CAMPO TÉCNICO DA INVENÇÃO
[1] A presente invenção refere-se aos novos derivados deindoloquinoxalina, aos métodos para prepará-los bem como ao seu uso farmacêutico. Em particular, a invenção refere-se aos novos derivados de indoloquinoxalina e ao uso deles no tratamento de infecções virais.
FUNDAMENTOS DA INVENÇÃO
[2] Como é bem conhecido, vírus são a causa etiológica de muitasdoenças, algumas vezes ameaçadoras da vida, de ambos humanos e animais. Por exemplo, vírus do herpes tais como vírus do herpes simples -1 (HSV-1), vírus do herpes simples 2 (HSV-2), citomegalovírus (CMV), vírus Epstein- Barr (EBV), vírus varicela-zoster (VZV) e vírus do herpes de humano 6 (HHV 6) estão associados com muitas doenças virais comuns.
[3] Infecção por CMV de Humano (HCMV) é uma aflição de vidalonga que pode resultar em morbidez e mortalidade. As patologias associadas com HMCV incluem microcefalia, hepatoesplenomegalia, icterícia, encefalite, infecções de infantes recém-nascidos ou de fetos no útero, e infecções de hospedeiros imunocomprometidos.
[4] Por várias razões, números crescentes de pessoas estão sob orisco de infecção por HCMV, e presentemente em estimativa 80% dos adultos nos Estados Unidos estão infectados com HCMV. Um grupo particularmente suscetível é daqueles de sistema imune enfraquecido, tais como pacientes com AIDS, onde infecção por HCMV causa retinite, gastrite e pneumonite. Também, hepatite e pneumonias induzidas por HCMV são freqüentes e complicações sérias de transplantes de medula óssea.
[5] Patente Européia EP 0.238.459 refere-se às indoloquinoxalinassubstituídas possuindo a fórmula geral
na qual Ri representa hidrogênio ou um ou vários, preferivelmente 1 a 4, substituintes similares ou diferentes nas posições 1-4 e/ou 7-10, selecionados de halogênio, preferivelmente Br, grupo alquila/alcóxi inferior possuindo não mais do que 4 átomos de carbono, grupo trifluorometila, triclorometila; X é um grupo -(CH2)n-R2, no qual R2 representa um resíduo básico contendo nitrogênio tal como NH2, NHR4 ou NRSRÓ, nos quais R4, Rs e RÓ independentemente são alquila inferior ou ciclo-alquila e n é um número inteiro 1 a 4 e R3 representa hidrogênio, grupo alquila inferior / ciclo-alquila possuindo não mais do que 4 átomos de carbono, e os produtos fisiologicamente aceitáveis dos compostos com ácidos e adutos de halogênio, preferivelmente adutos com iodo, monocloreto de iodo ou monobrometo de iodo.
[6] Contudo, está claro que ainda existe uma necessidade urgentede novos medicamentos possuindo eficácia antiviral, em particular contra vírus do herpes tal como HMCV, e um objetivo da presente invenção é proporcionar compostos que atendem esta necessidade.
SUMÁRIO DA INVENÇÃO
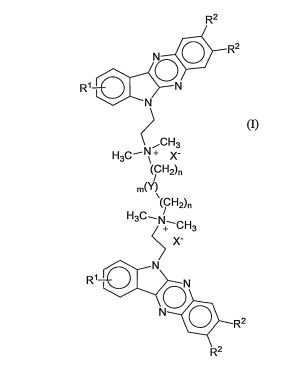
[7] De acordo com um primeiro aspecto a invenção proporcionaum composto de acordo com fórmula (I)
na qualR1 é selecionado de H, F, Cl, Br, CF3, CI-CÓ alcóxi e OH;R2 é selecionado de H e CI-CÓalquila;n é 1-12;m é 0 ou 1 ;eY é selecionado de CH2, NR3, (NR3R4)+X’, O e S;R3 e R4 são independentemente selecionados de H e C1-C4 alquila; eXé selecionado de ânions farmaceuticamente aceitáveis.
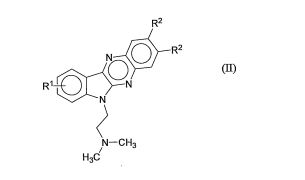
[8] De acordo com outro aspecto, é proporcionado um método depreparar um composto de acordo com fórmula (I), pelo tratamento de um composto de fórmula (II)
com um composto de fórmula (III) L(CH2)n(Y)m(CH2)nL (III)na qualR1, R2, Y, m e n são como definidos acima com respeito à fórmula (I); eL é um grupo de partida;em um solvente ou mistura de solventes.
[9] De acordo com ainda um outro aspecto a invenção proporcionauma composição farmacêutica compreendendo um composto de acordo com fórmula (I) em associação com pelo menos um excipiente farmaceuticamente aceitável.
[10] De acordo com um aspecto da invenção, a composiçãofarmacêutica é uma composição antiviral adequada para o tratamento de uma infecção virai.
[11] Outros aspectos da invenção bem como suas modalidades sãodefinidos nas reivindicações.
DESCRIÇÃO DETALHADA DA INVENÇÃO
[12] Em uma modalidade da presente invenção, R1 em fórmula (I) é selecionado de H, F, Cl, Br, CF3, OCH3 e OH.
[13] Ademais, em uma modalidade da invenção, R2 em fórmula (I)é selecionado de H e C1-C4 alquila, e.g. H e C1-C3 alquila, tais como H e CH3.
[14] O contra-íon X’ em fórmula (I) pode ser qualquer ânion farmaceuticamente aceitável, tal como Cf, Br, metanossulfonato, toluenossulfonato, acetato, citrato e maleato.
[15] O índice n em fórmula (I) pode ser selecionado de qualquervalor entre 1 e 12, tal como 2-10, ou 4-10, e.g. 4-8; ou 1-6, e.g. 1-3.
[16] O composto de fórmula (II), usado na preparação do compostoda invenção, pode ser ele mesmo preparado como geralmente ensinado em EP 0.238.459 bem como em Patente US 4.990.510 que são aqui incorporadas como referências.
[17] O composto de fórmula (III), a saber L(CH2)n(Y)m(CH2)nL,podem ser sintetizados por métodos bem conhecidos pela pessoa experiente na técnica, ou pode ser adquirido de fornecedores químicos.
[18] O grupo de partida L de fórmula (III) pode ser adequadamenteselecionado de e.g. Cl, Br, metanossulfonila e toluenossulfonila, embora a pessoa experiente entenderá que também outros grupos de saída podem ser contemplados.
[19] O sistema de solventes usado deve ser um no qual os reagentessão solúveis nas condições selecionadas de reação e deve adequadamente ser tal para favorecer a reação levando ao produto desejado. Como um exemplo, um ou vários solventes próticos ou apróticos polares podem ser selecionados, tais como acetonitrila, THF, metanol, etanol, isopropanol, acetato de etila acetato de metila. Está bem dentro do conhecimento da pessoa experiente a seleção de tal sistema de solventes bem como condições adequadas de reação.
[20] Os compostos da invenção são úteis como agentes antivirais eassim, de acordo com um aspecto da invenção, uma composição farmacêutica antiviral é proporcionada compreendendo um composto de fórmula (I) e pelo menos um excipiente farmaceuticamente aceitável.
[21] Em uma modalidade da invenção, a composição farmacêuticaé para o tratamento de um vírus selecionado de vírus do herpes, tais como vírus do herpes simples 1 (HSV-1), vírus do herpes simples 2 (HSV-2), citomegalovírus (CMV), vírus Epstein-Barr (EBV), vírus varicela-zoster (VZV) e vírus do herpes de humano 6 (HHV 6).
[22] Em uma modalidade da invenção, o vírus é umcitomegalovírus de humano.
[23] Os excipientes farmaceuticamente aceitáveis podem ser porexemplo, veículos, adjuvantes, agentes de transporte ou diluentes, tais como são bem conhecidos pela pessoa experiente na técnica e como descrito e.g. em Remington: The Science e Practice of Pharmacy, 21th ed., Mack Printing Company, Easton, Pennsylvania (2005). Ademais, é contemplado que a composição farmacêutica da invenção, em adição ao composto de fórmula (I), também pode conter outras substâncias terapeuticamente ativas, e.g. outros agentes antivirais.
[24] A composição farmacêutica da invenção pode ser administradaparenteral ou oralmente e pode ser usada em um tratamento antiviral local ou sistêmico de um vertebrado em necessidade de tal tratamento, e.g uma ave ou um mamífero, tal como um humano ou um animal tal como um animal doméstico ou um animal de fazenda. E contemplado que uma composição farmacêutica da invenção pode ser administrada junta com outras drogas compatíveis tal como outra droga antiviral em terapia de multi-drogas.
[25] Aqui abaixo a invenção é adicionalmente ilustrada porexemplos que contudo não devem ser entendidos como limitantes da invenção, cujo escopo é definido pelas reivindicações. E notado que a numeração de cada um dos sistemas de dois anéis é a mesma que para a fórmula geral da indoloquinoxalina substituída da Patente Européia EP 0.238.459, como mostrada aqui acima.
EXEMPLOS
Preparação de compostos da invenção
[26] Espectros de RMN foram registrados em soluções de DMSO-de na temperatura ambiente e usando o sinal de DMSO-Ó/Ô (1H: δ = 2,50 ppm;13C: δ = 39,5) como padrão interno, em um espectrômetro Bruker DPX 300 (300 MHz). Valores de δ são dados em ppm. Solventes foram de grau analítico e foram usados como recebidos do fornecedor.
EXEMPLO 1
Síntese de dímeros de alquileno
Procedimento geral (escala de 10 mmoles)
[27] B-220 (fórmula II, R' = H, R2= CH3, ou seus derivados), di-halo-alcano e acetonitrila foram aquecidos (sob refluxo ou a 70°C) por 15 h. O sólido assim formado foi isolado por filtração, lavado com acetonitrila e seco.la) R1 =H. R2= CH3, n= 3, m= 0, X = Br
[28] Rendimento: 70%; 'H-RMN δ: 8,34 (d, 1H), 7,94 (m, 2H),7,77 (m, 2H), 7,43 (t, 1H), 4,93 (br. s, 2H), 3,86 (br. s, 2H), 3,54 (br. s, 2H), 3,27 (s, 6H), 2,39 (s, 6H), 1,77 (br, s, 2H), 1,28 (br. s, 2H).lb) R' = H, R2= CH3, n = 5, m = 0, X- = Br
[29] Rendimento: 49 %; 'H-RMN δ: 8,35 (d, 1H), 8,00 (s, 1H),7,92 (d, 1H), 7,80 (m, 2H), 7,45 (t, 1 H), 4,91 (t, 2H), 3,85 (t, 2H), 3,49 (m, 2H), 3,24 (s, 6H), 2,48 (s, 3H), 2,45 (s, 3H), 1,69 (m, 2H), 1,17 (s, 6H).lc) R'=9-Br. R2=CH3, n=3, m=0, X = Br
[30] Rendimento: 73 %; 'H-RMN δ: 8,39 (s, 1H), 8,08-7,81 (m,3H), 7,73 (s, 1H), 5,16 (br. s, 2H), 3,69 (br. s, 2H), 3,43 (br. s, 2H), 3,25 (s, 6H), 2,39 (s, 3H), 2,37 (s, 3H), 1,88 (br. s, 2H), 1,32 (br. s, 2H).Id) RI =9-0, R2=H, n=3, m=0, X = Br
[31] 13C-RMN DMSO-Ó/6δ.- 21,6 (t), 25,2 (t), 35,3 (t), 50,8 (q), 59,0(t), 63,3 (t), 112,6 (d), 120,4 (s), 121,6 (d), 126,1 (s), 126,8 (d), 127,5 (d), 129,3 (d), 129,8 (d), 131,1 (d), 138,6 (s), 139,0 (s), 139,8 (s), 142,0 (s), 144,9 (s).le) R'=H. R2 =H, n = I , m = I, Y = CH2, X = Br
[32] 13C-RMN DMSO-ÓZ6δ: 17,0 (t), 35,0 (t), 50,9 (q), 59,9 (t), 60,5(t), 110,8 (d), 119,0 (s), 121,7 (d), 122,4 (d), 126,5 (d), 127,5 (d), 129,2 (d)*, 131,5 (d), 138,9 (s), 139,5 (s), 139,7 (s) 143,5 (s), 144,7 (s).* 1 sinal para dois carbonosIf) R1 = H, R2 = H, n = 3, m = 0, X = Br
[33] l3C-RMN DMSO-A δ: 21,7 (t), 25,4 (t), 35,0 (t), 50,8 (q),59,2(1), 63,2 (t), 110,7 (d), 119,1 (s), 121,8 (d), 122,5 (d), 126,6 (d), 127,4 (d), 129,2 (d), 129,3 (d), 131,6 (d), 139,0 (s), 139,6 (s), 139,8 (s), 143,6 (s), 144,8 (s).
EXEMPLO 2
Síntese de dímeros de éter
Procedimento geral (escala de 10 mmoles)
[34] B-220 (ou seus derivados), di-halo-alcano e acetonitrila foramaquecidos sob refluxo por 20 h. O sólido assim formado foi isolado por filtração, lavado com acetonitrila e seco.2a) R1 = H, R2= CH3, n = 2, Y = O, m = 1, X-= Br
[35] Rendimento: 58 %; 'H-RMN δ: 8,22 (d, 1H), 7,84 (s, 1H),7,72 (m, 2H), 7,59 (s, 1H), 7,47 (d, 1H), 7,38 (t, 1H), 7,08 (d, 1H), 4,85 (t, 2H), 4,09 (br. s, 2H), 3,93 (m, 4H), 3,29 (s, 6H), 2,35 (s, 3H), 2,26 (s, 3H), 2,24 (s, 3H).2b) R1 = 9-Br, R2 = CH3, n = 2. Y = O. ni = 1. X = BrRendimento: 91 %; 1H-RMN δ: 8,02 (d, 1H), 7,77-7,66 (m, 3H), 7,49 (s, 1H), 7,45 (d, 2H), 7,07 (d, 2H), 4,78 (t, 2H), 4,11 (br. s, 2H), 3,95-3,90 (m, 4H), 3,27 (s, 6H), 2,31 (s, 3H), 2,26 (s, 3H), 2,18 (s, 3H).
Teste biológico
[36] Testes de atividade antiviral contra citomegalovírus dehumano como descrito aqui abaixo foram realizados em um composto de acordo com a invenção, a saber o composto la de EXEMPLO 1. O composto de referência chamado B-220 é 2,3-dimetil-6-(N,N-dimetil-amino-etil)-6H- indolo(2,3-b)quinoxalina, descrito em Patente Européia 0.238.459.
Teste de efeitos inibitórios sobre infecção viral
[37] Com o objetivo de avaliar se seleção de proteínas viraisestruturais seria tão eficiente quanto seleção de transcrição virai, um ensaio de placa modificado foi configurado, no qual um dos novos agentes antivirais foi comparado com agentes antivirais já conhecidos que inibem quer transcrição de HCMV [GCV (Cymevene, Roche) e PFA (Foscavir, AstraZeneca)] quer infecção [IVIg (WIG CP, Biotest Pharma), um anticorpo].
[38] Em um experimento de 0 dpi (dias após infecção) os agentesantivirais e TB40/E foram simultaneamente adicionados, indicando deste modo quão bem os agentes inibem infecção. Os resultados deste experimento foram obtidos por comparação da quantidade de células infectadas das cavidades tratadas com aquela dos controles positivos, calculando assim a inibição da infecção alcançada pelos agentes em questão. O experimento foi repetido com a cepa AD-169 de HCMV e com um isolado clínico, respectivamente, com essencialmente os mesmos resultados.
[39] O efeito inibitório das substâncias testadas é mostrado emTabela 1, como inibição % de infecção. Estes dados são os resultados dos ensaios de placa usando cepa ADI69 e TB 40 de fibroblastos de pulmão de humano infectado com HCMV.
[40] Os resultados do teste indicam que os compostos da invençãopossuem excelente efeito inibitório sobre infecção viral.
Teste de inibição de Montagem e Egresso de HCMV
[41] Células de fibroblasto de pulmão de humano infectadas(células HL) foram tratadas com o agente antiviral B-220 e com outras substâncias de referência, como mostrado em Tabela 2, e com o composto la da invenção com o objetivo de avaliar o efeito dos compostos da invenção sobre infecção, montagem e egresso de HCMV.
[42] Os agentes antivirais foram adicionados a 3 ou 5 dias após ainfecção (dpi) nestes experimentos e deixados em cultura até 7 dpi. Depois o sobrenadante e as células trituradas foram transferidos para novas culturas de célula durante a noite e subseqüentemente corados para expressão de IE. Os resultados indicam quão bem as substâncias impedem a montagem e o egresso virai. Mais especificamente, a 3 dpi os capsídeos virais estão sendo montados em sua maioria no núcleo enquanto que a 5 dpi estão principalmente recebendo sua tegumentação no citoplasma e alguns possuem seu envelope secundário.
[43] Na Tabela 2 é mostrado o efeito inibitório das substânciasantivirais usando o sistema de ensaio de placa modificado. Várias substâncias mostraram inibição de 100% de expressão de IE como medida por coloração de TE e formação de capsídeo não foi observada por exame por microscopia eletrônica. O composto da invenção chamado de la mostrou resultados extremamente bons obtidos por Ganciclovir.
[44] Sem o desejo de se ligar a qualquer teoria de mecanismo deação dos compostos da invenção, é notado que o composto da invenção la mostra uma inibição muito clara de expressão de IE. Ademais, dados de microscópio eletrônico indicam a falha da montagem do vírus. De fato, a técnica de análise de imagem usada para identificar e quantificar partículas intermediárias estáveis de HCMV indicaram falha de ligação de proteína de tegumento na capsídeo. Juntos estes dados mostram um potencial alto para o uso dos compostos da invenção em terapia antiviral. Também, pelo uso dos compostos da invenção em combinação com pelo menos um outro agente antiviralmente ativo, tal como terapia de multi-drogas, um efeito sinérgico é esperado e o risco de aquisição de resistência à droga pode ser reduzido ou evitado.
Toxicidade
[45] Os compostos da invenção não mostraram qualquer toxicidadeconforme ensaiados por coloração de iodeto de propídeo de culturas celulares de fibroblastos de pulmão de humano não infectados e infectados. Uma concentração de compostos la 10 vezes a usada nos experimentos não mostrou toxicidade durante o período de tempo de 0-7 dpi. As concentrações de compostos usada nos experimentos virais estiveram no nível de uM. Toxicidade celular para B-220 tem sido mostrada para concentrações acima de 100 M.
Materiais e Métodos
Cultura Celular
[46] Os fibroblastos de pulmão de humano, células HL (MRC-5),usados nestes experimentos foram incubados a 37°C e CO2 5% em uma solução de MEM com Earle's e L-glutamina (de GIBCO) dentro da qual foram adicionados 10% de Soro Fetal Bovino (FCS) e 1% de Penicilina e Estreptomicina (Peste).
[47] Quando o experimento começou as células HL estavam sendomantidas em um frasco de cultura de células Falcon de 175 cm2. Tripsina e EDTA foram usados para soltar as células do frasco de cultura de células quando foram transferidas para multicavidades de 48-cavidades (Becton Dickinson) para infecção e incubação com os agentes antivirais.
[48] As células foram incubadas até que confluência de 50% fossealcançada sob as mesmas condições acima e foram usadas até a 26a passagem.
Infecção de Células com HCMV
[49] As células HL foram infectadas com HCMV, cepa virai TB40/E [um isolado clínico endotelialmente adaptado (URI814) gentilmente proporcionado por Prof. G. Jahn] e cepa viral AD-169, respectivamente, em uma multiplicidade de infecção (MOI) de 0,02 e incubadas até 3 ou 5 dias após a infecção (dpi) a 37°C e CO2 5% no mesmo meio que acima. Algumas células (para o experimento Odpi) foram simultaneamente expostas aos agentes anti virais (veja abaixo). Os controles negativos foram deixados não infectados.
Exposição das células aos inibidores e agentes antivirais
[50] O meio existente (nos experimentos de 3 e 5 dpi) foisubstituído e um meio novo adicionado, com inibidores e agentes antivirais em concentrações diferentes. Contudo isto foi feito simultaneamente à infecção no experimento Odpi e deixado incubar até 1 dpi. O meio contendo IVIg foi incubado por uma hora com o vírus sobre gelo antes de ser adicionado nas células.
Ensaio de Placa Modificado
[51] Nos experimentos de 3- e 5 dpi 0 sobrenadante de célulasMRC-5 foi transferido para células infectadas para avaliar a quantidade de vírus excretado. As células restantes receberam meio novo e foram trituradas com mármores de vidro por agitação das multicavidades em um IKA-VibraX’ VXR a 300 vascolejamentos pm por lOmin. Depois o fragmento celular foi transferido para células não infectadas para possibilitar a avaliação da quantidade de partículas virais intracelulares infecciosas.
[52] Após deixar as partículas virais infectarem as células novaspor aproximadamente uma hora o meio foi mudado, removendo assim por lavagem o fragmento celular. Nos experimentos Odpi as células foram imediatamente fixadas 1 dpi (de acordo com o procedimento explicado abaixo).
[53] Controles positivos (células infectadas não tratadas) e controlesnegativos (células na infectadas não tratadas), foram tratados, como acima.
Coloração Imunofluorescente das Células
[54] As novas células HL (nos experimentos de 3 e 5 dpi) foramfixadas no dia seguinte com para-formaldeído (PFA) 3% por 15 min na temperatura ambiente (RT). Para tornar as células permeáveis, Triton X 0,3% em Solução Salina Tampão Fosfato (PBS) foi usado por incubação de 15 min na RT seguido por bloqueio do fundo com bloqueio de fundo de DAKO por 20 min na RT, com uma quantidade apenas suficientemente grande para cobrir a superfície inteira. Depois de todas as multicavidades terem sido incubadas com anticorpos primários (camundongo), diluídos para 1:100, contra antígeno precoce imediato (IEA, Antígeno) por 45 min a 8°C. Subseqüentemente, as células foram incubadas com anticorpos secundários, de coelho anti FITC de camundongo (Dako Cytomation), diluídos para 1:100, por 45 min a 8°C e simultaneamente coradas com DAPI (Sigma), diluído para 1:250. DAPI é uma substância química que cora o núcleo das células.
[55] Os controles negativos e positivos, de ambos os tipos decélula, foram tratados, respectivamente, como acima.
Análise por Microscopia de Imunofluorescência
[56] As células foram analisadas por microscopia eletrônica usandoum Nikon Eclipse TE 2000-U. A quantidade de células expressando IEA, em duas partes diferentes da cavidade, foi contada a olho nu e comparada com a quantidade total de células (indicadas com DAPI), naquelas mesmas partes. Estes valores foram usados para estimar a percentagem de células infectadas em cada cavidade da qual foi calculada a quantidade de inibição alcançada pelas substâncias diferentes. Este método de cálculo de percentagem de células infectadas em duas partes de uma cavidade e então sua aplicação na cavidade inteira foi escolhido porque a quantidade total de células em uma cavidade seria impossível de contar manualmente.