WO2019221122A1 - β-ラクタマーゼ阻害剤 - Google Patents
β-ラクタマーゼ阻害剤 Download PDFInfo
- Publication number
- WO2019221122A1 WO2019221122A1 PCT/JP2019/019133 JP2019019133W WO2019221122A1 WO 2019221122 A1 WO2019221122 A1 WO 2019221122A1 JP 2019019133 W JP2019019133 W JP 2019019133W WO 2019221122 A1 WO2019221122 A1 WO 2019221122A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- hydrogen atom
- substituted
- amino group
- Prior art date
Links
- 0 C*([C@@](C(*)C(O*)=O)S(N(*)*)(=O)=O)=*I Chemical compound C*([C@@](C(*)C(O*)=O)S(N(*)*)(=O)=O)=*I 0.000 description 1
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/36—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/50—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to a ⁇ -lactamase inhibitor and the like.
- a treatment method is usually employed in which the causative bacteria are bacteriostatic or sterilized by the action of antibacterial drugs.
- an antibacterial agent drug-resistant bacteria against the antibacterial agent appear.
- ⁇ -lactam antibacterial agents there are resistant bacteria that produce an enzyme ( ⁇ -lactamase) having an activity of decomposing the same. So far, ⁇ -lactam antibacterial drugs with a new structure have been developed and used for newly emerging ⁇ -lactamase producing bacteria. Under such circumstances, carbapenem antibiotics are used as the newest ⁇ -lactam antibiotics.
- resistant bacteria that produce enzymes (class B ⁇ -lactamases, metallo- ⁇ -lactamases) having the activity of degrading the antimicrobial agents have emerged and become a problem.
- the resistant bacterium is resistant to many ⁇ -lactam antibacterials including carbapenem antibacterials.
- An object of the present invention is to provide a compound having ⁇ -lactamase inhibitory activity.
- an object of the present invention is to provide a compound having class B ⁇ -lactamase inhibitory activity.
- the present inventor is represented by the general formula (1) represented by a compound in which a specific position of a planar 5-membered ring is substituted with a sulfamoyl group and a carboxy group. It was found that these compounds have ⁇ -lactamase inhibitory activity, particularly class B ⁇ ⁇ ⁇ ⁇ -lactamase inhibitory activity. As a result of further research based on this knowledge, the present inventor completed the present invention. That is, the present invention includes the following aspects.
- R 1 represents —C (—R A ) —.
- R 2 represents —N (—R B ) n —, —O—, or —C (—R A ) —.
- R 3 represents —C (—R A ) — when R 2 is —N (—R B ) n — or —O—, and —S when R 2 is —C (—R A ) —.
- R A is the same or different and represents a hydrogen atom, a halogen atom, or a hydrocarbon group that may be substituted with an amino group.
- R B represents a hydrogen atom, a halogen atom, an amino group which may be substituted, or a hydrocarbon group which may be substituted with an amino group.
- n represents 0 or 1.
- R 6 represents a hydrogen atom or a hydrocarbon group.
- R 7 and R 8 are the same or different and each represents a hydrogen atom or a hydrocarbon group, or R 7 and R 8 are bonded to each other to form a ring with an adjacent nitrogen atom.
- a bond represented by a solid line and a dotted double line indicates a single bond or a double bond.
- a ⁇ -lactamase inhibitor comprising a compound represented by the formula: or a salt, hydrate or solvate thereof.
- Item 2 The inhibitor according to Item 1, wherein the ⁇ -lactamase is Class B ⁇ -lactamase.
- Item 3. The inhibitor according to Item 1 or 2, wherein the ⁇ -lactamase is class B1 ⁇ -lactamase.
- R 1 represents —C (—R A ) —.
- R 2 represents —N (—R B ) n —, —O—, or —C (—R A ) —.
- R 3 represents —C (—R A ) — when R 2 is —N (—R B ) n — or —O—, and —S when R 2 is —C (—R A ) —.
- R A is the same or different and represents a hydrogen atom, a halogen atom, or a hydrocarbon group that may be substituted with an amino group.
- R B represents a hydrogen atom, a halogen atom, an amino group which may be substituted, or a hydrocarbon group which may be substituted with an amino group.
- n represents 0 or 1.
- R 6 represents a hydrogen atom or a hydrocarbon group.
- R 7 and R 8 are the same or different and each represents a hydrogen atom or a hydrocarbon group, or R 7 and R 8 are bonded to each other to form a ring with an adjacent nitrogen atom.
- a bond represented by a solid line and a dotted double line indicates a single bond or a double bond.
- an antibacterial effect enhancer of a ⁇ -lactam antibacterial compound which comprises a compound represented by the formula: or a salt, hydrate or solvate thereof.
- R 1 represents —C (—R A ) —.
- R 2 represents —N (—R B ) n —, —O—, or —C (—R A ) —.
- R 3 represents —C (—R A ) — when R 2 is —N (—R B ) n — or —O—, and —S when R 2 is —C (—R A ) —.
- R A is the same or different and represents a hydrogen atom, a halogen atom, or a hydrocarbon group that may be substituted with an amino group.
- R B represents a hydrogen atom, a halogen atom, an amino group which may be substituted, or a hydrocarbon group which may be substituted with an amino group.
- n represents 0 or 1.
- R 6 represents a hydrogen atom or a hydrocarbon group.
- R 7 and R 8 are the same or different and each represents a hydrogen atom or a hydrocarbon group, or R 7 and R 8 are bonded to each other to form a ring with an adjacent nitrogen atom.
- a bond represented by a solid line and a dotted double line indicates a single bond or a double bond.
- a salt, hydrate or solvate thereof, and a ⁇ -lactam antibacterial compound is a salt, hydrate or solvate thereof, and a ⁇ -lactam antibacterial compound.
- R 1 represents —C (—R A ) —.
- R 2 represents —N (—R B ) n —, —O—, or —C (—R A ) —.
- R 3 represents —C (—R A ) — when R 2 is —N (—R B ) n — or —O—, and —S when R 2 is —C (—R A ) —.
- R A is the same or different and represents a hydrogen atom, a halogen atom, or a hydrocarbon group that may be substituted with an amino group.
- R B represents a hydrogen atom, a halogen atom, an amino group which may be substituted, or a hydrocarbon group which may be substituted with an amino group.
- n represents 0 or 1.
- R 6 represents a hydrogen atom or a hydrocarbon group.
- R 7 and R 8 are the same or different and each represents a hydrogen atom or a hydrocarbon group, or R 7 and R 8 are bonded to each other to form a ring with an adjacent nitrogen atom.
- a bond represented by a solid line and a dotted double line indicates a single bond or a double bond.
- R 2 represents —N (—R B ) — or —O—.
- R A is the same or different and is a linear or branched alkyl group optionally substituted with a hydrogen atom or an amino group (provided that when R 2 is —N (—R B ) —, the number of carbon atoms is 1 to And when R 2 is —O—, it has 2 to 5 carbon atoms), a cyclic alkyl group having 3 to 7 carbon atoms that may be substituted with an amino group, or an amino group.
- R B represents a hydrogen atom, an amino group which may be substituted with a linear or branched alkyl group, or an alkyl group which may be substituted with an amino group.
- R 6 represents a hydrogen atom.
- R 7 and R 8 are the same or different and represent a hydrogen atom or an alkyl group (except when both R 7 and R 8 are alkyl groups), or R 7 and R 8 are adjacent to each other. To form a ring with the nitrogen atom. ] Or a salt, hydrate or solvate thereof.
- Item 8 The compound according to Item 7, or a salt, hydrate or solvate thereof, wherein the alkyl group represented by R B is a linear alkyl group having 1 to 4 carbon atoms.
- Item 9. A pharmaceutical comprising the compound according to item 7 or 8, or a salt, hydrate or solvate thereof.
- Item 10 A reagent comprising the compound according to item 7 or 8, or a salt, hydrate or solvate thereof.
- a compound having ⁇ -lactamase inhibitory activity in particular, class B- ⁇ -lactamase inhibitory activity.
- class B- ⁇ -lactamase inhibitory activity By utilizing this, it is possible to provide an existing ⁇ -lactamase inhibitor having a ⁇ -lactam ring, an antibacterial effect enhancer of a ⁇ -lactam antibacterial compound, and the like. Further, since the compound of the present invention has relatively low toxicity, it can be used more safely.
- the outline of the test method of the drug sensitivity test (Test Example 1 etc.) is shown.
- the results of the ⁇ -lactamase inhibitory activity measurement test of Test Example 2 are shown.
- the vertical axis represents the imipenem residual degradation activity, and the horizontal axis represents the concentration of the test substance.
- the class B ⁇ -lactamase used is shown above each graph.
- the result of the drug sensitivity test of Test Example 3 is shown.
- the vertical axis represents MIC, and the horizontal axis represents the antibacterial agent used and the type (compound A) and concentration ( ⁇ g / mL) of the test substance.
- Each plot shows the data of each test strain.
- the result of the drug sensitivity test of Test Example 4 is shown.
- FIG. 2 shows a schematic diagram of a complex structure of IMP-1 and compound A obtained by X-ray crystal structure analysis of Test Example 5.
- FIG. 3 shows a schematic diagram of a complex structure of VIM-2 and compound A obtained by X-ray crystal structure analysis of Test Example 5.
- FIG. 2 shows a schematic diagram of a complex structure of NDM-1 and compound A obtained by X-ray crystal structure analysis of Test Example 5.
- FIG. 2 shows a schematic diagram of a complex structure of NDM-1 and Compound I obtained by X-ray crystal structure analysis of Test Example 5.
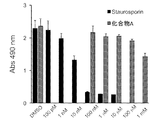
- the toxicity evaluation test result of Test Example 7 is shown.
- the vertical axis represents the absorbance reflecting live cells, and the horizontal axis represents the concentration of the test substance. In the horizontal axis, DMSO indicates the case where the test substance is not added.
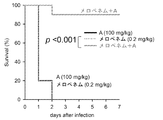
- the result of the animal test of Test Example 12 is shown.
- the vertical axis indicates the survival rate, and the horizontal axis indicates the number of days elapsed after infection with the experimental strain ( ⁇ -lactamase expression strain).
- A represents a test substance (compound A).
- the results when using an E. coli strain introduced with a plasmid encoding IMP-1 as an experimental strain are shown.
- the vertical axis indicates the survival rate, and the horizontal axis indicates the number of days elapsed after infection with the experimental strain ( ⁇ -lactamase expression strain).
- I represents a test substance (compound I).
- the vertical axis indicates the survival rate, and the horizontal axis indicates the number of days elapsed after infection with the experimental strain ( ⁇ -lactamase expression strain).
- I represents a test substance (compound I).
- the result of the animal test of Test Example 14 is shown.
- the vertical axis indicates the survival rate, and the horizontal axis indicates the number of days elapsed after infection with the experimental strain ( ⁇ -lactamase expression strain).
- X2d represents a test substance (compound X2d).
- R 2 represents —N (—R B ) n —, —O—, or —C (—R A ) —.
- R 3 represents —C (—R A ) — when R 2 is —N (—R B ) n — or —O—, and —S when R 2 is —C (—R A ) —.
- R 2 is preferably —N (—R B ) n —.
- R A is the same or different and represents a hydrogen atom, a halogen atom, or a hydrocarbon group that may be substituted with an amino group. In some embodiments of the present invention, preferably R A is a hydrocarbon group.
- Examples of the halogen atom represented by R A include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- the hydrocarbon group represented by R A is not particularly limited, and examples thereof include an alkyl group, an aryl group and the like, and a group in which these are arbitrarily combined (for example, an aralkyl group, an alkylaryl group, an alkylaralkyl group) and the like. Is mentioned.
- the hydrocarbon group is an alkyl group or an aryl group.
- the alkyl group represented by R A includes any of linear, branched, or cyclic (preferably linear or branched, more preferably linear).
- the number of carbon atoms of the alkyl group is not particularly limited, and is, for example, 1 to 12, preferably 1 to 8, more preferably 1 to 5, further preferably 1 to 4, Even more preferably, it is 1-2.
- the number of carbon atoms of the alkyl group (when cyclic) is not particularly limited, and is, for example, 3 to 7, preferably 4 to 6.
- the lower limit of the carbon number of the alkyl group is, for example, 2, 3, 4, 5, 6, 7, 8.
- alkyl group examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group, sec-butyl group, n-pentyl group, neopentyl group, n -Hexyl group, 3-methylpentyl group, n-heptyl group, n-octyl group, n-nonyl group, n-decyl group, n-undecyl group, n-dodecyl group and the like.
- the aryl group represented by R A is not particularly limited, but preferably has 6 to 12 carbon atoms, more preferably has 6 to 12 carbon atoms, and still more preferably has 6 to 8 carbon atoms.
- the aryl group may be monocyclic or polycyclic (for example, bicyclic, tricyclic, etc.), but is preferably monocyclic.
- the aryl group include a phenyl group, a naphthyl group, a biphenyl group, a pentaenyl group, an indenyl group, an anthranyl group, a tetracenyl group, a pentacenyl group, a pyrenyl group, a perylenyl group, a fluorenyl group, and a phenanthryl group.
- a phenyl group is mentioned.
- the aralkyl group represented by R A is not particularly limited, but for example, a linear or branched alkyl group having 1 to 6 (preferably 1 to 2) carbon atoms, such as 1 to 3, preferably 1 And an aralkyl group formed by substituting one hydrogen atom) with the aryl group.
- Specific examples of the aralkyl group include a benzyl group and a phenethyl group.
- the alkylaryl group represented by R A is not particularly limited, but, for example, the hydrogen atom of the aryl group (for example, 1 to 3, preferably 1 hydrogen atom) is a linear or branched carbon number of 1 to Examples include alkylaryl groups substituted by 6 (preferably 1 to 2) alkyl groups. Specific examples of the alkylaryl group include a tolyl group and a xylyl group.
- the alkylaralkyl group represented by R A is not particularly limited, but for example, a hydrogen atom (for example, 1 to 3, preferably 1 hydrogen atom) on the aromatic ring of the aralkyl group is linear or branched. Examples thereof include an alkylaralkyl group substituted with an alkyl group having 1 to 6 carbon atoms (preferably 1 to 2 carbon atoms).
- the amino group includes not only —NH 2 but also a substituted amino group in which a hydrogen atom of —NH 2 is substituted with a hydrocarbon group. Is included.
- the hydrocarbon group in the substituted amino group is the same as the hydrocarbon group represented by RA .
- R B represents a hydrogen atom, a halogen atom, an amino group which may be substituted, or a hydrocarbon group which may be substituted with an amino group.
- R B is a hydrogen atom or a hydrocarbon group, more preferably R B is a hydrocarbon group.
- Halogen atoms represented by R B, and the hydrocarbon group which may be substituted by an amino group represented by R B are the same as described above for R A.
- the amino group which may be substituted represented by R B for example -NH 2, the hydrogen atom of -NH 2 and the like and substituted amino group formed by substituted with a hydrocarbon group.
- the hydrocarbon group is the same as the hydrocarbon group represented by R A.
- N represents 0 or 1. In one embodiment of the present invention, n is preferably 1.
- the 5-membered ring in the partial structure represented by is not particularly limited.
- the 5-membered ring preferably has planarity.
- Specific examples of the 5-membered ring include furan, pyrrole and thiophene, more preferably furan and pyrrole, and still more preferably pyrrole.
- R 6 > R 6 represents a hydrogen atom or a hydrocarbon group. In one embodiment of the present invention, preferably R 6 is a hydrogen atom.
- the hydrocarbon group represented by R 6 is the same as described above for R A.
- R 7 and R 8 > R 7 and R 8 are the same or different and each represents a hydrogen atom or a hydrocarbon group, or R 7 and R 8 are bonded to each other to form a ring with an adjacent nitrogen atom.
- R 7 and R 8 are the same or different and each represents a hydrogen atom or a hydrocarbon group, more preferably, at least one of R 7 and R 8 is a hydrogen atom, More preferably, both R 7 and R 8 are hydrogen atoms.
- hydrocarbon group represented by R 7 and the hydrocarbon group represented by R 8 are the same as described above for R A.
- the ring formed by R 7 and R 8 bonded together and adjacent nitrogen atoms is not particularly limited, and is, for example, a monocyclic ring or a bicyclic ring.
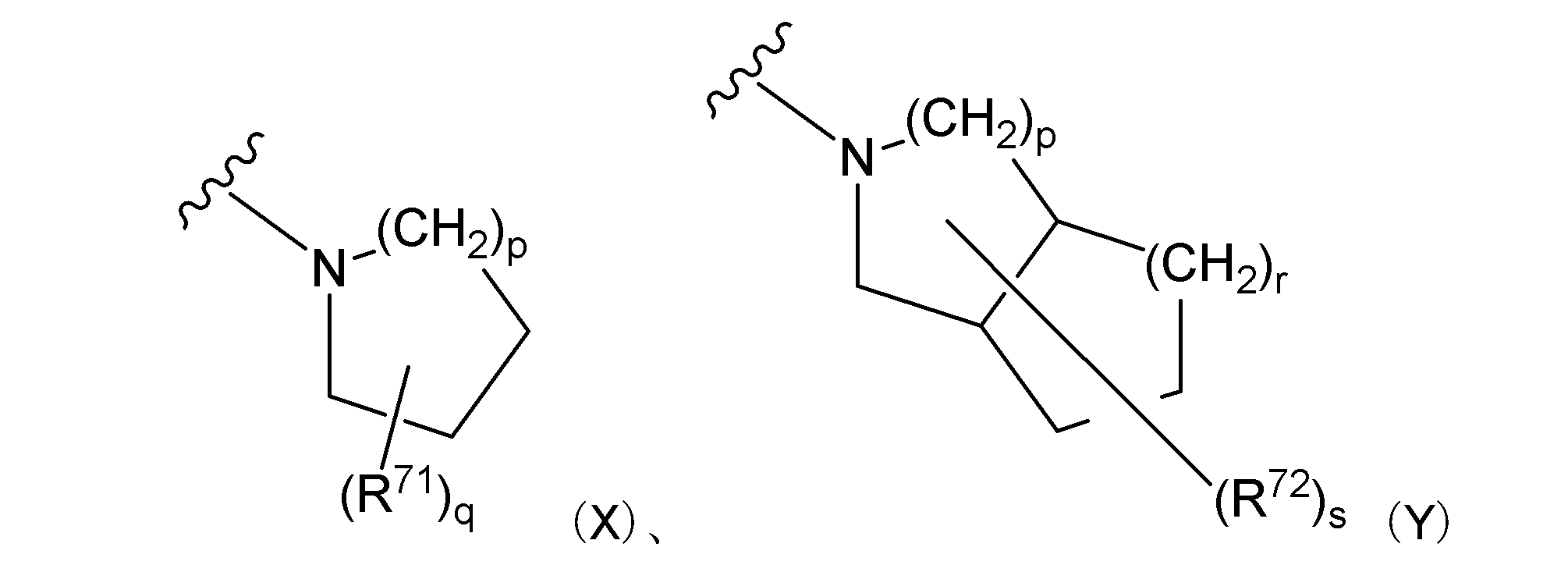
- Examples of the ring include general formula (X) and general formula (Y):
- R 71 and R 72 each represents an alkyl group. p and r each represents an integer of 1 to 3. q and s each represent 0 or an integer of 1 to 3. ]
- the alkyl group represented by R 71 and R 72 is the same as described above for R A. When a plurality of R 71 and R 72 are present, they may be bonded to the same carbon atom or may be bonded to different carbon atoms.
- R A , R B , n, R 6 , R 7 and R 8 are the same as defined above. ] Is mentioned.
- R A , R 6 , R 7 and R 8 are the same as defined above. ] Is mentioned.
- R 2 represents —N (—R B ) — or —O—.
- R A is the same or different and is a linear or branched alkyl group optionally substituted with a hydrogen atom or an amino group (provided that when R 2 is —N (—R B ) —, the number of carbon atoms is 1 to And when R 2 is —O—, it has 2 to 5 carbon atoms), a cyclic alkyl group having 3 to 7 carbon atoms that may be substituted with an amino group, or an amino group. Good phenyl group.
- R B represents a hydrogen atom, an amino group which may be substituted with a linear or branched alkyl group, or an alkyl group which may be substituted with an amino group.
- R 6 represents a hydrogen atom.
- R 7 and R 8 are the same or different and represent a hydrogen atom or an alkyl group (except when both R 7 and R 8 are alkyl groups), or R 7 and R 8 are adjacent to each other. To form a ring with the nitrogen atom.
- Specific examples of the compound represented by the general formula (1) include the following compounds.
- compound E compound E, compound I, compound X2d and the like are preferable.
- the compound represented by the general formula (1) includes stereoisomers and optical isomers, and these are not particularly limited.
- the salt of the compound represented by the general formula (1) is not particularly limited as long as it is a pharmaceutically acceptable salt.
- an acidic salt or a basic salt can be employed.
- acid salts include inorganic acid salts such as hydrochloride, hydrobromide, sulfate, nitrate, phosphate; acetate, propionate, tartrate, fumarate, maleate, malic acid
- Organic salts such as salts, citrates, methanesulfonates, paratoluenesulfonates and the like
- examples of basic salts include alkali metal salts such as sodium salts and potassium salts; and calcium salts, magnesium Alkaline earth metal salts such as salts; salts with ammonia; morpholine, piperidine, pyrrolidine, monoalkylamine, dialkylamine, trialkylamine, mono (hydroxyalkyl) amine, di (hydroxyalkyl) amine, tri (
- the compound represented by the general formula (1) may be a hydrate or a solvate.
- the solvent include pharmaceutically acceptable organic solvents (for example, ethanol, glycerol, acetic acid and the like).
- the compound represented by the general formula (1) can be synthesized by various methods.
- R 6 is a hydrocarbon group (R 6A ) and R 7 and R 8 are both hydrogen atoms
- the compound (compound 1d) is, for example, the following reaction formula:
- R 1 , R 2 , R 3 , R 7 and R 8 are the same as defined above.
- R 6A represents a hydrocarbon group.
- the compound (compound 1e) is, for example, R 6A ⁇ which is a protecting group for the carboxy group of the compound 1d. Can be obtained by deprotection using a strong base or the like.
- a compound in which at least one of R 7 and R 8 is a hydrocarbon group can be obtained by substituting the amino group of compound 1d or compound 1e with a hydrocarbon.
- the compound represented by the general formula (1) can also be synthesized using the Pearl-Kunol reaction. That is, when utilizing the Pearl-Kunol reaction, a coupling product (diketoester) of an appropriate ketoester and ketone is reacted in the presence of acid, ammonia, primary amine, etc. to furan or pyrrole (compound 1c). After that, the compound represented by the general formula (1) can be synthesized according to the above reaction formula.
- R A , R B, etc. for example, a compound (compound) in which the amino group on compound c is protected with an appropriate protecting group (for example, phthalimide, etc.) as necessary by the Pearl-Kunol reaction c ′) is obtained, and deprotection (for example, deprotection using hydrazine or the like) is performed at an appropriate timing during the reaction step, whereby the compound represented by the general formula (1) can be synthesized.
- an appropriate protecting group for example, phthalimide, etc.
- deprotection for example, deprotection using hydrazine or the like
- compound 1b can be obtained by reacting compound 1a, aluminum halide, and carbon dioxide.
- the aluminum halide is not particularly limited, and examples thereof include alkyl aluminum halide, preferably dimethyl aluminum chloride.
- the amount of aluminum halide used is usually preferably 0.5 to 1.5 mol, more preferably 0.8 to 1.2 mol, relative to 1 mol of compound 1a, from the viewpoints of yield, ease of synthesis, and the like.
- the amount of carbon dioxide used is usually preferably 1 to 15 moles per mole of compound 1a from the viewpoint of yield, ease of synthesis, and the like.
- This step is preferably performed in a solvent.
- a solvent for example, toluene etc. are mentioned.
- a solvent can be used individually by 1 type or in combination of 2 or more types.
- additives can be appropriately used within the range not impairing the effects of the present invention.
- the reaction atmosphere can usually be an inert gas atmosphere (argon gas atmosphere, nitrogen gas atmosphere, etc.).
- the reaction temperature can be any of heating, room temperature, and cooling, and it is usually preferably 0 to 100 ° C. (especially 15 to 40 ° C.).
- the reaction time is not particularly limited, and can usually be 3 to 48 hours, particularly 8 to 24 hours.
- purification can be performed according to a conventional method if necessary. Moreover, the following process can also be performed without performing a refinement
- compound 1c can be obtained by reacting compound 1b, R 6A —OH, and hydrogen halide.
- the amount of R 6A —OH used is usually preferably 3 to 15 parts by weight and more preferably 5 to 10 parts by weight with respect to 1 part by weight of compound 1b from the viewpoint of yield, ease of synthesis, and the like.
- the hydrogen halide is not particularly limited but preferably includes hydrogen chloride.
- the amount of hydrogen halide used is about 1 to 6 N, preferably 1.5 to 5 N in the reaction solution.
- R 6A —OH usually functions as a solvent, but a suitable solvent may be added.
- a solvent can be used individually by 1 type or in combination of 2 or more types.
- additives can be appropriately used within the range not impairing the effects of the present invention.
- the reaction atmosphere can usually be an inert gas atmosphere (argon gas atmosphere, nitrogen gas atmosphere, etc.).
- the reaction temperature can be any of heating, room temperature, and cooling, and it is usually preferably 0 to 100 ° C. (especially 15 to 40 ° C.).
- the reaction time is not particularly limited, and can usually be 3 to 48 hours, particularly 8 to 24 hours.
- purification can be performed according to a conventional method if necessary. Moreover, the following process can also be performed without performing a refinement
- compound 1d can be obtained by reacting compound 1c, halogenated sulfonyl acid, and ammonium hydroxide.
- This step can also be carried out in two steps, that is, by reacting compound 1c with a halogenated sulfonyl acid, and then reacting the resulting reactant with ammonium hydroxide.
- the halogenated sulfonyl acid is not particularly limited, but preferably a sulfonyl chloride is used.
- the amount of halogenated sulfonyl acid used is usually preferably 3 to 20 mol, more preferably 7 to 15 mol, relative to 1 mol of compound 1c, from the viewpoint of yield, ease of synthesis, and the like.
- the amount of ammonium hydroxide used is usually preferably 1 to 50 parts by weight and more preferably 3 to 20 parts by weight with respect to 1 part by weight of compound 1c from the viewpoint of yield, ease of synthesis, and the like.
- This step is preferably performed in a solvent.
- a solvent for example, acetonitrile etc. are mentioned.
- a solvent can be used individually by 1 type or in combination of 2 or more types.
- additives can be appropriately used within the range not impairing the effects of the present invention.
- the reaction atmosphere can usually be an inert gas atmosphere (argon gas atmosphere, nitrogen gas atmosphere, etc.).
- the reaction temperature can be any of heating, room temperature and cooling, and it is usually preferably 0 to 40 ° C. (especially 0 to 5 ° C.).
- the reaction time is not particularly limited, and can usually be 30 minutes to 10 hours, particularly 1 hour to 5 hours.
- purification can be performed according to a conventional method if necessary. Moreover, the following process can also be performed without performing a refinement
- the compound of the present invention has ⁇ -lactamase inhibitory activity, activity to enhance the antibacterial effect of ⁇ -lactam antibacterial compounds, and the like. Therefore, the compound of the present invention is more specifically used as an active ingredient of medicines, reagents and the like (in the present specification, sometimes referred to as “the drug of the present invention”), more specifically, ⁇ -lactamase inhibitors, ⁇ -It can be used as an active ingredient such as an antibacterial effect enhancer of a lactam antibacterial compound.
- an antibacterial agent containing the compound of the present invention and a ⁇ -lactam antibacterial compound using the ⁇ -lactamase inhibitory activity of the compound of the present invention, the antibacterial effect enhancing activity of the ⁇ -lactam antibacterial compound, and the like Contains a ⁇ -lactam antibacterial compound, an antibacterial agent used to be administered in combination with the compound of the present invention (these may also be referred to as “the drug of the present invention” in the present specification), and the like. Can also be provided.
- those containing the compound of the present invention are not particularly limited as long as they contain the compound of the present invention, and may further contain other components as necessary.
- Other components are not particularly limited as long as they are pharmaceutically acceptable components.
- additives include bases, carriers, solvents, dispersants, emulsifiers, buffers, osmotic pressure regulators, absorption promoters, stabilizers, excipients, binders, disintegrants, lubricants, thickeners. , Moisturizers, colorants, fragrances, chelating agents and the like.
- the ⁇ -lactamase to be inhibited by the compound of the present invention is not particularly limited as long as it is an enzyme derived from any source that catalyzes the opening of the ⁇ -lactam ring.
- ⁇ -lactamase (EC 3.5.2.6) is the enzyme most commonly produced by bacteria.
- ⁇ -lactamase catalyzes the hydrolytic ring-opening of ⁇ -lactam ring, penicillin, penum, penem, cephem, cephalosporin, carbacephem, cephamycin, monobactam, and carbapenem It is a cause of imparting bacterial resistance to ⁇ -lactam antibacterial compounds such as ⁇ -lactamases are classified into classes A to D, and among these, class B- ⁇ -lactamases are preferred as targets to be inhibited by the compounds of the present invention, and class B1 ⁇ -lactamases are more preferred.
- IMP-1, NDM-1, VIM-2, DIM-1, GIM-1, KHM-1, SIM-1, SPM-1, TMB-1, BcII, BlaB, CcrA, IND- 7 etc. are mentioned.
- SFH-1, GOB-1, etc. are mentioned as class B2 and B3, respectively.
- the ⁇ -lactam antibacterial compound that is an object of enhancing the antibacterial effect by the compound of the present invention is not particularly limited, and examples thereof include penicillin antibacterial compounds, cephem antibacterial compounds, carbapenem antibacterial compounds and the like.
- penicillin-based antibacterial compounds include benzylpenicillin, pheneticillin, cloxacillin, dicloxacillin, ampicillin, cyclacillin, amoxylin, tarampicillin, bacampicillin, lenampicillin, aspoxillin, piperacillin, sulbenicillin, sultipecilinciline , Propicillin, epicillin, ticarcillin, pirbenicillin, azurocillin, mezlocillin, and other known penicillin antibacterial compounds.
- cephem antibacterial compounds include cefaclor, cephazoline, cephatrizine, cefadroxyl, cephapillin, cefamandole nafate, cephalazine, cephalexin, cephalothin, cefepime, cefoxitin, cefuxime, cefdidim, cefditoren, cefdinir, cefdithrone, cefdiner Cefzoplan, ceftoxime, ceftazidime, ceftaroline, ceftiam, ceftizoxime, ceftibbutene, ceftezol, ceftetam, ceftriaxone, cefnicid, cefpyramide, cefpirom, cefbuperazone, cefprozil, cefperezone, cefpoxime, cefminoxef Fromoxef, Ceftrozan (C XA101, (6R, 7R)
- carbapenem antibacterial compounds include imipenem, panipenem, meropenem, biapenem, doripenem, ertapenem, and tevipenem.
- ⁇ -lactam antibacterial compounds other than penicillin antibacterial compounds, cephem antibacterial compounds, and carbapenem antibacterial compounds include aztreonam, carmonam, loracarbef, faropenem, ritipenem and the like.
- Gram-negative bacteria can be widely used as target bacteria of the drug of the present invention.
- Gram-negative bacteria include Enterobacteriaceae (eg, Escherichia, Klebsiella, Salmonella, Shigella, etc.), Acinetobacter, Pseudomonas (eg, Pseudomonas), Moraxella Bacteria, Helicobacter, Campylobacter, Aeromonas, Vibrio (for example, Vibrio cholerae, Vibrio parahaemolyticus), Haemophilus (for example, Haemophilus influenzae), Neisseria (for example, Neisseria gonorrhoeae, Neisseria meningitidis), Bacteroides Examples include genus bacteria.
- Gram-positive bacteria include, for example, staphylococci (eg, S. aureus, Staphylococcus epidermidis), enterococci (eg, Enterococcus), streptococci (eg, group A streptococci, group B streptococci, pneumococci) , Green streptococci), Bacillus (eg, Bacillus cereus, Bacillus anthracis), Clostridium (eg, tetanus, Clostridium botulinum, difficile), Corynebacterium (eg, Diphtheria), Listeria, Lactobacillus Examples include bacteria, Bifidobacterium, Propionibacterium (for example, acne causing acne), actinomycetes, and the like. Among these, Gram negative bacteria are preferable, and Enterobacteriaceae bacteria and Acinetobacter spp. Are more preferable.
- the agent of the present invention can exert its effect on a target bacterium that produces ⁇ -
- the usage mode of the drug of the present invention is not particularly limited, and an appropriate usage mode can be adopted depending on the type.
- the agent of the present invention can be used, for example, in vitro (for example, added to a culture medium of cultured cells) or in vivo (for example, administered to an animal) depending on its use. You can also.
- the target of application when the agent of the present invention is applied to animals or cells is not particularly limited, but in mammals, for example, humans, monkeys, mice, rats, dogs, cats, rabbits, pigs, horses, cows, sheep, goats. , Deer and the like.
- Examples of the cell include animal cells.
- the cell type is not particularly limited, for example, blood cells, hematopoietic stem cells / progenitor cells, gametes (sperm, ovum), fibroblasts, epithelial cells, vascular endothelial cells, neurons, hepatocytes, keratinocytes, muscle cells , Epidermal cells, endocrine cells, ES cells, iPS cells, tissue stem cells, cancer cells and the like.
- the drug of the present invention may be any dosage form suitable for pharmaceuticals, reagents, etc., for example, tablets (including orally disintegrating tablets, chewable tablets, effervescent tablets, troches, jelly-like drops, etc.), pills, granules Oral dosage forms such as fine granules, powders, hard capsules, soft capsules, dry syrups, liquids (including drinks, suspensions, syrups), jelly, and injectable preparations (eg, infusion) Preparations (for example, intravenous infusion preparations), intravenous injections, intramuscular injections, subcutaneous injections, intradermal injections), external preparations (for example, ointments, creams, poultices, lotions), suppository inhalation And parenteral preparations such as an ophthalmic preparation, an ophthalmic preparation, an eye ointment, a nasal drop, an ear drop, and a liposome.
- tablets including orally disintegrating tablets, chewable tablets, efferv
- the administration route of the drug of the present invention is not particularly limited as long as the desired effect is obtained, and enteral administration such as oral administration, tube feeding, enema administration, etc .; intravenous administration, transarterial administration, intramuscular administration, Examples include parenteral administration such as intracardiac administration, subcutaneous administration, intradermal administration, and intraperitoneal administration.
- the content of the active ingredient in the drug of the present invention depends on the use mode, application target, application target state, etc., and is not limited, but is, for example, 0.0001 to 100% by weight, preferably 0.001 to 50% by weight. %.
- the dose when administering the drug of the present invention to humans or animals is not particularly limited as long as it is an effective amount that exhibits a medicinal effect, and is usually the weight of the active ingredient, generally in the case of oral administration per day 0.1 to 1000 mg / kg body weight, preferably 0.5 to 500 mg / kg body weight per day.
- For parenteral administration 0.01 to 100 mg / kg body weight, preferably 0.05 to 50 mg / kg body weight per day It is.
- the above dose can be appropriately increased or decreased depending on the age, disease state, symptoms, etc. of the patient.
- the ⁇ -lactamase inhibitor of the present invention can also be used for detection of ⁇ -lactamase producing bacteria.
- a ⁇ -lactamase-producing bacterium can be detected based on the presence or absence of growth inhibition when the bacterium to be tested is cultured in the presence of a ⁇ -lactam antibacterial compound and the ⁇ -lactamase inhibitor of the present invention. That is, this inhibitor can be used for the disk method, the micro liquid dilution method, and the like.
- Test Example 1 Drug sensitivity test 1 For 96- or 384-well U-bottom plate, Mueller Hinton liquid medium, antibacterial drugs (ceftazidime [CAZ], imipenem [IPM], meropenem [MPM], doripenem [DPM], biapenem [BPM] for measurement of drug sensitivity test The plate was prepared and adjusted so that the maximum concentration of antibacterial drug was contained in well 1 at the left end of the 96-hole U-bottom plate A series, and right next well 2 had an antibacterial drug concentration halved. In order to achieve an antibacterial drug concentration of 1/4 in well 3, 1/2 antibacterial dilution series was prepared up to well 11. Well 12 was a blank well containing no antibacterial drug. A dilution series of antibacterial drugs was prepared, and the test substance (compound A) was added to the B and C series respectively at 10 ⁇ g / mL and 50 ⁇ g / mL (FIG. 1).
- CAZ ceftazidime
- IPM imipenem
- a test bacterium As a test bacterium, a bacterium (pBCSK (+) / E. Coli) into which a plasmid encoding class B ⁇ -lactamase (MBL (metallo- ⁇ -lactamase): IMP-1, NDM-1, or VIM-2) was introduced. coli DH5 ⁇ ) was used. The test bacteria were adjusted to 10 8 cfu / mL using Mueller Hinton medium. Furthermore, the test bacteria were diluted 10 times using Mueller Hinton medium (10 7 cfu / mL). 5 ⁇ l of this was collected and inoculated on a drug sensitivity test measurement plate. The cells were cultured overnight at 35 ° C, and the growth of the test bacteria was confirmed visually. Of the wells in which the growth of the test bacteria was not confirmed, the well containing the lowest antibacterial drug concentration was defined as the minimum inhibitory concentration value (FIG. 1).
- MBL metalo- ⁇ -lactamase
- Table 1 The results are shown in Table 1.
- the MBLs column indicates class B ⁇ -lactamase produced by the test bacterium, and Empty vector does not produce class B ⁇ -lactamase.
- Test Example 3 Drug sensitivity test 2 The test was carried out in the same manner as in Test Example 1 except that various strains of Enterobacteriaceae bacteria producing class B ⁇ -lactamase were used as test bacteria.
- Test Example 4 Drug sensitivity test 3 The test was carried out in the same manner as in Test Example 1 except that various strains of Acinetobacter producing class B ⁇ -lactamase were used as test bacteria.
- Test Example 5 X-ray crystal structure analysis ⁇ complex structure analysis of IMP-1 and test substance> Escherichia coli [pET9a- ⁇ IMP-1 / E. Coli BL21 (DE3)] producing IMP-1 was subjected to liquid culture, and the cells were collected by centrifugation. The bacterial cells were disrupted with ultrasonic waves, and then a soluble fraction and an insoluble fraction were separated by ultracentrifugation. The soluble fraction was passed through a cation exchange column, a hydrophobic interaction column, and a gel filtration column in this order to purify IMP-1. Using an ultrafiltration column, the purified IMP-1 was replaced with HEPES buffer and then concentrated.
- IMP-1 15 mg / mL
- reservoir solution [100 mM HEPES (pH 7.5), 200 mM Sodium acetate, 25% PEG3350] are mixed, and incubated at 20 ° C to crystallize IMP-1 Obtained.
- a complex crystal of IMP-1 and the test substance was prepared by infiltrating the IMP-1 crystal into the reservoir solution in which the test substance (compound A) was dissolved.
- the composite crystal was brought into the synchrotron and irradiated with X-ray radiation to collect diffraction data.
- the phase was determined by the molecular replacement method. Refmac5 was used for refinement, and coot was used for model construction.
- E. coli producing VIM-2 [pET29a-VIM-2 / E. Coli BL21 (DE3)] was subjected to liquid culture, and the cells were collected by centrifugation. The bacterial cells were disrupted with ultrasonic waves, and then a soluble fraction and an insoluble fraction were separated by ultracentrifugation. The soluble fraction was passed through an anion exchange column, a hydrophobic interaction column, and a gel filtration column in this order to purify VIM-2. Using an ultrafiltration column, the purified VIM-2 was replaced with Tris-HCl buffer and concentrated.
- VIM-2 Purified VIM-2 (15 mg / mL) and a reservoir solution [200 mM Magnesium Formate, 25% PEG3350] were mixed and incubated at 20 ° C. to obtain VIM-2 crystals.
- a complex crystal of VIM-2 and a test substance was prepared by infiltrating the VIM-2 crystal into the reservoir solution in which the test substance (compound A) was dissolved.
- the composite crystal was brought into the synchrotron and X-ray diffraction data was collected.
- the phase was determined by the molecular replacement method. Refmac5 was used for refinement, and coot was used for model construction.
- E. coli producing NDM-1 [pET28a-NDM-1 / E. Coli BL21 (DE3)] was subjected to liquid culture, and the cells were collected by centrifugation. The bacterial cells were disrupted with ultrasonic waves, and then a soluble fraction and an insoluble fraction were separated by ultracentrifugation. The soluble fraction was passed through a nickel column, an anion exchange column, and a gel filtration column in this order to purify NDM-1. Using an ultrafiltration column, the purified NDM-1 was replaced with Tris-HCl buffer, and then concentrated.
- NDM-1 Purified NDM-1 (40 mg / mL) and reservoir solution [100 mM Bis-Tris (pH 6.1), 200 mM Ammonium Sulfate, 25% PEG3350] were mixed and incubated at 20 ° C to Crystals were obtained.
- a complex crystal of NDM-1 and the test substance was prepared by infiltrating the NDM-1 crystal into the reservoir solution in which the test substance (compound A or compound I) was dissolved.
- the composite crystal was brought into the synchrotron and X-ray diffraction data was collected.
- the phase was determined by the molecular replacement method. Refmac5 was used for refinement, and coot was used for model construction.
- Test Example 6 Drug sensitivity test 4 and inhibition constant (Ki) measurement test 1 The drug sensitivity test was performed in the same manner as in Test Example 1 except that compounds A to C and E to I were used as test substances.
- the inhibition constant (Ki) measurement test was conducted as follows.
- the inhibition constant (Ki) was measured using various metallo- ⁇ -lactamases (final concentration 10 nM).
- concentration of imipenem as a substrate and the concentrations of test substances (compounds A to E) as inhibitors were varied, and the imipenem degradation activity ( ⁇ abs / min) at each point at an absorbance of 298 nm and 30 ° C. was measured.
- the measured value was fitted to the line Weber-Burk plot equation, and Ki was calculated.
- Test Example 7 Toxicity evaluation test HeLa cells were added with various concentrations of test substances (compound A) or staurosporine, and viable cells were counted using an MTT assay kit (Promega cat no. G4000).
- Test Example 8 Reverse mutation test (Ames test) Using the test substance (compound A) as the test substance, the Ames test was commissioned to the UBE Science Analysis Center. As a result, it was found that Compound A was Ames test negative.
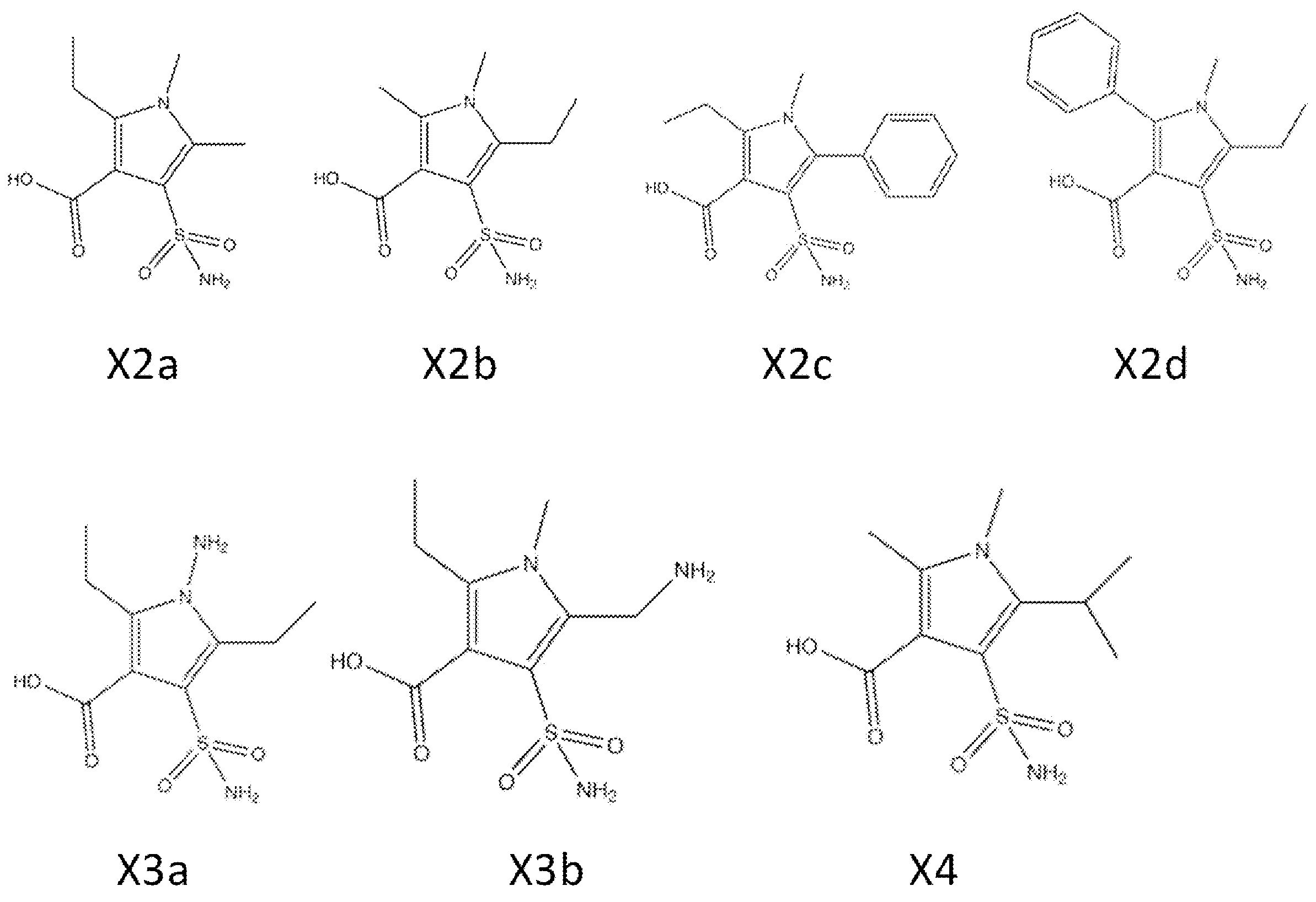
- Synthesis Example 7 Synthesis of Compounds X2a, X2b, X2c, X2d, and X4
- the target product 8 (compounds X2a, X2b, X2c, X2d, and X4) was synthesized by the following synthesis route.
- the specific structure of the target product 8 and raw materials is shown below.
- the number corresponds to the synthesis route.
- compounds having the same two-letter alphabet after the number indicate compounds on the same synthetic route. That is, the compound on the synthetic route of compound X2a (pyrrole 8ad) is a compound in which the two-letter alphabet shown after the number is “ad”.
- Pyrrole 4ad was synthesized from diketoester 3ad ⁇ ⁇ ⁇ ⁇ by Paal-Knorr pyrrole synthesis. That is, to a 100 mL mL one-necked flask equipped with a stirrer was added diketoester 3ad (2.50 g, 12.49 mmol), ammonium acetate (9.63 g, 124.9 mmol, 10.00 eq.) And acetic acid (40 mL), and the mixture was heated to 80 Was stirred at.
- Pyrrol 4ad ⁇ was alkylated with methyl iodide to synthesize N-methylpyrrole 5ad. Specifically, pyrrole 4ad (2.11 g, 11.63 mmol), methyl iodide (3.6 mL, 57.83 mmol, 4.97 eq.), And THF (45 mL) were added to a 100 mL one-necked flask equipped with a stir bar, and the mixture was ice-cooled. Next, sodium hydride (60% in liquid paraffin, 698 mg, 17.44 mmol, 1.50 eq.) was added to the obtained brown solution, and foaming was confirmed.
- Sulfonamide 7ad was synthesized by reacting sulfonic acid chloride 6ad with an ammonia / dioxane solution. That is, to a 100 mL mL one-necked flask equipped with a stir bar, add sulfonic acid chloride 6ad (1.75 g, 5.99 mmol) and 0.5 M ammonia / dioxane solution (36.00 mL, 18.00 mmol, 3.01 eq.) And stir at room temperature. did.
- the ester of sulfonamide 7ad was hydrolyzed under basic conditions to synthesize the target pyrrole compound 8ad (X 2a). That is, sulfonamide 7ad (Lot. 4188163, 1.41 g, 5.13 mmol) and 2 M potassium hydroxide / ethanol solution (50.0 mL, 100.0 mmol, 19.48 eq.) Were added to a 100 mL one-necked flask equipped with a stir bar, Stir at 80 ° C.
- Synthesis Example 7-3 To synthesize a compound X2c in the same manner as in Synthesis Example 7-1 Compound X2c. Compound X2c was obtained as a light brown solid (yield 13%, HPLC purity 96.0%).
- Diethyl ester 3 was synthesized by coupling ethyl 3-oxovalerate 1 and 1-bromo-2-butanone 2. That is, sodium hydride (60% in liquid paraffin, 139 mg, 3.48 mmol, 1.20 eq.) And THF (5.5 mL) were added to a 30 mL two-necked eggplant flask equipped with a thermometer and a stirring bar. Chilled on ice. Next, ethyl 3-oxovalerate 1 (418 mg, 2.90 mmol) was added dropwise to the resulting gray suspension. At this time, foaming and heat generation were confirmed (0.7 ° C ⁇ 12.5 ° C).
- Pa-Knorr pyrrole was synthesized using N-aminophthalimide 4 to synthesize phthalimide pyrrole 5. That is, to a 10 mL mL eggplant flask equipped with a stir bar and a Dimroth condenser, diketo ester 3 (Lot.5187301, 602 mg, 2.81 mmol) and N-aminophthalimide 4 (455 mg, 2.81 mmol, 1.00 eq) in a nitrogen atmosphere .) was added, and acetic acid (3.6 mL) was further added, followed by stirring at room temperature (N-aminophthalimide 4 insoluble).
- the oil bath temperature was set to around 130 ° C, and the reaction solution was refluxed (N-aminophthalimide 4 was completely dissolved at a bath temperature of about 80 ° C).
- TLC analysis was performed in the same manner, but diketoester 3 remained.
- a saturated aqueous sodium chloride solution (30 mL)
- sodium sulfate was added and dried.
- the filtrate was concentrated under reduced pressure to obtain 1.01 g of a yellow solid, and the resulting crude product was purified by a medium pressure preparative liquid chromatograph (silica gel) manufactured by Yamazen.
- Chlorosulfonation was performed using chlorosulfuric acid to synthesize sulfonic acid chloride 6. Specifically, chlorosulfuric acid (1.1 mL, 16.55 mmol, 10.09 eq.) was added to a 10 mL screw-cap test tube equipped with a stir bar and sufficiently purged with nitrogen, and stirred in an ice bath (fuming attention). Phthalimidopyrrole 5 (Lot.5188011, 596 mg, 1.75 mmol) was added little by little, and the mixture was sealed and heated to room temperature (orange transparent solution).
- the resulting sulfonic acid chloride 6 was reacted with an ammonia / dioxane solution to synthesize sulfonic acid amide 7. That is, a 30 mL mL eggplant flask equipped with a stir bar was charged with sulfonic acid chloride 6 (Lot.5187201, 369.3 mg, 0.84 mmol), and 0.5 mm ammonia / dioxane solution (7.3 mL, 3.64 mmol, 4.33 mmol) in an ice bath. .) was added and stirred at room temperature.
- the obtained fraction was concentrated and vacuum-dried at 40 ° C. for 1 hour to obtain a DMSO solution of N-aminopyrrole 8.
- Yamazen's medium pressure preparative liquid chromatograph equipped with an injection column (ethyl acetate / hexane 50% (3 minutes, gradient 13 minutes) ⁇ 100% (17 minutes) as eluent) was passed DMSO solution, and DMSO was added. By removal, 128 mg of N-aminopyrrole 8 was obtained as white crystals (yield 65%).
- N-aminopyrrole 8 was reacted with 2 M LiOH / H 2 O to synthesize compound 9 (X 3a). That is, N-aminopyrrole 8 (Lot.5189181, 96 mg, 0.33 mmol) was charged into a 10 mL screw mouth test tube equipped with a stirring bar. 2 M LiOH / H 2 O (717 ⁇ L, 1.433 mmol, 4.32 eq.) And ethanol (143 ⁇ L) were added, and the mixture was stirred at 100 ° C. After 1 hour, disappearance of N-aminopyrrole 8 was confirmed by HPLC analysis.
- reaction solution After cooling the reaction solution to room temperature, the pH was adjusted to about 4 to 4.5 with 1 M hydrochloric acid to precipitate a white precipitate.
- the reaction mixture was transferred to a 50 mL eggplant flask and concentrated under reduced pressure (pale yellow solid). Methanol (1 mL) was added to the system, and the solid was completely dissolved by ultrasonication. After adding chloroform (19 mL), the solution was allowed to stand overnight in a freezer (about ⁇ 10 ° C.). The precipitated solid was collected by suction filtration (10.1 mg). The mother liquor was concentrated under reduced pressure.
- 2-Bromopyrrole 1 (3.9 g, 15.85 mmol) and DMF (95 mL) were added to a 500 mL three-necked eggplant flask equipped with a stir bar and stirred at room temperature. Thereto were added copper (I) cyanide (7.1 g, 79.24 mmol, 5.0 eq.) And potassium iodide (184 mg, 1.11 mmol, 0.07 eq.), And the mixture was stirred at 120 ° C. Stir for 17 hours. After completion of the reaction, the reaction mixture was diluted with ethyl acetate (95 mL), distilled water (95 mL) was added, and the solid was filtered off through celite.
- 1,2-Dichloroethane (42 mL) and chlorosulfuric acid (7.23 mL, 12.73 mmol, 10.0 mL eq.) Were added to an 300 mL mL eggplant flask equipped with a stir bar, and the mixture was stirred in an ice bath. Thereto was added dropwise a solution of 2-cyanopyrrole 2 (2.1 g, 10.93 mmol) in 1,2-dichloroethane (13 mL), and the mixture was stirred at room temperature for 40 minutes.
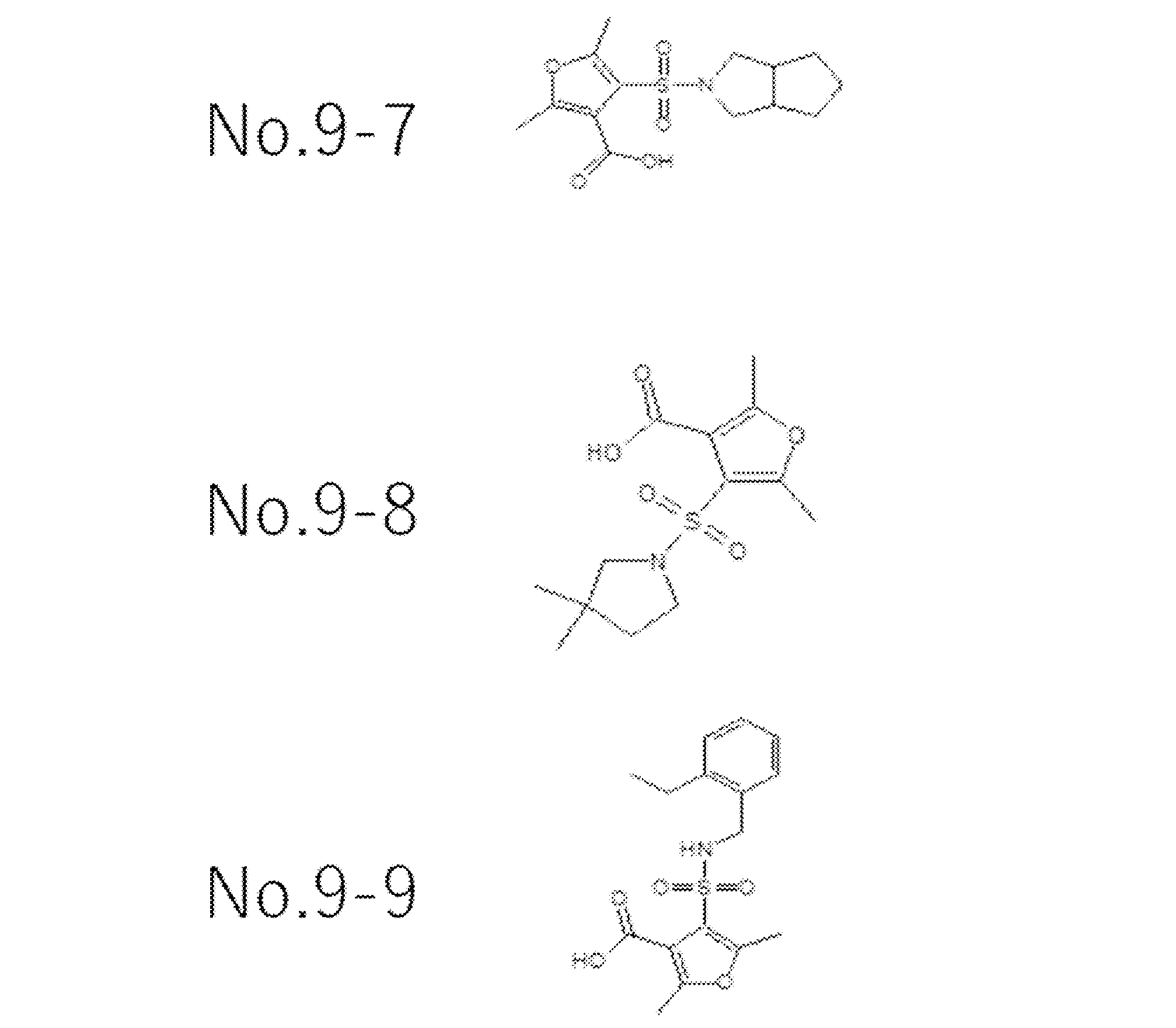
- Test Example 9 Enzyme activity measurement test Metallo- ⁇ -lactamase (IMP-1) (final concentration 10 nM) and imipenem (final concentration 100 ⁇ M) as a substrate, test substances (compound No. 9-7, The concentrations of No. 9-8 and No. 9-9) were changed (0 ⁇ M, 10 ⁇ M, and 100 ⁇ M), and the imipenem degradation activity ( ⁇ abs / min) at the absorbance of 298 nm and 30 ° C. at each point was measured. The activity in the case of the test substance 0 was defined as 100%, and the activity relative value when the test substance was added was calculated.
- IMP-1 final concentration 10 nM
- imipenem final concentration 100 ⁇ M
- test substances compound No. 9-7, The concentrations of No. 9-8 and No. 9-9) were changed (0 ⁇ M, 10 ⁇ M, and 100 ⁇ M), and the imipenem degradation activity ( ⁇ abs / min) at the absorbance of 298 nm and 30 ° C. at each point was
- Table 3 The results are shown in Table 3.
- the leftmost column shows a test substance that is an inhibitor.
- Test Example 10 Drug sensitivity test 5 The test was conducted in the same manner as in Test Example 1 except that X2a, X2b, X2c, X2d, X3a, and X4 were used as test substances to be inhibitors. The inhibitor concentration was 10 ⁇ g / mL.
- Table 4 The results are shown in Table 4.
- the leftmost column shows a test substance that is an inhibitor.
- Test Example 11 Inhibition constant (Ki) measurement test 2 The test was performed in the same manner as in Test Example 6 except that X2a, X2b, X2c, X2d, X3a, and X4 were used as test substances to be inhibitors.
- Table 5 The results are shown in Table 5.
- the leftmost column shows a test substance that is an inhibitor.
- Test Example 12 Animal test (mouse peritoneal infection model) 1 After infecting mice with an experimental strain (E. coli strain into which a plasmid encoding class B ⁇ -lactamase (IMP-1) was introduced), a test substance (compound A) was administered, and life and death were observed. Specifically, it was performed as follows.
- mice The experimental strain grown overnight on Mueller Hinton agar is scraped with a sterile cotton swab, suspended in physiological saline, adjusted to 2-5x10 8 CFU / mL, and mixed with an equal volume of 10% mucin solution (1 -2.5 ⁇ 10 8 CFU / mL), a bacterial solution was prepared. After measurement of the body weight of mice (Crl: CD1 (ICR), male, 4 weeks old (Charles River Japan)) acclimated for 4 days after arrival, 250 ⁇ L of the prepared bacterial solution was injected intraperitoneally per mouse for infection. .
- an antibacterial drug (meropenem trihydrate (WAKO 137-15674)) and a test substance were dissolved in physiological saline.
- the first antibacterial drug 0.2 mg / kg
- / or test substance 100 mg / kg
- the second administration was carried out in the same manner as the first administration (day 0 so far). From the next day (day 1), life and death were observed every day at regular times, and the survival rate was recorded.
- Test Example 13 Animal test (mouse abdominal cavity infection model) 2
- a bacterium K. pnuemoniae
- Compound I was administered as a test substance.
- the antibiotic dose was 0.8 mg / kg or 4 mg / kg, and the test substance dose was 10 mg / kg. Except for these, the same procedure as in Test Example 12 was performed.
- Test Example 14 Animal test (mouse abdominal infection model) 3 Bacteria (K. pnuemoniae) introduced with plasmids encoding NDM and VIM were used as experimental strains, and compound X2d was administered as a test substance.
- the antibiotic dose was 2 mg / kg or 8 mg / kg, and the test substance dose was 10 mg / kg. Except for these, the same procedure as in Test Example 12 was performed.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
β-ラクタマーゼ阻害活性を有する化合物を提供することを課題とする。該課題を、平面性を有する5員環の特定位置がスルファモイル基及びカルボキシ基に置換されてなる化合物に代表される一般式(1)で表される化合物により、解決する。
Description
本発明は、β-ラクタマーゼ阻害剤等に関する。
細菌感染症に対しては、通常、抗菌薬の作用により原因菌を静菌又は殺菌するという治療方法が採られる。ところが、抗菌薬を用いることによって、その抗菌薬に対する薬剤耐性菌が出現することとなる。例えば、β-ラクタム系抗菌薬に対しては、これを分解する活性を有する酵素(β-ラクタマーゼ)を産生する耐性菌が存在する。これまで、新たに出現したβ-ラクタマーゼ産生菌に対しては、新たな構造のβ-ラクタム系抗菌薬が開発され、使用されてきた。このような中で、カルバペネム系抗菌薬が、最も新しいβ-ラクタム系抗菌薬として使用されている。
しかし、カルバペネム系抗菌薬の使用により、該抗菌薬の分解活性を有する酵素(クラスB β-ラクタマーゼ、メタロ-β-ラクタマーゼ)を産生する耐性菌が出現し、問題になっている。該耐性菌は、カルバペネム系抗菌薬を含む多くのβ-ラクタム系抗菌薬に耐性を示す。
β-ラクタム系抗菌薬耐性菌に対しては、その耐性機構の原因酵素であるβ-ラクタマーゼの阻害剤を適当なβ-ラクタム系抗菌薬と併用することが、有効な対策の1つである(特許文献1~3)。しかし、クラスB β-ラクタマーゼに対しては、臨床的に用いることができる阻害剤は実用化されていない。
本発明は、β-ラクタマーゼ阻害活性を有する化合物を提供することを課題とする。好ましくは、本発明は、クラスB β-ラクタマーゼ阻害活性を有する化合物を提供することを課題とする。
本発明者は上記課題に鑑みて鋭意研究を進めた結果、平面性を有する5員環の特定位置がスルファモイル基及びカルボキシ基に置換されてなる化合物に代表される一般式(1)で表される化合物が、β-ラクタマーゼ阻害活性を有すること、特にクラスB β-ラクタマーゼ阻害活性を有することを見出した。本発明者は、この知見に基づいてさらに研究を進めた結果、本発明を完成させた。即ち、本発明は、下記の態様を包含する。
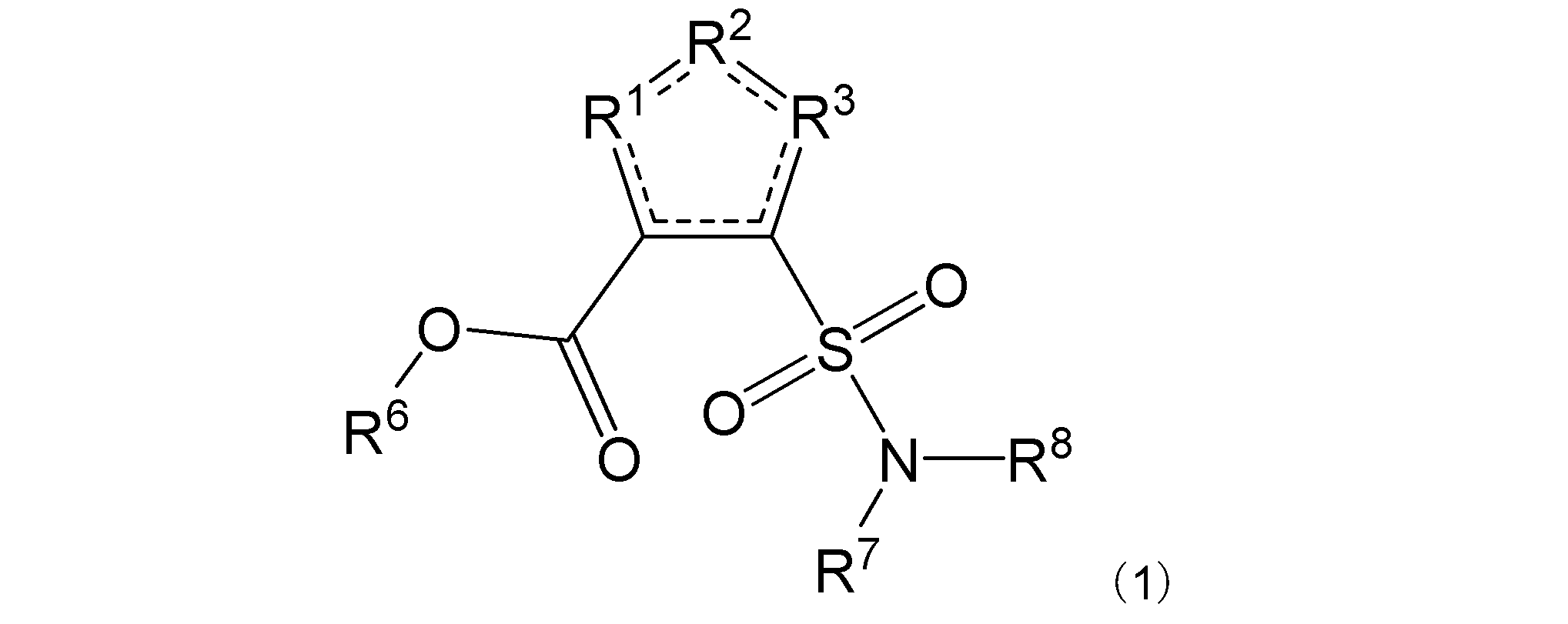
項1. 一般式(1):
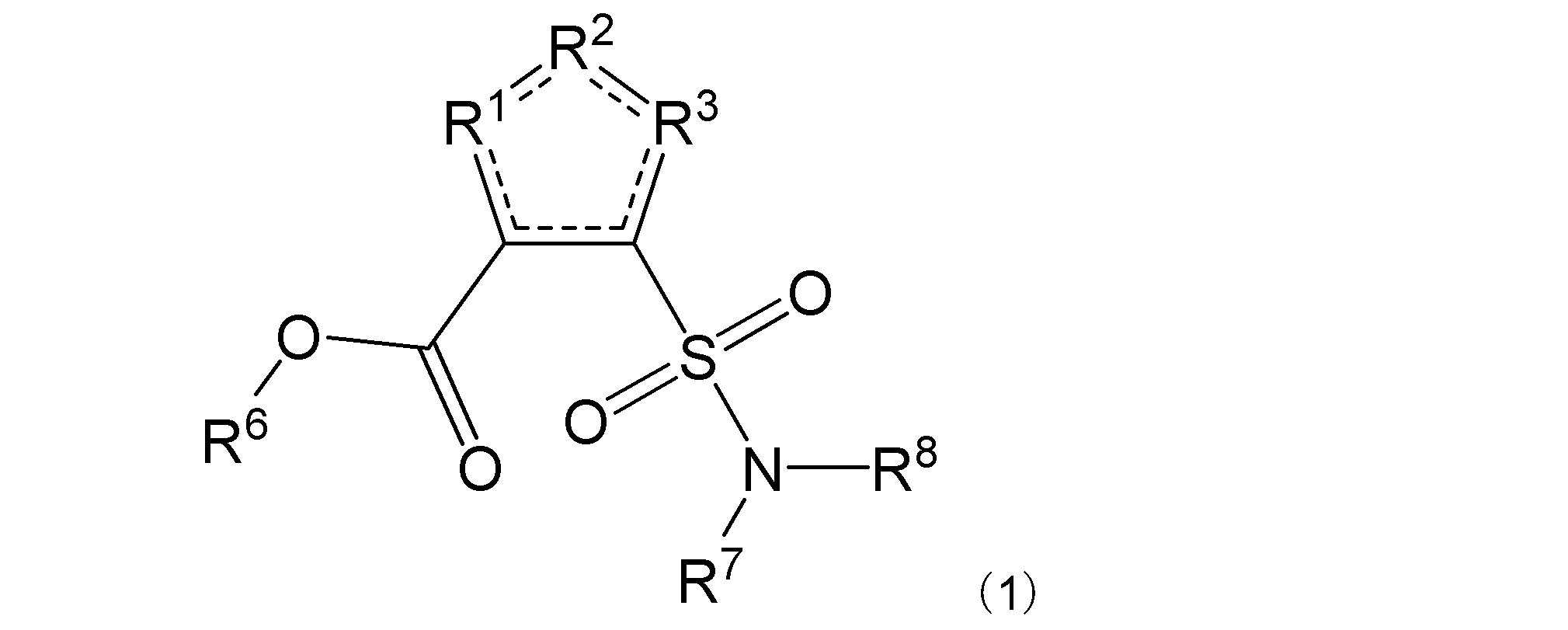
[式中:R1は-C(-RA)-を示す。R2は-N(-RB)n-、-O-、又は-C(-RA)-を示す。R3は、R2が-N(-RB)n-又は-O-の場合は-C(-RA)-を示し、R2が-C(-RA)-の場合は-S-を示す。RAは同一又は異なって、水素原子、ハロゲン原子、又はアミノ基で置換されていてもよい炭化水素基を示す。RBは水素原子、ハロゲン原子、置換されていてもよいアミノ基、又はアミノ基で置換されていてもよい炭化水素基を示す。nは、0又は1を示す。R6は水素原子又は炭化水素基を示す。R7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。実線と点線の二重線で表される結合は、単結合又は二重結合を示す。]
で表される化合物、又はその塩、水和物若しくは溶媒和物を含有する、β-ラクタマーゼ阻害剤。
で表される化合物、又はその塩、水和物若しくは溶媒和物を含有する、β-ラクタマーゼ阻害剤。
項2. 前記β-ラクタマーゼがクラスB β-ラクタマーゼである、項1に記載の阻害剤。
項3. 前記β-ラクタマーゼがクラスB1 β-ラクタマーゼである、項1又は2に記載の阻害剤。
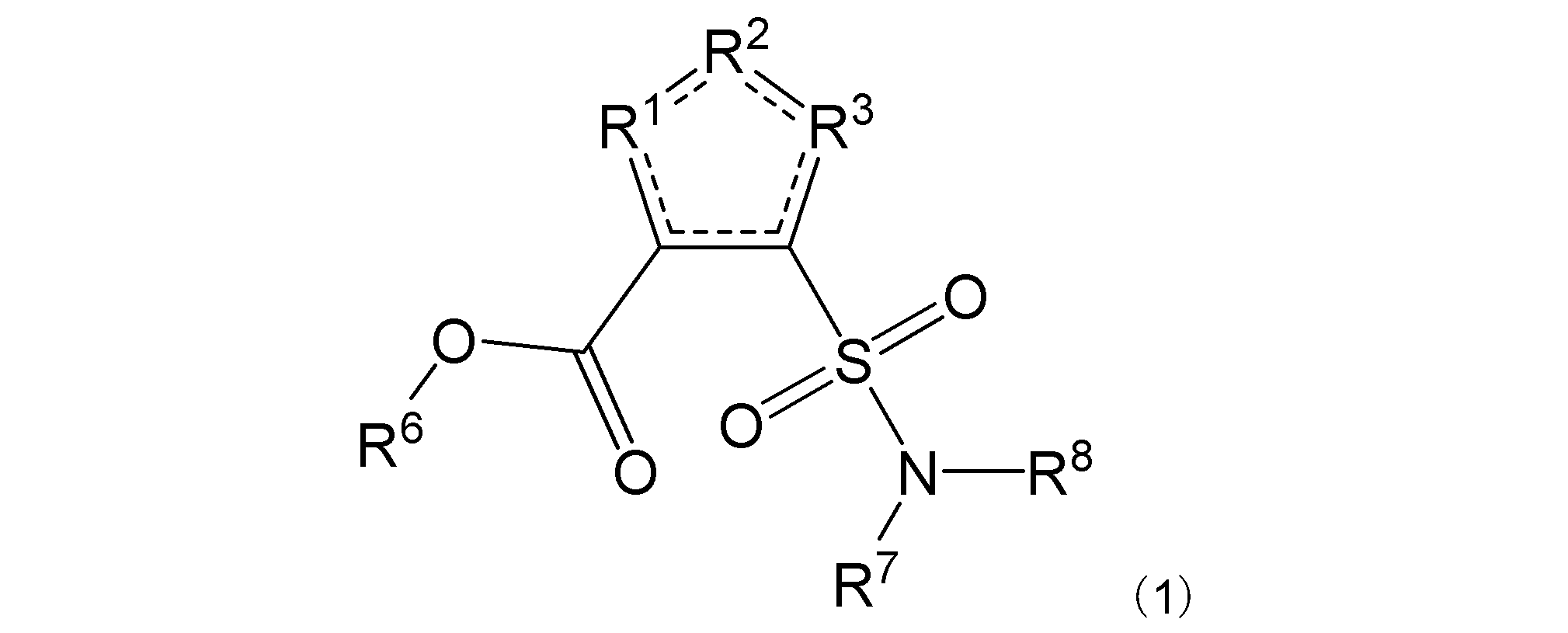
項4. 一般式(1):
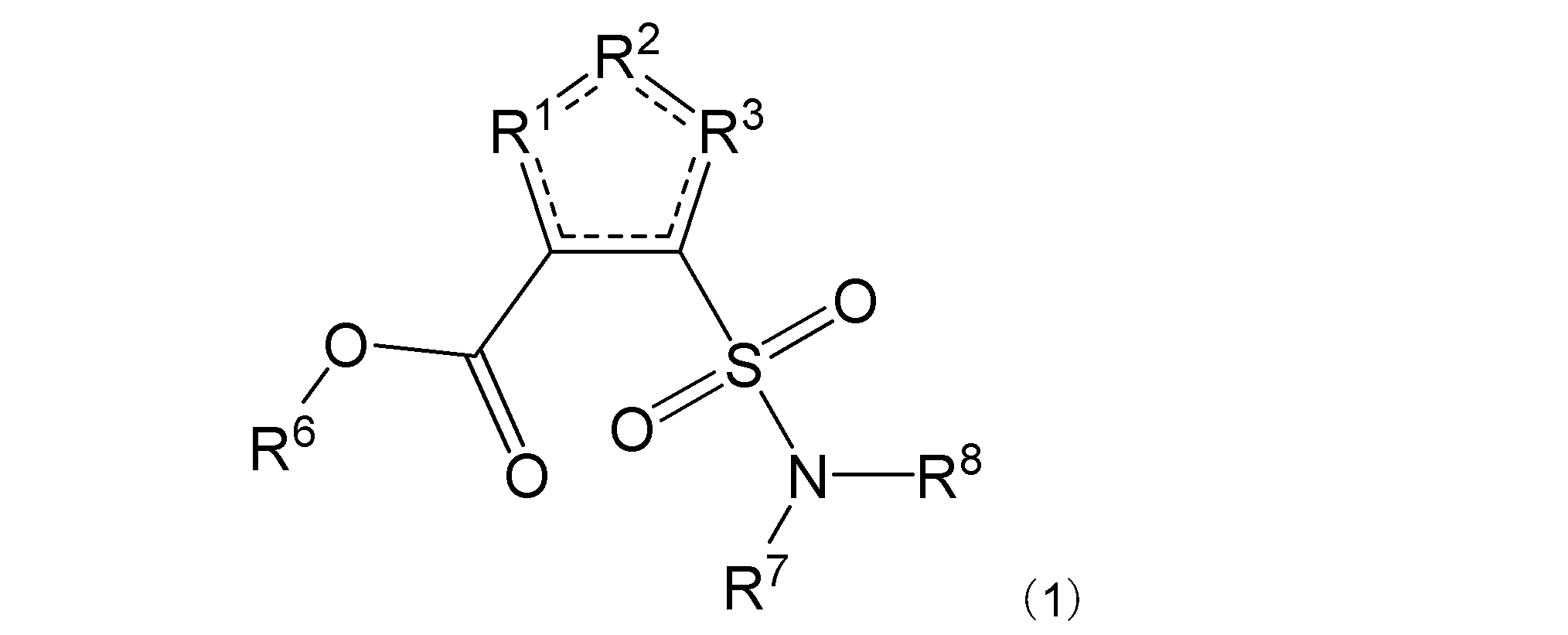
[式中:R1は-C(-RA)-を示す。R2は-N(-RB)n-、-O-、又は-C(-RA)-を示す。R3は、R2が-N(-RB)n-又は-O-の場合は-C(-RA)-を示し、R2が-C(-RA)-の場合は-S-を示す。RAは同一又は異なって、水素原子、ハロゲン原子、又はアミノ基で置換されていてもよい炭化水素基を示す。RBは水素原子、ハロゲン原子、置換されていてもよいアミノ基、又はアミノ基で置換されていてもよい炭化水素基を示す。nは、0又は1を示す。R6は水素原子又は炭化水素基を示す。R7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。実線と点線の二重線で表される結合は、単結合又は二重結合を示す。]
で表される化合物、又はその塩、水和物若しくは溶媒和物を含有する、β-ラクタム系抗菌化合物の抗菌効果の増強剤。
で表される化合物、又はその塩、水和物若しくは溶媒和物を含有する、β-ラクタム系抗菌化合物の抗菌効果の増強剤。
項5. 一般式(1):
[式中:R1は-C(-RA)-を示す。R2は-N(-RB)n-、-O-、又は-C(-RA)-を示す。R3は、R2が-N(-RB)n-又は-O-の場合は-C(-RA)-を示し、R2が-C(-RA)-の場合は-S-を示す。RAは同一又は異なって、水素原子、ハロゲン原子、又はアミノ基で置換されていてもよい炭化水素基を示す。RBは水素原子、ハロゲン原子、置換されていてもよいアミノ基、又はアミノ基で置換されていてもよい炭化水素基を示す。nは、0又は1を示す。R6は水素原子又は炭化水素基を示す。R7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。実線と点線の二重線で表される結合は、単結合又は二重結合を示す。]
で表される化合物、又はその塩、水和物若しくは溶媒和物、並びにβ-ラクタム系抗菌化合物を含有する、抗菌剤。
で表される化合物、又はその塩、水和物若しくは溶媒和物、並びにβ-ラクタム系抗菌化合物を含有する、抗菌剤。
項6. β-ラクタム系抗菌化合物を含有する、
一般式(1):
一般式(1):
[式中:R1は-C(-RA)-を示す。R2は-N(-RB)n-、-O-、又は-C(-RA)-を示す。R3は、R2が-N(-RB)n-又は-O-の場合は-C(-RA)-を示し、R2が-C(-RA)-の場合は-S-を示す。RAは同一又は異なって、水素原子、ハロゲン原子、又はアミノ基で置換されていてもよい炭化水素基を示す。RBは水素原子、ハロゲン原子、置換されていてもよいアミノ基、又はアミノ基で置換されていてもよい炭化水素基を示す。nは、0又は1を示す。R6は水素原子又は炭化水素基を示す。R7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。実線と点線の二重線で表される結合は、単結合又は二重結合を示す。]
で表される化合物、又はその塩、水和物若しくは溶媒和物と併用投与されるように用いられる抗菌剤。
で表される化合物、又はその塩、水和物若しくは溶媒和物と併用投与されるように用いられる抗菌剤。
項7. 一般式(1A):
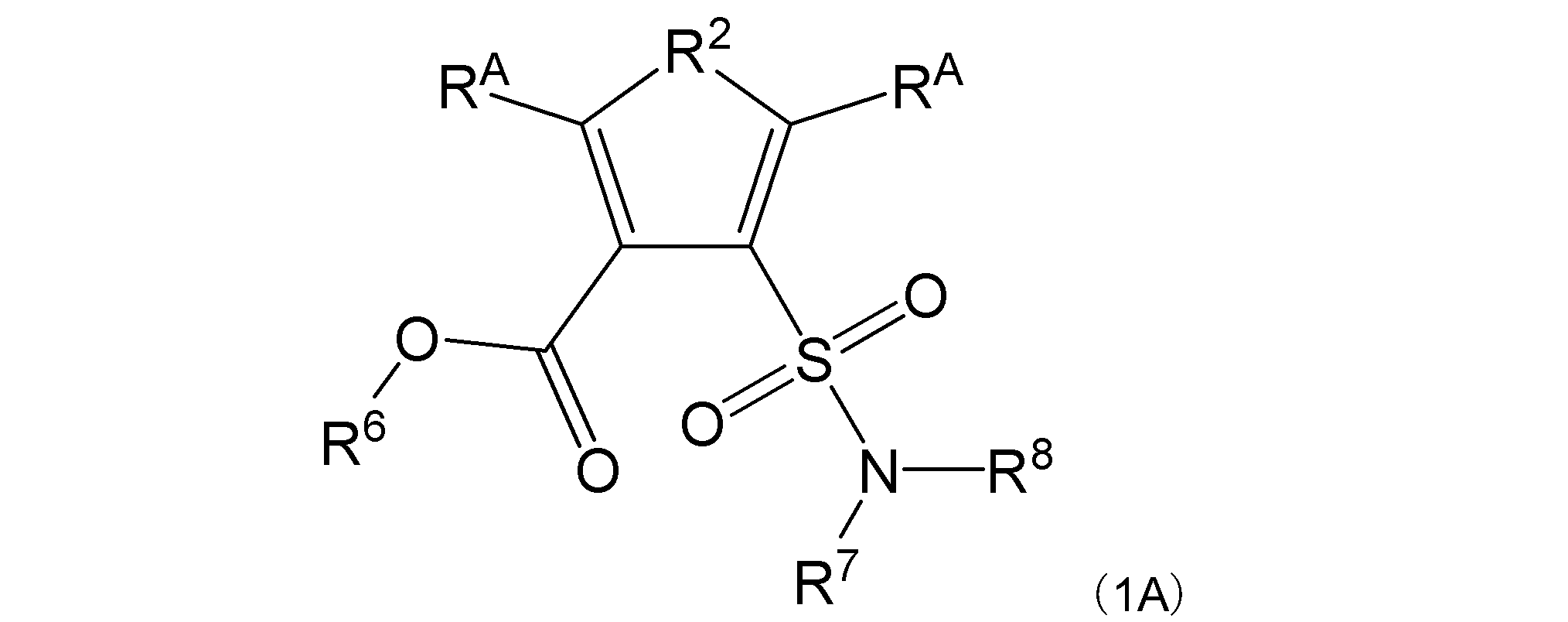
[式中:R2は、-N(-RB)-又は-O-を示す。RAは同一又は異なって、水素原子、アミノ基で置換されていてもよい直鎖状若しくは分岐鎖状アルキル基(但し、R2が-N(-RB)-の場合は炭素数1~5であり、R2が-O-の場合は炭素数2~5である)、アミノ基で置換されていてもよい炭素数3~7の環状アルキル基、又はアミノ基で置換されていてもよいフェニル基を示す。RBは水素原子、直鎖状若しくは分岐鎖状アルキル基で置換されてもよいアミノ基、又はアミノ基で置換されていてもよいアルキル基を示す。R6は水素原子を示す。R7及びR8は同一又は異なって、水素原子又はアルキル基を示し(但し、R7及びR8の両方がアルキル基である場合を除く)、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。]
で表される化合物、又はその塩、水和物若しくは溶媒和物。
で表される化合物、又はその塩、水和物若しくは溶媒和物。
項8. RBで示されるアルキル基が炭素数1~4の直鎖状アルキル基である、項7に記載の化合物、又はその塩、水和物若しくは溶媒和物。
項9. 項7又は8に記載の化合物、又はその塩、水和物若しくは溶媒和物を含有する、医薬。
項10. 項7又は8に記載の化合物、又はその塩、水和物若しくは溶媒和物を含有する、試薬。
本発明によれば、β-ラクタマーゼ阻害活性、特にクラスB β-ラクタマーゼ阻害活性を有する化合物を提供することができる。これを利用することにより、β-ラクタム環を有する既存のβ-ラクタマーゼ阻害剤、およびβ-ラクタム系抗菌化合物の抗菌効果の増強剤等を提供することができる。また、本発明の化合物は比較的毒性が低いので、より安全に使用することが可能である。
本明細書中において、「含有」及び「含む」なる表現については、「含有」、「含む」、「実質的にからなる」及び「のみからなる」という概念を含む。
1.化合物
本発明は、その一態様において、一般式(1):
本発明は、その一態様において、一般式(1):
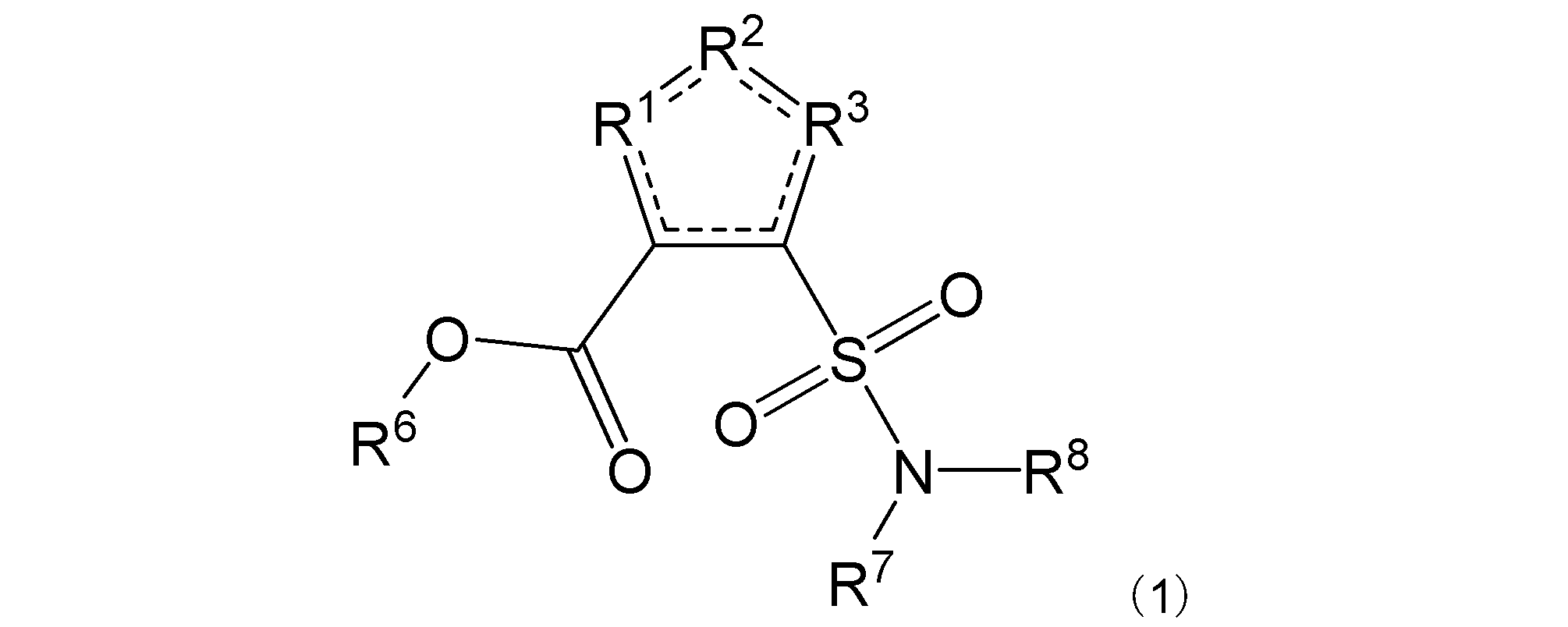
で表される化合物、又はその塩、水和物若しくは溶媒和物(本明細書において、これらを「本発明の化合物」と示すこともある。)に関する。以下にこれについて説明する。
<1-1.R1、R2及びR3>
R1は-C(-RA)-を示す。R2は-N(-RB)n-、-O-、又は-C(-RA)-を示す。R3は、R2が-N(-RB)n-又は-O-の場合は-C(-RA)-を示し、R2が-C(-RA)-の場合は-S-を示す。R2は好ましくは-N(-RB)n-である。
R1は-C(-RA)-を示す。R2は-N(-RB)n-、-O-、又は-C(-RA)-を示す。R3は、R2が-N(-RB)n-又は-O-の場合は-C(-RA)-を示し、R2が-C(-RA)-の場合は-S-を示す。R2は好ましくは-N(-RB)n-である。
RAは同一又は異なって、水素原子、ハロゲン原子、又はアミノ基で置換されていてもよい炭化水素基を示す。本発明のある態様においては、好ましくは、RAが炭化水素基である。
RAで示されるハロゲン原子としては、例えばフッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
RAで示される炭化水素基としては、特に制限されず、例えばアルキル基、アリール基等、さらにはこれらが任意に組み合わされてなる基(例えば、アラルキル基、アルキルアリール基、アルキルアラルキル基)等が挙げられる。本発明のある態様においては、好ましくは、炭化水素基がアルキル基又はアリール基である。この態様において、RAが2つ存在する場合、好ましくは、2つのRAが両方アリール基である場合は除かれる。 RAで示されるアルキル基には、直鎖状、分岐鎖状、又は環状(好ましくは直鎖状又は分枝鎖状、より好ましくは直鎖状)のいずれのものも包含される。該アルキル基(直鎖状又は分枝鎖状の場合)の炭素数は、特に制限されず、例えば1~12、好ましくは1~8、より好ましくは1~5、さらに好ましくは1~4、よりさらに好ましくは1~2である。該アルキル基(環状の場合)の炭素数は、特に制限されず、例えば3~7、好ましくは4~6である。一方で、本発明のある態様において、該アルキル基の炭素数の下限は、例えば2、3、4、5、6、7、8である。該アルキル基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、tert-ブチル基、sec-ブチル基、n-ペンチル基、ネオペンチル基、n-ヘキシル基、3-メチルペンチル基、n-ヘプチル基、n-オクチル基、n-ノニル基、n-デシル基、n-ウンデシル基、n-ドデシル基等が挙げられる。
RAで示されるアリール基は、特に制限されないが、炭素数が6~12のものが好ましく、6~12のものがより好ましく、6~8のものがさらに好ましい。該アリール基は、単環式又は多環式(例えば2環式、3環式等)のいずれでも有り得るが、好ましくは単環式である。該アリール基としては、具体的には、例えばフェニル基、ナフチル基、ビフェニル基、ペンタレニル基、インデニル基、アントラニル基、テトラセニル基、ペンタセニル基、ピレニル基、ペリレニル基、フルオレニル基、フェナントリル基等が挙げられ、好ましくはフェニル基が挙げられる。
RAで示されるアラルキル基は、特に制限されないが、例えば直鎖状又は分岐鎖状の炭素数1~6(好ましくは1~2)のアルキル基の水素原子(例えば1~3つ、好ましくは1つの水素原子)が上記アリール基に置換されてなるアラルキル基等が挙げられる。該アラルキル基としては、具体的には、例えばベンジル基、フェネチル基等が挙げられる。
RAで示されるアルキルアリール基は、特に制限されないが、例えば上記アリール基の水素原子(例えば1~3つ、好ましくは1つの水素原子)が、直鎖状又は分岐鎖状の炭素数1~6(好ましくは1~2)のアルキル基に置換されてなるアルキルアリール基等が挙げられる。該アルキルアリール基としては、具体的には、例えばトリル基、キシリル基等が挙げられる。
RAで示されるアルキルアラルキル基は、特に制限されないが、例えば上記アラルキル基の芳香環上の水素原子(例えば1~3つ、好ましくは1つの水素原子)が、直鎖状又は分岐鎖状の炭素数1~6(好ましくは1~2)のアルキル基に置換されてなるアルキルアラルキル基等が挙げられる。
RAで示される炭化水素基がアミノ基で置換されている場合、そのアミノ基には、-NH2のみならず、-NH2の水素原子が炭化水素基で置換されてなる置換アミノ基も包含される。置換アミノ基における炭化水素基については、RAで示される炭化水素基と同様である。
RBは水素原子、ハロゲン原子、置換されてもよいアミノ基、又はアミノ基で置換されていてもよい炭化水素基を示す。本発明のある態様においては、好ましくは、RBが水素原子又は炭化水素基であり、より好ましくはRBが炭化水素基である。RBで示されるハロゲン原子、及びRBで示されるアミノ基で置換されていてもよい炭化水素基については、RAについての上記記載と同様である。
RBで示される置換されていてもよいアミノ基としては、例えば-NH2、-NH2の水素原子が炭化水素基で置換されてなる置換アミノ基等が挙げられる。炭化水素基については、RAで示される炭化水素基と同様である。
nは、0又は1を示す。本発明のある態様においては、好ましくは、nが1である。
<1-2.5員環>
一般式(1)中、実線と点線の二重線で表される結合は、単結合又は二重結合を示す。該結合を含む、一般式(1)中の一般式(A):
一般式(1)中、実線と点線の二重線で表される結合は、単結合又は二重結合を示す。該結合を含む、一般式(1)中の一般式(A):

[式中:R1、R2、及びR3は前記に同じである。]
で表される部分構造中の5員環は、特に制限されない。該5員環は、好ましくは平面性を有するものである。該5員環の具体例としては、フラン、ピロール、チオフェン等が挙げられ、より好ましくはフラン、ピロール等が挙げられ、さらに好ましくはピロールが挙げられる。
で表される部分構造中の5員環は、特に制限されない。該5員環は、好ましくは平面性を有するものである。該5員環の具体例としては、フラン、ピロール、チオフェン等が挙げられ、より好ましくはフラン、ピロール等が挙げられ、さらに好ましくはピロールが挙げられる。
<1-3.R6>
R6は水素原子又は炭化水素基を示す。本発明のある態様においては、好ましくは、R6が水素原子である。R6で示される炭化水素基については、RAについての上記記載と同様である。
R6は水素原子又は炭化水素基を示す。本発明のある態様においては、好ましくは、R6が水素原子である。R6で示される炭化水素基については、RAについての上記記載と同様である。
<1-4.R7及びR8>
R7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。本発明のある態様においては、好ましくはR7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、より好ましくは、R7及びR8の内の少なくとも一方が水素原子であり、さらに好ましくはR7及びR8は共に水素原子である。
R7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。本発明のある態様においては、好ましくはR7及びR8は同一又は異なって、水素原子又は炭化水素基を示し、より好ましくは、R7及びR8の内の少なくとも一方が水素原子であり、さらに好ましくはR7及びR8は共に水素原子である。
R7で示される炭化水素基、及びR8で示される炭化水素基については、RAについての上記記載と同様である。
R7及びR8が互いに結合して隣接する窒素原子と共に形成する環は、特に制限されず、例えば単環又は二環である。該環としては、例えば一般式(X)、一般式(Y):
[式中:R71及びR72各々はアルキル基を示す。p及びr各々は1~3の整数を示す。q及びs各々は0又は1~3の整数を示す。]
R71及びR72で示されるアルキル基については、RAについての上記記載と同様である。R71及びR72各々について、複数存在する場合は、同一の炭素原子に結合していてもよいし、互いに異なる炭素原子に結合していてもよい。
R71及びR72で示されるアルキル基については、RAについての上記記載と同様である。R71及びR72各々について、複数存在する場合は、同一の炭素原子に結合していてもよいし、互いに異なる炭素原子に結合していてもよい。
<1-5.好ましい一般式(1)化合物>
本発明の一態様において、一般式(1)の中でも、好ましくは一般式(1A):
本発明の一態様において、一般式(1)の中でも、好ましくは一般式(1A):

[式中:RA、RB、n、R6、R7及びR8は前記に同じである。]
が挙げられる。
が挙げられる。
本発明の別の一態様において、一般式(1)の中でも、好ましくは一般式(1B):

[式中:RA、R6、R7及びR8は前記に同じである。]
が挙げられる。
が挙げられる。
一般式(1)、(1A)、(1B)において、好ましい態様は以下のとおりである:
R2は、-N(-RB)-又は-O-を示す。
RAは同一又は異なって、水素原子、アミノ基で置換されていてもよい直鎖状若しくは分岐鎖状アルキル基(但し、R2が-N(-RB)-の場合は炭素数1~5であり、R2が-O-の場合は炭素数2~5である)、アミノ基で置換されていてもよい炭素数3~7の環状アルキル基、又はアミノ基で置換されていてもよいフェニル基を示す。
RBは水素原子、直鎖状若しくは分岐鎖状アルキル基で置換されてもよいアミノ基、又はアミノ基で置換されていてもよいアルキル基を示す。
R6は水素原子を示す。
R7及びR8は同一又は異なって、水素原子又はアルキル基を示し(但し、R7及びR8の両方がアルキル基である場合を除く)、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。
R2は、-N(-RB)-又は-O-を示す。
RAは同一又は異なって、水素原子、アミノ基で置換されていてもよい直鎖状若しくは分岐鎖状アルキル基(但し、R2が-N(-RB)-の場合は炭素数1~5であり、R2が-O-の場合は炭素数2~5である)、アミノ基で置換されていてもよい炭素数3~7の環状アルキル基、又はアミノ基で置換されていてもよいフェニル基を示す。
RBは水素原子、直鎖状若しくは分岐鎖状アルキル基で置換されてもよいアミノ基、又はアミノ基で置換されていてもよいアルキル基を示す。
R6は水素原子を示す。
R7及びR8は同一又は異なって、水素原子又はアルキル基を示し(但し、R7及びR8の両方がアルキル基である場合を除く)、或いはR7及びR8は互いに結合して隣接する窒素原子と共に環を形成する。
一般式(1)で表される化合物として、具体的には、例えば以下の化合物が挙げられる。
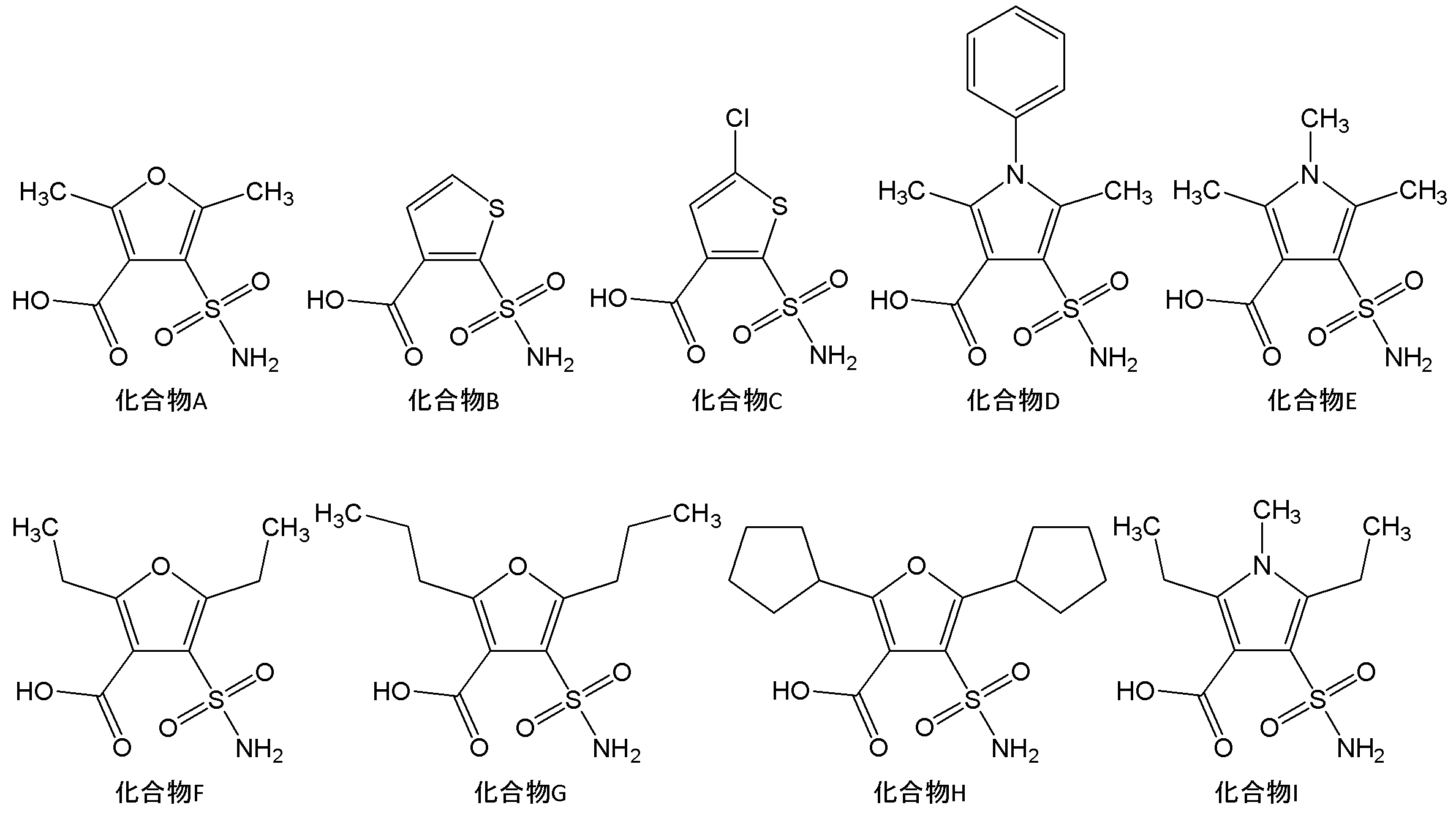
上記化合物の中でも、好ましくは化合物E、化合物I、化合物X2d等が挙げられる。
<1-6.異性体>
一般式(1)で表される化合物には、立体異性体及び光学異性体が含まれ、これらは特に限定されるものではない。
一般式(1)で表される化合物には、立体異性体及び光学異性体が含まれ、これらは特に限定されるものではない。
<1-7.塩、水和物、溶媒和物>
一般式(1)で表される化合物の塩は、薬学的に許容される塩である限り、特に制限されるものではない。該塩としては、酸性塩、塩基性塩のいずれも採用することができる。酸性塩の例としては、塩酸塩、臭化水素酸塩、硫酸塩、硝酸塩、リン酸塩等の無機酸塩; 酢酸塩、プロピオン酸塩、酒石酸塩、フマル酸塩、マレイン酸塩、リンゴ酸塩、クエン酸塩、メタンスルホン酸塩、パラトルエンスルホン酸塩等の有機酸塩が挙げられ、塩基性塩の例としては、ナトリウム塩、及びカリウム塩等のアルカリ金属塩; 並びにカルシウム塩、マグネシウム塩等のアルカリ土類金属塩; アンモニアとの塩; モルホリン、ピペリジン、ピロリジン、モノアルキルアミン、ジアルキルアミン、トリアルキルアミン、モノ(ヒドロキシアルキル)アミン、ジ(ヒドロキシアルキル)アミン、トリ(ヒドロキシアルキル)アミン等の有機アミンとの塩等が挙げられる。
一般式(1)で表される化合物の塩は、薬学的に許容される塩である限り、特に制限されるものではない。該塩としては、酸性塩、塩基性塩のいずれも採用することができる。酸性塩の例としては、塩酸塩、臭化水素酸塩、硫酸塩、硝酸塩、リン酸塩等の無機酸塩; 酢酸塩、プロピオン酸塩、酒石酸塩、フマル酸塩、マレイン酸塩、リンゴ酸塩、クエン酸塩、メタンスルホン酸塩、パラトルエンスルホン酸塩等の有機酸塩が挙げられ、塩基性塩の例としては、ナトリウム塩、及びカリウム塩等のアルカリ金属塩; 並びにカルシウム塩、マグネシウム塩等のアルカリ土類金属塩; アンモニアとの塩; モルホリン、ピペリジン、ピロリジン、モノアルキルアミン、ジアルキルアミン、トリアルキルアミン、モノ(ヒドロキシアルキル)アミン、ジ(ヒドロキシアルキル)アミン、トリ(ヒドロキシアルキル)アミン等の有機アミンとの塩等が挙げられる。
一般式(1)で表される化合物は水和物、溶媒和物とすることもできる。溶媒としては、例えば、薬学的に許容される有機溶媒(例えばエタノール、グリセロール、酢酸等)等が挙げられる。
2.製造方法
一般式(1)で表される化合物は、様々な方法で合成することができる。例えば、一般式(1)においてR6が炭化水素基(R6A)であり且つR7及びR8が共に水素原子である場合の化合物(化合物1d)は、例えば以下の反応式:
一般式(1)で表される化合物は、様々な方法で合成することができる。例えば、一般式(1)においてR6が炭化水素基(R6A)であり且つR7及びR8が共に水素原子である場合の化合物(化合物1d)は、例えば以下の反応式:

[式中、R1、R2、R3、R7及びR8は前記に同じである。R6Aは炭化水素基を示す。]
に従って又は準じて、合成することができる。
に従って又は準じて、合成することができる。
また、一般式(1)においてR6が水素原子であり且つR7及びR8が共に水素原子である場合の化合物(化合物1e)は、例えば化合物1dのカルボキシ基の保護基であるR6A-を強塩基等を用いて脱保護することにより、得ることができる。
さらに、R7及びR8の少なくとも一方が炭化水素基である化合物については、化合物1d又は化合物1eのアミノ基を炭化水素で置換することにより得ることができる。
また、これらの方法以外にも、一般式(1)で表される化合物は、パール・クノール反応を利用して合成することもできる。すなわち、パール・クノール反応を利用する場合、適当なケトエステルとケトンとのカップリング物(ジケトエステル)を酸又はアンモニア、第1級アミン等の存在下で反応させてフランやピロール(化合物1c)を合成し、その後は上記反応式に従って一般式(1)で表される化合物を合成することができる。
RA、RB等においてアミノ基が存在する場合は、例えば、パール・クノール反応により、化合物c上のアミノ基が必要に応じて適当な保護基(例えばフタルイミド等)で保護された化合物(化合物c’)を得て、反応工程中の適当なタイミングで脱保護(例えばヒドラジン等を用いて脱保護)することにより、一般式(1)で表される化合物を合成することができる。
<2-1.化合物1a→化合物1b>
本工程では、化合物1a、ハロゲン化アルミニウム、及び二酸化炭素を反応させることで、化合物1bを得ることができる。
本工程では、化合物1a、ハロゲン化アルミニウム、及び二酸化炭素を反応させることで、化合物1bを得ることができる。
ハロゲン化アルミニウムとしては、特に制限されないが、例えばハロゲン化アルキルアルミニウム、好ましくは塩化ジメチルアルミニウム等が挙げられる。
ハロゲン化アルミニウムの使用量は、収率、合成の容易さ等の観点から、通常、1モルの化合物1aに対して、0.5~1.5モルが好ましく、0.8~1.2モルがより好ましい。
二酸化炭素の使用量は、収率、合成の容易さ等の観点から、通常、1モルの化合物1aに対して、1~15モルが好ましい。
本工程は溶媒中で行うことが好ましい。溶媒としては、特に制限されないが、例えばトルエン等が挙げられる。溶媒は1種単独で、或いは2種以上を組み合わせて使用することができる。
本工程においては、上記成分以外にも、本発明の効果を損なわない範囲で、適宜添加剤を使用することもできる。
反応雰囲気は、通常、不活性ガス雰囲気(アルゴンガス雰囲気、窒素ガス雰囲気等)を採用し得る。反応温度は、加熱下、常温下及び冷却下のいずれでも行うことができ、通常、0~100℃(特に15~40℃)で行うことが好ましい。反応時間は特に制限されず、通常、3時間~48時間、特に8時間~24時間とすることができる。
反応終了後は、必要に応じて常法にしたがって精製処理をすることもできる。また、精製処理を施さずに次の工程を行うこともできる。
<2-2.化合物1b→化合物1c>
本工程では、化合物1b、R6A-OH、及びハロゲン化水素を反応させることで、化合物1cを得ることができる。
本工程では、化合物1b、R6A-OH、及びハロゲン化水素を反応させることで、化合物1cを得ることができる。
R6A-OHの使用量は、収率、合成の容易さ等の観点から、通常、1重量部の化合物1bに対して、3~15重量部が好ましく、5~10重量部がより好ましい。
ハロゲン化水素としては、特に制限されないが、好ましくは塩化水素が挙げられる。
ハロゲン化水素の使用量としては、反応液中、1~6規定、好ましくは1.5~5規定となる程度である。
本工程では、通常はR6A-OHが溶媒として機能するが、さらに適当な溶媒を追加してもよい。溶媒は1種単独で、或いは2種以上を組み合わせて使用することができる。
本工程においては、上記成分以外にも、本発明の効果を損なわない範囲で、適宜添加剤を使用することもできる。
反応雰囲気は、通常、不活性ガス雰囲気(アルゴンガス雰囲気、窒素ガス雰囲気等)を採用し得る。反応温度は、加熱下、常温下及び冷却下のいずれでも行うことができ、通常、0~100℃(特に15~40℃)で行うことが好ましい。反応時間は特に制限されず、通常、3時間~48時間、特に8時間~24時間とすることができる。
反応終了後は、必要に応じて常法にしたがって精製処理をすることもできる。また、精製処理を施さずに次の工程を行うこともできる。
<2-3.化合物1c→化合物1d>
本工程では、化合物1c、ハロゲン化スルホニル酸、及び水酸化アンモニウムを反応させることで、化合物1dを得ることができる。この工程は、2段階で、即ち、化合物1cとハロゲン化スルホニル酸を反応させた後、得られた反応物と水酸化アンモニウムとを反応させることにより実行することもできる。
本工程では、化合物1c、ハロゲン化スルホニル酸、及び水酸化アンモニウムを反応させることで、化合物1dを得ることができる。この工程は、2段階で、即ち、化合物1cとハロゲン化スルホニル酸を反応させた後、得られた反応物と水酸化アンモニウムとを反応させることにより実行することもできる。
ハロゲン化スルホニル酸としては、特に制限されないが、好ましくは塩化スルホニル酸等が挙げられる。
ハロゲン化スルホニル酸の使用量は、収率、合成の容易さ等の観点から、通常、1モルの化合物1cに対して、3~20モルが好ましく、7~15モルがより好ましい。
水酸化アンモニウムの使用量は、収率、合成の容易さ等の観点から、通常、1重量部の化合物1cに対して、1~50重量部が好ましく、3~20重量部がより好ましい。
本工程は溶媒中で行うことが好ましい。溶媒としては、特に制限されないが、例えばアセトニトリル等が挙げられる。溶媒は1種単独で、或いは2種以上を組み合わせて使用することができる。
本工程においては、上記成分以外にも、本発明の効果を損なわない範囲で、適宜添加剤を使用することもできる。
反応雰囲気は、通常、不活性ガス雰囲気(アルゴンガス雰囲気、窒素ガス雰囲気等)を採用し得る。反応温度は、加熱下、常温下及び冷却下のいずれでも行うことができ、通常、0~40℃(特に0~5℃)で行うことが好ましい。反応時間は特に制限されず、通常、30分間~10時間、特に1時間~5時間とすることができる。
反応終了後は、必要に応じて常法にしたがって精製処理をすることもできる。また、精製処理を施さずに次の工程を行うこともできる。
3.用途
本発明の化合物は、β-ラクタマーゼ阻害活性、β-ラクタム系抗菌化合物の抗菌効果の増強活性等を有する。このため、本発明の化合物は、医薬、試薬等(本明細書において、「本発明の薬剤」と示すこともある。)の有効成分として、より具体的には、β-ラクタマーゼ阻害剤、β-ラクタム系抗菌化合物の抗菌効果の増強剤等の有効成分としての利用が可能である。また、本発明の化合物のβ-ラクタマーゼ阻害活性、β-ラクタム系抗菌化合物の抗菌効果の増強活性等を利用して、本発明の化合物及びβ-ラクタム系抗菌化合物を含有する、抗菌剤、さらにはβ-ラクタム系抗菌化合物を含有する、本発明の化合物と併用投与されるように用いられる抗菌剤(これらも、本明細書において、「本発明の薬剤」と示すこともある。)、等を提供することもできる。
本発明の化合物は、β-ラクタマーゼ阻害活性、β-ラクタム系抗菌化合物の抗菌効果の増強活性等を有する。このため、本発明の化合物は、医薬、試薬等(本明細書において、「本発明の薬剤」と示すこともある。)の有効成分として、より具体的には、β-ラクタマーゼ阻害剤、β-ラクタム系抗菌化合物の抗菌効果の増強剤等の有効成分としての利用が可能である。また、本発明の化合物のβ-ラクタマーゼ阻害活性、β-ラクタム系抗菌化合物の抗菌効果の増強活性等を利用して、本発明の化合物及びβ-ラクタム系抗菌化合物を含有する、抗菌剤、さらにはβ-ラクタム系抗菌化合物を含有する、本発明の化合物と併用投与されるように用いられる抗菌剤(これらも、本明細書において、「本発明の薬剤」と示すこともある。)、等を提供することもできる。
本発明の薬剤の内、本発明の化合物を含有するものについては、本発明の化合物を含有する限りにおいて特に制限されず、必要に応じてさらに他の成分を含んでいてもよい。他の成分としては、薬学的に許容される成分であれば特に限定されるものではない。他の成分としては、薬理作用を有する成分のほか、添加剤も含まれる。添加剤としては、例えば基剤、担体、溶剤、分散剤、乳化剤、緩衝剤、浸透圧調整剤、吸収促進剤、安定剤、賦形剤、結合剤、崩壊剤、滑沢剤、増粘剤、保湿剤、着色料、香料、キレート剤等が挙げられる。
本発明の化合物による阻害対象であるβ-ラクタマーゼとしては、βーラクタム環の開環を触媒する、任意の供給源に由来する酵素である限り特に制限されない。β-ラクタマーゼ(EC3.5.2.6)は、最も一般的には細菌によって産生される酵素である。β-ラクタマーゼは、β-ラクタム環の加水分解性開環を触媒し、ペニシリン系、ペナム系、ペネム系、セフェム系、セファロスポリン系、カルバセフェム系、セファマイシン系、モノバクタム系、及びカルバペネム系などのβ-ラクタム系抗菌化合物に細菌耐性を付与する原因である。β-ラクタマーゼは、クラスA~Dに分類され、これらの中でも、本発明の化合物による阻害対象としては、クラスB β-ラクタマーゼが好ましく、クラスB1 β-ラクタマーゼがより好ましい。具体的には、例えばIMP-1、NDM-1、VIM-2、DIM-1、GIM-1、KHM-1、SIM-1、SPM-1、TMB-1、BcII、BlaB、CcrA、IND-7等が挙げられる。また、クラスB2やB3として、それぞれSFH-1やGOB-1等が挙げられる。
本発明の化合物による抗菌効果の増強対象であるβ-ラクタム系抗菌化合物としては、特に制限されず、例えばペニシリン系抗菌化合物、セフェム系抗菌化合物、カルバペネム系抗菌化合物等が挙げられる。
ペニシリン系抗菌化合物の具体的な例としては、ベンジルペニシリン、フェネチシリン、クロキサシリン、ジクロキサシリン、アンピシリン、シクラシリン、アモキシリン、タランピシリン、バカンピシリン、レナンピシリン、アスポキシリン、ピペラシリン、スルベニシリン、ピブメシリナム、スルタミシリン、フェノキシメチルペニシリン、カルベニシリン、アジドシリン、プロピシリン、エピシリン、チカルシリン、ピルベニシリン、アズロシリン、メズロシリン、並びに他の公知のペニシリン系抗菌化合物などが挙げられる。
セフェム系抗菌化合物の具体的な例としては、セファクロル、セファゾリン、セファトリジン、セファドロキシル、セファピリン、セファマンドール・ナフェート、セファラジン、セファレキシン、セファロチン、セフェピム、セフキシチン、セフキシム、セフジジム、セフジトレン、セフジニル、セフスロジン、セフセリス、セフゾプラン、セフタキシム、セフタジジム、セフタロリン、セフチアム、セフチゾキシム、セフチブテン、セフテゾール、セフテタム、セフトリアキソン、セフニシド、セフピラミド、セフピロム、セフブペラゾン、セフプロジル、セフペラゾン、セフポドキシム、セフミノクス、セフメタゾール、セフメノキシム、セフラジン、セフロキサジン、セフロキシム、ラタモキセフ、フロモキセフ、セフトロザン(CXA101、(6R,7R)-3-[5-アミノ-4-[3-(2-アミノエチル)ウレイド]-1-メチル-1H-ピラゾール-2-イウム-2-イルメチル]-7-[2-(5-アミノ-1,2,4-チアジアゾール-3-イル)-2-[(Z)-1-カルボキシ-1-メチルエトキシイミノ]アセタミド]-3-セフェム-4-カルボン酸 硫酸水素塩)ならびに他の公知のセフェム系抗菌化合物等が挙げられる。
カルバペネム系抗菌化合物の具体的な例としては、イミペネム、パニペネム、メロペネム、ビアペネム、ドリペネム、エルタペネム、テビペネムなどがある。
ペニシリン系抗菌化合物、セフェム系抗菌化合物、及びカルバペネム系抗菌化合物以外のβ-ラクタム系抗菌化合物の例としては、アズトレオナム、カルモナム、ロラカルベフ、ファロペネム、リチペネム等が挙げられる。
本発明の薬剤の対象菌としては、グラム陰性菌、グラム陽性菌等を広く採用することができる。グラム陰性菌としては、例えば、腸内細菌科細菌(例えば、エシェリヒア属菌、クレブシエラ属菌、サルモネラ属菌、赤痢菌属等)、アシネトバクター属菌、シュードモナス属菌(例えば緑膿菌)、モラクセラ属菌、ヘリコバクター属菌、カンピロバクター属菌、アエロモナス属菌、ビブリオ属菌(例えばコレラ菌、腸炎ビブリオ菌)、ヘモフィルス属菌(例えばインフルエンザ菌)、ナイセリア属菌(例えば淋菌、髄膜炎菌)、バクテロイデス属菌等が挙げられる。グラム陽性菌としては、例えば、ブドウ球菌属菌(例えば黄色ブドウ球菌、表皮ブドウ球菌)、腸球菌(例えばエンテロコッカス属菌)、レンサ球菌属菌(例えばA群連鎖球菌、B群連鎖球菌、肺炎球菌、緑色連鎖球菌)、バシラス属菌(例えばセレウス菌、炭疽菌)、クロストリジウム属菌(例えば破傷風菌、ボツリヌス菌、ディフィシル菌)、コリネバクテリウム属菌(例えばジフテリア菌)、リステリア属菌、ラクトバシラス属菌、ビフィドバクテリウム属菌、プロピオニバクテリウム属菌(例えばニキビの原因となるアクネ菌)、放線菌等が挙げられる。これらの中でも、好ましくはグラム陰性菌、より好ましくは腸内細菌科細菌、アシネトバクター属菌等が挙げられる。本発明の薬剤は、β-ラクタマーゼを産生する対象菌に対して、その効果を発揮することができる。
本発明の薬剤の使用態様は、特に制限されず、その種類に応じて適切な使用態様を採ることができる。本発明の薬剤は、その用途に応じて、例えばin vitroで使用する(例えば、培養細胞の培地に添加する。)こともできるし、in vivoで使用する(例えば、動物に投与する。)こともできる。
本発明の薬剤を動物又は細胞に適用する場合の適用対象は特に限定されないが、哺乳動物では、例えば、ヒト、サル、マウス、ラット、イヌ、ネコ、ウサギ、ブタ、ウマ、ウシ、ヒツジ、ヤギ、シカ等が挙げられる。また、細胞としては、動物細胞等が挙げられる。細胞の種類も特に制限されず、例えば血液細胞、造血幹細胞・前駆細胞、配偶子(精子、卵子)、線維芽細胞、上皮細胞、血管内皮細胞、神経細胞、肝細胞、ケラチン生成細胞、筋細胞、表皮細胞、内分泌細胞、ES細胞、iPS細胞、組織幹細胞、がん細胞等が挙げられる。
本発明の薬剤は、医薬、試薬等に適した任意の剤形、例えば錠剤(口腔内側崩壊錠、咀嚼可能錠、発泡錠、トローチ剤、ゼリー状ドロップ剤などを含む)、丸剤、顆粒剤、細粒剤、散剤、硬カプセル剤、軟カプセル剤、ドライシロップ剤、液剤(ドリンク剤、懸濁剤、シロップ剤を含む)、ゼリー剤などの経口製剤形態や、注射用製剤(例えば、点滴注射剤(例えば点滴静注用製剤等)、静脈注射剤、筋肉注射剤、皮下注射剤、皮内注射剤)、外用剤(例えば、軟膏剤、クリーム剤、パップ剤、ローション剤)、坐剤吸入剤、眼剤、眼軟膏剤、点鼻剤、点耳剤、リポソーム剤等の非経口製剤形態を採ることができる。
本発明の薬剤の投与経路としては、所望の効果が得られる限り特に制限されず、経口投与、経管栄養、注腸投与等の経腸投与; 経静脈投与、経動脈投与、筋肉内投与、心臓内投与、皮下投与、皮内投与、腹腔内投与等の非経口投与等が挙げられる。
本発明の薬剤中の有効成分の含有量は、使用態様、適用対象、適用対象の状態等に左右されるものであり、限定はされないが、例えば0.0001~100重量%、好ましくは0.001~50重量%とすることができる。
本発明の薬剤をヒトや動物に投与する場合の投与量は、薬効を発現する有効量であれば特に限定されず、通常は、有効成分の重量として、一般に経口投与の場合には一日あたり0.1~1000 mg/kg体重、好ましくは一日あたり0.5~500 mg/kg体重であり、非経口投与の場合には一日あたり0.01~100 mg/kg体重、好ましくは0.05~50 mg/kg体重である。上記投与量は、患者の年齢、病態、症状等により適宜増減することもできる。
また、本発明のβ-ラクタマーゼ阻害剤をβ-ラクタマーゼ産生菌の検出に用いることもできる。たとえば、β-ラクタム系抗菌化合物と本発明のβ-ラクタマーゼ阻害剤との共存下で検査対象の菌を培養したときの増殖阻害の有無でβ-ラクタマーゼ産生菌を検出することができる。すなわち、ディスク法や微量液体希釈法などに本阻害剤を用いることができる。
以下に、実施例に基づいて本発明を詳細に説明するが、本発明はこれらの実施例によって限定されるものではない。
1.化合物の準備1
以下に示される化合物A~Iを準備した。
以下に示される化合物A~Iを準備した。

化合物A~CはEnamine社から購入した。化合物D~Iは以下の合成例1~6に従って合成した。
合成例1.化合物Dの合成

上記反応式に従って、化合物Dを合成した。具体的には以下のようにして合成した。
乾燥トルエン(200mL)中の2,5-ジメチル-1-フェニル-1H-ピロール(D1、5.10g、29.8mmol)、塩化ジメチルアルミニウム(2.74g、29.8mmol)およびCO2(10.0g、227mmol)を一晩撹拌した。混合物を水(200mL)でクエンチし、ジクロロメタン(200mL×3)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して、2,5-ジメチル-1-フェニルピロール-3-カルボン酸エチル(D2)を黄色の油状物として得た(2.73g、42.6%)。
HCl/EtOH(20.0mL、4.00N)中の2,5-ジメチル-1-フェニルピロール-3-カルボン酸(D2、2.73g、12.7mmol)の溶液を一晩撹拌した。溶液を真空下で濃縮した。混合物を10.0%NaHCO3水溶液(100mL)でクエンチして、ジクロロメタン(100mL×3)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して、エチル2,5-ジメチル-1-フェニルピロール-3-カルボキシラート(D3)を黄色の油状物として得た(1.70g、55.2%)。
アセトニトリル(30.0mL)中の2,5-ジメチル-1-フェニルピロール-3-カルボン酸エチル(D3、1.70g、6.99mmol)の溶液に、0℃でゆっくりと塩化スルホニル酸(8.10g、69.8mmol)を添加した。溶液を2時間撹拌し、次いで水酸化アンモニウム(10.0mL)を添加した。溶液を1時間撹拌し、酢酸エチル(100mL×2)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して粗生成物を得て、これを分取HPLCで精製して、エチル2,5-ジメチル-1-フェニル-4-スルファモイルピロール-3-カルボキシラート(D4)を黄色油状物として得た(300mg、13.3%)。
MeOH(5.00mL)中のエチル2,5-ジメチル-1-フェニル-4-スルファモイルピロール-3-カルボキシラート(D4,300mg、0.932mmol)、NaOH(44.7mg、1.18mmol)および水(3.00mL)を16時間撹拌した。真空下でMeOHを除去した。残渣を酢酸エチル(10.0mL)および水(10.0mL)に加えた。混合物をHOAcでpH = 3~4に調整した。溶液を酢酸エチル(10.0mL×3)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して化合物Dを黄色固体として得た(20.5mg、7.48%)。
1H-NMR (400 MHz, DMSO_d6): δ 1.98-2.00 (m, 1 H), 2.14 (s, 6 H), 7.08 (s, 1 H), 7.33-7.36 (m, 2 H), 7.58-7.61 (m, 3 H). MS Calcd.: 294; MS Found: 295.1([M+1]+)。
1H-NMR (400 MHz, DMSO_d6): δ 1.98-2.00 (m, 1 H), 2.14 (s, 6 H), 7.08 (s, 1 H), 7.33-7.36 (m, 2 H), 7.58-7.61 (m, 3 H). MS Calcd.: 294; MS Found: 295.1([M+1]+)。
合成例2.化合物Eの合成

上記反応式に従って、合成例1と同様にして化合物Eを合成した。化合物Eを黄色固体として得た(収量20.0 mg, 収率18.7 %)。
1H-NMR (400 MHz, CD3OD): δ 2.35 (d, J= 1.6 Hz, 2 H), 2.48 (d, J= 8.0 Hz, 6 H), 3.46 (s, 3 H). MS Calcd.: 232; MS Found: 233.1([M+1]+)。
1H-NMR (400 MHz, CD3OD): δ 2.35 (d, J= 1.6 Hz, 2 H), 2.48 (d, J= 8.0 Hz, 6 H), 3.46 (s, 3 H). MS Calcd.: 232; MS Found: 233.1([M+1]+)。
合成例3.化合物Fの合成

上記反応式に従って、化合物Fを合成した。具体的には以下のようにして合成した。
フラン(F1、16.0g、0.235mol)およびTMEDA(60.0g、0.517mol)の溶液を0℃で冷却し、N-ブチルリチウム(ヘキサン中2.50M、207mL、0.517mol)を滴下した。溶液を室温に戻し、1時間攪拌した。テトラヒドロフラン(150mL)中のブロモエタン(76.8g、0.705mol)を滴下した。反応物を室温で一晩撹拌し、飽和塩化アンモニウム(300mL)でクエンチし、メチルtert-ブチルエーテル(500mL×2)で抽出した。有機層を1N HCl水溶液(100mL×3)で洗浄し、無水Na2SO4で乾燥させ、減圧下で濃縮して、2,5-ジエチルフラン(F2)を黄色の油状物として得た(18.9g、64.9%)。
乾燥トルエン(200mL)中の2,5-ジエチルフラン(F2、17.0g、0.137mol)、ジメチルアルミニウムクロリド(152mL、0.9M、0.137mol)およびCO2(20.0g、0.454mol)の溶液を一晩撹拌した。混合物を水(250mL)でクエンチし、ジクロロメタン(300mL×3)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して、赤褐色の固体として2,5-ジエチルフラン-3-カルボン酸(F3)を得た(3.6g、15.7%)。
HCl/EtOH(36.0mL、2.00N)中の2,5-ジエチルフラン-3-カルボン酸(F3、3.6g、0.021mol)の溶液を一晩撹拌した。溶液を真空下で濃縮した。混合物を10.0%NaHCO3水溶液(100mL)でクエンチし、ジクロロメタン(100mL×2)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して、エチル2,5-ジエチルフラン-3-カルボキシラート(F4)を得た(2.67g、63.6%)。
アセトニトリル(30.0mL)中のエチル2,5-ジエチルフラン-3-カルボキシレート(F4、2.50g、0.013mol)の溶液に、0℃でゆっくりとスルホクロリド酸(15.0g、0.130mol)を加えた。溶液を2時間撹拌し、次いで水酸化アンモニウム(50.0mL)を添加した。溶液を1時間撹拌した。溶液を酢酸エチル(100mL×2)で抽出した。有機層を無水Na2SO4で乾燥し、減圧下で濃縮して粗生成物を得、これをカラムで精製して、黄色固体として2,5-ジエチル-4-スルファモイルフラン-3-カルボン酸エチル(F5)を得た(880mg、25.1%)。
MeOH(10.0mL)中のエチル2,5-ジエチル-4-スルファモイルフラン-3-カルボキシレート(F5、880mg、3.20mmol)、NaOH(640mg、16.0mmol)および水(4.00mL)の溶液を、16時間撹拌した。真空下でMeOHを除去した。残渣に酢酸エチル(50.0mL)および水(20.0mL)を添加した。混合物を水性HCl(1N)でpH = 3~4に調整した。溶液を酢酸エチル(40.0mL×3)で抽出した。有機層を無水Na2SO4で乾燥させ、減圧下で濃縮して化合物Fを黄色固体として得た(120mg、15.2%)。
1H-NMR (400 MHz, CDCl3): δ 5.66 (s, 2 H), 3.00-3.04 (m, 4 H), 1.24-1.30 (m, 6 H). MS Calcd.: 247; MS Found: 248.1([M+1]+)。
1H-NMR (400 MHz, CDCl3): δ 5.66 (s, 2 H), 3.00-3.04 (m, 4 H), 1.24-1.30 (m, 6 H). MS Calcd.: 247; MS Found: 248.1([M+1]+)。
合成例4.化合物Gの合成
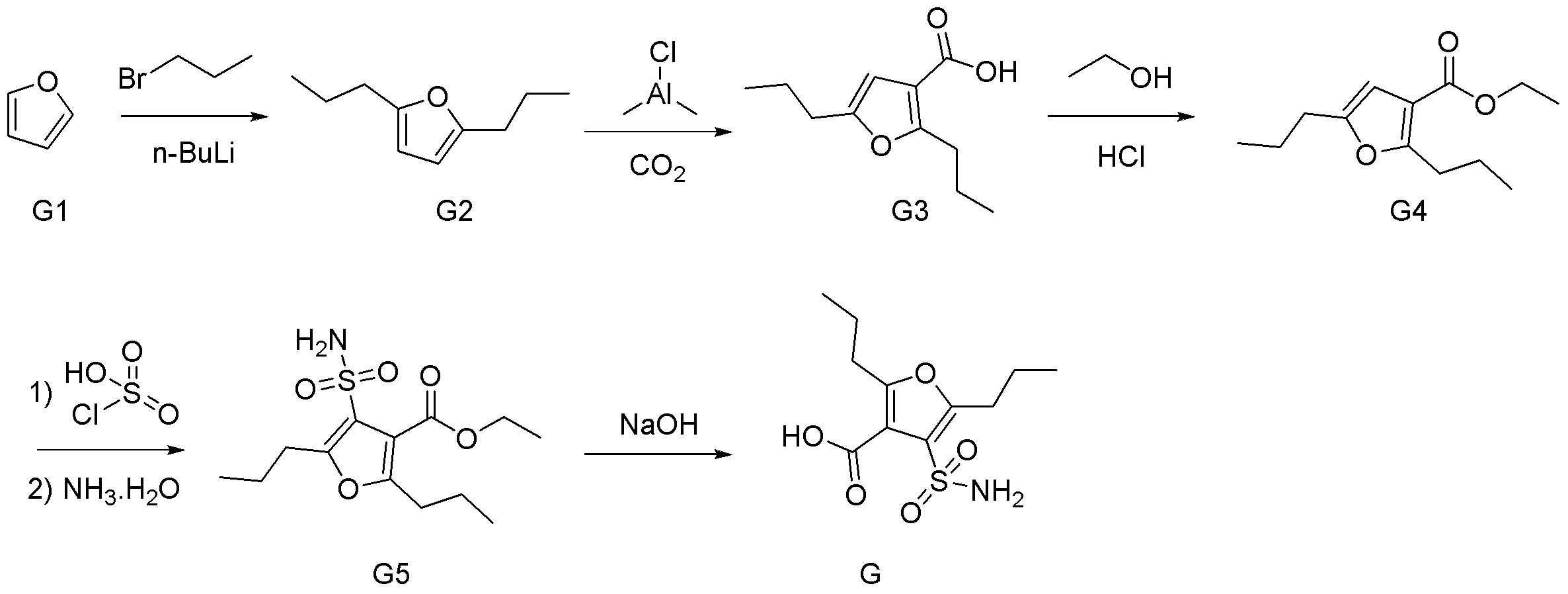
上記反応式に従って、合成例3と同様にして化合物Gを合成した。化合物Gを黄色固体として得た(収量0.130 g, 収率13.0 %)。
1H-NMR (400 MHz, CD3OD): δ 2.92-2.99 (m, 4 H), 1.66-1.76 (m, 4 H), 0.94-0.99 (m, 6 H). MS Calcd.: 275; MS Found: 276.0([M+1]+)。
1H-NMR (400 MHz, CD3OD): δ 2.92-2.99 (m, 4 H), 1.66-1.76 (m, 4 H), 0.94-0.99 (m, 6 H). MS Calcd.: 275; MS Found: 276.0([M+1]+)。
合成例5.化合物Hの合成
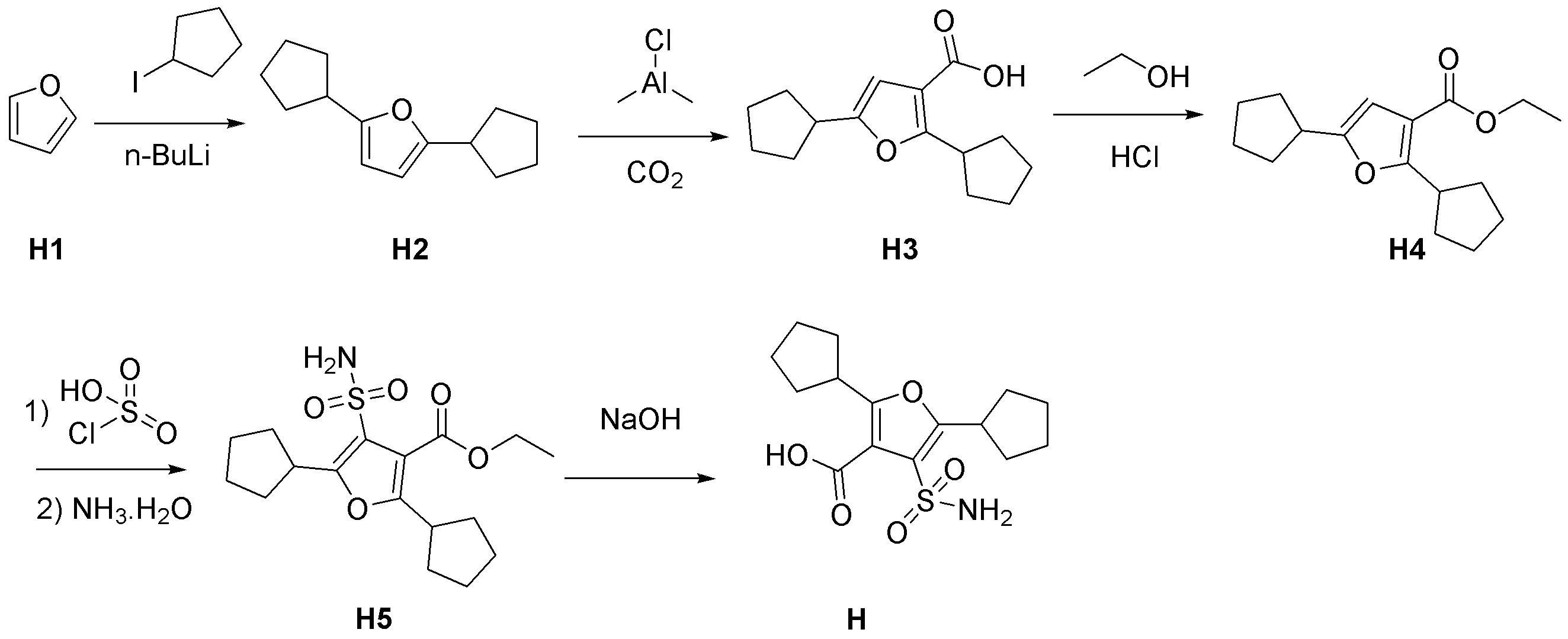
上記反応式に従って、合成例3と同様にして化合物Hを合成した。化合物Hを黄色固体として得た(収量0.135 mg, 収率18.3%)。
1H-NMR (400 MHz, CDCl3): δ 1.45-2.12 (m, 16 H), 3.83-3.95 (m, 2 H). MS Calcd: 327; MS Found: 326.0([M-1]-)。
1H-NMR (400 MHz, CDCl3): δ 1.45-2.12 (m, 16 H), 3.83-3.95 (m, 2 H). MS Calcd: 327; MS Found: 326.0([M-1]-)。
合成例6.化合物Iの合成
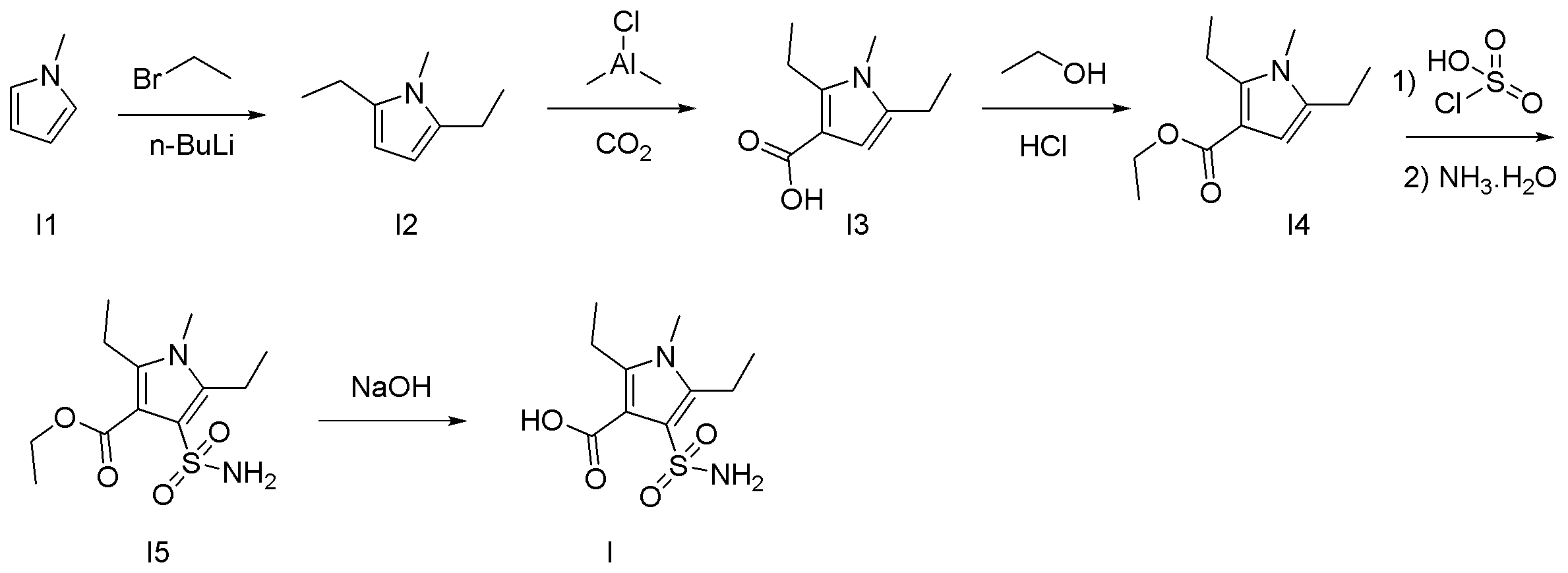
上記反応式に従って、合成例1と同様にして化合物Iを合成した。化合物Iを黄色固体として得た(収量110 mg, 収率11.1%)。
1H-NMR (400 MHz, CD3OD): δ 3.45 (s, 3 H), 2.88-2.94 (m, 4 H), 1.04-1.11 (m, 6 H). MS Calcd.: 260; MS Found: 261.1([M+1]+)。
1H-NMR (400 MHz, CD3OD): δ 3.45 (s, 3 H), 2.88-2.94 (m, 4 H), 1.04-1.11 (m, 6 H). MS Calcd.: 260; MS Found: 261.1([M+1]+)。
2.化合物の解析1
各化合物について、活性、複合体構造、毒性等を解析した(試験例1~7)。
各化合物について、活性、複合体構造、毒性等を解析した(試験例1~7)。
試験例1.薬剤感受性試験1
96穴あるいは384穴U底プレート、ミューラーヒントン液体培地、抗菌薬(セフタジジム[CAZ]、イミペネム[IPM]、メロペネム[MPM]、ドリペネム[DPM]、ビアペネム[BPM]を用いて、薬剤感受性試験測定用プレートを作製した。96穴等U底プレートA系列左端のウェル1に抗菌薬が最大濃度含まれるように調整し、右隣のウェル2は抗菌薬濃度が1/2に、さらにその右隣のウェル3は抗菌薬濃度1/4となるよう、1/2抗菌薬希釈系列をウェル11まで作製した。ウェル12は抗菌薬を含まないブランクウェルとした。B及びC系列にもA系列同様に抗菌薬の希釈系列を作製した。さらに、B及びC系列には、それぞれ被験物質(化合物A)を10 μg/mL、50 μg/mL添加した(図1)。
96穴あるいは384穴U底プレート、ミューラーヒントン液体培地、抗菌薬(セフタジジム[CAZ]、イミペネム[IPM]、メロペネム[MPM]、ドリペネム[DPM]、ビアペネム[BPM]を用いて、薬剤感受性試験測定用プレートを作製した。96穴等U底プレートA系列左端のウェル1に抗菌薬が最大濃度含まれるように調整し、右隣のウェル2は抗菌薬濃度が1/2に、さらにその右隣のウェル3は抗菌薬濃度1/4となるよう、1/2抗菌薬希釈系列をウェル11まで作製した。ウェル12は抗菌薬を含まないブランクウェルとした。B及びC系列にもA系列同様に抗菌薬の希釈系列を作製した。さらに、B及びC系列には、それぞれ被験物質(化合物A)を10 μg/mL、50 μg/mL添加した(図1)。
被検菌として、クラスB β-ラクタマーゼ(MBL(メタロ-β-ラクタマーゼ):IMP-1、NDM-1、又はVIM-2)をコードするプラスミドが導入された細菌(pBCSK (+) / E. coli DH5α)を用いた。ミューラーヒントン培地を用いて、被験菌を108 cfu/mLに調整した。さらに、ミューラーヒントン培地を用いて被験菌を10倍に希釈した(107 cfu/mL)。これを5 μl採取し、薬剤感受性試験測定用プレートに接種した。35℃で1晩培養し、目視にて被験菌の発育を確認した。被験菌の発育が確認されなかったウェルの中で、最も低い抗菌薬濃度を含むウェルを、最小発育阻止濃度(minimal inhibitory concentration)値とした(図1)。
結果を表1に示す。表1中、MBLsの列は、被検菌が産生するクラスB β-ラクタマーゼを示し、Empty vectorはクラスB β-ラクタマーゼを産生させていないことを示す。

表1に示されるように、化合物Aの添加により、β-ラクタマーゼ産生菌に対する抗菌薬のMICが低下することが分かった。
試験例2.β-ラクタマーゼ阻害活性測定試験
各種クラスB β-ラクタマーゼ(IMP-1、NDM-1、又はVIM-2:最終濃度 10 nM)、イミペネム(最終濃度 100μM)、被験物質(化合物A、化合物Z=下記に示す:最終濃度 0、1、10、100μM)を HEPES バッファー中で混和した。吸光度298 nm、30℃条件下におけるイミペネム分解活性(Δabs/min)を測定した。被験物質未添加時のイミペネム分解活性を100%とし、各々被験物質添加時のイミペネム残存分解活性をプロットした。
各種クラスB β-ラクタマーゼ(IMP-1、NDM-1、又はVIM-2:最終濃度 10 nM)、イミペネム(最終濃度 100μM)、被験物質(化合物A、化合物Z=下記に示す:最終濃度 0、1、10、100μM)を HEPES バッファー中で混和した。吸光度298 nm、30℃条件下におけるイミペネム分解活性(Δabs/min)を測定した。被験物質未添加時のイミペネム分解活性を100%とし、各々被験物質添加時のイミペネム残存分解活性をプロットした。
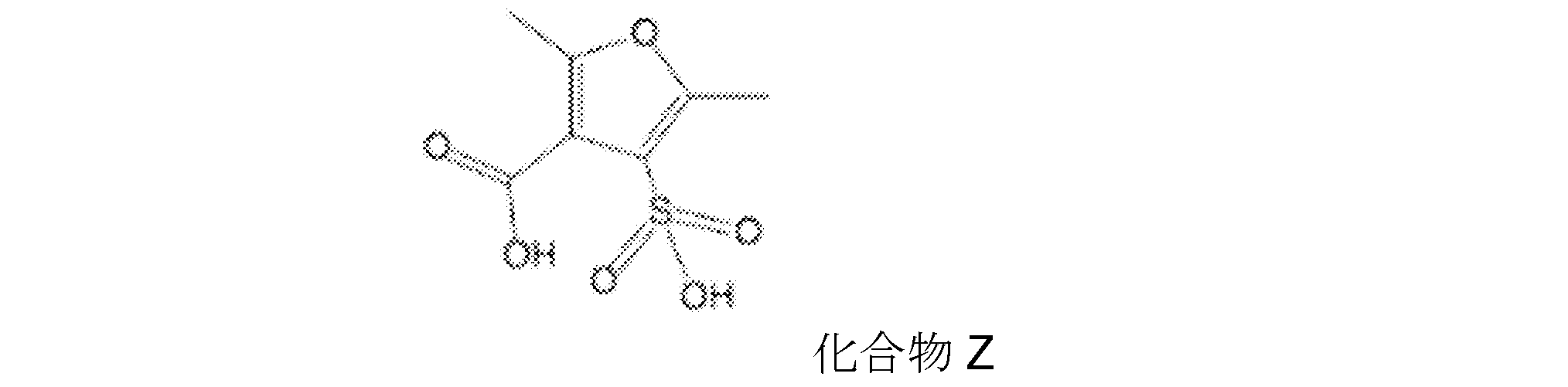
結果を図2に示す。図2に示されるように、化合物Aはβ-ラクタマーゼを阻害することが分かった。
試験例3.薬剤感受性試験2
被検菌として、クラスB β-ラクタマーゼを産生する腸内細菌科細菌の各種株を使用する以外は、試験例1と同様にして行った。
被検菌として、クラスB β-ラクタマーゼを産生する腸内細菌科細菌の各種株を使用する以外は、試験例1と同様にして行った。
結果を図3に示す。図3に示されるように、化合物Aの添加により、β-ラクタマーゼ産生腸内細菌科細菌に対する抗菌薬のMICが低下することが分かった。
試験例4.薬剤感受性試験3
被検菌として、クラスB β-ラクタマーゼを産生するアシネトバクターの各種株を使用する以外は、試験例1と同様にして行った。
被検菌として、クラスB β-ラクタマーゼを産生するアシネトバクターの各種株を使用する以外は、試験例1と同様にして行った。
結果を図4に示す。図4に示されるように、化合物Aの添加により、β-ラクタマーゼ産生アシネトバクターに対する抗菌薬のMICが低下することが分かった。
試験例5.X線結晶構造解析
<IMP-1と被験物質との複合体構造解析>
IMP-1を産生する大腸菌[pET9a-ΔIMP-1/E. coli BL21 (DE3)]を液体培養し、遠心操作より菌体を回収した。菌体を超音波にて破砕後、超遠心操作により可溶性画分と不溶性画分を分離した。可溶性画分を陽イオン交換カラム、疎水相互作用カラム、ゲル濾過カラムの順に通し、IMP-1を精製した。限外濾過カラムを使用し、精製したIMP-1をHEPES バッファーに置換の後、濃縮した。精製したIMP-1 (15 mg/mL)とリザーバー溶液[100 mM HEPES (pH 7.5)、200 mM Sodium acetate、25% PEG3350]を混和し、20℃でインキュベーションすることにより、IMP-1の結晶を得た。IMP-1結晶を、被験物質(化合物A)を溶解した上記リザーバー溶液に浸潤することで、IMP-1と被験物質との複合体結晶を作製した。複合体結晶をシンクロトロンに持ち込み、X線放射光を照射し回折データの回収を行った。公開されているIMP-1の構造情報を使用し、分子置換法にて位相を決定した。精密化にはRefmac5を、モデル構築にはcootを使用した。
<IMP-1と被験物質との複合体構造解析>
IMP-1を産生する大腸菌[pET9a-ΔIMP-1/E. coli BL21 (DE3)]を液体培養し、遠心操作より菌体を回収した。菌体を超音波にて破砕後、超遠心操作により可溶性画分と不溶性画分を分離した。可溶性画分を陽イオン交換カラム、疎水相互作用カラム、ゲル濾過カラムの順に通し、IMP-1を精製した。限外濾過カラムを使用し、精製したIMP-1をHEPES バッファーに置換の後、濃縮した。精製したIMP-1 (15 mg/mL)とリザーバー溶液[100 mM HEPES (pH 7.5)、200 mM Sodium acetate、25% PEG3350]を混和し、20℃でインキュベーションすることにより、IMP-1の結晶を得た。IMP-1結晶を、被験物質(化合物A)を溶解した上記リザーバー溶液に浸潤することで、IMP-1と被験物質との複合体結晶を作製した。複合体結晶をシンクロトロンに持ち込み、X線放射光を照射し回折データの回収を行った。公開されているIMP-1の構造情報を使用し、分子置換法にて位相を決定した。精密化にはRefmac5を、モデル構築にはcootを使用した。
<VIM-2と被験物質との複合体構造解析>
VIM-2を産生する大腸菌[pET29a-VIM-2/E. coli BL21 (DE3)]を液体培養し、遠心操作より菌体を回収した。菌体を超音波にて破砕後、超遠心操作により可溶性画分と不溶性画分を分離した。可溶性画分を陰イオン交換カラム、疎水相互作用カラム、ゲル濾過カラムの順に通し、VIM-2を精製した。限外濾過カラムを使用し、精製したVIM-2をTris-HCl バッファーに置換の後、濃縮した。精製したVIM-2 (15 mg/mL)とリザーバー溶液[200 mM Magnesium Formate、25% PEG3350]を混和し、20℃でインキュベーションすることにより、VIM-2の結晶を得た。VIM-2結晶を、被験物質(化合物A)を溶解した上記リザーバー溶液に浸潤することで、VIM-2と被験物質との複合体結晶を作製した。複合体結晶をシンクロトロンに持ち込み、X線回折データの回収を行った。公開されているVIM-2の構造情報を使用し、分子置換法にて位相を決定した。精密化にはRefmac5を、モデル構築にはcootを使用した。
VIM-2を産生する大腸菌[pET29a-VIM-2/E. coli BL21 (DE3)]を液体培養し、遠心操作より菌体を回収した。菌体を超音波にて破砕後、超遠心操作により可溶性画分と不溶性画分を分離した。可溶性画分を陰イオン交換カラム、疎水相互作用カラム、ゲル濾過カラムの順に通し、VIM-2を精製した。限外濾過カラムを使用し、精製したVIM-2をTris-HCl バッファーに置換の後、濃縮した。精製したVIM-2 (15 mg/mL)とリザーバー溶液[200 mM Magnesium Formate、25% PEG3350]を混和し、20℃でインキュベーションすることにより、VIM-2の結晶を得た。VIM-2結晶を、被験物質(化合物A)を溶解した上記リザーバー溶液に浸潤することで、VIM-2と被験物質との複合体結晶を作製した。複合体結晶をシンクロトロンに持ち込み、X線回折データの回収を行った。公開されているVIM-2の構造情報を使用し、分子置換法にて位相を決定した。精密化にはRefmac5を、モデル構築にはcootを使用した。
<NDM-1と被験物質との複合体構造解析>
NDM-1を産生する大腸菌[pET28a-NDM-1/E. coli BL21 (DE3)]を液体培養し、遠心操作より菌体を回収した。菌体を超音波にて破砕後、超遠心操作により可溶性画分と不溶性画分を分離した。可溶性画分をニッケルカラム、陰イオン交換カラム、ゲル濾過カラムの順に通し、NDM-1を精製した。限外濾過カラムを使用し、精製したNDM-1をTris-HClバッファーに置換の後、濃縮した。精製したNDM-1 (40 mg/mL)とリザーバー溶液[100 mM Bis-Tris (pH 6.1)、200 mM Ammonium Sulfate、25% PEG3350]を混和し、20℃でインキュベーションすることにより、NDM-1の結晶を得た。NDM-1結晶を、被験物質(化合物A、又は化合物I)を溶解した上記リザーバー溶液に浸潤することで、NDM-1と被験物質との複合体結晶を作製した。複合体結晶をシンクロトロンに持ち込み、X線回折データの回収を行った。公開されているNDM-1の構造情報を使用し、分子置換法にて位相を決定した。精密化にはRefmac5を、モデル構築にはcootを使用した。
NDM-1を産生する大腸菌[pET28a-NDM-1/E. coli BL21 (DE3)]を液体培養し、遠心操作より菌体を回収した。菌体を超音波にて破砕後、超遠心操作により可溶性画分と不溶性画分を分離した。可溶性画分をニッケルカラム、陰イオン交換カラム、ゲル濾過カラムの順に通し、NDM-1を精製した。限外濾過カラムを使用し、精製したNDM-1をTris-HClバッファーに置換の後、濃縮した。精製したNDM-1 (40 mg/mL)とリザーバー溶液[100 mM Bis-Tris (pH 6.1)、200 mM Ammonium Sulfate、25% PEG3350]を混和し、20℃でインキュベーションすることにより、NDM-1の結晶を得た。NDM-1結晶を、被験物質(化合物A、又は化合物I)を溶解した上記リザーバー溶液に浸潤することで、NDM-1と被験物質との複合体結晶を作製した。複合体結晶をシンクロトロンに持ち込み、X線回折データの回収を行った。公開されているNDM-1の構造情報を使用し、分子置換法にて位相を決定した。精密化にはRefmac5を、モデル構築にはcootを使用した。
<結果>
結果を図5~8に示す。図5~8に示されるように、被検物質は、IPM-1、VIM-2、NDM-1と、そのスルファモイル基及びカルボキシ基を介して相互作用することが分かった。一方、これらの基が連結されている側と反対側(一般式(1)のR1、R2及びR3側)と、IPM-1、VIM-2、及びNDM-1との間には一定の空間があり、この反対側の構造は自由度が高いと考えられた。
結果を図5~8に示す。図5~8に示されるように、被検物質は、IPM-1、VIM-2、NDM-1と、そのスルファモイル基及びカルボキシ基を介して相互作用することが分かった。一方、これらの基が連結されている側と反対側(一般式(1)のR1、R2及びR3側)と、IPM-1、VIM-2、及びNDM-1との間には一定の空間があり、この反対側の構造は自由度が高いと考えられた。
試験例6.薬剤感受性試験4及び阻害定数(Ki)測定試験1
薬剤感受性試験は、被検物質として化合物A~C及びE~Iを用いる以外は、試験例1と同様にして行った。
薬剤感受性試験は、被検物質として化合物A~C及びE~Iを用いる以外は、試験例1と同様にして行った。
阻害定数(Ki)測定試験は次のようにして行った。各種メタロ-β-ラクタマーゼ(最終濃度 10 nM)を用い、阻害定数(Ki)を測定した。基質であるイミペネムの濃度、阻害剤となる被験物質(化合物A~E)の濃度をそれぞれ変化させ、各ポイントにおける吸光度298 nm、30℃条件下のイミペネム分解活性(Δabs/min)を測定した。測定値をラインウェーバー・バークプロットの式にフィットし、Kiを算出した。
結果を表2に示す。表2中、最左列は、被験物質を示し、Noneは被検物質を添加しない場合を示す。

表2に示されるように、化合物A~C及びE~Iの添加により、β-ラクタマーゼ産生菌に対する抗菌薬のMICが低下すること、及びβ-ラクタマーゼ活性が阻害されることが分かった。
試験例7.毒性評価試験
HeLa細胞に各種濃度の被験物質(化合物A)又はスタウロスポリンを添加し、MTTアッセイキット(Promega cat no. G4000)を用いて生細胞をカウントした。
HeLa細胞に各種濃度の被験物質(化合物A)又はスタウロスポリンを添加し、MTTアッセイキット(Promega cat no. G4000)を用いて生細胞をカウントした。
結果を図9に示す。図9に示されるように、化合物Aの毒性は極めて低いことが分かった。
試験例8.復帰突然変異試験(Ames試験)
被検物質(化合物A)を被検物質として、Ames試験をUBE科学分析センター社に委託して行った。その結果、化合物AはAmes試験陰性であることが分かった。
被検物質(化合物A)を被検物質として、Ames試験をUBE科学分析センター社に委託して行った。その結果、化合物AはAmes試験陰性であることが分かった。
3.化合物の準備2
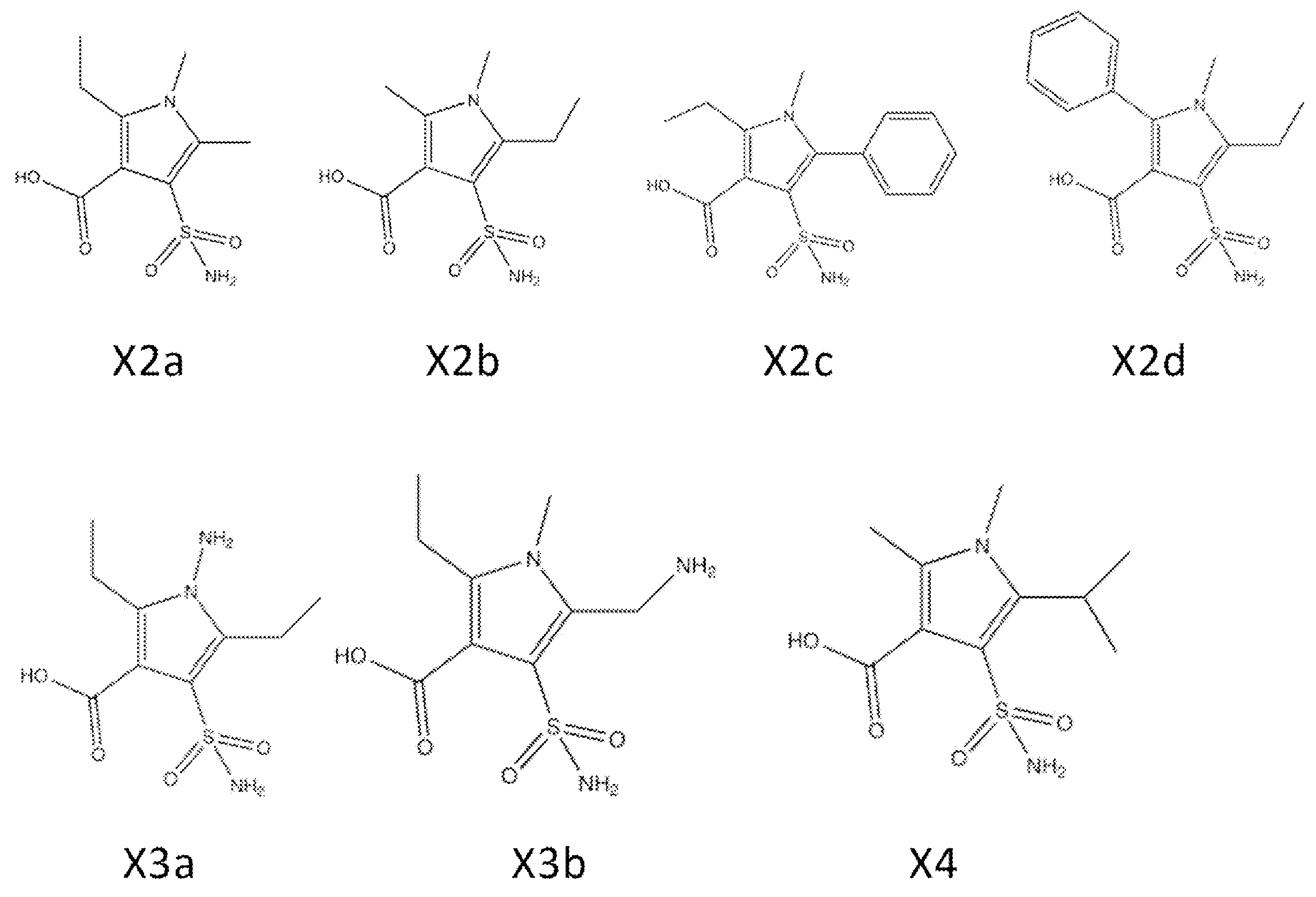

化合物X2a、X2b、X2c、X2d、X3a、及びX4を以下の合成例7~8に従って合成した。化合物X3bは以下の合成例9に従って合成することができる。化合物No.9-7、No.9-8、及びNo.9-9はナミキ商事株式会社から購入した。
合成例7.化合物X2a、X2b、X2c、X2d、及びX4の合成
下記の合成ルートにより目的物8(化合物X2a、X2b、X2c、X2d、及びX4)を合成した。
下記の合成ルートにより目的物8(化合物X2a、X2b、X2c、X2d、及びX4)を合成した。

目的物8、及び原料(ジケトエステル3等)の具体的な構造を以下に示す。番号は、上記合成ルートに対応する。また、番号の後に示される2文字のアルファベットが同じ化合物は、同一の合成ルート上の化合物であることを示す。すなわち、化合物X2a(ピロール8ad)の合成ルート上の化合物は、番号の後に示される2文字のアルファベットが「ad」である化合物である。
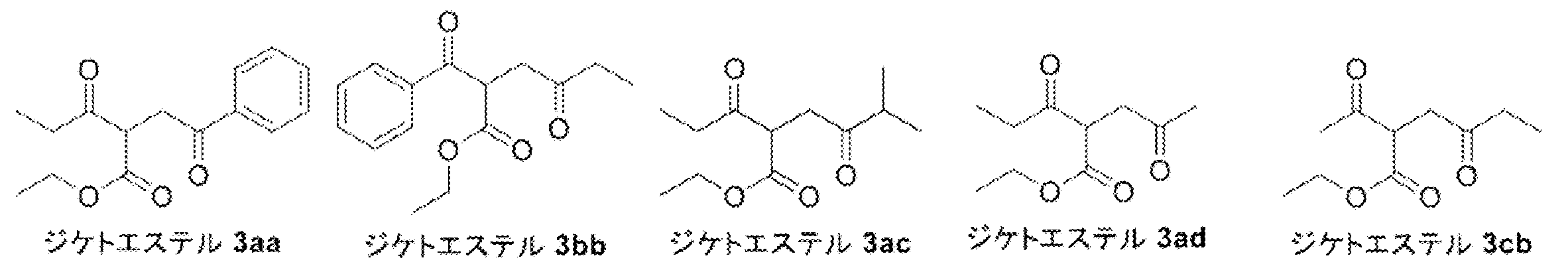
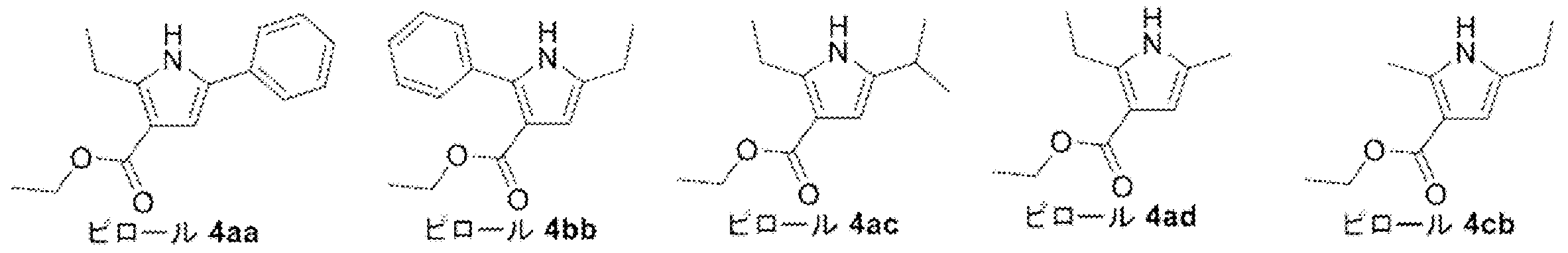

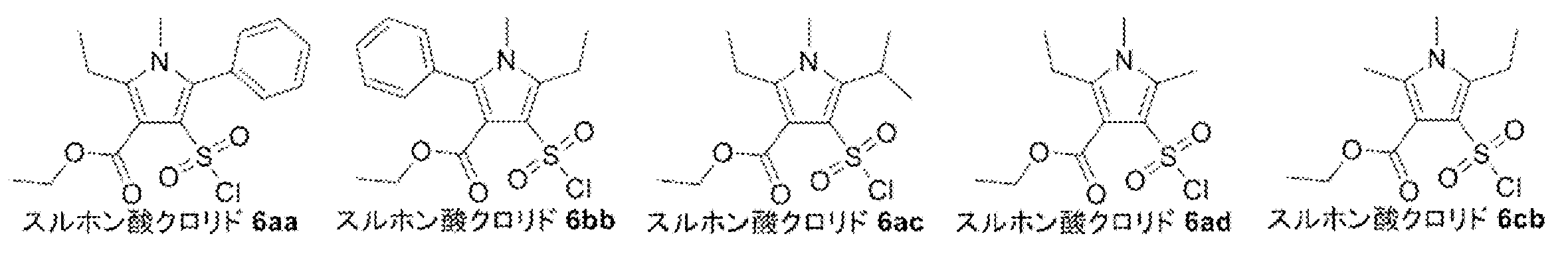
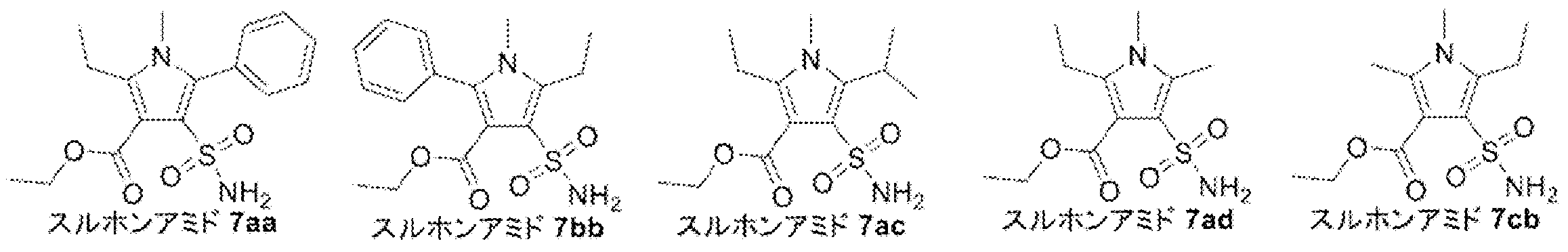
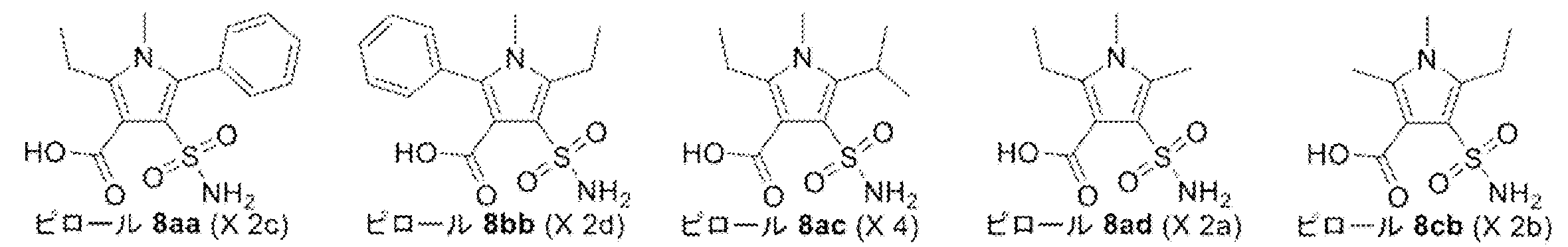
合成例7-1.化合物X2a(ピロール8ad)の合成
3-オキソ吉草酸エチル1a とブロモアセトン2d のカップリングを行い、ジケトエステル3ad を合成した。即ち、撹拌子を備えた50 mL 三口フラスコに、窒素雰囲気下で水素化ナトリウム(60% in 流動パラフィン, 960 mg, 24.00 mmol, 1.20 eq.)及びTHF(35 mL)を加え、氷冷した。次いで得られた灰色懸濁に、3-オキソ吉草酸エチル1a(2.85 mL, 20.00 mmol)をシリンジを用いてneat で滴下した。この時発泡を確認した。氷浴にてさらに30 分撹拌した後、ブロモアセトン2d(3.00 g, 22.00 mmol, 1.10 eq.)のTHF 溶液(15 mL)を素早く加えた。さらに1 時間後にTLC で分析(UV、リンモリブデン酸)したところ、原料1a の消失と1 つの新たなスポットの生成が確認された(展開溶媒:ヘキサン/酢酸エチル= 9/1、Rf 値:0.3)。そこで反応混合物に飽和塩化アンモニウム水溶液及び酢酸エチルを加えて反応を停止した。2 層を分離した後、水層を酢酸エチルで抽出し、合わせた有機層を飽和塩化ナトリウム水溶液で洗浄し、硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ橙色油状物質4.33 g が得られ、得られた粗生成物をシリカゲルカラムクロマトグラフィーにて精製した(計2 回、使用シリカゲル(1)(2)(各150 g)。溶出として、(1)ヘキサン/酢酸エチル= 9/1(2 L)→1/1(700 mL)、(2)ヘキサン/酢酸エチル= 4/1(2 L)を使用した。目的物のフラクションを濃縮した結果、目的のジケトエステル3ad を2.50 g(収率62%)、淡黄色液体で得た。
3-オキソ吉草酸エチル1a とブロモアセトン2d のカップリングを行い、ジケトエステル3ad を合成した。即ち、撹拌子を備えた50 mL 三口フラスコに、窒素雰囲気下で水素化ナトリウム(60% in 流動パラフィン, 960 mg, 24.00 mmol, 1.20 eq.)及びTHF(35 mL)を加え、氷冷した。次いで得られた灰色懸濁に、3-オキソ吉草酸エチル1a(2.85 mL, 20.00 mmol)をシリンジを用いてneat で滴下した。この時発泡を確認した。氷浴にてさらに30 分撹拌した後、ブロモアセトン2d(3.00 g, 22.00 mmol, 1.10 eq.)のTHF 溶液(15 mL)を素早く加えた。さらに1 時間後にTLC で分析(UV、リンモリブデン酸)したところ、原料1a の消失と1 つの新たなスポットの生成が確認された(展開溶媒:ヘキサン/酢酸エチル= 9/1、Rf 値:0.3)。そこで反応混合物に飽和塩化アンモニウム水溶液及び酢酸エチルを加えて反応を停止した。2 層を分離した後、水層を酢酸エチルで抽出し、合わせた有機層を飽和塩化ナトリウム水溶液で洗浄し、硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ橙色油状物質4.33 g が得られ、得られた粗生成物をシリカゲルカラムクロマトグラフィーにて精製した(計2 回、使用シリカゲル(1)(2)(各150 g)。溶出として、(1)ヘキサン/酢酸エチル= 9/1(2 L)→1/1(700 mL)、(2)ヘキサン/酢酸エチル= 4/1(2 L)を使用した。目的物のフラクションを濃縮した結果、目的のジケトエステル3ad を2.50 g(収率62%)、淡黄色液体で得た。
Paal-Knorr ピロール合成により、ジケトエステル3ad からピロール4ad を合成した。即ち、撹拌子を備えた100 mL 一口フラスコに、ジケトエステル3ad (2.50 g, 12.49 mmol)、酢酸アンモニウム(9.63 g, 124.9 mmol, 10.00 eq.)及び酢酸(40 mL)を加え、混合物を80 ℃にて撹拌した。3 時間後にTLC で分析(UV、リンモリブデン酸)したところ、原料3ad(展開溶媒:ヘキサン/酢酸エチル= 4/1、Rf 値:0.3)の消失及び原料の上に1 つの新たなスポット(Rf 値:0.4)の生成が確認された。そこで反応混合物を室温まで放冷し、減圧下濃縮した。次いで得られた褐色油状物質を酢酸エチル及び蒸留水で希釈し、2 層を分離した後、水層を酢酸エチルで抽出した。合わせた有機層を飽和塩化ナトリウム水溶液で洗浄し、硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別し、ろ液を減圧下濃縮したところ、目的のピロール4ad を2.11 g(収率93%)、褐色液体で得た。
ヨウ化メチルでピロール4ad をアルキル化し、N-メチルピロール5ad を合成した。即ち、撹拌子を備えた100 mL 一口フラスコに、ピロール4ad(2.11 g, 11.63 mmol)、ヨウ化メチル(3.6 mL, 57.83 mmol, 4.97 eq.)及びTHF (45 mL)を加え、氷冷した。次いで得られた褐色溶液に、水素化ナトリウム(60% in 流動パラフィン, 698 mg, 17.44 mmol, 1.50 eq.)を加えたところ、発泡を確認した。30 分後にTLC で分析(UV、リンモリブデン酸)したところ、原料4ad が残存していた。更に撹拌を続け、62 時間後にTLC 分析で消失を確認した。反応混合物に蒸留水を加えて反応を停止し、ジエチルエーテルで抽出した。合わせた有機層を飽和塩化ナトリウム水溶液で洗浄し、硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別し、ろ液を減圧下濃縮したところ、赤褐色油状物質2.80 g が得られ、得られた粗生成物をシリカゲルカラムクロマトグラフィー(50 g 使用、直径30 mm、高さ190 mm)にて精製した。溶出液として、ヘキサン/酢酸エチル= 9/1(650 mL)を使用した。目的物のフラクションを濃縮した結果、N-メチルピロール5ad を1.72 g(収率76%)、褐色液体で得た。
N-メチルピロール5ad のクロロスルホン化を行い、スルホン酸クロリド6ad を合成した。即ち、撹拌子を備え、十分に窒素置換した100 mL 一口フラスコに、クロロ硫酸(5.9 mL, 88.76 mmol, 10.05 eq.)を加えて氷浴で撹拌した(発煙注意)。そこにN-メチルピロール5ad(1.72 g, 8.83 mmol)を少しずつ加えた後、密閉して室温まで昇温した(褐色透明溶液)。1 時間30 分後、TLC で分析(UV、リンモリブデン酸)したところ、原料5ad(展開溶媒:ヘキサン/酢酸エチル=9/1、Rf 値:0.7)の消失と新たなスポット(Rf 値:0.35)の生成が確認された。氷冷した蒸留水に氷冷した反応混合物をゆっくり滴下したところ、淡褐色固体が析出した。生じた固体を塩化メチレンで溶解させ、2 層を分離後、水層を塩化メチレン抽出し、合わせた有機層に硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別し、ろ液を減圧下濃縮したところ、スルホン酸クロリド6ad を1.75 g(収率68%)、褐色固体で得た。
スルホン酸クロリド6ad をアンモニア/ジオキサン溶液と反応させ、スルホンアミド7ad を合成した。即ち、撹拌子を備えた100 mL 一口フラスコに、スルホン酸クロリド6ad (1.75g, 5.97 mmol)及び0.5 M アンモニア/ジオキサン溶液(36.00 mL, 18.00 mmol, 3.01 eq.)を加え、室温下にて撹拌した。26 時間30 分後、TLC で分析(UV、リンモリブデン酸)したところ、原料6ad(展開溶媒:ヘキサン/酢酸エチル=1/1、Rf 値:0.6)の消失と新たなスポット(Rf 値:0.3)の生成が確認された。減圧下濃縮し、得られた粗生成物を冷ヘキサン/酢酸エチル=1/1 でケーキ洗浄し、固体を回収して乾燥したところ、目的のスルホンアミド7ad を1.50 g(収率92%)、褐色固体で得た。
スルホンアミド7ad のエステルを塩基性条件で加水分解し、目的とするピロール化合物8ad(X 2a)を合成した。即ち、撹拌子を備えた100 mL 一口フラスコに、スルホンアミド7ad (Lot.4188163, 1.41 g, 5.13 mmol)及び2 M 水酸化カリウム/エタノール溶液(50.0 mL, 100.0 mmol, 19.48 eq.)を加え、80 ℃にて撹拌した。3 時間30 分後、TLC で分析(UV、リンモリブデン酸)したところ、原料7ad(展開溶媒:酢酸エチルのみ、Rf 値:0.75)の消失と新たなスポット(Rf 値:0.5)の生成が確認された。反応混合物を室温まで放冷し、酢酸(6 mL)加えて冷蔵庫に静置した。翌日析出した結晶を桐山ロートでろ過し、蒸留水(10 mL)でケーキ洗浄を行ったところ、固体が溶解したので再度減圧下濃縮し、析出した固体を蒸留水(10 mL)でケーキ洗浄を行った。残った固体を50 ℃にて減圧下乾燥したところ、褐色固体を687 mg 得た。得られた褐色固体をシリカゲルカラムクロマトグラフィー(シリカゲル157 g 使用)にて精製した。溶出液として、クロロホルム/メタノール= 9/1(2.1 L)⇒4/1(75 mL)⇒メタノールのみ(300 mL)を使用した。検討2.6.1 において目的物の極性が高く、収量が低かったため、ここではメタノール単独まで極性を上げた。それぞれのフラクションを濃縮した結果、低極性褐色固体を112 mg、高極性褐色固体を504 mg 得た。得られた固体の一部を重DMSO で希釈して1H 及び13C NMR を測定した結果、高極性褐色固体が目的のピロール8ad(X2a)であることを確認した(収率40%、HPLC 純度98.5%)。
1H-NMR (400 MHz, DMSO_d6): δ 1.06 (t, J = 6.0 Hz, 3H), 2.42 (s, 3H), 2.94 (q, J = 6.0Hz,2H), 3.42 (s, 3H), 3.5-9.0 (bs, 3H); 13C-NMR (100 MHz, DMSO-d6): 11.0, 14.3, 30.5, 48.0, 108.9, 121.4, 130.5, 140.4, 168.0; MS Calcd.: 245.06; MS Found: 245.03 ([M-1]-)。
1H-NMR (400 MHz, DMSO_d6): δ 1.06 (t, J = 6.0 Hz, 3H), 2.42 (s, 3H), 2.94 (q, J = 6.0Hz,2H), 3.42 (s, 3H), 3.5-9.0 (bs, 3H); 13C-NMR (100 MHz, DMSO-d6): 11.0, 14.3, 30.5, 48.0, 108.9, 121.4, 130.5, 140.4, 168.0; MS Calcd.: 245.06; MS Found: 245.03 ([M-1]-)。
合成例7-2.化合物X2bの合成
合成例7-1と同様にして化合物X2bを合成した。化合物X2bを淡褐色固体として得た(収率33%、HPLC 純度95.9%)。
1H-NMR (400 MHz, DMSO_d6): δ1.06 (t, J = 6.0 Hz, 3H), 2.41 (s, 3H), 2.92 (q, J = 6.0 Hz, 2H), 3.44 (s, 3H), 6.91 (bs, 2H, -SONH2); 13C-NMR (100 MHz, DMSO-d6): 11.4, 14.8, 17.3, 30.2, 109.8, 120.8, 135.1, 136.5, 165.4; MS Calcd.: 245.06; MS Found: 245.05 ([M-1]-)。
合成例7-1と同様にして化合物X2bを合成した。化合物X2bを淡褐色固体として得た(収率33%、HPLC 純度95.9%)。
1H-NMR (400 MHz, DMSO_d6): δ1.06 (t, J = 6.0 Hz, 3H), 2.41 (s, 3H), 2.92 (q, J = 6.0 Hz, 2H), 3.44 (s, 3H), 6.91 (bs, 2H, -SONH2); 13C-NMR (100 MHz, DMSO-d6): 11.4, 14.8, 17.3, 30.2, 109.8, 120.8, 135.1, 136.5, 165.4; MS Calcd.: 245.06; MS Found: 245.05 ([M-1]-)。
合成例7-3.化合物X2cの合成
合成例7-1と同様にして化合物X2cを合成した。化合物X2cを淡褐色固体として得た(収率13%、HPLC 純度96.0%)。
1H-NMR (400 MHz, DMSO-d6):δ 1.16 (t, J = 6.0 Hz, 3H), 2.95 (q, J = 6.0 Hz, 2H), 3.24(s, 3H), 6.84 (bs, 2H, -SONH2), 12.8 (bs, 1H, -COOH); 13C-NMR (100 MHz, DMSO-d6): 14.0, 18.1, 31.9, 108.6, 123.1, 128.1, 128.8, 131.3, 131,6, 135.1, 142.2, 166.6.; MS Calcd.: 307.08; MS Found: 307.09 ([M-1]-)。
合成例7-1と同様にして化合物X2cを合成した。化合物X2cを淡褐色固体として得た(収率13%、HPLC 純度96.0%)。
1H-NMR (400 MHz, DMSO-d6):δ 1.16 (t, J = 6.0 Hz, 3H), 2.95 (q, J = 6.0 Hz, 2H), 3.24(s, 3H), 6.84 (bs, 2H, -SONH2), 12.8 (bs, 1H, -COOH); 13C-NMR (100 MHz, DMSO-d6): 14.0, 18.1, 31.9, 108.6, 123.1, 128.1, 128.8, 131.3, 131,6, 135.1, 142.2, 166.6.; MS Calcd.: 307.08; MS Found: 307.09 ([M-1]-)。
合成例7-4.化合物X2dの合成
合成例7-1と同様にして化合物X2dを合成した。化合物X2dを淡黄色固体として得た(収率24%、HPLC 純度99.3%)。
1H-NMR (400 MHz, DMSO_d6):δ 1.14 (t, J = 6.0 Hz, 3H), 2.98 (q, J = 5.6 Hz, 2H), 3.27 (s, 3H), 7.10 (bs, 2H, -SONH2), 7.32 (m, 2H), 7.41 (m, 3H), 12.3 (bs, 1H, -COOH); 13C-NMR (100 MHz, DMSO-d6): 14.5, 31.8, 39.5, 121.3, 128.3, 128.4, 128.5, 131.1, 132.2, 136.7, 136.8, 160.0; MS Calcd.: 307.08; MS Found: 307.03 ([M-1]-)。
合成例7-1と同様にして化合物X2dを合成した。化合物X2dを淡黄色固体として得た(収率24%、HPLC 純度99.3%)。
1H-NMR (400 MHz, DMSO_d6):δ 1.14 (t, J = 6.0 Hz, 3H), 2.98 (q, J = 5.6 Hz, 2H), 3.27 (s, 3H), 7.10 (bs, 2H, -SONH2), 7.32 (m, 2H), 7.41 (m, 3H), 12.3 (bs, 1H, -COOH); 13C-NMR (100 MHz, DMSO-d6): 14.5, 31.8, 39.5, 121.3, 128.3, 128.4, 128.5, 131.1, 132.2, 136.7, 136.8, 160.0; MS Calcd.: 307.08; MS Found: 307.03 ([M-1]-)。
合成例7-5.化合物X4の合成
合成例7-1と同様にして化合物X4を合成した。化合物X4を淡黄色固体として得た(収率24%、HPLC 純度87.5%)。
1H-NMR (400 MHz, DMSO_d6):δ1.06 (t, J = 6.0 Hz, 3H), 1.27 (d, J = 6.0 Hz, 6H), 2,83 (q, J = 6.0 Hz, 2H), 2.90 (m, 1H), 3.60 (s, 3H), 4.27 (bs, 1H, -COOH), 6.95 (bs, 2H, -SONH2); MS Calcd.: 273.09; MS Found: 273.06 ([M-1]-)。
合成例7-1と同様にして化合物X4を合成した。化合物X4を淡黄色固体として得た(収率24%、HPLC 純度87.5%)。
1H-NMR (400 MHz, DMSO_d6):δ1.06 (t, J = 6.0 Hz, 3H), 1.27 (d, J = 6.0 Hz, 6H), 2,83 (q, J = 6.0 Hz, 2H), 2.90 (m, 1H), 3.60 (s, 3H), 4.27 (bs, 1H, -COOH), 6.95 (bs, 2H, -SONH2); MS Calcd.: 273.09; MS Found: 273.06 ([M-1]-)。
合成例8.化合物X3aの合成
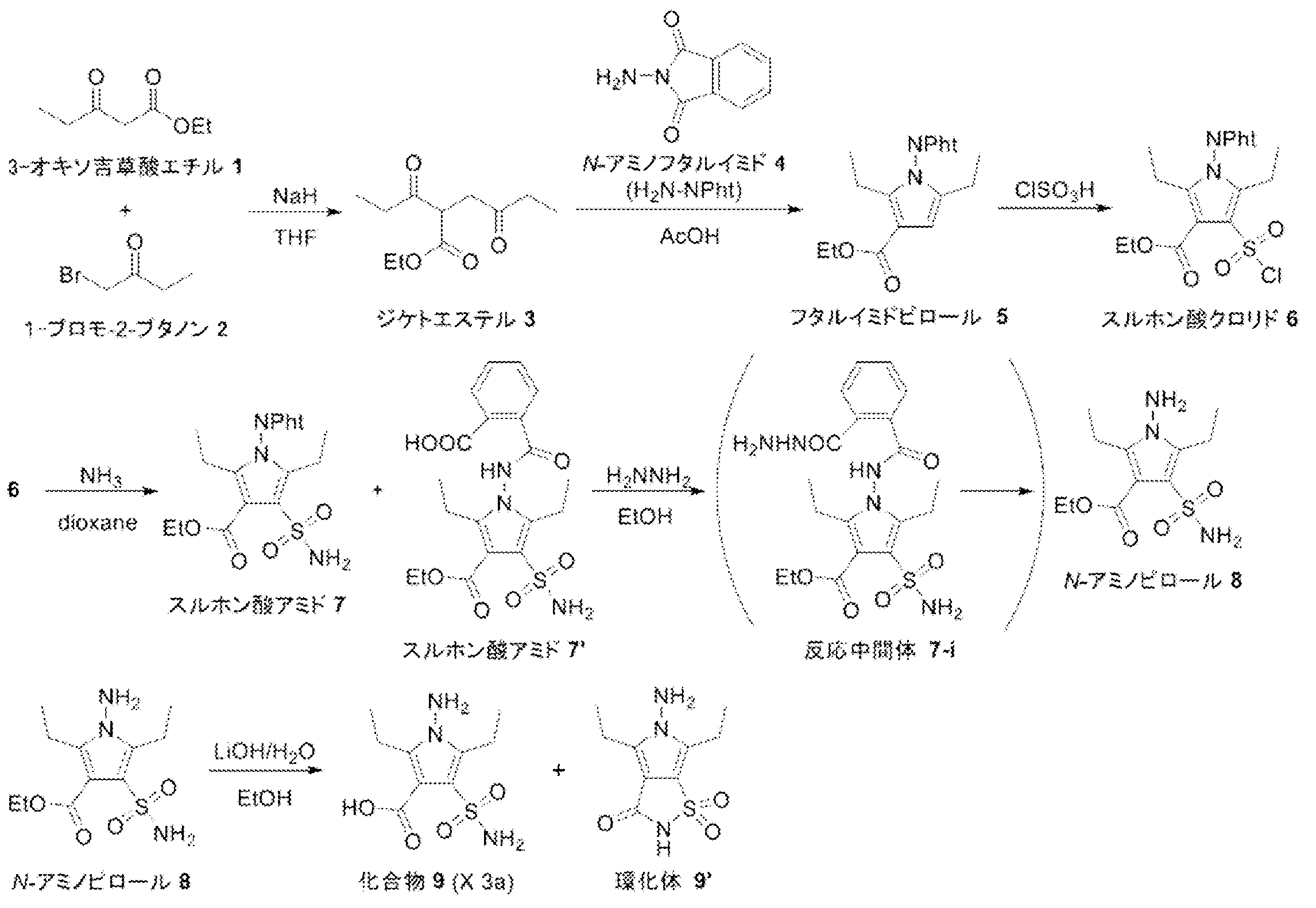
上記反応式に従って、化合物X3aを合成した。具体的には以下のようにして合成した。
3-オキソ吉草酸エチル1と1-ブロモ-2-ブタノン2のカップリングを行い、ジケトエステル3を合成した。即ち、温度計及び撹拌子を備えた30 mL二口ナスフラスコに、窒素雰囲気下で水素化ナトリウム(60% in 流動パラフィン, 139 mg, 3.48 mmol, 1.20 eq.)及びTHF(5.5 mL)を加え、氷冷した。次いで得られた灰色懸濁液に3-オキソ吉草酸エチル1(418 mg, 2.90 mmol)を滴下した。この時発泡及び発熱を確認(0.7 ℃→12.5 ℃)。氷浴にて30分撹拌することで透明溶液となった反応液に、1-ブロモ-2-ブタノン2(482 mg, 3.48 mmol, 1.10 eq.)のTHF溶液(2.0 mL)を滴下し、氷浴にて撹拌を続けた。反応液は透明溶液から白濁液となった。滴下2時間後にTLCで分析(UV、リンモリブデン酸)したところ、3-オキソ吉草酸エチル1の残存と新たなスポットが確認された(展開溶媒:ヘキサン/酢酸エチル=9/1、Rf値:0.3)。反応液を室温まで昇温し、さらに2.5時間撹拌した。同様にTLCで分析したが、3-オキソ吉草酸エチル1は室温昇温前と比べ、スポットの濃さから、反応は進行していると思われるが消失は確認されなかった。反応液を氷冷後、飽和塩化アンモニウム水溶液(20 mL)及び酢酸エチル(20 mL)を加え反応を停止した(発熱あり:0.8 ℃→15.7 ℃)。2層を分離した後、水層を酢酸エチル(30 mL)で2回抽出し、合わせた有機層を飽和塩化ナトリウム水溶液(40 mL)で洗浄し、硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ黄色油状物質894 mgが得られ、得られた粗生成物を山善製中圧分取液体クロマトグラフ(シリカゲル)にて精製した。溶出液として溶出液として酢酸エチル/ヘキサン0%(3分、グラジエント10分)⇒10%(27分)を使用した。目的物のフラクションを濃縮し、40 ℃で1時間真空乾燥することで、原料1が22.3 mg(3.6wt%)混在したジケトエステル3を624 mg、無色透明液体で得た(混合物中のジケトエステル3の重量602 mg、収率97%)。
N-アミノフタルイミド4を用いてPaal-Knorrピロール合成を行い、フタルイミドピロール5を合成した。即ち、撹拌子とジムロート冷却器を備えた10 mLナスフラスコに、窒素雰囲気下でジケトエステル3(Lot.5187301, 602 mg, 2.81 mmol)及びN-アミノフタルイミド4(455 mg, 2.81 mmol, 1.00 eq.)を加え、さらに酢酸(3.6 mL)を加えて室温で撹拌した(N-アミノフタルイミド4不溶)。オイルバス温度を130 ℃前後に設定し、反応液を還流させた(バス温度80 ℃程度でN-アミノフタルイミド4完溶)。1時間後、TLCで分析(UV、リンモリブデン酸)したところ、ジケトエステル3の残存と新たなスポットが確認された(展開溶媒:ヘキサン/酢酸エチル=4/1、Rf値:0.3)。さらに3時間還流を行った後、同様にTLC分析を行ったが、ジケトエステル3は残存したままだった。N-アミノフタルイミド4(91.1 mg, 0.56 mmol, 0.2 eq.)を追加し、30分還流した後に、TLC分析を行った結果、ジケトエステル3のスポットが薄くなったことから、さらに還流を18時間行った。反応液をTLCで分析(UV、リンモリブデン酸)したところ、ごく薄くジケトエステル3の残存が確認されたが、加熱を止めて反応を停止した。酢酸を減圧下留去し、酢酸エチル(40 mL)と蒸留水(30 mL)を加え、5分間撹拌した。2層を分離した後、水層を酢酸エチル(30 mL)で抽出し、合わせた有機層を飽和炭酸水素ナトリウム水溶液(40 mL)で洗浄し、pH試験紙にてpH = 9程度に調整した。飽和塩化ナトリウム水溶液(30 mL)で洗浄した後に、硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ黄色固体1.01 gが得られ、得られた粗生成物を山善製中圧分取液体クロマトグラフ(シリカゲル)にて精製した。溶出液として酢酸エチル/ヘキサン0%(3分、グラジエント10分)⇒10%(3分、グラジエント10分)⇒20%(16分)を使用した。目的物のフラクションを濃縮し、40 ℃で1時間真空乾燥することで、フタルイミドピロール5を596 mg、白色結晶で得た(収率62%)。
クロロ硫酸を用いてクロロスルホン化を行い、スルホン酸クロリド6を合成した。即ち、撹拌子を備え、十分に窒素置換した10 mLねじ口試験管に、クロロ硫酸(1.1 mL, 16.55 mmol, 10.09 eq.)を加えて氷浴で撹拌した(発煙注意)。そこへフタルイミドピロール5(Lot.5188011, 596 mg, 1.75 mmol)を少しずつ加えた後、密閉して室温まで昇温した(橙色透明溶液)。17時間後、TLCで分析(UV、リンモリブデン酸)したところ、フタルイミドピロール5の消失と新たなスポットが確認された(展開溶媒:ヘキサン/酢酸エチル=4/1、Rf値:0.2)。また、反応液を少量取り、重クロロホルムで1H NMRを測定してピロール環のプロトンの消失を確認した(褐色溶液)。氷冷した蒸留水(40 mL)に反応液をゆっくり滴下することで反応を停止した。水層を塩化メチレン(40 mL)で2回抽出した後、合わせた有機層に硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ淡褐色油状物質が得られ、40 ℃で1 時間真空乾燥することで、スルホン酸クロリド6 を410 mg、淡褐色アモルファス結晶で得た(収率72%)。
得られたスルホン酸クロリド6をアンモニア/ジオキサン溶液と反応させ、スルホン酸アミド7を合成した。即ち、撹拌子を備えた30 mLナスフラスコに、スルホン酸クロリド6(Lot.5187201, 369.3 mg、0.84 mmol)を仕込み、氷浴下で0.5 Mアンモニア/ジオキサン溶液(7.3 mL, 3.64 mmol, 4.32 eq.)を加えた後、室温で撹拌した。4時間後にTLCで分析(UV、リンモリブデン酸)したところ、新たに2種のスポットが確認された(展開溶媒:ヘキサン/酢酸エチル=1/1、Rf値:0.4及び0.02)。アンモニア/ジオキサン溶液を減圧留去し、得られた粗生成物を山善製中圧分取液体クロマトグラフ(シリカゲル)にて精製した。溶出液として溶出液として酢酸エチル/ヘキサン55%(3分、グラジエント10分)⇒76%(3分、グラジエント13分)⇒100%(11分)を使用した。目的物と思われたフラクションは得られず、代わりに主として得られたフラクションを濃縮し、40 ℃で1時間真空乾燥することで、スルホン酸アミド7が68.0 mg(収率19%)、スルホン酸アミド7’が170 mg(収率46%)双方、白色アモルファス結晶で得られた。
スルホン酸アミド7及び7’をヒドラジンと反応させ、N-アミノピロール8を合成した。即ち、撹拌子及び冷客管を備えた30 mLナスフラスコに、スルホン酸アミド7及び7’(Lot.5188232,合計295 mg, 0.68 mmol(すべて7’として計算))を仕込み、エタノール(2.4 mL)を加え撹拌した。室温下でヒドラジン一水和物(d = 1.03)(328 μL, 6.75 mmol, 10.0 eq.)を加えた後、100 ℃に加熱して撹拌した。 1時間後にTLCで分析(UV、リンモリブデン酸)したところ、スルホン酸アミド7及び7’と反応中間体の消失が確認され、2種のスポットが確認された(展開溶媒:ヘキサン/酢酸エチル = 1/1、Rf値:0.5及び0.03)。反応液を減圧留去し、得られた粗生成物を山善製中圧分取液体クロマトグラフ(シリカゲル)で精製した。チャージ時はDMSOを用いて粗生成物を溶解した。 溶出液として酢酸エチル/ヘキサン0%(16分、グラジエント13分)⇒ 55%(7分)を使用した。 得られたフラクションを濃縮、40 ℃で1時間真空乾燥し、N-アミノピロール8のDMSO溶液を得た。さらにインジェクトカラムを取り付けた山善製中圧分取液体クロマトグラフ(溶出液として酢酸エチル/ヘキサン50%(3分、グラジエント13分)⇒ 100%(17分))にDMSO溶液を通し、DMSOを除去することでN-アミノピロール8を128 mg、白色結晶で得た(収率65%)。
N-アミノピロール8を2 M LiOH/H2Oと反応させ、化合物9(X 3a)を合成した。 即ち、撹拌子を備えた10 mLねじ口試験管に、N-アミノピロール8(Lot.5189181, 96 mg, 0.33 mmol)を仕込んだ。2 M LiOH/ H2O(717μL, 1.433 mmol, 4.32 eq.)及びエタノール(143 μL)を加え、100 ℃で撹拌した。 1時間後、HPLC分析によりN-アミノピロール8の消失を確認した。反応液を室温まで冷却した後、1 M塩酸によりpH = 4~4.5程度に調製し、白色沈殿を析出させた。反応液を50 mLナスフラスコに移し、減圧下濃縮した(淡黄色固体)。 系内にメタノール(1 mL)を加え、超音波処理により、固体を完溶させた。クロロホルム(19 mL)を加えた後に溶液を冷凍庫(約-10℃)で一晩静置した。析出した固体を吸引ろ過により回収した(10.1 mg)。母液に関しては減圧下濃縮した。合一した粗生成物を、山善製中圧分取液体クロマトグラフ(溶出液としてメタノール/クロロホルム10%(5分、グラジエント5分)⇒ 25%(15分))に付し、精製を行った。化合物9が溶出したフラクションを合わせ、減圧濃縮及び50 ℃で2時間減圧乾燥することにより、化合物9を、淡黄色固体で得た(収率51%)。
1H-NMR (400 MHz, DMSO-d6): δ 1.09 (t, J = 6.0 Hz, 3H), 1.09 (t, J = 7.2 Hz, 3H), 2.91-2.98 (m, 4H), 5.64 (bs, 2H, -N-NH2), 7.24 (bs, 2H, -SONH2). 13C-NMR (100 MHz, DMSO-d6): δ 1.09。
1H-NMR (400 MHz, DMSO-d6): δ 1.09 (t, J = 6.0 Hz, 3H), 1.09 (t, J = 7.2 Hz, 3H), 2.91-2.98 (m, 4H), 5.64 (bs, 2H, -N-NH2), 7.24 (bs, 2H, -SONH2). 13C-NMR (100 MHz, DMSO-d6): δ 1.09。
合成例9.化合物X3bの合成
化合物X3bを以下のようにして合成することができる。
化合物X3bを以下のようにして合成することができる。
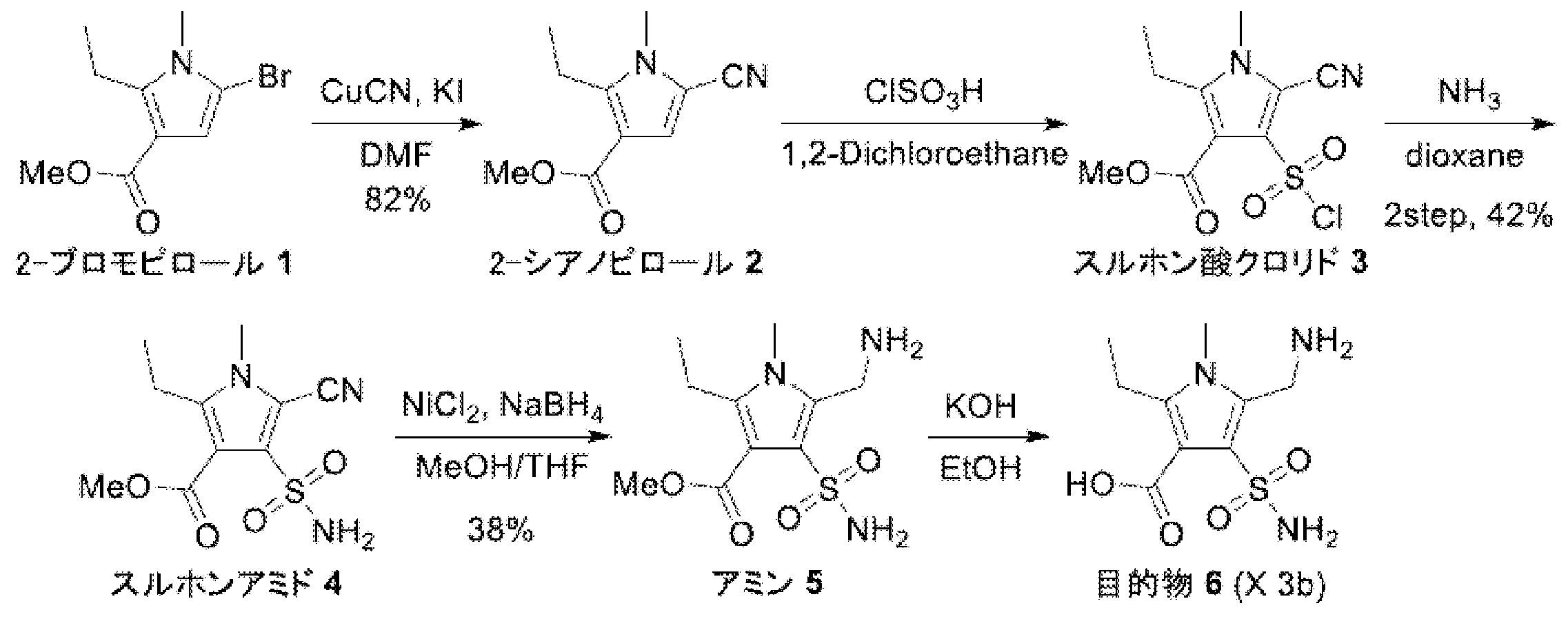
撹拌子を備えた500 mL 三口ナスフラスコに、2-ブロモピロール1(3.9 g, 15.85 mmol)及びDMF(95 mL)を加え、室温下にて撹拌した。そこへ、シアン化銅(I)(7.1 g, 79.24 mmol, 5.0 eq.)及びヨウ化カリウム(184 mg, 1.11 mmol, 0.07 eq.)を加え、120 ℃で撹拌した。17時間攪拌した。反応終了後、酢酸エチル(95 mL)で希釈後、蒸留水(95 mL)を加え、固体をセライトろ過でろ別した。ろ液を分液後に水層を酢酸エチル(100 mL)で抽出した。有機層を合わせ、5%塩化ナトリウム水溶液(300 mL)で3 回洗浄した。有機層に硫酸ナトリウムを加えて乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ、褐色油状の粗生成物3.12 g を得た。得られた粗生成物を山善製中圧分取液体クロマトグラフ(シリカゲル)にて精製した。溶出液として溶出液として酢酸エチル/ヘキサン10%(3 分、グラジエント10 分⇒31%、14 分)を使用した。フラクションを濃縮し、2-シアノピロール2(2.49 g、82%)で淡黄色固体として得た。
撹拌子を備えた300 mL ナスフラスコに、氷浴下、1,2-ジクロロエタン(42 mL)、クロロ硫酸(7.23 mL, 12.73 mmol, 10.0 eq.)を加え、氷浴下で撹拌した。そこへ、2-シアノピロール2(2.1 g, 10.93 mmol)の1,2-ジクロロエタン(13 mL)溶液を滴下し、室温で40分、撹拌した。反応終了後、反応液に氷冷下、五塩化リン(11.38 g, 54.63 mmol, 5.0 eq.)を加え、60 ℃で1時間加熱撹拌した。反応液を室温まで冷却後、氷冷した蒸留水(200 mL)中に滴下し、反応を停止した。分液後、水層をジクロロメタン200 mL で抽出し、有機層を合わせ、硫酸ナトリウムで乾燥した。乾燥剤をろ別後、ろ液を減圧下濃縮したところ、スルホン酸クロリド3の粗生成物(3.41 g、粗収率107%)を橙色油状物質として得た。
撹拌子を備えた300 mL ナスフラスコに、スルホン酸クロリド3の粗生成物(3.41 g, 11.73 mmol)へ、氷冷下、0.5 M アンモニア/ジオキサン溶液(101 mL, 50.67 mmol, 4.32 eq.)を加え、室温にて15時間撹拌した。反応終了後、ろ過にて不要物を除去し、スルホンアミド4(1.32 g、42%)、灰白色固体で得た。
撹拌子を備えた1000 mL ナスフラスコに、スルホンアミド4(933 mg, 3.442 mmol)、塩化ニッケル(II)(446.0 mg, 0.369 mmol, 1.0 eq.)、メタノール(177 mL)及びTHF(177 mL)を加え、室温にて撹拌した。そこへ、水素化ホウ素ナトリウム(390.6 mg, 10.32 mmol, 3.0 eq.)を加え、室温で3撹拌した。30分後、さらに水素化21 ホウ素ナトリウム(390.6 mg, 10.32 mmol, 3.0 eq.)を追加し、室温で撹拌した。1時間後、さらに塩化ニッケル(II)(446.0 mg, 0.369 mmol, 1.0 eq.)、水素化ホウ素ナトリウム(390.6 mg, 10.32 mmol, 3.0 eq.)を加え、室温で5時間攪拌した。蒸留水(100 mL)を加え、反応を停止した。不溶物を吸引ろ過でろ別した。ろ液を減圧下濃縮し、白色固体の粗生成物を得た。得られた粗生成物を中圧分取液体クロマトグラフ(溶出液:メタノール/ クロロホルム4%(3 分、グラジエント10 分)⇒11%(13 分)⇒30%(18 分))で精製しアミン5(355.6 mg、収率38%)を淡黄色固体として得た。

アミン5からX3bへの変換は、通常の塩基性条件下を用いると遊離のアミノ基とスルホンアミドとの環化反応が優先してしまうことが分かっている。そこで、DMSO中で塩化リチウムとともに加熱し中性条件下でメチルエステルをカルボン酸へと導くKrapcho反応が有用である(経路1)。また、遊離のアミン5にBoc基(t-ブトキシカルボニル基)を導入した後、メチルエステルを加水分解、Boc基を酸性条件下で脱保護することによって得られる(経路2)。いずれも最終的に塩酸塩とすることで、X3bを良好に合成可能である。
4.化合物の解析2
試験例9.酵素活性測定試験
メタロ-β-ラクタマーゼ(IMP-1)(最終濃度 10 nM)を使用し且つ基質としてイミペネム(最終濃度 100μM)を使用し、阻害剤となる被験物質(化合物No.9-7、No.9-8、No.9-9)の濃度をそれぞれ変化させ(0μM、10μM、100μM)、各ポイントにおける吸光度298 nm、30℃条件下のイミペネム分解活性(Δabs/min)を測定した。被検物質0の場合の活性を100%として、被検物質を添加した場合の活性相対値を算出した。
試験例9.酵素活性測定試験
メタロ-β-ラクタマーゼ(IMP-1)(最終濃度 10 nM)を使用し且つ基質としてイミペネム(最終濃度 100μM)を使用し、阻害剤となる被験物質(化合物No.9-7、No.9-8、No.9-9)の濃度をそれぞれ変化させ(0μM、10μM、100μM)、各ポイントにおける吸光度298 nm、30℃条件下のイミペネム分解活性(Δabs/min)を測定した。被検物質0の場合の活性を100%として、被検物質を添加した場合の活性相対値を算出した。
結果を表3に示す。表3中、最左列は、阻害剤である被験物質を示す。

表3に示されるように、化合物No.9-7、No.9-8、及びNo.9-9の添加により、β-ラクタマーゼ活性が阻害されることが分かった。
試験例10.薬剤感受性試験5
阻害剤となる被検物質としてX2a、X2b、X2c、X2d、X3a、及びX4を用いる以外は、試験例1と同様にして行った。阻害剤濃度は10μg/mLとした。
阻害剤となる被検物質としてX2a、X2b、X2c、X2d、X3a、及びX4を用いる以外は、試験例1と同様にして行った。阻害剤濃度は10μg/mLとした。
結果を表4に示す。表4中、最左列は、阻害剤である被験物質を示す。
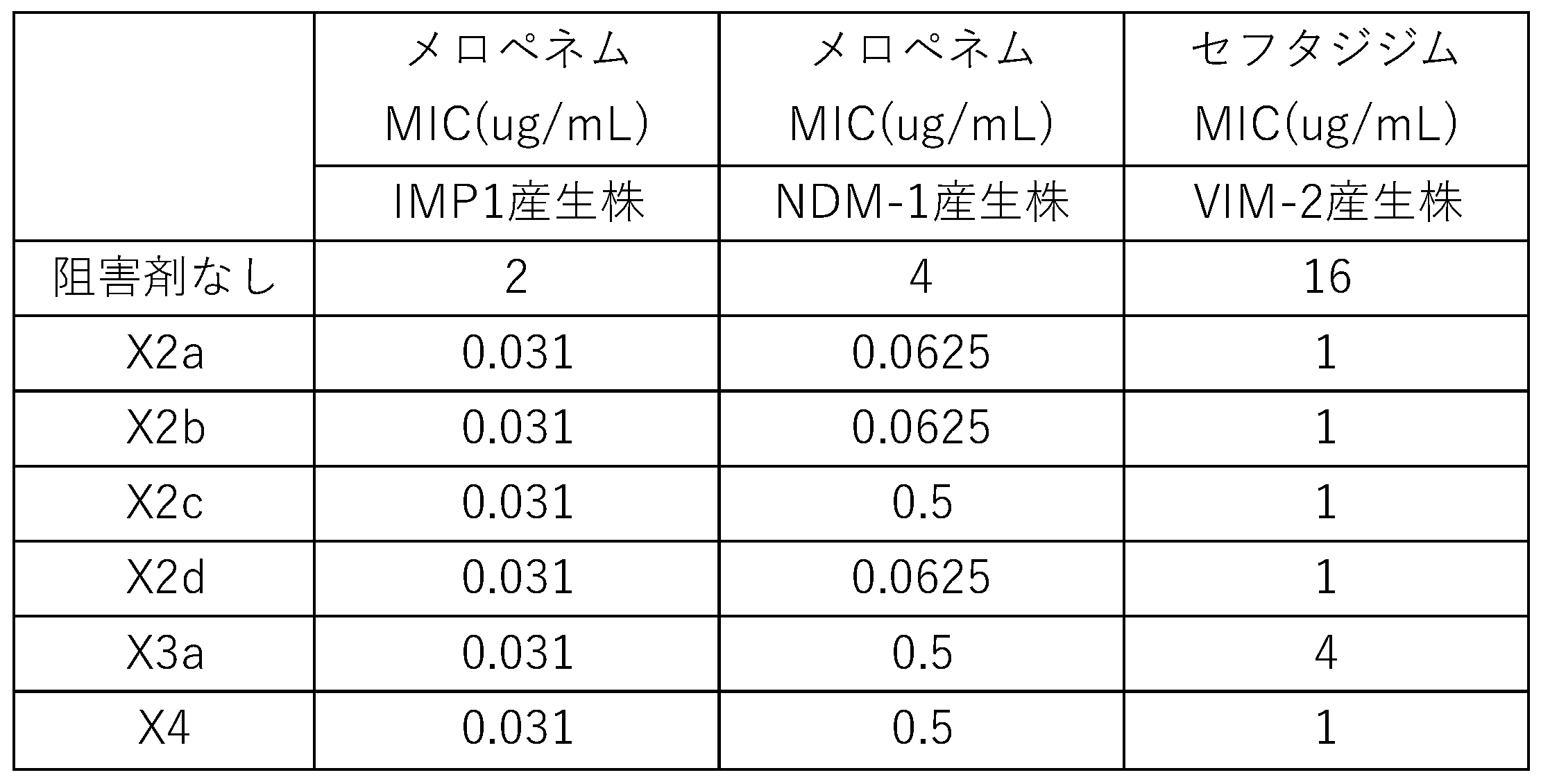
表4に示されるように、化合物X2a、X2b、X2c、X2d、X3a、及びX4の添加により、β-ラクタマーゼ産生菌に対する抗菌薬のMICが低下することが分かった。
試験例11.阻害定数(Ki)測定試験2
阻害剤となる被検物質としてX2a、X2b、X2c、X2d、X3a、及びX4を用いる以外は、試験例6と同様にして行った。
阻害剤となる被検物質としてX2a、X2b、X2c、X2d、X3a、及びX4を用いる以外は、試験例6と同様にして行った。
結果を表5に示す。表5中、最左列は、阻害剤である被験物質を示す。

表5に示されるように、化合物X2a、X2b、X2c、X2d、X3a、及びX4の添加により、β-ラクタマーゼ活性が阻害されることが分かった。
試験例12.動物試験(マウス腹腔感染モデル)1
実験用菌株(クラスB β-ラクタマーゼ(IMP-1)をコードするプラスミドが導入された大腸菌株)をマウスに感染させた後、被検物質(化合物A)を投与し、生死を観察した。具体的には以下のようにして行った。
実験用菌株(クラスB β-ラクタマーゼ(IMP-1)をコードするプラスミドが導入された大腸菌株)をマウスに感染させた後、被検物質(化合物A)を投与し、生死を観察した。具体的には以下のようにして行った。
ミューラーヒントン寒天培地上で1晩発育させた実験用菌株を滅菌綿棒でかきとり、生理食塩水に懸濁し、2-5x108 CFU/mLに調整後、10%ムチン溶液と等量混和して(1-2.5x108 CFU/mL)、菌液を作製した。入荷後に4日間馴化させたマウス(Crl:CD1(ICR)、雄、4週齢(日本チャールズリバー))の体重を測定後、作製した菌液をマウス1匹あたり250μL腹腔投与して感染させた。一方で、抗菌薬(メロペネム3水和物(WAKO 137-15674))及び被検物質を生理食塩水で溶解した。感染1時間後に、1回目の抗菌薬(0.2mg/kg)及び/又は被検物質(100mg/kg)の腹腔投与を行った。感染3時間後にも、1回目と同様に、2回目の投与を行った(ここまでday0)。翌日(day1)より、生死の観察を毎日、定時に行い、生存率を記録した。
結果を図10に示す。化合物Aの投与により、生存率が顕著に向上することが分かった。
試験例13.動物試験(マウス腹腔感染モデル)2
実験用菌株として、クラスB β-ラクタマーゼ(IMP-1)をコードするプラスミドが導入された大腸菌株に加え、NDM及びVIMをコードするプラスミドが導入された細菌(K.pnuemoniae)を使用し、被検物質として化合物Iを投与した。抗菌薬投与量は0.8mg/kg又は4mg/kg、被検物質投与量は10mg/kgとした。これら以外は、試験例12と同様にして行った。
実験用菌株として、クラスB β-ラクタマーゼ(IMP-1)をコードするプラスミドが導入された大腸菌株に加え、NDM及びVIMをコードするプラスミドが導入された細菌(K.pnuemoniae)を使用し、被検物質として化合物Iを投与した。抗菌薬投与量は0.8mg/kg又は4mg/kg、被検物質投与量は10mg/kgとした。これら以外は、試験例12と同様にして行った。
結果を図11及び12に示す。化合物Iの投与により、生存率が顕著に向上することが分かった。
試験例14.動物試験(マウス腹腔感染モデル)3
実験用菌株として、NDM及びVIMをコードするプラスミドが導入された細菌(K.pnuemoniae)を使用し、被検物質として化合物X2dを投与した。抗菌薬投与量は2mg/kg又は8mg/kg、被検物質投与量は10mg/kgとした。これら以外は、試験例12と同様にして行った。
実験用菌株として、NDM及びVIMをコードするプラスミドが導入された細菌(K.pnuemoniae)を使用し、被検物質として化合物X2dを投与した。抗菌薬投与量は2mg/kg又は8mg/kg、被検物質投与量は10mg/kgとした。これら以外は、試験例12と同様にして行った。
結果を図13に示す。化合物X2dの投与により、生存率が顕著に向上することが分かった。
Claims (10)
- 一般式(1):
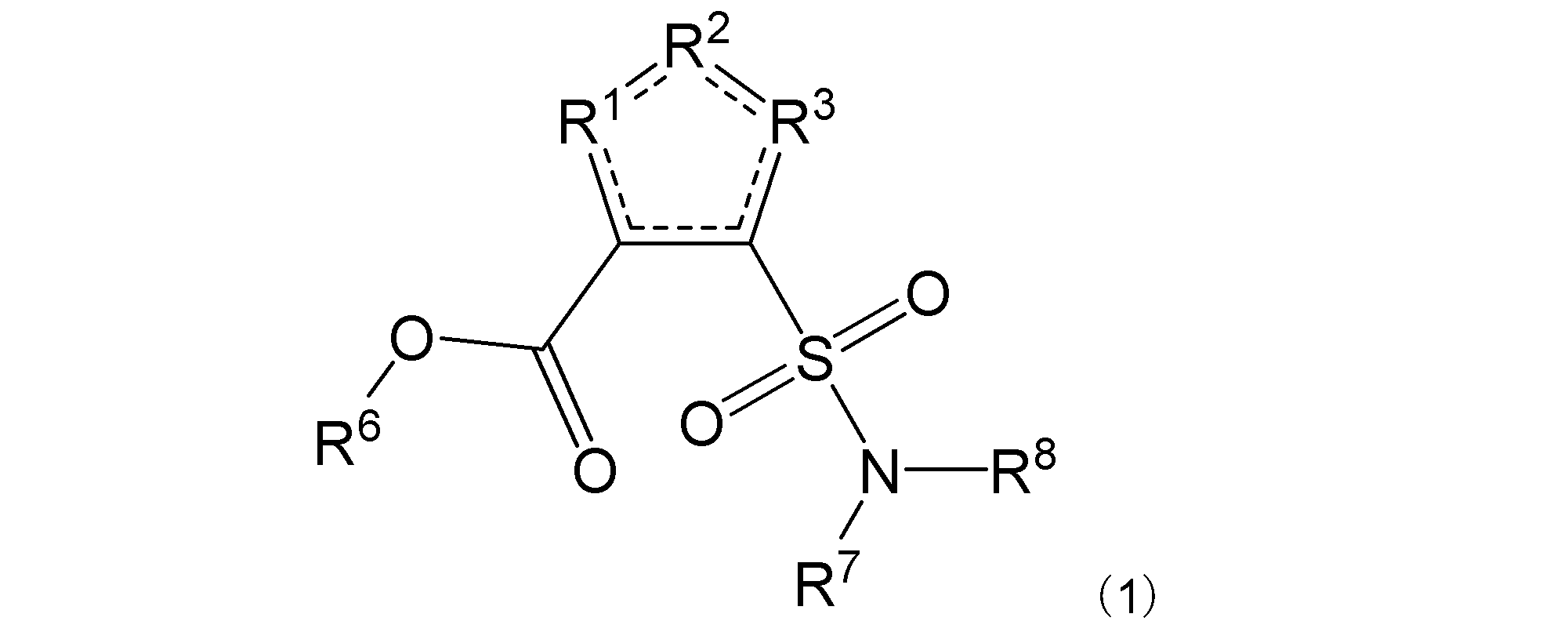
で表される化合物、又はその塩、水和物若しくは溶媒和物を含有する、β-ラクタマーゼ阻害剤。 - 前記β-ラクタマーゼがクラスB β-ラクタマーゼである、請求項1に記載の阻害剤。
- 前記β-ラクタマーゼがクラスB1 β-ラクタマーゼである、請求項1又は2に記載の阻害剤。
- 一般式(1):
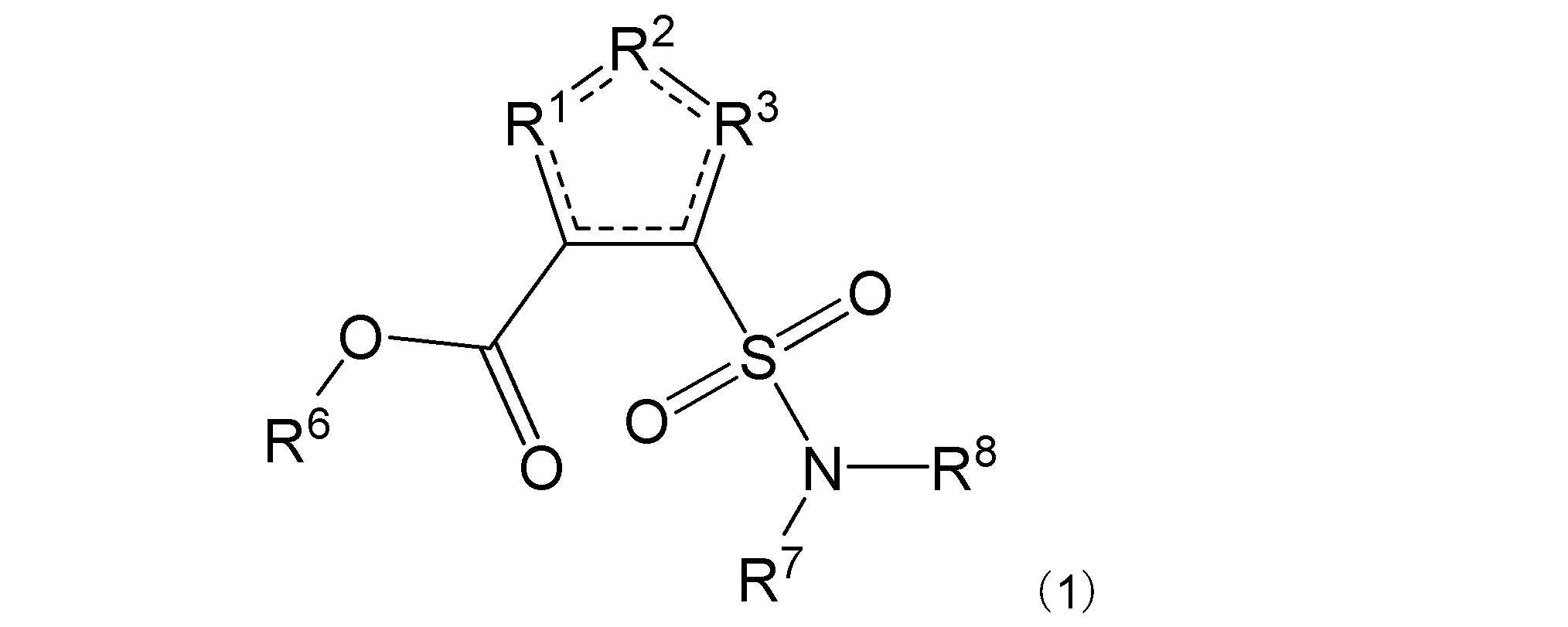
で表される化合物、又はその塩、水和物若しくは溶媒和物を含有する、β-ラクタム系抗菌化合物の抗菌効果の増強剤。 - 一般式(1):

で表される化合物、又はその塩、水和物若しくは溶媒和物、並びにβ-ラクタム系抗菌化合物を含有する、抗菌剤。 - β-ラクタム系抗菌化合物を含有する、
一般式(1):
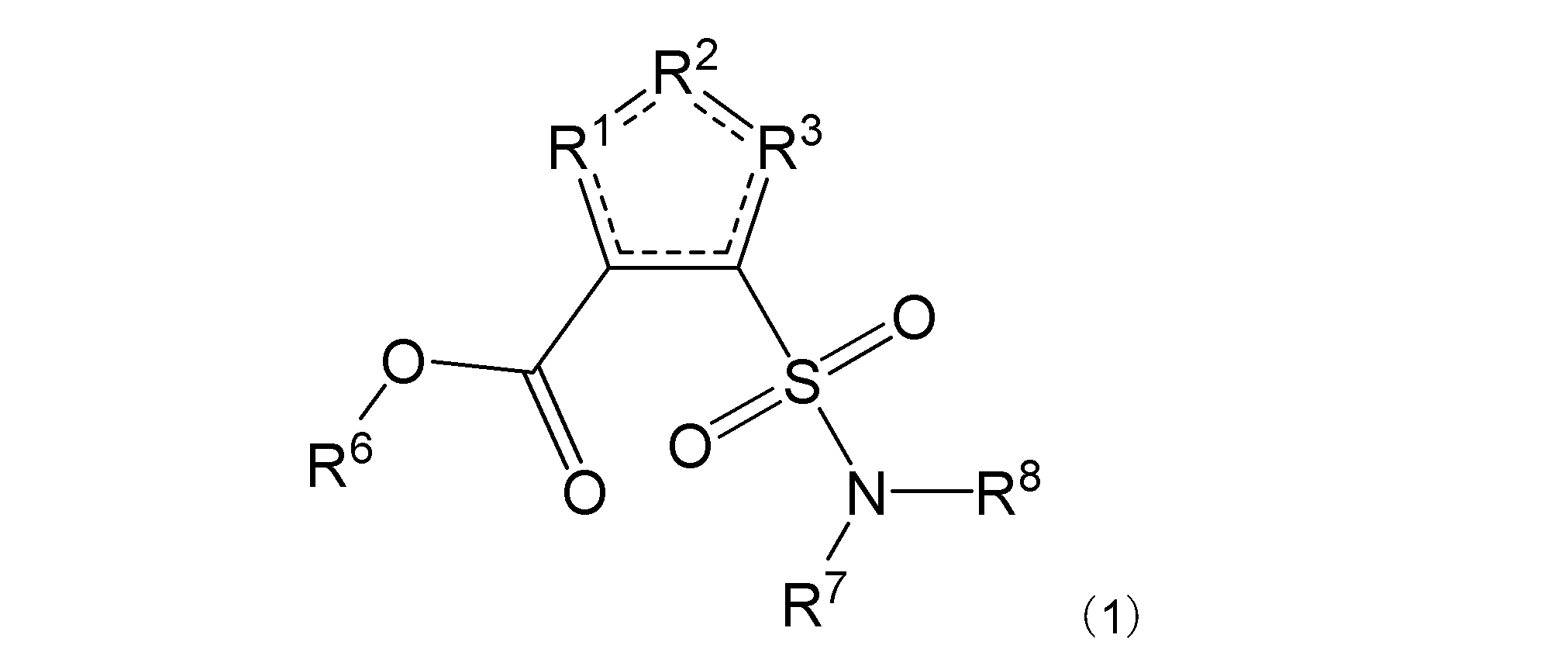
で表される化合物、又はその塩、水和物若しくは溶媒和物と併用投与されるように用いられる抗菌剤。 - 一般式(1A):
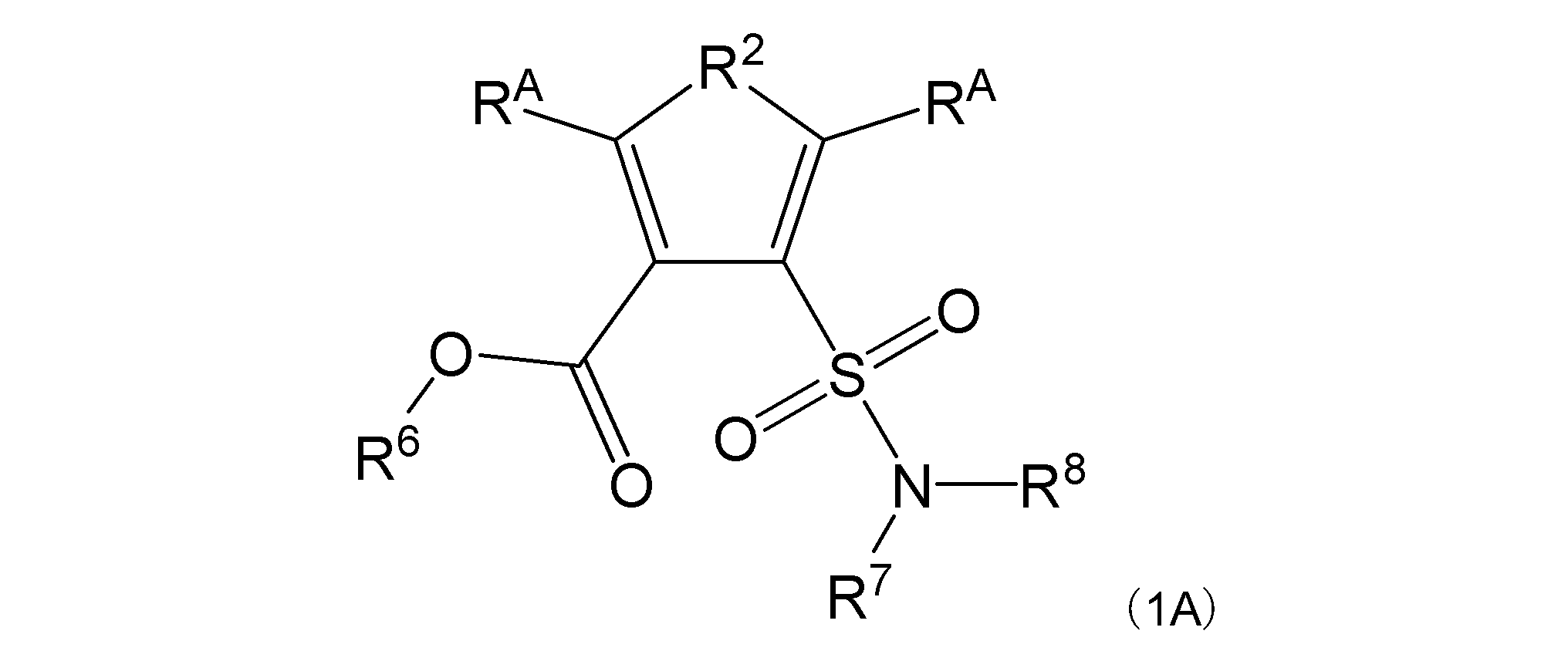
で表される化合物、又はその塩、水和物若しくは溶媒和物。 - RBで示されるアルキル基が炭素数1~4の直鎖状アルキル基である、請求項7に記載の化合物、又はその塩、水和物若しくは溶媒和物。
- 請求項7又は8に記載の化合物、又はその塩、水和物若しくは溶媒和物を含有する、医薬。
- 請求項7又は8に記載の化合物、又はその塩、水和物若しくは溶媒和物を含有する、試薬。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020519856A JP7318950B2 (ja) | 2018-05-14 | 2019-05-14 | β-ラクタマーゼ阻害剤 |
| CN201980032173.2A CN112165936B (zh) | 2018-05-14 | 2019-05-14 | β-内酰胺酶抑制剂 |
| EP19802755.9A EP3795149A4 (en) | 2018-05-14 | 2019-05-14 | BETA-LACTAMASE INHIBITOR |
| US17/055,006 US11497731B2 (en) | 2018-05-14 | 2019-05-14 | β-lactamase inhibitor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018093369 | 2018-05-14 | ||
| JP2018-093369 | 2018-05-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019221122A1 true WO2019221122A1 (ja) | 2019-11-21 |
Family
ID=68540045
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/019133 WO2019221122A1 (ja) | 2018-05-14 | 2019-05-14 | β-ラクタマーゼ阻害剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11497731B2 (ja) |
| EP (1) | EP3795149A4 (ja) |
| JP (1) | JP7318950B2 (ja) |
| CN (1) | CN112165936B (ja) |
| WO (1) | WO2019221122A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2602096A (en) * | 2020-12-17 | 2022-06-22 | Infex Therapeutics Ltd | Compounds |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003070682A1 (en) * | 2002-02-19 | 2003-08-28 | Northwestern University | NON-COVALENT INHIBITORS OF AmpC ß-LACTAMASE |
| WO2013180197A1 (ja) | 2012-05-30 | 2013-12-05 | Meiji Seikaファルマ株式会社 | 新規β-ラクタマーゼ阻害剤とその製造法 |
| JP2015512440A (ja) | 2012-04-02 | 2015-04-27 | アストラゼネカ アクチボラグ | β−ラクタマーゼ阻害剤としてのヘテロ二環式化合物 |
| JP2015155435A (ja) | 2007-10-09 | 2015-08-27 | ソファーミア,インコーポレイテッド | 広帯域β−ラクタマーゼ阻害薬 |
| JP2016520658A (ja) * | 2013-06-13 | 2016-07-14 | アンタビオ エスアーエス | 抗菌性チアゾールカルボン酸 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1192128A4 (en) * | 1999-06-15 | 2003-07-02 | Merck & Co Inc | THIOL DERIVATIVES AS METALLO-BETA-LACTAMASE INHIBITORS |
| US20120329842A1 (en) | 2010-12-23 | 2012-12-27 | Yongcheng Song | Small molecule compounds as broad-spectrum inhibitors of metallo-beta-lactamases |
| GB2533136A (en) * | 2014-12-11 | 2016-06-15 | Antabio Sas | Compounds |
| WO2016206101A1 (en) * | 2015-06-26 | 2016-12-29 | Merck Sharp & Dohme Corp. | Metallo-beta-lactamase inhibitors |
-
2019
- 2019-05-14 WO PCT/JP2019/019133 patent/WO2019221122A1/ja unknown
- 2019-05-14 US US17/055,006 patent/US11497731B2/en active Active
- 2019-05-14 CN CN201980032173.2A patent/CN112165936B/zh active Active
- 2019-05-14 JP JP2020519856A patent/JP7318950B2/ja active Active
- 2019-05-14 EP EP19802755.9A patent/EP3795149A4/en active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003070682A1 (en) * | 2002-02-19 | 2003-08-28 | Northwestern University | NON-COVALENT INHIBITORS OF AmpC ß-LACTAMASE |
| JP2015155435A (ja) | 2007-10-09 | 2015-08-27 | ソファーミア,インコーポレイテッド | 広帯域β−ラクタマーゼ阻害薬 |
| JP2015512440A (ja) | 2012-04-02 | 2015-04-27 | アストラゼネカ アクチボラグ | β−ラクタマーゼ阻害剤としてのヘテロ二環式化合物 |
| WO2013180197A1 (ja) | 2012-05-30 | 2013-12-05 | Meiji Seikaファルマ株式会社 | 新規β-ラクタマーゼ阻害剤とその製造法 |
| JP2016520658A (ja) * | 2013-06-13 | 2016-07-14 | アンタビオ エスアーエス | 抗菌性チアゾールカルボン酸 |
Non-Patent Citations (2)
| Title |
|---|
| RACHEL, A. ET AL.: "Structure-Based Discovery of a Novel", NONCOVALENT INHIBITOR OF AMPC BETA-LACTAMASE, STRUCTURE, vol. 10, no. 7, 2002, pages 1013 - 1023, XP009524466, DOI: 10.1016/S0969-2126(02)00799-2 * |
| See also references of EP3795149A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7318950B2 (ja) | 2023-08-01 |
| EP3795149A4 (en) | 2022-01-05 |
| CN112165936B (zh) | 2023-11-28 |
| EP3795149A8 (en) | 2021-05-26 |
| US11497731B2 (en) | 2022-11-15 |
| JPWO2019221122A1 (ja) | 2021-05-27 |
| EP3795149A1 (en) | 2021-03-24 |
| US20210236463A1 (en) | 2021-08-05 |
| CN112165936A (zh) | 2021-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9642869B2 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| US9101638B2 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| JP6672176B2 (ja) | ボロン酸誘導体およびその治療的使用 | |
| ES2287354T3 (es) | Antibioticos de accion dual. | |
| KR101933084B1 (ko) | 화합물 및 그의 용도 | |
| US9862729B2 (en) | Broad spectrum beta-lactamase inhibitors | |
| KR20120106720A (ko) | 제약 화합물의 다형성 및 가다형성 형태 | |
| KR101900249B1 (ko) | 자이라제 억제제 (r)1에틸3[6플루오로5[2(1하이드록시1메틸에틸)피리미딘5일]7(테트라하이드로푸란2일)1h벤즈이미다졸2일]우레아의 고체 형태 | |
| JP7329260B2 (ja) | ボロン酸誘導体およびその治療的使用 | |
| CN106715429A (zh) | 7‑氧代‑二氮杂二环[3.2.1]辛烷衍生物及其作为抗菌剂的用途 | |
| TW201925201A (zh) | 二氮雜二環辛烷衍生物 | |
| WO2019208797A1 (ja) | オキソ置換化合物 | |
| TW491849B (en) | Tricyclic amine derivatives and method for preparing quinolone compound | |
| WO2019221122A1 (ja) | β-ラクタマーゼ阻害剤 | |
| WO2018199291A1 (ja) | ヘテロ環誘導体 | |
| SU1015830A3 (ru) | Способ получени сложных эфиров 6-амидинопенициллановых кислот или их аддитивных солей с кислотами и его вариант | |
| WO2021049600A1 (ja) | 抗菌作用を有する医薬組成物 | |
| CN110840897B (zh) | 金属β-内酰胺酶抑制剂 | |
| JP3796612B2 (ja) | 抗菌剤 | |
| KR20060048259A (ko) | 1-베타메틸카바페넴 유도체 및 이의 제조방법 | |
| JP2020105148A (ja) | 非アリールヘテロ環置換芳香族化合物 | |
| Chang et al. | Novel anti-pseudomonas cephalosporins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19802755 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020519856 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019802755 Country of ref document: EP Effective date: 20201214 |