DE10330235A1 - Neues Eimeria Gen und Protein sowie deren Verwendung - Google Patents
Neues Eimeria Gen und Protein sowie deren Verwendung Download PDFInfo
- Publication number
- DE10330235A1 DE10330235A1 DE10330235A DE10330235A DE10330235A1 DE 10330235 A1 DE10330235 A1 DE 10330235A1 DE 10330235 A DE10330235 A DE 10330235A DE 10330235 A DE10330235 A DE 10330235A DE 10330235 A1 DE10330235 A1 DE 10330235A1
- Authority
- DE
- Germany
- Prior art keywords
- polynucleotide
- polypeptide
- etos22
- sequence
- protein
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/20—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans from protozoa
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/002—Protozoa antigens
- A61K39/012—Coccidia antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/44—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from protozoa
- C07K14/455—Eimeria
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biochemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Tropical Medicine & Parasitology (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Toxicology (AREA)
- Epidemiology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Polynucleotid
enthaltend:
a) die Sequenz gemäß SEQ ID NO:1 oder 3; oder
b) ein Polynucleotid, das zu dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 eine Identität von mehr als 50% aufweist; oder
c) ein Polynucleotid, die mit dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 unter stringenten Bedingungen hybridisiert; oder
d) ein Polynucleotid, welches für ein Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert; oder
e) ein Polynucleotid, das zu einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, eine Identität von mehr als 50% aufweist; oder
f) ein Polynucleotid, das mit einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, unter stringenten Bedingungen hybridisiert; oder
g) ein Polynucleotid, das von einem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 aufgrund der Degeneration des genetischen...
a) die Sequenz gemäß SEQ ID NO:1 oder 3; oder
b) ein Polynucleotid, das zu dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 eine Identität von mehr als 50% aufweist; oder
c) ein Polynucleotid, die mit dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 unter stringenten Bedingungen hybridisiert; oder
d) ein Polynucleotid, welches für ein Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert; oder
e) ein Polynucleotid, das zu einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, eine Identität von mehr als 50% aufweist; oder
f) ein Polynucleotid, das mit einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, unter stringenten Bedingungen hybridisiert; oder
g) ein Polynucleotid, das von einem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 aufgrund der Degeneration des genetischen...
Description
- Die Erfindung betrifft ein neues Oocysten Sporocysten Protein (EtOS22) des Parasiten der Art Eimeria tenella sowie das für dieses Protein kodierende Polynucleotid, Vektoren enthaltend dieses Polynucleotid, mit diesen Vektoren transformierte Zellen, Antikörper gerichtet gegen das Protein, Impfstoffe, die das Polynucleotid-, das Protein, Fragmente davon, die o.g. Vektoren oder Antikörper gerichtet gegen das Protein enthalten und die Verwendung des Polynucleotids oder des Polypeptids zum Auffinden von Wirkstoffen zur Behandlung einer Infektion mit Eimeria und Wirkstoffe, die zur Therapie einer Infektion mit Eimeria geeignet sind.
- Parasiten der Gattung Eimeria sind obligat intrazelluläre Protozoen, mit einem komplizierten Lebenszyklus, der in Abfolge geschlechtliche und ungeschlechtliche Entwicklungsstadien hervorbringt. Eimeria tenella lebt im Blinddarm des Haushuhns (Gallus domesticus) und ist mit den humanpathogenen Krankheitserregern Toxoplasma gondii, Plasmodium falciparum, Cryptosporidium parvum sowie bedeutenden tierpathogenen Gattungen Sarcocystis, Neospora, Babesia und Theileria eng verwandt. Vertreter dieser Gattungen gehören nach der systematischen Einordnung der Protozoen durch LEVINE (1980) zum Stamm der Apicomplexa.
- Eimeria tenella ist der Erreger der Geflügelkokzidiose, einer Erkrankung, die mit der intensiven Bodenhaltung von Küken und Hühnern zu einem ökonomisch bedeutsamen Problem wurde. Zur Pathologie einer Kokzidiose-Erkrankung gehören blutige Durchfälle, die durch eine verminderte Nahrungsaufnahme und einen Gewichtsverlust der Hühner große wirtschaftliche Schäden verursachen können. Neben Eimeria tenella sind sechs weitere Eimeria Arten für die Kokzidioseerkrankung im Haushuhn verantwortlich: Eimeria acervulina, Eimeria maxima, Eimeria brunetti, Eimeria necatrix, Eimeria praecox.
- Die Infektionsformen apikomplexer Parasiten (Sporozoiten und Merozoiten) sind durch besondere morphologische Eigenschaften gekennzeichnet, die sie von anderen Sporozoen eindeutig unterscheiden. Als wichtigstes Merkmal gilt ein „Apikalkomplex" am vorderen Zellpol, der sich aus drei sekretorisch tätigen Organellen (Rhoptrien, Mikronemen und Dichten Granula) sowie dem strukturbildenden Conoid mit Polringen und subpelliculären Mikrotubuli zusammensetzt.
- Eimeria tenella durchläuft eine monoxene Entwicklung im Haushuhn (Gallus gallus). Der Parasit ist streng wirtsspezifisch und obligat intrazellulär. Die Vermehrung erfolgt in epithelialen Zellen und in der Submucosa des Cecums. Das Haushuhn infiziert sich mit Eimeria tenella bei der Nahrungssuche. Nach oraler Aufnahme sporulierter Oocysten und ihrer mechanischen Bearbeitung im Kaumagen werden reife, ruhende Sporozoiten im Dünndarm unter dem Einfuß von Trypsin und Gallensalzen aus den Sporocysten am sogenannten Stiedakörper freigesetzt. Die Sporozoiten erlangen ihre Beweglichkeit und besiedeln Wirtszellen im Blinddarm unter Ausbildung einer parasitophoren Vakuole. Die parasitophore Vakuole schützt den intrazellulären Parasiten vor lysosomaler Verdauung. In ihrem Schutz bilden sich vielkernige Schizonten. Die Schizogonie (Merogonie) ist eine asexuelle Vermehrung des Parasiten. Bewegliche Merozoiten schnüren sich aus dem Schizonten rosettenartig ab. Nach ihrer Freisetzung gründen die Merozoiten bis zu 3 weitere Generationen von Schizonten in benachbarten Blinddarmzellen. Während einer akuten Infektion lösen die Schizogoniezyklen breitflächige Darmläsionen aus, die zu Darmblutungen, Gewichtsverlust und bei schwerer Infektion zum Tode des Wirtes führen können. Nach Ablauf der Schizogoniezyklen beginnt die Gamogonie unter Bildung vielkerniger männlicher Mikrogamonten und einkerniger weiblicher Makrogamonten, die zu Gameten reifen. Der Makrogamet enthält eosinophile „Wallforming" bodies, die nach der Befruchtung fusionieren und die Oocystenwand ausbauen. Nach Ablauf der Präpatenzzeit von 6 Tagen werden neue Oocysten ausgeschieden. Mit der Ausscheidung der Oocysten ist die Infektion abgeschlossen und der Wirt hat eine artspezifische Immunität erworben.
- Diagnostik
- Die genaue, schnelle und kostengünstige Identifikation der Kokzidien Art bei Hühnern ist für die Prophylaxe und die Behandlung einer Infektion von größter Bedeutung. Die derzeit routinemäßig angewandte Identifikation der sieben Eimeria Arten im Huhn erfolgt nach Long and Reid 1982 entsprechen der Oocysten Morphologie (mikroskopisch), der Wirtsspezifität, der Pathologie der Läsionen im Darm und der Präpatenzzeit. Daneben gibt es auch den Versuch einer biochemischen Charakterisierung über Isoenzymmuster. Hierbei dienen zumeist Enzyme des Zuckerstoffwechsels als genetische Marker zur Erstellung eines Zymograms (Johnston and Fernando 1997). Es zeigt sich, dass sowohl die herkömmliche morphologisch deskriptive Vorgehensweise, als auch die biochemischen Methoden eine genaue Art-Differenzierung nur unzureichend oder nur teilweise erzielt werden kann. Eine Charakterisierung der Art auf gentechnischer Ebene ist daher wünschenswert. Auf diesem Gebiet liegen bisher nur sehr wenige widersprechende Untersuchungsergebnisse vor (Comes and al. 1996). Die biologische Verschiedenheit von Eimeria Arten lässt vermuten, dass genetische Unterschiede in Form von DNA-Polymorphismen in den unterschiedlichen Arten vorliegen. Polymorphismen können durch Basenveränderungen (Deletion, Insertion) oder durch Chromosomenumlagerungen entstehen. In der ursprünglich für die Verwandtschaftsanalyse entwickelten Methode des DNA-Fingerprintings wird die variable DNA mit Hilfe von Restriktions-Endonzukleasen zerschnitten, mit radioaktiven DNA-Sonden hybridisiert und nach Gelelektrophorese und Southern blot in der Autoradiographie sichtbar gemacht. Mit Hilfe des so entstandenen genetischen Fingerabdrucks können Art und Stamm von Organismen eindeutig unterschieden werden. Eine Vereinfachung diese Ansatzes bietet die RAPD-PCR-Methode „Random Amplified Polymorphic DNA-Polymerase Kettenreaktion". Die Methode basiert auf der Amplifikation genomischer DNA mit einzelnen Primern einer zufälligen Nukleotidsequenz in einer Polymerase-Kettenraktion (PCR). Amplifizierte DNA-Segmente (RAPD-PCR Marker) ergeben nach Trennung auf einem Agarosegel und Färbung mit Ethidium-Bromid ein spezifisches Bandenmuster. Mit dieser Methode können jedoch nur Reinstämme voneinander unterschieden werden. Eine Identifikation einer Art in einem Feldisolat (Mischung verschiedener Eimeria Arten) ist mit diesen Techniken nicht möglich. Eine technisch einfachere und daher kostengünstigere Methode wäre das Auffinden einer spezifische Sonde basierend auf einer spezifischen Gensequenz. Bisher wurden hierfür nur bekannte Sequenzen ribosomaler DNA (Ellis and Bumstead 1990) aus dem Bereich Internal Transcriber Spacer ITS1 (Schnitzler et al. 1998) und ITS2 (Gasser et al. 2001) eingesetzt sowie ein Sporozoiten Antigen von Eimeria acervulina (EASZ 240/160) (Molloy et al. 1998).
- Therapie
- Zur Therapie dieser Erkrankung werden derzeit Antikokzidia im Wert von jährlich mindestens 300 Millionen US $. aufgewendet. Die chemotherapeutische Behandlung wird seit 1970 vor allem mit den Polyether-Ionophoren Monensin, Narasin, Salinomycin und Lasalocid durchgeführt. Daneben sind auch zahlreiche Wirkstoffe, welche die DNA-Synthese oder die Proteinsynthese der Parasitenstadien hemmen, auf dem Markt (Greif 2001). Die gegenwärtigen Therapien haben jedoch schwerwiegenden Probleme bzw. Nachteile. Neben der schwerwiegenden Medikamentenbelastung des Huhns (Rückstandsproblematik in essbaren Geweben), der ökotoxikobiolgischen Belastung (der Umwelt), gilt die Entstehung von Medikament-Resistenz als größtes Problem der Antikokzidia-Behandlung. Den entstehenden Resistenzen sucht man mit sogenannten Pendel- oder Rotationsprogrammen und der kostenaufwendigen Suche nach neuen Wirkungsmechanismen zu begegnen (Coombs 2002). Es besteht daher ein dringender Bedarf an verbesserten Wirkstoffen zur Behandlung von Eimeria-Infektionen und an Methoden zum Auffinden dieser Wirkstoffe.
- Impfung
- Eine weitaus bessere Alternative zur chemotherapeutischen Behandlung der Kokzidiose wäre die Immunprophylaxe (Impfung). Ein Erregerkontakt mit Eimeria Arten führt zur fast vollständigen Immunität gegen eine homologe Zweitinfektion. (Rose und Wakelin 1990). Eine kontinuierliche Gabe von Parasitenstadien über einen Zeitraum von 16-25 Tagen induziert in Eintagsküken eine natürliche Immunität gegen Eimeria tenella, Eimeria acervulina und Eimeria maxima (Stiff und Bafundo 1993). Die Immunisierung von Legehennen mit protektiven Gametocyten-Antigenen verbessert die Immunitätslage geschlüpfter Küken. Diese Strategie wurde als „Maternale Immunisierung" von Wallach entwickelt (Wallach 1992).
- Impfstoffprogramme mit vollvirulenten Eimeria Stämmen (Oocysten-Lebendimpfstoffe) werden derzeit mit den käuflichen Produkten Immucox® (Vetech Laboratories, Canada) und Coccivac® (Sterwin Laboratories, USA) durchgeführt. Die Produkte Paracox® (Schering Plough, England) und Livacox® (Williams 2002) basieren auf sogenannten attenuierten virulenten Stämmen. Seit kurzem sind auch polyetherresistente virulente Lebendvakzine auf dem Markt (Vermeulen 2001). Insgesamt sind derzeit 13 Oocysten enthaltende Lebendimpfstoffe für die Immunisierung gegen Hühner-Kokzidiose registriert (Chapman et al. 2002, Williams 2002).
- Alle diese auf dem Markt befindlichen Impfstoffe haben jedoch den wirtschaftlichen Nachteil hoher Produktionskosten sowie die notwendige Tierhaltung für die Passage der Oocysten. Ein zusätzliches Problem ist, dass die auf dem Markt befindlichen Lebendimpfstoffe zum pathogenen Ausgangstyp zurückzumutieren könnten.
- Es besteht daher ein dringender Bedarf verbesserte Impfstoffe zur Verfügung zu stellen, die vorzugsweise nicht-lebend Impfstoffe sind.
- Als „Traumziel" aller Immunisierungsverfahren gilt die gentechnologische Entwicklung eines rekombinanten Impfstoffes („subunit vaccine") auf der Grundlage sogenannter protektiver Antigene. Protektive Antigene sind Strukturverbindungen des Parasiten, die während der Parasit-Wirtszellinteraktion eine wichtige Funktion bei der Zellerkennung, der Zellanhaftung und der Zellinvasion haben sowie möglicherweise auch andere Proteine, deren Funktion bisher nicht bekannt wurde. Die bisherige Suche nach protektiven Eimeria tenella Antigenen erfasst Antigene der Oberfläche, der inneren Organellen sowie gradienten-isolierte Organell-Antigene von Oocysten, Sporozoiten und Merozoiten (Vermeulen 1998). Neben der gezielten Suche nach Gensequenzen bereits bekannter Proteine wird auch die zufällige Suche in EST-(expressed sequence tags) Datenbanken (Wang et al. 1999) oder Phagen Display Banken (Silva, A. et al 2002) zur Auffindung neuer Gene und Targets eingesetzt.
- Auf der Grundlage rekombinanter DNA-Techniken konnten Danforth et al. (1985) erstmals ein 60-70 kDa Antigen von Eimeria tenella in vitro herstellen. Seit diesem Versuch wurden einige wenige ausgewählte Eimeria tenella Oocysten-Antigene (Clark 1986, Crane et al. 1991, Bhogal et al 1992, Eschenbacher et al. 1996), Eimeria tenella Sporozoiten-Antigene (Files et al 1987, Miller et al. 1989) sowie Eimeria tenella Merozoiten Antigene (Ko et al. 1990, Binger et al. 1993) rekombinant hergestellt. Crane et al. (1991) konnten mit einem rekombinanten Eimeria tenella Antigen eine kreuzreagierende Protektion gegen vier Eimeria Arten des Huhns induzieren. Trotz vieler Immunisierungsversuche mit rekombinanten Antigenen wurden bis heute jedoch keine zufriedenstellenden Ergebnisse erzielt, sodass ein hoher Bedarf nach der Identifizierung neuer, bisher unbekannter Antigene und ihrer zugehörigen Gensequenzen besteht (Jenkins 1998, Vermeulen 2001).
- Beschreibung der Figuren
-
1 zeigt die DNA- und die abgeleitete Aminosäuresequenz der EtOS22-cDNA. Die Primer A17-f-length-64-up und A17-f-length-1176-lo sind durch Unterstriche gekennzeichnet. Das Signalpeptid ist hellgrau unterlegt. Die ursprüngliche Sequenz des im Phagen-Panning angereicherten Klons, von der aus die 5'- und 3'-RACE-PCR durchgeführt wurden, ist dunkelgrau unterlegt. -
2 zeigt die Bestimmung des Expressionsmusters von EtOS22 durch RT-PCR. Als DNA-Längenstandard (M) wurde jeweils 1 μg pUC-Mix Marker (MBI Fermentas, St. Leon-Rot) verwendet. Als Templat für die PCR-Reaktionen diente cDNA aus nicht-infizierten Hühnchenblinddärmen (n.i.), sowie aus infizierten Hühnchenblinddärmen 72, 137 und 148 h nach der Infektion (72 hpi, 137 hpi und 148 hpi) und aus sporulierten Oocysten. Die durch die Primer A17-22-up und A17-112-lo amplifizierten PCR-Produkte haben eine Größe von 91 bp (A), die durch die Primer EtACTIN-up und EtACTIN-lo amplifizierten PCR-Produkte sind 350 bp groß (B). In den mit (1) gekennzeichneten Spuren wurden jeweils Reaktionsansätze mit Reverser Transkriptase (RT) und mit RNA-Templat aufgetragen, in (2) Reaktionsansätze ohne RT und mit RNA-Templat und in (3) Reaktionsansätze mit RT aber ohne RNA-Templat. -
3 zeigt die Northern Blot Analyse für EtOS22. Gelelektrophoretisch aufgetrennt und geblottet wurde Gesamt-RNA aus sporulierten Oocysten (1), Gesamt-RNA aus infiziertem Hühnchenblinddarm 137 h (2), 148 h (3) nach der Infektion sowie Gesamt-RNA aus nicht-infiziertem Hühnchenblinddarm als Negativkontrolle (4). Hybridisiert wurde mit dem radioaktiv markierten, in Position 385 beginnenden, 3'-RACE-PCR-Produkt (816 bp). -
4 zeigt einen genomischen Southern Blot für EtOS22. Gelelektrophoretisch aufgetrennt und geblottet wurden in jeder Spur 10 μg genomische DNA von E. tenella. Diese wurde zuvor mit folgenden Restriktionsendonukleasen verdaut: BglI (1), ClaI (2), KpnI (3), AccI (4), BglII (5), DraI (6) und MvaI (7). Hybridisiert wurde mit dem radioaktiv markierten PCR-Produkt von Position 1 bis Position 1.106 (1106 bp). -
5.1 –5.3 zeigen Immunfluoreszenz gegen EtOS22 in E. tenella. Als Primärantikörper wurde der mAk E2E5 (IgG2a aus der Maus), als Sekundärantikörper ein Ziege anti-Maus IgG Konjugat Alexa 488 verwendet. Zu erkennen sind eine leere Oocystenschale (5.1 ), eine ruptierte Sporocyste (5.2 ) und eine intakte Sporocyste (5.3 ). Der Bereich des Stieda-Körpers ist mit einem Pfeil markiert. -
6 zeigt, dass EtOS22 ein Target zur Hemmung der Excystierung von E. tenella ist. Nach parallelen Excystierungsversuchen wurden im Versuchsansatz ohne Zugabe des mAk E2E5 (Kontrolle) sowie im Versuchsansatz mit Zugabe des mAk E2E5 die Anzahl freier Sporozoiten und die Anzahl von Sporocysten mit ungeschlüpften Sporozoiten bestimmt. - Beschreibung der Erfindung
- Gegenstand der Erfindung ist ein neues Oocysten Sporocysten Protein (EtOS22) des Parasiten der Art Eimeria tenella.
- Gegenstand der Erfindung ist auch das für dieses Protein kodierende Polynucleotid. SEQ ID NO:1 zeigt die "full-length" mRNA, die die für das neue Oocysten Sporocysten Protein von Eimeria tenella kodierende DNA-Sequenz enthält. Der offene Leserahmen (ORF), der für das Protein (SEQ ID NO:2) kodiert, wird in SEQ ID NO:3 gezeigt.
- Weiterhin basiert die Erfindung auf der Entdeckung, dass das neue Protein EtOS22 des Parasiten der Art Eimeria tenella an der Excystierung der Sporozoiten aus den Sporocysten beteiligt ist und damit für den Lebenszyklus des Parasiten essentiell ist. Die Excystierung kann mit Antikörpern gegen EtOS22 gehemmt werden.
- EtOS22 ist ein Intron-loses Gen, das aus einem einzigen codierenden Exon besteht. Das ORF des EtOS22 Gens mit einer Grösse von 594 bp findet sich in zwei Kopien im Genomklon 2257242.c007101021.Contig1 (71.864 bp, Stand: 03.03.2003).
- EtOS22 kommt aber vermutlich mit deutlich mehr als 2 Kopien im Genom von Eimeria tenella vor.
- Gegenstand der Erfindung ist auch:
- a) ein Polynucleotid, das zu dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 eine Identität von mehr als 50%, 60%, 70% oder 80%, bevorzugt mehr als 85% oder 90% und besonders bevorzugt mehr also 95% oder 97% aufweist;
- b) ein Polynucleotid, das mit dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 unter stringenten Bedingungen hybridisiert;
- c) ein Polynucleotid, das zu einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, eine Identität von mehr als 50%, 60%, 70% oder 80%, bevorzugt mehr als 85% oder 90% und besonders bevorzugt mehr also 95% oder 97% aufweist;
- d) ein Polynucleotid, das mit einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, unter stringenten Bedingungen hybridisiert;
- e) ein Polynucleotid, das von dem Polynucleotid nach SEQ ID NO:1 aufgrund der Degeneration des genetischen Codes abweicht; und
- f) ein Polynucleotid, das ein Fragment eines Polynucleotids nach a) bis e) darstellt und mindestens 6 Nucleotide oder 8 Nucleotide, bevorzugt mehr als 10 oder 20 Nukleotide, besonders bevorzugt mehr als 50 oder 100 Nucleotide und ganz besonders bevorzugt mehr als 200 oder mehr als 500 Nucleotide lang ist.
- Ein Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 sowie die vorstehend genannten Polynucleotide a) bis f) werden nachfolgend als EtOS22-Polynucleotide bezeichnet.
- Gegenstand der Erfindung ist weiterhin ein Polypeptid, das von einer Nukleinsäure gemäß a) bis f) kodiert wird und mindestens 8 Aminosäuren lang ist. Dieses Polypeptid sowie das Polypeptid gemäß SEQ ID NO:2 wird nachfolgend als EtOS22-Polypeptid bezeichnet.
- Gegenstand der Erfindung ist außerdem ein Expressionssystem oder Vektor enthaltend wenigstens eines der Polynucleotide gemäß a) bis f) und eine Expressionskontrollsequenz. Das Expressionssystem ermöglicht die Expression des erfindungsgemäßen EtOS22-Polypeptids.
- Bevorzugt steht dabei die Expression von EtOS22 unter Kontrolle des Cytomegalovirus (CMV) Promotors. Ein BGH- (bovine growth hormone) Polyadenylierungssignal widerum beendet die Transkription und sorgt für die Polyadenylierung der mRNA.
- Besonders bevorzugt als Expressions-Kontrollsequenz sind z.B. der frühe oder späte Promotor des SV40- oder Adenovirus, das lac-System, das trp System, das TAC System, das TRC System, die Haupt-Operator- und Promotorregionen des Phagen λ, die Kontrollregionen des fd-Hüllproteins, der Promotor der 3-Phospoglycerat Kinase, der Promotor der Sauren Phosphatase und der Promotor des α-Mating Faktors der Hefe.
- Gegenstand der Erfindung ist auch eine Wirtszelle, die den oben beschriebenen Vektor oder das Expressionssystem enthält.
- Bevorzugte Beispiele für die Wirtszelle sind: E. coli, Pseudomonas, Bacillus, Streptomyces, Hefe-, CHO-, R1.1-, B-W-, L-M-, COS 1-, COS 7-, BSC1-, BSC40- und BMT10-Zellen, Pflanzenzellen, Insektenzellen und Säugerzellen in Zellkultur. Die Expression in einem eukaryontischen System erfolgt besonders bevorzugt im Baculovirus-System, besonders in einem System, das das Einführen posttranslationaler Modifikationen gestattet.
- Die Erfindung betrifft ebenfalls Fusionsproteine, die ein EtOS22-Polypeptid wie oben beschrieben umfassen. Das Fusionsprotein kann dabei einen weiteren Polypeptidanteil enthalten, der für eine zusätzliche Aktivität des Fusionsproteins relevant ist [z.B β-Galactosidase, β-Glucuronidase, Green Fluorescent Protein (GFP), autofluoreszierende Proteine, wie blue fluorescent protein (BFP), Glutathione-S-Transferase (GST), Luciferase, Meerettich-Peroxidase (HRP), Chloramphenicol-Acetyltransferase (CAT)]. Zusätzlich oder alternativ können Epitop-Tags Teil des Fusionsproteins sein [z.B. His-Tags, FLAG-Tags, Influenza Hemagglutinin (HA)-Tags, Myc-Tags, VSV-G-Tags oder Thioredoxin (Trx)-Tags]. Fusionsproteine können auch Maltosebindingsprotein (MBP), S-Tags, Lex-DNA-Bindingsdomänen, GAL4-DNA-Bindingsdomänen oder Herpes Simplex Virus (HSV) BP16-Protein enthalten.
- Gegenstand der Erfindung ist auch ein Verfahren zur Herstellung eines EtOS22-Polypeptids oder eines Fusionsproteins wie oben beschrieben in passenden prokaryontischen oder eukaryontischen Expressionssystemen. Die Expression kann dabei permanent oder transient in einer jeweils entsprechenden Zelllinie bzw. entsprechenden Wirtszellen wie oben beschrieben erfolgen. Passende prokaryontische Expressionssysteme sind bekannte Wirts-Vektor-Systeme wie Bakterien (z.B. Streptomyces spp., Bacillus subtilis, Salmonella typhimurium, Serratia marcescens und besonders Escherichia coli).
- Gegenstand dieser Erfindung ist ebenfalls die Verwendung von EtOS22-Polynucleotiden zur Detektion von Polynucleotiden aus Parasiten der Gattung Eimeria, bevorzugt Eimeria acervulina, Eimeria maxima, Eimeria brunetti, Eimeria necatrix, Eimeria praecox und besonders bevorzugt Eimeria tenella. Die Erfindung bezieht sich dabei auf Polynucleotide, die mit Polynucleotiden der oben genannten Parasiten hybridisieren können. Die Erfindung bezieht sich insbesondere auf die Verwendung dieser Polynucleotide als:
- a) Proben in Northern- oder Southern-Blot-Assays,
- b) Polynucleotide oder Oligonucleotide, die auf Micro- oder Makroarrays gebunden sind,
- c) Primer für die PCR oder analoge Verfahren die für die Diagnostik der oben genannten Parasiten eingesetzt werden, wobei die DNA der betreffenden Parasiten spezifisch mit Hilfe der Primer und der PCR-Technik identifiziert und amplifiziert wird.
- Gegenstand dieser Erfindung sind ebenfalls Antikörper, die spezifisch mit einem Epitop eines EtOS22-Polypeptids reagieren.
- Gegenstand dieser Erfindung sind ebenfalls besonders monoklonale Antikörper, die spezifisch mit einem Epitop eines EtOS22-Polypeptids reagieren.
- Gegenstand dieser Erfindung ist ebenfalls die Verwendung der vorstehend genannten Antikörper als Parasitizide. Bevorzugt werden Antikörper zur Behandlung von Infektionen mit Eimeria und besonders bevorzugt zur Behandlung von Infektionen mit Eimeria tenella eingesetzt. Bevorzugt werden Infektionen von Geflügel und besonders bevorzugt Infektionen von Hühnern mit den vorstehend genannten Antikörpern behandelt.
- Diagnostik
- Ein weitere Gegenstand dieser Erfindung ist die Verwendung von EtOS22-Polynucleotiden oder den o.g. Antikörpern gegen EtOS22-Polypeptide zur Diagnostik von Eimeria-Infektionen und bevorzugt Infektionen mit Eimeria tenella.
- Ein Gegenstand der Erfindung ist auch ein Kit, enthaltend EtOS22-Polynucleotide oder Antikörper gegen EtOS22-Polypeptide sowie eine Anleitung zur Durchführung des diagnostischen Verfahrens.
- Impfstoffe
- Gegenstand der Erfindung ist ebenfalls ein Verfahren zur Herstellung einer immunogenen Zusammensetzung zur Immunisierung von Geflügel und bevorzugt Hühnern, bestehend aus mindestens einem der vorstehend genannten erfindungsgemäßen EtOS22-Polypeptide oder aus mindestens einem der vorstehend genannten Antikörper.
- Gegenstand der Erfindung ist ebenfalls die Verwendung der vorstehend beschriebenen Expressionsvektoren enthaltend eines der vorstehend genannten EtOS22-Polynucleotide zur Herstellung einer immunogene Zusammensetzung zur Verabreichung in einem Wirt zur Aktivierung einer protektiven Immunantwort in diesem Wirt, die auf das EtOS22-homologe Protein von Eimeria oder auf das EtOS22-Protein von Eimeria tenella gerichtet ist.
- Gegenstand dieser Erfindung ist auch die Verwendung der vorstehend genannten EtOS22-Polypeptide zur Herstellung von Impfstoffen gegen Kokzidiose.
- Gegenstand der Erfindung ist auch:
- 1. Ein inaktivierter Impfstoff enthaltend a) mindestens eines der vorstehend genannten EtOS22-Polypeptide, welches i. aus dem Parasitenstadien isoliert wird, oder ii. in vitro synthetisch hergestellt wird, oder iii. mit Hilfe rekombinanter DNA-Technologie hergestellt wird; oder b) ein Fusionsprotein wie oben genannt umfassend eines der vorstehend genannten EtOS22-Polypeptide, wobei das Polypeptid oder Fusionsprotein durch Amidierung, Carboxylierung oder Phosphorylierung in vivo oder in vitro verändert worden sein kann.
- 2. Ein Vektor-Impfstoff enthaltend: a) einen selbstreplizierenden Vektor (z.B. Bakterien, Pilze, Viren), der eines der vorstehend beschriebenen EtOS22-Polynucleotide enthält, welcher bevorzugt zur Langzeitsynthese eines EtOS22-Polypeptids und zur Antigen-Präsentation führt, wodurch das Immunsystem stimuliert wird; oder b) ein Plasmid, welches ein EtOS22-Polynucleotid enthält; oder c) ein reines EtOS22-Polynucleotid (nackte DNA).
- 3. Ein passiver Impfstoff enthaltend: a) Antikörpern, die gegen immunogene Epitope EtOS22-Polypeptids gerichtet sind; oder b) anti-idiotypische Antikörper, d.h. Antikörpern, die sich gegen den Idiotyp der Antikörper richten, die an ein EtOS22-Polypeptid binden.
- Screening-Verfahren
- Gegenstand dieser Erfindung ist ebenfalls ein Verfahren zum Identifizieren von Wirkstoffen, wie z.B. kleinen organischen Molekülen, Peptiden oder Antikörpern, die die Funktion des EtOS22-Polypeptids gemäß SEQ ID NO:2 modulieren und dadurch die Excystierung der Sporozoiten aus den Sporocysten von Eimeria modulieren. Der Grad der Modulation beträgt wenigstens 10%, bevorzugt wenigstens 20%, besonders bevorzugt wenigstens 30% und ganz besonders bevorzugt wenigstens 50%.
- Gegenstand der Erfindung ist auch ein Verfahren zum Auffinden von Wirkstoffen, die die Aktivität des EtOS22-Proteins bei der Excystierung von Sporozoiten aus Sporocysten modulieren bei dem man:
- a) den zu testenden Wirkstoff mit einem EtOS22-Polypeptid nach Anspruch 2 in Kontakt bringt, wobei die gewählten Bedingungen eine spezifische Bindung der Testsubstanz an das EtOS22-Polypeptid gestatten; und
- b) eine erfolgte spezifische Bindung an das Polypeptid detektiert;
- Gegenstand der Erfindung ist auch ein Verfahren zum Auffinden von Wirkstoffen, die die Aktivität des EtOS22-Proteins bei der Excystierung von Sporozoiten aus Sporocysten modulieren bei dem man:
- a) den zu testenden Wirkstoff mit einem EtOS22-Polypeptid nach Anspruch 2 in Kontakt bringt, wobei die gewählten Bedingungen eine spezifische Bindung der Testsubstanz an das EtOS22-Polypeptid gestatten; und
- b) eine Modulation der Aktivität des Polypeptids nach Anspruch 2 oder des EtOS22-Proteins nachweist;
- Gegenstand der Erfindung ist auch Verfahren zum Auffinden von Wirkstoffen zur Therapie der Kokzidiose, bei dem das EtOS22-Protein in seiner rekombinanten Form zum Screenen von Banken chemischer Verbindungen basierend auf Affinitätsselektion und Massenspektrometrie eingesetz wird. Zur Auffindung von Inhibitoren für das Targetprotein mit unbekannter, aber für das Überleben des Sporozoiten essentieller Funktion, können Screeningverfahren eingesetz werden, die Substanzbanken im Hinblick auf Affinität zum Protein testen. Eine Screening-Möglichkeit ist die Affinitätsselektion aus Substanzgemischen mit anschließender Detektion der Liganden im Massenspektrometer. Dazu müssen definierte Substanzgemische verwendet werden, aus denen mit Hilfe der Massendetektion einzelne Substanzen identifiziert werden können. Deshalb eigenen sich für diese Screeningmethode insbesondere Substanzgemische, die aus kombinatorischen Synthesen hergestellt worden sind.
- Substanzen, die in der Affinitätsselektion auffallen, werden weiteren Prüfungen wie dem Eimeria tenella in vitro-Test unterworfen.
- Gegenstand der Erfindung sind ebenfalls neue Wirkstoffe, die mit Hilfe der vorstehend beschriebenen Verfahren identifiziert werden und geeignet sind, die Excystierung der Sporozoiten aus den Sporocysten von Eimeria zu modulieren. Die neuen Wirkstoffe modulieren die Excystierung um wenigstens 10%, bevorzugt um wenigstens 20%, besonders bevorzugt um wenigstens 30% und ganz besonders bevorzugt um wenigstens 50%.
- Gegenstand der Erfindung sind ebenfalls neue Wirkstoffe, die die Excystierung der Sporozoiten aus den Sporocysten von Eimeria modulieren. Die neuen Wirkstoffe modulieren die Excystierung um wenigstens 10%, bevorzugt um wenigstens 20%, besonders bevorzugt um wenigstens 30% und ganz besonders bevorzugt um wenigstens 50%.
- Gegenstand der Erfindung ist ebenfalls die Verwendung von neuen Wirkstoffen, die mit einem der vorstehend beschriebenen Verfahren identifiziert wurden, zur Herstellung eines Medikamentes zur prophylaktischen oder therapeutischen Behandlung von Geflügel und bevorzugt Hühnern, die von Eimeria befallen werden können oder befallen wurden. Die erfindungsgemäßen Medikamente enthalten mindestens einen der mit einem der vorstehend beschriebenen Verfahren identifizierten Wirkstoffe und können nasal, dermal, parenteral oder enteral verabreicht werden.
- Gegenstand der Erfindung ist ebenfalls die Verwendung von neuen Wirkstoffen, die die Excystierung der Sporozoiten aus den Sporocysten von Eimeria um wenigstens 10%, bevorzugt um wenigstens 20%, besonders bevorzugt um wenigstens 30% und ganz besonders bevorzugt um wenigstens 50% modulieren, zur Herstellung eines Medikamentes zur prophylaktischen oder therapeutischen Behandlung von Kokzidiose. Bevorzugt werden die Wirkstoffe verwendet zur Herstellung eines Medikamentes zur Behandlung von Geflügel und besonders bevorzugt Hühnern, die von Eimeria befallen werden können oder befallen wurden. Die erfindungsgemäßen Medikamente enthalten mindestens eine der mit einem der vorstehend beschriebenen Verfahren identifizierten Wirkstoffen und können nasal, dermal, parenteral oder enteral verabreicht werden.
- Pharmazeutische Zusammensetzungen
- Die Anwendung kann sowohl prophylaktisch als auch therapeutisch erfolgen.
- Die Anwendung der Wirkstoffe erfolgt direkt oder in Form von geeigneten Zubereitungen enteral, parenteral, dermal, nasal.
- Die enterale Anwendung der Wirkstoffe geschieht z.B. oral in Form von Pulver, Zäpfchen, Tabletten, Kapseln, Pasten, Tränken, Granulaten, Drenchen, Boli, medikiertem Futter oder Trinkwasser. Die dermale Anwendung geschieht z.B. in Form des Tauchens (Dippen), Sprühens (Sprayen), Badens, Waschens, Aufgießens (pour-on and spot-on) und des Einpuderns. Die parenterale Anwendung geschieht z.B. in Form der Injektion (intramusculär, subcutan, intravenös, intraperitoneal) oder durch Implantate.
- Geeignete Zubereitungen sind: Lösungen wie Injektionslösungen, orale Lösungen, Konzentrate zur oralen Verabreichung nach Verdünnung, Lösungen zum Gebrauch auf der Haut oder in Körperhöhlen, Aufgußformulierungen, Gele; Emulsionen und Suspension zur oralen oder dermalen Anwendung sowie zur Injektion; Halbfeste Zubereitungen; Formulierungen, bei denen der Wirkstoff in einer Salbengrundlage oder in einer Öl in Wasser oder Wasser in Öl Emulsionsgrundlage verarbeitet ist; Feste Zubereitungen wie Pulver, Premixe oder Konzentrate, Granulate, Pellets, Tabletten, Boli, Kapseln; Aerosole und Inhalate, wirkstoffhaltige Formkörper.
- Injektionslösungen werden intravenös, intramuskulär und subcutan verabreicht. Injektionslösungen werden hergestellt, indem der Wirkstoff in einem geeigneten Lösungsmittel gelöst wird und eventuell Zusätze wie Lösungsvermittler, Säuren, Basen, Puffersalze, Antioxidantien, Konservierungsmittel zugefügt werden. Die Lösungen werden steril filtriert und abgefüllt.
- Als Lösungsmittel seien genannt: Physiologisch verträgliche Lösungsmittel wie Wasser, Alkohole wie Ethanol, Butanol, Benzylalkohol, Glycerin, Kohlenwasserstoffe, Propylenglykol, Polyethylenglykole, N-Methylpyrrolidon, sowie Gemische derselben.
- Die Wirkstoffe lassen sich gegebenenfalls auch in physiologisch verträglichen pflanzlichen oder synthetischen Ölen, die zur Injektion geeignet sind, lösen. Als Lösungsvermitler seien genannt: Lösungsmittel, die die Lösung des Wirkstoffs im Hauptlösungsmittel fördern oder sein Ausfallen verhindern. Beispiele sind Polyvinylpyrrolidon, polyoxyethyliertes Rhizinusöl, polyoxyethylierte Sorbitanester.
- Konservierungsmittel sind: Benzylalkohol, Trichlorbutanol, p-Hydroxybenzoesäureester, n-Butanol.
- Orale Lösungen werden direkt angewendet. Konzentrate werden nach vorheriger Verdünnung auf die Anwendungskonzentration oral angewendet. Orale Lösungen und Konzentrate werden, wie oben bei den Injektionslösungen beschrieben, hergestellt, wobei auf steriles Arbeiten verzichtet werden kann.
- Lösungen zum Gebrauch auf der Haut werden aufgeträufelt, aufgestrichen, eingerieben, aufgespritzt, aufgesprüht oder durch Tauchen (Dippen), Baden oder Waschen aufgebracht. Diese Lösungen werden, wie oben bei den Injektionslösungen beschrieben, hergestellt.
- Es kann vorteilhaft sein, bei der Herstellung Verdickungsmittel zuzufügen. Verdickungsmittel sind: Anorganische Verdickungsmittel wie Bentonite, kolloidale Kieselsäure, Aluminiummonostearat, organische Verdickungsmittel wie Cellulosederivate, Polyvinylalkohole und deren Copolymere, Acrylate und Methacrylate.
- Gele werden auf die Haut aufgetragen oder aufgestrichen oder in Körperhöhlen eingebracht. Gele werden hergestellt, indem Lösungen, die wie bei den Injektionslösungen beschrieben hergestellt worden sind, mit soviel Verdickungsmittel versetzt werden, daß eine klare Masse mit salbenartiger Konsistenz entsteht. Als Verdickungsmittel werden die weiter oben angegebenen Verdickungsmittel eingesetzt.
- Aufgießformulierungen werden auf begrenzte Bereiche der Haut aufgegossen oder aufgespritzt, wobei der Wirkstoff entweder die Haut durchdringt und systemisch wirkt oder sich auf der Körperoberfläche verteilt.
- Aufgießformulierungen werden hergestellt, indem der Wirkstoff in geeigneten hautverträglichen Lösungsmitteln oder Lösungsmittelgemischen gelöst, suspendiert oder emulgiert wird. Gegebenenfalls werden weitere Hilfsstoffe wie Farbstoffe, resorptionsfördernde Stoffe, Antioxidantien, Lichtschutzmittel, Haftmittel zugefügt.
- Als Lösungsmittel seien genannt: Wasser, Alkanole, Glycole, Polyethylenglycole, Polypropylenglycole, Glycerin, aromatische Alkohole wie Benzylalkohol, Phenylethanol, Phenoxyethanol, Ester wie Essigester, Butylacetat, Benzylbenzoat, Ether wie Alkylenglykolalkylether wie Dipropylenglykolmonomethylether, Diethylenglykolmono-butylether, Ketone wie Aceton, Methylethylketon, aromatische und/oder aliphatische Kohlenwasserstoffe, pflanzliche oder synthetische Öle, DMF, Dimethylacetamid, N-Methylpyrrolidon, 2-Dimethyl-4-oxy-methylen-1,3-dioxolan.
- Farbstoffe sind alle zur Anwendung am Tier zugelassenen Farbstoffe, die gelöst oder suspendiert sein können.
- Resorptionsfördernde Stoffe sind z.B. DMSO, spreitende Öle wie Isopropylmyristat, Dipropylenglykolpelargonat, Silikonöle, Fettsäureester, Triglyceride, Fettalkohole.
- Antioxidantien sind Sulfite oder Metabisulfite wie Kaliummetabisulfit, Ascorbinsäure, Butylhydroxytoluol, Butylhydroxyanisol, Tocopherol.
- Lichtschutzmittel sind z.B. Stoffe aus der Klasse der Benzophenone oder Novantisolsäure.
- Haftmittel sind z.B. Cellulosederivate, Stärkederivate, Polyacrylate, natürliche Polymere wie Alginate, Gelatine.
- Emulsionen können oral, dermal oder als Injektionen angewendet werden. Emulsionen sind entweder vom Typ Wasser in Öl oder von Typ Öl in Wasser. Sie werden hergestellt, indem man den Wirkstoff entweder in der hydrophoben oder in der hydrophilen Phase löst und diese unter Zuhilfenahme geeigneter Emulgatoren und gegebenenfalls weiterer Hilfsstoffe wie Farbstoffe, resorptionsfördernde Stoffe, Konservierungsstoffe, Antioxidantien, Lichtschutzmittel, viskositätserhöhende Stoffe, mit dem Lösungsmittel der anderen Phase homogenisiert.
- Als hydrophobe Phase (Öle) seien genannt: Paraffinöle, Silikonöle, natürliche Pflanzenöle wie Sesamöl, Mandelöl, Rizinusöl, synthetische Triglyceride wie Capryl/Caprinsäure-biglycerid, Triglyceridgemisch mit Pflanzenfettsäuren der Kettenlänge C8-12 oder anderen speziell ausgewählten natürlichen Fettsäuren, Partialglyceridgemische gesättigter oder ungesättigter, eventuell auch hydroxylgruppenhaltiger Fettsäuren, Mono- und Diglyceride der C8/C10-Fettsäuren; Fettsäureester wie Ethylstearat, Di-n-butyryl-adipat, Laurinsäurehexylester, Dipropylen-glykolpelargonat, Ester einer verzweigten Fettsäure mittlerer Kettenlänge mit gesättigten Fettalkoholen der Kettenlänge C16-C18, Isopropylmyristat, Isopropylpalmitat, Capryl/Caprinsäureester von gesättigten Fettalkoholen der Kettenlänge C12-C18, Isopropylstearat, Ölsäureoleylester, Ölsäuredecylester, Ethyloleat, Milchsäureethylester, wachsartige Fettsäureester wie Dibutylphthalat, Adipinsäurediisopropylester, letzterem verwandte Estergemische u.a. Fettalkohole wie Isotridecylalkohol, 2-Octyldodecanol, Cetylstearyl-alkohol, Oleylalkohol; Fettsäuren wie z.B. Ölsäure und ihre Gemische.
- Als hydrophile Phase seien genannt: Wasser, Alkohole wie z.B. Propylenglycol, Glycerin, Sorbitol und ihre Gemische.
- Als Emulgatoren seien genannt: nichtionogene Tenside, z.B. polyoxyethyliertes Rizinusöl, polyoxyethyliertes Sorbitan-monooleat, Sorbitanmonostearat, Glycerinmonostearat, Polyoxyethylstearat, Alkylphenylpolyglykolether; ampholytische Tenside wie Di-Na-N-lauryl-β-iminodipropionat oder Lecithin; anionaktive Tenside, wie Na-Laurylsulfat, Fettalkoholethersulfate, Mono/Dialkylpolyglykoletherorthophosphorsäureester-monoethanolaminsalz; kationaktive Tenside wie Cetyltrimethylammoniumchlorid.
- Als weitere Hilfsstoffe seien genannt: Viskositätserhöhende und die Emulsion stabilisierende Stoffe wie Carboxymethylcellulose, Methylcellulose und andere Cellulose- und Stärke-Derivate, Polyacrylate, Alginate, Gelatine, Gummi-arabicum, Polyvinylpyrrolidon, Polyvinylalkohol, Copolymere aus Methylvinylether und Maleinsäureanhydrid, Polyethylenglykole, Wachse, kolloidale Kieselsäure oder Gemische der aufgeführten Stoffe.
- Suspensionen können oral, dermal oder als Injektion angewendet werden. Sie werden hergestellt, indem man den Wirkstoff in einer Trägerflüssigkeit gegebenenfalls unter Zusatz weiterer Hilfsstoffe wie Netzmittel, Farbstoffe, resorptionsfördernde Stoffe, Konservierungsstoffe, Antioxidantien, Lichtschutzmittel suspendiert.
- Als Trägerflüssigkeiten seien alle homogenen Lösungsmittel und Lösungsmittelgemische genannt.
- Als Netzmittel (Dispergiermittel) seien die weiter oben angegebenen Tenside genannt.
- Als weitere Hilfsstoffe seien die weiter oben angegebenen genannt.
- Halbfeste Zubereitungen können oral oder dermal verabreicht werden. Sie unterscheiden sich von den oben beschriebenen Suspensionen und Emulsionen nur durch ihre höhere Viskosität.
- Zur Herstellung fester Zubereitungen wird der Wirkstoff mit geeigneten Trägerstoffen gegebenenfalls unter Zusatz von Hilfsstoffen vermischt und in die gewünschte Form gebracht.
- Als Trägerstoffe seien genannt alle physiologisch verträglichen festen Inertstoffe. Als solche dienen anorganische und organische Stoffe. Anorganische Stoffe sind z.B. Kochsalz, Carbonate wie Calciumcarbonat, Hydrogencarbonate, Aluminiumoxide, Kieselsäuren, Tonerden, gefälltes oder kolloidales Siliciumdioxid, Phosphate.
- Organische Stoffe sind z.B. Zucker, Zellulose, Nahrungs- und Futtermittel wie Milchpulver, Tiermehle, Getreidemehle und -schrote, Stärken.
- Hilfsstoffe sind Konservierungsstoffe, Antioxidantien, Farbstoffe, die bereits weiter oben aufgeführt worden sind.
- Weitere geeignete Hilfsstoffe sind Schmier- und Gleitmittel wie z.B. Magnesiumstearat, Stearinsäure, Talkum, Bentonite, zerfallsfördernde Substanzen wie Stärke oder quervernetztes Polyvinylpyrrolidon, Bindemittel wie z.B. Stärke, Gelatine oder lineares Polyvinylpyrrolidon sowie Trockenbindemittel wie mikrokristalline Cellulose.
- Homologe Sequenzen
- Gegenstand der Erfindung sind auch Polynucleotide und Polypeptide aus verwandten Organismen, die zu einer EtOS22-Nukleinsäure bzw. einem EtOS22-Polypeptid homolog sind und leicht mit Hilfe im Stand der Technik verfügbaren Methoden isoliert werden können.
- Zu diesen Methoden zählen: PCR mit degenerierten Primern, das Screening von Genbibliotheken mit EtOS22 als Sonde bei niedriger Stringenz und das Screenen von Expressionsbibliotheken mit dem monoklonalen Antikörper E2E5 (Sambrook and Russell, 2001).
- Gegenstand der Erfindung sind auch die oben beschriebenen Diagnostika, diagnostische Verfahren, Impfstoffe, Screening-Verfahren und Therapeutika, die in analoger Weise und für den Fachmann offensichtlich auf den homologen Polynucleotiden oder Polypeptiden beruhen.
- Definitionen
- Zum besseren Veständnis soll die Bedeutung bestimmter Wörter und Begriffe, die in der Beschreibung, den Beispielen und angefügten Ansprüchen verwendet werden, im Folgenden näher erläutert werden.
- Unter Polynucleotid oder Polynucleotiden soll verstanden werden doppel- sowie einzelsträngige DNA und doppel- sowie einzelsträngige RNA, cDNA, die sowohl als kodierender als auch als komplementärer Strang vorliegen können, Oligonucleotide, "small interfering RNA (siRNA)", Nukleinsäureanaloga wie z.B. "peptide nucleic acids (PNAs)", locked nucleic acids (LNAs)", "antisense" Oligonucleotide, die z.B. synthetisiert werden können durch kovalente Verbindung des 5'-Endes eines Nukleotids mit dem 3'-Ende eines anderen Nukleotids durch Nicht-Phosphodiester-Bindungen wie z.B. Alkylphosphonate, Phosphorothioate, Phosphorodithioate, Alkylphosphonothioate, Alkylphosphonate, Phosphoramidate, Phosphatester, Carbomate, Acetamidate, Carboxymethylester, Carbonate und Phosphattriester.
- Die Begriffe "Homologie", "Identität" oder "Ähnlichkeit" beziehen sich auf Sequenzähnlichkeiten zwischen zwei Peptiden oder zwischen zwei Nukleinsäuremolekülen bzw. Polynucleotiden. Homologie kann bestimmt werden, indem man jeweils eine Position in jeder Sequenz miteinander vergleicht. Ist eine Position in der verglichenen Sequenz von derselben Base oder Aminosäure besetzt, sind die beiden Moleküle an dieser Position homolog. Das Maß für Homologie zwischen Sequenzen ist eine Funktion der Anzahl der übereinstimmenden oder homologen Positionen, die die Sequenzen miteinander teilen. Eine "nicht homologe" Sequenz weist eine Identität von weniger als 40% auf, vorzugsweise allerdings weniger als 25% Identität. Eine Homologie oder Identität kann u.a. mit Hilfe von Computerprogrammen wie dem GCG-Programm [Devereux et al. (1983), Nucleic Acids Res. 12, 387-395] festgestellt werden.
- Eine "Homologie" besteht auch, wenn ein Polynucleotid-Abschnitt mit eines anderen Polynucleotids hybridisieren kann.
- Der Begriff "hybridisieren" oder "Hybridisierung" beschreibt den Vorgang, bei dem ein einzelsträngiges Polynucleotid mit einem komplementären DNA-Strang eine Basenpaarung eingeht, wobei die Fähigkeit eines einzelsträngigen Polynucleotids von der Stringenz der Hybridisierungsbedingungen abhängt.
- Der Begriff "Stringenz" bezieht sich auf die Hybridisierungsbedingungen. "Hohe Stringenz" ist dann gegeben, wenn eine Basenpaarung erschwert wird. "Niedrige Stringenz" ist dann gegeben, wenn eine Basenpaarung erleichtert wird.
- Stringente Hybridisierungsbedingungen sind dem Fachmann gut bekannt und werden z.B. beschrieben in Sambrook et al., MOLECULAR CLONING: A LABORATORY MANUAL., 2d ed., 1989, S. 9.50-9.51.
- Typischerweise sollte für stringente Hybridisierungsbedingungen die Kombination aus Temperatur und Salzkonzentration so gewählt werden, dass sie ungefähr 12-20°C unter der berechneten Schmelztemperatur Tm des Hybrids liegt. Es ist dem Fachmann bekannt, dass die Tm einer doppelsträngigen DNA um 1-1.5°C pro 1% Abnahme der Identität abnimmt [Bonner et al., J. Mol. Biol. 81, 123 (1973)]. Die Tm eines Hybrids aus einem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 und einem Polynucleotid, das wenigstens 50%, bevorzugt 60%, 70% 80%, 85%, 90%, 95% oder 97% identisch mit einem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 ist, kann z.B. durch die Gleichung von of Bolton und McCarthy berechnet werden [Proc. Natl. Acad. Sci. U.S.A. 48, 1390 (1962)]:
Tm = 81.5°C – 16.6(log10[Na+]) + 0.41(%G + C) – 0.63(% Formamid) – 600/l), worin l = Länge des Hybrids in Basenpaaren. - Stringente Waschbedingungen bei der Hybridisierung sind z.B. 4 X SSC bei 65°C oder 50% Formamid, 4 X SSC bei 42°C oder 0.5 X SSC, 0.1% SDS bei 65°C. Hochstringente Waschbedingungen sind z.B. 0,2 X SSC bei 65°C.
- Der Begriff "Plasmid" bezieht sich auf ein extrachromosomales genetisches Element. Die für die vorliegende Erfindung verwendeten Ursprungsplasmide sind entweder kommerziell erhältlich, frei zugänglich oder können von solchen Plasmiden nach bekannten Verfahren abgeleitet werden.
- Der Begriff "Vektor" beschreibt ein Polynucleotid, das zum Einbringen exogener Polynucleotide in Wirtszellen verwendet wird. Ein Vektor enthält eine Nucleotidsequenz, die für ein oder mehrere Polypeptide kodiert. Vektoren, die in der Lage sind, die Expression der Gene zu steuern, die sie enthalten, werden als "Expressionsvektoren" bezeichnet.
- Der Begriff "modulieren" bezieht sich sowohl auf eine Stimulation als auch auf eine Suppression oder Inhibition eines biochemischen Vorgangs. Im Rahmen der vorliegenden Erfindung bedeutet "modulieren" oder "Modulation" inhibieren oder eine Inhibition oder Suppression der Aktivität des EtOS22-Polypeptids, die für die Excystierung der Sporozoiten aus den Sporocysten wichtig ist.
- Bakterienstämme und Vektoren Bakterienstämme
-
- E. coli TOP10 chemisch-kompetent (Invitrogen, Groningen, NL)
- F– mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 deoR araD139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG
- E. coli TG1 elektro-kompetent (Stratagene, Heidelberg)
- supE thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM)5 (rk –mk –) [F' traD36 proAB lac1qZΔM15]
- Vektoren
-
- pG8SAET (Jacobsson und Frykberg, 1998; Zhang et al., 1999)
- pCR2.1-TOPO (Invitrogen, Groningen, NL)
- pcDNA3.1/VS-His-TOPO (Invitrogen, Groningen, NL)
- Beispiel 1
- Isolation von genomischer DNA aus Eimeria tenella
- Die DNA-Isolation erfolgte nach einer modifizierten Methode von Blin und Stafford (1976). 1 × 108 sporulierte Oocysten wurden bei 3.000 rpm für 10 min sedimentiert (Heraeus MULTIFUGE 3 L-R) und ein dem Sediment entsprechendes Volumen Glasperlen (∅ 0,45-0,5 mm) hinzugegeben. Durch Schütteln mit dem Vortex bei maximaler Geschwindigkeit für 2 min wurden Oocysten- und Sporocystenschalen aufgebrochen und mikroskopisch kontrolliert. Es folgte die Hinzugabe von 5 ml Extraktionspuffer (10 mM Tris-HCl pH 8,0; 0,1 M EDTA pH 8,0; 0,5 % SDS; 20 μg/ml RNaseA), eingestellt auf eine Proteinase K Konzentration von 100 μg/ml, und die Inkubation bei 56°C über Nacht. Die DNA wurde zweimal mit Roti-Phenol (Roth, Karlsruhe)/Chloroform und einmal mit Chloroform extrahiert, mit 2 Vol. absolutem Ethanol und 0,1 Vol. 3 M Natriumacetat gefällt und durch Zentrifugation bei 13.000 rpm im Beckman JS13.1 Rotor sedimentiert. Das Sediment wurde zweimal mit 70%igen Ethanol gewaschen, luftgetrocknet, in destilliertem H2O (dH2O) resuspendiert und die DNA-Konzentration in einem 1%igen TBE-Agarosegel durch den Vergleich mit 1 μg EcoRI/HindIII verdaute λ-DNA (MBI Fermentas, St. Leon-Rot) abgeschätzt.
- Beispiel 2 Isolation von Gesamt-RNA aus Eimeria tenella und Kükenblinddarm
- Die Isolation von Gesamt-RNA erfolgte aus 3 × 107 Oocysten bzw. 0,3 g Kükenblinddarm unter Verwendung des ,Invisorb RNA Kit II' (Invitek, Berlin-Buch). Sämtliche Arbeitsschritte wurden mit RNase-freiem Material und DEPC(Diethylpyrocarbonat)-behandelten Lösungen durchgeführt. Die Oocysten wurden in 500 μl Lysis-Lösung durch Schütteln mit dem Vortex mit Glasperlen aufgeschlossen, während das Darmgewebe in 2 ml Lysis-Lösung mittels Ultraturrax zerkleinert wurde. Der jeweilige Überstand wurde für die RNA-Isolation verwendet und alle weiteren Arbeitsschritte richteten sich nach den Angaben des Herstellers. Die in DEPC-H2O gelöste Gesamt-RNA wurde anschließend photometrisch bestimmt.
- Beispiel 3
- Präparation von Plasmid-DNA
- Die Präparation von Plasmid-DNA erfolgte aus stationären 3 ml bzw. 50 ml Kulturen von E. coli TG1- oder E. coli TOP10. Mit einer Einzelkolonie wurde LB-Medium unter Zusatz des entsprechenden Antibiotikums angeimpft und über Nacht bei 37°C und 280 rpm schüttelnd inkubiert. Zur Plasmid-Isolation wurden das ,NucleoSpin Plasmid Kit' (Macherey-Nagel, Düren) bzw. das ,Plasmid Midi Kit' (Qiagen, Hilden) verwendet. Die Plasmid-DNA wurde gemäß den Angaben des jeweiligen Herstellers aufgereinigt.
- Beispiel 4
- Bestimmung der Nukleinsäurekonzentration
- Die Bestimmung der Nukleinsäurekonzentration erfolgte photometrisch im Spektrophotometer DU 640 (Beckmann, München). Die Konzentration und Reinheit wurde nach Sambrook et al. (1989) berechnet. Restriktionsfragmente, die als Sonden für Southern- und Northern-Blot verwendet werden sollten, wurden in einem 1%igen TBE-Agarosegel durch den Vergleich mit 1 μg EcoRI/HindIII verdaute λ-DNA (MBI Fermentas, St. Leon-Rot) abgeschätzt.
- Beispiel 5
- Restriktion und elektrophoretische Auftrennung von DNA
- Die Restriktion von DNA durch Restriktionsendonukleasen wurde gemäß den Herstellerangaben und im empfohlenen Puffer für das jeweilige Enzym durchgeführt. In der Regel betrug die Inkubationszeit 3 h bei 37°C.
- Die elektrophoretische Auftrennung von DNA-Fragmenten erfolgte nach der Methode von Sambrook et al. (1989) in einer horizontalen Flachbettkammer. Dafür wurden 0,6-2%ige Agarosegele verwendet, die mit TBE- oder TAE-Puffer und unter Zugabe von 0,5 μg/ml Ethidiumbromid gegossen wurden. Die durch Ethidiumbromid angefärbten DNA-Moleküle wurden anschließend auf dem Transilluminator mit parallel aufgetrennten DNA-Längenstandards verglichen. Für Fragmente < 1 kb wurde 1 μg pUC-Mix Marker, für größere Fragmente 1 μg EcoRI/HindIII verdaute λ-DNA (beide MBI Fermentas, St. Leon-Rot) verwendet.
- Beispiel 6
- Isolation von DNA-Fragmenten aus Agarosegelen
- Die Isolation von DNA-Fragmenten aus Agarosegelen erfolgte entweder mittels Agarase (Roche Molecular Biochemicals, Mannheim) durch den Verdau von ,low melting point'-Agarose (Biozym, Hess. Oldendorf), die als Fenstergel in gleichprozentiges TAE-Agarosegel gegossen wurde, oder mittels des ,NucleoSpin Extract 2 in 1 Kits' (Macherey-Nagel, Düren). In beiden Fällen wurde das gewünschte Fragment unter langwelligem UV-Licht ausgeschnitten und gemäß den Angaben der Hersteller isoliert. Zur Konzentrationsbestimmung wurde 1/10 des Probenvolumens anschließend in einem Kontrollgel aufgetrennt und im Vergleich zum DNA-Längenstandard abgeschätzt.
- Beispiel 7
- Chemische Transformation von E. coli
- Die Transformation von chemisch-kompetenten E. coli TOP10 (Invitrogen, Groningen, NL) mit rekombinanten Plasmiden wurde nach Herstellerangaben durchgeführt. Nach dem Ausplattieren der transformierten Zellen auf Selektionsagar und über Nacht Inkubation bei 37°C konnten Transformanten isoliert und im Restriktionsverdau analysiert werden.
- Beispiel 8
- Konstruktion einer genomischen Expressionsbank von Eimeria tenella
- a) Fragmentierung genomischer DNA und ,blunt-end' Reaktion
- 25 μg genomische DNA von Eimeria tenella wurden in einem Volumen von 4 ml mit der Sonotrode MS73 bei 40%iger Geräteauslastung 10 × 30 s unter Kühlung im eiskalten Wasserbad fragmentiert. Diese Fragmente einer Größes zwischen 100 und 800 bp wurden anschließend präzipitiert, in 60 μl dH2O resuspendiert und in einem 1,2%igen Agarosegel mit gleichprozentigem ,low melting point'-Agarose Fenster ohne Ethidiumbromid aufgetrennt. Die durch Agarase-Verdau isolierten Fragmente wurden über ,S-400 HR Microspin Columns' (Amersham Pharamacia Biotech, Freiburg) nach Herstellerangaben aufgereinigt.
- Für die ,blunt-end' Reaktion wurde folgender Reaktionsansatz erstellt: 75 μl aufgereinigte DNA-Fragmente, 1 mM dNTP-Mix, 10 U AccuTherm DNA Polymerase (GeneCraft, Münster) und 1 × AccuTherm Puffer auf 100 μl dH2O. Dieser Reaktionsansatz wurde für 30 min bei 72°C inkubiert, anschließend Phenol-Chloroform extrahiert und die Fragmente in 100 μl dH2O resuspendiert. 2 μl davon wurden zur Konzentrationsabschätzung in einem Probegel aufgetrennt.
- b) Dephosphorylierung von pG8SAET
- 20 μg pG8SAET wurden mit 40 U SnaBI (Promega, Heidelberg) für 3 h bei 37°C inkubiert. Anschließend wurden dem Restriktionsansatz 4 U Alkalische Shrimp Phosphatase (USB, Bad Homburg) zugesetzt und über Nacht bei 37°C inkubiert. Nach 10 min Hitzeinaktivierung bei 65°C wurde der linearisierte und dephosphorylierte Vektor aus einem 0,8%igen ,low melting point'-Agarose Fenstergel durch Agarase-Verdau isoliert.
- c) Ligation von DNA
- Die Ligation von 8 μg genomischen DNA-Fragmenten und 5 μg linearisiertem und dephosphoryliertem pG8SAET erfolgte mittels 300 U T4 DNA Ligase HC (MBI Fermentas, St. Leon-Rot) in einem Gesamtvolumen von 100 μl bei 16°C für 48 h. Anschließend wurde die Ligase für 10 min bei 65°C hitzeinaktiviert und die rekombinanten Vektormoleküle unter Hinzugabe von 20 μg Glykogen präzipitiert. Das Sediment wurde in 100 μl dH2O resuspendiert, wovon jeweils 1,5 μl pro Elektrotransformation eingesetzt wurden.
- d) Elektrotransformation von E. coli
- Die Elektrotransformation von jeweils 50 μl E. coli TG1 mit 1,5 μl Ligationsansatz wurde in 0,1-cm Elektroporationskuvetten (BIO-RAD, München) bei einer Feldstärke von 17 kV/cm im Gene Pulser (BIO-RAD, München) bei 200 Ω und 25 μF nach Herstellerangaben durchgeführt. Die Anzahl der Transformanten wurde bestimmt und die rekombinanten Zellen bei –80°C in Form von Glycerolstocks konserviert.
- Es wurde eine repräsentative genomische DNA-Bank des Parasiten im Phagemidvektor pG8SAET konstruiert. Diese umfasst 4,7 × 106 unabhängige Klone (95% rekombinant) mit einer mittleren Insertgröße von 450 bp, woraus sich eine 7,3fache Repräsentation des Genoms von Eimeria tenella ergibt.
- Beispiel 9
- Phagen-Display und Phagen-Panning
- a) Präparation und Aufreinigung rekombinanter Phagemide
- 20 ml Ampicillin-haltiges LB-Medium (50 μg/ml) wurden mit 200 μl eines jeden Glycerolstock angeimpft und über Nacht bei 37°C und 280 rpm inkubiert. Mit 1 ml dieser Kultur wurden dann 100 ml Ampicillin-haltiges LB-Medium angeimpft, bis zu einer OD600 = 0,5 inkubiert und mit 500 μl (1 × 1011 pfu) Helferphagen R408 (Promega, Heidelberg) infiziert. Nach erneuter Inkubation bei 37°C und 280 rpm über Nacht wurden die Zellen für 10 min bei 5.000 rpm sedimentiert, der Überstand sterilfiltriert und die Phagemide über Vivaspin 20 Konzentratoren (Sartorius AG, Göttingen) gemäß Herstellerangaben aufkonzentriert.
- b) ,Coating' von DYNABEADS
- Für das 'Coating' von ,DYNABEADS Pan Mouse IgG' (Deutsche Dynal GmbH, Hamburg) wurde durch Ultrafiltration (100 kDa MWCO) 50fach konzentrierter Hybridoma-Kulturüberstand des monoklonalen Antikörpers (mAk) E2E5 verwendet (Mouafo et al., 2002). Pro mg DYNABEADS wurden 20 μg aufkonzentriertes Gesamtprotein bei einem Anteil von ca. 5% mAk E2E5 eingesetzt und über Nacht bei 4°C rotierend inkubiert. Durch 3maliges Waschen mit PBS (8 g NaCl; 0,2 g KCl; 1 g Na2HPO4 × 2 H2O; 0,15 g NaH2PO4 × H2O; 0,2 g KH2PO4 ad 11 H2O, pH 7,4)/0,1 % BSA wurden nicht-gebundene Proteine und Immunglobuline entfernt und die DYNABEADS anschließend in der Bindungsreaktion eingesetzt.
- c) Bindungsreaktion, Waschschritte und Elution
- In der Bindungsreaktion wurden 50 μl (2 × 107) DYNABEADS mit oder ohne mAk E2E5 auf der Oberfläche mit 200 μl Phagemidkonzentrat in einem Volumen von 400 μl in PBS/0,1% BSA über Nacht bei 4°C rotierend inkubiert. Nach 10maligem Waschen wurden schwach bindende Phagemide nach einer 15 minütigen rotierenden Inkubation in 400 μl Elutionspuffer (50 mM Natriumcitrat; 150 mM NaCl) pH 4,5 verworfen, und die im Elutionspuffer pH 1,8 eluierten Phagen mit 40 μl Neutralisationspuffer (2 M Tris-HCl, pH 8,6) versetzt und für die Titerbestimmung und Reinfektion verwendet.
- d) Titerbestimmung und Reinfektion
- Jedes Eluat wurde zur Reinfektion von 10 ml E. coli TG1, die sich in der logarithmischen Wachstumsphase befanden, verwendet. Nach Inkubation bei 37°C und 40 rpm für 0,5 h wurden die Zellen sedimentiert, in 400 μl Ampicillin-haltigem LB-Medium resuspendiert und zur Titerbestimmung bzw. Amplifikation auf Ampicillin-haltigen LB-Agarplatten ausgesät. Für eine weitere Runde Phagen-Panning wurden diese Platten nach 18 h mit Ampicillin-haltigem LB-Medium abgespült und in einer 50 ml Kultur mit 100 μl Helferphagen R408 infiziert. Nach Inkubation bei 37°C und 280 rpm über Nacht wurden die Phagemide wie beschrieben aufkonzentriert und für neue Bindungsreaktionen eingesetzt.
- e) Detektion E-tag-exprimierender Klone
- Zur Isolierung E-tag-exprimierender Klone wurden im Anschluss an mehrere Runden Phagen-Panning ca. 100 ,colony forming units' (cfu) auf Ampicillin-haltige Agarplatten ausgesät und auf Nitrozellulosemembran (Schleicher und Schuell, Dassel) transferiert. Die der Membran anhaftenden Zellen werden über Nacht in 6 ml Lysispuffer (100 mM Tris-HCl, pH 7,8; 150 mM NaCl; 5 mM MgCl2; 1,5% BSA; 1 μg/ml DNase I; 40 μg/ml Lysozym) lysiert, Zellreste durch 3faches Waschen mit PBS/0,05 % Tween-20 entfernt und unspezifische Bindungsstellen auf der Membran durch 1 h Blocken mit 1 × RotiBlock-Lösung (Roth, Karlsruhe) abgesättigt. Anschließend wurde die Membran für 2 h bei Raumtemperatur (22°C) mit dem Primärantikörper Maus-anti-E-Tag (Amersham Pharmacia, Freiburg), 1:500 verdünnt in 1 × RotiBlock, inkubiert. Nicht-gebundene Antikörper wurden durch 3maliges Waschen für 0,5 h entfernt. Als Sekundärantikörper wurde ein mit Alkalischer Phosphatase (AP-) gekoppelter Ziege-anti-Maus IgG in einer 1:2.000 Verdünnung eingesetzt. Nach 3 erneuten Waschschritten wurde die Membran für 2 min in Detektionspuffer (100 mM Tris-HCl, pH 9,5; 100 mM NaCl) equilibriert und gebundene Sekundärantikörper durch die Erzeugung von Chemilumineszenz mittels CDP-Star (Roche Molecular Biochemicals, Mannheim), 1:100 verdünnt in Detektionspuffer, nachgewiesen. Die Exposition erfolgte auf ECL Hyperfilm (Amersham Pharmacia, Freiburg) für 2 – 10 min bei 22°C.
- f) Phagen-Panning gegen mAk E2E5 – Anreicherung spezifisch-bindender Klone
- Der monoklonale Antikörper (mAk) E2E5 wurde an die Oberfläche von ,DYNABEADS Pan Mouse IgG' gebunden und im Phagen-Panning eingesetzt. Als Negativkontrolle dienten ,DYNABEADS Pan Mouse IgG' ohne weiteren Antikörper. Nach 3 Runden Phagen-Panning kam es zu einer 362fachen Anreicherung bindender Klone im Vergleich zur Negativkontrolle. Davon wurden E-tag-exprimierende Klone im Western Blot mit dem mAk E2E5 analysiert. Von den 62 isolierten, E-tag-exprimierenden Klonen wurden 6 (A14, A17, A45 – A47 und A62) im Western Blot vom mAk E2E5 erkannt. Sämtliche detektierten Fusionsproteine zeigten das gleiche Laufverhalten in der SDS-PAGE mit einem Molekulargewicht von ca. 14 kDa. Diese Fusionsproteine setzten sich aus 125 AS zusammen, wovon sich 48 AS auf das einklonierte Insert ,A17' zurückführen lassen.
- Beispiel 10
- Polymerase Kettenreaktion (PCR)
- Sämtliche PCRs wurden in einem PTC-200 Gradientencycler oder PTC-150 MiniCycler der Firma MJ Research (Biozym, Hess. Oldendorf) durchgeführt.
- Synthetische Oligonukleotide (Primer)
- Alle PCR-Primer wurden von der Firma MWG Biotech (Ebersbach) synthetisiert.
-

- a) RT-PCR
- Die RT-PCR umfasst die Reverse Transkription von Gesamt-RNA und anschließende PCR zur Amplifikation von DNA-Sequenzen mittels sequenz-spezifischer Primer. Der Reaktionsansatz für die RT setzte sich wie folgt in einem Gesamtvolumen von 50 μl zusammen: 3,5 μg Gesamt-RNA, 80 U RNasin Ribonuklease Inhibitor (Promega, Heidelberg), 0,4 mM dNTP-Mix, 50 U AMV Reverse Transkriptase, 1 × AMV-Puffer (alles Roche Molecular Biochemicals, Mannheim), 5 mM DTT und 2,5 μM Random Hexamer Primer als Startmoleküle der cDNA-Synthese. Auf eine Inkubation für 10 min bei 22°C folgte die cDNA-Synthese für jeweils 30 min bei 42°C und 55°C. Das Enzym wurde für 5 min bei 95°C hitzeinaktiviert. Für jede Reverse Transkription wurden zwei weitere Reaktionen ohne Reverse Transkriptase bzw. RNA-Templat als Negativkontrollen durchgeführt.
- Für die sich anschließende PCR wurde in einem Gesamtvolumen von 50 μl 1/10 der Reverse Transkriptase-Reaktionen als Templat eingesetzt. Zur Amplifikation wurden folgende PCR Systeme bei jeweils 0,4 μM der beiden sequenzspezifischen Primer gemäß den Herstellerangaben verwendet: ,Triple Master PCR System' (Eppendorf), ,Platinum Pfx DNA Polymerase' (Invitrogen, Groningen, NL) und ,High Fidelity PCR System' (Roche Molecular Biochemicals, Mannheim). Der intitialen Denaturierung für 2 min bei 94°C folgten 35 Zyklen aus 15 s Denaturierung bei 94°C, 30 s Annealing bei 63°C und 2 min Kettenverlängerung bei 72°C. Eine terminale Elongation für 10 min bei 72°C beendete die Reaktion. 1/5 dieser Reaktion wurde zur Kontrolle in einem entsprechend prozentigen TBE-Agarosegel aufgetrennt.
- b) 5'- und 3'-RACE-PCR
- sAls Ausgangsmaterial für die 5'- und 3'-RACE-PCR wurde Gesamt-RNA aus sporulierten Oocysten von Eimeria tenella unter Verwendung des ,5'/3' RACE Kits' (Roche Molecular Biochemicals, Mannheim) eingesetzt. Die cDNA-Synthese, Tailing-Reaktion (nur bei 5'-RACE) und Amplifikation der cDNA mit sequenzspezifischen Primern wurde nach Herstellerangaben durchgeführt. Darauf folgten bei der 5'-RACE eine, bei der 3'-RACE zwei weitere nested-PCRs um die Amplifikation der 5'- und 3'-Enden zu steigern. Als sequenzspezifische Primer wurden in der 5'-RACE A17-max-631-lo (cDNA-Synthese), A17-max-533-lo (Amplfikation der dA-tailed cDNA) und A17-112-lo (nested-PCR) verwendet, in der 3'- RACE A17-max-90-up (Amplifikation der cDNA), A17-max-150-up (1. nested-PCR) und A17-22-up (2. nested-PCR). Die im 2%igen Agarosegel aufgetrennten RACE-PCR-Produkte wurden nach der Methode von Chomczynski (1992) auf eine neutrale Hybond-N Nylonmembran (Amersham Pharmacia Biotech, Freiburg) transferiert, mit einer radioaktiv markierten Sonde hybridisiert und zur Belichtung von Kodak Biomax MS Röntgenfilmen verwendet. Die so identifizierten spezifischen RACE-PCR-Produkte wurden kloniert, isoliert und sequenziert.
- Beispiel 11
- Klonierung von PCR-Produkten
- Für die Klonierung von PCR-Produkten wurden das ,TOPO TA Cloning Kit' und das ,pcDNA3.1/V5-His TOPO TA Expression Kit' (Invitrogert, Groningen, NL) benutzt. Die PCR-Produkte wurden mittels ,NucleoSpin Extract 2 in 1 Kit' (Macherey-Nagel, Düren) aus Agarosegelen isoliert und anschließend mit 5 U Taq DNA Polymerase (Promega, Heidelberg), 1 × Taq DNA Polymerase Puffer und 0,4 mM dNTP-Mix für 0,5 h bei 72°C inkubiert. Die durch die Terminale-Transferase-Aktivität der Taq DNA Polymerase am 3' Ende adenylierten PCR-Produkte wurden ein weiteres Mal mittels ,NucleoSpin Extract 2 in 1 Kit' aufgereinigt und nach Herstellerangaben in der TOPO TA Klonierung eingesetzt.
- Beispiel 12
- DNA-Sequenzanalyse
- Die Sequenzierung von klonierter DNA erfolgte nicht-radioaktiv nach der Kettenabbruchmethode von Sanger et al. (1977) mit Hilfe des automatischen DNA-Sequenzierers LI-COR 4000 der Firma MWG Biotech (Ebersbach). Sequenziert wurde mit 5'-IRD-800-gekoppelten Primern für den Vektor pG8SAET (MWG Biotech, Ebersberg) sowie mit 5'-IRD-800-gekoppelten Standardprimern (LI-COR Bioscience, Bad Homburg).
- 5'-IRD-800-gekoppelte Primer

- a) Sequenzierung mit Thermo-Sequenase
- Für die Sequenzreaktion wurden das ,Thermo Sequenase Primer Cycle Sequencing Kit' (Amersham Pharmacia Biotech, Freiburg) sowie mit dem Infrarot-Fluoreszenzfarbstoff IRD-800-gekoppelte Primer (MWG Biotech, Ebersbach) verwendet. Pro Reaktion wurden dabei 1,5 μg Plasmid-DNA und 2 – 4 μl 5'-IRD-800-gekoppelter Primer (1 pmol/μl) in einem Gesamtvolumen von 13 μl gemischt und je 3 μl dieses Ansatzes zu je 3 μl des jeweiligen A, C-, G- oder T-Nukleotid-Mixes gegeben und anschließend mit 10 μl Mineralöl überschichtet. Die Sequenzreaktion erfolgte dann in einem PTC 100 Thermocycler (MJ Research, Biozym, Hess. Oldendorf). Dabei folgten auf eine 2minütige Denaturierung bei 94°C 30 Zyklen von Denaturierung (94°C für 30 s), Annealing (55°C für 30 s) und Strangsynthese (72°C für 1,5 min). Die Reaktionen wurden durch Zugabe von 6 μl Formamid-Ladungspuffer abgestoppt. Unmittelbar vor dem Auftragen der Sequenzreaktion auf das Sequenziergel wurden die Reaktionen für 3 min bei 72°C denaturiert und sofort danach lichtgeschützt auf Eis gelagert. Die fluoreszenzmarkierten Kettenabbruch-Fragmente wurden in 40 cm langen, 0,25 mm dünnen 6%igen Gelen aus modifiziertem Polyacrylamid (Ultra Pure Sequagel XR, National Diagnostics; Atlanta, USA) mit 8 M Harnstoff in 1 × TBE-Puffer bei 1.500 V und 50°C aufgetrennt und in Echtzeit über eine Laser-Photomultiplier-Einheit detektiert. Die Auswertung der Sequenzen erfolgte mit der Software Base ImagIR 4.0 von LI-COR (MWG Biotech, Ebersbach).
- b) Computeranalyse von Sequenzdaten
- Die durch Sequenzanalyse erhaltenen Daten wurden zunächst mit den Programmen ,Molecular BioComputing Suite' (Muller et al., 2001) und Sequenzen 3.0 aufgearbeitet und die deduzierten Proteinsequenzen ermittelt. Für die Datenbank-Recherche, also den Vergleich mit bereits bekannten Sequenzen in den Datenbanken EMBL, SwissProt oder mit den Daten des Eimeria tenella Genomprojekts (www.sanger.ac.uk/Projects/E_tenella/), wurden die Programme BLAST (Altschul et al., 1990) und omniBLAST benutzt. Das ,Alignment' von zwei oder mehreren DNA- oder Proteinsequenzen wurde mit Hilfe der Programme ,BLAST 2 sequences' (www.ncbi.nlm.nih.gov) (Tatusova und Madden, 1999), CLUSTALW (www.ebi.ac.uk) (Thompson et al., 1994) und DIALIGN (Morgenstern et al., 1998; 1999) durchgeführt. Weiterhin wurden die Programme Signale (www.cbs.dtu.dk/services/SignalP/) (Nielsen et al., 1997) zur Identifizierung von Signalpeptiden und Clone Manager 5 zur Planung von Klonierungen und Restriktionen und zur Suche nach offenen Leserastern genutzt.
- Beispiel 13
- Herstellung von Proteinextrakten aus E. coli
- 2 ml E. coli TG1 aus einer stationären über Nacht Kultur wurden sedimentiert, 1 × mit dH2O gewaschen und in 300 μl dH2O resuspendiert. 100 ul 4 × RotiLoad-Puffer (Roth, Karlsruhe) wurden hinzugegeben und die Probe für 5 min in kochendem Wasser denaturiert. Anschließend wurde die genomische DNA im Ansatz durch eine kurze Ultraschall-Behandlung fragmentiert und jeweils 10 ul dieser Probe auf ein SDS-Polyacrylamidgel aufgetragen.
- Beispiel 14
- Elektrophoretische Auftrennung von Proteinen in Polyacrylamid-gelen und Western Blot'
- a) SDS-PAGE
- Die Auftrennung von Proteinextrakten unter denaturierenden Bedingungen erfolgte in diskontinuierlichen Polyacrylamidgelen nach der Methode von Lämmli (1970). Dazu wurde die ,Mini-PROTEAN II Electrophoresis Cell Apparatur' (BIO-RAD, München) benutzt. Die Gelgröße beträgt in diesem System 8 × 10 cm. Es wurden Trenngele mit 15 % Polyacrylamid verwendet. Die Konzentration der Sammelgele betrug einheitlich 4,5 % Polyacrylamid. Die Auftrennung erfolgte bei 40 mA für ca. 2 – 2,5 h. Als Molekulargewichtsstandard wurden 5 μl des ,Prestained SDS Molecular Weight Marker Mix' (Sigma, Deisenhofen) verwendet.
- b) Proteintransfer auf Nitrozellulosemembran
- Die in der SDS-PAGE aufgetrennten Proteine wurden nach der Semi-Dry-Methode (Kyhse-Anderson, 1984) auf Protran BA 85 Nitrozellulosemembran (Schleicher und Schuell, Dassel) geblottet. Dabei wurde ein kontinuierliches Puffer-System (Lihme und Schafer-Nielsen, 1986) verwendet, bei dem nur die in Blotpuffer getränkten Lagen Whatman-Papier (Whatman Ltd., Maidstone, England) zwischen zwei Graphitplatten (Biometra Fast Blot, Göttingen) als Pufferreservoir dienten. Zwischen diesen Graphitplatten wurde ein homogenes elektrisches Feld erzeugt, worin die Proteine bei einer Stromstärke von 60 mA für 2 h aus dem SDS-Polyacrylamidgel auf die Nitrozellulosemembran übertragen wurden. Zur Kontrolle des Transfers wurde die Nitrozellulosemembran nach dem Blotten reversibel durch Ponceau S (0,2% Ponceau S in 3% Trichloressigsäure) angefärbt und mit dH2O wieder entfärbt.
- c) Immundetektion
- Die Nitrozellulosemembran wurde in ein 50 ml Zentrifugenröhrchen (Falcon, Becton Dickinson, Sunnyvale, CA, USA) eingerollt und rotierend für 1 h mit 10 ml 1 × RotiBlock-Lösung (Roth, Karlsruhe) inkubiert, um unspezifische Bindungsstellen abzusättigen. Anschließend wurde diese Block-Lösung gegen 50 μl 50fach aufkonzentrierten Hybridoma-Kulturüberstand des monoklonalen Antikörpers E2E5 (Primärantikörper) in 10 ml 1 × RotiBlock-Lösung ausgetauscht. Nach 2stündiger Inkubation bei 22°C wurde die Membran 3 × für insgesamt 0,5 h mit PBS/0,05 % Tween-20 gewaschen, um den nicht-gebundenden Überschuss an Primärantikörper zu entfernen. Dann wurde der Sekundärantikörper Ziege-anti-Maus IgG ,Horse Radish' Peroxidase- (HRP-) gekoppelt (Jackson Immuno Research Laboratories, West Grove, USA) 1:4.000 verdünnt in 10 ml 1 × RotiBlock-Lösung für 1 h hinzugegeben. Erneut wurde 3 × für insgesamt 0,5 h mit mit PBS/0,05 % Tween-20 gewaschen. Der Nachweis der gebundenen Antikörper erfolgte nun durch die Erzeugung von Chemilumineszenz mit Hilfe des ,ECL Western Blotting Detektionssystems' (Amersham Pharmacia, Freiburg) in Anlehnung an Roswell und White (1978). Gleiche Volumina der Detektionsreagenzien 1 und 2 wurden dafür gemischt und auf die Membran gegeben (0,125 ml/cm2). Nach 1 min wurde die Flüssigkeit abgenommen, die Membran 1 × kurz mit PBS/0,05 % Tween-20 gespült und anschließend luftblasenfrei zwischen zwei Overheadfolien gelegt. Die Exposition erfolgte auf ECL Hyperfilm (Amersham Pharmacia, Freiburg) für 2 – 5 min bei 22°C.
- Beispiel 15
- Radioaktive Markierung von DNA
- Die radioaktive Markierung von DNA war abhängig von der Größe der verwendeten Sonden. DNA-Fragmente > 800 bp wurden durch ,random priming' mit [α32P]dCTP in Anlehnung an die Methode von Feinberg und Vogelstein (1984) markiert. Für diese Markierungsreaktion wurde das ,Megaprime DNA Labelling Kit' (Amersham Pharmacia Biotech, Freiburg) gemäß den Herstellerangaben verwendet. Pro Ansatz wurden 40 ng DNA und 50 μCi [α32P]dCTP (10 μCi/μl, spez. Aktivität > 3.000 Ci/mmol) eingesetzt.
- Sehr kleine DNA-Fragmente, wie zum Beispiel Oligonukleotide, wurden mit [γ32P]-ATP radioaktiv markiert. Bei dieser Reaktion katalysiert die T4 Polynukleotid Kinase (MBI Fermentas, St. Leon-Rot) den Transfer des [γ32P]ATP auf die 5'-OH-Gruppe der DNA. Es wurden 20 ng Olignukleotide und 100 μCi [γ32P]ATP (10 μCi/μl, spez. Aktivität > 4.500 Ci/mmol) verwendet. Nach der Markierungsreaktion wurden nicht eingebaute Nukleotide mit dem ,NucleoSpin Extract 2 in 1 Kit' (Macherey-Nagel, Düren) abgetrennt. Die so markierte und gereinigte DNA wurde vor der Verwendung zur Hybridisierung für 10 min denaturiert.
- Beispiel 16
- ,Southern Blot': DNA-Transfer auf Membranen und Hybridisierung
- Mit dieser Technik wurden sowohl PCR-Produkte als auch genomische DNA auf eine neutrale Hybond-N Nylonmembran (Amersham Pharmacia Biotech, Freiburg) transferiert. Die genomische DNA wurde zuvor mit diversen Restriktionsendonukleasen verdaut (10 μg pro Restriktionsansatz) und über Nacht bei 20 mV in einem 0,6%igen, 14 cm langen Agarosegel aufgetrennt. Der ,Blot' erfolgte nach der Methode von Chomczynski (1992) durch abwärts gerichteten Kapillartransfer in alkalischem Transferpuffer (3 M NaCl, 8 mM NaOH, pH 11,40 – 11,45) für 2 h oder über Nacht. Vor dem Transfer wurde die DNA im Gel für 1 h in 1,5 M NaCl, 0,5 M NaOH denaturiert und anschließend für 10 min in Transferpuffer inkubiert. Im Anschluß an den Transfer wurde die Membran für 15 min mit 0,2 M Natriumphosphat-Puffer (pH 6,8) neutralisiert und für 20 min bei 80°C gebacken. Die DNA auf der Membran wurde nun zur Hybridisierung durch radioaktiv markierte Sonden genutzt. Zunächst wurde die Membran jedoch für 3h in Prähybridisierungslösung bei 60°C inkubiert. Anschließend wurde diese durch die Hybridisierungslösung ersetzt. Nach Zugabe der radioaktiv markierten Sonde erfolgte die Hybridisierung über Nacht bei 60°C. Für die Waschpuffer wurde eine 20 × SSC-Stammlösung (3 M NaCl; 0,3 M Natriumcitrat, pH 7,0 in H2O) verwendet. Die Membran wurde nacheinander für 30 min in 2 × SSC, 0,1 % SDS und für 30 min bis 2 hin 1 × SSC, 0,1 % SDS gewaschen. Die Exposition der Membran erfolgte auf Kodak Biomax MS Röntgenfilmen mit Verstärkerfolie bei –80°C.
- Beispiel 17
- Elektrophorese von RNA und ,Northern Blot'
- Sämtliche Arbeitsschritte zur Elektrophorese von RNA wurden unter RNase-freien Bedingungen mit 0,1 % DEPC-behandelten und anschließend autoklavierten Puffern durchgeführt. Die Denaturierung der RNA mit Glyoxal und DMSO sowie die anschließende elektrophoretische Auftrennung erfolgten nach Sambrook et al. (1989). 20 μg RNA in einem Volumen von 5,4 μl wurden mit 5,4 μl deionisiertem 6 M Glyoxal, 16,0 μl DMSO und 3 μl 0,1 M Natriumphosphat-Puffer (pH 7,0) versetzt und 1 h bei 50°C inkubiert. Anschließend wurden auf Eis 6 μl Glyoxalgel-Ladungspuffer (10 mM Natriumphosphat, pH 7,0; 50 % Glycerin; 0,25 % Bromphenolblau) zugegeben. Die Auftrennung erfolgte in einem 1,2%igen Agarosegel in 10 mM Natriumphosphat-Puffer (pH 7,0) bei 3 – 4 V/cm. Mit Hilfe einer abwärts gerichteten Kapillarblot-Technik unter Verwendung eines alkalischen Transferpuffers (3 M NaCl, 8 mM NaOH, pH 11,40 – 11,45) (Chomczynski, 1992) wurde die RNA auf eine neutrale Hybond-N Nylonmembran (Amersham Pharmacia Biotech, Freiburg) geblottet. Die Membran wurde 15 min in 0,2 M Natriumphosphat- Puffer (pH 6,8) neutralisiert und schließlich 20 min bei 80°C gebacken. Zur Größenabschätzung wurden 10 μg EcoRI/HindIII verdaute λ-DNA (MBI Fermentas, St. Leon-Rot) glyoxyliert und parallel zur RNA aufgetrennt. Die Spuren mit λ-DNA wurden nach der Elektrophorese vom Rest des Gels abgetrennt, zum Entfernen des Glyoxals für 20 min in 50 mM NaOH gewaschen, 15 min in 50 mM Natriumphosphat-Puffer (pH 7,0) neutralisiert und schließlich im gleichen Puffer mit 0,5 μg/ml Ethidiumbromid gefärbt. Die Hybridisierung erfolgte wie für den ,Southern Blot' beschrieben, jedoch unter stringenteren Bedingungen bei 65°C und mit 0,1 × SSC, 0,1 % SDS als zweitem Waschpuffer.
- Beispiel 18
- Sequenzierung der EtOS22-cDNA
- Die Phagenklone, deren Fusionsproteine im Western Blot von dem mAk E2E5 erkannt wurden, wurden durch DNA-Sequenzierung analysiert. Ausgehend von dieser bekannten Sequenz (dunkelgrau unterlegt) wurden durch 5'- und 3'-RACE-PCR das 3'-Ende dieses Gens sowie der größte Teil des 5'-Endes amplifiziert, wobei das 5'-Ende der cDNA durch 5'-RACE um 224 bp verlängert wurde. Das Leseraster war weiterhin durchgehend, es fehlte jedoch ein Startcodon (ATG) mit dem ein offenes Leseraster (ORF) beginnt. Um das vollständige ORF dieses Gens durch RT-PCR zu amplifizieren, wurden auf Basis von Daten aus dem Eimeria tenella Genomprojekt zwei neue Primer konstruiert: A17-f-length-64-up und A17-f-length-1176-lo, in der Sequenz durch Unterstriche gekennzeichnet, führten zu einem PCR-Produkt von 1.106 bp. So wurde das vollständige offene Leseraster amplifiziert. Die Primer hybridisieren im 5'- bzw. 3'-UTR der cDNA. Zwischen 5'-Primer und ATG-Startcodon befindet sich ein vorgeschaltetes Stop-Codon, wodurch sichergestellt ist, dass das PCR-Produkt das vollständige offene Leseraster enthält. Die EtOS22-cDNA weist ein ORF von 594 bp bzw. 198 AS auf und endet in Position 677 mit einem TAA-Stopcodon. Der 3'-UTR umfasst 506 bp. In
1 ist die vollständige Sequenz der cDNA für das EtOS22 aufgeführt, wobei die Länge des 5'-UTR noch nicht bestimmt wurde. - Beispiel 19
- Charakterisierung des EtOS22
- Das Programm Signale V1.1 (Nielsen et al., 1997) identifiziert am N-terminalen Ende des Proteins ein Signalpeptid von 18 AS (in der Sequenz hellgrau unterlegt). Die Spaltstelle zwischen Signalpeptid und reifem Protein liegt sehr wahrscheinlich zwischen Position 18 und 19 (A V A – A D). Somit liegt die Größe des reifen Proteins bei 180 AS. Daraus errechnet sich ein theoretisches Molekulargewicht von 21.039,7 Da ohne bzw. 22.830,9 Da mit Signalpeptid. Auffällig ist die Häufigkeit bestimmter AS im Vorläufer-Protein (bzw. im reifen Protein): Histidin (H) 23,2 % (25,6 %), Prolin (P) 17,2 % (18,9 %), Alanin (A) 8,6 % (6,7 %) und Glutamin (Q 7,6 % (8,3 %). Histidin und Prolin stellen zusammen über 40 % aller Aminosäuren im Protein.
- Beispiel 20
- Expression des EtOS22 in Eimeria tenella
- Um das Expressionsmuster des EtOS22 in Eimeria tenella durch den Nachweis des entsprechenden Transkripts zu untersuchen, wurden RT-PCRs und ,Northern Blots' mit sequenzspezifischen Primern bzw. radioaktiv-markierten Sonden durchgeführt. Für die RT-PCRs wurde cDNA von 4 verschiedenen Parasitenstadien verwendet: aus sporulierten Oocysten sowie aus intrazellulären Stadien 72 h, 137 h und 148 h nach der Infektion des Hühnchens, wobei für diese Stadien Gesamt-RNA aus infizierten Hühnchenblindärmen isoliert wurde. Die Amplifikation von RT-PCR-Produkten gelang 137 und 148 h nach der Infektion (Gamogonie) und im Oocystenstadium, nicht jedoch 72 h nach der Infektion (Schizogonie). Das Gen wird demnach 72 h nach der Infektion noch nicht transkribiert, die Transkription erfolgt dagegen spätestens ab 137 h nach der Infektion und auch noch in den sporulierten Oocysten (
2 ). - Im ,Northern Blot' zeigte sich ein sehr deutlicher Expressionspeak 137 h nach der Infektion. Nach 148 h und in den Oocysten ist das mRNA-Transkript des EtOS22 mit dieser Methode kaum bzw. nicht mehr nachweisbar (
3 ). Der ,Northern Blot' lieferte zudem Hinweise auf die Größe des vollständigen mRNA-Transkripts von ca. 1,1 kb. Dies stimmt sehr gut mit der Größe der klonierten cDNA überein. - Beispiel 21
- Lokalisation des Oocysten Sporocysten Proteins (EtOS22) in Eimeria tenella
- a) Immunfluoreszenz
- 3 × 107 sporulierte Oocysten wurden für 2 min bei 14.000 rpm sedimentiert, 1 × mit PBS gewaschen und anschließend mit einem dem Sediment entsprechenden Volumen Glasperlen (⌀ 0,45 – 0,5 mm) für 2 min heftig mit dem Vortex geschüttlelt, bis ein Teil der im Ansatz enthaltenen Oocysten und Sporocysten ruptiert war (mikroskopische Kontrolle). Diese Zellen und Zelltrümmer wurden sedimentiert, in – 20°C kaltem Methanol resuspendiert und für 10 min bei 22°C inkubiert. Nach einem weiteren Waschschritt erfolgte die Resuspension in PBS/0,1 % Triton X 100 für 10 min bei 22°C. Es folgte wiederholtes gründliches Waschen mit PBS, bevor unspezifische Bindungsstellen am Zellmaterial durch rotierende Inkubation in Blockpuffer (PBS/1% BSA) für 1 h bei 22°C abgesättigt wurden. Danach wurden 25 μl des 50fach aufkonzentrierten Hybridoma-Kulturüberstands des monoklonalen Antikörpers E2E5 (Primärantikörper) in 1 ml Blockpuffer hinzugegeben und für 2 h rotierend inkubiert. Durch 3maliges Waschen mit PBS für insgesamt 0,5 h wurde der Überschuss an Primärantikörper entfernt, bevor das Zellmaterial für 1 h rotierend und lichtgeschützt mit dem Sekundärantikörper Alexa Fluor 488 Ziege-anti-Maus IgG (H+L) (MoBiTec GmbH, Göppingen) inkubiert wurde. Nach 2fachem Waschen mit PBS wurde das Zellpellet in Mowiol (Polyscience Inc., Niles, IL, USA) resuspendiert, 15μl dieser Suspension auf Objektträger ausgebracht, mit einem Deckgläschen luftblasenfrei abgedeckt und lichtgeschützt bei 4°C aufbewahrt.
- b) Konfokale Laserscanning Mikroskopie
- Für die konfokale Laserscanning Mikroskopie wurde ein Zeiss IM 35 Mikroskop (Zeiss, Oberkochen) mit einem Leica CLSM-Aufsatz TCS NT (Leica Lasertechnik, Heidelberg), Version 1.5.451, verwendet. Die Fluoreszenz des Alexa 488 Farbstoffes wurde mit einem Argon-Laser bei einer Wellenlänge von 488 nm angeregt. Z-Serien von optischen Schnitten durch Oocysten und Sporocysten wurden mit einer Auflösung von 1.024 × 1.024 Pixeln gescannt. Die Auswertung erfolgte mit Adobe Photoshop 6.0 und Corel Draw 10.0 für Windows.
- Die Immunfluoreszenz gegen das EtOS22 in Oocysten (
5.1 ) und Sporocysten (5.2 und5.3 ) von Eimeria tenella bestätigte zunächst die Arbeiten von Mouafo et al. (2002). Die Anfärbung der Oocystenwand in ruptierten aber nicht in intakten Oocysten lässt auf eine Lokalisation an der inneren Wand schließen. Darüber hinaus zeigten sich distinkte Fluoreszenzsignale im Bereich des sogenannten ,Stieda-Körpers' der Sporocysten. Diese Struktur steht in engem Zusammenhang mit der Excystierung, dem Schlüpfen der beiden Sporozoiten aus der Sporocyste. Die Tatsache, dass nur bereits ruptierte Sporocysten diese Fluoreszenzsignale aufweisen, erlaubt die Schlussfolgerung, dass EtOS22 Bestandteil von Strukturen ist, die im Inneren der Sporocyste lokalisiert sind, und kein Bestandteil der äußeren Sporocystenschale ist. - Beispiel 22
- Excystierung von Sporozoiten
- Zur Gewinnung frischer Oocysten wurden 2 – 3 Wochen alte Hühnerküken mit ca. 5.000 sporulierten Oocysten von Eimeria tenella mittels einer Schlundsonde infiziert. Am 7. Tag nach der Infektion wurden die Tiere getötet und der Inhalt der Blinddärme in 2%iger Kaliumdichromatlösung gesammelt. Unter Rühren bei ca. 28°C sporulierten die Oocysten innerhalb von 48 h. Zur Sporocystengewinnung wurden die Oocysten mit einem Potter aufgebrochen. Dafür wurden in das Pottergefäß etwa 1,5 ml konzentrierte Oocystensuspension pipettiert und bei 1.300 rpm ruptiert, bis alle Oocysten aufgebrochen waren (mikroskopische Kontrolle).
- Die freigewordenen Sporocysten wurden in einem 50 ml Zentrifugenröhrchen (Falcon, Becton Dickinson, Sunnyvale, CA, USA) gesammelt und bei 2.000 rpm für 10 min zentrifugiert. Das Sediment wurde in 25 ml PBS resuspendiert und über Nacht unter Zusatz von 10 μg/ml Baytril (BAYER, Leverkusen) auf 4°C gelagert. Am nächsten Morgen wurde die Suspension sedimentiert und die Sporocysten in einem Gemisch aus 1 ml Gallensaft und 20 ml sterilfiltriertem PBS-Trypsin resuspendiert. Davon wurden für parallele Excystierungsversuche jeweils 2 ml Aliquots verwendet, ohne und unter Zugabe von 100 μl des 50fach aufkonzentrierten Hybridoma-Kulturüberstands des monoklonalen Antikörpers E2E5. Diese Ansätze wurden in einem 41,5°C-temperierten Brutschrank für 5 h inkubiert. Anschließend wurde die Anzahl freier Sporozoiten sowie nicht-geschlüpfter Sporozoiten in den Sporocysten für beide Ansätze mit einer Bürkerkammer bestimmt.
- Beispiel 23
- Hemmung der Excystierung von Sporozoiten
- Zur Untersuchung der Bedeutung von EtOS22 während der Excystierung wurden parallele Excystierungsansätze mit und ohne Zugabe des mAk E2E5 durchgeführt. Anschließend wurde die Anzahl der geschlüpften Sporozoiten und der Sporocysten mit nicht-geschlüpften Sporozoiten in beiden Ansätzen (je 2 ml) bestimmt und diese Daten miteinander verglichen. Die Anzahl geschlüpfter Sporozoiten betrug in der Kontrolle 9,6 × 106 bzw. 4,8 × 106 in Gegenwart des mAk. Die Anzahl von Sporocysten mit nicht-geschlüpften Sporozoiten stieg hingegen von 2,0 × 105 auf 2,2 × 106 (
6 ). Diese Reduzierung freier Sporozoiten um etwa die Hälfte bei gleichzeitiger Zunahme von Sporocysten mit nicht-geschlüpften Sporozoiten um etwa das 10fache zeigt, dass die Modulation der Aktivität von EtOS22 zur Hemmung der Excystierung von Eimeria tenella führt und damit eine Modulation der Aktivität von EtOS22 zur Therapie von Infektionen mit Eimeria geeignet sein kann. - Beispiel 24
- Testen von affinitätsisolierten Substanzen gegen Eimeria tenella in Zellkultur
- Die in vitro Testung erfolgt an primären Nierenzellkulturen. Hierfür wird Nierengewebe aus 12 Tage alten Legetypküken aseptisch präpariert und die daraus gewonnenen Nierenzellen für Monolayer-Gewebekulturen in 96 well-Platten angezüchtet. Als Nährmedium dient DMEM + 5% foetales Kälberserum + 2% Glutamin + 2% nichtessentielle Aminosäuren + 1 % HEPES + 1 % Natrium-Pyruvat. Nach zweitägigem Bebrüten bei 42°C und 5% CO2 werden die Gewebekulturen mit excystierten Sporozoiten von Eimeria tenella infiziert. Affinitätsisolierte Substanzen werden ausgehend von einer Stammkonzentration von 20 mg/ml Lösung in DMSO mit Nährmedium auf eine Endkonzentration von 10 ppm verdünnt und den infizierten Zellkulturen zugegeben. Am 5. Tag nach der Infektion wird die Kulturen mikroskopisch ausgewertet und der Zustand der Wirtszellen sowie die Anzahl der intakten Schizonten und freien Merozoiten (120 Stunden nach der Infektion) bestimmt. Die Wirksamkeit wirde wie folgt bewertet:

- Literatur:
-
- Altschul,S.F., Gish,W., Miller,W., Myers,E.W., Lipman,D.J., 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403-410.
- Blin,N., Stafford,D.W., 1976. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res. 3, 2303-2308.
- Chomczynski,P., 1992. One-hour downward alkaline capillary transfer for blotting of DNA and RNA. Anal. Biochem. 201, 134-139.
- Feinberg,A.P., Vogelstein,B., 1984. "A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity". Addendum. Anal. Biochem. 137, 266-267.
- Jacobsson,K., Frykberg,L., 1998. Gene VIII-based, phage-display vectors for selection against complex mixtures of ligands. Biotechniques 24, 294-301.
- Kyhse-Andersen,J., 1984. Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J.Biochem.Biophys.Methods 10, 203-209.
- Laemmli,U.K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685.
- Lihme,A., Schafer-Nielsen,C., Larsen,K.P., Muller,K.G., Bog-Hansen,T.C., 1986. Divinylsulphone-activated agarose. Formation of stable and non-leaking affinity matrices by immobilization of immunoglobulins and other proteins. J. Chromatogr. 376, 299-305.
- Morgenstern,B., Frech,K., Dress,A., Werner,T., 1998. DIALIGN: finding local similarities by multiple sequence alignment. Bioinformatics. 14, 290-294.
- Morgenstern,B., 1999. DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 15, 211-218.
- Mouafo,A.N., Weck-Heimann,A., Dubremetz,J.F., Entzeroth,R., 2002. Monoclonal antibodies specific for the two types of wall-forming bodies of Eimeria tenella macrogametes (Coccidia, Apicomplexa). Parasitol. Res. 88, 217-224.
- Muller,P.Y., Studer,E., Miserez,A.R., 2001. Molecular Biocomputing Suite: a word processor add-in for the analysis and manipulation of nucleic acid and protein sequence data. Biotechniques 31, 1306, 1308, 1310-1306, 1308, 1313.
- Nielsen,H., Engelbrecht,J., Brunak,S., von Heijne,G., 1997. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8, 581-599.
- Roswell,D.F., 1978. The chemiluminescence of luminol and related hydrazides. White, E. H. Methods Enzymol. 57, 409-423.
- Sambrook,J. Molecular cloning. A laboratory manual. Fritsch, E. F. Maniatis T., 1989. New York, Cold Spring Harbour Laboratory.
- Sanger,F., Nicklen,S., Coulson,A.R., 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A 74, 5463-5467.
- Tatusova,T.A., Madden,T.L., 1999. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174, 247-250.
- Thompson,J.D., Higgins,D.G., Gibson,T.J., 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673-4680.
- Zhang,L., Jacobsson,K., Strom,K., Lindberg,M., Frykberg,L., 1999. Staphylococcus aureus expresses a cell surface protein that binds both IgG and beta2-glycoprotein I. Microbiology 145 (Pt 1), 177-183.
- Bhogal, B.S. et al. (1992): Potential of a recombinant antigen as a prophylactic vaccine for day-old broiler chickens against Eimeria tenella infections. Vet. Immunol. Immunopathol. 31:323-335.
- Binger, M.H., Hug D., Weber, G., Schikdknecht, E., Hümbelin M., Pasamontes L. (1993): Cloning and characterization of a surface antigen of Eimeria tenella merozoites and expression using a recombinant vaccinia virus. Mol. Biochem. Parasitol. 61:179-188.
- Brake, D.A. (2002): Vaccinology for control of apicomplexan parasites: a simplified language of immune programming and ist use in vaccine design. Int. J. Parasitol. 32:509-515
- Chapman, H.D., Cherry T.E., Danforth, H.D., Richards G., Shirley M.W., Williams, R.B. (2002): Sustainable coccidiosis control in poultry production: the role of live vaccines. Int. J. Parasitol. 32:617-629.
- Clarke, L.E, Messer, L.I, Wisher, M.H. (1986): Antigens of Eimeria cloned and expressed in E. coli. J. Cell Biochem. 10:A 145.
- Coombs, G.H., Müller, S. (2002): Recent advances in the search for new anticoccidial drugs. Int. J. Parasitol 32:497-508.
- Crane, M.S.J. et al. (1991): Cross-protection against four species of chicken coccidia with a single recombinant antigen. Infect. Immun. 59:1271-1277.
- Danforth, H.D., Augustine P.C. (1985): Use of hybridoma antibodies and recombinant DNA technology in protozoan vaccine development. Avian Diseases 30:37-42.
- Eschenbacher, K.H., Eggli P., Wallach M., Braun R. (1996): Characterisation of a 14 kda oocyst wall protein of Eimeria tenella and Eimeria acervulina. Parasitol. 112(2):169-176.
- Files, J.G., Paul, L.S., Gabe, J.D. (1987): Identification and characterization of the gene for a major surface antigen of Eimeria tenella. In: Molecular strategies of parasite invasion. N. Agabian, H. Goodman and N. Noguiera (eds.). UCLA Symp. Mol. Cell. Biol., New Series, Vol. 32. Alan R. Liss, Inc., New York pp 713-723
- Greif, G., Harder, A., Haberkorn, A. (2001): Chemotherapeutic approaches to protozoa: Coccidiae – current level of knowledge and outlook. Parasitol Res 87:973–975
- Jenkins, M.C. (1998): Progress on developing a recombinant coccidiosis vaccine. Int. L. Parasitol. 28:1111-9
- Ko, C., Smith, C.K. II, McDonell M. (1990): Ifdentification and characterization of a target antigen of a monoclonal antibody directed against Eimeria tenella merozoites. Mol. Biochem. Parasitol. 41:53-64
- Levine, L.D. et al. (1980): A newly revised classification of Protozoa. J. Protozool. 27:37-58.
- Miller, G.A. et al. (1989): Characterization and vaccine potential of a novel recombinant coccidial antigen. Infect. Immun. 57:2014-2020.
- Pogonka, T., Klotz, C., Kovacs, F., Lucius R. (2003): A single dose of recombinant Salmonella typhimurium induces specific humoral immune responses against heterologous Eimeria tenella antigens in chicken. Int. J. Parasitol. 33:81-88
- Rose, M.E., Wakelin D. (1990): Immunity to coccidiosis. In: Coccidiosis of man and domestic animals, Long P.L. (ed.), CRC Press, pp 281-306.
- Sangster N., Batterham, P., Chapman H.D., Duraisingh, M., Jambre L.L., Shirley M., Upcroft, J., Upcroft, P. (2002): Resistance to antiparasitic drugs: the role of molecular diagnosis. Int. J. Parasitol. 32:637-653.
- Silva, A., Kawazoe, U., Freitas, F.F.T., Gatti, M.S.V., Dolder, H., Schumacher, R.I:, Juliano, M.A., Silva, M.J., Leite, A. (2002): Avian anticoccidial activity of a novel membrane-interactive peptide selected from phage diplay libraries. Molec. Biochem. Parasitol. 120:53-60.
- Stiff, M., Bafundo, K.W. (1993): Development of immunity in broilers continuously exposed to Eimeria sp. Avian Diseases 37:295-301.
- Vermeulen, A.N. (1998): Progress in recombinant vaccine development against coccidiosis. A review and prospects into the next millenium. Int. J. Parasitol. 28:1121-1130.
- Vermeulen, A.N., Schaap, D.C., Schetters P.M. (2001): Control of coccidiosis in chickens by vaccination. Vet. Parasitol. 100:13-20.
- Wallach, M., Halabi, A., Pillemer, G., Sar-Shalom, O., Mencher, D., Gilad, M., Bendheim, U., Danforth, H.D., Augustine, P.C. (1992): Maternal immunization with gametocyte antigens as a means of providing protective immunity against Eimeria maxima in chickens. Infection and Immunity 60:2036-2039.
- Williams, R.B. (2002): Anticoccidial vaccines for broiler chickens: pathway to success. Avian Pathology 31:317-353.
- Wan K-L., Chong S-P., Ng, S-T., Shirley, M.W., Tomley, F.M., Jangi M.S. (1999): A survey of genes in Eimeria tenella merozoites by EST sequencing. Int. J. Parasitol 29:1885-1892.
- Comes AM, Humbert JF, Cabaret J, Elard L (1996): Using molecular tools for diagnosis in veterinary parasitology. Vet Res 27 (4-5):333-42
- Ellis, J & Bumstead J (1990): Eimeria species: studies usnig rRNA and rDNA probes. Parasitology 101:1-6.
- Gasser RB, Woods WG, Wood JM, Ashdown L, Richards G, Whithear KG (2001): Automated, fluorescence-based approach for the specific diagnosis of chicken coccidiosis. Electrophoresis 22 (16):3546-50
- Johnston DA, Fernando MA (1997): Isoenzymes of eimeria from the domestic fowl: electrophorertic variants among species, strains and clones. Parasitol Res 83(5):464-70
- Long PL, and Reid WM (1982): A guide to the diagnosis of coccidiosis in chickens, Research Report 404, University of Georgia, Athens.
- Molloy JB, Eaves FW, Jeston PJ, Minchin CM, Stewart NP, Lew AE, Jorgensen WK (1998): Detection of Eimeria acervulina using the polymerase chain reaction. Avian Dis 42(1):119-23.
- Procunier JD, Fernando MA, Barta JR (1993): Species and strain differentiation of Eimeria spp. Of the domestic fowl using DNA polymorphisms amplified by arbitrary primers. Parasitol Res 79:98-102
- Schnitzler, BE, Thebo PL, Mattson, JG, Tomley, FM, Shirley, MW (1998): Development of a diagnostic PCR assay for the detection and discrimination of four pathogenic Eimeria species of the chicken. Avian Pathology 27:490-497.
- Sambrook, J and Russell DW (2001): Molecular Cloning. A laboratory Manual. Cold Spring Harbour Laboratory Press. New York.
- SEQUENCE LISTING

-

-

-
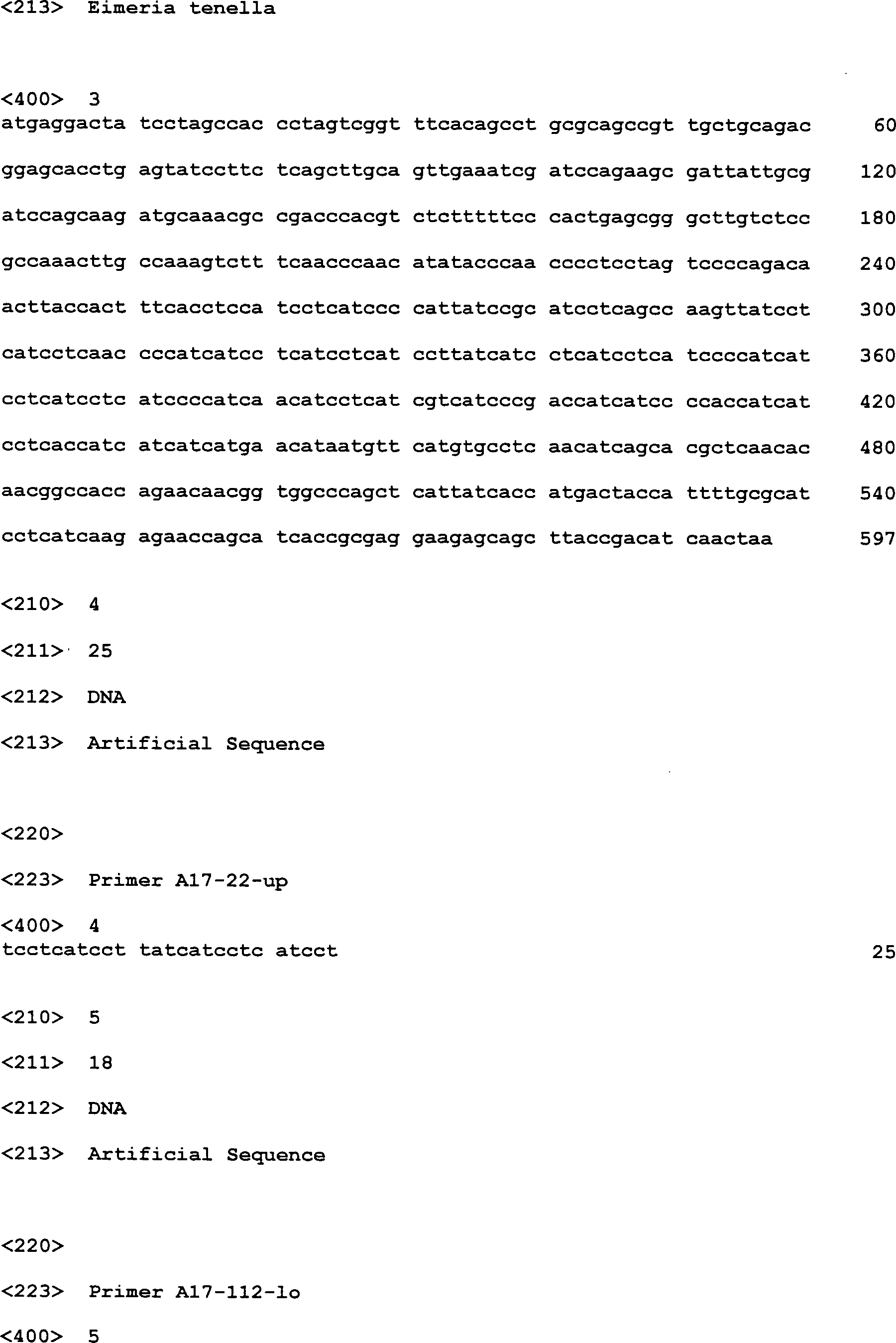
-

-

-

-

-
-

Claims (19)
- Polynucleotid enthaltend: a) die Sequenz gemäß SEQ ID NO:1 oder 3; oder b) ein Polynucleotid, das zu dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 eine Identität von mehr als 50% aufweist; oder c) ein Polynucleotid, die mit dem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 oder 3 unter stringenten Bedingungen hybridisiert; oder d) ein Polynucleotid, welches für ein Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert; oder e) ein Polynucleotid, das zu einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, eine Identität von mehr als 50% aufweist; oder f) ein Polynucleotid, das mit einem Polynucleotid, welches für das Polypeptid mit der Sequenz gemäß SEQ ID NO:2 kodiert, unter stringenten Bedingungen hybridisiert; oder g) ein Polynucleotid, das von einem Polynucleotid mit der Sequenz gemäß SEQ ID NO:1 aufgrund der Degeneration des genetischen Codes abweicht; h) ein Polynucleotid, das ein Fragment eines Polynucleotids nach a) bis g) darstellt und mindestens 6 Nucleotide lang ist.
- Polypeptid, das von einem Polynucleotid gemäß Anspruch 1 kodiert wird und mindestens 8 Aminosäuren lang ist.
- Vektor oder Expressionssystem enthaltend wenigstens eines der Polynucleotide nach Anspruch 1.
- Wirtszelle, enthaltend den Vektor oder das Expressionssystem nach Anspruch 3.
- Verfahren zur Herstellung eines Polypeptids nach Anspruch 2 mit Hilfe einer Wirtszelle nach Anspruch 4.
- Antikörper, dadurch gekennzeichnet, das er spezifisch an das Polypeptid nach Anspruch 2 bindet.
- Verwendung von Antikörpern nach Anspruch 6 als Parasitizide.
- Verfahren zum Nachweis eines Polynucleotids nach Anspruch 1, wobei ein Polynucleotid nach Anspruch 1 mit dem Nukleinsäurematerial aus einer biologischen Probe hybridisiert und die Hybridisierung nachgewiesen wird.
- Verfahren nach Anspruch 8, wobei der Nachweis der Hybridisierung durch Polymerase-Kettenreaktion erfolgt.
- Verfahren zum Nachweis eines Polypeptids nach Anspruch 2, wobei das Polypeptid durch Bindung des Antikörpers nach Anspruch 6 nachgewiesen wird.
- Zusammensetzung zum Nachweis eines Polynucleotids nach Anspruch 1 oder eines Polypeptids nach Anspruch 2 enthaltend ein Polynucleotid nach Anspruch 1 oder einen Antikörper nach Anspruch 6.
- Verwendung eines: a) Polynucleotids nach Anspruch 1; oder b) Polypeptids nach Anspruch 2; oder c) Vektors oder Expressionssystems nach Anspruch 3; oder d) Antikörpers nach Anspruch 6 zur Herstellung eines Impfstoffes.
- Impfstoff enthaltend: a) ein Polypeptid nach Anspruch 2; oder b) einen Vektor nach Anspruch 3; oder c) einen Antikörper nach Anspruch 6.
- Verfahren zum Auffinden von Wirkstoffen, die die Aktivität des EtOS22-Proteins bei der Excystierung von Sporozoiten aus Sporocysten modulieren bei dem man: a) den zu testenden Wirkstoff mit einem EtOS22-Polypeptid nach Anspruch 2 in Kontakt bringt, wobei die gewählten Bedingungen eine spezifische Bindung der Testsubstanz an das EtOS22-Polypeptid gestatten; und b) eine erfolgte spezifische Bindung an das Polypeptid detektiert; wobei ein Wirkstoff, der an das Polypeptid bindet, als potentieller Wirkstoff zur Therapie der Kokzidiose identifiziert wird.
- Verfahren zum Auffinden von Wirkstoffen, die die Aktivität des EtOS22-Proteins bei der Excystierung von Sporozoiten aus Sporocysten modulieren bei dem man: a) den zu testenden Wirkstoff mit einem EtOS22-Polypeptid nach Anspruch 2 in Kontakt bringt, wobei die gewählten Bedingungen eine spezifische Bindung der Testsubstanz an das EtOS22-Polypeptid gestatten; und b) eine Modulation der Aktivität des Polypeptids nach Anspruch 2 oder des EtOS22-Proteins nachweist; wobei ein Wirkstoff, der die Aktivität moduliert, als potentieller Wirkstoff zur Therapie der Kokzidiose identifiziert wird.
- Wirkstoff, auffindbar mit einem der Verfahren nach Anspruch 14 oder 15.
- Wirkstoff, der die Aktivität des EtOS22-Proteins bei der Excystierung von Sporozoiten aus Sporocysten moduliert.
- Pharmazeutische Zusammensetzung enthaltend den Wirkstoff nach Anspruch 16 oder 17 und einen pharmazeutisch zulässigen Träger.
- Verwendung eines Wirkstoffes, der die Aktivität des EtOS22-Proteins bei der Excystierung von Sporozoiten aus Sporocysten moduliert, zur Herstellung eines Medikamentes zur Behandlung von Kokzidiose.
Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10330235A DE10330235A1 (de) | 2003-07-04 | 2003-07-04 | Neues Eimeria Gen und Protein sowie deren Verwendung |
| AU2004255594A AU2004255594B2 (en) | 2003-07-04 | 2004-06-30 | Use of a novel eimeria gene and corresponding protein |
| PCT/EP2004/007080 WO2005005472A1 (en) | 2003-07-04 | 2004-06-30 | Use of a novel eimeria gene and corresponding protein |
| JP2006518068A JP4594304B2 (ja) | 2003-07-04 | 2004-06-30 | 新規eimeria遺伝子および対応タンパク質の使用 |
| EP10176984A EP2311859A1 (de) | 2003-07-04 | 2004-06-30 | Verwendungen von einem neuen Eimeria Gen und entsprechender Protein |
| CA002531161A CA2531161A1 (en) | 2003-07-04 | 2004-06-30 | Use of a novel eimeria gene and corresponding protein |
| BRPI0412323-9A BRPI0412323A (pt) | 2003-07-04 | 2004-06-30 | usos de um gene eimeria e proteìna correspondente |
| MXPA06000198A MXPA06000198A (es) | 2003-07-04 | 2004-06-30 | Usos de un nuevo gen de eimeria y proteina correspondiente. |
| EP04740461A EP1644407A1 (de) | 2003-07-04 | 2004-06-30 | Verwendungen von einem neuen eimeria gen und entsprechendes protein |
| NZ544451A NZ544451A (en) | 2003-07-04 | 2004-06-30 | Uses of a novel eimeria gene and corresponding protein |
| US10/562,324 US7691987B2 (en) | 2003-07-04 | 2004-06-30 | Use of a novel eimeria gene and corresponding protein |
| ARP040102341A AR045000A1 (es) | 2003-07-04 | 2004-07-02 | Gen y proteina de eimeria , y su uso |
| ZA200510465A ZA200510465B (en) | 2003-07-04 | 2005-12-23 | Use of a novel eimeria gene and corresponding protein |
| NO20060568A NO20060568L (no) | 2003-07-04 | 2006-02-03 | Anvendelse av et nytt eimeriagen og tilhorende protein |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10330235A DE10330235A1 (de) | 2003-07-04 | 2003-07-04 | Neues Eimeria Gen und Protein sowie deren Verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| DE10330235A1 true DE10330235A1 (de) | 2005-01-20 |
Family
ID=33521342
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE10330235A Withdrawn DE10330235A1 (de) | 2003-07-04 | 2003-07-04 | Neues Eimeria Gen und Protein sowie deren Verwendung |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US7691987B2 (de) |
| EP (2) | EP1644407A1 (de) |
| JP (1) | JP4594304B2 (de) |
| AR (1) | AR045000A1 (de) |
| AU (1) | AU2004255594B2 (de) |
| BR (1) | BRPI0412323A (de) |
| CA (1) | CA2531161A1 (de) |
| DE (1) | DE10330235A1 (de) |
| MX (1) | MXPA06000198A (de) |
| NO (1) | NO20060568L (de) |
| NZ (1) | NZ544451A (de) |
| WO (1) | WO2005005472A1 (de) |
| ZA (1) | ZA200510465B (de) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9072668B2 (en) | 2010-03-09 | 2015-07-07 | Janssen Biotech, Inc. | Non-aqueous high concentration reduced viscosity suspension formulations of antibodies |
| US20110223208A1 (en) * | 2010-03-09 | 2011-09-15 | Beth Hill | Non-Aqueous High Concentration Reduced Viscosity Suspension Formulations |
| EP2544721B1 (de) * | 2010-03-09 | 2017-12-06 | Janssen Biotech, Inc. | Nichtwässrige und hochkonzentrierte suspensionsformulierungen von reduzierter viskosität |
| EP2782458B1 (de) * | 2011-11-17 | 2018-09-12 | DSM IP Assets B.V. | VERWENDUNG VON SÄURESTABILEN PROTEASEN IN TIERFUTTER UM DIE LEISTUNGSFÄHIGKEIT VON GEGEN KOKzidien GEIMPFTEN MASTHÜHNERN ZU ERHÖHEN |
| JP2015508155A (ja) | 2012-01-24 | 2015-03-16 | ユニバーシティ オブ マサチューセッツ | 膵臓β細胞障害における可溶性MANF |
| CN103233017B (zh) * | 2013-05-06 | 2014-11-05 | 扬州大学 | 毒害艾美耳球虫的配子体抗原gam22基因及其应用 |
| CN111471107B (zh) * | 2020-04-27 | 2023-04-18 | 中国农业大学 | 抗鸡球虫多价重组蛋白卵黄抗体及其制备方法和应用 |
| CN112592961A (zh) * | 2021-01-07 | 2021-04-02 | 山东科硕生物技术有限公司 | 一种核酸样本保存液及其制备方法和应用 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5187080A (en) * | 1984-06-05 | 1993-02-16 | Solvay & Cie S.A. | DNA encoding an antigenic protein derived from Eimeria tenella and vaccines for prevention of coccidiosis caused by Eimeria tenella |
| EP0453054B1 (de) * | 1985-12-03 | 1996-07-03 | SOLVAY (Société Anonyme) | Antigene Proteine und diese enthaltende Impfstoffe zur Verhütung von Kokzidiose |
| ZA893740B (en) * | 1988-06-03 | 1990-02-28 | Hoffmann La Roche | Recombinant coccidiosis vaccines |
| JPH06504187A (ja) * | 1990-09-12 | 1994-05-19 | ジェネックス・コーポレーション | 遺伝子操作されたコクシジウム症ワクチン |
-
2003
- 2003-07-04 DE DE10330235A patent/DE10330235A1/de not_active Withdrawn
-
2004
- 2004-06-30 JP JP2006518068A patent/JP4594304B2/ja not_active Expired - Fee Related
- 2004-06-30 NZ NZ544451A patent/NZ544451A/en not_active IP Right Cessation
- 2004-06-30 EP EP04740461A patent/EP1644407A1/de not_active Ceased
- 2004-06-30 AU AU2004255594A patent/AU2004255594B2/en not_active Ceased
- 2004-06-30 MX MXPA06000198A patent/MXPA06000198A/es active IP Right Grant
- 2004-06-30 EP EP10176984A patent/EP2311859A1/de not_active Withdrawn
- 2004-06-30 US US10/562,324 patent/US7691987B2/en not_active Expired - Fee Related
- 2004-06-30 WO PCT/EP2004/007080 patent/WO2005005472A1/en active Application Filing
- 2004-06-30 BR BRPI0412323-9A patent/BRPI0412323A/pt not_active Application Discontinuation
- 2004-06-30 CA CA002531161A patent/CA2531161A1/en not_active Abandoned
- 2004-07-02 AR ARP040102341A patent/AR045000A1/es unknown
-
2005
- 2005-12-23 ZA ZA200510465A patent/ZA200510465B/xx unknown
-
2006
- 2006-02-03 NO NO20060568A patent/NO20060568L/no not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| CA2531161A1 (en) | 2005-01-20 |
| JP2008500003A (ja) | 2008-01-10 |
| US20070184054A1 (en) | 2007-08-09 |
| NZ544451A (en) | 2009-06-26 |
| AU2004255594A1 (en) | 2005-01-20 |
| ZA200510465B (en) | 2007-03-28 |
| BRPI0412323A (pt) | 2006-08-22 |
| NO20060568L (no) | 2006-03-31 |
| EP2311859A1 (de) | 2011-04-20 |
| US7691987B2 (en) | 2010-04-06 |
| AU2004255594B2 (en) | 2010-12-09 |
| AR045000A1 (es) | 2005-10-12 |
| WO2005005472A1 (en) | 2005-01-20 |
| MXPA06000198A (es) | 2006-03-21 |
| JP4594304B2 (ja) | 2010-12-08 |
| EP1644407A1 (de) | 2006-04-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Miller et al. | Characterization and vaccine potential of a novel recombinant coccidial antigen | |
| DE69729839T2 (de) | Attenuierter lebender Neospora Impfstoff | |
| JPH0699475B2 (ja) | コクシジウムのモノクロ−ナル抗体 | |
| US5028694A (en) | Antigenic proteins and vaccines containing them for prevention of coccidiosis caused by eimeria Eimeria necatrix and Eimeria tenella | |
| DE10330235A1 (de) | Neues Eimeria Gen und Protein sowie deren Verwendung | |
| DE69635472T2 (de) | Impfstoff gegen Geflügelkokzidose | |
| DE69737691T2 (de) | Impfstoff, der peroxiredoxin enthält | |
| JPH07196690A (ja) | コクシジウム症家禽ワクチン | |
| US8128921B2 (en) | Use of conditional plasmodium strains lacking nutrient transporters in malaria vaccination | |
| DE69931518T2 (de) | Chimäres Gen, das für die antigenischen Determinaten von vier Proteinen aus L. Infantum kodiert | |
| JPH08127593A (ja) | 家禽コクシジウム症ワクチン | |
| DE69934953T2 (de) | Impstoffe gegen Coccidiosis | |
| US7799330B2 (en) | Piroplasmid vaccine | |
| El-Ashram et al. | Protective potential of diclazuril-treated oocysts against coccidiosis in layer chicks | |
| DE602005003429T2 (de) | Kodierende DNS für ein Eimeria Apical Membran Antigen 1 und dessen Verwendungen | |
| DE60128569T2 (de) | Neospora caninum isolat | |
| US20190381159A1 (en) | Doubly attenuated late liver stage malaria parasites and related compositions and methods | |
| EP0283882A1 (de) | Klonierung von Malaria-spezifischen DNA-Sequenzen:Isolierung des Gens für das 140 kd Protein | |
| GREIF et al. | Patent 2531161 Summary | |
| EP0499834B1 (de) | Ein Plasmodium falciparum Blutstadien-Antigen, seine Herstellung und Verwendung | |
| US6207167B1 (en) | Chicken leucocytozoon vaccine | |
| Zhou | Molecular cloning and expression in Escherichia coli of a malaria exoantigen from Plasmodium falciparum, putative candidate for vaccine | |
| Fox | Regulation of TamS1 gene expression during differentiation to the merozoite in Theileria annulata | |
| Rojas | Biological, biochemical and molecular characterization of Flag-1, a Leishmania membrane-associated flagellar protein | |
| Bloomgarden | Characterization of PnudC: A plasmodial homolog of a protein essential for nuclear migration in the fungus Aspergillus nidulans |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8127 | New person/name/address of the applicant |
Owner name: BAYER ANIMAL HEALTH GMBH, 51373 LEVERKUSEN, DE |
|
| 8141 | Disposal/no request for examination |