CN1607962A - 聚谷氨酸-治疗剂结合物的生产方法 - Google Patents
聚谷氨酸-治疗剂结合物的生产方法 Download PDFInfo
- Publication number
- CN1607962A CN1607962A CNA008170290A CN00817029A CN1607962A CN 1607962 A CN1607962 A CN 1607962A CN A008170290 A CNA008170290 A CN A008170290A CN 00817029 A CN00817029 A CN 00817029A CN 1607962 A CN1607962 A CN 1607962A
- Authority
- CN
- China
- Prior art keywords
- therapeutic agent
- polyglutamic acid
- acid
- conjugate
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000003814 drug Substances 0.000 title claims abstract description 80
- 229940124597 therapeutic agent Drugs 0.000 title claims abstract description 71
- 238000004519 manufacturing process Methods 0.000 title description 8
- 238000000034 method Methods 0.000 claims abstract description 78
- 230000008569 process Effects 0.000 claims abstract description 14
- 229960001592 paclitaxel Drugs 0.000 claims description 76
- 229920002643 polyglutamic acid Polymers 0.000 claims description 76
- 229930012538 Paclitaxel Natural products 0.000 claims description 64
- 239000000243 solution Substances 0.000 claims description 63
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 52
- 108010020346 Polyglutamic Acid Proteins 0.000 claims description 51
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 49
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 41
- 239000007787 solid Substances 0.000 claims description 39
- 229920000642 polymer Polymers 0.000 claims description 37
- 238000002360 preparation method Methods 0.000 claims description 35
- 239000000203 mixture Substances 0.000 claims description 31
- -1 10,11-methylene Chemical group 0.000 claims description 30
- 235000002639 sodium chloride Nutrition 0.000 claims description 25
- 239000000725 suspension Substances 0.000 claims description 23
- 239000000376 reactant Substances 0.000 claims description 19
- 239000003153 chemical reaction reagent Substances 0.000 claims description 14
- 239000011780 sodium chloride Substances 0.000 claims description 14
- 239000002904 solvent Substances 0.000 claims description 14
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 13
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 12
- 239000012535 impurity Substances 0.000 claims description 12
- 150000003839 salts Chemical class 0.000 claims description 12
- 229960003668 docetaxel Drugs 0.000 claims description 11
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 10
- 239000003960 organic solvent Substances 0.000 claims description 10
- 238000005406 washing Methods 0.000 claims description 10
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 9
- 239000007864 aqueous solution Substances 0.000 claims description 9
- 229960002989 glutamic acid Drugs 0.000 claims description 9
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 8
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 8
- 229940073490 sodium glutamate Drugs 0.000 claims description 8
- UKIMCRYGLFQEOE-RLHMMOOASA-N (-)-Epothilone F Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(CO)sc2)/C)OC(=O)C[C@H](O)C1(C)C UKIMCRYGLFQEOE-RLHMMOOASA-N 0.000 claims description 6
- 241000790917 Dioxys <bee> Species 0.000 claims description 6
- UKIMCRYGLFQEOE-UHFFFAOYSA-N Epothilone F Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC2(C)OC2CC1C(C)=CC1=CSC(CO)=N1 UKIMCRYGLFQEOE-UHFFFAOYSA-N 0.000 claims description 6
- 239000002253 acid Substances 0.000 claims description 6
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 claims description 6
- UKIMCRYGLFQEOE-RGJAOAFDSA-N epothilone f Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(CO)=N1 UKIMCRYGLFQEOE-RGJAOAFDSA-N 0.000 claims description 6
- MZNMZWZGUGFQJP-UHFFFAOYSA-N 1-[11-(dodecylamino)-10-hydroxyundecyl]-3,7-dimethylpurine-2,6-dione Chemical compound O=C1N(CCCCCCCCCC(O)CNCCCCCCCCCCCC)C(=O)N(C)C2=C1N(C)C=N2 MZNMZWZGUGFQJP-UHFFFAOYSA-N 0.000 claims description 5
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 claims description 5
- 229930013356 epothilone Natural products 0.000 claims description 5
- 150000003883 epothilone derivatives Chemical class 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 239000002244 precipitate Substances 0.000 claims description 5
- 239000011734 sodium Substances 0.000 claims description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 4
- 229940024606 amino acid Drugs 0.000 claims description 4
- 150000001413 amino acids Chemical class 0.000 claims description 4
- 239000002246 antineoplastic agent Substances 0.000 claims description 4
- 230000008878 coupling Effects 0.000 claims description 4
- 238000010168 coupling process Methods 0.000 claims description 4
- 238000005859 coupling reaction Methods 0.000 claims description 4
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 4
- 229910052708 sodium Inorganic materials 0.000 claims description 4
- 159000000000 sodium salts Chemical class 0.000 claims description 4
- FUXVKZWTXQUGMW-FQEVSTJZSA-N 9-Aminocamptothecin Chemical compound C1=CC(N)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 FUXVKZWTXQUGMW-FQEVSTJZSA-N 0.000 claims description 3
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 claims description 3
- BEFZAMRWPCMWFJ-JRBBLYSQSA-N Epothilone C Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C=C\C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C BEFZAMRWPCMWFJ-JRBBLYSQSA-N 0.000 claims description 3
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 claims description 3
- 229950010817 alvocidib Drugs 0.000 claims description 3
- BEFZAMRWPCMWFJ-UHFFFAOYSA-N desoxyepothilone A Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC=CCC1C(C)=CC1=CSC(C)=N1 BEFZAMRWPCMWFJ-UHFFFAOYSA-N 0.000 claims description 3
- 229960004679 doxorubicin Drugs 0.000 claims description 3
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical group C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 claims description 3
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 claims description 3
- BEFZAMRWPCMWFJ-QJKGZULSSA-N epothilone C Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 BEFZAMRWPCMWFJ-QJKGZULSSA-N 0.000 claims description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 claims description 3
- 229960005420 etoposide Drugs 0.000 claims description 3
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 claims description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 3
- 229960005277 gemcitabine Drugs 0.000 claims description 3
- 230000035479 physiological effects, processes and functions Effects 0.000 claims description 3
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 claims description 3
- 229950009213 rubitecan Drugs 0.000 claims description 3
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 claims description 3
- 229960001278 teniposide Drugs 0.000 claims description 3
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 claims description 3
- NWXMGUDVXFXRIG-WESIUVDSSA-N (4s,4as,5as,6s,12ar)-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]4(O)C(=O)C3=C(O)C2=C1O NWXMGUDVXFXRIG-WESIUVDSSA-N 0.000 claims description 2
- YMKNDDWBMTZINE-UHFFFAOYSA-N 1-[11-[dodecyl(methyl)amino]-10-hydroxyundecyl]-3,7-dimethylpurine-2,6-dione Chemical compound O=C1N(CCCCCCCCCC(O)CN(C)CCCCCCCCCCCC)C(=O)N(C)C2=C1N(C)C=N2 YMKNDDWBMTZINE-UHFFFAOYSA-N 0.000 claims description 2
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 claims description 2
- 229930195573 Amycin Natural products 0.000 claims description 2
- XOZIUKBZLSUILX-SDMHVBBESA-N Epothilone D Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C(/C)=C/C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C XOZIUKBZLSUILX-SDMHVBBESA-N 0.000 claims description 2
- 125000003368 amide group Chemical group 0.000 claims description 2
- XOZIUKBZLSUILX-UHFFFAOYSA-N desoxyepothilone B Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC(C)=CCC1C(C)=CC1=CSC(C)=N1 XOZIUKBZLSUILX-UHFFFAOYSA-N 0.000 claims description 2
- XOZIUKBZLSUILX-GIQCAXHBSA-N epothilone D Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C(C)=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 XOZIUKBZLSUILX-GIQCAXHBSA-N 0.000 claims description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 claims 1
- 238000011161 development Methods 0.000 abstract description 4
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 45
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 37
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 33
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 22
- 238000003756 stirring Methods 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 239000000843 powder Substances 0.000 description 18
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 206010028980 Neoplasm Diseases 0.000 description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 229960002668 sodium chloride Drugs 0.000 description 12
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- 230000000903 blocking effect Effects 0.000 description 9
- 238000004108 freeze drying Methods 0.000 description 9
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 7
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 7
- 125000000524 functional group Chemical group 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 239000012141 concentrate Substances 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- OKDQKPLMQBXTNH-UHFFFAOYSA-N n,n-dimethyl-2h-pyridin-1-amine Chemical compound CN(C)N1CC=CC=C1 OKDQKPLMQBXTNH-UHFFFAOYSA-N 0.000 description 6
- 108700027936 paclitaxel poliglumex Proteins 0.000 description 6
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- 238000002835 absorbance Methods 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- 239000006227 byproduct Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N 1,3-di(propan-2-yl)urea Chemical compound CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- 208000035126 Facies Diseases 0.000 description 4
- 239000004471 Glycine Substances 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 239000007822 coupling agent Substances 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 4
- 238000011065 in-situ storage Methods 0.000 description 4
- 238000011031 large-scale manufacturing process Methods 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- 125000004423 acyloxy group Chemical group 0.000 description 3
- 125000005041 acyloxyalkyl group Chemical group 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- NBKZGRPRTQELKX-UHFFFAOYSA-N (2-methylpropan-2-yl)oxymethanone Chemical compound CC(C)(C)O[C]=O NBKZGRPRTQELKX-UHFFFAOYSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 2
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 2
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- MLIREBYILWEBDM-UHFFFAOYSA-N cyanoacetic acid Chemical compound OC(=O)CC#N MLIREBYILWEBDM-UHFFFAOYSA-N 0.000 description 2
- 229930182912 cyclosporin Natural products 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 230000006837 decompression Effects 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 229960000961 floxuridine Drugs 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 231100000682 maximum tolerated dose Toxicity 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- 238000012856 packing Methods 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 239000008213 purified water Substances 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000002798 spectrophotometry method Methods 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229960004559 theobromine Drugs 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229960001722 verapamil Drugs 0.000 description 2
- 238000009736 wetting Methods 0.000 description 2
- 229910052726 zirconium Inorganic materials 0.000 description 2
- HVWICSINEWOYNO-UHFFFAOYSA-N (2-oxo-1,3-oxazolidin-3-yl)phosphonic acid Chemical class OP(O)(=O)N1CCOC1=O HVWICSINEWOYNO-UHFFFAOYSA-N 0.000 description 1
- HJRJRUMKQCMYDL-UHFFFAOYSA-N 1-chloro-2,4,6-trinitrobenzene Chemical compound [O-][N+](=O)C1=CC([N+]([O-])=O)=C(Cl)C([N+]([O-])=O)=C1 HJRJRUMKQCMYDL-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- VRPJIFMKZZEXLR-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC(C)(C)OC(=O)NCC(O)=O VRPJIFMKZZEXLR-UHFFFAOYSA-N 0.000 description 1
- QKSGIGXOKHZCQZ-UHFFFAOYSA-N 2-chloro-2-phenylacetic acid Chemical compound OC(=O)C(Cl)C1=CC=CC=C1 QKSGIGXOKHZCQZ-UHFFFAOYSA-N 0.000 description 1
- XNINAOUGJUYOQX-UHFFFAOYSA-N 2-cyanobutanoic acid Chemical compound CCC(C#N)C(O)=O XNINAOUGJUYOQX-UHFFFAOYSA-N 0.000 description 1
- JDEFPFLTCXIVDH-UHFFFAOYSA-N 2-cyanopropanoic acid Chemical compound N#CC(C)C(O)=O JDEFPFLTCXIVDH-UHFFFAOYSA-N 0.000 description 1
- FVNIMHIOIXPIQT-UHFFFAOYSA-N 2-methoxybutane Chemical compound CCC(C)OC FVNIMHIOIXPIQT-UHFFFAOYSA-N 0.000 description 1
- SLAMLWHELXOEJZ-UHFFFAOYSA-N 2-nitrobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1[N+]([O-])=O SLAMLWHELXOEJZ-UHFFFAOYSA-N 0.000 description 1
- FDHRGQIRBRQMPF-UHFFFAOYSA-N 2h-pyridin-1-amine Chemical compound NN1CC=CC=C1 FDHRGQIRBRQMPF-UHFFFAOYSA-N 0.000 description 1
- QSIFOTQDNVCTTM-UHFFFAOYSA-N 3-methyl-1h-imidazol-3-ium;trifluoromethanesulfonate Chemical compound CN1C=CN=C1.OS(=O)(=O)C(F)(F)F QSIFOTQDNVCTTM-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- SJZRECIVHVDYJC-UHFFFAOYSA-N 4-hydroxybutyric acid Chemical compound OCCCC(O)=O SJZRECIVHVDYJC-UHFFFAOYSA-N 0.000 description 1
- WDJHALXBUFZDSR-UHFFFAOYSA-N Acetoacetic acid Natural products CC(=O)CC(O)=O WDJHALXBUFZDSR-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- LEUNVDPYYPEPEW-UHFFFAOYSA-N C(C)OCC(=O)O.ClC1=CC=CC=C1 Chemical compound C(C)OCC(=O)O.ClC1=CC=CC=C1 LEUNVDPYYPEPEW-UHFFFAOYSA-N 0.000 description 1
- WJKSDWSTJREQHM-UHFFFAOYSA-N C(C1=CC=CC=C1)#N.C(C)(=O)O.[O] Chemical compound C(C1=CC=CC=C1)#N.C(C)(=O)O.[O] WJKSDWSTJREQHM-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- 206010013183 Dislocation of vertebra Diseases 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 206010053759 Growth retardation Diseases 0.000 description 1
- 101000777550 Homo sapiens CCN family member 2 Proteins 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 238000003916 acid precipitation Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- ULELPEYHAFTGRE-UHFFFAOYSA-N bromobenzene formic acid Chemical compound C(=O)O.BrC1=CC=CC=C1 ULELPEYHAFTGRE-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- FOCAUTSVDIKZOP-UHFFFAOYSA-N chloroacetic acid Chemical compound OC(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-N 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- WPOPWLXGDCLQNP-UHFFFAOYSA-N chlorobenzene formic acid Chemical compound C(=O)O.ClC1=CC=CC=C1 WPOPWLXGDCLQNP-UHFFFAOYSA-N 0.000 description 1
- WBLIXGSTEMXDSM-UHFFFAOYSA-N chloromethane Chemical compound Cl[CH2] WBLIXGSTEMXDSM-UHFFFAOYSA-N 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- WOWBFOBYOAGEEA-UHFFFAOYSA-N diafenthiuron Chemical compound CC(C)C1=C(NC(=S)NC(C)(C)C)C(C(C)C)=CC(OC=2C=CC=CC=2)=C1 WOWBFOBYOAGEEA-UHFFFAOYSA-N 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- IMDXZWRLUZPMDH-UHFFFAOYSA-N dichlorophenylphosphine Chemical compound ClP(Cl)C1=CC=CC=C1 IMDXZWRLUZPMDH-UHFFFAOYSA-N 0.000 description 1
- INTMMHZKGCDQGT-UHFFFAOYSA-N diethyldiazene Chemical compound CCN=NCC INTMMHZKGCDQGT-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229920000370 gamma-poly(glutamate) polymer Polymers 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 231100000001 growth retardation Toxicity 0.000 description 1
- 102000047612 human CCN2 Human genes 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- JDNTWHVOXJZDSN-UHFFFAOYSA-N iodoacetic acid Chemical compound OC(=O)CI JDNTWHVOXJZDSN-UHFFFAOYSA-N 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 238000003913 materials processing Methods 0.000 description 1
- 229910000474 mercury oxide Inorganic materials 0.000 description 1
- UKWHYYKOEPRTIC-UHFFFAOYSA-N mercury(ii) oxide Chemical compound [Hg]=O UKWHYYKOEPRTIC-UHFFFAOYSA-N 0.000 description 1
- HNEGQIOMVPPMNR-NSCUHMNNSA-N mesaconic acid Chemical compound OC(=O)C(/C)=C/C(O)=O HNEGQIOMVPPMNR-NSCUHMNNSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- HNEGQIOMVPPMNR-UHFFFAOYSA-N methylfumaric acid Natural products OC(=O)C(C)=CC(O)=O HNEGQIOMVPPMNR-UHFFFAOYSA-N 0.000 description 1
- GQNZGCARKRHPOH-RQIKCTSVSA-N miocamycin Chemical compound C1[C@](OC(C)=O)(C)[C@@H](OC(=O)CC)[C@H](C)O[C@H]1O[C@H]1[C@H](N(C)C)[C@@H](O)[C@H](O[C@@H]2[C@H]([C@H](OC(=O)CC)CC(=O)O[C@H](C)C/C=C/C=C/[C@H](OC(C)=O)[C@H](C)C[C@@H]2CC=O)OC)O[C@@H]1C GQNZGCARKRHPOH-RQIKCTSVSA-N 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 238000000569 multi-angle light scattering Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 230000008634 non enzymatic mechanism Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000001374 small-angle light scattering Methods 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- RVEZZJVBDQCTEF-UHFFFAOYSA-N sulfenic acid Chemical compound SO RVEZZJVBDQCTEF-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- KOTXAHKUCAQPQA-AAUVPWARSA-N taxine Chemical compound C1([C@@H]([C@@H](O)C(=O)O[C@@H]2C=3/C[C@@](C([C@H](O)C4=C(C)[C@@H](OC(C)=O)CC(C4(C)C)[C@@H](OC(C)=O)\C=3)=O)(C)[C@@H](O)C2)N(C)C)=CC=CC=C1 KOTXAHKUCAQPQA-AAUVPWARSA-N 0.000 description 1
- 229930189271 taxine Natural products 0.000 description 1
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/645—Polycationic or polyanionic oligopeptides, polypeptides or polyamino acids, e.g. polylysine, polyarginine, polyglutamic acid or peptide TAT
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Abstract
本发明提供了制备用于临床开发和药物使用的聚谷氨酸-治疗剂结合物的新方法,以及由这些方法制备的聚谷氨酸-治疗剂结合物。
Description
发明领域
本发明涉及一种大规模生产聚谷氨酸治疗剂结合物以进行临床开发的方法。
发明背景
当给药于患肿瘤的宿主时,作为一种聚谷氨酸结合物的抗肿瘤剂紫杉醇与该药物的非结合形式相比,表现出效力提高且毒性降低(美国专利5,977,163;Li等人,Cancer Res.,58:2404,1998)。聚谷氨酸-紫杉醇结合物表现出水溶性提高,从体内清除较慢,并且增加在肿瘤内的聚集。希望聚谷氨酸和各种其它治疗剂的结合物能够提供目前所用配方的临床上有用的替代品。
出于研究的目的,聚谷氨酸-治疗剂结合物可以用上述Li等人公开的方法来生产。在该方法中,将此结合物制备成一种钠盐,渗析以除去低分子量的杂质和过量的盐,然后冻干。但是该方法并不适合于大规模生产大量结合物以进行临床开发和使用。具体地说,利用渗析来除去杂质太耗时间,并且最终产品的产率较低。此外,尽管许多药物当制备成盐时具有更好的性能(例如溶解性、储存性和加工性提高),但对于本发明的聚谷氨酸-治疗剂结合物来说并不是这样。此结合物的盐形式是带静电的固体,并非游离的流动粉末。与游离的流动粉末相比,它们更加难以包装,更容易受到粉尘污染的影响,并且更有可能用有毒试剂污染操作间。因此,需要一种改进的方法来生产聚谷氨酸-治疗剂结合物,该方法可以用来以高产率和改进材料加工和包装的方式生产几克到几百克数量的这种结合物。
发明概述
通过提供一种制备聚谷氨酸-治疗剂结合物的改进方法,本发明满足了这种需要,该方法能够以85%~98%或约85%~约98%的产率提供几克到几千克数量的药用级结合物。
在一个具体实施方案中,该方法包括:
(a)提供质子化形式的聚谷氨酸聚合物和一种用于与其结合的治疗剂;
(b)在一种惰性有机溶剂中将所述试剂共价结合到所述聚谷氨酸聚合物上,形成一种聚谷氨酸-治疗剂结合物;
(c)通过添加过量体积的水性盐溶液,将所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来;以及
(d)收集质子化固体形式的所述结合物。
附加的除去残留低分子量杂质的步骤可以在步骤(c)和步骤(d)之间或在步骤(d)之后进行。
在另一个目前是最优选的具体实施方案中,通过下列方法进行质子化聚谷氨酸-治疗剂结合物的原位生成,该方法包括:
(a)将聚谷氨酸聚合物的一种盐悬浮在一种惰性有机溶剂中;
(b)通过将一种无水的酸添加到所述悬浮液中使所述聚合物质子化,形成基于结合物的一种可溶盐;
(c)提供一种治疗剂,并将所述试剂共价结合到所述聚谷氨酸聚合物上,形成一种聚谷氨酸-治疗剂结合物;
(d)通过添加过量体积的水性盐溶液,将所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来;以及
(e)收集质子化固体形式的所述结合物。
在另一个具体实施方案中,用于大规模生产聚谷氨酸-2′紫杉醇结合物的方法包括:
(a)提供一种聚-L-谷氨酸钠盐的水溶液;
(b)将所述溶液酸化到pH为约2~4,由此将聚-L-谷氨酸的钠盐转化为质子化形式并从溶液中沉淀出来;
(c)收集聚-L-谷氨酸沉淀并用水洗涤;
(d)将聚-L-谷氨酸干燥到水含量介于约2~约7重量%,优选介于7~21重量%,最优选介于约7~约21重量%;
(e)使聚-L-谷氨酸和紫杉醇在标准的偶联条件下接触足够的时间,以通过在紫杉醇的2′-OH基团和聚-L-谷氨酸的羧基之间形成的酯键将紫杉醇结合到聚谷氨酸聚合物上;
(f)将反应混合物冷却到0℃~10℃或约0℃~10℃,同时将水性盐溶液缓慢地加入到反应混合物中;
(h)酸化得到的悬浮液;
(i)收集作为一种质子化固体的结合物;以及
(j)从质子化固体中萃取出杂质。
为了大规模生产聚谷氨酸-2′紫杉醇结合物,最优选的是用步骤(a′)和(b′)代替以上步骤(a)-(d):
(a′)提供一种聚-L-谷氨酸钠盐,或者聚-L-谷氨酸的锂盐、钾盐或季铵盐在惰性有机溶剂中的悬浮液;
(b′)加入约0.95当量的三氟乙酸或甲磺酸。
由此形成一种包含聚-L-谷氨酸三氟乙酸或聚谷氨酸甲磺酸的钠、锂、钾或季铵盐的溶液;以及
如上所述进行步骤(e)-(j)。
任何聚谷氨酸-治疗剂结合物都可以通过本文所述方法来制备。在一个优选的具体实施方案中,治疗剂是抗肿瘤剂,例如紫杉醇;docetaxel;依托泊苷;替尼泊苷;epothilone如epothilone A、epothilone B、epothilone C、epothilone、epothilone F和12,13-二氧代epothilone F;吉西他滨;20(S)(+)喜树碱;9-氨基喜树碱;9-硝基喜树碱;7-乙基-10-羟基喜树碱;9-二甲基氨基甲基-10-羟基喜树碱;10,11-亚甲基二氧喜树碱;7-methylpiperizinomethyl-10,11-亚乙基二氧喜树碱;flavopiridol;格尔德霉素;17-(烯丙基氨基)-17-去甲氧基格尔德霉素;海鞘素743;phthalascidin;CT-2584(1-(11-(十二烷基氨基)-10-羟基十一烷基)-3,7-二甲基黄嘌呤);CT-4582(1-(11-(N-甲基-N-十二烷基氨基)-10-羟基十一烷基)-3,7-二甲基黄嘌呤);多柔比星;阿霉素;苯丙氨酸氮芥;氟达拉滨;柔毛霉素;异搏定;5-氟尿嘧啶;氟尿苷(FUDR);环孢菌素;维生素A酸;7-(二甲基叔丁基甲硅烷氧基)-10-羟基喜树碱以及其它。
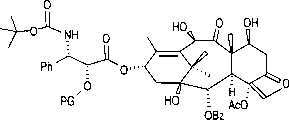
附图简述
图1.示范结合物
图2.聚-L-谷氨酸-紫杉醇结合物的生产流程
图3.聚-L-谷氨酸-紫杉醇结合物的质谱NMR扫描
图4.聚-L-谷氨酸-甘氨酰-20(S)喜树碱的制备
图5-7.I-III的反应流程
发明详述
定义
本文所使用的术语“聚谷氨酸”或“聚谷氨酸聚合物”包括聚(1-谷氨酸)、聚(d-谷氨酸)和聚(d1-谷氨酸)。优选聚谷氨酸聚合物包含至少50%其自身的氨基酸残基作为谷氨酸,且更优选100%。聚谷氨酸聚合物可以被天然存在或化学修饰的氨基酸取代最多到50%,优选亲水性氨基酸,假定当结合上治疗剂时,相对于未结合治疗剂,取代的聚谷氨酸聚合物具有改进的水溶性和/或改进的效力,优选治疗剂是非免疫原性的。
通过本文所述方法制备结合物所使用的聚谷氨酸聚合物的分子量通常大于5000道尔顿,优选15kd~80kd,更优选20kd~80kd,又更优选20kd~60kd,且最优选30kd~60kd(由粘度法测定)。在分子量的较低端,本发明聚谷氨酸聚合物的分子量为约10000、约11000、约12000、约13000、约14000、约15000、约16000、约17000、约18000、约19000、约20000、约21000、约22000、约23000、约24000、约25000、约26000、约27000、约28000、约29000到约30000道尔顿。在较高端,本发明聚谷氨酸聚合物的分子量为约31000、约32000、约33000、约34000、约35000、约36000、约37000、约38000、约39000、约40000、约41000、约42000、约43000、约44000、约45000、约46000、约47000、约48000、约49000、约50000、约51000、约52000、约53000、约54000、约55000、约56000、约57000、约58000、约59000、约60000、约61000、约62000、约63000、约64000、约65000、约66000、约67000、约68000、约69000、约70000、约71000、约72000、约73000、约74000、约75000、约76000、约77000、约78000、约79000到约80000道尔顿。本领域内技术人员明白当用其它方法测量时分子量的数值可以不同。这些其它方法包括,例如凝胶渗透法、小角光散射法、多角度激光散射、折射指数法和它们的组合。
本文所使用的术语“聚谷氨酸-治疗剂结合物”是指一种聚谷氨酸聚合物,它通过聚谷氨酸的一个羧酸残基和治疗剂的一个官能团之间的直接键合、或通过一个或多个双官能化连接体的间接键合而共价结合到治疗剂。优选的连接体是那些在循环中相对稳定地水解的连接体,当从结合物上切除时是可生物降解和无毒的。当然应该理解合适的连接体不会干扰结合物的抗肿瘤效力。连接体的范例包括氨基酸(例如甘氨酸、丙氨酸、亮氨酸、异亮氨酸)、羟基酸(例如γ-羟基丁酸)、二元醇、氨基硫醇、羟基硫醇、氨基醇以及它们的组合。治疗剂可以通过任何键合方法连接到聚合物或连接体上,从而产生可生理切除的键(即一种可以由酶或非酶机理切割的键,这取决于活体动物器官内的条件)。优选的键合物的例子包括酯、酰胺、氨基甲酸酯、碳酸酯、酰氧基烷基醚、酰氧基烷基硫醚、酰氧基烷基酯、酰氧基烷基酰胺、酰氧基烷氧羰基、酰氧基烷基胺、酰氧基烷基氨基甲酸酯、酰氧基烷基磺酰胺、缩酮、缩醛、二硫化物、硫醇酯、N-丙烯酰胺、烷氧基羰氧烷基、脲、和N-磺酰亚氨酸酯。
形成这些键的方法对有机化学合成领域内的技术人员来说是熟知的,并且例如可见于标准的教科书中,如J.March,Advanced OrganicChemistry(高等有机化学),Wiley Interscience,第四版。
生物活性治疗或诊断剂在聚合物上的承载程度(即“承载密度”)可以表示为每条聚谷氨酸聚合物链上承载的分子数或平均分子数,或者优选表示为结合物总重量的百分比(%)(“承载%”)。通过调节治疗剂和聚合物的比例,并在需要时优化其它试剂可以得到理想的承载百分比。对于给定的结合物和给定的用途,最佳承载密度基于结合物所需要的性能(例如水溶性、治疗效力、药代动力学性能、毒性和剂量需要)根据经验确定。对于此处所述具体的结合物,承载密度的范围是1%~约60%或约1%~约60%,优选5%~55%或约5%~约55%,更优选10%~45%或约10%~约45%。
承载百分比通常由四种方法来确定:(1)计算重量%法;(2)分光光度法,优选紫外分光光度法;(3)NMR比例法;和(4)水解法。
(1)计算重量%法基于聚谷氨酸起始原料的已知重量和治疗剂的重量。对于所有结合物,转化为结合物形式的比例是完全100%,由二氧化硅支持的TLC来测定。
(2)分光光度法,优选紫外分光光度法基于在适当波长的吸收(例如紫外吸收),或荧光(例如对紫杉醇-聚谷氨酸结合物)所测量的治疗剂重量%。结合物溶解于去离子水中(2.5或5mg/ml),在500g离心15分钟以除去可能存在的颗粒物,清澈溶液用去离子水稀释100倍到200倍。在特定波长处对稀释液测定吸光度,例如在228nm或260nm处对稀释液测定紫外吸收。用于制备结合物的等量聚谷氨酸溶液以与结合物相同的标称浓度溶解,然后在例如228nm或260nm处对稀释液测定其吸收。通过在例如228nm或260nm处测量溶于甲醇中的已知浓度紫杉醇溶液的吸光度作出一条线性校准曲线。为了计算承载百分比,从聚谷氨酸-紫杉醇的吸光度中减去聚谷氨酸溶液的吸光度(校准以符合聚谷氨酸-紫杉醇溶液中聚谷氨酸的理论承载百分比)。校准后的吸光度与紫杉醇标准曲线比较以得到结合物溶液中的紫杉醇浓度(w/v)。承载百分比是紫杉醇浓度与聚谷氨酸-紫杉醇结合物浓度的比例再乘以100。
(3)NMR比例法基于治疗剂的重量%,它是由聚合物得到的光谱中的峰相对于由治疗剂得到的峰的比例测量的。对于聚谷氨酸-紫杉醇结合物,说明如下:
约4.5ppm到约6.5ppm之间,优选4.5ppm到6.5ppm之间的面积相加并除以质子数(7)。然后该数值与约3.8ppm到约4.4ppm之间,优选3.8ppm到4.4ppm之间的聚合物骨架区比较,并由重叠的紫杉醇峰以2个质子数校准。考虑到紫杉醇和聚合物的分子量比较这两个面积/质子。
A=面积/质子(聚合物)÷面积/质子(紫杉醇)=21.36/1.98=10.79。
紫杉醇的Mw=837;聚谷氨酸单体的Mw=129。
承载%=(837/(10.79×129)+837)×100=37.6%。
本文所描述的方法通常用于制备聚谷氨酸和任何具有生物活性的治疗剂或诊断剂(如本文所述,适于官能化键合到聚谷氨酸上)的结合物。此处示范的结合物只是用来说明本发明,并不限制本发明的范围。
在一个优选的具体实施方案中,治疗剂包含可有效治疗癌症状况的药物,希望受益于这些结合物独特的药代动力学性能(例如增强肿瘤组织中的通透性和滞留性,延迟释放活性剂,与非结合的药剂相比有较长的生物半衰期,以及其它)。目前优选的药剂包括,举例来说,有紫杉碱类(例如紫杉醇;docetaxel);依托泊苷;替尼泊苷;epothilone如epothilone A、epothilone B、epothilone C、epothilone D、epothilone F和12,13-二氧代epothilone F;吉西他滨;20(S)(+)喜树碱;9-氨基喜树碱;9-硝基喜树碱;7-乙基-10-羟基喜树碱;9-二甲基氨基甲基-10-羟基喜树碱;10,11-亚甲基二氧喜树碱;7-methylpiperizinomethyl-10,11-亚乙基二氧喜树碱;flavopiridol;格尔德霉素;17-(烯丙基氨基)-17-去甲氧基格尔德霉素;海鞘素743;phthalascidin;CT-2584(1-(11-(十二烷基氨基)-10-羟基十一烷基)-3,7-二甲基黄嘌呤);CT-4582(1-(11-(N-甲基-N-十二烷基氨基)-10-羟基十一烷基)-3,7-二甲基黄嘌呤);多柔比星;阿霉素;苯丙氨酸氮芥;氟达拉滨;柔毛霉素;异搏定;5-氟尿嘧啶;氟尿苷(FUDR);环孢菌素;维生素A酸;7-二甲基-(叔丁基甲硅烷基)-10-羟基喜树碱以及其它。
治疗剂必需能够通过一个官能团结合到聚合物上,此官能团是本身分子中已经存在的或者是其它可以用有机化学合成中熟知的不改变试剂活性的方法引入的。在本文给出的实施例中,该试剂在非结合形式时是相对不溶于水的,并且在结合后表现出溶解性大大提高。但是,在水溶性药物与聚谷氨酸结合之后还希望它表现出多种优点(例如与非结合的试剂相比,改进的药代动力学和在作用位点的滞留性)。
在“标准偶联条件”下实施的反应在一种惰性溶剂中(例如DMF、DMSO、N-甲基吡咯烷酮),于-20℃~150℃或约-20℃~约150℃,优选0℃~70℃或约0℃~约70℃,更优选5℃~30℃或约5℃~约30℃的温度,在偶联剂和催化剂的存在下进行。当然,所用温度将取决于各种因素如治疗剂的稳定性和结合基团的反应性。合适的偶联剂是有机化学合成中所熟知的,并且包括但不限于碳化二亚胺、氯甲酸烷基酯和三乙基胺、吡啶鎓盐-三丁基胺、苯基二氯磷酸酯、2-氯-1,3,5-三硝基苯和吡啶、二-2-吡啶基碳酸酯、聚苯乙烯基二苯基膦、(三甲基甲硅烷基)乙氧基乙炔、1,1′-羰基双(3-甲基咪唑鎓)triflate、二乙基偶氮二羧酸酯和三苯基膦、N,N′-羰基二咪唑、甲烷磺酰氯、新戊酰氯、双(2-氧代-3-唑烷基)膦酸(“BOP-CI”)、2-氯甲基碘化吡啶(“CMPI”)等。对于醇偶联而言合适的催化剂包括有机碱,例如4-N,N-二甲基氨基吡啶和4-吡咯烷吡啶。
本文所使用的术语“惰性溶剂”是指在所述结合的反应条件下是惰性的一种溶剂,[包括,例如苯、甲苯、乙腈、四氢呋喃(THF)、二甲基甲酰胺(DMF)、氯仿(CHCl3)、氯化亚甲基(或二氯甲烷或“CH2Cl2”)、乙醚、乙酸乙酯、丙酮、甲基乙基酮、二氧杂环己烷、吡啶、二甲氧基乙烷、叔丁基甲基醚等]。除非另有具体说明,本发明反应中所使用的溶剂均为惰性溶剂。
如果在治疗剂中存在多个官能团,则该试剂的一个特定基团选择性结合到聚谷氨酸聚合物中就需要使用合适的保护基团。术语“保护基团”或“封闭基团”是指当结合到化合物的一个或多个羟基、巯基、氨基或羧基上时会防止在这些基团上发生反应的任何基团,并且保护基团可以用常规的化学或酶方法除去以重新恢复羟基、巯基、氨基或羧基。通常参见T.W.Greene & P.G.M.Wuts,“Protective Groups inOrganic Synthesis,”第三版,1999,John Wiley and Sons,N.Y。
所采用的可除去封闭基团并不重要,但优选的可除去羟基封闭基团包括常规的取代基,如烯丙基、苄基、乙酰基、氯代乙酰基、硫代苄基、亚苄基、苯甲酰甲基、叔丁基二苯基甲硅烷基、叔丁基二甲基甲硅烷基、三乙基甲硅烷基、MOM(甲氧基甲基)、MEM(2-甲氧基乙氧基甲基)、t-BOC(叔丁氧基羰基)、CBZ(苄氧基羰基)和任何其它基团,只要它能够通过化学方法引入到羟基官能团上,并且随后可以在与产物性质相容的温和条件下通过化学或酶方法选择性地除去。
优选的可除去氨基封闭基团包括常规的取代基,如叔丁氧基羰基(t-BOC)、苄氧基羰基(CBZ)、芴基甲氧基羰基(FMOC)、烯丙氧基羰基(ALOC)等,它们可以通过与产物性质相容的常规条件除去。
在另一个具体实施方案中,可以使用吡咯衍生的氨基封闭基团如吡咯谷氨酸。在一个特定的实施方案中,吡咯谷氨酸可以或不可以除去。
优选的羧基保护基团包括酯,优选酯含有烷基如甲基、乙基、丙基、叔丁基等,它们可以通过与产物性质相容的温和水解条件除去。
命名法
本文所述的按照本发明具体实施方案制备的示范结合物如图1所示。在以下实施例中的结合物按照与图1结合物相同的方式命名。
优选实施方案的描述
一般而言,以适用于临床开发和药物使用的规模来生产聚谷氨酸-治疗剂结合物的方法包括以下步骤:
(a)提供质子化形式的聚谷氨酸聚合物和一种用于与其结合的治疗剂;
(b)在一种惰性有机溶剂中将所述试剂共价结合到所述聚谷氨酸聚合物上,形成一种聚谷氨酸-治疗剂结合物;
(c)通过添加过量体积的水性盐溶液,将所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来;以及
(d)收集质子化固体形式的所述结合物。
步骤(a)中质子化形式的聚谷氨酸聚合物通过将一种用作原材料的含有聚谷氨酸盐的溶液酸化,然后将盐转化为其酸的形式来制取。离心分离固体之后,用水洗涤该固体(当在步骤(b)中使用二甲基氨基吡啶(“DMAP”)时,优选洗涤该固体直至水相pH值为3或更高)。然后在结合到所需治疗剂之前(步骤(b))干燥聚谷氨酸,优选采用冷冻干燥,并且优选干燥到包含约2%~约21%水,优选约7%~约21%水,更优选7%~21%水的恒重。
在结合前步骤(b)的治疗剂可能需要修饰,例如引入新的官能团、修饰已存在的官能团或者结合一个间隔分子。这样的修饰可能需要使用如上所述的保护基团。
反应方案I-III说明了用来将各种示范治疗剂直接或通过甘氨酸间隔分子键合到聚-L-谷氨酸(PG)上的方法。在这些方案中所示以及描述于实施例中的条件可以改变,可以容易地由有机化学合成领域中的技术人员理解。用来将一种具体治疗剂结合到聚谷氨酸上的具体条件可以基于治疗剂对反应条件的稳定性,键合基团的反应性,与生产工艺相关的其它因素(例如安全性和规范生产)等。如上所述,在制备结合物时可以使用各种类型的键,取决于治疗剂中可用的官能团以及如果使用连接体时的连接体分子。因此,治疗剂可以通过不是酯和酰胺键的键结合到聚谷氨酸和/或连接体分子上。也可以使用不是甘氨酸的连接体,不是本文所示的那些偶联剂的偶联剂。说明实施本发明具体方案的用于制备结合物的具体条件将在以下实施例中描述。
在步骤(c)中,将一种水性盐溶液加入到反应混合物中以将聚谷氨酸-治疗剂结合物从溶液中沉淀出来。任何水溶性无机盐都可以用于此目的,如钠、钾和铵盐,以及卤化物和硫酸盐(例如氯化钠、氯化钾、氯化铵、硫酸钠、硫酸铵等)。优选以1倍到4倍的体积使用10-15%的盐溶液。在一个优选的实施方案中,使用2.5倍体积的10%氯化钠。将盐溶液缓慢加入到反应混合物中,在添加过程中冷却。为了使结合物的产率最佳,温度保持在约0℃~约10℃,优选0℃~10℃。沉淀步骤从原材料和反应副产物中分离出聚谷氨酸-治疗剂结合物,其中副产物在用于沉淀结合物的条件下是完全或部分可溶的。
在步骤(d)中,收集作为质子化固体的结合物。优选酸化在步骤(c)中获得的悬浮液。取决于药物分子对酸条件的稳定性,pH值范围可以采用约pH为1~约pH为4,优选pH为1~4。但是为了制备聚谷氨酸-紫杉醇结合物,在pH低于2时酸化会导致紫杉醇分解,因此通常在pH约为2.5时进行酸化。优选地,在步骤(d)中采用除去碱如DMAP,以及酸如盐酸(HCl)的方法。该悬浮液可以过滤或离心以收集结合物,优选过滤。
在酸化以生产最终的质子化结合物之前或之后,可以除去未反应的原材料、副产物以及其它杂质(以下实施例2和3中,以及图2和4中作说明)。例如,加入盐溶液后,可以收集固体并重新溶解,然后过滤或用适当的溶剂萃取,在此溶剂中杂质是可溶的而结合物不可溶(例如乙酸乙酯、二氯甲烷、氯仿、己烷、庚烷、乙醚和二氧杂环己烷)。然后酸化该溶液并如上所述收集质子化形式的结合物。
此外,该固体可以冷冻干燥,然后用适当的溶剂或其混合物匀浆以从最终的质子化结合物产物中除去杂质,适当的溶剂如乙腈(MeCN);醚如乙醚、二氧杂环己烷、四氢呋喃;卤代溶剂如氯仿、二氯甲烷;酮如丙酮和甲基乙基酮(MEK);C1~C10的醇如叔丁醇、异丙醇、乙醇或甲醇。
在替代的一个优选实施方案中,以上步骤(c)用步骤(c′)代替,它包括:
(c′)从未反应的原材料和副产物中分离出所述的聚谷氨酸-治疗剂结合物,然后通过添加一种有机溶剂将所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来,在该溶剂中未反应的原材料和副产物是可溶的。
除了乙酸乙酯和乙腈之外,可以用来纯化结合物的其它溶剂的例子包括氯仿、四氢呋喃、二氧杂环己烷、甲苯、2-丁基甲基醚等。
一种替代方法是目前最优选的,其中通过下列方法进行质子化聚谷氨酸-治疗剂结合物的原位生成,该方法包括:
(a)将聚谷氨酸聚合物的一种盐悬浮在一种惰性有机溶剂中;
(b)通过将一种无水的酸添加到所述悬浮液中使所述聚合物质子化,形成基于结合物的一种可溶盐;
(c)提供一种治疗剂,并将所述试剂共价结合到所述聚谷氨酸聚合物上,形成一种聚谷氨酸-治疗剂结合物;
(d)通过添加过量体积的水性盐溶液,将所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来;以及
(e)收集质子化固体形式的所述结合物。
该原位工艺减少了制备质子化PG聚合物中的多个步骤,并且将整个加工时间减少最多达1周。此外,当由原位工艺制备的产物与本文所公开的其它方法相比时,该产物会更快地溶解在水溶液中。
在此工艺中,任何无水的酸都可以用于上述步骤(b),只要结合物基的盐在该工艺所选用的有机溶剂中是可溶的。合适的酸的例子包括三氟乙酸、氯代乙酸、溴代苯甲酸、氯代苯甲酸、氯代苯氧基乙酸、氯代苯基乙酸、氰基乙酸、氰基丁酸、氰基苯氧基乙酸、氰基丙酸、二氯乙酸、乙酰乙酸、富马酸、马尿酸、碘代乙酸、乳酸、丙二酸、甲基富马酸、萘甲酸、硝基苯甲酸、邻苯二甲酸、甲磺酸、HBr、HCl和HI。
步骤(c)、(d)和(e)如上所述按普通工艺进行。
表1表示对如以下实施例3中所述制备的聚-L-谷氨酸-紫杉醇结合物的代表性分析。表2表示对如以下实施例7中所述原位制备的聚-L-谷氨酸-紫杉醇结合物的代表性分析。
表1分析数据
| 重量%a | 总输出量b(克) | 承载%(UV)c | 承载%(NMR)d | 游离紫杉醇%e | 残留乙腈%f | 残留DMF%g | DIPU%h | ROIi |
| 93.6 | 87.80 | 42.0 | 34.0 | 0.128 | 0.15 | 0.27 | 0.160 | 0.87 |
a产率%;b结合物的重量(克);c紫杉醇的重量(克)/结合物重量(克),由紫外吸收法测定;d紫杉醇的重量(克)/结合物重量(克),由NMR法测定;e游离紫杉醇相对于结合物的wt%;f残留乙腈相对于结合物的wt%;g残留二甲基甲酰胺相对于结合物的wt%;h二异丙基脲相对于结合物的wt%;I燃烧残留物的wt%。
表2分析数据
| 重量%a | 总输出量b(克) | 承载%(NMR)d | 残留乙腈%f | 残留DMF%g | DIPU%h |
| 95.1 | 0.485 | 36.0 | 0-0.01 | 0.01-0.45 | 0 |
a产率%;b结合物的重量(克);d紫杉醇的重量(克)/结合物重量(克),由NMR法测定;f残留乙腈相对于结合物的wt%;f残留二甲基甲酰胺相对于结合物的wt%;h二异丙基脲相对于结合物的wt%。
通过以下实施例说明本发明,这些实施例不应理解为以任何方式限制本发明的范围。
实施例
在以下实施例中,结合物生产过程中的中间体用1HNMR来表征。用来制备如下所示结合物的聚谷氨酸(钠盐)的分子量范围是20kd~50kd,具体由供应商基于粘度测量法来确定(Sigma化学品公司,Milwaukee,WI)。结合物的平均承载密度是37%。
实施例1 制备聚-L-谷氨酸
聚-L-谷氨酸钠盐(85.9g)(Sigma化学品公司,由粘度法测量确定MW为37kd)溶解在USP纯化水中(534ml;6.2ml/g),然后将该溶液冷却到0℃~5℃。在剧烈搅拌并保持温度<10℃下滴加稀盐酸溶液(1M),直至pH介于2~2.5。在滴加过程中,聚-L-谷氨酸从溶液中分离出来。反应混合物升温到室温并搅拌1小时,悬浮液在2700×g离心10分钟,移去上面的水层,固体重新悬浮在560ml USP纯化水中,然后离心10分钟。移去上面的水层并测量pH值。如果需要的话继续洗涤,直至水层的pH值≥3.0。在LABCONCOTM冻干系统中冷冻干燥湿固体,直至得到恒重。由ICP测定,钠的重量%不超过7000ppm。
实施例2 制备聚-L-谷氨酸-2′-紫杉醇结合物
如以上实施例1所述制备的聚-L-谷氨酸(16.82g)悬浮在无水N,N-二甲基甲酰胺(180ml)、紫杉醇(9.923g,11.6mmol)和N,N-二甲基氨基吡啶(283mg,2.32mmol)中。反应混合物搅拌30分钟。用注射泵在3小时中加入N,N-二异丙基碳二亚胺(1.903g,15.08mmol)在N,N-二甲基甲酰胺(50ml)中的溶液。加入之后,搅拌直至反应完成(室温下约为4小时)。反应冷却到5℃~10℃,缓慢加入10%氯化钠溶液(345ml)以沉淀出聚-L-谷氨酸-紫杉醇结合物,通过将混合物移入离心管中并在1500g下将其离心来分离沉淀物。湿固体重新悬浮在水(150ml)中,在剧烈搅拌下缓慢加入1M的碳酸氢钠溶液(120ml),以调节溶液的pH值为7。反应物再搅拌1小时,然后通过0.2微米的过滤器过滤以除去杂质。滤液冷却到0℃~5℃,在剧烈搅拌下缓慢加入HCl溶液(1N),直至溶液的pH值调节到3,继续搅拌30分钟。沉淀出的固体在1500g离心,通过悬浮在水中(150ml)然后离心将湿固体洗涤两次。产物冷冻干燥,得到24g聚-L-谷氨酸-紫杉醇结合物(产率90%)。
在以上工序中,过滤步骤可以由用乙酸乙酯(250ml,2次)洗涤溶液以除去杂质来代替。
图3表示由如上所述相同方法制备的聚-L-谷氨酸-2′-紫杉醇结合物的一个代表性质谱NMR扫描,但具有较高的紫杉醇含量(即55%)。
实施例3 制备聚-L-谷氨酸-2′-紫杉醇结合物(生产工艺)
如以上实施例1所述制备的聚-L-谷氨酸(42g)加入到装配有机械搅拌器、进料漏斗和温度探针的三颈三升圆底烧瓶中。加入N,N-二甲基甲酰胺(350ml)并搅拌10分钟,加入紫杉醇(24.66g)和N,N-二甲基氨基吡啶(0.70g)并搅拌10分钟。在室温下用进料漏斗于1小时内加入N,N-二异丙基碳二亚胺(4.73g)在N,N-二甲基甲酰胺(143ml)中的溶液,并搅拌4小时。反应混合物冷却到5℃~10℃,然后用进料漏斗滴加10%的氯化钠冷溶液(1.2L),并通过在冰盐混合物中冷却烧瓶将温度保持在5℃~10℃。加入氯化钠溶液之后,滴加1N的盐酸溶液(35ml)直至反应物的pH值达到2.5。反应混合物在5℃~10℃搅拌30分钟,然后通过过滤收集沉淀出来的聚-L-谷氨酸-紫杉醇结合物。该固体用水洗涤3次,在冻干机中冷冻干燥24小时。用研钵和研杵将干燥后的固体粉碎成细小的粉末,该细小的粉末状聚-L-谷氨酸-紫杉醇结合物悬浮在乙腈中(1000ml)并搅拌2小时,然后过滤,并用2×200ml乙腈洗涤固体。固体在真空下干燥24小时以得到聚-L-谷氨酸-紫杉醇结合物(60g)。产率为90%。
实施例4 制备聚-L-谷氨酸-甘氨酸-紫杉醇结合物(反应方案II)
基本上如Mathew等人所述进行以下步骤1和2(Mathew,A.E.,Mejillano,M.R.,Nath,J.P.,Himes,R.H.,和Stella,V.J.,J.Med.Chem.,35:145-151,1992)。
步骤1 制备2′-(N-t-BOC-甘氨酰)紫杉醇
向N-t-BOC-L-甘氨酸(131mg,0.75mmol)和紫杉醇(640mg,0.75mmol)在二氯甲烷(20ml)的溶液中加入1,3-二异丙基碳二亚胺(124mg,0.98mmol),然后加入N,N-二甲基氨基吡啶(27mg,0.23mmol)。室温搅拌4小时后,在减压下将该混合物浓缩。采用硅胶快速色谱术纯化残留物,用1∶1(v/v)的乙酸乙酯/己烷洗脱,得到白色粉末状的2′-(N-t-BOC-甘氨酰)紫杉醇(720mg,产率95%)。
步骤2 制备2′-(甘氨酰)紫杉醇
2′-(N-t-BOC-甘氨酰)紫杉醇(245mg,0.242mmol)在甲酸中(2ml)的溶液搅拌30分钟。减压下浓缩之后,残留物悬浮在水中(15ml)。加入0.05M的碳酸氢钠冷溶液(45ml),然后用二氯甲烷(2×40ml)萃取该溶液(pH为8.0)。合并后的二氯甲烷萃取液用无水硫酸钠干燥,然后在减压下浓缩。采用硅胶快速色谱术纯化残留物,用4%甲醇/二氯甲烷洗脱,得到白色粉末状的2′-(甘氨酰)紫杉醇(161mg,产率73%)。
步骤3 制备聚-L-谷氨酸-2′-(甘氨酰)紫杉醇结合物
向聚-L-谷氨酸(275mg,1.87mmol)在无水二甲基甲酰胺(6ml)的搅拌悬浮液中加入2′-(甘氨酰)紫杉醇(161mg,0.177mmol)。在30分钟内将1,3-二异丙基碳二亚胺(29mg,0.23mmol)在二甲基甲酰胺(1.4ml)中的溶液加入到搅拌悬浮液中,在室温下搅拌3小时之后,在冰浴中冷却混合物,以调节冰浴温度为0℃~5℃,然后在30分钟内加入10%的氯化钠水溶液(7ml)以沉淀聚-L-谷氨酸-2′-(甘氨酰)紫杉醇结合物。得到的白色悬浮液在1500g离心15分钟。过滤后,通过悬浮在水中(10ml)然后离心将固体洗涤两次。粗产物悬浮在水中(6ml),然后在搅拌的同时缓慢加入1M的碳酸氢钠水溶液(2.3ml),以调节烧瓶中物质的pH值为7.6。再搅拌2小时之后,水层用乙酸乙酯(3×6ml)洗涤,然后通过加入1N的盐酸使pH值为2.8来酸化。沉淀的固体用离心分离,并用水洗涤(2×6ml)。冷冻干燥湿固体以得到白色粉末状的聚-L-谷氨酸-2′-(甘氨酰)紫杉醇结合物(315mg,,产率72%)。
采用类似工序,以上结合物可以被并非甘氨酸的其它氨基酸所取代。
实施例5 制备聚-L-谷氨酸-2′-docetaxel结合物(反应方案III)
步骤1 制备10-脱乙酰紫杉醇
基本上如Zheng,Q.Y.,Darbie,L.G.,Chen,X.,Murray,C.K.,Tetrahedron Letters.,36:2001-2004,1995和美国专利5629433中所述制备10-脱乙酰紫杉醇。
向紫杉醇(1.0g,1.17mmol)在四氢呋喃(20ml)的溶液中加入过氧化氢(30%,20ml),随后加入碳酸氢钠(1.92g,22.85mmol)。室温下搅拌18小时之后,混合物用二氯甲烷/水(1∶1(v/v),100ml)处理,有机相用水洗涤(2×30ml),用无水硫酸镁干燥,然后在真空下浓缩。采用硅胶快速色谱术纯化残留物,用3%甲醇/二氯甲烷(v/v)洗脱,得到白色粉末状的10-脱乙酰紫杉醇(890mg,产率93%)。
步骤2 制备2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇
如美国专利5629433中所述制备2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇。
室温下在氩气中于30分钟内向10-脱乙酰紫杉醇(850mg,1.05mmol)在无水吡啶(20ml)的溶液中加入氯化三乙基硅烷(2.72ml,20.1mmol)。搅拌17小时后,混合物用二氯甲烷(75ml)处理,用水(3×30ml)、用10%的硫酸铜水溶液(4×35ml)、用水(30ml)、以及用氯化钠的饱和水溶液(30ml)洗涤。有机相用无水硫酸镁干燥,然后在减压下浓缩以得到粉末状的2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇(980mg,产率90%)。
步骤3 制备2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇亚胺
如美国专利5629433中所述制备2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇亚胺。
向2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇(730mg,0.70mmol)在四氢呋喃(7.3ml)的溶液中加入氢氯化茂锆(543mg,2.11mmol)。室温下在氩气中搅拌15小时之后,将混合物倾入冷己烷中(75ml)。沉淀出的锆络合物通过过滤除去。滤液在减压下浓缩以得到白色粉末状的2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇亚胺(636mg,产率92%)。
步骤4 制备10-脱乙酰紫杉醇伯胺
按照美国专利5629433制备10-脱乙酰紫杉醇伯胺。
2′,7-双(三乙基甲硅烷基)-10-脱乙酰紫杉醇亚胺(636mg,0.621mmol)在1%(w/w)浓盐酸/95%乙醇(25ml)的溶液搅拌15小时,用水(65ml)处理,然后用己烷(2×30ml)洗涤。通过加入饱和碳酸氢钠水溶液来中和水层(pH值为7),并用二氯甲烷萃取(2×40ml)。合并后的萃取液用无水硫酸镁干燥,然后在减压下浓缩以得到白色粉末状的粗伯胺产物(405mg,产率92%)。该产物不需要进一步纯化就可用于下一步骤。
步骤5 制备docetaxel
按照美国专利5629433制备docetaxel。
向10-脱乙酰紫杉醇伯胺(405mg,0.57mmol)在乙酸乙酯(40ml)的溶液中加入饱和碳酸氢钠水溶液(40ml)。向此双相混合物中加入二碳酸二叔丁酯(225mg,1.03mmol),室温下搅拌15小时之后,加入乙酸乙酯(75ml)。有机相用水洗涤(2×30ml),用无水硫酸钠干燥,然后真空下浓缩。采用硅胶快速色谱术纯化残留物,用4%甲醇/二氯甲烷洗脱,以得到白色粉末状的docetaxel(351mg,产率76%)。
步骤6 制备聚-L-谷氨酸-2′-docetaxel结合物
向聚-L-谷氨酸(658mg,4.47mmol)在无水二甲基甲酰胺(10ml)的悬浮液中加入docetaxel(385mg,0.48mmol)和N,N-二甲基氨基吡啶(12mg,0.096mmol)。在20分钟内将1,3-二异丙基碳二亚胺(78.8mg,0.624mmol)在二甲基甲酰胺(3ml)中的溶液滴加到搅拌悬浮液中。搅拌15小时后,混合物在冰浴中冷却,并在30分钟内加入10%的氯化钠水溶液(20ml)。再搅拌1小时后,过滤固体,然后用水(4×50ml)洗涤过滤后的滤饼。将固体冷冻干燥到恒重,然后用乙腈(4×50ml)粉碎。在高真空下干燥15小时,得到白色粉末状的聚-L-谷氨酸-2′-docetaxel结合物(890mg,产率87%)。1HNMR(300MHz,DMSO-d6):δ12.10(s,-COOH),7.05-8.20(m,芳族质子),4.80-6.05(m),3.80-4.50(m),5.0-5.6(m,5-H2,7-H2),3.70-4.35(m),1.20-2.80(m),1.00(s)。
实施例6 制备聚-L-谷氨酸-甘氨酰-20(s)喜树碱(反应方案I)
如Greenwald,R.B.,Pendri,A.,Conover,C.D.,Lee,C.,Choe,Y.H.,Gillbert,C.,Martinez,A.,Xia,J.,Wu,D.,和Hsue,M.,Bioorg. & Med.Chem.,6:551-562,1998所述进行以下步骤1和2。
步骤1 制备20-(N-t-BOC-甘氨酰)-20(s)喜树碱
在0℃向N-t-BOC-甘氨酸(530mg,3.0mmol)在无水二氯甲烷(240ml)的溶液中加入1,3-二异丙基碳二亚胺(379mg,3.0mmol)、N,N-二甲基氨基吡啶(244mg,2mmol)和20(s)喜树碱(348mg,1.0mmol)。反应混合物升温到室温。搅拌18小时后,混合物用0.1N的盐酸水溶液(2×50ml)、水(2×50ml)、0.1M的碳酸氢钠水溶液(2×25ml)以及水(2×50ml)顺序洗涤。有机相用无水硫酸钠干燥,然后在减压下浓缩。残留物从甲醇中(7ml)结晶出来,得到黄色粉末状的20-(N-t-BOC-甘氨酰)-20(s)喜树碱(424mg,产率84%)。1HNMR(300MHz,CDCl3):δ8.35(s,1H),8.22(d,J=8.38Hz 1H),7.91(d,J=8.07 1H),7.76-7.85(m.1H),7.65(t,J=7.4Hz,1H),7.26(s,1H),5.70(d,J=17.25Hz,1H),5.40(d,J=17.25Hz,1H),5.25(s,2H),4.95(br,s,1H),3.98-4.25(m,2H),2.18-2.26(m,2H),1.38(s,9H),0.95(t,J=7.47Hz,3H)。
步骤2 制备20-甘氨酰-20(s)喜树碱三氟乙酸盐
20-(N-t-BOC-甘氨酰)-20(s)喜树碱(424mg,0.84mmol)在1∶1(v/v)的二氯甲烷/三氟乙酸(21ml)混合物的溶液在室温下搅拌1小时。减压下蒸发掉溶剂,从二氯甲烷/乙醚(3∶7(v/v),50ml)中结晶出黄色固体,得到微黄色粉末状的20-甘氨酰-20(s)喜树碱三氟乙酸盐(361mg,产率83%)。1H NMR(300MHz,DMSO-d6):δ8.78(s,1H),8.45(br,s,2H),8.20(d,J=8.2Hz,1H),7.70-7.95(m.2H),7.30(s,1H),5.55(s,2H),5.30(s,2H),4.35(d,J=17.9Hz,1H),4.15(d,J=17.9Hz,1H),2.10-2.30(m.2H),1.00(t,J=7.4Hz,3H)。
步骤3 制备聚-L-谷氨酸-20-甘氨酰-20(s)喜树碱结合物
20分钟内向20-甘氨酰-20(s)喜树碱三氟乙酸盐(351mg,0.68mmol)、聚-L-谷氨酸(465mg,3.16mmol)、和N,N-二甲基氨基吡啶(249mg,2.04mmol)在无水二甲基甲酰胺(13ml)的搅拌悬浮液中加入1,3-二异丙基碳二亚胺(111.6mg,0.88mmol)在二甲基甲酰胺(2ml)中的溶液。在氩气中搅拌2天后,混合物在冰浴中冷却,并在30分钟内加入10%的氯化钠水溶液(35ml)。再搅拌1小时后,通过加入1N的盐酸水溶液将悬浮液酸化到pH值为2.5。过滤收集黄色的沉淀物,用水洗涤(5×25ml),真空下干燥过夜,然后用乙腈粉碎(100ml)。高真空下干燥24小时后,得到黄色粉末状的聚-L-谷氨酸-20-甘氨酰-20(s)喜树碱结合物(703mg,产率95%)。1H NMR(300MHz,DMSO-d6):δ12.10(s,-COOH),7.05-8.74(m,7,9,10,11,12,& 14CH),5.0-5.6(m,5-CH2,7-CH2),3.70-4.35(m,-Gly-CH2,PG-N-CH-),1.42-2.62(m,18-CH2,PG-δCH2,δCH2),0.90(br,s,19-CH3)。1H NMR表明紫杉醇的含量为34%。
实施例7 原位法生产聚谷氨酸-紫杉醇
装有搅拌棒的100ml圆底烧瓶中加入聚-L-谷氨酸(钠盐,340mg,2.25mmol,11.3当量)和7ml干燥的二甲基甲酰胺。搅拌悬浮液并通过注射器恒速加入三氟乙酸(156μl,2.03mmol,10.2当量)。悬浮的固体在约5分钟内溶解,紫杉醇(170mg,0.199mmol,1.0当量)作为固体加入,然后加入4-(N,N-二甲基氨基)吡啶(10mg,0.082mmol,0.4当量)和二异丙基碳二亚胺(40μl,0.259mmol,1.3当量)。室温下搅拌溶液18小时,然后用冰浴冷却到0℃,在剧烈搅拌下缓慢加入10重量%的氯化钠水溶液,导致沉淀出细小的白色固体。用稀盐酸调节pH值为2.5,然后将悬浮液移到50ml的离心管中。旋转固体,滗出上清液,并将所得物质悬浮在35ml水中。悬浮液再次旋转、滗析、并重新悬浮在35ml水中。最终漂洗后,剩余的物质冷冻干燥以得到一种干燥粉末。该粉末用3×15ml的乙腈洗涤,然后在高真空下除去残留的溶剂,得到485mg白色粉末。1H NMR(d6 DMSO)表明紫杉醇的含量为38重量%。
实施例8 生物试验
用皮下移植有路易斯肺癌细胞(LL/2)的大鼠测试抗肿瘤活性。通过以0.25mlPBS+2%FBS的体积皮下注射2.5×105鼠路易斯肺(LL/2)癌细胞(ATTC CRL-1642)在右肩胛间区域的肌肉中产生肿瘤。在肿瘤细胞移植之后,当肿瘤生长到20±20mm3(230个肿瘤的平均值)时,腹膜内注射测试化合物和对照赋形剂7天。在0.1N Na2HPO4中的单剂量聚谷氨酸-治疗剂结合物以非结合剂最大耐受剂量的1到4倍给药,通常以0.75%盐水中的8.3%cremophore EL/8.3%乙醇给药。每个处理组由随机分配到每组中的10只大鼠组成。开始时每3到4天检测肿瘤生长,当肿瘤尺寸达到随机设定的2500mm3上限时,每天测定肿瘤的尺寸。根据公式:(长度×宽度×高度)/2来计算肿瘤的体积。肿瘤等于或大于2500mm3的大鼠用颈椎错位法处死,各种处理的效力表示为与非结合治疗剂的最大耐受剂量相比,肿瘤达到2500mm3体积的天数(即TGD,肿瘤生长延滞)。
测试以上实施例2、3、5和6中所述的聚谷氨酸-治疗剂结合物,并发现在本试验中有活性。
实施例9 制备聚-L-谷氨酸-CT2584
聚-L-谷氨酸(4.95g)悬浮在无水N,N-二甲基甲酰胺(120ml)中并加入CT2584(0.873g,1.634mmol)。搅拌下将反应混合物加热到50℃直至形成清澈溶液。反应混合物冷却回到室温,使用滴液漏斗在30分钟内加入N,N-二异丙基碳二亚胺(0.247g,1.96mmol)在N,N-二甲基甲酰胺(5ml)中的溶液。加入之后,在室温下搅拌反应物4小时,反应物冷却到5℃~10℃,缓慢加入10%的氯化钠溶液(200ml)以沉淀出聚-L-谷氨酸CT2584结合物。通过在1500×g下离心收集沉淀物。通过悬浮在水中(150ml)然后离心将湿固体洗涤2次。产物冻干,得到5.16g聚-L-谷氨酸CT2584结合物,产率=88.6%。
该产物用1H NMR表征,结果表明在3.9ppm和3.4ppm处有一个单峰,对应于在N3和N7上的甲基基团,以及在1.24ppm处有一个较宽的单峰,对应于烷基质子,以及在0.85ppm处有一个较宽的峰,对应于CT2584的末端甲基基团。此外NMR表明在1.5ppm-3.0ppm和3.5ppm-4.5ppm处有多重峰,对应于聚-L-谷氨酸骨架。
实施例10 制备聚-L-谷氨酸-喜树碱
20(s)-喜树碱(64mg,0.184mmol)、聚-L-谷氨酸(256mg,49.8kD)的混合物在真空下干燥6小时,然后溶解在无水二甲基甲酰胺中(15ml)。该溶液在冰/盐浴中冷却到-5℃,在氩气保护下向其中加入2-氯甲基碘化吡啶(85mg,0.33mmol)和N,N-二甲基氨基吡啶(81mg,0.66mmol)。反应混合物升温到室温过夜,4天后,反应烧瓶再次冷却到0℃,然后在25分钟内缓慢加入10%的氯化钠溶液(35ml)。该混合物用0.5N HCl(3.5ml)酸化到pH值为2.5,然后在室温再搅拌1小时,过滤所形成的黄色沉淀物,用水(4×30ml)洗涤,然后真空下干燥12小时。所得干燥的黄色滤饼研磨成细小的粉末,重新悬浮在2%MeOH/CH2Cl2(10ml)中并搅拌3小时。通过离心分离固体。重复该工艺4次以除去任何未反应的喜树碱。所得固体在真空下干燥2天,得到聚-L-谷氨酸-20(s)-喜树碱(295mg,产率97%,由基于回收喜树碱(13mg)的重量比确定)。1H NMR(300MHz,DMSO-d6):δ12.10(s,-COOH),6.90-8.80(m),5.15-5.8(m),3.10-4.35(m),1.42-2.62(m),0.90(br,s,19-CH3),紫杉醇承载%为16重量%。
尽管本发明已经参考其具体实施方案作了描述,但本领域内技术人员应该理解,可以作各种改动并可以替代等效物,只要不背离本发明的真实主旨和范畴。此外,可以作出各种修改以适应具体情况、材料、物质组成、工艺、工艺步骤或各步骤,以体现本发明的主旨和范畴。所有这些修改均在所附权利要求的范围之内。
在本申请中所引用的所有公开文献、专利申请和专利在此均以其相同范畴的整体内容引为参考文献,如同每个单独的公开文献、专利申请或专利均具体而单独地指出以其整体内容作为参考。
Claims (23)
1.一种制备聚谷氨酸和治疗剂的结合物的方法,该方法包括:
(a)提供质子化形式的聚谷氨酸聚合物和一种用于与其结合的治疗剂;
(b)在一种惰性有机溶剂中将所述试剂共价结合到所述聚谷氨酸聚合物上,形成一种聚谷氨酸-治疗剂结合物;
(c)通过添加过量体积的水性盐溶液,将所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来;以及
(d)收集质子化固体形式的所述结合物。
2.权利要求1的方法,其中步骤(a)进一步包括:
(a.1)提供一种聚-L-谷氨酸钠盐的水溶液;
(a.2)将该溶液酸化,从而使聚-L-谷氨酸钠盐转化为质子化形式并使其从溶液中沉淀出来;以及
(a.3)收集聚-L-谷氨酸沉淀物并用水洗涤所述沉淀物。
3.权利要求1的方法,其中步骤(a)中的治疗剂是一种抗肿瘤剂。
4.权利要求3的方法,其中该抗肿瘤剂选自紫杉醇;docetaxel;依托泊苷;替尼泊苷;epothilone;吉西他滨;20(S)(+)喜树碱;9-氨基喜树碱;9-硝基喜树碱;7-乙基-10-羟基喜树碱;9-二甲基氨基甲基-10-羟基喜树碱;10,11-亚甲基二氧喜树碱;7-methylpiperizinomethyl-10,11-亚乙基二氧喜树碱;flavopiridol;格尔德霉素;17-(烯丙基氨基)-17-去甲氧基格尔德霉素;海鞘素743;phthalascidin;CT-2584(1-(11-(十二烷基氨基)-10-羟基十一烷基)-3,7-二甲基黄嘌呤);CT-4582(1-(11-(N-甲基-N-十二烷基氨基)-10-羟基十一烷基)-3,7-二甲基黄嘌呤);多柔比星;7-(二甲基叔丁基甲硅烷氧基)-10-羟基喜树碱或阿霉素。
5.权利要求4的方法,其中epothilone是epothilone A、epothilone B、epothilone C、epothilone D、epothilone F和12,13-二氧代epothilone F。
6.权利要求4的方法,其中该治疗剂是紫杉醇或docetaxel。
7.权利要求1的方法,其中步骤(a)中所述聚谷氨酸按照粘度法测定的分子量为20kd~80kd。
8.权利要求1的方法,其中步骤(b)中所述试剂通过一个可生理断裂的键直接结合到所述聚谷氨酸的羧基上。
9.权利要求8的方法,其中所述键是一种酯键或酰胺键。
10.权利要求9的方法,其中所述键是一种酯键。
11.权利要求1的方法,其中步骤(b)中所述试剂通过一种连接体间接地结合到所述聚谷氨酸的羧基上,其中所述连接体通过可生理断裂的键结合到所述聚谷氨酸上并结合到所述试剂上。
12.权利要求11的方法,其中所述连接体是一种氨基酸。
13.权利要求1的方法,其中步骤(b)中所述聚谷氨酸-治疗剂结合物包含约5-55重量%的治疗剂。
14.权利要求13的方法,其中所述结合物包含约10-45重量%的治疗剂。
15.权利要求1的方法,其中步骤(c)中所述水性盐溶液包含氯化钠。
16.权利要求15的方法,其中所述水性盐溶液以反应混合物溶剂的1.5-4倍体积加入。
17.权利要求1的方法,步骤(c)进一步包含使反应混合物酸化的步骤。
18.权利要求1的方法,进一步包含从结合物中除去低分子量杂质的步骤,其中所述除去杂质的步骤可以在步骤(c)和(d)之间或在步骤(d)之后进行。
19.一种制备聚谷氨酸和治疗剂的结合物的方法,该方法包括:
(a)将聚谷氨酸聚合物的一种盐悬浮在一种惰性有机溶剂中;
(b)通过将一种无水的酸添加到所述悬浮液中使所述聚合物质子化,形成一种基于结合物的可溶盐;
(c)提供一种治疗剂,并使所述试剂共价结合到所述聚谷氨酸聚合物上,形成一种聚谷氨酸-治疗剂结合物;
(d)通过添加过量体积的水性盐溶液,使所述聚谷氨酸-治疗剂结合物从溶液中沉淀出来;以及
(e)收集质子化固体形式的所述结合物。
20.一种由聚-L-谷氨酸的钠盐和紫杉醇制备聚-L-谷氨酸-2′-紫杉醇结合物的方法,所述方法包括以下步骤:
(a)提供一种聚-L-谷氨酸钠盐的水溶液;
(b)将该溶液酸化到pH为约2~4,从而使聚-L-谷氨酸钠盐转化为质子化形式并使其从溶液中沉淀出来;
(c)收集聚-L-谷氨酸沉淀物并用水洗涤;
(d)将所述聚-L-谷氨酸干燥到水含量介于7重量%~21重量%;
(e)使聚-L-谷氨酸和紫杉醇在标准的偶联条件下接触足够的时间,以通过在紫杉醇的2′-OH基团和聚-L-谷氨酸的羧基之间形成的酯键将所述紫杉醇结合到所述聚谷氨酸聚合物上;
(f)将所述反应混合物冷却到0℃~10℃,同时将水性盐溶液缓慢地加入到该反应混合物中;
(h)将得到的悬浮液酸化;
(i)收集质子化固体形式的所述结合物;以及
(j)从所述质子化固体中萃取出杂质。
21.权利要求20的方法,其中步骤(a)-(d)用步骤(a′)和(b′)代替:
(a′)提供一种聚-L-谷氨酸钠盐在惰性有机溶剂中的悬浮液,以及
(b′)加入约0.95当量的三氟乙酸或甲磺酸,从而形成一种包含聚-L-谷氨酸三氟乙酸钠或聚谷氨酸甲磺酸钠的溶液;然后进行如权利要求20所述的步骤(e)-(j)。
22.由权利要求1的方法制备的聚谷氨酸-治疗剂结合物。
23.由权利要求19的方法制备的聚谷氨酸-治疗剂结合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15913599P | 1999-10-12 | 1999-10-12 | |
| US60/159,135 | 1999-10-12 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2009101276554A Division CN101507820A (zh) | 1999-10-12 | 2000-10-12 | 聚谷氨酸-治疗剂结合物的生产方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1607962A true CN1607962A (zh) | 2005-04-20 |
Family
ID=22571222
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2009101276554A Pending CN101507820A (zh) | 1999-10-12 | 2000-10-12 | 聚谷氨酸-治疗剂结合物的生产方法 |
| CNA008170290A Pending CN1607962A (zh) | 1999-10-12 | 2000-10-12 | 聚谷氨酸-治疗剂结合物的生产方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2009101276554A Pending CN101507820A (zh) | 1999-10-12 | 2000-10-12 | 聚谷氨酸-治疗剂结合物的生产方法 |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US20020077279A1 (zh) |
| EP (2) | EP1225917A2 (zh) |
| JP (1) | JP2003511423A (zh) |
| KR (1) | KR100821437B1 (zh) |
| CN (2) | CN101507820A (zh) |
| AU (1) | AU781735B2 (zh) |
| BR (1) | BRPI0014652B1 (zh) |
| CA (1) | CA2387611C (zh) |
| HU (1) | HUP0203123A3 (zh) |
| MX (1) | MXPA02003719A (zh) |
| NO (1) | NO20021701L (zh) |
| NZ (1) | NZ529789A (zh) |
| PL (1) | PL354623A1 (zh) |
| SG (1) | SG147286A1 (zh) |
| WO (1) | WO2001026693A2 (zh) |
| ZA (1) | ZA200202695B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106750272A (zh) * | 2016-12-05 | 2017-05-31 | 福建卫生职业技术学院 | 一种水溶性蛇葡萄素聚合物 |
Families Citing this family (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1294404A2 (en) | 2000-06-14 | 2003-03-26 | Corixa Corporation | Prodrug compounds with an oligopeptide having an isoleucine residue |
| US6872715B2 (en) | 2001-08-06 | 2005-03-29 | Kosan Biosciences, Inc. | Benzoquinone ansamycins |
| NZ564694A (en) | 2003-04-22 | 2009-11-27 | Sod Conseils Rech Applic | Peptide vectors |
| FR2855521B1 (fr) * | 2003-05-28 | 2005-08-05 | Flamel Tech Sa | Polyaminoacides fonctionnalises par au moins un groupement h ydrophobe et leurs applications notamment therapeutiques. |
| US7259156B2 (en) | 2004-05-20 | 2007-08-21 | Kosan Biosciences Incorporated | Geldanamycin compounds and method of use |
| WO2006020719A2 (en) | 2004-08-11 | 2006-02-23 | Arqule, Inc. | Aminoacid conjugates of beta - lapachone for tumor targeting |
| US8614228B2 (en) | 2004-08-11 | 2013-12-24 | Arqule, Inc. | Quinone prodrug compositions and methods of use |
| EP1792927B1 (en) | 2004-09-22 | 2013-03-06 | Nippon Kayaku Kabushiki Kaisha | Novel block copolymer, micelle preparation, and anticancer agent containing the same as active ingredient |
| RU2472812C2 (ru) | 2005-12-05 | 2013-01-20 | НИТТО ДЕНКО КОРПОРЭЙШН (Джэпэн/Джэпэн) | Конъюгаты полиглутамат-аминокислота и способы |
| US20070167349A1 (en) * | 2005-12-06 | 2007-07-19 | Cell Therapeutics, Inc. | Estrogen cancer therapy |
| US7671067B2 (en) * | 2006-02-09 | 2010-03-02 | Enzon Pharmaceuticals, Inc. | Treatment of non-hodgkin's lymphomas with multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamtothecin |
| US7462627B2 (en) * | 2006-02-09 | 2008-12-09 | Enzon Pharmaceuticals, Inc. | Multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin for treatment of breast, colorectal, pancreatic, ovarian and lung cancers |
| US8323669B2 (en) | 2006-03-28 | 2012-12-04 | Nippon Kayaku Kabushiki Kaisha | Polymer conjugate of taxane |
| WO2007135910A1 (ja) | 2006-05-18 | 2007-11-29 | Nippon Kayaku Kabushiki Kaisha | ポドフィロトキシン類の高分子結合体 |
| JP5503872B2 (ja) | 2006-11-06 | 2014-05-28 | 日本化薬株式会社 | 核酸系代謝拮抗剤の高分子誘導体 |
| WO2008056654A1 (en) | 2006-11-08 | 2008-05-15 | Nippon Kayaku Kabushiki Kaisha | Polymeric derivative of nucleic acid metabolic antagonist |
| CN101209350B (zh) * | 2006-12-30 | 2011-09-07 | 中国人民解放军军事医学科学院毒物药物研究所 | 以氨基酸为连接子的多聚谷氨酸-药物偶合物 |
| CN101605539B (zh) * | 2007-02-09 | 2013-01-02 | 安佐制药股份有限公司 | 用7-乙基-10-羟基喜树碱的多臂聚合缀合物治疗抵抗性或顽固性癌症 |
| US20080253969A1 (en) * | 2007-04-10 | 2008-10-16 | Nitto Denko Corporation | Multi-functional polyglutamate drug carriers |
| TWI401081B (zh) | 2007-04-30 | 2013-07-11 | Arqule Inc | 苯醌化合物的羥基磺酸鹽及其用途 |
| EP2155254B1 (en) * | 2007-05-09 | 2012-11-28 | Nitto Denko Corporation | Polymers conjugated with platinum drugs |
| ES2430380T3 (es) | 2007-05-09 | 2013-11-20 | Nitto Denko Corporation | Composiciones que incluyen un compuesto hidrófobo y un conjugado de poliaminoácido |
| WO2008141110A2 (en) * | 2007-05-09 | 2008-11-20 | Nitto Denko Corporation | Polyglutamate conjugates and polyglutamate-amino acid conjugates having a plurality of drugs |
| US8703878B2 (en) | 2007-09-28 | 2014-04-22 | Nippon Kayaku Kabushiki Kaisha | High-molecular weight conjugate of steroids |
| KR20100122510A (ko) * | 2008-03-06 | 2010-11-22 | 닛토덴코 가부시키가이샤 | 중합체 파클리탁셀 접합체 및 암 치료 방법 |
| US8920788B2 (en) | 2008-03-18 | 2014-12-30 | Nippon Kayaku Kabushiki Kaisha | High-molecular weight conjugate of physiologically active substances |
| US9149540B2 (en) | 2008-05-08 | 2015-10-06 | Nippon Kayaku Kabushiki Kaisha | Polymer conjugate of folic acid or folic acid derivative |
| US20100056555A1 (en) * | 2008-08-29 | 2010-03-04 | Enzon Pharmaceuticals, Inc. | Method of treating ras associated cancer |
| KR101705077B1 (ko) | 2008-10-07 | 2017-02-09 | 렉산 파마슈티컬스, 인코포레이티드 | Hpma-도세탁셀 또는 젬시타빈 컨쥬게이트 및 이의 용도 |
| KR20110074583A (ko) * | 2008-10-15 | 2011-06-30 | 닛토덴코 가부시키가이샤 | 폴리글루타메이트 접합체의 제조 방법 |
| WO2010048018A1 (en) * | 2008-10-21 | 2010-04-29 | Enzon Pharmaceuticals, Inc. | Treatment of neuroblastoma with multi-arm polymeric conjugates of 7-ethyl-10-hydroxycamptothecin |
| DE102008060549A1 (de) * | 2008-12-04 | 2010-06-10 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Wirkstoff-Peptid-Konstrukt zur extrazellulären Anreicherung |
| US8808749B2 (en) | 2009-05-15 | 2014-08-19 | Nippon Kayaku Kabushiki Kaisha | Polymer conjugate of bioactive substance having hydroxy group |
| CA2777397C (en) | 2009-10-13 | 2017-08-29 | Rexahn Pharmaceuticals, Inc. | Polymeric systems for the delivery of anticancer agents |
| US20110224148A1 (en) * | 2010-03-11 | 2011-09-15 | Nitto Denko Corporation | Carbohydrate-polyamino acid-drug conjugates |
| CN103221054A (zh) | 2010-11-17 | 2013-07-24 | 日本化药株式会社 | 新的胞苷类代谢拮抗剂的高分子衍生物 |
| CN102649810A (zh) * | 2011-05-19 | 2012-08-29 | 东北林业大学 | 喜树碱衍生物、其制备方法和用途 |
| US8691284B2 (en) | 2011-07-07 | 2014-04-08 | Empire Technology Development Llc | Fluorinated block co-polymers |
| PT2754682T (pt) | 2011-09-11 | 2017-08-29 | Nippon Kayaku Kk | Método para o fabrico de copolímero de bloco |
| US8383093B1 (en) | 2011-11-01 | 2013-02-26 | Serina Therapeutics, Inc. | Subcutaneous delivery of poly(oxazoline) conjugates |
| US8907045B2 (en) | 2012-04-03 | 2014-12-09 | Empire Technology Development Llc | Biocompatible adhesive polymers |
| EP2989116A4 (en) | 2013-04-26 | 2017-01-18 | Nitto Denko Corporation | Reduction of endotoxins from polyanionic polymer conjugates |
| KR101507119B1 (ko) * | 2013-05-31 | 2015-03-30 | 주식회사 바이오리더스 | 점막 점착성 폴리감마글루탐산 나노마이셀 및 이를 이용한 약물 전달체 |
| CN106267227A (zh) * | 2016-08-12 | 2017-01-04 | 北京蓝贝望生物医药科技股份有限公司 | 抗肿瘤药物 |
| CN110123748A (zh) * | 2019-04-22 | 2019-08-16 | 南京师范大学 | 一种叶酸介导的靶向双载药聚合物胶束及其制备方法和应用 |
| CN113773282A (zh) * | 2021-09-22 | 2021-12-10 | 无锡紫杉药业有限公司 | 一种10-乙酰紫杉醇的制备方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4356166A (en) * | 1978-12-08 | 1982-10-26 | University Of Utah | Time-release chemical delivery system |
| US5629433A (en) * | 1994-07-18 | 1997-05-13 | Hauser, Inc. | Selective process for the deacylation and deacetylation of taxol and taxanes |
| CA2192725C (en) * | 1995-12-28 | 2004-04-20 | Kenji Tsujihara | Camptothecin derivatives |
| US6441025B2 (en) | 1996-03-12 | 2002-08-27 | Pg-Txl Company, L.P. | Water soluble paclitaxel derivatives |
| KR100561788B1 (ko) * | 1996-03-12 | 2006-09-20 | 피지-티엑스엘 컴파니,엘.피. | 수용성파클리탁셀전구약물을포함하는조성물및이러한조성물을포함하는이식가능한의료장치 |
| KR20020082888A (ko) * | 2000-03-17 | 2002-10-31 | 쎌세러퓨틱스,인코포레이티드 | 폴리글루탐산-캄프토테신 컨주게이트 및 제조 방법 |
-
2000
- 2000-10-12 SG SG200401981-6A patent/SG147286A1/en unknown
- 2000-10-12 WO PCT/US2000/028109 patent/WO2001026693A2/en active IP Right Grant
- 2000-10-12 KR KR1020027004640A patent/KR100821437B1/ko not_active IP Right Cessation
- 2000-10-12 PL PL00354623A patent/PL354623A1/xx not_active IP Right Cessation
- 2000-10-12 CN CNA2009101276554A patent/CN101507820A/zh active Pending
- 2000-10-12 MX MXPA02003719A patent/MXPA02003719A/es active IP Right Grant
- 2000-10-12 CA CA2387611A patent/CA2387611C/en not_active Expired - Fee Related
- 2000-10-12 EP EP00972079A patent/EP1225917A2/en not_active Withdrawn
- 2000-10-12 EP EP04013703A patent/EP1466627A1/en not_active Withdrawn
- 2000-10-12 NZ NZ529789A patent/NZ529789A/en not_active IP Right Cessation
- 2000-10-12 HU HU0203123A patent/HUP0203123A3/hu unknown
- 2000-10-12 AU AU10793/01A patent/AU781735B2/en not_active Ceased
- 2000-10-12 BR BRPI0014652A patent/BRPI0014652B1/pt not_active IP Right Cessation
- 2000-10-12 JP JP2001529754A patent/JP2003511423A/ja active Pending
- 2000-10-12 CN CNA008170290A patent/CN1607962A/zh active Pending
-
2001
- 2001-10-09 US US09/971,657 patent/US20020077279A1/en not_active Abandoned
-
2002
- 2002-04-05 ZA ZA200202695A patent/ZA200202695B/en unknown
- 2002-04-11 NO NO20021701A patent/NO20021701L/no not_active Application Discontinuation
-
2003
- 2003-04-02 US US10/404,152 patent/US7399860B2/en not_active Expired - Fee Related
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106750272A (zh) * | 2016-12-05 | 2017-05-31 | 福建卫生职业技术学院 | 一种水溶性蛇葡萄素聚合物 |
| CN106750272B (zh) * | 2016-12-05 | 2019-05-28 | 福建卫生职业技术学院 | 一种水溶性蛇葡萄素聚合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20030224971A1 (en) | 2003-12-04 |
| WO2001026693A2 (en) | 2001-04-19 |
| CA2387611C (en) | 2011-03-29 |
| EP1225917A2 (en) | 2002-07-31 |
| PL354623A1 (en) | 2004-02-09 |
| JP2003511423A (ja) | 2003-03-25 |
| BRPI0014652B1 (pt) | 2016-08-09 |
| CN101507820A (zh) | 2009-08-19 |
| EP1466627A1 (en) | 2004-10-13 |
| BR0014652A (pt) | 2003-06-10 |
| CA2387611A1 (en) | 2001-04-19 |
| MXPA02003719A (es) | 2002-08-30 |
| AU781735B2 (en) | 2005-06-09 |
| NZ529789A (en) | 2005-04-29 |
| KR20020059616A (ko) | 2002-07-13 |
| AU1079301A (en) | 2001-04-23 |
| WO2001026693A3 (en) | 2001-12-27 |
| NO20021701D0 (no) | 2002-04-11 |
| SG147286A1 (en) | 2008-11-28 |
| KR100821437B1 (ko) | 2008-04-10 |
| US20020077279A1 (en) | 2002-06-20 |
| HUP0203123A2 (hu) | 2003-01-28 |
| NO20021701L (no) | 2002-05-23 |
| US7399860B2 (en) | 2008-07-15 |
| ZA200202695B (en) | 2004-02-25 |
| HUP0203123A3 (en) | 2004-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1607962A (zh) | 聚谷氨酸-治疗剂结合物的生产方法 | |
| CN1154512C (zh) | 喜树碱的聚合衍生物 | |
| CN1243779C (zh) | 具有y形分支的亲水性聚合物衍生物、其制备方法、与药物分子的结合物以及包含该结合物的药物组合物 | |
| CN101516969B (zh) | 靶向的聚赖氨酸树状聚体治疗剂改性的大分子2 | |
| CN1069099C (zh) | 阿霉素的二聚物和三聚物 | |
| CN109096170B (zh) | 近红外染料、其靶向成像剂、纳米载体和抗癌药物及应用 | |
| JPH10513187A (ja) | 高分子量のポリマーを基剤とするプロドラッグ | |
| CN1332640A (zh) | Dds化合物及其测定方法 | |
| JP2000517304A (ja) | 高分子量のポリマーを基剤とするプロドラッグ | |
| EP3459546B1 (en) | Multi-arm polymeric targeting anti-cancer conjugate | |
| CN105999299B (zh) | 一种小分子胶束纳米载药系统及其制备方法与应用 | |
| TWI480070B (zh) | 具礦質親和能力之多歧狀胜肽構型 | |
| JP2002518405A (ja) | ビタミンb12誘導体及びそれらの製造方法 | |
| CN111001012A (zh) | 一种亲水碳酸酯型抗体偶联药物 | |
| CN102911252B (zh) | 含肽类树状分子的阳离子脂质、转基因载体及其制备方法和应用 | |
| CN111097052B (zh) | 用于肿瘤主动靶向治疗的两亲性前药及其纳米颗粒的制备方法、应用 | |
| CN111848544A (zh) | 可荧光示踪的氨基酸衍生物及其制备方法和应用 | |
| CN1254496C (zh) | 制备分枝型聚乙二醇的方法 | |
| CN108752507A (zh) | 一种酶敏感和氧化还原敏感双重响应型共聚物及其制备方法和应用 | |
| CN113429559A (zh) | 一种末端异官能化的六臂聚乙二醇衍生物及其制备方法 | |
| CN1098106C (zh) | 可溶的合成聚合物结合的蒽环类化合物 | |
| CN1777610A (zh) | 用于光动力治疗的水溶性单-peg化四吡咯衍生物及其制备方法 | |
| CN113041355B (zh) | 一种可精准调控联用药物比率的共递送纳米药物及应用 | |
| CN1218983C (zh) | 叶酸-聚二醇类化合物及其药物复合物 | |
| CN110772643B (zh) | α-生育酚聚乙二醇琥珀酸酯修饰的强心苷类化合物前药 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |