WO2014081024A1 - 5-置換-2-フェニルアミノ-ベンズアミド類の製造方法 - Google Patents
5-置換-2-フェニルアミノ-ベンズアミド類の製造方法 Download PDFInfo
- Publication number
- WO2014081024A1 WO2014081024A1 PCT/JP2013/081588 JP2013081588W WO2014081024A1 WO 2014081024 A1 WO2014081024 A1 WO 2014081024A1 JP 2013081588 W JP2013081588 W JP 2013081588W WO 2014081024 A1 WO2014081024 A1 WO 2014081024A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- formula
- compound represented
- following formula
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 35
- -1 5-substituted-2-phenylaminobenzamide Chemical class 0.000 title claims description 85
- 238000002360 preparation method Methods 0.000 title abstract 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 93
- 238000006243 chemical reaction Methods 0.000 claims abstract description 51
- 150000003839 salts Chemical class 0.000 claims abstract description 46
- 125000000217 alkyl group Chemical group 0.000 claims description 71
- 125000001424 substituent group Chemical group 0.000 claims description 40
- 125000005843 halogen group Chemical group 0.000 claims description 31
- 229910052751 metal Inorganic materials 0.000 claims description 26
- 239000002184 metal Substances 0.000 claims description 26
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 24
- 125000000623 heterocyclic group Chemical group 0.000 claims description 19
- 239000002585 base Substances 0.000 claims description 18
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 14
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 13
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 13
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 13
- 125000002947 alkylene group Chemical group 0.000 claims description 13
- 125000004432 carbon atom Chemical group C* 0.000 claims description 13
- 125000003118 aryl group Chemical group 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- 125000003545 alkoxy group Chemical group 0.000 claims description 11
- 229910052783 alkali metal Inorganic materials 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 9
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 125000001153 fluoro group Chemical group F* 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 7
- 125000001072 heteroaryl group Chemical group 0.000 claims description 6
- 159000000003 magnesium salts Chemical class 0.000 claims description 5
- 125000006719 (C6-C10) aryl (C1-C6) alkyl group Chemical group 0.000 claims description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 4
- 229910052736 halogen Inorganic materials 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 229910052740 iodine Inorganic materials 0.000 claims description 4
- 229910003002 lithium salt Inorganic materials 0.000 claims description 4
- 159000000002 lithium salts Chemical class 0.000 claims description 4
- 125000004043 oxo group Chemical group O=* 0.000 claims description 4
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 claims description 3
- 239000002798 polar solvent Substances 0.000 claims description 3
- 150000003751 zinc Chemical class 0.000 claims description 3
- 230000015572 biosynthetic process Effects 0.000 abstract description 9
- 238000003786 synthesis reaction Methods 0.000 abstract description 9
- 239000003153 chemical reaction reagent Substances 0.000 abstract description 5
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 57
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 33
- 239000002904 solvent Substances 0.000 description 32
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- 239000000243 solution Substances 0.000 description 25
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 22
- 239000012046 mixed solvent Substances 0.000 description 20
- 239000000203 mixture Substances 0.000 description 19
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 18
- 239000012044 organic layer Substances 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 15
- 239000003960 organic solvent Substances 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- IOHPVZBSOKLVMN-UHFFFAOYSA-N 2-(2-phenylethyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1CCC1=CC=CC=C1 IOHPVZBSOKLVMN-UHFFFAOYSA-N 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 239000002253 acid Substances 0.000 description 11
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 11
- 230000035484 reaction time Effects 0.000 description 11
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 9
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 9
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 7
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 7
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 7
- 150000001408 amides Chemical class 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 239000003759 ester based solvent Substances 0.000 description 7
- 239000004210 ether based solvent Substances 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 6
- 150000002430 hydrocarbons Chemical group 0.000 description 6
- 238000002955 isolation Methods 0.000 description 6
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 6
- 229940011051 isopropyl acetate Drugs 0.000 description 6
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- DFTBJJRFIONUQU-UHFFFAOYSA-N 2,3,4-trifluoro-5-formylbenzoic acid Chemical compound OC(=O)C1=CC(C=O)=C(F)C(F)=C1F DFTBJJRFIONUQU-UHFFFAOYSA-N 0.000 description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 5
- 239000005456 alcohol based solvent Substances 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 125000000304 alkynyl group Chemical group 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 239000003638 chemical reducing agent Substances 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 229910052744 lithium Inorganic materials 0.000 description 5
- 238000001953 recrystallisation Methods 0.000 description 5
- 238000007363 ring formation reaction Methods 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 229910052723 transition metal Inorganic materials 0.000 description 5
- 150000003624 transition metals Chemical class 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 150000003863 ammonium salts Chemical class 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- ZWJINEZUASEZBH-UHFFFAOYSA-N fenamic acid Chemical class OC(=O)C1=CC=CC=C1NC1=CC=CC=C1 ZWJINEZUASEZBH-UHFFFAOYSA-N 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 4
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 0 *c(c(*)c(c(C=N***C(O)=O)c1)F)c1C(O)=O Chemical compound *c(c(*)c(c(C=N***C(O)=O)c1)F)c1C(O)=O 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- DELLTOPODWFJQY-CNHKJKLMSA-N 5-[(E)-(4-ethoxy-4-oxobutoxy)iminomethyl]-2,3,4-trifluorobenzoic acid Chemical compound CCOC(=O)CCCO/N=C/C1=CC(=C(C(=C1F)F)F)C(=O)O DELLTOPODWFJQY-CNHKJKLMSA-N 0.000 description 3
- RAUOOCHMVKJKFI-LIMNOBDPSA-N 5-[(E)-3-carboxypropoxyiminomethyl]-3,4-difluoro-2-(2-fluoro-4-iodoanilino)benzoic acid Chemical compound C1=CC(=C(C=C1I)F)NC2=C(C=C(C(=C2F)F)/C=N/OCCCC(=O)O)C(=O)O RAUOOCHMVKJKFI-LIMNOBDPSA-N 0.000 description 3
- PQTDWUKLFKNMPD-UHFFFAOYSA-N C1=C(C(=C(C(=C1C(=O)O)F)F)F)C=NOCCCC(=O)O Chemical compound C1=C(C(=C(C(=C1C(=O)O)F)F)F)C=NOCCCC(=O)O PQTDWUKLFKNMPD-UHFFFAOYSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 208000012839 conversion disease Diseases 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 229910052762 osmium Inorganic materials 0.000 description 3
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 159000000000 sodium salts Chemical class 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- LKUDPHPHKOZXCD-UHFFFAOYSA-N 1,3,5-trimethoxybenzene Chemical compound COC1=CC(OC)=CC(OC)=C1 LKUDPHPHKOZXCD-UHFFFAOYSA-N 0.000 description 2
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 2
- WWWTWPXKLJTKPM-UHFFFAOYSA-N 2-aminooxyethanol Chemical compound NOCCO WWWTWPXKLJTKPM-UHFFFAOYSA-N 0.000 description 2
- CUMTUBVTKOYYOU-UHFFFAOYSA-N 2-fluoro-4-iodoaniline Chemical compound NC1=CC=C(I)C=C1F CUMTUBVTKOYYOU-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- HJBLUNHMOKFZQX-UHFFFAOYSA-N 3-hydroxy-1,2,3-benzotriazin-4-one Chemical compound C1=CC=C2C(=O)N(O)N=NC2=C1 HJBLUNHMOKFZQX-UHFFFAOYSA-N 0.000 description 2
- HODXSJGOZRLWOK-UHFFFAOYSA-N 5-[(3-carboxypropoxyamino)methyl]-3,4-difluoro-2-(2-fluoro-4-iodoanilino)benzoic acid Chemical compound C1=CC(=C(C=C1I)F)NC2=C(C=C(C(=C2F)F)CNOCCCC(=O)O)C(=O)O HODXSJGOZRLWOK-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- MRRSRGWGMXREMY-UHFFFAOYSA-N C1C=CON(C1=O)CC2=CC(=C(C(=C2F)F)NC3=C(C=C(C=C3)I)F)C(=O)O Chemical compound C1C=CON(C1=O)CC2=CC(=C(C(=C2F)F)NC3=C(C=C(C=C3)I)F)C(=O)O MRRSRGWGMXREMY-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- FIMYFEGKMOCQKT-UHFFFAOYSA-N OCCONC(c(cc(CN1OCCCC1=O)c(F)c1F)c1Nc(c(F)c1)ccc1I)=O Chemical compound OCCONC(c(cc(CN1OCCCC1=O)c(F)c1F)c1Nc(c(F)c1)ccc1I)=O FIMYFEGKMOCQKT-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 150000001448 anilines Chemical class 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- NNTOJPXOCKCMKR-UHFFFAOYSA-N boron;pyridine Chemical compound [B].C1=CC=NC=C1 NNTOJPXOCKCMKR-UHFFFAOYSA-N 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- 238000009776 industrial production Methods 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Chemical compound [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 2
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 238000007248 oxidative elimination reaction Methods 0.000 description 2
- 239000003444 phase transfer catalyst Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 2
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 2
- 159000000001 potassium salts Chemical class 0.000 description 2
- 125000005505 thiomorpholino group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 238000006886 vinylation reaction Methods 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- VFTFKUDGYRBSAL-UHFFFAOYSA-N 15-crown-5 Chemical compound C1COCCOCCOCCOCCO1 VFTFKUDGYRBSAL-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- HPJYIPWVPDYSDU-UHFFFAOYSA-N 2,3,4-trifluoro-5-iodobenzoic acid Chemical compound OC(=O)C1=CC(I)=C(F)C(F)=C1F HPJYIPWVPDYSDU-UHFFFAOYSA-N 0.000 description 1
- WEPXLRANFJEOFZ-UHFFFAOYSA-N 2,3,4-trifluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C(F)=C1F WEPXLRANFJEOFZ-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- ZFFBIQMNKOJDJE-UHFFFAOYSA-N 2-bromo-1,2-diphenylethanone Chemical compound C=1C=CC=CC=1C(Br)C(=O)C1=CC=CC=C1 ZFFBIQMNKOJDJE-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- BMTZEAOGFDXDAD-UHFFFAOYSA-M 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium;chloride Chemical compound [Cl-].COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 BMTZEAOGFDXDAD-UHFFFAOYSA-M 0.000 description 1
- CSDQQAQKBAQLLE-UHFFFAOYSA-N 4-(4-chlorophenyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine Chemical compound C1=CC(Cl)=CC=C1C1C(C=CS2)=C2CCN1 CSDQQAQKBAQLLE-UHFFFAOYSA-N 0.000 description 1
- RVDLHGSZWAELAU-UHFFFAOYSA-N 5-tert-butylthiophene-2-carbonyl chloride Chemical compound CC(C)(C)C1=CC=C(C(Cl)=O)S1 RVDLHGSZWAELAU-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- RAUOOCHMVKJKFI-UHFFFAOYSA-N C1=CC(=C(C=C1I)F)NC2=C(C=C(C(=C2F)F)C=NOCCCC(=O)O)C(=O)O Chemical compound C1=CC(=C(C=C1I)F)NC2=C(C=C(C(=C2F)F)C=NOCCCC(=O)O)C(=O)O RAUOOCHMVKJKFI-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 239000012820 MEK1 Inhibitor Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 239000012327 Ruthenium complex Substances 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- CECABOMBVQNBEC-UHFFFAOYSA-K aluminium iodide Chemical compound I[Al](I)I CECABOMBVQNBEC-UHFFFAOYSA-K 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical group 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- HTZCNXWZYVXIMZ-UHFFFAOYSA-M benzyl(triethyl)azanium;chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC1=CC=CC=C1 HTZCNXWZYVXIMZ-UHFFFAOYSA-M 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 1
- SBGUYEPUJPATFD-UHFFFAOYSA-N bromo(tripyrrolidin-1-yl)phosphanium Chemical compound C1CCCN1[P+](N1CCCC1)(Br)N1CCCC1 SBGUYEPUJPATFD-UHFFFAOYSA-N 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 159000000006 cesium salts Chemical class 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- CDHICTNQMQYRSM-UHFFFAOYSA-N di(propan-2-yl)alumane Chemical compound CC(C)[AlH]C(C)C CDHICTNQMQYRSM-UHFFFAOYSA-N 0.000 description 1
- 125000005131 dialkylammonium group Chemical group 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- MVEAAGBEUOMFRX-UHFFFAOYSA-N ethyl acetate;hydrochloride Chemical compound Cl.CCOC(C)=O MVEAAGBEUOMFRX-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 150000004820 halides Chemical group 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical group [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 1
- 229910000103 lithium hydride Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- BLQJIBCZHWBKSL-UHFFFAOYSA-L magnesium iodide Chemical compound [Mg+2].[I-].[I-] BLQJIBCZHWBKSL-UHFFFAOYSA-L 0.000 description 1
- 229910001641 magnesium iodide Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003506 n-propoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000013110 organic ligand Substances 0.000 description 1
- 239000012285 osmium tetroxide Substances 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical class C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004346 phenylpentyl group Chemical group C1(=CC=CC=C1)CCCCC* 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 159000000005 rubidium salts Chemical class 0.000 description 1
- 125000005920 sec-butoxy group Chemical group 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- UOSKWYONTYQYHE-UHFFFAOYSA-J sodium trichloroalumane iodide Chemical compound [Na+].[I-].Cl[Al](Cl)Cl UOSKWYONTYQYHE-UHFFFAOYSA-J 0.000 description 1
- GVQPSSGWDYNZQT-UHFFFAOYSA-M sodium;chloro(trimethyl)silane;iodide Chemical compound [Na+].[I-].C[Si](C)(C)Cl GVQPSSGWDYNZQT-UHFFFAOYSA-M 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000005621 tetraalkylammonium salts Chemical class 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- HWCKGOZZJDHMNC-UHFFFAOYSA-M tetraethylammonium bromide Chemical compound [Br-].CC[N+](CC)(CC)CC HWCKGOZZJDHMNC-UHFFFAOYSA-M 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L31/00—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L31/18—Processes or apparatus specially adapted for the manufacture or treatment of these devices or of parts thereof
- H01L31/1884—Manufacture of transparent electrodes, e.g. TCO, ITO
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/04—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes
- C07C249/08—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes by reaction of hydroxylamines with carbonyl compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/02—1,2-Oxazines; Hydrogenated 1,2-oxazines
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L31/00—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L31/02—Details
- H01L31/0224—Electrodes
- H01L31/022408—Electrodes for devices characterised by at least one potential jump barrier or surface barrier
- H01L31/022425—Electrodes for devices characterised by at least one potential jump barrier or surface barrier for solar cells
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K30/00—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation
- H10K30/10—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation comprising heterojunctions between organic semiconductors and inorganic semiconductors
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L31/00—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L31/0248—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof characterised by their semiconductor bodies
- H01L31/0256—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof characterised by their semiconductor bodies characterised by the material
- H01L2031/0344—Organic materials
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K30/00—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation
- H10K30/80—Constructional details
- H10K30/81—Electrodes
- H10K30/82—Transparent electrodes, e.g. indium tin oxide [ITO] electrodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
Definitions
- the present invention relates to a method for producing 5-substituted-2-phenylamino-benzamides.
- a compound represented by the following formula (I), or a pharmaceutically acceptable salt thereof (hereinafter sometimes referred to as “2- (N-phenylamino) benzoic acid derivative of formula (I)”) is cancer, It is known to be useful as a selective MEK-1 inhibitor for the treatment of proliferative diseases including recurrent stenosis, psoriasis, and atherosclerosis (Patent Document 1).
- R 1 represents a halogen atom, C 2-6 alkenyl or C 2-6 alkynyl
- R 2 represents a halogen atom or C 1-6 alkyl, and the C 1-6 alkyl may have a hydroxyl group as a substituent
- R 3 represents a hydrogen atom or a halogen atom
- R 4 represents a hydrogen atom, C 1-6 alkyl, C 2-6 alkenyl or C 2-6 alkynyl, wherein the C 1-6 alkyl, the C 2-6 alkenyl, the C 2-6 alkynyl is —
- the 3-8 membered heterocycle and the 5-10 membered heteroaryl have 1 to 3 substituents selected from the group consisting of C 1-6 alkyl,
- X is a group represented by the following formula (i); (Where Y 1 or Y 2 are the same or different and each represents a single bond, —CO—, —COO—, —O—, —OCO—, —NR a —, or —SO 2 —; Z represents alkylene having 1 to 5 carbon atoms which may have 1 to 3 W as a substituent; W is the same or different and is C 1-6 alkyl, halogen atom, oxo, —OR a , —COOR a , —COOCOR a , —CO— [halogen atom], —OCOR a , —CONR a R b , — SR a , —SOR a , —SO 2 R a , —NR a R b , —NR a COR b , —NR a SO 2 R b , —SO 2 NR a R b , 3-8
- the vinylation step an organic transition metal catalyst is required, and a step of removing the organic transition metal complex and the organic ligand after the reaction is required, which is complicated and inefficient.
- the step of converting to a formyl group by oxidative cleavage reaction requires a reagent such as osmium tetroxide that is complicated to handle as an oxidizing agent, and is not suitable as an industrial production method.
- R 3 , Y 1 , Y 2 and Z are as defined in the above formula (I).
- R 5 represents a halogen atom or O-LG, where LG is SO 2 R c or P ( ⁇ O) (OR c ), R c is C 1-6 alkyl, or C 6-10 aryl It is.
- An object of the present invention is to provide a simple and efficient production method suitable for an industrial production method of a 2- (N-phenylamino) benzoic acid derivative, and a useful synthetic intermediate.
- the present inventors have taken the following production method to achieve the objective in a simple and high yield without using an organic transition metal catalyst or an osmium reagent. It has been found that a method for producing a compound is possible. Furthermore, in the coupling reaction between benzoic acid derivative (IV) and anilines, the reaction system can be made uniform by adding a metal salt, improving the reaction conversion rate and facilitating the reaction control. As a result, the present invention has been completed.
- R 1 represents a halogen atom, C 2-6 alkenyl or C 2-6 alkynyl
- R 2 represents a halogen atom or C 1-6 alkyl, and the C 1-6 alkyl may have a hydroxyl group as a substituent
- R 3 represents a hydrogen atom or a halogen atom
- R 4 represents a hydrogen atom, C 1-6 alkyl, C 2-6 alkenyl or C 2-6 alkynyl, wherein the C 1-6 alkyl, the C 2-6 alkenyl, the C 2-6 alkynyl is —
- the 3-8 membered heterocycle and the 5-10 membered heteroaryl have 1 to 3 substituents selected from
- X is a group represented by the following formula (i); (Where Y 1 or Y 2 are the same or different and each represents a single bond, —CO—, —COO—, —O—, —OCO—, —NR a —, or —SO 2 —; Z represents alkylene having 1 to 5 carbon atoms which may have 1 to 3 W as a substituent; W is the same or different and is C 1-6 alkyl, halogen atom, oxo, —OR a , —COOR a , —COOCOR a , —CO— [halogen atom], —OCOR a , —CONR a R b , — SR a , —SOR a , —SO 2 R a , —NR a R b , —NR a COR b , —NR a SO 2 R b , —SO 2 NR a R b , 3-8
- Step A includes the following formula (II) [Wherein R 5 represents a halogen atom or O-LG, where LG is SO 2 R c or P ( ⁇ O) (OR c ), and R c is C 1-6 alkyl, or C 6 A compound represented by the following formula (III): -10 aryl, R 3 is as defined above] [Wherein R 6 represents hydrogen, C 1-6 alkyl, C 6-10 aryl C 1-6 alkyl, or Si (R d ) 3 , and R d is the same or different, and C 1-6 alkyl And C 6-10 aryl, wherein Y 1 , Y 2 and Z are as defined above] and are further obtained when R 6 is a substituent other than a hydrogen atom
- the compound substituent —CO 2 R 6 is converted into a carboxyl group, and the following formula (IV) Wherein R 3 , R 5 , Y 1 , Y 2 and
- step C the compound represented by formula (VI) obtained in step B is subjected to reducing conditions, and the following formula (VII) Wherein R 1 , R 2 , R 3 and X are as defined above;
- step D the compound represented by formula (VII) obtained in step C is converted into the following formula (VIII). [Wherein R 4 is as defined above] is reacted with a compound represented by the formula (I) or a salt thereof.
- step B The method according to [1], wherein in step B, the metal salt is one or more selected from a lithium salt, a magnesium salt, and / or a zinc salt.
- the base is an alkali metal hexamethyldisilazide.
- R 5 is a fluorine atom.
- Step B the reaction is carried out in an aprotic polar solvent at ⁇ 78 ° C. to 25 ° C.
- R 1 , R 2 , and R 3 are each a halogen atom, R 4 is C 1-6 alkyl optionally substituted with a hydroxyl group, according to any one of [1] to [5] Method.
- R 4 is C 1-6 alkyl optionally substituted with a hydroxyl group, according to any one of [1] to [5] Method.
- Y 1 is a single bond
- Y 2 is —O—
- Z is alkylene having 1 to 5 carbon atoms.
- R 1 is an iodine atom
- R 2 and R 3 are each a fluorine atom
- R 4 is hydroxyethyl (CH 2 CH 2 OH)
- Y 1 is a single bond
- Y 2 is —O— Yes
- Z is alkylene having 3 carbon atoms
- the compound represented by the formula (I) is represented by the following formula (IX)
- the method of the present invention is a method for producing a compound represented by the following formula (I) (hereinafter sometimes referred to as “compound I”), or a pharmaceutically acceptable salt thereof, comprising the following step A, A method including Step B, Step C, and Step D is provided.
- compound I a compound represented by the following formula (I) (hereinafter sometimes referred to as “compound I”), or a pharmaceutically acceptable salt thereof, comprising the following step A, A method including Step B, Step C, and Step D is provided.
- R 1 represents a halogen atom, C 2-6 alkenyl or C 2-6 alkynyl
- R 2 represents a halogen atom or C 1-6 alkyl, and the C 1-6 alkyl may have a hydroxyl group as a substituent
- R 3 represents a hydrogen atom or a halogen atom
- R 4 represents a hydrogen atom, C 1-6 alkyl, C 2-6 alkenyl or C 2-6 alkynyl, wherein the C 1-6 alkyl, the C 2-6 alkenyl, the C 2-6 alkynyl is —
- the 3-8 membered heterocycle and the 5-10 membered heteroaryl have 1 to 3 substituents selected from the group consisting of C 1-6 alkyl,
- X is a group represented by the following formula (i).
- Y 1 or Y 2 are the same or different and each represents a single bond, —CO—, —COO—, —O—, —OCO—, —NR a —, or —SO 2 —
- Z represents alkylene having 1 to 5 carbon atoms which may have 1 to 3 W as a substituent; W is the same or different and is C 1-6 alkyl, halogen atom, oxo, —OR a , —COOR a , —COOCOR a , —CO— [halogen atom], —OCOR a , —CONR a R b , — SR a , —SOR a , —SO 2 R a , —NR a R b , —NR a COR b , —NR a SO 2 R b , —SO 2 NR a R b , 3-8
- alkyl in the present specification is a monovalent group derived by removing any one hydrogen atom from an aliphatic hydrocarbon. Does not contain heteroatoms or unsaturated carbon-carbon bonds in the skeleton. It has a subset of hydrocarbyl or hydrocarbon group structures containing hydrogen and carbon atoms. Alkyl includes linear and branched structures. Alkyl is preferably alkyl having 1 to 6 carbon atoms (referred to as “C 1-6 alkyl”, hereinafter “C pq ” means having p to q carbon atoms), C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl and the like can be mentioned.
- alkyl examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, s-butyl, t-butyl, pentyl, isopentyl, 2,3-dimethylpropyl, 3,3-dimethylbutyl, Examples include hexyl.
- alkenyl is a monovalent hydrocarbon group having at least one double bond (two adjacent SP2 carbon atoms). Depending on the arrangement of double bonds and substituents (if present), the geometry of the double bond can take an enthaneuve (E) or zanzanen (Z), or a cis or trans configuration. Examples of alkenyl include linear or branched ones, preferably C 2-6 alkenyl, and more preferably C 2-5 alkenyl.
- alkenyl examples include vinyl, allyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl (including cis and trans), 3-butenyl, pentenyl, hexenyl and the like.
- alkynyl is a monovalent hydrocarbon group having at least one triple bond (two adjacent SP carbon atoms). Examples include linear or branched alkynyl, preferably C 2-6 alkynyl, and more preferably C 2-5 alkynyl.
- alkynyl examples include ethynyl, 1-propynyl, propargyl, 3-butynyl, pentynyl, hexynyl, and preferably ethynyl.
- Alkenyl or alkynyl can have one or more double bonds or triple bonds, respectively. It can also have a double bond and a triple bond at the same time.
- cycloalkyl means a cyclic aliphatic hydrocarbon, preferably C 3-8 cycloalkyl and the like.
- Specific examples of cycloalkyl include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, and a cyclooctyl group.
- aryl means a monovalent aromatic hydrocarbon ring, preferably a C 6-10 aryl group. Specific examples of aryl include phenyl group, 1-naphthyl group, 2-naphthyl group and the like.
- heteroatom means a nitrogen atom (N), an oxygen atom (O), or a sulfur atom (S).
- heteroaryl means an aromatic heterocyclic group containing preferably 1 to 5 heteroatoms in the atoms constituting the ring. It may be a single ring or a condensed ring (for example, a bicyclic heteroaryl fused with a benzene ring or a monocyclic heteroaryl ring). The number of atoms constituting the ring is preferably 5 to 10 (5 to 10 membered heteroaryl).
- heteroaryl examples include furyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, triazolyl, tetrazolyl, Pyridyl group, pyrimidyl group, pyridazinyl group, pyrazinyl group, triazinyl group, benzofuranyl group, benzothienyl group, benzothiadiazolyl group, benzothiazolyl group, benzoxazolyl group, benzooxadiazolyl group, benzimidazolyl group, indolyl group, isoindolyl Group, indazolyl group, quinolyl group, isoquinolyl group, cinnolinyl group, quinazolinyl group, quinoxalinyl group, benzodio
- the “heterocycle” preferably has 3 to 8 atoms (3 to 8 membered heterocycles) constituting the ring, and includes 1 to 3 heteroatoms in the atoms constituting the ring,
- the ring may have a double bond, and means a non-aromatic monovalent ring.
- heterocycle examples include, for example, morpholino group, thiomorpholino group, piperidin-1-yl group, 4-substituted piperidin-1-yl group, piperazin-1-yl group, and 4-substituted piperazin-1-yl.
- morpholino group, thiomorpholino group, piperidin-1-yl group, 4-substituted piperidin-1-yl group, piperazin-1-yl group, 4-substituted piperazin-1-yl group, [1,3 A dioxolan-2-yl group or a [1,3] dioxane-2-yl group can be preferably used.
- Halogen atom in the present specification means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- alkoxy means an oxy group to which the above-defined “alkyl” is bonded, preferably a C 1-8 alkoxy group, more preferably a C 1-5 alkoxy group.
- arylalkyl means a group in which any hydrogen atom of the above-defined “alkyl” is substituted with “aryl” as defined above, for example, C 6-10 arylC 1-6 Alkyl is mentioned.
- C 6-10 aryl C 1-6 alkyl include phenylmethyl, phenylethyl, fanylpropyl, phenylbutyl, phenylpentyl, phenylhexyl and the like.
- amino refers to a monovalent group having two hydrogen atoms on a nitrogen atom (a group represented by —NH 2).
- the compound according to the present invention is included in the present invention even if it is a free form or a pharmaceutically acceptable salt.
- Such “salt” is particularly limited as long as it forms a salt with the compound represented by the formula (1) according to the present invention (sometimes referred to as a target compound) and is pharmaceutically acceptable.
- the acid salt which the target compound and the acid reacted, the base salt which the base reacted, etc. are mentioned.
- the acid used for preparing the pharmaceutically acceptable acid salt of the target compound is preferably one that reacts with the target compound to form a non-toxic acid salt.
- the acid salt include hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate, bisulfate, phosphate, acidic phosphate, acetate, lactate, citrate, Acid citrate, tartrate, bitartrate, succinate, maleate, fumarate, gluconate, saccharide, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate , P-toluenesulfonate, 1,1′-methylene-bis- (2-hydroxy-3-naphthoic acid) salt, and the like.
- the base used for preparing a pharmaceutically acceptable base salt of the target compound is preferably one that reacts with the target compound to form a non-toxic base salt.
- the base salt include alkali metal salts such as sodium salts and potassium salts, alkaline earth metal salts such as calcium salts and magnesium salts, water-soluble amine addition salts such as ammonium salts and N-methylglucamine salts, and lower alkanols. Mention may be made of ammonium salts, salts derived from other bases of pharmaceutically acceptable organic amines.
- Such a salt is also a salt of Compound I. Included in the invention.
- the target compound may absorb some other solvent and become a solvate, and such a salt is also included in the present invention as a salt of Compound I.
- Step A includes the following formula (II) [Wherein R 5 represents a halogen atom or O-LG, where LG is SO 2 Rc or P ( ⁇ O) (OR c ), R c is C 1-6 alkyl, or C 6- 10 aryl and R 3 is as defined above], a compound represented by the following formula (III) [Wherein R 6 represents hydrogen, C 1-6 alkyl, C 6-10 aryl C 1-6 alkyl, Si (R d ) 3 , and R d is the same or different, and C 1-6 alkyl and A compound selected from C 6-10 aryl, wherein Y 1 , Y 2 and Z are as defined above], and when R 6 is a substituent other than a hydrogen atom, In this step, the substituent —CO 2 R 6 of the compound is converted into a carboxyl group (—CO 2 H) to obtain a compound represented by the following formula (IV). [Wherein R 5 represents a halogen atom or O-LG, where LG is SO 2 Rc
- R 3 is preferably a halogen atom, more preferably a fluorine atom.
- R 5 is preferably a halogen atom, more preferably a fluorine atom.
- Y 1 is preferably a single bond.
- Y 2 is preferably —O—.
- Z is preferably alkylene having 3 carbon atoms.
- R 6 is preferably C 1-6 alkyl, more preferably C 2 alkyl.
- Examples of the compound of the formula (II) include 2,3,4-trifluoro-5-formylbenzoic acid.
- the compound of formula (II) can be synthesized based on the methods described in known literature.
- 2,3,4-trifluoro-5-formylbenzoic acid can be synthesized by the method described in (International Publication No. 2007/021001). It can also be synthesized by the method described in this example.
- reaction of the compound represented by the formula (II) and the compound represented by the formula (III) in the step A can be easily performed by adding and dissolving these compounds in a solvent, for example. be able to.
- Examples of the solvent in Step A include ether solvents (diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, t-butyl methyl ether, cyclopentyl methyl ether, dioxane, dimethoxyethane, etc.), hydrocarbon solvents (hexane, cyclohexane, benzene).
- ether solvents diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, t-butyl methyl ether, cyclopentyl methyl ether, dioxane, dimethoxyethane, etc.
- hydrocarbon solvents hexane, cyclohexane, benzene
- the reaction temperature in step A can be appropriately set based on the progress of the reaction and is not particularly limited.
- the reaction temperature is 0.1 to 48 hours, preferably 0.2 to 24 hours, more preferably 0.3 to 5 hours. You can select a range of time.
- the compound obtained after reacting the compound represented by the formula (II) and the compound represented by the formula (III) is usually preferably converted into a carboxyl group as it is. However, washing extraction, isolation (concentration, crystallization) Analysis), purification (recrystallization, column chromatography, etc.), and then conversion to a carboxyl group.
- the reaction for converting the substituent —CO 2 R 6 into a carboxyl group is carried out by, for example, converting the obtained compound into a hydroxide of an alkali metal (lithium, sodium, potassium, etc.), an alkali metal (lithium, Sodium, potassium, cesium, etc.) carbonate, trifluoroacetic acid, hydrochloric acid, bromic acid, Lewis acid (boron trichloride, magnesium iodide, sodium iodide-trimethylchlorosilane, sodium iodide-aluminum chloride, aluminum iodide, trimethylsilyl) Triflate), hydrogen-palladium, formic acid-palladium or the like in the presence of these.
- an alkali metal lithium, sodium, potassium, etc.
- an alkali metal lithium, Sodium, potassium, cesium, etc.
- Triflate triflate
- hydrogen-palladium formic acid-palladium or the like in the presence of these.
- the amount of these used is not particularly limited, and an appropriate amount usually used for hydrolysis may be used. For example, it is preferably 1000 to 0.001 equivalent, more preferably 100 to 0.001 equivalent to the compound to be hydrolyzed. It can adjust suitably in the range of 01 equivalent.
- Examples of the solvent for conversion to a carboxyl group include the same solvents as those used in the reaction of the compound represented by the above formula (II) and the compound represented by the above formula (III).
- ether solvents diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, t-butyl methyl ether, cyclopentyl methyl ether, dioxane, dimethoxyethane, etc.
- hydrocarbon solvents hexane, cyclohexane, benzene, toluene, xylene, etc.
- Alcohol solvents methanol, ethanol, n-propanol, isopropanol, etc.
- ester solvents methyl acetate, ethyl acetate, isopropyl acetate, etc.
- amide solvents for example, N, N-dimethylformamide, N, N— Dimethylacetamide, N-methylpyrrolidone Et
- phase transfer catalyst may be used to promote the reaction.
- the phase transfer catalyst used is, for example, tetrabutylammonium bromide, tetraethylammonium bromide, N-hexadecyl-N, N, N-trimethylammonium bromide, N-benzyl-N, N, N-triethylammonium chloride, tetrabutylammonium And hydroxide and 15-crown-5.
- the reaction time for conversion to carboxylic acid can be appropriately set based on the progress of the reaction and the like, and is not particularly limited.
- the reaction time is 0.1 to 48 hours, preferably 0.3 to 24 hours, more preferably.
- the reaction temperature for conversion to carboxylic acid can be appropriately set based on the progress of the reaction and is not particularly limited. For example, it is ⁇ 40 to 150 ° C., preferably ⁇ 20 to 100 ° C., more preferably 0 to It can be selected in the range of 50 ° C.
- Step B The mixture after completion of the reaction in Step A is usually preferably used as it is in Step B. However, after performing washing extraction, isolation (concentration, crystallization, etc.), purification (recrystallization, column chromatography, etc.), Step B is performed. You may use for.
- Step B is a step of obtaining a compound represented by the formula (VI) by reacting a compound represented by the formula (IV) with a compound represented by the formula (V) in the presence of a base and a metal salt.
- R 1 and R 2 are as defined above
- R 1 , R 2 , R 3 , Y 1 , Y 2 and Z are as defined above]
- R 1 is preferably a halogen atom, more preferably an iodine atom.
- R 2 is preferably a halogen atom, more preferably a fluorine atom.
- Examples of the base include, for example, lithium hexamethyldisilazide, sodium hexamethyldisilazide, group I metal cation hexamethyldisilazide including potassium hexamethyldisilazide or group II metal cation hexamethyldisilazide, hydrogen Group I metal cation hydrides including lithium hydride, sodium hydride, potassium hydride and calcium hydride, Group II metal cation hydrides, Group I metal cation dialkylamides or Group II metal cation dialkylamides including lithium diisopropylamide Group I metal cation amides including lithium amide, sodium amide, potassium amide, or Group II metal cation amides, and Group I metal cation alkoxides including sodium ethoxide, potassium tert-butoxide, magnesium ethoxide Sid or a group II metal cation alkoxide.
- Group I metal cation alkoxides
- the base is preferably a metal cation hexamethyldisilazide, more preferably an alkali metal hexamethyldisilazide, and particularly preferably lithium hexamethyldisilazide.
- the base is preferably used in an amount of 2 to 10 equivalents, more preferably 4 to 8 equivalents, relative to the compound of formula (V).
- the metal salt examples include a lithium salt, a magnesium salt, and a zinc salt.
- the metal salt is preferably a halide of lithium, magnesium or zinc, more preferably a lithium halide, and further preferably lithium bromide.
- the metal salt is preferably used in an amount of 2 to 10 equivalents, more preferably 4 to 8 equivalents, relative to the compound of formula (IV).
- Examples of the solvent in Step B include ether solvents (diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, t-butyl methyl ether, cyclopentyl methyl ether, dioxane, dimethoxyethane, etc.), hydrocarbon solvents (hexane, cyclohexane, benzene).
- ether solvents diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, t-butyl methyl ether, cyclopentyl methyl ether, dioxane, dimethoxyethane, etc.
- hydrocarbon solvents hexane, cyclohexane, benzene
- aprotic polar solvents are preferable, and for example, ether solvents, ester solvents, amide solvents, acetonitrile, and mixed solvents of these organic solvents (at least two) are preferable.
- the reaction time in the step B can be appropriately set based on the progress of the reaction and is not particularly limited.
- the reaction time is 0.1 to 48 hours, preferably 0.3 to 24 hours, more preferably 0.5 to It is performed in the range of 10 hours.
- the reaction temperature in step B can be appropriately set based on the progress of the reaction and the like, and is not particularly limited. For example, it is ⁇ 100 to 100 ° C., preferably ⁇ 80 to 50 ° C., more preferably ⁇ 78 to 25 ° C. The range of ⁇ 70 to 20 ° C. is particularly preferable.
- Step C The mixture after completion of the reaction in Step B is usually preferably used as it is in Step C. However, after performing washing extraction, isolation (concentration, crystallization, etc.), purification (recrystallization, column chromatography, etc.), Step C is performed. You may use for.
- the reaction system in the coupling reaction between the benzoic acid derivative (IV) and the anilines (V), the reaction system can be made uniform by adding a metal salt, and the reaction conversion rate can be dramatically improved. It is possible to improve the reaction and control the reaction easily.
- Step C is a step in which the compound represented by formula (VI) is subjected to reducing conditions to obtain a compound represented by formula (VII). More specifically, for example, in formula (VI) in a solvent, In this step, the compound represented by formula (VII) is cyclized to the group X represented by formula (i) after reacting the compound represented by a reducing agent.
- R 1 , R 2 , R 3 and X are as defined above]
- X is preferably 3-oxo [1,2] oxadinanyl.
- sodium borohydride, sodium cyanoborohydride, triethylsilane, trimethylsilane, lithium aluminum hydride, diisopropylaluminum hydride, borane-pyridine complex, decaborane, diborane, borane-dimethylsulfide complex, borane -THF complex, hydrogen, zinc borohydride and the like can be used.
- a metal catalyst for example, palladium on carbon, platinum, homogeneous palladium complex, homogeneous ruthenium complex, homogeneous port complex
- An acid or the like may be used to promote the reaction.
- Examples of the acid used include acetic acid, hydrochloric acid, trifluoroacetic acid, dichloroacetic acid, trifluoroboron-diethyl ether complex, trimethylsilyl triflate, aluminum chloride, trimethylsilyl chloride and the like.
- ether solvents diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentylmethyl ether, dioxane, dimethoxyethane, etc.
- alcohol solvents methanol, ethanol, n-propanol, isopropanol, etc.
- ester solvents etc.
- Solvent methyl acetate, ethyl acetate, isopropyl acetate, etc.
- amide solvent for example, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, etc.
- acetonitrile organic solvents thereof (at least 2 Seeds) and the like.
- water may be used, and a mixed solvent of water and the above organic solvent (may be a mixed solvent of at least two kinds of organic solvents) may be used.
- the reaction time in the step C can be appropriately set based on the progress of the reaction and is not particularly limited.
- the reaction time is 0.1 to 48 hours, preferably 0.3 to 24 hours, more preferably 0.5 to It is performed in the range of 10 hours.
- the reaction temperature in Step C can be selected, for example, in the range of ⁇ 80 to 200 ° C., preferably ⁇ 50 to 100 ° C., more preferably ⁇ 30 to 40 ° C.
- the reducing agent is preferably used in an amount of 0.2 to 250 equivalents, more preferably 0.5 to 120 equivalents, relative to the compound of formula (VI).
- the compound thus obtained is preferably cyclized as it is to the group X represented by the above formula (i) in the reaction system.
- washing extraction, isolation concentration, crystallization, etc.
- It may be cyclized after purification (recrystallization, column chromatography, etc.).
- Such a cyclization reaction can be easily carried out, for example, by treating with a reducing agent in Step C in a solvent and then stirring the reaction solution containing the obtained compound by raising the temperature as necessary. it can.
- ether solvents diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, cyclopentylmethyl ether, dioxane, dimethoxyethane, etc.
- Alcohol solvents methanol, ethanol, n-propanol, isopropanol, etc.
- ester solvents methyl acetate, ethyl acetate, isopropyl acetate, etc.
- amide solvents eg, N, N-dimethylformamide, N, N-dimethyl
- acetonitrile mixed solvents of these organic solvents (at least two).
- the cyclization reaction time in Step C can be appropriately set based on the progress of the reaction and is not particularly limited.
- the cyclization reaction time is 0.1 to 48 hours, preferably 1 to 24 hours, more preferably 3 to 10 hours. Done in a time range.
- the reaction temperature for cyclization in Step C can be selected, for example, in the range of 0 to 200 ° C., preferably 20 to 150 ° C., more preferably 30 to 100 ° C.
- Step D The mixture after completion of the reaction in Step C is usually preferably used as it is in Step D. However, after performing washing extraction, isolation (concentration, crystallization, etc.) and purification (recrystallization, column chromatography, etc.), Step D is performed. You may use for.
- Step D is a step of obtaining a compound represented by formula (I) or a salt thereof by subjecting a compound represented by formula (VII) to a condensation reaction with a compound represented by formula (VIII).
- Step D can be performed, for example, by adding an additive in a solvent in the presence of a condensing agent as necessary.
- R 4 is preferably C 1-6 alkyl substituted by OH, more preferably hydroxyethyl (CH 2 CH 2 OH).
- condensing agents in Step D include 1,3-dicyclohexylcarbodiimide (DCC), 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ), bromo-tris (pyrrolidino) -phosphonium hexafluorophos Fert (PyBrOP), 1-ethyl-3- (3′-dimethylaminopropyl) carbodiimide (EDC), (benzotriazolyloxy) tripyrrolidino-phosphonium hexafluorophosphate (PyBOP), O- (7-azabenzotriazole- 1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HATU), 2- (1H-benzotriazol-1-yl) -1,1,3,3-tetra Methyluronium hexafluorophosphate (HBTU), 2 (1H-benzotri
- Step D examples include hydroxybenzotriazole (HOBt), 3-hydroxy-4-oxo-3,4-dihydro-1,2,3-benzotriazine (HODhBt), 1-hydroxy-7-aza A benzotriazole (HOAt) etc. can be mentioned.
- HOBt hydroxybenzotriazole
- HODhBt 3-hydroxy-4-oxo-3,4-dihydro-1,2,3-benzotriazine
- HOAt 1-hydroxy-7-aza A benzotriazole
- Examples of the solvent in Step D include ether solvents (diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, t-butyl methyl ether, cyclopentyl methyl ether, dioxane, dimethoxyethane, etc.), alcohol solvents (methanol, ethanol, n- Propanol, isopropanol, etc.), ester solvents (methyl acetate, ethyl acetate, isopropyl acetate, etc.), amide solvents (eg, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, etc.), acetonitrile, And a mixed solvent of these organic solvents (at least two kinds). Further, water may be used, and a mixed solvent of water and the above organic solvent (may be a mixed solvent of at least two kinds of organic solvents) may be used.
- ether solvents diethyl ether,
- the reaction time in the step D can be appropriately set based on the progress of the reaction and is not particularly limited.
- the reaction time is 0.1 to 48 hours, preferably 0.3 to 24 hours, and more preferably 0.5 to It is performed in the range of 10 hours.
- the reaction temperature in step D can be selected, for example, in the range of ⁇ 40 to 200 ° C., preferably ⁇ 20 to 100 ° C., more preferably 0 to 50 ° C.
- the amount of the condensing agent to be used is preferably 1 equivalent to 20 equivalents, more preferably 1 equivalent to 10 equivalents, relative to the compound of formula (VII).
- the amount of additive used is preferably 1 equivalent to 20 equivalents, more preferably 1 equivalent to 10 equivalents, relative to the compound of formula (VII).
- a pharmaceutically acceptable salt of the compound of formula (I) can be produced by reacting the compound with a base that can be used in the production of a pharmaceutical product.
- salts include alkali metal salts (lithium salts, sodium salts, potassium salts, cesium salts, rubidium salts, etc.), alkaline earth metal salts (magnesium salts, calcium salts, etc.), ammonium salts (ammonium salts, Alkyl ammonium salt, dialkyl ammonium salt, trialkyl ammonium salt, tetraalkyl ammonium salt, etc.).
- alkali metal salts are preferable, and sodium salts are more preferable.
- all prior art documents cited in the present specification are incorporated herein by reference.
- NMR was measured using a nuclear magnetic resonance apparatus JNM-ECP-500 (manufactured by JEOL) or JNM-ECP-400 (manufactured by JEOL).
- Mass spectrometry was measured using a mass spectrometer LCT Premier XE (manufactured by Waters) or Agilent Technologies HP6890. Even when the product of one step was subjected to the next step without purification, the purified product was used for NMR analysis.
- reaction solution was added to ice-cooled 3N hydrochloric acid (100 ml), warmed to room temperature, and extracted with ethyl acetate (100 ml). The aqueous layer was extracted with ethyl acetate (50 ml). The organic layers were combined, washed with saturated brine (20 ml), dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. A mixed solvent of ethyl acetate (20 ml) and n-heptane (40 ml) was added to the obtained residue, and the mixture was suspended by heating to 60 ° C. Then, n-heptane (60 ml) was added at the same temperature and stirred for 10 minutes. did. It cooled to 0 degreeC and stirred for 30 minutes at the same temperature.
- the precipitated crystals were filtered, washed with a mixed solvent of ethyl acetate and n-heptane (1:10), and dried under reduced pressure to obtain the title compound (9.0 g, 73%).
- the mother liquor was evaporated under reduced pressure, ethyl acetate (3.0 ml) was added to the resulting residue, heated to 50 ° C. to dissolve, and n-heptane (15 ml) was added. After confirming solid precipitation, the mixture was cooled to 0 ° C. and stirred at the same temperature for 1 hour and 15 minutes.
- Step 2 Synthesis of (E) -2,3,4-trifluoro-5-[(4-ethoxy-4-oxobutoxyimino) methyl] benzoic acid
- a saturated aqueous sodium bicarbonate solution (70 ml) was added to the organic layer, and the aqueous layer was extracted.
- Saturated aqueous sodium hydrogen carbonate (20 ml) was again added to the organic layer, and the aqueous layer was extracted.
- the aqueous layers were combined and then washed with t-butyl methyl ether (50 ml) to obtain a solution containing the title compound.
- the product was used in the next step without further purification.
- Step 3 Synthesis of 2,3,4-trifluoro-5-[(3-carboxypropoxyimino) methyl] benzoic acid
- Step 4 Synthesis of (E) -3,4-difluoro-2- (2-fluoro-4-iodophenylamino) -5-[(3-carboxypropoxyimino) methyl] benzoic acid
- Step 5 Synthesis of 5-[(3-carboxypropoxyamino) methyl] -3,4-difluoro-2- (2-fluoro-4-iodophenylamino) benzoic acid
- Step 6 Synthesis of 3,4-difluoro-2- (2-fluoro-4-iodophenylamino) -5-[(3-oxo- [1,2] oxazin-2-yl) methyl] benzoic acid
- Step 7 Sodium (3,4-difluoro-2- (2-fluoro-4-iodophenylamino) -5-[(3-oxo- [1,2] oxazin-2-yl) methyl] benzoyl) (2 -Hydroxyethoxy) amide synthesis
- Step 8 3,4-Difluoro-2- (2-fluoro-4-iodophenylamino) -N- (2-hydroxyethoxy) 5-[(3-oxo- [1,2] oxazin-2-yl) Of methyl] benzamide
- the organic layer was washed with 10 wt% brine and dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure to obtain a residue.
- Acetonitrile (0.6 ml) was added to the obtained residue, and then the solvent was distilled off under reduced pressure.
- Acetonitrile (0.6 ml) was further added to the obtained residue, and the mixture was heated to 60 ° C. and dissolved. After cooling to room temperature, the mixture was stirred for 1 hour, and solid precipitation was confirmed. The mixture was cooled to 5 ° C. and further stirred for 1 hour. The precipitated solid was filtered, washed with acetonitrile (2 ml) cooled to 5 ° C., and dried under reduced pressure to give the title compound (21 mg, 44%).
- the raw material compound was dissolved in THF under the same conditions as described above except that the metal salt was not added.
- THF 1.0 M lithium hexamethyldisilazide in THF (290 ⁇ l, 0.29 mmol) was added dropwise, and the mixture was stirred at the same temperature for 3.5 hours. 2N HCl was added to this solution, and the temperature was raised to room temperature. Then, the organic layer was extracted with 2-methyltetrahydrofuran. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the title compound. The yield was calculated based on 1 H-NMR analysis of the obtained residue. (Internal standard substance: 1,3,5-trimethoxybenzene)
- Table 1 shows a comparison of yields due to the addition of metal salts.
- a simple and high-yield 5-substituted-2-phenylamino-benzamide compound composed of a homogeneous reaction that can be controlled without using an organic transition metal catalyst or an osmium reagent is used.
- a manufacturing method is provided.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Electromagnetism (AREA)
- Computer Hardware Design (AREA)
- Power Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- General Physics & Mathematics (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Sustainable Development (AREA)
- Sustainable Energy (AREA)
- Inorganic Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
本発明は、下記式(I)で表される化合物またはその薬理学的に許容される塩又はその合成中間体の製造に関し取り扱いが煩雑になる試薬等を使用することなく、また反応制御可能な均一系からなる反応で構成された簡便かつ高収率な新規な方法を提供する。(式中、R1、R2、R3、R4及びXは、特許請求の範囲に記載のR1、R2、R3、R4及びXと同意義である。)
Description
本発明は、5-置換-2-フェニルアミノ-ベンズアミド類の製造方法に関する。
下記式(I)で表わされる化合物、またはその医薬的に許容しうる塩(以下、「式(I)の2-(N-フェニルアミノ)安息香酸誘導体」ということがある。)は、癌、再発狭窄症、乾癬、及びアテローム性動脈硬化症を含む増殖性疾患の治療のための選択的MEK-1阻害剤として有用であることが知られている(特許文献1)。:

〔式中、R1はハロゲン原子、C2-6アルケニルまたはC2-6アルキニルを示し;
R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基;
 (式中、
(式中、
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基はお互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)〕。
R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基;
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基はお互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)〕。
前記式(I)の2-(N-フェニルアミノ)安息香酸誘導体の製造方法としては、例えば、下記スキーム1に示されるような方法が知られている(特許文献1の第103~104ページ(段落0188)の反応工程1)。〔下記式中、Lは脱離基を示し、R1、R2、R3、R4、およびXは前記式(I)において定義したものと同意義である。〕
スキーム1

スキーム1
特許文献1では、前記スキーム1、さらに下記スキーム2に記載のように2,3,4-トリフルオロ-5-ヨード安息香酸を出発原料として、ビニル化、酸化開裂反応を経て、2,3,4-トリフルオロ-5-ホルミル安息香酸を合成し、それを中間体として式(I)の2-(N-フェニルアミノ)安息香酸誘導体を製造する方法が記載されている。
スキーム2

スキーム2
しかしながら、ビニル化工程においては、有機遷移金属触媒を要し、反応後に有機遷移金属錯体および有機配位子を削除する工程が必要となり、操作が煩雑で効率的でない。また、酸化開裂反応によるホルミル基への変換工程は、酸化剤として四酸化オスミニウムなどの取り扱いが煩雑になる試薬を必要とし、工業的製造法として適していない。
また、下記(IV)の構造を有する安息香酸誘導体と、アルカリ金属ヘキサメチルジシラジドを用いたアニリン類とのカップリング反応では、反応系が不均一となり反応転化率が工業的には極めて好ましくない反応となることも本願発明の検討過程において見出された。

〔式中、R3、Y1、Y2およびZは前記式(I)にて定義した通りである。R5は、ハロゲン原子またはO-LGを表わし、ここでLGは、SO2RcまたはP(=O)(ORc)であり、RcはC1-6アルキル、またはC6-10アリールである。〕
本発明の目的は、2-(N-フェニルアミノ)安息香酸誘導体の工業的製法に適した簡便かつ効率的な製造方法、および有用な合成中間体を提供することである。
本発明の目的は、2-(N-フェニルアミノ)安息香酸誘導体の工業的製法に適した簡便かつ効率的な製造方法、および有用な合成中間体を提供することである。
本発明者らは、上記課題を解決するために鋭意検討を行った結果、下記の製造方法をとることにより、有機遷移金属触媒あるいはオスミウム試薬などを使用することなく、簡便かつ高収率で目的化合物の製造方法が可能となることを見出した。
さらに安息香酸誘導体(IV)とアニリン類とのカップリング反応においては、金属塩を添加することで反応系を均一とすることができ、反応転化率が向上することおよび反応制御が容易になることを見出して、本発明を完成するに至った。
さらに安息香酸誘導体(IV)とアニリン類とのカップリング反応においては、金属塩を添加することで反応系を均一とすることができ、反応転化率が向上することおよび反応制御が容易になることを見出して、本発明を完成するに至った。
すなわち、本発明は次の通りである。
〔1〕
下記式I:

〔式中、R1はハロゲン原子、C2-6アルケニルまたはC2-6アルキニルを示し;
R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基;
 (式中、
(式中、
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基は、互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)〕
で表わされる化合物、またはその医薬的に許容しうる塩を製造する方法であって、下記工程A、工程B、工程C、および工程Dを含む方法;
工程Aは、下記式(II)

〔式中、R5はハロゲン原子またはO-LGを表わし、ここでLGは、SO2RcまたはP(=O)(ORc)であり、RcはC1-6アルキル、またはC6-10アリールであり、R3は前記に定義した通りである〕で表わされる化合物を、下記式(III)

〔式中、R6は水素、C1-6アルキル、C6-10アリールC1-6アルキル、またはSi(Rd)3を表わし、Rdは、同一または異なって、C1-6アルキルおよびC6-10アリールから選択され、Y1、Y2およびZは前記にて定義した通りである〕である化合物と反応させ、R6が水素原子以外の置換基の場合はさらに得られた化合物の置換基-CO2R6をカルボキシル基へと変換して、下記式(IV)

〔式中、R3、R5、Y1、Y2およびZは前記にて定義した通りである〕で表わされる化合物を得る工程であり;
工程Bは、塩基および金属塩の存在下、工程Aで得られた式(IV)で表わされる化合物を下記式(V)

〔式中、R1、R2は前記にて定義した通りである〕で表わされる化合物と反応させ、下記式(VI)

〔式中、R1、R2、R3、Y1、Y2およびZは前記にて定義した通りである〕で表わされる化合物を得る工程であり;
工程Cは、工程Bで得られた式(VI)で表わされる化合物を還元条件に付し、下記式(VII)

〔式中、R1、R2、R3およびXは前記にて定義した通りである〕で表わされる化合物を得る工程であり;
工程Dは、前記工程Cで得られた式(VII)で表わされる化合物を下記式(VIII)

〔式中、R4は前記にて定義した通りである〕で表わされる化合物と反応させて、前記式(I)で表わされる化合物またはその塩を得る工程である。
〔2〕
工程Bにおいて、金属塩が、リチウム塩、マグネシウム塩、および/または亜鉛塩から選ばれる1種または2種以上である、〔1〕に記載の方法。
〔3〕
工程Bにおいて、塩基が、アルカリ金属ヘキサメチルジシラジドである、〔1〕または〔2〕に記載の方法。
〔4〕
R5がフッ素原子である、〔1〕~〔3〕のいずれか一項に記載の方法。
〔5〕
工程Bにおいて、反応が非プロトン性極性溶媒中で-78℃~25℃で行われる、〔1〕~〔4〕のいずれか一項に記載の方法。
〔6〕
R1、R2、およびR3はそれぞれハロゲン原子であり、R4は水酸基により置換されていてもよいC1-6アルキルである、〔1〕~〔5〕のいずれか一項に記載の方法。
〔7〕
Y1は単結合、Y2は-O-、および、Zが炭素原子1~5個のアルキレンである、〔1〕~〔6〕のいずれか一項に記載の方法。
〔8〕
R1がヨウ素原子であり、R2およびR3がそれぞれフッ素原子であり、R4がヒドロキシエチル(CH2CH2OH)であり、Y1が単結合であり、Y2が-O-であり、Zが炭素数3のアルキレンであり、式(I)で表される化合物が下記式(IX)

で表される化合物である、〔1〕~〔7〕のいずれか一項に記載の方法。
〔9〕
下記式(IV)

〔式中、R3、R5、Y1、Y2およびZは前記〔1〕にて定義した通りである〕で表わされる化合物を、塩基および金属塩の存在下、下記式(V)

〔式中、R1、およびR2は前記〔1〕にて定義した通りである〕で表わされる化合物と反応させる、下記式(VI)

〔式中、R1、R2、R3、Y1、Y2およびZは前記〔1〕にて定義した通りである〕で表わされる中間体化合物の製造方法である。
〔10〕
下記式(X)

〔式中、R5、R6、Y1、Y2およびZは〔1〕にて定義したR5、R6、Y1、Y2およびZとそれぞれ同意義を示す。〕で表わされる化合物又はその薬学上許容しうる塩。
〔1〕
下記式I:
R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基;
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基は、互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)〕
で表わされる化合物、またはその医薬的に許容しうる塩を製造する方法であって、下記工程A、工程B、工程C、および工程Dを含む方法;
工程Aは、下記式(II)
工程Bは、塩基および金属塩の存在下、工程Aで得られた式(IV)で表わされる化合物を下記式(V)
工程Cは、工程Bで得られた式(VI)で表わされる化合物を還元条件に付し、下記式(VII)
工程Dは、前記工程Cで得られた式(VII)で表わされる化合物を下記式(VIII)
〔2〕
工程Bにおいて、金属塩が、リチウム塩、マグネシウム塩、および/または亜鉛塩から選ばれる1種または2種以上である、〔1〕に記載の方法。
〔3〕
工程Bにおいて、塩基が、アルカリ金属ヘキサメチルジシラジドである、〔1〕または〔2〕に記載の方法。
〔4〕
R5がフッ素原子である、〔1〕~〔3〕のいずれか一項に記載の方法。
〔5〕
工程Bにおいて、反応が非プロトン性極性溶媒中で-78℃~25℃で行われる、〔1〕~〔4〕のいずれか一項に記載の方法。
〔6〕
R1、R2、およびR3はそれぞれハロゲン原子であり、R4は水酸基により置換されていてもよいC1-6アルキルである、〔1〕~〔5〕のいずれか一項に記載の方法。
〔7〕
Y1は単結合、Y2は-O-、および、Zが炭素原子1~5個のアルキレンである、〔1〕~〔6〕のいずれか一項に記載の方法。
〔8〕
R1がヨウ素原子であり、R2およびR3がそれぞれフッ素原子であり、R4がヒドロキシエチル(CH2CH2OH)であり、Y1が単結合であり、Y2が-O-であり、Zが炭素数3のアルキレンであり、式(I)で表される化合物が下記式(IX)
〔9〕
下記式(IV)
〔10〕
下記式(X)
本発明によれば、有機遷移金属触媒あるいはオスミウム試薬等を使用することなく、簡便かつ高収率な式(I)の2-(N-フェニルアミノ)安息香酸誘導体の製造方法を提供することができる。
以下、本発明の実施形態について説明する。
本発明の方法は、下記式(I)で表わされる化合物(以下、「化合物I」ということがある。)、またはその医薬的に許容しうる塩を製造する方法であって、下記工程A、工程B、工程C、および工程Dを含む方法を提供するものである。

式中、R1はハロゲン原子、C2-6アルケニルまたはC2-6アルキニルを示し;
R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基である。
 (式中、
(式中、
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基はお互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)
R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基である。
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基はお互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)
本明細書における「アルキル」は、脂肪族炭化水素から任意の水素原子を1個除いて誘導される1価の基である。骨格中にヘテロ原子または不飽和炭素-炭素結合を含有しない。水素および炭素原子を含有するヒドロカルビルまたは炭化水素基構造の部分集合を有する。アルキルは直鎖状および分枝鎖状の構造を含む。アルキルとしては、好ましくは炭素原子数1~6のアルキル(「C1-6アルキル」という。以下「Cp-q」とは炭素原子数がp~q個であることを意味する。)、C1-5アルキル、C1-4アルキル、C1-3アルキルなどが挙げられる。
アルキルとしては、具体的にはメチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、s-ブチル、t-ブチル、ペンチル、イソペンチル、2,3-ジメチルプロピル、3,3-ジメチルブチル、ヘキシルなどが挙げられる。
本明細書における「アルケニル」は、少なくとも1個の二重結合(2個の隣接SP2炭素原子)を有する1価の炭化水素基である。二重結合および置換分(存在する場合)の配置によって、二重結合の幾何学的形態は、エントゲーゲン(E)またはツザンメン(Z)、あるいはシス配置またはトランス配置をとることができる。アルケニルとしては、直鎖状または分岐鎖状のものが挙げられ、好ましくはC2-6アルケニル、さらに好ましくはC2-5アルケニルが挙げられる。
アルケニルとして、具体的には、たとえば、ビニル、アリル、1-プロペニル、2-プロペニル、1-ブテニル、2-ブテニル(シス、トランスを含む)、3-ブテニル、ペンテニル、ヘキセニルなどが挙げられる。
本明細書における「アルキニル」は、少なくとも1個の三重結合(2個の隣接SP炭素原子)を有する、1価の炭化水素基である。直鎖状または分枝鎖状のアルキニルが挙げられ、好ましくはC2-6アルキニル、更に好ましくはC2-5アルキニルが挙げられる。
アルキニルとしては具体的には、たとえば、エチニル、1-プロピニル、プロパルギル、3-ブチニル、ペンチニル、ヘキシニルなどが挙げられ、好ましくはエチニルが挙げられる。
アルケニルまたはアルキニルには、それぞれ1個または2個以上の二重結合または三重結合を有することができる。二重結合と三重結合とを同時に有することもできる。
本明細書における「シクロアルキル」は、環状の脂肪族炭化水素を意味し、好ましくは、C3-8シクロアルキルなどが挙げられる。シクロアルキルとしては具体的には、たとえば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基などが挙げられる。
本明細書における「アルキレン」は、-(CH2)n-で表わされる、2価の基を意味し、アルキレンとしては好ましくはC1-5アルキレン(n=1~5)、特に好ましくはC1-3アルキレン(n=1~3)が挙げられる。
本明細書における「アリール」は、1価の芳香族炭化水素環を意味し、好ましくはC6-10アリール基が挙げられる。アリールとしては具体的には、たとえば、フェニル基、1-ナフチル基、2-ナフチル基などが挙げられる。
本明細書における「ヘテロ原子」は、窒素原子(N)、酸素原子(O)または硫黄原子(S)を意味する。
本明細書における「ヘテロアリール」は、環を構成する原子中に好ましくは1~5個のヘテロ原子を含有する芳香族性の複素環基を意味する。単環でも縮合環(たとえば、ベンゼン環または単環へテロアリール環と縮合した2環式ヘテロアリール)でもよい。環を構成する原子の数は好ましくは5~10である(5~10員ヘテロアリール)。
ヘテロアリールとしては具体的には、たとえば、フリル基、チエニル基、ピロリル基、イミダゾリル基、ピラゾリル基、チアゾリル基、イソチアゾリル基、オキサゾリル基、イソオキサゾリル基、オキサジアゾリル基、チアジアゾリル基、トリアゾリル基、テトラゾリル基、ピリジル基、ピリミジル基、ピリダジニル基、ピラジニル基、トリアジニル基、ベンゾフラニル基、ベンゾチエニル基、ベンゾチアジアゾリル基、ベンゾチアゾリル基、ベンゾオキサゾリル基、ベンゾオキサジアゾリル基、ベンゾイミダゾリル基、インドリル基、イソインドリル基、インダゾリル基、キノリル基、イソキノリル基、シンノリニル基、キナゾリニル基、キノキサリニル基、ベンゾジオキソリル基、インドリジニル基、イミダゾピリジル基などが挙げられる。
本明細書における「ヘテロ環」は、環を構成する原子数が好ましくは3~8(3~8員ヘテロ環)であり、環を構成する原子中に1~3個のヘテロ原子を含み、環中に二重結合を有していてもよく、非芳香族性の1価の環を意味する。
ヘテロ環としては具体的には、たとえば、モルホリノ基、チオモルホリノ基、ピペリジン-1-イル基、4-置換ピペリジン-1-イル基、ピペラジン-1-イル基、4-置換ピペラジン-1-イル基、ピロリジン-1-イル基、ピロリニル基、イミダゾリジニル基、イミダゾリニル基、ピラゾリジニル基、ピラゾリニル基、[1,3]ジオキソラン-2-イル基、[1,3]ジオキサン-2-イル基などが挙げられる。これらのうちでは、モルホリノ基、チオモルホリノ基、ピペリジン-1-イル基、4-置換ピペリジン-1-イル基、ピペラジン-1-イル基、4-置換ピペラジン-1-イル基、[1,3]ジオキソラン-2-イル基、[1,3]ジオキサン-2-イル基を好ましく用いることができる。
本明細書における「ハロゲン原子」は、フッ素原子、塩素原子、臭素原子またはヨウ素原子を意味する。
本明細書における「アルコキシ」は、前記定義の「アルキル」が結合したオキシ基であることを意味し、好ましくはC1-8アルコキシ基、さらに好ましくはC1-5アルコキシ基である。具体的には、たとえば、メトキシ基、エトキシ基、1-プロポキシ基、2-プロポキシ基、n-ブトキシ基、i-ブトキシ基、sec-ブトキシ基、tert-ブトキシ基、1-ペンチルオキシ基、2-ペンチルオキシ基、3-ペンチルオキシ基、2-メチル-1-ブチルオキシ基、3-メチル-1-ブチルオキシ基、2-メチル-2-ブチルオキシ基、3-メチル-2-ブチルオキシ基、2,2-ジメチル-1-プロピルオキシ基、1-へキシルオキシ基、2-へキシルオキシ基、3-へキシルオキシ基、2-メチル-1-ペンチルオキシ基、3-メチル-1-ペンチルオキシ基、4-メチル-1-ペンチルオキシ基、2-メチル-2-ペンチルオキシ基、3-メチル-2-ペンチルオキシ基、4-メチル-2-ペンチルオキシ基、2-メチル-3-ペンチルオキシ基、3-メチル-3-ペンチルオキシ基、2,3-ジメチル-1-ブチルオキシ基、3,3-ジメチル-1-ブチルオキシ基、2,2-ジメチル-1-ブチルオキシ基、2-エチル-1-ブチルオキシ基、3,3-ジメチル-2-ブチルオキシ基、2,3-ジメチル-2-ブチルオキシ基、1-メチル-シクロプロピルメトキシ基などがあげられる。
本明細書における「アリールアルキル」は、前記定義の「アルキル」のいずれかの水素原子が、前記定義の「アリール」で置換された基を意味し、たとえば、C6-10アリールC1-6アルキルが挙げられる。C6-10アリールC1-6アルキルとしては、具体的には、たとえば、フェニルメチル、フェニルエチル、ファニルプロピル、フェニルブチル、フェニルペンチル、フェニルヘキシルなどが挙げられる。
本明細書における「アミノ」とは、窒素原子上に2つの水素原子を有する一価の基(-NH2であらわされる基)を示す。
本発明に係る化合物はフリー体であっても、医薬的に許容される塩であっても本発明に含まれる。このような「塩」とは、本発明に係る式(1)で表される化合物(目的化合物ということがある。)と塩を形成し、かつ医薬的に許容されるものであれば特に限定されず、たとえば、目的化合物と、酸とが反応した酸塩、塩基とが反応した塩基塩などが挙げられる。
目的化合物の薬剤学的に許容できる酸塩を調製するために用いる酸は、目的化合物と反応し、無毒の酸塩を形成するものが好ましい。酸塩としては、たとえば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硝酸塩、硫酸塩、重硫酸塩、リン酸塩、酸性リン酸塩、酢酸塩、乳酸塩、クエン酸塩、酸性クエン酸塩、酒石酸塩、重酒石酸塩、コハク酸塩、マレイン酸塩、フマル酸塩、グルコン酸塩、糖酸塩、安息香酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩、1,1'-メチレン-ビス-(2-ヒドロキシ-3-ナフトエ酸)塩などが挙げられる。
目的化合物の薬剤学的に許容できる塩基塩を調製するために用いる塩基は、目的化合物と反応し、無毒の塩基塩を形成するものが好ましい。塩基塩としては、たとえば、ナトリウム塩、カリウム塩などのアルカリ金属塩、カルシウム塩、マグネシウム塩などのアルカリ土類金属塩、アンモニウム塩、N-メチルグルカミン塩などの水溶性アミン付加塩、低級アルカノールアンモニウム塩、薬学的に許容することができる有機アミンの他の塩基から誘導される塩を挙げることができる。
また、目的化合物は、大気中に放置しておくことにより、水分を吸収し、吸着水が付いたり、水和物となったりする場合があり、そのような塩も、化合物Iの塩として本発明に包含される。
さらに、目的化合物は、他のある種の溶媒を吸収し、溶媒和物となる場合があるが、そのような塩も、化合物Iの塩として本発明に包含される。
以下、本発明の方法に含まれる、工程A~工程Dについて説明する。
工程A;
工程Aは、下記式(II)

〔式中、R5はハロゲン原子またはO-LGを表わし、ここでLGは、SO2RcまたはP(=O)(ORc)であり、RcはC1-6アルキル、またはC6-10アリールであり、R3は前記に定義した通りである〕で表わされる化合物を、下記式(III)

〔式中、R6は水素、C1-6アルキル、C6-10アリールC1-6アルキル、Si(Rd)3を表わし、Rdは、同一または異なって、C1-6アルキルおよびC6-10アリールから選択され、Y1、Y2およびZは前記にて定義した通りである〕で表される化合物と反応させ、R6が水素原子以外の置換基の場合はさらに得られた化合物の置換基-CO2R6をカルボキシル基(-CO2H)へと変換して、下記式(IV)で表わされる化合物を得る工程である。

〔式中、R3、R5、R6、Y1、Y2およびZは前記にて定義した通りである〕
工程A;
工程Aは、下記式(II)
R3は好ましくはハロゲン原子であり、さらに好ましくはフッ素原子である。
R5は好ましくはハロゲン原子であり、さらに好ましくはフッ素原子である。
Y1は好ましくは単結合である。
Y2は好ましくは-O-である。
Zは好ましくは炭素数が3のアルキレンである。
R6は好ましくはC1-6アルキルであり、更に好ましくはC2アルキルである
式(II)の化合物としては、例えば、2,3,4-トリフルオロ-5-ホルミル安息香酸が挙げられる。
式(II)の化合物は、公知文献に記載の方法に基づいて合成することができる。例えば、2,3,4-トリフルオロ-5-ホルミル安息香酸は、(国際公開第2007/021001号)に記載の方法により合成することができる。また、本実施例記載の方法によっても合成することができる。
工程Aにおける、前記式(II)で表される化合物と前記式(III)で表される化合物との反応は、たとえば、溶媒中で、これらの化合物を加えて溶解させるだけで、容易に行うことができる。
工程Aにおける溶媒の例としては、エーテル系溶媒(ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、t-ブチルメチルエーテル、シクロペンチルメチルエーテル、ジオキサン、ジメトキシエタンなど)、炭化水素系溶媒(ヘキサン、シクロヘキサン、ベンゼン、トルエン、キシレンなど)、アルコール系溶媒(メタノール、エタノール、n-プロパノール、イソプロパノールなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸イソプロピルなど)、アミド系溶媒(例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドンなど)、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒などが挙げられる。また、水を使用してもよく、水と上記有機溶媒(少なくとも2種の有機溶媒の混合溶媒であってもよい。)の混合溶媒を使用してもよい。反応促進のために酸を用いてもよい。使用される酸は、例えば、酢酸、塩酸、硫酸、トルエンスルホン酸、トリフルオロ酢酸などが挙げられる。
工程Aにおける反応時間、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、当該反応は、例えば-50~200℃、好ましくは-20~100℃、より好ましくは0~50℃の範囲で選択できる。
工程Aにおける反応温度は、反応の進行状況などに基づき適宜設定可能であり、特に限定されないが、例えば0.1~48時間、好ましくは0.2~24時間、より好ましくは0.3~5時間の範囲で選択できる。
式(III)で表わされる化合物のR6が水素以外の置換基である場合には、さらに、得られた化合物の置換基-CO2R6をカルボキシル基(-CO2H)へと変換することにより下記式(IV)で表わされる化合物を得ることができる。

〔式中、R3、R5、Y1、Y2およびZは前記にて定義した通りである〕
式(II)で表わされる化合物と式(III)で表わされる化合物を反応させた後に得られた化合物は通常、そのままカルボキシル基へと変換するのが好ましいが、洗浄抽出、単離(濃縮、晶析等)、精製(再結晶、カラムクロマトグラフィー等)を行ってからカルボキシル基へと変換させてもよい。
前記の置換基-CO2R6をカルボキシル基へと変換する反応は、たとえば、溶媒中、得られた化合物を、アルカリ金属(リチウム、ナトリウム、カリウムなど)の水酸化物、アルカリ金属(リチウム、ナトリウム、カリウム、セシウムなど)の炭酸塩、トリフルオロ酢酸、塩酸、臭素酸、ルイス酸(三塩化ホウ素、ヨウ化マグネシウム、ヨウ化ナトリウム-トリメチルクロロシラン、ヨウ化ナトリウム-塩化アルミニウム、ヨウ化アルミニウム、トリメチルシリルトリフラート)、水素-パラジウム、または蟻酸―パラジウムなどの存在下で、これらと反応させることにより容易に実施できる。
これらの使用量は特に限定されず、通常加水分解などに用いられる適量を用いればよく、たとえば、加水分解される化合物に対して、好ましくは1000~0.001当量、さらに好ましくは100~0.01当量の範囲で適宜調整できる。
カルボキシル基へと変換する際の溶媒の例としては、上記の前記式(II)で表される化合物と前記式(III)で表される化合物との反応で用いた溶媒と同様の溶媒が挙げられ、たとえば、エーテル系溶媒(ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、t-ブチルメチルエーテル、シクロペンチルメチルエーテル、ジオキサン、ジメトキシエタンなど)、炭化水素系溶媒(ヘキサン、シクロヘキサン、ベンゼン、トルエン、キシレンなど)、アルコール系溶媒(メタノール、エタノール、n-プロパノール、イソプロパノールなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸イソプロピルなど)、アミド系溶媒(例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドンなど)、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒などが挙げられる。また、水を使用してもよく、水と上記有機溶媒(少なくとも2種の有機溶媒の混合溶媒であってもよい。)の混合溶媒を使用してもよい。水を使用の場合は反応促進のために、相関移動触媒を用いてもよい。使用される相関移動触媒は、例えば、テトラブチルアンモニウムブロミド、テトラエチルアンモニウムブロミド、N-ヘキサデシル-N,N,Nートリメチルアンモニウムブロミド、N-ベンジル-N,N,Nートリエチルアンモニウムクロリド、テトラブチルアンモニウムヒドロキシド、15-クラウンー5などが挙げられる。
カルボン酸へと変換する際の反応時間は、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、例えば0.1~48時間、好ましくは0.3~24時間、より好ましくは0.5~10時間の範囲で選択できる。
カルボン酸へと変換する際の反応温度は、反応の進行状況などに基づき適宜設定可能であり、特に限定されないが、例えば-40~150℃、好ましくは-20~100℃、より好ましくは0~50℃の範囲で選択できる。
工程Aの反応終了後の混合物は通常、そのまま工程Bに供するのが好ましいが、洗浄抽出、単離(濃縮、晶析等)、精製(再結晶、カラムクロマトグラフィー等)を行ってから工程Bに、供してもよい。
工程B;
工程Bは、塩基および金属塩の存在下、式(IV)で表わされる化合物と式(V)で表わされる化合物とを反応させ、式(VI)で表わされる化合物を得る工程である。

〔式中、R1、R2は前記にて定義した通りである〕

〔式中、R1、R2、R3、Y1、Y2およびZは前記にて定義した通りである〕
工程Bは、塩基および金属塩の存在下、式(IV)で表わされる化合物と式(V)で表わされる化合物とを反応させ、式(VI)で表わされる化合物を得る工程である。
R1は好ましくはハロゲン原子であり、さらに好ましくはヨウ素原子である。
R2は好ましくはハロゲン原子であり、さらに好ましくはフッ素原子である。
塩基としては、例えば、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジドを包含するI族金属カチオンヘキサメチルジシラジド又はII族金属カチオンヘキサメチルジシラジド、水素化リチウム、水素化ナトリウム、水素化カリウムおよび水素化カルシウムを包含するI族金属カチオン水素化物又はII族金属カチオン水素化物、リチウムジイソプロピルアミドを包含するI族金属カチオンジアルキルアミド又はII族金属カチオンジアルキルアミド、リチウムアミド、ナトリウムアミド、カリウムアミドを包含するI族金属カチオンアミド又はII族金属カチオンアミド、並びにナトリウムエトキシド、カリウムtert-ブトキシド、マグネシウムエトキシドを包含するI族金属カチオンアルコキシド又はII族金属カチオンアルコキシドなどが挙げられる。
塩基としては、好ましくは金属カチオンヘキサメチルジシラジドが挙げられ、さらに好ましくはアルカリ金属ヘキサメチルジシラジドが挙げられ、特に好ましくはリチウムヘキサメチルジシラジドが挙げられる。
塩基は、式(V)の化合物に対して好ましくは2当量~10当量、さらに好ましくは4当量~8当量用いることが望ましい。
金属塩としては、例えば、リチウム塩、マグネシウム塩、または亜鉛塩が挙げられる。
金属塩は、好ましくは上記リチウム、マグネシウムまたは亜鉛のハライドであり、より好ましくはリチウムハライドであり、さらに好ましくはリチウムブロミドである。
金属塩は、好ましくは上記リチウム、マグネシウムまたは亜鉛のハライドであり、より好ましくはリチウムハライドであり、さらに好ましくはリチウムブロミドである。
金属塩は式(IV)の化合物に対して好ましくは2当量~10当量、さらに好ましくは4当量~8当量用いることが望ましい。
工程Bにおける溶媒の例としては、エーテル系溶媒(ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、t-ブチルメチルエーテル、シクロペンチルメチルエーテル、ジオキサン、ジメトキシエタンなど)、炭化水素系溶媒(ヘキサン、シクロヘキサン、ベンゼン、トルエン、キシレンなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸イソプロピルなど)、アミド系溶媒(例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドンなど)、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒などが挙げられる。これらのうちでは、非プロトン性極性溶媒が好ましく、たとえば、エーテル系溶媒、エステル系溶媒、アミド系溶媒、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒が好ましい。
工程Bにおける反応時間は、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、例えば0.1~48時間、好ましくは0.3~24時間、より好ましくは0.5~10時間の範囲で行われる。
工程Bにおける反応温度は、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、例えば-100~100℃、好ましくは-80~50℃、より好ましくは-78~25℃、特に好ましくは-70~20℃の範囲で選択できる。
工程Bの反応終了後の混合物は通常、そのまま工程Cに供するのが好ましいが、洗浄抽出、単離(濃縮、晶析等)、精製(再結晶、カラムクロマトグラフィー等)を行ってから工程Cに、供してもよい。
このような工程では、安息香酸誘導体(IV)とアニリン類(V)とのカップリング反応においては、金属塩を添加することにより反応系を均一とすることができ、反応転化率を飛躍的に向上させることができるとともに反応制御を容易に行うことができる。
工程C;
工程Cは、式(VI)で表わされる化合物を還元条件に付し、式(VII)で表される化合物を得る工程であり、より具体的には、たとえば、溶媒中、式(VI)で表わされる化合物を還元剤と反応させた後、前記式(i)で表される基Xへ環化し、式(VII)で表わされる化合物を得る工程である。
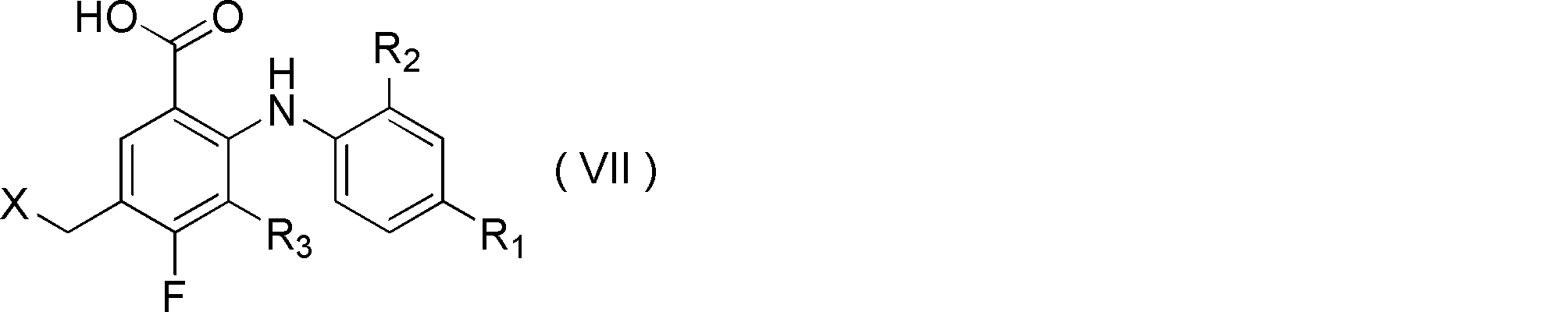
〔式中、R1、R2、R3およびXは前記にて定義した通りである〕
工程Cは、式(VI)で表わされる化合物を還元条件に付し、式(VII)で表される化合物を得る工程であり、より具体的には、たとえば、溶媒中、式(VI)で表わされる化合物を還元剤と反応させた後、前記式(i)で表される基Xへ環化し、式(VII)で表わされる化合物を得る工程である。
Xは好ましくは、3-オキソ[1,2]オキサジナニルである。
工程Cにおける還元剤として、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム、トリエチルシラン、トリメチルシラン、水素化リチウムアルミニウム、水素化ジイソプロピルアルミニウム、ボラン-ピリジンコンプレックス、デカボラン、ジボラン、ボラン-ジメチルスルフィドコンプレックス、ボラン-THFコンプレックス、水素、水素化ホウ素亜鉛などを用いることができる。また、水素雰囲気下での金属触媒(例えば、炭素担持パラジウム、白金、均一系パラジウム錯体、均一系ルテニウム錯体、均一系口ジウム錯体)を使用することができる。反応促進のために酸などを用いてもよい。使用される酸としては、酢酸、塩酸、トリフルオロ酢酸、ジクロロ酢酸、トリフルオロボロン-ジエチルエーテルコンプレックス、トリメチルシリルトリフラート、塩化アルミニウム、トリメチルシリルクロリドなどを挙げることができる。
工程Cにおける溶媒としては、エーテル系溶媒(ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、シクロペンチルメチルエーテル、ジオキサン、ジメトキシエタンなど)、アルコール系溶媒(メタノール、エタノール、n-プロパノール、イソプロパノールなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸イソプロピルなど)、アミド系溶媒(例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドンなど)、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒などが挙げられる。また、水を使用してもよく、水と上記有機溶媒(少なくとも2種の有機溶媒の混合溶媒であってもよい。)の混合溶媒を使用してもよい。
工程Cにおける反応時間は、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、例えば0.1~48時間、好ましくは0.3~24時間、より好ましくは0.5~10時間の範囲で行われる。
工程Cにおける反応温度は、例えば-80~200℃、好ましくは-50~100℃、より好ましくは-30~40℃の範囲で選択できる。
還元剤は、式(VI)の化合物に対して好ましくは0.2当量~250当量、さらに好ましくは0.5当量~120当量用いることが望ましい。
工程Cで還元剤で処理することにより、式(VII)で表される化合物に加え、下記化合物が得られる。

このようにして得られた化合物は通常、反応系中でそのまま前記式(i)で表される基Xへと環化することが好ましいが、洗浄抽出、単離(濃縮、晶析等)、精製(再結晶、カラムクロマトグラフィー等)を行ってから環化させてもよい。
このような環化反応は、たとえば、溶媒中、工程Cで還元剤で処理した後、得られた化合物を含む反応溶液を、必要に応じ昇温して攪拌等をすることにより、容易に実施できる。
工程Cにおける環化反応における溶媒としては、前記還元反応と同様のものを用いることができ、たとえば、エーテル系溶媒(ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、シクロペンチルメチルエーテル、ジオキサン、ジメトキシエタンなど)、アルコール系溶媒(メタノール、エタノール、n-プロパノール、イソプロパノールなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸イソプロピルなど)、アミド系溶媒(例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドンなど)、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒などが挙げられる。
工程Cにおける環化の反応時間は、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、例えば0.1~48時間、好ましくは1~24時間、より好ましくは3~10時間の範囲で行われる。
工程Cにおける環化の反応温度は、例えば0~200℃、好ましくは20~150℃、より好ましくは30~100℃の範囲で選択できる。
工程Cの反応終了後の混合物は通常、そのまま工程Dに供するのが好ましいが、洗浄抽出、単離(濃縮、晶析等)、精製(再結晶、カラムクロマトグラフィー等)を行ってから工程Dに、供してもよい。
工程D;
工程Dは、式(VII)で表わされる化合物を式(VIII)で表わされる化合物と縮合反応させることにより、式(I)で表わされる化合物またはその塩を得る工程である。工程Dは、たとえば、溶媒中、縮合剤の存在下、必要に応じ添加剤を添加して、実施することができる。

〔式中、R4は前記にて定義した通りである〕
工程Dは、式(VII)で表わされる化合物を式(VIII)で表わされる化合物と縮合反応させることにより、式(I)で表わされる化合物またはその塩を得る工程である。工程Dは、たとえば、溶媒中、縮合剤の存在下、必要に応じ添加剤を添加して、実施することができる。
R4は好ましくはOHにより置換されているC1-6アルキルであり、さらに好ましくはヒドロキシエチル(CH2CH2OH)である。
工程Dにおける縮合剤の例としては、1,3-ジシクロヘキシルカルボジイミド(DCC)、2-エトキシ-1-エトキシカルボニル-1,2-ジヒドロキノリン(EEDQ)、ブロモ-トリス(ピロリジノ)-ホスホニウム ヘキサフルオロホスファート(PyBrOP)、1-エチル-3-(3'-ジメチルアミノプロピル)カルボジイミド(EDC)、(ベンゾトリアゾールイルオキシ)トリピロリジノ-ホスホニウム ヘキサフルオロホスファート(PyBOP)、O-(7-アザベンゾトリ
アゾール-1-イル) -N,N,N',N'-テトラメチルウロニウムヘキサフルオロりん酸塩(HATU)、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム・ヘキサフルオロホスフェート(HBTU)、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム テトラフルオロボレート(TBTU)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリウムクロリド(DMT-MM)などを挙げることができる。
アゾール-1-イル) -N,N,N',N'-テトラメチルウロニウムヘキサフルオロりん酸塩(HATU)、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム・ヘキサフルオロホスフェート(HBTU)、2-(1H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム テトラフルオロボレート(TBTU)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリウムクロリド(DMT-MM)などを挙げることができる。
工程Dにおける添加剤の例としては、ヒドロキシベンゾトリアゾール(HOBt)、3-ヒドロキシ-4-オキソ-3,4-ジヒドロ-1,2,3-ベンゾトリアジン(HODhBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)などを挙げることができる。
工程Dにおける溶媒の例としては、エーテル系溶媒(ジエチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、t-ブチルメチルエーテル、シクロペンチルメチルエーテル、ジオキサン、ジメトキシエタンなど)、アルコール系溶媒(メタノール、エタノール、n-プロパノール、イソプロパノールなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸イソプロピルなど)、アミド系溶媒(例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドンなど)、アセトニトリル、及びこれらの有機溶媒(少なくとも2種)の混合溶媒などが挙げられる。また、水を使用してもよく、水と上記有機溶媒(少なくとも2種の有機溶媒の混合溶媒であってもよい。)の混合溶媒を使用してもよい。
工程Dにおける反応時間は、反応の進行状況などに基づき適宜設定可能であり、特には限定されないが、例えば0.1~48時間、好ましくは0.3~24時間、より好ましくは0.5~10時間の範囲で行われる。
工程Dにおける反応温度は、例えば-40~200℃、好ましくは-20~100℃、より好ましくは0~50℃の範囲で選択できる。
縮合剤の使用量は、式(VII)の化合物に対して好ましくは1当量~20当量、さらに好ましくは1当量~10当量用いることが望ましい。
添加剤の使用量は、式(VII)の化合物に対して好ましくは1当量~20当量、さらに好ましくは1当量~10当量用いることが望ましい。
式(I)の化合物の薬学上許容し得る塩は、該化合物と、医薬品の製造に使用可能な塩基とを、反応させることにより製造することができる。そのような塩としては、例えば、アルカリ金属塩(リチウム塩、ナトリウム塩、カリウム塩、セシウム塩、ルビジウム塩等)、アルカリ土類金属塩(マグネシウム塩、カルシウム塩等)、アンモニウム塩(アンモニウム塩、アルキルアンモニウム塩、ジアルキルアンモニウム塩、トリアルキルアンモニウム塩、テトラアルキルアンモニウム塩等)が挙げられる。中でも、アルカリ金属塩が好ましく、ナトリウム塩が更に好ましい。
なお、本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
なお、本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
以下、本発明の好適な実施例について詳細に説明する。
NMRは、核磁気共鳴装置 JNM-ECP-500(JEOL製)、またはJNM-ECP-400(JEOL製)を用いて測定した。質量分析は、質量分析装置LCT Premier XE(Waters製)あるいはAgilent Technologies HP6890を用いて測定した。ある工程の生成物を精製なしで次の工程に供する場合であっても、NMR分析には精製したものを使用した。
NMRは、核磁気共鳴装置 JNM-ECP-500(JEOL製)、またはJNM-ECP-400(JEOL製)を用いて測定した。質量分析は、質量分析装置LCT Premier XE(Waters製)あるいはAgilent Technologies HP6890を用いて測定した。ある工程の生成物を精製なしで次の工程に供する場合であっても、NMR分析には精製したものを使用した。
[実施例1]
工程1:2,3,4-トリフルオロ-5-ホルミル-安息香酸の合成

工程1:2,3,4-トリフルオロ-5-ホルミル-安息香酸の合成
窒素雰囲気下、2,2,6,6-テトラメチルピペリジン(21.2g、150mmol)をテトラヒドロフラン(120ml)に溶解した。-78℃に冷却し、この溶液にn-ブチルリチウムのn-ヘキサン溶液(2.67M、47ml、126mmol)を滴下後、同温にて35分間攪拌した。-78℃で2,3,4-トリフルオロ-安息香酸(10.6g、60.2mmol)のテトラヒドロフラン(60ml)溶液を滴下し、同温にて50分間攪拌した。DMF(9.0g、122mmol)を加えた後、さらに1時間攪拌した。
反応液を氷冷の3N塩酸(100ml)に加え、室温に昇温後、酢酸エチル(100ml)にて抽出した。水層を酢酸エチル(50ml)で抽出した。有機層を合わせ、飽和食塩水(20ml)で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下で溶媒を留去した。得られた残渣に酢酸エチル(20ml)およびn-ヘプタン(40ml)の混合溶媒を加え、60℃に加熱し懸濁した後、さらにn-ヘプタン(60ml)を同温にて加え、10分間攪拌した。0℃まで冷却し、同温にて30分間攪拌した。
析出した結晶をろ過し、酢酸エチルおよびn-ヘプタン(1:10)の混合溶媒で洗浄後、減圧下にて乾燥し、標題化合物(9.0g、73%)を得た。母液を減圧下で溶媒留去し、得られた残渣に酢酸エチル(3.0ml)を加え、50℃に加熱して溶解した後、n-ヘプタン(15ml)を加えた。固体析出を確認後、0℃に冷却し、同温にて1時間15分間攪拌した。析出した結晶をろ過し、酢酸エチルおよびn-ヘプタン(1:10)の混合溶媒で洗浄後、減圧下にて乾燥し、標題化合物(1.6g、13%)を得た。これら標題化合物を合わせて使用した(合計収率:86%)。
1H NMR(500 MHz, CDCl3) δ8.42 (1H,dt,J=2.0,7.5Hz),10.29(1H,s)
GC/MS(EI):203(M-H)-
GC/MS(EI):203(M-H)-
工程2:(E)-2,3,4-トリフルオロ-5-[(4-エトキシ-4-オキソブトキシイミノ)メチル]安息香酸の合成

公知文献(例えばJ.Med.Chem.1970,13,60、J.Med.Chem.1996,39,4058)に記載の方法に基づいて合成することができる4-(アミノオキシ)-ブタン酸エチルを含むt-ブチルメチルエーテル溶液に工程1で得られた2,3,4-トリフルオロ-5-ホルミル-安息香酸(9.1g、44.6mmol)を加え、25℃にて1時間攪拌した。この溶液に2M HCl(90ml)を加え、有機層を抽出した。有機層を再度、2M HCl(90ml)で洗浄し、有機層を抽出した。有機層に飽和重曹水(70ml)を加えて、水層を抽出した。有機層に再度、飽和重曹水(20ml)を加え、水層を抽出した。水層を合わせた後、t-ブチルメチルエーテル(50ml)で洗浄し、標題化合物を含む溶液を得た。生成物は、さらに精製することなく次の工程に使用した。
1H NMR(500MHz,CDCl3)δ1.27(3H,t,7.5Hz),2.04-2.09(2H,m),2.45(2H,t,J=7.5Hz),4.16(2H,q,J=7.5Hz),4.26(2H,t,J=6.0Hz),8.21(1H,s),8.29(1H,dt,J=2.5,7.5Hz)
LC/MS(ES+):356[M+Na]+
LC/MS(ES+):356[M+Na]+
工程3:2,3,4-トリフルオロ-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸の合成

前工程にて得られた(E)-2,3,4-トリフルオロ-5-[(4-エトキシ-4-オキソブトキシイミノ)メチル]安息香酸を含む溶液に2.5N NaOH(54ml)を加えて溶解した。この溶液を25℃で1時間攪拌した。反応液に2N HCl(130ml)を加えた後、35℃に昇温し、攪拌した。固体析出を確認後、析出した固体をろ過し、水で洗浄後、減圧下にて乾燥し、標題化合物(12.7g、2,3,4-トリフルオロ-5-ホルミル-安息香酸からの通算収率93%)を得た。
1H NMR(500MHz,DMSO-d6) δ1.85-1.91(2H,m),2.31(2H,t,J=7.0Hz),4.17(2H,t,J=6.0Hz),8.04(1H,dt,J=2.0,7.5Hz),8.33(1H,s)
LC/MS(ES+):328[M+Na]+
LC/MS(ES+):328[M+Na]+
工程4:(E)-3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸の合成

窒素雰囲気下、前工程にて得られた2,3,4-トリフルオロ-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸(1.0g、3.3mmol)、2-フルオロ-4-ヨードアニリン(1.1g、4.6mmol)、リチウムブロミド(1.9g、21.9mmol)にTHF(8.0ml)を加えて溶解した。-20℃に冷却し、1.0M リチウムヘキサメチルジシラジドのTHF溶液(14.5ml、14.5mmol)を滴下し、同温で3時間攪拌した。この溶液に2N HCl(20ml)を加え、室温に昇温後、2-メチルテトラヒドロフラン(10ml)にて有機層を抽出した。有機層を飽和食塩水(10ml)にて洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去した。得られた残渣に酢酸エチル(10ml)を加え、50℃に加熱した。0℃に冷却後、析出した固体をろ過し、酢酸エチルで洗浄後、減圧下にて乾燥し、標題化合物(1.3g、78%)を得た。
1H NMR(500MHz,DMSO-d6) δ1.85-1.90(2H,m),2.31(2H,t,J=7.5Hz),4.14(2H,t,J=6.5Hz),6.94(1H,dt,J=4.5,8.5Hz),7.45(1H,dd,J=2.0,8.5Hz),7.64(1H,dd,J=2.0,10.5Hz),8.11(1H,d,J=6.0Hz),8.27(s,1H)
LC/MS(ES-):521[M-H]-
LC/MS(ES-):521[M-H]-
工程5:5-[(3-カルボキシプロポキシアミノ)メチル]-3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)安息香酸の合成

(E)-3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸(15.0g、28.7mmol)をTHF(150ml)に溶解した。0℃に冷却し、ボラン-ピリジン(8.7ml、86.1mmol)および4M塩酸 酢酸エチル溶液(72ml、287mmol)を順次加え、同温にて1時間攪拌した。
反応液に2.5N水酸化ナトリウム水溶液(150ml)を加え、室温に昇温後、t-ブチルメチルエーテル(150ml)を加え、有機層を抽出した。有機層を飽和硫酸水素カリウム水溶液(100ml)および飽和食塩水(75ml)で順次洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去し標題化合物を含む油状物を得た。さらに水層に飽和硫酸水素カリウム水溶液(100ml)を加え、t-ブチルメチルエーテル(150ml)にて有機層を抽出し、飽和食塩水(75ml)で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去し標題化合物を含む油状物を得た。得られた標題化合物を含む油状物を合わせた(17.4g)。生成物は、さらに精製することなく次の工程に使用した。
1H NMR(500MHz,DMSO-d6) δ1.63-1.68(2H,m),2.17(2H,t,J=7.5Hz),3.55(2H,t,J=6.5Hz),3.98(2H,s),6.77(1H,dt,J=4.5,8.5Hz),7.42(1H,dd,J=1.5,8.5Hz),7.61(1H,dd,J=1.5,10.5Hz),7.89(1H,d,J=7.0Hz),9.03(1H,s)
LC/MS(ES-):523[M-H]-
LC/MS(ES-):523[M-H]-
工程6:3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-オキソ-[1,2]オキサジナン-2-イル)メチル]安息香酸の合成

前工程にて得られた5-[(3-カルボキシプロポキシアミノ)メチル]-3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)安息香酸を含む油状物(17.4g)をシクロペンチルメチルエーテル(150ml)に懸濁した。60℃に昇温した後、同温で7時間攪拌した。25℃に冷却後、減圧下で溶媒を留去した。再度、シクロペンチルメチルエーテル(75ml)を加え、懸濁した。60℃に昇温後、ジイソプロピルエーテル(150ml)を加え、同温にて30分間攪拌した。0℃に冷却後、20分間攪拌した。析出した固体をろ過し、シクロペンチルメチルエーテルおよびジイソプロピルエーテル(1:5)の混合溶媒で洗浄後、減圧下にて乾燥し、標題化合物(10.8g、(E)-3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸からの通算収率80%)を得た。
1H NMR(500MHz,DMSO-d6)δ2.00(2H,quintet, J=7.0Hz),2.42(2H,t,J=7.0Hz),3.97(2H,t,J=7.0Hz),4.77(2H,s),6.83(1H,dt,J=5.0,8.5Hz),7.42(1H,dd,J=1.5,8.5Hz),7.62(1H,dd,J=1.5,10.5Hz),7.78(1H,d,J=7.5Hz),9.09(1H,s)
LC/MS(ES-):505[M-H]-
LC/MS(ES-):505[M-H]-
工程7:ナトリウム(3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-オキソ-[1,2]オキサジナン-2-イル)メチル]ベンゾイル)(2-ヒドロキシエトキシ)アミドの合成

3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-オキソ-[1,2]オキサジナン-2-イル)メチル]安息香酸(99mg、0.20mmol)をDMF(0.10ml)とTHF(0.39ml)の混合溶媒に溶解した。1-ヒドロキシベンゾトリアゾール一水和物(33mg、0.22mmol)および1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(42mg、0.22mmol)を加え、室温にて3時間30分間攪拌した。
反応液に公知文献(例えばJ.Chem.Soc.,Perkin Trans.1,1987,2829)に記載の方法に基づいて合成することができる2-(アミノオキシ)エタノールのTHF溶液を加え、さらに1時間攪拌した。水(10ml)を加え、酢酸エチル(10ml)にて有機層を抽出した。有機層を3wt%炭酸水素ナトリウム水溶液(5ml)、0.1M塩酸水溶液(5ml)および飽和食塩水(5ml)で順次洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去し、残渣を得た。得られた残渣に酢酸エチル(1.7ml)およびエタノール(0.3ml)を加えて溶解した。5℃に冷却後、5N水酸化ナトリウム水溶液(37μl)を加え、同温にて1時間15分攪拌した。固体析出を確認後、析出した結晶をろ過し、酢酸エチル(5ml)で洗浄後、減圧下にて乾燥し、標題化合物(61mg、53%)を得た。
1H NMR(500MHz,DMSO-d6) δ2.00(2H,,quintet, J=7.0Hz),2.41(2H,t,J=7.0Hz),3.46(2H,q,J=5.0Hz),3.69(2H,t,J=5.0Hz),3.95(2H,t,J=7.0Hz),4.73(2H,s),5.19(1H,t,J=5.0Hz),6.49-6.54(1H,m),7.33(1H,dd,J=1.5,8.5Hz),7.53(1H,dd,J=1.5,11.0Hz),7.70(1H,d,J=5.5Hz),11.32(s,1H)
LC/MS(ES+):588[M+H]+
LC/MS(ES+):588[M+H]+
工程8:3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-N-(2-ヒドロキシエトキシ)5-[(3-オキソ-[1,2]オキサジナン-2-イル)メチル]ベンズアミドの合成

前工程にて得られたナトリウム(3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-オキソ-[1,2]オキサジナン-2-イル)メチル]ベンゾイル)(2-ヒドロキシエトキシ)アミド(50mg、0.09mmol)を酢酸エチル(0.4ml)に溶解した。1M塩酸水溶液(0.2ml)および20wt%食塩水(0.2ml)の混合溶媒を加え、室温にて81分間攪拌した。酢酸エチル(10ml)にて有機層を抽出した。有機層を10wt%食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去し、残渣を得た。得られた残渣にアセトニトリル(0.6ml)を加えた後、減圧下で溶媒を留去した。得られた残渣にさらにアセトニトリル(0.6ml)を加え、60℃に昇温し、溶解した。室温に冷却後、1時間攪拌し、固体析出を確認した。5℃に冷却し、さらに1時間攪拌した。析出した固体をろ過し、5℃に冷却したアセトニトリル(2ml)で洗浄後、減圧下にて乾燥し、標題化合物(21mg、44%)を得た。
1H NMR(500MHz,DMSO-d6) δ2.05(2H,quintet,J=7.0Hz),2.48(2H,t,J=7.0Hz),3.60(2H,s),3.86(2H,s),4.03(2H,t,J=7.0Hz),4.75(1H,s),4.82(2H,s),6.69(1H,dt,J=4.0,9.0Hz),7.39-7.40(2H,m),7.60(1H,dd,J=1.5,10.5Hz),8.63(1H,s),11.9(1H,s)
LC/MS(ES+):566[M+H]+
LC/MS(ES+):566[M+H]+
[実施例2]
工程Bにおける金属塩の添加効果について、以下に比較を行った。

工程Bにおける金属塩の添加効果について、以下に比較を行った。
(E)-3,4-ジフルオロ-2-(2-フルオロ-4-ヨードフェニルアミノ)-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸合成における金属塩の添加効果の検討
アルゴン雰囲気下、2,3,4-トリフルオロ-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸(20mg、0.066mmol)、2-フルオロ-4-ヨードアニリン(22mg、0.093mmol)および塩化リチウム、ヨウ化リチウムのいずれかの金属塩(0.43mmol)をTHF(0.15ml)に溶解した。比較対象としての金属塩を添加しない場合については、金属塩を添加しない点を除き前記と同様の条件で、原料化合物をTHFに溶解させた。
これら3種類の溶液をそれぞれ-20℃に冷却し、1.0M リチウムヘキサメチルジシラジドのTHF溶液(290μl、0.29mmol)を滴下し、同温で3.5時間攪拌した。この溶液に2N HClを加え、室温に昇温後、2-メチルテトラヒドロフランにて有機層を抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去し、標題化合物を得た。収率は得られた残渣の1H-NMR分析に基づいて算出した。(内部標準物質:1,3,5-トリメトキシベンゼン)
アルゴン雰囲気下、2,3,4-トリフルオロ-5-[(3-カルボキシプロポキシイミノ)メチル]安息香酸(20mg、0.066mmol)、2-フルオロ-4-ヨードアニリン(22mg、0.093mmol)および塩化リチウム、ヨウ化リチウムのいずれかの金属塩(0.43mmol)をTHF(0.15ml)に溶解した。比較対象としての金属塩を添加しない場合については、金属塩を添加しない点を除き前記と同様の条件で、原料化合物をTHFに溶解させた。
これら3種類の溶液をそれぞれ-20℃に冷却し、1.0M リチウムヘキサメチルジシラジドのTHF溶液(290μl、0.29mmol)を滴下し、同温で3.5時間攪拌した。この溶液に2N HClを加え、室温に昇温後、2-メチルテトラヒドロフランにて有機層を抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸マグネシウムで乾燥した後、減圧下で溶媒を留去し、標題化合物を得た。収率は得られた残渣の1H-NMR分析に基づいて算出した。(内部標準物質:1,3,5-トリメトキシベンゼン)
金属塩の添加による収率の比較を表1に示す。
以上の結果から、添加物なしの場合と比べ金属塩(特にリチウムハライド)を添加した場合には反応効率が大きく向上することが分かった。
本発明により、有機遷移金属触媒あるいはオスミウム試薬等を使用することなく、また反応制御可能な均一系からなる反応で構成された簡便かつ高収率な5-置換-2-フェニルアミノ-ベンズアミド類の製造方法が提供される。
Claims (10)
- 下記式I:

R2は、ハロゲン原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基を置換基に有していてもよく;
R3は、水素原子またはハロゲン原子を示し;
R4は、水素原子、C1-6アルキル、C2-6アルケニルまたはC2-6アルキニルを示し、該C1-6アルキル、該C2-6アルケニル、該C2-6アルキニルは、-ORa、-NRaRb、-NRaCORb、3-8員ヘテロ環および5-10員ヘテロアリールからなる群から選択される1~3個の置換基を有していてもよく、該3-8員ヘテロ環、該5-10員ヘテロアリールは、C1-6アルキル、-ORaおよび-NRaRbからなる群から選択される1~3個の置換基を有していてもよく;
Xは、下記式(i)で表される基;
(式中、
Y1またはY2は同一または異なって、単結合、-CO-、-COO-、-O-、-OCO-、-NRa-、または-SO2-を示し;
Zは、1~3個のWを置換基として有していてもよい炭素原子1~5個のアルキレンを示し;
Wは、同一または異なって、C1-6アルキル、ハロゲン原子、オキソ、-ORa、-COORa、-COOCORa、-CO-[ハロゲン原子]、-OCORa、-CONRaRb、-SRa、-SORa、-SO2Ra、-NRaRb、-NRaCORb、-NRaSO2Rb、-SO2NRaRb、3-8員ヘテロ環または5-10員ヘテロアリールを示し、3-8員ヘテロ環、5-10員ヘテロアリールはC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく、該C1-6アルキルは水酸基、C1-8アルコキシまたはアミノを置換基に有していてもよく、
オキソおよびハロゲンを除く前記置換基は、互いに結合してC3-10シクロアルキル、または3-8員ヘテロ環を形成してもよく、該C3-10シクロアルキルまたは該3-8員ヘテロ環は、-ORaで修飾されていてもよいC1-6アルキル、-ORaおよび-NRaRbからなる群から選択される置換基を有していてもよく;
それぞれのRa、それぞれのRbは、同一または異なって、水素原子またはC1-6アルキルを示し、該C1-6アルキルは水酸基、C1-8アルコキシおよびアミノから選ばれる基を1~3個有していてもよい。)〕
で表わされる化合物、またはその医薬的に許容しうる塩を製造する方法であって、下記工程A、工程B、工程C、および工程Dを含む方法;
工程Aは、下記式(II)



工程Bは、塩基および金属塩の存在下、工程Aで得られた式(IV)で表わされる化合物を下記式(V)


工程Cは、工程Bで得られた式(VI)で表わされる化合物を還元条件に付し、下記式(VII)

工程Dは、前記工程Cで得られた式(VII)で表わされる化合物を下記式(VIII)

- 工程Bにおいて、金属塩が、リチウム塩、マグネシウム塩、および/または亜鉛塩から選ばれる1種または2種以上である、請求項1に記載の方法。
- 工程Bにおいて、塩基が、アルカリ金属ヘキサメチルジシラジドである、請求項1または2に記載の方法。
- R5がフッ素原子である、請求項1-3のいずれか一項に記載の方法。
- 工程Bにおいて、反応が非プロトン性極性溶媒中で-78℃~25℃で行われる、請求項1-4のいずれか一項に記載の方法。
- R1、R2、およびR3はそれぞれハロゲン原子であり、R4は水酸基により置換されていてもよいC1-6アルキルである、請求項1-5のいずれか一項に記載の方法。
- Y1は単結合、Y2は-O-、および、Zが炭素原子1~5個のアルキレンである、請求項1-6のいずれか一項に記載の方法。
- R1がヨウ素原子であり、R2およびR3がそれぞれフッ素原子であり、R4がヒドロキシエチル(CH2CH2OH)であり、Y1が単結合であり、Y2が-O-であり、Zが炭素数3のアルキレンであり、式(I)で表される化合物が下記式(IX)

- 下記式(IV)



- 下記式(X)
〔式中、R5、R6、Y1、Y2およびZは請求項1において定義したR5、R6、Y1、Y2およびZとそれぞれ同意義を示す。〕で表わされる化合物又はその薬学上許容しうる塩。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012257598A JP2016034900A (ja) | 2012-11-26 | 2012-11-26 | 5−置換−2−フェニルアミノ−ベンズアミド類の製造方法 |
| JP2012-257598 | 2012-11-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014081024A1 true WO2014081024A1 (ja) | 2014-05-30 |
Family
ID=50776200
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/081588 WO2014081024A1 (ja) | 2012-11-26 | 2013-11-25 | 5-置換-2-フェニルアミノ-ベンズアミド類の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2016034900A (ja) |
| WO (1) | WO2014081024A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022018875A1 (ja) * | 2020-07-22 | 2022-01-27 | 中外製薬株式会社 | アリールアミド誘導体を含む細胞増殖性疾患の治療又は予防用医薬組成物 |
| US11964950B2 (en) | 2020-01-22 | 2024-04-23 | Chugai Seiyaku Kabushiki Kaisha | Arylamide derivative having antitumor activity |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2022312992A1 (en) * | 2021-07-21 | 2024-02-29 | Chugai Seiyaku Kabushiki Kaisha | Method for producing arylamide derivative |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002543055A (ja) * | 1999-04-21 | 2002-12-17 | ワーナー−ランバート・カンパニー | 2−(n−フェニルアミノ)安息香酸の製造方法 |
| WO2005028426A1 (ja) * | 2003-09-19 | 2005-03-31 | Chugai Seiyaku Kabushiki Kaisha | 新規4−フェニルアミノ−ベンズアルドオキシム誘導体並びにそのmek阻害剤としての使用 |
| JP2005515251A (ja) * | 2002-01-23 | 2005-05-26 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | N−(4−置換フェニル)−アントラニル酸ヒドロキサメートエステル |
| WO2006011466A1 (ja) * | 2004-07-26 | 2006-02-02 | Chugai Seiyaku Kabushiki Kaisha | Mek阻害物質としての5-置換-2-フェニルアミノ-ベンズアミド類 |
-
2012
- 2012-11-26 JP JP2012257598A patent/JP2016034900A/ja active Pending
-
2013
- 2013-11-25 WO PCT/JP2013/081588 patent/WO2014081024A1/ja active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002543055A (ja) * | 1999-04-21 | 2002-12-17 | ワーナー−ランバート・カンパニー | 2−(n−フェニルアミノ)安息香酸の製造方法 |
| JP2005515251A (ja) * | 2002-01-23 | 2005-05-26 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | N−(4−置換フェニル)−アントラニル酸ヒドロキサメートエステル |
| WO2005028426A1 (ja) * | 2003-09-19 | 2005-03-31 | Chugai Seiyaku Kabushiki Kaisha | 新規4−フェニルアミノ−ベンズアルドオキシム誘導体並びにそのmek阻害剤としての使用 |
| EP1674452A1 (en) * | 2003-09-19 | 2006-06-28 | Chugai Seiyaku Kabushiki Kaisha | Novel 4-phenylamino-benzaldoxime derivative and use thereof as mek inhibitor |
| US20070105859A1 (en) * | 2003-09-19 | 2007-05-10 | Yoshiaki Isshiki | 4-Phenylamino-benzaldoxime derivatives and uses thereof as mitogen-activated protein kinase kinase (mek) inhibitors |
| JP4931419B2 (ja) * | 2003-09-19 | 2012-05-16 | 中外製薬株式会社 | 新規4−フェニルアミノ−ベンズアルドオキシム誘導体並びにそのmek阻害剤としての使用 |
| WO2006011466A1 (ja) * | 2004-07-26 | 2006-02-02 | Chugai Seiyaku Kabushiki Kaisha | Mek阻害物質としての5-置換-2-フェニルアミノ-ベンズアミド類 |
| KR101153556B1 (ko) * | 2004-07-26 | 2012-06-11 | 추가이 세이야쿠 가부시키가이샤 | Mek 저해물질로서의5?치환?2?페닐아미노?벤즈아미드류 |
Non-Patent Citations (2)
| Title |
|---|
| JANIS LOUIE ET AL.: "Palladium-catalyzed synthesis of arylamines from aryl halides. Mechanistic studies lead to coupling in the absence of tin reagents", TETRAHEDRON LETTERS, vol. 36, no. 21, 1995, pages 3609 - 3612 * |
| LEE, DAE-YON ET AL.: "Zinc trimethylsilylamide as a mild ammonia equivalent and base for the amination of aryl halides and triflates", ORGANIC LETTERS, vol. 7, no. 6, 2005, pages 1169 - 1172 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11964950B2 (en) | 2020-01-22 | 2024-04-23 | Chugai Seiyaku Kabushiki Kaisha | Arylamide derivative having antitumor activity |
| WO2022018875A1 (ja) * | 2020-07-22 | 2022-01-27 | 中外製薬株式会社 | アリールアミド誘導体を含む細胞増殖性疾患の治療又は予防用医薬組成物 |
| KR20220012195A (ko) * | 2020-07-22 | 2022-02-03 | 추가이 세이야쿠 가부시키가이샤 | 아릴 아마이드 유도체를 포함하는 세포 증식성 질환의 치료 또는 예방용 의약 조성물 |
| JP2022145416A (ja) * | 2020-07-22 | 2022-10-04 | 中外製薬株式会社 | アリールアミド誘導体を含む細胞増殖性疾患の治療又は予防用医薬組成物 |
| JP7168734B2 (ja) | 2020-07-22 | 2022-11-09 | 中外製薬株式会社 | アリールアミド誘導体を含む細胞増殖性疾患の治療又は予防用医薬組成物 |
| KR102550455B1 (ko) | 2020-07-22 | 2023-06-30 | 추가이 세이야쿠 가부시키가이샤 | 아릴 아마이드 유도체를 포함하는 세포 증식성 질환의 치료 또는 예방용 의약 조성물 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016034900A (ja) | 2016-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6966590B2 (ja) | オメカムチブメカルビルの塩及び塩を調製するプロセス | |
| JP5154227B2 (ja) | N−(4−フルオロベンジル)−n−(1−メチルピペリジン−4−イル)−n’−(4−(2−メチルプロピルオキシ)フェニルメチル)カルバミドの塩及びその調製 | |
| EP2307377A1 (en) | Nicotinamide derivatives | |
| WO2012100722A1 (zh) | 一种制备消旋尼古丁的方法 | |
| AU2001277044A1 (en) | Process for making N-aryl-anthranilic acids and their derivatives | |
| JP2014516072A (ja) | アピキサバン製造方法 | |
| JP2009040783A5 (ja) | ||
| WO2008005423A1 (en) | Improved method of making sufentanil | |
| US20080262039A1 (en) | Vanilloid Receptor Ligands, Pharmaceutical Compositions Containing Them, Process For Making Them, and Use Thereof For Treating Pain and Other Conditions | |
| AU2002239761A1 (en) | Amidoalkyl-piperidine and amidoalkyl-piperazine derivatives for treating of nervous systems disorders | |
| WO2014081024A1 (ja) | 5-置換-2-フェニルアミノ-ベンズアミド類の製造方法 | |
| JP5680022B2 (ja) | 4−{3−[cis−ヘキサヒドロシクロペンタ[c]ピロール−2(1H)−イル]プロポキシ}ベンズアミド塩酸塩の合成方法及び結晶形態、ならびにそれを含む医薬組成物 | |
| CA2784933A1 (en) | Processes for the purification of lubiprostone | |
| MXPA03011728A (es) | Formas cristalinas novedosas del acido 4-[4-[4- (hidroxidifenilmetil) -1-piperidinil] -1-hidroxibutil] -alfa, alfa -dimetilbencen acetico y su clorohidrato. | |
| JP2020517674A (ja) | ボルチオキセチンHBr α型を製造するための方法 | |
| TWI443100B (zh) | 培美曲塞鹽的製法 | |
| CA2642452A1 (en) | Process for preparing 1-halo-2,7-naphthyridinyl derivatives | |
| US11242332B2 (en) | Method for producing benzimidazole derivative | |
| AU2013285078A1 (en) | Saxagliptin salts | |
| KR101123281B1 (ko) | 치환된 벤즈아미드 유도체 및 그의 약제학상 허용되는 염의제조방법 | |
| US20220411382A1 (en) | 3-((r)-2-(amino-2-phenylethyl)-1-(2-fluoro-6 trifluoromethyl benzyl)-5-iodo-6-methyl-1h-pyrimidine-2,4-dione or a salt thereof, process for its preparation, and its use in the synthesis of elagolix | |
| EP3448861B1 (en) | A process for the preparation of 2-pyrazolo[1,5-a]pyrazin-2-ylpyrido[1,2-a]pyrimidin-4-one | |
| JP4307996B2 (ja) | 4−メチルアミノ−4−フェニルピペリジンの製造方法 | |
| JP2018521061A (ja) | 2−アミノ−イソニコチン酸の6−置換又は5,6−二置換誘導体の一般的調製方法 | |
| JP2018070607A (ja) | アミノ基で置換されたオキサジアゾール誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13857630 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14434497 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13857630 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |