise isoliert. Exakte Masse, berechnet: 414,2328,
gefunden: 414,2313.
-
Beispiel 369
-
(S)-2-Methyl-1-[4-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2-yl]propan-2-ol
-
Auf ähnliche
Weise zu der in Beispiel 359 beschriebenen werden 2-Methyl-4,9-dihyro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(0,574 g, 2,33 mmol) und (S)-2-Methyl-1-piperazin-2-ylpropan-2-ol
(0,369 g, 2,33 mol) unter Bildung der Titelverbindung vereinigt:
Massenspektrum (APCI): m/z = 371,2 (M+1).
-
Beispiel 370
-
(S)-2-Methyl-1-[1-methyl-4-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2-yl]propan-2-oldihydrochlorid
-
Auf ähnliche
Weise zu der in Beispiel 360 beschriebenen und unter Verwendung
von (S)-2-Methyl-1-[4-(2-methyl-4H-3-thia-4,9-diaza-benzo[f]azulen-10-yl)piperazin-2-yl]propan-2-ol
wird die freie Base der Titelverbindung (0,080 g, 0,208 mmol, 16%)
als gelbes Öl
erhalten. Massenspektrum (APCI): m/z = 385,2 (M+1). Das reine Produkt
wird als das entsprechende Dihydrochlorid auf die in Beispiel 319
beschriebenen Weise isoliert. Exakte Masse, berechnet: 385,2062,
gefunden: 385,2053.
-
Beispiel 371
-
(S)-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(S)-oldihydrochlorid
und
(S)-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(R)-oldihydrochlorid
-
Auf ähnliche
Weise zu der für
(S)-10-[3-(2-Methoxyethyl)piperazin-2-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulen
beschriebenen, wird das diastereomere Gemisch aus 1-Piperazin-2(S)-yl-propan-2(S)ol und
1-Piperazin-2(s)-yl-propyn-2(R)-ol (477 mg, 3,3 mmol) mit 2-Methyl-4,9-dihydro-3-thia-4,9-diazabenzo[f]azulen-10-thion
(814 mg, 3,3 mmol) unter Bildung eines diastereomeren Gemisches
aus 1-[4-(2-Methyl-4H-3-thia-49-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(S)-oldihydrochlorid
und 1-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(R)-oldihydrochlorid
als braunes Öl
erhalten.
-
Beispiel 372
-
1-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(S)-oldihydrochlorid
-
-
Beispiel 373
-
1-[4-(2-Methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(R)-oldihydrochlorid
-
Mittels
eines Säulenchromatographieverfahrens
aus Beispiel 365 und Beispiel 366 werden die Gemisch des Diastereoisomere
aus Beispiel 371 mittels Säulenchromatographie
mittels eines Stufengradienten aus Dichlormethan bis zu 5% an 2
N Ammoniak-Methanol in Dichlormethan unter Bildung des Isomers 1
(184 mg, 16%) und des Isomers 2 (163 mg, 14%) als gelber Schaum
erhalten. Beide Isomere werden als die entsprechenden Isomere isoliert:
Isomer 1: Exakte Masse, berechnet: 357,1749, gefunden: 357,1729.
Isomer 2: Exakte Masse, berechnet: 357,1749, gefunden: 357,1743.
-
Beispiel 374
-
1-[1-Methyl-4H-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(S)-oldihydrochlorid (Isomer
1, diastereomerenrein)
-
Auf
eine Weise, die zu der in (S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
beschriebenen ähnlich
ist, wird das Isomer 1 von Beispiel 372 und Beispiel 373 (132 mg,
0,37 mmol) in das diastereomeren reine 1-[1-Methyl-4H-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(S)-ol
(Isomer 1, 75 mg, 56%) umgewandelt. Es wird als das Titeldihydrochlorid
isoliert: Isomer 1: Exakte Masse, berechnet: 371,1906, gefunden: 371,1916.
-
Beispiel 375
-
1-[1-Methyl-4-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2-oldihydrochlorid (Isomer
2, diasteromerenrein)
-
Auf
eine Weise, die zu der für
(S)-10-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-2-isopropyl-4H-3-thia-1,4,9-triazabenzo[f]azulendihydrochlorid
beschriebenen ähnlich
ist, wird das Isomer 2 von Beispiel 372 und Beispiel 373 (111 mg,
0,31 mmol) in das diastereomerenreine 1-[1-Methyl-4H-(2-methyl-4H-3-thia-4,9-diazabenzo[f]azulen-10-yl)piperazin-2(S)-yl]propan-2(S)-ol
(Isomer 2, 85 mg, 74%) umgewandelt. Es wird als das Titeldihydrochlorid
isoliert: Isomer 2: Exakte Masse, berechnet: 371,1906, gefunden: 371,1924.
-
Beispiel 380
-
(S)-5-[3-(2-Methoxyethyl)piperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diazadibenzo[a,f]azulen
-
8-Trifluormethyl-11H-12-thia-6,11-dibenzo[a,f]azulen-5-ylaminhydrochlorid
(0,75 g, 2,0 mmol) wird zu einer Lösung aus (S)-2-(2-Methoxyethyl)piperazin
(0,58 g, 4,1 mmol) in Dimethylsulfoxid : Toluol (1:8, 0,2 M) gegeben.
Diisopropylethylamin (1 Äquivalent)
wird zugegeben, es wird auf 110°C
erhitzt und gerührt.
Nach 45 Stunden wird auf Umgebungstemperatur gekühlt und mit Ethylacetat und
0,1 N NaOH verdünnt.
Die wässrige Phase
wird getrennt und mit Ethylacetat (3 ×) extrahiert. Alle organischen
Bestandteile werden mit einer gesättigten Lösung aus Natriumchlorid gewaschen
und dann getrocknet (Natriumsulfat), filtriert und unter verringertem
Druck konzentriert. Das Öl
wird durch Blitzchromatographie unter Elution mit einem Gradienten
aus einer 3% Lösung
aus 2 M Ammoniak in Methanol in Dichlormethan (0-100% in Dichlormethan)
eluiert. Das Material wird in Ethylacetat rekonstituiert und mit
einer gesättigten
Lösung
aus Natriumchlorid (2 ×)
unter Entfernung des restlichen Dimethylsufoxids gewaschen. Die
vereinigten wässrigen
Phasen werden mit Ethylacetat rückextrahiert.
Die organischen Phasen werden getrocknet (Natriumsulfat), filtriert
und dann unter verringertem Druck unter Bildung der Titelverbindung
(0,331 g) konzentriert. Massenspektrum (APCI+, m/e): 461 (M+1).
-
Beispiel 381
-
(S)-5-[3-(2-Methoxyethyl)piperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
-
Eine
Lösung
aus Acetylchlorid (0,078 ml, 1,1 mmol) in absolutem Ethanol wird
zu einer Lösung
aus (S)-5-[3-(2-Methoxyethyl)piperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,10 g, 0,22 mmol) in absolutem Ethanol gegeben und der ausgefallene
Feststoff wird durch Unterdruckfiltration isoliert. Der Feststoff
wird unter verringertem Druck unter Bildung der Titelverbindung
(0,095 g) getrocknet. Exaktes Massenspektrum (ES+, m/z, C23H23H3N4OS × 2
HCl): berechnet 461,1623 (M+1-2HCl),
gefunden 461,1613.
-
Beispiel 382
-
(S)-5-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Natriumtriacetoxyborhydrid
(0,154 g, 0,727 mmol) und wässriges
Formaldehyd (37% G/G, 0,047 ml, 0,62 mmol) werden zu einer Lösung aus
(S)-5-[3-(2-Methoxyethyl)piperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,223 g, 0,484 mmol) in Methanol : Dichlorethan (1:1, 8 ml) gegeben
und bei Umgebungstemperatur gerührt.
Nach einer Übernachtperiode
werden weitere 1,3 Äquivalente des
wässrigen
Formaldehyds (0,047 ml, 0,63 mmol) und 1,5 Äquivalente Natriumtriacetoxyborhydrid
(0,154 g, 0,727 mmol) zugegeben und mit Methanol gewaschen. Es wird
wenige Stunden bei Umgebungstemperatur gerührt und dann wird die Reaktion
mit einer gesättigten
Lösung
aus Natriumbicarbonat und Dichlormethan verdünnt. Die wässrige Phase wird abgetrennt
und mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt und mit einer gesättigten
Lösung
aus Natriumchlorid gewaschen. Es wird getrocknet (Natriumsulfat),
filtriert und die organischen Bestandteile werden unter verringertem
Druck zu einem Rückstand
konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Elution mit einem Gradienten
aus einer Lösung aus
2 M Ammoniak in Methanol und Dichlormethan in Dichlormethan unter
Bildung der Titelverbindung (0,225 g) gereinigt. Massenspektrum
(APCI+, m/e): 475 (M+1).
-
Beispiel 383
-
(S)-5-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-5-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-8-trifluormethyl-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,225 g, 0,474 mg) und einer Lösung
aus Acetylchlorid (0,169 ml, 2,37 mmol) in absolutem Ethanol bei
Umgebungstemperatur wird die Titelverbindung (0,238 g) erhalten.
Massenspektrum (APCI+, m/e): 475 (M+1-2HCl). Exaktes Massenspektrum (ES+,
m/e, C24H25F3N4OS × 2 HCl):
berechnet: 475,1779 (M+1-2 HCl), gefunden 475,1765.
-
Beispiel 384
-
(S)-5-[3-(2-Methoxyethyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Die
Herstellung erfolgt gemäß dem Verfahren
von Beispiel 380 unter Verwendung von 11H-12-Thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrochlorid
(0,40 g, 1,3 mmol) und (S)-2-(2-Methoxyethyl)piperazin (0,38 g,
2,7 mmmol) unter Rühren
bei 115°C
für 24
Stunden, Waschen der getrennten organischen Phase mit 0,1 N NaOH
(2 ×)
und Extraktion der vereinigten wässrigen
Phasen mit Ethylacetat. Die organischen Phasen werden vereinigt
und mit einer gesättigten
Lösung
aus Natriumchlorid (3 ×)
gewaschen und dann getrocknet (Natriumsulfat), filtriert und unter
verringertem Druck zu einem Rückstand
konzentriert. Der Rückstand
wird durch Blitzchromatographie unter Elution mit einem Gradienten
aus einer Lösung
aus 5% an 2 M Ammoniak in Methanol in Dichlormethan (in Dichlormethan)
unter Bildung der Titelverbindung (0,075 g) gereinigt. Massenspektrum
(APCI+, m/e)= 393 (M+1).
-
Beispiel 385
-
(S)-5-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Gemäß dem Verfahren
von Beispiel 272 und mittels (S)-5-[3-(2-Methoxyethyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,068 g, 0,18 mmol) wird eine teilweise Umwandlung zu der Titelverbindung
nach dem Rühren
bei Umgebungstemperatur für
5,5 Stunden erhalten. Weitere 1,3 Äquivalente an wässrigem
Formaldehyd (0,0171 g, 0,229 mmol) und 1,5 Äquivalente an Natriumtriacetoxyborhydrid
(0,0559 g, 0,264 mmol) mit Dichlormethan werden zugegeben, und es
wird gerührt.
Nach einer Übernachtperiode
wird die Reaktion mit einer gesättigten
wässrigen
Lösung
aus Natriumbicarbonat und Dichlormethan verdünnt und die Phasen werden getrennt.
Die wässrige
Phase wird mit Dichlormethan extrahiert, die organischen Phasen
werden vereinigt und mit einer gesättigten Lösung aus Natriumchlorid gewaschen.
Es wird getrocknet (Natriumsulfat), filtriert und die organischen
Anteile werden unter verringertem Druck zu einem Öl konzentriert.
Das Öl
wird durch Blitzchromatographie unter Elution mit einem Gradienten
aus einer Lösung
aus 5% an 2 M Ammoniak in Methanol in Dichlormethan (25-100% in
Dichlormethan über
43 Minuten) unter Bildung der Titelverbindung (0,043 g) gereinigt.
Massenspektrum (APCI+, m/e): 407 (M+1).
-
Beispiel 386
-
(S)-5-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
-
Mittels
des Verfahrens von Beispiel 273 und unter Verwendung von (S)-5-[3-(2-Methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,039 g, 0,096 mmol) und einer Lösung aus Acetylchlorid (0,0342
ml, 0,480 mol) in absolutem Ethanol bei Umgebungstemperatur wird
die Titelverbindung (0,042 g) erhalten. Massenspektrum (APCI+, m/e):
407 (M+1-2HCl). Exaktes Massenspektrum (ES+, m/e, C23H26F3N4OS × 2 HCl):
berechnet: 407,1906 (M+1-2 HCl), gefunden 407,1899.
-
Beispiel 387
-
(S)-8-Fluor-5-[3-(2-methoxyethyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Die
Herstellung erfolgt mittels des Verfahrens von Beispiel 380 und
unter Verwendung von 8-Fluor-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrochlorid
(0,40 g, 1,3 mmol) und (S)-2-(2-Methoxyethyl)piperazin
(0,36 g, 2,5 mmol), Rühren
bei 115°C
für 24
Stunden, Waschen der getrennten organischen Phase mit 0,1 N NaOH
(2 ×)
und Extraktion der vereinigten wässrigen
Phasen mit Ethylacetat. Die organischen Phasen werden vereinigt
und mit einer gesättigten
Lösung
aus Natriumchlorid (3 ×)
gewaschen und dann getrocknet (Natriumsulfat), filtriert und unter
verringertem Druck zu einem Rückstand
konzentriert. Der Rückstand wird
durch Blitzchromatographie unter Elution mit einem Gradienten aus
einer Lösung
aus 5% an 2 M Ammoniak in Methanol in Dichlormethan (in Dichlormethan)
unter Bildung der Titelverbindung (0,096 g) gereinigt. Massenspektrum
(APCI+, m/e)= 411 (M+1).
-
Beispiel 388
-
(S)-8-Fluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Gemäß dem Verfahren
von Beispiel 272 und mittels (S)-8-Fluor-5-[3-(2-methoxyethyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,091 g, 0,22 mmol) wird eine teilweise Umwandlung zu der Titelverbindung
nach dem Rühren
bei Umgebungstemperatur für
5,5 Stunden erhalten. Weitere 1,3 Äquivalente an wässrigem
Formaldehyd (0,0216 ml 0,288 mmol) und 1,5 Äquivalente an Natriumtriacetoxyborhydrid
(0,0705 g, 0,333 mmol) mit Dichlormethan werden zugegeben, und es
wird gerührt.
Nach einer Übernachtperiode
wird die Reaktion mit einer gesättigten
wässrigen
Lösung
aus Natriumbicarbonat und Dichlormethan verdünnt und die Phasen werden getrennt.
Die wässrige
Phase wird mit einer gesättigten
Lösung
aus Natriumbicarbonat gewaschen. Die wässrigen Phasen werden vereinigt
und mit Dichlormethan extrahiert. Die organischen Phasen werden
vereinigt, mit einer gesättigten
Lösung
aus Natriumchlorid gewaschen und getrocknet (Natriumsulfat), filtriert
und unter verringertem Druck zu einem Öl konzentriert. Das Öl wird durch
Blitzchromatographie unter Elution mit einem Gradienten aus einer
4% Lösung
an 2 M Ammoniak in Methanol in Dichlormethan (25-100% in Dichlormethan über 58 Minuten)
unter Bildung der Titelverbindung (0,047 g) gereinigt. Massenspektrum
(APCI+, m/e): 425 (M+1).
-
Beispiel 389
-
(S)-8-Fluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
-
Gemäß dem Verfahren
von Beispiel 273 und mittels (S)-8-Fluor-5-[3-(2-methoxyethyl)-4-piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(0,045 g, 0,11 mmol) und einer Lösung
aus Acetylchlorid (0,0378 ml, 0,530 mmol) in absolutem Ethanol bei
Umgebungstemperatur wird die Titelverbindung (0,050 g) erhalten.
Massenspektrum (APCI+, m/e): 425 (M+1-2HCl). Exaktes Massenspektrum
(ES+, m/e, C23H25FN4OS × 2
HCl): berechnet: 425,1811 (M+1-2 HCl), gefunden 425,1808.
-
Beispiel 390
-
(S)-9-Fluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
(S)-9-Fluor-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhyrogenchlorid
(510,2 mg, 1,6 mmol), (S)-2-(2-Methoxyethyl)piperazin (460,8 mg,
3,2 mmol) und Diisopropylethylamin (206 mg, 1,6 mmol) werden in
DMSO (0,75 ml) und Toluol (3,0 ml) vereinigt, gerührt und
auf 110°C
erhitzt. Nach 55 Stunden wird die Reaktion auf RT gekühlt, mit
CH2Cl2 verdünnt und
mit H2O und Kochsalzlösung gewaschen. Die organische
Phase wird mit Na2SO4 getrocknet.
Das rohe Material wird durch eine 5 g SCX Säule gegeben, der 0,2 N NH3/MeOH Eluent wird gesammelt und zu einem
Rückstand
konzentriert, der durch Chromatographie auf Silicagel, Gradient
(100% CH2Cl2 bis
100% CH2Cl2 : 2
N NH3/MeOH = 15:1) unter Bildung von 410
mg der Titelverbindung gereinigt wird. Massenspektrum: ACPI (m/e):
411,2 (M+1).
-
Beispiel 391
-
(S)-9-Fluor-5-[3-(3-methoxypropyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Mittels
des Verfahrens von (S)-11-[3-(3-Methoxypropyl)piperazin-1-yl]-2-trifluormethyl-5H-dibenzo[b,e][1,4]diazepin
und mittels 9-Fluor-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylamin
wird die Titelverbindung erhalten: Massenspektrum (m/e): 425,04
(M+1).
-
Beispiel 392
-
(S)-9-Fluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
-
(S)-9-Fluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen (300
mg, 0,73 mmol), Formaldehyd (37%, G/G, wässrig) (71 mg, 0,88 mmol) und
Natriumtriacetoxyborhydrid (232 mg, 1,09 mmol) in 5,0 ml an 1,2-Dichlorethan
werden vereinigt und bei RT für
5 Stunden gerührt.
Die Reaktion wird durch die Zugabe von Wasser gestoppt und dann
mit CH2Cl2 extrahiert
und die vereinigten organischen Lösemittel werden über Na2SO4 getrocknet.
Das rohe Material wird durch Blitzchromatographie auf Silicagel,
Gradient (100 CH2Cl2 bis
100% CH2Cl2 : 2
N NH3/MeOH = 30:1) über 55 Minuten unter Bildung
von 190 mg des gelben Schaums, (S)-9-Fluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
gereinigt. MS (APCI), (m/e): 425,2 (M+1). Das Dihydrochloridsalz
wird durch die Zugabe von 5 Äquivalenten
aus Acetylchlorid (175 mg, 2,25 mmol) zu der freien Base (190 mg,
0,45 mol) in Ethanol (5 ml) gebildet. Nach der Entfernung des Lösemittel
wird der Rückstand
in 10 ml gemischtem Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 200 mg einer bräunlichen Feststoffs lyophilisiert.
Massenspektrum (Elektrospray) (m/e): C23H25FN4OS × 2 HCl,
berechnete Masse: 425,1811 (M+1-2 HCl), gefunden 425,1818.
-
Beispiel 393
-
(S)-9-Fluor-5-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
Mittels
des Verfahrens von 222 wird die Titelverbindung erhalten. Massenspektrum
(m/e): 439,02 (M+1).
-
Beispiel 394
-
(S)-9-Fluor-5-[3-(3-methoxypropyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulenhydrochlorid
-
Mittels
des Verfahrens von 223 wird die Titelverbindung erhalten. Massenspektrum
(m/e): 439,02 (M+1).
-
Beispiel 395
-
(S)-8,9-Difluor-5-[3-(2-methoxyethyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
-
8,9-Difluor-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen-5-ylaminhydrogenchlorid
(506 mg, 1,5 mmol), (S)-2-(2-Methoxyethyl)piperazin (432 mg, 3,0
mmol) und Diisopropylamin (193 mg, 1,5 mmol) in DMSO (1,0 ml) und
Toluol (3 ml) werden vereinigt, gerührt und auf 110°C erhitzt.
Nach 48 Stunden wird die Reaktion auf RT gekühlt, mit CH2Cl2 verdünnt,
mit NaHCO3 (gesättigt) und Kochsalzlösung gewaschen.
Die organische Phase wird mit Na2SO4 getrocknet. Die organische Phase wird mit
Na2SO4 getrocknet.
Das rohe Material wird durch Chromatographie auf Silicagel, Gradient
(100% CH2Cl2 bis
100% CH2Cl2 : 2
N NH3t/MeOH = 20:1) unter Bildung von 137 mg der Titelverbindung
gereinigt.
-
Beispiel 396
-
(S)-8,9-Difluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
-
8,9-Difluor-5-[(S)-3-(2-methoxyethyl)piperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulen
(130 mg, 0,3 mmol), Formaldehyd (37%, G/G, wässrig) (29,5 mg, 0,38 mmol)
und Natriumtriacetoxyborhydrid (95 mg, 0,45 mmol) in 5,0 ml an 1,2-Dichlorethan
werden vereinigt und bei RT für
1 Stunde gerührt.
Die Reaktion wird durch die Zugabe von Wasser gestoppt und dann
mit CH2Cl2 extrahiert
und die vereinigten organischen Lösemittel werden über Na2SO4 getrocknet.
Das rohe Material wird durch Blitzchromatographie auf Silicagel, Gradient
(100% CH2Cl2 bis
100% CH2Cl2 : 2
N NH3/MeOH = 30:1) über 55 Minuten unter Bildung
von 125 mg des gelben Schaums, (S)-8,9-Difluor-5-[3-(2-methoxyethyl)-4-methylpiperazin-1-yl]-11H-12-thia-6,11-diaza-dibenzo[a,f]azulendihydrochlorid
gereinigt. MS (APCI), (m/e): 443,1 (M+1). Das Dihydrochloridsalz
wird durch die Zugabe von 5 Äquivalenten
aus Acetylchlorid (111 mg, 1,40 mmol) zu der freien Base (125 mg,
0,28 mmol) in Ethanol (5 ml) gebildet. Nach der Entfernung des Lösemittel
wird der Rückstand
in 10 ml gemischtem Lösemittel
aus CH3CN/H2O =
50/50 gelöst
und über
Nacht unter Bildung von 130 mg einer bräunlichen Feststoffs lyophilisiert.
Massenspektrum (Elektrospray) (m/e): C23H24F2N4OS × 2 HCl,
berechnete Masse: 443,1717 (M+1-2 HCl), gefunden 443,1729.
-
Rezeptorbindungstests
-
Serotonin-5-HT6,
Dopamin D2 und Histamin H1 Bindungstest
-
Die
verwendeten Testpuffer sind 50 mM Tris-HCl pH 7,4, 120 mM NaCl,
5 mM KCl, 5 mM MgCl2, 1 mM EDTA für den Dopamin
Des Rezeptorbindungstest. Der verwendete radioaktive Ligand ist
[125I]Iodspiperon von New England Nuclear
Katalognummer NEX284 – 2200
Ci/mmol. Die verwendeten Membranen stammen von Receptor Biology
(gehört
jetzt NEN), Katalognummer RBHD2CM für den D2 Rezeptor.
-
Die
Verbindungen werden als 10 mM Stammlösungen in 100% DMSO erhalten.
Sie werden in 100% DMSO durch die Zugabe von 180 μl DMSO auf
20 ml Stammlösung
in Platten mit 96 Vertiefungen mittels einer Multipipette gegeben.
Die 1 mM Stammlösungen
werden dann unter Bildung eines Konzentrationsbereichs mit 11 Punkten
von 125 μM
bis zu 1,25 nM in halblogarithmischen Stufen mittels 10% DMSO als
Verdünnungsmittel
verdünnt.
Dies wird mittels eines TECAN Roboters ausgeführt. Die DMSO Endkonzentration
beträgt
an dieser Stelle 10 bis 21,25% DMSO.
-
Der
radioaktive Ligand wird in Testpuffer unter Bildung von 0,1 nM für den D2 Test verdünnt. Jedes Röhrchen mit
Membranen wird bis zu 92 ml in Testpuffer verdünnt. Das schließliche Testvolumen
beträgt
250 μl und
besteht aus 210 μl
an verdünnten
Membrane, 20 μl
an Verbindung oder 10% an DMSO für
die Gesamtbindung und 20 μl
verdünntem
radioaktiven Liganden. Die Verbindungen werden aus Arzneimittelverdünnungsplatten
in Testplatten mit 96 Vertiefungen mittels einer Multimek Pipettenge räts für 96 Vertiefungen überführt. Der
radioaktive Ligand und die Membranen werden zu den Testplatten mittels
Multipipetten zugegeben. Die unspezifische Bindung wird in Vertiefungen
bestimmt, die eine Endkonzentration von 5 μM Haloperidol enthalten. Der
schließliche
Arzneimittelkonzentrationsbereich reicht von 10 μM in halblogarithmischen Stufen
bis zu 0,1 nM. Die Endkonzentration im Test beträgt 1 bis 1,7%.
-
Nach
der Zugabe von Arzneimittel, Membran und Ligand werden die Platten
für 2 Stunden
bei Raumtemperatur inkubiert. Während
dieser Zeit werden Millipore Filterplatten (MAFBNOB50) für mindestens
30 Minuten mit 200 μl
an 0,5% Polyethylenimin pro Vertiefung benetzt.
-
Die
0,5% PEI wird aus den Filterplattenvertiefungen mittels eines TiterTek
MAP Absaugegeräts
entfernt und 200 μl
des Inkubationsgemisches werden aus der Inkubationsplatte nach dem
Mischen auf die Filterplatte überführt. Dieser
Transfer wird mittels des Multimekpipettengeräts mit 96 Spitzen ausgeführt. Nach
dem Transfer auf die Filterpaltten werden die Filterplatten extrahiert
und zweimal mit 220 μl
pro Vertiefung an kaltem Puffer auf dem MAP Aspirator gewaschen.
Die abziehbaren Böden
werden aus den Filterplatten entfernt und 60 μl pro Vertiefung an Microscint
20 Scintillationsflüssigkeit
werden mittels einer Multipipette pro Vertiefung zugegeben. Die
Platten werden in stabile Halter gegeben und für 3 Stunden bei Raumtemperatur
stehengelassen und werden auf 3H entweder
auf einem Wallac Microbeta Zähler
oder auf einem Packard Topcount ausgezählt.
-
Protokoll für die [125I]DOI
SPA Bindung an 5-HT2A Rezeptor vom Rhesusaffen
-
Die
Inkubationen werden in einem Gesamtvolumen von 200 μl in Testplatten
mit 96 Vertiefungen ausgeführt.
50 μl [125I]DOI (NEN, 2200 Ci/mmol, Endkonzentration
= 0,075 nM) wird zu 50 μl
der in Wasser gelösten
Testverbindungen gegeben (± DMSO
und/oder Eisessig). 50 μl
Weizenkeimagglutinin (WGA) SPA Kügelchen
werden mit 1 mg/Vertiefung (Amersham Life Science) in Testpuffer
(67 mM Tris-HCl,
pHh 7,4, 13 mM MgCl2, 0,67 mM EDTA) zugegeben.
Membranhomogenat aus Zellen, die 5-HT2A vom
Rhesusaffen exprimieren, etwa 0,9 Millionen Zellen pro Vertiefung,
werden zuletzt zugegeben. Die Platten werden mit Verschlussfolie
verklebt (FasCal) und können
bei Raumtemperatur für
2 Stunden inkubieren. Die Platten werden dann bei etwa 200 × g für 10 Minuten
bei Raumtemperatur zentrifugiert. Die an die Membranen gebundene
Menge an 125I DOI, das heißt nahe
an die WGA SPA Kügelchen,
wird dann mittels eines Wallac MicroBeta Trilux Scintillationszählers bestimmt
(Wallac, Inc.).
-
Protokolle für die 3H
Pyrilamin SPA Bindung an humane Histamin 1 Rezeptoren
-
Die
Inkubationen werden in einem Gesamtvolumen von 200 μl in Testplatten
mit 96 Vertiefungen ausgeführt.
50 μl an 3H Pyrilamin (NEN, 20 Ci/mmol, Endkonzentration
= 3,0 nM) wird zu 50 μl
der Testverbindungen gegeben, die in Wasser gelöst sind (± DMSO und/oder Eisessig).
50 μl Weizenkeimagglutininkügelchen (WGA)
werden dann mit 1 mg/Vertiefung (Amersham Life Science) in Testpuffer
(67 mM TrisCl pH 7,4) zugegeben. Das Membranhomogenat aus den Zellen,
die humane Histamin 1 Rezeptoren exprimieren, etwa 700 000 Zellen
pro Vertiefung, wird zuletzt zugegeben. Die Platten werden mit Verschlussfolie
zugeklebt (FasCal) und können
für 2 Stunden
bei Raumtemperatur inkubieren. Die Platzen werden dann bei etwa
200 × g
für 10 Minuten
bei Raumtemperatur zentrifugiert. Die Menge an 3H
Pyrilamin, die an die Membranen gebunden ist, das heißt nahe
an die WGA Kügelchen,
wird dann mittels eines Wallac MicroBeta Trilux Scintillationszählers (Wallac,
Inc.) bestimmt.
-
Pharmazeutische Formulierungen
-
Kapsel
-
Es
wird eine Pulverformulierung durch Mischen des Wirkstoffs mit Silicon
und Stärke
hergestellt und in Hartgelatinekapseln gefüllt.
| | Pro
300 mg Kapsel |
| Verbindung
der Formel (I) | 5,0
mg |
| Silicon | 2,9
mg |
| Fließfähige Stärke | 292,1
mg |
-
Tablette
-
Es
wird eine Tablettenformulierung durch Granulierung des Wirkstoffs
mit einem geeigneten Verdünnungsmittel,
einem Gleitmittel, einem Zerfallshilfsmittel und einem Bindemittel
und Verpressen hergestellt.
| | pro
300 mg Tablette |
| Verbindung
der Formel (I) | 10,0
mg |
| Magnesiumstearat | 0,9
mg |
| Mikrokristalline
Cellulose | 75,0
mg |
| Povidone | 15,0
mg |
| Direkt
verpressbare Stärke | 199,1
mg |
-
Injektion
-
Es
wird eine wässrige
Injektion des Wirkstoffs als gefriergetrockneter Bodensatz zur Rekonstitution
vor der Verwendung in einem geeigneten sterilen Verdünnungsmittel
hergestellt (auf ein Gesamtvolumen von 10 ml).
| Verbindung
der Formel (I) | 20,0
mg |
| Mannit | 20,0
mg |
| 1
N Chlorwasserstoffsäure
und/oder 1 N Natriumhydroxid zur Einstellung des pH auf 5-5,5. | |
-
Kontrolliert freisetzende
Injektion
-
Eine
kontrolliert freisetzende Injektion zur intramuskulären Injektion
wird aus einer sterilen Suspension des mikronisierten Wirkstoffs
in einem ölartigen
Träger
gebildet.
| Verbindung
der Formel (I) | 65,0
mg |
| Aluminiumstearat | 0,04
mg |
| Sesamöl | 2
ml |
 für einen otional benzofusionierten fünf- oder sechsgliedrigen aromatischen Ring mit 0 bis 3 Heteroatomen steht, die unabhängig ausgewählt sind aus N, S und O,
für einen otional benzofusionierten fünf- oder sechsgliedrigen aromatischen Ring mit 0 bis 3 Heteroatomen steht, die unabhängig ausgewählt sind aus N, S und O,

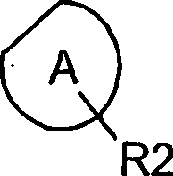

























 für einen Oxazolring steht, können durch Cyclisierung des Zwischenprodukts der Formel (XVIII) mit einem Dehydratisierungsmittel hergestellt werden, wie P2O5 oder PPh3/Tf2O.
für einen Oxazolring steht, können durch Cyclisierung des Zwischenprodukts der Formel (XVIII) mit einem Dehydratisierungsmittel hergestellt werden, wie P2O5 oder PPh3/Tf2O.


 für Pyrimidin steht, auch durch Umsetzung von Verbindungen der Formel (XX) jeweils mit einem substituierten Hydrazin oder einem Amidin hergestellt werden.
für Pyrimidin steht, auch durch Umsetzung von Verbindungen der Formel (XX) jeweils mit einem substituierten Hydrazin oder einem Amidin hergestellt werden.






















































































































































































































































































































































































































 für einen optional benzofusionierten fünf- oder sechsgliedrigen aromatischen Ring mit 0 bis 3 Hete- roatomen steht, die unabhängig ausgewählt sind aus N, S und O, Alk für (C1-C4)-Alkylen oder hydroxysubstituiertes (C1-C4)-Alkylen steht, X für Sauerstoff oder Schwefel steht, R1 für Wasserstoff, (C1-C6)-Fluoralkyl, (C3-C6)-Cycloalkyl oder (C1-C4)-Alkyl steht, worin das (C1-C4)-Alkyl unsubstituiert oder substituiert ist durch Hydroxy, Methoxy, Ethoxy, OCH2CH2OH oder -CN, R2 für H, Halogen, (C1-C6)-Fluoralkyl, (C3-C6)-Cycloalkyl, OR4, SR4, NO2, CN, COR4, C(O)OR4, CONR5R6, NR5R6, SO2NR5R6, NR5COR4, NR5SO2R4, das optional durch eine aromatische Gruppe substituiert ist, oder für (C1-C6)-Alkyl steht, worin das (C1-C6)-Alkyl unsubstituiert oder durch Hydroxy substituiert ist, R3 steht für Wasserstoff, (C1-C4)-Alkyl, (C3-C6)-Cycloalkyl, (C2-C6)-Alkenyl, Ar oder (C1-C4)-Alkyl-Ar, R4 für Wasserstoff, (C1-C6)-Alkyl, (C1-C6)-Fluoralkyl oder eine optional substituierte aromatische Gruppe steht, R5 und R6 unabhängig für Wasserstoff, (C1-C6)-Alkyl oder eine optional substituierte aromatische Gruppe stehen, R7 für Wasserstoff, (C1-C6)-Alkyl, (C1-C6)-Fluoralkyl oder eine optional substituierte aromatische Gruppe steht, R8 und R9 unabhängig für Wasserstoff, (C1-C6)-Alkyl oder eine optional substituierte aromatische Gruppe stehen, Ar für optional substituiertes Phenyl, Naphthyl, eine monocyclische heteroaromatische Gruppe oder eine bicyclische heteroaromatische Gruppe steht, Z1 und Z2 unabhängig ausgewählt sind aus Wasserstoff, Halogen, (C1-C6)-Alkyl, (C1-C6)-Fluoralkyl, OR7, SR7, NO2, CN, COR7, CONR8R9, NR8R9 und einer optional substituierten aromatischen Gruppe, und alle Salze, Solvate, optischen und geometrischen Isomeren und kristallinen Formen hiervon.
für einen optional benzofusionierten fünf- oder sechsgliedrigen aromatischen Ring mit 0 bis 3 Hete- roatomen steht, die unabhängig ausgewählt sind aus N, S und O, Alk für (C1-C4)-Alkylen oder hydroxysubstituiertes (C1-C4)-Alkylen steht, X für Sauerstoff oder Schwefel steht, R1 für Wasserstoff, (C1-C6)-Fluoralkyl, (C3-C6)-Cycloalkyl oder (C1-C4)-Alkyl steht, worin das (C1-C4)-Alkyl unsubstituiert oder substituiert ist durch Hydroxy, Methoxy, Ethoxy, OCH2CH2OH oder -CN, R2 für H, Halogen, (C1-C6)-Fluoralkyl, (C3-C6)-Cycloalkyl, OR4, SR4, NO2, CN, COR4, C(O)OR4, CONR5R6, NR5R6, SO2NR5R6, NR5COR4, NR5SO2R4, das optional durch eine aromatische Gruppe substituiert ist, oder für (C1-C6)-Alkyl steht, worin das (C1-C6)-Alkyl unsubstituiert oder durch Hydroxy substituiert ist, R3 steht für Wasserstoff, (C1-C4)-Alkyl, (C3-C6)-Cycloalkyl, (C2-C6)-Alkenyl, Ar oder (C1-C4)-Alkyl-Ar, R4 für Wasserstoff, (C1-C6)-Alkyl, (C1-C6)-Fluoralkyl oder eine optional substituierte aromatische Gruppe steht, R5 und R6 unabhängig für Wasserstoff, (C1-C6)-Alkyl oder eine optional substituierte aromatische Gruppe stehen, R7 für Wasserstoff, (C1-C6)-Alkyl, (C1-C6)-Fluoralkyl oder eine optional substituierte aromatische Gruppe steht, R8 und R9 unabhängig für Wasserstoff, (C1-C6)-Alkyl oder eine optional substituierte aromatische Gruppe stehen, Ar für optional substituiertes Phenyl, Naphthyl, eine monocyclische heteroaromatische Gruppe oder eine bicyclische heteroaromatische Gruppe steht, Z1 und Z2 unabhängig ausgewählt sind aus Wasserstoff, Halogen, (C1-C6)-Alkyl, (C1-C6)-Fluoralkyl, OR7, SR7, NO2, CN, COR7, CONR8R9, NR8R9 und einer optional substituierten aromatischen Gruppe, und alle Salze, Solvate, optischen und geometrischen Isomeren und kristallinen Formen hiervon.








