BR112012013325B1 - Composição farmacêutica e uso - Google Patents
Composição farmacêutica e uso Download PDFInfo
- Publication number
- BR112012013325B1 BR112012013325B1 BR112012013325-0A BR112012013325A BR112012013325B1 BR 112012013325 B1 BR112012013325 B1 BR 112012013325B1 BR 112012013325 A BR112012013325 A BR 112012013325A BR 112012013325 B1 BR112012013325 B1 BR 112012013325B1
- Authority
- BR
- Brazil
- Prior art keywords
- transnorsetraline
- mannitol
- hydrochloride
- disorder
- mixture
- Prior art date
Links
Images









Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/12—Drugs for genital or sexual disorders; Contraceptives for climacteric disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/33—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C211/39—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton
- C07C211/41—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton containing condensed ring systems
- C07C211/42—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton containing condensed ring systems with six-membered aromatic rings being part of the condensed ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Abstract
COMPOSIÇÃO FARMACÊUTICA, SAL CRISTALINO E MÉTODO DE TRATAMENTO, PREVENÇÃO OU CONTROLE DE UM DISTÚRBIO NEUROLÓGICO. São descritas na presente invenção, composições farmacêuticas compreendendo transnorsertralina, sais e formas polimórficas da transnorsertralina, métodos para preparo das composições, e métodos para seu uso no tratamento de distúrbios do sistema nervoso central, incluindo depressão.
Description
[0001] Esta aplicação reivindica o benefício do Pedido dePatente Provisório U.S. No. 61/266,64, depositado em 04 de dezembro de 2009, a totalidade deste é aqui incorporada por referência.
[0002] Estão fornecidas aqui composições farmacêuticascaracterizadas por compreender transnorsertralina, sais e formas polimórficas de transnorsertralina, os métodos de fabricação das composições e métodos para a sua utilização no tratamento de doenças do Sistema Nervoso Central (SNC), incluindo depressão.
[0003] A transnorsertralina, isto é, (1R,4S)-trans-4-(3,4-diclorofenil)-1,2,3,4-tetrahidro-1-naftalenamina e (1S,4R)- trans-4-(3,4-diclorofenil)-1,2,3,4-tetrahidro-1-naftalenaminasão descritas, por exemplo, na patente U.S. No. 7,087,785 B2 ("A patente '785’"; aqui incorporada por referência na sua totalidade), têm as seguintes estruturas químicas, respectivamente:
[0004] Usos da transnorsertralina no tratamento, prevenção ou controle de distúrbios afetivos e outros vários distúrbios do SNC são também revelados na patente '785. Esses distúrbios incluem, mas não estão limitados a, depressão, distúrbios de humor, distúrbios de ansiedade, distúrbios de comportamento, distúrbios alimentares, distúrbios de abuso de substâncias e distúrbios da função sexual.2.2 Sais e formas polimórficas
[0005] Se cristalino ou amorfo, potenciais formas sólidasde uma composição farmacêutica incluem sólidos de um único componente e de múltiplos componentes. Sólidos de um único componente consistem essencialmente no composto farmacêutico, na ausência de outros compostos. Variedade de materiais cristalinos de um único componente pode potencialmente surgir, por exemplo, a partir do fenômeno de polimorfismo, em que arranjos múltiplos tridimensionais existem para um composto farmacêutico particular (ver, por exemplo, S.R. Byrn et al., Solid State Chemistry de Drugs, (1999) SSCI, West Lafayette).
[0006] Formas sólidas, tais como sais, formas cristalinas,por exemplo, formas polimórficas de um composto, são conhecidas na arte farmacêutica para afetar, por exemplo, a solubilidade, a estabilidade, a fluidez, fractabilidade e compressibilidade do composto, bem como a segurança e eficácia de medicamentos baseados no composto, (ver, por exemplo, Knapman, K. Modern Drug Discoveries, 2000:53).
[0007] A importância de estudar polimorfos foi salientadapelo caso do ritonavir, um inibidor da protease do HIV que foi formulado como cápsulas de gelatina maleáveis. Cerca de dois anos após o produto ter sido lançado, a precipitação inesperada de um novo polimorfo menos solúvel na formulação necessitou da retirada do produto do mercado, até que uma formulação mais consistente pudesse ser desenvolvida (ver S. R. Chemburkar et al., Org. Process Res. Dev., (2000) 4:413417). Assim, a preparação de formas sólidas é de grande importância no desenvolvimento de um composto farmacêutico seguro, eficaz, estável e comercializável.
[0008] Novos sais e formas polimórficas de transnorsertralina podem promover o desenvolvimento de formulações para o tratamento, prevenção ou controle de doenças do SNC.a. Tratamento de Doenças Neurológicas
[0009] A serotonina, isto é, 5-HT, é conhecida por desempenhar um papel importante no tratamento de vários distúrbios do SNC. Entre outros, receptores 5-HT1A (serotonina 1A) fornecem um mecanismo importante para controlar a liberação de 5-HT no cérebro. Esses receptores estão localizados pré-sinapticamente nos núcleos rafe onde funcionam como autoreceptores para inibir a taxa de descarga de neurônios 5-HT. Receptores 5-HT1A são também localizados pós- sinapticamente em regiões corticolímbicas onde eles também reduzem a atividade de descarga de neurônios 5-HT. No início do tratamento com inibidores seletivos de recaptação de serotonina (SSRIs) ou inibidores de recaptação de serotonina norepinefrina (IRSNs), os autoreceptores 5-HT1A são ativados por 5-HT, levando a uma redução de descarga de 5-HT neuronal. Como os tratamentos SSRI ou IRSN continuam, no entanto, autoreceptores 5-HT1A tornam-se insensíveis e a atividade de descarga é restaurada.
[0010] Acredita-se que esta mudança adaptativa contribue, pelo menos em parte, com o atraso na eficácia de ISRS e IRSNs no tratamento de vários distúrbios neurológicos.
[0011] Portanto, existe uma necessidade para o tratamento, prevenção ou controle de várias desordens neurológicas, em que a dessensibilização dos receptores 5-HT pode ser minimizada e o aumento da descarga 5-HT neuronal pode ser mantida.
[0012] Estão aqui fornecidas composições farmacêuticas caracterizadas por compreender transnorsertralina, sais e formas polimórficas de transnorsertralina, os métodos de fabricação de composições com os sais e as formas polimórficas e métodos para a sua utilização no tratamento de doenças do SNC, incluindo depressão.
[0013] Em uma forma de concretização, aqui proporcionada, estão composições farmacêuticas estáveis e/ou formulações de transnorsertralina ou um sal farmaceuticamente aceitável ou solvato.
[0014] Em outra forma de concretização, aqui proporcionada, está sal de transnorsertralina selecionado entre o grupo consistindo de cloridrato, acetato, L-malato, besilato, benzoato, tosilato, fumarato, bromidrato, maleato, citrato, fosfato, succinato, L-tartarato, D-tartarato, S-mandelato e piroglutamato.
[0015] Em uma forma de concretização, o sal é o sal cloridrato. Em uma forma de concretização, o sal de cloridrato de transnorsertralina é um sólido anidro. Em outra forma de concretização, o sal de cloridrato de transnorsertralina existe como um mono-hidrato.
[0016] Em uma forma de concretização, o cloridrato de transnorsertralina é cloridrato de (1R,4S)-transnorsertralina, isto é, cloridrato de (1R,4S)-trans-4-(3,4-diclorofenil)- 1,2,3,4-tetrahidro-1-naftalenamina. Em outra concretização, o cloridrato de transnorsertralina é cloridrato de (1S,4R)- transnorsertralina, isto é, cloridrato de (1S,4R)-trans-4- (3,4-diclorofenil)-1,2,3,4-tetrahidro-1-naftalenamina.
[0017] Também aqui proporcionados estão métodos detratamento, prevenção ou controle de distúrbios neurológicos caracterizados por compreender a administração a um sujeito (por exemplo, o paciente) de uma formulação, sal ou polimorfo de transnorsertralina como aqui descrito. Distúrbiosneurológicos que podem ser tratados, prevenidos ou gerenciados através dos métodos aqui descritos, descritos em detalhe em outro local.
[0018] Em algumas realizações, a formulação, o sal oupolimorfo de transnorsertralina é administrado em combinação com um ou mais agentes terapêuticos adicionais ou sais farmaceuticamente aceitáveis, solvatos ou estereoisômeros do mesmo.
[0019] Fig. 1A ilustra o hábito forma cristalina docloridrato de transnorsertralina anidro.
[0020] Fig. 1B ilustra a forma cristalina do cloridrato detransnorsertralina monohidrato.
[0021] Fig. 2 ilustra o padrão de XRPD calculado docloridrato de transnorstertralina anidro.
[0022] Fig. 3 ilustra o padrão de XRPD experimental decloridrato de transnorstertralina anidro.
[0023] Fig. 4 ilustra o diagrama de ORTEP do cloridrato detransnorstertralina anidro.
[0024] Fig. 5 ilustra o padrão de XRPD calculado docloridrato de transnorstertralina monohidrato.
[0025] Fig. 6 ilustra o padrão de XRPD experimental docloridrato de transnorstertralina monohidrato.
[0026] Fig. 7 ilustra o diagrama de ORTEP do cloridrato detransnorstertralina monohidrato.
[0027] Fig. 8 é um cromatograma de HPLC típico docloridrato de transnorsertralina de comprimidos de 1 mg do exemplo 6.27.
[0028] Fig. 9 é uma sobreposição do cromatogramas de HPLC apartir dos estudos de estabilidade de exemplo 6.31.
[0029] Estão aqui fornecidas composições farmacêuticascaracterizadas por compreender transnorsertralina, sais e formas polimórficas de transnorsertralina, os métodos de fabricação de composições com os sais e as formas polimórficas e métodos para a sua utilização no tratamento de doenças do SNC, incluindo depressão.
[0030] Em uma forma de concretização, aqui fornecida, estãocomposições farmacêuticas estáveis e/ou formulações de transnorsertralina ou um sal farmaceuticamente aceitável ou solvato.
[0031] Em uma forma de concretização, as composiçõesfarmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos do que cerca de 3% em peso de um composto de fórmula (II):
[0032]Em outra forma de concretização, as composições farmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos do que cerca de 1,5% ou menos do que cerca de 1% em peso de um composto de fórmula (II).
[0033] Em outra forma de concretização, as composiçõesfarmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos do que cerca de 4% em peso de compostos de fórmula (III):
[0034] Em outra forma de concretização, as composições farmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos do que cerca de 2% ou menos do que cerca de 1% em peso de compostos de fórmula (III).
[0035] Em outra forma de concretização, as composições farmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos do que cerca de 3% em peso de um composto de fórmula (II) e menos do que cerca de 4% em peso dos compostos de fórmula (III).
[0036] Em outra forma de concretização, as composições farmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos do que cerca de 1,5% em peso de um composto de fórmula (II) e menos do que cerca de 2% em peso dos compostos de fórmula (III).
[0037] Em outra forma de concretização, as composiçõesfarmacêuticas estáveis e/ou formulações de transnorsertralina compreendem menos de menos de cerca de 1%, em peso, de cada um dos compostos de fórmulas (II) e (III).
[0038] Em certas concretizações, sem estar vinculado aqualquer teoria particular, acredita-se que o composto de fórmula (II) são adutos de transnorsertralina formados pela decomposição de transnorsertalina em uma forma de dosagem farmacêutica, por exemplo, um comprimido, na presença de manose.
[0039] Em certas concretizações, sem estar vinculado aqualquer teoria particular, acredita-se que os compostos de fórmula (III) são produtos de decomposição oxidativa de transnorsertralina formados pela decomposição detransnorsertalina em uma forma de dosagem farmacêutica, por exemplo, um comprimido, na presença de fosfato dicálcico (por exemplo, A-TAB).
[0040] Em uma forma de concretizações, as composiçõesfarmacêuticas estáveis fornecidas aqui estão em uma forma de dosagem de liberação imediata.
[0041] Em outra forma de concretizações, as composiçõesfarmacêuticas estáveis fornecidas aqui estão em uma forma de dosagem de liberação controlada.
[0042] Em uma forma de concretizações, a composiçãofarmacêutica é caracterizada por compreendertransnorsertralina ou um sal farmaceuticamente aceitável ou solvato destes e manitol, xilitol ou uma combinação destes. Em uma forma de concretização, a composição farmacêutica é caracterizada por compreender transnorsertralina ou um sal farmaceuticamente aceitável ou solvato destes e pelo menos cerca de 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97% ou 98% em peso de manitol ou xilitol.
[0043] Em outra forma de concretização, a composição farmacêutica é caracterizada por compreender transnorsertralina ou um sal farmaceuticamente aceitável ou solvato destes e manitol. Em uma forma de concretização, a composição farmacêutica é caracterizada por compreender transnorsertralina ou um sal farmaceuticamente aceitável ou solvato destes e pelo menos cerca de 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97% ou 98% em peso de manitol.
[0044] Em uma forma de concretizações, aqui fornecida está uma composição farmacêutica estável que é caracterizada por compreender transnorsertralina ou um sal farmaceuticamente aceitável ou solvato deste e manitol, em que a composição farmacêutica estável contém menos do que 1 μg a cerca de 100 μg de manose por 100 mg de manitol. Em outra forma de concretizações, a composição farmacêutica estável contém menos do que 10 μg a cerca de 100 μg de manose por 100 mg de manitol. Em outra forma de concretizações, a composição farmacêutica estável contém menos do que 1 μg a cerca de 50 μg de manose por 100 mg de manitol. Em outra forma de concretizações, a composição farmacêutica estável contém menos do que cerca de 1 μg a cerca de 20 μg de manose por 100 mg de manitol. Em outra forma de concretização, a composição farmacêutica estável contém menos do que cerca de 10 μg ou menos do que cerca de 5 μg de manose por 100 mg de manitol.
[0045] Em uma forma de concretizações, aqui fornecida,estão composições farmacêuticas que são estáveis durante pelomenos cerca de 5 a 30 semanas. Em outra forma de concretização, as composições são estáveis a uma temperatura de entre 20 °C a 50 °C durante pelo menos 5 a 30 semanas. Em outra forma de concretizações, as composições são estáveis a uma temperatura de entre cerca de 20 °C a 50 °C durante pelo menos cerca de 5 a 30 semanas a uma umidade relativa de cerca de 35% e 85%.
[0046] Em outra forma de concretizações, quando acomposição farmacêutica compreende manitol, a combinação de excipientes na composição, fora o ingrediente ativo, contém ou gera após armazenagem durante cerca de 5 a 30 semanas, a uma temperatura de cerca de 20°C até 50°C e a uma umidade relativa de cerca de 35% e 85% em uma embalagem selada, inferior a cerca de 0,05% de manose em relação ao peso de manitol. Em outra forma de concretização, o referido armazenamento é por cerca de 24 semanas. Em outra forma de concretização, a referida temperatura é de cerca de 30°C. Em outra forma de concretização, a referida temperatura é de cerca de 40°C. Em outra forma de concretização, a referida umidade relativa é de cerca de 65%. Em outra forma de concretização, a referida umidade relativa é de cerca de 75%. Em outra forma de concretização, a composição farmacêutica contém ou gera menos do que cerca de 0,02% de manose ou menos do que cerca de 0,01% de manose em relação ao peso de manitol.
[0047] Em uma forma de concretização, a composição farmacêutica é caracretizada por compreender ainda estearato de magnésio, estearato de cálcio, estearato de zinco ou ácido esteárico. Em uma forma de concretização, a composição farmacêutica é caracretizada por compreender ainda pelo menos 0,1%, 0,2%, 0,5%, 0,75%, 1%, 1,5%, 2%, 3% ou 5% em peso de estearato de magnésio, estearato de cálcio, estearato de zinco ou ácido esteárico.
[0048] Em uma forma de concretização, a composição farmacêutica é caracretizada por compreender ainda talco, caulim ou a bentonita. Em uma forma de concretização, a composição farmacêutica é caracretizada por compreender ainda pelo menos 0,5%, 1%, 2%, 3%, 5%, 10%, 15%, 20%, 30% ou 40% em peso de Talco, caulim ou a bentonita.
[0049] Em outra forma de concretização, aqui fornecida, está uma composição farmacêutica caracretizada por compreender transnorsertralina ou um sal farmaceuticamente aceitável ou solvato deste, manitol, estearato de magnésio, Talco e glicolato de amido de sódio.
[0050] Em outra forma de concretização, aqui fornecida, está uma composição farmacêutica caracretizada por compreender transnorsertralina ou um sal farmaceuticamente aceitável ou solvato deste, 10 a 98% em peso de manitol, estearato de magnésio, talco e glicolato de amido de sódio.
[0051] Em outra forma de concretização, a composição farmacêutica é caracretizada por compreender 50 a 98% em peso de manitol.
[0052] Em outra forma de concretização, a composição farmacêutica é caracretizada por compreender 80 a 98% em peso de manitol.
[0053] Em outra forma de concretização, a composição farmacêutica é caracretizada por compreender 85 a 98% em peso de manitol.
[0054] Em outra forma de concretização, a composição farmacêutica é caracretizada por compreender 86 e 98% em peso de manitol.
[0055] Em uma forma de concretização, a composição farmacêutica é uma cápsula compreendendo transnorsertralina ou um sal farmaceuticamente aceitável ou solvato deste, manitol, talco, glicolato de amido de sódio e estearato de magnésio em um invólucro da cápsula. A cápsula pode ser preparada a uma concentração de 0,5, 1,0 ou 2,0 mg de transnorsertralina. A cápsula pode ser preparada em cápsula peso de enchimento de 100, 150, 200 ou 300 mg.
[0056] Em outra forma de concretização, a composição farmacêutica é um comprimido compreendendo transnorsertralina ou um sal farmaceuticamente aceitável ou solvato deste, manitol, talco, glicolato de amido de sódio e estearato de magnésio. O comprimido pode ser revestido ou não revestido. O comprimido pode ser preparado a uma concentração de 0,5, 1,0 ou 2,0 mg de transnorsertralina. O comprimido pode ser preparado como um comprimido 100, 150, 200 ou 300 mg de peso.
[0057] Em certas concretizações, o manitol utilizado na preparação das composições aqui fornecidas é Pearlitol 160C.
[0058] Em certas concretizações, o glicolato de amido desódio usado na preparação das composições aqui fornecida é Primojel.
[0059] Também é fornecido um método de determinação daadequabilidade de um excipiente ou combinação de excipientes para uso em uma formulação de transnorsertralina aqui descrita. Em uma forma de concretização, o método é caracterizado por compreender a determinação do nível de manose em uma amostra de manitol ou uma formulação contendo manitol aqui fornecida, em que um nível de manose em manitol de menos do que ou igual a cerca de 0,1% em peso indica aptidão para utilização em uma formulação transnorsertralina estável.
[0060] Em outra concretização, um nível de manose emmanitol menor que ou igual a cerca de 0,05% em peso, indica aptidão para utilização em uma formulação detransnorsertralina estável.
[0061] Em outra concretização, um nível de manose emmanitol menor que ou igual a cerca de 0,02% em peso, indica aptidão para utilização em uma formulação detransnorsertralina estável.
[0062] Em outra concretização, um nível de manose emmanitol menor que ou igual a cerca de 0,01% em peso, indica aptidão para utilização em uma formulação detransnorsertralina estável.
[0063] Em uma forma de concretização, o método dedeterminação do nível de manose em manitol ou uma formulação contendo manitol, aqui fornecida, compreende a utilização de um instrumento de HPLC (cromatografia líquida de alta pressão). Em outra concretização, o instrumento de HPLC é compreende um detector de aerossol corona carregado.
[0064] Em outra concretização, o método de determinação do nível de manose em manitol ou uma formulação contendo manitol aqui fornecido compreende a utilização de íons de cromatografia (IC).
[0065] Também é descrito na presente invenção um sal de transnorsertralina selecionado entre o grupo consistindo de cloridrato, acetato, L-malato, besilato, benzoato, tosilato, fumarato, bromidrato, maleato, citrato, fosfato, succinato, L- tartarato, D-tartarato, S- mandelato e piroglutamato.
[0066] Em uma forma de concretização, o sal é o sal cloridrato. Em uma forma de concretização, o sal cloridrato de transnorsertralina é um sólido anidro. Em outra forma de concretização, o sal cloridrato de transnorsertralina existe como um mono-hidrato.
[0067] Em uma forma de concretização, o cloridrato de transnorsertralina é cloridrato de (1R,4S)-transnorsertralina, isto é, cloridrato de (1R,4S)-trans-4-(3,4-diclorofenil)- 1,2,3,4-tetrahidro-1-naftalenamina. Em outra concretização, o cloridrato de transnorsertralina é cloridrato de (1S,4R)- transnorsertralina, isto é, cloridrato de (1S,4R)-trans-4- (3,4-diclorofenil)-1,2,3,4-tetrahidro-1-naftalenamina.
[0068] Em uma forma de concretização, o sal cloridrato de transnorsertralina é essencialmente livre de água.
[0069] Em uma forma de concretização, o sal cloridrato de transnorsertralina é o anidrato cristalino.
[0070] Em uma forma de concretização, o sal cloridrato de anidrato transnorsertralina tem um padrão de difração raio-X do pó que compreende picos a cerca de 14.9, 17.8, 19.2, 23.3, 24.6 e 25.2 graus 2θ. Em outra forma de concretização, o sal cloridrato de anidrato de transnorsertralina tem um padrão de difração raio-X do pó que compreende ainda picos de cerca de 5.0 e 21.8 graus 2θ.
[0071] Em uma forma de concretização, o sal cloridrato de anidrato transnorsertralina tem um padrão de difração de raios-X do pó calculado que compreende picos de cerca de 5.0, 15.0, 18.0, 19.5, 22.0 23.5, 24.8 e 25.4 graus 2θ, com base em dados coletados a cerca de -100,15 °C (173K) em um único cristal.
[0072] Em uma forma de concretização, o sal cloridrato de anidrato transnorsertralina tem as seguintes dimensões aproximadas de células unitárias:a=16.8 Â, b=5.2 Â, c=19.1 Â, α=90.0°, β=113.1° e Y=90.0°.
[0073] Em outra forma de concretização, o sal cloridrato de anidrato de transnorsertralina tem as seguintes dimensões aproximadas de células unitárias, quando medidas a cerca de - 100,15 °C (173K):a=16.83Â, b=5.23Â, c=19.06Â, α=90.00°, β=113.10° e Y=90.00°.
[0074] Em outra forma de concretização, as dimensões aproximadas das células unitárias são:a=16.834Â, b=5.226Â, c=19.059Â, α=90.00°, β=113.10° e Y=90.00°.
[0075] Em uma forma de concretização, o sal cloridrato de anidrato transnorsertralina tem o espaço de grupo C2 (N°5).
[0076] Em uma forma de concretização, o sal cloridrato de anidrato transnorsertralina tem uma célula unitária que contém quatro cloridratos transnorsertralina (Z=4).
[0077] Em uma forma de concretização, o sal cloridrato de anidrato transnorsertralina tem uma densidade de cerca de 1,4 gcm-3.
[0078] Em uma forma de concretização, o sal cloridrato de transnorsertralina é um mono-hidrato.
[0079] Em outra forma de concretização, o sal cloridrato de mono-hidrato de transnorsertralina é cristalino.
[0080] Em uma forma de concretização, o sal cloridrato de mono-hidrato de transnorsertralina tem um padrão de difração de raio-X do pó que compreende picos a cerca de 12.1, 13.0, 16.8, 17.8, 20.4, 23.4, 24.2 e 27.1 graus 2θ. Em outra forma de concretização, o sal cloridrato de mono-hidrato de transnorsertralina tem um padrão de difração de raio-X do pó que compreende ainda picos a cerca de 20.9, 21.1 e 26.2 graus 2θ.
[0081] Em uma forma de concretização, o sal cloridrato de mono-hidrato de transnorsertralina tem um padrão de difração de raio-X do pó calculado que compreende picos a cerca de 12.1, 13.1, 16.9, 17.9, 20.5, 21.0, 21.3, 23.6, 24.3, 26.3 e 27.2 graus 2θ, com base em dados coletados a cerca de -123,15 °C (150K) em um único cristal.
[0082] Em uma forma de concretização, o sal cloridrato de mono-hidrato de transnorsertralina tem as seguintes dimensões aproximadas de células unitárias:a=7.3Â, b=7.6 Â, c=15.3 Â, a=90.0°, β=90.1° e Y=90.0°.
[0083] Em outra forma de concretização, o sal cloridrato de mono-hidrato de transnorsertralina tem as seguintes dimensões aproximadas de células unitárias, quando medida a cerca de - 123,15 °C (150K): a=7.30Â, b=7.56Â, c=15.29Â, α=90.00°, β=90.09° e Y=90.00°.
[0084] Em outra forma de concretização, as dimensõesaproximadas das células unitárias são:a=7.296Â, b=7.557Â, c=15.287Â, a=90.00°, β=90.09° e Y=90.00°.
[0085] Em uma forma de concretização, o sal cloridrato demono-hidrato de transnorsertralina tem o grupo de espaço P21 (N°4).
[0086] Em uma forma de concretização, o sal cloridrato demono-hidrato de transnorsertralina tem uma célula unitária que contém dois cloridratos transnorsertralina (Z=2).
[0087] Em uma forma de concretização, o sal cloridrato demono-hidrato de transnorsertralina tem uma densidade de cerca de 1,4 gcm-3.
[0088] Também é descrito na presente invençao um método detratamento, prevenção ou controle de distúrbio neurológico, compreendendo a administração a um paciente de uma quantidade terapeuticamente ou profilaticamente eficaz de um cloridrato de transnorsertralina ou um solvato farmaceuticamente aceitável ou estereoisómero do mesmo.
[0089] Em uma forma de concretização, é aqui descrito ummétodo de tratamento, prevenção ou controle de um distúrbio neurológico, compreendendo a administração a um paciente de uma composição aqui descrita, que compreende uma quantidade terapeuticamente ou profilaticamente eficaz detransnorsertralina ou um sal farmaceuticamente aceitável, solvato ou estereoisómero do mesmo.
[0090] Em uma modalidade, a distúrbio neurológico é adepressão, déficit cognitivo, fibromialgia, dor, distúrbio do sono relacionado, síndrome de fadiga crônica, distúrbio de déficit de atenção (DDA), déficit de atenção e hiperatividade (TDAH), síndrome das pernas inquietas, esquizofrenia, ansiedade, transtorno obsessivo compulsivo, transtorno de estresse pós-traumático, transtorno afetivo sazonal (SAD), disforia pré-menstrual, sintomas vasomotores na pós-menopausa, doença neurodegenerativa, condições de mania, transtorno distímico, transtorno ciclotímico, obesidade ou abuso de substâncias ou dependência.
[0091] Em uma forma de concretização, o método compreende a administração ao paciente de uma quantidade terapeuticamente ou profilaticamente eficaz de uma composição transnorsertralina aqui descrita como uma terapia adjuvante.
[0092] Em uma forma de concretização, o método é caracterizado por compreender ainda a administração ao paciente de uma quantidade terapeuticamente ou profilaticamente eficaz de um ou mais agentes ativos adicionais.5.1 Definições
[0093] Tal como é descrito na presente invenção, salvo indicação em contrário, os termos "tratar", "tratando" e "tratamento" referem-se à erradicação ou melhoria de uma doença ou distúrbio ou de um ou mais sintomas associados com a doença ou distúrbio. Em certas concretizações, os termos referem-se a minimização da propagação ou agravamento da doença ou distúrbio que resulta da administração de um ou mais agentes profiláticos ou terapêuticos a um sujeito com uma doença ou distúrbio. Em algumas concretizações, os termos referem-se à administração de um composto aqui descrito, com ou sem outro agente ativo adicional, após o início dos sintomas da doença particular.
[0094] Tal como é descrito na presente invenção, salvoindicação em contrário, os termos "prevenir" e "prevenção" referem-se à prevenção do aparecimento, recorrência ou a propagação de uma doença ou distúrbio ou de um ou mais sintomas dos mesmos. Em certas concretizações, os termos referem-se ao tratamento com ou administração de um composto aqui descrito, com ou sem outro composto adicional ativo, antes do início dos sintomas, particularmente para doentes em risco de doença ou distúrbio aqui descritos. Os termos englobam a inibição ou a redução de um sintoma da doença particular. Os pacientes com um histórico familiar de doença em particular, são candidatos a regimes preventivos em certas realizações. Além disso, os pacientes que têm um histórico de sintomas recorrentes também são candidatos potenciais para a prevenção. Neste sentido, o termo "prevenção" pode ser usado indistintamente com o termo "tratamento profilático".
[0095] Tal como é descrito na presente invenção, salvodisposição em contrário, os termos "gerenciar", "gestão" e "controle" referem-se a prevenção ou retardamento da progressão, disseminação ou agravamento de uma doença ou distúrbio ou de um ou mais sintomas da mesma. Muitas vezes, os efeitos benéficos em um sujeito deriva de um agente profilático e/ou terapêutico não resultando em uma cura da doença ou distúrbio. Neste caso, o termo "gestão" abrange o tratamento de um paciente que sofria da doença em particular, em uma tentativa de prevenir ou minimizar a recorrência da doença.
[0096] Tal como é descrito na presente invenção, salvo especificação em contrário, um "quantidade terapeuticamente eficaz" de um composto é uma quantidade suficiente para proporcionar um benefício terapêutico no tratamento ou controle de uma doença ou distúrbio ou para atrasar ou minimizar um ou mais sintomas associados à doença ou distúrbio. Uma quantidade terapeuticamente eficaz de um composto significa uma quantidade de agente terapêutico, sozinho ou em combinação com outras terapias, que proporciona um benefício terapêutico no tratamento ou controle da doença ou distúrbio. O termo "quantidade terapeuticamente eficaz" pode englobar uma quantidade que melhora a terapia, reduz ou evita os sintomas ou causas da doença ou distúrbio ou aumenta a eficácia terapêutica de outro agente terapêutico.
[0097] Tal como é descrito na presente invenção, salvo especificação em contrário, um "quantidade profilaticamente eficaz" de um composto é uma quantidade suficiente para prevenir uma doença ou distúrbio ou impedir a sua recorrência. Uma quantidade profilaticamente eficaz de um composto significa uma quantidade de agente terapêutico, sozinho ou em combinação com outros agentes, que proporciona um benefício profilático na prevenção da doença. A "quantidade profilaticamente eficaz" pode abranger uma quantidade que aumenta a profilaxia global ou aumenta a eficácia profilática de outro agente profilático.
[0098] Tal como é descrito na presente invenção, salvo especificação em contrário, o "sujeito" é aqui definido para incluir os animais, tais como mamíferos, incluindo, mas não se limitando a, primatas (seres humanos), vacas, ovelhas, cabras, cavalos, cães, gatos, coelhos, ratazanas, ratos e similares. Em realizações específicas, o sujeito é um ser humano.
[0099] Tal como é descrito na presente invenção, salvo especificação em contrário, o termo "estável" refere-se a um composto ou composição que não se decompõe facilmente ou altera a composição química ou estado físico. Uma composição estável ou formulação aqui descrita não se decompõe expressivamente sob fabricação normal ou condições de armazenamento.
[0100] Tal como é descrito na presente invenção, salvo indicação em contrário, o termo "sal farmaceuticamente aceitável" refere-se a sais preparados a partir de ácidos não- tóxicos farmaceuticamente aceitáveis, incluindo ácidos inorgânicos e ácidos orgânicos. Ácidos não tóxicos adequados incluem os ácidos inorgânicos e orgânicos tais como, mas não se limitam a, acético, algínico, antranílico, benzenossulfônico, benzóico, canforsulfônico, cítrico, etenosulfônico, fórmico, fumárico, furóico, glucônico, glutâmico, glucorenico, galacturônico, glicídico, bromídrico, clorídrico, isetiônico, láctico, maleico, málico (por exemplo, L-málico), mandélico (por exemplo, S-mandélico), metanossulfônico, múcico, nítrico, pamóico, pantoténico, fenilacético, fosfórico, propiônico, piroglutâmico, salicílico, esteárico, succínico, sulfanílico, ácido sulfúrico, tartárico (por exemplo, ácido L-tartárico e ácido D-tartárico), p-toluenossulfônico e semelhantes.
[0101] Tal como é descrito na presente invenção, salvo indicação em contrário, o termo "solvato" significa um composto aqui descrito ou um seu sal, que inclui ainda uma quantidade estequiométrica ou não-estequiométrica de solvente confinado por concentraçãos intermoleculares não covalentes. Quando o solvente é a água, o solvato é um hidrato.
[0102] Os termos "estado sólido, "formas sólidas" e termos relacionados, quando aqui usados, referem-se a uma forma física compreendendo transnorsertralina ou um seu sal, que não está em um estado líquido ou gasoso. Formas sólidas podem ser cristalina, amorfo, parcialmente cristalino e/ou parcialmente amorfa.
[0103] O termo "cristalinas" e termos relacionados aqui utilizados, quando usados para descrever uma substância, componente ou um produto, significa que a substância, componente ou um produto é substancialmente cristalino como determinado por difração de raio-X. Ver, por exemplo, Remington’s Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA, 173 (1990); The United States Pharmacopeia, 23rd ed., 1843-1844 (1995).
[0104] O termo "formas de cristal" e termos relacionados neste documento referem-se às várias modificações cristalinas que compõem uma dada substância, incluindo as formas de um único componente de cristal e múltiplas formas de componentes de cristal e incluindo, mas não limitado a, polimorfos, solvatos, hidratos, co-cristais e outros complexos moleculares, bem como sais, solvatos de sais, hidratos de sais, de outros complexos moleculares de sais e polimorfos do mesmo. Em certas realizações, uma forma de cristal de uma substância pode ser substancialmente livre de formas amorfas e/ou outras formas de cristais. Em outras formas de concretização, uma forma de cristal de uma substância pode conter cerca de 1%, cerca de 2%, cerca de 3%, cerca de 4%, cerca de 5%, cerca de 10%, cerca de 15%, cerca de 20%, cerca de 25%, cerca de 30 %, cerca de 35%, cerca de 40%, cerca de45% ou cerca de 50% de um ou mais formas amorfas e/ou outras formas de cristal sobre um peso e/ou base molar.
[0105] Formas cristalinas diferentes podem ter diferentespropriedades físicas, tais como, por exemplo, temperaturas de fusão, calor de fusão, solubilidades, taxas de dissolução e/ou espectro vibracional como um resultado do arranjo ou conformação das moléculas ou íons na estrutura de cristal. As diferenças nas propriedades físicas exibidas pelas formas cristalinas afetam os parâmetros farmacêuticos, tais como a estabilidade de armazenamento, compressibilidade e densidade (importante na formulação e na fabricação do produto) e velocidade de dissolução (um fator importante nabiodisponibilidade). As diferenças na estabilidade podem resultar a partir de alterações na reatividade química (por exemplo, oxidação diferencial, de modo que a forma de dosagem perde a cor mais rapidamente quando composta por um forma de cristal do que quando composta de uma outra forma de cristal) ou alterações mecânicas (por exemplo, os comprimidos desintegram-se em armazenamento quando uma forma de cristal converte-se em outra) ou ambos (por exemplo, comprimidos de uma forma de cristal são mais susceptíveis à degradação a uma umidade elevada). Como resultado das diferenças desolubilidade/dissolução, no caso extremo, algumas transições de forma de cristal podem resultar em falta de potência ou, no outro extremo, toxicidade. Além disso, as propriedades físicas da forma de cristal podem ser importantes no processamento. Por exemplo, uma forma de cristal pode ser mais susceptível a formar solvatos ou pode ser difícil de filtrar e lavar as impurezas (por exemplo, a forma de partículas e o tamanho de distribuição pode ser diferente entre as formas de cristal).
[0106] Formas cristalinas de uma substância podem serobtidas por uma série de métodos, tal como conhecidos na arte. Tais métodos incluem, mas não estão limitados a, recristalização por fusão, fusão e congelamento,recristalização do solvente, recristalização em espaços confinados tais como, por exemplo, em nanoporos ou capilares, recristalização em superfícies ou modelos, tais como, por exemplo, em polímeros, recristalização na presença de aditivos, tais como, por exemplo, co-cristal contra-moléculas, dessolvatação, desidratação evaporação rápida, rápidoarrefecimento, arrefecimento lento, difusão de vapor, sublimação, moagem, moagem de gota-solvente, precipitação induzida por microondas, precipitação induzida por sonicação, precipitação induzida por laser e precipitação a partir de um fluido supercrítico.
[0107] Técnicas para a caracterização de formas cristalinase formas amorfas incluem, mas não estão limitadas a, análise gravimétrica térmicas (TGA), calorimetria de varrimento diferencial (DSC), difractometria de raio-X de pó (XRPD), difractometria de raios-X de cristal único, espectroscopia vibracional, por exemplo, espectroscopia no infravermelho (IR) e Raman, espectroscopia por ressonância magnética nuclear em estado sólido (RMN), microscopia óptica, microscopia óptica de fase quente, microscopia electrônica de varrimento (MES), cristalografia de elétrons e análises quantitativas, análise de tamanho de partícula (ATP), análise de área de superfície, estudos de solubilidade e estudos de dissolução.
[0108] Os termos "polimorfo", "forma polimórfica" e termos aqui relacionados referem-se a uma forma de cristal que consiste na mesma molécula, moléculas e/ou íons como uma outra forma de cristal.
[0109] O termo "amorfo", "forma amorfa" e termos aqui relacionados utilizados significam que a substância, componente ou produto em questão não é substancialmente cristalino, conforme determinado por difração de raio-X. Em certas realizações, uma forma amorfa de uma substância pode ser substancialmente livre de outras formas amorfas e/ou formas de cristal. Em outras concretizações, uma forma amorfa de uma substância pode conter cerca de 1%, cerca de 2%, cerca de 3%, cerca de 4%, cerca de 5%, cerca de 10%, cerca de 15%, cerca de 20%, cerca de 25%, cerca de 30 %, cerca de 35%, cerca de 40%, cerca de 45% ou cerca de 50% de um ou mais outras formas amorfas e/ou formas de cristal em peso e/ou base molar. Formas amorfas de uma substância podem ser obtidas por uma série de métodos, tal como conhecidos na arte. Tais métodos incluem, mas não estão limitados a, aquecimento, derretimento com arrefecimento, derretimento com arrefecimento rápido, evaporação do solvente, evaporação rápida do solvente, dessolvatação, sublimação, trituração, moagem, crio-secagem e secagem por congelamento.
[0110] Tal como é descrito na presente invenção, salvo disposição em contrário, os termos "cerca de" e "aproximadamente", quando usado em conexão com doses, quantidades ou porcentagem de peso dos ingredientes de uma composição ou uma forma de dosagem, significa uma dose, quantidade ou porcentagem de peso que é reconhecido por aqueles com habilade na técncia para fornecer um efeito farmacológico equivalente ao obtido a partir da dose, quantidade ou porcentagem de peso especificada. Especificamente, os termos "cerca de" e "aproximadamente", quando usados neste contexto, contemplam uma dose, quantidade ou porcentagem de peso de 15%, mais especificamente dentro de 10%, mais especificamente dentro de 5%, da quantidade especificada da dose, quantidade ou porcentagem de peso.
[0111] Tal como é descrito na presente invenção, uma forma de cristal que é "essencialmente livre" de água e/ou solvente na rede cristalina tem uma quantidade de água e/ou solvente na rede cristalina, que é, em certas concretizações, aproximadamente perto do limite de detecção, em outras formas de concretização aproximadamente no limite de detecção e em outras formas de concretização, aproximadamente abaixo do limite de detecção para solvente e/ou água na rede cristalina, quando medido utilizando uma técnica analítica de estado sólido convencional, por exemplo, uma técnica aqui descrita. Em certas realizações, a técnica analítica de estado sólido utilizada para determinar a quantidade de água e/ou solvente na rede cristalina é a análise termogravimétrica. Em outras concretizações, a técnica analítica de estado sólido utilizada para determinar a quantidade de água e/ou solvente na rede cristalina é análise de Karl Fischer. Em outras formas de concretizações, uma forma de cristal que é "essencialmente livre" de água e/ou solvente na rede cristalina tem uma quantidade de água e/ou solvente é inferior a cerca de 5%, inferior a cerca de 4%, inferior a cerca de 3%, inferior a cerca de 2%, inferior a cerca de 1%, inferior a cerca de 0,9%, inferior a cerca de 0,8%, inferior a cerca de 0,7%, inferior a cerca de 0,6%, inferior a cerca de 0,5%, inferior a cerca de 0,4%, menos do que cerca de 0,3%, inferior a cerca de 0,2%, inferior a cerca de 0,1% ou inferior a cerca de 0,01% do peso total da forma cristalina.
[0112] Tal como é descrito na presente invenção, uma forma cristalina ou amorfa que é "pura", isto é, substancialmente livre de outras formas cristalinas ou amorfas, contém menos do que cerca de 10 por cento em peso de uma ou mais outra forma cristalina ou amorfa, de preferência inferior a cerca de 5 por cento em peso de uma ou mais outra forma cristalina ou amorfa, mais preferivelmente menos do que cerca de 3 por cento em peso de uma ou mais outra forma cristalina ou amorfa, mais preferivelmente menos do que cerca de 1 por cento em peso de uma ou mais outra forma cristalina ou amorfa.
[0113] Tal como é descrito na presente invenção, salvo indicação em contrário, uma composição que é "substancialmente livre" de um composto, significa que a composição contém menos do que cerca de 20 por cento em peso, mais preferivelmente menos do que cerca de 10 por cento em peso, ainda mais preferencialmente inferior a cerca de 5 por cento em peso e mais preferencialmente inferior a cerca de 3 por cento em peso do composto.
[0114] Tal como é descrito na presente invenção, a menos que especificado de outra forma, o termo "distúrbio neurológico" refere-se a qualquer condição do sistema nervoso central ou periférico de um mamífero. O termo "distúrbio neurológico" inclui, mas não está limitado a, doenças neurodegenerativas (por exemplo, doença de Alzheimer, doença de Parkinson e esclerose lateral amiotrófica), doenças neuropsiquiátricas (por exemplo, esquizofrenia e ansiedade, tais como distúrbio de ansiedade generalizada) e perturbações afetivas (por exemplo, depressão e transtorno de déficit de atenção). Exemplos de desordens neurológicas incluem, mas não estão limitados a, MLS (ataxia cerebelar), doença de Huntington, síndrome de Down, demência multi-enfarte, estado epilético, as lesões de contusão (por exemplo, lesão medular e traumatismo craniano), infecção viral que induzneurodegeneração (por exemplo, AIDS, encefalopatias),epilepsia, esquecimento benigno, traumatismo craniano fechado, distúrbios do sono, depressão, demências, distúrbios do movimento, psicoses, alcoolismo, pós-stress traumático e semelhantes. "Distúrbio neurológico" também inclui qualquer condição associada com o distúrbio. Por exemplo, um método de tratamento de um distúrbio neurodegenerativo inclui métodos detratamento de perda de memória e/ou perda de cognição associada com uma doença neurodegenerativa. Um método exemplarincluiria também tratamento ou prevenção de perda de função neuronal característica de distúrbio neurodegenerativo. "Distúrbio neurológico" também inclui qualquer doença ou condição que envolve, pelo menos em parte, modos de sinalização (por exemplo, doença cardiovascular) monoamina (por exemplo, norepinefrina,).
[0115] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "transtorno afetivo" inclui depressão, transtorno de déficit de atenção, transtorno de déficit de atenção com hiperatividade, condição bipolar e condição de mania (por exemplo, transtorno bipolar) e assim por diante. Os termos "transtorno de déficit de atenção" (TAD) e "transtorno de déficit de atenção com hiperatividade" (TADH) ou déficit de atenção/transtorno hiperatividade (DATH), são aqui utilizados de acordo com os significados aceitos como encontrado no Diagnostic and Statistical Manual of Mental Disorders, 4th Ed., American Psychiatric Association (1997)(DSM-IV™).
[0116] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "depressão" inclui todas as formas de depressão, incluindo mas não limitado a, Transtorno Depressivo Maior (TDM), transtorno afetivo sazonal (TAS) e distimia. "Transtorno depressivo maior" é aqui usado como sinônimo de "depressão unipolar" e "depressão maior". "Depressão" também pode inclui qualquer condição comumente associada à depressão, como todas as formas de fadiga (por exemplo, síndrome da fadiga crônica) e déficits cognitivos.
[0117] Tal como é descrito na presente invenção, a menosque especificado de outra forma, os termos "transtorno obsessivo-compulsivo", "abuso de substâncias", "síndrome pré- menstrual", "ansiedade", "distúrbios alimentares" e"enxaqueca" são usados aqui de uma forma consistente com os seus significados aceitos na técnica. Ver, por exemplo, a DSM- IV™. Por exemplo, o termo "distúrbio alimentar", como aqui utilizado, refere-se a compulsões anormais para evitar a alimentação ou impulsos incontroláveis para consumir anormalmente grandes quantidades de comida. Esses distúrbios podem afetar não só o bem-estar social, mas também o bem-estar físico dos pacientes. Exemplos de desordens alimentares incluem, mas não estão limitados a, anorexia nervosa, bulimia e compulsão alimentar.
[0118] Tal como é descrito na presente invenção, a menos que especificado de outra forma, o termo "dor" se refere a uma experiência sensorial e emocional desagradável. O termo "dor," como é aqui utilizado, refere-se a todas as categorias de dor, incluindo a dor, que é descrito em termos de estímulo do nervo ou de resposta, por exemplo, dor somática (resposta normal do nervo a um estímulo nocivo) e dor neuropática (resposta anormal de uma via sensorial ferida ou alterada, muitas vezes sem entrada nociva clara); dor que é classificada temporalmente, a dor, por exemplo, crônica e dor aguda; dor que é classificada em termos de sua gravidade, por exemplo, leve, moderada ou grave; e dor que é um sintoma ou um resultado de um estado de doença ou síndrome, por exemplo, dor inflamatória, dor de câncer, dor da AIDS, artropatia, enxaqueca, nevralgia trigeminal, isquémia cardíaca e dor diabética neuropática periférica. Ver, por exemplo, Harrison's Principles of Internal Medicine, pp. 93-98 (Wilson et al., eds., 12th ed. 1991); Williams et al., J. Med. Chem. 42: 14811485 (1999), aqui cada um incorporado por referência na sua totalidade. "Dor" também pretende incluir dor etiologia mista, dor de mecanismo duplo, alodinia, causalgia, dor central, hiperestesia, hiperpatia, disestesia e hiperalgesia. Além disso, o termo "dor" inclui dor resultante de disfunção do sistema nervoso: estados de dor orgânicos que compartilham características clínicas de dor neuropática e possíveis mecanismos de fisiopatologia comuns, mas não são iniciadas por uma lesão identificável em qualquer parte do sistema nervoso.
[0119] O termo "dor somática," tal como aqui utilizado, refere-se a uma resposta normal do nervo a um estímulo nocivo, tais como lesão ou doença, por exemplo, trauma, queimadura, infecção, inflamação ou processo de doença, tais como câncer e inclui tanto a dor cutânea (por exemplo, pele, músculo ou derivado articulações) e dor visceral (por exemplo, órgão derivado).
[0120] O termo "dor neuropática," como é aqui utilizado, refere-se a um grupo heterogêneo de doenças neurológicas que resultam de danos ao sistema nervoso. O termo também se refere a dor resultante de lesões ou disfunções das vias sensoriais periféricas e/ou centrais e de disfunções do sistema nervoso, onde a dor persiste ou muitas vezes ocorre sem uma entrada óbvio nociva. Isto inclui dor relacionada com as neuropatias periféricas, bem como a dor neuropática central. Os tipos mais comuns de dor neuropática periférica incluem a neuropatia diabética (também chamada de dor neuropática periférica diabética ou DN, DPN ou DPNP), neuralgia pós-herpética (PHN) e neuralgia trigêmea (TGN). Dor neuropática central, envolvendo danos ao cérebro ou medula espinhal, pode ocorrer após acidente vascular cerebral, lesão da medula espinhal e como resultado da esclerose múltipla e é também englobado pelo termo. Outros tipos de dor que se destinam a ser incluídos na definição de dor neuropática incluem, mas não estão limitados a, dor de dor neuropática de câncer, dor induzida de HIV/AIDS, dor do membro ilusória e síndrome da dor regional complexa.
[0121] O termo também engloba as características clínicascomuns de dor neuropática, incluindo, mas não limitado a, perda de sensibilidade, alodinia (estímulos não tóxicos que produzem dor), hiperalgesia e hiperpatia (percepção retardada, somatório e depois da sensação dolorosa). A dor é geralmente uma combinação de tipos nociceptiva e neuropática, por exemplo, dores na coluna mecânica e radiculopatia ou mielopatia.
[0122] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor aguda" refere-se a resposta fisiológica normal prevista a um produto químico nocivo, estímulo térmico ou mecânico tipicamente associado com procedimentos invasivos, trauma e doenças. É geralmente limitada no tempo e pode ser vista como uma resposta apropriada a um estímulo que ameaça e/ou produz lesões dos tecidos. O termo também se refere à dor, que é marcada pela curta duração ou início súbito.
[0123] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor crônica" abrange a dor que ocorre em uma ampla gama de distúrbios, por exemplo, trauma, neoplasias e doenças inflamatórias crônicas, tais como artrite reumatóide. A dor crônica pode durar mais de seis meses. Além disso, a intensidade da dor crônica pode ser desproporcional a intensidade do estímulo nocivo ou processo subjacente. O termo também se refere à dor associada à uma doença crônica ou dor que persiste além da resolução de um distúrbio subjacente ou cura de uma lesão e que é, muitas vezes, mais intenso do que o processo subjacente poderia prever. Pode ser sujeito a recorrência frequente.
[0124] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor inflamatória" é a dor em resposta a lesão do tecido e do processo inflamatório. Dor inflamatória é adaptável na medida em que provoca respostas fisiológicas que promovem a cura. No entanto, a inflamação pode também afetar a função neuronal. Mediadores inflamatórios, incluindo PGE2 induzida pela enzima COX2, bradicinina e outras substâncias, se ligam a receptores sobre neurônios transmissores de dor e alteram a sua função, aumentando a sua excitabilidade e, assim, aumentando a sensação de dor. Muitas dores crônicas tem um componente inflamatório. O termo também se refere a dor que é produzidacomo um sintoma ou um resultado de inflamação ou um distúrbiodo sistema imunitário.
[0125] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor visceral" refere-se à dor, que está localizada em um órgão interno.
[0126] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor etiologia mista" refere-se a dor que contém componentes inflamatórios e neuropáticos.
[0127] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor de mecanismo duplo" refere-se a dor que é amplificada e mantida por sensibilização periférica e central.
[0128] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "causalgia" refere-se a uma síndrome de queima sustentada, alodinia e hiperpatia após uma lesão traumática do nervo, muitas vezes combinados com disfunções vasomotoras e sudomotoras e alterações tróficas posteriores.
[0129] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor central" refere- se à dor iniciada por uma lesão primária ou disfunção do sistema nervoso central.
[0130] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "hiperestesia" refere-se ao aumento da sensibilidade à estimulação, excluindo os sentidos especiais.
[0131] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "hiperpatia" refere- se a uma síndrome dolorosa caracterizada por uma reação anormal dolorosa a um estímulo, especialmente um estímulo repetitivo, bem como um limite aumentado. Pode ocorrer com alodinia, hiperestesia, hiperalgesia, ou disestesia.
[0132] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "disestesia" refere- se a uma sensação anormal desagradável, espontânea ou evocada. Em certas realizações, disestesia inclue hiperalgesia e alodinia.
[0133] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "hiperalgesia" refere-se a um aumento da resposta a um estímulo que normalmente é doloroso. Este reflete um aumento da dor na estimulação supralimiar.
[0134] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "alodinia" refere-se a dor, devido a um estímulo que normalmente não provoca dor.
[0135] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor neuropática periférica diabética" (DPNP), também chamada neuropatia diabética, DN ou neuropatia periférica diabética), refere-se à dor crônica causada por neuropatia associada com diabetes mellitus. A apresentação clássica da DPNP é a dor ou formigamento nos pés que podem ser descritas, não apenas como "queimação" ou "shooting", mas também como dor severa. Menos comumente, os pacientes podem descrever a dor como coceira, lacrimejamento ou como uma dor de dente. A dor pode ser acompanhada por alodinia e hiperalgesia e uma ausência de sintomas, tais como dormência.
[0136] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "neuralgia pós- herpética", também chamado de "Neuralgia pós-herpética (PHN)", refere-se a uma condição dolorosa que afeta as fibras nervosas e a pele. Sem estar limitado por uma teoria particular, é uma complicação de herpes, um segundo ataque do vírus varicela- zoster (VZV), que inicialmente causa a varicela.
[0137] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor neuropática de câncer" refere-se a dor neuropática periférica, como resultado do câncer e pode ser causada diretamente pela infiltração ou compressão de um nervo por um tumor ou indiretamente por tratamentos contra o câncer, tais como terapia de radiação e quimioterapia (neuropatia induzida por quimioterapia).
[0138] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "HIV/AIDS neuropatia periférica" ou "neuropatia HIV/AIDS relacionada" refere-se a neuropatia periférica causada pelo HIV/AIDS, tais como neuropatia desmielinizante inflamatória aguda ou crônica (AIDP e PDIC, respectivamente), bem como a neuropatia periférica resultante de um efeito colateral de fármacos utilizados para tratar o HIV/ AIDS.
[0139] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "dor ilusória do membro" refere-se a dor que parece vir de onde um membro amputado estava. Dor ilusória do membro também pode ocorrer em membros após paralisia (por exemplo, na sequência de lesão da medula espinal). "Dor ilusória do membro" é geralmente de natureza crônica.
[0140] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "neuralgia trigeminal (NT)" refere-se a um distúrbio do quinto nervo (trigêmeo) cranial que provoca episódios de dor intensa, cortante, de choque elétrico, nas áreas do rosto onde as ramificações do nervo estão distribuídas (lábios, olhos, nariz, couro cabeludo, testa, queixo, parte superior e inferior da mandíbula). Ele também é conhecido como a "doença do suicídio".
[0141] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "Síndrome Dolorosa Complexa Regional (SDCR)," formalmente conhecida comoDistrofia Simpático Reflexa (DSR), refere-se a uma condição de dor crônica cujos principais sintomas é a dor contínua, intensa fora de proporção com a gravidade da lesão, que se agrava em vez de melhorar ao longo do tempo. O termo abrange SDRC do tipo 1, que inclui condições causadas por lesão do tecido que não seja do nervo periférico e SDRC do tipo 2, em que a síndrome é provocada por lesão do nervo principal e é por vezes chamada causalgia.
[0142] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "fibromialgia" refere-se a uma condição crônica caracterizada por músculo difuso ou específico, dores nas articulações ou osso, juntamente com a fadiga e uma gama de outros sintomas. Anteriormente, a fibromialgia era conhecida por outros nomes, como fibrosite síndrome de dor crônica muscular, reumatismo psicogênico e mialgias de tensão.
[0143] Tal como é descrito na presente invenção, a menosque especificado de outra forma, o termo "convulsão" se refere a um distúrbio neurológico e é usado como sinônimo de "apreensão", embora existam muitos tipos de apreensão, alguns dos quais têm sintomas sutis ou leve em vez de convulsões. Convulsões de todos os tipos podem ser causadas por atividade elétrica desorganizada e súbita do cérebro. Em algumas realizações, convulsão é uma agitação rápida e incontrolável durante o qual músculos se contraem e relaxam repetidamente.
[0144] As formas de concretização aqui descritas podem sercompreendidas de forma mais completa, por referência, à seguinte descrição detalhada e exemplos ilustrativos, que se destinam a exemplificar formas de concretização não limitativas.5.2 Composições farmacêuticas
[0145] Em uma forma de concretização aqui descrita,composições farmacêuticas são caracterizadas por compreender: transnorsertralina ou um sal farmaceuticamente aceitável ou solvato e um transportador ou excipiente farmaceuticamente aceitável.
[0146] As formas de dosagem sólidas de transnorsertralina ou sais farmaceuticamente aceitáveis ou solvatos dos mesmos, são desejadas pela facilidade de administração da dose para indivíduos e pacientes, bem como proporcionar fácil administração de formulações de dosagem out-of-clinic. Estas formas de dosagem devem ser fabricadas em equipamento automatizado e tem estabilidade química e física aceitável que pode ser superior a 1 ano. Estas formas de dosagem sólidas de transnorsertralina ou sais farmaceuticamente aceitáveis ou solvatos são desejadas para o desenvolvimento, usos clínicos, e comerciais.
[0147] Muitas misturas de excipiente com transnorsertralina ou um sal farmaceuticamente aceitável ou solvato não são quimicamente estáveis. Por exemplo, cápsulas de gelatina dura contendo cloridrato de transnorsertralina em combinação com os excipientes encontrados em comprimidos de Zoloft® (sertralina) resultaram em uma formulação com estabilidade química pobre e, em particular, com os produtos de múltiplas oxidações. Estes excipientes são di-hidrato de fosfato dibásico de cálcio, celulose microcristalina, glicolato de amido de sódio, estearato de magnésio, bem como outros excipientes que são susceptíveis ao revestimento desses comprimidos. Veja Physician’s Desk Reference entry for Zoloft® (sertraline).
[0148] Portanto, em certas realizações, os excipientes manitol ou xilitol podem ser utilizados em vez de outros excipientes sacarídeos comuns (por exemplo, lactose ou celulose), a fim de melhorar a estabilidade das composições transnorsertralina aqui descritas. A utilização de sacarídeos, com exceção de manitol ou xilitol, promove a degradação de composições farmacêuticas compreendendo transnorsertralina ou um sal farmaceuticamente aceitável ou solvato deste.
[0149] Em algumas concretizações, as composições farmacêuticas aqui descritas compreendem 10 a 98% em peso de manitol ou xilitol. Em outras realizações, excipientes adicionais utilizados nas composições farmacêuticas aqui descritas podem incluir estearato de magnésio, talco e glicolato de amido de sódio. Estearato de magnésio, talco e glicolato de amido de sódio foram descobertos para serem compatíveis com transnorsertralina ou um dos seus sais farmaceuticamente aceitáveis ou solvatos, tais que estes excipientes, além de manitol e xilitol, são os preferidos.
[0150] Formulações compreendendo transnorsertralina ou um dos seus sais farmaceuticamente aceitáveis ou solvatos dos mesmos e os excipientes acima descritos podem ser preparados de acordo com os seguintes processos.
[0151] Misturas para formulações em cápsulas contendo transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato, podem ser fabricadas usando um processo no qual cloridrato de transnorsertralina é primeiro misturado com talco; esta mistura é então misturada com manitol em diluição geométrica. O manitol restante e glicolato de amido de sódio são misturados com a mistura; por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0152] O processo pode ser modificado tal que a transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato é primeiro misturado com uma porção de Talco mais manitol; esta mistura é então misturada com manitol adicional. Em seguida, o manitol restante e glicolato de amido de sódio são misturados com a mistura; por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0153] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou um sal farmaceuticamente aceitável ou solvato com uma porção de talco mais manitol; esta mistura é, então, misturada com uma mistura de manitol mais glicolato de amido de sódio; por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0154] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol mais glicolato de amido de sódio; esta mistura é então misturada com os excipientes restantes (menos o estearato de magnésio). Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0155] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais glicolato de amido de sódio; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0156] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com talco; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0157] Outra modificação do processo pode ser realizada através de transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol; esta mistura é então misturada com o manitol restante. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0158] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com manitol; esta mistura é então misturada com uma mistura de talco mais manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semiautomática ou completamente automática.
[0159] Outra modificação do processo pode ser realizada por mistura de uma porção do estearato de magnésio com transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato, em cada um dos processos acima. Por último, o resto do estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0160] Misturas de formulações de comprimidos que contêm transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato podem ser fabricadas usando um processo em que transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato é primeiro misturada com talco; esta mistura é então misturada com manitol em diluição geométrica. Em seguida, o manitol restante e glicolato de amido de sódio são misturados com a mistura; por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0161] O processo para a fabricação de comprimidos não revestidos pode ser modificado, tal que a transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato é primeiro misturado com uma porção de talco mais manitol; esta mistura é então misturada com manitol adicional. Em seguida, o manitol restante e glicolato de amido de sódio são misturados com a mistura; por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa de ou máquina comprimidos.
[0162] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma porção de talco mais manitol; esta mistura é então misturada com uma mistura de manitol mais glicolato de amido de sódio; por último, estearato de magnésio é misturado com o anterior mistura. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0163] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol mais glicolato de amido de sódio; esta mistura é então misturada com os excipientes restantes (menos o estearato de magnésio). Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0164] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais glicolato de amido de sódio; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0165] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com talco; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0166] Outra modificação do processo pode ser ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol; esta mistura é então misturada com o manitol restante. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0167] Outra modificação do processo pode ser realizada através de mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com manitol; esta mistura é então misturada com uma mistura de talco mais manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0168] Outra modificação do processo pode ser realizada através de mistura de uma porção do estearato de magnésio com transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato, em cada um dos processos acima. Por último, o resto do estearato de magnésio pode ser misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0169] Cada um dos comprimidos descritos acima pode, também, ser fabricado como um comprimido revestido. O revestimento pode ser de um dos três tipos, que incluem revestimento por compressão, revestimento com filme ou revestimento de gelatina. Cada revestimento pode ou não conter um agente corante; estes agentes de coloração podem ser dióxido de titânio e/ou corantes solúveis, tais como corantes e/ou corantes insolúveis tais como pigmentos e/ou óxidos de ferro coloridos.
[0170] As formulações específicas de transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato em cápsula ou na forma de comprimido são descritas abaixo. As formulações de outros pesos para cápsulas ou comprimidos podem também ser preparados utilizando percentagens semelhantes ou variadas de excipientes.
[0171] Uma cápsula de 300,0mg pode ser preparada utilizando 1.125mg de cloridrato de anidrato transnorsertralina, 2.875mg de Talco, 275.0mg de Pearlitol 160C (manitol), 18.0mg de Primojel (glicolato de amido de sódio), 3.0mg de estearato de magnésio e uma cápsula laranja sueca tamanho #1 envoltório #4188.
[0172] Alternativamente, uma cápsula 300,0mg pode ser preparada sem Primojel, utilizando 1.125mg de cloridrato de anidrato de transnorsertralina, 2.875mg de talco, 293.0mg de Pearlitol 160C (manitol), 3.0mg de estearato de magnésio e uma cápsula laranja sueca tamanho #1 envoltório #4188.
[0173] Uma cápsula 150,0mg pode ser preparada utilizando 0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de talco, 137,5mg de Pearlitol 160C (manitol), 9,0mg de Primojel (glicolato de amido de sódio), 1,5mg de estearato de magnésio e uma cápsula laranja sueca tamanho #1 envoltório #4188.
[0174] Alternativamente, uma cápsula 150,0mg pode ser preparada sem Primojel, utilizando 0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 146,5mg de Pearlitol 160C (manitol), 1,5mg de estearato de magnésio e uma cápsula laranja sueca tamanho #1 envoltório #4188.
[0175] Uma cápsula 300,0mg pode ser preparada usando 2,25g de cloridrato de anidrato de transnorsertralina, 4,75mg de talco, 272,0mg de Pearlitol 160C (manitol), 18,0mg de Primojel (glicolato de amido de sódio), 3,0mg de estearato de magnésio e uma cápsula laranja sueca tamanho #1 envoltório #4188.
[0176] Alternativamente, a cápsula 300,0mg pode ser preparada sem Primojel, usando 2,25mg de cloridrato de anidrato de transnorsertralina, 4,75mg de Talco, 290,0mg de Pearlitol 160C (manitol), 3,0mg de estearato de magnésio e uma cápsula laranja sueca tamanho #1 envoltório #4188.
[0177] Cápsulas de enchimento de pesos 100.0, 150.0 e 200.0 mg com 0,5 mg transnorsertralina em tamanhos diferentes de envoltório de cápsula podem ser preparadas como se segue.
[0178] Uma cápsula 100,0 mg pode ser preparada utilizando 0,5625 mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de talco, 91,0 mg de manitol, 6,0 mg de Primojel (glicolato de amido de sódio), 1,0 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 4.
[0179] Uma cápsula 150,0 mg pode ser preparada utilizando 0,5625 mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de talco, 137,5 mg de manitol, 9,0 mg de Primojel (glicolato de amido de sódio), 1,5 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 3.
[0180] Uma cápsula 200,0 mg pode ser preparada utilizando 0,5625 mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de Talco, 184,0 mg de manitol, 12,0 mg de Primojel (glicolato de amido de sódio), 2,0 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 2.
[0181] Cápsulas de enchimento de pesos 100.0, 150.0 e 200.0 mg com 1,0 mg de transnorsertralina em tamanhos diferentes de envoltório de cápsula podem ser preparadas como se segue.
[0182] Uma cápsula 100,0 mg pode ser preparada utilizando 1,125 mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de Talco, 90,44 mg de manitol, 6,0 mg de Primojel (glicolato de amido de sódio), 1,0 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 4.
[0183] Uma cápsula 150,0 mg pode ser preparada utilizando 1,125 mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de Talco, 136,94 mg de manitol, 9,0 mg de Primojel (glicolato de amido de sódio), 1,5 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 3.
[0184] Uma cápsula 200,0 mg pode ser preparada utilizando 1,125 mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de Talco, 183,44 mg de manitol, 12,0 mg de Primojel (glicolato de amido de sódio), 2,0 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 2.
[0185] Cápsulas de enchimento de pesos 100.0, 150.0 e 200.0 mg com 2,0 mg de transnorsertralina em tamanhos diferentes de envoltório de cápsula podem ser preparadas como se segue.
[0186] Uma cápsula 100,0 mg pode ser preparada utilizando 2,25 mg de cloridrato de anidrato de transnorsertralina, 4,75 mg de Talco, 86,0 mg de manitol, 6,0 mg de Primojel (glicolato de amido de sódio), 1,0 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 4.
[0187] Uma cápsula 150,0 mg pode ser preparada utilizando 2,25 mg de cloridrato de anidratode transnorsertralina, 4,75 mg de Talco, 132,5 mg de manitol, 9,0 mg de Primojel (glicolato de amido de sódio), 1,5 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 3.
[0188] Uma cápsula 200,0 mg pode ser preparada utilizando2,25 mg de cloridrato de anidrato de transnorsertralina, 4,75 mg de Talco, 179,0 mg de manitol, 12,0 mg de Primojel (glicolato de amido de sódio), 2,0 mg de estearato de magnésio e um invólucro da cápsula de gelatina dura tamanho # 2.
[0189] Comprimidos de pesos 100.0, 150.0 e 200.0 mg com 0,5mg de transnorsertralina podem ser preparados como se segue.
[0190] Um comprimido de 100,0mg é preparado utilizando0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 91,0mg de manitol, 6,0mg de Primojel (glicolato de amido de sódio) e 1,0mg de estearato de magnésio.
[0191] Um comprimido de 100,0mg pode também ser preparadosem Primojel, utilizando 0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 97,0mg de manitol e 1,0mg de estearato de magnésio.
[0192] Um comprimido de 150,0 mg pode ser preparadoutilizando 0,5625mg de cloridrato de anidrato detransnorsertralina, 1,4375mg de Talco, 137,5mg de manitol, 9,0mg de Primojel (glicolato de amido de sódio) e 1,5mg de estearato de magnésio.
[0193] Um comprimido de 150,0mg pode também ser preparadosem Primojel, utilizando 0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 146,5mg de manitol e 1,5mg de estearato de magnésio.
[0194] Um comprimido de 200,0mg pode ser preparadoutilizando 0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 184,0mg de manitol, 12,0mg de Primojel (glicolato de amido de sódio) e 2,0 mg de estearato de magnésio.
[0195] Um comprimido de 200,0mg pode também ser preparadosem Primojel, utilizando 0,5625mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 196,0mg de manitol e 2,0mg de estearato de magnésio.
[0196] Comprimidos de pesos 100,0, 150,0 e 200,0mg com 1,0mg de transnorsertralina podem ser preparados como se segue.
[0197] Um comprimido 100,0 mg pode ser preparado utilizando1,125mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 90,44mg de manitol, 6,0mg de Primojel (glicolato de amido de sódio) e 1,0mg de estearato de magnésio.
[0198] Um comprimido de 100,0mg pode também ser preparadosem Primojel, utilizando 1,125mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 96,44mg de manitol e 1,0mg de estearato de magnésio.
[0199] Um comprimido de 150,0mg pode ser preparadoutilizando 1,125mg de cloridrato de anidrato detransnorsertralina, 1,4375mg de Talco, 136,94mg de manitol, 9,0mg de Primojel (glicolato de amido de sódio) e 1,5mg de estearato de magnésio.
[0200] Um comprimido de 150,0mg pode também ser preparadosem Primojel, utilizando 1,125mg de cloridrato de anidrato de transnorsertralina, 1,4375mg de Talco, 145,94mg de manitol e 1,5mg de estearato de magnésio.
[0201] Um comprimido de 200,0mg pode ser preparadoutilizando 1,125mg de cloridrato de anidrato de transnorsertralina, 1,4375 mg de Talco, 183,44 mg de manitol, 12,0 mg de Primojel (glicolato de amido de sódio) e 2,0 mg de estearato de magnésio.
[0202] Um comprimido de 200,0 mg pode também ser preparado sem Primojel, utilizando 1,125 mg de cloridrato de anidrato transnorsertralina, 1,4375 mg de Talco, 195,44 mg de manitol e 2,0 mg de estearato de magnésio.
[0203] Comprimidos de pesos 100,0, 150,0 e 200,0mg com 2,0 mg de transnorsertralina podem ser preparados como se segue.
[0204] Um comprimido 100,0 mg pode ser preparado utilizando 2,25 mg de cloridrato de anidrato transnorsertralina, 4,75 mg de Talco, 86,0 mg de manitol, 6,0 mg de Primojel (glicolato de amido de sódio) e 1,0 mg de estearato de magnésio.
[0205] Um comprimido de 100,0 mg pode também ser preparado sem Primojel, usando 2,25 mg de cloridrato de anidrato transnorsertralina, 4,75 mg de Talco, 92,0 mg de manitol e 1,0 mg de estearato de magnésio.
[0206] Um comprimido de 150,0 mg pode ser preparado usando 2,25 mg de cloridrato de anidrato transnorsertralina, 4,75 mg de Talco, 132,5 mg de manitol, 9,0 mg de Primojel (glicolato de amido de sódio) e 1,5 mg de estearato de magnésio.
[0207] Um comprimido de 150,0 mg pode também ser preparado sem Primojel, usando 2,25 mg de cloridrato de anidrato transnorsertralina, 4,75 mg de Talco, 141,5 mg de manitol e 1,5 mg de estearato de magnésio.
[0208] Um comprimido de 200,0 mg pode ser preparado usando 2,25 mg de cloridrato de anidrato transnorsertralina, 4,75 mg de Talco, 179,0 mg de manitol, 12,0 mg de Primojel (glicolato de amido de sódio) e 2,0 mg de estearato de magnésio.
[0209] Um comprimido de 200,0mg pode também ser preparadosem Primojel, usando 2,25 mg de cloridrato de anidrato transnorsertralina, 4,75 mg de Talco, 191,0 mg de manitol e 2,0 mg de estearato de magnésio.
[0210] Cápsulas e comprimidos de outros pesos podem serpreparados usando 10%-98% de manitol, 0,1% de estearato de magnésio 5%, 0,5%-40% de Talco e de 0%-10% de glicolato deamido de sódio.
[0211] Cápsulas e comprimidos de outros pesos também podemser preparados usando 5%-99% de manitol, 0,05%-15% de estearato de magnésio, 0%-50% de Talco e 0%-40% de glicolato de amido de sódio.
[0212] Cápsulas e comprimidos de outros pesos também podemser preparados usando 5%-99% de manitol, 0%-15% de estearato de magnésio, 0,5%-50% de Talco e de 0%-40% de glicolato de amido de sódio.
[0213] Em algumas conretizações, as composiçõesfarmacêuticas aqui descritas podem, opcionalmente, ser caracterizadas por compreender um ou mais outros agentes ativos. Exemplos de agentes adequados são descritos aqui em outro local.
[0214] Certas composições farmacêuticas são formasunitárias de dosagem adequadas para administração oral, por mucosa (por exemplo, nasal, sublingual, vaginal, bucal, traqueia, brônquios ou retal), parenteral (por exemplo, subcutânea, intravenosa, injeção de bolus, intramuscular ou intra-arterial) ou a administração transdérmica para um paciente. Exemplos de formas de dosagem incluem, mas não estão limitados a: comprimidos, cápsulas ovais, cápsulas, tais como cápsulas flexíveis elásticas ou de gelatina dura; hóstias; pastilhas; pastilhas; dispersões; supositórios; pomadas; cataplasmas; pastas; pós; frasco de soluções nebulizadas de dose unitária (UDV); pensos; cremes; emplastros; soluções; esparadrapos; aerossóis (por exemplo, sprays nasais ou inaladores); géis, formas de dosagem líquidas adequadas para administração oral ou na mucosa a um paciente, incluindo suspensões (por exemplo, aquoso ou suspensões não-aquosas de líquido, emulsões óleo-em-água ou uma emulsão líquida água-em- óleo), soluções e elixires; formas de dosagem líquidas adequadas para administração parenteral a um paciente; e sólidos estéril (por exemplo, sólidos cristalinos ou amorfos) que podem ser reconstituídos para fornecer formas de dosagem líquidas adequadas para administração parenteral a um paciente.
[0215] Em uma forma de concretização, a forma de dosagem éuma forma de dosagem oral. Em outra forma de concretização, a forma de dosagem oral é uma cápsula, comprimido ou xarope. Em outra forma de concretização, a forma de dosagem é uma forma de dosagem parenteral.
[0216] A formulação deve ser adequada ao modo deadministração. Por exemplo, a administração oral pode exigir revestimentos entéricos para proteger os compostosadministrados a partir de degradação no tratogastrointestinal. Em outro exemplo, os compostos podem ser administrados em uma formulação lipossomal para proteger os compostos a partir de enzimas degradativas, facilitando o transporte no sistema circulatório e entrega eficaz através das membranas celulares para sítios intracelulares.
[0217] A composição, forma e tipo de formas de dosagem irãotipicamente variar de acordo com a sua utilização. Por exemplo, uma forma de dosagem utilizada no tratamento de uma doença aguda pode conter quantidades maiores de um ou mais dos ingredientes ativos que a compõem do que uma forma de dosagem utilizada no tratamento crônico da mesma doença. Do mesmo modo, uma forma de dosagem parenteral podem conter quantidades menores de um ou mais dos ingredientes ativos do que o compõeuma forma de dosagem oral usada para tratar a mesma doença. Estes e outros modos em que as formas de dosagem específicasirão variar de uma para outra será prontamente evidente para os com habilidade na técnica. Ver, por exemplo, Remington’s Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA(1990).
[0218] O nível de dosagem selecionado e frequência deadministração das composições farmacêuticas aqui descritas dependerão de uma variedade de fatores, incluindo a via de administração, o tempo de administração, a taxa de excreção dos agentes terapêuticos, da duração do tratamento, outras drogas, os compostos e/ou materiais usados no paciente, a idade, sexo, peso, condição, estado geral de saúde e do histórico médico anterior do paciente a ser tratado e fatores semelhantes bem conhecidos nas artes médicas. Por exemplo, o regime de dosagem é susceptível a variar com as mulheres grávidas, lactantes e crianças com os adultos saudáveis. Um médico com pouco conhecimento na arte pode prontamente determinar e prescrever a quantidade terapeuticamente eficaz da composição farmacêutica requerida.
[0219] As composições farmacêuticas aqui descritas podem ainda ser caracterizadas por compreender um transportador farmaceuticamente aceitável. O termo "veículo farmaceuticamente aceitável" significa um ou mais excipientes farmaceuticamente aceitáveis. Exemplos de tais excipientes são bem conhecidos na arte e estão listados na USP (XXI)/NF (XVI), aqui incorporada na sua totalidade por referência e incluem, sem limitação, agentes aglutinantes, diluentes, agentes de enchimento, desintegrantes, super desintegrantes, lubrificantes, agentes tensoativos, antiaderentes, estabilizadores e outros semelhantes. O termo "aditivos" é sinônimo da expressão "excipientes", como aqui utilizado.
[0220] O termo "farmaceuticamente aceitável" é aqui utilizado para referir-se aos compostos, materiais, composições e/ou formas de dosagem que são, dentro do âmbito do julgamento médico, adequados para administração e para utilização em contato com os tecidos e fluidos de seres humano e animais sem toxicidade excessiva, irritação, reação alérgica ou outro problema ou complicação, compatíveis com uma razoável relação benefício/ risco.
[0221] Além disso, o termo "excipiente farmaceuticamente aceitável" é utilizado para significar que não há nenhuma substância nociva ou incompatibilidades físicas entre os ingredientes ativos e qualquer um dos componentes do excipiente de uma determinada forma de dosagem. Por exemplo, uma reação química inconveniente é aquela em que a potência de compostos utilizados nos métodos e composições aqui descritas é prejudicialmente reduzida ou aumentada devido à adição de um ou mais excipientes. Outro exemplo de uma reação química inconveniente é uma em que o sabor da forma de dosagem torna- se excessivamente doce, ácido ou semelhante, à medida em que a forma de dosagem torna-se intragável. Cada excipiente deve ser "aceitável", no sentido de ser compatível com os outros ingredientes da formulação e não prejudiciais para o paciente.
[0222] A incompatibilidade física refere-se a incompatibilidade entre os vários componentes da forma de dosagem e qualquer excipiente(s) da mesma. Por exemplo, a combinação do excipiente(s) e do ingrediente ativo(s) pode formar uma mistura excessivamente higroscópica ou uma mistura excessivamente segregada no grau que a forma desejada da forma de dosagem (por exemplo, comprimido, pastilha, etc), a sua estabilidade ou semelhante não pode ser suficientemente mantida para ser capaz de controlar a forma de dosagem de acordo com um regime de dosagem prescrita, como desejado.
[0223] Com a exceção do invólucro das cápsulas, é de notar que todos os excipientes utilizados nas composições farmacêuticas ou formas de dosagem aqui descritas, de preferência, satisfazem ou excedem os padrões para ingredientes farmacêuticos e suas combinações na USP/NF. O objetivo da USP/NF é proporcionar padrões e especificações autorizadas de materiais e substâncias e suas preparações que são utilizadas na prática da arte da cura. A USP/NF estabelece títulos, definições, descrições e os padrões de identidade, qualidade, concentração, pureza, embalagem e rotulagem e, também, se possível, fornece biodisponibilidade, estabilidade, procedimentos para manuseio e armazenamento e métodos para o seu exame e fórmulas para a fabricação ou preparação.
[0224] A estabilidade de um produto farmacêutico pode ser definida como a capacidade de uma formulação particular, em um recipiente específico, permanecer dentro da sua especificação física, química, microbiológica, terapêutica e toxicológica, embora haja exceções e de manter, pelo menos, cerca de 80%, de preferência cerca de 90%, mais preferivelmente cerca de 95% do nível de potência rotulada. Assim, por exemplo, a data de validade é definida como o tempo em que o produto farmacêutico irá se manter estável quando armazenado sob as condições recomendadas.
[0225] Muitos fatores afetam a estabilidade de um produto farmacêutico, incluindo a estabilidade do ingrediente terapêutico(s), o potencial interação entre ingredientes terapêuticos e inativos e semelhantes. Fatores físicos como calor, luz e umidade podem iniciar ou acelerar reações químicas.5.2.1 Dosagem das formas orais
[0226] As composições farmacêuticas aqui descritas, que são adequadas para administração oral, podem ser apresentadas como formas de dosagem discretas, tais como, mas não estão limitados a, comprimidos (por exemplo, comprimidos mastigáveis), cápsulas ovais, cápsulas e líquidos (por exemplo, xaropes aromatizados). Tais formas de dosagem contêm quantidades pré-determinadas de ingredientes ativos e podem ser preparados por métodos bem conhecidos de farmácia pelos peritos na arte. Ver em geral, Remington: The Science and Practice of Pharmacy, 20th Ed. (2000).
[0227] Típicas formas de dosagem oral são preparadas por combinação dos ingredientes ativos em mistura íntima com pelo menos um excipiente, de acordo com técnicas de composição farmacêuticas convencionais. Excipientes podem ter uma larga variedade de formas dependendo da forma de preparação desejada para administração.
[0228] Devido à sua facilidade de administração, os comprimidos e as cápsulas representam as mais vantajosas formas de dosagem orais unitárias, caso em que excipientes sólidos são empregados. Se desejado, os comprimidos podem ser revestidos por técnicas padrão aquosas ou não aquosas. Tais formas de dosagem podem ser preparadas por qualquer dos métodos farmacêuticos. Em geral, as composições farmacêuticas e formas de dosagem são preparadas por mistura uniforme e íntima dos ingredientes ativos com transportadores líquidos, veículos sólidos finamente divididos ou ambos e depois moldando o produto na apresentação desejada, se necessário.
[0229] Produção em larga escala de composições farmacêuticas ou formas de dosagem, de acordo com a presente divulgação, pode exigir, para além dos ingredientes de drogas terapêuticas, excipientes ou aditivos, incluindo, mas não limitado a, diluentes, ligantes, lubrificantes, desintegrantes, corantes, aromatizantes, agentes edulcorantes e semelhantes ou suas misturas. Pela incorporação destes e outros aditivos, uma variedade de formas de dosagem (por exemplo, comprimidos, cápsulas, cápsulas ovais, pastilhas e semelhantes) podem ser feitas. Estes incluem, por exemplo, cápsulas de gelatina, cápsulas ovais, comprimidos de açúcar revestidos, comprimidos com revestimento entérico para atrasar ação, múltiplos comprimidos, comprimidos de ação prolongada, comprimidos para solução, comprimidos efervescentes, comprimidos bucais e sublinguais, pastilhas e semelhantes.
[0230] Assim, as formas de dose unitária ou formulações de dosagem de uma composição farmacêutica aqui descritas, tal como um pastilha, um comprimido ou uma cápsula, pode ser formado pela combinação de uma quantidade desejada de cada um dos ingredientes ativos com um ou mais excipientes farmaceuticamente compatíveis ou aceitáveis, como descrito abaixo, em quantidades farmaceuticamente compatíveis para se obter uma dose de formulação de dosagem unitária com a quantidade desejada de cada ingrediente ativo. A forma de dose ou formulação de dosagem pode ser formada por métodos bem conhecidos na arte.
[0231] Os comprimidos são muitas vezes uma forma de dosagem preferida por causa das vantagens oferecidas tanto para o paciente (por exemplo, a precisão da dosagem, suavidade portabilidade, compacidade, gosto, bem como a facilidade de administração) e para o fabricante (por exemplo, a simplicidade e economia de preparação, estabilidade, bem como conveniência no transporte, embalagem e distribuição). Os comprimidos são formas sólidas de dosagem farmacêuticas que contêm substâncias de drogas terapêuticas, com ou sem aditivos adequados.
[0232] Os comprimidos são tipicamente feitos por moldagem, por compressão ou por métodos de conformação geralmente aceitos. Por conseguinte, os comprimidos compactados são geralmente preparados por métodos de produção em grande escala, enquanto os comprimidos moldados frequentemente envolvem operações de pequena escala. Por exemplo, existem três métodos gerais de preparação do comprimido: (1) o métodode granulação úmida, (2) o método de granulação a seco e (3) compressão direta. Estes métodos são bem conhecidos dos peritos na arte. Veja, Remington: The Science and Practice of Pharmacy, 20th Ed. (2000). Veja também, U.S. Pharmacopeia XXI,U.S. Pharmacopeial Convention, Inc., Rockville, Md. (1985).
[0233] Várias formulações de comprimidos podem ser feitasem conformidade com os métodos e composições aqui descritas. Estes incluem formas de dosagem de comprimidos, tais como comprimidos de açúcar revestidos, comprimidos revestidos por película e comprimidos com revestimento entérico, múltiplos comprimidos, comprimidos de ação prolongada e semelhantes. Comprimidos revestidos com açúcar (SCT) são comprimidos comprimidos contendo um revestimento de açúcar. Tais revestimentos podem ser coloridos e são benéficos no encobrimento substâncias de drogas que possuam gostos desagradáveis ou odores e na proteção de materiais sensíveis à oxidação. Comprimidos revestidos com película (FCT) são comprimidos comprimidos que são cobertos com uma camada fina ou película de um material solúvel em água. Certo número de substâncias poliméricas com propriedades de formação de película pode ser utilizado. O revestimento de película transmite as mesmas características gerais como revestimento de açúcar com a vantagem adicional de um período de tempo muito reduzido ser necessário para a operação de revestimento. Comprimidos entéricos revestidos são também adequados para utilização em métodos e composições aqui descritas. Comprimidos entéricos revestidos (ECT) são comprimidos comprimidos revestidos com substâncias que resistem dissolução em fluido gástrico, mas se desintegram no intestino. Revestimento entérico pode ser usado para os comprimidos que contêm substâncias de drogas que são inativadas ou destruídas no estômago, para aqueles que irritam a mucosa ou como um meio de liberação retardada da medicação.
[0234] Vários comprimidos prensados (MCT) são comprimidos prensados feitos por mais que um ciclo de compressão, tais como comprimidos em camadas ou comprimidos prensados revestidos. Os comprimidos em camadas são preparados por compressão da granulação adicional do comprimido sobre uma granulação previamente comprimida. A operação pode ser repetida para produzir comprimidos de várias camadas de dois, três ou mais camadas. Normalmente, prensas especiais de comprimido são necessárias para fazer comprimidos em camadas. Ver, por exemplo, Patente US No. 5,213,738, incorporada aqui na sua totalidade por referência.
[0235] Comprimidos prensados revestidos são outra forma de comprimidos de compressão múltipla. Tais comprimidos, também referidos como comprimidos revestidos secos, são preparados por alimentação de comprimidos anteriormente prensados em uma máquina de prensa e comprimindo uma outra camada de granulação em torno dos comprimidos pré-formados. Estes comprimidos têm todas as vantagens de comprimidos prensados, isto é, encaixamento, monograma, velocidade de desintegração, etc, embora mantendo os atributos de comprimidos revestidos com açúcar em mascaramento do sabor da substância medicamentosa no núcleo do comprimido. Comprimidos prensados revestidos podem também ser usados para separar substâncias medicamentosas incompatíveis. Além disso, eles podem ser usados para fornecer um revestimento entérico para os comprimidos centrais. Ambos os tipos de comprimidos (isto é, comprimidos em camadas e comprimidos prensados revestidos), podem ser utilizados, por exemplo, na concepção de formas de ação de dosagem prolongada.
[0236] As composições farmacêuticas ou formas de dosagemunitárias aqui descritas sob a forma comprimidos de ação prolongada podem compreender comprimidos prensados formulados para liberar a substância da droga, de modo a fornecer a medicação ao longo de um período de tempo. Há um número de tipos de comprimidos de ação retardada que incluem comprimidos em que a liberação da substância droga é impedida por um intervalo de tempo após a administração ou até que certas condições fisiológicas existam. Os comprimidos de ação repetida podem ser formados por periodicamente liberar uma dose completa da substância medicamentosa para os fluidos gastrointestinais. Além disso, os comprimidos de liberação prolongada continuamente liberam incrementos da substância da droga contida aos fluidos gastrointestinais podem ser formados.
[0237] A fim de substâncias medicinais ou ingredientesterapêuticos aqui descritos, com ou sem excipientes, sejam feitas em formas de dosagem sólida (por exemplo, comprimidos) com pressão, utilizando equipamento disponível, é necessário que o material, na forma cristalina ou em pó, possua um número de características físicas. Estas características podem incluir, por exemplo, a capacidade de fluir livremente, como um pó para aderir sobre a compactação e para ser facilmente liberado de ferramentas. Uma vez que a maioria dos materiais não têm nenhum ou apenas algumas destas propriedades, os métodos de formulação de comprimidos e de preparação têm sido desenvolvidos para conferir estas características desejáveis para o material que está a ser prensado em um comprimido ou forma de dosagem semelhante.
[0238] Como observado, além das drogas ou ingredientesterapêuticos, comprimidos e formas de dosagem semelhantes podem conter um número de referidos materiais como excipientes ou aditivos. Estes aditivos são classificados de acordo com o seu papel na formulação da forma de dosagem tal como um comprimido, uma cápsula oval, uma cápsula, um pastilha ou semelhante. Um grupo de aditivos inclui, mas não estão limitados a, aglutinante, diluentes (enchimentos),desintegrantes, lubrificantes e agentes tensoativos. Em uma concretização, diluente, aglutinante, desintegrante,lubrificante e não são as mesmas coisas.
[0239] Um aglutinante é utilizado para fornecer um pó defluxo livre a partir da mistura de ingredientes do comprimido,de modo a que o material fluirá quando usado em uma máquina decomprimidos. O aglutinante também fornece uma coesão do comprimido. Muito pouco aglutinante vai dar problemas de fluxo e produzir comprimidos que não mantenham a sua integridade, enquanto que em excesso pode afetar negativamente a liberação (taxa de dissolução) dos medicamentos ou ingredientes ativos a partir do comprimido. Assim, uma quantidade suficiente de agente aglutinante deve ser incorporada no comprimido para proporcionar uma mistura de fluxo livre dos ingredientes do comprimido sem afetar adversamente a taxa de dissolução dos ingredientes da droga a partir do comprimido. Com comprimidos de dose mais baixa, a necessidade de boa compressibilidade pode ser eliminada em certa medida, pela utilização de excipientes de diluição adequados chamados compressão aids. A quantidade de aglutinante utilizado varia de acordo com o tipo de formulação e modo de administração e é prontamente perceptível para os peritos na arte.
[0240] Os aglutinantes adequados para utilização comformulações de dosagem aqui descritas incluem, mas não estão limitados a, amido de milho, amido de batata ou outros amidos, gomas de gelatinas naturais e sintéticas tais como acácia, alginato de sódio, ácido algínico, outros alginatos, goma guar de tragacanto em pó, celulose e seus derivados (por exemplo, etil-celulose, acetato de celulose, carboximetil celulose de cálcio, carboximetil celulose de sódio), polivinil pirrolidona (povidona), metil celulose, amido pré-gelatinaizado,hidroxipropil metil celulose (por exemplo, N°s 2208, 2906,2910), celulose microcristalina ou suas misturas. As formas adequadas de celulose microcristalina podem incluir, por exemplo, os materiais vendidos como AVICEL PH-101, AVICEL PH103 e AVICEL PH-105 (disponível na FMC Corporation, American Viscose Division, Avicel vendas, Marcus Hook, PA, EUA).
[0241] Enchimentos ou diluentes são utilizados para seobter o pó (por exemplo, no comprimido ou cápsula) em grandes quantidades, de modo que um comprimido de tamanho aceitável, cápsula ou outra forma de dosagem desejável é produzido. Tipicamente, os ingredientes terapêuticos são formados em uma forma de dosagem conveniente de tamanho adequado pela incorporação de um diluente com a forma de dosagem. Tal como acontece com o aglutinante, a ligação do fármaco(s) ao material de enchimento pode ocorrer e afetar a biodisponibilidade. Por conseguinte, uma quantidade suficiente de material de enchimento deve ser utilizada para alcançar uma razão de diluição desejada, sem afetar prejudicialmente a liberação dos ingredientes da droga a partir da forma de dosagem contendo o material de enchimento. Além disso, um material de enchimento que é fisicamente e quimicamente compatível com o ingrediente terapêutico(s) da forma de dosagem deve ser utilizado. A quantidade de material de enchimento utilizado varia de acordo com o tipo de formulação e modo de administração e é prontamente perceptível para os peritos na arte. Exemplos de enchimentos incluem, mas não estão limitados a, lactose, glucose, sacarose, frutose, Talco, carbonato de cálcio (por exemplo, grânulos ou pó), celulose microcristalina, celulose em pó, dextratos, caulim, manitol, xilitol, ácido silícico, sorbitol, amido, amido pré- gelatinaizado, ou suas misturas.
[0242] Desintegrantes são utilizados para gerar adesistegração da forma de dose (por exemplo, comprimido) quando exposto a um ambiente aquoso. Grande quantidade de um desintegrante irá produzir comprimidos que podem se desintegrar na garrafa devido à umidade atmosférica. Pouca quantidde pode ser insuficiente para a desintegração ocorrer e pode, assim, alterar a taxa e a extensão da liberação da droga(s) ou ingrediente ativo(s) a partir da forma de dosagem. Assim, uma quantidade suficiente de agente de desintegração que não é nem tão pouco, nem demasiado para prejudicialmente alterar a liberação dos ingredientes da droga deve ser usado para formar as formas de dosagem aqui descritas. A quantidade de desintegrante utilizada varia com base no tipo de formulação e modo de administração e é prontamente perceptível pelos especialistas na matéria. Exemplos de desintegrantes incluem, mas não estão limitados a, ágar-ágar, ácido algínico, carbonato de cálcio, celulose microcristalina, croscarmelose de sódio, crospovidona, potássio polacrilina, glicolato de amido de sódio, amido de batata ou de tapioca, outros amidos, amido pré-gelatinaizado, argilas, outras alginas, outras celuloses, gomas, ou suas misturas.
[0243] Quando uma forma de dose que se dissolve muito rapidamente após a administração ao sujeito, por exemplo, no estômago do sujeito é desejado, um super desintegrante pode ser utilizado, tal como, mas não limitado a, croscarmelose de sódio ou glicolato de amido de sódio. O termo "super desintegrante", como aqui usado, significa um desintegrante, que resulta em rápida desintegração do fármaco ou do ingrediente ativo no estômago após administração oral. A utilização de um super desintegrante pode facilitar a absorção rápida do fármaco ou do ingrediente ativo(s), que pode resultar em um início de ação mais rápida.
[0244] A adesão dos ingredientes da forma de dosagem para misturar paredes, tremonhas, telas, recipientes de transferência e todas as superfícies do equipamento, incluindo, mas não limitado a, perfuração da máquina de fabricação (por exemplo, uma máquina de comprimidos) e doseadores da máquina de fabricação de cápsula deve ser minimizada ou idealmente evitada. A adesão é um problema particular para as composições aqui descritas. Por exemplo, quando a droga se acumula sobre as superfícies de perfurador, faz com que a superfície do comprimido se torne esburacada e, portanto, inaceitável. Além disso, fixar a droga ou excipientes desta forma exige, desnecessariamente, que as concentraçãos elevadas de ejeção, quando da remoção do comprimido a partir do molde. Excessivaas concentraçãos de ejeção podem conduzir a uma taxa de quebra elevada e aumentar o custo de produção, para não mencionar desgaste excessivo nos moldes. Na prática, é possível reduzir a aderência por aglomeração úmida ou pelo uso de lubrificantes, por exemplo, estearato de magnésio e outros excipientes antiaderentes. No entanto, a seleção de um sal da droga com boas propriedades anti-adesão também pode minimizar estes problemas.
[0245] Como observado, o lubrificante é usado para melhorar o fluxo da mistura de pó de comprimidos para a máquina de comprimidos e para evitar a aderência do comprimido no molde, após o comprimido ser comprimido. Pouca quantidade de lubrificante não permitirá que comprimidos satisfatórios sejam feitos e grande quantidade pode produzir um comprimido com um revestimento hidrofóbico impermeável a água, que pode se formar porque os lubrificantes são geralmente materiais hidrofóbicos, tais como ácido esteárico, estearato de magnésio, estearato de cálcio e semelhantes. Além disso, um revestimento hidrofóbico impermeável à água pode inibir a desintegração do comprimido e dissolução do ingrediente da droga(s). Assim, uma quantidade suficiente de lubrificante deve ser usada de modo que prontamente permita liberação do comprimido prensado a partir do molde, sem formar um revestimento hidrofóbico impermeável à água que prejudicialmente interfere na desintegração desejada e/ou dissolução do ingrediente da droga(s).
[0246] Exemplos de lubrificantes adequados para utilizaçãocom as composições aqui descritas incluem, mas não estão limitados a, estearato de cálcio, estearato de magnésio, óleo mineral, óleo mineral leve, glicerina, sorbitol, manitol, polietileno glicol, outros glicóis, ácido esteárico, lauril sulfato de sódio, Talco, óleo vegetal hidrogenado (por exemplo, óleo de amendoim, óleo de semente de algodão, óleo de girassol, óleo de sésamo, óleo de oliva, óleo de milho e óleo de soja), estearato de zinco, oleato de etila, laurato de etila, agar, ou suas misturas. Lubrificantes adicionais incluem, por exemplo, uma sílica gel Syloid (Aerosil 200, fabricado pela WR Grace Co. de Baltimore MD), um aerossol coagulado de sílica sintética (comercializado por Deaussa Co. de Plano, Texas), CAB-O-SIL (um produto de dióxido de silício pirogénico vendido por Cabot Co., de Boston, Massachusetts) ou suas misturas.
[0247] Os surfactantes são usados nas formas de dosagempara melhorar as características molhantes e/ou para melhorar a dissolução e são particularmente úteis em composições farmacêuticas ou formas de dosagem que contêm droga pouco solúvel ou insolúvel(s) ou ingredientes ativos. Exemplos de tensoativos incluem, mas não estão limitados a, ésteres de ácidos gordos de polioxietileno sorbitano, tais como aqueles disponíveis comercialmente como TWEENs (por exemplo, Tween 20 e Tween 80), polietileno glicóis, estearatos depolioxietileno, álcool polivinílico, polivinilpirrolidona, bloco de copolímeros poli (oxietileno)/poli (oxipropileno) tais como poloxâmeros (por exemplo, disponível comercialmente como PLURONICs) e copolímeros em bloco tetrafuncionais derivados de adição sequencial de óxido de propileno e óxido de etileno a etilenodiamina, tais como poliaminas (por exemplo, comercialmente como TETRONICs (BASF)), dextrano, lecitina, dialquilesteres de ácido sulfossuccínico de sódio, tais como Aerosol OT, lauril sulfato de sódio, alquil- sulfonatos de poliéter arilo ou álcoois, tais como TRITON X200 ou tiloxapol, p-isononilfenoxipoli (glicidol) (por exemplo Olin-10G ou surfactante 10-G (Olin Chemicals) ou suas misturas. Outros agentes tensoativos farmaceuticamente aceitáveis são bem conhecidos na arte e são descritos em detalhe no Handbook of Pharmaceutical Excipients.
[0248] Outras classes de aditivos para utilização com as composições farmacêuticas ou formas de dosagem aqui descritas incluem, mas não estão limitados a, agentes anti-aglomerantes ou antiaderentes, conservantes antimicrobianos, agentes de revestimento, corantes, dessecantes, sabores e perfumes, plastificantes, agentes de aumento da viscosidade, edulcorantes, agentes tamponantes, umectantes e semelhantes.
[0249] Exemplos de agentes anti-aglomerantes incluem, mas não estão limitados a, silicato de cálcio, silicato de magnésio, dióxido de silício, dióxido de silício coloidal, Talco ou suas misturas.
[0250] Exemplos de conservantes antimicrobianos incluem, mas não estão limitados a, solução de cloreto de benzalcónio, cloreto de benzetônio, ácido benzóico, álcool benzílico, butil parabeno, cloreto de cetilpiridínio, clorobutanol, cresol, ácido desidroacético, etilparabeno, metilparabeno, fenol, álcool feniletílico, acetato fenilmercúrico, nitrato fenilmercúrico, sorbato de potássio, propilparabeno, benzoato de sódio, dehidroacetato de sódio, propionato de sódio, ácido sórbico, Temporsol, timol ou suas misturas.
[0251] Exemplos de corantes para utilização com as composições aqui descritas incluem, mas não estão limitados a, corantes e pigmentos farmaceuticamente aceitáveis, caramelo, óxido férrico vermelho, óxido férrico amarelo ou suas misturas. Exemplos de dessecantes incluem, mas não estão limitados a, cloreto de cálcio, sulfato de cálcio, sílica gel ou suas misturas.
[0252] Sabores que podem ser utilizados incluem, mas não estão limitados a, acácia, tragacanto, óleo de amêndoa, anetol, óleo de anis, benzaldeído, alcaravia, óleo de alcaravia, cardamomo, óleo de semente de cardamomo, tintura composta cardamomo, sumo de cereja, canela, óleo de canela, óleo de cravo, cacau, óleo de coentros, eriodictyon, extrato fluido de eriodictyon, acetato de etila, etil vanilina, óleo de eucalipto, óleo de funcho, Glycyrrhiza, extrato puro de Glycyrrhiza, extrato fluido Glycyrrhiza, óleo de lavanda, óleo de limão, mentol, salicilato de metila, glutamato monossódico, óleo de noz-moscada, óleo de flor de laranja, água de flor de laranja, óleo de laranja, tintura doce de casca de laranja, solução alcoólica de composto de laranja, hortelã-pimenta, óleo de hortelã-pimenta, solução alcoólica de hortelã-pimenta, óleo de pinheiro, óleo de rosa, forte água de rosas, hortelã, óleo de hortelã, timol, tintura de tolu bálsamo, baunilha, tintura de baunilha e vanilina ou sua mistura.
[0253] Exemplos de agentes edulcorantes incluem, mas não estão limitados a, aspartame, dextratos, manitol, sacarina, sacarina de cálcio, sacarina de sódio, solução de sorbitol, sorbitol, ou suas misturas.
[0254] Exemplos de plastificantes para utilização com as composições aqui descritas incluem, mas não estão limitados a, óleo de rícino, monoglicéridos diacetilados, ftalato de dietila, glicerina, monoglicéridos mono e di acetilados, polietileno glicol, propileno glicol e triacetina ou suas misturas. Agentes de aumento de viscosidade adequados incluem, mas não estão limitados a, acácia, agar, ácido alamico, monoestearato de alumínio, bentonita, magma bentonita, carbómero 934, carboximetilcelulose de cálcio, carboximetilcelulose de sódio, carboximetilcelulose de sódio 12, carragenano, celulose, celulose microcristalina, gelatina, goma guar, hidroxietil celulose, hidroxipropil celulose, hidroxipropil metilcelulose (Nos. 2208; 2906; 2910), silicato de alumínio de magnésio, metilcelulose, pectina, álcool polivinílico, povidona, sílica gel, dióxido de silício coloidal, alginato de sódio, goma de tragacanto e xantana ou suas misturas .
[0255] Agentes tampão que podem ser utilizados nas composições aqui descritas incluem, mas não estão limitados a, hidróxido de magnésio, hidróxido de alumínio e semelhantes ou suas misturas. Exemplos de umectantes incluem, mas não estão limitados a, glicerol, outros umectantes ou suas misturas.
[0256] As formas de dosagem aqui descritas podem ainda incluir um ou mais dos seguintes: (1) agentes de retardamento de dissolução, tais como parafina; (2) aceleradores de absorção, tais como compostos de amônio quaternário, (3) agentes molhantes, tais como, por exemplo, álcool cetílico e o monoestearato de glicerol; (4) absorventes, tais como caulim e argila de bentonite, (5) antioxidantes, tais comoantioxidantes solúveis em água (por exemplo, ácido ascórbico, cloridrato de cisteína, bissulfato de sódio, metabisulfato de sódio, sulfito de sódio e semelhantes), solúveis em óleo antioxidantes (por exemplo, palmitato de ascorbilo,hidroxianisole (BHA), butilado de hidroxitolueno (BHT), lecitina, galato de propila, alfa-tocoferol e semelhantes); e (6) os agentes quelantes metálicos, tais como ácido cítrico, ácido de etilenodiamina tetracético (EDTA), sorbitol, ácido tartárico, ácido fosfórico e semelhantes.
[0257] As formas de dosagem aqui descritas, tais como umcomprimido ou cápsula, podem opcionalmente ser revestidas. Agentes de revestimento inertes compreendem tipicamente um agente formador de película inerte disperso em um solvente adequado, e pode ainda compreender outros adjuvantes farmaceuticamente aceitáveis, tais como corantes eplastificantes. Agentes de revestimento inertes adequados e métodos para revestimento, são bem conhecidos na arte, incluindo, sem limitação, técnicas de revestimento de película aquosas ou não-aquosas ou de microencapsulação. Exemplos de agentes de formação de película ou revestimento incluem, mas não estão limitados a, gelatina, esmalte farmacêutico, goma- laca, sacarose, dióxido de titânio, cera de carnaúba, cera microcristalina, celuloses, tais como metilcelulose,hidroximetil celulose, carboximetilcelulose, ftalato acetato de celulose, hidroxipropil metilcelulose (por exemplo, N°s: 2208, 2906, 2910), hidroxipropil celulose, ftalato dehidroxipropil metil celulose (por exemplo, N°s: 200, 731, 220, 824), hidroxietilcelulose, metilhidroxietilcelulose,etilcelulose, que pode opcionalmente ser reticulada, e carboximetil celulose de sódio; vinílicos, tais como pirrolidiona de polivinila, ftalato de acetato de polivinila; glicóis, tais como polietileno glicóis; acrílicos, tais como copolímero de éster de ácido metacrilato de metacrilato de dimetilaminoetil e copolímero de metilmetacrilato de etilacrilato e polímeros de hidratos de carbono, tais como maltodextrinas e polidextrose ou as suas misturas. A quantidade de agente de revestimento e do instrumento transportador (aquoso ou não aquoso) utilizado, varia de acordo com o tipo de formulação e modo de administração e é prontamente perceptível para os peritos na arte.
[0258] Um revestimento de um polímero formador de películapode, opcionalmente, ser aplicado a um comprimido ou cápsula (por exemplo, um comprimido em forma de cápsula) usando um dos vários tipos de aparelhos, tais como uma auto-clave de revestimento convencional, coluna de suspensão em ar Accelacota, High-Cola ou Worster. Tal equipamento tem, tipicamente, um sistema de escape para remover a poeira e os vapores do solvente ou da água para facilitar a secagem rápida. Pistolas de pulverização ou outro equipamento de atomização adequado pode ser introduzido na auto-clave de revestimento para gerar padrões de pulverização conducentes a cobertura rápida e uniforme do leito dos comprimidos.Normalmente, o ar de secagem aquecido ou frio é introduzido sobre o leito comprimido de forma contínua ou alternada com um ciclo de pulverização para acelerar a secagem da solução de revestimento de filme.
[0259] A solução de revestimento pode ser pulverizadausando deslocamento positivo pneumático ou sistemas de bomba peristáltica em um ciclo de pulverização a seco contínuo ou intermitente. O tipo particular de aplicação por pulverização é selecionado dependendo da eficiência de secagem da autoclave de revestimento. Na maioria dos casos, o material de revestimento é pulverizado até que os comprimidos sejam uniformemente revestidos com a espessura desejada e a desejada aparência do comprimido seja alcançada. Muitos tipos diferentes de revestimentos podem ser aplicados, tais como entéricos, revestimentos de liberação lenta ou revestimentos do tipo de dissolução rápida para uma rápida atuação dos comprimidos. De preferência, revestimentos do tipo de dissolução rápida são utilizados para permitir a liberação mais rápida dos ingredientes ativos, resultando no surgimento acelerado. A espessura do revestimento do polímero formador da película aplicado a um comprimido, por exemplo, pode variar. No entanto, é preferível que a espessura simule a aparência, percepção (tátil e boca) e a função de uma cápsula de gelatina. Onde liberação mais rápida ou retardada do agente terapêutico(s) é desejada, um perito na arte seiria facilmente reconhecer o tipo de filme e espessura, se for o caso, com base em características, tais como, os níveis sanguíneos pretendidos de ingrediente ativo, a taxa de liberação, a solubilidade do ingrediente ativo e desempenho desejado da forma de dosagem.
[0260] Um número de agentes formadores de películaadequados para a utilização em revestimento de uma forma de dosagem final, tais como comprimidos incluem, por exemplo, metilcelulose, hidroxipropil metil celulose (Pharmacoat 606 6 cps), polivinilpirrolidona (povidona), etilcelulose (Ethocel 10 cps), vários derivados de ácidos metacrílico e ésteres de ácido metacrílico, ftalato de acetato de celulose ou suas misturas.
[0261] O método de preparação e os excipientes ou aditivos a serem incorporadoas na forma de dosagem (tal como um comprimido ou cápsula) são selecionados a fim de dar a formulação de comprimidos as características físicas desejáveis, permitindo simultaneamente a facilidade de fabricação (por exemplo, a compressão rápida dos comprimidos). Depois da fabricação, a forma de dose, de preferência, deve ter um número de atributos adicionais, por exemplo, para comprimidos, tais atributos incluem aparência, a dureza capacidade de desintegração e uniformidade, que são influenciados tanto pelo método de preparação quanto pelos aditivos presentes no comprimido formulação.
[0262] Além disso, é de notar que os comprimidos ou outras formas de dosagem das composições farmacêuticas aqui descritas devem manter a sua origem de tamanho, forma, peso e cor sob a manipulação normal e condições de armazenamento ao longo da sua vida de prateleira. Assim, por exemplo, pó excessivo ou partículas sólidas na parte inferior do recipiente, fissuras ou chips na superfície de um comprimido ou o aparecimento de cristais na superfície dos comprimidos ou em paredes do recipiente são indicativos de instabilidade física dos comprimidos não revestidos. Assim, o efeito de agitação suave, uniforme e reprodutível e caimento de comprimidos deve ser realizado para assegurar que os comprimidos têm estabilidade física suficiente. Dureza dos comprimidos pode ser determinada por durômetros comercialmente disponíveis. Além disso, a disponibilidade in vitro dos ingredientes ativos não deve alterar apreciavelmente com o tempo.
[0263] Os comprimidos e outras formas de dosagem das composições farmacêuticas aqui descritas, tais como dragas, cápsulas, pílulas e grânulos, podem opcionalmente ser marcados ou preparados com revestimentos e invólucros, tais como revestimentos entéricos e outros revestimentos bem conhecidos na arte da formulação farmacêutica.5.2.2 Formas de Dosagem parenteral
[0264] As formas de dosagem parenteral podem ser administradas aos pacientes por várias vias, incluindo, mas não se limitam a, subcutânea, intravenosa (incluindo injeção de pílula grande), intramuscular e intrarterial. Devido a sua administração tipicamente contornar as defesas naturais dos pacientes contra contaminantes, formas de dosagem parenterais são de preferência estéril ou capaz de ser esterilizada antes da administração a um paciente. Exemplos de formas de dosagem parenterais incluem, mas não estão limitados a, soluções prontas para injeção, produtos secos prontos para serem dissolvidos ou suspensos em um veículo farmaceuticamente aceitável para injeção, suspensões prontas para injeção e emulsões.
[0265] Veículos adequados que podem ser usados para fornecer formas de dosagem parenterais aqui descritos são bem conhecidos dos peritos na arte. Os exemplos incluem, mas não estão limitados a: água para injeção USP; dispositivos aquosos, tais como, mas não limitado a, injeção de cloreto de sódio, injeção de Ringer, injeção de dextrose, injeção de dextrose e cloreto de sódio e injeção de lactato de Ringer; veículos miscíveis em água, tais como, mas não se limitam a, álcool etílico, polietileno glicol e polipropileno glicol, e veículos não-aquosos, tais como, mas não se limitam a, óleo de milho, óleo de semente de algodão, óleo de amendoim, óleo de sésamo, oleato de etila, miristato de isopropila e benzoato de benzila.
[0266] Os compostos que aumentam a solubilidade de um ou mais ingredientes ativos (isto é, os compostos utilizados nos métodos e composições aqui descritas) aqui revelados podem também ser incorporados nas formas de dosagem parenterais.5.2.3 Formas de Dosagem transdérmica, tópica e mucosal
[0267] As formas de dosagem transdérmica, tópica e mucosal descritas aqui incluem, mas não estão limitados a, soluções oftálmicas, sprays, aerossóis, cremes, loções, pomadas, géis, soluções, emulsões, suspensões ou outras formas conhecidas de um perito na arte. Ver, por exemplo, Remington’s Pharmaceutical Sciences, 16th and 18th eds., Mack Publishing, Easton PA (1980 & 1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). As formas de dosagem transdérmica incluem esparadrapos "tipo reservatório" ou "tipo de matriz", que podem ser aplicadas à pele e usadas por um período específico de tempo para permitir a penetração de uma quantidade desejada de ingredientes ativos.
[0268] Os excipientes adequados (por exemplo, transportadores e diluentes) e outros materiais que podem ser usados para fornecer formas de dosagem transdérmica, tópica e mucosal aqui descritas são bem conhecidos para os peritos nas artes farmacêuticas e dependem do tecido particular para o qual uma dada forma de composição farmacêutica ou de dosagem irá ser aplicada.
[0269] Dependendo do tecido específico a ser tratado, componentes adicionais podem ser utilizados antes, em conjunto com ou subsequentemente ao tratamento com ingredientes ativos aqui descritos. Por exemplo, promotores de penetração podem ser usados para ajudar na entrega dos ingredientes ativos para o tecido.
[0270] O pH de uma composição farmacêutica ou forma de dosagem ou do tecido para o qual a composição farmacêutica ou forma de dosagem é aplicada, pode também ser ajustado para melhorar a entrega de um ou mais ingredientes ativos. Do mesmo modo, a polaridade de um transportador de solvente, a sua concentração iônica ou tonicidade pode ser ajustada para melhorar a entrega. Compostos, tais como estearatos, também podem ser adicionados a composições farmacêuticas ou formas de dosagem para vantajosamente alterar a hidrofilicidade ou lipofilicidade de um ou mais ingredientes ativos, de modo a melhorar a entrega. A este respeito, estearatos podem servir como um dispositivo lipídico para a formulação, como um agente emulsionante ou agente tensoativo e como um agente acentuador de entrega ou acentuador de penetração. Diferentes sais ou solvatos (por exemplo, hidratos) dos ingredientes ativos podem ser usados para ajustar mais as propriedades da composição resultante.5.2.4 Composições com estabilidade acentuada
[0271] A adequação de um excipiente particular podedepender também dos ingredientes ativos específicos sob a forma de dosagem. Por exemplo, a decomposição de um ingrediente ativo, por exemplo, transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato, pode ser acelerada por certos excipientes. Determinados sacarideos, particularmente mono ou di sacarídeos, podem acelerar a decomposição do ingrediente ativo de uma composição aqui descrita. Por exemplo, composições compreendendo transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato devem conter pouca, se alguma, lactose, manose, xilose ou celulosemicrocristalina.
[0272] Além disso, composições farmacêuticas anidras eformas de dosagem descritas são caracterizadas por compreender ingredientes ativos, uma vez que a água pode facilitar a degradação de alguns compostos. Por exemplo, a adição de água (por exemplo, 5%) é amplamente aceita nas artes farmacêuticas como um meio de simulação de armazenamento em longo prazo, a fim de determinar características, tais como vida de prateleira ou estabilidade de formulações ao longo do tempo. Ver, por exemplo, Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995,pp. 379-80. Com efeito, a água e o calor aceleram a decomposição de alguns compostos. Assim, o efeito da água em uma formulação pode ser de grande importância, visto que a umidade e/ou umidade são vulgarmente encontradas durante a fabricação, manuseamento, embalagem, armazenagem, transporte e uso de formulações.
[0273] Composições farmacêuticas anidras e formas de dosagem aqui descritas podem ser preparadas utilizando ingredientes anidros ou que contenham baixa umidade e condições de baixa umidade. As composições farmacêuticas e formas de dosagem aqui descritas podem ser anidras, se substancial contato com o orvalho e/ou umidade durante fabricação, embalagem e/ou armazenamento é esperado.
[0274] Uma composição farmacêutica anidra deve ser preparada e armazenada tal que a sua natureza anidra seja mantida. Assim, as composições anidras são, de preferência, embaladas utilizando materiais conhecidos para evitar a exposição à água, de tal modo que eles podem ser incluídos em kits de fórmulas adequados. Exemplos de embalagens adequadas incluem, mas não estão limitados a, películas hermeticamente fechadas, plásticos, recipientes de dose unitária (por exemplo, frascos), pacotes de blisters e pacotes de tiras.
[0275] Também aqui descritas estão composições farmacêuticas e formas de dosagem caracterizadas por compreender um ou mais compostos que reduzem a taxa à qual um ingrediente ativo irá decompor. Tais compostos, que são aqui referidos como "estabilizadores," incluem, mas não estão limitados a, antioxidantes tais como ácido ascórbico, tampões de pH, ou tampões de sal.
[0276] Exemplos não limitativos específicos de composições farmacêuticas estáveis são aqui descritas nos Exemplos 6.1 a 6.13.
[0277] Tal como as quantidades e tipos de excipientes, as quantidades e tipos específicos de ingredientes ativos em uma forma de dosagem podem variar dependendo de fatores tais como, mas não limitado a, via pela qual é para ser administrado a pacientes.5.2.5 Formas de Dosagem de liberação lenta
[0278] Os ingredientes ativos utilizados em métodos ecomposições aqui descritos podem ser administrados por meios de liberação controlada ou por dispositivos de entrega que são bem conhecidos de qualquer perito na arte. Os exemplos incluem, mas não estão limitados a, aqueles descritos nas Patentes U.S. N°s: 3,845,770; 3,916,899; 3,536,809; 3,598,123;e 4,008,719, 5,674,533, 5,059,595, 5,591,767, 5,120,548,5,073,543, 5,639,476, 5,354,556, e 5,733,566, cada uma dasquais é incorporada aqui por referência. Tais formas de dosagem podem ser usadas para fornecer liberação lenta ou controlada de um ou mais ingredientes ativos, utilizando, por exemplo, hidroxipropilmetil celulose, outras matrizes de polímero, géis, membranas permeáveis, sistemas osmóticos, revestimentos de multicamadas, micropartículas, lipossomas, microesferas ou uma combinação dos mesmos para fornecer o perfil de liberação desejado em proporções variáveis. Formulações de liberação controlada adequadas, conhecidas de qualquer perito na arte, incluindo os aqui descritos, podem ser prontamente selecionadas para utilização com os compostos utilizados nos métodos e composições aqui descritos. Assim, aqui descritas estão formas de dosagem unitárias adequadas para administração oral tais como, mas não se limitam a, comprimidos, cápsulas, gelcaps e cápsulas ovais que estão adaptados para liberação controlada.
[0279] Todos os produtos farmacêuticos de liberaçãocontrolada têm um objetivo comum de melhorar a terapia medicamentosa acima do atingido pelas suas cópias não controladas. Idealmente, o uso de uma preparação de liberação controlada concebida no tratamento médico é caracterizada pelo emprego mínimo de substância de fármaco a ser utilizada para curar ou controlar a condição em uma quantidade mínima de tempo. Vantagens das formulações de liberação controlada incluem atividade extendida da droga, reduzida frequência de dosagem e aumento da flexibilidade do paciente. Além disso, formulações de liberação controlada podem ser utilizadas para afetar o tempo de início de ação ou outras características, tais como os níveis sanguíneos da droga e pode, assim, influenciar a ocorrência de outros efeitos (por exemplo, adverso).
[0280] A maioria das formulações de liberação controlada éconcebida para liberar, inicialmente, uma quantidade de droga (ingrediente ativo) que prontamente produz o efeitoterapêutico desejado e, gradualmente e continuamente, libera outros montantes da droga para manter esse nível de efeito terapêutico ou profiláctico durante um período prolongado de tempo. A fim de manter esse nível de fármaco constante no corpo, a droga deve ser liberada a partir da forma de dosagem a uma taxa que irá substituir a quantidade de droga a ser metabolizada e excretada do corpo. Ingrediente ativo de liberação controlada pode ser estimulado por várias condições, incluindo, mas não se limitando a, pH, temperatura, enzimas, água ou outras condições fisiológicas ou compostos.5.2.6 Kits
[0281] Em alguns casos, os ingredientes ativos utilizadosnos métodos e composições aqui descritos são, de preferência, não administrados a um paciente ao mesmo tempo ou pela mesma via de administração. Por conseguinte, descritos na presente invenção estão kits que, quando utilizado pelo médico, podem simplificar a administração de quantidades apropriadas de ingredientes ativos a um paciente.
[0282] Em uma forma de concretização, o kit é caracterizadopor compreender uma unidade de forma de dosagem única dos compostos utilizados nos métodos e composições aqui descritas ou seu sal farmaceuticamente aceitável, solvato ou seu estereoisômero e uma unidade de forma de dosagem única de outro agente que possa ser usado em combinação com esses compostos. Kits aqui descritos podem ainda compreender dispositivos que são usados para controlar os ingredientes ativos. Exemplos de tais dispositivos incluem, mas não estão limitados a, seringas, sacos de gotejamento, emplastros e inaladores.
[0283] Kits descritos na presente invenção podem aindacompreender veículos farmaceuticamente aceitáveis que podem ser utilizados para controle de um ou mais ingredientes ativos. Por exemplo, se um ingrediente ativo é descrito em uma forma sólida que tem de ser reconstituído para administração parenteral, o kit pode compreender um recipiente selado de um dispositivo adequado, em que o ingrediente ativo pode ser dissolvido para formar uma solução estéril livre de partículas, que é adequada para administração parenteral. Exemplos de veículos farmaceuticamente aceitáveis incluem, mas não estão limitados a: água para injeção USP; veículosaquosos, tais como, mas não limitado a, injeção de cloreto de sódio, injeção de Ringer, injeção de dextrose, injeção de dextrose e cloreto de sódio e injeção de lactato de Ringer, veículos miscíveis em água, tais como, mas não limitados a, álcool etílico, polietileno glicol e polipropileno glicol, e dispositivos não aquosos, tais como, mas não limitados a, óleo de milho, óleo de semente de algodão, óleo de amendoim, óleo de sésamo, oleato de etila, miristato de isopropila, benzoato de benzila.
[0284] Certas realizações são exemplificadas nos exemplosnão limitativos a seguir. Será evidente para os peritos na arte que muitas modificações, tanto de materiais quanto de métodos, podem ser praticadas sem se afastar do sentido e âmbito da presente divulgação.5.3 Métodos de Prevenção, Tratamento e Controle
[0285] Em uma forma de concretização descrita na presenteinvenção está um método de tratamento, prevenção ou controle um distúrbio do sistema nervoso central caracterizado por compreender a administração a um sujeito (por exemplo, o paciente) de uma quantidade terapeuticamente ouprofilaticamente eficaz de uma formulação de sal ou polimorfo de transnorsertralina como aqui revelado.
[0286] Em uma forma de concretização descrita na presenteinvenção está um método de gerar um efeito anti-depressor. O método é caracterizado por compreender a administração a um sujeito de uma quantidade terapeuticamente eficaz de uma formulação, sal ou polimorfo de transnorsertralina como aqui revelado. Efeitos anti-depressivos semelhantes podem ser medidos utilizando um modelo animal da doença, tais como aqueles conhecidos na arte e aqueles aqui descritos.
[0287] Em outras modalidades, um distúrbio neurológica é:depressão (por exemplo, transtorno depressivo maior,transtorno bipolar, transtorno unipolar, distimia e transtorno afetivo sazonal); déficits cognitivos, fibromialgia, dor (por exemplo, dor neuropática); distúrbios do sono relacionados (por exemplo, apnéia ao dormir, insônia, narcolepsia, cataplexia), incluindo os transtornos do sono que são produzidos por distúrbios psiquiátricos, síndrome da fadiga crônica; transtorno de déficit de atenção (ADD), déficit de atenção e hiperatividade (TDAH), síndrome de pernas inquietas; esquizofrenia; ansiedades (por exemplo, ansiedade geral, transtorno de ansiedade social, transtorno do pânico); transtorno obsessivo compulsivo; transtorno de estresse pós- traumático; transtorno afetivo sazonal (SAD); disforia pré- menstrual, sintomas pós-menopausa vasomotores (por exemplo, afrontamentos, suores noturnos), doença neurodegenerativa (por exemplo, doença de Parkinson, doença de Alzheimer e esclerose lateral amiotrófica), condições de maníacos; transtorno distímico, transtorno ciclotímico, obesidade e abuso de substâncias ou dependência (por exemplo, o vício em cocaína, dependência da nicotina). Em outra concretização, os compostos aqui descritos são úteis para o tratamento de duas ou mais condições/distúrbios, que são comórbida, tais como déficit cognitivo e depressão.
[0288] Em certas realizações, distúrbios neurológicosincluem distúrbios da função cerebral, incluindo, sem limitação, demência senil, demência tipo de Alzheimer, cognição, perda de memória, amnésia/síndrome amnésica, epilepsia, perturbações de consciência, coma, diminuição da atenção, perturbações da fala, síndrome de Lennox, autismo e síndrome hipercinética.
[0289] A dor neuropática inclui, sem limitação a, neuralgia pós herpética (ou pós-zona), distrofia/causalgia reflexo simpático ou trauma de nervo, dor do membro aparente, síndrome do túnel do carpo e neuropatia periférica (como a neuropatia diabética ou neuropatia decorrente do uso crônico de álcool).
[0290] Outros exemplos de doenças e condições que podem ser tratados, impedidos e/ou controlados com os métodos e/ou composições aqui descritos incluem, mas não estão limitados a: obesidade; enxaqueca ou dor de cabeça com enxaqueca; incontinência urinária, incluindo, sem limitação micção involuntária de urina, drible ou perda de urina, estresse incontinência urinária (IUE), incontinência urgente, incontinência de esforço urinária, incontinência reflexa, incontinência passiva e incontinência por transbordamento e disfunção sexual, em homens ou mulheres, incluindo, sem limitação de disfunção sexual causada por violência psicológica e/ou fatores fisiológicos, disfunção erétil, ejaculação precoce, ressecamento vaginal, falta de excitação sexual, incapacidade de obter orgasmo e psico-disfunção sexual, incluindo, sem limitação, inibição do desejo sexual, excitação sexual inibida, inibição do orgasmo feminino, inibição do orgasmo masculino, dispareunia funcional, vaginismo funcional e disfunção psicossexual atípica.
[0291] Em uma forma de concretização, o distúrbio neurológico é a depressão. Em outra concretização, o distúrbio neurológico é distúrbio de ansiedade. Em outra concretização, o distúrbio neurológico é a dor. Em outra concretização, o distúrbio neurológico é a dor neuropática. Em outra concretização, a dor neuropática é neuropatia diabética.
[0292] Em uma forma de concretização, o distúrbio neurológico é uma doença neurodegenerativa. Em uma forma de concretização, a doença neurodegenerativa é a doença de Parkinson. Em outra forma de concretização, a doença neurodegenerativa é a doença de Alzheimer.
[0293] Em uma forma de concretização, o distúrbio neurológico é a incontinência, por exemplo, a incontinência urinária. Em outra concretização, o distúrbio neurológico é a disfunção sexual.
[0294] Em uma forma de concretização, o distúrbio neurológico é a obesidade e a quantidade terapeuticamente eficaz do composto para fornecer a um paciente é suficiente para que o referido paciente sinta-se saciado.
[0295] Em uma forma de concretização, os compostos aqui descritos tratam, previnem e/ou controlam um distúrbio do nervo central, sem causar vício de compostos referidos.
[0296] Em algumas realizações, os métodos aqui descritos podem, opcionalmente, ser caracterizados por compreender a administração de um ou mais outros agentes ativos. Tais outros agentes incluem, mas não estão limitados a, fármacos ou terapias convencionalmente utilizados para o tratamento, prevenção e/ou controle de distúrbios neurológicos aqui descritos.
[0297] Qualquer via de administração adequada pode ser empregada para fornecer ao paciente uma dose terapeuticamente ou profilaticamente eficaz de um ingrediente ativo. Por exemplo, oral, mucosal (por exemplo, nasal, sublingual, bucal, retal, vaginal), parenteral (por exemplo, intravenosa, intramuscular), transdérmica e subcutânea podem ser empregados. Exemplos de rotas de administração incluem oral, transdermal e mucosas. Formas de dosagem adequadas para tais rotas incluem, mas não estão limitados a, sistemas transdérmicos, soluções oftálmicas, sprays e aerossóis. As composições transdérmicas podem também tomar a forma de cremes, loções e/ou emulsões, que podem ser incluídos em um adesivo adequado para aplicação na pele ou pode ser incluído em um sistema transdérmico do tipo matriz ou reservatório como são convencionais na arte para esta finalidade. Uma forma de dosagem transdérmica exemplar é um esparadrapo "tipo reservatório" ou "tipo matriz", que é aplicado à pele e usado por um período específico de tempo para permitir a penetração de uma quantidade desejada de ingrediente ativo. O adesivo pode ser substituído por um esparadrapo novo quando necessário, para proporcionar a administração constante do ingrediente ativo para o paciente.
[0298] A quantidade a ser administrada a um sujeito (por exemplo, o paciente) para tratar, prevenir e/ou controlar as desordens descritas na presente invenção dependerá de uma variedade de fatores incluindo a atividade do composto particular utilizado, a via de administração, o tempo de administração, a taxa de excreção ou metabolismo do composto particular a ser empregado, a duração do tratamento, outras drogas, compostos e/ou materiais usados em combinação com o composto particular empregado, a idade, sexo, peso, condição, saúde geral e histórico médico anterior do paciente a ser tratado e fatores semelhantes bem conhecidos nas artes médicas.
[0299] Um médico ou veterinário com conhecimentos na artepodem prontamente determinar e prescrever a quantidade eficaz necessária. Por exemplo, o médico ou veterinário pode iniciar doses dos compostos empregados em níveis mais baixos do que o requerido para atingir o efeito terapêutico desejado e aumentar gradualmente a dosagem até que o efeito desejado seja alcançado.
[0300] Em geral, uma dose diária adequada de um compostodescrito na presente invenção será a quantidade do composto que é a dose eficaz mais baixa para produzir um efeito terapêutico ou profilático. Tal dose eficaz dependerá geralmente dos fatores descritos acima. Geralmente, as doses orais, intravenosas, intracerebroventricular e subcutânea dos compostos aqui descritos para um paciente variará entre cerca de 0,005mg por kg a cerca de 5mg por kg de peso corporal por dia. Em uma forma de concretização, a dose oral de um composto aqui descrito irá variar entre cerca de 0,05mg a cerca de 5g por dia. Em uma forma de concretização, a dose oral de um composto aqui descrito irá variar entre cerca de 0,1mg a cerca de 3g por dia. Em uma forma de concretização, a dose oral de um composto aqui descrito irá variar entre cerca de 0,25mg a cerca de 2g por dia. Em uma forma de concretização, a dose oral de um composto aqui descrito irá variar entre cerca de 0,5mg a cerca de 1g por dia. Em uma forma de concretização, a dose oral de um composto aqui descrito irá variar entre cercade 1mg a cerca de 500mg por dia. Em outra concretização, a dose oral de um composto aqui descrito irá variar entre cerca de 2mg a cerca de 250mg por dia. Em outra concretização, a dose oral de um composto aqui descrito irá variar de cerca de 3mg a cerca de 300mg por dia. Em uma forma de concretização, a dose oral de um composto aqui descrito irá variar entre cerca de 5mg a cerca de 300mg por dia. Em outra concretização, a dose oral de um composto aqui descrito irá variar de cerca de 10mg a cerca de 100mg por dia. Em outra concretização, a dose oral de um composto aqui descrito irá variar de cerca de 25mg a cerca de 50mg por dia. Em outra concretização, a dose oral de um composto aqui descrito irá variar de cerca de 30mg a cerca de 200mg por dia. Cada uma das faixas de dosagem acima citadas podem ser formuladas como uma formulação de unidade de dosagem única ou múltipla.6. EXEMPLOS1 .1 Formulações estáveis de Transnorsertralina
[0301] As formas de dosagem sólidas de transnorsertralina ou sais farmaceuticamente aceitáveis ou solvatos dos mesmos, são desejadas pela facilidade de administração da dose aos indivíduos e pacientes, bem como por proporcionar fácil controle das formulações de dosagem fora da clínica. Estas formas de dosagem devem ser fabricadas em equipamento automatizado e devem ter química e estabilidade física aceitável que pode ser superior a 1 ano.
[0302] Misturas excipiente múltiplas com transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato foram preparadas e avaliadas quanto à estabilidade química e viabilidade de fabricação. Estes excipientes incluem vários diluentes: fosfato anidro dibásico de cálcio, di-hidrato de fosfato dibásico de cálcio, amido pré-gelatinaizado, celulose microcristalina, lactose e manitol; desintegrantes: croscarmelose sódica, amido pré-gelatinaizado, glicolato de amido e sódio; deslizantes: Talco, sílica coloidal e sílica vaporizada; e vários lubrificantes: ácido esteárico, fumarato estearil de sódio, óleo vegetal hidrogenado e estearato de magnésio. A maioria das combinações era inaceitável devido a pobre estabilidade química; algumas combinações foram também inaceitáveis devido aos pobres atributos de fabricação, incluindo pobre homogeneidade da mistura, baixo teor de fármaco em cápsulas e variável teor de fármaco em cápsulas.
[0303] Por exemplo, quando cápsulas de gelatina dura foram formuladas, verificou-se que a combinação de cloridrato de transnorsertralina com os excipientes encontrados em comprimidos de Zoloft® (sertralina) resultou em uma formulação com estabilidade química pobre e, em particular, com múltiplos produtos de oxidação. Estes excipientes são di-hidrato fosfato dibásico de cálcio, celulose microcristalina, glicolato de amido de sódio, estearato de magnésio, bem como outros excipientes que são prováveis no revestimento desses comprimidos. Veja publicação no Physician’s Desk Reference para Zoloft® (sertralina).
[0304] Além disso, a maioria das combinações testadas utilizando os excipientes acima eram inaceitáveis devido a estabilidade química pobre. Algumas combinações foram também inaceitáveis devido aos pobres atributos de fabricação, incluindo pobre homogeneidade da mistura, baixo teor de fármaco em cápsula e teor variável de fármaco em cápsulas. Combinações estáveis incluem manitol, glicolato de amido de sódio, Talco e estearato de magnésio em um invólucro da cápsula clara ou colorida de gelatina dura.Resultados do Estudo de Estabilidade
[0305] As seguintes misturas ou misturas em cápsulas forampreparadas durante o desenvolvimento de uma formulação de cápsulas de cloridrato de transnorsertralina estável. Em alguns casos, o comprimento do teste varia. No entanto, métodos analíticos uniformes foram utilizados para todas as amostras. Degradação é relatada como impurezas totais com base em uma % de área a partir de análise das formulações em HPLC, como são comuns quando relatado tais resultados, quando uma mudança é anterior a uma caracterização completa da degradação. Percentual de degradação foi medido após armazenamento a 40°C/75% umidade relativa, uma condição de armazenamento típica e necessária.Tabela 1: Estabilidade da mistura de excipiente HCl deTransnorsertralina
 2 cloridrato de transnorsertralina anidro.3 fosfato de cálcio dibásico, anidro.4 fumarato de estearilo de sódio.5 di-hidrato de fosfato de cálcio dibásico.6 amido pré-gelatinaizado.7 celulose microcristalina.8 croscarmelose sódica9 Percentual de Degradação, quando armazenado a 40°C/75% de umidade relativa.10 mono-hidrato de cloridrato de transnorsertralina.
2 cloridrato de transnorsertralina anidro.3 fosfato de cálcio dibásico, anidro.4 fumarato de estearilo de sódio.5 di-hidrato de fosfato de cálcio dibásico.6 amido pré-gelatinaizado.7 celulose microcristalina.8 croscarmelose sódica9 Percentual de Degradação, quando armazenado a 40°C/75% de umidade relativa.10 mono-hidrato de cloridrato de transnorsertralina.
[0306] Como resultado do estudo de estabilidade, a seguinteformulação foi selecionada. Formulações em cápsula de cloridrato de transnorsertralina foram preparadas a 1,0mg concentração (com base na base livre) por cápsula. As formulações em cápsula incluem manitol, glicolato de amido de sódio, Talco e estearato de magnésio em um invólucro da cápsula de gelatina dura colorida.11 2 Formulações de cápsulas de concentração de 1.0mg de cloridrato Transnorsertralina anidro
[0307] Cápsulas estáveis de concentração de 1,0 mg(baseadas em base livre) de cloridrato de anidrato transnorsertralina (1,125mg de sal de HCl) foram preparadas de acordo com a formulação 1. A formulação foi inicialmente preparada com a mão, mostrando mistura aceitável e cápsula homogênea; um estudo de estabilidade mostrou estabilidade química melhorada destas cápsulas em comparação com outras formulações. Viabilidade de fabricação foi demonstrada quando um lote, de acordo com a formulação 1, foi fabricado em equipamento farmacêutica típico; mistura aceitável e homogeneidade da cápsula, bem como a estabilidade química melhorada foi mostrado para este lote.
[0308] Viabilidade de fabricação foi demonstrada em umtamanho maior de mistura quando um lote, de acordo com a formulação 1, foi fabricado em equipamento farmacêutico típico; mistura aceitável e homogeneidade da cápsula, bem como a estabilidade química melhorada, têm sido mostrados para este lote. Outra cápsula pode ser preparada de acordo com a formulação 2, sem que nenhum glicolato de amido de sódio esteja presente.12 3 Formulações de cápsulas de Concentração 0,5 mg em Cloridrato anidro de Transnorsertralina
[0309] Cápsulas estáveis de concentração 0,5 mg (baseadas em base livre) de cloridrato de anidrato de transnorsertralina (0,5625 mg de sal de HCl) foram preparados de acordo com a formulação 1. Viabilidade de fabricação foi demonstrada em um grande tamanho de mistura quando um lote de formulação 1 foi fabricado em equipamento farmacêutica típico; mistura aceitável e homogeneidade da cápsula, bem como a estabilidade química melhorada, têm sido mostrados para este lote. Outra cápsula pode ser preparada de acordo com a formulação 2, sem que nenhum glicolato de amido de sódio esteja presente. 6.4 Formulações de cápsulas de concentração 2.0mg em cloridrato anidro Transnorsertralina
[0310] Cápsulas estáveis de concentração 2,0mg (com base em base livre) de cloridrato de anidrato transnorsertralina (2,25 mg de sal de HCl) foram preparadas de acordo com a formulação 1. Viabilidade de fabricação foi demonstrada em um grande tamanho de mistura quando um lote de formulação 1 foi fabricado em equipamento farmacêutica típico; mistura aceitável e homogeneidade da cápsula, bem como a estabilidade química melhorada, têm sido mostrados para este lote. Outra cápsula pode ser preparada de acordo com a formulação 2, sem que nenhum glicolato de amido de sódio esteja presente.13 5 Formulações de cápsulas de concentração 0,5 mg de cloridrato anidro Transnorsertralina em Cápsula de Vários Tamanhos
[0311] Cápsulas representativas de concentração de 0,5mg(com base em base livre) de cloridrato de anidrato de transnorsertralina (0,5625 mg) podem ser preparada em três pesos de enchimento de 200,0mg, 150,0mg e 100,0mg em cápsulasde vários tamanhos, como mostrado acima. 6.6 Formulação de cápsulas de Concentração 1.0 mg em de cloridrato anidro de Transnorsertralina em Cápsula de Vários Tamanhos
[0312] Cápsulas representativas de concentração 1,0mg (com base em base livre) de cloridrato de anidrato transnorsertralina (1,125 mg de sal de HCl) podem ser preparadas em três pesos de enchimento de 200,0mg, 150,0mg e 100,0mg de cápsulas de vários tamanhos, como mostrado acima. 6.7 Formulações de cápsulas de Concentração 2.0 mg de cloridrato anidro de Transnorsertralina em Cápsula de Vários Tamanhos
[0313] Cápsulas representativas de concentração 2,0mg (com base em base livre) de cloridrato de anidrato de transnorsertralina (2,25 mg de sal de HCI) podem ser preparadas em três pesos de enchimento de 200,0mg, 150,0mg e 100,0mg de cápsulas de vários tamanhos, como mostrado acima.
[0314] Nos exemplos acima, se monohidrato de cloridrato de transnorsertralina é usado no lugar do anidrato cloridrato de transnorsertralina, um fator de conversão de 1,186mg de cloridrato mono-hidratado de transnorsertralina equivalentes a 1,0 mg de base livre de transnorsertralina pode ser aplicada a cada formulação.14 8 Processos de Fabricação de formulações em cápsula de Transnorsertralina
[0315] Misturas para formulações de cápsulas contendo transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato podem ser fabricadas usando um processo no qual cloridrato de transnorsertralina é primeiro misturado com talco; esta mistura é então misturada com manitol em diluição geométrica. O manitol restante e glicolato de amido de sódio são misturados com a mistura, por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0316] O processo pode ser modificado tal que transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato é primeiro misturado com uma porção de talco mais manitol; esta mistura é então misturada com manitol adicional. Em seguida, o manitol restante e glicolato de amido de sódio são misturados com a mistura, por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0317] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma porção de talco mais manitol; esta mistura é então misturada com uma mistura de manitol mais glicolato de amido de sódio, por último, estearato de magnésio é misturado com o anterior mistura. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0318] Outra modificação do processo pode ser realizada por mistura transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol mais glicolato de amido de sódio; esta mistura é então misturada com os excipientes restantes (menos o estearato de magnésio). Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0319] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais glicolato de amido de sódio; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semiautomática ou completamente automática.
[0320] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com talco; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semiautomática ou completamente automática.
[0321] Outra modificação do processo pode ser realizada por transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol; esta mistura é então misturada com o manitol restante. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0322] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com manitol; esta mistura é então misturada com uma mistura de talco mais manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática.
[0323] Outra modificação do processo pode ser realizada por mistura de uma porção do estearato de magnésio com transnorsertralina ou um seu sal farmaceuticamente aceitável ou solvato, em cada um dos processos acima. Por último, o resto do estearato de magnésio é misturado com a mistura anterior. A mistura pode ser encapsulada em uma máquina ou dispositivo de enchimento de cápsula manual, semi-automática ou completamente automática. 6.9 Formulações de comprimidos de concentração 0,5mg de cloridrato anidro de Transnorsertralina de comprimidos de vários tamanhos
[0324] Comprimidos representativos de concentração de 0,5mg (com base em base livre) de cloridrato de anidrato de transnorsertralina (0,5625 mg de sal de HCI) podem ser preparados em três tamanhos, como mostrado acima, com ou sem o uso de glicolato de amido de sódio (Primojel).6.10 Formulações de comprimidos de Concentração 1.0 mg de cloridrato anidro de Transnorsertralina de comprimidos de vários tamanhos

[0325] Comprimidos representativos de concentração de 1.0mg (com base em base livre) de cloridrato de anidrato de transnorsertralina (1,125 mg de sal de HCl) podem ser preparados em três tamanhos, como mostrado acima, com ou sem o uso de glicolato de amido de sódio (Primojel).15 11 Formulações de comprimidos de Concentração 2.0 mg de cloridrato anidro de Transnorsertralina de comprimidos de vários tamanhos

[0326] Comprimidos representativos de concentração de 2.0 mg (com base em base livre) de cloridrato de anidrato de transnorsertralina (2,25 mg de sal de HCI) podem ser preparados em três tamanhos, como mostrado acima, com ou sem o uso de glicolato de amido de sódio (Primojel).
[0327] Nos exemplos acima, se monohidrato de cloridrato de transnorsertralina é usado no lugar do anidrato cloridrato de transnorsertralina, um fator de conversão de 1,186 mg de cloridrato mono-hidratado de transnorsertralina equivalentes a 1,0 mg de base livre transnorsertralina pode ser aplicado.16 12 Processos de Fabricação para Formulações de Comprimidos não revestidos de Transnorsertralina
[0328] Misturas de formulações de comprimidos que contêm transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato podem ser fabricadas usando um processo em que transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato é primeiro misturado com talco; esta mistura é então misturada com manitol em diluição geométrica. Em seguida, o manitol restante e glicolato de amido de sódio são misturados com a mistura, por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0329] O processo para a fabricação de comprimidos não revestidos pode ser modificado tal que transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato é primeiro misturado com uma porção de talco mais manitol; esta mistura é então misturada com manitol adicional. Em seguida, o manitol restante e glicolato de amido de sódio são misturados com a mistura, por último, estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0330] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma porção de talco mais manitol; esta mistura é então misturada com uma mistura de manitol mais glicolato de amido de sódio, por último, estearato de magnésio é misturado com o anterior mistura. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0331] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol mais glicolato de amido de sódio; esta mistura é então misturada com os excipientes restantes (menos o estearato de magnésio). Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0332] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais glicolato de amido de sódio; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0333] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com talco; esta mistura é então misturada com o manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0334] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com uma mistura de talco mais manitol; esta mistura é então misturada com o manitol restante. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0335] Outra modificação do processo pode ser realizada por mistura de transnorsertralina ou sal farmaceuticamente aceitável ou solvato com manitol; esta mistura é então misturada com uma mistura de talco mais manitol. Finalmente, o estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.
[0336] Outra modificação do processo pode ser realizada por mistura de uma porção do estearato de magnésio com transnorsertralina ou seu sal farmaceuticamente aceitável ou solvato, em cada um dos processos acima. Por último, o resto do estearato de magnésio é misturado com a mistura anterior. A mistura pode ser comprimida em uma prensa ou máquina de comprimidos.17 13 Processos de Fabricação para Comprimidos revestidos de Transnorsertralina
[0337] Cada um dos comprimidos descritos acima pode também ser fabricado como um comprimido revestido. O revestimento pode ser um dos três tipos, que incluem revestimento porcompressão, revestimento com filme ou revestimento degelatina. Cada revestimento pode ou não conter um agentecorante; estes agentes de coloração podem ser dióxido detitânio e/ou corantes solúveis, tais como corantes, e/oucorantes insolúveis tais como pigmentos e/ou óxidos de ferrocoloridos.6.14 Formas Sólidas de Sais de Transnorsertralina
[0338] Dezesseis sais de transnorsertralina foraminvestigados utilizando microscopia de luz polarizada (PSM), a fim de identificar os sais de transnorsertralina na forma cristalina: cloridrato, citrato, fumarato, maleato, fosfato,succinato, sulfato, L-tartarato, besilato, tosilato, L-malato,
[0339] S-mandelato, acetato, benzoato, bromidrato epiroglutamato.
[0340] As amostras foram observadas através do microscópiode polarização de luz Nikon Microphot. As amostras foram preparadas em líquido Cargille com um índice de refração de 1,600. As amostras foram observadas utilizando luz polarizada cruzada e fotografada utilizando luz polarizada cruzada com uma quarta placa de onda. Determinação inicial de cristalinidade de sais de transnorsertralina foi realizada por observação direta sob luz polarizada cruzada (Tabela 2). Qualquer sal testado que continha o material sólido deficiente birrefração quando observados sob luz polarizada cruzada, indicando sólidos amorfos ou parcialmente amorfos, foi rejeitado.Tabela 2. PLM Observações de Sais de Transnorsertralina
[0341] Os termos “Fino ou Finos” sãodefinidos neste relatório como partículas tendo larguras <10 μm.
[0342] Cada sal, com a exceção do sal succinato, exibiu boabirrefração sob polarização de luz cruzada, indicando um sólido cristalino. Formas de cristais variaram de agulhas finas a grandes placas (Tabela 2).6.15 Propriedades Térmicas de Sais de Transnorsertralina
[0343] Cada um dos sais de transnorsertralina do Exemplo6,14 foi analisado utilizando calorimetria de varredura direta (DSC) ou Hotstage. Todas as análises DSC foram realizadas utilizando Calorímetro de Varredura Diferencial Perkin Elmer DSC 7. Cada amostra foi analisada em uma autoclave cravada comum furo, aqueceu-se sob uma purga de azoto a uma taxa de 10°C/min, a partir de uma temperatura inicial de 25°C até umatemperatura final de 325°C. As amostras Hotstage foram analisadas usando microscópio de luz polarizada Nikon Microphot equipado com um Linkam Hotstage THMs 600. Cada amostra foi colocada em um pedaço de cobertura, situada na fornalha hotstage, isolados a acima por 2 camadas (2 lamelas com espaço de ar entre as camadas) e a tampa hotstage e aqueceu-se a uma taxa de 10°C/min. Os resultados de DSC e hotstage são mostrados na Tabela 3.Tabela 3. Resultados para Sais Transnorsertralina por DSC eHotstage

 6.16 Conteúdo de umectação e Higroscopicidade de sais de Transnorsertralina
6.16 Conteúdo de umectação e Higroscopicidade de sais de Transnorsertralina
[0344] Os dezesseis sais de transnorsertralina do Exemplo6,14 foram analisados quanto ao teor de umidade e higroscopicidade. Cada sal foi analisado por titulação coulométrica usando um titulador EM Scientific Aquastar C3000 para determinar o conteúdo de água. O tamanho da amostra foi de 18mg a 134mg. Cada sal foi analisado utilizando um Analizador Termogravimétrico (TGA) Perkin Elmer TGA 7. As amostras foram aquecidas a partir de uma temperatura inicial de 25°C a 325°C a uma taxa de 10°C/min. Isotermas de sorção de umidade para cada sal foram geradas usando o Analizador de sorção de Vapor VTI SGA-100 Symmetric. As amostras foram executadas como recebidas sem pré-análise de secagem. Critérios de equilíbrio tiveram menos de 0,01% de alteração em peso em 5 minutos ou de 180 minutos em cada passo de umidade relativa (UR). A temperatura foi fixada em 25°C e os passos de umidade relativa (25 a 95% a 25%) em incrementos de 5%. A análise foi repetida para cada amostra em análises consecutivas (amostra não foi removida do analisador). As dimensões das amostras foram de 18 mg a 35 mg.
[0345] Dados de isotérma de umidade VTI, teor de umidade(KF) e dados de TGA estão resumidos na Tabela 4.Tabela 4. Resultados de sais de Transnorsertralina por KF, TGA e VTI
 n.m. = não mensurado
n.m. = não mensurado
[0346] VTI mostrou que o citrato, fosfato, succinato,sulfato, L-tartarato, S-mandelato e sais de piroglutamato detransnorsertralina exibiram significativa absorção de umidade(2,7 a 17,3%) de 25 a 95% RH (Tabela 4).6.17 Solubilidade de água em sais de Transnorsertralina
[0347] Doze sais de transnorsertralina foram investigados para solubilidade em água: cloridrato, maleato, fumarato, fosfato, succinato, sulfato, L-tartarato, acetato de besilato, tosilato, L-malato e benzoato de metila. Para cada sal, soluções saturadas com sólidos em excesso em água desionizada foram preparadas em frascos de vidro transparente de cintilação de 20 mL com tampa de rosca. Todas as amostras foram agitadas a 300 rpm em condições ambientais até nove dias até que o equilíbrio foi alcançado. Solubilidade foi determinada utilizando um método de HPLC (Tabela 5).Tabela 5. Solubilidade de sais de Transnorsertralina em água deionizada.

[0348] O cloridrato, maleato, fosfato, succinato, besilato,L-malato e acetato de estavam entre os sais testados que exibiram solubilidade adequada em água (0,99-5,49 mgA/mL).6.18 Solubilidade dos sais Transnorsertralina em tampão aquoso
[0349] Os doze sais de transnorsertralina do Exemplo 6,4foram investigados para a sua solubilidade nos seguintes sistemas de tampão aquoso: fluido gástrico simulado (SGF),0,05M de tampão de acetato (pH 4,5) e fluido intestinal simulado (SIF). Soluções saturadas com sólidos em excesso foram preparadas em frascos de cintilação de vidro transparente de 20 ml. Fluido gástrico simulado (pH 1,0, ~0,1NHCl, 0,03M NaCl, sem enzimas), fluido intestinal simulado (pH 6,8, 0,05M de KH2PO4, ~0,02N NaOH, sem enzimas) e tampão deacetato (pH 4,5, 0,02M de sódio acetato, ácido acético 0,03M)foram preparadas em conformidade com USP28 (USP28 Test Solutions p2855, Volumetric Solutions p2863). Todas as amostras foram agitadas a 300 rpm em condições ambientais por nove dias até que o equilíbrio foi atingido. Solubilidade foi determinada utilizando um método de HPLC. (Tabela 6).Tabela 6. Solubilidade de sais de Transnorsertralina Salts em sistemas tampão aquoso. a enzymas não foram incluídas no tampãob equilíbrio não atingido depois de 9 dias
a enzymas não foram incluídas no tampãob equilíbrio não atingido depois de 9 dias 
 a enzymas não inclusas no tampão6.19 Caracterização de Sais Transnorsertralina recuperados a partir de experimentos de solubilidade
a enzymas não inclusas no tampão6.19 Caracterização de Sais Transnorsertralina recuperados a partir de experimentos de solubilidade
[0350] Os sólidos recuperados a partir de suspensões de experiência de solubilidade (Exemplos 6.17 e 6.18) foram filtrados a vácuo e seco a 40°C durante a noite. Cada amostra foi analisada utilizando um calorímetro diferencial de varrimento Perkin Elmer DSC 7. Cada amostra foi aquecida em uma autoclave cravada com um furo sob uma purga de azoto a uma taxa de 10°C/min, a partir de uma temperatura inicial de 25°C até uma temperatura final de 325°C. Ver Tabela 7.
[0351] Como mostrado na Tabela 7, o sal de cloridrato de transnorsertralina pareceu converter a uma forma mono- hidratada, quando os sólidos foram equilibrados em água deionizada (DI) e SGF. A DSC para o sal de mono-hidrato de cloridrato de mostrou uma endotermia a cerca de 100°C, seguido de uma fusão do anidro sublimado a ~300°C (confirmado por hotstage). Esta hidratação também foi marcada por uma mudança de forma do cristal das hastes de placas. Veja as figuras 1A e 1B. Sais adicionais testados pareceram converter para o mono- hidrato de HCl durante as experiências de solubilidade em SGF (Tabela 7). Esta conversão não era inesperada, uma vez que SGF contém ácido clorídrico suficiente (0,23 m) para formar o sal cloridrato, que por sua vez pode converter para o mono- hidrato. Sólidos recuperados a partir de experiências de solubilidade em tampão de acetato não pareceu alterar a partir de sua forma de sal original. Parece que alguns dos sais (maleato, acetato, besilato e L-malato) foram convertidos para uma forma semelhante em SIF (Tabela 7). A DSC para esta forma desconhecida mostra uma endotermia única em torno de 100°C com um calor de fusão pequena (29-49 J/g).Tabela 7. Resultados do DSC para sólidos recuperados de experimentos de solubilidade
 a: Fluido gástrico simulado (“SGF”), USP, pH 0.9, sem pepsina b: Fluido intestinal simulado (“SIF”), USP, pH 6.8, sempancreatinac: Sólidos recuperados da água e SGF tendo 4.8% água (KF) ec.9 % perda de peso (TGA), que é consistente com o monohidrato n.m.: não mensuradon.o.: não observadoc.20 Experimentos Repetidos para os Sais cloridrato de acetato e L-malato
a: Fluido gástrico simulado (“SGF”), USP, pH 0.9, sem pepsina b: Fluido intestinal simulado (“SIF”), USP, pH 6.8, sempancreatinac: Sólidos recuperados da água e SGF tendo 4.8% água (KF) ec.9 % perda de peso (TGA), que é consistente com o monohidrato n.m.: não mensuradon.o.: não observadoc.20 Experimentos Repetidos para os Sais cloridrato de acetato e L-malato
[0352] As experiências seguintes foram repetidas. Lotesadicionais dos sais de acetato, cloridrato e L-malato de transnorsertralina foram testados para (i) consistência das propriedades térmicas por DSC e/ou hotstage e (ii) as consistência das propriedades de umidade por dados de KF, TGA e VTI.
[0353] Um segundo lote do sal de cloridrato detransnorsertralina sublimou a 166°C e o sublimado se fundiu a 249°C medida por hotstage de acordo com o procedimento do Exemplo 6.2 acima. Estes resultados estão em boa concordância com as do primeiro lote (sublimado a 170°C, o sublimado se fundiu a 250°C).
[0354] Os segundo e terceiro lotes de acetato detransnorsertralina demonstraram propriedades térmicassemelhantes como o primeiro lote de acetato como medido por DSC, de acordo com o procedimento do Exemplo 6.15 acima. (Tabela 8).Tabela 8. Resultados do Acetato de Transnorsertralina por DSC
 n.m.: não mensurado
n.m.: não mensurado
[0355] O segundo lote de L-malato de transnorsertralinademonstrou propriedades térmicas semelhantes como o primeiro lote de acetato, como medido por DSC. No entanto, o terceiro lote fundiu a uma temperatura aproximadamente 8°C mais baixa do que outros lotes (Tabela 9). Todas as experiências foram realizadas de acordo com o procedimento do Exemplo 6.15 acima.Tabela 9. Resultados do L-Malato de Transnorsertralina por DSC n.m.: não mensurado
n.m.: não mensurado
[0356] Um segundo lote do sal cloridrato de transnorsertralina foi analisado para higroscopicidade por VTI, de acordo com o procedimento do Exemplo 6.16 acima. Os resultados foram semelhantes em comparação com o primeiro lote de cloridrato (adsorção VTI 0,01% de ganho de peso, 25% a 95% de umidade relativa; dessorção VTI 0,01% de perda de peso, a partir de 95% a 25% de umidade relativa).
[0357] Os segundo e terceiro lotes dos sais de acetato e L- malato de transnorsertralina também foram analisados e comparados com os resultados do primeiro lote, de acordo com o procedimento do Exemplo 6.3 acima. Os resultados são mostrados na Tabela 10. Todos os segundo e terceiro lotes testados tiveram isotermas de umidade semelhantes como primeiros lotes, com exceção do lote 3 do L-malato, que adsorveu umidade > 5% amais do que outros lotes de L-malato de 25 a 95% de umidade relativa.Tabela 10. Dados de KF, TGA e VTI para L-Malato deTransnorsertralinae Acetato de Transnorsertralina n.m.: não mensuradoc.21 Estabilidade do Sólido de Sais de Transnorsertralina
n.m.: não mensuradoc.21 Estabilidade do Sólido de Sais de Transnorsertralina
[0358] Sais de transnorsertralina foram testados quanto àestabilidade do sólido sob várias condições. As amostras sólidas do sal HCl foram colocados em recipientes de polietileno duplo revestido de polietileno de alta densidade alta (HDPE) fechados com tampas de PEAD e armazenadas a 25°C/60% de umidade relativa ou 40°C/75% de umidade relativa. As amostras foram analisadas por HPLC. Anidrato cloridrato de Transnorsertralina era estável em ambas as condições durante pelo menos 6 meses e em 25°C/60% de umidade relativa durante 2 anos, exibindo menos de 0,05% e menos de 0,1% de impurezas, respectivamente.c.22 Estudo de Conversão polimórfica de cloridrato de Transnorsertralina
[0359] Cloridrato Transnorsertralina existe em pelo menosduas formas cristalinas. Forma A é um material cristalino anidro e Forma B é um monohidrato cristalino. Imagens de microscopia de luz polarizada mostram que as duas formas cristalinas têm formas de cristal distintas. A forma anidra exibe longas lâminas finas (Figura 1A), enquanto que a forma de hidrato mostra placas aproximadamente quadradas finas (Figura 1B). Ambas as amostras apresentam birrefração e extinção sob polarizadores cruzados quando da rotação.
[0360] O estudo da conversão do cloridrato detransnorsertralina anidro para a forma de hidrato em meios aquosos foi investigado, utilizando monitoramento in situ Raman. O sistema foi mostrado por ser adequado para o monitoramento Raman, com um limite de detecção da forma hidrato, em uma suspensão anidro/hidrato em água a 70 mg/mL abaixo de 5,7%.
[0361] A espectroscopia Raman foi realizada utilizando umKaiser Optical Systems Inc. dispersiva RamanRXN3 para a monitorização da reação em linha ou in-situ. O sistema de RamanRXN3 utiliza um comprimento de onda de excitação de 785 nm, com uma cavidade externa estabilizada, laser de diodo. Todos os espectros foram adquiridos utilizando um M" de sonda de imersão com cerca de 100mW de potência de laser na ponta da sonda. Diferentes tempos de exposição e números de acumulações de espectro foram utilizados para a análise das duas amostras secas. Um tempo de exposição de 4 segundos com 2 acumulações foi utilizado para o controle de todas as formas de conversão das experiências. Calibração do comprimento de onda e do comprimento de onda do laser foram realizadas utilizando um padrão interno de neon e padrão de deslocamento diamante Raman, respectivamente. A intensidade da calibração foi realizada utilizando um acessório de calibração Kaiser Raman.
[0362] Os espectros de Raman adquiridos nas duas formas nas regiões 2850-3150cm-1 e 200-1600cm-1 mostraram que as duas formas podem ser diferenciadas por Raman. Regiões 660-715 cm-1 e 1430-1490cm-1, em particular, mostram pouca sobreposição dos picos característicos de cada forma. Os resultados experimentais confirmaram que nenhum pico nas regiões
[0363] 660-720 cm-1 e 1430-1490 cm-1 eram susceptíveis a sobrepor com picos de interesse para seguir a conversão entre as duas formas cristalinas de cloridrato de transnorsertralina.
[0364] A conversão do cloridrato de transnorsertralina anidro para a forma monohidrato foi monitorada em água. A razão de pico I (677 cm-1)/I (695 cm-1) foi observada para uma suspensão de cloridrato de transnorsertralina anidro em água. Com base na intensidade da razão de pico I (677 cm-1)/I (695 cm-1), um tempo de indução de cerca de 1,1 horas foi observada antes do início da conversão. O fim da conversão foi estimada em cerca de 2 horas a partir do início da suspensão. A região de Raman 660-710 cm-1 mostrou o aparecimento de um picocaracterístico da forma de hidrato e o desaparecimento de um pico característico da forma anidra do cloridrato de transnorsertralina. A análise de XRPD dos sólidos coletados no final do monitoramento Raman da conversão (após cerca de 2h 10min) foi consistente com a forma de hidrato, com uma pequena quantidade detectível de forma anidra. A pequena quantidade de forma anidra pode ser devido a sólidos presentes sobre as paredes do vaso, que não foram convertidos. Além disso, o limite de detecção Raman da forma anidra na mistura não foi estimado.
[0365] A conversão do cloridrato de transnorsertralinaanidro para a forma de hidrato em água, fluido gástrico simulado (SGF) e fluido intestinal simulado (FIS), sem enzimas e HCl 0,1N foi monitorada a 37°C. A conversão da forma em água, SGF e HCl 0,1N foi mostrado para começar após cerca de 1,3 horas (água e SGF) a 2 horas (0,1N HCl) e ser completada dentro de 3 a 4 horas. A conversão da forma era significativamente mais lenta em SIF, que começou a cerca de10 horas em pequena escala e 19 horas em uma escala maior eterminou após cerca de 12,5 horas (em pequena escala) a 36 horas (em grande escala).
[0366] Em geral, resultados semelhantes foram obtidos emágua e fluido gástrico simulado, com o início da conversão da forma detectada em aproximadamente 1,3 horas, com uma conversão ligeiramente mais rápida em fluido gástrico simulado em comparação com a água. A conversão completa foi estimada a ocorrer dentro de 3 a 4 horas nos dois meios. Tempos de indução ligeiramente mais longos foram observados em 0,1N HCl, aproximadamente 2 a 2,3 horas em maior escala. A conversão completa foi observada em cerca de 4 horas. Os resultados sugerem que a conversão da forma de cloridratotransnorsertralina em fluido intestinal simulado é muito lenta, estimado em 10 horas em pequena escala e 19 horas à escala maior e que termina após cerca de 12,5 horas (em pequena escala) e 36 horas (em grande escala). Análises em XPRD dos sólidos coletados no final de cada experiências eram consistentes com a forma de hidrato, com ou sem alguma forma anidra presente. A pequena quantidade de forma anidra pode ser devido a sólidos residuais sobre as paredes do vaso, no momento da suspensão.c.23 Caracterização de cloridrato de Transnorsertralina cristalina anidra
[0367] Uma amostra de hidrocloreto de transnorsertralinaanidra (Forma A) foi submetida a análise de estrutura de cristal único. A estrutura foi determinada por uma difração de raios-X de único cristal. A coleção de dados, redução e determinação da estrutura não foram realizadas de acordo com as especificações de cGMP.Experimento
[0368] Uma agulha fina incolor de C16H16Cl3N tendo dimensõesaproximadas de 0,29 x 0,08 x 0,02 mm foi revestida com óleo Paratone N, suspenso em uma presilha de fibra pequena e colocada em uma corrente arrefecida de gás azoto em uma orientação aleatória. Exames preliminares e coleta de dados foram realizados com radiação Cu Ka (À = 1.54178 Â) em umdifratômetro de tubo selado APEX D8 Bruker II CCD.
[0369] A coleta de dados, indexação e refinamentos de células iniciais foram todas realizadas com APEX II. Veja APEX II, 2005, Bruker AXS, Inc., Analytical X-ray Systems, 5465 East Cheryl Parkway, Madison WI 53711-5373. Integração de quadro e refinamentos de célula final foram realizadas utilizando software SAINT. Refinamentos foram realizados em um PC usando SHELXTL. Veja SAINT Versão 6.45A, 2003, Bruker AXS, Inc., Analytical X-ray Systems, 5465 East Cheryl Parkway, Madison WI 53711-5373; SHELXTL V6.12, 2002, Bruker AXS, Inc., Analytical X-ray Systems, 5465 East Cheryl Parkway, Madison WI 53711-5373.
[0370] Os parâmetros celulares finais e uma matriz de orientação para coleta de dados foram determinados a partir refinamento por mínimos quadrados em 1553 reflexões na faixa de 5,26°<θ<58,04°. O grupo espacial foi determinado como sendo C2 (N°5) pelo programa XPREP. Veja Bruker, XPREP in SHELXTL version 6.12, Bruker AXS Inc., Madison, Wisconsin, USA, 2002.
[0371] Os dados foram coletados usando uma série de combinações de varredura phi e omega com 30 exposições segundo quadro e larguras de quadro 0.5o a uma temperatura de -100,15 ± -271,15 °C (173 ± 2K) . Os dados foram coletados a um valor máximo de 116,08° 2θ.
[0372] Um total de 2910 reflexões foi coletado, dos quais 1533 foram únicas. Correções de Lorentz e de polarização foram aplicadas aos dados. O coeficiente de absorção linear é 52,75 cm-1 para radiação Ka Cu. Uma correção de absorção empírica usando SADABS foi aplicada. Veja Blessing, R. H., SADABS, Program for absorption correction using Siemens CCD. Based on Blessing R. Acta Cryst. 1995, A51, 33. Coeficientes de transmissão variaram de 0,3099 a 0,9018. Intensidades de reflexões equivalentes foram calculadas. O fator de acordo para a média foi de 4,1% com base na intensidade.
[0373] A estrutura foi resolvida por métodos diretos,utilizando SHELXS-97. Veja Sheldrick, GM SHELX97, A Program for the Solution of Crystal Structure, University of Gottingen, Germany, 1997. Átomos de hidrogênio foram colocados nas suas posições químicas esperadas utilizando o comando HFIX estavam localizadas em uma diferencial final de Fourier e foram incluídos nos ciclos finais de mínimos quadrados com Uij’s isotrópicos relacionados ao átomo ao qual eles estão ligados. Todos os átomos não hidrogenados foram refinados anisotropicamente. A estrutura foi refinada em uma matriz total de quadrados mínimos, minimizando a função:
[0374] O peso w é definido como 1/[G2(FQ2) + (0.0450P)2 +(0.3158P)], onde P = (FO2+2FC2)/3. Fatores de espalhamento foram retirados das "International Tables for Crystallography." International Tables for Crystallography, Vol. C, Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992, tabelas 4.2.6.8 e 6.1.1.4. Das 3171 reflexões utilizadas nos aperfeiçoamentos, apenas as reflexões com FQ2>2G(FQ2) foramutilizadas no cálculo de R. Um total de 1553 reflexões foram utilizadas no cálculo. q ciclo final de refinamento incluiuparâmetros variáveis e convergentes (a maior mudança de parâmetro era essencialmente igual ao seu desvio padrãoestimado) com fatores de acordo não ponderados e ponderados de:
[0375] O desvio padrão de uma observação da unidade de pesofoi de 1,074. O pico mais alto na última diferencial deFourier tinha uma altura de 1,115e/Â3. O pico negativo mínimotinha uma altura de -0,288e/Â3. O fator para a determinação daestrutura absoluta refinada para 0,04 (4). Ver Flack, H. D.Acta Cryst. 1983, A39, 876.Resultados
[0376] Os parâmetros de célula monoclínica e volumecalculado são: a=16,834(3), b=5,2264(9), c=19,059(3)Â, α=90,00,β=113,103 (6), Y=90, 00°, V= 1542,4(4)Â3. O peso da fórmula para transnorsertralina é 328,65 g/mol com Z=4 e uma densidade calculada de 1,415 gcm-3. O grupo espacial foi determinado como sendo C2 (N°5). Um resumo dos dados do cristal e parâmetroscoletados de dados cristalográficos são descritos na Tabela11.
[0377] A qualidade da estrutura obtida é consideradamoderada a elevada, tal como indicado pelo valor de R de 0,0566 (5,66%). O valor R para esta estrutura é apenas dentroda faixa de valor de R de 0,02 a 0,06 que são citados para as estruturas mais confiáveis determinadas. Glusker, Jenny Pickworth; Trueblood, Kenneth N. Crystal Structure Analysis: A Primer, 2nd ed.; Oxford University press: New York, 1985; p.87.Tabela 11. Dados do cristal e parâmetros de coleta de dados para Cloridrato Transnorsertralina anidro


[0378] Um padrão de difração de raios-X de pó calculado(XRPD) foi gerado para a radiação Cu usando PowderCell 2.3 e as coordenadas atômicas, grupo espacial e os parâmetros da célula unitária, a partir dos dados de cristal único. Veja PowderCell for Windows Version 2.3 Kraus, W.; Nolze, G. Federal Institute for Materials Research and Testing, Berlin Germany, EU, 1999.
[0379] O padrão de XRPD calculado do cloridrato detransnorstertralina anidro é mostrado na Figura 2. O padrão de XRPD experimental é mostrado na Figura 3. Todos os picos nos padrões experimentais estão representados no padrão XRPD calculado, indicando que o material a granel é provável uma única fase. As diferenças nas intensidades calculadas e observadas nos padrões de XRPD são, provavelmente, devido a orientação preferencial. Orientação preferencial é a tendência de cristais, geralmente placas ou agulhas, para alinhar-se com algum grau de ordem. Orientação preferencial pode afetar as intensidades de pico, mas não as posições de pico, nos padrões de XRPD. As pequenas mudanças na posição do pico são, provavelmente, o resultado de pequenas mudanças nos parâmetros da célula unitária como uma função da temperatura. Os padrõesde XRPD calculados gerados a partir dos dados de único cristalforam coletados a 173K, enquanto que o padrão de pó experimental foi coletado à temperatura ambiente. A coleta de dados a baixa temperatura é tipicamente usada na análise de cristal único para melhorar a qualidade da estrutura. ORTEP e diagramas de embalagem
[0380] O diagrama de ORTEP foi preparado usando ORTEP III. Veja Johnson, C. K. ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, U.S.A. 1996. OPTEP-3 for Windows V1.05,. Farrugia, L.J., J. Appl. Cryst. 1997, 30, 565. Átomos são representados por 50% de probabilidade de elipsóides térmicos anisotrópicos. Diagramas de embalagem foram preparados utilizando software de modelagem CAMERON. Veja Watkin, D.J; Prout, C K.; Pearce, L.J.CAMERON, Chemical Crystallography Laboratory, University of Oxford, Oxford, 1996. Valores adicionais foram gerados usando o software de modelagem Mercury 1.3. Veja Bruno, I.J.Cole, J.C.Edgington, P.R.Kessler, M.K.Macrae, C.F.McCabe, P.Pearson, J. and Taylor, R. Acta Crystallogr., 2002 B58, 389. A ligação de hidrogênio é representada como linhas tracejadas.
[0381] Um desenho ORTEP de cloridrato de transnorsertralina anidro é mostrado na Figura 4. A unidade assimétrica mostrada na Figura 4 contém uma única molécula transnorsertralina protonada e um anion de cloreto.Configuração absoluta
[0382] A configuração absoluta de cloridrato de transnorsertralina anidro pode ser determinada por análise de dispersão de raios-X anómala pelo cristal. As diferenças de intensidade do espalhamento anômalo são, então, comparadas com intensidades de espalhamento calculadas para cada enantiômero. Estas intensidades medidas e calculadas podem, então, estar aptas a um parâmetro, o fator de Flack. Ver Flack,H.D.; Bernardinelli,G. Acta Cryst. 1999, A55, 908; Flack,H.D.; Bernardinelli,G.J. Appl. Cryst. 2000, 33, 1143. Depois de uma estrutura ser dissolvida, a qualidade dos dados é avaliada quanto à sua potência de inversão distinta, isto é, feito através de um exame da incerteza do padrão do parâmetro Flack. Para cloridrato de transnorsertralina anidro, a incerteza padrão, (u), é igual a 0,07, que é classificada como poder distinto enantiopuros suficiente. O fator de Flack medido para a estrutura cristalina do cloridrato de transnorsertralina anidro mostrado na Figura 4 é -0,13, com um padrão de incerteza de 0,04. A molécula contém dois centros quirais localizados em C7 e C14 (consulte a Figura 4), os quais foram atribuídos com configurações S e R, respectivamente.6.24 Análise de difração por Raio-X de pó de cloridrato de Transnorsertralina anidro
[0383] Análises de difração por Raio-X (XRPD) de pó decloridrato de Transnorsertralina anidro foram realizadas utilizando um difratômetro Inel XRG-3000 equipado com um detector CPS (Curved Position Sensitive) com um intervalo 2θ de 120°. Dados em tempo real foram coletados por meio de radiação Cu-Kα a partir de aproximadamente 4° 2θ a uma resolução de0.03°2θ. A voltagem do tubo e amperagem foram definidos para 40 kV e 30 mA, respectivamente. A fenda do monocromador foi de 5 mm por 160 μm. O padrão é exibido 2.5 a 40 °2θ. As amostrasforam preparadas para análise embalando-os em capilares de vidro de paredes finas. Cada capilar foi montado sobre uma cabeça goniômetro, que é motorizado para permitir a fiação do capilar durante a aquisição de dados. As amostras foram analisadas por 300 segundos. A calibração do instrumento foi realizada utilizando um padrão de referência de silício. Os padrões experimentais XRPD foram coletados de acordo com de cGMP. A Tabela 12 mostra os picos XRPDcloridrato transnorsertralina anidro. Tabela 12. Picos XRPD observados para cloridratotransnorsertralina anidro˚2θd space (Å) Intensity (%)
[0384] A Tabela 13 mostra picos XRPD proeminentes paracloridrato transnorsertralina anidro. As diferenças entre ospicos calculados e experimentais são devido a orientaçãopreferencial e efeitos estatísticos de partículas Tabela 13. Dados de XRPD Prominentes Data cloridratotransnorsertralina anidro. 6.25 Caracterização do mono-hidrato cristalino de CloridratoTransnorsertralina
6.25 Caracterização do mono-hidrato cristalino de CloridratoTransnorsertralina
[0385] Uma amostra de mono-hidrato de cloridrato detransnorsertralina (Forma B) foi submetida a análise de estrutura de cristal único. A coleta de dados do cristal único, a solução da estrutura e refinamento não foram realizados de acordo com as especificações de cGMP.Experimental
[0386] Uma agulha incolor de mono-hidrato de cloridrato detransnorsertralina, C16H18Cl3NO [Cl, C16H16Cl2N,H2O], tendo as dimensões aproximadas de 0,60 x 0,40 x 0,07 mm, foi montada sobre uma fibra de vidro em orientação aleatória. Exame preliminar e coleta de dados foram realizados com radiação Mo Kα de (À = 0.71073 Â) em um difratômetro Nonius KappaCCDequipado com um cristal de grafite, monocromador de feixe incidente. Refinamentos foram realizados em um PC com Linux utilizando SHELX97. Veja Sheldrick, G. M. SHELX97, A Program for Crystal Structure Refinement, University of Gottingen, Germany, 1997.
[0387] Constantes de célula e uma matriz de orientação para a coleta de dados foram obtidas a partir do método dos mínimos quadrados, utilizando ângulos de definição de 6712 reflexões na faixa de 3°<θ<27°. O mosaico refinado de DENZO/SCALEPACK foi de 0,47°, indicando boa qualidade de cristal. Ver Otwinowski,Z.; Minor,W. Methods Enzymol. 1997, 276, 307. O grupo espacial foi determinado pelo programa ABSEN. Veja McArdle,P.C.J. Appl. Cryst. 1996, 29, 306. A partir da presença sistemática da seguinte condição: 0k0 k=2n e de subsequente refinamento por mínimos quadrados, o grupo espacial foi determinado como sendo P21 (N°4). Este é um grupo espacial quiral. Os dados foram coletados para um valor máximo de 54,92 °2θ, a uma temperatura de 150 ± 1K.
[0388] Quadros foram integrados com DENZO-SMN. Ver Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307. Um total de 6712 reflexões foi coletado, dos quais 3171 foram únicas. Correções de Lorentz e de polarização foram aplicadas aos dados. O coeficiente de absorção linear é 0,543 mm-1 para radiação Mo Ka. Uma correção de absorção empírica usando SCALEPACK foi aplicada. Coeficientes de transmissão de Id. variou de 0,892 a 0,963. Intensidades de reflexões equivalentes foram calculadas. O fator de acordo para a média foi de 4,5%, com base na intensidade.Solução de Estrutura e Refinamento
[0389] A estrutura foi resolvida por métodos diretos,utilizando SIR2004. Ver Burla et al., J. Appl. Cryst. 2005, 38, 381. Os átomos remanescentes foram localizados emsucessivas sínteses de diferença de Fourier. Átomos de hidrogênio foram incluídos no refinamento, mas impedido de montar sobre o átomo ao qual eles estão ligados. A estrutura foi refinada na matriz total de mínimos quadrados, minimizando a função:
[0390] O peso w é definido como 1/[G2(FQ2) + (0.0450P)2 +(0.3158P)], onde P =(FO2 + 2 FC2)/3. Fatores de espalhamentoforam retirados das “International Tables forCrystallography”. International Tables for Crystallography, Vol. C, Kluwer Academic Publishers: Dordrecht, TheNetherlands, 1992, Tabelas 4.2.6.8 e 6.1.1.4. Das 3171 reflexões utilizadas nos aperfeiçoamentos, apenas as reflexões com FQ2>2G(FQ2) foram utilizados no cálculo de R. Um total de2757 reflexões foram utilizados no cálculo. O ciclo final de refinamento incluiu 210 parâmetros variáveis e convergentes (a maior mudança de parâmetro era essencialmente igual ao seu desvio padrão estimado) com fatores de acordo não ponderados e ponderados de:
[0391] O desvio padrão de uma observação da unidade de pesofoi de 1,04. O pico mais alto na última diferença de Fouriertinha uma altura de 0,35 e/Å3. O pico negativo mínimo tinha umaaltura de -0,37 e/Å3. O fator para a determinação da estruturaabsoluta refinada para 0,13 (7). Ver Flack,H.D. Acta Cryst.1983, A39, 876.
[0392] Os parâmetros da célula monoclínica e volumecalculado são: a = 7,2962(2)Â, b = 7,5569(2)Â, c =15,2870(5)Â,α = 90, 00, β = 90, 0852(14), y = 90, 00°, V = 842,87(4)Â3. Parao mono-hidrato, o peso da fórmula é 346,69 g/mol com Z = 2resultando em uma densidade calculada de 1,366 g/cm-3. O grupo espacial foi determinado como sendo P21 (No.4), que é um grupoespacial quiral. Um resumo dos dados do cristal e parâmetros cristalográficos coletados é descrito na Tabela 14. A qualidade da estrutura obtida é elevada, tal como indicado pelo valor de R de 0,041 (4,1%). Normalmente valores de R nointervalo de 0,02 a 0,06 são citados para as estruturas determinadas de forma mais confiável. Glusker, Jenny Pickworth; Trueblood, Kenneth N. Crystal Structure Analysis: A Primer, 2nd ed.; Oxford University press: New York, 1985; p.87.Tabela 14. Dados do cristal e parâmetros cristalográficos coletados para o monohidrato do cloridrato deTransnorsertralina Determinação da estrutura absolutaFlack parâmetrob(-0.13( 7))a Otwinowski Z. & Minor, W. Methods Enzymol., 1997, 276, 307.b Flack,H. D. Acta Cryst., 1983 A39, 876.Padrão de difração de raios-X de pó calculado
Determinação da estrutura absolutaFlack parâmetrob(-0.13( 7))a Otwinowski Z. & Minor, W. Methods Enzymol., 1997, 276, 307.b Flack,H. D. Acta Cryst., 1983 A39, 876.Padrão de difração de raios-X de pó calculado
[0393] Um padrão de difração de raios-X (XRPD) de pócalculado foi gerado para a radiação Cu usando PowderCell 2.3 e as coordenadas atômicas, grupo espacial e parâmetros da célula unitária, a partir dos dados de cristal único. Ver PowderCell for Windows Version 2.3 Kraus, W.; Nolze, G. Federal Institute for Materials Research and Testing, Berlin Germany, EU, 1999.
[0394] O padrão de XRPD calculado de monohidrato decloridrato de transnorstertralina é mostrado na Figura 5. O padrão de XRPD experimental é mostrado na Figura 6. Todos os picos dos padrões experimentais estão representados no padrão calculado XRPD, indicando que o material granel éprovávelmente uma única fase. As pequenas mudanças na posição do pico são, provavelmente, o resultado de pequenas mudanças nos parâmetros da célula unitária como uma função da temperatura. Os padrões de XRPD calculados gerados a partir dos dados de cristal único foram coletados a 150K, enquanto que o padrão de pó experimental foi coletado à temperatura ambiente. A coleta de dados a baixa temperatura é tipicamente usado na análise de cristal único para melhorar a qualidade da estrutura.Diagramas ORTEP e de embalagem
[0395] O diagrama de ORTEP foi preparado usando ORTEP III.Veja Johnson, C. K. ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, U.S.A. 1996. OPTEP-3 for Windows V1.05,. Farrugia, L.J., J. Appl. Cryst. 1997, 30, 565. Átomos são representados por 50% de probabilidade de elipsóides térmicas anisotrópicas. Diagramas de embalagem foram preparados utilizando software de modelagem CAMERON. Veja Watkin, D. J.; Prout, C .K.; Pearce, L. J. CAMERON, Chemical Crystallography Laboratory, University of Oxford, Oxford, 1996. Figuras adicionais foram geradas usando Mercury 1.4.1. Veja Bruno, I. J. Cole, J. C. Edgington, P. R. Kessler, M. K. Macrae, C. F. McCabe, P. Pearson, J. and Taylor, R. Acta Crystallogr., 2002 B58, 389. Ligação de hidrogênio é representada como linhas tracejadas.
[0396] Um desenho ORTEP de monohidrato de cloridrato de transnorsertralina é mostrado na Figura 7. A unidade assimétrica mostrada contém uma única molécula de mono-hidrato cloridrato de transnorsertralina protonada, um ânion cloreto e uma água de hidratação totalmente ocupada. A formação de sal foi confirmada por localização dos átomos de hidrogênio na amina primária e da molécula de água diretamente a partir do mapa de Fourier.Configuração absoluta
[0397] A configuração absoluta de mono-hidrato de cloridrato de transnorsertralina pode ser determinada por análise de dispersão de raios-X anômala do cristal. As diferenças de intensidade do espalhamento anômalo são, então, comparadas com intensidades de espalhamento calculadas para cada enantiômero. Estas intensidades medidas e calculadas podem, então, estar aptas a um parâmetro, o fator de Flack. Ver Flack, H. D.; Bernardinelli, G. Acta Cryst. 1999, A55, 908; Flack, H. D.; Bernardinelli, G. J. Appl. Cryst. 2000, 33,1143. Depois de uma estrutura ser dissolvida, a qualidade dos dados é avaliada quanto à sua potência de inversão distinta, isto é feito através de um exame do padrão de incerteza do parâmetro Flack. O fator de Flack medido para a estrutura cristalina do mono-hidrato de cloridrato de transnorsertralina mostrado na Figura 7 é -0,13 com um padrão de incerteza de 0,07. A incerteza padrão (u) é igual a 0.07, que é classificada como potência distinta suficiente deenantiopuros. Um erro desta magnitude significa que a priori uma evidência biológica química, física ou é requerida para mostrar que o composto é verdadeiramente um enantiômero puro e para demonstrar que a determinação da estrutura absoluta é válida. Enquanto o fator Flack medido estiver fora da faixa para permitir a validação baseada unicamente nos dados cristalográficos, a configuração absoluta pode ser confirmada por comparação com a molécula de cloridrato detransnorsertralina a partir da estrutura de cristal anidro. Portanto, a configuração absoluta do modelo da Figura 7 está correta. A molécula de mono-hidrato de cloridrato de transnorsertralina contém dois centros quirais localizados em C1 e C4 (referem-se a Figura 7), os quais foram atribuídos com configuração S e R, respectivamente.6.26 Análise de difração por Raio-X do pó de cloridrato monohidratado de Transnorsertralina
[0398] Análises de difração por Raio-X (XRPD) do pó decloridrato monohidratado de Transnorsertralina foramrealizadas utilizando um difratômetro Inel XRG-3000 equipado com um detector CPS (Curved Position Sensitive) com um intervalo 2θ de 120°. Dados em tempo real foram coletados por meio de radiação Cu-Kα a partir de aproximadamente 4 °2θ a uma resolução de 0.03°2θ. A voltagem e amperagem do tubo foram definidas para 40 kV e 30 mA, respectivamente. A fenda do monocromador foi de 5 mm por 160 μm. O padrão é exibido 2,5 a 40 °2θ. As amostras foram preparadas para análise por embalá- los em capilares de vidro de paredes finas. Cada capilar foi montado sobre uma cabeça goniômetro que é motorizada para permitir a fiação do capilar durante a aquisição de dados. As amostras foram analisadas por 300 segundos. A calibração do instrumento foi realizada utilizando um padrão de referência de silício. Os padrões experimentais XRPD foram coletados de acordo com especificações de cGMP. A Tabela 15 mostra os picos observados XRPD para mono-hidrato de cloridrato de transnorsertralina.Tabela 15. Picos XRPD observados para o monohidrato cloridrato de Transnorsertralina

[0399] Tabela 16 mostra picos XRPD proeminentes paramonohidrato de cloridrato de transnorsertralina. As diferençasentre os picos calculados e experimentais são devido àorientação preferencial e efeitos estatísticos de partículas.Tabela 16. Dados XRPD proeminentes para o monohidratocloridrato de Transnorsertralina 6.27 Estudos adicionais de estabilidade de cloridrato de Transnorsertralina
6.27 Estudos adicionais de estabilidade de cloridrato de Transnorsertralina
[0400] Em um estudo de estabilidade típico, a misturaexcipiente ou forma de dosagem completa foi preparada com a droga ativa. O material foi armazenado em um recipiente selado, de preferência uma garrafa de polietileno de alta densidade (HDPE) selada com uma película de indução de calor. O material foi colocado em um forno com umidade controlada de tal modo que as amostras foram expostas a cerca de 40°C e cerca de 75% de umidade relativa (UR) durante um período de cerca de 2 semanas a cerca de 6 meses.
[0401] A Tabela 17 mostra os dados de ensaio e as impurezaspara os comprimidos através de seis meses de armazenamento utilizando HPLC. Um cromatograma de HPLC típico é mostrado na Figura 8. No t0, o ensaio foi de ~93%, enquanto que as impurezas totais foram 0.89%, do qual 0,17% pode ser atribuída a tetralona. Uma impureza desconhecida (RT ~18 minutos, 0,67%) foi também detectada durante análise t0. De um valor inicial de 93,4%, valores de ensaio para comprimidos armazenados a 30°C/65% UR e 25°C/60% UH permaneceu acima de 90% através do ponto de tempo de seis meses, enquanto os armazenados a 40°C/75% UR caiu para 80%. A 40°C/75% UR, o degradante principal era tetralona; níveis aumentaram de um valor inicial de 0,17% a 4,63% em seis meses. Em contraste, os níveis de impurezas desconhecidas a 25°C/60% UR aumentaram de um valor inicial de 0,67% a 1,84% dentro de um mês, subsequentemente caindo para 0,23% no ponto de tempo de seis meses.Tabela 17. Ensaio e dados de impureza do comprimido de 1 mg de cloridrato de Transnorsertralina
[0402] A estrutura do degradante foi confirmada pelo seutempo de retenção no HPLC, espectro de UV e resultado LC-MScomo possuindo a fórmula II. O degradante tem um pesomolecular de 454 e a sua estrutura química é mostrada abaixo:
[0403] Os dados de estudos adicionais indicaram que 0,005%de manose em manitol resultou em uma formação degradante conjugada de cerca de 0,12% em duas semanas e que 0,01% de manose em manitol resultou em uma formação degradante conjugada de cerca de 0,25% em duas semanas. A quantidade de degradante conjugada nivelou-se em cerca de duas semanas e depois diminuiu ligeiramente à medida que aumentou tempo de estresse. A formação do degradante de fórmula II também depende da temperatura. A temperatura mais elevada (por exemplo, > 35°C), o degradante de fórmula II foi submetido adecomposição.6.28 Análise da pureza do Excipiente Manitol
[0404] Estudos de estabilidade adicionais sobre as cápsulastêm mostrado que as misturas na forma de comprimidos embalados em Aclar® (película policlorotrifloroetileno, Honeywell Int'l Inc.) são mais estáveis (aproximadamente 4 vezes) do que as misturas em condições de embalagem aberto. A presença de manose em manitol foi determinada usando Detector HPLC-Corona charged Aerosol (HPLC-CAD) e métodos de cromatografia de íons (CI) como mostrados na Tabela 18:Tabela 18: Análise da pureza do Manitol Método HPLC-CAD. Neste método, ascondições/instrumentos foram utilizadas:Coluna: Sugar SZ5532Tamanho Coluna: 6 mm ID X 150 mm LTemperatura da Coluna: 65°CDetector: Corona CAD Fase móvel: 80% Acetonitrila em águaVazão: 1 mL/minVolume injetado: 100 μL
Método HPLC-CAD. Neste método, ascondições/instrumentos foram utilizadas:Coluna: Sugar SZ5532Tamanho Coluna: 6 mm ID X 150 mm LTemperatura da Coluna: 65°CDetector: Corona CAD Fase móvel: 80% Acetonitrila em águaVazão: 1 mL/minVolume injetado: 100 μL
[0405] O eluente da coluna de HPLC é nebulizado com azoto eas gotículas são secas, produzindo partículas de analito. Um fluxo de azoto secundário torna-se carregado positivamente e transfere a carga para partículas de analito. A carga é então medida, gerando um sinal em proporção direta com a quantidade de analito presente.Método de Cromatografia de Íons (CI): Neste método, asseguintes condições/instrumentos foram utilizadas:
[0406] O programa de gradiente usado para o método IC: 100% de fase móvel A a 0 min; Fase B 100% móvel em 18 min; 100% fase móvel B em 30 min.
[0407] Forma de Onda potencial aplicada, o método de IC:
[0408] Embora os resultados de cada método tenham sidogeralmente comparáveis, o método CI foi selecionado para análise posterior, devido à maior sensibilidade em relação ao método de HPLC-CAD (amostra concentração de 1 mg/mL vs
[0409] 100 mg/mL de manitol). O limite de quantificação(LQ) do método de Cromatografia de Íons (IC) é de 0,005% demanose em manitol. Vários lotes de manitol (ver Tabela 18) foram analisados e os valores de manose são dados abaixo:
 * Embora o LQ do método CI seja de 0,005% de manose em manitol, a fim de estimar a quantidade de manose, a % de manose em manitol foi calculada usando a área do pico de manose observada versus aquela de padrão manose externa.
* Embora o LQ do método CI seja de 0,005% de manose em manitol, a fim de estimar a quantidade de manose, a % de manose em manitol foi calculada usando a área do pico de manose observada versus aquela de padrão manose externa.
[0410] Dos 2 lotes de manitol utilizados a partir da tabelaacima, o lote 8 spray seco (0,0115% de manose) formou o degradante conjugado, ao passo que o lote 1 de manitol cristalino (0,0009% de manose, <LQ) não formou o degradante conjugado. A quantidade de manose em manitol parecia ser o fator de controle, não o tipo de manitol utilizado na formulação de cloridrato de transnorsertralina (isto é, cristalina vs seco). Isto foi posteriormente confirmado a partir dos resultados obtidos usando amostras recristalizadas de manitol dopada com manose no estudo de estabilidade de formulações HCl de transnorsertralina.6.29 Estudo de estabilidade da cápsula de cloridrato de Transnorsertralina
[0411] A mistura de excipiente ou forma de dosagem completa foi preparada com a droga ativa. O material foi armazenado em um frasco selado de HDPE com uma folha de indução de calor. O material foi colocado em um forno com umidade controlada de tal modo que as amostras são expostas a cerca de 40°C e cerca de 75% de UR durante um período de 8 Semanas. As amostras de cápsula foram preparadas como se segue:
[0412] Diluente da amostra foi preparado como se segue.Para cada litro preparado, medida com precisão de 600 mL de água, 200 mL de THF e 200 mL de acetonitrila em um recipiente adequado. Adicionar 0,5 mL de ácido trifluoroacético e homogeneizar.HPLC método analítico. A Zorban SB-CN 5 μM, 25 cm x 4,6 mm de coluna analítica usando um ácido trifluoroacético a 0,05% em água 80:20, solução de acetonitrila (fase móvel A) seguida por um ácido trifluoroacético a 0,05% em água 15:85; solução de acetonitrila (fase móvel B) foi usada. Temperatura da coluna: 30°C. A vazão: 1,0 mL/min. Volume de injeção: 50 mL. Comprimento de onda: 220 nm. Tempo mínimo de funcionamento: 50 min (15 min de atraso). Gradiente de fase móvel:
[0413] Para preparação da amostra para HPLC, os conteúdosforam transferidos para um balão volumétrico de 50 mL. Uma mistura de água/acetonitrila/THF/ácido trifluoroacético na razão 60/20/20/0.05 foi utilizada como diluente de amostra. Após o enchimento de ^ do balão com diluente de amostra, foi vigorosamente agitado à mão e então submetido à ação de agitação durante 30 minutos, sonicação durante 20 minutos e agitação novamente durante 30 minutos. Após arrefecimento até à temperatura ambiente, o volume foi completado até 50 ml usando diluente de amostra, bem misturada e filtrada usando osobrenadante 25 mm de filtro de seringa de diâmetro de 0,45 μmPTFE GD/X em um frasco de HPLC e analisados usando as condições de HPLC mostradas acima. A concentração de substância ativa nas amostras foi de 0,14 mg/mL.
[0414] Tabela 19 mostra que os mesmos produtos dedegradação são formados em diferentes quantidades em cápsulas de resistência diferentes. Cápsulas de 1 mg continham mais degradantes totais do que 5 e/ou 10 mg de concentração.Tabela 19: Estabilidade de protótipos de Cápsulas deTransnorsertralina * Impureza em ou acima de 0.05% estão listadasRRF Presumido (fator de resposta relativo) de impurezas = 1PVDC = cloreto de polivinilideno
* Impureza em ou acima de 0.05% estão listadasRRF Presumido (fator de resposta relativo) de impurezas = 1PVDC = cloreto de polivinilideno
[0415] Produtos de degradação semelhantes foram observados utilizando condições de prato aberto. Recipientes de prato aberto de boca larga (~20 frascos de cintilação mL) foram submetidos a 40°C e cerca de 75% de UR durante 3 Semanas.
[0416] 7,88 mg de HCl de transnorsertralina e a quantidade apropriada de excipiente(s) (ativa a razão de excipiente de 1:1, 1:124 e/ou 1:372) foram pesados nos recipientes.Tabela 20: Estudo de prato Aberto * Impureza em ou acima de 0.05% estão listadasRRF Presumido (fator de resposta relativo) de impurezas= 1
* Impureza em ou acima de 0.05% estão listadasRRF Presumido (fator de resposta relativo) de impurezas= 1
[0417] A comparação dos resultados apresentados nas Tabelas19 e 20 revelaram que os mesmos produtos de degradação seformaram em níveis ligeiramente diferentes e que a degradaçãoem prato aberto é mais rápida. Cromatogramas de HPLC obtidospara estudo de estabilidade da cápsula e estudo de pratoaberto são mostrados na Figura 9.
[0418] Uma vez que as cápsulas de 1 mg produziram o maiornúmero de impurezas/produtos de degradação, a razão detransnorsertralina (ativo) para excipientes nas amostras forammantidas semelhantes àqueles em 1 mg Cápsulas (1 mg detransnorsertralina é equivalente a 1,125 mg de cloridrato detransnorsertralina):Tabela 21: Composições de 1 mg HCl Transnorsertralina emCápsula
[0419] Os seguintes lotes de excipientes foram utilizadoscom vários lotes de Ativos (por exemplo, "Ativo 1"):
 Conclusões: O estudo de degradação de compatibilidade do HClde transnorsertralina e seus excipientes demonstraram que quanto maior área da superfície do ativo, maior a quantidade de degradação. Dos 5 excipientes presentes em cápsulas 1.0, 5.0 e 10.0 mg, apenas a utilização de um A-TAB permitido para a degradação do HCl de transnorsertralina, semelhante ao observado quando cápsulas de desenvolvimento per se foram estressadas e a extensão da degradação causada por um A-TAB dependia do lote utilizado. O mecanismo que leva à degradação do ativo, aparentemente exacerbada pela presença de um A-TAB, não foi determinado. Produtos de degradação individuais podem ser isolados, por exemplo, pela colocação de uma mistura de 2 g de Ativo 3 e 248 g de um A-TAB 2 em um prato aberto durante 3 Semanas a 40°C/75% UR. Os produtos de degradação individuais foram isolados em quantidades aproximadamente de 5 a 10 mg.6.30 Exemplos de estudos de estabilidade de pratos abertos
Conclusões: O estudo de degradação de compatibilidade do HClde transnorsertralina e seus excipientes demonstraram que quanto maior área da superfície do ativo, maior a quantidade de degradação. Dos 5 excipientes presentes em cápsulas 1.0, 5.0 e 10.0 mg, apenas a utilização de um A-TAB permitido para a degradação do HCl de transnorsertralina, semelhante ao observado quando cápsulas de desenvolvimento per se foram estressadas e a extensão da degradação causada por um A-TAB dependia do lote utilizado. O mecanismo que leva à degradação do ativo, aparentemente exacerbada pela presença de um A-TAB, não foi determinado. Produtos de degradação individuais podem ser isolados, por exemplo, pela colocação de uma mistura de 2 g de Ativo 3 e 248 g de um A-TAB 2 em um prato aberto durante 3 Semanas a 40°C/75% UR. Os produtos de degradação individuais foram isolados em quantidades aproximadamente de 5 a 10 mg.6.30 Exemplos de estudos de estabilidade de pratos abertos
[0420] HCl de Transnorsertralina (1) (“Ativo 1”) foimisturado com cada excipiente presente em cápsula de concentração de 1mg, respectivamente. As amostras continham duas proporções diferentes (1:1 e como nas Cápsulas 1mg) de ativo e excipientes.Definição da matriz 1a da amostraAmostras colocadas a 40°C/75 UR por 3 Semanas* * A proporção de ativo para excipiente é de 1:1Definição da matriz 1b da amostraAmostras colocadas a 40°C/75 UR por 3 Semanas
* A proporção de ativo para excipiente é de 1:1Definição da matriz 1b da amostraAmostras colocadas a 40°C/75 UR por 3 Semanas  * A proporção de ativo para excipiente é como nascápsulas de 1 mg
* A proporção de ativo para excipiente é como nascápsulas de 1 mg
[0421] Os resultados obtidos para experiências do grupo 1são apresentados na Tabelas 6 e 7.Tabela 22: Estabilidade de amostras definidas 1a * A proporção de ativo para excipiente é 1:1
* A proporção de ativo para excipiente é 1:1
[0422] Como mostrado na Tabela 22, quando a proporção de substância ativa 1 para excipiente é de 1:1, nenhuma impureza apreciável é formada devido à presença de vários excipientes. Apenas uma pequena quantidade de tetralona foi observada.
[0423] Quando a relação foi alterada para cápsulas 1mg, excipientes tais como amido, formulações contendo Ac-Di-Sol, estearato de magnsésio demonstraram um pequeno crescimento da impureza tetralona (RRT 1,64, Tabela 7). A-TAB 1 pareceu causar a maior degradação. As impurezas/produtos de degradação desconhecidas em RRT 0.61, 0.68, 0.78, 0.80, 1.34, 1.53 e 1.70 estão presentes em ou acima de níveis de 0,10% nesta amostra e alguns produtos de degradação aproximaram-se do nível de 1,0%. Impurezas conhecidas como impureza sintética 1, o cis- diastereoisômero e tetralona cresceram 3-4 vezes mais com A- TAB 1 do que com ativo 1 sozinho.Tabela 23: Estabilidade definida da amostra 1 b
 * A proporção de ativo para excipiente é como nas cápsulasde 1 mg
* A proporção de ativo para excipiente é como nas cápsulasde 1 mg
[0424] Para experimentos da definição 2, 3 lotes diferentesde ativos foram misturados com 3 lotes diferentes cada de um
[0425] A-TAB separadamente e em 3 diferentes proporções(1:124, 1:372), como mostrado na definição 2a e 2b,respectivamente.Definição da Matrix da amostra 2aPratos Abertos - Amostras colocadas a 40°C/75 UR por 2Semanas*
 A proporção de ativo para excipiente é 1:124Definição da Matrix da amostra 2bPratos Abertos - Amostras colocadas a 40°C/75 UR por 2Semanas*
A proporção de ativo para excipiente é 1:124Definição da Matrix da amostra 2bPratos Abertos - Amostras colocadas a 40°C/75 UR por 2Semanas* A proporção de ativo para excipiente é 1:372
A proporção de ativo para excipiente é 1:372
[0426] Os resultados das amostras definidas em 2a contendoativo e excipiente na razão 1:124 estão indicados na Tabela 24.Tabela 24: Estabilidade das amostras definidas em 2a*

[0427] Os resultados da Tabela 24 mostram que variando a quantidade de A-TAB, enquanto se mantém a quantidade ativo fixa, resultou na maior parte da degradação. Variando a quantidade de ativo, enquanto mantém a quantidade de A-TAB resultou no ativo 3, fornecendo a maioria das impurezas. A combinação de Ativo 3 e A-TAB 2 forneceu a maior quantidade de impurezas totais/degradação. A área de superfície dos lotes do ativo diminuiu do Ativo 3 para Ativo 4 para Ativo 2 para Ativo 1. Assim, a área de superfície do Ativo pode desempenhar um papel na susceptibilidade de Ativo para degradação.
[0428] A-TAB 4, o lote em pó, fornecido apenas em uma pequena quantidade de tetralona. Quando a proporção de substância ativa para excipiente foi aumentada para 1:372, três vezes mais do que isso presente em Cápsulas de 1 mg, as impurezas totais formadas permaneceram quase a mesma que a encontrada em relação 1:124. Ver Tabelas 24 e 25. Parece que as impurezas totais formadas atingiram um platô a uma certa proporção de substância ativa para o excipiente.Tabela 25: Estabilidade definida das amostras 2b*
 6.31: Isolamento e Identificação de produtos de degradação da fórmula (III)
6.31: Isolamento e Identificação de produtos de degradação da fórmula (III)
[0429] As amostras usadas para a preparação e isolamento de produtos de degradação de fórmula (III) foram preparadas como se segue: para um frasco de 500 ml de vidro castanho, 2,25 g de ativo e 278 g de A-TAB foram misturados à mão durante 1 minuto. A mistura resultante foi passada através de peneira de 35 mesh, duas vezes, de tal modo que o material remanescente na peneira foi mínimo. A amostra foi misturada novamente em um misturador Turbula a 22 rpm durante 20 minutos. A mistura foi então transferida para um prato de cristalização e o recipiente aberto foi colocado a 40°C/75% UR durante 3 semanas. Uma alíquota da mistura foi analisada em 2 e 3 semanas. A amostra foi removida da câmara de aquecimento depois de 3 Semanas e armazenada em refrigeração.
[0430] A partir da amostra degradada, 62 g da mistura foi pesada para um balão de Erlenmeyer de 1-L. Adicionou-se um volume de 500 mL de metanol e agitou-se à temperatura ambiente utilizando uma barra de agitação magnética durante 1 hora. A mistura agitada foi deixada em repouso durante 30 minutos. O sobrenadante foi filtrada a vácuo usando filtro MAGNA de Nylon suportado, liso de 0,45 μm. A pequena quantidade de pó sobre o papel de filtro foi lavada com metanol e rinsagem combinada. O filtrado foi analisado utilizando o método de HPLC descrito abaixo. O diluente de amostra foi metanol. A área total obtida pela adição das áreas dos picos de ativo e impurezas foi quantificada utilizando uma solução padrão de ativo. A quantidade de ativo e, assim, todas as impurezas extraídas era 369,2 mg. O sólido no Erlenmeyer foi tratado novamente com outro 500 mL de metanol.
[0431] Ao repetir o mesmo procedimento descrito acima, a quantidade de ativo e todas as impurezas extraídas da segunda vez era de 38,5 mg. Os dois filtrados foram combinados e metanol foi removido utilizando um evaporador rotativo. Um sólido amarelo laranja permaneceu no fundo do balão. Uma mistura de 6,5 mL de metanol e 3 mL de água foi adicionada. O sólido dissolveu formando uma solução amarela laranja. Esta solução foi centrifugada à temperatura ambiente e o sobrenadante transparente foi transferido para um frasco de vidro de 10 mL e armazenado sob refrigeração.Separação por HPLC dos 4 degadantes principais. As seguintes condições semi-preparativas de HPLC foram desenvolvidas e usadas para separar as 4 principais impurezas a partir do solução de amostra por degradadação forçada em prato aberto preparada como descrito acima.Coluna HPLC:Zorbax SB-CN (Agilent), 9.2 mm x 250 mm, 5 μmFase móvel A:0.1% formic acid in waterFase móvel B:0.1% ácido fórmico em acetonitrilaComprimento de onda: 220 nmVolume injetado: 10 μLVazão: 4 mL/minTempo de corrida: 44 minutosGradiente usado:
[0432] Apenas as 4 principais frações impurezas foramcoletadas. Impurezas ativas e outras foram lavadas para fora da coluna. Cerca de 35 injeções foram feitas e as frações de cada degradante foram agrupadas separadamente. Cada uma das 4 frações foi então analisada utilizando HPLC. A separação por coluna analítica das 4 frações é mostrada na Figura 9. Impurezas 1 e 3 possuiam, cada, 100% de pureza. Impureza 2 possuiu 98% de pureza. Fração 4 foi caracterizada ser uma mistura de impurezas 4 e 5 presentes na razão de 63:37.Identificação estrutural de Degradantes de fórmula (III).
[0433] Produtos de degradação do ativo foram isoladosusando HPLC semi-preparativa (Zorbax SB-SN, 5 um, 9,2 x 250 mm; 20% ACN/H2O com 0,1% de ácido fórmico como fase móvel, 4 ml/min) com a amostra de degradação prato aberto acimamencionado. As frações contendo estas impurezas foram neutralizadas com 0,1 M NH4OAc, antes da secagem a vácuo, para evitar possível decomposição.
[0434] Todas as impurezas foram inicialmente analisadas porLC-MS, análises de fragmentação e alta resolução MS. Produtos de degradação III-A e III-b apresentaram características de massas espectrais quase idênticos. [M + H+] em m/z 308 para ambos os compostos foi observada, enquanto o padrão isotópico confirmou a natureza bis-cloro das moléculas, indicando a sua origem na transnorsertralina. A diferença de 16 unidades de massa, em peso molecular de transnorsertralina versus aquela de produtos de degradação III-A e III-b sugeriu que ambas as impurezas podem ser produtos de oxidação de transnorsertralina sob a forma de grupo hidroxila. A perda de H2O (-18 mu) em fragmentação MS sugeriu que o grupo hidroxila pode residir no anel alifático.
[0435] Esforço de isolamento para degradante III-a produziuuma pequena quantidade de composto relativamente puro. O espectro de 1H NMR de degradante Ill-a em ACN-d3 revelou que (a) os anéis aromáticos de transnorsertralina não foram alterados como evidenciado pelos sinais de prótons aromáticos e padrões e (b) um dos prótons benzílicos de transnorsertralina foi substituído quando apenas um foi observado a 4,16 ppm. Como os dados espectrais de massa confirmaram que o grupo amino não foi alterado nesta impureza, a outra única posição para um grupo hidroxila benzilico é a posição 4. Portanto, degradante III-a foi identificado como 4- hidroxi transnorsertralina.
[0436] Degradante III-b isolado apresentou um padrão muito semelhante no seu espectro de 1H NMR, isto é, anéis aromáticos inalterados e o desaparecimento de um próton benzílico. Esta informação, juntamente com os seus dados espectrais de massa, levou à conclusão de que as impurezas 1 e 2 eram um par diastereoisomérico de 4-hidroxi transnorsertralina.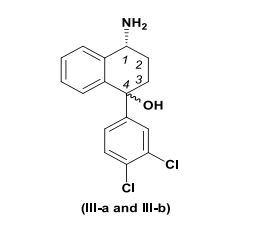
[0437] As impurezas 3 e 4 partilham o mesmo peso molecularde 323 com o padrão de fragmentação MS quase idêntica. Após o processo de isolamento e secagem, foi notado que as impurezas 3 e 4 foram convertidas para as impurezas 1 e 2, respectivamente (por retenção de Tempo HPLC, dados MS e espectro 1H NMR). Como notado acima, a fim de minimizar a possível decomposição durante o processo de isolamento, NH4OAc foi utilizado para neutralizar o ácido fórmico nas frações coletadas, que podem ter resultado na conversão.
[0438] Experiências adicionais mostraram que frações recentemente isoladas de degradante-III-c e III-d sem NH4OAc eram relativamente estáveis mesmo depois de alguns dias à temperatura ambiente. No entanto, na presença de NH4OAc, quase 50% dos compostos foram convertidos após 24 horas. Além disso, observou-se que após a secagem a vácuo, produtos de degradação III-c e III-d foram convertidos em produtos de degradação III- a e III-b independente da presença de NH4OAc. Como a diferença da fórmula molecular entre produtos de degradação III-c e III- a foi um átomo de oxigênio tal como medido por UR-MS, concluiu-se que os produtos de degradação II-c e III-d eram um par diasteroisômero de 4-hidroperoxi transnorsertralina. As conversões de impureza 3 e degradante III-d para produtos de degradação III-A e III-b podem ser por um processo de decomposição de hidroperóxidos. 6.32: Estudos de Estabilidade de Transnorsertralina com Manitol
6.32: Estudos de Estabilidade de Transnorsertralina com Manitol
[0439] Três lotes de manitol recristalizado dopados com manose foram selecionados para um estudo mais aprofundado com base em seus valores % de manose. Manitol cristalino e manitol spray seco foram utilizados como controle. O lote de manitol spray seco recristalizado também foi utilizado no estudo para determinar se o tipo de manitol utilizado (cristalino vs spray seco) foi significativo. Misturas de HCl de Transnorsertralina foram feitas usando os lotes selecionados de manitol e colocados a 30°C/65% UR em condições de prato aberto e inicialmente analisados e depois de 2, 4 e 6 Semanas usando HPLC. Os resultados são mostrados na Tabela 26.Tabela 26: Quantidade de degradante II em TransnorsertralinaHCl Misturas Todas as patentes, pedidos de patentes e publicações referidas no presente pedido estão aqui incorporadas na sua totalidade. Além disso, a citação ou identificação de qualquer referência no presente pedido não constitui uma admissão de que essa referência está disponível como técnica anterior.
Todas as patentes, pedidos de patentes e publicações referidas no presente pedido estão aqui incorporadas na sua totalidade. Além disso, a citação ou identificação de qualquer referência no presente pedido não constitui uma admissão de que essa referência está disponível como técnica anterior.
Claims (7)
1. Composição farmacêutica caracterizada por compreender como seu único ingrediente ativo, cloridrato de transnorsertralina ou solvato farmaceuticamente do mesmo, e manitol, em que compreende pelo menos 10% em massa de manitol, o dito manitol contendo menos que 0,1% em massa de manose.
2. Composição farmacêutica de acordo com a reivindicação 1, caracterizada por compreender adicionalmente talco, caulim ou bentonita.
3. Composição farmacêutica de acordo com as reivindicações 1 ou 2, caracterizada por compreender adicionalmente estearato de magnésio, estearato de cálcio, estearato de zinco ou ácido esteárico.
4. Composição farmacêutica de acordo com a reivindicação 1, caracterizada por compreender um sal de ácido clorídrico de transnorsertralina ou um hidrato do mesmo, manitol, estearato de magnésio, talco e glicolato de amido sódico.
5. Composição farmacêutica de acordo com a reivindicação 1, caracterizada por estar na forma de tablete ou cápsula.
6. Uso de uma composição conforme definida em qualquer uma das reivindicações de 1-5 caracterizado pelo fato do dito uso ser para preparação de um medicamento para tratar, prevenir ou controlar um distúrbio neurológico.
7. Uso de acordo com a reivindicação 6, caracterizadopor o distúrbio neurológico ser depressão, déficit cognitivo, fibromialgia, dor, distúrbio de sono, síndrome de fadiga crônica, distúrbio de déficit de atenção (ADD), distúrbio de hiperatividade de déficit de atenção (ADHD), síndrome das pernas inquietas, esquizofrenia, ansiedade, distúrbio obsessivo compulsivo, stress pós-traumático, distúrbio afetivo sazonal (SAD), disforia pré-menstrual, sintomas vasomotores pós- menopausa, doença neurodegenerativa, condições maníacas, distimia, ciclotimia, obesidade, distúrbio alimentar ou abuso de substâncias ou dependência.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26686409P | 2009-12-04 | 2009-12-04 | |
| US61/266,864 | 2009-12-04 | ||
| PCT/US2010/058831 WO2011069032A2 (en) | 2009-12-04 | 2010-12-03 | Formulations, salts and polymorphs of transnorsertraline and uses thereof |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BR112012013325A2 BR112012013325A2 (pt) | 2017-09-05 |
| BR112012013325B1 true BR112012013325B1 (pt) | 2021-05-18 |
| BR112012013325B8 BR112012013325B8 (pt) | 2021-05-25 |
Family
ID=44115510
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR112012013325A BR112012013325B8 (pt) | 2009-12-04 | 2010-12-03 | composição farmacêutica e uso |
Country Status (20)
| Country | Link |
|---|---|
| US (3) | US8957114B2 (pt) |
| EP (2) | EP2937083B1 (pt) |
| JP (2) | JP5766713B2 (pt) |
| KR (1) | KR101755743B1 (pt) |
| CN (2) | CN102740842B (pt) |
| AU (2) | AU2010325894B2 (pt) |
| BR (1) | BR112012013325B8 (pt) |
| CA (1) | CA2782240A1 (pt) |
| DK (1) | DK2506842T3 (pt) |
| ES (1) | ES2531312T3 (pt) |
| HK (2) | HK1216986A1 (pt) |
| IL (2) | IL219963A (pt) |
| MX (2) | MX339619B (pt) |
| NZ (1) | NZ600107A (pt) |
| PL (1) | PL2506842T3 (pt) |
| PT (1) | PT2506842E (pt) |
| RU (2) | RU2578956C2 (pt) |
| SG (1) | SG181466A1 (pt) |
| WO (1) | WO2011069032A2 (pt) |
| ZA (1) | ZA201203621B (pt) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8134029B2 (en) | 2002-09-16 | 2012-03-13 | Sunovion Pharmaceuticals Inc. | Treatment of CNS disorders with trans 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-napthalenamine |
| CN101553217A (zh) | 2005-07-06 | 2009-10-07 | 塞普拉科公司 | 艾司佐匹克隆与反式4-(3,4-二氯苯基)-1,2,3,4-四氢-n-甲基-1-萘胺或反式4-(3,4-二氯苯基)-1,2,3,4-四氢-1-萘胺的组合以及治疗绝经期和心境障碍、焦虑症和认知障碍的方法 |
| JP6226471B2 (ja) * | 2014-02-12 | 2017-11-08 | 株式会社リガク | 構造精密化装置、方法およびプログラム |
| CN106660936A (zh) * | 2014-05-13 | 2017-05-10 | 赛诺维信制药公司 | 用于治疗adhd的方法和达斯曲林(dasotraline)组合物 |
| US10076503B2 (en) | 2014-05-13 | 2018-09-18 | Sunovion Pharmaceuticals Inc. | Dosage of dasotraline and method for treatment of ADHD |
| WO2019143920A1 (en) * | 2018-01-19 | 2019-07-25 | Sunovion Pharmaceuticals Inc. | Oral dosage forms |
| CN110310711B (zh) * | 2019-06-26 | 2023-06-27 | 河南省人造金刚石微粉质量监督检验中心 | 一种金刚石微粉杂质含量分析结果的不确定度评定方法 |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3536809A (en) | 1969-02-17 | 1970-10-27 | Alza Corp | Medication method |
| US3598123A (en) | 1969-04-01 | 1971-08-10 | Alza Corp | Bandage for administering drugs |
| US3845770A (en) | 1972-06-05 | 1974-11-05 | Alza Corp | Osmatic dispensing device for releasing beneficial agent |
| US3916899A (en) | 1973-04-25 | 1975-11-04 | Alza Corp | Osmotic dispensing device with maximum and minimum sizes for the passageway |
| US4008719A (en) | 1976-02-02 | 1977-02-22 | Alza Corporation | Osmotic system having laminar arrangement for programming delivery of active agent |
| IE58110B1 (en) | 1984-10-30 | 1993-07-14 | Elan Corp Plc | Controlled release powder and process for its preparation |
| US5073543A (en) | 1988-07-21 | 1991-12-17 | G. D. Searle & Co. | Controlled release formulations of trophic factors in ganglioside-lipsome vehicle |
| IT1229203B (it) | 1989-03-22 | 1991-07-25 | Bioresearch Spa | Impiego di acido 5 metiltetraidrofolico, di acido 5 formiltetraidrofolico e dei loro sali farmaceuticamente accettabili per la preparazione di composizioni farmaceutiche in forma a rilascio controllato attive nella terapia dei disturbi mentali organici e composizioni farmaceutiche relative. |
| US5120548A (en) | 1989-11-07 | 1992-06-09 | Merck & Co., Inc. | Swelling modulated polymeric drug delivery device |
| US5733566A (en) | 1990-05-15 | 1998-03-31 | Alkermes Controlled Therapeutics Inc. Ii | Controlled release of antiparasitic agents in animals |
| US5213738A (en) | 1990-05-15 | 1993-05-25 | L. Perrigo Company | Method for making a capsule-shaped tablet |
| US5580578A (en) | 1992-01-27 | 1996-12-03 | Euro-Celtique, S.A. | Controlled release formulations coated with aqueous dispersions of acrylic polymers |
| US5591767A (en) | 1993-01-25 | 1997-01-07 | Pharmetrix Corporation | Liquid reservoir transdermal patch for the administration of ketorolac |
| IT1270594B (it) | 1994-07-07 | 1997-05-07 | Recordati Chem Pharm | Composizione farmaceutica a rilascio controllato di moguisteina in sospensione liquida |
| JP2003518487A (ja) | 1999-12-23 | 2003-06-10 | ファイザー・プロダクツ・インク | ヒドロゲル駆動型積層薬物製剤 |
| US20060057207A1 (en) * | 2001-11-30 | 2006-03-16 | Pfizer Inc | Fast-disintegrating dosage forms of 5,8,14-triazatetracyclo[10.3.1.02,11.04,9]-hexadeca-2(11),3,5,7,9-pentaene |
| CN100584818C (zh) * | 2002-09-16 | 2010-01-27 | 塞普拉科公司 | 用反式4-(3,4-二氯苯基)-1,2,3,4-四氢-1-萘胺及其甲酰胺治疗cns病症 |
| JP2006504693A (ja) | 2002-09-16 | 2006-02-09 | セプラコア インコーポレーテッド | トランス4−(3,4−ジクロロフェニル)−1,2,3,4−テトラヒドロ−n−メチル−1−ナフタレンアミンによるcns障害の治療 |
| EP1542961B1 (en) * | 2002-09-16 | 2013-09-11 | Sepracor, Inc. | Trans 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-napthalenamine for use in treatment of cns disorders |
| CN101553217A (zh) * | 2005-07-06 | 2009-10-07 | 塞普拉科公司 | 艾司佐匹克隆与反式4-(3,4-二氯苯基)-1,2,3,4-四氢-n-甲基-1-萘胺或反式4-(3,4-二氯苯基)-1,2,3,4-四氢-1-萘胺的组合以及治疗绝经期和心境障碍、焦虑症和认知障碍的方法 |
| CN101421228B (zh) * | 2006-03-31 | 2014-05-21 | 塞普拉柯公司 | 手性酰胺和胺的制备 |
| CA2653498A1 (en) | 2006-05-31 | 2007-12-13 | Sepracor Inc. | Treatment of pain disorders with trans 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro-1-naphthalenamine and its formamide |
| US7711807B2 (en) * | 2006-07-27 | 2010-05-04 | Intel Corporation | Selective filtering of exception data units |
| US20060257475A1 (en) * | 2006-08-17 | 2006-11-16 | Boehringer Ingelheim International Gmbh | Stable Sertraline Hydrochloride Formulation and Method |
| CN102573823B (zh) | 2009-05-13 | 2015-01-07 | 桑诺维恩药品公司 | 包含反式去甲舍曲林和血清素受体1a激动剂/拮抗剂的组合物及其用途 |
-
2010
- 2010-12-03 SG SG2012040366A patent/SG181466A1/en unknown
- 2010-12-03 DK DK10835166.9T patent/DK2506842T3/en active
- 2010-12-03 CN CN201080063074.XA patent/CN102740842B/zh active Active
- 2010-12-03 KR KR1020127017065A patent/KR101755743B1/ko active IP Right Grant
- 2010-12-03 RU RU2012127777/15A patent/RU2578956C2/ru active
- 2010-12-03 RU RU2016104396A patent/RU2016104396A/ru not_active Application Discontinuation
- 2010-12-03 NZ NZ600107A patent/NZ600107A/en unknown
- 2010-12-03 EP EP15150813.2A patent/EP2937083B1/en active Active
- 2010-12-03 US US13/513,170 patent/US8957114B2/en active Active
- 2010-12-03 MX MX2012006226A patent/MX339619B/es active IP Right Grant
- 2010-12-03 PL PL10835166T patent/PL2506842T3/pl unknown
- 2010-12-03 BR BR112012013325A patent/BR112012013325B8/pt active IP Right Grant
- 2010-12-03 EP EP10835166.9A patent/EP2506842B1/en active Active
- 2010-12-03 WO PCT/US2010/058831 patent/WO2011069032A2/en active Application Filing
- 2010-12-03 AU AU2010325894A patent/AU2010325894B2/en active Active
- 2010-12-03 ES ES10835166T patent/ES2531312T3/es active Active
- 2010-12-03 CN CN201510354186.5A patent/CN105125528A/zh active Pending
- 2010-12-03 PT PT108351669T patent/PT2506842E/pt unknown
- 2010-12-03 MX MX2016006875A patent/MX367327B/es unknown
- 2010-12-03 JP JP2012542211A patent/JP5766713B2/ja active Active
- 2010-12-03 CA CA2782240A patent/CA2782240A1/en not_active Abandoned
-
2012
- 2012-05-17 ZA ZA2012/03621A patent/ZA201203621B/en unknown
- 2012-05-23 IL IL219963A patent/IL219963A/en active IP Right Grant
-
2013
- 2013-03-04 HK HK16104266.1A patent/HK1216986A1/zh unknown
- 2013-03-04 HK HK13102688.8A patent/HK1175692A1/zh unknown
-
2015
- 2015-01-26 US US14/605,334 patent/US20150196502A1/en not_active Abandoned
- 2015-06-17 JP JP2015121651A patent/JP2015199763A/ja active Pending
- 2015-10-28 AU AU2015249065A patent/AU2015249065B2/en active Active
-
2016
- 2016-09-27 IL IL248077A patent/IL248077B/en active IP Right Grant
-
2017
- 2017-06-23 US US15/631,909 patent/US20180071230A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2015249065B2 (en) | Formulations, salts and polymorphs of transnorsertraline and uses thereof | |
| ES2261234T5 (es) | Metabolitos de bupropion y metodos de sintesis y uso. | |
| US20120077818A1 (en) | Compositions comprising transnorsertraline and serotonin receptor 1a agonists/antagonists and uses thereof | |
| JP2010506846A (ja) | フェニルアルキルカルバメート組成物 | |
| BR112019011640B1 (pt) | Compostos conjugados de d-metilfenidato, composições que compreendem os mesmos e kit farmacêutico que compreende as ditas composições | |
| KR102235994B1 (ko) | 환상 아민 유도체의 결정 및 그 의약 용도 | |
| DK2961391T3 (en) | Formulation comprising a benzothiazolone compound | |
| CN107108560A (zh) | 化合物、组合物及其方法 | |
| US20210094961A1 (en) | Form of ponatinib | |
| BR112020010135A2 (pt) | formas polimórficas de maleato de trimebutina e método de usar as mesmas | |
| EA045716B1 (ru) | Композиция конденсированных трициклических производных гамма-аминокислоты для лечения и/или предупреждения механической боли и её приготовление |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B08F | Application dismissed because of non-payment of annual fees [chapter 8.6 patent gazette] | ||
| B08G | Application fees: restoration [chapter 8.7 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] |
Free format text: NOTIFICACAO DE ANUENCIA RELACIONADA COM O ART 229 DA LPI |
|
| B06T | Formal requirements before examination [chapter 6.20 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 03/12/2010, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 03/12/2010 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |