CN1241953C - 烯烃聚合催化剂体系、聚合方法以及由其制备的聚合物 - Google Patents
烯烃聚合催化剂体系、聚合方法以及由其制备的聚合物 Download PDFInfo
- Publication number
- CN1241953C CN1241953C CN 99814533 CN99814533A CN1241953C CN 1241953 C CN1241953 C CN 1241953C CN 99814533 CN99814533 CN 99814533 CN 99814533 A CN99814533 A CN 99814533A CN 1241953 C CN1241953 C CN 1241953C
- Authority
- CN
- China
- Prior art keywords
- group
- fraction
- polyolefine
- catalyzer
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65912—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an organoaluminium compound
Abstract
本发明涉及一种烯烃聚合催化剂体系,含有至少一种活化剂和以下组分中的一种:a)至少两种基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂,如由式(I)、(II)、(III)或(IV)表示;b)活化剂与基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂的组合产物,如式(III)或(IV)所示,可以在将其引入反应器之前反应优选至少15分钟,和/或c)添加剂与基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂的组合产物,由式(I)或(II)所示。本发明进一步涉及由该催化剂制备的聚合物,特别是独特的聚乙烯树脂,更优选由本发明制备的双模高密度聚乙烯树脂。
Description
发明领域
本发明涉及含有活化剂和基于过渡金属化合物的催化剂的烯烃聚合催化剂体系,该过渡金属化合物含有双齿配体,该配体包含吡啶或喹啉部分及其混合物,涉及使用这种催化剂体系的聚合方法以及由其制备的聚合物。
发明背景
金属茂聚烯烃催化剂的深入工业化已引起对非金属茂、均相催化剂设计的广泛关注。该领域更是一种学术关注,因为新型非金属茂催化剂可提供制备现有产品的更容易的方法,并还可提供超过金属茂催化剂能力的产品和工艺的机会。另外,特定的非环戊二烯基配体是更经济的,因为各种取代的类似物更易于合成。
所以,在本领域中需要新型烯烃聚合催化剂以及由这些催化剂制备的独特聚合物。还需要催化剂体系可容易地通过向单一催化剂中加入添加剂进行改性,以形成双模产物。另外,需要确定能制备单模和双模聚烯烃的催化剂。本发明确定一类独特的催化剂,其能单独使用或组合使用,以制备独特的聚烯烃,特别是双模聚烯烃。
WO96/23101、WO97/02298、WO96/33202和Furhmann等人,Inorg.Chem.35:6742-6745(1996)都公开了含氮的单点催化剂体系。
USSN09/103620,以WO99/01460于1999年1月14日公开,公开了使用含有双齿配体的过渡金属化合物,该配体包括吡啶或喹啉部分及其混合物,与活化剂一起使用来聚合烯烃。特别是[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基与改性甲基铝氧烷在气相中组合,以制备乙烯己烯聚合物。我们已发现USSN09/103620可通过直接加入添加剂来改进,以从单催化剂制备双模产物。
1998年11月5日公开的WO98/49208公开了一种含氮的金属配合物及其在1-烯烃聚合中的应用。EP-A1-0 619 325公开了一种使用至少两种金属茂制备具有多模或至少双模分子量分布的烯烃的方法,其中一种金属茂已桥接,一种金属茂未桥接。
为了US目的,提及以下参考文献:US4845067;US4999327;JP1126111;US4508842;和UK1015054。
发明概述
本发明涉及一种烯烃聚合催化剂体系,含有至少一种活化剂和以下组分中的一种:
a)至少两种基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂,如由下式I、II、III或IV表示,
b)活化剂与基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂的组合产物,如式III或IV所示,可以在将其引入反应器之前反应优选至少15分钟,和/或
c)添加剂与基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂的组合产物,如下式I或II所示。
本发明进一步涉及由该催化剂体系制备的聚合物,特别是独特的聚乙烯树脂,更优选由本发明制备的双模高密度聚乙烯树脂。
附图简述
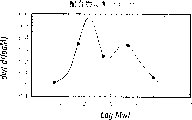
图1-5是重均分子量的log值与dwt/d(logM)的关系曲线,用于衡量表1实验的分子量分布。
图6和7是实施例11和12中实验的体积排除色谱(SEC)图。
图8是实施例14中制得的聚合物的SEC图。
图9是1998年6月23日递交的USSN09/103620(公开为WO99/01460)中实施例14的SEC图。
发明详述
A.本发明部分涉及烯烃聚合催化剂体系,该体系含有至少一种活化剂和至少两种基于含吡啶或喹啉部分的双齿配体的过渡金属催化剂。该活化剂可以是任何公知的催化剂活化剂,在一个实施方案中是烷基铝、铝氧烷、改性铝氧烷、聚铝氧烷、非配位阴离子、路易斯酸、硼烷或其混合物。
有各种制备铝氧烷和改性铝氧烷的方法,非限定性例子描述于美国专利4665208、4952540、5091352、5206199、5204419、4874734、4924018、4908463、4968827、5308815、5329032、5248801、5235081、5157137、5103031、5391793、5391529、5693838、5731253和5731451以及欧洲专利申请EP-A-0 561 476、EP-B1-0 279 586和EP-A-0594-218,以及PCT公开WO94/10180,将其全部引入本文以供参考。甲基铝氧烷、改性甲基铝氧烷、三异丁基铝氧烷、和聚甲基铝氧烷是优选的活化剂。
离子化合物(非配位阴离子)可包含活性质子或与离子化合物的剩余离子缔合但未配位或仅仅弱配位的一些其它阳离子。这种化合物及其类似物描述于欧洲专利申请EP-A-0 570 982、EP-A-0 520 732、EP-A-0 495 375、EP-A-0 426 637、EP-A-500 944、EP-A-0 277 003和EP-A-0 277 004,以及美国专利5153157、5198401、5066741、5206197、5241025、5387568、5384299和5502124,以及1994年8月3日递交的美国专利中请系列号08/285380,将其全部引入本文以供参考。其它活化剂包括PCT公开WO98/07515所述的那些,例如三(2,2’,2”-九氟二苯基)氟铝酸盐,将其全部引入本文以供参考。活化剂的组合也可考虑用于本发明,例如铝氧烷与离子化活化剂的组合,参见例如PCT公开WO94/07928和WO95/14044以及美国专利5153157和5453410,将其全部引入本文以供参考。同样,活化方法例如使用辐射等也可考虑在本发明中用于活化剂。
在优选的实施方案中,活化剂选自以下物质:三(2,2’,2”-九氟二苯基)氟铝酸盐、铝氧烷、三苯基硼、三乙基硼、三正丁基铵四乙基硼酸盐、三芳基硼烷、三正丁基铵四(五氟苯基)硼或三全氟苯基硼,或二乙基氯化铝。
在一个实施方案中,基于含有吡啶或喹啉部分的双齿配体的过渡金属催化剂化合物由下式表示;
((Z)XAt(YJ))qMQn (I)其中M是选自元素周期表的3-13族或镧系和锕系的金属;Q键接于M上,且每个Q是单价、二价或三价阴离子;X和Y键接于M上;X和Y独立地是碳或杂原子,前提是X和Y中的至少一个是杂原子,优选X和Y都是杂原子;Y含于杂环J中,其中J含有2-50个非氢原子,优选2-30个碳原子;Z键接于X上,其中Z含有1-50个非氢原子,优选1-50个碳原子,或甲硅烷基,烷基甲硅烷基例如三烷基甲硅烷基,优选Z是含3-50个原子、优选3-30个碳原子的环状基团;t是0或1;当t是1时,A是桥接基团,连接X、Y或J中的至少一个,优选X和J;q是1或2;如果Q是单价阴离子,则n是M的氧化态减去q,如果Q是二价阴离子,则n是(M的氧化态-q)/2,或如果Q是三价阴离子,则n是(M的氧化态-q)/3,通常n是1-4的整数,取决于M的氧化态。
在一个实施方案中,如果X是氧或硫,则Z是任选的。在另一个实施方案中,如果X是氮或磷,则Z是存在的。在一个实施方案中,Z优选是芳基,更优选是取代的芳基。
在另一个实施方案中,过渡金属催化剂化合物由下式表示:
((R’mZ)XA(YJR”p))qMQn (II)其中M是选自元素周期表的3-13族的金属,优选4-12族过渡金属,更优选4、5或6族过渡金属,甚至更优选4族过渡金属,例如钛、锆或铪,最优选锆;
每个Q键接于M上,且每个Q是单价、二价或三价阴离子。优选每个Q独立地选自卤素、氢、烷基、芳基、链烯基、烷基芳基、芳基烷基、氢化羧基(hydrocarboxy)或具有1-20个碳原子的苯氧基。每个Q可以是氨基、磷化物、硫化物、甲硅烷基烷基、二酮根和羧酸根。任选地,每个Q可以含有一个或多个杂原子,更优选每个Q选自卤素、烷基和芳基烷基。最优选,每个Q选自芳基烷基,例如苄基。的至少一个是杂原子,优选X和Y都是杂原子,更优选X和Y独立地选自氮、氧、硫和磷,甚至更优选氮或磷,最优选氮;
Y含于杂环或环体系J中。J含有2-30个碳原子,优选2-7个碳原子,更优选3-6个碳原子,最优选5个碳原子。任选地,含Y的杂环J可以含有其它杂原子。J可被R”基团所取代,R”基团独立地选自氢或直链、支链、环状的烷基或链烯基、炔基、烷氧基、芳基或芳氧基。同样,两个或更多的R”基团可以连接形成环状部分,例如脂族或芳族环。优选R”是氢或芳基,最优选R”是氢。当R”是芳基且Y是氮时,形成喹啉基团。任选地,R”可以与A连接;
Z是与X连接的烃基,优选Z是含有1-50个碳原子的烃基,优选Z是含有3-30个碳原子的环状基团,优选Z是含3-30个碳原子的取代或未取代的环状基团,任选地包含一个或多个杂原子,更优选Z是芳基,最优选取代的芳基,在另一个实施方案中Z可以是甲硅烷基或烷基甲硅烷基,优选三烷基甲硅烷基;
Z可以被R’基团取代,其中R’基团独立地选自氢或直链、支链烷基或环烷基、链烯基、炔基或芳基。同样,两个或更多的R’基团可以连接形成环状部分,例如脂族或芳族环。优选R’是含1-20个碳原子的烷基,更优选R’是甲基、乙基、丙基、丁基、戊基等,包括其异构体,更优选R’是甲基,或伯、仲或叔烃基,包括异丙基、叔丁基等,最优选R’是异丙基。任选地,R’基团可以与A连接。优选至少一个R’处于X的邻位;
A是与X和J中的至少一个、优选两者连接的桥接基团。桥接基团A含有一个或多个元素周期表的13-16族元素。更优选,A含有一个或多个14族元素,最优选A是取代的碳基团、二取代的碳基团或乙烯基;和
在式(II)中,m和p独立地是0-5的整数,优选m是2;如果Q是单价阴离子,则n是M的氧化态减去q,如果Q是二价阴离子,则n是(M的氧化态-q)/2,或如果Q是三价阴离子,则n是(M的氧化态-q)/3,优选n是1-4的整数;q是1或2,且当q是2时,式(II)的两个((R’mZ)XA(YJR”m))经由桥接基团互相桥接,优选含14族元素的桥接基团。
在优选实施方案中,当式I或II中的n是2或3时,则除了一个Q基团是氢化羧基、硼酸根或氨基,第二种催化剂与第一种催化剂相同。在特别优选的实施方案中,当式I或II中的n是2或3时,则除了一个Q基团是烷氧基、苯氧基、乙酰丙酮根、羧酸根、环戊二烯基、芴基或茚基,第二种催化剂与第一种催化剂相同。在另一个特别优选的实施方案中,当式I或II中的n是2或3时,则第二种催化剂与第一种催化剂相同,除了第二种催化剂的一个Q基团是在第一种催化剂上的类似Q基团的氢化羧基加合物,优选烷氧基加合物、硼酸根、苯氧基加合物、乙酰丙酮根加合物、羧酸根加合物、氨基加合物、环戊二烯基加合物、芴基加合物或茚基加合物。
在优选的实施方案中,至少一种过渡金属催化剂化合物由下式表示:
((Z)XAt(YJ))qMQmTs (III)其中M是选自元素周期表的3-13族或镧系和锕系的金属;T键接于M上,并是13-16族元素,优选氧、硼、氮、硅、磷、硫或铝,T还可以与任选含一个或多个杂原子的一个或多个C1至C50基团连接,优选T是氢化羧基、硼酸根或氨基,优选烷氧基、苯氧基、乙酰丙酮根,或羧酸根或类环戊二烯基基团例如环戊二烯基、芴基和茚基,Q键接于M上,且每个Q是单价、二价或三价阴离子;X和Y键接于M上;X和Y独立地是碳或杂原子,前提是X和Y中的至少一个是杂原子,优选X和Y都是杂原子;Y含于杂环J中,其中J含有2-50个非氢原子,优选2-30个碳原子;Z键接于X上,其中Z含有1-50个非氢原子,优选1-50个碳原子,优选Z是含3-50个原子、优选3-30个碳原子的环状基团,另外Z可以是甲硅烷基,优选烷基甲硅烷基;t是0或1;当t是1时,A是桥接基团,连接X、Y或J中的至少一个,优选X和J;q是1或2;如果Q是单价阴离子,则m是M的氧化态减去q减去s,如果Q是二价阴离子,则m是(M的氧化态-q-s)/2,或如果Q是三价阴离子,则m是(M的氧化态-q-s)/3,优选m是1-3的整数,s是1、2或3,优选1或2。在一个实施方案中,当X是氧或硫时,则Z是任选的。在另一个实施方案中,如果X是氮或磷,则Z是存在的。在一个优选实施方案中,T是氧,并与烷基、芳基或烷芳基连接。
在另一个实施方案中,至少一种过渡金属催化剂化合物由下式表示:
(R’mZ)XA(YJR”p))qMQnTs (IV)其中M是选自元素周期表的3-13族的金属,优选4-12族过渡金属,更优选4、5或6族过渡金属,甚至更优选4族过渡金属,例如钛、锆或铪,最优选锆;
T键接于M上,并是13-16族元素,优选氧、硼、氮、硅、磷、硫或铝,T还可以与任选含一个或多个杂原子的一个或多个C1至C50基团连接,T优选是氢化羧基、硼酸根或氨基,优选烷氧基、苯氧基、乙酰丙酮根,或羧酸根或类环戊二烯基基团例如环戊二烯基、芴基和茚基。
每个Q键接于M上,且每个Q是单价、二价或三价阴离子。优选每个Q独立地选自卤素、氢、烷基、芳基、链烯基、烷基芳基、芳基烷基、氢化羧基或具有1-20个碳原子的苯氧基。每个Q也可以是氨基、磷化物、硫化物、甲硅烷基烷基、二酮根和羧酸根。任选地,每个Q可以含有一个或多个杂原子,更优选每个Q选自卤素、烷基和芳基烷基。最优选,每个Q选自芳基烷基,例如苄基。
X和Y独立地是C或杂原子,前提是X和Y中的至少一个是杂原子,优选X和Y都是杂原子,更优选独立地选自氮、氧、硫和磷,甚至更优选氮或磷,最优选氮;
Y含于杂环或环体系J中。J含有2-30个碳原子,优选2-7个碳原子,更优选3-6个碳原子,最优选5个碳原子。任选地,含Y的杂环J可以含有其它杂原子。J可被R”基团所取代,R”基团独立地选自氢或直链、支链、环状的烷基或链烯基、炔基、烷氧基、芳基或芳氧基。同样,两个或更多的R”基团可以连接形成环状部分,例如脂族或芳族环。优选R”是氢或芳基,最优选R”是氢。当R”是芳基且Y是氮时,形成喹啉基团。任选地,R”可以与A连接;
Z是与X连接的烃基,优选Z是含有1-50个碳原子的烃基,优选Z是含有3-30个碳原子的环状基团,优选Z是含3-30个碳原子的取代或未取代的环状基团,任选地包含一个或多个杂原子,Z可以是甲硅烷基、烷基甲硅烷基或三烷基,在另一个实施方案中Z不是芳基;
Z可以被R’基团取代,其中R’基团独立地选自氢或直链、支链烷基或环烷基、链烯基或炔基。同样,两个或更多的R’基团可以连接形成环状部分,例如脂族或芳族环。优选R’是含1-20个碳原子的烷基,更优选R’是甲基、乙基、丙基、丁基、戊基等,包括其异构体,更优选R’是甲基,或伯、仲或叔烃基,包括异丙基、叔丁基等,最优选R’是异丙基。任选地,R’基团可以与A连接。优选至少一个R’处于X的邻位;
A是与X和J中的至少一个、优选两者连接的桥接基团。桥接基团A含有一个或多个元素周期表的13-16族元素。更优选,A含有一个或多个14族元素,最优选A是取代的碳基团、二取代的碳基团或乙烯基;和
在式(IV)中,m和p独立地是0-5的整数,优选m是2;s是1-3的整数;q是1或2,如果Q是单价阴离子,则n是M的氧化态减去q减去s,如果Q是二价阴离子,则n是(M的氧化态-q-s)/2,或如果Q是三价阴离子,则n是(M的氧化态-q-s)/3,且当q是2时,式(IV)的两个((R’mZ)XA(YJR”m))经由桥接基团互相桥接,优选含14族元素的桥接基团。
在一个实施方案中,J是任何上式中的吡啶。
过渡金属化合物可通过本领域公知的任何方法来制备。例如USSN09/103620,1998年6月23日递交,由1997年7月2日递交的前申请号60/051581要求优先权,公开为WO99/01460,公开了制备这些化合物的方法。
在优选实施方案中,过渡金属化合物是[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基。在另一个优选实施方案中,[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基与铝氧烷、优选甲基铝氧烷、更优选改性甲基铝氧烷在气相或淤浆相反应器中组合使用,以制备聚乙烯,优选高密度聚乙烯。在另一个优选实施方案中,非配位阴离子,例如三正丁基铵四(五氟苯基)硼或三全氟苯基硼,与[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基在气相或淤浆相反应器中组合使用。在另一个实施方案中,活化剂选自以下物质:三(2,2’,2”-九氟二苯基)氟铝酸盐、铝氧烷、三苯基硼、三乙基硼、三正丁基铵四乙基硼酸盐、三芳基硼酸盐、三正丁基铵四(五氟苯基)硼,或三全氟苯基硼,或二乙基氯化铝。
在一个实施方案中,在本发明实践中,选择两种或更多的催化剂来制备期望的产物。两种或更多的催化剂选自上式中的任何催化剂。例如可选择来自式(I)的两种不同催化剂,或可以组合来自式I或II的化合物和来自式III或IV的化合物。相似地,在式IV定义范围内的两种不同的催化剂可以组合。在优选实施方案中,来自式I或II的化合物与至少一种来自式III或IV的化合物一起使。有可能通过选择已知能制备不同分子量的催化剂来获得双模产物。
在另一个实施方案中,将两种催化剂分别加入反应器中。
在优选实施方案中,选择由式I或II表示的第一种催化剂,其中至少一个Q基团不是含氧加合物,第二种催化剂与第一种催化剂体系相同,除了一个、两个或全部三个Q基团是与第一种催化剂中存在的相同Q基团的含氧加合物。例如如果选择[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基作为第一种催化剂,则[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基可以是第二种催化剂。
为了本发明的目的和权利要求,含氧加合物定义为O-R,其中O是氧,R是任选可含有一个或多个杂原子的C1至C50基团。优选的R基团包括叔丁基、叔戊基、叔己基、异丙基、2-[2-甲基-1-苯基-丙基]、2-[2-苄基-丁基]、3-[3-苄基-戊基]。其它可能的R基团包括苄基、甲基苄基、乙基苄基等。在另一个实施方案中,含氧加合物可以由式O-B-R表示,其中O是氧,B是硼,R是任选可含有一个或多个杂原子的C1至C50基团。优选的R基团包括叔丁基、叔戊基、叔己基、异丙基、2-[2-甲基-1-苯基-丙基]、2-[2-苄基-丁基]、3-[3-苄基-戊基]。其它可能的R基团包括苄基、甲基苄基、乙基苄基等。
在优选实施方案中,两种催化剂,[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基和[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基,与铝氧烷、优选甲基铝氧烷、更优选改性甲基铝氧烷在气相或淤浆反应器中组合使用,以制备聚乙烯,优选高密度聚乙烯或低密度聚乙烯。在另一个优选实施方案中,非配位阴离子,例如三正丁基铵四(五氟苯基)硼或三全氟苯基硼可以与两种催化剂,即[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基和[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基,在气相或淤浆相反应器中组合使用,以制备聚烯烃,优选聚乙烯。
在优选的实施方案中,两种催化剂化合物通常按照0.001∶1至约10000∶1的比率组合,优选0.5∶1至1000∶1。在优选实施方案中,第一种催化剂的存在量约为0.5-99.5重量%,第二种催化剂的存在量约为99.5-0.5重量%,以两种催化剂重量、而不是活化剂或载体的重量为基准,优选5-95重量%第一种催化剂和95-5重量%第二种催化剂,优选10-90重量%第一种催化剂和90-10重量%第二种催化剂。
在优选实施方案中,第一种催化剂的存在量约为0.5-99.5重量%,第二种和第三种催化剂的存在量约为99.5-0.5重量%,以三种催化剂重量、而不是活化剂或载体的重量为基准,优选5-95重量%第一种催化剂,优选10-90重量%第一种催化剂。
B.本发明进一步涉及烯烃聚合催化剂体系,该体系含有至少一种活化剂以及活化剂(如上所述)与由式III或IV表示的过渡金属催化剂(如上所述)的组合产物,在将其引入反应器之前使其优选反应至少15分钟。在这种实施方案中,由式III或IV表示的单过渡金属化合物可用于制备独特的、优选双模聚烯烃。已观察到,温度显然对由式III或IV表示的催化剂有影响,并显然制得其它物质。虽然不希望受限于任何理论,但是看来该转化提供了制备双模产物的能力。因此,我们注意到,在较高的温度下从催化剂体系制得更多的双模产物,其中一种过渡金属化合物用于催化剂的制备。相似地已观察到,使过渡金属化合物与活化剂反应一段时间,也可提供能制备双模或多模聚合物的体系。因此,在优选实施方案中,使过渡金属化合物和活化剂在与烯烃接触之前反应至少15分钟,优选至少30分钟,优选至少1小时,优选至少4小时。但是,我们还注意到,恰好在将[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基喷到反应器中之前对其进行分别第二种活化,制得十分活泼的催化剂体系,该体系也显然制得双模树脂。在另一个实施方案中,聚合优选在至少80℃的温度下进行,优选至少85℃,优选至少90℃,优选至少100℃。
C.本发明进一步涉及烯烃聚合催化剂体系,含有至少一种活化剂(如上所述)以及添加剂与由式I或II表示的基于含吡啶或喹啉部分的双齿配体的过渡金属化合物(如上所述)的组合产物。
该添加剂优选是烷氧基化合物。烷氧基化合物定义为由式R=O表示的化合物,其中R是C1至C100基团,氧可以在沿着R基团的任何点上连接。R基团除1-100个碳原子以外还可以含有杂原子。优选的烷氧基化合物包括酮和醛。特别优选的烷氧基化合物包括丙酮、二苯甲酮、甲基乙基酮、二乙基酮、甲基异丁基酮、甲基异丙基酮、二异丙基酮、甲基叔丁基酮、苯乙酮、环己酮、环戊酮、苯甲醛、新戊醛、乙基正丙基酮、乙基异丙基酮等。
在优选的实施方案中,添加剂与过渡金属催化剂化合物组合,用量为0.5重量%至约90重量%,以过渡金属催化剂化合物和添加剂的重量为基准,而不是任何活化剂或载体的重量为基准,优选1重量%至约80重量%,更优选10-60重量%。添加剂可与过渡金属催化剂化合物(含有或不含活化剂)在加入聚合反应器之前组合。在一个实施方案中,将添加剂加入注射管中的在线过渡金属催化剂化合物中。
在优选实施方案中,使活化剂与过渡金属催化剂化合物(已与添加剂反应)在与烯烃组合之前反应至少5分钟,优选10分钟,更优选15分钟。已注意到,如果过渡金属催化剂化合物简单地与添加剂组合,然后直接加入反应器中,则获得较少的双模产物。如果使添加剂和过渡金属化合物反应一段时间,则可获得较多的双模产物。
在优选实施方案中,使添加剂和过渡金属催化剂化合物在与活化剂组合之前组合。在另一个实施方案中,过渡金属催化剂、烯烃和活化剂已存在于聚合反应器中,并加入添加剂。在实施方案中,其中在活化剂和过渡金属催化剂化合物已组合之后加入添加剂,且活化剂是铝氧烷,则可能需要额外量的添加剂。
不同的添加剂可用于获得对所得聚合物的不同影响。例如与使用甲基乙基酮作为添加剂相比,使用二乙基酮作为添加剂制得具有较高分子量的聚合物。相似地,与使用丙酮作为添加剂相比,使用甲基乙基酮作为添加剂制得具有较高分子量的聚合物。
在优选实施方案中,过渡金属催化剂化合物[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基和添加剂丙酮与铝氧烷、优选甲基铝氧烷、更优选改性甲基铝氧烷在气相或淤浆反应器中组合使用,以制备聚乙烯,优选高密度聚乙烯。在另一个优选实施方案中,非配位阴离子,例如三正丁基铵四(五氟苯基)硼或三全氟苯基硼,与过渡金属催化剂化合物[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基和丙酮在气相或淤浆相反应器中组合使用。
当将活化剂在过渡15金属化合物置于反应器之前或之后加入其中时,添加剂可以存在或不存在。在另一个优选的实施方案中,两种不同的过渡金属化合物与活化剂和添加剂在同一反应器中组合使用。在另一个优选的实施方案中,将过渡金属催化剂化合物和活化剂独立于添加剂加入反应器中。虽然不希望受限于任何理论,但看来添加剂与过渡金属催化剂化合物反应,以提供另一种活性催化剂物质。在本发明的实施方案中,已注意到,温度显然影响两种形式催化剂之间的平衡。看来较高的温度可以促进过渡金属催化剂化合物在添加剂的存在下向第二种催化剂物质转化。因此,通过选择添加剂的量及其组合和/或使用的温度,可选择所期望的最终产物。
在优选实施方案中,能制得较低分子量的催化剂组分的存在量是10ppm至70重量%,以所有催化剂的重量为基准,而不是活化剂或载体的重量,优选100ppm至8重量%,甚至更优选1000ppm至5重量%。在另一个实施方案中,能制得较低分子量的化合物的存在量是30-70重量%,以所有催化剂的重量为基准,而不是活化剂或载体的重量,优选40-60重量%,甚至更优选45-55重量%。
在另一个实施方案中,能制备低分子量部分的组分的存在量将能制备20-70重量%的最终聚合物产物。
如果使用多种催化剂,则两种或更多的催化剂可在相同或不同的时间被活化,在进入反应器之前或之后,以及在置于载体上之前或之后。在一个实施方案中,多种催化剂被同样的活化剂在置于反应器中之前所活化。在另一个实施方案中,一种催化剂在置于反应器中之前被活化,加入第二种催化剂,任选地不含活化剂、相同的活化剂或不同的活化剂。在另一个实施方案中,催化剂载附于相同的载体上,然后被同样的活化剂在置于反应器中之前活化。在另一个实施方案中,两种催化剂被相同或不同的活化剂活化,然后在置于反应器中之前置于载体上。
相似地,一种或多种催化剂体系或组分可以载附于有机或无机载体上。通常载体可以是任何固体、多孔载体。通常的载体材料包括滑石;无机氧化物,例如二氧化硅、氯化镁、氧化铝、二氧化硅-氧化铝;聚合物载体,例如聚乙烯、聚丙烯、聚苯乙烯;等。优选的载体包括二氧化硅、粘土、滑石、氯化镁等。优选载体以细分形式使用。在使用之前,载体优选部分或完全脱水。脱水可通过物理煅烧进行,或通过化学转化全部或部分活性羟基来进行。关于怎样载附催化剂的更详细信息,请参见US4808561,其中教导了怎样载附金属茂催化剂体系。其中所用的技术通常可用于本发明中。
催化剂可置于分别的载体上,或可置于相同的载体上。相似地,活化剂可置于与催化剂相同的载体上,或可置于分别的载体上。催化剂/催化剂体系和/或其组分不需要以同样的方式加入反应器中。例如,一种催化剂或其组分可以在载体上淤浆化到反应器中,而其它催化剂或组分可以溶液形式提供。
在优选实施方案中,将催化剂体系以溶液或淤浆形式加入反应器中。烃类用于溶液或淤浆。例如溶液可以是甲苯、己烷、异戊烷或其组合,例如甲苯和异戊烷或甲苯和戊烷。典型的溶液将是0.02-0.05摩尔催化剂在烃载体中,优选异戊烷或己烷。
在另一个实施方案中,用于催化剂体系或其组分的载体是超临界流体,例如乙烷或丙烷。关于超临界流体作为催化剂加料剂的更详细信息参见EP 0 764 665 A2。
在另一个优选实施方案中,一种或全部催化剂与以催化剂、任何载体和硬脂酸盐重量为基准的最多6重量%硬脂酸金属盐组合(优选硬脂酸铝,更优选二硬脂酸铝),优选2-3重量%。在另一个实施方案中,将硬脂酸金属盐的溶液加入反应器中。这些试剂可以用催化剂进行干法滚抛,或可以含有或不含催化剂体系或其组分的溶液形式加入反应器中。在优选实施方案中,与活化剂组合的催化剂用1重量%二硬脂酸铝和/或2重量%抗静电剂如甲氧基化胺来滚抛,例如来自ICISpecialties,Bloomington Delaware的Witco的Kemamine AS-990。硬脂酸金属盐和/或抗静电剂可以在矿物油中淤浆化到反应器中,研磨成粉末,然后悬浮于矿物油中,再加入反应器中,或作为粉末直接吹入反应器中。
关于使用硬脂酸铝型添加剂的更详细信息可参见1998年7月10日递交的USSN09/113216,将其引入本文以供参考。
本发明的聚合方法
上述催化剂和催化剂体系适用于溶液、气相或淤浆聚合方法或其组合,最优选气相或淤浆相聚合方法。
在一个实施方案中,本发明涉及溶液、淤浆或气相聚合反应,包括一种或多种含2-30个碳原子、优选2-12个碳原子、更优选2-8个碳原子的单体进行聚合。优选的单体包括乙烯、丙烯、丁烯-1、戊烯-1、4-甲基-戊烯-1、己烯-1、辛烯-1、癸烯-1、3-甲基-戊烯-1和环烯烃或其组合中的一种或多种。其它单体可包括乙烯基单体,二烯烃例如二烯类,多烯,降冰片烯,降冰片二烯,乙烯基降冰片烯,亚乙基降冰片烯单体。优选制得乙烯的均聚物。在另一个实施方案中,制得乙烯与一种或多种上述单体的共聚物。
在另一个实施方案中,乙烯或丙烯与至少两种不同的共聚单体聚合形成三元共聚物。优选的共聚单体是含4-10个碳原子、更优选4-8个碳原子的α-烯烃单体任选地与至少一种二烯单体的组合。优选的三元共聚物包括组合,例如乙烯/丁烯-1/己烯-1,乙烯/丙烯/丁烯-1,丙烯/乙烯/己烯-1,乙烯/丙烯/降冰片烯等。
在特别优选的实施方案中,本发明方法涉及乙烯与至少一种含4-8个碳原子、优选4-7个碳原子的共聚单体进行聚合。特别是,共聚单体是丁烯-1、4-甲基-戊烯-1、3-甲基-戊烯-1、己烯-1和辛烯-1,最优选己烯-1、丁烯-1和辛烯-1。
通常在气相聚合方法中,使用连续循环,其中在反应器体系循环的一部分中,循环气流,也称为再循环气流或流动介质,在反应器中通过聚合热进行加热。通过反应器外部的冷却体系从另一部分循环中的循环组合物中除去该热量。通常,在用于制备聚合物的气相流化床方法中,使含有一种或多种单体的气流在催化剂存在下在反应条件下从流化床连续循环。从流化床排出气流,并循环返回到反应器中。同时,从反应器排出聚合物产物,并加入新鲜单体以代替已聚合的单体。(参见例如美国专利4543399、4588790、5028670、5317036、5352749、5405922、5436304、5453471、5462999、5616661和5668228,将其全部引入本文以供参考)。
气相方法中的反应器压力可从约10psig(69千帕)至约500psig(3448千帕)变化,优选从约100psig(690千帕)至约500psig(3448千帕),优选在约200psig(1379千帕)至约400psig(2759千帕)的范围内,更优选在约250psig(1724千帕)至约350psig(2414千帕)的范围内。
气相方法中的反应器温度可以从约30℃至约120℃变化,优选从约60℃至约115℃,更优选在约70℃至约110℃的范围内,最优选在约70℃至约95℃的范围内。在另一个实施方案中,当需要高密度聚乙烯时,反应器温度通常在70-105℃之间。
催化剂或催化剂体系在气相体系中的产率受主要单体分压的影响。优选的主要单体乙烯或丙烯、优选乙烯的摩尔百分率是约25-90摩尔%,共聚单体的分压在约20psia(138千帕)至约300psia(517千帕)的范围内,优选约75psia(517千帕)至约300psia(2069千帕),这是气相聚合方法中的典型条件。在一些体系中存在共聚单体也能提高产率。
在优选实施方案中,用于本发明的反应器是有能力的,本发明方法制备大于500lbs聚合物/小时(227千克/小时)至约200,000lbs/小时(90,900千克/小时)或更高的聚合物,优选大于1000lbs/小时(455千克/小时),更优选大于10,000lbs/小时(4540千克/小时),甚至更优选大于25,000lbs/小时(11300千克/小时),进一步更优选大于35,000lbs/小时(15,900千克/小时),进一步更优选大于50,000lbs/小时(22700千克/小时),并优选大于65,000lbs/小时(29,000千克/小时)至大于100,000lbs/小时(45,500千克/小时),最优选大于100,0001bs/小时(45,500千克/小时)。
可考虑用于本发明的其它气相方法包括美国专利5627242、5665818和5677375中所述的那些,以及欧洲专利申请EP-A-0 794 200、EP-A-0802 202和EP-B-634 421,将其全部引入本文以供参考。
淤浆聚合方法通常使用在约1-50大气压(15-735psi,103-5068千帕)范围内的压力或甚至更大,温度在0℃至约120℃的范围内。在淤浆聚合中,固体粒子聚合物的悬浮液在液体聚合稀释剂介质中形成,向其中加入乙烯和共聚单体以及催化剂。将包含稀释剂的悬浮液间歇或连续从反应器中移出,其中从聚合物中分离挥发性组分,并任选地在蒸馏之后循环到反应器中。在聚合介质中所用的液体稀释剂通常是含3-7个碳原子的烷烃,优选支化烷烃。所用的介质应该在聚合条件下是液体,并是较惰性的。当使用丙烷介质时,该方法必须在反应稀释剂的临界温度和压力以上进行操作。优选使用己烷或异丁烷介质。
在一个实施方案中,优选的本发明聚合技术指粒子形式的聚合,或淤浆方法,其中使温度保持低于聚合物成为溶液的温度。该技术是本领域公知的,描述于例如美国专利3248179,将其全部引入本文以供参考。在粒子形式的方法中,优选温度在约185°F(85℃)至约230°F(110℃)的范围内。两种用于淤浆法的优选聚合方法是使用环管反应器和多个串联、平行连接的搅拌反应器,或其组合。淤浆法的非限定性例子包括连续环管或搅拌釜方法。淤浆法的其它例子还描述于美国专利4613484中,将其全部引入本文以供参考。
在另一个实施方案中,淤浆法在环管反应器中连续操作。将在异丁烷中作为淤浆的催化剂或作为自由流动干粉末的催化剂有规律地注入反应器环管中,该环管本身填充有正在增长的聚合物粒子在含单体和共聚单体的异丁烷稀释剂中的循环淤浆。氢气任选地可作为分子量控制剂加入。反应器保持在约525psig至625psig(3620千帕至4309千帕)的压力和在约140°F至约220°F(约60℃至约104℃)范围内的温度下,取决于所需的聚合物密度。反应热量通过环管壁除去,因为大多数反应器是双夹套管的形式。使淤浆在有规律的间隔内从反应器排出或连续排到经加热的低压闪蒸器、旋转干燥器和氮气吹扫塔内,以除去异丁烷稀释剂和所有未反应的单体和共聚单体。然后将所得的烃自由粉末配混,以用于各种用途。
在另一个实施方案中,用于本发明淤浆法的反应器是有能力的,且本发明方法制备大于2000lbs聚合物/小时(907千克/小时),更优选大于5000lbs/小时(2268千克/小时),最优选大于10,000lbs/小时(4540千克/小时)。在另一个实施方案中,用于本发明方法的淤浆反应器制备大于15,000lbs聚合物/小时(6804千克/小时),优选大于25,000lbs/小时(11,340千克/小时)至约100,000lbs/小时(45,500千克/小时)。
在另一个实施方案中,本发明的淤浆法中总反应器压力在400psig(2758千帕)至800psig(5516千帕)的范围内,优选450psig(3103千帕)至约700psig(4827千帕),更优选500psig(3448千帕)至约650psig(4482千帕),最优选约525psig(3620千帕)至625psig(4309千帕)。
在另一个实施方案中,本发明的淤浆法中乙烯在反应器液体介质中的浓度在约1-10重量%范围内,优选约2-7重量%,更优选约2.5-6重量%,最优选约3-6重量%。
本发明的另一种方法是该方法,优选淤浆或气相方法在不存在或基本上没有任何清除剂下操作,例如三乙基铝、三甲基铝、三异丁基铝和三正己基铝和二乙基氯化铝,二丁基锌等。该方法描述于PCT公开WO96/08520和美国专利5712352中,将其全部引入本文以供参考。
在另一个实施方案中,该方法在清除剂的存在下进行。典型的清除剂包括三甲基铝、三异丁基铝和过量的铝氧烷或改性铝氧烷。
进料催化剂溶液组分的比例可以变化,以改变分子量和其它性能。例如,改变催化剂比率将改变流动指数、熔融指数、熔体流动速率和/或密度。例如,在由式I表示的催化剂与由式IV表示的催化剂组合的体系中,如果提高由式IV表示的催化剂的比例,则制得更多的较低分子量的材料,进而提高流动指数,改变分子量分布。在优选实施方案中,能制得较低分子量组分的催化剂的存在量使得能制备45-65重量%的最终聚合物产物。对于一些用途,例如薄膜,已发现55-35重量%(A)[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基与45-65重量%的(B)[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基的组合是有效的。
另一种改变分子量的方法是向体系中加入氢气提高氢气乙烯比率。控制密度的方法是改变共聚单体含量。
本文也提供控制分子量分布(Mw/Mn)、流动指数和/或密度的方法,包括在工业规模气相反应器(即,具有1500立方英尺(42.5米3)或更大的体积)中在线改变反应温度,和/或在均质混合的催化剂溶液中的催化剂比率,和/或氢气浓度,和/或活化剂对过渡金属的比率例如铝/锆比率。
注射和混合温度也提供改变产物性能的手段,因为温度影响活化和/或溶剂蒸发,并因此改变催化剂组成,进而改变最终产物。
活化的顺序和时机也提供改变催化剂组成的机会,进而改变最终产物。例如在含(A)[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基和(B)[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基的体系中,较高浓度的甲基铝氧烷将改变由这两种催化剂所得产物的平衡。这包括在活化和/或混合和/或运输和/或喷入反应器期间的较高浓度。相似地,我们已注意到增加在催化剂进料中的烃载体,可以提高所得较低分子量级分的量。
也可通过改变反应温度来改变产物。我们已注意到提高反应温度可以增加较高分子量组分的量,并在体积排除色谱图中的两种模式不寻常地互相靠近(即,当在较低温度下与同样体系比较时,Mw/Mn变低)。
也可通过改变反应器温度,在催化剂体系进入反应器之前改变催化剂体系的温度,改变催化剂对活化剂的比率,改变载体的体积和/或使过渡金属组分在被活化剂活化之前与溶剂接触来改变分子量分布。
在优选的实施方案中,第一种催化剂对第二种或其它催化剂的比率是5∶95至95∶5,优选25∶75至75∶25,甚至更优选40∶60至60∶40。
在另一个优选实施方案中,催化剂体系是液体形式,并被引入反应器内的树脂粒子贫区中。关于怎样将液体催化剂体系引入流化床聚合中粒子贫区的信息,请参见US5693727,将其引入本文以供参考。
典型的聚合例子是:在烃例如己烷或异戊烷(约1-10%铝)中的改性甲基铝氧烷与10-30重量%的[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基和70-90重量%的[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基悬浮在溶液中,然后干燥,用2重量%二硬脂酸铝滚抛,然后淤浆化到温度保持为约85-100℃的流化床气相反应器中,其中Al对Zr的比率为约500∶1至约1000∶1,将乙烯气体喷入反应器中,以保持70-100psi(0.5-0.7兆帕)的分压,然后使反应器运行约30分钟。回收具有0.01-10dg/分钟熔融指数的聚乙烯。
在优选实施方案中,所回收的聚烯烃通常具有在ASTM D-1238,条件E,在190℃下检测为1克/10分钟或更小的熔融指数。在优选实施方案中,聚烯烃是乙烯均聚物或共聚物。共聚单体优选是C3至C20直链、支链或环状单体,在一个实施方案中是C3至C12直链或支链α-烯烃,优选丙烯、己烯、戊烯、己烯、庚烯、辛烯、壬烯、癸烯、十二烯、4-甲基-戊烯-1、3-甲基-戊烯-1、3,5,5-三甲基己烯-1等。
在优选实施方案中,上述催化剂体系用于制备高密度聚乙烯,其密度为0.925-0.965克/厘米3(由ASTM 2839检测),和/或其熔融指数为1.0克/10分钟或更小(由ASTM D-1238,条件E,在190℃下检测)。在另一个实施方案中,上述催化剂体系用于制备0.85-0.924克/厘米3的聚乙烯。
在另一个实施方案中,由本发明制备的聚合物具有至少为10的分子量分布(Mw/Mn),优选至少15,优选至少20,甚至更优选至少为30。
另外,虽然不希望受限于任何理论,据信,由本发明制备的聚合物具有独特的优点,即两种聚合物产物均质共混,以致在聚合物粒子内的两种聚合物在从反应器排出时平均分布。未经加工、未经处理的粒状聚合物称为纯聚合物。然后将纯聚合物通过根据ASTM D 1921塑性材料的粒径(筛网分析)、方法A或PEG法507的标准筛网尺寸分离成级分。
| 筛网尺寸 | 收集的级分 | 级分名称 |
| 10目18目35目60目120目盘 | >2000微米2000-1000微米<1000-500微米<500-250微米<250-125微米<125微米 | 级分1级分2级分3级分4级分5级分6 |
然后检测个别级分(级分1、4、6)的物理性能。熔融指数根据ASTM1238,条件E,190℃检测。结晶度用下面实施例13所述的X射线衍射来检测。
本发明制备的聚合物的独特特性在于,不同级分的熔融指数不显著变化。在优选实施方案中,级分1、4和6的熔融指数的变化不大于20%相对值,优选不大于15%相对值,优选不大于10%相对值,优选不大于8%相对值,优选不大于6%相对值,优选不大于4%相对值。相对值指相对于级分1、4和6的平均值。
本发明制备的聚合物的另一个独特特性在于,不同级分的结晶度百分率不显著变化。在优选实施方案中,级分1、4和6的结晶度百分率的变化不大于6%相对值,优选不大于5%相对值,优选不大于4%相对值,优选不大于3%相对值,优选不大于2%相对值。相对值指相对于级分1、4和6的平均值。
本发明制备的聚合物的另一个独特特性在于,不同级分的Mw/Mn不显著变化。在优选实施方案中,级分1、4、5和6的Mw/Mn的变化不大于20%相对值,优选不大于10%相对值,优选不大于8%相对值,优选不大于6%相对值,优选不大于4%相对值,优选不大于2%相对值。相对值指相对于级分1、4和6的平均值。Mn和Mw通过凝胶渗透色谱在配有差示折光指数检测仪的waters150℃GPC仪器上检测。GPC柱通过实验一系列窄聚苯乙烯标准物来校正,用宽聚乙烯标准国家标准局1496来计算所述聚合物的分子量。
在另一个实施方案中,本发明制备的聚合物具有8或更大的Mw/Mn,优选10或更大,优选12或更大。
在另一个实施方案中,所得聚合物的参数是微晶形态学随粒径而变化。级分1优选具有0.5-1.5的纵横比,级分4具有1.2-4的纵横比,级分6具有约1.75-5的纵横比,前提是级分的差别至少为0.3,优选至少0.5,更优选至少1.0。在一个实施方案中,级分1的纵横比<级分4的纵横比<级分6的纵横比。纵横比是[<110>/<011>],如下面
实施例13所述。
在另一个优选实施方案中,根据本发明制备的聚合物含有10-90重量%的低分子量聚合物(低达50000或更低,优选40000或更低),优选20-80重量%,更优选40-60重量%,以聚合物的重量为基准。
在另一个实施方案中,发现所制备的聚烯烃具有至少两种分子量,以聚合物重量为基准大于20重量%存在。
在本发明的另一个实施方案中,所制得的聚合物是双模或多模的(在SEC图上)。双模或多模指聚合物的SEC图具有两个或更多的正斜率,两个或更多的负斜率,和三个或更多的拐点(拐点是第二条衍生曲线变为负值时的点),或者该图至少具有一个正斜率,一个负斜率,一个拐点,且正和/或负斜率的变化大于改变前斜率的20%。在另一个实施方案中,SEC图具有一个正斜率,一个负斜率,一个拐点,且Mw/Mn为10或更大,优选15或更大,更优选20或更大。SEC图由凝胶渗透色谱在配有差示折光指数检测仪的waters150℃ GPC仪器得到。该柱通过实验一系列窄聚苯乙烯标准物来校正,并用Mark Houwink系数来计算所述聚合物的分子量。
聚烯烃可制成薄膜、模塑制品、片材、管材等。薄膜可通过本领域公知的任何传统技术形成,包括挤出、共挤出、层合、吹塑和流延。薄膜可通过平膜或管式法获得,该方法可随后在膜平面上的单轴方向或在两个互相垂直方向上取向。用于聚合物成膜的特别优选方法包括在吹塑和流延薄膜线上挤出或共挤出。
所得的薄膜可进一步含有添加剂,例如润滑剂、防结块剂、抗氧化剂、颜料、填料、防雾剂、紫外光稳定剂、抗静电剂、高分子加工助剂、中和剂、润滑剂、表面活性剂、颜料、染料和成核剂。优选的添加剂包括二氧化硅、合成二氧化硅、二氧化钛、聚二甲基硅氧烷、碳酸钙、硬脂酸金属盐、硬脂酸钙、硬脂酸锌、滑石、BaSO4、硅藻土、蜡、碳黑、阻燃添加剂、低分子量树脂、玻璃珠等。添加剂可以本领域公知的常用有效量存在,例如0.001-10重量%。
用本发明聚合物所制备的薄膜具有极其优秀的外观性能。该薄膜具有低的凝胶含量和/或具有良好的浊度和光泽度。在优选实施方案中,1密耳薄膜(1.0密耳=0.25微米)的45°光泽度为7或更大,优选8或更大,由ASTM D 2745来检测。在优选的实施方案中,1密耳薄膜(1.0密耳=0.25微米)的浊度为75或更小,优选70或更小,由ASTM D 1003,条件A来检测。
实施例
I2和I21由ASTM 1238,条件E和F检测。
MFR熔体流动速率由ASTM 1238检测。
BBF(丁基支化率/1000碳原子)由红外光谱检测,如美国专利5527752所述。
PDI(多分散性指数)等于Mw/Mn,由体积排除色谱检测。
Mn和Mw由凝胶渗透色谱在配有差示折光指数检测仪的waters150℃GPC仪器上检测。该GPC柱通过实验一系列窄聚苯乙烯标准物来校正,并用Mark Houwink系数计算所述聚合物的分子量。
熔融指数(MI)通过根据ASTM 1238,条件E的步骤来检测。
熔融指数比率(MIR)是I21对I2的比率,根据ASTM D 1238的步骤检测。
密度根据ASTM D 1505检测。
实施例1
[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基]胺的制备

在干燥箱内,将22.45毫摩尔(6.34克)2-乙酰基吡啶(2,6-二异丙基苯基亚胺)加入配备有搅拌棒和隔膜的250毫升圆底烧瓶中。将该烧瓶密封,从干燥箱中移出,并置于氮气吹扫下。加入无水甲苯(50毫升),并搅拌溶解该配体。将该容器在湿冰浴中骤冷到0℃。在10分钟内滴加三甲基铝(Aldrich,2.0M在甲苯中)。反应温度不得超过10℃。当完成加入三甲基铝时,使该混合物缓慢加热到室温。然后将其置于油浴中,并加热到40℃25分钟。从油浴移开该容器,并置于冰浴中。将含100毫升5%KOH的滴液漏斗连接到烧瓶上。在1小时内将碱性物质滴加入反应中。将该混合物转移到另外的漏斗中。除去含水层。溶剂层用100毫升水洗涤,然后用100毫升盐水洗涤。红棕色液体产物用Na2SO4干燥,真空汽提,并在高真空下放置过夜。将80毫升红棕色液体转移到配备有搅拌棒的200毫升Schlenk烧瓶中。将带有干冰冷凝器的蒸馏头连接到烧瓶上。将该混合物进行真空蒸馏,得到约70克深黄色粘性液体产物。
实施例2
[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄
基的制备
在暗房和暗的干燥箱中,将实施例1中制得的5.0毫摩尔(1.45克)配体加入配备有搅拌棒的100毫升Schlenk管中。使配体溶解于5毫升甲苯中。向配备有搅拌棒的第二容器中加入5.5毫摩尔(2.5克)四苄基锆和10毫升甲苯。
将配体溶液转移到四苄基锆溶液中。该容器用铝箔覆盖,并使其在干燥箱中在室温下搅拌。在室温下6小时后将80毫升无水己烷加入反应溶液中,并使其搅拌过夜。将反应混合物从中等孔隙率的玻璃料中过滤,收集约2克浅黄色固体。
实施例3
[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基][2-甲
基-1-苯基-2-丙氧基]锆合二苄基的制备

向经烘箱干燥、冷却、吹扫和密封的GC管形瓶中加入0.10毫升无水丙酮。将GC管形瓶密封在壳形管形瓶中,并放入干燥箱中。在暗房和暗的干燥箱中,将实施例2中制得的2.0毫摩尔(1.3克)物质和9毫升甲苯加入配备有搅拌棒的100毫升Schlenk烧瓶中。向第二GC管形瓶中加入2.0毫摩尔(146微升)丙酮和1.0毫升甲苯。由吸管将丙酮/甲苯溶液逐滴加入搅拌下的[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基溶液中。该容器用铝箔覆盖,并使其在干燥箱中在室温下搅拌过夜。将反应溶液真空汽提成粘性的橙色残余物。加入无水己烷(20毫升),并将残余物剧烈搅拌,然后再次真空汽提成黄色-橙色玻璃。再加入己烷,并剧烈搅拌。将容器置于冷冻器(-24℃)中约2小时。将该混合物从中等孔隙率的玻璃料中过滤。收集浅黄色固体(0.8克)。缓慢小心地加入丙酮,且良好的混合是最好的。
实施例4
在实验室规模淤浆相反应器中制备一系列双模乙烯/己烯共聚物,使用由根据本发明实施例2和实施例3制备的配合物与改性甲基铝氧烷(MMAO)助催化剂类型3A(购自Akzo化学公司,商品名为改性甲基铝氧烷类型3A,公开为专利号US5041584)的混合催化剂组合物。
在每种情况下,催化剂组合物通过准备来自实施例2和实施例3的配合物在甲苯中的混合物,然后与MMAO溶液(7.0重量%Al在庚烷中)在0.1毫升1-己烯存在下接触来制备。聚合反应条件是85℃,85psi(586千帕)乙烯,43毫升1-己烯,0.5微摩尔Zr,且MMAO/Zr的摩尔比是1000∶1。配合物比率由实施例3制备的配合物对实施例2制备的配合物的摩尔比来表示。结果如下表1所示。
表1
| 配合物比率 | 活性克PF/毫摩尔催化剂/100psiC2/小时 | I2dg/分钟 | MFR | BBF每1000C’s | PDI |
| 100∶090∶1080∶2060∶4050∶50 | 139765291765175529235765189647 | 162.213.490.050.00850.012 | 1.8261.661,027317.4173.1 | 6.2810.087.549.9211.01 | 11.1825.8423.7428.2430.25 |
对表1中制备的树脂进行体积排除色谱检测。结果清楚地证明,随着来自实施例2的配合物的浓度升高,高分子量组分增加。来自实施例3和实施例2的配合物的相对量分别代表在这些双模树脂中的低和高分子量组分。这表明两种催化剂是高度相容性的。
实施例5
乙烯己烯共聚物在14英寸(35.6厘米)中试工厂气相反应器中制备,在85℃、220psi(1517千帕)下操作,该反应器具有水冷却式热交换器。乙烯以约55磅乙烯/小时(25千克/小时)的速率加入,己烯以1.4磅/小时(0.64千克/小时)加入,氢气以0.021磅/小时(0.01千克/小时)的速率加入,以制备约35磅/小时(15.9千克/小时)的聚合物。总反应器压力是350psi(2413千帕)。氮气以约3-7磅/小时(1.4-3.2千克)存在。反应器配备有压力通风系统,设置为1800磅/小时(818.2千克/小时),带有内径为0.055英寸(0.14厘米)的单孔自来喷嘴注射器。(压力通风系统是用于在流化床气相反应器中创造粒子贫区的设备。关于压力通风系统应用的更详细信息参见美国专利5693727)重复该步骤,并如表2所示改变反应温度、Al/Zr比率、反应温度、注射温度或烃进料载体中的一种或多种。
表2
| 实施例 | Rxn温度 | 重量%低Mw | 催化剂 | Mw/Mn | Al∶Zr比率 |
| A | 85 | 60 | 3 | 14 | 350∶1∷Al∶Zr |
| B | 90 | 57 | 3 | 16 | 360∶1∷Al∶Zr |
| C | 95 | 51 | 3 | 12 | 350∶1∷Al∶Zr |
| D | 105 | 35 | 3 | 11 | 350∶1∷Al∶Zr |
| E | 85 | 22 | 60/40 2∶3 | 450∶1∷Al∶Zr | |
| F | 85 | 70 | 3 | 72∶1∷Al∶Zr |
“重量%低Mw”是低分子量物质的重量%,由体积排除色谱用正态高斯解卷积log值表征。
实施例6
用实施例2和3制得的催化剂重复以上实施例,不同的是聚合条件是85℃,220psi(1517千帕)C2,500∶1Al/Zr,催化剂进料10毫升/小时,MMAO进料300毫升/小时(2.3重量%Al在己烷中)。变化两种催化剂的比率。
表3
| 催化剂比率2/3 | 活性(克PE/毫摩尔Zr/小时) | I2dg/分钟 | MFR | 密度克/厘米3 |
| 60/4040/6020/800/10080/20 | 1565117929169051606140048 | 0.1960.1500.1650.1670.150 | 51.4759.8963.4576.2752.08 | 0.94550.94750.94980.9510.9422 |
实施例7
两种乙烯己烯共聚物在8英尺(2.4米)直径的气相反应器(体积约为2000立方英尺)中制备,该反应器具有床高度为38英尺(11.6米)。乙烯进料速率是约8000-9000磅/小时(3636-4090千克/小时)。己烯进料速率是约200-230磅/小时(90.0-104.5千克/小时)。氢气进料速率是约1-2磅/小时(2.2-4.4千克/小时)。共聚物以8000-9000磅/小时(3636-4090千克/小时)制备。将30-60磅/小时(13.6-27.3千克/小时)氮气加入反应器中。反应器配备有压力通风系统,设置为50000磅/小时(22727千克/小时),以及直径为0.125英寸(0.32厘米)缩减到直径为0.05英寸(0.13厘米)中心孔的三孔喷嘴,以及距离垂直于气流的喷嘴端0.30英寸(0.76厘米)和5/64英寸(0.20厘米)宽的两个其它孔。循环气体速率是约2-2.2英尺/秒(60-67厘米/秒)。注射温度是第一次实验为22℃,第二次实验为80℃。催化剂是实施例3制备的催化剂与2重量%改性甲基铝氧烷3A以Al∶Zr比率为150∶1组合。第一次实验制得具有44重量%较低分子量部分的乙烯己烯共聚物,第二次实验制得具有36重量%较低分子量部分的乙烯己烯共聚物。
实施例8
实施例2和3中所得化合物在甲苯中的五种0.02摩尔溶液以80/20、60/40、40/60、20/80和0/100的比率制备。根据实施例5的步骤用改性甲基铝氧烷3A作为助催化剂将其聚合。床温度保持为85℃。乙烯分压是220psi(1537千帕),Al∶Zr比率为500∶1。
表4
| 80/20 | 60/40 | 40/60 | 20/80 | 0/100 | |
| C6/C2比率(×10-3) | 8.1-9.1 | 6.6-7.7 | 6.1-6.7 | 5.4-5.6 | 5.4-5.7 |
| H2/C2比率(×10-3) | 23.7-25.5 | 17.5-18.6 | 14.0 | 12.3-12.6 | 12.1-12.2 |
| 生产速率(pph) | 28 | 26 | 26 | 26 | 27 |
| 活性克PE/毫摩尔Zr/小时 | 19000 | 17500 | 15000 | 16100 | 17500 |
| 熔融指数dg/分钟 | 0.15-0.26 | 0.17-0.26 | 0.13-0.34 | 0.12-0.17 | 0.16-0.21 |
| 流动指数dg/分钟 | 8.07-12.3 | 0.95-12.88 | 8.81-14.07 | 9.68-10.95 | 12.53-14.50 |
| 熔体流速MFR | 39.87-55.63 | 46.37-64.39 | 32.96-65.75 | 61.24-81.87 | 70.73-77.83 |
| 密度克/毫升 | 0.942-0.944 | 0.945-0.947 | 0.947-0.950 | 0.950-0.951 | 0.9951-0.952 |
实施例9
[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基][3-苄基-
3-戊氧基]锆合二苄基的合成
将二乙基酮(40毫摩尔,4.0毫升,Aldrich,3-戊酮,99.5%,[86.13])溶解于96毫升无水甲苯中。将二乙基酮溶液缓慢加入实施例2所得配合物的搅拌溶液(400毫升,0.125M在甲苯中)中。将所得溶液搅拌过夜。
实施例10
[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基][2-苄基-
2-丁氧基]锆合二苄基的合成
将甲基乙基酮(40毫摩尔,3.6毫升,Aldrich,2-丁酮,99.5%)溶解于100毫升无水甲苯中。将甲基乙基酮溶液缓慢加入实施例2所得配合物的搅拌溶液(400毫升,0.125M在甲苯中)中。将所得溶液搅拌过夜。
实施例11
在干燥箱中,将1-己烯(0.1毫升,氧化铝干燥)加入经烘箱干燥的4英钱玻璃管形瓶中。将来自实施例2的配合物(0.25微摩尔,2.0微升,在甲苯中的0.125M溶液),和实施例9制备的配合物(0.25微摩尔,3.7微升,在氘化苯中的0.067M溶液)加入1-己烯中,得到浅黄色溶液。然后将MMAO型3A(0.25毫摩尔)加入管形瓶中,得到浅黄色反应溶液。将反应溶液加入含600毫升正己烷、43毫升1-己烯和0.13毫升(0.25毫摩尔)MMAO型3A的反应器中,并在70℃、85psi乙烯和10psi氢气下操作30分钟。该反应得到26.3克聚乙烯树脂(活性=123765克聚乙烯/毫摩尔Zr/小时/100psi乙烯,I2=28.49,I21=838,MFR=29.4,BBF=7.63)。体积排除色谱(SEC)显示以下分子量结果:Mn=12611,Mw=50585,PDI=4.01。 SEC图如图6所示。
实施例12
在于燥箱中,将1-己烯(0.1毫升,氧化铝干燥)加入经烘箱干燥的4英钱玻璃管形瓶中。将来自实施例2的配合物(0.25微摩尔,在甲苯中的0.125M溶液),和来自实施例10的配合物(0.25微摩尔,在甲苯中的0.080M溶液)加入1-己烯中,得到浅黄色溶液。然后将MMAO型3A(0.25毫摩尔)加入管形瓶中,得到浅黄色反应溶液。将反应溶液加入含600毫升正己烷、43毫升1-己烯和0.13毫升(0.25毫摩尔)MMAO型3A的1升淤浆反应器中,并在70℃、85psi乙烯和10psi氢气下操作30分钟。该反应得到30.7克树脂(活性=144471克聚乙烯/毫摩尔Zr/小时/100psi乙烯,I2=11.47,I21=468,MFR=40.8,BBF=7.53)。体积排除色谱(SEC)显示以下分子量结果:Mn=12794,Mw=62404,PDI=4.88。SEC图如图7所示。
实施例13
根据实施例5的步骤制备密度为约0.946克/毫升的乙烯己烯共聚物(A),不同的是来自实施例3的催化剂以9毫升/小时的速率加入,来自实施例2的催化剂以1毫升/小时的速率加入(90∶10比率)。己烯以约0.8-0.9lbs(0.36-0.41千克)/小时的速率加入。乙烯以约41-43磅(18.5-19.4千克)/小时的速率加入。氢气以约17-20毫磅(7.7-9.1克)/小时的速率加入,己烷载体以约100毫升/小时的速率加入。乙烯的分压是120psi(827千帕),反应器温度是75℃。铝锆比率是Al∶Zr∷300∶1。
用同样的步骤制备另一种乙烯己烯共聚物(B),不同的是仅仅将来自实施例3的催化剂加入反应器中(10毫升/小时),反应器温度是95℃,氢气以12-14毫磅(5.4-6.4克)/小时的速率加入,乙烯以41-45磅(18.5-20.3千克)/小时的速率加入,乙烯的分压是220psi(827千帕)。共聚物的密度是0.951克/毫升。
用同样的步骤制备另一种乙烯己烯共聚物(C),不同的是仅仅将来自实施例3的催化剂加入反应器中(12毫升/小时),反应器温度是95℃,氢气以12-13毫磅(5.4-5.9克)/小时的速率加入,乙烯以44-47磅(19.9-21.2千克)/小时的速率加入,乙烯的分压是220psi(827千帕),己烷载体以70毫升/小时的速率加入。共聚物的密度是0.951克/毫升。
然后用以下筛网将这些共聚物分级:
| 筛网尺寸 | 收集的级分 | 级分名称 |
| 10目18目35目60目120目盘 | >2000微米2000-1000微米<1000-500微米<500-250微米<250-125微米<125微米 | 级分1级分2级分3级分4级分5级分6 |
然后如表A、B、C和D中所报告表征这些级分。用于X射线衍射数据收集的实验步骤和用于结晶度百分率和微晶形态学分析的技术概括
X射线衍射数据收集:
全部数据收集的条件是,Siemens GADDS X射线衍射(XRD)装置配备有CopperX射线管源(λ=1.54056_),在40kV和40mA的电源设置下操作,石墨单色仪,在样品前的0.3毫米准直管,以及多线路通用区域衍射检测器(GADD),样品-检测器的距离是15厘米。将大的个别粒子粘在细玻璃棒上,这使粒子悬浮并在电子束中旋转,且不受玻璃棒或胶的影响。细粒子置于用于分析的细二氧化硅毛细管中。所有数据收集在透射模式为电子束在样品后3厘米处停止下进行。在三种不同的衍射计设置下收集光谱,检测器覆盖以下2θ区域,4°-34°,32°-58°和56°-76°,收集时间通常是600秒/区。这三个不同的区域进行校正(变形校正),然后合并在一起形成覆盖4°-76°2θ的单个光谱。在样品-检测器距离15厘米处的GADDS体系提供Debye环的120°弧度视野。这清楚地显示沿着环的均匀强度,表示完全无规均匀的结构,显示不存在优选的取向或纹理。
曲线拟合:
由GADDS体系提供的全部分布曲线拟合程序用于对光谱的峰形拟合。对于大粒子,在同样条件下的空气散射花纹在拟合之前从实验数据中减去,在小粒子的情况下,结合空白二氧化硅毛细管和空气散射的光谱在拟合之前从实验数据中减去。四种组分用于拟合衍射数据;(i)衍射峰用具有混合参数为1的假-voigt轮廓-形状-函数来拟合,(ii)无定形峰用具有峰的FWHM(总半宽最大值)和基于无定形散射模拟的固定峰位的Lorentzian轮廓-形状-函数来拟合,(iii)次晶组分用具有指数50的Pearson VII轮廓-形状-函数来拟合和(iv)用于康普顿散射的线性拟合。
在康普顿散射从数据中减掉后,在图案中的总衍射强度与三种组分拟合,(1)无定形强度Iam,(2)次晶组分Ipara和(3)晶体衍射峰Ixal。以下表达式用于计算结晶度百分率:
结晶度百分率=Ixal/(Iam+Ipara+Ixal)×100
微晶尺寸:
微晶尺寸用在粒状聚乙烯树脂的X射线衍射图案中{110}和{011}反射的在最大强度一半下的峰宽来计算。这两个值使得可以用Scherrer方程式计算在给定晶体学方向中微晶的尺寸:
微晶尺寸(t)=0.9λ/B’cosθB
B’=B检测值-B仪器变宽其中λ是入射X射线的波长,B’是在最大强度一半下校正的峰宽弧度(仪器电子束增宽组分用国家标准局SRM 660 Lanthanum Hexaboride样品检测),θB是峰位,t是在基于反射方面的晶体学方向上的晶体尺寸。<110>反射提供在<110>方向上的微晶尺寸信息,该方向位于PE正交晶体的ab平面内,相似地,{011}反射提供在<011>方向上的微晶尺寸数据,该方向位于具有c-轴组分的bc平面内,从而得到关于微晶厚度的信息。
所以,在这两个方向上的微晶尺寸大小可通过定义为<110>/<011>的形状因子来提供关于微晶形态学的信息。当该比率大时,晶体在ab平面内是大的,但在bc平面内薄,所以它们是“片状的”,当该比率达到1时,两种尺寸相似,从而微晶形态学更多是立方形或球形的。
表A提供所有三种研究样品的X射线衍射数据。发现结晶度不随粒径而变化,其中小粒子具有与较大粒子相同的结晶度百分率。
观察到在ab平面内的微晶尺寸随着两种样品粒径的增加而单调下降。相反,观察到在bc平面(c轴组分的估计值)内的微晶尺寸随着粒径的增加而单调增加。
这些结果表明,微晶形状随着粒径而变化。在小粒子中的微晶形状更多是“片状”,纵横比[<110>/<011>]≌2.2-3.5,随着粒子变大,纵横比向≌1-1.5的方向移动,表明微晶更多是立方形/球形的。因此,随着粒子尺寸从PAN(<125微米)增加到10目(>2000微米),包埋于粒状树脂中的微晶相畴的形状从“片状”向立方形/球形变化。粒子可以视为具有分级型微观结构。
在该催化剂体系中观察到的性质正是分级微观结构的例子。相反的结构可在中心形成立方形/球形微晶,然后转变成更为“片状”的形态。另一种微晶形态学可以是“棒状”,其中形状因子变为<1。不同微晶形态学的分布可引起宽范围的可能的分级微观结构,其可通过催化剂类型来控制。
表A-实施例13样品的X射线衍射数据
| 样品 | %Xal | %Para | %Am | FWHM(110)° | FWHM(011)° | Xal尺寸(110)_ | Xal尺寸(011)_ |
| A | |||||||
| 10-目-1 | 55.8 | 4.3 | 39.9 | 0.4086 | 0.5883 | 197.7 | 143.4 |
| 10-目-2 | 56.2 | 4.7 | 39.1 | 0.3926 | 0.5883 | 205.8 | 143.4 |
| 10-目-3 | 56.1 | 5.3 | 38.6 | 0.4091 | 0.5883 | 197.5 | 143.4 |
| 60-目-1 | 55.6 | 6.5 | 37.8 | 0.3557 | 0.6647 | 227.2 | 126.9 |
| PAN-1 | 53.7 | 3.9 | 42.4 | 0.3510 | 0.8433 | 230.2 | 100.0 |
| PAN-2 | 55.1 | 5.6 | 39.4 | 0.3450 | 0.7190 | 234.2 | 117.3 |
| Av.(10目) | 56.0 | 4.8 | 39.2 | 0.40347 | 0.5883 | 200.4 | 143.4 |
| Stdev(10) | 0.2 | 0.50 | 0.6 | 0.0094 | 0.0000 | 4.7 | 0.0 |
| Av.(60/盘) | 54.8 | 5.3 | 39.9 | 0.3506 | 0.7423 | 230.5 | 114.7 |
| Stdev(60/盘) | 1.0 | 1.3 | 2.3 | 0.0054 | 0.0916 | 3.5 | 13.6 |
| C | |||||||
| 10-目-1 | 61.9 | 4.1 | 34.0 | 0.3028 | 0.4678 | 266.9 | 180.3 |
| 10-目-2 | 60.8 | 3.1 | 36.1 | 0.2975 | 0.4678 | 271.6 | 180.3 |
| 10-目-3 | 60.0 | 3.3 | 36.7 | 0.2970 | 0.4695 | 272.1 | 179.6 |
| 60-目-1 | 61.5 | 5.3 | 33.2 | 0.2672 | 0.5274 | 302.4 | 159.9 |
| PAN-1 | 59.3 | 3.8 | 36.9 | 0.2393 | 0.5477 | 337.7 | 154.0 |
| PAN-2 | 59.9 | 6.9 | 33.2 | 0.2517 | 0.5875 | 321.0 | 143.6 |
| Av.(10目) | 60.92 | 3.5 | 35.6 | 0.2991 | 0.4684 | 270.2 | 180.1 |
| Stdev(10) | 1.0 | 0.5 | 1.4 | 0.0032 | 0.0010 | 2.9 | 0.4 |
| Av.(60/盘) | 60.2 | 5.3 | 34.4 | 0.2527 | 0.5542 | 320.4 | 152.5 |
| Stdev(60/盘) | 1.1 | 1.5 | 2.1 | 0.0140 | 0.0306 | 17.6 | 8.3 |
| B | |||||||
| 10-目-1 | 63.4 | 7.9 | 28.6 | 0.2931 | 0.3638 | 275.9 | 229.2 |
| 10-目-2 | 65.2 | 6.8 | 28.0 | 0.2278 | 0.3638 | 355.0 | 229.2 |
| 10-目-3 | 63.6 | 6.8 | 29.7 | 0.2518 | 0.5895 | 321.2 | 143.3 |
| 60-目-1 | 62.7 | 6.1 | 31.2 | 0.1800 | 0.6451 | 449.3 | 131.0 |
| PAN-1 | 64.7 | 6.7 | 28.6 | 0.1768 | 0.6451 | 457.4 | 131.0 |
| PAN-2 | 65.9 | 6.9 | 28.2 | 0.1754 | 0.6451 | 461.1 | 131.0 |
| Av.(10目) | 64.1 | 7.2 | 28.8 | 0.2576 | 0.4422 | 317.4 | 200.6 |
| Stdev(10) | 1.00 | 0.7 | 0.8 | 0.0330 | 0.1275 | 39.7 | 49.6 |
| Av.(60/盘) | 64.1 | 6.6 | 29.3 | 0.1774 | 0.6451 | 455.9 | 131.0 |
| Stdev(60/盘) | 1.2 | 0.4 | 1.6 | 0.0024 | 0.0000 | 6.0 | 0.0 |
对于表B、C和D,CHMS指高分子量物质(大于500,000),CLMS指低分子量物质(小于3000),VLD指很低的密度,LD指低密度,HD指高密度,CCLDI指可结晶链长度分布指数,并在美国专利5698427中定义。CDI(50)指用于衡量多少聚合物在中值任一侧25%内的组成分布指数。CDI(100)用于衡量多少聚合物在中值任一侧50%内的组成分布指数。两种组成分布用温度升高洗提分级(TREF)技术检测,如Wild等(J.Polym.Sci.Phys.Ed.第20卷,p441-445(1982))所述。将聚合物在溶剂例如1,2,4-三氯苯中的洗提溶液在高温下装入填充柱内。使该柱以0.1℃/分钟缓慢冷却到环境温度。在缓慢冷却过程中,乙烯聚合物结晶到填充物上,以便随温度降低而增加支化(降低结晶度)。冷却后,将该柱以0.7℃/分钟再加热,使恒定溶剂从该柱流过,并用红外浓度检测仪监测流出液。
CDI(100)和CDI(50)指数与“组成分布宽度指数”(CDBI)相似。CDBI定义为,具有在中值总摩尔共聚单体含量的50%(±25%)内的共聚单体含量的共聚物链重量%(参见美国专利5470811)。CDBI与CDI(50)指数之间的不同在于CDI(50)使用平均支化率(或共聚单体含量)代替中值共聚单体含量。CDI(50)如下由TREF数据通过将洗提温度转化成支化率来确定:
支化率表示为沿聚合物主链的支链之间的平均距离(在CH2单元中),或表示为可结晶链长度(L),其中,
使用与分子量分布相似的分布矩,可确定Li的数均(Ln)和重均(Lw)矩,其中:
Ln=1/∑i(wi/Li) 和Lw=∑iwiLiwi是聚合物组分i的重量分数,该组分在两个相邻支化点之间具有平均主链间隔Li。那么,组成分布指数或可结晶链长度分布指数(CCLDI)定义为:
CCLDI=Lw/Ln
平均支化率由下式计算:
其中Wi和bi分别是TREF色谱图的每片i的重量分数和支化率。CDI(50)然后通过确定在BF的±25%内所含的累积重量分数来计算。CDI(100)定义为在BF的±50%内所含的累积重量分数。
表B聚合物A的表征数据
| 级分 | 1 | 4 | 5 | 6 |
| Mn | 10,604 | 8,320 | 8,815 | 8,831 |
| Mw | 230,395 | 227,935 | 234,524 | 247,901 |
| Mw/Mn | 19.2 | 27.4 | 26.6 | 28.1 |
| CHMS(%) | 11.3 | 13.1 | 13.7 | 14.5 |
| CLMS(%) | 1.3 | 2.2 | 2.0 | 2.0 |
| VLD(%) | 3.5 | 5.7 | 3.6 | 7 |
| LD(%) | 5.8 | 7.6 | 4.9 | 8.8 |
| HD(%) | 66.1 | 62 | 67.7 | 64.1 |
| CCLDI | 10.9 | 12.4 | 10.6 | 13.9 |
| CDI(50)(%) | 9.8 | 10.2 | 8.7 | 10 |
| CDL(100)(%) | 19.5 | 20.2 | 17.1 | 19.7 |
表C聚合物B的表征数据
| 级分 | 1 | 3 | 4 | 5 | 6 |
| Mn | 12,335 | 11,486 | 10,957 | 10,408 | 10,400 |
| Mw | 285,402 | 236,008 | 249,496 | 244,990 | 248,000 |
| Mw/Mn | 23.1 | 22.9 | 22.8 | 23.5 | 23.8 |
| CHMS(%) | 17.1 | 15.9 | 15.3 | 15.0 | 15.2 |
| CLMS(%) | 0.5 | 0.6 | 0.7 | 0.7 | 0.73 |
| VLD(%) | 4.2 | 5.9 | 2.5 | 3.8 | 2.3 |
| LD(%) | 5.7 | 7.6 | 3.8 | 5.5 | 3.8 |
| HD(%) | 65.5 | 60.9 | 63.8 | 58 | 62.6 |
| CCLDI | 11 | 11.7 | 9.7 | 10.5 | 9.7 |
| CDI(50)(%) | 11 | 13.9 | 12.5 | 15.5 | 14.1 |
| CDL(100)(%) | 20.6 | 26.5 | 23.5 | 28.8 | 25.3 |
表D聚合物C的表征数据
| 级分 | 1 | 4 | 5 | 6 |
| Mn | 13,114 | 13,081 | 11,451 | 11.328 |
| Mw | 243,241 | 236,908 | 219,370 | 217,120 |
| Mw/Mn | 18.5 | 18.2 | 19.2 | 19.2 |
| CHMS(%) | 13.7 | 13.6 | 12.6 | 12.5 |
| CLMS(%) | 0.7 | 0.6 | 0.9 | 1.0 |
| VLD(%) | 1.5 | 2.5 | 1.8 | 3.9 |
| LD(%) | 1.9 | 3.3 | 2.9 | 4.5 |
| HD(%) | 84.3 | 77 | 78.6 | 75.4 |
| CCLDI | 6.6 | 8.4 | 8.1 | 9.2 |
| CDI(50)(%) | 4.1 | 9.2 | 8.3 | 11.8 |
| CDL(100)(%) | 9.3 | 17.7 | 16.11 | 21.5 |
实施例14
在实验室规模淤浆相反应器中制备双模乙烯/己烯共聚物,使用根据本发明实施例3制备的配合物与MMAO型3A助催化剂(购自Akzo化学公司,商品名为改性甲基铝氧烷类型3A,公开为专利号US5,041,584)。在该实验中,使MMAO3A在进行聚合反应之前在甲苯中与实施例3制备的配合物反应4小时。
在干燥箱中,将甲苯(0.4毫升,氧化铝干燥)加入经烘箱干燥的4英钱玻璃管形瓶中。将实施例3制备的配合物(2.0微摩尔,0.067毫升在氘化苯中的0.03OM溶液)加入甲苯中,得到浅黄色溶液。然后将MMAO型3A(1.0毫摩尔,0.52毫升,1.89M,7.0重量%在庚烷溶剂中)加入管形瓶中,得到0.002026M浅黄色反应溶液。然后用铝箔密封管形瓶。在4.0小时后,将0.25毫升(0.5微摩尔ZR,0.25毫摩尔MMAO)反应溶液加入含600毫升正己烷、43毫升1-己烯和0.13毫升MMAO型3A(0.25毫摩尔,1.89M,7.0重量%在庚烷溶剂中)的反应器中。反应器在85℃和85psi(0.6兆帕)乙烯下操作30分钟。该反应得到44.3克聚乙烯树脂(活性=208471克聚乙烯/毫摩尔Zr/小时/100psi乙烯(将psi换算成兆帕时,将psi值乘以0.0068948),I2=20.9,I21=824.1,BBF=9.5丁基支链/1000CH2。体积排除色谱(SEC)显示以下分子量性质:Mn=9123,Mw=104,852,MW/Mn=11.49。SEC图如图8所示。
实施例15
在干燥箱中,将MMAO型3A(5.8毫升,10毫摩尔,1.74M,6.42重量%在庚烷中)加入经烘箱干燥的4英钱玻璃管形瓶中。将2-甲基-1-苯基-2-丙醇(15.5微升,0.1毫摩尔)在搅拌的同时逐滴加入MMAO中,得到透明溶液。
在干燥箱中,将甲苯(0.1毫升,氧化铝干燥)加入经烘箱干燥的4英钱玻璃管形瓶中。将实施例2制备的配合物(0.5微摩尔,6.3微升在甲苯中的0.08OM溶液)加入1-己烯中,得到浅黄色溶液。然后将上段制备的MMAO/2-甲基-1-苯基-2-丙醇溶液(0.25毫摩尔,0.13毫升)加入管形瓶中,得到浅黄色反应溶液。将管形瓶在油浴中于50℃加热5分钟,得到红棕色反应溶液。
将反应溶液加入含600毫升正己烷、43毫升1-己烯和0.13毫升(0.25毫摩尔)MMAO/2-甲基-1-苯基-2-丙醇溶液的1升淤浆反应器中,并在85℃和85psi(0.6兆帕)乙烯下操作30分钟。该反应得到16.3克聚乙烯树脂(活性=76706克聚乙烯/毫摩尔Zr/小时/100psi(0.7兆帕)乙烯,I2=0.069,I21=2.15,MFR=31.1,BBF=7.71)。体积排除色谱(SEC)显示以下分子量性质:Mn=54637,Mw=292411,PDI=5.35。
实施例16
在干燥箱中,将MMAO型3A(5.8毫升,10毫摩尔,1.74M,6.42重量%在庚烷中)加入经烘箱干燥的4英钱玻璃管形瓶中。将2-甲基-1-苯基-2-丙醇(15.5微升,0.1毫摩尔)在搅拌的同时逐滴加入MMAO中,得到透明溶液。
在干燥箱中,将甲苯(0.1毫升,氧化铝干燥)加入经烘箱干燥的4英钱玻璃管形瓶中。将实施例2制备的配合物(0.5微摩尔,6.3微升在甲苯中的0.08OM溶液)加入甲苯中,得到浅黄色溶液。然后将上段制备的MMAO/2-甲基-1-苯基-2-丙醇溶液(0.25毫摩尔,0.13毫升)加入管形瓶中,得到浅黄色反应溶液。将管形瓶在油浴中于50℃加热15分钟,得到红棕色反应溶液。
将反应溶液加入含600毫升正己烷、43毫升1-己烯和0.13毫升(0.25毫摩尔)MMAO/2-甲基-1-苯基-2-丙醇溶液的1升淤浆反应器中,并在85℃和85psi(0.6兆帕)乙烯下操作30分钟。该反应得到13.2克聚乙烯树脂(活性=62118克聚乙烯/毫摩尔Zr/小时/100psi(0.7兆帕)乙烯,I2=0.248,I21=7.85,MFR=31.6,BBF=6.30)。体积排除色谱(SEC)显示以下分子量性质:Mn=42411,Mw=205990,PDI=4.86。
将上述所有文献引入本文以供参考,包括任何优先权文件和/或检测方法。从上述一般描述和具体实施方案可见,虽然已说明和描述了本发明的形式,但可在不超出本发明精神和范围的情况下进行各种改进。因此,本发明不受其限制。
Claims (28)
1.一种烯烃聚合催化剂组合物,含有至少一种活化剂和下列组分中的至少一种:
a)至少两种不同的过渡金属化合物,各自由式I表示:
((Z)XAt(YJ))qMQn (I)
其中M是选自元素周期表的4、5或6族的金属;Q键接于M上,且每个Q是单价、二价或三价阴离子;X和Y键接于M上;X和Y独立地是碳或杂原子,前提是X和Y中的至少一个是杂原子,Y含于杂环J中,其中J含有2-50个非氢原子;Z键接于X上,其中Z含有1-50个非氢原子;t是0或1;当t是1时,A是桥接基团,连接X、Y或J中的至少一个;q是1或2;如果Q是单价阴离子,则n是M的氧化态减去q,如果Q是二价阴离子,则n是(M的氧化态-q)/2,或如果Q是三价阴离子,则n是(M的氧化态-q)/3,或
b)由上式I表示的过渡金属化合物与添加剂的产物,或
c)至少两种不同的化合物,其中每种化合物由下式表示:
((Z)XAt(YJ))qMQmTs
其中M是选自元素周期表的4、5或6族的金属;T键接于M上,并是13-16族元素,其中T还可以与一个或多个C1至C50基团或含一个或多个杂原子的一个或多个C1至C50基团连接,Q键接于M上,且每个Q是单价、二价或三价阴离子;X和Y键接于M上;X和Y独立地是碳或杂原子,前提是X和Y中的至少一个是杂原子,Y含于杂环J中,其中J含有2-50个非氢原子,Z含有1-50个非氢原子,t是0或1;当t是1时,A是桥接基团,连接X、Y或J中的至少一个;q是1或2;如果Q是单价阴离子,则m是M的氧化态减去q减去s,如果Q是二价阴离子,则m是(M的氧化态-q-s)/2,或如果Q是三价阴离子,则m是(M的氧化态-q-s)/3,s是1、2或3,其中所述活化剂和所述化合物已经反应至少15分钟;或
d)(a)中表示的一种化合物和(c)中表示的一种化合物的组合,
其中所述添加剂是由式R=O表示的烷氧化合物,其中R是C1至C100基团,且氧可以在沿R基团的任何点上连接,R基团除1-100个碳原子以外还可含有杂原子,
其中每个Q独立地选自硼酸根、卤素、氢、烷基、芳基、链烯基、烷基芳基、芳基烷基、具有1-20个碳原子的氢化羧基或苯氧基、氨基、磷化物、硫化物、甲硅烷基烷基、二酮根和羧酸根,和
其中所述活化剂是铝氧烷、非配位阴离子或改性甲基铝氧烷。
2.权利要求1的组合物,其中t是1,Z与一个或多个R’基团连接,其中R’基团独立地选自氢或直链、支链烷基或环烷基、链烯基、炔基或芳基,两个或更多的R’基团可以连接形成环状部分;J被两个或更多的R”基团所取代,其中R”基团独立地选自氢或直链、支链、环状的烷基,或链烯基、炔基、烷氧基、芳基或芳氧基,且两个或更多的R”基团可以连接形成环状部分。
3.权利要求2的组合物,其中R’或R”,或R’和R”二者与A连接。
4.权利要求1的组合物,其中T是氢化羧基、硼酸根、氨基或类环戊二烯基基团。
5.权利要求1的组合物,其中T是烷氧基、乙酰丙酮根、羧酸根、苯氧基或其组合。
6.权利要求1的组合物,其中n是2或3,且除了一个Q基团是氢化羧基、硼酸根或氨基外,第二种催化剂化合物与第一种催化剂相同。
7.权利要求1的组合物,其中n是2或3,且除了一个Q基团是烷氧基、苯氧基、乙酰丙酮根、羧酸根、环戊二烯基、芴基或茚基外,第二种催化剂化合物与第一种催化剂化合物相同。
8.权利要求1的组合物,其中n是2或3,且除了第二种催化剂化合物中的一个Q基团是第一种催化剂上类似Q基团的氢化羧基加合物外,第二种催化剂化合物与第一种催化剂化合物相同。
9.权利要求8的组合物,其中所述氢化羧基加合物是烷氧基加合物、硼酸根或氨基加合物、苯氧基加合物、乙酰丙酮根加合物、或羧酸根加合物。
10.权利要求1的组合物,其中所述添加剂包括丙酮、二苯甲酮、甲基乙基酮、二乙基酮、甲基异丁基酮、甲基异丙基酮、二异丙基酮、甲基叔丁基酮、苯乙酮、环己酮、环戊酮、苯甲醛、新戊醛、乙基正丙基酮和乙基异丙基酮中的一种或多种。
11.权利要求1的组合物,其中M是钛、锆或铪。
12.权利要求1的组合物,其中
X和Y独立地是氮、氧、硫或磷,和
Z是芳基,和
J是吡啶。
13.权利要求1的组合物,其中所述第一种催化剂化合物是[1-(2-吡啶基)N-1-甲基乙基][1-N-2,6-二异丙基苯基氨基]锆合三苄基,第二种催化剂化合物是[[1-(2-吡啶基)N-1-甲基乙基]-[1-N-2,6-二异丙基苯基氨基]][2-甲基-1-苯基-2-丙氧基]锆合二苄基。
14.一种聚合烯烃的方法,包括使一种或多种烯烃与根据权利要求1-13中任一项的催化剂组合物进行接触。
15.权利要求14的方法,其中所述烯烃包括一种或多种具有2-30个碳原子的单体。
16.权利要求14的方法,其中所述烯烃包括单独的乙烯或乙烯与丙烯、丁烯-1、戊烯-1、4-甲基-戊烯-1、己烯-1、辛烯-1、癸烯-1以及3-甲基-戊烯-1中的一种或多种的组合。
17.权利要求14、15或16的方法,其中第一种催化剂与第二种催化剂的比率是5∶95至95∶5。
18.一种通过权利要求14的方法制备的聚烯烃,其Mw/Mn为10或更大,并且其中聚烯烃级分1、4和6的熔融指数相对于级分1、4和6熔融指数的平均值的变化不大于20%,且其中级分1、4和6通过使用以下筛网获得:
筛网尺寸
收集的级分
级分名称
10目18目35目60目120目盘
>2000微米2000-1000微米1000-500微米<500-250微米<250-125微米<125微米
级分1级分2级分3级分4级分5级分6
19.权利要求18的聚烯烃,其中该聚烯烃是乙烯均聚物或乙烯与C3至C20直链、支链或环状烯烃的共聚物。
20.权利要求18的聚烯烃,其中该聚烯烃是乙烯与丙烯、丁烯、己烯、戊烯、庚烯、辛烯、壬烯、癸烯、4-甲基-戊烯-1、3-甲基-戊烯-1、3,5,5-三甲基己烯-1和十二烯中的一种或多种的共聚物。
21.权利要求18的聚烯烃,其中
a)聚烯烃级分1、4和6的结晶度百分率相对于级分1、4和6平均值的变化不大于6%,或
b)聚烯烃级分1、4和6的Mw/Mn相对于级分1、4和6平均值的变化不大于20%,或
c)级分1的纵横比为0.5-1.5,级分4的纵横比为1.2-4,级分6的纵横比为1.75-5,前提是级分的纵横比相差至少0.3,
d)上述a),b)和c)的任意组合。
22.权利要求21的聚烯烃,其中所述级分的纵横比相差至少0.5。
23.权利要求20的聚烯烃,其中Mw/Mn为20或更大。
24.权利要求21的聚烯烃,其中SEC图具有一个正斜率、一个负斜率、一个拐点且Mw/Mn为20或更大。
25.权利要求21的聚烯烃,其中SEC图具有两个或更多的正斜率、两个或更多的负斜率、三个或更多的拐点且Mw/Mn为10或更大。
26.权利要求21的聚乙烯,其中SEC图具有一个或多个正斜率、一个或多个负斜率、一个或多个拐点,Mw/Mn为10或更大,并且与变化前的斜率相比,任何斜线上斜率的变化为20%或更大。
27.一种通过权利要求20的聚烯烃进行吹塑、挤出或流延制得的薄膜。
28.一种通过权利要求20的聚烯烃进行吹塑或挤塑制得的模塑制品。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/216,215 | 1998-12-18 | ||
| US09/213,627 | 1998-12-18 | ||
| US09/216,163 | 1998-12-18 | ||
| US09/216,215 US6303719B1 (en) | 1998-12-18 | 1998-12-18 | Olefin polymerization catalyst system |
| US09/215,706 | 1998-12-18 | ||
| US09/215,706 US6268447B1 (en) | 1998-12-18 | 1998-12-18 | Olefin polymerization catalyst |
| US09/213,627 US6320002B1 (en) | 1997-07-02 | 1998-12-18 | Olefin polymerization catalyst |
| US09/216,163 US6265513B1 (en) | 1997-07-02 | 1998-12-18 | Polyolefin |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1335860A CN1335860A (zh) | 2002-02-13 |
| CN1241953C true CN1241953C (zh) | 2006-02-15 |
Family
ID=27498942
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 99814533 Expired - Lifetime CN1241953C (zh) | 1998-12-18 | 1999-11-19 | 烯烃聚合催化剂体系、聚合方法以及由其制备的聚合物 |
Country Status (12)
| Country | Link |
|---|---|
| EP (1) | EP1144465A2 (zh) |
| JP (1) | JP2002533483A (zh) |
| CN (1) | CN1241953C (zh) |
| AR (1) | AR025281A1 (zh) |
| AU (1) | AU756833B2 (zh) |
| BR (1) | BR9916313A (zh) |
| CA (1) | CA2350451A1 (zh) |
| CZ (1) | CZ20012224A3 (zh) |
| MX (1) | MXPA01006185A (zh) |
| NO (1) | NO20012982L (zh) |
| PL (1) | PL348825A1 (zh) |
| WO (1) | WO2000037511A2 (zh) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6900321B2 (en) | 2000-11-07 | 2005-05-31 | Symyx Technologies, Inc. | Substituted pyridyl amine complexes, and catalysts |
| KR100445185B1 (ko) * | 2000-11-20 | 2004-09-13 | 충남대학교산학협력단 | 올레핀 중합용 촉매 |
| AU2002337850A1 (en) * | 2001-11-15 | 2003-06-10 | Exxonmobil Chemical Patents Inc. | Polymerization monitoring and control using leading indicators |
| US7094848B2 (en) | 2003-05-13 | 2006-08-22 | Exxonmobil Chemical Patents Inc. | Olefin polymerization catalyst system |
| US7288596B2 (en) | 2003-12-22 | 2007-10-30 | Univation Technologies, Llc | Polyethylene compositions having improved tear properties |
| EP2112175A1 (en) | 2008-04-16 | 2009-10-28 | ExxonMobil Chemical Patents Inc. | Activator for metallocenes comprising one or more halogen substituted heterocyclic heteroatom containing ligand coordinated to an alumoxane |
| TW200936619A (en) | 2007-11-15 | 2009-09-01 | Univation Tech Llc | Polymerization catalysts, methods of making, methods of using, and polyolefin products made therefrom |
| EP2470573A1 (en) * | 2009-12-18 | 2012-07-04 | Univation Technologies, LLC | Methods for making polyolefin products having different shear thinning properties and haze |
| CN102453157B (zh) * | 2010-10-22 | 2014-12-10 | 中国石油化工股份有限公司 | 用于在单一反应器中制备宽/双峰聚乙烯的催化剂体系 |
| AT510547B1 (de) | 2011-01-03 | 2012-05-15 | Aba Hoertnagl Gmbh | Schnalle |
| US9637567B2 (en) * | 2011-05-13 | 2017-05-02 | Univation Technologies, Llc | Spray-dried catalyst compositions and polymerization processes employing the same |
| CN103917292B (zh) | 2011-11-08 | 2016-11-23 | 尤尼威蒂恩技术有限责任公司 | 制备催化剂体系的方法 |
| AT512540B1 (de) | 2012-06-28 | 2013-09-15 | Aba Hoertnagl Gmbh | Klemmvorrichtung zum Festklemmen zumindest eines Gurtes |
| AT512541B1 (de) | 2012-06-28 | 2013-09-15 | Aba Hoertnagl Gmbh | Spannvorrichtung |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT619325E (pt) * | 1993-04-07 | 2002-02-28 | Atofina Res | Processo e catalisadores para a producao de olefinas |
| GB9708487D0 (en) * | 1997-04-25 | 1997-06-18 | Bp Chem Int Ltd | Novel catalysts for olefin polymerisation |
| US6103657A (en) * | 1997-07-02 | 2000-08-15 | Union Carbide Chemicals & Plastics Technology Corporation | Catalyst for the production of olefin polymers |
-
1999
- 1999-11-19 WO PCT/US1999/027502 patent/WO2000037511A2/en not_active Application Discontinuation
- 1999-11-19 AU AU20272/00A patent/AU756833B2/en not_active Ceased
- 1999-11-19 CA CA002350451A patent/CA2350451A1/en not_active Abandoned
- 1999-11-19 PL PL99348825A patent/PL348825A1/xx not_active Application Discontinuation
- 1999-11-19 EP EP99963936A patent/EP1144465A2/en not_active Withdrawn
- 1999-11-19 MX MXPA01006185A patent/MXPA01006185A/es unknown
- 1999-11-19 BR BR9916313-6A patent/BR9916313A/pt not_active IP Right Cessation
- 1999-11-19 JP JP2000589580A patent/JP2002533483A/ja active Pending
- 1999-11-19 CN CN 99814533 patent/CN1241953C/zh not_active Expired - Lifetime
- 1999-11-19 CZ CZ20012224A patent/CZ20012224A3/cs unknown
- 1999-12-17 AR ARP990106514 patent/AR025281A1/es unknown
-
2001
- 2001-06-15 NO NO20012982A patent/NO20012982L/no not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| PL348825A1 (en) | 2002-06-17 |
| EP1144465A2 (en) | 2001-10-17 |
| MXPA01006185A (es) | 2005-04-19 |
| CA2350451A1 (en) | 2000-06-29 |
| WO2000037511A2 (en) | 2000-06-29 |
| CN1335860A (zh) | 2002-02-13 |
| BR9916313A (pt) | 2001-10-02 |
| NO20012982D0 (no) | 2001-06-15 |
| CZ20012224A3 (cs) | 2001-09-12 |
| AU756833B2 (en) | 2003-01-23 |
| AU2027200A (en) | 2000-07-12 |
| JP2002533483A (ja) | 2002-10-08 |
| NO20012982L (no) | 2001-08-14 |
| AR025281A1 (es) | 2002-11-20 |
| WO2000037511A3 (en) | 2000-11-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1053676C (zh) | 生产宽分子量分布的聚烯烃的方法 | |
| CN1283672C (zh) | 用于催化剂组分的离子化合物、包含其的分散体和固体加聚催化剂 | |
| CN1222548C (zh) | 膦亚胺-Cp载体催化剂 | |
| CN1206247C (zh) | 聚合方法 | |
| CN1185270C (zh) | 多成分催化剂溶液供料 | |
| CN1210312C (zh) | 含酮酰亚胺配位体的催化剂 | |
| CN1184242C (zh) | 催化剂体系及其在聚合反应方法中的应用 | |
| CN1054859C (zh) | 使用可溶性无载体催化剂进行气相聚合反应 | |
| CN1241953C (zh) | 烯烃聚合催化剂体系、聚合方法以及由其制备的聚合物 | |
| CN1275985C (zh) | 用非单中心/单中心催化剂结合物制备的乙烯/α-烯烃共聚物及其制备方法和用途 | |
| US20100160581A1 (en) | Catalyst Composition for Polymerization of Olefins, Polymerization Process Using the Same, and Method for Its Preparation | |
| CN1784431A (zh) | 高活性烯烃聚合催化剂和方法 | |
| CN100339403C (zh) | 用于烯烃聚合的卤代催化剂体系 | |
| CN1105674A (zh) | 乙烯聚合物的制备方法以及由乙烯聚合物制得的产物 | |
| CN1270595A (zh) | 改性铝氧烷催化剂活化剂 | |
| CN1968980A (zh) | 可控制组成分布的制备聚合物的方法 | |
| CN1487907A (zh) | 低聚或聚合乙烯的方法 | |
| CN1307597A (zh) | 催化剂组合物及其制备方法以及其在聚合方法中的应用 | |
| CN1535295A (zh) | 作为抗冲改性剂的茂金属生产的极低密聚乙烯或线型低密度聚乙烯 | |
| CN1288161C (zh) | 连接金属茂配合物,催化剂体系和使用该催化剂体系的烯烃聚合方法 | |
| EP2225251B1 (en) | Transition metal complexes, catalysts composition containing the same, and process for preparing ethylene homopolymers or copolymers of ethylene and alpha-olefins using the same | |
| JP5348421B2 (ja) | 遷移金属触媒系及びこれを用いたエチレン単独重合体又はエチレンとオレフィンとの共重合体の製造方法 | |
| CN1319995C (zh) | 反应器壁涂层及其形成方法 | |
| CN1649910A (zh) | 聚合反应催化剂 | |
| CN1264866C (zh) | 聚乙烯树脂、其制备方法及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| C10 | Entry into substantive examination | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CX01 | Expiry of patent term | ||
| CX01 | Expiry of patent term |
Granted publication date: 20060215 |