制备器官型细胞培养物的方法
本发明涉及细胞和组织培养。更具体地,本发明提供一种利用从动物器官获得的解离的细胞或微外植块制备器官型培养物的方法。
组织培养是先体内再体外地维持源自动物或植物生物体器官或组织的细胞。几十年来,已经开发和改良了组织培养的方法。
培养从植物或动物器官或组织直接获得的细胞称为原代培养。按照一种原代培养的方法,将器官外植块置于合适的无菌培养基中,所述培养基装于提供具有合适组成的无菌空气的合适的培养容器中,因此细胞从外植块的边缘生长。原代培养的另一种方法是通过蛋白水解酶,如胰蛋白酶的处理解离器官或组织的细胞,其分解引起组成器官或组织的细胞间粘附的蛋白质。也可以使用机械方法解离器官或组织细胞,或可以使用机械和酶方法的组合。然后可以在如上所述的适合环境中培养解离的细胞。在需要培养特殊细胞种群的情形中,可以将解离的细胞分级,例如通过密度梯度离心。然后在培养基中重新悬浮分离的目标细胞种群,由此使细胞解离,并在适合的环境中培养属于那个种群的解离的细胞。
动物细胞原代培养的一个缺点是细胞仅具有有限的寿命。原代培养中的细胞可以经历细胞分裂,但它们在经历被称为衰老的细胞死亡形式前,通常仅进行有限次数的分裂。基于外植块的动物细胞原代培养的另一个缺点是培养的细胞通常丧失许多源器官细胞典型的体内特征,除非进行特殊的用于阻止所述特征丧失的步骤。体内特征的丧失通过3中不同的途径发生。第一,所述器官都是多种细胞类型的混合物,且在原代培养中一种细胞类型可以比其他细胞类型生长的更快,从而在该培养中占优势。第二,一种或多种该成分细胞类型可以去分化到一定程度,从而丧失其许多特殊特征。第三,培养中的一种或多种去分化或部分去分化的细胞可以成为无限增殖的,无法衰老,且生长成为永久细胞品系。可以无限地培养该无限增殖细胞品系,且存在数千种来源于人和试验动物,如大鼠和小鼠的近乎 每种器官的无限增殖细胞品系的实例。
基于外植块或基于解离细胞的原代培养过程中的体内特征的丧失,和无限增殖细胞系的出现对生物和医药研究以及产品开发具有重要的含意,其原因在于它意味着原代细胞培养不能准确预测体内应答。作为结果,必须在完整动物体内进行许多生物检验,从而评估候选药物的安全性和功效。所述在完整动物中的检验很昂贵,这导致更高的卫生保健成本,并且该检验会损害动物的健康。因此,多年来,存在着对开发更准确预测体内应答的体外检验相当大的动力。
器官培养是先体外再体内地维持动物器官的整体或部分,其在将器官的生命和功能维持一段时期的条件下进行。例如,存在培养肝脏(Wicks W.,1968),心脏(Wildenthal K.,1971)和肠(Corradino R.,1973)的确定程序。器官培养具有超越基于外植块原代细胞培养和细胞系的重要优点,在于维持了该器官大部分或全部生理性质。然而,器官培养的通量受到从宿主手术摘除器官和建立培养体系所必需的操作的限制。此外,从每个供体动物仅能获得一个,二个或很少的培养物。这些限制致使器官培养对于药物筛选和药靶筛选,以及许多在生物学研究中的其他应用来说,太过缓慢和昂贵。
在组织培养领域中的一个重要进展是引入器官和组织切片的器官型培养。在切片保持其源器官的细胞组成,形态学和所述生理性质的条件下培养动物器官的薄切片(50-500μm)。培养器官切片的条件对完成器官型培养是至关重要的。将器官切片培养在多孔膜的上表面上,并从该多孔膜的下表面供给营养,由此器官切片不完全浸没,而是仅仅被一薄层培养基所覆盖(Stoppini L.等,1991)。为了吸收氧气和去除二氧化碳,向切片传送的气体,比在按照外植块培养方法将切片完全浸没在培养基中时更有效得多。另外,器官型切片培养不具有与上述基于外植块和基于解离细胞原代培养有关的缺点。例如,来自啮齿动物脑海马区域的解离细胞的原代培养引起神经元的损失和被胶质细胞替换,且仅可以用于进行神经研究2-3天。相反,海马培养的器官型切片方法导致胶质细胞向与所述膜接触的切片表面的最初迁移,但保留神经元,及具体地保留神经元间和胶质细胞—神经元的连通性。已经显示出新突触在器官型脑切片培养中形成(Buchs P.等,1993),且通过轴突突出和突触形成可以至少部分地修复对器官型脑切 片培养中神经元连接的损伤(Stoppini L等,1993)。基于相同的原理,存在着许多其他组织切片的器官型培养实例,且存在着对改进培养单一器官型切片的方法的建议。例如,Giehl(2002)公开了应用吸引术降低来自老年动物的器官型切片的表面湿度。
器官型切片培养比器官培养显著地更快且更灵活,但是它对于药物发现所需的大规模筛选仍然太缓慢且昂贵。为从动物解剖器官或处理手术后人类材料所使用的程序是劳动密集的,且在大多数实验室中仅可能平行地进行数10个培养。对于药物筛选,平行地提供几千、几万或几十万个培养将更加有效。此外,器官切片对于转染或转导载体对转基因或小干扰RNA(siRNA)表达的应用或寡核糖核苷酸直接对基因表达的siRNA脱离的应用,是相对难控制的。通常只能有效地转染或转导切片的表面层,且总转染或转导效率通常在切片中细胞的10-30%的范围内。这经常不足以评估转基因表达的生物作用或siRNA诱导的基因表达的脱离。
考虑到与器官型切片培养有关的缺点,已经作出开发利用来自具体器官而非器官切片的解离细胞制备器官型培养物的方法的努力。
一种从解离细胞形成器官型培养物的方法一直是使用细胞外基质蛋白来促使解离细胞迁移到适合的层或区中,并在适合的生长因子存在下重新形成功能性细胞—细胞相互作用。已在器官型肝培养物的制备使用了该方法(Michalopoulos G.等,2001;Michalopoulos G.和Bowen W.,2004)。相似地,已将人工基质用作混悬液中神经细胞生长组构的“支架”,从而允许细胞团块的生长,所述细胞团块作为植入物潜在地对治疗疾病有效(Rochkind S.等,2002;Shahar A.等,2001)。上皮组织一直是研究的特殊焦点,且已显示出蛋白质基质可以用于促使解离的肠上皮细胞形成器官型培养物(Kalabis J.等,2003;Herlyn M.,2004)。存在着许多相似的皮肤上皮的器官型培养的方法,其对于皮肤损伤后的移植很重要。然而,这些方法不理想,原因在于需要在创建该培养前建立基质蛋白的3D结构,它们依赖于昂贵和难以使用的基质蛋白。
已经开发了两种不依赖于使用基质蛋白的培养方法。例如,已经发现旋转介导的解离神经细胞的聚集可以用于形成所谓的“神经球”(Honegger,P.,和Monnet-Tschudi,F.,2001)。还发现了可以利用特殊生长因子诱导来自 分层上皮基底层的干细胞以逐步的进程分化,从而创造分层或角质化上皮细胞,其中作为其增殖能力的结果,所述干细胞通过培养从解离上皮分离而来(Wille J.,1998)。然而,该过程特异于分层上皮,且要求使用适合于分层上皮分化的纯化生长因子。
尽管至今已实施了广泛的研究,但仍然存在着对用于培养器官型组织的简单、便宜和灵活方法的需要,其适合于广泛多种组织,并合适于产生数千个平行培养物,以进行高通量筛选。
发明概述
按照第一方面,本发明提供制备器官型培养物的方法,所述方法包括在表面上培养来自器官的细胞,所述方法的特征在于所述细胞是压缩的。
通过“器官型培养”意指细胞以这样的方式联合,所述方式是尽可能接近地复制该细胞起源器官的生化和生理性质。
优选地,所述细胞是解离细胞,微外植块或外植块。用于本文时,术语“解离细胞”指从器官分离的单细胞。术语“微外植块”指从器官分离的多达500个细胞的小组。术语“外植块”指从器官分离的多于500个细胞的较大的组。本发明的方法涉及压缩和培养多于一个解离细胞,或多于一个微外植块,或多于一个外植块。优选地,本发明的方法涉及压缩和培养从器官分离的许多解离细胞,或许多微外植块,或许多外植块。
优选地,在其上培养所述细胞的表面是膜,优选地是多孔膜。当在多孔膜的一个表面上培养压缩的细胞时,所述方法优选地包括向该膜的对侧表面供给营养。优选地,供给到该膜对侧表面的营养处于液体形式,且所述方法由此涉及向该膜的对侧表面供给液体培养基。在这个实施方案中,如例如Stoppini L.等(1991)所述,该膜上的细胞不浸没在液体培养基中,而是仅被培养基薄层覆盖,从而容许培养基和细胞间更好的气体交换。因此所述膜优选地对液体培养基具有充分的多孔性,从而使液体培养基穿透该膜并到达位于相反表面上的压缩的细胞。本文提供了合适的膜的实例。优选地,通过毛细作用保持液体培养基与膜的对侧表面相接触。文本描述了能够通过毛细作用保持一定体积液体培养基的合适装置。
令人震惊地,发现了本领域中所描述的从器官切片制备器官型培养物 的培养条件(参见,例如,Stoppini L.等,1991)可以用于从来自器官的细胞,特别地解离细胞、微外植块和外植块制备器官型培养物,只要压缩所述细胞使其紧密地压缩在一起。压缩的细胞随着时间将自发地重新组织为复杂的3-D功能性实质细胞。例如,压缩的脑细胞将引起组织状结构的形成,其中存在完整中枢神经系统区域的许多适合的突触电路,生理学和神经递质受体的分布。所述培养物中神经元的功能活性与它们在脑和在器官型切片培养物中的对应物相似。
与从解离细胞制备器官型培养物的现有技术方法不同,本发明的方法不要求使用生物分子和合成支架。此外,由大多数现有技术方法所产生的培养物仅表现器官型特征3-5天。相反,按照本发明的方法所产生的器官型培养物表现器官型特征数周或数月。
由此本发明提供利用来自广泛多种器官的细胞,特别是解离细胞、微外植块或外植块,制备器官型培养物的简单方法,所述培养物可以保持数周或数月。易于产生和维持器官型培养物的事实使它们理想地用于构建来自单一器官的数千个平行培养物,从而进行药物候选物的高通量筛选。此外,可以在创建器官型培养前对解离细胞进行有效地遗传学操作,从而通过在合适载体中的转染或转导的方法,或通过引入作为寡核苷酸或从合适的载体表达的siRNA,引入一个或多个转基因。
用于本文时,术语“压缩的”意指对细胞施加将它们挤压在一起的压缩力,由此使它们被紧密地压缩。尽管本申请人不希望受到理论限制,但仍相信是由压缩所导致的细胞—细胞接触诱导了器官型生长。
活细胞不表现为刚体,而为基本不可压缩体。它们变形并使它们的表面适合于互相粘附,但是除非它们损失细胞液,否则它们的体积保持基本恒定。如果应用了充足的压缩力,则将获得100%的紧密压缩,即该细胞将被完全紧密地压缩,其所有细胞膜均与相邻细胞的膜相接触。100%的紧密压缩是在不将细胞挤压到使它们损失细胞液的程度下对于给定细胞尺寸最大的细胞数量/单位体积。已发现如果组成培养物的元件间的平均紧密压缩度是至少约10%,更优选地5%,则按照本发明的细胞、微外植块或外植块的培养起到器官型培养物的作用。
优选地,压缩本发明的方法中的细胞,以获得按照本文所述的紧密压 缩定义的5%-100%间的紧密压缩。优选地,压缩所述细胞,以获得大于5%的紧密压缩,优选地,大于10%,20%,30%,40%,50%,60%,70%,75%,80%,85%,90%,95%,97%,98%或99%的如本文所定义的紧密压缩。
当本发明的方法使用解离细胞时,可以利用以下公式计算近似的紧密压缩度:
紧密压缩度=(Ne×Ve/Vcm)×100% (公式1)
其中Ne是细胞的数量,
Ve是细胞的平均体积,和
Vcm培养物中的细胞团块总体积的测量体积。
由此,如果测量的培养物中总细胞体积是平均细胞体积总和体积的2倍,则获得50%的紧密压缩。如果测量的细胞团块总体积是平均细胞体积总和体积的10倍,则获得10%的紧密压缩。以下更加详细地描述了确定细胞的数量(Ne),细胞平均体积(Ve)和细胞团块总体积的体积的方法。
当分离自器官的解离细胞用于本发明的方法时,公式1提供了紧密压缩度的很好近似值。然而,公式1不提供这么好的外植块和微外植块紧密压缩度的近似值。外植块和微外植块的尺寸可以在从几个细胞到数千细胞间变化。组成每个外植块和微外植块的细胞已为100%和近似100%紧密压缩的,但是压缩后相邻的外植块和微外植块表面上的细胞间的细胞表面接触度可以显著地小于100%。外植块和微外植块越大,则这些表面接触对按照公式1测量的紧密压缩度影响越小,尽管它们对外植块和微外植块表现为单一器官型培养物的影响很深刻。
克服这个问题的一种途径可能是测量微外植块和外植块间的细胞表面接触,从而确定这些元件间的紧密压缩度。然而,测量细胞表面接触的方法是困难和不精确的,其依赖于高放大率显微镜检验和主观判断。因此,改为使用培养物团块和成分外植块或微外植块体积总和之间的体积差异作为细胞表面间空间的有效测量,其中所述细胞表面是按照本发明被压缩力所移除的。测量体积差异,以确定外植块或微外植块的压缩度十分方便,原因在于,与细胞表面接触不同,可以相当简单和快速地获得外植块或微外植块数量和体积的测量,如下所述。
因此通过表达外植块或微外植块中细胞的体积可以估算外植块和微 外植块的紧密压缩度,所述外植块或微外植块能够制造新接触,其为实际占培养的细胞团块总空间的一定比例。由此可以通过以下公式测量微外植块或外植块的紧密压缩度
紧密压缩度=
((Nnc×Vnc×Pnc)/((Nnc×Vmc×Pnc)+(Vcm-(Ne×Ve)))×100%(公式2)
其中,Nnc是能够制造新接触的细胞数量,
Vnc是能够制造新接触的培养物中细胞的平均体积,
Pnc是能够制造新接触的细胞表面积的平均比例,其中所述新接触实际上可以用于制造新接触,
Vcm是测量的培养物中细胞团块的总体积,
Ne是培养物(细胞,外植块或微外植块)中元件的数量,和
Ve是培养物(细胞,外植块或微外植块)中元件的平均体积。
在解离细胞的情形中,培养物中所有细胞都能够在它们的整个表面上制造新接触,且因此Nnc=Ne,Vnc=Ve和Pnc=1。由此,在解离细胞的情形中,公式2等价于公式1。
公式1和2可以用于约计由任意尺寸的细胞、外植块或微外植块组成的培养物中培养元件的紧密压缩度。
100%紧密压缩意指组成细胞团块的元件间不存在空间,无论这些元件是细胞、外植块或微外植块。
50%紧密压缩意指组成细胞团块的元件间空间的总体积等于能够制造新接触的细胞相应比例体积的总和。在解离细胞的情形中,所有细胞能够在它们的整个表面上制造新接触,且因此能够制造新接触的细胞体积的总和等价于组成细胞团块的细胞体积的总和。当培养物元件是外植块或微外植块时,只有在外植块或微外植块表面处的细胞能够制造新接触,且仅在它们约50%的表面上。因此,50%紧密压缩度意指外植块或微外植块间空间的总体积等于能够制造新接触的细胞的体积总和的一半。
10%紧密压缩度意指组成细胞团块的元件间的空间总体积是能够制造新接触的细胞相应比例体积总和的10倍。在解离细胞的情形中,这等价于组成细胞团块的细胞的体积总和的10倍。当培养物元件是外植块或微外植块时,10%紧密接触意指外植块或微外植块间的空间总体积是能够制 造新接触的细胞的体积总和的5倍。
按照公式1和2计算近似紧密压缩度所需要的经验测量值是Ne,Ve 和Vcm。公式2另外需要经验测量值Nnc,Vnc和Pnc。
对于接触的细胞,可以利用Beckman Coulter Inc销售的Coulter计数器测量细胞数量(Ne)和细胞体积(Ve)。这个仪器使用电感应区技术来测量细胞体积和细胞数量。可以方便地在压缩前在混悬液中测量解离细胞的平均体积(Ve)和数量(Ne)。则压缩后培养物(Vcm)中总细胞团块体积的测量容许按照公式1直接计算紧密压缩度。
为了计算Vcm,可以模型化实施例中所描述的培养物的形状。按照本发明的典型器官型培养物是直径5000μm和深100μm的球冠形状。球冠是由一个平面切割下来的球的一部分,且可以按照公式(π/6)(3r2+h2)h计算球冠的体积,其中r=底面半径,且h=高。由此,典型培养物具有总体积9.8×108μm3。测量培养物中细胞团块总体积的简便方法是利用具有倒置显微镜的分度目镜在低放大率下测量培养物样品的直径,并且通过计数跨越培养物完整深度所需要的已知深度的共焦切片的数量来测量它们的深度。然后可以使用上述公式计算球冠的体积。
可以使用的测量培养物中元件数量(Ne)、培养物中元件平均体积(Ve)和细胞团块总体积(Vcm)的其他方法涉及分度显微镜目镜的应用和基于像素计数的图像分析的应用。共焦显微术的应用还允许在多个已知深度区域内的精确计数。
基于显微术和图像分析的方法可以用于确定解离细胞,或外植块或微外植块中的细胞的数量和平均体积,以及外植块和微外植块自身的数量和体积。检验单细胞所需的放大率显然通常高于检验外植块和微外植块所需的放大率。细胞体积可以在细胞群中的不同单独细胞间变化,甚至是在这些细胞属于相同类型时,因此给定显微镜视野面积内的细胞的数量和体积可以变化。相似地,当检验一种制备物时,外植块和微外植块的数量和尺寸可以在不同视野间及不同制备物间变化。优选地,在压缩前,可以利用在合适放大率下的具有倒置显微镜的分度目镜和用于操作的塑料或玻璃针测量外植块或微外植块的数量和体积。在随机选择的已知尺寸显微镜视野中测量适合的外植块或微外植块样品的数量和体积。于是计算已知体积 液体或细胞团块中的细胞或细胞组的平均数量和尺寸成为简单的事情。
当使用不同解离细胞的混合物时,将100%紧密压缩定义为细胞混合物中所有细胞体积的总和。100%紧密压缩的细胞的混和群的总体积等于平均细胞体积乘以细胞数量。由此可以通过测量适合细胞样品的尺寸和计算每个细胞的体积获得平均细胞体积。当小比例的非典型尺寸细胞可以对计算平均体积具有实质影响时,测量充分大的细胞样本的尺寸是必要的,从而确保这些非典型尺寸细胞不受取样误差的排斥。
如果不能通过电感应区技术或其他直接方法直接测量细胞或细胞团块的体积,则可以从它的尺寸计算细胞、微外植块或外植块的体积。为了本发明的目的,可以充分地估计细胞、外植块或微外植块的形状。例如,如果细胞或细胞组的形状近似立方体,则可以按照公式Vcm=L1×L2×L3 来计算所述细胞或细胞组的体积,其中L1,L2和L3是沿着近似立方体细胞或细胞组的3个原则相互正交轴测量的尺寸。作为进一步的实例,如果椭圆体细胞类型的尺寸是长度12μm,宽度8μm和深度5μm,则按照众所周知的计算椭圆体体积的公式:4/3×π×L1×L2×L3,所述细胞的体积应该是2010μm3,其中L1,L2和L3是沿着近似椭圆体细胞的3个原则相互正交轴测量的尺寸。对于复杂形状的细胞或细胞组,存在由多维计算体积的计算机算法式。然后可以计算适合数量的细胞、外植块或微外植块的平均体积。
应该理解,对于解离细胞Ne=Nnc,Ve=Vnc和Pnc=1,而且可以通过上述方法直接测量或间接测量及计算Ne和Ve。对于外植块或微外植块,可以通过上述方法直接测量或间接测量及计算Ne和Ve。还可以从外植块或微外植块中细胞的平均尺寸简单计算外植块或微外植块表面处细胞的近似平均体积Vnc,其中所述外植块或微外植块是那些能够制造新细胞—细胞连接的。
外植块和微外植块的Pnc,即能够制造新接触的细胞表面积的平均比例,是外植块或微外植块表面处细胞形状和它们可用于形成新细胞—细胞接触的表面积比例的函数,所述新接触是可实际用于制造新接触的。可以通过显微法确定这个数值。基于对按照本发明所产生的培养物的观察,估算Pnc通常是0.5,即能够制造新接触的细胞表面积的大约一半,所述新接 触是可实际用于制造新接触的。
可以从Vnc和外植块和微外植块的平均尺寸估算外植块和微外植块的Nnc,即平均体积的外植块和微外植块表面处细胞的数量。例如假设外植块或微外植块以及组成它们的细胞近似立方体,且测量和计算平均体积的外植块或微外植块具有长度Lx1,宽度Lx2和厚度Lx3的尺寸,以及测量和计算了平均体积的成分细胞具有长度Ly1,宽度Ly2和厚度Ly3的尺寸,则估算的外植块或微外植块表面处细胞的平均数量是估算的外植块或微外植块表面积除去作为估算的细胞表层中各个细胞平均接触表面积的一部分的细胞表层。对于该近似法,从外植块或微外植块尺寸中减去表层上细胞的厚度,从而导出除去表层的估算的表面积。然后可以按照以下公式计算Nnc,即平均体积外植块或微外植块表面处的细胞数量:
Nnc=((2×((Lx1-2Ly3)×(Lx2-2Ly3)))+(2×((Lx1-2Ly3)×(Lx3-Ly3)))+
(2×((Lx2-2Ly3)×(Lx3-Ly3))))/((Ly1×Ly2)
公式3
对于本领域中的技术人员,显然还可以通过使用多种测量设备和应用适合的公式或计算机算法式计算不同尺寸和形状的细胞的体积和不同尺寸和形状的培养物的体积。
按照我们的测量法,器官型培养的压缩度可以在约6%和100%之间。按照本发明制造的含有10,000个具有平均细胞体积520μm3的解离细胞的功能器官型培养可以占大至9×107μm3的空间。这等于5.8%的压缩。按照本发明制造的含有20,000个具有平均细胞体积14,000μm3的解离细胞的功能器官型培养可以占小至28.1×107μm3的空间。这等于99.6%的压缩。
可以通过任意已知方法压缩细胞,优选地为解离细胞,微外植块或外植块,从而获得优选的紧密压缩度。所施加的压力足够在不引起细胞损伤的情况下导致细胞紧密接触,从而达到所需要的紧密压缩水平。优选地,施加于解离细胞或外植块或微外植块内细胞的压力小于2×10-3达因/细胞,从而避免损伤细胞,更优选地,在10-5达因/细胞和5×10-4达因/细胞之间。
优选地,通过重力、流体动力或流体静力压缩细胞。优选地,通过离心所施加的重力场压缩细胞。还可以通过抽吸压缩细胞。还可以使用多于一种压缩机制的组合。例如,可以通过离心及随后通过抽吸压缩细胞。
当通过离心压缩细胞时,混悬液中的细胞,不论该细胞处于解离细胞、微外植块或外植块的形式,均优选地通过100-5000g的离心进行压缩,其施加了在10-5达因/细胞至5×10-4达因/细胞的优选范围内的力。
当通过抽吸压缩细胞时,可将混悬液中的细胞置于表面的一侧上,且施加于该表面相对侧的吸力导致细胞逼近所述表面。放置细胞的表面必须适合于容许施加于该表面一侧的吸力有效地压缩在该表面另一侧的细胞。可以使用泵施加流体动力,以推动流体流经该表面,且在该情形中,应该将所述泵置于该表面的与待压缩细胞相同的一侧。
可以在将细胞转移到在其上培养它们的表面上之前或之后,对它们进行压缩。按照本发明第一方面的第一实施方案,在将细胞转移到进行培养的表面上之前,压缩该细胞。由此提供了制备器官型培养物的方法:
i)压缩来自器官的细胞、外植块或微外植块,其中所述细胞优选地为解离细胞;
ii)将所述压缩的细胞转移到表面上;和
iii)在合适的条件下培养所述压缩的细胞,从而形成器官型培养物。
如上所述,可以通过离心、抽吸或任何其他技术人员已知的方法进行细胞的压缩。优选地,如上所述通过离心压缩细胞。这个方法区别于现存的在正常通路中收获胰蛋白酶化的细胞的方法,原因在于现存方法要求在对其进行涂布前,将离心的细胞再悬浮于培养基中,并由此解离。在本发明第一方面的这个第一实施方案中,将由离心细胞所产生的沉淀在离心后转移到用于培养的表面。按照本发明第一方面的第二实施方案,通过离心压缩细胞和将所述细胞转移到所述表面是同时进行的。在这个第二实施方案中,有利的是提供保持细胞悬液处于正确方向的导管,所述正确方向是相对于通过重力场强度进行压缩的表面而言的。最佳方向是所述表面从最小重力场强度的位点远离细胞悬液。在离心机中产生重力场的情形中,将该表面置于与细胞悬液相比远离离心机转子旋转轴的位置。优选地,在导管和表面的连接处提供密封,密封位于表面和与表面相邻的导管边缘之间,从而确保将细胞有效地转移到表面,而不从导管中损失。
本发明第一方面的第一和第二实施方案的方法可以包括在将被压缩的细胞转移到所述表面后进一步对其进行压缩的步骤。可以通过上述任一 机制进行这个进一步的压缩步骤。例如,可以将被压缩了的细胞转移到表面的一侧,然后通过对该表面的另一侧施加抽吸力进行进一步的压缩。
优选地,在将细胞转移到表面后,通过由毛细作用产生的流体静力进一步压缩细胞。按照这个优选的实施方案,在其上培养细胞的表面优选地是多孔膜,且向该膜的对侧表面供给液体培养基,培养基与膜保持接触。由此液体培养基通过毛细作用流过所述膜中的孔,且由膜中的孔所施加的毛细作用表现出通过流体静力进一步挤压细胞。由此,保持在膜的对侧上的液体培养基所施加的毛细作用进一步压缩细胞。优选地,液体培养基通过毛细作用保持在所述膜的对侧。
在另一实施方案中,可以仅在将细胞转移到表面后,压缩该细胞。按照本发明第一方面的第二实施方案,提供生产器官型培养的方法,其包括:
i)将来自器官的细胞、外植块或微外植块转移到表面上,其中所述细胞优选地为解离细胞;
ii)在所述表面上压缩所述细胞;和
iii)在合适的条件下培养所述压缩的细胞,从而形成器官型培养物。
如上所述,可以通过离心或抽吸,或其他任何基质压缩细胞。按照这个实施方案的方法可以包括多个压缩步骤。例如,可以通过在表面上的离心和随后通过抽吸压缩被转移到该表面上的解离细胞。
在将压缩的细胞转移到其上的表面是多孔膜时,所述进一步的压缩步骤优选地受到毛细作用的影响。优选地,所述进一步的压缩步骤受到由保持在膜的与压缩的细胞相对侧上的液体施加的毛细作用的影响。优选地,所述液体是液体培养基,其含有用于细胞器官型生长所需要的营养。优选地,将压缩的细胞转移到多孔膜的一个表面,将液体培养基供给到该多孔膜的对侧表面,且该液体通过毛细作用保持在膜的对侧表面,这导致进一步压缩位于膜相反侧上的细胞,从而确保该细胞粘附于并被压平在该膜上。
按照本发明的优选方法,通过离心压缩来自器官的细胞,将其转移到多孔膜的一侧,并通过在所述多孔膜对侧供给的液体培养基所产生的毛细作用对其进行进一步的压缩。本文中描述了适合于将液体培养基保持在多 孔膜对侧,以通过毛细作用进一步压缩细胞的装置。按照本发明进一步优选的方法,通过离心直接将细胞压缩在多孔膜的表面上,并通过在多孔膜对侧供给的液体培养基所产生的毛细作用对其进行进一步的压缩。本文描述了在离心之前或过程中适合用于将细胞悬液保持在多孔膜一侧上,及用于将液体培养基保持在多孔膜对侧上的装置,该装置用于通过毛细作用进一步压缩细胞。
优选地,本发明的方法进一步包括从器官分离细胞的预备步骤。
本领域中已知用于从器官分离解离细胞的方法。可以通过机械或酶解离组织,或全部二者,从目标器官分离解离的细胞。例如,可以通过利用无钙和镁的Hank’s平衡盐溶液(HBSS)中0.25%(w/w)的蛋白水解酶胰蛋白酶解离器官,以获得解离的细胞。在加入胰蛋白酶抑制剂停止酶解离后,可以在混悬液中短暂培养该细胞,以容许未解离的细胞下沉到底部,从而将解离的细胞保留在混悬液中。
然后可以如上所述压缩这些解离的细胞。例如,可以将混悬液中的解离细胞吸入新的离心管中,并通过在200-1000g离心1-5分钟进行压缩。在抽吸上清液后,利用合适的工具,例如一次性吸移管管尖(pipette tip)从该管中移出含有压缩的解离细胞的细胞沉淀,并直接将其置于用于培养的表面,如多孔膜上。备选地,可以将混悬液中的解离细胞直接置于合适的表面上,并离心所述表面和细胞,从而压缩细胞。在这个实施方案中,有利的是提供保持细胞悬液处于正确位置和方向的导管,所述正确的位置和方向是相对用于通过离心进行压缩的表面而言的。将该表面置于与容纳细胞悬液的导管相比远离离心机转子旋转轴的位置。在优选的实施方案中,在导管的连接处提供了密封,其位于该表面和与表面连接的导管之间,从而确保将细胞有效地转移到所述表面,且不从导管中损失。
可以通过将目的器官机械地缩小为小的组织碎片,来获得用于本发明的方法中的微外植块和外植块。例如,通常可以通过在一次性吸移管管尖中重复抽吸出生后的组织或通过用手术刀片的离析作用获得微外植块。优选地,所述组织是新生组织。
本发明的方法可以用于制备来自广泛多种器官的器官型培养物,且用于此过程中的细胞的性质将依赖于需要的器官型培养物。优选地,从中获 得细胞的器官是动物器官,优选地是哺乳动物器官,优选地是人的器官。
可以从动物的任意器官获得细胞,所述器官包括但不仅限于中枢神经系统,骨髓,血液(例如,单核细胞),脾,胸腺心脏,乳腺,肝,胰腺,甲状腺,骨骼肌,肾,肺,肠,卵巢,膀胱,睾丸,子宫或结缔组织。优选地,解离细胞、外植块或微外植块来自于中枢神经系统,心脏,肝或肾。当解离细胞、外植块或微外植块来自于中枢神经系统时,它们可以来自脑或来自脊髓。优选地,所述细胞来自于脑,优选地来自于海马或皮质。当使用解离细胞时,它们还可以是干细胞。干细胞是多能性细胞,其可以受诱导进行分化,且一些干细胞能够分化为多细胞谱系。胚胎干细胞可以原则上分化为任意细胞类型。当使用胚胎干细胞时,它们不来自于人类胚胎干细胞。
可以从器官的特殊区域获得细胞。例如,当所述器官是脑时,可以从海马或从皮质获得细胞。如本文的实施例中所示范的,来自于皮质区的解离细胞可以用于制备器官型培养物,所述器官型培养表现出海马的典型细胞组成和细胞间连接。当所述器官是心脏时,可以从心肌获得细胞。如实施例中所示,按照本发明的方法培养的解离肌细胞形成器官型培养,其具有紧密的细胞间连接和许多心脏组织的生理特征,包括节奏协调的收缩,其类似于搏动心脏的节奏。
可以从多于一个器官获得细胞,并将它们在一起培养。例如,细胞可以来源于2,3,4或多个不同器官。获自多于一个器官的细胞的共培养容许产生来源于不同器官的组织的相互作用模型。优选地,当使用来自多于一个器官的细胞时,所述器官在体内应该是天然接触存在的,由此来自于这些器官的细胞共培养所引起的器官型培养物应该为体内情形提供模型。例如,免疫细胞,具体地,白细胞,可以与来自于多种器官的细胞共培养,从而研究炎症。肿瘤细胞也可以与来自于多种器官的细胞共培养,从而研究癌症发展。干细胞可以与其他细胞类型共培养,从而产生混和的培养物。骨骼肌细胞可以与来自于中枢神经系统,包括海马、皮质、小脑和脊髓的细胞共培养,从而产生神经-肌肉连接的模型。血管内衬的内皮细胞可以与脑细胞共培养,从而形成血-脑屏障模型。
当共同培养获自多种器官的细胞时,本发明的方法可以涉及压缩来自 于不同器官的细胞(无论它们是解离细胞、微外植块或外植块)的混合物,并培养该压缩的混合物。在特殊情形中,可能有利的是确保不同细胞类型随机分布于培养物中。在其他情形中,可能有利的是形成不同细胞类型层,由此每种细胞类型处于基本上分离的层中。当有利的是形成不同细胞类型层时,在一些进一步的情形中可能有利的是确保一层或多层一种或多种细胞类型处于多孔膜的一侧,且另外一层或多层一种或多种细胞类型处于多孔膜的对侧。适合于举例说明这个原则的一个实例是血脑屏障模型,其包括与中枢神经系统细胞层共同培养的血管内皮细胞层。在一些情形中,有利的是在多孔膜的一侧培养这两个细胞层,例如在多孔膜的与培养基对侧的一侧上。在这些情形中,可以有利的是将内皮细胞层培养在邻近多孔膜处,将中枢神经系统细胞层培养在邻近内皮细胞层。如果然后在中枢神经系统细胞层中检测到引入到培养基的分子,则可以推断出它们已经越过内皮细胞层,只要培养物的形式不允许所述分子通过直接越过多孔膜的运动绕过内皮细胞层到达中枢神经系统细胞层。相似的在多孔膜一侧上的内皮细胞层的培养和在多孔膜对侧上的中枢神经系统细胞层的培养应该确保引入与内皮细胞层相邻的培养基中的分子越过了内皮细胞层,从而到达中枢神经系统细胞层,只要培养物的形式不允许所述分子通过直接越过多孔膜的运动绕过内皮细胞层到达中枢神经系统细胞层。然而,在后一种情形中,内皮细胞和中枢神经系统细胞间的直接接触将受到多孔膜的阻止。为了确保培养基的形式不允许分子绕过一层到达远离多孔膜的另一层,本发明提供装置,其允许组装相同尺寸的层。这些装置包括在压缩,例如通过离心的压缩之前或过程中保持细胞悬液的导管。通过将不同细胞类型连续引入导管和连续压缩每种细胞类型层,将细胞类型组装到层中。可以将导管的形状设计为确保后来的细胞层具有不大于先前细胞层的直径。
备选地,可以在培养前分别压缩来自不同器官的细胞,并将其置于用于培养的表面上的不同位置,由此它们在生长过程中相互作用。例如,可以将来自一个器官的压缩细胞置于所述表面上,其与来自另一个器官的压缩细胞相距数毫米。在这种情形中,来自这两个器官的压缩细胞优选地被小于5mm的空间分开,更优选地,小于3mm。本发明的方法还可以涉及在表面上的不同位置共培养多于一组来自相同器官的压缩细胞,从而提供 这些细胞相互作用的模型。这些压缩的细胞可以来自于一个器官的不同区域,或可以来自于一个器官的相同区域。实施例1描述了来自于脑皮质区的两组压缩细胞的制备,将所述两组压缩细胞置于多孔膜的不同区域,且通过3mm分开,从而创建了当这两组细胞相互作用时的胼胝体模型。
在一些情形中,有利的是将不同的压缩细胞培养物置于多孔膜的不同区域,这是通过在离心后将沉淀转移该表面的不同区域进行的,其中所述沉淀是通过离心每种类型的解离细胞产生的。在其他情形中,有利的是同时通过离心压缩不同细胞类型,和将不同细胞类型的压缩细胞转移到所述表面上。在这些后面的情形中,有利的是提供单独的导管,其保持每种细胞悬液处于正确的方向,所述正确的方向是根据用于通过离心进行压缩的表面而言的。可以提供数个所述的导管,从而将每个单独的培养物置于表面的不同区域上。每个所述的单独培养物可以含有一种细胞类型或多种细胞类型。如果所述单独培养物含有多于一种细胞类型,则这些细胞类型可以随机地混合,或可以通过连续向导管引入不同细胞类型和连续压缩每种细胞类型将这些细胞组装到层中。
用于本发明第一方面的方法中的细胞可以来源于健康器官或来源于患病器官。更详细地描述如下,本发明第一方面的方法的快速简便制备器官型培养物的能力意味着该方法应该在用于研究疾病联系和药物筛选的器官型培养物的生产中具有广泛的应用。通过本发明的方法,从健康器官和患病器官获得器官型培养物的比较将促进目前疾病状态的知识,并容许识别指示疾病状态的生物标志和药靶。
可以基因改造用于本发明的方法的细胞。例如,可以基因改造所述细胞,从而调节药靶或生物标志的表达。生物标志是分子标志,其以一定水平或以一定分子形式的存在说明了患病状态的存在。药靶是分子种类,其可以受到调节从而影响疾病的过程,即药物通过其发挥作用的分子。改变药靶的性质或功能水平必须对疾病效果具有正面的影响,且该目标应该属于服从调节的分子类型。在许多情形中,从遗传或其他生物研究获得有关药靶的信息,且可以获得已知与那些目标相互作用的化合物种类。经常需要调节这些生物标志和药靶在生物系统中的水平和研究其生物学结果。
备选地,可以遗传性地改变细胞,从而表达容许在视觉上跟踪该细胞 的可视标志,如荧光标志。
本领域中已知表达克隆基因和消除克隆或内源基因的技术。这些技术可以用于增加或减少标志,如药靶或生物标志在本发明的方法中所使用的细胞中的表达。例如,在压缩细胞和制备器官型培养物前,可以在选择的解离细胞中调节药靶的表达。该方法比试图在最终器官型培养物中改变药靶更加有效得多,原因在于可以更容易地对单一解离细胞进行操作。
增加克隆或内源基因表达的技术基于以募集细胞表达系统的形式将外源DNA引入,并且许多不同的方法为本领域中的那些技术人员所熟知。在一些情形中,裸露DNA可以与亲脂性转染试剂一起使用,所述DNA包括与待表达基因共线性的强启动子和允许引入DNA的细胞质复制的复制起点。在其他情形中,病毒载体可以用于增加DNA的引入效率。相似地,本领域中那些技术人员所熟知的消除基因表达的方法包括反义DNA寡核苷酸,肽核酸和双链RNA干扰。在一些情形中,可以使用裸露的核酸。在其他情形中,特别是对于使用小干扰RNA,表达载体可以用于在自装配发夹形式中表达分子。还显示出可以直接将蛋白质引入细胞中,只要它们不附着于促使从细胞外向细胞内运输的本体。人类免疫缺陷病毒(HIV)的Tat蛋白是一个这样的本体,并且可以产生待转移的蛋白质作为与HIV-Tat的融合蛋白,并将其引入到细胞中(Becker-Hapak M.等,2001)。
还对本领域中的那些技术人员显然的是,代替如上所述的转化或转染细胞用于本发明的方法的细胞可以来自于转基因动物。例如,细胞可以来自于表达可视标志,如荧光标志的转基因动物,或来自于这样的转基因动物,所述动物中增加或减低了特殊药靶或生物标志的表达。
优选地,本发明的方法中的在其上培养压缩细胞的表面是光学透明的,从而容许使用具有物镜的显微镜方法,以观察来自该表面任意侧的细胞。当在其上培养压缩细胞的表面是多孔膜时,它优选地是亲水性聚四氟乙烯(PTFE,也作为DuPont商业名称Teflon已知)膜,所述聚四氟乙烯膜是光学透明的。为了在本发明的方法中使用的备选膜的实例可以由聚碳酸酯,PET(聚对苯二甲酸乙二醇酯),或AnoporeTM(无机氧化铝,WhatmanCorp的商标)制成。
在提供对器官型生长必要的营养培养基的存在中,培养压缩的细胞。 优选地,所述培养基是液体培养基。例如,在Stoppini L.等(1991)和Muller等(2001)中描述了合适的液体培养基的实例。
按照本发明第一方面进一步的实施方案,本发明的方法可以进一步包括低温贮藏由此引起的器官型培养物的步骤。低温贮藏容许培养物的积累和储存,从而用于筛选的目的。典型地,低温贮藏是通过在液氮温度冷冻完成的。
与需要由特殊材料组成的人工基质的现有技术方法不同,本发明的方法不需要任何特殊材料,并因此可以大规模进行,从而容许了多重器官型培养物的平行制备。因此本发明提供了用于制备大量器官型培养物的高通量方法,所述方法包括按照本发明第一方面的方法平行制备多重器官型培养物。优选地,利用大量产生的预制培养装置和机器人分配进行本发明的高通量方法。
优选地,在这样的装置中进行本发明第一方面的方法,所述装置容许多重平行培养/装置,优选地,2,4,8,16,24,96,384,1536或更多平行培养/装置。
按照本发明第二方面,提供了用于进行本发明第一方面的方法的装置。优选地,所述装置足够小到容许通过平行的培养装置,同时制备和维持数千个培养物,优选地数万个器官型培养物。可将每个培养物维持在单独的表面上或大表面的单独区域上,并分别供给营养。优选地,所述培养装置为了测量器官型培养物中所产生的电生理反应,进一步引入电极。在欧洲专利EP1133691中描述了合适的装置。
如上所述,当本发明第一方面的方法涉及在多孔膜上培养压缩的细胞时,该方法优选地包括对膜的对侧表面供给营养,优选地以液体培养基的形式。优选地,通过毛细作用将所述液体培养基保持在膜的表面处,从而导致进一步压缩膜上的细胞。本发明还因此提供一种装置,其适合于进行本发明第一方面的这个优选的方法。按照本发明第二方面优选的实施方案,因此提供了进行本发明第一方面方法的装置,所述装置包括:
导管,其具有一个开口末端和一个由与其横跨熔合的多孔膜封闭的末端;和
框架,其保持所述导管处于基本垂直的方向;
其中所述导管适合于通过毛细作用容许将充足体积的液体培养基保留在所述导管中,从而与所述多孔膜表面接触,并由此向可以在所述多孔膜上生长的细胞供给营养。
用于维持导管中培养基的毛细作用力应用允许通过处于竖立或倒置方向的吸移步骤移除或替换培养基。当供给培养基时,应该使吸移管管尖位于可实施的最靠近膜表面的位置。
优选地,所述导管适合于这样:当所述装置处于竖立或倒置位置时,其通过毛细作用保持充足体积的液体培养基,从而维持所述导管内的多孔膜表面和培养基之间的接触。
通过竖立位置意指框架保持导管基本上垂直,其由多孔膜密封的末端位于最上方,由此当使用该装置时,器官型培养物在膜的上表面上生长。通过倒置位置意指框架保持导管基本上垂直,其开口末端位于最上方且由多孔膜封闭的末端位于最下方,由此当使用该装置时,器官型培养物位于膜的下表面中。与本领域中存在的装置相反,本发明的装置由此容许孵育器官型培养物并用处于竖立或倒置位置的装置改变用于器官型培养的培养基。这种在培养和装置方向中的灵活性意味着具有面向上的物镜镜头的显微镜或具有面向下的物镜镜头的显微镜均可互换地用于研究该培养,并且液体输送装置可以用于任何方向,以添加或去除培养基。
优选地,所述导管是圆柱体。所述导管还可以是矩形或不对称横截面的。以这样的方式选择所述导管的精确尺寸和组成,使得在器官型培养过程中,其通过毛细作用保持充足体积的液体培养基,以维持导管内多孔膜表面和培养基之间的接触,优选地无论所述装置是处于竖立和倒置的位置。保持的液体体积是充足的,从而在使用中,在无需不合理地常常更换培养基的条件下,向器官型培养供给足量的营养。
毛细作用依赖于若干参数。毛细作用力是圆柱体管直径和矩形截面导管宽度(width)或宽度(breadth)的反函数。对水性溶液的毛细作用力还依赖于受该力控制的溶液的表面张力,该力可以通过在溶液中存在表面活性剂如清洁剂来减弱。毛细作用受到液体分子和表面分子之间吸引力强度的影响。在水性液体的情形中,毛细作用受到导管表面亲水性程度的影响。影响液体培养基在导管中保留的另一个因素是培养基的体积。因此需要考 虑这些因素,从而确保本发明的装置可以通过毛细作用保持一定体积的液体培养基与多孔膜表面接触。
在所述装置中,两种不同的毛细作用力发生作用,从而保持导管中的液体培养基与多孔膜接触。一种力是通过液体培养基和管之间的吸引力所施加的毛细作用力。另一种力是通过液体培养基和膜的孔壁之间的吸引力所施加的。如果其足够强,则前者将会抵消重力,从而在不考虑竖立或倒置的前提下,保持导管中的液体,并且后者将会保持该液体与膜接触。在某个阈值,培养基上的重力将会超过毛细作用力,且不受其他力控制的培养基将会从导管中落下。
当导管是圆柱体时,保持在该圆柱体中的液体的质量和由此从该圆柱体中移除所述液体所作用的重力与该圆柱体半径的平方成正比,而保持该圆柱体中液体所作用的毛细作用力与半径成反比。因此,对于给定的液体和圆柱体长度,存在着最大半径,如果超过该最大半径,将不能对抗重力而保持圆柱体中的液体,其中所述圆柱体的表面组成是给定的,但是不存在比其低时将不对抗重力而保持液体的最小半径。
优选地,所述导管是圆柱体,其具有0.5cm或更小的半径,优选地,0.3cm,0.25cm,0.2cm,0.15cm或更小。优选地,所述圆柱体具有约0.3cm,0.15cm或0.075cm的半径。已经发现具有0.5cm或更小半径的圆柱形导管适合于维持1cm柱的标准液体培养基,如Dulbecco′s极限必需培养基与导管中多孔膜表面的接触,这无需考虑该装置是处于竖立或倒置的位置。
优选地,所述优选为圆柱体的导管,长度约为1cm,从而容许其保持1cm的液体柱。优选地,所述导管在长度上稍微大于1cm,优选地,长度约为1.1cm或1.2cm。
优选地,所述导管由亲水性材料制成,优选地是亲水性聚合物,从而当液体培养基处于所述导管中时,增加对其施加的毛细作用力。亲水性聚合物已为本领域技术人员所已知。可以进一步增强制造导管的聚合物的亲水性,例如,通过包含聚乙二醇基。
本发明第二方面的装置不受具有小于0.5cm的半径的圆柱体的限制,原因在于在技术人员能力范围内,可以很好地确定可以用于所述装置的其他导管的尺寸。特别地,技术人员应该能够计算施加于不同尺寸导管中给 定体积的液体培养基上的毛细作用力和重力,并从而确定哪种导管尺寸应该用于所述装置,以确保毛细作用力超过重力,从而将该液体保持在导管中。此外,导管中可以包括缩颈(constriction)、平台或其它障碍,从而增加对从导管中移除培养基所作用的重力的对抗。
按照拉普拉斯-开尔文(Laplace-kelvin)方程,
毛细作用力=表面张力/(R1-R2),
其中R1=以cm为单位的管或孔的半径(在该情形中是导管),且R2=与管或孔壁相接触的弯液层的厚度。
1达因是在1cm秒-2时加速1克所需要的力。除非包括表面活性剂如清洁剂,水性培养基的表面张力是约73达因cm-2。将清洁剂引入培养基中不是惯例操作,但是蛋白质也可以影响表面张力,且在培养基中,特别是血清形式的培养基中通常包括蛋白质。通常,液体培养基的表面张力是至少50达因cm-2。施加于给定体积液体培养基上的总重力是98×(以cm3 为单位的体积)达因。当为了本发明的目的计算毛细作用力时,通常无需考虑弯液层的厚度(R2)。当R2很小时,它对毛细作用力的作用可以忽略,且随着R2接近R1,毛细作用力变得更大。由于为了本发明的目的,对于给定的导管和水性培养基,仅需要确定是否达到了最小毛细作用力的要求,所以R2的测量是不必要的。然而,如果需要更精确地计算毛细作用力,测量R2当然是可能的。
对于长1cm和半径0.5cm的圆柱体,应该由此要求至少为77达因的总毛细作用力,从而在倒置时,通过毛细作用对抗重力并维持导管中表面张力为50达因cm-2的1cm的液体柱。如果该圆柱体表面的亲水性足够高,则所述毛细作用力可以对该液体柱施加大于100达因的力。
对于长1cm和半径0.3cm的圆柱体,应该因此要求至少为28达因的总毛细作用力,从而在倒置时,对抗重力和维持导管中表面张力为50达因cm-2的1cm的液体柱。如果该圆柱体表面的亲水性足够高,则所述毛细作用力可以对该液体柱施加大于170达因的力。
只要不移动或振动装置,当倒置时,这些毛细作用力足够保持所述液体柱,其原因在于由移动或振动所引起的加速改变所述液体柱的动量,并可以克服控制力。优选地,导管的尺寸是这样的,即合理的动量改变,如 可以由标准手工或机器人操作所导致的动量改变,不引起从导管中损失液体。
优选地,以这样的方式选择导管的尺寸,即为在多孔膜表面保持给定体积的液体培养基的所作用的毛细作用力是为释放该培养基的所作用的重力的至少6倍。已经发现是所述重力6倍的毛细作用力足以确保在标准操作下,液体培养基保留在装置导管中,甚至是当该培养基含有减小该培养基表面张力的蛋白质成分,如血清中的那些蛋白质时。
优选地,所述多孔膜通过胶粘,通过热封或通过超声焊接与导管的一个末端横跨熔合。按照拉普拉斯-开尔文方程(见上),多孔膜向导管中的液体施加毛细作用力,其依赖于该膜中的孔的半径和表面成分。由该膜施加的毛细作用力应该足以浸湿该膜并保持液体与膜的接触。优选地,本发明的装置中的多孔膜包括具有尺寸~0.4μm的孔。适合用于本发明装置的膜包括但不限于由Millipore Corporation生产的光学透明的亲水性聚四氟乙烯(PTEE,也已知为DuPont商标Teflon)膜,由聚碳酸酯,PET(聚对苯二甲酸乙二醇酯),或AnoporeTM(无机氧化铝,Whatman Corp的商标)制成的膜。
优选地,所述多孔膜是光学透明的。这个特征使该测试培养能够在所有时刻接受显微镜检验和取样,以进行生化测定。优选地,所述多孔膜在用于激发的波长处,通常在400-750nm的范围内产生低背景荧光。优选地,所述多孔膜由亲水性聚四氟乙烯(PTEE)膜组成。
优选地,所述框架保持导管处于垂直的方向,这样由膜封闭的导管末端或导管的开口末端均不与任何表面接触。优选地,所述装置进一步包括密封圈,其确保将所述框架坚固地保持为与导管接触。优选地,所述装置包括两个密封圈。所述装置可以进一步包括另外的工具,以确保坚固地保持框架与导管接触,因此当倒置时不释放该导管。所述另外的工具可以包括,例如,摩擦工具,如框架和导管之间的弹簧。
优选地,所述装置进一步包括腔室,其围绕导管的开口末端。所述腔室可以形成保持导管处于垂直方向的框架的一部分。当使用所述装置时,该腔室容纳具有合适气体组成的空气,其与导管中的培养基接触,从而维持该培养基中的最佳酸度和氧气水平。优选地,该腔室是密封的,以确保 在使用过程中液体培养基不暴露于外界空气。所述腔室可以进一步包括气体入口和气体出口,以容许控制腔室内的空气条件。
优选地,所述装置进一步包括一个或多个导管,优选地为可除去的导管,以容纳被重力场压缩前或过程中的细胞,所述导管或可除去的导管位于膜的与装有培养基的导管对侧上。在压缩细胞之前或过程中使用多于一个导管来容纳细胞允许在所述膜区域内不同位点上建立多于一个的培养,所述膜与每个培养基导管接触。如果容纳细胞的导管是不能除去的,则它们可以通过胶粘或或通过热封或通过超声焊接与多孔膜永久地密封。如果容纳细胞的导管是能除去的,则它们与多孔膜非永久地密封。非永久密封的实例包括集中压力的定型边缘和硅氧烷以及其他可压缩物质,所述硅氧烷以及其他可压缩物质可以组成导管的全部或导管的密封垫边缘。那些熟悉离心法的人应该理解重力场对包含靠近多孔膜的导管的装置的应用会将密封垫边缘压向多孔膜,其中所述靠近多孔膜的导管是相对离心转子的旋转轴而言的。在可除去的导管的情形中,这提高所述密封边缘的密封特性。在设计用于重力场中的装置的领域中的那些技术人员应该理解承重表面必须通过足够强度的结构来支持,从而避免损坏。例如,本发明的多孔膜即使在弱重力场中,仍不具有用于在不损坏该多孔膜的条件下,支持导管的足够强度。因此,已经采取了一些步骤,其用于确保本发明的导管在所述装置的框架中受到足够强度的结构的支持。如果在导管和多孔膜之间使用可压缩的密封垫,则在所有情形中重力场影响下的压缩程度受到在所述装置框架中导管和足够强度结构间的接触的限制。这些考虑对于设计用于重力场中的装置领域中的那些技术人员是显而易见的。
优选地,所述密封的腔室进一步包括容许改变培养基的开口。优选地,将所述开口设计为在改变培养基时,最小化培养基向空气的暴露。所述开口可以通过隔膜或阀密封,所述隔膜或阀一般是密封的,但可以被吸移管管尖穿透,从而取出培养基并引入新的培养基。所述隔膜可以由橡胶或氯丁橡胶制成。所述开口可以用于将特殊成分,如生长因子或抗生素或毒素,引入到现存培养基中,而不是完全地改变培养基。优选地,所述吸移步骤是在不使所述培养在流体静压方面显著改变的条件下进行的。
应该对手工和机器人吸移构造领域中的技术人员显而易见的是,为了 从导管中取出液体,必须施加比将液体保持在导管中的压力更大的负压。避免吸移管管尖对多孔膜的破坏应该是很重要的,且由于这个原因,所述吸移管管尖不应该前移到与所述膜接触。因此,可能不能通过单一的吸移步骤从导管移除全部的液体培养基。液体可能被保留在导管中,其保留在吸移管管尖到达的最远端和所述膜之间的导管区域内。所述液体通过毛细作用力在导管中的滞留很可能与非常小的圆柱体半径有直接关系,尽管其还依赖于吸移管管尖和液体的精确特性。如果确实发生液体滞留,则在大多数情形中,它不会影响该培养物的健康。
然而,在一些情形中,例如如果测试该培养物暴露于毒素物质,液体的滞留可以潜在地影响试验数据。在这个情形中,液体的移除和用新鲜液体的替换的吸移步骤可以重复必要多次,从而通过稀释去除毒素物质。例如,如果所述圆柱体是1cm长,且吸移管管尖可以安全地前移到距所述膜0.1cm的范围内,则最多可在该圆柱体中保留体积的10%。向该全部1cm的长度加入新鲜液体会将毒素稀释到其初始浓度的10%。重复该过程的会将毒素稀释到其初始浓度的1%。吸移步骤的时间规划应该考虑对容许毒素平衡,从而最大化由稀释引起的去除功效的需要。
优选地,所述装置进一步包括盖子,其覆盖导管外的多孔膜表面。该盖子覆盖这样的多孔膜表面,即当使用所述装置时培养物位于所述多孔膜表面上。如果导管用于在压缩前或过程中容纳细胞且该导管是永久固定的,则所述盖子覆盖该导管。如果导管用于在压缩前或过程中容纳细胞且该导管不是永久固定的,则可以在固定盖子前去除所述导管。当所述装置包括盖子时,所述腔室和框架优选地包括额外的端口,以容许气体在腔室和盖子所围绕的膜上方的空间之间流动,这容许了在超过几周或更长的时期内控制培养物周围的空气。
本发明第二方面的装置优选地适合用于高通量方法中,该方法涉及同时制备和维持多个器官型培养物。因此按照本发明这个方面的第二实施方案,提供了用于高通量器官型培养的装置,其包括多个按照本发明第一方面的装置。优选地,用于高通量器官型培养的装置包括96,384,1536或更多个按照本发明第二方面的装置。
高通量装置可以由此含有上千个导管,可以独立地用培养基供给其中 的每个导管,且为此可以独立地改变培养基。优选地,通过上述多通道吸移管或机器人进行培养基的改变。
优选地,高通量装置包括单一的盖子,其覆盖所述装置中所有的独立导管。
优选地,在高通量装置中的包含每个导管开口末端的腔室通过开口连接,这容许气体在腔室间流动,由此可以通过在高通量装置中单一的气流入口和出口控制流到所述装置中所有腔室中的气体。
可以将高生产量装置中的多个装置装配为单一单位。备选地,可以提供所述高生产量装置作为独立装置,每个所述独立装置含有单一的导管,所述导管可以由使用者装配到含有所需数量导管的高生产量装置中。还可以提供所述高生产量装置作为单独装置的条带,例如,在2,4,8,或12组中,任选地,可以由使用者将其装配到含有所需数量的导管的高生产量装置中。尽管其不用于器官型培养,包含条带的高生产量装置在细胞培养领域中时众所周知的,其中所述条带含有固定数量的孔。Dynatech在ThomeA.(1979)的美国专利4,154,795中描述了这种类型的多孔装置。
优选地,对于高生产量装置,该装置的总尺寸和该装置中单独导管的位置应该与标准微滴定板匹配,从而允许将为标准微滴定板设计的机器人技术使用于该装置。例如,在包括96个装置的高生产量装置中,优选地,将所述装置安排为8×12个装置的列阵,其类似于标准96孔微滴定板。优选地,在所述由96个装置组成的高生产量装置中的导管是圆柱体。优选地,每个圆柱体具有约0.3cm的半径,即标准96孔微滴定板中的孔的半径。已经在上文描述了作用于这样的圆柱体的毛细作用和重力。
在包括384个装置的高生产量装置中,每个装置中的导管优选地是圆柱体,且所述圆柱体的半径优选地是约0.15cm,即标准384孔微滴定板的孔半径。同样1cm长的该圆柱体中的液体的重量仅仅是具有圆柱体直径0.3cm的相应重量的25%,但是与上述较大的圆柱体相比,其毛细作用力加倍。在包括1536个装置的高生产量装置中,每个装置中的导管优选地是圆柱体,且所述圆柱体的半径优选地是约0.075cm,即标准1536孔微滴定板的孔半径。在这种情形中,同样1cm长的该圆柱体中的液体的重量仅仅是具有圆柱体直径0.3cm的相应重量的6.25%,但是其毛细作用力高4 倍。因此,按照本发明标准微滴定板总尺寸制成的96,384或1536个圆柱体的装置都将液体保持在处于倒置位置的圆柱体中,且都可以包含永久和可除去的细胞悬液导管。
按照本发明的第三方面,提供了通过本发明第一方面的方法获得的器官型培养物或大量器官型培养物。尽管通过本发明第一方面的方法产生的来自压缩细胞的器官型培养物保持与器官型器官切片培养物相同的功能特征,它们在其解剖结构方面与器官型器官切片培养物不同。
器官型器官切片培养物保持来源器官的解剖特性,只要那些特性发生在被切下用于创建该切片的器官区域内。例如,由脑的海马区域制备的横截切片保持表征海马的典型细胞排列,例如CA1,CA2和CA3区域的空间排列。按照本发明第一方面的方法制备的器官型培养物与来自切片的器官型培养物不同,原因在于通过器官内细胞的解离或通过将器官切割为多个外植块或微外植块丧失了其解剖特性。
令人震惊地,尽管丧失该解剖特性,按照本发明第一方面的方法制备的器官型培养物具有器官型特性,即它们保持器官的功能特征。例如,在图1中可见按照本发明第一方面的方法制备的来自于压缩的解离细胞的培养物没有解剖上的分化,其中所述解离细胞来自于大脑皮质。然而,培养过程中在该培养中创建的细胞间连接发生在通常在体内形成连接的细胞之间。这些连接的准确性和适当性的原因似乎是每个细胞产生其应该在体内产生的化学信号,以及其他细胞对那些信号适当的应答。
对所述信号的应答应该依赖于细胞的正常功能。在一些情形中,对该信号应答的细胞可以用延长细胞突起,如神经元的轴突突起进行应答。在其他情形中,应答的细胞可以用细胞突起进行应答,其中所述细胞突起导致穿过该培养物的,朝向或背离产生信号的单个细胞或多个细胞的细胞移动。备选地,应答的细胞可以用细胞分裂进行应答,或者可以抑制它们否则可能经历的细胞分裂。应答的细胞还可以通过产生其他信号进行应答,对该信号应答的细胞不直接应答第一个细胞产生的信号。在该方法中,在培养过程中可以在该培养物中发生多种细胞数量、功能和分布的改变,且这些改变导致该培养物的器官型特性。
如对有技术经验的读者显然易见地,存在大量对本发明第三方面的器 官型培养物的应用,所述器官型培养物是通过本发明第一方法的方法获得的。例如,该器官型培养物可以用于研究给定健康器官中的细胞功能和组成,以及细胞间通讯的效果。这些功能和效果通常在疾病状态中改变,且该器官型培养物可以用于研究患病组织和正常组织,以及外源因子,包括优选药物对这些疾病状态的影响。
如上所述,生物标志是分子标志,其在某种水平或在某种分子形式中指示患病状态的存在。药靶是可以受到调节而影响疾病过程的分子种类。本发明器官型培养物的一种应用是在生物标志和药靶的识别中。
可以使用表达疾病状态或相应的非疾病状态的器官型培养中若干分子分类,如蛋白质和脂质的筛选识别生物标志。目前使用确认的生物标志识别疾病状态的载体和监视它们向正常发展,该发展可以受到治疗方案如药物的辅助。有必要在候选生物标志和疾病状态间建立统计学上显著的联系,从而为了临床试验中的应用确认生物标志。本发明的器官型培养由于以下事实理想地适合于生物标志的发现和确认,所述事实是它们复制器官功能和生理机能,以及可以通过本发明的方法快速简便地产生,因此它们适合于高通量检验。由此本发明的器官型培养可以比目前用于生物标志识别和确认的完整动物更加迅速和便宜地进行使用。
按照本发明进一步的方面,由此提供了用于识别和确认生物标志和药靶的方法,其包括筛选本发明第三方面的器官型培养。用于识别生物标志和药靶的检验包括使用转录概况分析,蛋白质组学,质谱测定法,凝胶电泳,气相色谱分析和本领域技术人员已知的其他用于分子概况分析的方法。
替代标志是可以用于评估疾病状态存在和发展的生物标志的子集,但是不直接测量疾病的临床效果。本发明的器官型培养物可以以与其他生物标志相同的方法用于识别和确认替代标志。
本发明的器官型培养物不仅在识别与疾病状态有关的生物标志和药靶中有效,而且还在筛选候选药物中有效,所述药物缓解这些疾病状态。器官型培养物特别有效用于筛选候选药物,原因在于对于该筛选来说很重要的是目标培养物具有这样的生化和生理性质,所述性质尽可能接近地匹配其目标器官的那些体内特征。然而,必然可能的是以高通量使用的器官 型培养物允许为了成功识别先导药物的高概率筛选充分大量药物候选物。另外大规模检验对在临床前和临床药物开发程序中确认先导药物的内含物经常是必要的。
本发明第一方面的方法可以用于同时产生数千个器官型培养物,且由此对于每个培养物独特地适合于涉及多重检验的高通量应用。在一个实施方案中,用于按照本发明的第一方面制备器官型培养物的方法进一步包括在筛选和临床前确认候选药物的方法中利用由此产生的器官型培养进行筛选的步骤。如上所述,本发明方法的一个特别有效的方面是其促进器官型培养物的高通量形成,其中已经基因改造了细胞,从而调节生物标志或药靶的表达。这些改造的器官型培养物还应该在筛选候选药物中有效。
毒物学领域是本发明的进一步的应用领域,其应该很大地受益于由本发明的方法提供的增强的灵活性和通量。器官型应答在该领域中是至关重要的,原因在于不同组织在它们对毒素的应答中非常不同,其具有不同的临床后果。不同组织可以含有不同的酶系统,特别地细胞色素P450种类,其代谢不同种类的外源化合物。化合物代谢的程度和类型可以深深地影响其毒性。在广泛多种组织中的大规模毒性筛选在目前是非常昂贵的,以至于许多常用的化学药品未经过充分的测试。增加潜在毒性意识已经将压力引到以能接受的成本在不用进行该测试的方法的情况下进行该测试。因此本发明还包括利用本发明的器官型培养物评估化学药品毒性的方法。
现在将通过实施例更加详细地描述本发明的多个方面和实施方案。应该理解可以在不背离本发明范围的情况下进行细节的修改。
附图简述
图1:按照本发明的方法由解离小鼠皮质细胞制成的典型器官型培养物的显微照片。
图2:在小鼠皮质的器官型培养物中5周后的功能性神经元和神经胶质细胞。
A. 用抗-ChAT染色的胆碱能神经元。
B. 用抗-GFAP检测到的星形胶质细胞。
C. 用抗-β微管蛋白III检测到的神经元。
图3:按照本发明的方法制备的器官型培养物为了进行电生理测量覆盖在多电极装置上。
图4:自发性场电位在图3的装置上的8对电极间改变,其中在所述装置上覆盖了器官型培养物,所述培养物是按照本发明的方法由解离小鼠皮质细胞制成的。
图5:成对脉冲刺激诱导来自器官型培养物的诱发场电位(EEP),其中所述器官型培养物是按照本发明的方法由解离的小鼠皮质细胞制成的。
图6:共培养两个器官型培养物的示意图,其中所述器官型培养物是按照本发明的方法由来自小鼠皮质(表示中枢神经系统(CNS))的解离细胞制成的,其中所述小鼠皮质安排来模仿胼胝体。
图7:器官型培养10天后,两个THY-1 GFP小鼠皮质培养物间的轴突生长。
A.在亮视野(左侧)和荧光(右侧)图像中,两个培养物和水凝胶桥的低放大率视图。
B.在所述水凝胶中轴突的高放大率视图。
图8:用于整合于多孔膜中的电生理测量的多电极装置。
图9:由冷冻和解冻的解离细胞制成的培养物的电生理应答,所述解离细胞来自于小鼠皮质。
A.对脉冲诱发场电位。
B.自发活动。
图10:在复制小鼠皮质培养物中的蛋白质浓度。
图11:来自于图8的装置上的8对电极的自发场电位,所述装置位于器官型培养物下方,所述器官型培养物按照本发明由解离小鼠心脏肌肉细胞制成。
图12:小鼠胚胎肝脏的器官型培养。
A.亮视野
B.用抗-平滑肌α肌动蛋白染色
C.用Hoechst(细胞核)染色
图13.人胎儿胰腺的器官型培养:
A.亮视野
B.用抗-胰岛素抗体染色
C.用抗-SOX9抗体染色
D.用DAPI(细胞核)染色
图14:在用表达EGFP的lentiviral载体转导后24小时的解离P0小鼠脑细胞的器官型培养。
图15:用于器官型培养的装置的横截面。
图16:在装置中制备器官型培养物的过程的示意性描述,所述装置包括用于在压缩之前和过程中容纳细胞的可除去型导管。
图17:用于器官型培养的装置的横截面,所述装置包括在压缩之前和过程中容纳细胞的导管。
图18:用于器官型培养的高通量装置的横截面。
图19:高通量装置和用于高通量装置的可除去型导管,所述高通量装置包括4个在压缩之前和过程中容纳细胞的导管/孔。
图20:用于器官型培养的改良装置的横截面,其具有固定导管处于合适位置的额外部件。
实施例
实施例1:来自于小鼠胚胎皮质(E-18)的解离细胞的器官型培养
从小鼠胚胎摘除胚胎的脑,并分离两个半球。摘除了小脑以及丘脑和基底神经节后,小心地摘除软膜,并用Hepes缓冲的盐溶液(HBSS)中0.1%的胰蛋白酶消化来自于5个脑的皮质区。在重力下容许细胞团块沉降,并且用吸移管轻轻地抽吸混悬液中覆盖在上面的解离细胞,并置于新的离心管中。
通过在1000×g旋转10分钟来压缩解离的细胞,并直接用无菌的吸移管管尖移除2-5μl的由此产生的细胞沉淀,并置于Millipore-CM装置的Biopore亲水性PTFE膜的中心上。
将皮质培养基(10%Ham′s F12(Sigma),8%FBS,2%马血清,10mMHepes(Gibco),2mM L-谷氨酰胺(Gibco),50单位Pen/strep在DMEM(+葡萄糖)中制成近似于1x的培养基)加入到该膜的下侧。还可以使用其他含有血清的培养基。该培养物通过毛细作用进一步压缩达到液体水平的平 衡。
在图1中显示了按照本发明从解离的小鼠皮质细胞制成的典型的培养物。该培养物不表现出总体的解剖学特性,所述特性是切片培养物的典型特性,但是其具有均匀的表面。在3-5天内,这些培养物表现出器官型特性,且保持至少5周。例如,可以通过用标记的抗体染色来证明适合的细胞类型的存在,其中所述标记的抗体识别细胞类型特异性抗原。在图2中显示了用3种所述抗体染色典型培养物,所述典型培养物是按照本发明从解离小鼠皮质细胞制成的。图2A显示了用由Chemicon(目录编号AB144P)提供的山羊抗胆碱乙酰转移酶染色的活性胆碱能神经元,其以1∶100的稀释度使用。用Cy3-缀合的亲和力-纯化的驴抗-山羊IgG抗体(杰克逊免疫研究实验室公司(Jackson ImmunoResearch Laboratories,Inc))检测该山羊抗体。图2B显示了用来自Sigma(目录编号G9269)的兔抗-胶质细胞原纤维酸性蛋白(GFAP)抗体检测星形胶质细胞,以1∶200的稀释度使用该抗体。用Alexa FluorTM568标记的山羊抗兔IgG抗体(分子探针(MolecularProbes),Invitrogen)检测兔抗体。图2C显示了表达神经元特异性β微管蛋白III的神经元,其通过以1∶200的稀释度使用的Covance单克隆抗β微管蛋白III抗体(目录编号MMS-435P)进行检测。用Alexa FluorTM488标记的山羊抗小鼠IgG抗体(分子探针(Molecular Probes),Invitrogen)检测小鼠的单克隆抗体。这些图像证明甚至在培养5周后,仍存在健康功能性神经元,且神经元和星形胶质细胞的标记均表现出适当的星形胶质细胞—神经元连接。如果该培养物不是器官型的,则几天后神经胶质细胞将控制该培养。按照本发明利用解离细胞制成的培养物表现出神经元间的连接,所述连接对器官型脑培养物是典型的,但是当通过其他程序培养解离细胞时,没有发现该连接。
通过培养物的电生理活动显示了器官型活性的进一步证据。图3中所示,器官型培养物被多电极阵列(M.E.A.)覆盖,所述多电极阵列是常与器官型切片培养物共同使用的电生理装置。本发明人在Ep1133691和美国专利6,689,594中描述了整合在所述膜中的合适的M.E.A.。该装置不干扰细胞的压缩。
可以记录来自M.E.A.不同电极的自发活动和诱发场电位。在图4中显 示了在上述按照本发明产生的皮质培养物中的自发电生理活动。也可利用M.E.A.测量该培养物对任意对电极间所施加电位差的影响。图5中显示了在上述按照本发明产生的皮质培养物中所激活的电生理活动。这些诱发的电位是脑器官型培养物所典型的,并且指出按照本方面发明制备的培养物令人震惊地具有器官型培养物的典型特征。该解离细胞或大量外植块或微外植块能够进行高通量培养,而器官型切片培养物不适于高通量的应用。
在记录的进一步的试验中,将两个如上述由来自小鼠皮质的解离细胞制备的相同的培养物置于相同的Biopore亲水性PTFE膜上,以3mm宽的空间分开。图6中示意性地显示了该培养物的安排。图7中显示了两个这样相邻的培养物的实例,其具有位于所述两个培养物之间的水凝胶斑(BDTM PuraMatrixTM肽水凝胶,BD生物科学(BD Biosciences)目录编号354250)。也可以在PuraMatrixTM水凝胶的位置使用BD生物科学MatrigelTM 或琼脂糖。在这个情形中,可以使用来自THY-1 GFP转基因小鼠(Feng G.等,2000)的皮质,原因在于来自这些小鼠的神经元表达绿色荧光蛋白且可以直接成像。图7显示按照本发明利用来自小鼠皮质的解离细胞制备的培养物可以超过该培养物的边缘生长轴突,其朝向位于相同多孔膜上的第二个培养物。轴突突起向两个方向延伸,其原因在于趋化性信号的扩散,并且在培养物间形成新的神经元连接。图8中举例说明的M.E.A的设计用于论证神经元连接的完整性。此外,已经证明了神经元连接的完整性在国际空中旅行之前和之后出现在相同的组织中,这说明了本发明方法的稳定性和有效性。
通过图9所示的数据提供了对本发明方法的有效性和灵活性进一步的证明。通过本发明的方法可以在器官型培养之前利用完全建立的程序冷冻和储存解离细胞。图9显示了按照本发明从冷冻和解冻的解离细胞制成的培养物的电生理应答,其中所述解离细胞来自小鼠皮质。图9A中所示的成对脉冲诱发场电位和图9B中所示的自发活动对其他器官型脑培养物和器官型脑切片培养物是典型的,其中所述其他器官型脑培养物是按照本发明制成的。
通过图10中所示的不同培养物间蛋白质浓度的微小变化例证了本发明培养方法的高再现性。通过BCA检验(Smith P.等.1985)测量蛋白质浓 度。
我们还证明了小鼠神经元干细胞的生长和分化,所述干细胞处于按照本发明与来自小鼠皮质的解离细胞的共培养中。其他共培养实例包括当按照本发明器官型地与解离脑细胞共培养时,巨噬细胞向小胶质细胞的分化。
实施例2:来自大鼠心脏的解离细胞的器官型培养
如Ren等(1998)所描述的方法分离解离的新生大鼠心室肌细胞。简要地,对动物进行安乐死,并迅速地摘除和用氧化的Krebs-Henseleit重碳酸盐(KHB)缓冲剂灌注它们的心脏。随后用名义上的无Ca2+KHB缓冲剂灌注心脏2-3分钟,直至自发性收缩停止,随后用含有0.5%w/v的II型胶原酶(Invitrogen目录号17101)和0.1mg/mL透明质酸酶(Sigma-Aldrich目录号H 4272)的无Ca2+KHB缓冲剂灌注20分钟。灌注后,摘除、切碎、并用新鲜酶溶液(含有0.5%w/v的II型胶原酶的无Ca2+KHB)温育左心室3-5分钟。在经尼龙筛孔(300μm)过滤前,用0.2%胰蛋白酶进一步消化该细胞。在重力下容许细胞团块沉降,并用吸移管轻轻地抽吸混悬液中覆盖在上面的解离细胞,并将其置于新的离心管中。
通过在1000×g旋转10分钟来压缩解离的细胞,并直接用无菌的吸移管管尖移除2-5μl由此产生的细胞沉淀,并将其置于Millipore-CM培养装置的Biopore亲水性PTFE膜的中央。
将增补了Gibco(Invitrogen目录号17504-044)的神经基部培养基(Invitrogen目录号21103-049)加入到该膜的下侧。通过利用毛细作用的液体水平平衡进一步压缩培养物。
由此产生的培养物不表现出全部对切片培养物典型的解剖特性,但是具有均匀的表面。10天内,这些培养物表现出器官型特性。图11显示了自发间歇性电生理活动,其于心脏搏动相当。显示出的搏动率为每2.6秒1次搏动,这是在缺乏神经支配条件下的心肌细胞培养中他人获得的典型搏动活动。然而,在本发明的情形中,与以前用于解离细胞培养的方法不同,所述培养的器官型特性稳定数周或数月,而不是对解离细胞的其他培养物典型的3-5天。通过本发明获得的长期器官型培养物的稳定性容许 具有转基因和siRNA稳定表达的试验及在筛选试验中对长时期的药物应答的测量,而其他解离细胞培养物仅稳定几天,且当稳定的转基因表达最佳时不再是器官型。
实施例3:来自人心脏的解离细胞的器官型培养
如Mummery C.等(2002)所述,从组织分离解离的人胎儿心肌细胞,所述组织是由南安普敦大学(University of Southampton)在许可下提供的。简要地,在经尼龙筛孔(300μm)过滤前,用0.2%的胰蛋白酶浸软和消化胎儿心房或心室。容许细胞团块在重力下沉降,并用吸移管轻轻地抽吸在混悬液中覆盖在上面的解离细胞,并将其置于新的离心管中。通过在1000xg旋转5分钟来压缩解离的细胞,并直接用无菌的吸移管管尖移除5-10μl由此产生的细胞沉淀,并将其置于Millipore-CM培养装置的Biopore亲水性PTFE膜的中央。将增补了Gibco(Invitrogen目录号17504-044)的神经基部培养基(Invitrogen目录号21103-049)加入到该膜的下侧。通过利用毛细作用的液体水平平衡进一步压缩培养物。
如实施例2中,在10天内建立搏动的培养物。在心房培养物的情形中,搏动率是140-150搏动/分钟。心室培养物搏动较慢,大约30搏动/分钟。这些实施例证明了本发明的方法不特异于啮齿动物器官,且其他实施例举例说明了其对一定范围组织的应用。
实施例4:来自小鼠胚胎肝脏的解离细胞的器官型培养(E-16-18)
从小鼠胚胎摘除胚胎肝脏,并将其置于冰冷的磷酸缓冲盐溶液中。然后用手术刀将肝脏切成小块,再将其转移到加热到37℃的Earle′s平衡盐溶液(EBSS)中。然后用3个火焰抛光的具有减小尺寸孔径的Pasteur吸移管研磨肝脏块,从而产生细胞悬浮液。容许细胞团块在重力下沉降,并用吸移管轻轻地抽吸在混悬液中覆盖在上面的解离细胞,并将其置于新的离心管中。通过在1500xg离心3分钟来压缩解离的细胞。再将剩余沉淀重新悬浮到浓度约为20,000细胞/μl,并直接用无菌的吸移管管尖移除5-10μl由此产生的细胞沉淀,并将其置于Millipore-CM培养装置的Biopore亲水性PTFE膜的中央。将肝脏培养基(Verrill C.等(2002))加入到该膜的 下侧。通过利用毛细作用的液体水平平衡进一步压缩培养物。体外维持该肝脏培养物至少10天。
图12显示用单克隆抗平滑肌α肌动蛋白(SMA)抗体克隆1A4纯化小鼠免疫球蛋白对按照本发明制成的小鼠胚胎肝脏培养物的染色。用AlexaFluorTM488标记的山羊抗小鼠IgG抗体(分子探针(Molecular Probes),Invitrogen)检测结合的抗体。SMA存在于活化的肝星形细胞中,并且为活性的肝脏再生提供标志。通过本发明的方法产生的肝培养物具有器官型的表现。这为利用肝细胞的高通量药物开发检验提供了非常重要的优势。由本发明提供的从解离肝细胞创建器官型培养物的能力允许容易地开发高通量检验,且具有适当的细胞间连接的器官型培养物与具有非器官型培养的解离细胞的检验相比,是对药物或毒素的体内应答的更加可靠的指示剂,其中所述从解离肝细胞创建器官型培养物可以大量地获得。由于肝脏在药物代谢和毒性中至关重要的作用,器官型肝脏培养物是重要的商业机会。
实施例5:来自人胰腺的解离细胞的器官型培养
如Turnpenny L.等(2003)所述,从人胎儿胰腺分离解离的细胞。简要地,在经尼龙筛孔(300μm)过滤前,用0.2%的胰蛋白酶消化胎儿胰腺。容许细胞团块在重力下沉降,并用吸移管轻轻地抽吸在混悬液中覆盖在上面的解离细胞,并将其置于新的离心管中。通过在1000xg离心5分钟来压缩解离的细胞,并直接用无菌的吸移管管尖移除5-10μl由此产生的细胞沉淀,并将其置于Millipore-CM培养装置的Biopore亲水性PTFE膜的中央。将增补了Gibco(Invitrogen目录号17504-044)的神经基部培养基(Invitrogen目录号21103-049)加入到该膜的下侧。通过利用毛细作用的液体水平平衡进一步压缩培养物。
图13表明本发明的方法从解离的胰腺细胞产生了器官型培养物,其保持胰腺体内的重要特性。在培养中11天后,用抗胰岛素抗体的染色(图13B)显示出胰腺的胰岛素表达特征。胰腺的发育伴随着SOX9同源域转录因子的表达(Piper K.等2002)。如图13C所示,通过抗SOX9抗体(来自Chemicon的多克隆兔抗SOX9抗体,通过生物素标记的(生物素化的) 抗兔及随后的链酶抗生物素得克萨斯红检测,二者均来自Vector Labs)揭示了SOX9在按照本发明制成的器官型胰腺培养物中的表达。图13D显示用DAPI染色的细胞核。
实施例6:来自小鼠脑的转染的解离细胞的器官型培养
本发明方法的关键特性是在器官型培养物产生前,将用于过表达的基因或用于消除表达的siRNA引入到解离细胞中的能力。转基因或siRNA的引入在解离细胞中比在器官型切片培养物中更加有效,原因在于每个解离细胞的全部表面都可接近用于引入的亲脂性复合物或病毒载体。图14显示从来自P0小鼠脑的解离细胞产生的器官型培养物。在即将培养前,用用于增强绿色荧光蛋白(EGFP)的基因转导该细胞。
摘除新生脑,并分离两个半球。摘除了小脑以及丘脑和基底神经节后,小心地摘除软膜,并用Hepes缓冲的盐溶液(HBSS)中0.2%的胰蛋白酶消化来自于脑的皮质区。在重力下容许细胞团块沉降,并且用吸移管轻轻地抽吸混悬液中覆盖在上面的解离细胞,并置于新的离心管中。通过在1000xg旋转5分钟来压缩解离的细胞,并直接用无菌的吸移管管尖移除5μl的由此产生的细胞沉淀。将2μl浓度为1.811物质粒子/ml的lentiviral构建体rHIV1-cPPT-SYN1-GFP-WPRE加入到每5μl离心的制备物中,其中所述lentiviral构建体rHIV1-cPPT-SYN1-GFP-WPRE由疱疹性口腔炎病毒血清型G(VSVG)包膜所包封,随后将其置于Millipore-CM培养装置的Biopore亲水性PTFE膜的中央。将皮质培养基(10%Ham′s F12(Sigma),8%FBS,2%马血清,10mM Hepes(Gibco),2mM L-谷氨酰胺(Gibco),50单位Pen/strep在DMEM(+葡萄糖)中制成近似于1x的培养基)加入到该膜的下侧。通过利用毛细作用的液体水平平衡进一步压缩培养物。7天后,获取数字图像。
实施例7:器官型培养的装置
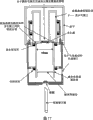
如图15中所示,本发明优选的装置包括圆柱体和多孔膜,以及框架,其中所述多孔膜胶粘或热封或超声焊接于所述圆柱体的一个末端,所述框架保持所述圆柱体垂直并创建围绕所述圆柱体开口末端的腔室。
所述圆柱体含有一定体积的液体培养基,其通过毛细作用保持在该圆柱体中,由此与多孔膜下表面接触。显示了所述一定体积液体培养基的液体弯液层。
该装置进一步包括盖子,其封闭所述导管的膜密封末端,并提供对器官型培养物周围的空气的控制,其中可能进行气体交换,但防止微生物污染。
如图16和17中所示,本发明进一步优选的装置包括图15装置的改良,因此另外的圆柱体密封于多孔膜,该圆柱体位于所述多孔膜的与图15中所示的圆柱体的对侧。如图16中本发明方法的这个方面的示意描述所示,另外的圆柱体用作压缩前或压缩过程中容纳细胞悬液的导管。在例如通过离心或抽吸压缩之后,可以移除或在适当位置保留容纳细胞悬液的圆柱体。如将其保留在适当的位置,则在培养过程中用盖子覆盖它,从而提供对器官型培养物周围的空气的控制。离心后,使用不用于容纳细胞悬液的圆柱体来容纳一定体积的液体培养基,所述液体培养基通过毛细作用保持。用于容纳细胞悬液的圆柱体可以具有比用于容纳液体培养基的圆柱体更小的内部横截面,从而确保该培养的全部面积都可以被穿过多孔膜的培养基接近。将用于容纳细胞悬液的圆柱体设计为确保圆柱体受到足够的支持,且圆柱体在重力场的影响下不损伤多孔膜。例如,圆柱体外部横截面和其内部横截面的尺寸差异足够在离心过程中通过所述装置的框架完全支持该圆柱体重量。
在用于高通量的装置集合的情形中,盖子优选地应该覆盖整个组件。盖子可以是松散匹配的,从而容许其他扩散,可以具有与所述组件的边缘重叠的边缘,从而在微小的空气涡流的存在下最小化污染。应该最小化空气涡流是组织培养的实践者所熟知的。备选地,盖子可以紧密匹配,在该情形中,可以在框架中提供端口,由此气体可以在腔室和盖子与膜间的空间之间循环。
所述腔室优选地包括预制的塑料型材,其将两个定位腔室的密封圈坚固地合并在所述圆柱体底部周围。优选地,密封垫是由氯丁橡胶制造的密封圈,其在所述圆柱体底部周围形成气密的密封。
所述腔室的底部包括隔膜,其可以被吸移管穿透,从而在不将培养基 暴露于周围环境的条件下帮助腔室内培养基的交换。
所述腔室的内部优选地对气体扩散和灌注是易接近的,且腔室包括两个用于其他流动的孔或端口,从而控制空气条件。所述腔室和框架优选地包括另外的端口,以容许在腔室和膜上方由盖子覆盖的空间之间流动的气体流动。
如图18和19所示,按照本发明用于多重器官型培养的优选的高通量装置包括多个如图15和17中所示的用于单一器官型培养的装置。高通量装置中每个圆柱体含有其各自的液体培养基供应,液体培养基通过毛细作用保持与膜接触。然而,位于圆柱体底部的腔室是连通的,且腔室间的气体流动是可能的,由此容许通过位于装置各个末端的气体流动端口控制所有腔室中的空气条件。此外,通过框架中的端口将盖子和培养物之间的空间与腔室连接起来,由此容许控制膜上培养物周围的空气条件。
如图19所示,单一插入物附件可以包括多个用于高通量装置的可除去的导管,从而在压缩之前或过程中容纳细胞悬液。对于每个用于容纳液体培养基的固定导管可以存在一个或多个这样的可除去的导管。在所示的实施例中,对每个固定导管存在4个这样的导管。
如图20所示,图15和17的装置可以进一步包括元件,其保持所述导管相对于所述框架来说处于适当的位置,诸如例如在导管和框架之间的弹簧。
参考文献
Becker-Hapak M.等,2001,方法,第24卷,第247-56页,TAT介导的蛋白质向哺乳动物细胞中的转导(Methods,vol 24 pp247-56 TAT-mediatedprotein transduction into mammalian cells).
Buchs P.等,1993,脑研究发展脑研究,第71卷,第81-91页,大鼠海马趾器官型培养的区域CA1中与突触发育有关的结构修饰(Brain Res.Dev.Brain.Res.Vol 71 pp81-91,Structural modifications associated with synapticdevelopment in area CAl of rat hippocampal organotypic cultures)Corradino R.,1973,J.细胞生物学,第58卷,第64-78页,器官培养中的胚胎鸡肠。用于研究肠内钙吸收机制的独特系统(Cell Biol.Vol 58,pp64-78, Embryonic chick intestine in organ culture.A unique system for the study ofthe intestinal calcium absorptive mechanism).
Feng G.等,2000,神经元,第28卷,第41-51页,成像表达GFP的多频变体的转基因小鼠中的神经元子集(Neuron Vol 28,pp41-51,Imaging neuronalsubsets in transgenic mice expressing multiple spectral variants of GFP).Giehl,2002,EP1205541,用于长期培养脑和其他出生后,特别是成年哺乳动物组织的器官型切片的程序(Procedure for the long-term cultivation oforganotypic slices of brain and other postnatal tissues,in particular of adultmammals).
Herlyn M.,2004,美国专利申请20040175367,器官型肠培养及其应用方法(Organotypic intestinal culture and methods of use thereof).
Honegger,P.,和Monnet-Tschudi,F.,2001,聚集神经细胞培养,第199-218页,在神经细胞培养方案(Fedoroff,S.,和Richardson,A.,eds)中,第三版,(Aggregating Neural Cell Cultures,ppl99-218 in Protocols for Neural CellCulture(Fedoroff,S.,and Richardson,A.,eds)Third Edition)Humana Press,Totowa,NJ,USA,ISBN 1-59259-207-4).
Kalabis J.等,(2003),FASEB杂志,第17卷,第1115-7页,通过白血病抑制因子对人结肠上皮细胞的刺激依赖于器官型培养中胶原蛋白包埋的成纤维细胞(FASEB J.vol 17,ppl115-7,Stimulation of human colonicepithelial cells by leukemia inhibitory factor is dependent oncollagen-embedded fibroblasts in organotypic culture).
Michalopoulos G.等,2001,病理学Am杂志,第159卷,第1877-87页,肝细胞类器官培养中的组织构造(Am J Pathol,vol 159 ppl877-87,Histological organization in hepatocyte organoid cultures)Michalopoulos G.和Bowen W.,2004,美国专利申请20040151729,新型长期三维培养系统(Novel long-term three-dimensional culture system).Muller等(2001)神经细胞培养方案,第三版,第13-27页(Protocols forNeural Cell Culture,3rd Ed.Pp13-27),S.Fedoroff和A.Richardson eds,Humana Press,Inc.,Totowa,NJ.器官型海马趾切片的界面培养(Interface器官型Hippocampal Slice Cultures).
Mummery C.等(2002),解剖杂志,第1200卷,第233-42页,小鼠和人胚胎干细胞的心肌细胞分化(J Anat.vol200 pp233-42,Cardiomyocytedifferentiation of mouse and human embryonic stem cells).Piper K(2002),,Mech.Dev.,第116卷,第223-6页,人胰腺发育过程中的新型SOX9的表达与Campomelic发育异常中的异常性有关(Vol 116pp223-6.Novel SOX9 expression during human pancreas developmentcorrelates to abnormalities in Campomelic dysplasia).Ren等(1998)生理学Am杂志,第275卷,第H823-H830页,对糖尿病大鼠心脏中的胰岛素状生长因子I改变的的变力应答:细胞内Ca2+和氧化氮的影响(Am J Physiol,vol 275 ppH823-H830 Altered inotropic response toinsulin-like growth factor I in diabetic rat heart:influence of intracellular Ca2+ and nitric oxide)
Rochkind S.等,2002,神经性研究,第24卷,第355-60页,在生物可降解的微载体上培养的脊髓神经细胞的移植及随后的低能量激光照射,其用于治疗大鼠外伤瘫痪(Neurol Res.vol 24 pp355-60.Transplantation ofembryonal spinal cord nerve cells cultured on biodegradable microcarriersfollowed by low power laser irradiation for the treatment of traumaticparaplegia in rats).
Shahar A.等,2001,欧洲专利申请EP1073420,为再造受损中枢神经系统特别设计的神经元植入(Specially devised neuronal implants forreconstruction of damaged central nervous system).
Smith P.等,1985,分析生物化学,第150卷,第76-85页,利用bicinchoninic酸测量蛋白质(Anal.Biochem.Vol 150 pp76-85,Measurement of proteinusing bicinchoninic acid).
Stoppini L.等,1991,神经科学方法,第37卷,第173-82页,用于神经组织器官型培养的简单方法(Neurosci.Methods,Vol 37 pp173-82,A simplemethod for organotypic cultures of nervous tissue).
Stoppini L等,1993,神经科学,第57卷,第985-94页,在海马趾器官型培养中由病灶诱导的轴突的萌发和突触的形成(Neuroscience,Vol 57,pp985-94,Lesion-induced neurite sprouting and synapse formation in hippocampal organotypic cultures).
Turnpenny L等,2003,干细胞,第21卷,第598-609页,人胚胎生殖细胞的来源:多能性干细胞的可选择性来源(Stem Cells,Vol 21 pp598-609.
Derivation of human embryonic germ cells:an alternative source of pluripotentstem cells).
Verrill C等,2002,药物毒物学方法杂志,第48卷,第103-10页,利用新生大鼠或成年人组织在流动空气界面中的器官型肝脏培养—体外纤维化的模型(J Pharmacol Toxicol Methods,Vol 48 ppl03-10.器官型liver culturein a fluid-air interface using slices of neonatal rat and adult human tissue-amodel of fibrosis in vitro).
Wicks W.,1968,J.生物化学,第243卷,第900-6页,胎儿大鼠肝脏中酪氨酸α酮戊二酸酯转氨酶的诱导(Biol.Chem.,Vol 243,pp900-6,Inductionof tyrosine-alpha-ketoglutarate transaminase in fetal rat liver).Wildenthal K.,1971,J.应用生理学,第30卷,第53-7页器官培养中自发搏动的小鼠心脏的长期维持(Appl.Physiol.,Vol 30,ppl 53-7,Long-termmaintenance of spontaneously beaing mouse hearts in organ culture).Wille J.,1998,美国专利5,834,312,用于人上皮细胞生长的程序和培养基(Process and media for the growth of human epithelia).