CN1053065A - 氮杂羟基吲哚衍生物 - Google Patents
氮杂羟基吲哚衍生物 Download PDFInfo
- Publication number
- CN1053065A CN1053065A CN91100034A CN91100034A CN1053065A CN 1053065 A CN1053065 A CN 1053065A CN 91100034 A CN91100034 A CN 91100034A CN 91100034 A CN91100034 A CN 91100034A CN 1053065 A CN1053065 A CN 1053065A
- Authority
- CN
- China
- Prior art keywords
- formula
- phenyl
- alkyl
- compound
- azaoxindole
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Immunology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本发明涉及新的4-、5-、6-和7-氮杂羟基吲哚
衍生物。该化合物是消炎和止痛剂,并且是一种或多
种前列腺素H2合成酶,5-脂氧合酶及白细胞介素-1
生物合成的抑制剂。可将它们用于治疗慢性炎症、变
应性疾病、牛皮癣、各种骨疾病以及免疫功能障碍,例
如,系统性红斑狼疮。
Description
本发明涉及氮杂羟基吲哚类化合物,具体涉及4-,5-,6-,和7-氮杂羟基吲哚衍生物,这类化合物的药用组合物和采用这类化合物的治疗方法。
美国专利4,569,942公开了某些下式所示2-羟基吲哚-1-甲酰氨类化合物
式中,实际上,X是H、氟、氯、溴、(C1-4)烷基、(C3-7)环烷基、(C1-4)烷氧基、(C1-4)烷硫基、三氟甲基、(C1-4)烷基亚磺酰基、(C1-4)烷基磺酰基、硝基、苯基、(C2-4)烷酰基、苯甲酰基、噻吩甲酰基、(C1-4)烷基酰氨基、苯甲酰胺基或在每个烷基上含有1至3个碳原子的N,N-二烷基氨磺酰基;Y是H、氟、氯、溴、(C1-4)烷基、(C3-7)环烷基、(C1-4)烷氧基、(C1-4)烷硫基和三氟甲基;R1是(C1-6)烷基、(C3-7)环烷基、(C4-7)环烯基、苯基、取代苯基,在所说烷基中含有1至3个碳原子的苯基烷基、在所说烷基中含有1-3个碳原子的(取代苯基)烷基,在所说烷基中含有1-3个碳原子的(取代苯氧基)烷基、在所述烷基中含有1-3个碳原子的(苯硫基)烷基、萘基、双环〔2,2,1〕庚烷-2-基、双环〔2,2,1〕庚-5-烯-2-基或-(CH2)n-Q-R0;n是0,1或2;Q是来自下述杂环的两价基团:呋喃、噻吩、吡咯、吡唑、咪唑、噻唑、异噻唑、噁唑、异噁唑、1,2,3-噻二唑、1,3,4-噻二唑、1,2,5-噻二唑、四氢呋喃、四氢噻吩、四氢吡喃、四氢噻喃、吡啶、嘧啶、吡嗪、苯并〔b〕呋喃和苯并〔b〕噻吩;R0是H或(C1-3)烷基;和R2是(C1-6)烷基、(C3-7)环烷基、苄基、噻吩基、吡啶基、或
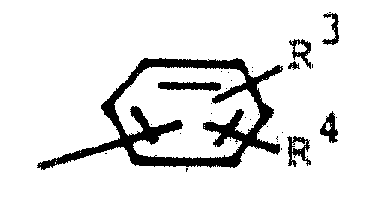
式中R3和R4各自为氢、氟、氯、(C1-4)烷基、(C1-4)烷氧基或三氟甲基。
该专利还公开了:所述2-羟基吲哚-1-甲酰氨类化合物是环氧合酶和脂氧合酶的抑制剂,在哺乳动物体内具有止痛作用,并可用于治疗疼痛,缓解慢性疾病(如:炎症)的症状,以及缓解风湿性关节炎及骨关节炎的疼痛。
美国专利4,556,672公开了某些下式所示3-酰基取代的-2-羟基吲哚-1-甲酰胺类化合物:

式中,X、Y和R1的定义与前述美国专利4,569,942中所述化合物中的定义相同。美国专利4,556,672所公开的化合物所具有的作用与前述美国专利4,569,942所讨论的化合物相同。
1988年4月13日提交的,同时转让给受让人的申请号为181,131的美国专利申请公开了下式化合物及其药物上可接受的碱式盐的用途,

式中X是H、Cl或F,Y是H或Cl,R是苄基或噻吩基,该化合物用于抑制白细胞介素-1(IL-1)的生物合成,并用于治疗由白细胞介素诱发的疾病和机能障碍。
1988年10月18日提交的同时转让给受让人的申请号为PCT/US88/03658的PCT专利申请介绍了下式所示的非甾体消炎药:
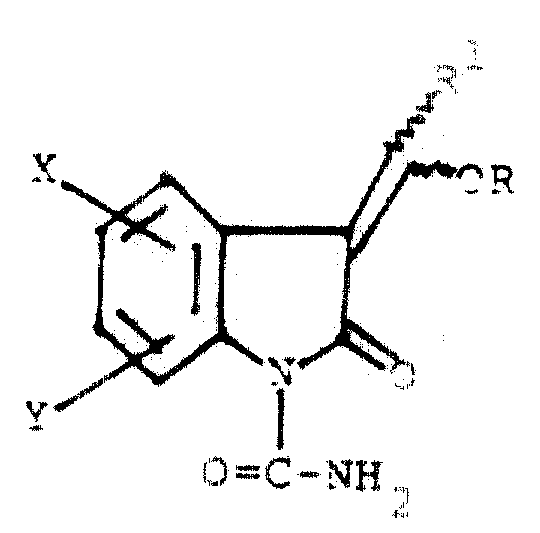
式中,X和Y各自是氢、氟或氯;R1是2-噻吩基或苄基;R是含有2-10个碳原子的烷酰基、含有5-7个碳原子的环烷基羰基,含有7-10个碳原子的苯烷酰基、氯代苯甲酰基、甲氧基苯甲酰基、噻吩甲酰基、ω-烷氧羰基烷酰基、所说烷氧基含有1-3个碳原子,所说烷酰基含有3-5个碳原子;含有2-10个碳原子的烷氧羰基;苯氧羰基;1-(酰氧)烷基,所说酰基含有1-4个碳原子,所说烷基含有2-4个碳原子;1-(烷氧羰基氧基)烷基,所说烷氧基含有2-5个碳原子,所说烷基含有1-4个碳原子;含有1-3个碳原子的烷基;甲基苯基磺酰基或二烷基磷酸酯基,每个所说烷基含有1-3个碳原子。
本发明化合物是消炎和止痛剂,并且是一种或多种前列腺素H2合成酶、5-脂氧合酶、及白细胞介素-1生物合成的抑制剂。前列腺素H2合成酶及5-脂氧合酶催化分别称之为前列腺素和白细胞介素类化合物的体内合成,这两种化合物均是多种炎症疾病的诱发因素。例如,已知在哺乳动物之关节炎的病因学中特别涉及前列腺素H2合成酶。还已知白细胞介素是下述疾病的诱发因素:气喘、关节炎、牛皮癣、胃溃疡、中风、心肌炎、过敏性肠炎。
由于本发明化合物能够抑制前列腺素H2合成酶及5-脂氧合酶,由此抑制前列腺素和白细胞介素的合成,这使得可将本发明化合物用于预防和治疗前列腺素诱发的疾病以及白细胞介素诱发的疾病。
式Ⅰ化合物抑制IL-1生物合成的能力使得可将它们用于治疗IL-1诱发的疾病及哺乳动物的免疫机能障碍。IL-1诱发的疾病包括(但不限于)骨以及连接组织代谢疾病,例如,骨质疏松、牙周病、和组织疤痕。IL-1诱发的免疫机能障碍包括(但不限于):过敏症、牛皮癣、系统性红斑狼疮。
式Ⅰ化合物的止痛作用使得可将它们用于给哺乳动物止痛,例如,用于治疗术后疼痛和创伤疼痛。它们的止痛作用也使得可将它们长期给哺乳动物服用,借此缓解慢性病的症状(例如,用于风湿性关节炎),和由骨关节炎及其他肌肉-骨骼疾病引起的疼痛。
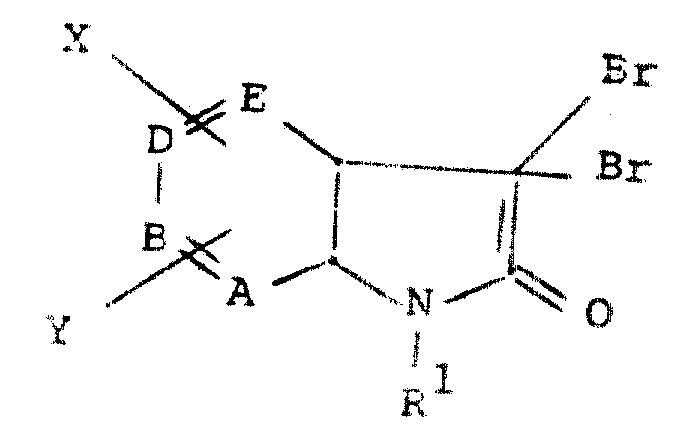
本发明涉及式Ⅰ化合物

式中A、B、D和E中之一是N,其他的是CH;X和Y分别选自氢、OR3、羟基、(C1-6)烷基、CF3、COR3、卤素(氟、氯、溴或碘)、COOR3、CONR3R3、CN、NO2、SR3、SOR3、SO2R3、和SO2NR3R3;R1是(C1-6)烷基或CONHR4;R2是(C1-8)烷基(优选C3-8环烷基)、(CH2)nR5式中n是0或1,或NHR6;R3是(C1-6)烷基、苯基、苄基、烯丙基或氢,其中所述苯基及所述苄基中的苯基部分可任意地被1个或多个分别选自下述基团的取代基取代,所述基团包括:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、CF3;R4是氢、(C1-6)烷基、(C2-6)羟烷基、(C3-8)环烷基、COR3(式中R3的定义如前),苯基、取代苯基、杂芳基或取代杂芳基,其中所述杂芳基及取代杂芳基中的杂芳基部分选自噻吩和呋喃,并且,其中每个所述取代苯基和取代杂芳基是由1或2个下述取代基取代的,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基和CF3;R5是(C3-8)环烷基,氢、苯基、取代苯基、杂芳基和取代杂芳基,其中每一所述杂芳基和取代杂芳基的杂芳基部分选自噻吩和呋喃,每一所述取代苯基和取代杂芳基被1个或2个取代基取代,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、和三氟甲基;R6是苯基、噻吩和呋喃、其中所述苯基、噻吩和呋喃可任意地由1个或多个取代基取代,这些取代基分别选自:氟、氯、溴、碘、羟基、(C1-6)烷基、(C1-3)烷氧基、三氟甲基;W是氢、(C2-10)烷酰基、(C5-7)环烷酰基、(C7-10)苯基烷酰基、氯代苯甲酰基、噻吩甲酰基、ω-(C2-4)烷氧羰基(C3-5)烷酰基、(C2-10)烷氧羰基、苯氧羰基、1-〔(C1-4)酰氧〕-(C2-4)烷基、1-〔(C2-5)烷氧羰基氧〕-(C1-4)烷基,(C1-3)烷基磺酰基、(C1-3)烷基、甲苯基磺酰基和二-(C1-3)烷基磷酸酯基;其前提是(a)如果E是氮,那么X和Y中至少有一个不是氢;(b)如果R2是NHR6或者如果R1是(C1-6)烷基,那么W是氢。
本发明还涉及式Ⅰ化合物的药物上可以接受的酸成盐或碱成盐。适宜酸成盐的实例是乙酸盐、乳酸盐、琥珀酸盐、马来酸盐、酒石酸盐、柠檬酸盐、葡萄酸盐、抗坏血酸盐、苯甲酸盐、肉桂酸盐、富马酸盐、硫酸盐、磷酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、氨磺酸盐、磺酸盐(如:甲磺酸盐和苯甲磺酸盐)以及其他酸的盐。优选者是磷酸盐。经典的可以制得的式Ⅰ化合物的碱成盐是伯、仲和叔胺盐、及碱金属或碱土金属盐,特别有价值的是乙醇胺盐、二乙醇胺盐和三乙醇胺盐。
本发明的优选方案涉及式中B或E是氮、X和Y中至少有一个是氯、R2是(CH2)nR5,n是0、R5是未取代杂芳基,R1是CONHR4,而R4是氢或(C1-6)烷基的式Ⅰ化合物。
特别优选的式Ⅰ化合物是:
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺;
5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氯-3-(3-糠甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
其他具体的式Ⅰ化合物是:
5-氯-3-(3-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
3-(2-糠酰基)-5-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
3-(2-噻吩甲酰基)-6-三氟甲基-4-氮杂羟基吲哚-1-甲酰胺;
6-氯-3-(3-糠酰基)-5-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-乙酰基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氰基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
3-(2-糠酰基)-5-三氟甲基-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺;
5-氯-3-苯基乙酰基-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺。
本发明还涉及下式化合物

式中A、B、D、E、X和Y的定义如式Ⅰ所述。这些化合物是合成式Ⅰ化合物的中间体。
本发明还涉及用于预防或治疗哺乳动物(包括人)下述疾病的药用组合物:例如,慢性炎症,如:气喘、牛皮癣、风湿性关节炎、骨性关节炎,和免疫机能障碍,如:系统性红斑狼疮、该组合物包括预防或缓解上述疾病有效量的式Ⅰ化合物或其药物上可以接受的盐,以及药物上可以接受的载体。
本发明还涉及用于预防或治疗哺乳动物(包括人)疼痛的药用组合物,该组合物包括预防或缓解疼痛有效量的式Ⅰ化合物或其药物上可以接受的盐,以及药物上可以接受的载体。
本发明还涉及预防或治疗哺乳动物(包括人)体下述慢性炎症的方法:例如,气喘、牛皮癣、风湿性关节炎、骨性关节炎、以及免疫机能障碍,例如,系统性红斑狼疮,该方法包括给所述哺乳动物服用预防或缓解上述疾病有效量的式Ⅰ化合物或其药物上可以接受的盐。
本发明还涉及防止或治疗哺乳动物(包括人)疼痛的方法,该方法包括给所述哺乳动物服用预防或缓解疼痛有效量的式Ⅰ化合物或其药物上可以接受的盐。
本发明还涉及用于在哺乳动物(包括人)体内抑制5-脂氧合酶或白细胞介素-1合成的药用组合物,该组合物包括抑制5-脂氧合酶或抑制白细胞介素-1合成有效量的式Ⅰ化合物或其药物上可以接受的盐,和药物上可以接受的载体。
本发明还涉及在哺乳动物(包括人)体内抑制5-脂氧合酶或抑制白细胞介素-1合成的方法,该方法包括给所述哺乳动物服用抑制5-脂氧合酶或抑制白细胞介素-1合成有效量的式Ⅰ化合物或其药物上可以接受的盐。
本发明还涉及在哺乳动物(包括人)体内抑制前列腺素H2合成酶合成的物用组合物,该组合物包括抑制这类合成有效量的式Ⅰ化合物或其药物上可以接受的盐和药物上可以接受的载体。
本发明还涉及在哺乳动物(包括人)体内抑制前列腺素H2合成酶合成的方法,该方法包括给所述哺乳动物服用抑制这类合成有效量的式Ⅰ化合物或其药物上可以接受的盐。
本文采用的术语“烷基”意指具有直链、支链或环化部分或其组合的饱和单价烃基。
式中W不是氢的式Ⅰ化合物是式中W是氢的式Ⅰ化合物的前体药物。术语“前体药物”意指作为前体药物的化合物经哺乳动物服用吸收,经代谢过程在体内释放出药物。
由于羰基碳处于氮杂吲哚环的2-位,以及酰基碳连接于该环的3-位碳,所以式Ⅰ化合物存在有多种互变异构体。这类化合物还可以互变异构结构的几何异构体存在,其中。双键处于该环1-位氮原子和2-位碳原子之间。本发明涉及式Ⅰ化合物的所有互变异构形式及几何异构体。
按下文反应路线1-5所示。可以制得式中W是氢的式Ⅰ化合物。
除特别指明外,在反应路线及下文讨论中出现的A、B、D、E、X、Y、R1、R2、R3、R4、R5和R6的定义如前所述。
反应路线1

反应路线2
反应路线4

反应路线5

根据反应路线1,按如下方式可以制得式Ⅰ化合物。在非质子极性溶剂(如:二甲基甲酰胺或1,2-二甲氧基乙烷)中,于约-30至约50℃,式中Q是卤素的式Ⅱ化合物与式R7O2CCH2CO2R8丙二酸二烷基酯(式中R7和R8相同或不同,选自(C1-6)烷基和苄基)反应,或者与式NCCH2CO2R7氰基乙酸酯反应。优选的溶剂是1,2-二甲氧基乙烷,优选的温度是25℃。由此制得式中G是CO2R8或CN,R7和R8定义如前的式Ⅲ硝基化合物。
然后,将式Ⅲ化合物还原,制得相应的式中R7和G定义如前的式Ⅳ硝基化合物,或者制得式中G的定义如前的式Ⅳ′氮杂吲哚(可以形成其中1个或所有两个产物)。一般在氢气中,在适宜的溶剂中,采用金属催化剂,于大约0°-70℃(最好是环境温度,大约20℃)进行该反应。适宜的溶剂包括:甲醇、乙醇、丙醇、乙酸乙酯和二甲基甲酰胺。乙醇是优选的溶剂,优选的催化剂是阮尼镍。反应的氢气压力应保持在约1个大气压和约5个大气压之间,最好是约3个大气压。滤除催化剂,除去溶剂即可得到式Ⅳ和Ⅳ′化合物中的一个或两者。另外,也可采用金属(如:锌、铁或锡)和酸(如:含水盐酸或乙酸)还原式Ⅲ硝基化合物。该反应也可产生式中R7和G定义如前的式Ⅳ和Ⅳ′化合物中的一个或两者。适宜的温度是0℃左右至大约120℃,为方便起见,最好采用室温。
由相应的前述反应产生的式Ⅳ或Ⅳ′化合物(式中R7和G的定义如前)可以制得式Ⅴ氮杂吲哚,具体方法是:分离式Ⅳ或Ⅳ′化合物,并使它们与稀盐酸、氢溴酸或硫酸反应,反应温度为约50℃至约反应混合物的回流温度,优选回流温度。
采用本文所述式Ⅴ氮杂吲哚环,可按如下方式制备式Ⅰ氮杂吲哚-1-甲酰胺。
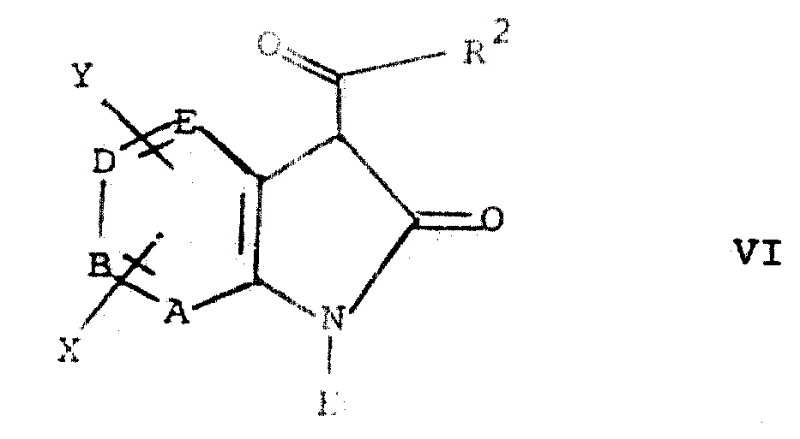
第一步包括连接3-酰基取代基。采用常规方法,在低级烷醇的碱金属盐(如:乙醇钠)存在下,以低级烷醇为溶剂(如:乙醇),使式Ⅴ化合物与适宜的式R2COOH酸反应,即可完成上述酰化反应。可采用的典型的式R2COOH酸衍生物包括酰氯、式R2COOCOR2酸酐以及式R2COO-(C1-6烷基)酯。
可采用过量的所述酸衍生物,以该衍生物计,醇盐的用量可以是1至7摩尔当量。优选是:每2当量简单烷酯采用5当量醇盐、每2当量酰氯或酸酐采用7当量醇盐。
通常在约0-25℃开始式Ⅴ化合物与式R2COOH酸衍生物之间的反应。然后一般在约50-130℃(最好是约80℃)加热该反应混合物,以使反应完成。在这类反应中,通常所采用的反应时间是几小时(如:2小时)至几天(如:2天)。然后将该反应混合物冷却,用过量的水稀释、酸化。
然后过滤收集式Ⅵ酰化产物,或者采用常规溶剂提取法收集之。
由此形成的式Ⅵ化合物可以与氯磺酰异氰酸酯反应,制得除R1是CONHSO2Cl外其他均与式Ⅰ相同的化合物(下文称之为式Ⅶ)。该反应在反应惰性溶剂中进行,即,该溶剂不与氯磺酰异氰酸酯或式ⅦN-氯磺酰-2-羟基吲哚-1-甲酰胺产物反应。具有代表性的溶剂是二烷醚类,例如,乙醚;环醚类,例如,二噁烷和四氢呋喃;芳香烃类,例如,苯,二甲苯,和甲苯;氯代烃类,例如,二氯甲烷,氯仿;乙腈;以及它们的混合物。
一般在常温至溶剂回流温度下进行该反应,最好是约25-110℃,如有必要,该反应温度可以低于20℃,例如,低至-70℃。但是,由于反应温度低于0℃会降低产率,出于经济原因,实际上应避免使用。
式Ⅵ化合物与氯磺酰异氰酸酯反应的摩尔比是等摩尔至氯磺酰异氰酸酯过量30%,即为1∶1至1∶1.3。采用更多的氯磺酰异氰酸酯似乎并无益处,故出于经济原因不必再增加其用量。
如有必要可以分离由此产生的式Ⅶ氯磺酰衍生物,或者也可以不经分离,在同一反应器中通过水解直接转化为式中R1是CONH2的式Ⅰ化合物。采用本技术领域普通技术人员熟知的方法,例如,过滤或蒸除溶剂,即可分离式Ⅶ中间体-氯磺酰化合物。
用水(最好是冰水)、含水酸或含水碱处理经过分离或未经分离的式Ⅶ氯磺酰衍生物,即可将后者水解。一般只用水或含水酸是有利的,既使该水解步骤涉及到两相系时也是如此。尽管该水解反应速度之快以至要解决溶解度的问题,但是,就大规模反应而言,单用水要比其他水解方法更便宜。
对于该水解来说,酸的用量并不严格,其范围可以是从低于摩尔当量到高于摩尔当量。所采用的酸浓度也不严格。一般在使用含水酸进行该水解时,每摩尔式Ⅶ化合物用约0.1-3.0摩尔的酸。为了易于操作,一般采用的酸浓度是约1-6摩尔。通常在分离式Ⅶ中间体并期望形成单相水解混合物时才采用含水酸。具有代表性的酸是盐酸、硫酸、硝酸、磷酸、乙酸、甲酸、柠檬酸和苯甲酸。
通常优选的水解方法是:在大约室温下,在开放式反应器中,简单地搅拌N-氯磺酰甲酰胺即可。较高的温度(高于50℃)会导致产物水解。可以采用低至DMSO冻结点的较低温度,但是会导致反应速率降低。以DMSO-d6作溶剂,采用1H NMR可以很容易地跟踪该反应。待反应完成后,将该混合物倒入过量的水中,过滤分离粗产物。也可采用除DMSO之外的其他溶剂,适宜的例子包括氯仿和二甲基甲酰胺。
按照反应路线1,使式Ⅵ化合物或式Ⅶ化合物与式R4-N=C=O异氰酸酯反应,可以制得式中W是氢、R1是CONHR4(R4不是氢)的式Ⅰ化合物。最普通的反应方法是:在惰性溶剂中,在约25℃至约150℃,优选约80℃-130℃,使基本上等摩尔当量的上述反应物接触即可。就此而言,惰性溶剂是至少能溶解一种反应物,而且既不会对反应物也不会对产物产生有害影响的溶剂。可采用的典型溶剂包括:脂烃类,例如,辛烷、壬烷、癸烷和萘烷;芳烃类,例如,苯,氯苯,甲苯、二甲苯和1,2,3,4-四氢化萘;氯代烃类,例如,1,2-氯乙烷;醚类,例如,四氢呋喃、二噁烷,1,2-二甲氧基乙烷和二(2-甲氧基乙基)醚;极性非质子溶剂,例如,N,N-二甲基甲酰胺,N,N-二甲基乙酰胺、N-甲基吡咯烷酮和亚砜类。反应时间随反应温度而异,但是,通常采用100-130℃的反应温度,数小时(如:5-10小时)的反应时间。
在式Ⅵ化合物与式R4-N=C=O异氰酸酯反应时,如果使用极性比较低的反应溶剂,在反应结束将反应混合物冷却至室温时,通常从溶液中析出产物。在这些情况下,通常采用过滤法回收产物。但是,如果采用极性比较强的溶剂,反应结束时产物不会从溶液中析出,因此,可以通过蒸除溶剂回收产物。另外,如果采用可与水混溶的溶剂,用水稀释反应介质则可使产物沉淀,再通过过滤回收之,该反应产物可采用常规方法(如:重结晶法)使之纯化。
加入下述碱可以使式Ⅵ化合物与式R4-N=C=O异氰酸酯之间的反应加速,所述碱包括:例如,叔胺,如:三甲胺,三乙胺、三丁胺、N-甲基哌啶、N-甲基吗啉或N,N-二甲基苯胺。通常加入约1-4当量的上述碱性试剂。该反应的反应温度是约20-100℃。在反应结束时,必须中和(或酸化)反应介质,然后按前述方法分离产物。
用于制备式中R1是COHNR4的式ⅠA化合物的优选条件是:溶剂-DMSO;温度-80℃~100℃;碱-三乙胺(2当量);异氰酸酯-1.5当量;时间-3-6小时。
式R4-N=C=O异氰酸酯可按常规方法制得。(Sandler和Daro,<有机官能团的制备>Ⅰ部,第二版,Academic Press,Inc.,New York,N.Y.,第12章,364-369页(1983))。另一有用的方法是由适宜的式R4-NH2胺与光气反应来制备之:
R4-NH2+COCl2→R4-N=C=O+2HCl
反应路线1示出的用于制备式中X和Y均为氢的式Ⅴ化合物(即,未取代的4和6-氮杂羟基吲哚)之各种方法在文献中已有介绍。(参见:Finch et al.,Journal of Organic Chemisfry,37,51(1972);Daisley and Hanbali,Synthetic Communications,11,743(1981),Parrick et al.,Journal of the Chemical Society,1531(1974))。
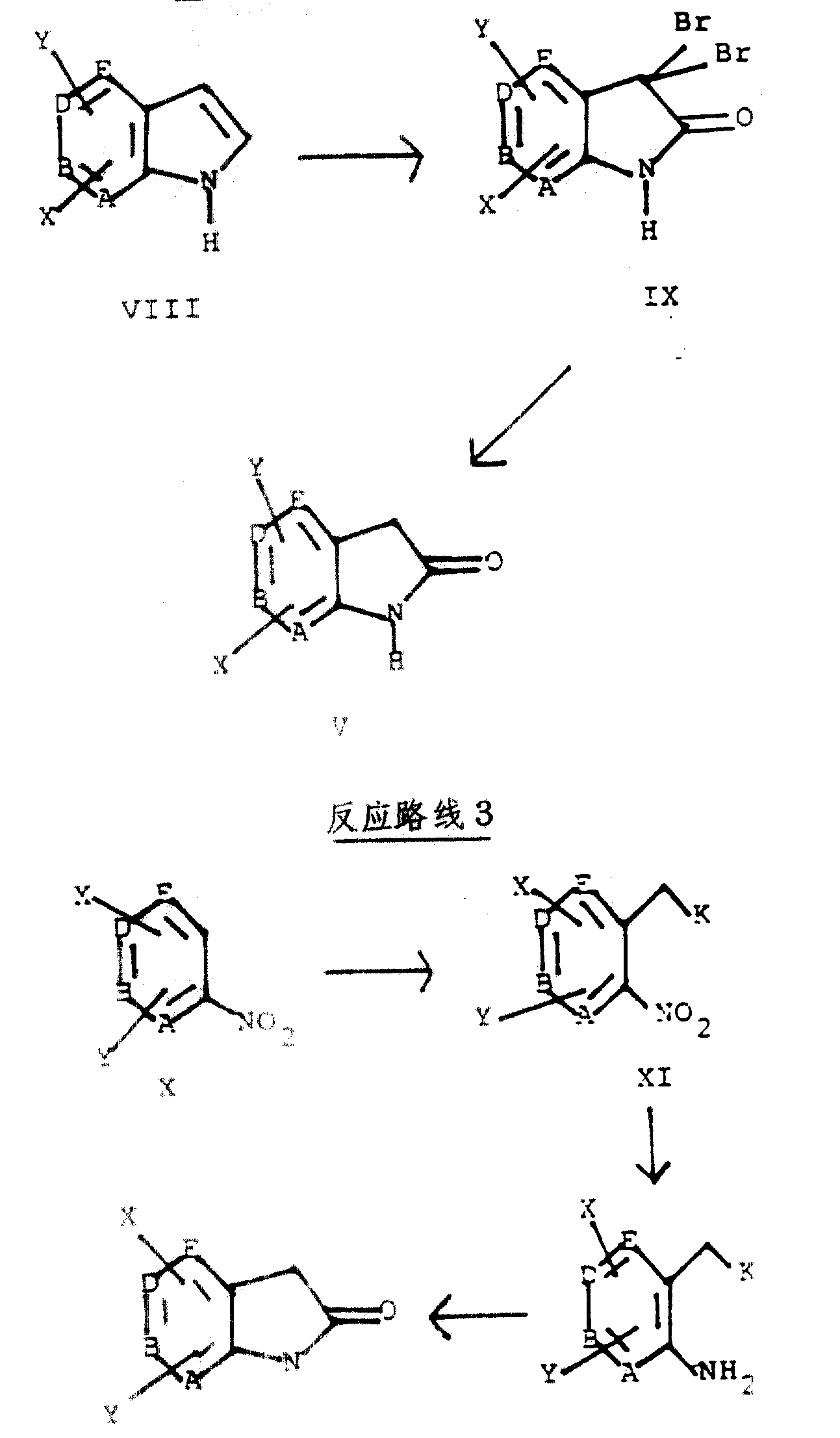
反应路线2和3说明了制备式Ⅴ氮杂羟基吲哚的其他方法。
按照反应路线2,由式Ⅷ氮杂吲哚与3当量的溴反应,制得式Ⅸ二溴化合物。可例举的适宜试剂是溴,溴化吡啶鎓盐过溴化物,和N-溴琥珀酰胺。该反应在极性的惰性溶剂(如:叔丁醇/水或叔丁醇,最好是叔丁醇/水)中进行,pH值在1-7之间。适宜的反应温度是约0-50℃,优选25℃。然后,与氢气反应将由此得到的式Ⅸ化合物还原,得到式Ⅴ氮杂羟基吲哚。一般在10%钯-炭催化剂存在下,于约25-50℃,并在约1至5个大气压下进行该还原反应。优选的反应温度是摄氏25℃,优选的压力是3个大气压。
按照反应路线2,采用与前述程序不同的方法,可以制得式中D是氮原子的氮杂羟基吲哚(5-氮杂羟基吲哚)。即,由式中b是氮原子的式Ⅷ化合物与4当量(而不是3当量)溴反应,通过慢慢地将混合物的pH调至约6.5-7,得到式中X是溴的式Ⅸ化合物,并且该X连接于氮杂羟基吲哚环的“7”位碳原子上。然后将碳-溴键氢解,得到式中b是氮原子的式Ⅴ化合物。一般采用10%的钯-炭,在大约1-5个大气压下(优选3个大气压),进行后一反应。
文献中例举了按照反应路线2合成式中A是氮原子的式Ⅴ化合物的方法,参见:Marfat和Carta,<四面体通讯>28,4027(1987)。
反应路线3解释了用于制备式Ⅴ化合物(和式Ⅰ化合物)的另一途径。按照反应路线3,由式Ⅹ化合物与2-(4-氯苯氧基)乙腈反应,得到式中K是CN的式Ⅺ化合物。一般在强碱存在下,在适宜的溶剂中进行该反应。(参见:Makosza,et al.,(Liebigs Ann.Chem.,1988,203))。适宜的碱包括:叔醇钠或叔醇钾。优选叔丁醇钾。适宜的溶剂包括:四氢呋喃、乙醚、和二甲基甲酰胺。优选四氢呋喃。反应温度为约-78℃至约25℃,优选-10℃。采用无机酸(优选稀盐酸)将反应混合物中和,并采用乙酸乙酯、乙醚或二氯甲烷(优选乙醚)进行常规提取分离,可以纯化前述所得式Ⅺ化合物。然后将来自提取液的有机残留物还原,形成式中K是CN的式Ⅻ化合物。一般在金属催化剂存在下,在约0°-70℃,优选常温(约20℃),于适宜的溶剂中,在氢气中进行该反应。适宜的溶剂包括:甲醇、乙醇、丙醇、乙酸乙酯、二甲基甲酰胺、优选乙醇。优选的催化剂是阮尼镍。该反应的氢气压应保持在约1个气压至约5个大气压之间,优选约3个大气压。滤除催化剂,除去溶剂,即可得到式Ⅶ化合物。
然后将由此形成的式Ⅻ化合物在含水无机酸中进行水解,使之环合,得到式Ⅴ氮杂羟基吲哚。适用酸的例子是:硫酸水溶液,盐酸水溶液及氢溴酸水溶液。反应温度为约25-150℃,优选150℃。
下述方法是反应路线3所示方法的变异。式Ⅹ化合物在强碱存在下、于适宜溶剂中,与苯硫基乙酸叔丁酯反应。(参见:Makosza和Winiarski,J.Org.Chem.,49,(1984))。适宜的碱包括:氢化钠和氢氧化钠,优选氢氧化钠。适宜的溶剂包括:二甲亚砜、液氨、和吡啶,优选二甲亚砜。该反应温度为约-78°至约50℃,优选约25℃。该反应产生式中K是CO2tBu的式Ⅺ化合物,然后,按照对前述式中K是CN的式Ⅺ化合物所述方法将其纯化。继之,将式中K是CO2tBu的式Ⅺ化合物还原,形成式中K是CO2tBu的式Ⅻ化合物。按照前文对式中K是CN的式Ⅺ化合物所述的催化氢化法,或者使式中K是CO2tBu的式Ⅺ化合物与金属(如:锌、铁或锡)在酸(如:盐酸或乙酸水溶液)中进行反应,即可完成上述还原反应。在分离式中K是CO2tBu的式Ⅻ化合物后,通过在惰性溶剂中用酸处理,即可将Ⅻ环化成相应的式Ⅴ氮杂羟基吲哚。适宜的酸包括:盐酸、三氟乙酸和对甲苯磺酸。适宜的溶剂包括:二氯甲烷、苯和甲苯、优选苯。反应温度为约25℃至约150℃、优选80℃。在某些情况下,例如,在酸中采用金属还原式中K是CO2tBu的式Ⅺ化合物,式中K是CO2tBu的式Ⅻ化合物可就地环合。为此就地环合,优选温度为100℃,优选酸为乙酸。
Daisley等人(Synthetic Communictions,5(1)53-57(1975))介绍了式中X和Y一个是甲基而另一个是氢的某些式Ⅴ化合物的合成方法。
按照反应路线4示出的程序,也可以制得式中R1是CONHR4,W是氢的式Ⅰ化合物。该方法是反应路线1方法的变异,它包括:在将R1取代基加到“1”位之前,酰化氮杂羟基吲哚环的“3”位。为实施这一方法,在非质子溶剂(如:1,2-二甲氧基乙烷或N,N-二甲基甲酰胺)中,在碱如氢化钠存在下,在约-30℃至约50℃下,使式Ⅱ化合物与式中Bz为苄基的式CH2(CO2Bz)2酯反应。该反应产生式ⅩⅣ化合物,然后,按照前文反应路线1所示由式Ⅲ酯形成式Ⅴ氮杂羟基吲哚的方法,将式ⅩⅣ转化为相应的式ⅩⅤ胺和式ⅩⅥ氮杂羟基吲哚。继之,按照前文反应路线1所示由式Ⅵ化合物制备式Ⅰ化合物的方法,使式ⅩⅥ氮杂羟基吲哚与式R4-N=C=O异氰酸酯反应,取代式ⅩⅥ的1-位,形成式中R1是CONHR4的式ⅩⅦ氮杂羟基吲哚。
用氢将式ⅩⅦ酯还原得到相应的式ⅩⅧ羧酸。一般在10%钯-炭催化剂存在下,在大约25-50℃,约1至5个大气压下进行该还原反应。然后,在约50℃至约200℃下加热式ⅩⅧ酸,产生式ⅩⅨ化合物,其中氮杂羟基吲哚环的“3”位未被取代。
然后,将由此形成的式ⅩⅨ化合物与酰氯或式R2COOCOR2酸酐反应,在“3”位进行再酰化,形成式Ⅰ化合物。一般在碱如三乙胺或4-二甲基氨基吡啶的存在下,在下述极性非质子溶剂中进行该反应:N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、二甲亚砜或二氯甲烷、优选的溶剂是N,N-二甲基甲酰胺。将酰氯或酸酐缓慢地加到含有式ⅩⅨ化合物、溶剂和碱的溶液中,在此期间,一般将反应混合物的温度冷至0℃左右。然后使之温热至25℃左右,并在该温度下继续该反应。反应时间一般为约30分钟至约2小时。在反应结束时,将反应混合物酸化,例如,通过过滤回收产物。可以洗涤回收产物、干燥,用常规方法(例如,重结晶法)进一步使之纯化。
下述方法是用于由式ⅩⅨ化合物制备式中R2是NHR6的式Ⅰ化合物之优选方法。式ⅩⅨ化合物与式R6NCO化合物反应。该反应一般在反应惰性溶剂中进行,优选极性非质子溶剂,例如,二甲基甲酰胺、二乙基甲酰胺、N-甲基-2-吡咯烷酮、或二甲亚砜。此外,该反应优选在碱存在下进行。这类碱包括碱金属或碱土金属氢化物和有机叔胺。优选氢化钠。
实际上,将异氰酸酯加到于适宜溶剂中的羟基吲哚衍生物和碱中。优选采用约1摩尔当量的异氰酸酯和碱,但使它们各自过量50%时,可取得最好的结果。一般优选在冷却到-10至0℃左右的条件下将各试剂混合,然后使该反应混合物温热至室温。在大约室温至约45℃,根据异氰酸酯的反应活性,在约数分钟至约18小时内完成该反应。待反应完成后,通过将反应混合物加到冰/水中用足量的酸处理之,使pH降至约2~5,由此分离产物。通过过滤,或用与水不混溶的溶剂提取该产物。
反应路线5解释了由式Ⅷ氮杂吲哚出发,合成式中R1是(C1-6)烷基的式Ⅰ化合物的方法。式Ⅷ氮杂吲哚与式R10Br或R10I化合物(其中R10是C1-6烷基)反应,得到式ⅩⅩN-取代的氮杂吲哚。一般在反应惰性溶剂中、在碱存在下进行该反应。适宜的碱包括:氢氧化钠或氢氧化钾、以及氢化钠或氢化钾。如果采用氢氧化物作为碱,适宜的溶剂包括丙酮和二甲亚砜,优选丙酮。如果采用氢化物作为碱,适宜的溶剂包括二甲亚砜和N,N-二甲基甲酰胺。该反应的温度为约0~150℃,优选约25℃。
下述两个方法可用于由式ⅩⅩ氮杂吲哚制备式中R1是(C1-6)烷基的式ⅩⅨN-取代氮杂吲哚。第一个方法类似于反应路线2所示由式Ⅷ氮杂吲哚制备式ⅤN-取代氮杂羟基吲哚的方法,即通过形成成类似于式Ⅸ的二溴氮杂羟基吲哚中间体进行反应,其不同之处是由(C1-6)烷基取代“1”位氮。
第二个方法包括用N-氯琥珀酰亚胺处理式ⅩⅩ化合物,得到式ⅩⅪ3-氯氮杂吲哚。一般在反应惰性溶剂(如:二氯甲烷、氯仿或叔丁醇)中进行该反应。二氯甲烷是优选溶剂。适宜的反应温度为约0~80℃。
使式ⅩⅪ3-氯氮杂吲哚与强酸(如:磷酸、硫酸或高氯酸)反应,采用冰乙酸作溶剂,可将式ⅩⅪ转化为式ⅩⅨ氮杂羟基吲哚。适宜的反应温度为约25℃至约120℃,优选约60℃。优选的酸是磷酸。根据反应物及所采用的温度,反应时间可在约1小时至7天的范围内变化。
按照前文讨论反应路线4,用于由式ⅩⅨ化合物制备式Ⅰ化合物时,可以将式中R1是(C1-6)烷基的式ⅩⅨ氮杂羟基吲哚衍生为式中R1是(C1-6)烷基的式Ⅰ化合物。
采用下述两个方法,从适宜的3-酰基-3-氮杂羟基吲哚-1-甲酰胺(或N-取代甲酰胺)出发,可以制备式中W不是氢的式Ⅰ化合物,即,式中W为氢之化合物的前药。第一个方法包括:在约-10至约10℃,优选0℃左右,用稍过量的所需酰氯、氯甲酸酯、  盐或烷化剂处理于反应惰性溶剂(如:氯仿或四氢呋喃)中的适宜3-酰基-氮杂羟基吲哚-1-甲酰胺(或N-取代甲酰胺)和等摩尔量三乙胺的溶液。使该反应物温热至室温,并在该温度下维持大约2-3小时。如果羟基吲哚原料没有完全反应,再将混合物冷却,补加酰化剂或烷化剂并按比例加入三乙胺,重复上述方法,直到所有起始羟基吲哚消耗完了为止。在经1N的盐酸洗涤,并用饱和NaHCO3溶液抽提后,从反应溶剂中分离产物。将减压除去溶剂后的残留物经重结晶或色谱纯化。在某些情况下,通过过滤反应混合物收集不溶性产物来直接分离产物。
盐或烷化剂处理于反应惰性溶剂(如:氯仿或四氢呋喃)中的适宜3-酰基-氮杂羟基吲哚-1-甲酰胺(或N-取代甲酰胺)和等摩尔量三乙胺的溶液。使该反应物温热至室温,并在该温度下维持大约2-3小时。如果羟基吲哚原料没有完全反应,再将混合物冷却,补加酰化剂或烷化剂并按比例加入三乙胺,重复上述方法,直到所有起始羟基吲哚消耗完了为止。在经1N的盐酸洗涤,并用饱和NaHCO3溶液抽提后,从反应溶剂中分离产物。将减压除去溶剂后的残留物经重结晶或色谱纯化。在某些情况下,通过过滤反应混合物收集不溶性产物来直接分离产物。
第二个方法包括:在无水反应惰性溶剂(如:丙酮)中,使下述成份相接触:适宜的3-酰基-氮杂羟基吲哚-1-甲酰胺(或N-取代的甲酰胺),三倍摩尔过量的所需α-氯烷基碳酸酯、五倍摩尔过量的NaI,以及两倍摩尔过量的无水碳酸钾,将该反应混合物在溶剂的回流温度下加热大约16小时。然后用水稀释反应混合物,用与水不混溶的溶剂(如:乙醚或氯仿)提取产物。浓缩含有产物的溶剂,得到粗产物,后者可经重结晶和/或色谱纯化。
就上述各反应而言,压力并不严格,一般适宜的压力是约0.5~50个大气压,最方便的是通常采用的1个大气压。
按照常规方法,用大约1个化学当量的药物适用酸处理游离碱(Ⅰ)的溶液或混悬液,可以制得式Ⅰ化合物的酸成盐。在分离该盐时,采用常用的浓缩和重结晶技术。按常规方法,用大约1个化学当量的有机或无机碱处理这类式Ⅰ化合物可以制得式Ⅰ化合物的碱成盐。
采用下述测试方法,可以确定式Ⅰ化合物对白细胞介素1生物合成的抑制能力。
将C3H/HeN小鼠(Charles River,Wilmington,Massachusetts)颈部脱位处死,并用70%乙醇喷洗腹部以避免后期细胞制备的污染,在每只小鼠腹膜内注入8ml RPMI1内含5%FCS2、青霉素、链霉素(100单位/ml-100μg/ml)、谷氨酸(2mM)。揉挤腹膜以助分离细胞,而后切开腹部皮肤,暴露其下的肌层。采用20号针头,斜下进针,经暴露的肌层,恰至胸骨骨下方,吸除腹腔液。将六只小鼠的腹腔液收集于一个塑料园锥形管中,显微镜下检查细菌污染情况。未污染液体经大约600×g离心六分钟,倾出上清液,混合来自五~六只管的细胞团块并重新悬浮于总体积20ml的RPMI-FCS3中,用血细胞计数器确定细胞数;并经台盼蓝染色后,也用血细胞计数器测定细胞存活度,而后用RPMI-FCS将细胞稀释至3×106/ml。将上述1ml的细胞悬液加入35毫米孔板的各孔中,在5%CO2条件下,37℃孵育2小时,以使巨噬细胞贴附于各孔的壁上;强力漩震各孔,倾倒弃之上清液;用RPMI-SF4洗贴壁细胞(如巨噬细胞)两次;在含有贴壁细胞的孔中加入1ml受试化合物,其在RPMI-SF中的浓度范围为0.1~100μg/ml或以1ml RPMI-SF做为对照,而后将在RPMI-SF中的100μl LPS5(1mg/5ml)置5%CO2条件下24小时,取出上清液,或者立即进行白细胞介素-1分析或者冷冻以后分析。
按下述受体结合试验对上清液进行白细胞介素-1定量分析。标准曲线制作如下:将EL4-6.1鼠胸腺瘤细胞〔10-15×106细胞数于0.4ml结合缓冲液(RPMI 1640,5% FCS,25mM HEPES,0.01% NaN3,pH7.3)中〕加到不同量的未标记鼠rIL-1〔根据发表的白细胞介素-1的115-270氨基酸序列,从大肠杆菌中制备的重组白细胞介素-1,Lomeclico,P.M.,等,Nature,312,458-462(1984)〕中(0.5ml缓冲液中4pg~40ng),4℃孵育1小时,持续振动,而后加入0.8ng(0.1ml)人125碘-γIL-1(New England Nuclear,Boston,Massachusetts),持续振荡,继续3小时。采用Yeda装置(Linca Co.,Tel-Aviv,Isyeal)样品经Wbatman GF/C2.4cm玻璃纤维滤膜(已用0.5%奶粉,37℃封闭2小时)过滤,再用3ml冰冷缓冲液洗一次。将滤膜置于Searle计数器中计数,将非特异性结合作为在200ng未标记rIL-1中的化合物。坡度标准曲线的构成是:绘制Log(Y/100-Y)比Log C,在此Y代表对照125碘-rIL-1结合率,C代表未标记rIL-1的浓度,在Y值20%-80%之间添上线性最小平方线,然后按上述定量悬液中IL-1的水平,在上述步骤中,稀释的悬液取代rIL-1,根据标准坡形图所测百分结合值用以确定IL-1浓度,每一稀释液分析两次,通常只用在20-80%的Y值稀释液计算平均IL-1水平。
经下列分析程序可测定式Ⅰ化合物对前列腺素H2合成酶和5-脂氧合酶的抑制能力。根据这一程序,可测定细胞的前列腺素合成酶和5-脂氧合酶已知产物的水平,具体方法是用受试化合物处理细胞,通过上述已知产物量的减少或缺失证明前列腺素H2合成酶和/或5-脂氧合酶受到抑制。
依据Jakschik,B.A.,等人方法〔Nature 287:51-52(1980)〕,使呈单层的RBL-1细胞在Spinner培养物于Minium Essential Medium(Eagle)〔内含Earle′S盐加15%胎牛血清,并加抗生素/抗霉菌溶液(Gibco)〕中生长1-2天,然后洗涤细胞两次并以4×106细胞/ml的细胞密度再悬浮于冷RPMI 1640中,继之将0.25ml等份的受试化合物以所需浓度于RPMI 1640中,37℃平衡5分钟。平衡后的部分加入0.25ml预温的细胞悬液,此混合物于37℃孵育5分钟,再加入10μl(内含14碳-花生四烯酸和A-23187(钙离子载体,Sigma Chemical)的溶液在37℃下将混合物再孵育5分钟,而后加入267μl乙腈/0.3%乙酸,混合液置冰上30分钟,旋震含混合物的管,经离心(3000rpm,10分钟)分离,倾出上清液,在微量离心机中高速再离心两分钟。然后在Perkin Elmer-HS(3微米)柱上采用HPLC法分析100μl等份上清液,以含0.1%三氟乙酸的乙腈/水进行梯度洗脱,流速为2毫升/分。采用Berthold LB504放射活性监测仪测定放射活性,该监测仪装有洗脱柱,并带有每分钟混合2.4毫升Omniflour的800μl流式细胞混合器(Trademark of New England Nuclear,Boston,Massachusetts)。采用Spectra Physics SP 4200 Omputing积分仪定量洗脱出的放射活性。将由此获得的数据用于数据约分程序,在该程序中,根据总积分单位的百分率并与平均对照水平相比较,计算出每一产物的积分单位。
根据Siegmud等人的方法<Proc.Soc.Exp.Biol.Med.,95,729-731(1957)>,按Milne和Twomey所述改进为高输出法<Agents and Actions 10,31-37(1980)>先给小鼠施用2-苯基-1,4-苯醌(PBQ)诱发腹肌紧张,然后根据式Ⅰ化合物表现出的对该紧张的阻断作用来测定式Ⅰ化合物的止痛作用。
根据标准的角叉菜胶诱发大鼠脚部水肿试验来证实式Ⅰ化合物的抗炎作用<Winter等,Proc Soc.Exp.Biol.Med.,111,544(1963)>。
注:
1 RPMI-1640培养基(Hazelton Research Products Inc.,Lenexa,Kansas)。
2 在每个孔中加入事先筛选过的胎牛血清,其目的是为了在胸腺嘧啶测试中(hyclone Laboratories,Logan,Utah)对IL-1出现良好的反应并在没有IL-1时出现低自发性增殖。培养皿于37℃保温。
3 RPMI-1640培养基含有5%胎牛血清。
4 RPMI含有青霉素-链霉素(100单位/毫升-10μg/ml)及谷酰胺(2mM)。
5 由Salmonella minnesota精心纯制的脂多糖,事先经检查确定C3H/HeJ小鼠对它无反应。
在将式Ⅰ化合物或其药物上可以接受的盐用作IL-1抑制剂、止痛剂或消炎剂时,既可以给哺乳类受试物单独服用式Ⅰ化合物或其盐,或者最好是按标准制药实践,将其与药物上可以接受的载体或稀释剂配伍,以药用组合物的形式服用。该化合物既可以口服,又可以经胃肠道外给药。胃肠道外给药包括:静脉、肌内、腹腔、皮下或局部给药。
在包括式Ⅰ化合物或其药物上可接受盐的药用组合物中,载体与活性成份的重量比一般是1∶4至4∶1,最好是1∶2至2∶1。但是,无论在任何给定的情况下,该比例的选择取决于下述因素:如活性成份的溶解度,所规划的剂量和具体的给药途径。
就口服本发明的式Ⅰ化合物而言,可以例如片剂或胶囊剂,或含水溶液或混悬液的形式给药。在口服用片剂中,通常采用的载体包括:乳糖和玉米淀粉。通常加入润滑剂例如硬脂酸镁。就口服胶囊剂型而言,有用的稀释剂是乳糖和干燥玉米淀粉。在需要口服含水混悬液时,将活性成份与乳化剂及悬浮剂混合。如有必要,可以加入某些增甜和/或香味剂。就肌肉,腹腔、皮下及静脉给药而言,通常制备活性成份的无菌溶液,并应适当地调整和缓冲溶液的pH值。就用于静脉而言,应控制溶质的总浓度以使之为等渗制剂。
在将式Ⅰ化合物或其盐用于人体时,一般由经治医生确定日剂量。此外,该剂量可随下述因素改变:年龄,体重,患者的个体反应,以及患者症状的严重程度和所服用具体化合物的效力。但是,就应急服药缓解疼痛而言,在大多数情况下,有效止痛起始剂量根据需要(例如每4-24小时)约为5mg至500mg。为减轻炎症及疼痛或为抑制IL-1生物合成而采用长期服药法时,在大多数情况下,无论是每日一次或分次给药,其有效剂量是约5mg-1.0g/天,最好是50mg-500mg/天。
下列实施例解释本发明。实施例中的所有熔点未经校正。除特别指出外,所有的反应均在氮气氛中进行。
实施例1
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.5-氯-4-氮杂羟基吲哚
在装有通氮导管和机械搅拌器的无水烧瓶中放入氢化钠(60%油混悬体,12.8g,0.32mol)。用己烷洗两次除去大部分油,然后将剩下的氢化钠固体混悬于无水1,2-二甲氧基乙烷(DME)(350ml)中。搅拌下,将丙二酸二乙酯(49.3ml,0.325mol)的DME(175ml)溶液滴加到上述浆液中。将该混合物在室温下搅拌1小时,然后加入市售2,6-二氯-3-硝基吡啶(25g,0.13mol)的DME(175ml)溶液,得到一深红色溶液。在室温下连续搅拌过夜,然后将该反应混合物倒入水中。用6N的盐酸溶液将反应物酸化至pH3,用醚提取该混合物。用盐水洗涤醚相,用MgSO4干燥,减压浓缩,得到一黄色油。(在60℃下高真空加热除去大部分过量的丙二酸二乙酯)。1H NMR表明:该残留物由2-二(乙氧基羰基)甲基-6-氯-3-硝基吡啶,不希望的异构体6-二(乙氧基羰基)甲基-2-氯-3-硝基吡啶的2∶1的混合物组成,后者是在起始物6位置换氯原子而生成的,该残留物中还含有一些残留的丙二酸二乙酯。该混合物经在硅胶上进行闪层析纯化,用4∶1己烷/乙酸乙酯洗脱,合并所有含有所期产物的级份,浓缩,得到40.5g含有2-二(乙氧基羰基)甲基-6-氯-3-硝基吡啶,6-二(乙氧基羰基)甲基-2-氯-3-硝基吡啶及丙二酸二乙酯(近似摩尔比为10∶4∶3)的油状物。由此计算,2-二(乙氧基羰基)甲基-6-氯-3-硝基吡啶的得量近似26g(63%)。
将2-二(乙氧基羰基)甲基-6-氯-3-硝基吡啶、6-二(乙氧基羰基)甲基-2-氯-3-硝基吡啶及丙二酸二乙酯的混合物溶解在乙醇(300ml)中,然后将其加到50%阮尼镍于水(26g)的混悬液中,用乙醇稀释至700ml。将该混合物在帕尔振荡器中于3个大气压下氢化4小时,然后通过硅藻土过滤,除去催化剂。减压蒸除溶剂,剩下3-氨基-6-二(乙氧基羰基)甲基-6-氯吡啶、3-氨基-6-二(乙氧基羰基)甲基-2-氯吡啶-不希望的异构体、及丙二酸二乙酯的混合物,为蜡样固体(35.7g)。
将上述所得混合物移至6N的HCl溶液(700ml)中,加热回流5小时,减压下除去含水酸后,将残物移至水中,再次浓缩,经空气干燥后,得到棕色固体状5-氯-4-氮杂羟基吲哚。得量为7.04g(按2,6-二氯-3-硝基吡啶计总收率为32%),经用异丙醇重结晶,制得分析样品;m.p.250-254℃(分解)。
B.5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚
将金属钠颗粒(2.0g,87.0mmol)加到于干燥园底烧瓶中的50ml无水乙醇中。待金属钠完全溶解后,加入5-氯-4-氮杂羟基吲哚(3.0g,17.8mmol),然后加入噻吩羧酸乙酯(4.8ml,40mmol)。在充氮下,将该混合物加热回流过夜。在此期间,出现沉淀。将该混合物冷却、倒入冰/水中,用6N的盐酸酸化至pH3,过滤用水和醚洗涤,收集固体产物(3.7g,75%,m.p.235-238℃)。从母液中结晶,又收集到第二批产物(375mg,8%,m.p.240-241℃)。
C.5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
依次将三乙胺(0.54ml,3.87mmol)和异氰酸叔丁酯(0.3ml,2.63mmol)加到5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(500mg,1.79mmol)的无水DMSO(10ml)溶液中。在氮气氛下,于油浴中,在85℃将所得混合物加热3小时。冷至室温后,将溶液倒入冰/水中,用6N盐酸酸化至pH2,过滤收集不溶性固体,在两个硅胶柱上连续进行闪层析,第一个柱用乙酸乙酯洗脱,第二柱用20%己烷/氯仿洗脱。层析后的产物用乙腈重结晶,得到题目化合物(300mg,44%),为黄色固体;m.p.150-152℃。
1H NMR(DMSO-d6):δ9.00(s,1H),8.83(d,J=3.6Hz,1H),8.35(d,J=8.1Hz,1H),8.00(d,J=4.9Hz,1H),7.28-7.25(m,1H),7.24(d,J=8.1Hz,1H),1.40(s,9H).IR(KBr disc)1713,1647,1582,1461cm-1.MS m/e(相对百分比)379(4),377(18),280(23),278(70),196(38),194(100),111(12).
元素分析计算值C17H16ClN3O3S:C 54.04,H,4.27,N 11.12.实测值:C 53.59,H 4.01,N 10.92.
实施例2
6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
A.6-氯-4-氮杂羟基吲哚
按照<化学文摘>70,68286y(1969)的方法,由2-羟基-3-硝基-5-氯吡啶制备2,5-二氯-3-硝基吡啶。将市售2-羟基-5-硝基吡啶硝化(H2SO4/HNO3/60°)制备2-羟基-3-硝基-5-氯吡啶。与已发表的涉及将2-氨基-5-氯吡啶硝化的方法(<化学文摘>70,68286y(1969))相比,这一途径更简单、产率更高。
在装有氮气导管和机械搅拌器的干燥烧瓶中加入60%氢化钠油混悬体(4.0g,0.10mol),用己烷洗涤两次除去大部分油。然后将剩下的固体氢化钠混悬于无水1,2-二甲氧基乙烷(DME)(125ml)中。搅拌下,将丙二酸二乙酯(15.7ml,0.10mol)的DME(50ml)溶液滴加到前述所得浆液中。将该混合物在室温下搅拌1小时,然后加入2,5-二氯-3-硝基吡啶(10g,51.8mmol)的DME(75ml)溶液。在室温下连续搅拌过夜,然后用水稀释该反应混合物。先用1N盐酸溶液将其酸化至pH2,然后用醚提取该混合物。用盐水洗涤醚相,用MgSO4干燥,减压浓缩,得到一红色的油,后者经硅胶闪层析纯化,用9∶1己烷/乙酸乙酯洗脱。合并含有所期产物的所有级份,浓缩,得到油状2-二(乙氧基羰基)甲基-5-氯-3-硝基吡啶(13.6g,82%)。
将该二酯的乙醇(200ml)溶液加到用乙醇(300ml)稀释的50%阮尼镍-水(8.8g)的混悬液中。在帕尔振荡器中,在3个大气压下将该混合物氢化4小时,然后通过硅藻土滤除催化剂,减压除去溶剂,得到淡黄色固体状3-氨基-2-二(乙氧基羰基)甲基-5-氯吡啶(12.6g)。
将3-氨基-2-二(乙氧基羰基)甲基-5-氯吡啶和6N盐酸(325ml)的混合物加热回流4小时。减压除去含水酸后,将残留物移至水中,过滤除去少量黑色不溶物。用固体NaHCO3将滤液的pH调至6.5,过滤收集以棕黄色固体沉淀出的6-氯-4-氮杂羟基吲哚(2.6g)。用乙酸乙酯提取滤液,合并提取液,用MgSO4干燥,浓缩,得到另一部分6-氯-4-氮杂羟基吲哚(3.6g)。因此,总得量为6.2g(按2,5-二氯-3-硝基吡啶计,总收率为71%)。
B.6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚
在干燥的园底烧瓶中,将金属钠颗粒(3.4g,0.15mol)加到无水乙醇(90ml)中。待金属钠完全溶解后,加入固体状6-氯-4-氮杂羟基吲哚(5.0g,29.7mmol),然后加入2-噻吩羧酸乙酯(8ml,55mmol)。在氮气氛下,将该混合物加热回流过夜,在此期间出现沉淀。将该混合物冷却,倒入冰/水中,用6N盐酸酸化至pH3。过滤收集氮杂羟基吲哚固体,用水和醚洗涤,并在真空条件下加热干燥(7.8g,94%,m.p.250℃)。
C.6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
将6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(3.3g,11.8mmol)和无水乙腈(100ml)的混合物冷却至0℃,然后用N-氯磺酰异氰酸酯(1.5ml,17.2mmol)处理。将该混合物在室温下搅拌6小时,然后倒入冰/水中。过滤收集固体,用水洗涤,然后于100℃在水中搅拌20分钟,过滤收集产物,干燥,用乙酸重结晶,得到分析纯的6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺,m.p.>250℃。
1H NMR(DMSO-d6):δ8.78(d,J=3Hz,1H),8.57(br s,1H),8.39(s,1H),7.92-7.89(m,3H),7.23-7.21(m,1H).IR(KBr disc)1721,1602,1420cm-1.MS m/e(相对百分比)321(13),280(11),278(36),196(40),194(100),111(333).元素分析计算值:C13H8ClN3O3S:C 48.53,H 2.51,N 13.06.实测值C 48.58,H 2.42,N 12.95.
实施例3
5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.(5-氨基-2-氯-4-吡啶基)乙腈
将叔丁醇钾(24.69g,220mmol,2.2当量)溶于无水四氢呋喃(150ml)中,在氮气氛下,于-50℃,搅拌下滴加2-氯-5-硝基吡啶(15.85g,100mmol)及(4-氯苯氧基)乙腈(E.Grochowski et al.,Bull.Acad.Pol.Sci.Ser.Sci.Chim.,11,443,(1963))(18.44g,110mmol,1.1当量)的无水四氢呋喃(150ml)溶液,在用干冰/丙酮浴冷却下,滴加速度应使反应温度保持在-40℃至-50℃。然后在氮气氛下,于-78℃,将所得紫色反应混合物搅拌1小时,此时,在该反应混合物中加入冰乙酸(20ml,0.35mol,3.5当量),使该混合物温热至室温。将5%HCl(100ml)溶液加到该反应混合物中,先用乙醚(100ml)、再用二氯甲烷(2×100ml)提取该含水混合物。合并提取液,用MgSO4干燥,使之通过硅胶滤层(约150g),然后用二氯甲烷(1200ml)洗涤,减压蒸发该滤液,残油经硅胶(约300g)层析,用25%己烷/二氯甲烷液洗脱,得到一油状物(Rf=0.52,展开剂:二氯甲烷),后者在冷却无水醚中研磨,得到白色固体结晶状6-氯-3-硝基-2-吡啶基乙腈(1.37g,7%);m.p.121.5-123.5℃。进一步洗脱,得到另一油状物(Rf=0.48,展开剂:二氯甲烷),将其在冷却无水乙醚中研制,得到(2-氯-5-硝基-4-吡啶基)乙腈(1.87g,9%),为白色固体结晶,m.p.87-89℃,IR(KBr)3080,2240,1600,1545,1520,1450,1390,1340,1135cm-1。
将(2-氯-5-硝基-4-吡啶基)乙腈的乙醇(100ml)溶液加到用乙醇(150ml)稀释的50%阮尼镍-水(3.2g)混悬液中。在帕尔振荡器中,于3个大气压下,将该混合物氢化2.5小时,然后经硅藻土(CeliteR)过滤,除去催化剂。减压蒸除溶剂,剩下一深色的油,后者经硅胶闪层析纯化,用3∶1乙酸乙酯/己烷洗脱,合并只含有题目化合物的级份,浓缩,得到一油状物(850mg,32%)。也将纯度差的级份合并,浓缩,得到一主要成份是题目化合物(约75%)的油状物(600mg)。
B.5-氯-6-氮杂羟基吲哚
将(5-氨基-2-氯-4-吡啶基)乙腈(1.40g,8.4mmol)移至6N HCl溶液(100ml)中,在50-100℃加热2小时,经冷却后,通过加入固体NaHCO3将溶液调至pH7,用乙酸乙酯提取,合并乙酸乙酯提取液,用盐水洗涤,用MgSO4干燥,减压浓缩,残留物经硅胶闪层析纯化,用乙酸乙酯洗脱,(采用一些甲醇有助于溶解固体)。合并含有所期产物的级份,浓缩,得到固体状题目化合物(650mg,46%),m.p.230℃(分解)。核磁共振谱表明:该物质除含有题目化合物外,还含有少量的副产物-2氨基-5-氯-6-氮杂羟基吲哚。尽管如此,该物质不经进一步纯化即可用于下一步反应。
C.5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚
将金属钠小颗粒(232mg,10mmol)加到于干燥园底烧瓶中的无水乙醇(10ml)中。待钠完全溶解后,加入固体5-氯-6-氮杂羟基吲哚(340mg,2.0mmol),然后加入2-噻吩甲酸乙酯(0.54ml,4.0mmol)。充氮下,将该混合物加热回流过夜,在此期间出现沉淀。将该混合物冷却,倒入冰/水中,用6N盐酸酸化至pH4。过滤收集固体产物(475mg),用水洗涤,空气干燥,将该产物用甲醇重结晶,得到题目化合物(190mg,34%)。
1H NMR(DMSO-d6):δ10.62(br s,1H),8.79(d,J=3.2Hz,1H),7.92(s,1H),7.77(d,J=5Hz,1H),7.65(s,1H),7.17-7.14(m,1H).
D.5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-N-丁基甲酰胺
将5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚(190mg,0.68mmol)溶于无水DMSO(3ml),依次加入三乙胺(0.20ml,1.44mmol)和异氰酸叔丁酯(0.11ml,0.95mmol)。充氮下,将所得混合物在85℃油浴中加热4小时。将该溶液冷却至室温,然后倒入冰/水中,用1N的盐酸酸化至pH2。过滤收集不溶的固体,空气干燥,用甲醇重结晶,得到所期产物(175mg,68%,m.p.224℃(分解)。1H NMR(DMSO-d6):δ9.45(s,1H),8.70(s,1H),8.43(dd,J=1.2,3.4Hz,1H),8.06(s,1H),7.82(dd,J=1.2,4.9Hz,1H),7.18(dd,J=3.4,4.9Hz,1H),1.40(s,9H).IR(KBr disc)1723,1660,1624,1586,1552,1474cm-1.Ms m/e(相对百分比)377(2),280(21),278(59),196(41),194(100),111(41).元素分析 计算值C17H16ClN3O3S:C 54.04,H 4.27,N 11.12.实测值C 53.88,H 4.21,N 11.04.
实施例4
3-(2-噻吩甲酰基)-5-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.3,3,7-三溴-5-氮杂羟基吲哚
用于合成3,3,7-三溴-5-氮杂羟基吲哚的起始物是5-氮杂吲哚,制备方法见美国专利No.4,625,033。另外,该原料也可按下述文献方法制得:Yamanaka等<Chem.Pharm.Bull.,35,1823(1987)>,或Okuda和Robison<J.Org.Chem.,24,1008(1959)>。将5-氮杂吲哚(1.5g,12.7mmol)溶于叔丁醇(100ml)和水(100ml)中,室温搅拌下,用20分钟滴加纯净的溴(2.6ml,50.5mmol)。滴加完成后,该混合物的pH大约是1。用半小时小心地慢慢加入饱和NaHCO3溶液,由此,将混合物的pH调至6.5-7。在此期间,出现大量的沉淀。将该混合物过滤,收集沉淀,用水洗涤,空气干燥,得到3.7g(79%)黄色固体,m.p.250℃。用乙酸乙酯提取滤液,又得到一部分题目化合物(700mg,15%);但是经TLC测定,后一部分样品纯度稍差,合并几份不纯的级份,经硅胶闪层析纯化,用10%甲醇/CHCl3洗脱。
B.5-氮杂羟基吲哚
将10%的钯-炭(3.2g)加到3,3,7-三溴-5-氮杂羟基吲哚(6.4g,17.3mmol)的乙醇(1200ml)溶液中。采用帕尔振荡器,在3个大气压的氢气下,将所得混合物氢化3小时。使该混合物通过硅藻土垫过滤除去催化剂,用乙醇充分洗涤。除去溶剂,剩下一棕色固体(主要是所期产物的氢溴酸盐),3.5g。将该固体溶于水中,用活性炭处理,经硅藻土过滤,加入饱和NaHCO3水溶液将滤液调至pH7.5。然后用正丁醇将该混合物提取3次。合并正丁醇提取液,用盐水洗涤,用MgSO4干燥,减压浓缩,剩下一固体。将该固体与丁醇一起研制,过滤收集褐色固体状5-氮杂羟基吲哚,将母液所得固体用甲醇重结晶,又得到一部分题目化合物(50mg),m.p.>250℃。
C.3-(2-噻吩甲酰基)-5-氮杂羟基吲哚
将金属钠小颗粒(1.15g,50mmol)加到于干燥园底烧瓶中的30ml无水乙醇中。待金属钠完全溶解后,加入固体状5-氮杂羟基吲哚(1.40mg,10.4mmol),然后加入2-噻吩甲酸乙酯(2.7ml,20.1mmol)。充氮下,将该混合物加热回流1小时。此时,通过常压蒸馏乙醇,将反应物体积减少到50%,将该混合物冷却,倒入冰/水中,将所得溶液过滤,除去少量不溶物,并将后者用水充分洗涤,用6N盐酸将滤液酸化至pH7,产物沉淀而出。过滤收集产物,用水洗涤,空气干燥,得到黄/棕色固体状题目化合物(2.0g,83%)。
D.(2-噻吩甲酰基)-5-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将3-(2-噻吩甲酰基)-5-氮杂羟基吲哚(500mg,2.05mmol)溶于无水DMSO(10ml),依次加入三乙胺(0.6ml,4.3mmol),和异氰酸叔丁酯(0.35ml,3.06mmol)。充氮下,在85℃油浴中,将该混合物加热5小时。冷却至室温后,将该溶液倒入冰/水中,用6N盐酸酸化至pH2。过滤收集不溶的固体,用水洗涤,空气干燥,该粗产物经硅胶闪层析纯化,用氯仿/甲醇(9∶1)洗脱。合并含有所期产物的级份,浓缩后剩下固体,该固体用甲醇/氯仿/乙腈重结晶,然后用氯仿/甲醇重结晶,得到题目化合物(210mg,31%),m.p.>250℃。
1H NMR(DMSO-d6):δ9.85(s,1H),9.18(s,1H),8.70(dd,J=1.6,3.5Hz,1H),8.41(d,J=6.2Hz,1H)8.26(d,J=6.2Hz,1H),7.73(dd,J=1.6,4.9Hz,1H),7.16(dd,J=3.5,4.9Hz,1H),1.41(s,9H).IR(KBr disc)1723,1653,1615,1549,1474,1427cm-1.MS m/e(相对百分比)343(2),244(30),160(90),111(28),84(100).元素分析计算值C17H17N3O3S:C 59.46,H 4.99,N 12.24.实测值C 58.68,H 4.87,N 11.54.
实施例5
5-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.5-氟-2-羟基-3-硝基吡啶
按照Nesnow和Heidelberger<J.Heterocyclic Chem.,10,779(1973)>所述方法,由市售5-氨基-2-氟吡啶制备5-氟-2-羟基吡啶(5-氟-2-吡啶酮),不同之处仅在于:在最后一步2-氟-5-甲氧基吡啶的水解中采用了48%氢溴酸回流反应,而前述文献方法是在密封玻璃管中采用25%盐酸热至145℃反应。在0℃下,将5-氟-2-羟基吡啶(11.16g,98.7mmol)分次加入浓H2SO4(90ml)中,将该反应混合物温热至室温,然后在55-60℃加热3小时。将该混合物冷却至室温,然后倒入冰/水中。过滤收集黄色产物,用水洗涤,空气干燥,得到8.24g(53%)题目化合物。加入固体NaHCO3将滤液调至pH2,用乙酸乙酯提取。合并提取液,用MgSO4干燥,浓缩,又得到一部分题目化合物(1.71g),收率11%。
B.2-氯-5-氟-3-硝基吡啶
在60℃,将5-氟-2-羟基-3-硝基吡啶(6.5g,41.1mmol)分批加入五氯化磷(9.41g,45.2mmol)和三氯氧磷(4.2ml,45.1mmol)的混合物中。充氮下,将该混合物在100℃的油浴中加热过夜,冷却至室温,倒入冰/水中。再加入一部分水和乙酸乙酯,该混合物经硅藻土过滤除去深色不溶物,用盐水洗涤有机相,再次过滤又除去一部分深色物质,用MgSO4干燥,浓缩。残留物经硅胶闪层析纯化,用氯仿洗脱。合并含题目化合物的级份,浓缩后得到一黄色油(3.51g,48%),该油在5℃放置过液固化。
C.6-氟-4-氮杂羟基吲哚
在干燥烧瓶中放入3.1g60%氢化钠油分散体(77.5mmol)。用己烷洗涤两次除去大部分油。然后将留下的氢化钠固体混悬于无水DMF(100ml)中,冷却0℃。然后在搅拌下滴加丙二酸二乙酯(11.8ml,77.7mmol)。将该混合物在0℃搅拌1小时,然后加入2-氯-5-氟-3-硝基吡啶(5.21g,29.5mmol)的DMF(40ml)溶液。于室温下连继搅拌过夜,然后将该反应混合物倒入冰/水中。用6N盐酸将其酸化至pH3,然后用乙酸乙酯提取该混合物,用盐水洗涤有机相,用MgSO4干燥,减压浓缩,剩下一红色的油,后者经硅胶闪层析纯化,用3∶7乙酸乙酯/己烷洗脱。合并所有含所期产物的级份,浓缩后得到11.5g油状物,该油含有2-二(乙氧羰基)甲基-5-氟-3-硝基吡啶和丙二酸二乙酯,其摩尔比大约是11∶9。经计算,2-二(乙氧羰基)甲基-5-氟-3-硝基吡啶的得量大约是8g(90%)。
将2-二(乙氧羰基)甲基-5-氟-3-硝基吡啶和丙二酸二乙酯的混合物溶解在乙醇(100ml)中,并将其加到用150ml乙醇稀释的7.8g50%阮尼镍-水混悬液中。在帕尔振荡器中,于3个大气压下将该混合物氢化3小时,经硅藻土过滤除去催化剂,减压除去溶剂剩下3-氨基-2-二(乙氧羰基)甲基-5-氟吡啶和丙二酸二乙酯的油状混合物。将该混合物移至6N盐酸(280ml)中,加热回流3小时。减压除去含水酸,残留物溶于水,再次浓缩,剩下一固体,将后者溶于无水乙醇中,浓缩两次,得到淡绿色固体状题目化合物(4.07g),用热乙酸乙酯研制该固体,空气干燥。尽管核磁共振谱显示该物质稍有不纯,但可不经进一步纯化用于下一步反应。
D.5-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚
将颗粒状金属钠(0.75g,32.6mmol)加到于干燥园底烧瓶中的无水乙醇(30ml)中。待金属钠完全溶解后,加入固体状6-氟-4-氮杂羟基吲哚(1.0g,6.57mmol),然后加入2-噻吩甲酸乙酯(4.8ml,13.4mmol)。充氮下,将该混合物加热回流2小时,在此期间,出现黄色沉淀。将该混合物冷却,倒入冰/水中,用6N盐酸酸化至pH2,过滤收集题目化合物,经用水和醚洗涤,得量为854mg(50%),滤液结晶,又得到少量的第二批产物(34mg,2%)。
E.5-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将5-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(450mg,1.72mmol)溶于无水DMSO(15ml)中,依次加入三乙胺(0.50ml,3.59mmol)和异氰酸叔丁酯(0.30ml,2.62mmol)。充氮下,将所得混合物在85℃油浴中加热过夜。使该溶液冷却至室温,然后倒入冰/水中,加入1N盐酸将pH调至2。过滤收集不溶性绿色固体,空气干燥,经硅胶闪层析纯化,用乙酸乙酯洗脱。(为溶解固体,需加入一些乙腈)。合并含有所期产物的级份,浓缩。所得固体经乙酸乙酯/乙腈重结晶,得到题目化合物,为绿色针状结晶(181mg,29%)。
1H NMR(DMSO-d6):δ14.00(br s,1H),9.28(s,1H),8.77(d,J=4Hz,1H),8.44(dd,J=1.9,9.5Hz,1H),7.98-7.96(m,1H),7.89(d,J=4.9Hz,1H),7.24(dd,J=4,4.49Hz,1H),1.40(s,9H).IR(KBr disc)1721,1609,1552,1423cm-1.MS m/e(相对百分比)361(4),262(40),178(100),111(13).元素分析计算值C17H16FN3O3S:C 56.50,H 4.46,N 11.63.实测值:C 55.86,H 4.48,N 11.41.
实施例6
5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.5,6-二氯-4-氮杂羟基吲哚
按<Helv.Chim.Acta,59,190(1976)>所述方法,由市售五氯吡啶制得了3-硝基-2,5,6-三氯吡啶。第一步所得到的是所期2,5,6-三氯吡啶和少量三种四氯吡啶异构体的混合物。按前述参考文献方法将所得混合物硝化,得到3-硝基-2,5,6-三氯吡啶及少量2,5,6-三氯吡啶和四氯吡啶异构体(经实验很难分离)的混合物。在干燥的烧瓶中放入60%氢化钠油分散体(7.92g,198mmol),将后者混悬于无水DMF(90ml)中。然后,在搅拌下滴加丙二酸二乙酯。将该混合物于室温下搅拌0.25小时,冷却至0℃。将3-硝基-2,5,6-三氯吡啶(12.5g,55mmol),2,5,6-三氯吡啶(1.6g)及各四氯吡啶异构体(总量为6.4g)溶于DMF(40ml)中,将该溶液冷却,然后将其滴加到前述混合物中,将该混合物于0℃搅拌0.25小时,移至水中,并采用6N盐酸酸化。
经用醚提取后,合并醚提取层,用盐水洗涤,用MgSO4干燥,浓缩,剩下一油状物。使该油通过一厚的硅胶垫,先用己烷洗脱(除去油和三氯及四氯吡啶),然后用乙酸乙酯洗脱,洗脱出产物混合物。除去溶剂后,该混合物经硅胶闪层析纯化,用19∶1己烷/乙酸乙酯洗脱,合并所有含所期产物的级份,浓缩后剩下一油状物,其组成如下:所期望的〔2-二(乙氧基羰基)甲基-5,6-二氯-3-硝基吡啶〕(5.2g,收率:27%),2-二(乙氧基羰基)甲基-3,6-二氯-5-硝基吡啶(10.5g,收率:54%)和丙二酸二乙酯。
将上述所得混合物溶解在100ml乙醇中,然后将其加到用10ml乙醇稀释过的50%阮尼镍-水(30g)混悬液中。在帕尔振荡器中,于3个大气压下将该混合物氢化5小时,然后通过硅藻土过滤除去催化剂,减压下除去溶剂,得到一油状物,后者经硅胶闪层析纯化,用4∶1己烷/乙酸乙酯洗脱,分别将各级份浓缩,采用1H NMR(CDCl3)检测残留物,在洗脱出丙二酸二乙酯后,洗脱出所期产物3-氨基-2-二(乙氧基羰基)甲基-5,6-二氯吡啶,紧随其后的是不希望的异构体5-氨基-2-二(乙氧基羰基)甲基-3,6-二氯吡啶。尽管可以很干净地分离掉丙二酸二乙酯,但大量的所期产物和异构体以混合级份洗脱而出。将只含有3-氨基-2-二(乙氧基羰基)甲基-5,6-二氯吡啶的先导级份与含有至少10%该物质的混合级份合并,得到一其组成如下的固体:3-氨基-2-二(乙氧基羰基)甲基-5,6-二氯吡啶(3.17g)和不期望的异构体3-氨基-2-二(乙氧基羰基)甲基-3,6-二氯吡啶(4.03g)。将该混合物移至6N盐酸(120ml)中,加热回流3小时。冷却至室温后,减压除去挥发物。加入乙醇,然后蒸发帮助除去水。重复这一操作。所得棕色固体经硅胶闪层析纯化,用9∶1氯仿/甲醇洗脱。合并所有含所期产物的级份,浓缩后得到一固体,将后者用甲醇研制,得到5,6-二氯-4-氮杂羟基吲哚(1.42g),按3-氨基-2-二(乙氧基羰基)甲基-5,6-二氯吡啶计,收率为71%;按3-硝基-2,5,6-三氯吡啶计,总收率为13%,m.p.230-233℃(分解)。
B.5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚
将颗粒状金属钠(0.29g,12.6mmol)加到于干燥园底烧瓶中的无水乙醇(10ml)中。待金属钠完全溶解后,加入固体状5,6-二氯-4-氮杂羟基吲哚(500mg,2.46mmol),然后加入2-噻吩甲酸乙酯(0.67ml,5.0mmol),充氮下,将该混合物加热回流1天,将该混合物冷却,倒入冰/水中,用6N盐酸酸化至pH3,过滤收集题目化合物,真空干燥,得到607mg(79%)产物。
C.5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(300mg,0.96mmol)溶解在无水DMSO(8ml)中,依次加入三乙胺(0.20ml,1.43mmol),和异氰酸叔丁酯(0.16ml,1.4mmol),将所得混合物在充氮下,于85℃油浴中加热5小时,将该溶液倒入冰/水中,加入1N盐酸酸化至pH2,过滤收集不溶的固体,干燥,经硅胶闪层析纯化,用99∶1氯仿/甲醇洗脱,合并含所期产物的级份,浓缩,将所得固体用氯仿/甲醇重结晶,得到207mg题目化合物,m.p.189-190℃。1H NMR
1H NMR(DMSO-d6):δ9.66(d,J=4Hz,0.4H),9.02(d,J=4Hz,0.6H),8.84(br s,0.6H),8.70(s,0.4H),8.58(s,0.6H),8.44(br s,0.4H),7.83(d,J=5Hz,0.4H),7.76(d,J=5Hz,0.6H),7.30(dd,J=4.5Hz,0.4H),7.23(dd,J=4.5Hz,0.6H),1.47(s,9H).IR(KBr disc)1712,1640,1579,1534cm-1.Ms m/e(相对百分比)413(1),411(2),314(30),312(38),230(75),228(100),111(38)。元素分析计算值C17H15Cl2N3O3S:C 49.52,H 3.67,N 10.19;实测值C 49.45,H 3.58;N 9.91。
实施例7
3-(2-噻吩甲酰基)-6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.2-氯-3-硝基-5-三氟甲基吡啶
按英国专利1421619所述方法由市售2-氯-5-三氟甲基吡啶制备了起始原料2-羟基-3-硝基-5-三氟甲基吡啶。
将2-羟基-3-硝基-5-三氟甲基吡啶(8.8g,42.3mmol)在60℃加到三氯氧磷(4.2ml,45.9mmol)和五氯化磷(9.6g,46.1mmol)的混合物中。然后,充氮下,将该混合物于80℃加热过夜。使所得深色产物混合物冷却至室温,倒入冰/水中,用醚提取该混合物,用水和盐水洗涤醚提取液,经用MgSO4干燥后,除去溶剂,剩下一深色油,后者经硅胶闪层析纯化,用氯仿洗脱,合并只含有所期产物的级份,浓缩后,剩下一棕色油(5.0g,52%),1H NMR(DMSO-d6):δ9.20(s,1H),9.07(s,1H)。
B.2-二(苄氧羰基)甲基-3-硝基-5-三氟甲基吡啶
在干燥烧瓶中加入氢化钠(60%油分散体)(800mg,2.0mmol),用己烷洗涤两次除去大部分油,然后将留下的氢化钠混悬于无水1,2-二甲氧基乙烷(DME)(20ml)中,搅拌下滴加丙二酸二苄基酯(5.0ml,2.0mmol)的DME(15ml)溶液,将该混合物在室温下搅拌半小时,然后加入2-氯-3-硝基-5-三氟甲基吡啶(2.3g,10.2mmol)的DME(15ml)溶液,于室温下连续搅拌过夜,将该混合物倒入冰/水中,先用1N盐酸酸化至pH3,然后用乙酸乙酯提取该混合物,用盐水洗涤有机相,用MgSO4干燥,减压浓缩,得到一油状物。后者经硅胶闪层析纯化,用甲苯洗脱,合并所有含所期产物的级份,浓缩后得到一褐色固体;3.8g,(79%):m.p.82-84℃。
C.3-苄氧羰基-6-三氟甲基-4-氮杂羟基吲哚
在机械搅拌下,将2-二(苄氧羰基)甲基-3-硝基-5-三氟甲基吡啶(1.2g,2.5mmol),铁粉(495mg,8.9mmol),冰乙酸(50ml)的混合物加热回流2小时,冷却后,将该混合物倒入冰/水中,过滤收集白色固体沉淀,空气干燥,然后真空干燥过夜,得量:780mg(93%);m.p.>250℃。
D.3-苄氧羰基-6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将3-苄氧羰基-6-三氟甲基-4-氮杂羟基吲哚(750mg,2.23mmol)溶于无水DMSO(15ml)中,依次加入三乙胺(0.6ml,4.3mmol)和异氰酸叔丁酯(0.38ml,3.33mmol)。充氮下,将所得混合物在90-100℃油浴中加热5小时。冷却至室温后,将溶液倒入冰/水中,加入1N盐酸将其酸化至pH3。过滤收集不溶的固体,将其溶于氯仿中,用MgSO4干燥,减压浓缩,残留物经硅胶闪层析纯化,用氯仿洗脱,合并含有所期产物的级份,浓缩,剩下白色固体,得量:830mg(86%);m.p.>250℃。
E.6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将3-苄氧羰基-6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺(1.10g,2.53mmol),10%钯-炭(300mg),及乙醇(100ml)的混合物在帕尔振荡器中于3个大气压氢化2小时,使该混合物通过硅藻土过滤除去催化剂,蒸掉溶剂得到一灰色固体(6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺-3-羧酸)(850mg)。将该固体移至乙醇(100ml)中,加热回流1小时,冷却至室温后,减压除去溶剂,残留物经硅胶闪层析纯化,先用氯仿,再用10%甲醇/氯仿洗脱,合并含有所期产物的级份,浓缩,得到米色固体状产物;610mg(80%)。
F.3-(2-噻吩甲酰基)-6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺(350mg,1.16mmol)和4-二甲氨基吡啶(317mg,2.59mmol)溶于无水DMF(5ml)中,加入噻吩-2-甲酰氯(0.14ml,1.3mmol)。在室温下将该混合物搅拌1.5小时,然后倒入冰/水中,用1N盐酸将pH调至2左右,过滤收集所得沉淀,用水洗涤,空气干燥,然后用乙腈重结晶2次,得到一黄色固体,160mg(32%);m.p.>250℃。
1H NMR(CDCl3):δ14.12(br s,1H),9.18(s,1H),8.74(d,J=3Hz,1H),8.52(s,1H),8.14(s,1H),7.94(d,J=5Hz,1H),7.25(dd,J=3.5Hz,1H),1.41(s,9H).MS m/e(相对百分比)411(2),312(23),228(100),111(27).IR(KBr disc)1725,1675,1645,1605,1535,1520,1500,1415cm-1.元素分析计算值C18H16O3N3F3S:C 52.55;H,3.92;N 10.21.实测值C,52.52;H 3.84;N 10.12.
实施例8
3-(2-呋喃甲酰基)-6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
按实施例7F所述方法,从6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺开始,采用300mg(1.0mmol)6-三氟甲基-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺,0.11ml(1.1mmol)2-呋喃甲酰氯,244mg(2.0mmol)4-N,N-二甲氨基吡啶及10ml DMF制得了题目化合物。用甲醇研制粗产物,用乙酸重结晶,再用甲醇研制,得到题目化合物。得量:230mg(58%),m.p.>250℃。
1H NMR(DMSO d6)δ9.22(s,1H),8.52(d,δ=1.7Hz,1H),9.18-8.17(m,2H),8.00(d,δ=1.7Hz,1H),6.74-6.72(m,1H),1.41(s,9H).MS m/e(相对百分比)395(3),296(53),228(100).IR(KBr disc)1720,1670,1640,1615,1540,1515,1460,1425cm-1.元素分析计算值C18H16F3N3O4iH2O C 54.07,H 4.16,N 10.51.实测值:C 53.89,H 3.97,N 10.41.
实施例9
5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.(3-氨基-6-异丙氧基-2-吡啶基)乙腈
将叔丁醇钾(12.34g,110mmol)溶于无水DMF(30ml)中,冷却至-10℃,充氮,搅拌下滴加(4-氯苯氧基)乙腈(9.22g,55mmol)和2-异丙氧基-5-硝基吡啶(按Eriedman等人的方法制备<J.Am.Chem.Soc.69,1204(1947)>)(9.11g,50mmol)的DMF(30ml)溶液。在0至10℃下,将所得紫色溶液保温1小时,加入盐酸水溶液(80ml,5%),使所得混合物温热至室温,将该混合物用二氯甲烷提取两次,合并提取液,用MgSO4干燥,减压浓缩,留下一油状物,使后者通过一个厚硅胶垫,用1∶1二氯甲烷/己烷洗脱,减压蒸发滤液,将含有所期(6-异丙氧基-3-硝基-2-吡啶基)乙腈的残油溶解在6∶1乙醇/乙酸混合物中,在该混合物中加入5%钯-炭(0.8g)。在帕尔振荡器中,于3个大气压下,将该混合物氢化5小时,使该混合物经硅藻土过滤除去催化剂,减压浓缩滤液,将残油移至水中,加入NaCO3将pH调至10,将该混合物用二氯甲烷提取两次,合并提取液,用MgSO4干燥,浓缩。残留物经硅胶闪层析纯化。依次用1∶2醚/己烷,1∶1醚/己烷和乙酸乙酯洗脱,合并用乙酸乙酯洗脱出的含所期产物的级份,蒸除溶剂,得到米黄色固体状(3-氨基-6-异丙氧基-2-吡啶基)乙腈(5.60g,59%);m.p.83-85℃。
B.5-异丙氧基-4-氮杂羟基吲哚
将(3-氨基-6-异丙氧基-2-吡啶基)乙腈(4.5g,23.5mmol)溶于3N盐酸溶液中,并在50-55℃下加热过夜。冷却至0℃后,缓慢地加入浓NaOH溶液使该混合物碱化。将该混合物用乙酸乙酯提取两次,合并乙酸乙酯提取液,用盐水洗涤,用MgSO4干燥,浓缩后得到一固体。该固体经硅胶闪层析纯化,用9∶1氯仿/甲醇洗脱,将含产物(5-异丙氧基)-4-氮杂羟基吲哚的级份浓缩,得到一褐色固体(1.0g,22%)。
C.5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚
将颗粒状金属钠(250mg,11mmol)加到于干燥园底烧瓶中的无水乙醇(10ml)中,待金属钠完全溶解后,加入固体状5-异丙氧基-4-氮杂羟基吲哚(419mg,2.22mmol),然后加入2-噻吩甲酸乙酯(0.59ml,688mg),将该混合物加热回流过夜,然后冷却至室温,倒入冰/水中,然后用1N盐酸将该混合物酸化,用乙酸乙酯提取,合并乙酸乙酯提取液,用盐水洗涤,用MgSO4干燥,减压浓缩,残留物经硅胶闪层析纯化,依次用氯仿和49∶1氯仿/甲醇洗脱,合并含所期产物的所有级份,浓缩,残留物再次经硅胶闪层析纯化,用氯仿洗脱,合并只含有产物的级份,浓缩,得到5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(黄色凝胶)(300mg,45%)。
D.5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(300mg,1.0mmol)溶于无水DMSO(7ml)中,依次加入三乙胺(0.3ml,2.2mmol)和异氰酸叔丁酯(0.17ml,1.5mmol),在80℃油浴中将所得混合物加热4小时,待冷却至室温后,将该溶液倒入冰/水中,并用1N盐酸溶液酸化,过滤收集沉淀出的固体,空气干燥,将该物质溶解在醚中,用活性炭处理该醚溶液,使该混合物通过硅藻土过滤,减压浓缩滤液,得到一黄色固体;将5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺粗品用环己烷重结晶,得到纯产物,为淡黄色结晶固体(105mg,26%);m.p.160-163℃。
1H NMR(DMSO d6):δ9.20(br s,1H),8.87(d,J=3Hz,1H),8.43(d,J=8.5Hz,1H),7.90(d,J=5Hz,1H),7.23(dd,J=3,5Hz,1H),6.71(d,J=8.5Hz,1H),4.90(heptet,J=6Hz,1H),1.40(s,9H),1.38(d,J=6H,6H).MS m/e(相对百分比)401(12),302(61),176(100),148(18),111(59).IR(KBr disc)1703,1654,1624,1604,1547,1518,1472,1421cm-1.元素分析计算值C20H23H3O4S:C 59.83,H 5.77,N 10.47.实测值:C 59.59,H 5.62,N 10.46.
实施例10
5-苯硫基-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺
A.(3-硝基-6-苯硫基-2-吡啶基)乙酸叔丁酯
将2-氟-5-硝基吡啶(按Finger和Starr所述方法制备<J.Am.Chem.Soc.,81,2674(1959)>)(5.7g,40mmol)和(苯硫基)乙酸叔丁酯(9.0g,40mmol)的DMSO(75ml)溶液滴加到机械搅拌下的粉状氢氧化钠(16.0g,400mmol)的DMSO(75ml)浆液中,同时将反应温度维持在30℃以下,在室温下将该混合物搅拌过夜,然后倒入冰/水中,用1N盐酸将其pH调至2左右,然后用乙酸乙酯提取该混合物,合并有机提取液,用MgSO4干燥,减压浓缩,残油经硅胶闪层析纯化,用2∶1氯仿/己烷洗脱,合并含有所期产物的级份,浓缩,得到黄色固体,用醚研制,得到题目化合物(1.5g,11%),m.p.104-107℃。
B.5-苯硫基-6-氮杂羟基吲哚
将(3-硝基-6-苯硫基-2-吡啶基)乙酸叔丁酯(1.04g,3.0mmol)溶于含有铁粉(600mg,10.7mmol)的冰乙酸中,加热回流5小时,冷却至室温后,将该混合物倒入冰/水中,用氯仿提取,合并氯仿提取液,用盐水洗涤,干燥(MgSO4),浓缩,得到题目化合物,为淡黄色固体(560mg,77%),m.p.186-189℃。
C.5-苯硫基-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚
将颗粒状金属钠(264mg,11.5mmol)加到于干燥园序烧瓶中的无水乙醇(10ml)中,待金属钠完全溶解后,加入5-苯硫基-6-氮杂羟基吲哚(560mg,2.3mmol)的乙醇(5ml)浆液,将该混合物温热至50℃,此时加入2-噻吩甲酸乙酯(0.55ml,4.6mmol)。然后将该混合物加热回流30小时,待冷却至室温后,将该混合物倒入冰/水中,加入6N盐酸将其pH调至1,过滤收集沉淀出的固体,干燥,经硅胶闪层析纯化,用9∶1氯仿/甲醇洗脱,合并含有产物5-苯硫基-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚的级份,减压蒸除溶剂,得到一金黄色固体(620mg,76%);m.p.248-252℃(分解)。
D.5-苯硫基-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺
将5-苯硫基-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚(255mg,0.72mmol)溶于DMSO(5ml)中,依次加入三乙胺(0.2ml,1.4mmol)和异氰酸叔丁酯(0.12ml,1.1mmol),将该溶液在80℃加热过夜,待冷却后将该混合物倒入冰/水中,加入1N的盐酸将其酸化至pH2.5左右,过滤收集固体,空气干燥,用氯仿,以及甲醇/氯仿重结晶,得到题目化合物(70mg,22%);m.p.>250℃。
1H NMR(DMSO d6)δ9.885(s,1H),8.95(s,1H),8.52-8.50(m,1H),7.92(s,1H),7.64-7.62(m,1H),7.42-7.30(m,5H),7.12-7.07(m,1H),1.39(s,9H).IR(KBr disc)1706,1619,1587,1554,1465,1427cm-1.元素分析计算值C23H21O3N3S21/3CHCl3:C 57.04,H 4.38,N 8.55.实测值:C 56.74,H 4.60,N 8.23.
实施例11
6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
按照实施例1C所述方法,从6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例2B)开始,制得了题目化合物,具体采用6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(450mg,1.6mmol),异氰酸叔丁酯(0.78ml,2.4mmol),三乙胺(0.49ml,3.5mmol)和DMSO(10ml)。粗产物经甲醇/氯仿重结晶后得到330mg(55%)产物。MS m/e(相对百分比)379(3),377(10),280(24),278(71),196(40),194(100),111(20).IR(KBr disc)1717,1659,1597,1424cm1.元素分析计算值C17H16ClN3O3S:C 54.04,H 4.27,N 11.12;实测值:C 53.64,H 4.14,N 10.99.
实施例12
6-氯-3-(2-呋喃甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
按照实施例2B所述方法,先制得了6-氯-3-(2-呋喃甲酰基)-4-氮杂羟基吲哚,具体采用6-氯-4-氮杂羟基吲哚(1.0g,5.9mmol),钠(678mg,29.5mmol),2-糠酸乙酯(1.65ml,11.8mmol)及乙醇(30ml)。用热乙醇研制粗产物。得量:825mg(53%),m.p.250℃。
然后按实施例1C方法,由6-氯-3-(2-呋喃甲酰基)-4-氮杂羟基吲哚制备题目化合物,具体采用6-氯-3-(2-呋喃甲酰基)-4-氮杂羟基吲哚(400mg,1.5mmol),异氰酸叔丁酯(0.26mg,2.2mmol)及三乙胺(0.41ml,3.0mmol)。反应时间为4小时,粗产物经氯仿重结晶,得量:190mg(35%)
元素分析计算值C17H16ClN3O4:C 56.44,H 4.46,N 11.61;实测值C 56.17,H 4.26,N 11.20.M.p.>250℃.
实施例13
6-氯-3-(3-呋喃甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
首先按实施例2B方法制得了6-氯-3-(3-呋喃甲酰基)-4-氮杂羟基吲哚,采用6-氯-4-氮杂羟基吲哚(1.5g,8.9mmol),钠(1g,44.5mmol),3-糠酸乙酯(2.4ml,17.8mmol),和乙醇(40ml)。用热乙醇研制粗产物,得量:1.0g(43%),m.p.250℃。
然后按实施例2C方法,从6-氯-3-(3-糠酰)-4-氮杂羟基吲哚开始制备题目化合物,具体采用6-氯-3-(3-糠酰)-4-氮杂羟基吲哚(500mg,1.9mmol),N-氯磺酰异氰酸酯(0.25ml,2.8mmol),和乙腈(20ml),粗产物经乙酸重结晶,得量:175mg(30%),元素分析计算值:C13H8ClN3O4:C 51.08,H 2.64,N 13.75;实测值C 51.04,H 2.41,N 13.46.M.p.>250°。
实施例14
3-苯甲酰基-6-氯-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例2B方法,制备了3-苯甲酰基-6-氯-4-氮杂羟基吲哚,具体采用6-氯-4-氮杂羟基吲哚(1.5g,8.9mmol),钠(1g,44.5mmol),苯甲酸乙酯(2.5ml,17.5mmol)及乙醇(40ml)。用热甲醇研制粗产物,得量:1.2g(49%)。
然后按实施例1C方法由前述产物制备了题目化合物,具体采用3-苯甲酰基-6-氯-4-氮杂羟基吲哚(500mg,1.83mmol),异氰酸叔丁酯(0.3ml,2.62mmol),三乙胺(0.5ml,3.59mmol)和DMSO(15ml)。粗产物经甲醇/氯仿重结晶。
1H NMR(DMSO-d6):δ9.10(s,1H),8.43(d,J=1.4Hz,1H),7.93(d,J=1.4Hz,1H),7.73(d,J=7Hz,2H),7.52-7.42(m,3H),1.35(s,9H).元素分析计算值C19H18ClN3O3:C 61.38,H 4.88,N 11.30;实测值C 61.19,H 4.51,N 10.99.M.p.>260℃.
实施例15
6-氯-3-(2-糠酰)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,从6-氯-3-(2-糠酰)-4-氮杂羟基吲哚开始,制备了题目化合物,具体采用400mg(1.5mmol)6-氯-3-(2-糠酰)-4-氮杂羟基吲哚,0.19ml(2.25mmol)N-氯磺酰异氰酸酯和15ml乙腈。在通大气的烧瓶中将N-氯磺酰甲酰胺粗品在DMSO(5ml)中搅拌2小时使之水解。用水稀释,过滤分离产物,并用乙酸重结晶,得量:160mg(35%)。
元素分析计算值C13H8ClN3O4:C 51.08,H 2.64,N 13.75;实测值C 51.24,H 2.55,N 13.44.M.p.>250°.
实施例16
6-氯-3-(4-氯-2-噻吩甲酰)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例2B方法制备6-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚,具体采用:6-氯-4-氮杂羟基吲哚(1.5g,8.9mmol)、钠(1.0g,44.5mmol),4-氯-2-噻吩甲酸乙酯(3.3g,17.8mmol)、和乙醇(40ml),得量:1.8g(64%),m.p.>250℃。
然后按实施例1C方法,从6-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚开始,制备了题目化合物,具体采用6-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚(900mg,2.8mmol)、异氰酸叔丁酯(0.49ml,4.3mmol)、三乙胺(0.77ml,5.6mmol)及DMSO(25ml)。粗产物经甲醇重结晶,得量:140mg(12%)。
元素分析计算值C17H15Cl2N3O3S:C 49.52,H 3.67,N 10.19;实测值C 49.18,H 3.31,N 10.00.M.p.>250°.
实施例17
6-氯-3-(3-糠酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
按实施例1C的方法,从6-氯-3-(3-糠酰基)-4-氮杂羟基吲哚(实施例13)制得了题目化合物,具体采用6-氯-3-(3-糠酰基)-4-氮杂羟基吲哚(500mg,1.9mmol)、异氰酸叔丁酯(0.32ml,2.8mmol)、三乙胺(0.52ml,3.8mmol)和DMSO(15ml)。粗产物经连续硅胶层析纯化,用氯仿洗脱,经环己烷重结晶,再经硅胶闪层析用1∶1乙酸乙酯/己烷洗脱,最后用环己烷重结晶。得量:160mg(23%)。
元素分析计算值C17H16ClN3O4:C 56.44,H 4.46,N 11.61;实测值C 56.38,H 4.45,N 10.67.M.p.250°.
实施例18
3-苯甲酰基-6-氯-4-氮杂羟基吲哚-1-甲酰胺
按照实施例2C的方法,由3-苯甲酰基-6-氯-4-氮杂羟基吲哚(实施例14)制得了题目化合物,具体采用3-苯甲酰基-6-氯-4-氮杂羟基吲哚(500mg,1.83mmol)、N-氯磺酰基异氰酸酯(0.24ml,2.76mmol)和乙腈(20ml)。粗产物用乙腈/氯仿重结晶,得量:121mg(21%)。
元素分析计算值C15H10ClN3O3:1/4CHCl3:C 53.01,H 2.98,N 12.16;实测值C 53.14,H 2.78,N 11.92.M.P.260°.
实施例19
6-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按照实施例2C方法,由6-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚制备了题目化合物,具体采用6-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚(850mg,2.7mmol)、N-氯磺酰基异氰酸酯(0.35ml,4.0mmol)及乙腈(30ml)。粗产物用乙酸重结晶,得量:280mg(29%)。
1H NMR(DMSO-d6):δ8.75(d,J=1.2Hz,1H),8.49(br s,1H),8.38(d,J=1.6Hz,1H),7.92(m,3H).MS m/e(相对百分比)355(5),314(8),312(14),196(21),194(72),145(30).IR(KBr disc)1730,1680,1600,1510,1415cm-1.元素分析计算值C13H7Cl2N3O3S:C 43.84,H 1.98,N 11.80;实测值C 43.90,H 2.01,N 11.23.M.p.>250°.
实施例20
6-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按照修改的实施例2B方法,制得了6-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚,具体采用6-氯-4-氮杂羟基吲哚(1.3g,7.71mmol)、钠(1.75g,49.7mmol)、4-甲基噻吩-2-甲酰氯(1.93g,12.0mmol)和乙醇(40ml)。将该酰氯加到乙醇钠溶液中就地制得了4-甲基噻吩-2-甲酸乙酯。加入所述氮杂羟基吲哚并且按实施例1B所述进行反应。得量:1.64g(46%)m.p.>250℃。
按照实施例1C的方法,由6-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚制得了题目化合物,具体采用6-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚(470mg,1.6mmol)、异氰酸叔丁基酯(0.27ml,2.4mmol)、三乙胺(1.1ml,3.2mmol)和DMSO(30ml)。粗产物经连续硅胶闪层析纯化,用氯仿/乙醇重结晶。得量:300mg(48%),m.p.>250℃。
1H NMR(DMSO d6)δ9.19(5,1H),8.57(s,1H),8.42(s,1H),7.91(s,1H),7.50(s,1H),2.27(s,3H),1.41(s,9H).MS m/e(相对百分比)393(1),391(3),294(10),292(27),196(33),194(100),125(23).IR(KBR disc)1725,1710,1660,1630,1600,1560,1525,1500cm-1.元素分析计算值C18H18ClN3O3S:C 55.17,H 4.63,N 10.72.实测值C 55.17,H 4.34,N 10.51.
实施例21
6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-苯基甲酰胺
按照实施例1B的方法,由6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例2B)制备了题目化合物,具体采用6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(1.0g,3.6mmol)、异氰酸苯酯(0.5ml,5.4mmol)、三乙胺(1.0ml,7.2mmol)和DMSO(35ml)。粗产物先用乙酸,再用DMSO重结晶,通过用甲醇研制除去痕量的DMSO。得量:515mg(36%),m.p.>250℃。
MS m/e(相对百分比)399(8),397(23),280(37),278(100),196(28),194(86),119(93).IR(KBr disc)1720,1680,1630,1605,1580,1500,1425,1405cm-1.元素分析计算值C19H12ClN3O3S:C 57.36,H 3.04,N 10.56.实测值C 56.58,H 2.95,N 10.27.
实施例22
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制得了题目化合物,具体采用5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(500mg,1.79mmol)、N-氯磺酰基异氰酸酯(0.18ml,2.15mmol)和乙腈(15ml)。在与大气相通的烧瓶中,将N-氯磺酰基甲酰胺粗品在DMSO(1.5ml)中搅拌1小时使之水解。加入水沉淀出产物,过滤收集沉淀物,用乙酸重结晶,得量:36mg(6%)。
MS m/e(相对百分比)323(7),321(17),280(9),278(24),196(22),194(62),170(25),168(100).元素分析计算值C13H8 35ClN3O3S:320.9975;实测值320.9977.IR(KBr disc)1724,1623,1570,1512,1415cm-1.M.p.222-224℃.
实施例23
5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例1B方法制备5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚,具体采用5-氯-4-氮杂羟基吲哚(1.0g,5.9mmol)、钠(0.68g,29.6mmol)、乙醇(30ml)、2-糠酸乙酯(1.65g,11.8mmol)。得量:500mg(33%),m.p.>250℃。
然后按实施例1C方法制备题目化合物,具体采用5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚(500mg,1.90mmol)、异氰酸叔丁酯(0.33ml,2.9mmol)、三乙胺(0.53ml,3.8mmol)和DMSO(10ml)。粗产物用甲醇重结晶。得量:240mg(35%),m.p.194-195℃。
1H NMR(DMSO-d6):δ8.96(s,1H),8.38(s,1H),8.35(d,J=8.3Hz,1H),8.06(s,1H),7.24(d,J=8.3Hz,1H),6.80-6.78(m,1H),1.40(s,9H).IR(KBr disc)1725,1590,1569,1541cm-1.MS m/e(相对百分比)361(10),262(13),196(37),194(100),95(4).元素分析计算值C17H16ClN3O4:C 56.44,H,4.46,N 11.61.实测值C 56.18,H 4.43,N 11.56.
实施例24
5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚(实施例23)制得了题目化合物,具体采用5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚(200mg,0.76mmol)、N-氯磺酰基异氰酸酯(0.10ml,1.1mmol)和乙腈(8ml)。加入水使上述反应淬灭,并将该混合物在室温下搅拌过夜,以此使N-氯磺酰基甲酰胺水解。过滤收集产物,用DMSO进行重结晶。得量:75mg(32%)。
FAB MS m/e 306 m.p.248-260℃。
实施例25
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-苯基甲酰胺
按实施例1C方法,由5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制得了题目化合物,具体采用5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(500mg,1.8mmol)、异氰酸苯酯(0.29ml,2.7mmol)、三乙胺(0.54ml,3.9mmol)、DMSO(10ml)。粗产物经乙酸乙酯重结晶。准确计算其分子量为
C19H12 35ClN3S:397.0287;实测值为397.0295.IR(KBr disc)1728,1622,1603,1582,1563,1412cm-1.元素分析计算值C19H12ClN3O3S:C 57.36,H 3.04,N 10.56;实测值C 56.84,H 2.87,N 10.52.M.p.226-228℃.
实施例26
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-环己基甲酰胺
按实施例1C方法,由5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制得了题目化合物,具体采用5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(560mg,2.0mmol)、异氰酸环己基酯(0.38ml,3.0mmol)、三乙胺(0.56ml,4.0mmol)和DMSO(10ml)。用甲醇研制粗产物,并用甲醇/氯仿重结晶。得量:91mg(11%)。
M.P.169-170°.元素分析计算值C19H18Cl3O3S 1/2 H2O:C 55.27,H 4.64,N 10.17.实测值C 55.17,H 4.34,N 9.87.
MS m/e(相对百分比)405(1),403(3),280(22),278(56),196(33),194(100),111(14).IR(KBr disc)1712,1625,1585,1518,1417cm-1.
实施例27
5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
按照实施例20的方法,首先制备了5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚,具体采用5-氯-4-氮杂羟基吲哚(1.0g,5.93mmol)、钠(0.95g,41.3mmol)、乙醇(25ml)、4-氯噻吩-2-甲酰氯(2.2g,11.3mmol)。得量:1.68g(90%)。
然后,按实施例1C方法,由5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚制备了题目化合物,具体采用5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚(0.75g,2.39mmol)、异氰酸叔丁酯(0.4ml,3.5mmol)、三乙胺(0.65ml,4.7mmol)和DMSO(20ml)。粗产物经连续硅胶闪层析纯化,用乙酸乙酯洗脱,并用甲醇/氯仿重结晶。得量:438mg(44%)。
元素分析计算值C17H15Cl2N3O3S:C 49.53,H 3.67,N 10.19;实测值C 49.58,H 3.39,N 9.94.M.p.208-209℃.
实施例28
5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C的方法,由5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例27)制备了题目化合物,具体采用:5-氯-3-(4-氯-2-噻吩甲酰基)-4-氮杂羟基吲哚(0.75g,2.39mmol)、N-氯磺酰基异氰酸酯(0.31ml,3.56mmol)和乙腈(12ml)。在与大气相通的烧瓶中,于室温下,将N-氯磺酰基甲酰胺在DMSO(8ml)中搅拌4小时使之水解。用水稀释该混合物,然后过滤收集产物,最后用乙酸重结晶。得量:352mg(41%)。
元素分析计算值C13H7Cl2N3O3S:C 43.84,H 1.98,N 11.80;实测值C 43.52,H 1.93,N 11.52.M.p.>250℃.
实施例29
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-(2,4-二氯苯基)甲酰胺
按实施例1C方法,由5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制备了题目化合物,具体采用5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(580mg,2.08mmol)、三乙胺(0.58ml,4.15mmol)、异氰酸2,4-二氯苯基酯(0.57g,3.1mmol)和DMSO(10ml)。粗产物用甲醇/氯仿重结晶。得量:231mg(24%)。
元素分析计算值C19H10Cl3N3O3S:C 48.90,H 2.16,N 9.00;实测值C 48.73,H 1.95,N 8.97.M.p.244.5-245℃.
实施例30
5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例27方法,从5-氯-4-氮杂羟基吲哚开始,制备了5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚,具体采用5-氯-4-氮杂羟基吲哚(1.45g,8.60mmol)、钠(1.3g,56.5mmol)、乙醇(40ml)和4-甲基噻吩-2-甲酰氯(2.38g,14.8mmol)。得量:1.71g(76%)。反应时间:过夜。
然后按实施例1C方法,由5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚制备了题目化合物,具体采用5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚(0.8g,2.73mmol),异氰酸叔丁酯(0.5ml,4.36mmol)、三乙胺(0.8ml,5.74mmol)和DMSO(25ml)。粗产物先经硅土闪层析纯化,用乙酸乙酯洗脱,然后用甲醇/二氯甲烷重结晶,得量:600mg(56%)。
1H NMR(DMSO-d6):δ9.06(s,1H),8.63(s,1H),8.37(d,J=8.2Hz,1H),7.61(s,1H),7.23(d,J=8.2Hz,1H),2.31(s,3H),1.42(s,9H).MS m/e(相对百分比)393(1),391(4),294(16),292(43),196(33),194(100),125(11).IR(KBr disc)1720,1670,1585,1540,1415cm-1.元素分析计算值C18H18ClN3O3S:C 51.17,H 4.63,N 10.72;实测值C 55.14,H 4.38,N 10.57.M.p.167-169℃.
实施例31
5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例30)制得了题目化合物,具体采用:5-氯-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚(0.90g,3.07mmol)、N-氯磺酰基异氰酸酯(0.40ml,4.60mmol)和乙腈(15ml)。在与大气相通的烧瓶中,将N-氯磺酰基异氰酸酯粗品在DMSO中搅拌,使之水解。用水稀释,过滤分离产物,并用乙酸重结晶,得量:190mg(18%),m.p.227-228℃。
元素分析计算值C14H10ClN3O3S:C 50.08,H 3.00,N 12.5.
实测值C 49.88,H 2.96,H 12.39.
1H NMR(DMSO-d6)δ:8.71(s,1H),8.46(br s,1H),8.35(d,J=8.5Hz,1H),7.84(br s,1H),7.61(s,1H),7.24(d,J=8.5Hz,1H),2.31(s,3H).MS m/z(相对百分比)337(33),336(42),335(100).IR(KBr disc)1730,1630,1580,1430cm-1.
实施例32
3-苯甲酰基-5-氯-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例1B方法制备了3-苯甲酰基-5-氯-4-氮杂羟基吲哚,具体采用:5-氯-4-氮杂羟基吲哚(1.5g,8.9mmol)、钠(1.0g,44.4mmol)、乙醇(40ml)和苯甲酸乙酯(2.5ml,17.5mmol)。得量:1.6g(66%)。
然后按照实施例1C方法,由3-苯甲酰基-5-氯-4-氮杂羟基吲哚制得了题目化合物,具体采用:3-苯甲酰基-5-氯-4-氮杂羟基吲哚(800mg,2.93mmol)、三乙胺(0.8ml,5.74mmol)、异氰酸叔丁酯(0.5ml,4.36mmol)和DMSO(25ml)。
粗产物经硅胶闪层析纯化,用乙酸乙酯洗脱,然后用己烷重结晶,得量:320mg(29%)。
M.p.107-111℃.元素分析计算值C19H18ClN3O3:C 61.38,H 4.88,N 11.30.实测值C 62.00,H 5.11,N 10.75.
1H NMR(DMSO-d6)δ9.03(s,1H),8.36(d,J=8.2Hz,1H),7.78(d,J=6.9Hz,2H),7.59-7.46(m,3H),7.20(d,J=8.2Hz,1H),1.35(s,9H).MS m/e(相对百分比)373(1),371(3),274(34),272(100),194(44),105(55).IR(KBr disc)1730,1720,1590,1550,1455cm-1.
实施例33
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-甲基甲酰胺
按照与实施例1C稍有不同的方法,从5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制得了题目化合物。在55℃进行该反应,采用干冰冷凝器以防丢失异氰酸甲酯。反应时间:5小时。所使用的反应物量如下:5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(560mg,2.0mmol)、异氰酸甲酯(0.18ml,3.0mmol)、三乙胺(0.56ml,4.0mmol),和DMSO(10ml)。
粗产物经甲醇/氯仿重结晶,得量:151mg(23%),m.p.179-180℃。
1H NMR(CDCl3)δ8.98-8.94(m,2H),8.45(d,J=8.5Hz,1H),7.70(d,J=5Hz,1H),7.22(dd,J=3,5Hz,1H),7.02(d,J=8.5Hz,1H),3.01(s,0.5H),2.99(s,0.5H).
元素分析计算值C14H10ClN3O3S 1/2 H2O:C 47.88 H 3.21,N 12.18.实测值C 49.00,H 2.84,N 12.05.精确计算分子量为C14H10 35ClN3O3S:335.0121.实测值335.0012.
实施例34
5-氯-3-(3-糠酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例1B方法制备了5-氯-3-(3-糠酰基)-4-氮杂羟基吲哚,具体采用:5-氯-4-氮杂羟基吲哚(1.0g,5.9mmol)、钠(6.82mg,29.6mmol)、3-糠酸乙酯(1.5ml,11.8mmol)和乙醇(25ml)。得量:1.2g(80%)。
按照实施例1C的方法,从5-氯-3-(3-糠酰基)-4-氮杂羟基吲哚制备了题目化合物,具体采用:5-氯-3-(3-糠酰基)-4-氮杂羟基吲哚(1.2g,4.5mmol)、异氰酸叔丁酯(0.78ml,6.8mmol)、三乙胺(1.2ml,9.0mmol)和DMSO(45ml)。
粗产物经硅胶闪层析纯化,用4∶1乙酸乙酯/己烷洗脱,处理含有所期产物的级份得到一固体,后者用甲醇研制,并用乙腈重结晶,得量:740mg(76%),m.p.182-184℃。
1H NMR(DMSO-d6)δ9.13(br s,1H),8.77(br s,1H),8.32(d,J=8.6Hz,1H),7.86(s,1H),7.25(d,J=8.6Hz,1H),7.12(s,1H),1.40(s,9H).MS m/e(相对百分比)363(5),361(16),264(34),262(100),247(6),245(19),236(17),234(41),194(24).IR(KBr disc)1725,1490,1545cm-1.元素分析计算值C17H16ClN3O4:C 56.44,H 4.46,N 11.61.实测值C 56.33,H 4.17,N 11.68.
实施例35
6-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按照实施例2C方法,由6-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例5)制备了题目化合物,具体采用6-氟-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(419mg,1.60mmol)、N-氯磺酰基异氰酸酯(0.2ml,2.3mmol)和乙腈(8ml)。反应时间是3天。在与大气相通的烧瓶中,搅拌处于DMSO中的N-氯磺酰基甲酰胺粗品,使之水解,用水稀释,过滤收集产物,用乙酸重结晶,得量:210mg(43%)。
元素分析计算值C13H8FN3O3S:C 51.15,H 2.64,N 13.76;实测值C 50.90,H 2.46,N 13.45.M.p.265℃.
实施例36
6-氟-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按照实施例20的方法制备了6-氟-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚,具体采用:6-氟-4-氮杂羟基吲哚(1.0g,657mmol)、钠(1.05g,45.6mmol)、乙醇(30ml)和4-甲基噻吩-2-甲酰氯(1.86g,11.6mmol)。得量:1.17g(64%)。
然后按实施例5E方法,由6-氟-3-(4-甲基-2-噻吩甲酰基)氮杂羟基吲哚制备了题目化合物。经层析纯化后,将产物用甲醇重结晶,得量:234mg(31%)。
1H NMR(DMSO-d6):δ14.0(br s,1H),9.24(s,1H),8.60(s,1H),8.45(dd,J=2.3,9.4Hz 1H),7.99-7.96(m,1H),7.50(s,1H),2.28(s,3H),1.42(s,9H).MS m/e(相对百分比)375(15),276(67),178(100),125(12).IR(KBr disc)1720,1670,1610,1560,1530,1425cm-1.元素分析计算值C18H18FN3O3S:C 57.59,H 4.83,N 11.19;实测值C 57.37,H 4.73,N 11.33.M.p.275℃.
实施例37
6-氟-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由6-氟-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例36)制得了题目化合物,具体采用:6-氟-3-(4-甲基-2-噻吩甲酰基)-4-氮杂羟基吲哚(614mg,2.22mmol)、N-氯磺酰基异氰酸酯(0.30ml,3.45mmol)和乙腈(10ml)。在与大气相通的烧瓶中将上述N-氯磺酰基甲酰胺粗品在DMSO(4ml)中搅拌过夜使之水解,用水稀释,过滤收集产物,并用乙酸重结晶,得量:249mg(35%),m.p.>250℃。
元素分析计算值C14H10FN3O3S:C 52.66,H 3.16,N 13.16.实测值C 52.16,H 3.00,N 13.03.
1H NMR(DMSO-d6)δ13.96(br s,1H),8.64(s,1H),8.60(br s,1H),8.39(dd,J=2.3,9.5Hz,1H),7.96(dd,J=2.3,3.5Hz,1H),7.89(br s,1H),7.49(s,1H),2.26(s,3H).Ms m/e(相对百分比)319(7),276(13),178(100),125(19).IR(KBr disc)1725,1610,1590,1510,1430cm-1.
实施例38
3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例1B方法制备了3-(2-噻吩甲酰基)-6-氮杂羟基吲哚,具体采用6-氮杂羟基吲哚(2.8g,20.9mmol)、钠(2.4g,104mmol)、乙醇(45ml)和噻吩-2-甲酸乙酯(55ml,40.9mmol)。得量:4.05g(79%),m.p.>280℃。
然后按实施例1C方法,由3-(2-噻吩甲酰基)-6-氮杂羟基吲哚制备了题目化合物,具体采用:3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(500mg,2.0mmol)、异氰酸叔丁酯(0.35ml,3.0mmol)、三乙胺(0.6ml,4.4mmol)和DMSO(10ml)。
粗产物经闪层析纯化,用9∶1氯仿/甲醇洗脱,合并只含有所期产物的级份,浓缩后得到一固体,后者用丙酮/甲醇/氯仿重结晶。得量:390mg(57%)。
1H NMR(DMSO-d6):δ9.57(s,1H),8.91(s,1H),8.43(d,J=3.5Hz,1H),8.15(d,J=6.4Hz,1H),8.09(d,J=6.4Hz,1H),7.80(d,J=5Hz,1H)),7.17(dd,J=3.5,5Hz,1H),1.40(s,9H).MS m/e(相对百分比)343(0.4),244(6),160(27),111(9),84(100).IR(KBr disc)1715,1593,1539,1408cm-1.元素分析计算值C17H17N3O3S:C 59.46,H 4.99,N 12.24;实测值C 58.99,H 4.85,N 12.10.M.p.250-252℃.
实施例39
5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-甲酰胺
按实施例2C的方法,由5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚(实施例3C)制得了题目化合物,具体采用:5-氯-3-(2-噻吩甲酰基)-6-氮杂羟基吲哚(140mg,0.5mmol)、N-氯磺酰基异氰酸酯(65ml,0.75mmol)和乙腈(5ml)。在与大气相通的烧瓶中,将上述N-氯磺酰基甲酰胺粗品在DMSO中搅拌2小时使其水解。加入水沉淀出产物,过滤收集,固体用甲醇重结晶,得量:27mg(17%)。
1H NMR(DMSO-d6):δ8.73(br s,1H),8.61(s,1H),8.42(d,J=4Hz,1H),8.03(s,1H),7.80(d,J=5Hz,1H),7.76(br s,1H),7.14(dd,J=4,5Hz,1H).FAB MS m/e 322.M.p.252℃.
实施例40
3-(2-噻吩甲酰基)-7-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先按实施例1B方法制备了3-(2-噻吩甲酰基)-7-氮杂羟基吲哚,具体采用:7-氮杂羟基吲哚(1.5g,11.2mmol)、钠(1.3g,56.5mmol)、乙醇(25ml)和噻吩-2-甲酸乙酯(3ml,22.3mmol)。得量:2.54g(93%)。
然后按实施例1C方法,由3-(2-噻吩甲酰基)-7-氮杂羟基吲哚制备了题目化合物,具体采用:3-(2-噻吩甲酰基)-7-氮杂羟基吲哚(500mg,2.0mmol)、异氰酸叔丁酯(0.35ml,3.0mmol)、三乙胺(0.6ml,4.4mmol)和DMSO(10ml)。粗产物经硅胶闪层析纯化,用9∶1氯仿/甲醇洗脱,并且用甲醇/氯仿重结晶。得量:70mg(25%)。
1H NMR(DMSO-d6):δ9.34(s,1H),8.81(d,J=3.5Hz,1H),8.54(d,J=8.0Hz,1H),7.91-7.87(m,2H),7.25-7.18(m,2H),1.41(s,9H).MS m/e(相对百分比)343(35),244(81),160(100),111(9).IR(KBr disc)1718,1654,1629,1607,1554,1534,1497,1433cm-1.元素分析计算值C17H17N3O3S:C 59.46,H 4.99,N 12.24;实测值C 59.24,H 4.77,N 12.14.M.p.>250℃.
实施例41
5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例6B)制得了题目化合物,具体采用:5,6-二氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(194mg,0.62mmol)、N-氯磺酰基异氰酸酯(81ml,0.93mmol)、和乙腈(10ml),反应温度为50℃。在与大气相通的烧瓶中,将上述N-氯磺酰基甲酰胺粗品在DMSO中搅拌使之水解,用水稀释后,过滤收集固体,并且用乙酸重结晶。得量:129mg(58%)。
1H NMR(DMSO-d6):δ8.66(d,J=3.5Hz,1H),8.58(br s,1H),8.36(s,1H),7.95(d,J=5Hz,1H),7.78(br s,1H),7.26(dd,J=3.5,5Hz,1H).IR(DMSO)1710,1550,1515,1455cm-1.元素分析计算值C13H7Cl2N3O3S:C 43.84,H 1.98,N 11.80;实测值C 43.65,H 1.87,N 11.68.M.p.237-239.5℃.
实施例42
3-(2-噻吩甲酰基)-6-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由3-(2-噻吩甲酰基)-6-氮杂羟基吲哚(实施例38)制得了题目化合物,具体采用:3-(2-噻吩甲酰基)-6-氮杂羟基吲哚(3.09g,12.6mmol)、N-氯磺酰基异氰酸酯(1.2ml,13.8mmol)和乙腈(60ml)。反应时间为3.5小时。
在与大气相通的烧瓶中,将上述N-氯磺酰基甲酰胺粗品在DMSO(30ml)中搅拌过夜使之水解。用水稀释后,过滤收集固体,干燥。然后将该固体用乙酸重结晶两次,用甲·乙酮重结晶一次。得量:163mg(5%),m.p.213-215℃。
1H NMR(DMSO-d6)δ8.86(br s,2H),8.45(d,J=3.6Hz,1H),8.16-8.09(m,2H),8.80(d,J=4.3Hz,1H),7.76(br s,1H),7.16(dd,J=3.6,4.3Hz,1H).IR(KBr disc)1733,1708,1630,1559,1517,1479,1418cm-1.元素分析计算值C13H9N3O3S:C 54.35,H 3.16,N 14.63.实测值C 54.04,H 3.24,N 14.16.
实施例43
3-苯基乙酰基-6-氮杂羟基吲哚-1-甲酰胺
首先按实施例1B的方法,由6-氮杂羟基吲哚制得了3-苯基乙酰基-6-氮杂羟基吲哚,具体采用6-氮杂羟基吲哚(1g,7.4mmol)、钠(257mg,11.2mmol)、苯基乙酸乙酯(2.3ml,14.9mmol)和乙醇(30ml)。反应时间为4.5小时。反应后处理包括将该混合物倒入冰/水中,并将该混合物调至pH6左右。过滤收集产物,用乙酸乙酯洗涤。得量:1.23g(66%)。m.p.250℃。
按实施例2C方法,由3-苯基乙酰基-6-氮杂羟基吲哚制备了题目化合物,具体采用3-苯基乙酰基-6-氮杂羟基吲哚(1.0g,3.9mmol)、N-氯磺酰基异氰酸酯(0.41ml,4.7mmol)和乙腈(40ml)。反应时间为4小时。在与大气相通的烧瓶中,将上述N-氯磺酰基甲酰胺粗品在DMSO(6ml)中搅拌1小时使之水解,经用水稀释后,过滤收集产物,干燥,用乙酸重结晶,得量:172mg(15%),m.p.131-133℃(分解)。
1H NMR(DMSO-d6)δ13.28(s,1H),8.86(br s,1H),8.80-8.78(m,1H),8.16(br s,2H),7.74(br s,1H),7.26-7.12(m,5H),4.21(s,2H).MS m/e(相对百分比)252(30),161(100).FAB MS精确计算分子量为C16H14N3O3(M+1):296.1035.实测值296.1021.
实施例44
5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例9C)制得了题目化合物,具体采用:5-异丙氧基-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(70mg,0.23mmol)、N-氯磺酰基异氰酸酯(25ml,0.29mmol)和乙腈(1.5ml)。反应时间为4小时。将上述N-氯磺酰基甲酰胺粗品在氯仿中搅拌3天使之水解。减压除去溶剂后,产物经硅胶闪层析纯化,用2∶1乙酸乙酯/己烷洗脱,并用乙醚研制,得量:30mg(37%),m.p.194-196℃。
1H NMR(DMSO-d6)δ13.04(s,1H),9.08(br s,1H),8.99-8.98(m,1H),8.48(d,J=8.6Hz,1H),7.62-7.60(m,1H),7.19-7.16(m,1H),6.26(d,J=8.6Hz,1H),5.29(br s,1H),4.74(heptet,J=6.4Hz,1H),1.42(d,J=6.4Hz,1H).IR(KBr disc)1720,1607,1565cm-1.FAB MS精确计算分子量为C16H16H3O4S(M+1):346.0862.实测值346.0844.
实施例45
3-(4-氯-2-噻吩甲酰基)-5-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先,按实施例20方法,由5-氮杂羟基吲哚(实施例4B)制得了3-(4-氯-2-噻吩甲酰基)-5-氮杂羟基吲哚,具体采用:5-氮杂羟基吲哚(1.0g,7.45mmol)、钠(1.21g,52.6mmol)、4-氯噻吩-2-甲酰氯(2.72g,15.0mmol)和乙醇(40ml)。反应时间为3天。用甲醇研制粗产物,得量:427mg(21%),m.p.250℃。
然后,按实施例1C方法,由3-(4-氯-2-噻吩甲酰基)-5-氮杂羟基吲哚制得了题目化合物,具体采用:3-(4-氯-2-噻吩甲酰基)-5-氮杂羟基吲哚(427mg,1.53mmol)、异氰酸叔丁酯(0.26ml,2.3mmol)、三乙胺(0.43ml,3.1mmol)和DMSO(10ml)。反应时间:过夜。粗产物经连续闪层析纯化,用乙酸乙酯洗脱,用乙腈重结晶。
得量:172mg(30%),m.p.250℃。
1H NMR(DMSO-d6)δ9.72(s,1H),9.19(s,1H),8.73(s,1H),8.44(d,J=7Hz,1H),8.31(d,J=7Hz,1H),7.81(s,1H),1.43(s,9H).元素分析计算值C17H16Cl3N3O3S:C 54.04,H 4.27,N 11.12.实测值C 53.76,H 3.93,N 10.98.
实施例46
3-(2-噻吩甲酰基)-5-氮杂羟基吲哚-1-甲酰胺
按实施例2C方法,由3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例4C),制备了题目化合物,具体采用:3-(2-噻吩甲酰基)-5-氮杂羟基吲哚(500mg,2.0mmol)、N-氯磺酰基异氰酸酯(0.26g,3.0mmol)和乙腈(15ml)。反应时间为2.25小时。在与大气相通的烧瓶中,将上述N-氯磺酰基甲酰胺粗品在DMSO(1.5ml)中搅拌1.5小时使之水解。加入乙醚形成两相混合物,然后加入甲醇,形成均相溶液。放置片刻,出现绿色沉淀,过滤除去之。将滤液放置过夜,在此期间从该溶液中结晶出产物。过滤收集,得量:39mg(7%),m.p.250℃。
1H NMR 14.21(br s,1H),9.15(s,1H),9.12(br s,1H),8.68(d,-J=4Hz,1H),8.37(d,J=7Hz,1H),8.24(d,J=7H,1H),7.85(br s,1H),7.72(d,J=4.5Hz,1H),7.14(dd,J=4,4.5Hz,1H).IR(KBr disc)1741,1476,1433cm-1.FAB MS精确计算分子量为C13H10N3O3S(M+1):288.0443.实测值288.0439.
实施例47
6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺的乙酰基前药。
在6-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-甲酰胺(500mg,1.55mmol)的四氢呋喃(THF)(60ml)混悬液中依次加入三乙胺(0.4ml,2.87mmol)和乙酰氯(0.2ml,2.81mmol)。将该混合物在室温下搅拌过夜。补加三乙胺(0.4ml,2.87mmol)和乙酰氯(0.2ml,2.81mmol)。于室温下再搅拌3天后,将该混合物过滤收集产物,依次用氯仿、水和甲醇洗涤产物,剩下黄色固体(390mg,69%)。过滤滤液,用氯仿洗涤得到第二批产物(109mg,19%)。合并产物样品,用氯仿重结晶,得到黄色固体(289mg,51%),m.p.>250℃。
1H NMR(DMSO-d6)δ8.51(d,J=4Hz,1H),8.43(d,J=2Hz,1H),8.36(d,J=2Hz,1H),8.23(d,J=5Hz,1H),8.05(br s,2H),7.37(dd,J=4,5Hz,1H).
由于在2.50处的DMSO吸收使得乙酰基中CH3吸收峰模糊不清。
元素分析计算值C15H10ClN3O4S:C 49.53 H 2.77,N 11.55.实测值C 49.23,H 2.53,N 11.52.
实施例48
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-乙基甲酰胺
按实施例1C方法,由5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制得了题目化合物,具体采用5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(1.25g,4.89mmol)、三乙胺(3.2ml,23mmol)、异氰酸乙酯(1.77ml,22.4mmol)和DMSO(30ml)。反应时间为6小时。先用己烷研制粗产物,然后用同一溶剂重结晶。得量:1.09g(70%)。
元素分析计算值C15H12ClN3O3S:C 51.51,H 3.46,N 12.01.实测值C 51.56,H 3.21,N 11.90.M.p.153-154℃.
1H NMR(CDCl3)δ9.04(br s,1H),9.01(dd,J=1,4Hz,1H),8.47(d,J=8.2Hz,1H),7.71(dd,J=1,4.9Hz,1H),7.22(dd,J=4,4.9Hz,1Hz),7.03(d,J=8.2Hz,1H),3.51-3.41(m,2H),1.28(t,J=7.3Hz,3H).
IR(KBr disc)1720,1605,1585,1540,1415cm-1.MS m/e(相对百分比)351(2),349(5),280(10),278(29),196(32),194(100),111(26).
实施例49
5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚-1-N-异丙基甲酰胺
按实施例1C方法,由5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(实施例1B)制得了题目化合物,具体采用:5-氯-3-(2-噻吩甲酰基)-4-氮杂羟基吲哚(1.25g,4.49mmol)、三乙胺(3.2ml,23mmol)、异氰酸异丙酯(2.2ml,22.4mmol)和DMSO(30ml)。反应时间为6小时。粗产物用己烷重结晶。得量:1.30g(80%)。
元素分析计算值C16H14ClN3O3S:C 52.82,H 3.88,N 11.55.实测值C 52.93,H 3.65,N 11.31.M.p.163-165℃.
1H NMR(CDCl3)δ9.02(dd,J=1,4Hz,1H),8.96(br d,1H),8.46(d,J=8.3Hz,1H),7.71(dd,J=1,5Hz,1H),7.22(J=4,5Hz,1H),7.02(d,J=8.3Hz,1H),4.19-4.08(m,1H),1.30(d,J=6.6Hz,6H).
IR(KBr disc)1710,1605,1585,1540,1520,1420cm-1.MS m/e(相对百分比)365(3),363(14),280(17),278(45),196(32),194(100),111(18).
实施例50
3-苯甲酰基-5-氯-4-氮杂羟基吲哚-1-甲酰胺
按实施例25方法,由3-苯甲酰基-5-氯-4-氮杂羟基吲哚(实施例32)制得了题目化合物,具体采用3-苯甲酰基-5-氯-4-氮杂羟基吲哚(2.73g,10.0mmol)、N-氯磺酰基异氰酸酯(1.3ml,15mmol)和乙腈(80ml)。反应时间是20小时。将上述N-氯磺酰基甲酰胺粗品在DMSO(15ml)中搅拌20小时使之水解,粗产物经乙酸重结晶,用甲醇洗涤。得量:0.36g(11%)。
元素分析计算值C15H10ClN3O31/4H2O:C 56.26,H 3.30,N 13.12.实测值C 56.26,H 2.92,N 13.25.M.p.220℃.
1H NMR(DMSO-d6)δ8.41(br s,1H),8.33(d,J=8.3Hz,1H),7.80(d,J=7.3Hz,2H),7.72(br s,1H),7.58-7.45(m,3H),7.20(d,J=8.3Hz,1H).
IR(KBr disc)1750,1660,1620,1600,1590,1575,1390cm-1.
实施例51
5,6-二氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-叔丁基甲酰胺
首先,按实施例1B方法制备5,6-二氯-3-(2-糠酰基)-4-氮杂羟基吲哚,具体采用:5,6-二氯-4-氮杂羟基吲哚(763mg,3.76mmol)、钠(0.43g,18.8mmol)、2-糠酸乙酯(1.05g,7.5mmol)和乙醇(25ml)。得量:0.98(88%)。
然后,按实施例1C方法,由5,6-二氯-3-(2-糠酰基)-4-氮杂羟基吲哚制得了题目化合物,具体采用:5,6-二氯-3-(2-糠酰基)-4-氮杂羟基吲哚(721mg,2.43mmol)、三乙胺(1.8ml,15.4mmol)、异氰酸叔丁酯(1.4ml,12.3mmol)和DMSO(20ml)。反应时间为22小时。用甲醇研制粗产物,并用己烷重结晶。得量:218mg(23%)。
元素分析计算值C17H15Cl2N3O4:C 51.53,H 3.82,N 10.60.实测值C 51.70,H 3.81,N 10.57.M.p.205-206℃.
1H NMR(DMSO-d6)δ9.37(br s,1H),8.32(s,1H),7.91(s,1H),7.85(d,J=3.7Hz,1H),6.69(d,J=3.7Hz,1H),1.38(s,9H).
IR(KBr disc)1730,1620,1605,1590,1555,1535cm-1.MS m/e(相对百分比)397(0.5),395(2),298(21),296(33),230(62),228(100),95(40).
实施例52
5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-(1-羟基-2-甲基)丙-2-基甲酰胺
A.2-氨基-1-苄氧基-2-甲基丙烷
将2-氨基-2-甲基-1-丙醇(7.1mmol)的四氢呋喃(THF)(25ml)溶液滴加到60%氢化钠油分散体(3g,75mmol)的THF(75ml)浆液中。将该混合物在室温下搅拌1小时,然后在冰浴中冷却。滴加苄基溴(5.9ml,50mmol)。在0℃搅拌2小时后,将该反应混合物倒入冰/水中,用乙酸乙酯提取,用盐水洗涤有机相,干燥(MgSO4),浓缩得到一油状物,后者经硅胶闪层析纯化,用5%甲醇/氯仿洗脱,合并含所期产物的级份,浓缩,得到一油状物,得量:4.2g(47%)。
B.(1-苄氧基-2-甲基)丙-2-基异氰酸酯
将三光气(989mg,3.3mmol)分三次加到冰冷却的2-氨基-1-苄氧基-2-甲基丙烷(2.1g,10mmol)和三乙胺(4.5ml,32mmol)的二氯甲烷(50ml)溶液中,将该反应混合物在0℃搅拌0.25小时,然后在室温下搅拌4小时,减压除去挥发物,用乙醚研制残留物得到油状所期产物。得量:1.9g(95%)。
C.5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-(1-苄氧基-2-甲基)丙-2-基甲酰胺
按实施例1C方法,由5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚(实施例23)制得了题目化合物,具体采用:5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚(1.90g,7.2mmol)、三乙胺(2.8ml,21mmol)、(1-苄氧基-2-甲基)丙-2-基异氰酸酯(2.2g,10.7mmol)和DMSO(75ml)。反应过夜,粗产物经硅胶闪层析纯化,用氯仿洗脱,合并含有所期产物的级份,浓缩,得到一黄色油状物。得量:2.2g(65%)。
D.5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-(1-羟基-2-甲基)丙-2-基甲酰胺
将浓度为1M的三溴化硼(BBr3)的二氯甲烷溶液(3ml,3mmol)滴加到冰冷却的5-氯-3-(2-糠酰基)-4-氮杂羟基吲哚-1-N-(1-苄氧基-2-甲基)丙-2-基甲酰胺(1.0g,2.1mmol)的二氯甲烷(25ml)溶液中。将该反应混合物在0℃搅拌2小时,然后补加1M BBr3的二氯甲烷溶液(0.5ml,0.5mmol),再于0℃反应1小时后,将该混合物倒入冰/水中,用乙酸乙酯提取,用盐水洗涤有机相,干燥(MgSO4),浓缩,得到一黄色固体,先将该固体研制,然后用甲醇重结晶,得量:552mg(70%)。
元素分析计算值C17H16ClN3O5:C 54.05,H 4.27,N 11.12.实测值C 53.82,H 4.05,N 10.92.M.p.185-186℃。
1H NMR(CDCl3)δ9.3(br s,1H),8.49(dd,J=1.3,3.6Hz,1H),8.38(d,J=8.2Hz,1H),7.70(dd,J=1.3,1.6Hz,1H),6.98(d,J=8.2Hz,1H),6.61(dd,J=1.6,3.6Hz,1H),3.73(br s,1H),3.71(s,2H),1.41(s,6H).
IR(KBr disc)1730,1720,1665,1630,1595,1540,1460,1445,1420cm-1.MS m/e(相对百分比)380(1),379(3),378(5),377(9),265(18),264(50),263(53),262(100),197(12),196(60),195(33),194(97),95(25).
实施例53
5-氯-1-乙基-3-(2-噻吩甲酰基)-7-氮杂羟基吲哚
A.1-乙基-7-氮杂吲哚
在室温下,将10g(0.0846mol)7-氮杂吲哚(Aldrich(商标))溶解在200ml试剂级丙酮中,并用10g(0.178mol)KOH粉末处理之。约2~3分钟后,用5-10分钟加入67ml(0.846mol)碘乙烷,将该反应混合物在室温搅拌30-40分钟,薄层层析(展开剂:95∶5二氯甲烷/乙酸乙酯)显示起始物全部耗尽,并且形成了单一的弱极性产物。将该反应混合物减压浓缩,残留物分配于水和二氯甲烷(350ml)之间。分离有机层,用水和盐水洗涤,干燥(Na2SO4),减压浓缩有机提取物,得到一棕黄色油状物,后者经硅胶柱层析纯化,用95∶5二氯甲烷/乙酸乙酯洗脱,得到总量为11.15g(90%)的纯净终产物(淡黄色油)。
60MgHz1H NMR(CDCl3)δ:1.35-1.65(t,3H);4.20-4.60(q,2H);6.35-8.45(m,5H).
B.3,3-二溴-1-乙基-7-氮杂羟基吲哚
在30℃下,将溴代吡啶鎓过溴化物(27.2g,85mmol)分次加到1-乙基-7-氮杂吲哚(5.4g,34mmol)的叔丁醇(200ml)溶液中。将该反应混合物在室温下搅拌过夜,然后倒入冰/水中,搅拌0.5小时后,用乙酸乙酯提取该混合物,用水洗涤有机提取液,干燥(MgSO4),浓缩后得到一棕色油状物,后者经硅胶柱层析纯化,用氯仿洗脱,合并只含有所期产物的级份,减压浓缩,得到一黄色油状物,5.30g(49%)。
C.5-氯-3,3-二溴-1-乙基-7-氮杂羟基吲哚
在装有干冰冷凝器的三颈烧瓶中,将3,3-二溴-1-乙基-7-氮杂羟基吲哚(5.3g,16.5mmol)溶于DMF中,于冰浴中冷却至0℃。在该溶液中通入氮气气泡,时间为4分钟使之饱和。将该反应混合物在0℃搅拌2小时,然后倒入冰/水中。搅拌半小时后,用乙酸乙酯提取该混合物,用水洗涤有机提取液,干燥(MgSO4),浓缩,得到一黄色油状物,后者经硅胶柱层析,用乙酸乙酯洗脱,合并含有所期产物的级份,减压浓缩,得到4.69g(80%)黄色固体。
D.5-氯-1-乙基-7-氮杂羟基吲哚
将锌粉(2.5g,39mmol)分次加入5-氯-3,3-二溴-1-乙基-7-氮杂羟基吲哚(4.60g,13.0mmol)的冰乙酸(75ml)溶液中。立即发生放热反应,尽管如此,在室温下将反应物连续搅拌1小时。将该混合物倒入冰/水中,然后用乙酸乙酯提取,用水洗涤有机提取液,干燥(MgSO4),蒸发后得到一油状物,后者经硅胶柱层析纯化,用氯仿洗脱,合并含有所期产物的级份,减压浓缩,得到1.70g(68%)米黄色固体,m.p.78-82℃。
E.5-氯-1-乙基-3-(2-噻吩甲酰基)-7-氮杂羟基吲哚
在0℃下将噻吩-2-甲酰氯(0.24ml,2.24mmol)和4-二甲氨基吡啶(537mg,4.4mmol)的DMF(10ml)溶液中。将该反应混合物在室温下搅拌2小时,然后倒入冰/水中,用乙酸乙酯提取该混合物,用水和盐水洗涤有机层,干燥(MgSO4)减压浓缩。残留物经硅胶柱层析纯化,用氯仿洗脱。合并只含有所期产物的级份,浓缩后经己烷重结晶得到固体状题目化合物,所得重结晶物重量为29mg(50%)。
M.p.111-112℃.元素分析计算值C14H11ClN2O2S:C 54.82,H 3.61,N 9.13.实测值C 54.45,H 3.34,N 8.80.MS m/z(相对百分比)308(6),306(19),224(32),222(100),209(8),207(24),196(24),194(77),111(77).
实施例54
5-氯-1-乙基-3-(2-糠酰基)-7-氮杂羟基吲哚
按实施例53E的方法,由5-氯-1-乙基-7-氮杂羟基吲哚(实施例53D)制得了题目化合物,具体采用:5-氯-1-乙基-7-氮杂羟基吲哚(405mg,2.06mmol)、4-二甲氨基吡啶(500mg,4.09mmol)、2-糠酰氯(0.22ml,2.23mmol)和DMF(10ml)。将层析后所得固体产物用己烷重结晶。得到110mg(18%)产物。
M.p.162-163℃.元素分析计算值C14H11ClN2O3:C 57.84,H 3.81,N,9.64.实测值C,57.59,H 3.54,N 9.49.
1H NMR(CDCl3)δ8.43(d,J=1.7Hz,1H),8.11(s,1H),7.86(s,1H),7.38(d,J=3.5Hz,1H),6.72(dd,J=1.7,3.5Hz,1H),4.04(q,J=7.1Hz,2H),1.35(t,J=7.1Hz,3H).
IR(KBr disc)1645,1620,1535,1470,1440cm-1.MS m/z(相对百分比)292(8),290(27),224(32),222(100),209(8),207(27),196(28),194(86),95(70).
实施例55
5-氯-1-乙基-7-氮杂羟基吲哚-3-N-(4-氟苯基)甲酰胺
按实施例53E的方法,由5-氯-1-乙基-7-氮杂羟基吲哚(实施例53D)制得了题目化合物,具体采用:5-氯-1-乙基-7-氮杂羟基吲哚(400mg,2.03mmol)、4-二甲氨基吡啶(537mg,4.4mmol)、异氰酸4-氟苯基酯(0.25ml,2.2mmol)和DMF(10ml)。与所引用方法的唯一差别是:将反应混合物倒入冰/水中,然后用6N盐酸溶液酸化至pH3。将经层析后得到的固体产物用乙醚/己烷重结晶。得量为95mg(15%)。
M.p.155-157℃(dec.).元素分析计算值C16H13ClFN3O2:C 57.58,H 3.93,N 12.59.实测值C 57.50,H 3.64,N 12.33.
1H NMR(CDCl3)δ9.42(br s,1H),8.22(s,1H),8.05(s,1H),7.53(dd,J=5,7Hz,1H),7.02(t,J=7Hz,1H),4.43(s,1H),3.90(q,J=7Hz,2H),1.29(t,J=7H,3H).IR(KBr disc)1725,1665,1605,1580,1550,1510,1470,1435cm-1.MS m/z(相对百分比)335(18),333(52),198(33),196(100),170(10),168(30),111(21).
实施例56
5-氯-1-乙基-7-氮杂羟基吲哚-3-N-苯基甲酰胺
按实施例55的方法,由5-氯-1-乙基-7-氮杂羟基吲哚(实施例53D)制得了题目化合物,具体采用:5-氯-1-乙基-7-氮杂羟基吲哚(451mg,2.29mmol)、4-二甲氨基吡啶(607mg,4.97mmol)、异氰酸苯基酯(0.27ml,2.48mmol)和DMF(11ml)。先将所得反应混合物倒入水中,酸化,过滤收集所得固体,将该固体用乙酸乙酯研制,得到一白色固体(不是所期产物)。然后将母液浓缩,得到一棕色固体,后者用冷乙酸乙酯研制,并将所得黄色固体用环己烷重结晶,得到38mg(5%)白色固体状题目化合物。
M.p.157-158℃.元素分析计算值C16H14ClN3O2:C 60.86,H 4.47,N 13.31.实测值C 60.83,H 4.27,N 13.13.
1H NMR(CDCl3)δ9.43(br s,1H),8.21(s,1H),8.04(s,1H),7.48-7.54(m,2H),7.35-7.24(m,2H),7.15-7.10(m,1H),4.41(s,1H),3.91(q,J=7.3Hz,2H),1.31(t,J=7.3Hz,3H).IR(KBr disc)1730,1660,1605,1580,1550,1470,1445cm-1.MS m/z(相对百分比)317(3),315(10),198(33),196(100),183(3),181(10),170(11),168(36),93(22),77(20).
Claims (10)
1、制备下式Ⅰ(a)化合物的方法

式中A、B、D和E中之一是N,其他的是CH;X和Y分别选自氢、OR3、羟基、(C1-6)烷基、CF3、COR3、卤素、COOR3、CONR3R3、CN、NO2、SR3、SOR3、SO2R3、和SO2NR3R3;R1是CONH2;R2是(C1-8)烷基、(CH2)nR5,式中n是0或1,或NHR6;R3是(C1-6)烷基、苯基、苄基、烯丙基或氢,其中所述苯基及所述苄基中的苯基部分可被1个或多个分别选自下述基团的取代基取代,所述基团包括:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、CF3;R5是(C3-8)环烷基、氢、苯基、取代苯基、杂芳基和取代杂芳基,其中每一所述杂芳基和取代杂芳基的杂芳基部分选自噻吩和呋喃,每一所述取代苯基和取代杂芳基被1个或2个取代基取代,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、和三氟甲基;
R6是苯基、噻吩或呋喃、其中所述苯基、噻吩和呋喃可由1个或多个取代基取代,这些取代基分别选自:氟、氯、溴、碘、羟基、(C1-6)烷基、(C1-3)烷氧基、三氟甲基;W是氢;其前提是如果E是氮,那么X和Y中至少有一个不是氢;
该方法包括(a)下式Ⅵ化合物与氯磺酰基异氰酸酯反应,式Ⅵ为
式中A、B、D、E、X、Y和R2的定义如前,和(b)将该反应的产物水解。
2、按照权利要求1的方法,其中,用式R2COOH的适宜酸的衍生物将下式Ⅴ化合物酰化,得到所述式Ⅵ化合物,式Ⅴ为
式中A、B、D、E、X、Y和R2的定义同权利要求1所述。
3、制备下式Ⅰ(b)化合物的方法

式中A、B、D和E中之一是N,其他的是CH;X和Y分别选自氢、OR3、羟基、(C1-6)烷基、CF3、COR3、卤素、COOR3、CONR3R3、CN、NO2、SR3、SOR3、SO2R3、和SO2NR3R3;R1是CONHR4;R2是(C1-8)烷基、(CH2)nR5,式中n是0或1,或NHR6;R3是(C1-6)烷基、苯基、苄基、烯丙基或氢,其中所述苯基及所述苄基中的苯基部分可被1个或多个分别选自下述基团的取代基取代,所述基团包括:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、CF3;R4是(C1-6)烷基、(C2-6)羟烷基、(C3-8)环烷基、COR3(式中R3的定义如前)、苯基、取代苯基、杂芳基或取代杂芳基,其中所述杂芳基及取代杂芳基中的杂芳基部分选自噻吩和呋喃,并且,其中每个所述取代苯基和取代杂芳基是由1或2个下述取代基取代的,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基和CF3;R5是(C3-8)环烷基,氢、苯基、取代苯基、杂芳基和取代杂芳基,其中每一所述杂芳基和取代杂芳基的杂芳基部分选自噻吩和呋喃,每一所述取代苯基和取代杂芳基被1个或2个取代基取代,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、和三氟甲基;R6是苯基、噻吩或呋喃,其中所述苯基、噻吩和呋喃可由1个或多个取代基取代,这些取代基分别选自:氟、氯、溴、碘、羟基、(C1-6)烷基、(C1-3)烷氧基、三氟甲基;W是氢,其前提是如果E是氮,那么X和Y中至少有一个不是氢;
该方法包括使式中R4定义如前的式R4-N=C=O异氰酸酯与下式Ⅵ化合物反应
式中A、B、D、E、X、Y和R2的定义同前。
4、制备下式Ⅰ(c)化合物的方法
式中A、B、D和E中之一是N,其他的是CH;X和Y分别选自氢、OR3、羟基、(C1-6)烷基、CF3、COR3、卤素、COOR3、CONR3R3、CN、NO2、SR3、SOR3、SO2R3、和SO2NR3R3;R1是CONHR4;R2是(C1-8)烷基、(CH2)nR5,式中n是0或1,或NHR6;R3是(C1-6)烷基、苯基、苄基、烯丙基或氢,其中所述苯基及所述苄基中的苯基部分可被1个或多个分别选自下述基团的取代基取代,所述基团包括:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、CF3;R4是氢、(C1-6)烷基、(C2-6)羟烷基、(C3-8)环烷基、COR3(式中R3的定义如前)、苯基、取代苯基、杂芳基或取代杂芳基,其中每个所述杂芳基及取代杂芳基中的杂芳基部分选自噻吩和呋喃,并且,其中每个所述取代苯基和取代杂芳基是由1或2个下述取代基取代的,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基和CF3;R5是(C3-8)环烷基,氢、苯基、取代苯基、杂芳基和取代杂芳基,其中每一所述杂芳基和取代杂芳基的杂芳基部分选自噻吩和呋喃,每一所述取代苯基和取代杂芳基被1个或2个取代基取代,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、和三氟甲基;R6是苯基、噻吩或呋喃,其中所述苯基、噻吩和呋喃可由1个或多个取代基取代,这些取代基分别选自:氟、氯、溴、碘、羟基、(C1-6)烷基、(C1-3)烷氧基、三氟甲基;W是氢;其前提是,如果E是氢,那么X和Y中至少有一个不是氢;
该方法包括:用式中R2定义如前的式R2COOCOR2酸酐或酰氯将式中A、B、D、E、Y、X和R1定义如前的下式ⅩⅨ化合物酰化

5、按照权利要求4的方法,其中,将相应的式中A、B、D、E、X和Y定义如权利要求4所述的下式ⅩⅧ羧酸加热,得到所说的式ⅩⅨ化合物

6、制备下式Ⅰ(d)化合物的方法

式中A、B、D和E中之一是N,其他的是CH;X和Y分别选自氢、OR3、羟基、(C1-6)烷基、CF3、COR3、卤素、COOR3、CONR3R3、CN、NO2、SR3、SOR3、SO2R3、和SO2NR3R3;R1是(C1-6)烷基;R2是(C1-8)烷基、(CH2)nR5,式中n是0或1,或NHR6;R3是(C1-6)烷基、苯基、苄基、烯丙基或氢,其中所述苯基及所述苄基中的苯基部分可被1个或多个分别选自下述基团的取代基取代,所述基团包括:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、CF3;R4是氢、(C1-6)烷基、(C2-6)羟烷基、(C3-8)环烷基、COR3(式中R3的定义如前)、苯基、取代苯基、杂芳基或取代杂芳基,其中每个所述杂芳基及取代杂芳基中的杂芳基部分选自噻吩和呋喃,并且,其中每个所述取代苯基和取代杂芳基是由1或2个下述取代基取代的,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基和CF3;R5是(C3-8)环烷基,氢、苯基、取代苯基、杂芳基和取代杂芳基,其中每一所述杂芳基和取代杂芳基的杂芳基部分选自噻吩和呋喃,每一所述取代苯基和取代杂芳基被1个或2个取代基取代,所述取代基分别选自:氟、氯、溴、碘、羟基、(C1-3)烷基、(C1-3)烷氧基、和三氟甲基;R6是苯基、噻吩或呋喃,其中所述苯基、噻吩和呋喃可由1个或多个取代基取代,这些取代基分别选自:氟、氯、溴、碘、羟基、(C1-6)烷基、(C1-3)烷氧基、三氟甲基;W是氢、(C2-10)烷酰基、(C5-7)环烷酰基、(C7-10)苯基烷酰基、氯代苯甲酰基、噻吩甲酰基、ω-(C2-4)烷氧羰基(C3-5)烷酰基、(C2-10)烷氧羰基、苯氧羰基、1-〔(C1-4)酰氧〕-(C2-4)烷基、1-〔(C2-5)烷氧羰基氧〕-(C1-4)烷基、(C1-3)烷基磺酰基、(C1-3)烷基、甲苯基磺酰基和二-(C1-3)烷基膦酸酯基;其前提是如果E是氮,那么X和Y中至少有一个不是氢;
该方法包括:用式中R2的定义如前的式R2COOCOR2的酰氯或酸酐将下式ⅩⅨ化合物酰化,

式中A、B、D、E、Y、X和R1的定义如前所述。
7、按照权利要求6的方法,其中,将下式ⅩⅩ化合物与溴反应,

产生下式二溴化合物
然后将所述二溴化合物还原,由此得到所述式ⅩⅨ化合物。
8、按照权利要求6的方法,其中,使下式ⅩⅪ化合物与强酸反应
由此得到所述式ⅩⅨ化合物。
9、制备下述化合物的药用盐的方法,所述化合物是:(a)权利要求1所述式Ⅰ(a)化合物,(b)权利要求3所述式Ⅰ(b)化合物,(c)权利要求4所述式Ⅰ(c)化合物或(d)权利要求6所述式Ⅰ(d)化合物,该方法包括使上述化合物与药物上可以接受的酸或药物上可以接受的碱反应。
10、按照权利要求1-5中任一项的方法,其中还包括使所述方法的产物与三乙胺和所需酰氯、氯甲酸酯、锌盐或烷化剂反应,产生除W基团外与所述产物相同的化合物,W选自:(C2-10)烷酰基、(C5-7)环烷羰基、(C7-10)苯基烷酰基、氯代苯甲酰基、噻吩甲酰基、ω-(C2-4)烷氧羰基-(C3-5)烷酰基、(C2-10)烷氧羰基、苯氧羰基、1-〔(C1-4)酰氧基〕-(C2-4)烷基、1-〔(C2-5)烷氧基-羰基氧基〕-(C1-4)烷基、(C1-3)烷基磺酰基、(C1-3)烷基、甲基苯磺酰基和二-(C1-3)烷基膦酸酯。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90/000,107 | 1990-01-05 | ||
| USPCT/US90/00107 | 1990-01-05 | ||
| PCT/US1990/000107 WO1991009598A1 (en) | 1990-01-05 | 1990-01-05 | Azaoxindole derivatives |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1053065A true CN1053065A (zh) | 1991-07-17 |
| CN1031053C CN1031053C (zh) | 1996-02-21 |
Family
ID=22220612
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN91100034A Expired - Fee Related CN1031053C (zh) | 1990-01-05 | 1991-01-04 | 氮杂羟基吲哚衍生物的制备方法 |
Country Status (19)
| Country | Link |
|---|---|
| EP (1) | EP0436333A3 (zh) |
| JP (1) | JPH0826018B2 (zh) |
| KR (1) | KR930005447B1 (zh) |
| CN (1) | CN1031053C (zh) |
| AU (1) | AU635593B2 (zh) |
| BR (1) | BR9100046A (zh) |
| CA (1) | CA2033531C (zh) |
| CS (1) | CS991A2 (zh) |
| FI (1) | FI922347A0 (zh) |
| IE (1) | IE910022A1 (zh) |
| IL (1) | IL96819A0 (zh) |
| MY (1) | MY109722A (zh) |
| NO (1) | NO922634D0 (zh) |
| NZ (1) | NZ236582A (zh) |
| PL (2) | PL165653B1 (zh) |
| PT (1) | PT96411B (zh) |
| WO (1) | WO1991009598A1 (zh) |
| YU (1) | YU47728B (zh) |
| ZA (1) | ZA9172B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100338062C (zh) * | 2002-03-28 | 2007-09-19 | 卫材株式会社 | 作为c-Jun N-末端激酶抑制剂的氮杂吲哚类化合物 |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU1625192A (en) * | 1991-05-31 | 1992-12-03 | Zeneca Limited | Heterocyclic derivatives |
| GB9226855D0 (en) * | 1992-12-23 | 1993-02-17 | Erba Carlo Spa | Vinylene-azaindole derivatives and process for their preparation |
| FR2722195B1 (fr) * | 1994-07-07 | 1996-08-23 | Adir | Nouveaux derives de 1,3-dihydro-2h-pyrrolo(2,3-b) pyridin-2-ones et oxazolo(4,5-b) pyridin-2(3h)-ones, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| FR2722196B1 (fr) * | 1994-07-07 | 1996-08-23 | Adir | Nouveaux derives amines de 1,3-dihydro-2h-pyrrolo(2,3-b) pyridin-2-ones et oxazolo(4,5-b) pyridin-2 (3h)-ones, leur procede de preparation et les compositions pharmaceutique qui les contiennent |
| US5618819A (en) * | 1994-07-07 | 1997-04-08 | Adir Et Compagnie | 1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one and oxazolo[4,5-b]pyridin-2-(3H)-one compounds |
| US5939069A (en) * | 1996-08-23 | 1999-08-17 | University Of Florida | Materials and methods for detection and treatment of immune system dysfunctions |
| GB9718913D0 (en) | 1997-09-05 | 1997-11-12 | Glaxo Group Ltd | Substituted oxindole derivatives |
| US6153634A (en) * | 1998-12-17 | 2000-11-28 | Hoffmann-La Roche Inc. | 4,5-azolo-oxindoles |
| TR200101860T2 (tr) | 1998-12-17 | 2001-12-21 | F.Hoffmann-La Roche Ag | Sikline bağlı kinaz inhibitörleri olarak 4-alkenil (ve alkinil) oksidoller |
| JP2002532493A (ja) | 1998-12-17 | 2002-10-02 | エフ.ホフマン−ラ ロシュ アーゲー | Jnkプロテインキナーゼ阻害剤としての4−アリールオキシインドール |
| JP2002532503A (ja) | 1998-12-17 | 2002-10-02 | エフ.ホフマン−ラ ロシュ アーゲー | プロテインキナーゼ阻害剤としての4,5−ピラジノオキシンドール |
| US6624171B1 (en) | 1999-03-04 | 2003-09-23 | Smithkline Beecham Corporation | Substituted aza-oxindole derivatives |
| US6492398B1 (en) | 1999-03-04 | 2002-12-10 | Smithkline Beechman Corporation | Thiazoloindolinone compounds |
| GB9904933D0 (en) | 1999-03-04 | 1999-04-28 | Glaxo Group Ltd | Compounds |
| US6313310B1 (en) | 1999-12-15 | 2001-11-06 | Hoffmann-La Roche Inc. | 4-and 5-alkynyloxindoles and 4-and 5-alkenyloxindoles |
| AR029423A1 (es) | 1999-12-21 | 2003-06-25 | Sugen Inc | Compuesto derivado de pirrolo-[pirimidin o piridin]-6-ona, metodo de preparacion de dichos compuestos, composiciones farmaceuticas que los comprenden, un metodo para regular, modular o inhibir la actividad de la proteina quinasa y un metodo de tratar o prevenir una enfermedad de mamiferos |
| US6620818B1 (en) | 2000-03-01 | 2003-09-16 | Smithkline Beecham Corporation | Method for reducing the severity of side effects of chemotherapy and/or radiation therapy |
| DE10053275A1 (de) * | 2000-10-27 | 2002-05-02 | Dresden Arzneimittel | Neue 7-Azaindole, deren Verwendung als Inhibitoren der Phosphodiesterase 4 und Verfahren zu deren Herstellung |
| TWI262920B (en) | 2000-10-27 | 2006-10-01 | Elbion Ag | New 7-azaindoles, their use as inhibitors of phosphodiesterase 4, and a method for synthesizing them |
| EP1370527A1 (en) | 2001-03-06 | 2003-12-17 | AstraZeneca AB | Indolone derivatives having vascular-damaging activity |
| DE602005010698D1 (de) | 2004-06-09 | 2008-12-11 | Glaxo Group Ltd | Pyrrolopyridinderivate |
| WO2006042102A2 (en) * | 2004-10-05 | 2006-04-20 | Neurogen Corporation | Pyrrolo-pyridine, pyrrolo-pyrimidine and related heterocyclic compounds |
| WO2009147476A1 (en) * | 2008-06-02 | 2009-12-10 | Matrix Laboratories Ltd. | Novel pde inhibitors, pharmaceutical compositions containing them and processes for their preparation |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4556672A (en) * | 1984-03-19 | 1985-12-03 | Pfizer Inc. | 3-Substituted 2-oxindole-1-carboxamides as analgesic and anti-inflammatory agents |
| US4569942A (en) * | 1984-05-04 | 1986-02-11 | Pfizer Inc. | N,3-Disubstituted 2-oxindole-1-carboxamides as analgesic and antiinflammatory agents |
| DE3681358D1 (de) * | 1985-07-09 | 1991-10-17 | Pfizer | Substituierte oxindol-3-carboxamine als entzuendungshemmendes und schmerzstillendes mittel. |
| AU3776589A (en) * | 1988-06-14 | 1990-01-12 | Schering Corporation | Heterobicyclic compounds having antiinflammatory activity |
-
1990
- 1990-01-05 WO PCT/US1990/000107 patent/WO1991009598A1/en active Application Filing
- 1990-12-14 EP EP19900313669 patent/EP0436333A3/en not_active Withdrawn
- 1990-12-20 NZ NZ236582A patent/NZ236582A/xx unknown
- 1990-12-25 JP JP2419301A patent/JPH0826018B2/ja not_active Expired - Fee Related
- 1990-12-28 IL IL96819A patent/IL96819A0/xx unknown
- 1990-12-31 MY MYPI90002304A patent/MY109722A/en unknown
-
1991
- 1991-01-03 AU AU68606/91A patent/AU635593B2/en not_active Ceased
- 1991-01-03 PL PL91288598A patent/PL165653B1/pl not_active IP Right Cessation
- 1991-01-03 PL PL91303916A patent/PL166512B1/pl not_active IP Right Cessation
- 1991-01-03 PT PT96411A patent/PT96411B/pt not_active IP Right Cessation
- 1991-01-03 CA CA002033531A patent/CA2033531C/en not_active Expired - Fee Related
- 1991-01-04 KR KR1019910000020A patent/KR930005447B1/ko not_active IP Right Cessation
- 1991-01-04 CN CN91100034A patent/CN1031053C/zh not_active Expired - Fee Related
- 1991-01-04 IE IE002291A patent/IE910022A1/en unknown
- 1991-01-04 ZA ZA9172A patent/ZA9172B/xx unknown
- 1991-01-04 CS CS919A patent/CS991A2/cs unknown
- 1991-01-04 YU YU791A patent/YU47728B/sh unknown
- 1991-01-07 BR BR919100046A patent/BR9100046A/pt not_active Application Discontinuation
-
1992
- 1992-05-22 FI FI922347A patent/FI922347A0/fi not_active Application Discontinuation
- 1992-07-03 NO NO1992922634A patent/NO922634D0/no unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100338062C (zh) * | 2002-03-28 | 2007-09-19 | 卫材株式会社 | 作为c-Jun N-末端激酶抑制剂的氮杂吲哚类化合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1031053C (zh) | 1996-02-21 |
| NO922634L (no) | 1992-07-03 |
| YU47728B (sh) | 1996-01-08 |
| KR910014375A (ko) | 1991-08-31 |
| IE910022A1 (en) | 1991-07-17 |
| IL96819A0 (en) | 1991-09-16 |
| MY109722A (en) | 1997-05-31 |
| FI922347A (fi) | 1992-05-22 |
| YU791A (sh) | 1994-01-20 |
| PL288598A1 (en) | 1991-12-02 |
| JPH04210981A (ja) | 1992-08-03 |
| WO1991009598A1 (en) | 1991-07-11 |
| PT96411A (pt) | 1991-10-15 |
| AU6860691A (en) | 1992-03-05 |
| PL165653B1 (pl) | 1995-01-31 |
| FI922347A0 (fi) | 1992-05-22 |
| CS991A2 (en) | 1991-09-15 |
| NZ236582A (en) | 1993-10-26 |
| KR930005447B1 (ko) | 1993-06-22 |
| CA2033531A1 (en) | 1991-07-06 |
| AU635593B2 (en) | 1993-03-25 |
| PL166512B1 (pl) | 1995-05-31 |
| BR9100046A (pt) | 1991-10-22 |
| NO922634D0 (no) | 1992-07-03 |
| JPH0826018B2 (ja) | 1996-03-13 |
| CA2033531C (en) | 1998-06-23 |
| EP0436333A3 (en) | 1992-05-27 |
| ZA9172B (en) | 1992-10-28 |
| PT96411B (pt) | 1999-04-30 |
| EP0436333A2 (en) | 1991-07-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1031053C (zh) | 氮杂羟基吲哚衍生物的制备方法 | |
| CN1022832C (zh) | 3-取代的-2-羟吲哚衍生物的制备方法 | |
| CN1186324C (zh) | 稠合杂芳基衍生物 | |
| CN1263755C (zh) | 作为选择性cox-2抑制剂的吡唑并吡啶衍生物 | |
| CN1274690C (zh) | 三杂环化合物和包括其作为活性组分的药物 | |
| CN1055182A (zh) | N-(吡咯并《2,3-d》嘧啶-3-基酰基)-谷氨酸衍生物 | |
| CN1009831B (zh) | 杂环氧代-2,3-二氮杂萘基乙酸的制备方法 | |
| CN1314899A (zh) | 视黄酸类相关的受体机能调节剂 | |
| CN1216531A (zh) | 用作内皮缩血管肽受体拮抗剂的吲哚衍生物 | |
| CN1173497A (zh) | 含杂环碳酸衍生物 | |
| CN1934097A (zh) | 炔基化哌嗪化合物及其作为代谢型谷氨酸受体拮抗剂的应用 | |
| CN1094028A (zh) | 选择性的磷酸二酯酶(ⅳ)抑制剂儿茶酚二醚类 | |
| CN1060841A (zh) | 喹唑啉衍生物及其制备方法 | |
| CN1109059A (zh) | 新的噻吩并噻嗪衍生物 | |
| CN1291095A (zh) | 化学化合物 | |
| CN1030252C (zh) | 四氢苯并咪唑衍生物的制备方法 | |
| CN1274685C (zh) | 苯并咪唑衍生物调节趋化因子受体 | |
| CN1918160A (zh) | 用作趋化因子受体活性调节剂的新颖三环螺环衍生物 | |
| CN101058564A (zh) | 芳基烷基氨基甲酸酯衍生物的生产及其在治疗中的用途 | |
| CN1402710A (zh) | 抗细小核糖核酸病毒化合物和组合物,其药用,和其合成原料 | |
| CN1063870A (zh) | 环取代的2-氨基-1,2,3,4,-四氢萘类和3-氨基苯并二氢吡喃类化合物 | |
| CN1138583A (zh) | 9-取代的2-(2-正烷氧基苯基)-嘌呤-6-酮类化合物 | |
| CN85108120A (zh) | 三环吡啶衍生物的制备及其应用 | |
| CN1092059A (zh) | 取代苯甲酸 | |
| CN87108111A (zh) | 一种新颖的咪唑衍生物及其制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |