WO2021106912A1 - 樹脂発泡体 - Google Patents
樹脂発泡体 Download PDFInfo
- Publication number
- WO2021106912A1 WO2021106912A1 PCT/JP2020/043761 JP2020043761W WO2021106912A1 WO 2021106912 A1 WO2021106912 A1 WO 2021106912A1 JP 2020043761 W JP2020043761 W JP 2020043761W WO 2021106912 A1 WO2021106912 A1 WO 2021106912A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin foam
- resin
- thickness
- polyolefin
- compression
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
- C08J9/122—Hydrogen, oxygen, CO2, nitrogen or noble gases
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/32—Layered products comprising a layer of synthetic resin comprising polyolefins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/18—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by features of a layer of foamed material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0014—Use of organic additives
- C08J9/0023—Use of organic additives containing oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0061—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof characterized by the use of several polymeric components
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0066—Use of inorganic compounding ingredients
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0095—Mixtures of at least two compounding ingredients belonging to different one-dot groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/20—Adhesives in the form of films or foils characterised by their carriers
- C09J7/22—Plastics; Metallised plastics
- C09J7/26—Porous or cellular plastics
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/10—Homopolymers or copolymers of propene
- C08J2323/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2423/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2423/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2423/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2423/10—Homopolymers or copolymers of propene
- C08J2423/12—Polypropene
Definitions
- the present invention relates to a resin foam.
- Resin foam is often used as a cushioning material for screen protection of electronic devices and protection of substrates.
- the cushion material is required to exhibit high shock absorption even if it is arranged in a place having a narrow clearance, in accordance with the tendency of electronic devices to become thinner.
- dust resistance may be required to protect the peripheral members.
- Cushion materials are often repeatedly compressed, and the resulting thinning contributes to a decrease in dust resistance.
- the cushion material may be repeatedly compressed due to a device falling, vibrating, touching the screen, or the like, and as a result, the thickness of the cushion material may become thin. When the thickness of the cushion material becomes thin, a gap is formed in the peripheral member, and the dustproof property is lowered.
- Patent Document 1 discloses a foam having excellent waterproof properties, but does not disclose or suggest any provision of dust resistance. Further, although Patent Document 2 discloses a foam having excellent dust resistance, no disclosure or suggestion is made regarding the dust resistance after repeated compression.
- An object of the present invention is to provide a resin foam which is thin, has excellent shock absorption, and has excellent dust resistance.
- the resin foam of the present invention has a 50% compression load of 5 N / cm 2 or more, a thickness change rate of 10% or less due to repeated compression measured in the following test, and has a bubble structure.
- ⁇ Repeated compression test> A disk jig with a flat tip (contact area: 4.9 cm 2 ) is pressed against the resin foam, and the rate of change in thickness when 50% compression / 10% compression (thickness standard) cycle is performed 5000 times is measured.
- the resin foam has a 50% compression load of 30 N / cm 2 or less.
- the resin foam has a compressive elastic modulus of 200 kPa or more.
- the resin foam has a non-foam bending stress of 5 MPa or more.
- the resin foam has an apparent density of 0.02 g / cm 3 to 0.50 g / cm 3 . In one embodiment, the resin foam has a thickness recovery rate of 70% or more. In one embodiment, the aspect ratio of bubbles contained in the resin foam is 1.5 or more. In one embodiment, the resin foam has an average cell diameter of 10 ⁇ m to 200 ⁇ m. In one embodiment, the resin foam has a bubble ratio of 30% or more. In one embodiment, the resin foam has a coefficient of variation of bubble diameter of 0.5 or less. In one embodiment, the resin foam has a bubble wall thickness of 0.1 ⁇ m to 10 ⁇ m. In one embodiment, the resin foam comprises a polyolefin-based resin.
- the polyolefin-based resin is a mixture of a polyolefin other than the polyolefin-based elastomer and a polyolefin-based elastomer.
- the resin foam has a hot melt layer on one or both sides.
- a foamed member is provided. This foamed member has a resin foamed layer and an adhesive layer arranged on at least one side of the resin foamed layer, and the resin foamed layer is the resin foam.
- the resin foam of the present invention has a 50% compression load of 5 N / cm 2 or more and a thickness change rate of 28% or less due to repeated compression.
- the resin foam of the present invention has a bubble structure (cell structure).
- the cell structure (cell structure) include a closed cell structure, an open cell structure, and a semi-continuous semi-closed cell structure (a bubble structure in which a closed cell structure and an open cell structure are mixed).
- the cell structure of the resin foam is preferably a continuous cell structure or a semi-continuous semi-closed cell structure, and more preferably a semi-continuous semi-closed cell structure.
- the resin foam of the present invention is obtained by foaming the resin composition.
- the resin composition is a composition containing at least the resin constituting the resin foam.
- the 50% compressive load of the resin foam of the present invention is 5 N / cm 2 or more. Within such a range, it is possible to obtain a resin foam having an appropriate hardness and less likely to undergo compressive deformation even when subjected to a high impact force. Such a resin foam is hard to be crushed, can prevent a gap with a peripheral member when used as a cushioning material, and is excellent in dust resistance.
- the 50% compressive load of the resin foam is preferably 7 N / cm 2 or more, more preferably larger than 9 N / cm 2 , and further preferably 10 N / cm 2 or more. Within such a range, the above effect becomes remarkable.
- the upper limit of the 50% compressive load of the resin foam is preferably 50 N / cm 2, more preferably 40N / cm 2, more preferably from 30 N / cm 2, particularly preferably at 25 N / cm 2 ..
- the repulsive force when applied to a gap can be appropriately suppressed, and an adverse effect on peripheral members can be prevented.
- the thickness change rate of the resin foam of the present invention due to repeated compression is 10% or less. Within such a range, thinning due to crushing is unlikely to occur, gaps with peripheral members can be prevented when used as a cushioning material, and a resin foam having excellent dust resistance can be obtained.
- the thickness change rate due to repeated compression of the resin foam is more preferably 8% or less, further preferably 5% or less, particularly preferably 3% or less, and most preferably 1% or less. Within such a range, the above effect becomes remarkable.
- the smaller the rate of change in thickness due to repeated compression of the resin foam the more preferable, but the lower limit thereof is, for example, 0.5% (preferably 0.3%, more preferably 0.1%, still more preferably 0.05%). Is.
- the rate of change in thickness due to repeated compression of the resin foam is such that a disk jig with a flat tip (contact area: 4.9 cm 2 ) is pressed against the resin foam, and 50% compression / 10% compression (thickness standard) cycles are performed 5000 times. It means the rate of change in thickness when performed.
- the rate of change is calculated by the formula ⁇ (thickness before test)-(thickness after test) ⁇ / (thickness before test) x 100.
- the resin foam of the present invention has a feature of being excellent in shock absorption while having a predetermined hardness by setting the thickness change rate due to a 50% compression load and repeated compression within the above range. Therefore, it is unlikely to be crushed even when it receives a high impact force, the thickness is maintained for a long period of time, and it is excellent in dust resistance.
- the apparent density of the resin foam is preferably 0.02 g / cm 3 to 0.5 g / cm 3 . Within such a range, a resin foam having excellent flexibility and stress dispersibility can be obtained.
- the apparent density of the resin foam is more preferably 0.04 g / cm 3 to 0.4 g / cm 3 , still more preferably 0.06 g / cm 3 to 0.3 g / cm 3, and even more preferably 0.06 g / cm 3 to 0.3 g / cm 3. It is 0.08 g / cm 3 to 0.25 g / cm 3 , particularly preferably 0.1 g / cm 3 to 0.2 g / cm 3 , and most preferably 0.13 g / cm 3 to 0.2 g / cm. It is 3. Within such a range, the above effect becomes remarkable. The method for measuring the apparent density will be described later.
- the 25% compressive load of the resin foam is preferably 3 N / cm 2 or more. Within such a range, the effect of the present invention becomes remarkable.
- 25% compressive load of the resin foam is preferably 3.2 N / cm 2 or more, more preferably 3.5 N / cm 2 or more, still more preferably 4N / cm 2 or more.
- the upper limit of the 25% compressive load of the resin foam is, for example, 10 N / cm 2 (preferably 20 N / cm 2 ).
- the method for measuring the 25% compressive load conforms to the method for measuring the compressive hardness of the foam described in JIS K 6767.
- the compressive elastic modulus of the resin foam is preferably 200 kPa or more. Within such a range, it is possible to obtain a resin foam that is less likely to undergo compressive deformation even when subjected to a high impact force and is not easily crushed.
- the compressive elastic modulus of the resin foam is preferably 220 kPa or more, more preferably 250 kPa or more, and further preferably 300 kPa or more. Within such a range, the above effect becomes remarkable.
- the upper limit of the compressive elastic modulus of the resin foam is preferably 1000 kPa, more preferably 800 kPa, and even more preferably 600 kPa.
- the compressibility of the resin foam is determined by measuring the compressibility (%) and compressibility (kPa) of the resin foam based on the section of the compression test of JIS K 6767, and the compressibility when the compressibility is 5%. Read F (5%) and the repulsive force F (15%) at the time of compression when the compression ratio is 15%, and from F (5%) and F (15%), (F (15%) -F (5). %))) / (15-5) ⁇ 100 is calculated by the formula.
- the elastic strain energy of the resin foam during compression is preferably 10 kPa or more.
- "Elastic strain energy during compression” means the total amount of compression repulsive force when the resin foam is compressed by 10%. Specifically, the “elastic strain energy during compression” was determined by a compression test (test temperature: 23 ° C., sample size: 10 mm x 10 mm, compression rate: 10 mm / min) according to JIS K 6767. When the compression rate (%) and the compression repulsive force (kPa) are measured, it is obtained from the compression ss curve with the x-axis as the compression rate (%) and the y-axis as the compression repulsive force (kPa), and the compression rate is 0%.
- the elastic strain energy of the resin foam during compression is more preferably 20 kPa or more, further preferably 30 kPa or more, still more preferably 50 kPa or more, still more preferably 60 kPa or more, still more preferably 80 kPa or more.
- the upper limit of the elastic strain energy at the time of compression of the resin foam is, for example, 500 kPa (preferably 800 kPa).
- the non-foaming bending stress of the resin foam is preferably 5 MPa or more, more preferably 6 MPa or more, further preferably 9 MPa or more, and particularly preferably 10 MPa or more.
- a large amount of energy is required to deform the bubble wall (cell wall) of the resin foam, and a resin foam having excellent shock absorption can be obtained.
- the above-mentioned resin foam makes it possible to improve impact resistance (impact absorption) by appropriately reducing the flexibility, which has been regarded as important in cushioning materials, and balancing it with other characteristics.
- the resin foam that exhibits shock absorption due to an action different from the conventional one is particularly useful in a configuration in which the impact propagates in a narrow range (a configuration in which the impact does not easily spread in the plane direction). It is particularly useful when applied to flexible members (eg, members made of resin).
- the upper limit of the non-foam bending stress is preferably 20 MPa, more preferably 15 MPa. Within such a range, a resin foam having more excellent flexibility and stress dispersibility can be obtained.
- the “non-foaming bending stress” means the bending stress of the resin molded product a that has been returned to the non-foaming state (bulk) without bubbles by heat-pressing the resin foam.
- the density of the resin molded body a can be made equal to the density of the resin molded body b before foaming formed by the resin composition described later.
- the bending stress of the resin molded body a (non-foamed bending stress of the resin foam) can be the same as that of the resin molded body b. The method for measuring the bending stress will be described later.
- the impact absorption rate of the resin foam is preferably 25% or more, more preferably 30% or more, still more preferably 35% or more.
- the shock absorption rate is measured as follows. -A resin foam, double-sided tape (product number: No. 5603W, manufactured by Nitto Denko), and PET film (product number: diamond foil MRF75, manufactured by Mitsubishi resin) were arranged in this order on the impact force sensor to form a test body. From a height of 50 cm above the PET film, 66 g of an iron ball is dropped onto the test piece, and the impact force F1 is measured. -In addition, the impact force F0 of the blank is measured by dropping the iron ball directly onto the impact force sensor as described above. -From F1 and F0, the shock absorption rate (%) is calculated by the formula (F0-F1) / F0 ⁇ 100.
- the thickness recovery rate of the resin foam is preferably 70% or more, more preferably 75% or more, and further preferably 80% or more.
- the thickness recovery rate of the foamed layer is defined by the following formula.
- the thickness recovery rate of this foam layer is a recovery rate measured by applying a load to a foam sheet with a certain area and compressing it, and is a so-called dent recovery rate measured by locally applying a load and denting only a part of the foam sheet. Is different.
- Thickness recovery rate (%) ⁇ (thickness 0.5 seconds after decompression) / (initial thickness) ⁇ x 100
- Initial thickness The thickness of the resin foam before applying a load.
- Thickness 0.5 seconds after decompression Maintained for 120 seconds with a load of 1000 g / cm 2 applied to the resin foam, decompressed, 0.5 seconds after decompression Thickness of resin foam.
- the tensile elastic modulus of the resin foam at 23 ° C. is preferably 0.6 MPa or more, more preferably 0.7 MPa to 5 MPa, and even more preferably 1 MPa to 4 MPa. Within such a range, it is possible to obtain a resin foam having excellent stress dispersibility and capable of exhibiting excellent shock absorption even in a thin film.
- the tensile elastic modulus is obtained by fixing a sample (size: 10 mm ⁇ 80 mm) at a distance between chucks of 40 mm and performing a tensile test at a tensile speed of 500 mm / min to obtain a tensile strain-tensile strength curve, which is the origin of this curve. It is obtained from the slope of a straight line connecting the tensile strength when the tensile strain is 10%.
- the elongation at break of the resin foam at 25 ° C. is preferably 120% or less, more preferably 110% or less, further preferably 100% or less, and particularly preferably 90% or less. Within such a range, a resin foam having excellent flexibility and stress dispersibility can be obtained. If the elongation at break is small, the deformation of the cell wall of the resin foam becomes small when a load is applied to the resin foam. For example, when a filler is added, the resin foam constitutes the resin foam. Slip is likely to occur at the interface between the resin and the filler, and the load can be further relaxed. On the other hand, if the breaking elongation is too large, the deformation of the cell wall of the resin foam becomes large, and it may be difficult to relax the load. The elongation at break can be measured according to JIS K 6767.
- the stress retention rate of the resin foam is preferably 60% or more, more preferably 63% to 100%, and further preferably 63% to 95%. Within such a range, it is possible to obtain a resin foam having excellent stress dispersibility and capable of exhibiting excellent shock absorption even in a thin film.
- the stress retention rate refers to the tensile strength immediately after stretching and 12 hours after stretching by stretching a resin foam (width 10 mm ⁇ length 100 mm) at a rate of 300 m / min in the length direction by 20%. The ratio to the tensile strength after holding (tensile strength after 12 hours / tensile strength immediately after stretching ⁇ 100).
- the thickness of the resin foam is preferably 30 ⁇ m to 5000 ⁇ m, more preferably 35 ⁇ m to 4000 ⁇ m, further preferably 40 ⁇ m to 3000 ⁇ m, further preferably 45 ⁇ m to 2000 ⁇ m, and particularly preferably 50 ⁇ m to 1000 ⁇ m. Most preferably, it is 55 ⁇ m to 500 ⁇ m.
- the resin foam of the present invention exhibits excellent impact resistance even though it is a thin layer. Further, when the thickness of the resin foam is within the above range, a fine and uniform bubble structure can be formed, which is advantageous in that excellent shock absorption can be exhibited.
- the average cell diameter (average cell diameter) of the resin foam is preferably 10 ⁇ m to 200 ⁇ m, more preferably 15 ⁇ m to 180 ⁇ m, still more preferably 20 ⁇ m to 150 ⁇ m, still more preferably 23 ⁇ m to 120 ⁇ m. It is particularly preferably 25 ⁇ m to 100 ⁇ m, and most preferably 30 ⁇ m to 80 ⁇ m. Within such a range, it is possible to obtain a resin foam that can exhibit high shock absorption without being excessively deformed when subjected to a shock. Further, it can be a resin foam having an appropriate hardness and suitable as a cushioning material for filling a gap. The method for measuring the average cell diameter will be described later.
- the aspect ratio of bubbles contained in the resin foam is preferably 1.5 or more. Within such a range, a resin foam having excellent thickness recovery can be obtained.
- the aspect ratio of the bubbles constituting the resin foam is preferably 2.0 or more, and more preferably 2.5 or more. Within such a range, the above effect becomes remarkable.
- the upper limit of the aspect ratio of the bubbles constituting the resin foam is preferably 5, more preferably 4, and even more preferably 3.5. Within such a range, a resin foam having excellent shock absorption can be obtained.
- the "aspect ratio of bubbles contained in the resin foam” is the aspect ratio of individual bubbles existing in a predetermined area (3 mm 2) in the cross section of the resin foam at randomly selected locations. Means the average value.
- the specific method for obtaining the "aspect ratio of bubbles contained in the resin foam” is as follows. -The resin foam is cut using a razor blade in the TD (direction orthogonal to the flow direction) and in the direction perpendicular to the main surface of the resin foam (thickness direction), and the cut surface is cut with a microscope (for example, in the thickness direction). Using a Keyence "VHX-2000”) , observe a predetermined area (3 mm 2 ) range at a magnification of 100 times.
- the length of one bubble in the thickness direction and the length of TD are measured. -Similar measurement is performed for all bubbles existing in a predetermined area. -The aspect ratio of bubbles is calculated by the length in the TD direction ⁇ the length in the thickness direction, the same calculation is performed for all bubbles, and the average value is defined as the "aspect ratio of bubbles possessed by the resin foam".
- the coefficient of variation of the bubble diameter (cell diameter) of the resin foam is preferably 0.5 or less, more preferably 0.48 or less, still more preferably 0.45 or less, and particularly preferably 0. It is 43 or less, and most preferably less than 0.4. Within such a range, it is possible to obtain a resin foam in which deformation due to impact becomes uniform, local stress load is prevented, stress dispersibility is excellent, and impact resistance is particularly excellent.
- the bubble ratio (cell ratio) of the resin foam is preferably 30% or more, more preferably 50% or more. Within such a range, a resin foam having a small repulsive stress during compression can be obtained. Such a resin foam can reduce the stress applied to other members when the resin foam is applied by compressing it to a place having a narrow clearance. For example, when the resin foam is applied to the display member, the stress applied to the display member can be relaxed and dispersed, which is useful from the viewpoint of reducing color unevenness and protecting the member.
- the upper limit of the bubble ratio is preferably 99% or less, more preferably 95% or less, and further preferably 90% or less. Within such a range, a resin foam having an appropriate hardness and a small amount of deformation when subjected to an impact can be obtained, and therefore a resin foam having high impact absorption can be obtained. The method for measuring the bubble ratio will be described later.
- the thickness of the bubble wall (cell wall) of the resin foam is preferably 0.1 ⁇ m to 10 ⁇ m, more preferably 0.3 ⁇ m to 8 ⁇ m, still more preferably 0.5 ⁇ m to 5 ⁇ m, and particularly preferably. It is 0.7 ⁇ m to 4 ⁇ m, most preferably 1 ⁇ m to 3 ⁇ m. Within such a range, it is possible to obtain a resin foam that can exhibit high shock absorption without being excessively deformed when subjected to a shock. Further, it can be a resin foam having an appropriate hardness and suitable as a cushioning material for filling a gap. The method for measuring the thickness of the bubble wall will be described later.
- the proportion of the closed cell structure in the cell is preferably 40% or less, more preferably 30% or less. Within such a range, a resin foam having an appropriate hardness and suitable as a cushioning material for filling a gap can be obtained.
- the self-foaming ratio of the resin foam is, for example, in an environment of a temperature of 23 ° C. and a humidity of 50%, the object to be measured is submerged in water, the mass thereof is measured thereafter, and then in an oven at 80 ° C. After sufficiently drying, the mass is measured again to obtain the determination. Further, since open cells can retain water, the mass thereof can be measured and obtained as open cells.
- any appropriate shape can be adopted depending on the purpose.
- Such a shape is typically a sheet shape.
- the resin foam may have a hot melt layer on one side or both sides thereof.
- the resin foam having the hot melt layer is, for example, a resin foam (or a precursor of the resin foam) using a pair of heating rolls heated to a temperature equal to or higher than the melting temperature of the resin composition constituting the resin foam. Can be obtained by rolling.
- the resin foam can be formed by any suitable method as long as the effects of the present invention are not impaired.
- a typical example of such a method is a method of foaming a resin composition containing a resin material (polymer).
- the resin foam of the present invention can be typically obtained by foaming a resin composition.
- the resin composition comprises any suitable resin material (polymer).
- polymer examples include acrylic resin, silicone resin, urethane resin, polyolefin resin, ester resin, rubber resin and the like.
- the polymer may be used alone or in combination of two or more.
- the content ratio of the polymer is preferably 30 parts by weight to 95 parts by weight, more preferably 35 parts by weight to 90 parts by weight, and further preferably 40 parts by weight to 80 parts by weight with respect to 100 parts by weight of the resin composition. It is a part, particularly preferably 40 parts by weight to 60 parts by weight. Within such a range, a resin foam having more excellent flexibility and stress dispersibility can be obtained.
- a polyolefin resin is used as the polymer.
- the content ratio of the polyolefin resin is preferably 50 parts by weight to 100 parts by weight, more preferably 70 parts by weight to 100 parts by weight, and further preferably 90 parts by weight to 100 parts by weight with respect to 100 parts by weight of the polymer. It is a part by weight, particularly preferably 95 parts by weight to 100 parts by weight, and most preferably 100 parts by weight.
- the polyolefin-based resin preferably includes at least one selected from the group consisting of polyolefins and polyolefin-based elastomers, and more preferably, polyolefins and polyolefin-based elastomers are used in combination.
- polyolefins and polyolefin-based elastomers are used in combination.
- the polyylene and the polyolefin-based elastomer only one type may be used alone, or two or more types may be used in combination.
- the term "polyolefin” does not include "polyolefin-based elastomer”.
- the weight ratio of the polyolefin and the polyolefin-based elastomer is preferably 1/99 to 99/1, more preferably 10/90 to It is 90/10, more preferably 20/80 to 80/20, and particularly preferably 30/70 to 70/30. Within such a range, the effect of the present invention becomes remarkable.
- any suitable polyolefin can be adopted as long as the effect of the present invention is not impaired.
- examples of such polyolefins include linear polyolefins and branched-chain (branched-chain) polyolefins.
- a branched-chain polyolefin is used as the polyolefin-based resin.
- the polyolefin only the branched polyolefin may be used, or the branched polyolefin and the linear polyolefin may be used in combination.
- the content ratio of the branched polyolefin is preferably 30 parts by weight to 100 parts by weight, and more preferably 80 parts by weight to 120 parts by weight with respect to 100 parts by weight of the polyolefin.
- polystyrene resin examples include polymers containing a structural unit derived from ⁇ -olefin.
- the polyolefin may be composed of only a structural unit derived from an ⁇ -olefin, or may be composed of a structural unit derived from an ⁇ -olefin and a structural unit derived from a monomer other than the ⁇ -olefin.
- any suitable copolymerization form can be adopted as the copolymerization form thereof. For example, random copolymers, block copolymers and the like can be mentioned.
- ⁇ -olefins examples include ⁇ -olefins having 2 to 8 carbon atoms (preferably 2 to 6, more preferably 2 to 4) (for example, ethylene, propylene, 1-butene, 1-pentene). , 1-Hexene, 4-methyl-1-pentene, 1-heptene, 1-octene, etc.) are preferred.
- the ⁇ -olefin may be only one kind or two or more kinds.
- Examples of the monomer other than the ⁇ -olefin constituting the polyolefin include ethylenically unsaturated monomers such as vinyl acetate, acrylic acid, acrylic acid ester, methacrylic acid, methacrylic acid ester, and vinyl alcohol.
- the monomer other than the ⁇ -olefin may be only one kind or two or more kinds.
- polystyrene resin examples include low-density polyethylene, medium-density polyethylene, high-density polyethylene, linear low-density polyethylene, polypropylene (propylene homopolymer), a copolymer of ethylene and propylene, and ethylene and other than ethylene.
- Examples include a copolymer with a body.
- a polypropylene-based polymer having a propylene-derived structural unit is used as the polyolefin.
- the polypropylene-based polymer include polypropylene (propylene homopolymer), a copolymer of ethylene and propylene, a copolymer of propylene and an ⁇ -olefin other than propylene, and preferably polypropylene (propylene homopolymer). ).
- the polypropylene-based polymer only one type may be used alone, or two or more types may be used in combination.
- the melt flow rate (MFR) of the polyolefin at a temperature of 230 ° C. is preferably 0.2 g / 10 min to 10 g / 10 min, and more preferably 0.25 g / 10 in that the effects of the present invention can be more exhibited. Minutes to 5 g / 10 minutes, more preferably 0.3 g / 10 minutes to 3 g / 10 minutes, and particularly preferably 0.35 g / 10 minutes to 1.5 g / 10 minutes.
- the melt flow rate (MFR) refers to an MFR measured at a temperature of 230 ° C. and a load of 2.16 kgf based on ISO1133 (JIS-K-7210).
- melt flow rate (MFR) at a temperature of 230 ° C. is preferably 0.2 g / 10 minutes or more and less than 0.7 g / 10 minutes (more preferably 0.2 g / 10 minutes to 0.65 g / 10 minutes).
- the polyolefin and melt flow rate (MFR) at a temperature of 230 ° C. are preferably 0.7 g / 10 min to 10 g / 10 min (more preferably 0.7 g / 10 min to 5 g / 10 min, and even more preferably 0.
- the melt flow rate (MFR) at a temperature of 230 ° C. is preferably 0.
- the polyolefin of 2 g / 10 minutes or more and less than 0.7 g / 10 minutes (more preferably 0.2 g / 10 minutes to 0.65 g / 10 minutes) and the melt flow rate (MFR) at a temperature of 230 ° C. are preferably 0.7 g.
- the weight ratio of 10 minutes to 1.5 g / 10 minutes, most preferably 0.7 g / 10 minutes to 1.3 g / 10 minutes) with the polyolefin is preferably 1/99 to 99/1. , More preferably 10/90 to 90/10, further preferably 20/80 to 80/20, particularly preferably 30/70 to 70/30, and most preferably 40/60 to 60/40. Is.
- polystyrene resin As the polyolefin, a commercially available product may be used, for example, “E110G” (manufactured by Prime Polymer Co., Ltd.), “EA9” (manufactured by Japan Polypropylene Corporation), “EA9FT” (manufactured by Japan Polypropylene Corporation), “E-”. Examples thereof include “185G” (manufactured by Prime Polymer Co., Ltd.), "WB140HMS” (manufactured by Borealis), and "WB135HMS” (manufactured by Borealis).
- any suitable polyolefin-based elastomer can be adopted as long as the effects of the present invention are not impaired.
- examples of such polyolefin-based elastomers include ethylene-propylene copolymers, ethylene-propylene-diene copolymers, ethylene-vinyl acetate copolymers, polybutene, polyisobutylene, chlorinated polyethylene, and polyolefin components and rubber components.
- thermoplastic olefin-based elastomer such as an elastomer in which is physically dispersed, or an elastomer having a structure in which a polyolefin component and a rubber component are microphase-separated; a resin component A (olefin) forming a matrix.
- a mixture containing a system resin component A) and a rubber component B forming a domain is dynamically heat-treated in the presence of a cross-linking agent, and cross-linked rubber particles are contained in the resin component A which is a matrix (sea phase).
- TPV dynamically crosslinked thermoplastic olefin-based elastomer
- TPV dynamically crosslinked thermoplastic olefin-based elastomer
- the polyolefin-based elastomer preferably contains a rubber component.
- rubber components include JP-A-08-302111, JP-A-2010-241934, JP-A-2008-024882, JP-A-2000-007858, JP-A-2006-052277, and JP-A.
- Examples thereof include those described in Japanese Patent Application Laid-Open No. 2012-072306, Japanese Patent Application Laid-Open No. 2012-057068, Japanese Patent Application Laid-Open No. 2010-2418997, Japanese Patent Application Laid-Open No. 2009-067969, and Japanese Patent Application Laid-Open No. 03/002654.
- the elastomer having a structure in which the polyolefin component and the olefin rubber component are microphase-separated include polypropylene resin (PP) and an elastomer composed of polypropylene resin (PP) and ethylene-propylene rubber (EPM). Examples thereof include an elastomer composed of ethylene-propylene-diene rubber (EPDM).
- the weight ratio of the polyolefin component and the olefin rubber component is preferably 90/10 to 10/90, and more preferably 80/20 to 20/80.
- the dynamically crosslinked thermoplastic olefin elastomer generally has a higher elastic modulus and a smaller compression set than the non-crosslinked thermoplastic olefin elastomer (TPO). As a result, the recoverability is good, and when a foam is used, excellent recoverability can be exhibited.
- the dynamically crosslinked thermoplastic olefin elastomer is a cross-linking agent containing a mixture containing a resin component A (olefin resin component A) forming a matrix and a rubber component B forming a domain. It is a multiphase polymer having a sea-island structure in which crosslinked rubber particles are finely dispersed as domains (island phases) in the resin component A which is a matrix (sea phase) obtained by dynamically heat-treating in the presence. ..
- thermoplastic olefin elastomer examples include JP-A-2000-007858, JP-A-2006-052277, JP-A-2012-072306, JP-A-2012-057068, and Japanese Patent Application Laid-Open No. 2012-057068. Examples thereof include those described in Japanese Patent Application Laid-Open No. 2010-2418997, Japanese Patent Application Laid-Open No. 2009-067969, and Re-Table 03/002654.
- thermoplastic olefin elastomer a commercially available product may be used, for example, “Zeotherm” (manufactured by Zeon Corporation), “Thermoran” (manufactured by Mitsubishi Chemical Corporation), “Sarlink 3245D”. (Manufactured by Toyobo Corporation) and the like.
- the melt flow rate (MFR) of the polyolefin-based elastomer at a temperature of 230 ° C. is preferably 2 g / 10 min to 15 g / 10 min, more preferably 3 g / 10 min to 10 g / 10 min, and even more preferably 3. It is 5 g / 10 minutes to 9 g / 10 minutes, particularly preferably 4 g / 10 minutes to 8 g / 10 minutes, and most preferably 4.5 g / 10 minutes to 7.5 g / 10 minutes.
- the melt tension (190 ° C., at break) of the polyolefin-based elastomer is preferably less than 10 cN, and more preferably 5 cN to 9.5 cN.
- the JIS A hardness of the polyolefin-based elastomer is preferably 30 ° to 95 °, more preferably 35 ° to 90 °, further preferably 40 ° to 88 °, and particularly preferably 45 ° to 85 °. Yes, most preferably 50 ° to 83 °.
- the JIS A hardness is measured based on ISO7619 (JIS K6253).
- the resin foam (ie, the resin composition) may further comprise a filler.
- a filler By containing the filler, it is possible to form a resin foam that requires a large amount of energy to deform the bubble wall (cell wall), and the resin foam exhibits excellent shock absorption. Further, by containing the filler, a fine and uniform bubble structure can be formed, which is also advantageous in that excellent shock absorption can be exhibited.
- the filler only one kind may be used alone, or two or more kinds may be used in combination.
- the content ratio of the filler is preferably 10 parts by weight to 150 parts by weight, more preferably 30 parts by weight to 130 parts by weight, still more preferably, with respect to 100 parts by weight of the polymer constituting the resin foam. It is 50 parts by weight to 100 parts by weight. Within such a range, the above effect becomes remarkable.
- the filler is an inorganic substance.
- the material constituting the filler which is an inorganic substance include aluminum hydroxide, magnesium hydroxide, calcium carbonate, magnesium carbonate, calcium silicate, magnesium silicate, calcium oxide, magnesium oxide, aluminum oxide, aluminum nitride, and boric acid.
- examples thereof include aluminum whisker, silicon nitride, boron nitride, crystalline silica, amorphous silica, metals (for example, gold, silver, copper, aluminum, nickel), carbon, graphite and the like.
- the filler is organic.
- the material constituting the filler which is an organic substance include polymethyl methacrylate (PMMA), polyimide, polyamideimide, polyetheretherketone, polyetherimide, polyesterimide and the like.
- a flame retardant may be used as the filler.
- the flame retardant include a bromine-based flame retardant, a chlorine-based flame retardant, a phosphorus-based flame retardant, an antimony-based flame retardant, and the like.
- a non-halogen-non-antimony flame retardant is used.
- non-halogen-non-antimony flame retardant examples include compounds containing aluminum, magnesium, calcium, nickel, cobalt, tin, zinc, copper, iron, titanium, boron and the like.
- examples of such a compound (inorganic compound) include hydrated metal compounds such as aluminum hydroxide, magnesium hydroxide, magnesium oxide / nickel oxide hydrate, and magnesium oxide / zinc oxide hydrate. ..
- the above-mentioned filler may be subjected to any appropriate surface treatment.
- the surface treatment include a silane coupling treatment and a stearic acid treatment.
- the bulk density of the filler is preferably 0.8 g / cm 3 or less, more preferably 0.6 g / cm 3 or less, still more preferably 0.4 g / cm 3 or less, and particularly preferably 0. .3 g / cm 3 or less.
- the filler can be contained with good dispersibility, and the effect of adding the filler can be sufficiently exhibited while reducing the content of the filler.
- a resin foam having a low filler content is advantageous in that it is highly foamed, flexible, and has excellent stress dispersibility and appearance.
- the lower limit of the bulk density of the filler is, for example, 0.01 g / cm 3, preferably 0.05 g / cm 3, more preferably 0.1 g / cm 3.
- the number average particle size (primary particle size) of the filler is preferably 5 ⁇ m or less, more preferably 3 ⁇ m or less, and further preferably 1 ⁇ m or less. Within such a range, the filler can be contained with good dispersibility and a uniform bubble structure can be formed. As a result, a resin foam having excellent stress dispersibility and appearance can be obtained.
- the lower limit of the number average particle size of the filler is, for example, 0.1 ⁇ m.
- the number average particle size of the filler can be measured using a particle size distribution meter (MicrotracII, Microtrac Bell Co., Ltd.) using a suspension prepared by mixing 100 g of water with 1 g of the filler as a sample. ..
- the specific surface area of the filler is preferably 2 m 2 / g or more, more preferably 4 m 2 / g or more, and further preferably 6 m 2 / g or more. Within such a range, the filler can be contained with good dispersibility and a uniform bubble structure can be formed. As a result, a resin foam having excellent stress dispersibility and appearance can be obtained.
- the upper limit of the specific surface area of the filler is, for example, 20 m 2 / g.
- the specific surface area of the filler can be measured by the BET method, that is, by adsorbing a molecule having a known adsorption area to the surface of the filler at a low temperature of liquid nitrogen and measuring the adsorption amount.
- the resin composition may contain any suitable other components as long as the effects of the present invention are not impaired.
- Such other components may be only one kind or two or more kinds.
- Such other components include, for example, rubber, resins other than polymers blended as resin materials, softeners, aliphatic compounds, antioxidants, antioxidants, light stabilizers, weather resistant agents, and ultraviolet absorption.
- Agents dispersants, plastics, carbons, antistatic agents, surfactants, cross-linking agents, thickeners, rust preventives, silicone compounds, tension modifiers, shrinkage inhibitors, fluidity modifiers, gelling Agents, hardeners, reinforcing agents, foaming agents, foaming nucleating agents, coloring agents (pigments, dyes, etc.), pH adjusting agents, solvents (organic solvents), thermal polymerization initiators, photopolymerization initiators, lubricants, crystal nucleating agents, Examples thereof include crystallization accelerators, solubilizers, surface treatment agents, and dispersion aids.
- the resin foam of the present invention is typically obtained by foaming a resin composition.
- foaming method bubble forming method
- a method usually used for foam molding such as a physical method or a chemical method
- the resin foam may be typically a foam (physical foam) formed by foaming by a physical method, or a foam formed by foaming by a chemical method (physical foam). It may be a chemical foam).
- the physical method is generally one in which a gas component such as air or nitrogen is dispersed in a polymer solution to form bubbles by mechanical mixing (mechanical foam).
- the chemical method is generally a method of forming a cell by a gas generated by thermal decomposition of a foaming agent added to a polymer base to obtain a foam.
- the resin composition to be subjected to foam molding can be, for example, a component of any suitable melt-kneading device, for example, an open mixing roll, a non-open Banbury mixer, a single-screw extruder, a twin-screw extruder, or a continuous type. It may be prepared by mixing using any suitable means such as a kneader and a pressure kneader.
- an emulsion resin composition (an emulsion containing a resin material (polymer) or the like) is mechanically foamed to foam (step A).
- the foaming device include a high-speed shearing device, a vibration device, a pressurized gas discharge device, and the like.
- a high-speed shearing device is preferable from the viewpoint of reducing the bubble diameter and producing a large capacity.
- This one embodiment 1 for forming the resin foam is applicable to the formation from any resin composition.
- the solid content concentration of the emulsion is preferably high from the viewpoint of film forming property.
- the solid content concentration of the emulsion is preferably 30% by weight or more, more preferably 40% by weight or more, still more preferably 50% by weight or more.
- the bubbles when foamed by mechanical stirring are those in which a gas is incorporated into the emulsion.
- a gas any suitable gas can be adopted as long as it is inert to the emulsion, as long as the effects of the present invention are not impaired. Examples of such a gas include air, nitrogen, carbon dioxide and the like.
- the resin foam of the present invention can be obtained by subjecting the emulsion resin composition foamed by the above method (bubble-containing emulsion resin composition) to a substrate and drying it (step B). it can.
- the base material include a peel-treated plastic film (peeling-treated polyethylene terephthalate film and the like), a plastic film (polyethylene terephthalate film and the like), and the like.
- Step B any appropriate method can be adopted as the coating method and the drying method as long as the effects of the present invention are not impaired.
- Step B includes a preliminary drying step B1 in which the bubble-containing emulsion resin composition coated on the substrate is dried at 50 ° C. or higher and lower than 125 ° C., and then a main drying step B2 in which the bubble-containing emulsion resin composition is further dried at 125 ° C. or higher and 200 ° C. or lower. It is preferable to have.
- the pre-drying step B1 and the main drying step B2 it is possible to prevent the coalescence of bubbles and the bursting of bubbles due to a rapid temperature rise.
- bubbles are united and burst due to a rapid rise in temperature, so it is of great significance to provide the pre-drying step B1.
- the temperature in the pre-drying step B1 is preferably 50 ° C. to 100 ° C.
- the time of the pre-drying step B1 is preferably 0.5 minutes to 30 minutes, more preferably 1 minute to 15 minutes.
- the temperature in the main drying step B2 is preferably 130 ° C. to 180 ° C. or lower, and more preferably 130 ° C. to 160 ° C.
- the time of the main drying step B2 is preferably 0.5 minutes to 30 minutes, more preferably 1 minute to 15 minutes.
- ⁇ Embodiment 2 for forming a resin foam there is a mode in which the resin composition is foamed with a foaming agent to form a foam.
- a foaming agent those usually used for foam molding can be used, and from the viewpoint of environmental protection and low pollution to the foam to be foamed, it is preferable to use a high-pressure inert gas.
- any suitable inert gas can be adopted as long as it is inert to the resin composition and can be impregnated.
- examples of such an inert gas include carbon dioxide, nitrogen gas, and air. These gases may be mixed and used. Of these, carbon dioxide is preferable from the viewpoint of a large amount of impregnation into the resin material (polymer) and a high impregnation rate.
- the inert gas is preferably in a supercritical state. That is, it is particularly preferable to use carbon dioxide in a supercritical state. In the supercritical state, the solubility of the inert gas in the resin composition is further increased, a high concentration of the inert gas can be mixed, and the inert gas becomes high concentration at the time of a sudden pressure drop. Since the number of nuclei generated increases and the density of bubbles formed by the growth of the bubble nuclei becomes higher than in other states even if the porosity is the same, fine bubbles can be obtained.
- the critical temperature of carbon dioxide is 31 ° C.
- the critical pressure is 7.4 MPa.
- a gas impregnation step of impregnating a resin composition containing a resin material (polymer) with an inert gas under high pressure for example, a gas impregnation step of impregnating a resin composition containing a resin material (polymer) with an inert gas under high pressure, the same.
- a decompression step of lowering the pressure after the step to foam the resin material (polymer) for example, a method of forming the resin material (polymer) through a heating step of growing bubbles by heating if necessary.
- a preformed unfoamed molded product may be impregnated with an inert gas, or the molten resin composition is impregnated with an inert gas under a pressurized state and then subjected to molding when the pressure is reduced.
- You may.
- These steps may be performed by either a batch method or a continuous method. That is, after the resin composition is previously molded into an appropriate shape such as a sheet to form an unfoamed resin molded product, the unfoamed resin molded product is impregnated with a high-pressure gas to release the pressure for foaming. It may be a batch method in which the resin composition is kneaded together with a high-pressure gas under pressure, and the pressure is released at the same time as molding, and molding and foaming may be performed at the same time.
- a resin sheet for foam molding is produced by extruding the resin composition using an extruder such as a single-screw extruder or a twin-screw extruder.
- the resin composition is uniformly kneaded using a kneader equipped with blades such as a roller, a cam, a kneader, and a bambari type, and pressed to a predetermined thickness using a hot plate press or the like.
- a hot plate press or the like.
- the unfoamed resin molded body thus obtained is placed in a high-pressure container, and a high-pressure inert gas (carbon dioxide or the like in a supercritical state) is injected to impregnate the unfoamed resin molded body with the inert gas.
- a high-pressure inert gas carbon dioxide or the like in a supercritical state
- the pressure is released (usually up to atmospheric pressure) to generate bubble nuclei in the resin.
- the bubble nuclei may be grown as they are at room temperature, but in some cases, they may be grown by heating.
- known or conventional methods such as a water bath, an oil bath, a hot roll, a hot air oven, far infrared rays, near infrared rays, and microwaves can be adopted.
- the non-foamed resin molded product to be foamed is not limited to a sheet-like product, and various shapes can be used depending on the intended use. Further, the non-foamed resin molded product to be used for foaming can be produced by extrusion molding, press molding, or other molding method such as injection molding.
- a heating step of growing bubbles by heating may be provided, if necessary. After the bubbles are grown in this way, the shape may be fixed by rapidly cooling with cold water or the like, if necessary. Further, the high-pressure gas may be introduced continuously or discontinuously. Further, in the kneading impregnation step and the molding depressurization step, for example, an extruder or an injection molding machine can be used.
- the heating method for growing the bubble nuclei include any suitable method such as a water bath, an oil bath, a hot roll, a hot air oven, far infrared rays, near infrared rays, and microwaves. Any suitable shape may be adopted as the shape of the foam. Examples of such a shape include a sheet shape, a prismatic shape, a cylindrical shape, and a modified shape.
- the amount of the gas mixed when the resin composition is foam-molded is, for example, preferably 2% by weight to 10% by weight, based on the total amount of the resin composition, in that a highly foamed foam can be obtained. It is preferably 2.5% by weight to 8% by weight, more preferably 3% by weight to 6% by weight.
- the pressure when the resin composition is impregnated with the inert gas can be appropriately selected in consideration of operability and the like.
- a pressure is, for example, preferably 6 MPa or more (for example, 6 MPa to 100 MPa), more preferably 8 MPa or more (for example, 8 MPa to 50 MPa).
- the pressure when carbon dioxide in the supercritical state is used is preferably 7.4 MPa or more from the viewpoint of maintaining the supercritical state of carbon dioxide.
- the pressure is lower than 6 MPa, the bubble growth during foaming is remarkable, the bubble diameter becomes too large, and a preferable average cell diameter (average cell diameter) may not be obtained.
- the temperature in the gas impregnation step varies depending on the inert gas used, the type of component in the resin composition, etc., and can be selected in a wide range. Considering operability and the like, the temperature is preferably 10 ° C to 350 ° C.
- the impregnation temperature is preferably 10 ° C. to 250 ° C., more preferably 40 ° C. to 230 ° C. in the batch type.
- the impregnation temperature is preferably 60 ° C. to 350 ° C. in the continuous type.
- the temperature at the time of impregnation is preferably 32 ° C. or higher, more preferably 40 ° C. or higher, in order to maintain the supercritical state.
- the decompression rate is preferably 5 MPa / sec to 300 MPa / sec in order to obtain uniform fine bubbles.
- the heating temperature in the heating step is preferably 40 ° C. to 250 ° C., more preferably 60 ° C. to 250 ° C.
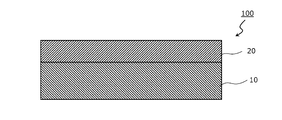
- FIG. 1 is a schematic cross-sectional view of a foaming member according to one embodiment of the present invention.
- the foaming member 100 has a resin foaming layer 10 and an adhesive layer 20 arranged on at least one side of the resin foaming layer 10.
- the resin foam layer 10 is composed of the resin foam.
- the thickness of the pressure-sensitive adhesive layer is preferably 5 ⁇ m to 300 ⁇ m, more preferably 6 ⁇ m to 200 ⁇ m, further preferably 7 ⁇ m to 100 ⁇ m, and particularly preferably 8 ⁇ m to 50 ⁇ m.
- the foamed member can exhibit excellent shock absorption.
- the pressure-sensitive adhesive layer a layer made of any suitable pressure-sensitive adhesive can be adopted.
- the pressure-sensitive adhesive constituting the pressure-sensitive adhesive layer include rubber-based pressure-sensitive adhesives (synthetic rubber-based pressure-sensitive adhesives, natural rubber-based pressure-sensitive adhesives, etc.), urethane-based pressure-sensitive adhesives, acrylic urethane-based pressure-sensitive adhesives, acrylic-based pressure-sensitive adhesives, and silicone-based pressure-sensitive adhesives.
- examples thereof include pressure-sensitive adhesives, polyester-based pressure-sensitive adhesives, polyamide-based pressure-sensitive adhesives, epoxy-based pressure-sensitive adhesives, vinyl alkyl ether-based pressure-sensitive adhesives, fluorine-based pressure-sensitive adhesives, and rubber-based pressure-sensitive adhesives.
- the pressure-sensitive adhesive constituting the pressure-sensitive adhesive layer is preferably at least one selected from an acrylic pressure-sensitive adhesive, a silicone-based pressure-sensitive adhesive, and a rubber-based pressure-sensitive adhesive. Such an adhesive may be only one kind or two or more kinds.
- the pressure-sensitive adhesive layer may be one layer or two or more layers.
- the adhesives are, for example, emulsion type adhesives, solvent type adhesives, ultraviolet crosslinked (UV crosslinked) adhesives, electron beam crosslinked (EB crosslinked) adhesives, and heat melt type adhesives. Agents (hot melt type adhesives) and the like can be mentioned.
- Such an adhesive may be only one kind or two or more kinds.
- Steam moisture permeability of the adhesive layer is preferably not more than 50 (g / (m 2 ⁇ 24 hr)), more preferably 30 (g / (m 2 ⁇ 24 hr)) or less, more preferably 20 (g / (m 2 ⁇ 24 hr)) or less, particularly preferably 10 (g / (m 2 ⁇ 24 hr)) or less.
- the foamed sheet can stabilize the shock absorption without being affected by moisture.
- the water vapor permeability can be measured, for example, by a method according to JIS Z 0208 under test conditions of 40 ° C. and a relative humidity of 92%.
- the pressure-sensitive adhesive constituting the pressure-sensitive adhesive layer may contain any suitable other component as long as the effect of the present invention is not impaired.
- Other components include, for example, other polymer components, softeners, antioxidants, hardeners, plasticizers, fillers, antioxidants, thermal polymerization initiators, photopolymerization initiators, UV absorbers, light stabilizers.
- Colorants pigments, dyes, etc.
- solvents organic solvents
- surfactants eg, ionic surfactants, silicone-based surfactants, fluorine-based surfactants, etc.
- cross-linking agents eg, polyisocyanate-based
- Cross-linking agents silicone-based cross-linking agents, epoxy-based cross-linking agents, alkyl etherified melamine-based cross-linking agents, etc.
- the thermal polymerization initiator and the photopolymerization initiator may be included in the material for forming the polymer component.
- the foamed member can be manufactured by any suitable method.
- the foamed member is manufactured, for example, by a method of laminating a resin foam layer and a pressure-sensitive adhesive layer, or by laminating a material for forming a pressure-sensitive adhesive layer and a resin foam layer and then forming a pressure-sensitive adhesive layer by a curing reaction or the like. How to do it.
- ⁇ Thickness change rate due to repeated compression> Using a micro servo (MMT-250NV-10, manufactured by Shimadzu Corporation), use a disk jig with a flat tip (compression attachment disk type compression jig PC-5040, contact area: 4.9 cm 2 , Imada Co., Ltd.). The jig was repeatedly displaced so as to achieve 50% compression and 10% compression on the foam, and compression was repeated 5000 times (initial load 0.15 N, down speed 5 min / mm, up speed 5 min / mm). The thickness after the test was measured using a digital microscope (trade name "VHX-500", manufactured by KEYENCE CORPORATION). The thickness before the test was Z0, the thickness after the test was Z1, and the rate of change in thickness due to repeated compression was calculated by the formula (Z0-Z1) / Zo ⁇ 100.
- the density (apparent density) of the resin foam was calculated as follows.
- VHX-2000 Keyence's "VHX-2000” was used to observe a predetermined area (3 mm 2 ) range at a magnification of 100 times, and the length of one bubble in the thickness direction and the length of TD were measured. -Similar measurements were made for all bubbles present within a predetermined area. The aspect ratio of the bubbles was calculated by dividing the length of the TD by the length in the thickness direction, and the same calculation was performed for all the bubbles, and the average value was taken as the "aspect ratio of the bubbles of the resin foam".
- ⁇ Bubble ratio (cell ratio)> The measurement was carried out in an environment of a temperature of 23 ° C. and a humidity of 50%. Resin foam obtained in Examples and Comparative Examples with a 100 mm x 100 mm punching blade type (two processing blades (trade name "NCA07", thickness 0.5 mm, cutting edge angle 45 °, manufactured by Nakayama Co., Ltd.)) was punched out, and the dimensions of the punched sample were measured. Further, the thickness was measured with a 1/100 dial gauge having a diameter ( ⁇ ) of 20 mm of the measurement terminal. From these values, the volumes of the resin foams obtained in Examples and Comparative Examples were calculated. Next, the weight of the resin foams obtained in Examples and Comparative Examples was measured with a precision balance having a minimum scale of 0.01 g or more. From these values, the bubble ratio (cell ratio) of the resin foams obtained in Examples and Comparative Examples was calculated.
- ⁇ Thickness of bubble wall (cell wall)> Using a razor blade, the resin foam is cut in the TD (direction orthogonal to the flow direction) and in the direction perpendicular to the main surface of the resin foam (thickness direction), and a digital microscope (commodity) is used as a measuring instrument. Using the name "VHX-500" (manufactured by Keyence Co., Ltd.), an enlarged image of the bubble part of the resin foam is captured, and the image is analyzed using the analysis software of the same measuring instrument to analyze the bubble wall (cell wall). Thickness ( ⁇ m) was determined. The number of bubbles in the captured enlarged image was about 400.
- Non-foam bending stress The resin foam is pressed for 5 minutes at a temperature of (melting point + 70 ° C.) and a pressure of 15 MPa using a vacuum press molding machine (IVM-70: Iwaki Kogyo Co., Ltd.). Got The resin molded body a was cut into a sample having a width of 6 mm and a length of 50 mm, and this sample was placed on a three-point bending tool having a distance between fulcrums of 25 mm. A pushing test (manufactured by Shimadzu Corporation, trade name "AG-Xplus”) was performed at 5 mm / min, and the load (g) when the sample was pushed 5 mm was defined as a non-foam bending stress.
- Thickness recovery rate ⁇ (thickness 0.5 seconds after decompression) / (initial thickness) ⁇ x 100
- Black acrylic plate on the aluminum plate side 22 is an evaluation sample (frame-shaped foam sealant), 23 is an aluminum plate, 24 is a base plate, 25 is a powder supply unit, 26 is a screw, 27 is a foam compression plate, Reference numeral 28 denotes a cover plate fixing bracket.
- Example 1 Polypropylene (melt flow rate (MFR) (230 ° C): 0.40 g / 10 min) 65 parts by weight, polyolefin-based elastomer (melt flow rate (MFR): 6 g / 10 min, JIS A hardness: 79 °) 35 parts by weight, water 80 parts by weight of magnesium oxide (trade name "KISUMA 5P” manufactured by Kyowa Chemical Industry Co., Ltd.), 10 parts by weight of carbon (trade name "Asahi # 35" manufactured by Asahi Carbon Co., Ltd.), and 1 part by weight of stearate monoglyceride It was kneaded at a temperature of 200 ° C.
- the sheet-shaped molded product was thinned using a slicer to obtain a foamed structure a1 having a thickness of 0.3 mm. Further, the resin foam a1 is passed between the rolls (the gap between the rolls) in the pair of rolls in which one roll is heated to 200 ° C. to form the resin foam A having a thickness of 0.1 mm. Obtained. The gap between the rolls was set so that the resin foam A having a thickness of 0.1 mm could be obtained. The obtained resin foam A was subjected to the above evaluation. The results are shown in Table 1.
- Example 2 Polypropylene (melt flow rate (MFR) (230 ° C): 0.40 g / 10 min) 65 parts by weight, polyolefin-based elastomer (melt flow rate (MFR): 6 g / 10 min, JIS A hardness: 79 °) 35 parts by weight, water 100 parts by weight of magnesium oxide (trade name "KISUMA 5P” manufactured by Kyowa Chemical Industry Co., Ltd.), 10 parts by weight of carbon (trade name "Asahi # 35" manufactured by Asahi Carbon Co., Ltd.), and 1 part by weight of stearate monoglyceride It was kneaded at a temperature of 200 ° C.
- the sheet-shaped molded product was thinned using a slicer to obtain a foamed structure b1 having a thickness of 0.3 mm. Further, the resin foam b1 is passed between the rolls (the gap between the rolls) in the pair of rolls in which one roll is heated to 200 ° C. to obtain the resin foam B having a thickness of 0.1 mm. Obtained. The gap between the rolls was set so that the resin foam B having a thickness of 0.1 mm could be obtained. The obtained resin foam B was subjected to the above evaluation. The results are shown in Table 1.
- Example 3 Polypropylene (melt flow rate (MFR) (230 ° C): 0.40 g / 10 min) 65 parts by weight, polyolefin-based elastomer (melt flow rate (MFR): 6 g / 10 min, JIS A hardness: 79 °) 35 parts by weight, water 120 parts by weight of magnesium oxide (trade name "KISUMA 5P” manufactured by Kyowa Chemical Industry Co., Ltd.), 10 parts by weight of carbon (trade name "Asahi # 35" manufactured by Asahi Carbon Co., Ltd.), and 1 part by weight of stearate monoglyceride It was kneaded at a temperature of 200 ° C.
- MFR melt flow rate
- MFR polyolefin-based elastomer
- the sheet-shaped molded product was thinned using a slicer to obtain a foamed structure c1 having a thickness of 0.3 mm. Further, the resin foam c1 is passed between the rolls (the gap between the rolls) in the pair of rolls in which one roll is heated to 200 ° C. to obtain the resin foam C having a thickness of 0.1 mm. Obtained. The gap between the rolls was set so that the resin foam C having a thickness of 0.1 mm could be obtained. The obtained resin foam C was subjected to the above evaluation. The results are shown in Table 1.
- the sheet-shaped molded product was thinned using a slicer to obtain a foamed structure d1 having a thickness of 0.4 mm. Further, the resin foam d1 is passed between the rolls (gap between the rolls) in the pair of rolls in which one roll is heated to 200 ° C. to form a resin foam D having a thickness of 0.1 mm. Obtained. The gap between the rolls was set so that the resin foam D having a thickness of 0.1 mm could be obtained. The obtained resin foam D was subjected to the above evaluation. The results are shown in Table 1.
- the sheet-shaped molded product was thinned using a slicer to obtain a foamed structure e1 having a thickness of 0.4 mm. Further, the resin foam e1 is passed between the rolls (gap between the rolls) in the pair of rolls in which one roll is heated to 200 ° C. to form a resin foam E having a thickness of 0.1 mm. Obtained. The gap between the rolls was set so that the resin foam E having a thickness of 0.1 mm could be obtained. The obtained resin foam E was subjected to the above evaluation. The results are shown in Table 1.
- the sheet-shaped molded product was thinned using a slicer to obtain a foamed structure f1 having a thickness of 0.3 mm. Further, the resin foam f1 is passed between the rolls (the gap between the rolls) in the pair of rolls in which one roll is heated to 200 ° C. to obtain the resin foam F having a thickness of 0.1 mm. Obtained. The gap between the rolls was set so that the resin foam G having a thickness of 0.1 mm could be obtained. The obtained resin foam F was subjected to the above evaluation. The results are shown in Table 1.
- the resin foam of the present invention can be suitably used, for example, as a cushioning material for electronic devices.
- Foam member 10 Resin foam layer 20 Adhesive layer 2 Evaluation container equipped with an evaluation sample (evaluation container for dynamic dustproof evaluation) 211 Black acrylic plate (black acrylic plate on the cover plate side) 212 Black acrylic plate (black acrylic plate on the aluminum plate side) 22 Evaluation sample (frame-shaped foam sealant) 23 Aluminum plate 24 Base plate 25 Powder supply part 26 Screw 27 Foam compression plate 28 Cover plate fixing bracket
Abstract
薄く、衝撃吸収性に優れ、かつ、潰れにくい樹脂発泡体を提供する。 本発明の樹脂発泡体は、50%圧縮荷重が5N/cm2以上であり、下記試験で測定される繰り返し圧縮による厚み変化率が10%以下であり、気泡構造を有する。 <繰り返し圧縮試験> 先端が平坦な円盤治具(接触面積:4.9cm2)を樹脂発泡体に押し付け、50%圧縮/10%圧縮(厚み基準)サイクルを5000回行った際の厚みの変化率を測定する。 1つの実施形態においては、上記樹脂発泡体は、50%圧縮荷重が30N/cm2以下である。1つの実施形態においては、上記樹脂発泡体は、圧縮弾性率が、200kPa以上である。
Description
本発明は、樹脂発泡体に関する。
電子機器の画面保護や基板の保護等のクッション材として、樹脂発泡体が多用されている。近年、クッション材は、電子機器の薄型化の傾向に応じて、クリアランスの狭い箇所に配置されても、高い衝撃吸収性を発揮し得ることが求められている。このようなクッション材においては、周辺部材を保護するために防塵性が求められることがある。クッション材は、繰り返しの圧縮がかけられることが多く、これによる薄化が、防塵性低下の一因となる。例えば、スマートフォンにおいては、機器の落下、振動の他、画面タッチ等により、クッション材に繰り返し圧縮がかかり、その結果、クッション材の厚みが薄くなることがある。クッション材の厚みが薄くなると、周辺部材に対して隙間が生じ、防塵性が低下する。
特許文献1には、防水に優れた発泡体が開示されているが、防塵性の付与については何ら開示も示唆もされていない。また、特許文献2には、防塵性に優れた発泡体が開示されているが、繰り返し圧縮を受けた後の防塵性については何ら開示も示唆もされていない。
本発明の課題は、薄く、衝撃吸収性に優れ、かつ、防塵性に優れる樹脂発泡体を提供することにある。
本発明の樹脂発泡体は、50%圧縮荷重が5N/cm2以上であり、下記試験で測定される繰り返し圧縮による厚み変化率が10%以下であり、気泡構造を有する。
<繰り返し圧縮試験>
先端が平坦な円盤治具(接触面積:4.9cm2)を樹脂発泡体に押し付け、50%圧縮/10%圧縮(厚み基準)サイクルを5000回行った際の厚みの変化率を測定する。
1つの実施形態においては、上記樹脂発泡体は、50%圧縮荷重が30N/cm2以下である。
1つの実施形態においては、上記樹脂発泡体は、圧縮弾性率が、200kPa以上である。
1つの実施形態においては、上記樹脂発泡体は、非発泡曲げ応力が、5MPa以上である。
1つの実施形態においては、上記樹脂発泡体は、みかけ密度が、0.02g/cm3~0.50g/cm3である。
1つの実施形態においては、上記樹脂発泡体は、厚み回復率が、70%以上である。
1つの実施形態においては、上記樹脂発泡体が有する気泡のアスペクト比が、1.5以上である。
1つの実施形態においては、上記樹脂発泡体は、平均気泡径が10μm~200μmである。
1つの実施形態においては、上記樹脂発泡体は、気泡率が、30%以上である。
1つの実施形態においては、上記樹脂発泡体は、気泡径の変動係数が、0.5以下である。
1つの実施形態においては、上記樹脂発泡体は、気泡壁の厚みが、0.1μm~10μmである。
1つの実施形態においては、上記樹脂発泡体は、ポリオレフィン系樹脂を含む。
1つの実施形態においては、上記ポリオレフィン系樹脂が、ポリオレフィン系エラストマー以外のポリオレフィンとポリオレフィン系エラストマーの混合物である。
1つの実施形態においては、上記樹脂発泡体は、片面または両面に、熱溶融層を有する。
本発明の別の局面によれば、発泡部材が提供される。この発泡部材は、樹脂発泡層と、該樹脂発泡層の少なくとも一方の側に配置された粘着剤層を有し、該樹脂発泡層が、上記樹脂発泡体である。
<繰り返し圧縮試験>
先端が平坦な円盤治具(接触面積:4.9cm2)を樹脂発泡体に押し付け、50%圧縮/10%圧縮(厚み基準)サイクルを5000回行った際の厚みの変化率を測定する。
1つの実施形態においては、上記樹脂発泡体は、50%圧縮荷重が30N/cm2以下である。
1つの実施形態においては、上記樹脂発泡体は、圧縮弾性率が、200kPa以上である。
1つの実施形態においては、上記樹脂発泡体は、非発泡曲げ応力が、5MPa以上である。
1つの実施形態においては、上記樹脂発泡体は、みかけ密度が、0.02g/cm3~0.50g/cm3である。
1つの実施形態においては、上記樹脂発泡体は、厚み回復率が、70%以上である。
1つの実施形態においては、上記樹脂発泡体が有する気泡のアスペクト比が、1.5以上である。
1つの実施形態においては、上記樹脂発泡体は、平均気泡径が10μm~200μmである。
1つの実施形態においては、上記樹脂発泡体は、気泡率が、30%以上である。
1つの実施形態においては、上記樹脂発泡体は、気泡径の変動係数が、0.5以下である。
1つの実施形態においては、上記樹脂発泡体は、気泡壁の厚みが、0.1μm~10μmである。
1つの実施形態においては、上記樹脂発泡体は、ポリオレフィン系樹脂を含む。
1つの実施形態においては、上記ポリオレフィン系樹脂が、ポリオレフィン系エラストマー以外のポリオレフィンとポリオレフィン系エラストマーの混合物である。
1つの実施形態においては、上記樹脂発泡体は、片面または両面に、熱溶融層を有する。
本発明の別の局面によれば、発泡部材が提供される。この発泡部材は、樹脂発泡層と、該樹脂発泡層の少なくとも一方の側に配置された粘着剤層を有し、該樹脂発泡層が、上記樹脂発泡体である。
薄く、衝撃吸収性に優れ、かつ、潰れにくい樹脂発泡体を提供することができる。
A.樹脂発泡体
本発明の樹脂発泡体は、50%圧縮荷重が5N/cm2以上であり、繰り返し圧縮による厚み変化率が28%以下である。本発明の樹脂発泡体は、気泡構造(セル構造)を有する。気泡構造(セル構造)としては、独立気泡構造、連続気泡構造、半連続半独立気泡構造(独立気泡構造と連続気泡構造が混在している気泡構造)などが挙げられる。樹脂発泡体の気泡構造は、連続気泡構造、または半連続半独立気泡構造が好ましく、半連続半独立気泡構造がより好ましい。本発明の樹脂発泡体は、樹脂組成物を発泡させることにより得られる。上記樹脂組成物は、樹脂発泡体を構成する樹脂を少なくとも含有する組成物である。
本発明の樹脂発泡体は、50%圧縮荷重が5N/cm2以上であり、繰り返し圧縮による厚み変化率が28%以下である。本発明の樹脂発泡体は、気泡構造(セル構造)を有する。気泡構造(セル構造)としては、独立気泡構造、連続気泡構造、半連続半独立気泡構造(独立気泡構造と連続気泡構造が混在している気泡構造)などが挙げられる。樹脂発泡体の気泡構造は、連続気泡構造、または半連続半独立気泡構造が好ましく、半連続半独立気泡構造がより好ましい。本発明の樹脂発泡体は、樹脂組成物を発泡させることにより得られる。上記樹脂組成物は、樹脂発泡体を構成する樹脂を少なくとも含有する組成物である。
上記のとおり、本発明の樹脂発泡体の50%圧縮荷重は、5N/cm2以上である。このような範囲であれば、適度な硬さを有し、高い衝撃力を受けた際にも圧縮変形が生じ難い樹脂発泡体を得ることができる。このような樹脂発泡体は、潰れにくく、クッション材として使用する際に周辺部材との隙間を防止することができ、防塵性に優れる。樹脂発泡体の50%圧縮荷重は、好ましくは7N/cm2以上であり、より好ましくは9N/cm2より大きく、さらに好ましくは10N/cm2以上である。このような範囲であれば、上記効果が顕著となる。樹脂発泡体の50%圧縮荷重の上限は、好ましくは50N/cm2であり、より好ましくは40N/cm2であり、さらに好ましくは30N/cm2であり、特に好ましくは25N/cm2である。このような範囲であれば、隙間(特に、クリアランスの狭い隙間)に適用した際の反発力が適度に抑えられ、周辺部材への悪影響を防止することができる。
上記のとおり、本発明の樹脂発泡体の繰り返し圧縮による厚み変化率は、10%以下である。このような範囲であれば、潰れによる薄化が生じがたく、クッション材として使用する際に周辺部材との隙間を防止することができ、防塵性に優れる樹脂発泡体を得ることができる。樹脂発泡体の繰り返し圧縮による厚み変化率は、より好ましくは8%以下であり、さらに好ましくは5%以下であり、特に好ましくは3%以下であり、最も好ましくは1%以下である。このような範囲であれば、上記効果が顕著となる。樹脂発泡体の繰り返し圧縮による厚み変化率は小さいほど好ましいが、その下限は、例えば、0.5%(好ましくは0.3%、より好ましくは0.1%、さらに好ましくは0.05%)である。樹脂発泡体の繰り返し圧縮による厚み変化率は、先端が平坦な円盤治具(接触面積:4.9cm2)を樹脂発泡体に押し付け、50%圧縮/10%圧縮(厚み基準)サイクルを5000回行った際の厚みの変化率を意味する。当該変化率は、{(試験前の厚み)-(試験後の厚み)}/(試験前の厚み)×100の式により算出される。
本発明の樹脂発泡体は、50%圧縮荷重および繰り返し圧縮による厚み変化率を上記範囲とすることにより、所定の硬さを有しながらも、衝撃吸収性に優れるという特徴を有する。そのため、高い衝撃力を受けた際にも潰れが生じ難く、厚みが長期に維持され、防塵性に優れる。
上記樹脂発泡体の見かけ密度は、好ましくは0.02g/cm3~0.5g/cm3である。このような範囲であれば、柔軟性および応力分散性に優れる樹脂発泡体を得ることができる。上記樹脂発泡体の見かけ密度は、より好ましくは0.04g/cm3~0.4g/cm3であり、さらに好ましくは0.06g/cm3~0.3g/cm3であり、さらに好ましくは0.08g/cm3~0.25g/cm3であり、特に好ましくは0.1g/cm3~0.2g/cm3であり、最も好ましくは0.13g/cm3~0.2g/cm3である。このような範囲であれば、上記効果が顕著となる。見かけ密度の測定方法は、後述する。
上記樹脂発泡体の25%圧縮荷重は、好ましくは3N/cm2以上である。このような範囲であれば、上記本発明の効果は顕著となる。樹脂発泡体の25%圧縮荷重は、好ましくは3.2N/cm2以上であり、より好ましくは3.5N/cm2以上であり、さらに好ましくは4N/cm2以上である。樹脂発泡体の25%圧縮荷重の上限は、例えば、10N/cm2(好ましくは20N/cm2)である。25%圧縮荷重の測定方法は、JIS K 6767に記載されている発泡体の圧縮硬さ測定方法に準ずる。
上記樹脂発泡体の圧縮弾性率は、好ましくは200kPa以上である。このような範囲であれば、高い衝撃力を受けた際にも圧縮変形が生じ難く、潰れがたい樹脂発泡体を得ることができる。樹脂発泡体の圧縮弾性率は、好ましくは220kPa以上であり、より好ましくは250kPa以上であり、さらに好ましくは300kPa以上である。このような範囲であれば、上記効果が顕著となる。樹脂発泡体の圧縮弾性率の上限は、好ましくは1000kPaであり、より好ましくは800kPaであり、さらに好ましくは600kPaである。樹脂発泡体の圧縮弾性率は、JIS K 6767の圧縮試験の項に基づき、樹脂発泡体の圧縮率(%)、圧縮反発力(kPa)を測定し、圧縮率5%の時の圧縮反発力F(5%)と圧縮率15%の時の圧縮時の反発力F(15%)をそれぞれ読み取り、F(5%)、F(15%)から、(F(15%)-F(5%))/(15-5)×100の式により、算出される。
上記樹脂発泡体の圧縮時の弾性歪エネルギーは、好ましくは10kPa以上である。「圧縮時の弾性歪エネルギー」は、樹脂発泡体を10%圧縮させる際の圧縮反発力の総量を意味する。具体的には、「圧縮時の弾性歪エネルギー」は、JIS K 6767に準じた圧縮試験(試験温度:23℃、サンプルサイズ:10mm×10mm、圧縮速度:10mm/min)により、樹脂発泡体の圧縮率(%)および圧縮反発力(kPa)を測定した際の、x軸を圧縮率(%)としy軸を圧縮反発力(kPa)とする圧縮ss曲線から求められ、圧縮率が0%~10%である範囲における、当該ss曲線と当該x軸とから規定される領域の面積である。樹脂発泡体の圧縮時の弾性歪エネルギーが、上記範囲であれば、衝撃吸収性に優れる樹脂発泡体を得ることができる。より詳細には、上記範囲の弾性歪エネルギーを有する樹脂発泡体は、衝撃が加わった際に、当該樹脂発泡体の変形に多くのエネルギーが消費されるため、強い衝撃に対しても、これを良好に吸収することができる。上記樹脂発泡体の圧縮時の弾性歪エネルギーは、より好ましくは20kPa以上であり、さらに好ましくは30kPa以上であり、さらに好ましくは50kPa以上であり、さらに好ましくは60kPa以上であり、さらに好ましくは80kPa以上であり、特に好ましくは100kPa以上であり、最も好ましくは150kPa以上である。このような範囲であれば、上記効果が顕著となる。上記樹脂発泡体の圧縮時の弾性歪エネルギーの上限は、例えば、500kPa(好ましくは800kPa)である。
上記樹脂発泡体の非発泡曲げ応力は、好ましくは5MPa以上であり、より好ましくは6MPa以上であり、さらに好ましくは9MPa以上であり、特に好ましくは10MPa以上である。このような範囲であれば、樹脂発泡体の気泡壁(セル壁)を変形させるのに大きなエネルギーが必要とされ、優れた衝撃吸収性を有する樹脂発泡体を得ることができる。上記樹脂発泡体は、従来、クッション材で重要視されていた柔軟性を適度に減じ、他の特性とのバランスをとることにより、耐衝撃性(衝撃吸収性)の向上を可能としている。このように従来と異なる作用により、衝撃吸収性を発現する上記樹脂発泡体は、狭い範囲で衝撃が伝搬するような構成(衝撃が面方向に拡がり難い構成)において特に有用であり、例えば、可撓性を有する部材(例えば、樹脂から構成される部材)に適用した際に、特に有用である。当該非発泡曲げ応力の上限は、好ましくは20MPaであり、より好ましくは15MPaである。このような範囲であれば、柔軟性および応力分散性により優れる樹脂発泡体を得ることができる。「非発泡曲げ応力」とは、樹脂発泡体を加熱プレスすることで、気泡のない非発泡状態(バルク)に戻した樹脂成形体aの曲げ応力を意味する。樹脂成形体aの密度は、後述の樹脂組成物により形成された発泡させる前の樹脂成形体bの密度と同等にされ得る。なお、樹脂成形体aの曲げ応力(樹脂発泡体の非発泡曲げ応力)は、樹脂成形体bと同等となり得る。曲げ応力の測定方法は、後述する。
上記樹脂発泡体の衝撃吸収率は、好ましくは25%以上であり、より好ましくは30%以上であり、さらに好ましくは35%以上である。衝撃吸収率は、以下のようにして測定される。
・衝撃力センサー上に、樹脂発泡体、両面テープ(品番:No.5603W、日東電工製)、PETフィルム(品番:ダイヤホイルMRF75、三菱樹脂製)をこの順に配置して試験体を形成した。PETフィルム上方50cmの高さから、66gの鉄球を試験体に落下させて、衝撃力F1を測定する。
・また、衝撃力センサーに直接、上記のように鉄球を落下させて、ブランクの衝撃力F0を測定する。
・F1、F0から、(F0-F1)/F0×100の式により、衝撃吸収率(%)を算出する。
・衝撃力センサー上に、樹脂発泡体、両面テープ(品番:No.5603W、日東電工製)、PETフィルム(品番:ダイヤホイルMRF75、三菱樹脂製)をこの順に配置して試験体を形成した。PETフィルム上方50cmの高さから、66gの鉄球を試験体に落下させて、衝撃力F1を測定する。
・また、衝撃力センサーに直接、上記のように鉄球を落下させて、ブランクの衝撃力F0を測定する。
・F1、F0から、(F0-F1)/F0×100の式により、衝撃吸収率(%)を算出する。
上記樹脂発泡体の厚み回復率は、好ましくは70%以上であり、より好ましくは75%以上であり、さらに好ましくは80%以上である。なお、発泡層の厚み回復率は、下記式で定義される。この発泡層の厚み回復率は、ある程度の面積をもって発泡シートに荷重をかけて圧縮して測定される回復率であり、局所的に荷重をかけて一部分のみを凹ませて測定するいわゆる凹み回復率とは異なる。
厚み回復率(%)={(圧縮状態を解除してから0.5秒後の厚み)/(初期厚み)}×100
初期厚み:荷重を加える前の樹脂発泡体の厚み。
圧縮状態を解除してから0.5秒後の厚み:樹脂発泡体に1000g/cm2の荷重を加えた状態で120秒間維持し、圧縮を解除し、解除してから0.5秒後の樹脂発泡体の厚み。
厚み回復率(%)={(圧縮状態を解除してから0.5秒後の厚み)/(初期厚み)}×100
初期厚み:荷重を加える前の樹脂発泡体の厚み。
圧縮状態を解除してから0.5秒後の厚み:樹脂発泡体に1000g/cm2の荷重を加えた状態で120秒間維持し、圧縮を解除し、解除してから0.5秒後の樹脂発泡体の厚み。
上記樹脂発泡体の23℃における引張弾性率は、好ましくは0.6MPa以上であり、より好ましくは0.7MPa~5MPaであり、より好ましくは1MPa~4MPaである。このような範囲であれば、応力分散性に優れ、薄膜でも優れた衝撃吸収性を発揮し得る樹脂発泡体を得ることができる。上記引張弾性率は、チャック間距離40mmでサンプル(サイズ:10mm×80mm)を固定し、引張速度500mm/minで引張試験を行って得られる引張歪-引張強度曲線を得、この曲線の原点と引張歪10%の時の引張強度を結んだ直線の傾きより、求められる。
上記樹脂発泡体の25℃における破断伸びは、好ましくは120%以下であり、より好ましくは110%以下であり、さらに好ましくは100%以下であり、特に好ましくは90%以下である。このような範囲であれば、柔軟性および応力分散性に優れる樹脂発泡体を得ることができる。なお、破断伸びが小さいと、樹脂発泡体に荷重が加わったときに、当該樹脂発泡体のセル壁の変形が小さくなり、例えば、充填材が添加されている場合、当該樹脂発泡体を構成する樹脂と当該充填材との界面で滑りが発生しやすくなり、荷重をより緩和し得る。一方、破断伸びが大き過ぎると、樹脂発泡体のセル壁の変形が大きくなり、荷重を緩和しにくくなるおそれがある。破断伸びは、JIS K 6767に準じて測定することができる。
上記樹脂発泡体の応力保持率は、好ましくは60%以上であり、より好ましくは63%~100%であり、さらに好ましくは63%~95%である。このような範囲であれば、応力分散性に優れ、薄膜でも優れた衝撃吸収性を発揮し得る樹脂発泡体を得ることができる。本明細書において、上記応力保持率とは、樹脂発泡体(幅10mm×長さ100mm)を長さ方向に300m/minの速度で20%延伸し、延伸直後の引張強度と、延伸後12時間保持した後の引張強度との比率(12時間後の引張強度/延伸直後の引張強度×100)である。
上記樹脂発泡体の厚みは、好ましくは30μm~5000μmであり、より好ましくは35μm~4000μmであり、さらに好ましくは40μm~3000μmであり、さらに好ましくは45μm~2000μmであり、特に好ましくは50μm~1000μmであり、最も好ましくは55μm~500μmである。上記のとおり、本発明の樹脂発泡体は薄層でありながらも優れた耐衝撃性を発揮する。また、樹脂発泡体の厚みが上記範囲内であれば、微細かつ均一な気泡構造を形成することができ、優れた衝撃吸収性を発現し得る点で有利である。
上記樹脂発泡体の平均気泡径(平均セル径)は、好ましくは10μm~200μmであり、より好ましくは15μm~180μmであり、さらに好ましくは20μm~150μmであり、さらに好ましくは23μm~120μmであり、特に好ましくは25μm~100μmであり、最も好ましくは30μm~80μmである。このような範囲であれば、衝撃を受けた際に、過度に変形することがなく、高い衝撃吸収性を発現し得る樹脂発泡体を得ることができる。また、適度な硬さを有し、隙間を埋めるクッション材として好適な樹脂発泡体とすることができる。平均気泡径の測定方法は、後述する。
上記樹脂発泡体が有する気泡のアスペクト比は、好ましくは1.5以上である。このような範囲であれば、厚み回復性に優れる樹脂発泡体を得ることができる。上記樹脂発泡体を構成する気泡のアスペクト比は、好ましくは2.0以上であり、より好ましくは2.5以上である。このような範囲であれば、上記効果は顕著となる。また、上記樹脂発泡体を構成する気泡のアスペクト比の上限は、好ましくは5であり、より好ましくは4であり、さらに好ましくは3.5である。このような範囲であれば、衝撃吸収性に優れる樹脂発泡体を得ることができる。
なお、本明細書において、「樹脂発泡体が有する気泡のアスペクト比」は、無作為に選択した箇所における樹脂発泡体断面中の所定面積(3mm2)範囲に存在する個々の気泡のアスペクト比の平均値を意味する。「樹脂発泡体が有する気泡のアスペクト比」の具体的な求め方は、以下のとおりである。
・樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、切断面をマイクロスコープ(例えば、キーエンス製「VHX-2000」)を用いて、所定面積(3mm2)範囲を倍率100倍で観察する。気泡一個の厚み方向の長さとTDの長さを測定する。
・同様の測定を所定面積内に存在する全ての気泡に対して行う。
・気泡のアスペクト比は、TD方向の長さ÷厚み方向の長さで計算され、全ての気泡で同様の計算を行い、平均した値を「樹脂発泡体が有する気泡のアスペクト比」とする。
・樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、切断面をマイクロスコープ(例えば、キーエンス製「VHX-2000」)を用いて、所定面積(3mm2)範囲を倍率100倍で観察する。気泡一個の厚み方向の長さとTDの長さを測定する。
・同様の測定を所定面積内に存在する全ての気泡に対して行う。
・気泡のアスペクト比は、TD方向の長さ÷厚み方向の長さで計算され、全ての気泡で同様の計算を行い、平均した値を「樹脂発泡体が有する気泡のアスペクト比」とする。
上記樹脂発泡体の気泡径(セル径)の変動係数は、好ましくは0.5以下であり、より好ましくは0.48以下であり、さらに好ましくは0.45以下であり、特に好ましくは0.43以下であり、最も好ましくは0.4未満である。このような範囲であれば、衝撃による変形が均一になり、局所的な応力負荷が防止され、応力分散性に優れ、かつ、耐衝撃性に特に優れる樹脂発泡体を得ることができる。当該変動係数は、小さいほど好ましいがその下限は、例えば、0.2(好ましくは0.15、より好ましくは0.1、さらに好ましくは0.01)である。気泡径の変動係数の測定方法は、後述する。
上記樹脂発泡体の気泡率(セル率)は、好ましくは30%以上であり、より好ましくは50%以上である。このような範囲であれば、圧縮時の反発応力が小さい樹脂発泡体を得ることができる。このような樹脂発泡体は、クリアランスの狭い箇所に多少圧縮して上記樹脂発泡体を適用する場合において、他の部材へかかる応力を低減することができる。例えば、樹脂発泡体を表示部材に適用する場合は、当該表示部材にかかる応力を緩和・分散することができるため、色ムラ低減、部材保護の観点から有用である。当該気泡率の上限は、好ましくは99%以下であり、より好ましくは95%以下であり、さらに好ましくは90%以下である。このような範囲であれば、適度な硬さを有して、衝撃を受けた際の変形量が小さく、そのため、高い衝撃吸収性を有する樹脂発泡体とすることができる。気泡率の測定方法は、後述する。
上記樹脂発泡体の気泡壁(セル壁)の厚みは、好ましくは0.1μm~10μmであり、より好ましくは0.3μm~8μmであり、さらに好ましくは0.5μm~5μmであり、特に好ましくは0.7μm~4μmであり、最も好ましくは1μm~3μmである。このような範囲であれば、衝撃を受けた際に、過度に変形することがなく、高い衝撃吸収性を発現し得る樹脂発泡体を得ることができる。また、適度な硬さを有し、隙間を埋めるクッション材として好適な樹脂発泡体とすることができる。気泡壁の厚みの測定方法は、後述する。
上記樹脂発泡体の気泡構造が半連続半独立気泡構造である場合、その中の独立気泡構造の割合は、好ましくは40%以下であり、より好ましくは30%以下である。このような範囲であれば、適度な硬さを有し、隙間を埋めるクッション材として好適な樹脂発泡体とすることができる。本明細書において、樹脂発泡体の独泡率は、例えば、温度23℃、湿度50%の環境下で、測定対象を水分中に沈め、その後の質量を測定し、その後、80℃のオーブンで十分に乾燥させた後、再度質量を測定して求められる。また、連続気泡であれば水分を保持できるため、その質量分を連続気泡として測定して求められる。
上記樹脂発泡体の形状としては、目的に応じて、任意の適切な形状を採用し得る。このような形状としては、代表的には、シート状である。
上記樹脂発泡体は、その片面または両面に熱溶融層を有していてもよい。熱溶融層を有する樹脂発泡体は、例えば、樹脂発泡体を構成する樹脂組成物の溶融温度以上に加温された一対の加熱ロールを用いて、樹脂発泡体(または樹脂発泡体の前駆体)を圧延することにより、得られ得る。
上記樹脂発泡体は、本発明の効果を損なわない範囲で、任意の適切な方法によって形成することができる。このような方法としては、代表的には、樹脂材料(ポリマー)を含む樹脂組成物を発泡させる方法が挙げられる。
A-1.樹脂組成物
本発明の樹脂発泡体は、代表的には、樹脂組成物を発泡させて得られ得る。樹脂組成物は、任意の適切な樹脂材料(ポリマー)を含む。
本発明の樹脂発泡体は、代表的には、樹脂組成物を発泡させて得られ得る。樹脂組成物は、任意の適切な樹脂材料(ポリマー)を含む。
上記ポリマーとしては、例えば、アクリル系樹脂、シリコーン系樹脂、ウレタン系樹脂、ポリオレフィン系樹脂、エステル系樹脂、ゴム系樹脂などが挙げられる。上記ポリマーは、1種のみを単独で用いてもよく、2種以上を組み合わせて用いてもよい。
ポリマーの含有割合は、樹脂組成物100重量部に対して、好ましくは30重量部~95重量部であり、より好ましくは35重量部~90重量部であり、さらに好ましくは40重量部~80重量部であり、特に好ましくは40重量部~60重量部である。このような範囲であれば、柔軟性および応力分散性により優れる樹脂発泡体を得ることができる。
1つの実施形態においては、上記ポリマーとして、ポリオレフィン系樹脂が用いられる。
ポリオレフィン系樹脂の含有割合は、上記ポリマー100重量部に対して、好ましくは50重量部~100重量部であり、より好ましくは70重量部~100重量部であり、さらに好ましくは90重量部~100重量部であり、特に好ましくは95重量部~100重量部であり、最も好ましくは100重量部である。
ポリオレフィン系樹脂としては、好ましくは、ポリオレフィンおよびポリオレフィン系エラストマーからなる群から選ばれる少なくとも1種が挙げられ、より好ましくは、ポリオレフィンとポリオレフィン系エラストマーとが併用される。ポリイレフンおよびポリオレフィン系エラストマーはそれぞれ、1種のみを単独で用いてもよく、2種以上を組み合わせて用いてもよい。なお、本明細書において、「ポリオレフィン」と称する場合には、「ポリオレフィン系エラストマー」は含まれないものとする。
ポリオレフィン系樹脂としてポリオレフィンとポリオレフィン系エラストマーを併用する場合、ポリオレフィンとポリオレフィン系エラストマーの重量比率(ポリオレフィン/ポリオレフィン系エラストマー)は、好ましくは1/99~99/1であり、より好ましくは10/90~90/10であり、さらに好ましくは20/80~80/20であり、特に好ましくは30/70~70/30である。このような範囲であれば、本発明の効果は顕著となる。
ポリオレフィンとしては、本発明の効果を損なわない範囲で、任意の適切なポリオレフィンを採用し得る。このようなポリオレフィンとしては、例えば、直鎖状のポリオレフィン、分岐鎖状の(分岐鎖を有する)ポリオレフィンなどが挙げられる。1つの実施形態においては、ポリオレフィン系樹脂として、分岐鎖状のポリオレフィンが用いられる。この実施形態においては、ポリオレフィンとして、分岐状のポリオレフィンのみを用いてもよく、分岐状のポリオレフィンと直鎖状のポリオレフィンとを併用して用いてもよい。分岐状のポリオレフィンを用いることにより、平均気泡径が小さく、耐衝撃性に優れる樹脂発泡体を得ることができる。分岐状のポリオレフィンの含有割合は、ポリオレフィン100重量部に対して、好ましくは30重量部~100重量部であり、より好ましくは80重量部~120重量部である。
上記ポリオレフィンとしては、例えば、α-オレフィン由来の構成単位を含むポリマーが挙げられる。ポリオレフィンは、α-オレフィン由来の構成単位のみから構成されていてもよく、α-オレフィン由来の構成単位と、α-オレフィン以外のモノマー由来の構成単位とから構成されていてもよい。ポリオレフィンが共重合体である場合、その共重合形態としては、任意の適切な共重合形態を採用し得る。例えば、ランダムコポリマー、ブロックコポリマーなどが挙げられる。
ポリオレフィンを構成し得るα-オレフィンとしては、例えば、炭素数2~8(好ましくは2~6、より好ましくは2~4)のα-オレフィン(例えば、エチレン、プロピレン、1-ブテン、1-ペンテン、1-ヘキセン、4-メチル-1-ペンテン、1-へプテン、1-オクテンなど)が好ましい。α-オレフィンは、1種のみであってもよいし、2種以上であってもよい。
ポリオレフィンを構成するα-オレフィン以外のモノマーとしては、例えば、酢酸ビニル、アクリル酸、アクリル酸エステル、メタクリル酸、メタクリル酸エステル、ビニルアルコールなどのエチレン性不飽和単量体が挙げられる。α-オレフィン以外のモノマーは、1種のみであってもよいし、2種以上であってもよい。
ポリオレフィンとしては、具体的には、例えば、低密度ポリエチレン、中密度ポリエチレン、高密度ポリエチレン、直鎖状低密度ポリエチレン、ポリプロピレン(プロピレンホモポリマー)、エチレンとプロピレンとの共重合体、エチレンとエチレン以外のα-オレフィンとの共重合体、プロピレンとプロピレン以外のα-オレフィンとの共重合体、エチレンとプロピレンとエチレンおよびプロピレン以外のα-オレフィンとの共重合体、プロピレンとエチレン性不飽和単量体との共重合体などが挙げられる。
1つの実施形態においては、ポリオレフィンとして、プロピレン由来の構成単位を有するポリプロピレン系重合体が用いられる。ポリプロピレン系重合体としては、例えば、ポリプロピレン(プロピレンホモポリマー)、エチレンとプロピレンとの共重合体、プロピレンとプロピレン以外のα-オレフィンとの共重合体などが挙げられ、好ましくはポリプロピレン(プロピレンホモポリマー)である。ポリプロピレン系重合体は、1種のみを単独で用いてもよく、2種以上を組み合わせて用いてもよい。
ポリオレフィンの温度230℃におけるメルトフローレート(MFR)は、本発明の効果をより発現させ得る点で、好ましくは0.2g/10分~10g/10分であり、より好ましくは0.25g/10分~5g/10分であり、さらに好ましくは0.3g/10分~3g/10分であり、特に好ましくは0.35g/10分~1.5g/10分である。なお、本明細書において、上記メルトフローレート(MFR)は、ISO1133(JIS-K-7210)に基づき、温度230℃、荷重2.16kgfで測定されたMFRをいうものとする。
1つの実施形態においては、温度230℃におけるメルトフローレート(MFR)が上記の範囲内で異なる2種以上のポリオレフィンが併用される。この場合、温度230℃におけるメルトフローレート(MFR)が、好ましくは0.2g/10分以上0.7g/10分未満(より好ましくは0.2g/10分~0.65g/10分)のポリオレフィンと、温度230℃におけるメルトフローレート(MFR)が好ましくは0.7g/10分~10g/10分(より好ましくは0.7g/10分~5g/10分であり、さらに好ましくは0.7g/10分~3g/10分であり、特に好ましくは0.7g/10分~1.5g/10分であり、最も好ましくは0.7g/10分~1.3g/10分である)のポリオレフィンとが併用され得る。このようにすれば、平均気泡径が小さく、耐衝撃性に優れる樹脂発泡体を得ることができる。
ポリオレフィンとして、温度230℃におけるメルトフローレート(MFR)が上記の範囲内で異なる2種以上のポリオレフィンを併用する場合、例えば、上記の温度230℃におけるメルトフローレート(MFR)が、好ましくは0.2g/10分以上0.7g/10分未満(より好ましくは0.2g/10分~0.65g/10分)のポリオレフィンと、温度230℃におけるメルトフローレート(MFR)が好ましくは0.7g/10分~10g/10分(より好ましくは0.7g/10分~5g/10分であり、さらに好ましくは0.7g/10分~3g/10分であり、特に好ましくは0.7g/10分~1.5g/10分であり、最も好ましくは0.7g/10分~1.3g/10分である)のポリオレフィンとの重量比率は、好ましくは1/99~99/1であり、より好ましくは10/90~90/10であり、さらに好ましくは20/80~80/20であり、特に好ましくは30/70~70/30であり、最も好ましくは40/60~60/40である。
ポリオレフィンとしては、市販品を用いてもよく、例えば、「E110G」(株式会社プライムポリマー製)、「EA9」(日本ポリプロ株式会社製)、「EA9FT」(日本ポリプロ株式会社製)、「E-185G」(株式会社プライムポリマー製)、「WB140HMS」(ボレアリス社製)、「WB135HMS」(ボレアリス社製)などが挙げられる。
ポリオレフィン系エラストマーとしては、本発明の効果を損なわない範囲で、任意の適切なポリオレフィン系エラストマーを採用し得る。このようなポリオレフィン系エラストマーとしては、例えば、エチレン-プロピレン共重合体、エチレン-プロピレン-ジエン共重合体、エチレン-酢酸ビニル共重合体、ポリブテン、ポリイソブチレン、塩素化ポリエチレン、ポリオレフィン成分とゴム成分とが物理的に分散したエラストマー、ポリオレフィン成分とゴム成分とがミクロ相分離した構造を有したエラストマーなどの、いわゆる非架橋型の熱可塑性オレフィン系エラストマー(TPO);マトリックスを形成する樹脂成分A(オレフィン系樹脂成分A)およびドメインを形成するゴム成分Bを含む混合物を、架橋剤の存在下、動的に熱処理することにより得られ、マトリックス(海相)である樹脂成分A中に、架橋ゴム粒子がドメイン(島相)として細かく分散した海島構造を有する多相系のポリマーである動的架橋型熱可塑性オレフィン系エラストマー(TPV);などが挙げられる。
ポリオレフィン系エラストマーは、好ましくは、ゴム成分を含む。このようなゴム成分としては、特開平08-302111号公報、特開2010-241934号公報、特開2008-024882号公報、特開2000-007858号公報、特開2006-052277号公報、特開2012-072306号公報、特開2012-057068号公報、特開2010-241897号公報、特開2009-067969号公報、再表03/002654号公報などに記載のものが挙げられる。
ポリオレフィン成分とオレフィン系ゴム成分とがミクロ相分離した構造を有したエラストマーとしては、具体的には、ポリプロピレン樹脂(PP)とエチレン-プロピレンゴム(EPM)とからなるエラストマー、ポリプロピレン樹脂(PP)とエチレン-プロピレン-ジエンゴム(EPDM)とからなるエラストマーなどが挙げられる。ポリオレフィン成分とオレフィン系ゴム成分の重量比(ポリオレフィン成分/オレフィン系ゴム)は、好ましくは90/10~10/90であり、より好ましくは80/20~20/80である。
動的架橋型熱可塑性オレフィン系エラストマー(TPV)は、一般的に、非架橋型の熱可塑性オレフィン系エラストマー(TPO)より、弾性率が高く、かつ圧縮永久歪みも小さい。これにより、回復性が良好であり、発泡体とした場合に優れた回復性を示し得る。
動的架橋型熱可塑性オレフィン系エラストマー(TPV)とは、上述したように、マトリックスを形成する樹脂成分A(オレフィン系樹脂成分A)およびドメインを形成するゴム成分Bを含む混合物を、架橋剤の存在下、動的に熱処理することにより得られ、マトリックス(海相)である樹脂成分A中に、架橋ゴム粒子がドメイン(島相)として細かく分散した海島構造を有する多相系のポリマーである。
動的架橋型熱可塑性オレフィン系エラストマー(TPV)としては、例えば、特開2000-007858号公報、特開2006-052277号公報、特開2012-072306号公報、特開2012-057068号公報、特開2010-241897号公報、特開2009-067969号公報、再表03/002654号等に記載のものなどが挙げられる。
動的架橋型熱可塑性オレフィン系エラストマー(TPV)としては、市販品を用いてもよく、例えば、「ゼオサーム」(日本ゼオン社製)、「サーモラン」(三菱化学社製)、「サーリンク3245D」(東洋紡績株式会社製)などが挙げられる。
ポリオレフィン系エラストマーの温度230℃におけるメルトフローレート(MFR)は、好ましくは2g/10分~15g/10分であり、より好ましくは3g/10分~10g/10分であり、さらに好ましくは3.5g/10分~9g/10分であり、特に好ましくは4g/10分~8g/10分であり、最も好ましくは4.5g/10分~7.5g/10分である。
ポリオレフィン系エラストマーの溶融張力(190℃、破断時)は、好ましくは10cN未満であり、より好ましくは5cN~9.5cNである。
ポリオレフィン系エラストマーのJIS A硬度は、好ましくは30°~95°であり、より好ましくは35°~90°であり、さらに好ましくは40°~88°であり、特に好ましくは45°~85°であり、最も好ましくは50°~83°である。なお、JIS A硬度とは、ISO7619(JIS K6253)に基づいて測定される。
1つの実施形態においては、上記樹脂発泡体(すなわち、樹脂組成物)は、充填材をさらに含み得る。充填材を含有させることにより、気泡壁(セル壁)を変形させるのに大きなエネルギーを必要とする樹脂発泡体を形成することができ、当該樹脂発泡体は、優れた衝撃吸収性を発揮する。また、充填材を含有させることにより、微細かつ均一な気泡構造を形成することができ、優れた衝撃吸収性を発現し得る点でも有利である。充填材は、1種のみを単独で用いてもよく、2種以上を組み合わせて用いてもよい。
上記充填材の含有割合は、樹脂発泡体を構成するポリマー100重量部に対して、好ましくは10重量部~150重量部であり、より好ましくは30重量部~130重量部であり、さらに好ましくは50重量部~100重量部である。このような範囲であれば、上記効果が顕著となる。
1つの実施形態においては、上記充填材は無機物である。無機物である充填材を構成する材料としては、例えば、水酸化アルミニウム、水酸化マグネシウム、炭酸カルシウム、炭酸マグネシウム、ケイ酸カルシウム、ケイ酸マグネシウム、酸化カルシウム、酸化マグネシウム、酸化アルミニウム、窒化アルミニウム、ホウ酸アルミニウムウィスカ、窒化ケイ素、窒化ホウ素、結晶質シリカ、非晶質シリカ、金属(例えば、金、銀、銅、アルミニウム、ニッケル)、カーボン、グラファイト等が挙げられる。
1つの実施形態においては、上記充填材は有機物である。有機物である充填材を構成する材料としては、例えば、ポリメタクリル酸メチル(PMMA)、ポリイミド、ポリアミドイミド、ポリエーテルエーテルケトン、ポリエーテルイミド、ポリエステルイミド等が挙げられる。
上記充填材として、難燃剤を用いてもよい。難燃剤としては、例えば、臭素系難燃剤、塩素系難燃剤、リン系難燃剤、アンチモン系難燃剤などが挙げられる。好ましくは、安全性の観点から、ノンハロゲン-ノンアンチモン系難燃剤が用いられる。
ノンハロゲン-ノンアンチモン系難燃剤としては、例えば、アルミニウム、マグネシウム、カルシウム、ニッケル、コバルト、スズ、亜鉛、銅、鉄、チタン、ホウ素等を含む化合物が挙げられる。このような化合物(無機化合物)としては、例えば、水酸化アルミニウム、水酸化マグネシウム、酸化マグネシウム・酸化ニッケルの水和物、酸化マグネシウム・酸化亜鉛の水和物等の水和金属化合物などが挙げられる。
上記充填材は、任意の適切な表面処理が施されていてもよい。表面処理としては、例えば、シランカップリング処理、ステアリン酸処理などが挙げられる。
上記充填材のかさ密度は、好ましくは0.8g/cm3以下であり、より好ましくは0.6g/cm3以下であり、さらに好ましくは0.4g/cm3以下であり、特に好ましくは0.3g/cm3以下である。このような範囲であれば、分散性よく充填材を含有させることができ、充填材の含有量を少なくしながらも、充填材添加効果が十分に発揮され得る。充填材の含有量が少ない樹脂発泡体は、高発泡、柔軟、かつ応力分散性および外観に優れる点で有利である。充填材のかさ密度の下限値は、例えば、0.01g/cm3であり、好ましくは0.05g/cm3であり、より好ましくは0.1g/cm3である。
上記充填材の数平均粒子径(1次粒子径)は、好ましくは5μm以下であり、より好ましくは3μm以下であり、さらに好ましくは1μm以下である。このような範囲であれば、分散性よく充填材を含有させることができ、かつ、均一な気泡構造を形成することができる。その結果、応力分散性および外観に優れる樹脂発泡体を得ることができる。充填材の数平均粒子径の下限値は、例えば、0.1μmである。充填材の数平均粒子径は、水100gに充填剤1gを混合して調製した懸濁液をサンプルとして、粒度分布計(MicrtracII、マイクロトラック・ベル株式会社)を用いて、測定することができる。
上記充填材の比表面積は、好ましくは2m2/g以上であり、より好ましくは4m2/g以上であり、さらに好ましくは6m2/g以上である。このような範囲であれば、分散性よく充填材を含有させることができ、かつ、均一な気泡構造を形成することができる。その結果、応力分散性および外観に優れる樹脂発泡体を得ることができる。充填材の比表面積の上限値は、例えば、20m2/gである。充填材の比表面積は、BET法で、すなわち、吸着占有面積が既知である分子を、液体窒素による低温下で、充填材表面に吸着させ、その吸着量から測定することができる。
樹脂組成物には、本発明の効果を損なわない範囲で、任意の適切な他の成分が含まれていてもよい。このような他の成分は、1種のみであってもよいし、2種以上であってもよい。このような他の成分としては、例えば、ゴム、樹脂材料として配合されているポリマー以外の樹脂、軟化剤、脂肪族系化合物、老化防止剤、酸化防止剤、光安定剤、耐候剤、紫外線吸収剤、分散剤、可塑剤、カーボン、帯電防止剤、界面活性剤、架橋剤、増粘剤、防錆剤、シリコーン系化合物、張力改質剤、収縮防止剤、流動性改質剤、ゲル化剤、硬化剤、補強剤、発泡剤、発泡核剤、着色剤(顔料や染料等)、pH調整剤、溶剤(有機溶剤)、熱重合開始剤、光重合開始剤、滑剤、結晶核剤、結晶化促進剤、加硫剤、表面処理剤、分散助剤などが挙げられる。
A-2.樹脂発泡体の形成
本発明の樹脂発泡体は、代表的には、樹脂組成物を発泡させて得られる。発泡の方法(気泡の形成方法)としては、物理的方法や化学的方法など、発泡成形に通常用いられる方法が採用できる。すなわち、上記樹脂発泡体は、代表的には、物理的方法により発泡して形成された発泡体(物理発泡体)であってもよいし、化学的方法により発泡して形成された発泡体(化学発泡体)であってもよい。物理的方法は、一般的に、空気や窒素等のガス成分をポリマー溶液に分散させて、機械的混合により気泡を形成させるもの(機械発泡体)である。化学的方法は、一般的に、ポリマーベースに添加された発泡剤の熱分解により生じたガスによりセルを形成し、発泡体を得る方法である。
本発明の樹脂発泡体は、代表的には、樹脂組成物を発泡させて得られる。発泡の方法(気泡の形成方法)としては、物理的方法や化学的方法など、発泡成形に通常用いられる方法が採用できる。すなわち、上記樹脂発泡体は、代表的には、物理的方法により発泡して形成された発泡体(物理発泡体)であってもよいし、化学的方法により発泡して形成された発泡体(化学発泡体)であってもよい。物理的方法は、一般的に、空気や窒素等のガス成分をポリマー溶液に分散させて、機械的混合により気泡を形成させるもの(機械発泡体)である。化学的方法は、一般的に、ポリマーベースに添加された発泡剤の熱分解により生じたガスによりセルを形成し、発泡体を得る方法である。
発泡成形に付す樹脂組成物は、例えば、構成成分を、任意の適切な溶融混練装置、例えば、開放型のミキシングロール、非開放型のバンバリーミキサー、1軸押出機、2軸押出機、連続式混練機、加圧ニーダーなど、任意の適切な手段を用いて混合することにより調製すればよい。
<樹脂発泡体を形成させる実施形態1>
上記樹脂発泡体を形成させる一つの実施形態1としては、例えば、エマルション樹脂組成物(樹脂材料(ポリマー)などを含むエマルション)を機械的に発泡させて起泡化させる工程(工程A)を経て樹脂発泡体を形成する形態が挙げられる。起泡装置としては、例えば、高速せん断方式の装置、振動方式の装置、加圧ガスの吐出方式の装置などが挙げられる。これらの起泡装置の中でも、気泡径の微細化、大容量作製の観点から、高速せん断方式の装置が好ましい。上記樹脂発泡体を形成させるこの一つの実施形態1は、どのような樹脂組成物からの形成にも適用可能である。
上記樹脂発泡体を形成させる一つの実施形態1としては、例えば、エマルション樹脂組成物(樹脂材料(ポリマー)などを含むエマルション)を機械的に発泡させて起泡化させる工程(工程A)を経て樹脂発泡体を形成する形態が挙げられる。起泡装置としては、例えば、高速せん断方式の装置、振動方式の装置、加圧ガスの吐出方式の装置などが挙げられる。これらの起泡装置の中でも、気泡径の微細化、大容量作製の観点から、高速せん断方式の装置が好ましい。上記樹脂発泡体を形成させるこの一つの実施形態1は、どのような樹脂組成物からの形成にも適用可能である。
エマルションの固形分濃度は、成膜性の観点から高い方が好ましい。エマルションの固形分濃度は、好ましくは30重量%以上、より好ましくは40重量%以上、さらに好ましくは50重量%以上である。
機械的撹拌により起泡した際の気泡は、気体(ガス)がエマルション中に取り込まれたものである。ガスとしては、エマルションに対して不活性であれば、本発明の効果を損なわない範囲で任意の適切なガスを採用し得る。このようなガスとしては、例えば、空気、窒素、二酸化炭素などが挙げられる。
上記方法により起泡化したエマルション樹脂組成物(気泡含有エマルション樹脂組成物)を基材上に塗工して乾燥する工程(工程B)を経ることによって、本発明の樹脂発泡体を得ることができる。基材としては、例えば、剥離処理したプラスチックフィルム(剥離処理したポリエチレンテレフタレートフィルム等)、プラスチックフィルム(ポリエチレンテレフタレートフィルム等)等が挙げられる。
工程Bにおいて、塗工方法、乾燥方法としては、本発明の効果を損なわない範囲で任意の適切な方法を採用できる。工程Bは、基材上に塗布した気泡含有エマルション樹脂組成物を50℃以上125℃未満で乾燥する予備乾燥工程B1と、その後さらに125℃以上200℃以下で乾燥する本乾燥工程B2を含んでいることが好ましい。
予備乾燥工程B1と本乾燥工程B2を設けることにより、急激な温度上昇による気泡の合一化、気泡の破裂を防止できる。特に、厚みの小さい発泡シートでは温度の急激な上昇により気泡が合一化、破裂するので、予備乾燥工程B1を設ける意義は大きい。予備乾燥工程B1における温度は、好ましくは50℃~100℃である。予備乾燥工程B1の時間は、好ましくは0.5分~30分であり、より好ましくは1分~15分である。本乾燥工程B2における温度は、好ましくは130℃~180℃以下であり、より好ましくは130℃~160℃である。本乾燥工程B2の時間は、好ましくは0.5分~30分であり、より好ましくは1分~15分である。
<樹脂発泡体を形成させる実施形態2>
上記樹脂発泡体を形成させる一つの実施形態2としては、樹脂組成物を発泡剤により発泡させて発泡体を形成する形態が挙げられる。発泡剤としては、発泡成形に通常用いられるものを使用でき、環境保護及び被発泡体に対する低汚染性の観点から、高圧の不活性ガスを用いることが好ましい。
上記樹脂発泡体を形成させる一つの実施形態2としては、樹脂組成物を発泡剤により発泡させて発泡体を形成する形態が挙げられる。発泡剤としては、発泡成形に通常用いられるものを使用でき、環境保護及び被発泡体に対する低汚染性の観点から、高圧の不活性ガスを用いることが好ましい。
不活性ガスとしては、樹脂組成物に対して不活性で且つ含浸可能なものであれば、任意の適切な不活性ガスを採用し得る。このような不活性ガスとしては、例えば、二酸化炭素、窒素ガス、空気などが挙げられる。これらのガスは混合して用いてもよい。これらのうち、樹脂材料(ポリマー)への含浸量が多く、含浸速度の速いという観点から、二酸化炭素が好ましい。
不活性ガスは超臨界状態であることが好ましい。すなわち、超臨界状態の二酸化炭素を用いることが特に好ましい。超臨界状態では、樹脂組成物への不活性ガスの溶解度がより増大し、不活性ガスの高濃度の混入が可能であるとともに、急激な圧力降下時に不活性ガスが高濃度となるため、気泡核の発生が多くなり、その気泡核が成長してできる気泡の密度が、気孔率が同じであっても他の状態の場合より大きくなるため、微細な気泡を得ることができる。なお、二酸化炭素の臨界温度は31℃、臨界圧力は7.4MPaである。
樹脂組成物に高圧の不活性ガスを含浸させることにより発泡体を形成する方法としては、例えば、樹脂材料(ポリマー)を含む樹脂組成物に不活性ガスを高圧下で含浸させるガス含浸工程、該工程後に圧力を低下させて樹脂材料(ポリマー)を発泡させる減圧工程、および、必要に応じて加熱により気泡を成長させる加熱工程を経て形成する方法などが挙げられる。この場合、予め成形した未発泡成形体を不活性ガスに含浸させてもよく、また、溶融した樹脂組成物に不活性ガスを加圧状態下で含浸させた後に減圧の際に成形に付してもよい。これらの工程は、バッチ方式、連続方式のいずれの方式で行ってもよい。すなわち、予め樹脂組成物を、シート状などの適宜な形状に成形して未発泡樹脂成形体とした後、この未発泡樹脂成形体に、高圧のガスを含浸させ、圧力を解放することにより発泡させるバッチ方式であってもよく、樹脂組成物を加圧下、高圧のガスと共に混練し、成形すると同時に圧力を解放し、成形と発泡を同時に行う連続方式であってもよい。
バッチ方式で発泡体を製造する例を以下に示す。例えば、樹脂組成物を単軸押出機、2軸押出機等の押出機を使用して押し出すことにより、発泡体成形用樹脂シートを作製する。あるいは、樹脂組成物を、ローラ、カム、ニーダー、バンバリ型等の羽根を設けた混練機を使用して均一に混練しておき、熱板のプレスなどを用いて所定の厚みにプレス加工することにより、未発泡樹脂成形体を作製する。こうして得られた未発泡樹脂成形体を高圧容器中に入れて、高圧不活性ガス(超臨界状態の二酸化炭素など)を注入し、未発泡樹脂成形体中に不活性ガスを含浸させる。十分に不活性ガスを含浸させた時点で圧力を解放し(通常、大気圧まで)、樹脂中に気泡核を発生させる。気泡核はそのまま室温で成長させてもよいが、場合によっては加熱することによって成長させてもよい。加熱の方法としては、ウォーターバス、オイルバス、熱ロール、熱風オーブン、遠赤外線、近赤外線、マイクロ波などの公知や慣用の方法を採用できる。このようにして気泡を成長させた後、冷水などにより急激に冷却し、形状を固定化することにより発泡体を得ることができる。なお、発泡に供する未発泡樹脂成形体はシート状物に限らず、用途に応じて種々の形状のものを使用できる。また、発泡に供する未発泡樹脂成形体は押出成形、プレス成形のほか、射出成形等の他の成形法により作製することもできる。
連続方式で発泡体を製造する例を以下に示す。例えば、樹脂組成物を、単軸押出機、二軸押出機等の押出機を使用して混練しながら、高圧のガス(特に不活性ガス、さらには二酸化炭素)を注入(導入)し、十分に高圧のガスを樹脂組成物に含浸させる混練含浸工程、押出機の先端に設けられたダイスなどを通して樹脂組成物を押し出すことにより圧力を解放し(通常、大気圧まで)、成形と発泡を同時に行う成形減圧工程により発泡成形する。また、連続方式での発泡成形の際には、必要に応じて、加熱することによって気泡を成長させる加熱工程を設けてもよい。このようにして気泡を成長させた後、必要により冷水などにより急激に冷却し、形状を固定化してもよい。また、高圧のガスの導入は連続的に行ってもよく不連続的に行ってもよい。さらに、混練含浸工程および成形減圧工程では、例えば、押出機や射出成形機を用い得る。なお、気泡核を成長させる際の加熱の方法としては、ウォーターバス、オイルバス、熱ロール、熱風オーブン、遠赤外線、近赤外線、マイクロ波などの任意の適切な方法が挙げられる。発泡体の形状としては、任意の適切な形状を採用し得る。このような形状としては、例えば、シート状、角柱状、円筒状、異型状などが挙げられる。
樹脂組成物を発泡成形する際のガスの混合量は、高発泡な発泡体を得られ得る点で、例えば、樹脂組成物全量に対して、好ましくは2重量%~10重量%であり、より好ましくは2.5重量%~8重量%であり、さらに好ましくは3重量%~6重量%である。
不活性ガスを樹脂組成物に含浸させるときの圧力は、操作性等を考慮して適宜選択できる。このような圧力は、例えば、好ましくは6MPa以上(例えば、6MPa~100MPa)であり、より好ましくは8MPa以上(例えば、8MPa~50MPa)である。なお、超臨界状態の二酸化炭素を用いる場合の圧力は、二酸化炭素の超臨界状態を保持する観点から、好ましくは7.4MPa以上である。圧力が6MPaより低い場合には、発泡時の気泡成長が著しく、気泡径が大きくなりすぎて、好ましい平均セル径(平均気泡径)を得ることができない場合がある。これは、圧力が低いとガスの含浸量が高圧時に比べて相対的に少なく、気泡核形成速度が低下して形成される気泡核数が少なくなるため、1気泡あたりのガス量が逆に増えて気泡径が極端に大きくなるからである。また、6MPaより低い圧力領域では、含浸圧力を少し変化させるだけで気泡径、気泡密度が大きく変わるため、気泡径及び気泡密度の制御が困難になりやすい。
ガス含浸工程における温度は、用いる不活性ガスや樹脂組成物中の成分の種類等によって異なり、広い範囲で選択できる。操作性等を考慮した場合、好ましくは10℃~350℃である。未発泡成形体に不活性ガスを含浸させる場合の含浸温度は、バッチ式では、好ましくは10℃~250℃であり、より好ましくは40℃~230℃である。また、ガスを含浸させた溶融ポリマーを押し出して発泡と成形とを同時に行う場合の含浸温度は、連続式では、好ましくは60℃~350℃である。なお、不活性ガスとして二酸化炭素を用いる場合には、超臨界状態を保持するため、含浸時の温度は、好ましくは32℃以上であり、より好ましくは40℃以上である。
減圧工程において、減圧速度としては、均一な微細気泡を得るため、好ましくは5MPa/秒~300MPa/秒である。
加熱工程における加熱温度は、好ましくは40℃~250℃であり、より好ましくは60℃~250℃である。
B.発泡部材
図1は、本発明の1つの実施形態による発泡部材の概略断面図である。発泡部材100は、樹脂発泡層10と、樹脂発泡層10の少なくとも一方の側に配置された粘着剤層20とを有する。樹脂発泡層10は、上記樹脂発泡体により構成される。
図1は、本発明の1つの実施形態による発泡部材の概略断面図である。発泡部材100は、樹脂発泡層10と、樹脂発泡層10の少なくとも一方の側に配置された粘着剤層20とを有する。樹脂発泡層10は、上記樹脂発泡体により構成される。
粘着剤層の厚さは、好ましくは5μm~300μmであり、より6μm~200μmであり、さらに好ましくは7μm~100μmであり、特に好ましくは8μm~50μmである。粘着剤層の厚さが上記範囲内にあることによって、上記発泡部材は、優れた衝撃吸収性を発揮できる。
粘着剤層としては、任意の適切な粘着剤からなる層を採用し得る。粘着剤層を構成する粘着剤としては、例えば、ゴム系粘着剤(合成ゴム系粘着剤、天然ゴム系粘着剤など)、ウレタン系粘着剤、アクリルウレタン系粘着剤、アクリル系粘着剤、シリコーン系粘着剤、ポリエステル系粘着剤、ポリアミド系粘着剤、エポキシ系粘着剤、ビニルアルキルエーテル系粘着剤、フッ素系粘着剤、ゴム系粘着剤などが挙げられる。粘着剤層を構成する粘着剤としては、好ましくは、アクリル系粘着剤、シリコーン系粘着剤、ゴム系粘着剤から選ばれる少なくとも1種である。このような粘着剤は、1種のみであってもよいし、2種以上であってもよい。粘着剤層は、1層であってもよいし、2層以上であってもよい。
粘着剤としては、粘着形態で分類すると、例えば、エマルジョン型粘着剤、溶剤型粘着剤、紫外線架橋型(UV架橋型)粘着剤、電子線架橋型(EB架橋型)粘着剤、熱溶融型粘着剤(ホットメルト型粘着剤)などが挙げられる。このような粘着剤は、1種のみであってもよいし、2種以上であってもよい。
粘着剤層の水蒸気透湿度は、好ましくは50(g/(m2・24時間))以下であり、より好ましくは30(g/(m2・24時間))以下であり、さらに好ましくは20(g/(m2・24時間))以下であり、特に好ましくは10(g/(m2・24時間))以下である。粘着剤層の水蒸気透湿度が上記範囲内にあれば、上記発泡シートは、水分による影響を受けずに衝撃吸収性を安定化させることができる。なお、水蒸気透湿度は、例えば、JIS Z 0208に準じた方法により、40℃、相対湿度92%の試験条件で測定することができる。
粘着剤層を構成する粘着剤には、本発明の効果を損なわない範囲で、任意の適切な他の成分を含んでいてもよい。他の成分としては、例えば、他のポリマー成分、軟化剤、老化防止剤、硬化剤、可塑剤、充填材、酸化防止剤、熱重合開始剤、光重合開始剤、紫外線吸収剤、光安定剤、着色剤(顔料や染料など)、溶剤(有機溶剤)、界面活性剤(例えば、イオン性界面活性剤、シリコーン系界面活性剤、フッ素系界面活性剤など)、架橋剤(例えば、ポリイソシアネート系架橋剤、シリコーン系架橋剤、エポキシ系架橋剤、アルキルエーテル化メラミン系架橋剤など)などが挙げられる。なお、熱重合開始剤や光重合開始剤は、ポリマー成分を形成するための材料に含まれ得る。
上記発泡部材は、任意の適切な方法によって製造し得る。上記発泡部材は、例えば、樹脂発泡層と粘着剤層とを積層して製造する方法や、粘着剤層の形成材料と樹脂発泡層を積層した後に硬化反応等によって粘着剤層を形成させて製造する方法などが挙げられる。
以下、実施例により本発明を具体的に説明するが、本発明はこれら実施例になんら限定されるものではない。なお、実施例等における、試験および評価方法は以下のとおりである。なお、「部」と記載されている場合は、特記事項がない限り「重量部」を意味し、「%」と記載されている場合は、特記事項がない限り「重量%」を意味する。
[評価方法]
<50%圧縮荷重>
JIS K 6767に記載されている発泡体の圧縮硬さ測定方法に準じて測定した。具体的には、実施例・比較例で得られた樹脂発泡体を30mm×30mmサイズに切り出して試験片とし、圧縮速度10mm/minで圧縮率が50%となるまで圧縮したときの応力(N)を単位面積(1cm2)当たりに換算して、25%圧縮荷重(N/cm2)、50%圧縮荷重(N/cm2)、とした。
<50%圧縮荷重>
JIS K 6767に記載されている発泡体の圧縮硬さ測定方法に準じて測定した。具体的には、実施例・比較例で得られた樹脂発泡体を30mm×30mmサイズに切り出して試験片とし、圧縮速度10mm/minで圧縮率が50%となるまで圧縮したときの応力(N)を単位面積(1cm2)当たりに換算して、25%圧縮荷重(N/cm2)、50%圧縮荷重(N/cm2)、とした。
<繰り返し圧縮による厚み変化率>
マイクロサーボ(MMT-250NV-10、島津製作所製)を用いて、先端が平坦の円盤治具(圧縮用アタッチメント円盤型圧縮治具 PC-5040、接触面積:4.9cm2、株式会社イマダ)を発泡体に50%圧縮、10%圧縮となるように繰り返し治具を変位させ、5000回繰り返し圧縮を行った(初期荷重0.15N、ダウンスピード5min/mm、アップスピード5min/mm)。試験後の厚みをデジタルマイクロスコープ(商品名「VHX-500」、キーエンス株式会社製)を用いて測定した。試験前の厚みZ0、試験後の厚みをZ1とし、(Z0-Z1)/Zo×100 の式により、繰り返し圧縮による厚み変化率を算出した。
マイクロサーボ(MMT-250NV-10、島津製作所製)を用いて、先端が平坦の円盤治具(圧縮用アタッチメント円盤型圧縮治具 PC-5040、接触面積:4.9cm2、株式会社イマダ)を発泡体に50%圧縮、10%圧縮となるように繰り返し治具を変位させ、5000回繰り返し圧縮を行った(初期荷重0.15N、ダウンスピード5min/mm、アップスピード5min/mm)。試験後の厚みをデジタルマイクロスコープ(商品名「VHX-500」、キーエンス株式会社製)を用いて測定した。試験前の厚みZ0、試験後の厚みをZ1とし、(Z0-Z1)/Zo×100 の式により、繰り返し圧縮による厚み変化率を算出した。
<見かけ密度>
樹脂発泡体の密度(見かけ密度)は、以下のように算出した。実施例・比較例で得られた樹脂発泡体を20mm×20mmサイズに打ち抜いて試験片とし、試験片の寸法をノギスで測定した。次に、試験片の重量を電子天秤にて測定した。そして、次式により算出した。
見かけ密度(g/cm3)=試験片の重量/試験片の体積
樹脂発泡体の密度(見かけ密度)は、以下のように算出した。実施例・比較例で得られた樹脂発泡体を20mm×20mmサイズに打ち抜いて試験片とし、試験片の寸法をノギスで測定した。次に、試験片の重量を電子天秤にて測定した。そして、次式により算出した。
見かけ密度(g/cm3)=試験片の重量/試験片の体積
<平均気泡径(平均セル径)、気泡径(セル径)の変動係数>
樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)、かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、計測器としてデジタルマイクロスコープ(商品名「VHX-500」、キーエンス株式会社製)を用い、樹脂発泡体の気泡部の拡大画像を取り込み、同計測器の解析ソフトを用いて、画像解析することにより、数平均気泡径(平均セル径)(μm)を求めた。なお、取り込んだ拡大画像の気泡数は400個程度であった。また、セル径の全データから標準偏差を計算し、以下の式を用いて変動係数を算出した。
変動係数=標準偏差/平均気泡径(平均セル径)
樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)、かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、計測器としてデジタルマイクロスコープ(商品名「VHX-500」、キーエンス株式会社製)を用い、樹脂発泡体の気泡部の拡大画像を取り込み、同計測器の解析ソフトを用いて、画像解析することにより、数平均気泡径(平均セル径)(μm)を求めた。なお、取り込んだ拡大画像の気泡数は400個程度であった。また、セル径の全データから標準偏差を計算し、以下の式を用いて変動係数を算出した。
変動係数=標準偏差/平均気泡径(平均セル径)
<気泡のアスペクト比>
計測器としてデジタルマイクロスコープ(商品名「VHX-2000、キーエンス株式会社製)を用い、下記の方法で、実施例・比較例で得られた樹脂発泡体が有する気泡のスペクト比を測定した。
・樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)、かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、切断面をマイクロスコープ(例えば、キーエンス製「VHX-2000」)を用いて、所定面積(3mm2)範囲を倍率100倍で観察し、気泡一個の厚み方向の長さとTDの長さを測定した。
・同様の測定を所定面積内に存在する全ての気泡に対して行った。
・気泡のアスペクト比は、TDの長さ÷厚み方向の長さで計算され、全ての気泡で同様の計算を行い、平均した値を「樹脂発泡体が有する気泡のアスペクト比」とした。
計測器としてデジタルマイクロスコープ(商品名「VHX-2000、キーエンス株式会社製)を用い、下記の方法で、実施例・比較例で得られた樹脂発泡体が有する気泡のスペクト比を測定した。
・樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)、かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、切断面をマイクロスコープ(例えば、キーエンス製「VHX-2000」)を用いて、所定面積(3mm2)範囲を倍率100倍で観察し、気泡一個の厚み方向の長さとTDの長さを測定した。
・同様の測定を所定面積内に存在する全ての気泡に対して行った。
・気泡のアスペクト比は、TDの長さ÷厚み方向の長さで計算され、全ての気泡で同様の計算を行い、平均した値を「樹脂発泡体が有する気泡のアスペクト比」とした。
<気泡率(セル率)>
温度23℃、湿度50%の環境下で測定を行った。100mm×100mmの打抜き刃型(2枚の加工刃(商品名「NCA07」、厚さ0.5mm、刃先角度45°、ナカヤマ社製))にて実施例・比較例で得られた樹脂発泡体を打抜き、打抜いた試料の寸法を測定した。また、測定端子の直径(φ)20mmである1/100ダイヤルゲージにて厚みを測定した。これらの値から実施例・比較例で得られた樹脂発泡体の体積を算出した。次に、実施例・比較例で得られた樹脂発泡体の重量を最小目盛り0.01g以上の上皿天秤にて測定した。これらの値より、実施例・比較例で得られた樹脂発泡体の気泡率(セル率)を算出した。
温度23℃、湿度50%の環境下で測定を行った。100mm×100mmの打抜き刃型(2枚の加工刃(商品名「NCA07」、厚さ0.5mm、刃先角度45°、ナカヤマ社製))にて実施例・比較例で得られた樹脂発泡体を打抜き、打抜いた試料の寸法を測定した。また、測定端子の直径(φ)20mmである1/100ダイヤルゲージにて厚みを測定した。これらの値から実施例・比較例で得られた樹脂発泡体の体積を算出した。次に、実施例・比較例で得られた樹脂発泡体の重量を最小目盛り0.01g以上の上皿天秤にて測定した。これらの値より、実施例・比較例で得られた樹脂発泡体の気泡率(セル率)を算出した。
<気泡壁(セル壁)の厚み>
樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)、かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、計測器としてデジタルマイクロスコープ(商品名「VHX-500」、キーエンス株式会社製)を用いて、樹脂発泡体の気泡部の拡大画像を取り込み、同計測器の解析ソフトを用いて、画像解析することにより、気泡壁(セル壁)の厚み(μm)を求めた。なお、取り込んだ拡大画像の気泡数は400個程度であった。
樹脂発泡体を、カミソリ刃を用いて、TD(流れ方向に直交する方向)、かつ、樹脂発泡体の主面に対して垂直方向(厚み方向)に切断し、計測器としてデジタルマイクロスコープ(商品名「VHX-500」、キーエンス株式会社製)を用いて、樹脂発泡体の気泡部の拡大画像を取り込み、同計測器の解析ソフトを用いて、画像解析することにより、気泡壁(セル壁)の厚み(μm)を求めた。なお、取り込んだ拡大画像の気泡数は400個程度であった。
<非発泡曲げ応力>
樹脂発泡体を、真空プレス成型機(IVM-70:岩城工業社)を用いて、(融点+70℃)の温度、15MPaの圧力で5分間、プレスすることにより、非発泡状態の樹脂成形体aを得た。
樹脂成形体aを、幅6mm、長さ50mmに切り出してサンプルとし、このサンプルを支点間距離25mmの3点曲げ冶具の上に置き、23℃×50%RHの環境下で、押し込み速度0.5mm/minで押し込み試験(島津製作所社製、商品名「AG-Xplus」)を行い、当該サンプルを5mm押し込んだときの荷重(g)を非発泡曲げ応力とした。
樹脂発泡体を、真空プレス成型機(IVM-70:岩城工業社)を用いて、(融点+70℃)の温度、15MPaの圧力で5分間、プレスすることにより、非発泡状態の樹脂成形体aを得た。
樹脂成形体aを、幅6mm、長さ50mmに切り出してサンプルとし、このサンプルを支点間距離25mmの3点曲げ冶具の上に置き、23℃×50%RHの環境下で、押し込み速度0.5mm/minで押し込み試験(島津製作所社製、商品名「AG-Xplus」)を行い、当該サンプルを5mm押し込んだときの荷重(g)を非発泡曲げ応力とした。
<圧縮弾性率>
JIS K 6767の圧縮試験の項に基づいて、樹脂発泡体の圧縮率(%)、圧縮反発力(kPa)を測定し、圧縮率5%の時の圧縮反発力F(5%)と圧縮率15%の時の圧縮j反発力F(15%)をそれぞれ読み取り、F(5%)、F(15%)から、(F(15%)-F(5%))/(15-5)×100の式により、圧縮弾性率を算出した。
JIS K 6767の圧縮試験の項に基づいて、樹脂発泡体の圧縮率(%)、圧縮反発力(kPa)を測定し、圧縮率5%の時の圧縮反発力F(5%)と圧縮率15%の時の圧縮j反発力F(15%)をそれぞれ読み取り、F(5%)、F(15%)から、(F(15%)-F(5%))/(15-5)×100の式により、圧縮弾性率を算出した。
<厚み回復率>
樹脂発泡体に、樹脂発泡体に1000g/cm2の荷重を加えた状態で120秒間維持し、圧縮を解除し、解除してから0.5秒後の樹脂発泡体の厚み(圧縮状態を解除してから0.5秒後の厚み)を測定した。「圧縮状態を解除してから0.5秒後の厚み」と、荷重を加える前の樹脂発泡体の厚み(初期厚み)とから、下記の式により、厚み回復率を求めた。
厚み回復率(%)={(圧縮状態を解除してから0.5秒後の厚み)/(初期厚み)}×100
樹脂発泡体に、樹脂発泡体に1000g/cm2の荷重を加えた状態で120秒間維持し、圧縮を解除し、解除してから0.5秒後の樹脂発泡体の厚み(圧縮状態を解除してから0.5秒後の厚み)を測定した。「圧縮状態を解除してから0.5秒後の厚み」と、荷重を加える前の樹脂発泡体の厚み(初期厚み)とから、下記の式により、厚み回復率を求めた。
厚み回復率(%)={(圧縮状態を解除してから0.5秒後の厚み)/(初期厚み)}×100
<動的防塵性>
樹脂発泡体を、図2に示す額縁状(窓枠状)(幅:2mm)に打ち抜き、評価用サンプルを作製した。
この評価用サンプルを、図3に示すように、評価容器2(後述の動的防塵性評価用の評価容器、図3及び図4参照)に装着した。図3に示すように、評価用サンプル22は、フォーム圧縮板27と、ベース板24に固定されたアルミニウム板上の黒色アクリル板212との間に設けられている。評価用サンプル22を装着した評価容器2では、評価用サンプル22により、内部の一定領域が閉じられた系となっている。
図3に示すように、評価用サンプル22を評価容器2に装着後、粉末供給部25に粉塵としてのコーンスターチを0.1g入れて、評価容器をドラム式落下試験器(回転式落下装置)に入れ、3rpmの速度で回転させた。
そして、10回転させた後、粉末供給部から、樹脂発泡体を通過して、アルミニウム板上の黒色アクリル板212及びカバー板としての黒色アクリル板211に付着した粒子を、マイクロスコープで観察した。アルミニウム板側の黒色アクリル板212及びカバー板側の黒色アクリル板211について静止画像を作成し、画像解析ソフトを用いて2値化処理を行い、粒子の個数として粒子総面積を計測した。なお、観察は、空気中の浮遊粉塵の影響を少なくするためクリーンベンチ内で行った。
アルミニウム板側の黒色アクリル板に付着している粒子及びカバー板側の黒色アクリル板に付着している粒子を合わせた粒子総面積が、1500[Pixel×Pixel]未満である場合を良好(〇)と判定し、1500~2000[Pixel×Pixel]である場合をやや不良(△)と判定し、2000[Pixel×Pixel]を越える場合を不良(×)と判定した。
なお、図3及び4において、2は評価用サンプルを装着した評価容器(動的防塵性評価用の評価容器)、211は黒色アクリル板(カバー板側の黒色アクリル板)、212は黒色アクリル板(アルミニウム板側の黒色アクリル板)、22は評価用サンプル(額縁状の発泡シール材)、23はアルミニウム板、24はベース板、25は粉末供給部、26はネジ、27はフォーム圧縮板、28はカバー板固定金具を示す。
樹脂発泡体を、図2に示す額縁状(窓枠状)(幅:2mm)に打ち抜き、評価用サンプルを作製した。
この評価用サンプルを、図3に示すように、評価容器2(後述の動的防塵性評価用の評価容器、図3及び図4参照)に装着した。図3に示すように、評価用サンプル22は、フォーム圧縮板27と、ベース板24に固定されたアルミニウム板上の黒色アクリル板212との間に設けられている。評価用サンプル22を装着した評価容器2では、評価用サンプル22により、内部の一定領域が閉じられた系となっている。
図3に示すように、評価用サンプル22を評価容器2に装着後、粉末供給部25に粉塵としてのコーンスターチを0.1g入れて、評価容器をドラム式落下試験器(回転式落下装置)に入れ、3rpmの速度で回転させた。
そして、10回転させた後、粉末供給部から、樹脂発泡体を通過して、アルミニウム板上の黒色アクリル板212及びカバー板としての黒色アクリル板211に付着した粒子を、マイクロスコープで観察した。アルミニウム板側の黒色アクリル板212及びカバー板側の黒色アクリル板211について静止画像を作成し、画像解析ソフトを用いて2値化処理を行い、粒子の個数として粒子総面積を計測した。なお、観察は、空気中の浮遊粉塵の影響を少なくするためクリーンベンチ内で行った。
アルミニウム板側の黒色アクリル板に付着している粒子及びカバー板側の黒色アクリル板に付着している粒子を合わせた粒子総面積が、1500[Pixel×Pixel]未満である場合を良好(〇)と判定し、1500~2000[Pixel×Pixel]である場合をやや不良(△)と判定し、2000[Pixel×Pixel]を越える場合を不良(×)と判定した。
なお、図3及び4において、2は評価用サンプルを装着した評価容器(動的防塵性評価用の評価容器)、211は黒色アクリル板(カバー板側の黒色アクリル板)、212は黒色アクリル板(アルミニウム板側の黒色アクリル板)、22は評価用サンプル(額縁状の発泡シール材)、23はアルミニウム板、24はベース板、25は粉末供給部、26はネジ、27はフォーム圧縮板、28はカバー板固定金具を示す。
〔実施例1〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)65重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)35重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)80重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して4.5重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体a1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体a1を通過させて、厚みが0.1mmの樹脂発泡体Aを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Aが得られるように設定した。
得られた樹脂発泡体Aを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)65重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)35重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)80重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して4.5重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体a1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体a1を通過させて、厚みが0.1mmの樹脂発泡体Aを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Aが得られるように設定した。
得られた樹脂発泡体Aを上記評価に供した。結果を表1に示す。
〔実施例2〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)65重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)35重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)100重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して3.5重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体b1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体b1を通過させて、厚みが0.1mmの樹脂発泡体Bを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Bが得られるように設定した。 得られた樹脂発泡体Bを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)65重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)35重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)100重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して3.5重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体b1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体b1を通過させて、厚みが0.1mmの樹脂発泡体Bを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Bが得られるように設定した。 得られた樹脂発泡体Bを上記評価に供した。結果を表1に示す。
〔実施例3〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)65重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)35重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)120重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して3.0重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体c1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体c1を通過させて、厚みが0.1mmの樹脂発泡体Cを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Cが得られるように設定した。
得られた樹脂発泡体Cを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)65重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)35重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)120重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して3.0重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体c1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体c1を通過させて、厚みが0.1mmの樹脂発泡体Cを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Cが得られるように設定した。
得られた樹脂発泡体Cを上記評価に供した。結果を表1に示す。
〔実施例4〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)15重量部、ポリプロピレン(メルトフローレート(MFR)(230℃):1.1g/10min)15重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)75重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)80重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して2.8重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.4mmの発泡構造体d1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体d1を通過させて、厚みが0.1mmの樹脂発泡体Dを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Dが得られるように設定した。
得られた樹脂発泡体Dを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)15重量部、ポリプロピレン(メルトフローレート(MFR)(230℃):1.1g/10min)15重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)75重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)80重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して2.8重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.4mmの発泡構造体d1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体d1を通過させて、厚みが0.1mmの樹脂発泡体Dを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Dが得られるように設定した。
得られた樹脂発泡体Dを上記評価に供した。結果を表1に示す。
〔実施例5〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)15重量部、ポリプロピレン(メルトフローレート(MFR)(230℃):1.1g/10min)15重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)75重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)60重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して2.6重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.4mmの発泡構造体e1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体e1を通過させて、厚みが0.1mmの樹脂発泡体Eを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Eが得られるように設定した。
得られた樹脂発泡体Eを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)15重量部、ポリプロピレン(メルトフローレート(MFR)(230℃):1.1g/10min)15重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)75重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)60重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して2.6重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.4mmの発泡構造体e1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体e1を通過させて、厚みが0.1mmの樹脂発泡体Eを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Eが得られるように設定した。
得られた樹脂発泡体Eを上記評価に供した。結果を表1に示す。
〔比較例1〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)45重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)55重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)10重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して5.5重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体f1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体f1を通過させて、厚みが0.1mmの樹脂発泡体Fを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Gが得られるように設定した。
得られた樹脂発泡体Fを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)45重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)55重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)10重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して5.5重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.3mmの発泡構造体f1を得た。
さらに、一方のロールが200℃に加熱された一対のロールにおけるロール間(ロールとロールの間の隙間)に、上記樹脂発泡体f1を通過させて、厚みが0.1mmの樹脂発泡体Fを得た。なお、ロール間のギャップ(隙間)は、厚みが0.1mmの樹脂発泡体Gが得られるように設定した。
得られた樹脂発泡体Fを上記評価に供した。結果を表1に示す。
〔比較例2〕
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)15重量部、ポリプロピレン(メルトフローレート(MFR)(230℃):1.1g/10min)15重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)75重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)80重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して2.8重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.1mmの樹脂発泡体Gを得た。
得られた樹脂発泡体Gを上記評価に供した。結果を表1に示す。
ポリプロピレン(メルトフローレート(MFR)(230℃):0.40g/10min)15重量部、ポリプロピレン(メルトフローレート(MFR)(230℃):1.1g/10min)15重量部、ポリオレフィン系エラストマー(メルトフローレート(MFR):6g/10min、JIS A硬度:79°)75重量部、水酸化マグネシウム(商品名「KISUMA 5P」協和化学工業製)80重量部、カーボン(商品名「旭♯35」旭カーボン株式会社製)10重量部、およびステアリン酸モノグリセリド1重量部を、日本製鋼所(JSW)社製の二軸混練機にて、200℃の温度で混練した後、ストランド状に押出し、水冷後ペレット状に成形した。このペレットを、日本製鋼所社製の単軸押出機に投入し、220℃の雰囲気下、13MPa(注入後12MPa)の圧力で、二酸化炭素ガスを注入した。二酸化炭素ガスは、樹脂100重量部に対して2.8重量部の割合で注入した。二酸化炭素ガスを十分飽和させた後、発泡に適した温度まで冷却後、ダイから押出して、シート状成形体を得た。当該シート状成形体をスライサーを用いて薄膜化し、厚みが0.1mmの樹脂発泡体Gを得た。
得られた樹脂発泡体Gを上記評価に供した。結果を表1に示す。

本発明の樹脂発泡体は、例えば、電子機器用のクッション材として好適に利用できる。
100 発泡部材
10 樹脂発泡層
20 粘着剤層
2 評価用サンプルを装着した評価容器(動的防塵性評価用の評価容器)
211 黒色アクリル板(カバー板側の黒色アクリル板)
212 黒色アクリル板(アルミニウム板側の黒色アクリル板)
22 評価用サンプル(額縁状の発泡シール材)
23 アルミニウム板
24 ベース板
25 粉末供給部
26 ネジ
27 フォーム圧縮板
28 カバー板固定金具
10 樹脂発泡層
20 粘着剤層
2 評価用サンプルを装着した評価容器(動的防塵性評価用の評価容器)
211 黒色アクリル板(カバー板側の黒色アクリル板)
212 黒色アクリル板(アルミニウム板側の黒色アクリル板)
22 評価用サンプル(額縁状の発泡シール材)
23 アルミニウム板
24 ベース板
25 粉末供給部
26 ネジ
27 フォーム圧縮板
28 カバー板固定金具
Claims (15)
- 50%圧縮荷重が5N/cm2以上であり、
下記試験で測定される繰り返し圧縮による厚み変化率が10%以下であり、
気泡構造を有する、
樹脂発泡体;
<繰り返し圧縮試験>
先端が平坦な円盤治具(接触面積:4.9cm2)を樹脂発泡体に押し付け、50%圧縮/10%圧縮(厚み基準)サイクルを5000回行った際の厚みの変化率を測定する。 - 50%圧縮荷重が30N/cm2以下である、請求項1に記載の樹脂発泡体。
- 圧縮弾性率が、200kPa以上である、請求項1または2に記載の樹脂発泡体。
- 非発泡曲げ応力が、5MPa以上である、請求項1から3のいずれかに記載の樹脂発泡体。
- みかけ密度が、0.02g/cm3~0.50g/cm3である、請求項1から4のいずれかに記載の樹脂発泡体。
- 厚み回復率が、70%以上である、請求項1から5のいずれかに記載の樹脂発泡体。
- 前記樹脂発泡体が有する気泡のアスペクト比が、1.5以上である、請求項1から6のいずれかに記載の樹脂発泡体。
- 平均気泡径が10μm~200μmである、請求項1から7のいずれかに記載の樹脂発泡体。
- 気泡率が、30%以上である、請求項1から8のいずれかに記載の樹脂発泡体。
- 気泡径の変動係数が、0.5以下である、請求項1から9のいずれかに記載の樹脂発泡体。
- 気泡壁の厚みが、0.1μm~10μmである、請求項1から10のいずれかに記載の樹脂発泡体。
- ポリオレフィン系樹脂を含む、請求項1から11のいずれかに記載の樹脂発泡体。
- 前記ポリオレフィン系樹脂が、ポリオレフィン系エラストマー以外のポリオレフィンとポリオレフィン系エラストマーの混合物である、請求項12に記載の樹脂発泡体。
- 片面または両面に、熱溶融層を有する、請求項1から13のいずれかに記載の樹脂発泡体。
- 樹脂発泡層と、該樹脂発泡層の少なくとも一方の側に配置された粘着剤層を有し、
該樹脂発泡層が、請求項1から14のいずれかに記載の樹脂発泡体である、
発泡部材。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202080075112.7A CN114641521A (zh) | 2019-11-25 | 2020-11-25 | 树脂发泡体 |
| JP2021561443A JPWO2021106912A1 (ja) | 2019-11-25 | 2020-11-25 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-212436 | 2019-11-25 | ||
| JP2019212436 | 2019-11-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021106912A1 true WO2021106912A1 (ja) | 2021-06-03 |
Family
ID=76129410
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/043760 WO2021106911A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
| PCT/JP2020/043761 WO2021106912A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
| PCT/JP2020/043763 WO2021106914A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
| PCT/JP2020/043762 WO2021106913A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
| PCT/JP2020/043759 WO2021106910A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/043760 WO2021106911A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/043763 WO2021106914A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
| PCT/JP2020/043762 WO2021106913A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
| PCT/JP2020/043759 WO2021106910A1 (ja) | 2019-11-25 | 2020-11-25 | 樹脂発泡体 |
Country Status (4)
| Country | Link |
|---|---|
| JP (5) | JPWO2021106911A1 (ja) |
| KR (1) | KR20220106752A (ja) |
| CN (5) | CN114616278A (ja) |
| WO (5) | WO2021106911A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023139878A1 (ja) * | 2022-01-18 | 2023-07-27 | 日東電工株式会社 | 発泡部材 |
| WO2023139879A1 (ja) * | 2022-01-18 | 2023-07-27 | 日東電工株式会社 | 発泡部材 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2023176984A1 (ja) * | 2022-03-18 | 2023-09-21 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001348452A (ja) * | 2000-06-05 | 2001-12-18 | Nitto Denko Corp | ポリオレフィン系樹脂発泡体及びその製造方法 |
| WO2015146756A1 (ja) * | 2014-03-26 | 2015-10-01 | 日東電工株式会社 | 樹脂発泡体、発泡部材、及び、タッチパネル搭載機器 |
| JP2016088977A (ja) * | 2014-10-31 | 2016-05-23 | 日東電工株式会社 | 樹脂発泡体及び発泡部材 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5670102A (en) | 1993-02-11 | 1997-09-23 | Minnesota Mining And Manufacturing Company | Method of making thermoplastic foamed articles using supercritical fluid |
| JP5701508B2 (ja) * | 2009-03-04 | 2015-04-15 | 日東電工株式会社 | 導電性を有する樹脂発泡体 |
| JP2010242061A (ja) * | 2009-03-19 | 2010-10-28 | Nitto Denko Corp | 難燃性樹脂発泡体及び難燃性発泡部材 |
| JP5914141B2 (ja) * | 2011-07-29 | 2016-05-11 | 日東電工株式会社 | ポリオレフィン系樹脂発泡体用樹脂組成物、ポリオレフィン系樹脂発泡体、発泡体製造方法、及び発泡シール材 |
| WO2013168798A1 (ja) * | 2012-05-11 | 2013-11-14 | 日東電工株式会社 | 樹脂発泡体及び発泡シール材 |
| JP6358825B2 (ja) * | 2013-04-10 | 2018-07-18 | 日東電工株式会社 | 樹脂発泡複合体 |
| JP5833213B2 (ja) | 2014-10-31 | 2015-12-16 | 日東電工株式会社 | 衝撃吸収材 |
| WO2016143684A1 (ja) * | 2015-03-06 | 2016-09-15 | 積水化成品工業株式会社 | ポリプロピレン系樹脂発泡シート、ポリプロピレン系樹脂発泡シートの製造方法及び粘着シート |
| JP6534645B2 (ja) | 2016-03-30 | 2019-06-26 | 積水化成品工業株式会社 | 緩衝材用発泡体及び緩衝材 |
| JP6962317B2 (ja) * | 2017-03-08 | 2021-11-05 | 東レ株式会社 | 発泡体及びその製造方法 |
| JP6473846B1 (ja) * | 2017-08-28 | 2019-02-20 | 日東電工株式会社 | 樹脂シートおよび粘着剤層付樹脂シート |
| CN113646368B (zh) * | 2019-04-10 | 2023-09-15 | 日东电工株式会社 | 阻燃发泡体及发泡构件 |
-
2020
- 2020-11-25 JP JP2021561442A patent/JPWO2021106911A1/ja active Pending
- 2020-11-25 JP JP2021561441A patent/JPWO2021106910A1/ja active Pending
- 2020-11-25 WO PCT/JP2020/043760 patent/WO2021106911A1/ja active Application Filing
- 2020-11-25 WO PCT/JP2020/043761 patent/WO2021106912A1/ja active Application Filing
- 2020-11-25 JP JP2021561444A patent/JPWO2021106913A1/ja active Pending
- 2020-11-25 CN CN202080075113.1A patent/CN114616278A/zh active Pending
- 2020-11-25 CN CN202080081928.0A patent/CN114761474A/zh active Pending
- 2020-11-25 WO PCT/JP2020/043763 patent/WO2021106914A1/ja active Application Filing
- 2020-11-25 CN CN202080075112.7A patent/CN114641521A/zh active Pending
- 2020-11-25 KR KR1020227017000A patent/KR20220106752A/ko unknown
- 2020-11-25 JP JP2021561443A patent/JPWO2021106912A1/ja active Pending
- 2020-11-25 WO PCT/JP2020/043762 patent/WO2021106913A1/ja active Application Filing
- 2020-11-25 JP JP2021561445A patent/JPWO2021106914A1/ja active Pending
- 2020-11-25 WO PCT/JP2020/043759 patent/WO2021106910A1/ja active Application Filing
- 2020-11-25 CN CN202080074465.5A patent/CN114585670A/zh active Pending
- 2020-11-25 CN CN202080081924.2A patent/CN114761473A/zh active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001348452A (ja) * | 2000-06-05 | 2001-12-18 | Nitto Denko Corp | ポリオレフィン系樹脂発泡体及びその製造方法 |
| WO2015146756A1 (ja) * | 2014-03-26 | 2015-10-01 | 日東電工株式会社 | 樹脂発泡体、発泡部材、及び、タッチパネル搭載機器 |
| JP2016088977A (ja) * | 2014-10-31 | 2016-05-23 | 日東電工株式会社 | 樹脂発泡体及び発泡部材 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023139878A1 (ja) * | 2022-01-18 | 2023-07-27 | 日東電工株式会社 | 発泡部材 |
| WO2023139879A1 (ja) * | 2022-01-18 | 2023-07-27 | 日東電工株式会社 | 発泡部材 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114585670A (zh) | 2022-06-03 |
| WO2021106911A1 (ja) | 2021-06-03 |
| JPWO2021106910A1 (ja) | 2021-06-03 |
| CN114761474A (zh) | 2022-07-15 |
| JPWO2021106912A1 (ja) | 2021-06-03 |
| JPWO2021106914A1 (ja) | 2021-06-03 |
| JPWO2021106913A1 (ja) | 2021-06-03 |
| CN114616278A (zh) | 2022-06-10 |
| WO2021106913A1 (ja) | 2021-06-03 |
| JPWO2021106911A1 (ja) | 2021-06-03 |
| WO2021106910A1 (ja) | 2021-06-03 |
| KR20220106752A (ko) | 2022-07-29 |
| CN114641521A (zh) | 2022-06-17 |
| WO2021106914A1 (ja) | 2021-06-03 |
| CN114761473A (zh) | 2022-07-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021106912A1 (ja) | 樹脂発泡体 | |
| WO2020209353A1 (ja) | 樹脂発泡体および発泡部材 | |
| KR101852221B1 (ko) | 전기 또는 전자 기기용 발포 적층체 | |
| US20110262744A1 (en) | Resin foam and foamed member | |
| KR102097950B1 (ko) | 수지 발포체 시트 및 수지 발포 복합체 | |
| JP6473846B1 (ja) | 樹脂シートおよび粘着剤層付樹脂シート | |
| KR20170065514A (ko) | 발포 복합 시트 | |
| JP6110213B2 (ja) | 熱可塑性樹脂発泡体、発泡シール材及び熱可塑性樹脂発泡体の製造方法 | |
| WO2013187372A1 (ja) | 樹脂発泡体及び発泡シール材 | |
| WO2013168798A1 (ja) | 樹脂発泡体及び発泡シール材 | |
| WO2022074899A1 (ja) | 樹脂発泡体 | |
| WO2022210956A1 (ja) | 樹脂発泡体および発泡部材 | |
| JP7288994B2 (ja) | 樹脂発泡体 | |
| WO2022209767A1 (ja) | 樹脂発泡体 | |
| CN116997600A (zh) | 树脂发泡体 | |
| WO2023140251A1 (ja) | 発泡部材 | |
| WO2023139878A1 (ja) | 発泡部材 | |
| WO2023139879A1 (ja) | 発泡部材 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20892777 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021561443 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20892777 Country of ref document: EP Kind code of ref document: A1 |