WO2012133490A1 - 末端に複数の水酸基を有するポリオキシエチレン誘導体 - Google Patents
末端に複数の水酸基を有するポリオキシエチレン誘導体 Download PDFInfo
- Publication number
- WO2012133490A1 WO2012133490A1 PCT/JP2012/058069 JP2012058069W WO2012133490A1 WO 2012133490 A1 WO2012133490 A1 WO 2012133490A1 JP 2012058069 W JP2012058069 W JP 2012058069W WO 2012133490 A1 WO2012133490 A1 WO 2012133490A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- bond
- compound
- reaction
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
- C08G65/2603—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen
- C08G65/2606—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds the other compounds containing oxygen containing hydroxyl groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/333—Polymers modified by chemical after-treatment with organic compounds containing nitrogen
- C08G65/33331—Polymers modified by chemical after-treatment with organic compounds containing nitrogen containing imide group
- C08G65/33337—Polymers modified by chemical after-treatment with organic compounds containing nitrogen containing imide group cyclic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/02—Applications for biomedical use
Definitions
- the present invention relates to a polyoxyethylene derivative having a plurality of hydroxyl groups at its terminals, which is used for modifying biological materials.
- Non-patent Documents 1 and 2 ABC (accelerated blood clearance) phenomenon, in which the blood half-life after the second administration is reduced when repeated administration of liposomes and nanoparticles modified with polyoxyethylene to the same individual, compared to the first administration. has been reported to occur. This is thought to be due to the development of antibodies against polyoxyethylene modified with liposomes and nanoparticles, which recognize various sites such as polyoxyethylene chain ends and polyoxyethylene repeat structures. It is said that.
- liposomes and nanoparticles modified with some hydrophilic polymers such as polyglycerin hardly cause ABC phenomenon.
- these hydrophilic polymers other than polyoxyethylene cannot provide sufficient retention in blood, and are rarely used in clinical practice, and are not sufficient as an alternative to polyoxyethylene.
- Patent Document 1 describes a bio-related substance modified with a polyoxyethylene derivative having one hydroxyl group at the terminal.
- a polyoxyethylene derivative having a hydroxyl group at the end is used, data in which antigenicity is lower than that of a polyoxyethylene derivative having an alkoxy group at the end has been obtained.
- arranging a hydroxyl group at the terminal of the polyoxyethylene derivative is considered to be one improvement measure that contributes to a decrease in the antigenicity of the polyoxyethylene.
- the polyoxyethylene derivative described in this document is purified using reverse phase chromatography, the yield is greatly reduced, and industrial production is unsuitable.
- drugs with improved blood retention have been developed, and it is necessary to further reduce antigenicity.
- Patent Documents 2 and 3 describe target-targeting preparations in which monosaccharides or polysaccharides having a plurality of hydroxyl groups are introduced to the ends of hydrophilic polymers and bound to drugs. ing.
- this saccharide is for imparting target directivity by a sugar chain recognition mechanism in vivo, and is not intended to improve antigenicity.
- Patent Documents 4 and 5 describe a hydrophobic polyoxyalkylene having a polyglycerin derivative having a large number of hydroxyl groups. Such hydrophobic polyoxyalkylene is a surfactant that utilizes the hydrophilicity of polyglycerol. In these documents, only examples of hydrophobic polyoxyalkylene are shown, and it is difficult to obtain a high-purity polyoxyethylene derivative suitable for modification of a biological substance by the production method described.
- Patent Document 6 describes a copolymer of polyoxyethylene and polyglycidol.
- polyglycidol becomes a branched polymer having a plurality of branches, and a mixture of polymers having various structures.
- a raw material for pharmaceuticals a high-purity product having a single structure is required, and a mixture is not preferable.
- the mixture is very difficult because it is necessary to clarify the composition ratio of the ingredients in the application for registration of the pharmaceutical raw material.
- it is difficult to control the number of hydroxyl groups in the polymer it is difficult to control the number of hydroxyl groups in the polymer, and there is a problem that the viscosity of the polymer solution increases as the number of hydroxyl groups increases.
- An object of the present invention is to provide a novel polyoxyethylene derivative having a plurality of hydroxyl groups at its terminals. More specifically, an object of the present invention is to provide a polyoxyethylene derivative having a plurality of hydroxyl groups at its terminals, which can be effectively used for modifying bio-related substances and can be industrially produced.
- the present inventors have completed a polyoxyethylene derivative having a plurality of hydroxyl groups at the terminals and having the following constitution.
- the present invention is as follows.
- L 1 , L 2 and L 3 are each independently an alkylene group, a phenylene group or an ester.
- X represents a functional group capable of reacting with a biological substance,
- Y represents a residue of xylitol or boremitol, or 3 to
- Z represents a residue of a compound having 2 to 5 active hydrogens
- b and c are 1 ⁇ b ⁇ 4, 1 ⁇ c ⁇ 4 and 2 ⁇ b + c ⁇ 5, and
- d and e are each independently 0 or 1.
- L 1 , L 2 and L 3 are each independently an alkylene group, a phenylene group or an ester.
- X represents a functional group capable of reacting with a biological substance
- Z has 2 to 5 active hydrogens A residue of the compound, wherein a is 1 or 2.
- b and c are 1 ⁇ b ⁇ 4, 1 ⁇ c ⁇ 4, and 2 ⁇ b + c ⁇ 5, and d and e are 0 or 1.
- Polyoxyethylene derivatives represented by [3] A polyoxyethylene derivative represented by the following formula (3), wherein Z in the formula (2) is an ethylene glycol residue, b is 1, c is 1, d is 0, and e is 1. The polyoxyethylene derivative according to the above [2].
- L 1 and L 3 each independently represent an alkylene group, a phenylene group, an ester bond, an amide bond, an ether bond, a urethane bond, a carbonate bond, a secondary amino group or a bond thereof.
- X represents a functional group capable of reacting with a biological substance, a is 1 or 2, and n1 is 11 to 3650.
- [4] which is a polyoxyethylene derivative represented by the following formula (4), wherein Z in the formula (2) is a glycerin residue, b is 2, c is 1, and d is 0 Polyoxyethylene derivative.
- L 1 and L 3 each independently represent an alkylene group, a phenylene group, an ester bond, an amide bond, an ether bond, a urethane bond, a carbonate bond, a secondary amino group, or a bond thereof.
- X is a living body.
- a functional group capable of reacting with a related substance a being 1 or 2, e being 0 or 1.
- n2 being 11 to 1825.
- X is an active ester group, active carbonate group, aldehyde group, isocyanate group, isothiocyanate group, epoxy group, carboxyl group, thiol group, maleimide group, substituted maleimide group, hydrazide group, dithiol.
- X is an active ester group, an active carbonate group, an aldehyde group, an isocyanate group, an isothiocyanate group, an epoxy group, a carboxyl group, a thiol group, a maleimide group, a substituted maleimide group, a hydrazide group, a dithiol.
- X is an active ester group, an active carbonate group, an aldehyde group, an isocyanate group, an isothiocyanate group, an epoxy group, a carboxyl group, a thiol group, a maleimide group, a substituted maleimide group, a hydrazide group, a dithiol.
- X is an active ester group, active carbonate group, aldehyde group, isocyanate group, isothiocyanate group, epoxy group, carboxyl group, thiol group, maleimide group, substituted maleimide group, hydrazide group, dithiol.
- the polyoxyethylene derivative of the present invention has a plurality of hydroxyl groups (preferably 4 or more hydroxyl groups) at the terminal, when the bio-related substance is modified, it is large due to strong hydrogen bonding around the bio-related substance. A hydration layer will be formed. For this reason, the modified bio-related substance reduces the interaction with opsonin and the cell surface that constitutes each tissue in the body, and the transition to each tissue is reduced, thereby improving the blood half-life. .
- hydrophilic molecule having a plurality of hydroxyl groups derived from sugar alcohols such as xylitol and boremitol or polyglycerin of 3 to 31 mer is bonded to the polyoxyethylene terminal, conventional polyoxyethylene derivatives of this type The expression of the antibody that has recognized the alkoxy group at the terminal of the polyoxyethylene chain can be effectively suppressed. Furthermore, since these hydrophilic molecules exhibit strong hydration ability at the end of the polyoxyethylene chain, they can be used stably even under high salt concentration conditions. Moreover, since it can manufacture efficiently with high purity, it can manufacture industrially.
- the polyoxyethylene derivative according to the present invention has the formula (1):
- L 1 , L 2 and L 3 are each independently an alkylene group, a phenylene group or an ester.
- X represents a functional group capable of reacting with a biological substance,
- Y represents a residue of xylitol or boremitol, or 3 to
- Z represents a residue of a compound having 2 to 5 active hydrogens,
- b and c are 1 ⁇ b ⁇ 4, 1 ⁇ c ⁇ 4 and 2 ⁇ b + c ⁇ 5.
- D and e are each independently 0 or 1.
- the molecular weight of the polyoxyethylene derivative of the formula (1) is usually 500 to 160,000, preferably 1,000 to 80,000, more preferably 2,000 to 40,000.
- L 1 , L 2 , and L 3 are respectively a linker that connects a hydrophilic group Y having a plurality of hydroxyl groups and polyoxyethylene by a covalent bond, polyoxyethylene and 2 to 5 Linker that connects covalent residues between residues Z of compounds with active hydrogen, covalently connects residues Z of compounds with 2 to 5 active hydrogens and functional groups X that can react with biological substances The linker is shown.
- linkers are not particularly limited as long as they are groups capable of forming a covalent bond, but are preferably alkylene groups, phenylene groups, ester bonds, amide bonds, ether bonds, urethane bonds, carbonate bonds, secondary amino groups, or these. More preferably an alkylene group, a phenylene group, or an ester bond, an amide bond, an ether bond, a urethane bond, a carbonate bond, or a bond between a secondary amino group and one or two alkylene groups. Particularly preferred embodiments are those shown in the following group (I).
- s in the formula represents an integer of 1 to 10, preferably an integer of 1 to 6, and more preferably an integer of 1 to 3.
- two s in the formulas may be the same or different, but the same is preferable.
- L 1 is —OCO—NH—, —O—, — (CH 2 ) s—CO—NH—.
- the “functional group capable of reacting with a biological substance” represented by X in the formula (1) is a functional group such as an amino group, mercapto group, aldehyde group, carboxyl group, unsaturated bond or azide group that the biological substance has.
- the functional group is not particularly limited as long as it is a functional group capable of chemically bonding to.
- examples thereof include a vinyl sulfone group, an amino group, an oxyamino group, an iodoacetamide group, an alkylcarbonyl group, an alkenyl group, an alkynyl group, and an azide group.
- the functional group X can be classified into the following group (II), group (III), group (IV), group (V), group (VI) and group (VII).
- Group (II) a functional group capable of reacting with an amino group of a biological substance (a), (b), (c), (d), (e), (f), (i)
- Group (III) Functional group capable of reacting with mercapto group contained in biological material (a), (b), (c), (d), (e), (f), (g), (h ), (I), (j)
- Group (IV) Functional groups capable of reacting with aldehydes in biological materials The following (g), (k), (l), (m)
- Group (V) Functional group capable of reacting with a carboxyl group of a biological substance The following (g), (k), (l), (m)
- Group (VI) Functional groups capable of reacting with unsaturated bonds of biological materials The following (g), (k), (n)
- W in the formula represents a halogen atom such as a chlorine atom (Cl), a bromine atom (Br) or an iodine atom (I), preferably Br, I, more preferably I.
- a halogen atom such as a chlorine atom (Cl), a bromine atom (Br) or an iodine atom (I), preferably Br, I, more preferably I.
- Y 1 and Y 3 in the formula each independently represent a hydrogen atom or a hydrocarbon group having 1 to 5 carbon atoms, preferably 1 carbon atom. Is a hydrocarbon group of .about.5. Specific examples of the hydrocarbon group having 1 to 5 carbon atoms include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, and a tertiary butyl group. A methyl group and an ethyl group are preferred.
- Y 2 in the formula represents a hydrocarbon group having 1 to 10 carbon atoms which may contain a fluorine atom, specifically a methyl group, an ethyl group, a propyl group, Isopropyl group, butyl group, tertiary butyl group, hexyl group, nonyl group, vinyl group, phenyl group, benzyl group, 4-methylphenyl group, trifluoromethyl group, 2,2,2-trifluoroethyl group, 4- (Trifluoromethoxy) phenyl group etc. are mentioned.
- a methyl group, a vinyl group, a 4-methylphenyl group, and a 2,2,2-trifluoroethyl group are preferred.
- the active ester group is an ester group obtained by condensing a carboxyl group with an alkoxy group having a high leaving ability.
- Examples include esters of a carboxyl group with nitrophenol, N-hydroxysuccinimide, pentafluorophenol, and the like, and an ester group obtained by condensing a carboxyl group with N-hydroxysuccinimide is preferable.
- the active carbonate group is a carbonate group having an alkoxy group with high leaving ability.
- the alkoxy group having high leaving ability include nitrophenol, N-hydroxysuccinimide, and pentafluorophenol, and a carbonate group bonded to nitrophenol and N-hydroxysuccinimide is preferable.
- the substituted maleimide group is a maleimide group in which a hydrocarbon group is bonded to one carbon atom of the double bond of the maleimide group.
- a hydrocarbon group include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, and a tertiary butyl group, and a methyl group and an ethyl group are preferable.
- the substituted sulfonate group is a sulfonate group in which a hydrocarbon group which may contain a fluorine atom is bonded to a sulfur atom of the sulfonate group.
- a hydrocarbon group which may contain a fluorine atom
- the hydrocarbon group that may contain a fluorine atom include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tertiary butyl group, a hexyl group, a nonyl group, a vinyl group, and a phenyl group.
- a methyl group, a vinyl group, a 4-methylphenyl group, and a 2,2,2-trifluoroethyl group are preferred.
- the “residue of a compound having 2 to 5 active hydrogens” represented by Z in the formula (1) is a polyhydric alcohol having 2 to 5 hydroxyl groups (ethylene glycol, glycerin, diglycerin, pentaerythritol). Residues obtained by removing the hydroxyl group from lysine, residues obtained by removing one active hydrogen of the amino group from lysine, residues obtained by removing the OH of the carboxy group from aspartic acid, residues obtained by removing the OH of the carboxy group from glutamic acid Groups and the like. Preferred are ethylene glycol, glycerin, pentaerythritol, xylitol, and lysine residues, and more preferred are ethylene glycol and glycerin residues.
- b represents the number of polyoxyethylene chains to which a hydrophilic group Y having a plurality of hydroxyl groups is bonded
- c represents the number of functional groups X capable of reacting with a biological substance.
- b and c are 1 ⁇ b ⁇ 4, 1 ⁇ c ⁇ 4, and 2 ⁇ b + c ⁇ 5, preferably 1 ⁇ b ⁇ 2, 1 ⁇ c ⁇ 2, and 2 ⁇ b + c ⁇ 4. is there.
- d and e indicate the presence or absence of a linker. When d is 0, there is no linker, and when d is 1, it has a linker.
- Y in the formula (1) represents a hydrophilic group having a plurality of hydroxyl groups each consisting of a residue of xylitol or boremitol or a residue of polyglycerol of 3 to 31 mer.
- “residue of xylitol or boremitol or residue of polyglycerol of 3 to 31 mer” means a binding reaction with a polyoxyethylene chain in xylitol or boremitol or triglycerin of 3 to 31 mer. A residue from which the contributed hydroxyl group has been removed.
- hydrophilic group Y in the formula (1) is a residue of xylitol or boremitol
- xylitol and boremitol may be bonded at any one of the hydroxyl groups at the 1-position or 3-position to the polyoxyethylene chain.
- the hydrophilic group Y in the formula (1) is polyglycerin
- the polyglycerin may be linear or dendrimer, but a dendrimer is preferable, and a trimer, heptamer, 15mer, 31mer Are preferred, more preferably trimers and heptamers.
- the following formula (2) shows a polyoxyethylene derivative of a preferred embodiment in which the hydrophilic group Y having a plurality of hydroxyl groups is a residue of xylitol or boremitol, and when a in the formula is 1, the xylitol structure , A is 2, it is a boremitol structure.
- the total molecular weight of the polyoxyethylene derivative is 500 to 160,000, and n is 5 to 3650.
- L 1 , L 2 , L 3 , X, Z, a, b, c, d and e are as defined above.
- Z in the formula is an ethylene glycol residue
- b is 1
- c is 1
- d is 0, and e is 1.
- (3) shows such a more preferred polyoxyethylene derivative.
- n1 is the average number of added moles of oxyethylene group, and n1 is usually 11 to 3650, preferably 22 to 1825, and more preferably 44 to 910.
- Z in the formula is a glycerin residue
- b is 2
- c is 1
- d is 0.
- the following formula (4) is The polyoxyethylene derivative of such a more preferable aspect is shown.
- n2 is an average addition mole number of oxyethylene groups, and n2 is usually 11 to 1825, preferably 22 to 1370, more preferably 44 to 925.
- the polyoxyethylene derivative of the present invention can be produced, for example, by the route shown in the following process diagram (process diagram (I)).
- Step (A) is a step of protecting even-numbered hydroxyl groups of polyhydric alcohol by cyclic acetalization.
- Step (B) is a step of polymerizing 11 to 3650 moles of ethylene oxide to the remaining hydroxyl group of the polyhydric alcohol derivative protected in step (A).
- Step (C) is a step of functionalizing the terminal hydroxyl group of the polyoxyethylene derivative. Depending on the type of the functional group, further functionalization can be performed after the deacetalization in the step (D). Depending on the type of functional group, the deacetalization in the step (D) can be performed simultaneously with the functionalization.
- Step (D) is a step of cutting the cyclic acetal structure.
- the polyoxyethylene derivative (10) can be produced by the route shown in the following process diagram (process diagram II).
- R 1 , R 2 and R 3 each independently represents a hydrocarbon group having 1 to 10 carbon atoms, W represents a halogen atom, and POE, L 3 and X are as defined above.
- Process (A) consists of the following three processes (A1), (A2), and (A3).
- Step (A1) is a step of converting the hydroxyl group of the polyhydric alcohol into a cyclic acetal.
- the four hydroxyl groups of xylitol are cyclic acetalized, and 1,2,3,4-diisopropylidenexylitol represented by the formula (5) and 1,2,4,5-didiform represented by the formula (6) are obtained.
- a mixture of isomers of isopropylidene xylitol is obtained.
- the method for acetalization is not particularly limited as long as it is a general hydroxyl protecting method as described in PROTECTIVEECTGROUPS IN ORGANIC SYNTHESIS (THEODORA W.GREENE et al), etc.
- an acid catalyst such as hydrochloric acid, phosphoric acid, p-toluenesulfonic acid, and p-toluenesulfonic acid monohydrate, the formulas (5) and (6) Is obtained in a molar ratio of about 9: 1.
- the amount of the acid used is preferably 5 ⁇ 10 ⁇ 6 to 5 ⁇ 10 ⁇ 3 equivalents relative to xylitol, and more preferably 5 ⁇ 10 ⁇ 5 to 5 ⁇ 10 ⁇ 4 equivalents.
- the amount of 2,2-dimethoxypropane used is preferably 2.0 to 4.0 equivalents, more preferably 2.5 to 3.5 equivalents, based on xylitol.
- the reaction can be performed in a solvent or without a solvent. When a solvent is used, for example, dimethylformamide, dichloromethane or the like can be used, but no solvent is preferable.
- the reaction temperature is usually 0 to 90 ° C., preferably 30 to 80 ° C.
- the reaction time is preferably 1 to 24 hours. If the reaction time is short, the reaction becomes insufficient.
- Purification is not particularly limited, and column chromatography, extraction, distillation, supercritical extraction, and the like can be performed. Suitably, it can be carried out by distillation under normal pressure.
- Step (A2) is a step in which only one of the structural isomers is selectively protected and separated from the other. Only the primary hydroxyl group of 1,2,3,4-diisopropylidenexylitol represented by formula (5) is selectively protected, and 1,2,4,5-diisopropylidenexylitol represented by formula (6) And separate.
- a mixture of compounds represented by formula (5) and formula (6) is reacted using a silicon compound represented by formula (11) and a tertiary amine, and only the primary hydroxyl group of compound (5) is converted to silyl ether. By doing this, the compound shown by Formula (7) is obtained.
- the reaction for silyl etherification is preferably carried out in a reaction solvent because stirring efficiency is lowered due to high viscosity in the absence of a solvent and the silyl etherification rate is lowered.
- the solvent species is not particularly limited, and examples thereof include aprotic solvents such as tetrahydrofuran, dimethyl ether, dichloromethane, chloroform, dimethylformamide, toluene, and benzene, with dichloromethane and chloroform being more preferable.
- the amount of the solvent used is 1 to 40 times by weight, preferably 2 to 20 times by weight, more preferably 3 to 10 times by weight of the mixture of the compounds represented by formula (5) and formula (6).
- the halogen atom represented by W includes Cl, Br, and I, and is preferably Cl.
- R 1 , R 2 and R 3 are the same or different hydrocarbon groups having 1 to 10 carbon atoms.
- the hydrocarbon group include a linear or branched alkyl group having 1 to 10 carbon atoms, a linear or branched alkenyl group having 2 to 10 carbon atoms, and a linear or branched alkynyl group having 2 to 10 carbon atoms.
- aryl group having 6 to 10 carbon atoms linear or branched arylalkyl group having 7 to 10 carbon atoms, linear or branched aryl alkenyl group having 8 to 24 carbon atoms, linear chain having 8 to 24 carbon atoms Or a branched aryl alkynyl group, a C7-10 linear or branched alkylaryl group, etc. are mentioned.
- silicon compound (11) examples include trimethylsilane chloride, triethylsilane chloride, triisopropylpropylsilane chloride, dimethylisopropylsilane chloride, dimethylethylsilane chloride, tert-butyldimethylsilane chloride, and tert-butyldiphenyl chloride.
- Silane, triphenylsilane chloride, and the like can be mentioned, and tert-butyldimethylsilane chloride, tert-butyldiphenylsilane chloride, triphenylsilane chloride, and the like are more preferable, and tert-butyldiphenylsilane chloride is still more preferable.
- the amount of the silicon compound (11) used is 0.8 to 20 molar equivalents, preferably 0.9 to 10 molar equivalents, more preferably a mixture of the compounds represented by the formulas (5) and (6). Is 1.0 to 5 molar equivalent times.
- Tertiary amines include dimethylaminopyridine (DMAP), 1,8-diazadibicyclo [5,4,0] undec-7-ene (DBU), 1,1,3,3-tetramethylguanidine, 1,5 -Preferably used alone or selected from the group consisting of diazabicyclo [4,3,0] non-5-ene (DABCO) and ethyldiisopropylamine, or mixed with triethylamine or pyridine. More preferably, DMAP or DBU alone or a mixture of DMAP or DBU and triethylamine is preferable, and a mixture of DMAP and triethylamine is particularly preferable.
- the ratio of DBU or DMAP in the mixed base is preferably 5 to 100 mol%, more preferably 5 to 80 mol%, and further preferably 5 to 50 mol%.
- the amount of the tertiary amine used is 0.9 to 20 molar equivalents, preferably 1.0 to 10 molar equivalents, more preferably the mixture of the compounds represented by formulas (5) and (6). 1.1 to 5 molar equivalent times. If the tertiary amine is insufficient, the acid generated as a by-product with the progress of the reaction cannot be efficiently trapped, and the reaction rate may be lowered.
- the reaction temperature for silyl etherification is usually ⁇ 20 to 80 ° C., preferably ⁇ 10 to 60 ° C.
- the reaction time is preferably 30 minutes to 24 hours.
- the mixture after completion of the reaction contains an unreacted compound represented by the formula (6).
- the compound represented by formula (6) remains, it becomes an impurity having the same molecular weight as that of the target product in the polymerization of ethylene oxide in the step (B). Therefore, it is preferable to separate and purify at this stage.
- the purification method is not particularly limited, but it is preferable to separate the unreacted compound represented by formula (4) by a purification means such as column chromatography, distillation, extraction, supercritical extraction, etc. More preferably.
- a purification means such as column chromatography, distillation, extraction, supercritical extraction, etc. More preferably.
- purifying by distillation it is preferable to separate the compound represented by the formula (6) at 80 to 160 ° C. and a vacuum degree of 10 mmHg or less. When the temperature is higher than 160 ° C., an impurity in which the acetal group is eliminated may be generated at a high temperature.
- Step (A3) is a step of deprotecting the compound represented by formula (7) protected in step (A2). There is obtained 1,2,3,4-diisopropylidenexylitol of formula (5) without structural isomers.
- the compound represented by the formula (7) is deprotected.
- the conditions for the deprotection reaction are not particularly limited, but the compound represented by the formula (5) can be obtained by a desilylation reaction with a desilylating agent.
- the reaction solvent is not particularly limited as long as it is an aprotic solvent, and preferably tetrahydrofuran, dimethyl ether, dichloromethane, chloroform, dimethylformamide, toluene, benzene, and the like, more preferably tetrahydrofuran.
- the viscosity of the compound represented by the formula (7) is high, the stirring efficiency is lowered, the proportion of desilylation is lowered, and the compound represented by the formula (7) may remain.
- the amount of the solvent used is 0.4 to 30 times, preferably 0.6 to 20 times, more preferably 0.8 to 10 times the weight of the compound represented by the formula (7).
- anhydrous tetrabutylammonium fluoride is preferably used, but a commercially available mixed solution of tetrabutylammonium fluoride / tetrahydrofuran may be used.
- a commercially available mixed solution of tetrabutylammonium fluoride / tetrahydrofuran may be used.
- the catalytic action of tetrabutylammonium fluoride is hindered, and the desilylation does not proceed and the compound represented by the formula (7) may remain.
- acid catalysts such as hydrochloric acid and acetic acid are not preferable because deacetalization occurs along with desilylation.
- the amount of the desilylating agent used is 1.0 to 20 molar equivalents, preferably 1.1 to 10 molar equivalents, more preferably 1.2 to 5 moles, relative to the compound represented by the formula (7). Equivalent times. When the desilylating agent is insufficient, the reaction does not proceed completely, and the compound represented by the formula (7) remains.
- the reaction temperature is preferably 60 ° C. or lower in order to suppress side reactions, and is preferably ⁇ 20 ° C. or higher in order to suppress an increase in the viscosity of the reaction solution.
- the reaction time is preferably 30 minutes to 24 hours. If it is shorter than 30 minutes, the reaction rate may be low, and if it is longer than 24 hours, a side reaction may occur.
- the method for purifying the compound represented by the formula (5) after completion of the reaction is not particularly limited, but it is preferable to perform column chromatography, distillation, extraction, supercritical extraction, etc., more preferably column chromatography, Extraction. If the tetrabutylammonium fluoride or tetrabutylammonium salt of the desilylating agent contained in the formula (5) remains, the catalyst used in the next step may be inhibited, and the reaction rate may decrease. If the compound represented by 7) remains, it decomposes during the polymerization of ethylene oxide in step (B), consumes ethylene oxide as a monomer, and may become a polyoxyethylene impurity, so it must be removed.
- Process (B) consists of the following two processes (B1) and (B2).
- Step (B1) is a step of converting the compound of formula (5) to an alcoholate, and either step (B1-1) or step (B1-2) may be used.
- Step (B1-1) uses metallic sodium or metallic potassium as a catalyst.
- Step (B1-2) uses sodium methoxide, potassium t-butoxide, potassium methoxide or the like as a catalyst.
- Step (B2) is a step of addition polymerization of ethylene oxide at a reaction temperature of 50 to 130 ° C.
- step (B1-1) metallic sodium or metallic potassium is used as a catalyst, preferably metallic sodium is used, and the catalyst amount is dissolved at 5 to 50 mol% at 10 to 50 ° C.
- the amount of catalyst in the step (B1-1) is less than 5 mol%, the polymerization reaction rate of ethylene oxide becomes slow, and impurities such as terminal vinyl ethers are generated due to a high temperature reaction for a long time. This is advantageous in producing a high molecular weight product.
- the catalyst exceeds 50 mol%, the viscosity of the reaction solution increases or solidifies during the alcoholation reaction, and the stirring efficiency tends to decrease, and the alcoholation tends not to be promoted. Moreover, when it solidifies, there exists a tendency for handling to worsen and it becomes a cause of moisture absorption. When the alcoholate is absorbed, a polyalkylene glycol derivative derived from moisture is generated and mixed as an undesirable impurity for pharmaceutical use.
- This high molecular weight impurity is an impurity capable of reacting with a plurality of biological substances because a plurality of functional groups are introduced in the functionalization in the next step (C). Polyoxyethylene derivatives having these impurities are undesirable for pharmaceutical applications where high purity products are required.
- the solvent used in the alcoholation reaction is not particularly limited as long as it is an aprotic solvent such as toluene, benzene, xylene, acetonitrile, tetrahydrofuran, dimethyl sulfoxide, dimethylformamide, dimethylacetamide, but preferably toluene or no solvent.
- the reaction time is preferably 1 to 24 hours. If it is shorter than 1 hour, the catalyst may not be completely dissolved. If it is longer than 24 hours, the above-described decomposition reaction may occur.
- step (B1-2) sodium methoxide, potassium t-butoxide or potassium methoxide is added as a catalyst, preferably sodium methoxide in an amount of 5 to 50 mol%, and reacted at 20 to 80 ° C. At this time, a decompression operation may be performed so that the exchange reaction is promoted.
- the amount of catalyst is preferably 5 to 50 mol% for the reasons described above.
- the reaction temperature is lower than 20 ° C.
- the reaction rate of the exchange reaction decreases, alcohol such as methanol remains, and an impurity having the same molecular weight as the target product is generated through addition polymerization of ethylene oxide.
- it is higher than 80 ° C., a decomposition reaction occurs.
- the reaction time is preferably 1 to 3 hours.
- the reaction solvent is not particularly limited as long as it is an aprotic solvent, but is preferably toluene or no solvent.
- Step (B2) is an addition polymerization of ethylene oxide at a reaction temperature of 50 to 130 ° C. to obtain a compound of formula (8) (polyoxyethylene derivative (8)).
- the reaction temperature is lower than 50 ° C.
- the rate of the polymerization reaction is slow, and the quality of the compound of the formula (8) tends to be lowered.
- the temperature is higher than 130 ° C.
- side reactions such as terminal vinyl etherification occur during polymerization, and the quality of the target product tends to deteriorate.
- an aprotic solvent preferably toluene, may be added as appropriate.
- Step (C) is a step of functionalizing the terminal hydroxyl group of the compound of formula (8) (polyoxyethylene derivative (8)).
- the deacetalization in the step (D) can be performed simultaneously with the functionalization.
- polyoxyethylene derivative (8) By using the terminal hydroxyl group of the compound of formula (8) (polyoxyethylene derivative (8)), group (II), group (III), group (IV), group (V), group (VI) and group ( The polyoxyethylene derivative represented by the formula (9) can be produced by modifying the functional groups shown in VII).
- a compound having each functional group of group (II), group (III), group (IV), group (V), group (VI) and group (VII) is used as an intermediate, and other compounds and Functionalization can be carried out by reaction.
- an intermediate having the functional group (k) can be used as a raw material to obtain the functional group (a) or (d).
- the ratio of the organic base and inorganic base used is not particularly limited, but is preferably equimolar or more with respect to the compound (8).
- An organic base may be used as a solvent.
- W 2 in the formula (b1) and the formula (e1) is a halogen atom selected from Cl, Br, and I, preferably Cl.
- the ratio of compound (b1) and compound (e1) used is not particularly limited, but is preferably equimolar or more, more preferably equimolar to 50-fold molar relative to compound (8).
- the reaction temperature is preferably 0 to 300 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the produced compound may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction or the like.
- W 2 represents a halogen atom selected from Cl, Br and I.
- Y 2 represents a hydrocarbon group which may contain a fluorine atom having 1 to 10 carbon atoms.
- the reaction temperature is preferably 0 to 200 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- an organic base such as triethylamine, pyridine, dimethylaminopyridine, or an inorganic base such as sodium carbonate, sodium hydroxide, sodium hydrogen carbonate, sodium acetate, potassium carbonate, potassium hydroxide may be used as a catalyst.
- the proportion of the catalyst used is preferably 0.1 to 50% by weight, more preferably 0.5 to 20% by weight.
- the carboxyl body (f) thus produced may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction, or used as a raw material for the condensation reaction. In that case, it may be used as it is.
- a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction, or used as a raw material for the condensation reaction. In that case, it may be used as it is.
- the carboxyl form (f) can be obtained by reacting the compound (8) with a halogenated alkyl ester such as ethyl 6-bromohexanoate or ethyl 7-bromoheptanoate.
- a halogenated alkyl ester such as ethyl 6-bromohexanoate or ethyl 7-bromoheptanoate.
- the etherification reaction between the compound (8) and the halogenated alkyl ester is carried out in the above-mentioned aprotic solvent or in the absence of a solvent.
- the ratio of the halogenated alkyl ester to be used is not particularly limited, but is preferably equimolar or more, more preferably equimolar to 30 times molar to the compound (8).
- the reaction temperature is preferably 0 to 200 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- an organic base such as triethylamine, pyridine, dimethylaminopyridine, or an inorganic base such as sodium carbonate, sodium hydroxide, sodium hydrogen carbonate, sodium acetate, potassium carbonate, potassium hydroxide may be used as a catalyst.
- the proportion of the catalyst used is preferably 0.1 to 500% by weight, more preferably 0.5 to 300% by weight.
- hydrolysis of the ester is performed by adding an aqueous solution such as sodium hydroxide or potassium hydroxide in the case of an organic base, and water in the case of an inorganic base.
- the reaction temperature is preferably 0 to 100 ° C, more preferably 20 to 100 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- neutralization is performed with hydrochloric acid or sulfuric acid.
- the carboxyl body (f) thus produced may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction, or used as a raw material for the condensation reaction. In that case, it may be used as it is.
- Carboxyl compound (f) is subjected to a condensation reaction with N-hydroxysuccinimide in the presence of a condensing agent such as dicyclohexylcarbodiimide (DCC) or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (EDC).
- a condensing agent such as dicyclohexylcarbodiimide (DCC) or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (EDC).
- DCC dicyclohexylcarbodiimide
- EDC 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride
- a condensation reaction is carried out in the above-mentioned aprotic solvent or in the absence of a solvent.
- a condensing agent Preferably it is DCC.
- the proportion of DCC used is preferably equimolar or more, more preferably equimolar to 5-fold molar to the carboxyl body (f).
- the use ratio of N-hydroxysuccinimide is preferably equimolar or more, more preferably equimolar to 5-fold molar to the carboxyl body (f).
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced succinimide (a) may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, or supercritical extraction.
- the succinimide body (a) can also be obtained by reacting the compound (8) with N, N′-disuccinimide carbonate.
- the reaction is carried out in an aprotic solvent or in the absence of a solvent as in the above reaction.
- the ratio of N, N′-disuccinimide carbonate to be used is preferably equimolar or more, more preferably equimolar to 5-fold molar to the compound (8).
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced compound may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction or the like.
- Compound (8) is obtained by adding acrylonitrile or the like in a solvent such as water or acetonitrile, using an inorganic base such as sodium hydroxide or potassium hydroxide as a catalyst, to obtain a nitrile body, and then under a nickel or palladium catalyst in an autoclave.
- an amine body (k) having a functional group (k) can be obtained.
- the ratio of the inorganic base used in obtaining the nitrile body is not particularly limited, but is preferably 0.01 to 50% by weight based on the compound (8).
- the use ratio of acrylonitrile and the like is not particularly limited, but is preferably 0.5 to 5 times the weight, more preferably 1 to 4 times the weight of the compound (8). . Further, acrylonitrile may be used as a solvent.
- the reaction temperature is preferably ⁇ 50 to 100 ° C., more preferably ⁇ 20 to 60 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the reaction solvent in the subsequent hydrogenation reaction of the nitrile body is not particularly limited as long as it is a solvent that does not participate in the reaction, but is preferably toluene.
- the proportion of nickel or palladium catalyst used is not particularly limited, but is 0.05 to 30% by weight, preferably 0.5 to 20% by weight, based on the nitrile body.
- the reaction temperature is preferably 20 to 200 ° C, more preferably 50 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the hydrogen pressure is preferably 2 to 10 MPa, more preferably 3 to 8 MPa.
- ammonia may be added to the reaction system.
- the ammonia pressure when adding ammonia is not particularly limited, but is 0.1 to 10 MPa, more preferably 0.3 to 2 MPa.
- the resulting compound may be purified by the aforementioned purification means.
- the amine body (k) can also be obtained by reacting the compound (e) with aqueous ammonia.
- the reaction is carried out in aqueous ammonia, and the ammonia concentration is not particularly limited, but is preferably in the range of 10 to 40%.
- the proportion of ammonia water used is preferably 1 to 300 times the weight of the compound (e).
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 72 hours, more preferably 1 to 36 hours.
- the amine body (k) can also be obtained by reacting the compound (e) with ammonia in an autoclave.
- limiting in particular about the reaction solvent Preferably methanol and ethanol are mentioned.
- the amount of ammonia is preferably 10 to 300% by weight, more preferably 20 to 200% by weight, based on the compound (e).
- the reaction temperature is preferably 50 to 200 ° C, more preferably 80 to 150 ° C.
- the reaction time is preferably 10 minutes to 24 hours, more preferably 30 minutes to 12 hours.
- the resulting compound may be purified by the aforementioned purification means.
- the amine body (k) can also be obtained by combining compound (8) and phthalimide by a Mitsunobu reaction in an aprotic solvent and deprotecting with a polyvalent amine.
- the reaction conditions for the Mitsunobu reaction are not particularly limited, but the reaction solvent is preferably chloroform or dichloromethane.
- triphenylphosphine is equimolar or more, preferably equimolar to 50-fold molar with respect to compound (8)
- diisopropyl azodicarboxylate is equimolar or more, preferably equimolar to 50-fold molar with respect to compound (8). It is preferred to use.
- the reaction temperature is preferably 0 to 100 ° C., more preferably 10 to 50 ° C.
- the reaction time is preferably 10 minutes to 72 hours, more preferably 30 minutes to 6 hours.
- hydrazine or a polyvalent amine such as ethylenediamine in an amount of equimolar or more, preferably equimolar to 500-fold molar to compound (8).
- Methanol is preferable.
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 72 hours, more preferably 1 to 10 hours.
- the produced compound may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction or the like.
- Method of introducing functional group (d) After reacting the amino group of the amine body (k) obtained by the above-described method with maleic anhydride in the above-mentioned aprotic solvent or in the absence of a solvent to obtain a maleimide body, acetic anhydride and sodium acetate are catalyzed. As described above, a maleimide body (d) having a functional group (d) introduced therein can be obtained by ring-closing reaction.
- the ratio of maleic anhydride to be used in the maleimidation reaction is not particularly limited, but is preferably equimolar or more, more preferably equimolar to 5-fold molar to the amine (k).
- the reaction temperature is preferably 0 to 200 ° C, more preferably 20 to 120 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced maleimide body (d) may be purified by the above-described purification means, or may be used as it is for the next ring closure reaction.
- the reaction solvent in the subsequent ring closure reaction is not particularly limited, but an aprotic solvent or acetic anhydride is preferable.
- the ratio of sodium acetate to be used is not particularly limited, but is preferably equimolar or more, more preferably equimolar to 50 times molar with respect to the maleimide body (d).
- the reaction temperature is preferably 0 to 200 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the resulting compound may be purified by the aforementioned purification means.
- the maleimide body (d) can also be obtained by reacting the compound (d1) represented by the following formula (d1) with the amino group of the amine body (k) described above.
- the reaction is carried out in the above-mentioned aprotic solvent or in the absence of a solvent, and the compound (d1) is added in an equimolar amount or more with respect to the amino group of the amine (k) and reacted.
- the proportion of the compound (d1) used is preferably equimolar or more, more preferably equimolar to 5-fold molar to the amino group of the amine (k).
- the reaction temperature is preferably 0 to 200 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours. Light may be shielded during the reaction.
- the resulting compound may be purified by the aforementioned purification means.
- Q represents a hydrocarbon group having 1 to 9 carbon atoms.
- Y 1 represents a hydrogen atom or a hydrocarbon having 1 to 5 carbon atoms.
- the functional group (i) can be obtained by reacting the amine of the amine body (k) obtained by the above-described method with iodoacetic anhydride in the above-mentioned aprotic solvent or in the absence of a solvent.
- the ratio of iodoacetic anhydride to be used is not particularly limited, but is preferably equimolar or more, more preferably equimolar to 5-fold molar to the amino group of the amine (k).
- the reaction temperature is preferably 0 to 200 ° C, more preferably 20 to 120 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced compound (i) having the functional group (i) may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, or supercritical extraction.
- the functional group (i) can be obtained by subjecting the amine body (k) to a condensation reaction with iodoacetic acid in the presence of a condensing agent such as DCC or EDC. Similarly, the condensation reaction is carried out in the aforementioned aprotic solvent or in the absence of a solvent.
- a condensing agent Preferably it is DCC.
- the proportion of DCC used is preferably equimolar or more, more preferably equimolar to 5-fold molar to the amine compound (k).
- the ratio of iodoacetic acid to be used is preferably equimolar or more, more preferably equimolar to 5-fold molar to the amine compound (k).
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the resulting compound may be purified by the aforementioned purification means.
- the proportion of compound (l1) used is preferably equimolar or more, more preferably equimolar to 20 molar relative to the carbonate body (b).
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced compound may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction or the like, or may be proceeded to the next step without purification.
- Q represents a hydrocarbon group having 1 to 9 carbon atoms.
- An oxyphthalimide body can also be obtained by combining compound (8) and hydroxyphthalimide by a Mitsunobu reaction in an aprotic solvent and deprotecting with a polyvalent amine.
- the reaction conditions for the Mitsunobu reaction are not particularly limited, but the reaction solvent is preferably chloroform or dichloromethane.
- triphenylphosphine is used in an equimolar amount or more, preferably equimolar to 50-fold mol with respect to compound (8), and diisopropyl azodicarboxylate is equimolar or more, preferably equimolar to 50-fold mol with respect to compound (8). It is preferred to use.
- the reaction temperature is preferably 0 to 100 ° C., more preferably 10 to 50 ° C.
- the reaction time is preferably 10 minutes to 72 hours, more preferably 30 minutes to 6 hours.
- the oxyamine (1) having the functional group (l) introduced can be obtained.
- the reaction solvent is not particularly limited, but methanol, dichloromethane and water are preferable.
- the ratio of the polyvalent amine is not particularly limited, but is preferably equimolar or more, more preferably equimolar to 50 times molar to the oxyphthalimide compound.
- the reaction temperature is preferably 0 to 100 ° C, more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced compound may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction or the like.
- Compound (c1) can be prepared from the corresponding alcohol using sodium metal, metal potassium, sodium hydride, potassium hydride, sodium methoxide, potassium t-butoxide and the like.
- the reaction temperature is preferably 0 to 300 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the compound represented by the following (c3) compound (c3)
- An acetal body can be obtained by reacting the compound (c3) with b), (e) or (f)).
- the solvent for the reaction is not particularly limited, but it is preferably carried out in the aprotic solvent described above.
- the charge ratio of compound (c3) to compound (a), (b), (e) or (f) is preferably equimolar or more, more preferably equimolar to 10-fold molar.
- the reaction temperature is preferably ⁇ 30 to 200 ° C., more preferably 0 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- a condensing agent such as DCC or EDC may be used as appropriate. Any acetalization reaction may be carried out in the dark.
- the acetal thus obtained may be purified by the above-described purification means, or may be used as it is in the next aldehyde reaction without being purified.
- the aldehyde (c) can be obtained by making the acetal form an aqueous solution of 0.1 to 50% and hydrolyzing it in an aqueous solution adjusted to pH 1 to 4 with an acid such as acetic acid, phosphoric acid, sulfuric acid or hydrochloric acid. it can.
- the reaction temperature is preferably ⁇ 20 to 100 ° C., more preferably 0 to 80 ° C.
- the reaction time is preferably 10 minutes to 24 hours, more preferably 30 minutes to 10 hours.
- the reaction may be carried out in the dark.
- the resulting compound may be purified by the aforementioned purification means.
- the deacetal of a process (D) can also be performed simultaneously.
- R 4 and R 5 each independently represent a hydrocarbon group having 1 to 3 carbon atoms, and may be the same or different from each other.
- M is sodium or potassium
- W 2 is a halogen atom selected from Cl, Br, and I
- t is an integer of 1 to 5.
- the mercapto compound (compound (g)) having the functional group (g) can be obtained by reacting the compound (e) with a thialating agent such as thiourea.
- a thialating agent such as thiourea.
- the production of compound (e) is as described above.
- the thialation reaction is performed in a solvent such as water, alcohol, acetonitrile, or in the absence of a solvent.
- the use ratio of thiourea is preferably equimolar or more with respect to compound (e), more preferably in the range of equimolar to 50 times molar.
- the reaction temperature is preferably 0 to 300 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the resulting thiazolium salt can be hydrolyzed with alkali to obtain a mercapto compound.
- the resulting compound may be purified by the aforementioned purification means.
- the deacetal of a process (D) can also be performed simultaneously in pH adjustment after hydrolysis.
- the mercapto compound can also be obtained by reacting the compound (e) with a compound represented by the following formula (g1) (compound (g1)) and decomposing it with a primary amine.
- the reaction between (e) and (g1) is carried out in the above-mentioned aprotic solvent or in the absence of a solvent.
- the use ratio of the compound (g1) is preferably equimolar or more with respect to the compound (e), more preferably in the range of equimolar to 50 times molar.
- the reaction temperature is preferably 0 to 300 ° C, more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the subsequent alkali decomposition with a primary amine is carried out in the aprotic solvent described above or in the absence of a solvent.
- primary amine Preferably, ammonia, methylamine, ethylamine, propylamine, butylamine, pentylamine, hexylamine, cyclohexylamine, ethanolamine, propanolamine, butanolamine, etc. are mentioned. Of course, these primary amines may be used as a solvent.
- the resulting compound may be purified by the aforementioned purification means.
- the compound having the functional group (h) (compound (h)) can be obtained by reacting the compound (g) with 2,2-dipyridyl disulfide.
- the solvent is not particularly limited, but is preferably carried out in alcohol.
- the charging ratio of 2,2-dipyridyl disulfide to compound (g) is preferably equimolar or more, more preferably equimolar to 50 times molar.
- the reaction temperature is preferably ⁇ 30 to 100 ° C., more preferably 0 to 60 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the acetal thus obtained may be purified by the above-described purification means.
- the reaction temperature is preferably 0 to 200 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the produced compound (m) may be purified by the aforementioned purification means. Further, when the Boc group is deprotected, deacetalization in the step (D) can be performed at the same time.
- Acetylene having a functional group (j) is obtained by reacting the compound (a), (b), (c) or (e) with an acetylene compound (compound (j1)) represented by the following formula (j1).
- a compound (compound (j)) can be obtained.
- the acetylene reaction is carried out in a protic solvent or in the absence of a solvent in an amount of equimolar or more, preferably equimolar to 50-fold molar of the compound (j1) with respect to the compound (a), (b), (c) or (e).
- the reaction temperature is preferably 0 to 300 ° C, more preferably 20 to 150 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 24 hours.
- the resulting compound may be purified by the aforementioned purification means.
- t is an integer of 1 to 5.
- Y 3 represents a hydrogen atom or a hydrocarbon group having 1 to 5 carbon atoms.
- the proportion of compound (n1) to be used is preferably equimolar or more, more preferably equimolar to 5-fold molar to compound (k).
- the reaction temperature is preferably 0 to 100 ° C., more preferably 20 to 80 ° C.
- the reaction time is preferably 10 minutes to 48 hours, more preferably 30 minutes to 12 hours.
- the resulting compound may be purified by the aforementioned purification means.
- Q represents a hydrocarbon group having 1 to 9 carbon atoms.
- Step (D) is a deprotection step of cleaving the cyclic acetal structure of the polyoxyethylene derivative represented by the formula (9) having a functional group (hereinafter also referred to as “compound (9)”). Depending on the type of the functional group, further functionalization can be performed after the deacetalization in the step (D).
- the method for deprotecting the cyclic acetal is not particularly limited as long as it is a general deprotection method as described in PROTECTIVE GROUPS IN ORGANIC SYNTHESIS (THEODORA W.GREENE et al), etc.
- Deprotection can be performed in the presence of an acid catalyst.
- the acid catalyst include acetic acid, hydrochloric acid, phosphoric acid, p-toluenesulfonic acid and the like, preferably hydrochloric acid and phosphoric acid, and more preferably phosphoric acid.
- the amount of acid used is preferably 0.05 to 2 times by weight, more preferably 0.1 to 1 times by weight with respect to the compound (9).
- the solvent used for the deprotection reaction is water, methanol, ethanol, acetonitrile, tetrahydrofuran, dioxane, dimethyl sulfoxide, dimethylformamide, dimethylacetamide, and preferably water or methanol.
- the amount of the solvent to be used is 1 to 50 times by weight, preferably 2 to 35 times by weight, more preferably 5 to 20 times by weight of the compound (8).
- the reaction time is preferably 1 to 24 hours. When it is shorter than 1 hour, the deprotection reaction becomes insufficient. If it is longer than 24 hours, there is a risk of oxidative decomposition of polyoxyethylene by acid or deactivation of functional groups.
- the reaction temperature is usually 0 to 60 ° C., preferably 10 to 40 ° C.
- Compound (9) After deprotection, it may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, supercritical extraction, etc., preferably by performing recrystallization and drying the crystal under reduced pressure, Compound (9) can be obtained.
- a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, supercritical extraction, etc.
- step (D) Further functionalization can be performed after the deacetalization in step (D).
- functionalization For functional groups that may react and decompose under deacetal conditions, it is desirable to perform functionalization after step (D).
- the polyoxyethylene derivative (1) of the present invention can also be produced by the production method shown in the following process diagram (process diagram III).
- R represents a functional group protected by a protecting group or an optionally protected hydroxyl group
- X 2 represents an amino group, an active group, and an active group. (A carbonate group or an active sulfonate group is shown.)
- Step (E) is a step of converting the remaining hydroxyl group of the protected polyhydric alcohol derivative into a functional group.
- Step (F) is a step of binding the polyhydric alcohol derivative functionalized in step (E) and the polyoxyethylene derivative by reaction.
- Step (G) is a step of functionalization by deprotecting R which is a protecting group. If necessary, functionalization can be carried out in accordance with the step (C) in the above-mentioned process diagrams I and II. Depending on the type of R protecting group at the end of polyoxyethylene, deacetalization in the next step can be performed simultaneously with deprotection.
- Step (H) is a step of cutting the cyclic acetal structure.
- further functionalization can be performed after the step (H).
- a polyoxyethylene derivative (polyoxyethylene derivative (2b)) represented by the formula (2b) is produced by performing the above-described process steps (E), (F), (G), and (H).
- the polyoxyethylene derivative (15) can be produced by the route shown in the following process diagram (process diagram IV).
- R 1 , R 2 and R 3 represent a hydrocarbon group having 1 to 10 carbon atoms, W represents a halogen atom.
- POE, L 3 and X are as defined above.
- R, X 2 , and X are as defined above.
- Step (E) is a step for functionalizing a mixture of compounds represented by formula (5) and formula (6) to obtain a compound of formula (12) that does not contain structural isomers.
- step (E) either the following step (E1) or step (E2) may be used.
- Process (E1) consists of the following two processes (E1-1) and (E1-2).
- Step (E1-1) is a step in which only one of the structural isomers is selectively phthalimidized and separated from the other. Only the primary hydroxyl group of 1,2,3,4-diisopropylidenexylitol represented by formula (5) is selectively phthalimidated, and 1,2,4,5-diisopropylidenexylitol represented by formula (6) To separate.
- Step (E1-2) is a phthalimide group deprotection step.
- step (E1-1) a mixture of the compounds represented by the formulas (5) and (6) is reacted with phthalimide, and only the primary hydroxyl group of the compound (5) is phthalimidized, whereby the formula (17) The compound shown is obtained.
- the phthalimidation is preferably azeotropically dehydrated before the reaction to remove water in the reaction system
- the solvent used is not particularly limited as long as it is an aprotic solvent capable of azeotropic dehydration, but preferably toluene, xylene Or it is cyclohexene, More preferably, it is toluene.
- the amount of the solvent is 1 to 10 times by weight of the mixture, preferably 2 to 6 times by weight, more preferably 3 to 5 times by weight.
- the solvent amount is 5 to 75% by weight, preferably 10 to 50% by weight of the amount of the organic solvent charged. Reflux and evaporate at boiling temperature or higher for 30 minutes or longer and within 3 hours. If the amount of distillation is small or the reflux time is shorter than 30 minutes, dehydration becomes insufficient, and there is a possibility that the residual moisture will cause a side reaction in the reaction, resulting in a decrease in purity.
- the reaction solvent is preferably an organic solvent, and is not particularly limited as long as it is an aprotic solvent, but is preferably a dehydrated solvent, preferably chloroform, dichloromethane, tetrahydrofuran, acetonitrile, dimethyl sulfoxide, and more preferably dichloromethane. , Chloroform.
- the amount of the organic solvent is 1 to 50 times by weight of the mixture, preferably 2 to 30 times by weight, more preferably 3 to 20 times by weight. The reason why a solvent having a low water content is used is to suppress the side reaction described above.
- the amount of phthalimide used for phthalimidation is 1 to 10 molar equivalents, preferably 1.01 to 5 molar equivalents, more preferably 1 to the mixture of the compounds represented by formula (5) and formula (6). 0.02 to 3 molar equivalent times.
- azo reagents used for phthalimidation 1,1′-azobis (N, N-dimethylformamide), 1,1 ′-(azodicarbonyl) dipiperidine, dibenzyl azodicarboxylate, diethyl azodicarboxylate, azodicarboxylic acid Diisopropyl, dimethyl azodicarboxylate, 1,1′-azobis (N, N-diisopropylformamide), 1,6-dimethyl-1,5,7-hexahydro-1,4,6,7-tetrazocine-2,5- Dione and the like can be mentioned, preferably diethyl azodicarboxylate and diisopropyl azodicarboxylate, more preferably diisopropyl azodicarboxylate.
- the amount of the azo reagent is 1 to 10 mole equivalents, preferably 1.01 to 5 mole equivalents, more preferably 1.02 to moles, with respect to the mixture of the compounds represented by
- phosphine reagents used for phthalimidation include dicyclohexylphenylphosphine, diethylphenylphosphine, 4- (dimethylamino) phenyldiphenylphosphine, diphenyl-2-pyridylphosphine, isopropyldiphenylphosphine, triisobutylphosphine, tri-n-butylphosphine, Examples include tri-t-butylphosphine, tricyclohexylphosphine, tri-n-hexylphosphine, tri-n-octylphosphine, and triphenylphosphine, with triphenylphosphine being preferred.
- the amount of the phosphine reagent is 1 to 10 mole equivalents, preferably 1.01 to 5 mole equivalents, more preferably 1.02 to moles, relative to the mixture of the compounds represented by formula (5) and formula (6). 3 molar equivalent times.
- the reaction temperature is not particularly limited, but is preferably room temperature.
- the reaction time is preferably 5 minutes or more, and if it is less than 5 minutes, the reaction rate may be lowered.
- the reaction solution after completion of the reaction contains an unreacted substance represented by the formula (6).
- the method for removing this is not particularly limited, but it is preferable to separate the unreacted substance represented by the formula (6) by a purification means such as column chromatography, distillation, extraction, recrystallization, supercritical extraction, It is more preferable to purify by recrystallization.
- examples of the good solvent include toluene, ethyl acetate, methanol, ethanol, acetonitrile, etc., preferably toluene, ethyl acetate, ethanol, and more preferably ethyl acetate. These solvents can be used alone or in combination of two or more.
- examples of the poor solvent include hexane, diethyl ether, methyl t-butyl ether, and the like, and preferably hexane.
- the amount of the good solvent is 1 to 50 times by weight of the mixture, preferably 2.5 to 35 times by weight, more preferably 5 to 20 times by weight.
- the amount of the poor solvent is 0.5 to 30 times by weight, preferably 1 to 20 times by weight, more preferably 2 to 10 times by weight.
- the recrystallization temperature is ⁇ 20 to 30 ° C., preferably ⁇ 10 to 20 ° C. If the temperature exceeds 30 ° C., the crystals may dissolve and the yield may decrease.
- the recrystallization time is preferably 15 minutes or longer. If the time is less than 15 minutes, the removal of impurities may be insufficient. Recrystallization is performed repeatedly to increase purification efficiency, and the number of times is not particularly limited, but is preferably 1 to 5 times, more preferably 2 to 4 times.
- the crystals of the obtained compound (17) are dried under reduced pressure.
- Step (E1-2) is a deprotection step of compound (17) obtained in step (E1-1).
- the deprotection method is not particularly limited as long as it is a general phthalimide deprotection method as described in PROTECTIVE GROUPS IN ORGANIC SYNTHESIS (THEODORA W.GREENE et al), etc., but a deprotection reagent having an amino group Is preferably used.
- Examples of the solvent used in the deprotection step include dichloromethane, chloroform, methanol, ethanol and the like, preferably chloroform and ethanol.
- the amount of the solvent is 1 to 50 times by weight, preferably 2 to 30 times by weight, more preferably 3 to 20 times by weight of the compound represented by the formula (17).
- the deprotecting reagent used in the step (E1-2) is not particularly limited as long as it is a low molecular weight amine compound having a primary amino group, and specific examples include hydrazine, ethylenediamine, trimethylenediamine, diethylenetriamine, and the like. Preferred are hydrazine and ethylenediamine.
- the amount of the deprotecting reagent is 1 to 30 molar equivalents, preferably 2 to 20 molar equivalents, more preferably 3 to 10 molar equivalents, relative to the compound represented by the formula (17).
- the reaction temperature is not particularly limited, but is preferably 10 to 80 ° C, more preferably 20 to 60 ° C.
- the reaction time is 1 hour or more, and if it is shorter than 1 hour, the reaction rate may be low.
- the purification method after completion of the reaction is not particularly limited, but it is preferable to separate a compound such as a deprotection reagent by a purification means such as column chromatography, distillation, extraction, recrystallization, or supercritical extraction. More preferably, it is purified.
- examples of the organic solvent include toluene, dichloromethane, chloroform, methanol, and the like, preferably dichloromethane. These solvents can be used alone or in combination of two or more.
- the amount of the organic solvent is 1 to 20 times by weight, preferably 2 to 10 times by weight of the compound represented by the formula (17).
- the aqueous solution used is an aqueous solution of 1 to 25% by weight of an alkali metal inorganic salt, and the alkali metal inorganic salt is preferably an alkali metal halogen salt, more preferably sodium chloride.
- the amount of the aqueous solution is 1 to 20 times by weight, preferably 2 to 10 times by weight that of the compound represented by the formula (17).
- the mixing and separation time in the extraction step is not particularly limited, but is preferably 1 minute to 6 hours, and more preferably 10 minutes to 3 hours.
- the extraction temperature is 10 to 80 ° C., preferably 20 to 60 ° C. Purification efficiency is improved by repeated extraction. The number of times is not particularly limited, but is preferably 1 to 4 times, more preferably 2 to 3 times.
- dehydration is preferably performed using a dehydrating agent. After removing the dehydrating agent by filtration, the solvent is distilled off to obtain the compound represented by the formula (16).
- Process (E2) consists of the following two processes (E2-1) and (E2-2).
- Step (E2-1) is a step in which both structural isomers are activated carbonate or sulfonate.
- Step (E2-2) is a purification step for separating structural isomers by utilizing slight differences in physical properties of functionalized structural isomers.
- the compound (19) corresponds to the compound represented by the formula (12) in the process diagram IV.
- step (E2-1) a mixture of compounds represented by formula (5) and formula (6), a compound represented by the following formula (b1) and formula (e1) (compound (b1), compound (e1) ) Is caused to react with each other to introduce an activated carbonate group and an activated sulfonate group, respectively.
- the activated sulfonate group the compounds represented by formula (19) and formula (20) are obtained.
- an activated carbonate group or an activated sulfonate group it can be produced by substantially the same production method. Therefore, the introduction of the activated sulfonate group will be described below.
- reaction solvent used in the step (E2-1) examples include aprotic solvents such as toluene, acetonitrile, ethyl acetate, tetrahydrofuran, chloroform, and dichloromethane, or in the absence of solvent, preferably toluene and chloroform.
- the amount of the solvent is 1 to 50 times by weight, preferably 2 to 30 times by weight, more preferably 3 to 20 times by weight of the mixture of the compounds represented by formula (5) and formula (6).
- W 2 in the compound represented by the general formula (e1) used in the step (E2-1) is a halogen atom selected from Cl, Br, and I, and preferably Cl.
- the amount of the compound represented by (e1) is not particularly limited, but is 1 to 10 mole equivalents, preferably 1.01 to 5 moles, with respect to the mixture of the compounds represented by formula (5) and formula (6). Equivalent times, more preferably 1.02 to 3 molar equivalent times.
- the base used in the reaction is an organic base such as triethylamine, pyridine, 4-dimethylaminopyridine, or an inorganic base such as sodium carbonate, sodium hydroxide, sodium hydrogen carbonate, sodium acetate, potassium carbonate, potassium hydroxide, Triethylamine is preferred.
- the amount of the base used is not particularly limited, but is 1 to 15 molar equivalents, preferably 1.1 to 10 molar equivalents, more preferably 1.2 to 5 molar equivalents, relative to the mixture.
- the reaction temperature is not particularly limited, but is preferably 0 to 80 ° C, more preferably 20 to 60 ° C.
- the reaction time is 1 hour or more, and if it is shorter than 1 hour, the reaction rate may be low.
- Step (E2-2) is a step of separating and purifying a mixture of the compounds represented by formula (19) and formula (20) generated in step (E2-1).
- the purification method is not particularly limited, but it is preferable to separate the compound represented by the formula (20) by a purification means such as column chromatography, distillation, extraction, recrystallization, supercritical extraction, etc. More preferably, it is purified.
- examples of the good solvent include toluene, ethyl acetate, methanol, ethanol, acetonitrile, etc., preferably toluene, ethyl acetate, ethanol, and more preferably ethyl acetate. These solvents can be used alone or in combination of two or more.
- examples of the poor solvent include hexane, diethyl ether, and methyl-t-butyl ether, and hexane is preferable.
- the amount of the good solvent is 1 to 50 times by weight of the mixture, preferably 2.5 to 35 times by weight, more preferably 5 to 20 times by weight.
- the amount of the poor solvent is 0.5 to 30 times by weight, preferably 1 to 20 times by weight, more preferably 2 to 10 times by weight.
- the recrystallization temperature is ⁇ 20 to 30 ° C., preferably ⁇ 10 to 20 ° C. If the temperature exceeds 30 ° C., the crystals may dissolve and the yield may decrease.
- the recrystallization time is preferably 15 minutes or longer. If the time is less than 15 minutes, the removal of impurities may be insufficient. Recrystallization is performed repeatedly to increase purification efficiency, and the number of times is not particularly limited, but is preferably 1 to 5 times, more preferably 2 to 4 times.
- the obtained crystals of compound (15) are dried under reduced pressure.
- step (F) the polyhydric alcohol derivative (12) functionalized in step (E) and the polyoxyethylene derivative (21) are reacted as shown in the following process diagram (process diagram V). It is the process of combining.
- R represents a functional group protected by a protecting group or an optionally protected hydroxyl group.
- L 4 represents a linker, and X 3 represents A functional group capable of reacting with X 2 is shown.
- linker L 4 are the same as those given as specific examples of the linkers L 1 to L 3 described above.
- the polyoxyethylene derivative represented by the above formula (21) used in the step (F) has a functional group X 3 capable of reacting with X 2 of the compound represented by the formula (12).
- X 3 is not particularly limited as long as X 3 is a functional group that reacts with an amino group, and examples thereof include an active ester group, an active carbonate group, an aldehyde group, a substituted sulfonate group, and a carboxyl group.
- An active ester group and an active carbonate group are preferred.
- X 3 is not particularly limited as long as it is a functional group that reacts with the active carbonate group or sulfonate group, but is preferably an amino group or an alkoxide group.
- R represents a functional group protected by a protecting group or an optionally protected hydroxyl group.
- protected functional groups include amino groups, carboxyl groups, aldehyde groups, and thiol groups.
- Specific examples of the amino-protecting group include a t-butyl carbamate group, a benzyl group, and a trityl group, and a t-butyl carbamate group is preferable.
- Specific examples of the protecting group for a carboxyl group include a t-butyl group and a benzyl group, with a benzyl group being preferred.
- protecting group for the aldehyde group examples include an acetal group containing 3 to 9 carbon atoms, and a diethyl acetal group is preferable.
- protecting groups for thiol groups include t-butyl group, benzyl group, trityl group, t-butyldimethylsilyl group, t-butyldiphenylsilyl group, etc., preferably t-butyl group, benzyl group More preferably a t-butyl group.
- hydroxyl-protecting group examples include a t-butyl group, a benzyl group, a trityl group, a t-butyldimethylsilyl group, a t-butyldiphenylsilyl group, and the like, preferably a t-butyl group, a benzyl group. And more preferably a benzyl group.
- the amount of the compound represented by the formula (12) used in the step (F) is not particularly limited, but is 1 to 20 molar equivalents, preferably 1 with respect to the polyoxyethylene derivative represented by the formula (21). 0.5 to 15 molar equivalent times, more preferably 2 to 10 molar equivalent times.
- reaction solvent used in the step (F) examples include aprotic solvents such as toluene, acetonitrile, ethyl acetate, tetrahydrofuran, chloroform, and dichloromethane, preferably toluene and chloroform.
- the amount of the solvent is 1 to 50 times, preferably 2 to 25 times, more preferably 3 to 10 times the weight of the compound represented by the formula (21).
- the reaction temperature is not particularly limited, but is preferably 0 to 100 ° C, and more preferably 20 to 80 ° C.
- the reaction time is 1 hour or more, and if it is shorter than 1 hour, the reaction rate may be low.
- purification method is not particularly limited, purification means such as column chromatography, distillation, extraction, recrystallization, supercritical extraction and the like can be mentioned, and purification by recrystallization is more preferable.
- examples of good solvents include toluene, ethyl acetate, methanol, ethanol, acetonitrile, and the like, preferably toluene and ethyl acetate, and more preferably ethyl acetate. These solvents can be used alone or in combination of two or more.
- examples of the poor solvent include hexane, diethyl ether, and methyl-t-butyl ether, and hexane is preferable.
- the amount of the good solvent is 1 to 50 times by weight of the mixture, preferably 2.5 to 35 times by weight, more preferably 5 to 20 times by weight.
- the amount of the poor solvent is 0.5 to 30 times by weight, preferably 1 to 20 times by weight, more preferably 2 to 10 times by weight.
- the recrystallization temperature is ⁇ 20 to 30 ° C., preferably ⁇ 10 to 20 ° C. If the temperature exceeds 30 ° C., the crystals may dissolve and the yield may decrease.
- the recrystallization time is preferably 15 minutes or longer. If the time is less than 15 minutes, the removal of impurities may be insufficient. Recrystallization is performed repeatedly to increase purification efficiency, and the number of times is not particularly limited, but is preferably 1 to 5 times, more preferably 2 to 4 times.
- the crystals of the obtained compound (13) are dried under reduced pressure.
- Step (G) is a step in which a functional group or a hydroxyl group appears by deprotection of the protecting group R of the polyoxyethylene derivative represented by the formula (13) obtained in Step (F).
- a functional group is introduced by the same production method as in step (C) of the above-mentioned process diagram II.
- step (C) the type of R protecting group at the end of the polyoxyethylene, there may be a case where deprotection in the next step can be performed simultaneously with deprotection.
- deprotection can be performed using a general deprotection method as described in PROTECTIVE GROUPS IN ORGANIC SYNTHESIS (THEODORA W. GREENE et al). After deprotection, it may be purified by a purification means such as extraction, recrystallization, adsorption treatment, reprecipitation, supercritical extraction, etc., preferably by performing recrystallization and drying the crystal under reduced pressure, A compound represented by the formula (13) can be obtained.
- Step (H) is a deprotection step for cutting the cyclic acetal structure of the polyoxyethylene derivative represented by the formula (14) obtained in Step (G). Depending on the type of the functional group at the terminal of polyoxyethylene, further functionalization can be performed after the step (H).
- Process (H) is the same process as process (D). By performing the step (D), the compound represented by the formula (15) which is the target product can be obtained.
- a polyoxyethylene derivative (1) having a plurality of hydroxyl groups at its terminals can be industrially produced with high purity and efficiency.
- the polyoxyethylene derivative (1) obtained by the present invention has the advantage of improving the blood half-life and antigenicity as compared with the conventional polyoxyethylene derivative, and can be used to modify biological materials. Useful.
- Example 1-1 Compounds (5) and (6): Synthesis of diisopropylidenexylitol
- Example 1-2 Compound (7): Synthesis of 1,2,3,4-diisopropylidene-5- (t-butyldiphenylsilyl) xylitol
- the crystals were collected by filtration, dissolved in 800 g of ethyl acetate at 40 ° C., cooled to room temperature, and crystallized by adding 600 g of n-hexane. The crystals collected by filtration were washed with 1.0 kg of n-hexane. The crystals were collected by filtration and dried under vacuum to obtain 184 g of the following compound (p2).
- the crystals were collected by filtration, dissolved in 100 g of ethyl acetate at 40 ° C., cooled to room temperature, and crystallized by adding 50 g of n-hexane. Crystal dissolution and crystallization steps were repeated once more. The crystals collected by filtration were washed with 50 g of n-hexane. The crystals were collected by filtration and dried under vacuum to obtain 9 g of the following compound (p4).
- 100 g (2.5 mmol) of the above-mentioned compound (p5), which is a bifurcated PEG of 1,000, and 500 g of toluene were added and dissolved by heating to 60 ° C. while stirring and blowing nitrogen. The temperature was raised to 110 ° C., and about 100 g of a fraction was withdrawn while being azeotroped with toluene, followed by dehydration.
- the crystals were collected by filtration, dissolved in 500 g of ethyl acetate at 40 ° C., cooled to room temperature and crystallized by adding 200 g of n-hexane. The crystal dissolution and crystallization steps were repeated two more times. The crystals collected by filtration were washed with 200 g of n-hexane. The crystals were collected by filtration and dried under vacuum to obtain 44 g of the following compound (p7).
- the crystals were collected by filtration, dissolved in 210 g of ethyl acetate at 40 ° C., cooled to room temperature and crystallized by adding 90 g of n-hexane. The crystal dissolution and crystallization steps were repeated two more times. The crystals collected by filtration were washed with 90 g of n-hexane. The crystals were collected by filtration and dried under vacuum to obtain 26 g of the following compound (p9).
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Polyethers (AREA)
Abstract
末端に複数の水酸基を有する新規ポリオキシエチレン誘導体の提供、特に、生体関連物質を修飾する用途に効果的に用いることができ、且つ工業的に製造することができる、末端に複数の水酸基を有するポリオキシエチレン誘導体を提供する。式(1):(式中、ポリオキシエチレン誘導体の全体の分子量は500~160,000であり、nは5~3650である。L1、L2、L3はそれぞれ独立して、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合を示す。Xは生体関連物質と反応可能な官能基を示す。Yはキシリトールもしくはボレミトールの残基または3~31量体のポリグリセリンの残基からなる複数の水酸基を有する親水性基を示す。Zは2~5個の活性水素を持つ化合物の残基を示す。bおよびcは1≦b≦4、1≦c≦4、かつ2≦b+c≦5である。dおよびeはそれぞれ独立して0または1である。)で表されるポリオキシエチレン誘導体。
Description
本発明は、生体関連物質を修飾する用途に使用される、末端に複数の水酸基を有するポリオキシエチレン誘導体に関する。
近年、ホルモンやサイトカインなどの細胞間情報伝達物質、抗体、酵素などの生体関連物質を用いた医薬品の開発が行われている。これらの生体関連物質は、通常生体内へ投与されると腎臓における糸球体濾過や肝臓や脾臓などにおけるマクロファージの取り込みにより生体内から排出されてしまう。そのため血中半減期が短く、十分な薬理効果を得ることが難しい。この問題を解決するため、リポソームやポリマーミセル中へ生体関連物質を封入することや、生体関連物質を糖鎖やポリオキシエチレンなどの両親媒性高分子やアルブミンなどによって化学修飾する試みがなされている。これらの試みにより、分子量の増大や水和層の形成などにより生体関連物質の生体内挙動が改善される。また、ポリオキシエチレンでの修飾により、毒性や抗原性の低下や難水溶性薬剤の溶解性向上などの効果が得られることも知られている。
非特許文献1、2には、ポリオキシエチレンで修飾したリポソームやナノ粒子を同一個体に繰り返し投与すると、初回投与時に比べ、二回目以降の血中半減期が低下するABC(accelerated blood clearance)現象が起きる場合があることが報告されている。これはリポソームやナノ粒子を修飾したポリオキシエチレンに対する抗体が発現するためと考えられており、その抗体はポリオキシエチレン鎖の末端やポリオキシエチレンの繰り返し構造等、様々な部位を認識しているといわれている。一方、ポリグリセリン等の一部の親水性ポリマーで修飾したリポソームやナノ粒子はABC現象をほとんど起こさないという報告例がある。しかし、これらポリオキシエチレン以外の親水性ポリマーでは十分な血中滞留性を得ることができない他、臨床での使用例も乏しく、ポリオキシエチレンの代替としては十分なものとはいえない。
一方、特許文献1には、一つの水酸基を末端に有したポリオキシエチレン誘導体で修飾された生体関連物質に関する記載がなされている。水酸基を末端に有したポリオキシエチレン誘導体を用いると、アルコキシ基を末端に有したポリオキシエチレン誘導体よりも、抗原性が低下するデータが得られている。このようにポリオキシエチレン誘導体の末端に水酸基を配することはポリオキシエチレンの抗原性の低下に寄与する一つの改善策と考えられる。しかしながら、この文献に記載されているポリオキシエチレン誘導体は逆相クロマトグラフィーを用いた精製を行っていることから、歩留まりが大きく低下するため、工業的な生産は不向きである。また、近時においては、血中滞留性をより向上させた薬剤の開発が行われており、さらに抗原性を低下させる必要がある。
ポリオキシエチレンの末端に複数の水酸基を持たせることはキャリアの周囲により強固で大きい水和層を形成することにつながるため、オプソニンとの相互作用を低下させて、結果としてさらに抗原性を低減し得ると考えられる。複数の水酸基を有したポリオキシエチレンとしては以下のような文献がある。
特許文献2、3を含む多くの文献には、複数の水酸基を有する単糖類または多糖類を親水性ポリマーの末端に導入し、薬物と結合させた標的指向化(ターゲッティング)型製剤に関する記載がなされている。しかしながら、この糖類は生体内にある糖鎖認識機構によって標的指向性を付与するためのものであり、抗原性の改善を意図したものではない。
特許文献4、5には多数の水酸基を有するポリグリセリン誘導体を有する疎水性ポリオキシアルキレンに関する記載がなされている。かかる疎水性ポリオキシアルキレンはポリグリセリンの親水性を利用した界面活性剤である。これら文献では疎水性ポリオキシアルキレンの例のみが示されており、また記載の製造方法では生体関連物質の修飾に適した高純度なポリオキシエチレン誘導体を得ることは難しい。
特許文献6にはポリオキシエチレンとポリグリシドールのコポリマーに関する記載がなされている。文献記載のランダムまたはブロックポリマーの製造方法ではポリグリシドールは枝分かれを複数持った分岐ポリマーとなり、様々な構造のポリマーの混合物となる。医薬品の原料としては単一な構造の高純度品が求められており混合物は好ましくない。さらに混合物は医薬原料の登録申請において含有成分の組成比等を明確にする必要があり非常に困難である。またグリシドールの重合ではポリマー中の水酸基の数を制御することは難しく、水酸基の数が増えるとポリマー溶液の粘度が大きくなる問題もある。
以上のように、生体関連物質の修飾に適した、血中半減期、抗原性を改善することができる複数の水酸基を片末端に有した、工業的に生産が可能なポリオキシエチレン誘導体は得られていないのが現状である。
T.Ishida, H.Kiwada, et al.,J.control.Release.122, 349-355(2007)
W.Jiskoot, R.M.F.van Schie, et al.,Pharmaceutical Research, 26, 6, 1303-1314 (2009)
本発明の課題は、末端に複数の水酸基を有する新規ポリオキシエチレン誘導体を提供することにある。より具体的には、生体関連物質を修飾する用途に効果的に用いることができ、且つ工業的に製造することができる、末端に複数の水酸基を有するポリオキシエチレン誘導体を提供することにある。
本発明者らは、上記課題を解決するために鋭意検討した結果、下記の構成からなる末端に複数の水酸基を有するポリオキシエチレン誘導体を完成した。
即ち、本発明は以下の通りである。
[1] 式(1):

(式中、ポリオキシエチレン誘導体の全体の分子量は500~160,000であり、nは5~3650である。L1、L2、L3はそれぞれ独立して、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合を示す。Xは生体関連物質と反応可能な官能基を示す。Yはキシリトールもしくはボレミトールの残基または3~31量体のポリグリセリンの残基からなる複数の水酸基を有する親水性基を示す。Zは2~5個の活性水素を持つ化合物の残基を示す。bおよびcは1≦b≦4、1≦c≦4、かつ2≦b+c≦5である。dおよびeはそれぞれ独立して0または1である。)で表されるポリオキシエチレン誘導体。
[2] 式(2):
[2] 式(2):

(式中、ポリオキシエチレン誘導体の全体の分子量は500~160,000であり、nは5~3650である。L1、L2、L3はそれぞれ独立して、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合を示す。Xは生体関連物質と反応可能な官能基を示す。Zは2~5個の活性水素を持つ化合物の残基を示す。aは1または2である。bおよびcは1≦b≦4、1≦c≦4、かつ2≦b+c≦5である。dおよびeは0または1である。)で表されるポリオキシエチレン誘導体。
[3] 式(2)中のZがエチレングリコール残基、bが1、cが1、dが0、eが1である、下記式(3)で表されるポリオキシエチレン誘導体である、上記[2]記載のポリオキシエチレン誘導体。
[3] 式(2)中のZがエチレングリコール残基、bが1、cが1、dが0、eが1である、下記式(3)で表されるポリオキシエチレン誘導体である、上記[2]記載のポリオキシエチレン誘導体。

(式中、L1、L3はそれぞれ独立してそれぞれ独立して、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合を示す。Xは生体関連物質と反応可能な官能基を示す。aは1または2であり、n1は11~3650である。)
[4] 式(2)中のZがグリセリン残基、bが2、cが1、dが0である、下記式(4)で表されるポリオキシエチレン誘導体である、上記[2]記載のポリオキシエチレン誘導体。
[4] 式(2)中のZがグリセリン残基、bが2、cが1、dが0である、下記式(4)で表されるポリオキシエチレン誘導体である、上記[2]記載のポリオキシエチレン誘導体。

(式中、L1、L3はそれぞれ独立して、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合を示す。Xは生体関連物質と反応可能な官能基を示す。aは1または2であり、eは0または1である。n2は11~1825である。)
[5] 式(1)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[1]に記載のポリオキシエチレン誘導体。
[6] 式(2)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[2]に記載のポリオキシエチレン誘導体。
[7] 式(3)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[3]に記載のポリオキシエチレン誘導体。
[8] 式(4)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[4]に記載のポリオキシエチレン誘導体。
[6] 式(2)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[2]に記載のポリオキシエチレン誘導体。
[7] 式(3)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[3]に記載のポリオキシエチレン誘導体。
[8] 式(4)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である上記[4]に記載のポリオキシエチレン誘導体。
本明細書中の化学式において、

の部分は、1、2-ジヒドロキシエチレン基を繰り返し単位として規定するものであり、

と同義である。
本発明のポリオキシエチレン誘導体は末端に複数の水酸基(好適には4個以上の水酸基)を有しているため、生体関連物質を修飾した場合、生体関連物質の周囲に強固な水素結合による大きな水和層を形成することになる。このため、修飾された生体関連物質は体内にてオプソニンや各組織を構成する細胞表面との相互作用が低下し、各組織への移行性が減少することで、血中半減期が改善される。またポリオキシエチレン末端にキシリトール、ボレミトール等の糖アルコールまたは3~31量体のポリグリセリン由来の複数の水酸基を有する親水性分子が結合しているため、従来のこの種のポリオキシエチレン誘導体でのポリオキシエチレン鎖の末端のアルコキシ基を認識していた抗体の発現を効果的に抑制することができる。さらに、それら親水性分子がポリオキシエチレン鎖の末端にて強力な水和能を発揮することから、高塩濃度条件でも安定に使用することができる。また、高純度に効率良く製造できるため、工業的に製造することができる。
本発明に係るポリオキシエチレン誘導体は、式(1):

(式中、ポリオキシエチレン誘導体の全体の分子量は500~160,000であり、nは5~3650である。L1、L2、L3はそれぞれ独立して、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合を示す。Xは生体関連物質と反応可能な官能基を示す。Yはキシリトールもしくはボレミトールの残基または3~31量体のポリグリセリンの残基からなる複数の水酸基を有する親水性基を示す。Zは2~5個の活性水素を持つ化合物の残基を示す。bおよびcは1≦b≦4、1≦c≦4、かつ2≦b+c≦5である。dおよびeはそれぞれ独立して0または1である。)で表されるポリオキシエチレン誘導体(以下、「本発明のポリオキシエチレン誘導体(1)」とも称す。)である。
かかる式(1)のポリオキシエチレン誘導体の分子量は、通常500~160,000であり、好ましくは1,000~80,000であり、更に好ましくは2,000~40,000である。
式(1)中の、L1、L2、L3は、それぞれ、複数の水酸基を有する親水性基Yとポリオキシエチレン間を共有結合にて繋ぐリンカー、ポリオキシエチレンと2~5個の活性水素を持つ化合物の残基Z間を共有結合にて繋ぐリンカー、2~5個の活性水素を持つ化合物の残基Zと生体関連物質と反応可能な官能基X間を共有結合にて繋ぐリンカーを示す。
これらのリンカーは共有結合を形成しうる基であれば特に制限は無いが、好ましくは、アルキレン基、フェニレン基、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合、2級アミノ基またはこれらの結合であり、より好ましくは、アルキレン基、フェニレン基、または、エステル結合、アミド結合、エーテル結合、ウレタン結合、カーボネート結合もしくは2級アミノ基と1個または2個のアルキレン基との結合であり、特に好ましい態様は、下記の群(I)に示されるものである。
群(I):

(式中、sは0または1~10の整数である。)
式(式(z1)~式(z6))において、式中のsは1~10の整数を示し、好ましくは1~6の整数、更に好ましくは1~3の整数を示す。また、式(z3)および式(z6)において、式中の2個のsは同一でも、異なっていてもよいが、同一が好ましい。
L1の特に好ましい態様は、-OCO-NH-、-O-、-(CH2)s-CO-NH-である。
式(1)中のXで示される「生体関連物質と反応可能な官能基」は、生体関連物質が有するアミノ基、メルカプト基、アルデヒド基、カルボキシル基、不飽和結合またはアジド基などの官能基と化学結合可能な官能基であれば特に制限されない。具体的には、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基、アジド基等が挙げられる。
好適な実施形態において、かかる官能基Xは、下記の群(II)、群(III)、群(IV)、群(V)、群(VI)および群(VII)に分類することができる。
群(II):生体関連物質が有するアミノ基と反応可能な官能基
下記の (a)、(b)、(c)、(d)、(e)、(f)、(i)
下記の (a)、(b)、(c)、(d)、(e)、(f)、(i)
群(III):生体関連物質が有するメルカプト基と反応可能な官能基
下記の(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)、(j)
下記の(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)、(j)
群(IV):生体関連物質が有するアルデヒドと反応可能な官能基
下記の(g)、 (k)、(l)、(m)
下記の(g)、 (k)、(l)、(m)
群(V):生体関連物質が有するカルボキシル基と反応可能な官能基
下記の(g)、(k)、(l)、(m)
下記の(g)、(k)、(l)、(m)
群(VI):生体関連物質が有する不飽和結合と反応可能な官能基
下記の(g)、(k)、(n)
下記の(g)、(k)、(n)
群(VII):生体関連物質が有するアジド基と反応可能な官能基
下記の(j)
下記の(j)

官能基(i)において、式中のWは塩素原子(Cl)、臭素原子(Br)またはヨウ素原子(I)などのハロゲン原子を示し、好ましくはBr、I、より好ましくはIである。
また、官能基(d)及び官能基(j)において、式中のY1、Y3は、それぞれ独立して、水素原子または炭素数1~5の炭化水素基を示し、好ましくは炭素数1~5の炭化水素基である。炭素数1~5の炭化水素基としては、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、第三ブチル基などが挙げられる。好ましくはメチル基、エチル基である。
また、官能基(e)において、式中のY2はフッ素原子を含んでいてもよい炭素数が1~10の炭化水素基を示し、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、第三ブチル基、ヘキシル基、ノニル基、ビニル基、フェニル基、ベンジル基、4-メチルフェニル基、トリフルオロメチル基、2,2,2-トリフルオロエチル基、4-(トリフルオロメトキシ)フェニル基等が挙げられる。好ましくはメチル基、ビニル基、4-メチルフェニル基、2,2,2-トリフルオロエチル基である。
活性エステル基とは、カルボキシル基を脱離能の高いアルコキシ基と縮合させたエステル基である。カルボキシル基とニトロフェノール、N-ヒドロキシスクシンイミド、ペンタフルオロフェノールなどとのエステルが挙げられ、好ましくはカルボキシル基をN-ヒドロキシスクシンイミドと縮合させたエステル基である。
活性カーボネート基とは、脱離能の高いアルコキシ基を有したカーボネート基である。脱離能の高いアルコキシ基はニトロフェノールやN-ヒドロキシスクシンイミド、ペンタフルオロフェノール等が挙げられ、好ましくはニトロフェノール、N-ヒドロキシスクシンイミドと結合したカーボネート基である。
置換マレイミド基とは、マレイミド基の二重結合の片方の炭素原子に炭化水素基が結合しているマレイミド基である。炭化水素基は、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、第三ブチル基などが挙げられ、好ましくはメチル基、エチル基である。
置換スルホネート基とは、スルホネート基の硫黄原子にフッ素原子を含んでいてもよい炭化水素基が結合しているスルホネート基である。フッ素原子を含んでいてもよい炭化水素基としては、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、第三ブチル基、ヘキシル基、ノニル基、ビニル基、フェニル基、ベンジル基、4-メチルフェニル基、トリフルオロメチル基、2,2,2-トリフルオロエチル基、4-(トリフルオロメトキシ)フェニル基等が挙げられる。好ましくはメチル基、ビニル基、4-メチルフェニル基、2,2,2-トリフルオロエチル基である。
式(1)中のZで示される「2~5個の活性水素を持つ化合物の残基」とは、2~5個の水酸基を有する多価アルコール(エチレングリコール、グリセリン、ジグリセリン、ペンタエリスリトール、キシリトールなど)から水酸基を除いた残基、リジンからアミノ基の一つの活性水素を除いた残基、アスパラギン酸からカルボキシ基のOHを除いた残基、グルタミン酸からカルボキシ基のOHを除いた残基等が挙げられる。好ましくは、エチレングリコール、グリセリン、ペンタエリスリトール、キシリトール、リジンの残基であり、さらに好ましくはエチレングリコール、グリセリンの残基である。
式(1)中のbは、複数の水酸基を有する親水性基Yが結合したポリオキシエチレン鎖の数を示し、cは、生体関連物質と反応可能な官能基Xの数を示す。bおよびcは、1≦b≦4、1≦c≦4、かつ、2≦b+c≦5であり、好ましくは、1≦b≦2、1≦c≦2、かつ、2≦b+c≦4である。
また、式(1)中の、dおよびeは、リンカーの有無を示し、dが0の場合、リンカーは無く、dが1の場合、リンカーを有する。
式(1)中のYは、キシリトールもしくはボレミトールの残基または3~31量体のポリグリセリンの残基からなる複数の水酸基を有する親水性基を示す。ここで「キシリトールもしくはボレミトールの残基または3~31量体のポリグリセリンの残基」とは、キシリトールもしくはボレミトール、または、3~31量体のポリグリセリンにおける、ポリオキシエチレン鎖との結合反応に寄与した水酸基が除かれた残基のことである。
式(1)中の親水性基Yがキシリトールまたはボレミトールの残基の場合、キシリトールおよびボレミトールはこれらの1位、3位のいずれの水酸基がポリオキシエチレン鎖と結合してもよいが、好ましくは1位の水酸基である。また、式(1)中の親水性基Yがポリグリセリンの場合、ポリグリセリンは直鎖でもデンドリマーでもよいが、デンドリマーが好ましく、また、3量体、7量体、15量体、31量体が好ましく、より好ましくは3量体、7量体である。
以下の式(2)は、複数の水酸基を有する親水性基Yがキシリトールまたはボレミトールの残基からなる、好適態様のポリオキシエチレン誘導体を示しており、式中のaが1の場合はキシリトール構造、aが2の場合はボレミトール構造である。

(式中、ポリオキシエチレン誘導体の全体の分子量は500~160,000であり、nは5~3650である。また、L1、L2、L3、X、Z、a、b、c、d、eは前記と同義である。)
また、かかる式(2)のポリオキシエチレン誘導体においては、式中のZがエチレングリコール残基、bが1、cが1、dが0、eが1であるのがより好ましく、以下の式(3)は、そのようなより好ましい態様のポリオキシエチレン誘導体を示している。

(式中、L1、L3、X、aは前記と同義であり、n1は11~3650である。)
式(3)中のn1はオキシエチレン基の平均付加モル数であり、n1は通常11~3650、好ましくは22~1825、さらに好ましくは44~910である。
また、式(2)のポリオキシエチレン誘導体においては、式中のZがグリセリン残基、bが2、cが1、dが0であるのがより好ましく、下記の式(4)は、そのようなより好ましい態様のポリオキシエチレン誘導体を示している。

(式中、L1、L3、X、a、eは前記と同義であり、n2は11~1825である。)
式(4)中のn2はオキシエチレン基の平均付加モル数であり、n2は通常11~1825、好ましくは22~1370、さらに好ましくは44~925である。
本発明のポリオキシエチレン誘導体は、例えば次のような工程図(工程図(I))に示すルートにより製造することができる。

(式中、POEはポリオキシエチレン鎖を示し、L3、X、aは前記と同義である。)
工程(A)は、多価アルコールの偶数個の水酸基を環状アセタール化して保護する工程である。
工程(B)は、工程(A)で保護された多価アルコール誘導体の残存した水酸基へ、エチレンオキシドを11~3650モル重合する工程である。
工程(C)は、ポリオキシエチレン誘導体の末端の水酸基を官能基化する工程である。官能基の種類によっては、工程(D)の脱アセタール後にさらに官能基化を行うこともできる。また官能基の種類によっては、官能基化の際に同時に工程(D)の脱アセタールを行うことができる。
工程(D)は、環状アセタール構造を切断する工程である。a=1の場合は4個の水酸基を出現させ、a=2の場合は6個の水酸基を出現させる。
上記の工程(A)(B)(C)(D)を行うことで、式(2a)で表されるポリオキシエチレン誘導体(ポリオキシエチレン誘導体(2a))が製造される。
以下に、かかるポリオキシエチレン誘導体(2a)の製造方法の好適な具体例をさらに説明するが、a=1と、a=2のどちらの場合においても、同様の製法にて製造することができるため、a=1の誘導体、すなわち、以下の式(10)で表されるポリオキシエチレン誘導体(ポリオキシエチレン誘導体(10))について説明を行う。

(式中、POE、L3、Xは前記と同義である。)
ポリオキシエチレン誘導体(10)は、次のような工程図(工程図II)に示すルートにより製造することができる。

(式中、R1、R2、R3はそれぞれ独立して炭素数1~10の炭化水素基を示し、Wはハロゲン原子を示す。POE、L3、Xは前記と同義である。)
工程(A)は以下の(A1)、(A2)、(A3)の3つの工程から成る。
工程(A1)は多価アルコールの水酸基を環状アセタール化する工程である。この工程ではキシリトールの4つの水酸基を環状アセタール化して、式(5)で示される1,2,3,4-ジイソプロピリデンキシリトールと式(6)で示される1,2,4,5-ジイソプロピリデンキシリトールの異性体の混合物を得る。
アセタール化の方法としては、PROTECTIVE GROUPS IN ORGANIC SYNTHESIS(THEODORA W.GREENE et al)等に記載があるような一般的な水酸基の保護法であれば特に制限はないが、具体的には、酢酸、塩酸、リン酸、p-トルエンスルホン酸、p-トルエンスルホン酸一水和物などの酸触媒存在下、キシリトールに2,2-ジメトキシプロパンを反応させることにより、式(5)と式(6)で示される化合物の混合物がモル比約9:1で得られる。
酸の使用量は、キシリトールに対して5×10-6~5×10-3当量が好ましく、5×10-5~5×10-4当量がより好ましい。
2,2-ジメトキシプロパンの使用量は、キシリトールに対して2.0~4.0当量が好ましく、2.5~3.5当量がより好ましい。
当該反応は、溶媒中または無溶媒で行うことができる。溶媒を用いる場合は、例えば、ジメチルホルムアミド、ジクロロメタンなどを用いることができるが、無溶媒が好ましい。
反応温度は、通常0~90℃、好ましくは、30~80℃である。反応時間は、1~24時間が好ましい。反応時間が短いと反応が不十分となる。
2,2-ジメトキシプロパンの使用量は、キシリトールに対して2.0~4.0当量が好ましく、2.5~3.5当量がより好ましい。
当該反応は、溶媒中または無溶媒で行うことができる。溶媒を用いる場合は、例えば、ジメチルホルムアミド、ジクロロメタンなどを用いることができるが、無溶媒が好ましい。
反応温度は、通常0~90℃、好ましくは、30~80℃である。反応時間は、1~24時間が好ましい。反応時間が短いと反応が不十分となる。
反応で副生したアセタール化されていない不純物またはキシリトール分子間がアセタールで結合された不純物などは、精製除去を行うのが好ましい。精製は、特に制限されないが、カラムクロマトグラフィー、抽出、蒸留、超臨界抽出等を行うことができる。好適には、常圧下、蒸留で行うことができる。
工程(A2)は構造異性体の片方のみを選択的に保護し、もう一方と分離する工程である。式(5)で示される1,2,3,4-ジイソプロピリデンキシリトールの1級水酸基のみを選択的に保護し、式(6)で示される1,2,4,5-ジイソプロピリデンキシリトールと分離する。
工程(A1)のアセタール化で得られた式(5)と式(6)で示される化合物の混合物から、式(5)で示される化合物を単離するには、蒸留、カラムクロマトグラフィーなどを利用することができる。しかし、これら構造異性体は沸点や分子極性等の物理的性質が類似している。そのため物理的性質を利用した蒸留やカラムクロマトグラフィーは、効率良く分離することができず、低収率となるためスケールアップに不向きである。一方、式(5)と式(6)で示される化合物の混合物をシリルエーテル化すると、式(7)と式(8)で示される化合物の混合物が得られる。これらは水酸基とシリルエーテル基の違いにより、分子極性が大きく異なるものになる。沸点などの物理的性質が大幅に変わることで、式(6)で示される化合物との分離が容易になり、効率的な精製が可能になる。
式(5)と式(6)で示される化合物の混合物を式(11)で示されるケイ素化合物と第三級アミンを用いて反応を行い、化合物(5)の1級水酸基のみをシリルエーテル化することにより、式(7)で示される化合物が得られる。
シリルエーテル化の反応は、無溶媒中では高粘度のため攪拌効率が低下し、シリルエーテル化率が低下するため、反応溶媒中で行うのが好ましい。溶媒種については、特に制限はないが、テトラヒドロフラン、ジメチルエーテル、ジクロロメタン、クロロホルム、ジメチルホルムアミド、トルエン、ベンゼンなど非プロトン性溶媒が挙げられるが、より好ましくはジクロロメタン、クロロホルムである。溶媒の使用量は、式(5)と式(6)で示される化合物の混合物の1~40重量倍、好ましくは2~20重量倍、さらに好ましくは3~10重量倍である。
シリルエーテル化の反応は、無溶媒中では高粘度のため攪拌効率が低下し、シリルエーテル化率が低下するため、反応溶媒中で行うのが好ましい。溶媒種については、特に制限はないが、テトラヒドロフラン、ジメチルエーテル、ジクロロメタン、クロロホルム、ジメチルホルムアミド、トルエン、ベンゼンなど非プロトン性溶媒が挙げられるが、より好ましくはジクロロメタン、クロロホルムである。溶媒の使用量は、式(5)と式(6)で示される化合物の混合物の1~40重量倍、好ましくは2~20重量倍、さらに好ましくは3~10重量倍である。
式(11)で示されるケイ素化合物において、Wで示されるハロゲン原子は、Cl、Br、Iが挙げられ、好ましくはClである。R1、R2およびR3は、同一または異なった炭素数1~10の炭化水素基である。当該炭化水素基としては、炭素数1~10の直鎖または分岐鎖のアルキル基、炭素数2~10の直鎖または分岐鎖のアルケニル基、炭素数2~10の直鎖または分岐鎖のアルキニル基、炭素数6から10のアリール基、炭素数7から10の直鎖または分岐鎖のアリールアルキル基、炭素数8から24の直鎖または分岐鎖のアリールアケニル基、炭素数8から24の直鎖または分岐鎖のアリールアルキニル基、炭素数7から10の直鎖または分岐鎖のアルキルアリール基等が挙げられる。
ケイ素化合物(11)としては、具体的には、塩化トリメチルシラン、塩化トリエチルシラン、塩化トリイソプルピルシラン、塩化ジメチルイソプロピルシラン、塩化ジメチルエチルシラン、塩化tert-ブチルジメチルシラン、塩化tert-ブチルジフェニルシラン、塩化トリフェニルシラン等が挙げられるが、より好ましくは、塩化tert-ブチルジメチルシラン、塩化tert-ブチルジフェニルシラン、塩化トリフェニルシラン等であり、更に好ましくは塩化tert-ブチルジフェニルシランである。
ケイ素化合物(11)の使用量は、式(5)と式(6)で示される化合物の混合物に対して0.8~20モル当量倍、好ましくは0.9~10モル当量倍、さらに好ましくは1.0~5モル当量倍である。
第三級アミンには、ジメチルアミノピリジン(DMAP)、1,8-ジアザジビシクロ[5,4,0]ウンデカ-7-エン(DBU)、1,1,3,3-テトラメチルグアニジン、1,5-ジアザビシクロ[4,3,0]ノン-5-エン(DABCO)およびエチルジイソプロピルアミンからなる群から選択されるいずれか単独で使用するか、あるいは、トリエチルアミンまたはピリジンと混合させても用いるのが好ましく、より好ましくはDMAPまたはDBUの単独か、あるいは、DMAPまたはDBUとトリエチルアミンとの混合が好ましく、特に好ましくはDMAPとトリエチルアミンの混合である。混合塩基中のDBUまたはDMAPの割合は、5~100モル%が好ましく、より好ましくは5~80モル%であり、さらには5~50モル%が好ましい。
第三級アミンの使用量は、式(5)と式(6)で示される化合物の混合物に対して0.9~20モル当量倍、好ましくは1.0~10モル当量倍、さらに好ましくは1.1~5モル当量倍である。第三級アミンが不足すると反応の進行に伴って副生する酸を効率よくトラップできないため反応率が低くなる可能性がある。
シリルエーテル化の反応温度は、通常-20~80℃、好ましくは、-10~60℃である。反応時間は、30分~24時間が好ましい。
反応終了後の混合物には、未反応の式(6)で示される化合物が含まれている。式(6)で示される化合物が残存した場合、工程(B)のエチレンオキシドの重合にて目的物と同分子量の不純物となる。したがって、この段階で分離精製するのが好ましい。精製する方法は、特に制限はないが、カラムクロマトグラフィー、蒸留、抽出、超臨界抽出等の精製手段にて未反応の式(4)で示される化合物を分離するのが好ましく、蒸留にて精製するのがさらに好ましい。
蒸留にて精製する場合、80~160℃、真空度10mmHg以下にて式(6)で示される化合物を分離することが好ましい。160℃より高い場合、高温によりアセタール基が脱離した不純物が生じる恐れがある。
蒸留にて精製する場合、80~160℃、真空度10mmHg以下にて式(6)で示される化合物を分離することが好ましい。160℃より高い場合、高温によりアセタール基が脱離した不純物が生じる恐れがある。
工程(A3)は工程(A2)にて保護された式(7)で示される化合物の脱保護工程。構造異性体の無い、式(5)の1,2,3,4-ジイソプロピリデンキシリトールを得る。
式(7)で示される化合物の脱保護反応を行う。脱保護反応の条件は、特に制限されないが、脱シリル化剤による脱シリル化反応により、式(5)で示される化合物を得ることができる。
反応溶媒は、非プロトン性溶剤であれば特に制限されないが、好ましくはテトラヒドロフラン、ジメチルエーテル、ジクロロメタン、クロロホルム、ジメチルホルムアミド、トルエン、ベンゼンなど挙げられるが、より好ましくはテトラヒドロフランである。無溶媒では、式(7)で示される化合物の粘性が高く、攪拌効率が低下し、脱シリル化の割合が低下し、式(7)で示される化合物が残存してしまう恐れがある。溶媒の使用量は、式(7)で示される化合物の0.4~30重量倍、好ましくは0.6~20重量倍、さらに好ましくは0.8~10重量倍である。
脱シリル化剤には、フッ化テトラブチルアンモニウムの無水物が好ましく用いられるが、市販のフッ化テトラブチルアンモニウム/テトラヒドロフランの混合溶液を利用してもよい。フッ化テトラブチルアンモニウムの水和物では、フッ化テトラブチルアンモニウムの触媒作用を阻害し、脱シリル化が進行せずに式(7)で示される化合物が残存してしまう恐れがある。また、塩酸や酢酸等の酸触媒では、脱シリル化と共に脱アセタール化が起こるため好ましくない。
脱シリル化剤の使用量は、式(7)で示される化合物に対して、1.0~20モル当量倍、好ましくは1.1~10モル当量倍、さらに好ましくは1.2~5モル当量倍である。脱シリル化剤が不足すると、反応が完全に進行しなくなり、式(7)で示される化合物が残存してしまう。
反応温度については、副反応を抑制するためには60℃以下が好ましく、また反応液の粘性上昇を抑制するために-20℃以上が好ましい。反応時間については、30分~24時間が好ましい。30分より短いと反応率が低い恐れがあり、24時間より長いと副反応が起る恐れがある。
反応終了後、式(5)で示される化合物を精製する方法は、特に制限はないが、カラムクロマトグラフィー、蒸留、抽出、超臨界抽出等を行うことが好ましく、さらには好ましくはカラムクロマトグラフィー、抽出である。式(5)に含まれている脱シリル化剤のフッ化テトラブチルアンモニウムやテトラブチルアンモニウム塩は残存すると、次工程に使用する触媒を阻害し、反応率が低下する可能性があり、式(7)で示される化合物が残存すると、工程(B)のエチレンオキシドの重合の際に分解し、モノマーであるエチレンオキシドを消費し、ポリオキシエチレン不純物となる可能性があるため、除去する必要がある。
工程(B)は以下の(B1)、(B2)の2つの工程から成る。
工程(B1)は、式(5)の化合物をアルコラート化する工程であり、工程(B1-1)または工程(B1-2)のどちらを用いてもよい。
工程(B1-1)は触媒として金属ナトリウム又は金属カリウム等を用いる。
工程(B1-2)は触媒としてナトリウムメトキシド、カリウムt-ブトキシドまたはカリウムメトキシド等を用いる。
工程(B2)は50~130℃の反応温度でエチレンオキシドを付加重合する工程である。
工程(B1-1)は触媒として金属ナトリウム又は金属カリウムを用い、好ましくは金属ナトリウムを用い、触媒量を5~50モル%、10~50℃で溶解させる。
工程(B1-1)における触媒量は、5モル%未満だとエチレンオキシドの重合反応速度が遅くなり、長時間の高温反応により末端ビニルエーテル体等の不純物が生じるため、5モル%以上が高品質の高分子量体を製造する上で有利である。触媒が50モル%を超えると、アルコラート化反応の際に反応液の粘性が高まり、あるいは固化してしまい、攪拌効率が低下し、アルコラート化が促進されない傾向がある。また固化した場合はハンドリングが悪くなる傾向があり、吸湿の原因となる。アルコラート化物が吸湿してしまうと、水分由来のポリアルキレングリコール体が生成し、医薬品用途としては望ましくない不純物として混入してしまう。
溶解時の温度が50℃より高いと、分解反応がおき、メタノールやキシリトールが生成する。メタノールが生成した場合、目的物と同様にエチレンオキシドとの付加重合が起き、目的物と同分子量を有する不純物が生成する。メタノール由来の不純物が生成した場合は、目的物と同様に次工程(C)の官能基化にて官能基が導入されるので、生体関連物質と反応可能な不純物となる。また、キシリトールが生成した場合も同様に、エチレンオキシドとの付加重合が起き、目的物の5倍の分子量を有する高分子量不純物が生成する。この高分子量不純物は、次工程(C)の官能基化にて複数の官能基が導入されるので、生体関連物質と複数反応可能な不純物となる。これらの不純物を有したポリオキシエチレン誘導体は、高純度品が求められる医薬品用途には望ましくないものとなる。
10℃より低い温度で溶解する場合、触媒量が50モル%より多い場合と同様、アルコラート化反応の際に反応液の粘性が高まり、あるいは固化してしまい、ハンドリングが悪くなる傾向があり、また吸湿の原因となる。
アルコラート化反応に使用する溶媒については、トルエン、ベンゼン、キシレン、アセトニトリル、テトラヒドロフラン、ジメチルスルホキシド、ジメチルホルムアミド、ジメチルアセトアミド等の非プロトン性溶媒であれば特に制限はないが、トルエンあるいは無溶媒が好ましい。反応時間については、1~24時間が好ましい。1時間より短いと触媒が完全に溶解しない恐れがある。24時間より長いと、前述の分解反応が起きる恐れがある。
工程(B1-2)は触媒としてナトリウムメトキシド、カリウムt-ブトキシド又はカリウムメトキシドを、好ましくはナトリウムメトキシドを5~50モル%の量で添加し、20~80℃で反応させる。このとき交換反応が促進するように、減圧操作を行ってもよい。
触媒量は前述の理由で5~50モル%の量が好ましい。反応温度については、20℃より低いと、交換反応の反応率が低下し、メタノール等のアルコールが残存し、エチレンオキシドの付加重合を経て、目的物と同分子量を有する不純物が生成する。80℃より高いと分解反応が起きる。このアルコラート化反応は、分解反応が起き易いため、反応時間は1~3時間とするのが望ましい。反応溶媒においては、非プロトン性溶媒であれば特に制限はないが、好ましくはトルエン、あるいは無溶媒である。
工程(B2)は、50~130℃の反応温度でエチレンオキシドを付加重合し、式(8)の化合物(ポリオキシエチレン誘導体(8))を得る。反応温度は、50℃より低いと重合反応の速度が遅く、式(8)の化合物の品質が低下する傾向がある。また、130℃より高いと、重合中に末端のビニルエーテル化等の副反応が起き、目的物の品質が低下する傾向がある。重合中、分子量が大きくなるにつれ、反応液の粘度が上がるため、適宜非プロトン性溶剤、好ましくはトルエンを加えてもよい。
工程(C)は、式(8)の化合物(ポリオキシエチレン誘導体(8))の末端の水酸基を官能基化する工程である。官能基の種類によっては、官能基化の際に同時に工程(D)の脱アセタールを行うことができる。
式(8)の化合物(ポリオキシエチレン誘導体(8))の末端の水酸基を用いて、群(II)、群(III)、群(IV)、群(V)、群(VI)および群(VII)に示した各種官能基へ変性させることで、式(9)で示されるポリオキシエチレン誘導体を製造することができる。
また、群(II)、群(III)、群(IV)、群(V)、群(VI)および群(VII)の各官能基を有する化合物を中間体として、これらに更に他の化合物と反応させて官能基化を行うことができる。例えば、(k)の官能基を有する中間体を原料とし、(a)や(d)の官能基を得ることができる。
また、群(II)、群(III)、群(IV)、群(V)、群(VI)および群(VII)の各官能基を有する化合物を中間体として、これらに更に他の化合物と反応させて官能基化を行うことができる。例えば、(k)の官能基を有する中間体を原料とし、(a)や(d)の官能基を得ることができる。
以下、式(9)で示されるポリオキシエチレン誘導体(ポリオキシエチレン誘導体(9))の合成方法を詳細に説明する。
[官能基(b)、(e)の導入方法]
化合物(8)とトルエン、ベンゼン、キシレン、アセトニトリル、酢酸エチル、ジエチルエーテル、t-ブチルメチルエーテル、テトラヒドロフラン、クロロホルム、ジクロロメタン、ジメチルスルホキシド、ジメチルホルムアミド、ジメチルアセトアミド等の非プロトン性溶媒、もしくは無溶媒中、トリエチルアミン、ピリジン、4-ジメチルアミノピリジン等の有機塩基、もしくは炭酸ナトリウム、水酸化ナトリウム、炭酸水素ナトリウム、酢酸ナトリウム、炭酸カリウム、水酸化カリウム等の無機塩基と、下記の式(b1)、式(e1)で示される化合物(化合物(b1)、化合物(e1))のいずれかとを反応させることで、それぞれ官能基(b)、(e)を導入することができる(官能基(b)、(e)が導入された化合物(b)(e)が得られる)。有機塩基、無機塩基の使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましい。また、有機塩基を溶媒として用いてもよい。式(b1)及び式 (e1)におけるW2はCl、Br、Iより選択されるハロゲン原子であり、好ましくはClである。化合物(b1)及び化合物(e1)の使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましく、更に好ましくは等モル~50倍モルの範囲で反応させるのが好ましい。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。生成した化合物は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
化合物(8)とトルエン、ベンゼン、キシレン、アセトニトリル、酢酸エチル、ジエチルエーテル、t-ブチルメチルエーテル、テトラヒドロフラン、クロロホルム、ジクロロメタン、ジメチルスルホキシド、ジメチルホルムアミド、ジメチルアセトアミド等の非プロトン性溶媒、もしくは無溶媒中、トリエチルアミン、ピリジン、4-ジメチルアミノピリジン等の有機塩基、もしくは炭酸ナトリウム、水酸化ナトリウム、炭酸水素ナトリウム、酢酸ナトリウム、炭酸カリウム、水酸化カリウム等の無機塩基と、下記の式(b1)、式(e1)で示される化合物(化合物(b1)、化合物(e1))のいずれかとを反応させることで、それぞれ官能基(b)、(e)を導入することができる(官能基(b)、(e)が導入された化合物(b)(e)が得られる)。有機塩基、無機塩基の使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましい。また、有機塩基を溶媒として用いてもよい。式(b1)及び式 (e1)におけるW2はCl、Br、Iより選択されるハロゲン原子であり、好ましくはClである。化合物(b1)及び化合物(e1)の使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましく、更に好ましくは等モル~50倍モルの範囲で反応させるのが好ましい。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。生成した化合物は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。

(式中、W2はCl、Br、Iより選択されるハロゲン原子を示す。Y2は炭素数1~10のフッ素原子を含んでいてもよい炭化水素基を示す。)
[官能基(f)の導入方法]
化合物(8)や後述するアミン(k)を無水コハク酸や無水グルタル酸等のジカルボン酸無水物と反応させることで、官能基(f)が導入されたカルボキシル体(f)を得ることができる。化合物(8)やアミン(k)とジカルボン酸無水物との反応は、上述の非プロトン性溶媒、もしくは無溶媒中で行う。ジカルボン酸無水物の使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。反応にはトリエチルアミン、ピリジン、ジメチルアミノピリジン等の有機塩基や炭酸ナトリウム、水酸化ナトリウム、炭酸水素ナトリウム、酢酸ナトリウム、炭酸カリウム、水酸化カリウム等の無機塩基を触媒として用いてもよい。触媒の使用割合は0.1~50重量%が好ましく、さらに好ましくは0.5~20重量%である。このようにして生成したカルボキシル体(f)は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよいし、縮合反応の原料として用いる場合は、そのまま用いてもよい。
化合物(8)や後述するアミン(k)を無水コハク酸や無水グルタル酸等のジカルボン酸無水物と反応させることで、官能基(f)が導入されたカルボキシル体(f)を得ることができる。化合物(8)やアミン(k)とジカルボン酸無水物との反応は、上述の非プロトン性溶媒、もしくは無溶媒中で行う。ジカルボン酸無水物の使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。反応にはトリエチルアミン、ピリジン、ジメチルアミノピリジン等の有機塩基や炭酸ナトリウム、水酸化ナトリウム、炭酸水素ナトリウム、酢酸ナトリウム、炭酸カリウム、水酸化カリウム等の無機塩基を触媒として用いてもよい。触媒の使用割合は0.1~50重量%が好ましく、さらに好ましくは0.5~20重量%である。このようにして生成したカルボキシル体(f)は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよいし、縮合反応の原料として用いる場合は、そのまま用いてもよい。
化合物(8)を、6-ブロモヘキサン酸エチルや7-ブロモヘプタン酸エチル等のハロゲン化アルキルエステルと反応させることで、カルボキシル体(f)を得ることができる。化合物(8)とハロゲン化アルキルエステルとのエーテル化反応は、上述の非プロトン性溶媒、もしくは無溶媒中で行う。ハロゲン化アルキルエステルの使用割合は、特に制限はないが、化合物(8)に対して等モル以上が好ましく、更に好ましくは等モル~30倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。反応にはトリエチルアミン、ピリジン、ジメチルアミノピリジン等の有機塩基や炭酸ナトリウム、水酸化ナトリウム、炭酸水素ナトリウム、酢酸ナトリウム、炭酸カリウム、水酸化カリウム等の無機塩基を触媒として用いてもよい。触媒の使用割合は0.1~500重量%が好ましく、さらに好ましくは0.5~300重量%である。エーテル化後、有機塩基の場合は水酸化ナトリウムや水酸化カリウム等の水溶液を、無機塩基の場合は、水を追加してエステルの加水分解を行う。反応温度としては、0~100℃が好ましく、更に好ましくは、20~100℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。反応後、塩酸や硫酸等で中和を行う。このようにして生成したカルボキシル体(f)は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよいし、縮合反応の原料として用いる場合は、そのまま用いてもよい。
[官能基(a)の導入方法]
カルボキシル体(f)を、ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC)等の縮合剤存在下、N-ヒドロキシコハク酸イミドと縮合反応させることで、官能基(a)が導入されたコハク酸イミド体(a)を得ることができる。かかる縮合反応は、上述の非プロトン性溶媒、もしくは無溶媒中で行う。縮合剤としては、特に制限は無いが、好ましくはDCCである。DCCの使用割合は、カルボキシル体(f)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。N-ヒドロキシコハク酸イミドの使用割合はカルボキシル体(f)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成したコハク酸イミド体(a)は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
カルボキシル体(f)を、ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC)等の縮合剤存在下、N-ヒドロキシコハク酸イミドと縮合反応させることで、官能基(a)が導入されたコハク酸イミド体(a)を得ることができる。かかる縮合反応は、上述の非プロトン性溶媒、もしくは無溶媒中で行う。縮合剤としては、特に制限は無いが、好ましくはDCCである。DCCの使用割合は、カルボキシル体(f)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。N-ヒドロキシコハク酸イミドの使用割合はカルボキシル体(f)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成したコハク酸イミド体(a)は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
また、コハク酸イミド体(a)は、化合物(8)とN,N'-ジスクシンイミドカーボネートとを反応させることでも得ることができる。反応は、上記の反応と同様に非プロトン性溶媒中、もしくは無溶媒中で行う。N,N'-ジスクシンイミドカーボネートの使用割合は化合物(8)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
[官能基(k)の導入方法]
化合物(8)を水、アセトニトリル等の溶媒中、水酸化ナトリウム、水酸化カリウム等の無機塩基を触媒とし、アクリロニトリル等を付加させてニトリル体を得たあと、オートクレーブ中でニッケルやパラジウム触媒下でニトリル基の水添反応を行うことで、官能基(k)を有するアミン体(k)を得ることができる。ニトリル体を得る際の無機塩基の使用割合は、特に制限はないが、化合物(8)に対して0.01~50重量%が好ましい。アクリロニトリル等の使用割合は、特に制限はないが、化合物(8)の重量に対して0.5~5倍重量が好ましく、更に好ましくは1~4倍重量の範囲で反応させるのが、より好ましい。また、アクリロニトリルを溶媒として用いてもよい。反応温度としては、-50~100℃が好ましく、更に好ましくは、-20~60℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。続くニトリル体の水添反応における反応溶媒は、反応に関与しない溶媒であれば特に制限は無いが、好ましくはトルエンである。ニッケル、もしくはパラジウム触媒の使用割合は、特に制限は無いが、ニトリル体に対して0.05~30重量%であり、好ましくは0.5~20重量%である。反応温度は20~200℃が好ましく、更に好ましくは50~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。水素圧は2~10MPaが好ましく、更に好ましくは3~8MPaである。また、2量化を防ぐために反応系中にアンモニアを加えてもよい。アンモニアを加える場合のアンモニア圧は特に制限はないが、0.1~10MPaであり、更に好ましくは0.3~2MPaである。生成した化合物は前述の精製手段にて精製してもよい。
化合物(8)を水、アセトニトリル等の溶媒中、水酸化ナトリウム、水酸化カリウム等の無機塩基を触媒とし、アクリロニトリル等を付加させてニトリル体を得たあと、オートクレーブ中でニッケルやパラジウム触媒下でニトリル基の水添反応を行うことで、官能基(k)を有するアミン体(k)を得ることができる。ニトリル体を得る際の無機塩基の使用割合は、特に制限はないが、化合物(8)に対して0.01~50重量%が好ましい。アクリロニトリル等の使用割合は、特に制限はないが、化合物(8)の重量に対して0.5~5倍重量が好ましく、更に好ましくは1~4倍重量の範囲で反応させるのが、より好ましい。また、アクリロニトリルを溶媒として用いてもよい。反応温度としては、-50~100℃が好ましく、更に好ましくは、-20~60℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。続くニトリル体の水添反応における反応溶媒は、反応に関与しない溶媒であれば特に制限は無いが、好ましくはトルエンである。ニッケル、もしくはパラジウム触媒の使用割合は、特に制限は無いが、ニトリル体に対して0.05~30重量%であり、好ましくは0.5~20重量%である。反応温度は20~200℃が好ましく、更に好ましくは50~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。水素圧は2~10MPaが好ましく、更に好ましくは3~8MPaである。また、2量化を防ぐために反応系中にアンモニアを加えてもよい。アンモニアを加える場合のアンモニア圧は特に制限はないが、0.1~10MPaであり、更に好ましくは0.3~2MPaである。生成した化合物は前述の精製手段にて精製してもよい。
アミン体(k)は、化合物(e)をアンモニア水と反応させることでも得ることができる。反応は、アンモニア水中で行い、アンモニアの濃度は特に制限は無いが、好ましくは10~40%の範囲である。アンモニア水の使用割合は、化合物(e)の重量に対して1~300倍であるのが好ましい。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~72時間が好ましく、更に好ましくは1~36時間である。また、アミン体(k)は、オートクレーブ中、化合物(e)をアンモニアと反応させても得ることができる。反応溶剤については、特に制限はないが、好ましくはメタノール、エタノールが挙げられる。アンモニア量は化合物(e)に対して10~300重量%が好ましく、更に好ましくは20~200重量%である。反応温度としては、50~200℃が好ましく、80~150℃が更に好ましい。反応時間については、10分~24時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は前述の精製手段にて精製してもよい。
また、アミン体(k)は非プロトン性溶媒中、光延反応で化合物(8)とフタルイミドを結合させ、多価アミンで脱保護することで得ることもできる。光延反応の反応条件は特に制限は無いが、反応溶媒としては、クロロホルム、ジクロロメタンが好ましい。また、トリフェニルホスフィンを化合物(8)に対して等モル以上、好ましくは等モル~50倍モル、アゾジカルボン酸ジイソプロピルを化合物(8)に対して等モル以上、好ましくは等モル~50倍モル使用するのが好ましい。反応温度としては、0~100℃が好ましく、更に好ましくは、10~50℃である。反応時間は10分~72時間が好ましく、更に好ましくは30分~6時間である。
脱保護については、ヒドラジン、もしくはエチレンジアミンのような多価アミンを化合物(8)に対して等モル以上、好ましくは等モル~500倍モル使用するのが好ましい。反応溶媒としては特に制限は無いが、メタノールが好ましい。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~72時間が好ましく、更に好ましくは1~10時間である。生成した化合物は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
[官能基(d)の導入方法]
前述の手法により得られたアミン体(k)のアミノ基を、前述の非プロトン性溶媒、もしくは無溶媒中、無水マレイン酸と反応させてマレイミド体を得たあと、無水酢酸及び酢酸ナトリウムを触媒として、閉環反応させることで、官能基(d)が導入されたマレイミド体(d)を得ることができる。マレイミド化反応における無水マレイン酸の使用割合は、特に制限はないが、アミン体(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~120℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成したマレイミド体(d)は、前述の精製手段にて精製してもよいし、そのまま次の閉環反応に用いてもよい。
前述の手法により得られたアミン体(k)のアミノ基を、前述の非プロトン性溶媒、もしくは無溶媒中、無水マレイン酸と反応させてマレイミド体を得たあと、無水酢酸及び酢酸ナトリウムを触媒として、閉環反応させることで、官能基(d)が導入されたマレイミド体(d)を得ることができる。マレイミド化反応における無水マレイン酸の使用割合は、特に制限はないが、アミン体(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~120℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成したマレイミド体(d)は、前述の精製手段にて精製してもよいし、そのまま次の閉環反応に用いてもよい。
続く閉環反応における反応溶媒は特に限定されないが、非プロトン性溶媒または無水酢酸が好ましい。酢酸ナトリウムの使用割合は、特に制限はないが、マレイミド体(d)に対して等モル以上が好ましく、更に好ましくは等モル~50倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は前述の精製手段にて精製してもよい。
上記マレイミド体(d)は、下記の式(d1)に示される化合物(d1)と、上述のアミン体(k)のアミノ基を反応させることでも得ることができる。反応は、前述の非プロトン性溶媒、もしくは無溶媒中で行い、化合物(d1)をアミン体(k)のアミノ基に対して等モル以上加えて反応させる。化合物(d1)の使用割合はアミン体(k)のアミノ基に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。反応時は遮光してもよい。生成した化合物は前述の精製手段にて精製してもよい。

(式中、Qは炭素数1~9の炭化水素基を示す。Y1は水素原子または炭素数1~5の炭化水素を示す。)
[官能基(i)の導入方法]
前述の手法により得られたアミン体(k)のアミンを、前述の非プロトン性溶媒、もしくは無溶媒中、ヨード酢酸無水物と反応させることで官能基(i)を得ることができる。ヨード酢酸無水物の使用割合は、特に制限はないが、アミン体(k)のアミノ基に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~120℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した官能基(i)を有する化合物(i)は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
前述の手法により得られたアミン体(k)のアミンを、前述の非プロトン性溶媒、もしくは無溶媒中、ヨード酢酸無水物と反応させることで官能基(i)を得ることができる。ヨード酢酸無水物の使用割合は、特に制限はないが、アミン体(k)のアミノ基に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~120℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した官能基(i)を有する化合物(i)は、抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
また、官能基(i)は、アミン体(k)を、DCC、EDC等の縮合剤存在下、ヨード酢酸と縮合反応させることで、得ることができる。縮合反応も同様に前述の非プロトン性溶媒中、もしくは無溶媒中で行う。縮合剤としては、特に制限は無いが、好ましくはDCCである。DCCの使用割合はアミン体(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。ヨード酢酸の使用割合はアミン体(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は前述の精製手段にて精製してもよい。
[官能基(l)の導入方法]
カーボネート体(b)を、トリエチルアミンやピリジン等のアルカリ触媒存在下、下記の式(l1)で示される化合物(化合物(l1))と反応させることで、オキシフタルイミド基が導入されたオキシフタルイミド体を得ることができる。反応は無溶媒下もしくは極性溶媒下で行うことができ、溶媒は特に制限はないが、好ましくはメタノールである。アルカリ触媒の使用割合は、特に制限はないが、カーボネート体(b)に対して等モル以上が好ましく、更に好ましくは等モル~20倍モルである。化合物(l1)の使用割合は、カーボネート体(b)に対して等モル以上が好ましく、更に好ましくは等モル~20モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよいし、精製せず次の工程に進めてもよい。
カーボネート体(b)を、トリエチルアミンやピリジン等のアルカリ触媒存在下、下記の式(l1)で示される化合物(化合物(l1))と反応させることで、オキシフタルイミド基が導入されたオキシフタルイミド体を得ることができる。反応は無溶媒下もしくは極性溶媒下で行うことができ、溶媒は特に制限はないが、好ましくはメタノールである。アルカリ触媒の使用割合は、特に制限はないが、カーボネート体(b)に対して等モル以上が好ましく、更に好ましくは等モル~20倍モルである。化合物(l1)の使用割合は、カーボネート体(b)に対して等モル以上が好ましく、更に好ましくは等モル~20モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよいし、精製せず次の工程に進めてもよい。

(式中、Qは炭素数1~9の炭化水素基を示す。)
オキシフタルイミド体は非プロトン性溶媒中、光延反応で化合物(8)とヒドロキシフタルイミドを結合させ、多価アミンで脱保護することで得ることもできる。光延反応の反応条件は特に制限は無いが、反応溶媒としては、クロロホルム、ジクロロメタンが好ましい。また、トリフェニルホスフィンを化合物(8)に対して等モル以上、好ましくは等モル~50倍モル、アゾジカルボン酸ジイソプロピルを化合物(8)に対して等モル以上、好ましくは等モル~50倍モル使用するのが好ましい。反応温度としては、0~100℃が好ましく、更に好ましくは、10~50℃である。反応時間は10分~72時間が好ましく、更に好ましくは30分~6時間である。
これらの方法で得られたオキシフタルイミド体を、ヒドラジンやエチレンジアミン等の多価アミン存在下で反応させることで、官能基(l)が導入されたオキシアミン体(l)を得ることができる。
反応溶媒は、特に制限はないが、メタノール、ジクロロメタン、水が好ましい。多価アミンの使用割合は、特に制限はないが、オキシフタルイミド体に対して等モル以上が好ましく、更に好ましくは等モル~50倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は抽出、再結晶、吸着処理、再沈殿、カラムクロマトグラフィー、超臨界抽出等の精製手段にて精製してもよい。
[官能基(c)の導入方法]
化合物(e)を下記の式(c1)で示されるアセタール化合物(化合物(c1))と反応させてアセタール体を得た後、酸性条件にて加水分解を行うことで、官能基(c)を有するアルデヒド体(c)を得ることができる。アセタール化反応は前述の非プロトン性溶媒中、もしくは無溶媒中、化合物(e)と等モル以上、好ましくは等モル~50倍モルの化合物(c1)を反応させることで得ることができる。化合物(c1)は相当するアルコールから、金属ナトリウム、金属カリウム、水素化ナトリウム、水素化カリウム、ナトリウムメトキシド、カリウムt-ブトキシド等を用いて調製することができる。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。
化合物(e)を下記の式(c1)で示されるアセタール化合物(化合物(c1))と反応させてアセタール体を得た後、酸性条件にて加水分解を行うことで、官能基(c)を有するアルデヒド体(c)を得ることができる。アセタール化反応は前述の非プロトン性溶媒中、もしくは無溶媒中、化合物(e)と等モル以上、好ましくは等モル~50倍モルの化合物(c1)を反応させることで得ることができる。化合物(c1)は相当するアルコールから、金属ナトリウム、金属カリウム、水素化ナトリウム、水素化カリウム、ナトリウムメトキシド、カリウムt-ブトキシド等を用いて調製することができる。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。
下記の式(c2)で示される化合物(化合物(c2))を用いる場合、化合物(8)の水酸基を上述の方法でアルコラートとした後、前述の非プロトン性溶媒中、もしくは無溶媒中、化合物(c2)を等モル以上、好ましくは等モル~100倍モルの割合で反応させることでアセタール体を得ることができる。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、特に好ましくは30分~24時間である。
下記の(c3)で示される化合物(化合物(c3))を用いる場合、前述の官能基(a)、(b)、(e)もしくは(f)が導入された化合物(化合物(a)、(b)、(e)もしくは(f))と該化合物(c3)を反応させることでアセタール体を得ることができる。該反応の溶媒は特に制限されないが、好ましくは前述の非プロトン性溶媒中で行う。化合物(a)、(b)、(e)もしくは(f)に対する化合物(c3)の仕込み割合は、等モル以上が好ましく、更に好ましくは等モル~10倍モルである。反応温度としては、-30~200℃が好ましく、更に好ましくは、0~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。化合物(f)を用いる場合は、適宜DCC、EDC等の縮合剤を用いてもよい。いずれのアセタール化反応も遮光して行ってもよい。このようにして得られたアセタール体は前述の精製手段にて精製してもよいし、精製を行わずにそのまま次のアルデヒド化反応に用いてもよい。
アセタール体を0.1~50%の水溶液とし、酢酸、リン酸、硫酸、塩酸等の酸にてpH1~4に調整した水溶液中で加水分解させることで、アルデヒド体(c)を得ることができる。反応温度としては、-20~100℃が好ましく、更に好ましくは、0~80℃である。反応時間は10分~24時間が好ましく、更に好ましくは30分~10時間である。反応は遮光して行ってもよい。生成した化合物は前述の精製手段にて精製してもよい。また、このアルデヒド化においては、同時に工程(D)の脱アセタールを行うこともできる。

(式中、R4、R5はそれぞれ独立して炭素数1~3の炭化水素基を示し、互いに同一であっても、異なっていてもよい。また、相互に環を形成していてもよい。Mはナトリウムもしくはカリウムを示し、W2はCl、Br、Iから選択されるハロゲン原子であり、tは1~5の整数である。)
[官能基(g)の導入方法]
官能基(g)を有するメルカプト体(化合物(g))は、化合物(e)とチオウレア等のチア化剤と反応させることで得ることができる。化合物(e)の製造は前述の通りである。チア化反応は水、アルコール、アセトニトリル等の溶媒中もしくは無溶媒中で行う。チオウレアの使用割合は、化合物(e)に対して等モル以上が好ましく、更に好ましくは等モルから50倍モルの範囲である。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。反応後、生成したチアゾリウム塩をアルカリ加水分解し、メルカプト体を得ることができる。生成した化合物は前述の精製手段にて精製してもよい。また、このメルカプト化においては、加水分解後のpH調製において、同時に工程(D)の脱アセタールを行うこともできる。
官能基(g)を有するメルカプト体(化合物(g))は、化合物(e)とチオウレア等のチア化剤と反応させることで得ることができる。化合物(e)の製造は前述の通りである。チア化反応は水、アルコール、アセトニトリル等の溶媒中もしくは無溶媒中で行う。チオウレアの使用割合は、化合物(e)に対して等モル以上が好ましく、更に好ましくは等モルから50倍モルの範囲である。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。反応後、生成したチアゾリウム塩をアルカリ加水分解し、メルカプト体を得ることができる。生成した化合物は前述の精製手段にて精製してもよい。また、このメルカプト化においては、加水分解後のpH調製において、同時に工程(D)の脱アセタールを行うこともできる。
また、上記メルカプト体は、化合物(e)を下記の式(g1)で示される化合物(化合物(g1))と反応させ、1級アミンにて分解させることでも得ることができる。(e)と(g1)との反応は、前述の非プロトン性溶媒中、もしくは無溶媒中で行う。化合物(g1)の使用割合は、化合物(e)に対して等モル以上が好ましく、更に好ましくは等モルから50倍モルの範囲である。反応温度としては、0~300℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。続く1級アミンによるアルカリ分解は、前述の非プロトン性溶媒、もしくは無溶媒中で行う。用いる1級アミンとしては特に制限は無いが、好ましくはアンモニア、メチルアミン、エチルアミン、プロピルアミン、ブチルアミン、ペンチルアミン、ヘキシルアミン、シクロヘキシルアミン、エタノールアミン、プロパノールアミン、ブタノールアミン等が挙げられる。当然、これらの1級アミンを溶媒として用いてもよい。生成した化合物は前述の精製手段にて精製してもよい。

[官能基(h)の導入方法]
官能基(h)を有する化合物(化合物(h))は、化合物(g)を2,2-ジピリジルジスルフィドと反応させることで得ることができる。反応では、溶媒は特に制限されないが、好ましくはアルコール中で行う。化合物(g)に対する2,2-ジピリジルジスルフィドの仕込み割合は、等モル以上が好ましく、更に好ましくは等モル~50倍モルである。反応温度としては、-30~100℃が好ましく、更に好ましくは、0~60℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。このようにして得られたアセタール体は前述の精製手段にて精製してもよい。
官能基(h)を有する化合物(化合物(h))は、化合物(g)を2,2-ジピリジルジスルフィドと反応させることで得ることができる。反応では、溶媒は特に制限されないが、好ましくはアルコール中で行う。化合物(g)に対する2,2-ジピリジルジスルフィドの仕込み割合は、等モル以上が好ましく、更に好ましくは等モル~50倍モルである。反応温度としては、-30~100℃が好ましく、更に好ましくは、0~60℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。このようにして得られたアセタール体は前述の精製手段にて精製してもよい。
[官能基(m)の導入方法]
前述の化合物(a)、(b)、(c)もしくは(e)を、前述の非プロトン性溶媒、もしくは無溶媒中、カルバジン酸tert-ブチルと反応させ、tert-ブトキシカルボニル基(Boc基)を脱保護することで、官能基(m)を有する化合物(化合物(m))を得ることができる。カルバジン酸tert-ブチルの使用割合は、特に制限はないが、化合物(a)、(b)、(c)もしくは(e)に対して等モル以上が好ましく、更に好ましくは等モル~10モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物(m)は、前述の精製手段にて精製してもよい。また、このBoc基を脱保護する際に、同時に工程(D)の脱アセタール化を行うこともできる。
前述の化合物(a)、(b)、(c)もしくは(e)を、前述の非プロトン性溶媒、もしくは無溶媒中、カルバジン酸tert-ブチルと反応させ、tert-ブトキシカルボニル基(Boc基)を脱保護することで、官能基(m)を有する化合物(化合物(m))を得ることができる。カルバジン酸tert-ブチルの使用割合は、特に制限はないが、化合物(a)、(b)、(c)もしくは(e)に対して等モル以上が好ましく、更に好ましくは等モル~10モルである。反応温度としては、0~200℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物(m)は、前述の精製手段にて精製してもよい。また、このBoc基を脱保護する際に、同時に工程(D)の脱アセタール化を行うこともできる。
[官能基(j)の導入方法]
前述の化合物(a)、(b)、(c)もしくは(e)を、下記式(j1)で示されるアセチレン化合物(化合物(j1))と反応させることで、官能基(j)を有するアセチレン化合物(化合物(j))を得ることができる。アセチレン化反応はプロトン性溶媒中、もしくは無溶媒中、化合物(a)、(b)、(c)もしくは(e)に対して等モル以上、好ましくは等モル~50倍モルの化合物(j1)を反応させることで得ることができる。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。生成した化合物は前述の精製手段にて精製してもよい。
前述の化合物(a)、(b)、(c)もしくは(e)を、下記式(j1)で示されるアセチレン化合物(化合物(j1))と反応させることで、官能基(j)を有するアセチレン化合物(化合物(j))を得ることができる。アセチレン化反応はプロトン性溶媒中、もしくは無溶媒中、化合物(a)、(b)、(c)もしくは(e)に対して等モル以上、好ましくは等モル~50倍モルの化合物(j1)を反応させることで得ることができる。反応温度としては、0~300℃が好ましく、更に好ましくは、20~150℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~24時間である。生成した化合物は前述の精製手段にて精製してもよい。

(式中、tは1~5の整数である。Y3は水素原子または炭素数1~5の炭化水素基を示す。)
[官能基(n)の導入方法]
前述の通りの手法で得られたアミン体(k)を、DCC、EDC等の縮合剤存在下、下記の式(n1)で示される化合物(化合物(n1))と反応させることで、官能基(n)を有するアジド化合物(化合物(n))を得ることができる。縮合反応は前述の非プロトン性溶媒中、もしくは無溶媒中で行う。縮合剤としては、特に制限は無いが、好ましくはDCCである。DCCの使用割合はアミン体(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。化合物(n1)の使用割合は化合物(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は前述の精製手段にて精製してもよい。
前述の通りの手法で得られたアミン体(k)を、DCC、EDC等の縮合剤存在下、下記の式(n1)で示される化合物(化合物(n1))と反応させることで、官能基(n)を有するアジド化合物(化合物(n))を得ることができる。縮合反応は前述の非プロトン性溶媒中、もしくは無溶媒中で行う。縮合剤としては、特に制限は無いが、好ましくはDCCである。DCCの使用割合はアミン体(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。化合物(n1)の使用割合は化合物(k)に対して等モル以上が好ましく、更に好ましくは等モル~5倍モルである。反応温度としては、0~100℃が好ましく、更に好ましくは、20~80℃である。反応時間は10分~48時間が好ましく、更に好ましくは30分~12時間である。生成した化合物は前述の精製手段にて精製してもよい。

(式中、Qは炭素数1~9の炭化水素基を示す。)
工程(D)は、官能基を有した式(9)で示されるポリオキシエチレン誘導体(以下、「化合物(9)」とも称す。)の環状アセタール構造を切断する脱保護工程である。官能基の種類によっては、工程(D)の脱アセタール後にさらに官能基化を行うこともできる。
環状アセタールの脱保護の方法としては、 PROTECTIVE GROUPS IN ORGANIC SYNTHESIS(THEODORA W.GREENE et al)等に記載があるような一般的な脱保護法であれば特に制限はないが、具体的には、酸触媒存在下、脱保護することができる。酸触媒は酢酸、塩酸、リン酸、p-トルエンスルホン酸等が挙げられ、好ましくは塩酸、リン酸であり、より好ましくはリン酸である。
酸の使用量は、化合物(9)に対して0.05~2重量倍が好ましく、0.1~1重量倍がより好ましい。脱保護反応に使用する溶媒については、水、メタノール、エタノール、アセトニトリル、テトラヒドロフラン、ジオキサン、ジメチルスルホキシド、ジメチルホルムアミド、ジメチルアセトアミドであり、水あるいはメタノールが好ましい。溶媒の使用量は、化合物(8)の1~50重量倍、好ましくは2~35重量倍、さらに好ましくは5~20重量倍である。
反応時間は、1~24時間が好ましい。1時間より短いと脱保護反応が不十分となる。24時間より長いと、酸によるポリオキシエチレンの酸化分解や官能基の失活が起こる恐れがある。反応温度は、通常0~60℃、好ましくは、10~40℃である。
脱保護後、抽出、再結晶、吸着処理、再沈殿、超臨界抽出等の精製手段にて精製してもよく、好適には、再結晶を行い、結晶を減圧下にて乾燥することで、化合物(9)を得ることができる。
工程(D)の脱アセタール後にさらに官能基化を行うこともできる。脱アセタール条件にて反応、分解してしまう恐れのある官能基については、工程(D)後に官能基化を行うことが望ましい。
本発明のポリオキシエチレン誘導体(1)は以下の工程図(工程図III)に示す製法によっても製造することができる。
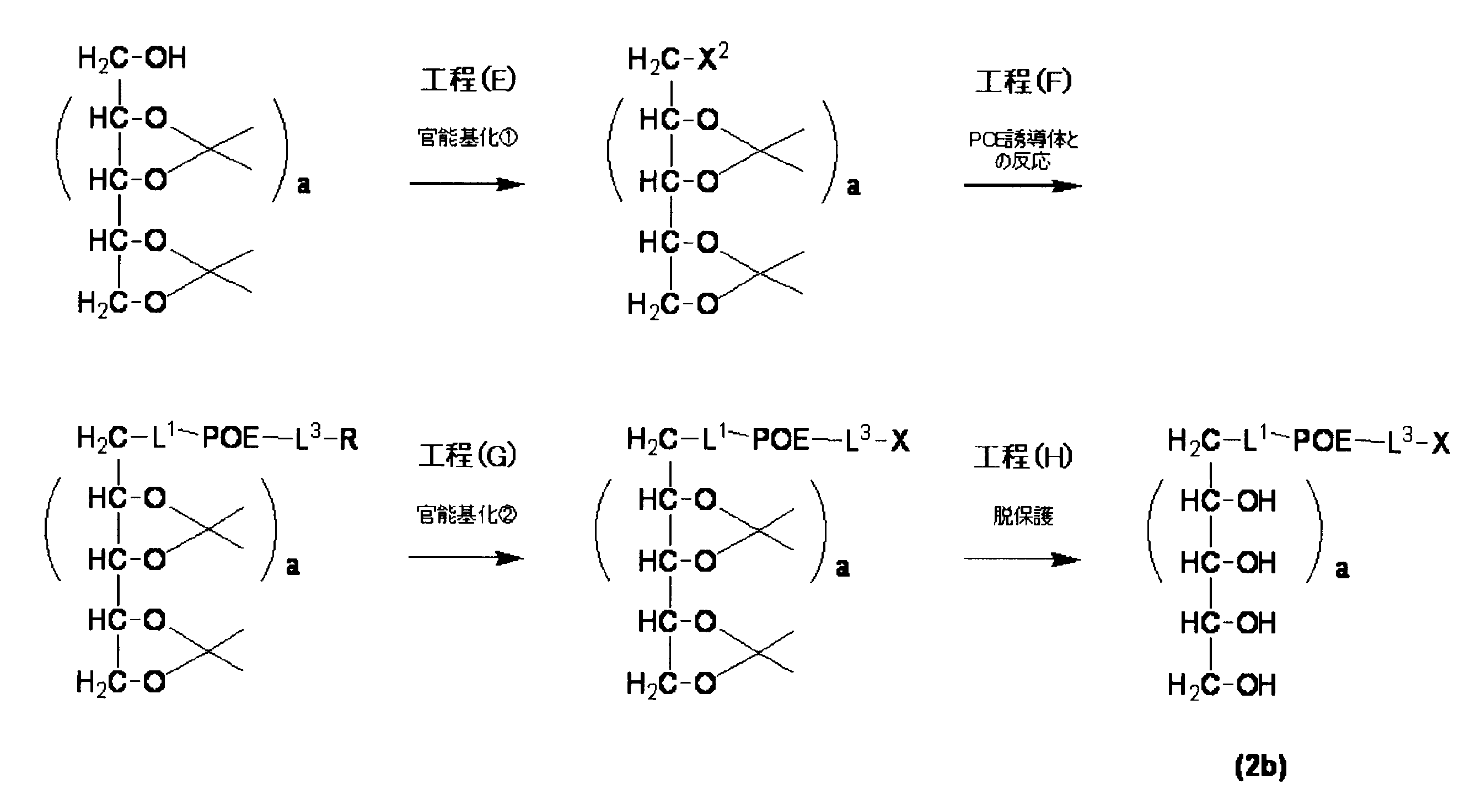
(式中、POE、L1、L3、X、aは前記と同義である。Rは保護基で保護された官能基または保護されていてもよい水酸基を示し、X2はアミノ基、活性カーボネート基または活性スルホネート基を示す。)
工程(E)は、保護された多価アルコール誘導体の残存した水酸基を官能基に変換する工程である。
工程(F)は、工程(E)で官能基化された多価アルコール誘導体と、ポリオキシエチレン誘導体を反応にて結合する工程である。
工程(G)は保護基であるRの脱保護により、官能基化を行う工程である。必要であれば、前述の工程図I、IIにおける工程(C)にならって官能基化を行うこともできる。ポリオキシエチレン末端のRの保護基の種類によっては、脱保護と同時に、次工程の脱アセタールを行うことができる。
工程(H)は、環状アセタール構造を切断する工程である。a=1の場合は4個の水酸基を出現させ、a=2の場合は6個の水酸基を出現させる。ポリオキシエチレン末端の官能基Xの種類によっては、工程(H)後にさらに官能基化を行うこともできる。
上記の工程工程(E)(F)(G)(H)を行うことで、式(2b)で表されるポリオキシエチレン誘導体(ポリオキシエチレン誘導体(2b))が製造される。
以下に、かかるポリオキシエチレン誘導体(2b)の製造方法の好適な具体例をさらに説明するが、a=1と、a=2のどちらの場合においても、同様の製法にて製造することができるため、a=1の誘導体、すなわち、以下の式(15)で表されるポリオキシエチレン誘導体(ポリオキシエチレン誘導体(15))について説明を行う。

(式中、POE、L1、L3、Xは前記と同義である。)
ポリオキシエチレン誘導体(15)は、次のような工程図(工程図IV)に示すルートにより製造することができる。

(式中、R1、R2、R3は炭素数1~10の炭化水素基を示し、Wはハロゲン原子を示す。POE、L3、Xは前記と同義である。)
(式中、POE、L1、L3、R、X2、Xは前記と同義である。)
(式中、POE、L1、L3、R、X2、Xは前記と同義である。)
上記工程図において、化合物(15)は、式(2b)で示される化合物に相当する。
工程(E)は、式(5)と式(6)で示される化合物の混合物を官能基化し、構造異性体を含まない式(12)の化合物を得るための工程である。工程(E)は、以下の工程(E1)または工程(E2)のどちらを用いてもよい。
工程(E1)は、以下の(E1-1)、(E1-2)の2つの工程から成る。
工程(E1-1)は、構造異性体の片方のみを選択的にフタルイミド化し、もう一方と分離する工程。式(5)で示される1,2,3,4-ジイソプロピリデンキシリトールの1級水酸基のみ選択的にフタルイミド化し、式(6)で示される1,2,4,5-ジイソプロピリデンキシリトールと分離する。
工程(E1-2)は、フタルイミド基の脱保護工程である。

上記工程図において、化合物(17)は、式(12)で示される化合物に相当する。
工程(E1-1)は、式(5)と式(6)で示される化合物の混合物をフタルイミドと反応させ、化合物(5)の1級水酸基のみをフタルイミド化することにより、式(17)で示される化合物を得る。
フタルイミド化は、反応前に共沸脱水し、反応系中の水分を除去することが好ましく、使用する溶媒は共沸脱水できる非プロトン性溶媒であれば特に制限されないが、好ましくは、トルエン、キシレンまたはシクロヘキセンであり、さらに好ましくはトルエンである。溶媒量は、混合物の1~10重量倍、好ましくは2~6重量倍、さらに好ましくは3~5重量倍である。
式(5)と式(6)で示される化合物の混合物を共沸脱水できる有機溶媒に溶解後、仕込んだ有機溶媒量の5~75重量%、好ましくは10~50重量%の溶媒量を共沸温度以上で30分以上、3時間以内で還流、留去する。留去量が少ないか、または還流時間が30分より短いと脱水が不十分となり、反応にて残存水分が副反応を起し、純度が低下する恐れがある。
脱水後、反応に適した反応溶媒を添加する。反応溶媒としては有機溶媒が好ましく、非プロトン性溶剤であれば特に制限されないが、脱水処理された溶媒が好ましく、その中でも好ましくはクロロホルム、ジクロロメタン、テトラヒドロフラン、アセトニトリル、ジメチルスルホキシドであり、更に好ましくはジクロロメタン、クロロホルムである。有機溶媒の量は、混合物の1~50重量倍、好ましくは2~30重量倍、さらに好ましくは3~20重量倍である。含水量が低い溶媒を使用するのは前述した副反応を抑制するためである。
フタルイミド化に用いられるフタルイミドの量は、式(5)と式(6)で示される化合物の混合物に対して1~10モル当量倍、好ましくは1.01~5モル当量倍、さらに好ましくは1.02~3モル当量倍である。
フタルイミド化に用いられるアゾ系試薬としては1,1’-アゾビス(N,N-ジメチルホルムアミド)、1,1’-(アゾジカルボニル)ジピペリジン、アゾジカルボン酸ジベンジル、アゾジカルボン酸ジエチル、アゾジカルボン酸ジイソプロピル、アゾジカルボン酸ジメチル、1,1’-アゾビス(N,N-ジイソプロピルホルムアミド)、1,6-ジメチル-1,5,7-ヘキサヒドロ-1,4,6,7-テトラゾシン-2,5-ジオン、などが挙げられ、好ましくはアゾジカルボン酸ジエチル、アゾジカルボン酸ジイソプロピルであり、さらに好ましくはアゾジカルボン酸ジイソプロピルである。アゾ系試薬の量は、式(5)と式(6)で示される化合物の混合物に対して1~10モル当量倍、好ましくは1.01~5モル当量倍、さらに好ましくは1.02~3モル当量倍である。
フタルイミド化に用いられるホスフィン系試薬としてはジシクロヘキシルフェニルホスフィン、ジエチルフェニルホスフィン、4-(ジメチルアミノ)フェニルジフェニルホスフィン、ジフェニル-2-ピリジルホスフィン、イソプロピルジフェニルホスフィン、トリイソブチルホスフィン、トリ-n-ブチルホスフィン、トリ-t-ブチルホスフィン、トリシクロヘキシルホスフィン、トリ-n-ヘキシルホスフィン、トリ-n-オクチルホスフィン、トリフェニルホスフィンなどが挙げられ、好ましくはトリフェニルホスフィンである。ホスフィン系試薬の量は、式(5)と式(6)で示される化合物の混合物に対して1~10モル当量倍、好ましくは1.01~5モル当量倍、さらに好ましくは1.02~3モル当量倍である。
フタルイミド、ホスフィン系試薬を投入し、最後にアゾ系試薬をゆっくり仕込み反応を行う。反応温度については特に制限されないが、好ましくは室温である。また反応時間は5分以上が好ましく、5分未満であると反応率が低くなる恐れがある。
反応終了後の反応液には、式(6)で示される未反応物が含まれている。これを除去する方法は、特に制限はないが、カラムクロマトグラフィー、蒸留、抽出、再結晶、超臨界抽出等の精製手段にて式(6)で示される未反応物を分離するのが好ましく、再結晶にて精製するのがさらに好ましい。
再結晶にて精製する場合、良溶媒はトルエン、酢酸エチル、メタノール、エタノール、アセトニトリルなどが挙げられ、好ましくはトルエン、酢酸エチル、エタノールであり、さらに好ましくは酢酸エチルである。これらの溶媒は1種単独で使用することもでき、2種以上を組み合せて使用することもできる。また貧溶媒はヘキサン、ジエチルエーテル、メチルt-ブチルエーテルなどが挙げられ、好ましくはヘキサンである。良溶媒の量は、混合物の1~50重量倍、好ましくは2.5~35重量倍、さらに好ましくは5~20重量倍である。また貧溶媒の量は、0.5~30重量倍、好ましくは1~20重量倍、さらに好ましくは2~10重量倍である。
再結晶の温度は-20~30℃であり、好ましくは-10~20℃である。温度が30℃を超えると結晶が溶解して歩留まりが低下する恐れがある。また再結晶時間は15分以上が好ましい。時間が15分未満であると不純物の除去が不十分になる恐れがある。再結晶は繰り返し行うことで精製効率が上がり、回数は特に制限されないが、好ましくは1~5回、さらに好ましくは2~4回である。得られた化合物(17)の結晶は減圧にて乾燥する。
工程(E1-2)は、工程(E1-1)で得られた化合物(17)の脱保護工程である。脱保護方法は、PROTECTIVE GROUPS IN ORGANIC SYNTHESIS(THEODORA W.GREENE et al)等に記載があるような一般的なフタルイミドの脱保護法であれば特に制限はないが、アミノ基を有した脱保護試薬を用いることが好ましい。
脱保護工程に使用する溶媒としてはジクロロメタン、クロロホルム、メタノール、エタノールなどが挙げられ、好ましくはクロロホルム、エタノールである。溶媒の量は、式(17)で示される化合物の1~50重量倍、好ましくは2~30重量倍、さらに好ましくは3~20重量倍である。
工程(E1-2)で用いられる脱保護試薬は1級のアミノ基を有するアミン低分子化合物であれば特に制限されないが、具体的にはヒドラジン、エチレンジアミン、トリメチレンジアミン、ジエチレントリアミンなどが挙げられ、好ましくはヒドラジン、エチレンジアミンである。脱保護試薬の量は、式(17)で示される化合物に対して、1~30モル当量倍、好ましくは2~20モル当量倍、さらに好ましくは3~10モル当量倍である。
反応温度については特に制限されないが、好ましくは10~80℃であり、さらに好ましくは20~60℃である。また反応時間は1時間以上であり、1時間より短いと反応率が低い恐れがある。
反応温度については特に制限されないが、好ましくは10~80℃であり、さらに好ましくは20~60℃である。また反応時間は1時間以上であり、1時間より短いと反応率が低い恐れがある。
反応終了後の精製方法は、特に制限はないが、カラムクロマトグラフィー、蒸留、抽出、再結晶、超臨界抽出等の精製手段にて脱保護試薬等の化合物を分離するのが好ましく、抽出にて精製するのがさらに好ましい。
抽出にて精製する場合、有機溶媒はトルエン、ジクロロメタン、クロロホルム、メタノールなどが挙げられ、好ましくはジクロロメタンである。これらの溶媒は1種を単独で使用することもでき、2種以上を組み合せて使用することもできる。有機溶媒の量は、式(17)で示される化合物の1~20重量倍、好ましくは2~10重量倍である。また使用する水溶液は1~25重量%のアルカリ金属無機塩の水溶液であり、アルカリ金属無機塩は好ましくはアルカリ金属ハロゲン塩、より好ましくは塩化ナトリウムである。水溶液の量は、式(17)で示される化合物の1~20重量倍、好ましくは2~10重量倍である。
抽出工程の混合、分層時間は特に制限されないが、好ましくは1分~6時間であり、より好ましくは10分~3時間である。また抽出温度は10~80℃、好ましくは20~60℃である。抽出は繰り返し行うことで精製効率が向上する。回数は特に制限されないが、好ましくは1~4回、さらに好ましくは2~3回である。抽出後、好ましくは脱水剤を用いた脱水を行う。脱水剤をろ別後、溶媒を留去し、式(16)で示される化合物を得ることができる。
抽出工程の混合、分層時間は特に制限されないが、好ましくは1分~6時間であり、より好ましくは10分~3時間である。また抽出温度は10~80℃、好ましくは20~60℃である。抽出は繰り返し行うことで精製効率が向上する。回数は特に制限されないが、好ましくは1~4回、さらに好ましくは2~3回である。抽出後、好ましくは脱水剤を用いた脱水を行う。脱水剤をろ別後、溶媒を留去し、式(16)で示される化合物を得ることができる。
工程(E2)は、以下の(E2-1)、(E2-2)の2つの工程から成る。
工程(E2-1)は、構造異性体の両方を活性化カーボネートまたはスルホネート化する工程である。
工程(E2-2)は、官能基化された構造異性体のわずかな物性の違いを利用して、構造異性体を分離する精製工程である。

上記工程図において、化合物(19)は、前記工程図IV中の式(12)で示される化合物に相当する。
工程(E2-1)は、式(5)と式(6)で示される化合物の混合物と、下記の式(b1)、式(e1)で示される化合物(化合物(b1)、化合物(e1))のいずれかとを反応させることで、それぞれ活性化カーボネート基、活性スルホネート基を導入する工程である。例えば、活性化スルホネート基においては、式(19)と式(20)で示される化合物を得る。活性化カーボネート基、活性スルホネート基、どちらを導入する場合においても、ほぼ同様の製法にて製造することができるため、以下、活性化スルホネート基の導入について説明を行う。

(式中、W2、Y2は前記と同義である。)
工程(E2-1)に使用する反応溶媒は、トルエン、アセトニトリル、酢酸エチル、テトラヒドロフラン、クロロホルム、ジクロロメタン等の非プロトン性溶媒が挙げられ、もしくは無溶媒中であり、好ましくはトルエン、クロロホルムである。溶媒の量は、式(5)と式(6)で示される化合物の混合物の1~50重量倍、好ましくは2~30重量倍、さらに好ましくは3~20重量倍である。
工程(E2-1)に使用する上記一般式(e1)で示される化合物におけるW2はCl、Br、Iより選択されるハロゲン原子であり、好ましくはClである。(e1)で示される化合物の使用量は特に制限はないが、式(5)と式(6)で示される化合物の混合物に対して1~10モル当量倍、好ましくは1.01~5モル当量倍、さらに好ましくは1.02~3モル当量倍である。
反応に使用する塩基としては、トリエチルアミン、ピリジン、4-ジメチルアミノピリジン等の有機塩基、もしくは炭酸ナトリウム、水酸化ナトリウム、炭酸水素ナトリウム、酢酸ナトリウム、炭酸カリウム、水酸化カリウム等の無機塩基であり、好ましくはトリエチルアミンである。塩基の使用量は、特に制限はないが、混合物に対して1~15モル当量倍、好ましくは1.1~10モル当量倍、さらに好ましくは1.2~5モル当量倍である。
反応温度については特に制限されないが、好ましくは0~80℃であり、さらに好ましくは20~60℃である。また反応時間は1時間以上であり、1時間より短いと反応率が低い恐れがある。
反応温度については特に制限されないが、好ましくは0~80℃であり、さらに好ましくは20~60℃である。また反応時間は1時間以上であり、1時間より短いと反応率が低い恐れがある。
工程(E2-2)は、工程(E2-1)にて生成した式(19)と式(20)で示される化合物の混合物を分離精製する工程である。
精製法は、特に制限はないが、カラムクロマトグラフィー、蒸留、抽出、再結晶、超臨界抽出等の精製手段にて、式(20)で示される化合物を分離するのが好ましく、再結晶にて精製するのがさらに好ましい。
再結晶にて精製する場合、良溶媒はトルエン、酢酸エチル、メタノール、エタノール、アセトニトリルなどが挙げられ、好ましくはトルエン、酢酸エチル、エタノールであり、さらに好ましくは酢酸エチルである。これらの溶媒は1種を単独で使用することもでき、2種以上を組み合せて使用することもできる。また貧溶媒はヘキサン、ジエチルエーテル、メチル-t-ブチルエーテルなどが挙げられ、好ましくはヘキサンである。良溶媒の量は、混合物の1~50重量倍、好ましくは2.5~35重量倍、さらに好ましくは5~20重量倍である。また貧溶媒の量は、0.5~30重量倍、好ましくは1~20重量倍、さらに好ましくは2~10重量倍である。
再結晶の温度は-20~30℃であり、好ましくは-10~20℃である。温度が30℃を超えると結晶が溶解して歩留まりが低下する恐れがある。また再結晶時間は15分以上が好ましい。時間が15分未満であると不純物の除去が不十分になる恐れがある。再結晶は繰り返し行うことで精製効率が上がり、回数は特に制限されないが、好ましくは1~5回、さらに好ましくは2~4回である。得られた化合物(15)の結晶は減圧にて乾燥する。
工程(F)は、以下の工程図(工程図V)に示されるように、工程(E)で官能基化された多価アルコール誘導体(12)とポリオキシエチレン誘導体(21)を反応にて結合する工程である。

(式中、POE、L1、L3、X2は前記と同義である。Rは保護基で保護された官能基または保護されていてもよい水酸基を示す。L4はリンカー、X3はX2と反応可能な官能基を示す。)
リンカーL4の具体例は前述のリンカーL1~L3の具体例として挙げたものと同じである。
工程(F)に使用される、上記の式(21)で示されるポリオキシエチレン誘導体は、式(12)で示される化合物のX2と反応可能な官能基X3を有する。X2がアミノ基の場合、X3はアミノ基と反応する官能基であれば特に制限はないが、例えば、活性エステル基、活性カーボネート基、アルデヒド基、置換スルホネート基、カルボキシル基などであり、好ましくは活性エステル基、活性カーボネート基である。X2が活性カーボネート基またはスルホネート基の場合、X3は活性カーボネート基またはスルホネート基と反応する官能基であれば特に制限はないが、好ましくはアミノ基、アルコキシド基である。
Rは保護基で保護された官能基または保護されていてもよい水酸基を示す。保護されている官能基としては、アミノ基、カルボキシル基、アルデヒド基、チオール基が挙げられる。具体的なアミノ基の保護基としてはt-ブチルカーバーメート基、ベンジル基、トリチル基等が挙げられるが、好ましくはt-ブチルカーバーメート基である。具体的なカルボキシル基の保護基としてはt-ブチル基、ベンジル基等が挙げられるが、好ましくはベンジル基である。具体的なアルデヒド基の保護基としては3~9個の炭素原子を含むアセタール基等が挙げられるが、好ましくはジエチルアセタール基である。具体的なチオール基の保護基としてはt-ブチル基、ベンジル基、トリチル基、t-ブチルジメチルシリル基、t-ブチルジフェニルシリル基等が挙げられるが、好ましくはt-ブチル基、ベンジル基であり、さらに好ましくはt-ブチル基である。
また、水酸基の具体的な保護基としてはt-ブチル基、ベンジル基、トリチル基、t-ブチルジメチルシリル基、t-ブチルジフェニルシリル基等が挙げられるが、好ましくはt-ブチル基、ベンジル基であり、さらに好ましくはベンジル基である。
工程(F)にて使用する式(12)で示される化合物の使用量は特に制限はないが、式(21)で示されるポリオキシエチレン誘導体に対して1~20モル当量倍、好ましくは1.5~15モル当量倍、さらに好ましくは2~10モル当量倍である。
工程(F)に使用する反応溶媒は、トルエン、アセトニトリル、酢酸エチル、テトラヒドロフラン、クロロホルム、ジクロロメタン等の非プロトン性溶媒が挙げられ、好ましくはトルエン、クロロホルムである。溶媒の量は、式(21)で示される化合物の1~50重量倍、好ましくは2~25重量倍、さらに好ましくは3~10重量倍である。
反応温度については特に制限されないが、好ましくは0~100℃であり、さらに好ましくは20~80℃である。また反応時間は1時間以上であり、1時間より短いと反応率が低い恐れがある。
精製法は、特に制限はないが、カラムクロマトグラフィー、蒸留、抽出、再結晶、超臨界抽出等の精製手段が挙げられ、好ましくは、再結晶にて精製するのがさらに好ましい。
再結晶にて精製する場合、良溶媒はトルエン、酢酸エチル、メタノール、エタノール、アセトニトリルなどが挙げられ、好ましくはトルエン、酢酸エチルであり、さらに好ましくは酢酸エチルである。これらの溶媒は1種を単独で使用することができ、2種以上を組み合せて使用することもできる。また貧溶媒はヘキサン、ジエチルエーテル、メチル-t-ブチルエーテルなどが挙げられ、好ましくはヘキサンである。良溶媒の量は、混合物の1~50重量倍、好ましくは2.5~35重量倍、さらに好ましくは5~20重量倍である。また貧溶媒の量は、0.5~30重量倍、好ましくは1~20重量倍、さらに好ましくは2~10重量倍である。
再結晶の温度は-20~30℃であり、好ましくは-10~20℃である。温度が30℃を超えると結晶が溶解して歩留まりが低下する恐れがある。また再結晶時間は15分以上が好ましい。時間が15分未満であると不純物の除去が不十分になる恐れがある。再結晶は繰り返し行うことで精製効率が上がり、回数は特に制限されないが、好ましくは1~5回、さらに好ましくは2~4回である。得られた化合物(13)の結晶は減圧にて乾燥する。
工程(G)は、工程(F)で得られた式(13)で示されるポリオキシエチレン誘導体の保護基Rの脱保護により、官能基または水酸基を出現させる工程である。水酸基を出現させた場合は、続けて前述の工程図IIの工程(C)と同製法にて官能基の導入を行う。ポリオキシエチレン末端のRの保護基の種類によっては、脱保護と同時に、次工程の脱アセタールを行うことができるものもある。
これらの保護基の脱保護の方法としては、PROTECTIVE GROUPS IN ORGANIC SYNTHESIS(THEODORA W.GREENE et al)等に記載があるような一般的な脱保護法を用いて脱保護を行うことができる。
脱保護後、抽出、再結晶、吸着処理、再沈殿、超臨界抽出等の精製手段にて精製してもよく、好適には、再結晶を行い、結晶を減圧下にて乾燥することで、式(13)で示される化合物を得ることができる。
脱保護後、抽出、再結晶、吸着処理、再沈殿、超臨界抽出等の精製手段にて精製してもよく、好適には、再結晶を行い、結晶を減圧下にて乾燥することで、式(13)で示される化合物を得ることができる。
工程(H)は、工程(G)で得られた式(14)で示されるポリオキシエチレン誘導体の環状アセタール構造を切断する脱保護工程である。ポリオキシエチレン末端の官能基の種類によっては、工程(H)後にさらに官能基化を行うこともできる。
工程(H)は、工程(D)と同工程である。工程(D)を行うことで目的物である式(15)で示される化合物を得ることができる。
本発明により、末端に複数の水酸基を有するポリオキシエチレン誘導体(1)を高純度で効率よく、工業的に製造することができる。
また、本発明により得られるポリオキシエチレン誘導体(1)は、従来のポリオキシエチレン誘導体に比べ、血中半減期、抗原性を改善できる利点を有しており、生体関連物質を修飾するのに有用である。
また、本発明により得られるポリオキシエチレン誘導体(1)は、従来のポリオキシエチレン誘導体に比べ、血中半減期、抗原性を改善できる利点を有しており、生体関連物質を修飾するのに有用である。
以下、実施例に基づいて本発明をさらに具体的に説明する。なお、例中の化合物の分析、同定には1H-NMR、GPCを用いた。
<1H-NMRの分析方法>
1H-NMR分析では、日本電子データム(株)製JNM-ECP400を用いた。NMR測定値における積分値は理論値である。
1H-NMR分析では、日本電子データム(株)製JNM-ECP400を用いた。NMR測定値における積分値は理論値である。
<GPC分析の分析方法>
GPC分析は、下記条件にて測定を行った。
装置:島津LC-10Avp
カラム:PL gel MIXED-D×2(ポリマーラボラトリー)
展開溶媒:ジメチルホルムアミド
流速:0.7ml/min
カラム温度:65℃
検出器:RI
サンプル量:1mg/g、100μl
分子量はピークトップ分子量Mpである。
GPC分析は、下記条件にて測定を行った。
装置:島津LC-10Avp
カラム:PL gel MIXED-D×2(ポリマーラボラトリー)
展開溶媒:ジメチルホルムアミド
流速:0.7ml/min
カラム温度:65℃
検出器:RI
サンプル量:1mg/g、100μl
分子量はピークトップ分子量Mpである。
(実施例1)
ポリオキシエチレン誘導体(1)の合成
(L1=-O-、L3=-CH2CH2-NHCO-CH2CH2-、X=マレイミド基、Z=エチレングリコール残基、a=1、b=1、c=1、d=0、e=1、分子量約20000の場合)
(実施例1-1)
化合物(5)(6):ジイソプロピリデンキシリトールの合成
ポリオキシエチレン誘導体(1)の合成
(L1=-O-、L3=-CH2CH2-NHCO-CH2CH2-、X=マレイミド基、Z=エチレングリコール残基、a=1、b=1、c=1、d=0、e=1、分子量約20000の場合)
(実施例1-1)
化合物(5)(6):ジイソプロピリデンキシリトールの合成
温度計、窒素吹き込み管、撹拌機を付した5L丸底フラスコへキシリトール1000g、2,2-ジメトキシプロパン1916g、およびp-トルエンスルホン酸・1水和物37.5mgを入れ、窒素を吹き込みながら65℃で反応を行った。反応液の溶媒を留去し、蒸留精製(b.p.108℃/0.15mmHg)し、1,2,3,4-ジイソプロピリデンキシリトール(式(5))と1,2,4,5-ジイソプロピリデンキシリトール(式(6))の異性体混合物を得た。1H-NMR(CDCl3, 内部標準TMS)δ(ppm): 1.37-1.44(12H,m,-C(CH
3)2) 、3.59-3.65(1H,m,-CH-O-)、3.81-3.90(2H,m,-CH
2-O-)、3.98-4.01(1H,m,-CH-O-)、4.04-4.10(2H,m,-CH
2-O-) 4.11-4.23(1H, m, -CH-O-)
(実施例1-2)
化合物(7):1,2,3,4-ジイソプロピリデン-5-(t-ブチルジフェニルシリル)キシリトールの合成
化合物(7):1,2,3,4-ジイソプロピリデン-5-(t-ブチルジフェニルシリル)キシリトールの合成
温度計、窒素吹き込み管、撹拌機を付した2L丸底フラスコに、1-1で精製したジイソプロピリデンキシリトール(異性体混合物)250g、ジクロロメタン1000g、4-ジメチルアミノピリジン26g、およびトリエチルアミン109gを入れ、窒素を吹き込みながら室温で溶解させ、10℃以下に冷却後、t-ブチルクロロジフェニルシラン297gを滴下した。滴下後室温に戻して2時間反応後、飽和炭酸水素ナトリウム水溶液を加えて洗浄し、硫酸マグネシウムで脱水後、溶媒を留去し、135℃、減圧下(0.2mmHg)で1,2,4,5-ジイソプロピリデンキシリトールを除去し、1,2,3,4-ジイソプロピリデン-5-(t-ブチルジフェニルシリル)キシリトール(式(7))を200g得た。1H-NMR(CDCl3, 内部標準TMS) δ(ppm): 1.06(9H,m, -Si-C-(CH
3
)
3 ) 1.37、1.42、1.43(12H,s, -O-C-CH
3 ) 3.72-3.82(1H,m,-CH-O-、-CH
2 -O-)、3.95(1H,dd, -CH-O-)
3.99-4.06(2H,m,-CH 2 -O-) 4.11-4.15(1H,m,-CH-O-) 7.36-7.54(6H,m,Ph-Si(-Ph)-O-)
7.66-7.70(4H,m,Ph-Si(-Ph)-O-)
3.99-4.06(2H,m,-CH 2 -O-) 4.11-4.15(1H,m,-CH-O-) 7.36-7.54(6H,m,Ph-Si(-Ph)-O-)
7.66-7.70(4H,m,Ph-Si(-Ph)-O-)

(実施例1-3)
化合物(5):1,2,3,4-ジイソプロピリデンキシリトールの合成
化合物(5):1,2,3,4-ジイソプロピリデンキシリトールの合成
温度計、窒素吹き込み管、撹拌機を付した2L丸底フラスコに、1-2で得られた1,2,3,4-ジイソプロピリデン-5-(t-ブチルジフェニルシリル)キシリトール500gと脱水テトラヒドロフラン440gを入れ、窒素を吹き込みながら室温で均一化させ、20℃以下に冷却後、テトラブチルアンモニウムフルオリド(1mol/Lテトラヒドロフラン溶液)1270mlを滴下した。滴下後室温に戻して2時間反応後、減圧下で溶媒を留去した。酢酸エチル2000gで溶解後、精製水で酢酸エチル層を水洗し、硫酸マグネシウムで脱水後、溶媒を留去し、クロロホルムとメタノールを溶媒、充填剤にシリカゲルを用いたカラムクロマトグラフィーにて1,2,3,4-ジイソプロピリデンキシリトール(式(5))を250g得た。1H-NMR(CDCl3 , 内部標準TMS) δ(ppm): 1.39、1.44(12H,s,-CH
3 ) 、3.62(1H,dd,-CH-O-)、3.08-3.89(2H,m,-CH
2-O-)、3.98-4.08(1H,m,-CH-O-,2H,m,-CH
2 O-)、4.18-4.23(1H,m,-CH-O-)

(実施例1-4)
化合物(p1):α-ジイソプロピリデンキシリトール ポリオキシエチレン(分子量20,000)の合成
化合物(p1):α-ジイソプロピリデンキシリトール ポリオキシエチレン(分子量20,000)の合成
1,2,3,4-ジイソプロピリデンキシリトール(5)を100g(0.43mol)、脱水トルエンを200g、ナトリウムメトキシド28%メタノール溶液10.8gを5Lオートクレーブへ仕込み、系内を窒素置換後、50℃に昇温し、減圧にてトルエンとメタノールを留去した。100~150℃、1MPa以下の圧力でエチレンオキシド4205g(95.6mol)を加えた後、更に1時間反応を続け、内容物の半量2150gを抜き取った。続いて100~150℃、1MPa以下の圧力でエチレンオキシド2150g(48.9mol)を加えた後、更に1時間反応を続けた。減圧にて未反応のエチレンオキシドガスを除去後、下記化合物(p1)を得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.37-1.44(12H,m,-C(CH 3)2)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)mH)
分子量(GPC/Mp):20678(m=約470)
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.37-1.44(12H,m,-C(CH 3)2)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)mH)
分子量(GPC/Mp):20678(m=約470)

(実施例1-5)
化合物(p2):α-ジイソプロピリデンキシリトール ω-アミン ポリオキシエチレン(分子量20,000)の合成
化合物(p2):α-ジイソプロピリデンキシリトール ω-アミン ポリオキシエチレン(分子量20,000)の合成
温度計、窒素吹き込み管、撹拌機、Dean-Stark管及び冷却管を付した1L四つ口フラスコに、α-ジイソプロピリデンキシリトール ポリオキシエチレン(p1)を200g(10mmol)、トルエン600gを加え、撹拌、窒素吹込みをしながら60℃に加温して溶解した。110℃に昇温し、トルエンと共沸させながら約300gの留分を抜き取り、脱水を行った。40℃まで冷却し、脱水アセトニトリル1.0kgを加え、フタルイミド2.2g(15mmol)、トリフェニルホスフィン3.9g(15mmol)を加えた後、ジイソプロピルアゾジカルボキシレート3.0g(15mmol)を加え、室温で2時間反応した。
反応後、減圧にて溶剤を留去し、メタノール400g、エチレンジアミン30g(0.5mol)を加え、60℃で4時間反応した。これにジクロロメタン1.0kgで希釈し、25%食塩水500gで2回抽出を行った。40℃、微減圧下で約1.5kgの留分を抜き取り、その後室温まで冷却し、これに酢酸エチル600gを加え、硫酸マグネシウムを添加し脱水を行った。硫酸マグネシウムを濾別後、濾液にn-へキサン600gを加えて結晶化した。結晶を濾取した後、酢酸エチル800gに40℃で溶解し、室温に冷却後n-へキサン600gを加えて結晶化した。濾取した結晶をn-へキサン1.0kgで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p2)184gを得た。
1H-NMR(D2O)δ(ppm):1.37-1.44(12H,m,-C(CH 3)2)、2.84-2.88(2H,t,-CH 2-NH2)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)m-CH 2O-)
1H-NMR(D2O)δ(ppm):1.37-1.44(12H,m,-C(CH 3)2)、2.84-2.88(2H,t,-CH 2-NH2)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)m-CH 2O-)

(実施例1-6)
化合物(p3):α-キシリトール ω-アミン ポリオキシエチレン(分子量20,000)の合成
化合物(p3):α-キシリトール ω-アミン ポリオキシエチレン(分子量20,000)の合成
温度計、撹拌機を付した3L三つ口フラスコに、α-ジイソプロピリデンキシリトール ω-アミン ポリオキシエチレン(p2)を100g(5mmol)、イオン交換水1.8kgを加え、撹拌、窒素吹込みをしながら溶解した。85%リン酸を滴下しながらpH1.40になるように添加し、室温で8時間反応した。
反応後、10N水酸化ナトリウム水溶液を加えて中和し、食塩360gを添加後、更に10N水酸化ナトリウム水溶液を加えてpH12.0に調製した。そこにトルエンを500g加え、50℃で2回抽出を行った。減圧にて溶剤を留去し、酢酸エチル500gを加え、硫酸マグネシウムを添加し脱水を行った。硫酸マグネシウムを濾別後、濾液にn-へキサン400gを加えて結晶化した。濾取した結晶をn-へキサン400gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p3)90gを得た。
1H-NMR(D2O)δ(ppm): 2.84-2.88(2H,t,-CH2-NH2)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)m-CH 2-)
1H-NMR(D2O)δ(ppm): 2.84-2.88(2H,t,-CH2-NH2)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)m-CH 2-)

(実施例1-7)
化合物(p4)α-キシリトール ω-マレイミド ポリオキシエチレン(分子量20,000)の合成
化合物(p4)α-キシリトール ω-マレイミド ポリオキシエチレン(分子量20,000)の合成
温度計、窒素吹き込み管、撹拌機及び冷却管を付した100mL四つ口フラスコに、α-キシリトール ω-アミン ポリオキシエチレン(p3)を10g(0.5mmol)、トルエンを50g仕込み、40℃に加温して溶解した。遮光後、N-スクシンイミジルマレイミドプロピオン酸を160mg(0.6mmol)添加し、40℃で4時間反応した。
反応後、ろ過し、酢酸エチル30gを加え希釈し、n-へキサン40gを加えて結晶化した。結晶を濾取した後、酢酸エチル100gに40℃で溶解し、室温に冷却後n-へキサン50gを加えて結晶化した。結晶の溶解、結晶化工程をさらに1回繰り返した。濾取した結晶をn-へキサン50gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物
(p4)9gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):2.49-2.54(2H,t, -NHCOCH 2CH2-)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)m-CH 2-,-CH 2NHCO-)、6.70(2H, s, -CH=CH-)
反応後、ろ過し、酢酸エチル30gを加え希釈し、n-へキサン40gを加えて結晶化した。結晶を濾取した後、酢酸エチル100gに40℃で溶解し、室温に冷却後n-へキサン50gを加えて結晶化した。結晶の溶解、結晶化工程をさらに1回繰り返した。濾取した結晶をn-へキサン50gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物
(p4)9gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):2.49-2.54(2H,t, -NHCOCH 2CH2-)、3.40-3.90(約1880H, m, -CH 2O(CH 2 CH 2O)m-CH 2-,-CH 2NHCO-)、6.70(2H, s, -CH=CH-)

(実施例2)
ポリオキシエチレン誘導体(1)の合成
(L1=-OCO-NH-、X=p-ニトロフェニルカーボネート基、Z=グリセリン残基、a=1、b=2、c=1、d=0、e=0、分子量約40000の場合)
ポリオキシエチレン誘導体(1)の合成
(L1=-OCO-NH-、X=p-ニトロフェニルカーボネート基、Z=グリセリン残基、a=1、b=2、c=1、d=0、e=0、分子量約40000の場合)
(実施例2-1)
化合物(17)の合成
化合物(17)の合成
温度計、窒素吹き込み管、撹拌機、Dean-Stark管及び冷却管を付した1L四つ口フラスコに、ジイソプロピリデンキシリトール(5)(6)を50g(0.22mol)、トルエン100gを加え、撹拌、窒素吹込みをしながら110℃に昇温し、トルエンと共沸させながら約80gの留分を抜き取り、脱水を行った。40℃まで冷却し、脱水アセトニトリル500gを加え、ヒドロキシフタルイミド31.7g(0.22mol)、トリフェニルホスフィン56.5g(0.22mol)を加えた後、ジイソプロピルアゾジカルボキシレート43.5g(0.22mol)をゆっくり滴下し、室温で2時間反応した。
反応後、減圧にて溶剤を留去し、酢酸エチル500g、エタノール300g、n-へキサン200gを加えて、10℃以下に冷却し結晶化した。結晶を濾取して真空下で乾燥し、下記化合物(17)50gを得た。
1H-NMR(CDCl3 , 内部標準TMS)δ(ppm): 1.34-1.44(12H,m, -C(CH
3)2) 、3.80-3.90(2H,m,N-CH
2-CH)、3.93-4.02(2H,m,-CH
2-O-)、4.07-4.12(1H,m,-CH-O-)、4.23-4.32(2H,m,-CH-O-)、7.71-7.89(4H, m, Ph)

(実施例2-2)化合物(18)の合成
温度計、窒素吹き込み管、撹拌機及び冷却管を付した1L四つ口フラスコに、化合物(15)を25g(69mmol)、クロロホルム125g、エチレンジアミン20.8g
(0.345mol)を加え、60℃で4時間反応した。これに25%食塩水100gを加え、2回抽出し、硫酸マグネシウムを添加し脱水を行った。硫酸マグネシウムを濾別後、減圧にて溶剤を留去し、真空下で乾燥して粘性液体である下記化合物(18)12.5gを得た。
(0.345mol)を加え、60℃で4時間反応した。これに25%食塩水100gを加え、2回抽出し、硫酸マグネシウムを添加し脱水を行った。硫酸マグネシウムを濾別後、減圧にて溶剤を留去し、真空下で乾燥して粘性液体である下記化合物(18)12.5gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm): 1.36-1.44(12H,m,-C(CH
3)2) 、2.78-2.96(2H,m, -CH
2-NH2)、3.81-3.86(2H,m,-CH
2-O-)、3.95-3.99(1H,m,-CH-O-)、4.03-4.06(1H,m,-CH-O-)、4.15-4.19(1H,m,-CH-O-)

(実施例2-3)
ポリオキシエチレン誘導体(p6)(分子量40,000)の合成
ポリオキシエチレン誘導体(p6)(分子量40,000)の合成

温度計、窒素吹き込み管、撹拌機、Dean-Stark管及び冷却管を付した1L四つ口フラスコに、日本国特許公開:特開2004-197077号公報の実施例16に準じて合成した分子量40,000の2分岐型PEGである上記化合物(p5)を100g(2.5mmol)、トルエン500gを加え、撹拌、窒素吹込みをしながら60℃に加温して溶解した。110℃に昇温し、トルエンと共沸させながら約100gの留分を抜き取り、脱水を行った。60℃まで冷却し、トリエチルアミンを1.5g(15.0mmol)、p-ニトロフェニルクロロホルメートを2.5g(12.5mmol)を加え、60℃で6時間反応した。
これにトルエン300gを加え希釈し、濾過後、n-へキサン300gを加えて結晶化した。結晶を濾取した後、酢酸エチル700gに40℃で溶解し、室温に冷却後n-へキサン300gを加えて結晶化した。結晶の溶解、結晶化工程をさらに2回繰り返した。濾取した結晶をn-へキサン500gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p6)94gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-、-CH 2 -O-CH2-Ph)、4.44(4H, t, -OCH2 CH 2 -O-CO-O-)、4.54(2H, s, -O-CH 2 -Ph)、7.39(4H, d, -Ph-NO2)、8.28(4H, d, -Ph-NO2)分子量(GPC/Mp):42273(m=約480)
これにトルエン300gを加え希釈し、濾過後、n-へキサン300gを加えて結晶化した。結晶を濾取した後、酢酸エチル700gに40℃で溶解し、室温に冷却後n-へキサン300gを加えて結晶化した。結晶の溶解、結晶化工程をさらに2回繰り返した。濾取した結晶をn-へキサン500gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p6)94gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-、-CH 2 -O-CH2-Ph)、4.44(4H, t, -OCH2 CH 2 -O-CO-O-)、4.54(2H, s, -O-CH 2 -Ph)、7.39(4H, d, -Ph-NO2)、8.28(4H, d, -Ph-NO2)分子量(GPC/Mp):42273(m=約480)

(実施例2-4)
ポリオキシエチレン誘導体(p7)(分子量40,000)の合成
ポリオキシエチレン誘導体(p7)(分子量40,000)の合成
温度計、窒素吹き込み管、撹拌機及び冷却管を付した500mL四つ口フラスコに、化合物(p11)を50g(1.25mmol)、トルエン250gを加え、撹拌、窒素吹込みをしながら40℃に加温して溶解した。化合物(18)を1.2g(5.0mmol)加え、40℃で4時間反応した。
反応後、酢酸エチル250gで希釈し、室温に冷却後n-へキサン200gを加えて結晶化した。結晶を濾取した後、酢酸エチル500gに40℃で溶解し、室温に冷却後n-へキサン200gを加えて結晶化した。結晶の溶解、結晶化工程をさらに2回繰り返した。濾取した結晶をn-へキサン200gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p7)44gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.36-1.44(24H, m, -C(CH 3)2)、3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-、-CH 2 -O-CH2-Ph)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-, -OCH2 CH 2 -O-CO-NH-)、4.54(2H, s, -O-CH 2 -Ph)
反応後、酢酸エチル250gで希釈し、室温に冷却後n-へキサン200gを加えて結晶化した。結晶を濾取した後、酢酸エチル500gに40℃で溶解し、室温に冷却後n-へキサン200gを加えて結晶化した。結晶の溶解、結晶化工程をさらに2回繰り返した。濾取した結晶をn-へキサン200gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p7)44gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.36-1.44(24H, m, -C(CH 3)2)、3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-、-CH 2 -O-CH2-Ph)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-, -OCH2 CH 2 -O-CO-NH-)、4.54(2H, s, -O-CH 2 -Ph)

(実施例2-5)
ポリオキシエチレン誘導体(p8)(分子量40,000)の合成
ポリオキシエチレン誘導体(p8)(分子量40,000)の合成
温度計、撹拌機を付した500mL三つ口フラスコに、化合物(p7)を40g(1.0mmol)、5%パラジウムカーボン(50%含水品)20gを仕込み、窒素置換後、メタノール400mL、シクロヘキセン67mLを加えて昇温し、52~55℃で緩やかに還流させ、3時間反応させた。室温まで冷却後、パラジウムカーボンを濾別し、濾液を濃縮した。濃縮液にトルエン350g、n-ヘキサン250gを加えて結晶化した。濾取した結晶をn-へキサン200gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p8)36gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.36-1.44(24H, m,-C(CH 3)2)、3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-、-CH 2 -OH)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-,-OCH2 CH 2 -O-CO-NH-)
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.36-1.44(24H, m,-C(CH 3)2)、3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-、-CH 2 -OH)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-,-OCH2 CH 2 -O-CO-NH-)

(実施例2-6)
ポリオキシエチレン誘導体(p9)(分子量40,000)の合成
ポリオキシエチレン誘導体(p9)(分子量40,000)の合成
温度計、窒素吹き込み管、撹拌機及び冷却管を付した300mL四つ口フラスコに、化合物(p8)を30g(0.75mmol)、トルエン150gを加え、撹拌、窒素吹込みをしながら60℃に加温して溶解した。トリエチルアミンを228mg(2.25mmol)、p-ニトロフェニルクロロホルメートを378mg(1.88mmol)加え、60℃で4時間反応した。
これにトルエン150gを加え希釈し、濾過後、n-へキサン120gを加えて結晶化した。結晶を濾取した後、酢酸エチル210gに40℃で溶解し、室温に冷却後n-へキサン90gを加えて結晶化した。結晶の溶解、結晶化工程をさらに2回繰り返した。濾取した結晶をn-へキサン90gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p9)26gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.36-1.44(24H, m, -C(CH 3)2)、3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-, -OCH2 CH 2 -O-CO-NH-)、4.32-4.50(2H, m, -CH 2 -O-CO-O-Ph-NO2)、 7.39(2H, d, -Ph-NO2)、8.28(2H, d, -Ph-NO2)
これにトルエン150gを加え希釈し、濾過後、n-へキサン120gを加えて結晶化した。結晶を濾取した後、酢酸エチル210gに40℃で溶解し、室温に冷却後n-へキサン90gを加えて結晶化した。結晶の溶解、結晶化工程をさらに2回繰り返した。濾取した結晶をn-へキサン90gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p9)26gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):1.36-1.44(24H, m, -C(CH 3)2)、3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-, -OCH2 CH 2 -O-CO-NH-)、4.32-4.50(2H, m, -CH 2 -O-CO-O-Ph-NO2)、 7.39(2H, d, -Ph-NO2)、8.28(2H, d, -Ph-NO2)

(実施例2-7)
ポリオキシエチレン誘導体(p10)(分子量40,000)の合成
ポリオキシエチレン誘導体(p10)(分子量40,000)の合成
温度計、撹拌機を付した500mL三つ口フラスコに、化合物(p9)を25g(0.63mmol)、イオン交換水450gを加え、撹拌、窒素吹込みをしながら溶解した。85%リン酸を滴下しながらpH1.0になるように添加し、室温で3時間反応した。
反応後、クロロホルムを250g加え、室温で2回抽出を行った。硫酸マグネシウムを添加し脱水を行い、硫酸マグネシウムを濾別後、減圧にて溶剤を留去した。そこに酢酸エチル150gを加え、n-へキサン100gを加えて結晶化した。濾取した結晶をn-へキサン100gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p10)20gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-, -OCH2 CH 2 -O-CO-NH-)、4.32-4.50(2H, m, -CH 2 -O-CO-O-Ph-NO2)、7.39(2H, d, -Ph-NO2)、8.28(2H, d, -Ph-NO2)
反応後、クロロホルムを250g加え、室温で2回抽出を行った。硫酸マグネシウムを添加し脱水を行い、硫酸マグネシウムを濾別後、減圧にて溶剤を留去した。そこに酢酸エチル150gを加え、n-へキサン100gを加えて結晶化した。濾取した結晶をn-へキサン100gで洗浄した。結晶を濾取して真空下で乾燥して下記化合物(p10)20gを得た。
1H-NMR(CDCl3, 内部標準TMS)δ(ppm):3.40-3.80(約3840H, m, -CH 2(OCH 2 CH 2)mOCH 2 CH2-O-CO-, -CH(OCH 2 CH 2)mOCH 2 CH2-O-CO-)、4.02-4.09(4H, m, -CH-O-)、4.15-4.25(6H, m, -NH-CH2-CH-O-, -OCH2 CH 2 -O-CO-NH-)、4.32-4.50(2H, m, -CH 2 -O-CO-O-Ph-NO2)、7.39(2H, d, -Ph-NO2)、8.28(2H, d, -Ph-NO2)

本発明を特定の態様を参照して詳細に説明したが、本発明の精神と範囲を離れることなく様々な変更および修正が可能であることは、当業者にとって明らかである。
なお、本出願は、2011年3月30日付で出願された日本特許出願(特願2011-076682)に基づいており、その全体が引用により援用される。また、ここに引用されるすべての参照は全体として取り込まれる。
なお、本出願は、2011年3月30日付で出願された日本特許出願(特願2011-076682)に基づいており、その全体が引用により援用される。また、ここに引用されるすべての参照は全体として取り込まれる。
Claims (8)
- 式(1):

- 式(2):

- 式(2)中のZがエチレングリコール残基、bが1、cが1、dが0、eが1である、下記式(3)で表されるポリオキシエチレン誘導体である、請求項2記載のポリオキシエチレン誘導体。

- 式(2)中のZがグリセリン残基、bが2、cが1、dが0である、下記式(4)で表されるポリオキシエチレン誘導体である、請求項2記載のポリオキシエチレン誘導体。

- 式(1)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である請求項1記載のポリオキシエチレン誘導体。
- 式(2)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である請求項2記載のポリオキシエチレン誘導体。
- 式(3)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である請求項3記載のポリオキシエチレン誘導体。
- 式(4)において、Xが、活性エステル基、活性カーボネート基、アルデヒド基、イソシアネート基、イソチオシアネート基、エポキシ基、カルボキシル基、チオール基、マレイミド基、置換マレイミド基、ヒドラジド基、ジチオピリジン基、置換スルホネート基、ビニルスルホン基、アミノ基、オキシアミノ基、ヨードアセトアミド基、アルキルカルボニル基、アルケニル基、アルキニル基またはアジド基である請求項4記載のポリオキシエチレン誘導体。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201280016523.4A CN103492458B (zh) | 2011-03-30 | 2012-03-28 | 末端具有多个羟基的聚氧乙烯衍生物 |
| KR1020137025831A KR101811911B1 (ko) | 2011-03-30 | 2012-03-28 | 폴리옥시에틸렌 유도체 말단에 복수 개의 하이드록실기를 포함하는 폴리옥시에틸렌 유도체 |
| DK12764912T DK2692771T3 (da) | 2011-03-30 | 2012-03-28 | Polyoxyethylen-derivat med flere terminale hydroxyl-grupper |
| EP12764912.7A EP2692771B1 (en) | 2011-03-30 | 2012-03-28 | Polyoxyethylene derivative having a plurality of hydroxyl groups at end |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-076682 | 2011-03-30 | ||
| JP2011076682 | 2011-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012133490A1 true WO2012133490A1 (ja) | 2012-10-04 |
Family
ID=46931199
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/058069 WO2012133490A1 (ja) | 2011-03-30 | 2012-03-28 | 末端に複数の水酸基を有するポリオキシエチレン誘導体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8952125B2 (ja) |
| EP (1) | EP2692771B1 (ja) |
| JP (1) | JP5949037B2 (ja) |
| KR (1) | KR101811911B1 (ja) |
| CN (2) | CN107573498B (ja) |
| DK (1) | DK2692771T3 (ja) |
| WO (1) | WO2012133490A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013147015A1 (ja) * | 2012-03-30 | 2013-10-03 | 日油株式会社 | マルチアーム型ポリエチレングリコール誘導体、その中間体及び製造方法 |
| WO2017188372A1 (ja) * | 2016-04-28 | 2017-11-02 | 日油株式会社 | 分岐型ポリオキシエチレン化合物およびコンタクトレンズ |
| JP2018062655A (ja) * | 2016-10-07 | 2018-04-19 | 国立大学法人東京工業大学 | 分岐型ヘテロ単分散ポリエチレングリコール、その製造方法、及びその結合体 |
| WO2018180917A1 (ja) * | 2017-03-31 | 2018-10-04 | 日油株式会社 | 末端に複数の水酸基を有するポリオキシエチレン誘導体の製造方法 |
| WO2019189431A1 (ja) * | 2018-03-27 | 2019-10-03 | 日油株式会社 | マルチアーム型ポリエチレングリコール誘導体の製造方法 |
| CN111936550A (zh) * | 2018-03-29 | 2020-11-13 | 日油株式会社 | 分解性聚乙二醇衍生物 |
| WO2022131098A1 (ja) * | 2020-12-15 | 2022-06-23 | 日油株式会社 | マレイミドポリエチレングリコール誘導体の製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104350084B (zh) * | 2012-05-31 | 2016-10-12 | 株式会社钟化 | 具有有多个反应性硅基的末端结构的聚合物及其制造方法和利用 |
| CN104530126B (zh) * | 2014-12-18 | 2016-08-24 | 江苏苏博特新材料股份有限公司 | 一种季磷盐及其应用 |
| JP6874762B2 (ja) * | 2016-04-28 | 2021-05-19 | 日油株式会社 | 末端に多水酸基を有するポリオキシエチレン化合物およびコンタクトレンズ |
| CA3095644A1 (en) * | 2018-03-29 | 2019-10-03 | Nof Corporation | Degradable polyethylene glycol conjugate |
| JP2020041139A (ja) * | 2018-09-11 | 2020-03-19 | 国立大学法人神戸大学 | エーテル誘導体の製造方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004197077A (ja) | 2002-11-20 | 2004-07-15 | Nof Corp | 修飾された生体関連物質、その製造方法および中間体 |
| WO2005037911A2 (en) | 2003-08-26 | 2005-04-28 | Smithkline Beecham Corporation | Heterofunctional copolymers of glycerol and polyethylene glycol, their conjugates and compositions |
| WO2006028129A1 (ja) | 2004-09-10 | 2006-03-16 | Toray Industries, Inc. | 医薬品製剤 |
| JP2006510601A (ja) | 2002-09-30 | 2006-03-30 | マウンテン ビュー ファーマシューティカルズ,インコーポレイテッド | 抗原性が低下したポリマー結合体、その調製方法および使用方法 |
| JP2007031554A (ja) | 2005-07-26 | 2007-02-08 | Nof Corp | ポリグリセリン誘導体及びこれを含む界面活性剤 |
| WO2008096904A1 (ja) | 2007-02-08 | 2008-08-14 | Tokyo Metropolitan Organization For Medical Research | マンノース6-リン酸-ポリエチレングリコール結合体 |
| JP2008188557A (ja) | 2007-02-07 | 2008-08-21 | Nof Corp | ポリグリセリン誘導体を含む界面活性剤 |
| WO2008105514A1 (ja) * | 2007-02-28 | 2008-09-04 | Nof Corporation | 多分岐鎖ポリオキシアルキレン誘導体 |
| WO2010114074A1 (ja) * | 2009-03-31 | 2010-10-07 | 日油株式会社 | 多分岐鎖ポリオキシアルキレン化合物、およびその製造方法 |
| JP2010235450A (ja) * | 2009-03-30 | 2010-10-21 | Nof Corp | 脂質誘導体 |
| JP2010254978A (ja) * | 2009-03-31 | 2010-11-11 | Nof Corp | 高分子量ポリオキシアルキレン誘導体の精製方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3700250B2 (ja) * | 1996-05-15 | 2005-09-28 | 日本油脂株式会社 | ポリオキシアルキレントリグリセリルエーテル化合物及び変性シリコーン化合物並びにそれらの製造方法 |
| JP5136849B2 (ja) * | 2008-05-20 | 2013-02-06 | 日油株式会社 | ポリオキシアルキレン変性オルガノポリシロキサン化合物 |
-
2012
- 2012-03-27 JP JP2012071102A patent/JP5949037B2/ja active Active
- 2012-03-28 KR KR1020137025831A patent/KR101811911B1/ko active IP Right Grant
- 2012-03-28 CN CN201610833860.2A patent/CN107573498B/zh active Active
- 2012-03-28 EP EP12764912.7A patent/EP2692771B1/en active Active
- 2012-03-28 WO PCT/JP2012/058069 patent/WO2012133490A1/ja unknown
- 2012-03-28 CN CN201280016523.4A patent/CN103492458B/zh active Active
- 2012-03-28 DK DK12764912T patent/DK2692771T3/da active
- 2012-03-30 US US13/435,556 patent/US8952125B2/en active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006510601A (ja) | 2002-09-30 | 2006-03-30 | マウンテン ビュー ファーマシューティカルズ,インコーポレイテッド | 抗原性が低下したポリマー結合体、その調製方法および使用方法 |
| JP2004197077A (ja) | 2002-11-20 | 2004-07-15 | Nof Corp | 修飾された生体関連物質、その製造方法および中間体 |
| WO2005037911A2 (en) | 2003-08-26 | 2005-04-28 | Smithkline Beecham Corporation | Heterofunctional copolymers of glycerol and polyethylene glycol, their conjugates and compositions |
| WO2006028129A1 (ja) | 2004-09-10 | 2006-03-16 | Toray Industries, Inc. | 医薬品製剤 |
| JP2007031554A (ja) | 2005-07-26 | 2007-02-08 | Nof Corp | ポリグリセリン誘導体及びこれを含む界面活性剤 |
| JP2008188557A (ja) | 2007-02-07 | 2008-08-21 | Nof Corp | ポリグリセリン誘導体を含む界面活性剤 |
| WO2008096904A1 (ja) | 2007-02-08 | 2008-08-14 | Tokyo Metropolitan Organization For Medical Research | マンノース6-リン酸-ポリエチレングリコール結合体 |
| WO2008105514A1 (ja) * | 2007-02-28 | 2008-09-04 | Nof Corporation | 多分岐鎖ポリオキシアルキレン誘導体 |
| JP2010235450A (ja) * | 2009-03-30 | 2010-10-21 | Nof Corp | 脂質誘導体 |
| WO2010114074A1 (ja) * | 2009-03-31 | 2010-10-07 | 日油株式会社 | 多分岐鎖ポリオキシアルキレン化合物、およびその製造方法 |
| JP2010254978A (ja) * | 2009-03-31 | 2010-11-11 | Nof Corp | 高分子量ポリオキシアルキレン誘導体の精製方法 |
Non-Patent Citations (3)
| Title |
|---|
| T. ISHIDA; H. KIWADA ET AL., J. CONTROL. RELEASE, vol. 122, 2007, pages 349 - 355 |
| THEODORA W. GREENE, PROTECTIVE GROUPS IN ORGANIC SYNTHESIS |
| W. JIS KOOT; R.M.F. VAN SCHIE ET AL., PHARMACEUTICAL RESEARCH, vol. 26, no. 6, 2009, pages 1303 - 1314 |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9908971B2 (en) | 2012-03-30 | 2018-03-06 | Nof Corporation | Multi-arm polyethylene glycol derivative, intermediate thereof, and method for producing same |
| US9428609B2 (en) | 2012-03-30 | 2016-08-30 | Nof Corporation | Multi-arm polyethylene glycol derivative, intermediate thereof, and method for producing same |
| WO2013147015A1 (ja) * | 2012-03-30 | 2013-10-03 | 日油株式会社 | マルチアーム型ポリエチレングリコール誘導体、その中間体及び製造方法 |
| JPWO2017188372A1 (ja) * | 2016-04-28 | 2019-04-04 | 日油株式会社 | 分岐型ポリオキシエチレン化合物およびコンタクトレンズ |
| WO2017188372A1 (ja) * | 2016-04-28 | 2017-11-02 | 日油株式会社 | 分岐型ポリオキシエチレン化合物およびコンタクトレンズ |
| JP2018062655A (ja) * | 2016-10-07 | 2018-04-19 | 国立大学法人東京工業大学 | 分岐型ヘテロ単分散ポリエチレングリコール、その製造方法、及びその結合体 |
| JP7042053B2 (ja) | 2016-10-07 | 2022-03-25 | 国立大学法人東京工業大学 | 分岐型ヘテロ単分散ポリエチレングリコール、その製造方法、及びその結合体 |
| US11613607B2 (en) | 2016-10-07 | 2023-03-28 | Tokyo Institute Of Technology | Branched type hetero monodispersed polyethylene glycol, production method thereof, and conjugate thereof |
| WO2018180917A1 (ja) * | 2017-03-31 | 2018-10-04 | 日油株式会社 | 末端に複数の水酸基を有するポリオキシエチレン誘導体の製造方法 |
| US10947343B2 (en) | 2017-03-31 | 2021-03-16 | Nof Corporation | Production method of polyoxyethylene derivative having plurality of hydroxyl groups at terminal |
| WO2019189431A1 (ja) * | 2018-03-27 | 2019-10-03 | 日油株式会社 | マルチアーム型ポリエチレングリコール誘導体の製造方法 |
| US11905366B2 (en) | 2018-03-27 | 2024-02-20 | Nof Corporation | Method for producing multi-arm type polyethylene glycol derivative |
| CN111936550A (zh) * | 2018-03-29 | 2020-11-13 | 日油株式会社 | 分解性聚乙二醇衍生物 |
| CN111936550B (zh) * | 2018-03-29 | 2023-11-28 | 日油株式会社 | 分解性聚乙二醇衍生物 |
| WO2022131098A1 (ja) * | 2020-12-15 | 2022-06-23 | 日油株式会社 | マレイミドポリエチレングリコール誘導体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20120322955A1 (en) | 2012-12-20 |
| KR20140038377A (ko) | 2014-03-28 |
| EP2692771A4 (en) | 2015-12-23 |
| EP2692771A1 (en) | 2014-02-05 |
| JP5949037B2 (ja) | 2016-07-06 |
| CN103492458B (zh) | 2016-10-12 |
| KR101811911B1 (ko) | 2017-12-22 |
| CN103492458A (zh) | 2014-01-01 |
| CN107573498A (zh) | 2018-01-12 |
| CN107573498B (zh) | 2020-04-24 |
| EP2692771B1 (en) | 2019-09-04 |
| JP2012214747A (ja) | 2012-11-08 |
| US8952125B2 (en) | 2015-02-10 |
| DK2692771T3 (da) | 2019-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5949037B2 (ja) | 末端に複数の水酸基を有するポリオキシエチレン誘導体 | |
| JP5589503B2 (ja) | 多分岐鎖ポリオキシアルキレン化合物、その製造方法および中間体 | |
| JP5418360B2 (ja) | ポリオキシアルキレン誘導体の製造方法 | |
| JP5866789B2 (ja) | 多官能性ポリオキシアルキレン化合物、その製造方法及び中間体 | |
| JP6935060B2 (ja) | 末端に複数の水酸基を有するポリオキシエチレン誘導体の製造方法 | |
| JP5945896B2 (ja) | 分岐型ヘテロ多官能性ポリオキシアルキレン化合物、及びその中間体 | |
| US11905366B2 (en) | Method for producing multi-arm type polyethylene glycol derivative | |
| CA3094055A1 (en) | Branched monodispersed polyethylene glycol, intermediate and methods for producing same | |
| JP7342757B2 (ja) | ヘテロ型単分散ポリエチレングリコール誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12764912 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20137025831 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |