CN114058664A - 一种天然胶原的n端精准接枝改性方法 - Google Patents
一种天然胶原的n端精准接枝改性方法 Download PDFInfo
- Publication number
- CN114058664A CN114058664A CN202111352994.XA CN202111352994A CN114058664A CN 114058664 A CN114058664 A CN 114058664A CN 202111352994 A CN202111352994 A CN 202111352994A CN 114058664 A CN114058664 A CN 114058664A
- Authority
- CN
- China
- Prior art keywords
- collagen
- solution
- terminal
- grafting
- dialyzing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 108010035532 Collagen Proteins 0.000 title claims abstract description 156
- 102000008186 Collagen Human genes 0.000 title claims abstract description 156
- 229920001436 collagen Polymers 0.000 title claims abstract description 156
- 238000002715 modification method Methods 0.000 title claims abstract description 8
- 125000003277 amino group Chemical group 0.000 claims abstract description 37
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 29
- 238000000034 method Methods 0.000 claims abstract description 26
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 25
- 230000004048 modification Effects 0.000 claims abstract description 21
- 238000012986 modification Methods 0.000 claims abstract description 21
- 102000004190 Enzymes Human genes 0.000 claims abstract description 11
- 108090000790 Enzymes Proteins 0.000 claims abstract description 11
- 238000007112 amidation reaction Methods 0.000 claims abstract description 7
- 239000000243 solution Substances 0.000 claims description 50
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 42
- 238000000502 dialysis Methods 0.000 claims description 38
- 239000003153 chemical reaction reagent Substances 0.000 claims description 20
- 102000057297 Pepsin A Human genes 0.000 claims description 13
- 108090000284 Pepsin A Proteins 0.000 claims description 13
- 229940111202 pepsin Drugs 0.000 claims description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 12
- 239000008055 phosphate buffer solution Substances 0.000 claims description 12
- 229940088598 enzyme Drugs 0.000 claims description 10
- MFGALGYVFGDXIX-UHFFFAOYSA-N 2,3-Dimethylmaleic anhydride Chemical compound CC1=C(C)C(=O)OC1=O MFGALGYVFGDXIX-UHFFFAOYSA-N 0.000 claims description 9
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 claims description 9
- 229940014800 succinic anhydride Drugs 0.000 claims description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 8
- 239000004365 Protease Substances 0.000 claims description 6
- 238000004108 freeze drying Methods 0.000 claims description 6
- 108091005804 Peptidases Proteins 0.000 claims description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 4
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 4
- 230000001681 protective effect Effects 0.000 claims description 4
- 102000012422 Collagen Type I Human genes 0.000 claims description 3
- 108010022452 Collagen Type I Proteins 0.000 claims description 3
- 150000001875 compounds Chemical class 0.000 claims description 3
- 239000006184 cosolvent Substances 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 2
- 102000004142 Trypsin Human genes 0.000 claims description 2
- 108090000631 Trypsin Proteins 0.000 claims description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 claims description 2
- PQLVXDKIJBQVDF-UHFFFAOYSA-N acetic acid;hydrate Chemical compound O.CC(O)=O PQLVXDKIJBQVDF-UHFFFAOYSA-N 0.000 claims description 2
- 210000001361 achilles tendon Anatomy 0.000 claims description 2
- 238000004090 dissolution Methods 0.000 claims description 2
- 230000007071 enzymatic hydrolysis Effects 0.000 claims description 2
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 claims description 2
- 239000003960 organic solvent Substances 0.000 claims description 2
- -1 t-butoxycarbonyl compounds Chemical class 0.000 claims description 2
- 239000012588 trypsin Substances 0.000 claims description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 3
- 239000007864 aqueous solution Substances 0.000 claims 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 claims 1
- 239000002994 raw material Substances 0.000 abstract description 4
- 238000010511 deprotection reaction Methods 0.000 abstract description 3
- 238000001976 enzyme digestion Methods 0.000 abstract description 3
- 239000012620 biological material Substances 0.000 abstract description 2
- 238000007306 functionalization reaction Methods 0.000 abstract description 2
- 230000007062 hydrolysis Effects 0.000 abstract description 2
- 238000006460 hydrolysis reaction Methods 0.000 abstract description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 20
- 241000252230 Ctenopharyngodon idella Species 0.000 description 13
- 238000006243 chemical reaction Methods 0.000 description 13
- 125000000524 functional group Chemical group 0.000 description 11
- 239000000463 material Substances 0.000 description 11
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 10
- 239000007853 buffer solution Substances 0.000 description 10
- 238000003756 stirring Methods 0.000 description 8
- 238000005303 weighing Methods 0.000 description 8
- 229920003172 poly (isopropyl acrylamide) Polymers 0.000 description 5
- ATXASKQIXAJYLM-UHFFFAOYSA-N 1-hydroxypyrrolidine-2,5-dione;prop-2-enoic acid Chemical compound OC(=O)C=C.ON1C(=O)CCC1=O ATXASKQIXAJYLM-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 108010045569 atelocollagen Proteins 0.000 description 4
- 239000000515 collagen sponge Substances 0.000 description 4
- 230000003301 hydrolyzing effect Effects 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- 241001597062 Channa argus Species 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000003999 initiator Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229920000139 polyethylene terephthalate Polymers 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 229920002799 BoPET Polymers 0.000 description 2
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 description 2
- 230000009134 cell regulation Effects 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000001502 gel electrophoresis Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- DWFKOMDBEKIATP-UHFFFAOYSA-N n'-[2-[2-(dimethylamino)ethyl-methylamino]ethyl]-n,n,n'-trimethylethane-1,2-diamine Chemical compound CN(C)CCN(C)CCN(C)CCN(C)C DWFKOMDBEKIATP-UHFFFAOYSA-N 0.000 description 2
- QNILTEGFHQSKFF-UHFFFAOYSA-N n-propan-2-ylprop-2-enamide Chemical compound CC(C)NC(=O)C=C QNILTEGFHQSKFF-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 235000019419 proteases Nutrition 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000001338 self-assembly Methods 0.000 description 2
- NHJVRSWLHSJWIN-UHFFFAOYSA-N 2,4,6-trinitrobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O NHJVRSWLHSJWIN-UHFFFAOYSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000010382 chemical cross-linking Methods 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 description 1
- 239000003292 glue Substances 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000002211 ultraviolet spectrum Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/06—Preparation of peptides or proteins produced by the hydrolysis of a peptide bond, e.g. hydrolysate products
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/78—Connective tissue peptides, e.g. collagen, elastin, laminin, fibronectin, vitronectin or cold insoluble globulin [CIG]
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08H—DERIVATIVES OF NATURAL MACROMOLECULAR COMPOUNDS
- C08H1/00—Macromolecular products derived from proteins
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biophysics (AREA)
- General Chemical & Material Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Materials Engineering (AREA)
- Polymers & Plastics (AREA)
- Peptides Or Proteins (AREA)
Abstract
本发明属于生物材料技术领域,具体涉及一种天然胶原的N端精准接枝改性方法。本发明采用“全氨基保护→酶切端肽→N端精准接枝→脱保护”的新型策略,以具端肽结构的天然胶原为原料,首先采用保护基团保护胶原表面所有的活性氨基,进而采用酶水解法去除胶原的端肽结构,在胶原N端重新获得活性α‑氨基,通过酰胺化反应可以实现胶原N端氨基的精准接枝改性(侧链氨基已经被保护基保护),最后脱除胶原侧链氨基的保护基即可获得N端精准接枝改性胶原。该方法简单有效,实现了对天然胶原的精准接枝改性,为胶原的进一步功能化提供了技术支持,拓展了改性胶原的应用领域。
Description
技术领域
本发明属于生物材料技术领域,具体涉及一种天然胶原的N端精准接枝改性方法。
背景技术
胶原,一种广泛存在于脊椎动物体内的结构性蛋白质,其典型结构特征为由3条肽链相互缠绕形成的三螺旋结构,直径约1.5nm,长度约300nm。胶原因具有良好的生物相容性、可生物降解性、优异的机械性能等优势而被广泛的应用于生物医学和组织工程材料领域。随着组织工程、临床医学、医学美容等领域对胶原基材料的性能指标和功能多样化的要求越来越高,胶原固有的物性、成胶性能以及生物稳定性等已不能完全满足实践应用需求。因此,通过对胶原固有结构和性能进行改性成为胶原基材料领域发展的必由之路。
目前,胶原改性领域的研究主要分为以下几个方面:(1)通过化学交联剂进行交联改性,利用简单的化学反应,如酰胺化、醛类交联等实现胶原表面官能团之间的共价键结合,可以有效的提高胶原基材料的热稳定性、耐酶降解性和机械性能,但是无法赋予其多样化的功能;(2)采用小分子化合物或聚合物对胶原侧链官能团进行接枝改性,通过共价键将多样化的分子接枝到胶原侧链活性基团(氨基、羧基等)上,可以赋予胶原基材料额外的性能,有利于实现胶原基材料功能的多样化。此外,牢固稳定的共价键连接方式有效避免了相分离问题,因此,该方法在胶原材料领域备受关注。
但是,常规的胶原接枝改性策略仍存在接枝位点和接枝密度无法精准控制、改性胶原产物分子结构不均一等问题。胶原的侧链官能团与其固有自组装性能和细胞调控性能息息相关,无法控制接枝位点的常规接枝改性策略无疑会对胶原的固有性能带来极大的负面影响。已有研究表明,胶原肽链两端的官能团(N端的氨基和C端的羧基)不会对其自组装行为和细胞调控性能等产生明显影响。因此,实现对胶原分子末端官能团的精准改造对于新颖的改性胶原基材料开发无疑具有重要的科学价值和实践意义。目前,胶原接枝改性领域最常用的官能团为氨基,主要包含N端α-氨基和侧链赖氨酸的ε-氨基,虽然二者的pKa值不同,但因实际反应活性相差甚微而难以实现N端氨基的选择性接枝。
发明内容
为了解决上述问题,本发明提供了一种温和、简便、有效的天然胶原N端精准接枝改性方法,这种方法采用“全氨基保护→酶切端肽→N端精准接枝→脱保护”的策略,采用具端肽结构的天然胶原为原料,首先以氨基保护试剂保护胶原表面所有的活性氨基,进而采用酶水解法去除胶原的端肽结构,在胶原N端重新形成活性α-氨基,通过酰胺化反应可以实现胶原N端氨基的精准接枝改性(侧链氨基已经被保护基保护),最后脱除胶原侧链氨基的保护基即可获得N端精准接枝改性胶原。该方法获得的改性胶原不仅能够具有接枝分子的性能,而且因侧链官能团未受影响而不会影响胶原固有性能。
本发明采用如下的技术方案:
一种天然胶原的N端精准接枝改性方法,包括如下步骤:
1)在低温条件下,用醋酸水溶液溶解天然胶原,得到胶原浓度为1~10mg/mL的胶原溶液;
2)将步骤1所得胶原溶液用pH为8.0~10.0的磷酸盐缓冲溶液(即PBS)中透析12~48h,除去醋酸分子;
3)将氨基保护试剂滴加到步骤2所得溶液中,摇匀后,低温反应12~48h;
4)将步骤3所得溶液透析除去未反应的氨基保护试剂,得到侧链保护的改性胶原;
5)在步骤4所得侧链保护改性胶原溶液中,加入蛋白酶,在低温条件下水解除去胶原端肽结构,形成具有末端氨基/侧链保护的改性胶原;
6)将步骤5所得溶液透析纯化,得到具有末端氨基/侧链保护的改性胶原;
7)通过酰胺化反应,将接枝改性试剂加入步骤6所得溶液中,摇匀后,低温反应12~48h;
8)对将步骤7所得溶液依次采用酸性溶液和纯水透析、冻干后,待用。
进一步地,所述步骤1)中,所述天然胶原是从哺乳动物、鱼类、两栖动物的皮肤、跟腱组织中提取并分离纯化的具有完整三螺旋分子结构和端肽结构的天然Ⅰ型胶原。低温条件为溶解温度小于20℃(温度过高会导致胶原蛋白变性)。
进一步地,所述步骤2)中,将步骤1所得胶原溶液置于截留分子量为12000~15000道尔顿的透析袋,以pH为8.0~10.0的磷酸盐缓冲溶液为透析液,在低于20℃下透析12~48h,直至透析液pH不再降低为止,每隔3~4h更换一次透析液;
进一步地,所述步骤3)中,氨基保护试剂为能与氨基进行反应的常用保护试剂,包括但不限于:2,3-二甲基马来酸酐、丁二酸酐、叔丁氧羰基(Boc)。(若试剂水溶性不够,可适量添加有机溶剂,如乙醇、丙酮、二甲亚砜、1,4-二氧六环、N,N-二甲基甲酰胺等作为助溶剂),氨基保护试剂与胶原的质量比为(0.5~2):1,低温条件为溶解温度小于20℃。
进一步地,所述步骤4)中,所述透析操作采用截留分子量为12000~15000道尔顿的透析袋,以pH为7.0~10.0的PBS为透析液,在低于20℃下透析12~48 h,每隔3~4 h更换一次透析液。
进一步地,所述步骤5)中,所述蛋白酶为可用于胶原端肽去除但不影响胶原三螺旋结构的酶,包括但不限于:胃蛋白酶、胰蛋白酶、组织蛋白酶、木瓜蛋白酶、枯草杆菌蛋白酶以及复合酶,酶的浓度为10~50 Unit/g,低温条件为温度小于20℃,根据所使用的酶,选择合适的pH条件;
进一步地,所述步骤6)中,所述透析操作采用截留分子量为12000~15000道尔顿的透析袋,以pH为7.0~10.0的PBS为透析液,在低于20℃下透析12~48 h,每隔3~4 h更换一次透析液。
进一步地,所述步骤7)中,接枝改性试剂为可与氨基进行酰胺化反应的化学试剂,包括但不限于:末端为羧基或者活化状态羧基的化合物(脂肪酸、N-羟基琥珀酰亚胺丙烯酸酯);
进一步地,所述步骤8)中,所述透析操作采用截留分子量为12000~15000道尔顿的透析袋,在低于20℃下使用0.5mol/L的醋酸溶液透析12~48h,每隔3~4h更换一次透析液,再以纯水为透析液,透析12~48h,每隔3~4h更换一次透析液。
本发明的有益效果在于:
本发明采用“全氨基保护→酶切端肽→N端精准接枝→脱保护”的新型策略,以具端肽结构的天然胶原为原料,首先采用保护基团保护胶原表面所有的活性氨基,进而采用酶水解法去除胶原的端肽结构,在胶原N端重新获得活性α-氨基,通过酰胺化反应可以实现胶原N端氨基的精准接枝改性(侧链氨基已经被保护基保护),最后脱除胶原侧链氨基的保护基即可获得N端精准接枝改性胶原。该方法实现了对天然胶原N端氨基的精准接枝改性,为胶原的进一步功能化提供了技术支持,拓展了改性胶原的应用领域。
本发明方法获得的改性胶原不仅具有N端接枝分子的性能,而且因侧链官能团未被占用而不会影响胶原的固有性能。该方法可以实现胶原N端氨基接枝分子结构的多样化,所得产品在生物医学材料领域具有广泛应用。
附图说明
图1是本方法的制备过程示意图;
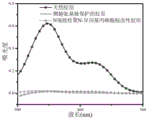
图2为三种胶原的UV图谱;
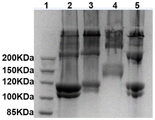
图3为胶原酶切端肽前后的凝胶电泳图;
图4为胶原修饰前后纳米孔道跨膜离子电流变化图;
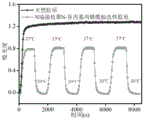
图5为胶原改性前后温度响应图。
具体实施方式
下面通过具体的实施例、对照例进一步说明本发明,但是,应当理解为,这些实施例仅仅是用于更详细具体地说明之用,而不应理解为用于以任何形式限制本发明。
实施例1
草鱼皮胶原的N端精准接枝改性方法,步骤如下:
(1)称取200mg草鱼皮胶原海绵样品,用0.5mol/L醋酸溶解配制浓度为2mg/mL的样品100mL;
(2)将配置好的胶原置于分子量为14000道尔顿的透析袋中,使用pH为9.0的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,得到透析好的天然草鱼皮胶原溶液;
(3)称取2,3-二甲基马来酸酐330mg溶解于1.2mL的二甲基亚砜中并分4次加入到天然草鱼皮胶原溶液中,4℃下搅拌反应24h;
(4)反应结束后将体系转移至分子量为14000道尔顿的透析袋中,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,透析完成后得到2,3-二甲基马来酸酐保护的胶原溶液;
(5)在上述体系中加入猪胃蛋白酶(20Unit/g),在10℃下反应12h,水解除去端肽结构,获得末端氨基,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液。
(6)透析完成后加入自主合成的自由基引发剂,4℃下搅拌反应24h后采用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液;
(7)透析完成后,加入100mgN-异丙基丙烯酰胺、189.42mg的溴化亚铜、0.37mL 1,1,4,7,10,10-六甲基三亚乙基四胺,在氮气保护下,20℃反应24h,获得N端接枝聚异丙基丙烯酰胺的改性胶原。反应结束后将体系转移至分子量为14000道尔顿的透析袋中,使用0.5mol/L的醋酸溶液在4℃下透析至体系蓝色褪去,每隔4h更换一次透析液,再使用纯水在4℃下透析至体系pH变成中性,每隔4h更换一次透析液;
(8)经透析后的溶液置于冷冻干燥机中,抽真空,在5分钟内系统绝对压力降低至20Pa,继续冷冻干燥48小时后得到冷冻干燥的N端接枝聚异丙基丙烯酰胺的改性胶原,待用。
实施例2
草鱼皮胶原的N端精准接枝改性方法(以丁二酸酐保护氨基),步骤如下:
(1)称取200mg草鱼皮胶原海绵样品,用0.5mol/L醋酸溶解配制浓度为2mg/mL的样品100mL;
(2)将配置好的胶原置于分子量为14000道尔顿的透析袋中,使用pH为9.0的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,得到透析好的天然草鱼皮胶原溶液;
(3)称取丁二酸酐330mg溶解于1.2mL的二甲基亚砜中并分4次加入到天然草鱼皮胶原溶液中,4℃下搅拌反应24h,反应结束后将体系转移至分子量为14000道尔顿的透析袋中,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,透析完成后得到丁二酸酐保护的胶原溶液;
(4)在上述体系中加入猪胃蛋白酶(20 Unit/g),在10℃下反应12h,水解除去端肽结构,获得末端氨基,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液;
(5)透析完成后加入自主合成的自由基引发剂,4℃下搅拌反应24h后采用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液;
(6)透析完成后,加入100mg N-异丙基丙烯酰胺、189.42mg的溴化亚铜、0.37mL 1,1,4,7,10,10-六甲基三亚乙基四胺,在氮气保护下,20℃反应24h,获得N端接枝聚异丙基丙烯酰胺的改性胶原。反应结束后将体系转移至分子量为14000道尔顿的透析袋中,使用0.5mol/L的醋酸溶液在4℃下透析至体系蓝色褪去,每隔4h更换一次透析液,再使用纯水在4℃下透析至体系pH变成中性,每隔4h更换一次透析液;
(7)经透析后的溶液置于冷冻干燥机中,抽真空,在5分钟内系统绝对压力降低至20Pa,继续冷冻干燥48小时后得到冷冻干燥的N端接枝聚异丙基丙烯酰胺的改性胶原,待用。
实施例3
草鱼皮胶原的N端精准接枝N-羟基琥珀酰亚胺丙烯酸酯的方法,步骤如下:
(1)称取200mg草鱼皮胶原海绵样品,用0.5mol/L醋酸溶解配制浓度为2mg/mL的样品100mL;
(2)将配置好的胶原置于分子量为14000道尔顿的透析袋中,使用pH为9.0的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,得到透析好的天然草鱼皮胶原溶液;
(3)称取丁二酸酐330mg溶解于1.2mL的二甲基亚砜中并分4次加入到天然草鱼皮胶原溶液中,4℃下搅拌反应24h,反应结束后将体系转移至分子量为14000道尔顿的透析袋中,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,透析完成后得到丁二酸酐保护的胶原溶液;
(4)在上述体系中加入猪胃蛋白酶(20 Unit/g),在10℃下反应12h,水解除去端肽结构,获得末端氨基,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液;
(5)透析完成后加入N-羟基琥珀酰亚胺丙烯酸酯(终浓度为50mg/mL),4℃下搅拌反应24h后使用0.5mol/L的醋酸溶液透析24h,每隔4h更换一次透析液,再使用纯水在4℃下透析至体系pH变成中性,每隔4h更换一次透析液;
(6)经透析后的溶液置于冷冻干燥机中,抽真空,在5分钟内系统绝对压力降低至20Pa,继续冷冻干燥48小时后得到冷冻干燥的N端丙烯酰基接枝改性胶原,待用。
实施例4
乌鳢鱼皮胶原的N端精准接枝改性方法,步骤如下:
(1)称取200mg乌鳢鱼皮胶原海绵样品,用0.5mol/L醋酸溶解配制浓度为2mg/mL的样品100mL;
(2)将配置好的胶原置于分子量为14000道尔顿的透析袋中,使用pH为9的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,得到透析好的天然乌鳢鱼皮胶原溶液;
(3)称取丁二酸酐330mg溶解于1.2mL的二甲基亚砜中并分4次加入到天然草鱼皮胶原溶液中,4℃下搅拌反应24h,反应结束后将体系转移至分子量为14000道尔顿的透析袋中,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液,透析完成后得到丁二酸酐保护的胶原溶液;
(4)在上述体系中加入猪胃蛋白酶(20 Unit/g),在10℃下反应12h,水解除去端肽结构,获得末端氨基,使用pH8.5的磷酸缓冲溶液在4℃下透析24h,每隔4h更换一次透析液;
(5)透析完成后加入N-羟基琥珀酰亚胺丙烯酸酯(终浓度为50mg/mL),4℃下搅拌反应24h后使用0.5mol/L的醋酸溶液透析24h,每隔4h更换一次透析液,再使用纯水在4℃下透析至体系pH变成中性,每隔4h更换一次透析液;
(6)经透析后的溶液置于冷冻干燥机中,抽真空,在5分钟内系统绝对压力降低至20Pa,继续冷冻干燥48小时后得到冷冻干燥的N端丙烯酰基接枝改性胶原,待用。
实施例5
游离氨基的接枝率实验,具体如下:
通过2,4,6-三硝基苯磺酸比色法测定天然胶原、2,3-二甲基马来酸酐保护侧链氨基的胶原、N端接枝聚异丙基丙烯酰胺/侧链保护的胶原中游离氨基的数量,胶原样品中游离氨基酸量越多,则在346nm处的吸收峰越强,以天然胶原的游离氨基数量为100%,则可推算接枝改性胶原中氨基的接枝率,如图2所示,侧链氨基被保护的胶原和N端接枝聚异丙基丙烯酰胺/侧链保护的胶原的氨基接枝率均为~97.1%。
实施例6
酶解效果实验,具体如下:
为了证实酶切全氨基保护胶原端肽,采用凝胶电泳表征酶切端肽前后的胶原的分子量变化。如图3所示,条带2和3分别为缺端肽胶原和具端肽胶原,二者均在~110kDa处具有I型胶原独特的α1链和α2链,由于端肽的影响,具端肽胶原的分子量略大于缺端肽胶原;条带4为氨基被2,3-二甲基马来酸酐保护的具端肽胶原,由于氨基被2,3-二甲基马来酸酐保护,故其肽链的分子量大于天然具端肽胶原;采用胃蛋白酶处理氨基被2,3-二甲基马来酸酐保护的具端肽胶原可切除端肽从而使得其分子量变小(条带5)。上述结果初步表明,胃蛋白酶处理可以将全氨基保护胶原的端肽成功切除。
实施例7
为了证实酶切端肽产生全新活性氨基,采用皮安级电流计测定改性胶原样本修饰前后纳米孔道跨膜离子电流变化。纳米孔道的跨膜离子电流与其有效截面积密切相关,若胶原含有的特定官能团可通过共价键接枝到孔道内,则孔道的有效截面积变小,其跨膜离子电流也相应变小。基于上述原理,皮安级电流计测定改性胶原样本修饰前后纳米孔道跨膜离子电流变化的方法可表征改性胶原表面官能团情况。首先,采用PET膜(孔道表面具有羧基官能团)为基底,分别将具N端氨基/侧链保护胶原(prot-Col)和侧链保护的具端肽胶原(prot-telo-Col)(对照组)在EDC/NHS存在条件下与纳米孔道膜反应,充分洗涤避免物理吸附后,采用皮安计测定PET膜的跨膜离子电流,结果如图4所示。与空白PET膜相比,prot-Col修饰PET膜的跨膜离子电流明显变小,而prot-telo-Col修饰PET的跨膜离子电流几乎不变(图4中),表明prot-Col因端肽切除而新形成的N端氨基可以与孔道表面羧基反应(图4左),而prot-telo-Col因所有活性氨基均被保护而无法与孔道表面羧基反应(图4右),从而证实了选择性获得胶原N端游离氨基的可行性。
实施例8
为了证实胃蛋白酶切除胶原端肽的效果,分别以胃蛋白酶处理前后的2,3-二甲基马来酸酐保护侧链氨基的具端肽胶原为原料,通过接枝自由基引发剂和随后的自由基聚合反应制备N端接枝温敏聚合物(聚异丙基丙烯酰胺)改性胶原。采用浊度法表征其在不同温度条件下的响应性能。结果显示(图5),胃蛋白酶处理后的2,3-二甲基马来酸酐保护侧链氨基的具端肽胶原经接枝反应可以具有温度响应性,而未经胃蛋白处理的样品不具有温度响应性,从而表明,(1)胃蛋白酶切除胶原端肽可以特异性的获得N端活性氨基;(2)N端精准接枝改性胶原可以具有接枝分子的独特性能。
Claims (10)
1.一种天然胶原的N端精准接枝改性方法,包括如下步骤:
以醋酸水溶液溶解天然胶原,得到胶原溶液;
胶原溶液透析,除去醋酸分子,得溶液A;
滴加氨基保护试剂至溶液A,反应12~48h,得溶液B;
溶液B透析,除去未反应的氨基保护试剂,得到侧链保护的改性胶原;
向侧链保护改性胶原溶液中加入蛋白酶,以酶水解法除去胶原端肽结构,得到具有末端氨基/侧链保护的改性胶原溶液;
具有末端氨基/侧链保护的改性胶原溶液透析纯化,得到溶液C;
将接枝改性试剂加入溶液C中,摇匀,反应12~48h,得到溶液D;
溶液D透析、冻干,得到N端精准接枝改性的天然胶原。
2.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(1)中,醋酸水溶液在低于20℃的条件下溶解天然胶原;
其中,天然胶原是从脊椎动物的皮肤或跟腱组织中提取并分离纯化的具有完整三螺旋分子结构和端肽结构的天然Ⅰ型胶原;
胶原溶液浓度为1~10mg/mL。
3.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(2)中,胶原溶液用pH为8.0~10.0的磷酸盐缓冲溶液透析12~48h,除去醋酸分子。
4.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(3)中,氨基保护试剂为任选的,能与氨基进行反应的常用保护试剂,包括2,3-二甲基马来酸酐、丁二酸酐、叔丁氧羰基类化合物、氯甲酸苄酯类化合物中的一种;
氨基保护试剂与胶原的质量比为(0.5~2):1。
5.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(3)中,滴加氨基保护试剂至溶液A,添加助溶剂促进氨基保护试剂的溶解;其中助溶剂为任选的有机溶剂,包括乙醇、丙酮、二甲亚砜、1,4-二氧六环、N,N-二甲基甲酰胺中的一种。
6.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(4)中,透析具体操作为:采用截留分子量为12000~15000道尔顿的透析袋,以pH为7.0~10.0的磷酸盐缓冲溶液为透析液,在低于20℃下透析12~48h,每隔3~4h更换一次透析液。
7.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(5)中,蛋白酶为可用于胶原端肽去除但不影响胶原三螺旋结构的酶,包括胃蛋白酶、胰蛋白酶及其复合酶中的一种;蛋白酶的浓度为10~50Unit/g,酶解条件为小于20℃。
8.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(6)中,透析具体操作为:采用截留分子量为12000~15000道尔顿的透析袋,以pH为7.0~10.0的磷酸盐缓冲溶液为透析液,在低于20℃下透析12~48h,每隔3~4h更换一次透析液。
9.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(6)中,接枝改性试剂为任选的,可与氨基进行酰胺化反应的化学试剂,包括末端为羧基或活化状态羧基的化合物。
10.如权利要求1所述的一种天然胶原的N端精准接枝改性方法,其特征在于,(6)中,透析具体操作为:采用截留分子量为12000~15000道尔顿的透析袋,在低于20℃下使用0.5mol/L的醋酸溶液透析12~48h,每隔3~4h更换一次透析液,再以纯水为透析液,透析12~48h,每隔3~4h更换一次透析液。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111352994.XA CN114058664B (zh) | 2021-11-16 | 2021-11-16 | 一种天然胶原的n端精准接枝改性方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111352994.XA CN114058664B (zh) | 2021-11-16 | 2021-11-16 | 一种天然胶原的n端精准接枝改性方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN114058664A true CN114058664A (zh) | 2022-02-18 |
| CN114058664B CN114058664B (zh) | 2023-05-26 |
Family
ID=80272720
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111352994.XA Active CN114058664B (zh) | 2021-11-16 | 2021-11-16 | 一种天然胶原的n端精准接枝改性方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN114058664B (zh) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4883864A (en) * | 1985-09-06 | 1989-11-28 | Minnesota Mining And Manufacturing Company | Modified collagen compound and method of preparation |
| CN105936671A (zh) * | 2016-06-06 | 2016-09-14 | 陕西科技大学 | 一种乙烯基胶原蛋白的制备方法 |
| CN108192941A (zh) * | 2018-03-07 | 2018-06-22 | 广州创尔生物技术股份有限公司 | 一种具有生物活性的胶原的质量控制方法 |
-
2021
- 2021-11-16 CN CN202111352994.XA patent/CN114058664B/zh active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4883864A (en) * | 1985-09-06 | 1989-11-28 | Minnesota Mining And Manufacturing Company | Modified collagen compound and method of preparation |
| CN105936671A (zh) * | 2016-06-06 | 2016-09-14 | 陕西科技大学 | 一种乙烯基胶原蛋白的制备方法 |
| CN108192941A (zh) * | 2018-03-07 | 2018-06-22 | 广州创尔生物技术股份有限公司 | 一种具有生物活性的胶原的质量控制方法 |
Non-Patent Citations (1)
| Title |
|---|
| CHARLOTTE H. JENSEN: "Quantification of the N-terminal propeptide of human procollagen type I (PINP): Comparison of ELISA and RIA with respect to different molecular forms", CLINICA CHIMICA ACTA * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114058664B (zh) | 2023-05-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3772207B2 (ja) | 生分解性生体高分子材料、その製造方法、およびこの高分子材料からなる機能性素材 | |
| CA2277896C (en) | Preparation of collagen | |
| US7208171B2 (en) | Injectable and bioadhesive polymeric hydrogels as well as related methods of enzymatic preparation | |
| US5436135A (en) | New preparation of placenta collagen, their extraction method and their applications | |
| US10195314B2 (en) | Fractionation of charged keratin | |
| JP5979673B2 (ja) | タンパク質水溶液 | |
| BRPI0110312B1 (pt) | método para o preparo de colágeno bi-orientado, preparação de colágeno bi-orientado e matriz de colágeno bi-orientado fibrilar | |
| CN107630060B (zh) | 自组装胶原蛋白及其制备方法 | |
| AU2007343561B2 (en) | Multiple modified derivatives of gelatin and crosslinked material thereof | |
| CN1997408A (zh) | 包含角蛋白的伤口护理产品 | |
| CN114601958A (zh) | 一种透明质酸/丝素蛋白双交联可注射水凝胶及其制备方法 | |
| Rauterberg | The C-terminal non-helical portion of the collagen molecule. | |
| WO2004020470A1 (ja) | システインプロテアーゼ処理コラーゲンの製造方法およびシステインプロテアーゼ処理コラーゲン | |
| CN107630059B (zh) | 新型自组装胶原蛋白及制备方法 | |
| CN110464870A (zh) | 一种基于改性胶原的软组织粘合剂及其制备方法 | |
| CN107630058B (zh) | 一种新型自组装胶原蛋白及制备方法 | |
| CN108912284A (zh) | 一种具有纤维化性能的丙烯酸接枝天然胶原蛋白及其制备方法 | |
| CN114058664A (zh) | 一种天然胶原的n端精准接枝改性方法 | |
| US3984391A (en) | Modified gelatin with a reduced gel-melting point | |
| JPH06292595A (ja) | 低分子量フィブロインの製造方法 | |
| CN108384788A (zh) | 具有黏附能力的表皮细胞生长因子及其编码基因、制备方法和应用 | |
| JP3862361B2 (ja) | 医療用手当材およびそれに用いる新規なペプチド | |
| JP6496484B2 (ja) | 創傷治癒剤 | |
| US11505587B2 (en) | Method of preparing a keratin-based biomaterial and keratin-based biomaterial formed thereof | |
| KR20030072512A (ko) | 유기용매를 이용한 가용성 콜라겐의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |