WO2013145455A1 - 希土類元素の回収方法 - Google Patents
希土類元素の回収方法 Download PDFInfo
- Publication number
- WO2013145455A1 WO2013145455A1 PCT/JP2012/081855 JP2012081855W WO2013145455A1 WO 2013145455 A1 WO2013145455 A1 WO 2013145455A1 JP 2012081855 W JP2012081855 W JP 2012081855W WO 2013145455 A1 WO2013145455 A1 WO 2013145455A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- rare earth
- earth element
- earth elements
- recovering
- leachate
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B59/00—Obtaining rare earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B09—DISPOSAL OF SOLID WASTE; RECLAMATION OF CONTAMINATED SOIL
- B09B—DISPOSAL OF SOLID WASTE
- B09B3/00—Destroying solid waste or transforming solid waste into something useful or harmless
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F17/00—Compounds of rare earth metals
- C01F17/10—Preparation or treatment, e.g. separation or purification
- C01F17/17—Preparation or treatment, e.g. separation or purification involving a liquid-liquid extraction
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F17/00—Compounds of rare earth metals
- C01F17/20—Compounds containing only rare earth metals as the metal element
- C01F17/206—Compounds containing only rare earth metals as the metal element oxide or hydroxide being the only anion
- C01F17/212—Scandium oxides or hydroxides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F17/00—Compounds of rare earth metals
- C01F17/20—Compounds containing only rare earth metals as the metal element
- C01F17/206—Compounds containing only rare earth metals as the metal element oxide or hydroxide being the only anion
- C01F17/218—Yttrium oxides or hydroxides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F17/00—Compounds of rare earth metals
- C01F17/20—Compounds containing only rare earth metals as the metal element
- C01F17/206—Compounds containing only rare earth metals as the metal element oxide or hydroxide being the only anion
- C01F17/224—Oxides or hydroxides of lanthanides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01F—COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS
- C01F17/00—Compounds of rare earth metals
- C01F17/20—Compounds containing only rare earth metals as the metal element
- C01F17/247—Carbonates
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B3/00—Extraction of metal compounds from ores or concentrates by wet processes
- C22B3/04—Extraction of metal compounds from ores or concentrates by wet processes by leaching
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B3/00—Extraction of metal compounds from ores or concentrates by wet processes
- C22B3/20—Treatment or purification of solutions, e.g. obtained by leaching
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B3/00—Extraction of metal compounds from ores or concentrates by wet processes
- C22B3/20—Treatment or purification of solutions, e.g. obtained by leaching
- C22B3/26—Treatment or purification of solutions, e.g. obtained by leaching by liquid-liquid extraction using organic compounds
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B3/00—Extraction of metal compounds from ores or concentrates by wet processes
- C22B3/20—Treatment or purification of solutions, e.g. obtained by leaching
- C22B3/44—Treatment or purification of solutions, e.g. obtained by leaching by chemical processes
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B7/00—Working up raw materials other than ores, e.g. scrap, to produce non-ferrous metals and compounds thereof; Methods of a general interest or applied to the winning of more than two metals
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22B—PRODUCTION AND REFINING OF METALS; PRETREATMENT OF RAW MATERIALS
- C22B7/00—Working up raw materials other than ores, e.g. scrap, to produce non-ferrous metals and compounds thereof; Methods of a general interest or applied to the winning of more than two metals
- C22B7/006—Wet processes
- C22B7/007—Wet processes by acid leaching
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P10/00—Technologies related to metal processing
- Y02P10/20—Recycling
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02W—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO WASTEWATER TREATMENT OR WASTE MANAGEMENT
- Y02W30/00—Technologies for solid waste management
- Y02W30/50—Reuse, recycling or recovery technologies
Definitions
- the present invention relates to a solid residue (hereinafter referred to as “bauxite residue”) which is a by-product in a buyer process in which the aluminum component in bauxite is separated and collected from the bauxite and is red when Fe 2 O 3 is the main component.
- bauxite residue a solid residue
- red mud rare earth elements
- Rare earth elements are widely used for applications such as high-strength Al alloys, phosphors, magnetic materials, optical glasses, and catalysts.
- rare earth elements are added to transition elements in magnetic materials.
- Patent Document 1 discloses a material for a permanent magnet having an excellent maximum energy product and residual magnetic flux density of an Nd—Fe—B system.
- Patent Document 2 discloses a technique for improving the thermal stability of magnetic properties, which is a disadvantage of the magnet, by replacing part of Nd of the Nd-Fe-B permanent magnet with Dy. ing.
- Such rare earth elements are, for example, ores such as monazite, bastonite, xenotime, and ion-adsorbed clay minerals. From these ores, rare earth elements such as mineral acids such as sulfuric acid are used. Although the elements are leached and separated from the obtained leachate, these ore resources are unevenly distributed on the earth, and the abundance ratio of each element in the rare earth elements varies greatly for each ore.
- heavy rare earth elements having atomic numbers of 64 to 71 have few mines that can extract mines that are miningly profitable, and Dy, whose demand is particularly high, is concerned about the depletion of resources.
- Sc is not produced as a single profit-based ore, but is produced only from tailings such as U ore, which is a raw material for nuclear fuel, and its output is extremely small.
- bauxite which is abundant in resources than ores such as monazite, bust nesite, xenotime, and ion-adsorbed clay minerals, also contains rare earth elements in bauxite, which is an ore resource of aluminum.
- bauxite which is an ore resource of aluminum.
- the main components of bauxite are aluminum oxide and iron oxide, but in the buyer process in which the aluminum component in the raw material bauxite is separated from the raw material and collected, the aluminum oxide in the bauxite is used using an alkaline aqueous sodium hydroxide solution. Is dissolved as aluminum hydroxide, leached and separated to collect the aluminum component in the raw material. Moreover, the main component in the bauxite residue by-produced at this time is iron oxide that does not react with the sodium hydroxide aqueous solution, but when the rare earth element is contained in the bauxite, the rare earth element is not contained in the sodium hydroxide aqueous solution.
- the rare earth element should be concentrated by the amount of leaching of the aluminum component with the sodium hydroxide solution in the buyer process.
- the bauxite residue contains about three times as much rare earth elements on average as compared with the content of rare earth elements in the bauxite. Since it is an industrial waste produced as a by-product in the production of aluminum from bauxite and is readily available, it is expected to be used as a raw material for rare earth elements.
- Patent Document 5 when the above-mentioned Patent Document 5 is examined in detail, as shown in Examples 1 and 2, 52.0% Fe 2 O 3 and 6.5% TiO 2 in a dry state. , 18.0% loss on ignition, 12.9% Al 2 O 3 , 2.4% SiO 2 , 1.6% Na 2 O, 5.0% CaO, 0.6% P 2 Using a bauxite residue containing O 5 as a raw material, a leaching operation (or digestion) at 10 to 70 ° C. is repeated 2 to 3 times using a sulfite solution having a high pH value to a sulfite solution having a low pH value.
- rare earth elements are leached while keeping the elution of Fe and Ti contained in the bauxite residue low, and further separated and recovered by a solvent extraction method.
- the leaching amount of rare earth elements can be almost saturated without increasing the elution amount of Fe.
- Y leaches out about 65% of the content contained in the bauxite residue but the leaching rate of Nd is only about 58% lower than Y (patent document) 5 column 7, lines 32 to 36, Tables 1 to 3 and FIG. 2).
- Example 1 see Table IV-1) and Example 2 (Table 3), the total leaching rate in Example 1 is higher in the first and second times.
- the digestion was performed twice as 4: 1 and 10: 1.
- the extractant required for the separation / recovery process for separating and recovering rare earth elements from the leachate by a solvent extraction method is also commensurate with the amount of leachate.
- Large amounts are required and high as extractant Since expensive EHEHPA or the like is used, there is a problem that the cost for the separation and recovery process is increased.
- the present inventors used 0.102 kg of bauxite residue having the same composition as that used in the examples described later, and also used a sulfite aqueous solution as the acid aqueous solution, and the solid-liquid ratio (L / S) 5.0 and Table 1 shows the result of the additional trial of Example 1 of Patent Document 5 in which the same extraction operation is repeated three times under the conditions of a temperature of 30 ° C., a pressure of 0.1 MPa, and a time of 15 minutes. Then, the leaching rate of Y remained at 5% by mass or less, and the total leaching rate of Y by the second and third leaching operations was 52% by mass. However, the leaching rates of Nd and Dy were 41% by mass, respectively. And only 43% by mass, which is only a lower value than the leaching rate of Y.
- Non-Patent Document 1 shows the leaching rates of Sc, Y and Fe when the pH at the end of leaching is about 0.15 to 0.44 using 0.6N HNO 3 .
- the leaching rate of Sc and Y rapidly decreases as the pH at the end of leaching increases, and the leaching rate of Y is about 38% when the pH at the end of leaching is 0.44 (see Figure 4).
- Patent Document 5 in Non-Patent Document 1, it is necessary to repeat the leaching operation 2 to 3 times in order to increase the leaching rate. Further, a higher solid / liquid ratio of 50 to 100 is required. Further, since the pH of the leachate is low, the elution rate of Fe is as high as 2 to 4%.
- Non-Patent Document 1 when a rare earth element containing Sc is leached from a bauxite residue as a raw material and recovered, Sc is less soluble in acid than lanthanoid, so 0.6N nitric acid is used.
- the leaching operation is performed under the conditions of a solid-liquid ratio (S / L) of 0.1 to 0.01 and a leaching time of 0.5 to 3 hours (see Table 2).
- S / L solid-liquid ratio
- the leaching rate of Sc and Nd are 68.0% and 53.8%, respectively (see Table 3).
- the amount of Fe in the leaching solution in this case is 146.0 ⁇ 10 3
- disadvantages such as being 100 mg or more of the rare earth element and having a solid-liquid ratio of 0.01.
- JP 59-046,008 JP-A 62-165,305 JP 09-176,757 A Japanese Patent Laid-Open No. 09-184,028 United States Patent No. 5,030,424
- the present inventors first studied to improve the leaching rate when leaching rare earth elements from bauxite residues.
- mineral acids such as sulfuric acid, hydrochloric acid, nitric acid, and sulfurous acid as the acid for leaching rare earth elements from bauxite residues improves the leaching rate.
- the pH is less than 0.5
- the leaching rate of impurities Fe and Al is increased, and the amount of the pH adjusting agent required for pH adjustment in the process after leaching is increased. The cost was found to increase.
- the present inventors next lowered the leaching rate of rare earth elements, particularly Sc, Nd, and Dy, which is observed when the pH of the leaching treatment liquid in Non-Patent Document 1 is increased to 0.44. Further investigation was made on the cause of the above and the following findings were obtained.
- Crystalline particles and / or porous particles and aggregates thereof (hereinafter simply referred to as “fine particles”), coarse crystalline particles of 50 to 1000 ⁇ m (hereinafter referred to as “coarse particles”), polygons, And relatively large dense crystalline particles (hereinafter referred to as “crystal particles”), pretreatment methods and conditions such as bauxite ore and mining, heating and drying, and aluminum components in the buyer process.
- the specific surface area of the fine particles usually at approximately 35m 2 / g or more, and coarse particles, Daiasupoa, boehmite, silica, rutile, f It is a crystalline oxide such as tight or goethite.
- the crystal particles are Ca, Ti, newly generated in the buyer process of calcium titanate, calcium aluminate, sodalite, etc. having a perovskite (ABX3) type structure. It is a crystalline oxide containing Fe and O.
- the main components of bauxite are aluminum oxide and iron oxide, which are thought to have been formed by weathering of igneous rocks such as granite and limestone.
- igneous rocks such as granite mainly composed of aluminosilicate minerals (feldspars) and limestone mainly composed of calcium carbonate (calcite) are exposed to high temperature and heavy rain environment, and alkali metals among the above principal components
- the remainder of the aluminum oxide or iron oxide from which calcium, silicon oxide, etc. are eluted is the main component of bauxite.
- the concentration of rare earth elements in bauxite is about 10 times the concentration of rare earth elements in igneous rocks such as granite and limestone before weathering.
- the coarse particles in the bauxite residue are coarse particles that did not change in the buyer process, and calcium aluminate and sodalite produced in the buyer process.
- the rare earth elements in the bauxite are Not concentrated.
- rare earth elements are concentrated at a relatively high concentration in the fine particles of the bauxite residue, and are also taken into crystal particles such as calcium titanate having a perovskite structure newly formed in the buyer process.
- the ratio between the fine particles in the bauxite residue and newly formed crystal particles is determined by the amount of titanium oxide in the bauxite, the operating conditions of the buyer process, particularly the processing temperature, and removal of impurities such as Si and P.
- the processing temperature of the buyer process is less than 160 ° C.
- the generation of new crystal particles is small, and as a result, rare earth elements are fine particles.
- the rare earth element contained in such fine particles can be efficiently leached. This is because fine particles have a high specific surface area and a large reaction area with the leaching treatment solution, and newly generated crystal particles are chemically stable perovskite (ABX3) type titanate. It is considered that it is calcium [CaTi (Fe) O 3 ] and the like and is difficult to dissolve in mineral acids. Even if the processing temperature in the buyer process is 160 ° C.
- the CaO content in the bauxite residue is less than 4% by mass, the generation of crystal particles in the buyer process is small, so that rare earth elements are efficiently leached. be able to.
- the processing temperature of the buyer process is 230 ° C. or higher, crystal grains are generated, and even fine particles mainly composed of Fe 2 O 3 are almost completely from incomplete crystal structures. Since the specific surface area is reduced and leaching of rare earth elements is difficult, the rare earth elements are hardly leached when the processing conditions in the buyer process are 230 ° C. or higher.
- the present inventors used a bauxite having a specific surface area of 26 m 2 / g or more as a raw material in the buyer process, and a raw material bauxite residue obtained by adopting processing conditions of a temperature of less than 160 ° C., or By using a bauxite residue having a CaO content of less than 4% by mass as a raw material bauxite residue obtained by adopting treatment conditions of a treatment temperature of less than 230 ° C., or containing fine particles, coarse particles and crystal particles By using a high specific surface area fraction obtained by fractionating bauxite residue as a raw material bauxite residue, a leaching treatment of pH 0.5 to 2.2 capable of suppressing the leaching rate of impurities Fe and Al from this raw material bauxite residue The inventors have found that the rare earth elements can be efficiently recovered using the liquid, and completed the present invention.
- an object of the present invention is to provide a method for recovering a rare earth element that can efficiently recover the rare earth element in the bauxite residue using the bauxite residue containing the rare earth element as a raw material.
- the present invention provides a bauxite residue by-produced in Bayer process for collecting separated aluminum components from bauxite as a raw material, a method for recovering a rare earth element to recover rare earth elements from the raw material, specific surface area 35m 2 as the starting material
- a leaching treatment solution consisting of an aqueous solution of one or more mineral acids selected from sulfuric acid, hydrochloric acid, nitric acid, and sulfurous acid is added to this raw material bauxite residue, and the solid-liquid ratio is 2
- a slurry having a pH of 30 to 30 and a pH of 0.5 to 2.2 is prepared, and a rare earth element is leached under a temperature condition of room temperature to 160 ° C. Then, the slurry after the leaching is solid-liquid separated from the leachate obtained.
- a method for recovering rare earth elements comprising separating and recovering rare earth elements.
- the present invention provides a bauxite obtained from a buyer process in which the raw material bauxite residue is bauxite having a specific surface area of 26 m 2 / g or more and the treatment temperature is 160 ° C. or less in the above rare earth element recovery method.
- This is a method for recovering a rare earth element as a residue, and is obtained from a buyer process using bauxite having a specific surface area of 26 m 2 / g or more as a raw material at a processing temperature of less than 230 ° C., and has a CaO content of 4 mass.
- the rare earth element is a method for recovering rare earth elements that are less than% bauxite residue and is a high specific surface area fraction mainly composed of fine particles having a specific surface area of 35 m 2 / g or more obtained by fractionating the bauxite residue. This is an element recovery method.
- the term “rare earth element” is a generic term for Sc having an atomic number of 21, Sc, Y having an atomic number of 39, and La to Lu having atomic numbers of 57 to 71 (hereinafter referred to as “lanthanoids”). Although used, this does not deny that Ac to Lr of atomic numbers 89 to 103 are leached, separated and recovered by the method of the present invention.
- the particle size distribution of the bauxite residue obtained from the buyer process is examined, as shown in FIG. 3, the particle size distribution is usually 86 to 93% by mass at 38 ⁇ m or less, and 2 to 4 at 38 to 75 ⁇ m.
- the mass%, 75 to 300 ⁇ m is 1 to 4 mass%, and 300 ⁇ m or more is 3 to 6 mass%.
- the bauxite having a specific surface area of 26 m 2 / g or more according to the method of the present invention is used as a raw material, and the raw material bauxite residue obtained from the buyer process performed under the condition of the processing temperature of 160 ° C. or less, the specific surface area of 26 m 2 / g.
- the specific surface area is 51.5 m in the case of fine line hatching in FIG. 3 / g, fine particles having a high specific surface area of 40.7 m 2 / g in the case of thick line hatching in FIG. 3, and high leaching of rare earth elements by the method of the present invention using these raw material bauxite residues can be recovered at a rate, whereas in the case of FIG. 3 dots a specific surface area of 17.9m 2 / g, even by the method of the present invention performs the recovery of rare earth elements It is not possible to achieve a high leaching rate.
- fine particles are selectively selected mainly by fractionation from bauxite residues having a mixture of fine particles having a relatively high specific surface area and coarse particles having a relatively low specific surface area and a specific surface area of less than 35 m 2 / g.
- classification is performed using a sieve having a sieve size of 38 to 400 ⁇ m, preferably 38 to 300 ⁇ m. The method of doing can be illustrated.
- the fractionation treatment using the sieve having a mesh size of 38 to 400 ⁇ m may be wet or dry, and a high specific surface area fraction suitable as the raw material bauxite residue of the present invention can be obtained. .
- this fractionation treatment in addition to coarse particles caused by bauxite as a raw material, coarse particles of crystal particles generated in the buyer process can be removed. For this reason, when the bauxite residue contains a large amount of coarse calcium aluminate and sodalite as crystal particles and the neutralization treatment described later is performed, the amount of mineral acid aqueous solution used in the neutralization treatment is suppressed. can do.
- the fractionation treatment as described above can be omitted.
- the method may be adopted if particles having a specific surface area of 35 m 2 / g or more in the bauxite residue by-produced in the buyer process can be selectively separated by a method other than image processing. .
- the content of the rare earth element in the raw material bauxite residue is not particularly limited. However, from the viewpoint of leaching efficiency in the leaching treatment, it is preferably dried at 110 ° C. for 2 hours.
- the obtained solid component preferably contains Sc, Y and lanthanoid oxides in a total amount of 1500 to 10000 ppm. If the total content of the rare earth elements is less than 1500 ppm, the profitability may decrease due to the low content.
- Ca content is less than 4 mass% as CaO. This is because, as described above, when the Ca content is 4% by mass or more as CaO, the newly formed crystal particles are not easily dissolved in mineral acid at 160 ° C. or higher, and the calcium titanate having a perovskite (ABX3) type structure is used.
- ABX3 perovskite
- a Ca compound coating layer is easily formed on the particle surface having a large specific surface after aluminum oxide is removed in the buyer process as described above. This is because the compound coating layer prevents leaching of rare earth elements.
- the method of the present invention by using a raw material bauxite residue having a specific surface area as a raw material, it is possible to easily separate and recover rare earth elements, particularly Sc, Y, and lanthanoids at a high leaching rate.
- rare earth elements particularly Sc, Y, and lanthanoids
- it is possible to eliminate many concerns such as uneven distribution of rare earth element ore, variation in the ratio of each element in rare earth elements for each ore, and resource depletion. .
- FIG. 1 is an observation photograph when a bauxite residue by-produced in the buyer process is observed with an optical microscope (the arrows in FIG. 1 indicate coarse crystalline particles).
- FIG. 2 is an observation photograph of the bauxite residue by-produced in the buyer process when observed with a scanning electron microscope (the arrows in FIG. 2 indicate dense crystals).
- FIG. 3 is a graph showing the particle size distribution of bauxite residue obtained from the buyer process.
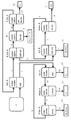
- FIG. 4 is a flowchart showing impurity element removal and rare earth element concentration of the leachate by the two-stage solvent extraction method according to Example 59 of the present invention.
- the raw material bauxite residue mainly composed of fine particles with a specific surface area of 35 m 2 / g or more is recovered from a buyer process performed using bauxite having a specific surface area of 26 m 2 / g or more as a raw material and under a processing temperature of less than 160 ° C.
- a leaching treatment liquid composed of a predetermined mineral acid aqueous solution is added to the raw material bauxite residue so as to have a predetermined pH at a predetermined solid-liquid ratio, mixed to prepare a slurry, and leached at a predetermined temperature.
- a leaching treatment liquid composed of a predetermined mineral acid aqueous solution
- a slurry preparation one kind selected from sulfuric acid, hydrochloric acid, nitric acid and sulfurous acid can be used alone, or two or more kinds can be mixed and used as the mineral acid.
- the solid-liquid ratio (L / S) between the solid component (S) and the liquid component (L) of the prepared slurry is 2 or more and 30 or less, preferably 4 or more and 10 or less.
- the solid-liquid ratio (L / S) is lower than 2, the viscosity of the slurry will increase, making it difficult to handle in the subsequent solid-liquid separation process. As a result, the recovery rate of the leachate will be reduced. Even if the solid-liquid ratio (L / S) of the slurry is higher than 30, not only does the saturation rate of rare earth elements saturate and improve, but also the amount of water used increases and the amount of leachate increases, so that the leaching treatment
- the equipment used for solid-liquid separation to obtain a leachate later and the separation and recovery for recovering rare earth elements from this leachate is increased in size, and the equipment is increased in size, the chemicals used, and the cost of waste disposal. The demerit that becomes.
- the pH value of the slurry in this leaching treatment is 0.5 to 2.2, preferably 0.7 to 2.0.
- the pH value of this slurry is higher than 2.2, the leaching rate of rare earth elements decreases.
- the pH value is less than 0.5, the separation of rare earth elements becomes difficult due to the increase in the amounts of Al, Fe and Ti during the leaching process. As a result, the recovery cost of rare earth elements increases.
- sulfuric acid is used as the leaching treatment liquid, the leaching rate of Y and lanthanoid is high immediately after the start of leaching, but decreases with time.
- the leaching treatment liquid has a predetermined solid-liquid ratio (L / S) and a predetermined pH value.
- L / S solid-liquid ratio
- a mineral acid aqueous solution having a predetermined concentration and amount that can be achieved is prepared in advance, and this leaching treatment liquid is preferably added to the raw material bauxite residue and mixed. According to such a method, the prepared slurry is solidified.
- the liquid ratio (L / S) and pH value can be easily adjusted to the desired values, and it is possible to prevent elution of impurities such as Fe due to local concentration of mineral acid into the leachate. it can.
- the leaching treatment Prior to the leaching treatment, it is preferable to remove elements such as Al, Si, and Ca that are eluted with the decomposition of calcium aluminate and sodalite in the crystal particles generated in the buyer process. Therefore, prior to the leaching treatment, it is preferable to neutralize the raw material bauxite residue within a pH range of 3.5 to 5 where the rare earth elements do not elute, and this neutralization treatment facilitates the separation and recovery of the rare earth elements from the later leaching solution. become.
- the mineral acid aqueous solution used in this neutralization treatment it is preferable to use sulfurous acid having a high Ca elution rate, but it is also possible to use waste acid discharged during the rare earth element separation and recovery described later. is there.
- the oxidizing agent Prior to the leaching treatment, the oxidizing agent is added in an equivalent amount of 0.1 to 0.3, preferably 0.15 to 0.25, based on the Fe component in the raw material bauxite residue.
- the Fe 2+ ions contained from the components in the raw material bauxite residue are converted to Fe 3+ ions, and Fe and Al are precipitated during the subsequent separation and recovery of the rare earth elements. Separation and recovery processing can be facilitated.
- the oxidizing agent added for this purpose include preferably a hydrogen peroxide solution and a perchloric acid aqueous solution, and more preferably 30% by mass-hydrogen peroxide solution and 70% by mass-perchloric acid aqueous solution. It is. If the addition amount of this oxidant is less than 0.1 equivalent, Fe 2+ ions remain in the leachate until the pH is high, and conversely, the effect does not change even if it exceeds 0.3 eq. Is wasted.
- the treatment temperature during the leaching treatment is in the range of room temperature (20 ° C.) to 160 ° C., preferably 50 ° C. to 105 ° C.
- the leaching rate of the rare earth element tends to increase as the processing temperature increases, but it is preferable to select and determine the leaching rate in consideration of the balance between the energy cost, the type of rare earth element to be recovered and the leaching rate. For example, when the leaching rates of Examples 5 to 11 described later are observed (see Table 4), the leaching rate of Sc decreases from 36% by mass at 100 ° C. to 11.2% to 28.8% by mass at 25 ° C. However, the leaching rate of Y decreased from 61.5% by mass at 100 ° C.
- the leaching rate of La of the lanthanoid was 89.6% by mass at 100 ° C. From 25.degree. C. to 65.7% by mass to 79.8% by mass, and the leaching rate of Nd of the lanthanoid is 82.2% by mass at 100.degree. C. to 66.9% by mass to 77.1% by mass at 25.degree.
- the lanthanoid Dy leaching rate decreases from 69.1% by mass at 100 ° C. to 50.8% by mass or 68.7% by mass at 25 ° C.
- the treatment temperature exceeds 160 ° C., the leaching rate of Sc is maintained at a high value, but the leaching rate of other rare earth elements is significantly lowered.
- the holding time at the above-mentioned temperature in the leaching process is 1 second or more and 180 hours or less. Preferably, it is 30 minutes to 180 hours for Sc leaching and 1 second to 7 minutes for lanthanoid leaching. It is preferable to stir the slurry during the leaching process because a leaching rate with little variation is obtained. When the holding time is less than 1 second, there arises a problem that variation in the leaching rate increases. Moreover, when leaching the lanthanoid, it is also preferable to dilute with water having a temperature equal to or greater than the amount of the slurry and equal to or less than 50 ° C. immediately after holding for 1 second to 7 minutes.
- the holding time of the leaching process at the leaching temperature can be easily controlled.
- Sc is leached again with a holding time of 30 minutes or more and 180 hours or less, and both Sc and the lanthanoid are efficiently removed. It can be leached and collected.
- the pH adjuster used for this purpose is not particularly limited, sodium hydroxide, potassium hydroxide, calcium hydroxide, ammonia, bauxite residue and the like are preferably used.
- raw material bauxite residue is used as a pH adjuster, the pH adjuster necessary for pH adjustment is saved, and leaching of rare earth elements from the added bauxite residue is added, so the concentration of rare earth elements increases.
- the amount of mineral acid used in the leaching process can also be saved.
- the solid-liquid separation process can be reduced because the solid-liquid separation operation for separating the Fe and Al hydroxides and bauxite residues precipitated by pH adjustment from the leachate is sufficient.
- the slurry after the leaching treatment is then subjected to solid-liquid separation by means of filtration, centrifugation, sedimentation separation, decantation, etc., and a leachate containing rare earth elements is recovered.
- the solid residue produced by this solid-liquid separation is preferably washed with washing water, the leachate adhering to the solid residue is washed, transferred to water and recovered, and obtained by solid-liquid separation first. Together with the leachate, the leachate is used to separate and recover the next rare earth element. If the amount of washing water used for washing the solid residue is too small, the leachate adhering to the solid residue cannot be sufficiently recovered. Conversely, if it is too much, the next rare earth element is separated and recovered. Since the load increases, the solid-liquid ratio (L / S) between the solid residue (S) and the washing water (L) is usually in the range of 2 to 30.
- the leachate obtained by the above-described solid-liquid separation treatment is then transferred to the rare earth element separation and recovery for separating and collecting Sc, Y and the lanthanoid rare earth elements.
- the separation and recovery of rare earth elements from the leachate can be performed by a known method, and as a separation method, a hydroxide precipitation method, an oxalate precipitation method, a carbonate method, a solvent extraction method, an ion exchange method, or the like is used. .
- the leachate in which the amount of Fe and Ti eluted is small, the leachate can be directly treated by the oxalate precipitation method or the solvent extraction method, but Al and / or under conditions where the pH of the leach treatment is low and the temperature is high.
- the pretreatment for concentrating the leachate may also be performed by a known method.
- a method for evaporating and concentrating the leachate obtained by the solid-liquid separation treatment, a concentration method using an RO membrane concentrator, and a solvent are used.
- a method of extracting and separating the rare earth elements Sc, Y and lanthanoid by the solvent extraction method and concentrating them simultaneously A method of obtaining a rare earth element compound solid by solid-liquid separation of rare earth elements as a rare earth hydroxide, rare earth oxalate, rare earth carbonate solid by a hydroxide precipitation method, an oxalate precipitation method, a carbonate method, and the like; Further, in order to reduce the amount of the drug used in the above-described method, there is a method (pH adjustment method) that combines a method in which Fe and Al are removed from the leachate by solid-liquid separation in advance by pH adjustment.
- the pH adjustment method used in combination with other methods usually has a pH value of 1 to 3 in the leachate, so first add a pH adjuster to the leachate to adjust the pH value to 4-6.
- the Fe and Al hydroxides precipitated by this pH adjustment are separated and removed by solid-liquid separation, and the impurity concentration of Fe and Al is lowered, whereby the purity of the rare earth compound can be increased.
- the pH adjuster used for this purpose is not particularly limited, sodium hydroxide, potassium hydroxide, calcium hydroxide, ammonia, bauxite residue and the like are preferably used. In this way, a leachate having a high rare earth element concentration or a solid rare earth element compound is obtained by rough purification and concentration of the rare earth element.
- an oxidizing agent as necessary to oxidize the Fe 2+ ions in the leachate to Fe 3+ ions, so that insoluble Fe (OH) 3 becomes stable, Fe can be easily separated and removed.
- the oxidizing agent for example, air blowing, hydrogen peroxide, perchloric acid, permanganic acid, hypochlorous acid or the like can be suitably used.
- hydrogen peroxide is used as the oxidizing agent, the concentration of the oxidizing agent only affects the solid-liquid ratio, so an appropriate concentration can be selected from the handling and cost.
- both 30% by mass-hydrogen peroxide solution and 70% by mass-perchloric acid aqueous solution are used in an amount of 0.1 to the Fe component in the bauxite residue. It is preferably 0.5 equivalent.
- the hydroxide precipitation method in order to separate rare earth elements as Sc, Y and lanthanoids from the leachate as hydroxides, the leachate obtained by the above solid-liquid separation treatment or the pH of the leachate is adjusted to Fe. And Al are precipitated as hydroxides, and a liquid obtained by solid-liquid separation is added with a pH adjuster to adjust the pH value to 7 or more, and rare earth elements are precipitated as their hydroxides.
- the rare earth element hydroxide is separated into solid and liquid and recovered as a roughly recovered product.
- the pH adjuster is preferably sodium hydroxide, potassium hydroxide, calcium hydroxide, ammonia or the like, and the rare earth element is precipitated as a hydroxide.
- This is solid-liquid separated and recovered as a rare earth element hydroxide, or for the purpose of lowering the concentration of Al, which is an impurity, to the rare earth element hydroxide deposited, at least five times the equivalent of Al. It is also preferable to add a sodium hydroxide solution to dissolve and remove the Al content as aluminate ions.
- the leachate obtained by the above solid-liquid separation treatment, or the liquid obtained by adjusting the pH of the leachate and precipitating Fe and Al as hydroxides and solid-liquid separation An insoluble rare earth oxalate is formed by adding 1.3 to 6 equivalents of oxalic acid in the molar amount of all rare earth elements present in the liquid, and this rare earth oxalate compound is converted into a crude rare earth element compound ( It is recovered as a crude recovered product).
- carbonic acid or sodium carbonate is added as a pH adjuster to the leachate obtained by the above solid-liquid separation treatment, and the pH is adjusted to 4 to 5 to precipitate rare earth elements as carbonates. Liquid-separated and recovered as a rare earth element crude recovery.
- the solvent extraction method may be a known method, but as an extractant, phosphate ester (DEHPA, EHPA), phosphonate ester (PC88A), phosphinate ester (Cyanex 272 , Cyanex 30), non-polar organic solvents such as hexane, aliphatic hydrocarbons such as benzene, toluene, alcohols such as octanol, and petroleum fractions such as kerosene.
- phosphate ester DEHPA, EHPA
- PC88A phosphonate ester
- Cyanex 272 Cyanex 30
- non-polar organic solvents such as hexane, aliphatic hydrocarbons such as benzene, toluene, alcohols such as octanol, and petroleum fractions such as kerosene.
- Those diluted with a solvent can be suitably used. It is also preferable to carry out the recovery of the roughly recovered product by the solvent extraction method in two or more stages. According to the recovery of the roughly recovered product by the solvent extraction method over two or more stages, it becomes possible to separate the rare earth element into each element.
- the leaching raw material is a solid residue (bauxite residue) after elution of aluminum hydroxide from bauxite by the Bayer method.
- a crude rare earth element compound obtained by the above solid-liquid separation treatment.
- the solvent is recovered as it is or after being adjusted again to pH 1.2 to 2.5. It is preferable to extract.
- an emulsion or suspension hereinafter referred to as “emulsion” formed between the organic phase and the aqueous phase during solvent extraction or the like. Occurrence can be prevented. If the emulsion occurs, it can be removed by filtration. If the pH of the aqueous phase at the time of solvent extraction is less than 1.2, the recovery rate of rare earth elements decreases, which is not preferable.
- Such pH adjustment is preferably performed by adding bauxite residue. If the pH is adjusted by adding bauxite residue, the amount of alkaline chemicals used can be reduced, and the bauxite residue is a by-product in the buyer process when producing aluminum from bauxite. The cost can be reduced. In addition, when the pH is adjusted by the addition of bauxite residue, the rare earth elements contained in the added bauxite residue are leached into the leachate. Therefore, the acid aqueous solution used for the leaching treatment should be used effectively.
- rare earth elements leached from the added bauxite residue can be recovered, and at this time, Ca and Ti are precipitated together with Fe, and the concentration of these elements in the leachate is lowered, resulting in efficiency. Rare earth elements can be recovered.
- the extraction rate of the rare earth element can be kept low, and as a result, the concentration of the rare earth element to be separated and recovered can be increased.
- the extraction time is preferably 5 minutes or less, and more preferably 0.5 to 3 minutes.
- the extraction rate of Al can be kept low, and as a result, the concentration of the rare earth element to be separated and recovered can be increased.
- the extraction time exceeds 5 minutes, the extraction rate of Al increases, and as a result, the concentration of rare earth elements to be separated and recovered decreases.
- Sc is separated into the pre-extracted organic phase, but Sc can be recovered as a solid hydroxide from the pre-extracted organic phase by back-extracting an alkaline aqueous solution having a pH of 7.5 or more as a back extractant.
- an alkaline aqueous solution having a pH of 7.5 or more as a back extractant.
- emulsion may occur between the organic phase and the aqueous phase during solvent extraction. When the emulsion occurs, the precipitate can be removed by filtration.
- a 2N to 8N hydrochloric acid aqueous solution or a 30 to 70% by weight sulfuric acid aqueous solution is preferably used as the back extraction agent.
- the back extraction time is preferably 5 minutes or less, and more preferably 0.5 to 3 minutes.
- the back extraction time is 0.5 to 3 minutes, the extraction rate of Al can be kept low, and as a result, the concentration of the rare earth element to be separated and recovered can be increased.
- the back extraction time exceeds 5 minutes the extraction rate of Al increases, and as a result, the concentration of the rare earth element to be separated and recovered decreases.
- the rare earth element when an aqueous sulfuric acid solution having a concentration of 30 to 70% by mass is used as the back extractant, the rare earth element is precipitated as a solid sulfate, so that the volume can be made very small.
- the back extraction time is preferably 5 minutes or less, and more preferably 0.5 to 3 minutes.
- the extraction rate of Al can be kept low, and as a result, the concentration of the rare earth element to be separated and recovered can be increased.
- the back extraction time exceeds 5 minutes the extraction rate of Al increases, and as a result, the concentration of the rare earth element to be separated and recovered decreases.
- the rare earth element precipitated as solid sulfate can be recovered by solid-liquid separation.
- Al in the organic phase can be recovered as aluminum sulfate by performing back extraction for 120 minutes or more using a 30 to 70% by mass sulfuric acid aqueous solution as a back extractant. it can.
- Sc, Ti, Th accumulated in the used extractant is reduced by back extraction using a 2N-8N hydrochloric acid aqueous solution or an alkaline aqueous solution as a back extractant to regenerate. It can be reused as an extractant.
- the separation of the crude recovery product into each element includes phosphate esters, phosphonate esters, phosphinate esters, thiophosphinate esters, and the like.
- Esters selected from a mixture of the following esters with tributyl phosphate and / or trioctylphosphine oxide, aliphatic hydrocarbons such as hexane, aromatic hydrocarbons such as benzene and toluene
- the separation by such a solvent extraction method is preferably by a countercurrent multistage solvent extraction method.
- the pH value of the leachate is adjusted to 4 to 6, and Fe and Al precipitated by this pH adjustment.
- the solid hydroxide is removed by solid-liquid separation, and then a pH adjuster is added to adjust the pH value to 7 or more, and the precipitated rare earth hydroxides Sc, Y and lanthanoids are solid-liquid separated.
- a pH adjuster is added to adjust the pH value to 7 or more, and the precipitated rare earth hydroxides Sc, Y and lanthanoids are solid-liquid separated.
- oxalate precipitation method Fe and Al are precipitated as a hydroxide directly or by adjusting the pH as in the hydroxide method, and after solid-liquid separation, oxalic acid is added to add Sc, Y and lanthanoids.
- the rare earth element is precipitated as oxalate and recovered as a oxalic acid compound of Sc, Y and a lanthanoid rare earth element, which is then treated with caustic soda to form a crude hydroxide of Sc, Y and a lanthanoid rare earth element.
- the recovered material or the oxalic acid compound of the rare earth element which is Sc, Y and the lanthanoid is calcined, and the crude recovered material is recovered as an oxide of the rare earth element which is Sc, Y and the lanthanoid.
- Sc, Y and lanthanoids are collected as rare earth carbonate compounds from the leachate, and this is treated with caustic soda to obtain a coarsely recovered product as Sc, Y and lanthanoid rare earths hydroxide.
- a rare-earth carbonate compound of Sc, Y, and a lanthanoid is calcined, and a crude recovery product is recovered as an oxide of Sc, Y, and a lanthanoid-rare earth element.
- this crude recovered product is dissolved in sulfuric acid, hydrochloric acid or nitric acid, and then solvent extraction is performed using the extractant, so that the amount of expensive extractant used can be reduced as much as possible. There is.
- Bauxite was pulverized using a ball mill, and the obtained bauxite having a specific surface area of 24 to 35 m 2 / g shown in Table 2 was used.
- the processing temperature shown in Table 2 was 105 to 250 ° C. and the CaO addition amount was 0.0 to 3
- the buyer process was carried out under the condition of 5% by mass, and the bauxite residue was recovered from the buyer process. Thereafter, a part of the recovered bauxite residue is used as a raw material bauxite residue as it is, and for the other part, 500 parts by weight of water is added to 100 parts by weight of the bauxite residue, and the slurry is once mixed.
- Examples 1 to 11 and Comparative Example 1 First, the raw material bauxite residue of sample No. 1 is used, and a 2N sulfuric acid aqueous solution is used as the leaching treatment liquid, and the slurry solid-liquid ratio (L / S), slurry pH (initial), treatment shown in Table 4 A leaching process for recovering the rare earth element was carried out under the conditions of temperature (° C.) and holding time (minutes). Then, after the leaching process was completed, the slurry was subjected to solid-liquid separation by filtration, and the leachate was recovered.
- the mass of the dried product obtained by drying under the drying conditions of 110 ° C. and 2 hours was measured, and the mass was determined as the solid weight (S) of the raw material bauxite residue. It was.
- Example 6 except that sulfurous acid gas was blown into the slurry prepared with the raw material bauxite residue and water, the water in this slurry was used as a sulfite aqueous solution, and this sulfite aqueous solution was used as a leaching treatment solution.
- the leaching treatment was performed in the same manner as in Example 1, and the leaching solution was recovered.
- the obtained leachates of Examples 1 to 11 and Comparative Example 1 are Sc, Y, lanthanoids La, Nd, and Dy and impurities by ICP-AES (inductively coupled plasma emission spectroscopy) analysis. About Al, Fe, Ca, Si, and Ti, the element content was measured and the leaching rate of each element was calculated
- Examples 12 to 17 and Comparative Examples 2 to 6 Using the raw material bauxite residue of sample No. shown in Table 5, and using the leaching treatment liquid shown in Table 5, the slurry solid-liquid ratio (L / S), slurry pH (initial), treatment temperature (° C.) shown in Table 5 ) And holding time (minutes), except that the leaching treatment for collecting the rare earth elements was carried out, and the leachate was collected in the same manner as in Examples 1 to 11 above. 17 and the leaching solutions of Comparative Examples 2 to 6, the element contents of Sc, Y, lanthanoids La, Nd and Dy and impurities Al, Fe, Ca, Si and Ti were measured. The leaching rate of each element was determined. The results are shown in Table 5.
- Comparative Example 1 where the temperature of the leaching treatment exceeds 160 ° C.
- Comparative Example 5 using phosphoric acid as the mineral acid of the leaching treatment liquid
- Comparative Example 6 where the pH value of the slurry in the leaching treatment exceeds 2.2
- 50% by mass or more of the lanthanoids La, Nd and Dy contained in the raw material bauxite residue could not be leached.
- the leaching rate of Sc and Y the higher the leaching rate of Sc and Y, but the leaching rate of lanthanoids (La, Nd and Dy)
- the lower the pH value of the slurry the higher.
- the higher the temperature within the processing temperature range of 20 to 160 ° C. the higher the leaching rate tended to be in a short time.
- the leaching rate of lanthanoid has a minimum value between 5 and 60 minutes, and the leaching rate tends to increase as the retention time is shorter and shorter as the retention time is shorter. It was.
- Example 18 Using the raw material bauxite residue of sample No. shown in Table 6, and using the leaching treatment liquid shown in Table 6, the slurry solid-liquid ratio (L / S), slurry pH (initial), treatment temperature (° C.) shown in Table 5 ) And holding time (minutes), except that the leaching treatment for collecting the rare earth elements was carried out, and Sc and the leaching solution collected in the same manner as in Examples 1 to 17 and Comparative Examples 2 to 6 above. , Y, lanthanoids Nd and Dy, and impurities Al, Fe, Ca, Si, and Ti, their element contents were measured, and the leaching rate of each element was determined. The results are shown in Table 6.
- Example 19 In the same method as Example 18, Sc, Y, lanthanoids Nd and Dy, and impurities Al, Fe, Ca, Si, and the like were recovered after neutralization with bauxite residue after leaching. About Ti, the element content was measured and the leaching rate of each element was calculated
- Example 19 neutralized with bauxite residue, the concentrations of impurities Al, Fe, Si, and Ti were decreased compared to Example 18, whereas Sc and Y It can be seen that the concentrations of lanthanoid Nd and Dy are increased. As a result, the amount of the mineral acid aqueous solution used relative to the recovered amounts of Sc, Y, and lanthanoids Nd and Dy could be reduced.
- Example 20 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a solvent extraction method.
- this solvent extraction method first, the pH of the leachate is once set to 3.0, the deposited precipitate is removed, adjusted to 1.5, and then DEHPA is diluted to 0.8 M with kerosene. , The leachate and the extractant were agitated and contacted at a liquid ratio of 1: 1 for 3 minutes, and then separated into an extracted organic phase and an aqueous phase after extraction (water phase after extraction) by liquid-liquid separation. .
- the extracted organic phase 6N-hydrochloric acid aqueous solution was used as the back extractant, and the extracted organic phase and the back extractant were contacted with stirring at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation and back extraction.
- the organic phase after completion (the organic phase after back extraction) and the back extracted water phase were separated, and the rare earth elements in the extracted organic phase were transferred to the back extracted water phase, separated and recovered.
- the organic phase after back extraction is made by using 0.02N-hydrochloric acid aqueous solution as a back extractant, contacting the organic phase after back extraction with the back extractant at a liquid ratio of 1: 1 for 3 minutes, and then separating the liquid and liquid.
- DEHPA can be reused cyclically as an extractant diluted with kerosene to a concentration of 0.8M. Table 8 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Example 21 to 24 In the same method as in Example 20, the contact time between the leachate and the extractant was 0.5 minutes, 1 minute, 5 minutes, and 10 minutes, and the other conditions were the same as in Example 20, and the rare earth element was back-extracted water. The phase was separated and recovered. Table 8 shows the rare earth element recovery rate and impurity concentration recovered by this solvent extraction method.
- Example 25 to 29 In the same method as in Example 20, the contact time between the extracted organic phase and the back extractant was 0.5 minutes, 1 minute, 5 minutes, 10 minutes, and 15 minutes, and the other conditions were the same as in Example 20. Rare earth elements were transferred to the back-extracted aqueous phase and separated and recovered. Table 8 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Example 30 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a solvent extraction method.
- this solvent extraction method first, the pH of the leachate is once adjusted to 1.75, and then an extractant in which DEHPA is diluted to 0.8 M with kerosene is used, and the leachate and the extractant are mixed at a liquid ratio of 1: 1 for 3 minutes. After stirring and contacting, liquid-liquid separation was performed to separate the extracted organic phase and the aqueous phase after extraction. During the solvent extraction, an emulsion was formed between the organic phase and the aqueous phase. The emulsion was separated to the organic phase side during liquid-liquid separation, and then the organic phase was removed by filtration through a filter.
- the extracted organic phase 6N-hydrochloric acid aqueous solution was used as a back extractant, and the extracted organic phase and the back extractant were stirred and contacted at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation and back extraction.
- the organic phase and the back-extracted aqueous phase were separated, and the rare earth element was transferred from the extracted organic phase to the back-extracted aqueous phase to be separated and recovered.
- the organic phase after back extraction is made by using 0.02N-hydrochloric acid aqueous solution as a back extractant, contacting the organic phase after back extraction with the back extractant at a liquid ratio of 1: 1 for 3 minutes, and then separating the liquid and liquid.
- DEHPA can be reused cyclically as an extractant diluted with kerosene to a concentration of 0.8M. Table 8 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Example 31 The other implementation methods and conditions were the same as in Example 20 except that the pH was adjusted by addition of the same bauxite residue as used in Example 4 instead of addition of the aqueous sodium hydroxide solution, and the rare earth elements were back-extracted water. The phase was separated and recovered. At this time, the amount of the added bauxite residue was 0.115 kg with respect to 0.1 kg of the bauxite residue which was the leaching raw material.
- Table 8 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method. However, in the calculation of the recovery rate, the recovery rate for 2.15 times the amount of the bauxite residue which was the leaching raw material is shown in consideration of the rare earth element contained in the bauxite residue used for pH adjustment.
- Y which has the lowest recovery rate
- a recovery rate exceeding 75% by mass can be obtained with a back extraction time of 1 minute, and the longer the extraction time and back extraction time, the more the impurities such as Al. It turns out that a recovery rate becomes high.
- Example 30 when the emulsion was generated between the organic phase and the aqueous phase during solvent extraction, the recovery rate of rare earth elements was compared with Example 20 in which the extraction time and the back extraction time were the same. Can be seen to be slightly lower.
- the rare earth element which eluted from the bauxite residue added at the time of pH adjustment is also collect
- the recovery rate is recovery from the bauxite residue which was the leaching raw material. Since the recovery rate is lower than that of Example 20 because it is not as high as the rate, it can be seen that Ca and Ti are precipitated together with Fe, and the concentrations of these elements are greatly reduced.
- the bauxite residue is a by-product in the buyer process when producing aluminum from bauxite, resulting in cost reduction.
- Example 32 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a solvent extraction method.
- this solvent extraction method first, the pH of the leachate is once set to 3.0, the deposited precipitate is removed, adjusted to 1.0, and then an extractant in which DEHPA is diluted to 0.8 M with kerosene is added.
- the leachate and the extractant were used and agitated at a liquid ratio of 1: 1 for 3 minutes and then contacted, followed by liquid-liquid separation to separate the extracted organic phase and the extracted aqueous phase.
- the extracted organic phase 6N-hydrochloric acid aqueous solution was used as a back extractant, and the extracted organic phase and the back extractant were stirred and contacted at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation and back extraction.
- the organic phase and the back-extracted aqueous phase were separated, and the rare earth element was transferred from the extracted organic phase to the back-extracted aqueous phase to be separated and recovered.
- the organic phase after back extraction is 0.02N-hydrochloric acid aqueous solution as a back extractant, the organic phase after back extraction and the back extractant are stirred at a liquid ratio of 1: 1 for 3 minutes and then purified by liquid-liquid separation. It can be reused cyclically as an extractant with DEHPA diluted to a concentration of 0.8M with kerosene. Table 9 shows the recovery rate of rare earth elements recovered by this solvent extraction method and the concentration of impurities.
- Examples 33 and 34 In the same method as in Example 32, an extractant in which DEHPA was diluted to 1.2 M with kerosene and an extractant in which DEHPA was diluted to 1.5 M with kerosene were used, and the other conditions were the same as in Example 32.
- the rare earth elements were transferred to the back-extracted water phase and separated and recovered. Table 9 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Examples 35 and 36 In the same method as in Example 32, the pH of the leachate was once set to 3.0, the deposited precipitate was removed, the pH was adjusted again to 1.5 or 2.0, and other conditions were performed. As in Example 32, the rare earth element was transferred to the back-extracted aqueous phase, separated and recovered. Table 9 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Example 37 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a solvent extraction method.
- this solvent extraction method first, the pH of the leachate was once set to 3.0, the deposited precipitate was removed, the pH was adjusted to 2.0 again, and PC88A was diluted to 0.8 M with kerosene.
- the extractant Using the extractant, the leachate and the extractant were stirred and brought into contact with each other at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation to separate the extracted organic phase and the extracted aqueous phase.
- the extracted organic phase 6N-hydrochloric acid aqueous solution was used as a back extractant, and the extracted organic phase and the back extractant were stirred and contacted at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation and back extraction.
- the organic phase and the back-extracted aqueous phase were separated, and the rare earth element was transferred from the extracted organic phase to the back-extracted aqueous phase to be separated and recovered.
- the organic phase after back extraction is made by using 0.02N-hydrochloric acid aqueous solution as a back extractant, contacting the organic phase after back extraction with the back extractant at a liquid ratio of 1: 1 for 3 minutes, and then separating the liquid and liquid. If purified, it can be reused cyclically as an extractant in which PC88A is diluted to a concentration of 0.8M with kerosene. Table 9 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Examples 38 to 40 In the same method as in Example 37, an extractant obtained by diluting PC88A with kerosene to a concentration of 0.5 to 1.5 M was used, and other conditions were the same as in Example 37, and the rare earth element was transferred to the back-extracted aqueous phase. Separated and recovered. Table 9 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Example 41 to 43 In the same method as in Example 37, the pH of the leachate was once set to 3.0, the deposited precipitate was removed, the pH was adjusted again to 1.5 to 3.0, and other conditions were performed. As in Example 37, the rare earth element was separated into the back-extracted aqueous phase and recovered. Table 9 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- DEHPA has a higher rare earth element recovery rate than PC88A, a lower Al recovery rate, and the extractant is DEHPA.
- the Sc recovery rate is 0%, and in order to recover Sc, it is necessary to perform pre-extraction described later.
- Examples 44 to 49 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a solvent extraction method including pre-extraction. In this method, first, the pH of the leachate is once set to 3.0, the deposited precipitate is removed, the pH is adjusted to 1.0 or 1.25 again, and then PC88A is made 0.01 to 0 with kerosene. Using a pre-extractant diluted to a concentration of 0.02 M, the leachate and the pre-extractant were brought into contact with stirring at a liquid ratio of 1: 1 for 3 minutes, and then liquid-liquid separation was performed. Separated.
- the pre-extracted organic phase 1M-sodium carbonate aqueous solution was used as the back extractant, and the pre-extracted organic phase and the back extractant were stirred and contacted at a liquid ratio of 1: 1 for 3 minutes, and then liquid-liquid separated again. After back extraction, it was separated into an organic phase and a back extraction water phase, and the rare earth element was transferred from the extraction organic phase to the back extraction water phase to be separated and recovered.
- 6N-hydrochloric acid aqueous solution was used as a back extractant, and the extracted organic phase and the back extractant were stirred and contacted at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation and back extraction.
- the organic phase and the back-extracted aqueous phase were separated, and the rare earth element was transferred from the extracted organic phase to the back-extracted aqueous phase to be separated and recovered.
- the organic phase after back extraction is made by using 0.02N-hydrochloric acid aqueous solution as a back extractant, contacting the organic phase after back extraction with the back extractant at a liquid ratio of 1: 1 for 3 minutes, and then separating the liquid and liquid.
- DEHPA can be reused cyclically as an extractant diluted with kerosene to a concentration of 0.8M.
- Table 10 shows the recovery rates of rare earth elements and impurities recovered from the extracted organic phase by this solvent extraction method, and the recovery rates of rare earth elements and impurities recovered from the pre-extracted organic phase.
- the recovery rates of the rare earth elements and impurities of Examples 44 to 49 shown in Table 10 the recovery rates of the rare earth elements other than Sc are kept substantially the same as in Example 32, but the impurities It can be seen that the recovery rate of Ca and Ti is significantly reduced.
- the recovery rate of rare earth elements and impurities from the pre-extracted organic phase it can be seen that 90% or more of Sc can be recovered separately from other rare earth elements.
- Examples 50 to 58 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a solvent extraction method.
- this solvent extraction method first, the pH of the leachate was once set to 3.0, the deposited precipitate was removed, the pH was adjusted to 1.0 again, and DEHPA was diluted to 0.8 M with kerosene.
- the extractant Using the extractant, the leachate and the extractant were stirred and brought into contact with each other at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation to separate the extracted organic phase and the extracted aqueous phase.
- the extracted organic phase 50% by mass-sulfuric acid aqueous solution was used as the back extractant, and the extracted organic phase and the back extractant were stirred and contacted at a liquid ratio of 1: 1 to 180 minutes. Since an element containing a rare earth element was precipitated as a solid sulfate, the solid sulfate containing the rare earth element was recovered by solid-liquid separation.
- the organic phase after back extraction is prepared by bringing the organic phase after back extraction and the back extractant into contact with each other at a liquid ratio of 10: 1 by stirring for 3 minutes, followed by liquid-liquid separation.
- DEHPA can be reused cyclically as an extractant diluted with kerosene to a concentration of 0.8M. Table 11 shows the recovery rates of rare earth elements and impurities recovered by this solvent extraction method.
- Example 59 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by a two-stage solvent extraction method shown in FIG.
- a description will be given with reference to FIG.
- the pH of the leachate (1) is adjusted to 2.0, and then an extraction agent obtained by diluting DEHPA to 0.02M with hexane is used in the extraction operation A (Ext. A).
- the leachate (1) and the extractant were agitated and brought into contact with each other at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation to separate the extracted organic phase A (2) and the aqueous phase A (3) after extraction.
- Y and Dy are contained in the extracted organic phase A (2), and rare earth elements up to La-Nd are contained in the aqueous phase A (3) after extraction.
- aqueous phase A (3) after adjusting the pH to 2 and using an extractant obtained by diluting DEHPA to a concentration of 0.8M with hexane, after extraction in extraction operation C (Ext. C)
- the aqueous phase A (3) and the extractant are stirred and brought into contact with each other at a liquid ratio of 1: 1 for 3 minutes, followed by liquid-liquid separation to obtain an extracted organic phase C (12) and an aqueous phase C (13) after extraction.
- aqueous phase C (13 was the waste liquid (14).
- the extracted organic phase C (12) a 0.1N hydrochloric acid aqueous solution was used as a back extractant, and the liquid ratio of the extracted organic phase C (12) and the back extractant was 1 in the back extraction operation C (R-Ext. C). : After stirring for 5 minutes at 1 minute, liquid-liquid separation is performed, and after back extraction, organic phase C (15) and back extracted aqueous phase C (16) are obtained, and Ca is removed from the extracted organic phase C (12). At the same time, the back-extracted aqueous phase C (16) containing Ca was used as the waste liquid (17).
- Example 60 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were carried out by oxalate precipitation.
- the leaching solution of Example 3 was added with about 1.5 times the chemical equivalent of oxalic acid contained in this solution to precipitate only the rare earth element as oxalate, The rare earth element oxalate was recovered by liquid separation.
- Table 12 shows the recovery rate of rare earth elements recovered by this oxalate precipitation method and the concentration of impurities.
- Example 61 Using the leachate having the composition shown in Table 7 obtained in Example 3, removal of impurity elements and concentration of rare earth elements were performed by hydroxide precipitation.
- this hydroxide precipitation method first, the leachate of Example 3 was adjusted to pH 4.5 where the solubility of Al ions and Fe ions was small and the solubility of rare earth element ions was large, and Al and Fe were converted to hydroxides. After removing the precipitated Al and Fe hydroxides by solid-liquid separation, the pH is raised to 11 by adding caustic soda solution to precipitate rare earth ions as hydroxides, and solid-liquid separation Thus, the rare earth element hydroxide was recovered. Table 12 shows the recovery rate of rare earth elements and the concentration of impurities by this hydroxide precipitation method.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Geology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Environmental & Geological Engineering (AREA)
- Mechanical Engineering (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Metallurgy (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Inorganic Chemistry (AREA)
- Geochemistry & Mineralogy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Analytical Chemistry (AREA)
- Manufacture And Refinement Of Metals (AREA)
- Compounds Of Alkaline-Earth Elements, Aluminum Or Rare-Earth Metals (AREA)
- Treatment Of Sludge (AREA)
Abstract
Description

先ず、比表面積26m2/g以上のボーキサイトを原料とし、かつ、処理温度160℃未満の処理条件で実施したバイヤー工程から比表面積35m2/g以上の微細粒子を主とする原料ボーキサイト残渣を回収するか、又は、処理温度230℃の処理条件で実施したバイヤー工程からCaO含有量が4質量%未満であって、かつ、比表面積35m2/g以上の微細粒子を主とする原料ボーキサイト残渣を回収するか、若しくは、比較的高比表面積の微細粒子と比較的低比表面積の粗大粒子とが混在して比表面積が35m2/g未満であるボーキサイト残渣から、分画処理により選択的に微細粒子を主とする比表面積35m2/g以上の高比表面積画分の原料ボーキサイト残渣を回収する。
溶媒抽出法による粗回収物の回収を2段階以上に亘って実施することも好ましい。2段階以上に亘る溶媒抽出法による粗回収物の回収によれば、希土類元素の各元素への分離も可能となる。
逆抽出剤として2N~8Nの塩酸水溶液を用いる場合、逆抽出時間は、5分間以下とすることが好ましく、0.5~3分間とすることがさらに好ましい。逆抽出時間を0.5~3分間とすると、Alの抽出率を低く保つことができ、結果的に分離回収する希土類元素の濃度を高くすることができる。逆抽出時間が5分間を超えるとAlの抽出率が高くなり、結果的に分離回収する希土類元素の濃度が低下する。
使用済みの抽出剤については、2N~8Nの塩酸水溶液又はアルカリ水溶液を逆抽出剤とする逆抽出を行うことにより、前記使用済みの抽出剤中に蓄積したSc、Ti、Thを低減させ、再生抽出剤として再利用することができる。
このような溶媒抽出法による分離は、向流多段溶媒抽出法によることが好適である。
ボールミルを用いてボーキサイトを粉砕し、得られた表2に示す比表面積24~35m2/gのボーキサイトを用い、また、表2に示す処理温度105~250℃及びCaO添加量0.0~3.5質量%の条件でバイヤー工程を実施し、このバイヤー工程からボーキサイト残渣を回収した。その後、回収されたボーキサイト残渣の一部のものについてはそのまま原料ボーキサイト残渣とし、また、他のものについては、ボーキサイト残渣100重量部に対して水500重量部を添加し、混合して一旦スラリーを調製し、次いで篩目38μmの篩を用いて水中で分級し、篩目38μmの篩上と篩下とに分画して篩下の高比表面積画分を原料ボーキサイト残渣とした。
結果を表2に示す。
なお、表2中にはバイヤー工程の条件及び分画処理が同一ながら比表面積の異なるデータが存在するが、これはボーキサイトが異なることに由来するものである。

試料No.1の原料ボーキサイト残渣について、その100重量部に対して水500重量部を添加し、混合して一旦スラリーを調製し、次いで篩目38μm又は300μmの篩を用いて水中で分級し、篩目38μmの篩上及び篩下と篩目300μmの篩上及び篩下とに分画し、各分画処理後の篩上及び篩下についてそれぞれ比表面積と希土類元素含有量の測定を行った。
結果を表3に示す。

先ず、原料ボーキサイト残渣として試料No.1のものを用い、また、浸出処理液として濃度2Nの硫酸水溶液を用い、表4に示すスラリー固液比(L/S)、スラリーpH(初期)、処理温度(℃)及び保持時間(分)の条件で希土類元素を回収するための浸出処理を実施し、次いで、浸出処理が終了した後、スラリーを濾過により固液分離し、浸出液を回収した。ここで、スラリー固液比及び浸出率の計算においては、110℃及び2時間の乾燥条件で乾燥させて得られた乾燥物の質量を測定し、当該質量を原料ボーキサイト残渣の固体重量(S)とした。
これらの結果を表4に示す。

表5に示す試料No.の原料ボーキサイト残渣を用い、また、表5に示す浸出処理液を用い、表5に示すスラリー固液比(L/S)、スラリーpH(初期)、処理温度(℃)及び保持時間(分)の条件で希土類元素を回収するための浸出処理を実施した以外は、上記実施例1~11の場合と同様にして浸出液を回収し、得られた各実施例12~17及び比較例2~6の浸出液について、Scと、Yと、ランタノイドであるLa、Nd及びDyと、不純物であるAl、Fe、Ca、Si及びTiとについて、その元素含有量を測定し、各元素の浸出率を求めた。
結果を表5に示す。

表6に示す試料No.の原料ボーキサイト残渣を用い、また、表6に示す浸出処理液を用い、表5に示すスラリー固液比(L/S)、スラリーpH(初期)、処理温度(℃)及び保持時間(分)の条件で希土類元素を回収するための浸出処理を実施した以外は、上記実施例1~17及び比較例2~6の場合と同様にして回収した浸出液の、Scと、Yと、ランタノイドであるNd及びDyと、不純物であるAl、Fe、Ca、Si及びTiとについて、その元素含有量を測定し、各元素の浸出率を求めた。
結果を表6に示す。
実施例18と同一の方法において、浸出処理後にボーキサイト残渣により中和してから回収した浸出液の、Scと、Yと、ランタノイドであるNd及びDyと、不純物であるAl、Fe、Ca、Si及びTiとについて、その元素含有量を測定し、各元素の浸出率を求めた。
結果を表6に示す。

実施例3で得られた表7に示す組成の浸出液を用い、溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。この溶媒抽出法では、先ず、浸出液のpHを一旦3.0とし、析出した析出物を除去してから、1.5に調整し、その後、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤を用い、浸出液と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相と抽出終了後の水相(抽出後水相)とに分離した。

逆抽出後有機相は、0.02N-塩酸水溶液を逆抽出剤として、逆抽出後有機相と逆抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて精製すれば、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤として循環的に再利用することができる。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表8に示す。
実施例20と同一の方法において、浸出液と抽出剤との接触時間を0.5分、1分、5分及び10分とし、他の条件は実施例20と同一として、希土類元素を逆抽出水相へと移行させて分離し、回収した。
この溶媒抽出法で回収された希土類元素の回収率と不純物の濃度を表8に示す。
実施例20と同一の方法において、抽出有機相と逆抽出剤との接触時間を0.5分、1分、5分、10分及び15分とし、他の条件は実施例20と同一として、希土類元素を逆抽出水相へと移行させて分離し、回収した。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表8に示す。
実施例3で得られた表7に示す組成の浸出液を用い、溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。この溶媒抽出法では、先ず、浸出液のpHを一旦1.75とした後、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤を用い、浸出液と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相と抽出後水相とに分離した。溶媒抽出時に有機相と水相の中間に乳濁が生成したが、該乳濁は、液液分離時に有機相側に分離し、その後に有機相をフィルターで濾過して除去した。
逆抽出後有機相は、0.02N-塩酸水溶液を逆抽出剤として、逆抽出後有機相と逆抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて精製すれば、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤として循環的に再利用することができる。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表8に示す。
水酸化ナトリウム水溶液の添加に代えて、実施例4に用いたものと同じボーキサイト残渣の添加によりpH調整をした以外、他の実施方法及び条件は実施例20と同一として、希土類元素を逆抽出水相へと移行させて分離し、回収した。このとき、添加したボーキサイト残渣の量は、浸出原料であったボーキサイト残渣0.1kgに対し、0.115kgであった。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表8に示す。ただし回収率の計算にあたっては、pH調整に使用したボーキサイト残渣中に含有されていた希土類元素を考慮し、浸出原料であったボーキサイト残渣の2.15倍の量に対する回収率を示している。

実施例30によれば、溶媒抽出時に有機相と水相の中間に乳濁が生成した場合には、抽出時間及び逆抽出時間が同一である実施例20と比較して、希土類元素の回収率は、わずかに低いことがわかる。
実施例3で得られた表7に示す組成の浸出液を用い、溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。この溶媒抽出法では、先ず、浸出液のpHを一旦3.0として、析出した析出物を除去してから、1.0に調整した後、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤を用い、浸出液と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相と抽出後水相とに分離した。
逆抽出後有機相は、0.02N-塩酸水溶液を逆抽出剤として、逆抽出後有機相と逆抽出剤とを液比1:1で3分間撹拌した後、液液分離させて精製すれば、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤として循環的に再利用することができる。
この溶媒抽出法で回収された希土類元素の回収率と不純物の濃度を表9に示す。
実施例32と同一の方法において、ケロシンでDEHPAを1.2M濃度に希釈した抽出剤、及びケロシンでDEHPAを1.5M濃度に希釈した抽出剤を用い、他の条件は実施例32と同一として、希土類元素を逆抽出水相へと移行させて分離し、回収した。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表9に示す。
実施例32と同一の方法において、浸出液のpHを一旦3.0として、析出した析出物を除去してから、再びpHの調整を行って1.5又は2.0とし、他の条件は実施例32と同一として、希土類元素を逆抽出水相へと移行させて分離し、回収した。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表9に示す。
実施例3で得られた表7に示す組成の浸出液を用い、溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。この溶媒抽出法では、先ず、浸出液のpHを一旦3.0とし、析出した析出物を除去してから、再びpHを2.0に調整した後、ケロシンでPC88Aを0.8M濃度に希釈した抽出剤を用い、浸出液と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相と抽出後水相とに分離した。
逆抽出後有機相は、0.02N-塩酸水溶液を逆抽出剤として、逆抽出後有機相と逆抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて精製すれば、ケロシンでPC88Aを0.8M濃度に希釈した抽出剤として循環的に再利用することができる。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表9に示す。
実施例37と同一の方法において、ケロシンでPC88Aを0.5~1.5M濃度に希釈した抽出剤を用い、他の条件は実施例37と同一として、希土類元素を逆抽出水相へと移行させて分離し、回収した。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表9に示す。
実施例37と同一の方法において、浸出液のpHを一旦3.0として、析出した析出物を除去してから、再びpHの調整を行って1.5~3.0とし、他の条件は実施例37と同一として、希土類元素を逆抽出水相へと分離、回収した。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表9に示す。

実施例3で得られた表7に示す組成の浸出液を用い、前抽出を含む溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。この方法では、先ず、浸出液のpHを一旦3.0として、析出した析出物を除去してから、再びpHを1.0又は1.25に調整した後、ケロシンでPC88Aを0.01~0.02M濃度に希釈した前抽出剤を用い、浸出液と前抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて前抽出有機相と抽出後水相とに分離した。続いて、回収された抽出後水相について、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤を用い、抽出後水相と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相と抽出後水相とに分離した。
抽出有機相については、6N-塩酸水溶液を逆抽出剤として、抽出有機相と逆抽出剤とを液比1:1で3分間撹拌して接触させた後、再び液液分離させて逆抽出後有機相と逆抽出水相とに分離し、希土類元素を抽出有機相から逆抽出水相へと移行させて分離し、回収した。
逆抽出後有機相は、0.02N-塩酸水溶液を逆抽出剤として、逆抽出後有機相と逆抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて精製すれば、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤として循環的に再利用することができる。
この溶媒抽出法で抽出有機相から回収された希土類元素と不純物の回収率、及び前抽出有機相から回収された希土類元素と不純物の回収率を表10に示す。

実施例3で得られた表7に示す組成の浸出液を用い、溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。この溶媒抽出法では、先ず、浸出液のpHを一旦3.0として、析出した析出物を除去してから、再びpHを1.0に調整した後、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤を用い、浸出液と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相と抽出後水相とに分離した。
逆抽出後有機相は、0.02N-塩酸水溶液を逆抽出剤として、逆抽出後有機相と逆抽出剤とを液比10:1で3分間撹拌して接触させた後、液液分離させて精製すれば、ケロシンでDEHPAを0.8M濃度に希釈した抽出剤として循環的に再利用することができる。
この溶媒抽出法で回収された希土類元素と不純物の回収率を表11に示す。

実施例3で得られた表7に示す組成の浸出液を用い、図4に示す2段階の溶媒抽出法により不純物元素の除去と希土類元素の濃縮を実施した。以下、図4を参照しながら説明する。
この2段階溶媒抽出法では、先ず、浸出液(1)のpHを2.0に調整した後、ヘキサンでDEHPAを0.02M濃度に希釈した抽出剤を用い、抽出操作A(Ext.A)において浸出液(1)と抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて抽出有機相A(2)と抽出後水相A(3)とに分離した。
このとき、YとDyは抽出有機相A(2)に、La-Ndまでの希土類元素は抽出後水相A(3)に、それぞれ含有される。
逆抽出後有機相A(4)については、2N-塩酸水溶液を逆抽出剤として、精製操作(P)において逆抽出後有機相A(4)と逆抽出剤とを液比1:1で3分間撹拌して接触させた後、液液分離させて精製すれば、後述のScの逆抽出工程を経て、ヘキサンでDEHPAを0.02M濃度に希釈した抽出剤として循環的に再利用され、また、使用済みの逆抽出剤は廃液(W)として廃棄される。
逆抽出後有機相B(9)は、図示外の上記精製操作(P)と同様にして、ヘキサンでDEHPAを0.02M濃度に希釈した抽出剤として循環的に再利用される。
逆抽出後有機相D(18)は、図示外の上記精製操作(P)と同様にして、ヘキサンでDEHPAを0.8M濃度に希釈した抽出剤として循環的に再利用される。
この2段階溶媒抽出法で回収された希土類元素の回収率と不純物の濃度を表12に示す。
実施例3で得られた表7に示す組成の浸出液を用い、蓚酸塩析出法により不純物元素の除去と希土類元素の濃縮を実施した。この蓚酸塩析出法においては、実施例3の浸出液について、この液中に含まれる希土類元素イオンの約1.5倍の化学等量の蓚酸を加えて希土類元素のみを蓚酸塩として沈殿させ、固液分離して希土類元素蓚酸塩を回収した。
この蓚酸塩析出法で回収された希土類元素の回収率と不純物の濃度を表12に示す。
実施例3で得られた表7に示す組成の浸出液を用い、水酸化物析出法により不純物元素の除去と希土類元素の濃縮を実施した。この水酸化物析出法においては、先ず、実施例3の浸出液について、AlイオンとFeイオンの溶解度が小さく、希土類元素イオンの溶解度が大きなpH4.5に調整して、AlとFeを水酸化物として沈殿させ、固液分離して沈殿したAl及びFeの水酸化物を除去した後、更に苛性ソーダ液を加えてpHを11まで上昇させ、希土類元素イオンを水酸化物として沈殿させ、固液分離して希土類元素水酸化物を回収した。
この水酸化物析出法による希土類元素の回収率と不純物の濃度を表12に示す。

Claims (30)
- ボーキサイトからアルミニウム成分を分離し採取するバイヤー工程で副生するボーキサイト残渣を原料とし、この原料から希土類元素を回収する希土類元素の回収方法であり、
前記原料として比表面積35m2/g以上のボーキサイト残渣を用い、
この原料ボーキサイト残渣に硫酸、塩酸、硝酸、及び亜硫酸から選ばれた1種又は2種以上の鉱酸の水溶液からなる浸出処理液を添加し、固液比2~30及びpH0.5~2.2のスラリーを調製し、室温~160℃の温度条件で希土類元素の浸出処理を行い、
次いで、浸出処理後のスラリーを固液分離して得られた浸出液から希土類元素を分離回収することを特徴とする希土類元素の回収方法。 - 前記原料ボーキサイト残渣が、比表面積26m2/g以上のボーキサイト粉末を原料とし、処理温度160℃以下の条件で行ったバイヤー工程から得られたボーキサイト残渣である請求項1記載の希土類元素の回収方法。
- 前記原料ボーキサイト残渣が、比表面積26m2/g以上のボーキサイト粉末を原料とし、処理温度230℃未満の条件で行ったバイヤー工程から得られたボーキサイト残渣であり、かつ、CaO含有量が4質量%未満である請求項1記載の希土類元素の回収方法。
- 前記原料ボーキサイト残渣が、ボーキサイト残渣を分画処理して得られた微細粒子を主とする高比表面積画分である請求項1~3のいずれかに記載の希土類元素の回収方法。
- 前記分画処理して得られた高比表面積画分が、ボーキサイト残渣を篩目38~400μmの大きさの篩で分級し、篩上粒子を除去して得られたボーキサイト残渣である請求項4に記載の希土類元素の回収方法。
- 前記浸出処理に先駆けて、ボーキサイト残渣中のFe成分に対して0.1~0.3当量の割合で酸化剤を添加する請求項1~5のいずれかに記載の希土類元素の回収方法。
- 前記酸化剤が、30質量%-過酸化水素水又は70質量%-硝酸水溶液である請求項6記載の希土類元素の回収方法。
- 前記浸出処理を終えたスラリーにpH調整剤を添加してpH2.5~6に調整し、このpH調整後のスラリーを固液分離して得られた浸出液から希土類元素を分離回収する請求項1~7のいずれかに記載の希土類元素の回収方法。
- 前記pH調整剤として、原料ボーキサイト残渣を用いる請求項8に記載の希土類元素の回収方法。
- 前記浸出液からの希土類元素の分離回収に先駆けて、浸出液にpH調整剤を添加してpH4~6に調整し、このpH調整で析出したFe及びAlの水酸化物を固液分離して除去して得られた液から希土類元素を分離回収する請求項1~9のいずれかに記載の希土類元素の回収方法。
- 前記浸出液にpH調整剤を添加してpH4~6に調整するpH調整の際に、過酸化水素、過塩素酸、過マンガン酸、及び次亜塩素酸から選ばれた酸化剤を添加し、抽出液中のFe2+イオンを酸化してFe3+イオンにする請求項10に記載の希土類元素の回収方法。
- 前記希土類元素の分離回収においては、前記固液分離処理で得られた浸出液、又は、該浸出液をpH調整してFe及びAlを水酸化物として沈殿させ、固液分離して得られた液に、pH調整剤を添加してpH7以上に調整し、このpH調整で析出したCa及び希土類元素の水酸化物を固液分離して粗回収物として回収する請求項1~11のいずれかに記載の希土類元素の回収方法。
- 前記希土類元素の分離回収においては、前記固液分離処理で得られた浸出液、又は、該浸出液をpH調整してFe及びAlを水酸化物として沈殿させ、固液分離して得られた液に存在する希土類元素の化学当量以上の蓚酸を加えて希土類元素を蓚酸塩として析出させ、更に該蓚酸塩を固液分離して前記希土類元素を粗回収物として回収する、請求項1~11のいずれかに記載の希土類元素の回収方法。
- 前記希土類元素の分離回収においては、前記固液分離処理で得られた浸出液、又は、該浸出液をpH調整してFe及びAlを水酸化物として沈殿させ、固液分離して得られた液に、リン酸エステル類、ホスホン酸エステル類、ホスフィン酸エステル類、チオホスフィン酸エステル類、及びこれらのエステル類とリン酸トリブチル及び/又はトリオクチルホスフィンオキサイドとの混合物から選ばれたエステル類を、ヘキサンなどの脂肪族炭化水素、ベンゼン、トルエンなどの芳香族炭化水素、オクタノールなどのアルコール、及び石油分留物であるケロシンから選ばれた溶媒で希釈して得られた抽出剤を加えて溶媒抽出法により希土類元素を粗回収物として分離、回収する、請求項1~11のいずれかに記載の希土類元素の回収方法。
- 前記溶媒抽出法による分離回収に先駆けて、浸出液のpH調整時に発生した乳濁を予め濾過により除去する、請求項14に記載の希土類元素の回収方法。
- 前記溶媒抽出法による分離回収に先駆けて、浸出液をpH2.5~3.5に調整し、析出した析出物を除去する、請求項14に記載の希土類元素の回収方法。
- 前記溶媒抽出法による分離回収に先駆けて行うpH調整は、ボーキサイト残渣の添加により行われる、請求項16に記載の希土類元素の回収方法。
- 前記溶媒抽出法の抽出剤がDEHPAである、請求項14~17のいずれかに記載の希土類元素の回収方法。
- 前記溶媒抽出法の抽出剤としてのDEHPAの濃度が0.1~1.5Mである、請求項18に記載の希土類元素の回収方法。
- 前記溶媒抽出法の抽出時間が5分間以下である、請求項14~19のいずれかに記載の希土類元素の回収方法。
- 前記溶媒抽出法の抽出時間が0.5~3分間である、請求項20に記載の希土類元素の回収方法。
- 前記DEHPAを抽出剤として用いる溶媒抽出法に先駆けて、PC88A、リン酸トリブチル又はナフテン酸を前抽出剤として用いる浸出液の前抽出を行い、この浸出液からFe、Sc及びTiを分離する、請求項18~21のいずれかに記載の希土類元素の回収方法。
- 前記溶媒抽出法は、逆抽出剤が2N~8Nの塩酸水溶液であるとともに、逆抽出時間が5分間以下である、請求項14~22のいずれかに記載の希土類元素の回収方法。
- 前記溶媒抽出法の逆抽出時間が0.5~3分間である、請求項23に記載の希土類元素の回収方法。
- 前記溶媒抽出法で用いる逆抽出剤が濃度30~70質量%の硫酸水溶液であり、希土類元素を固体硫酸塩として回収する、請求項14~22のいずれかに記載の希土類元素の回収方法。
- 前記溶媒抽出法の逆抽出時間が5分間以下である、請求項25に記載の希土類元素の回収方法。
- 前記溶媒抽出法において、使用済みの抽出剤に対して、2N~8Nの塩酸水溶液又はアルカリ水溶液を逆抽出剤とする逆抽出を行い、前記使用済みの抽出剤中に蓄積したSc、Ti、Thを低減させ、再生抽出剤として再利用する、請求項14~26のいずれかに記載の希土類元素の回収方法。
- 前記粗回収物から各元素への分離は、粗回収物を酸水溶液に溶解し、次いでリン酸エステル類、ホスホン酸エステル類、ホスフィン酸エステル類、チオホスフィン酸エステル類、及びこれらのエステル類とリン酸トリブチル(tributyl phosphate)及び/又はトリオクチルホスフィンオキサイド(trioctylphosphine oxide)との混合物から選ばれたエステル類を、ヘキサンなどの脂肪族炭化水素、ベンゼン、トルエンなどの芳香族炭化水素、及び石油分留物であるケロシンから選ばれた溶媒で希釈して得られた抽出剤を用いる溶媒抽出法で行なう請求項14~27のいずれかに記載の希土類元素の回収方法。
- 前記粗回収物から各元素への分離の溶媒抽出法が向流多段溶媒抽出法である請求項28に記載の希土類元素の回収方法。
- 前記原料ボーキサイト残渣は、110℃及び2時間の乾燥条件で乾燥して得られた固体成分中に希土類元素のSc、Y、及びランタノイドをその酸化物として合計で1500~10000ppmの割合で含有する請求項1~29のいずれかに記載の希土類元素の回収方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014507326A JP5598631B2 (ja) | 2012-03-30 | 2012-12-07 | 希土類元素の回収方法 |
| CA2868962A CA2868962C (en) | 2012-03-30 | 2012-12-07 | Method of recovering rare-earth elements |
| US14/389,304 US9228248B2 (en) | 2012-03-30 | 2012-12-07 | Method of recovering rare-earth elements |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-081228 | 2012-03-30 | ||
| JP2012081228 | 2012-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013145455A1 true WO2013145455A1 (ja) | 2013-10-03 |
Family
ID=49258788
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/081855 WO2013145455A1 (ja) | 2012-03-30 | 2012-12-07 | 希土類元素の回収方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9228248B2 (ja) |
| JP (1) | JP5598631B2 (ja) |
| CA (1) | CA2868962C (ja) |
| WO (1) | WO2013145455A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103290235A (zh) * | 2013-06-26 | 2013-09-11 | 中国地质科学院矿产综合利用研究所 | 一种含锶稀土矿的综合利用工艺 |
| JP2015098621A (ja) * | 2013-11-18 | 2015-05-28 | Jx日鉱日石金属株式会社 | 有機溶媒中のリン酸エステル系抽出剤の除去方法 |
| JP5889455B1 (ja) * | 2015-02-27 | 2016-03-22 | 日本軽金属株式会社 | 希土類元素の回収方法 |
| JP2016108583A (ja) * | 2014-12-03 | 2016-06-20 | Dowaメタルマイン株式会社 | 貴金属製錬スラグからの希土類元素回収方法 |
| JP2017061710A (ja) * | 2015-09-23 | 2017-03-30 | 三菱マテリアル株式会社 | 希土類元素の回収方法 |
| JP2018150588A (ja) * | 2017-03-13 | 2018-09-27 | Jx金属株式会社 | レアメタル回収方法 |
| WO2019022320A1 (ko) * | 2017-07-24 | 2019-01-31 | 한국전력공사 | 석탄회로부터 희소 금속 원소 추출 방법 및 희소 금속 원소 추출 장치 |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI568672B (zh) * | 2016-01-12 | 2017-02-01 | 光宇材料股份有限公司 | 廢矽泥的使用方法及其所得產品 |
| JP6641593B2 (ja) * | 2016-01-12 | 2020-02-05 | 三菱マテリアル株式会社 | 希土類元素と鉄の分離方法 |
| EP3386932B1 (de) * | 2016-03-15 | 2020-09-30 | Fluorchemie GmbH Frankfurt | Zusammensetzung enthaltend modifizierten, chromatarmen rotschlamm sowie verfahren zu deren herstellung |
| MA44488A (fr) * | 2016-03-25 | 2019-01-30 | Fakon Vallalkozasi Kft | Procédé de traitement de boue rouge et de production de sels de métal de terres rares |
| SE539918C2 (en) * | 2016-05-17 | 2018-01-16 | Separation of rare earth elements from other elements | |
| IT201600101809A1 (it) * | 2016-10-11 | 2018-04-11 | Ecotec Gestione Impianti S R L | Procedimento per la preparazione di un concentrato contenente metalli, metalli rari e terre rare da residui generati nella filiera di produzione dell’allumina tramite il processo Bayer, o da materiali di composizione chimica similare, e affinamento del concentrato così ottenibile. |
| US10669610B2 (en) | 2017-03-17 | 2020-06-02 | University Of North Dakota | Rare earth element extraction from coal |
| US11155897B2 (en) | 2017-11-09 | 2021-10-26 | University Of Kentucky Research Foundation | Low-cost selective precipitation circuit for recovery of rare earth elements from acid leachate of coal waste |
| KR102091804B1 (ko) * | 2018-03-16 | 2020-03-20 | 한국화학연구원 | 폐 re:yag 결정으로부터 희토류 성분의 회수방법 |
| KR102067606B1 (ko) * | 2018-12-20 | 2020-01-17 | 전남대학교산학협력단 | 폐 모바일폰 카메라모듈 침출액에 함유된 디스프로슘 및 네오디뮴의 선택적 분리 및 회수방법 |
| EP4013726A4 (en) | 2019-08-13 | 2023-09-06 | California Institute of Technology | PROCESS FOR MANUFACTURING CALCIUM OXIDE OR ORDINARY PORTLAND CEMENT FROM CALCIUM-CONTAINING ROCKS AND MINERALS |
| US10988386B1 (en) | 2020-05-27 | 2021-04-27 | Winston-Salem State University | Separation and purification of rare-earth elements by chemical reduction in aqueous solutions |
| CN111850296B (zh) * | 2020-07-16 | 2022-02-25 | 乐山盛和稀土股份有限公司 | 稀土矿中回收制备高纯锶化物的方法 |
| US11913090B2 (en) * | 2020-11-12 | 2024-02-27 | University Of North Dakota | Method for leaching rare earth elements and critical minerals from organically associated materials |
| US11447397B1 (en) | 2021-03-19 | 2022-09-20 | Lynas Rare Earths Limited | Materials, methods and techniques for generating rare earth carbonates |
| CN113104888B (zh) * | 2021-03-31 | 2023-06-06 | 河南科技大学 | 一种赤泥的全回收利用方法 |
| CN114166600B (zh) * | 2021-12-02 | 2024-03-26 | 中国科学院过程工程研究所 | 一种icp检测固体废弃物中重金属的制样方法 |
| CN115466858B (zh) * | 2022-09-01 | 2023-08-22 | 江苏南方永磁科技有限公司 | 一种氧化铕两级萃取生产方法 |
| CN117025985B (zh) * | 2023-08-15 | 2024-04-05 | 中国科学院大学 | 一种利用嗜酸嗜热红藻从赤泥中回收稀土元素的方法及应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000313928A (ja) * | 1999-04-26 | 2000-11-14 | Taiheiyo Kinzoku Kk | 酸化鉱石からニッケルとスカンジウムを回収する方法 |
| JP2005120420A (ja) * | 2003-10-16 | 2005-05-12 | Jfe Engineering Kk | ボーキサイト溶解残渣の処理方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2835554A (en) * | 1954-12-23 | 1958-05-20 | Gulf Research Development Co | Utilization of anhydrous aluminum chloride sludge |
| JPS5946008A (ja) | 1982-08-21 | 1984-03-15 | Sumitomo Special Metals Co Ltd | 永久磁石 |
| JPS62165305A (ja) | 1986-01-16 | 1987-07-21 | Hitachi Metals Ltd | 熱安定性良好な永久磁石およびその製造方法 |
| US5030424A (en) | 1989-04-03 | 1991-07-09 | Alcan International Limited | Recovery of rare earth elements from Bayer process red mud |
| BR9605956A (pt) | 1995-12-13 | 1998-08-18 | Cytec Tech Corp | Processo para recuperar um elemento de terra rara a partir de uma soluçao ácida |
| US5622679A (en) | 1995-12-13 | 1997-04-22 | Cytec Technology Corp. | Extraction of rare earth elements using alkyl phosphinic acid or salt/tetraalkylammonium salt as extractant |
| BRPI0417199A (pt) * | 2003-12-24 | 2007-03-06 | Mt Aspiring Geochemistry Consu | material particulado poroso para tratamento de fluido, composição cimentosa e método de manufatura deste |
-
2012
- 2012-12-07 WO PCT/JP2012/081855 patent/WO2013145455A1/ja active Application Filing
- 2012-12-07 US US14/389,304 patent/US9228248B2/en not_active Expired - Fee Related
- 2012-12-07 JP JP2014507326A patent/JP5598631B2/ja active Active
- 2012-12-07 CA CA2868962A patent/CA2868962C/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000313928A (ja) * | 1999-04-26 | 2000-11-14 | Taiheiyo Kinzoku Kk | 酸化鉱石からニッケルとスカンジウムを回収する方法 |
| JP2005120420A (ja) * | 2003-10-16 | 2005-05-12 | Jfe Engineering Kk | ボーキサイト溶解残渣の処理方法 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103290235A (zh) * | 2013-06-26 | 2013-09-11 | 中国地质科学院矿产综合利用研究所 | 一种含锶稀土矿的综合利用工艺 |
| CN103290235B (zh) * | 2013-06-26 | 2014-07-09 | 中国地质科学院矿产综合利用研究所 | 一种含锶稀土矿的综合利用工艺 |
| JP2015098621A (ja) * | 2013-11-18 | 2015-05-28 | Jx日鉱日石金属株式会社 | 有機溶媒中のリン酸エステル系抽出剤の除去方法 |
| JP2016108583A (ja) * | 2014-12-03 | 2016-06-20 | Dowaメタルマイン株式会社 | 貴金属製錬スラグからの希土類元素回収方法 |
| JP5889455B1 (ja) * | 2015-02-27 | 2016-03-22 | 日本軽金属株式会社 | 希土類元素の回収方法 |
| US10246759B2 (en) | 2015-02-27 | 2019-04-02 | Nippon Light Metal Company, Ltd. | Method of recovering rare-earth elements |
| JP2017061710A (ja) * | 2015-09-23 | 2017-03-30 | 三菱マテリアル株式会社 | 希土類元素の回収方法 |
| JP2018150588A (ja) * | 2017-03-13 | 2018-09-27 | Jx金属株式会社 | レアメタル回収方法 |
| WO2019022320A1 (ko) * | 2017-07-24 | 2019-01-31 | 한국전력공사 | 석탄회로부터 희소 금속 원소 추출 방법 및 희소 금속 원소 추출 장치 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5598631B2 (ja) | 2014-10-01 |
| CA2868962A1 (en) | 2013-10-03 |
| JPWO2013145455A1 (ja) | 2015-12-10 |
| US9228248B2 (en) | 2016-01-05 |
| US20150086449A1 (en) | 2015-03-26 |
| CA2868962C (en) | 2018-02-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5598631B2 (ja) | 希土類元素の回収方法 | |
| JP5545418B2 (ja) | 希土類元素の回収方法 | |
| Jorjani et al. | The production of rare earth elements group via tributyl phosphate extraction and precipitation stripping using oxalic acid | |
| RU2595178C2 (ru) | Способ извлечения редкоземельных элементов и редких металлов | |
| EP3060690B1 (en) | Deriving high value products from waste red mud | |
| US9945009B2 (en) | Processes for recovering rare earth elements from aluminum-bearing materials | |
| JP5894262B2 (ja) | 種々の鉱石から希土類元素を回収する方法 | |
| da Silva et al. | Selective removal of impurities from rare earth sulphuric liquor using different reagents | |
| JPH02277730A (ja) | 希土類元素鉱石の処理方法 | |
| WO2017100933A1 (en) | Rare earth ore processing methods by acid mixing, sulphating and decomposing | |
| JP5889455B1 (ja) | 希土類元素の回収方法 | |
| EP3526353B1 (en) | Process for the preparation of a concentrate of metals, rare metals and rare earth metals from residues of alumina production by bayer process or from materials with a chemical composition similar to said residues, and refinement of the concentrate so obtained | |
| EP2910654B1 (de) | Verfahren zur Rückgewinnung und gegebenenfalls Trennung von Lanthaniden in Form ihrer Chloride oder Oxide aus mineralischen Abfällen und Reststoffen | |
| US20220403482A1 (en) | Dissolution process | |
| US4964996A (en) | Liquid/liquid extraction of rare earth/cobalt values | |
| US4964997A (en) | Liquid/liquid extraction of rare earth/cobalt values | |
| CN115404360A (zh) | 从废硫酸钒催化剂中选择性回收钒及铯的方法及通过该方法制备的高品质钒水溶液和铯矾 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12873070 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014507326 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2868962 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14389304 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12873070 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |