CN102432594B - 一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法 - Google Patents
一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法 Download PDFInfo
- Publication number
- CN102432594B CN102432594B CN 201110383990 CN201110383990A CN102432594B CN 102432594 B CN102432594 B CN 102432594B CN 201110383990 CN201110383990 CN 201110383990 CN 201110383990 A CN201110383990 A CN 201110383990A CN 102432594 B CN102432594 B CN 102432594B
- Authority
- CN
- China
- Prior art keywords
- phenyl
- solvent
- methylpiperazine
- ether
- ethyl acetate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Abstract
本发明涉及医药中间体领域,特别公开了一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法。该药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法,其特征在于:由以下步骤依次制成:(1)将1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪与醚类溶剂混合反应;(2)向混合反应液中缓慢加入硼氢化钠;(3)再缓慢加入无水三氯化铝;(4)温度缓缓升至0-100℃并保持反应;(5)减压蒸除溶剂,冷却,用溶剂提取,洗涤,干燥,浓缩,重结晶即得。本发明的有益效果是:后处理方法简便,提高生产效率,产品的收率均在80%以上,产品的纯度在99%以上。
Description
(一) 技术领域
本发明涉及医药中间体领域,特别涉及一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法。
(二) 背景技术
1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪(化合物Ⅰ)是制备米氮平的关键中间体。米氮平(英文名称为mirtazapine,化合物Ⅱ)是一种有效的抗抑郁药物(US4062848),它是选择性5-羟色胺再摄取抑制剂,此药物于1994年上市销售,于1996年获得美国FDA认可,目前已经在全世界许多国家临床上广泛应用。
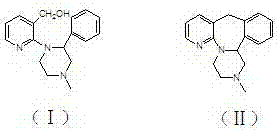
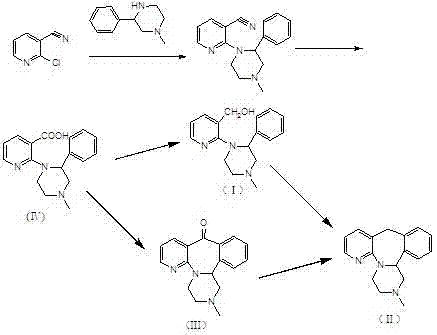
根据US4062848,米氮平的合成路线如式1所示:
式1
路线中如果通过化合物(Ⅲ)来制备米氮平,化合物(Ⅲ)的还原涉及到高温等苛刻的反应条件,导致难于操作、副产物多、能耗高等问题。如果通过化合物(Ⅰ)来制备,根据专利描述,1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪(化合物Ⅳ)还原为(Ⅰ)需用大量的还原剂氢化铝锂以及大量的溶剂四氢呋喃,缺点是四氢铝锂易燃,难于操作,不适合工业化生产,大量溶剂的使用也造成成本的大大提高。
根据文献的查阅表明,目前所有的米氮平的合成几乎均以1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪为中间体,如式2所示:
式2
由式2可以看出,化合物(Ⅰ)是合成米氮平的关键的中间体,它是一种苄醇化合物,而其制备均是从其相应的羧酸(Ⅳ)还原而来,根据US4062848,在四氢呋喃溶剂中,用大量的四氢铝锂在回流条件下还原制备,此种方法所用的试剂昂贵、易燃、不安全,产物难以纯化等缺点,在实际的生产过程中存在很多的问题,不适合大规模的工业生产。
针对相应的羧酸还原为1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪,张庆文等(中国医药工业杂志Chinese Journal of Pharmaceuticals 2006,37(10))进行了改进,用二硼烷在乙二醇二甲醚中在氮气保护条件下进行还原,其中涉及使用无水氯化氢的乙二醇二甲醚的溶液及乙醚三氟化硼的复合物,此类试剂或价格昂贵,或者制作成本很高,难以大量使用与操作。
(三) 发明内容
本发明为了弥补现有技术的不足,提供了一种低成本、高收率、操作方便的药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法。
本发明是通过如下技术方案实现的:
一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法,其特征在于:由以下步骤依次制成:
(1)将1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪与醚类溶剂混合反应,醚类溶剂的用量为500-5000ml/mol1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪;
(2)向步骤(1)所得的混合反应液中缓慢加入硼氢化钠,硼氢化钠的用量为1-3mol/mol1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪,控制温度为0-50℃,搅拌时间为0-10小时;
(3)再缓慢加入无水三氯化铝,无水三氯化铝的用量为三分之一摩尔/摩尔硼氢化钠,保持温度为0-50℃;
(4)加毕,温度缓缓升至0-100℃,并于0-100℃下保持反应;
(5)反应完成后,减压蒸除溶剂,冷却,加入水,调节PH为12,用溶剂S1提取,洗涤,干燥,浓缩,用溶剂S2重结晶即得。
步骤(1)中醚类溶剂为乙二醇二甲醚、乙醚、四氢呋喃或二氧六环。
步骤(2)中硼氢化钠的用量优选为2-2.5mol/mol1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪;控制温度优选为0-25℃,搅拌时间优选为0-2小时。
步骤(4)中优选为温度缓缓升至60-80℃,并于60-80℃下保持反应。
步骤(5)中溶剂S1为乙酸乙酯、甲苯、乙醚、二氯甲烷或氯仿,优选乙酸乙酯;溶剂S2为乙酸乙酯、石油醚、乙醚、正己烷、环己烷、甲苯、甲醇、乙醇、异丙醇中的一种或几种的组合,优选乙酸乙酯与石油醚的混合溶剂,乙酯乙酯:石油醚的体积比为1:2。
本发明药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法的有益效果是:摒弃以往技术中使用的昂贵、易燃易爆试剂,而改用相对价格低廉、温和、易于操作的硼氢化钠,降低生产成本、提高生产安全性,并且后处理方法简便,产品纯度更高;能方便的放大,扩大生产规模,提高生产效率,产品的收率均在80%以上,产品的纯度在99%以上(HPLC外标)。
(四) 具体实施方式
为了对本发明做更好的阐述,下面以具体的实施例做进一步的说明。
实施例1:
反应瓶中加入1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪29.7g(0.1mol,按US4062848制得)及900ml四氢呋喃,搅拌,缓慢加入7.6g硼氢化钠(0.2mol),控制内温不高于25℃,加毕,搅拌1小时,冷却条件下缓慢加入无水三氯化铝8.9g(0.067mol),控制温度不高于25℃,加毕,缓慢升温至回流状态反应,以TLC检测反应终点,稍冷,减压蒸除700ml溶剂,缓慢加入300ml水,搅拌1小时,用质量分数为40%的氢氧化钠溶液调节PH到12,加入乙酸乙酯200ml提取两次,合并有机相,用质量分数为5%的碳酸钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩得粗品,用乙酸乙酯/石油醚混合溶剂重结晶(乙酯乙酯:石油醚=1:2)得精品23.5g(收率83.0%);HPLC99.2%。
实施例2:
1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪59.4g(0.2mol)及1200ml二氧六环混合搅拌,冷却,缓慢加入15.2g硼氢化钠(0.4mol),控制内温低于25℃,加完后搅拌1小时,冷却条件下缓慢加入无水三氯化铝17.8g(0.134mol),控制温度不高于25℃,加毕,升温至70℃,以TLC检测反应终点,稍冷,减压蒸除900ml溶剂,缓慢加入400ml水,搅拌1.5小时,用质量分数为40%的氢氧化钠溶液调节PH到12,加入乙酸乙酯400ml提取两次,合并有机相,用质量分数为5%的碳酸钠溶液洗涤,无水硫酸镁干燥,过滤,浓缩,用乙酸乙酯/石油醚混合溶剂重结晶(乙酯乙酯:石油醚=1:2)得精品46.0g(收率81.3%);HPLC99.6%。
本发明摒弃以往技术中使用的昂贵、易燃易爆试剂,而改用相对价格低廉、温和、易于操作的硼氢化钠,降低生产成本、提高生产安全性,并且后处理方法简便,产品纯度更高;能方便的放大,扩大生产规模,提高生产效率,产品的收率均在80%以上,产品的纯度在99%以上(HPLC外标)。
Claims (4)
1.一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法,其特征在于:由以下步骤依次制成:
(1)将1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪与醚类溶剂混合反应,醚类溶剂的用量为500-5000ml/mol1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪;醚类溶剂为乙二醇二甲醚、乙醚、四氢呋喃或二氧六环;
(2)向步骤(1)所得的混合反应液中缓慢加入硼氢化钠,硼氢化钠的用量为1-3mol/mol1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪,控制温度为0-25℃,搅拌时间为0-2小时;
(3)再缓慢加入无水三氯化铝,无水三氯化铝的用量为三分之一摩尔/摩尔硼氢化钠,保持温度为0-50℃;
(4)加毕,温度缓缓升至60-80℃,并于60-80℃下保持反应;
(5)反应完成后,减压蒸除溶剂,冷却,加入水,调节pH为12,用溶剂S1提取,洗涤,干燥,浓缩,用溶剂S2重结晶即得;溶剂S1为乙酸乙酯、甲苯、乙醚、二氯甲烷或氯仿;溶剂S2为乙酸乙酯、石油醚、乙醚、正己烷、环己烷、甲苯、甲醇、乙醇、异丙醇中的一种或几种的组合。
2.根据权利要求1所述的药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法,其特征在于:步骤(2)中硼氢化钠的用量为2-2.5mol/mol1-(3-羧基吡啶-2-基)-2-苯基-4-甲基哌嗪。
3.根据权利要求1所述的药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法,其特征在于:步骤(5)中溶剂S1为乙酸乙酯。
4.根据权利要求1所述的药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法,其特征在于:步骤(5)中溶剂S2为乙酸乙酯与石油醚的混合溶剂,乙酸乙酯:石油醚的体积比为1:2。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201110383990 CN102432594B (zh) | 2011-11-28 | 2011-11-28 | 一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 201110383990 CN102432594B (zh) | 2011-11-28 | 2011-11-28 | 一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102432594A CN102432594A (zh) | 2012-05-02 |
| CN102432594B true CN102432594B (zh) | 2013-09-11 |
Family
ID=45980983
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 201110383990 Active CN102432594B (zh) | 2011-11-28 | 2011-11-28 | 一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102432594B (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015174853A (ja) * | 2014-03-17 | 2015-10-05 | 株式会社トクヤマ | 2−(4−メチル−2−フェニルピペラジン−1−イル)ピリジン−3−メタノールの製造方法 |
| JP6433809B2 (ja) * | 2015-02-20 | 2018-12-05 | 株式会社トクヤマ | 1−(3−ヒドロキシメチルピリジル−2−)−2−フェニル−4−メチルピペラジンの製造方法 |
| CN105367571B (zh) * | 2015-11-30 | 2017-11-28 | 北京哈三联科技有限责任公司 | 一种米氮平的合成方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4062848A (en) * | 1975-04-05 | 1977-12-13 | Akzona Incorporated | Tetracyclic compounds |
| CN1521166A (zh) * | 2003-02-13 | 2004-08-18 | 上海医药工业研究院 | 1-(3-羟甲基吡啶基-2)-2-苯基-4-甲基哌嗪的制备方法 |
-
2011
- 2011-11-28 CN CN 201110383990 patent/CN102432594B/zh active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4062848A (en) * | 1975-04-05 | 1977-12-13 | Akzona Incorporated | Tetracyclic compounds |
| CN1521166A (zh) * | 2003-02-13 | 2004-08-18 | 上海医药工业研究院 | 1-(3-羟甲基吡啶基-2)-2-苯基-4-甲基哌嗪的制备方法 |
Non-Patent Citations (7)
| Title |
|---|
| 张涛 等.抗抑郁药米氮平的合成.《华东理工大学学报(自然科学版)》.2006,第32卷(第3期),第318-320,326页. * |
| 张润虎 等.米氮平的合成进展.《化工中间体》.2008,(第12期),第24-28页. |
| 抗抑郁药米氮平的新合成方法;陈升 等;《中国新药杂志》;20071231;第16卷(第2期);第137-139页 * |
| 硼氢化钠体系作还原剂对羰基的还原;罗枝伟 等;《广东化工》;20051231(第3期);第12-14页 * |
| 米氮平的合成进展;张润虎 等;《化工中间体》;20081231(第12期);第24-28页 * |
| 罗枝伟 等.硼氢化钠体系作还原剂对羰基的还原.《广东化工》.2005,(第3期),第12-14页. |
| 陈升 等.抗抑郁药米氮平的新合成方法.《中国新药杂志》.2007,第16卷(第2期),第137-139页. |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102432594A (zh) | 2012-05-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103242216A (zh) | N- Boc -六氢-5-氧代环戊[C]并吡咯的合成方法 | |
| CN102432594B (zh) | 一种药物中间体1-(3-羟甲基吡啶-2-基)-2-苯基-4-甲基哌嗪的制备方法 | |
| CN102898279B (zh) | 一种固体金属醇盐的制备方法 | |
| CN103420855A (zh) | 一种反式-4-氨基环己基甲醇盐酸盐及其制备方法 | |
| CN104478719A (zh) | 一种4-甲氧基乙酰乙酸甲酯的制备方法 | |
| CN103833560A (zh) | (S)-5-氯-α-环丙炔基-2-氨基-α-三氟甲基苯甲醇的制备方法 | |
| CN102766061A (zh) | 脱氢枞酸基二芳胺类化合物及其合成方法和应用 | |
| CN103044468A (zh) | N-(2-吡嗪羰基)-l-苯丙氨酸-l-亮氨酸硼酸的制备方法 | |
| CN102718662A (zh) | 一种制备盐酸西那卡塞的方法 | |
| CN103497138B (zh) | 一种利用氯化锌、硼氢化钾制备顺式全氢异吲哚的方法 | |
| CN103508934A (zh) | 一种格列齐特的制备方法 | |
| CN106749116B (zh) | 一种3-氨基甲基四氢呋喃的制备方法 | |
| CN109438373A (zh) | 一种n-甲基高哌嗪的合成方法 | |
| CN110862421B (zh) | 含氮杂环二茂铁衍生物的合成方法 | |
| CN103922943B (zh) | 一种制备盐酸芬戈莫德的方法 | |
| CN105418507A (zh) | 一种1-(3-甲基-1-苯基-1h-吡唑-5-基)哌嗪的制备方法 | |
| CN103848756B (zh) | 特立氟胺及其中间体的制备方法 | |
| CN106008363B (zh) | 2-甲基-4-氨基-5-氰基嘧啶的制备方法 | |
| CN102070513A (zh) | 1-叔丁氧羰基-4-哌啶酮的合成方法 | |
| CN105439978A (zh) | 阿考替胺中间体的制备方法 | |
| CN103214496A (zh) | 一种双氢青蒿素的简单快速制备工艺 | |
| CN105017137B (zh) | 一种由苹果酸制备维生素b6的方法 | |
| CN103319358B (zh) | 一种7-氨基庚酸的制备方法 | |
| CN104788482B (zh) | 一种制备2-氨基嘧啶-5-硼酸频那醇酯的方法 | |
| CN105001149A (zh) | 一种琥珀酸多西拉敏中间体2-吡啶基苯基甲基甲醇的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant |