WO2022203086A1 - 積層体 - Google Patents
積層体 Download PDFInfo
- Publication number
- WO2022203086A1 WO2022203086A1 PCT/JP2022/014876 JP2022014876W WO2022203086A1 WO 2022203086 A1 WO2022203086 A1 WO 2022203086A1 JP 2022014876 W JP2022014876 W JP 2022014876W WO 2022203086 A1 WO2022203086 A1 WO 2022203086A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin layer
- bis
- cured resin
- laminate
- layer
- Prior art date
Links
- 229920005989 resin Polymers 0.000 claims abstract description 155
- 239000011347 resin Substances 0.000 claims abstract description 155
- 239000010410 layer Substances 0.000 claims abstract description 149
- 239000000178 monomer Substances 0.000 claims abstract description 56
- 229920000642 polymer Polymers 0.000 claims abstract description 55
- 238000010438 heat treatment Methods 0.000 claims abstract description 49
- 239000012790 adhesive layer Substances 0.000 claims abstract description 48
- 239000011342 resin composition Substances 0.000 claims abstract description 32
- 238000005259 measurement Methods 0.000 claims abstract description 8
- 229920001721 polyimide Polymers 0.000 claims description 49
- 239000009719 polyimide resin Substances 0.000 claims description 41
- 230000009477 glass transition Effects 0.000 claims description 8
- 238000000034 method Methods 0.000 description 55
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 45
- -1 aromatic diamine compound Chemical class 0.000 description 45
- 239000002904 solvent Substances 0.000 description 36
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 32
- 238000000576 coating method Methods 0.000 description 31
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 27
- 239000011248 coating agent Substances 0.000 description 27
- 238000006243 chemical reaction Methods 0.000 description 26
- 239000000758 substrate Substances 0.000 description 20
- 229920005575 poly(amic acid) Polymers 0.000 description 16
- GTDPSWPPOUPBNX-UHFFFAOYSA-N ac1mqpva Chemical compound CC12C(=O)OC(=O)C1(C)C1(C)C2(C)C(=O)OC1=O GTDPSWPPOUPBNX-UHFFFAOYSA-N 0.000 description 15
- 239000000463 material Substances 0.000 description 14
- 239000003960 organic solvent Substances 0.000 description 14
- 230000001070 adhesive effect Effects 0.000 description 13
- 238000006116 polymerization reaction Methods 0.000 description 13
- 239000000243 solution Substances 0.000 description 13
- 239000000126 substance Substances 0.000 description 13
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 238000001035 drying Methods 0.000 description 12
- 239000003505 polymerization initiator Substances 0.000 description 12
- 125000006158 tetracarboxylic acid group Chemical group 0.000 description 12
- 229920005992 thermoplastic resin Polymers 0.000 description 12
- 239000000853 adhesive Substances 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 11
- 125000000962 organic group Chemical group 0.000 description 11
- 229920001230 polyarylate Polymers 0.000 description 11
- 239000003999 initiator Substances 0.000 description 10
- 238000004519 manufacturing process Methods 0.000 description 10
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 9
- 125000001153 fluoro group Chemical group F* 0.000 description 9
- 238000010030 laminating Methods 0.000 description 9
- 239000004642 Polyimide Substances 0.000 description 8
- 150000001875 compounds Chemical class 0.000 description 8
- 229920006122 polyamide resin Polymers 0.000 description 8
- 125000002723 alicyclic group Chemical group 0.000 description 7
- 239000000155 melt Substances 0.000 description 7
- 230000003287 optical effect Effects 0.000 description 7
- 229920000139 polyethylene terephthalate Polymers 0.000 description 7
- 239000005020 polyethylene terephthalate Substances 0.000 description 7
- 238000009835 boiling Methods 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 239000000123 paper Substances 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- 239000004925 Acrylic resin Substances 0.000 description 5
- 229920000178 Acrylic resin Polymers 0.000 description 5
- 230000001588 bifunctional effect Effects 0.000 description 5
- 239000011810 insulating material Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 125000004433 nitrogen atom Chemical group N* 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- TXBCBTDQIULDIA-UHFFFAOYSA-N 2-[[3-hydroxy-2,2-bis(hydroxymethyl)propoxy]methyl]-2-(hydroxymethyl)propane-1,3-diol Chemical class OCC(CO)(CO)COCC(CO)(CO)CO TXBCBTDQIULDIA-UHFFFAOYSA-N 0.000 description 4
- 239000004721 Polyphenylene oxide Substances 0.000 description 4
- 125000000217 alkyl group Chemical group 0.000 description 4
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 4
- 238000005266 casting Methods 0.000 description 4
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 4
- 238000010894 electron beam technology Methods 0.000 description 4
- 230000001678 irradiating effect Effects 0.000 description 4
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 4
- 229920000570 polyether Polymers 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 238000011282 treatment Methods 0.000 description 4
- NVKGJHAQGWCWDI-UHFFFAOYSA-N 4-[4-amino-2-(trifluoromethyl)phenyl]-3-(trifluoromethyl)aniline Chemical group FC(F)(F)C1=CC(N)=CC=C1C1=CC=C(N)C=C1C(F)(F)F NVKGJHAQGWCWDI-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 3
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 229920005601 base polymer Polymers 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 229920001971 elastomer Polymers 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000005060 rubber Substances 0.000 description 3
- 238000012719 thermal polymerization Methods 0.000 description 3
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- MXPYJVUYLVNEBB-UHFFFAOYSA-N 2-[2-(2-carboxybenzoyl)oxycarbonylbenzoyl]oxycarbonylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(=O)OC(=O)C1=CC=CC=C1C(=O)OC(=O)C1=CC=CC=C1C(O)=O MXPYJVUYLVNEBB-UHFFFAOYSA-N 0.000 description 2
- YCPMSWJCWKUXRH-UHFFFAOYSA-N 2-[4-[9-[4-(2-prop-2-enoyloxyethoxy)phenyl]fluoren-9-yl]phenoxy]ethyl prop-2-enoate Chemical compound C1=CC(OCCOC(=O)C=C)=CC=C1C1(C=2C=CC(OCCOC(=O)C=C)=CC=2)C2=CC=CC=C2C2=CC=CC=C21 YCPMSWJCWKUXRH-UHFFFAOYSA-N 0.000 description 2
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 2
- UVUCUHVQYAPMEU-UHFFFAOYSA-N 3-[2-(3-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]aniline Chemical compound NC1=CC=CC(C(C=2C=C(N)C=CC=2)(C(F)(F)F)C(F)(F)F)=C1 UVUCUHVQYAPMEU-UHFFFAOYSA-N 0.000 description 2
- DOAYUKLNEKRLCQ-UHFFFAOYSA-N 3-[2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]aniline Chemical compound C1=CC(N)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=CC(N)=C1 DOAYUKLNEKRLCQ-UHFFFAOYSA-N 0.000 description 2
- GBUNNYTXPDCASY-UHFFFAOYSA-N 3-[3-[2-[3-(3-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(C=CC=2)C(C=2C=C(OC=3C=C(N)C=CC=3)C=CC=2)(C(F)(F)F)C(F)(F)F)=C1 GBUNNYTXPDCASY-UHFFFAOYSA-N 0.000 description 2
- MFTFTIALAXXIMU-UHFFFAOYSA-N 3-[4-[2-[4-(3-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(=CC=2)C(C=2C=CC(OC=3C=C(N)C=CC=3)=CC=2)(C(F)(F)F)C(F)(F)F)=C1 MFTFTIALAXXIMU-UHFFFAOYSA-N 0.000 description 2
- VPWNQTHUCYMVMZ-UHFFFAOYSA-N 4,4'-sulfonyldiphenol Chemical compound C1=CC(O)=CC=C1S(=O)(=O)C1=CC=C(O)C=C1 VPWNQTHUCYMVMZ-UHFFFAOYSA-N 0.000 description 2
- BEKFRNOZJSYWKZ-UHFFFAOYSA-N 4-[2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]aniline Chemical compound C1=CC(N)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(N)C=C1 BEKFRNOZJSYWKZ-UHFFFAOYSA-N 0.000 description 2
- WJMZKMOQJDOAIE-UHFFFAOYSA-N 4-[3-[2-[3-(4-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(C(C=2C=C(OC=3C=CC(N)=CC=3)C=CC=2)(C(F)(F)F)C(F)(F)F)=C1 WJMZKMOQJDOAIE-UHFFFAOYSA-N 0.000 description 2
- LEUQLXVKRVZUEX-UHFFFAOYSA-N 4-[3-[3-(4-aminophenoxy)phenyl]sulfanylphenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(SC=2C=C(OC=3C=CC(N)=CC=3)C=CC=2)=C1 LEUQLXVKRVZUEX-UHFFFAOYSA-N 0.000 description 2
- RIMWCPQXJPPTHR-UHFFFAOYSA-N 4-[4-[2-[3-[2-[4-[4-amino-2-(trifluoromethyl)phenoxy]phenyl]propan-2-yl]phenyl]propan-2-yl]phenoxy]-3-(trifluoromethyl)aniline Chemical compound C=1C=CC(C(C)(C)C=2C=CC(OC=3C(=CC(N)=CC=3)C(F)(F)F)=CC=2)=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=C(N)C=C1C(F)(F)F RIMWCPQXJPPTHR-UHFFFAOYSA-N 0.000 description 2
- HHLMWQDRYZAENA-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=C(C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)(C(F)(F)F)C(F)(F)F)C=C1 HHLMWQDRYZAENA-UHFFFAOYSA-N 0.000 description 2
- PJWQLRKRVISYPL-UHFFFAOYSA-N 4-[4-amino-3-(trifluoromethyl)phenyl]-2-(trifluoromethyl)aniline Chemical group C1=C(C(F)(F)F)C(N)=CC=C1C1=CC=C(N)C(C(F)(F)F)=C1 PJWQLRKRVISYPL-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- SDDLEVPIDBLVHC-UHFFFAOYSA-N Bisphenol Z Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)CCCCC1 SDDLEVPIDBLVHC-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 239000004593 Epoxy Substances 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- MHABMANUFPZXEB-UHFFFAOYSA-N O-demethyl-aloesaponarin I Natural products O=C1C2=CC=CC(O)=C2C(=O)C2=C1C=C(O)C(C(O)=O)=C2C MHABMANUFPZXEB-UHFFFAOYSA-N 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- LFOXEOLGJPJZAA-UHFFFAOYSA-N [(2,6-dimethoxybenzoyl)-(2,4,4-trimethylpentyl)phosphoryl]-(2,6-dimethoxyphenyl)methanone Chemical compound COC1=CC=CC(OC)=C1C(=O)P(=O)(CC(C)CC(C)(C)C)C(=O)C1=C(OC)C=CC=C1OC LFOXEOLGJPJZAA-UHFFFAOYSA-N 0.000 description 2
- 239000006096 absorbing agent Substances 0.000 description 2
- 125000003670 adamantan-2-yl group Chemical group [H]C1([H])C(C2([H])[H])([H])C([H])([H])C3([H])C([*])([H])C1([H])C([H])([H])C2([H])C3([H])[H] 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 2
- 239000012965 benzophenone Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 150000001805 chlorine compounds Chemical class 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- ZQMIGQNCOMNODD-UHFFFAOYSA-N diacetyl peroxide Chemical compound CC(=O)OOC(C)=O ZQMIGQNCOMNODD-UHFFFAOYSA-N 0.000 description 2
- 150000004985 diamines Chemical class 0.000 description 2
- MLCHBQKMVKNBOV-UHFFFAOYSA-M dioxido(phenyl)phosphanium Chemical compound [O-]P(=O)C1=CC=CC=C1 MLCHBQKMVKNBOV-UHFFFAOYSA-M 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000005401 electroluminescence Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000005227 gel permeation chromatography Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 2
- HJOVHMDZYOCNQW-UHFFFAOYSA-N isophorone Chemical compound CC1=CC(=O)CC(C)(C)C1 HJOVHMDZYOCNQW-UHFFFAOYSA-N 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- OTLDLKLSNZMTTA-UHFFFAOYSA-N octahydro-1h-4,7-methanoindene-1,5-diyldimethanol Chemical compound C1C2C3C(CO)CCC3C1C(CO)C2 OTLDLKLSNZMTTA-UHFFFAOYSA-N 0.000 description 2
- 150000001451 organic peroxides Chemical class 0.000 description 2
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 239000002985 plastic film Substances 0.000 description 2
- 229920006255 plastic film Polymers 0.000 description 2
- 239000004014 plasticizer Substances 0.000 description 2
- 229920000058 polyacrylate Polymers 0.000 description 2
- 229920006267 polyester film Polymers 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 229920000098 polyolefin Polymers 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- SMGZBBKJFKNDSC-UHFFFAOYSA-N propyl 2-(2-propoxycarbonylprop-2-enoxymethyl)prop-2-enoate Chemical compound CCCOC(=O)C(=C)COCC(=C)C(=O)OCCC SMGZBBKJFKNDSC-UHFFFAOYSA-N 0.000 description 2
- 239000004065 semiconductor Substances 0.000 description 2
- 238000004528 spin coating Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- HJIAMFHSAAEUKR-UHFFFAOYSA-N (2-hydroxyphenyl)-phenylmethanone Chemical compound OC1=CC=CC=C1C(=O)C1=CC=CC=C1 HJIAMFHSAAEUKR-UHFFFAOYSA-N 0.000 description 1
- SZCWBURCISJFEZ-UHFFFAOYSA-N (3-hydroxy-2,2-dimethylpropyl) 3-hydroxy-2,2-dimethylpropanoate Chemical compound OCC(C)(C)COC(=O)C(C)(C)CO SZCWBURCISJFEZ-UHFFFAOYSA-N 0.000 description 1
- WXPWZZHELZEVPO-UHFFFAOYSA-N (4-methylphenyl)-phenylmethanone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=CC=C1 WXPWZZHELZEVPO-UHFFFAOYSA-N 0.000 description 1
- HCNHNBLSNVSJTJ-UHFFFAOYSA-N 1,1-Bis(4-hydroxyphenyl)ethane Chemical compound C=1C=C(O)C=CC=1C(C)C1=CC=C(O)C=C1 HCNHNBLSNVSJTJ-UHFFFAOYSA-N 0.000 description 1
- HSLFISVKRDQEBY-UHFFFAOYSA-N 1,1-bis(tert-butylperoxy)cyclohexane Chemical compound CC(C)(C)OOC1(OOC(C)(C)C)CCCCC1 HSLFISVKRDQEBY-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- RTTZISZSHSCFRH-UHFFFAOYSA-N 1,3-bis(isocyanatomethyl)benzene Chemical compound O=C=NCC1=CC=CC(CN=C=O)=C1 RTTZISZSHSCFRH-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- UICXTANXZJJIBC-UHFFFAOYSA-N 1-(1-hydroperoxycyclohexyl)peroxycyclohexan-1-ol Chemical compound C1CCCCC1(O)OOC1(OO)CCCCC1 UICXTANXZJJIBC-UHFFFAOYSA-N 0.000 description 1
- XSZYESUNPWGWFQ-UHFFFAOYSA-N 1-(2-hydroperoxypropan-2-yl)-4-methylcyclohexane Chemical compound CC1CCC(C(C)(C)OO)CC1 XSZYESUNPWGWFQ-UHFFFAOYSA-N 0.000 description 1
- VLDPXPPHXDGHEW-UHFFFAOYSA-N 1-chloro-2-dichlorophosphoryloxybenzene Chemical compound ClC1=CC=CC=C1OP(Cl)(Cl)=O VLDPXPPHXDGHEW-UHFFFAOYSA-N 0.000 description 1
- VKQJCUYEEABXNK-UHFFFAOYSA-N 1-chloro-4-propoxythioxanthen-9-one Chemical compound S1C2=CC=CC=C2C(=O)C2=C1C(OCCC)=CC=C2Cl VKQJCUYEEABXNK-UHFFFAOYSA-N 0.000 description 1
- 239000012956 1-hydroxycyclohexylphenyl-ketone Substances 0.000 description 1
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 1
- XLPJNCYCZORXHG-UHFFFAOYSA-N 1-morpholin-4-ylprop-2-en-1-one Chemical compound C=CC(=O)N1CCOCC1 XLPJNCYCZORXHG-UHFFFAOYSA-N 0.000 description 1
- YIYBRXKMQFDHSM-UHFFFAOYSA-N 2,2'-Dihydroxybenzophenone Chemical class OC1=CC=CC=C1C(=O)C1=CC=CC=C1O YIYBRXKMQFDHSM-UHFFFAOYSA-N 0.000 description 1
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 description 1
- KWVGIHKZDCUPEU-UHFFFAOYSA-N 2,2-dimethoxy-2-phenylacetophenone Chemical compound C=1C=CC=CC=1C(OC)(OC)C(=O)C1=CC=CC=C1 KWVGIHKZDCUPEU-UHFFFAOYSA-N 0.000 description 1
- YRTNMMLRBJMGJJ-UHFFFAOYSA-N 2,2-dimethylpropane-1,3-diol;hexanedioic acid Chemical compound OCC(C)(C)CO.OC(=O)CCCCC(O)=O YRTNMMLRBJMGJJ-UHFFFAOYSA-N 0.000 description 1
- BRKORVYTKKLNKX-UHFFFAOYSA-N 2,4-di(propan-2-yl)thioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC(C(C)C)=CC(C(C)C)=C3SC2=C1 BRKORVYTKKLNKX-UHFFFAOYSA-N 0.000 description 1
- UXCIJKOCUAQMKD-UHFFFAOYSA-N 2,4-dichlorothioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC(Cl)=CC(Cl)=C3SC2=C1 UXCIJKOCUAQMKD-UHFFFAOYSA-N 0.000 description 1
- AVTLBBWTUPQRAY-UHFFFAOYSA-N 2-(2-cyanobutan-2-yldiazenyl)-2-methylbutanenitrile Chemical compound CCC(C)(C#N)N=NC(C)(CC)C#N AVTLBBWTUPQRAY-UHFFFAOYSA-N 0.000 description 1
- VXHYVVAUHMGCEX-UHFFFAOYSA-N 2-(2-hydroxyphenoxy)phenol Chemical class OC1=CC=CC=C1OC1=CC=CC=C1O VXHYVVAUHMGCEX-UHFFFAOYSA-N 0.000 description 1
- BLDLRWQLBOJPEB-UHFFFAOYSA-N 2-(2-hydroxyphenyl)sulfanylphenol Chemical class OC1=CC=CC=C1SC1=CC=CC=C1O BLDLRWQLBOJPEB-UHFFFAOYSA-N 0.000 description 1
- XSVZEASGNTZBRQ-UHFFFAOYSA-N 2-(2-hydroxyphenyl)sulfinylphenol Chemical class OC1=CC=CC=C1S(=O)C1=CC=CC=C1O XSVZEASGNTZBRQ-UHFFFAOYSA-N 0.000 description 1
- QUWAJPZDCZDTJS-UHFFFAOYSA-N 2-(2-hydroxyphenyl)sulfonylphenol Chemical class OC1=CC=CC=C1S(=O)(=O)C1=CC=CC=C1O QUWAJPZDCZDTJS-UHFFFAOYSA-N 0.000 description 1
- XMNIXWIUMCBBBL-UHFFFAOYSA-N 2-(2-phenylpropan-2-ylperoxy)propan-2-ylbenzene Chemical compound C=1C=CC=CC=1C(C)(C)OOC(C)(C)C1=CC=CC=C1 XMNIXWIUMCBBBL-UHFFFAOYSA-N 0.000 description 1
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 1
- BOQSYAZCZOTYFO-UHFFFAOYSA-N 2-(prop-2-enoxymethyl)prop-2-enoic acid Chemical compound OC(=O)C(=C)COCC=C BOQSYAZCZOTYFO-UHFFFAOYSA-N 0.000 description 1
- GJKGAPPUXSSCFI-UHFFFAOYSA-N 2-Hydroxy-4'-(2-hydroxyethoxy)-2-methylpropiophenone Chemical compound CC(C)(O)C(=O)C1=CC=C(OCCO)C=C1 GJKGAPPUXSSCFI-UHFFFAOYSA-N 0.000 description 1
- SPSNALDHELHFIJ-UHFFFAOYSA-N 2-[(1-cyano-1-cyclopropylethyl)diazenyl]-2-cyclopropylpropanenitrile Chemical compound C1CC1C(C)(C#N)N=NC(C)(C#N)C1CC1 SPSNALDHELHFIJ-UHFFFAOYSA-N 0.000 description 1
- PFHOSZAOXCYAGJ-UHFFFAOYSA-N 2-[(2-cyano-4-methoxy-4-methylpentan-2-yl)diazenyl]-4-methoxy-2,4-dimethylpentanenitrile Chemical compound COC(C)(C)CC(C)(C#N)N=NC(C)(C#N)CC(C)(C)OC PFHOSZAOXCYAGJ-UHFFFAOYSA-N 0.000 description 1
- WYGWHHGCAGTUCH-UHFFFAOYSA-N 2-[(2-cyano-4-methylpentan-2-yl)diazenyl]-2,4-dimethylpentanenitrile Chemical compound CC(C)CC(C)(C#N)N=NC(C)(C#N)CC(C)C WYGWHHGCAGTUCH-UHFFFAOYSA-N 0.000 description 1
- SICQTWWJCGGHIX-UHFFFAOYSA-N 2-[2-[2-[2-(4-benzoylphenoxy)ethoxy]ethoxy]ethoxy]ethyl prop-2-enoate Chemical compound C1=CC(OCCOCCOCCOCCOC(=O)C=C)=CC=C1C(=O)C1=CC=CC=C1 SICQTWWJCGGHIX-UHFFFAOYSA-N 0.000 description 1
- FGTYTUFKXYPTML-UHFFFAOYSA-N 2-benzoylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C(=O)C1=CC=CC=C1 FGTYTUFKXYPTML-UHFFFAOYSA-N 0.000 description 1
- VLLZXMSTOYKVPY-UHFFFAOYSA-N 2-bromo-4-[(3-bromo-4-hydroxyphenyl)-phenylmethyl]phenol Chemical compound C1=C(Br)C(O)=CC=C1C(C=1C=C(Br)C(O)=CC=1)C1=CC=CC=C1 VLLZXMSTOYKVPY-UHFFFAOYSA-N 0.000 description 1
- RLGGIBKAEYMHOI-UHFFFAOYSA-N 2-bromo-4-[1-(3-bromo-4-hydroxyphenyl)-1-phenylethyl]phenol Chemical compound C=1C=C(O)C(Br)=CC=1C(C=1C=C(Br)C(O)=CC=1)(C)C1=CC=CC=C1 RLGGIBKAEYMHOI-UHFFFAOYSA-N 0.000 description 1
- ZCDADJXRUCOCJE-UHFFFAOYSA-N 2-chlorothioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC(Cl)=CC=C3SC2=C1 ZCDADJXRUCOCJE-UHFFFAOYSA-N 0.000 description 1
- CKSAKVMRQYOFBC-UHFFFAOYSA-N 2-cyanopropan-2-yliminourea Chemical compound N#CC(C)(C)N=NC(N)=O CKSAKVMRQYOFBC-UHFFFAOYSA-N 0.000 description 1
- QZUITNVOBZTUAD-UHFFFAOYSA-N 2-fluoro-4-[(3-fluoro-4-hydroxyphenyl)-(4-fluorophenyl)methyl]phenol Chemical compound C1=C(F)C(O)=CC=C1C(C=1C=C(F)C(O)=CC=1)C1=CC=C(F)C=C1 QZUITNVOBZTUAD-UHFFFAOYSA-N 0.000 description 1
- BFZPKFLWSACAMJ-UHFFFAOYSA-N 2-fluoro-4-[(3-fluoro-4-hydroxyphenyl)-phenylmethyl]phenol Chemical compound C1=C(F)C(O)=CC=C1C(C=1C=C(F)C(O)=CC=1)C1=CC=CC=C1 BFZPKFLWSACAMJ-UHFFFAOYSA-N 0.000 description 1
- MIRQGKQPLPBZQM-UHFFFAOYSA-N 2-hydroperoxy-2,4,4-trimethylpentane Chemical compound CC(C)(C)CC(C)(C)OO MIRQGKQPLPBZQM-UHFFFAOYSA-N 0.000 description 1
- WFUGQJXVXHBTEM-UHFFFAOYSA-N 2-hydroperoxy-2-(2-hydroperoxybutan-2-ylperoxy)butane Chemical compound CCC(C)(OO)OOC(C)(CC)OO WFUGQJXVXHBTEM-UHFFFAOYSA-N 0.000 description 1
- PCKZAVNWRLEHIP-UHFFFAOYSA-N 2-hydroxy-1-[4-[[4-(2-hydroxy-2-methylpropanoyl)phenyl]methyl]phenyl]-2-methylpropan-1-one Chemical compound C1=CC(C(=O)C(C)(O)C)=CC=C1CC1=CC=C(C(=O)C(C)(C)O)C=C1 PCKZAVNWRLEHIP-UHFFFAOYSA-N 0.000 description 1
- XMLYCEVDHLAQEL-UHFFFAOYSA-N 2-hydroxy-2-methyl-1-phenylpropan-1-one Chemical compound CC(C)(O)C(=O)C1=CC=CC=C1 XMLYCEVDHLAQEL-UHFFFAOYSA-N 0.000 description 1
- OMIGHNLMNHATMP-UHFFFAOYSA-N 2-hydroxyethyl prop-2-enoate Chemical compound OCCOC(=O)C=C OMIGHNLMNHATMP-UHFFFAOYSA-N 0.000 description 1
- LWRBVKNFOYUCNP-UHFFFAOYSA-N 2-methyl-1-(4-methylsulfanylphenyl)-2-morpholin-4-ylpropan-1-one Chemical compound C1=CC(SC)=CC=C1C(=O)C(C)(C)N1CCOCC1 LWRBVKNFOYUCNP-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- MYISVPVWAQRUTL-UHFFFAOYSA-N 2-methylthioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC(C)=CC=C3SC2=C1 MYISVPVWAQRUTL-UHFFFAOYSA-N 0.000 description 1
- KTALPKYXQZGAEG-UHFFFAOYSA-N 2-propan-2-ylthioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC(C(C)C)=CC=C3SC2=C1 KTALPKYXQZGAEG-UHFFFAOYSA-N 0.000 description 1
- WOFFQWIGWBMFPL-UHFFFAOYSA-N 2-tert-butyl-4-[(3-tert-butyl-4-hydroxyphenyl)-phenylmethyl]phenol Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C=2C=CC=CC=2)C=2C=C(C(O)=CC=2)C(C)(C)C)=C1 WOFFQWIGWBMFPL-UHFFFAOYSA-N 0.000 description 1
- OCEQSAGTCSHWKF-UHFFFAOYSA-N 2-tert-butyl-4-[1-(3-tert-butyl-4-hydroxyphenyl)-1-phenylethyl]phenol Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C)(C=2C=CC=CC=2)C=2C=C(C(O)=CC=2)C(C)(C)C)=C1 OCEQSAGTCSHWKF-UHFFFAOYSA-N 0.000 description 1
- BIISIZOQPWZPPS-UHFFFAOYSA-N 2-tert-butylperoxypropan-2-ylbenzene Chemical compound CC(C)(C)OOC(C)(C)C1=CC=CC=C1 BIISIZOQPWZPPS-UHFFFAOYSA-N 0.000 description 1
- NUIURNJTPRWVAP-UHFFFAOYSA-N 3,3'-Dimethylbenzidine Chemical group C1=C(N)C(C)=CC(C=2C=C(C)C(N)=CC=2)=C1 NUIURNJTPRWVAP-UHFFFAOYSA-N 0.000 description 1
- FRIBMENBGGCKPD-UHFFFAOYSA-N 3-(2,3-dimethoxyphenyl)prop-2-enal Chemical compound COC1=CC=CC(C=CC=O)=C1OC FRIBMENBGGCKPD-UHFFFAOYSA-N 0.000 description 1
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 1
- ZBMISJGHVWNWTE-UHFFFAOYSA-N 3-(4-aminophenoxy)aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(N)=C1 ZBMISJGHVWNWTE-UHFFFAOYSA-N 0.000 description 1
- ZMPZWXKBGSQATE-UHFFFAOYSA-N 3-(4-aminophenyl)sulfonylaniline Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=CC(N)=C1 ZMPZWXKBGSQATE-UHFFFAOYSA-N 0.000 description 1
- CKOFBUUFHALZGK-UHFFFAOYSA-N 3-[(3-aminophenyl)methyl]aniline Chemical compound NC1=CC=CC(CC=2C=C(N)C=CC=2)=C1 CKOFBUUFHALZGK-UHFFFAOYSA-N 0.000 description 1
- FGWQCROGAHMWSU-UHFFFAOYSA-N 3-[(4-aminophenyl)methyl]aniline Chemical compound C1=CC(N)=CC=C1CC1=CC=CC(N)=C1 FGWQCROGAHMWSU-UHFFFAOYSA-N 0.000 description 1
- DVXYMCJCMDTSQA-UHFFFAOYSA-N 3-[2-(3-aminophenyl)propan-2-yl]aniline Chemical compound C=1C=CC(N)=CC=1C(C)(C)C1=CC=CC(N)=C1 DVXYMCJCMDTSQA-UHFFFAOYSA-N 0.000 description 1
- HIYRIYOUSQLJHP-UHFFFAOYSA-N 3-[2-(4-aminophenyl)propan-2-yl]aniline Chemical compound C=1C=CC(N)=CC=1C(C)(C)C1=CC=C(N)C=C1 HIYRIYOUSQLJHP-UHFFFAOYSA-N 0.000 description 1
- DKKYOQYISDAQER-UHFFFAOYSA-N 3-[3-(3-aminophenoxy)phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(OC=3C=C(N)C=CC=3)C=CC=2)=C1 DKKYOQYISDAQER-UHFFFAOYSA-N 0.000 description 1
- FJWUJUIPIZSDTR-UHFFFAOYSA-N 3-[3-[2-[3-(3-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=CC(OC=2C=C(N)C=CC=2)=CC=1C(C)(C)C(C=1)=CC=CC=1OC1=CC=CC(N)=C1 FJWUJUIPIZSDTR-UHFFFAOYSA-N 0.000 description 1
- GZBHMJRTCUJCBO-UHFFFAOYSA-N 3-[3-[3-(3-aminophenoxy)phenoxy]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(OC=3C=C(OC=4C=C(N)C=CC=4)C=CC=3)C=CC=2)=C1 GZBHMJRTCUJCBO-UHFFFAOYSA-N 0.000 description 1
- SABXTRNPHKCTFO-UHFFFAOYSA-N 3-[3-[3-(3-aminophenoxy)phenyl]sulfanylphenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(SC=3C=C(OC=4C=C(N)C=CC=4)C=CC=3)C=CC=2)=C1 SABXTRNPHKCTFO-UHFFFAOYSA-N 0.000 description 1
- YLTIRYJAWQHSQS-UHFFFAOYSA-N 3-[3-[3-(3-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(C=CC=2)S(=O)(=O)C=2C=C(OC=3C=C(N)C=CC=3)C=CC=2)=C1 YLTIRYJAWQHSQS-UHFFFAOYSA-N 0.000 description 1
- OLFCXXUMDWEXKG-UHFFFAOYSA-N 3-[3-[[3-(3-aminophenoxy)phenyl]methyl]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(CC=3C=C(OC=4C=C(N)C=CC=4)C=CC=3)C=CC=2)=C1 OLFCXXUMDWEXKG-UHFFFAOYSA-N 0.000 description 1
- LBPVOEHZEWAJKQ-UHFFFAOYSA-N 3-[4-(3-aminophenoxy)phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(OC=3C=C(N)C=CC=3)=CC=2)=C1 LBPVOEHZEWAJKQ-UHFFFAOYSA-N 0.000 description 1
- NYRFBMFAUFUULG-UHFFFAOYSA-N 3-[4-[2-[4-(3-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=C(OC=2C=C(N)C=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=CC(N)=C1 NYRFBMFAUFUULG-UHFFFAOYSA-N 0.000 description 1
- OSYIROQVSVAEAH-UHFFFAOYSA-N 3-[4-[3-(3-aminophenoxy)phenyl]phenoxy]aniline Chemical group NC1=CC=CC(OC=2C=CC(=CC=2)C=2C=C(OC=3C=C(N)C=CC=3)C=CC=2)=C1 OSYIROQVSVAEAH-UHFFFAOYSA-N 0.000 description 1
- NQZOFDAHZVLQJO-UHFFFAOYSA-N 3-[4-[4-(3-aminophenoxy)phenoxy]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(OC=3C=CC(OC=4C=C(N)C=CC=4)=CC=3)=CC=2)=C1 NQZOFDAHZVLQJO-UHFFFAOYSA-N 0.000 description 1
- JERFEOKUSPGKGV-UHFFFAOYSA-N 3-[4-[4-(3-aminophenoxy)phenyl]sulfanylphenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(SC=3C=CC(OC=4C=C(N)C=CC=4)=CC=3)=CC=2)=C1 JERFEOKUSPGKGV-UHFFFAOYSA-N 0.000 description 1
- YSMXOEWEUZTWAK-UHFFFAOYSA-N 3-[4-[[4-(3-aminophenoxy)phenyl]methyl]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(CC=3C=CC(OC=4C=C(N)C=CC=4)=CC=3)=CC=2)=C1 YSMXOEWEUZTWAK-UHFFFAOYSA-N 0.000 description 1
- BQFSURDUHGBXFL-UHFFFAOYSA-N 3-methylthioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC=C(C)C=C3SC2=C1 BQFSURDUHGBXFL-UHFFFAOYSA-N 0.000 description 1
- ICNFHJVPAJKPHW-UHFFFAOYSA-N 4,4'-Thiodianiline Chemical compound C1=CC(N)=CC=C1SC1=CC=C(N)C=C1 ICNFHJVPAJKPHW-UHFFFAOYSA-N 0.000 description 1
- YBRVSVVVWCFQMG-UHFFFAOYSA-N 4,4'-diaminodiphenylmethane Chemical compound C1=CC(N)=CC=C1CC1=CC=C(N)C=C1 YBRVSVVVWCFQMG-UHFFFAOYSA-N 0.000 description 1
- VWGKEVWFBOUAND-UHFFFAOYSA-N 4,4'-thiodiphenol Chemical compound C1=CC(O)=CC=C1SC1=CC=C(O)C=C1 VWGKEVWFBOUAND-UHFFFAOYSA-N 0.000 description 1
- QYIMZXITLDTULQ-UHFFFAOYSA-N 4-(4-amino-2-methylphenyl)-3-methylaniline Chemical group CC1=CC(N)=CC=C1C1=CC=C(N)C=C1C QYIMZXITLDTULQ-UHFFFAOYSA-N 0.000 description 1
- NEQFBGHQPUXOFH-UHFFFAOYSA-N 4-(4-carboxyphenyl)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=C(C(O)=O)C=C1 NEQFBGHQPUXOFH-UHFFFAOYSA-N 0.000 description 1
- ZGRIVKAIJRLAIC-UHFFFAOYSA-N 4-(4-hydroxy-3-methylphenoxy)-2-methylphenol Chemical compound C1=C(O)C(C)=CC(OC=2C=C(C)C(O)=CC=2)=C1 ZGRIVKAIJRLAIC-UHFFFAOYSA-N 0.000 description 1
- IBNFPRMKLZDANU-UHFFFAOYSA-N 4-(4-hydroxy-3-methylphenyl)sulfanyl-2-methylphenol Chemical compound C1=C(O)C(C)=CC(SC=2C=C(C)C(O)=CC=2)=C1 IBNFPRMKLZDANU-UHFFFAOYSA-N 0.000 description 1
- JWXGVUCKVZVSQE-UHFFFAOYSA-N 4-(4-hydroxy-3-methylphenyl)sulfinyl-2-methylphenol Chemical compound C1=C(O)C(C)=CC(S(=O)C=2C=C(C)C(O)=CC=2)=C1 JWXGVUCKVZVSQE-UHFFFAOYSA-N 0.000 description 1
- XTEGBRKTHOUETR-UHFFFAOYSA-N 4-(4-hydroxy-3-methylphenyl)sulfonyl-2-methylphenol Chemical compound C1=C(O)C(C)=CC(S(=O)(=O)C=2C=C(C)C(O)=CC=2)=C1 XTEGBRKTHOUETR-UHFFFAOYSA-N 0.000 description 1
- NZGQHKSLKRFZFL-UHFFFAOYSA-N 4-(4-hydroxyphenoxy)phenol Chemical compound C1=CC(O)=CC=C1OC1=CC=C(O)C=C1 NZGQHKSLKRFZFL-UHFFFAOYSA-N 0.000 description 1
- RQCACQIALULDSK-UHFFFAOYSA-N 4-(4-hydroxyphenyl)sulfinylphenol Chemical compound C1=CC(O)=CC=C1S(=O)C1=CC=C(O)C=C1 RQCACQIALULDSK-UHFFFAOYSA-N 0.000 description 1
- HLBLWEWZXPIGSM-UHFFFAOYSA-N 4-Aminophenyl ether Chemical compound C1=CC(N)=CC=C1OC1=CC=C(N)C=C1 HLBLWEWZXPIGSM-UHFFFAOYSA-N 0.000 description 1
- MDEQXFQEJZFWNA-UHFFFAOYSA-N 4-[(4-bromophenyl)-(4-hydroxyphenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(Br)=CC=1)C1=CC=C(O)C=C1 MDEQXFQEJZFWNA-UHFFFAOYSA-N 0.000 description 1
- IJSGQDFHZUWQES-UHFFFAOYSA-N 4-[(4-chlorophenyl)-(4-hydroxyphenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(Cl)=CC=1)C1=CC=C(O)C=C1 IJSGQDFHZUWQES-UHFFFAOYSA-N 0.000 description 1
- YOGHLYZODNECHR-UHFFFAOYSA-N 4-[(4-fluorophenyl)-(4-hydroxy-3,5-dimethylphenyl)methyl]-2,6-dimethylphenol Chemical compound CC1=C(O)C(C)=CC(C(C=2C=CC(F)=CC=2)C=2C=C(C)C(O)=C(C)C=2)=C1 YOGHLYZODNECHR-UHFFFAOYSA-N 0.000 description 1
- AHUGQNPASAGFGP-UHFFFAOYSA-N 4-[(4-fluorophenyl)-(4-hydroxyphenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(F)=CC=1)C1=CC=C(O)C=C1 AHUGQNPASAGFGP-UHFFFAOYSA-N 0.000 description 1
- UEVIXTQYIIKFSM-UHFFFAOYSA-N 4-[(4-hydroxy-2,3,6-trimethylphenyl)-phenylmethyl]-2,3,5-trimethylphenol Chemical compound CC1=CC(O)=C(C)C(C)=C1C(C=1C(=C(C)C(O)=CC=1C)C)C1=CC=CC=C1 UEVIXTQYIIKFSM-UHFFFAOYSA-N 0.000 description 1
- UBBWYWBCXCFATG-UHFFFAOYSA-N 4-[(4-hydroxy-2,6-dimethylphenyl)-phenylmethyl]-3,5-dimethylphenol Chemical compound CC1=CC(O)=CC(C)=C1C(C=1C(=CC(O)=CC=1C)C)C1=CC=CC=C1 UBBWYWBCXCFATG-UHFFFAOYSA-N 0.000 description 1
- PNCYFKUQOKAIEA-UHFFFAOYSA-N 4-[(4-hydroxy-3-methylphenyl)-phenylmethyl]-2-methylphenol Chemical compound C1=C(O)C(C)=CC(C(C=2C=CC=CC=2)C=2C=C(C)C(O)=CC=2)=C1 PNCYFKUQOKAIEA-UHFFFAOYSA-N 0.000 description 1
- MIFGCULLADMRTF-UHFFFAOYSA-N 4-[(4-hydroxy-3-methylphenyl)methyl]-2-methylphenol Chemical compound C1=C(O)C(C)=CC(CC=2C=C(C)C(O)=CC=2)=C1 MIFGCULLADMRTF-UHFFFAOYSA-N 0.000 description 1
- SNSZIOPICMBKJK-UHFFFAOYSA-N 4-[(4-hydroxy-3-phenylphenyl)-phenylmethyl]-2-phenylphenol Chemical compound OC1=CC=C(C(C=2C=CC=CC=2)C=2C=C(C(O)=CC=2)C=2C=CC=CC=2)C=C1C1=CC=CC=C1 SNSZIOPICMBKJK-UHFFFAOYSA-N 0.000 description 1
- BATCUENAARTUKW-UHFFFAOYSA-N 4-[(4-hydroxyphenyl)-diphenylmethyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 BATCUENAARTUKW-UHFFFAOYSA-N 0.000 description 1
- RSSGMIIGVQRGDS-UHFFFAOYSA-N 4-[(4-hydroxyphenyl)-phenylmethyl]phenol Chemical compound C1=CC(O)=CC=C1C(C=1C=CC(O)=CC=1)C1=CC=CC=C1 RSSGMIIGVQRGDS-UHFFFAOYSA-N 0.000 description 1
- MMNLWVVVDFQANH-UHFFFAOYSA-N 4-[1-(4-hydroxy-3-methylphenyl)-1-phenylethyl]-2-methylphenol Chemical compound C1=C(O)C(C)=CC(C(C)(C=2C=CC=CC=2)C=2C=C(C)C(O)=CC=2)=C1 MMNLWVVVDFQANH-UHFFFAOYSA-N 0.000 description 1
- XDGXPHFWSPGAIB-UHFFFAOYSA-N 4-[1-(4-hydroxy-3-methylphenyl)ethyl]-2-methylphenol Chemical compound C=1C=C(O)C(C)=CC=1C(C)C1=CC=C(O)C(C)=C1 XDGXPHFWSPGAIB-UHFFFAOYSA-N 0.000 description 1
- GZEBECRWRATTQK-UHFFFAOYSA-N 4-[1-(4-hydroxy-3-phenylphenyl)-1-phenylethyl]-2-phenylphenol Chemical compound C=1C=C(O)C(C=2C=CC=CC=2)=CC=1C(C=1C=C(C(O)=CC=1)C=1C=CC=CC=1)(C)C1=CC=CC=C1 GZEBECRWRATTQK-UHFFFAOYSA-N 0.000 description 1
- XCDFWULUIVXACG-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)-1-(4-nitrophenyl)ethyl]phenol Chemical compound C=1C=C(O)C=CC=1C(C=1C=CC(=CC=1)[N+]([O-])=O)(C)C1=CC=C(O)C=C1 XCDFWULUIVXACG-UHFFFAOYSA-N 0.000 description 1
- CNMNEYFYZTUKLS-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)-1-phenylpropyl]phenol Chemical compound C=1C=C(O)C=CC=1C(C=1C=CC(O)=CC=1)(CC)C1=CC=CC=C1 CNMNEYFYZTUKLS-UHFFFAOYSA-N 0.000 description 1
- UMPGNGRIGSEMTC-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)-3,3,5-trimethylcyclohexyl]phenol Chemical compound C1C(C)CC(C)(C)CC1(C=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 UMPGNGRIGSEMTC-UHFFFAOYSA-N 0.000 description 1
- APXJLYIVOFARRM-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(C(O)=O)C(C(O)=O)=C1 APXJLYIVOFARRM-UHFFFAOYSA-N 0.000 description 1
- ZYEDGEXYGKWJPB-UHFFFAOYSA-N 4-[2-(4-aminophenyl)propan-2-yl]aniline Chemical compound C=1C=C(N)C=CC=1C(C)(C)C1=CC=C(N)C=C1 ZYEDGEXYGKWJPB-UHFFFAOYSA-N 0.000 description 1
- QHJPJZROUNGTRJ-UHFFFAOYSA-N 4-[2-(4-hydroxyphenyl)octan-2-yl]phenol Chemical compound C=1C=C(O)C=CC=1C(C)(CCCCCC)C1=CC=C(O)C=C1 QHJPJZROUNGTRJ-UHFFFAOYSA-N 0.000 description 1
- WUPRYUDHUFLKFL-UHFFFAOYSA-N 4-[3-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(OC=2C=CC(N)=CC=2)=C1 WUPRYUDHUFLKFL-UHFFFAOYSA-N 0.000 description 1
- WVIGQQBEFCXWRW-UHFFFAOYSA-N 4-[3-[3-(4-aminophenoxy)phenyl]phenoxy]aniline Chemical group C1=CC(N)=CC=C1OC1=CC=CC(C=2C=C(OC=3C=CC(N)=CC=3)C=CC=2)=C1 WVIGQQBEFCXWRW-UHFFFAOYSA-N 0.000 description 1
- VZZOONBAZHZSEB-UHFFFAOYSA-N 4-[3-[3-(4-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(S(=O)(=O)C=2C=C(OC=3C=CC(N)=CC=3)C=CC=2)=C1 VZZOONBAZHZSEB-UHFFFAOYSA-N 0.000 description 1
- IXZCKKLBICAHRA-UHFFFAOYSA-N 4-[3-[[3-(4-aminophenoxy)phenyl]methyl]phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(CC=2C=C(OC=3C=CC(N)=CC=3)C=CC=2)=C1 IXZCKKLBICAHRA-UHFFFAOYSA-N 0.000 description 1
- JCRRFJIVUPSNTA-UHFFFAOYSA-N 4-[4-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC(C=C1)=CC=C1OC1=CC=C(N)C=C1 JCRRFJIVUPSNTA-UHFFFAOYSA-N 0.000 description 1
- KMKWGXGSGPYISJ-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=C(OC=2C=CC(N)=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=C(N)C=C1 KMKWGXGSGPYISJ-UHFFFAOYSA-N 0.000 description 1
- LDFYRFKAYFZVNH-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenoxy]phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC(C=C1)=CC=C1OC(C=C1)=CC=C1OC1=CC=C(N)C=C1 LDFYRFKAYFZVNH-UHFFFAOYSA-N 0.000 description 1
- HYDATEKARGDBKU-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenyl]phenoxy]aniline Chemical group C1=CC(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)C=C1 HYDATEKARGDBKU-UHFFFAOYSA-N 0.000 description 1
- SXTPNMJRVQKNRN-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenyl]sulfanylphenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC(C=C1)=CC=C1SC(C=C1)=CC=C1OC1=CC=C(N)C=C1 SXTPNMJRVQKNRN-UHFFFAOYSA-N 0.000 description 1
- UGVRJVHOJNYEHR-UHFFFAOYSA-N 4-chlorobenzophenone Chemical compound C1=CC(Cl)=CC=C1C(=O)C1=CC=CC=C1 UGVRJVHOJNYEHR-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- OPPHXULEHGYZRW-UHFFFAOYSA-N 4-methoxy-2,4-dimethyl-2-phenyldiazenylpentanenitrile Chemical compound COC(C)(C)CC(C)(C#N)N=NC1=CC=CC=C1 OPPHXULEHGYZRW-UHFFFAOYSA-N 0.000 description 1
- VGVHNLRUAMRIEW-UHFFFAOYSA-N 4-methylcyclohexan-1-one Chemical compound CC1CCC(=O)CC1 VGVHNLRUAMRIEW-UHFFFAOYSA-N 0.000 description 1
- IKVYHNPVKUNCJM-UHFFFAOYSA-N 4-propan-2-ylthioxanthen-9-one Chemical compound S1C2=CC=CC=C2C(=O)C2=C1C(C(C)C)=CC=C2 IKVYHNPVKUNCJM-UHFFFAOYSA-N 0.000 description 1
- VQVIHDPBMFABCQ-UHFFFAOYSA-N 5-(1,3-dioxo-2-benzofuran-5-carbonyl)-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(C(C=2C=C3C(=O)OC(=O)C3=CC=2)=O)=C1 VQVIHDPBMFABCQ-UHFFFAOYSA-N 0.000 description 1
- YWFPGFJLYRKYJZ-UHFFFAOYSA-N 9,9-bis(4-hydroxyphenyl)fluorene Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2C2=CC=CC=C21 YWFPGFJLYRKYJZ-UHFFFAOYSA-N 0.000 description 1
- BKQXUNGELBDWLS-UHFFFAOYSA-N 9,9-diphenylfluorene Chemical group C1=CC=CC=C1C1(C=2C=CC=CC=2)C2=CC=CC=C2C2=CC=CC=C21 BKQXUNGELBDWLS-UHFFFAOYSA-N 0.000 description 1
- RSWGJHLUYNHPMX-UHFFFAOYSA-N Abietic-Saeure Natural products C12CCC(C(C)C)=CC2=CCC2C1(C)CCCC2(C)C(O)=O RSWGJHLUYNHPMX-UHFFFAOYSA-N 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- VOWWYDCFAISREI-UHFFFAOYSA-N Bisphenol AP Chemical compound C=1C=C(O)C=CC=1C(C=1C=CC(O)=CC=1)(C)C1=CC=CC=C1 VOWWYDCFAISREI-UHFFFAOYSA-N 0.000 description 1
- HTVITOHKHWFJKO-UHFFFAOYSA-N Bisphenol B Chemical compound C=1C=C(O)C=CC=1C(C)(CC)C1=CC=C(O)C=C1 HTVITOHKHWFJKO-UHFFFAOYSA-N 0.000 description 1
- GIXXQTYGFOHYPT-UHFFFAOYSA-N Bisphenol P Chemical compound C=1C=C(C(C)(C)C=2C=CC(O)=CC=2)C=CC=1C(C)(C)C1=CC=C(O)C=C1 GIXXQTYGFOHYPT-UHFFFAOYSA-N 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- BLDKQZUPYCQRTI-UHFFFAOYSA-N C(C)[P]C(C1=C(C=C(C=C1C)C)C)=O Chemical compound C(C)[P]C(C1=C(C=C(C=C1C)C)C)=O BLDKQZUPYCQRTI-UHFFFAOYSA-N 0.000 description 1
- FPLWETRGFUTPIH-UHFFFAOYSA-N C1=CC=CC=2SC3=CC=CC=C3C(C12)=O.CC1=CC=2C(C3=CC=CC=C3SC2C(=C1)C)=O Chemical compound C1=CC=CC=2SC3=CC=CC=C3C(C12)=O.CC1=CC=2C(C3=CC=CC=C3SC2C(=C1)C)=O FPLWETRGFUTPIH-UHFFFAOYSA-N 0.000 description 1
- SXNICUVVDOTUPD-UHFFFAOYSA-N CC1=CC(C)=CC(C)=C1C(=O)P(=O)C1=CC=CC=C1 Chemical compound CC1=CC(C)=CC(C)=C1C(=O)P(=O)C1=CC=CC=C1 SXNICUVVDOTUPD-UHFFFAOYSA-N 0.000 description 1
- WOMMDHPMBLLCIT-UHFFFAOYSA-N CCCCCCOC(=O)C(=C)COCC(=C)C(=O)OCCCCCC Chemical compound CCCCCCOC(=O)C(=C)COCC(=C)C(=O)OCCCCCC WOMMDHPMBLLCIT-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- YIVJZNGAASQVEM-UHFFFAOYSA-N Lauroyl peroxide Chemical compound CCCCCCCCCCCC(=O)OOC(=O)CCCCCCCCCCC YIVJZNGAASQVEM-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- BWPYBAJTDILQPY-UHFFFAOYSA-N Methoxyphenone Chemical compound C1=C(C)C(OC)=CC=C1C(=O)C1=CC=CC(C)=C1 BWPYBAJTDILQPY-UHFFFAOYSA-N 0.000 description 1
- NQSMEZJWJJVYOI-UHFFFAOYSA-N Methyl 2-benzoylbenzoate Chemical compound COC(=O)C1=CC=CC=C1C(=O)C1=CC=CC=C1 NQSMEZJWJJVYOI-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 239000004820 Pressure-sensitive adhesive Substances 0.000 description 1
- KHPCPRHQVVSZAH-HUOMCSJISA-N Rosin Natural products O(C/C=C/c1ccccc1)[C@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 KHPCPRHQVVSZAH-HUOMCSJISA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- SLINHMUFWFWBMU-UHFFFAOYSA-N Triisopropanolamine Chemical compound CC(O)CN(CC(C)O)CC(C)O SLINHMUFWFWBMU-UHFFFAOYSA-N 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- AFCITVNGOYKNAF-UHFFFAOYSA-N [4-(4-methylbenzoyl)-2,3-diphenylphenyl]-phenylmethanone Chemical class C1=CC(C)=CC=C1C(=O)C(C(=C1C=2C=CC=CC=2)C=2C=CC=CC=2)=CC=C1C(=O)C1=CC=CC=C1 AFCITVNGOYKNAF-UHFFFAOYSA-N 0.000 description 1
- GUCYFKSBFREPBC-UHFFFAOYSA-N [phenyl-(2,4,6-trimethylbenzoyl)phosphoryl]-(2,4,6-trimethylphenyl)methanone Chemical compound CC1=CC(C)=CC(C)=C1C(=O)P(=O)(C=1C=CC=CC=1)C(=O)C1=C(C)C=C(C)C=C1C GUCYFKSBFREPBC-UHFFFAOYSA-N 0.000 description 1
- KYIKRXIYLAGAKQ-UHFFFAOYSA-N abcn Chemical compound C1CCCCC1(C#N)N=NC1(C#N)CCCCC1 KYIKRXIYLAGAKQ-UHFFFAOYSA-N 0.000 description 1
- VEBCLRKUSAGCDF-UHFFFAOYSA-N ac1mi23b Chemical compound C1C2C3C(COC(=O)C=C)CCC3C1C(COC(=O)C=C)C2 VEBCLRKUSAGCDF-UHFFFAOYSA-N 0.000 description 1
- 229920006243 acrylic copolymer Polymers 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 229920000180 alkyd Polymers 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- LLEMOWNGBBNAJR-UHFFFAOYSA-N biphenyl-2-ol Chemical class OC1=CC=CC=C1C1=CC=CC=C1 LLEMOWNGBBNAJR-UHFFFAOYSA-N 0.000 description 1
- TUQQUUXMCKXGDI-UHFFFAOYSA-N bis(3-aminophenyl)methanone Chemical compound NC1=CC=CC(C(=O)C=2C=C(N)C=CC=2)=C1 TUQQUUXMCKXGDI-UHFFFAOYSA-N 0.000 description 1
- KWKBGEYSGDAPGY-UHFFFAOYSA-N bis(4-hydroxy-3-methylphenyl)methanone Chemical compound C1=C(O)C(C)=CC(C(=O)C=2C=C(C)C(O)=CC=2)=C1 KWKBGEYSGDAPGY-UHFFFAOYSA-N 0.000 description 1
- MQDJYUACMFCOFT-UHFFFAOYSA-N bis[2-(1-hydroxycyclohexyl)phenyl]methanone Chemical compound C=1C=CC=C(C(=O)C=2C(=CC=CC=2)C2(O)CCCCC2)C=1C1(O)CCCCC1 MQDJYUACMFCOFT-UHFFFAOYSA-N 0.000 description 1
- QDVNNDYBCWZVTI-UHFFFAOYSA-N bis[4-(ethylamino)phenyl]methanone Chemical compound C1=CC(NCC)=CC=C1C(=O)C1=CC=C(NCC)C=C1 QDVNNDYBCWZVTI-UHFFFAOYSA-N 0.000 description 1
- WKDNYTOXBCRNPV-UHFFFAOYSA-N bpda Chemical compound C1=C2C(=O)OC(=O)C2=CC(C=2C=C3C(=O)OC(C3=CC=2)=O)=C1 WKDNYTOXBCRNPV-UHFFFAOYSA-N 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- NHBHTFOUKROSBB-UHFFFAOYSA-N butyl 2-(2-butoxycarbonylprop-2-enoxymethyl)prop-2-enoate Chemical compound CCCCOC(=O)C(=C)COCC(=C)C(=O)OCCCC NHBHTFOUKROSBB-UHFFFAOYSA-N 0.000 description 1
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- SPTHWAJJMLCAQF-UHFFFAOYSA-M ctk4f8481 Chemical compound [O-]O.CC(C)C1=CC=CC=C1C(C)C SPTHWAJJMLCAQF-UHFFFAOYSA-M 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- KOMDZQSPRDYARS-UHFFFAOYSA-N cyclopenta-1,3-diene titanium Chemical compound [Ti].C1C=CC=C1.C1C=CC=C1 KOMDZQSPRDYARS-UHFFFAOYSA-N 0.000 description 1
- FOTKYAAJKYLFFN-UHFFFAOYSA-N decane-1,10-diol Chemical compound OCCCCCCCCCCO FOTKYAAJKYLFFN-UHFFFAOYSA-N 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- 239000012933 diacyl peroxide Substances 0.000 description 1
- 238000007607 die coating method Methods 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- POSWICCRDBKBMH-UHFFFAOYSA-N dihydroisophorone Natural products CC1CC(=O)CC(C)(C)C1 POSWICCRDBKBMH-UHFFFAOYSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- VFHVQBAGLAREND-UHFFFAOYSA-N diphenylphosphoryl-(2,4,6-trimethylphenyl)methanone Chemical compound CC1=CC(C)=CC(C)=C1C(=O)P(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 VFHVQBAGLAREND-UHFFFAOYSA-N 0.000 description 1
- 239000003759 ester based solvent Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- VXYKEAZMHQVHRY-UHFFFAOYSA-N ethyl 2-(2-ethoxycarbonylprop-2-enoxymethyl)prop-2-enoate Chemical compound CCOC(=O)C(=C)COCC(=C)C(=O)OCC VXYKEAZMHQVHRY-UHFFFAOYSA-N 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- RMBPEFMHABBEKP-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2C3=C[CH]C=CC3=CC2=C1 RMBPEFMHABBEKP-UHFFFAOYSA-N 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000011086 glassine Substances 0.000 description 1
- 238000007756 gravure coating Methods 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 239000012775 heat-sealing material Substances 0.000 description 1
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 238000007602 hot air drying Methods 0.000 description 1
- 150000002432 hydroperoxides Chemical class 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 239000005453 ketone based solvent Substances 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 229940018564 m-phenylenediamine Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- VOTNBIJNPNEGJA-UHFFFAOYSA-N methyl 2-(2-methoxycarbonylprop-2-enoxymethyl)prop-2-enoate Chemical compound COC(=O)C(=C)COCC(=C)C(=O)OC VOTNBIJNPNEGJA-UHFFFAOYSA-N 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- DFFZOPXDTCDZDP-UHFFFAOYSA-N naphthalene-1,5-dicarboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1C(O)=O DFFZOPXDTCDZDP-UHFFFAOYSA-N 0.000 description 1
- RXOHFPCZGPKIRD-UHFFFAOYSA-N naphthalene-2,6-dicarboxylic acid Chemical compound C1=C(C(O)=O)C=CC2=CC(C(=O)O)=CC=C21 RXOHFPCZGPKIRD-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N o-biphenylenemethane Natural products C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 125000000963 oxybis(methylene) group Chemical group [H]C([H])(*)OC([H])([H])* 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- XNLICIUVMPYHGG-UHFFFAOYSA-N pentan-2-one Chemical compound CCCC(C)=O XNLICIUVMPYHGG-UHFFFAOYSA-N 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- HPAFOABSQZMTHE-UHFFFAOYSA-N phenyl-(2,4,6-trimethylphenyl)methanone Chemical compound CC1=CC(C)=CC(C)=C1C(=O)C1=CC=CC=C1 HPAFOABSQZMTHE-UHFFFAOYSA-N 0.000 description 1
- LYXOWKPVTCPORE-UHFFFAOYSA-N phenyl-(4-phenylphenyl)methanone Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1C(=O)C1=CC=CC=C1 LYXOWKPVTCPORE-UHFFFAOYSA-N 0.000 description 1
- 229920003207 poly(ethylene-2,6-naphthalate) Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920001707 polybutylene terephthalate Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000011112 polyethylene naphthalate Substances 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920001289 polyvinyl ether Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 229920005573 silicon-containing polymer Polymers 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229940014800 succinic anhydride Drugs 0.000 description 1
- 238000010345 tape casting Methods 0.000 description 1
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 1
- 150000000000 tetracarboxylic acids Chemical class 0.000 description 1
- 230000000930 thermomechanical effect Effects 0.000 description 1
- YRHRIQCWCFGUEQ-UHFFFAOYSA-N thioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC=CC=C3SC2=C1 YRHRIQCWCFGUEQ-UHFFFAOYSA-N 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- KHPCPRHQVVSZAH-UHFFFAOYSA-N trans-cinnamyl beta-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OCC=CC1=CC=CC=C1 KHPCPRHQVVSZAH-UHFFFAOYSA-N 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/20—Adhesives in the form of films or foils characterised by their carriers
- C09J7/22—Plastics; Metallised plastics
- C09J7/25—Plastics; Metallised plastics based on macromolecular compounds obtained otherwise than by reactions involving only carbon-to-carbon unsaturated bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/02—Physical, chemical or physicochemical properties
- B32B7/027—Thermal properties
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2479/00—Presence of polyamine or polyimide
- C09J2479/08—Presence of polyamine or polyimide polyimide
- C09J2479/086—Presence of polyamine or polyimide polyimide in the substrate
Definitions
- the present invention relates to a laminate including a cured resin layer and an adhesive layer.
- organic insulating materials have come to be used frequently as an insulating layer or the like between them.
- organic insulating materials contribute to weight reduction and flexibility as compared with inorganic insulating materials, but are inferior in heat resistance.
- the element or the like inside the electronic device is affected by the exposure, and the performance of the device is deteriorated due to the occurrence of a short circuit or the like, or the life of the device is shortened. Further, when the underlying layer has unevenness, flattening the uneven surface has not been sufficient.
- an organic insulating layer is formed as an organic insulating material such as acrylic resin or the like. It is disclosed that the polyimide resin is formed by a spin coating method or the like.
- the unevenness of the inorganic insulating layer can be embedded with the liquid organic insulating material, the unevenness is reflected on the surface of the obtained organic insulating layer opposite to the inorganic insulating layer side on the pixel portion. As a result, a short circuit or disconnection may occur between the wirings of the display element, the touch panel, or the like.
- the loss tangent has a peak in a specific temperature range, and after heating at a specific temperature, the loss tangent peak is shifted to the specific temperature range.
- the above problem is solved by forming a laminate including a cured resin layer made of a cured product of a curable resin composition containing a polymer component (A) and a curable monomer (B), and an adhesive layer.
- the present invention was completed after discovering that the problem could be solved. That is, the present invention provides the following [1] to [5].
- a laminate comprising a cured resin layer and an adhesive layer, wherein the cured resin layer is a cured resin composition containing a polymer component (A) and a curable monomer (B) It has a loss tangent peak in the temperature range of 100 to 200 ° C. in dynamic viscoelasticity measurement measured at a temperature increase rate of 3 ° C./min from 25 ° C. After heating at 180 ° C. for 1 hour, A laminate having a loss tangent peak at 220° C. or higher.
- a laminate including a cured resin layer and an adhesive layer, which excels in flattening uneven surfaces and suppresses short circuits and disconnections.
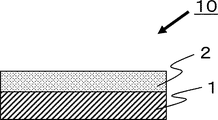
- BRIEF DESCRIPTION OF THE DRAWINGS It is a cross-sectional schematic diagram which shows the basic composition of the laminated body of this invention.
- BRIEF DESCRIPTION OF THE DRAWINGS It is a cross-sectional schematic diagram which shows 1st Embodiment of the laminated body of this invention. It is a cross-sectional schematic diagram which shows 2nd Embodiment of the laminated body of this invention.
- the definition of being preferred can be arbitrarily selected, and it can be said that a combination of the definitions of being preferred is more preferred.
- the description "XX to YY” means “XX or more and YY or less”.
- the lower and upper limits described stepwise can be independently combined. For example, from the statement “preferably 10 to 90, more preferably 30 to 60", combining "preferred lower limit (10)” and “more preferred upper limit (60)” to "10 to 60” can also In this specification, for example, "(meth)acrylic acid” indicates both “acrylic acid” and “methacrylic acid”, and the same applies to other similar terms.
- the laminate of the present invention is a laminate comprising a cured resin layer and an adhesive layer, wherein the curable resin layer comprises a curable resin containing a polymer component (A) and a curable monomer (B). It is a layer made of a cured product of a resin composition, and has a loss tangent peak in the temperature range of 100 ° C. to 200 ° C. in dynamic viscoelasticity measurement measured from 25 ° C. at a heating rate of 3 ° C./min. It is characterized by having a loss tangent peak at 220°C or higher after heating at 1 hour.
- the cured resin layer used in the present invention has a loss tangent peak at each temperature condition specified above, so that it melts at a temperature of, for example, over 180 ° C., and is used for wiring of electronic devices and other layers and other coatings.
- the uneven surface of a base material such as a body (hereinafter sometimes simply referred to as "uneven substrate") is embedded and then heated at 180° C. for 1 hour to complete the curing reaction.
- the uneven shape does not appear on the surface of the adhesive layer, and the surface of the adhesive layer opposite to the uneven surface side can be flattened.
- the laminate of the present invention has adhesiveness, the surface of the adhesive layer opposite to the uneven surface can be easily adhered (laminated) to another adherend, layer, or the like.
- the uneven shape, depth, width, etc. of the "uneven substrate” are not particularly limited as long as the uneven shape can be embedded.
- the material of the uneven substrate is not particularly limited as long as it can maintain the adhesiveness with the resin.
- the laminate of the present invention comprises a cured resin layer and an adhesive layer.
- FIG. 1 is a schematic cross-sectional view showing the basic structure of the laminate of the present invention.
- a laminate 10 is obtained by laminating an adhesive layer 2 on a cured resin layer 1 .
- FIG. 2 is a schematic cross-sectional view showing a first embodiment of the laminate of the present invention.
- the laminate 20 is obtained by laminating the process film 13 on the surface of the cured resin layer 11 opposite to the adhesive layer 12 side, for example.
- FIG. 3 is a schematic cross-sectional view showing a second embodiment of the laminate of the present invention.
- the laminated body 30 is obtained by laminating the concave-convex substrate 23 on the surface of the cured resin layer 21 opposite to the adhesive layer 22 side, for example.
- the cured resin layer used in the present invention is a layer made of a cured product of a curable resin composition containing a polymer component (A) and a curable monomer (B).
- the cured resin layer has a loss tangent peak in the temperature range of 100 to 200 ° C. in dynamic viscoelasticity measurement measured at a temperature increase rate of 3 ° C./min from 25 ° C. After heating at 180 ° C. for 1 hour, the loss tangent has a peak at 220° C. or higher.
- the dynamic viscoelasticity measurement of the cured resin layer measured from 25° C. at a heating rate of 3° C./min, there may be a plurality of loss tangent peaks in the temperature range of 100 to 200° C. If there are a plurality of loss tangent peaks, at least one loss tangent peak should be in the temperature range of 100 to 200.degree.
- the loss tangent has a peak only at less than 100° C., for example, the cured resin layer melts and adheres to the uneven substrate when heated at 190° C., but the loss tangent peak position does not change even after heating at 180° C. for 1 hour.
- the loss tangent peak remains below 100° C., making it unsuitable for processes involving heat treatments at higher temperatures, for example.
- it has a loss tangent peak above 200°C, the cured resin layer will not melt even when heated to 190°C, for example, and cannot be adhered to an adherend or the like.
- the peak loss tangent is preferably 120 to 200°C, more preferably 140 to 200°C, still more preferably 160 to 200°C. When the peak of the loss tangent falls within this range, the cured resin layer melts and is easily adhered to the uneven substrate.
- the loss tangent peak after heating at 180° C. for 1 hour exists only below 220° C., for example, it is difficult to obtain sufficient heat resistance in a process involving heat treatment at a higher temperature.
- the peak of the loss tangent after heating at 180°C for 1 hour exists only at 220°C or higher and does not exist below 220°C.
- the loss tangent peak after heating at 180° C. for 1 hour is preferably 240° C. or higher, more preferably 250° C. or higher, and still more preferably 260° C. or higher.
- the peak of loss tangent after heating at 180° C. for 1 hour is within this range, for example, high heat resistance is exhibited in a process involving heat treatment at a higher temperature.
- the cured resin layer has a loss tangent peak at 200° C. or less, for example, it becomes a rubber state during high-temperature heat treatment (for example, 190° C. for 10 minutes), and easily melts and adheres to the uneven substrate, resulting in unevenness. can be embedded. Further, after melt bonding to the uneven substrate, for example, by heating at 180 ° C. for 1 hour, the curing reaction of the curable monomer (B) proceeds and the loss tangent peak before curing disappears. Post exhibits excellent heat resistance. This provides high heat resistance in processes involving heat treatments at higher temperatures. Furthermore, by providing an adhesive layer on the surface of the cured resin layer opposite to the uneven surface side, the uneven shape does not appear on the surface of the adhesive layer, and the adhesive layer is formed on the opposite side to the uneven surface side. can be flattened.
- the cured resin layer has excellent solvent resistance. Because of its excellent solvent resistance, for example, even when an organic solvent is used to form an adhesive layer on the cured resin layer, the surface of the cured resin layer is hardly dissolved. In this case, since the components of the cured resin layer are less likely to mix into the adhesive layer, the adhesiveness is less likely to deteriorate.
- the gel fraction of the cured resin layer is preferably 80% or higher, more preferably 85% or higher, even more preferably 87% or higher, and particularly preferably 90% or higher.
- a cured resin layer having a gel fraction of 80% or more has excellent solvent resistance, so even when an organic solvent is used, the surface of the cured resin layer hardly dissolves, resulting in a laminate with excellent solvent resistance. can be easily obtained.
- the gel fraction is within this range, a laminate having an adhesive layer formed by direct coating can be obtained.
- the adhesive layer is formed from a coating film, the cured resin layer is not affected by the intrusion of the organic solvent contained in the adhesive layer and deformed, and the adhesive layer has sufficient inherent adhesiveness. can be expressed.
- a coating film is a film obtained by coating a coating material on a base material or an object and, if necessary, performing a treatment such as drying or curing by heating.
- the gel fraction is obtained by, for example, performing the following operations (a), (b), and (c), and measuring the weight of the dried structure before immersion in MEK (methyl ethyl ketone) solvent. calculated by dividing by the weight of (a)
- the cured resin layer was wrapped with a mesh ( ⁇ _UX SCREEN 150-035/380TW manufactured by NBC Meshtec) and stapled to form a structure, and the weight of the structure was measured.
- MEK Methyl ethyl ketone
- the cured resin layer is preferably colorless and transparent. Since the cured resin layer is colorless and transparent, the laminate having the adhesive layer of the present invention can be preferably used for optical applications.
- the cured resin layer has a low birefringence and excellent optical isotropy.
- the in-plane retardation of the cured resin layer is usually 2.0 nm or less, preferably 1.0 nm or less.
- the retardation in the thickness direction is usually ⁇ 500 nm or less, preferably ⁇ 450 nm or less.
- the value (birefringence) obtained by dividing the in-plane retardation by the thickness of the cured resin layer is usually 100 ⁇ 10 ⁇ 5 or less, preferably 20 ⁇ 10 ⁇ 5 or less.
- a laminate having a low birefringence and excellent optical isotropy can be obtained.
- a laminate having an agent layer can be preferably used for optical applications.
- the breaking elongation of the cured resin layer is preferably 2.5% or more, more preferably 3.0% or more, and still more preferably 3.5% or more.
- the breaking elongation of the cured resin layer is 2.5% or more, it becomes easy to adjust the breaking elongation of the laminate to about 2% or more, and as a result, a laminate having excellent flexibility can be easily obtained.
- the thickness of the cured resin layer is appropriately adjusted and determined according to the shape and dimensions of the uneven surface of the uneven substrate.
- the thickness of the cured resin layer is usually 0.1 to 500 ⁇ m, preferably 0.1 to 100 ⁇ m, more preferably 0.1 to 20 ⁇ m, still more preferably 0.1 to 15 ⁇ m, particularly preferably 0.2 to 0.2 ⁇ m. 10 ⁇ m.
- a thermoplastic resin is preferable, and an amorphous thermoplastic resin is more preferable.
- an amorphous thermoplastic resin By using an amorphous thermoplastic resin, it becomes easy to obtain a cured resin layer excellent in optical isotropy, and it becomes easy to obtain a laminate excellent in transparency.
- amorphous thermoplastic resins are generally easily dissolved in organic solvents, the cured resin layer can be efficiently formed using a solution casting method, as will be described later.
- the amorphous thermoplastic resin means a thermoplastic resin whose melting point is not observed in differential scanning calorimetry.
- the Tg of the polymer component (A) is preferably 250°C or higher, more preferably 290°C or higher, still more preferably 320°C or higher.
- Tg is within this range, a laminate having an adhesive layer with sufficiently excellent heat resistance can be obtained.
- the adhesive layer is formed from a coating film, the cured resin layer is not affected by heating during coating of the coating film, causing deformation or peeling. It is possible to express sufficient adhesiveness possessed.
- a coating film is a film obtained by coating a coating material on a base material or an object and, if necessary, performing a treatment such as drying or curing by heating.
- the curable resin composition is applied to an object to be coated such as a process film, and curing treatment is performed by either or both of drying, heating, irradiation of active energy rays, etc. It is a coating obtained by performing
- Tg is the peak temperature of tan ⁇ (loss modulus/storage modulus) obtained by viscoelasticity measurement (measurement in tensile mode in the range of 0 to 250°C at a temperature increase rate of 3°C/min at a frequency of 10Hz).
- the weight average molecular weight (Mw) of the polymer component (A) is preferably 100,000 to 5,000,000, more preferably 150,000 to 4,000,000, still more preferably 200,000 to 350,000. is in the range of Also, the molecular weight distribution (Mw/Mn) is preferably in the range of 1.0 to 5.0, more preferably 1.2 to 3.0.
- a weight average molecular weight (Mw) and a molecular weight distribution (Mw/Mn) are polystyrene equivalent values measured by a gel permeation chromatography (GPC) method. By setting Mw to 100,000 or more, it becomes easy to increase the breaking elongation of the cured resin layer.
- the polymer component (A) is particularly preferably soluble in low-boiling general-purpose organic solvents such as benzene and methyl ethyl ketone (MEK). If it is soluble in a general-purpose organic solvent, it becomes easy to form a curable resin layer by coating.
- general-purpose organic solvents such as benzene and methyl ethyl ketone (MEK). If it is soluble in a general-purpose organic solvent, it becomes easy to form a curable resin layer by coating.
- a particularly preferable polymer component (A) is an amorphous thermoplastic resin having a Tg of 250°C or higher, which is soluble in low-boiling general-purpose organic solvents such as benzene and methyl ethyl ketone.
- the polymer component (A) is preferably a thermoplastic resin having a ring structure such as an aromatic ring structure or an alicyclic structure, more preferably a thermoplastic resin having an aromatic ring structure.
- polymer component (A) examples include polyimide resins and polyarylate resins preferably having a Tg of 250°C or higher. These resins generally have a high Tg and excellent heat resistance, and since they are amorphous thermoplastic resins, they can be formed into a coating film by a solution casting method. Among these, polyimide resins are preferred because they have a high Tg and excellent heat resistance, and are easy to obtain those that are soluble in general-purpose organic solvents while exhibiting good heat resistance.
- the polyimide resin is not particularly limited as long as it does not impair the effects of the present invention.
- aromatic polyimide resin aromatic (carboxylic acid component) - cycloaliphatic (diamine component) polyimide resin, cyclic Group (carboxylic acid component)-aromatic (diamine component) polyimide resins, cycloaliphatic polyimide resins, fluorinated aromatic polyimide resins, and the like can be used.
- a polyimide resin having a fluoro group in the molecule is particularly preferred.
- a polyimide resin obtained through polymerization to polyamic acid and chemical imidization reaction using an aromatic diamine compound and a tetracarboxylic dianhydride is preferred.
- the aromatic diamine compound is a polyimide that is soluble in a common solvent (for example, N,N-dimethylacetamide (DMAC)) and has a certain level of transparency by reacting with the tetracarboxylic dianhydride that is used together.
- a common solvent for example, N,N-dimethylacetamide (DMAC)
- Any aromatic diamine compound can be used as long as it provides the aromatic diamine compound.
- aromatic diamine compounds may be used alone, or two or more aromatic diamine compounds may be used.
- preferable aromatic diamine compounds include 2,2-bis(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropane, 2,2 -bis(3-aminophenyl)-1,1,1,3,3,3-hexafluoropropane, 2-(3-aminophenyl)-2-(4-aminophenyl)-1,1,1,3 ,3,3-hexafluoropropane, 2,2-bis[4-(3-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropane, 2,2-bis[4- (4-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropane, 2,2-bis[3-(3-aminophenoxy)phenyl]-1,1,1,3, 3,3-hexafluoropropane,
- tetracarboxylic dianhydride a tetracarboxylic acid that is soluble in a common solvent (eg, N,N-dimethylacetamide (DMAC)) and gives a polyimide having a predetermined transparency, like the aromatic diamine compound.
- a common solvent eg, N,N-dimethylacetamide (DMAC)
- Any dianhydride can be used, and specifically, 4,4′-(1,1,1,3,3,3-hexafluoropropane-2,2-diyl)diphthalic acid di anhydride, pyromellitic dianhydride, 3,3',4,4'-benzophenonetetracarboxylic dianhydride, 1,4-hydroquinonedibenzoate-3,3',4,4'-tetracarboxylic acid Examples include acid dianhydride, 3,3′,4,4′-biphenyltetracarboxylic dianhydride, and 3,3′,4,4′-diphenylethertetracarboxylic dianhydride.
- tetracarboxylic dianhydrides may be used alone, or two or more kinds of tetracarboxylic dianhydrides may be used.
- 4,4′-(1,1,1,3,3,3-hexafluoropropane-2,2-diyl)diphthalic dianhydride and the like from the viewpoint of transparency, heat resistance and solubility in solvents preferably a tetracarboxylic dianhydride having at least one fluoro group.
- Polymerization into polyamic acid can be carried out by reacting the above aromatic diamine compound and tetracarboxylic dianhydride while dissolving in a solvent in which the resulting polyamic acid is soluble.
- Solvents used for polymerization to polyamic acid include solvents such as N,N-dimethylacetamide, N,N-dimethylformamide, N-methyl-2-pyrrolidone, 1,3-dimethyl-2-imidazolidinone, and dimethylsulfoxide. can be used.
- the polymerization reaction to polyamic acid is preferably carried out while stirring in a reaction vessel equipped with a stirring device.
- a method of dissolving a predetermined amount of an aromatic diamine compound in the above solvent, adding tetracarboxylic dianhydride while stirring to react, and obtaining polyamic acid, dissolving the tetracarboxylic dianhydride in the solvent is mentioned.
- the temperature of the polymerization reaction to polyamic acid is not particularly limited, but it is preferably carried out at a temperature of 0 to 70°C, more preferably 10 to 60°C, and even more preferably 20 to 50°C. By performing the polymerization reaction within the above range, it is possible to obtain a high-molecular-weight polyamic acid with little coloration and excellent transparency.
- the aromatic diamine compound and the tetracarboxylic dianhydride used for polymerization to polyamic acid are generally used in equimolar amounts. It is also possible to change the molar amount of /aromatic diamine compound (molar ratio) within the range of 0.95 to 1.05.
- the molar ratio of the tetracarboxylic dianhydride and the aromatic diamine compound is preferably in the range of 1.001-1.02, more preferably 1.001-1.01.
- the degree of polymerization of the resulting polyamic acid can be stabilized, and the unit derived from the tetracarboxylic dianhydride can be It can be placed at the end of the polymer, and as a result, it is possible to obtain a polyimide with little coloration and excellent transparency.
- the concentration of the polyamic acid solution to be produced is preferably adjusted to an appropriate concentration (for example, about 10 to 30% by mass) so that the viscosity of the solution can be properly maintained and handling in the subsequent steps can be facilitated.
- An imidizing agent is added to the resulting polyamic acid solution to carry out a chemical imidization reaction.
- the imidizing agent carboxylic acid anhydrides such as acetic anhydride, propionic anhydride, succinic anhydride, phthalic anhydride, and benzoic anhydride can be used. Preference is given to using acetic acid.
- the equivalent of the imidizing agent to be used is at least the equivalent of the amide bond of the polyamic acid that performs the chemical imidization reaction, preferably 1.1 to 5 times the equivalent of the amide bond, and 1.5 to 4 times. is more preferable. By using the imidizing agent in a slightly excess amount with respect to the amide bond in this way, the imidization reaction can be efficiently carried out even at a relatively low temperature.
- aliphatic, aromatic or heterocyclic tertiary amines such as pyridine, picoline, quinoline, isoquinoline, trimethylamine and triethylamine can be used as imidization accelerators.
- imidization accelerators such as pyridine, picoline, quinoline, isoquinoline, trimethylamine and triethylamine.
- the chemical imidization reaction temperature it is preferably carried out at 10°C or higher and lower than 50°C, more preferably at 15°C or higher and lower than 45°C.
- a poor solvent for polyimide is added to the polyimide solution obtained by the chemical imidization reaction to precipitate polyimide to form powder, which is powderization and drying.
- the polyimide resin it is preferable that it is compatible with low boiling point organic solvents such as benzene and methyl ethyl ketone. In particular, it is preferably soluble in methyl ethyl ketone. When it is soluble in methyl ethyl ketone, a layer of the curable resin composition can be easily formed by coating and drying.
- a polyimide resin containing a fluoro group is particularly preferable from the viewpoint that it is easily dissolved in a general-purpose organic solvent having a low boiling point such as methyl ethyl ketone and that a curable resin layer is easily formed by a coating method.
- the polyimide resin having a fluoro group is preferably an aromatic polyimide resin having a fluoro group in the molecule, and preferably has a skeleton represented by the following chemical formula in the molecule.
- a polyimide resin having a skeleton represented by the above chemical formula has an extremely high Tg exceeding 300°C due to the high rigidity of the skeleton. Therefore, the heat resistance of the cured resin layer can be greatly improved.
- the skeleton is linear and has relatively high flexibility, which facilitates increasing the breaking elongation of the cured resin layer.
- the polyimide resin having the above skeleton can dissolve in general-purpose low-boiling organic solvents such as methyl ethyl ketone due to the presence of fluoro groups. Therefore, the coating can be performed using a solution casting method to form a curable resin layer as a coating film, and the solvent can be easily removed by drying.
- the polyimide resin having a skeleton represented by the above chemical formula includes 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl and 4,4′-(1,1,1,3,3,3 -Hexafluoropropane-2,2-diyl)diphthalic dianhydride can be obtained by polymerization and imidization reaction of the polyamic acid described above.
- a polyarylate resin is a resin made of a polymer compound obtained by reacting an aromatic diol with an aromatic dicarboxylic acid or its chloride.
- Polyarylate resins also have relatively high Tg and relatively good elongation properties.
- the polyarylate resin is not particularly limited, and known ones can be used.
- aromatic diols include bis(4-hydroxyphenyl)methane [bisphenol F], bis(3-methyl-4-hydroxyphenyl)methane, 1,1-bis(4′-hydroxyphenyl)ethane, 1, 1-bis(3'-methyl-4'-hydroxyphenyl)ethane, 2,2-bis(4'-hydroxyphenyl)propane [bisphenol A], 2,2-bis(3'-methyl-4'-hydroxy bis(hydroxyphenyl)alkanes such as phenyl)propane, 2,2-bis(4'-hydroxyphenyl)butane, 2,2-bis(4'-hydroxyphenyl)octane; 1,1-bis(4'- bis(hydroxyphenyl)cyclopentane, 1,1-bis(4'-hydroxyphenyl)cyclohexane [bisphenol Z], 1,1-bis(4'-hydroxyphenyl)-3,3,5-trimethylcyclohexane, etc.
- bisphenol F bis(4-hydroxyphenyl)methan
- phenyl)cycloalkanes bis(4-hydroxyphenyl)phenylmethane, bis(3-methyl-4-hydroxyphenyl)phenylmethane, bis(2,6-dimethyl-4-hydroxyphenyl)phenylmethane, bis(2, 3,6-trimethyl-4-hydroxyphenyl)phenylmethane, bis(3-t-butyl-4-hydroxyphenyl)phenylmethane, bis(3-phenyl-4-hydroxyphenyl)phenylmethane, bis(3-fluoro- 4-hydroxyphenyl)phenylmethane, bis(3-bromo-4-hydroxyphenyl)phenylmethane, bis(4-hydroxyphenyl)-4-fluorophenylmethane, bis(3-fluoro-4-hydroxyphenyl)-4- Fluorophenylmethane, bis(4-hydroxyphenyl)-4-chlorophenylmethane, bis(4-hydroxyphenyl)-4
- aromatic dicarboxylic acids or chlorides thereof include phthalic acid, isophthalic acid, terephthalic acid, 4,4′-biphenyldicarboxylic acid, diphenoxyethanedicarboxylic acid, diphenyl ether 4,4′-dicarboxylic acid, 4,4′- diphenylsulfonedicarboxylic acid, 1,5-naphthalenedicarboxylic acid, 2,6-naphthalenedicarboxylic acid, chlorides thereof, and the like.
- the polyarylate-based resin to be used may be a modified polyarylate-based resin.
- the polyarylate-based resin is preferably a resin composed of a polymer compound obtained by reacting 2,2-bis(4'-hydroxyphenyl)propane with isophthalic acid.
- the polymer component (A) can be used singly or in combination of two or more. Also, the polymer component (A) and the polymer component (A') having a glass transition temperature of less than 250°C may be used in combination. Examples of the polymer component (A′) include polyamide resins and polyarylate resins having a Tg of less than 250° C. Polyamide resins are preferred.
- the polyamide resin one that is soluble in an organic solvent is preferable, and a rubber-modified polyamide resin is preferable.
- a rubber-modified polyamide resin for example, those described in JP-A-2004-035638 can be used.
- polymer component (A) and the polymer component (A') one using a single type of polyimide resin, one using a plurality of different types of polyimide resin, and polyimide resin with polyamide resin and polyarylate resin It is preferable to add at least one of them from the viewpoint of adjusting the elongation characteristics and the viewpoint of solvent resistance.
- the amount of the resin to be added is 100 parts by mass of the polyimide resin from the viewpoint of imparting moderate flexibility while maintaining a high Tg. , preferably 100 parts by mass or less, more preferably 70 parts by mass or less, even more preferably 50 parts by mass or less, still more preferably 30 parts by mass or less, and preferably 1 part by mass or more, more preferably It is 3 parts by mass or more.
- a curable monomer (B) is a monomer having a polymerizable unsaturated bond, and is a monomer that can participate in a polymerization reaction or a polymerization reaction and a cross-linking reaction.
- curing means a broad concept including this "polymerization reaction of monomers” or “polymerization reaction of monomers and subsequent cross-linking reaction of polymers”.
- the cured resin layer By making the cured resin layer a layer made of a cured product of a curable resin composition containing the above-described polymer component (A) and the above-described curable monomer (B), excellent heat resistance and thin curing can be obtained. It becomes easy to form a resin layer. In addition, the use of such a material eliminates optical problems caused by materials having anisotropic molecular orientation, such as polyester films generally used as base materials for laminates. When the Tg of the polymer component is 250° C. or higher, the cured resin layer has heat resistance, and when the adhesive layer is formed from the coating film, the cured resin layer is damaged by heat during coating of the coating film. Deterioration of adhesive performance due to deformation or the like is suppressed.
- the molecular weight of the curable monomer (B) is generally 3,000 or less, preferably 150-2,000, more preferably 150-1,000.
- the number of polymerizable unsaturated bonds in the curable monomer (B) is not particularly limited.
- the curable monomer (B) is a monofunctional monomer having one polymerizable unsaturated bond, or a multifunctional monomer such as a bifunctional or trifunctional monomer having a plurality of polymerizable unsaturated bonds. There may be.
- Examples of the monofunctional monomers include monofunctional (meth)acrylic acid derivatives.
- the monofunctional (meth)acrylic acid derivative is not particularly limited, and known compounds can be used. Examples include monofunctional (meth)acrylic acid derivatives having a nitrogen atom, monofunctional (meth)acrylic acid derivatives having an alicyclic structure, and monofunctional (meth)acrylic acid derivatives having a polyether structure. .
- Examples of monofunctional (meth)acrylic acid derivatives having a nitrogen atom include compounds represented by the following formula.
- R 1 represents a hydrogen atom or an alkyl group having 1 to 6 carbon atoms
- R 2 and R 3 each independently represent a hydrogen atom or an organic group having 1 to 12 carbon atoms
- R 2 and R 3 may combine to form a ring structure
- R 4 represents a divalent organic group.
- the alkyl group having 1 to 6 carbon atoms represented by R 1 include a methyl group, an ethyl group, a propyl group and the like, and a methyl group is preferred.
- Examples of organic groups having 1 to 12 carbon atoms represented by R 2 and R 3 include alkyl groups having 1 to 12 carbon atoms such as methyl group, ethyl group and propyl group; cycloalkyl groups having 3 to 12 carbon atoms; aromatic groups having 6 to 12 carbon atoms such as phenyl, biphenyl and naphthyl groups; These groups may have a substituent at any position. Also, R 2 and R 3 may combine to form a ring, and the ring may further have a nitrogen atom or an oxygen atom in its skeleton. Divalent organic groups represented by R 4 include groups represented by -(CH 2 ) m - and -NH-(CH 2 ) m -. Here, m is an integer of 1-10.
- the (meth)acryloylmorpholine represented by the following formula is preferred as the monofunctional (meth)acrylic acid derivative having a nitrogen atom.
- a cured resin layer with more excellent heat resistance can be formed.
- Examples of monofunctional (meth)acrylic acid derivatives having an alicyclic structure include compounds represented by the following formulas.
- R 1 has the same meaning as above, and R 5 is a group having an alicyclic structure.
- the group having an alicyclic structure represented by R 5 includes cyclohexyl group, isobornyl group, 1-adamantyl group, 2-adamantyl group, tricyclodecanyl group and the like.
- monofunctional (meth)acrylic acid derivatives having an alicyclic structure include isobornyl (meth)acrylate, cyclohexyl (meth)acrylate, 1-adamantyl (meth)acrylate, 2-adamantyl (meth)acrylate and the like. mentioned.
- a cured resin layer with more excellent optical properties can be formed.
- the monofunctional (meth)acrylic acid derivative having a polyether structure includes compounds represented by the following formula.
- R 1 has the same meaning as above, and R 6 represents an organic group having 1 to 12 carbon atoms.
- the organic group having 1 to 12 carbon atoms represented by R 6 include alkyl groups having 1 to 12 carbon atoms such as methyl group, ethyl group and propyl group; A cycloalkyl group; an aromatic group having 6 to 12 carbon atoms such as a phenyl group, a biphenyl group and a naphthyl group; j represents an integer from 2 to 20;
- monofunctional (meth)acrylic acid derivatives having a polyether structure include ethoxylated o-phenylphenol (meth)acrylate, methoxypolyethyleneglycol (meth)acrylate, phenoxypolyethyleneglycol (meth)acrylate, and the like. .
- a cured resin layer with excellent toughness can be formed.
- polyfunctional monomers examples include polyfunctional (meth)acrylic acid derivatives.
- the polyfunctional (meth)acrylic acid derivative is not particularly limited, and known compounds can be used. Examples thereof include di- to hexa-functional (meth)acrylic acid derivatives.
- Bifunctional (meth)acrylic acid derivatives include compounds represented by the following formulas.
- R 1 has the same meaning as above, and R 7 represents a divalent organic group.
- R 7 represents a divalent organic group. Examples of the divalent organic group represented by R 7 include groups represented by the following formulas.
- bifunctional (meth)acrylic acid derivative represented by the above formula examples include tricyclodecanedimethanol di(meth)acrylate, polyethylene glycol di(meth)acrylate, propoxylated ethoxylated bisphenol A di(meth)acrylate. , ethoxylated bisphenol A di(meth)acrylate, 1,10-decanediol di(meth)acrylate, 1,6-hexanediol di(meth)acrylate, 9,9-bis[4-(2-acryloyloxyethoxy) phenyl]fluorene and the like.
- the divalent organic group represented by R7 has a tricyclodecane skeleton, propoxy
- the divalent organic group represented by R7 has a bisphenol skeleton, 9,9-bis [4-(2-Acryloyloxyethoxy)phenyl]fluorene, etc., in which the divalent organic group represented by R 7 in the above formula preferably has a 9,9-bisphenylfluorene skeleton.
- bifunctional (meth)acrylic acid derivatives other than these include neopentyl glycol adipate di(meth)acrylate, neopentylglycol hydroxypivalate di(meth)acrylate, caprolactone-modified dicyclopentenyl di(meth)acrylate, Ethylene oxide-modified phosphoric acid di(meth)acrylate, di(acryloxyethyl)isocyanurate, allylated cyclohexyl di(meth)acrylate and the like can be mentioned.
- Trifunctional (meth)acrylic acid derivatives include trimethylolpropane tri(meth)acrylate, pentaerythritol tri(meth)acrylate, propionic acid-modified dipentaerythritol tri(meth)acrylate, propylene oxide-modified trimethylolpropane tri(meth)acrylate. ) acrylate, tris(acryloxyethyl) isocyanurate, and the like.
- Examples of tetrafunctional (meth)acrylic acid derivatives include pentaerythritol tetra(meth)acrylate.
- Pentafunctional (meth)acrylic acid derivatives include propionic acid-modified dipentaerythritol penta(meth)acrylate.
- Examples of hexafunctional (meth)acrylic acid derivatives include dipentaerythritol hexa(meth)acrylate and caprolactone-modified dipentaerythritol hexa(meth)acrylate.
- a cyclization-polymerizable monomer may be used as the curable monomer (B).
- a cyclization-polymerizable monomer is a monomer that has the property of being radically polymerized while being cyclized.
- Cyclopolymerizable monomers include non-conjugated dienes, for example, ⁇ -allyloxymethyl acrylic acid-based monomers can be used, 2-allyloxymethyl acrylic acid alkyl esters having 1 to 4 carbon atoms, Cyclohexyl 2-(allyloxymethyl)acrylate is preferred, C 1-4 alkyl esters of 2-allyloxymethylacrylic acid are more preferred, and methyl 2-(allyloxymethyl)acrylate is even more preferred.
- the curable monomer (B) can be used singly or in combination of two or more.
- the curable monomer (B) is preferably a polyfunctional monomer because a curable resin layer having excellent heat resistance and solvent resistance can be obtained.
- the polyfunctional monomer a bifunctional (meth)acrylic acid derivative is preferable from the viewpoints that it is easily mixed with the polymer component (A) and that curing shrinkage of the polymer hardly occurs and curling of the cured product can be suppressed.
- the curable monomer (B) contains a polyfunctional (meth)acrylate compound and a cyclopolymerizable monomer.
- the content is preferably 70% by mass or less, more preferably 50% by mass or less, of the total amount of the curable monomer (B). preferable.
- the curable resin composition used to form the curable resin layer contains the polymer component (A), the curable monomer (B), and if desired, the polymerization initiator and other components described later. It can be prepared by dissolving or dispersing in a suitable solvent.
- the total content of the polymer component (A) and the curable monomer (B) in the curable resin composition is preferably 40 to 99 with respect to the total mass of the curable resin composition excluding the solvent. .5% by mass, more preferably 60 to 99% by mass, still more preferably 80 to 98% by mass.
- the mass ratio of the polymer component (A): curable monomer (B) is in such a range, the flexibility of the resulting cured resin layer is more likely to be improved, and curing The solvent resistance of the resin layer tends to be easily maintained.
- the content of the curable monomer (B) in the curable resin composition is within the above range, for example, when the curable resin layer is obtained by a solution casting method or the like, the solvent can be removed efficiently. Therefore, the problem of deformation such as curling and waviness caused by a prolonged drying process can be solved.
- the polymer component (A) when using a combination of a plurality of resins with different solvent solubility, such as a combination of the polyimide resin described above and a polyamide resin or a polyarylate resin, first, the resin is added to a solvent suitable for each. After dissolving, it is preferable to add a solution in which another resin is dissolved to the low boiling point organic solvent in which the resin is dissolved.
- the curable resin composition can optionally contain a polymerization initiator.
- Any polymerization initiator can be used without particular limitation as long as it initiates the curing reaction. Examples thereof include thermal polymerization initiators and photopolymerization initiators.
- Thermal polymerization initiators include organic peroxides and azo compounds.
- organic peroxides include dialkyl peroxides such as di-t-butyl peroxide, t-butylcumyl peroxide and dicumyl peroxide; diacyl peroxides such as acetyl peroxide, lauroyl peroxide and benzoyl peroxide.
- ketone peroxides such as methyl ethyl ketone peroxide, cyclohexanone peroxide, 3,3,5-trimethylcyclohexanone peroxide and methyl cyclohexanone peroxide
- peroxyketals such as 1,1-bis(t-butylperoxy)cyclohexane ; t-butyl hydroperoxide, cumene hydroperoxide, 1,1,3,3-tetramethylbutyl hydroperoxide, p-menthane hydroperoxide, diisopropylbenzene hydroperoxide, 2,5-dimethylhexane-2, Hydroperoxides such as 5-dihydroperoxide; esters; and the like.
- Azo compounds include 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile), 2,2′-azobis(2-cyclopropylpropionitrile), 2,2′-azobis(2 ,4-dimethylvaleronitrile), azobisisobutyronitrile, 2,2′-azobis(2-methylbutyronitrile), 1,1′-azobis(cyclohexane-1-carbonitrile), 2-(carbamoylazo ) isobutyronitrile, 2-phenylazo-4-methoxy-2,4-dimethylvaleronitrile and the like.
- Photoinitiators include 2,2-dimethoxy-1,2-diphenylethan-1-one, 1-hydroxy-cyclohexyl-phenylketone, 2-hydroxy-2-methyl-1-phenyl-propan-1-one , 1-[4-(2-hydroxyethoxy)-phenyl]-2-hydroxy-2-methyl-1-propan-1-one, 2-hydroxy-1-[4-[4-(2-hydroxy-2 -methyl-propionyl)-benzyl]phenyl]-2-methyl-propan-1-one, 2-methyl-1-(4-methylthiophenyl)-2-morpholinopropan-1-one, 2-benzyl-2-dimethyl Amino-1-(4-morpholinophenyl)-butanone-1,2-(dimethylamino)-2-[(4-methylphenyl)methyl]-1-[4-(4-morpholinyl)phenyl]-1-butanone Alkylphenone-based photopolymerization initiators such as; -Phosphorus-based
- 2,4,6-trimethylbenzoyl-diphenylphosphine oxide bis(2,4,6-trimethylbenzoyl)-phenylphosphine oxide, ethyl(2,4,6-trimethylbenzoyl)- Phosphorus-based photopolymerization initiators such as phenylphosphinate and bis(2,6-dimethoxybenzoyl)-2,4,4-trimethyl-pentylphosphine oxide are preferred.
- the polymer component (A) is a thermoplastic resin having an aromatic ring
- the polymer component (A) absorbs ultraviolet rays, and as a result, the curing reaction may be difficult to occur.
- a polymerization initiator can be used individually by 1 type or in combination of 2 or more types.
- the content of the polymerization initiator is preferably 0.05 to 15% by mass, more preferably 0.05 to 10% by mass, and still more preferably 0.05 to 5% by mass, relative to the entire curable resin composition.
- the curable resin composition contains a polymer component (A), a curable monomer (B), and a polymerization initiator, as well as triisopropanolamine and photopolymerization such as 4,4′-diethylaminobenzophenone. It may contain an initiation aid.
- the solvent used for preparing the curable resin composition is not particularly limited, and examples thereof include aliphatic hydrocarbon solvents such as n-hexane and n-heptane; aromatic hydrocarbon solvents such as toluene and xylene; and dichloromethane.
- ethylene chloride, chloroform, carbon tetrachloride, 1,2-dichloroethane, monochlorobenzene and other halogenated hydrocarbon solvents methanol, ethanol, propanol, butanol, propylene glycol monomethyl ether and other alcohol solvents; acetone, methyl ethyl ketone, 2 - ketone solvents such as pentanone, isophorone and cyclohexanone; ester solvents such as ethyl acetate and butyl acetate; cellosolve solvents such as ethyl cellosolve; ether solvents such as 1,3-dioxolane;
- the content of the solvent in the curable resin composition is not particularly limited, but is usually 0.1 to 1,000 g, preferably 1 to 50 g, per 1 g of polymer component (A). By appropriately adjusting the amount of the solvent, the viscosity of the curable resin composition can be appropriately adjusted.
- the curable resin composition may further contain known additives such as plasticizers, antioxidants, and ultraviolet absorbers within a range that does not impair the objects and effects of the present invention.
- the method for curing the curable resin composition can be appropriately determined according to the type of polymerization initiator and curable monomer used. The details will be described in the section of the manufacturing method of the laminate to be described later.
- the adhesive layer has adhesive properties and, in the present invention, plays a role of flattening the uneven surface of the uneven substrate with the cured resin layer interposed therebetween.
- the adhesive may be a pressure sensitive adhesive (adhesive) or a heat sealing material.
- Adhesives include acrylic polymers, epoxy polymers, silicone polymers, polyesters, polyurethanes, polyamides, polyvinyl ethers, vinyl acetate/vinyl chloride copolymers, modified polyolefins, epoxy polymers, fluorine polymers, rubber polymers, etc.
- an adhesive having an acrylic polymer as a base polymer or an adhesive having a rubber polymer as a base polymer can also be preferably used as adhesives.
- the material constituting the adhesive layer may contain other optional components as long as the effects of the laminate of the present invention are not impaired.
- Optional components contained in the adhesive include, for example, organic solvents, conductive materials, high thermal conductivity materials, flame retardants, tackifiers, ultraviolet absorbers, antioxidants, preservatives, plasticizers, antifoaming agents, and A wettability modifier and the like can be mentioned.
- the adhesive layer may be a single layer or may be composed of multiple layers.
- the thickness of the adhesive layer is appropriately adjusted and determined depending on the shape and dimensions of the uneven surface of the uneven substrate. 25 ⁇ m, more preferably 3 to 20 ⁇ m, more preferably 5 to 20 ⁇ m.
- the thickness of the laminate is also appropriately adjusted and determined according to the shape and dimensions of the uneven surface of the uneven substrate.
- the substantial thickness of the laminate is preferably 0.1 to 30 ⁇ m, more preferably 0.3 to 20 ⁇ m, more preferably 0.5 to 15 ⁇ m, particularly preferably 0.7 to 10 ⁇ m, from the viewpoint of handleability. is.
- substantially thickness means the thickness in the state of use. That is, the laminate may have a process film, etc. as described above, but the thickness of the portion (process film, etc.) that is removed during use is not included in the "substantial thickness". do not have.
- Laminate manufacturing method A method for manufacturing a laminate including a process film, which is the first embodiment of the present invention, will be described. By using the process film, the laminate can be produced efficiently and easily.
- the method for producing a laminate of the present invention includes the following (Step 1) to (Step 3).
- Step 1) Step of forming a curable resin layer on a process film using a curable resin composition containing a polymer component (A) and a curable monomer (B)
- Step 2) Step Step of curing the curable resin layer obtained in 1 to form a cured resin layer
- step 3) Step of forming an adhesive layer on the cured resin layer obtained in step 2
- the method of applying the curable resin composition onto the process film is not particularly limited, and may be spin coating, spray coating, bar coating, knife coating, roll coating, blade coating, die coating, or gravure coating.
- a known coating method such as a method can be used.
- the method for drying the obtained coating film is not particularly limited, and conventionally known drying methods such as hot air drying, hot roll drying, and infrared irradiation can be used.
- the drying temperature of the coating film is usually 30 to 150°C, preferably 50 to 130°C.
- the curable resin layer obtained in step 1 is cured to form a cured resin layer.
- a method for curing the curable resin layer is not particularly limited, and a known method can be employed.
- the curable resin layer can be cured by heating the curable resin layer. .
- the heating temperature is usually 30 to 150°C, preferably 50 to 130°C.
- the curable resin layer is cured by irradiating the curable resin layer with an active energy ray.
- Active energy rays can be applied using a high-pressure mercury lamp, an electrodeless lamp, a xenon lamp, or the like.
- the wavelength of the active energy ray is preferably 200-400 nm, more preferably 350-400 nm.
- the illuminance of the active energy ray is usually in the range of 50-1,000 mW/cm 2 , preferably 100-600 mW/cm 2 .
- the amount of active energy rays is in the range of 50 to 5,000 mJ/cm 2 , preferably 300 to 4,000 mJ/cm 2 .
- the irradiation time is usually 0.1 to 1,000 seconds, preferably 1 to 500 seconds, more preferably 10 to 100 seconds. In consideration of the heat load of the light irradiation process, the irradiation may be performed multiple times in order to satisfy the aforementioned light amount.
- a filter that absorbs light of wavelengths unnecessary for the curing reaction is interposed, and the active energy rays are applied.
- the curable resin composition may be irradiated.
- the filter absorbs light of a wavelength that is unnecessary for the curing reaction and degrades the polymer component (A). It becomes easy to obtain a resin.
- a resin film such as a polyethylene terephthalate film can be used as the filter.
- the resin film is usually peeled off after step 2.
- the curable resin layer can also be cured by irradiating the curable resin layer with an electron beam.
- the curable resin layer can be cured usually without using a photopolymerization initiator.
- an electron beam accelerator or the like can be used.
- the irradiation dose is usually in the range of 10 to 1,000 krad.
- the irradiation time is usually 0.1 to 1,000 seconds, preferably 1 to 500 seconds, more preferably 10 to 100 seconds.
- the curing of the curable resin layer may be performed under an inert gas atmosphere such as nitrogen gas, if necessary. Curing in an inert gas atmosphere makes it easier to prevent oxygen, moisture, and the like from interfering with curing.
- a desired adhesive layer is formed on the cured resin layer obtained in step 2.
- the coating method described above can be appropriately adopted.
- the laminate When the laminate has a process film, the laminate may have the process film on one side or both sides. In the latter case, it is preferable to use two types of process films and make the process film to be peeled off first easier to peel.
- the process film is preferably sheet-like or film-like.
- sheet-like or film-like includes not only a long one but also a short flat plate-like one.
- Process films include paper base materials such as glassine paper, coated paper, and fine paper; laminated paper obtained by laminating these paper base materials with thermoplastic resins such as polyethylene and polypropylene; Those subjected to filling treatment with alcohol, acrylic-styrene resin, etc.; plastic films such as polyester films such as polyethylene terephthalate, polybutylene terephthalate and polyethylene naphthalate, and polyolefin films such as polyethylene and polypropylene; and glass. Further, the process film may be a paper substrate or a plastic film having a release agent layer provided thereon from the viewpoint of ease of handling.
- the release layer can be formed using conventionally known release agents such as silicone release agents, fluorine release agents, alkyd release agents, and olefin release agents.
- the thickness of the release agent layer is not particularly limited, but is usually 0.02 to 2.0 ⁇ m, more preferably 0.05 to 1.5 ⁇ m.
- the thickness of the process film is preferably 1 to 500 ⁇ m, more preferably 5 to 300 ⁇ m, from the viewpoint of ease of handling.
- the process film is usually peeled off in a predetermined process depending on the use of the laminate.
- the manufacturing method including the above (Step 1) to (Step 3) forms a cured resin layer using a process film, and the laminate obtained by this method has a process film. may or may not have.
- a method for manufacturing a laminate according to the second embodiment of the present invention includes the following (steps A) to (steps C).
- Step A) The surface of the cured resin layer in the laminate (cured resin layer/adhesive layer) obtained in step 3, opposite to the adhesive layer side, faces the uneven surface side of the uneven substrate.
- step B) A step of heating the laminated body having the stuck uneven substrate at more than 180 ° C. and 250 ° C. or less to melt the cured resin layer, and embedding the melt in the uneven part of the uneven substrate
- step C) A step of laminating at 180° C. for 1 hour from the side of the adhesive layer opposite to the side of the cured resin layer, and curing the cured resin layer.
- the method of heating the laminate and the method of laminating in (Step B) and (Step C) can be performed by known methods.
- this manufacturing method it is possible to manufacture a laminate including an uneven substrate in which the uneven surface is embedded with a cured resin layer interposed and the surface of the adhesive layer opposite to the uneven surface side is flattened.
- the laminate according to the first embodiment and the second embodiment, including the laminate having the basic configuration of the present invention can be efficiently, continuously, and easily manufactured. be able to.
- the peak temperature of the loss tangent of the cured resin layer prepared in the example and the polyimide resin film and acrylic resin film used in the comparative example, the heat resistance of each after heat treatment, and the adhesion between the cured resin layer and the adherend Evaluation was performed by the following methods.
- the heating temperature at the convex portion of the loss tangent (loss modulus/storage modulus) curve was taken as the loss tangent peak temperature.
- the laminate was heated at 180° C. for 1 hour using a heating oven (manufactured by ESPEC, model name “SPHH-202”) to complete the curing reaction.
- the loss tangent of the cured resin layer after heat treatment was measured in the same manner as described above.
- the heat resistance after heat treatment was evaluated from the loss tangent peak temperature ((glass transition temperature in Comparative Examples 1 and 2)).
- B The loss tangent peak temperature exists at less than 200 ° C.
- Example 1 Formation of cured resin layer A curable resin composition was prepared as follows.
- PI polyimide resin
- MEK methyl ethyl ketone
- curable monomer (B) tricyclodecanedimethanol diacrylate (manufactured by Shin-Nakamura Chemical Co., Ltd., A-DCP) 120 parts by mass, and as a polymerization initiator, (2,4, 6-Trimethylbenzoyl)-phenylphosphine oxide (manufactured by BASF, Irgacure 819) of 5 parts by mass was added and mixed to prepare a curable resin composition.
- the curable monomer (B) and the polymerization initiator used in this example do not contain a solvent and are all raw materials having a solid content of 100%.
- PET polyethylene terephthalate
- PET100A-4100 thickness 100 ⁇ m
- a curable resin composition was applied to the surface, and the resulting coating film was dried by heating at 90° C. for 2 minutes.
- the illuminance at a light wavelength of 365 nm is 130 mW/cm 2 and the light amount is 700 mJ/cm 2 (manufactured by Heraus, ultraviolet light meter, Under the conditions of UV Power Puck (registered trademark) II), a curing reaction was performed by irradiating ultraviolet rays in a nitrogen atmosphere to form a cured resin layer having a thickness of 5 ⁇ m.
- BA butyl acrylate
- MMA methyl methacryl
- Example 1 (Comparative example 1) Instead of the cured resin layer of Example 1, a polyimide resin film (Mitsubishi Gas Chemical Co., Ltd., L-3430, glass transition temperature 300° C., thickness 50 ⁇ m) was used. The peak temperature of the loss tangent of the polyimide resin film, the heat resistance after the heat treatment, and the adhesion were determined from the physical properties (glass transition temperature) of the polyimide resin film used. Table 1 shows the results.
- Comparative example 2 An acrylic resin film (manufactured by Mitsubishi Chemical Corporation, HBS010P, glass transition temperature 120° C., thickness 50 ⁇ m) was used instead of the cured resin layer of Example 1. The peak temperature of the loss tangent of the acrylic resin film, the heat resistance after the heat treatment, and the adhesion were determined from the physical properties (glass transition temperature) of the acrylic resin film used. Table 1 shows the results.
- Example 1 since the loss tangent peak is at 200 ° C. or less before heat treatment, the resin layer melts when heated at 190 ° C., and adheres to the polyimide resin film that is the adherend. do.
- the curing reaction is completed by heating at 180 ° C. for 1 hour, whereby the loss tangent peak is only 260 ° C. Therefore, for example, even if it is put into a heat treatment process under high temperature, it has high heat resistance. It can be seen that the In Comparative Example 1, since it has a loss tangent peak at 300° C. before heat treatment, the resin layer does not melt when heated to 190° C., and cannot be adhered to the adherend, the polyimide resin film.
- the laminate of the present invention since it is possible to flatten the uneven substrate, it can be expected to be used as an insulating layer for wiring in display devices and semiconductor devices. At the same time, since it has adhesiveness, it can be used for easily laminating another layer on the side of the adhesive layer opposite to the uneven side.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Laminated Bodies (AREA)
Abstract
Description
しかし、一般に有機絶縁材料は、無機絶縁材料に比べて、軽量化及びフレキシブル化に寄与するものの、耐熱性に劣り、例えば、前記電子デバイス等の絶縁層として用いる際に、機械的な損傷等を受けることにより、電子デバイス内部の素子等に作用し、短絡の発生等によりデバイス性能が低下したり、寿命が短くなるという問題があった。また、下地層が凹凸を有する場合には、凹凸面を平坦化することが十分ではなかった。
例えば、特許文献1には、表示素子やタッチパネル等の配線を絶縁、封止する目的で、画素部上の凹凸を有する無機絶縁層上に、例えば、有機絶縁層を有機絶縁材料としてアクリル樹脂やポリイミド樹脂を用いスピンコート法等により形成することが開示されている。
すなわち、本発明は、以下の[1]~[5]を提供するものである。
[1]硬化樹脂層と接着剤層とを含む積層体であって、前記硬化樹脂層は、重合体成分(A)及び硬化性単量体(B)を含有する硬化性樹脂組成物の硬化物からなる層であり、25℃から昇温速度3℃/分で測定した動的粘弾性測定において、100~200℃の温度範囲に損失正接のピークを有し、180℃1時間加熱後、損失正接のピークを220℃以上に有する、積層体。
[2]前記重合体成分(A)のガラス転移温度が250℃以上である、上記[1]に記載の積層体。
[3]前記重合体成分(A)の重量平均分子量が100,000以上である、上記[1]又は[2]に記載の積層体。
[4]前記重合体成分(A)がポリイミド樹脂を含む、上記[1]~[3]のいずれかに記載の積層体。
[5]前記硬化樹脂層の厚さが20μm以下である、上記[1]に記載の積層体。
本明細書において、「XX~YY」との記載は、「XX以上YY以下」を意味する。
本明細書において、好ましい数値範囲(例えば、含有量等の範囲)について、段階的に記載された下限値及び上限値は、それぞれ独立して組み合わせることができる。例えば、「好ましくは10~90、より好ましくは30~60」という記載から、「好ましい下限値(10)」と「より好ましい上限値(60)」とを組み合わせて、「10~60」とすることもできる。
本明細書において、例えば、「(メタ)アクリル酸」とは、「アクリル酸」と「メタクリル酸」の双方を示し、他の類似用語も同様である。
本発明の積層体は、硬化樹脂層と接着剤層とを含む積層体であって、前記硬化性樹脂層は、重合体成分(A)及び硬化性単量体(B)を含有する硬化性樹脂組成物の硬化物からなる層であり、25℃から昇温速度3℃/分で測定した動的粘弾性測定において、100℃~200℃の温度範囲に損失正接のピークを有し、180℃1時間加熱後、損失正接のピークを220℃以上に有することを特徴とする。
本発明に用いる硬化樹脂層は、上記に規定した各温度条件において損失正接のピークを有することにより、例えば、180℃超の温度で溶融し、電子デバイスの配線等に係る層や他の被着体等の基材等(以降、まとめて単に、「凹凸基板」ということがある。)に備わる凹凸面を埋め込み、次いで180℃1時間加熱することにより硬化反応が完了する。同時に、硬化樹脂層の、凹凸面側とは反対側の面上に接着剤層を備えることで、凹凸形状が接着剤層表面に現れることなく、接着剤層の、凹凸面側とは反対側の面を平坦化することができる。加えて本発明の積層体は接着性を有することから、接着剤層の、凹凸面側とは反対側の面をさらに他の被着体、層等と容易に接着(積層)することができる。
なお、本明細書において、「凹凸基板」の凹凸形状、深さ、幅等は、凹凸形状を埋め込むことができれば、特に制限されない。また、凹凸基板の材料については、樹脂との接着性が維持できるものであれば特に制限されない。
図1において、積層体10は、硬化樹脂層1上に接着剤層2を積層したものである。
図2において、積層体20は、例えば、硬化樹脂層11の、接着剤層12側とは反対側の面に工程フィルム13を積層したものである。
図3において、積層体30は、例えば、硬化樹脂層21の、接着剤層22側とは反対側の面に凹凸基板23を積層したものである。
本発明に用いる硬化樹脂層は、重合体成分(A)及び硬化性単量体(B)を含有する硬化性樹脂組成物の硬化物からなる層である。
硬化樹脂層の25℃から昇温速度3℃/分で測定した動的粘弾性測定において、100~200℃の温度範囲にある損失正接のピークは、複数あってもよい。損失正接のピークが複数ある場合には、少なくとも1つの損失正接のピークを100~200℃の温度範囲に有していればよい。
損失正接のピークは、好ましくは120~200℃、より好ましくは140~200℃、さらに好ましくは160~200℃である。
損失正接のピークがこの範囲にあると、硬化樹脂層が溶融し、凹凸基板に接着し易くなる。
180℃1時間加熱後の損失正接のピークは、好ましくは240℃以上、より好ましくは250℃以上、さらに好ましくは260℃以上である。180℃1時間加熱後の損失正接のピークがこの範囲にあると、例えば、より高い温度における熱処理をともなうプロセスにおいて、高い耐熱性を示す。
なお、塗膜とは、塗布材料を基材や対象物上に塗布し、必要に応じて乾燥や加熱等による硬化等の処理を施して得られる被膜である。
(a)硬化樹脂層を、メッシュ(NBCメッシュテック社製、α_UX SCREEN 150―035/380TW)で包み、ホチキスで止めた構成体とし当該構成体の重量を測定
(b)メチルエチルケトン(MEK)溶媒を満たしたビンに、構成体を浸漬した後、密閉し、25℃で36時間放置
(c)構成体を溶媒から取り出し、100℃で60分間の乾燥を行い、乾燥後の構成体の重量を測定
硬化樹脂層は、複屈折率が低く光学等方性に優れる。前記硬化樹脂層の面内の位相差は、通常、2.0nm以下であり、1.0nm以下が好ましい。厚さ方向の位相差は、通常、-500nm以下であり、-450nm以下が好ましい。また、面内の位相差を硬化樹脂層の厚さで割った値(複屈折率)は、通常、100×10-5以下であり、好ましくは20×10-5以下である。
硬化樹脂層の面内の位相差、厚さ方向の位相差、複屈折率が上記の範囲内であれば、複屈折率が低く光学等方性に優れる積層体が得られ、本発明の接着剤層を有する積層体を光学用途に好ましく用いることができる。
重合体成分(A)としては、熱可塑性樹脂が好ましく、非晶性熱可塑性樹脂がより好ましい。非晶性熱可塑性樹脂を用いることで、光学等方性に優れた硬化樹脂層を得られ易くなり、また、透明性に優れる積層体が得られ易くなる。また、非晶性熱可塑性樹脂は概して有機溶剤に溶け易いため、後述するように、溶液キャスト法を利用して、効率よく硬化樹脂層を形成することができる。
ここで、非晶性熱可塑性樹脂とは、示差走査熱量測定において、融点が観測されない熱可塑性樹脂をいう。
重合体成分(A)のTgは好ましくは250℃以上であり、より好ましくは290℃以上、さらに好ましくは320℃以上である。Tgがこの範囲にあると、耐熱性に十分優れる接着剤層を有する積層体を得ることができる。例えば、接着剤層を塗膜から形成する場合に、塗膜の塗工時の加熱によって硬化樹脂層が影響を受けて変形や剥がれ等を生じることがなく、結果的に、接着剤層が本来有する十分な接着性を発現させることができる。
なお、塗膜とは、塗布材料を基材や対象物上に塗布し、必要に応じて乾燥や加熱等による硬化等の処理を施して得られる被膜である。また、硬化樹脂層を塗膜とする場合は、硬化性樹脂組成物を工程フィルム等の被塗布体に塗布し、乾燥及び加熱や活性エネルギー線の照射等のいずれか一方のみ又は両方による硬化処理を行って得られる被膜である。
ここでTgは、粘弾性測定(周波数10Hz、昇温速度3℃/分で0~250℃の範囲で引張モードによる測定)により得られたtanδ(損失弾性率/貯蔵弾性率)のピーク温度をいう。
フルオロ基を有するポリイミド樹脂としては、分子内にフルオロ基を有する芳香族ポリイミド樹脂が好ましく、分子内に以下の化学式で示す骨格を有するものが好ましい。

硬化性単量体(B)は、重合性不飽和結合を有する単量体であって、重合反応、又は、重合反応及び架橋反応に関与し得る単量体である。なお、本明細書において、「硬化」とは、この「単量体の重合反応」、又は、「単量体の重合反応及び引き続く重合体の架橋反応」を含めた広い概念を意味する。硬化性単量体(B)を用いることで、耐溶剤性に優れる積層体を得ることができる。
硬化樹脂層を、上述した重合体成分(A)と上記硬化性単量体(B)とを含有する硬化性樹脂組成物の硬化物からなる層とすることで、耐熱性に優れ、薄い硬化樹脂層を形成することが容易になる。また、このような材料を用いれば、積層体の基材として一般的に用いられるポリエステル系フィルムのような、異方性の分子配向を有する材料に起因した光学上の問題が生じない。重合体成分のTgは、250℃以上であることで、硬化樹脂層が耐熱性を有し、接着剤層を塗膜から形成する場合に、塗膜の塗工時の熱による硬化樹脂層の変形等により、接着性の性能が低下することが抑制される。
硬化性単量体(B)中の重合性不飽和結合の数は特に制限されない。硬化性単量体(B)は、重合性不飽和結合を1つ有する単官能型の単量体であっても、複数有する2官能型や3官能型等の多官能型の単量体であってもよい。
単官能の(メタ)アクリル酸誘導体としては、特に限定されず、公知の化合物を用いることができる。例えば、窒素原子を有する単官能の(メタ)アクリル酸誘導体、脂環式構造を有する単官能の(メタ)アクリル酸誘導体、ポリエーテル構造を有する単官能の(メタ)アクリル酸誘導体等が挙げられる。

R1で表される炭素数1~6のアルキル基としては、メチル基、エチル基、プロピル基等が挙げられ、メチル基が好ましい。
R2及びR3で表される炭素数1~12の有機基としては、メチル基、エチル基、プロピル基等の、炭素数1~12のアルキル基;シクロペンチル基、シクロへキシル基等の、炭素数3~12のシクロアルキル基;フェニル基、ビフェニル基、ナフチル基等の、炭素数6~12の芳香族基;が挙げられる。これらの基は、任意の位置に置換基を有していてもよい。また、R2とR3が一緒になって環を形成してもよく、該環は、骨格中に更に窒素原子や酸素原子を有していてもよい。
R4で表される2価の有機基としては、-(CH2)m-、-NH-(CH2)m-で表される基が挙げられる。ここで、mは、1~10の整数である。


R5で表される脂環式構造を有する基としては、シクロへキシル基、イソボルニル基、1-アダマンチル基、2-アダマンチル基、トリシクロデカニル基等が挙げられる。

多官能の(メタ)アクリル酸誘導体としては、特に限定されず、公知の化合物を用いることができる。例えば、2~6官能の(メタ)アクリル酸誘導体が挙げられる。
2官能の(メタ)アクリル酸誘導体としては、下記式で示される化合物が挙げられる。


4官能の(メタ)アクリル酸誘導体としては、ペンタエリスリトールテトラ(メタ)アクリレート等が挙げられる。
5官能の(メタ)アクリル酸誘導体としては、プロピオン酸変性ジペンタエリスリトールペンタ(メタ)アクリレート等が挙げられる。
6官能の(メタ)アクリル酸誘導体としては、ジペンタエリスリトールヘキサ(メタ)アクリレート、カプロラクトン変性ジペンタエリスリトールヘキサ(メタ)アクリレート等が挙げられる。
また、ジメチル -2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート、ジエチル-2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート、ジ(n-プロピル)-2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート、ジ(i-プロピル)-2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート、ジ(n-ブチル)-2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート、ジ(n-ヘキシル)-2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート、ジシクロヘキシル-2,2’-[オキシビス(メチレン)]ビス-2-プロペノエート等の環化重合性モノマーを用いることもできる。
これらの中でも、硬化性単量体(B)は、耐熱性及び耐溶剤性により優れる硬化性樹脂層が得られることから、多官能型の単量体が好ましい。多官能の単量体としては、重合体成分(A)と混ざりやすく、かつ、重合物の硬化収縮が起こりにくく硬化物のカールが抑制できるという観点から、2官能(メタ)アクリル酸誘導体が好ましい。
硬化性単量体(B)として、多官能(メタ)アクリレート化合物と、環化重合性モノマーとが含まれることがより好ましい。
硬化性単量体(B)が多官能型の単量体を含む場合、その含有量は、硬化性単量体(B)の全量中、70質量%以下が好ましく、50質量%以下がより好ましい。
硬化性樹脂層を形成するのに用いる硬化性樹脂組成物は、重合体成分(A)、硬化性単量体(B)、及び所望により、後述する重合開始剤やその他の成分を混合し、適当な溶媒に溶解又は分散させることにより調製することができる。
硬化性樹脂組成物において、重合体成分(A):硬化性単量体(B)の質量比がこのような範囲にあることで、得られる硬化樹脂層の柔軟性がより向上しやすく、硬化樹脂層の耐溶剤性も保たれやすい傾向がある。さらに、上記成分割合であれば、硬化樹脂層中に、所定量の硬化性単量体が残存するものと推定され、100~200℃の温度範囲に損失正接のピークを有し、180℃1時間加熱後、損失正接のピークを220℃以上有する硬化樹脂層を得やすくなる。
有機過酸化物としては、ジ-t-ブチルパーオキサイド、t-ブチルクミルパーオキサイド、ジクミルパーオキサイド等のジアルキルパーオキサイド類;アセチルパーオキサイド、ラウロイルパーオキサイド、ベンゾイルパーオキサイド等のジアシルパーオキサイド類;メチルエチルケトンパーオキサイド、シクロヘキサノンパーオキサイド、3,3,5-トリメチルシクロヘキサノンパーオキサイド、メチルシクロヘキサノンパーオキサイド等のケトンパーオキサイド類;1,1-ビス(t-ブチルパーオキシ)シクロヘキサン等のパーオキシケタール類;t-ブチルヒドロパーオキサイド、クメンヒドロパーオキサイド、1,1,3,3-テトラメチルブチルヒドロパーオキサイド、p-メンタンヒドロパーオキサイド、ジイソプロピルベンゼンヒドロパーオキサイド、2,5-ジメチルヘキサン-2,5-ジヒドロパーオキサイド等のヒドロパーオキサイド類;t-ブチルパーオキシアセテート、t-ブチルパーオキシ-2-エチルヘキサノエート、t-ブチルパーオキシベンゾエート、t-ブチルパーオキシイソプロピルカーボネート等のパーオキシエステル類;等が挙げられる。
アゾ系化合物としては、2,2’-アゾビス(4-メトキシ-2,4-ジメチルバレロニトリル)、2,2’-アゾビス(2-シクロプロピルプロピオニトリル)、2,2’-アゾビス(2,4-ジメチルバレロニトリル)、アゾビスイソブチロニトリル、2,2’-アゾビス(2-メチルブチロニトリル)、1,1’-アゾビス(シクロヘキサン-1-カルボニトリル)、2-(カルバモイルアゾ)イソブチロニトリル、2-フェニルアゾ-4-メトキシ-2,4-ジメチルバレロニトリル等が挙げられる。
重合体成分(A)が芳香族環を有する熱可塑性樹脂である場合、重合体成分(A)が紫外線を吸収する結果、硬化反応が起こりにくいことがある。しかしながら、上記のリン系光重合開始剤を用いることで、上記重合体成分(A)に吸収されない波長の光を利用して硬化反応を効率よく進行させることができる。
重合開始剤は1種単独で、あるいは2種以上を組み合わせて用いることができる。
接着剤層は、接着性を有すると同時に、本発明にあっては、硬化樹脂層を介在し、凹凸基板の凹凸面を平坦化する役割を担う。
接着剤層を構成する材料においては、本発明の積層体の効果を損なわない限り、その他の任意成分が含まれていてもよい。接着剤に含まれる任意成分としては、例えば、有機溶媒、導電性材料、高熱伝導性材料、難燃剤、粘着付与剤、紫外線吸収剤、酸化防止剤、防腐剤、可塑剤、消泡剤、及び濡れ性調整剤等が挙げられる。
接着剤層の厚さは、凹凸基板の凹凸面の形状や寸法により、適宜、調整し決定されるが、通常、接着性能を向上させつつ、凹凸面を平坦化させる観点から、好ましくは1~25μm、より好ましくは3~20μm、さらに好ましくは5~20mである。
なお、「実質的な厚さ」とは、使用状態における厚さをいう。すなわち、上記積層体は、前述したように工程フィルム等を有していてもよいが、使用時に除去される部分(工程フィルム等)の厚さは、「実質的な厚さ」には含まれない。
本発明の第1の実施形態である工程フィルムを含む積層体の製造方法を説明する。工程フィルムを用いることで、積層体を効率よく、かつ、容易に製造することができる。
(工程1):工程フィルム上に、重合体成分(A)及び硬化性単量体(B)を含有する硬化性樹脂組成物を用いて硬化性樹脂層を形成する工程
(工程2):工程1で得られた硬化性樹脂層を硬化させて、硬化樹脂層を形成する工程
(工程3):工程2で得られた硬化樹脂層上に、接着剤層を形成する工程
塗膜の乾燥温度は、通常、30~150℃、好ましくは、50~130℃である。
硬化性樹脂層を硬化する方法としては、特に限定されず、公知の方法が採用できる。例えば、硬化性樹脂層が、熱重合開始剤を含有する硬化性樹脂組成物を用いて形成されたものである場合、硬化性樹脂層を加熱することで硬化性樹脂層を硬化させることができる。加熱温度は、通常、30~150℃、好ましくは、50~130℃である。
また、硬化性樹脂層が、光重合開始剤を含有する硬化性樹脂組成物を用いて形成されたものである場合、硬化性樹脂層に活性エネルギー線を照射することで硬化性樹脂層を硬化させることができる。活性エネルギー線は、高圧水銀ランプ、無電極ランプ、キセノンランプ等を用いて照射することができる。
フィルタとしては、ポリエチレンテレフタレートフィルム等の樹脂フィルムを利用することができる。樹脂フィルムを用いる場合、工程1と工程2の間に、硬化樹脂層上にポリエチレンテレフタレートフィルム等の樹脂フィルムを積層させる工程を設けることが好ましい。なお、樹脂フィルムは、通常は、工程2の後に剥離される。
接着剤層を形成する方法としては、先に説明した塗布方法を適宜採用することができる。
工程フィルムは、シート状またはフィルム状のものが好ましい。シート状またはフィルム状とは、長尺のものに限らず、短尺の平板状のものも含まれる。
工程フィルムとしては、グラシン紙、コート紙、上質紙等の紙基材;これらの紙基材にポリエチレンやポリプロピレン等の熱可塑性樹脂をラミネートしたラミネート紙;上記紙基材に、セルロース、デンプン、ポリビニルアルコール、アクリル-スチレン樹脂等で目止め処理を行ったもの;あるいはポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンナフタレート等のポリエステルフィルムやポリエチレンやポリプロピレン等のポリオレフィンフィルム等のプラスチックフィルム;ガラス等が挙げられる。
また、工程フィルムは、取り扱い易さの点から、紙基材や、プラスチックフィルム上に剥離剤層を設けたものであってもよい。剥離層は、シリコーン系剥離剤、フッ素系剥離剤、アルキッド系剥離剤、オレフィン系剥離剤等、従来公知の剥離剤を用いて形成することができる。
剥離剤層の厚さは、特に制限されないが、通常、0.02~2.0μm、より好ましくは0.05~1.5μmである。
工程フィルムの厚さは、取り扱い易さの点から、1~500μmが好ましく、5~300μmがより好ましい。
なお、工程フィルムは、通常は、積層体の用途等に応じて、所定の工程において剥離される。
(工程A):工程3で得られた積層体(硬化樹脂層/接着剤層)における硬化樹脂層の、接着剤層側の面とは反対側の面を、凹凸基板の凹凸面側に対向させ貼付する工程
(工程B):貼付された凹凸基板を有する積層体を180℃超、250℃以下で加熱し硬化樹脂層を溶融させ、該溶融物を凹凸基板の凹凸部に埋め込む工程
(工程C):接着剤層の、硬化樹脂層側の面とは反対側の面から、180℃1時間にてラミネートし、硬化樹脂層を硬化する工程
当該製造方法により、硬化樹脂層を介在し凹凸面が埋め込まれた、接着剤層の、凹凸面側とは反対側の面が平坦化された凹凸基板を含む積層体を製造することができる。
実施例で得られた、硬化樹脂層の両面の工程フィルムに相当するポリエチレンテレフタレート(PET)フィルムを剥離除去した硬化樹脂層を、8枚積層して40μmの厚さの積層体とした。次に、5mm×30mmの試験片に裁断し、熱機械分析装置(NETZSCH Japan社製、製品名「DMA242」)を用いて、チャック間距離15mmに設定して、前記硬化樹脂層の積層体を把持した。次いで、当該硬化樹脂層の積層体を昇温速度3℃/minで25℃から280℃まで昇温させた。そして、25℃から280℃までの温度範囲において、損失正接(損失弾性率/貯蔵弾性率)曲線の凸部における加熱温度を損失正接ピーク温度とした。次に、前記積層体に対して加熱オーブン(ESPEC 社製、型名「SPHH-202」)を使用して180℃にて1時間加熱し、硬化反応を完了させた。その後、加熱処理後の硬化樹脂層に対して上記と同様に損失正接の測定を行った。
加熱処理後の耐熱性については損失正接ピーク温度((比較例1、2ではガラス転移温度))から評価した。
A:損失正接ピーク温度が200℃以上にのみ存在している
B:損失正接ピーク温度が200℃未満に存在している
(2)密着性評価
硬化樹脂層を被着体としてのポリイミド樹脂フィルム(三菱瓦斯化学社製、L-3430、厚さ50μm)に積層し、加熱オーブン(ESPEC社製、型名「SPHH-202」)を使用して、190℃にて10分加熱することで、硬化樹脂層とポリイミド樹脂フィルムとの積層体を得た。その後、得られた積層体に対して加熱オーブンを使用して180℃にて1時間加熱し硬化反応を完了させた。硬化樹脂層とポリイミド樹脂フィルムとの密着性については、以下の基準で目視を行った。
A:硬化樹脂層とポリイミド樹脂フィルムが接合している
B:硬化樹脂層とポリイミド樹脂フィルムが接合していない
・硬化樹脂層の形成
硬化性樹脂組成物を以下のように調製した。
重合体成分(A)として、ポリイミド樹脂(PI)のペレット(河村産業社製、製品名「KPI-MX300F」、Tg=354℃、重量平均分子量280,000)100質量部をメチルエチルケトン(MEK)に溶解して、ポリイミド樹脂の15質量%溶液を調製した。次いで、この溶液に、硬化性単量体(B)として、トリシクロデカンジメタノールジアクリレート(新中村化学工業社製、A-DCP)120質量部、及び重合開始剤として、(2,4,6-トリメチルベンゾイル)-フェニルホスフィンオキサイド(BASF社製、Irgacure819)5質量部を添加、混合して、硬化性樹脂組成物を調製した。なお、本実施例において使用した硬化性単量体(B)及び重合開始剤は溶媒を含まず、全て固形分100%の原料である。
次に、工程フィルムとして、片面に易接着層を有するポリエチレンテレフテレート(PET)フィルム(東洋紡社製、PET100A-4100、厚さ100μm)を使用し、このPETフィルムの易接着層面とは反対の面に、硬化性樹脂組成物を塗布し、得られた塗膜を90℃で2分間加熱して乾燥した。
次に、高圧水銀ランプ(アイグラフィクス社製、製品名「H04-L41」)を用いて、光線波長365nmの照度が130mW/cm2、光量が700mJ/cm2(Heraus社製、紫外線光量計、UV Power Puck(登録商標)II)の条件で、窒素雰囲気下にて紫外線照射して硬化反応を行い、厚さ5μmの硬化樹脂層を形成した。
・接着剤層の形成
アクリル系共重合体(n-ブチルアクリレート(BA)/メチルメタクリレート(MMA)/酢酸ビニル(VAc)/2-ヒドロキシエチルアクリレート(2HEA)=80.0/10.0/9.0/1.0(質量比)、重量平均分子量:100万、溶剤:酢酸エチル、固形分濃度15質量%)100質量部(固形分比)に、粘着付与剤として、ロジン系樹脂(荒川化学工業社製、製品名「KE-359」、軟化点:94~104℃)25質量部(固形分比)、架橋剤として、イソシアネート系架橋剤(三井化学社製、製品名「タケネート D-110N」)1.62質量部(固形分比)を配合して混合し、均一に攪拌して、接着組成物の酢酸エチル溶液を調製した。
接着剤組成物の酢酸エチル溶液を、得られた硬化樹脂層の一方の面に積層し、乾燥させることで、厚さ5μmの接着剤層と硬化樹脂層からなる積層体を作成した。
得られた硬化樹脂層の損失正接のピーク温度、加熱処理後の耐熱性及び密着性評価を行った。結果を表1に示す。
実施例1の硬化樹脂層の代わりにポリイミド樹脂フィルム(三菱瓦斯化学社製、L-3430、ガラス転移温度300℃、厚さ50μm)を用いた。
ポリイミド樹脂フィルムの損失正接のピーク温度、加熱処理後の耐熱性及び密着性評価は用いたポリイミド樹脂フィルムの物性値(ガラス転移温度)から判断した。結果を表1に示す。
実施例1の硬化樹脂層の代わりにアクリル樹脂フィルム(三菱ケミカル社製、HBS010P、ガラス転移温度120℃、厚さ50μm)を用いた。
アクリル樹脂フィルムの損失正接のピーク温度、加熱処理後の耐熱性及び密着性評価は用いたアクリル樹脂フィルムの物性値(ガラス転移温度)から判断した。結果を表1に示す。


比較例1では、そもそも加熱処理前に300℃に損失正接のピークを有するために、190℃の加熱時に樹脂層が溶融せず、被着体であるポリイミド樹脂フィルムに接着できないことがわかる。
比較例2では、加熱処理前に200℃以下に損失正接のピークを有するために、190℃の加熱時に樹脂層が溶融し、被着体であるポリイミド樹脂フィルムと接着する。しかしながら、180℃にて1時間の加熱後も損失正接のピーク位置が変化せず、損失正接のピークが120℃のままであるため、例えば、高温下の熱処理プロセスに投入できないことがわかる。
1,11,21:硬化樹脂層
2,12,22:接着剤層
13:工程フィルム
23:凹凸基板
Claims (5)
- 硬化樹脂層と接着剤層とを含む積層体であって、前記硬化樹脂層は、重合体成分(A)及び硬化性単量体(B)を含有する硬化性樹脂組成物の硬化物からなる層であり、25℃から昇温速度3℃/分で測定した動的粘弾性測定において、100~200℃の温度範囲に損失正接のピークを有し、180℃1時間加熱後、損失正接のピークを220℃以上に有する、積層体。
- 前記重合体成分(A)のガラス転移温度が250℃以上である、請求項1に記載の積層体。
- 前記重合体成分(A)の重量平均分子量が100,000以上である、請求項1又は2に記載の積層体。
- 前記重合体成分(A)がポリイミド樹脂を含む、請求項1~3のいずれか1項に記載の積層体。
- 前記硬化樹脂層の厚さが20μm以下である、請求項1に記載の積層体。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202280024366.5A CN117062716A (zh) | 2021-03-26 | 2022-03-28 | 层叠体 |
| KR1020237032775A KR20230164674A (ko) | 2021-03-26 | 2022-03-28 | 적층체 |
| JP2023509354A JPWO2022203086A1 (ja) | 2021-03-26 | 2022-03-28 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021054132 | 2021-03-26 | ||
| JP2021-054132 | 2021-03-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022203086A1 true WO2022203086A1 (ja) | 2022-09-29 |
Family
ID=83397489
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/014876 WO2022203086A1 (ja) | 2021-03-26 | 2022-03-28 | 積層体 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JPWO2022203086A1 (ja) |
| KR (1) | KR20230164674A (ja) |
| CN (1) | CN117062716A (ja) |
| TW (1) | TW202306773A (ja) |
| WO (1) | WO2022203086A1 (ja) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006117848A (ja) * | 2004-10-22 | 2006-05-11 | Kaneka Corp | 熱硬化性樹脂組成物およびその利用 |
| WO2013161645A1 (ja) * | 2012-04-27 | 2013-10-31 | リンテック株式会社 | 熱電変換材料及びその製造方法 |
| JP2014215440A (ja) * | 2013-04-25 | 2014-11-17 | 日立化成株式会社 | 感光性樹脂組成物、フィルム状接着剤、接着シート、接着剤パターン、接着剤層付半導体ウェハ、及び半導体装置 |
| WO2020138207A1 (ja) * | 2018-12-27 | 2020-07-02 | リンテック株式会社 | ガスバリア性積層体 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6906397B2 (ja) | 2017-08-10 | 2021-07-21 | 株式会社ジャパンディスプレイ | 表示装置 |
-
2022
- 2022-03-28 JP JP2023509354A patent/JPWO2022203086A1/ja active Pending
- 2022-03-28 TW TW111111730A patent/TW202306773A/zh unknown
- 2022-03-28 CN CN202280024366.5A patent/CN117062716A/zh active Pending
- 2022-03-28 KR KR1020237032775A patent/KR20230164674A/ko unknown
- 2022-03-28 WO PCT/JP2022/014876 patent/WO2022203086A1/ja active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006117848A (ja) * | 2004-10-22 | 2006-05-11 | Kaneka Corp | 熱硬化性樹脂組成物およびその利用 |
| WO2013161645A1 (ja) * | 2012-04-27 | 2013-10-31 | リンテック株式会社 | 熱電変換材料及びその製造方法 |
| JP2014215440A (ja) * | 2013-04-25 | 2014-11-17 | 日立化成株式会社 | 感光性樹脂組成物、フィルム状接着剤、接着シート、接着剤パターン、接着剤層付半導体ウェハ、及び半導体装置 |
| WO2020138207A1 (ja) * | 2018-12-27 | 2020-07-02 | リンテック株式会社 | ガスバリア性積層体 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202306773A (zh) | 2023-02-16 |
| JPWO2022203086A1 (ja) | 2022-09-29 |
| CN117062716A (zh) | 2023-11-14 |
| KR20230164674A (ko) | 2023-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7225530B2 (ja) | 無溶剤型樹脂組成物 | |
| JP7401463B2 (ja) | ガスバリア性積層体 | |
| JP6082354B2 (ja) | 硬化性樹脂組成物、硬化性樹脂成形体、硬化樹脂成形体およびそれらの製造方法、並びに積層体 | |
| KR101903217B1 (ko) | 경화성 수지 조성물, 경화성 수지 성형체, 경화 수지 성형체 및 그들의 제조 방법, 그리고 적층체 | |
| WO2021261195A1 (ja) | 光学用フィルム、光学用フィルムの製造方法、透明導電フィルム、及び、ガスバリアフィルム | |
| JP7222976B2 (ja) | ガスバリア性積層体 | |
| WO2022203086A1 (ja) | 積層体 | |
| JP7398394B2 (ja) | ガスバリア性積層体 | |
| WO2023054528A1 (ja) | 積層体 | |
| WO2022203071A1 (ja) | 硬化性樹脂組成物及びそれを用いた硬化樹脂層 | |
| WO2022203067A1 (ja) | 硬化性樹脂組成物及びそれを用いた硬化樹脂層 | |
| JP2023104378A (ja) | 構成体 | |
| JP2023086574A (ja) | 硬化樹脂層 | |
| WO2021193889A1 (ja) | 透明導電フィルム用積層体、透明導電フィルム、及び、透明導電フィルムの製造方法 | |
| JP2023151639A (ja) | 積層体、透明導電フィルム、及び、透明導電フィルムの製造方法 | |
| JP2023114821A (ja) | 光学フィルム | |
| JP2023131120A (ja) | コーティング接着性が改善されたポリアミド系フィルムおよびその製造方法 | |
| JP2022071695A (ja) | フィルム積層体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22775867 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2023509354 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280024366.5 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22775867 Country of ref document: EP Kind code of ref document: A1 |