WO2014125700A1 - 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ - Google Patents
改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ Download PDFInfo
- Publication number
- WO2014125700A1 WO2014125700A1 PCT/JP2013/082325 JP2013082325W WO2014125700A1 WO 2014125700 A1 WO2014125700 A1 WO 2014125700A1 JP 2013082325 W JP2013082325 W JP 2013082325W WO 2014125700 A1 WO2014125700 A1 WO 2014125700A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- rubber
- natural rubber
- modified natural
- mass
- latex
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C1/00—Treatment of rubber latex
- C08C1/02—Chemical or physical treatment of rubber latex before or during concentration
- C08C1/04—Purifying; Deproteinising
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C4/00—Treatment of rubber before vulcanisation, not provided for in groups C08C1/00 - C08C3/02
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L15/00—Compositions of rubber derivatives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
- C08L7/02—Latex
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/045—Fullerenes
Definitions
- the present invention relates to a modified natural rubber, a method for producing the modified natural rubber, a rubber composition for a tire using the modified natural rubber, and a pneumatic tire using the rubber composition.
- Patent Document 1 discloses a method of adding a surfactant to a natural rubber latex and performing a cleaning process as a fuel economy improvement by reforming the natural rubber.
- protein and gel components can be reduced to some extent by this method, it is not a sufficient level, and it is desired to further reduce tan ⁇ .
- rubbers for tires are also required to have performance such as heat aging resistance, but in the method of Patent Document 1, heat resistance is insufficient and improvement in coexistence of low fuel consumption and heat aging resistance is also desired. There is.
- Natural rubber on the other hand, has a high Mooney viscosity and poor processability compared to other synthetic rubbers, and is usually used after masticating by adding a peptizing agent to reduce the Mooney viscosity. Bad sex. Furthermore, the molecular chains of natural rubber are broken by mastication, so that the characteristics (such as good abrasion resistance, low fuel consumption performance, rubber strength, heat aging resistance, etc.) of high molecular weight polymers inherently possessed by natural rubber are lost. There is also a problem.
- the present invention solves the above-mentioned problems and improves the low fuel consumption, heat aging resistance, processability and fracture resistance in a well-balanced manner, a method for producing the same, and a tire using the same.
- An object of the present invention is to provide a rubber composition and a pneumatic tire.
- the inventors of the present invention conducted intensive studies to remove non-rubber components and the like in the rubber to achieve high purification, and to use a modified natural rubber whose pH has been adjusted to 2 to 7, a conventional natural rubber.
- the present invention has been accomplished by finding that the balance of performances of fuel economy, heat aging resistance, processability and fracture resistance can be remarkably improved.
- the present invention relates to a modified natural rubber which is highly purified and whose pH is adjusted to 2 to 7.
- a modified natural rubber having a Mooney viscosity ML (1 + 4) 130 ° C. measured according to JIS K 6300: 2001-1 of 75 or less, and having a heat aging resistance index of 75 to It is preferably 120%.
- the present invention relates to a modified natural rubber having a pH of 2 to 7 obtained by treatment with an acidic compound after removing the non-rubber component of natural rubber.
- the heat aging resistance index is preferably 75 to 120%.
- the modified natural rubber preferably has a phosphorus content of 200 ppm or less.
- the modified natural rubber preferably has a nitrogen content of 0.15% by mass or less. It is preferable that the said modified
- the present invention relates to a modified natural rubber obtained by washing and further treating a saponified natural rubber latex with an acidic compound and having a pH of 2 to 7.
- the heat aging resistance index is preferably 75 to 120%.
- the washing is preferably carried out until the phosphorus content in the rubber is 200 ppm or less.
- the present invention relates to a modified natural rubber obtained by washing a deproteinized natural rubber latex and further treating it with an acidic compound and having a pH of 2 to 7.
- the heat aging resistance index is preferably 75 to 120%.
- the washing is preferably carried out until the nitrogen content in the rubber is 0.15% by mass or less.
- the pH is obtained by cutting the modified natural rubber to a size within 2 mm square of each side, immersing in distilled water, extracting at 90 ° C. for 30 minutes while irradiating microwaves, and measuring the immersion water using a pH meter It is preferable that the value is
- the present invention comprises the aforementioned modified natural rubber comprising a step 1-1 of saponifying a natural rubber latex, a step 1-2 of washing a saponified natural rubber latex, and a step 1-3 of treating with an acidic compound.
- the present invention comprises the above-mentioned modified natural rubber comprising a step 2-1 of deproteinizing natural rubber latex, a step 2-2 of washing the deproteinized natural rubber latex, and a step 2-3 of treating with an acidic compound.
- the present invention relates to a rubber composition for a tire comprising a rubber component and carbon black and / or a white filler, wherein the content of the above-mentioned modified natural rubber is 5% by mass or more in 100% by mass of the rubber component.
- the radius of inertia of the carbon black cluster is preferably 300 nm or less.
- the white filler is preferably silica, and the radius of inertia of the cluster of silica is preferably 600 nm or less.
- the method of measuring the radius of inertia is preferably X-ray scattering measurement or neutron scattering measurement.
- the present invention also relates to a pneumatic tire produced using the rubber composition.
- the modified natural rubber is highly purified and the pH is adjusted to 2 to 7, the performance balance of low fuel consumption, heat aging resistance, processability and fracture resistance is significantly improved. Be done.
- the modified natural rubber of the present invention is one which has been highly purified and its pH has been adjusted to 2 to 7.
- non-rubber components such as proteins and phospholipids being removed and highly purified
- the pH of the rubber is controlled to an appropriate value
- fuel economy and processability are improved.
- the removal of non-rubber components and the basic character or strong acidity of the rubber make the deterioration of the rubber easy to progress, but adjusting the pH of the rubber to a predetermined range suppresses the decrease in molecular weight during storage Good heat aging resistance can be obtained. Therefore, the performance balance of fuel economy, heat aging resistance, processability and fracture resistance can be significantly improved.
- purification means removing impurities such as phospholipids and proteins other than natural polyisoprenoid components.
- Natural rubber has a structure in which the isoprenoid component is coated with the above-mentioned impurity component, and by removing the component, the structure of the isoprenoid component is changed to change the interaction with the compounding agent, and energy It is presumed that the loss can be reduced, the durability can be improved, and a better modified natural rubber can be obtained.
- the modified natural rubber of the present invention which is highly purified and whose pH is adjusted to 2 to 7, the modified natural rubber which is highly purified by reducing the amount of non-rubber component and which has a rubber pH of 2 to 7
- the rubber is not particularly limited as long as it is a rubber, and specifically, (1) a modified natural rubber having a pH of 2 to 7 obtained by treating with an acidic compound after removing the non-rubber component of the natural rubber 2) A saponified natural rubber latex is washed and further treated with an acidic compound to obtain a modified natural rubber having a pH of 2 to 7, and (3) a deproteinized natural rubber latex is washed and further treated with an acidic compound And modified natural rubber having a pH of 2 to 7, and the like.
- the modified natural rubber can be prepared by a method of washing saponified natural rubber latex or deproteinized natural rubber latex with distilled water or the like and further treating with an acidic compound, but the distilled water used for washing with water It is important to lower the pH value by shifting to the acidic side by the treatment of the acidic compound as compared to the pH value of.
- the pH of distilled water is not 7.00, and is about 5 to 6. In this case, it is important to reduce the pH value to an acid side more than 5 to 6 by treatment with an acidic compound. Become. Specifically, it is preferable to lower the pH value by 0.2 to 2 by the treatment of the acidic compound than the pH value of water used for washing.
- the pH of the modified natural rubber of the present invention is 2 to 7, preferably 3 to 6, and more preferably 4 to 6.
- the pH of the modified natural rubber is obtained by cutting the rubber to a size within 2 mm square of each side, immersing in distilled water, extracting for 30 minutes at 90 ° C. while irradiating microwaves, and using the pH meter for immersion water Specifically, it is measured by the method described in the examples described later.
- the water-soluble component can not be completely extracted from the inside of the rubber, so the internal pH can not be accurately known.
- the present inventors have found that extraction makes it possible to know the substance of rubber.
- the modified natural rubber of the present invention is purified by various methods such as the above (1) to (3), and for example, the phosphorus content in the modified natural rubber is preferably 200 ppm or less, more preferably Preferably it is 150 ppm or less. If it exceeds 200 ppm, the Mooney viscosity may increase during storage to deteriorate the processability, or tan ⁇ may increase to fail to improve the low fuel consumption.
- the phosphorus content can be measured by a conventional method such as ICP emission analysis. Phosphorus is considered to be derived from phospholipids contained in natural rubber.
- the nitrogen content after immersion in acetone at room temperature (25 ° C.) for 48 hours is preferably 0.15% by mass or less, 0 It is more preferable that the content be 1% by mass or less. If the content is more than 0.15% by mass, the Mooney viscosity may increase during storage to deteriorate the processability, or the improvement in fuel economy may not be sufficiently obtained.
- the highly purified natural rubber is likely to be deteriorated by long-term storage since the natural anti-aging agent component originally said to be possessed by natural rubber is removed. Therefore, artificial anti-aging agents may be added.
- the said nitrogen content is a measured value after removing the artificial anti-aging agent in rubber
- the nitrogen content can be measured by a conventional method such as Kjeldahl method or a trace nitrogen meter. Nitrogen is derived from proteins and amino acids.
- the modified natural rubber preferably has a Mooney viscosity ML (1 + 4) 130 ° C., measured according to JIS K 6300: 2001-1, of 75 or less, more preferably 40 to 75, still more preferably 45 to 75. , Particularly preferably 50 to 70, most preferably 55 to 65.
- 75 or less the mastication normally required before rubber kneading becomes unnecessary. Therefore, the said modified natural rubber produced without passing through the process of mastication can be used conveniently as a compounding material of a rubber composition.
- it exceeds 75 mastication is required before use, and there is a tendency for exclusive use of equipment, loss of electricity and heat energy, and the like to occur.
- the modified natural rubber is preferably a rubber having a heat aging resistance index of 75 to 120% represented by the following formula with respect to the Mooney viscosity ML (1 + 4) 130 ° C.
- the heat aging resistance index represented by the above formula is more preferably 80 to 115%, still more preferably 85 to 110%.
- Various methods have been reported for evaluating the heat aging resistance of rubber, but tire manufacturing is performed using the method of evaluating the change rate before and after heat treatment at 80 ° C. for 18 hours at 130 ° C. Mooney viscosity ML (1 + 4). It is possible to accurately evaluate the heat aging resistance at the time of use or when using a tire. Here, if it is in the above-mentioned range, excellent heat aging resistance can be obtained, and the performance balance of low fuel consumption and heat aging resistance can be remarkably improved.
- the modified natural rubber of the present invention which has been highly purified such as (1) to (3) and whose pH is adjusted to 2 to 7 is (Process 1) a step of saponifying natural rubber latex 1-1 And a step 1-2 of washing the saponified natural rubber latex, and a step 1-3 of treating with an acidic compound, (Production method 2) a step 2-1 of deproteinizing the natural rubber latex, It can be prepared by a production method including the step 2-2 of washing the protein natural rubber latex, and the step 2-3 of treating with an acidic compound.
- Step 1-1 the natural rubber latex is saponified.
- phospholipids and proteins in the rubber are decomposed to prepare a saponified natural rubber latex in which non-rubber components are reduced.
- Natural rubber latex is collected as a sap of natural rubber trees such as Hevea trees and contains not only rubber but also water, protein, lipids, inorganic salts etc.
- the gel in the rubber is based on the complex presence of various impurities It is considered to be a thing.
- raw latex field latex
- concentrated latex concentrated by centrifugal separation method or creaming method refined latex, high ammonia with ammonia added by ordinary method
- Latex such as LATZ latex stabilized with zinc flower and TMTD and ammonia can be used.
- the saponification treatment can be carried out by adding an alkali and, if necessary, a surfactant to the natural rubber latex and leaving it to stand at a predetermined temperature for a certain period of time, and stirring or the like may be performed as necessary.
- alkali used for a saponification process although sodium hydroxide, potassium hydroxide, etc. are preferable, it is not limited to these.
- the surfactant is not particularly limited, and known anionic surfactants such as polyoxyethylene alkyl ether sulfuric acid ester salt, nonionic surfactants and amphoteric surfactants may be mentioned, but the rubber is not coagulated and favorably From the viewpoint of saponification, anionic surfactants such as polyoxyethylene alkyl ether sulfates are preferred.
- the amounts of alkali and surfactant added, and the temperature and time of the saponification treatment may be set appropriately.
- step 1-2 the saponified natural rubber latex obtained in step 1-1 is washed.
- the washing removes non-rubber components such as proteins.
- step 1-2 for example, after coagulating the saponified natural rubber latex obtained in the step 1-1 to prepare a flocculated rubber, the obtained flocculated rubber is treated with a basic compound and further washed.
- the non-rubber component can be removed by diluting the water and transferring the water-soluble component to the aqueous layer and removing the water, and further treating it with a basic compound after aggregation.
- the non-rubber component trapped in the rubber can be redissolved at the time of aggregation. In this way, non-rubber components such as proteins strongly adhered to the aggregated rubber can be removed.
- Examples of the aggregation method include a method in which an acid such as formic acid, acetic acid, sulfuric acid or the like is added to adjust the pH, and a polymer coagulant is further added as needed.
- an acid such as formic acid, acetic acid, sulfuric acid or the like
- a polymer coagulant is further added as needed.
- the pH is preferably adjusted to a range of 3.0 to 5.0, more preferably 3.5 to 4.5.
- a cationic polymer coagulant such as a polymer of methyl chloride quaternary salt of dimethylaminoethyl (meth) acrylate, an anionic polymer coagulant such as a polymer of acrylate, an acrylamide polymer And nonionic polymeric flocculants, and amphoteric polymeric flocculants such as methyl chloride quaternary salt copolymer of dimethylaminoethyl (meth) acrylate, and copolymers thereof.
- the addition amount of the polymer coagulant can be appropriately selected.
- the obtained aggregated rubber is subjected to a treatment with a basic compound.
- the basic compound is not particularly limited, but a basic inorganic compound is preferable from the viewpoint of the removal performance of a protein or the like.
- Examples of basic inorganic compounds include metal hydroxides such as alkali metal hydroxides and alkaline earth metal hydroxides; metal carbonates such as alkali metal carbonates and alkaline earth metal carbonates; alkali metal hydrogencarbonates etc.
- Examples of the alkali metal hydroxide include lithium hydroxide, sodium hydroxide and potassium hydroxide.
- Examples of alkaline earth metal hydroxides include magnesium hydroxide, calcium hydroxide and barium hydroxide.
- Examples of the alkali metal carbonate include lithium carbonate, sodium carbonate and potassium carbonate.
- Examples of the alkaline earth metal carbonate include magnesium carbonate, calcium carbonate and barium carbonate.
- Examples of the alkali metal hydrogen carbonate include lithium hydrogen carbonate, sodium hydrogen carbonate and potassium hydrogen carbonate.
- Examples of the alkali metal phosphate include sodium phosphate and sodium hydrogen phosphate.
- Examples of alkali metal acetates include sodium acetate and potassium acetate.
- Examples of alkali metal hydrides include sodium hydride and potassium hydride.
- metal hydroxides, metal carbonates, metal hydrogencarbonates, metal phosphates and ammonia are preferable, alkali metal carbonates, alkali metal hydrogencarbonates and ammonia are more preferable, and sodium carbonate and sodium hydrogencarbonate are further preferable.
- the above basic compounds may be used alone or in combination of two or more.
- the method for treating the aggregated rubber with the basic compound is not particularly limited as long as the aggregated rubber is brought into contact with the above basic compound, and for example, a method of immersing the aggregated rubber in an aqueous solution of a basic compound, base on aggregated rubber By spraying an aqueous solution of the organic compound.
- An aqueous solution of a basic compound can be prepared by diluting and dissolving each basic compound with water.
- the content of the basic compound in 100% by mass of the aqueous solution is preferably 0.1% by mass or more, and more preferably 0.3% by mass or more. If the amount is less than 0.1% by mass, the protein may not be removed sufficiently.
- the content is preferably 10% by mass or less, more preferably 5% by mass or less. If it exceeds 10% by mass, the amount of proteolysis does not increase although a large amount of basic compound is required, and the efficiency tends to be poor.
- the pH of the aqueous solution of the basic compound is preferably 9 to 13, and more preferably 10 to 12 from the viewpoint of treatment efficiency.
- the treatment temperature may be selected as appropriate, but is preferably 10 to 50 ° C., more preferably 15 to 35 ° C.
- the treatment time is usually 1 minute or more, preferably 10 minutes or more, and more preferably 30 minutes or more. If it is less than one minute, the effects of the present invention may not be obtained well.
- the upper limit is not limited, but is preferably 48 hours or less, more preferably 24 hours or less, still more preferably 16 hours or less from the viewpoint of productivity.
- the washing treatment makes it possible to sufficiently remove non-rubber components such as proteins confined in the rubber at the time of aggregation, and at the same time sufficiently remove not only the surface of the aggregated rubber but also the basic compound present inside. .
- the basic compound remaining in the whole rubber in the washing step it becomes possible to sufficiently apply the treatment with the acidic compound described later to the whole rubber, and not only the surface of the rubber but also the internal pH 2 It can be adjusted to ⁇ 7.
- a means capable of sufficiently removing the non-rubber component and the basic compound contained in the whole rubber can be suitably used, for example, a method in which the rubber component is diluted with water and washed and then centrifuged. There is a method in which the rubber is allowed to float and float, and only the aqueous phase is discharged to take out the rubber content.
- the number of washings may be any number that can reduce the amount of non-rubber components such as proteins and basic compounds to a desired amount, but washing with adding 1000 mL of water to 300 g of dry rubber and dewatering after stirring If it is the method of repeating a cycle, 3 times (3 cycles) or more are preferable, 5 times (5 cycles) or more are more preferable, and 7 times (7 cycles) or more are still more preferable.
- the washing treatment is preferably carried out until the phosphorus content in the rubber is 200 ppm or less and / or the nitrogen content is 0.15 mass% or less. Fuel efficiency and processability are improved by sufficient removal of phospholipids and proteins by washing treatment.
- Step 1-3 the washed rubber obtained in step 1-2 is treated with an acidic compound.
- the pH of the entire rubber is adjusted to 2 to 7 by performing the treatment, and a modified natural rubber excellent in the various performances can be provided.
- the heat aging resistance tends to decrease due to the treatment of the basic compound and the like, but by further treating with an acidic compound, such problems can be prevented and good heat aging resistance can be obtained.
- the acidic compound is not particularly limited, and inorganic acids such as hydrochloric acid, nitric acid, sulfuric acid, phosphoric acid, polyphosphoric acid, metaphosphoric acid, boric acid, boronic acid, sulfanilic acid, sulfamic acid; formic acid, acetic acid, glycolic acid, oxalic acid, propione Acid, malonic acid, succinic acid, adipic acid, maleic acid, malic acid, tartaric acid, citric acid, benzoic acid, phthalic acid, isophthalic acid, glutaric acid, gluconic acid, lactic acid, aspartic acid, glutamic acid, salicylic acid, methanesulfonic acid, Itaconic acid, benzenesulfonic acid, toluenesulfonic acid, naphthalenedisulfonic acid, trifluoromethanesulfonic acid, styrenesulfonic acid, trifluoroacetic acid, barb
- the method for treating the aggregated rubber with acid is not particularly limited as long as the aggregated rubber is brought into contact with the above acidic compound, for example, a method of immersing the aggregated rubber in an aqueous solution of the acidic compound, an aqueous solution of the acidic compound in the aggregated rubber The method of spraying etc. are mentioned.
- An aqueous solution of an acidic compound can be prepared by diluting and dissolving each acidic compound with water.
- the content of the acidic compound in 100% by mass of the aqueous solution is not particularly limited, but the lower limit is preferably 0.1% by mass or more, more preferably 0.3% by mass or more, and the upper limit is preferably 15% by mass or less. More preferably, it is 10% by mass or less, still more preferably 5% by mass or less. When the content is in the above range, good heat aging resistance can be obtained.
- the treatment temperature may be selected as appropriate, but is preferably 10 to 50 ° C., more preferably 15 to 35 ° C.
- the treatment time is usually preferably 3 seconds or more, more preferably 10 seconds or more, and further preferably 30 seconds or more. If it is less than 3 seconds, the neutralization can not be sufficiently performed, and the effects of the present invention may not be obtained well.
- the upper limit is not limited, but is preferably 24 hours or less, more preferably 10 hours or less, still more preferably 5 hours or less from the viewpoint of productivity.
- the pH is adjusted to 6 or less.
- Such neutralization provides excellent heat aging resistance.
- the upper limit of the pH is more preferably 5 or less, still more preferably 4.5 or less.
- the lower limit is not particularly limited, and although depending on the immersion time, when the acid is too strong, the rubber may be deteriorated or the waste water treatment may be complicated, and therefore it is preferably 1 or more, more preferably 2 or more.
- the immersion treatment can be carried out by leaving the aggregated rubber in an aqueous solution of an acidic compound.
- washing treatment of the agglomerated rubber after treatment may be appropriately carried out.
- the washing treatment the same method as described above may be mentioned.
- the non-rubber component may be further reduced by repeating the washing, and the content may be adjusted to a desired content.
- the coagulated rubber after the treatment of the acidic compound may be squeezed with a roll-type squeezer or the like to form a sheet or the like.
- drying is not specifically limited, For example, it can implement using normal dryers, such as a trolley type dryer, a vacuum dryer, an air dryer, a drum dryer, etc. which are used in order to dry TSR.
- normal dryers such as a trolley type dryer, a vacuum dryer, an air dryer, a drum dryer, etc. which are used in order to dry TSR.
- step 2-1 natural rubber latex is deproteinized.
- a deproteinized natural rubber latex from which non-rubber components such as proteins are removed can be prepared.
- Examples of the natural rubber latex used in step 2-1 include the same as described above.
- a known method capable of removing a protein can be adopted without particular limitation, and for example, a method of degrading a protein by adding a proteolytic enzyme to natural rubber latex and the like can be mentioned.
- the proteolytic enzyme used for deproteinization is not particularly limited, and may be any of bacterial origin, filamentous fungus origin and yeast origin. Specifically, protease, peptidase, cellulase, pectinase, lipase, esterase, amylase and the like can be used alone or in combination.
- the amount of the proteolytic enzyme added is preferably 0.005 parts by mass or more, more preferably 0.01 parts by mass or more, still more preferably 0.05 parts by mass or more, based on 100 parts by mass of the solid content in the natural rubber latex. It is. Below the lower limit, there is a risk that the protein degradation reaction will be insufficient.
- a surfactant may be added together with the proteolytic enzyme.
- anionic, cationic, nonionic, amphoteric surfactants and the like can be mentioned.
- step 2-2 the deproteinized natural rubber latex obtained in step 2-1 is washed.
- the washing removes non-rubber components such as proteins.
- the step 2-2 can be carried out, for example, by coagulating the deproteinized natural rubber latex obtained in the step 2-1 to produce a coagulated rubber and then washing the obtained coagulated rubber. In this way, non-rubber components such as proteins strongly adhered to the aggregated rubber can be removed.
- the aggregation method can be carried out in the same manner as in step 1-2 above. Furthermore, if necessary, it may be treated with a basic compound as described above. After preparation of the coagulated rubber, a washing process is performed. The washing treatment can be carried out in the same manner as in step 1-2 above, whereby non-rubber components such as proteins and basic compounds can be removed. The washing treatment is preferably carried out until the phosphorus content in the rubber is 200 ppm or less and / or the nitrogen content is 0.15 mass% or less for the same reason as described above.
- Step 2-3 the washed rubber obtained in step 2-2 is treated with an acidic compound.
- an enzyme having an optimum pH in the alkaline region is used as a proteolytic enzyme because it can be deproteinized suitably, and the enzyme reaction is carried out under alkaline conditions according to the optimum pH.
- the acidic compound those similar to the above-mentioned step 1-3 can be mentioned.
- the method for treating the aggregated rubber with an acid is not particularly limited as long as the aggregated rubber is brought into contact with the above acidic compound, for example, a method of immersing the aggregated rubber in an aqueous solution of the acidic compound The method of spraying an aqueous solution etc. are mentioned.
- An aqueous solution of an acidic compound can be prepared by diluting and dissolving each acidic compound with water.
- the content of the acidic compound in 100% by mass of the aqueous solution is not particularly limited, but the lower limit is preferably 0.01% by mass or more, more preferably 0.03% by mass or more, and the upper limit is preferably 15% by mass or less. More preferably, it is 10% by mass or less, still more preferably 5% by mass or less. When the content is in the above range, good heat aging resistance can be obtained.
- the treatment temperature and treatment time may be appropriately selected, and the same temperature as in the step 1-3 may be employed. Further, in the treatment such as immersion of the acidic compound in the aqueous solution, it is preferable to adjust the pH to the same value as in the step 1-3.
- washing treatment of the agglomerated rubber after treatment may be appropriately performed.
- the washing treatment the same method as described above may be mentioned.
- the non-rubber component may be further reduced by repeating the washing, and the content may be adjusted to a desired content.
- drying is performed to obtain the modified natural rubber of the present invention.
- drying is not specifically limited, The above-mentioned method etc. are employable.
- the rubber composition for a tire of the present invention contains a rubber component and carbon black and / or a white filler, and the rubber component contains a predetermined amount of the above-mentioned modified natural rubber.
- the content of the modified natural rubber in 100% by mass of the rubber component is 5% by mass or more, preferably 50% by mass or more, and more preferably 80% by mass. If it is less than 5% by mass, excellent fuel economy may not be obtained.
- NR natural rubber
- EMR epoxidized natural rubber
- IR isoprene rubber
- BR butadiene rubber
- SBR styrene butadiene rubber
- SIBR styrene isoprene butadiene rubber
- EPDM ethylene propylene diene rubber
- CR chloroprene rubber
- NBR acrylonitrile butadiene rubber
- the rubber composition of the present invention contains carbon black and / or a white filler. Thereby, a reinforcing effect is obtained.
- the nitrogen adsorption specific surface area (N 2 SA) of carbon black is preferably not less than 70m 2 / g, 100m 2 / g or more is more preferable. If it is less than 70 m 2 / g, there is a tendency that a sufficient reinforcing effect can not be obtained.
- N 2 SA of carbon black is preferably 300 meters 2 / g or less, more preferably 250m 2 / g. If it exceeds 300 m 2 / g, fuel economy tends to be reduced.
- the nitrogen adsorption specific surface area of carbon black is calculated
- white filler those generally used in the rubber industry, such as silica, calcium carbonate, mica such as sericite, aluminum hydroxide, magnesium oxide, magnesium hydroxide, clay, talc, alumina, titanium oxide Etc. can be used. Among them, silica is particularly preferable in terms of low fuel consumption.
- the silica is not particularly limited, and examples thereof include dry method silica (anhydrous silicic acid), wet method silica (hydrous silicic acid) and the like, but wet method silica is preferable because it has many silanol groups.
- the nitrogen adsorption specific surface area (N 2 SA) of silica is preferably 40 m 2 / g or more, more preferably 80 m 2 / g or more, and still more preferably 100 m 2 / g or more. If it is less than 40 m 2 / g, the fracture resistance after vulcanization tends to decrease. Further, nitrogen adsorption specific surface area (N 2 SA) is preferably from 300 meters 2 / g or less, more preferably 250m 2 / g, 200m 2 / g or less is more preferable. If it exceeds 300 m 2 / g, fuel economy and rubber processability tend to be reduced.
- the N 2 SA of silica is a value measured by the BET method according to ASTM D3037-81.
- the content of carbon black is preferably 10 parts by mass or more, more preferably 30 parts by mass or more, with respect to 100 parts by mass of the rubber component.
- the content is preferably 150 parts by mass or less, more preferably 100 parts by mass or less.
- Favorable fuel consumption is obtained as it is in the said range.
- the content of silica is preferably 10 parts by mass or more, more preferably 30 parts by mass or more, with respect to 100 parts by mass of the rubber component.
- the content is preferably 150 parts by mass or less, more preferably 100 parts by mass or less.
- Favorable fuel consumption is obtained as it is in the said range.
- the total content of carbon black and white filler is preferably 10 parts by mass or more, more preferably 30 parts by mass or more, with respect to 100 parts by mass of the rubber component.
- the content is preferably 150 parts by mass or less, more preferably 100 parts by mass or less.
- Favorable fuel consumption is obtained as it is in the said range.
- a vulcanization accelerator sulphenamide type, guanidine type vulcanization accelerator, etc.
- the inertial radius of the carbon black cluster contained is preferably 300 nm or less, more preferably 280 nm or less, and still more preferably 270 nm or less And particularly preferably 260 nm or less.
- the inertial radius is 300 nm or less, both fuel economy and fracture resistance are compatible.
- the lower limit is not particularly limited.
- the inertial radius of the cluster of the contained silica is preferably 600 nm or less, more preferably 580 nm or less, still more preferably 570 nm or less, 560 nm It is particularly preferred that When the inertial radius is 600 nm or less, both fuel economy and destruction resistance are compatible.
- the lower limit is not particularly limited.
- X-ray scattering measurement or neutron scattering measurement in which a rubber composition for measuring X-rays or neutrons is irradiated.
- SAXS Small-Angle X-ray Scattering
- structural information of a substance can be obtained by measuring X-rays scattered onto the substance by irradiating X-rays with a small scattering angle, and microphase separation structure of polymer material, etc. , Can analyze regular structures at the level of several nanometers.
- the X-rays emitted from the synchrotron have a luminance of at least 10 10 (photons / s / mrad 2 / mm 2 /0.1% bw) or more.
- bw indicates the band width of the X-ray emitted from the synchrotron.
- the brightness (photons / s / mrad 2 / mm 2 /0.1% bw) of the above-mentioned X-ray is preferably 10 10 or more, more preferably 10 12 or more.
- the upper limit is not particularly limited, but it is preferable to use an X-ray intensity not more than a level that causes no radiation damage.
- the photon number (photons / s) of the above-mentioned X-ray is preferably 10 7 or more, more preferably 10 9 or more.
- the upper limit is not particularly limited, but it is preferable to use an X-ray intensity not more than a level that causes no radiation damage.
- SANS Small-Angle Neutron Scattering small angle neutron scattering (scattering angle: usually 10 degrees or less)) measurement in which a polymer material is irradiated with a neutron beam and the scattering intensity is measured
- small-angle neutron scattering neutron beams are irradiated to a substance and scattered neutrons are measured to measure the one with a small scattering angle to obtain structural information on the substance. It can analyze the regular structure at meter level.
- the polymer material can be swollen with a deuterated solvent, and the polymer material in an equilibrium state in the deuterium solvent can be irradiated with a neutron beam to measure the scattering intensity. .
- deuterated solvent for swelling the polymer material heavy water, deuterated hexane, deuterated toluene, deuterated chloroform, deuterated methanol, heavy DMSO ((D 3 C) 2 S SO And deuterated tetrahydrofuran, deuterated acetonitrile, deuterated dichloromethane, deuterated benzene, deuterated N, N-dimethylformamide and the like.
- Neutron beams used for neutron scattering measurement such as SANS are obtained using beam line SANS-J of JRR-3 research reactor owned by Japan Atomic Energy Agency.
- the neutron flux intensity (neutrons / cm 2 / s) of the above-mentioned neutron beam is preferably 10 3 or more, more preferably 10 4 or more, since a high S / N ratio neutron scattering profile is obtained. It is above. Although the upper limit is not particularly limited, it is preferable to use a neutron flux intensity not exceeding radiation damage.
- q represented by the following (Formula 1) is 10 nm using the above-mentioned X-ray and neutron beam from the point that it is necessary to measure a finer molecular structure of the polymer material. It is preferable to measure in the region of -1 or less.
- the q (nm- 1 ) region is desirable from the viewpoint that smaller information can be obtained as the numerical value becomes larger, so the q region is more preferably 20 nm- 1 or less.
- X-rays scattered in SAXS measurement are detected by an X-ray detector, and an image is generated by an image processor or the like using X-ray detection data from the X-ray detector.
- X-ray detector for example, a two-dimensional detector (X-ray film, nuclear plate, X-ray imaging tube, X-ray fluorescence intensifier, X-ray image intensifier, X-ray imaging plate, X-ray CCD , Amorphous materials for X-rays, etc., line sensor one-dimensional detectors can be used.
- the X-ray detector may be selected as appropriate depending on the type and state of the polymer material to be analyzed.
- the image processing apparatus one capable of generating a normal X-ray scattering image based on X-ray detection data by the X-ray detection apparatus can be used as appropriate.
- SANS measurement can be performed by the same principle as SAXS measurement, and the scattered neutron beam is detected by a neutron beam detector, and an image is generated by an image processor or the like using the neutron beam detection data from the neutron beam detector. Be done.
- the neutron beam detection device a known two-dimensional detector or one-dimensional detector can be used, and as the image processing device, any one capable of generating a known neutron beam scattering image can be used. Good.
- the obtained scattering intensity curve is set to the following method
- R g radius of inertia
- Curve fitting is performed on the scattering intensity curve I (q) obtained by SAXS measurement and SANS measurement as shown in FIGS. 1 and 2 using the following (Equation 2) to (Equation 3), and the least squares fitting parameter is used Ask for.
- the inertial radius R g of the molecular structure with a size of 1 nm to 100 ⁇ m is the inertial radius of the cluster formed by the aggregation of metal atoms and the cluster formed by the aggregation of the filler is R g It is estimated that it corresponds.
- the correlation between the inertial radius R g and the energy loss is high, and the energy loss is smaller as the R g is smaller, it is considered that the R g has a great influence on the energy loss.
- evaluation of the energy loss of the polymer material is carried out by performing X-ray scattering measurement such as SAXS or neutron beam scattering measurement such as SANS, and determining R g by curve fitting using (Equation 2) to (Equation 3) Is possible.
- the largest inertial radius R g is taken as the inertial radius in the present invention.
- the above-mentioned components are kneaded using a rubber kneading apparatus such as an open roll or a Banbury mixer and then vulcanized. It can be manufactured.
- a rubber kneading apparatus such as an open roll or a Banbury mixer
- the rubber composition of the present invention comprises a cap tread, a base tread, an undertread, clinch apex, bead apex, side wall, breaker, edge band, full band, breaker cushion rubber, rubber for carcass cord coating, run flat reinforcing layer It can be suitably used for tire components such as insulation, chafers, inner liners, belts, rolls and the like.
- the tire of the present invention is manufactured by the usual method using the above rubber composition. That is, according to the shape of each member (tread, etc.) of the tire at the unvulcanized stage, the rubber composition containing various materials according to necessity is extruded and processed by the usual method on the tire molding machine After an unvulcanized tire is formed by molding, it can be manufactured by heating and pressing in a vulcanizer.
- Examples of the tire according to the present invention include pneumatic tires and airless (solid) tires. Among them, pneumatic tires are preferable.
- Field latex Field latex Emar E-27C (surfactant) obtained from Muhiba Latex Co., Ltd .: Emar E-27 C (polyoxyethylene lauryl ether sodium sulfate, active ingredient 27% by mass) manufactured by Kao Corporation NaOH: NaOH manufactured by Wako Pure Chemical Industries, Ltd.
- Wingstay L (anti-aging agent): Wingstay L manufactured by ELIOKEM (compound obtained by butylated a condensate of ⁇ -cresol and dicyclopentadiene)
- Emulbin W (surfactant): Emulbin W (aromatic polyglycol ether) manufactured by LANXESS Tamol NN 9104 (surfactant): Tamol NN 9104 manufactured by BASF (a sodium salt of naphthalene sulfonic acid / formaldehyde)
- Van gel B (surfactant): Van gel B (hydrate of magnesium aluminum silicate) manufactured by Vanderbilt TSR: NR (TSR) Carbon black 1: Diablack I (ISAF class) manufactured by Mitsubishi Chemical Corporation (N 2 SA: 114 m 2 / g) Carbon black 2: Diablack LH (N 326, N 2 SA: 84 m 2 / g) manufactured by Mitsubishi Chemical Corporation Silica: Ultrasil VN3 (N 2 SA: 175 m 2 / g)
- stearic acid Beads made by NOF Co., Ltd. beads stearic acid oil of perilla oil: Idemitsu Kosan Co., Ltd.
- Diana Process NH-70S Anti-aging agent 1 Noclac 6C (N-phenyl-N '-(1,3-dimethylbutyl) -p-phenylenediamine) (6PPD) manufactured by Ouchi Emerging Chemical Industry Co., Ltd.
- Anti-aging agent 2 Noclac RD (poly (2,2,4-trimethyl-1,2-dihydroquinoline)) manufactured by Ouchi Emerging Chemical Industry Co., Ltd.
- Insoluble sulfur 1 Seimi sulfur (oil content: 10%) manufactured by Nippon Denryo Kogyo Co., Ltd.
- Insoluble sulfur 2 Cristex HSOT20 (oil content: 20%) manufactured by Flexis.
- Vulcanization accelerator TBBS (NS) Noccellar NS manufactured by Ouchi Emerging Chemical Industry Co., Ltd.
- An anti-aging agent dispersion was prepared by mixing 12.5 g of Emarbin W, 12.5 g of Tamol NN 9104, 12.5 g of Vangel B, and 500 g of Wingstay L (total 1000 g) with 462.5 g of water in a ball mill for 16 hours.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 5 mm.
- the obtained aggregate was taken out and immersed in 1000 ml of a 2% by mass aqueous solution of sodium carbonate at normal temperature for 4 hours, and then the rubber was taken out. To this, 2000 ml of water was added and stirred for 2 minutes, and the work of removing water as much as possible was repeated seven times. Thereafter, 500 ml of water was added, 2% by mass formic acid was added until pH 4 and left for 15 minutes. Furthermore, after removing water as much as possible, adding water again and repeating the operation of stirring for 2 minutes three times, squeeze the water with a water squeezing roll into a sheet, and then dry at 90 ° C. for 4 hours to obtain solid rubber. Obtained.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in 1000 ml of a 2% by mass aqueous solution of sodium carbonate at normal temperature for 4 hours, and then the rubber was taken out. To this, 1000 ml of water was added and stirred for 2 minutes, and the work of removing water as much as possible was repeated seven times. Thereafter, 500 ml of water was added, and 2% by mass formic acid was added until pH 4 and stirred for 30 minutes. The rubber was sheeted while being watered by a scraper, and dried at 90 ° C. for 4 hours to obtain a solid rubber.
- Production Example 1-9 An aggregate obtained by adding a cationic polymer flocculant in Production Example 1-6 was immersed in 1000 ml of a 1% by mass aqueous sodium carbonate solution for 30 minutes at normal temperature, and then the rubber was taken out, and 1000 ml of water was added to this. A solid rubber was obtained by the same procedure except that the mixture was additionally stirred for 2 minutes and the work of removing water as much as possible was repeated seven times.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 3 to 15 mm.
- the obtained aggregate was taken out and immersed in 1000 ml of a 2% by mass aqueous solution of sodium carbonate at normal temperature for 4 hours, and then the rubber was taken out. To this, 1000 ml of water was added and stirred for 2 minutes, and the work of removing water as much as possible was performed once. Thereafter, 500 ml of water was added, and 2% by mass formic acid was added until pH 4 and stirred for 15 minutes. Furthermore, after removing water as much as possible, the operation of adding water again and stirring for 2 minutes was repeated three times, and then dried at 90 ° C. for 4 hours to obtain a solid rubber.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in 1000 ml of a 2% by mass aqueous solution of sodium carbonate at normal temperature for 4 hours, and then the rubber was taken out. To this, 1000 ml of water was added and stirred for 2 minutes, and the work of removing water as much as possible was performed once. Thereafter, 500 ml of water was added, and 2% by mass formic acid was added until pH 4 and stirred for 15 minutes. Further, water was removed as much as possible, and work of adding water again and stirring for 2 minutes was repeated three times, then sheeted with a scraper and dried at 90 ° C. for 4 hours to obtain a solid rubber.
- Comparative Production Example 1-4 In Comparative Production Example 1-3, after immersing in 1000 ml of 2% by mass aqueous sodium carbonate solution for 4 hours at normal temperature, remove the rubber, add 1000 ml of water to this, stir for 2 minutes, repeat the work of removing water as much as possible 7 times. Then, a solid rubber was obtained by the same procedure except for sheeting with a scraper and drying at 90 ° C. for 4 hours.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- the obtained creamy fraction was dispersed in distilled water to prepare a deproteinized rubber latex having a solid rubber content of 60%.
- the latex was adjusted to pH 4.0 by adding formic acid while stirring slowly, and then a cationic polymer flocculant was added and stirred for 2 minutes for aggregation.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in 1000 ml of a 2% by mass aqueous solution of sodium carbonate at normal temperature for 4 hours, and then the rubber was taken out.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, manufactured by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, manufactured by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, manufactured by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, manufactured by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- the obtained creamy fraction was dispersed in distilled water to prepare a deproteinized rubber latex having a solid rubber content of 30%.
- the latex was adjusted to pH 4.0 by adding formic acid while stirring slowly, and then a cationic polymer flocculant was added and stirred for 2 minutes for aggregation.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in a 1% by mass aqueous ammonia solution at normal temperature for 2 hours, and then the work of removing water as much as possible was repeated twice.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- the obtained creamy fraction was dispersed in distilled water to prepare a deproteinized rubber latex having a solid rubber content of 30%.
- a deproteinized rubber latex having a solid rubber content of 30%.
- the cationic polymer surfactant was added, and the mixture was stirred for 2 minutes for aggregation.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in a 2% by mass aqueous sodium carbonate solution at normal temperature for 2 hours, and then the work of removing water as much as possible was repeated twice.
- the latex was adjusted to pH 4.0 by adding formic acid while stirring slowly, and then a cationic polymer flocculant was added and stirred for 2 minutes for aggregation.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in a 2% by mass aqueous sodium carbonate solution at normal temperature for 2 hours, and then the work of removing water as much as possible was repeated twice. Thereafter, 1000 ml of water was added, and the mixture was stirred for 2 minutes, and the work of removing water as much as possible was repeated seven times. Thereafter, 500 ml of water was added, and 2% by mass formic acid was added until pH 3 and stirred for 30 minutes. Then, it was sheeted with a scraper and dried at 90 ° C. for 4 hours to obtain a solid rubber.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- Comparative Production Example 2-2 The rubber solidified in Comparative Production Example 2-1 was taken out, immersed in a 0.5 mass% aqueous solution of sodium carbonate for 1 hour, then sheeted while being washed with water with a scraper, and then dried at 90 ° C. for 4 hours , Solid rubber was obtained by the same procedure.
- a 1% aqueous solution of a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m. Centrifuged for 30 minutes at a rotational speed of Next, the cream-like fraction produced by centrifugation is dispersed in a 1% aqueous solution of the above-mentioned emalgen 810 to adjust the rubber concentration to 8%, and again 11,000 r. p. m.
- a nonionic surfactant [Emulgen 810, trade name by Kao Corporation] was added to the latex having undergone the enzyme treatment to adjust the rubber concentration to 8%, and 11,000 r. p. m.
- the obtained creamy fraction was dispersed in distilled water to prepare a deproteinized rubber latex having a solid rubber content of 30%.
- the latex was adjusted to pH 4.0 by adding formic acid while stirring slowly, and then a cationic polymer flocculant was added and stirred for 2 minutes for aggregation.
- the diameter of the aggregate (flocculated rubber) thus obtained was about 0.5 to 3 mm.
- the obtained aggregate was taken out and immersed in 1000 ml of a 2% by mass aqueous solution of sodium carbonate at normal temperature for 4 hours, and then the rubber was taken out. To this, 1000 ml of water was added and stirred for 2 minutes, and the work of removing water as much as possible was repeated seven times.
- the rubber was sheeted with a scraper and dried at 90 ° C. for 4 hours to obtain a solid rubber.
- Comparative Production Example 2-4 After coagulating the rubber in Comparative Production Example 2-3, the aggregate was not treated with an aqueous solution of sodium carbonate, and 1000 ml of water was added as it is, and the operation was stirred for 2 minutes, and the work of removing water as much as possible was repeated seven times. The rubber was sheeted with a scraper and dried at 90 ° C. for 4 hours to obtain a solid rubber.
- the nitrogen content was determined by decomposing and gasifying each acetone-extracted sample obtained above using the trace nitrogen-carbon measurement device “SUMIGRAPH NC 95A (manufactured by Sumika Chemical Analysis Service Co., Ltd.)”, and converting the gas into a gas
- the nitrogen content was quantified by analyzing with a gas chromatograph "GC-8A (manufactured by Shimadzu Corp.)".
- the phosphorus content was determined using an ICP emission analyzer (P-4010, manufactured by Hitachi, Ltd.).
- a modified natural rubber having a rubber pH in the range of 2 to 7 and having a pre-heat-treated Mooney viscosity, an amount of nitrogen or an amount of phosphorus, and an appropriate heat aging resistance index is The heat aging resistance was superior to that of the outer rubber.
- Comparative examples 1-5, 2-5, 3-1, 3-2, 4-1, and 4-2 were used as comparative examples.
- the unvulcanized rubber composition obtained by the said preparation method is shape
- the unvulcanized rubber composition obtained by the above-mentioned production method was molded into a sheet of 1.1 mm and used as a 1 mm vulcanized rubber composition.
- the combination of the two curves is performed by fixing the scattering intensity curve obtained from BL03XU on the wide angle side and shifting the scattering intensity curve obtained from BL20XU on the small angle side to obtain the scattering intensity curve I (q) by SAXS measurement
- the scattering intensity curve I (q) obtained here is based on carbon black or silica contained in the vulcanized rubber composition.
- the inertial radius Rg (in nm) which is the largest among the inertial radii Rg obtained by curve fitting with (Expression 2) and (Expression 3), was determined. The smaller the value, the better the dispersibility of carbon black and silica.
- SAXS device SAXS: SAXS measuring equipment attached to the beamline BL03XU of the large synchrotron radiation facility SPring-8 owned by the High Intensity Optical Science Research Center (measurement conditions)
- X-ray intensity 5 ⁇ 10 12 photons / s / mrad 2 / mm 2 /0.1% bw Number of photons of X-ray: 2 ⁇ 10 9 photons / s
- X-ray energy 8 keV (BL03XU), 23 keV (BL20 XU)
- Distance from sample to detector 3 m (BL03XU), 160 m (BL20 XU) (Detector)
- Two-dimensional detector imaging intensifier and CCD camera
- SANS device SANS: SANS measurement device attached to beamline SANS-J of JRR-3 research reactor owned by Japan Atomic Energy Agency (National Institute of Nuclear Research and Development) (measurement conditions)
- Neutron beam wavelength 6.5 ⁇
- Neutron flux intensity of neutron beam 9.9 ⁇ 10 7 neutrons / cm 2 / s
- Distance from sample to detector 2.5 m, 10 m
- Detector 2-dimensional detector (3 the He two-dimensional detector and the two-dimensional photomultiplier + ZnS / 6 LiF detectors)
- the obtained unvulcanized rubber composition was measured at 130 ° C. in accordance with the measuring method of Mooney viscosity in accordance with JIS K6300.
- the Mooney viscosity ML (1 + 4) of the reference comparative example was set to 100, and the index was displayed by the following formula. The larger the index, the lower the Mooney viscosity and the better the processability.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Tires In General (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description

前記改質天然ゴムは、窒素含有量が0.15質量%以下であることが好ましい。
前記改質天然ゴムは、トルエン不溶分として測定されるゲル含有率が20質量%以下であることが好ましい。
ここで、前記耐熱老化性指数が75~120%であることが好ましい。
前記洗浄は、ゴム中のリン含有量が200ppm以下になるまで洗浄するものであることが好ましい。
ここで、前記耐熱老化性指数が75~120%であることが好ましい。
前記洗浄は、ゴム中の窒素含有量が0.15質量%以下になるまで洗浄するものであることが好ましい。
ここで、前記カーボンブラックのクラスターの慣性半径が300nm以下であることが好ましい。
前記白色充填剤がシリカであり、前記シリカのクラスターの慣性半径が600nm以下であることが好ましい。
ここで、前記測定において、下記(式1)で表されるqが10nm-1以下の領域で測定することが好ましい。

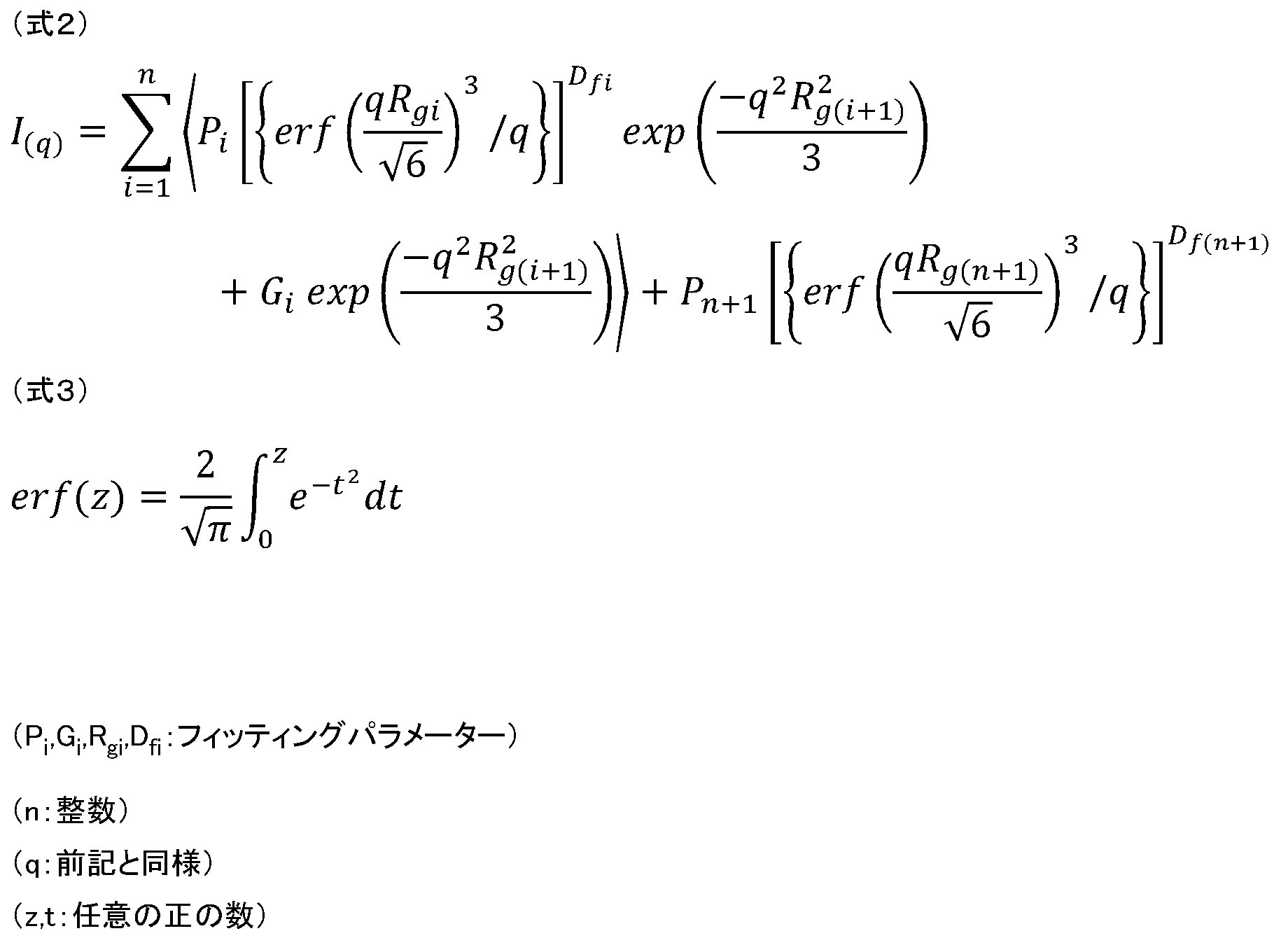
本発明の改質天然ゴムは、高純度化され、かつpHが2~7に調整されたものである。
タンパク質、リン脂質などの非ゴム成分を除去して高純度化するとともに、ゴムのpHを適切な値にコントロールした改質天然ゴムであるため、低燃費性、加工性が改善される。また、非ゴム成分の除去やゴムが塩基性又は強酸性となることで、ゴムの劣化が進行し易くなるが、ゴムのpHを所定範囲に調整することで、保存中の分子量の低下が抑制されるので、良好な耐熱老化性が得られる。従って、低燃費性、耐熱老化性、加工性及び耐破壊性能の性能バランスを顕著に改善できる。

(工程1-1)
工程1-1では、天然ゴムラテックスをケン化処理する。これにより、ゴム中のリン脂質やタンパク質が分解され、非ゴム成分が低減されたケン化天然ゴムラテックスが調製される。
工程1-2では、前記工程1-1で得られたケン化天然ゴムラテックスを洗浄する。該洗浄により、タンパク質などの非ゴム成分を除去する。
工程1-3では、工程1-2で得られた洗浄後のゴムに酸性化合物による処理が施される。前記のとおり、当該処理を施すことでゴム全体のpHが2~7に調整され、前記各種性能に優れた改質天然ゴムを提供できる。なお、塩基性化合物の処理などに起因して耐熱老化性が低下する傾向があるが、更に酸性化合物で処理することで、そのような問題を防止し、良好な耐熱老化性が得られる。
(工程2-1)
工程2-1では、天然ゴムラテックスを脱蛋白処理する。これにより、タンパク質などの非ゴム成分が除去された脱蛋白天然ゴムラテックスが調製できる。工程2-1で使用する天然ゴムラテックスとしては、前記と同様のものが挙げられる。
工程2-2では、前記工程2-1で得られた脱蛋白天然ゴムラテックスを洗浄する。該洗浄により、タンパク質などの非ゴム成分を除去する。
工程2-3では、工程2-2で得られた洗浄後のゴムに酸性化合物による処理が施される。塩基性化合物での処理はもちろん、酸凝集においても酸量が少ない場合、最終的に得られたゴムを水で抽出した際、アルカリ性~中性になることに起因して耐熱老化性が低下する傾向がある。一般的に、好適に脱蛋白できるという理由から、蛋白質分解酵素として、アルカリ領域に至適pHを有する酵素が使用されており、当該酵素反応は、至適pHに合わせてアルカリ条件下で行われることが多く、最終的なゴムのpHを2~7に調整するために、工程2-1における天然ゴムラテックスの脱蛋白処理は、pH8.5~11で実施することが好ましい。その後、凝集の時に酸性下で凝固されるが、そのゴムを水洗しただけでは、後述する抽出でpHが抽出液よりも上がり、この場合に特に耐熱老化性の低下が大きかった。これに対して、凝固後、必要に応じて塩基性化合物で処理後に、酸性化合物で処理することで、そのような問題を防止し、良好な耐熱老化性が得られる。
本発明のタイヤ用ゴム組成物は、ゴム成分とカーボンブラック及び/又は白色充填剤とを含み、該ゴム成分中に上記改質天然ゴムを所定量含む。

金属原子を含みかつ金属配位能を有する官能基を含む高分子材料や充填剤を含む高分子材料について、SAXS測定やSANS測定を実施した場合、例えば、得られた散乱強度曲線を以下の方法で解析することにより、1nm~100μmのクラスター(散乱体)の慣性半径(Rg)を求めることができる。

なお、得られる慣性半径Rgのうち、最大の慣性半径Rgを本発明における慣性半径とした。
以下に、実施例で用いた各種薬品について説明する。
フィールドラテックス:ムヒバラテックス社から入手したフィールドラテックス
エマールE-27C(界面活性剤):花王(株)製のエマールE-27C(ポリオキシエチレンラウリルエーテル硫酸ナトリウム、有効成分27質量%)
NaOH:和光純薬工業(株)製のNaOH
Wingstay L(老化防止剤):ELIOKEM社製のWingstay L(ρ-クレゾールとジシクロペンタジエンとの縮合物をブチル化した化合物)
エマルビンW(界面活性剤):LANXESS社製のエマルビンW(芳香族ポリグリコールエーテル)
タモールNN9104(界面活性剤):BASF社製のタモールNN9104(ナフタレンスルホン酸/ホルムアルデヒドのナトリウム塩)
Van gel B(界面活性剤):Vanderbilt社製のVan gel B(マグネシウムアルミニウムシリケートの水和物)
TSR:NR(TSR)
カーボンブラック1:三菱化学(株)製のダイアブラックI(ISAFクラス)(N2SA:114m2/g)
カーボンブラック2:三菱化学(株)製のダイアブラックLH(N326、N2SA:84m2/g)
シリカ:エボニックデグサ社製のウルトラシルVN3(N2SA:175m2/g)
酸化亜鉛:三井金属鉱業(株)製の酸化亜鉛2種
ステアリン酸:日油(株)製のビーズステアリン酸つばき
オイル:出光興産(株)製のダイアナプロセスNH-70S
老化防止剤1:大内新興化学工業(株)製のノクラック6C(N-フェニル-N’-(1,3-ジメチルブチル)-p-フェニレンジアミン)(6PPD)
老化防止剤2:大内新興化学工業(株)製のノクラックRD(ポリ(2,2,4-トリメチル-1,2-ジヒドロキノリン))
不溶性硫黄1:日本乾溜工業(株)製のセイミ硫黄(オイル分:10%)
不溶性硫黄2:フレキシス社製のクリステックスHSOT20(オイル分:20%)
加硫促進剤TBBS(NS):大内新興化学工業(株)製のノクセラーNS
(老化防止剤分散体の調製)
水 462.5gにエマルビンW 12.5g、タモールNN9104 12.5g、Van gel B 12.5g、Wingstay L 500g(合計1000g)をボールミルで16時間混合し、老化防止剤分散体を調製した。
フィールドラテックスの固形分濃度(DRC)を30%(w/v)に調整した後、該ラテックス1000gに、10%エマールE-27C水溶液25gと25%NaOH水溶液60gを加え、室温で24時間ケン化反応を行い、ケン化天然ゴムラテックスを得た。次いで、老化防止剤分散体6gを添加し、2時間撹拌した後、更に水を添加してゴム濃度15%(w/v)となるまで希釈した。次いで、ゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~5mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。これに、水2000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、15分間放置した。更に、水を極力取り除き、再度水を添加して2分間撹拌する作業を3回繰返した後、水しぼりロールで水を絞ってシート状にした後、90℃で4時間乾燥して固形ゴムを得た。
製造例1-1においてpH1になるまで2質量%のギ酸を添加したほかは、同様の手順で固形ゴムを得た。
製造例1-1においてpH2になるまで2質量%のギ酸を添加したほかは、同様の手順で固形ゴムを得た。
製造例1-1においてpH3になるまで2質量%のギ酸を添加したほかは、同様の手順で固形ゴムを得た。
製造例1-1においてpH5になるまで2質量%のギ酸を添加した添加したほかは、同様の手順で固形ゴムを得た。
フィールドラテックスの固形分濃度(DRC)を30%(w/v)に調整した後、該ラテックス1000gに、10%エマールE-27C水溶液25gと25%NaOH水溶液60gを加え、室温で24時間ケン化反応を行い、ケン化天然ゴムラテックスを得た。次いで、老化防止剤分散体6gを添加し、2時間撹拌した後、更に水を添加してゴム濃度15%(w/v)となるまで希釈した。次いで、ゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、30分間撹拌した。このゴムをクレーパーにより水をかけながらシート化し、90℃で4時間乾燥して固形ゴムを得た。
製造例1-6において水1000mlを加えて2分間撹拌し、極力水を取り除く作業を3回繰り返したほかは、同様の手順で固形ゴムを得た。
製造例1-6において水1000mlを加えて2分間撹拌し、極力水を取り除く作業を5回繰り返したほかは、同様の手順で固形ゴムを得た。
製造例1-6においてカチオン系高分子凝集剤を添加して得られた凝集物を1質量%の炭酸ナトリウム水溶液1000mlに、常温で30分浸漬した後、ゴムを取出し、これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返したほかは、同様の手順で固形ゴムを得た。
製造例1-6においてカチオン系高分子凝集剤を添加して得られた凝集物を炭酸ナトリウム水溶液で処理せず、そのまま水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返したほかは、同様の手順で固形ゴムを得た。
フィールドラテックスの固形分濃度(DRC)を30%(w/v)に調整した後、該ラテックス1000gに、10%エマールE-27C水溶液25gと25%NaOH水溶液60gを加え、室温で24時間ケン化反応を行い、ケン化天然ゴムラテックスを得た。次いで、老化防止剤分散体6gを添加し、2時間撹拌した後、更に水を添加してゴム濃度15%(w/v)となるまで希釈した。次いで、ゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は3~15mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を1回行った。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、15分間撹拌した。更に、水を極力取り除き、再度水を添加して2分間撹拌する作業を3回繰返した後、90℃で4時間乾燥して固形ゴムを得た。
製造例1-1において炭酸ナトリウム水溶液で処理し、水洗を7回繰り返した後、2質量%ギ酸による酸処理をすることなく、水しぼりロールで水を絞ってシート状にしたほかは、同様の手順で固形ゴムを得た。
フィールドラテックスの固形分濃度(DRC)を30%(w/v)に調整した後、該ラテックス1000gに、10%エマールE-27C水溶液25gと25%NaOH水溶液60gを加え、室温で24時間ケン化反応を行い、ケン化天然ゴムラテックスを得た。次いで、老化防止剤分散体6gを添加し、2時間撹拌した後、更に水を添加してゴム濃度15%(w/v)となるまで希釈した。次いで、ゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を1回行った。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、15分間撹拌した。更に、水を極力取り除き、再度水を添加して2分間撹拌する作業を3回繰り返した後、クレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
比較製造例1-3において2質量%炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出し、これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返し、次いでクレーパーでシート状にし、90℃で4時間乾燥したほかは、同様の手順で固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810〕の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作を2回繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分60%の脱蛋白ゴムラテックスを調製した。
このラテックスに2質量%ギ酸をpH4になるまで添加し、更にカチオン系高分子凝集剤を添加して0.5~5mmのゴム粒を得た。これの水を極力取り除き、水をゴム分10gに対して50g添加の上、2質量%ギ酸をpH3になるまで添加した。30分後ゴムを引き上げ、クレーパーでシート化した後、90℃で4時間乾燥し、固形ゴムを得た。
製造例2-1において2質量%ギ酸をpH1になるまで添加したほかは、同様の手順で固形ゴムを得た。
製造例2-1において2質量%ギ酸をpH2になるまで添加したほかは、同様の手順で固形ゴムを得た。
製造例2-1において、0.5~5mmのゴム粒を得た後、2質量%の炭酸ナトリウム水溶液1000mlに30分間浸漬し、このゴムを引き上げ、乾燥ゴム100gに対して水を350gの割合で足して2分間撹拌後静置して水層を極力捨てる作業を5回繰り返し、その後、水をゴム分10gに対して50g添加の上、2質量%ギ酸をpH3になるまで添加したほかは、同様の手順で固形ゴムを得た。
製造例2-1において2質量%ギ酸をpH5になるまで添加したほかは、同様の手順で固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810〕の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作を2回繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分60%の脱蛋白ゴムラテックスを調製した。
このラテックスをゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。
これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を2回繰り返した。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、30分間撹拌した。その後、このゴムをクレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810]の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作を2回繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分30%の脱蛋白ゴムラテックスを調製した。
このラテックスをゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、これに水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、30分間撹拌した。その後、クレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にp Hを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810]の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作を3回繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分30%の脱蛋白ゴムラテックスを調製した。
このラテックスをゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、1質量%のアンモニア水溶液に常温で2時間浸漬した後、極力水を取り除く作業を2回繰り返した。その後、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後、水500mlを添加し、pH4になるまで2質量%ギ酸を添加し、30分間撹拌した。その後、クレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%]を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810〕の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作を3回繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分30%の脱蛋白ゴムラテックスを調製した。
このラテックスをゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子疑集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液に常温で2時間浸漬した後、極力水を取り除く作業を2回繰り返した。その後、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後水500mlを添加し、pH2になるまで2質量%ギ酸を添加し、30分間撹拌した。その後クレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカフーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、アニオン系界面活性剤〔花王社製の商品名エマールE27C〕の0.2%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマールE27Cの0.2%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作を3回繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分30%の脱蛋白ゴムラテックスを調製した。
このラテックスをゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液に常温で2時間浸漬した後、極力水を取り除く作業を2回繰り返した。その後、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。その後、水500mlを添加し、pH3になるまで2質量%ギ酸を添加し、30分間撹拌した。その後、クレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810〕の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作をもう一度繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分60%の脱蛋白ゴムラテックスを調製した。
このラテックスにゴムが固まるまで50質量%ギ酸を添加し、凝固したゴムを取り出した。このゴムをクレーパーで水で洗いながらシート化した後、90℃で4時間乾燥し、固形ゴムを得た。
比較製造例2-1において凝固したゴムを取り出した後、0.5質量%炭酸ナトリウム水溶液に1時間浸漬し、次いでクレーパーで水で洗いながらシート化した後、90℃で4時間乾燥したほかは、同様の手順で固形ゴムを得た。
市販のハイアンモニアラテックス〔マレイシアのムヒバラテックス社製、固形ゴム分62.0%〕を、0.12%のナフテン酸ソーダ水溶液で希釈して、固形ゴム分を10%にし、更に燐酸二水素ナトリウムを添加してpHを9.2に調整した。そしてゴム分10gに対して、蛋白質分解酵素(アルカラーゼ2.0M)を0.87gの割合で添加し、更にpHを9.2に再調整した後、37℃で24時間維持した。
次に、酵素処理を完了したラテックスに、ノニオン系界面活性剤〔花王社製の商品名エマルゲン810〕の1%水溶液を加えてゴム分濃度を8%に調整し、11,000r.p.m.の回転速度で30分間遠心分離した。次に、遠心分離により生じたクリーム状留分を、上記エマルゲン810の1%水溶液に分散して、ゴム分濃度が8%になるように調整した後、再度、11,000r.p.m.の回転速度で30分間遠心分離した。この操作をもう一度繰り返した後、得られたクリーム状留分を蒸留水に分散して、固形ゴム分30%の脱蛋白ゴムラテックスを調製した。
このラテックスをゆっくり撹拌しながらギ酸を添加してpHを4.0に調整した後、カチオン系高分子凝集剤を添加し、2分間撹拌し、凝集させた。これにより得られた凝集物(凝集ゴム)の直径は0.5~3mm程度であった。得られた凝集物を取り出し、2質量%の炭酸ナトリウム水溶液1000mlに、常温で4時間浸漬した後、ゴムを取出した。これに、水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。このゴムをクレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
比較製造例2-3においてゴムを凝集させた後、その凝集物を炭酸ナトリウム水溶液で処理をせず、そのまま水1000mlを加えて2分間撹拌し、極力水を取り除く作業を7回繰り返した。このゴムをクレーパーでシート状にして、90℃で4時間乾燥して固形ゴムを得た。
得られたゴム5gを5mm以下(約1~2×約1~2×約1~2(mm))に切断して100mlビーカーに入れ、常温の蒸留水50mlを加えて2分間で90℃に昇温し、その後90℃に保つように調整しながらマイクロ波(300W)を13分(合計15分)照射した。次いで、浸漬水をアイスバスで冷却して25℃とした後、pHメーターを用いて、浸漬水のpHを測定した。
80℃で18時間熱処理した前後の固形ゴムのムーニー粘度ML(1+4)130℃をJIS K6300:2001-1に準拠して測定し、更に前記式により耐熱老化性指数を算出した。熱処理前のムーニー粘度が50~70の範囲、特に50~65の範囲であれば物性が良く、素練りの必要もなく、優れている。低すぎるとゴム物性が悪い。また、耐熱老化性指数が大きいほど、耐熱老化性が優れている。
(アセトン抽出(試験片の作製))
各固形ゴムを1mm角に細断したサンプルを約0.5g用意した。サンプルをアセトン50g中に浸漬して、室温(25℃)で48時間後にゴムを取出し、乾燥させ、各試験片(老化防止剤抽出済み)を得た。
(測定)
得られた試験片の窒素含有量を以下の方法で測定した。
窒素含有量は、微量窒素炭素測定装置「SUMIGRAPH NC95A((株)住化分析センター製)」を用いて、上記で得られたアセトン抽出処理済みの各試験片を分解、ガス化し、そのガスをガスクロマトグラフ「GC-8A((株)島津製作所製)」で分析して窒素含有量を定量した。
ICP発光分析装置(P-4010、(株)日立製作所製)を使用してリン含有量を求めた。
1mm×1mmに切断した生ゴムのサンプル約70mgを正確に計り、これに35mLのトルエンを加え1週間冷暗所に静置した。次いで、遠心分離に付してトルエンに不溶のゲル分を沈殿させ上澄みの可溶分を除去し、ゲル分のみをメタノールで固めた後、乾燥し質量を測定した。次の式によりゲル含有率(%)を求めた。
ゲル含有率(質量%)=[乾燥後の質量mg/最初のサンプル質量mg]×100


表3~8に示す配合処方に従って、1.7Lバンバリーを用いて、硫黄及び加硫促進剤以外の薬品を混練りした。次に、ロールを用いて、得られた混練り物に硫黄及び加硫促進剤を添加して練り込み、未加硫ゴム組成物を得た。得られた未加硫ゴム組成物を150℃で12分間プレス加硫して加硫物を得た。得られた加硫物を下記により評価し、結果を表3~4に示した。なお、混練りは、前記製造例、比較製造例で作製した10倍量のゴムを用いて行った。基準比較例を比較例1-5、2-5、3-1、3-2、4-1、4-2とした。
なお、表5~8に示すゴム組成物については、上記作製方法で得られた未加硫ゴム組成物を2.1mmのシート状に成形し、150℃で30分間加硫し、2mmの加硫ゴム組成物として使用した。また、放射光測定用については、上記作製方法で得られた未加硫ゴム組成物を1.1mmのシート状に成形し、1mmの加硫ゴム組成物として使用した。
粘弾性スペクトロメーターVES((株)岩本製作所製)を用いて、温度70℃、初期歪み10%、動歪み1%、周波数10Hzの条件下で、各配合(加硫物)の損失正接(tanδ)を測定し、基準比較例の転がり抵抗指数を100として、下記計算式により算出した。転がり抵抗指数が小さいほど、転がり抵抗が低減され、好ましいことを示す。
(転がり抵抗指数)=(各配合のtanδ)/(基準比較例のtanδ)×100
財団法人高輝度光科学研究センター所有の大型放射光施設SPring-8のビームラインBL03XU及びBL20XUにて測定を行った。トルエン中で12時間膨潤させた厚み約1mmのシート状の加硫ゴム組成物をサンプルホルダーに取り付け、室温にて試料にX線を照射した。下記BL03XUでの測定から得られた散乱強度曲線と、BL20XUでの測定から得られた散乱強度曲線を最小2乗法にて結合させた。2つの曲線の結合は、広角側のBL03XUから得られる散乱強度曲線を固定し、小角側のBL20XUから得られる散乱強度曲線をシフトさせることにより行い、SAXS測定による散乱強度曲線I(q)を得た。なお、ここで得られる散乱強度曲線I(q)は、加硫ゴム組成物に含まれるカーボンブラック又はシリカによるものである。得られた散乱強度曲線I(q)に対し、(式2)、(式3)でカーブフィッティングして得られる慣性半径Rgのうち、最大となる慣性半径Rg(単位nm)を求めた。
値が小さいほどカーボンブラック及びシリカの分散性が良いことを示す。
SAXS:財団法人高輝度光科学研究センター所有の大型放射光施設SPring-8のビームラインBL03XU及びBL20XU付属のSAXS測定装置
(測定条件)
X線の輝度:5×1012photons/s/mrad2/mm2/0.1%bw
X線の光子数:2×109photons/s
X線のエネルギー:8keV(BL03XU)、23keV(BL20XU)
試料から検出器までの距離:3m(BL03XU)、160m(BL20XU)
(検出器)
2次元検出器(イメージング・インテンシファイアー及びCCDカメラ)
独立行政法人日本原子力研究開発機構所有のJRR-3研究炉のビームラインSANS-Jにて、厚み約1mmのシート状の加硫ゴム組成物を重水素化溶媒で平衡膨潤させた状態でサンプルホルダーに取り付け、室温にて試料に中性子線を照射した。中性子線の波長は6.5Åとした。試料から検出器までの距離は、2.5m、10mとした。中性子線散乱強度の検出器には、2次元検出器を用いた。なお、ここで得られる散乱強度曲線I(q)は、加硫ゴム組成物に含まれるカーボンブラック又はシリカによるものである。得られた散乱強度曲線I(q)に対し、(式2)、(式3)でカーブフィッティングして得られる慣性半径Rgのうち、最大となる慣性半径Rg(単位nm)を求めた。
値が小さいほどカーボンブラック及びシリカの分散性が良いことを示す。
SANS:独立行政法人日本原子力研究開発機構所有のJRR-3研究炉のビームラインSANS-J付属のSANS測定装置
(測定条件)
中性子線の波長:6.5Å
中性子線の中性子束強度:9.9×107neutrons/cm2/s
試料から検出器までの距離:2.5m、10m(なお、更に小角側の情報を得るために試料から検出器までの距離10mの条件下、フォーカシングレンズを用いた測定を行った。)
(検出器)
2次元検出器(3He 2次元検出器及び2次元フォトマル+ZnS/6LiF検出器)
ASTM D2663 B法に基づき、各加硫ゴム組成物のカーボンブラック又はシリカの分散性を測定した。加硫ゴム組成物の試験片(約3mm幅×8mm長×2mm厚)を採取し、ミクロトームの試料台に貼り付け、液体窒素又はドライアイスで冷却し硬化させた。ガラスナイフを装着したミクロトームで2μm前後の薄片を作製し、薄片をソルベントナフサに浸漬し膨潤させた。膨潤後の薄片を顕微鏡のプレートガラス上に広げ、接眼レンズに10×10μm、横目100目(計10000目)の格子状スケールを置き、全倍率75~100倍にして1目の1/2以上の未分散塊の個数を測定した。分散度を下記式により算出し、基準比較例を100として指数表示した。指数が大きいほど、分散性が良好であることを示す。
分散度(%)=100-S×U/L
S:カーボンブラック及びシリカ未分散塊の占める全格子数
U:測定試料の膨潤ファクター(膨潤後の測定試料の面積/膨潤前の測定試料の面積)
L:コンパウンド中のカーボンブラック及びシリカの容積分率(%)(下記式)

得られた未加硫ゴム組成物について、JIS K6300に準拠したムーニー粘度の測定方法に従い、130℃で測定した。基準比較例のムーニー粘度ML(1+4)を100とし、下記計算式により指数表示した。指数が大きいほどムーニー粘度が低く、加工性に優れる。
得られた加硫ゴム組成物について、JIS K6251に準じて3号ダンベルを用いて引張試験を実施し、破断強度(TB)及び破断時伸び(EB)(%)を測定した。そして、TB×EB/2の数値を破壊強度とし、基準比較例の破壊強度を100として各配合の破壊強度を下記計算式により指数表示した。指数が大きいほど耐破壊性能に優れる。
(耐破壊性能指数)=(各配合のTB×EB/2)/(基準比較例のTB×EB/2)×100

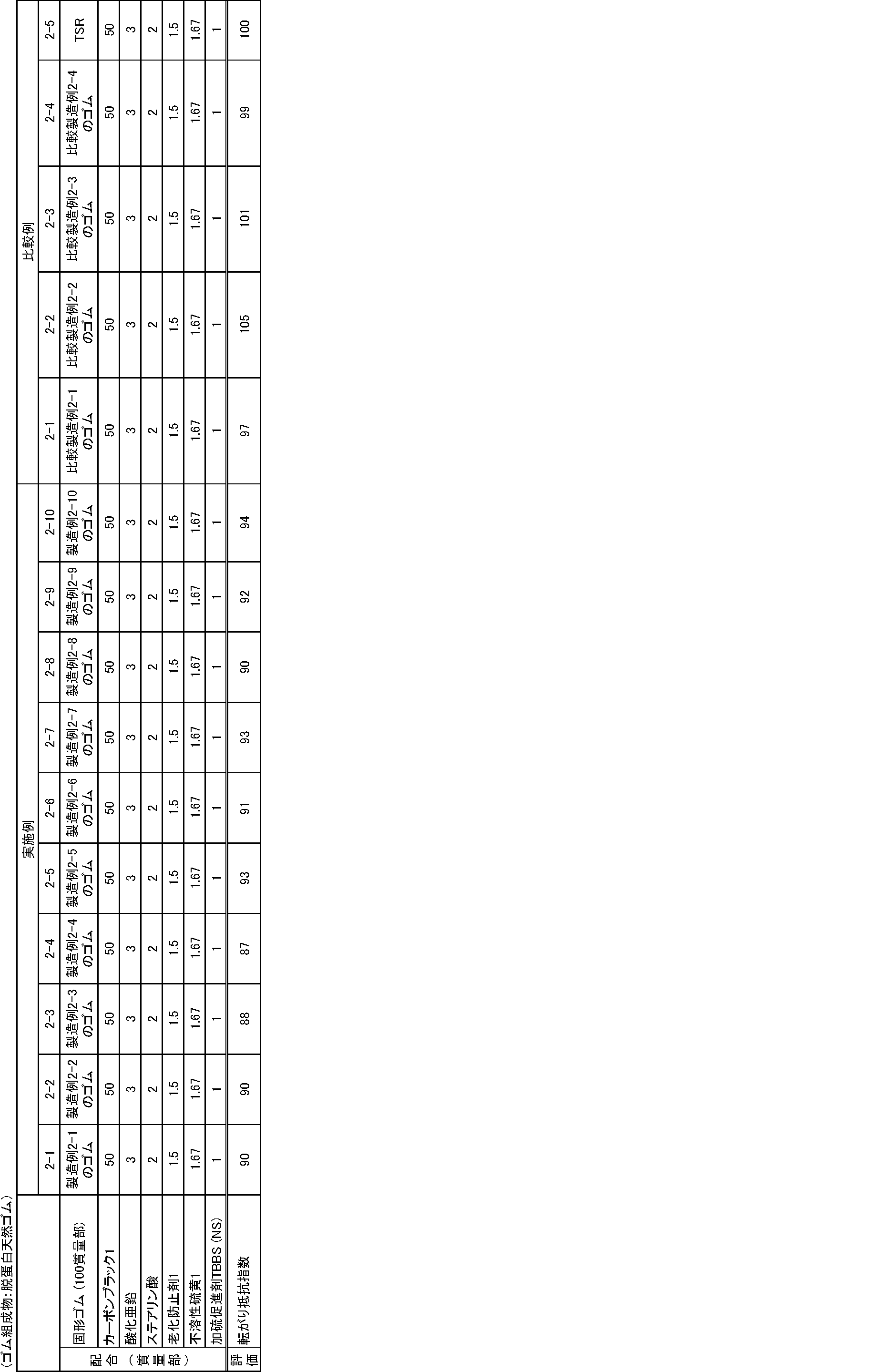
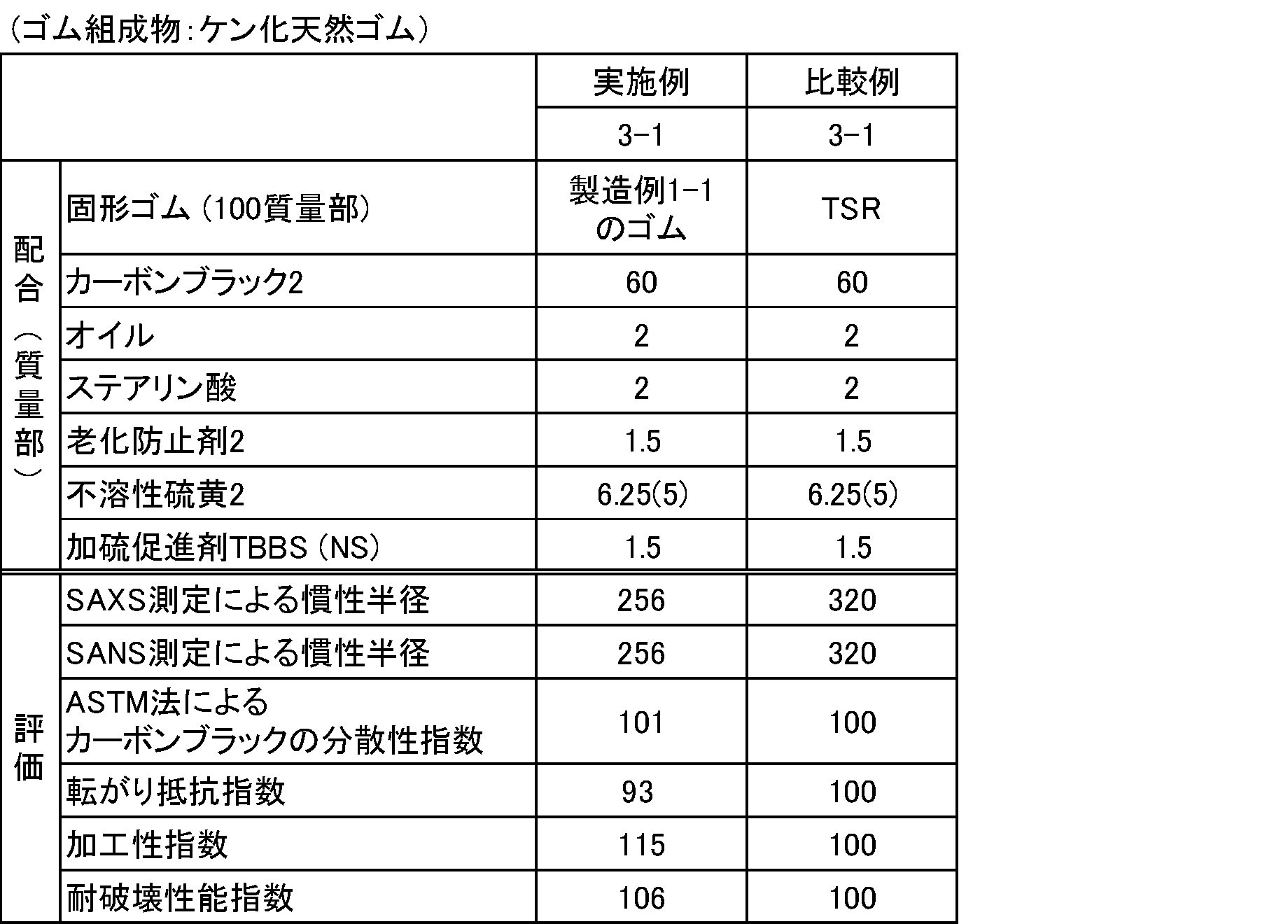
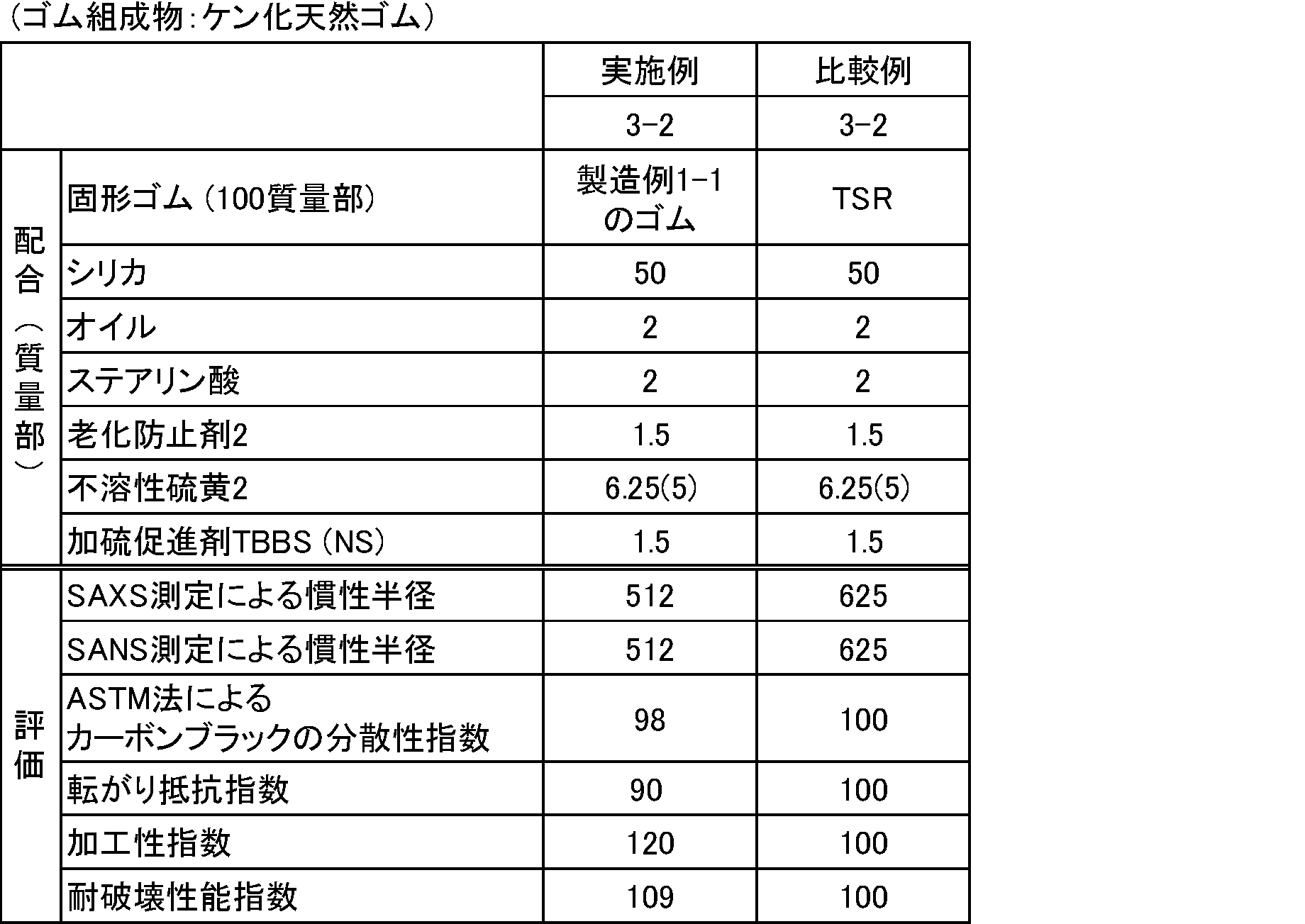


Claims (23)
- 高純度化され、かつpHが2~7に調整された改質天然ゴム。
- JIS K 6300:2001-1に準拠して測定したムーニー粘度ML(1+4)130℃が75以下である改質天然ゴムであって、
下記式で表される耐熱老化性指数が75~120%である請求項1記載の改質天然ゴム。

- 天然ゴムの非ゴム成分を除去した後、酸性化合物で処理して得られ、pHが2~7である改質天然ゴム。
- 前記耐熱老化性指数が75~120%である請求項3記載の改質天然ゴム。
- リン含有量が200ppm以下である請求項1~4のいずれかに記載の改質天然ゴム。
- 窒素含有量が0.15質量%以下である請求項1~5のいずれかに記載の改質天然ゴム。
- トルエン不溶分として測定されるゲル含有率が20質量%以下である請求項1~6のいずれかに記載の改質天然ゴム。
- ケン化天然ゴムラテックスを洗浄し、更に酸性化合物で処理して得られ、pHが2~7である改質天然ゴム。
- 前記耐熱老化性指数が75~120%である請求項8記載の改質天然ゴム。
- 前記洗浄は、ゴム中のリン含有量が200ppm以下になるまで洗浄するものである請求項8又は9記載の改質天然ゴム。
- 脱蛋白天然ゴムラテックスを洗浄し、更に酸性化合物で処理して得られ、pHが2~7である改質天然ゴム。
- 前記耐熱老化性指数が75~120%である請求項11記載の改質天然ゴム。
- 前記洗浄は、ゴム中の窒素含有量が0.15質量%以下になるまで洗浄するものである請求項11又は12記載の改質天然ゴム。
- 前記pHは、前記改質天然ゴムを各辺2mm角以内の大きさに切って蒸留水に浸漬し、マイクロ波を照射しながら90℃で30分間抽出し、浸漬水をpHメーターを用いて測定された値である請求項1~13のいずれかに記載の改質天然ゴム。
- 天然ゴムラテックスをケン化処理する工程1-1と、ケン化天然ゴムラテックスを洗浄する工程1-2と、酸性化合物で処理する工程1-3とを含む請求項1~14のいずれかに記載の改質天然ゴムの製造方法。
- 天然ゴムラテックスを脱蛋白処理する工程2-1と、脱蛋白天然ゴムラテックスを洗浄する工程2-2と、酸性化合物で処理する工程2-3とを含む請求項1~14のいずれかに記載の改質天然ゴムの製造方法。
- ゴム成分とカーボンブラック及び/又は白色充填剤とを含み、前記ゴム成分100質量%中、請求項1~14のいずれかに記載の改質天然ゴムの含有量が5質量%以上であるタイヤ用ゴム組成物。
- 前記カーボンブラックのクラスターの慣性半径が300nm以下である請求項17記載のタイヤ用ゴム組成物。
- 前記白色充填剤がシリカであり、前記シリカのクラスターの慣性半径が600nm以下である請求項17記載のタイヤ用ゴム組成物。
- 前記慣性半径の測定方法が、X線散乱測定又は中性子散乱測定である請求項18又は19記載のタイヤ用ゴム組成物。
- 前記測定において、下記(式1)で表されるqが10nm-1以下の領域で測定する請求項20記載のタイヤ用ゴム組成物。

- 前記慣性半径が、前記測定により得られた散乱強度曲線I(q)に対し、下記(式2)~(式3)でカーブフィッティングして得られる慣性半径Rgのうち、最大となる慣性半径Rgである請求項18~21のいずれかに記載のタイヤ用ゴム組成物。

- 請求項17~22のいずれかに記載のタイヤ用ゴム組成物を用いて作製した空気入りタイヤ。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/437,758 US10131763B2 (en) | 2013-02-15 | 2013-12-02 | Modified natural rubber, process for producing same, rubber composition for tire, and pneumatic tire |
| CN201380065368.XA CN104854136B (zh) | 2013-02-15 | 2013-12-02 | 改性天然橡胶及其制造方法,轮胎用橡胶组合物及充气轮胎 |
| BR112015018474A BR112015018474A2 (pt) | 2013-02-15 | 2013-12-02 | borracha natural modificada, processo para produzir a mesma, composição de borracha para pneu e pneumático |
| EP13874978.3A EP2896633B1 (en) | 2013-02-15 | 2013-12-02 | Modified natural rubber, process for producing same, rubber composition for tire, and pneumatic tire |
| JP2014519116A JP5667327B1 (ja) | 2013-02-15 | 2013-12-02 | 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-028202 | 2013-02-15 | ||
| JP2013028202 | 2013-02-15 | ||
| JP2013-028203 | 2013-02-15 | ||
| JP2013028203 | 2013-02-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014125700A1 true WO2014125700A1 (ja) | 2014-08-21 |
Family
ID=51353719
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/082325 WO2014125700A1 (ja) | 2013-02-15 | 2013-12-02 | 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10131763B2 (ja) |
| EP (1) | EP2896633B1 (ja) |
| JP (1) | JP5667327B1 (ja) |
| CN (1) | CN104854136B (ja) |
| BR (1) | BR112015018474A2 (ja) |
| WO (1) | WO2014125700A1 (ja) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105504100A (zh) * | 2014-10-08 | 2016-04-20 | 住友橡胶工业株式会社 | 轮胎用橡胶组合物以及充气轮胎 |
| JP2016079272A (ja) * | 2014-10-15 | 2016-05-16 | 住友ゴム工業株式会社 | 天然ゴムの処理方法 |
| JP2016094558A (ja) * | 2014-11-14 | 2016-05-26 | 住友ゴム工業株式会社 | 加硫ゴム組成物の製造方法、加硫ゴム組成物およびそれを用いたスタッドレスタイヤ |
| EP3075775A4 (en) * | 2013-12-26 | 2017-08-16 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tires and pneumatic tire |
| JP2018035250A (ja) * | 2016-08-31 | 2018-03-08 | 住友ゴム工業株式会社 | 天然ゴム−白色充填剤複合体の製造方法およびタイヤ |
| JP2018091636A (ja) * | 2016-11-30 | 2018-06-14 | 住友ゴム工業株式会社 | 散乱強度の評価方法 |
| EP3421528A1 (en) | 2017-04-19 | 2019-01-02 | Sumitomo Rubber Industries, Ltd. | Composite and pneumatic tire |
| KR20190055067A (ko) * | 2016-08-19 | 2019-05-22 | 베르살리스 에스.피.에이. | 폴리쿼터너리 중합체에 의한 천연 고무 라텍스의 고형화 |
| EP4052927A1 (en) * | 2021-03-04 | 2022-09-07 | Sumitomo Rubber Industries, Ltd. | Tire |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014188901A1 (ja) | 2013-05-23 | 2014-11-27 | 住友ゴム工業株式会社 | 空気入りタイヤ及びスタッドレスタイヤ、並びに該空気入りタイヤの製造方法及び該スタッドレスタイヤの製造方法 |
| JP7378196B2 (ja) * | 2017-03-06 | 2023-11-13 | 住友ゴム工業株式会社 | 空気入りタイヤ |
| JP6881042B2 (ja) * | 2017-06-01 | 2021-06-02 | 住友ゴム工業株式会社 | ゴム・コード複合体及び接着性評価方法 |
| JP7192329B2 (ja) * | 2018-09-12 | 2022-12-20 | 住友ゴム工業株式会社 | トレッドゴム組成物及び空気入りタイヤ |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3294901B2 (ja) | 1993-05-20 | 2002-06-24 | 花王株式会社 | ゴム組成物 |
| JP2006213752A (ja) * | 2005-02-01 | 2006-08-17 | Bridgestone Corp | 天然ゴムを含むゴム組成物 |
| JP2006213753A (ja) * | 2005-02-01 | 2006-08-17 | Bridgestone Corp | タイヤトレッド用ゴム組成物 |
| JP2010138359A (ja) | 2008-12-15 | 2010-06-24 | Sumitomo Rubber Ind Ltd | 天然ゴム、その製造方法、ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2010174169A (ja) | 2009-01-30 | 2010-08-12 | Sumitomo Rubber Ind Ltd | トレッド又はカーカスコード被覆用ゴム組成物及びタイヤ |
| JP2012116970A (ja) * | 2010-12-01 | 2012-06-21 | Sumitomo Rubber Ind Ltd | 改質天然ゴム、タイヤ用ゴム組成物及び空気入りタイヤ |
| JP2012149134A (ja) * | 2011-01-17 | 2012-08-09 | Sumitomo Rubber Ind Ltd | 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ |
| JP2012241066A (ja) * | 2011-05-17 | 2012-12-10 | Sumitomo Rubber Ind Ltd | 高純度化天然ゴム及びその製造方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1324053C (zh) | 2002-12-06 | 2007-07-04 | 株式会社普利司通 | 天然胶乳,天然橡胶,含有该物质的橡胶组合物及轮胎 |
| CN102245644B (zh) * | 2008-12-15 | 2015-01-07 | 住友橡胶工业株式会社 | 天然橡胶、其制造方法、橡胶组合物及使用它的充气轮胎、改性天然橡胶及其制造方法、以及胎面或包覆胎体帘线用橡胶组合物及使用它们的充气轮胎 |
| JP4810568B2 (ja) * | 2008-12-15 | 2011-11-09 | 住友ゴム工業株式会社 | 改質天然ゴムおよびその製造方法 |
| CN102115554B (zh) * | 2010-01-04 | 2014-09-10 | 住友橡胶工业株式会社 | 轮胎用橡胶组合物以及无钉防滑轮胎 |
| EP2390088B1 (en) * | 2010-05-28 | 2013-09-11 | Sumitomo Rubber Industries, Ltd. | Rubber composition for breaker and pneumatic tire |
| JP5411214B2 (ja) * | 2011-07-28 | 2014-02-12 | 住友ゴム工業株式会社 | トレッド用ゴム組成物、その製造方法及び重荷重用タイヤ |
| CN102268106B (zh) * | 2011-08-26 | 2013-05-01 | 中国热带农业科学院农产品加工研究所 | 一种高纯度天然橡胶的制备方法 |
| WO2014188901A1 (ja) * | 2013-05-23 | 2014-11-27 | 住友ゴム工業株式会社 | 空気入りタイヤ及びスタッドレスタイヤ、並びに該空気入りタイヤの製造方法及び該スタッドレスタイヤの製造方法 |
| JP6199733B2 (ja) * | 2013-12-26 | 2017-09-20 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及び空気入りタイヤ |
| JP6199736B2 (ja) * | 2013-12-27 | 2017-09-20 | 住友ゴム工業株式会社 | タイヤ用ゴム組成物及び空気入りタイヤ |
-
2013
- 2013-12-02 WO PCT/JP2013/082325 patent/WO2014125700A1/ja active Application Filing
- 2013-12-02 BR BR112015018474A patent/BR112015018474A2/pt not_active Application Discontinuation
- 2013-12-02 JP JP2014519116A patent/JP5667327B1/ja active Active
- 2013-12-02 CN CN201380065368.XA patent/CN104854136B/zh active Active
- 2013-12-02 EP EP13874978.3A patent/EP2896633B1/en active Active
- 2013-12-02 US US14/437,758 patent/US10131763B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3294901B2 (ja) | 1993-05-20 | 2002-06-24 | 花王株式会社 | ゴム組成物 |
| JP2006213752A (ja) * | 2005-02-01 | 2006-08-17 | Bridgestone Corp | 天然ゴムを含むゴム組成物 |
| JP2006213753A (ja) * | 2005-02-01 | 2006-08-17 | Bridgestone Corp | タイヤトレッド用ゴム組成物 |
| JP2010138359A (ja) | 2008-12-15 | 2010-06-24 | Sumitomo Rubber Ind Ltd | 天然ゴム、その製造方法、ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2010174169A (ja) | 2009-01-30 | 2010-08-12 | Sumitomo Rubber Ind Ltd | トレッド又はカーカスコード被覆用ゴム組成物及びタイヤ |
| JP2012116970A (ja) * | 2010-12-01 | 2012-06-21 | Sumitomo Rubber Ind Ltd | 改質天然ゴム、タイヤ用ゴム組成物及び空気入りタイヤ |
| JP2012149134A (ja) * | 2011-01-17 | 2012-08-09 | Sumitomo Rubber Ind Ltd | 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ |
| JP2012241066A (ja) * | 2011-05-17 | 2012-12-10 | Sumitomo Rubber Ind Ltd | 高純度化天然ゴム及びその製造方法 |
Non-Patent Citations (4)
| Title |
|---|
| See also references of EP2896633A4 * |
| SEIICHI KAWAHARA, PROTEIN-FREE NATURAL RUBBER, COLLOID POLYM SCI, vol. 290, 2012, pages 331 - 338, XP055253317 * |
| SEIICHI KAWAHARA: "Preparation and characterization of protein-free natural rubber", POLYM. ADV. TECHNOL., vol. 23, 2012, pages 825 - 828, XP055253323 * |
| SEIICHI KAWAHARA: "Removal of proteins from natural rubber with urea", POLYM. ADV. TECHNOL., vol. 15, 2004, pages 181 - 184, XP001221488 * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10066090B2 (en) | 2013-12-26 | 2018-09-04 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tires and pneumatic tire |
| EP3075775A4 (en) * | 2013-12-26 | 2017-08-16 | Sumitomo Rubber Industries, Ltd. | Rubber composition for tires and pneumatic tire |
| CN105504100A (zh) * | 2014-10-08 | 2016-04-20 | 住友橡胶工业株式会社 | 轮胎用橡胶组合物以及充气轮胎 |
| CN105504100B (zh) * | 2014-10-08 | 2019-04-09 | 住友橡胶工业株式会社 | 轮胎用橡胶组合物以及充气轮胎 |
| JP2016079272A (ja) * | 2014-10-15 | 2016-05-16 | 住友ゴム工業株式会社 | 天然ゴムの処理方法 |
| JP2016094558A (ja) * | 2014-11-14 | 2016-05-26 | 住友ゴム工業株式会社 | 加硫ゴム組成物の製造方法、加硫ゴム組成物およびそれを用いたスタッドレスタイヤ |
| KR20190055067A (ko) * | 2016-08-19 | 2019-05-22 | 베르살리스 에스.피.에이. | 폴리쿼터너리 중합체에 의한 천연 고무 라텍스의 고형화 |
| KR102314199B1 (ko) | 2016-08-19 | 2021-10-19 | 베르살리스 에스.피.에이. | 폴리쿼터너리 중합체에 의한 천연 고무 라텍스의 고형화 |
| JP2018035250A (ja) * | 2016-08-31 | 2018-03-08 | 住友ゴム工業株式会社 | 天然ゴム−白色充填剤複合体の製造方法およびタイヤ |
| JP2018091636A (ja) * | 2016-11-30 | 2018-06-14 | 住友ゴム工業株式会社 | 散乱強度の評価方法 |
| JP7024181B2 (ja) | 2016-11-30 | 2022-02-24 | 住友ゴム工業株式会社 | 散乱強度の評価方法 |
| EP3421528A1 (en) | 2017-04-19 | 2019-01-02 | Sumitomo Rubber Industries, Ltd. | Composite and pneumatic tire |
| EP4052927A1 (en) * | 2021-03-04 | 2022-09-07 | Sumitomo Rubber Industries, Ltd. | Tire |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5667327B1 (ja) | 2015-02-12 |
| US20150291765A1 (en) | 2015-10-15 |
| CN104854136B (zh) | 2017-03-08 |
| CN104854136A (zh) | 2015-08-19 |
| EP2896633B1 (en) | 2017-05-31 |
| US10131763B2 (en) | 2018-11-20 |
| EP2896633A4 (en) | 2016-07-20 |
| JPWO2014125700A1 (ja) | 2017-02-02 |
| EP2896633A1 (en) | 2015-07-22 |
| BR112015018474A2 (pt) | 2017-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014125700A1 (ja) | 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP5457378B2 (ja) | 改質天然ゴム、その製造方法、タイヤ用ゴム組成物及び空気入りタイヤ | |
| WO2011155561A1 (ja) | 改質天然ゴム、その製造方法、ゴム組成物及び空気入りタイヤ | |
| JP5650796B2 (ja) | スタッドレスタイヤ用ゴム組成物及びスタッドレスタイヤ | |
| WO2015098417A1 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| WO2015098419A1 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP6297894B2 (ja) | 空気入りタイヤ | |
| JP5020353B2 (ja) | 天然ゴム、その製造方法、ゴム組成物及び空気入りタイヤ | |
| JP2014227494A (ja) | インナーライナー用ゴム組成物及び空気入りタイヤ | |
| JP6215694B2 (ja) | 空気入りタイヤ | |
| JP5650799B2 (ja) | プライトッピング用ゴム組成物及び空気入りタイヤ | |
| JP5650803B2 (ja) | アンダートレッド用ゴム組成物及び空気入りタイヤ | |
| JP7378196B2 (ja) | 空気入りタイヤ | |
| JP5492254B2 (ja) | 天然ゴム、その製造方法、ゴム組成物及び空気入りタイヤ | |
| JP5650794B2 (ja) | サイドウォール用ゴム組成物及び空気入りタイヤ | |
| JP6006170B2 (ja) | キャップトレッドゴム組成物及び重荷重用タイヤ | |
| JP6006171B2 (ja) | キャップトレッドゴム組成物及び重荷重用タイヤ | |
| JP6006168B2 (ja) | ベーストレッドゴム組成物及び重荷重用タイヤ | |
| JP5650798B2 (ja) | ブレーカートッピング用ゴム組成物及び空気入りタイヤ | |
| JP5650795B2 (ja) | ベーストレッド用ゴム組成物及び空気入りタイヤ | |
| JP6006169B2 (ja) | サイドウォールゴム組成物及び重荷重用タイヤ | |
| JP6006172B2 (ja) | キャップトレッドゴム組成物及び重荷重用タイヤ | |
| JP5650797B2 (ja) | トレッド用ゴム組成物及び空気入りタイヤ | |
| WO2014188992A1 (ja) | 重荷重用タイヤ及び該重荷重用タイヤの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2014519116 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13874978 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013874978 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013874978 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14437758 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015018474 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112015018474 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150731 |