BRPI0710195B1 - Formulação de medicamento de liberação retardada para dispensação de um medicamento ao cólon, uso de uma formulação, e, métodos para preparar uma formulação de medicamento de liberação retardada - Google Patents
Formulação de medicamento de liberação retardada para dispensação de um medicamento ao cólon, uso de uma formulação, e, métodos para preparar uma formulação de medicamento de liberação retardada Download PDFInfo
- Publication number
- BRPI0710195B1 BRPI0710195B1 BRPI0710195-3A BRPI0710195A BRPI0710195B1 BR PI0710195 B1 BRPI0710195 B1 BR PI0710195B1 BR PI0710195 A BRPI0710195 A BR PI0710195A BR PI0710195 B1 BRPI0710195 B1 BR PI0710195B1
- Authority
- BR
- Brazil
- Prior art keywords
- drug
- formulation
- formulation according
- coating
- fact
- Prior art date
Links
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5036—Polysaccharides, e.g. gums, alginate; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
- A61K31/606—Salicylic acid; Derivatives thereof having amino groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/167—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction with an outer layer or coating comprising drug; with chemically bound drugs or non-active substances on their surface
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5036—Polysaccharides, e.g. gums, alginate; Cyclodextrin
- A61K9/5042—Cellulose; Cellulose derivatives, e.g. phthalate or acetate succinate esters of hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2121/00—Preparations for use in therapy
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
<b>formulação de medicamento de liberação retardada uso de uma formulação, e, metodos para tratar uma doença, para prevenir carcinoma, para preparar uma formulação de medicamento de liberação retardada, e para alvejar um medicamento ao colon.<d> um revestimento de liberação retardada compreendendo uma mistura de um primeiro material selecionado de amido; amilose; amilopectina; quitosano; sulfato de condroitina; ciclodextrina; dextrano; pululano; carragenano; escleroglucano; quitina; curdulano e levano, e um segundo material que tem um limiar de ph em tomo do ph 5 ou acima, é usado para alvejar a liberação de um medicamento de um núcleo ao intestino, particularmente o cólon.
Description
[0001] A presente invenção diz respeito a uma formulação de liberação retardada com um núcleo compreendendo um medicamento e um revestimento de liberação retardada. Em particular, ela diz respeito a uma formulação de liberação retardada para um medicamento para liberar ao cólon.
[0002] O alvejamento de medicamentos ao cólon pode ser utilizado como um meio de obter terapia local ou tratamento sistêmico. O cólon é suscetível a vários estados de doença, incluindo doença inflamatória intestinal, síndrome do intestino irritável, constipação, diarréia, infecção e carcinoma. Em tais condições, o alvejamento do medicamento ao cólon maximizaria a eficácia terapêutica do tratamento. O cólon também pode ser utilizado como um portal para a entrada de medicamentos na circulação sistêmica. Várias formulações foram desenvolvidas para a liberação colônica de medicamento, incluindo pró medicamentos assim como formas de dosagem formuladas, com as últimas sendo mais populares visto que o conceito uma vez provado pode ser aplicado a outros medicamentos.
[0003] A população bacteriana mais alta no cólon também foi explorada no desenvolvimento de formas de dosagem de liberação colônica de medicamento através do uso, como materiais carregadores, de polissacarídeos complexos que ocorrem naturalmente que constituem substratos para as numerosas enzimas das bactérias colônicas residentes. Estes materiais são capazes de passar através das regiões gastrintestinais superiores intactas, mas são digeridos na entrada no cólon. Aqueles estudados até agora incluem amilose amorfa, pectina, quitosano e galactomanana.
[0004] A amilose amorfa é resistente à digestão pelas enzimas do trato gastrintestinal superior. Ela é, entretanto, fermentada no cólon por enzimas a-amilase produzidas por mais da metade das 400 espécies de bactérias residentes no cólon.
[0005] Uma principal atração de usar polissacarídeos neste acesso de enzima bacteriana para a liberação colônica de medicamento é que materiais usados são de grau alimentício e assim seriam seguros para o uso em seres humanos. Eles são usualmente aplicados como revestimentos ou incorporados no material do núcleo como um carregador matriz, e sua digestão na entrada no cólon pelas enzimas bacterianas colônicas leva à liberação da carga de medicamento. Um exemplo de uma tal formulação, que utiliza um revestimento de amilose, é divulgado na EP-A-0343993 (BTG International Limited).
[0006] Uma maior limitação com estes materiais que ocorrem naturalmente, entretanto, é que eles intumescem excessivamente em meios aquosos levando a lixiviação da carga de medicamento nas regiões gastrintestinais superiores. Para evitar este problema, eles foram utilizados em uma mistura com materiais impermeáveis (por exemplo, amilose amorfa misturada com o polímero insolúvel em água etilcelulose). Entretanto, o problema com tais modificações/misturas está em encontrar o equilíbrio correto entre hidrofobicidade e hidrofilicidade que impediriam a liberação inoportuna de medicamento nas regiões gastrintestinais superiores, mas que também permitiriam ao mesmo tempo o acesso da enzima ao substrato de polissacarídeo e garantiriam a liberação do medicamento em uma taxa adequada no cólon.
[0007] Uma tentativa para resolver o problema do intumescimento excessivo da amilose é divulgado na EP-A-0502032 (British Technology Group Ltd). Esta utiliza um revestimento externo compreendendo um material polimérico de celulose ou acrilato formador de película e amilose amorfa para um tablete compreendendo um composto ativo. Uma forma de realização tem o composto ativo revestido primeiro com um revestimento interno de amilose e depois um revestimento externo separado do material polimérico de celulose ou acrilato. Uma outra forma de realização tem um revestimento externo que é uma mistura de amilose e um polímero de celulose ou acrilato. A referência torna claro que a degradação dos materiais de celulose in vivo é, em geral, não dependente do pH e é preferido que isto também seja verdade para os materiais de acrilato. Cada exemplo divulgado na referência é de um polímero celulósico ou de acrilato independente do pH.
[0008] Um artigo no Journal of Controlled Release (Milojevic et al; 38; (1996); 75-84) relata os resultados de investigações com respeito à incorporação de uma faixa de polímeros insolúveis em um revestimento de amilose de modo a controlar o intumescimento de amilose. Uma faixa de copolímeros com base em celulose e acrilato é avaliada, e um etil celulose comercialmente disponível (Ethocel®) é descoberto controlar o intumescimento o mais efetivamente. Um outro revestimento de camada única que é investigado é uma mistura de amilose e dois polímeros acrílicos independentes de pH, isto é, Eudragit® RS e RL, mas este revestimento é descoberto não fornecer tais resultados eficazes. Um revestimento solúvel dependente do pH de Eudragit® LI 00 é utilizado, mas apenas em um sistema de camada múltipla compreendendo um bioativo revestido com um revestimento interno de amilose e depois um revestimento externo de Eudragit® LI 00.
[0009] Uma outra composição de revestimento com base em amilose é divulgada na WO-A-99/21536 (BTG International Limited). A composição de revestimento compreende uma mistura de amilose e um polímero formador de película insolúvel em água que é formado a partir de um material polimérico celulósico ou de acrilato insolúvel em água. Como com a EP-A- 0502032, está claro que a degradação dos materiais de celulose in vivo é, em geral, não dependente do pH e é preferido que isto também seja verdade para os materiais de acrilato. Deve parecer que a especificação da PCT contenha um erro tipográfico, porque é desnecessário dizer que uma forma preferida do material de acrilato é o “Eudragit L cuja degradação é independente e pH”. É acreditado que esta deve se referir ao “Eudragit® RL” cuja degradação é de fato independente do pH. Esta não pode ser intencionada a se referir ao Eudragit® L, ao passo que a degradação deste polímero é dependente do pH.
[00010] A WO-A-99/25325 (BTG International Limited) também divulga um revestimento de liberação retardada que compreende amilose e (preferivelmente) etil celulose ou alternativamente um polímero de acrilato cuja degradação é independente do pH. A composição de revestimento também inclui um plastificador e o método encontra uma aplicação particular na preparação das formas de dosagem as quais compreendem os materiais ativos que são instáveis em temperaturas em excesso de 60° C, ao passo que a composição é formada em temperaturas mais baixas do que isto. Deve ser notado que esta referência também inclui o erro tipográfico relativo ao Eudragit® L descrito acima.
[00011] Os inventores observam que as formulações divulgadas nas referências da BTG divulgadas acima usam nos revestimentos amilose ao invés de amido e que a liberação das formulações é prolongada ao longo de uma porção do intestino.
[00012] A WO-A-03/068196 (Alizyme Therapeutics Ltd) divulga um revestimento de liberação retardada específico para a metassulfobenzoato de sódio de prednisolona bioativo que compreende amilose vítrea, etil celulose e sebacato de dibutila.
[00013] O uso dos polissacarídeos outros que não amilose amorfa em um revestimento de liberação retardada é divulgado na GB-A-2367002 (British Sugar PLC). Os exemplos incluem goma guar, goma karaya, goma tragacanto e goma xantana. As micropartículas destes polissacarídeos são dispersas em uma polímero matriz formadora de película insolúvel em água formada, por exemplo, a partir de um derivado de celulose, um polímero acrílico ou um lignina.
[00014] A WO-A-Ol/76562 (Tampereen Patenttitoimisto Oy) divulga uma formulação farmacêutica perorai que contem um medicamento e um quitosano (um polissacarídeo obtido a partir de quitina) para controlar sua liberação. O medicamento e o quitosano são misturados em uma mistura de pó mecânica homogênea que é granulada e depois opcionalmente tabletizada. A granulação pode ser realizada com um polímero entérico (tal como um copolímero do ácido metacrílico) ou os grânulos podem ser fornecidos com um revestimento entérico poroso.
[00015] A WO-A-2004/052339 (Salvona LLC) divulga um sistema de liberação do medicamento dependente do pH que é um pó de fluxo livre das nano-esferas hidrofóbicas sólidas que compreende um medicamento encapsulado em uma micro-esfera sensível ao pH. As nano-esferas são formadas a partir do medicamento em combinação com um material ceroso, e a micro-esfera sensível ao pH formada a partir de um polímero sensível ao pH (tal como um polímero Eudragit®) em combinação com um material sensível a água tal como um polissacarídeo. O presente requerente acredita, entretanto que os tamanhos de partícula muito pequenos envolvidos nesta referência não atrasariam em prática a liberação do núcleo bioativo, além do estômago ou duodeno.
[00016] Um artigo no European Journal of Pharmaceutical Sciences (Akhgari et al; 28 de março de 2006; 307-314) relata os resultados das investigações no uso de certos polímeros de polimetacrilato para, entre outras coisas, controlar o intumescimento da inulina. Os polímeros de polimetacrilato testados foram Eudragit® RS; Eudragit® RL; misturas de 1 : 1 de Eudragit® RS e Eudragit® RL; Eudragit® FS; e misturas de 1 : 1 de Eudragit® RS e Eudragit® S. Os resultados indicaram que as composições poliméricas que compreendem os polimetacrilatos de liberação sustentada (Eudragit® RS e Eudragit® RL; polímeros não sensíveis ao pH) com perfis de intumescimento apresentados com inulina indicando uma adequabilidade para o uso como revestimentos para a liberação colônica. Entretanto, outros resultados indicaram que as composições poliméricas que compreendem inulina com Eudragit® FS ou misturas 1 : 1 de Eudragit® RS e Eudragit® S (polímeros dependente do pH) não serão adequadas para tal uso devido aos perfis de intumescimento indesejáveis.
[00017] A US-A-5422121 (Rohm GmbH) divulga uma forma de dosagem oral que contem pelo menos um ingrediente ativo incluso dentro de um material de revestimento que compreende um polissacarídeo que decompõe no cólon. Os material de revestimento contêm um polímero formador de película em uma mistura com o polissacarídeo. A razão em peso do polissacarídeo para o polímero formador de película é de 1 : 2 a 5 : 1, preferivelmente de 1 : 1 a 4 : 1. Os exemplos de polissacarídeos adequados incluem aqueles polissacarídeos que são decomponível pelas enzimas glicosídicas. Os polissacarídeos que contêm quantidades consideráveis, preferivelmente cerca de 20% em peso a 100% em peso, de galactose e as unidades de manose são particularmente adequadas com goma de alfarroba e goma guar sendo preferidas. Os polímeros formadores de película preferidos incluem os polímeros de acrilato que são independentes do pH (insolúvel por todo o trato GI) e dependente do pH (insolúvel no fluido estomacal mas solúvel no fluido intestinal no pH 5,5 ou acima). A referência exemplifica o uso de uma mistura de goma guar com Eudragit RL 30 D (em um razão de 4:1), Eudragit® L 30 D (em um razão de 3 : 1) ou Eudragit® S 100 (em um razão de 2,5 : 1) como um revestimento de tablete.
[00018] Um artigo no European Journal of Pharmaceutical Sciences (Krogars et al; 17; (2002); 23-30) divulga o uso de Hylon® VII (um amido de milho rico em amilose (-70% em peso); National Starch, Alemanha) como um revestimento de película para tabletes que contêm um medicamento (teofilina). A dissolução dos tabletes em um meio ácido foi rápida com mais do que 75% do medicamento sendo dissolvido dentro de 15 minutos. O revestimento não conteve um segundo polímero formador de película.
[00019] De acordo com um primeiro aspecto da presente invenção, é fornecido uma formulação de medicamento de liberação retardada que compreende uma partícula com um núcleo e um revestimento para o núcleo, o núcleo compreendendo um medicamento e o revestimento compreendendo uma mistura de um primeiro material que é suscetível ao ataque por bactérias colônicas e um segundo material que tem um limiar de solubilidade em torno do pH 5 ou acima, em que o primeiro material compreende um polissacarídeo selecionado do grupo que consiste de amido; amilose; amilopectina; quitosano; sulfato de condroitina; ciclodextrina; dextrano; pululano; carragenano; escleroglucano; quitina; curdulano e levano.
[00020] O primeiro material compreende um polissacarídeo, preferivelmente contendo um grande número de unidades de glicose. Preferivelmente o polissacarídeo é amido, amilose ou amilopectina, mais preferivelmente amido.
[00021] Foi surpreendentemente descoberto que o intumescimento desvantajoso dos materiais suscetíveis ao ataque por bactérias colônicas, por exemplo, amilose, pode ser controlado por um material dependente do pH que é solúvel no pH 5 ou acima. Além disso, os inventores descobriram que, inesperadamente, os revestimentos que contêm grandes proporções de amilopectina também podem funcionar para fornecer a liberação colônica de um medicamento a partir de uma formulação oral de dosagem.
[00022] Uma outra vantagem técnica da presente invenção (comparado, por exemplo, à formulação divulgada na WO-A-Ol/76562) é que substancialmente nenhum medicamento é liberado por um período estendido (isto é, enquanto o revestimento está sendo dissolvido), a seguir de que o medicamento é liberado de um modo relativamente rápido. Isto está em contraste com os tabletes homogêneos a partir dos quais o perfil de liberação do medicamento é gradual a partir do início ao invés de retardada depois pulsátil.
[00023] A pessoa habilitada na técnica é capaz de determinar se um material é suscetível ao ataque por bactérias colônicas usando as técnicas que compreendem parte do conhecimento geral comum. Por exemplo, uma quantidade pré-determinada de um dado material pode ser exposta a um ensaio que contem uma enzima a partir de uma bactéria encontrada no cólon e a mudança em peso do material durante o tempo pode ser medida.
[00024] O polissacarídeo é preferivelmente amido. Os amidos são usualmente extraídos de fontes naturais tais como cereais; legumes; e tubérculos. Os amidos adequados para o uso na presente invenção são tipicamente amidos de grau alimentício e incluem amido de arroz; amido de trigo; amido de milho; amido de ervilha; amido de batata; amido de batata doce; amido de tapioca; amido de sorgo; amido de sagu; e amido de araruta. O uso de amido do milho é exemplificado abaixo.
[00025] O amido é atualmente uma mistura de dois polissacarídeos diferentes, isto é, amilose e amilopectina. Os amidos diferentes podem ter proporções diferentes destes dois polissacarídeos. Os amidos de milho mais naturais (não modificados) têm de cerca de 20% em peso a cerca de 30% em peso de amilose com o restante sendo pelo menos substancialmente feito de amilopectina. Os amidos adequados para o uso na presente invenção tipicamente têm pelo menos 0,1% em peso, por exemplo, pelo menos 10% ou 15%, preferivelmente pelo menos 35% em peso, de amilose. Os amidos de “amilose alta”, são os amidos tendo pelo menos 50% em peso de amilose. Os amidos particularmente adequados têm de cerca de 65% em peso a cerca de 75% em peso, por exemplo, cerca de 70% em peso de amilose.
[00026] Os amidos adequados para o uso na presente invenção podem ter até 100% de amilopectina, mais tipicamente de cerca de 0,1% em peso a cerca de 99.9% em peso de amilopectina. Os amidos de “amilose baixa”, isto é, os amidos tendo não mais do que 50% em peso de amilose e pelo menos 50% em peso de amilopectina, por exemplo, até 75% em peso de amilopectina e ainda tanto quanto até 99% em peso de amilopectina, são adequados. O amido pode ser, por exemplo, amido de milho ceroso não modificado. Isto tipicamente compreende cerca de 100% de amilopectina. O amido de “amilose baixa” não foi esperado ser adequado, visto que o amido de amilose baixa é tipicamente degradado pelas enzimas pancreáticas no intestino delgado. Os amidos preferidos não têm mais do que 50% em peso de amilopectina. Os amidos particularmente adequados têm de cerca de 25% em peso a cerca de 35% em peso de amilopectina, por exemplo, cerca de 30% em peso de amilopectina.
[00027] A pessoa habilitada na técnica é capaz de determinar as proporções relativas de amilose e amilopectina em qualquer amido dado. Por exemplo, a espectroscopia próxima ao infravermelho (“NIR”) pode ser usada para determinar o teor de amilose e amilopectina de um amido usando as curvas de calibração obtidas por NIR usando as misturas produzidas em laboratório de quantidades conhecidas destes dois componentes. Além disso, o amido pode ser hidrolisado à glicose usando amiloglucosidase. Uma série de reações de fosforilação e oxidação catalisadas pelas enzimas resultam na formação do fosfato de nicotinamida adenina dinucleotídica reduzido (“NADPH”). A quantidade de NADPH formado é estequiométrico com o teor de glicose original. Os kits de testes adequados para este procedimento são disponíveis (por exemplo, da R-Biopharm GmbH, Alemanha). Um outro método que pode ser usado envolve submeter o revestimento à digestão através de enzimas bacterianas, por exemplo, a-amilase, para produzir ácidos graxos de cadeia curta (“SCFA”) que podem ser quantificados através da cromatografia gasosa-líquida usando uma coluna capilar.
[00028] Os amidos preferidos têm amilose na sua forma vítrea embora a amilose na sua forma amorfa também pode ser usada em conjunção com a presente invenção.
[00029] Os amidos preferidos são amidos “fora da prateleira”, isto é, amidos que não necessitam de nenhum processamento antes do uso no contexto da presente invenção. Os exemplos de amidos de “amilose alta” particularmente adequados incluem Hylon® VII (National Starch, Alemanha) ou Eurylon® 7 (Roquette, Lestrem, França) ou Amylogel 03003 (Cargill, Minneapolis, EUA) todos os quais são exemplos de um amido de milho tendo cerca de 70% em peso de amilose.
[00030] A presente invenção envolve o uso de um segundo material que dissolve em um maneira dependente do pH. O segundo material tem um “limiar de pH” que é o pH abaixo do qual este é insolúvel e no ou acima do qual este é solúvel. O pH do meio circundante deflagra a dissolução do segundo material. Deste modo, nenhum (ou essencialmente nenhum) do segundo material dissolve abaixo do limiar do pH. Uma vez que o pH do meio circundante alcança (ou excede) o limiar do pH, o segundo material se torna solúvel. Por “insolúvel” queremos dizer que 1 g do segundo material necessita mais do que 10.000 ml de solvente (meio circundante) para dissolver em um dado pH. Por “solúvel”, queremos dizer que 1 g do segundo material necessita mais menos do que 10.000 ml, preferivelmente menos do que 5.000 ml, mais preferivelmente menos do que 1000 ml, ainda mais preferivelmente menos do que 100 ml ou 10 ml do solvente para dissolver em um dado pH. Meio circundante preferivelmente significa o meio no trato gastrintestinal, tal como o fluido gástrico ou o fluido intestinal. Alternativamente, o meio circundante pode ser o equivalente in vitro do meio no trato gastrintestinal.
[00031] O pH normal do fluido gástrico é usualmente na faixa de 1 a 3. O segundo material é insolúvel abaixo do pH 5 e solúvel em torno do pH 5 ou acima e solúvel em torno do pH 5 ou acima e, deste modo, é usualmente insolúvel em fluido gástrico. Um tal material pode ser referido como um material “entérico”.
[00032] O segundo material é solúvel no pH 5 ou acima, por exemplo, no fluido intestinal. O pH do fluido intestinal aumenta de modo gradual de cerca de 6 no duodeno a cerca de 7 a 8 no cólon. O segundo material é preferivelmente insolúvel abaixo do pH 6,5 (e solúvel em torno do pH 6,5 ou acima) e, mais preferivelmente, é insolúvel abaixo do pH 7 (e solúvel em torno do pH 7 ou acima).
[00033] O limiar do pH no qual um material se torna solúvel pode ser determinado por uma técnica de titulação simples que seve ser parte do conhecimento geral comum para a pessoa habilitada na técnica.
[00034] O segundo material é tipicamente um material polimérico formador de película tal como um polímero de acrilato, um polímero de celulose ou um polímero com base em polivinila. Os exemplos de polímeros de celulose adequados incluem ftalato de acetato de celulose (“CAP”); trimetilato de acetato de celulose (“CAT”); e succinato de acetato de hidropropilmetilcelulose. Os exemplos de polímeros adequados com base em polivinila incluem ftalato de acetato de polivinila (“PVAP”).
[00035] O segundo material é preferivelmente um co-polímero de um ácido (met)acrílico e um ácido éster de alquila C1-4 do ácido (met)acrílico, ou exemplo, um copolímero do ácido metacrílico e éster metílico do ácido metacrílico. Um tal polímero é conhecido como um co-polímero de poli(ácido metacrílico/metacrilato de metila). Os exemplos adequados de tais copolímeros são usualmente aniônicos e polimetacrilatos de liberação não sustentada. A razão dos ácidos de grupo carbóxi para os grupos de éster metílico (a “razão ácido:éster”) nestes co-polímeros determina o pH no qual que o co-polímero é solúvel. A razão ácido:éster pode ser de cerca de 2:1 para cerca de 1:3, por exemplo, cerca de 1:1 ou, preferivelmente, cerca de 1:2. O peso molecular (“MW”) de co-polímeros aniônicos preferidos é usualmente de cerca de 120.000 a 150.000, preferivelmente de cerca de 135.000.
[00036] Os co-polímeros aniônicos do poli(ácido metacrílico/ metacrilato de metila) preferidos incluem Eudragit® L (razão ácido:éster de cerca de 1:1; MW a cerca de 135.000; limiar de pH de cerca de 6,0); Eudragit® S (razão ácido:éster de cerca de 1:2; MW a cerca de 135.000; limiar do pH de cerca de 7); e Eudragit® FS (um poli(acrilato de metila/metacrilato de metila/ácido metacrílico); razão ácido:éster de cerca de 1:10; MW cerca de 220.000; limiar de pH de cerca de 7.)
[00037] O segundo material pode ser um copolímero de ácido metacrílico e acrilato de etila. O Eudragit® LI 00-55 poli(ácido metacrílico/ acrilato de etil); razão de ácido:éster de cerca de 1:1; MW cerca de 250.000; limiar de pH de cerca de 5,5 é adequado. Os co-polímeros de Eudragit® são fabricados e/ou distribuídos pela Degussa AG, Darmstadt, Alemanha.
[00038] As misturas de materiais poliméricos formadores de película podem ser usados como apropriado. Um exemplo de uma mistura adequada podem incluir uma mistura, por exemplo, uma mistura 1:1, de Eudragit® L e Eudragit® S. Entretanto, o uso de um material polimérico formador de película particular, por exemplo, um co-polímero de poli(ácido metacrílico/metacrilato de metila), sozinho é preferido.
[00039] O uso de Eudragit® S sozinho como o segundo material é particularmente preferido.
[00040] Em uma forma de realização preferida, foi descoberto que uma mistura de dois polímeros adequados em uma razão apropriada, aplicado como um revestimento de película em um núcleo, pelo menos minimiza, e pode substancialmente eliminar, a liberação do medicamento no estômago e no intestino delgado. A liberação subseqüente do medicamento no cólon é acreditado ocorrer pelos deflagradores fisiológicos ativos combinados: isto é pela dissolução do segundo material.; particularmente Eudragit® S, e digestão do primeiro material, por exemplo, amido ou amilose.
[00041] A proporção do primeiro material ao segundo material é tipicamente menos do que 99:1 e pode em algumas circunstâncias ser até 50:50. A proporção é usualmente até 35:65 e é preferivelmente de 15:85 a 35:65, por exemplo, 15:85 a 30:70. O inventor descobriu que uma razão do primeiro material ao segundo material de cerca de 25:75 a cerca de 35:65, por exemplo, cerca de 30:70, é particularmente adequado para alvejar a liberação do medicamento ao cólon, particularmente se o primeiro material é amido e o segundo material é Eudragit® S. A mistura do primeiro e segundo materiais é preferivelmente substancialmente homogênea.
[00042] Opcionalmente, os excipientes convencionais tais como os plastificadores para a formação de uma película (por exemplo, citrato de trietila) e agentes anti-pegajosidade (tais como monoestearato de glicerila) podem ser incluídos em quantidades de até 30% em peso da composição final da preparação de revestimento polimérico.
[00043] A espessura do revestimento da partícula é tipicamente de cerca de 10 pm a cerca de 150 pm. A espessura de um revestimento específico, entretanto, dependerá da composição do revestimento. Por exemplo, a espessura do revestimento é diretamente proporcional à quantidade de polissacarídeo no revestimento. Deste modo, em formas de realização onde o revestimento compreende amido de amilose alta e Eudragit® S em uma razão de cerca de 30 : 70, a espessura do revestimento pode ser de cerca de 70 pm a cerca de 130 pm, e preferivelmente de cerca de 90 pm a cerca de 110 pm. A espessura (em pm) para uma dada composição de revestimento é independente do tamanho do núcleo.
[00044] A espessura do revestimento também pode ser medida pelo “ganho de peso teórico” (“TWG”) da formulação revestida. O TWG para a presente formulação dependerá de vários fatores incluindo a composição do revestimento e o tamanho do núcleo a ser revestido. Por exemplo, em formas de realização onde o núcleo é um pequeno tablete (por exemplo, tendo um diâmetro de cerca de 8 mm) e o revestimento compreende amido de amilose alta e Eudragit® S (por exemplo, em um razão de cerca de 30 : 70), o TWG é tipicamente de cerca de 4% a cerca de 12%, por exemplo, de cerca de 5% a cerca de 10%, preferivelmente de cerca de 8% a cerca de 9%. Em formas de realização onde o núcleo é uma pelota (por exemplo, tendo um diâmetro de cerca de 1 mm) tendo o mesmo revestimento, o TWG pode ser de cerca de 15% a cerca de 35%, por exemplo, de cerca de 20% a cerca de 30%, preferivelmente cerca de 25%.
[00045] Dizendo-se que o revestimento compreende uma mistura do primeiro e segundo materiais, é intencionado excluir a forma de dosagem de múltiplas camadas conhecida (divulgada por exemplo em Milojevic et al. descrita acima) em que um núcleo ativo é revestido primeiro com um revestimento interno de amilose e depois com um revestimento externo de Eudragit® LI 00. No contexto da presente invenção, uma tal forma de dosagem de múltiplas camadas não compreende uma mistura de amido e Eudragit® LI 00. O revestimento é preferivelmente uma camada única de uma mistura do primeiro e segundo materiais, preferivelmente uma mistura homogênea.
[00046] A formulação da presente invenção pode entretanto ter uma camada adicional entre o núcleo ativo e a camada que compreende a composição de liberação retardada da presente invenção e/ou um revestimento de camada externa da camada de composição de liberação retardada da presente invenção. Por exemplo, se a camada de composição de liberação retardada compreende uma mistura de Eudragit® L e amido, a adição de uma camada externa de um material de revestimento de liberação dependente do pH tendo um limiar de pH de cerca de 7, por exemplo, Eudragit® S, pode ser preferível. Nas formas de realização preferidas, o revestimento de liberação retardada da presente invenção é aplicado diretamente ao núcleo ativo, isto é não existe nenhuma camada adicional entre este revestimento e o núcleo ativo. O revestimento de liberação retardada da presente invenção é preferivelmente o revestimento externo da formulação. Vantajosamente, foi descoberto que nenhuma camada externa adicional é necessária para assegurar que a composição seja uma composição de liberação retardada.
[00047] A composição preferivelmente forma um revestimento em torno do bioativo que é mais preferivelmente uma mistura do amido e Eudragit® S. O “bioativo” é usualmente o núcleo que compreende o medicamento.
[00048] A formulação compreende pelo menos uma partícula com um núcleo e um revestimento para o núcleo. A formulação pode compreender qualquer forma de dosagem oral revestida adequada incluindo cápsulas; tabletes; mini-tabletes; pelotas; grânulos; e cristais.
[00049] O diâmetro mínimo de cada partícula é tipicamente pelo menos cerca de IO-4 m, usualmente pelo menos cerca de 5 x 10-4m e, preferivelmente, pelo menos cerca de 10-3 m. o diâmetro máximo é usualmente de não mais do que 30 mm, tipicamente não mais do que 20 mm e, preferivelmente, não mais do que 10 mm. Em formas de realização preferidas, a partícula tem um diâmetro de cerca de 0,2 mm a cerca de 15 mm, preferivelmente de cerca de 1 mm a cerca de 4 mm (por exemplo, para pelotas ou mini-tabletes) ou de cerca de 6 mm a cerca de 12 mm (por exemplo, para certos tabletes ou cápsulas). O termo “diâmetro” se refere à dimensão linear mais larga através da partícula.
[00050] A formulação pode compreende uma pluralidade de partículas de modo a fornecer uma dose única do(s) medicamento(s), particularmente em formas de realização em que a partícula é “pequena”, por exemplo, tendo um diâmetro de menos do que 5 mm. As formas de dosagem de múltiplas unidades que compreendem as partículas tendo um diâmetro de menos do que 3 mm são preferidas.
[00051] A presente invenção tem aplicação em uma formulação de liberação do medicamento de múltiplas fases que compreende pelo menos duas multiplicidades de partículas, por exemplo, pelotas revestidas, na mesma forma de dosagem, por exemplo, uma cápsula, em que as partículas de uma multiplicidade são diferenciadas das partículas de ou cada outra multiplicidade pelo revestimento. Os revestimentos podem diferir-se de uma multiplicidade para a próxima em termos de espessura de revestimento ou composição, por exemplo, a razão e/ou identidade dos componentes. As formulações de liberação do medicamento de múltiplas fases devem ser particularmente adequadas para aqueles que sofrem da doença de Crohn que afeta as diferentes regiões ao longo do intestino.
[00052] O “núcleo” é usualmente um corpo sólido único. O núcleo pode consistir do(s) medicamento(s) sozinho(s) ou pode ser uma pérola de material comestível, por exemplo, açúcar, que é revestido com uma camada que compreende o(s) medicamento(s). Mais usualmente, entretanto, o núcleo consiste de uma mistura do(s) medicamento(s) com uma carga ou material diluente, por exemplo, material de lactose ou celulose tal como celulose microcristalina; um aglutinante, por exemplo, polivinilpirrolidona (“PVP”); um desintegrante, por exemplo, Ac-Di-Sol® (isto é, croscarmelose sódica); e/ou um lubrificante, por exemplo, estearato de magnésio. O núcleo pode ser um granulado comprimido que compreende pelo menos alguns destes materiais.
[00053] A liberação das formulações de acordo com a presente invenção é retardada até o intestino e preferivelmente ao cólon. A liberação de certas formulações também podem ser sustentadas. Entretanto, em formulações preferidas, a liberação é pulsátil.
[00054] Uma formulação é usualmente definida como resistente gástrico se existe menos do que 10% em peso de liberação do medicamento em meio ácido depois de 2 horas. As formulações de acordo com a presente invenção tipicamente demonstram muito menos do que 10% em peso de liberação do medicamento em meio ácido e podem ser consideradas ser resistentes gástricos. As formulações usualmente demonstram menos do que 1 % em peso de liberação do medicamento em meio ácido e, tipicamente, não demonstram substancialmente nenhuma liberação do medicamento em meio ácido. Quando o amido é combinado com um material de acrilato formador de película para formar o revestimento para o núcleo, tipicamente menos do que 5% de liberação do medicamento ocorre durante as 5 horas em condições que simulam o estômago e o intestino delgado. Na combinação de amido com um material celulósico formador de película para o revestimento para o núcleo, tipicamente menos do que 10% de liberação do medicamento ocorre durante as 5 horas em condições que simulam o estômago e o intestino delgado.
[00055] O tempo entre a exposição inicial às condições adequadas para a liberação do medicamento e o começo da liberação do medicamento é conhecida como a “tempo de retardo”. A “tempo de retardo” depende de vários fatores incluindo a espessura do revestimento e a composição. As formulações de acordo com a presente invenção usualmente demonstram um tempo de retardo em condições colônicas de pelo menos 30 minutos. Na maioria das formas de realização da presente invenção, o tempo de retardo é de cerca de 30 minutos a cerca de 3 horas e, em formulações preferidas, o tempo de retardo é preferivelmente de cerca de 45 minutos a cerca de 2 horas.
[00056] O tempo entre a exposição inicial às condições adequadas para a liberação do medicamento e a liberação completa do medicamento também depende de vários fatores incluindo a composição de revestimento e a natureza do medicamento. Na maioria das formas de realização da presente invenção, este tempo é usualmente não mais do que 5 horas. Em formas de realização preferidas, este tempo é usualmente não mais do que 4 horas.
[00057] Por via de um exemplo, em formas de realização em que um núcleo de tablete é revestido a uma espessura de 8% a 9% de TWG com um revestimento compreende um amido de amilose alta e Eudragit S (30:70), o tempo entre a liberação inicial e a liberação completa pode ser menos do que cerca de 2 horas, preferivelmente menos do que cerca de 1,5 horas.
[00058] Em uma forma de realização preferida, o núcleo é uma pelota tendo um diâmetro de cerca de 1 mm. Em uma outra forma de realização, o núcleo é um tablete tendo um diâmetro de cerca de 8 mm. Em ambos os casos, o revestimento é preferivelmente uma mistura de 30:70 de amido de amilose alta, por exemplo, Eurylon® 7, e um polímero acrílico, por exemplo, Eudragit® S. Em ambas as formas de realização preferidas, o núcleo é revestido a uma espessura de cerca de 100 pm que é de cerca de 8% a cerca de 9% de TWG para o tablete e de cerca de 27% a cerca de 32% em peso para a pelota.
[00059] De acordo com um segundo aspecto da presente invenção, é fornecido uma formulação de acordo com o primeiro aspecto para o uso em um método de tratamento médico do corpo humano ou animal através da terapia.
[00060] O núcleo compreende pelo menos um medicamento. A formulação é usualmente usada para administrar um medicamento simples como o componente terapeuticamente ativo único. Entretanto, mais do que um medicamento pode ser administrado em uma formulação única.
[00061] A formulação da presente invenção é intencionada para administrar uma ampla faixa de medicamentos. Os medicamentos adequados incluem aqueles medicamentos que são conhecidos para a administração intestinal usando as formulações oral de liberação retardada conhecidas. A presente invenção pode ser usada para administrar os medicamentos tendo um local ou um efeito sistêmico.
[00062] A formulação da presente invenção tem aplicação particular na administração intestinal de um medicamento que compreende pelo menos um grupo ácido tal como um, grupo do ácido carboxilico. Tais medicamentos podem ser medicamentos ácidos ou medicamentos zwitteriônicos. Um exemplo de um tal medicamento é o ácido 5-amino-salicílico (“5-ASA”).
[00063] A identidade do(s) medicamento(s) na formulação obviamente depende da condição a ser tratada. Em relação a isto, a formulação tem aplicação particular no tratamento da IBD (incluindo doença de Crohn e colite ulcerativa); IBS; constipação; diarréia; infecção; e carcinoma, particularmente câncer colônico ou colo-retal).
[00064] Para o tratamento ou prevenção da IBD, a formulação pode compreender pelo menos um medicamento selecionado do grupo que consiste de agente anti-inflamatório (por exemplo, 5-ASA); esteróides (por exemplo, prednisolona; budesonida ou fluticasona); imunossupressores (por exemplo, azatioprina; cicloesporina; e metotrexato); e antibióticos.
[00065] Para o tratamento ou prevenção do câncer, a formulação pode compreender pelo menos um agente antineoplásico. Os agentes antineoplásicos adequados incluem fluorouracila; metotrexato; dactinomicina; bleomicina; etoposídeo; taxol; vincristina; doxorubicina; cisplatina; daunorubicina; VP-16; raltitrexed; oxaliplatina; e derivados e sais farmacologicamente aceitáveis deste. Para a prevenção do câncer colônico ou câncer colo-retal, primeiramente em pacientes que sofrem de colite, a formulação pode compreender o agente anti-inflamatório, 5-ASA.
[00066] Para o tratamento ou prevenção da IBS, constipação, diarréia ou infecção, a formulação pode compreender pelo menos um agente ativo adequado para o tratamento ou prevenção destas condições.
[00067] Os derivados e/ou sais do medicamentos farmacologicamente aceitáveis também podem ser usados na formulação. Um exemplo de um sal adequado de prednisolona é succinato de metil prednisolona sódico. Um outro exemplo é propionato de fluticasona.
[00068] A presente invenção tem aplicação particular no tratamento da IBD (particularmente, colite ulcerativa) ou na prevenção do câncer colônico ou câncer colo-retal (primeiramente em pacientes de colite), ambos usando 5- ASA. Esta também tem aplicação como um portal de entrada de medicamentos na circulação sistêmica por intermédio do cólon. Isto é particularmente vantajoso para os medicamentos peptídeos e protéicos que são instáveis no trato gastrintestinal superior. A presente invenção também pode ser utilizada para o propósito da cronoterapia.
[00069] Em um terceiro aspecto da invenção, é fornecido um método de alvejar um medicamento ao cólon que compreende administrar a um paciente uma formulação como definido acima.
[00070] Em um quarto aspecto da invenção, é fornecido o uso de uma formulação como definido acima na fabricação de um medicamento para o tratamento ou prevenção da IBD (particularmente colite ulcerativa); IBS; constipação; diarréia; infecção; e câncer.
[00071] Também é fornecido o uso de pelo menos um medicamento selecionado de agente anti-inflamatórios e esteróides na fabricação de um medicamento que compreende uma formulação como definido acima para o uso no tratamento da IBD. Além disso, também é fornecido o uso de pelo menos um agente antineoplásico na fabricação de um medicamento que compreende uma formulação como definido acima para o uso no tratamento do carcinoma. Além disso, também é fornecido o uso de 5-ASA na fabricação de um medicamento que compreende uma formulação como definida acima para o uso na prevenção do câncer colônico ou câncer colo-retal.
[00072] De acordo com um quinto aspecto da presente invenção, é fornecido um método de tratamento médico ou prevenção de IBD ou carcinoma que compreende administrar a um paciente uma quantidade terapêutica de uma formulação como definido acima.
[00073] A formulação tipicamente compreenderá uma quantidade terapeuticamente eficaz do ou de cada medicamento que pode ser de cerca de 0,01% em peso a cerca de 99% em peso, com base no peso total da formulação. A dosagem real seria determinada pelo técnico habilitado usando seu conhecimento geral comum. Entretanto, por via de exemplo, as formulações de “baixa” dosagem tipicamente compreendem não mais do que cerca de 20% em peso do medicamento, e preferivelmente compreendem de cerca de 1% em peso a cerca de 10% em peso, por exemplo, cerca de 5% em peso, do medicamento. As formulações de “alta” dosagem tipicamente compreendem pelo menos 40% em peso do medicamento, e preferivelmente de cerca de 45% em peso a cerca de 85% em peso, por exemplo, cerca de 50% em peso ou cerca de 80% em peso.
[00074] De acordo com um sexto aspecto da presente invenção, é fornecido um método de preparar uma formulação de medicamento de liberação retardada de acordo com o primeiro aspecto, o dito método que compreende:
[00075] formar um núcleo que compreende pelo menos um medicamento; e
[00076] revestir o núcleo com uma preparação de revestimento polimérico que compreende uma mistura de um primeiro material que é suscetível ao ataque por bactérias colônicas e um segundo material que tem um limiar de pH em torno do pH 5 ou acima, em que o primeiro material compreende um polissacarídeo selecionado do grupo que consiste de amido; amilose; amilopectina; quitosano; sulfato de condroitina; ciclodextrina; dextrano; pululano; carragenano; escleroglucano; quitina; curdulano e levano. Os polissacarídeos preferidos são como detalhados acima. O núcleo é preferivelmente revestido por pulverização com a dita preparação de revestimento polimérico.
[00077] Em formas de realização em que o núcleo é formado a partir de um granulado comprimido, o método preferivelmente compreende:
[00078] misturar a seco o(s) medicamento(s) com pelo menos um excipiente para formar uma mistura seca;
[00079] granular a úmido pelo menos uma porção da dita mistura seca para formar um granulado úmido; comprimir pelo menos uma porção do dito granulado úmido para formar o dito núcleo; e
[00080] revestir por pulverização o dito núcleo com a dita preparação de revestimento polimérico para formar a dito formulação medicamentosa de liberação retardada. Uma máquina de revestimento por pulverização de leito fluidizado é preferivelmente usado para revestir o(s) núcleo(s) com a preparação de revestimento polimérico para formar a(s) partícula(s) da formulação.
[00081] Em formas de realização preferidas, o método compreende:
[00082] formar uma dispersão aquosa que compreende o dito primeiro material;
[00083] formar uma solução alcoólica ou aquosa que compreende o dito segundo material; e
[00084] adicionar, preferivelmente às gotas, pelo menos uma porção da dita dispersão aquosa do dito primeiro material a pelo menos uma porção da dita solução alcoólica ou aquosa do dito segundo material para formar a dita preparação de revestimento polimérico.
[00085] O primeiro material é usualmente dispersado em pelo menos um álcool, preferivelmente um álcool de Ci a CÓ, por exemplo, metanol; etanol; propan-l-ol; propan-2-ol; butan-l-ol; butan-2-ol; e misturas destes, particularmente butan-l-ol sozinho, e depois água é usualmente de modo subseqüente adicionada com boa agitação. A dispersão aquosa resultante é usualmente aquecida até a ebulição e depois esfriada com agitação durante a noite. O propósito do(s) álcool(is) é solvatar o primeiro material pronto para formar a dispersão aquosa. Alternativamente, o material pode ser disperso diretamente em água.
[00086] O segundo material, é tipicamente dissolvido em pelo menos um solvente, por exemplo água ou um solvente orgânico. O solvente orgânico pode ser um álcool, por exemplo, metanol; etanol; propan-2-ol; metil glicol; butil glicol; acetona; acetato de metil glicol; e misturas destes tais como acetona e álcool isopropílico (por exemplo, em um razão de cerca de 4:6). O segundo material é preferivelmente dissolvido em etanol (preferivelmente de 85 a 98%), sob agitação de alta velocidade.
[00087] A preparação do revestimento polimérico é preferivelmente formada adicionando-se uma quantidade apropriada da dispersão aquosa à solução alcoólica, às gotas sob agitação rápida. O(s) outro(s) excipiente(s) tais como um plastificador (por exemplo, citrato de trietila) e/ou um lubrificante (por exemplo, monoestearato de glicerila) é usualmente adicionado à preparação durante a agitação.
[00088] Várias formas de realização preferidas da presente invenção serão agora descritas com referência às figuras, em que: A figura 1 é um gráfico que representa os perfis de dissolução dos tabletes prednisolona revestidos com uma película misturada a 5% de TWG e tabletes revestidos de Eudragit® S a 5% de TWG em um tampão de pH7,0; A figura 2 é um gráfico que representa os perfis de dissolução dos tabletes revestidos com uma película misturada como para a figura 1 mas com 6% de TWG; • A figura 3 é um gráfico que representa os perfis de dissolução dos tabletes revestidos com uma película misturada como para a figura 1 mas com 7,4% de TWG; A figura 4 é um gráfico que representa os perfis de dissolução dos tabletes revestidos com uma película misturada como para a figura 1 mas com 8.3% de TWG; A figura 5 é um gráfico que representa os perfis de dissolução dos tabletes de prednisolona revestidos com 30% de amido : 70% de Eudragit S em vários ganhos de peso poliméricos e tabletes revestidos com Eudragit® S com 5% de TWG; A figura 6 é um gráfico que representa o perfil de dissolução dos tabletes de prednisolona revestidos com 30% amido : 70% Eudragit® S em um tampão de pH 6,8, com e sem pancreatina; A figura 7 é um gráfico que representa a liberação do medicamento a partir de tabletes de prednisolona revestidos a 8,3% de TWG em um tampão de pH 6,8 que contem 50 ll/ml de amilase; A figura 8 é um gráfico que representa o perfil de dissolução dos tabletes de 5-ASA revestidos com 30% amido : 70% Eudragit® S para 8,3% de TWG em um tampão de pH 6,8; A figura 9 é um gráfico que representa os perfis de dissolução dos tabletes de 5-ASA revestidos com 30% amido : 70% Eudragit® S em vários ganhos de peso poliméricos em um tampão de pH 6,8 que contem 50 U/ml de amilase; A figura 10 é um gráfico que representa os perfis de dissolução dos tabletes de prednisolona revestidos com 70% Eudragit® S : 30% em peso de amido tendo 70% em peso ou 27% em peso de amilose em um tampão de pH7; A figura 11 é um gráfico que representa os perfis de dissolução dos tabletes de prednisolona como para a figura 10 em um tampão de pH 6,8; A figura 12 é um gráfico que representa os perfis de dissolução dos tabletes de prednisolona como para a figura 10 em um tampão de pH 6,8 o qual contem 50 U/ml de amilase; A figura 13 é um gráfico que representa o perfil de dissolução dos tabletes de prednisolona revestidos com 70% de Eudragit® L : 30% de amido a 8.3% de TWG em um tampão de pH 5,5; A figura 14 é um gráfico que representa o perfil de dissolução dos tabletes de prednisolona como para a figura 13 em um tampão de pH 5.5 que contem 50 U/ml de amilase; A figura 15 é um gráfico que representa o perfil de dissolução dos tabletes de prednisolona revestidos com 70% de HPMCAS-HG : 30% de amido em um tampão de pH 6,5; A figura 16 é um gráfico que representa o perfil de dissolução dos tabletes de prednisolona como para a figura 15 em um tampão de pH 6,5 o qual contem 50 U/ml de amilase; e A figura 17 é um gráfico que representa o perfil de dissolução dos tabletes de prednisolona como para a figura 15 em um tampão de pH 6,8.
[00089] Os tabletes de prednisolona (200 mg de peso, diâmetro de 8 mm e padrão bi-côncavo) foram preparados de acordo com a seguinte fórmula:
[00090] A prednisolona foi misturada a seco com os excipientes e depois granulada a úmido. Os grânulos com uma fração de tamanho de 500 a 710 pm foram comprimidos usando uma máquina de tabletagem de compressão única (Manesty, RU). Formulação para a Dispersão Aquosa de Amido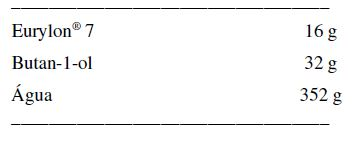
[00091] O amido Eurylon® 7 foi bem dispersado em butan-l-ol e foi água subseqüentemente adicionada com boa agitação. A dispersão resultante foi depois aquecida até a ebulição, e esfriada com agitação durante a noite. Os teores de sólidos em% da dispersão resfriada foi calculada com base no peso final da dispersão (permitindo a evaporação durante o aquecimento).
[00092] A solução de Eudragit® S foi preparada através da dissolução do polímero de Eudragit® S 100 em 96% de etanol sob agitação de alta velocidade. A solução final continha aproximadamente 6% de sólidos poliméricos.
[00093] As quantidades apropriadas da dispersão de amido e solução de Eudragit® foram misturadas para fornecer as razões requeridas estabelecidas como a substância polimérica seca. O amido foi sempre adicionado à solução de Eudragit® às gotas sob agitação rápida. As dispersões resultantes foram deixadas agitar por duas horas antes da adição dos excipientes e por outras duas horas depois de adicionar os excipientes. Os excipientes adicionados foram:
[00094] A preparação de revestimento polimérico misturado final foi revestida com uma película nos tabletes usando um máquina de revestimento por pulverização de leito fluidizado. A espessura do revestimento foi avaliada como aumento de% em peso dos tabletes a seguir do revestimento (% de TWG)
[00095] Os parâmetros do revestimento por pulverização foram como segue:
[00096] A dispersão de amido foi preparada a partir de Eurylon 7, um amido de “amilose alta”, e misturada com uma solução de Eudragit® S em etanol. A composição e o método de preparação para as dispersões de revestimento são como descritos acima. Várias combinações de amido/ Eudragit® S foram preparadas as quais contêm 15%, 20%, 25%, 30% e 35% de amido. As dispersões de revestimento de Eudragit®/amido misturadas foram depois revestidas com uma película em tabletes de prednisolona preparados de acordo com o método descrito acima. Os tabletes foram revestidos em várias espessuras, calculados como ganho de peso total no polímero, para determinar também a espessura de revestimento ótima. A mistura de revestimentos produziu películas de boa qualidade até uma razão de 30% de amido.
[00097] Os tabletes revestidos foram depois testados in vitro quanto a liberação do medicamento em uma solução tampão de pH variado. A razão de amido/Eudragit® S e ganho de peso do revestimento ótimos era primeiramente com base na comparação do perfil de dissolução com tabletes revestidos com Eudragit® S convencionais.
[00098] Os resultados são apresentados nas figuras de 1 a 7.
[00099] Muito surpreendentemente, estes tabletes revestidos com uma película misturada foram capazes de resistir à liberação do medicamento em HC1 de pH 1,2 simulando o meio gástrico (ver o lado esquerdo dos gráficos das figuras de 1 a 6).
[000100] Também não houve nenhuma liberação do medicamento de qualquer um dos tabletes revestidos por até 12 horas em pH 6,8 simulando o meio do intestino delgado (ver a figura 6). Estudos in vitro prévios usando polímeros de amilose/acrilato misturados com base nos produtos de Eudragit® RL e RS insolúveis em água apresentaram o intumescimento incontrolável e liberação rápida do medicamento em ácido e tampão (Milojevic et al., 1996).
[000101] A liberação dos perfis de medicamentos a partir dos tabletes revestidos em um meio de tampão de pH 7,0 é apresentada nas Figuras de 1 a 5. Com base em uma análise dos perfis de dissolução, os tabletes revestidos com uma mistura de amido/Eudragit® a 30% para uma espessura de película equivalente a um TWG de 8,3% foram julgados ser ótimos, e também foram testados para avaliar a capacidade de digestão do componente de amido da película.
[000102] Os tabletes foram testados quanto a dissolução em um tampão de pH 6,8 o qual contem 50 U (unidades)Zml de a-amilase derivada de B. licheniformis (ver a figura 7). Um teste de dissolução também foi realizado em um meio de pH 6,8 com pancreatina para testar se o amido é digestível pela a-amilase pancreática (ver a figura 6).
[000103] Os resultados dos testes de dissolução na presença das enzimas apresentaram que o componente de amido da película não é digestível na presença da pancreatina (o que sugere a resistência no intestino delgado), mas a liberação do medicamento ocorreu dentro de três horas na presença de a- amilase da B. licheniformis. Estes resultados fornecem a evidência de que a película misturada resiste à liberação do medicamento em condições simuladas do trato gastrintestinal superior mas é digestível na presença de enzimas bacterianas (mesmo em um pH menos do que o limiar do pH do polímero de Eudragit® S para a dissolução).
[000104] Seguindo o sucesso surpreendente dos estudos in vitro com os tabletes revestidos com uma película de amido/Eudragit® S misturada; o desempenho da forma de dosagem foi testado em pacientes humanos saudáveis. Os tabletes foram radio-rotulados com índio-111 e administrados à oito voluntários do sexo masculino saudáveis em três ocasiões separadas. O trânsito e o sítio de desintegração do tablete no trato gastrintestinal foi seguido usando uma câmera de gama.
[000105] O tempo e o sítio de desintegração destes tabletes de película misturada podem ser vistos na Tabela 1. Os resultados surpreendentemente mostram um excelente alvejamento colônico, com a desintegração do tablete ocorrendo primeiramente no cólon.
[000106] Os resultados dos estudos com voluntários saudáveis fornecem a evidência de que o amido e Eudragit® misturados em uma proporção de 30% de amido a 70% de Eudragit® S e revestidos em tabletes em aproximadamente 8,3% de TWG, é capaz de resistir à desintegração do tablete no estômago e intestino delgado mas deflagrar a desintegração no cólon.
[000107] A tabela 1 apresenta o sítio e tempo de desintegração dos tabletes revestidos de 30% de amido e 70% de Eudragit® S em oito voluntários do sexo masculino saudáveis em três ocasiões separadas.
[000108] Chave para a Tabela 1: “Em jejum” - tablete dado em um estômago vazio; “Pré-alimentação” - tablete dado em estômago vazio, mas a alimentação é administrada 30 minutos após a dose; e “Alimentado” - tablete dado depois do café da manhã. “ICJ” - junção ileocolônica; “AC” - cólon ascendente; “HF” - flexão hepática; e “SF” - flexão esplênica.
Claims (24)
1. Formulação de medicamento de liberação retardada para dispensação de um medicamento ao cólon, caracterizada pelo fato de que compreende uma partícula com um núcleo e um revestimento para o núcleo, o núcleo compreendendo um medicamento e o revestimento compreendendo uma mistura de um primeiro material que é suscetível ao ataque por bactérias colônicas e um segundo material que tem um limiar de pH em 6,5 ou acima, em que o primeiro material compreende um polissacarídeo selecionado do grupo que consiste de amido; amilopectina; quitosano; sulfato de condroitina; ciclodextrina; dextrano; e carragenano, em que a liberação do medicamento é retardada até o cólon e em que o limite de pH do segundo material é o pH abaixo do qual este é insolúvel e em ou acima do qual é solúvel.
2. Formulação de acordo com a reivindicação 1, caracterizada pelo fato de que o polissacarídeo é amido compreendendo pelo menos 35% de amilose.
3. Formulação de acordo com reivindicação 1 ou reivindicação 2, caracterizada pelo fato de que o segundo material é um material polimérico formador de película.
4. Formulação de acordo com reivindicação 3, caracterizada pelo fato de que o segundo material é um polímero de acrilato.
5. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que o segundo material é um copolímero aniônico de ácido (met)acrílico e éster alquílico CM do ácido (met)acrílico.
6. Formulação de acordo com a reivindicação 5, caracterizada pelo fato de que o segundo material é um copolímero aniônico de ácido metacrílico e éster metílico do ácido metacrílico, em que a proporção de ácido metacrílico para éster metílico do ácido metacrílico é 1:2.
7. Formulação de acordo com qualquer uma das reivindicações de 1 a 3, caracterizada pelo fato de que o segundo material é um polímero de celulose.
8. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que a proporção do primeiro material para o segundo material é até 50:50.
9. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que a proporção do primeiro material para o segundo material é até 35:65.
10. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que o diâmetro mínimo da partícula é pelo menos 5 x 10'4 m.
11. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que a espessura do revestimento como medido pelo ganho de peso teórico (“TWG”) da formulação revestida é de 5 a 10%.
12. Formulação de acordo com qualquer uma das reivindicações de 1 a 10, caracterizada pelo fato de que a espessura do revestimento como medido pelo ganho de peso teórico (“TWG”) da formulação revestida édel5a35%.
13. Formulação de acordo com qualquer uma das reivindicações de 1 a 10, caracterizada pelo fato de que o revestimento tem uma espessura de cerca de 10 pm a cerca de 150 pm.
14. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que o medicamento compreende pelo menos um grupo ácido.
15. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que o medicamento é um agente anti-inflamatório.
16. Formulação de acordo com qualquer uma das reivindicações de 1 a 13, caracterizada pelo fato de que o medicamento é um esteróide.
17. Formulação de acordo com qualquer uma das reivindicações de 1 a 13, caracterizada pelo fato de que o medicamento é um agente antineoplásico.
18. Formulação de acordo com qualquer uma das reivindicações precedentes, caracterizada pelo fato de que é para tratamento do corpo humano ou animal.
19. Uso de uma formulação como definida na reivindicação 15 ou reivindicação 16, caracterizado pelo fato de ser na fabricação de um medicamento para o tratamento de doença inflamatória intestinal.
20. Uso de uma formulação de acordo com a reivindicação 17, caracterizado pelo fato de ser na fabricação de um medicamento para o tratamento de carcinoma.
21. Uso de uma formulação de acordo com a reivindicação 15, caracterizado pelo fato de ser na fabricação de um medicamento para a profilaxia de carcinoma.
22. Método para preparar uma formulação de medicamento de liberação retardada como definida na reivindicação 1, caracterizado pelo fato de que compreende: formar um núcleo compreendendo um medicamento; e revestir o núcleo com uma preparação de revestimento polimérico compreendendo uma mistura de um primeiro material que é suscetível ao ataque por bactérias colônicas e um segundo material que tem um limiar de pH em 6,5 ou acima, em que o primeiro material compreende um polissacarídeo selecionado do grupo que consiste de amido; amilopectina; quitosano; sulfato de condroitina; ciclodextrina; dextrano; e carragenano.
23. Método de acordo com a reivindicação 22, caracterizado pelo fato de que o núcleo é revestido por pulverização com a dita preparação de revestimento polimérico.
24. Método de acordo com a reivindicação 22 ou reivindicação 23, caracterizado pelo fato de que compreende: formar um dispersão aquosa do dito primeiro material; formar uma solução alcoólica ou aquosa do dito segundo material; e adicionar pelo menos uma porção da dita dispersão aquosa do dito primeiro material a pelo menos uma porção da dita solução alcoólica ou aquosa do dito segundo material para formar a dita preparação de revestimento polimérico.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0607534.5 | 2006-04-13 | ||
| GBGB0607534.5A GB0607534D0 (en) | 2006-04-13 | 2006-04-13 | Colonic drug delivery formulation |
| PCT/GB2007/001354 WO2007122374A2 (en) | 2006-04-13 | 2007-04-13 | Colonic drug delivery formulation |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0710195A2 BRPI0710195A2 (pt) | 2011-08-09 |
| BRPI0710195B1 true BRPI0710195B1 (pt) | 2020-08-04 |
| BRPI0710195B8 BRPI0710195B8 (pt) | 2021-05-25 |
Family
ID=36571851
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0710195A BRPI0710195B8 (pt) | 2006-04-13 | 2007-04-13 | formulação de medicamento de liberação retardada para dispensação de um medicamento ao cólon, uso de uma formulação, e, método para preparar uma formulação de medicamento de liberação retardada |
Country Status (26)
| Country | Link |
|---|---|
| US (6) | US9023368B2 (pt) |
| EP (1) | EP2018159B2 (pt) |
| JP (5) | JP5176017B2 (pt) |
| KR (1) | KR101360122B1 (pt) |
| CN (1) | CN101448488B (pt) |
| AU (1) | AU2007242648B8 (pt) |
| BR (1) | BRPI0710195B8 (pt) |
| CA (2) | CA2648658C (pt) |
| CY (1) | CY1113307T1 (pt) |
| DK (1) | DK2018159T4 (pt) |
| ES (1) | ES2390106T5 (pt) |
| GB (1) | GB0607534D0 (pt) |
| HK (1) | HK1128627A1 (pt) |
| HR (1) | HRP20120766T4 (pt) |
| IL (2) | IL194632A (pt) |
| MX (1) | MX2008013036A (pt) |
| NO (1) | NO344029B1 (pt) |
| NZ (2) | NZ778023A (pt) |
| PL (1) | PL2018159T5 (pt) |
| PT (1) | PT2018159E (pt) |
| RS (1) | RS52434B2 (pt) |
| RU (2) | RU2646825C2 (pt) |
| SA (1) | SA07280292B1 (pt) |
| SI (1) | SI2018159T2 (pt) |
| WO (1) | WO2007122374A2 (pt) |
| ZA (1) | ZA200808677B (pt) |
Families Citing this family (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0607534D0 (en) * | 2006-04-13 | 2006-05-24 | Univ London Pharmacy | Colonic drug delivery formulation |
| ES2425762T3 (es) * | 2008-10-27 | 2013-10-17 | Roquette Freres | Polímero insoluble en agua: revestimientos de película a base de derivados de almidón modificado para la liberación dirigida al colon |
| MX2011005042A (es) | 2008-11-13 | 2011-08-17 | Giuliani Int Ltd | Composiciones antisentido, y metodos para obtener y usar las mismas. |
| US9314444B2 (en) | 2009-01-12 | 2016-04-19 | Biokier, Inc. | Composition and method for treatment of NASH |
| US9006288B2 (en) | 2009-01-12 | 2015-04-14 | Biokier, Inc. | Composition and method for treatment of diabetes |
| HUE030062T2 (en) | 2010-11-08 | 2017-04-28 | Albireo Ab | IBAT inhibitors for the treatment of liver diseases |
| WO2013030726A1 (en) | 2011-08-26 | 2013-03-07 | Wockhardt Limited | Programmed drug delivery |
| LT2659881T (lt) * | 2012-04-30 | 2018-02-12 | Tillotts Pharma Ag | Uždelsto atpalaidavimo vaisto forma |
| US9682096B2 (en) | 2012-05-02 | 2017-06-20 | Janssen R & D Ireland | Polyinosinic-polycytidylic acid (poly (I:C)) formulations for the treatment of upper respiratory tract infections |
| TN2016000119A1 (en) | 2013-10-29 | 2017-10-06 | Tillotts Pharma Ag | A delayed release drug formulation |
| SI2946774T1 (sl) * | 2014-05-19 | 2020-07-31 | Tillotts Pharma Ag | Obložene kapsule s prirejenim sproščanjem |
| KR20220082931A (ko) | 2014-06-25 | 2022-06-17 | 이에이 파마 가부시키가이샤 | 고형 제제 및 그의 착색 방지 또는 착색 감소 방법 |
| EP3012252A1 (en) | 2014-10-24 | 2016-04-27 | Ferring BV | Crystal modifications of elobixibat |
| WO2017040864A1 (en) | 2015-09-01 | 2017-03-09 | First Wave Biopharma | Methods and compositions for treating conditions associated with an abnormal inflammatory responses |
| US10786529B2 (en) | 2016-02-09 | 2020-09-29 | Albireo Ab | Oral cholestyramine formulation and use thereof |
| US10441604B2 (en) | 2016-02-09 | 2019-10-15 | Albireo Ab | Cholestyramine pellets and methods for preparation thereof |
| US10441605B2 (en) | 2016-02-09 | 2019-10-15 | Albireo Ab | Oral cholestyramine formulation and use thereof |
| US10588864B2 (en) | 2016-03-11 | 2020-03-17 | Gateway Pharmaceuticals LLC | Pharmaceutical compositions for colon-specific delivery |
| EP3429573A4 (en) | 2016-03-17 | 2019-10-30 | Thiogenesis Therapeutics, Inc. | COMPOSITIONS FOR THE CONTROLLED RELEASE OF CYSTEAMINE AND FOR SYSTEMIC TREATMENT OF CYSTEAMIN SENSITIVE DISEASES |
| EP3257501A1 (en) | 2016-06-14 | 2017-12-20 | Tillotts Pharma AG | Multiple unit dosage form comprising a core with individual core units covered by a mucoadhesive material, and an enteric core coating |
| CN107540761B (zh) * | 2016-06-27 | 2020-08-11 | 中国科学院微生物研究所 | 一种耐酸型普鲁兰多糖衍生物及其制备方法 |
| CN106380636B (zh) * | 2016-08-30 | 2019-05-14 | 华南理工大学 | 一种壳聚糖接枝的抗消化淀粉载体材料及其制备方法和应用 |
| EP3409688A1 (en) | 2017-05-31 | 2018-12-05 | Tillotts Pharma Ag | Topical treatment of inflammatory bowel disease using anti-tnf-alpha antibodies and fragments thereof |
| CA3071285A1 (en) | 2017-08-09 | 2019-02-14 | Albireo Ab | Cholestyramine granules, oral cholestyramine formulations and use thereof |
| EP3459529A1 (en) | 2017-09-20 | 2019-03-27 | Tillotts Pharma Ag | Preparation of sustained release solid dosage forms comprising antibodies by spray drying |
| ES2938608T3 (es) | 2017-09-20 | 2023-04-13 | Tillotts Pharma Ag | Método para preparar una forma farmacéutica sólida que comprende anticuerpos mediante granulación en húmedo, extrusión y esferonización |
| JP7208982B2 (ja) | 2017-09-20 | 2023-01-19 | チオジェネシス セラピューティクス, インコーポレイテッド | システアミン感受性障害の治療方法 |
| EP3459528B1 (en) | 2017-09-20 | 2022-11-23 | Tillotts Pharma Ag | Preparation of solid dosage forms comprising antibodies by solution/suspension layering |
| US10793534B2 (en) | 2018-06-05 | 2020-10-06 | Albireo Ab | Benzothia(di)azepine compounds and their use as bile acid modulators |
| PE20210182A1 (es) | 2018-06-20 | 2021-01-29 | Albireo Ab | Modificaciones de cristales de odexibat |
| US11007142B2 (en) | 2018-08-09 | 2021-05-18 | Albireo Ab | Oral cholestyramine formulation and use thereof |
| US10722457B2 (en) | 2018-08-09 | 2020-07-28 | Albireo Ab | Oral cholestyramine formulation and use thereof |
| US11549878B2 (en) | 2018-08-09 | 2023-01-10 | Albireo Ab | In vitro method for determining the adsorbing capacity of an insoluble adsorbant |
| MX2021002686A (es) | 2018-09-06 | 2021-08-11 | Fachhochschule Nordwestschweiz | Formulacion controlada de liberacion de farmacos. |
| EP3662900A1 (en) | 2018-12-07 | 2020-06-10 | Tillotts Pharma AG | Colonic drug delivery formulation |
| WO2020114616A1 (en) | 2018-12-07 | 2020-06-11 | Tillotts Pharma Ag | Topical treatment of immune checkpoint inhibitor induced diarrhoea, colitis or enterocolitis using antibodies and fragments thereof |
| EP3662895A1 (en) | 2018-12-07 | 2020-06-10 | Tillotts Pharma AG | A process for manufacturing reducing sugar-free 5-asa tablet cores |
| EP3662901A1 (en) * | 2018-12-07 | 2020-06-10 | Tillotts Pharma AG | Delayed release drug formulation comprising an outerlayer with an enzymaticyaaly degradable polymer, its composition and its method of manufacturing |
| EP3662902A1 (en) * | 2018-12-07 | 2020-06-10 | Tillotts Pharma AG | Colonic drug delivery formulation |
| GB201905940D0 (en) | 2019-04-29 | 2019-06-12 | Intract Pharma Ltd | 5-aminolevulinic acid for the local treatment of inflammatory bowel disease |
| JP2022533610A (ja) | 2019-05-15 | 2022-07-25 | エボニック オペレーションズ ゲーエムベーハー | カプセル充填機を用いて、(メタ)アクリレートコポリマー系のコーティングを有する、充填されたハードシェルカプセルを製造する方法 |
| GB201906917D0 (en) | 2019-05-16 | 2019-07-03 | Intract Pharma Ltd | Novel compositions |
| GB201906906D0 (en) | 2019-05-16 | 2019-07-03 | Intract Pharma Ltd | Novel compositions |
| KR102104507B1 (ko) * | 2019-08-23 | 2020-04-24 | 브릿지바이오테라퓨틱스(주) | 팔미토일-l-프롤릴-l-프롤릴-글리실-l-타이로신 나트륨을 포함하는 약제학적 제제 및 이의 제조방법 |
| CA3154308A1 (en) | 2019-09-13 | 2021-03-18 | Mark Smith | Compositions and methods for treating autism spectrum disorder |
| AU2020366529A1 (en) | 2019-10-18 | 2022-05-19 | Finch Therapeutics Holdings Llc | Compositions and methods for delivering a bacterial metabolite to a subject |
| WO2021097288A1 (en) | 2019-11-15 | 2021-05-20 | Finch Therapeutics Holdings Llc | Compositions and methods for treating neurodegenerative diseases |
| WO2021142238A1 (en) | 2020-01-10 | 2021-07-15 | First Wave Bio, Inc. | Deuterated niclosamide |
| WO2021142353A1 (en) | 2020-01-10 | 2021-07-15 | Finch Therapeutics Holdings Llc | Compositions and methods for treating hepatitis b (hbv) and hepatitis d (hdv) |
| WO2021142358A1 (en) | 2020-01-10 | 2021-07-15 | Finch Therapeutics Holdings Llc | Compositions and methods for treating hepatic encephalopathy (he) |
| WO2021142347A1 (en) | 2020-01-10 | 2021-07-15 | Finch Therapeutics Holdings Llc | Compositions and methods for non-alcoholic steatohepatitis (nash) |
| WO2021188564A1 (en) | 2020-03-16 | 2021-09-23 | First Wave Bio, Inc. | Methods of treating covid-19 with a niclosamide compound |
| US10980756B1 (en) | 2020-03-16 | 2021-04-20 | First Wave Bio, Inc. | Methods of treatment |
| US20230201265A1 (en) | 2020-03-31 | 2023-06-29 | Finch Therapeutics Holdings Llc | Compositions comprising non-viable fecal microbiota and methods of use thereof |
| US11045434B1 (en) | 2020-04-01 | 2021-06-29 | UNION therapeutics A/S | Niclosamide formulations for treating disease |
| GB202017863D0 (en) | 2020-11-12 | 2020-12-30 | Intract Pharma Ltd | Novel compositions |
| WO2022178294A1 (en) | 2021-02-19 | 2022-08-25 | Finch Therapeutics Holdings Llc | Compositions and methods for providing secondary bile acids to a subject |
| CA3236808A1 (en) * | 2021-11-03 | 2023-05-11 | Jeffrey Hubbell | Prodrug copolymers and polymeric micelles thereof for the delivery of short-chain fatty acids, the promotion of gut health, and the treatment of immune and/or inflammatory conditions and food allergy |
| WO2023102491A1 (en) * | 2021-12-01 | 2023-06-08 | Invea Therapeutics, Inc. | Methods for treating gastrointestinal inflammatory disease |
| GB202201708D0 (en) | 2022-02-10 | 2022-03-30 | Intract Pharma Ltd | Compositions for oral delivery of biotherapeutics |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA825384B (en) * | 1981-07-31 | 1983-05-25 | Tillott J B Ltd | Orally administrable pharmaceutical compositions |
| GB8812490D0 (en) | 1988-05-26 | 1988-06-29 | Agricultural & Food Res | Delayed release formulations |
| IT1230576B (it) * | 1988-10-20 | 1991-10-28 | Angeli Inst Spa | Formulazioni farmaceutiche per via orale a liberazione selettiva nel colon |
| GB8926639D0 (en) * | 1989-11-24 | 1990-01-17 | Agricultural & Food Res | Delayed release formulations |
| WO1991016881A1 (en) * | 1990-05-04 | 1991-11-14 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | Colonic drug delivery system |
| US5422121A (en) * | 1990-11-14 | 1995-06-06 | Rohm Gmbh | Oral dosage unit form |
| SK281042B6 (sk) * | 1992-09-18 | 2000-11-07 | Yamanouchi Pharmaceutical Co., Ltd | Prípravok hydrogélového typu s ustáleným uvoľňovaním |
| WO1995028963A1 (fr) * | 1994-04-22 | 1995-11-02 | Yamanouchi Pharmaceutical Co., Ltd. | Systeme de liberation de medicament specifique au colon |
| GB9412394D0 (en) * | 1994-06-21 | 1994-08-10 | Danbiosyst Uk | Colonic drug delivery composition |
| US6248362B1 (en) * | 1997-03-26 | 2001-06-19 | Meiji Seika Kaisha, Ltd. | Large intestinal delivery composite |
| EA001906B1 (ru) * | 1997-07-01 | 2001-10-22 | Пфайзер Продактс Инк. | Лекарственные формы с замедленным высвобождением сертралина |
| GB9722426D0 (en) * | 1997-10-23 | 1997-12-24 | Univ London Pharmacy | Controlled release formulations |
| GB9724186D0 (en) | 1997-11-14 | 1998-01-14 | British Tech Group | Low temperature coatings |
| US6251430B1 (en) | 1998-02-04 | 2001-06-26 | Guohua Zhang | Water insoluble polymer based sustained release formulation |
| EP1063977A1 (en) * | 1998-03-27 | 2001-01-03 | Valtion Teknillinen Tutkimuskeskus | Coated starch capsules and a process for producing them |
| JP4659316B2 (ja) * | 1999-08-09 | 2011-03-30 | 大日本住友製薬株式会社 | キトサン粉末含有固形製剤およびその製造方法 |
| FI20000780A (fi) | 2000-04-03 | 2001-10-04 | Novasso Oy | Peroraalinen lääkemuoto lääkeaineen hallituksi vapauttamiseksi |
| GB2367002A (en) | 2000-09-25 | 2002-03-27 | British Sugar Plc | Coating composition |
| GB0203421D0 (en) | 2002-02-13 | 2002-04-03 | Alizyme Therapeutics Ltd | Composition |
| CN1201736C (zh) * | 2002-07-19 | 2005-05-18 | 安徽省药物研究所 | 甲硝唑结肠定位缓释制剂 |
| US7670627B2 (en) | 2002-12-09 | 2010-03-02 | Salvona Ip Llc | pH triggered targeted controlled release systems for the delivery of pharmaceutical active ingredients |
| EA200600598A1 (ru) * | 2003-09-19 | 2006-08-25 | Пенвест Фармасьютикалз Ко. | Лекарственные формы замедленного высвобождения |
| CZ296459B6 (cs) * | 2004-09-14 | 2006-03-15 | Pliva-Lachema A. S. | Perorální farmaceutická kompozice pro cílený transport komplexu platiny do kolorektální oblasti, zpusob její prípravy a tato kompozice pro pouzití jako lécivo |
| EP1896002A4 (en) * | 2005-06-27 | 2009-11-25 | Biovail Lab Int Srl | BUPROPIONAL SALT FORMULATIONS WITH MODIFIED RELEASE |
| US11744803B2 (en) * | 2005-07-29 | 2023-09-05 | Stichting Groningen Centre for Drug Rese | PH-controlled pulsatile delivery system, methods for preparation and use thereof |
| GB0607534D0 (en) * | 2006-04-13 | 2006-05-24 | Univ London Pharmacy | Colonic drug delivery formulation |
| JP6148210B2 (ja) * | 2014-06-17 | 2017-06-14 | 東京エレクトロン株式会社 | 現像方法及びコンピュータ読み取り可能な記録媒体 |
-
2006
- 2006-04-13 GB GBGB0607534.5A patent/GB0607534D0/en not_active Ceased
-
2007
- 2007-04-13 RU RU2012153207A patent/RU2646825C2/ru active
- 2007-04-13 DK DK07732397.0T patent/DK2018159T4/da active
- 2007-04-13 SI SI200731022T patent/SI2018159T2/sl unknown
- 2007-04-13 WO PCT/GB2007/001354 patent/WO2007122374A2/en active Application Filing
- 2007-04-13 JP JP2009504820A patent/JP5176017B2/ja active Active
- 2007-04-13 CA CA2648658A patent/CA2648658C/en active Active
- 2007-04-13 US US11/735,248 patent/US9023368B2/en active Active
- 2007-04-13 PT PT07732397T patent/PT2018159E/pt unknown
- 2007-04-13 AU AU2007242648A patent/AU2007242648B8/en active Active
- 2007-04-13 KR KR1020087027321A patent/KR101360122B1/ko active IP Right Grant
- 2007-04-13 BR BRPI0710195A patent/BRPI0710195B8/pt active IP Right Grant
- 2007-04-13 NZ NZ778023A patent/NZ778023A/en unknown
- 2007-04-13 NZ NZ572760A patent/NZ572760A/xx unknown
- 2007-04-13 CA CA2825758A patent/CA2825758A1/en not_active Abandoned
- 2007-04-13 MX MX2008013036A patent/MX2008013036A/es active IP Right Grant
- 2007-04-13 ES ES07732397T patent/ES2390106T5/es active Active
- 2007-04-13 CN CN200780018366XA patent/CN101448488B/zh active Active
- 2007-04-13 RS RS20120404A patent/RS52434B2/sr unknown
- 2007-04-13 RU RU2008144700/15A patent/RU2478372C2/ru active
- 2007-04-13 EP EP07732397.0A patent/EP2018159B2/en active Active
- 2007-04-13 PL PL07732397T patent/PL2018159T5/pl unknown
- 2007-06-06 SA SA7280292A patent/SA07280292B1/ar unknown
-
2008
- 2008-10-07 IL IL194632A patent/IL194632A/en active IP Right Grant
- 2008-10-10 ZA ZA2008/08677A patent/ZA200808677B/en unknown
- 2008-10-14 NO NO20084292A patent/NO344029B1/no unknown
-
2009
- 2009-07-27 HK HK09106896.3A patent/HK1128627A1/xx unknown
-
2012
- 2012-09-24 CY CY20121100871T patent/CY1113307T1/el unknown
- 2012-09-27 HR HRP20120766TT patent/HRP20120766T4/hr unknown
- 2012-10-19 JP JP2012231560A patent/JP2013047250A/ja not_active Withdrawn
-
2015
- 2015-03-18 JP JP2015054167A patent/JP2015134806A/ja not_active Withdrawn
- 2015-04-01 US US14/676,225 patent/US9993435B2/en active Active
-
2017
- 2017-01-24 JP JP2017009975A patent/JP6273387B2/ja active Active
- 2017-02-19 IL IL250674A patent/IL250674B/en active IP Right Grant
- 2017-12-07 US US15/834,436 patent/US10369112B2/en active Active
-
2018
- 2018-01-05 JP JP2018000411A patent/JP2018076362A/ja active Pending
-
2019
- 2019-03-15 US US16/354,683 patent/US10668023B2/en active Active
-
2020
- 2020-02-24 US US16/798,676 patent/US10758491B2/en active Active
- 2020-04-17 US US15/929,260 patent/US10857103B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10857103B2 (en) | Colonic drug delivery formulation | |
| EP2659881B1 (en) | A delayed release drug formulation | |
| BR112021010914A2 (pt) | Formulação de fármaco de liberação retardada que compreende uma camada externa com um polímero enzimaticamente degradável, sua composição e seu método de fabricação | |
| AU2012247013B2 (en) | Colonic drug delivery formulation | |
| BR112021010913A2 (pt) | Formulação para entrega de fármacos colônicos | |
| BR112021010915A2 (pt) | Formulação para entrega de fármacos colônicos | |
| NZ748404A (en) | Colonic drug delivery formulation | |
| NZ748404B2 (en) | Colonic drug delivery formulation | |
| NZ785519B2 (en) | Colonic drug delivery formulation | |
| NZ767015B2 (en) | Colonic drug delivery formulation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B25A | Requested transfer of rights approved |
Owner name: UNIVERSITY COLLEGE LONDON (GB) |
|
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 04/08/2020, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 13/04/2007 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |