WO2018147368A1 - オキシイミノ化合物の幾何異性体の製造方法 - Google Patents
オキシイミノ化合物の幾何異性体の製造方法 Download PDFInfo
- Publication number
- WO2018147368A1 WO2018147368A1 PCT/JP2018/004391 JP2018004391W WO2018147368A1 WO 2018147368 A1 WO2018147368 A1 WO 2018147368A1 JP 2018004391 W JP2018004391 W JP 2018004391W WO 2018147368 A1 WO2018147368 A1 WO 2018147368A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- solvent
- mass
- formula
- oxyimino
- Prior art date
Links
- RXYVTMQTZRXYQK-KDPHDJQDSA-N CC(C)O/N=C(\C[C@@H](C)C(c1c[n](C)nc1C(F)F)=[U])/c(ncc(C#CC1CC1)c1)c1Cl Chemical compound CC(C)O/N=C(\C[C@@H](C)C(c1c[n](C)nc1C(F)F)=[U])/c(ncc(C#CC1CC1)c1)c1Cl RXYVTMQTZRXYQK-KDPHDJQDSA-N 0.000 description 1
- 0 CC(C)ON=C(CC[C@@](C)C(c1c[n](C)nc1C(*)F)=O)c(ncc(C#CC1CC1)c1)c1Cl Chemical compound CC(C)ON=C(CC[C@@](C)C(c1c[n](C)nc1C(*)F)=O)c(ncc(C#CC1CC1)c1)c1Cl 0.000 description 1
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/09—Geometrical isomers
Definitions
- oximino compounds are known to be useful as agricultural chemicals and pharmaceuticals (see Patent Documents 1 to 5).
- an oxyimino compound has an oximino group, and therefore has a configuration of E [hereinafter abbreviated as E form. ]
- a compound having a configuration of Z hereinafter abbreviated as Z-form.
- Patent Documents 1 and 2 For example, a method for producing a Z form from an E form by irradiating light (see Patent Documents 1 and 2), a method for producing a Z form from an E form by using an acidic compound (see Patent Document 6), A method for producing an E-form from a Z-form by using an acidic compound (see Patent Documents 4, 5, 7 and 8) and a method for producing a mixture of the E-form and the Z-form from the E-form by irradiating light (See Patent Document 3).
- an oximino compound when manufacturing an oximino compound, it is obtained as a mixture with E body and Z body. Since the E-form and Z-form are very close to each other, only the E-form or only the Z-form is selectively reduced from the mixture of the E-form and the Z-form by a normal purification operation such as column chromatography. It is very difficult to do.
- one geometric isomer when used as a medicine, agricultural chemical, or the like, the other geometric isomer is mixed into the product as an impurity, which adversely affects the quality and performance of the product. Therefore, development of a method for producing a novel oximino compound for producing a desired geometric isomer from an undesired geometric isomer is desired.
- Formula (EZ) -1 A mixture of geometric isomers of oximino compounds represented by formula (E) -1; An E-form of an oxyimino compound represented by the formula (Z) -1; The Z-form of the oximino compound represented by the above formula (Z) -1 is produced using the E-form of the oximino compound represented by the above formula (E) -1 as a starting material.
- solvent one or more solvents selected from the group consisting of toluene, ortho-xylene, cyclopentyl methyl ether, tertiary butyl methyl ether, dimethoxyethane, diethylene glycol dimethyl ether, methyl ethyl ketone, ethyl acetate and 1,2-dichloroethane are used.
- the oxyimino according to any one of the above [1] to [8], which comprises a step of adding one or more solvents selected from aliphatic hydrocarbon solvents after mixing a starting material and an acidic compound in a solvent.
- a stereoselective method for producing a compound [10] The stereoselective method for producing an oxyimino compound according to the above [9], wherein the aliphatic hydrocarbon solvent is normal heptane. [11] The stereoselective method for producing an oxyimino compound according to any one of the above [1] to [10], wherein the precipitated crystal, salt or solvate thereof, or solvate is separated from the reaction system.
- an oximino compound useful as a pharmaceutical or agrochemical can be produced in a high yield and with a high stereoselectivity, and a production method useful for industrial production can be provided.
- each of the two types of isomers is referred to as an E isomer in which the oximino group has an E configuration, and an isomer having a Z configuration in an oximino group as a Z isomer.
- the mixing ratio of E-form and Z-form can be qualitatively analyzed and calculated by any measurement method such as high performance liquid chromatography, gas chromatography, nuclear magnetic resonance spectrum and the like.
- n- represents normal, i- represents iso, s- represents secondary, and tert- or t- represents tertiary, o- represents ortho, m- represents meta, and p- represents para.
- examples of the basic compound include inorganic bases and organic bases.
- examples of the inorganic base include lithium hydroxide, sodium hydroxide, potassium hydroxide, cesium hydroxide, magnesium hydroxide, calcium hydroxide, barium hydroxide, lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate, and sodium bicarbonate.
- Potassium hydrogen carbonate cesium hydrogen carbonate, ammonium hydrogen carbonate, sodium acetate, potassium acetate, cesium acetate, calcium acetate, barium acetate, sodium phosphate, potassium phosphate, disodium hydrogen phosphate, dipotassium hydrogen phosphate, etc. It is done.
- organic base examples include ammonia, ethylamine, diethylamine, triethylamine, diisopropylethylamine, tributylamine, pyridine, 2-methylpyridine, 3-methylpyridine, 4-methylpyridine, 2,3-lutidine, 2,4-lutidine, Examples include 2,6-lutidine, 3,5-lutidine, diazabicycloundecene, 1,4-diazabicyclo [2.2.2] octane, 1,1,3,3-tetramethylguanidine.
- the compound represented by the formula (E) -1 of the present invention [hereinafter abbreviated as compound (E) -1. ]
- a compound represented by the formula (EZ) -1 [hereinafter abbreviated as mixture (EZ) -1.
- a compound represented by formula (Z) -1 [hereinafter abbreviated as compound (Z) -1. ] From the mixture (EZ) -1, a method for producing the compound (E) -1 from the compound (Z) -1, and a method for producing the compound (Z) -1 from the compound (E) -1 Will be described in detail.
- the amount of the acidic compound used in the production of compound (E) -1 can be 0.00001 to 0.1 equivalents relative to the starting material [mixture (EZ) -1 or compound (Z) -1], preferably Can be used in an amount of 0.001 to 0.1 equivalent, more preferably 0.01 to 0.07 equivalent.
- the amount of the acidic compound used in the production of compound (Z) -1 can be 0.7 to 10 equivalents relative to the starting material [mixture (EZ) -1 or compound (E) -1], preferably Can be used in an amount of 0.7 to 5 equivalents, more preferably 0.7 to 2 equivalents.
- Examples of the acidic compound to be used include hydrogen halide, inorganic acid, carboxylic acid, sulfonic acid and the like.
- Examples of the hydrogen halide include hydrogen fluoride, hydrogen chloride, hydrogen bromide, hydrogen iodide, and the like.
- Examples of inorganic acids include nitric acid, sulfuric acid, phosphoric acid, chloric acid, boric acid, perchloric acid and the like.
- carboxylic acids include formic acid, acetic acid, trifluoroacetic acid, trichloroacetic acid, propionic acid, butyric acid, valeric acid, pivalic acid, isovaleric acid, caproic acid, enanthic acid, caprylic acid, pelargonic acid, caprylic acid, lauric acid , Myristic acid, palmitic acid, stearic acid, acrylic acid, methacrylic acid, tiglic acid, oleic acid, linolenic acid, linoleic acid, arachidonic acid, oxalic acid, malonic acid, succinic acid, glutaric acid, adipic acid, fumaric acid, maleic acid Acid, malic acid, lactic acid, ascorbic acid, citric acid, mandelic acid, tartaric acid, pyruvic acid, benzoic acid, o-methylbenzoic acid, m-methylbenzoic acid, p-methylbenzoic acid, o
- sulfonic acid examples include methanesulfonic acid, ethanesulfonic acid, 10-camphorsulfonic acid, trifluoromethanesulfonic acid, benzenesulfonic acid, o-toluenesulfonic acid, m-toluenesulfonic acid, p-toluenesulfonic acid, o- Trifluoromethylbenzenesulfonic acid, m-trifluoromethylbenzenesulfonic acid, p-trifluoromethylbenzenesulfonic acid, o-chlorobenzenesulfonic acid, m-chlorobenzenesulfonic acid, p-chlorobenzenesulfonic acid, o-nitrobenzenesulfonic acid, m -Nitrobenzenesulfonic acid, p-nitrobenzenesulfonic acid and the like.
- acidic compound hydrogen halide, sulfuric acid and methanesulfonic acid are preferable, and hydrogen chloride, hydrogen bromide and sulfuric acid are more preferable.
- acidic compounds may contain water. These acidic compounds can be used in combination of two or more.
- Examples of the solvent used as the solvent A include aromatic hydrocarbon solvents, halogenated hydrocarbon solvents, alcohol solvents, ether solvents, ester solvents, amide solvents, nitrile solvents, ketone solvents, dimethyl sulfoxide and the like.
- Examples of the aromatic hydrocarbon solvent include benzene, toluene, o-xylene, m-xylene, p-xylene, chlorobenzene, 1,2-dichlorobenzene, 1,3-dichlorobenzene, 1,4-dichlorobenzene, and nitrobenzene.
- Examples of the halogenated hydrocarbon solvent include chloroform, dichloromethane, dichloroethane and the like.
- Examples of the alcohol solvent include methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, t-butyl alcohol and the like.
- Examples of the ether solvent include diethyl ether, tetrahydrofuran, cyclopentyl methyl ether, tertiary butyl methyl ether, 1,4-dioxane, dimethoxyethane, diethylene glycol dimethyl ether and the like.
- Examples of the ester solvent include methyl acetate, ethyl acetate, butyl acetate, methyl propionate, and the like.
- the amide solvent examples include N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone and the like.
- the nitrile solvent examples include acetonitrile and propionitrile
- examples of the ketone solvent include methyl ethyl ketone and methyl isobutyl ketone.
- the solvent A is preferably an aromatic hydrocarbon solvent, a halogenated hydrocarbon solvent, an ether solvent, an ester solvent, or a ketone solvent.
- toluene, o-xylene, 1,2-dichloroethane, cyclopentyl methyl ether, t-butyl methyl ether, dimethoxyethane, diethylene glycol dimethyl ether, ethyl acetate, and methyl ethyl ketone are used. More preferred are 1,2-dichloroethane and ethyl acetate. These solvents can be used not only as one kind but also as a mixture of two or more kinds.
- the amount of the solvent to be used can be 0.01 to 100 parts by weight, preferably 0.1 to 50 parts by weight, based on the starting material. More preferably, 0.5 to 20 parts by mass can be used, and further preferably 1 to 10 parts by mass can be used.
- the reaction temperature when the starting material and acidic compound are mixed and reacted in a solvent is usually ⁇ 20 to 150 ° C., preferably 0 to 100 ° C., more preferably 10 to 60 ° C.
- the temperature at which the crystals are precipitated is usually -20 to 150 ° C, preferably -10 to 80 ° C, more preferably 10 to 50 ° C.
- the reaction time when the starting material and acidic compound are mixed and reacted in a solvent is 1 minute to 1000 hours, preferably 5 minutes to 100 hours, more preferably 10 minutes to 48 hours.
- the time for depositing the crystals varies depending on the concentration of the reaction substrate and the reaction temperature, but is usually 1 minute to 1000 hours, preferably 5 minutes to 500 hours, more preferably 10 minutes to 100 hours.
- the treatment method after the reaction is not particularly limited, but the target product can be obtained by separating the target product from the reaction system by filtering the precipitated crystals.
- the oximino compound in the obtained filtrate can be used as a starting material for the production method of the present invention after being concentrated as necessary.
- the obtained crystals can be purified by an optional purification method such as recrystallization as necessary to obtain a target product with higher purity.
- the target product produced by the reaction is obtained as a salt with the acidic compound used in the reaction in some cases. Even when obtained as a salt, it can be obtained by the same method as above, but if necessary, water and then an organic solvent are added and dissolved in the organic solvent as necessary. Or you may neutralize by adding basic compounds, such as the aqueous solution.
- the target product produced by the reaction may be obtained as a solvate containing a solvent (hereinafter abbreviated as a solvate) by solvating with the solvent used in the reaction in some cases. Also when obtained as a solvate, it can be obtained by the same method as described above, but if necessary, after dissolving or suspending the obtained solvate in a non-solvated solvent, if necessary
- the desired product can be obtained by performing usual post-treatment such as addition of water, liquid separation, and concentration as necessary.
- the target product produced by the reaction may be obtained as a solvate of the salt of the target product depending on the acidic compound and solvent used in the reaction. Also in this case, it can be obtained by the same method as described above, but the desired product can be obtained by combining the post-treatments as necessary.
- the mixture (EZ) -1 was reacted in a solvent (hereinafter abbreviated as solvent A) in the presence of an acidic compound, and then a solvent (hereinafter abbreviated as solvent B) was further added to achieve the purpose.
- solvent A a solvent
- solvent B a solvent
- the product can be obtained as crystals.
- by controlling the amount of the acidic compound to be used either one of the geometric isomers of the compound (E) -1 or the compound (Z) -1 can be selectively produced.
- compound (E) -1 can be selectively produced from compound (Z) -1 by controlling the amount of acidic compound used, and compound (Z) -1 can be produced from compound (E) -1.
- ) -1 can be selectively produced.
- the amount and type of the acidic compound to be used are the same as described in Production Method 1.
- Examples of the solvent used as the solvent A include the solvents described in Production Method 1. These solvents can be used not only as one kind but also as a mixture of two or more kinds. The amount of the solvent used is the same as described in Production Method 1.
- Examples of the solvent used as the solvent B include aliphatic hydrocarbon solvents such as n-pentane, n-hexane, cyclohexane, n-heptane, n-octane and i-octane. Preferred are n-pentane, n-hexane and n-heptane. More preferred is n-heptane. These solvents can be used not only as one kind but also as a mixture of two or more kinds.
- the amount of solvent B (when using a mixture of two or more solvents) may be 0.01 to 1000 parts by weight, preferably 0.1 to 100 parts by weight, based on the starting material. More preferably, 0.5 to 30 parts by mass can be used, and further preferably 1 to 10 parts by mass can be used.
- a dropping method is desirable. It can be added to the starting material at a rate of 0.001 to 100 parts by mass per hour, preferably added dropwise at a rate of 0.01 to 10 parts by mass per hour, more preferably 1 hour. It can be dropped at a rate of 0.1 to 3 parts by mass per unit.
- the notation “quantitative yield” described in the examples is calculated from the sum of the contents of the Z-form and E-form, and the notation “Z-form / E-form content ratio” is also the Z-form and E-form. It calculated from content of.
- “4.4 mass% hydrogen chloride / ethyl acetate solution” is a solution of ethyl acetate containing 4.4 mass% of hydrogen chloride (manufactured by Fuji Junyaku), “15.5 mass% hydrogen chloride / cyclopentyl methyl ether solution” or “15.5 mass “% Hydrogen chloride / CPME solution” is a cyclopentyl methyl ether solution (made by Watanabe Chemical Co., Ltd.) containing 15.5% by mass of hydrogen chloride.
- Example 1 In accordance with the method described in Production Method 2, hydrogen chloride was used as the acidic compound, and the equivalent amount of hydrogen chloride was changed.
- Example 1-1 (E) -N- [2- [3-Chloro-5- (cyclopropylethynyl) pyridin-2-yl] -2- (isopropoxyimino) ethyl] -3- (difluoromethyl) -1-methyl-1H -Pyrazole-4-carboxamide [hereinafter abbreviated as compound (E) -1.
- Example 1-2 (Z) -N- [2- [3-Chloro-5- (cyclopropylethynyl) pyridin-2-yl] -2- (isopropoxyimino) ethyl] -3- (difluoromethyl) -1-methyl-1H -Pyrazole-4-carboxamide hydrochloride 0.5 (1,2-dichloroethane) solvate [hereinafter abbreviated as Compound (Z) -2.
- Examples 1-3 to 1-9 According to the method described in Examples 1-1 and 1-2, the reaction was carried out by changing the amount of 4.5 mass% hydrogen chloride / ethyl acetate solution used. The amount of ethyl acetate used was adjusted to 2.5 parts by mass with respect to the raw material.
- Equivalent hydrogen chloride referred to as “HCl equivalent” in the table
- quantitative yield of compound (Z) -1 or compound (E) -1 contained in the obtained crystals in the table, “quantitative yield”).
- the purity of the Z and E isomers confirmed by qualitative analysis using HPLC (denoted as purity in the table), and the Z isomer calculated from the results of qualitative analysis using HPLC. And the ratio of E isomers (described as “Z isomer / E isomer ratio” in the table) are shown in Table 2 below.
- Example 2 In accordance with the method described in Production Method 1, the type of solvent, the type of acidic compound, and the equivalent amount were changed.
- CPME cyclopentyl Methyl ether
- Example 3 In accordance with the description in Production Method 1, two types of solvents were used as the solvent, and concentrated sulfuric acid was used as the acidic compound. In the following Examples 3-1 to 3-8, toluene was used as the first solvent A.
- Example 3-1 Method for producing compound (E) -1 using t-butyl methyl ether as second solvent A
- TBME Mass parts of t-butyl methyl ether
- Example 3--7 Method for producing sulfate of compound (Z) -1 using t-butyl methyl ether as second solvent
- Example 3-10 Method for producing sulfate of compound (Z) -1
- Example 4 The type of solvent A and the type of acidic compound were changed according to the method described in the above production method 2.
- Example 4-1 Method for producing compound (E) -1 using cyclopentyl methyl ether as solvent A and hydrogen chloride as acidic compound
- CPME cyclopentyl methyl ether
- EZ hydrogen chloride
- Example 4-2 Method for producing compound (E) -1 using toluene as solvent A and sulfuric acid as acidic compound
- the precipitated crystals were filtered and washed with a mixed solution of 2 parts by mass of toluene and 1 part by mass of n-heptane to obtain a sulfate of compound (Z) -1 as pale yellow crystals.
- the sum of the relative area percentages of the Z-form and E-form peaks was 98.4%.
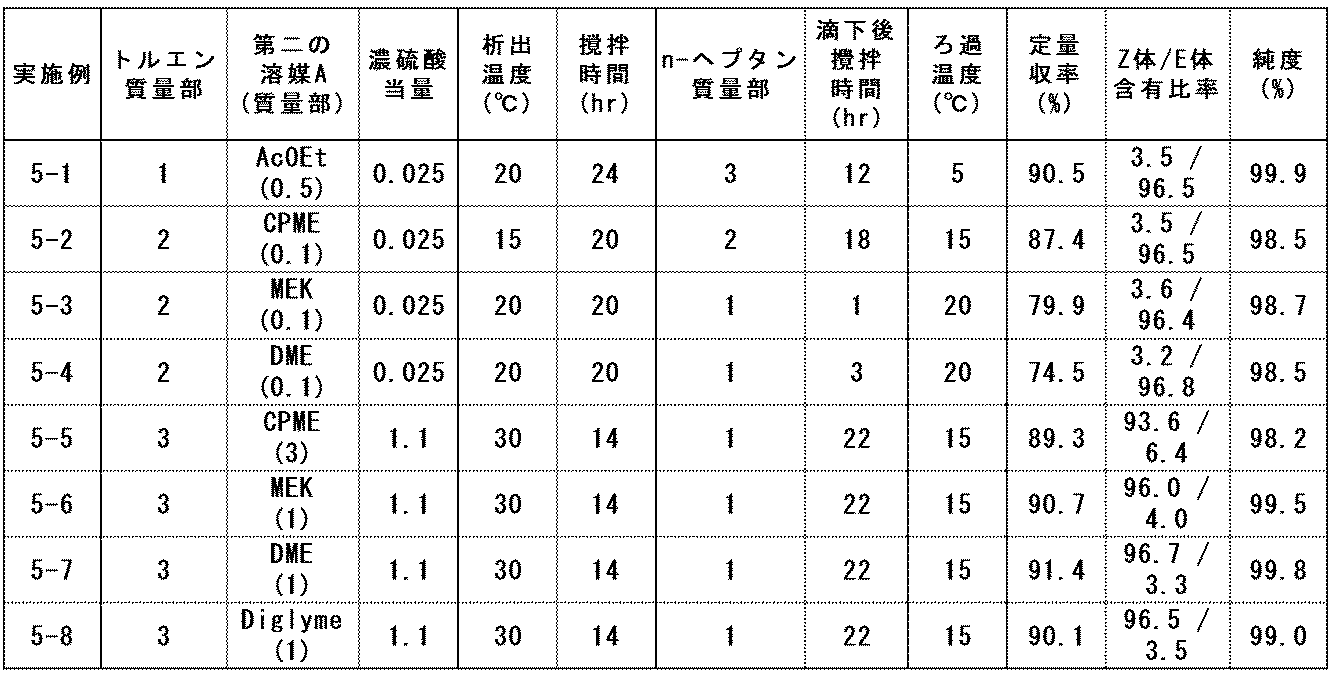
- Example 5 In accordance with the method described in the above production method 2, two solvents were used as the solvent A, and the equivalent amount of the acidic compound was changed. In Examples 5-1 to 5-8 below, one of the solvents A was toluene, n-heptane was used as the solvent B, and concentrated sulfuric acid was used as the acidic compound.
- Example 5-1 Compound (E) -1 Production Method Using Ethyl Acetate as Second Solvent A
- the mixed solution was stirred at 20 ° C. for 24 hours to precipitate crystals.
- 3 parts by mass of n-heptane was added dropwise to the suspension at the same temperature over 6 hours, followed by stirring for 12 hours.
- the suspension was cooled to 5 ° C. and stirred for 2 hours.
- the precipitated crystals were filtered and washed with a mixed solution of 2 parts by mass of toluene and 1 part by mass of n-heptane to obtain pale yellow crystals.
- the sum of the relative area percentages of the Z-form and E-form peaks was 99.9%.
- Example 5-2 to 5-4 were conducted according to the method described in Example 5-1, and Examples 5-6 to 5-8 were conducted according to the method described in Example 5-5. And the equivalent of sulfuric acid was changed.
- the reaction conditions and results are listed in Table 7.
- “second solvent” describes the type of the second solvent A out of the two solvents used as the solvent A, and the mass part of the solvent is described in parentheses.
- “AcOEt” represents ethyl acetate (the same applies hereinafter), and “MEK” represents methyl ethyl ketone.
- Examples 5-9 and 5-10 below use 1,2-dichloroethane and ethyl acetate as solvent A, n-heptane as solvent B, hydrogen chloride as the acidic compound, hydrogen chloride The equivalent weight of was changed.
- Example 5-9 Production method of compound (E) -1
- Example 9 In accordance with the method described in the above production method 1, the kind of solvent, the kind of acidic compound and the equivalent amount were changed, and the reaction was performed using a small amount of the salt of compound (Z) -1 in order to precipitate crystals. .
- Example 9-1 Compound (Z) -1 hydrochloride using CPME as a solvent and hydrogen chloride as an acidic compound [hereinafter abbreviated as compound (Z) -3.
- the compound (EZ) -1 (Z isomer / E isomer 48.8 / 51.2) 2.0 g and 1.45 parts by mass of CPME were mixed at 50 ° C.
- Example 9-2 and 9-3 According to the method described in Example 9-1, the reaction was carried out by changing the type of solvent and the type and equivalent of the acidic compound.
- the reaction conditions and results are listed in Table 9.
- the type of the solvent is described in “Solvent”, and the mass part of the solvent is described in parentheses.
- the kind of acidic compound is described in “Acid compound”, and the equivalent amount of the acidic compound is described in parentheses.
- Example 10 According to the method described in the above production method 2, the type of solvent, the type and equivalent of the acidic compound were changed, and the reaction was performed using a small amount of the salt of compound (Z) -1 in order to precipitate crystals. .
- Example 10-1 and Example 10-2 The reaction conditions and results of Example 10-1 and Example 10-2 are shown in Table 10.
- solvent describes the type of the solvent A, and the mass part of the solvent is described in parentheses.
- the kind of acidic compound used was described in “acidic compound”, and the equivalent amount of acidic compound was described in parentheses.
- Example 11 Method for producing compound (Z) -3 using compound (E) -1 as a raw material
- 7.39 g of a 4.5 mass% hydrogen chloride / ethyl acetate solution (1.14 equivalent as hydrogen chloride) was added, and the mixture was stirred at the same temperature for 2 hours.
- 36 mg of compound (Z) -3 was added to the mixed solution and stirred for 2 hours, crystals were precipitated.
- 14.4 g of n-heptane was added dropwise over 16 hours, followed by stirring at 30 ° C. for 5 hours.
- Example 12 Method for producing compound (Z) -1 using compound (EZ) -1 as a raw material
- 8.93 g of a 4.4 mass% hydrogen chloride / ethyl acetate solution (1.2 equivalents as hydrogen chloride) was added, followed by stirring at the same temperature for 30 minutes.
- 2.0 mg of compound (Z) -3 was added to the mixed solution and stirred for 6 hours, crystals were precipitated.
- 18.2 g of n-heptane was added dropwise over 8 hours, followed by stirring at 25 ° C. for 9 hours.
- the suspension was cooled to 0 ° C. and stirred for 1 hour. After adding 20.2 g of water dropwise to the suspension, 18.2 g of ethyl acetate was added. To the reaction solution, 6.74 g of 8.0 mass% sodium hydroxide aqueous solution was added dropwise at 0 ° C., and the mixture was stirred at the same temperature for 30 minutes. After completion of the stirring, the organic layer was separated at 25 ° C. The obtained organic layer was washed with 12.1 g of water, and the solvent was distilled off under reduced pressure. After the obtained residue and 12.1 g of toluene were mixed, the solvent was distilled off under reduced pressure.
- Example 13 Method for producing compound (Z) -1 using compound (EZ) -1 as a raw material
- the mixed solution was cooled to 40 ° C., 5.0 mg of compound (Z) -1 was added at the same temperature, and the mixture was stirred for 30 minutes to precipitate crystals.
- the suspension was cooled to 0 ° C. and stirred for 13 hours.
- the precipitated crystals were filtered, and the obtained crystals were washed with a mixed solution of 1 part by mass of toluene and 0.5 parts by mass of n-heptane to obtain 3.96 g of compound (Z) -1 as pale yellow crystals. (Yield 78.6%).
- the area ratio of the Z body and the E body was 97.1 / 2.9, and the sum of the relative area percentages of the two peaks was 99.2%.
- Example 14 The reaction was carried out according to the method according to Example 1, and the precipitated crystals were filtered, and then the oxyimino compound in the obtained filtrate was used as a starting material.
- the peak with a retention time of 10.8 minutes is the Z-form used as a raw material, and the peak with a retention time of 12.1 minutes is confirmed to be the E-form from the results of mass spectrometry by LC / MS and the 1 H-NMR measurement results did. From the above, in the qualitative analysis using HPLC, it can be seen that the retention time of the Z form is 10.8 minutes and the retention time of the E form is 12.1 minutes.
- the sum of the relative area percentages of the Z-form and E-form peaks was 99.8%.
- the obtained crystals (50 mg) and methanol (5.0 mL) were mixed at room temperature.
- 0.005 mol / L sodium hydroxide aqueous solution was added.
- the content of oxalate ion was 17.5% by mass and the molar ratio of compound (Z) -1 to sulfuric acid was 1: 1.
- FIG. 2 shows a chart of the obtained powder X-ray diffraction spectrum. The following peak values were obtained as characteristic peaks in powder X-ray diffraction. The error of the peak value of powder X-ray diffraction can usually be ⁇ 0.2.
- Ethyl acetate was added to the obtained residue at room temperature to prepare 420 g of an ethyl acetate solution.
- 20 mL of a 4.5 mass% hydrogen chloride / ethyl acetate solution (0.056 equivalents as hydrogen chloride) was added at 35 ° C.
- 252 g of n-heptane was added dropwise to the reaction solution at the same temperature over 1 hour, crystal precipitation was observed.
- the reaction mixture was stirred at the same temperature for 1 hour.
- 252 g of n-heptane was added dropwise over 1 hour, and the reaction mixture was stirred for 1 hour.
- the method for producing a geometric isomer according to the present invention is extremely useful as an efficient method for producing an oximino compound useful as a pharmaceutical or agrochemical.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
医薬・農薬として有用なオキシイミノ化合物のE体及びZ体の幾何異性体を作り分ける新規な製造方法の提供。 式(EZ)-1で表されるオキシイミノ化合物の幾何異性体の混合物及び酸性化合物を混合することにより、式(E)-1で表されるオキシイミノ化合物のE体、又は式(Z)-1で表されるオキシイミノ化合物のZ体を高収率かつ高立体選択的に製造する方法。
Description
本発明は、医農薬として有用なオキシイミノ化合物の幾何異性体を製造する方法に関するものである。
ある種のオキシイミノ化合物は農薬や医薬として有用な化合物であることが知られている(特許文献1乃至5参照)。一般的にオキシイミノ化合物は、オキシイミノ基を有することから、Eの立体配置の化合物[以下、E体と略称する。]とZの立体配置の化合物[以下、Z体と略称する。]の2種類の幾何異性体が存在し、E体からZ体へ、Z体からE体へ又はそれぞれよりE体とZ体の混合物へ変換する方法が知られている。例えば、光を照射することによるE体よりZ体を製造する方法(特許文献1及び2参照。)、酸性化合物を用いることによるE体よりZ体を製造する方法(特許文献6参照。)、酸性化合物を用いることによるZ体よりE体を製造する方法(特許文献4、5、7及び8参照。)及び、光を照射することによるE体よりE体及びZ体の混合物を製造する方法(特許文献3参照。)が知られている。
通常オキシイミノ化合物を製造する場合、E体及びZ体との混合物として得られる。E体及びZ体は、お互いに非常に近い構造である為、例えばカラムクロマトグラフィーなどの通常の精製操作により、E体及びZ体の混合物から、E体のみ又はZ体のみを選択的に低減することは非常に困難である。また、一方の幾何異性体を医薬、農薬等として使用する場合、もう一方の幾何異性体は不純物として製品に混入することとなり、製品の品質及び性能に悪影響を及ぼす原因となる。
従って、望まない幾何異性体から望む幾何異性体を製造する新規なオキシイミノ化合物を製造する方法の開発が望まれている。
従って、望まない幾何異性体から望む幾何異性体を製造する新規なオキシイミノ化合物を製造する方法の開発が望まれている。
本発明者は上記の課題を解決すべく鋭意検討した結果、E体及びZ体の混合物である式(EZ)-1で表されるオキシイミノ化合物に、酸性化合物を作用させ異性化反応を行う際、用いる酸性化合物の添加量を制御することにより、E体及びZ体を作り分けられることを見出し、本発明を完成させた。
すなわち、本発明は下記〔1〕乃至〔11〕に記載の発明である。
すなわち、本発明は下記〔1〕乃至〔11〕に記載の発明である。
〔1〕
式(EZ)-1;

で表されるオキシイミノ化合物の幾何異性体の混合物を出発物質として式(E)-1;

で表されるオキシイミノ化合物のE体若しくは式(Z)-1;

で表されるオキシイミノ化合物のZ体を製造する、上記式(E)-1で表されるオキシイミノ化合物のE体を出発物質として上記式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、又は、上記式(Z)-1で表されるオキシイミノ化合物のZ体を出発物質として上記式(E)-1で表されるオキシイミノ化合物のE体を製造する、オキシイミノ化合物の立体選択的な製造方法であって、
(i)式(EZ)-1で表される化合物の幾何異性体の混合物又は式(Z)-1で表されるオキシイミノ化合物のZ体を出発物質とし、溶媒中で該出発物質及び該出発物質に対して0.1当量以下の酸性化合物を混合して、式(E)-1で表されるオキシイミノ化合物のE体を製造するか、又は
(ii)式(EZ)-1で表される化合物の幾何異性体の混合物又は式(E)-1で表されるオキシイミノ化合物のE体を出発物質とし、溶媒中で該出発物質及び該出発物質に対して0.7当量以上の酸性化合物を混合して、式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、
ことを特徴とするオキシイミノ化合物の立体選択的な製造方法。
式(EZ)-1;
で表されるオキシイミノ化合物の幾何異性体の混合物を出発物質として式(E)-1;
で表されるオキシイミノ化合物のE体若しくは式(Z)-1;
で表されるオキシイミノ化合物のZ体を製造する、上記式(E)-1で表されるオキシイミノ化合物のE体を出発物質として上記式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、又は、上記式(Z)-1で表されるオキシイミノ化合物のZ体を出発物質として上記式(E)-1で表されるオキシイミノ化合物のE体を製造する、オキシイミノ化合物の立体選択的な製造方法であって、
(i)式(EZ)-1で表される化合物の幾何異性体の混合物又は式(Z)-1で表されるオキシイミノ化合物のZ体を出発物質とし、溶媒中で該出発物質及び該出発物質に対して0.1当量以下の酸性化合物を混合して、式(E)-1で表されるオキシイミノ化合物のE体を製造するか、又は
(ii)式(EZ)-1で表される化合物の幾何異性体の混合物又は式(E)-1で表されるオキシイミノ化合物のE体を出発物質とし、溶媒中で該出発物質及び該出発物質に対して0.7当量以上の酸性化合物を混合して、式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、
ことを特徴とするオキシイミノ化合物の立体選択的な製造方法。
〔2〕
出発物質が、式(EZ)-1で表される化合物の幾何異性体の混合物である、上記〔1〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔3〕
出発物質を溶媒に溶解させた後、酸性化合物を添加する、上記〔1〕又は〔2〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔4〕
出発物質に対して0.01当量以上0.07当量以下の酸性化合物を用いて、式(E)-1で表されるオキシイミノ化合物のE体を製造する、上記〔1〕乃至〔3〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔5〕
出発物質に対して1.0当量以上2.0当量以下の酸性化合物を用いて、式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、上記〔1〕乃至〔3〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
出発物質が、式(EZ)-1で表される化合物の幾何異性体の混合物である、上記〔1〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔3〕
出発物質を溶媒に溶解させた後、酸性化合物を添加する、上記〔1〕又は〔2〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔4〕
出発物質に対して0.01当量以上0.07当量以下の酸性化合物を用いて、式(E)-1で表されるオキシイミノ化合物のE体を製造する、上記〔1〕乃至〔3〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔5〕
出発物質に対して1.0当量以上2.0当量以下の酸性化合物を用いて、式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、上記〔1〕乃至〔3〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔6〕
酸性化合物が、ハロゲン化水素、硫酸又はメタンスルホン酸である、上記〔1〕乃至〔5〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔7〕
溶媒として、芳香族炭化水素溶媒、エーテル溶媒、ケトン溶媒、エステル溶媒及びハロゲン化炭化水素溶媒からなる群から選ばれる1種以上での溶媒を用いる、上記〔1〕乃至〔6〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔8〕
溶媒として、トルエン、オルトキシレン、シクロペンチルメチルエーテル、ターシャリーブチルメチルエーテル、ジメトキシエタン、ジエチレングリコールジメチルエーテル、メチルエチルケトン、酢酸エチル及び1,2-ジクロロエタンからなる群から選ばれる1種以上の溶媒を用いる、上記〔7〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔9〕
出発物質及び酸性化合物を溶媒中で混合した後、脂肪族炭化水素溶媒から選ばれる1種以上の溶媒を添加する工程を含む、上記〔1〕乃至〔8〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔10〕
脂肪族炭化水素溶媒がノルマルヘプタンである、上記〔9〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔11〕
析出した、結晶、塩若しくはその溶媒和物、又は溶媒和物を反応系から分離する、上記〔1〕乃至〔10〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
酸性化合物が、ハロゲン化水素、硫酸又はメタンスルホン酸である、上記〔1〕乃至〔5〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔7〕
溶媒として、芳香族炭化水素溶媒、エーテル溶媒、ケトン溶媒、エステル溶媒及びハロゲン化炭化水素溶媒からなる群から選ばれる1種以上での溶媒を用いる、上記〔1〕乃至〔6〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔8〕
溶媒として、トルエン、オルトキシレン、シクロペンチルメチルエーテル、ターシャリーブチルメチルエーテル、ジメトキシエタン、ジエチレングリコールジメチルエーテル、メチルエチルケトン、酢酸エチル及び1,2-ジクロロエタンからなる群から選ばれる1種以上の溶媒を用いる、上記〔7〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔9〕
出発物質及び酸性化合物を溶媒中で混合した後、脂肪族炭化水素溶媒から選ばれる1種以上の溶媒を添加する工程を含む、上記〔1〕乃至〔8〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
〔10〕
脂肪族炭化水素溶媒がノルマルヘプタンである、上記〔9〕に記載のオキシイミノ化合物の立体選択的な製造方法。
〔11〕
析出した、結晶、塩若しくはその溶媒和物、又は溶媒和物を反応系から分離する、上記〔1〕乃至〔10〕のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
本発明によれば、医薬・農薬として有用なオキシイミノ化合物を、高収率且つ高立体選択的に製造することができ、工業的生産に有益な製造方法を提供することができる。
以下、本発明について詳細に説明する。
本発明に包含されるオキシイミノ化合物には、オキシイミノ基の存在に起因する2種類の立体配置を有する異性体が存在する。本明細書においては2種類の異性体を各々、オキシイミノ基がEの立体配置を有する異性体をE体、オキシイミノ基がZの立体配置を有する異性体をZ体と表す。
また、本発明に包含される前記E体の幾何異性体の混合比は、E体/Z体=90/10~100/0であり、本発明に包含される前記Z体の幾何異性体の混合比は、E体/Z体=10/90~0/100であり、本発明に包含される前記E体と前記Z体の混合物はE体/Z体=10/90を超えかつ90/10未満である。E体及びZ体の混合比は、高速液体クロマトグラフィー、ガスクロマトグラフィー、核磁気共鳴スペクトル等の任意の測定方法によって定性分析、算出することができる。
本発明に包含されるオキシイミノ化合物には、オキシイミノ基の存在に起因する2種類の立体配置を有する異性体が存在する。本明細書においては2種類の異性体を各々、オキシイミノ基がEの立体配置を有する異性体をE体、オキシイミノ基がZの立体配置を有する異性体をZ体と表す。
また、本発明に包含される前記E体の幾何異性体の混合比は、E体/Z体=90/10~100/0であり、本発明に包含される前記Z体の幾何異性体の混合比は、E体/Z体=10/90~0/100であり、本発明に包含される前記E体と前記Z体の混合物はE体/Z体=10/90を超えかつ90/10未満である。E体及びZ体の混合比は、高速液体クロマトグラフィー、ガスクロマトグラフィー、核磁気共鳴スペクトル等の任意の測定方法によって定性分析、算出することができる。
本明細書における式[例えば、式(EZ)-1]の記載においては、オキシイミノ基の波線の結合は、E体及びZ体の幾何異性体の混合物であることを意味する。

本明細書において、n-はノルマル、i-はイソ、s-はセカンダリー、及びtert-又はt-はターシャリーを各々意味し、o-はオルト、m-はメタ、p-はパラを各々意味する。
本明細書において、塩基性化合物としては、無機塩基や有機塩基が挙げられる。
無機塩基としては、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化マグネシウム、水酸化カルシウム、水酸化バリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素セシウム、炭酸水素アンモニウム、酢酸ナトリウム、酢酸カリウム、酢酸セシウム、酢酸カルシウム、酢酸バリウム、リン酸ナトリウム、リン酸カリウム、リン酸水素二ナトリウム、リン酸水素二カリウム等が挙げられる。
有機塩基としては、例えば、アンモニア、エチルアミン、ジエチルアミン、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、ピリジン、2-メチルピリジン、3-メチルピリジン、4-メチルピリジン、2,3-ルチジン、2,4-ルチジン、2,6-ルチジン、3,5-ルチジン、ジアザビシクロウンデセン、1,4-ジアザビシクロ[2.2.2]オクタン、1,1,3,3-テトラメチルグアニジン等が挙げられる。
本明細書において、塩基性化合物としては、無機塩基や有機塩基が挙げられる。
無機塩基としては、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化マグネシウム、水酸化カルシウム、水酸化バリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素セシウム、炭酸水素アンモニウム、酢酸ナトリウム、酢酸カリウム、酢酸セシウム、酢酸カルシウム、酢酸バリウム、リン酸ナトリウム、リン酸カリウム、リン酸水素二ナトリウム、リン酸水素二カリウム等が挙げられる。
有機塩基としては、例えば、アンモニア、エチルアミン、ジエチルアミン、トリエチルアミン、ジイソプロピルエチルアミン、トリブチルアミン、ピリジン、2-メチルピリジン、3-メチルピリジン、4-メチルピリジン、2,3-ルチジン、2,4-ルチジン、2,6-ルチジン、3,5-ルチジン、ジアザビシクロウンデセン、1,4-ジアザビシクロ[2.2.2]オクタン、1,1,3,3-テトラメチルグアニジン等が挙げられる。
次に本発明の、式(E)-1で表される化合物[以下、化合物(E)-1と略称する。]を式(EZ)-1で表される化合物[以下、混合物(EZ)-1と略称する。]より製造する方法、式(Z)-1で表される化合物[以下、化合物(Z)-1と略称する。]を混合物(EZ)-1より製造する方法、化合物(E)-1を化合物(Z)-1より製造する方法、及び、化合物(Z)-1を化合物(E)-1より製造する方法について詳細に説明する。
〔製造方法1〕
反応式1

反応式1
混合物(EZ)-1と酸性化合物とを溶媒(以下、溶媒Aと略称する。)中で混合し、反応させた後、目的物を結晶として析出させることにより得ることができる。この際、使用する酸性化合物の量を制御することにより、化合物(E)-1又は化合物(Z)-1のいずれか一方の幾何異性体を選択的に製造することができる。同様に、使用する酸性化合物の量を制御することにより、化合物(Z)-1から化合物(E)-1を選択的に製造することができ、また、化合物(E)-1から化合物(Z)-1を選択的に製造することができる。
化合物(E)-1を製造する場合に使用する酸性化合物の量は、出発物質[混合物(EZ)-1又は化合物(Z)-1]に対して0.00001~0.1当量を用いることができ、好ましくは0.001~0.1当量を用いることができ、より好ましくは0.01~0.07当量を用いることができる。
化合物(Z)-1を製造する場合に使用する酸性化合物の量は、出発物質[混合物(EZ)-1又は化合物(E)-1]に対して0.7~10当量を用いることができ、好ましくは0.7~5当量を用いることができ、より好ましくは0.7~2当量を用いることができる。
化合物(E)-1を製造する場合に使用する酸性化合物の量は、出発物質[混合物(EZ)-1又は化合物(Z)-1]に対して0.00001~0.1当量を用いることができ、好ましくは0.001~0.1当量を用いることができ、より好ましくは0.01~0.07当量を用いることができる。
化合物(Z)-1を製造する場合に使用する酸性化合物の量は、出発物質[混合物(EZ)-1又は化合物(E)-1]に対して0.7~10当量を用いることができ、好ましくは0.7~5当量を用いることができ、より好ましくは0.7~2当量を用いることができる。
使用する酸性化合物としては、ハロゲン化水素、無機酸、カルボン酸、スルホン酸等が挙げられる。
ハロゲン化水素としては、例えば、フッ化水素、塩化水素、臭化水素、ヨウ化水素等が挙げられる。
無機酸としては、例えば、硝酸、硫酸、リン酸、塩素酸、ホウ酸、過塩素酸等が挙げられる。
カルボン酸としては、例えば、ギ酸、酢酸、トリフルオロ酢酸、トリクロロ酢酸、プロピオン酸、酪酸、吉草酸、ピバル酸、イソ吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリル酸、ラウリル酸、ミリスチン酸、パルミチン酸、ステアリン酸、アクリル酸、メタクリル酸、チグリン酸、オレイン酸、リノレン酸、リノール酸、アラキドン酸、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、フマル酸、マレイン酸、リンゴ酸、乳酸、アスコルビン酸、クエン酸、マンデル酸、酒石酸、ピルビン酸、安息香酸、o-メチル安息香酸、m-メチル安息香酸、p-メチル安息香酸、o-トリフルオロメチル安息香酸、m-トリフルオロメチル安息香酸、p-トリフルオロメチル安息香酸、o-クロロ安息香酸、m-クロロ安息香酸、p-クロロ安息香酸、o-ニトロ安息香酸、m-ニトロ安息香酸、p-ニトロ安息香酸、フタル酸、イソフタル酸、テレフタル酸、サリチル酸、没食子酸、メリト酸、ケイ皮酸等が挙げられる。
スルホン酸としては、例えば、メタンスルホン酸、エタンスルホン酸、10-カンファースルホン酸、トリフルオロメタンスルホン酸、ベンゼンスルホン酸、o-トルエンスルホン酸、m-トルエンスルホン酸、p-トルエンスルホン酸、o-トリフルオロメチルベンゼンスルホン酸、m-トリフルオロメチルベンゼンスルホン酸、p-トリフルオロメチルベンゼンスルホン酸、o-クロロベンゼンスルホン酸、m-クロロベンゼンスルホン酸、p-クロロベンゼンスルホン酸、o-ニトロベンゼンスルホン酸、m-ニトロベンゼンスルホン酸、p-ニトロベンゼンスルホン酸等が挙げられる。
酸性化合物としては、好ましくはハロゲン化水素、硫酸及びメタンスルホン酸が挙げられ、より好ましくは塩化水素、臭化水素及び硫酸が挙げられる。
これらの酸性化合物は水を含んでいてもよい。また、これらの酸性化合物は2種以上を混合して使用することもできる。
ハロゲン化水素としては、例えば、フッ化水素、塩化水素、臭化水素、ヨウ化水素等が挙げられる。
無機酸としては、例えば、硝酸、硫酸、リン酸、塩素酸、ホウ酸、過塩素酸等が挙げられる。
カルボン酸としては、例えば、ギ酸、酢酸、トリフルオロ酢酸、トリクロロ酢酸、プロピオン酸、酪酸、吉草酸、ピバル酸、イソ吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリル酸、ラウリル酸、ミリスチン酸、パルミチン酸、ステアリン酸、アクリル酸、メタクリル酸、チグリン酸、オレイン酸、リノレン酸、リノール酸、アラキドン酸、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、フマル酸、マレイン酸、リンゴ酸、乳酸、アスコルビン酸、クエン酸、マンデル酸、酒石酸、ピルビン酸、安息香酸、o-メチル安息香酸、m-メチル安息香酸、p-メチル安息香酸、o-トリフルオロメチル安息香酸、m-トリフルオロメチル安息香酸、p-トリフルオロメチル安息香酸、o-クロロ安息香酸、m-クロロ安息香酸、p-クロロ安息香酸、o-ニトロ安息香酸、m-ニトロ安息香酸、p-ニトロ安息香酸、フタル酸、イソフタル酸、テレフタル酸、サリチル酸、没食子酸、メリト酸、ケイ皮酸等が挙げられる。
スルホン酸としては、例えば、メタンスルホン酸、エタンスルホン酸、10-カンファースルホン酸、トリフルオロメタンスルホン酸、ベンゼンスルホン酸、o-トルエンスルホン酸、m-トルエンスルホン酸、p-トルエンスルホン酸、o-トリフルオロメチルベンゼンスルホン酸、m-トリフルオロメチルベンゼンスルホン酸、p-トリフルオロメチルベンゼンスルホン酸、o-クロロベンゼンスルホン酸、m-クロロベンゼンスルホン酸、p-クロロベンゼンスルホン酸、o-ニトロベンゼンスルホン酸、m-ニトロベンゼンスルホン酸、p-ニトロベンゼンスルホン酸等が挙げられる。
酸性化合物としては、好ましくはハロゲン化水素、硫酸及びメタンスルホン酸が挙げられ、より好ましくは塩化水素、臭化水素及び硫酸が挙げられる。
これらの酸性化合物は水を含んでいてもよい。また、これらの酸性化合物は2種以上を混合して使用することもできる。
本発明で使用する混合物(EZ)-1は、特開2016-011286号(JP2016-011286)に記載の既知の方法に準じて合成することができ、E体とZ体との混合比は前記の比率の範囲であればよい。
溶媒Aとして使用する溶媒としては、芳香族炭化水素溶媒、ハロゲン化炭化水素溶媒、アルコール溶媒、エーテル溶媒、エステル溶媒、アミド溶媒、ニトリル溶媒、ケトン溶媒、ジメチルスルホキシド等が挙げられる。
芳香族炭化水素溶媒としては、例えば、ベンゼン、トルエン、o-キシレン、m-キシレン、p-キシレン、クロロベンゼン、1,2-ジクロロベンゼン、1,3-ジクロロベンゼン、1,4-ジクロロベンゼン、ニトロベンゼン等が挙げられる。ハロゲン化炭化水素溶媒としては、例えば、クロロホルム、ジクロロメタン、ジクロロエタン等が挙げられる。アルコール溶媒としては、例えば、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、t-ブチルアルコール等が挙げられる。エーテル溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、シクロペンチルメチルエーテル、ターシャリーブチルメチルエーテル、1,4-ジオキサン、ジメトキシエタン、ジエチレングリコールジメチルエーテル等が挙げられる。エステル溶媒としては、例えば、酢酸メチル、酢酸エチル、酢酸ブチル、プロピオン酸メチル等が挙げられる。アミド溶媒としては、例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン等が挙げられる。ニトリル溶媒としては、例えば、アセトニトリル、プロピオニトリル等が挙げられ、ケトン溶媒としては、例えば、メチルエチルケトン、メチルイソブチルケトン等が挙げられる。
溶媒Aとしては、好ましくは、芳香族炭化水素溶媒、ハロゲン化炭化水素溶媒、エーテル溶媒、エステル溶媒、ケトン溶媒が挙げられる。より好ましくは、トルエン、o-キシレン、1,2-ジクロロエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル、ジメトキシエタン、ジエチレングリコールジメチルエーテル、酢酸エチル、メチルエチルケトンが挙げられる。さらに好ましくは1,2-ジクロロエタン、酢酸エチルが挙げられる。
これらの溶媒は1種で使用するだけでなく、2種以上を混合して使用することもできる。
芳香族炭化水素溶媒としては、例えば、ベンゼン、トルエン、o-キシレン、m-キシレン、p-キシレン、クロロベンゼン、1,2-ジクロロベンゼン、1,3-ジクロロベンゼン、1,4-ジクロロベンゼン、ニトロベンゼン等が挙げられる。ハロゲン化炭化水素溶媒としては、例えば、クロロホルム、ジクロロメタン、ジクロロエタン等が挙げられる。アルコール溶媒としては、例えば、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、t-ブチルアルコール等が挙げられる。エーテル溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、シクロペンチルメチルエーテル、ターシャリーブチルメチルエーテル、1,4-ジオキサン、ジメトキシエタン、ジエチレングリコールジメチルエーテル等が挙げられる。エステル溶媒としては、例えば、酢酸メチル、酢酸エチル、酢酸ブチル、プロピオン酸メチル等が挙げられる。アミド溶媒としては、例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン等が挙げられる。ニトリル溶媒としては、例えば、アセトニトリル、プロピオニトリル等が挙げられ、ケトン溶媒としては、例えば、メチルエチルケトン、メチルイソブチルケトン等が挙げられる。
溶媒Aとしては、好ましくは、芳香族炭化水素溶媒、ハロゲン化炭化水素溶媒、エーテル溶媒、エステル溶媒、ケトン溶媒が挙げられる。より好ましくは、トルエン、o-キシレン、1,2-ジクロロエタン、シクロペンチルメチルエーテル、t-ブチルメチルエーテル、ジメトキシエタン、ジエチレングリコールジメチルエーテル、酢酸エチル、メチルエチルケトンが挙げられる。さらに好ましくは1,2-ジクロロエタン、酢酸エチルが挙げられる。
これらの溶媒は1種で使用するだけでなく、2種以上を混合して使用することもできる。
使用する溶媒の量(溶媒を2種以上を混合して使用する場合はそれらの総和量)は、出発物質に対して0.01~100質量部を用いることができ、好ましくは0.1~50質量部を用いることができ、より好ましくは0.5~20質量部を用いることができ、更に好ましくは1~10質量部を用いることができる。
出発物質及び酸性化合物を溶媒中で混合し、反応させる時の反応温度は、通常-20~150℃であり、好ましくは0~100℃、より好ましくは10~60℃である。また、結晶を析出させる時の温度は、通常-20~150℃であり、好ましくは-10~80℃、より好ましくは10~50℃である。
出発物質及び酸性化合物を溶媒中で混合し、反応させる時の反応時間は、1分~1000時間、好ましくは5分~100時間、より好ましくは10分~48時間である。また、結晶を析出させる時の時間は、反応基質の濃度、反応温度によって変化するが、通常1分~1000時間、好ましくは5分~500時間、より好ましくは10分~100時間である。
出発物質及び酸性化合物を溶媒中で混合し、反応させる時の反応温度は、通常-20~150℃であり、好ましくは0~100℃、より好ましくは10~60℃である。また、結晶を析出させる時の温度は、通常-20~150℃であり、好ましくは-10~80℃、より好ましくは10~50℃である。
出発物質及び酸性化合物を溶媒中で混合し、反応させる時の反応時間は、1分~1000時間、好ましくは5分~100時間、より好ましくは10分~48時間である。また、結晶を析出させる時の時間は、反応基質の濃度、反応温度によって変化するが、通常1分~1000時間、好ましくは5分~500時間、より好ましくは10分~100時間である。
反応後の処理方法は特に制限はないが、析出した結晶をろ過することにより反応系から目的物を分離して、目的物を得ることができる。得られたろ液中のオキシイミノ化合物は、必要に応じて濃縮した後、本発明の製造方法の出発物質として使用することができる。得られた結晶は、必要に応じて再結晶等の任意の精製方法によって精製することにより、より高い純度の目的物を得ることができる。
反応により生成した目的物は、場合によっては反応に用いた酸性化合物との塩として得られる。塩として得られた場合にも、上記と同様の方法により得ることができるが、必要に応じて水、次いで有機溶媒を添加し、必要に応じて有機溶媒に溶解させた後、例えば水酸化ナトリウム又はその水溶液等の塩基性化合物を添加することにより中和してもよい。中和後、必要に応じて水を添加、分液、必要に応じて濃縮等の通常の後処理を行ない、目的物を得ることができる。
反応により生成した目的物は、場合によっては反応に用いた溶媒と溶媒和することにより、溶媒を含む和物(以下、溶媒和物と略称する。)として得られる。溶媒和物として得られた場合にも、上記と同様の方法により得ることができるが、必要に応じて得られた溶媒和物を溶媒和しない溶媒に溶解又は懸濁させた後、必要に応じて水を添加、分液、必要に応じて濃縮等の通常の後処理を行ない、目的物を得ることができる。
反応により生成した目的物は、場合によっては反応に用いた酸性化合物及び溶媒により、目的物の塩の溶媒和物として得られる。この場合にも、上記と同様の方法により得ることができるが、必要に応じて上記の後処理を組み合わせることにより、目的物を得ることができる。
反応により生成した目的物は、場合によっては反応に用いた酸性化合物との塩として得られる。塩として得られた場合にも、上記と同様の方法により得ることができるが、必要に応じて水、次いで有機溶媒を添加し、必要に応じて有機溶媒に溶解させた後、例えば水酸化ナトリウム又はその水溶液等の塩基性化合物を添加することにより中和してもよい。中和後、必要に応じて水を添加、分液、必要に応じて濃縮等の通常の後処理を行ない、目的物を得ることができる。
反応により生成した目的物は、場合によっては反応に用いた溶媒と溶媒和することにより、溶媒を含む和物(以下、溶媒和物と略称する。)として得られる。溶媒和物として得られた場合にも、上記と同様の方法により得ることができるが、必要に応じて得られた溶媒和物を溶媒和しない溶媒に溶解又は懸濁させた後、必要に応じて水を添加、分液、必要に応じて濃縮等の通常の後処理を行ない、目的物を得ることができる。
反応により生成した目的物は、場合によっては反応に用いた酸性化合物及び溶媒により、目的物の塩の溶媒和物として得られる。この場合にも、上記と同様の方法により得ることができるが、必要に応じて上記の後処理を組み合わせることにより、目的物を得ることができる。
〔製造方法2〕
反応式2

反応式2
混合物(EZ)-1を溶媒(以下、溶媒Aと略称する。)中、酸性化合物の存在下で反応させた後、更に溶媒(以下、溶媒Bと略称する。)を添加することにより、目的物を結晶として得ることができる。この際、使用する酸性化合物の量を制御することにより、化合物(E)-1又は化合物(Z)-1のいずれか一方の幾何異性体を選択的に製造することができる。同様に、使用する酸性化合物の量を制御することにより、化合物(Z)-1から化合物(E)-1を選択的に製造することができ、また、化合物(E)-1から化合物(Z)-1を選択的に製造することができる。
使用する酸性化合物の量及び種類としては、製造方法1の記載と同様である。
使用する酸性化合物の量及び種類としては、製造方法1の記載と同様である。
溶媒Aとして使用する溶媒としては、例えば、製造方法1に記載した溶媒が挙げられる。これらの溶媒は1種で使用するだけでなく、2種以上を混合して使用することもできる。また、使用する溶媒の量は製造方法1の記載と同様である。
溶媒Bとして使用する溶媒は、例えば、n-ペンタン、n-ヘキサン、シクロヘキサン、n-ヘプタン、n-オクタン、i-オクタン等の脂肪族炭化水素溶媒が挙げられる。好ましくはn-ペンタン、n-ヘキサン、n-ヘプタンが挙げられる。より好ましくはn-ヘプタンが挙げられる。これらの溶媒は1種で使用するだけでなく、2種以上を混合して使用することもできる。
溶媒Bの量(溶媒を2種以上を混合して使用する場合はそれらの総和量)は、出発物質に対して0.01~1000質量部を用いることができ、好ましくは0.1~100質量部を用いることができ、より好ましくは0.5~30質量部を用いることができ、更に好ましくは1~10質量部を用いることができる。
溶媒Bの添加方法としては、滴下する方法が望ましい。出発物質に対して1時間あたり0.001質量部~100質量部の速度で添加することができ、好ましくは1時間あたり0.01質量部~10質量部の速度で滴下することができ、より好ましくは1時間あたり0.1質量部~3質量部の速度で滴下することができる。
溶媒Bとして使用する溶媒は、例えば、n-ペンタン、n-ヘキサン、シクロヘキサン、n-ヘプタン、n-オクタン、i-オクタン等の脂肪族炭化水素溶媒が挙げられる。好ましくはn-ペンタン、n-ヘキサン、n-ヘプタンが挙げられる。より好ましくはn-ヘプタンが挙げられる。これらの溶媒は1種で使用するだけでなく、2種以上を混合して使用することもできる。
溶媒Bの量(溶媒を2種以上を混合して使用する場合はそれらの総和量)は、出発物質に対して0.01~1000質量部を用いることができ、好ましくは0.1~100質量部を用いることができ、より好ましくは0.5~30質量部を用いることができ、更に好ましくは1~10質量部を用いることができる。
溶媒Bの添加方法としては、滴下する方法が望ましい。出発物質に対して1時間あたり0.001質量部~100質量部の速度で添加することができ、好ましくは1時間あたり0.01質量部~10質量部の速度で滴下することができ、より好ましくは1時間あたり0.1質量部~3質量部の速度で滴下することができる。
反応後の処理方法は特に制限はなく、製造方法1に記載した処理方法により、より高い純度の目的物を得ることができる。
反応により生成した目的物は、製造方法1の記載と同様に、場合によっては反応に用いた酸性化合物との塩として、場合によっては反応に用いた溶媒との溶媒和物として、場合によっては反応に用いた酸性化合物及び溶媒により塩の溶媒和物として得られる。必要に応じて、製造方法1に記載の後処理を行うことにより、目的物を得ることができる。
本発明の製造法を用いることにより、オキシイミノ化合物の工業的生産に有益な製造方法を提供することができる。工業的生産の観点では高収率や高立体選択的の観点は重要であるが、例えば副生物の低減、反応時間の短縮、ろ過などの簡便な操作で目的物が得られるなどの観点も重要である。
反応により生成した目的物は、製造方法1の記載と同様に、場合によっては反応に用いた酸性化合物との塩として、場合によっては反応に用いた溶媒との溶媒和物として、場合によっては反応に用いた酸性化合物及び溶媒により塩の溶媒和物として得られる。必要に応じて、製造方法1に記載の後処理を行うことにより、目的物を得ることができる。
本発明の製造法を用いることにより、オキシイミノ化合物の工業的生産に有益な製造方法を提供することができる。工業的生産の観点では高収率や高立体選択的の観点は重要であるが、例えば副生物の低減、反応時間の短縮、ろ過などの簡便な操作で目的物が得られるなどの観点も重要である。
以下に本発明の合成例を実施例として具体的に述べることで、本発明をさらに詳しく説明するが、本発明はこれらによって限定されるものではない。
実施例及び参考例に記載の1H-NMRとの表記はプロトン核磁気共鳴スペクトルを表し、以下同様に、HPLCは高速液体クロマトグラフィーを、LC/MSは液体クロマトグラフィー質量分析計を、ICはイオンクロマトグラフィーを、Rtは保持時間を表す。
1H-NMRは、基準物質としてテトラメチルシランを用い、重ジメチルスルフィド溶媒中で、300MHzにて測定した。また、後述の1H-NMRデータの記号は、下記の意味を表す。
s:シングレット、d:ダブレット、t:トリプレット、sep:セプテット、m:マルチプレット。
実施例及び参考例に記載の1H-NMRとの表記はプロトン核磁気共鳴スペクトルを表し、以下同様に、HPLCは高速液体クロマトグラフィーを、LC/MSは液体クロマトグラフィー質量分析計を、ICはイオンクロマトグラフィーを、Rtは保持時間を表す。
1H-NMRは、基準物質としてテトラメチルシランを用い、重ジメチルスルフィド溶媒中で、300MHzにて測定した。また、後述の1H-NMRデータの記号は、下記の意味を表す。
s:シングレット、d:ダブレット、t:トリプレット、sep:セプテット、m:マルチプレット。
〔定性分析〕
HPLCを用いた定性分析は、以下に記載する測定条件を用いて実施した。
カラム:Inertsil ODS-SP 250mm 4.6mmφ 5μm(ジーエルサイエンス社製)
流速:1.0mL/min
カラム温度:40℃
検出波長:UV 254nm
溶離液:アセトニトリル/水/トリフルオロ酢酸=600/400/1(体積比)
サンプル溶解液:アセトニトリル/トリエチルアミン=98/2(体積比)
HPLCを用いた定性分析は、以下に記載する測定条件を用いて実施した。
カラム:Inertsil ODS-SP 250mm 4.6mmφ 5μm(ジーエルサイエンス社製)
流速:1.0mL/min
カラム温度:40℃
検出波長:UV 254nm
溶離液:アセトニトリル/水/トリフルオロ酢酸=600/400/1(体積比)
サンプル溶解液:アセトニトリル/トリエチルアミン=98/2(体積比)
〔定量分析〕
HPLCを用いた定量分析には、4-メチルビフェニルを内標準物質とした内部標準法を用い、測定は定性分析と同様の測定条件で実施した。測定用のサンプルは、サンプルが塩基性になるように2.0体積%トリエチルアミン/アセトニトリル溶液を添加し、調整した。
特に記載がない限り、製造により得られた結晶中のZ体及びE体の含有量は、目的物については上記の定量分析により算出し、逆の幾何異性体については、前記の目的物の含有量及びHPLC測定結果のピーク面積の比率により算出した。このとき、Z体及びE体のUV吸収の感度(Z体/E体=1.097)を考慮した。また、実施例に記載の「定量収率」との表記は上記Z体及びE体の含有量の和より算出し、「Z体/E体含有比率」との表記も上記Z体及びE体の含有量より算出した。
HPLCを用いた定量分析には、4-メチルビフェニルを内標準物質とした内部標準法を用い、測定は定性分析と同様の測定条件で実施した。測定用のサンプルは、サンプルが塩基性になるように2.0体積%トリエチルアミン/アセトニトリル溶液を添加し、調整した。
特に記載がない限り、製造により得られた結晶中のZ体及びE体の含有量は、目的物については上記の定量分析により算出し、逆の幾何異性体については、前記の目的物の含有量及びHPLC測定結果のピーク面積の比率により算出した。このとき、Z体及びE体のUV吸収の感度(Z体/E体=1.097)を考慮した。また、実施例に記載の「定量収率」との表記は上記Z体及びE体の含有量の和より算出し、「Z体/E体含有比率」との表記も上記Z体及びE体の含有量より算出した。
〔質量分析〕
LC/MSによる質量分析の条件を以下に記載する。
カラム:SunFire C18 50mm 2.1mmφ 2.5μm(Waters社製)
流速:0.3mL/min
カラム温度:40℃
検出波長:UV 254nm
溶離液:

LC/MSによる質量分析の条件を以下に記載する。
カラム:SunFire C18 50mm 2.1mmφ 2.5μm(Waters社製)
流速:0.3mL/min
カラム温度:40℃
検出波長:UV 254nm
溶離液:
〔X線回折〕
粉末X線結晶回析は以下に記載する測定条件で実施した。
測定装置:X’pert Pro MPD(PANalytical社製)
X線源:Cu
電圧:45kV
電流:40mA
データ範囲:2.0191~39.7471°2Th.
スキャン軸:Gonio
ステップサイズ:0.0260°2Th.
スキャンステップ時間:47.9400sec
スキャンの種類:Continuous
PSDモード:Scanning
PSD距離:3.35°2Th.
発散スリットタイプ:自動
輻射幅:10.00mm
試料幅:10.00mm
測定温度:25.00℃
K-Alpha1:1.54060A
K-Alpha2:1.54443A
K-Beta:1.39225A
K-A2/K-A1比:0.50000
ゴニオメーター半径:240.00mm
フォーカス-DS間の距離:100.00mm
粉末X線結晶回析は以下に記載する測定条件で実施した。
測定装置:X’pert Pro MPD(PANalytical社製)
X線源:Cu
電圧:45kV
電流:40mA
データ範囲:2.0191~39.7471°2Th.
スキャン軸:Gonio
ステップサイズ:0.0260°2Th.
スキャンステップ時間:47.9400sec
スキャンの種類:Continuous
PSDモード:Scanning
PSD距離:3.35°2Th.
発散スリットタイプ:自動
輻射幅:10.00mm
試料幅:10.00mm
測定温度:25.00℃
K-Alpha1:1.54060A
K-Alpha2:1.54443A
K-Beta:1.39225A
K-A2/K-A1比:0.50000
ゴニオメーター半径:240.00mm
フォーカス-DS間の距離:100.00mm
実施例に記載した「質量部」との記載は、特に記載のない限り原料に対する質量部であり、「当量」との記載は、特に記載のない限り原料に対する当量である。
実施例に記載した「4.5質量%塩化水素/酢酸エチル溶液」との記載は塩化水素を4.5質量%含む酢酸エチルの溶液を表し、富士純薬製又は東京化成工業製を使用した。同様に「4.4質量%塩化水素/酢酸エチル溶液」は塩化水素を4.4質量%含む酢酸エチルの溶液(富士純薬製)であり、「15.5質量%塩化水素/シクロペンチルメチルエーテル溶液」又は「15.5質量%塩化水素/CPME溶液」は塩化水素を15.5質量%含むシクロペンチルメチルエーテル溶液(渡辺化学工業製)である。
実施例に記載した「4.5質量%塩化水素/酢酸エチル溶液」との記載は塩化水素を4.5質量%含む酢酸エチルの溶液を表し、富士純薬製又は東京化成工業製を使用した。同様に「4.4質量%塩化水素/酢酸エチル溶液」は塩化水素を4.4質量%含む酢酸エチルの溶液(富士純薬製)であり、「15.5質量%塩化水素/シクロペンチルメチルエーテル溶液」又は「15.5質量%塩化水素/CPME溶液」は塩化水素を15.5質量%含むシクロペンチルメチルエーテル溶液(渡辺化学工業製)である。
〔実施例1〕
前記の製造方法2に記載した方法に準じて、酸性化合物として塩化水素を用い、塩化水素の当量を変更した。
〔実施例1-1〕
(E)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド[以下、化合物(E)-1と略称する。]の製造方法

(EZ)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド[以下、化合物(EZ)-1と略称する。](Z体/E体=51.7/48.3)5.40g、原料に対して1質量部の1,2-ジクロロエタン5.40g、4.5質量%塩化水素/酢酸エチル溶液0.243g(塩化水素として0.025当量)及び原料に対して2.5質量部となるように酢酸エチル13.7gを、25℃にて混合した。該混合溶液を同温度にて30分間撹拌した。該反応溶液にn-ヘプタン27.0gを10時間かけて滴下すると、結晶が析出した。該懸濁液を25℃にて8時間撹拌した。該懸濁液を5℃に冷却し、同温度にて3時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン10.8g及び酢酸エチル2.7gの混合溶液で洗浄することにより、目的物4.02gを淡黄色結晶として得た[化合物(E)-1として定量収率74.5%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が0.6/99.4(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は99.6パーセントであった。
融点:113~114℃
実施例1-1で得られた化合物(E)-1の1H-NMRを以下に記す。
1H-NMR: δ8.42(d, J=1.8Hz, 1H), 8.30(t, J=5.7Hz, 1H), 8.03(s, 1H), 7.91(d, J=1.8Hz, 1H), 7.10(t, J=54.3Hz, 1H), 4.42(d, J=5.7Hz, 2H), 4.35(sep, J=6.6Hz, 1H), 3.83(s, 3H), 1.60-1.50(m, 1H), 1.23(d, J=6.6Hz, 6H), 0.95-0.85(m, 2H), 0.80-0.73(m, 2H).
前記の製造方法2に記載した方法に準じて、酸性化合物として塩化水素を用い、塩化水素の当量を変更した。
〔実施例1-1〕
(E)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド[以下、化合物(E)-1と略称する。]の製造方法
融点:113~114℃
実施例1-1で得られた化合物(E)-1の1H-NMRを以下に記す。
1H-NMR: δ8.42(d, J=1.8Hz, 1H), 8.30(t, J=5.7Hz, 1H), 8.03(s, 1H), 7.91(d, J=1.8Hz, 1H), 7.10(t, J=54.3Hz, 1H), 4.42(d, J=5.7Hz, 2H), 4.35(sep, J=6.6Hz, 1H), 3.83(s, 3H), 1.60-1.50(m, 1H), 1.23(d, J=6.6Hz, 6H), 0.95-0.85(m, 2H), 0.80-0.73(m, 2H).
〔実施例1-2〕
(Z)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド塩酸塩0.5(1,2-ジクロロエタン)和物[以下、化合物(Z)-2と略称する。]の製造方法

化合物(EZ)-1(Z体/E体=51.7/48.3)5.40g、原料に対して1質量部の1,2-ジクロロエタン5.40g及び4.5質量%塩化水素/酢酸エチル溶液14.6g(塩化水素として1.5当量:酢酸エチルの量は原料に対して2.5質量部である。)を25℃にて混合した。該混合溶液を同温度にて4時間撹拌すると結晶が析出した。該懸濁液にn-ヘプタン27.0gを10時間かけて滴下し、25℃にて8時間撹拌した。該懸濁液を5℃に冷却し、同温度にて3時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン10.8g及び酢酸エチル2.70gの混合溶液で洗浄することにより、目的物6.12gを淡黄色結晶として得た[化合物(Z)-1として定量収率95.3%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.0/2.0(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は99.2パーセントであった。得られた結晶を、下記の解析条件にて単結晶X線構造解析を行った結果、オキシイミノ基の立体配置はZであり、前記の結晶は化合物(Z)-2であることを確認した。
化合物(Z)-2の1H-NMRを以下に記す。
1H-NMR: δ8.52(t, J=6.0Hz, 1H), 8.44(d, J=1.8Hz, 1H), 8.14(s, 1H), 7.88(d, J=1.8Hz, 1H), 7.11(t, J=54.3Hz, 1H), 4.35-4.05(m, 6H), 3.85(s, 2H, 0.5分子の1,2-ジクロロエタン), 1.60-1.50(m, 1H), 1.05(d, J=6.3Hz, 6H), 0.95-0.85(m, 2H), 0.75-0.70(m, 2H).
[解析条件]
装置:SMART APEXII ULTRA(ブルカーエイエックスエス社製)
X線:CuKα(50kV, 50mA)
測定温度:-50℃
(Z)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド塩酸塩0.5(1,2-ジクロロエタン)和物[以下、化合物(Z)-2と略称する。]の製造方法
化合物(Z)-2の1H-NMRを以下に記す。
1H-NMR: δ8.52(t, J=6.0Hz, 1H), 8.44(d, J=1.8Hz, 1H), 8.14(s, 1H), 7.88(d, J=1.8Hz, 1H), 7.11(t, J=54.3Hz, 1H), 4.35-4.05(m, 6H), 3.85(s, 2H, 0.5分子の1,2-ジクロロエタン), 1.60-1.50(m, 1H), 1.05(d, J=6.3Hz, 6H), 0.95-0.85(m, 2H), 0.75-0.70(m, 2H).
[解析条件]
装置:SMART APEXII ULTRA(ブルカーエイエックスエス社製)
X線:CuKα(50kV, 50mA)
測定温度:-50℃
〔実施例1-3~1-9〕
実施例1-1及び1-2に記載した方法に準じて、4.5質量%塩化水素/酢酸エチル溶液の使用量を変更して反応を行った。尚、酢酸エチルは原料に対して2.5質量部となるように使用量を調整した。
塩化水素の当量(表中、「HCl当量」と記載する。)、得られた結晶中に含まれる化合物(Z)-1又は化合物(E)-1の定量収率(表中、「定量収率」と記載する。)、HPLCを用いた定性分析で確認したZ体及びE体の合わせた純度(表中、純度と記載する。)、HPLCを用いた定性分析の結果より算出したZ体及びE体の比(表中、「Z体/E体比」と記載する)を以下の表2に記載する。
実施例1-1及び1-2に記載した方法に準じて、4.5質量%塩化水素/酢酸エチル溶液の使用量を変更して反応を行った。尚、酢酸エチルは原料に対して2.5質量部となるように使用量を調整した。
塩化水素の当量(表中、「HCl当量」と記載する。)、得られた結晶中に含まれる化合物(Z)-1又は化合物(E)-1の定量収率(表中、「定量収率」と記載する。)、HPLCを用いた定性分析で確認したZ体及びE体の合わせた純度(表中、純度と記載する。)、HPLCを用いた定性分析の結果より算出したZ体及びE体の比(表中、「Z体/E体比」と記載する)を以下の表2に記載する。

〔実施例2〕
前記の製造方法1に記載した方法に準じて、溶媒の種類、酸性化合物の種類及び当量を変更した。
〔実施例2-1〕
溶媒としてシクロペンチルメチルエーテルを、酸性化合物としてメタンスルホン酸を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g及び3質量部のシクロペンチルメチルエーテル(以下、CPMEと略称する。)を40℃にて混合した。該混合溶液に0.02当量のメタンスルホン酸(85mg)を添加した後、20℃にて60時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を5℃に冷却した後、5時間撹拌した。析出した結晶をろ過し、得られた結晶を、反応溶媒と同じであるCPMEを1質量部用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は73.1%、Z体及びE体含有比率はZ/E=0.4/99.6であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
前記の製造方法1に記載した方法に準じて、溶媒の種類、酸性化合物の種類及び当量を変更した。
〔実施例2-1〕
溶媒としてシクロペンチルメチルエーテルを、酸性化合物としてメタンスルホン酸を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g及び3質量部のシクロペンチルメチルエーテル(以下、CPMEと略称する。)を40℃にて混合した。該混合溶液に0.02当量のメタンスルホン酸(85mg)を添加した後、20℃にて60時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を5℃に冷却した後、5時間撹拌した。析出した結晶をろ過し、得られた結晶を、反応溶媒と同じであるCPMEを1質量部用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は73.1%、Z体及びE体含有比率はZ/E=0.4/99.6であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
〔実施例2-6〕
溶媒としてシクロペンチルメチルエーテルを、酸性化合物として硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び4質量部のCPMEを40℃にて混合した。該混合溶液に0.5当量の濃硫酸(218mg)を添加した後、同温度にて1.5時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.6当量の濃硫酸(264mg)を添加し、更に20時間撹拌した。該懸濁液を5℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、得られた結晶を、反応溶媒と同じであるCPME(2mL)で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(E)-1の定量収率は87.9%、Z体及びE体含有比率はZ/E=94.6/5.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.8%であった。
溶媒としてシクロペンチルメチルエーテルを、酸性化合物として硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び4質量部のCPMEを40℃にて混合した。該混合溶液に0.5当量の濃硫酸(218mg)を添加した後、同温度にて1.5時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.6当量の濃硫酸(264mg)を添加し、更に20時間撹拌した。該懸濁液を5℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、得られた結晶を、反応溶媒と同じであるCPME(2mL)で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(E)-1の定量収率は87.9%、Z体及びE体含有比率はZ/E=94.6/5.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.8%であった。
〔実施例2-7〕
溶媒としてトルエンを、酸性化合物として硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)1.5g及び4質量部のトルエンを30℃にて混合した。該混合溶液に0.55当量の75質量%硫酸水溶液(240mg)を添加した後、同温度にて2時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に同温度にて0.55当量の75質量%硫酸水溶液(240mg)を添加し、更に16時間撹拌した。該懸濁液を0℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、得られた結晶を、反応溶媒と同じであるトルエンを2質量部用いて洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は80.4%、Z体及びE体含有比率はZ/E=95.1/4.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は97.9%であった。
溶媒としてトルエンを、酸性化合物として硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)1.5g及び4質量部のトルエンを30℃にて混合した。該混合溶液に0.55当量の75質量%硫酸水溶液(240mg)を添加した後、同温度にて2時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に同温度にて0.55当量の75質量%硫酸水溶液(240mg)を添加し、更に16時間撹拌した。該懸濁液を0℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、得られた結晶を、反応溶媒と同じであるトルエンを2質量部用いて洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は80.4%、Z体及びE体含有比率はZ/E=95.1/4.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は97.9%であった。
〔実施例2-2~2-5〕
実施例2-1に記載した方法に準じて、溶媒及び酸性化合物の種類を変更して反応を行った。反応条件と結果を表3に記載する。表中、「溶媒」には溶媒の種類を記載し、溶媒の質量部を括弧内に記載した。また、「酸性化合物」には酸性化合物の種類を記載し、酸性化合物の当量を括弧内に記載した。
実施例2-1に記載した方法に準じて、溶媒及び酸性化合物の種類を変更して反応を行った。反応条件と結果を表3に記載する。表中、「溶媒」には溶媒の種類を記載し、溶媒の質量部を括弧内に記載した。また、「酸性化合物」には酸性化合物の種類を記載し、酸性化合物の当量を括弧内に記載した。

〔実施例3〕
前記の製造方法1に記載に準じて、溶媒として2種類の溶媒を用い、酸性化合物として濃硫酸を用いた。下記、実施例3-1~3-8において第一の溶媒Aとしてはトルエンを用いた。
〔実施例3-1〕
第二の溶媒Aにt-ブチルメチルエーテルを用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、2質量部のトルエン及び3質量部のt-ブチルメチルエーテル(以下、TBMEと略称する。)を40℃にて混合した。該混合溶液に0.05当量の濃硫酸(22mg)を添加した後、15℃で20時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を5℃に冷却し、24時間撹拌した。析出した結晶をろ過し、得られた結晶をトルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、白色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は70.0%、Z体及びE体含有比率はZ/E=6.2/93.8であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.9%であった。
前記の製造方法1に記載に準じて、溶媒として2種類の溶媒を用い、酸性化合物として濃硫酸を用いた。下記、実施例3-1~3-8において第一の溶媒Aとしてはトルエンを用いた。
〔実施例3-1〕
第二の溶媒Aにt-ブチルメチルエーテルを用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、2質量部のトルエン及び3質量部のt-ブチルメチルエーテル(以下、TBMEと略称する。)を40℃にて混合した。該混合溶液に0.05当量の濃硫酸(22mg)を添加した後、15℃で20時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を5℃に冷却し、24時間撹拌した。析出した結晶をろ過し、得られた結晶をトルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、白色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は70.0%、Z体及びE体含有比率はZ/E=6.2/93.8であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.9%であった。
〔実施例3-2〕
第二の溶媒Aにシクロペンチルメチルエーテルを用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、1質量部のトルエン及び0.05質量部のCPMEを40℃にて混合した。該混合溶液に0.025当量の濃硫酸(11mg)を添加した後、20℃で24時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を10℃に冷却し、1時間撹拌した。析出した結晶をろ過し、得られた結晶をトルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、白色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は83.6%、Z体及びE体含有比率はZ/E=0/100であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.7%であった。
第二の溶媒Aにシクロペンチルメチルエーテルを用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、1質量部のトルエン及び0.05質量部のCPMEを40℃にて混合した。該混合溶液に0.025当量の濃硫酸(11mg)を添加した後、20℃で24時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を10℃に冷却し、1時間撹拌した。析出した結晶をろ過し、得られた結晶をトルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、白色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は83.6%、Z体及びE体含有比率はZ/E=0/100であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.7%であった。
〔実施例3-5〕
第二の溶媒Aに1,2-ジメトキシエタンを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g、3質量部のトルエン及び0.5質量部の1,2-ジメトキシエタン(以下、DMEと略称する。)を30℃にて混合した。該混合溶液に0.5当量の濃硫酸(172mg)を添加した後、4時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.6当量の濃硫酸(207mg)を添加し、72時間撹拌した。該懸濁液を5℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエンで洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は87.3%、Z体及びE体含有比率はZ/E=93.5/6.5であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は96.7%であった。
第二の溶媒Aに1,2-ジメトキシエタンを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g、3質量部のトルエン及び0.5質量部の1,2-ジメトキシエタン(以下、DMEと略称する。)を30℃にて混合した。該混合溶液に0.5当量の濃硫酸(172mg)を添加した後、4時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.6当量の濃硫酸(207mg)を添加し、72時間撹拌した。該懸濁液を5℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエンで洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は87.3%、Z体及びE体含有比率はZ/E=93.5/6.5であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は96.7%であった。
〔実施例3-7〕
第二の溶媒Aにt-ブチルメチルエーテルを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)1.5g、3質量部のトルエン及び3質量部のTBMEを40℃にて混合した。該混合溶液に1.1当量の濃硫酸(379mg)を添加し、30℃で42時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を20℃に冷却し、1時間撹拌した。析出した結晶をろ過した後、得られた結晶を2質量部のトルエンで洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率81.5%、Z体及びE体含有比率はZ/E=96.0/4.0であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は96.5%であった。
第二の溶媒Aにt-ブチルメチルエーテルを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)1.5g、3質量部のトルエン及び3質量部のTBMEを40℃にて混合した。該混合溶液に1.1当量の濃硫酸(379mg)を添加し、30℃で42時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を20℃に冷却し、1時間撹拌した。析出した結晶をろ過した後、得られた結晶を2質量部のトルエンで洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率81.5%、Z体及びE体含有比率はZ/E=96.0/4.0であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は96.5%であった。
〔実施例3-8〕
第二の溶媒Aにシクロペンチルメチルエーテルを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)3.0g、2質量部のトルエン及び4.1質量部のCPMEを40℃にて混合した。該混合溶液に0.5当量の濃硫酸(344mg)を添加し、8時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.8当量の濃硫酸(551mg)を添加した後、30℃で40時間撹拌した。該懸濁液を0℃に冷却し、2時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエン及び2質量部のCPMEの混合溶液で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率89.0%、Z体及びE体含有比率はZ/E=93.7/6.3であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.4%であった。
第二の溶媒Aにシクロペンチルメチルエーテルを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)3.0g、2質量部のトルエン及び4.1質量部のCPMEを40℃にて混合した。該混合溶液に0.5当量の濃硫酸(344mg)を添加し、8時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.8当量の濃硫酸(551mg)を添加した後、30℃で40時間撹拌した。該懸濁液を0℃に冷却し、2時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエン及び2質量部のCPMEの混合溶液で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率89.0%、Z体及びE体含有比率はZ/E=93.7/6.3であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.4%であった。
〔実施例3-3、3-4及び3-6〕
実施例3-2に記載した方法に準じて実施例3-3及び実施例3-4を、実施例3-5に記載した方法に準じて実施例3-6を、第二の溶媒Aの種類を変更して反応を行った。反応条件と結果を表4に記載する。表中、「第二溶媒」に第二の溶媒Aの種類を記載した。「Diglyme」との記載はジエチレングリコールジメチルエーテルを表す(以下、同じである。)。
実施例3-2に記載した方法に準じて実施例3-3及び実施例3-4を、実施例3-5に記載した方法に準じて実施例3-6を、第二の溶媒Aの種類を変更して反応を行った。反応条件と結果を表4に記載する。表中、「第二溶媒」に第二の溶媒Aの種類を記載した。「Diglyme」との記載はジエチレングリコールジメチルエーテルを表す(以下、同じである。)。

下記、実施例3-9及び実施例3-10においては第一の溶媒Aとしてはo-キシレンを、第二の溶媒Aとしてはシクロペンチルメチルエーテルを用い、用いる濃硫酸の当量を変更した。
〔実施例3-9〕
化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、1質量部のo-キシレン及び0.5質量部のCPMEを50℃にて混合した。該混合溶液に0.025当量の濃硫酸(11mg)を添加し、20℃で24時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を5℃に冷却し、1時間撹拌した。析出した結晶をろ過し、得られた結晶を1質量部のCPMEで洗浄することにより、白色結晶を得た。定量分析の結果、化合物(E)-1の定量収率80.7%、Z体及びE体含有比率はZ/E=1.2/98.8であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
〔実施例3-9〕
化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、1質量部のo-キシレン及び0.5質量部のCPMEを50℃にて混合した。該混合溶液に0.025当量の濃硫酸(11mg)を添加し、20℃で24時間撹拌し結晶を析出させた。結晶析出後、該懸濁液を5℃に冷却し、1時間撹拌した。析出した結晶をろ過し、得られた結晶を1質量部のCPMEで洗浄することにより、白色結晶を得た。定量分析の結果、化合物(E)-1の定量収率80.7%、Z体及びE体含有比率はZ/E=1.2/98.8であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
〔実施例3-10〕
化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g、3質量部のo-キシレン及び1質量部のCPMEを30℃にて混合した。該混合溶液に0.5当量の濃硫酸(172mg)を添加し、14時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.6当量の濃硫酸(207mg)を添加し、44時間撹拌した。該懸濁液を20℃に冷却した後、1時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のo-キシレンで洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率82.7%、Z体及びE体含有比率はZ/E=93.1/6.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は95.8%であった。
上記、実施例3-9及び実施例3-10の反応条件及び結果を表5に記載する。表中、「o-キシレン/CPME質量部」との記載は、o-キシレン及びCPMEの質量部を表し、例えば、「1/0.5」との記載は、o-キシレンを1質量部、CPMEを0.5質量部用いたことを表す。

化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g、3質量部のo-キシレン及び1質量部のCPMEを30℃にて混合した。該混合溶液に0.5当量の濃硫酸(172mg)を添加し、14時間撹拌し結晶を析出させた。結晶析出後、該懸濁液に0.6当量の濃硫酸(207mg)を添加し、44時間撹拌した。該懸濁液を20℃に冷却した後、1時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のo-キシレンで洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率82.7%、Z体及びE体含有比率はZ/E=93.1/6.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は95.8%であった。
上記、実施例3-9及び実施例3-10の反応条件及び結果を表5に記載する。表中、「o-キシレン/CPME質量部」との記載は、o-キシレン及びCPMEの質量部を表し、例えば、「1/0.5」との記載は、o-キシレンを1質量部、CPMEを0.5質量部用いたことを表す。
〔実施例4〕
前記の製造方法2に記載した方法に準じて、溶媒Aの種類及び酸性化合物の種類を変更した。
〔実施例4-1〕
溶媒Aとしてシクロペンチルメチルエーテルを、酸性化合物として塩化水素を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び3質量部のCPMEを45℃にて混合した。該混合溶液に0.05当量の15.5質量%塩化水素/CPME溶液(0.054g)を添加した。該混合溶液を25℃に冷却し、2質量部のn-ヘプタンを2時間かけて滴下すると結晶が析出した。該懸濁液を同温度で24時間撹拌した。析出した結晶をろ過し、得られた結晶をCPME及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は67.7%、Z体及びE体含有比率はZ/E=0.4/99.6であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.3%であった。
前記の製造方法2に記載した方法に準じて、溶媒Aの種類及び酸性化合物の種類を変更した。
〔実施例4-1〕
溶媒Aとしてシクロペンチルメチルエーテルを、酸性化合物として塩化水素を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び3質量部のCPMEを45℃にて混合した。該混合溶液に0.05当量の15.5質量%塩化水素/CPME溶液(0.054g)を添加した。該混合溶液を25℃に冷却し、2質量部のn-ヘプタンを2時間かけて滴下すると結晶が析出した。該懸濁液を同温度で24時間撹拌した。析出した結晶をろ過し、得られた結晶をCPME及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は67.7%、Z体及びE体含有比率はZ/E=0.4/99.6であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.3%であった。
〔実施例4-2〕
溶媒Aとしてトルエンを、酸性化合物として硫酸を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g及び2質量部のトルエンを50℃にて混合した。該混合溶液に0.03当量の85質量%硫酸水溶液(15mg)を添加した。該混合溶液を20℃で14時間撹拌すると結晶が析出した。該懸濁液に同温度にて1質量部のn-ヘプタンを1時間かけて滴下した後、22時間撹拌した。該懸濁液を5℃に冷却した後、2時間撹拌した。析出した結晶をろ過し2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は90.1%、Z体及びE体含有比率はZ/E=7.1/92.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
溶媒Aとしてトルエンを、酸性化合物として硫酸を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g及び2質量部のトルエンを50℃にて混合した。該混合溶液に0.03当量の85質量%硫酸水溶液(15mg)を添加した。該混合溶液を20℃で14時間撹拌すると結晶が析出した。該懸濁液に同温度にて1質量部のn-ヘプタンを1時間かけて滴下した後、22時間撹拌した。該懸濁液を5℃に冷却した後、2時間撹拌した。析出した結晶をろ過し2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は90.1%、Z体及びE体含有比率はZ/E=7.1/92.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
〔実施例4-3〕
溶媒Aとしてトルエンを、酸性化合物としてp-トルエンスルホン酸を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び4質量部のトルエンを40℃にて混合した。該混合溶液に0.05当量のp-トルエンスルホン酸1水和物(42mg)を添加した。該反応溶液を15℃に冷却した後、20時間撹拌すると、結晶が析出した。該懸濁液に20℃で1質量部のn-ヘプタンを1時間かけて滴下し、同温度で24時間撹拌した。析出した結晶をろ過し、トルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は75.6%、Z体及びE体含有比率はZ/E=1.8/98.2であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.5%であった。
溶媒Aとしてトルエンを、酸性化合物としてp-トルエンスルホン酸を用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び4質量部のトルエンを40℃にて混合した。該混合溶液に0.05当量のp-トルエンスルホン酸1水和物(42mg)を添加した。該反応溶液を15℃に冷却した後、20時間撹拌すると、結晶が析出した。該懸濁液に20℃で1質量部のn-ヘプタンを1時間かけて滴下し、同温度で24時間撹拌した。析出した結晶をろ過し、トルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は75.6%、Z体及びE体含有比率はZ/E=1.8/98.2であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.5%であった。
〔実施例4-4〕
溶媒Aとしてトルエンを、酸性化合物として硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g及び4質量部のトルエンを30℃にて混合した。該混合溶液に1.1当量の85質量%硫酸水溶液(423mg)を添加した後、16時間撹拌すると、結晶が析出した。該懸濁液に同温度で2質量部のn-ヘプタンを3時間かけて滴下した後、16時間撹拌した。該懸濁液を0℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は82.4%、Z体及びE体含有比率はZ/E=96.6/3.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.4%であった。
溶媒Aとしてトルエンを、酸性化合物として硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g及び4質量部のトルエンを30℃にて混合した。該混合溶液に1.1当量の85質量%硫酸水溶液(423mg)を添加した後、16時間撹拌すると、結晶が析出した。該懸濁液に同温度で2質量部のn-ヘプタンを3時間かけて滴下した後、16時間撹拌した。該懸濁液を0℃に冷却した後、3時間撹拌した。析出した結晶をろ過し、2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は82.4%、Z体及びE体含有比率はZ/E=96.6/3.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.4%であった。
上記、実施例4-1~4-4の反応条件と結果を表6に記載する。表中、「酸性化合物」に酸性化合物の種類を記載し、酸性化合物の当量は「当量」に記載する。

〔実施例5〕
前記の製造方法2に記載した方法に準じて、溶媒Aとして2種の溶媒を用い、酸性化合物の当量を変更した。下記、実施例5-1~5-8は、溶媒Aのうち1種はトルエンを用い、溶媒Bとしてはn-ヘプタンを用い、酸性化合物としては濃硫酸を用いた。
〔実施例5-1〕
第二の溶媒Aとして酢酸エチルを用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、1質量部のトルエン及び0.5質量部の酢酸エチルを40℃にて混合した。該混合溶液に0.025当量の濃硫酸(12mg)を添加した。該混合溶液を20℃で24時間撹拌し結晶を析出させた。該懸濁液に同温度で3質量部のn-ヘプタンを6時間かけて滴下した後、12時間撹拌した。該懸濁液を5℃に冷却した後、2時間撹拌した。析出した結晶をろ過し、2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は90.5%、Z体及びE体含有比率はZ/E=3.5/96.5であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
前記の製造方法2に記載した方法に準じて、溶媒Aとして2種の溶媒を用い、酸性化合物の当量を変更した。下記、実施例5-1~5-8は、溶媒Aのうち1種はトルエンを用い、溶媒Bとしてはn-ヘプタンを用い、酸性化合物としては濃硫酸を用いた。
〔実施例5-1〕
第二の溶媒Aとして酢酸エチルを用いた化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、1質量部のトルエン及び0.5質量部の酢酸エチルを40℃にて混合した。該混合溶液に0.025当量の濃硫酸(12mg)を添加した。該混合溶液を20℃で24時間撹拌し結晶を析出させた。該懸濁液に同温度で3質量部のn-ヘプタンを6時間かけて滴下した後、12時間撹拌した。該懸濁液を5℃に冷却した後、2時間撹拌した。析出した結晶をろ過し、2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は90.5%、Z体及びE体含有比率はZ/E=3.5/96.5であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
〔実施例5-5〕
第二の溶媒Aとしてシクロペンチルメチルエーテルを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g、3質量部のトルエン及び3質量部のCPMEを30℃にて混合した。該混合溶液に1.1当量の濃硫酸(379mg)を添加し、14時間撹拌し結晶を析出させた。該懸濁液に同温度で1質量部のn-ヘプタンを2時間かけて滴下した後、22時間撹拌した。該懸濁液を15℃に冷却した後、2時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は89.3%、Z体及びE体含有比率はZ/E=93.6/6.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.2%であった。
第二の溶媒Aとしてシクロペンチルメチルエーテルを用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)1.5g、3質量部のトルエン及び3質量部のCPMEを30℃にて混合した。該混合溶液に1.1当量の濃硫酸(379mg)を添加し、14時間撹拌し結晶を析出させた。該懸濁液に同温度で1質量部のn-ヘプタンを2時間かけて滴下した後、22時間撹拌した。該懸濁液を15℃に冷却した後、2時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、化合物(Z)-1の硫酸塩を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は89.3%、Z体及びE体含有比率はZ/E=93.6/6.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.2%であった。
〔実施例5-2~5-4、5-6~5-8〕
実施例5-1に記載した方法に準じて実施例5-2~5-4を、実施例5-5に記載した方法に準じて実施例5-6~5-8を行い、溶媒の種類及び硫酸の当量を変更した。反応条件と結果を表7に記載する。表中、「第二溶媒」に溶媒Aとして用いた2種の溶媒のうち第二の溶媒Aの種類を記載し、溶媒の質量部を括弧内に記載した。表中、「AcOEt」との記載は酢酸エチルを表し(以下、同様である。)、「MEK」との記載はメチルエチルケトンを表す。
実施例5-1に記載した方法に準じて実施例5-2~5-4を、実施例5-5に記載した方法に準じて実施例5-6~5-8を行い、溶媒の種類及び硫酸の当量を変更した。反応条件と結果を表7に記載する。表中、「第二溶媒」に溶媒Aとして用いた2種の溶媒のうち第二の溶媒Aの種類を記載し、溶媒の質量部を括弧内に記載した。表中、「AcOEt」との記載は酢酸エチルを表し(以下、同様である。)、「MEK」との記載はメチルエチルケトンを表す。
下記、実施例5-9及び実施例5-10は、溶媒Aとして1,2-ジクロロエタン及び酢酸エチルを用い、溶媒Bとしてはn-ヘプタンを用い、酸性化合物としては塩化水素を用い、塩化水素の当量を変更した。
〔実施例5-9〕
化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)100g、0.5質量部の1,2-ジクロロエタン及び2.0質量部の酢酸エチルを25℃にて混合した。該混合溶液に0.042当量の塩化水素ガス(340mg)を吹き込み、同温度で1時間撹拌した。該混合溶液に同温度で5質量部のn-ヘプタンを12時間かけて滴下すると、結晶が析出した。該懸濁液を同温度で16時間撹拌した。該懸濁液を5℃に冷却した後、2時間撹拌した。析出した結晶をろ過し、得られた結晶を3質量部のn-ヘプタン及び1質量部の酢酸エチルの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は90.5%、Z体及びE体含有比率はZ/E=1.1/98.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.8%であった。
〔実施例5-9〕
化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)100g、0.5質量部の1,2-ジクロロエタン及び2.0質量部の酢酸エチルを25℃にて混合した。該混合溶液に0.042当量の塩化水素ガス(340mg)を吹き込み、同温度で1時間撹拌した。該混合溶液に同温度で5質量部のn-ヘプタンを12時間かけて滴下すると、結晶が析出した。該懸濁液を同温度で16時間撹拌した。該懸濁液を5℃に冷却した後、2時間撹拌した。析出した結晶をろ過し、得られた結晶を3質量部のn-ヘプタン及び1質量部の酢酸エチルの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は90.5%、Z体及びE体含有比率はZ/E=1.1/98.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.8%であった。
〔実施例5-10〕
化合物(Z)-2の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)5.0g、0.5質量部の1,2-ジクロロエタン及び2.0質量部の酢酸エチルを25℃にて混合した。該混合溶液に1.17当量の塩化水素ガス(475mg)を吹き込み、同温度で5時間撹拌すると結晶が析出した。該懸濁液に同温度で5質量部のn-ヘプタンを8時間かけて滴下した後、8時間撹拌した。該懸濁液を0℃に冷却した後、5時間撹拌した。析出した結晶をろ過し、得られた結晶を3質量部のn-ヘプタン及び1質量部の酢酸エチルの混合溶液で洗浄することにより、化合物(Z)-2を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は94.4%、Z体及びE体含有比率はZ/E=97.6/2.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.6%であった。
化合物(Z)-2の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)5.0g、0.5質量部の1,2-ジクロロエタン及び2.0質量部の酢酸エチルを25℃にて混合した。該混合溶液に1.17当量の塩化水素ガス(475mg)を吹き込み、同温度で5時間撹拌すると結晶が析出した。該懸濁液に同温度で5質量部のn-ヘプタンを8時間かけて滴下した後、8時間撹拌した。該懸濁液を0℃に冷却した後、5時間撹拌した。析出した結晶をろ過し、得られた結晶を3質量部のn-ヘプタン及び1質量部の酢酸エチルの混合溶液で洗浄することにより、化合物(Z)-2を淡黄色結晶として得た。定量分析の結果、化合物(Z)-1の定量収率は94.4%、Z体及びE体含有比率はZ/E=97.6/2.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.6%であった。
上記、実施例5-9及び実施例5-10の反応条件と結果を表8に記載する。表中、「溶媒」に溶媒Aの種類を記載し、溶媒の質量部を括弧内に記載した。

〔実施例5-11〕
化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、2質量部のトルエン及び0.1質量部のDiglymeを40℃にて混合した。該混合溶液に0.025当量の濃硫酸(11mg)を添加した後、20℃で20時間撹拌すると結晶が析出した。該懸濁液に同温度で1質量部のn-ヘプタンを1時間かけて滴下し、30℃で100時間撹拌した。該懸濁液を20℃に冷却した後、1時間撹拌した。析出した結晶をろ過し、得られた結晶をトルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は74.5%、Z体及びE体含有比率はZ/E=4.0/96.0であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は98.4%であった。
化合物(E)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、2質量部のトルエン及び0.1質量部のDiglymeを40℃にて混合した。該混合溶液に0.025当量の濃硫酸(11mg)を添加した後、20℃で20時間撹拌すると結晶が析出した。該懸濁液に同温度で1質量部のn-ヘプタンを1時間かけて滴下し、30℃で100時間撹拌した。該懸濁液を20℃に冷却した後、1時間撹拌した。析出した結晶をろ過し、得られた結晶をトルエン及びn-ヘプタンの混合溶液(体積比1:1)を2mL用いて洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(E)-1の定量収率は74.5%、Z体及びE体含有比率はZ/E=4.0/96.0であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は98.4%であった。
〔実施例6〕
化合物(Z)-1を原料とした化合物(E)-1の製造方法
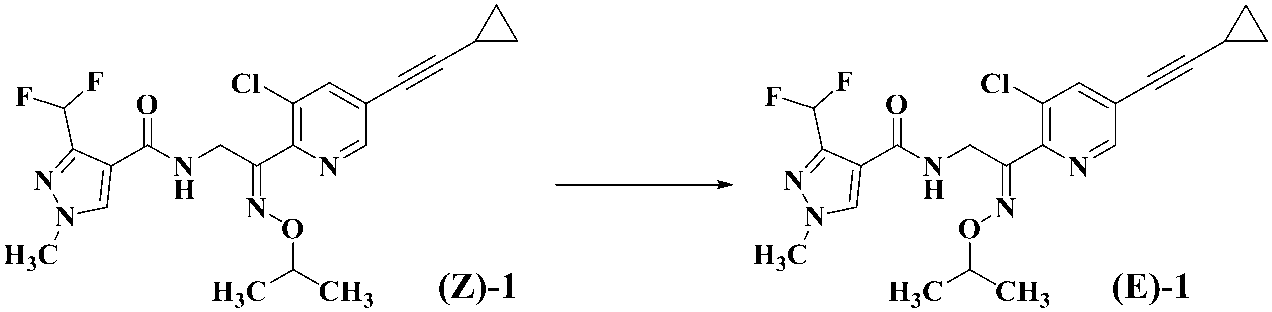
参考例1で得られた化合物(Z)-1(Z体/E体=99.7/0.3)50g及び酢酸エチル75gを室温にて混合し、該混合溶液に4.5質量%塩化水素/酢酸エチル溶液2.25g(塩化水素として0.025当量)を室温にて添加した。該混合溶液を40℃にて24時間撹拌した後、同温度にてn-ヘプタン250gを5時間かけて滴下した。この際、n-ヘプタン100gを滴下した段階で結晶の析出が認められた。n-ヘプタン滴下終了後、該反応混合物を40℃にて4時間撹拌し、次いで室温にて16時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン40g及び酢酸エチル10gの混合溶液で洗浄することにより、目的物46.7gを淡黄色結晶として得た[化合物(E)-1として定量収率93.4%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が6.8/93.2(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は97.6パーセントであった。
化合物(Z)-1を原料とした化合物(E)-1の製造方法
〔実施例7〕
化合物(E)-1を原料とした化合物(Z)-2の製造方法

(E)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド(Z体/E体=6.4/93.6)3.60g及び1,2-ジクロロエタン7.20gを30℃にて混合した。該混合溶液に4.5質量%塩化水素/酢酸エチル溶液7.22g(塩化水素として1.11当量)を添加した後、同温度にて6時間撹拌すると、結晶が析出した。該懸濁液にn-ヘプタン14.4gを16時間かけて滴下し、30℃にて16時間撹拌した。該懸濁液を15℃に冷却し、同温度にて3時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン7.20g及び酢酸エチル1.80gの混合溶液で洗浄することにより、目的物3.97gを淡黄色結晶として得た[化合物(Z)-1として定量収率89.4%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.1/1.9(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は97.5パーセントであった。
化合物(E)-1を原料とした化合物(Z)-2の製造方法
〔実施例8〕
化合物(E)-1を原料とした化合物(Z)-1の製造方法

化合物(E)-1(Z体/E体=0.1/99.9)3.60g及び1,2-ジクロロエタン7.20gを30℃にて混合した。該混合溶液に4.5質量%塩化水素/酢酸エチル溶液7.22g(塩化水素として1.11当量)を添加した後、同温度にて4時間撹拌すると、結晶が析出した。該懸濁液にn-ヘプタン14.4gを8時間かけて滴下し、30℃にて8時間撹拌した。該懸濁液を0℃に冷却し、同温度にて2時間撹拌した。該懸濁液に水10.8gを、0℃にて滴下した後、2.0mol/L炭酸カリウム水溶液6.0mLを滴下した。該反応液を0℃にて2時間撹拌した後、析出した結晶をろ過し、得られた結晶を水10.8g、次いで水2.40g及びエタノール4.80gの混合溶液で洗浄することにより、目的物2.93gを白色結晶として得た(収率79.6%)。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が99.0/1.0(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は98.5パーセントであった。
化合物(E)-1を原料とした化合物(Z)-1の製造方法
化合物(E)-1(Z体/E体=0.1/99.9)3.60g及び1,2-ジクロロエタン7.20gを30℃にて混合した。該混合溶液に4.5質量%塩化水素/酢酸エチル溶液7.22g(塩化水素として1.11当量)を添加した後、同温度にて4時間撹拌すると、結晶が析出した。該懸濁液にn-ヘプタン14.4gを8時間かけて滴下し、30℃にて8時間撹拌した。該懸濁液を0℃に冷却し、同温度にて2時間撹拌した。該懸濁液に水10.8gを、0℃にて滴下した後、2.0mol/L炭酸カリウム水溶液6.0mLを滴下した。該反応液を0℃にて2時間撹拌した後、析出した結晶をろ過し、得られた結晶を水10.8g、次いで水2.40g及びエタノール4.80gの混合溶液で洗浄することにより、目的物2.93gを白色結晶として得た(収率79.6%)。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が99.0/1.0(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は98.5パーセントであった。
〔実施例9〕
前記の製造方法1に記載した方法に準じて、溶媒の種類、酸性化合物の種類及び当量を変更し、結晶を析出させるために少量の化合物(Z)-1の塩を用いて反応を行った。
〔実施例9-1〕
溶媒としてCPMEを、酸性化合物として塩化水素を用いた化合物(Z)-1の塩酸塩[以下、化合物(Z)-3と略称する。]の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び1.45質量部のCPMEを50℃にて混合した。該混合溶液に50℃にて1.2当量の15.5質量%塩化水素/CPME溶液(1.3g)を添加した。該混合溶液に同温度にて、参考例3で製造した化合物(Z)-3を2.0mg添加した後、20分間撹拌することにより結晶を析出させた。該懸濁液を25℃に冷却した後、20時間撹拌した。析出した結晶をろ過し、得られた結晶を反応溶媒と同じCPMEを2mL用いて洗浄することにより、薄赤色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は73.0%、Z体及びE体含有比率はZ/E=90.8/9.2であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は97.8%であった。
前記の製造方法1に記載した方法に準じて、溶媒の種類、酸性化合物の種類及び当量を変更し、結晶を析出させるために少量の化合物(Z)-1の塩を用いて反応を行った。
〔実施例9-1〕
溶媒としてCPMEを、酸性化合物として塩化水素を用いた化合物(Z)-1の塩酸塩[以下、化合物(Z)-3と略称する。]の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び1.45質量部のCPMEを50℃にて混合した。該混合溶液に50℃にて1.2当量の15.5質量%塩化水素/CPME溶液(1.3g)を添加した。該混合溶液に同温度にて、参考例3で製造した化合物(Z)-3を2.0mg添加した後、20分間撹拌することにより結晶を析出させた。該懸濁液を25℃に冷却した後、20時間撹拌した。析出した結晶をろ過し、得られた結晶を反応溶媒と同じCPMEを2mL用いて洗浄することにより、薄赤色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は73.0%、Z体及びE体含有比率はZ/E=90.8/9.2であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は97.8%であった。
〔実施例9-2、9-3〕
実施例9-1に記載した方法に準じて、溶媒の種類並びに酸性化合物の種類及び当量を変更して反応を行った。反応条件と結果を表9に記載する。表中、「溶媒」に溶媒の種類を記載し、溶媒の質量部を括弧内に記載した。「酸性化合物」に酸性化合物の種類を記載し、酸性化合物の当量を括弧内に記載した。

実施例9-1に記載した方法に準じて、溶媒の種類並びに酸性化合物の種類及び当量を変更して反応を行った。反応条件と結果を表9に記載する。表中、「溶媒」に溶媒の種類を記載し、溶媒の質量部を括弧内に記載した。「酸性化合物」に酸性化合物の種類を記載し、酸性化合物の当量を括弧内に記載した。
〔実施例10〕
前記の製造方法2に記載した方法に準じて、溶媒の種類、酸性化合物の種類及び当量を変更し、結晶を析出させるために少量の化合物(Z)-1の塩を用いて反応を行った。
〔実施例10-1〕
溶媒AとしてCPMEを、酸性化合物として塩化水素を用いた化合物(Z)-3の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び原料に対して2.45質量部のCPMEを45℃にて混合した。該混合溶液に1.2当量の15.5質量%塩化水素/CPME溶液(1.3g)を添加した。該混合溶液に同温度にて、参考例3で製造した化合物(Z)-3を2.0mg添加した後、20分間撹拌することにより結晶を析出させた。該懸濁液に2質量部のn-ヘプタンを2時間かけて滴下した後、24時間撹拌した。析出した結晶をろ過し、得られた結晶をCPME(2mL)で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は79.1%、Z体及びE体含有比率はZ/E=95.6/4.4であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は99.2%であった。
前記の製造方法2に記載した方法に準じて、溶媒の種類、酸性化合物の種類及び当量を変更し、結晶を析出させるために少量の化合物(Z)-1の塩を用いて反応を行った。
〔実施例10-1〕
溶媒AとしてCPMEを、酸性化合物として塩化水素を用いた化合物(Z)-3の製造方法
化合物(EZ)-1(Z体/E体=48.8/51.2)2.0g及び原料に対して2.45質量部のCPMEを45℃にて混合した。該混合溶液に1.2当量の15.5質量%塩化水素/CPME溶液(1.3g)を添加した。該混合溶液に同温度にて、参考例3で製造した化合物(Z)-3を2.0mg添加した後、20分間撹拌することにより結晶を析出させた。該懸濁液に2質量部のn-ヘプタンを2時間かけて滴下した後、24時間撹拌した。析出した結晶をろ過し、得られた結晶をCPME(2mL)で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は79.1%、Z体及びE体含有比率はZ/E=95.6/4.4であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は99.2%であった。
〔実施例10-2〕
溶媒Aとしてトルエン及び酢酸エチルを、酸性化合物として濃硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、2質量部のトルエン及び3質量部の酢酸エチルを30℃にて混合した。該混合溶液に1.1当量の濃硫酸(507mg)を添加した。混合溶液に同温度にて、参考例4で製造した化合物(Z)-1の硫酸塩を2.0mg添加した後、14時間撹拌することにより結晶を析出させた。該懸濁液に同温度で1質量部のn-ヘプタンを24時間かけて滴下した後、18時間撹拌した。該懸濁液を0℃に冷却した後、4時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は88.3%、Z体及びE体含有比率はZ/E=97.0/3.0であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.8%であった。
溶媒Aとしてトルエン及び酢酸エチルを、酸性化合物として濃硫酸を用いた化合物(Z)-1の硫酸塩の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)2.0g、2質量部のトルエン及び3質量部の酢酸エチルを30℃にて混合した。該混合溶液に1.1当量の濃硫酸(507mg)を添加した。混合溶液に同温度にて、参考例4で製造した化合物(Z)-1の硫酸塩を2.0mg添加した後、14時間撹拌することにより結晶を析出させた。該懸濁液に同温度で1質量部のn-ヘプタンを24時間かけて滴下した後、18時間撹拌した。該懸濁液を0℃に冷却した後、4時間撹拌した。析出した結晶をろ過し、得られた結晶を2質量部のトルエン及び1質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は88.3%、Z体及びE体含有比率はZ/E=97.0/3.0であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.8%であった。
上記、実施例10-1及び実施例10-2の反応条件と結果を表10に記載する。表中、「溶媒」に溶媒Aの種類を記載し、溶媒の質量部を括弧内に記載した。用いた酸性化合物の種類を「酸性化合物」に記載し、酸性化合物の当量を括弧内に記載した。

〔実施例11〕
化合物(E)-1を原料とした化合物(Z)-3の製造方法

化合物(E)-1(Z体/E体=0.1/99.9)3.60g及びトルエン7.20gを30℃にて混合した。該混合溶液に4.5質量%塩化水素/酢酸エチル溶液7.39g(塩化水素として1.14当量)を添加した後、同温度にて2時間撹拌した。該混合溶液に36mgの化合物(Z)-3を添加して2時間撹拌すると結晶が析出した。該懸濁液にn-ヘプタン14.4gを16時間かけて滴下し、30℃にて5時間撹拌した。該懸濁液を15℃に冷却し、3時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン7.20g及び酢酸エチル1.80gの混合溶液で洗浄することにより、目的物3.72gを淡黄色結晶として得た[化合物(Z)-1として定量収率92.2%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.1/1.9(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は97.6パーセントであった。
得られた結晶7.5mg及びトルエン5.0mLを室温にて混合し、0.005mol/L水酸化ナトリウム水溶液6.0mLを添加した。同温度にて30分間撹拌した後、水層と有機層を分離した。有機層に0.005mol/L水酸化ナトリウム水溶液3.0mLを添加し、30分間撹拌した後、水層と有機層を分離した。得られた水層を混合した。ICで分析した結果、塩化物イオンの含有量は7.1質量%であり、化合物(Z)-1と塩化水素のモル比が1:1であることを確認した。
化合物(E)-1を原料とした化合物(Z)-3の製造方法
化合物(E)-1(Z体/E体=0.1/99.9)3.60g及びトルエン7.20gを30℃にて混合した。該混合溶液に4.5質量%塩化水素/酢酸エチル溶液7.39g(塩化水素として1.14当量)を添加した後、同温度にて2時間撹拌した。該混合溶液に36mgの化合物(Z)-3を添加して2時間撹拌すると結晶が析出した。該懸濁液にn-ヘプタン14.4gを16時間かけて滴下し、30℃にて5時間撹拌した。該懸濁液を15℃に冷却し、3時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン7.20g及び酢酸エチル1.80gの混合溶液で洗浄することにより、目的物3.72gを淡黄色結晶として得た[化合物(Z)-1として定量収率92.2%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.1/1.9(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は97.6パーセントであった。
得られた結晶7.5mg及びトルエン5.0mLを室温にて混合し、0.005mol/L水酸化ナトリウム水溶液6.0mLを添加した。同温度にて30分間撹拌した後、水層と有機層を分離した。有機層に0.005mol/L水酸化ナトリウム水溶液3.0mLを添加し、30分間撹拌した後、水層と有機層を分離した。得られた水層を混合した。ICで分析した結果、塩化物イオンの含有量は7.1質量%であり、化合物(Z)-1と塩化水素のモル比が1:1であることを確認した。
〔実施例12〕
化合物(EZ)-1を原料とした化合物(Z)-1の製造方法

化合物(EZ)-1(Z体/E体=42.4/57.6)4.04g及びトルエン8.08gを25℃にて混合した。該混合溶液に4.4質量%塩化水素/酢酸エチル溶液8.93g(塩化水素として1.2当量)を添加した後、同温度にて30分間撹拌した。該混合溶液に2.0mgの化合物(Z)-3を添加して6時間撹拌すると結晶が析出した。該懸濁液にn-ヘプタン18.2gを8時間かけて滴下し、25℃にて9時間撹拌した。該懸濁液を0℃に冷却し、1時間撹拌した。該懸濁液に水20.2gを滴下した後、酢酸エチル18.2gを添加した。該反応液に0℃にて、8.0質量%水酸化ナトリウム水溶液6.74gを滴下し、同温度にて30分間撹拌した。撹拌終了後、25℃にて有機層を分液した。得られた有機層を水12.1gで洗浄し、減圧下にて溶媒を留去した。得られた残留物とトルエン12.1gとを混合した後、減圧下にて溶媒を留去した。得られた残留物とトルエン16.16gとを室温にて混合した後、70℃に加熱した。該混合溶液にn-ヘプタン8.08gを添加した後、同温度にて30分間撹拌した。該混合溶液を40℃に冷却し、同温度にて4.0mgの化合物(Z)-1を添加した。該混合溶液を20℃に冷却すると結晶が析出した。該懸濁液を同温度にて12時間撹拌した後、0℃に冷却した。該懸濁液を同温度にて5時間撹拌した後、析出した結晶をろ過し、得られた結晶をトルエン4.04g及びn-ヘプタン2.02gの混合溶液で洗浄することにより、目的物3.36gを淡黄色結晶として得た(収率83.3%)。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が97.3/2.7(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は98.2パーセントであった。
化合物(EZ)-1を原料とした化合物(Z)-1の製造方法
化合物(EZ)-1(Z体/E体=42.4/57.6)4.04g及びトルエン8.08gを25℃にて混合した。該混合溶液に4.4質量%塩化水素/酢酸エチル溶液8.93g(塩化水素として1.2当量)を添加した後、同温度にて30分間撹拌した。該混合溶液に2.0mgの化合物(Z)-3を添加して6時間撹拌すると結晶が析出した。該懸濁液にn-ヘプタン18.2gを8時間かけて滴下し、25℃にて9時間撹拌した。該懸濁液を0℃に冷却し、1時間撹拌した。該懸濁液に水20.2gを滴下した後、酢酸エチル18.2gを添加した。該反応液に0℃にて、8.0質量%水酸化ナトリウム水溶液6.74gを滴下し、同温度にて30分間撹拌した。撹拌終了後、25℃にて有機層を分液した。得られた有機層を水12.1gで洗浄し、減圧下にて溶媒を留去した。得られた残留物とトルエン12.1gとを混合した後、減圧下にて溶媒を留去した。得られた残留物とトルエン16.16gとを室温にて混合した後、70℃に加熱した。該混合溶液にn-ヘプタン8.08gを添加した後、同温度にて30分間撹拌した。該混合溶液を40℃に冷却し、同温度にて4.0mgの化合物(Z)-1を添加した。該混合溶液を20℃に冷却すると結晶が析出した。該懸濁液を同温度にて12時間撹拌した後、0℃に冷却した。該懸濁液を同温度にて5時間撹拌した後、析出した結晶をろ過し、得られた結晶をトルエン4.04g及びn-ヘプタン2.02gの混合溶液で洗浄することにより、目的物3.36gを淡黄色結晶として得た(収率83.3%)。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が97.3/2.7(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は98.2パーセントであった。
〔実施例13〕
化合物(EZ)-1を原料とした化合物(Z)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)5.04g、3質量部のトルエン及び1質量部のDMEを30℃にて混合した。該混合溶液に1.1当量の濃流酸(1.27g)を添加した後、同温度にて30分間撹拌した。該混合溶液に1.5mgの化合物(Z)-1の硫酸塩を添加して20.5時間撹拌すると結晶が析出した。該懸濁液を0℃に冷却し、2.5時間撹拌した。該懸濁液に5質量部の水を滴下した後、3質量部のトルエンを添加した。該反応液に0℃にて、2.2当量の8.0質量%水酸化ナトリウム水溶液(13.99g)を滴下し、同温度にて30分間撹拌した。撹拌終了後、25℃にて有機層を分液した後、得られた有機層を3質量部の水で2回洗浄した。得られた有機層を減圧下にて溶媒を留去した。得られた残留物、4質量部のトルエン及び2質量部のn-ヘプタンを70℃にて混合した。該混合溶液を40℃に冷却し、同温度にて5.0mgの化合物(Z)-1を添加し、30分間撹拌することにより結晶を析出させた。該懸濁液を0℃に冷却し、13時間撹拌した。析出した結晶をろ過し、得られた結晶を1質量部のトルエン及び0.5質量部のn-ヘプタンの混合溶液で洗浄することにより、3.96gの化合物(Z)-1を淡黄色結晶として得た(収率78.6%)。定性分析の結果、Z体とE体の面積比が97.1/2.9であり、2つのピークの相対面積百分率の和は99.2%であった。
化合物(EZ)-1を原料とした化合物(Z)-1の製造方法
化合物(EZ)-1(Z体/E体=50.4/49.6)5.04g、3質量部のトルエン及び1質量部のDMEを30℃にて混合した。該混合溶液に1.1当量の濃流酸(1.27g)を添加した後、同温度にて30分間撹拌した。該混合溶液に1.5mgの化合物(Z)-1の硫酸塩を添加して20.5時間撹拌すると結晶が析出した。該懸濁液を0℃に冷却し、2.5時間撹拌した。該懸濁液に5質量部の水を滴下した後、3質量部のトルエンを添加した。該反応液に0℃にて、2.2当量の8.0質量%水酸化ナトリウム水溶液(13.99g)を滴下し、同温度にて30分間撹拌した。撹拌終了後、25℃にて有機層を分液した後、得られた有機層を3質量部の水で2回洗浄した。得られた有機層を減圧下にて溶媒を留去した。得られた残留物、4質量部のトルエン及び2質量部のn-ヘプタンを70℃にて混合した。該混合溶液を40℃に冷却し、同温度にて5.0mgの化合物(Z)-1を添加し、30分間撹拌することにより結晶を析出させた。該懸濁液を0℃に冷却し、13時間撹拌した。析出した結晶をろ過し、得られた結晶を1質量部のトルエン及び0.5質量部のn-ヘプタンの混合溶液で洗浄することにより、3.96gの化合物(Z)-1を淡黄色結晶として得た(収率78.6%)。定性分析の結果、Z体とE体の面積比が97.1/2.9であり、2つのピークの相対面積百分率の和は99.2%であった。
〔実施例14〕
実施例1に準じた方法により反応を行い、析出した結晶をろ過した後、得られたろ液中のオキシイミノ化合物を出発物質として使用した。
〔実施例14-1〕
化合物(E)-1の製造方法
実施例1-1に準じた方法により、化合物(EZ)-1(Z体/E体=48.8/51.2)10g、1質量部の1,2-ジクロロエタン、0.025当量の4.5質量%塩化水素/酢酸エチル溶液0.450g及び酢酸エチル25.4gを用い、実施例1-1に準じた操作を行うことにより、淡黄色結晶(8.45g)を得た。定量分析の結果、化合物(E)-1の定量収率は82.8%であった。定性分析した結果、Z体とE体の面積比はZ/E=1.4/98.6であり、2つのピークの相対面積百分率の和は99.9%であった。一方、得られたろ液を、定量分析した結果、1.62gのZ体とE体の混合物(Z体及びE体含有比率はZ/E=67.8/32.2)が含まれていた。
この化合物(EZ)-1(Z体/E体=67.8/32.2)1.62gを含むろ液を減圧下にて濃縮し、1質量部の1,2-ジクロロエタン、0.025当量の4.5質量%塩化水素/酢酸エチル溶液(0.073g)及び酢酸エチル4.11gを用いて、再度上記に準じた操作を行うことにより1.31gの淡黄色結晶を得た。定量分析の結果化合物(E)-1の定量収率は12.5%[10gの化合物(EZ)-1より算出]であり、Z体及びE体含有比率はZ/E=1.8/98.2であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は99.7パーセントであった。
以上のようにろ液を使用し反応を繰り返すことにより、合計収率95.3%で化合物(E)-1を得た。
実施例1に準じた方法により反応を行い、析出した結晶をろ過した後、得られたろ液中のオキシイミノ化合物を出発物質として使用した。
〔実施例14-1〕
化合物(E)-1の製造方法
実施例1-1に準じた方法により、化合物(EZ)-1(Z体/E体=48.8/51.2)10g、1質量部の1,2-ジクロロエタン、0.025当量の4.5質量%塩化水素/酢酸エチル溶液0.450g及び酢酸エチル25.4gを用い、実施例1-1に準じた操作を行うことにより、淡黄色結晶(8.45g)を得た。定量分析の結果、化合物(E)-1の定量収率は82.8%であった。定性分析した結果、Z体とE体の面積比はZ/E=1.4/98.6であり、2つのピークの相対面積百分率の和は99.9%であった。一方、得られたろ液を、定量分析した結果、1.62gのZ体とE体の混合物(Z体及びE体含有比率はZ/E=67.8/32.2)が含まれていた。
この化合物(EZ)-1(Z体/E体=67.8/32.2)1.62gを含むろ液を減圧下にて濃縮し、1質量部の1,2-ジクロロエタン、0.025当量の4.5質量%塩化水素/酢酸エチル溶液(0.073g)及び酢酸エチル4.11gを用いて、再度上記に準じた操作を行うことにより1.31gの淡黄色結晶を得た。定量分析の結果化合物(E)-1の定量収率は12.5%[10gの化合物(EZ)-1より算出]であり、Z体及びE体含有比率はZ/E=1.8/98.2であった。定性分析の結果、Z体とE体のピークの相対面積百分率の和は99.7パーセントであった。
以上のようにろ液を使用し反応を繰り返すことにより、合計収率95.3%で化合物(E)-1を得た。
〔実施例14-2~14-3〕
実施例14-1に記載した方法に準じて、塩化水素/酢酸エチル溶液の使用量を変更して反応を行った。表中の表記表2と同様である。

実施例14-1に記載した方法に準じて、塩化水素/酢酸エチル溶液の使用量を変更して反応を行った。表中の表記表2と同様である。
〔参考例1〕
化合物(Z)-1の製造方法
特開2016-011286号に記載の合成例27の方法により得られた結晶を、HPLCを用いて定性分析した結果、保持時間10.8分のピークが観測された。
融点:105~107℃
1H-NMR:δ8.55-8.45(m, 2H), 8.14(s, 1H), 7.95-7.90(m, 1H), 7.16(t, J=54.3Hz, 1H), 4.26(d, J=6.0Hz, 2H), 4.30-4.20(m, 1H), 3.88(s, 3H), 1.65-1.55(m, 1H), 1.23(d, J=6.6Hz, 6H), 0.95-0.85(m, 2H), 0.80-0.73(m, 2H).
この結晶1mg及びクロロホルム1mLを混合し、n-ヘキサンを用いた蒸気拡散法にて柱状結晶を得た。得られた結晶を、下記の解析条件にて単結晶X線構造解析を行った結果、オキシイミノ基の立体配置はZであり、前記の結晶は化合物(Z)-1であることを確認した。
[解析条件]
装置:SMART APEXII ULTRA(ブルカーエイエックスエス社製)
X線:CuKα(50kV, 24mA)
測定温度:-75℃
化合物(Z)-1の製造方法
特開2016-011286号に記載の合成例27の方法により得られた結晶を、HPLCを用いて定性分析した結果、保持時間10.8分のピークが観測された。
融点:105~107℃
1H-NMR:δ8.55-8.45(m, 2H), 8.14(s, 1H), 7.95-7.90(m, 1H), 7.16(t, J=54.3Hz, 1H), 4.26(d, J=6.0Hz, 2H), 4.30-4.20(m, 1H), 3.88(s, 3H), 1.65-1.55(m, 1H), 1.23(d, J=6.6Hz, 6H), 0.95-0.85(m, 2H), 0.80-0.73(m, 2H).
この結晶1mg及びクロロホルム1mLを混合し、n-ヘキサンを用いた蒸気拡散法にて柱状結晶を得た。得られた結晶を、下記の解析条件にて単結晶X線構造解析を行った結果、オキシイミノ基の立体配置はZであり、前記の結晶は化合物(Z)-1であることを確認した。
[解析条件]
装置:SMART APEXII ULTRA(ブルカーエイエックスエス社製)
X線:CuKα(50kV, 24mA)
測定温度:-75℃
〔参考例2〕
化合物(EZ)-1の製造方法

参考例1で得られた化合物(Z)-1(Z体/E体=99.7/0.3)120g及び酢酸エチル180gを室温にて混合した。該混合溶液に4.5質量%塩化水素/酢酸エチル溶液5.40g(0.025当量)を室温にて添加した後、同温度にて48時間撹拌した。反応終了後、該反応溶液に室温にて水180g及び炭酸カリウム369g(0.1当量)を添加し、同温度にて30分間撹拌した。有機層を分離した後、飽和食塩水(100mLx2)次いで無水硫酸ナトリウムの順で脱水・乾燥、減圧下にて濃縮した。残留物にn-ヘプタン1800g及び酢酸エチル180gを室温にて添加し、同温度にて24時間撹拌した。析出した結晶をろ過し、n-ヘプタン200g及び酢酸エチル20gの混合溶液で洗浄することにより、乳白色結晶を114.7g得た。得られた結晶を、LC/MSを用いて質量分析した結果、2つのピーク(保持時間3.5分及び4.0分)が観測され、2つのピークのMSが共にm/z:450(M+)であることを確認した。また、得られた結晶を、HPLCを用いて定性分析した結果、2つのピークが観測され、2つのピークの相対面積百分率の和は99.8パーセントであった。また、2つのピークのうち、1つのピークは保持時間が10.8分(相対面積百分率51.6%)であり、もう1つのピークは保持時間が12.1分(相対面積百分率48.2%)であった。保持時間10.8分のピークは原料として用いたZ体であり、保持時間12.1分のピークは、上記LC/MSによる質量分析の結果及び1H-NMRの測定結果により、E体であることを確認した。以上によりHPLCを用いた定性分析において、Z体の保持時間は10.8分、E体の保持時間は12.1分であることが分かる。
(E)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミドの1H-NMRを以下に記す。
1H-NMR:δ8.42(d, J=1.8Hz, 1H), 8.30(t, J=5.7Hz, 1H), 8.03(s, 1H), 7.91(d, J=1.8Hz, 1H), 7.10(t, J=54.3Hz, 1H), 4.42(d, J=5.7Hz, 2H), 4.35(sep, J=6.6Hz, 1H), 3.83(s, 3H), 1.60-1.50(m, 1H), 1.23(d, J=6.6Hz, 6H), 0.95-0.85(m, 2H), 0.80-0.73(m, 2H).
化合物(EZ)-1の製造方法
(E)-N-[2-[3-クロロ-5-(シクロプロピルエチニル)ピリジン-2-イル]-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミドの1H-NMRを以下に記す。
1H-NMR:δ8.42(d, J=1.8Hz, 1H), 8.30(t, J=5.7Hz, 1H), 8.03(s, 1H), 7.91(d, J=1.8Hz, 1H), 7.10(t, J=54.3Hz, 1H), 4.42(d, J=5.7Hz, 2H), 4.35(sep, J=6.6Hz, 1H), 3.83(s, 3H), 1.60-1.50(m, 1H), 1.23(d, J=6.6Hz, 6H), 0.95-0.85(m, 2H), 0.80-0.73(m, 2H).
〔参考例3〕
化合物(Z)-1の塩酸塩[化合物(Z)-3]の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)1.80g及びトルエン3.60gを25℃にて混合した。該混合溶液に塩化水素の酢酸エチル溶液(4.5質量%)3.61g(塩化水素として1.11当量)を添加した後、同温度にて30分間撹拌した。該混合溶液に化合物(Z)-1の塩酸塩0.5(1,2-ジクロロエタン)和物18mgを加えて16時間撹拌すると結晶の析出が認められた。該懸濁液を15℃に冷却し、4時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン2.70g及び酢酸エチル0.90gの混合溶液で洗浄することにより、目的物0.86gを淡黄色結晶として得た[化合物(Z)-1として定量収率40.7%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.4/1.6(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は96.3パーセントであった。
化合物(Z)-1の塩酸塩[化合物(Z)-3]の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)1.80g及びトルエン3.60gを25℃にて混合した。該混合溶液に塩化水素の酢酸エチル溶液(4.5質量%)3.61g(塩化水素として1.11当量)を添加した後、同温度にて30分間撹拌した。該混合溶液に化合物(Z)-1の塩酸塩0.5(1,2-ジクロロエタン)和物18mgを加えて16時間撹拌すると結晶の析出が認められた。該懸濁液を15℃に冷却し、4時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン2.70g及び酢酸エチル0.90gの混合溶液で洗浄することにより、目的物0.86gを淡黄色結晶として得た[化合物(Z)-1として定量収率40.7%]。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.4/1.6(Rt=10.8分/12.1分)であり、2つのピークの相対面積百分率の和は96.3パーセントであった。
〔参考例4〕
化合物(Z)-1の硫酸塩の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)5.0g及び5質量部の酢酸エチルを0℃にて混合した。該混合溶液に1.0当量の濃硫酸(1.13g)を添加し、同温度にて30分間撹拌すると結晶が析出した。析出した結晶をろ過し、得られた結晶を2質量部の酢酸エチル及び2質量部のn-ヘプタンの混合溶液で洗浄することにより、白色結晶6.00gを得た。定量分析の結果、化合物(Z)-1の定量収率94.6%、Z体及びE体含有比率はZ/E=98.6/1.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
得られた結晶1.0mg及びトルエン2.0mLを室温にて混合し、0.005mol/L水酸化ナトリウム水溶液10mLを添加した。同温度にて30分間撹拌した後、水層と有機層を分離した。得られた水層をICで分析した結果、硫酸イオンの含有量は17.9質量%であり、化合物(Z)-1と硫酸のモル比が1:1であることを確認した。
1H-NMR: δ8.51(d, J=6.0Hz, 1H), 8.50(t, J=1.5Hz, 1H), 8.16(s, 1H), 7.95(t, J=1.5Hz, 1H), 7.41(brs, 2H, 硫酸), 7.17(t, J=54.6Hz, 1H), 4.30-4.20(m, 3H), 3.90(s, 3H), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
化合物(Z)-1の硫酸塩の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)5.0g及び5質量部の酢酸エチルを0℃にて混合した。該混合溶液に1.0当量の濃硫酸(1.13g)を添加し、同温度にて30分間撹拌すると結晶が析出した。析出した結晶をろ過し、得られた結晶を2質量部の酢酸エチル及び2質量部のn-ヘプタンの混合溶液で洗浄することにより、白色結晶6.00gを得た。定量分析の結果、化合物(Z)-1の定量収率94.6%、Z体及びE体含有比率はZ/E=98.6/1.4であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.9%であった。
得られた結晶1.0mg及びトルエン2.0mLを室温にて混合し、0.005mol/L水酸化ナトリウム水溶液10mLを添加した。同温度にて30分間撹拌した後、水層と有機層を分離した。得られた水層をICで分析した結果、硫酸イオンの含有量は17.9質量%であり、化合物(Z)-1と硫酸のモル比が1:1であることを確認した。
1H-NMR: δ8.51(d, J=6.0Hz, 1H), 8.50(t, J=1.5Hz, 1H), 8.16(s, 1H), 7.95(t, J=1.5Hz, 1H), 7.41(brs, 2H, 硫酸), 7.17(t, J=54.6Hz, 1H), 4.30-4.20(m, 3H), 3.90(s, 3H), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
〔参考例5〕
化合物(Z)-1のメタンスルホン酸塩の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)20g及び3質量部の酢酸エチルを0℃にて混合した。該混合溶液に1.0当量のメタンスルホン酸(4.27g)を添加し、同温度にて1時間撹拌すると、結晶が析出した。析出した結晶をろ過し、得られた結晶を2質量部の酢酸エチル及び2質量部のn-ヘプタンの混合溶液で洗浄することにより、白色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は88.4%、Z体及びE体含有比率はZ/E=99.1/0.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.2%であった。
1H-NMR: δ8.90(brs, 1H, メタンスルホン酸), 8.54(d, J=6.0Hz, 1H), 8.50(t, J=1.5Hz, 1H), 8.17(s, 1H), 7.96(d, J=1.5Hz, 1H), 7.17(t, J=54.0Hz, 1H), 4.30-4.20(m, 3H), 3.90(s, 3H), 2.40(s, 3H, メタンスルホン酸), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
化合物(Z)-1のメタンスルホン酸塩の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)20g及び3質量部の酢酸エチルを0℃にて混合した。該混合溶液に1.0当量のメタンスルホン酸(4.27g)を添加し、同温度にて1時間撹拌すると、結晶が析出した。析出した結晶をろ過し、得られた結晶を2質量部の酢酸エチル及び2質量部のn-ヘプタンの混合溶液で洗浄することにより、白色結晶を得た。定量分析の結果、化合物(Z)-1の定量収率は88.4%、Z体及びE体含有比率はZ/E=99.1/0.9であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.2%であった。
1H-NMR: δ8.90(brs, 1H, メタンスルホン酸), 8.54(d, J=6.0Hz, 1H), 8.50(t, J=1.5Hz, 1H), 8.17(s, 1H), 7.96(d, J=1.5Hz, 1H), 7.17(t, J=54.0Hz, 1H), 4.30-4.20(m, 3H), 3.90(s, 3H), 2.40(s, 3H, メタンスルホン酸), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
〔参考例6〕
化合物(Z)-1のp-トルエンスルホン酸塩の製造方法
p-トルエンスルホン酸1水和物11.05g及び10質量部のトルエンを室温にて混合した。該混合溶液を還流下で2時間撹拌し、Dean-Stark装置を用いて共沸脱水した後、該混合溶液の重量が100gとなるようトルエンを添加した。
化合物(Z)-1(Z体/E体=99.7/0.3)3.0g及び2質量部の酢酸エチルを0℃して混合した。該混合溶液に、12.63gの上記混合溶液(1.1当量のp-トルエンスルホン酸を含む。)を添加し、同温度にて30分間撹拌すると、結晶が析出した。析出した結晶をろ過し、得られた結晶を2質量部の酢酸エチル及び2質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶2.15gを得た。定量分析の結果、化合物(Z)-1の定量収率は45.0%、Z体及びE体含有比率はZ/E=95.5/4.5であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.7%であった。
1H-NMR: δ12.20(brs, 1H, p-トルエンスルホン酸), 8.54(d, J=6.0Hz, 1H), 8.50(t, J=1.5Hz, 1H), 8.19(s, 1H), 7.96(t, J=1.5Hz, 1H), 7.52(d, J=8.1Hz, 2H, p-トルエンスルホン酸), 7.18(t, J=54.9Hz, 1H), 7.15(d, J=8.1Hz, 2H, p-トルエンスルホン酸), 4.30-4.20(m, 3H), 3.90(s, 3H), 2.30(s, 3H, p-トルエンスルホン酸), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
化合物(Z)-1のp-トルエンスルホン酸塩の製造方法
p-トルエンスルホン酸1水和物11.05g及び10質量部のトルエンを室温にて混合した。該混合溶液を還流下で2時間撹拌し、Dean-Stark装置を用いて共沸脱水した後、該混合溶液の重量が100gとなるようトルエンを添加した。
化合物(Z)-1(Z体/E体=99.7/0.3)3.0g及び2質量部の酢酸エチルを0℃して混合した。該混合溶液に、12.63gの上記混合溶液(1.1当量のp-トルエンスルホン酸を含む。)を添加し、同温度にて30分間撹拌すると、結晶が析出した。析出した結晶をろ過し、得られた結晶を2質量部の酢酸エチル及び2質量部のn-ヘプタンの混合溶液で洗浄することにより、淡黄色結晶2.15gを得た。定量分析の結果、化合物(Z)-1の定量収率は45.0%、Z体及びE体含有比率はZ/E=95.5/4.5であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は98.7%であった。
1H-NMR: δ12.20(brs, 1H, p-トルエンスルホン酸), 8.54(d, J=6.0Hz, 1H), 8.50(t, J=1.5Hz, 1H), 8.19(s, 1H), 7.96(t, J=1.5Hz, 1H), 7.52(d, J=8.1Hz, 2H, p-トルエンスルホン酸), 7.18(t, J=54.9Hz, 1H), 7.15(d, J=8.1Hz, 2H, p-トルエンスルホン酸), 4.30-4.20(m, 3H), 3.90(s, 3H), 2.30(s, 3H, p-トルエンスルホン酸), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
〔参考例7〕
化合物(Z)-1のシュウ酸塩の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)1.0g及び3質量部のトルエンを0℃にて混合した。該混合溶液に1.0当量のシュウ酸(200mg)を添加し、同温度にて20時間撹拌すると、結晶が析出した。析出した結晶をろ過し、得られた結晶を1質量部のトルエンで洗浄することにより、白色結晶989mgを得た。定量分析の結果、化合物(Z)-1の定量収率は80.0%、Z体及びE体含有比率はZ/E=99.3/0.7)であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.8%であった。
得られた結晶50mg及びメタノール5.0mLを室温にて混合した。該混合溶液に、0.005mol/L水酸化ナトリウム水溶液を添加した。得られた混合溶液をICで分析した結果、シュウ酸イオンの含有量は17.5質量%であり、化合物(Z)-1と硫酸のモル比が1:1であることを確認した。
1H-NMR: δ8.55-8.45(m, 2H), 8.16(s, 1H), 8.00-7.95(m, 1H), 7.18(t, J=54.3Hz, 1H), 4.30-4.20(m, 3H), 3.90(s, 3H), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
化合物(Z)-1のシュウ酸塩の製造方法
化合物(Z)-1(Z体/E体=99.7/0.3)1.0g及び3質量部のトルエンを0℃にて混合した。該混合溶液に1.0当量のシュウ酸(200mg)を添加し、同温度にて20時間撹拌すると、結晶が析出した。析出した結晶をろ過し、得られた結晶を1質量部のトルエンで洗浄することにより、白色結晶989mgを得た。定量分析の結果、化合物(Z)-1の定量収率は80.0%、Z体及びE体含有比率はZ/E=99.3/0.7)であった。定性分析の結果、Z体及びE体のピークの相対面積百分率の和は99.8%であった。
得られた結晶50mg及びメタノール5.0mLを室温にて混合した。該混合溶液に、0.005mol/L水酸化ナトリウム水溶液を添加した。得られた混合溶液をICで分析した結果、シュウ酸イオンの含有量は17.5質量%であり、化合物(Z)-1と硫酸のモル比が1:1であることを確認した。
1H-NMR: δ8.55-8.45(m, 2H), 8.16(s, 1H), 8.00-7.95(m, 1H), 7.18(t, J=54.3Hz, 1H), 4.30-4.20(m, 3H), 3.90(s, 3H), 1.65-1.55(m, 1H), 1.10(d, J=6.0Hz, 6H), 1.00-0.90(m, 2H), 0.85-0.75(m, 2H).
〔参考例8〕
化合物(E)-1の粉末X線回折
実施例1-1で得られた化合物(E)-1の粉末X線回折を行った。粉末X線回折スペクトルのチャートを図1に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.35、8.00、12.83、14.62、15.26、15.93、16.35、16.64、18.00、18.22、18.61、20.66、22.07、22.64、23.15、23.40、23.96、24.77、25.55、25.75及び28.24。
化合物(E)-1の粉末X線回折
実施例1-1で得られた化合物(E)-1の粉末X線回折を行った。粉末X線回折スペクトルのチャートを図1に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.35、8.00、12.83、14.62、15.26、15.93、16.35、16.64、18.00、18.22、18.61、20.66、22.07、22.64、23.15、23.40、23.96、24.77、25.55、25.75及び28.24。
〔参考例9〕
化合物(Z)-2の粉末X線回折
実施例1-2で得られた化合物(Z)-2の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図2に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.02、8.85、13.96、15.39、15.71、17.45、18.00、18.34、20.03、20.62、20.83、21.05、21.67、22.66、22.86、23.40、23.61、23.86、25.13、25.98及び28.14。
化合物(Z)-2の粉末X線回折
実施例1-2で得られた化合物(Z)-2の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図2に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.02、8.85、13.96、15.39、15.71、17.45、18.00、18.34、20.03、20.62、20.83、21.05、21.67、22.66、22.86、23.40、23.61、23.86、25.13、25.98及び28.14。
〔参考例10〕
化合物(Z)-3の粉末X線回折
実施例11で得られた化合物(Z)-3の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図3に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.05、10.48、14.03、14.21、15.79、17.47、18.47、20.02、20.70、21.09、21.56、21.85、22.26、22.83、23.42、23.80、25.17及び25.76。
化合物(Z)-3の粉末X線回折
実施例11で得られた化合物(Z)-3の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図3に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.05、10.48、14.03、14.21、15.79、17.47、18.47、20.02、20.70、21.09、21.56、21.85、22.26、22.83、23.42、23.80、25.17及び25.76。
〔参考例11〕
化合物(Z)-1の粉末X線回折
実施例12で得られた化合物(Z)-1の粉末X線回折を行った。得られた粉末X線回折チャートを図4に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.54、8.91、9.67、14.07、14.50、15.00、15.88、17.42、17.95、18.14、20.74、21.01、21.77、22.22、22.54、23.26、25.49、25.86及び26.70。
化合物(Z)-1の粉末X線回折
実施例12で得られた化合物(Z)-1の粉末X線回折を行った。得られた粉末X線回折チャートを図4に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.54、8.91、9.67、14.07、14.50、15.00、15.88、17.42、17.95、18.14、20.74、21.01、21.77、22.22、22.54、23.26、25.49、25.86及び26.70。
〔参考例12〕
化合物(Z)-1の硫酸塩の粉末X線回折
参考例4で得られた化合物(Z)-1の硫酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図5に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):5.45、10.81、12.02、14.29、15.45、15.70、16.31、19.44、21.37、22.07、22.43、22.75、23.74、24.41、24.92及び25.59。
化合物(Z)-1の硫酸塩の粉末X線回折
参考例4で得られた化合物(Z)-1の硫酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図5に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):5.45、10.81、12.02、14.29、15.45、15.70、16.31、19.44、21.37、22.07、22.43、22.75、23.74、24.41、24.92及び25.59。
〔参考例13〕
化合物(Z)-1のメタンスルホン酸塩の粉末X線回折
参考例5で得られた化合物(Z)-1のメタンスルホン酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図6に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):5.39、8.03、10.68、11.17、12.50、12.73、15.86、16.22、19.07、20.28、20.57、21.32、21.72、22.27、22.53、23.99、25.03、25.31、25.56及び25.79。
化合物(Z)-1のメタンスルホン酸塩の粉末X線回折
参考例5で得られた化合物(Z)-1のメタンスルホン酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図6に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):5.39、8.03、10.68、11.17、12.50、12.73、15.86、16.22、19.07、20.28、20.57、21.32、21.72、22.27、22.53、23.99、25.03、25.31、25.56及び25.79。
〔参考例14〕
化合物(Z)-1のp-トルエンスルホン酸塩の粉末X線回折
参考例6で得られた化合物(Z)-1のp-トルエンスルホン酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図7に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.56、9.99、11.64、12.37、14.54、15.04、15.24、17.14、18.55、19.97、20.42、22.04、22.61、22.84、23.34、24.15、25.26、26.52及び26.97。
化合物(Z)-1のp-トルエンスルホン酸塩の粉末X線回折
参考例6で得られた化合物(Z)-1のp-トルエンスルホン酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図7に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):7.56、9.99、11.64、12.37、14.54、15.04、15.24、17.14、18.55、19.97、20.42、22.04、22.61、22.84、23.34、24.15、25.26、26.52及び26.97。
〔参考例15〕
化合物(Z)-1のシュウ酸塩の粉末X線回折
参考例7で得られた化合物(Z)-1のシュウ酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図8に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):6.60、9.26、10.76、13.17、14.55、14.78、15.67、18.97、19.75、20.02、21.37、22.92、23.82、25.45、27.09及び28.03。
化合物(Z)-1のシュウ酸塩の粉末X線回折
参考例7で得られた化合物(Z)-1のシュウ酸塩の粉末X線回折を行った。得られた粉末X線回折スペクトルのチャートを図8に示す。粉末X線回折において下記のピーク値が特徴的なピークとして得られた。尚、粉末X線回折のピーク値の誤差としては、通常±0.2を取りえる。
回折角度(2θ):6.60、9.26、10.76、13.17、14.55、14.78、15.67、18.97、19.75、20.02、21.37、22.92、23.82、25.45、27.09及び28.03。
〔参考例16〕
(Z)-N-[2-(3,5-ジクロロピリジン-2-イル)-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミドの製造方法

国際公開第2014/010737号(WO2014/010737)に記載の既知の方法に従って合成した(EZ)-N-[2-(3,5-ジクロロピリジン-2-イル)-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミド(Z体/E体=15.2/84.8)168g及び酢酸エチル672gを室温にて混合し、該混合溶液に4.5質量%塩化水素/酢酸エチル溶液20mL(塩化水素として0.056当量)を35℃にて添加した。該混合溶液を同温度にて1時間撹拌した後、減圧下にて溶媒を留去した。得られた残留物に酢酸エチルを室温にて加え、酢酸エチル溶液420gを調整した。該酢酸エチル溶液に4.5質量%塩化水素/酢酸エチル溶液20mL(塩化水素として0.056当量)を35℃にて添加した。同温度にて該反応液にn-ヘプタン252gを1時間かけて滴下すると、結晶の析出が認められた。n-ヘプタン滴下終了後、該反応混合物を同温度にて1時間撹拌した。撹拌終了後、n-ヘプタン252gを1時間かけて滴下し、該反応混合物を1時間撹拌した。撹拌終了後、n-ヘプタン252gを1時間かけて滴下し、該反応混合物を1時間撹拌した。次いで、該反応混合物を室温にて24時間撹拌した。析出した結晶をろ過し、得られた結晶をn-ヘプタン268.8g及び酢酸エチル67.2gの混合溶液で洗浄することにより、目的物144.5gを淡黄色結晶として得た(収率86.0%)。得られた結晶を、HPLCを用いて定性分析した結果、Z体とE体の面積比が98.2/1.8であり、2つのピークの相対面積百分率の和は98.6パーセントであった。
融点:126.0~127.0℃
1H-NMR: δ8.61(d, J=2.1Hz, 1H), 8.55(t, J=6.3Hz, 1H), 8.25(d, J=2.1Hz, 1H), 8.14(s, 1H), 7.15(t, J=54.6Hz, 1H), 4.35-4.20(m, 3H), 3.89(s, 3H), 1.10(d, J=6.3Hz, 6H).
(Z)-N-[2-(3,5-ジクロロピリジン-2-イル)-2-(イソプロポキシイミノ)エチル]-3-(ジフルオロメチル)-1-メチル-1H-ピラゾール-4-カルボキサミドの製造方法
融点:126.0~127.0℃
1H-NMR: δ8.61(d, J=2.1Hz, 1H), 8.55(t, J=6.3Hz, 1H), 8.25(d, J=2.1Hz, 1H), 8.14(s, 1H), 7.15(t, J=54.6Hz, 1H), 4.35-4.20(m, 3H), 3.89(s, 3H), 1.10(d, J=6.3Hz, 6H).
本発明に係る幾何異性体の製造方法は、医薬・農薬として有用なオキシイミノ化合物の効率的な製造方法として極めて有用である。
なお、2017年02月08日に出願された日本特許出願2017-021298号及び2017年12月15日に出願された日本特許出願2017-241013号の明細書、特許請求の範囲及び要約書の全内容をここに引用し、本発明の明細書の開示として、取り入れるものである。
なお、2017年02月08日に出願された日本特許出願2017-021298号及び2017年12月15日に出願された日本特許出願2017-241013号の明細書、特許請求の範囲及び要約書の全内容をここに引用し、本発明の明細書の開示として、取り入れるものである。
Claims (11)
- 式(EZ)-1;


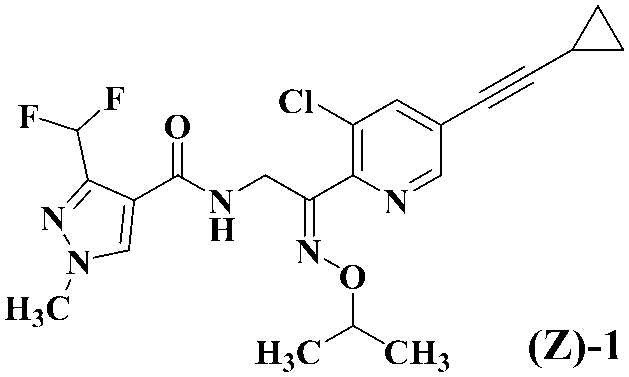
(i)式(EZ)-1で表される化合物の幾何異性体の混合物又は式(Z)-1で表されるオキシイミノ化合物のZ体を出発物質とし、溶媒中で該出発物質及び該出発物質に対して0.1当量以下の酸性化合物を混合して、式(E)-1で表されるオキシイミノ化合物のE体を製造するか、又は
(ii)式(EZ)-1で表される化合物の幾何異性体の混合物又は式(E)-1で表されるオキシイミノ化合物のE体を出発物質とし、溶媒中で該出発物質及び該出発物質に対して0.7当量以上の酸性化合物を混合して、式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、
ことを特徴とするオキシイミノ化合物の立体選択的な製造方法。 - 出発物質が、式(EZ)-1で表される化合物の幾何異性体の混合物である、請求項1に記載のオキシイミノ化合物の立体選択的な製造方法。
- 出発物質を溶媒に溶解させた後、酸性化合物を添加する、請求項1又は請求項2に記載のオキシイミノ化合物の立体選択的な製造方法。
- 出発物質に対して0.01当量以上0.07当量以下の酸性化合物を用いて、式(E)-1で表されるオキシイミノ化合物のE体を製造する、請求項1乃至請求項3のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
- 出発物質に対して1.0当量以上2.0当量以下の酸性化合物を用いて、式(Z)-1で表されるオキシイミノ化合物のZ体を製造する、請求項1乃至請求項3のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
- 酸性化合物が、ハロゲン化水素、硫酸又はメタンスルホン酸である、請求項1乃至請求項5のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
- 溶媒として、芳香族炭化水素溶媒、エーテル溶媒、ケトン溶媒、エステル溶媒及びハロゲン化炭化水素溶媒からなる群から選ばれる1種以上の溶媒を用いる、請求項1乃至請求項6のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
- 溶媒として、トルエン、オルトキシレン、シクロペンチルメチルエーテル、ターシャリーブチルメチルエーテル、ジメトキシエタン、ジエチレングリコールジメチルエーテル、メチルエチルケトン、酢酸エチル及び1,2-ジクロロエタンからなる群から選ばれる1種以上の溶媒を用いる、請求項7に記載のオキシイミノ化合物の立体選択的な製造方法。
- 出発物質及び酸性化合物を溶媒中で混合した後、脂肪族炭化水素溶媒から選ばれる1種以上の溶媒を添加する工程を含む、請求項1乃至請求項8のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
- 脂肪族炭化水素溶媒がノルマルヘプタンである、請求項9に記載のオキシイミノ化合物の立体選択的な製造方法。
- 析出した、結晶、塩若しくはその溶媒和物、又は溶媒和物を反応系から分離する、請求項1乃至請求項10のいずれか1項に記載のオキシイミノ化合物の立体選択的な製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/484,337 US10710977B2 (en) | 2017-02-08 | 2018-02-08 | Method for producing geometrical isomer of oximino compound |
| CN201880004553.0A CN109996793B (zh) | 2017-02-08 | 2018-02-08 | 肟基化合物的几何异构体的制造方法 |
| JP2018567488A JP7021643B2 (ja) | 2017-02-08 | 2018-02-08 | オキシイミノ化合物の幾何異性体の製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017021298 | 2017-02-08 | ||
| JP2017-021298 | 2017-02-08 | ||
| JP2017-241013 | 2017-12-15 | ||
| JP2017241013 | 2017-12-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018147368A1 true WO2018147368A1 (ja) | 2018-08-16 |
Family
ID=63107584
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/004391 WO2018147368A1 (ja) | 2017-02-08 | 2018-02-08 | オキシイミノ化合物の幾何異性体の製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10710977B2 (ja) |
| JP (1) | JP7021643B2 (ja) |
| CN (1) | CN109996793B (ja) |
| WO (1) | WO2018147368A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2018147368A1 (ja) * | 2017-02-08 | 2019-12-12 | 日産化学株式会社 | オキシイミノ化合物の幾何異性体の製造方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5551049A (en) * | 1978-08-16 | 1980-04-14 | Mobil Oil | Manufacture of e isomer of over 98 * purity of arylalkylketoxime |
| JPH03200754A (ja) * | 1989-12-28 | 1991-09-02 | Sumitomo Chem Co Ltd | オキシムエーテルの異性化方法 |
| JPH06219986A (ja) * | 1992-11-02 | 1994-08-09 | Shionogi & Co Ltd | (e)−アルコキシイミノまたはヒドロキシイミノアセトアミド系化合物の製造法およびその製造用中間体 |
| JPH09143138A (ja) * | 1995-11-28 | 1997-06-03 | Sumitomo Chem Co Ltd | (e)−2’,4’−ジクロロアセトフェノンオキシムの製造法 |
| JPH10195064A (ja) * | 1997-01-13 | 1998-07-28 | Katayama Seiyakushiyo:Kk | 5−アミノ−1,2,4−チアジアゾール酢酸誘導体(シン異性体)の製造方法 |
| WO2011093423A1 (ja) * | 2010-01-29 | 2011-08-04 | 田辺三菱製薬株式会社 | (e)-ヒドロキシイミノフェニル酢酸誘導体の製法 |
| JP2016011286A (ja) * | 2014-01-15 | 2016-01-21 | 日産化学工業株式会社 | オキシム置換アミド化合物及び有害生物防除剤 |
| JP2017100972A (ja) * | 2015-11-30 | 2017-06-08 | 日産化学工業株式会社 | オキシイミノ誘導体の幾何異性体の製造方法 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4434182A (en) | 1982-11-01 | 1984-02-28 | Fmc Corporation | Insecticidal substituted-biphenylmethyl oxime ethers |
| EP0741125A1 (en) | 1992-11-02 | 1996-11-06 | Shionogi Seiyaku Kabushiki Kaisha | Benzyl compounds and process for producing same |
| AR036881A1 (es) * | 2001-10-15 | 2004-10-13 | Schering Corp | Un proceso para la preparacion de 4-(piperidil)(2-piridil)metanona-(e)-o-metiloxima y sales |
| AR036882A1 (es) * | 2001-10-15 | 2004-10-13 | Schering Corp | Sintesis de (4-bromofenil)(4-piperidil)metanona-(z)-o-etiloxima y sales |
| US20110105794A1 (en) | 2008-06-30 | 2011-05-05 | Basf Se | Process for the Isomerization of Semicarbazone Compounds |
| AU2013233473B2 (en) | 2012-03-13 | 2015-07-09 | Nippon Soda Co., Ltd. | Compound, method for producing compound, and method for purifying compound |
| AU2013287590B2 (en) | 2012-07-12 | 2017-02-09 | Nissan Chemical Corporation | Oxime-substituted amide compound and pest control agent |
| BR112016017430B1 (pt) | 2014-02-07 | 2021-07-13 | Nissan Chemical Industries Ltd | Composição fungicida, inseticida, nematicida ou bactericida e método para controlar pragas, insetos nocivos, nematódeos ou bactérias |
| US10029986B2 (en) | 2014-02-18 | 2018-07-24 | Nissan Chemical Industries, Ltd. | Alkynyl pyridine-substituted amide compound and pesticide |
| JP2017137283A (ja) | 2015-05-15 | 2017-08-10 | 日産化学工業株式会社 | 5−アルキニルピリジン化合物及びその製造方法 |
| WO2018003924A1 (ja) | 2016-06-29 | 2018-01-04 | 日産化学工業株式会社 | アルキニルピリジン置換アミド化合物及び有害生物防除剤 |
| JP7021643B2 (ja) | 2017-02-08 | 2022-02-17 | 日産化学株式会社 | オキシイミノ化合物の幾何異性体の製造方法 |
| CN110606840A (zh) | 2018-06-15 | 2019-12-24 | 日产化学株式会社 | 5-炔基吡啶化合物的制造方法 |
-
2018
- 2018-02-08 JP JP2018567488A patent/JP7021643B2/ja active Active
- 2018-02-08 CN CN201880004553.0A patent/CN109996793B/zh active Active
- 2018-02-08 WO PCT/JP2018/004391 patent/WO2018147368A1/ja active Application Filing
- 2018-02-08 US US16/484,337 patent/US10710977B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5551049A (en) * | 1978-08-16 | 1980-04-14 | Mobil Oil | Manufacture of e isomer of over 98 * purity of arylalkylketoxime |
| JPH03200754A (ja) * | 1989-12-28 | 1991-09-02 | Sumitomo Chem Co Ltd | オキシムエーテルの異性化方法 |
| JPH06219986A (ja) * | 1992-11-02 | 1994-08-09 | Shionogi & Co Ltd | (e)−アルコキシイミノまたはヒドロキシイミノアセトアミド系化合物の製造法およびその製造用中間体 |
| JPH09143138A (ja) * | 1995-11-28 | 1997-06-03 | Sumitomo Chem Co Ltd | (e)−2’,4’−ジクロロアセトフェノンオキシムの製造法 |
| JPH10195064A (ja) * | 1997-01-13 | 1998-07-28 | Katayama Seiyakushiyo:Kk | 5−アミノ−1,2,4−チアジアゾール酢酸誘導体(シン異性体)の製造方法 |
| WO2011093423A1 (ja) * | 2010-01-29 | 2011-08-04 | 田辺三菱製薬株式会社 | (e)-ヒドロキシイミノフェニル酢酸誘導体の製法 |
| JP2016011286A (ja) * | 2014-01-15 | 2016-01-21 | 日産化学工業株式会社 | オキシム置換アミド化合物及び有害生物防除剤 |
| JP2017100972A (ja) * | 2015-11-30 | 2017-06-08 | 日産化学工業株式会社 | オキシイミノ誘導体の幾何異性体の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| MARUZEN KK: "Synthesis and Reactions of Organic Compounds III", EXPERIMENTAL CHEMISTRY, vol. 14, 20 February 1978 (1978-02-20), pages 1331 - 1332 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2018147368A1 (ja) * | 2017-02-08 | 2019-12-12 | 日産化学株式会社 | オキシイミノ化合物の幾何異性体の製造方法 |
| JP7021643B2 (ja) | 2017-02-08 | 2022-02-17 | 日産化学株式会社 | オキシイミノ化合物の幾何異性体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109996793B (zh) | 2022-02-25 |
| JPWO2018147368A1 (ja) | 2019-12-12 |
| US10710977B2 (en) | 2020-07-14 |
| CN109996793A (zh) | 2019-07-09 |
| JP7021643B2 (ja) | 2022-02-17 |
| US20190367479A1 (en) | 2019-12-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10196373B2 (en) | Substituted 2-hydroxy-4-(2-(phenylsulfonamido)acetamido)benzoic acid analogs as inhibitors of STAT protein | |
| JP4996786B2 (ja) | 結晶質の化合物ビス[(e)−7−[4−(4−フルオロフェニル)−6−イソプロピル−2−[メチル(メチルスルホニル)アミノ]ピリミジン−5−イル](3r,5s)−3,5−ジヒドロキシ−6−ヘプテン酸]カルシウム塩 | |
| JP2022502385A (ja) | Shp2の活性を阻害するための化合物の製造方法 | |
| JP2008510778A5 (ja) | ||
| WO2013047738A1 (ja) | グルホシネートp遊離酸の製造方法 | |
| JP2016523256A (ja) | ヒストン脱メチル化酵素阻害剤としての置換(e)−n’−(1−フェニルエチリデン)ベンゾヒドラジド類似体 | |
| SK15892000A3 (sk) | Spôsob syntézy cox-2 inhibítorov a medziprodukty | |
| MXPA05004661A (es) | Procedimientos para preparar oxazolidinonas n-ariladas mediante una reaccion de acoplamiento cruzado catalizada por cobre. | |
| KR20110126694A (ko) | Dhodh 억제제로서의 히드록실 및/또는 카르복실 기를 함유하는 아민의 아미노 니코틴산 유도체와의 부가염 | |
| MX2008012398A (es) | Proceso para la preparacion de inhibidores de proteasa de vih. | |
| WO2018147368A1 (ja) | オキシイミノ化合物の幾何異性体の製造方法 | |
| Sasaki et al. | Synthesis of α, α-difluoro-β-hydroxy ketone via the La (OTf) 3-catalyzed aldol reaction of carbonyl compounds with difluoroenol O-Boc esters | |
| JP2003064033A (ja) | 核フッ素化芳香族の改良製法 | |
| US11542254B2 (en) | Methods and composition of 4-substituted benzoylpiperazine-1-substituted carbonyls as beta-catenin/B-cell lymphoma 9 inhibitors | |
| ES2364632T3 (es) | Procedimiento de producción de éster de ácido oxoheptenoico ópticamente activo. | |
| JP2023545129A (ja) | (6r,10s)-10-{4-[5-クロロ-2-(4-クロロ-1h-1,2,3-トリアゾール-1-イル)フェニル]-6-オキソ-1(6h)-ピリミジニル}- 1-(ジフルオロメチル)-6-メチル-1,4,7,8,9,10-ヘキサヒドロ-11,15-(メタノ)ピラゾロ[4,3-b][1,7]ジアザシクロテトラデシン-5(6h)-オンの製造方法 | |
| JPS59104364A (ja) | 新規ピリミジン誘導体およびその製造法 | |
| WO2018159846A1 (ja) | α,β-不飽和酸エステルまたはα-ハロエステルの製造方法 | |
| JP2007332049A (ja) | 光学活性ベンジルオキシピロリジン誘導体塩酸塩粉体及びその製造法 | |
| JP2017100972A (ja) | オキシイミノ誘導体の幾何異性体の製造方法 | |
| JP2003300952A (ja) | スルフィド誘導体の製法 | |
| WO2021023993A1 (en) | Molecular complexes | |
| US20080096973A1 (en) | Z-stilbenes derivatives and the pharmaceutical composition thereof | |
| FENG | CHIRAL CYCLIC SELENIUM CATALYZED ENANTIOSLECTIVE HALOCYLIZATION REACTIONS | |
| WO2011114212A1 (en) | Ammonium, calcium and tris salts of fosamprenavir |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18751442 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018567488 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 18751442 Country of ref document: EP Kind code of ref document: A1 |