WO2019176916A1 - 体内における薬物動態を制御する組成物 - Google Patents
体内における薬物動態を制御する組成物 Download PDFInfo
- Publication number
- WO2019176916A1 WO2019176916A1 PCT/JP2019/009919 JP2019009919W WO2019176916A1 WO 2019176916 A1 WO2019176916 A1 WO 2019176916A1 JP 2019009919 W JP2019009919 W JP 2019009919W WO 2019176916 A1 WO2019176916 A1 WO 2019176916A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- drug
- blood
- administered
- composition according
- hours
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 72
- 150000001768 cations Chemical class 0.000 claims abstract description 126
- 239000004480 active ingredient Substances 0.000 claims abstract description 9
- 239000003814 drug Substances 0.000 claims description 132
- 229940079593 drug Drugs 0.000 claims description 124
- 210000004369 blood Anatomy 0.000 claims description 117
- 239000008280 blood Substances 0.000 claims description 117
- 229920006317 cationic polymer Polymers 0.000 claims description 83
- 210000004185 liver Anatomy 0.000 claims description 67
- 229920001223 polyethylene glycol Polymers 0.000 claims description 61
- 239000002202 Polyethylene glycol Substances 0.000 claims description 59
- 210000000056 organ Anatomy 0.000 claims description 45
- 210000002889 endothelial cell Anatomy 0.000 claims description 42
- 210000003734 kidney Anatomy 0.000 claims description 36
- 210000000952 spleen Anatomy 0.000 claims description 34
- 238000012377 drug delivery Methods 0.000 claims description 31
- 210000001519 tissue Anatomy 0.000 claims description 27
- 230000014759 maintenance of location Effects 0.000 claims description 24
- 230000029142 excretion Effects 0.000 claims description 18
- 229920001477 hydrophilic polymer Polymers 0.000 claims description 14
- 230000009467 reduction Effects 0.000 claims description 8
- 229920001400 block copolymer Polymers 0.000 claims description 6
- 238000000034 method Methods 0.000 abstract description 22
- 102000039446 nucleic acids Human genes 0.000 description 64
- 108020004707 nucleic acids Proteins 0.000 description 64
- 150000007523 nucleic acids Chemical class 0.000 description 64
- 108020004999 messenger RNA Proteins 0.000 description 60
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 50
- 239000000693 micelle Substances 0.000 description 49
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 45
- 239000008103 glucose Substances 0.000 description 45
- 239000000243 solution Substances 0.000 description 39
- 229920000642 polymer Polymers 0.000 description 36
- 238000009825 accumulation Methods 0.000 description 35
- 125000002091 cationic group Chemical group 0.000 description 32
- 230000000694 effects Effects 0.000 description 24
- 210000004204 blood vessel Anatomy 0.000 description 21
- 229920001897 terpolymer Polymers 0.000 description 20
- 230000001965 increasing effect Effects 0.000 description 19
- 238000006116 polymerization reaction Methods 0.000 description 19
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 18
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 17
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 17
- 235000001014 amino acid Nutrition 0.000 description 17
- 210000002216 heart Anatomy 0.000 description 17
- 108020004414 DNA Proteins 0.000 description 15
- 102000053602 DNA Human genes 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 230000014509 gene expression Effects 0.000 description 15
- 230000002218 hypoglycaemic effect Effects 0.000 description 15
- 230000001939 inductive effect Effects 0.000 description 15
- 108090000765 processed proteins & peptides Proteins 0.000 description 15
- 210000003556 vascular endothelial cell Anatomy 0.000 description 15
- 208000013016 Hypoglycemia Diseases 0.000 description 14
- 150000001413 amino acids Chemical class 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 210000004556 brain Anatomy 0.000 description 13
- 241000699670 Mus sp. Species 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 241000700605 Viruses Species 0.000 description 12
- 229920001577 copolymer Polymers 0.000 description 12
- 238000002474 experimental method Methods 0.000 description 12
- 230000002209 hydrophobic effect Effects 0.000 description 12
- 102000058063 Glucose Transporter Type 1 Human genes 0.000 description 11
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- 108091006296 SLC2A1 Proteins 0.000 description 11
- 230000003247 decreasing effect Effects 0.000 description 11
- 239000000178 monomer Substances 0.000 description 11
- 229920002477 rna polymer Polymers 0.000 description 11
- 102000009027 Albumins Human genes 0.000 description 10
- 108010088751 Albumins Proteins 0.000 description 10
- 239000013598 vector Substances 0.000 description 10
- 239000004472 Lysine Substances 0.000 description 9
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 230000008859 change Effects 0.000 description 9
- 238000010586 diagram Methods 0.000 description 9
- 150000002632 lipids Chemical class 0.000 description 9
- 239000002479 lipoplex Substances 0.000 description 9
- 235000018977 lysine Nutrition 0.000 description 9
- 239000011241 protective layer Substances 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- GXCJLVVUIVSLOQ-UHFFFAOYSA-N 2-propyl-4,5-dihydro-1,3-oxazole Chemical compound CCCC1=NCCO1 GXCJLVVUIVSLOQ-UHFFFAOYSA-N 0.000 description 7
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 7
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 7
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- 238000009826 distribution Methods 0.000 description 7
- 239000002502 liposome Substances 0.000 description 7
- 229960003104 ornithine Drugs 0.000 description 7
- 239000002245 particle Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 108060001084 Luciferase Proteins 0.000 description 6
- 239000005089 Luciferase Substances 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- 108010039918 Polylysine Proteins 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 238000001647 drug administration Methods 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 230000004060 metabolic process Effects 0.000 description 6
- 239000013612 plasmid Substances 0.000 description 6
- 229920000656 polylysine Polymers 0.000 description 6
- 238000006722 reduction reaction Methods 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 229920000208 temperature-responsive polymer Polymers 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 239000012024 dehydrating agents Substances 0.000 description 5
- 210000002865 immune cell Anatomy 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 229920002714 polyornithine Polymers 0.000 description 5
- 108010055896 polyornithine Proteins 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 238000011282 treatment Methods 0.000 description 5
- 229920000428 triblock copolymer Polymers 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 210000005098 blood-cerebrospinal fluid barrier Anatomy 0.000 description 4
- 210000001759 blood-nerve barrier Anatomy 0.000 description 4
- 230000004378 blood-retinal barrier Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 231100000371 dose-limiting toxicity Toxicity 0.000 description 4
- 210000003038 endothelium Anatomy 0.000 description 4
- 230000036737 immune function Effects 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000012139 lysis buffer Substances 0.000 description 4
- 229920001515 polyalkylene glycol Polymers 0.000 description 4
- 230000000379 polymerizing effect Effects 0.000 description 4
- -1 polymethacrylamide Polymers 0.000 description 4
- CSDQQAQKBAQLLE-UHFFFAOYSA-N 4-(4-chlorophenyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine Chemical compound C1=CC(Cl)=CC=C1C1C(C=CS2)=C2CCN1 CSDQQAQKBAQLLE-UHFFFAOYSA-N 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 241000702421 Dependoparvovirus Species 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 3
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- 239000012505 Superdex™ Substances 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000000427 antigen Substances 0.000 description 3
- 102000036639 antigens Human genes 0.000 description 3
- 108091007433 antigens Proteins 0.000 description 3
- 229920006187 aquazol Polymers 0.000 description 3
- 239000012861 aquazol Substances 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 235000003704 aspartic acid Nutrition 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 210000004155 blood-retinal barrier Anatomy 0.000 description 3
- 210000005013 brain tissue Anatomy 0.000 description 3
- 239000011557 critical solution Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 208000019622 heart disease Diseases 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 239000003999 initiator Substances 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 229940023041 peptide vaccine Drugs 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 2
- FVEZUCIZWRDMSJ-UHFFFAOYSA-N 2-propan-2-yl-4,5-dihydro-1,3-oxazole Chemical compound CC(C)C1=NCCO1 FVEZUCIZWRDMSJ-UHFFFAOYSA-N 0.000 description 2
- 108090000331 Firefly luciferases Proteins 0.000 description 2
- 229930091371 Fructose Natural products 0.000 description 2
- 239000005715 Fructose Substances 0.000 description 2
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 2
- 108010074032 HLA-A2 Antigen Proteins 0.000 description 2
- 102000025850 HLA-A2 Antigen Human genes 0.000 description 2
- 108010013476 HLA-A24 Antigen Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- 108020004682 Single-Stranded DNA Proteins 0.000 description 2
- 108020004566 Transfer RNA Proteins 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 230000002490 cerebral effect Effects 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- 239000000412 dendrimer Substances 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 229920000736 dendritic polymer Polymers 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 238000007865 diluting Methods 0.000 description 2
- 230000036267 drug metabolism Effects 0.000 description 2
- 210000003989 endothelium vascular Anatomy 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000010419 fine particle Substances 0.000 description 2
- 150000004676 glycans Chemical class 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 108091027963 non-coding RNA Proteins 0.000 description 2
- 102000042567 non-coding RNA Human genes 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 229920003213 poly(N-isopropyl acrylamide) Polymers 0.000 description 2
- 229920001282 polysaccharide Polymers 0.000 description 2
- 239000005017 polysaccharide Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 150000003141 primary amines Chemical group 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- LMBVCSFXFFROTA-UHFFFAOYSA-N prop-2-ynyl 4-methylbenzenesulfonate Chemical compound CC1=CC=C(S(=O)(=O)OCC#C)C=C1 LMBVCSFXFFROTA-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 108020004418 ribosomal RNA Proteins 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 229920006027 ternary co-polymer Polymers 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 210000000689 upper leg Anatomy 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- DEPMSUUWSGUYKQ-IWXIMVSXSA-N (2r,3s,4r,5r)-2,3,4,5-tetrahydroxy-6-[(4-nitro-2,1,3-benzoxadiazol-7-yl)amino]hexanal Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CNC1=CC=C([N+]([O-])=O)C2=NON=C12 DEPMSUUWSGUYKQ-IWXIMVSXSA-N 0.000 description 1
- GBGCQZQJCOQSLB-JEDNCBNOSA-N (2s)-2,6-diaminohexanoic acid;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.NCCCC[C@H](N)C(O)=O GBGCQZQJCOQSLB-JEDNCBNOSA-N 0.000 description 1
- VRYALKFFQXWPIH-PBXRRBTRSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)CC=O VRYALKFFQXWPIH-PBXRRBTRSA-N 0.000 description 1
- VZPBLPQAMPVTFO-NKWOADHPSA-N (4ar,6s,7r,8r,8as)-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxine-6,7,8-triol Chemical compound O1[C@H](O)[C@H](O)[C@@H](O)[C@@H]2OC(C)OC[C@H]21 VZPBLPQAMPVTFO-NKWOADHPSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical group NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- RMTFNDVZYPHUEF-XZBKPIIZSA-N 3-O-methyl-D-glucose Chemical compound O=C[C@H](O)[C@@H](OC)[C@H](O)[C@H](O)CO RMTFNDVZYPHUEF-XZBKPIIZSA-N 0.000 description 1
- 241001655883 Adeno-associated virus - 1 Species 0.000 description 1
- 241000702423 Adeno-associated virus - 2 Species 0.000 description 1
- 241000202702 Adeno-associated virus - 3 Species 0.000 description 1
- 241000580270 Adeno-associated virus - 4 Species 0.000 description 1
- 241001634120 Adeno-associated virus - 5 Species 0.000 description 1
- 241000972680 Adeno-associated virus - 6 Species 0.000 description 1
- 241001164823 Adeno-associated virus - 7 Species 0.000 description 1
- 241000649045 Adeno-associated virus 10 Species 0.000 description 1
- 239000012110 Alexa Fluor 594 Substances 0.000 description 1
- 239000012099 Alexa Fluor family Substances 0.000 description 1
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 1
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical group NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- KWHLDPLUSVYYCT-ULVCCGIGSA-N N-[1,3-bis[[(2R,3S,4R,5S)-1,2,3,4,5-pentahydroxy-6-oxohexan-3-yl]oxy]propan-2-yl]benzamide Chemical compound O=C[C@@H](O)[C@@H](O)[C@](O)([C@H](O)CO)OCC(CO[C@@](O)([C@H](O)CO)[C@H](O)[C@H](O)C=O)NC(=O)C1=CC=CC=C1 KWHLDPLUSVYYCT-ULVCCGIGSA-N 0.000 description 1
- 108091036407 Polyadenylation Proteins 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 108091034057 RNA (poly(A)) Proteins 0.000 description 1
- 229940022005 RNA vaccine Drugs 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 238000003800 Staudinger reaction Methods 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- PMMURAAUARKVCB-UHFFFAOYSA-N alpha-D-ara-dHexp Natural products OCC1OC(O)CC(O)C1O PMMURAAUARKVCB-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 235000013361 beverage Nutrition 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical class [H]OC(*)=O 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229920001940 conductive polymer Polymers 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 238000001476 gene delivery Methods 0.000 description 1
- 150000002303 glucose derivatives Chemical class 0.000 description 1
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 229920000578 graft copolymer Polymers 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 229920000831 ionic polymer Polymers 0.000 description 1
- 238000003670 luciferase enzyme activity assay Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 108700021021 mRNA Vaccine Proteins 0.000 description 1
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- QARBMVPHQWIHKH-KHWXYDKHSA-N methanesulfonyl chloride Chemical class C[35S](Cl)(=O)=O QARBMVPHQWIHKH-KHWXYDKHSA-N 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- 239000002679 microRNA Substances 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000013618 particulate matter Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 229920000948 poly(ethylene glycol)–block-poly(l-lysine) Polymers 0.000 description 1
- 229920002946 poly[2-(methacryloxy)ethyl phosphorylcholine] polymer Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000003505 polymerization initiator Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 239000002924 silencing RNA Substances 0.000 description 1
- 235000021309 simple sugar Nutrition 0.000 description 1
- 239000004055 small Interfering RNA Substances 0.000 description 1
- 235000010378 sodium ascorbate Nutrition 0.000 description 1
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 1
- 229960005055 sodium ascorbate Drugs 0.000 description 1
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
Images














Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/25—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids with polyoxyalkylated alcohols, e.g. esters of polyethylene glycol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/549—Sugars, nucleosides, nucleotides or nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/645—Polycationic or polyanionic oligopeptides, polypeptides or polyamino acids, e.g. polylysine, polyarginine, polyglutamic acid or peptide TAT
- A61K47/6455—Polycationic oligopeptides, polypeptides or polyamino acids, e.g. for complexing nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6905—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion
- A61K47/6907—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a colloid or an emulsion the form being a microemulsion, nanoemulsion or micelle
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
Definitions
- the present invention relates to a composition for controlling pharmacokinetics in the body (for example, a composition for changing the distribution of a drug, suppressing the metabolism of the drug, or suppressing the excretion of the drug), in particular, the similar endothelium of the liver
- a composition for controlling pharmacokinetics in the body for example, a composition for changing the distribution of a drug, suppressing the metabolism of the drug, or suppressing the excretion of the drug
- the present invention relates to an agent for suppressing clearance of a drug (for example, a pharmaceutically active ingredient or a carrier for drug delivery) from blood by cells.
- the present invention also relates to an inhibitor of clearance of a drug from the blood by the kidney.
- the invention also relates to a composition for increasing drug delivery to the spleen.
- Blood drug concentration is important in order to exert the effect of a pharmaceutical product.
- Blood drug concentration is determined by four processes: drug absorption, distribution, metabolism, and excretion.
- drug metabolism and drug excretion are important determinants of blood drug concentration.
- Non-patent Document 1 a terpolymer comprising a hydrophilic block, a temperature-responsive block, and a polycationic block.
- This terpolymer forms micelles when mixed with plasmid DNA at low temperature.
- the plasmid DNA forms a complex with the polycationic block of the terpolymer.
- the temperature-responsive block changes from hydrophilic to hydrophobic, thereby forming a hydrophobic intermediate layer covering the plasmid DNA, which is used as a DNA protective layer.
- a micelle preparation technology that greatly improves the blood stability of mRNA has not yet been known.
- a method in which a copolymer of a hydrophilic segment and a cationic segment and a nucleic acid are combined to neutralize the charge Patent Document 1.
- the present invention relates to a composition for controlling pharmacokinetics (for example, a composition for changing the distribution of a drug, suppressing the metabolism of the drug, or suppressing the excretion of the drug).
- a composition for controlling pharmacokinetics for example, a composition for changing the distribution of a drug, suppressing the metabolism of the drug, or suppressing the excretion of the drug.
- an agent for suppressing the excretion of drugs for example, pharmaceutically active ingredients and carriers for drug delivery
- the present invention also provides an inhibitor of the ability of the kidney to excrete drugs from the blood.
- polyvalent cations especially polyvalent cations modified with polyethylene glycol to enhance biocompatibility
- polyethylene glycol especially polyethylene glycol to enhance biocompatibility
- multivalent cations increase the amount of drug delivery to various organs or tissues including the spleen. The present invention is based on such knowledge.
- a composition comprising a polyvalent cation as an active ingredient for use in controlling pharmacokinetics in the body ⁇ wherein the polyvalent cation may be a cationic polymer ⁇ .
- (5A) The composition according to any one of (1A) to (4A) above, wherein the control of pharmacokinetics is control of clearance from blood.
- (6A) The composition according to any one of the above (1A) to (5A), wherein the control of pharmacokinetics is a reduction in the ability of the liver's homologous endothelial cells to excrete the drug from the blood.
- (7A) The composition according to any one of (1A) to (5A) above, wherein the control of pharmacokinetics is a reduction in the ability of the kidney to excrete the drug from the blood.
- (8A) The composition according to any one of (1A) to (6A) above, wherein the control of pharmacokinetics is an increase in the amount delivered to a target organ or tissue.
- the present invention also provides the following inventions.
- a cationic polymer as an active ingredient.
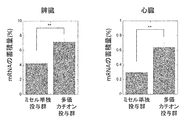
- FIG. 1 is a graph showing the effect of a cationic polymer on carrier accumulation (ion complex) in the liver and kidney.
- FIG. 2 shows the effect of multivalent cations on drug accumulation in the brain.
- FIG. 2A is a diagram showing that mRNA accumulation increases in organs other than the liver and kidney.
- FIG. 3 shows the effect of multivalent cations on carrier accumulation (lipoplex) in the liver (upper panel) and kidney (lower panel).
- FIG. 3A shows that mRNA accumulation increases in organs other than the liver and kidney.
- FIG. 4 is a diagram showing accumulation of polyvalent cations on the inner surface of blood vessels of liver-like endothelial cells. In particular, it is shown that polyvalent cations coat the inner surface of similar endothelial cells.
- FIG. 4 is a diagram showing accumulation of polyvalent cations on the inner surface of blood vessels of liver-like endothelial cells. In particular, it is shown that polyvalent cations coat the inner surface of similar endo
- FIG. 5 is a diagram showing accumulation of polyvalent cations on the inner surface of blood vessels of peripheral (ear) vascular endothelial cells. Substantial multivalent cation coverage or accumulation is not observed in peripheral vascular endothelial cells.
- FIG. 6 is a diagram showing the effect of a polyvalent cation on the retention in blood of a nucleic acid-containing carrier.
- FIG. 7 is a graph showing the effect of polyvalent cations on the accumulation of nucleic acid-encapsulating carriers on the blood vessel inner surface of liver endothelial cells.
- FIG. 8 is a diagram showing that the multivalent cation of the present invention does not substantially interact with albumin.
- FIG. 9 is a graph showing the effect of polyvalent cations on the accumulation of mRNA in the spleen of lipoplexes.
- FIG. 10 is a diagram showing blood retention of bPEG-PLL, PEG-PLL, and bPEG.
- FIG. 11 is a diagram showing the change over time of the abundance of bPEG-PLL in the luminal wall of the hepatoid endothelial cells.
- FIG. 12 is a diagram showing the influence of multivalent cations on the accumulation properties of virus particles in each organ.
- FIG. 13 is a diagram showing the influence of multivalent cations on the accumulation properties of virus particles in each organ.
- subject refers to mammals including humans.
- the subject may be a healthy subject or a subject suffering from some disease.
- the subject is a mammal, eg, a human, and in particular can be a mammal, eg, a human, for whom administration of micelles of the invention is beneficial.
- “for drug delivery” means being biocompatible and capable of encapsulating a drug in a carrier.
- “for drug delivery” means a use for prolonging the blood remaining time of a drug in comparison with the blood remaining time of a naked drug, or a delivery amount of a drug to a predetermined tissue. It may mean an improved use.
- carrier refers to fine particles or hollow fine particles that can contain a substance.
- the carrier preferably has a biocompatible shell or modification.
- a liposome and a micelle are mentioned, Especially the liposome comprised by a phospholipid etc. is mentioned.
- micelle means a carrier formed by aggregation of molecules such as polymers.
- examples of micelles include micelles formed from amphiphilic molecules such as surfactants, and micelles formed from polyion complexes (PIC micelles).
- the micelle is preferably modified on the outer surface with an uncharged hydrophilic chain from the viewpoint of improving bioavailability.
- average molecular weight means number average molecular weight unless otherwise specified.
- degree of polymerization means the number of monomer units in the polymer
- average degree of polymerization means the number average degree of polymerization unless otherwise specified.
- the “cationic block” and the “cationic polymer” are obtained by polymerizing a monomer unit including a cationic monomer, respectively.
- the cationic polymer include a homocationic polymer, a polymer in which a homocationic polymer and an uncharged hydrophilic chain are linked, and the like.
- the cationic polymer portion may be referred to as a cationic block.
- a cationic polymer is a pharmaceutically acceptable cationic polymer.
- the “multivalent cation” refers to a molecule having a plurality of groups having a cationic property among the molecules that are cationic.
- a “multivalent cation” can be cationic as a whole molecule in the blood environment.
- Multivalent cations include molecules that are cationic in the blood environment, such as cationic polymers and cationic dendrimers.
- the polyvalent cation is a polyvalent cation having biocompatibility.
- “dendrimer” refers to a molecule having a plurality of stages of branching from one core atom.
- the “hydrophilic block” means a polymer chain that is soluble in an aqueous medium, and is sometimes referred to as a hydrophilic polymer block.
- the uncharged hydrophilic chain is a pharmaceutically acceptable uncharged hydrophilic chain.
- Such hydrophilic chains include polyethylene glycol (PEG) and poly (2-ethyl-2-oxazoline).
- the non-charged hydrophilic chain may have a polar atom as long as the charge is neutralized locally and entirely.
- the hydrophilic block may or may not contain a branch. If the hydrophilic block has branch points, the number of branch points may be one or more.
- temperature-responsive block and “temperature-responsive polymer” mean a polymer block and a polymer that can change from hydrophilic to hydrophobic depending on temperature, respectively.
- Various substances that change from hydrophilic to hydrophobic depending on temperature are known.
- a temperature-responsive polymer has a lower critical solution temperature (LCST), is hydrophilic below LCST, and hydrophobic above LCST.
- the temperature responsive polymer comprises a temperature responsive block.
- the temperature responsive polymer includes a temperature responsive block and a hydrophilic block.
- the temperature responsive polymer does not have a cationic block.
- the temperature-responsive polymer may be a polymer consisting essentially of a temperature-responsive block or a polymer consisting of a temperature-responsive block.
- the lower critical solution temperature (LCST) can be preferably 4 ° C. or higher and 40 ° C. or lower, and can be lower than the body temperature of a subject such as a human.
- ternary copolymer or “triblock copolymer” means a block copolymer containing three different polymer blocks. Each block may be connected via a linker or a spacer.
- a terpolymer may be denoted as “ABC” if it contains three different blocks, A, B and C, in this order. The symbol “-” can be a bond or a linker or spacer.
- the terpolymer may contain other different polymer blocks as long as it contains three different polymer blocks.
- the terpolymer may consist essentially of three different polymer blocks “ABC” or may consist of “ABC”.
- outer shell means a protective layer that wraps RNA.
- the outer shell does not necessarily mean to exist in the outermost shell.
- the “protective layer” can protect RNA from degradation by a degrading enzyme such as RNase as compared to the case where it does not exist.
- “comprise” is used to mean “consisting of” and “essentially consistent”. “Contains” means that a component other than the target component may be included, and “consists of” does not include any component other than the target component. means. In this specification, “consisting essentially of” means that a component other than the target component is not included in a mode that exhibits a special function (such as a mode that completely loses the effect of the invention). To do.
- administering A and B separately means that A and B are administered separately in time, and that A and B are not mixed together at the same time. Used to mean administration.
- “clearance” refers to drug metabolism and drug excretion.
- clearance can be a decrease in drug due to metabolism or a decrease in drug excreted from the blood. It is known that the drug reduces the retention in the blood by clearance.
- target organ or tissue refers to an organ or tissue to which a drug to be administered is to be delivered.
- the target organ or tissue will vary depending on the type of drug being administered and the type of patient being administered.
- target organ or tissue is not meant to mean delivering a drug only to the organ or tissue, but to any organ or tissue other than the organ or tissue as long as the drug is delivered to the target organ or tissue. It shall mean that the drug may be delivered. Delivery of the drug to the “target organ or tissue” is preferably selective to the target organ or tissue.
- selective means that the drug accumulates more in the target organ or tissue than in other organs or tissues.
- the polyvalent cation represented by the cationic polymer can be used to control the pharmacokinetics in the body.
- the cationic polymer can also be used to control pharmacokinetics in the body.
- control of pharmacokinetics is improvement or change of pharmacokinetics.
- Control of pharmacokinetics includes control of clearance. More specifically, the control of pharmacokinetics includes a reduction in the ability of the liver to excrete the drug by the same endothelial cells. Control of pharmacokinetics also includes reducing the ability of the kidneys to excrete drugs from the blood. Control of pharmacokinetics further includes increasing the amount delivered to the target organ. Control of pharmacokinetics further includes increasing the amount of drug delivered to the spleen. Further control of pharmacokinetics includes an increase in drug retention in the blood.
- a polyvalent cation such as a cationic polymer
- the metabolism of blood drug by the liver's homologous endothelial cells or the excretion through the same is suppressed, and the metabolism or excretion by the kidney is suppressed.
- a polyvalent cation such as a cationic polymer controls the dynamics in the body of other pharmaceutically active ingredients (drugs) administered simultaneously or separately.
- a polyvalent cation such as a cationic polymer need not be a pharmaceutically active ingredient.
- a polyvalent cation such as a cationic polymer does not need to form a complex with a pharmaceutically active ingredient (that is, it may be in a free form).
- the polyvalent cation may be modified with a hydrophilic polymer from the viewpoint of improving the bioavailability of the cation itself.
- the cationic polymer may be, for example, in the form of a copolymer of a cationic block and a hydrophilic polymer block.
- hydrophilic polymer block for example, an uncharged hydrophilic polymer block can be used.
- Uncharged hydrophilic polymer blocks are pharmaceutically acceptable polymers.
- Such a polymer is not particularly limited.
- non-charged hydrophilic polymer polyalkylene glycol and poly (2-oxazoline) are preferably used, and polyalkylene glycol can be particularly preferably used.
- polyalkylene glycol polyethylene glycol (PEG) can be preferably used.
- the average molecular weight of the PEG portion can be, for example, 10 kD or more, 15 kD or more, 20 kD or more, 30 kD or more, or 40 kD or more (for example, 80 kD or more).
- it may be 70 kD or less, 60 kD or less, or 50 kD or less), preferably 20 kD or more, more preferably 30 kD or more.
- the cationic polymer may have an average degree of polymerization of 15 or more, 20 or more, 30 or more, or 40 or more (for example, 80 or less, 70 or less, 60 or less Or may be 50 or less).
- the PEG moiety may have a high average molecular weight, for example, 40 kD or more, 50 kD or more, 60 kD or more, or 70 kD or more (eg, 80 kD or less, 70 kD or less, 60 kD or less, or 50 kD or less.
- a branched PEG having a plurality of can also be used. From the viewpoint of increasing the bulk of the PEG moiety, branched PEG can be preferably used.
- the copolymer comprising a cationic polymer moiety and a PEG moiety is a branched PEG wherein the PEG moiety has multiple PEG chains of 10 kD or more, 15 kD or more, 20 kD or more, 30 kD or more, or 40 kD or more,
- the cationic polymer portion can have an average degree of polymerization of 15 or more, 20 or more, 30 or more, or 40 or more.
- the copolymer comprising a cationic polymer and PEG is a branched PEG in which the PEG moiety has multiple PEG chains of 20 kD or greater, 30 kD or greater, or 40 kD or greater, and the cationic polymer moiety is an average polymerized
- the degree can be 20 or more, 30 or more, or 40 or more.
- the copolymer comprising a cationic polymer moiety and a PEG moiety is a single-chain PEG having a PEG moiety of 40 kD or more, and the cationic polymer moiety has an average degree of polymerization of 15 or more, 20 or more, 30 or more, It may be 40 or more, 50 or more, 60 or more, or 70 or more (for example, 80 or less, 70 or less, 60 or less, or 50 or less.
- the higher the average molecular weight the more the blood stays. The effect of improving the sex is great).
- the cationic polymer block has an average degree of polymerization of 15-30, the cationic polymer block is linked to a branched PEG, and the branched PEG has a total average molecular weight of the PEG moiety of 40 kD to 100 kD, It can be 50 kD to 90 kD, or 60 kD to 80 kD.
- a branched PEG has one branch, and the average molecular weight of each PEG chain extending from the branch can be independently, for example, 20 kD to 60 kD, 25 kD to 50 kD, or 30 kD to 40 kD.
- such a copolymer of a branched PEG and a cationic polymer can be administered as the polyvalent cation of the present invention.
- the cationic polymer block has an average degree of polymerization of 15 to 30, the cationic polymer block is linked to a branched PEG, and the branched PEG has one branch and extends from the branch.
- the average molecular weight of each PEG chain can independently be, for example, 20 kD to 60 kD, 25 kD to 50 kD, or 30 kD to 40 kD.
- a copolymer of such a branched PEG and a cationic polymer Can be administered as the multivalent cation of the present invention.
- the cationic polymer or cationic polymer moiety includes, for example, cationic natural amino acids and cationic unnatural amino acids, for example, cationic natural amino acids such as histidine, tryptophan, ornithine, arginine and lysine, and / or A group represented by — (NH— (CH 2 ) 2 ) p —NH 2 , wherein p is an integer of 1 to 5; ⁇
- a side chain for example, a cationic non-natural amino acid polymer block having the cationic side chain, for example, a cationic non-natural amino acid such as aspartic acid or glutamic acid having the cationic side chain. Examples include polymer blocks.
- the polycation block is a group represented by — (NH— (CH 2 ) 2 ) p —NH 2 , where p is an integer of 1 to 5.
- p is an integer of 1 to 5.
- the cationic natural amino acid is preferably histidine, tryptophan, ornithine, arginine and lysine, more preferably arginine, ornithine and lysine, still more preferably ornithine and lysine, and still more preferably. Includes lysine.
- the cationic polymer or cationic polymer portion can be polylysine or polyornithine.
- the polycation block In the polycation block, a cationic amino acid and an amino acid having a cationic side chain may be mixed. That is, in one embodiment of the present invention, the polycation block is a polymer of monomer units containing a cationic natural amino acid, a cationic unnatural amino acid, or a cationic natural amino acid and a cationic unnatural amino acid. In some embodiments of the invention, the bond between monomer units in the polycation block is a peptide bond.
- the cationic unnatural amino acid is a group represented by — (NH— (CH 2 ) 2 ) p —NH 2 as a side chain ⁇ where p is an integer of 1 to 5 ⁇ It is an amino acid having
- the polycation block includes a cationic natural amino acid and a group represented by — (NH— (CH 2 ) 2 ) p —NH 2 , where p is an integer of 1 to 5. is there.
- 40%, 50%, 60%, 70%, 80%, 90%, 95%, 98%, or 100% of the monomer units in the polymer have-(NH- (CH 2 2 ) a group represented by p -NH 2 (where p is an integer of 1 to 5). ⁇ .
- polylysine or polyornithine having a branched PEG wherein the PEG moiety is a branched PEG having two PEGs having an average molecular weight of 20 to 50 kD
- ornithine include polylysine or polyornithine having a branched PEG having an average degree of polymerization of 20 to 70, or 30 to 60.
- the PEG or branched PEG can be linked to the end of the cationic polymer.
- Another embodiment is a polymer having a main skeleton of a cationic monomer such as lysine and ornithine, and 5% to 80%, or 20% to 50% of the side chain of the monomer unit is hydrophilic such as PEG or branched PEG.
- the polymer may be modified with a conductive polymer (that is, a graft copolymer).
- the polyvalent cation of the present invention can reduce the clearance function of the endothelial cells themselves by coating the blood vessel inner surface of the liver similar endothelial cells and the blood vessel inner surface of the kidney vascular endothelial cells.
- the multivalent cation of the present invention can be administered before, simultaneously with, or after administration of various drugs. In either case, when the multivalent cation and the drug are administered separately, the pharmacokinetics can be controlled when both are present in the blood simultaneously ⁇ the multivalent cation prior to drug administration.
- the multivalent cation is administered while the drug remains in the blood ⁇ .
- the multivalent cation can be administered, for example, immediately prior to drug administration, for example, can be administered 30 seconds or more before, 1 minute or more, 2 minutes or more, 3 minutes or more, 4 minutes or more. Or 5 minutes or more before.
- the multivalent cation can be administered, for example, within 60 minutes, within 50 minutes, within 40 minutes, within 30 minutes, within 20 minutes, or within 10 minutes prior to drug administration.
- the drug can be administered 10 minutes, 5 minutes, 4 minutes, 3 minutes, 2 minutes, 1 minute, 30 seconds, or just before the administration of the multivalent cation, A short period from administration to administration of polyvalent cation is preferred.
- the drug and polyvalent cation can be mixed in the infusion bag and administered.
- the cationic polymer of the present invention can reduce the clearance function of the endothelial cells themselves by coating the blood vessel inner surface of the liver similar endothelial cells and the blood vessel inner surface of the kidney vascular endothelial cells.
- the cationic polymers of the present invention can be administered before, simultaneously with, or after administration of various drugs.
- the pharmacokinetics can be controlled when both are present in the blood simultaneously ⁇ the cationic polymer prior to drug administration.
- the cationic polymer When the cationic polymer is retained in the blood or while remaining on the blood vessel inner surface of the similar endothelial cell of the liver or while remaining on the blood vessel inner surface of the renal endothelial cell, When a drug is administered and a cationic polymer is administered after the drug is administered, the cationic polymer is administered while the drug remains in the blood ⁇ .
- the cationic polymer can be administered, for example, immediately prior to drug administration, for example, can be administered 30 seconds or more before, 1 minute or more, 2 minutes or more, 3 minutes or more, 4 minutes or more. Or 5 minutes or more before.
- the cationic polymer can be administered, for example, within 60 minutes, within 50 minutes, within 40 minutes, within 30 minutes, within 20 minutes, or within 10 minutes prior to drug administration.
- the drug can be administered 10 minutes, 5 minutes, 4 minutes, 3 minutes, 2 minutes, 1 minute, 30 seconds, or just prior to administration of the cationic polymer, A shorter time from administration to administration of the cationic polymer is better.
- simultaneous administration for example, the drug and the cationic polymer can be mixed in the infusion bag and administered.
- the multivalent cation of the present invention can be administered at a dosage and frequency so that the clearance of the administered drug is reduced by a necessary time in combination with (or before and after) the administration time of the drug. Can be determined. In certain embodiments of the invention, the multivalent cation of the invention may be administered in a single dose or multiple doses to reduce the clearance of the administered drug by the required time in conjunction with (or before or after) the administration of the drug. Can be administered. In certain embodiments of the invention, depending on the purpose, the administration can be bolus administration or infusion administration.
- the drug delivery carriers (micelles and liposomes) encapsulating nucleic acids are reduced in the accumulation in the liver's homologous endothelial cells and in the kidney's endothelial cells by the polyvalent cations. Therefore, the polyvalent cation of the present invention can be used particularly for controlling the pharmacokinetics of a drug delivery carrier encapsulating a nucleic acid. According to the present invention, the drug delivery carriers (micelles and liposomes) encapsulating nucleic acids have reduced accumulation in liver similar endothelial cells and kidney endothelial cells by the cationic polymer.
- the cationic polymer of the present invention can be used particularly for controlling the pharmacokinetics of a drug delivery carrier encapsulating a nucleic acid.
- nucleic acids include deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), modified nucleic acids such as Locked nucleic acid (LNA) and cross-linked nucleic acid (BNA), or hybrids thereof.
- LNA Locked nucleic acid
- BNA cross-linked nucleic acid
- DNA include, but are not limited to, linear double-stranded DNA, linear single-stranded DNA, circular double-stranded DNA, and circular single-stranded DNA.
- RNA examples include, but are not limited to, siRNA, shRNA, micro RNA, messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), non-coding RNA (ncRNA), and double-stranded RNA, and These RNA derivatives are mentioned.
- Multivalent cations can be present in free form when administered with drug delivery carriers (micelles or liposomes) encapsulating nucleic acids. That is, it is known that a polyvalent cation can form a complex directly with a nucleic acid. However, in the present invention, the polyvalent cation coats the vascular endothelial cells of the liver and kidney. Is preferably present in free form (i.e., not in complex with nucleic acid).
- the drug delivery carrier encapsulating the nucleic acid may be a lipid complex such as a lipoplex instead of an ion complex.
- the cationic polymer can be present in a free form when administered together with a drug delivery carrier (micelle or liposome) encapsulating nucleic acid.
- a cationic polymer can directly form a complex with a nucleic acid.
- the cationic polymer coats vascular endothelial cells of the liver and kidney.
- the cationic polymer can be present in free form (ie, not complexed with nucleic acid).
- a cationic polymer different from the cationic polymer used for producing a drug delivery carrier encapsulating a nucleic acid can be used.
- the drug delivery carrier encapsulating the nucleic acid may be a lipid complex such as a lipoplex instead of an ion complex.
- a virus may be used as a carrier in order to increase the efficiency of introducing a nucleic acid into a cell.
- Carriers for increasing the efficiency of introducing nucleic acid into cells include retroviral vectors, lentiviral vectors, adenoviral vectors, adeno-associated viral vectors (AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, and Virus vectors such as AAV10), Sendai virus vectors, measles virus vectors, vaccinia virus vectors, herpes virus vectors and the like can be used.
- These vectors may have, for example, an expression unit (for example, a nucleic acid operably linked to a promoter or an enhancer) that expresses the nucleic acid to be delivered in the cell.
- a method for administering a drug to a subject comprising administering a multivalent cation to the subject and administering the drug to the subject.
- the multivalent cation and RNA are administered separately.
- the multivalent cation can be administered to the subject simultaneously with the drug, or prior to or after the drug.
- the multivalent cation may be administered at a dose that is biocompatible but does not exhibit dose limiting toxicity (DLT).
- a method of administering a drug to a subject comprising administering a cationic polymer to the subject and administering a drug to the subject.
- the cationic polymer and RNA are administered separately.
- the cationic polymer can be administered to the subject simultaneously with the drug, or prior to or after the drug.
- Cationic polymers can be administered at doses that are biocompatible but do not exhibit dose limiting toxicity (DLT).
- a method for administering a drug delivery carrier encapsulating a nucleic acid in a subject comprising administering a multivalent cation to the subject and administering a drug delivery carrier encapsulating the nucleic acid in the subject.
- a method of delivering a nucleic acid to a tissue of a subject comprising administering a polyvalent cation to the subject and administering a drug delivery carrier encapsulating the nucleic acid to the subject.
- the multivalent cation can be administered to the subject simultaneously with the drug delivery carrier encapsulating the nucleic acid, or before or after the drug.
- a method for administering a drug delivery carrier encapsulating a nucleic acid to a subject comprising administering a cationic polymer to the subject and administering a drug delivery carrier encapsulating the nucleic acid to the subject.
- a method for delivering a nucleic acid to a tissue of a subject comprising administering a cationic polymer to the subject and administering a drug delivery carrier encapsulating the nucleic acid to the subject.
- the cationic polymer can be administered to the subject simultaneously with, or before or after the drug delivery carrier encapsulating the nucleic acid.
- micelles formed by terpolymers comprising a hydrophilic block, a temperature responsive block and a cationic block dramatically improve the blood stability of nucleic acids, eg, brain tissue It was found that nucleic acids can be delivered in large quantities. Therefore, according to the present invention, for example, an organ or tissue comprising a nucleic acid, a terpolymer comprising a hydrophilic block, a temperature-responsive block, and a cationic block, and a micelle whose surface is modified with glucose.
- Compositions are provided for use in delivering nucleic acids, particularly to the brain.
- Such micelles can be prepared by the following procedure.
- a terpolymer containing a hydrophilic block, a temperature-responsive block and a cationic block, and RNA are mixed in an aqueous solution at a temperature lower than LCST so that micelles can be formed.
- the temperature of the aqueous solution is raised above the LCST to change the temperature responsive block from hydrophilic to hydrophobic.
- the micelles thus obtained are considered to have a state in which the nucleic acid is encapsulated and has a hydrophobic protective layer as its outer shell, and the encapsulated nucleic acid can be stably maintained in the blood. it can. That is, in this micelle, the nucleic acid is covered with an outer shell formed of a temperature-responsive block that has changed to hydrophobicity.
- the LCST of the terpolymer is preferably 35 ° C. or lower. By doing so, the micelle state is maintained in the blood after administration.
- LCST is preferably 5 ° C. or higher. Note that it is preferable to set an appropriate LCST in consideration of various other situations.
- the LCST can be 5 ° C. to 35 ° C., 10 ° C. to 32 ° C., or 25 ° C. to 32 ° C.
- water soluble polymers have LCST.
- any polymer having LCST within the above temperature range can be appropriately used as a temperature-responsive block.
- the hydrophilic block of the terpolymer can be polyethylene glycol or poly (2-ethyl-2-oxazoline).
- the average molecular weight of the hydrophilic block can be, for example, 10 kD or more, 15 kD or more, 20 kD or more, 30 kD or more, or 40 kD or more.
- the hydrophilic block can also have an average molecular weight of, for example, 20 kD or less, 30 kD or less, 40 kD or less, or 50 kD or less.
- the temperature responsive block of the terpolymer is poly (N-isopropylacrylamide), poly (2-n-propyl-2-oxazoline), or poly (2-isopropyl-2oxazoline). It can be.
- the average molecular weight of the temperature-responsive block can be, for example, 3 kD or more, 5 kD or more, 7 kD or more, or 10 kD or more.
- the average molecular weight of the temperature responsive block can also be, for example, 10 kD or less, 15 kD or less, or 20 kD or less.
- the polycationic block of the terpolymer may be a peptide containing a natural or non-natural cationic amino acid as a monomer unit.
- the polycationic block of the terpolymer can be, for example, a peptide containing natural cationic amino acids such as lysine and ornithine as monomer units, such as polylysine or polyornithine.
- the polycationic block of the terpolymer is, for example, a peptide containing, for example, aspartic acid or glutamic acid having a carboxyl group modified with diethylenetriamine or triethylenetetraamine as a monomer unit, such as a homopolymer.
- the average degree of polymerization of the polycationic block can be, for example, 30 or more, 40 or more, 50 or more, 60 or more, or 70 or more.
- the average degree of polymerization of the polycationic block can also be 70 or less, 80 or less, 90 or less, or 100 or less.
- the terpolymer has a hydrophilic block of polyethylene glycol or poly (2-ethyl-2-oxazoline) and a temperature-responsive block of poly (2-n-propyl-2- Oxazoline) and the polycationic block is polylysine or polyornithine.
- the terpolymer has a hydrophilic block of polyethylene glycol, the temperature-responsive block is poly (2-n-propyl-2-oxazoline), and the polycationic block is Polylysine.
- the drug delivery carrier encapsulating the nucleic acid may be a micelle in which the nucleic acid encapsulated by the hydrophobic protective layer, such as the terpolymer, is stabilized.
- the carrier for drug delivery including the nucleic acid also includes vesicles, for example, lipid-based vesicles, for example, liposomes, for example, lipid-based vesicles such as Invivofectamine TM (for example, lipoplexes). ) Can be used. These vesicles preferably have a hydrophilic polymer coating on their surface.
- a carrier whose outer surface is coated with a GLUT1 ligand such as glucose can be used (for details, see WO2015 / 075942A1).
- This carrier improves brain accumulation when administered according to the following regimen. That is, the dosing regimen includes administering the composition to a subject that has been fasted or has induced hypoglycemia and inducing an increase in blood glucose levels in the subject.
- the nucleic acid delivered to the spleen is not particularly limited, and examples thereof include a nucleic acid encoding a factor that enhances the function of the spleen (for example, immune function).
- nucleic acids encoding factors that enhance immune function include nucleic acids encoding peptides and proteins that serve as antigens of immune cells.
- nucleic acids encoding peptides and proteins that serve as antigens of immune cells include, for example, peptide vaccines, particularly peptides used as tumor-specific peptide vaccines (for example, HLA-restricted peptides such as HLA-A24 and HLA-A2 And a nucleic acid encoding a restriction peptide (for example, HLA-A2: 01 and HLA-A24: 01 restriction peptide). Many such peptides have already been developed and many have undergone clinical studies, and those skilled in the art will be able to select them appropriately and use them in the present invention.
- AAV8 for delivery of nucleic acids to the spleen.
- AAV8 in the manufacture of a medicament for use in delivering nucleic acids to the spleen.
- the use of a combination of AAV8 and a multivalent cation for the delivery of nucleic acids to the spleen there is provided the use of a combination of AAV8 and a multivalent cation in the manufacture of a medicament for use in delivering a nucleic acid to the spleen.
- the use of a multivalent cation for use in delivering AAV8 to the spleen there is provided the use of a multivalent cation for use in delivering AAV8 to the spleen.
- a multivalent cation in the manufacture of a medicament for use in delivering AAV8 to the spleen there is provided the use of a medicament for use in delivering AAV8 to the spleen comprising a multivalent cation is provided.
- the heart disease or disorder to be treated includes, for example, myocardial infarction, but is not limited thereto.
- a method of treating these heart diseases or disorders in a subject in need thereof comprising administering AAV used in combination with a multivalent cation of the present invention.
- AAV9 can be used as AAV.
- AAV9 may have a gene encoding a factor having a cardiac regeneration effect such as vascular endothelial growth factor (VEGF).
- VEGF vascular endothelial growth factor
- the use of a combination of AAV9 and a multivalent cation in the manufacture of a medicament for use in delivering a nucleic acid to the heart According to the present invention there is provided the use of AAV9 for delivery of nucleic acids to the heart. According to the present invention there is provided the use of a combination of AAV9 and a multivalent cation in the manufacture of a medicament for use in delivering a nucleic acid to the heart. According to the present invention there is provided the use of a multivalent cation for use in delivering AAV9 to the heart. According to the present invention there is provided the use of a multivalent cation in the manufacture of a medicament for use in delivering AAV9 to the heart. According to the present invention, a medicament for use in delivering AAV9 to the heart comprising a multivalent cation is provided.
- GLUT1 ligand means a substance that specifically binds to GLUT1.
- Various ligands are known as GLUT1 ligands, including, but not limited to, molecules such as glucose and hexose, both of which are used in the present invention for the preparation of carriers or conjugates instead of glucose. be able to.
- the GLUT1 ligand preferably has an affinity for GLUT1 that is equal to or greater than that of glucose.
- inducing hypoglycemia means lowering the blood glucose level in the subject than the blood glucose that should have been shown if the treatment was not performed.
- examples of a method for inducing hypoglycemia include administration of a diabetic drug.
- in inducing hypoglycemia as long as the purpose of inducing hypoglycemia is achieved, for example, taking other drugs or drinking a drink such as water is acceptable.
- Inducing hypoglycemia may involve other treatments that do not substantially affect blood glucose.
- fasting means fasting a subject, for example, 3 hours or more, 4 hours or more, 5 hours or more, 6 hours or more, 7 hours or more, 8 hours or more, 9 hours or more, 10 hours or more, 11 hours or more, 12 hours or more, 13 hours or more, 14 hours or more, 15 hours or more, 16 hours or more, 17 hours or more, 18 hours or more, 19 hours or more, 20 hours or more, 21 hours or more, 22 hours or more, 23 hours Or more, 24 hours or more, 25 hours or more, 26 hours or more, 27 hours or more, 28 hours or more, 29 hours or more, 30 hours or more, 31 hours or more, 32 hours or more, 33 hours or more, 34 hours or more, 35 hours or more, 36 hours or more, 37 hours or more, 38 hours or more, 39 hours or more, 40 hours or more, 41 hours or more, 42 hours or more, 43 hours or more, 44 hours or more, 45 hours or more, 46 hours or more, 47 hours It means to the upper or fasted at least 48 hours.
- the fasting period is determined by a doctor or the like in view of the health condition of the subject. For example, it is preferable that the fasting period be a period of time longer than the time for the subject to reach fasting blood glucose.
- the fasting period may be, for example, a period of time beyond which the expression of GLUT1 on the intravascular surface of cerebral vascular endothelial cells increases or reaches a plateau.
- the fasting period can be, for example, the above period that is 12 hours or more, 24 hours or more, or 36 hours or more. Fasting may also involve other treatments that do not substantially affect blood glucose levels or the expression of GLUT1 on the intravascular surface.
- “inducing an increase in blood glucose level” means increasing the blood glucose level in a subject in which hypoglycemia is induced or in a subject in which a hypoglycemic state is maintained.
- the blood glucose level can be increased by various methods well known to those skilled in the art.
- administration of an agent that induces an increase in blood glucose level for example, an increase in blood glucose level such as glucose, fructose (fructose), galactose, etc.
- administration of a simple sugar administration of a polysaccharide that induces an increase in blood sugar level such as maltose, intake of a carbohydrate that induces an increase in blood sugar level such as starch, or diet.
- blood glucose manipulation refers to inducing hypoglycemia in a subject and then raising the blood glucose level. After inducing hypoglycemia in the subject, the subject's blood glucose level can be maintained at the hypoglycemia.
- the time for maintaining the subject's blood sugar level at low blood sugar is, for example, 0 hour or more, 1 hour or more, 2 hours or more, 3 hours or more, 4 hours or more, 5 hours or more, 6 hours or more, 7 hours or more, 8 hours or more 9 hours or more, 10 hours or more, 11 hours or more, 12 hours or more, 13 hours or more, 14 hours or more, 15 hours or more, 16 hours or more, 17 hours or more, 18 hours or more, 19 hours or more, 20 hours or more, 21 More than time, more than 22 hours, more than 23 hours, more than 24 hours, more than 25 hours, more than 26 hours, more than 27 hours, more than 28 hours, more than 29 hours, more than 30 hours, more than 31 hours, more than 32 hours, more than 33 hours 34 hours or more, 35 hours or more, 36 hours or more, 37 hours or more, 38 hours or more, 39 hours or more, 40 hours or more, 41 hours or more, 42 hours or more, 43 hours or more, 44 hours or more, 45 hours or less , 46 hours or more
- the blood sugar level can be raised.
- “maintaining blood glucose” is permitted to take other drugs or drink beverages such as water, for example, as long as the purpose of maintaining hypoglycemia in the subject is achieved. Inducing hypoglycemia may involve other treatments that do not substantially affect blood glucose.
- the composition may be administered to the subject simultaneously, sequentially or sequentially with the induction of an increase in blood glucose level in the subject.
- the dosing schedule may or may not have an interval between administration of the composition to the subject and induction of an increase in blood glucose level in the subject.
- the composition When the composition is administered concurrently with inducing an increase in blood glucose level in the subject, the composition may be administered to the subject in a form mixed with an agent that causes an increase in blood glucose level.
- the agent may be administered in a form other than the drug that induces an increase in blood glucose level in the subject.
- the composition also induces an increase in blood glucose level in the subject and, if administered continuously or sequentially to the subject, the composition precedes the induction of an increase in blood glucose level in the subject.
- the composition can be administered to the subject prior to inducing an increase in blood glucose levels in the subject.
- the composition is administered to the subject within 15 minutes, within 10 minutes.
- the composition is administered to the subject within 6 hours, within 4 hours, within 2 hours.
- the above regimen cycle may be performed more than once.
- the context of glucose administration and sample administration can be determined by the timing of passage through the blood brain barrier.
- the micelle in which the nucleic acid encapsulated by the hydrophobic protective layer, such as the terpolymer of the present invention is stabilized can be used for delivery to cerebral vascular endothelial cells.
- the role of glucose in the present invention is also the same in the blood nerve barrier, blood retinal barrier and blood cerebrospinal fluid barrier.
- GLUT1 is also expressed in vascular endothelial cells during hypoglycemia at the blood nerve barrier, blood retina barrier and blood cerebrospinal fluid barrier.
- the above-described ternary copolymer or the like of the present invention which is a stabilized micelle of a nucleic acid encapsulated by a hydrophobic protective layer, is used to pass through the blood nerve barrier, blood retinal barrier and blood cerebrospinal fluid barrier.
- Micelles stabilized by nucleic acid encapsulated by a hydrophobic protective layer, such as the above terpolymers of the present invention are also delivered to vascular endothelial cells present at the blood nerve barrier, blood retinal barrier and blood cerebrospinal fluid barrier It can also be used to do.
- Example 1 Effect of multivalent cation administration on accumulation of drug delivery micelles in liver
- micelles having glucose modification on the surface of triblock polymer micelles of PEG (molecular weight 11k) -b-PnPrOx (molecular weight 8k) -b-PLys (polymerization degree 43) were used.
- Surface glucose modification is a modification for facilitating delivery of micelles to the brain.
- glucose-modified micelles are administered to a fasted subject, and the blood glucose level of the subject before and after this is administered. As the blood glucose level rises, the micelles are remarkably taken up by the brain.
- DIG-PEG glucose derivative DIG at the PEG side end
- a copolymer of DIG-PEG, PnPrOx, and PLys was obtained by linking PnPrOx-b-PLys synthesized according to the description in European Polymer Journal, 2017, 88, 553-561 and Glc-PEG.
- the synthesis scheme of the triblock copolymer was as shown in Schemes 1 to 4 below.
- Alkyne-PnPrOx-b-PLys was synthesized as follows. First, the polymerization initiator propargyl p-toluenesulfonate (61 mg, 0.29 mmol) was dissolved in 7 mL of acetonitrile, and 2-n-propyl-2-oxazoline (2.5 g, 22 mmol) was added. After reacting the reaction solution at 42 ° C.
- Acetonitrile as a reaction solvent was purchased from wako, and used after being purified by distillation using calcium hydride as a dehydrating agent. Sodium azide was purchased from Wako and used as it was. After the polymerization reaction, the reaction solution was dialyzed 5 times against water and freeze-dried to obtain alkyne-PnPrOx-N 3 having an azide end. The obtained PnPrOx was found to have a molecular weight of 8.3 k from analysis by MALDI-TOF MS (UltraFlextreme, Bruker) and 1 H-NMR (ESC400, JEOL).
- alkyne-PnPrOx-N 3 was converted to the amine end by the Staudinger reaction.
- alkyne-PnPrOx-N 3 (830 mg, 0.10 mmol) was dissolved in 20 mL of methanol, triphenylphosphine (530 mg, 2.0 mmol) was added, and the mixture was stirred at 40 ° C. for 3 hours.
- 20 mL of pure water was added and ice-cooled, and unreacted triphenylphosphine and the like were removed by filtration. The obtained solution was dialyzed 3 times against pure water and then recovered by freeze-drying.
- alkyne-PnPrOx-NH 2 was further purified using an open column using CM-Sephadex C50 purchased from GE-health care as a column filler. Methanol and triphenylphosphine used here were purchased from Sigma-Aldrich and Tokyo Kasei, respectively.
- Lys (TFA) -NCA was polymerized from alkyne-PnPrOx-NH 2 purified and recovered as described above, and then alkyne-PnPrOx-b-PLys was obtained by deprotecting the TFA group with a base.
- Alkyne-PnPrOx-NH 2 (330 mg, 0.040 mmol) was lyophilized using dioxane.
- Lys (TFA) -NCA 480 mg, 1.8 mmol was measured in a flask in an Ar bag and dissolved in 6 mL of DMF (1M TU). The prepared Lys (TFA) -NCA solution was added to the initiator solution under an Ar atmosphere, and the polymerization reaction was carried out by stirring at 25 ° C. for 3 days.
- the reaction solution was dialyzed 5 times against water and then freeze-dried to obtain PnPrOx-b-Poly (L-Lysine) (TFA) (PLys (TFA)). Subsequently, 500 mg of the obtained alkyne-PnPrOx-b-PLys (TFA) was measured and dissolved in 25 mL of methanol. To this, 7.5 mL of 1M NaOH solution was added and reacted at 35 ° C. for 12 hours. After the reaction, dialysis was performed 3 times against 0.01 M HCl and then 3 times against water, and PnPrOx-b-PLys was recovered by lyophilization.
- the recovered product was confirmed to have a unimodal molecular weight distribution by SEC (AKTAexplorer, GE Helthcere) using GE Healthcare column Superdex 200.
- the obtained polymer was found to have Lys of 43 from analysis by 1 H-NMR (ESC400, JEOL).
- Lys (TFA) -NCA was synthesized by the Fuchs-Farthing method according to non-patent literature (J. Polym. Sci. Part A Polym. Chem. 2003, 41 1167-1187).
- Dioxane was purchased from Wako.
- DMF (1M TU) was prepared by dissolving thiourea purchased from Sigma-Aldrich in a dehydrated solvent purchased from Kanto Chemical.
- a 1M NaOH solution was prepared by diluting a 5M NaOH solution purchased from Nakarai with pure water. 0.01 M HCl was prepared by diluting concentrated hydrochloric acid purchased from Inusha with pure water.
- the OH terminal of DIG-PEG-OH was converted to an azide group.
- DIG-PEG-OH 550 mg, 0.050 mmol was lyophilized with benzene. This was dissolved in 20 mL of THF, and triethylamine (25 ⁇ L, 0.20 mmol) was added to form a PEG solution.
- methanesulfonyl chloride (16 ⁇ L, 0.20 mmol) was measured and diluted with 5 mL of THF. Under water cooling, the above-prepared PEG solution was added to this methanesulfonyl chloride diluted solution and allowed to react overnight. The reaction was performed in an Ar atmosphere. The reaction product was recovered by reprecipitation with 500 mL of diethyl ether. From the analysis by 1 H-NMR (ESC400, JEOL), it was found that the collected sample was DIG-PEG-Ms whose end was mesylated.
- DIG-PEG-Ms (440 mg, 0.044 mmol) was dissolved in 20 mL of DMF, sodium azide (286 mg, 4.4 mmol) was added, and the mixture was stirred at 50 ° C. for 3 days.
- ultra-dehydrated solvent purchased from Kanto Chemical was used.
- Triethylamine was purchased from Wako and used after being purified by distillation using calcium hydride as a dehydrating agent.
- Mesyl chloride was purchased from Nacalai and distilled using diphosphorus pentoxide as a dehydrating agent.
- Sodium azide purchased from Wako was used as it was.
- Alkyne-PnPrOx-PLys and DIG-PEG-N 3 were coupled by Click Chemistry.
- Alkyne-PnPrOx-PLys 31 mg, 0.0020 mmol was dissolved in 2 mL of water, and 20 ⁇ L each of 1M CuSO 4 solution and 1M sodium ascorbate solution were added and stirred.
- DIG-PEG-N 3 110 mg, 0.01 mmol was dissolved in 2 mL of water and added to the Alkyne-PnPrOx-PLys solution. This was left at -20 ° C overnight and melted at 4 ° C over 2 hours.
- the product was recovered by dialyzing the reaction solution 5 times against water and lyophilizing.
- the recovered material contains Glc-PEG-N 3 added in excess to react all of the Alkyne-PnPrOx-PLys and unreacted Alkyne-PnPrOx-PLys. . Focusing on the difference in molecular weight between these unreacted products and the resulting triblock copolymer, purification was performed by SEC. Finally, as described in WO2015075942A1, DIG was converted to glucose using TFA to obtain Glc-PEG-b-PnPrOx-b-PLys.
- a cationic polymer was synthesized in which hydrophilic blocks were connected to improve biocompatibility.
- branched poly (ethylene glycol) -b-poly (L-lysine) polymerization degree 20
- PEG-b-PLL branched poly (ethylene glycol) -b-poly (L-lysine)
- PEG-b-PLL block cationomer having branched PEG (molecular weight 37000 ⁇ 2)
- Non-patent literature J. Polym. Sci. Part A Polym. Chem.
- branched polyethylene glycol (molecular weight 37000 ⁇ 2) with a primary amine structure purchased from NOF as an initiator.
- bPEG branched polyethylene glycol
- -1187 is obtained by polymerizing Lysine (TFA) -NCA synthesized by the Fuchs-Farthing method and then treating with a base.
- bPEG-PLL first, bPEG having a primary amine (740 mg, 0.010 mmol) was freeze-dried with benzene purchased from Nacalai.
- DMF (1M TU) prepared by dissolving thiourea (TU) purchased from Sigma Aldrich in dehydrated N, N-dimethylformamide (DMF) purchased from Kanto Chemical Co. as a reaction solvent.
- Lyophilized bPEG was dissolved in 10 mL DMF (1M TU).
- Lys (TFA) -NCA was measured in a 59 mg (0.22 mmol) flask in an Ar bag and dissolved in 2 mL of DMF.
- the prepared Lys (TFA) -NCA solution was added to the bPEG solution under an Ar atmosphere and stirred at 25 ° C. for 3 days.
- the reaction solution was titrated against 300 mL of a 1: 9 mixed solution of methanol (Sigma Aldrich) and diethyl ether (Showa Ether) to reprecipitate the copolymer bPEG-PLL (TFA).
- the resulting bPEG-PLL (TFA) was collected by filtration and dried in vacuo. 500 mg of the obtained bPEG-PLL (TFA) was measured and dissolved in 25 mL of methanol. Subsequently, 7.5 mL of 1M NaOH was added to this bPEG-PLL (TFA) solution and stirred at 35 ° C. for 12 hours to deprotect the TFA group.
- bPEG-PLL was recovered by lyophilization.
- SEC LC2000 system, JASCO Corporation
- 1 H-NMR ESC400, JEOL
- Polyplex micelles were formed from the obtained triblock polymer and mRNA. Specifically, the mixture was mixed with mRNA at a charge ratio of 2 at 4 ° C., and then allowed to stand at 37 ° C. to form a hydrophobic layer so as to wrap the mRNA in the triblock polymer. Polyplex micelles having a protective layer for mRNA were obtained.
- mRNA is prepared by in vitro transcription of template DNA using mMESSAGE mMACHINE T7 Ultra Kit (Ambion) and poly (A) modification using poly (A) tail kit (Ambion) did.
- template DNA pCMV-Gluc control plasmid purchased from New England Biolabs was used.
- pDNA pCAG-Luc2 encoded by luciferase provided by RIKEN BioResource Center and having a CAG promoter was used.
- the whole collected organ is ground using Lysis buffer (promega) and multi-bead shocker (Yasui Kikai), and Cy5 fluorescence of this suspension is measured with a plate reader (Infinite M100 Pro, TECAN).
- the organ distribution was evaluated. The total dose was 100%, and the amount (%) accumulated in the organ was determined. The results were as shown in FIGS.
- the amount of mRNA accumulated in the liver was less than 30% in the micelle-administered group, whereas the amount accumulated in the polyvalent cation-administered group was about 10%. It was shown that the amount accumulated in the liver was reduced to less than half. As shown in FIG. 1, the amount of mRNA accumulated in the kidney was about 7% in the micelle-administered group, but about 5% in the polyvalent cation-administered group. It has been shown that the amount accumulated in has decreased to about 70%.
- the accumulated amount of mRNA in other organs such as the spleen and the heart was evaluated, as shown in FIG. 2A, in the polyvalent cation administration group, the accumulated amount of mRNA in the spleen was also accumulated in the heart. The amount also increased significantly.
- the decrease in the amount of mRNA accumulated in the liver and kidney was related to the excretion ability of mRNA from the blood in the liver and kidney. Therefore, the polyvalent cation has the effect of decreasing the mRNA excretion ability from the blood in the liver and kidney, thereby increasing the distribution of mRNA, that is, the drug to organs other than the liver and kidney. Became clear.
- blood glucose manipulation fasting and glucose administration
- micelle coating with glucose were performed to promote selective accumulation in the brain. Therefore, in such a system aimed at selective delivery of mRNA, a decrease in the excretion ability of mRNA and an increase in retention in blood enhanced the accumulation of selective mRNA in the brain.
- Invivofectamine TM is a reagent for systemic administration of lipid-based mRNA and is particularly suitable for delivery of mRNA to the liver.
- Invivofectamine TM and mRNA were mixed according to the manufacturer's protocol to form a lipid-based mRNA complex.
- the mRNA used in this experiment was fluorescently labeled in advance with Label IT Tracker Cy5 Kit (Mirus Bio).
- mice were intravenously administered with 100 ⁇ L of a PEGasus-PLL solution adjusted to a concentration of 12.5 mg / mL. Five minutes later, 200 ⁇ L of the in vivofectamine solution prepared above was administered iv. After 30 minutes, whole blood was collected and perfused with PBS, and then the mouse was dissected to remove organs (liver and kidney). As a negative control group for observing the effect of polymer addition, mice were compared with a group in which 200 ⁇ L of the solution prepared above was intravenously administered without prior administration of the polymer additive.
- the collected organs were ground using Lysis buffer (promega) and multi-bead shocker (Yasui Kikai), and the Cy5 fluorescence of this suspension was measured with a plate reader (Infinite M100 Pro, TECAN). Distribution was evaluated.
- the amount of mRNA accumulated in other organs such as the spleen and heart was evaluated. As shown in FIG. 3A, the amount of mRNA accumulated in the spleen was also increased in the spleen in the polyvalent cation administration group. The amount of mRNA accumulated in the heart and the amount of mRNA accumulated in the heart increased significantly. From this, it was suggested that the delivery of mRNA, that is, a drug to various organs was promoted by the decrease in the ability to excrete mRNA from blood.
- Example 2 Pharmacokinetics of polyvalent cations
- the pharmacokinetics of polyvalent cations were elucidated in order to investigate the cause of the above function.
- mice were intravenously injected with 100 ⁇ L of PEGasus-PLL (37 ⁇ 2-20) at a concentration of 12.5 ⁇ mg / mL, which was fluorescently labeled using Alexa Fluor 594-NHS Ester purchased from Thermo-Fisher Scientific. Thereafter, the blood vessels of the liver were observed with an in vivo confocal microscope. Intravascular polymer accumulation can be assessed by fluorescence.
- an ear blood vessel was observed as an example of a peripheral blood vessel.
- FIG. 5 it was confirmed that the polyvalent cation flows in the blood vessel, but accumulation of the ear vascular endothelial cells on the inner surface of the blood vessel was not observed.
- liver and kidney are organs that greatly affect the ability of drugs to be excreted from the blood in pharmacokinetics.
- Multivalent cations adsorb to the surface of similar endothelial cells and physically coat the inner surface of blood vessels to reduce the excretion of blood from similar endothelial cells of the liver and kidneys, and drugs from the blood It was found that it has an action of lowering the excretion ability.
- This coating was also specific for liver homologous endothelial cells and kidney vascular endothelial cells.
- Example 3 Effect of Improving Micellar Retention in Blood by Multivalent Cations
- the polyvalent cation has an action of decreasing the ability of the liver to excrete drugs from the blood by similar endothelial cells and kidneys. It has been made clear. In this example, it was confirmed whether the decrease in the excretion ability contributes to the improvement of the retention of the drug in the blood.
- mice were intravenously administered with 100 ⁇ L of a PEGasus-PLL solution adjusted to a concentration of 12.5 mg / mL as a polyvalent cation.
- 200 ⁇ L of plasmid DNA (pDNA) -encapsulating polymer micelle solution prepared by the method of Biomaterial, 2017, 126, 31-38 was administered.
- the pDNA used in this experiment was fluorescently labeled in advance with Label IT Tracker Cy5 Kit (Mirus Bio). Thereafter, the retention of pDNA in blood was confirmed. The maximum value of fluorescence intensity was set to 100%, and the retention of pDNA in blood was evaluated by the change in fluorescence intensity over time.
- As a control group a group in which 200 ⁇ L of the solution prepared above was intravenously administered without prior administration of the polyvalent cation to mice was also observed.
- the cause of the decrease in the drug excretion ability from the blood observed in Examples 1 and 2 is that the polyvalent cation coats the inner surface of the blood vessel of the liver's similar endothelial cells. It was considered that the accumulation of the drug in the cells was inhibited, and that the excretion of the drug via the glomeruli was inhibited by covering the inner surface of the vascular endothelial cells of the kidney with the cationic polymer. In this way, the polyvalent cation has the effect of reducing the ability of the drug to be excreted from the blood and improving its retention in the blood, even when administered alone (ie, in free form). Thus, it was considered that the amount of drug delivered to the target organ could be improved.
- the interaction between the polyvalent cation and albumin was confirmed. Specifically, the amount of heat generated when albumin and PEGasus-PLL were mixed was measured using Malvern's isothermal titration calorimeter, Microcal® PEAQ-ITC. 200 ⁇ L of an albumin solution in which albumin was dissolved in PBS at a concentration of 33.3 ⁇ M was prepared as a cell side sample, and a PEGasus-PLL solution in which PEGasus-PLL was dissolved in PBS at a concentration of 313 ⁇ M was prepared as a syringe side sample.
- Example 4 Improvement of gene expression in spleen of lipoplex by polyvalent cations
- Invivofectamine TM is commercially available from Invitrogen as a reagent for delivering RNA to the liver.
- Invivofectamine TM is a reagent for systemic administration of lipid-based mRNA, and is said to be particularly suitable for delivery of mRNA to the liver.
- Invivofectamine TM and mRNA were mixed according to the manufacturer's protocol to form a lipid-based mRNA complex (lipoplex).
- Mice were intravenously administered with 100 ⁇ L of PEGasus-PLL solution adjusted to a concentration of 12.5 mg / mL as a polyvalent cation. Five minutes later, 200 ⁇ L of the in vivofectamine solution prepared above was administered iv.
- mice were compared with a group in which 200 ⁇ L of the solution prepared above was administered intravenously without prior administration of a polyvalent cation.
- the collected organ was ground using a Lysis buffer (promega) and a multi-bead shocker (Yasui Kikai). From this suspension, the Luciferase assay kit purchased from promega was used according to the manufacturer's protocol, and the luciferase luminescence was measured with a luminometer (Lumat LB9507, Berthold) to evaluate the gene expression level in the liver and spleen did. The result was as shown in FIG.
- gene expression in the spleen of Lipoplex could be increased 100 times or more by adding polyvalent cations.
- the addition of a polyvalent cation suppresses the uptake of the drug by the liver's similar endothelial cells and the kidney's endothelial cells and enhances the drug's retention in the blood, thereby allowing the drug to be used in organs other than the liver and kidney. Proved to increase the accumulation of.
- FIG. 9 and FIG. 3A is that in FIG. 3A, the amount of Cy5 fluorescence is evaluated, whereas in FIG. 9, the expression level of the administered mRNA is evaluated.
- mRNA can be effectively delivered to the spleen.
- the spleen is an organ from which immune cells are produced, and achieving delivery and expression of mRNA into the spleen is a major achievement that leads to the development of vaccines such as RNA vaccines and peptide vaccines.
- Example 5 Discharge characteristics of bPEG-PLL from the body In this example, the discharge of polyvalent cations from the body was examined.
- bPEG-PLL (TFA) and PEG-PLL (TFA) were converted to bPEG-PLL and PEG-PLL fluorescently labeled by deprotecting the TFA protecting group with a base as described above.
- bPEG-PLL and PEG-PLL to which lysine having a polymerization degree of 20 which is a polyvalent cation is bound, have a higher blood retention than bPEG having no cationic block.
- bPEG-PLL it was almost excreted from the blood after 10 hours (FIG. 10). From this, it was suggested that the cationic block has an action of reducing the blood retention of bPEG.
- bPEG-PLL when administered alone in free form, coats the luminal inner wall of endothelial cells for a sufficient amount of time to deliver the drug while reducing drug clearance. It was thought that it could disappear after a while.
- the polyvalent cation of the present invention is used in a single time so that the clearance of the administered drug is reduced by a necessary time together with (or before and after) the administration time of the drug. It was suggested that it could be administered or administered multiple times.
- the polyvalent cation of the present invention can be administered in such a manner that it is excreted from the body after the drug is delivered (administration frequency, dosage, etc.). It has been suggested.
- Example 6 Delivery Experiment Using Virus Particles
- the effect of multivalent cation administration on gene delivery by virus particles was examined.
- AAV8 adeno-associated virus
- AAV8 was purchased from Vector Biolabs with a gene encoding firefly luciferase under the CMV promoter (Catalog #: VB1473).
- AAV8 (1 ⁇ 10 10 viral genome / mouse) was administered from the tail vein of a mouse (Balb / c, female, 6 weeks old). Twenty-one days after administration, mice were sacrificed and organ (liver, pancreas, quadriceps, thigh) tissues were collected, and 100 mg of tissue was homogenized in 1 mL of passive lysis buffer.
- the homogenate was centrifuged at 18,000 g for 10 minutes at 4 ° C., and luminescence from luciferase was detected in 20 ⁇ L of the supernatant.
- a BCA assay was performed to determine protein concentration. Luciferase activity was expressed in relative optical units (RLU) normalized to protein content. The result was as shown in FIG.
- the polyvalent cation decreased the luciferase introduction efficiency into the liver by AAV8 and enhanced the luciferase expression in the pancreas, quadriceps, and thigh tissues. It is assumed that the accumulation in the liver was decreased and the accumulation in multiple organs was increased. From this, it was confirmed that the effect of changing the kinetics of the administered drug and reducing the clearance by the polycation of the present invention is also exerted on the virus particles.
- AAV9 capable of carrying a gene to the myocardium was used instead of AAV8.
- AAV9 was purchased from SignaGen Laboratories (Catalog #: SL101494) with a gene encoding firefly luciferase under the CMV promoter.
- the dose was 5 ⁇ 10 10 viral genome / mouse, and the mice were sacrificed 2 days after administration.
- the polyvalent cation decreased the accumulation of AAV9 in the liver and decreased the gene transfer efficiency into the liver, while increasing the accumulation in the heart, Increased gene transfer efficiency to the heart.
- the cationic polymer blocks the surface of the liver's homologous endothelial cells. This will reduce the clearance in the liver of particulate matter such as virus particles, not limited to micelles, and as a result, or at the same time, enhance the organ or tissue specificity of the virus itself It became clear to do.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Dispersion Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Emergency Medicine (AREA)
- Nanotechnology (AREA)
- Dermatology (AREA)
- Biophysics (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
本発明は、薬物動態を制御する組成物を提供する。具体的には、本発明では、多価カチオンを有効成分として含む、薬物動態を制御するための組成物、および多価カチオンを用いる薬物動態を制御する方法が提供される。
Description
本発明は、体内における薬物動態を制御する組成物(例えば、薬物の分布を変更し、薬物の代謝を抑制し、または薬物の排泄を抑制するための組成物)、特に、肝臓の類同内皮細胞による薬剤(例えば、医薬活性成分や薬剤送達用のキャリア)の血中からのクリアランスの抑制剤に関する。本発明はまた、腎臓による薬物の血中からのクリアランスの抑制剤に関する。本発明はまた、脾臓へ薬物送達量を増加させるための組成物に関する。
体内での薬物動態は、医薬品開発において重要な意味を有する。例えば、血中薬物濃度が、医薬品の効果を発揮させるために重要であることは言うまでも無い。血中薬物濃度は、薬物の吸収、分布、代謝、排泄の4つの過程によって決定される。特に、薬物の代謝および薬物の排泄(クリアランス)は、血中薬物濃度の重要な決定因子である。
ところで、プラスミドDNAの血中安定性を高める技術として、親水性ブロック-温度応答性ブロック-ポリカチオン性ブロックからなる三元共重合体が開発されている(非特許文献1)。この三元共重合体は、プラスミドDNAと低温で混合するとミセルを形成する。このミセルでは、プラスミドDNAは前記三元共重合体のポリカチオン性ブロックと複合体を形成する。その後、温度を上昇させると、温度応答性ブロックが親水性から疎水性に変化し、これによりプラスミドDNAを覆う疎水性の中間層を形成させ、これをDNAの保護層として用いる技術である。しかし、mRNAの血中安定性が大きく改善されたミセル製剤技術はいまだ知られていない。また、親水性セグメントとカチオン性セグメントとの共重合体と核酸を、電荷を中和するように複合体とする方法が開示されている(特許文献1)。
Osawa S. et al., Biomacromolecule, 17(1):354-361, 2016
本発明は、薬物動態を制御するための組成物(例えば、薬物の分布を変更し、薬物の代謝を抑制し、または薬物の排泄を抑制するための組成物)、特に、肝臓の類同内皮細胞による薬剤(例えば、医薬活性成分や薬剤送達用のキャリア)の排泄能の抑制剤を提供する。本発明はまた、腎臓による薬物の血中からの排泄能の抑制剤を提供する。
本発明者らは、多価カチオン(特に、生体適合性を高めるためにポリエチレングリコールによって修飾した多価カチオン)が、肝臓の類同内皮細胞の血管内面および腎臓の血管内皮の血管内面に局在すること、および、血中からの薬物の排泄能を低減させることを見出した。本発明者らはさらにまた、多価カチオンが脾臓を含む様々な臓器または組織への薬物送達量を増加させることを見出した。
本発明は、このような知見に基づくものである。
本発明は、このような知見に基づくものである。
すなわち、本発明によれば、以下の発明が提供される。
(1A)多価カチオンを有効成分として含む、体内の薬物動態を制御することに用いるための組成物{ここで、多価カチオンはカチオン性ポリマーであってもよい}。
(2A)多価カチオンが、親水性ポリマーブロックとの結合体の形態である、上記(1A)に記載の組成物。
(3A)多価カチオンが、2本以上の親水性ポリマー鎖を有するカチオン性ポリマーである、上記(1A)に記載の組成物。
(4A)多価カチオンが、カチオン性ポリマーブロックと分岐ポリエチレングリコールとのブロック共重合体である、上記(2A)に記載の組成物。
(5A)薬物動態の制御が、血中からのクリアランスの制御である、上記(1A)~(4A)のいずれかに記載の組成物。
(6A)薬物動態の制御が、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減である、上記(1A)~(5A)のいずれかに記載の組成物。
(7A)薬物動態の制御が、腎臓による血中からの薬物の排泄能の低減である、上記(1A)~(5A)のいずれかに記載の組成物。
(8A)薬物動態の制御が、標的臓器または組織への送達量の増加である、上記(1A)~(6A)のいずれかに記載の組成物。
(9A)薬物動態の制御が、脾臓への送達量の増加である、上記(1A)~(6A)のいずれかに記載の組成物。
(10A)薬物動態の制御が、薬物の血中滞留性の増大である、上記(1A)~(4A)のいずれかに記載の組成物。
(11A)動態が制御される薬物よりも先に投与される、上記(1A)~(10A)のいずれかに記載の組成物。
(12A)動態が制御される薬物と同時に投与される、上記(1A)~(10A)のいずれかに記載の組成物。
(13A)動態が制御される薬物よりも後に投与される、上記(1A)~(10A)のいずれかに記載の組成物{但し、前記投与は、前記薬物が血中に滞留している間に行われることを条件とする}。
(14A)多価カチオンが、遊離形態である、または遊離形態で投与される、上記(1A)~(13A)のいずれかに記載の組成物。
(15A)動態が制御される薬物が、薬物送達用キャリアに内包された薬物である、上記(1A)~(14A)のいずれかに記載の組成物。
(16A)(1A)~(15A)のいずれかに記載の組成物を使用することを特徴とする薬物動態の制御方法。
(2A)多価カチオンが、親水性ポリマーブロックとの結合体の形態である、上記(1A)に記載の組成物。
(3A)多価カチオンが、2本以上の親水性ポリマー鎖を有するカチオン性ポリマーである、上記(1A)に記載の組成物。
(4A)多価カチオンが、カチオン性ポリマーブロックと分岐ポリエチレングリコールとのブロック共重合体である、上記(2A)に記載の組成物。
(5A)薬物動態の制御が、血中からのクリアランスの制御である、上記(1A)~(4A)のいずれかに記載の組成物。
(6A)薬物動態の制御が、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減である、上記(1A)~(5A)のいずれかに記載の組成物。
(7A)薬物動態の制御が、腎臓による血中からの薬物の排泄能の低減である、上記(1A)~(5A)のいずれかに記載の組成物。
(8A)薬物動態の制御が、標的臓器または組織への送達量の増加である、上記(1A)~(6A)のいずれかに記載の組成物。
(9A)薬物動態の制御が、脾臓への送達量の増加である、上記(1A)~(6A)のいずれかに記載の組成物。
(10A)薬物動態の制御が、薬物の血中滞留性の増大である、上記(1A)~(4A)のいずれかに記載の組成物。
(11A)動態が制御される薬物よりも先に投与される、上記(1A)~(10A)のいずれかに記載の組成物。
(12A)動態が制御される薬物と同時に投与される、上記(1A)~(10A)のいずれかに記載の組成物。
(13A)動態が制御される薬物よりも後に投与される、上記(1A)~(10A)のいずれかに記載の組成物{但し、前記投与は、前記薬物が血中に滞留している間に行われることを条件とする}。
(14A)多価カチオンが、遊離形態である、または遊離形態で投与される、上記(1A)~(13A)のいずれかに記載の組成物。
(15A)動態が制御される薬物が、薬物送達用キャリアに内包された薬物である、上記(1A)~(14A)のいずれかに記載の組成物。
(16A)(1A)~(15A)のいずれかに記載の組成物を使用することを特徴とする薬物動態の制御方法。
本発明によればまた、以下の発明が提供される。
(1B)カチオン性ポリマーを有効成分として含む、体内の薬物動態を制御することに用いるための組成物。
(2B)カチオン性ポリマーが、親水性ポリマーブロックとの結合体(例えば、共重合体)の形態である、上記(1B)に記載の組成物。
(3B)カチオン性ポリマーが、2本以上の親水性ポリマー鎖を有するカチオン性ポリマーである、上記(1B)に記載の組成物。
(4B)カチオン性ポリマーが、カチオン性ポリマーブロックと分岐ポリエチレングリコールとのブロック共重合体である、上記(2B)に記載の組成物。
(5B)薬物動態の制御が、血中からのクリアランスの制御である、上記(1B)~(4B)のいずれかに記載の組成物。
(6B)薬物動態の制御が、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減である、上記(1B)~(5B)のいずれかに記載の組成物。
(7B)薬物動態の制御が、腎臓による血中からの薬物の排泄能の低減である、上記(1B)~(5B)のいずれかに記載の組成物。
(8B)薬物動態の制御が、標的臓器または組織への送達量の増加である、上記(1B)~(6B)のいずれかに記載の組成物。
(9B)薬物動態の制御が、脾臓への送達量の増加である、上記(1B)~(6B)のい
ずれかに記載の組成物。
(10B)薬物動態の制御が、薬物の血中滞留性の増大である、上記(1B)~(4B)のいずれかに記載の組成物。
(11B)動態が制御される薬物よりも先に投与される、上記(1B)~(10B)のいずれかに記載の組成物。
(12B)動態が制御される薬物と同時に投与される、上記(1B)~(10B)のいずれかに記載の組成物。
(13B)動態が制御される薬物よりも後に投与される、上記(1B)~(10B)のいずれかに記載の組成物{但し、前記投与は、前記薬物が血中に滞留している間に行われることを条件とする}。
(14B)カチオン性ポリマーが、遊離形態である、または遊離形態で投与される、上記(1B)~(13B)のいずれかに記載の組成物。
(15B)動態が制御される薬物が、薬物送達用キャリアに内包された薬物である、上記(1B)~(14B)のいずれかに記載の組成物。
(16B)(1B)~(15B)のいずれかに記載の組成物を使用することを特徴とする薬物動態の制御方法。
(1B)カチオン性ポリマーを有効成分として含む、体内の薬物動態を制御することに用いるための組成物。
(2B)カチオン性ポリマーが、親水性ポリマーブロックとの結合体(例えば、共重合体)の形態である、上記(1B)に記載の組成物。
(3B)カチオン性ポリマーが、2本以上の親水性ポリマー鎖を有するカチオン性ポリマーである、上記(1B)に記載の組成物。
(4B)カチオン性ポリマーが、カチオン性ポリマーブロックと分岐ポリエチレングリコールとのブロック共重合体である、上記(2B)に記載の組成物。
(5B)薬物動態の制御が、血中からのクリアランスの制御である、上記(1B)~(4B)のいずれかに記載の組成物。
(6B)薬物動態の制御が、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減である、上記(1B)~(5B)のいずれかに記載の組成物。
(7B)薬物動態の制御が、腎臓による血中からの薬物の排泄能の低減である、上記(1B)~(5B)のいずれかに記載の組成物。
(8B)薬物動態の制御が、標的臓器または組織への送達量の増加である、上記(1B)~(6B)のいずれかに記載の組成物。
(9B)薬物動態の制御が、脾臓への送達量の増加である、上記(1B)~(6B)のい
ずれかに記載の組成物。
(10B)薬物動態の制御が、薬物の血中滞留性の増大である、上記(1B)~(4B)のいずれかに記載の組成物。
(11B)動態が制御される薬物よりも先に投与される、上記(1B)~(10B)のいずれかに記載の組成物。
(12B)動態が制御される薬物と同時に投与される、上記(1B)~(10B)のいずれかに記載の組成物。
(13B)動態が制御される薬物よりも後に投与される、上記(1B)~(10B)のいずれかに記載の組成物{但し、前記投与は、前記薬物が血中に滞留している間に行われることを条件とする}。
(14B)カチオン性ポリマーが、遊離形態である、または遊離形態で投与される、上記(1B)~(13B)のいずれかに記載の組成物。
(15B)動態が制御される薬物が、薬物送達用キャリアに内包された薬物である、上記(1B)~(14B)のいずれかに記載の組成物。
(16B)(1B)~(15B)のいずれかに記載の組成物を使用することを特徴とする薬物動態の制御方法。
本明細書では、「対象」とは、ヒトを含む哺乳動物である。対象は、健常の対象であってもよいし、何らかの疾患に罹患した対象であってもよい。対象は、哺乳動物、例えば、ヒトであり、特に、本発明のミセルの投与が有益である哺乳動物、例えば、ヒトであり得る。
本明細書では、「薬剤送達用の」とは、生体適合性であること、および、薬剤をキャリアに内包できることを意味する。本明細書では、「薬剤送達用の」とは、薬剤の血中残存時間を、裸の薬剤の血中残存時間と比べて長期化する用途、または、所定の組織への薬剤の送達量を向上させる用途を意味することがある。
本明細書では、「キャリア」は、物質を内包できる微粒子や中空微粒子を指す。キャリアは、好ましくは生体適合性の外殻または修飾を有する。キャリアとしては、特に限定されないが例えば、リポソームおよびミセルが挙げられ、特にリン脂質等によって構成されるリポソームが挙げられる。
本明細書では、「ミセル」とは、高分子などの分子が集合して形成されるキャリアを意味する。ミセルとしては、界面活性剤などの両親媒性分子により形成されるミセル、および、ポリイオンコンプレックスにより形成されるミセル(PICミセル)が挙げられる。ミセルは、生物学的利用能の改善の観点では、その外表面を非電荷親水性鎖で修飾することが好ましい。
本明細書では、「平均分子量」は、特に断りの無い限り、数平均分子量を意味する。
本明細書では、「重合度」は、ポリマーにおける単量体単位の数を意味し、「平均重合度」は、特に断りのない限り数平均重合度を意味する。
本明細書では、「カチオン性ブロック」および「カチオン性ポリマー」とはそれぞれ、カチオン性の単量体を含む単量体単位を重合させて得られ、全体としてカチオン性であるポリマーブロックおよびポリマーを意味する。カチオン性ポリマーには、ホモカチオン性ポリマー、ホモカチオン性ポリマーと非電荷親水性鎖とが連結したポリマー等が挙げられる。カチオン性ポリマーが他のポリマーとブロック共重合体を形成しているとき、カチオン性ポリマー部分は、カチオン性ブロックと呼ばれることがある。本明細書では、カチオン性ポリマーは、薬学的に許容されるカチオン性ポリマーである。本明細書では、「多価カチオン」とは、カチオン性である分子のうち、カチオンの性質を示す基を分子内に複数有する分子をいう。本明細書では、「多価カチオン」は、血液環境において分子全体としてカチオン性を有しうる。多価カチオンとしては、カチオン性ポリマー、カチオン性デンドリマーなどの、血液環境においてカチオン性である分子が挙げられる。多価カチオンは、生体適合性を有する多価カチオンである。本明細書では、「デンドリマー」とは、コアとなる1つの原子から複数段階の分岐を有する分子をいう。
本明細書では、「親水性ブロック」とは、水性媒体に対して溶解性を示すポリマー鎖を意味し、親水性ポリマーブロックということがある。する。本発明では、非電荷親水性鎖は、薬学的に許容される非電荷親水性鎖である。このような親水性鎖としては、ポリエチレングリコール(PEG)やポリ(2-エチル-2-オキサゾリン)が挙げられる。非電荷親水性鎖は、局所的にも全体的にも電荷が中和されている限り、極性原子を有していてもよい。親水性ブロックは、分岐を含んでいても、含んでいなくてもよい。親水性ブロックが分岐点を有する場合には、分岐点の数は、1つまたはそれ以上であり得る。
本明細書では、「温度応答性ブロック」および「温度応答性重合体」とはそれぞれ、温度に依存して親水性から疎水性に変化することができるポリマーブロックおよびポリマーを意味する。温度に依存して親水性から疎水性に変化する物質は様々知られており、例えば、ポリ(N-イソプロピルアクリルアミド)やポリ(2-n-プロピル-2-オキサゾリン)、ポリ(2-イソプロピル-2-オキサゾリン)が挙げられる。温度応答性のポリマーは、下限臨界共溶温度(LCST)を有し、LCST未満では親水性であり、LCST以上では疎水性である。温度応答性重合体は、温度応答性ブロックを含んでなる。ある態様では、温度応答性重合体は、温度応答性ブロックと親水性ブロックとを含む。ある態様では、温度応答性重合体は、カチオン性ブロックを有しない。温度応答性重合体は、温度応答性ブロックから本質的になる重合体であっても、温度応答性ブロックからなる重合体であってもよい。下限臨界共溶温度(LCST)は、好ましくは、4℃以上40℃以下であり得、特にヒト等の対象の体温よりも低くすることができる。
本明細書では、「三元共重合体」または「トリブロック共重合体」とは、異なるポリマーブロックを3つ含むブロック共重合体を意味する。各ブロックは、リンカーやスペーサーを介して連結していてよい。三元共重合体は、3つの異なるブロック、A、BおよびCをこの順番で含む場合には、「A-B-C」と表記することができる。記号「-」は、結合、またはリンカー若しくはスペーサーであり得る。三元共重合体は、3つの異なるポリマーブロックを含んでいる限り、他の異なるポリマーブロックを含んでいてもよい。三元共重合体は、3つの異なるポリマーブロック「A-B-C」から本質的になる、または「A-B-C」からなるものであってもよい。
本明細書では、「外殻」とは、RNAを包む保護層を意味する。外殻は、必ずしも最外殻に存在することを意味するものではない。本明細書では、「保護層」は、それが無い場合と比較して、RNase等の分解酵素による分解からRNAを保護することができる。
本明細書では、「を含んでなる」(comprise)とは、「からなる」(consist of)および「から本質的になる」(essentially consist of)を含む意味で用いられる。「を含んでなる」とは、対象となる構成要素以外の構成要素を含んでいてもよいことを意味し、「からなる」とは、対象となる構成要素以外の構成要素を含まないことを意味する。本明細書では、「から本質的になる」とは、対象となる構成要素以外の構成要素を特別な機能を発揮する態様(発明の効果を完全に喪失させる態様など)では含まないことを意味する。
本明細書では、「AとBとを別々に投与する」またはその類似表現は、AとBとを時間的に別々に投与すること、および、AとBとを混合せずにそれぞれを同時に投与することを含む意味で用いられる。
本明細書では、「クリアランス」とは、薬物の代謝および薬物の排泄をいう。例えば、クリアランスは、薬物が代謝により減少すること、または血中から薬物が排泄されて減少することであり得る。クリアランスにより薬物はその血中滞留性を低下させることが知られている。
本明細書では、「標的臓器または組織」とは、投与する薬物を送達させたい臓器または組織をいう。投与する薬物の種類、投与対象の患者の種類によって、標的臓器または組織は異なるであろう。「標的臓器または組織」という用語は、当該臓器または組織だけに薬物を送達することを意味する用語ではなく、標的臓器または組織に薬物が送達される限り、当該臓器または組織以外の臓器または組織に薬物が送達されてもよいことを意味するものとする。「標的臓器または組織」への薬物の送達は、好ましくは、当該標的臓器または組織に選択的であることが好ましい。ここで「選択的である」とは、当該標的臓器または組織に対して、それ以外の臓器または組織に対するよりも多く薬物が集積することを意味する。
後述される実施例に示されたように、多価カチオンを血中に投与すると、多価カチオンは肝臓の類同内皮細胞や腎臓の血管内皮細胞の管腔側内壁に集積し、血管内壁を被覆した。また、この結果として、多価カチオンは、肝臓の類同内皮細胞や腎臓の血管内皮細胞による薬物のクリアランスを低下させ、薬物の血中滞留性を向上させた。従って、本発明によれば、カチオン性ポリマーに代表される多価カチオンは、体内の薬物動態を制御することに用い得る。
本発明によればまた、カチオン性ポリマーは、体内の薬物動態を制御することに用いることができる。
ここで、薬物動態の制御とは、薬物動態の改善または変更である。薬物動態の制御としては、クリアランスの制御が挙げられる。薬物動態の制御は、より具体的には、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減が挙げられる。薬物動態の制御としてはまた、腎臓による血中からの薬物の排泄能の低減が挙げられる。薬物動態の制御としてはさらに、標的臓器への送達量の増加が挙げられる。薬物動態の制御としてはさらにまた、脾臓への薬物の送達量の増加が挙げられる。薬物動態の制御としてはさらにまた、薬物の血中滞留性の増大が挙げられる。
このように、本発明では、カチオン性ポリマーなどの多価カチオンを用いることにより、血中の薬物の肝臓の類同内皮細胞による代謝またはそれを通しての排泄を抑制し、腎臓による代謝または排泄を抑制し、薬物の血中滞留性を高め、および/または、標的臓器若しくは組織(例えば、脳、肺、心臓、および脾臓など臓器または組織、並びに腫瘍など)への薬物の送達量を向上させることができる。すなわち、本発明によれば、カチオン性ポリマーなどの多価カチオンは、同時にまたは別々に投与される他の医薬有効成分(薬物)の体内の動態を制御するものである。従って、本発明によれば、カチオン性ポリマーなどの多価カチオンは医薬有効成分である必要がない。また、カチオン性ポリマーなどの多価カチオンは、医薬有効成分と複合体を形成している必要は無い(すなわち、遊離形態でよい)。
本発明によればまた、カチオン性ポリマーは、体内の薬物動態を制御することに用いることができる。
ここで、薬物動態の制御とは、薬物動態の改善または変更である。薬物動態の制御としては、クリアランスの制御が挙げられる。薬物動態の制御は、より具体的には、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減が挙げられる。薬物動態の制御としてはまた、腎臓による血中からの薬物の排泄能の低減が挙げられる。薬物動態の制御としてはさらに、標的臓器への送達量の増加が挙げられる。薬物動態の制御としてはさらにまた、脾臓への薬物の送達量の増加が挙げられる。薬物動態の制御としてはさらにまた、薬物の血中滞留性の増大が挙げられる。
このように、本発明では、カチオン性ポリマーなどの多価カチオンを用いることにより、血中の薬物の肝臓の類同内皮細胞による代謝またはそれを通しての排泄を抑制し、腎臓による代謝または排泄を抑制し、薬物の血中滞留性を高め、および/または、標的臓器若しくは組織(例えば、脳、肺、心臓、および脾臓など臓器または組織、並びに腫瘍など)への薬物の送達量を向上させることができる。すなわち、本発明によれば、カチオン性ポリマーなどの多価カチオンは、同時にまたは別々に投与される他の医薬有効成分(薬物)の体内の動態を制御するものである。従って、本発明によれば、カチオン性ポリマーなどの多価カチオンは医薬有効成分である必要がない。また、カチオン性ポリマーなどの多価カチオンは、医薬有効成分と複合体を形成している必要は無い(すなわち、遊離形態でよい)。
多価カチオンは、当該カチオン自体の生物学的利用能を向上させる観点で、親水性ポリマーにより修飾を受けていてもよい。
カチオン性ポリマーは、当該ポリマー自体の生物学的利用能を向上させる観点で、例えば、カチオン性ブロックと親水性ポリマーブロックとの共重合体の形態であってもよい。
このようにすることで、カチオン性ポリマーの生物学的利用能が向上し、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減効果、または腎臓による血中からの薬物の排泄能の低減効果が増大することが期待できる。
カチオン性ポリマーは、当該ポリマー自体の生物学的利用能を向上させる観点で、例えば、カチオン性ブロックと親水性ポリマーブロックとの共重合体の形態であってもよい。
このようにすることで、カチオン性ポリマーの生物学的利用能が向上し、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減効果、または腎臓による血中からの薬物の排泄能の低減効果が増大することが期待できる。
親水性ポリマーブロックは、例えば、非電荷親水性ポリマーブロックを用いることができる。非電荷親水性ポリマーブロックは、薬学的に許容可能なポリマーである。このようなポリマーとしては、特に限定されないが、例えば、ポリアルキレングリコール、ポリ(2-オキサゾリン)、ポリサッカライド、ポリビニルアルコール、ポリビニルピロリドン、ポリアクリルアミド、ポリメタクリルアミド、ポリアクリル酸エステル、ポリメタクリル酸エステル、ポリ(2-メタクロイルオキシエチルホスホリルコリン)挙げられる。非電荷親水性ポリマーとしては、ポリアルキレングリコール、ポリ(2-オキサゾリン)が好ましく用いられ、ポリアルキレングリコールが特に好ましく用いられ得る。ポリアルキレングリコールとしては、ポリエチレングリコール(PEG)が好ましく用いられ得る。
上記カチオン性ポリマー部分とPEG部分とを含む共重合体においては、PEG部分の平均分子量は、例えば、10kD以上、15kD以上、20kD以上、30kD以上、または40kD以上とすることができ(例えば、80kD以下、70kD以下、60kD以下、または50kD以下であり得る)、好ましくは、20kD以上であり、より好ましくは30kD以上である。上記カチオン性ポリマー部分とPEG部分とを含む共重合体においては、カチオン性ポリマーは、平均重合度15以上、20以上、30以上、または40以上であり得る(例えば、80以下、70以下、60以下、または50以下であり得る)。PEG部分の嵩を増す観点では、PEG部分は、平均分子量の大きな、例えば、40kD以上、50kD以上、60kD以上、または70kD以上(例えば、80kD以下、70kD以下、60kD以下、または50kD以下であり得る)の一本鎖PEGとすることもできるし、10kD以上、15kD以上、20kD以上、30kD以上、または40kD以上(例えば、80kD以下、70kD以下、60kD以下、または50kD以下であり得る)のPEG鎖を複数本有する分岐PEGとすることもできる。PEG部分の嵩を増加させる観点では、分岐PEGは好ましく用いられ得る。
ある態様では、カチオン性ポリマー部分とPEG部分とを含む共重合体は、PEG部分が、10kD以上、15kD以上、20kD以上、30kD以上、または40kD以上のPEG鎖を複数本有する分岐PEGであり、カチオン性ポリマー部分が平均重合度15以上、20以上、30以上、または40以上であり得る。この特定の態様において、カチオン性ポリマーとPEGを含む共重合体は、PEG部分が、20kD以上、30kD以上、または40kD以上のPEG鎖を複数本有する分岐PEGであり、カチオン性ポリマー部分が平均重合度20以上、30以上、または40以上であり得る。
ある態様では、カチオン性ポリマー部分とPEG部分とを含む共重合体は、PEG部分が、40kD以上の一本鎖PEGであり、カチオン性ポリマー部分が平均重合度15以上、20以上、30以上、40以上、50以上、60以上または70以上であり得る(例えば、80以下、70以下、60以下、または50以下であり得る。特に一本鎖PEGの場合は、平均分子量が大きいほど血中滞留性の改善効果が大きい)。
ある特定の態様では、カチオン性ポリマーブロックの平均重合度が15~30であり、カチオン性ポリマーブロックが分岐PEGと連結しており、分岐PEGは、PEG部分の総平均分子量が、40kD~100kD、50kD~90kD、または60kD~80kDであり得る。また、例えば、分岐PEGは1箇所の分岐を有し、当該分岐から伸びるそれぞれのPEG鎖の平均分子量が独立して、例えば、20kD~60kD、25kD~50kD、または30kD~40kDであり得る。本発明によれば、このような分岐PEGとカチオン性ポリマーとの共重合体を本発明の多価カチオンとして投与することができる。
さらなる特定の態様では、カチオン性ポリマーブロックの平均重合度が15~30であり、カチオン性ポリマーブロックが分岐PEGと連結しており、分岐PEGは、1箇所の分岐を有し、当該分岐から伸びるそれぞれのPEG鎖の平均分子量が独立して、例えば、20kD~60kD、25kD~50kD、または30kD~40kDであり得、本発明によれば、このような分岐PEGとカチオン性ポリマーとの共重合体を本発明の多価カチオンとして投与することができる。
ある態様では、カチオン性ポリマー部分とPEG部分とを含む共重合体は、PEG部分が、10kD以上、15kD以上、20kD以上、30kD以上、または40kD以上のPEG鎖を複数本有する分岐PEGであり、カチオン性ポリマー部分が平均重合度15以上、20以上、30以上、または40以上であり得る。この特定の態様において、カチオン性ポリマーとPEGを含む共重合体は、PEG部分が、20kD以上、30kD以上、または40kD以上のPEG鎖を複数本有する分岐PEGであり、カチオン性ポリマー部分が平均重合度20以上、30以上、または40以上であり得る。
ある態様では、カチオン性ポリマー部分とPEG部分とを含む共重合体は、PEG部分が、40kD以上の一本鎖PEGであり、カチオン性ポリマー部分が平均重合度15以上、20以上、30以上、40以上、50以上、60以上または70以上であり得る(例えば、80以下、70以下、60以下、または50以下であり得る。特に一本鎖PEGの場合は、平均分子量が大きいほど血中滞留性の改善効果が大きい)。
ある特定の態様では、カチオン性ポリマーブロックの平均重合度が15~30であり、カチオン性ポリマーブロックが分岐PEGと連結しており、分岐PEGは、PEG部分の総平均分子量が、40kD~100kD、50kD~90kD、または60kD~80kDであり得る。また、例えば、分岐PEGは1箇所の分岐を有し、当該分岐から伸びるそれぞれのPEG鎖の平均分子量が独立して、例えば、20kD~60kD、25kD~50kD、または30kD~40kDであり得る。本発明によれば、このような分岐PEGとカチオン性ポリマーとの共重合体を本発明の多価カチオンとして投与することができる。
さらなる特定の態様では、カチオン性ポリマーブロックの平均重合度が15~30であり、カチオン性ポリマーブロックが分岐PEGと連結しており、分岐PEGは、1箇所の分岐を有し、当該分岐から伸びるそれぞれのPEG鎖の平均分子量が独立して、例えば、20kD~60kD、25kD~50kD、または30kD~40kDであり得、本発明によれば、このような分岐PEGとカチオン性ポリマーとの共重合体を本発明の多価カチオンとして投与することができる。
本発明では、カチオン性ポリマーまたはカチオン性ポリマー部分としては、例えば、カチオン性天然アミノ酸およびカチオン性非天然アミノ酸、例えば、ヒスチジン、トリプトファン、オルニチン、アルギニンおよびリジンなどのカチオン性天然アミノ酸、および/または、-(NH-(CH2)2)p-NH2で表される基{ここで、pは1~5の整数である。}を側鎖として有するポリマーブロック、例えば、上記カチオン性の側鎖を有するカチオン性非天然アミノ酸のポリマーブロック、例えば、上記カチオン性の側鎖を有するアスパラギン酸またはグルタミン酸などのカチオン性非天然アミノ酸のポリマーブロックが挙げられる。本発明のある態様では、ポリカチオンブロックは、-(NH-(CH2)2)p-NH2で表される基{ここで、pは1~5の整数である。}を側鎖として有するポリマーブロックである。ここで、カチオン性天然アミノ酸としては、好ましくはヒスチジン、トリプトファン、オルニチン、アルギニンおよびリジンが挙げられ、より好ましくはアルギニン、オルニチンおよびリジンが挙げられ、さらに好ましくはオルニチンおよびリジンが挙げられ、さらにより好ましくはリジンが挙げられる。本発明のある態様では、カチオン性ポリマーまたはカチオン性ポリマー部分は、ポリリジンまたはポリオルニチンとすることができる。
ポリカチオンブロックには、カチオン性アミノ酸およびカチオン性の側鎖を有するアミノ酸が混在していてもよい。すなわち、本発明のある態様では、ポリカチオンブロックは、カチオン性天然アミノ酸、カチオン性非天然アミノ酸、または、カチオン性天然アミノ酸およびカチオン性非天然アミノ酸を含むモノマー単位の重合体である。本発明のある態様では、ポリカチオンブロック中のモノマー単位間の結合はペプチド結合である。本発明の好ましい態様では、カチオン性非天然アミノ酸が、側鎖として-(NH-(CH2)2)p-NH2で表される基{ここで、pは1~5の整数である}を有するアミノ酸である。また、本発明のある態様では、ポリカチオンブロックは、カチオン性天然アミノ酸並びに-(NH-(CH2)2)p-NH2で表される基{ここで、pは1~5の整数である。}で修飾されたアスパラギン酸およびグルタミン酸が任意の順番で重合してなるポリカチオンブロックとすることができる。本発明のある態様では、ポリマー中のモノマー単位の40%、50%、60%、70%、80%、90%、95%、98%または100%が側鎖として-(NH-(CH2)2)p-NH2で表される基{ここで、pは1~5の整数である。}を有する。
例えば、本発明のカチオン性ポリマーの一態様として、分岐PEGを有するポリリジンまたはポリオルニチンであって、PEG部分は、20~50kDの平均分子量を有するPEGを二本有する分岐PEGであり、ポリリジンまたはポリオルニチンは、平均重合度が20~70、または30~60である、分岐PEGを有するポリリジンまたはポリオルニチンが挙げられる。好ましいある態様では、PEGまたは分岐PEGは、カチオン性ポリマーの末端に連結し得る。他の一態様として、リジンおよびオルニチンなどのカチオン性モノマーを主骨格とするポリマーであり、そのモノマーユニットの側鎖の5%~80%、または20%~50%がPEGまたは分岐PEG等の親水性ポリマーにより修飾されたもの(すなわち、グラフト共重合体)であり得る。
本発明の多価カチオンは、肝臓の類同内皮細胞の血管内面や腎臓の血管内皮細胞の血管内面を被覆することにより、内皮細胞自体のクリアランス機能を低下させ得る。従って、本発明の多価カチオンは、様々な薬物の投与の前に、同時に、または、後に投与することができる。いずれの場合であっても、多価カチオンと薬物とを別々に投与する場合には、双方が同時に血中に存在するときに薬物動態の制御が可能である{薬物投与の前に多価カチオンを投与する場合は、カチオン性ポリマーが血中に滞留している間または肝臓の類同内皮細胞の血管内面に残存している間若しくは腎臓の内皮細胞の血管内面に残存している間に前記薬物を投与し、薬物の投与後に多価カチオンを投与する場合は、前記薬物が血中に滞留している間に多価カチオンを投与する}。ある態様では、多価カチオンは、例えば、薬物投与の直前に投与することができ、例えば、30秒以上前に投与することができ、1分以上、2分以上、3分以上、4分以上、または5分以上前に投与することができる。ある態様では、多価カチオンは、例えば、薬物投与前60分以内、50分以内、40分以内、30分以内、20分以内、または10分以内に投与することができる。ある態様では、薬物は、多価カチオンの投与10分前、5分前、4分前、3分前、2分前、1分前、30秒前、または直前に投与することができ、薬物投与から多価カチオン投与までは短いと好ましい。同時に投与する場合には、例えば、輸液バックに薬物と多価カチオンを混入させて投与することができる。
本発明のカチオン性ポリマーは、肝臓の類同内皮細胞の血管内面や腎臓の血管内皮細胞の血管内面を被覆することにより、内皮細胞自体のクリアランス機能を低下させ得る。従って、本発明のカチオン性ポリマーは、様々な薬物の投与の前に、同時に、または、後に投与することができる。いずれの場合であっても、カチオン性ポリマーと薬物とを別々に投与する場合には、双方が同時に血中に存在するときに薬物動態の制御が可能である{薬物投与の前にカチオン性ポリマーを投与する場合は、カチオン性ポリマーが血中に滞留している間または肝臓の類同内皮細胞の血管内面に残存している間若しくは腎臓の内皮細胞の血管内面に残存している間に前記薬物を投与し、薬物の投与後にカチオン性ポリマーを投与する場合は、前記薬物が血中に滞留している間にカチオン性ポリマーを投与する}。ある態様では、カチオン性ポリマーは、例えば、薬物投与の直前に投与することができ、例えば、30秒以上前に投与することができ、1分以上、2分以上、3分以上、4分以上、または5分以上前に投与することができる。ある態様では、カチオン性ポリマーは、例えば、薬物投与前60分以内、50分以内、40分以内、30分以内、20分以内、または10分以内に投与することができる。ある態様では、薬物は、カチオン性ポリマーの投与10分前、5分前、4分前、3分前、2分前、1分前、30秒前、または直前に投与することができ、薬物投与からカチオン性ポリマーの投与までは短い方がよい。同時に投与する場合には、例えば、輸液バックに薬物とカチオン性ポリマーを混入させて投与することができる。
本発明のカチオン性ポリマーは、肝臓の類同内皮細胞の血管内面や腎臓の血管内皮細胞の血管内面を被覆することにより、内皮細胞自体のクリアランス機能を低下させ得る。従って、本発明のカチオン性ポリマーは、様々な薬物の投与の前に、同時に、または、後に投与することができる。いずれの場合であっても、カチオン性ポリマーと薬物とを別々に投与する場合には、双方が同時に血中に存在するときに薬物動態の制御が可能である{薬物投与の前にカチオン性ポリマーを投与する場合は、カチオン性ポリマーが血中に滞留している間または肝臓の類同内皮細胞の血管内面に残存している間若しくは腎臓の内皮細胞の血管内面に残存している間に前記薬物を投与し、薬物の投与後にカチオン性ポリマーを投与する場合は、前記薬物が血中に滞留している間にカチオン性ポリマーを投与する}。ある態様では、カチオン性ポリマーは、例えば、薬物投与の直前に投与することができ、例えば、30秒以上前に投与することができ、1分以上、2分以上、3分以上、4分以上、または5分以上前に投与することができる。ある態様では、カチオン性ポリマーは、例えば、薬物投与前60分以内、50分以内、40分以内、30分以内、20分以内、または10分以内に投与することができる。ある態様では、薬物は、カチオン性ポリマーの投与10分前、5分前、4分前、3分前、2分前、1分前、30秒前、または直前に投与することができ、薬物投与からカチオン性ポリマーの投与までは短い方がよい。同時に投与する場合には、例えば、輸液バックに薬物とカチオン性ポリマーを混入させて投与することができる。
本発明のある態様では、本発明の多価カチオンは、薬物の投与時間と合わせて(またはその前後に合わせて)投与薬物のクリアランスを必要な時間だけ低下させるように、投与量および投与回数を決定することができる。本発明のある態様では、本発明の多価カチオンは、薬物の投与時間と合わせて(またはその前後に合わせて)投与薬物のクリアランスを必要な時間だけ低下させるように、単回投与または複数回投与され得る。本発明のある態様では、目的に応じて、投与は、ボーラス投与または輸液投与とすることができる。
本発明によれば、核酸を内包した薬物送達用キャリア(ミセルやリポソーム)が、多価カチオンによって肝臓の類同内皮細胞への蓄積や腎臓の内皮細胞への蓄積が低減された。従って、本発明の多価カチオンは、特に核酸を内包した薬物送達用キャリアの薬物動態の制御に用いることができる。
本発明によれば、核酸を内包した薬物送達用キャリア(ミセルやリポソーム)が、カチオン性ポリマーによって肝臓の類同内皮細胞への蓄積や腎臓の内皮細胞への蓄積が低減された。従って、本発明のカチオン性ポリマーは、特に核酸を内包した薬物送達用キャリアの薬物動態の制御に用いることができる。
核酸としては、例えば、デオキシリボ核酸(DNA)およびリボ核酸(RNA)、並びにLocked核酸(LNA)および架橋核酸(BNA)などの修飾核酸、またはこれらのハイブリッドが挙げられる。DNAとしては、特に限定されないが、直鎖状二本鎖DNA、直鎖状一本鎖DNA、環状二本鎖DNAおよび環状一本鎖DNAが挙げられる。RNAとしては、特に限定されないが、例えば、siRNA、shRNA、マイクロRNA、メッセンジャーRNA(mRNA)、トランスファーRNA(tRNA)、リボソームRNA(rRNA)、ノンコーディングRNA(ncRNA)、および二本鎖RNA、並びにこれらのRNAの誘導体が挙げられる。
本発明によれば、核酸を内包した薬物送達用キャリア(ミセルやリポソーム)が、カチオン性ポリマーによって肝臓の類同内皮細胞への蓄積や腎臓の内皮細胞への蓄積が低減された。従って、本発明のカチオン性ポリマーは、特に核酸を内包した薬物送達用キャリアの薬物動態の制御に用いることができる。
核酸としては、例えば、デオキシリボ核酸(DNA)およびリボ核酸(RNA)、並びにLocked核酸(LNA)および架橋核酸(BNA)などの修飾核酸、またはこれらのハイブリッドが挙げられる。DNAとしては、特に限定されないが、直鎖状二本鎖DNA、直鎖状一本鎖DNA、環状二本鎖DNAおよび環状一本鎖DNAが挙げられる。RNAとしては、特に限定されないが、例えば、siRNA、shRNA、マイクロRNA、メッセンジャーRNA(mRNA)、トランスファーRNA(tRNA)、リボソームRNA(rRNA)、ノンコーディングRNA(ncRNA)、および二本鎖RNA、並びにこれらのRNAの誘導体が挙げられる。
多価カチオンは、核酸を内包した薬物送達用キャリア(ミセルやリポソーム)と一緒に投与する場合には、遊離形態で存在させ得る。すなわち、多価カチオンは、核酸と直接複合体を形成し得ることが知られているが、本発明では、多価カチオンは肝臓や腎臓の血管内皮細胞を被覆するものであるから、多価カチオンは、遊離形態(すなわち、核酸と複合体を形成していない状態)で存在させることが好ましい。核酸を内包した薬物送達用キャリアは、イオンコンプレックスではなく、リポプレックスなどの脂質複合体としてもよい。
カチオン性ポリマーは、核酸を内包した薬物送達用キャリア(ミセルやリポソーム)と一緒に投与する場合には、遊離形態で存在させ得る。すなわち、カチオン性ポリマーは、核酸と直接複合体を形成し得ることが知られているが、本発明では、カチオン性ポリマーは肝臓や腎臓の血管内皮細胞を被覆するものであると考えられることから、カチオン性ポリマーは、遊離形態(すなわち、核酸と複合体を形成していない状態)で存在させることができる。カチオン性ポリマーとしては、核酸を内包した薬物送達用キャリアを製造するときに用いるカチオン性ポリマーとは異なるカチオン性ポリマーを用いることができる。核酸を内包した薬物送達用キャリアは、イオンコンプレックスではなく、リポプレックスなどの脂質複合体としてもよい。
本発明のある態様では、細胞への核酸導入効率を高めるためにキャリアとしてウイルスを用いてもよい。細胞への核酸導入効率を高めるためにキャリアとしては、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター(AAV1、AAV2、AAV3、AAV4、AAV5、AAV6、AAV7、AAV8、AAV9、およびAAV10など)、センダイウイルスベクター、麻疹ウイルスベクター、ワクシニアウイルスベクター、ヘルペスウイルスベクター等のウイルスベクターを用い得る。これらのベクターは、例えば、送達する核酸を細胞内で発現させる発現ユニット(例えば、プロモーターまたはエンハンサーに核酸が作動可能に連結されたもの)を有してもよい。
カチオン性ポリマーは、核酸を内包した薬物送達用キャリア(ミセルやリポソーム)と一緒に投与する場合には、遊離形態で存在させ得る。すなわち、カチオン性ポリマーは、核酸と直接複合体を形成し得ることが知られているが、本発明では、カチオン性ポリマーは肝臓や腎臓の血管内皮細胞を被覆するものであると考えられることから、カチオン性ポリマーは、遊離形態(すなわち、核酸と複合体を形成していない状態)で存在させることができる。カチオン性ポリマーとしては、核酸を内包した薬物送達用キャリアを製造するときに用いるカチオン性ポリマーとは異なるカチオン性ポリマーを用いることができる。核酸を内包した薬物送達用キャリアは、イオンコンプレックスではなく、リポプレックスなどの脂質複合体としてもよい。
本発明のある態様では、細胞への核酸導入効率を高めるためにキャリアとしてウイルスを用いてもよい。細胞への核酸導入効率を高めるためにキャリアとしては、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター(AAV1、AAV2、AAV3、AAV4、AAV5、AAV6、AAV7、AAV8、AAV9、およびAAV10など)、センダイウイルスベクター、麻疹ウイルスベクター、ワクシニアウイルスベクター、ヘルペスウイルスベクター等のウイルスベクターを用い得る。これらのベクターは、例えば、送達する核酸を細胞内で発現させる発現ユニット(例えば、プロモーターまたはエンハンサーに核酸が作動可能に連結されたもの)を有してもよい。
本発明のある側面では、対象に薬物を投与する方法であって、対象に多価カチオンを投与することと、対象に薬物を投与することとを含む、方法が提供される。本発明のある態様では、多価カチオンとRNAとは別々に投与される。本発明のある態様では、多価カチオンは、薬物と同時に、または薬物よりも先に若しくは後に対象に投与され得る。多価カチオンは、生体適合性を有するが、用量制限毒性(DLT)を奏さないような用量で投与され得る。
本発明のある側面では、対象に薬物を投与する方法であって、対象にカチオン性ポリマーを投与することと、対象に薬物を投与することとを含む、方法が提供される。本発明のある態様では、カチオン性ポリマーとRNAとは別々に投与される。本発明のある態様では、カチオン性ポリマーは、薬物と同時に、または薬物よりも先に若しくは後に対象に投与され得る。カチオン性ポリマーは、生体適合性を有するが、用量制限毒性(DLT)を奏さないような用量で投与され得る。
本発明のある側面では、対象に薬物を投与する方法であって、対象にカチオン性ポリマーを投与することと、対象に薬物を投与することとを含む、方法が提供される。本発明のある態様では、カチオン性ポリマーとRNAとは別々に投与される。本発明のある態様では、カチオン性ポリマーは、薬物と同時に、または薬物よりも先に若しくは後に対象に投与され得る。カチオン性ポリマーは、生体適合性を有するが、用量制限毒性(DLT)を奏さないような用量で投与され得る。
本発明のある側面では、対象に核酸を内包した薬物送達用キャリアを投与する方法であって、対象に多価カチオンを投与することと、対象に核酸を内包した薬物送達用キャリアを投与することとを含む、方法が提供される。本発明のある側面では、対象の組織に核酸を送達する方法であって、対象に多価カチオンを投与することと、対象に核酸を内包した薬物送達用キャリアを投与することとを含む、方法が提供される。本発明のある態様では、多価カチオンは、核酸を内包した薬物送達用キャリアと同時に、または薬物よりも先に若しくは後に対象に投与され得る。
本発明のある側面では、対象に核酸を内包した薬物送達用キャリアを投与する方法であって、対象にカチオン性ポリマーを投与することと、対象に核酸を内包した薬物送達用キャリアを投与することとを含む、方法が提供される。本発明のある側面では、対象の組織に核酸を送達する方法であって、対象にカチオン性ポリマーを投与することと、対象に核酸を内包した薬物送達用キャリアを投与することとを含む、方法が提供される。本発明のある態様では、カチオン性ポリマーは、核酸を内包した薬物送達用キャリアと同時に、または薬物よりも先に若しくは後に対象に投与され得る。
本発明のある側面では、対象に核酸を内包した薬物送達用キャリアを投与する方法であって、対象にカチオン性ポリマーを投与することと、対象に核酸を内包した薬物送達用キャリアを投与することとを含む、方法が提供される。本発明のある側面では、対象の組織に核酸を送達する方法であって、対象にカチオン性ポリマーを投与することと、対象に核酸を内包した薬物送達用キャリアを投与することとを含む、方法が提供される。本発明のある態様では、カチオン性ポリマーは、核酸を内包した薬物送達用キャリアと同時に、または薬物よりも先に若しくは後に対象に投与され得る。
本発明者らはまた、親水性ブロックと温度応答性ブロックとカチオン性ブロックを含む三元共重合体により形成されるミセルが、核酸の血中安定性を劇的に向上させ、例えば、脳組織に核酸を大量に送達できることを見出した。したがって、本発明によれば、例えば、核酸と、親水性ブロックと温度応答性ブロックとカチオン性ブロックを含む三元共重合体とを含み、表面がグルコースで修飾されたミセルを含む、臓器または組織(特に脳)に核酸を送達することに用いる組成物が提供される。
このようなミセルは、以下の手順で調製することができる。すなわち、親水性ブロックと温度応答性ブロックとカチオン性ブロックを含む三元共重合体と、RNAとをミセルを形成できるようにLCST未満の温度の水溶液中で混合する。ミセルが得られたら、水溶液の温度をLCST以上に上昇させ、温度応答性ブロックを親水性から疎水性に変化させる。このようにして得られるミセルは、核酸を内部に内包してその外殻として疎水性の保護層を有する状態となっていると考えられ、内包した核酸を血中で安定的に維持することができる。すなわち、このミセルでは、核酸は、疎水性に変化した温度応答性ブロックで形成される外殻によって覆われている。
このようなミセルは、以下の手順で調製することができる。すなわち、親水性ブロックと温度応答性ブロックとカチオン性ブロックを含む三元共重合体と、RNAとをミセルを形成できるようにLCST未満の温度の水溶液中で混合する。ミセルが得られたら、水溶液の温度をLCST以上に上昇させ、温度応答性ブロックを親水性から疎水性に変化させる。このようにして得られるミセルは、核酸を内部に内包してその外殻として疎水性の保護層を有する状態となっていると考えられ、内包した核酸を血中で安定的に維持することができる。すなわち、このミセルでは、核酸は、疎水性に変化した温度応答性ブロックで形成される外殻によって覆われている。
血液の温度がヒトの場合、36℃~37℃程度であることを考慮すると、三元共重合体のLCSTは、35℃以下であることが好ましい。このようにすることで投与後に血中でミセルの状態が維持される。また、mRNAの調製を0℃~4℃程度で行うことを考慮すると、LCSTは、5℃以上であることが好ましい。なお、LCSTは、その他の様々な状況を考慮して適切なLCSTを設定することが好ましい。例えば、LCSTは、5℃~35℃とすることができ、10℃~32℃とすることができ、または25℃~32℃とすることができる。一般に水溶性ポリマーは、LCSTを有する。本発明では、上記の温度範囲内にLCSTを有するポリマーであれば適宜温度応答性ブロックとして用いることができる。
本発明のある態様では、三元共重合体の親水性ブロックは、ポリエチレングリコールまたはポリ(2-エチル-2-オキサゾリン)であり得る。親水性ブロックの平均分子量は、例えば、10kD以上、15kD以上、20kD以上、30kD以上、または40kD以上とすることができる。親水性ブロックはまた、例えば、20kD以下、30kD以下、40kD以下、または50kD以下の平均分子量とすることができる。
本発明のある態様では、三元共重合体の温度応答性ブロックは、ポリ(N-イソプロピルアクリルアミド)やポリ(2-n-プロピル-2-オキサゾリン)、またはポリ(2-イソプロピル-2オキサゾリン)であり得る。温度応答性ブロックの平均分子量は、例えば、3kD以上、5kD以上、7kD以上、または10kD以上であり得る。温度応答性ブロックの平均分子量はまた、例えば、10kD以下、15kD以下、または20kD以下であり得る。
本発明のある態様では、三元共重合体のポリカチオン性ブロックは、天然または非天然のカチオン性アミノ酸を単量体単位として含むペプチドであり得る。三元共重合体のポリカチオン性ブロックは、例えば、リジンおよびオルニチンなどの天然のカチオン性アミノ酸を単量体単位として含むペプチド、例えば、ポリリジンまたはポリオルニチンであり得る。本発明のある態様では、三元共重合体のポリカチオン性ブロックは、例えば、ジエチレントリアミンまたはトリエチレンテトラアミンでカルボキシル基を修飾したアスパラギン酸やグルタミン酸を単量体単位として含むペプチド、例えば、ホモポリマーであり得る。ポリカチオン性ブロックの平均重合度は、例えば、30以上、40以上、50以上、60以上、または70以上であり得る。ポリカチオン性ブロックの平均重合度はまた、70以下、80以下、90以下、または100以下であり得る。
本発明のある態様では、三元共重合体は、親水性ブロックがポリエチレングリコールまたはポリ(2-エチル-2-オキサゾリン)であり、温度応答性ブロックは、ポリ(2-n-プロピル-2-オキサゾリン)であり、ポリカチオン性ブロックは、ポリリジンまたはポリオルニチンである。本発明のある態様では、三元共重合体は、親水性ブロックがポリエチレングリコールであり、温度応答性ブロックは、ポリ(2-n-プロピル-2-オキサゾリン)であり、ポリカチオン性ブロックは、ポリリジンである。
本発明において、核酸を内包する薬物送達用キャリアとしては、上記、三元共重合体等による、疎水性保護層により内包された核酸が安定化されたミセルを用いることができる。本発明においてはまた、核酸を内包する薬物送達用キャリアとしては、小胞、例えば、脂質ベースの小胞、例えば、リポソーム、例えば、Invivofectamine(商標)などの脂質ベースの小胞(例えば、リポプレックス)を用いることができる。これらの小胞は、その表面に親水性ポリマーの被覆を有することが好ましい。
本発明において、核酸を脳に送達する際に用いる薬物送達用キャリアとしては、外表面をグルコースなどのGLUT1リガンドにより被覆したキャリアを用いることができる(詳細の原理は、WO2015/075942A1参照)。このキャリアは、以下の投与計画に従って投与すると脳への蓄積が向上する。すなわち、投与計画は、絶食させるか、または低血糖を誘発させた対象に該組成物を投与することおよび該対象において血糖値の上昇を誘発させることを含むものである。
本発明において、脾臓に送達する核酸としては、特に限定されないが例えば、脾臓の機能(例えば、免疫機能)を高める因子をコードする核酸が挙げられる。免疫機能を高める因子をコードする核酸としては、免疫細胞の抗原となるペプチドやタンパク質をコードする核酸が挙げられる。このようにすることで、特定の抗原に対する免疫機能が高まり、感染や、がんなどに対する予防効果または治療効果が得られ得る。免疫細胞の抗原となるペプチドやタンパク質をコードする核酸としては、例えば、ペプチドワクチン、特に腫瘍特異的なペプチドワクチンとして用いられるペプチド(例えば、HLA拘束性のペプチド、例えば、HLA-A24やHLA-A2拘束性のペプチド、例えば、HLA-A2:01やHLA-A24:01拘束性のペプチド)をコードする核酸が挙げられる。このようなペプチドは、既に多数開発されており、臨床研究がなされているものも多く、当業者であれば適宜選択して本発明で用いることができるであろう。脾臓には、樹状細胞が存在し、これらの樹状細胞にペプチドが発現することにより当該ペプチドが免疫細胞に提示される。免疫細胞は脾臓において当該ペプチドに応答性になり、当該ペプチドに対する免疫機能が高まって、当該ペプチドを細胞表面に発現するがん細胞を攻撃できるようになる。これにより、最終的には、抗腫瘍効果が高まると期待される。本発明によれば、脾臓への核酸の送達のためのAAV8の使用が提供される。本発明によれば、脾臓へ核酸を送達することに用いるための医薬の製造におけるAAV8の使用が提供される。本発明によれば、脾臓への核酸の送達のためのAAV8と多価カチオンの組合せの使用が提供される。本発明によれば、脾臓へ核酸を送達することに用いるための医薬の製造におけるAAV8と多価カチオンとの組合せの使用が提供される。本発明によれば、AAV8を脾臓へ送達することに用いるための多価カチオンの使用が提供される。本発明によれば、AAV8を脾臓に送達することに用いるための医薬の製造における多価カチオンの使用が提供される。本発明によれば、多価カチオンを含む、AAV8を脾臓に送達することに用いるための医薬が提供される。
本発明において、治療対象となる心疾患または障害は、例えば、心筋梗塞が挙げられるが、これらに限定されない。本発明によれば、これらの心疾患または障害をそれを必要とする対象において処置する方法であって、本発明の多価カチオンと組み合わせて用いられるAAVを投与することを含む方法が提供される。ここでAAVとしては、例えば、AAV9を用いることができる。AAV9は例えば、血管内皮増殖因子(VEGF)などの心再生作用を有する因子をコードする遺伝子を有し得る。本発明によれば、心臓への核酸の送達のためのAAV9の使用が提供される。本発明によれば、心臓へ核酸を送達することに用いるための医薬の製造におけるAAV9と多価カチオンの組合せの使用が提供される。本発明によれば、心臓への核酸の送達のためのAAV9の使用が提供される。本発明によれば、心臓へ核酸を送達することに用いるための医薬の製造におけるAAV9と多価カチオンの組合せの使用が提供される。本発明によれば、AAV9を心臓へ送達することに用いるための多価カチオンの使用が提供される。本発明によれば、AAV9を心臓に送達することに用いるための医薬の製造における多価カチオンの使用が提供される。本発明によれば、多価カチオンを含む、AAV9を心臓に送達することに用いるための医薬が提供される。
本明細書では、「GLUT1リガンド」とは、GLUT1と特異的に結合する物質を意味する。GLUT1リガンドとしては、様々なリガンドが知られ、特に限定されないが例えば、グルコースおよびヘキソースなどの分子が挙げられ、GLUT1リガンドは、いずれも本発明でグルコースの代わりにキャリアまたはコンジュゲートの調製に使用することができる。GLUT1リガンドは、好ましくはGLUT1に対してグルコースと同等またはそれ以上の親和性を有する。2-N-4-(1-アジ-2,2,2-トリフルオロエチル)ベンゾイル-1,3-ビス(D-マンノース-4-イルオキシ)-2-プロピルアミン(ATB-BMPA)、6-(N-(7-ニトロベンズ-2-オキサ-1,3-ジアゾール-4-イル)アミノ)-2-デオキシグルコース(6-NBDG)、4,6-O-エチリデン-α-D-グルコース、2-デオキシ-D-グルコースおよび3-O-メチルグルコースもGLUT1と結合することが知られ、これらの分子もGLUT1リガンドとして本発明に用いることができる。
本明細書では、「低血糖を誘発させる」とは、対象において、その処置がされなければ示したはずの血糖よりも血糖値を低下させることをいう。低血糖を誘発させる方法としては、糖尿病薬の投与などが挙げられる。例えば、低血糖を誘発させる際に、低血糖を誘発させるという目的を達する限りにおいて、例えば、他の薬剤を摂取し、または水などの飲料を飲むことは許容される。低血糖を誘発させることは、血糖に実質的に影響しない他の処置を伴ってもよい。
本明細書では、「絶食させる」とは、対象に絶食、例えば、3時間以上、4時間以上、5時間以上、6時間以上、7時間以上、8時間以上、9時間以上、10時間以上、11時間以上、12時間以上、13時間以上、14時間以上、15時間以上、16時間以上、17時間以上、18時間以上、19時間以上、20時間以上、21時間以上、22時間以上、23時間以上、24時間以上、25時間以上、26時間以上、27時間以上、28時間以上、29時間以上、30時間以上、31時間以上、32時間以上、33時間以上、34時間以上、35時間以上、36時間以上、37時間以上、38時間以上、39時間以上、40時間以上、41時間以上、42時間以上、43時間以上、44時間以上、45時間以上、46時間以上、47時間以上または48時間以上の絶食をさせることを意味する。絶食により対象は低血糖を引き起こす。絶食期間は、対象の健康状態に鑑みて医師等により決定され、例えば、対象が空腹時血糖に達する時間以上の期間とすることが好ましい。絶食期間は、例えば、脳血管内皮細胞の血管内表面でのGLUT1の発現が増大する、またはプラトーに達する以上の時間としてもよい。絶食期間は、例えば、12時間以上、24時間以上または36時間以上である上記期間とすることができる。また、絶食は、血糖値やGLUT1の血管内表面での発現に実質的に影響しない他の処置を伴ってもよい。
本明細書では、「血糖値の上昇を誘発させる」とは、低血糖を誘発させた対象、または、低血糖状態を維持させた対象において血糖値を上昇させることをいう。血糖値は、当業者に周知の様々な方法により上昇させることができるが、例えば、血糖値の上昇を誘発するものの投与、例えば、グルコース、フルクトース(果糖)、ガラクトースなどの血糖値の上昇を誘発する単糖の投与、マルトースなどの血糖値の上昇を誘発する多糖の投与、若しくは、デンプンなどの血糖値の上昇を誘発する炭水化物の摂取、または、食事により上昇させることができる。
本明細書では、「血糖操作」とは、対象に対して、低血糖を誘発させ、その後、血糖値を上昇させることをいう。対象に対して低血糖を誘発させた後は、対象の血糖値を低血糖に維持することができる。対象の血糖値を低血糖に維持する時間は、例えば、0時間以上、1時間以上、2時間以上、3時間以上、4時間以上、5時間以上、6時間以上、7時間以上、8時間以上、9時間以上、10時間以上、11時間以上、12時間以上、13時間以上、14時間以上、15時間以上、16時間以上、17時間以上、18時間以上、19時間以上、20時間以上、21時間以上、22時間以上、23時間以上、24時間以上、25時間以上、26時間以上、27時間以上、28時間以上、29時間以上、30時間以上、31時間以上、32時間以上、33時間以上、34時間以上、35時間以上、36時間以上、37時間以上、38時間以上、39時間以上、40時間以上、41時間以上、42時間以上、43時間以上、44時間以上、45時間以上、46時間以上、47時間以上、48時間以上とすることができる。その後、血糖値を上昇させることができる。本明細書では、「血糖を維持する」とは、対象において低血糖を維持するという目的を達する限りにおいて、例えば、他の薬剤を摂取し、または水などの飲料を飲むことは許される。低血糖を誘発させることは、血糖に実質的に影響しない他の処置を伴ってもよい。
本発明による投与計画では、該組成物は、該対象における血糖値の上昇の誘発と、同時に、連続してまたは逐次的に該対象に投与され得る。投与計画は、該対象への組成物の投与と該対象における血糖値の上昇の誘発との間にインターバルを有してもよいし、有さなくてもよい。該組成物が該対象における血糖値の上昇の誘発と同時に投与される場合には、該組成物は、血糖値の上昇の誘発を引き起こす薬剤と混合した形態で該対象に投与してもよいし、該対象における血糖値の上昇の誘発を引き起こす薬剤とは別の形態で投与してもよい。また、該組成物は、該対象における血糖値の上昇の誘発と、連続してまたは逐次的に該対象に投与される場合には、該組成物は該対象における血糖値の上昇の誘発より前に該対象に投与してもよいし、後に投与してもよいが、好ましくは、該組成物は該対象における血糖値の上昇の誘発より前に該対象に投与することができる。該対象への該組成物の投与よりも先に該対象において血糖値の上昇を誘発させる場合には、該対象において血糖値の上昇を誘発させてから、1時間以内、45分以内、30分以内、15分以内または10分以内に該対象に該組成物を投与することが好ましい。また、該対象への該組成物の投与よりも後に該対象において血糖値の上昇を誘発させる場合には、該対象に該組成物を投与してから、6時間以内、4時間以内、2時間以内、1時間以内、45分以内、30分以内、15分以内または10分以内に該対象において血糖値の上昇を誘発させることが好ましい。上記の投与計画のサイクルは、2回以上行なってもよい。グルコース投与とサンプル投与の前後関係は、血液脳関門を通過させるタイミングにより決定することができる。
本発明の上記三元共重合体等による、疎水性保護層により内包された核酸が安定化されたミセルは、脳血管内皮細胞に送達することに用いることができる。また、本発明におけるグルコースの役割は、血液神経関門、血液網膜関門および血液髄液関門においても、同様である。特に、血液神経関門、血液網膜関門および血液髄液関門においても、低血糖時の血管内皮細胞にはGLUT1が発現する。従って、本発明の上記、三元共重合体等による、疎水性保護層により内包された核酸が安定化されたミセルは、血液神経関門、血液網膜関門および血液髄液関門を通過させるために用いることができる。本発明の上記三元共重合体等による、疎水性保護層により内包された核酸が安定化されたミセルはまた、血液神経関門、血液網膜関門および血液髄液関門に存在する血管内皮細胞に送達することに用いることもできる。
実施例1:薬剤送達用ミセルの肝臓への蓄積に対する多価カチオン投与の効果
[材料]
薬剤送達用ミセルとしては、PEG(分子量11k)-b-PnPrOx(分子量8k)-b-PLys(重合度43)のトリブロック重合体ミセルの表面にグルコース修飾を有するミセルを用いた。表面のグルコース修飾は、ミセルを脳へ送達し易くするための修飾であり、WO2015075942A1の開示によれば、絶食状態の対象にグルコース修飾をしたミセルを投与し、これに前後して対象の血糖値を上昇させると、血糖値の上昇に伴って当該ミセルが顕著に脳に取り込まれる。
グルコース誘導体DIGをPEG側末端に有するポリマー(DIG-PEG)の合成は、WO2015075942A1に記載の通りに行った。また、DIG-PEGとPnPrOx、PLysの共重合体は、European Polymer Journal, 2017, 88, 553-561の記載に従って合成したPnPrOx-b-PLysと、Glc-PEGとを
連結させて得た。トリブロック共重合体の合成スキームは以下スキーム1~4に示される通りであった。
薬剤送達用ミセルとしては、PEG(分子量11k)-b-PnPrOx(分子量8k)-b-PLys(重合度43)のトリブロック重合体ミセルの表面にグルコース修飾を有するミセルを用いた。表面のグルコース修飾は、ミセルを脳へ送達し易くするための修飾であり、WO2015075942A1の開示によれば、絶食状態の対象にグルコース修飾をしたミセルを投与し、これに前後して対象の血糖値を上昇させると、血糖値の上昇に伴って当該ミセルが顕著に脳に取り込まれる。
グルコース誘導体DIGをPEG側末端に有するポリマー(DIG-PEG)の合成は、WO2015075942A1に記載の通りに行った。また、DIG-PEGとPnPrOx、PLysの共重合体は、European Polymer Journal, 2017, 88, 553-561の記載に従って合成したPnPrOx-b-PLysと、Glc-PEGとを
連結させて得た。トリブロック共重合体の合成スキームは以下スキーム1~4に示される通りであった。




具体的には、温度応答性鎖であるPoly(2-n-propyl-2-oxazoline) (PnPrOx)とカチオン性鎖Poly(L-Lysine) (PLys)からなりかつalkyne末端を有するブロック共重合体alkyne-PnPrOx-b-PLysは以下のように合成した。
まず重合開始剤p-トルエンスルホン酸プロパルギル(61 mg, 0.29 mmol)をアセトニトリル 7 mLに溶解させ、2-n-プロピル-2-オキサゾリン(2.5 g, 22 mmol)を加えた。反応溶液を42℃で6日間反応させた後、アジ化ナトリウム(380 mg, 5.8 mmol)を加えて70℃で一時間撹拌して、重合反応を停止させた。上記のプロセスは全てAr雰囲気化で行った。ここで用いたp-トルエンスルホン酸プロパルギルは東京化成より購入し、Wako社より購入した五酸化ニリンを脱水剤として用いて蒸留精製して使用した。2-n-プロピル-2-オキサゾリンは東京化成より購入し、シグマアルドリッチ社より購入したカルシウムハイドライドを脱水剤として用いて蒸留精製して使用した。反応溶媒のアセトニトリルはwako社より購入し、カルシウムハイドライドを脱水剤として用いて蒸留精製して使用した。アジ化ナトリウムはWako社より購入し、そのまま用いた。重合反応後、反応溶液を水に対して5回透析し、凍結乾燥することでアジド末端を持つalkyne-PnPrOx-N3を得た。得られたPnPrOxは、MALDI-TOF MS(UltraFlextreme, Bruker社)と1H-NMR(ESC400, JEOL)による解析から、分子量が8.3kであることが分かった。
続いてalkyne-PnPrOx-N3のアジド末端をスタウディンガー反応により、アミン末端に変換した。alkyne-PnPrOx-N3(830 mg, 0.10 mmol)をメタノール20 mLに溶解し、トリフェニルホスフィン(530 mg, 2.0 mmol)加えて40℃で三時間撹拌した。続いて純水20 mLを加えて氷冷し、未反応のトリフェニルホスフィン等を濾過で取り除いた。得られた溶液は純水に対して3回透析した後、凍結乾燥により回収した。得られたalkyne-PnPrOx-NH2はGE-health care社より購入したCM-Sephadex C50をカラム充填剤としたオープンカラムを用いて更に精製した。ここで用いたメタノール、トリフェニルホスフィンは、それぞれシグマアルドリッチ社、東京化成より購入した。
上述で精製・回収したalkyne-PnPrOx-NH2よりLys(TFA)-NCAを重合し、その後塩基によるTFA基の脱保護により、alkyne-PnPrOx-b-PLysを得た。alkyne-PnPrOx-NH2 (330 mg, 0.040 mmol )を、ジオキサンを用いて凍結乾燥した。これを1Mの濃度でチオウレアを溶かしたDMF (DMF (1M TU)) 2 mLに溶解させて、開始剤溶液とした。別途Arバック中でLys(TFA)-NCA(480 mg, 1.8 mmol)をフラスコに測りとり、6 mLのDMF(1M TU)に溶解させた。調製したLys(TFA)-NCA溶液を、Ar雰囲気下で開始剤溶液に加え、25℃で3日間撹拌して重合反応を行った。反応溶液を水に対して5回透析したのち、凍結乾燥することでPnPrOx-b-Poly(L-Lysine)(TFA) (PLys(TFA))を得た。続いて得られたalkyne-PnPrOx-b-PLys(TFA)を500 mg測りとり、25 mLのメタノールに溶解させた。これに対して、7.5 mLの1M NaOH溶液を加え、35℃で12時間反応させた。反応後は、0.01 M HClに対して3回透析を行った後、水に対して3回透析を行い、凍結乾燥によりPnPrOx-b-PLysを回収した。回収物はGEヘルスケア社のカラムSuperdex 200を用いたSEC(AKTAexplorer, GE Helthcere)によって単峰性の分子量分布を持つことが確認された。また得られたポリマーは、1H-NMR(ESC400, JEOL)による解析から、Lysが43であることが分かった。ここでLys(TFA)-NCAは非特許文献(J. Polym. Sci. Part A Polym. Chem. 2003, 41 1167-1187)に則りFuchs-Farthing法により合成した。ジオキサンはWako社より購入したものを用いた。DMF(1M TU)は関東化学より購入した脱水溶媒にシグマアルドリッチ社より購入したチオウレアを溶解させて調整した。1M NaOH溶液はナカライ社より購入した5M NaOH溶液を純水で薄めて用意した。0.01 M HClは犬印社より購入した濃塩酸を純水で薄めて用意した。
また文献Bioconjugate, 2007, 18, 2191-2196にある通り、DIG-PEG-OHのOH末端をアジド基に変換した。まず、DIG-PEG-OH(550 mg, 0.050 mmol)をベンゼン凍結乾燥した。これを20 mLのTHFに溶かし、トリエチルアミン(25 μL, 0.20 mmol)加えてPEG溶液とした。別のフラスコに、メタンスルホニルクロリド(16 μL, 0.20 mmol)を測りとり、THF 5 mLで希釈した。水冷下で、このメタンスルホニルクロリド希釈液に上述で調整したPEG溶液を加え、一晩反応させた。反応はAr雰囲気下で行った。反応物は、ジエチルエーテル 500 mLにより再沈殿することで回収した。1H-NMR(ESC400, JEOL)による解析から、回収したサンプルは末端がメシル化されたDIG-PEG-Msであることが分かった。得られたDIG-PEG-Ms(440 mg, 0.044 mmol)をDMFに20 mLに溶かし、アジ化ナトリウム(286 mg, 4.4 mmol)を加え、50℃で三日間撹拌した。THFおよびDMFは関東化学より購入した超脱水溶媒を用いた。トリエチルアミンはWako社より購入し、カルシウムハイドライドを脱水剤として用いて蒸留精製して使用した。メシルクロライドはナカライ社より購入し、五酸化二リンを脱水剤として蒸留したものを用いた。アジ化ナトリウムはWako社より購入したものをそのまま用いた。
続いて、Alkyne-PnPrOx-PLysとDIG-PEG-N3をClick Chemistryによりカップリングした。Alkyne-PnPrOx-PLys (31 mg, 0.0020 mmol)を水2 mLに溶解し、1M のCuSO4溶液と1Mのアスコルビン酸ナトリウム溶液を20 μLずつ加え、撹拌した。別途、DIG-PEG- N3(110 mg, 0.01 mmol)を水2 mLに溶解し、Alkyne-PnPrOx-PLys溶液に加えた。これを-20℃で一晩静置し、4℃で2時間かけて融解させた。生成物は、反応溶液を水に対して5回透析し、凍結乾燥することで回収した。ここでの回収物はトリブロック共重合体の他に、Alkyne-PnPrOx-PLysを全て反応させるために過剰に加えたGlc-PEG-N3および未反応のAlkyne-PnPrOx-PLysも含まれている。これら未反応物と生成物のトリブロック共重合体の分子量の違いに着目し、SECにより精製作業を行った。最後にWO2015075942A1に記載の通りにTFAを用いてDIGをグルコースに変換し、Glc-PEG-b-PnPrOx-b-PLysとした。
カップリングの進行の確認や、SECによる精製作業はGEヘルスケア社のカラムSuperdex 200を用いたSEC(AKTAexplorer, GE Helthcere)を用いた。
本トリブロック共重合体の下限臨界共溶温度(LCST)は約30℃であった。
まず重合開始剤p-トルエンスルホン酸プロパルギル(61 mg, 0.29 mmol)をアセトニトリル 7 mLに溶解させ、2-n-プロピル-2-オキサゾリン(2.5 g, 22 mmol)を加えた。反応溶液を42℃で6日間反応させた後、アジ化ナトリウム(380 mg, 5.8 mmol)を加えて70℃で一時間撹拌して、重合反応を停止させた。上記のプロセスは全てAr雰囲気化で行った。ここで用いたp-トルエンスルホン酸プロパルギルは東京化成より購入し、Wako社より購入した五酸化ニリンを脱水剤として用いて蒸留精製して使用した。2-n-プロピル-2-オキサゾリンは東京化成より購入し、シグマアルドリッチ社より購入したカルシウムハイドライドを脱水剤として用いて蒸留精製して使用した。反応溶媒のアセトニトリルはwako社より購入し、カルシウムハイドライドを脱水剤として用いて蒸留精製して使用した。アジ化ナトリウムはWako社より購入し、そのまま用いた。重合反応後、反応溶液を水に対して5回透析し、凍結乾燥することでアジド末端を持つalkyne-PnPrOx-N3を得た。得られたPnPrOxは、MALDI-TOF MS(UltraFlextreme, Bruker社)と1H-NMR(ESC400, JEOL)による解析から、分子量が8.3kであることが分かった。
続いてalkyne-PnPrOx-N3のアジド末端をスタウディンガー反応により、アミン末端に変換した。alkyne-PnPrOx-N3(830 mg, 0.10 mmol)をメタノール20 mLに溶解し、トリフェニルホスフィン(530 mg, 2.0 mmol)加えて40℃で三時間撹拌した。続いて純水20 mLを加えて氷冷し、未反応のトリフェニルホスフィン等を濾過で取り除いた。得られた溶液は純水に対して3回透析した後、凍結乾燥により回収した。得られたalkyne-PnPrOx-NH2はGE-health care社より購入したCM-Sephadex C50をカラム充填剤としたオープンカラムを用いて更に精製した。ここで用いたメタノール、トリフェニルホスフィンは、それぞれシグマアルドリッチ社、東京化成より購入した。
上述で精製・回収したalkyne-PnPrOx-NH2よりLys(TFA)-NCAを重合し、その後塩基によるTFA基の脱保護により、alkyne-PnPrOx-b-PLysを得た。alkyne-PnPrOx-NH2 (330 mg, 0.040 mmol )を、ジオキサンを用いて凍結乾燥した。これを1Mの濃度でチオウレアを溶かしたDMF (DMF (1M TU)) 2 mLに溶解させて、開始剤溶液とした。別途Arバック中でLys(TFA)-NCA(480 mg, 1.8 mmol)をフラスコに測りとり、6 mLのDMF(1M TU)に溶解させた。調製したLys(TFA)-NCA溶液を、Ar雰囲気下で開始剤溶液に加え、25℃で3日間撹拌して重合反応を行った。反応溶液を水に対して5回透析したのち、凍結乾燥することでPnPrOx-b-Poly(L-Lysine)(TFA) (PLys(TFA))を得た。続いて得られたalkyne-PnPrOx-b-PLys(TFA)を500 mg測りとり、25 mLのメタノールに溶解させた。これに対して、7.5 mLの1M NaOH溶液を加え、35℃で12時間反応させた。反応後は、0.01 M HClに対して3回透析を行った後、水に対して3回透析を行い、凍結乾燥によりPnPrOx-b-PLysを回収した。回収物はGEヘルスケア社のカラムSuperdex 200を用いたSEC(AKTAexplorer, GE Helthcere)によって単峰性の分子量分布を持つことが確認された。また得られたポリマーは、1H-NMR(ESC400, JEOL)による解析から、Lysが43であることが分かった。ここでLys(TFA)-NCAは非特許文献(J. Polym. Sci. Part A Polym. Chem. 2003, 41 1167-1187)に則りFuchs-Farthing法により合成した。ジオキサンはWako社より購入したものを用いた。DMF(1M TU)は関東化学より購入した脱水溶媒にシグマアルドリッチ社より購入したチオウレアを溶解させて調整した。1M NaOH溶液はナカライ社より購入した5M NaOH溶液を純水で薄めて用意した。0.01 M HClは犬印社より購入した濃塩酸を純水で薄めて用意した。
また文献Bioconjugate, 2007, 18, 2191-2196にある通り、DIG-PEG-OHのOH末端をアジド基に変換した。まず、DIG-PEG-OH(550 mg, 0.050 mmol)をベンゼン凍結乾燥した。これを20 mLのTHFに溶かし、トリエチルアミン(25 μL, 0.20 mmol)加えてPEG溶液とした。別のフラスコに、メタンスルホニルクロリド(16 μL, 0.20 mmol)を測りとり、THF 5 mLで希釈した。水冷下で、このメタンスルホニルクロリド希釈液に上述で調整したPEG溶液を加え、一晩反応させた。反応はAr雰囲気下で行った。反応物は、ジエチルエーテル 500 mLにより再沈殿することで回収した。1H-NMR(ESC400, JEOL)による解析から、回収したサンプルは末端がメシル化されたDIG-PEG-Msであることが分かった。得られたDIG-PEG-Ms(440 mg, 0.044 mmol)をDMFに20 mLに溶かし、アジ化ナトリウム(286 mg, 4.4 mmol)を加え、50℃で三日間撹拌した。THFおよびDMFは関東化学より購入した超脱水溶媒を用いた。トリエチルアミンはWako社より購入し、カルシウムハイドライドを脱水剤として用いて蒸留精製して使用した。メシルクロライドはナカライ社より購入し、五酸化二リンを脱水剤として蒸留したものを用いた。アジ化ナトリウムはWako社より購入したものをそのまま用いた。
続いて、Alkyne-PnPrOx-PLysとDIG-PEG-N3をClick Chemistryによりカップリングした。Alkyne-PnPrOx-PLys (31 mg, 0.0020 mmol)を水2 mLに溶解し、1M のCuSO4溶液と1Mのアスコルビン酸ナトリウム溶液を20 μLずつ加え、撹拌した。別途、DIG-PEG- N3(110 mg, 0.01 mmol)を水2 mLに溶解し、Alkyne-PnPrOx-PLys溶液に加えた。これを-20℃で一晩静置し、4℃で2時間かけて融解させた。生成物は、反応溶液を水に対して5回透析し、凍結乾燥することで回収した。ここでの回収物はトリブロック共重合体の他に、Alkyne-PnPrOx-PLysを全て反応させるために過剰に加えたGlc-PEG-N3および未反応のAlkyne-PnPrOx-PLysも含まれている。これら未反応物と生成物のトリブロック共重合体の分子量の違いに着目し、SECにより精製作業を行った。最後にWO2015075942A1に記載の通りにTFAを用いてDIGをグルコースに変換し、Glc-PEG-b-PnPrOx-b-PLysとした。
カップリングの進行の確認や、SECによる精製作業はGEヘルスケア社のカラムSuperdex 200を用いたSEC(AKTAexplorer, GE Helthcere)を用いた。
本トリブロック共重合体の下限臨界共溶温度(LCST)は約30℃であった。
次に、多価カチオンとして、親水性ブロックを連結させて生体適合性を高めたカチオン性ポリマーを合成した。具体的には、分岐PEG(分子量37000×2)を有するブロックカチオマーである、分岐ポリ(エチレングリコール)-b-ポリ(L-リジン) (重合度20) (以下、「bPEG-b-PLL」、「PEGasus-PLL」、または「PEGasus-PLL(37×2-20)」とも称する)を合成した。
日油より購入した末端に一級アミンの構造を持つ分岐ポリエチレングリコール(bPEG)(分子量37000×2)を開始剤として、非特許文献(J. Polym. Sci. Part A Polym. Chem. 2003, 41 1167-1187)に則りFuchs-Farthing法により合成したLysine(TFA)-NCAを重合した後、塩基で処理することで得られる。
具体的には、bPEG-PLLの合成では、まず一級アミンを持つbPEG(740 mg, 0.010mmol)をナカライ社より購入したベンゼンにより凍結乾燥した。続いて反応溶媒として、シグマアルドリッチ社より購入したチオウレア(TU)を関東化学社より購入した脱水N,N-ジメチルホルムアミド(DMF)に溶解させて1Mの濃度に調製したDMF(1M TU)を用意した。凍結乾燥させたbPEGは10 mLのDMF (1M TU)に溶解させた。続いて、Lys(TFA)-NCAをArバック中で59 mg (0.22 mmol)フラスコに測り、DMF 2 mLに溶解させた。調製したLys(TFA)-NCA溶液をAr雰囲気下でbPEG溶液に加え、25℃で3日間撹拌した。反応溶液は、メタノール(シグマアルドリッチ社)とジエチルエーテル(昭和エーテル社)の1:9混合溶液300 mLに対して滴定し、共重合体bPEG-PLL(TFA)を再沈殿させた。 得られたbPEG-PLL(TFA)を濾過によって回収し、真空乾燥した。得られたbPEG-PLL(TFA)を500 mg測りとり、25 mLのメタノールに溶解させた。続いてこのbPEG-PLL(TFA)溶液に1M NaOH を7.5 mL加えて35℃で12時間撹拌し、TFA基の脱保護を行った。反応後は、0.01 M HClに対して3回透析を行った後、水に対して3回透析を行い、凍結乾燥によりbPEG-PLLを回収した。得られたbPEG-PLLはGEヘルスケア社のカラムsuperdex 200を用いた SEC(LC2000 system, 日本分光社)、1H-NMR(ESC400, JEOL)により解析した。
日油より購入した末端に一級アミンの構造を持つ分岐ポリエチレングリコール(bPEG)(分子量37000×2)を開始剤として、非特許文献(J. Polym. Sci. Part A Polym. Chem. 2003, 41 1167-1187)に則りFuchs-Farthing法により合成したLysine(TFA)-NCAを重合した後、塩基で処理することで得られる。
具体的には、bPEG-PLLの合成では、まず一級アミンを持つbPEG(740 mg, 0.010mmol)をナカライ社より購入したベンゼンにより凍結乾燥した。続いて反応溶媒として、シグマアルドリッチ社より購入したチオウレア(TU)を関東化学社より購入した脱水N,N-ジメチルホルムアミド(DMF)に溶解させて1Mの濃度に調製したDMF(1M TU)を用意した。凍結乾燥させたbPEGは10 mLのDMF (1M TU)に溶解させた。続いて、Lys(TFA)-NCAをArバック中で59 mg (0.22 mmol)フラスコに測り、DMF 2 mLに溶解させた。調製したLys(TFA)-NCA溶液をAr雰囲気下でbPEG溶液に加え、25℃で3日間撹拌した。反応溶液は、メタノール(シグマアルドリッチ社)とジエチルエーテル(昭和エーテル社)の1:9混合溶液300 mLに対して滴定し、共重合体bPEG-PLL(TFA)を再沈殿させた。 得られたbPEG-PLL(TFA)を濾過によって回収し、真空乾燥した。得られたbPEG-PLL(TFA)を500 mg測りとり、25 mLのメタノールに溶解させた。続いてこのbPEG-PLL(TFA)溶液に1M NaOH を7.5 mL加えて35℃で12時間撹拌し、TFA基の脱保護を行った。反応後は、0.01 M HClに対して3回透析を行った後、水に対して3回透析を行い、凍結乾燥によりbPEG-PLLを回収した。得られたbPEG-PLLはGEヘルスケア社のカラムsuperdex 200を用いた SEC(LC2000 system, 日本分光社)、1H-NMR(ESC400, JEOL)により解析した。
得られたトリブロック重合体とmRNAとからポリプレックスミセルを形成させた。具体的には、4℃においてmRNAと電荷比2となるよう混合し、その後、37℃で静置することによりトリブロック重合体にmRNAを包み込むように疎水性層を形成させ、疎水性層をmRNAの保護層として持つポリプレックスミセルを得た。
mRNAは、テンプレートのDNAをmMESSAGE mMACHINE T7 Ultra Kit (Ambion社) を用いてin vitroトランスクリプションする、またpoly(A) tail kit(Ambion社)を用いてpoly(A)修飾を施すことで調製した。またテンプレートのDNAとしては、New England Biolabs社より購入したpCMV-Gluc control plasmidを用いた。
pDNAは理研バイオリソースセンターより提供されたルシフェラーゼがコードされCAGプロモーターを持つpCAG-Luc2を用いた。
pDNAは理研バイオリソースセンターより提供されたルシフェラーゼがコードされCAGプロモーターを持つpCAG-Luc2を用いた。
(1)多価カチオンによるポリプレックスミセルの体内動態の変化
[実験]
実験では、絶食マウスに対して、上記グルコース修飾ポリプレックスミセルを単独で投与した場合(ミセル単独投与群)と、同量の上記グルコース修飾ポリプレックスミセルに加えてカチオン性ポリマー(PEGasus-PLL)と投与した場合(多価カチオン投与群)とで、mRNAの体内動態を比較した。この実験で用いるmRNAはあらかじめLabel IT Tracker Cy5 Kit(Mirus Bio社)で蛍光ラベル化した。
具体的には、一晩マウスを絶食し、飢餓状態にした。このマウスに対して、20%グルコース溶液をi.p.投与した。30分後、ミセル単独投与群には、200 ng/μLのmRNA濃度に調整したポリプレックスミセル溶液200 μLに100 μLのHEPES bufferを加えた300 μLを投与し、多価カチオン投与群には、200 ng/μLのmRNA濃度に調整したポリプレックスミセル溶液200 μLに加えて、投与するmRNAに対して電荷比3となるよう濃度を調整したPEGasus-PLL溶液を100 μL(計300 μL)を投与した。投与30分後に全血を回収してPBSにより灌流した後、マウスを解剖して、臓器(肝臓、腎臓および脳)を摘出した。回収した臓器の全体はLysis buffer (promega社)とマルチビーズショッカー(安井器械社)を用いてすりつぶし、この懸濁液のCy5蛍光量をプレートリーダー (Infinite M100 Pro, TECAN社) で測定し、ミセルの臓器分布を評価した。全投与量を100%とし、臓器に蓄積した量(%)を求めた。結果は、図1および2に示される通りであった。
[実験]
実験では、絶食マウスに対して、上記グルコース修飾ポリプレックスミセルを単独で投与した場合(ミセル単独投与群)と、同量の上記グルコース修飾ポリプレックスミセルに加えてカチオン性ポリマー(PEGasus-PLL)と投与した場合(多価カチオン投与群)とで、mRNAの体内動態を比較した。この実験で用いるmRNAはあらかじめLabel IT Tracker Cy5 Kit(Mirus Bio社)で蛍光ラベル化した。
具体的には、一晩マウスを絶食し、飢餓状態にした。このマウスに対して、20%グルコース溶液をi.p.投与した。30分後、ミセル単独投与群には、200 ng/μLのmRNA濃度に調整したポリプレックスミセル溶液200 μLに100 μLのHEPES bufferを加えた300 μLを投与し、多価カチオン投与群には、200 ng/μLのmRNA濃度に調整したポリプレックスミセル溶液200 μLに加えて、投与するmRNAに対して電荷比3となるよう濃度を調整したPEGasus-PLL溶液を100 μL(計300 μL)を投与した。投与30分後に全血を回収してPBSにより灌流した後、マウスを解剖して、臓器(肝臓、腎臓および脳)を摘出した。回収した臓器の全体はLysis buffer (promega社)とマルチビーズショッカー(安井器械社)を用いてすりつぶし、この懸濁液のCy5蛍光量をプレートリーダー (Infinite M100 Pro, TECAN社) で測定し、ミセルの臓器分布を評価した。全投与量を100%とし、臓器に蓄積した量(%)を求めた。結果は、図1および2に示される通りであった。
図1に示されるように、肝臓へのmRNAの蓄積量は、ミセル単独投与群では、30%弱であったのに対して、多価カチオン投与群では、蓄積量は10%強程度であり、肝臓への蓄積量が半分以下に低下していることが示された。また、図1に示されるように腎臓へのmRNAの蓄積量は、ミセル単独投与群では、約7%であったのに対して、多価カチオン投与群では、約5%程度であり、腎臓への蓄積量が7割程度に低下していることが示された。
次に、脳へのmRNAの送達量を、脳組織へのmRNAの蓄積量で評価したところ、図2に示されるように、ミセル単独投与の場合であっても0.05%もの大量のmRNAの脳組織への蓄積が確認された。これは、ポリプレックスミセル自体がmRNAを極めて顕著に安定化したことを意味する(なお、mRNAは血中では不安定であり、数秒で検出限界以下になる)。また、これに対して、多価カチオン投与群では、脳へのmRNAの送達量がさらに5倍以上も向上していた。
さらに、脾臓、心臓など他の臓器へのmRNAの蓄積量を評価したところ、図2Aに示されるように、多価カチオン投与群では、脾臓へのmRNAの蓄積量も、心臓へのmRNAの蓄積量も有意に増加した。
また、肝臓や腎臓へのmRNAの蓄積量の低下は、肝臓や腎臓での血中からのmRNAの排泄能と関連すると考えられた。従って、多価カチオンは、肝臓や腎臓での血中からのmRNAの排泄能を低下させ、これにより肝臓や腎臓以外の臓器へのmRNA、すなわち薬物の分布を増加させる作用を有していることが明らかとなった。上記実施例では、脳への送達実験では、脳への選択的な蓄積を促すために血糖操作(絶食およびグルコース投与)およびグルコースによるミセルの被覆を行った。従って、このような選択的なmRNAの送達を狙った系において、mRNAの排泄能の低下と血中滞留性の増加が、脳への選択的なmRNAの蓄積を増強させた。一方で、上記実施例では、脾臓や心臓に対しては、これらの臓器への選択的な蓄積のための特別な操作は行っていない。しかし、それでも脾臓や心臓などの臓器に対してmRNAの蓄積量が向上していることから、カチオン性ポリマーは、mRNAの血中滞留性を向上させる効果を通じて、肝臓や腎臓以外の臓器への薬物の蓄積を促進する効果を有していると考えられた。
(2)多価カチオンによる脂質ベースmRNA複合体の体内動態の変化
肝臓にRNAを送達する試薬としてInvivofectamine(商標)がインビトロジェン社より市販されている。Invivofectamine(商標)は脂質ベースのmRNAを全身投与するための試薬であり、特に肝臓へのmRNAの送達に適している。
ここでは、Invivofectamine(商標)とmRNAを製造者プロトコルに従って混合し、脂質ベースのmRNA複合体を形成させた。この実験で用いるmRNAはあらかじめLabel IT Tracker Cy5 Kit(Mirus Bio社)で蛍光ラベル化した。
マウスに対して、12.5 mg/mLの濃度に調整したPEGasus-PLL溶液100 μLを静脈投与した。5分後に、上述で調整したinvivofectamine溶液200 μLをi.v.投与した。30分後に全血を回収してPBSにより灌流した後、マウスを解剖して臓器(肝臓および腎臓)を摘出した。ポリマー添加の効果を見る陰性対照の群として、マウスに対してポリマー添加剤の先行投与を行わずに上述で調整した溶液200 μLを静脈投与した群と比較した。回収した臓器はLysis buffer (promega社)とマルチビーズショッカー(安井器械社)を用いてすりつぶし、この懸濁液のCy5蛍光量をプレートリーダー (Infinite M100 Pro, TECAN社) で測定し、ミセルの臓器分布を評価した。
肝臓にRNAを送達する試薬としてInvivofectamine(商標)がインビトロジェン社より市販されている。Invivofectamine(商標)は脂質ベースのmRNAを全身投与するための試薬であり、特に肝臓へのmRNAの送達に適している。
ここでは、Invivofectamine(商標)とmRNAを製造者プロトコルに従って混合し、脂質ベースのmRNA複合体を形成させた。この実験で用いるmRNAはあらかじめLabel IT Tracker Cy5 Kit(Mirus Bio社)で蛍光ラベル化した。
マウスに対して、12.5 mg/mLの濃度に調整したPEGasus-PLL溶液100 μLを静脈投与した。5分後に、上述で調整したinvivofectamine溶液200 μLをi.v.投与した。30分後に全血を回収してPBSにより灌流した後、マウスを解剖して臓器(肝臓および腎臓)を摘出した。ポリマー添加の効果を見る陰性対照の群として、マウスに対してポリマー添加剤の先行投与を行わずに上述で調整した溶液200 μLを静脈投与した群と比較した。回収した臓器はLysis buffer (promega社)とマルチビーズショッカー(安井器械社)を用いてすりつぶし、この懸濁液のCy5蛍光量をプレートリーダー (Infinite M100 Pro, TECAN社) で測定し、ミセルの臓器分布を評価した。
結果は、図3に示される通りであった。図3上パネルに示されるように、肝臓へのmRNAの蓄積量は、複合体単独投与の場合、7%強であったのに対して、多価カチオンを事前に投与した場合、蓄積量は0.5%以下であった。このことから、多価カチオンの事前投与により、mRNA複合体の肝臓への蓄積を劇的に減少させることが明らかとなった。
また、図3下パネルに示されるように、腎臓へのmRNAの蓄積量は、単独投与の場合8%弱であったのに対して、多価カチオンを事前に投与した場合には蓄積量は4%弱であった。このことから、多価カチオンは、mRNA複合体の腎臓への蓄積も減少させることが明らかとなった。
また、図3下パネルに示されるように、腎臓へのmRNAの蓄積量は、単独投与の場合8%弱であったのに対して、多価カチオンを事前に投与した場合には蓄積量は4%弱であった。このことから、多価カチオンは、mRNA複合体の腎臓への蓄積も減少させることが明らかとなった。
また、リポプレックスについても、脾臓、心臓など他の臓器へのmRNAの蓄積量を評価したところ、図3Aに示されるように、多価カチオン投与群では、脾臓へのmRNAの蓄積量も、肺へのmRNAの蓄積量も、心臓へのmRNAの蓄積量も有意に増加した。このことから、血中からのmRNAの排泄能の低下により、各種臓器へのmRNA、すなわち薬物の送達が促進されたことが示唆された。
実施例2:多価カチオンの体内動態
本実施例では、上記機能が発揮される原因を追及するため、多価カチオンの体内動態を解明した。
本実施例では、上記機能が発揮される原因を追及するため、多価カチオンの体内動態を解明した。
マウスに対して、Thermo Fisher scientific社より購入したAlexa Fluor 594 NHS Esterを用いて蛍光標識した12.5 mg/mLの濃度のPEGasus-PLL(37×2-20)を100 μL静注した。その後、肝臓の血管をin vivo共焦点顕微鏡で観察した。血管内のポリマー蓄積は、蛍光により評価できる。
結果は図4に示される通りであった。図4に示されるように、多価カチオンは、投与1分後から類同内皮細胞の血管内面に吸着し、蓄積するようすが観察された。
次に、末梢血管の一例として、耳の血管を観察した。結果は図5に示されるように、多価カチオンが血管内を流れることが確認できたが、耳の血管内皮細胞の血管内面への蓄積は観察されなかった。
このように、多価カチオンは、肝臓の類同内皮細胞の血管内面には蓄積する一方で、末梢血管の血管内面への蓄積は認められなかった。
肝臓や腎臓は、薬物動態学において、血中からの薬物の排泄能に大きな影響を与える臓器であることが知られている。多価カチオンは、類同内皮細胞の表面に吸着し、血管内面を物理的に被覆することによって、肝臓の類同内皮細胞や腎臓による血中からの排泄能を低減させ、血中からの薬物の排泄能の低下作用を有していることが分かった。また、この被覆は、肝臓の類同内皮細胞や腎臓の血管内皮細胞に対して特異的であった。
実施例3:多価カチオンによる、ミセルの血中滞留性の向上効果
上記実施例では、多価カチオンが、肝臓の類同内皮細胞や腎臓による血中からの薬物の排泄能の低下作用を有することを明らかにしてきた。
本実施例では、これら排泄能の低下が、薬剤の血中滞留性の向上に寄与しているかを確認した。
上記実施例では、多価カチオンが、肝臓の類同内皮細胞や腎臓による血中からの薬物の排泄能の低下作用を有することを明らかにしてきた。
本実施例では、これら排泄能の低下が、薬剤の血中滞留性の向上に寄与しているかを確認した。
マウスに対して、多価カチオンとして12.5 mg/mLの濃度に調整したPEGasus-PLL溶液100μLを静脈投与した。5分後に、Biomaterial, 2017, 126, 31-38の方法で作成したプラスミドDNA(pDNA)内包高分子ミセル溶液200 μLを投与した。この実験で用いるpDNAはあらかじめLabel IT Tracker Cy5 Kit(Mirus Bio社)で蛍光ラベル化した。その後、pDNAの血中滞留性を確認した。蛍光強度の最大値を100%とし、時間経過に伴う蛍光強度の変化により、pDNAの血中滞留性を評価した。対照の群として、マウスに対して多価カチオンの先行投与を行わずに、上述で調整した溶液200 μLを静脈投与した群についても観察した。
結果は、図6に示される通りであった。図6に示されるように、多価カチオンを先行投与しなかった群では、急速にpDNAの血中残存量が低下していくのに対して、多価カチオンを先行投与した群では、その血中滞留性が顕著に増大することが明らかとなった。
また、一本鎖PEGを用いた実験では、PEG部分の平均分子量は大きい方が血中滞留性は向上した(例えば、50kDより60kD、70kDのほうが血中滞留性が向上)。
また、一本鎖PEGを用いた実験では、PEG部分の平均分子量は大きい方が血中滞留性は向上した(例えば、50kDより60kD、70kDのほうが血中滞留性が向上)。
次に、肝臓へのミセルの蓄積をin vivo共焦点顕微鏡で観察した。図7上パネルに示されるように、多価カチオンの先行投与をしない群では、pDNAが肝臓の類同内皮細胞の血管内面に蓄積するようすが確認できる。これに対して、図7下パネルに示されるように、多価カチオンの先行投与をした群では、pDNAの肝臓の類同内皮細胞の血管内面への蓄積が顕著に低下した。
これらの結果から、実施例1や2において観察された血中からの薬物の排泄能の低下の原因は、肝臓の類同内皮細胞の血管内面を多価カチオンが被覆することにより、類同内皮細胞への薬物の集積が阻害されること、および腎臓の血管内皮細胞の血管内面をカチオン性ポリマーが被覆することにより、糸球体を介した薬物の排泄が阻害されることによると考えられた。このようにして、多価カチオンは、単独で(すなわち、遊離形態で)投与しても、血中からの薬剤の排泄能を低下させ、その血中滞留性を向上させる効果を有し、これにより、標的臓器への薬物送達量を改善することができると考えられた。
次に、多価カチオンとアルブミンとの相互作用を確認した。具体的には、マルバーンの等温滴定型カロリメーター、Microcal PEAQ-ITCを用いて、アルブミンとPEGasus-PLLを混合した際に発生する熱量を測定した。セル側サンプルとしてPBSに33.3 μMの濃度でアルブミンを溶解したアルブミン溶液を200 μL、シリンジ側サンプルとしてPBSに313 μMの濃度でPEGasus-PLLを溶解したPEGasus-PLL溶液を用意した。撹拌スピード750 rpmのもと、アルブミン溶液に37 μLのPEGasus-PLL溶液を滴下し、熱変化を観察した。コントロール実験として、PBSにPEGasus-PLLを滴下したときの熱変化を観察した。装置条件は、Filter periodを5秒間、Feedback modeをhighとして行った。結果は図8に示される通りであった。
図8に示されるように、熱変化はアルブミン存在下および非存在下でほとんど同等であり、有意な相違は認められなかった。この結果は、PEGasus-PLLとアルブミンの間に相互作用がない、すなわち、PEGasus-PLLとアルブミンは結合しないことを示している。PEGasus-PLLは、アルブミンによる薬物の排除効果ではなく、肝臓の類同内皮細胞を被覆することによるクリアランス低下作用により、薬物の血中滞留性を向上させていると考えられる。
実施例4:多価カチオンによる、lipoplexの脾臓での遺伝子発現性向上
肝臓にRNAを送達する試薬としてInvivofectamine(商標)がインビトロジェン社より市販されている。Invivofectamine(商標)は脂質ベースのmRNAを全身投与するための試薬であり、特に肝臓へのmRNAの送達に適していると言われている。
ここでは、Invivofectamine(商標)とmRNAを製造者プロトコルに従って混合し、脂質ベースのmRNA複合体(lipoplex)を形成させた。
マウスに対して、多価カチオンとして12.5 mg/mLの濃度に調整したPEGasus-PLL溶液100μLを静脈投与した。5分後に、上述で調整したinvivofectamine溶液200 μLをi.v.投与した。24時間後に臓器(肝臓および脾臓)を摘出した。ポリマー添加の効果を見る陰性対照の群として、マウスに対して多価カチオンの先行投与を行わずに上述で調製した溶液200 μLを静脈投与した群と比較した。回収した臓器はLysis buffer (promega社)とマルチビーズショッカー(安井器械社)を用いてすりつぶした。この懸濁液より、promega社より購入したLuciferase assay kitを製造者プロトコルに従って用いて、luciferaseの発光量をルミノメーター (Lumat LB9507, ベルトールド社) で測定し、肝臓と脾臓での遺伝子発現量を評価した。結果は、図9に示される通りであった。
ここでは、Invivofectamine(商標)とmRNAを製造者プロトコルに従って混合し、脂質ベースのmRNA複合体(lipoplex)を形成させた。
マウスに対して、多価カチオンとして12.5 mg/mLの濃度に調整したPEGasus-PLL溶液100μLを静脈投与した。5分後に、上述で調整したinvivofectamine溶液200 μLをi.v.投与した。24時間後に臓器(肝臓および脾臓)を摘出した。ポリマー添加の効果を見る陰性対照の群として、マウスに対して多価カチオンの先行投与を行わずに上述で調製した溶液200 μLを静脈投与した群と比較した。回収した臓器はLysis buffer (promega社)とマルチビーズショッカー(安井器械社)を用いてすりつぶした。この懸濁液より、promega社より購入したLuciferase assay kitを製造者プロトコルに従って用いて、luciferaseの発光量をルミノメーター (Lumat LB9507, ベルトールド社) で測定し、肝臓と脾臓での遺伝子発現量を評価した。結果は、図9に示される通りであった。
図9に示されるように、多価カチオン添加により、Lipoplexの脾臓での遺伝子発現を100倍以上高めることが出来た。このように、多価カチオンの添加は、肝臓の類同内皮細胞や腎臓の内皮細胞による薬剤の取込みを抑制し、薬剤の血中滞留性を高めることにより、肝臓や腎臓以外の臓器への薬物の蓄積を高めることが実証された。図9と図3Aとの相違は、図3AではCy5の蛍光量を評価しているのに対して、図9は投与したmRNAの発現量が評価されているという点である。これらの実施例により、脾臓でのmRNAの発現量を向上させるためには多価カチオンの投与が有効であることが明らかとなった。
ところで、本実施例によれば、脾臓にmRNAを効果的に送達することができる。脾臓は、免疫細胞が産生される臓器であり、脾臓へのmRNAの送達および発現の達成は、RNAワクチンやペプチドワクチンなどのワクチンの開発につながる大きな成果である。
また、多価カチオン添加を行った場合、上記実施例で示されたのと同様に、肝臓での遺伝子発現は若干の減少が見られた。これは、先打ちしたPEGasus-b-PLLがlipoplexの肝臓の類洞内皮への集積を抑えたためであると考えられる。特に肝臓での遺伝子発現は、PEGasus-b-PLLでその大部分が抑制されたことから、Lipoplexは主に肝臓の類同内皮細胞に蓄積していたことが分かる。一方で、両者の肝臓での遺伝子発現量およびその差は劇的に大きなものではないことから、肝臓の類洞内皮にLipoplexを集積させたとしてもそれだけでは肝臓での高い遺伝子発現は得られないことを示している。また、これらの結果から、核酸の肝臓類洞内皮への集積は、血中からの核酸キャリアの除去に寄与すると共に、核酸の失活にも繋がっていると考えられる。
実施例5:bPEG-PLLの体内からの排出特性
本実施例では、多価カチオンの体内からの排出を調べた。
本実施例では、多価カチオンの体内からの排出を調べた。
具体的には、Thermofisher社より購入したAlexa Fluor 594 NHS esterを用いて、bPEG及び前述のようにLys(TFA)-NCAを重合することで合成したbPEG(分子量40k×2)-PLL(TFA)(重合度20)とPEG(分子量80k)-PLL(TFA)(重合度20)を蛍光ラベル化した。bPEG-PLL(TFA)とPEG-PLL(TFA)を前述のように塩基によってTFA保護基を脱保護して蛍光ラベル化されたbPEG-PLL、PEG-PLLとした。12.5 mg/mLの濃度に調節したこれらの溶液を100 μLマウスに対して尾静脈投与し、その血中滞留性をin vivo共焦点顕微鏡で観察した。結果は、図10に示される通りであった。
また、bPEG-PLL についてはin vivo共焦点顕微鏡で肝臓の類同内皮細胞の管腔側内壁を観察した。結果は、図11に示される通りであった。
また、bPEG-PLL についてはin vivo共焦点顕微鏡で肝臓の類同内皮細胞の管腔側内壁を観察した。結果は、図11に示される通りであった。
図10に示されるように、多価カチオンである重合度20のリシンが結合したものであるbPEG-PLLとPEG-PLLは、カチオン性ブロックを有しないbPEGと比較して血中滞留性がより低く、特にbPEG-PLLにおいては10時間後にはほとんど血中から排出された(図10)。このことから、カチオン性ブロックは、bPEGの血中滞留性を低下させる作用を有していることが示唆された。
また、図11に示されるように、類同内皮細胞の管腔側内壁を被覆したbPEG-PLLの存在量を経時的に観察したところ、投与後少なくとも1時間以上にわたって内壁を被覆し続けることができるが、投与6時間後には内壁からほとんどが消失した(図11)。
このことから、bPEG-PLLは、遊離形態における単独投与によって、薬物が送達されるに十分な時間は内皮細胞の管腔側内壁を被覆して薬物のクリアランスを低下させる一方で、薬物が送達された後には消失し得るものであると考えられた。また、bPEG-PLLを用いた実験から、本発明の多価カチオンは、薬物の投与時間と合わせて(またはその前後に合わせて)投与薬物のクリアランスを必要な時間だけ低下させるように、単回投与または複数回投与することが可能であることが示唆された。さらに、bPEG-PLLを用いた実験から、本発明の多価カチオンは、薬物が送達された後には体内から排出されるように投与する投与方式(投与頻度および投与量等)が可能であり得ることが示唆された。
実施例6:ウイルス粒子を用いた送達実験
本実施例では、多価カチオンの投与が、ウイルス粒子による遺伝子の送達に及ぼす影響を調べた。
本実施例では、多価カチオンの投与が、ウイルス粒子による遺伝子の送達に及ぼす影響を調べた。
多価カチオンとしては、PEGasus-b-PLLを用いた。また、ウイルス粒子としては、アデノ随伴ウイルス(AAV8)を用いた。AAV8には、CMVプロモーター下にホタルルシフェラーゼをコードする遺伝子を組込んだものをVector Biolabs社より購入した(Catalog #: VB1473)。マウス(Balb/c、雌、6週齢)の尾静脈からAAV8(1×1010ウイルスゲノム/マウス)を投与した。投与21日後にマウスを犠牲にして、臓器(肝臓、膵臓、四頭筋、大腿)の組織を採取し、パッシブ溶解緩衝液1mL中で組織100mgをホモジェナイズした。ホモジェネートを18,000gで10分間4℃にて遠心し、上清20μLについてルシフェラーゼよる発光を検出した。タンパク質濃度を決定するためにBCAアッセイを行った。ルシフェラーゼ活性は、プロテイン含量に対して正規化した相対的光学単位(RLU)で示された。結果は、図12に示される通りであった。
図12に示されるように、多価カチオンは、AAV8による、肝臓へのルシフェラーゼ導入効率を低下させ、膵臓、四頭筋、大腿の組織におけるルシフェラーゼ発現を増強させた。肝臓への蓄積性を減少させ、多臓器への集積性を増大させたことが想定される。このことから、本願発明のポリカチオンによって、投与する薬物の動態を変化させ、クリアランスを減少させる効果は、ウイルス粒子に対しても発揮されることが確認された。
次に、AAV8の代わりに心筋に遺伝子を運ぶことができるAAV9を用いる以外は同様に上記実験を行った。AAV9には、CMVプロモーター下にホタルルシフェラーゼをコードする遺伝子を組込んだものをSignaGen Laboratories社より購入した(Catalog #: SL101494)。但し、用量は、5×1010ウイルスゲノム/マウスとし、投与2日後にマウスを犠牲にした。その結果、図13に示されるように、多価カチオンは、AAV9の肝臓に対しては蓄積性を減少させ、肝臓への遺伝子導入効率を低下させた一方で、心臓に対する集積性を増強させ、心臓に対する遺伝子導入効率を高めた。
このように、ウイルスの種類や血清型によってウイルスの組織特異性には相違が存在するが、本発明によれば、カチオン性ポリマー(ポリカチオン)は、肝臓の類同内皮細胞の表面をブロックすることによって、ミセルに限られず、ウイルス粒子などの粒子状物の肝臓でのクリアランスを低下させるであろうこと、および、結果として、または同時に、ウイルス自身が有している臓器もしくは組織特異性を増強することが明らかとなった。
Claims (15)
- 多価カチオンを有効成分として含む、体内の薬物動態を制御することに用いるための組成物。
- 多価カチオンが、親水性ポリマーブロックとの結合体の形態である、請求項1に記載の組成物。
- 多価カチオンが、2本以上の親水性ポリマー鎖を有するカチオン性ポリマーである、請求項1に記載の組成物。
- 多価カチオンが、カチオン性ポリマーブロックと分岐ポリエチレングリコールとのブロック共重合体である、請求項2に記載の組成物。
- 薬物動態の制御が、血中からのクリアランスの制御である、請求項1~4のいずれか一項に記載の組成物。
- 薬物動態の制御が、肝臓の類同内皮細胞による血中からの薬物の排泄能の低減である、請求項1~5のいずれか一項に記載の組成物。
- 薬物動態の制御が、腎臓による血中からの薬物の排泄能の低減である、請求項1~5のいずれか一項に記載の組成物。
- 薬物動態の制御が、標的臓器または組織への送達量の増加である、請求項1~6のいずれか一項に記載の組成物。
- 薬物動態の制御が、脾臓への送達量の増加である、請求項1~6に記載の組成物。
- 薬物動態の制御が、薬物の血中滞留性の増大である、請求項1~4のいずれか一項に記載の組成物。
- 動態が制御される薬物よりも先に投与される、請求項1~10のいずれか一項に記載の組成物。
- 動態が制御される薬物と同時に投与される、請求項1~10のいずれか一項に記載の組成物。
- 動態が制御される薬物よりも後に投与される、請求項1~10のいずれか一項に記載の組成物{但し、前記投与は、前記薬物が血中に滞留している間に行われることを条件とする}。
- 多価カチオンが、遊離形態である、または遊離形態で投与される、請求項1~13のいずれか一項に記載の組成物。
- 動態が制御される薬物が、薬物送達用キャリアに内包された薬物である、請求項1~14のいずれか一項に記載の組成物。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980018952.7A CN111918673A (zh) | 2017-10-05 | 2019-03-12 | 控制体内的药物动态的组合物 |
| JP2020506543A JP7434145B2 (ja) | 2017-10-05 | 2019-03-12 | 体内における薬物動態を制御する組成物 |
| EP19768480.6A EP3766519A4 (en) | 2017-10-05 | 2019-03-12 | COMPOSITION FOR THE CONTROL OF PHARMACOKINETICS IN THE BODY |
| US16/980,063 US11957708B2 (en) | 2017-10-05 | 2019-03-12 | Composition controlling pharmacokinetics in the body |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017195061 | 2017-10-05 | ||
| JP2018-043880 | 2018-03-12 | ||
| JP2018043880A JP2019069933A (ja) | 2017-10-05 | 2018-03-12 | 体内における薬物動態を制御する組成物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019176916A1 true WO2019176916A1 (ja) | 2019-09-19 |
Family
ID=66441009
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/009919 WO2019176916A1 (ja) | 2017-10-05 | 2019-03-12 | 体内における薬物動態を制御する組成物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11957708B2 (ja) |
| EP (1) | EP3766519A4 (ja) |
| JP (2) | JP2019069933A (ja) |
| CN (1) | CN111918673A (ja) |
| WO (1) | WO2019176916A1 (ja) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013162041A1 (ja) | 2012-04-27 | 2013-10-31 | 国立大学法人 東京大学 | 核酸デリバリー用ユニット構造型医薬組成物 |
| WO2015075942A1 (ja) | 2013-11-22 | 2015-05-28 | 国立大学法人 東京大学 | 薬剤送達用のキャリア、コンジュゲートおよびこれらを含んでなる組成物並びにこれらの投与方法 |
| WO2016178431A1 (ja) * | 2015-05-07 | 2016-11-10 | 国立大学法人 東京大学 | ポリイオンコンプレックス型ポリマーソームを用いたナノリアクタとその製造方法 |
| WO2018038155A1 (ja) * | 2016-08-23 | 2018-03-01 | 国立大学法人東京大学 | ミセル及びその使用 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008201673A (ja) * | 2005-05-16 | 2008-09-04 | Kyushu Univ | Rna含有組成物 |
| CA2636599C (en) * | 2006-01-20 | 2014-07-15 | Starpharma Pty Limited | Modified macromolecule |
| WO2007099660A1 (ja) * | 2006-03-01 | 2007-09-07 | The University Of Tokyo | 核酸内包高分子ミセル複合体 |
| EP2087912B1 (en) * | 2006-11-22 | 2017-05-10 | The University of Tokyo | Sirna carrier using disulfide-bridged polymeric micelle |
| AU2009224286B2 (en) * | 2008-03-10 | 2014-06-12 | The University Of Tokyo | Copolymer including uncharged hydrophilic block and cationic polyamino acid block having hydrophobic group in part of side chains, and use thereof |
| WO2010106700A1 (ja) * | 2009-03-17 | 2010-09-23 | 国立大学法人東京大学 | タンパク質の電荷調節剤、及びタンパク質内包高分子ミセル複合体 |
| JP5463531B2 (ja) * | 2009-07-22 | 2014-04-09 | 国立大学法人 東京大学 | Phd2発現抑制物質搭載ポリイオンコンプレックス |
| WO2012005376A1 (ja) | 2010-07-09 | 2012-01-12 | 国立大学法人 東京大学 | 核酸送達用組成物及び担体組成物、それを用いた医薬組成物、並びに核酸送達方法 |
| CN102727907B (zh) | 2011-04-13 | 2015-03-11 | 苏州瑞博生物技术有限公司 | 一种小干扰rna药物的给药系统和制剂 |
| WO2014133172A1 (ja) * | 2013-03-01 | 2014-09-04 | 独立行政法人科学技術振興機構 | 物質内包ベシクル及びその製造方法 |
| EP3106177B1 (en) * | 2014-02-12 | 2022-07-27 | NanoCarrier Co., Ltd. | Polyion complex comprising mrna for therapy |
| EP3299020A4 (en) | 2015-05-21 | 2019-01-23 | The University Of Tokyo | MICROM COMPOSITION FOR NUCLEIC ACID RELIEF WITH TEMPERATURE-PERMANENT POLYMER AND METHOD FOR THE PRODUCTION THEREOF |
| WO2017086467A1 (ja) | 2015-11-19 | 2017-05-26 | 公立大学法人名古屋市立大学 | 抗腫瘍性ドラッグデリバリー製剤 |
-
2018
- 2018-03-12 JP JP2018043880A patent/JP2019069933A/ja active Pending
-
2019
- 2019-03-12 WO PCT/JP2019/009919 patent/WO2019176916A1/ja unknown
- 2019-03-12 US US16/980,063 patent/US11957708B2/en active Active
- 2019-03-12 EP EP19768480.6A patent/EP3766519A4/en active Pending
- 2019-03-12 CN CN201980018952.7A patent/CN111918673A/zh active Pending
- 2019-03-12 JP JP2020506543A patent/JP7434145B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013162041A1 (ja) | 2012-04-27 | 2013-10-31 | 国立大学法人 東京大学 | 核酸デリバリー用ユニット構造型医薬組成物 |
| WO2015075942A1 (ja) | 2013-11-22 | 2015-05-28 | 国立大学法人 東京大学 | 薬剤送達用のキャリア、コンジュゲートおよびこれらを含んでなる組成物並びにこれらの投与方法 |
| JP2017105802A (ja) * | 2013-11-22 | 2017-06-15 | 国立大学法人 東京大学 | 薬剤送達用のキャリア、コンジュゲートおよびこれらを含んでなる組成物並びにこれらの投与方法 |
| WO2016178431A1 (ja) * | 2015-05-07 | 2016-11-10 | 国立大学法人 東京大学 | ポリイオンコンプレックス型ポリマーソームを用いたナノリアクタとその製造方法 |
| WO2018038155A1 (ja) * | 2016-08-23 | 2018-03-01 | 国立大学法人東京大学 | ミセル及びその使用 |
Non-Patent Citations (8)
| Title |
|---|
| "Part A Polym. Chem.", J. POLYM. SCI., vol. 41, 2003, pages 1167 - 1187 |
| BIOCONJUGATE, vol. 18, 2007, pages 2191 - 2196 |
| BIOMATERIAL, vol. 126, 2017, pages 31 - 38 |
| EUROPEAN POLYMER JOURNAL, vol. 88, 2017, pages 553 - 561 |
| MINCHIN, RODNEY F. ET AL.: "Polyinosinic Acid and Polycationic Liposomes Attenuate the Hepatic Clearance of Circulating Plasmid DNA", THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 296, 2001, pages 1006 - 1012, XP055636124 * |
| OSAWA S. ET AL., BIOMACROMOLECULE, vol. 17, no. 1, 2016, pages 354 - 361 |
| See also references of EP3766519A4 |
| ZHENG, MENGYAO ET AL.: "Enhancing in vivo circulation and siRNA deli very with biodegradable polyethylenimine-graft-polycaprolactone-block- poly(ethylene glycol) copolymers", BIOMATERIALS, vol. 33, 2012, pages 6551 - 6558, XP028401114 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2019069933A (ja) | 2019-05-09 |
| US11957708B2 (en) | 2024-04-16 |
| JPWO2019176916A1 (ja) | 2021-02-25 |
| EP3766519A1 (en) | 2021-01-20 |
| JP7434145B2 (ja) | 2024-02-20 |
| US20210038634A1 (en) | 2021-02-11 |
| EP3766519A4 (en) | 2021-12-01 |
| CN111918673A (zh) | 2020-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Miyata et al. | Rational design of smart supramolecular assemblies for gene delivery: chemical challenges in the creation of artificial viruses | |
| Chen et al. | Polycations for gene delivery: dilemmas and solutions | |
| Wagner | Polymers for siRNA delivery: inspired by viruses to be targeted, dynamic, and precise | |
| Sarett et al. | Hydrophobic interactions between polymeric carrier and palmitic acid-conjugated siRNA improve PEGylated polyplex stability and enhance in vivo pharmacokinetics and tumor gene silencing | |
| Ewe et al. | A novel tyrosine-modified low molecular weight polyethylenimine (P10Y) for efficient siRNA delivery in vitro and in vivo | |
| Liu et al. | A novel galactose-PEG-conjugated biodegradable copolymer is an efficient gene delivery vector for immunotherapy of hepatocellular carcinoma | |
| Kano et al. | Grafting of poly (ethylene glycol) to poly-lysine augments its lifetime in blood circulation and accumulation in tumors without loss of the ability to associate with siRNA | |
| Fan et al. | Recent progress of crosslinking strategies for polymeric micelles with enhanced drug delivery in cancer therapy | |
| Ji et al. | Development of self-assembled multi-arm polyrotaxanes nanocarriers for systemic plasmid delivery in vivo | |
| JP2015515530A (ja) | インビボ核酸送達のためのポリ(アクリラート)ポリマー | |
| KR20160040524A (ko) | Rna를 세포에 도입하기 위한 조성물 | |
| US20230157969A1 (en) | POLYMERIC NANOPARTICLES PROVIDING NUCLEIC ACIDS ENCODING TNF-a | |
| JP6853924B2 (ja) | 温度感受性重合体を用いた核酸の送達用ミセル組成物およびその製造方法 | |
| US11793764B2 (en) | siRNA nanocapsule and preparation method and use thereof | |
| Sarisozen et al. | Lipid-based siRNA delivery systems: challenges, promises and solutions along the long journey | |
| Kozlu et al. | An aquaporin 4 antisense oligonucleotide loaded, brain targeted nanoparticulate system design | |
| Mogoşanu et al. | Natural and synthetic polymers for drug delivery and targeting | |
| Schwerdt et al. | Hyperthermia-induced targeting of thermosensitive gene carriers to tumors | |
| Casper et al. | Polyethylenimine (PEI) in gene therapy: Current status and clinical applications | |
| US8791086B2 (en) | Polyion complex comprising PHD2 expression inhibiting substance | |
| US20200171169A1 (en) | LONG-CIRCULATING ZWITTERIONIC POLYPLEXES FOR siRNA DELIVERY | |
| US9579394B2 (en) | Polyanion nanocomplexes for therapeutic applications | |
| JP7471373B2 (ja) | 血中で安定なmRNA内包ミセル | |
| JP7434145B2 (ja) | 体内における薬物動態を制御する組成物 | |
| US11147827B2 (en) | Conjugation of lipophilic albumin-binding moiety to RNA for improved carrier-free in vivo pharmacokinetics and gene silencing |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19768480 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020506543 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019768480 Country of ref document: EP Effective date: 20201012 |