-
Die
vorliegende Erfindung betrifft das Gebiet chemotherapeutischer Mittel
und betrifft insbesondere bestimmt substituierte Polyketide und
die Verwendung der Polyketide bei der Behandlung von Tumoren.
-
HINTERGRUND
DER ERFINDUNG
-
Krebs
ist ein schwerwiegendes Gesundheitsproblem in der ganzen Welt. Die
Krebsneuerkrankung oder Krebsinzidenz in den USA hat z.B. während der
letzten 30 Jahre um 30% zugenommen, und es wird erwartet, dass sie
kontinuierlich zunimmt im nächsten
Jahrhundert. Dies ist der zunehmenden Prävalenz des Zigarettenrauchens
bei Frauen im Vergleich zu Männern
zuzuschreiben, dem allgemeinen Altern der Bevölkerung und verbesserten Diagnosemöglichkeiten
sowie potenziellen Abnahmen in der Mortalität oder Sterblichkeit aus anderen
Gründen.
Als Ergebnis wurde eine umfangreiche Anzahl von Forschungsanstrengungen
in einem Bemühen
unternommen, geeignete Therapien zu entwickeln für die Behandlung und Verminderung oder
Linderung von Krebs bei Menschen.
-
Auf
dem chemotherapeutischen Gebiet wurde eine Forschung durchgeführt, um
Antitumormittel zu entwickeln, die wirksam sind gegen verschiedene
Typen von Krebs. Oftmals sind Antitumormittel, die entwickelt worden
sind und von denen gefunden wurde, dass sie gegen Krebszellen wirksam
sind, leider auch gegenüber
normalen Zellen toxisch. Diese Toxizität offenbart sich in Gewichtsverlust, Übelkeit
oder Brechreiz, Erbrechen, Haarverlust, Erschöpfung, Juckreiz, Halluzinationen,
Appetitverlust, etc. bei der Verabreichung des Antitumormittels
an einen Patienten, der einer Krebschemotherapie bedarf.
-
Außerdem haben
herkömmlich
verwendete chemotherapeutische Mittel nicht die gewünschte Wirksamkeit
oder sind nicht so breit wirksam gegen verschiedene Typen von Krebs
wie erwünscht.
Als Ergebnis existiert ein großer
Bedarf an chemotherapeutischen Mitteln, die nicht nur wirksamer
sind gegen alle Typen von Krebs, sondern die einen höheren Grad
an Selektivität
zum Töten
von Krebszellen haben mit keiner oder nur geringen Wirkung auf normale
gesunde Zellen. Außerdem
sind hoch wirksame und selektive Antitumormittel erwünscht, insbesondere
gegen Krebse des Kolons, der Blase, der Prostata, des Magens, des
Pankreas, der Brust, der Lunge, der Leber, des Gehirns, der Hoden,
des Eierstocks, des Gebärmutterhalses,
der Haut, der Vulva und des Dünndarms.
Außerdem
ist eine Antitumorwirksamkeit gegen Kolon-, Brust-, Lungen- und Prostatakrebs
sowie Melanome besonders gewünscht
wegen des Mangels irgendeiner wirksamen Therapie zum gegenwärtigen Zeitpunkt.
-
(+)-Discodermolid
ist ein neues Polyketidnaturprodukt, das isoliert wurde aus Extrakten
des Meeresschwamms Discodermia dissoluta durch Forscher an der Harbor
Branch Oceanographic Institution (HBOI) (SP Gunasekera, M Gunasekera,
RE Longley, GK Schulte, Discodermolide: a new bioactive polyhydroxylated
lactone from the marine sponge Discodermia dissoluta, [veröffentlichte
Berichtigung erfolgt in J. Org. Chem. 1991; 56: 1346], J. Org. Chem.
1990; 55: 4912-4915). Die WO 01/42179 A offenbart Discodermolidderivate
und ihre Verwendung als antiproliferatives Mittel. Discodermolid
fehlt eine ersichtliche strukturelle Ähnlichkeit zu Paclitaxel, dennoch
teilt es mit Paclitaxel (der wirksamen Substanz in dem Arzneimittel
Taxol) die Fähigkeit
Mikrotubuli zu stabilisieren. Bei Mechanismus-basierenden Untersuchungen
ist Discodermolid wirksamer als Paclitaxel. Tatsächlich ist von den Handvoll
Verbindungen, die bekannt sind, eine Polymerisation von gereinigtem Tubulin
zu induzieren, Discodermolid die potenteste oder wirksamste. Mikrotubuli
jedoch, die Hauptstrukturbestandteile in Zellen, sind nicht einfach
Gleich gewichtspolymere von Tubulin. Sie existieren als regulierte GTP-getriebene
dynamische Komponenten von Heterodimeren von α- und β-Tubulin. Obwohl die Dynamiken relativ
gering sind in Interphasenzellen, nimmt beim Eintreten der Mitose
die Rate des Wachstums und des Verkürzens auf das 20- bis 100-fache
zu – der
mittlere Mikrotubulus setzt sich alle 10 Sekunden um die Hälfte der
Tubulinuntereinheiten um. Diese Veränderung in der Rate oder Geschwindigkeit
ermöglicht
es dem cytosketalen Mikrotubulusnetzwerk, sich abzubauen, und einer
bipolaren Spindel-geformten Anordnung von Mikrotubuli aufzubauen.
Die Spindel haftet sich an Chromosome und bewegt sie auseinander.
Die Antwort auf eine vollständige
Unterdrückung
oder Suppression der Mikrotubulusdynamiken in Zellen ist der Tod.
Mitotische Zellen sind jedoch empfindlicher oder sensitiver, und
die Toleranzschwelle scheint Zelltyp-spezifisch zu sein. Moleküle, wie
Paclitaxel, das mit hoher Affinität an Mikrotubuli bindet, unterbrechen
die dynamischen Prozesse in Tumorzellen mit tödlichen Ergebnissen, selbst
wenn das Verhältnis
von gebundenem Arzneimittel zu Tubulin sehr gering ist. Discodermolid
bindet an Tubulin kompetitiv mit Paclitaxel. Da von Paclitaxel gezeigt
wurde, dass es nützlich
ist bei der Behandlung von einigen Krebsen, können andere Verbindungen der
gleichen mechanistischen Klasse Nützlichkeit gegen hyperproliferative
Störungen
aufweisen.
-
Die
Entwicklung von Discodermolid oder strukturell verwandten Analoga
wird behindert durch den Mangel einer verlässlichen natürlichen
Quelle der Verbindung oder einen brauchbaren oder praktikablen Syntheseweg.
Natürlich
vorkommendes Discodermolid ist knapp und die Ernte der produzierenden
Organismen stellt logistische Probleme dar. Es gibt einen immer
wachsenden Bedarf für
verbesserte Synthesen, die die Herstellung von mehreren Grammmengen
von Discodermolid und strukturell verwandte Analoga ermöglichen.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
vorliegende Erfindung stellt neue Antitumormittel bereit, die wirksam
sind gegen eine Vielzahl von Krebszellen. Insbesondere betrifft
die vorliegende Erfindung bestimmt substituierte Polyketide, die
einen höheren
Grad an Selektivität
beim Töten
von Krebszellen aufweisen. Außerdem
stellt die vorliegende Erfindung pharmazeutische Zusammensetzungen
bereit, die nützlich
sind bei der Behandlung von Tumoren, die eine therapeutisch wirksame
Menge von bestimmt substituiertem Polyketid umfassen. Außerdem stellt
die vorliegende Erfindung ein Verfahren zur Behandlung von Tumoren
bereit, umfassend eine Verabreichung einer therapeutisch wirksamen
Menge eines bestimmt substituierten Polyketids an einen Säuger, der
davon befallen ist oder davon heimgesucht wird.
-
DETAILLIERTE
BESCHREIBUNG DER ERFINDUNG
-
Das
Wesentliche der vorliegenden Erfindung ist die Entdeckung, dass
bestimmt substituierte Polyketide nützlich sind bei der Behandlung
von Tumoren. Bei einer Ausführungsform
stellt die vorliegende Erfindung neue Antitumormittel der Formel
I bereit:
wobei
A steht für -CH
2N(R
2)C(O)-, -C(O)N(R
2)CH
2-, -CH
2N(R
2)CH
2-,
-CH
2N(CO
2R
3)CH
2- oder -CH
2N(COR
2)CH
2-;
B steht für -CH(R
1)CH=CHCH=CH
2, -CH(R
2)R
1, -CH(R
1)CH=CHR
2, -CH(R
1)CH=CHC(O)OR
2, -CH(R
1)CH=CHC(O)N(R
1)R
2, -CH(R
1)CH
2OR
2 oder
Ar;
C steht für
H, -C(O)N(R
1)R
2,
-C(O)NHCH
2(CH
2)
nN(CH
3)
2 oder
-C(O)NHCH
2(CH
2)
n-4-morpholino;
R
1 steht
für H oder
(C
1-C
6)-Alkyl;
R
2 steht für
H, (C
1-C
6)-Alkyl,
(C
2-C
6)-Alkenyl,
(C
2-C
6)-Alkinyl,
(C
1-C
6)-Alkyl-Ar
oder Ar;
R
3 steht für (C
1-C
6)-Alkyl, (C
1-C
6)-Alkyl-Ar oder Ar;
Ar für einen
aromatischen oder heteroaromatischen Ring steht, der ausgewählt ist
aus
R
4 und R
5 unabhängig
stehen
für H,
(C
1-C
6)-Alkyl, OH,
O(C
1-C
6)-Alkyl,
OCH
2(CH
2)
nOH, O(CH
2)
nCO
2H, OCH
2(CH
2)
nN(CH
3)
2, OCH
2(CH
2)
n-4-morpholino,
F, Cl, Br oder CF
3; und
n für 1 oder
2 steht;
oder ein Säure-
oder Baseadditionssalz davon, wenn dies möglich ist. Bevorzugte Verbindungen
sind solche der Formel Ia:
wobei
A' steht für -CH
2N(R
2')C(O)-, -C(O)N(R
2')CH
2-, -CH
2N(CO
2R
3')CH
2-
oder -CH
2N(COR
2')CH
2-;
B' steht für -CH(R
1')CH=CHCH=CH
2, -CH(R
2')R
1', -CH(R
1')CH=CHR
2', -CH(R
1')CH
2OR
2' oder
Ar';
C' steht für H, -C(O)N(R
1')R
2',
-C(O)NHCH
2(CH
2)
nN(CH
3)
2 oder
-C(O)NHCH
2(CH
2)
n-4-morpholino;
R
1' steht für H oder
(C
1-C
6)-Alkyl;
R
2' steht
für H,
(C
1-C
6)-Alkyl, (C
2-C
6)-Alkenyl, (C
2-C
6)-Alkinyl, (C
1-C
6)-Alkyl-Ar' oder Ar';
R
3' steht für (C
1-C
6)-Alkyl, (C
1-C
6)-Alkyl-Ar' oder Ar';
Ar' für einen
aromatischen oder heteroaromatischen Ring steht, der ausgewählt ist
aus
R
4' und
R
5'
unabhängig
stehen
für H,
(C
1-C
6)-Alkyl, OH,
O(C
1-C
6)-Alkyl,
OCH
2(CH
2)
nOH, O(CH
2)
nCO
2H, OCH
2(CH
2)
nN(CH
3)
2, OCH
2(CH
2)
n-4-morpholino,
F, Cl, Br oder CF
3; und
n für 1 oder
2 steht;
oder ein Säure-
oder Baseadditionssalz davon, wenn dies möglich ist.
-
Noch
bevorzugter sind solche Verbindungen der Formel Ib:
wobei
A'' steht für -CH
2N(R
2'')C(O)- oder -C(O)N(R
2'')CH
2-;
B'' steht für -CH(R
1'')CH=CHCH=CH
2,
-CH(R
2'')R
1'', -CH(R
1'')CH=CHR
2'', -CH(R
1'')CH
2OR
2'' oder Ar'';
C'' steht
für H,
-C(O)N(R
1'')R
2'', -C(O)NHCH
2(CH
2)
nN(CH
3)
2 oder -C(O)NHCH
2(CH
2)
n-4-morpholino;
R
1'' steht für H oder
-CH
3;
R
2'' steht für H, (C
1-C
6)-Alkyl, (C
2-C
6)-Alkenyl, (C
2-C
6)-Alkinyl, (C
1-C
6)-Alkyl-Ar'' oder
Ar'';
Ar'' für
einen aromatischen oder heteroaromatischen Ring steht, der ausgewählt ist
aus
R
4'' und R
5''
unabhängig
stehen für H, (C
1-C
6)-Alkyl, OH,
O(C
1-C
6)-Alkyl,
OCH
2(CH
2)
nOH, O(CH
2)
nCO
2H, OCH
2(CH
2)
nN(CH
3)
2, OCH
2(CH
2)
n-4-morpholino,
F, Cl, Br oder CF
3; und
n für 1 oder
2 steht;
oder ein Säure-
oder Baseadditionssalz davon, wenn dies möglich ist.
-
Darüber hinaus
bevorzugtere Verbindungen sind solche der Formel Ic:
wobei
A''' steht
für -CH
2N(R
2''')C(O)-
oder -C(O)N(R
2''')CH
2-;
B''' steht
für -CH(R
1''')CH=CHCH=CN
2,
-CN(R
2''')R
1''',
-CH(R
1''')CH=CHR
2''',
-CH(R
1''')CH
2OR
2''' oder Ar''';
C''' steht
für H oder
-C(O)N(R
1''')R
2''';
R
1''' steht für H oder CH
3;
R
2''' steht für H, (C
1-C
6)-Alkyl, (C
2-C
6)-Alkenyl, (C
2-C
6)-Alkinyl, (C
1-C
6)-Alkyl-Ar''' oder Ar''';
Ar''' für einen
aromatischen Ring steht, der die folgende Formel aufweist:
R
4''' und
R
5'''
unabhängig
stehen für H, (C
1-C
6)-Alkyl, OH,
O(C
1-C
6)-Alkyl,
F, Cl, Br oder CF
3;
oder ein Säure- oder
Baseadditionssalz davon, wenn dies möglich ist.
-
Bei
einer weiteren Ausführungsform
stellt die vorliegende Erfindung pharmazeutische Zusammensetzungen
bereit, die nützlich
sind bei der Behandlung von Tumoren, wobei die Zusammensetzungen
einen pharmazeutisch annehmbaren oder akzeptablen Träger oder
ein pharmazeutisch annehmbares oder akzeptables Verdünnungsmittel
und eine therapeutisch wirksame Menge einer Verbindung der obigen
Formel I oder eines pharmazeutisch annehmbaren oder akzeptablen
Säure-
oder Baseadditionssalzes davon, wenn dies möglich ist, umfasst, vorzugsweise
einer Verbindung der Formel Ia oben oder eines pharmazeutisch annehmbaren Säure- oder
Baseadditionssalzes davon, wenn dies möglich ist, noch bevorzugter
einer Verbindung der Formel Ib oben oder eines pharmazeutisch annehmbaren
Säure- oder Basesalzes
davon, wenn dies möglich
ist, und sogar noch bevorzugter einer Verbindung der Formel Ic oben
oder eines pharmazeutisch annehmbaren Säure- oder Baseadditionssalz
davon, wenn dies möglich
ist.
-
In
den obigen Definitionen gilt: 1) Die Alkylgruppen, die 1 bis 6 Kohlenstoffatome
enthalten, sind entweder gerade oder verzweigte Ketten oder ein
Cycloalkan, von denen Beispiele Isopropyl, Isobutyl, tert.-Butyl, Isopentyl,
Neopentyl, Isohexyl, 3-Methylpentyl, 2,2,-Dimethylbutyl, 2,3-Dimethylbutyl,
1,1,2,2-Tetramethylethyl,
Cyclopentyl und Cyclohexyl einschließen.
-
Obwohl
die pharmazeutisch annehmbaren Säure-
oder Baseadditionssalze bevorzugt sind, sollte verstanden werden,
dass es beabsichtigt ist, dass alle der Säure- oder Baseadditionssalze
der Verbindungen der Formel I in den Schutzbereich der vorliegenden
Erfindung eingeschlossen sind.
-
Die
Säureadditionssalze
der Verbindungen der Formel 1 können
solche von pharmazeutisch annehmbaren organischen oder anorganischen
Säuren
sein. Obwohl die bevorzugten Säureadditionssalze
solche sind von Chlorwasserstoff- und Methansulfonsäure, können auch
Salze von Schwefel-, Phosphor-, Citronen-, Furmar-, Malein-, Benzoe-,
Benzolsulfon-, Bernstein-, Wein-, Milch- und Essigsäure verwendet
werden.
-
In
gleicher Weise können
die Baseadditionssalze der Verbindungen der Formel I solche von
pharmazeutisch annehmbaren organischen oder anorganischen Basen
sein. Bevorzugte Baseadditionssalze sind solche, die abgeleitet
sind von pharmazeutisch annehmbaren anorganischen Basen, besonders
bevorzugt Ammoniumhydroxid oder einem Alkali- oder Erdalkalimetallhydroxid,
z.B. Lithiumhydroxid, Natriumhydroxid, Kaliumhydroxid, Calciumhydroxid,
Magnesiumhydroxid und Manganhydroxid.
-
Die
substituierten Polyketide der Formel I können wie unten gezeigt hergestellt
werden. In dem Fall, dass die Gruppen A-F freie Hydroxygruppen enthalten,
dann gibt die Kennzeichnung mit dem Stern (z.B. A*) an, dass diese
Gruppen geschützt
sind mit säurelabilen
Schutzgruppen (z.B. TBS). Alle säurelabilen
Schutzgruppen, die abgedeckt werden durch den Stern, werden in der
letzten Stufe entfernt (HCI). Schema
1
-
Hinsichtlich
der einzelnen Stufen in Schema 1 schließt die Stufe A die Addition
eines Ketons der Formel 2 an einen Aldehyd der Formel 1 ein, um
ein Hydroxyketon der Formel 3 zu erhalten. Die Addition erfordert zwischen
1 und 20 Äquivalente
an 2, bezogen auf den Aldehyd 1, vorzugsweise zwischen 5 und 15 Äquivalente
an 2, bezogen auf den Aldehyd 1. Die Kupplung wird durchgeführt in Gegenwart
von: 1) einem Dialkylborhalogenid oder Triflat, vorzugsweise einem
chiralen Borchlorid oder Triflat, besonders bevorzugt B-Chlordiisopinocampheylboran;
2) einer Base, vorzugsweise einem Amin, besonders bevorzugt Triethylamin;
und 3) einem polaren organischen Lösemittel, vorzugsweise einem
Ether, besonders bevorzugt Diethylether, bei einer Temperatur von
zwischen -100°C
und 20°C,
vorzugsweise zwischen -78°C
und -20°C, über einen
Zeitraum von zwischen 2 und 72 Stunden, vorzugsweise 16 Stunden
lang.
-
Die
Stufe B betrifft die Reduktion des Hydroxyketons der Formel 3, um
eine 1,3-Diolverbindung der Formel 4 zu erhalten. Die Reduktion
wird durchgeführt
in Gegenwart von: 1) einem Ketonreduktionsmittel, vorzugsweise einem
Borhydrid, wie Tetramethylammoniumtriacetoxyborhydrid; 2) einem
polaren organischen Lösemittel,
vorzugsweise Acetonitril; und 3) einem protischen Lösemittel,
vorzugsweise einer Carbonsäure,
wie Essigsäure,
bei einer Temperatur von zwischen -78°C und 20°C, vorzugsweise zwischen -40°C und -10°C, über einen
Zeitraum von zwischen 2 und 72 Stunden, vorzugsweise 16 Stunden
lang.
-
Die
Stufe C betrifft die Hydrolyse und Cyclisierung der 1,3-Diolverbindung
4 zu einem substituierten Polyketid der Formel 5. Die Hydrolysereaktion
wird durchgeführt
in Gegenwart von: 1) einer protischen Säure, vorzugsweise einer wässrigen
protischen Säurelösung, vorzugsweise
einer wässrigen
Halogenwasserstofflösung,
wie wässrigem
Chlorwasserstoff; und 2) einem polaren organischen Lösemittel,
vorzugsweise einem Gemisch von polaren organischen Lösemitteln,
vorzugsweise einem Gemisch eines aliphatischen Alkohols und eines
Ethers, wie Methanol und Tetrahydrofuran, bei einer Temperatur von
zwischen -20°C
und 40°C,
vorzugsweise von zwischen 20°C
und 25°C über einen
Zeitraum von 8 Stunden und 7 Tagen, vorzugsweise zwischen 16 und
72 Stunden, besonders bevorzugt zwischen 24 und 48 Stunden. Schema
2
-
Hinsichtlich
der einzelnen Stufen in Schema 2 schließt die Stufe A die oxidative
Hydrolyse eines para-Methoxybenzylethers der Formel 1 zu einem Diol
der Formel 2 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2), Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
Die
Stufe B schließt
die Oxidation eines Alkohols der Formel 2 ein, um einen Aldehyd
der Formel 3 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart
von: 1) einem Oxidationsreagens, vorzugsweise einem milden Oxidationsreagens
oder Oxidationsmittel, wie den Kombinationen von Oxalylchlorid,
DMSO und Triethylamin; einem Schwefeltrioxid-Pyridinkomplex, DMSO
und Triethylamin; und dem freien Radikal 2,2,6,6-Tetramethyl-1-piperidinyloxy
und Diacetoxyiodbenzol; und 2) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise von -20°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe C schließt
die Olefinierung eines Aldehyds der Formel 3 mit einem Olefinierungsreagens
ein, vorzugsweise (CF3CH2O)2P(O)CH2CO2CH3, um ein Olefin
der Formel 4 zu erhalten. Die Olefinierung wird durchgeführt in Gegenwart
von: 1) einer starken Base, vorzugsweise einem Alkalimetallsalz,
wie Kaliumhexamethyldisilazid oder Butyllithium; und 2) einem inerten
organischen Lösemittel,
vorzugsweise einem Kohlenwasserstoff, wie Toluol, oder einem Ether,
wie Tetrahydrofuran, bei einer Temperatur von zwischen -78°C und 25°C, vorzugsweise
bei 0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3 Stunden
lang.
-
Die
Stufe D betrifft die Carbamoylierung des Olefins der Formel 4 mit
einem Isocyanat von entweder der Formel C*NCO oder Cl3C(O)NCO,
um ein Carbamat der Formel 5 zu ergeben. Im Falle der Verwendung von
C*NCO wird die Carbamoylierung in Gegenwart einer Lewis-Säure, wie
Bu2Sn(OAc)2 oder
einer schwachen Base, wie Triethylamin, in einem polaren aprotischen
Lösemittel,
vorzugsweise einem halogenierten Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -20°C und 100°C, vorzugsweise zwischen 0°C und 50°C, über einen
Zeitraum von zwischen 5 Minuten und 72 Stunden, vorzugsweise zwischen
1 Stunde und 24 Stunden, durchgeführt. Im Fall der Verwendung
von Cl3C(O)NCO, das substituierte Polyketide
der Formel 1 erzeugt, wobei C=H gilt, wird die Carbamoylierung in
Gegenwart eines polaren aprotischen Lösemittels durchgeführt, vorzugsweise
eines halogenierten Lösemittels,
wie Methylenchlorid, bei einer Temperatur von zwischen -20°C und 100°C, vorzugsweise
bei 25°C, über einen
Zeitraum von zwischen 5 Minuten und 72 Stunden, vorzugsweise zwischen
1 Stunde und 8 Stunden; die Aufarbeitung dieser Stufe wird durchgeführt in Gegenwart
eines protischen organischen Lösemittels,
vorzugsweise eines Alkohols, wie Methanol, in Gegenwart einer Base,
z.B. einem Carbonat, wie Kaliumcarbonat, bei einer Temperatur von
zwischen 0°C
und 100°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 5 Minuten und 72 Stunden, vorzugsweise zwischen
1 Stunde und 8 Stunden.
-
Die
Stufe E schließt
die Reduzierung eines Carbamats der Formel 5 ein, um einen Alkohol
der Formel 6 zu erhalten. Die Reduktion wird durchgeführt in Gegenwart
von: 1) einem Reduktionsmittel, vorzugsweise einem Aluminiumhydridreagens,
wie Diisopropylaluminiumhydrid; und 2) einem inerten organischen
Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -100°C und 0°C, vorzugsweise bei -78°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe F schließt
die Oxidation eines Alkohols der Formel 6 ein, um einen Aldehyd
der Formel 7 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart
von: 1) einem Oxidationsreagens oder Oxidationsmittel, vorzugsweise
einem milden Oxidationsreagens, wie dem Dess-Martin-Periodinanreagens,
oder den Kombinationen von Oxalylchlorid, DMSO und Triethylamin;
einem Schwefeltrioxid-Pyridinkomplex,
DMSO und Triethylamin; und dem freien Radikal 2,2,6,6-Tetramethyl-1-piperidinyloxy
und Diacetoxyiodbenzol; und 2) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise von -20°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise über einen
Zeitraum von zwischen 1 und 3 Stunden.
-
-
In
Bezug auf die einzelnen Stufen in Schema 3 schließt die Stufe
A die Reduktion eines cyclischen para-Methoxyphenylacetals der Formel
1 ein, um einen Alkohol der Formel 2 zu erhalten. Die Reduktion
wird durchgeführt
in Gegenwart von: 1) einem Metallhydrid, vorzugsweise einem Aluminiumhydrid,
wie Diisobutylaluminiumhydrid; und 2) einem aprotischen organischen
Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -100°C
und 10°C,
vorzugsweise von -78°C
bis 0°C, über einen Zeitraum
von zwischen 10 Minuten und 8 Stunden, vorzugsweise 2 Stunden lang.
-
Die
Stufe B schließt
die Veretherung eines Alkohols der Formel 2 ein, um einen Ether
der Formel 3 zu erhalten. Die Veretherung wird durchgeführt in Gegenwart
von: 1) einem Alkohol der Formel R2*OH,
wobei R2* so wie oben beschrieben ist; 2)
einem Kupplungsreagens, wie Diethylazodicarboxylat; 3) ei nem Phosphin,
wie Triphenylphosphin; und 4) einem polaren organischen Lösemittel,
wie Tetrahydrofuran, bei einer Temperatur von zwischen -78°C und 60°C, vorzugsweise
zwischen -20°C
und 40°C, über einen
Zeitraum von zwischen 2 und 72 Stunden, vorzugsweise 16 Stunden
lang. Alternativ oder bei einer weiteren Ausführungsform wird R2*OH
ersetzt durch ein R2*Halogenid oder R2*Sulfonat. In diesem Fall wird die Veretherung
durchgeführt
in Gegenwart von: 1) einer Base, vorzugsweise einer Alkalimetallbase,
wie Natriumhydrid; 2) einem polaren organischen Lösemittel,
wie N,N-Dimethylformamid; und 3) einer optionalen katalytischen
Menge eines Iodidsalzes, wie Kaliumiodid, bei einer Temperatur von
zwischen -78°C
und 60°C,
vorzugsweise zwischen -20°C
und 40°C, über einen
Zeitraum von zwischen 2 und 72 Stunden, vorzugsweise 16 Stunden
lang.
-
Die
Stufe C schließt
die oxidative Hydrolyse eines Ethers der Formel 3 zu einem Diol
der Formel 4 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 4 schließt die Stufe
A die reduktive Aminierung eines Aldehyds der Formel 1 ein, um ein
Amin der Formel 2 zu erhalten. Die reduktive Aminierung wird durchgeführt in Gegenwart
von: 1) einem Amin der Formel R5NH2, wobei R5 so wie
oben definiert ist; 2) einem Reduktionsmittel, vorzugsweise einem
Hydrid, besonders bevorzugt einem Borhydridsalz, wie Natriumborhydrid;
und 3) einem polaren organischen Lösemittel, vorzugsweise einem
protischen organischen Lösemittel,
wie Ethanol, bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise von 5°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
Die
Stufe B schließt
die Acylierung eines Amins der Formel 2 ein, um ein Amid der Formel
4 zu erhalten. Die Acylierung wird durchgeführt in Gegenwart von: 1) einer
Carbonsäure
der Formel 3; 2) einem Carbonsäurekupplungsmittel,
vorzugsweise einem Diimid, wie 1-(3-Dimethylaminopropyl)-3- ethylcarbodiimidhydrochlorid,
und einem geeigneten Aktivierungsmittel, das üblich ist für Diimidkupplungsreaktionen,
wie 1-Hydroxybenzotriazol; und 3) einem polaren organischen Lösemittel,
vorzugsweise einem Amid mit niedrigem Molekulargewicht, wie DMF,
bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise bei 25°C, über einen Zeitraum
von zwischen 1 und 24 Stunden.
-
Die
Stufe C schließt
die oxidative Hydrolyse eines Amids der Formel 4 zu einem Diol der
Formel 5 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 5 schließt die Stufe
A die Oxidation eines Aldehyds der Formel 1 ein, um eine Carbonsäure der
Formel 2 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart von:
1) einem oxidativen Mittel oder Oxidationsmittel, wie Natriumchlorit;
2) einem Phosphatsalz, vorzugsweise Natriumdihydrogenphosphat; 3)
einem protischen organischen Lösemittel,
vorzugsweise einem Alkohol, wie tert.-Butanol; und 4) einem Alken,
vorzugsweise 2-Methylpropen, bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise
bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 1 Stunde
lang.
-
Die
Stufe B schließt
die Acylierung eines Amins der Formel 3 ein, um ein Amid der Formel
4 zu erhalten. Die Acylierung wird durchgeführt in Gegenwart von: 1) einer
Carbonsäure
der Formel 2; 2) einem Carbonsäurekupplungsmittel,
vorzugsweise einem Diimid, wie 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid,
und einem geeigneten Aktivierungsmittel, das üblich ist für Diimidkupplungsreaktionen,
wie 1-Hydroxybenzotriazol; und 3) einem polaren organischen Lösemittel,
vorzugsweise einem Amid mit niedrigem Molekulargewicht, wie DMF,
bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise bei 25°C, über einen Zeitraum
von zwischen 1 Stunde und 24 Stunden.
-
Die
Stufe C schließt
die oxidative Hydrolyse eines Amids der Formel 4 zu einem Diol der
Formel 5 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 6 schließt die Stufe
A die reduktive Aminierung eines Aldehyds der Formel 1 ein, um ein
Amin der Formel 3 zu erhalten. Die reduktive Aminierung wird durchgeführt in Gegenwart
von: 1) einem Amin der Formel 2; 2) einem Reduktionsmittel, vorzugsweise
einem Hydrid, besonders bevorzugt einem Borhydridsalz, wie Natriumborhydrid;
und 3) einem polaren organischen Lösemittel, vorzugsweise einem
Niederalkanol, wie Ethanol, bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise
von 5°C
bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16 Stunden
lang.
-
Die
Stufe B schließt
die Acylierung eines Amins der Formel 3 ein, um ein Amid der Formel
4 zu erhalten. Die Acylierung wird durchgeführt in Gegenwart von: 1) einer
Carbonsäure
der Formel R2*CO2H,
wobei R2* wie oben definiert ist; 2) einem
Carbonsäurekupplungsreagens,
vorzugsweise einem Diimid, wie 1-(3-Dimethylaminopropyl)-2-ethylcarbodiimidhydrochlorid,
und einem geeigneten Aktivierungsmittel, das üblich ist für Diimidkupplungsreaktionen,
wie 1-Hydroxybenzotriazol; und 3) einem polaren organischen Lösemittel,
vorzugsweise einem Amid mit niedrigem Molekulargewicht, wie DMF,
bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 24 Stunden.
-
Die
Stufe C schließt
die oxidative Hydrolyse eines Amids der Formel 4 zu einem Diol der
Formel 5 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 7 schließt die Stufe
A die Acylierung eines Amins der Formel 1 ein, um ein Carbamat der
Formel 2 zu erhalten. Die Acylierung wird durchgeführt in Gegenwart
von: 1) einem Chlorformiat der Formel ClCO2R3*, wobei R3* so
wie oben definiert ist; 2) einer schwachen Base, vorzugsweise einem
Amin, wie Triethylamin; und 3) einem polaren organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise bei 5°C, über einen
Zeitraum von zwischen 1 Stunde und 24 Stunden.
-
Die
Stufe B schließt
die oxidative Hydrolyse eines Carbamats der Formel 2 zu einem Diol
der Formel 3 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-bezochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 8 schließt die Stufe
A die Olefinierung eines Aldehyds der Formel 1 mit einem Phosphoniumsalz
der Formel 2 ein, um ein Alken der Formel 3 zu erhalten. Die Olefinierung
wird durchgeführt
in Gegenwart von: 1) einer starken Base, vorzugsweise einem Alkalimetallsalz,
wie Kaliumhexamethyldisilazid oder Butyllithium; und 2) einem inerten
organischen Lösemittel,
vorzugsweise einem Kohlenwasserstoff, wie Toluol, oder einem Ether,
wie Tetrahydrofuran, bei einer Temperatur von zwischen -78°C und 25°C, vorzugsweise
bei 0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe B schließt
die oxidative Hydrolyse eines Alkens der Formel 3 zu einem Diol
der Formel 4 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 9 schließt die Stufe
A die Olefinierung eines Aldehyds der Formel 1 mit einem Phosphonat
der Formel 2 ein, um ein Olefin der Formel 3 zu erhalten. Die Olefinierung wird
durchgeführt
in Gegenwart von: 1) einer starken Base, vorzugsweise einem Kaliumsalz,
wie Kaliumhexamethyldisilazid; 2) einem Kronenether, wie 18-Krone-6;
und 3) einem inerten organischen Lösemittel, vorzugsweise einem
Kohlenwasserstoff, wie Toluol, bei einer Temperatur von zwischen
-78°C und
25°C, vorzugsweise bei
0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe B schließt
die oxidative Hydrolyse eines Alkens der Formel 3 zu einem Diol
der Formel 4 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 10 schließt die Stufe
A die Olefinierung eines Aldehyds der Formel 1 mit einem Phosphonat
der Formel 2 ein, um ein Olefin der Formel 3 zu erhalten. Die Olefinierung wird
durchgeführt
in Gegenwart von: 1) einer starken Base, vorzugsweise einem Kaliumsalz,
wie Kaliumhexamethyldisilazid; 2) einem Kronenether, wie 18-Krone-6;
und 3) einen inerten organischen Lösemittel, vorzugsweise einem
Kohlenwasserstoff, wie Toluol, bei einer Temperatur von zwischen
-78°C und
25°C, vorzugsweise bei
0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe B schließt
die oxidative Hydrolyse eines Alkens der Formel 3 an ein Diol der
Formel 4 ein. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
-
Die
in Schema 11 beschriebenen Synthesen können angewendet werden, wenn
B* nicht für -CH(R1)CH=CH-CH=CH2 oder
-CH(R1)CH=CH2 steht.
In Bezug auf die einzelnen Stufen in Schema 11 schließt die Stufe
A die Addition einer Butengruppe an einen Aldehyd der Formel 1 ein,
um einen Alkohol der Formel 2 zu erhalten. Die Addition wird durchgeführt in Gegenwart
von: 1) einem Crotylborreagens, vorzugsweise einem chiralen Crotylborreagens,
besonders bevorzugte einem Z-Crotylboronat, abgeleitet von einem
Diisopropyltartrat; 2) einem optionalen oder gegebenenfalls einem
Trocknungsmittel, wie Molekularsieben; und 3) einem inerten organischen
Lösemittel,
vorzugsweise einem Kohlenwasserstoff, wie Toluol, bei einer Temperatur von
zwischen -100°C
und 5°C,
vorzugsweise bei -78°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe B schließt
die Alkylierung eines Alkohols der Formel 2 ein, um einen Alkohol
der Formel 3 zu erhalten. Die Alkylierung wird durchgeführt in Gegenwart
von: 1) einem reaktiven Benzylierungsmittel, vorzugsweise einem
reaktiven para-Methoxybenzylierungsreagens, wie p-Methoxybenzyl-2,2,2-trichloracetimidat;
2) einer Protonenquelle, vorzugsweise einer Sulfonsäure, wie
Pyridin-p-toluolsulfonat;
und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -78°C
und 25°C,
vorzugsweise bei 0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe C schließt
die zweistufige oxidative Spaltung eines Alkohols der Formel 3 ein,
um einen Aldehyd der Formel 4 zu erhalten. Die erste Stufe der oxidativen
Spaltung wird durchgeführt
in Gegenwart von: 1) einem Dihydroxylierungsreagens, vorzugsweise
einem Osmiumreagens, wie Osmium tetroxid; 2) einem Cooxidationsmittel,
wie N-Morpholin-N-oxid; und 3) einem Gemisch von aprotischen polaren
und protischen Lösemitteln,
wie einem Gemisch von Aceton, Wasser und tert.-Butanol, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang. Die zweite Stufe der oxidativen Spaltung wird durchgeführt in Gegenwart
von: 1) einem Periodatsalz, wie Natriumperiodat; und 2) einem Gemisch
von aprotischen polaren und protischen Lösemitteln, wie einem Gemisch
von Tetrahydrofuran und Wasser, bei einer Temperatur von zwischen
-20°C und 40°C, vorzugsweise
bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe D schließt
die Addition einer Butengruppe an einen Aldehyd der Formel 4 ein,
um einen Alkohol der Formel 5 zu erhalten. Die Addition wird durchgeführt in Gegenwart
von: 1) einem Crotyladditionsreagens, vorzugsweise einem Crotylzinnreagens,
wie Crotyltributylzinn; 2) einer Lewis-Säure, wie Bortrifluoridetherat;
und 3) einem inerten organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -100°C
und 5°C,
vorzugsweise bei -78°C, über einen Zeitraum
von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2 Stunden lang.
-
Die
Stufe E schließt
die Silylierung eines Alkohols der Formel 5 ein, um einen Silylether
der Formel 6 zu erhalten. Die Silylierung wird durchgeführt in Gegenwart
von: 1) einem Silylierungsreagens, vorzugsweise einem tert.-Butyldimethylsilylierungsreagens,
wie tert.-Butyldimethylsilyltriflat; 2) einer schwachen Base, vorzugsweise
einer Stickstoff-enthaltenden Base, besonders bevorzugt einer Pyridinbase,
wie 2,6-Lutidin; und 3) einem inerten organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -100°C und 5°C, vorzugsweise bei -20°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
-
Die
in Schema 12 beschriebenen Synthesen können angewendet werden, wenn
B* nicht für -CH(R1)CH=CH-CH=CH2 oder
-CH(R1)CH=CH2 steht.
In Bezug auf die einzelnen Stufen in Schema 12 schließt die Stufe
A die zweistufige oxidative Spaltung eines Alkens der Formel 1 ein,
um einen Aldehyd der Formel 2 zu erhalten. Die erste Stufe der oxidativen
Spaltung wird durchgeführt
in Gegenwart von: 1) einem Dihydroxylierungsreagens, vorzugsweise
einem Osmiumreagens, wie Osmiumtetroxid; 2) einem Cooxidationsmittel,
wie N-Morpholin-N-oxid; und 3) einem Gemisch von aprotischen polaren
und protischen Lösemitteln,
wie einem Gemisch von Aceton, Wasser und tert.-Butanol, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang. Die zweite Stufe der oxidativen Spaltung wird durchgeführt in Gegenwart
von: 1) einem Periodatsalz, wie Natriumperiodat; und 2) einem Gemisch
von aprotischen polaren und protischen Lösemitteln, wie einem Gemisch von
Tetrahydrofuran und Wasser, bei einer Temperatur von zwischen -20°C und 40°C, vorzugsweise
bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe B schließt
die Reduktion eines Aldehyds der Formel 2 ein, um einen Alkohol
der Formel 3 zu erhalten. Die Reduktion wird durchgeführt in Gegenwart
von: 1) einem Hydridreduktionsmittel, vorzugsweise einem Aluminiumhydrid,
wie Lithiumaluminiumhydrid oder Diisobutylaluminiumhydrid, oder
einem Borhydrid, wie Natriumborhydrid; und 2) einem polaren organischen
Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -100°C
und 40°C,
vorzugsweise von -20°C
bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe C schließt
die Iodierung eines Alkohols der Formel 3 ein, um ein Iodid der
Formel 4 zu erhalten. Die Iodierung wird durchgeführt in Gegenwart
von: 1) einem Iodierungsreagens, wie I2;
2) einer Phosphor enthaltenden Verbindung, wie Triphenylphosphin;
3) einer schwachen Base, vorzugsweise einer schwachen Stickstoff
enthaltenden Base, wie Imidazol; und 4) einem polaren organischen
Lösemittel,
vorzugsweise einem Ester, wie Ethylacetat; bei einer Temperatur
von zwischen -10°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe D schließt
die zweistufige Hydroxylierung eines Alkens der Formel 1 ein, um
einen Alkohol der Formel 5 zu erhalten. Die erste Stufe der Hydroxylierung
wird durchgeführt
in Gegenwart von: 1) einem Boran, wie 9-Borabicylo[3.3.1]nonan;
und 2) einem polaren organischen Lösemittel, vorzugsweise einem Ether,
wie Tetrahydrofuran, bei einer Temperatur von zwischen -10°C und 40°C, vorzugsweise
bei 0°C, über einen
Zeitraum von zwischen 1 Stunde und 48 Stunden, vorzugsweise 24 Stunden
lang. Die zweite Stufe der Hydroxylierung wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem Peroxid, wie
Wasserstoffperoxid; 2) einer starken Alkalibase, vorzugsweise einer
Hydroxidbase, wie Natriumhydroxid; und 3) einem polaren organischen
Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -10°C
und 40°C,
vorzugsweise bei 0°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 1 Stunde
lang.
-
Die
Stufe E schließt
die Iodierung eines Alkohols der Formel 5 ein, um ein Iodid der
Formel 6 zu erhalten. Die Iodierung wird durchgeführt in Gegenwart
von: 1) einem Iodierungsreagens, wie I2;
2) einer Phosphor enthaltenden Verbindung, wie Triphenylphosphin;
3) einer schwachen Base, vorzugsweise einer Stickstoff enthaltenden
Base, wie Imidazol; und 4) einem polaren organischen Lösemittel,
vorzugsweise einem Ester, wie Ethylacetat; bei einer Temperatur
von zwischen -10°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe F schließt
die zweistufige Iodierung eines Alkens der Formel 1 ein, um ein
Iodid der Formel 6 zu erhalten. Die erste Stufe der Iodierung wird
durchgeführt
in Gegenwart von: 1) einem Boran, wie 9-Borabicyclo[3.3.1]nonan;
und 2) einem polaren organischen Lösemittel, vorzugsweise einem
Ether, wie Tetrahydrofuran, bei einer Temperatur von zwischen -10°C und 40°C, vorzugsweise
bei 0°C, über einen
Zeitraum von zwischen 1 Stunde und 48 Stunden, vorzugsweise 24 Stunden
lang. Die zweite Stufe der Iodierung wird durchgeführt in Gegenwart
von I2; und 2) einem polaren organischen
Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -10°C
und 40°C,
vorzugsweise bei 0°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden.
-
Die
Stufe G schließt
die Phosphinadditionsreaktion eines Iodids der Formel 6 ein, um
ein Phosphoniumiodidsalz der Formel 7 zu erhalten. Die Phosphinadditionsreaktion
wird durchgeführt
in Gegenwart von: 1) einem Phosphorreagens, wie Triphenylphosphin;
2) einer Base, vorzugsweise einer Aminbase, wie Diisopropylethylamin;
und 3) einem organischen Lösemittel,
vorzugsweise einem polaren aprotischen Lösemittel, wie Acetonitril,
bei einer Temperatur von zwischen 25°C und 150°C, vorzugsweise bei 90°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 18 Stunden
lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 13 schließt die Stufe
A die Alkylierung eines Alkohols der Formel 1 ein, um einen Ether
der Formel 2 zu erhalten. Die Alkylierung wird durchgeführt in Gegenwart
von: 1) einem reaktiven Benzylierungsreagens, vorzugsweise einem
reaktiven para-Methoxybenzylierungsreagens,
wie p-Methoxybenzyl-2,2,2-trichloracetimidat; 2) einer Protonenquelle,
vorzugsweise einer Sulfonsäure, wie
Pyridinium-p-toluolsulfonat; und 3) einem polaren organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 25°C, vorzugsweise bei 0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
stunden lang.
-
Die
Stufe B schließt
die Reduktion eines Ethers der Formel 2 ein, um einen Alkohol der
Formel 3 zu erhalten. Die Reduktion wird durchgeführt in Gegenwart
von: 1) einem Metallhydrid, vorzugsweise einem Aluminiumhydrid,
wie Lithiumaluminiumhydrid oder Diisobutylaluminiumhydrid; und 2)
einem polaren organischen Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -100°C
und 10°C,
vorzugsweise von -78°C
bis 0°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 2 Stunden
lang.
-
Die
Stufe C schließt
die Alkylierung eines Alkohols der Formel 3 ein, um einen Ether
der Formel 4 zu erhalten. Die Alkylierung wird durchgeführt in Gegenwart
von: 1) einem Alkohol der Formel A*OH, wobei A* so wie oben definiert
ist; 2) einem Kupplungsreagens, wie Diethylazodicarboxylat; 3) einem
Phosphin, wie Triphenylphosphin; und 4) einem polaren organischen
Lösemittel,
wie Tetrahydrofuran, bei einer Temperatur von zwischen -78°C und 60°C, vorzugsweise
zwischen -20°C
und 40°C, über einen
Zeitraum von zwischen 2 Stunden und 72 Stunden, vorzugsweise 16
Stunden lang.
-
Die
Stufe D schließt
die oxidative Hydrolyse eines Ethers der Formel 4 ein zu einem Alkohol
der Formel 5. Die oxidative Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vor zugsweise einem Chinon, wie 2,3-Dichlor-5,6-dicyano-1,4-benzochinon;
2) Wasser; und 3) einem polaren organischen Lösemittel, vorzugsweise einem
halogenierten Kohlenwasserstoff, wie Methylenchlorid, bei einer
Temperatur von zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 1 Stunde und 72 Stunden, vorzugsweise 1 Stunde
lang.
-
Die
Stufe E schließt
die Oxidation eines Alkohols der Formel 5 ein, um einen Aldehyd
der Formel 6 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart
von: 1) einem Oxidationsreagens, vorzugsweise einem milden Oxidationsreagens,
wie den Kombinationen von Oxalylchlorid, DMSO und Triethylamin;
einem Schwefeltrioxid-Pyridin-Komplex, DMSO und Triethylamin; und
dem freiem Radikal 2,2,6,6-Tetramethyl-1-piperidinyloxy
und Diacetoxyiodbenzol; und 2) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise von -20°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 14 schließt die Stufe
A die Oxidation eines Aldehyds der Formel 1 ein, um eine Carbonsäure der
Formel 2 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart von:
1) einem Oxidationsmittel, wie Natriumchlorit; 2) einem Phosphatsalz,
vorzugsweise Natriumdihydrogenphosphat; 3) einem protischen organischen
Lösemittel,
vorzugsweise einem Alkohol, wie tert.-Butanol; und 4) einem Alken,
vorzugsweise 2-Methylpropen, bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise bei
25°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 1 Stunde
lang.
-
Die
Stufe B schließt
die reduktive Aminierung eines Aldehyds der Formel 1 ein, um ein
Amin der Formel 3 zu erhalten. Die reduktive Aminierung wird durchgeführt in Gegenwart
von: 1) einem Amin der Formel R5NH2, wobei R5 so wie
oben definiert ist; 2) einem Reduktionsmittel, vorzugsweise einem
Hydrid, besonders bevorzugt einem Borhydridsalz, wie Natriumborhydrid;
und 3) einem polaren organischen Lösemittel, vorzugsweise einem
protischen organischen Lösemittel,
wie Ethanol, bei einer Temperatur von zwischen 0°C und 40°C, vorzugsweise von 5°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
Die
Stufe C schließt
die Azidierung eines Iodids der Formel 4 ein, um ein Azid der Formel
5 zu erhalten. Die Azidierung wird durchgeführt in Gegenwart von: 1) einem
Azidsalz, wie Natriumazid; und 2) einem polaren organischen Lösemittel,
wie DMF, bei einer Temperatur von zwischen 25°C und 150°C, vorzugsweise bei 90°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
Die
Stufe D schließt
die Reduktion eines Azids der Formel 5 ein, um ein Amin der Formel
6 zu erhalten. Die Azidierung wird durchgeführt in Gegenwart von: 1) einem
Reduktionsmittel, vorzugsweise einem Phosphin, wie Triphenylphosphin,
in Gegenwart von Wasser; und 2) einem polaren organischen Lösemittel,
wie Tetrahydrofuran, bei einer Temperatur von zwischen 0°C und 100°C, vorzugsweise
von 25°C
bis 60°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 15 schließt die Stufe
A die Reduktion eines Aldehyds der Formel 1 ein, um einen Alkohol
der Formel 2 zu erhalten. Die Reduktion wird durchgeführt in Gegenwart
von: 1) einem Hydridreduktionsmittel, vorzugsweise einem Aluminiumhydrid,
wie Lithiumaluminiumhydrid oder Diisobutylaluminiumhydrid, oder
einem Borhydrid, wie Natriumborhydrid; und 2) einem polaren organischen
Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -100°C
und 40°C,
vorzugsweise von -20°C
bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe B schließt
die Silylierung eines Alkohols der Formel 2 ein, um einen Silylether
der Formel 3 zu erhalten. Die Silylierung wird durchgeführt in Gegenwart
von: 1) einem Silylierungsreagens, vorzugsweise einem tert.-Butyldimethylsilylierungsreagens,
wie tert.-Butyldimethylsilyltriflat; 2) einer schwachen Base, vorzugsweise
einer Stickstoff enthaltenden Base, besonders bevorzugt einer Pyridinbase,
wie 2,6-Lutidin; und 3) einem inerten organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -100°C und 5°C, vorzugs weise bei -20°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe C schließt
die reduktive Hydrolyse eines Silylethers der Formel 3 ein, um einen
Alkohol der Formel 4 zu erhalten. Die reduktive Hydrolyse wird durchgeführt in Gegenwart
von: 1) einem Lewissäurehydrid, vorzugsweise
einem Aluminiumhydrid, wie Diisobutylaluminiumhydrid; und 2) einem
polaren organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -100°C und 5°C, vorzugsweise bei -78°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 1
Stunde lang.
-
Die
Stufe D schließt
die Oxidation eines Alkohols der Formel 4 ein, um einen Aldehyd
der Formel 5 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart
von: 1) einem Oxidationsreagens, vorzugsweise einem milden Oxidationsreagens,
wie den Kombinationen von Oxalylchlorid, DMSO und Triethylamin;
Schwefeltrioxid-Pyridin-Komplex, DMSO und Triethylamin; und dem
freien Radikal 2,2,6,6-Tetramethyl-1-piperidinyloxy und
Diacetoxyiodbenzol; und 2) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise von -20°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe E schließt
die Olefinierung eines Aldehyds der Formel 5 ein, um ein Dien der
Formel 6 zu erhalten. Die Olefinierung wird durchgeführt in Gegenwart
von: 1) einem halogenierten Silylpropen, wie 1-Brom-1-trimethylsilyl-2-propen;
2) einem Chrom(II)-reagens, wie Chrom(II)-chlorid; und 3) einem
polaren organischen Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -100°C
und 40°C,
vorzugsweise bei 20°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe F schließt
die Hydrolyse eines Diens der Formel 6 ein, um einen Alkohol der
Formel 7 zu erhalten. Die Hydrolyse wird durchgeführt in Gegenwart
von: 1) einer protischen Säure,
vorzugsweise einem Wasserstoffhalogenid, wie Chlorwasserstoffsäure; und
2) einem polaren organischen Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -10°C
und 40°C,
vorzugsweise bei 20°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 1
Stunde lang.
-
Die
Stufe G schließt
die Oxidation eines Alkohols der Formel 7 ein, um einen Aldehyd
der Formel 8 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart
von: 1) einem Oxidationsreagens, vorzugsweise einem milden Oxidationsreagens,
wie den Kombinationen von Oxalylchlorid, DMSO und Triethylamin;
einem Schwefeltrioxid-Pyridin-Komplex, DMSO und Triethylamin; und
dem freien Radikal 2,2,6,6-Tetramethyl-1-piperidinyloxy
und Diacetoxyiodbenzol; und 2) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise von -20°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe H schließt
die Propionatadditionsreaktion eines Aldehyds der Formel 8 ein,
um ein Imid der Formel 10 zu erhalten. Die Propionatadditionssreaktion
wird durchgeführt
in Gegenwart von: 1) einem Propanimid der Formel 9; 2) einer Lewis-Säure, vorzugsweise
einer Bor enthaltenden Lewis-Säure,
wie Dibutylbortriflat; 3) einer schwachen Base, vorzugsweise einer
Aminbase, wie Triethylamin; und 4) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -100°C und 20°C, vorzugsweise von -78°C bis 0°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
Die
Stufe I schließt
die Silylierung eines Alkohols der Formel 10 ein, um einen Silylester
der Formel 11 zu erhalten. Die Silylierung wird durchgeführt in Gegenwart
von: 1) einem Silylierungsreagens, vorzugsweise einem tert.-Butyldimethylsilylierungsreagens,
wie tert.-Butyldimethylsilyltriflat; 2) einer schwachen Base, vorzugsweise
einer Stickstoff enthaltenden Base, besonders bevorzugt einer Pyridinbase,
wie 2,6-Lutidin; und 3) einem inerten organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -100°C und 5°C, vorzugsweise bei -20°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 2
Stunden lang.
-
-
In
Bezug auf die einzelnen Stufen in Schema 16 schließt die Stufe
A die Hydrolyse eines Imids der Formel 1 ein, um eine Carbonsäure der
Formel 2 zu erhalten. Die Hydrolyse wird durchgeführt in Gegenwart von:
1) einer starken Base, vorzugsweise einem Hydroxidsalz, wie Lithiumhydroxid;
2) einem Oxidationsmittel, vorzugsweise einem Peroxid, wie Wasserstoffperoxid;
und 3) einem polaren organischen Lösemittel, vorzugsweise einem
Ether, wie Tetrahydrofuran, bei einer Temperatur von zwischen -20°C und 40°C, vorzugsweise von
-10°C bis
25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 18
Stunden lang.
-
Die
Stufe B schließt
die Reduktion eines Imids der Formel 1 ein, um einen Alkohol der
Formel 3 zu erhalten. Die Reduktion wird durchgeführt in Gegenwart
von: 1) einem Hydridreduktionsmittel, wie Lithiumborhydrid; 2) einem
protischen organischen Lösemittel,
vorzugsweise einem Niederalkanol, wie Ethanol; und 3) einem polaren
organischen Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur von
zwischen -20°C
und 40°C,
vorzugsweise bei 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 18
Stunden lang.
-
Die
Stufe C schließt
die Acylierung eines Imids der Formel 1 ein, um ein Amid der Formel
4 zu erhalten. Die Acylierung wird durchgeführt in Gegenwart von: 1) N,O-Dimethylhydroxylaminhydrochlorid;
2) einem aluminiumorganischen Reagens, wie Trimethylaluminium; und
3) einem polaren organischen Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -78°C
und 40°C,
vorzugsweise bei -20°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 2 Stunden
lang.
-
Die
Stufe D schließt
die Oxidation eines Alkohols der Formel 3 ein, um einen Aldehyd
der Formel 5 zu erhalten. Die Oxidation wird durchgeführt in Gegenwart
von: 1) einem Oxidationsmittel, vorzugsweise einem milden Oxidationsreagens,
wie den Kombinationen von Oxalylchlorid, DMSO und Triethylamin;
einem Schwefeltrioxid-Pyridin-Komplex, DMSO und Triethylamin; und
dem freien Radikal 2,2,6,6-Tetramethyl-1-piperidinyloxy
und Diacetoxyiodbenzol und 2) einem inerten organischen Lösemittel,
vorzugsweise einem polaren organischen Lösemittel, wie Methylenchlorid,
bei einer Temperatur von zwischen -78°C und 40°C, vorzugsweise von -20°C bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 3
Stunden lang.
-
Die
Stufe E schließt
die Reduktion eines Amids der Formel 4 ein, um einen Aldehyd der
Formel 5 zu erhalten. Die Reduktion wird durchgeführt in Gegenwart
von: 1) einem Metallhydrid, vorzugsweise einem Aluminiumhydrid,
wie Lithiumaluminiumhydrid oder Diisobutylaluminiumhydrid und 2)
einem polaren organischen Lösemittel,
vorzugsweise einem Ether, wie Tetrahydrofuran, bei einer Temperatur
von zwischen -100°C
und 10°C,
vorzugsweise von -78°C
bis 0°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 2 Stunden
lang.
-
Die
Stufe F schließt
die reduktive Aminierung eines Aldehyds der Formel 5 ein, um ein
Amin der Formel 6 zu erhalten. Die reduktive Aminierung wird durchgeführt in Gegenwart
von: 1) einem Amin der Formel R5NH2, wobei R5 wie oben
definiert ist; 2) einem Reduktionsmittel, vorzugsweise einem Hydrid,
besonders bevorzugt einem Borhydridsalz, wie Natriumborhydrid und
3) einem polaren organischen Lösemittel,
vorzugsweise einem protischen organischen Lösemittel, wie Ethanol, bei
einer Temperatur von zwischen 0°C
und 40°C, vorzugsweise
von 5°C
bis 25°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
Die
Stufe G schließt
die Mesylierung eines Alkohols der Formel 3 ein, um ein Mesylat
der Formel 7 zu erhalten. Die Mesylierung wird durchgeführt in Gegenwart
von: 1) Methansulfonylchlorid; 2) einer Base, vorzugsweise einer
Aminbase, wie Triethylamin; und 3) einem polaren organischen Lösemittel,
vorzugsweise einem halogenierten Kohlenwasserstoff, wie Methylenchlorid,
bei einer Temperatur von zwischen -20°C und 40°C, vorzugsweise von 0°C bis 5°C, über einen
Zeitraum von zwischen 10 Minuten und 8 Stunden, vorzugsweise 2 Stunden
lang.
-
Die
Stufe H schließ die
Azidierung eines Mesylats der Formel 7 ein, um ein Azid der Formel
8 zu erhalten. Die Azidierung wird durchgeführt in Gegenwart von: 1) einem
Azidsalz, wie Natriumazid; und 2) einem polaren organischen Lösemittel,
wie DMF, bei einer Temperatur von zwischen 25°C und 150°C, vorzugsweise bei 90°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
Die
Stufe I schließt
die Reduktion eines Azids der Formel 8 ein, um ein Amin der Formel
9 zu erhalten. Die Azidierung wird durchgeführt in Gegenwart von: 1) einem
Reduktionsmittel, vorzugsweise einem Phosphin, wie Triphenylphosphin,
in Gegenwart von Wasser; und 2) einem polaren organischen Lösemittel,
wie Tetrahydrofuran, bei einer Temperatur von zwischen 0°C und 100°C, vorzugsweise
von 25°C
bis 60°C, über einen
Zeitraum von zwischen 10 Minuten und 48 Stunden, vorzugsweise 16
Stunden lang.
-
Obwohl
das Produkt von jeder oben beschriebenen Reaktion gereinigt werden
kann, wenn dies gewünscht
wird, durch herkömmliche
Techniken, wie Chromatographie oder Umkristallisation (wenn es ein
Feststoff ist), wird das Rohprodukt einer Reaktion vorteilhaft in
der folgenden Reaktion ohne Reinigung eingesetzt.
-
Wie
den Leuten vom Fach ersichtlich ist, enthalten die Verbindungen
der Formel I asymmetrische Kohlenstoffatome. Es sollte daher verstanden
werden, dass die einzelnen Stereoisomere unter den Schutzbereich dieser
Erfindung fallend betrachtet werden.
-
Insbesondere
erstreckt sich die vorliegende Erfindung auf ein Verfahren zur Herstellung
einer Verbindung der Formel I
wobei
A steht für -CH
2N(R
2)CH
2-,
-CH
2N(R
2)C(O)-,
-C(O)N(R
2)CH
2-,
-CH
2N(CO
2R
3)CH
2- oder -CH
2N(COR
2)CH
2-;
B steht für -CH(R
1)CH=CHCH=CH
2, -CH(R
2)R
1, -CH(R
1)CH=CHR
2, -CH(R
1)CH=CHC(O)OR
2, -CH(R
1)CH=CHC(O)N(R
1)R
2, -CH(R
1)CH
2OR
2 oder
Ar;
C steht für
H, -C(O)N(R
1)R
2,
-C(O)NHCH
2(CH
2)
nN(CH
3)
2 oder
-C(O)NHCH
2(CH
2)
n-4-morpholino;
R
1 steht
für H oder
(C
1-C
6)-Alkyl;
R
2 steht für
H, (C
1-C
6)-Alkyl,
(C
2-C
6)-Alkenyl,
(C
2-C
6)-Alkinyl,
(C
1-C
6)-Alkyl-Ar
oder Ar;
R
3 steht für (C
1-C
6)-Alkyl, (C
1-C
6)-Alkyl-Ar oder Ar;
Ar für einen
aromatischen oder heteroaromatischen Ring steht, der ausgewählt ist
aus
R
4 und R
5 unabhängig
stehen
für H,
(C
1-C
6)-Alkyl, OH,
O(C
1-C
6)-Alkyl,
OCH
2(CH
2)
nOH, O(CH
2)
nCO
2H, OCH
2(CH
2)
nN(CH
3)
2, OCH
2(CH
2)
n-4-morpholino,
F, Cl, Br oder CF
3; und
n für 1 oder
2 steht;
wobei eine Verbindung der Formel II
-
-
wobei
die Gruppen A*, B* und C*, den Gruppen A, B und C entsprechen, wie
für die
Verbindung der Formel 1 definiert, mit der Ausnahme, dass in dem
Fall, dass diese Gruppen freie Hydroxygruppen enthalten, dann die
Kennzeichnung mit dem Stern (z.B. A*) angibt, dass diese Gruppen
geschützt
sind mit säurelabilen Schutzgruppen
(z.B. TBS), und PG für
eine Schutzgruppe steht, z.B. TBS, hydrolysiert und cyclisiert wird
durch Umsetzung mit einer protischen Säure, vorzugsweise einer wässrigen
protischen Säurelösung, vorzugsweise einer
wässrigen
Halogenwasserstofflösung,
wie wässrigem
Chlorwasserstoff, in einem polaren organischen Lösemittel, vorzugsweise einem
Gemisch von polaren organischen Lösemitteln, vorzugsweise einem
Gemisch eines aliphatischen Alkohols und eines Ethers, wie Methanol
und Tetrahydrofuran, bei einer Temperatur von zwischen -20°C und 40°C, vorzugsweise
zwischen 20°C
bis 25°C, über einen
Zeitraum von 8 Stunden und 7 Tagen, vorzugsweise zwischen 16 und
72 Stunden, besonders bevorzugt zwischen 24 und 48 Stunden; und wobei
jegliche Schutzgruppen in einem geschützten Derivat von einer Verbindung
der Formel I anschießend entfernt
werden.
-
Wie
oben angegeben, sind alle Verbindungen der Formel I Antitumormittel
und sind daher nützlich
bei der Hemmung oder Inhibierung des Wachstums von verschiedenen
Lymphomen, Sarkomen, Karzinomen, Myelomen und Leukämiezelllinien.
Die Antitumoraktivität
oder -wirksamkeit der Verbindung der Formel I kann gezeigt werden
durch den Einsatz des Anchorage Dependent Growth Monolayer Assays
(ADGMA), der den Wachstumshemmeffekt von Testverbindungen auf die
Proliferation von adhärenten
oder anhaftenden Zellmonoschichten misst. Dieser Assay oder Test
wurde an den 60 Zelllinientest, der von dem National Cancer Institute
(NCI) verwendet wird, mit den folgenden Modifikationen angepasst: 1)
Vier Zelllinien stellvertretend für die wichtigen Tumortypen,
nämlich
das MIP 101 Kolonkarzinom, HCT 116 Kolonkarzinom, 1A9 Ovariarkarzinom und
1A9PTX22 Oariarkarzinom, wurden eingesetzt; und 2) ein Tetrazliumderivat,
nämlich
MTT, wurde verwendet, um die Zelldichte zu bestimmen.
-
Der
ADGMA vergleicht die Anzahl von lebensfähigen Zellen nachdem sie 3
Tage lang einer Testverbindung ausgesetzt wurden in Bezug auf die
Anzahl von Zellen, die zu dem Zeitpunkt vorliegen als die Testverbindung
zugegeben wurde. Die Zellenlebensfähigkeit wird gemessen unter
Verwendung eines Tetrazoliumderivats, nämlich 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl-tetrazoliumbromid
(MTT), das metabolisch in Gegenwart eines Elektronenkupplungsagenzes
(PMS; Phenazinmethosulfat) reduziert wird durch lebensfähige Zellen
zu einem wasserlöslichen
Formazanderivat. Die Absorption bei 540 nm (A540) des Formazanderivats ist
proportional zu der Anzahl von lebenden Zellen. Der IC50-Wert
für eine
Testverbindung ist die Konzentration einer Verbindung, die benötigt wird,
um die Zellzahl am Schluss auf 50% der Endkontrollzellanzahl zu
reduzieren. Wenn die Zellproliferation inhibiert oder gehemmt wird,
definiert der Assay oder Test die Verbindungen als cytostatisch
(Zellenanzahl nach 3 Tagen Verbindungsinkubation > Zellenanzahl zum Zeitpunkt
der Zugabe der Verbindung) oder cytotoxisch (Zellenanzahl nach 3
Tagen Verbindungsinkubation < Zellenanzahl
zum Zeitpunkt der Zugabe der Verbindungen).
-
Die
HCT 116 Kolonkarzinomzelllinie wurde erhalten von der Amerikanischen
Kultursammlung (ATCC (American Type Culture Collection), Rockville,
MD). Die MIP 101 Kolonkarzinomzelllinie wurde erhalten von Dr. Robert
Kramer (Bristol Meyers Squibb) und wurde vorgehend beschrieben (RM
Niles, SA Wilhelm, GD JR Steele, B Burke, T Christensen, D Dexter,
MJ O'Brien, P Thomas,
N Zamcheck, Isolation and characterization of an undifferentiated
human colon carcinoma cell line (MIP-101), Cancer Invest. 1987;
5(6): 545-552.). Die 1A9 und die 1A9PTX22 Ovariartumorzelllinien
wurden erhalten von Dr. Tito Fojo, Medicine Branch, Division of Clinical
Sciences, National Cancer Institute, National Institutes of Health,
Bethesda, MD 20892. Die 1A9 Zelllinie ist ein Klon der Ovariarkarzinomzelllinie
A2780 (P Giannakakou, DL Sackett, Y-K Kang, Z Zhan, JTM Buters,
T Fojo, MS Poruchynsky, Paclitaxel-resistant human ovarian cancer
cells have mutant δ-tubulins
that impaired paclitaxel-driven polymerization, J. Biol. Chem. 1997,
272(4): 17118-17125). Die 1A9PTX22 Unterlinie wurde isoliert als
ein einzelner Klon aus der 1A9 Zelllinie in einer Einzelstufenselektion
indem sie gegen 5 ng/ml Paclitaxel in Gegenwart von 5 μg/ml Verapamil
ausgesetzt wurde. Alle Zelllinien wurden verwendet zwischen Durchgängen 4-20
nach einem Tauen. Die MIP-101 Kolonkarzinom-, HCT 116 Kolonkarzinom-,
1A9 Ovariarkarzinom- und 1A9PTX22 Ovariarkarzinomzelllinien werden
erhalten und plattiert in RPMI 1640 Medium, das 10% fetales Rinderserum
enthielt.
-
Die
Zellen werden trypsiniert und gezählt unter Verwendung eines
Hemacytometers, um Zellkonzentrationen zu bestimmen. Die Zellen
wurden dann plattiert in ihren jeweiligen Erhaltungsmedien (200 μl/Vertiefung)
in Platten mit 96 Vertiefungen bei den folgenden Konzentrationen:
MIP-101, 2000 Zellen/Vertiefung; HCT 116, 2000 Zellen/Vertiefung;
1A9, 10000 Zellen/Vertiefung und 1A9PTX22, 10000 Zellen/Vertiefung.
Die Anzahl von Zellen/Vertiefung wurde in vorhergehenden Experimenten
bestimmt und führte
zu 75-90% Konfluenz am Tag 4 nach dem Plattieren. Die Zelldichten
am Anfang, einen Tag nach dem Plattieren getestet, weisen ungefähr 0,10-0,20
A540 größere Absorptionseinheiten
auf als die bloßen
Medien oder Blindmedien. Platten mit 96 Vertiefungen werden am Tag
0 beimpft, und die Test verbindungen werden am Tag 1 zugegeben. Eine Platte „Zeit 0" wurde erzeugt, die
nur Medien in der Reihe A erhielt und eine Zelllinie/Reihe in den
Reihen B-E. Die Platte „Zeit
0" wurde 24 Stunden
nach dem Plattieren verarbeitet (zu der Zeit, als die Arzneimittel
zu den experimentellen Platten gegeben wurden), wie folgt: 5 μl der MTT
Stammlösung
(0,5 mg/ml in PBS) wurden zu jeder Vertiefung gegeben und dann 3
Stunden lang bei 37°C
inkubiert, 5% CO2, in einer befeuchteten
Umgebung. Die Medien wurden dann vorsichtig und vollständig entfernt.
Die Platten ließ man
im Dunkeln trocknen. DMSO (Dimethylsulfoxid) wurde zu jeder Vertiefung
(100 μl/Vertiefung)
zugegeben, und die Platten wurden 2 Stunden lang auf einen Orbitalschüttler gegeben.
Die Platten wurden eingelesen in den Leser für eine Platte mit 96 Vertiefungen
bei 540 nm in einem Plattenleser von Molecular Devices unter Verwendung
der Softmax Version 2.35 im Absorptionsmodus-Endpoint L-1, unter
Verwendung von DMSO als Leerwert oder Blindwert. Ein Tag nach dem
Plattieren wurden die Testverbindungen (in einer Endverdünnung von
1:10) zu den Testplatten gegeben und nachfolgend 10 Mal reihenverdünnt. Die
Kontrollplatte erhielt nur eine 1:10 Verdünnung des Lösemittels (10% DMSO/90% RPMI
1640). 3 Tage nach der Zugabe der Testverbindungen wurden alle experimentellen
Platten und die Kontrollplatte verarbeitet, wie oben beschrieben
für die
Platte „Zeit
0". Die IC50-Werte wurden bestimmt aus graphischen
Auftragungen des prozentuellen Nettowachstums als Funktion einer
Verbindungskonzentration. Das prozentuelle Nettowachstum wird berechnet
als (Zelle + Arzneimittel A540 – Anfangs-540/Zelle
+ Arzneimittelträger
540 – Anfangs-540).
-
Die
folgenden IC
50-Werte (Mittelwert von 2 oder
mehr getrennten Experimenten) in μM
wurden erhalten:
-
Die
genaue Dosierung der Verbindungen der Formel I, die eingesetzt wird
zum Inhibieren oder Hemmen von Tumoren, hängt von mehreren Faktoren ab,
die den Patienten, die Natur und den Schwerheitsgrad des Zustands,
der behandelt wird, die Art oder den Weg der Verabreichung und die
besondere eingesetzte Verbindung einschließen. Im Allgemeinen wird jedoch
eine zufrieden stellende Inhibierung oder Hemmung von Tumoren erhalten,
wenn eine Verbindung der Formel I parenteral verabreicht wird, z.B.
intraperitoneal, intravenös,
intramuskulär,
subkutan, intratumoral oder rektal oder enteral, wie z.B. oral,
vorzugsweise intravenös oder
oral, besonders bevorzugt intravenös mit einer Einzeldosierung
von 1-300 mg/kg Körpergewicht
pro Zyklus (Zyklus = 3-6 Wochen) oder für die meisten größeren Primaten,
in einer Einzeldosierung von 50-5000 mg pro Behandlungszyklus. Eine
bevorzugte intravenöse
Einzeldosierung pro 3-6 Wochen Behandlungszyklus beträgt 1-75
mg/kg Körpergewicht
oder für
die meisten größeren Primaten
eine tägliche
Dosierung von 50-1500 mg. Eine typische intravenöse Dosierung beträgt 45 mg/kg
einmal alle 3 Wochen.
-
In
der Regel wird eine geringe Dosis am Anfang verabreicht, und die
Dosierung wird schrittweise erhöht,
bis die optimale Dosierung für
den Patienten unter der Behandlung bestimmt ist. Die obere Grenze
der Dosierung ist die, die erhalten wird durch Nebeneffekte, und
kann bestimmt werden durch Versuche an dem Patienten, der behandelt
wird.
-
Die
Verbindungen der Formel I können
kombiniert werden mit ein oder mehreren pharmazeutisch annehmbaren
Trägern,
und, gegebenenfalls, ein oder mehreren anderen herkömmlichen
pharmazeutischen Hilfsmitteln und enteral verabreicht werden, z.B.
oral, in Form von Tabletten, Kapseln, Caplets, etc., oder parenteral,
z.B. intraperitoneal oder intravenös, in der Form von sterilen
injizierbaren Lösungen
oder Suspensionen. Die enteralen oder parenteralen Zusammensetzungen
können
hergestellt werden durch herkömmliche Mittel
oder Maßnahmen.
-
Die
Verbindungen der Formel I können
formuliert werden in enterale und parenterale pharmazeutische Zusammensetzungen,
die eine Menge der wirksamen Substanz oder des Wirkstoffs enthalten,
die wirksam ist zum Inhibieren oder Hemmen von Tumoren, wobei solche
Zusammensetzungen in Einheitsdosierungsform und solchen Zusammensetzungen
einen pharmazeutisch annehmbaren Träger umfassen.
-
Die
Verbindungen der vorliegenden Erfindung können allein verabreicht werden
oder zusammen oder in Kombination mit ein oder mehreren anderen
therapeutischen Mitteln, wobei eine mögliche Kombinationstherapie,
die Form von fixen Kombinationen einnimmt oder die Verabreichung
einer Verbindung der Erfindung und ein oder mehrerer anderer therapeutischer
Mittel, die abgestuft gegeben werden oder unabhängig voneinander gegeben werden,
oder die kombinierte Verabreichung von fixen Kombinationen und ein
oder mehreren anderen therapeutischen Mitteln. Die Verbindungen
der Erfindung können
insbesondere verabreicht werden z.B. im Fall einer Tumortherapie
in Kombination mit einer Chemotherapie, Strahlentherapie, Immuntherapie,
eines operativen Eingriffs oder einer Kombination von diesen. Eine
Langzeittherapie oder langfristige Therapie ist gleichfalls möglich wie
eine Unterstützungstherapie
im Zusammenhang mit anderen Behandlungsstrategien, wie oben beschrieben.
Andere mögliche
Behandlungen sind eine Therapie, um den Status des Patienten beizubehalten
nach einer Tumorregression oder sogar chemopräventiven Therapie, wie z.B.
bei Patienten, bei denen ein Risiko besteht.
-
Therapeutische
Mittel für
eine mögliche
Kombination sind insbesondere ein oder mehrere antiproliferative,
cytostatische oder cytotoxische Verbindungen, z.B. ein chemotherapeutisches
Mittel, oder mehrere Mittel, die ausgewählt sind aus der Gruppe, die
einschließt
aber nicht beschränkt
ist auf einen Inhibitor der Polyaminbiosynthese, einen Inhibitor
einer Proteinkinase, insbesondere einer Serin/Threoninproteinkinase,
wie Proteinkinase C, oder einer Tyrosinproteinkinase, wie der EGF-Rezeptortyrosinkinase,
z.B. PKI166, der VEGF Rezeptortyrosinkinase, z.B. PTK787, oder den
PDGF Rezeptortyrosinkinase, z.B. STI571, ein Cytokin, einen negativen
Wachstumsregulator, wie TGF-β oder
IFN-β, einen
Aromataseinhibitor, z.B. Letrozol oder Anastrozol, einen Inhibitor
der Wechselwirkung einer SH2 Domäne
mit einem phosphorylierten Protein, Antiöstrogene, Topoisomerase I Inhibitoren,
wie Irinotecan, Topoisomerase II Inhibitoren, Microtubuli-wirksame
Mittel, z.B. Paclitaxel, (+)-Discodermolid oder ein Epothilon, wie
Epothilon B, Alkylierungsmittel, antineoplastische Antimetaboliten,
wie Gemcitabin oder Capecitabin, Platinverbindungen, wie Carboplatin
oder Cisplatin, anti-angiogene Verbindungen, Gonado relinagonisten,
Anti-Androgene, Bisphosphonate, z.B. AREDIA® oder
ZOMETA®,
und Trastuzumab. Die Struktur der wirksamen Mittel, die durch Codenummern
identifiziert werden, generische oder Handelsnamen, können entnommen
werden aus der aktuellen Ausgabe des Standardwerks "The Merck Index" oder aus Datenbanken,
z.B. Patents International (z.B. IMS World Publications).
-
Außerdem betrifft
die vorliegende Erfindung die Verwendung einer Verbindung der Formel
I, Ia, Ib oder Ic bei der Behandlung des tierischen oder menschlichen
Körpers
oder bei der Herstellung einer pharmazeutischen Zubereitung für die Behandlung
einer Tumorerkrankung.
-
Die
folgenden Beispiele zeigen stellvertretende Verbindungen, die durch
diese Erfindung umfasst werden, und deren Synthese. Es sollte jedoch
klar verstanden werden, dass dies nur zu Zwecken der Veranschaulichung
dient.
-
Beispiel 1: Synthese von (2R,3S,4R,5S,7S,8Z)-13-[[(2R,3S,4S,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-hydroxy-2,4,6-timethyl-1-oxo-7,9-decadienyl]methylamino]-3,5,7,11-tetrahydroxy-2,4,10,12-tetramethyl-8-tridecensäure-δ-lacton.
-
a) Herstellung von 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-5-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-1-(methylamino)-L-arabinitol.
-
Eine
Lösung
von 2,4-Dideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-5-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-arabinose
(2,8 g, 7,37 mmol) in THF (10 ml) wird langsam zu einer gerührten Lösung von Methylamin
(14,7 ml, 29,5 mmol) in THF bei Raumtemperatur gegeben. Nach einem
Rühren
bei Raumtemperatur über
einen Zeitraum von 20 Minuten werden Natriumtriacetoxyborhydrid
(2,3 g, 11,05 mmol), gefolgt von Essigsäure (10 Tropfen) bei Raumtemperatur
zugegeben. Nach einem Rühren
bei Raumtemperatur über Nacht
wird das Reaktionsgemisch mit EtOAc (100 ml) verdünnt und
mit gesättigtem
NaHCO3 (2 × 30 ml), H2O (50
ml), Salzlösung
(50 ml) gewaschen und über
Na2SO4 getrocknet.
Nach Entfernung des Lösemittels
im Vakuum wird der Rückstand
an Kieselgel mit MeOH/CH2Cl2 (1/99)
chromatographiert, um die gewünschte
Verbindung als blasses viskoses Öl
zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 7,25
(d, J = 8,24 Hz, 2H), 6,87 (d, J = 8,70 Hz, 2H), 4,42 (dd, J = 18,76, 11,60
Hz, 2H), 3,80 (s, 3H), 3,62 (dd, J = 6,11, 2,45 Hz, 1H), 3,51 (dd,
J = 9,16, 5,04 Hz, 1H), 3,24 (dd, J = 9,00, 7,47 Hz, 1H), 2,53 (dd,
J = 11,59, 6,86 Hz, 1H), 2,40 (m, 1H), 2,63 (s, 3H), 1,96 (m, 1H),
1,80 (m, 1H), 0,98 (d, J = 7,02 Hz, 1H), 0,95 (d, J = 6,86 Hz, 3H),
0,88 (s, 9H), 0,87 (d, J = 6,80, 3H), 0,04 (s, 3H), 0,03 (s, 3H).
-
b) Herstellung von 2,4-Dideoxy-3,5-O-[(4-methoxyphenyl)methylen]-2,4-dimethyl-L-arabinitol.
-
Eine
Suspension von LiAlH4 (5,33 g, 37,95 mmol)
in THF (250 ml) wird auf -74°C
gekühlt.
2,4-Dideoxy-3,5-O-[(4-methoxyphenyl)methylen]-2,4-dimethyl-L-arabinose
(23,2 g, 87,88 mmol) in 50 ml THF wird tropfenweise über einen
Zeitraum von 30 Minuten bei -70°C
bis -74°C
zugegeben. Die erhaltene Lösung
wird bei diesem Temperaturbereich 1,5 Stunden lang gerührt und
dann auf 0°C
erwärmt.
Die Reaktion wird gestoppt oder gequencht mit 6 ml Wasser in 90
ml THF, 6ml 15% NaOH und 30 ml Wasser bei 0°C bis 10°C. Die organische Phase wird
getrennt, und die wässrige
Phase wird mit Ethylacetat (3 × 200
ml) extrahiert. Die vereinigten organischen Phasen werden mit Wasser
(2 × 300
ml), Salzlösung
(2 × 300
ml) gewaschen, über
Na2SO4 getrocknet
und konzentriert. Das gewünschte
Produkt wird als ein klares Öl
erhalten.
-
1H NMR (300 MHz, CDCl3), δ 7,38 (d,
J = 8,7 Hz, 2H), 6,88 (d, J = 8,7 Hz, 2H), 5,48 (s, 1H), 4,11 (m, 1H),
3,8-3,65 (m, 3H), 3,80 (s, 3H), 3,52 (sichtbar t, J = 11,3 Hz, 1H),
2,17-1,92 (m, 3H), 1,05 (d, J = 7,16 Hz, 3H), 0,77 (d, J = 6,8 Hz,
3H).
-
c) Herstellung von 2,4-Dideoxy-1-O-[(1,1-dimethylethyl)dimethylsilyl]-3,5-O-[(4-methoxyphenyl)methylen]-2,4-dimethyl-L-arabinitol.
-
Eine
Lösung
von 2,4-Dideoxy-3,5-O-[(4-methoxyphenyl)methylen]-2,4-dimethyl-L-arabinitol
(24,5 g, 92,48 mmol), 2,6-Lutidin (17,84 g, 166,46 mmol) und Methylenchlorid
(400 ml) wird auf -20°C
gekühlt.
TBSOTf (36,67 g, 138,72 mmol) wird während 30 Minuten zugegeben.
Nach einem Rühren
für weitere
2 Stunden bei 0°C
wird das Gemisch mit Ether (400 ml) verdünnt, mit wässrigem NaHSO4 Salzlösung gewaschen,
und konzentriert. Eine Flash-Chromatographie ergibt das gewünschte Produkt
(30 g, 86%) als weißen
Feststoff.
-
1H NMR (300 MHz, CDCl3), δ 7,37 d,
J = 8,7 Hz, 2H), 6,85 (d, J = 8,7 Hz, 2H), 5,38 (s, 1H), 4,10 (dd, J
= 11,3, 4,9, 1H), 3,77 (s, 3H), 3,66-3,60 (m, 2H), 3,51-3,44 (m,
2H), 2,07-1,88 (m, 2H), 0,90 (d, J = 7,54 Hz, 3H), 0,87 (s, 9H),
0,72 (d, J = 6,8 Hz, 3H), 0,004 (s, 6H).
-
d) Herstellung von 2,4-Dideoxy-1-O-((1,1-dimethylethyl)dimethylsilyl]-3-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-arabinitol.
-
Eine
Lösung
von 2,4-Dideoxy-1-O-[(1,1-dimethylethyl)dimethylsilyl]-3,5-O-[(4-methoxyphenyl)methylen]-2,4-dimethyl-L-arabinitol
(29 g, 76,3 mmol) und Methylenchlorid (500 ml) wird auf -73°C gekühlt, und
DIBAL (1,0 M in Hexan, 763 ml, 763 mmol) wird tropfenweise über einen
Zeitraum von 1,5 Stunden zugegeben. Nach einer weiteren Stunde bei
-78°C wird
das Reaktionsgemisch auf 0°C
erwärmt.
Die Reaktion wird sehr langsam gestoppt mit gesättigtem Rochelle-Salz. Die
Lösung
wird getrennt. Die wässrige
Phase wird mit Ethylacetat (2 × 200
ml) extrahiert. Die kombinierte oder vereinigte organische Phase
wird mit Salzlösung
(2 × 200 ml)
gewaschen und konzentriert. Eine Flash-Chromatographie ergibt das gewünschte Produkt
als klares Öl.
-
1H NMR (300 MHz, CDCl3), δ 7,22 (d,
J = 7,9 Hz, 2H), 6,82 (d, J = 8,7 Hz, 2H), 4,50 (dd, J = 26,4, 10,9, 2H),
3,74 (s, 3H), 3,59-3,44 (m, 5H), 2,75 (sichtbar t, J = 5,80, 1H),
1,92-1,76 (m, 1H), 0,87-0,84 (m, 3H), 0,85 (s, 9H), 0,825 (d, J
= 1,9 Hz, 3H), 0,00 (s, 6H).
-
e) Herstellung von 2,4-Dideoxy-5-O-[(1,1-dimethylethyl)dimethylsilyl]-3-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-lyxose.
-
Eine
Lösung
von 2,4-Dideoxy-1-O-[(1,1-dimethylethyl)dimethylsilyl]-3-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-arabinitol
(10,77 g, 28,19 mmol), DMSO (30 ml), NEt3 (11,39
g, 112,76 mmol) und Methylenchlorid wird auf -10°C gekühlt und mit SO3-Pyr
(13,45 g, 84,58 mmol) in DMSO (60 ml) 40 Minuten lang behandelt.
Nach zusätzlichen
2 Stunden bei 0°C
wird das Gemisch mit Ether (300 ml) verdünnt, mit 1 M wässrigem
NaHSO4 (100 ml) Salzlösung (100 ml) gewaschen, und
konzentriert. Eine Flash-Chromatographie (5% Ethylacetat in Hexan)
ergibt das gewünschte
Produkt als ein farbloses Öl.
-
1H NMR (300 MHz, CDCl3), δ 9,70 (d,
J = 2,26 Hz, 1H), 7,17 (d, J = 8,3 Hz, 2H), 6,81 (d, J = 8,3 Hz, 2H),
4,45 (dd, J = 20,7, 10,9 Hz, 2H), 3,79 (dd, J = 7,91, 3,76, 1H),
3,74 (s, 3H), 3,56-3,44 (m, 2H), 2,71-2,59 (m, 1H), 1,89-1,76 (m,
1H), 0,98 (d, J = 7,16 Hz, 3H), 0,86 (d, J = 8 Hz, 3H), 0,85 (s,
9H), 0,002 (s, 6H).
-
f) Herstellung des (1,1-Dimethylethyl)[[(2S,3S,4S,5Z)-3-[(4-methoxyphenyl)methoxy]-2,4-dimethyl-5,7-octadienyl]oxy]dimethyl-silans.
-
Eine
Suspension, bestehend aus CrCl2 (12,93 g,
105,38 mmol) und 700 ml wasserfreiem THF wird auf 0°C gekühlt. 2,4-Dideoxy-5-O-[(1,1-dimethylethyl)dimethylsilyl]-3-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-lyxose
(8,9 g, 23,42 mmol) in 40 ml THF wird tropfenweise zugegeben. (1-Bromoallyl)trimethylsilan (22,61,
117,1 mmol) wird pur zugegeben. Nach 3 Stunden bei Raumtemperatur
wird das Reaktionsgemisch auf 10°C
gekühlt.
Methanol (270 ml) und 6 N wässriges
KOH (550 ml) werden nacheinander zugegeben, wobei die Reaktionstemperatur
unterhalb von 20°C
gehalten wird während
der Zugabe. Das Gemisch wird über Nacht
bei Raumtemperatur gerührt.
Die organische Phase wird getrennt. Die wässrige Phase wird mit Ether
(3 × 400
ml) extrahiert. Die vereinigte organische Phase wird mit Salzlösung gewaschen, über NaSO4 getrocknet und konzentriert. Eine Flash-Chromatographie
ergibt die gewünschte
Verbindung als klares Öl.
-
1H NMR (300 MHz, CDCl3), δ 7,20 (d,
J = 8,7 Hz, 2H), 6,81 (d, J = 8,7 Hz, 2H), 6,61 (dt, J = 16,96, 10,92
Hz, 1H), 5,97 (t, J = 10,92 Hz, 1H), 5,46 (t, J = 10,55 Hz, 1H),
5,09 (dd, J = 33,5, 17,0 Hz, 2H), 4,45 (dd, J = 27,5, 9,4 Hz, 2H),
3,76 (s, 3H), 3,51-3,32 (m, 3H), 2,97-2,82 (m, 1H), 1,85-1,76 (m,
1H), 0,97 (d, J = 7,16 Hz, 3H), 0,88 (d, J = 7,9 Hz, 3H), 0,87 (s,
9H), 0,001 (s, 6H).
-
g) Herstellung von (2S,3S,4S,5Z)-3-[(4-Methoxyphenyl)methoxy]-2,4-dimethyl-5,7-octadien-1-ol.
-
Eine
Lösung
von (1,1-Dimethylethyl)[[(2S,3S,4S,5Z)-3-[(4-methoxyphenyl)methoxy]-2,4-dimethyl-5,7-octadienyl]oxy]dimethylsilan
(6,74 g, 16,68 mmol) und THF (70 ml) wird auf 0°C gekühlt. 4 N wässrige HCl (70 ml) wird zu
der Lösung
tropfenweise gegeben, um die Reaktionstemperatur bei weniger als
5°C zu halten.
Das Gemisch wird bei 0°C
1 Stunde lang gerührt.
Das Gemisch wird gequencht oder die Reaktion gestoppt durch die
Zugabe von gesättigter
wässriger
NaCO3. Die organische Phase wird abgetrennt.
Die wässrige
Phase wird mit Ethylacetat (5 × 100
ml) extrahiert. Die vereinigte organische Phase wird mit Salzlösung gewaschen, über NaSO4 getrocknet und konzentriert. Eine Flash-Chromatographie
ergibt die gewünschte
Verbindung als klares Öl.
-
1H NMR (300 MHz, CDCl3), δ 7,25 (d,
J = 8,7 Hz, 2H), 6,86 (d, J = 8,7 Hz, 2H), 6,67 (dt, J = 21,5, 11,3 Hz,
1H), 6,04 (t, J = 11,31 Hz, 1H), 5,55 (t, J = 10,55 Hz, 1H), 5,16
(dd, J = 34,3, 16,95 Hz, 2H), 4,53 (dd, J = 36,6, 10,9, 2H), 3,80
(s, 3H), 3,65-3,49 (m, 2H), 3,40 (dd, J = 5,7, 3,8 Hz, 1H), 3,05-2,95
(m, 1H), 2,03-1,90 (m, 1H), 1,83 (t, J = 5,7 Hz, 1H), 1,02 (d, J
= 6,8 Hz, 3H), 0,95 (d, J = 6,8 Hz, 3H).
-
h) Herstellung von (2R,3S,4S,5Z)-3-[(4-Methoxyphenyl)methoxy]-2,4-dimethyl-5,7-octadienal.
-
Eine
Lösung
von (2S,3S,4S,5Z)-3-[(4-Methoxyphenyl)methoxy]-2,4-dimethyl-5,7-octadien-1-ol
(2,0 g, 6,9 mmol), DMSO (2,1 ml), NEt3 (2,79
g, 27,59 mmol) und Methylenchlorid (20 ml) wird auf 0°C gekühlt und mit
SO3-Pyr (3,29 g, 20,69 mmol) und DMSO (8
ml) 10 Minuten lang behandelt. Nach weiteren 2 Stunden bei 0°C wird das
Gemisch mit Ether (200 ml) verdünnt,
mit 1 M wässriger
NaHSO4 (100 ml), Salzlösung (100 ml) gewaschen und
konzentriert. Eine Flash-Chromatographie (1 % bis 10% Ethylacetat
in Hexan) ergibt die gewünschte
Verbindung als farbloses Öl.
-
1H NMR (300 MHz, CDCl3), δ 9,69 (d,
J = 1,1 Hz, 1H), 7,22 (d, J = 8,7 Hz, 2H), 6,86 (d, J = 8,7 Hz, 2H),
6,55 (dt, J = 17,0, 10,2 Hz, 1H), 6,05 (t, J = 10,9 Hz, 1H), 5,45
(t, J = 10,55 Hz, 1H), 5,16 (dd, J = 30,9, 16,95 Hz, 2H), 4,475
(dd, J = 18,5,10,9, 2H), 3,80 (s, 3H), 3,71 (t, J = 4,9 Hz, 1H),
3,03-2,90 (m, 1H), 2,64-2,55 (m, 1H), 1,16 (d, J = 7,2 Hz, 3H),
1,06 (d, J = 6,9 Hz, 3H).
-
i) Herstellung von (4R)-3-[(2R,3S,4S,5S,6S,7Z)-3-Hydroxy-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]-4-(phenylmethyl)-2-oxazolidinon.
-
Eine
Lösung
von (R)-4-Benzyl-3-propionyl-2-oxazolidinon (2,18 g, 9,34 mmol)
und Methylenchlorid (15 ml) wird auf -20°C gekühlt. (n-Bu)2BOTf
(1,0 M in DCM, 8,7 ml, 8,7 mmol) wird tropfenweise zugegeben. Die Reaktion
lässt man
auf 0°C
erwärmen.
NEt3 (1,15 g, 11,34 mmol) wird zugegeben,
und das Gemisch wird 1 Stunde lang bei 0°C gerührt, dann auf -78°C gekühlt. Eine
Lösung
von (2R,3S,4S,5Z)-3-[(4-Methoxyphenyl)methoxy]-2,4-dimethyl-5,7-octadienal
(1,95 g, 6,77 mmol) in DCM (10 ml) wird bei -70°C 10 Minuten lang tropfenweise
zugegeben. Nach 1 Stunde bei -74°C
wird das Gemisch auf 0°C
erwärmt.
Die Reaktion wird gequencht oder gestoppt mit einem Puffer bei pH
7 (15 ml). Das Gemisch wird langsam mit einer Lösung von 30% H2O2 in MeOH (1:2, 15 ml) bei 0°C behandelt,
40 Minuten lang bei Raumtemperatur gerührt und konzentriert. Der Rückstand
wird mit Ethylacetat (3 × 100
ml) extrahiert. Die vereinigte organische Phase wird mit gesättigter
wässriger
NaHCO3, Salzlösung gewaschen und konzentriert.
Eine Flash-Chromatographie ergibt die gewünschte Verbindung als klares Öl.
-
1H NMR (300 MHz, CDCl3), δ 7,38-7,16
(m, 7H), 6,82 (d, J = 8,7 Hz, 2H), 6,69 (dt, J = 17,0, 10,2 Hz, 1H),
6,06 (t, J = 11,3 Hz, 1H), 5,50 (t, J = 10,55 Hz, 1H), 5,17 (dd,
J = 30,1, 16,95 Hz, 2H), 4,53 (dd, J = 98,7, 10,9, 2H), 4,63-4,56
(m, 1H), 4,23-4,10 (m, 2H), 4,02-3,96 (m, 1H), 3,83 (t, J = 6,4
Hz, 1H), 3,78 (s, 3H), 3,38-3,33 (m, 1H), 3,25-3,15 (m, 2H), 3,07-2,95
(m, 1H), 2,74 (dd, J = 13,6, 9,8 Hz, 1H), 1,84-1,73 (m, 1H), 1,29 (d, J = 7,2 Hz, 3H),
1,02 (d, J = 6,8 Hz, 3H), 0,98 (d, J = 6,8 Hz, 3H); 13C
NMR (CDCl3, 75 MHz), δ 176,8, 159,0, 152,7, 135,6,
135,1, 132,5, 130,7, 129,5, 129,4, 129,3, 128,9, 127,4, 117,7, 113,6,
86,3, 74,3, 73,8, 66,0, 55,3, 55,0, 40,6, 37,8, 37,7, 35,8, 18,14,
13,28.
-
j) Herstellung von ((4R)-3-[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]-4-(phenylmethyl)-2-oxazolidinon.
-
Eine
Lösung
von (4R)-3-[(2R,3S,4S,5S,6S,7Z)-3-Hydroxy-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]-4-(phenylmethyl)-2-oxazolidinon
(3,4 g, 6,53 mmol), 2,6-Lutidin (3,5 g, 32,65 mmol) und Methylenchlorid
(100 ml) wird auf 0°C
gekühlt.
TBSOTf (7,12 g, 26,94 mmol) wird über einen Zeitraum von 30 Minuten
zugegeben, und das erhaltene Gemisch lässt man über Nacht bei 0°C reagieren.
Das Gemisch wird mit Ether (300 ml) verdünnt, mit wässrigem NaHSO4,
Salzlösung
gewaschen und konzentriert. Eine Flash-Chromatographie ergibt die
gewünschte
Verbindung als Öl.
-
1H NMR (300 MHz, CDCl3), δ 7,25-7,05
(m, 7H), 6,74 (d, J = 8,7 Hz, 2H), 6,58 (dt, J = 16,5, 10,2 Hz, 1H),
5,86 (t, J = 11,3 Hz, 1H), 5,49 (t, J = 10,55 Hz, 1H), 5,10-5,01
(m, 3H), 4,44-4,38 (m, 1H), 4,41 (s, 2H), 4,00-3,87 (m, 4H), 3,69
(s, 3H), 3,26 (dd, J = 7,5, 3 Hz, 1H), 2,96-2,84 (m, 1H), 2,59 (dd,
J = 13,6, 9,8 Hz, 1H), 1,56-1,46 (m, 1H), 1,13 (d, J = 6,4 Hz, 3H),
1,01 (d, J = 7,2 Hz, 3H), 0,92 (d, J = 7,2 Hz, 3H), 0,86 (s, 9H),
0,035 (d, J = 6,4 Hz, 6H).
-
k) Herstellung von (2R,3S,4R,5S,6S,7Z)-3-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-7,9-decadiensäure.
-
Eine
Lösung
von (4R)-3-[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]-4-(phenylmethyl)-2-oxazolidinon),
THF (15 ml) und Wasser (5 ml) wird auf 10°C gekühlt. Wasserstoffperoxid (50%,
20 ml) wird tropfenweise zugegeben, gefolgt von der Zugabe von LiOH (0,575
g). Das Gemisch wird bei 10°C über Nacht
gelagert. Die Reaktion wird gequencht oder gestoppt durch die Zugabe
von gesättigtem
wässrigem
Na2SO3. Das Gemisch
wird angesäuert
durch die Zugabe von wässriger
HCl (12 M) bis zu pH 1, dann mit Ether extrahiert. Die vereinigte
organische Phase wird mit Salzlösung gewaschen, über Na2SO4 getrocknet und
konzentriert. Eine Flash-Chromatographie ergibt die gewünschte Verbindung
als einen weißen
Feststoff.
-
δ 7,26 (d,
J = 8,7, 2H), 6,85 (d, J = 8,7 Hz, 2H), 6,69 (dt, J = 17,0, 10,9
Hz, 1H), 6,02 (t, J = 11,3 Hz, 1H), 5,50 (t, J = 10,55 Hz, 1H),
5,21 (d, J = 16,5 Hz, 1H), 5,12 (d, J = 10,2 Hz, 1H), 4,50 (dd,
J = 29,4, 10,2 Hz, 2H), 4,07 (t, J = 4,5 Hz, 1H), 3,79 (s, 3H),
3,25 (t, J = 5,3 Hz, 1H), 3,05-2,94 (m, 1H), 2,74-2,65 (m, 1H), 1,90-1,79
(m, 1H), 1,11 (d, J = 7,2 Hz, 3H), 1,06 (d, J = 6,8 Hz, 3H), 1,00
(d, J = 6,8 Hz, 3H), 0,93 (s, 9H), 0,10 (d, J = 7,9 Hz, 6H).
-
l) Herstellung von 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-1-[[(2R,3S,4R,-5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-5-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-arabinitol.
-
Zu
einer Lösung
von 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-5-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-1-(methylamino)-L-arabinitol
(1,23 g, 3,1 mmol) und (2R,3S,4R,5S,6S,7Z)-3-([(1,1-Dimethylethyl)dimethylsilyl]oxy]-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-7,9-decadiensäure (1,14
g, 2,4 mmol) in DMF (20 ml) bei Raumtemperatur wurden Benzotriazol-1-yl-oxyl-tris(dimethylamino)phosphoniumhexafluorphosphat
(BOP) (2,12 g, 4,8 mmol) und DIEA (1,86 g, 14,4 mmol) gegeben. Nach
einem Rühren
bei Raumtemperatur über
Nacht wird das Reaktionsgemisch durch Säulenchromatogiaphie an Kieselgel
mit Hexan/EtOAc (90/10) gereinigt, um das gewünschte Produkt als ein blasses
viskoses Öl
zu ergeben.
-
1H NMR (CDCl3, 300
MHz), δ 7,22-7,17
(m, 4H), 6,83-6,76 (m, 4H), 6,70-6,59 (m, 1H), 5,96 (t, J = 10,20 Hz,
1H), 5,58-5,45 (m, 1H), 5,16-5,05 (m, 2H), 4,57-4,46 (m, 1H), 4,35-4,34
(m, 2H), 4,16 (dd, J = 9,04, 4,90 Hz, 1H), 3,75 (s, 3H), 3,74 (s,
3H), 3,54 (t, J = 5,65 Hz, 1H), 3,47-3,39 (m, 3H), 3,25-3,17 (m,
2H), 3,14-3,03 (m, 1H), 3,00-2,90 (m, 2H), 2,55 (s, 3H), 1,94-1,81
(m, 2H), 1,78-1,73 (m, 1H), 1,06 (t, J = 6,03 Hz, 3H), 0,99 (d,
J = 7,16 Hz, 3H), 0,95-0,88 (m, 12H), 0,85-0,81 (m, 12H), 0,72 (d,
J = 6,78 Hz, 3H), 0,11 (s, 3H), 0,06 (s, 3H), 0,00 (s, 3H), -0,04
(s, 3H).
-
m) Herstellung von 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-1-[[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-hydroxy-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-2,4-dimethyl-L-arabinitol.
-
Zu
einer Lösung,
bestehend aus 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-1-[[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-[(4-methoxyphenyl)methoxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-5-O-[(4-methoxyphenyl)methyl]-2,4-dimethyl-L-arabinitol
(1,9 g, 2,2 mmol), CH2Cl2 (40
ml) und H2O (1 ml) bei Raumtemperatur wird
2,3-Dichlor-5,6-dicyano-1,4- benzochinon
(DDQ) (2,53 g, 11,1 mmol) gegeben. Nach 10 Minuten rühren bei
Raumtemperatur wird das Reaktionsgemisch durch Säulenchromatographie an Kieselgel
mit Hexan/EtOAc (90/10) gereinigt, um das gewünschte Produkt als weißen Feststoff
zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,57-6,45
(m, 1H), 6,03 (t, J = 11,24 Hz, 1H), 5,20 (d, J = 10,93 Hz, 1H), 5,13
(d, J = 16,95 Hz, 1H), 5,03 (d, J = 9,78 Hz, 1H), 4,04-3,91 (m,
1H), 3,54-3,45 (m, 4H), 3,22-3,16 (m, 1H), 3,07 (t, J = 7,16 Hz,
1H), 2,93 (s, 3H), 2,67-2,59 (m, 1H), 2,45 (s, 1H), 2,10-1,96 (m,
1H), 1,80-1,72 (m,
1H), 1,52 (s, 1H), 0,98 (d, J = 7,16 Hz, 3H), 0,96 (d, J = 7,16
Hz, 3H), 0,84 (d, J = 6,77 Hz, 3H), 0,83-0,77 (m, 21H), 0,01 (s,
3H), 0,00 (s, 3H), 0,00 (s, 3H), -0,01 (s, 3H).
-
n) Herstellung von 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-1-[[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxyj-5-hydroxy-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-2,4-dimethyl-L-arabinitol.
-
Zu
einer Lösung,
bestehend aus 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-1-[((2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-hydroxy-2,4,6-trimethyl-1-oxo-7,9-decadienylJmethylamino]-2,4-dimethyl-L-arabinitol
(1,8 g, 2,94 mmol), 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) (86 mg, 0,57
mmol) und CH2Cl2 (10
ml) bei Raumtemperatur wird Iodbenzoldiacetat (BAIB) (1,13 g, 3,51
mmol) gegeben. Das Gemisch wird bei Raumtemperatur gerührt, bis
ein DC (Hexan/EtOAc 95:5) den gesamten Verbrauch des Ausgangsmaterials
anzeigt. Das Gemisch wird verdünnt
mit CH2Cl2 (10 ml)
und nacheinander mit gesättigter
Na2S2O3 (20
ml), 1 M NaHCO3 (20 ml), dann Salzlösung (20
ml) gewaschen. Die Lösung
wird getrocknet (Na2SO4)
und im Vakuum konzentriert. Das gewünschte Produkt wird als ein Öl erhalten
und wird in der nächsten
Stufe ohne weitere Reinigung verwendet.
-
1H NMR (CDCl3, 300
MHz), δ 9,62
(d, J = 2,62 Hz, 1H), 6,52 (td, J = 10,93, 16,95 Hz, 1H), 6,03 (t,
J = 10,92 Hz, 1H), 5,25-5,02 (m, 3H), 4,03 (q, J = 3,76 Hz, 1H),
3,72 (q, J = 5,65 Hz, 1H), 3,46 (d, J = 7,91 Hz, 1H), 3,19-3,03
(m, 2H), 2,92 (s, 3H), 2,63 (q, J = 7,53 Hz, 1H), 2,46 (td, J =
2,63, 5,27 Hz, 1H), 2,01-1,90 (m, 1H), 1,79-1,75 (m, 1H), 1,48 (s,
1H), 1,19-1,47 (m, 4H), 1,02-0,96 (m, 6H), 0,83-0,73 (m, 24H), 0,00-0,05
(m, 12H),
-
o) Herstellung von (2Z,4S,5S,6S)-5-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-7-[[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyljoxy]-5-hydroxy-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-4,6-dimethyl-2-heptensäuremethylester.
-
-
Zu
einer Lösung,
bestehend aus Bis(2,2,2-trifluorethyl)-2,2,5-(methoxycarbonylmethyl)phosphonat (715
mg, 2,25 mmol), 18-Krone-6 (453 mg, 1,7 mmol) und Toluol (5 ml)
bei -20°C
wird langsam eine Lösung vom
Kaliumbis(trimethylsilyl)amid (0,5 M in Toluol, 4,5 ml, 2,25 mmol)
gegeben. Nach 20 Minuten Rühren
bei 0°C
wird das Gemisch auf -20°C
gekühlt,
und eine Lösung
von 1,2,4-Trideoxy-3-O-[(1,1-dimethylethyl)dimethylsilyl]-1-[[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-hydroxy- 2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-2,4-dimethyl-L-arabinitol
(711 mg, 0,85 mmol) in Toluol (3 ml) wird langsam zugegeben. Das
Gemisch wird 3 Stunden lang bei 0°C
gerührt,
woraufhin das Gemisch mit gesättigter
NH4Cl (10 ml) gequencht oder gestoppt wurde
und mit EtOAc (3 × 20
ml) extrahiert wird. Die vereinigten organischen Phasen werden dann
getrocknet (Na2SO4),
konzentriert und durch Säulenchromatographie
an Kieselgel mit Hexan/EtOAc (90/10) gereinigt, um die gewünschte Verbindung
als dickes farbloses Öl
zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,60-6,47
(m, 1H), 6,31 (t, J = 12,06 Hz, 1H), 6,04 (t, J = 10,92 Hz, 1H), 5,69
(dd, J = 11,30, 3,01 Hz, 1H), 5,24 (t, J = 10,55 Hz, 1H), 5,13 (d,
J = 7,32 Hz, 1H), 5,04 (d, J = 10,17 Hz, 1H), 2H), 4,05 (dd, J =
8,29, 4,52 Hz, 1H), 3,93 (dd, J = 8,24, 4,14 Hz, 1H), 3,60 (s, 3H),
3,52 (d, J = 7,91 Hz, 1H), 3,45-3,39 (m, 3H), 3,09-3,02 (m, 1H),
2,88 (s, 3H), 2,71-2,61 (m, 2H), 1,89-1,76 (m, 2H), 1,60 (dd, J
= 14,32, 3,77 Hz, 1H), 1,17 (d, J = 3,01 Hz, 1H), 0,99 (d, J = 7,06
Hz, 3H), 0,96 (d, J = 4,14 Hz, 3H), 0,94 (d, J = 4,03 Hz, 3H), 0,83
(s, 18H), 0,79-0,74 (m, 6H), 0,03-0,00 (m, 12H).
-
p) Herstellung von (2Z,4S,5S,6S)-7-[[(2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4,6-dimethyl-2-heptensäuremethylester.
-
Zu
einer Lösung
von (2Z,4S,5S,6S)-5-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-7-[[(2R,3S,4R,5S,6S,7Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5-hydroxy-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-4,6-dimethyl-2-heptensäuremethylester
(1,8 g, 2,7 mmol) in CH2Cl2 (10
ml) bei Raumtemperatur wird Trichloracetylisocyanat (763 mg, 4,05
mmol) in einer Portion gegeben. Das Gemisch wird bei Raumtemperatur
1 Stunde lang gerührt
und im Vakuum konzentriert. Der Rückstand wird in MeOH (10 ml)
gelöst,
und dann wird K2CO3 (0,4
g) zugegeben. Nach einem Rühren
bei Raumtemperatur über
einen Zeitraum von 2 Stunden wird das Gemisch im Vakuum konzentriert, dann
durch Säulenchromatographie
mit Kieselgel, mit Hexan/EtOAc (70/30) eluiert, gereinigt, um die
gewünschte
Verbindung (1,77 g, 92,4%) als farblosen glasigen Feststoff zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,65
(td, J = 10,87, 16,71 Hz, 1H), 6,46-6,36 (m, 1H), 6,01 (t, J = 10,87 Hz,
1H), 5,80 (dd, J = 11,67, 8,04 Hz, 1H), 5,42 (q, J = 11,11 Hz, 1H),
5,21-5,51 (m, 2H), 4,94 (t, J = 5,51 Hz, 1H), 4,83 (dd, J = 8,51,
3,46 Hz, 1H), 4,60 (s, 1H), 4,52 (s, 1H), 4,08 (dd, J = 7,88, 3,15
Hz, 1H), 3,97 (dd, J = 7,56, 1,42 Hz, 1H), 3,71 (s, 3H), 3,58-3,57
(m, 1H), 3,54 (t, J = 3,63 Hz, 1H), 3,42-3,31 (m, 2H), 3,16-3,09
(m, 1H), 3,01-2,96 (m, 1H), 2,93 (s, 1H), 2,87 (t, J = 7,10 Hz,
1H), 2,79-2,75 (m, 1H), 2,71 (s, 1H), 1,95-1,87 (m, 1H), 1,09 (d,
J = 3,62 Hz, 3H), 1,08 (d, J = 3,74 Hz, 3H), 1,05 (d, J = 8,36 Hz,
3H), 1,10 (d, J = 6,94 Hz, 3H), 0,94 (s, 18H), 0,86 (d, J = 7,25
Hz, 3H), 0,83 (d, J = 6,94 Hz, 3H), 0,12 (s, 3H), 0,12 (s, 3H),
0,11 (s, 3H), 0,11 (s, 3H).
-
q) Herstellung von (2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-N-[(2S,3S,4S,5Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-hydroxy-2,4-dimethyl-5-heptenyl]-N,2,4,6-tetramethyl-7,9-decadienamid.
-
Zu
einer Lösung
von (2Z,4S,5S,6S)-7-[[(2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-4,6-dimethyl-2-heptensäuremethylester
(1,77 g, 2,5 mmol) und CH2Cl2 (15
ml), gekühlt
auf -78°C,
wurde eine Lösung
gegeben, bestehend aus Diisobutylaluminumhydrid (1,0 M in Hexan,
7,5 ml, 7,5 mmol). Nach Rühren
bei -40°C über einen
Zeitraum von 4 Stunden wird das Gemisch gequencht oder die Reaktion
gestoppt durch Zugabe von gesättigtem
Kaliumnatriumtartrat (10 ml). Das Gemisch wird dann extrahiert mit
CH2Cl2 (3 × 20 ml).
Die vereinigten organischen Phasen werden dann getrocknet (Na2SO4), in Vakuum
konzentriert und chromatographiert unter Verwendung von Kieselgel,
mit Hexan/EtOAc (70/30) eluiert, um die gewünschte Verbindung als farblosen glasigen
Feststoff zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,68-6,60
(m, 1H), 6,06-6,01 (m, 1H), 5,67-5,60 (m, 2H), 5,42 (t, J = 9,77 Hz,
1H), 5,22 (d, J = 6,86 Hz, 1H), 5,13 (d, J = 5,83 Hz, 1H), 4,94
(t, J = 5,83 Hz, 1H), 4,47 (s, 1H), 4,41-4,36 (m, 2H), 4,06 (dd,
J = 7,41, 3,47 Hz, 1H), 4,01-3,96 (m, 1H), 3,69 (dd, J = 13,24,
11,83 Hz, 1H), 3,53 (dd, J = 8,82, 4,25 Hz, 1H), 3,48 (dd, J = 5,36,
2,36 Hz, 1H), 3,07 (dd, J = 13,04, 4,25 Hz, 1H), 3,00-2,97 (m, 1H), 2,95 (s,
3H), 2,89-2,81 (m, 2H), 2,06-2,01 (m, 1H), 1,94-1,91 (m, 1H), 1,07
(d, J = 6,94 Hz, 3H), 1,01 (d, J = 6,78 Hz, 3H), 0,99 (d, J = 6,94
Hz, 3H), 0,94 (s, 18H), 0,86 (d, J = 7,25 Hz, 3H), 0,85 (d, J =
7,05 Hz, 3H), 0,13 (s, 3H), 0,11 (s, 3H), 0,09 (s, 6H).
-
r) Herstellung von (2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-N-[(2S,3S,4S,5Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-7-oxo-5-heptenyl]-N,2,4,6-tetramethyl-7,9-decadienamid.
-
Zu
einer Lösung
von Dess-Martin-Periodinan (199 mg, 0,47 mmol) und CH2Cl2 (4 ml) bei Raumtemperatur wird eine Lösung gegeben
von (2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-N-[(2S,3S,4S,5Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-hydroxy-2,4-dimethyl-5-heptenyl]-N,2,4,6-tetramethyl-7,9-decadienamid
(200 mg, 0,29 mmol) und CH2Cl2 (1
ml). Nach Rühren
bei Raumtemperatur über
einen Zeitraum von 1 Stunde wird das Gemisch gequencht durch Zugabe
von gesättigtem
NaHCO3 (1 ml), gefolgt von gesättigtem Na2S2O3 (1
ml). Dieses Gemisch wird dann bei Raumtemperatur 30 Minuten lang
gerührt
und dann mit CH2Cl2 (4 × 10 ml)
extrahiert. Die vereinigten organischen Phasen werden getrocknet
(Na2SO4), im Vakuum
konzentriert und dann chromatographiert (Kieselgel, eluiert mit
Hexan/EtOAc 90/10), um die gewünschte
Verbindung als einen farblosen glasigen Feststoff zu ergeben.
-
-
1H NMR (CDCl3, 300
MHz), δ 6,61-6,47
(m, 2H), 5,94-5,83 (m, 1H), 5,30 (t, J = 9,80 Hz, 1H), 5,11-5,00 (m,
2H), 4,81 (t, J = 5,66 Hz, 1H), 4,49 (s, 1H), 3,95 (dd, J = 7,54,
3,02 Hz, 1H), 3,44 (t, J = 3,76 Hz, 1H), 3,16-3,11 (m, 1H), 2,88
(m, 1H), 2,86 (s, 3H), 2,77 (t, 2,77 Hz, 1H), 2,64 (s, 1H), 1,94-1,72
(m, 2H), 1,20-1,13 (m, 2H), 1,05-0,97 (m, 6H), 0,89 (d, J = 6,78
Hz, 3H), 0,83 (s, 9H), 0,81 (s, 9H), 0,78-0,74 (m, 6H), 0,03 (s, 3H), 0,01 (s,
3H), 0,00 (s, 3H), -0,04 (s, 3H).
-
s) Herstellung von (2R,3S,4R,7S,8Z,10S,11S,12S)-13-[[(2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-3,11-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-hydroxy-N-methoxy-N,2,4,10,12-pentamethyl-5-oxo-8-tridecenamide.
-
Zu
einer Lösung
von (+)-β-Chlordiisopinocampheylboran
(502 mg, 1,52 mmol) und Ether (1,3 ml), gekühlt auf -6°C, wird Et3N
(0,23 ml, 1,67 mmol) gegeben. Zu diesem Gemisch wird eine Lösung von (2R,3S,4R)-3-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-N-methoxy-N-methyl-2,4-trimethyl-5-oxo-hexanamid
und Ether (1,5 ml) gegeben. Nach 2 Stunden Rühren bei 0°C wird das Gemisch auf -78°C gekühlt. Zu
diesem Gemisch wird eine Lösung
von (2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-N-[(2S,3S,4S,5Z)-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4-dimethyl-7-oxo-5-heptenyl]-N,2,4,6-tetramethyl-7,9-decadienamid (103
mg, 0,152 mmol) und Ether gegeben. Das Gemisch wird 3 Stunden lang
bei -78°C
gerührt
und dann bei -30°C über Nacht.
Das Reaktionsgemisch wird gequencht oder die Reaktion gestoppt durch
die nachfolgende Zugabe von MeOH (4 ml), einem Phosphatpuffer (pH
= 7, 1,5 ml), dann H2O2 (30%,
1,0 ml) bei -78°C.
Das Gemisch wird auf Raumtemperatur erwärmt und 2 Stunden lang gerührt. Das
Gemisch wird mit CH2Cl2 (4 × 10 ml)
extrahiert. Die vereinigten organischen Phasen werden dann getrocknet
(Na2SO4), im Vakuum
konzentriert und dann chromatographiert (Kieselgel, eluiert mit
Hexan/EtOAc 80/20), um die gewünschte
Verbindung als einen farblosen, glasigen Feststoff zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,69-6,61
(m, 1H), 6,03 (t, J = 11,42 Hz, 1H), 5,58 (t, J = 10,55 Hz, 1H), 5,44-5,40
(m, 2H), 5,22-5,19 (m, 1H), 5,17-5,13 (m, 1H), 4,95 (t, J = 6,06
Hz 1H), 4,84-4,80 (m, 1H), 4,66-4,45 (m, 2H), 4,35 (dd, J = 8,24,
4,12 Hz, 1H), 4,09 (dd, J = 7,62, 3,20 Hz, 1H), 3,74 (s, 3H), 3,49-3,38 (m, 3H), 3,11 (s,
3H), 3,07-3,02 (m, 1H), 3,00-2,94 (m, 3H), 2,89-2,76 (m, 4H), 2,69-2,56
(m, 1H), 2,02-1,97 (M, 1H), 1,91-1,88 (m, 1H), 1,72-1,66 (m, 1H),
1,27 (s, 3H), 1,15-1,11 (m, 6H), 1,10-1,08 (m, 6H), 1,03-1,00 (m,
6H), 0,95 (s, 9H), 0,93 (s, 9H), 0,92 (s, 9H), 0,14 (s, 3H), 0,13
(s, 3H), 0,12 (s, 3H), 0,12 (s, 3H), 0,12 (s, 3H), 0,11 (s, 3H).
-
t) Herstellung von (2R,3S,4S,5S,7S,8Z,10S,11S,12S)-13-[[(2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[((1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-3,11-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5,7-dihydroxy-N-methoxy-N,2,4,10,12-pentamethyl-8-tridecenamid.
-
Tetramethylammoniumtriacetoxyborhydrid
(182 mg, 0,69 mmol) wird zuerst in einer gemischten Lösung von
THF/AcOH (1:1, 0,5 ml) gelöst.
Nach 1 Stunde Rühren
bei Raumtemperatur wird das Gemisch auf -30°C gekühlt und dann wird langsam eine
Lösung
von (2R,3S,4R,7S,8Z,10S,11S,12S)-13-[[(2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-3,11-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-hydroxy-N-methoxy-N,2,4,10,12-pentamethyl-5-oxo-8-tridecenamid
(50 mg, 0,049 mmol) in THF/AcOH (1:1, 0,5 ml) langsam zugegeben.
Das Reaktionsgemisch wird 3 Stunden lang bei -30°C und über Nacht bei 0°C gerührt. Das
Reaktionsgemisch wird gequencht oder die Reaktion gestoppt durch
Zugabe von gesättigtem Natriumkaliumtartrat
(0,5 ml). Das Gemisch wird mit CH2Cl2 (3 × 10
ml) extrahiert. Die vereinigten organischen Phasen werden neutralisiert
durch Zugabe von gesättigtem
NaHCO3, getrocknet (Na2SO4), im Vakuum konzentriert, dann chromatographiert
(Kieselgel, eluiert mit Hexan/EtOAc 95/5), um die gewünschte Verbindung als
einen farblosen, glasigen Feststoff zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,58-6,49
(m, 1H), 5,93-5,88 (m, 1H), 5,40-5,30 (m, 3H), 5,10-5,01 (m, 2H),
4,81 (t, J = 5,52 Hz 1H), 4,71 (dd, J = 8,51, 2,84 Hz, 1H), 4,59
(s, 1H), 4,55-4,41 (m, 3H), 4,10 (d, J = 9,45, 1H), 3,96 (dd, J
= 7,56, 2,99 Hz, 1H), 3,86 (s, 1H), 3,83 (d, J = 7,72 Hz, 1H), 3,63
(s, 3H), 3,33 (s, 1H), 3,30-3,23 (m, 4H), 3,08 (s, 3H), 3,03-2,94
(m, 2H), 2,86 (s, 3H), 2,77-2,69 (m, 2H), 2,63-2,58 (m, 2H), 1,86-1,81
(m, 2H), 1,80-1,76 (m, 2H), 1,68-1,62 (m, 2H), 1,57-1,48 (m, 2H),
1,43-1,39 (m, 1H), 1,07 (dd, J = 6,94, 2,21 Hz, 3H), 0,98 (t, J
= 7,56 Hz, 3H), 0,93-0,88 (m, 6H), 0,83-0,80 (m, 18H), 0,75-0,69 (m, 9H), 0,06
bis -0,05 (m, 18H).
-
u) Herstellung von (2R,3S,4R,5S,7S,8Z)-13-[[(2R,3S,4S,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-hydroxy-2,4,6-trimethyl-1-oxo-7,9-decadienyl]methylamino]-3,5,7,11-tetrahydroxy-2,4,10,12-tetramethyl-8-tridecensäure-δ-lacton
-
-
(2R,3S,4S,5S,7S,8Z,10S,11S,12S)-13-[[(2R,3S,4R,5S,6S,7Z)-5-[(Aminocarbonyl)oxy]-3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2,4,6-trimethyl-1-oxo-7,9-decadienyljmethylamino]-3,11-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-5,7-dihydroxy-N-methoxy-N,2,4,10,12-pentamethyl-8-tridecenamid
(115 mg, 0,114 mmol) wird gelöst
in einer Lösung,
bestehend aus Isopropanol (15 ml) und HCl (4 N,12 ml) bei Raumtemperatur.
Nach 60 Stunden Rühren
bei Raumtemperatur wird das Gemisch auf -15°C gekühlt, und mit EtOAc (50 ml)
verdünnt.
Die Lösung wird
neutralisiert durch Zugabe von festem NaHCO3 in
sehr kleinen Portionen bei -15°C,
und wird dann extrahiert mit EtOAc (6 × 30 ml). Die vereinigten organischen
Phasen werden getrocknet (Na2SO4),
im Vakuum konzentriert, dann durch HPLC auf einer Umkehrphasensäule (Nova-Pak
C18, Waters), mit CH3CN/H2O
(32:68) eluiert, gereinigt, um die gewünschte Verbindung als einen
farblosen glasigen Feststoff zu ergeben.
-
1H NMR (CDCl3, 500
MHz), δ 6,64
(dd, J = 16,86, 10,40 Hz, 1H), 6,05 (t, J = 10,72 Hz, 1H), 5,52
(dd, J = 11,03, 7,56 Hz, 1H), 5,31-5,22 (m, 2H), 5,15 (d, J = 10,14
Hz, 1H), 4,90 (s, 1H), 4,76 (s, 1H), 4,66-4,62 (m, 1H), 4,60 (d,
J = 9,62 Hz, 1H), 4,52 (m, 1H), 4,03-3,96 (m, 2H), 3,75 (s, 1H),
3,63 (d, J = 8,83 Hz, 1H), 3,28 (d, J = 7,09 Hz, 1H), 3,07 (s, 3H),
3,02 (d, J = 6,62 Hz, 1H), 2,91 (d, J = 4,26 Hz, 1H), 2,83-2,78
(m, 1H), 2,69-2,61 (m, 4H), 2,35 (s, 1H), 1,99-1,91 (m, 3H), 1,89-1,85
(m, 2H), 1,35 (d, J = 7,26 Hz, 3H), 1,16 (d, J = 7,10 Hz, 3H), 1,13
(d, J = 6,77 Hz, 3H), 1,10 (d, J = 6,94 Hz, 3H), 1,01 (d, J = 6,78
Hz, 3H), 0,94 (dd, J = 10,87, 6,62 Hz, 2H), 0,90 (d, J = 6,47 Hz,
3H), 0,87 (d, J = 6,78 Hz, 3H); 13C NMR
(CDCl3, 500 MHz), δ 180,0, 174,5, 157,3, 135,8, 134,4,
132,7, 130,7, 118,3, 73,5, 73,2, 72,7, 72,3, 65,5, 51,6, 43,5, 41,5,
36,6, 36,3, 35,6, 35,1, 35,0, 33,5, 32,7, 30,1, 18,0, 17,3, 15,8,
12,8, 10,9, 10,5, 9,7.

























 R4 und R5
R4 und R5

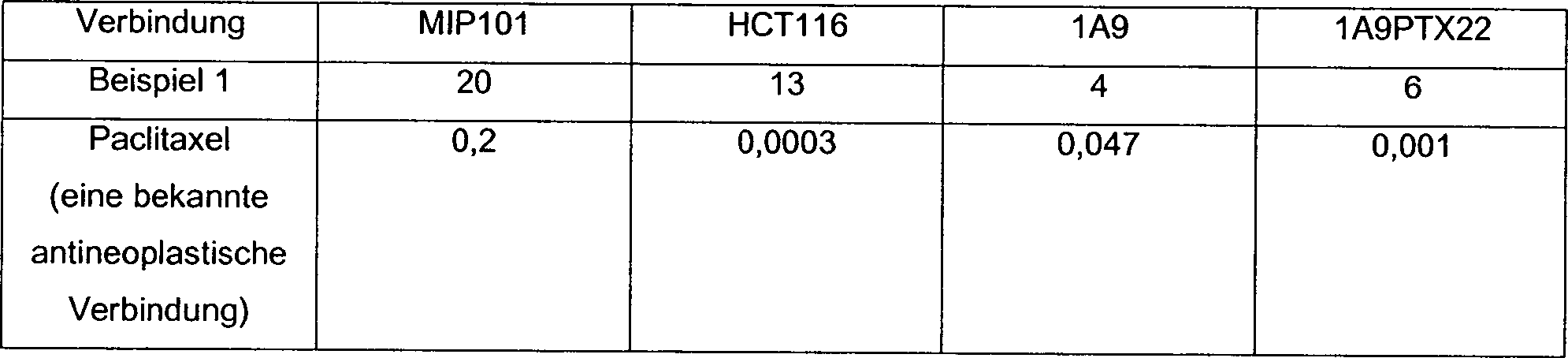



 R4 und R5 unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
R4 und R5 unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
 R4' und R5' unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
R4' und R5' unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
 R4'' und R5'' unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
R4'' und R5'' unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
 R4''' und R5''' unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, F, Cl, Br oder CF3; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
R4''' und R5''' unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, F, Cl, Br oder CF3; oder ein Säure- oder Baseadditionssalz davon, wenn dies möglich ist.
 R4 und R5 unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; wobei eine Verbindung der Formel II
R4 und R5 unabhängig stehen für H, (C1-C6)-Alkyl, OH, O(C1-C6)-Alkyl, OCH2(CH2)nOH, O(CH2)nCO2H, OCH2(CH2)nN(CH3)2, OCH2(CH2)n-4-morpholino, F, Cl, Br oder CF3; und n für 1 oder 2 steht; wobei eine Verbindung der Formel II wobei die Gruppen A*, B* und C*, den Gruppen A, B und C entsprechen, wie für die Verbindung der Formel I definiert mit der Ausnahme, dass in dem Fall, dass diese Gruppen freie Hydroxygruppen enthalten, dann die Kennzeichnung mit dem Stern (z.B. A*) angibt, dass diese Gruppen geschützt sind mit säurelabilen Schutzgruppen, und PG für eine Schutzgruppe steht, hydrolysiert und cyclisiert wird durch Umsetzung mit einer protischen Säure in einem polaren organischen Lösemittel; und wobei jegliche Schutzgruppen in einem geschützten Derivat von einer Verbindung der Formel I anschießend entfernt werden.
wobei die Gruppen A*, B* und C*, den Gruppen A, B und C entsprechen, wie für die Verbindung der Formel I definiert mit der Ausnahme, dass in dem Fall, dass diese Gruppen freie Hydroxygruppen enthalten, dann die Kennzeichnung mit dem Stern (z.B. A*) angibt, dass diese Gruppen geschützt sind mit säurelabilen Schutzgruppen, und PG für eine Schutzgruppe steht, hydrolysiert und cyclisiert wird durch Umsetzung mit einer protischen Säure in einem polaren organischen Lösemittel; und wobei jegliche Schutzgruppen in einem geschützten Derivat von einer Verbindung der Formel I anschießend entfernt werden.