CN1028362C - 脱乙酰基秋水仙素衍生物的制备方法 - Google Patents
脱乙酰基秋水仙素衍生物的制备方法 Download PDFInfo
- Publication number
- CN1028362C CN1028362C CN91112823A CN91112823A CN1028362C CN 1028362 C CN1028362 C CN 1028362C CN 91112823 A CN91112823 A CN 91112823A CN 91112823 A CN91112823 A CN 91112823A CN 1028362 C CN1028362 C CN 1028362C
- Authority
- CN
- China
- Prior art keywords
- deacetylcolchicine
- acid
- compound
- acid amides
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- HFPMXDMZJUJZBX-AWEZNQCLSA-N Deacetylcolchicine Chemical class C1([C@@H](N)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC HFPMXDMZJUJZBX-AWEZNQCLSA-N 0.000 title claims abstract description 33
- 238000000034 method Methods 0.000 title claims description 14
- 238000002360 preparation method Methods 0.000 title description 5
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical class C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 claims abstract description 32
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 7
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims abstract description 5
- -1 saccharide carboxylic acids Chemical class 0.000 claims description 31
- 229960001338 colchicine Drugs 0.000 claims description 15
- 238000006243 chemical reaction Methods 0.000 claims description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 12
- 229940097043 glucuronic acid Drugs 0.000 claims description 11
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 8
- 229950006191 gluconic acid Drugs 0.000 claims description 7
- 239000011541 reaction mixture Substances 0.000 claims description 5
- 150000002148 esters Chemical class 0.000 claims description 4
- 125000002252 acyl group Chemical group 0.000 claims description 3
- 229940059260 amidate Drugs 0.000 claims description 3
- 150000001408 amides Chemical class 0.000 claims description 3
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 claims description 3
- 239000012453 solvate Substances 0.000 claims description 3
- 125000004036 acetal group Chemical group 0.000 claims 2
- 210000004881 tumor cell Anatomy 0.000 abstract description 7
- 239000002246 antineoplastic agent Substances 0.000 abstract description 4
- 229940045695 antineooplastic colchicine derivative Drugs 0.000 abstract 1
- 150000001735 carboxylic acids Chemical class 0.000 abstract 1
- 230000002401 inhibitory effect Effects 0.000 abstract 1
- 231100000053 low toxicity Toxicity 0.000 abstract 1
- 230000035755 proliferation Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 42
- 150000001875 compounds Chemical class 0.000 description 40
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 20
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 239000000203 mixture Substances 0.000 description 14
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 12
- 206010028980 Neoplasm Diseases 0.000 description 11
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 11
- 239000000243 solution Substances 0.000 description 11
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- 239000012141 concentrate Substances 0.000 description 7
- 235000008504 concentrate Nutrition 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- 239000003814 drug Substances 0.000 description 6
- 238000010898 silica gel chromatography Methods 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 239000003513 alkali Substances 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 208000006268 Sarcoma 180 Diseases 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- TXHIDIHEXDFONW-UHFFFAOYSA-N benzene;propan-2-one Chemical compound CC(C)=O.C1=CC=CC=C1 TXHIDIHEXDFONW-UHFFFAOYSA-N 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 229940125898 compound 5 Drugs 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- GVOISEJVFFIGQE-YCZSINBZSA-N n-[(1r,2s,5r)-5-[methyl(propan-2-yl)amino]-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 GVOISEJVFFIGQE-YCZSINBZSA-N 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- HBENZIXOGRCSQN-VQWWACLZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-[(2S)-2-hydroxy-3,3-dimethylpentan-2-yl]-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol Chemical compound N1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@H]3[C@@]5(OC)CC[C@@]2([C@@]43CC1)C[C@@H]5[C@](C)(O)C(C)(C)CC)CC1CC1 HBENZIXOGRCSQN-VQWWACLZSA-N 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- 229920004449 Halon® Polymers 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical compound ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 3
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 229940034982 antineoplastic agent Drugs 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 150000001720 carbohydrates Chemical group 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical group FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 229940095064 tartrate Drugs 0.000 description 3
- RBNPOMFGQQGHHO-UHFFFAOYSA-N -2,3-Dihydroxypropanoic acid Natural products OCC(O)C(O)=O RBNPOMFGQQGHHO-UHFFFAOYSA-N 0.000 description 2
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 2
- RBNPOMFGQQGHHO-UWTATZPHSA-N D-glyceric acid Chemical compound OC[C@@H](O)C(O)=O RBNPOMFGQQGHHO-UWTATZPHSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 210000000683 abdominal cavity Anatomy 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- KGYGBOORGRYDGQ-UHFFFAOYSA-N benzene;methanol Chemical compound OC.C1=CC=CC=C1 KGYGBOORGRYDGQ-UHFFFAOYSA-N 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 238000000151 deposition Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000011010 flushing procedure Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 235000017550 sodium carbonate Nutrition 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000006200 vaporizer Substances 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FEWJPZIEWOKRBE-LWMBPPNESA-L D-tartrate(2-) Chemical compound [O-]C(=O)[C@@H](O)[C@H](O)C([O-])=O FEWJPZIEWOKRBE-LWMBPPNESA-L 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical group COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- MJOQJPYNENPSSS-XQHKEYJVSA-N [(3r,4s,5r,6s)-4,5,6-triacetyloxyoxan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1CO[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O MJOQJPYNENPSSS-XQHKEYJVSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 231100000215 acute (single dose) toxicity testing Toxicity 0.000 description 1
- 238000011047 acute toxicity test Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 229910052728 basic metal Inorganic materials 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- WQPDQJCBHQPNCZ-UHFFFAOYSA-N cyclohexa-2,4-dien-1-one Chemical compound O=C1CC=CC=C1 WQPDQJCBHQPNCZ-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 231100000086 high toxicity Toxicity 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000012204 lemonade/lime carbonate Nutrition 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C225/00—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones
- C07C225/24—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones the carbon skeleton containing carbon atoms of quinone rings
- C07C225/26—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones the carbon skeleton containing carbon atoms of quinone rings having amino groups bound to carbon atoms of quinone rings or of condensed ring systems containing quinone rings
- C07C225/32—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones the carbon skeleton containing carbon atoms of quinone rings having amino groups bound to carbon atoms of quinone rings or of condensed ring systems containing quinone rings of condensed quinone ring systems formed by at least three rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
- C07H13/02—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids
- C07H13/04—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids having the esterifying carboxyl radicals attached to acyclic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/14—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/02—Acyclic radicals
- C07H7/033—Uronic acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
由式(I)表示的脱乙酰基秋水仙素衍生物:
其中R为从C3-C7糖羧酸中除去COOH后得到的残基,在这些残基中存在的羟基可用羟基保护基团适当保护,该脱乙酰基秋水仙素衍生物具有较低毒性并对抑制肿瘤细胞增生具有较强作用,预计可作抗肿瘤剂使用。
Description
本发明涉及新的脱乙酰基秋水仙素衍生物。更准确地说,本发明涉及由下式所表示的脱乙酰基秋水仙素衍生物:
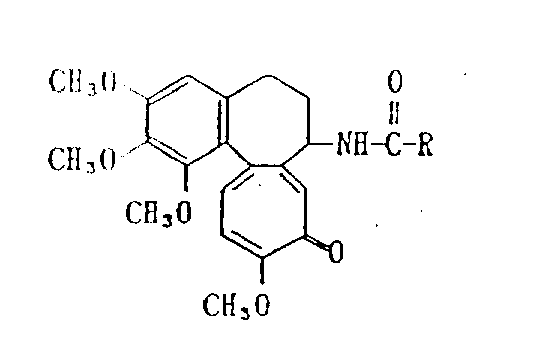
其中R为从C3-C7糖羧酸中除去COOH所得到的残基,在该残基中的羟基可用羟基的保护基
团适当地保护。
已知由下式
所表示的秋水仙素对肿瘤细胞、痛风等有药物活性。[参见《在农业、医药、生物学和化学中的秋水仙素》(Jowa Stage College Press Amis,Jowa,19550]
然而,秋水仙素具有相当高的毒性,随着后来发现的脱甲秋水仙素(脱乙酰基-N-甲基秋水仙素)的出现[(Chem.Engng.News,37,No.41,67(1959)],秋水仙素被完全忽视了。
本发明人为此做了不懈地研究以寻找具有低毒性和较好的抗肿瘤活性的秋水仙素衍生物。因此,发现了由式(Ⅰ)所表示的脱乙酰基秋水仙素衍生物对抑制肿瘤细胞增生显示出高活性,并且预计将作为抗肿瘤剂使用。这一发现使本发明得以完成。
由R所代表的“从C3-C7糖羧酸中除去COOH所得的残基”,包括从C3-C7单糖羧酸如甘油酸、核糖羧酸、葡糖醛酸、葡糖酸或葡庚糖酸中除去COOH所得到的单价残基(下文称为糖残基)。这些残基的例子如下所示:

存在于糖残基的多个羟基中的至少一部分可以用羟基的保护基团适当地保护。这些保护基团的例子为酰基,如乙酰基、丙酰基、丁酰基、新戊酰基和苯甲酰基;和用下式表示的乙缩醛和酮缩醇基:

本发明的化合物可以通过,例如,用下式表示的糖羧酸:
其中R的定义如上或它们的反应的衍生物使下式表示的脱乙酰基秋水仙素酰胺化来制备。
采用肽化学领域中已知的酰胺化反应,使式(Ⅲ)表示的糖羧酸或它的反应的衍生物(如:卤化物和活性酯)和脱乙酰基秋水仙素进行酰胺化反应。
例如,在碱存在下,脱乙酰基秋水仙素与式(Ⅲ)的糖羧酸卤化物反应制备本发明的化合物。上述反应通常可在约0℃-30℃温度下进行,优选约0℃至室温。卤化物的量无严格限制,通常对每摩尔脱乙酰基秋水仙素为1-1.5摩尔,优选1-1.2摩尔。碱的例子为叔胺,如三乙基胺和吡啶;和碱金属(氢)碳酸盐,如碳酸钠、碳酸氢钠、碳酸钾和碳酸氢钾。碱的量通常为,对摩尔脱乙酰基秋水仙素1-1.5摩尔,优选1-1.2摩尔。
上述反应通常在惰性溶剂中进行,溶剂的例子为卤化烃,如二氯甲烷、氯仿、四氯化碳、二氯乙烯和三氯乙烯;脂肪醚,如乙醚、甲氧基乙醇;和芳香烃,如苯和甲苯。
在缩合剂如二环己基碳化二亚胺(DDC)存在下,脱乙酰基秋水仙素与糖羧酸直接反应或脱乙酰基秋水仙素与式(Ⅲ)的糖羧酸的酯(如甲酯、乙酯或丁酯)反应制备本发明的化合物。
本发明所得到的化合物可用已知方法分离和纯化,如:萃取、色谱、结晶或其组合。
本发明的化合物,其羟基保护基团存在于由
R代表的糖残基中的情况下,可通过脱保护基反应(如所需要的水解反应)把保护基脱去。
在前述的反应中,作为起始物的脱乙酰基秋水仙素本身是已知化合物[参见J.Am.Chem.Soc.,75,5292(1953)],可用已知方法制备,或者可由下式表示的秋水仙素:
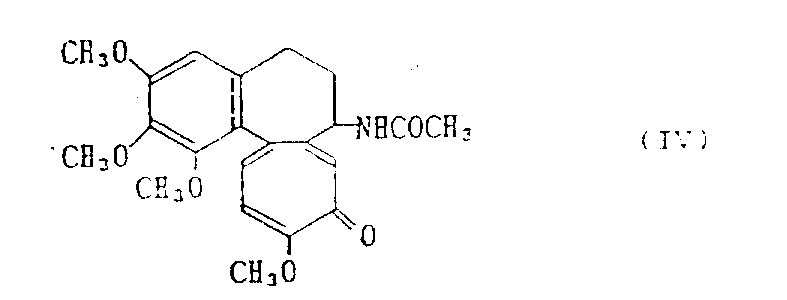
与三烷基氧鎓氟硼酸盐反应,然后按本发明人另外开发的方法用水处理反应混合物而制备。
秋水仙素与三烷基氧鎓氟硼酸盐的反应可在惰性有机溶剂中,在约0℃至30℃温度,优选约0℃至室温下进行。惰性有机溶剂的例子为卤化烃如二氯甲烷、氯仿、四氯化碳、二氯乙烯和三氯乙烯;脂肪醚,如乙醚和甲氧基乙醇;芳香烃,如苯和甲苯。
与秋水仙素反应的三烷基氧鎓氟硼酸盐是由下式表示的化合物:
(R′)3O+.BF- 4(Ⅴ)
其中R′为烷基。
实际上三乙基氧鎓氟硼酸盐作为米尔文剂是较好的。
对每摩尔秋水仙素,三烷基氧鎓氟硼酸盐的量通常为1-2摩尔,优选1-1.5摩尔。
推测秋水仙素与三烷基氧鎓氟硼酸盐反应生成下式表示的化合物:
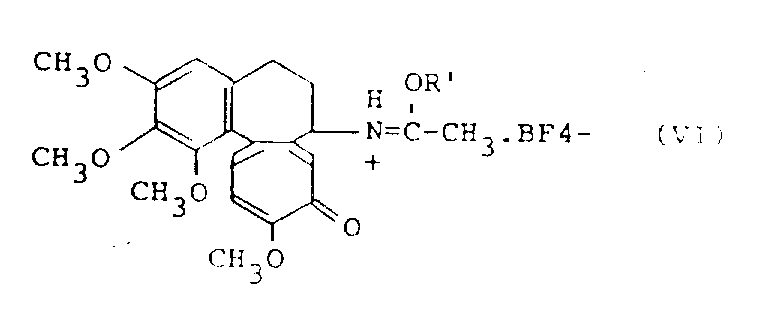
其中R′的定义如上。
如用水处理式(Ⅵ)的化合物可以形成式(Ⅱ)的脱乙酰基秋水仙素。用水处理通常在约0℃-30℃温度下,特别是在室温下,通过搅拌30分钟至3小时来完成,对每摩尔起始物秋水仙素,水的量至少1摩尔,通常为过量。
因此,脱乙酰基秋水仙素是以溶于水相的状态形成的,可以从水相中分离出来并用已知方法精制。例如,用加入碱而形成的pH范围9-10的碱性水层来分离脱乙酰基秋水仙素。这些碱包括如:氢氧化钠、氢氧化钾、碳酸钠或碳酸氢钠。然后,用有机溶剂萃取,有机溶剂包括例如:卤化烃,如二氯甲烷、氯仿、四氯化碳、二氯乙烯或三氯乙烯;脂肪醚,如乙醚或甲氧基乙醇;或芳烃,如苯或甲苯。
这样分离出的脱乙酰基秋水仙素可被精制,例如,把它转化成酒石酸盐或苹果酸盐。
本发明式(Ⅰ)的脱乙酰基秋水仙素衍生物具有极好的抗肿瘤活性。这一点在以下所描述的对肿瘤细胞的体外或体内实验结果中很明显的看出。
实施例1:体外抑制肿瘤细胞增生实验
抗鼠白血病细胞P388/ADR的2×105亚德里亚霉素被悬浮在含有10%的牛胎儿血清的RPMI1640培养介质中,在实验化合物(该实验化合物被溶解在二甲基亚砜中,其浓度为1mg/ml,这是用磷酸盐缓冲溶液稀释的)存在下培养2天。测试对细胞增生的影响,抑制50%增生的浓度:测定IC50(μg/ml)。结果列于表1中。
表1
试验化合物 IC50
化合物1a 0.036
化合物1b 0.066
化合物4 0.034
化合物5 0.037
*在以下实施例中说明化合物编号的意义
实验例2:
内肉瘤180腹水肿瘤移植至鼠体的体内实验。
1×106肉瘤180细胞移植入六周大的ddy雄性鼠的腹部空腔中。每组由6只鼠组成。在植入肿瘤细胞第1天后的7天内,实验化合物(溶于丙二醇,并用磷酸盐缓冲液稀释,最后丙二醇的浓度为20%或更低)给药于鼠的腹部空腔中,每日一次。计算存活日的平均数和延长的百分数*。结果列表2中。
*延长百分数= ((在给药组中-(在对照组中存存活日的平均数)活日的平均数))/((在对照组中存活日的平均数)) ×100
(表2见文后)
实施例3:
移植体内的肉瘤180固体肿瘤鼠的体内实验2×106肉瘤180细胞被移植于六周大的ddy雌性鼠的背皮下,每组由6只鼠组成。在肿瘤植入后从第6天起的10天之内,每天一次,连续把实验化合物(溶于丙二醇,用磷酸盐缓冲溶液稀释,最后丙二醇的浓度为20%或更低)给药于腹空腔中。在肿瘤植入30天后,取出肿瘤,测定其重量,计算平均肿瘤重和抑制百分率,结果列表3中。
*抑制百分数= ((在对照组中-(在给药组中的平均肿瘤重)平均肿瘤重))/((在对照组中平均肿瘤重)) ×100
(表3见文后)
实施例4:急性毒性实验
实验化合物给药于每只5周大的ddy雄性鼠体内,观察一周的死亡率。根据每组中鼠的死亡数目,采用Litchfield-wilcoxon方法来计算50%的致死量(LD50)。实验化合物(化合物(a))溶在10%的丙二醇中。从最大浓度80mg/kg,比率为1.2来制备十份稀释梯度溶液。然后进行实验。
表4
给药方法 LD50(mg/kg)
腹膜内的 42(可信范围37.0-47.7)
静脉注射 38(可信范围34.3-42.2)
从以上实验结果,可知本发明的化合物对肿瘤细胞具有很高的抑制活性,并预计作为抗肿瘤剂使用。
本发明的化合物作为抗肿瘤剂处理和治疗肿瘤时,该化合物可通过口服或非肠道(例如:静脉、肌肉、皮下或直肠内的)给药。依据病情、性别、病人体重,给药途径、医生诊断等情况,化合物剂量可在较宽范围内变化,通常为1-20mg/kg。在口服情况下。适合的剂量为5-10mg/kg;在静脉注射的情况下,其量为2-4mg/kg。
本发明化合物可制剂成片剂、颗粒、粉剂、胶囊、糖浆、注射剂、滴剂或栓剂。根据已知方法,把本发明的化合物与药物可接受的载体或稀释剂混合来进行配制。载体或稀释剂的例子为水、乙醇、淀粉、乳糖、蔗糖、葡萄糖、甘露糖醇、硅石、羧甲基纤维素、藻酸盐、明胶、聚乙烯吡咯烷酮、甘油、琼脂、碳酸钙、石蜡、高岭土、滑石、硬脂酸钙、硬脂酸镁和聚乙二醇。
通过以下实施例更具体说明本发明。
实施例1
脱乙酰基秋水仙素的制备
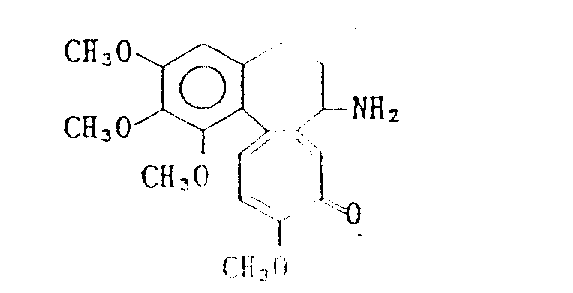
4g(10mmol)秋水仙素溶解在无水的二氯甲烷中并冷却至0℃,逐滴加入15mmol的三乙基氧鎓氟硼酸盐
(Meerwein试剂)的二氯甲烷溶液。溶液在0℃搅拌1小时,进一步在室温下搅拌5小时。往反应混合物中加入30ml水,得到的溶液搅拌1小时。搅拌后,水层用漏斗分离,用50ml的水进一步萃取二氯甲烷层五次。二氯甲烷层用硫酸镁干燥并用来回收未反应的秋水仙素。用1N的氢氧化钠来调节水层的pH至10,并用氯仿萃取。氯仿层用硫酸镁干燥,然后用蒸发器浓缩。剩余物溶解在30ml乙醇中,加入1g D-酒石酸,然后加热混合物1小时。混合物冷却到室温后,过滤沉淀。所得的酒石酸盐用干燥器干燥。(在熔点219-220℃分解)。
酒石酸盐溶解在50ml水中,用1N的氢氧化钠重新调节pH至10,并用氯仿萃取。萃取物用硫酸镁干燥,在减压下用蒸发器浓缩,获得1.38g油状的脱乙酰基秋水仙素。产率为39%。
未反应的秋水仙素通过原来的二氯甲烷层(1.71g)的硅胶色谱柱,从被苯一丙酮洗脱的部分中回收。去掉这部分所得脱乙酰基秋水仙素产率为61%。
实施例2
脱乙酰基秋水仙素-甘油酸丙酮化物酰胺(化合物1)

把甘油酸钾丙酮化物(3.60g,20mmol)悬浮在30ml无水乙醚中,往悬浮液中逐滴加入2.4g(20mmol)亚硫酰氯乙醚(5ml)溶液。加完之后,混合物回流3小时。混合物被冷却到室温后,用抽吸法过滤沉淀物,在减压下浓缩滤液。往剩余物中加入无水二氯甲烷并使其溶解。
其间,把2.96g(8.3mmol)脱乙酰基秋水仙素和2.02g(20mmol)三乙基胺溶解在30ml二氯甲烷中混合物冷却到0℃,逐滴加入上述的甘油酰氯的二氯甲烷溶液。在0℃搅拌3小时后,用碳酸氢钠水溶液洗涤二氯甲烷溶液。二氯甲烷层用硫酸镁干燥,然后在减压下浓缩,剩余物通过硅胶柱色谱分离,从苯-丙酮(5∶1)冲洗的部分中得到1.11g产物(化合物1a:L-异构体)。产率为28%,m.p.251-253℃(分解)。
另外,从苯-丙酮(5∶2)冲洗的部分中得到第二种产物(化合物1b∶D-异构体)0.58g,产率为14%。
化合物1a:IR(KBr):3250cm-1(NH),1670cm-1(C=0),1250cm-1(-O-);
NMR(DCDl3):=1.40(3H,s),1.60(3H,s),1.69-2.67(4H,m),3.62(3H,s),3.87(3H,s),3.91(3H,s),3.93(3H,s),4.00-4.50(4H,s),6.49-7.29(4H,m)。
[0046]
化合物1b:IR(KBr):3250cm-1(NH),1670cm-1(C=0),1250cm-1(-O-);
NMR(DCDl3):=1.37(3H,s),1.46(3H,s),1.69-2.67(4H,m),3.62(3H,s),3.86(3H,s),3.91(3H,s),3.97(3H,s),4.00-4.50(4H,s),6.49-7.29(4H,m)。
实施例3
脱乙酰基秋水仙素-甘油酸酰胺
[N-(5,6,7,9-四氢-1,2,3,10-四甲氧基-4-氧代苯并[a]庚间三烯并庚间三烯-7-基)甘油酰胺](化合物2)

10ml 5%的盐酸加入0.94g(2mmol)实施例2中得到的脱乙酰基秋水仙素甘油酸丙酮酰胺(化合物1)的30ml甲醇溶液中,在室温下搅拌5小时。搅拌后,加入200ml氯仿,混合物用碳酸氢钠水溶液和饱和Nacl水溶液洗涤。氯仿层用硫酸镁干燥,然后在减压下浓缩。剩余物用硅胶柱色谱分离,用苯-丙酮(1∶2)溶剂洗脱,得到0.45g上述标题化合物(化合物2);D,L-混合物)。产率为52%,m.p.48-50℃。
IR(KBr):3350cm-1(OH),1660cm-1(C=0),1280cm-1(-O-);
NMR(DCDl3):=1.87-2.64(4H,m),3.62(3H,s),3.87(3H,s),3.91(3H,s),3.96(3H,s),3.56-4.84(6H,m),6.51-7.58(4H,m),7.9(1H,brs)。
实施例4
脱乙酰基秋水仙素-葡糖醛酸四-乙酰基酰胺(化合物3)

把亚硫酰氯(1.19g,10mmol)加入1.81g(5mmol)的葡糖醛酸四乙酸酯的30ml氯仿溶液
中,混合物回流3小时。待混合物冷却到室温后,在减压下除去溶剂和过量的亚硫酰氯。剩余的酰基氯溶解在10ml二氯甲烷中。其间,1.78g(5mmol)的脱乙酰基秋水仙素和0.60g(6mmol)的三乙胺溶解在30ml的二氯甲烷中并冷却到0℃。上述酰基氯加入混合物中。在0℃搅拌1.5小时,并在室温下搅拌1.5小时。反应混合物用碳酸氢钠水溶液洗涤,然后用硫酸镁干燥。溶剂浓缩后,剩余物用硅胶柱色谱分离。用苯-丙酮(11∶3)溶剂洗脱。得到1.30g上述标题化合物(化合物3),产率为37%,m.p.145-147℃(分解)
IR(KBr):1750cm-1(OH),1680cm-1(C=O);
NMR(DCDl3):=1.91(3H,s),1.96(3H,s),2.00(2H,s),2.09(3H,s),2.10-2.64(4H,m),3.58(3H,s),3.87(3H,s),3.89(6H,s),4.00-4.22(1H,m),5.00-5.38(4H,m),5.80-5.89(1H,m),6.47-7.53(4H,m)。
实施例5
脱乙酰基秋水仙素-葡糖醛酸二乙酰基酰胺(化合物4)

(可能的结构式)
上述标题化合物按实施例4方法制备,只是使用3.62g(13mmol)的葡糖醛酸二乙酸酯,1.71g(15mmol)亚硫酰氯,2.89(8mmol)脱乙酰基秋水仙素,和1.52g(15mmol)三乙胺。产量为1.67g,产率为35%。
NMR(DCDl3):=2.15(6H,s),2.26-2.71(4H,m),3.64(3H,s),3.89(6H,s),3.98(3H,s),3.37-4.48(8H,m),6.53-7.81(4H,m)。
实施例6
脱乙酰基秋水仙素-葡糖醛酸酰胺[N-(5,6,7,9-四氢-1,2,3,10-四甲氧基-9-氧代苯并[a]庚间三烯并庚间三烯-7-基)葡糖醛酸酰胺](化合物5)
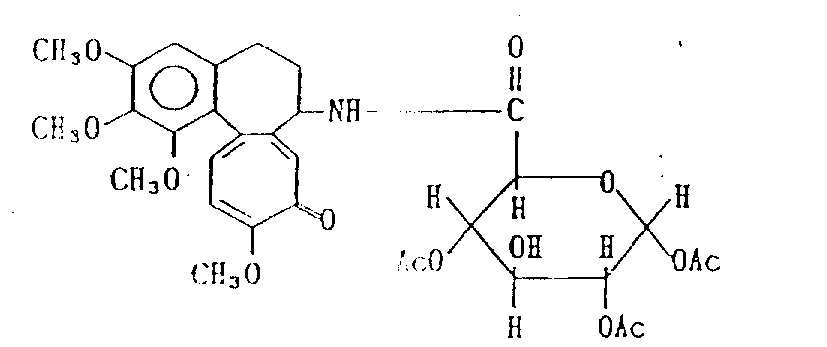
在实施例5中得到的脱乙酰基秋水仙素-葡糖醛酸二乙酰基酰胺(化合物4)(1.52g,2.5mmol)溶解在30mmol甲醇中并冷却到0℃。逐滴加入5ml 1N氢氧化钠,混合物在0℃搅拌1小时。用稀盐酸调节混合物pH为至7,并在减压下浓缩。往剩余物中加入氯仿,过滤沉淀,然后,用硅胶柱色谱分离滤液。结果从被苯-甲醇(5∶1)洗脱的部分中得到0.93克上述标题化合物(化合物5)。产率为73%,m.p.53-57℃。
NMR(DCDl3):=2.22-2.67(4H,m),2.96-3.27(4H,m),3.62(3H,s),3.87(6H,s),3.93(3H,s),3.50-4.22(6H,m),6.48-7.78(4H,m)。
实施例7
脱乙酰基秋水仙素-葡糖酸五乙酰基酰胺(化合物6)
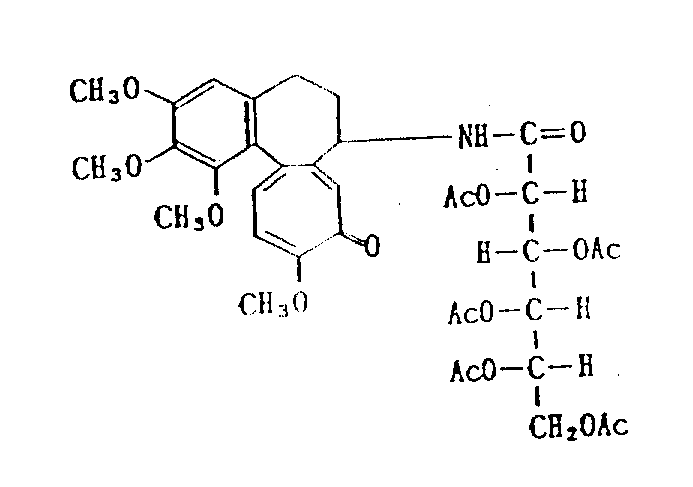
把亚硫酰氯(1.50g,13mmol)加入含有2.57g(6.3mmol)葡糖酸五乙酸酯的30ml氯仿溶液中,回流3小时。待混合物冷却到室温后,在减压下除去溶剂和过量亚硫酰氯。剩余的酰基氯溶解在10ml二氯甲烷。其间,1.53g(4.3mmol)脱乙酰基秋水仙素和1.00g(10mmol)三乙胺溶解在40ml二氯甲烷中并冷却到0℃。上述的酰基氯逐滴加入混合物中,在0℃搅拌1.5小时并在室温搅
拌1.5小时,反应混合物用碳酸氢钠水溶液洗涤,然后用硫酸镁干燥。待溶剂浓缩后,剩余物用硅胶柱色谱分离。用苯-丙酮溶剂(11∶3)洗脱,得到1.83g上述标题化合物(化合物6)。产率为57%。
实施例8
脱乙酰基秋水仙素-葡糖酸酰胺(化合物7)

把实施例7中得到的脱乙酰基秋水仙素-葡糖酸五乙酰基酰胺(化合物6)(3.73g,5mmol)溶解在30ml甲醇中并冷却到0℃,逐滴加入5ml 1N氢氧化钠,在0℃搅拌1小时,用稀盐酸调节混合物pH至7,在减压下浓缩。往剩余物中加入氯仿,过滤沉淀物。浓缩滤液,剩余物用硅胶柱色谱分离。结果,从苯-甲醇(5∶1)洗脱的部分中得到1.12g上述标题化合物(化合物7)7)。产率为42%。
表2
实验 剂量(mg/ 存活日的平均数 延长百分数 治愈例
化合物 kgi.P) (平均值±SD) (%)
2.5 54.2±11.6 333 4
化合物 1.25 47.2±11.5 277 2
1a 0.63 47.7±11.1 281 1
0.31 36.0±8.5 188
化合物 5 57.0±5.3 356 4
1b 2.5 57.0±7.3 356 5
1.25 42.5±15.4 240 1
(20%丙二 14.3±3.3 14.7 0
醇)
(对照:未 12.5±4.8 0
处理)
表3
实验化合物 剂量(μg/kgi.p) 平均肿瘤重 抑制百分率
(平均值±SD,g) (%)
化合物1a 5.0 0.60±0.52 57.4
2.5 0.63±0.51 55.3
1.25 0.88±0.78 47.6
0.625 0.71±0.38 49.6
对照 (20%丙二醇) 1.41±0.58 -
Claims (4)
1、制备由下式表示的脱乙酰基秋水仙素衍生物的方法,
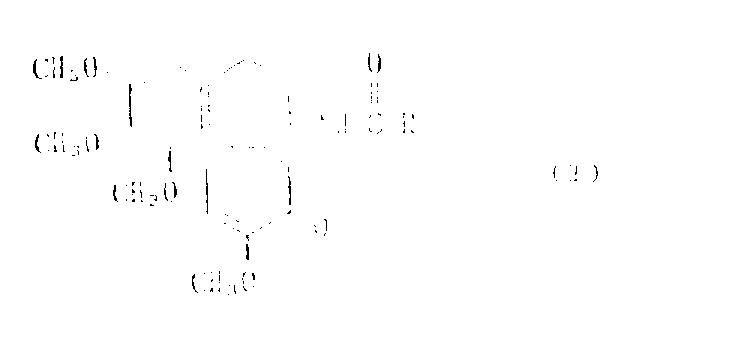
其中R为从C3-C7糖羧酸中除去COOH后得到的残基,在该残基中存在的羟基可用选自酰基、乙缩醛基和酮缩醇基的羟基保护基团适当保护,所述方法包括使式(Ⅱ)表示的脱乙酰基秋水仙素

与下式表示的糖羧酸或它们的酰卤或活性酯进行酰胺化反应,
其中R定义同权利要求1。
2、权利要求1的方法,其中R为:

存在于这些基中的羟基可用酰基、乙缩醛基或酮缩醇基保护。
3、权利要求1的方法,其中式(Ⅰ)的脱乙酰基秋水仙素衍生物选自脱乙酰基秋水仙素-甘油酸丙酮化物酰胺、脱乙酰基秋水仙素-甘油酸酰胺、脱乙酰基秋水仙素-葡糖醛酸四乙酰基酰胺、脱乙酰基秋水仙素-葡糖醛酸二乙酰基酰胺、脱乙酰基秋水仙素-葡糖醛酸酰胺、脱乙酰基秋水仙素-葡糖酸五乙酰基酰胺和脱乙酰基秋水仙素-葡糖酸酰胺。
4、权利要求1的方法,其中由式(Ⅱ)表示的脱乙酰基秋水仙素衍生物如下制备,
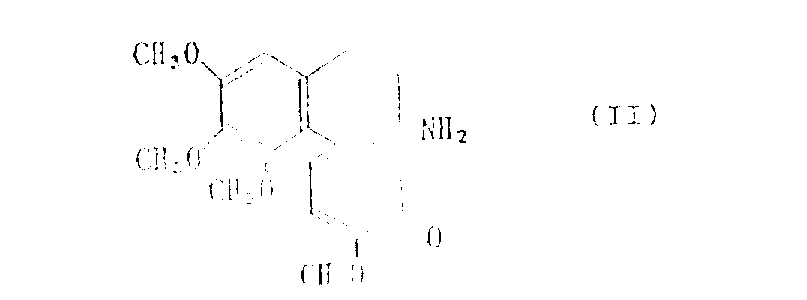
由下式表示的秋水仙素
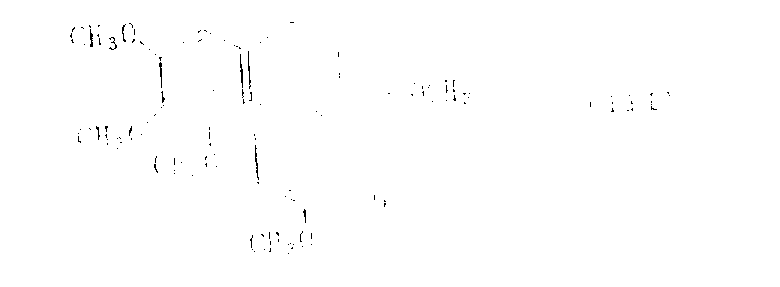
与三烷基氧鏻氧硼酸盐反应,然后用水处理反应混合物。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP413683/90 | 1990-12-25 | ||
| JP41368390A JP2894366B2 (ja) | 1990-12-25 | 1990-12-25 | デアセチルコルヒチンの製造方法 |
| JP419162/90 | 1990-12-25 | ||
| JP2419162A JPH04224550A (ja) | 1990-12-25 | 1990-12-25 | デアセチルコルヒチン誘導体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1063486A CN1063486A (zh) | 1992-08-12 |
| CN1028362C true CN1028362C (zh) | 1995-05-10 |
Family
ID=26583027
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN91112823A Expired - Fee Related CN1028362C (zh) | 1990-12-25 | 1991-12-24 | 脱乙酰基秋水仙素衍生物的制备方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5220002A (zh) |
| EP (1) | EP0493064B1 (zh) |
| KR (1) | KR920012011A (zh) |
| CN (1) | CN1028362C (zh) |
| AT (1) | ATE118477T1 (zh) |
| AU (1) | AU634921B2 (zh) |
| CA (1) | CA2058060A1 (zh) |
| DE (1) | DE69107431T2 (zh) |
| DK (1) | DK0493064T3 (zh) |
| ES (1) | ES2071241T3 (zh) |
| RU (1) | RU2065863C1 (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06199749A (ja) * | 1992-12-28 | 1994-07-19 | Bigen Kenkyusho:Kk | N−メチルデアセチルコルヒセインアミド誘導体 |
| JPH06211762A (ja) * | 1993-01-20 | 1994-08-02 | Bigen Kenkyusho:Kk | N−メチルデアセチルコルヒセインアミド誘導体 |
| DE4330701A1 (de) * | 1993-09-10 | 1995-03-16 | Boehringer Ingelheim Kg | Neues Verfahren zur Herstellung von 1,2-5,6-Diaceton-D-glucose |
| US6825236B2 (en) * | 2003-04-14 | 2004-11-30 | California Pacific Medical Center | Colchicine derivatives |
| ITMI20031144A1 (it) * | 2003-06-06 | 2004-12-07 | Indena Spa | Analoghi del colchicoside. |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1175407A (fr) * | 1955-06-10 | 1959-03-26 | Roussel Uclaf | Nouveaux dérivés des n-désacétyl alcoylthiocolchicéines et leur procédé d'obtention |
| US4533675A (en) * | 1984-04-17 | 1985-08-06 | The United States Of America As Represented By The Department Of Health And Human Services | Carbamates of colchicine for treatment of gout |
| US4692463A (en) * | 1985-02-26 | 1987-09-08 | The United States Of America As Represented By The Department Of Health And Human Services | Antiinflammatory 2,3-didemethylcolchicine and additional derivatives |
| US4904697A (en) * | 1987-04-09 | 1990-02-27 | Merrell Dow Pharmaceuticals Inc. | Controlling the growth of certain tumor tissue with chalcone derivatives |
| AU633867B2 (en) * | 1989-02-02 | 1993-02-11 | Eli Lilly And Company | Delivery of cytotoxic agents |
-
1991
- 1991-12-17 AU AU89820/91A patent/AU634921B2/en not_active Ceased
- 1991-12-19 CA CA002058060A patent/CA2058060A1/en not_active Abandoned
- 1991-12-20 US US07/810,883 patent/US5220002A/en not_active Expired - Fee Related
- 1991-12-23 EP EP91311942A patent/EP0493064B1/en not_active Expired - Lifetime
- 1991-12-23 AT AT91311942T patent/ATE118477T1/de active
- 1991-12-23 DE DE69107431T patent/DE69107431T2/de not_active Expired - Fee Related
- 1991-12-23 DK DK91311942.6T patent/DK0493064T3/da active
- 1991-12-23 ES ES91311942T patent/ES2071241T3/es not_active Expired - Lifetime
- 1991-12-24 KR KR1019910024165A patent/KR920012011A/ko not_active Ceased
- 1991-12-24 CN CN91112823A patent/CN1028362C/zh not_active Expired - Fee Related
- 1991-12-24 RU SU915010441A patent/RU2065863C1/ru active
Also Published As
| Publication number | Publication date |
|---|---|
| DK0493064T3 (da) | 1995-06-06 |
| ATE118477T1 (de) | 1995-03-15 |
| DE69107431T2 (de) | 1995-06-14 |
| KR920012011A (ko) | 1992-07-25 |
| CN1063486A (zh) | 1992-08-12 |
| RU2065863C1 (ru) | 1996-08-27 |
| DE69107431D1 (de) | 1995-03-23 |
| US5220002A (en) | 1993-06-15 |
| AU634921B2 (en) | 1993-03-04 |
| CA2058060A1 (en) | 1992-06-26 |
| AU8982091A (en) | 1992-07-02 |
| EP0493064B1 (en) | 1995-02-15 |
| ES2071241T3 (es) | 1995-06-16 |
| EP0493064A1 (en) | 1992-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1145619C (zh) | 1,4-苯并硫杂吖庚因衍生物、其制备方法、含有该化合物的药物及其用途 | |
| CN1357000A (zh) | 位阻醇或酚的水溶性前药 | |
| JP2002523495A (ja) | 高い水溶性を有するトリプトリドプロドラッグ | |
| CN1202105A (zh) | 2-烷基吡咯烷化合物 | |
| CN1182430A (zh) | 与糖类结合的生物活性化合物 | |
| CN1136224C (zh) | 具有类胰岛素作用的肌醇聚糖 | |
| CN1568328A (zh) | 新的糖脂及其作为有效成分的用于自体免疫疾病的药物 | |
| CN1028362C (zh) | 脱乙酰基秋水仙素衍生物的制备方法 | |
| CN1042911A (zh) | 心脏保护生育酚类似物 | |
| CA2770793C (en) | Azetidinone compounds and medical use thereof | |
| FR2723093A1 (fr) | Esters d'acyle d'acides amines achiraux du glanciclovir | |
| CN1027264C (zh) | 粟籽豆精胺苷的制备方法 | |
| CN1060846A (zh) | 含四氢吡喃环的大环化合物制备方法 | |
| CN1224626C (zh) | β-D-5-硫代木糖衍生物、其制备方法及医疗用途 | |
| CN1023896C (zh) | 羧基酰胺衍生物的制备方法 | |
| CN1099034A (zh) | 椭圆玫瑰树碱衍生物及其制备方法 | |
| CN1305486A (zh) | 从α-D-木糖衍生的新化合物,制备方法和治疗用途 | |
| CN1108297C (zh) | 维甲酰香豆素类化合物及其制备方法和含该化合物的药物组合物 | |
| CN1781932A (zh) | 阿霉素的衍生物及其制备方法和用途 | |
| CN1131239C (zh) | 治疗或预防前列腺癌的组合物 | |
| CN1343121A (zh) | 新颖的免疫抑制剂 | |
| CN1649600A (zh) | 氟西汀及类似物的糖醛酰胺、苷和原酸酯苷及其应用 | |
| CN1090266A (zh) | N-甲基脱乙酰基秋水仙裂碱氨化物衍生物 | |
| CN1013491B (zh) | 氟化二氨基炔衍生物的制备方法 | |
| CN1201447A (zh) | 乳酸的丁酸酯前药 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |