WO2019013260A1 - 腸溶性硬質カプセル - Google Patents
腸溶性硬質カプセル Download PDFInfo
- Publication number
- WO2019013260A1 WO2019013260A1 PCT/JP2018/026216 JP2018026216W WO2019013260A1 WO 2019013260 A1 WO2019013260 A1 WO 2019013260A1 JP 2018026216 W JP2018026216 W JP 2018026216W WO 2019013260 A1 WO2019013260 A1 WO 2019013260A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- component
- hard capsule
- enteric
- mass
- enteric hard
- Prior art date
Links
Images








Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4891—Coated capsules; Multilayered drug free capsule shells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4816—Wall or shell material
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- enteric hard capsules Disclosed herein are enteric hard capsules, enteric hard capsule preparations, methods of preparing enteric hard capsule preparations, and methods of preparing enteric hard capsules.
- Enteric is one of the dosage forms of an orally administered formulation and generally refers to a formulation characteristic that is less soluble in the stomach.
- the preparation has the property of being easily dissolved after transfer to the intestine.
- An enteric preparation does not release the drug active ingredient in the stomach under strong acid environment, but releases the drug active ingredient after the preparation moves into the intestine.
- enteric-coated preparations are mainly used for the purpose of protecting a drug active ingredient from gastric acid or enzymes in the stomach, and for the purpose of sustained release of the drug active ingredient utilizing the time when the preparation travels from the stomach to the small intestine Be done.
- enteric formulation satisfying the above requirements has been prepared by coating the tablet with a so-called enteric polymer (Non-patent Document 1, Chapters 9 and 10).
- the dosage form is a hard capsule
- preparing an enteric hard capsule formulation by coating the same enteric polymer as the tablet on the hard capsule filled with contents coating method
- the enteric coating is applied to the non-enteric soluble capsule before release from the immersion pin by the immersion method from before (patent documents 1 to 6, non-patent documents 2 and 3).
- Patent Documents 12 and 13 Partially using polymers, such as conventional water-soluble, high film-forming gelatin and water-soluble cellulose, mainly comprising a poorly water-soluble acid-resistant enteric polymer (Patent Documents 12 and 13); (4) Salifying almost all the acid groups (especially carboxyl groups) of the enteric polymer to obtain a water-soluble derivative containing a poorly water-soluble enteric polymer, or basic neutralization of a non-chloride polymer Agents must be at least partially neutralized and dissolved in water, or use of non-chlorinated emulsion dispersions (Patent Documents 12 to 20); and (5) need to solubilize the polymer, such as injection molding Using an alternative technology that does not Etc. are made.
- polymers such as conventional water-soluble, high film-forming gelatin and water-soluble cellulose, mainly comprising a poorly water-soluble acid-resistant enteric polymer (Patent Documents 12 and 13); (4) Salifying almost all the acid groups (especially carboxyl
- Hard capsules are usually prepared in the dipping (immersion) method. Specifically, in the immersion method, the capsule shell polymer material is dissolved into an aqueous solution, a molding pin (generally a molding pin made of stainless steel) is immersed in the polymer aqueous solution, and the molding pin is pulled up from the immersion liquid. The forming pin is inverted, and the aqueous polymer solution attached to the surface of the forming pin is dried to form a film having a thickness of about 100 ⁇ m. The dried capsule shell is then removed from the molding pin and cut to the desired length before filling the contents, assembling the cap and body, printing on the surface of the hard capsule and packaging the hard capsule.
- immersion method the capsule shell polymer material is dissolved into an aqueous solution
- a molding pin generally a molding pin made of stainless steel
- the forming pin is inverted, and the aqueous polymer solution attached to the surface of the forming pin is dried to form a film having a thickness of about 100 ⁇ m.
- the polymer which is the main component of the hard capsule film is water-soluble, or most of it is an aqueous solution, or some of them are very It is desirable to form a dispersion containing fine colloidal or solid particles.
- the polymer when pulling up the molding pin immersed in the preparation liquid, the polymer has a property of causing gelation and rapid viscosity increase with rapid increase or decrease of temperature, that is, having a cold gelation or thermal gelation ability. Is desirable.
- the preparation liquid for immersion can suppress dripping immediately after pulling up the molding pin, and it is required that the solid content by subsequent evaporation of water is eventually solidified to form a film having sufficient hardness and toughness as a hard capsule.
- enteric polymers enteric base
- Enteric polymers marketed for tablet coating can function as a film on the solid surface of the tablet, but do not have film forming properties and strength that can be self-supporting as a film alone. Therefore, even if a film is formed of an enteric polymer, the film alone can not be used as a hard capsule.
- the prior art includes the following problems.
- the moldability of the hard capsule shell is improved, the acid resistance is insufficient.
- the pH of the aqueous solution containing the polymer, or the cation and the ionic group of the enteric polymer There is a problem that the stability of the polymer aqueous solution or dispersion and the cold gelation performance of the gelling agent are impaired by the interaction of
- the acid group of the enteric polymer is chlorinated, or the enteric polymer is almost completely neutralized (or salified) in order to obtain a preparation liquid for immersion.
- these treatments give the formed hard capsule shell itself an undesirable water sensitivity.
- the stability of the polymer aqueous solution or dispersion and the cold gelling performance of the gelling agent are impaired due to the pH of the aqueous solution containing the polymer or the interaction between the cation and the ionic group of the enteric polymer.
- the hard capsule mainly composed of the enteric polymer to which the above-mentioned treatment is added is stored under severe conditions of high temperature because the neutralizing agent (for example, the alkaline agent) is excessive. It may come out, so-called salting out, and may turn yellow in appearance.
- the neutralizing agent for example, the alkaline agent
- the general manufacturing apparatus by the immersion method can not be used at all.
- mold a capsule using the thermoplasticity of a polymer in injection molding the heat denaturation of the polymer itself by heat-processing about 100 degreeC in a formation process is concerned.
- the film is subjected to excessive stress due to heat contraction, and there is a concern that the capsule after molding may be cracked.
- the film of the hard capsule currently in circulation generally has a thickness of about 100 ⁇ m, and the contents are filled by a capsule filling machine.
- An object of the present invention is to provide a hard capsule formed of a hard capsule shell having enteric properties, which can be molded by a cold gel method.
- An enteric cellulose compound comprising a nonionic water-soluble cellulose compound and an enteric methacrylic acid copolymer having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s
- Hard capsules comprising a film comprising at least one component selected from the group consisting of a water-insoluble (meth) acrylic acid alkyl ester copolymer, and a polyvinyl alcohol copolymer, a plasticizer, and a surfactant have enteric properties Found out.
- an enteric hard capsule preparation containing the above-mentioned components can be used to prepare hard capsules by the cold gel method.
- An enteric hard capsule comprising a film comprising a first component and a second component and further comprising at least one component of a third component, a fourth component, and a fifth component
- the first component is a nonionic water-soluble cellulose compound having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s
- the second component is an enteric methacrylic acid copolymer
- the third component is an enteric cellulose compound
- the fourth component is a water insoluble (meth) acrylic acid alkyl ester copolymer
- the fifth component is at least one selected from the group consisting of polyvinyl alcohol, a plasticizer, and a surfactant, Enteric hard capsule.
- Item 2 The enteric hard capsule according to item 1, wherein the non-ionic water-soluble cellulose compound is at least one selected from the group consisting of hydroxypropyl methylcellulose, methyl cellulose and hydroxypropyl cellulose.
- Item 3 The item 1 or 2, wherein the enteric methacrylic acid copolymer is at least one selected from the group consisting of a copolymer of methacrylic acid and methyl methacrylate and methyl acrylate, or a copolymer of methacrylic acid and ethyl acrylate Enteric hard capsule as described.
- Item 5 The enteric hard according to any one of Items 1 to 4, wherein the enteric cellulose compound is at least one selected from the group consisting of hydroxypropyl methylcellulose phthalate, hydroxypropyl methylcellulose acetate succinate, and cellulose acetate phthalate. capsule.
- the enteric hard capsule according to any one of Items 1 to 5, wherein the (meth) acrylic acid alkyl ester copolymer is a copolymer of methyl methacrylate and ethyl acrylate.
- Item 7 The total mass of the first component, the second component, the third component, the fourth component, and the fifth component contained in the film is 100 mass%, and the ratio of the first component is ⁇ mass%, the ratio of the second component Is ⁇ mass%, the ratio of the third component is ⁇ mass%, the ratio of the fourth component is ⁇ %, and the ratio of the fifth component is ⁇ , 0.5 ⁇ ( ⁇ + ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ) ⁇ 0 Item 9.
- Item 8 The total mass of the first component, the second component, the third component, the fourth component, and the fifth component contained in the film is 100 mass%, and the ratio of the first component is ⁇ mass%, the ratio of the second component Is ⁇ mass%, the ratio of the third component is ⁇ mass%, the ratio of the fourth component is ⁇ %, and the ratio of the fifth component is ⁇ , 0.05 ⁇ ⁇ / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ) ⁇ 0.5
- the total mass of the first component, the second component, the third component, the fourth component, and the fifth component contained in the film is 100% by mass, and the ratio of the second component is ⁇ % by mass and that of the third component Item 9.
- the ratio of the first component when the total of the mass of the first component, the second component, the third component, the fourth component, and the fifth component contained in the film is 100 mass%, ⁇ mass%, the second component
- the ratio of ⁇ ⁇ mass%
- the ratio of the fourth component is ⁇ %
- At least a portion of the second component is contained as a pharmaceutically or food additive acceptable salt, and / or at least a third component is a pharmaceutically or food additive acceptable salt.
- the content of the carboxyl group forming the salt Item 12.
- Item 13 Item 13
- Item 16 The enteric hard capsule according to any one of items 1 to 15, wherein the film of the enteric hard capsule comprises a sea-island structure, and the island phase substantially consists of the first component.
- Item 17. The enteric hard capsule according to item 16, wherein the minor axis of the island phase is 0.1 ⁇ m or more and less than 30 ⁇ m.
- Item 19 The enteric hard capsule according to item 18, wherein the dissolution rate of the enteric hard capsule in the dissolution test is 10% or less.
- Item 20. An enteric composition comprising an ith component, an ii component, a basic neutralizing agent acceptable as a pharmaceutical or food additive, and a solvent, and further comprising at least one of a iii component, an iv component and a v component.
- the i-th component is a nonionic water-soluble cellulose compound having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s
- the second component is an enteric methacrylic acid copolymer
- the third component is an enteric cellulose compound
- the fourth component is a water insoluble (meth) acrylic acid alkyl ester copolymer
- the v component is at least one selected from the group consisting of polyvinyl alcohol, a plasticizer, and a surfactant, Enteric hard capsule preparation.
- Item 23 The enteric hard capsule preparation liquid according to item 20, wherein the ith component is dispersed as solid particles.
- the total mass of the ith component, the ii component, the iii component, the iv component, and the v component contained in the enteric hard capsule preparation liquid is 100 mass%, and the ratio of the i component is ⁇ 'mass%
- the ratio of the ii component is ⁇ ' mass%
- the ratio of the iv component is ⁇ 'mass%
- the ratio of the v component is ⁇ ' mass% 0.5 ⁇ ( ⁇ ′ + ⁇ ′ + ⁇ ′) / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) ⁇ 0.9, and 0.4 ⁇ ( ⁇ ′ + ⁇ ′) / ( ⁇ ′ + ⁇
- the enteric hard capsule preparation liquid according to any one of items 20 to 30, which is '+ ⁇ ').
- the total mass of the i-th component, the ii-th component, the iii-th component, the iv-th component, and the v-th component contained in the enteric hard capsule preparation liquid is 100 mass%, and the ratio of the i-th component is ⁇ 'mass %, The proportion of the ii component is ⁇ 'mass%, the proportion of the third component is ⁇ ' mass%, the proportion of the iv component is ⁇ 'mass%, and the proportion of the v component is ⁇ ' mass% Item 33.
- the enteric hard capsule preparation liquid according to any one of Items 20 to 31, wherein 0.05 ⁇ ⁇ ′ / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) ⁇ 0.5. Item 33.
- the ratio of the ii component is ⁇ Item 33.
- Soluble hard capsule preparation Item 34.
- the enteric hard capsule preparation liquid according to item 33 wherein 0.3 ⁇ ⁇ ′ / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) ⁇ 0.7.
- Item 35 The enteric hard capsule preparation liquid according to item 34, wherein the degree of neutralization of the second component by the basic neutralizing agent is 2 to 20%.
- Item 36 The enteric hard capsule preparation liquid according to any one of items 20 to 35, wherein the basic neutralizing agent is at least one selected from the group consisting of sodium hydroxide, potassium hydroxide and calcium hydroxide .
- Item 38. Item 31. The total amount of the i-th component, the ii-component, the iii-component, the iv-component and the v-component is 10 to 30% by mass, based on 100% by mass of the enteric hard capsule preparation liquid.
- 37. The enteric hard capsule preparation liquid according to any one of to 37.
- a method for preparing an enteric hard capsule preparation liquid, wherein the i-th component and the ii-th component are mixed under the condition that a pharmaceutically or food additive acceptable basic neutralizing agent is present in a solvent The preparation method, wherein the component i is a nonionic water-soluble cellulose compound having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s, and the component ii is an enteric methacrylic acid copolymer. Item 41.
- the method for preparing an enteric hard capsule preparation liquid according to Item 40 wherein the nonionic water-soluble cellulose compound is at least one selected from the group consisting of hydroxypropyl methylcellulose, methyl cellulose and hydroxypropyl cellulose.
- Method of preparation. Item 44.
- Step A preparing a neutralization solution of the component iii
- Step B adding the i-th component to the neutralized solution containing the iii-th component to prepare a partially dissolved solution of the i-th component
- Step C neutralizing or partially dissolving the dispersion of the second component Mixing with liquid
- the method for preparing an enteric hard capsule preparation liquid according to item 45 wherein the enteric cellulose compound is at least one selected from the group consisting of hydroxypropyl methylcellulose phthalate, hydroxypropyl methylcellulose acetate succinate, and cellulose acetate phthalate.
- the step A is a step of preparing a neutralization solution in which the component iii is at least partially neutralized and dissolved in a solvent with a basic neutralizing agent that is acceptable as a pharmaceutically or food additive,
- Item 48 is a step of preparing a neutralization solution in which the component iii is at least partially neutralized and dissolved in a solvent with a basic neutralizing agent that is acceptable as a pharmaceutically or food additive.
- Step B is a partially dissolved solution in which the i-th component is partially dissolved in a neutralized solution containing the above-mentioned component iii, or a mixed solution of a neutralized solution of the above-mentioned component iii and a dispersion liquid of the above component ii. It is a process of preparing, and the process of preparing a partial solution is the neutralization liquid containing the iii component or the iii component at the first temperature T1 of the i th component at the first temperature T1 or more of the cloud point T0 of the i component 46.
- a process of preparing a dispersion in which the i-th component is partially dissolved at a second temperature T2 lower than the cloud point, added to the mixed solution of the neutralization liquid and the dispersion of the component ii, A method for preparing an enteric hard capsule preparation liquid according to any one of the preceding claims.
- Preparation method of capsule preparation liquid Item 50.
- the method for preparing an enteric hard capsule preparation liquid according to Item 49 wherein the water-insoluble (meth) acrylic acid alkyl ester copolymer is a copolymer of methyl methacrylate and ethyl acrylate.
- Item 51 The method according to any one of items 45 to 50, further comprising a step E of maintaining the solution obtained in the step B, C or D at a third temperature T3 lower than the cloud point of the i-th component.
- Method of preparing enteric hard capsule preparation liquid Item 52.
- Step A ′ preparing a partially neutralized solution of the component ii
- Step B ′ preparing a partial solution of the i-th component
- step C ′ mixing the dispersion of the i-th component with the solution prepared in step A or B
- a method of preparing enteric hard capsule preparation liquid according to item 40 The iv component is a water insoluble (meth) acrylic acid alkyl ester copolymer Method of preparation.
- the step A ′ is a step of preparing a neutralization solution in which the component ii is at least partially neutralized and dissolved in a solvent with a basic neutralizing agent acceptable as a pharmaceutically or food additive.
- the step B ′ is a step of preparing a partial solution in which the i-th component is partially dissolved in a neutralization solution containing the ii-th component
- the step of preparing the partial solution comprises mixing the i-th component with the neutralization liquid containing the ii-th component or the neutralization liquid of the ii-th component at a first temperature T1 above the cloud point T0 of the i-th component. 50.
- a process of preparing a dispersion in which the i-th component is partially dissolved at a second temperature T2 lower than the cloud point, which is added to the mixture of the dispersion of the iv-th component, any one of items 52 to 54 A method for preparing an enteric hard capsule preparation liquid according to Item.
- Process for the preparation of enteric hard capsules comprising the following steps: Item 40.
- Item 66. Item 20.
- a hard capsule preparation comprising the enteric hard capsule according to any one of Items 1 to 19 in a hard capsule which can be dissolved under acidic conditions.
- the present invention can provide a hard capsule made of a hard capsule shell having enteric properties, which can be molded by a cold gel method. Also, according to the present invention, enteric capsules can be prepared without using a gelling agent. Furthermore, the hard capsule can be filled with contents using a capsule filling machine conventionally used.
- FIG. 1 is a view showing a schematic view of the dynamic viscoelastic behavior in the process of lowering the temperature of the enteric hard capsule preparation liquid.
- T0 indicates the cloud point or the melting start temperature.
- T1, T2 and T3 represent the first temperature, the second temperature and the third temperature described in the specification, respectively.
- T4 shows a rapid viscosity rise start temperature.
- T5 represents room temperature (20 ° C. to 25 ° C.).
- FIG. 2 is a view showing a scanning electron microscope image of the cross section of the capsule coating prepared as Example 2-2.
- FIG. 3 shows an optical microscope image of the capsule preparation liquid (55 ° C.) prepared as Example 2-2.
- FIG. 4 is a diagram showing dynamic viscoelastic behavior at the time of temperature decrease of the capsule preparation liquid prepared as Example 2-2.
- 1.00E + 2 on the vertical axis indicates 100 and 1.00E + 3 indicates 1000.
- FIG. 5 shows an example of a typical tensile stress-elongation (strain,%) curve in a tensile test, and an explanation of elastic modulus (Young's modulus) and elongation at break.
- the modulus of elasticity on the vertical axis indicates the slope in the low stress elastic region.
- 1.00E-02 on the vertical axis indicates 0.01, and 1.00E-1 indicates 0.1.
- FIG. 6 is a view showing a scanning electron microscope image of a cross section of a capsule coating prepared as Example 6-2.
- FIG. 7 is a view showing an optical microscope image of a capsule preparation liquid (55 ° C.) prepared as Example 6-2.
- FIG. 8 is a diagram showing the dissolution characteristics of a double capsule in which the enteric hard capsule of the present disclosure is used.
- a "hard capsule” is an empty capsule for filling the manufactured capsule shell with the contents.
- a hard capsule consists of a cap portion and a body portion and is also called a hard capsule or a two-piece capsule.
- the “hard capsule” in the present disclosure can be given the same or similar shape as a commercially available conventional hard capsule intended for oral administration to human or animal subjects.
- a content is filled between two films, a soft capsule is produced by bonding the films, and the content is produced by dropping the content together with the coating solution.
- Seamless capsules and microcapsules prepared by incorporating the active ingredient inside by precipitation or emulsification of a substrate are not included.
- an empty hard capsule is simply referred to as a hard capsule or capsule, and one filled with contents is referred to as a "hard capsule formulation”.
- enterric hard capsule refers to a hard capsule having “enteric” characteristics in which the coating of the capsule body itself meets the following conditions. That is, “enteric” means a characteristic that satisfies at least the following (i).
- the test subject is immersed in the first liquid at 37 ° C ⁇ 0.5 ° C for 2 hours
- the dissolution rate of the contents when done is 25% or less, preferably 10% or less.
- the pH of the first solution is about 1.2.
- the first solution can be prepared, for example, by adding 7.0 ml of hydrochloric acid and water to 2.0 g of sodium chloride to make 1000 ml.
- the term "enteric” preferably satisfies the conditions of (ii) below, in addition to the conditions of (i) above.
- the contents are eluted when the test subject is immersed in the second liquid at 37 ° C. ⁇ 0.5 ° C.
- the pH of the second solution is about 6.8.
- the second solution is prepared, for example, by dissolving 3.40 g of potassium dihydrogen phosphate and 3.55 g of anhydrous disodium hydrogen phosphate in water to make 1000 mL and adding 1 volume of water to 1 volume of phosphate buffer. Can.
- the time for measuring the dissolution rate of the contents in the second solution is not limited.
- the dissolution rate after 30 minutes after immersing the test subject in the second solution is 50%, preferably 70% or more, More preferably, it is 80% or more.
- the dissolution rate after 45 minutes is 75% or more, preferably 80% or more, and more preferably 90% or more.
- the dissolution rate after 1 hour is 75% or more, preferably 80%, more preferably 90% or more.
- the dissolution test is carried out according to the dissolution test method defined in the 17th station (the 17th station, 6.10-1.2 paddle method (50 revolutions per minute of paddle rotation), and Fig. 6.10-2a. It can be tested according to the corresponding sinker use).
- the contents used for the dissolution test are not limited as long as the contents themselves are rapidly dissolved in the test solution and can be quantified by known methods. For example, acetaminophen can be mentioned.
- nonionic water-soluble cellulose compound does not have an ionic group in the molecule, and is non-ionic hydrophilic such as -OH, OO, etc. It is a cellulose compound (polymer) that becomes water-soluble by having a group, and refers to a water-soluble cellulose ether in which part of hydroxyl groups of the glucose ring of cellulose is etherified.
- water-soluble cellulose ethers in which a hydrogen atom of a cellulose hydroxy group is substituted with at least one of an alkyl group or a hydroxyalkyl group.
- the "alkyl group” as referred to the above alkyl group or hydroxyalkyl group is a linear or branched lower alkyl group having 1 to 6, preferably 1 to 4 carbon atoms, specifically a methyl group, an ethyl group, Mention may be made of butyl and propyl.
- nonionic water-soluble cellulose compounds include lower alkyl celluloses such as methyl cellulose (MC); hydroxy lower alkyl celluloses such as hydroxyethyl cellulose (HEC) and hydroxypropyl cellulose (HPC); and hydroxyethyl methylcellulose, hydroxyethyl ethyl cellulose And hydroxy lower alkyl alkyl celluloses such as hydroxypropyl methyl cellulose (also referred to herein as hypromellose or HPMC) and the like. It is commercially available methylcellulose, hydroxypropyl cellulose, hydroxypropyl cellulose, which is particularly suitable for pharmaceutical and food applications.
- MC methyl cellulose
- HEC hydroxyethyl cellulose
- HPC hydroxypropyl cellulose
- HPMC hypromellose
- the degree of substitution of the water-soluble cellulose ether is not particularly limited, and hydroxypropyl methylcellulose, methylcellulose, hydroxypropyl cellulose and the like defined in the Japanese Pharmacopoeia are used.
- the degree of substitution of methoxy group in hydroxypropyl methylcellulose is preferably 16.5 to 30.0% by mass, more preferably 19.0 to 30.0% by mass, and particularly preferably 28.0 to 30.0% by mass.
- the degree of substitution of the hydroxypropoxy group is preferably 4.0 to 32.0% by mass, more preferably 4.0 to 12.0% by mass, and particularly preferably 7.0 to 12.0% by mass.
- the degree of substitution of the methoxy group of methylcellulose is preferably 26.0 to 33.0% by mass, more preferably 28.0 to 31.0% by mass.
- these substitution degrees can be measured by the method based on the measuring method of the substitution degree of hydroxypropyl methylcellulose, methylcellulose, and hydroxypropyl cellulose as described in 17th aspect.
- hydroxypropyl methylcellulose represented by the following formula is an optimum cellulose compound in that it is excellent in film formability and mechanical strength under low moisture.
- hydroxypropyl methylcelluloses used in the present disclosure include hypromellose of the substitution degree type (degree of substitution grade) 2910, 2906, 2208 as defined in the 17th Act.
- hydroxypropyl methylcellulose includes hypromellose having the following molecular weight which has been recognized for use as a food additive in Japan.
- Molecular weight Non-substituted structural unit 162.14
- Substituted structural unit about 180 (degree of substitution 1.19), about 210 (degree of substitution 2.37)
- As commercially available high "viscosity value" hydroxypropyl cellulose mention may be made of Ashland's Klucel (R) series, Nippon Soda's NISSO HPC.
- Ashland's Klucel (R) series Nippon Soda's NISSO HPC.
- the indicated viscosity types correspond to those of G, M, H.
- nonionic water-soluble cellulose compounds are usually supplied as finely divided solid particles in the range of about 0.1 to about 100 ⁇ m. Moreover, it is preferable that it is “non-chloride.” "Non-chlorinated” means that most of the free acid residues of the cellulose compound are not chlorinated, except for trace amounts of chloride which are present as impurities or contaminatingly contained in the manufacturing process of the cellulose compound. .
- a nonionic water-soluble cellulose compound in which the “viscosity value” of a 2% by mass aqueous solution at 20 ° C. is 100 mPa ⁇ s or more.
- the value of the viscosity may be simply referred to as "viscosity value”.
- the method of measuring the "viscosity value” is measured in accordance with the section of methyl cellulose and hypromellose formulated on the basis of the international harmonization plan from the 15th Act.
- the “viscosity value” refers to the value (mPa ⁇ s) of the viscosity of a 2% by mass aqueous solution of water-soluble cellulose at 20 ° C. ⁇ 0.1 ° C.
- “viscosity value” if “viscosity value” is less than 600 mPa ⁇ s, use “Method 1 of the general test method 2.53 viscosity measurement method (Ubbelohde method)” and “viscosity value” of 600 mPa ⁇ s or more
- general test method 2.53 method 2 of viscometry method 2.1.2 single cylindrical rotary viscometer (Brookfield viscometer) is used.
- viscosity value it is also possible to adopt a display viscosity (also referred to as a viscosity grade value) by a compound maker.
- a display viscosity also referred to as a viscosity grade value
- the display viscosity and the width of the display viscosity for example, in the METOLOSE series of Shin-Etsu Chemical, when the display viscosity is less than 600 mPa ⁇ s, the display viscosity is 80 to 120% and the display viscosity is 600 mPa ⁇ s or more. , 75-140% of the indicated viscosity.
- the lower limit value 100 mPa ⁇ s in the present disclosure the indicated viscosity can be used as it is as the “viscosity value” as long as the purpose of the present disclosure is not impaired.
- the viscosity value of an aqueous solution having a concentration of 2% by mass at 20 ° C. is also displayed using the Urobede method in ASTM D1347 or D2363.
- the relationship between viscosity (viscosity grade) and number average molecular weight and weight average molecular weight is substantially compatible with the above-mentioned local value. Any indication viscosity can be used as it is as the "viscosity value" as long as the purpose of the present disclosure is not impaired.
- the lower limit value of the preferable “viscosity value” is 100 mPa ⁇ s, more preferably 200 mPa ⁇ s, and still more preferably 400 mPa ⁇ s.
- the preferable “viscosity value” is 100,000 mPa ⁇ s, which is the upper limit value of practically available cellulose compounds.
- the number average molecular weight (g / Mol) corresponding to the “viscosity value” of 100 to 200,000 mPa ⁇ s is approximately 30,000 to 300,000.
- the weight average molecular weight (g / Mol) is about 100,000 to 1,000,000 (from the catalog values of Shin-Etsu Chemical METOLOSE (registered trademark) series, Dow Chemical's METOCEL (registered trademark) series).
- the non-ionic water-soluble cellulose compound in the solid state is usually solid particles having a particle size of the order of 1 to 100 ⁇ m.
- the compound is characterized by having a low critical solution temperature (LCST), that is, T0.
- LCST is a temperature at which the polymer in the solution undergoes gelation or phase separation when the water temperature becomes higher than T0 in the temperature rising process, if the water temperature becomes lower than T0 in the temperature lowering process.
- the solution becomes transparent when the water-soluble cellulose compound is completely dissolved in a solvent (such as water) at around room temperature.
- a solvent such as water
- gelation at T0 or phase separation with a solvent is referred to as cloud point because it is observed as turbidity of the aqueous solution.
- undissolved water-soluble cellulose particles usually 1 to 100 ⁇ m in diameter
- they are first dispersed at a cloud point T0 or higher, and then the water temperature is lowered to dissolve the particles. However, it does not dissolve completely, and maintains the dispersed state of the solid fine particles.
- the temperature is further lowered to about room temperature to obtain a complete solution.
- MC and HPMC form a gel in which water molecules are incorporated into a network of cellulose polymers, and HPC separates into a solid phase of cellulose polymers and a water phase.
- the lower limit critical solution temperature hereinafter, also referred to as "dissolution temperature”
- cloud point are names that focus on the temperature lowering process or the temperature rising process, respectively, and when there is a slight deviation due to the history of the temperature lowering or temperature rising process. But there is a general agreement. The following description is treated as equivalent.
- the cloud point of the non-ionic water-soluble cellulose compound depends on the pH of the aqueous solution, etc., but is usually in the range of 40 to 70 ° C. (High Polymer Journal, Vol. 38 (1981), p. 133-137 J. Polym. Sci. C, Vol. 36 (1971), p. 491-508).
- HPMC which is a typical nonionic water-soluble cellulose compound, is about 60 ° C.
- MC is about 40 ° C.
- HPC is about 40 ° C.
- enteric cellulose compound is an acid-resistant cellulose compound (polymer). Specifically, it refers to a compound in which a hydrogen atom of a hydroxy group of cellulose is etherified with a phthalic acid, an acetic acid, a succinic acid or the like containing a carboxyl group.
- enteric cellulose compounds include hydroxypropyl methylcellulose phthalate (HPMCP), hydroxypropyl methylcellulose acetate succinate (HPMCAS), and cellulose acetate phthalate (CAP).
- HPMCP is also called hypromellose phthalate, and is obtained by further reacting a carboxybenzoyl group (-COC 6 H 4 COOH) by reacting HPMC (hypromellose) with phthalic anhydride as a catalyst using anhydrous sodium acetate as a catalyst. It is. While the carboxybenzoyl group contains a carboxyl group and is itself hydrophobic and shows acid resistance, it dissolves in the weakly acidic to neutral region by dissociation of the carboxybenzoyl group. Therefore, depending on the binding amount of the carboxybenzoyl group, it is possible to change the dissolution pH, that is, the pH which becomes a threshold value at which the dissolution generally starts.
- HP-55 substitution type 200731
- HP-50 substitution type 220824
- HP-55S is available that is high and has excellent film strength.
- the dissolution pH of HP50 and HP55 is approximately pH 5.0 and pH 5.5, respectively.
- HPMCAS is also called hypromellose acetate ester succinate ester, hypromellose acetate succinate, and by reacting HPMC (hypromellose) with acetic anhydride, succinic anhydride, etc., acetyl group (-COCH 3 ) and succinoyl ( It is one into which a “succinyl” or “succinyl” group (—COC 2 H 4 COOH) is introduced.
- the -COOH group (carboxyl group) in the succinoyl group is important for the expression of the enteric function.
- the content of substituents of HPMCAS is not particularly limited, but is preferably 12 to 28% by mass, more preferably 20 to 26% by mass, and preferably 4 to 23% by mass, more preferably 5 to 28% by mass.
- the acetyl group is preferably 2 to 16% by mass, more preferably 5 to 14% by mass, and the succinoyl group is 2 to 20% by mass, more preferably 4 to 18% by mass.
- Examples of products are, for example, available from Shin-Etsu Chemical Co., Ltd. as AQOAT (registered trademark) series products.
- AS-L, AS-M and AS-H are classified into three grades of substitution degree depending on the substitution degree of succinoyl group and acetyl group. While the succinoyl group, and thus the carboxyl group content, increases in the order of grade (L, M or H), the acetyl group content is controlled to decrease and the dissolution pH is set to increase.
- the dissolved pH of AS-L, M and H is approximately pH 5.0, pH 5.5 and pH 6.0, respectively.
- CAP is also referred to as ceracephate (British Pharmacopoeia), cellulose acetate phthalate (Japanese Pharmacopoeia), Celeria cetus Phthalas (European Pharmacopoeia), and Ceracephate (US Pharmacopoeia). It is obtained by reacting cellulose acetate (acetylated cellulose) with phthalic anhydride using anhydrous sodium acetate as a catalyst and introducing a carboxybenzoyl group (-COC 6 H 4 COOH). Commercially, it is available from FMC's Aquateric® series products, or Eastman chemical.
- enteric cellulose compounds are insoluble in water in the non-neutralized state and are solubilized by at least partial neutralization with a basic neutralizing agent.
- the non-neutralized state means that free acid residues (eg, carboxylic acid residues of phthalic acid, succinic acid and acetic acid moieties present in the molecule) are not neutralized.
- Methacrylic acid copolymer is also referred to as “methacrylate copolymer”.
- Methacrylic acid copolymers are polymers that contain methacrylic acid monomer units in the backbone.
- the methacrylic acid copolymer is composed of methacrylic acid monomer units which are anionic groups, and alkyl ester monomer units of acrylic acid or methacrylic acid which is neutral.
- alkyl esterified with acrylic acid or methacrylic acid include alkyl having 1 to 4 carbon atoms, preferably alkyl having 1 to 3 carbon atoms. More specifically, as the alkyl ester of acrylic acid or methacrylic acid, at least one selected from the group consisting of methyl methacrylate, ethyl methacrylate, butyl methacrylate, methyl acrylate, ethyl acrylate and butyl acrylate It can be mentioned.
- the methacrylic acid copolymer is preferably enteric. More preferably, as an enteric methacrylic acid copolymer, a copolymer (copolymer) of the following methacrylic acid (formula (I)), methyl methacrylate (formula (II)) and methyl acrylate (formula (III)), Or the copolymer (copolymer) of methacrylic acid (formula (I)) and ethyl acrylate (formula (IV)) can be mentioned. (Non-Patent Document 1, Chapter 9)
- the total content (total number of units or total number of groups) of the monomers forming the copolymer is 100, containing at least 5%, preferably 5 to 70%, particularly 8 to 60% of methacrylic acid monomer units, more preferably And preferably 30 to 60%.
- the ratio of each monomer unit can be easily converted into mass% using the molecular weight of each monomer unit.
- Preferred methacrylic acid copolymers are 40 to 60% by weight of methacrylic acid (molecular weight 86.04), 60 to 40% by weight of methyl methacrylate (molecular weight 100.05), or 60 to 40% ethyl acrylate (molecular weight 100.05). It is a polymer consisting of% by mass (for example, EUDRAGIT (registered trademark) L100 or EUDRAGIT (registered trademark) L100-55). EUDRAGIT® L 100-55 is particularly suitable, which is a copolymer consisting of 50% by weight of methacrylic acid and 50% by weight of ethyl acrylate. EUDRAGIT® L30D-55 is an aqueous dispersion containing approximately 30% by weight of EUDRAGIT® L100-55. These methacrylic acid copolymers are set to dissolve at a pH of about 5.5 or more.
- Another preferable example is a polymer comprising 5 to 15% by mass of methacrylic acid, 10 to 30% by mass of methyl methacrylate, and 50 to 70% by mass of methyl acrylate (molecular weight: 86.04). More specifically, it is EUDRAGIT® FS, which is a copolymer consisting of 10% by weight of methacrylic acid, 25% by weight of methyl methacrylate and 65% by weight of methyl acrylate. EUDRAGIT® FS 30 D is a dispersion containing approximately 30% by weight of EUDRAGIT® FS. This methacrylic acid copolymer is set to dissolve at a pH of about 7 or more, and may be used when colon delivery is intended, which is a higher pH environment.
- the above-mentioned enteric methacrylic acid copolymer is generally produced in advance by an emulsion polymerization process from the monomer level through a copolymerization process in an aqueous solution to produce an aqueous emulsion containing very small colloidal particles. Therefore, an aqueous dispersion of very fine colloidal particles having an average particle size of less than 1 ⁇ m can be obtained without going through the dissolution step by neutralization of the solid polymer component with a basic neutralizing agent.
- aqueous dispersion equivalent to EUDRGIT Leeds (Evonik) L30D-55, and equivalent commercialized methacrylic acid copolymers mention is also made of Kollicoat series (BASF) MAE 30D / DP, Poly Kid series (Sanyo Chemical Industries) PA-30
- Kollicoat series BASF
- MAE 30D / DP Poly Kid series
- PA-30 anyo Chemical Industries
- these aqueous dispersions usually contain less than 0.3% of residual monomers, and trace amounts of polysorbate 80 and sodium lauryl sulfate for the production process and stabilization thereof, but the present disclosure
- Such a hard capsule shell, and an unavoidable impurity contained in the hard capsule preparation can be accepted.
- the “(meth) acrylic acid alkyl ester copolymer” is a substantially neutral (meth) acrylic acid copolymer, and is mainly composed of alkyl ester neutral monomer units of methacrylic acid or acrylic acid.
- alkyl esterified with acrylic acid or methacrylic acid include alkyl having 1 to 4 carbon atoms, preferably alkyl having 1 to 3 carbon atoms. More specifically, as the alkyl ester of acrylic acid or methacrylic acid, at least one selected from the group consisting of methyl methacrylate, ethyl methacrylate, butyl methacrylate, methyl acrylate, ethyl acrylate and butyl acrylate It can be mentioned.
- the proportion of neutral monomers is, for example, more than 95% by weight, more than 98% by weight, more than 99% by weight, or 100% by weight.
- the presence of ionic groups in the polymer is not completely excluded, and the content of ionic groups, in particular anionic groups, is less than 5% by mass, preferably less than 2% by mass, preferably less than 1% by mass
- a methacrylic acid copolymer may be included.
- the (meth) acrylic acid alkyl ester copolymer is preferably water insoluble.
- EUDRAGIT® NE is suitable, which is a copolymer consisting of 70% by weight of ethyl acrylate and 30% by weight of methyl methacrylate. In any case, it may contain less than 5% by weight, preferably less than 2% by weight, and preferably less than 1% by weight of methacrylic acid (molecular weight 86.04).
- These water-insoluble (meth) acrylic acid alkyl ester copolymers have a glass transition temperature of less than 100.degree. C., or a minimum film-forming temperature (MFT) of less than 50.degree.
- MFT minimum film-forming temperature
- the dispersion containing the colloidal particles of the acid copolymer is dried to form a film, the adhesion between the particles is promoted to obtain a transparent and hard-to-break dry film.
- the water-insoluble (meth) acrylic acid alkyl ester copolymer has an advantage that the acid resistance is not impaired at a proper addition amount.
- the above-mentioned water-insoluble (meth) acrylic acid alkyl ester copolymer can also be produced in advance by an emulsion polymerization process from the monomer level through the copolymerization process in an aqueous solution to form an aqueous emulsion containing very small colloidal particles. Therefore, an aqueous dispersion of very fine colloidal particles having an average particle size of less than 1 ⁇ m can be obtained without going through the dissolution step by neutralization of the solid polymer component with a basic neutralizing agent.
- Polyvinyl alcohol is a polymer obtained by saponifying polyvinyl acetate, and generally has a degree of saponification of 97% or more and a completely saponified product represented by the following formula (1); There is a partial saponification represented by the following formula (2) at ⁇ 96%.
- any of the above fully saponified products and partially saponified products can be used.
- partial saponification having a degree of saponification, n / (n + m) of about 78 to 90%, particularly about 87 to 90% is preferably used.
- the average degree of polymerization (n) of PVA is not particularly limited as long as it can exhibit film forming ability, but it is usually preferably 400 to 3300, and particularly preferably about 1000 to 3000.
- the weight average molecular weight of the PVA is about 18,000 to about 200,000 when calculated from the average degree of polymerization and the degree of saponification, but it is not particularly limited thereto.
- the addition of PVA can provide the capsule shell with adequate mechanical strength (elastic modulus and cracking resistance) while maintaining entericity.
- PVA and a PVA copolymer may be used in combination.
- a PVA copolymer the PVA copolymer obtained by copolymerizing a polymerizable vinyl monomer with PVA mentioned above can be mentioned.
- the PVA copolymer is a polymer copolymer obtained by copolymerizing acrylic acid and methyl methacrylate with the partially saponified PVA described above as a skeleton.
- POVACOAT registered trademark
- Nisshin Kasei Co., Ltd. can be exemplified.
- the enteric hard capsule shell according to the present disclosure further comprises a plasticizer, surfactant (emulsifier), base (excluding nonionic water-soluble cellulose compound) and binder, which are acceptable as pharmaceutical and food additives. (Excluding PVA), a coating agent, etc. may be included. In addition, it may contain a controlled release agent, a solubilizing agent, a solubilizing agent, and the like for controlling the solubility, in particular, the elution characteristic in the neutral pH range.
- additive acceptable as a pharmaceutical additive for example, the additive described in the Pharmaceutical Additive Dictionary, 2016 edition (Edited by the Japan Pharmaceutical Additives Association, Pharmaceutical Jichi Nipponsha Co., Ltd.) according to the application can be used. Is not limited to these. In addition, these additives may be classified redundantly to a plurality of uses.
- the plasticizer is not necessarily limited to the specific substance shown in the above-mentioned Dictionary of Pharmaceutical Additives, and can be used in a pharmaceutical or food composition and is not particularly limited as long as it can be added to a capsule film to impart flexibility.
- Suitable substances are in general those having a molecular weight (Mw) of 100 to 20,000 and having one or more hydrophilic groups, such as hydroxyl groups, ester groups or amino groups in one molecule.
- Surfactants are used as solubilizers, suspending agents, emulsifying agents, dispersing agents, solubilizing agents, stabilizers and the like.
- benzalkonium chloride benzethonium chloride polyoxyethylene (40) monostearate (polyoxyl stearate 40 *), sorbitan sesquioleate (sorbitan sesquioleate *), polyoxyethylene (20) sorbitan monoole Examples include aate (polysorbate 80 *), glyceryl monostearate (glyceryl monostearate *), sodium lauryl sulfate, polyoxyethylene lauryl ether (lauromacrogol *) and the like.
- the enteric hard capsule shell according to the present disclosure may further contain a lubricant, a sequestering agent, a coloring agent, a light shielding agent, a binder, and the like at most about 5% by mass.
- a sequestering agent ethylenediaminetetraacetic acid, acetic acid, boric acid, citric acid, gluconic acid, lactic acid, phosphoric acid, tartaric acid, or salts thereof, metaphosphate, dihydroxyethyl glycine, lecithin, ⁇ -cyclodextrin, or these Combinations can be mentioned.
- the lubricant is not particularly limited as long as it can be used for pharmaceuticals or food compositions.
- calcium stearate, magnesium stearate, sodium stearyl fumarate, carnauba wax, starch, sucrose fatty acid ester, light anhydrous silicic acid, macrogol, talc, hydrogenated vegetable oil and the like can be mentioned.
- a sequestering agent ethylenediaminetetraacetic acid, acetic acid, boric acid, citric acid, gluconic acid, lactic acid, phosphoric acid, tartaric acid, or salts thereof, metaphosphate, dihydroxyethyl glycine, lecithin, ⁇ -cyclodextrin, or these Combinations can be mentioned.
- the coloring agent and the light shielding agent are not particularly limited as long as they can be used for pharmaceuticals or food compositions.
- a coloring agent for example, asenyakutannin powder, turmeric extract, methyl rosanilin chloride, yellow iron oxide, yellow iron trioxide, opaspray K-1-24904, orange essence, brown iron oxide, carbon black, caramel, carmine, carotene liquid , ⁇ -carotene, photosensitive element No. 201, licorice extract, gold foil, black extract, black iron oxide, light anhydrous silicic acid, gaskets, zinc oxide, titanium oxide, ferric oxide, disazo yellow, edible blue No. 1 and its aluminum lake Food blue No. 2 and its aluminum lake, food yellow No.
- titanium oxide and / or a calcium compound may be added as a light shielding agent to prevent deterioration of the contents due to ultraviolet light and the like.
- the calcium-containing compound include calcium carbonate, inorganic calcium salts such as calcium hydrogen carbonate, calcium hydroxide, calcium oxide, calcium complexes such as dolomite and hydroxyapatite, and compounds containing other calcium elements.
- the first aspect of the present disclosure relates to a hard enteric capsule.
- a first component which is a nonionic water-soluble cellulose compound having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s
- a second component which is an enteric methacrylic acid copolymer
- a third component which is a soluble cellulose compound and a fourth component which is a water insoluble (meth) acrylic acid alkyl ester copolymer, a polyvinyl alcohol, a plasticizer, and a fifth component which is at least one selected from the group consisting of surfactants
- An enteric hard capsule comprising a film containing at least one component of Among them, the second and third components impart an enteric function, and the first component mainly assists the film formation to become a self-supporting capsule shape, and the fourth and fifth components mainly A self-supporting capsule shell is used to obtain mechanical strength suitable as a hard capsule while maintaining the solubility function.
- hypromellose hard capsules for oral administration in which the solubility without delay is important, regardless of pH, use 3 to 15 mPa ⁇ s as the indication viscosity (viscosity grade) value of water-soluble cellulose (Japanese Patent Application Laid-Open Nos. 08-208458, 2001-506692, 2010-270039, 2011-500871).
- almost 100% in the film gelling agent, gelling aid, light shielding agent, coloring agent, etc., may contain about 0 to 5% by mass and 0 to 10% by mass of residual moisture
- the dissolution rate is almost independent of pH, and is determined by the molecular weight of the water-soluble cellulose and hence the viscosity value, and usually pH 1.2 test solution, 6.8 test solution And, in pure water, 100% of the acetaminophen in the inside is eluted within 30 minutes.
- the viscosity value is 100 mPa ⁇ s or more, elution tends to be delayed, and it has hardly been used as a fast-dissolving capsule film material.
- an enteric polymer having a very high viscosity that is, a very high molecular weight nonionic water-soluble cellulose compound having a viscosity value of 100 mPa ⁇ s or more, as compared to the prior art. It is thought that the following characteristics can be realized. Although not restricted by theory, it is considered that nonionic water-soluble cellulose compounds generally function as a filler for relatively brittle enteric polymers. Furthermore, since it has a very high molecular weight, in the test liquid (the first liquid) having a pH of 1.2, swelling due to entry of water is appropriately suppressed, and the acid resistance function of the enteric polymer which is the main component is impaired. There is no
- the enteric polymer promotes rapid dissolution, so dissolution delay does not easily occur even if water-soluble cellulose having a viscosity value of 100 mPa ⁇ s or more is contained.
- the enteric methacrylic acid copolymer which is the second component in the present disclosure, is an essential component to realize the enteric hard capsule according to the present disclosure.
- enteric methacrylic acid copolymer As an intrinsic property of enteric base, enteric methacrylic acid copolymer is very stable (NPL 6, especially Figure 3) and has a low water vapor transmission rate compared to enteric cellulose compounds in long-term storage That is, there is an advantage that the moisture resistance of the film is excellent (Non-Patent Document 6, particularly Table 2).
- enteric cellulose compounds (third component) can be mixed.
- the third component can achieve the desired mechanical strength as a hard capsule shell while ensuring sufficient entericity.
- pH dependency can be more flexibly controlled by blending the third component in addition to the second component. That is, it is possible to control the elution characteristic of the intermediate pH region of about pH 4 to 5.
- part or all of the enteric cellulose compound can be replaced with a fourth component, a water-insoluble (meth) acrylic acid alkyl ester copolymer.
- the fourth component can improve mechanical strength, in particular, cracking resistance, without deteriorating the acid resistance performance. Further, unlike the third component, in order to obtain a completely dissolved or finely divided dispersion, it is not necessary to neutralize, so the concentration of residual salt in the film can be suppressed.
- At least one selected from the group consisting of PVA, a plasticizer and a surfactant may be added as a fifth component.
- the fifth component is preferable because it can maintain the transparency of the film, in addition to the effects of imparting appropriate hardness and cracking resistance.
- Some plasticizers and surfactants such as TriEthyl Citrate (TEC), Polyethylene Glycol (PEG), Propylene Glycol (PG), etc. are also useful to refine and stabilize the particle size in dispersions of enteric polymers. is there.
- PVA has the effect of increasing the hardness of the film.
- the total mass of the first component, the second component, the third component, the fourth component, and the fifth component contained in the film is 100% by mass
- the ratio of the second component is ⁇ % by mass
- the ratio of the third component is ⁇ % by mass
- the ratio of the fourth component is ⁇ %
- the ratio of the fifth component is ⁇
- the ratio of the total of the soluble polymer (the second component and the third component) and the fourth component, that is, ( ⁇ + ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ) is preferably 0.5 or more.
- the value of ( ⁇ + ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ) is more preferably 0.55 or more, still more preferably 0.6 or more.
- ( ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ ) is preferably 0.4 or more and more preferably 0.5 or more.
- the upper limit of ( ⁇ + ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ) is 0.9 or less, preferably 0.8 or less, in order to maintain appropriate hardness and crack resistance of the capsule shell.
- a water-soluble cellulose compound having a viscosity value of 100 to 1000 mPa ⁇ s When it is required to move into the intestine for quick dissolution, it is preferable to use a water-soluble cellulose compound having a viscosity value of 100 to 1000 mPa ⁇ s.
- the ratio ⁇ is preferably less than 30% by mass, and more preferably less than 20% by mass.
- the enteric methacrylic acid copolymer preferably has a ⁇ / ( ⁇ + ⁇ ) of 0.1 or more, more preferably 0.2 or more. More preferably, it is 0.4 or more.
- enteric methacrylic acid copolymer is more chemically stable than enteric cellulose compound, and free carboxylic acid by decomposition of carboxyl group in storage under high humidity for a long period of time There is almost no generation of
- the enteric methacrylic acid copolymer also has the advantage of low water vapor permeability, that is, excellent moisture resistance of the film.
- ⁇ in order to reduce the ratio of the third component, ⁇ , part or all of the enteric cellulose compound of the third component is replaced with the water-insoluble (meth) acrylic acid alkyl ester copolymer of the fourth component.
- ⁇ can be 0.
- the water-insoluble (meth) acrylic acid alkyl ester copolymer has the effect of improving the mechanical strength of the film, in particular the cracking resistance, without deteriorating the acid resistance.
- ⁇ 0, ⁇ / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ), that is, ⁇ / ( ⁇ + ⁇ + ⁇ + ⁇ ) is preferably 0.3 or more, and more preferably 0.4 or more.
- the enteric methacrylic acid copolymer is a material that easily makes the capsule shell brittle
- the upper limit of ⁇ / ( ⁇ + ⁇ + ⁇ + ⁇ ) is preferably 0.7 or less, more preferably 0.65 or less. It is preferable that ( ⁇ + ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ + ⁇ + ⁇ ), that is, ( ⁇ + ⁇ ) / ( ⁇ + ⁇ + ⁇ + ⁇ ) be 0.5 or more and 0.9 or less as described above. Furthermore, in order to maintain appropriate hardness in cracking, it is preferable to set ⁇ / ( ⁇ + ⁇ + ⁇ + ⁇ ), that is, ⁇ / ( ⁇ + ⁇ + ⁇ + ⁇ ) to 0.2 or more.
- the ratio of the fifth component is preferably 0.15 or less, more preferably 0.1 or less, in any of the above component ratios.
- a mixture of plural types of nonionic water-soluble cellulose compounds having different viscosity values or substitution degree types having viscosity values of 100 mPa ⁇ s or more may be used, and those viscosity values are 100 mPa ⁇ s or more.
- the amount of the nonionic water-soluble cellulose as a whole can be regarded as the first component, and the ratio can be ⁇ mass%.
- the same applies to the second, third, and fourth components and when plural types of enteric methacrylic acid copolymers are used, the total amount thereof is regarded as the second component, and the ratio thereof is defined as ⁇ mass%.
- the total amount is regarded as the third component, and the ratio is considered as ⁇ mass%, and when plural types of water-insoluble (meth) acrylic acid alkyl ester copolymers are used, The entire amount is regarded as the fourth component, and the ratio is taken as ⁇ mass%.
- the fifth component when at least two selected from the group consisting of PVA, a plasticizer and a surfactant are simultaneously used, the total amount thereof is regarded as the fifth component, and the ratio is taken as ⁇ mass%.
- a lubricant, a metal sequestering agent, a colorant, a light shielding agent, and residual moisture can be included.
- the total mass of the first component, the second component, the third component, the fourth component and the fifth component contained in the film is X, and the mass total of the lubricant, the sequestering agent, the colorant and the light shielding agent is ⁇ .
- ⁇ / X can be in the range of 0.2 or less, more preferably 0.1 or less, and even more preferably 0.05 or less.
- the presence of a salt by at least partial neutralization of an enteric polymer consisting of an enteric methacrylic acid copolymer and / or an enteric cellulose compound, and the neutralization of other coating components associated therewith Can tolerate the presence of objects.
- the salts include at least one salt selected from the group consisting of alkali metal salts, alkaline earth metal salts, and ammonium salts.
- the salt can include at least one salt selected from the group consisting of sodium (Na) salt and potassium (K) salt. Particularly preferred is the Na salt.
- the carboxyl group of the enteric cellulose compound is neutralized by a metal ion such as Na and may be stably present in the solid film as a group such as -COONa.
- the ratio of these neutralized acid (such as carboxylic acid) residues is 50% or less, for example, when the number of moles (number of groups) of carboxyl residues before neutralization contained in the enteric polymer is 100%. Is preferably 30% or less, more preferably 20% or less. This is referred to as the degree of neutralization (a detailed definition of the degree of neutralization will be described in the second embodiment described later).
- the presence of excess salt is not preferable because the film may be cracked, the film may be deteriorated due to salting out, or may be broken due to excessive penetration of water.
- the presence of a suitable salt assists the penetration and swelling of the capsule shell containing the enteric polymer by water. Swelling of the capsule shell causes the gap between the cap and the body to be in close contact, which has the effect of preventing the dissolution completely.
- the degree of neutralization is preferably 2%, more preferably 5% or more.
- the salt contained in the capsule shell is a Na salt
- it is preferably 0.1% by mass or more, more preferably 0. It is 2 mass%.
- the content is preferably 5% by mass or less, more preferably 2% by mass or less, and still more preferably 1% by mass or less.
- ⁇ 0, it can be 2% by mass or less.
- the capsule shell according to the present disclosure preferably contains 2 to 10% by mass of residual water content to maintain the resistance to cracking.
- An appropriate amount of water content acts as a plasticizer with little effect on the solubility of the capsule.
- the water content depends on the environmental humidity at the time of capsule storage, but in a relative humidity range of about 20 to 60%, it changes reversibly in proportion to the environmental humidity.
- the water content value of the capsule shell is a saturation value after storage (conditioning) for several days at a constant relative humidity of 43% at room temperature.
- the moisture content after conditioning can be measured by the loss on drying method as follows.
- a sample (hard capsule or film) is placed in a constant atmosphere containing potassium carbonate saturated salt, sealed, and conditioned at 25 ° C. for 1 week.
- the following saturated salt (aqueous solution) is used for humidity control. That is, in the presence of potassium acetate saturated salt, potassium carbonate saturated salt and ammonium nitrate saturated salt, an atmosphere having a relative humidity of about 22%, 43% and 60%, respectively, can be created.
- the sample is then dried by heating at 105 ° C. for 2 hours, and the mass (dry mass) of the sample is measured again.
- the water content at room temperature and 43% relative humidity is preferably at least 2% or more, more preferably 3% or more, and still more preferably 4% or more. If it is less than 2%, it is easily broken. On the other hand, if the water content is too high, it may react with the drug loaded inside when stored for a long period of time, so it is preferably 10% or less, more preferably 8% or less, More preferably, it is 6% or less.
- the enteric hard capsule according to the present disclosure has the same or similar shape and mechanical strength (hardness and hardness) as conventional hard capsules marketed for oral administration to human or animal subjects. Is desirable.
- the commercially available hard capsules to be referred to are gelatin or HPMC (hypromellose) capsules. Therefore, the thickness of the film of the capsule is 50 ⁇ m or more, preferably 60 ⁇ m or more, and more preferably 70 ⁇ m or more.
- the upper limit is 250 ⁇ m or less, preferably 200 ⁇ m or less, and more preferably 150 ⁇ m or less. In particular, the range of 70 to 150 ⁇ m is suitable for use as it is in commercial filling machines.
- Non-Patent Document 1 Chapter 4
- the mechanical strength of the film depending on the composition of each component of the hard capsule is prepared by casting a film by a casting method using a preparation having the same composition as each composition of each composition of the hard capsule preparation. It can be used for evaluation.
- the cast film is placed on a glass surface kept at room temperature or on a PET film, and a metallic applicator is placed, and a preparation liquid of 50 ° C. to 60 ° C. is poured and moved at a constant speed to produce a uniform film of 100 ⁇ m. Thereafter, drying is carried out at room temperature to 30 ° C. for about 10 hours.
- an applicator having a gap of 0.4 mm to 1.5 mm may be properly used.
- the produced film is cut into, for example, a 5 mm ⁇ 75 mm dumbbell shape (specified by JIS K-7161-2-1BA), and then a tensile test is performed using, for example, a small bench test machine (SHIMAZU CORPORATION EZ-LX) be able to.
- a small bench test machine SHIMAZU CORPORATION EZ-LX
- both ends of the film are set in a holder (gap length 60 mm), tensile speed, tension at 10 mm / min, film elongation and stress (tensile stress) -elongation (strain) curve generated in the film are shown.
- FIG. 5 shows the typical elongation-tensile stress test results.
- the modulus of elasticity which is an index of hardness
- the elongation at break can be obtained as the elongation at break (%) (Non-Patent Document 1, Chapter 4) .
- the mechanical strength be maintained under an environment of normal use conditions (temperature about 5 to 30 ° C., relative humidity about 20 to 60%).
- a tensile test may be performed to evaluate the mechanical strength. it can.
- the tensile test is preferably performed under a temperature and humidity environment of 25 ° C. and a relative humidity of 22%.
- a tensile test is performed to evaluate the mechanical strength.
- the tensile test is preferably performed under the same temperature and humidity environment as the humidity control condition.
- the elastic modulus which is an index of hardness, is preferably 1 to 5 GPa, and more preferably 2 to 4 GPa.
- the elongation at break which is an indicator of the degree of cracking resistance evaluated by a tensile test, is preferably about 2 to 30%, and more preferably about 3 to 30%.
- the hardness and hardness of the enteric hard capsule shell according to the present disclosure are often in a trade-off relationship within this range.
- Coating films and soft capsule films are often softer and have a high breaking elongation. For example, coatings with a breaking elongation of more than 30% are usually too soft and often not suitable as free standing hard capsule coatings.
- the elongation at break is less than 2%, it becomes significantly susceptible to cracking even in normal handling.
- the moisture present in the capsule film at a few percent or so can usually affect the mechanical strength, particularly the cracking, as a plasticizer.
- the relative humidity is low
- the water content decreases, for example, when it is about 2 to 3%, it tends to be broken, that is, the breaking elongation tends to decrease.
- the high humidity side the water content tends to increase and the elastic modulus tends to decrease. After all, the elongation at break becomes a problem on the low humidity side, and the elastic modulus becomes a problem on the high humidity side.
- humidity control is performed under an environment of relatively low humidity 22% relative humidity and a temperature of 25 ° C.
- a tensile test to obtain a film having a breaking elongation of 2 to 30%.
- a humidity control and a tensile test are conducted under a relatively high humidity environment of 60% relative temperature and a temperature of 25 ° C., and a film having an elastic modulus of 1 to 5 GPa can be obtained.
- an elastic modulus in the range of 1 to 5 Gpa and an elongation at break of 3 to 30% can be obtained in most relative humidity and temperature ranges at room conditions. More preferably, the elastic modulus is in the range of 2 to 5 GPa, and the elongation at break is in the range of 3 to 10%.
- the enteric hard capsule film according to the first aspect exhibits a structure in which a phase comprising a nonionic water-soluble cellulose compound as a main component is dispersed in a phase consisting essentially of other components.
- the said structure considers the phase which has a nonionic water-soluble cellulose compound as a main component a "island" phase, and the phase which substantially consists of other components as a "sea" phase, and calls it a sea island structure.
- the island phase consists essentially of the first component.
- “substantially” means that the island phase may contain other components, particularly the enteric cellulose polymer which is the third component, while the sea phase partially dissolves. Meaning that the first component can be included.
- the sea phase also contains a second component, such as methacrylic acid copolymer, plasticizer, surfactant (emulsifier), lubricant, binder, light shielding agent, pigment, pigment, lubricant, and the like.
- a second component such as methacrylic acid copolymer, plasticizer, surfactant (emulsifier), lubricant, binder, light shielding agent, pigment, pigment, lubricant, and the like.
- the “sea-island structure” can be confirmed by observing the cross section of the hard capsule film with a scanning electron microscope, as shown in the examples below. Such a “sea-island structure” is considered to be difficult to form by injection molding or extrusion using the thermoplasticity of the film component polymer because it needs to undergo a kind of dispersion equilibrium state in the solution state. Also, it is presumed that the island phase is not formed even when the first component is exposed to a low temperature near room temperature and completely dissolved in the preparation process of the capsule preparation
- each island phase depends on the size of solid particles of the non-ionic water-soluble cellulose compound used to prepare the hard capsule.
- the island phase in the hard capsule film preferably has a minor axis of 0.1 ⁇ m or more and less than 30 ⁇ m. More preferably, the island phase has a minor axis of 0.2 ⁇ m or more and less than 20 ⁇ m.
- the present invention relates to a preparation for preparing the enteric hard capsule described in the above.
- the hard enteric capsule according to the present disclosure comprises a film obtained by drying the preparation liquid of the present embodiment and removing the solvent.
- the i-th component is a nonionic water-soluble cellulose compound having a viscosity value of preferably 100 mPa ⁇ s to 100,000 mPa ⁇ s in the “viscosity value” of a 2% aqueous solution at 20 ° C .;
- Component ii which is an enteric methacrylic acid copolymer, a basic neutralizing agent, and a solvent, and further, component iii, which is an enteric cellulose, and a water insoluble (meth) acrylic acid alkyl ester copolymer, iv
- the solvent used for the preparation liquid is mainly composed of water, and in particular, it is preferable that it is purified water.
- purified water in the dissolution process to obtain a dispersion from a solid powder of a nonionic water-soluble cellulose compound, an enteric cellulose compound, and / or an enteric methacrylic acid copolymer, water; and at least one selected from ethanol and absolute ethanol; A mixed solvent of can be used.
- Most of this ethanol evaporates during the preparation of the preparation liquid according to the present disclosure or in the immersion step, so that the preparation liquid during immersion actually has a water content of 80% by mass, and more preferably It is 90 mass% or more.
- Substantially 100% purified water can be used except for unavoidable impurities.
- the enteric methacrylic acid copolymer as the component ii and the enteric cellulose compound as the component iii are used alone or together as an enteric polymer.
- These enteric polymers are substantially insoluble in neutral water, because their solubility depends on the pH of the solvent, and can be at least partially dissolved in the presence of a basic neutralizing agent. It is desirable to use it as a dispersion of fine particles of about 10 ⁇ m, preferably about 1 ⁇ m or less, by dissolving it. If the particle size is larger than this, the surface irregularities of the capsule coating and the strength of the capsule coating may be adversely affected.
- neutralizing solution or “partially neutralizing solution” is also referred to below including the case where at least a portion is neutralized and dissolved.
- the “neutralizing solution” may be a suspension containing undissolved fine particles in a dispersed state.
- the basic neutralizing agent is not limited as long as it is a pharmaceutically or food additive acceptable compound.
- the basic neutralizing agent include at least one selected from the group consisting of alkali metal salts, alkaline earth metal salts, and ammonium salts. Preferably, it is at least one selected from the group consisting of sodium salts and ammonium salts.
- the basic neutralizing agent at least one selected from the group consisting of sodium hydroxide, potassium hydroxide, calcium hydroxide, ammonia and ammonium carbonate can be mentioned. More preferably, the basic neutralizing agent is sodium hydroxide, optionally at least one selected from the group consisting of ammonia and ammonium carbonate.
- the basic neutralizing agent is ammonia
- ammonia is used as the basic neutralizing agent, it is preferable to use HPMCAS as the enteric cellulose compound because it is easy to volatilize the ammonia.
- a solid powder of an enteric cellulose compound neutralized product neutralized in advance by a basic neutralizing agent may be used by dissolving or dispersing it in a solvent.
- a basic neutralizing agent capable of neutralizing at least a part thereof is added to dissolve the enteric cellulose compound in the solvent.
- the amount of basic neutralizing agent required to neutralize the enteric polymer (component ii or component iii) can be defined as follows.
- the enteric polymer In order to completely neutralize the enteric polymer, it is achieved by adding the cation derived from the basic neutralizing agent to be equal to or more than 1 mole of the carboxyl group contained in the enteric polymer. Be done. When the cation derived from the basic neutralizing agent is divalent or higher, it is replaced by 1 / valent number. The case where the cation derived from the basic neutralizing agent is dissolved in a solvent so as to be substantially equivalent to the carboxyl group contained in the enteric polymer is called complete neutralization.
- equivalent number of moles of cation ie, “equivalent amount (equimolar amount)” is, for example, sequestered by 100% neutralization of the number (number of moles) of carboxyl residues before neutralization contained in the enteric methacrylic acid copolymer It is the number of moles of cation that can be done.
- the weight of KOH (molecular weight: 56.10) necessary for neutralizing 1 g of the target enteric polymer, and (KOH) mg / g can be prescribed (KOH equivalent).
- the degree of neutralization is defined as the ratio of the mass of the basic neutralizing agent actually added to the equivalent of the basic neutralizing agent required for complete neutralization.
- the basic neutralizing agent is sodium hydroxide NaOH (molecular weight 40.00), and calcium hydroxide Ca (OH) 2 (molecular weight 74.09), ammonia NH 3 (molecular weight 17.03), ammonium carbonate (NH 4 ) 2 CO
- the equivalent in the case of 3 (molecular weight 96.09) is
- the equivalent of the basic neutralizing agent necessary for complete neutralization can be displayed by the manufacturer with a tolerance of the degree of substitution of the carboxyl group with a range of about ⁇ 10 to 20%.
- a more accurate neutralization equivalent can be determined by a common titration method.
- the component ii is Eudragit, L30D55, L100-55 and L100 manufactured by Evonik
- its KOH equivalent is 301.2 mg / g
- the basic neutralizing agent is sodium hydroxide
- ammonia it is 91.4 mg / g.
- the component ii is Evonik Eudragit, FS30D
- its KOH equivalent is 56.7 mg / g
- the basic neutralizing agent is sodium hydroxide
- 40.4 mg / g when it is ammonia Is 17.2 mg / g.
- the KOH equivalents thereof are 79.0 to 101.6 mg / g and 101.6 to 131.7 mg / g, respectively.
- the basic neutralizing agent is sodium hydroxide, it is 56.3 to 72.4 mg / g and 72.4 to 93.9 mg / g, respectively.
- ammonia it is 24.0 to 30.8 mg / g and 30.8 to 40.0 mg / g, respectively.
- the component iii is Shin-Etsu Chemical HPMCAS, AQUAT AS-H, AS-M, or AS-L
- its KOH equivalent is 22.2-44.4, 55.5-77.7, 77, respectively.
- the basic neutralizing agent is sodium hydroxide, it becomes 15.8 to 31.7 mg / g, 39.6 to 55.4 mg / g, and 55.4 to 71.2 mg / g, respectively.
- ammonia it is 6.7 to 13.5 mg / g, 16.8 to 23.6 mg / g, and 23.6 to 30.3, respectively.
- the degree of neutralization is defined as the mass ratio of the basic neutralizing agent actually added to the amount of the basic neutralizing agent corresponding to the neutralization equivalent.
- the degree of neutralization is, at the same time, equal to the number of moles of residues which are blocked by pouring out of the number of moles of carboxylic acid residues: .
- the degree of neutralization is: /(0.2418 ⁇ ) ⁇ 100 (%), Become.
- the degree of neutralization will be g / (0.065 x m) x 100 (%).
- a median of 65 mg / g of neutralization equivalents of 56.3 to 72.4 for NaOH of HP50 is applied.
- an enteric methacrylic acid copolymer L30D55 when used as an enteric methacrylic acid copolymer L30D55, a mixture of g1 (g) and an enteric cellulose compound HP50, ⁇ 2 (g) and neutralized with ⁇ (g) of NaOH, an enteric polymer
- the degree of neutralization of the whole can be calculated as E / (0.2418 ⁇ ⁇ 1 + 0.065 ⁇ ⁇ 2) ⁇ 100 (%).
- the enteric cellulose compound which is the third component iii, controls the molecular weight of large cellulose blocks (solid particles) originally contained in the raw material pulp by hydrolysis or chemical decomposition by enzymes, and mechanical milling such as mechanical milling. It is crushed by a method to obtain solid particles of about 10 to 100 ⁇ m. In order to further refine this into a dispersion of fine particles of about 10 ⁇ m or less or to completely dissolve it, the degree of neutralization of the enteric cellulose compound may be completely dissolved or partially dissolved. Also in order to make the size of the fine particles contained in the dispersion approximately 10 ⁇ m or less, the size is preferably 50% or more. The upper limit is 100%, and excessive neutralization exceeding 100% is not preferable because salting out of the salt remaining in the film after drying and the like occur.
- the enteric methacrylic acid polymer which is the component ii very small colloidal particles having a diameter of more than about 0.01 ⁇ m and less than 1 ⁇ m through the copolymerization process from the monomer level in the aqueous solution by the emulsion polymerization process.
- the resulting acidic dispersion (aqueous emulsion) is obtained directly.
- the dispersion is provided as a dispersion of very fine colloidal particles having an average particle size of less than 1 ⁇ m without going through a dissolution step by neutralization with a solid-base neutralizing agent.
- the above-mentioned Evonik's L30D55 etc. may be mentioned.
- the pH of the colloidal dispersion of L30D55 is about 2.5.
- enteric methacrylic acid copolymer powder (specifically, Evonik Co., Ltd. and L10055 etc.) which has been synthesized by emulsion polymerization in solution and then dried and solidified into solid fine particles is re-dispersed in water to obtain a base. It is also possible to obtain an aqueous dispersion which is partially neutralized with an acid neutralizing agent to form a fine particle. In such a case, an aqueous dispersion having a sufficiently finely divided particle size can be obtained even with a degree of neutralization of about 2 to 20%.
- the i-th component nonionic water-soluble cellulose compound which does not inherently have cold gelation ability alone alone, the ii-component enteric methacrylic acid copolymer, and the iii-component enteric It is found that cold-gelling ability can be imparted to enteric hard capsule preparation liquid by mixing the three components in the presence of a basic neutralizing agent for enteric hard capsule preparation liquid containing a cellulose compound.
- a basic neutralizing agent for enteric hard capsule preparation liquid containing a cellulose compound The
- the importance of the interaction between the high viscosity nonionic water-soluble cellulose compound which is the i-th component and the enteric methacrylic acid copolymer which is the ii-th component in the presence of an appropriate amount of a basic neutralizing agent is seen. It came out.
- the enteric hard capsule preparation liquid according to the present disclosure has a temperature lower than the cloud point T0 (cloud point or dissolution start temperature) of the nonionic water-soluble cellulose compound which is the i-th component, as shown in FIG.
- T0 cloud point or dissolution start temperature
- the storage and loss elastic modulus rapidly increase at a fourth temperature T4 (rapid viscosity rise start temperature), which is preferably lower than the second temperature T2 or the third temperature T3, and around room temperature Enteral hard capsule preparation liquid in the gel state, ie, storage elastic modulus G ′> loss elastic modulus G ′ ′.
- the rapid increase in viscosity near T4 in the cooling process of the capsule preparation liquid according to the present disclosure does not usually occur in the enteric methacrylic acid copolymer dispersion of the second component and the neutralization liquid of the enteric cellulose compound of the third component. Therefore, it is presumed that the rapid increase in viscosity near T4 is mainly caused by the structural viscosity of the partially dissolved nonionic water-soluble cellulose compound which is the i-th component.
- the viscosity tends to increase rapidly by one digit or more in the temperature range T4 of about 30 to 50 ° C in the cooling process. Becomes noticeable.
- the ratio of the i-th component contained in the preparation liquid is 0.05 ⁇ ⁇ ′ / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) ⁇ It is preferable that it is 0.5.
- the viscosity increase tends to be moderate, and if it is more than 0.5, the viscosity is too high and it tends to be difficult to form by the immersion method described later. More preferably, 0.07 ⁇ ⁇ ′ / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) ⁇ 0.4.
- ⁇ ′ / ( ⁇ ′ + ⁇ ′) is larger than 0, preferably 0.1 or more, more preferably 0.2 or more, and still more preferably 0.4 or more.
- concentration with respect to the solvent of solid content of a component i and a component ii is 10 mass% or more.
- Non-patent Document 5 Such cold gelation properties are undesirable because the gelled product causes clogging in the spray nozzle and the like in the coating such as spray coating, and therefore, high concentration and high viscosity non-ionic water solubility Selective combination of a cellulose compound and an enteric methacrylic acid copolymer is not usually performed (Non-patent Document 5).
- component iii, ⁇ ′ in order to reduce the ratio of component iii, ⁇ ′, it is possible to replace part or all of component iii with a water-insoluble (meth) acrylic acid alkyl ester copolymer as component iv.
- the amount of enteric polymer required, and hence the total amount of the basic neutralizing agent, can be further reduced without deteriorating the acid resistance, which is preferable.
- Water-insoluble (meth) acrylic acid alkyl ester copolymer dispersions can directly produce aqueous dispersions of colloids by an emulsion polymerization process. Therefore, it is preferable to use a water-insoluble (meth) acrylic acid alkyl ester copolymer dispersion without using an organic solvent for solubilization in water.
- the enteric polymer comprises an enteric cellulose compound and an enteric methacrylic acid copolymer
- the enteric polymer dispersion solution uses, for example, a basic neutralizing agent in an amount substantially or completely neutralized upon dissolution of the enteric cellulose compound. If, as the enteric methacrylic acid copolymer, a colloidal dispersion liquid by emulsion polymerization which does not require neutralization is used, the amount of the basic neutralizing agent used can be extremely reduced with respect to the total amount of these enteric polymers. As a whole, it is possible to make a dispersion of finely divided particles in which only a small proportion of enteric polymer has been neutralized (partially neutralized).
- This also has the advantage of being able to reduce the amount of residual salt in the capsule shell after drying without using volatile basic neutralizing agents such as ammonia.
- ⁇ ′ / ( ⁇ ′ + ⁇ ′) is 0.2 or more, the degree of neutralization can be 50% or less for the entire enteric polymer component including the enteric cellulose compound and the enteric methacrylic acid copolymer preferable.
- (beta) '/ ((beta)' + (gamma) ') is 0.4 or more, as an enteric polymer part whole, the degree of neutralization can be made into 30% or less and is more preferable.
- the degree of neutralization may be 20% or less, which is more preferable.
- the lower limit of the degree of neutralization is preferably 2% or more, and more preferably 5% or more, because of limitations in the preparation method of the capsule preparation liquid described later.
- adding at least one selected from the group consisting of PVA, a plasticizer and a surfactant as a v component is It can be used to adjust the viscosity of the capsule preparation. Alternatively, it can be used to stabilize the dispersion state of the colloid or solid fine particles of the capsule preparation liquid.
- ⁇ 'mass of the proportion of the ith component when the total mass of the ith component, the ii component, the iii component, the iv component, and the v component contained in the enteric hard capsule preparation liquid is 100% by mass %
- the proportion of the component ii is ⁇ 'mass%
- the proportion of the component iii is ⁇ ' mass%
- the proportion of the iv component is ⁇ 'mass%
- the proportion of the v component is ⁇ ' mass%
- the ratio is almost the same as the ratio of each component in the hard capsule film obtained by drying the preparation liquid.
- component ratios which are preferred as capsule films can be applied.
- the mass of each component is the mass of solid content.
- the ratio of the total of enteric polymer (components ii and iii) and component iv, ( ⁇ ′ + ⁇ ′ + ⁇ ′) / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) is 0.5 or more Is preferred.
- the value of ( ⁇ ′ + ⁇ ′ + ⁇ ′) / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) is more preferably 0.55 or more, and still more preferably 0.6 or more.
- ( ⁇ ′ + ⁇ ′) / ( ⁇ ′ + ⁇ ′ + ⁇ ′) is preferably 0.4 or more and more preferably 0.5 or more.
- the upper limit value of ( ⁇ ′ + ⁇ ′ + ⁇ ′) / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) is 0.9, preferably, in order to maintain appropriate hardness and crack resistance of the capsule shell. It is assumed to be 0.8.
- a more preferred embodiment is a composition in which all enteric cellulose compounds of the iii component are water insoluble (meth) acrylic acid alkyl ester copolymers of the iv component, as in the film component. That is, the composition is such that ⁇ ′ / ( ⁇ ′ + ⁇ ′ + ⁇ ′ + ⁇ ′) is 0.3 or more, more preferably 0.4 or more, when ⁇ ′ is 0%.
- the upper limit is preferably 0.7 or less, more preferably 0.65 or less.
- the ratio of the v component, ⁇ '/ ( ⁇ ' + ⁇ '+ ⁇ ' + ⁇ '+ ⁇ '), in any of the above cases, is preferably 0.15 or less, and 0.1 or less More preferable.
- a lubricant In addition to the i-th component, the ii-component, the iii-component, the iv-component, and the v-th component, a lubricant, a sequestering agent, a coloring agent, a light shielding agent and the like can be included.
- the sum of the mass of the ith component, the ii component, the iii component, the iv component, and the v component contained in the film is X ′, a plasticizer, a lubricant, a sequestering agent, a coloring agent, and a light shielding agent
- ⁇ ′ / X ′ can be in the range of 0.2 or less, more preferably in the range of 0.1 or less.
- the solid content of the i-th component, the ii-component, the iii-component, the iv-component and the v-component contained in the intestinal positive hard capsule preparation liquid is not limited as long as the hard capsule preparation liquid can be prepared.
- the enteric hard capsule preparation liquid is 100% by mass
- the total solid content of the ith component, the ii component, the iii component, the iv component and the v component is 10 to 30% by mass It is. More preferably, it is 13 to 25% by mass.
- the total amount thereof is 6% by mass or less, preferably 3% by mass or less, based on 100% by mass of the enteric hard capsule preparation liquid. More preferably, it is 2 mass% or less, More preferably, it is 1 mass% or less.
- the solid content dissolved or dispersed in addition to the components i to v is present in the capsule film with the ratio of the components being substantially maintained.
- part of the water in the solvent may remain in the film.
- a third aspect of the present disclosure relates to a method of preparing the enteric hard capsule preparation liquid of the second aspect.
- the third aspect is a method for preparing an enteric hard capsule preparation liquid, wherein the i-th component and the ii-th component are mixed under the condition that a basic neutralizing agent is present in a solvent,
- the i-th component is a nonionic water-soluble cellulose compound having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s
- the ii-component is an enteric methacrylic acid copolymer.
- enteric methacrylic acid copolymers it is preferred to use a colloidal dispersion.
- the i-th component having a viscosity value in the range of 100 mPa ⁇ s to 100,000 mPa ⁇ s. It is important to use together the ionic water-soluble cellulose compound and the enteric methacrylic acid copolymer, which is the second component, but when both are directly mixed, aggregation occurs immediately and a stable dispersion is obtained. It may be necessary to mix under conditions where a basic neutralizing agent is present in the solvent.
- the degree of neutralization with respect to the component ii is preferably 2% or more, more preferably 5% or more.
- the upper limit is 20% or less, more preferably 15% or less. If it exceeds 20%, the cold gelation performance tends to be impaired.
- the degree of neutralization of the enteric polymer relative to the whole is preferably 10% or more, more preferably 20% or more.
- the upper limit is preferably 50% or less, more preferably 30% or less.
- the third embodiment is divided into an embodiment (third embodiment) including the enteric cellulose compound of the component iii as an enteric polymer and an embodiment not including the enteric cellulose compound (third embodiment).
- the third aspect is Step A: preparing a neutralized solution of the component iii, step B: adding the component i to the neutralized solution containing the component iii, preparing a partial solution of the component i, and step C: mixing the dispersion of component ii with the solution prepared in step A or step B,
- the present invention relates to a method for preparing an enteric hard capsule preparation liquid containing a film component containing the i-th component, the ii-component and the iii-component and a solvent.
- each component may be added to a solvent, or the component may be added or mixed to the solution prepared in the previous step that already contains other components.
- the transition temperature between the steps or the temperature control at the time of mixing the solution prepared in each step can be appropriately set in accordance with the following requirements.
- step A as the neutralization liquid of the component iii, a solution obtained by dissolving the neutralized product of the component iii may be used, but after the non-neutralized component iii is dispersed in a solvent, the basic neutralizing agent is used. It may be prepared by adding Preferably, step A comprises dispersing the unneutralized component iii in a solvent and then adding and at least partially neutralizing the pharmaceutically or food additive acceptable basic neutralizing agent described above. It is the process of preparing the neutralization liquid to be dissolved in a solvent.
- the non-neutralized component iii is introduced into a solvent such as purified water in an amount of about 5 times the total amount of all components excluding the solvent, and dispersed uniformly so as not to be dull. Thereafter, a basic neutralizing agent is added to dissolve the component iii. In order to completely neutralize the component iii, as described above, the cation derived from the basic neutralizing agent is equal to or more than 1 mol of the carboxyl group contained in the component iii. It is achieved by adding.
- enteric cellulose compounds have a large mass of pulp as a starting point, and their molecular weight is controlled by hydrolysis or chemical decomposition by enzymes, and they are crushed by mechanical methods such as mechanical milling to obtain 10 to 10 It is a solid particle of the order of 100 ⁇ m. In order to miniaturize it to make the particle size smaller than about 1 ⁇ m or dissolve it, it is preferable to set the degree of neutralization when neutralizing only the iii component to 50% or more of the equivalent amount. The state in which the degree of neutralization is not completely neutralized is called partial neutralization. That is, the neutralization liquid in step A preferably has a degree of neutralization of 50% or more, or may be a complete neutralization liquid.
- the pH of the completely neutralized enteric cellulose solution is preferably set to a pH near the dissociation point of the enteric cellulose compound.
- the pH is preferably 4.5 to 7.0, and more preferably 5.0 to 6.5 around the dissociation point of the enteric cellulose compound. If the pH is less than 4.5, the enteric cellulose compound may not dissolve, and if the pH exceeds 7.0, the excess cation may neutralize to a methacrylic acid copolymer to be added later. .
- the dissociation point of the enteric cellulose compound is, for example, in the case of HPMCP, a pH at which the carboxyl group of the carboxybenzoyl group is ionized in the neutralization titration to dissolve HPMCP and neutralize the solution.
- HPMCAS the carboxyl group of the succinoyl group and acetyl group is ionized in the neutralization titration
- the pH is the pH at which the HPMCAS is dissolved and the solution is neutralized.
- the pH near the dissociation point of HPMCP and HMCAS is pH 5-7.
- Step B specifically prepares a partially dissolved solution in which the i-th component is partially dissolved in a neutralized solution containing the iii component or a mixed solution of the neutralized solution of the iii component and the dispersion of the ii component.
- the step of preparing the partial solution, the i-th component at a first temperature T1 above the cloud point T0 of the i-th component, the neutralization solution containing the iii-component or the iii-component This is a step of preparing a dispersion in which the i-th component is partially dissolved at a second temperature T2 lower than the cloud point, added to the mixture of the neutralization liquid and the dispersion of the component ii.
- the first temperature T1 is not limited as long as it is a cloud point T0 or more and a temperature lower than the boiling point of the solvent.
- the temperature can be in the range of 60 ° C. to 90 ° C.
- the temperature T1 can be in the range of 70 ° C to 90 ° C.
- the temperature T1 is preferably in the range of 60 ° C to 80 ° C. Dispersing above the cloud point is to prevent the formation of soot before being uniformly dispersed.
- the second temperature T2 is preferably higher than room temperature (20 ° C. to 25 ° C.) and lower than the cloud point T0.
- the temperature is preferably in the range of 30 ° C. to 60 ° C.
- the temperature T2 can be in the range of 30 ° C to 60 ° C.
- the temperature T2 is preferably in the range of 30 ° C to 40 ° C.
- the viscosity of the dispersion in which the non-ionic water-soluble cellulose compound is not dissolved and dispersed at a temperature of T0 or more is very low and is generally less than 100 mPa ⁇ s.
- T0 is passed in the temperature lowering process.
- T0 is passed in the temperature lowering process.
- a dispersion in which solid particles of undissolved water-soluble cellulose are stably present is obtained.
- the temperature T2 be equal to or lower than T0 and not lower than T4. Specifically, it is preferable not to fall below 30 ° C., and it is more preferable to set the temperature to 35 ° C. or more. Further, T2 is preferably 60 ° C. or less, more preferably 55 ° C. or less.
- the i-th component is obtained by suspending the nonionic water-soluble cellulose compound which is the i-th component in the neutralized solution of the iii-component at a temperature T1 above the cloud point T0 and lowering the temperature to the second temperature T2.
- a partially dissolved dispersion can be prepared.
- the methacrylic acid copolymer as the second component is mixed with the neutralization solution containing the enteric cellulose compound as the aqueous dispersion,
- the enteric polymer as a whole including the enteric cellulose compound and the methacrylic acid copolymer can be in a partially neutralized state. Therefore, when mixing the i-th component and the ii-th component, it is preferable that the basic neutralizing agent is already present in the solvent.
- the degree of neutralization of the combined enteric cellulose compound and methacrylic acid copolymer with respect to the entire enteric base can be 50% or less, and can be 30% or less. This is effective in the dissolution test to suppress the dissolution of the capsule film in the intermediate region of pH 4-5.
- This embodiment 3-1 may include the step D of mixing the solution prepared in the step A, B or C with the water-insoluble (meth) acrylic acid alkyl ester copolymer as the iv component. Further, the method may further include the step E of maintaining the solution obtained in the step B, C or D at a third temperature T3 lower than the cloud point of the i-th component.
- the temperature may be in the range of 30 ° C. to 65 ° C.
- the temperature T3 can be in the range of 40 ° C. to 65 ° C.
- the temperature T3 can be in the range of 45 ° C. to 60 ° C., and more preferably, in the range of 50 to 60 ° C.
- a temperature difference of about 10 ° C. or more can be secured between the temperature T4 at which the viscosity jump due to cold gelation starts, and it is preferable to keep the prepared solution stable.
- the temperature T3 is preferably in the range of 30 ° C to 50 ° C.
- the 3-1 aspect of the present disclosure further includes 2 aspects of the 3-1a aspect and the 3-1b aspect.
- the step a1 a step of preparing a neutralized solution of the component iii
- the step b1 a step of partially dissolving the component i in the neutralized solution of the component iii
- a step c1 Mixing the dispersion of the component ii with the solution obtained in the step b1), an enteric hard capsule preparation liquid comprising a film component comprising the component i, the component ii, and the component iii, and a solvent Relates to the preparation method of
- the description of the neutralization liquid of component iii, the cloud point T0, the temperature T1, the temperature T2 and the temperature T3 in the above 3-1 aspect is incorporated herein by reference.
- the step a1 is a step of preparing a neutralization liquid of the iii component
- the step a1 conforms to the step A in the third aspect.
- step b1 the i-th component is dispersed in the liquid prepared in step a1 at a first temperature T1 higher than its cloud point T0 and then cooled, and partially dissolved at a second temperature T2 lower than the cloud point To prepare a dispersion. That is, a dispersion liquid in which the i-th component is partially dissolved can be prepared by dispersing the i-th component in the liquid prepared in step a1 at a temperature above the cloud point T0 and lowering the temperature to the second temperature T2. .
- step c1 the partial solution of the i-th component containing the third component iii obtained in step b1 and the dispersion of the ii component are mixed.
- the mixing method is not limited as long as the neutralization liquid of the iii component and the dispersion liquid of the ii component can be mixed.
- the water-insoluble (meth) alkyl ester copolymer is added to the solution obtained in step c1 after step c1 in step d1, and further, the solution after step b1, c1 or d1 is the i-th component
- the method may further include the step e1 of maintaining the third temperature T3 lower than the cloud point of
- the step a2 preparing a neutralized solution of the component iii
- the step c2 mixing the dispersed solution of the component ii with the neutralized solution of the component iii
- a step b2 partially dissolving the i-th component in the solution obtained in the step c2; preparing an enteric hard capsule preparation liquid comprising a film component and a solvent comprising the i-th component, the ii-component and the iii-component On the way.
- the step a2 is a step of preparing a neutralization liquid of the iii component, and the step a2 conforms to the step A in the third aspect.
- step c2 the neutralization liquid of the iii component and the dispersion liquid of the ii component are mixed.
- the mixing method is not limited as long as the neutralization liquid of the iii component and the dispersion liquid of the ii component can be mixed.
- step b2 the i-th component is dispersed in the liquid prepared in step c2 at a first temperature T1 higher than its cloud point T0 and then cooled, and partially dissolved at a second temperature T2 lower than the cloud point To prepare a dispersion. That is, by suspending the i-th component in the liquid prepared in step c2 at a temperature above the cloud point T0 and lowering the temperature to the second temperature T2, a dispersion in which the i-th component is partially dissolved can be prepared. .
- the method may further include the step e2 of maintaining at a third temperature T3 lower than the cloud point of the i component.
- at least one selected from the group consisting of polyvinyl alcohol which is the v component, a plasticizer, and a surfactant is maintained at temperature T3 in step E after raising the solution temperature to temperature T1 in step B. It is preferable to add in the process until it is carried out.
- the step A ′ preparing a partially neutralized solution of the component ii
- Step B ′ preparing a partial solution of the ith component
- Step C ′ mixing the dispersion of the iv component with the solution prepared in step A ′ or B ′
- the iv component is a dispersion of a water insoluble (meth) acrylic acid alkyl ester copolymer.
- the methacrylic acid copolymer which is the second component it is preferable to use a colloidal dispersion in which the additional dispersing step by neutralization can be omitted.
- a colloidal dispersion in which the additional dispersing step by neutralization can be omitted.
- step A ′ a basic neutralizing agent which accepts a component ii as a pharmaceutically or food additive is added in advance to prepare a partially neutralized solution of the component ii.
- the degree of neutralization is relatively low, preferably 2 to 20%. More preferably, it is 5 to 15%.
- step B ′ the i-th component is added to the solution containing the basic neutralizing agent and the second component to prepare a partially dissolved solution.
- the i-th component is added to the neutralized solution containing the ii-component or the mixed solution of the neutralized solution of the ii-component and the dispersion of the iv-component at a first temperature T1 higher than the cloud point T0 of the i-th component Preparing a dispersion in which the i-th component is partially dissolved at a second temperature T2 lower than the cloud point.
- the present embodiment 3-2 may further include the step E ′ of maintaining the solution obtained in the step B ′ or C ′ at a third temperature T3 lower than the cloud point of the i-th component.
- the third temperature T3 is preferably a temperature higher than T2 and not lower than the cloud point T0 by 10 ° C. or more.
- the temperature may be in the range of 30 ° C. to 605 ° C.
- the temperature T3 can be in the range of 40 ° C to 65 ° C.
- the temperature T3 can be in the range of 45 ° C. to 60 ° C., and more preferably, in the range of 50 to 60 ° C.
- a temperature difference of about 10 ° C. or more can be secured between the temperature T4 at which the viscosity jump due to cold gelation starts, and it is preferable to keep the prepared solution stable.
- the temperature T3 can preferably be in the range of 30 ° C to 40 ° C.
- the (meth) acrylic acid alkyl ester copolymer as the iv component substantially interacts with any of the i th component and the ii component.
- the present embodiment may further include, after step C ′, step E ′ of maintaining the solution obtained in step C ′ at a third temperature T3 lower than the cloud point of the i-th component.
- the temperature of the solution is raised to the temperature T1 in the step B ′ and then the temperature T3 in the step E ′. It is preferable to add it in the process until it is retained.
- the fourth aspect of the present disclosure relates to a method of preparing enteric hard capsule.
- capsule preparation machines that prepare other hard capsules can be used to prepare enteric hard capsules.
- the enteric hard capsule according to the present disclosure is characterized by being formed by a dipping method, in particular a "cold pin dipping method".
- the “cold pin immersion method” is characterized in that the surface temperature of the molding pin at the time of immersion is lower than the temperature of the capsule preparation liquid.
- the method for preparing (forming) the enteric hard capsule is not particularly limited as long as it includes the step of preparing the capsule using the enteric hard capsule preparation liquid according to the present disclosure.
- enteric hard capsules are prepared by immersing mold pins (caps molding pins) which become molds of the capsules in enteric hard capsule preparation liquid, and curing and drying the film adhering when pulled up. To obtain the desired capsule shape and thickness (dipping method).
- the enteric hard capsule is prepared by preparing the enteric hard capsule preparation liquid by the method described above or purchasing the enteric hard capsule preparation liquid, and the like. After immersing the mold pin in the preparation liquid, the mold pin is pulled up, the mold pin is turned upside down, and the solution adhered to the mold pin is dried.
- the enteric hard capsule used in the present disclosure can be manufactured through the following molding process. (1) A step of immersing the mold pin in the enteric hard capsule preparation liquid (immersion step), (2) a step of pulling up the mold pin from the enteric hard capsule preparation liquid (immersion liquid) and drying the enteric hard capsule preparation liquid adhering to the outer surface of the mold pin (drying step); (3) A step of detaching the dried capsule film (film) from the capsule molding pin (elimination step).
- the enteric hard capsule preparation liquid is a temperature lower than the cloud point of the nonionic water-soluble cellulose compound when the mold pin is immersed, and a temperature T5 higher than room temperature (20 ° C. to 25 ° C.) Is held by.
- T5 is preferably a temperature not lower by 10 ° C. or more than the cloud point T0, and more preferably higher than T2.
- T3 and T5 can be the same. Thereby, the partial dissolution state of the i-th component can be stably maintained.
- T5 is preferably in the range of 30 ° C.
- the temperature T5 is preferably in the range of 30 ° C to 40 ° C.
- the viscosity at the time of immersion of the capsule preparation liquid is preferably 100 mPa ⁇ s or more at its holding temperature T5, more preferably 500 mPa ⁇ s or more, and still more preferably 1000 mPa ⁇ s or more.
- the viscosity at the time of immersion of the capsule preparation liquid is preferably 10000 mPa ⁇ s or less at its holding temperature T5, more preferably 5000 mPa ⁇ s or less, and still more preferably 3000 mPa ⁇ s or less.
- the viscosity of the capsule preparation liquid can be measured using a single cylindrical rotational viscometer (Brookfield viscometer, B-type viscometer). For example, after preparing a capsule preparation in a 1-liter beaker (volume: 600 ml), add M3 rotor (measurement range: 0 to 10,000 mPa ⁇ s) to the capsule preparation maintained at T5, and the rotor rotation speed , 12 r. p. m. , Measurement time can be measured in 50 seconds.
- Brookfield viscometer B-type viscometer
- the surface temperature T6 of the mold pin at the time of immersion is lower than the liquid temperature T5 of the enteric hard capsule preparation liquid, and further lower than the temperature T4 which causes a sharp viscosity increase due to cold gelation.
- it is in the range of 20 ° C. to 30 ° C., more preferably in the range of 20 to 28 ° C.
- the drying step (2) can be carried out at room temperature (20 to 30 ° C.), although it is not particularly limited. It is usually performed by blowing air at room temperature.
- the capsule coating thus prepared can be provided as an enteric hard capsule in a state in which the body portion and the cap portion are fitted or not fitted after being cut and adjusted to a predetermined length. .
- the film thickness of the enteric hard capsule is usually in the range of 50 to 250 ⁇ m.
- the thickness of the side wall portion of the capsule is usually 75 to 150 ⁇ m, more preferably 80 to 120 ⁇ m, for capsules currently on the market.
- the size of the enteric hard capsule there are 00, 0, 1, 2, 3, 4, 5 and the like, but according to the present disclosure, enteric hard capsules of any size may be prepared. it can.
- enteric-coated hard capsule preparation The enteric-coated hard capsule according to the present disclosure is filled with general food, health functional food (functional display food, nutritional function food, food for specified health use), quasi-drugs, quasi-drugs, etc. It can be done.
- components raw plants, partially dried microorganisms, or completely dried plants, plant extracts, plant extracts, etc.
- plants including unicellular green algae
- microorganisms bacteria, yeast, etc.
- Midorimu etc. or components derived from the above-mentioned microorganisms (raw microorganisms, partially dried microorganisms, or completely dried microorganisms, processed products of microorganisms, microorganism extracts etc.)
- nourishing tonic health agents antipyretic analgesic anti-inflammatory agents, Psychotropic, Anxiolytic, Antidepressant, Hypnosis and Sedative, Anticonvulsant, Central Nervous Agents, Cerebral Metabo
- fillers are not particularly limited, and a wide variety of known fillers can be mentioned. These components can be used alone or in combination with other components.
- the filler may be in the form of solid, powder, granules, crushed material, liquid, gel and the like. In addition, these components are appropriately filled with a known appropriate amount determined according to the condition, age, etc. of the administration subject.
- vitamin A for example, vitamin A, vitamin D, vitamin E (such as d- ⁇ -tocopherol acetate), vitamin B1 (such as dibenzoyl thiamine or flusultiamine hydrochloride), vitamin B2 (such as riboflavin butyrate), vitamin Vitamins such as B6 (pyridoxine hydrochloride etc.), vitamin C (ascorbic acid, sodium L-ascorbate etc.), vitamin B12 (hydroxocobalamin acetate, cyanocobalamin etc.), minerals such as calcium, magnesium and iron, proteins, amino acids, oligosaccharides, Crude drug etc. are included.
- vitamin B1 such as dibenzoyl thiamine or flusultiamine hydrochloride
- vitamin B2 such as riboflavin butyrate
- vitamin Vitamins such as B6 (pyridoxine hydrochloride etc.)
- vitamin C ascorbic acid, sodium L-ascorbate etc.
- vitamin B12 hydroxocobala
- antipyretic analgesic anti-inflammatory agents include aspirin, acetaminophen, etendamide, ibuprofen, diphenhydramine hydrochloride, dl-chlorpheniramine maleate, dihydrocodeine phosphate, noscapine, methylephedrine hydrochloride, phenylpropanolamine hydrochloride, caffeine, caffeine anhydride And serapeptase, lysozyme chloride, tolfenamic acid, mefenamic acid, diclofenac sodium, flufenamic acid, salicylamide, aminopyrine, ketoprofen, indomethacin, bucolome, pentazocine and the like. However, it is not limited to these.
- the enteric hard capsule is highly useful when it is likely to have side effects on the stomach when it is dissolved in the stomach.
- it may be acid labile and need to be absorbed in the intestine without dissolving in the stomach. That is, the preparation which can reduce the efficacy of the active ingredient by gastric acid can protect the active ingredient from gastric acid by the enteric hard capsule preparation according to the present disclosure, effectively pass through the stomach and can be delivered to the intestine. , Especially useful.
- aspirin is known to have side effects that cause gastric ulcer-like symptoms, for example, when administered in large amounts in naked granules, and is one of the representative drugs for which the application of enteric hard capsules is desired.
- examples of the acid labile active ingredient include omeprazole, lansoprazole, rabeprazole sodium, esomeprazole magnesium hydrate, etc. which are known as proton pump inhibitors (PPIs).
- PPI proton pump inhibitors
- PPI travels in the bloodstream and reaches parietal cells, and is activated in contact with high concentrations of hydrogen ions in the parietal secretory tubules of parietal cells.
- PPI is an extremely unstable drug in an acidic environment and can not exert its full effect if exposed to acid before reaching parietal cells. For this reason, in order to exert the strong acid secretion inhibitory power of PPI, it is usually formulated into an enteric formulation.
- Duloxetine which is one of the antidepressants called serotonin / noradrenaline reuptake inhibitors, is also acid-sensitive, and so is an example of a drug substance where enteric formulation is desirable.
- Fucoidan, heme iron, polyphenols, peptides and amino acids for example, as a general food, health functional food (functional display food, nutritional function food, food for specified health)) for enteric hard capsules according to the present disclosure
- Royal jelly, ornithine, citrulline, aminolevulinic acid, black vinegar, or hydrophobic amino acids such as methionine, valine, leucine, isoleucine, proteins (milk proteins such as lactoferrin, collagen, placenta, etc.), glycoproteins, Enzyme-fermented foods (such as nattokinase), coenzymes (such as coenzyme Q10), vitamins (such as ⁇ -carotene), minerals, living microorganisms (such as green algae, chlorella, yeast, lactic acid bacteria, bifidobacteria), plant extracts (such as Herbal medicine, herbs such as turmeric extract, ginseng , Plum extract, ginkgo leaf extract, blueberry extract, etc. cha
- Such contents may be filled into enteric hard capsules by a capsule filling machine known per se, such as a fully automatic capsule filling machine (model name: LIQFILsuper 80/150, manufactured by Qualicaps Co., Ltd.), a capsule filling and sealing machine Model name: LIQFILsuperFS, manufactured by Qualicaps Co., Ltd., etc. can be used.
- a capsule filling machine known per se, such as a fully automatic capsule filling machine (model name: LIQFILsuper 80/150, manufactured by Qualicaps Co., Ltd.), a capsule filling and sealing machine Model name: LIQFILsuperFS, manufactured by Qualicaps Co., Ltd., etc.
- the body portion and the cap portion of the hard capsule thus obtained are filled with the contents in the body portion, and then the body portion is covered with the cap portion and the body portion and the cap portion are joined by fitting them together.
- the filled capsules can then be tamper-proofed by using an appropriate technique to permanently seal the seam.
- sealing sealing or banding (hereinafter referred to as sealing) techniques may be used, wherein these techniques are well known to those skilled in the art of capsules.
- the sealing agent for the polymer solution hereinafter referred to as “the polymer solution sealing agent in the circumferential direction of the body portion and the cap portion on the surface of the body portion and the surface of the cap portion with a constant width around the edge portion of the cap portion
- the fitting portion can be sealed by applying the seal preparation solution (one or two times), preferably once or twice, to obtain an enteric hard capsule preparation.
- the polymer solution may be a dilute aqueous solution of an enteric polymer used for capsule film, or a solution dissolved in water / ethanol or water / isopropanol solvent. When a dilute aqueous solution or a solution dissolved in water / ethanol or water / isopropanol solvent is used, it may be partially neutralized and dissolved with the above-mentioned basic neutralizing agent.
- the polymer contained in the seal preparation liquid preferably comprises the same enteric polymer or nonionic water-soluble cellulose compound as contained in the enteric hard capsule shell to which the seal is applied. As well as being excellent in adhesion to the capsule shell, it also prevents unnecessary additive components from being included in the capsule preparation.
- the viscosity value of the nonionic water-soluble cellulose compound may be 100 mPa ⁇ s.
- the seal preparation can be generally used at room temperature or under heating. From the viewpoint of liquid leakage prevention of the hard capsule, it is desirable to use a seal preparation liquid within a temperature range of preferably about 23-45 ° C., more preferably about 23-35 ° C., and most preferably about 25-35 ° C.
- the temperature adjustment of the seal preparation liquid can be carried out by a method known per se, such as a panel heater or a hot water heater, for example, a circulating hot water heater or a seal pan unit of the integrated capsule filling and sealing machine. It is preferable to adjust the temperature of the heating water heater type, because the temperature range can be finely adjusted.
- the enteric hard capsule preparation according to the present disclosure thus obtained exhibits acid resistance in the stomach when administered and ingested in the human or animal body, and mainly transits to the intestines to dissolve the capsule coat, resulting in contents Is designed to be released. For this reason, it is suitable as a preparation filled with medicines and food which release in the stomach is not desirable.
- the capsule coating is externally coated with an additional one or more polymer layers to enhance enteric function, to control additional drug delivery control functions, or to control gas or moisture permeability. It may be
- a functional polymer layer means a layer containing a functional polymer that imparts certain mechanical or chemical properties to the coated capsule coating.
- the functional polymer is an enteric polymer and / or colon releasable polymer conventionally used to coat pharmaceutical solid dosage forms (ie, used to disintegrate coated dosage forms in the colon area of a subject) Polymers, etc.).
- Hard capsule preparation As a novel application using the enteric hard capsule according to the present disclosure, a hard capsule preparation characterized by including the enteric hard capsule according to the present disclosure in the inside of the hard capsule which can be dissolved under acidic conditions Can be mentioned.
- Hard capsules that can be dissolved under acidic conditions include, but are not limited to, gelatin capsules and hypromellose capsules, or pullulan capsules.
- the hypromellose hard capsule one having a display viscosity (viscosity grade) value of water-soluble cellulose of 3 to 15 mPa ⁇ s is used (Japanese Patent Application Laid-Open Nos. 08-208458, 2001-506692 and 2010 -270039, JP-A-2011-500871).
- the enteric hard capsule according to the present disclosure is filled with the active ingredient B beforehand, and the medicinal active ingredient A and the filled enteric hard capsule are filled inside the hard capsule which can be dissolved under acidic conditions.
- Such a double capsule formulation enables delivery of selective and different active ingredients at multiple sites so that the active ingredient A is released in the stomach and the active ingredient B is released after reaching the intestine.
- the active ingredient A and the active ingredient B are the above-mentioned 5.
- the active ingredient as described in can be mentioned.
- Nonionic Water-Soluble Cellulose Compounds Methylcellulose (MC) and hydroxypropyl methylcellulose (HPMC) can be used as the hydroxypropyl cellulose (HPC) using the METOLOSE® series or the TC-5® series of Shin-Etsu Chemical Co., Ltd. ) Used the NISSO HPC series of Nippon Soda Co., Ltd.
- Specific product names and substitution degree types, “viscosity value”, (indicated viscosity or viscosity grade) are as shown in Table 2.
- HP50 hydroxypropyl cellulose phthalate
- HPMCAS-MF Hydroxypropyl methylcellulose acetate succinate
- HP50 0.065 g / g
- HMPCAS-MF or HMPCAS-MG 0.048 g / g
- L30D55 0.215 g / g
- FS30D 0.215 g / g
- HP 50 was used as completely neutralized as possible because the particle size was relatively large and the film was rough.
- HPMCAS-MF has a relatively small particle size, an almost transparent solution is obtained when the degree of neutralization is 50% or more, more preferably 80% or more.
- the equivalent weight of neutralization to ammonia is 0.0274 g / g for HP50, 0.0202 g / g for HMPCAS-MF or HMPCAS-MG, and 0.0914 g / g for L30D55.
- FS30D it is 0.0172 g / g.
- ammonia due to the volatilization in the preparation liquid and the resulting pucell preparation process, it was added in excess over the equivalent.
- Methacrylic Acid Copolymer Evonik Industries AG, EUDRAGIT (registered trademark) series L30D55 and FS30D were used. All are aqueous dispersions having a solid content of 30% by mass.
- L30D55 was dried and pulverized, and after being dispersed in purified water and stirred, NaOH (10% aqueous solution) was added so as to have a predetermined degree of neutralization. As a result, a micronized aqueous dispersion was obtained although it was somewhat coarser than the L30D55 colloidal particles.
- Capsule Dissolution Test In principle, the dissolution test in the 17th revised Japanese Pharmacopoeia was applied to the present disclosure. However, since the Japanese Pharmacopoeia does not necessarily define the solubility of the empty hard capsule itself, in the present disclosure, the solubility of the capsule itself is evaluated by evaluating the dissolution of fast-dissolving acetaminophen. Characteristics) were evaluated.
- acetaminophen mixed powder 20 mg of sodium starch glycolate (hereinafter referred to as "acetaminophen mixed powder"), and the obtained enteric hard capsule preparation is defined in the Japanese Pharmacopoeia Aceto was tested according to the specified dissolution method (The 17th edition, 6.10-1.2 paddle method (paddle rotation speed: 50 rpm) and the use of a sinker corresponding to 6.10-2 a in the same figure). The time change of the dissolution rate of aminophen was measured. A Distek bath type dissolution tester Model 2100 was used for the dissolution test. Separately, the entire volume of acetaminophen is dissolved in the solution in the dissolution tester bath.
- the absorbance at 244 nm is 100%, and the dissolution tester bath rises with the dissolution of acetaminophen from the capsule.
- the elution rate was determined from the absorbance at 244 nm of the solution contained therein.
- the following aqueous solution was used as a 1st liquid and a 2nd liquid here. In all cases, the temperature of the solution in the bath was 37.degree.
- First solution 7.0 mL of hydrochloric acid and water were added to 2.0 g of sodium chloride to dissolve, and the solution was adjusted to 1000 mL (pH is about 1.2, hereinafter may be referred to as an acidic solution).
- Liquid 2 Dissolve 3.40 g of potassium dihydrogen phosphate and 3.55 g of anhydrous disodium hydrogen phosphate in water, and prepare 1000 mL by adding 1 volume of water to 1 volume of phosphate buffer (pH is about 6.8, hereinafter sometimes referred to as neutral solution).
- the dynamic viscoelasticity of the capsule preparation liquid was measured using a rheometer (MCR102) manufactured by Anton Paar.
- MCR102 rheometer
- a double cylindrical tube measurement jig (model number CC27 / T200 / SS) and a temperature control system C-PTD200 were used.
- the volume was about 19 mL.
- about 1 mL of cottonseed oil was dropped on the outermost surface of the sample solution in the cylindrical tube.
- the temperature dependence was measured from 60 ° C. to 20 ° C. at 1 ° C./min and at the same time linearly decreasing the strain swing angle from 1 to 0.1%.
- the angular frequency ⁇ (rad / sec) is 2 ⁇ / sec.
- storage elastic modulus G ′ (Pa) loss elastic modulus G ′ ′ (Pa), complex viscosity
- / ⁇ ⁇ (G′2 + G′′2) / ⁇
- the values of (Pa ⁇ s) and viscosity '' ′ ′ G ′ ′ / ⁇ (Pa ⁇ s) were measured.
- Viscosity of Capsule Preparation Liquid The viscosity of the capsule preparation liquid (55 ° C.) was measured using a Brookfield viscometer (TVB-10M (Toki Sangyo)). An M3 rotor (measurement range: 0 to 10,000 mPa ⁇ s) was used for measurement. The number of revolutions of the rotor was determined by preparing a capsule preparation solution (liquid volume: 600 ml) with a 12 rpm, 1 L beaker, then placing the rotor in the beaker and measuring the measurement time for 50 seconds.
- a Brookfield viscometer Brookfield viscometer
- a scanning electron microscope (SEM) and microscopic Raman were used to observe the film structure.
- SEM The scanning electron microscope used Carl Zeiss Ultra 55.
- the prepared capsule coating is cut into circular cut pieces, embedded in an epoxy resin, and then cut with a microtome to cut it for observation (approximately 300 to 400 ⁇ m square with a thickness of 2 to 3 ⁇ m) Made. Sections were evaporated with PtPd. The electron beam was irradiated at an acceleration voltage of 3 kV to scan the section.
- the preparation solution was observed using an optical microscope (BX53 manufactured by Olympus Corporation) having a stage temperature control function. Use an eyepiece of 10x and an objective lens of 10x. The transmission was observed.
- the 55 ° C. preparation was dropped onto a preheated glass slide, also on the 55 ° C. stage, and then covered with a coverslip, also preheated to 55 ° C.
- the salt (sodium) in the capsule film was quantified by atomic absorption spectrophotometry (AAS) after dry ashing treatment according to the following procedure.
- AAS atomic absorption spectrophotometry
- the sample was precisely weighed in a platinum crucible and heated to 650 ° C. in an electric furnace until the organic matter disappeared after addition of concentrated sulfuric acid.
- the remaining ash was dissolved in dilute hydrochloric acid, diluted appropriately, and quantified with an atomic absorption spectrometer (SpectrAA-220 manufactured by VARIAN).
- the cast film was placed on a glass surface kept at room temperature or on a PET film with a metallic applicator, and the preparation liquid at 50 ° C. to 60 ° C. was poured and moved at a constant speed to produce a uniform film of 100 ⁇ m. Thereafter, drying was carried out at room temperature to 30 ° C. for about 10 hours. In order to obtain a film having a uniform film thickness of 100 ⁇ m, an applicator having a gap of 0.4 mm to 1.5 mm was properly used.
- the produced film was cut into a dumbbell shape of 5 mm ⁇ 75 mm (specified by JIS K-7161-2-1BA), and then subjected to a tensile test using a small bench test machine (EZ-LX manufactured by Shimadzu Corporation). Both ends of the film were set in a holder (gap length 60 mm), tensile rate, tension at 10 mm / min, film elongation and stress (tensile stress) -elongation (strain) curve generated in the film were determined. From the inclination of the elastic deformation area at the time of low stress in FIG. 5, the modulus of elasticity, which is an index of hardness, is obtained, and the elongation at break is taken as the elongation at break (%) (Non-Patent Document 1, Chapter 4).
- a capsule preparation liquid was prepared according to the following procedure. All operations were performed while stirring the solution.
- the solid content of the i-th component is referred to as polymer solid content.
- the total solution mass is added to purified water which is a solvent, and becomes the total mass of polymer solid content, basic neutralizing agent, and other solid content (plasticizer, light shielding agent, etc.).
- the polymer solid concentration means the ratio (% by mass) of the total mass of the polymer solid content to the total solution mass.
- the amount (equivalent amount) necessary to just completely neutralize the enteric cellulose compound was used, unless otherwise specified.
- c. The temperature of this solution was raised to 83 ° C., and then a nonionic water-soluble cellulose compound was added, and a suspension was prepared by uniformly dispersing the solution so that it could not be scalded.
- d. The temperature of the dispersion of the nonionic water-soluble cellulose compound was lowered, the temperature was lowered to a temperature T2 below the melting temperature (cloud point), and the nonionic water-soluble cellulose compound was partially dissolved to prepare a dispersion.
- the partial melting temperature T2 was properly adjusted between 30 and 55.degree. e. d.
- the dispersion prepared in the above was maintained at preparation solution temperature T3 (30 to 50.degree. C. for MC, 45 to 60.degree. C. for HPMC, and 30 to 40.degree. C. for HPC).
- the viscosity in the Brookfield viscometer was in the range of approximately 1,000 to 3,000 mPa ⁇ s.
- the final concentration of polymer solids was finely adjusted by addition of warm pure water and evaporation so that the viscosity was in this range.
- the dispersion of methacrylic acid copolymer is b. After neutralization, or e.
- the partial solution of nonionic water-soluble cellulose compound was added at any stage after completion of the solution.
- an aqueous dispersion is prepared in advance, and c. Were introduced before the operation. In all the above steps, stirring is performed at 100 to 1,000 rpm using a propeller stirring blade.
- a predetermined amount of methacrylic acid copolymer dispersion was poured into the purified water at room temperature, and then sodium hydroxide (NaOH) was added as a basic neutralizing agent to prepare a partially neutralized solution.
- NaOH sodium hydroxide
- This partially neutralized solution is heated to 83 ° C., and then the titanium oxide dispersion is charged, and after sufficiently stirring with a three-one motor, a non-ionic water-soluble cellulose compound is charged, so that no defects can occur.
- the suspension was prepared by dispersing and degassing.
- the viscosity in the Brookfield viscometer was in the range of approximately 1,000 to 3,000 mPa ⁇ s.
- the final total solid content concentration was finely adjusted by addition of warm pure water and evaporation so that the viscosity was in this range.
- stirring was performed at 100 to 1,000 rpm using a propeller blade.
- a hard capsule was prepared by a cold pin dipping method using the capsule preparation prepared in 1. above.
- the holding temperature T5 is approximately the same as T3, and after immersing the mold pin (size 2 size) left at room temperature (about 25 ° C.) for several seconds in the capsule preparation liquid kept at a substantially constant temperature, I pulled it up.
- the molding pin to which the capsule preparation liquid was attached was turned upside down and dried at room temperature for 2 to 10 hours or more.
- the immersion time of the mold pin, the pulling speed, and the like were appropriately adjusted so that the film thickness of the cylindrical capsule side surface was about 100 ⁇ m.
- the capsule part was pulled out of the mold pin and cut so that the length of the cylindrical part was a predetermined length. The above operation was performed for each of the cap and the body.
- ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, ⁇ ′, which are the composition of the preparation liquid component, are the same as the composition of the film component are the composition ⁇ , ⁇ , It used as what was the same as (gamma), (sigma), (phi), (delta), (epsilon).
- Preparation Example V-1 Preparation Example (Aspect 3-1 of Preparation Method)
- a capsule preparation liquid was prepared according to Preparation Example III-1 (embodiment 3-1 of the preparation method), and molding was performed according to molding method IV.
- the mass ratio of the basic neutralizing agent (NaOH) and the titanium oxide (light shielding agent) to the total mass of the solid content of the polymer was ⁇ (%) and ⁇ (%), respectively.
- the mass ratio of the solid content of the components i to iii in the total mass of the purified water as the solvent and the solid content of the components i to iii was taken as the polymer solid content concentration (%).
- Tables 3 to 7 show specific compositions of each.
- the degree of neutralization is the degree of neutralization of the neutralization dissolution of the component iii in step A of the preparation method. Basically, the degree of neutralization of the component iii in step A is 100%, and is completely neutralized. In the case of Examples 2-10 where the basic neutralizing agent is ammonia, an excess of ammonia is added in consideration of the volatility, but the residual content in the final film is significantly less than 100%. It is estimated to be.
- the degree of neutralization is the degree of neutralization of the enteric polymer as a whole of the components ii and iii.
- a dispersion of an enteric methacrylic acid copolymer is used as the second component, and the basic neutralizing agent is added only in the neutralization process of the third component of step A in the preparation process, so the whole preparation liquid Apparent neutralization degree for enteric polymers of
- the degree of neutralization of the whole enteric polymer could be less than 50% by mixing the component iii with the aqueous dispersion. This can prevent harmful effects due to excess residual salt in the capsule shell.
- Example 1 Using methyl cellulose (MC) having a “viscosity value” of 100 mPa ⁇ s or more as the nonionic water-soluble cellulose compound, the above-mentioned capsule preparation liquid having the composition of Examples 1-1 to 1-7 shown in Table 3 III-1.
- the capsule was prepared by the following procedure. All the capsule preparation liquids were in the form of a cloudy (suspended) dispersion liquid at the temperature T5 when the molding pin was immersed. In addition, it was separately confirmed that the dispersion became a white turbidity or translucent dispersion even before the addition of titanium oxide.
- a hard capsule of size 2 was made by the method of
- the dissolution test for the first and second solutions was conducted according to the following.
- the dissolution rate after 2 hours after immersing each capsule in the first solution was less than 10% in each case, indicating poor solubility in an acidic solution.
- the dissolution rate after 30 minutes after immersing each capsule in the second solution was 70% or more for all capsules. Both capsules were shown to be readily soluble in neutral solutions.
- Example 1-3 Although the hard capsule of Example 1-3 has good dissolution characteristics during the dissolution test, the degree of neutralization with respect to the total enteric polymer is as high as 50% or more, and not only the solubility in the neutral test solution In particular, the solubility in pure water was high. In addition, yellowing of the film during storage was also a concern.
- Example 2 A capsule preparation liquid having the composition of Examples 2-1 to 2-10 shown in Table 4 using hydroxypropyl methylcellulose (HPMC) having a “viscosity value” of 100 mPa ⁇ s or more as the nonionic water-soluble cellulose compound Using the above III-1.
- HPMC hydroxypropyl methylcellulose
- the capsule was prepared by the following procedure. All of the capsule preparation liquids in Example 2 became a cloudy dispersion at a temperature of 55 ° C. when the molding pin was immersed.
- a hard capsule of size 2 was made by the method of
- the dissolution test for the first and second solutions was conducted according to the following.
- the dissolution rate of each of the capsules prepared from the capsule preparation solutions of Examples 2-1 to 2-3 and 2-5 to 2-10 after immersing in the first solution was less than 10% for 2 hours.
- the dissolution rate of the capsule prepared from the capsule preparation of Example 2-4 was 16.4%.
- the dissolution rate in the second liquid of the capsule prepared from the capsule preparation liquids of Examples 2-1 to 2-4, 2-6 and 2-10 was 70% or more within 30 minutes.
- the properties of Examples 2-1 to 2-4, 2-10 are desirable if it is desirable to disintegrate immediately after reaching the intestine.
- the characteristics of Examples 2-7 to 2-9 can be appropriately selected.
- Example 2-2 the temperature of the preparation liquid used in Example 2-2 is dropped onto a slide glass on a stage maintained at 55 ° C., and further enclosed with a cover glass preheated to 55 ° C., and then observed with an optical microscope
- the transmission image is shown in FIG.
- the whiteish parts in the figure are solid particles of HPMC partially dissolved.
- the dark area in the periphery is an aqueous solution based on an enteric polymer, which looks dark because it contains titanium oxide.
- FIG. 4 shows changes in storage elastic modulus G ′ (Pa) and loss elastic modulus G ′ ′ (Pa) when the prepared liquid used in Example 2-2 is cooled from temperature T1 to room temperature.
- the storage elastic modulus G '(Pa) exceeded the loss elastic modulus G' '(Pa) between 40 ° C and 35 ° C, which indicated that the preparation liquid was suitable for preparation of hard capsules by the cold gel method .
- Example 3 Using hydroxypropyl cellulose (HPC) having a “viscosity value” of 100 mPa ⁇ s or more as the nonionic water-soluble cellulose compound, the capsule preparation liquid of the composition of Example 3-1 shown in Table 5 is used. 1. The capsule was prepared by the following procedure. The capsule preparation liquid in Example 3 was a cloudy dispersion at a temperature of 55 ° C. when the molding pin was immersed. Furthermore, the above IV. A hard capsule of size 2 was made by the method of
- the dissolution test for the first and second solutions was conducted according to the following.
- the dissolution rate after 2 hours of immersion of the capsule prepared from the capsule preparation liquid of Example 3-1 in the first liquid was 1.4%.
- the dissolution rate in the second solution is It was 100% in 30 minutes.
- the capsules prepared from the preparation of Example 3-1 were shown to be readily soluble in neutral solutions.
- Example 4 and Comparative Example 4 Using hydroxypropyl methylcellulose (HPMC) having various “viscosity values” as the nonionic water-soluble cellulose compound, it is possible to use Examples 4-1 to 4-3 and Comparative Examples 4-1 to 4-4 shown in Table 6.
- Table 6 shows the composition of the capsule preparation, the preparation viscosity at T5 (Brookfield viscometer), the result of the dynamic viscoelasticity measurement at the time of temperature decrease, that is, the presence or absence of gelation around room temperature (G '> in rheometer measurement) In case of G ′ ′, it is judged as gelation), and shows the presence or absence of a sharp viscosity increase at about 30 to 50 ° C.
- the viscosity value of HPMC is 100 mPa ⁇ s or more (Examples 4-1, 4-2, 4-3)
- the viscosity of the preparation liquid is about 1,000 to 3,000 mPa ⁇ s, and 30 to It met the requirement of rapid viscosity increase at 50 ° C., ie gelation around room temperature.
- the “viscosity value” of HPMC is less than 100 mPa ⁇ s (comparative examples 4-1, 4-2, 4-3), the viscosity increase at about 30 to 50 ° C. is slow and is around room temperature. It was shown that no gelation was observed.
- Example 5 and Reference Example 1 In order to confirm that the i-th component, the ii-component and the neutralized iii-component are necessary for the preparation method of the preparation liquid of the embodiment 3-1 according to the present disclosure and the preparation liquid of the cold pin immersion method, The various components were removed, and the mass of the component was simply replaced with purified water to prepare various solutions, and the suitability as a capsule preparation liquid was confirmed.
- Table 7 shows the composition of the preparation (in any case, it does not contain titanium oxide), the results of dynamic viscoelasticity measurement at temperature drop with a rheometer, ie, the presence or absence of gelation around room temperature (G by rheometer measurement) If it is '>G', it is judged as gelation and it is judged as “o”.
- G ′ is very small, In fact, when it can not be solidified, it indicates the presence or absence of an abrupt increase in viscosity at about 30 to 50 ° C. and represented by “x”. Moreover, it was evaluated whether the film which became self-supporting by the cast method could be obtained as "stand-alone dry film formation.” This evaluation indicates whether a self-supporting film can be formed without any other support, and also whether it has adequate mechanical strength as an empty hard capsule film.
- the polymer solids concentration may be appropriately adjusted while maintaining the ratio between the polymer components.
- the thing which the film which became independent in Table 7 could be formed is shown by (circle). Even if the polymer solid content is slightly adjusted in the casting method, those which are too brittle or too soft when peeled off from the substrate to which the preparation liquid is applied, and which are difficult to peel as a self-supporting film are indicated by x.
- a capsule preparation liquid according to the present disclosure which includes all three components of HPMC, dispersion of Eudragit (L30D55) as component ii, and HP50 (neutralized with NaOH) as component iii (Example 5) (Example 5)
- Dynamic visco-elastic behavior at the time of temperature drop was compared for the solution of Reference Examples 1-1 to 1-8 in which any component is missing.
- Example 5 in which all the components are contained is substantially the same as the case where titanium oxide is removed in Example 2-2.
- the components to be excluded based on those obtained by removing titanium oxide from Example 2-2 were simply replaced with water of the same mass as Reference Examples 1-1 to 1-8.
- the i-th component, the ii-component and the neutralized iii-component were all present as a capsule preparation liquid for preparing the enteric hard capsule.
- the basic neutralizing agent used to neutralize the third component was effective in preventing aggregation of the dispersion.
- the preferred cold gelation properties were lost once the entire enteric polymer had been neutralized (and thus completely neutralized to not only the iii component but also the ii component).
- the polymer solid content concentration (%) was defined as the mass ratio of the solid content of the i-th, ii, iv, v component in the total mass of the solid content of the purified water as the solvent and the i, ii, iv, v component. .
- the degree of neutralization is the degree of neutralization of the basic neutralizing agent added to the L30D55 dispersion in the preparation step A ′ with respect to the mass of L30D55 solid content.
- the basic neutralizing agent here is added for the purpose of preventing immediate aggregation caused by the mixing of the i-th component and the ii-th component, and it is not for the purpose of micronization by dissolution of the ii-th component itself.
- the degree of neutralization can be sufficiently lowered to about 8%.
- Example 6-8 in place of the colloidal dispersion of L30D55, fine particles in which L10055 which was dried and powdered into a solid was neutralized and dissolved at a degree of neutralization of about 8% also in step A ′. Dispersion was used.
- Example 6 The capsule preparation liquid of the composition of Examples 6-1 to 6-10 shown in Table 8 using hydroxypropyl methylcellulose (HPMC) having a “viscosity value” of 100 mPa ⁇ s or more as a nonionic water-soluble cellulose compound Above III-2.
- HPMC hydroxypropyl methylcellulose
- the capsule preparation was prepared by the following procedure. All the capsule preparation solutions were in the form of a cloudy dispersion at temperature T5 when the molding pin was immersed. In addition, it was separately confirmed that the dispersion became a white suspension (suspension) or a translucent dispersion even before the addition of titanium oxide.
- the dissolution test for the first and second solutions was conducted according to the following.
- the dissolution rate after 2 hours after immersing each capsule in the first solution is less than 10% in all cases except for the case of 10.1% of Example 6-4 containing 10% by mass or more of the fifth component PVA. It showed poor solubility in acidic solution.
- the dissolution rate 45 minutes after immersing each capsule in the second solution was 90% or more for all capsules. Both capsules were shown to be readily soluble in neutral solutions.
- Example 6 and 7 show an optical microscopic image of the capsule preparation liquid in Example 6-2 and a scanning electron microscopic image of a cross section of the capsule coating, respectively.
- the capsule preparation liquid was confirmed to be a dispersion liquid in which fine solid particles of HPMC, which is the i-th component, were dispersed.
- the capsule capsule after drying has a sea-island structure including an island phase mainly composed of HPMC.
- the composition of each polymer component in the preparation liquid was substantially maintained in the capsule film.
- Example 7 and Reference Example 2 In order to confirm that it is necessary for the preparation liquid of the cold pin immersion method according to the present disclosure, it is necessary that the i-th component and the ii-component, the iv-component, and the basic neutralizing agent are all present.
- various solutions were prepared by removing any component and simply replacing the mass thereof with purified water, and the suitability as a capsule preparation liquid was confirmed.
- Table 9 shows the composition of the preparation (in any case, it does not contain titanium oxide), the results of dynamic viscoelasticity measurement at temperature drop with a rheometer, ie, presence or absence of gelation around room temperature, and about 30 to 50 ° C. Indicates the presence or absence of a sharp increase in viscosity. In addition, we also showed the possibility of “standing dry film formation”.
- a capsule preparation liquid according to the present disclosure (Example 7) according to the present disclosure, which includes, as the i-th component, Eudragit, a dispersion of L30D55 as the ii-component, a combination of two types as the iv-component, and a basic neutralizing agent.
- the dynamic visco-elastic behavior during temperature decrease was compared with the solutions of Reference Examples 2-1 to 2-16 lacking any of the components.
- Example 7 in which all the components are contained is the same as in Example 6-2 except that titanium oxide is removed.
- the components excluded from Example 6-2 except for titanium oxide were simply replaced with water of the same mass as Reference Examples 2-1 to 2-16.
- the dispersion in which the i-th component was partially dissolved (Reference Example 2-1) showed a viscosity increase at 30 to 50 ° C., but did not show gelation (G ′> G ′ ′) at around room temperature .
- the second component dispersion (Reference Example 2-2), the iv component dispersion (Reference Example 2-3), and the v-th component solution (Reference Example 2-4) are each almost completely liquid in all temperature ranges. Behavior was very small, and both G ′ and G ′ ′ were approximately less than 100 mPa ⁇ s from 55 ° C. to room temperature. That is, neither a suitable viscosity rise in the temperature lowering process nor a cold gelling ability at around room temperature was shown.
- the i-th component is HPMC and the ii-component is FS30D according to Reference Examples 2-12, 2-13, and 2-14
- the i-th component is MC or HPC and the ii-component is L30D55.
- the degree of neutralization is zero, it is possible to immediately aggregate by mixing the i-th component and the ii-th component (colloid dispersion liquid), and it is possible to confirm that cold gel performance is not exhibited when the degree of neutralization is 100%. The That is, it is necessary to appropriately adjust the degree of neutralization within the range of 2 to 20% in order to obtain the cold gel performance of the capsule preparation liquid.
- Example 8 Examples of enteric hard capsules containing the first to fourth components or all of the first to fifth components are shown in Table 10 as Examples 8-1 and 8-2, respectively.
- a capsule preparation liquid was prepared according to Preparation Example III-1 (Aspect 3-1 of Preparation Method), and molding was performed according to molding method IV.
- the total solid mass of the ith component (first component), the ii component (second component), the iii component (third component), the iv component (fourth component), and the v component (fifth component) Each mass% when (polymer solid content mass total) is 100 mass% was made into (alpha), (beta), (gamma), (sigma), (phi).
- the mass ratio of the basic neutralizing agent (NaOH) and the titanium oxide (light shielding agent) to the total mass of the solid content of the polymer was ⁇ (%) and ⁇ (%), respectively. Further, the mass ratio of the solid content of the components i to v in the total mass of the purified water as the solvent and the solid content of the components i to iii was taken as the polymer solid content concentration (%).
- Table 10 shows each specific composition.
- the degree of neutralization (relative to the component iii) is the degree of neutralization of the neutralization dissolution of the component iii in step A of the preparation method. Basically, the degree of neutralization of the component iii in step A is 100%, and is completely neutralized.
- Example 8-1 in which the basic neutralizing agent is ammonia, an excess of ammonia is added in consideration of the volatility, but the residual content in the final film is significantly less than 100%. It is estimated to be. In all cases, they had sufficient dissolution characteristics and mechanical strength as enteric hard capsules.
- the elastic modulus was confirmed to be in the range of 2 to 5 GPa at a relative humidity of 60%.
- the elongation rate is in the range of 3 to 10% even at 22% relative humidity conditions on the low humidity side, and has mechanical strength that does not show any problems such as large deformation or cracking in normal handling.
- the fourth component has an effect of increasing the elongation at break of the capsule coating and improving the crackability.
- PVA of the fifth component has an effect of improving the hardness and the elongation at break, that is, the cracking, of the capsule coating, particularly in the range where the relative humidity is lower than 50%.
- the plasticizer PEG 35000 or PG
- the elongation at break is increased and the crackability is improved, but the relative humidity is 60%.
- capsules that show a dissolution rate of about 10% two hours after immersion in solution 1 may show a drop in dissolution rate of about 1 to 2% due to the application of a band seal, which is more reliable. It was considered effective to apply a band seal when it is necessary to suppress elution.
- Example 9 In an enteric hard capsule (size 2) according to the present disclosure of Example 1-1, a capsule preparation filled with acetaminophen mixed powder was prepared and used as an inner capsule. A capsule preparation having a double capsule structure in which hypromellose capsule (Quali-V (registered trademark), size 00) and caffeine 100 mg and the inner capsule were filled was prepared. After conducting a dissolution test for 2 hours in the first solution, a dissolution test was conducted in the second solution. The time change of the dissolution rate of caffeine and acetaminophen is shown in FIG.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Pain & Pain Management (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
(1)耐酸性腸溶性ポリマーの代わりに、又は併用して、ジェランガムのような耐酸性を付与できるゲル化剤を使用し、ゲル化性、皮膜性能を改善しつつ、耐酸性を維持すること(特許文献7~10);
(2)水ベースの溶液の代わりに溶媒ベースの浸漬溶液を用いること(特許文献11 );
(3)難水溶性の耐酸性腸溶ポリマーを主成分として、従来の水溶性かつ皮膜形成能の高いゼラチンや水溶性セルロースなどのポリマーを部分的に使用すること(特許文献12,13);
(4)難水性の腸溶性ポリマーを含む水溶性誘導体を得るために、腸溶性ポリマーのほぼ全ての酸基(特にカルボキシル基)を塩化(salifying)する、あるいは、非塩化ポリマーを塩基性中和剤で少なくとも部分的に中和して水に溶解すること、あるいは、非塩化のエマルジョン分散液を利用すること(特許文献12~20); 及び
(5)射出成形など、ポリマーの可溶化を必要としない代替技術を用いること(特許文献21~25、非特許文献4)
等がなされている。
このような事情から、硬質カプセルの皮膜そのものが腸溶性であることが望まれる。
上記(1)の従来技術は、硬質カプセル皮膜の成型性は改善されるものの、耐酸性は不十分である。さらに、ゲル化剤を使用してポリマーをゲル化させる場合、特にゲル化助剤としてのカチオンを必要とする冷ゲル法において、ポリマーを含む水溶液のpH、又はカチオンと腸溶性ポリマーのイオン基との相互作用により、ポリマー水溶液もしくは分散液の安定性、ゲル化剤の冷ゲル化性能が損なわれるという問題がある。
粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物と腸溶性メタクリル酸コポリマーを含み、腸溶性セルロース化合物、
水不溶性(メタ)アクリル酸アルキルエステルコポリマー、及び、ポリビニルアルコール共重合体、可塑剤、及び界面活性剤よりなる群から選択される少なくとも一成分を含む皮膜からなる硬質カプセルが腸溶性特性を有することを見出した。さらに前記成分を含む腸溶性硬質カプセル調製液は、冷ゲル法によって硬質カプセルを調製できることを見出した。
項1.第1成分及び第2成分を含み、さらに第3成分、第4成分、及び第5成分の少なくとも一成分を含む皮膜からなる腸溶性硬質カプセルであって、
第1成分は、粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物であり、
第2成分は、腸溶性メタクリル酸コポリマーであり、
第3成分は、腸溶性セルロース化合物であり、
第4成分は、水不溶性(メタ)アクリル酸アルキルエステルコポリマーであり、及び、
第5成分は、ポリビニルアルコール、可塑剤、及び界面活性剤よりなる群から選択される少なくとも一種である、
腸溶性硬質カプセル。
項2.前記非イオン性水溶性セルロース化合物が、ヒドロキシプロピルメチルセルロース、メチルセルロース、及びヒドロキシプロピルセルロースからなる群から選択される少なくとも一種である、項1に記載の腸溶性硬質カプセル。
項3.前記腸溶性メタクリル酸コポリマーが、メタクリル酸とメタクリル酸メチル及びアクリル酸メチルとのコポリマー、又は、メタクリル酸とアクリル酸エチルとのコポリマーからなる群から選択される少なくとも一種である、項1又は2に記載の腸溶性硬質カプセル。
項4.前記腸溶性メタクリル酸コポリマーが、メタクリル酸40~60質量%とアクリル酸エチル60~40質量%とからなるコポリマーであることを特徴とする、請求項1~3のいずれか一項に記載の腸溶性硬質カプセル。
項5.前記腸溶性セルロース化合物が、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、及びセルロースアセテートフタレートからなる群から選択される少なくとも一種である、項1~4のいずれか一項に記載の腸溶性硬質カプセル。
項6.前記(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、項1~5のいずれか一項に記載の腸溶性硬質カプセル。
項7.前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とし、第1成分の割合をα質量%、第2成分の割合をβ質量%、第3成分の割合をγ質量%、第4成分の割合をσ%、及び第5成分の割合をφとした場合に、0.5≦(β+γ+σ)/(α+β+γ+σ+φ)≦0.9であり、かつ、0.4≦(β+γ)/(β+γ+σ)である、項1~6のいずれか一項に記載の腸溶性硬質カプセル。
項8.前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とし、第1成分の割合をα質量%、第2成分の割合をβ質量%、第3成分の割合をγ質量%、第4成分の割合をσ%、及び第5成分の割合をφとした場合に、0.05≦α/(α+β+γ+σ+φ)≦0.5である、項1~7のいずれか一項に記載の腸溶性硬質カプセル。
項9.前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とし、第2成分の割合をβ質量%、及び第3成分の割合をγ質量%とした場合に、0.1≦β/(β+γ)≦1である、項1~8のいずれか一項に記載の腸溶性硬質カプセル。
項10.前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とした場合の第1成分の割合をα質量%、第2成分の割合をβ質量%、第4成分の割合をσ%、及び第5成分の割合をφとした場合に、γ=0であり、かつ、0.3≦β/(α+β+γ+σ+φ)≦0.7である、項9に記載の腸溶性硬質カプセル。
項11.前記第2成分の少なくとも一部がその薬学的に又は食品添加物として許容される塩として含まれる、及び/又は第3成分の少なくとも一部がその薬学的に又は食品添加物として許容される塩として含まれる、項1~10のいずれか一項に記載の腸溶性硬質カプセル。
項12.前記皮膜に含まれる前記第2成分及び第3成分における塩を形成したカルボキシル基と塩を形成していないカルボキシル基のモル数の合計を100モル%とした場合、塩を形成したカルボキシル基の含有量が2~50モル%である、項11に記載の腸溶性硬質カプセル。
項13.前記皮膜の厚みが50~250μmである、項1~12のいずれか一項に記載の腸溶性硬質カプセル。
項14.前記皮膜の25℃、相対湿度60%における弾性率が1GPa~5GPaである、項13に記載の腸溶性硬質カプセル。
項15.前記皮膜の25℃、相対湿度22%における破断伸び率が2%~30%である、項13又は14に記載の腸溶性硬質カプセル。
項16.前記腸溶性硬質カプセルの皮膜が海島構造を含み、島相が実質的に第1成分からなる、項1~15のいずれか一項に記載の腸溶性硬質カプセル。
項17.前記島相の短径が0.1μm以上、かつ30μm未満である、項16に記載の腸溶性硬質カプセル。
項18.pH1.2を有する溶液を用いた溶出試験において、2時間後の前記腸溶性硬質カプセルの溶出率が、25%以下である、項1~17のいずれか一項に記載の腸溶性硬質カプセル。
項19.前記溶出試験における腸溶性硬質カプセルの溶出率が、10%以下である、項18に記載の腸溶性硬質カプセル。
項20.第i成分、第ii成分、薬学的又は食品添加物として許容される塩基性中和剤、及び溶媒を含み、さらに第iii成分、第iv成分及び第v成分の少なくとも一成分を含む腸溶性硬質カプセル調製液であって、
第i成分は、粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物であり、
第ii成分は、腸溶性メタクリル酸コポリマーであり、
第iii成分は、腸溶性セルロース化合物であり、
第iv成分は、水不溶性(メタ)アクリル酸アルキルエステルコポリマーであり、及び、
第v成分は、ポリビニルアルコール、可塑剤、及び界面活性剤よりなる群から選択される少なくとも一種である、
腸溶性硬質カプセル調製液。
項21.前記第i成分が、固体粒子として分散されている、項20に記載の腸溶性硬質カプセル調製液。
項22.前記、第ii成分の一部及び/又は第iii成分の一部が、前記塩基性中和剤によって部分中和されている、項20又は21に記載の腸溶性硬質カプセル調製液。
項23.前記部分中和の中和度が第ii及び第iii成分の完全中和に必要なモル数に対して、2~50%である、項22に記載の腸溶性硬質カプセル調製液。
項24.前記第ii成分が、コロイド粒子として分散されている、項20~23のいずれか一項に記載の腸溶性硬質カプセル調製液。
項25.前記非イオン性水溶性セルロース化合物が、ヒドロキシプロピルメチルセルロース、メチルセルロース、及びヒドロキシプロピルセルロースからなる群から選択される少なくとも一種である、項20~24のいずれか一項に記載の腸溶性硬質カプセル調製液。
項26.前記腸溶性セルロース化合物が、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、及びセルロースアセテートフタレートからなる群から選択される少なくとも一種である、項20~25のいずれか一項に記載の腸溶性硬質カプセル調製液。
項27.前記腸溶性メタクリル酸コポリマーが、メタクリル酸とメタクリル酸メチル及びアクリル酸メチルとのコポリマー、又は、メタクリル酸とアクリル酸エチルとのコポリマーからなる群から選択される少なくとも一種である、項20~26のいずれか一項に記載の腸溶性硬質カプセル調製液。
項28.前記腸溶性セルロース化合物の一部又は全部を、第iv成分である水不溶性(メタ)アクリル酸アルキルエステルコポリマーで置換したことを特徴とする、項20~27のいずれか一項に記載の腸溶性硬質カプセル調製液。
項29.前記水不溶性(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、項20~28のいずれか一項に記載の腸溶性硬質カプセル調製液。
項30.前記第iv成分が、コロイド粒子として分散されている、項28又は29のいずれか一項に記載の腸溶性硬質カプセル調製液。
項31.前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とし、第i成分の割合をα’質量%とし、第ii成分の割合をβ’質量%、第iii成分の割合をγ’質量%、第iv成分の割合をσ’質量%、及び第v成分の割合をφ’質量%とした場合に、0.5≦(β’+γ’+σ’)/(α’+β’+γ’+σ’+φ’)≦0.9であり、かつ、0.4≦(β’+γ’)/(β’+γ’+σ’)である、項20~30項のいずれか一項に記載の腸溶性硬質カプセル調製液。
項32.前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分及び、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とし、第i成分の割合をα’質量%、第ii成分の割合をβ’質量%、第iii成分の割合をγ’質量%、第iv成分の割合をσ’質量%、及び第v成分の割合をφ’質量%とした場合に、0.05≦α’/(α’+β’+γ’+σ’+φ’)≦0.5である、項20~31のいずれか一項に記載の腸溶性硬質カプセル調製液。
項33.前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とした場合の、第ii成分の割合をβ’質量%、第iii成分の割合をγ’質量%とした場合に、0.1≦β’/(β’+γ’)≦1である、項20~32のいずれか一項に記載の腸溶性硬質カプセル調製液。
項34.前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とした場合の第i成分の割合をα’質量%、第ii成分の割合をβ’質量%、第iv成分の割合をσ’質量%、及び第v成分の割合をφ’質量%とした場合に、γ’=0であり、かつ、0.3≦β’/(α’+β’+γ’+σ’+φ’)≦0.7である項33に記載の腸溶性硬質カプセル調製液。
項35.前記塩基性中和剤による第ii成分の中和度が、2~20%である項34に記載の腸溶性硬質カプセル調製液。
項36.前記塩基性中和剤が、水酸化ナトリウム、水酸化カリウム、及び水酸化カルシウムからなる群から選択される少なくとも一種である、項20~35のいずれか一項に記載の腸溶性硬質カプセル調製液。
項37.前記塩基性中和剤が、アンモニア及び炭酸アンモニウムからなる群から選択される少なくとも一種である、項20~35のいずれか一項に記載の腸溶性硬質カプセル調製液。
項38.腸溶性硬質カプセル調製液を100質量%としたときに、前記第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の合計量が10~30質量%である、項31~37のいずれか一項に記載の腸溶性硬質カプセル調製液。
項39.粘度が、100~10,000mPa・sである、項20~38のいずれか一項に記載の腸溶性硬質カプセル調製液。
項40.溶媒中に薬学的又は食品添加物として許容される塩基性中和剤が存在する条件下で、第i成分と第ii成分が混合される腸溶性硬質カプセル調製液の調製方法であって、第i成分は、粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物であり、第ii成分は、腸溶性メタクリル酸コポリマーである、調製方法。項41.
前記非イオン性水溶性セルロース化合物が、ヒドロキシプロピルメチルセルロース、メチルセルロース、及びヒドロキシプロピルセルロースからなる群から選択される少なくとも一種である、項40に記載の腸溶性硬質カプセル調製液の調製方法。
項42.前記腸溶性メタクリル酸コポリマーが、メタクリル酸とメタクリル酸メチル及びアクリル酸メチルとのコポリマー、又は、メタクリル酸とアクリル酸エチルとのコポリマーからなる群から選択される少なくとも一種である、項40又は41のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項43.前記塩基性中和剤が、水酸化ナトリウム、水酸化カリウム、水酸化カルシウムからなる群から選択される少なくとも一種である、項40~42のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項44.前記塩基性中和剤が、アンモニア及び炭酸アンモニウムからなる群から選択される少なくとも一種である、項40~42のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項45.工程A:第iii成分の中和液を準備する工程、
工程B:前記第iii成分を含む中和液に第i成分を加え、第i成分の部分溶解液を準備する工程、及び
工程C:前記第ii成分の分散液を、中和液もしくは部分溶解液と混合する工程、
を順不同で含む、
腸溶性硬質カプセル調製液の調製方法であって、第iii成分は、腸溶性セルロース化合物である、項40~44のいずれか一項に記載の調製方法。
項46.前記腸溶性セルロース化合物が、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、及びセルロースアセテートフタレートからなる群から選択される少なくとも一種である、項45に記載の腸溶性硬質カプセル調製液の調製方法。
項47.前記工程Aが、前記第iii成分を薬学的に又は食品添加物として許容される塩基性中和剤により、少なくとも部分的に中和して溶媒に溶解させる中和液を調製する工程であり、その中和度が50%以上又は、完全に中和されている、項45又は46に記載の腸溶性硬質カプセル調製液の調製方法。
項48.前記工程Bが、前記第iii成分を含む中和液、又は前記第iii成分の中和液と前記第ii成分の分散液の混合液に、前記第i成分を部分溶解させた部分溶解液を調製する工程であり、部分溶解液を調製する工程が、第i成分を、第i成分の曇点T0以上の第1の温度T1で、第iii成分を含む中和液、又は第iii成分の中和液と第ii成分の分散液の混合液に添加し、前記曇点よりも低い第2の温度T2で第i成分を部分溶解させた分散液を調製する工程である、項45~47のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項49.前記工程A、B、もしくはCで準備された溶液と第iv成分である水不溶性(メタ)アクリル酸エステルコポリマーを混合する工程Dを含む項45~48のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項50.前記水不溶性(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、項49に記載の腸溶性硬質カプセル調製液の調製方法。
項51.前記工程B、C又はDで得られた溶液を、前記第i成分の曇点よりも低い第3の温度T3に保持する工程Eをさらに含む、項45~50のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項52.工程A’:第ii成分の部分中和液を準備する工程、
工程B’:第i成分の部分溶解液を準備する工程、及び
工程C’:第iv成分の分散液を、工程AもしくはBで準備された溶液と混合する工程、
を順不同で含む、
項40記載の腸溶性硬質カプセル調製液の調製方法であって、
第iv成分は、水不溶性(メタ)アクリル酸アルキルエステルコポリマーである、
調製方法。
項53.前記水不溶性(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、項52に記載の腸溶性硬質カプセル調製液の調製方法。
項54.前記工程A’が、前記第ii成分を薬学的に又は食品添加物として許容される塩基性中和剤により、少なくとも部分的に中和して溶媒に溶解させる中和液を調製する工程であり、その中和度が2~20%である、項52又は53に記載の腸溶性硬質カプセル調製液の調製方法。
項55.前記工程B’が、前記第ii成分を含む中和液に、前記第i成分を部分溶解させた部分溶解液を調製する工程であり、
前記部分溶解液を調製する工程が、第i成分を、第i成分の曇点T0以上の第1の温度T1で、第ii成分を含む中和液、又は第ii成分の中和液と前記第iv成分の分散液の混合液に添加し、前記曇点よりも低い第2の温度T2で第i成分を部分溶解させた分散液を調製する工程である、項52~54のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項56.前記工程B’又はC’で得られた溶液を、前記第i成分の曇点よりも低い第3の温度T3に保持する工程E’をさらに含む、項55に記載の腸溶性硬質カプセル調製液の調製方法。
項57.前記第3の温度の範囲T3が、40℃~60℃である、項51又は56に記載の腸溶性硬質カプセル調製液の調製方法。
項58.前記第1の温度T1の範囲が、60℃~90℃である、項48~51及び55~57のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項59.前記第2の温度T2の範囲が、30℃~60℃である、項48~51及び55~57のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項60.前記腸溶性硬質カプセル調製液の粘度が、100~10,000mPa・sである、項40~59のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
項61.下記工程を含む、腸溶性硬質カプセルの調製方法:
項20~39のいずれか一項に記載の腸溶性硬質カプセル調製液、又は項40~60のいずれか一項に記載の調製方法により得られた腸溶性硬質カプセル調製液の中に、前記腸溶性硬質カプセル調製液の温度よりも低い表面温度を有するモールドピンを浸漬する第1工程;及び
前記腸溶性硬質カプセル調製液からモールドピンを引き上げて、モールドピンに付着した腸溶性硬質カプセル調製液を乾燥させる第2工程。
項62.前記腸溶性硬質カプセル調製液の温度が、40~60℃である、項61に記載の腸溶性硬質カプセルの調製方法。
項63.前記調製液に浸漬する前のモールドピンの表面温度が、5~40℃である、項62又は61又は62に記載の腸溶性硬質カプセルの調製方法。
項64.モールドピンに付着した腸溶性硬質カプセル調製液を乾燥する温度が、40℃未満である、項61~63のいずれか一項に記載の腸溶性硬質カプセルの調製方法。
項65.項1~19のいずれか一項に記載の腸溶性硬質カプセルに対し、腸溶性メタクリル酸コポリマー及び腸溶性セルロース化合物よりなる群から選択される少なくとも一種の腸溶性ポリマーの少なくとも部分中和された希釈水溶液、あるいは、水/エタノール又は水/イソプロパノール溶剤に溶解した液からなるシール液によってシールされたことを特徴とする腸溶性硬質カプセル製剤。
項66.酸性条件で溶解可能な硬質カプセルの内部に項1~19のいずれか一項に記載の腸溶性硬質カプセルを内包することを特徴とする硬質カプセル製剤。
はじめに、本明細書、及び特許請求の範囲等で使用される用語及び材料について説明する。本開示に関する用語及び材料は、特に記載がない限り、本項の説明に従う。
また、本開示では、空の硬質カプセルを単に硬質カプセルもしくはカプセルと呼び、内容物を充填したものを「硬質カプセル製剤」と呼ぶ。
すなわち、「腸溶性」とは、少なくとも下記(i)の条件を満たす特性をいう。
ここで、第2液中での内容物の溶出率を測定する時間に制限はない。例えば、腸に到達後、比較的速やかに溶出することが求められる場合には、第2液に被験対象を浸漬してから、30分後の溶出率が、50%、好ましくは70%以上、さらに好ましくは80%以上である。また、例えば、第2液に被験対象を浸漬してから、45分後の溶出率が、75%以上、好ましくは80%より好ましくは90%以上である。さらに、例えば、第2液に被験対象を浸漬してから、1時間後の溶出率が、75%以上、好ましくは80%、より好ましくは90%以上である。
溶出試験に使用する内容物は、それ自身が試験溶液中で速やかに溶解される内容物であって、公知の方法によって定量できるものである限り制限されない。例えば、アセトアミノフェンを挙げることができる。


また、本開示に係るヒドロキシプロピルメチルセルロースには、日本国で食品添加剤としての使用が認められている下記分子量を有するヒプロメロースが含まれる。
<分子量>
非置換構造単位:162.14
置換構造単位:約180(置換度1.19)、約210(置換度2.37)
重合体:約13,000(n=約70)~約200,000(n=約1000)。

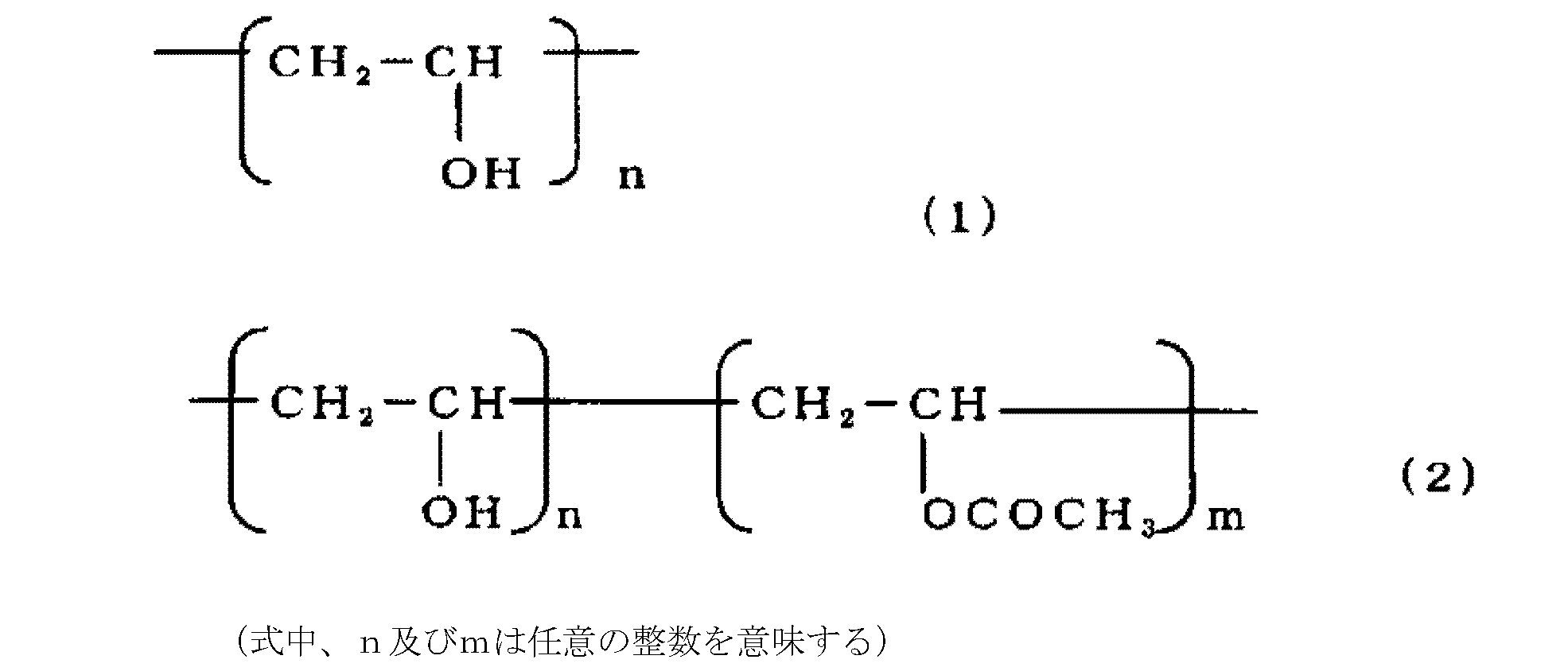
金属封鎖剤としては、エチレンジアミン四酢酸、酢酸、ホウ酸、クエン酸、グルコン酸、乳酸、リン酸、酒石酸、又はこれらの塩、メタホスフェート、ジヒドロキシエチルグリシン、レシチン、β-シクロデキストリン、又はこれらの組み合わせを挙げることができる。
本開示に係る第1の態様は、腸溶性硬質カプセルに関する。
デシケーターに、炭酸カリウム飽和塩を入れて恒湿状態とした雰囲気中に試料(硬質カプセル、又はフィルム)を入れ密閉し、25℃で1週間調湿する。なお、調湿には、以下の飽和塩(水溶液)を用いる。すなわち、酢酸カリウム飽和塩、炭酸カリウム飽和塩、硝酸アンモニウム飽和塩の存在下では、それぞれ、相対湿度約22%、43%、60%の雰囲気を作成することができる。調湿後の試料の質量(湿質量)を測定した後、次いで当該試料を105℃で2時間加熱乾燥し、再度試料の質量(乾燥質量)を測定する。乾燥前の質量(湿質量)と乾燥後の質量(乾燥質量)の差から、下式にしたがって、105℃で2時間加熱乾燥することによって減少する水分量の割合(含水率)を算出し、これを含有水分量(質量%)とする。

本開示に係る第2の態様は、上記2.に記載の腸溶性硬質カプセルを調製するための調製液に関する。本開示に係る硬質腸溶性カプセルは、本態様調製液を乾燥して溶媒を除去して得られる皮膜からなる。


工程A:第iii成分の中和液を準備する工程、工程B:第iii成分を含む中和液に第i成分を加え、第i成分の部分溶解液を準備する工程、及び工程C:第ii成分の分散液を、工程Aもしくは工程Bで準備された溶液と混合する工程、を含む、
第i成分、第ii成分、及び第iii成分を含む皮膜成分及び溶媒を含む腸溶性硬質カプセル調製液の調製方法に関する。
上記第3-1の態様における第iii成分の中和液、曇点T0、温度T1、温度T2及び温度T3の説明は、ここに援用される。
工程a1は、第iii成分の中和液を準備する工程であり、工程a1は、上記第3の態様における工程Aに準ずる。
また、本態様は、工程c1の後に、工程c1で得られた溶液に、水不溶性(メタ)アルキルエステルコポリマーを工程d1、さらには、工程b1、c1、又はd1の後で溶液を第i成分の曇点よりも低い第3の温度T3に保持する工程e1をさらに含んでいてもよい。
工程B’:第i成分の部分溶解液を準備する工程、及び
工程C’:第iv成分の分散液を、工程A’もしくはB’で準備された溶液と混合する工程、
を含む、第i成分、第ii成分、及び第iv成分を含む皮膜成分及び溶媒を含む腸溶性硬質カプセル調製液の調製方法に関する。本カプセル調製液の調製方法は、特に、γ’=0、すなわち、腸溶性ポリマーが、腸溶性メタクリル酸コポリマーのみである場合に適している。
その後、工程B’において、塩基性中和剤と第ii成分を含む溶液に、第i成分を添加して部分溶解させた部分溶解液を調製する。第i成分を、第i成分の曇点T0以上の第1の温度T1で、第ii成分を含む中和液、又は第ii成分の中和液と第iv成分の分散液の混合液に添加し、前記曇点よりも低い第2の温度T2で第i成分を部分溶解させた分散液を調製する工程である、
本開示における第4の態様は、腸溶性硬質カプセルの調製方法に関する。本開示によれば、他の硬質カプセルを調製するカプセル調製機を使用して、腸溶性硬質カプセルを調製することができる。本開示に係る腸溶性硬質カプセルは、浸漬法、中でも「コールドピン浸漬法」によって形成されることを特徴とする。「コールドピン浸漬法」は、浸漬時の成型ピンの表面温度が、カプセル調製液の温度よりも低いことを特徴とする。
(1)腸溶性硬質カプセル調製液に、モールドピンを浸漬する工程(浸漬工程)、
(2)腸溶性硬質カプセル調製液調製液(浸漬液)からモールドピンを引き上げて、モールドピンの外表面に付着した腸溶性硬質カプセル調製液を乾燥する工程(乾燥工程)、
(3)乾燥したカプセルフィルム(皮膜)をカプセル成型用ピンから脱離する工程(脱離工程)。
乾燥工程(2)は、特に制限されるものではないが、室温(20~30℃)で行うことができる。通常、室温の空気を送風することによって行なわれる。
本開示に係る腸溶性硬質カプセルには、一般食品、保健機能食品(機能性表示食品、栄養機能食品、特定保健用食品)、医薬部外品、医薬品等の充填物が充填され得る。充填物として例えば、植物(単細胞緑藻類を含む)に由来する成分(生の植物、一部乾燥された微生物、又は完全乾燥された植物、植物加工品、植物抽出物等)、微生物(細菌、酵母、ミドリムシ等)又は前記微生物に由来する成分(生の微生物、一部乾燥された微生物、又は完全乾燥された微生物、微生物加工品、微生物抽出物等)、滋養強壮保健剤、解熱鎮痛消炎剤、向精神剤、抗不安剤、抗うつ剤、催眠鎮静剤、鎮痙剤、中枢神経作用剤、脳代謝改善剤、脳循環改善剤、抗てんかん剤、交感神経興奮剤、胃腸剤、制酸剤、抗潰瘍剤、鎮咳去痰剤、鎮吐剤、呼吸促進剤、気管支拡張剤、抗アレルギー剤、歯科口腔用剤、抗ヒスタミン剤、強心剤、不整脈用剤、利尿剤、血圧降下剤、血管収縮剤、冠血管拡張剤、末梢血管拡張剤、高脂血症用剤、利胆剤、抗生物質、化学療法剤、糖尿病用剤、骨粗しょう症用剤、抗リウマチ剤、骨格筋弛緩剤、鎮けい剤、ホルモン剤、アルカロイド系麻薬、サルファ剤、痛風治療剤、血液凝固阻止剤、抗悪性腫瘍剤等の有効成分、又は前記有効成分を含む組成物を挙げることができる。なお、これらの充填物は、特に制限されず公知のものを広く挙げることができる。これらの成分は単独又は他の成分との合剤として使用することができる。充填物は、固形、粉末、顆粒、粉砕物、液体、ジェル等のいずれの形態であってもよい。また、これらの成分は、投与対象者の状態、年齢等に応じて適宜、定められた公知の適量が充填される。
本開示に係る腸溶性硬質カプセルを用いた新規な応用例として、酸性条件で溶解可能な硬質カプセルの内部に本開示に係る腸溶性硬質カプセルを内包することを特徴とする硬質カプセル製剤が挙げられる。酸性条件で溶解可能な硬質カプセルとしては、ゼラチンカプセル及びヒプロメロースカプセル、あるいは、プルランカプセルが挙げられるが、これらに限定されるものではない。特に、ヒプロメロース硬質カプセルでは、水溶性セルロースの表示粘度(粘度グレード)値として、3~15mPa・sのものが用いられている(特開平08-208458号公報、2001-506692号公報、特開2010-270039号公報、特開2011-500871号公報)。これらにおいては、皮膜中のほぼ100%(ゲル化剤、ゲル化助剤、遮光剤、着色料等、0~5質量%程度及び、0~10質量%程度の残留水分を含む場合がある)が水溶性セルロース、特にHPMCである。本開示に係る腸溶性硬質カプセルにあらかじめ有効成分Bを充填しておき、酸性条件で溶解可能な硬質カプセルの内部に、薬効成分A及び該充填済腸溶性硬質カプセルを、充填する。このような二重カプセル製剤は、胃において有効成分Aを放出させ、腸に達してから薬効成分Bを放出させるような、複数部位に選択的かつ異なる薬効成分の送達を可能にする。有効成分A及び有効成分Bは、上記5.に記載の有効成分を挙げることができる。
実施例、参考例、比較例に用いる材料は下記の通りである。
1.非イオン性水溶性セルロース化合物
メチルセルロース(MC)、ヒドロキシプロピルメチルセルロース(HPMC)は信越化学工業(株)のMETOLOSE(登録商標)シリーズもしくはTC-5(登録商標)シリーズを使用し、ヒドロキシプロピルセルロース(HPC)は日本曹達(株)のNISSO HPCシリーズを使用した。具体的な製品名と置換度タイプ、「粘度値」、(表示粘度もしくは粘度グレード)は表2の通りである。

(1)ヒドロキシプロピルセルロースフタレート(HPMCP)
信越化学工業(株) HPMCP(登録商標)シリーズのHP50グレードを使用した(以下、「HP50」と表記する)。
これら非塩化腸溶性セルロース化合物の塩基性中和剤による中和は、メーカーにより推奨されたNaOH当量範囲のその中心値を用いることで、ほぼ完全中和状態とした。完全中和の塩基性中和剤は、これら当量に対し、100±1%未満の誤差範囲内となるようにした。その際の溶液のpHは、5~7程度であった。
具体的には、HP50に対しては、0.065g/g、HMPCAS-MFもしくはHMPCAS-MGに対しては、0.048g/g、L30D55に対しては、0.215g/g、FS30Dに対しては、0.0404g/gとなる。特にHP50は、比較的粒径が大きく皮膜がざらついてしまうので、可能な限り完全中和して用いた。他方、HPMCAS-MFは、比較的粒径が小さいので、中和度が50%以上、より好ましくは80%以上であれば、ほとんど透明な溶解液が得られた。
また、同様にアンモニア対する中和の当量は、HP50に対しては、0.0274g/g、HMPCAS-MFもしくはHMPCAS-MGに対しては、0.0202g/g、L30D55に対しては、0.0914g/g、FS30Dに対しては、0.0172g/gとなる。アンモニアの場合は、調製液及び成果プセル調製過程での揮発があるので、当量より過剰に加えた。
Evonik Industries AG社、EUDRAGIT(登録商標)シリーズのL30D55及びFS30Dを使用した。いずれも固形分含有量30質量%の水分散液である。また、L30D55を乾燥微粉末化したL10055については、精製水中に分散させて撹拌したのち、NaOH(10%水溶液)を所定の中和度となるように添加した。これにより、L30D55のコロイド粒子よりはやや粗いものの、微粒子化された水分散液が得られた。
Evonik Industries AG社、Eudragit(登録商標)シリーズのNE30Dを使用した。固形分含有量30質量%の水分散液として供される。
日本合成化学工業株式会社のゴーセノール(登録商標)シリーズEG48Pを使用した。けん化度は、86.5~89.0%であり、推定重合度は2500である。PEG35000(ポリエチレングリコール)は、シグマアルドリッチ社から購入した。PG(プロピレングリコール)は和光純薬社から購入した。
水酸化ナトリウム(粒状 試薬特級)及びアンモニア水(28% 試薬特級)は和光純薬工業株式会社から購入した。酸化チタン(タイペークA-100)は石原産業株式会社から購入した。
1.カプセルの溶出試験
本開示においては、原則、第17改正日本薬局方における溶出試験を適用した。但し、日本薬局方は、空の硬質カプセル自体の溶解性を規定しているわけではないので、本開示では、速溶性のアセトアミノフェンの溶出を評価することによって、カプセル自体の溶解性(溶出特性)を評価した。1カプセルあたり、アセトアミノフェン40 mg、乳糖140 mg、デンプングリコール酸ナトリウム20 mg(以下、「アセトアミノフェン混合末」という)を充填し、得られた腸溶性硬カプセル製剤を日本薬局方に定められた溶出試験法(第17局方、6.10-1.2パドル法(パドル回転数50回転/分)、及び、同図6.10-2aに対応するシンカー使用)に従い試験し、アセトアミノフェンの溶出率の時間変化を測定した。溶出試験にはDistek社製バス型溶出試験器Model 2100を用いた。同容量のアセトアミノフェンを別途、全量、溶出試験器バス内の溶液に溶解させたときの244 nmにおける吸光度を100 %とし、カプセルからのアセトアミノフェンの溶出に伴って上昇する溶出試験器バス内の溶液の244 nmにおける吸光度から溶出率を求めた。n数に関しては、n=1~6とした。なお、ここで第1液及び第2液として、下記の水溶液が使用した。いずれもバス内の溶液の温度は37℃とした。
第2液:リン酸二水素カリウム3.40g及び無水リン酸水素二ナトリウム3.55gを水に溶かし、1000 mLとしたリン酸塩緩衝液1容量に水1容量を加えて調製した(pHは、約6.8、以下中性溶液と称することがある)。
カプセル調製液の動的粘弾性はAntonPaar社製、レオメーター(MCR102)を用いて測定した。測定には二重円筒管測定治具(型番CC27/T200/SS)と温度制御システムC-PTD200を使用した。液量は約19 mLとした。また、測定中の水分蒸発を防ぐため、円筒管中のサンプル液の最表面に1 mL程度の綿実油を垂らした。温度依存性は、60℃から20℃まで、1℃/分で低下させ、同時にひずみの振り角を1から0.1%まで線形に低下させながら測定した。角周波数ω(rad/sec)は、2π/秒である。動的粘弾性として、貯蔵弾性率G’(Pa)、損失弾性率G’’(Pa)、複素粘度|η*|=|G*|/ω=√(G’2+G’’2)/ω(Pa・s)、及び、粘度η’’=G’’/ω(Pa・s)の値を測定した。
カプセル調製液(55℃)の粘度は、ブルックフィールド粘度計(TVB-10M(東機産業))を使用して測定した。測定にはM3ロータ(測定範囲 0~10,000 mPa・s)を使用した。ロータ回転数は、12 r.p.m.、1Lビーカーでカプセル調製液を調製(液量は600 ml)したのち、該ビーカーにロータを入れて測定時間50秒で測定した。
皮膜構造の観察には、走査型電子顕微鏡(SEM)、及び顕微ラマンを使用した。
(1)SEM
走査型電子顕微鏡は、Carl Zeiss社製Ultra55を使用した。
カプセル被膜の断面を観察するため、調製したカプセル皮膜を輪切りにした小片に切り出し、エポキシ樹脂に包埋後、ミクロトームで薄切し観察用の切片(およそ300~400μm四方で2~3μm厚み)を作製した。切片にPtPdで蒸着処理した。電子線は、加速電圧3kVで照射し、切片をスキャンした。
顕微ラマン装置にはThermo Fisher Scientific製Nicolet Almega XRを使用した。励起波長は、532nm、分解能は、約10/cm(10カイザー)、照射径は、1μmφ(100倍対物レンズ、25μmピンホール:平面方向 1μmφ × 深さ方向(=切片厚み) 2μmの円柱状内部の情報が得られる。)、励起出力は、100%(10mW以下@試料位置)、露光時間×積算回数は10sec×2回とした。
輪切りにしたカプセル小片をエポキシ樹脂に包埋後、ミクロトームで薄切し、厚み2μmの切片を作成した。切片を金属板上にのせ、観察した。
調製液の観察は、ステージの温度調節機能を有する光学顕微鏡(オリンパス社製BX53) を使用して行った。接眼レンズは10倍、対物レンズは10倍のものを使用し、
透過観察した。55℃の調製液をやはり55℃のステージ上で予熱したスライドガラス上に滴下し、さらにその上をやはり55℃に予熱したカバーガラスで覆った。
カプセル皮膜中の塩(ナトリウム)は以下の手順で乾式灰化処理後、原子吸光光度法(AAS)で定量した。試料を白金坩堝に精秤し、濃硫酸を添加後650℃の電気炉で有機物がなくなるまで加熱した。残った灰分を希塩酸に溶解し、適宜希釈して原子吸光度計(VARIAN社製SpectrAA-220)で定量した。
<乾燥減量法によるカプセル皮膜中の含水率の測定方法>
デシケーターに、炭酸カリウム飽和水溶液を入れて恒湿状態とした雰囲気中に試料(硬質カプセル、又はフィルム)を入れ密閉し、25℃で1週間調湿した。なお、調湿には、以下の飽和塩(水溶液)を用いた。すなわち、酢酸カリウム飽和塩、炭酸カリウム飽和塩、硝酸アンモニウム飽和塩の存在下で、それぞれ、相対湿度約22%、43%、60%の雰囲気を作成した。調湿後の試料の質量(湿質量)を測定した後、次いで当該試料を105℃で2時間加熱乾燥し、再度試料の質量(乾燥質量)を測定した。乾燥前の質量(湿質量)と乾燥後の質量(乾燥質量)の差から、下式にしたがって、105℃で2時間加熱乾燥することによって減少する水分量の割合(含水率)を算出し、含有水分量(質量%)とした。

硬質カプセルの皮膜の機械強度を評価する場合、被験皮膜の厚みをそろえて比較することが重要である。このため、硬質カプセルの各成分組成に依存する皮膜の機械強度は、ディッピング法によって成形された硬質カプセルのかわりに、硬質カプセル調製液の各成分組成と同一成分組成である調製液を用いて、キャスト法によりフィルムを作製し、当該キャストフィルムを用いて評価した。当該フィルムは、厚みの均一性、評価の再現性に優れており、かつカプセル皮膜としての機械強度をよく反映するものである。
100μmの均一な膜厚のフィルムを得るため、ギャップが0.4 mm~1.5 mmのアプリケーターを適宜使い分けた。
以下手順にしたがって、カプセル調製液を調製した。操作はすべて溶液を撹枠しながら行った。以下においては、第i~v成分の固形分をポリマー固形分と称する。また、全溶液質量は、溶媒である精製水に加え、ポリマー固形分、塩基性中和剤、その他の固形分(可塑剤、遮光剤など)合計質量となる。ポリマー固形分濃度とは、前記ポリマー固形分合計質量の全溶液質量に対する比率(質量%)をいう。
a.後に加えるメタクリル酸コポリマーの水分散液(固形分濃度30質量%)、及び遮光剤である酸化チタン(濃度22質量%)の分散液の水分量を考慮し、最終的に、ポリマー固形分濃度が、所定濃度(20%程度)となるような量の室温の精製水を用意した。
b.室温にて、精製水に腸溶性セルロース化合物を投入し、だま、ができないように均一に分散させた。その後塩基性中和剤を投入し、腸溶性セルロースを溶解させた。塩基性中和剤は、特に断らない限り、以下の例においては、腸溶性セルロース化合物をちょうど完全中和するのに必要な分量(当量)を用いた。
c.この溶液を83℃にまで昇温させたのち、非イオン性水溶性セルロース化合物を投入し、だま、ができないように均一に分散させ懸濁液を調製した。
d.非イオン性水溶性セルロース化合物の分散液の温度を下げ、溶解温度(曇点)以下の温度T2まで降温し、非イオン性水溶性セルロース化合物を部分溶解させ分散液を調製した。部分溶解温度T2 は30~55℃の間で適宜調整した。
e.d.で調製された分散液を調製液温度T3(MCの場合は30~50℃、HPMCの場合は45~60℃、HPCの場合は30~40℃)で保持した。結果、ブルックフィールド粘度計での粘度が、ほぼ1,000~3,000 mPa・sの範囲となった。なお、最終的なポリマー固形分濃度は、粘度がこの範囲になるよう、温純水の追加、蒸発による微調整を行った。
f.メタクリル酸コポリマーの分散液は、b.の中和後、もしくはe.の非イオン性水溶性セルロース化合物の部分溶解液完成後のいずれかの段階で加えた。さらに、酸化チタンを入れる場合は、あらかじめ水分散液を作成したうえ、c.の操作の前に投入した。なお、上記すべての工程で、プロペラ撹拌翼を用いて、100~1,000 rpmで撹拌を行っている。
a.後に加えるメタクリル酸コポリマーの水分散液(固形分濃度30質量%)、(メタ)アクリル酸アルキルエステルコポリマー分散液(固形分濃度30質量%)及び遮光剤である酸化チタンの分散液(濃度22質量%)の水分量を考慮し、ポリマー固形分濃度が、所定濃度(20%程度)となるような量の室温の精製水を用意した。
b.室温にて、メタクリル酸コポリマー分散液を所定量上記精製水に投入し、その後塩基性中和剤として水酸化ナトリウム(NaOH)を投入し、部分中和液を調製した。NaOHは、特に断らない限り、以下の例においては、メタクリル酸コポリマーのカルボキシル基の約8%を部分中和するのに相当する分量を用いた。
c.この部分中和溶液を83℃にまで昇温させたのち、酸化チタン分散液を投入しスリーワンモターで十分撹拌した後、非イオン性水溶性セルロース化合物を投入し、だま、ができないように均一に分散させ懸濁液を調製し、脱泡した。その後、さらにPVA又は可塑剤を投入し溶解させた。
d.NaOHの存在下で、非イオン性水溶性セルロース化合物、メタクリル酸コポリマーが混合されたの分散液(さらに、酸化チタンとPVAを含んでいる溶液)の温度を下げ、非イオン性水溶性ポリマーの溶解温度(曇点)以下の温度T2まで降温し、非イオン性水溶性セルロース化合物を部分溶解させた分散溶液を調製した。T2は、30~55℃の間で適宜調整した。e.d.で調製された分散液を調製液温度T3(MCの場合は、35~40℃、HPMCの場合は、30~65℃)で保持しながら、NE30D分散液を投入した。結果、ブルックフィールド粘度計での粘度が、ほぼ1,000~3,000 mPa・sの範囲となった。なお、最終的な全固形分濃度は、粘度がこの範囲になるよう、温純水の追加、蒸発による微調整を行った。また、上記すべての工程で、プロペラ翼を用いて、100~1,000 rpmで撹拌を行った。
上記III.で調製されたカプセル調製液を用いて、コールドピン浸漬法により硬質カプセルを調製した。保持温度T5は、ほぼT3と同じとして、ほぼ一定温度に保ったカプセル調製液中に、室温(25℃程度)に放置したモールドピン(サイズ2号)を数秒間浸漬させたのち、大気中に引き上げた。カプセル調製液が付着した成型ピンを上下反転させ、室内雰囲気温度で2~10時間以上乾燥させた。筒状のカプセル側面の膜厚は、約100μmとなるようモールドピンの浸漬時間、引き上げ速度などを適宜調整した。その後、モールドピンから、カプセル部分を引き抜き、筒状部分の長さが所定の長さとなるようにカットした。以上の操作を、キャプ及びボディそれぞれで行った。
V-1.調製例(調製方法の態様3-1)
以下の実施例1~5、比較例においては、調製例III-1(調製方法の態様3-1)にしたがってカプセル調製液を調製し、成型方法IVによって成型を行った。第i成分(第1成分)、第ii成分(第2成分)、及び第iii成分(第3成分)の固形分質量合計(ポリマー固形分質量合計)を100質量%としたときのそれぞれの質量%を、α、β、γとした。塩基性中和剤(NaOH)、酸化チタン(遮光剤)の、上記ポリマー固形分質量合計に対する質量比をそれぞれ、δ(%)、ε(%)とした。また、溶媒である精製水と第i~iii成分の固形分の合計質量における、第i~iii成分の固形分の質量比をポリマー固形分濃度(%)とした。表3~7にそれぞれの具体的な組成を示した。また、これらの表中において中和度(対第iii成分)とは、調製方法の工程Aにおける、第iii成分の中和溶解の中和度である。基本的に、工程Aでの第iii成分の中和度は100%で完全中和としている。塩基性中和剤がアンモニアである実施例2-10の場合のみ、揮発性を考慮して過剰のアンモニアを加えているが、最終的な皮膜内の残留分は、100%より大幅に少ないものと推定される。
非イオン性水溶性セルロース化合物として、「粘度値」100 mPa・s以上のメチルセルロース(MC)を用いて、表3に示す実施例1-1~1-7の組成のカプセル調製液を用いて上記III-1.の手順でカプセルを調製した。各カプセル調製液は、成型ピン浸漬時の温度T5において、すべて、白濁(懸濁)した分散液となっていた。また、酸化チタン投入前であっても、白濁、もしくは半透明の分散液となっていることは別途確認した。

非イオン性水溶性セルロース化合物として、「粘度値」100 mPa・s以上のヒドキシプロピルメチルセルロース(HPMC)を用いて、表4に示す実施例2-1~2-10の組成のカプセル調製液を用いて上記III-1.の手順でカプセルを調製した。実施例2におけるカプセル調製液は、成型ピン浸漬時の温度55℃において、すべて、白濁した分散液となっていた。

表4において、「-」は、測定していないことを表す。
非イオン性水溶性セルロース化合物として、「粘度値」100 mPa・s以上のヒドロキシプロピルセルロース(HPC)を用いて、表5に示す実施例3-1の組成のカプセル調製液を用いて上記III-1.の手順でカプセルを調製した。実施例3におけるカプセル調製液は、成型ピン浸漬時の温度55℃において、白濁した分散液となっていた。
さらに上記IV.の方法によりサイズ2号の硬質カプセルを作成した。
30分で100%であった。実施例3-1の調製液から調製されたカプセルは、中性溶液に対しては易溶性であることが示された。

非イオン性水溶性セルロース化合物として、種々の「粘度値」のヒドロキシプロピルメチルセルロース(HPMC)を用いて、表6に示す実施例4-1~4-3及び比較例4-1~4-4の組成のカプセル調製液を用いて上記III-1.の手順でカプセルを調製した。
各カプセル調製液について、ブルックフィールド粘度計による粘度、及び、レオメーターによる降温時における動的粘弾性挙動を前記の装置及び手順で測定した。評価する特性は、i.図Cにおける調製液保持(浸漬)温度T3=T5=約55℃における粘度が好ましい範囲にあるか、ii.冷却時において図CのT4=約30~50℃の範囲での構造粘性もしくは冷ゲル化の開始による急激な粘度増加があるか、そして、iii.最終的に、室温近傍(20~30℃)の乾燥温度で、G’>G’’となってゲル化しているか、の3点である。表6にカプセル調製液の組成、T5における調製液粘度(ブルックフィールド粘度計)、降温時の動的粘弾性測定結果、すなわち、室温付近でのゲル化の有無(レオメーター測定で、G’>G’’ならばゲル化と判断)、約30~50℃での急激な粘度増加の有無を示す。HPMCの「粘度値」が、100 mPa・s以上の場合(実施例4-1、4-2、4-3)に、調製液の粘度が1,000~3,000 mPa・s程度となり、また、30~50℃での急激な粘度増加、つまり室温近傍でのゲル化の要件を満たした。他方、HPMCの「粘度値」が、100 mPa・s未満の場合(比較例4-1、4-2、4-3)、約30~50℃での粘度増加が緩やかであり室温付近でのゲル化も見られないことが示された。

第i成分、第ii成分及び中和された第iii成分が、本開示に係る態様3-1の調製液の調製方法及びコールドピン浸漬法の調製液に必要なことを確認するため、いずれかの成分を抜き、その分の質量を単純に精製水で置き換えた各種溶液を作成し、カプセル調製液としての適性を確認した。表7に調製液の組成(いずれの場合も酸化チタンは含まない)、レオメーターでの降温時の動的粘弾性測定結果、すなわち、室温付近でのゲル化の有無(レオメーター測定で、G’>G’’ならばゲル化と判断し、「○」で表す。G’<G’’であるか、見かけ上G’>G’’であっても、G’が非常に小さくて、実際上固体化不能である場合は、「×」で表す)及び約30~50℃での急激な粘度増加の有無を示す。また、「自立乾燥皮膜形成」として、キャスト法により自立した皮膜が得られるかどうかを評価した。この評価は、他の支持材によらずに、自立した皮膜が形成できるかどうか、さらには、空の硬質カプセル皮膜として適当な機械強度を有るかどうかを示している。この場合、100μm程度の厚みのキャストフィルムを得るために、単に特性成分を水で置換するだけでなく、ポリマー成分間の比率は保ったまま、ポリマー固形分濃度を適宜調製している場合がある。表7において自立した皮膜が形成できたものは○で示す。キャスト法において多少ポリマー固形分濃度を調製しても、調製液を塗布する基板からはがす際に脆すぎたり、柔らかすぎたりして、自立可能な皮膜として剥離困難なものは×で示す。

以下の実施例6~7においては、調製例III-2(調製方法の態様3-2)にしたがってカプセル調製液を調製し、成型方法IVによって、成型を行った。第i成分(第1成分)、第ii成分(第2成分)、第iv成分(第4成分)、及び第v成分(第5成分)の固形分質量合計(ポリマー固形分質量合計)を100質量%としたときのそれぞれの質量%を、α、γ、σ、φとした。塩基性中和剤、酸化チタン(遮光剤)の、上記ポリマー固形分質量合計に対する質量比をそれぞれ、δ(%)、ε(%)とした。また、溶媒である精製水と第i、ii、iv、v成分の固形分の合計質量における、第i、ii、iv、v成分の固形分の質量比をポリマー固形分濃度(%)とした。表8~9において、中和度は、調製の工程A’において、L30D55分散液に添加する塩基性中和剤の、L30D55固形分質量に対する中和度である。ここでの塩基性中和剤は、第i成分と第ii成分の混合によって直ちに凝集が生じることを防ぐ目的で添加されており、第ii成分自体の溶解による微粒子化を目的としたものではなく、その中和度は、8%程度と十分低くできる。
非イオン性水溶性セルロース化合物として、「粘度値」100 mPa・s以上のヒドロキシプロピルメチルセルロース(HPMC)を用いて、表8に示す実施例6-1~6-10の組成のカプセル調製液を用いて上記III-2.の手順でカプセル調製液を調製した。各カプセル調製液は、成型ピン浸漬時の温度T5において、すべて、白濁した分散液となっていた。また、酸化チタン投入前であっても、白濁(懸濁)、もしくは半透明の分散液となっていることは別途確認した。

第i成分、及び第ii成分、第iv成分、及び塩基性中和剤がすべてそろっていることが、本開示に係るコールドピン浸漬法の調製液に必要なことを確認するため、調製方法の態様3-2において、いずれかの成分を抜き、その分の質量を単純に精製水で置き換えた各種溶液を作成し、カプセル調製液としての適性を確認した。表9に調製液の組成(いずれの場合も酸化チタンは含まない)、レオメーターでの降温時の動的粘弾性測定結果、すなわち、室温付近でのゲル化の有無、及び約30~50℃での急激な粘度増加の有無を示す。また、「自立乾燥皮膜形成」の可否も示した。

第1~4成分、もしくは第1~5成分のすべてを含む腸溶性硬質カプセルの例を表10にそれぞれ、実施例8-1及び8-2として示した。調製例III-1(調製方法の態様3-1)にしたがってカプセル調製液を調製し、成型方法IVによって成型を行った。第i成分(第1成分)、第ii成分(第2成分)、第iii成分(第3成分)、第iv成分(第4成分)、及び第v成分(第5成分)の固形分質量合計(ポリマー固形分質量合計)を100質量%としたときのそれぞれの質量%を、α、β、γ、σ、φとした。塩基性中和剤(NaOH)、酸化チタン(遮光剤)の、上記ポリマー固形分質量合計に対する質量比をそれぞれ、δ(%)、ε(%)とした。また、溶媒である精製水と第i~iii成分の固形分の合計質量における、第i~v成分の固形分の質量比をポリマー固形分濃度(%)とした。表10にそれぞれの具体的な組成を示した。また、これらの表中において中和度(対第iii成分)とは、調製方法の工程Aにおける、第iii成分の中和溶解の中和度である。基本的に、工程Aでの第iii成分の中和度は100%で完全中和としている。塩基性中和剤がアンモニアである実施例8-1の場合のみ、揮発性を考慮して過剰のアンモニアを加えているが、最終的な皮膜内の残留分は、100%より大幅に少ないものと推定される。
いずれの場合も、腸溶性硬質カプセルとして十分な溶解特性及び機械的強度を有していた。

実施例1~7における硬質カプセル化された皮膜の硬さは、空の硬質カプセル皮膜として安定な形状を保つに十分な機械的強度があった。
実施例1~8における溶出試験において、バンドシール(例えば、HPMCAS-MFを水とエタノール2:8の比率の溶媒に溶解させた溶液からなるシール液をサイズ2号カプセルの胴体部のキャップとボディを嵌合した部位に5 mm程度の幅で帯状に塗布後乾燥した)を適用した場合、適用しない場合に比べて、溶出率にはほとんど影響がなかった。本開示に係る腸溶性硬質カプセルは、第1液中で若干膨潤する性質があり、これが、キャップとボディの隙間を効果的にふさぐためと考えられる。但し、第1液に浸漬してから2時間後の溶出率が10%程度を示すカプセルでは、バンドシールの適用により、1~2%程度溶出率の低下がみられる場合もあり、より確実な溶出抑制が必要な場合には、バンドシールを適用することが有効であると考えられた。
実施例1-1の本開示に係る腸溶性硬質カプセル(サイズ2号)に、アセトアミノフェン混合末を充填したカプセル製剤を用意しこれを内部カプセルとした。ヒプロメロースカプセル(Quali-V(登録商標)、サイズ00号)にカフェイン100 mgと、前記内部カプセルを充填した2重カプセル構造を有するカプセル製剤を用意した。第1液中で2時間溶出試験を行った後、第2液中で溶出試験を行った。カフェイン及びアセトアミノフェンの溶出率の時間変化を図8に示す。第1液中ではpH依存性のないヒプロメロースカプセルのみが溶解し、中身のカフェインのみが短時間でほぼ100%溶出したが、内側の本開示に係る腸溶性硬質カプセルは溶解せず、アセトアミノフェンの溶出はほぼゼロであった。第2液中に移行してから、速やかに溶解が始まり、アセトミノフェンが約30分で100%溶出していることが示された。
Claims (66)
- 第1成分及び第2成分を含み、さらに第3成分、第4成分、及び第5成分の少なくとも一成分を含む皮膜からなる腸溶性硬質カプセルであって、
第1成分は、粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物であり、
第2成分は、腸溶性メタクリル酸コポリマーであり、
第3成分は、腸溶性セルロース化合物であり、
第4成分は、水不溶性(メタ)アクリル酸アルキルエステルコポリマーであり、及び、
第5成分は、ポリビニルアルコール、可塑剤、及び界面活性剤よりなる群から選択される少なくとも一種である、
腸溶性硬質カプセル。 - 前記非イオン性水溶性セルロース化合物が、ヒドロキシプロピルメチルセルロース、メチルセルロース、及びヒドロキシプロピルセルロースからなる群から選択される少なくとも一種である、請求項1に記載の腸溶性硬質カプセル。
- 前記腸溶性メタクリル酸コポリマーが、メタクリル酸とメタクリル酸メチル及びアクリル酸メチルとのコポリマー、又は、メタクリル酸とアクリル酸エチルとのコポリマーからなる群から選択される少なくとも一種である、請求項1又は2に記載の腸溶性硬質カプセル。
- 前記腸溶性メタクリル酸コポリマーが、メタクリル酸40~60質量%とアクリル酸エチル60~40質量%とからなるコポリマーであることを特徴とする、請求項1~3のいずれか一項に記載の腸溶性硬質カプセル。
- 前記腸溶性セルロース化合物が、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、及びセルロースアセテートフタレートからなる群から選択される少なくとも一種である、請求項1~4のいずれか一項に記載の腸溶性硬質カプセル。
- 前記(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、請求項1~5のいずれか一項に記載の腸溶性硬質カプセル。
- 前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とし、第1成分の割合をα質量%、第2成分の割合をβ質量%、第3成分の割合をγ質量%、第4成分の割合をσ%、及び第5成分の割合をφとした場合に、0.5≦(β+γ+σ)/(α+β+γ+σ+φ)≦0.9であり、かつ、0.4≦(β+γ)/(β+γ+σ)である、請求項1~6のいずれか一項に記載の腸溶性硬質カプセル。
- 前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とし、第1成分の割合をα質量%、第2成分の割合をβ質量%、第3成分の割合をγ質量%、第4成分の割合をσ%、及び第5成分の割合をφとした場合に、0.05≦α/(α+β+γ+σ+φ)≦0.5である、請求項1~7のいずれか一項に記載の腸溶性硬質カプセル。
- 前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とし、第2成分の割合をβ質量%、及び第3成分の割合をγ質量%とした場合に、0.1≦β/(β+γ)≦1である、請求項1~8のいずれか一項に記載の腸溶性硬質カプセル。
- 前記皮膜に含まれる第1成分、第2成分、第3成分、第4成分、及び第5成分の質量の合計を100質量%とした場合の第1成分の割合をα質量%、第2成分の割合をβ質量%、第4成分の割合をσ%、及び第5成分の割合をφとした場合に、γ=0であり、かつ、0.3≦β/(α+β+γ+σ+φ)≦0.7である、請求項9に記載の腸溶性硬質カプセル。
- 前記第2成分の少なくとも一部がその薬学的に又は食品添加物として許容される塩として含まれる、及び/又は第3成分の少なくとも一部がその薬学的に又は食品添加物として許容される塩として含まれる、請求項1~10のいずれか一項に記載の腸溶性硬質カプセル。
- 前記皮膜に含まれる前記第2成分及び第3成分における塩を形成したカルボキシル基と塩を形成していないカルボキシル基のモル数の合計を100モル%とした場合、塩を形成したカルボキシル基の含有量が2~50モル%である、請求項11に記載の腸溶性硬質カプセル。
- 前記皮膜の厚みが50~250μmである、請求項1~12のいずれか一項に記載の腸溶性硬質カプセル。
- 前記皮膜の25℃、相対湿度60%における弾性率が1GPa~5GPaである、請求項13に記載の腸溶性硬質カプセル。
- 前記皮膜の25℃、相対湿度22%における破断伸び率が2%~30%である、請求項13又は14に記載の腸溶性硬質カプセル。
- 前記腸溶性硬質カプセルの皮膜が海島構造を含み、島相が実質的に第1成分からなる、請求項1~15のいずれか一項に記載の腸溶性硬質カプセル。
- 前記島相の短径が0.1μm以上、かつ30μm未満である、請求項16に記載の腸溶性硬質カプセル。
- pH1.2を有する溶液を用いた溶出試験において、2時間後の前記腸溶性硬質カプセルの溶出率が、25%以下である、請求項1~17のいずれか一項に記載の腸溶性硬質カプセル。
- 前記溶出試験における腸溶性硬質カプセルの溶出率が、10%以下である、請求項18に記載の腸溶性硬質カプセル。
- 第i成分、第ii成分、薬学的又は食品添加物として許容される塩基性中和剤、及び溶媒を含み、さらに第iii成分、第iv成分及び第v成分の少なくとも一成分を含む腸溶性硬質カプセル調製液であって、
第i成分は、粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物であり、
第ii成分は、腸溶性メタクリル酸コポリマーであり、
第iii成分は、腸溶性セルロース化合物であり、
第iv成分は、水不溶性(メタ)アクリル酸アルキルエステルコポリマーであり、及び、
第v成分は、ポリビニルアルコール、可塑剤、及び界面活性剤よりなる群から選択される少なくとも一種である、
腸溶性硬質カプセル調製液。 - 前記第i成分が、固体粒子として分散されている、請求項20に記載の腸溶性硬質カプセル調製液。
- 前記、第ii成分の一部及び/又は第iii成分の一部が、前記塩基性中和剤によって部分中和されている、請求項20又は21に記載の腸溶性硬質カプセル調製液。
- 前記部分中和の中和度が第ii及び第iii成分の完全中和に必要なモル数に対して、2~50%である、請求項22に記載の腸溶性硬質カプセル調製液。
- 前記第ii成分が、コロイド粒子として分散されている、請求項20~23のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記非イオン性水溶性セルロース化合物が、ヒドロキシプロピルメチルセルロース、メチルセルロース、及びヒドロキシプロピルセルロースからなる群から選択される少なくとも一種である、請求項20~24のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性セルロース化合物が、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、及びセルロースアセテートフタレートからなる群から選択される少なくとも一種である、請求項20~25のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性メタクリル酸コポリマーが、メタクリル酸とメタクリル酸メチル及びアクリル酸メチルとのコポリマー、又は、メタクリル酸とアクリル酸エチルとのコポリマーからなる群から選択される少なくとも一種である、請求項20~26のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性セルロース化合物の一部又は全部を、第iv成分である水不溶性(メタ)アクリル酸アルキルエステルコポリマーで置換したことを特徴とする、請求項20~27のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記水不溶性(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、請求項20~28のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記第iv成分が、コロイド粒子として分散されている、請求項28又は29のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とし、第i成分の割合をα’質量%とし、第ii成分の割合をβ’質量%、第iii成分の割合をγ’質量%、第iv成分の割合をσ’質量%、及び第v成分の割合をφ’質量%とした場合に、0.5≦(β’+γ’+σ’)/(α’+β’+γ’+σ’+φ’)≦0.9であり、かつ、0.4≦(β’+γ’)/(β’+γ’+σ’)である、請求項20~30項のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分及び、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とし、第i成分の割合をα’質量%、第ii成分の割合をβ’質量%、第iii成分の割合をγ’質量%、第iv成分の割合をσ’質量%、及び第v成分の割合をφ’質量%とした場合に、0.05≦α’/(α’+β’+γ’+σ’+φ’)≦0.5である、請求項20~31のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とした場合の、第ii成分の割合をβ’質量%、第iii成分の割合をγ’質量%とした場合に、0.1≦β’/(β’+γ’)≦1である、請求項20~32のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記腸溶性硬質カプセル調製液に含まれる第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の質量の合計を100質量%とした場合の第i成分の割合をα’質量%、第ii成分の割合をβ’質量%、第iv成分の割合をσ’質量%、及び第v成分の割合をφ’質量%とした場合に、γ’=0であり、かつ、0.3≦β’/(α’+β’+γ’+σ’+φ’)≦0.7である請求項33に記載の腸溶性硬質カプセル調製液。
- 前記塩基性中和剤による第ii成分の中和度が、2~20%である請求項34に記載の腸溶性硬質カプセル調製液。
- 前記塩基性中和剤が、水酸化ナトリウム、水酸化カリウム、及び水酸化カルシウムからなる群から選択される少なくとも一種である、請求項20~35のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 前記塩基性中和剤が、アンモニア及び炭酸アンモニウムからなる群から選択される少なくとも一種である、請求項20~35のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 腸溶性硬質カプセル調製液を100質量%としたときに、前記第i成分、第ii成分、第iii成分、第iv成分、及び第v成分の合計量が10~30質量%である、請求項31~37のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 粘度が、100~10,000mPa・sである、請求項20~38のいずれか一項に記載の腸溶性硬質カプセル調製液。
- 溶媒中に薬学的又は食品添加物として許容される塩基性中和剤が存在する条件下で、第i成分と第ii成分が混合される腸溶性硬質カプセル調製液の調製方法であって、第i成分は、粘度値が100mPa・s~100,000mPa・sの範囲である非イオン性水溶性セルロース化合物であり、第ii成分は、腸溶性メタクリル酸コポリマーである、調製方法。
- 前記非イオン性水溶性セルロース化合物が、ヒドロキシプロピルメチルセルロース、メチルセルロース、及びヒドロキシプロピルセルロースからなる群から選択される少なくとも一種である、請求項40に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記腸溶性メタクリル酸コポリマーが、メタクリル酸とメタクリル酸メチル及びアクリル酸メチルとのコポリマー、又は、メタクリル酸とアクリル酸エチルとのコポリマーからなる群から選択される少なくとも一種である、請求項40又は41のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記塩基性中和剤が、水酸化ナトリウム、水酸化カリウム、水酸化カルシウムからなる群から選択される少なくとも一種である、請求項40~42のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記塩基性中和剤が、アンモニア及び炭酸アンモニウムからなる群から選択される少なくとも一種である、請求項40~42のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 工程A:第iii成分の中和液を準備する工程、
工程B:前記第iii成分を含む中和液に第i成分を加え、第i成分の部分溶解液を準備する工程、及び
工程C:前記第ii成分の分散液を、中和液もしくは部分溶解液と混合する工程、
を順不同で含む、
腸溶性硬質カプセル調製液の調製方法であって、第iii成分は、腸溶性セルロース化合物である、請求項40~44のいずれか一項に記載の調製方法。 - 前記腸溶性セルロース化合物が、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、及びセルロースアセテートフタレートからなる群から選択される少なくとも一種である、請求項45に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記工程Aが、前記第iii成分を薬学的に又は食品添加物として許容される塩基性中和剤により、少なくとも部分的に中和して溶媒に溶解させる中和液を調製する工程であり、その中和度が50%以上又は、完全に中和されている、請求項45又は46に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記工程Bが、前記第iii成分を含む中和液、又は前記第iii成分の中和液と前記第ii成分の分散液の混合液に、前記第i成分を部分溶解させた部分溶解液を調製する工程であり、部分溶解液を調製する工程が、第i成分を、第i成分の曇点T0以上の第1の温度T1で、第iii成分を含む中和液、又は第iii成分の中和液と第ii成分の分散液の混合液に添加し、前記曇点よりも低い第2の温度T2で第i成分を部分溶解させた分散液を調製する工程である、請求項45~47のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記工程A、B、もしくはCで準備された溶液と第iv成分である水不溶性(メタ)アクリル酸エステルコポリマーを混合する工程Dを含む請求項45~48のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記水不溶性(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、請求項49に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記工程B、C又はDで得られた溶液を、前記第i成分の曇点よりも低い第3の温度T3に保持する工程Eをさらに含む、請求項45~50のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 工程A’:第ii成分の部分中和液を準備する工程、
工程B’:第i成分の部分溶解液を準備する工程、及び
工程C’:第iv成分の分散液を、工程AもしくはBで準備された溶液と混合する工程、
を順不同で含む、
請求項40記載の腸溶性硬質カプセル調製液の調製方法であって、
第iv成分は、水不溶性(メタ)アクリル酸アルキルエステルコポリマーである、
調製方法。 - 前記水不溶性(メタ)アクリル酸アルキルエステルコポリマーが、メタクリル酸メチルとアクリル酸エチルとのコポリマーである、請求項52に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記工程A’が、前記第ii成分を薬学的に又は食品添加物として許容される塩基性中和剤により、少なくとも部分的に中和して溶媒に溶解させる中和液を調製する工程であり、その中和度が2~20%である、請求項52又は53に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記工程B’が、前記第ii成分を含む中和液に、前記第i成分を部分溶解させた部分溶解液を調製する工程であり、
前記部分溶解液を調製する工程が、第i成分を、第i成分の曇点T0以上の第1の温度T1で、第ii成分を含む中和液、又は第ii成分の中和液と前記第iv成分の分散液の混合液に添加し、前記曇点よりも低い第2の温度T2で第i成分を部分溶解させた分散液を調製する工程である、請求項52~54のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。 - 前記工程B’又はC’で得られた溶液を、前記第i成分の曇点よりも低い第3の温度T3に保持する工程E’をさらに含む、請求項55に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記第3の温度の範囲T3が、40℃~60℃である、請求項51又は56に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記第1の温度T1の範囲が、60℃~90℃である、請求項48~51及び55~57のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記第2の温度T2の範囲が、30℃~60℃である、請求項48~51及び55~57のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 前記腸溶性硬質カプセル調製液の粘度が、100~10,000mPa・sである、請求項40~59のいずれか一項に記載の腸溶性硬質カプセル調製液の調製方法。
- 下記工程を含む、腸溶性硬質カプセルの調製方法:
請求項20~39のいずれか一項に記載の腸溶性硬質カプセル調製液、又は請求項40~60のいずれか一項に記載の調製方法により得られた腸溶性硬質カプセル調製液の中に、前記腸溶性硬質カプセル調製液の温度よりも低い表面温度を有するモールドピンを浸漬する第1工程;及び
前記腸溶性硬質カプセル調製液からモールドピンを引き上げて、モールドピンに付着した腸溶性硬質カプセル調製液を乾燥させる第2工程。 - 前記腸溶性硬質カプセル調製液の温度が、40~60℃である、請求項61に記載の腸溶性硬質カプセルの調製方法。
- 前記調製液に浸漬する前のモールドピンの表面温度が、5~40℃である、請求項62又は61又は62に記載の腸溶性硬質カプセルの調製方法。
- モールドピンに付着した腸溶性硬質カプセル調製液を乾燥する温度が、40℃未満である、請求項61~63のいずれか一項に記載の腸溶性硬質カプセルの調製方法。
- 請求項1~19のいずれか一項に記載の腸溶性硬質カプセルに対し、腸溶性メタクリル酸コポリマー及び腸溶性セルロース化合物よりなる群から選択される少なくとも一種の腸溶性ポリマーの少なくとも部分中和された希釈水溶液、あるいは、水/エタノール又は水/イソプロパノール溶剤に溶解した液からなるシール液によってシールされたことを特徴とする腸溶性硬質カプセル製剤。
- 酸性条件で溶解可能な硬質カプセルの内部に請求項1~19のいずれか一項に記載の腸溶性硬質カプセルを内包することを特徴とする硬質カプセル製剤。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019529765A JP7198205B2 (ja) | 2017-07-11 | 2018-07-11 | 腸溶性硬質カプセル |
| EP18832265.5A EP3653203A4 (en) | 2017-07-11 | 2018-07-11 | ENTERAL HARD CAPSULE |
| CN201880045888.7A CN110891552B (zh) | 2017-07-11 | 2018-07-11 | 肠溶性硬质胶囊 |
| US16/628,742 US20200375910A1 (en) | 2017-07-11 | 2018-07-11 | Enteric hard capsule |
| KR1020207001752A KR20200026901A (ko) | 2017-07-11 | 2018-07-11 | 장용성 경질 캡슐 |
| BR112020000011-6A BR112020000011A2 (pt) | 2017-07-11 | 2018-07-11 | cápsula dura entérica |
| CA3069396A CA3069396A1 (en) | 2017-07-11 | 2018-07-11 | Enteric hard capsule |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017135666 | 2017-07-11 | ||
| JP2017-135666 | 2017-07-11 | ||
| JP2018039184 | 2018-03-06 | ||
| JP2018-039184 | 2018-03-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019013260A1 true WO2019013260A1 (ja) | 2019-01-17 |
Family
ID=65001930
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/026216 WO2019013260A1 (ja) | 2017-07-11 | 2018-07-11 | 腸溶性硬質カプセル |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20200375910A1 (ja) |
| EP (1) | EP3653203A4 (ja) |
| JP (1) | JP7198205B2 (ja) |
| KR (1) | KR20200026901A (ja) |
| CN (1) | CN110891552B (ja) |
| BR (1) | BR112020000011A2 (ja) |
| CA (1) | CA3069396A1 (ja) |
| TW (1) | TWI793141B (ja) |
| WO (1) | WO2019013260A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019245031A1 (ja) * | 2018-06-22 | 2019-12-26 | クオリカプス株式会社 | 腸溶性硬質カプセル |
| JP2020138926A (ja) * | 2019-02-28 | 2020-09-03 | アピ株式会社 | ハードカプセル及びその製造方法 |
| WO2021024930A1 (ja) | 2019-08-02 | 2021-02-11 | クオリカプス株式会社 | タグを含むバンドシールにより封緘された硬質カプセル製剤 |
| CN112546016A (zh) * | 2020-12-15 | 2021-03-26 | 浙江万里学院 | 一种肠溶空心纤维素胶囊及其制备方法 |
| JP2022525180A (ja) * | 2019-03-14 | 2022-05-11 | エボニック オペレーションズ ゲーエムベーハー | コア-シェルポリマーとセルロースとを含むカプセルシェル |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI676684B (zh) * | 2017-06-15 | 2019-11-11 | 財團法人食品工業發展研究所 | 乳酸桿菌、使用其製備色素之方法、乳酸桿菌培養物與包括其之色素組成物 |
| CN117159489B (zh) * | 2023-10-31 | 2024-02-23 | 潍坊医学院 | 一种肠溶空心胶囊及其制备方法 |
Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US630966A (en) | 1896-06-15 | 1899-08-15 | Electro Gas Company | Carbid-furnace. |
| US2196768A (en) | 1938-03-11 | 1940-04-09 | Eastman Kodak Co | Enteric coating |
| US2718667A (en) | 1952-05-01 | 1955-09-27 | Eastman Kodak Co | Method of preparing enteric capsules |
| JPS473547U (ja) | 1971-01-30 | 1972-09-05 | ||
| US3826666A (en) | 1972-07-20 | 1974-07-30 | Parke Davis & Co | Enteric capsules |
| US3927195A (en) | 1974-01-31 | 1975-12-16 | Lilly Industries Ltd | Production of capsules |
| JPS5352619A (en) | 1976-10-25 | 1978-05-13 | Shin Etsu Chem Co Ltd | Enteric capsule |
| US4138013A (en) | 1976-08-27 | 1979-02-06 | Parke, Davis & Company | Enteric capsules |
| JPS55136061A (en) | 1979-04-12 | 1980-10-23 | Freunt Ind Co Ltd | Sheath capsule |
| JPS5732230A (en) * | 1980-07-18 | 1982-02-20 | Parke Davis & Co | Instetine-soluble capsule |
| US4365060A (en) | 1979-04-28 | 1982-12-21 | Shin-Etsu Chemical Co. Ltd. | Enterosoluble capsules |
| JPH08208458A (ja) | 1994-12-01 | 1996-08-13 | Nippon Eranko Kk | カプセル用皮膜組成物 |
| JP2001506692A (ja) | 1996-12-17 | 2001-05-22 | ワーナー−ランバート・カンパニー | カプセル用のポリマーフィルム組成物 |
| US20030104047A1 (en) * | 2001-12-03 | 2003-06-05 | Gan-Lin Chen | Method for manufacturing hard non-gelatin pharmaceutical capsules |
| JP2003325642A (ja) | 2002-05-09 | 2003-11-18 | Sansho Pharmaceutical Co Ltd | 腸溶性皮膜付き硬質空カプセルと、腸溶性皮膜付き硬質空カプセルの製造方法と、腸溶性皮膜付き硬質カプセル剤 |
| JP2004522746A (ja) | 2001-01-30 | 2004-07-29 | スミスクライン ビーチャム パブリック リミテッド カンパニー | 医薬製剤 |
| JP2006016372A (ja) | 2004-07-05 | 2006-01-19 | Shionogi Qualicaps Co Ltd | 腸溶性硬カプセル剤 |
| JP2006052819A (ja) | 2004-08-16 | 2006-02-23 | Cik Giken Kk | 止水構造及び既設管と更生管の止水方法 |
| US7094425B2 (en) | 1998-09-28 | 2006-08-22 | Warner-Lambert Company | Enteric and colonic delivery using HPMC capsules |
| JP2006528197A (ja) * | 2003-07-21 | 2006-12-14 | スミスクライン ビーチャム パブリック リミテッド カンパニー | 製薬用処方 |
| JP2009196961A (ja) | 2008-02-25 | 2009-09-03 | Qualicaps Co Ltd | 腸溶性カプセル |
| JP2010202550A (ja) | 2009-03-02 | 2010-09-16 | Qualicaps Co Ltd | 腸溶性カプセル |
| JP2010270039A (ja) | 2009-05-20 | 2010-12-02 | Qualicaps Co Ltd | 溶解性または硬度が改善された硬質カプセル |
| JP2011500871A (ja) | 2006-10-27 | 2011-01-06 | ファイザー・プロダクツ・インク | ヒドロキシプロピルメチルセルロース硬カプセルおよび製造方法 |
| JP2011503048A (ja) | 2007-11-08 | 2011-01-27 | グラクソ グループ リミテッド | 医薬製剤 |
| WO2011036601A1 (en) | 2009-09-24 | 2011-03-31 | Pfizer Inc. | Acid resistant capsules |
| JP2013500293A (ja) | 2009-07-29 | 2013-01-07 | エボニック レーム ゲゼルシャフト ミット ベシュレンクテル ハフツング | カプセル片の浸漬被覆用被覆剤 |
| JP2013504565A (ja) | 2009-09-11 | 2013-02-07 | 三星精密化学株式会社 | 腸溶性硬質カプセル用水性組成物、腸溶性硬質カプセルの製造方法及び腸溶性硬質カプセル |
| JP2013540149A (ja) | 2010-10-21 | 2013-10-31 | 三星精密化学株式会社 | 腸溶性硬質カプセル用組成物、及び該組成物を使用して調製された腸溶性硬質カプセル |
| JP2013540806A (ja) | 2010-10-26 | 2013-11-07 | キャプシュゲル・ベルジウム・エヌ・ヴィ | バルク腸溶カプセルシェル |
| JP2015515962A (ja) | 2012-05-02 | 2015-06-04 | キャプシュゲル・ベルジウム・エヌ・ヴィ | 放出制御ポリマーの水性分散液並びにそのシェル及びカプセル |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS473547Y1 (ja) | 1968-02-21 | 1972-02-07 | ||
| KR20060052819A (ko) | 2003-08-07 | 2006-05-19 | 마츠시타 덴끼 산교 가부시키가이샤 | 광 정보 기록매체 및 그 제조 방법 |
| TW201010745A (en) * | 2008-06-13 | 2010-03-16 | Glaxo Group Ltd | Pharmaceutical formulations |
| EP2283830A1 (en) * | 2009-07-23 | 2011-02-16 | Actogenix N.V. | Aqueous enteric capsule coating |
| EP2946774B1 (en) * | 2014-05-19 | 2020-04-22 | Tillotts Pharma AG | Modified release coated capsules |
| US20190000768A1 (en) * | 2015-08-04 | 2019-01-03 | Fuji Capsule Co., Ltd. | Enteric capsule |
-
2018
- 2018-07-11 CA CA3069396A patent/CA3069396A1/en active Pending
- 2018-07-11 WO PCT/JP2018/026216 patent/WO2019013260A1/ja unknown
- 2018-07-11 EP EP18832265.5A patent/EP3653203A4/en active Pending
- 2018-07-11 US US16/628,742 patent/US20200375910A1/en active Pending
- 2018-07-11 TW TW107123960A patent/TWI793141B/zh active
- 2018-07-11 BR BR112020000011-6A patent/BR112020000011A2/pt not_active Application Discontinuation
- 2018-07-11 KR KR1020207001752A patent/KR20200026901A/ko active IP Right Grant
- 2018-07-11 JP JP2019529765A patent/JP7198205B2/ja active Active
- 2018-07-11 CN CN201880045888.7A patent/CN110891552B/zh active Active
Patent Citations (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US630966A (en) | 1896-06-15 | 1899-08-15 | Electro Gas Company | Carbid-furnace. |
| US2196768A (en) | 1938-03-11 | 1940-04-09 | Eastman Kodak Co | Enteric coating |
| US2718667A (en) | 1952-05-01 | 1955-09-27 | Eastman Kodak Co | Method of preparing enteric capsules |
| JPS473547U (ja) | 1971-01-30 | 1972-09-05 | ||
| US3826666A (en) | 1972-07-20 | 1974-07-30 | Parke Davis & Co | Enteric capsules |
| US3927195A (en) | 1974-01-31 | 1975-12-16 | Lilly Industries Ltd | Production of capsules |
| US4138013A (en) | 1976-08-27 | 1979-02-06 | Parke, Davis & Company | Enteric capsules |
| JPS5352619A (en) | 1976-10-25 | 1978-05-13 | Shin Etsu Chem Co Ltd | Enteric capsule |
| JPS55136061A (en) | 1979-04-12 | 1980-10-23 | Freunt Ind Co Ltd | Sheath capsule |
| US4365060A (en) | 1979-04-28 | 1982-12-21 | Shin-Etsu Chemical Co. Ltd. | Enterosoluble capsules |
| JPS5732230A (en) * | 1980-07-18 | 1982-02-20 | Parke Davis & Co | Instetine-soluble capsule |
| JPH08208458A (ja) | 1994-12-01 | 1996-08-13 | Nippon Eranko Kk | カプセル用皮膜組成物 |
| JP2001506692A (ja) | 1996-12-17 | 2001-05-22 | ワーナー−ランバート・カンパニー | カプセル用のポリマーフィルム組成物 |
| US7094425B2 (en) | 1998-09-28 | 2006-08-22 | Warner-Lambert Company | Enteric and colonic delivery using HPMC capsules |
| JP2004522746A (ja) | 2001-01-30 | 2004-07-29 | スミスクライン ビーチャム パブリック リミテッド カンパニー | 医薬製剤 |
| US20030104047A1 (en) * | 2001-12-03 | 2003-06-05 | Gan-Lin Chen | Method for manufacturing hard non-gelatin pharmaceutical capsules |
| JP2003325642A (ja) | 2002-05-09 | 2003-11-18 | Sansho Pharmaceutical Co Ltd | 腸溶性皮膜付き硬質空カプセルと、腸溶性皮膜付き硬質空カプセルの製造方法と、腸溶性皮膜付き硬質カプセル剤 |
| JP2006528197A (ja) * | 2003-07-21 | 2006-12-14 | スミスクライン ビーチャム パブリック リミテッド カンパニー | 製薬用処方 |
| JP2006016372A (ja) | 2004-07-05 | 2006-01-19 | Shionogi Qualicaps Co Ltd | 腸溶性硬カプセル剤 |
| JP2006052819A (ja) | 2004-08-16 | 2006-02-23 | Cik Giken Kk | 止水構造及び既設管と更生管の止水方法 |
| JP2011500871A (ja) | 2006-10-27 | 2011-01-06 | ファイザー・プロダクツ・インク | ヒドロキシプロピルメチルセルロース硬カプセルおよび製造方法 |
| JP2011503048A (ja) | 2007-11-08 | 2011-01-27 | グラクソ グループ リミテッド | 医薬製剤 |
| JP2009196961A (ja) | 2008-02-25 | 2009-09-03 | Qualicaps Co Ltd | 腸溶性カプセル |
| JP2010202550A (ja) | 2009-03-02 | 2010-09-16 | Qualicaps Co Ltd | 腸溶性カプセル |
| JP2010270039A (ja) | 2009-05-20 | 2010-12-02 | Qualicaps Co Ltd | 溶解性または硬度が改善された硬質カプセル |
| JP2013500293A (ja) | 2009-07-29 | 2013-01-07 | エボニック レーム ゲゼルシャフト ミット ベシュレンクテル ハフツング | カプセル片の浸漬被覆用被覆剤 |
| JP2013504565A (ja) | 2009-09-11 | 2013-02-07 | 三星精密化学株式会社 | 腸溶性硬質カプセル用水性組成物、腸溶性硬質カプセルの製造方法及び腸溶性硬質カプセル |
| WO2011036601A1 (en) | 2009-09-24 | 2011-03-31 | Pfizer Inc. | Acid resistant capsules |
| JP2013540149A (ja) | 2010-10-21 | 2013-10-31 | 三星精密化学株式会社 | 腸溶性硬質カプセル用組成物、及び該組成物を使用して調製された腸溶性硬質カプセル |
| JP2013540806A (ja) | 2010-10-26 | 2013-11-07 | キャプシュゲル・ベルジウム・エヌ・ヴィ | バルク腸溶カプセルシェル |
| JP2015515962A (ja) | 2012-05-02 | 2015-06-04 | キャプシュゲル・ベルジウム・エヌ・ヴィ | 放出制御ポリマーの水性分散液並びにそのシェル及びカプセル |
| JP2015518005A (ja) | 2012-05-02 | 2015-06-25 | キャプシュゲル・ベルジウム・エヌ・ヴィ | ヒドロキシプロピルメチルセルロースアセテートスクシネート(hpmcas)の水性分散液 |
Non-Patent Citations (11)
| Title |
|---|
| "Aqueous Polymeric Coating For Pharmaceutical Dosage Forms", 2017, CRC PRESS |
| "Drug Targeting Technology", 2001, CRC PRESS, pages: 1 - 29 |
| "Japanese Pharmaceutical Excipients Directory", 2016 |
| COLE, E. T. ET AL.: "Enteric coated HPMC capsules designed to achieve intestinal targeting", INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 231, no. 1, 1 January 2002 (2002-01-01), pages 83 - 95, XP002560462, Retrieved from the Internet <URL:https://doi.org/10.1016/S0378-5173(01)00871-7> * |
| COLLECTION OF PAPERS ON POLYMER, vol. 38, 1981, pages 133 - 137 |
| DRUG DEV. IND. PHARM., vol. 27, 2011, pages 1131 - 1140 |
| INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 231, 2002, pages 83 - 95 |
| INTERNATIONAL JOURNAL OF PHARMACEUTICS, vol. 440, 2013, pages 264 - 272 |
| J. POLYM. SCI. C, vol. 36, 1971, pages 491 - 508 |
| REPORTS OF THE MIE PREFECTURE INDUSTRIAL RESEARCH INSTITUTE, vol. 33, 2009, pages 59 - 64 |
| See also references of EP3653203A4 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019245031A1 (ja) * | 2018-06-22 | 2019-12-26 | クオリカプス株式会社 | 腸溶性硬質カプセル |
| CN112566668A (zh) * | 2018-06-22 | 2021-03-26 | 快力胶囊股份有限公司 | 肠溶性硬胶囊 |
| JPWO2019245031A1 (ja) * | 2018-06-22 | 2021-07-08 | クオリカプス株式会社 | 腸溶性硬質カプセル |
| EP3811976A4 (en) * | 2018-06-22 | 2022-07-06 | Qualicaps Co., Ltd. | GASTRO-RESISTANT HARD CAPSULE |
| JP7366893B2 (ja) | 2018-06-22 | 2023-10-23 | クオリカプス株式会社 | 腸溶性硬質カプセル |
| JP2020138926A (ja) * | 2019-02-28 | 2020-09-03 | アピ株式会社 | ハードカプセル及びその製造方法 |
| JP2022525180A (ja) * | 2019-03-14 | 2022-05-11 | エボニック オペレーションズ ゲーエムベーハー | コア-シェルポリマーとセルロースとを含むカプセルシェル |
| WO2021024930A1 (ja) | 2019-08-02 | 2021-02-11 | クオリカプス株式会社 | タグを含むバンドシールにより封緘された硬質カプセル製剤 |
| CN112546016A (zh) * | 2020-12-15 | 2021-03-26 | 浙江万里学院 | 一种肠溶空心纤维素胶囊及其制备方法 |
| CN112546016B (zh) * | 2020-12-15 | 2022-04-22 | 浙江万里学院 | 一种肠溶空心纤维素胶囊及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3653203A1 (en) | 2020-05-20 |
| JPWO2019013260A1 (ja) | 2020-05-21 |
| CN110891552B (zh) | 2023-06-02 |
| CN110891552A (zh) | 2020-03-17 |
| BR112020000011A2 (pt) | 2020-07-21 |
| CA3069396A1 (en) | 2019-01-17 |
| KR20200026901A (ko) | 2020-03-11 |
| TWI793141B (zh) | 2023-02-21 |
| JP7198205B2 (ja) | 2022-12-28 |
| TW201912150A (zh) | 2019-04-01 |
| US20200375910A1 (en) | 2020-12-03 |
| EP3653203A4 (en) | 2020-06-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7198205B2 (ja) | 腸溶性硬質カプセル | |
| JP5248739B2 (ja) | 腸溶性調製物 | |
| TWI472323B (zh) | 用於腸溶性硬膠囊的水性組合物、腸溶性硬膠囊的製備方法以及以後者方法製備而得的硬膠囊 | |
| JP6317425B2 (ja) | シルク系カプセル剤 | |
| US20090010975A1 (en) | Non-transparent composition for film | |
| JP5489471B2 (ja) | Peg充填硬質カプセル剤のバンドシール | |
| JP5289429B2 (ja) | 硬質カプセル剤 | |
| JP2022123274A (ja) | 腸溶性硬質カプセル | |
| EP3811976B1 (en) | Enteric hard capsule | |
| JP5328768B2 (ja) | 硬質カプセル剤 | |
| JPWO2020071393A1 (ja) | 強度が改善された硬質カプセル、及びその製造方法 | |
| HUE026267T2 (en) | Composition comprising shellac and / or its salt and sodium starch glycolate | |
| EP4008320A1 (en) | Hard capsule formulation sealed with band seal including tag | |
| WO2024019133A1 (ja) | 多層構造腸溶性硬質カプセル | |
| JPWO2020071395A1 (ja) | 強度が改善された硬質カプセル、及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18832265 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019529765 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3069396 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020000011 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20207001752 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018832265 Country of ref document: EP Effective date: 20200211 |
|
| ENP | Entry into the national phase |
Ref document number: 112020000011 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200102 |