WO2015131405A1 - 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 - Google Patents
用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 Download PDFInfo
- Publication number
- WO2015131405A1 WO2015131405A1 PCT/CN2014/073079 CN2014073079W WO2015131405A1 WO 2015131405 A1 WO2015131405 A1 WO 2015131405A1 CN 2014073079 W CN2014073079 W CN 2014073079W WO 2015131405 A1 WO2015131405 A1 WO 2015131405A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- intermediate compound
- compound
- preparation
- reaction
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 81
- LALFOYNTGMUKGG-BGRFNVSISA-L rosuvastatin calcium Chemical compound [Ca+2].CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O.CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O LALFOYNTGMUKGG-BGRFNVSISA-L 0.000 title claims abstract description 43
- 229960004796 rosuvastatin calcium Drugs 0.000 title claims abstract description 42
- 238000000034 method Methods 0.000 title claims abstract description 20
- 238000002360 preparation method Methods 0.000 claims abstract description 34
- 159000000007 calcium salts Chemical class 0.000 claims abstract description 12
- 125000006239 protecting group Chemical group 0.000 claims abstract description 11
- 238000007239 Wittig reaction Methods 0.000 claims abstract description 10
- 238000006460 hydrolysis reaction Methods 0.000 claims abstract description 10
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 8
- 230000007062 hydrolysis Effects 0.000 claims abstract description 7
- 239000002994 raw material Substances 0.000 claims abstract description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 42
- 238000006243 chemical reaction Methods 0.000 claims description 31
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 18
- 239000003960 organic solvent Substances 0.000 claims description 18
- 239000002904 solvent Substances 0.000 claims description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 16
- -1 isopropyl Sodium phenylamide Chemical compound 0.000 claims description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 12
- 230000008569 process Effects 0.000 claims description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 9
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 9
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 9
- 239000011734 sodium Substances 0.000 claims description 9
- 229910052708 sodium Inorganic materials 0.000 claims description 9
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 8
- 239000002253 acid Substances 0.000 claims description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 8
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 7
- 238000000605 extraction Methods 0.000 claims description 7
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 claims description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 claims description 6
- 239000012046 mixed solvent Substances 0.000 claims description 6
- 238000002425 crystallisation Methods 0.000 claims description 5
- 230000008025 crystallization Effects 0.000 claims description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical group [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 4
- JCIVHYBIFRUGKO-UHFFFAOYSA-N lithium;2,2,6,6-tetramethylpiperidine Chemical compound [Li].CC1(C)CCCC(C)(C)N1 JCIVHYBIFRUGKO-UHFFFAOYSA-N 0.000 claims description 4
- GAWSJNQKYLRPPD-UHFFFAOYSA-N lithium;phenyl(propan-2-yl)azanide Chemical compound [Li+].CC(C)[N-]C1=CC=CC=C1 GAWSJNQKYLRPPD-UHFFFAOYSA-N 0.000 claims description 4
- 150000003839 salts Chemical class 0.000 claims description 4
- 239000012312 sodium hydride Substances 0.000 claims description 4
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical group C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- 239000012043 crude product Substances 0.000 claims description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 claims description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 238000001816 cooling Methods 0.000 claims description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 claims 1
- MYGSPKVPRYSNFE-UHFFFAOYSA-L [Mg+2].[Cl-].CC1(NC(CCC1)(C)C)C.[Cl-] Chemical compound [Mg+2].[Cl-].CC1(NC(CCC1)(C)C)C.[Cl-] MYGSPKVPRYSNFE-UHFFFAOYSA-L 0.000 claims 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims 1
- 229910052744 lithium Inorganic materials 0.000 claims 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims 1
- 239000000543 intermediate Substances 0.000 description 48
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 26
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 26
- 239000007864 aqueous solution Substances 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 19
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- OBTZDIRUQWFRFZ-UHFFFAOYSA-N 2-(5-methylfuran-2-yl)-n-(4-methylphenyl)quinoline-4-carboxamide Chemical compound O1C(C)=CC=C1C1=CC(C(=O)NC=2C=CC(C)=CC=2)=C(C=CC=C2)C2=N1 OBTZDIRUQWFRFZ-UHFFFAOYSA-N 0.000 description 17
- 238000003756 stirring Methods 0.000 description 17
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 15
- 239000008213 purified water Substances 0.000 description 14
- 239000002585 base Substances 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- 239000000243 solution Substances 0.000 description 11
- 239000008346 aqueous phase Substances 0.000 description 10
- 239000012065 filter cake Substances 0.000 description 10
- 239000000203 mixture Substances 0.000 description 9
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 8
- 229920002554 vinyl polymer Polymers 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 239000007791 liquid phase Substances 0.000 description 6
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 235000019439 ethyl acetate Nutrition 0.000 description 5
- 238000004448 titration Methods 0.000 description 5
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 4
- 229940126062 Compound A Drugs 0.000 description 4
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 150000001336 alkenes Chemical class 0.000 description 4
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 description 4
- 239000001639 calcium acetate Substances 0.000 description 4
- 229960005147 calcium acetate Drugs 0.000 description 4
- 235000011092 calcium acetate Nutrition 0.000 description 4
- 239000001110 calcium chloride Substances 0.000 description 4
- 229910001628 calcium chloride Inorganic materials 0.000 description 4
- ZCCIPPOKBCJFDN-UHFFFAOYSA-N calcium nitrate Chemical compound [Ca+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O ZCCIPPOKBCJFDN-UHFFFAOYSA-N 0.000 description 4
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 4
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 238000000967 suction filtration Methods 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- COXYEVPGUANEQY-UHFFFAOYSA-L [Mg+2].[Cl-].[Cl-].C1CCNCC1 Chemical compound [Mg+2].[Cl-].[Cl-].C1CCNCC1 COXYEVPGUANEQY-UHFFFAOYSA-L 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 2
- 238000005352 clarification Methods 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000000265 homogenisation Methods 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- QISVKQXLGLNOTM-ANMDKAQQSA-N methyl (e,3r,5s)-7-[4-(4-fluorophenyl)-5-(hydroxymethyl)-2,6-di(propan-2-yl)pyridin-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound COC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C(C)C)N=C(C(C)C)C(CO)=C1C1=CC=C(F)C=C1 QISVKQXLGLNOTM-ANMDKAQQSA-N 0.000 description 2
- 238000006772 olefination reaction Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- BDBRXPCTVVZHBA-UHFFFAOYSA-N C(C)(C)[N-]C1=CC=CC=C1.[Na+] Chemical compound C(C)(C)[N-]C1=CC=CC=C1.[Na+] BDBRXPCTVVZHBA-UHFFFAOYSA-N 0.000 description 1
- 229910004261 CaF 2 Inorganic materials 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 208000030673 Homozygous familial hypercholesterolemia Diseases 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 206010049287 Lipodystrophy acquired Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 1
- UCUJUFDOQOJLBE-UHFFFAOYSA-N [Cl].[Ca] Chemical compound [Cl].[Ca] UCUJUFDOQOJLBE-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229940066901 crestor Drugs 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 208000006132 lipodystrophy Diseases 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000012450 pharmaceutical intermediate Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 150000004714 phosphonium salts Chemical group 0.000 description 1
- TYRDEZUMAVRTEO-UHFFFAOYSA-N pyrimidin-5-ylmethanol Chemical compound OCC1=CN=CN=C1 TYRDEZUMAVRTEO-UHFFFAOYSA-N 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 238000007039 two-step reaction Methods 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/645—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having two nitrogen atoms as the only ring hetero atoms
- C07F9/6509—Six-membered rings
- C07F9/6512—Six-membered rings having the nitrogen atoms in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the invention relates to the field of synthesis of pharmaceuticals and pharmaceutical intermediates, in particular to an intermediate compound which can be used for preparing rosuvastatin calcium, and a method for preparing rosuvastatin calcium from the intermediate compound. Background technique
- Rosuvastatin calcium is a selective HMG-CoA reductase inhibitor developed by AstraZeneca and has been marketed in the United States, Japan, Europe, China and other countries and regions.
- Rosuvastatin calcium is a highly effective drug for lowering blood lipids.
- Patent WO 2004103977A reports that a trans-critical intermediate compound 3 can be obtained with a selectivity of about 50: 1 by olefination of compound 1 with a side chain aldehyde under the action of a base.
- Compound 3 is subjected to removal of a protecting group, hydrolysis and calcium salt to obtain rosuvastatin calcium.
- the rivastatin calcium patent US 2005/0124639 A1 reports the olefination reaction by quaternary phosphonium salt compound 4 and the aldehyde side chain 5 to give the trans key intermediate compound 6 with moderate selectivity.
- Compound 6 is subjected to removal of a protecting group, hydrolysis and calcium salt to obtain rosuvastatin calcium, wherein the R 1 , R 2 and R 3 groups of the quaternary phosphonium salt may be a mercapto group and an aromatic group, and the anion may be a halogen.
- Patent EP 0521471 A1 reports the preparation of rosuvastatin calcium by the steps of Wittig reaction, removal of protecting groups, selective reduction, hydrolysis, and the like by pyrimidine aldehyde compound 8 and compound 9. Patent one step improvement.
- an object of the present invention is to provide an intermediate for preparing rosuvastatin calcium. Another object of the present invention is to provide a process for the preparation of rosuvastatin calcium.
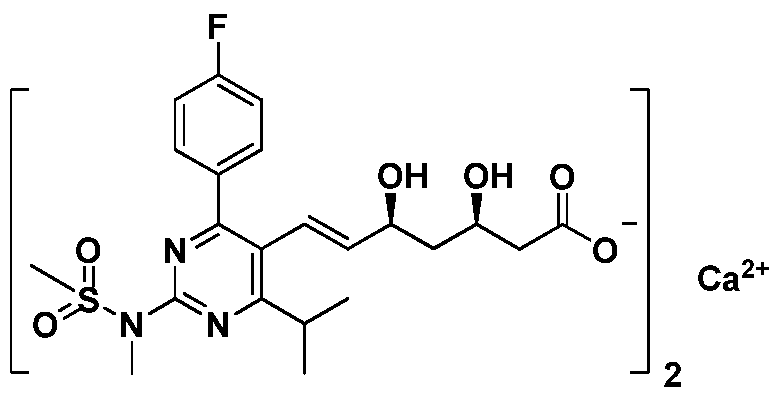
- the intermediate compound provided by the present invention for preparing rosuvastatin calcium has the structure of formula (I).
- R 1 represents a TBS, TES or TIPS protecting group, or two OR 1 groups form an O' R1 , O structure, in which case R 1 represents an isopropyl group; and R 2 represents a C4 group. ⁇ C10 sulfhydryl.
- R 2 represents a tert-butyl group, a tert-amyl group, a cyclopentyl group or a cyclohexyl group.
- the Wittig reaction process is: adding the intermediate compound of claim 1 to an organic solvent, cooling to -80 to - 20 ° C, adding a base, and dropping at -80 to - 20 ° C
- a solution of the compound of the formula (II) after completion of the dropwise addition, is reacted at -80 to - 20 ° C for 1 to 3 hours, and the temperature is returned to -45 to 25 ° C until the reaction is complete, quenching reaction, extraction, extraction
- the liquid is concentrated, and the solvent is crystallized by adding a solvent to the obtained crude product to obtain an intermediate compound of the formula ( ⁇ ).
- the base is selected from the group consisting of sodium hydride, n-butyl lithium, lithium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, 2, 2, 6, 6-tetra Piperidine magnesium chloride (Cas: 215863-85-7), 2, 2, 6, 6-tetramethylpiperidine lithium, hexamethyldisilazide potassium, isopropylphenyl amide lithium or cumene Sodium amide; the molar ratio of the base to the intermediate compound of the formula (I) described in the above technical scheme is from 0.9 to 2.0:1, preferably 1.1:1.
- the organic solvent is selected from the group consisting of 1,4-dioxane, tetrahydrofuran, 2-methyltetrahydrofuran, N,N-dimethylformamide, methyl tert-butyl ether, and ethylene glycol dimethyl ether. Or a mixed solvent of any two ratios, preferably tetrahydrofuran; the organic solvent is used in an amount of 5 to 20 mL/go relative to the intermediate compound of the formula (I) described in the above technical scheme.
- the molar ratio of the intermediate compound of the formula (I) to the compound of the formula ( ⁇ ) in the above technical means is 1: 1 to 2, preferably 1: 1.2.
- the solvent used for the extraction is selected from the group consisting of ethyl acetate, diethyl ether, methyl tert-butyl ether, n-glyoxime, toluene, dichloromethane, or a mixed solvent of any ratio, preferably n-glycol Or toluene; the solvent is used in an amount of 5 to 40 mL/g based on the intermediate compound of the formula (I) described in the above technical scheme.
- the solvent used for the crystallization is selected from the group consisting of methanol, ethanol or isopropanol, preferably methanol; and the solvent is used in an amount of 2 to 20 mL/g based on the intermediate compound of the formula (I) described in the above technical scheme.
- the acid is hydrochloric acid having a mass percentage concentration of 0.02 to 10%
- the alkali is selected from one or more of sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate and potassium t-butoxide.
- Aqueous solution is selected from one or more of sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate and potassium t-butoxide.
- the calcium salt is selected from the group consisting of calcium chloride, calcium nitrate or calcium acetate in a mass percentage concentration of 3 ⁇ 20% aqueous solution.
- the stereoselectivity and yield of the olefin product obtained by using the intermediate compound of the present invention that is, the intermediate represented by the formula (III) can be greatly improved, and the stereoselectivity E/Z > 99 : 1, purity and yield can also be significantly improved, and can also effectively simplify the purification step. Further, the preparation method of the intermediate compound of the present invention is simple, low in cost and easy to obtain.
- the preparation method of rosuvastatin calcium provided by the invention has obvious improvement in stereoselectivity, purity and yield of the obtained intermediate product by using the above intermediate compound, and the overall process is simple, the cost is greatly reduced, and the industrial application prospect is large-scale. . detailed description
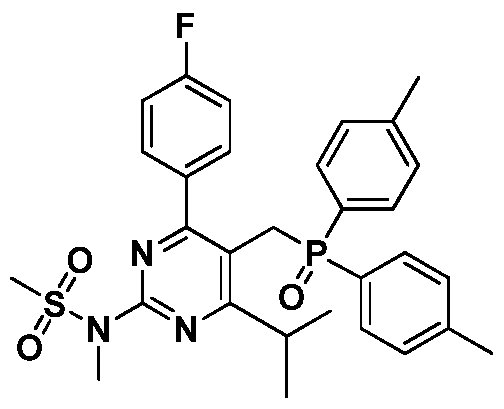
- One aspect of the present invention provides an intermediate compound for the preparation of rosuvastatin calcium having the structure of formula (I).
- the chemical name of the intermediate compound is N-(5-((4, 4'-methylphenylphosphoryl)-methyl)-4-(4-fluorophenyl)-6-isopropylpyrimidine-2 -N-methylmethanesulfonamide
- the olefin product obtained using the intermediate of the present invention is compared to the existing similar intermediate compound due to the superconjugation effect of the methyl group and the phenyl group and the change in steric hindrance Both yield and stereoselectivity are significantly improved.
- Another aspect of the present invention provides a method for preparing rosuvastatin calcium, which has the above formula
- the intermediate compound of the structure shown is a raw material, and is first subjected to Wittig reaction with a compound of the formula ( ⁇ ) to form an intermediate of the formula (III), and the intermediate represented by the formula (III) is subjected to removal of a protecting group and The hydrolysis step, and the salt forming step, yields rosuvastatin calcium.
- R 1 represents a TBS, TES or TIPS protecting group, or two OR 1 groups form a 0' R1 , 0 structure, in which case R 1 represents an isopropyl group;
- R 2 represents a fluorenyl group of C4 to C10.
- R 2 may represent tert-butyl, tert-amyl, cyclopentyl or cyclohexyl.
- the preparation method of the present invention comprises the following steps:
- the step (1) is as follows: Adding N-(5-((4,4'-methylphenylphosphoryl)-methyl)-4-(4-fluorophenyl)- to the organic solvent 6-isopropylpyrimidin-2-yl)-N-methylmethanesulfonamide, stir until clarified to -80-20 ° C, add alkali, stir at -80-20 °C for 2 hours, then add dropwise After the dropwise addition, the solution of the compound of the formula ( ⁇ ) is reacted at -80-20 ° C for 1 to 3 hours, and the temperature is returned to -45 to 25 ° C until the reaction is complete, the reaction is quenched, and the organic solvent is used. The aqueous phase is extracted, and the organic phase is combined and concentrated. The solvent is crystallized from the crude product obtained by concentration to give the intermediate compound of formula (III).
- the organic solvent used to dissolve the reactants is selected from the group consisting of 1,4-dioxane, tetrahydrofuran, 2-methyltetrahydrofuran, N, N-dimethylformamide, methyl tert-butyl ether, ethylene glycol dimethyl ether, a mixed solvent of any ratio of two, preferably tetrahydrofuran, the amount of organic solvent relative to compound I is 5 ⁇ 20 mL/g.
- the base used in the reaction is selected from the group consisting of sodium hydride, n-butyl lithium, lithium diisopropylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, 2,2,6,6-tetramethyl.
- the molar ratio of I is from 0.9 to 2.0:1, preferably 1. 1:1.
- the solvent used for the extraction is selected from one or more mixed solvents of any ratio of ethyl acetate, diethyl ether, methyl tert-butyl ether, n-heptane, toluene, and dichloromethane, preferably n-glycol or toluene; extraction solvent
- the amount of the compound I is 5 to 40 mL/g.
- the solvent used for the crystallization is selected from the group consisting of methanol, ethanol or isopropanol, preferably methanol; and the amount of the crystallization solvent relative to the compound I is 2 to 20 mL/g.
- Step (2) The process is as follows: Compound III is added to the organic solvent, and the acid is added after stirring, and the reaction is completed until the consumption of the compound III is complete. Then, the alkali solution is added to the reaction system and stirred, and the disappearance of the dihydroxy ester intermediate is stopped. After stirring, the reaction system is dehydrated to remove the organic solvent, and then purified water is added to make the system clear. The aqueous phase is extracted with an organic solvent for 1 to 3 times, and the obtained aqueous phase is removed under reduced pressure to obtain an aqueous solution of compound IV.
- the organic solvent to be reacted is selected from one or more of a mixture of any of methanol, ethanol, acetonitrile, isopropanol or acetone, preferably acetonitrile.
- the reaction temperature after the addition of the acid is 10 to 50 ° C, preferably 35 to 40 ° C.
- the acid to be added to the reaction is selected from hydrochloric acid having a mass percentage concentration of 0.02 to 10%, preferably a dilute hydrochloric acid having a mass fraction of 0.06%; and the molar ratio of the added acid to the compound III is 0.001:1 to 5:1, preferably 0.02. : 1.
- the reaction time after acid addition may be from 1 to 24 hours, preferably from 4 to 5 hours.
- the base to be added is selected from one or more aqueous solutions of any ratio of sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate and potassium t-butoxide, preferably a 4% by mass aqueous solution of sodium hydroxide.
- the reaction temperature after the addition of the base is 10 to 50 ° C, preferably 20 to 25 ° C.
- the molar ratio of the added base to the compound III is 0.01:1 to 10:1, preferably 1.1:1.
- the reaction time after the addition of the base may be from 1 to 24 hours, preferably from 6 to 7 hours.
- the post-reaction extraction solvent is selected from one or more of a mixture solvent of toluene, ethyl acetate, diethyl ether, methyl tert-butyl ether, n-heptane, and xylene, preferably methyl tert-butyl ether.
- Step (3) The procedure is as follows: To (-7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino]pyrimidin-5-yl]-(3R , a water-soluble calcium salt is added dropwise to an aqueous solution of 5S)-3, 5-dihydroxy-6-heptenoate (Compound IV), reacted for several hours, suction filtered, and the filter cake is washed once with purified water, and drained. The filter cake was dried under vacuum to give rosuvastatin calcium.
- the water-soluble calcium salt to be added dropwise is selected from the group consisting of calcium chloride, calcium nitrate or calcium acetate in an aqueous solution having a concentration of 3 to 20% by mass, preferably a calcium chloride aqueous solution having a mass percentage of 10%.
- the molar ratio of calcium ion to compound IV in the added calcium salt is from 0.5 to 3:1, preferably 0.6:1.
- the temperature at which the water-soluble calcium salt is added dropwise is 20 to 80 ° C, preferably 35 to 45 ° C.
- the reaction time after the addition of the water-soluble calcium salt may be from 1 to 24 hours, preferably from 2 to 3 hours.
- Tetrahydrofuran 2 L (8 mL/g) and N-(5-((4, 4'-methylphenylphosphoryl)-methyl)-4-(4-fluorobenzene) were sequentially added to a 5 L four-necked flask.
- the filter cake was beaten with purified water 2 L (10 mL/g), drained, and the filter cake was vacuum dried to obtain rosuvastatin calcium.
- the liquid phase purity of the product was >99.5%, and the yield was 87%.
- the reaction system was added to 800 mL (4 mL/g) of purified water to be quenched, and the liquid phase was treated with n-glycol 1 L (5 mL/g).
- the extract was extracted twice, and the organic phase was combined, washed with 600 mL (3 mL / g) of brine, concentrated to dryness, and 1 L (5 mL/g) methanol was added to the concentrated system to obtain a compound.
- the purity was 99%
- the stereoselectivity 99.7: 0.3
- the yield was 77%.
- a 11.0 g (1.1 mol) aqueous solution of 4% by mass of sodium hydroxide was added dropwise to the reaction system, and stirred for 2.5 hours until the dihydroxyester intermediate formed in the first stage disappeared. Then, 2 L (10 mL/g) of purified water was added to the system after acetonitrile was concentrated, stirred and clarified, and extracted with methyl t-butyl ether 300 mL (3 mL/g) three times, and the aqueous phase was further concentrated.
- the filter cake was beaten with purified water 2 L (10 mL/g), drained, and the filter cake was baked at 50 ° C to obtain rosuvastatin calcium.
- the liquid phase purity of the product was >99.5%, and the yield was 83. %.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Molecular Biology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description











Claims


Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES14885058.9T ES2655062T3 (es) | 2014-03-07 | 2014-03-07 | Compuesto intermedio para la preparación de rosuvastatina cálcica y procedimiento para la preparación de rosuvastatina cálcica a partir del mismo |
| PCT/CN2014/073079 WO2015131405A1 (zh) | 2014-03-07 | 2014-03-07 | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 |
| EP14885058.9A EP3115367B1 (en) | 2014-03-07 | 2014-03-07 | Intermediate compound for preparing rosuvastatin calcium and method for preparing rosuvastatin calcium therefrom |
| HUE14885058A HUE037976T2 (hu) | 2014-03-07 | 2014-03-07 | A Rozuvasztatin-kalcium elõállítására szolgáló intermedier vegyület és az elõállítás módszer |
| JP2016572868A JP6240346B2 (ja) | 2014-03-07 | 2014-03-07 | ロスバスタチンカルシウムを製造するための中間体化合物、及びそれを用いてロスバスタチンカルシウムを製造する方法 |
| PT148850589T PT3115367T (pt) | 2014-03-07 | 2014-03-07 | Composto intermediário para preparar rosuvastatina cálcica e método para preparar rosuvastatina cálcica a partir deste |
| US15/124,083 US9926283B2 (en) | 2014-03-07 | 2014-03-07 | Intermediate compound for preparing rosuvastatin calcium and method for preparing rosuvastatin calcium therefrom |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2014/073079 WO2015131405A1 (zh) | 2014-03-07 | 2014-03-07 | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015131405A1 true WO2015131405A1 (zh) | 2015-09-11 |
Family
ID=54054408
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/073079 WO2015131405A1 (zh) | 2014-03-07 | 2014-03-07 | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9926283B2 (zh) |
| EP (1) | EP3115367B1 (zh) |
| JP (1) | JP6240346B2 (zh) |
| ES (1) | ES2655062T3 (zh) |
| HU (1) | HUE037976T2 (zh) |
| PT (1) | PT3115367T (zh) |
| WO (1) | WO2015131405A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106674281A (zh) * | 2016-12-31 | 2017-05-17 | 安徽美诺华药物化学有限公司 | 一种瑞舒伐他汀中间体化合物、制备方法及其用途 |
| CN111548312A (zh) * | 2020-06-01 | 2020-08-18 | 雅本化学股份有限公司 | 一种瑞舒伐他汀钙片及其制备工艺 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019008593A1 (en) * | 2017-07-05 | 2019-01-10 | Melody Healthcare Pvt. Ltd | PROCESS FOR THE PRODUCTION OF AMORPHOUS CALCIUM ROSUVASTATIN |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0521471A1 (en) * | 1991-07-01 | 1993-01-07 | Shionogi Seiyaku Kabushiki Kaisha | Pyrimidine derivatives as HMG-CoA reductase inhibitors |
| WO2004103977A2 (en) * | 2003-05-21 | 2004-12-02 | Ciba Specialty Chemicals Holding Inc. | Process for the preparation of pyrimidine derivatives |
| US20050124639A1 (en) * | 2003-12-04 | 2005-06-09 | Narendra Joshi | Process for the preparation of pyrimidine derivatives |
| CN103804414A (zh) * | 2014-03-07 | 2014-05-21 | 凯莱英医药集团(天津)股份有限公司 | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9903472D0 (en) * | 1999-02-17 | 1999-04-07 | Zeneca Ltd | Chemical process |
| CN102351901A (zh) * | 2011-08-15 | 2012-02-15 | 上海皓元化学科技有限公司 | 25-羟基维生素d2系列药物侧链及其制备方法 |
| CN102643302A (zh) * | 2012-04-06 | 2012-08-22 | 上海皓元化学科技有限公司 | 25-羟基维生素D2和1α,25-二羟基维生素D2合成中间体的制备方法 |
-
2014
- 2014-03-07 PT PT148850589T patent/PT3115367T/pt unknown
- 2014-03-07 HU HUE14885058A patent/HUE037976T2/hu unknown
- 2014-03-07 EP EP14885058.9A patent/EP3115367B1/en not_active Not-in-force
- 2014-03-07 WO PCT/CN2014/073079 patent/WO2015131405A1/zh active Application Filing
- 2014-03-07 ES ES14885058.9T patent/ES2655062T3/es active Active
- 2014-03-07 US US15/124,083 patent/US9926283B2/en not_active Expired - Fee Related
- 2014-03-07 JP JP2016572868A patent/JP6240346B2/ja not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0521471A1 (en) * | 1991-07-01 | 1993-01-07 | Shionogi Seiyaku Kabushiki Kaisha | Pyrimidine derivatives as HMG-CoA reductase inhibitors |
| WO2004103977A2 (en) * | 2003-05-21 | 2004-12-02 | Ciba Specialty Chemicals Holding Inc. | Process for the preparation of pyrimidine derivatives |
| US20050124639A1 (en) * | 2003-12-04 | 2005-06-09 | Narendra Joshi | Process for the preparation of pyrimidine derivatives |
| CN103804414A (zh) * | 2014-03-07 | 2014-05-21 | 凯莱英医药集团(天津)股份有限公司 | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106674281A (zh) * | 2016-12-31 | 2017-05-17 | 安徽美诺华药物化学有限公司 | 一种瑞舒伐他汀中间体化合物、制备方法及其用途 |
| CN111548312A (zh) * | 2020-06-01 | 2020-08-18 | 雅本化学股份有限公司 | 一种瑞舒伐他汀钙片及其制备工艺 |
Also Published As
| Publication number | Publication date |
|---|---|
| HUE037976T2 (hu) | 2018-09-28 |
| JP6240346B2 (ja) | 2017-11-29 |
| JP2017508790A (ja) | 2017-03-30 |
| PT3115367T (pt) | 2018-01-04 |
| EP3115367A4 (en) | 2017-01-11 |
| EP3115367A1 (en) | 2017-01-11 |
| US9926283B2 (en) | 2018-03-27 |
| ES2655062T3 (es) | 2018-02-16 |
| EP3115367B1 (en) | 2017-11-01 |
| US20170022169A1 (en) | 2017-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090036680A1 (en) | Salts of hmg-coa reductase inhibitors and use thereof | |
| CN105153010B (zh) | HMG-CoA还原酶抑制剂及其中间体的制备方法 | |
| BRPI0612851A2 (pt) | processos para a fabricação de um composto, e para formar um composto ou um sal farmaceuticamente aceitável do mesmo, e, composto | |
| JP2007509119A (ja) | ロスバスタチン(e)−7−’4−(4−フルオロフェニル)−6−イソプロピル−2−’メチル(メチルスルホニル)アミノ!ピリミジン−5−イル!(3r,5s)−3,5−ジヒドロキシヘプタ−6−エン酸のカルシウム塩、および、それらの結晶質の中間体の製造方法 | |
| WO2006076845A1 (fr) | Procede de production de la rosuvastatine calcique, intermediaire pour la preparer et procede de production de l'intermediaire | |
| EP1902036B1 (en) | Process for the preparation of rosuvastatin and intermediates | |
| JP6596417B2 (ja) | インドール化合物の製造方法 | |
| JP5558492B2 (ja) | ロスバスタチンまたは医薬として許容されるこの塩の合成のための主要中間体 | |
| WO2015131405A1 (zh) | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 | |
| JP5968900B2 (ja) | ロスバスタチン塩の製法 | |
| WO2009143776A1 (zh) | 瑞舒伐他汀钙盐的制备方法及其中间体 | |
| CN103804414B (zh) | 用于制备瑞舒伐他汀钙的中间体化合物以及由其制备瑞舒伐他汀钙的方法 | |
| KR101063146B1 (ko) | 피타바스타틴 중간체의 제조방법 및 이를 이용한 피타바스타틴 헤미 칼슘염의 제조방법 | |
| JPWO2014051077A1 (ja) | 高純度の含窒素複素環化合物の製造方法 | |
| JP2007517797A (ja) | 高化学的r−5−(2−(2−エトキシフェノキシエチルアミノ)プロピル)−2−メトキシベンゼンスルホンアミド塩酸塩の調製 | |
| CN106674281A (zh) | 一种瑞舒伐他汀中间体化合物、制备方法及其用途 | |
| JP5796836B2 (ja) | ピタバスタチンまたはその塩の中間体の製造方法 | |
| JP2022547113A (ja) | ピリミジニルビピリジン化合物の製造方法及びそのための中間体 | |
| JP6034888B2 (ja) | 新規なスタチン中間体、並びにこれを用いたピタバスタチン、ロスバスタチン、セリバスタチン及びフルバスタチンの製造方法 | |
| JP3467265B2 (ja) | アゼチジノン化合物の結晶 | |
| JP6059157B2 (ja) | モンテルカスト中間体のカンファースルホン酸塩 | |
| CN104744256B (zh) | 制备2-(烷氧基亚烷基)-3-氧代羧酸酯、嘧啶化合物的方法及铁作为催化剂的用途 | |
| KR20110134249A (ko) | 피타바스타틴 또는 그의 염의 중간체의 제조방법 | |
| WO2007086559A1 (ja) | テトラヒドロピラン化合物の製造方法 | |
| JP2002105053A (ja) | ピリジン化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14885058 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016572868 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15124083 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014885058 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014885058 Country of ref document: EP |