WO2012020584A1 - 新規な含ケイ素脂環式ポリイミド樹脂及びポリアミド酸樹脂並びにそれらの製造方法 - Google Patents
新規な含ケイ素脂環式ポリイミド樹脂及びポリアミド酸樹脂並びにそれらの製造方法 Download PDFInfo
- Publication number
- WO2012020584A1 WO2012020584A1 PCT/JP2011/059209 JP2011059209W WO2012020584A1 WO 2012020584 A1 WO2012020584 A1 WO 2012020584A1 JP 2011059209 W JP2011059209 W JP 2011059209W WO 2012020584 A1 WO2012020584 A1 WO 2012020584A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- norbornane ring
- polyimide resin
- formula
- exo
- resin
- Prior art date
Links
- 0 CCCC[C@](CC(C1[N+](*(C)=C)[O-])[S@@](C)=*(C)NOC=*)C1*(*(C(C)(C)*)=C)=O Chemical compound CCCC[C@](CC(C1[N+](*(C)=C)[O-])[S@@](C)=*(C)NOC=*)C1*(*(C(C)(C)*)=C)=O 0.000 description 4
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1057—Polyimides containing other atoms than carbon, hydrogen, nitrogen or oxygen in the main chain
- C08G73/106—Polyimides containing other atoms than carbon, hydrogen, nitrogen or oxygen in the main chain containing silicon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/42—Polyamides containing atoms other than carbon, hydrogen, oxygen, and nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1046—Polyimides containing oxygen in the form of ether bonds in the main chain
- C08G73/105—Polyimides containing oxygen in the form of ether bonds in the main chain with oxygen only in the diamino moiety
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1075—Partially aromatic polyimides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1075—Partially aromatic polyimides
- C08G73/1078—Partially aromatic polyimides wholly aromatic in the diamino moiety
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/42—Block-or graft-polymers containing polysiloxane sequences
- C08G77/452—Block-or graft-polymers containing polysiloxane sequences containing nitrogen-containing sequences
- C08G77/455—Block-or graft-polymers containing polysiloxane sequences containing nitrogen-containing sequences containing polyamide, polyesteramide or polyimide sequences
Definitions
- the present invention relates to a novel silicon-containing alicyclic polyimide resin and polyamic acid resin, and methods for producing them.
- polyimide resin is known as a high-performance polymer used in electronic devices, and aromatic polyimide resin has been used so far from the viewpoint of heat resistance.
- aromatic polyimide resin is limited because it has absorption in the above wavelength region. Therefore, development of non-aromatic polyimide resin with high heat resistance is required.
- heat-resistant transparent materials applied in the optoelectronics field require not only mechanical properties such as tensile strength and elongation at break, but also properties such as adhesion to silicon and glass substrates and solubility in general-purpose solvents. Has been.
- Patent Document 1 a polyimide using a tetracarboxylic dianhydride represented by the following chemical formula (4) synthesized from 5-norbornene-2,3-dicarboxylic anhydride and hydrosilyl-type dimethylsiloxane at both ends A resin is disclosed.
- Non-Patent Document 1 a tetracarboxylic dianhydride represented by the following chemical formula (5) synthesized from 5-norbornene-endo-2,3-dicarboxylic anhydride and hexamethyltrisiloxane was used.
- a polyimide resin is disclosed, and it is reported that this polyimide resin is transparent at a wavelength of 350 nm or more.
- Non-Patent Document 2 a unit derived from tetracarboxylic dianhydride represented by the following chemical formula (6), which has a pentamethyldisiloxane structure at both ends by a very special polymerization method (disiloxane equilibration reaction).
- a polyimide resin having the same repeating unit structure is disclosed.
- the tetracarboxylic dianhydride disclosed in Non-Patent Document 1 is a tetracarboxylic dianhydride having a terminal end norbornanoic acid anhydride moiety represented by the chemical formula (5) having an endo structure.
- this tetracarboxylic dianhydride has the following formula (5a) depending on the position where two silicon atoms are bonded to the norbornane ring.
- the R—S form is a meso form
- the R—R form is a meso form
- the R—R form is a racemate
- the SS form form a racemate and can be separated into a meso form having a melting point of 143.5 to 144.5 ° C. and a racemate having a melting point of 119.5 to 121.5 ° C.
- Non-Patent Document 1 only describes that a polyimide resin was synthesized using tetracarboxylic dianhydride having a melting point of 133.4 to 133.9 ° C.
- tetracarboxylic dianhydrides disclosed in Patent Document 1 and Non-Patent Document 1 are both tetracarboxylic dianhydrides having an end structure at both terminal norbornanic acid anhydride moieties.
- Non-Patent Document 2 synthesis of a polyimide resin from tetracarboxylic dianhydride having both ends of norbornanic anhydride in chemical formula (7) in the following reaction formula (1) and an end structure, and m-phenylenediamine, As a result, it is described that a polyimide resin having the same structure of the repeating unit portion as that of the reaction formula (1) was synthesized by the method represented by the following reaction formula (2). Moreover, it is reported that the polyimide resin synthesized by the reaction formula (1) has a lower molecular weight than that of the polyimide resin synthesized by the reaction formula (2) and a cyclic oligomer is formed.
- an exo-polyimide resin having a norbornane structure is synthesized by the same method as in reaction formula (2), and this polyimide resin is reported to have high intrinsic viscosity and little cyclic oligomer formation.
- the repeating unit structure of the polyimide resin synthesized by the following reaction formula (3) is the same as that of the polyimide resin synthesized from tetracarboxylic dianhydride of chemical formula (6) and m-phenylenediamine.
- the method for synthesizing the polyimide resin represented by the reaction formula (3) is not easy to synthesize and purify the precursor, and the types are limited.
- One of the features of the polyimide resin is that it can be synthesized by combining various tetracarboxylic dianhydrides and diamines according to the purpose of use and required properties, and further copolymerizing other tetracarboxylic dianhydrides and diamines. Although it is possible to improve the characteristics by doing so, such flexibility is lacking. In addition, half of pentamethyldisiloxane added to the precursor is removed at the time of polymerization, which is not an industrial polyimide resin synthesis method.
- the present invention uses a tetracarboxylic dianhydride having an exo structure in the steric structure of both terminal norbornanic anhydride moieties, so that it is soluble in a general-purpose solvent, has good transparency, has a high molecular weight, has a high tensile strength, It aims at providing the polyamic acid resin which is the polyimide resin which is excellent also in mechanical characteristics, such as elongation, and its precursor.
- the present invention is a polyimide resin having a repeating unit of the following general formula (1) synthesized using a tetracarboxylic dianhydride represented by the following general formula (3) and a precursor thereof:
- a polyamic acid resin having a repeating unit of the general formula (2) is provided.
- m represents an integer of 2 to 30.
- all silicon atoms bonded to the norbornane ring are exo-configured with respect to the norbornane ring, and all acid anhydride groups bonded to the norbornane ring are exo-configured with respect to the norbornane ring. Yes.
- R represents a diamine or diisocyanate residue, and m represents an integer of 2 to 30.
- m represents an integer of 2 to 30.
- all silicon atoms bonded to the norbornane ring are arranged in an exo configuration with respect to the norbornane ring, and all imide rings bonded to the norbornane ring are arranged in an exo configuration with respect to the norbornane ring.
- R represents a diamine residue, and m represents an integer of 2 to 30.
- a polyimide resin having good transparency, a high molecular weight, excellent mechanical properties such as tensile strength and elongation, and soluble in a general-purpose solvent and a polyamic acid resin as a precursor thereof can be obtained.
- FIG. 1 is a 1 H-NMR spectrum of polyimide resins of Examples 1, 2, and 3.
- FIG. It is an ultraviolet-visible transmittance spectrum of the polyimide resin of Example 1 and Comparative Example 1.
- 3 is a 1 H-NMR spectrum of the polyimide resin of Example 4.
- 2 is an FT-IR spectrum of the polyimide resin of Example 1.
- 4 is an FT-IR spectrum of the polyamic acid resin of Example 5.
- 2 is a 1 H-NMR spectrum of polyimide resins of Comparative Examples 1 and 2.
- the polyimide resin according to this embodiment has a repeating unit structure represented by the following general formula (1). That is, all the silicon atoms bonded to the norbornane ring are arranged in an exo configuration with respect to the norbornane ring, and all the imide rings bonded to the norbornane ring are arranged in an exo configuration with respect to the norbornane ring.
- m represents a number from 2 to 30, preferably from 2 to 10, and more preferably from 2 to 5.
- m of a plurality of repeating units (1) may be a polyimide resin having a single value, or may be a polyimide resin composed of mixed repeating units (1) having different values of m.
- R represents a diamine or diisocyanate residue used to synthesize a polyimide resin.
- the polyimide resin according to the present embodiment is also synthesized by dehydrating and cyclizing a polyamic acid resin having a repeating unit structure represented by the following general formula (2).
- this polyamic acid resin all of the silicon atoms bonded to the norbornane ring are arranged in an exo configuration with respect to the norbornane ring, and both the amide group and the carboxyl group bonded to the norbornane ring are arranged in an exo configuration with respect to the norbornane ring.
- m represents a number from 2 to 30, preferably 2 to 10, and more preferably 2 to 5.
- m of the plurality of repeating units (2) may be a polyamic acid resin having a single value, or may be a polyamic acid resin composed of mixed repeating units (2) having different values of m.
- R represents the residue of the diamine used for synthesizing the polyamic acid resin.
- the tetracarboxylic dianhydride that is a raw material for synthesizing the polyimide resin and the polyamic acid resin according to the present embodiment is represented by the following general formula (3). That is, any silicon atom bonded to the norbornane ring is exo-configured with respect to the norbornane ring, and all acid anhydride groups bonded to the norbornane ring are exo-configured with respect to the norbornane ring.
- m represents a number from 2 to 30, preferably from 2 to 10, and more preferably from 2 to 5. Note that a tetracarboxylic dianhydride having a single value for m may be used, or a mixed tetracarboxylic dianhydride having a different value for m may be used.
- This tetracarboxylic dianhydride includes 5-norbornene-exo-2,3-dicarboxylic acid anhydride represented by the following chemical formula (8), a hydrosilyl-type dimethylsiloxane compound represented by the following general formula (9), It can obtain by the method of including the hydrosilylation reaction process of these.
- the hydrosilylation reaction is well known, and those skilled in the art can easily select the reaction conditions, and therefore the details thereof are omitted.
- m represents a number from 2 to 30, preferably from 2 to 10, and more preferably from 2 to 5. It should be noted that a both-end hydrosilyl-type dimethylsiloxane compound having a single value for m may be used, or a mixed-end-end hydrosilyl-type dimethylsiloxane compound having a different value for m may be used.
- the tetracarboxylic dianhydride represented by the general formula (3) is an isomer mixture of tetracarboxylic dianhydrides represented by the following general formulas (3a), (3b) and (3c), respectively.
- This tetracarboxylic dianhydride of the general formula (10) is also a stereoisomer, that is, a mixture of RS, RR and SS, but at least m is a tetramer having 2,3,4. Carboxylic dianhydride crystallizes at room temperature. However, the tetracarboxylic dianhydride of the general formula (3) according to this example remains in a liquid state at room temperature even after 2 years have passed since synthesis.
- Optional tetracarboxylic dianhydrides that can be used for copolymerization with the tetracarboxylic dianhydride of general formula (3) include aromatic tetracarboxylic dianhydrides and alicyclic tetracarboxylic dianhydrides. .
- Arbitrary tetracarboxylic dianhydrides have different UV-visible absorption spectra when made into polyimides depending on their structures and diamines used, but they can be used up to 50 mol% in the total tetracarboxylic dianhydrides. .
- aromatic tetracarboxylic dianhydride examples include, in addition to pyromellitic dianhydride and naphthalene tetracarboxylic dianhydride, tetracarboxylic dianhydrides represented by the following general formula (11).
- X is selected from the following formula (12).
- Y is selected from the following formula (13).
- the bonding position on the benzene ring of —X— and —Y is not particularly limited as long as the effect of the present invention is not impaired.
- the alicyclic tetracarboxylic dianhydride can be appropriately selected from, for example, the following group of compounds.
- the configuration of each acid anhydride group and the relative configuration of the two acid anhydride groups are not particularly limited.
- diamine which reacts with tetracarboxylic dianhydride to form a polyamic acid resin various diamines such as aromatic diamine and alicyclic diamine are used.
- aromatic diamine examples include diamines represented by the following general formula (14).
- Z is selected from the following formula (15).
- Y is selected from the above equation (13).
- the bonding position on the benzene ring of —NH 2 , —Y and —Z— is not particularly limited as long as the effect of the present invention is not impaired.
- Examples of the alicyclic diamine include the following compounds.
- the polyimide resin of the present invention is usually tetracarboxylic dianhydride (tetracarboxylic dianhydride of formula (3), and optionally used aromatic tetracarboxylic dianhydride and alicyclic tetracarboxylic dianhydride). It is obtained by a two-stage synthesis method in which a polyamic acid resin is synthesized from diamine and diamine, and the resulting polyamic acid resin is dehydrocyclized. Further, by using diisocyanate instead of diamine, a polyimide resin can be synthesized by a one-step synthesis method.
- reaction conditions of the two-step synthesis method and the one-step synthesis method are selected from the reaction conditions of the conventionally known two-step synthesis method and one-step synthesis method except that the tetracarboxylic dianhydride of the formula (3) is used. It is. A preferred embodiment will be described below.
- the tetracarboxylic dianhydride and the diamine are preferably polymerized in approximately equimolar amounts.
- a diamine is dissolved in a polar solvent such as N-methyl-2-pyrrolidinone, N, N-dimethylacetamide, 1,2-dichlorobenzene, N-cyclohexylpyrrolidone, methacresol, and then tetracarboxylic dianhydride is added.
- a polar solvent such as N-methyl-2-pyrrolidinone, N, N-dimethylacetamide, 1,2-dichlorobenzene, N-cyclohexylpyrrolidone, methacresol
- the total concentration of tetracarboxylic dianhydride and diamine in the reaction system is preferably 10 to 30% by mass, more preferably 15 to 20% by mass.
- concentration is lower than the above range, the degree of polymerization does not increase.
- concentration is high, problems such as a Weisenberg effect may occur.
- the reaction of tetracarboxylic dianhydride and diamine is preferably carried out at room temperature (23 ° C.) to 100 ° C. for 0.5 to 5 hours until the reaction solution becomes uniform and transparent.
- the polyamic acid resin is usually dehydrated and cyclized in a solution state to obtain a polyimide resin. However, if necessary, the obtained polyamic acid resin can be recovered by pouring it into water.
- the polyamic acid resin is obtained by cyclization using a polyamic acid resin solution obtained as described above or a solution obtained by dissolving the isolated polyamic acid resin in the polar solvent to obtain a polyimide resin.
- concentration of the polyamic acid resin in the solution is preferably 5 to 50% by mass, more preferably 10 to 30% by mass, since the molecular weight of the polyimide resin is sufficiently large and the uniformity of the solution by stirring is easily maintained. .
- a method of dehydrating cyclization there are a heating method or a chemical method of dehydrating cyclization in the presence of a dehydrating agent such as acetic anhydride and a catalyst such as pyridine.
- a dehydrating agent such as acetic anhydride and a catalyst such as pyridine.
- the polyamic acid resin solution dissolved in a high-boiling solvent azeotropic with water such as 1,2-dichlorobenzene, N-cyclohexylpyrrolidone, and methacresol is heated, and the water generated by dehydration cyclization is removed from the system together with the solvent.
- a method of obtaining a polyimide resin while removing azeotropically is also preferable.
- the dehydration cyclization temperature of the polyamic acid resin is preferably the boiling point of the solvent.
- the solvent is 1,2-dichlorobenzene
- the boiling point is 179 ° C.
- the solvent is metacresol
- the boiling point is 203 ° C.
- the reaction time is preferably 3 to 8 hours. If the reaction time is short, dehydration cyclization may not be performed completely, which will adversely affect the properties of the resulting polyimide resin.
- Dehydration cyclization is preferably carried out in the presence of a catalyst such as benzoic acid, parahydroxybenzoic acid, isoquinoline, 2-hydroxypyridine, etc.
- the catalyst amount is preferably 1 to 5% by mass of the total amount of tetracarboxylic dianhydride and diamine.
- a polyimide resin can be synthesized in one step from tetracarboxylic dianhydride and diisocyanate.
- Diisocyanates such as 4,4′-methylenedi (phenyl isocyanate) and 2,4-tolylene diisocyanate and tetracarboxylic dianhydride are heated to about 130 ° C. in the above polar solvent such as N, N-dimethylacetamide.
- a polyimide resin is produced while desorbing carbon dioxide.
- the reaction of tetracarboxylic dianhydride and diisocyanate is known, and those skilled in the art can easily select the reaction conditions, so the details are omitted.
- the weight average molecular weights of the polyimide resin represented by the general formula (1) and the polyamide resin represented by the general formula (2) are determined in terms of the mechanical properties of the film, particularly the elongation value and the viscosity of the varnish. From the viewpoint, it is preferably 30,000 to 200,000, more preferably 50,000 to 180,000, and particularly preferably 80,000 to 160,000.
- the ratio of the weight average molecular weight to the number average molecular weight, that is, the degree of dispersion is preferably 1.3 to 3.0, more preferably 1.3 to 2.0, and 1.3 to 1. 6 is particularly preferred.
- the number average molecular weight and the weight average molecular weight are values measured by gel permeation chromatography (GPC) and converted using a standard polystyrene calibration curve.
- GPC measurement conditions for polyimide resin The measurement conditions of GPC used for the molecular weight measurement of the polyimide resin in this example are shown below.
- NMR measurement conditions The measurement conditions of NMR used for the measurement of the average number of silicon of the tetracarboxylic dianhydride and the confirmation of the structure of the polyimide resin in this example are shown below.
- Apparatus Bruker AV400M type superconducting Fourier transform nuclear magnetic resonance apparatus Resonance frequency: 1 H-NMR 400.23 MHz
- Solvent Deuterated chloroform Sample concentration: 15 mg / 0.75 mL Measurement temperature: Room temperature (24 ° C) Integration count: 16 times
- thermogravimetric analysis of polyamic acid resin The measurement conditions for thermogravimetric analysis of the polyamic acid resin in this example are shown below. Apparatus: Seiko Instruments Inc. TG / DTA-6200 type differential thermogravimetric simultaneous measurement apparatus Sample amount: 10 mg Temperature increase rate: 10 ° C / min Atmosphere: air, 10 mL / min
- Synthesis example 1 “Synthesis of tetracarboxylic dianhydride in which m is 2 in the general formula (3)”
- the catalyst Platinum (0) -1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex (xylene solution having a platinum concentration of 2%) manufactured by Aldrich was used. 184 g (1.214 ⁇ 10 ⁇ 4 gram atom as platinum metal) was added, and 31.27 g (0.1500 mol) of 1,1,3,3,5,5-hexamethyltrisiloxane was gradually added from the dropping funnel into the flask. It was dripped in. Since the reaction temperature increased with the dropwise addition, the reaction was continued for 1 hour while keeping the temperature in the flask at 70 ° C.
- Synthesis example 2 “Synthesis of tetracarboxylic dianhydride in which m is 3 in the general formula (3)”
- the amount of 5-norbornene-exo-2,3-dicarboxylic anhydride was changed to 39.61 g (0.2413 mol) and the amount of catalyst was changed to 0.9516 g, and 1,1,3,3,5,5-hexamethyl
- the reaction and post-treatment operations were performed in the same manner as in Synthesis Example 1 except that 33.93 g (0.1201 mol) of 1,1,3,3,5,5,7,7-octamethyltetrasiloxane was used instead of trisiloxane. went.
- Synthesis example 3 “Synthesis of tetracarboxylic dianhydride in which m is 4 in the general formula (3)”
- the amount of 5-norbornene-exo-2,3-dicarboxylic acid anhydride was changed to 41.26 g (0.2513 mol) and the amount of catalyst was changed to 0.9745 g, and 1,1,3,3,5,5-hexamethyl
- the reaction was conducted in the same manner as in Synthesis Example 1 except that 44.59 g (0.1250 mol) of 1,1,3,3,5,5,7,7,9,9-decamethylpentasiloxane was used instead of trisiloxane.
- a post-processing operation was performed.
- Synthesis example 4 “Synthesis of tetracarboxylic dianhydride in which m is an average of 4.11 and m is 2 to 10 in the general formula (3)”
- the amount of 5-norbornene-exo-2,3-dicarboxylic acid anhydride was changed to 39.61 g (0.2413 mol) and the amount of catalyst was changed to 0.9480 g, and 1,1,3,3,5,5-hexamethyl
- 49.90 g of both-end hydrosilyl-type dimethylsiloxane mixed oligomer in which the number n of silicon (m + 1 in the formula (3)) is distributed from 3 to 11 is involved in the hydrosilylation reaction.
- Synthesis example 5 "Synthesis of a meso form of tetracarboxylic dianhydride of formula (5)" To a 1 L four-necked flask equipped with a stirrer, dropping funnel, condenser and thermometer, 200 g of toluene and 82.08 g of 5-norbornene-endo-2,3-dicarboxylic acid anhydride having a melting point of 166 to 167 ° C. 0.5000 mol) was charged, and heating and stirring were started. 5.
- the catalyst Platinum (0) -1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex (xylene solution having a platinum concentration of 2%) manufactured by Aldrich. 4731 g (6.64 ⁇ 10 ⁇ 4 gram atom as platinum metal) was added, and 52.12 g (0.2500 mol) of 1,1,3,3,5,5-hexamethyltrisiloxane was gradually added from the dropping funnel into the flask. It was dripped in. Since the reaction temperature increased with the dropwise addition, the reaction was continued for another 4 hours while maintaining the temperature in the flask at 100 ° C.
- Synthesis Example 6 Synthesis of racemic tetracarboxylic dianhydride of formula (5)
- the filtrate stored in Synthesis Example 5 there is a racemic form of tetracarboxylic dianhydride of the formula (5), and the meso form remains.
- GPC analysis it was found that unreacted 5-norbornene-endo-2,3-dicarboxylic anhydride and a small amount of a compound with unknown structure were mixed. Therefore, after removing toluene using a rotary evaporator, it was recrystallized with diethyl ether.
- the melting point was 104 to 114 ° C., and as a result of GPC analysis, it was found that unreacted 5-norbornene-endo-2,3-dicarboxylic anhydride and a small amount of a compound with unknown structure were removed.
- the obtained crystals were recrystallized with a mixed solvent of 75% by mass of toluene / 25% by mass of hexane.
- the melting point of the produced crystal was 136 to 139 ° C., and the crystal was meso form. Hexane was added so that the solvent composition in the filtrate was 50% by mass of toluene / 50% by mass of hexane to precipitate crystals.
- this crystal is shown in the unit cell as an RR form represented by the following steric structure (5b) and a steric structure (5c). It has been confirmed that the S—S form is a racemate (endo-NB-TriSXDA racemate) that exists in a pair.
- Synthesis example 7 "Synthesis of tetracarboxylic dianhydride represented by chemical formula (6)" Into a 1 L four-necked flask equipped with a stirrer, dropping funnel, condenser and thermometer, 240 g of toluene and 5-norbornene-exo-2,3-dicarboxylic acid anhydride having a melting point of 148-149.5 ° C. 123. 20 g (0.7505 mol) was charged and heating and stirring were started. 1.
- the catalyst Platinum (0) -1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex (xylene solution having a platinum concentration of 2%) manufactured by Aldrich. 99 g (3.07 ⁇ 10 ⁇ 4 gram atom as platinum metal) was added, and 50.17 g (0.3735 mol) of 1,1,3,3-tetramethyldisiloxane was gradually dropped into the flask from the dropping funnel. Since the reaction temperature increased with the dropwise addition, the reaction was continued for another 1 hour while keeping the temperature in the flask at 90 ° C. after dropwise addition over 1 hour taking care to maintain the temperature in the flask at 90 ° C.
- the extraction side flask of the continuous single extraction apparatus was charged with 86.8 g of a viscous, colorless and transparent liquid, 169.4 g of hexane, and 23.9 g of toluene, 101.3 g of hexane in the distillation side flask, and 60.0 g of hexane in the dropping funnel. Prepared. Extraction was performed for 10 hours with the water bath temperature on the extraction side set to 65 ° C. and the oil bath temperature on the distillation side set to 115 ° C. The lower layer of the extraction side flask was recovered, the contained solvent was removed, and the weight was determined to be 62.0 g. Further, the following chemical formula (16b), which is another by-product, was removed by distillation while blowing a small amount of nitrogen under a reduced pressure of 70 Pa.
- Synthesis example 8 “Synthesis of tetracarboxylic dianhydride in which m is 3 in the general formula (10)” To a 300 mL four-necked flask equipped with a stirrer, dropping funnel, condenser and thermometer, 116 g of toluene and 60.12 g of 5-norbornene-endo-2,3-dicarboxylic anhydride having a melting point of 166 to 167 ° C. 0.3662 mol) was charged and heating and stirring were started. 1.
- the catalyst Platinum (0) -1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex (xylene solution having a platinum concentration of 2%) manufactured by Aldrich. 58 g (2.65 ⁇ 10 ⁇ 4 gram atom as platinum metal) was added, and 51.50 g (0.1822 mol) of 1,1,3,3,5,5,7,7-octamethyltetrasiloxane was added from the dropping funnel. The solution was gradually dropped into the flask. Since the reaction temperature increased with the dropwise addition, the dropwise addition was performed over 1 hour while taking care to maintain the temperature in the flask at 100 ° C.
- the reaction solution was sampled and analyzed by GPC. Thereafter, while maintaining the temperature in the flask at 100 ° C., the reaction solution was sampled and subjected to GPC analysis at 2 hours after the start of the reaction. As a result, no change was observed in the reaction solution composition. Therefore, an additional 1.28 g of catalyst was added 3 hours after the start of the reaction. Thereafter, the reaction solution was analyzed, and 1.13 g of an additional catalyst was added at 5.5 hours after the start of the reaction, and 1.12 g of an additional catalyst was added at 7.5 hours. -Since the remaining amount of endo-2,3-dicarboxylic anhydride was 5.9% of the charged amount, the reaction was terminated.
- the total amount of catalyst used in the reaction was 6.11 g (6.26 ⁇ 10 ⁇ 4 gram atom as platinum metal). Thereafter, the temperature was lowered to 50 ° C., 12 g of activated carbon was added and stirred at room temperature for 30 minutes, and then the activated carbon was removed by filtration. The toluene in the filtrate was removed using a rotary evaporator to obtain 89.05 g of white crystals having a melting point of 87.5-94.5 ° C. This is a mixture of meso form and racemic form (endo-NB-TetraSXDA), and the yield is 80.0%.
- crystals were recrystallized with a mixed solvent of toluene and hexane in a weight ratio of 2: 1 to obtain crystals having a melting point of 99.0 to 100.0 ° C.
- This crystal had a purity of 99.7% according to GPC analysis.
- Example 1 Polyimide resin synthesized from exo-NB-TriSXDA synthesized in Synthesis Example 1 and oxydianiline
- a polyimide resin was synthesized as follows using exo-NB-TriSXDA synthesized in Synthesis Example 1. To a 100 mL four-necked flask equipped with a stirring blade, a thermometer, a Dean-Stark fractionation tube and a nitrogen introduction tube, 5.11812 g (9.651 mmol) of 4,4′-oxydianiline and exo-NB-TriSXDA were added.
- FIG. 1 shows the 1 H-NMR spectrum. It can be confirmed that the spectrum reflects the repeating unit structure.
- the number average molecular weight in terms of standard polystyrene was 30,400, and the weight average molecular weight was 56,200.
- the solubility of this polyimide resin was examined. About 50 mg of polyimide resin was precisely weighed, 10 times the amount of solvent was added thereto, and the dissolution was observed at room temperature. As a result, it was dissolved in tetrahydrofuran, acetone, N-methyl-2-pyrrolidinone and N, N-dimethylacetamide, but with toluene, the solution became cloudy and separated into two layers after standing. A film was made to clarify the characteristics of this polyimide resin.
- the creation procedure is as follows.
- N, N-dimethylacetamide solution having a resin concentration of 25% by mass was prepared. Filter using 5B filter paper. Cast and apply the filtered solution to a glass plate so that the dry film thickness is 20-30 ⁇ m, leave it in a 65 ° C. drying oven for 30 minutes, and then increase the temperature to 165 ° C. over 20 minutes. , Held at this temperature for 15 minutes. Then, the glass plate was immersed in warm water for 10 minutes, and the film was peeled from the glass plate. Table 1 summarizes the results of the measurement of the glass transition point under tensile conditions and the tensile test using a thermomechanical analyzer. Further, the transmittance spectrum obtained by the ultraviolet visible spectrophotometer is shown in FIG. 2, and the FT-IR spectrum obtained by the infrared spectrophotometer is shown in FIG.
- Example 2 Polyimide resin synthesized from exo-NB-TetraSXDA synthesized in Synthesis Example 2 and oxydianiline] Using exo-NB-TetraSXDA synthesized in Synthesis Example 2, a polyimide resin was synthesized as follows.
- the spectrum reflects the repeating unit structure.
- the number average molecular weight in terms of standard polystyrene was 40,500, and the weight average molecular weight was 68,400.
- the solubility of this polyimide resin was examined by the same method as in Example 1. As a result, it was dissolved in tetrahydrofuran, acetone, toluene, N-methyl-2-pyrrolidinone and N, N-dimethylacetamide, and all became uniform transparent solutions.
- a film was prepared by the method described in Example 1, and the glass transition point was measured and a tensile test was performed. The results are also shown in Table 1.
- Example 3 Polyimide resin synthesized from exo-NB-PentaSXDA synthesized in Synthesis Example 3 and oxydianiline
- a polyimide resin was synthesized as follows using exo-NB-PentaSXDA synthesized in Synthesis Example 3.
- Example 1 1.9349 g (9.6629 mmol), 0.2564 g of 2-hydroxypyridine as a catalyst and 59.87 g of 1,2-dichlorobenzene were charged and reacted in exactly the same manner as in Example 1 to precipitate the resin.
- the mass of the obtained resin was 6.75 g, which was 82.2% of the theoretical yield of charging.
- FIG. 1 also shows the 1 H-NMR spectrum. It can be confirmed that the spectrum reflects the repeating unit structure.
- the number average molecular weight in terms of standard polystyrene was 59,900, and the weight average molecular weight was 106,800.
- the solubility of this polyimide resin was examined by the same method as in Example 1.
- Example 4 Polyimide resin synthesized from exo-NB-MixSXDA synthesized in Synthesis Example 4 and oxydianiline
- a polyimide resin was synthesized as follows using exo-NB-MixSXDA synthesized in Synthesis Example 4.
- 6.9907 g (10.084 mmol) of exo-NB-MixSXDA and 4,4′-oxydianiline were added.
- Example 2 2.0167 g (10.071 mmol), 0.2725 g of 2-hydroxypyridine as a catalyst and 63.08 g of 1,2-dichlorobenzene were charged and reacted in exactly the same manner as in Example 1 to precipitate the resin.
- the mass of the obtained resin was 6.80 g, which was 78.7% of the theoretical yield of charging.
- FIG. 3 shows the 1 H-NMR spectrum. It can be confirmed that the spectrum reflects the repeating unit structure.
- the number average molecular weight in terms of standard polystyrene was 31,900, and the weight average molecular weight was 50,400.
- the solubility of this polyimide resin was examined by the same method as in Example 1.
- Example 5 Polyamic acid resin synthesized from exo-NB-TriSXDA synthesized in Synthesis Example 1 and oxydianiline
- 6.3208 g (11.775 mmol) of exo-NB-TriSXDA synthesized in Synthesis Example 1 and 4,4′-oxydianiline were added.
- 2.3577 g (11.774 mmol) and 49.21 g of N-methyl-2-pyrrolidinone were charged, and stirring was started at room temperature (28 ° C.).
- Example 6 Polyimide resin synthesized from exo-NB-TriSXDA synthesized in Synthesis Example 1 and oxydianiline
- the raw material concentration of the reaction system total concentration of tetracarboxylic dianhydride and diamine
- a higher molecular weight polyimide resin was synthesized as follows.
- 14.5792 g (27.16 mmol) of exo-NB-TriSXDA and 4,4′-oxydianiline were added.
- the upper layer was removed, and 45 g of methanol was added to the lower layer to precipitate a gummy mass.
- the solvent was removed by placing in a 90 ° C. dryer for 4 hours.
- the mass of the obtained resin was 10.28 g, which was 51.3% of the theoretical yield of charging.
- the number average molecular weight in terms of standard polystyrene was 55,900, and the weight average molecular weight was 88,200.
- a film was prepared by the method described in Example 1, and the glass transition point was measured and a tensile test was performed. The glass transition point was 138 ° C., the tensile strength was 43 MPa, the elastic modulus was 1.54 GPa, and the elongation at break was 22%.
- Example 7 Polyimide resin synthesized from exo-NB-PentaSXDA synthesized in Synthesis Example 3 and oxydianiline
- the raw material concentration of the reaction system total concentration of tetracarboxylic dianhydride and diamine
- a higher molecular weight polyimide resin was synthesized as follows.
- the upper layer was removed, and 40 g of methanol was added to the lower layer to precipitate a gummy mass. This precipitate was dissolved in 50 g of acetone, and 23 g of methanol was added dropwise to separate into two layers. The upper layer was removed, and 40 g of methanol was added to the lower layer to precipitate a gummy mass. The solvent was removed by placing in a 90 ° C. dryer for 4 hours. The mass of the obtained resin was 10.98 g, which was 66.3% of the theoretical yield of charging.
- the number average molecular weight in terms of standard polystyrene was 89,800, and the weight average molecular weight was 141,000.
- a film was prepared by the method described in Example 1, and the glass transition point was measured and a tensile test was performed.
- the glass transition point was 88 ° C.
- the tensile strength was 29 MPa
- the elastic modulus was 1.23 GPa
- the elongation at break was 127%.
- Comparative Example 1 Polyimide resin synthesized from endo-NB-TriSXDA meso form and oxydianiline obtained in Synthesis Example 5] A polyimide resin was synthesized as follows using the endo-NB-TriSXDA meso form obtained in Synthesis Example 5. In a 100 mL four-necked flask equipped with a stirring blade, a thermometer, a Dean-Stark fractionation tube and a nitrogen introduction tube, 6.0239 g (11.222 mmol) of 4,4′-oxydi was added to the endo-NB-TriSXDA meso body.
- the mass of the obtained resin was 2.85 g, which was 36.2% of the theoretical yield of charging.
- the number average molecular weight in terms of standard polystyrene was 19,800, and the weight average molecular weight was 31,900.
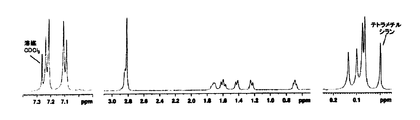
- FIG. 6 shows the 1 H-NMR spectrum. It can be confirmed that the spectrum reflects the repeating unit structure. In particular, since the two methyl groups bonded to the silicon at the center of the trisiloxane were clearly split into two (0.029 ppm and 0.040 ppm), it was confirmed that the meso structure was retained. The solubility of this polyimide resin was examined by the same method as in Example 1.
- this polyimide resin was dissolved in N, N-dimethylacetamide to prepare a film.
- this polyimide resin was made into fine powder with an agate mortar and the glass transition point was measured with a differential scanning calorimeter. The results are also shown in Table 1.
- the polyimide resin of Example 1 has a large transparency in the region of about 290 to 350 nm and good transparency as compared with the polyimide resin of Comparative Example 1.
- the resin was reprecipitated in the same manner as in Comparative Example 1 to obtain a resin having a number average molecular weight of 15,700 in terms of standard polystyrene and a weight average molecular weight of 27,500.
- the mass of the obtained resin was 6.65 g, which was 50.0% of the theoretical yield of charging.
- FIG. 6 shows the 1 H-NMR spectrum. It can be confirmed that the spectrum reflects the repeating unit structure. The two methyl groups bonded to the center silicon of trisiloxane are equivalent and observed as one peak (0.035 ppm), confirming that the racemic structure is retained.
- this polyimide resin was dissolved in N, N-dimethylacetamide to prepare a film.
- Comparative Example 3 Polyimide resin synthesized from exo-NB-DiSXDA and oxydianiline obtained in Synthesis Example 7] To a 100 mL four-necked flask equipped with a stirring blade, a thermometer, a Dean-Stark fractionation tube, and a nitrogen introduction tube, 4.3735 g (9.453 mmol) of exo-NB-DiSXDA obtained in Synthesis Example 7, 4, 4 1.8929 g (9.453 mmol) of '-oxydianiline, 0.1882 g of 2-hydroxypyridine as a catalyst, and 43.88 g of 1,2-dichlorobenzene were charged, and heating and stirring were started.
- the total concentration of exo-NB-DiSXDA and 4,4′-oxydianiline in the reaction system was 12.5%.
- the synthesis reaction of polyimide resin was performed in the same manner as in Example 1, and the same resin purification as in Example 6 was performed.
- the mass of the obtained resin was 2.69 g, which was 43.0% of the theoretical yield of charging.
- the number average molecular weight in terms of standard polystyrene was 25,700, and the weight average molecular weight was 35,700.
- the solubility of this polyimide resin was examined by the same method as in Example 1.
- a film was prepared by the method described in Example 1, and the glass transition point was measured and a tensile test was performed. The glass transition point was 178 ° C., the tensile strength was 41 MPa, the elastic modulus was 1.86 GPa, and the elongation at break was 2.6%.
- Comparative Example 4 Polyimide resin synthesized from endo-NB-TetraSXDA and oxydianiline obtained in Synthesis Example 8] To a 100 mL four-necked flask equipped with a stirring blade, a thermometer, a Dean-Stark fractionation tube, and a nitrogen introduction tube, 5.9255 g (9.699 mmol) of endo-NB-TetraSXDA obtained in Synthesis Example 8 was used. 1.9419 g (9.698 mmol) of '-oxydianiline, 0.237 g of 2-hydroxypyridine as a catalyst, and 55.08 g of 1,2-dichlorobenzene were charged, and heating and stirring were started.
- the total concentration of endo-NB-TetraSXDA and 4,4′-oxydianiline in the reaction system was 12.5%.
- the synthesis reaction of the polyimide resin was performed in the same manner as in Example 1, and the same resin purification as in Example 1 was performed. However, the mass of the resin obtained was only 0.69 g, which was 8.8% of the theoretical yield of charging.
- the number average molecular weight in terms of standard polystyrene was 17,600, and the weight average molecular weight was 28,200.
- the solubility of this polyimide resin was examined by the same method as in Example 1.
- a polyimide resin having good transparency, a high molecular weight, excellent mechanical properties such as tensile strength and elongation, and soluble in a general-purpose solvent and a polyamic acid resin as a precursor thereof can be obtained.
- the polyimide resin or polyamic acid resin of the present invention has characteristics as a material used in the field of optoelectronics, and can be suitably used.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Abstract
Description




特開2010-254602号に記載したように,本発明者らの検討によれば,このテトラカルボン酸二無水物は,2つのケイ素原子がノルボルナン環に結合している位置によって,下記式(5a)~(5c)で表されるR-S体,R-R体,S-S体の3つの異性体が存在することが判明し,R-S体がメソ体であり,R-R体とS-S体が対になってラセミ体を形成しており,融点が143.5~144.5℃のメソ体と融点が119.5~121.5℃のラセミ体に分離できる。




















本実施例におけるテトラカルボン酸二無水物の粘度測定条件を以下に示す。
装置:東京計器製EH型粘度計
測定範囲:0.0512~102.4 Pa・s
測定温度:25℃または40℃
本実施例におけるテトラカルボン酸二無水物の屈折率測定条件を以下に示す。
装置:株式会社アタゴ製DR-S1型アッベ屈折計
測定温度:25℃
本実施例におけるテトラカルボン酸二無水物の融点測定条件を以下に示す。
装置:株式会社石井商店製精密微量融点測定器
毛細管:一端封じ形,内径1.05mm,外径1.55mm
昇温速度:2℃/分
本実施例におけるテトラカルボン酸二無水物の純度分析に用いたGPCの測定条件を以下に示す。
カラム:日立化成工業株式会社製 Gelpack GL-A110 500mm2本
を直列に結合
溶離液:テトラヒドロフラン
流量:1.0mL/分
検出器:株式会社日立製作所製 L-3300RI型示差屈折計
測定温度:25℃
本実施例におけるポリイミド樹脂の分子量測定に用いたGPCの測定条件を以下に示す。
カラム:日立化成工業株式会社製 Gelpack GL-R440,GL-R450,およびGL-400M の3種類のカラムを直列に結合
溶離液:テトラヒドロフラン
流量:2.05mL/分
検出器:株式会社日立製作所製 L-3300RI型示差屈折計
測定温度:25℃
本実施例におけるテトラカルボン酸二無水物のケイ素の平均個数の測定並びにポリイミド樹脂の構造確認に用いたNMRの測定条件を以下に示す。
装置:Bruker社製 AV400M型 超伝導フーリエ変換核磁気共鳴装置
共鳴周波数:1H-NMR 400.23MHz
溶媒:重クロロホルム
試料濃度:15mg/0.75mL
測定温度:室温(24℃)
積算回数:16回
本実施例におけるポリイミド樹脂のガラス転移点の測定条件を以下に示す。
装置:セイコーインスツルメント株式会社製 TMA/SS-6200型熱機械的分析
装置
測定方式:引張り,引張り加重5g
試料形状:長さ10.0mm,幅2.0mm,厚さ20~25μm
昇温速度:10℃/分
雰囲気:空気
本比較例におけるポリイミド樹脂のガラス転移点の測定条件を以下に示す。
装置:セイコーインスツルメント株式会社製 DSC-6200型示差走査熱量分析計
試料量:10mg
昇温速度:10℃/分
雰囲気:空気
本実施例におけるポリイミド樹脂の機械特性評価のために行った引張り試験(引張強さ、弾性率、破断伸びの3項目)の条件を以下に示す。
装置:島津製作所製AGS-5KNG型オートグラフ
試験片形状:厚さ20~25μm,幅10.0mm
つかみ具間距離:20.0mm
引張速度:5mm/分
測定温度:23℃
本実施例におけるポリイミド樹脂の透過率測定条件を以下に示す。
装置:株式会社日立製作所製 U-3310型紫外可視分光光度計
走査速度:300nm/分
本実施例におけるポリアミド酸樹脂の熱重量分析の測定条件を以下に示す。
装置:セイコーインスツルメント株式会社製 TG/DTA-6200型示差熱熱重量同時測定装置
試料量:10mg
昇温速度:10℃/分
雰囲気:空気,10mL/分
装置:バリアン社製 3100型フーリエ変換赤外分光光度計
測定法:KBr法
「一般式(3)においてmが2であるテトラカルボン酸二無水物の合成」
攪拌子,滴下ロート,冷却管及び温度計を取り付けた300mLの4つ口フラスコに,トルエン115g及び融点が148~149.5℃の5-ノルボルネン-エキソ-2,3-ジカルボン酸無水物49.50g(0.3015mol)を仕込み,加熱攪拌を開始した。フラスコ内の温度が65℃になった時点で,触媒のアルドリッチ社製Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex(白金濃度2%のキシレン溶液)1.184g(白金金属として1.214×10-4グラム原子)を加え,1,1,3,3,5,5-ヘキサメチルトリシロキサン31.27g(0.1500mol)を滴下ロートからフラスコ内に徐々に滴下した。滴下に伴って反応温度が上昇するので,フラスコ内の温度が70℃を維持するよう注意しながら1時間かけて滴下した後,フラスコ内温度を70℃に保ちながら更に1時間反応を続けた。
その後,冷却し,活性炭8gを加え室温で30分間攪拌し,濾過操作で活性炭を除いた。ロータリーエバポレーターを使って濾液を減圧濃縮し,次いで残存する微量の5-ノルボルネン-エキソ-2,3-ジカルボン酸無水物を,70Paの減圧下で窒素を微量吹き込みながら除去した。
このようにして,標記のテトラカルボン酸二無水物,すなわち5,5'-エキソ-(1,1,3,3,5,5-ヘキサメチルトリシロキサン-1,5-ジイル)ビスビシクロ[2.2.1]ヘプタン-エキソ-2,3-ジカルボン酸無水物(exo-NB-TriSXDA)を得た。このもののGPC分析による純度は99.5%であり,粘度は,40℃で38.4Pa・sであった。
「一般式(3)においてmが3であるテトラカルボン酸二無水物の合成」
5-ノルボルネン-エキソ-2,3-ジカルボン酸無水物の量を39.61g(0.2413mol),触媒量を0.9516gに変更し、1,1,3,3,5,5-ヘキサメチルトリシロキサンの代わりに1,1,3,3,5,5,7,7-オクタメチルテトラシロキサンを33.93g(0.1201mol)用いた以外は合成例1と同様に反応と後処理操作を行った。
このようにして,標記のテトラカルボン酸二無水物,すなわち5,5'-エキソ-(1,1,3,3,5,5,7,7-オクタメチルテトラシロキサン-1,7-ジイル)ビスビシクロ[2.2.1]ヘプタン-エキソ-2,3-ジカルボン酸無水物(exo-NB-TetraSXDA)を得た。このもののGPC分析による純度は98.7%,25℃での粘度は22.5Pa・sであり,屈折率は1.4825であった。
「一般式(3)においてmが4であるテトラカルボン酸二無水物の合成」
5-ノルボルネン-エキソ-2,3-ジカルボン酸無水物の量を41.26g(0.2513mol),触媒量を0.9745gに変更し、1,1,3,3,5,5-ヘキサメチルトリシロキサンの代わりに1,1,3,3,5,5,7,7,9,9-デカメチルペンタシロキサンを44.59g(0.1250mol)用いた以外は合成例1と同様に反応と後処理操作を行った。
このようにして,標記のテトラカルボン酸二無水物,すなわち5,5'-エキソ-(1,1,3,3,5,5,7,7,9,9-デカメチルペンタシロキサン-1,9-ジイル)ビスビシクロ[2.2.1]ヘプタン-エキソ-2,3-ジカルボン酸無水物(exo-NB-PentaSXDA)を得た。
このもののGPC分析による純度は99.7%,25℃での粘度は5.84Pa・sであり,屈折率は1.4735であった。
「一般式(3)においてmが平均4.11でありmが2から10の混合組成であるテトラカルボン酸二無水物の合成」
5-ノルボルネン-エキソ-2,3-ジカルボン酸無水物の量を39.61g(0.2413mol),触媒量を0.9480gに変更し、1,1,3,3,5,5-ヘキサメチルトリシロキサンの代わりにケイ素の数n(式(3)のm+1)が3から11に分布している両末端ヒドロシリル型ジメチルシロキサン混合オリゴマーを49.90g(この混合オリゴマー中にはヒドロシリル化反応に関与しない環状オリゴマーが10.8%含まれているので,これを補正した値で0.1193mol)を用いた以外は合成例1と同様に反応と後処理操作を行った。
1H-NMRスペクトルからケイ素の平均個数nを求め,標記のテトラカルボン酸二無水物(exo-NB-MixSXDA)であることを確認した。このもののGPC分析による純度は99.8%,25℃での粘度は4.11Pa・sであり,屈折率は1.4706であった。
「式(5)のテトラカルボン酸二無水物のメソ体の合成」
攪拌子,滴下ロート,冷却管及び温度計を取り付けた1Lの4つ口フラスコに,トルエン200g及び融点が166~167℃の5-ノルボルネン-エンド-2,3-ジカルボン酸無水物82.08g(0.5000mol)を仕込み,加熱攪拌を開始した。フラスコ内の温度が95℃になった時点で,触媒のアルドリッチ社製Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex(白金濃度2%のキシレン溶液)6.4731g(白金金属として6.64×10-4グラム原子)を加え,1,1,3,3,5,5-ヘキサメチルトリシロキサン52.12g(0.2500mol)を滴下ロートからフラスコ内に徐々に滴下した。滴下に伴って反応温度が上昇するので,フラスコ内の温度が100℃を維持するよう注意しながら1時間かけて滴下した後,フラスコ内温度を100℃に保ちながら更に4時間反応を続けた。
その後,50℃に温度を下げ,活性炭12gを加え室温で30分間攪拌した後,濾過操作で活性炭を除いた。別途合成していた融点が141.5~142.5℃の結晶を種結晶として極微量を濾液に添加して結晶の析出を促した。1晩静置したところ微細な結晶が析出していた。濾過操作で結晶を取出し,結晶を熱風乾燥した。なお,このときの濾液中にはラセミ体が存在しているので,保存しておいた。結晶の融点は136~140℃と低かったので,再度トルエンで再結晶して融点が143~144℃の結晶を得た。この結晶のGPCによる純度は99.6%であった。
1H-NMRスペクトルで,トリシロキサンの中央のケイ素に結合している2つのメチル基が2本に分裂する(0.036ppmと0.038ppm)ことから下記の立体構造(5a)で示されるメソ体(endo-NB-TriSXDAメソ体)であることを確認した。

「式(5)のテトラカルボン酸二無水物のラセミ体の合成」
合成例5において保存しておいた濾液中には,式(5)のテトラカルボン酸二無水物のラセミ体が存在し,メソ体も残存している。さらには,GPC分析の結果,未反応の5-ノルボルネン-エンド-2,3-ジカルボン酸無水物及び少量の構造不明化合物が混在していることが分かった。
そこで,ロータリーエバポレーターを使ってトルエンを除去した後,ジエチルエーテルで再結晶した。融点は104~114℃であり,GPC分析の結果,未反応の5-ノルボルネン-エンド-2,3-ジカルボン酸無水物及び少量の構造不明化合物は除去されていることが分かった。
得られた結晶をトルエン75質量%/ヘキサン25質量%の混合溶媒で再結晶した。生成した結晶の融点は136~139℃であり,結晶はメソ体であった。濾液中の溶媒組成がトルエン50質量%/ヘキサン50質量%になるようにヘキサンを加えて,結晶を析出させた。得られた結晶の融点は113~118℃であったので,更にトルエン50質量%/ヘキサン50質量%の混合溶媒で再結晶して,融点が117~120℃の結晶を得た。この結晶のGPCによる純度は,98.4%であった。なお,特開2010-254602号に記したように,X線構造解析の結果,この結晶は単位胞中に下記の立体構造(5b)で示されるR-R体と立体構造(5c)で示されるS-S体が1対で存在するラセミ体(endo-NB-TriSXDAラセミ体)であることが確認されている。

「化学式(6)で表されるテトラカルボン酸二無水物の合成」
攪拌子,滴下ロート,冷却管及び温度計を取り付けた1Lの4つ口フラスコに,トルエン240g及び融点が148~149.5℃の5-ノルボルネン-エキソ-2,3-ジカルボン酸無水物123.20g(0.7505mol)を仕込み,加熱攪拌を開始した。フラスコ内の温度が80℃になった時点で,触媒のアルドリッチ社製Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex(白金濃度2%のキシレン溶液)2.99g(白金金属として3.07×10-4グラム原子)を加え,1,1,3,3-テトラメチルジシロキサン50.17g(0.3735mol)を滴下ロートからフラスコ内に徐々に滴下した。滴下に伴って反応温度が上昇するので,フラスコ内の温度が90℃を維持するよう注意しながら1時間かけて滴下した後,フラスコ内温度を90℃に保ちながら更に1時間反応を続けた。
その後,冷却し,活性炭15gを加え室温で2間攪拌し,濾過操作で活性炭を除いた。ロータリーエバポレーターを使って真空加熱下に無色透明の濾液を濃縮し,粘性のある無色透明の液体166.4gを得た。GPC分析の結果,このものには溶媒のトルエンが含まれていないことを確認した。
次いで,1,1,3,3-テトラメチルジシロキサンを用いる場合にだけ特異的に副生する成分である下記化学式(16a)で表される化合物を除去するために抽出精製を行った。連続単抽出装置の抽出側フラスコに,粘性のある無色透明の液体86.8g,ヘキサン169.4g及びトルエン23.9gを仕込み,蒸留側フラスコにヘキサン101.3g及び滴下ロートにヘキサンを60.0g仕込んだ。抽出側のウオーターバス温度を65℃,蒸留側のオイルバス温度を115℃に設定して10時間抽出した。抽出側フラスコの下層を回収し,含まれる溶媒を除去して重量を求めたところ,62.0gであった。さらに,副生するもう1つの成分である下記化学式(16b)を70Paの減圧下で窒素を微量吹き込みながら蒸留で除去した。

「一般式(10)においてmが3であるテトラカルボン酸二無水物の合成」
攪拌子,滴下ロート,冷却管及び温度計を取り付けた300mLの4つ口フラスコに,トルエン116g及び融点が166~167℃の5-ノルボルネン-エンド-2,3-ジカルボン酸無水物60.12g(0.3662mol)を仕込み,加熱攪拌を開始した。フラスコ内の温度が95℃になった時点で,触媒のアルドリッチ社製Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex(白金濃度2%のキシレン溶液)2.58g(白金金属として2.65×10-4グラム原子)を加え,1,1,3,3,5,5,7,7-オクタメチルテトラシロキサン51.50g(0.1822mol)を滴下ロートからフラスコ内に徐々に滴下した。滴下に伴って反応温度が上昇するので,フラスコ内の温度が100℃を維持するよう注意しながら1時間かけて滴下した。反応液をサンプリングし,GPC分析した。その後もフラスコ内温度を100℃に保ちながら,反応開始後2時間目に反応液をサンプリングしGPC分析したところ,反応液組成に変化が見られなかった。そのため,反応開始後3時間目に触媒を1.28g追加投入した。その後も反応液を分析して,反応開始後5.5時間目に1.13g更に7.5時間目に1.12gの触媒を追加投入し,反応開始後9時間目に原料の5-ノルボルネン-エンド-2,3-ジカルボン酸無水物の残存量が仕込み量の5.9%となったので,反応を終了した。反応に用いた触媒の合計量は6.11g(白金金属として6.26×10-4グラム原子)となった。
その後,50℃に温度を下げ,活性炭12gを加え室温で30分間攪拌した後,濾過操作で活性炭を除いた。ロータリーエバポレーターを使って濾液中のトルエンを除去して,融点が87.5~94.5℃の白色の結晶89.05gを得た。これは,メソ体とラセミ体の混合物(endo-NB-TetraSXDA)であり,収率は80.0%である。その後,この結晶をトルエンとヘキサンの重量比2:1の混合溶媒で再結晶し,融点が99.0~100.0℃の結晶を得た。この結晶は,GPC分析での純度が99.7%であった。
[合成例1で合成したexo-NB-TriSXDAとオキシジアニリンから合成されるポリイミド樹脂]
合成例1で合成したexo-NB-TriSXDAを用いてポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,exo-NB-TriSXDAを5.1812g(9.651mmol),4,4'-オキシジアニリンを1.9328g(9.652mmol),触媒である2-ヒドロキシピリジンを0.2220g及び1,2-ジクロロベンゼンを50.38g仕込み,加熱攪拌を開始した。
100℃で30分間加熱攪拌して反応液が均一透明液になったことを確認したのち,30分掛けて1,2-ジクロロベンゼンの沸点である179℃に昇温した。この温度で4時間反応を続けた。この間,ディーンスターク分留管上部に水が混じって白濁した1,2-ジクロロベンゼンが溜まったので,これを抜き出し,同量の1,2-ジクロロベンゼンをフラスコに仕込んだ。
4時間経過後,加熱を止めて温度を下げフラスコ内温度が65℃になったときに,アセトン35gを加え,この溶液を600gのメタノールに注ぎ込んだ。ビーカーの底にガム状の塊が沈殿した。この塊を60gのアセトンに溶解させ,このアセトン溶液を600gの新たなメタノールに注ぎ込んだ。ビーカーの底に生じたガム状の塊を取出し,90℃の乾燥器に4時間置いて溶媒を除去した。得られた樹脂の質量は5.51gであり,仕込みの理論収量の81.4%であった。
図1に1H-NMRスペクトルを示す。繰り返し単位構造を反映したスペクトルであることが確認できる。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,30,400であり,重量平均分子量は56,200であった。
このポリイミド樹脂の溶解性を調べた。ポリイミド樹脂約50mgを精秤し,これに10倍量の溶媒を加えて,室温で溶解の様子を観察した。その結果,テトラヒドロフラン,アセトン,N-メチル-2-ピロリジノン及びN,N-ジメチルアセトアミドに溶解したが,トルエンでは溶液が白濁し静置後に2層に分離した。
このポリイミド樹脂の特性を明らかにするために,フィルムを作成した。作成手順は次のとおりである。
樹脂濃度が25質量%のN,N-ジメチルアセトアミド溶液を調製し,この溶液を東洋ろ紙製のNo.5B濾紙を用いて濾過した。乾燥膜厚が20~30μmとなるように,ガラス板に濾過した溶液を流延・塗布し,65℃の乾燥炉に30分間静置した後,20分を要して165℃に昇温し,この温度で15分間保持した。その後,ガラス板ごと温水に10分間浸漬し,フィルムをガラス板から剥離した。
熱機械的分析装置による引張り条件でのガラス転移点の測定,引張試験の結果を表1にまとめて記す。また,紫外可視分光光度計による透過率スペクトルを図2に、赤外分光光度計によるFT-IRスペクトルを図4に示した。
[合成例2で合成したexo-NB-TetraSXDAとオキシジアニリンから合成されるポリイミド樹脂]
合成例2で合成したexo-NB-TetraSXDAを用いてポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,exo-NB-TetraSXDAを7.5602g(純度補正した値は7.4619g,12.214mmol),4,4'-オキシジアニリンを2.4457g(12.270mmol),触媒である2-ヒドロキシピリジンを0.2965g及び1,2-ジクロロベンゼンを69.37g仕込み,実施例1とまったく同じ操作で反応させ,樹脂を沈殿させた。
得られた樹脂の質量は7.11gであり,仕込みの理論収量の75.1%であった。
図1に1H-NMRスペクトルを併せて示す。繰り返し単位構造を反映したスペクトルであることが確認できる。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,40,500であり,重量平均分子量は68,400であった。
このポリイミド樹脂の溶解性を実施例1と同じ方法で調べた。その結果,テトラヒドロフラン,アセトン,トルエン,N-メチル-2-ピロリジノン及びN,N-ジメチルアセトアミドに溶解し,いずれも均一透明溶液となった。
実施例1に記載した方法でフィルムを作成し,ガラス転移点の測定,引張試験を行った。結果を表1に併せて示した。
[合成例3で合成したexo-NB-PentaSXDAとオキシジアニリンから合成されるポリイミド樹脂]
合成例3で合成したexo-NB-PentaSXDAを用いてポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,exo-NB-PentaSXDAを6.6203g(9.6633mmol),4,4'-オキシジアニリンを1.9349g(9.6629mmol),触媒である2-ヒドロキシピリジンを0.2564g及び1,2-ジクロロベンゼンを59.87g仕込み,実施例1とまったく同じ操作で反応させ,樹脂を沈殿させた。
得られた樹脂の質量は6.75gであり,仕込みの理論収量の82.2%であった。
図1に1H-NMRスペクトルを併せて示す。繰り返し単位構造を反映したスペクトルであることが確認できる。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,59,900であり,重量平均分子量は106,800であった。
このポリイミド樹脂の溶解性を実施例1と同じ方法で調べた。その結果,テトラヒドロフラン,アセトン,トルエン,N-メチル-2-ピロリジノン及びN,N-ジメチルアセトアミドに溶解し,いずれも均一透明溶液となった。
実施例1に記載した方法でフィルムを作成し,ガラス転移点の測定,引張試験を行った。結果を表1に併せて示した。
[合成例4で合成したexo-NB-MixSXDAとオキシジアニリンから合成されるポリイミド樹脂]
合成例4で合成したexo-NB-MixSXDAを用いてポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,exo-NB-MixSXDAを6.9907g(10.084mmol),4,4'-オキシジアニリンを2.0167g(10.071mmol),触媒である2-ヒドロキシピリジンを0.2725g及び1,2-ジクロロベンゼンを63.08g仕込み,実施例1とまったく同じ操作で反応させ,樹脂を沈殿させた。
得られた樹脂の質量は6.80gであり,仕込みの理論収量の78.7%であった。
図3に1H-NMRスペクトルを示す。繰り返し単位構造を反映したスペクトルであることが確認できる。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,31,900であり,重量平均分子量は50,400であった。
このポリイミド樹脂の溶解性を実施例1と同じ方法で調べた。その結果,テトラヒドロフラン,アセトン,トルエン,N-メチル-2-ピロリジノン及びN,N-ジメチルアセトアミドに溶解し,いずれも均一透明溶液となった。
実施例1に記載した方法でフィルムを作成し,ガラス転移点の測定,引張試験を行った。結果を表1に併せて示した。
[合成例1で合成したexo-NB-TriSXDAとオキシジアニリンから合成されるポリアミド酸樹脂]
攪拌羽根,温度計及び窒素導入管を取り付けた100mLの4つ口フラスコに,合成例1で合成したexo-NB-TriSXDAを6.3208g(11.775mmol),4,4'-オキシジアニリンを2.3577g(11.774mmol)及びN-メチル-2-ピロリジノンを49.21g仕込み,室温(28℃)で攪拌を開始した。
3時間経過後,ポリアミド酸溶液をビーカーに移し,この溶液に精製水100mLを加えたところ白色の塊が沈殿した。この塊を50gのアセトンに溶解させ,300mLの精製水に激しく撹拌しながらこのアセトン溶液を徐々に注ぎ込んだ。生成した微細な樹脂を濾過して取出し,室温で真空乾燥した。得られた樹脂の質量は5.34gであり,仕込みの理論収量の61.5%であった。
得られた樹脂がポリアミド酸樹脂であることを確認するために,熱重量分析を行った。120~180℃において4.5%の重量減少を示した(脱水環化の理論値4.9%)。また、赤外分光光度計によるFT-IRスペクトルを図5に示した。
このポリアミド酸樹脂の溶解性を調べた。樹脂約40mgを精秤し,これに10倍量の溶媒を加えて,室温で溶解の様子を観察した。その結果,メタノール,テトラヒドロフラン,アセトン及びN-メチル-2-ピロリジノンに溶解したが,トルエンには溶解しなかった。
[合成例1で合成したexo-NB-TriSXDAとオキシジアニリンから合成されるポリイミド樹脂]
実施例1に比べて反応系の原料濃度(テトラカルボン酸二無水物とジアミンの合計濃度)を高め,より高分子量のポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた200mLの4つ口フラスコに,exo-NB-TriSXDAを14.5792g(27.16mmol),4,4'-オキシジアニリンを5.4386g(27.16mmol),触媒である2-ヒドロキシピリジンを0.5986g及び1,2-ジクロロベンゼンを113.36g仕込み,加熱攪拌を開始した。反応系のexo-NB-TriSXDAと4,4'-オキシジアニリンの合計濃度は15.0%であった。
100℃で30分間加熱攪拌して反応液が均一透明液になったことを確認したのち,30分掛けて1,2-ジクロロベンゼンの沸点である179℃に昇温した。この温度で4時間反応を続けた。この間,ディーンスターク分留管上部に水が混じって白濁した1,2-ジクロロベンゼンが溜まったので,これを抜き出し,同量の1,2-ジクロロベンゼンをフラスコに仕込んだ。
4時間経過後,加熱を止めて温度を下げフラスコ内温度が65℃になったときに,アセトン27gを加え,この溶液を400gのメタノールに注ぎ込んだ。ビーカーの底にガム状の塊が沈殿した。この塊を100gのアセトンに溶解させ,このアセトン溶液にメタノールを徐々に滴下した。15gのメタノールを滴下したとき,2層に分離した。上層を除去し,下層に45gのメタノールを加えてガム状の塊を析出させた。90℃の乾燥器に4時間置いて溶媒を除去した。得られた樹脂の質量は10.28gであり,仕込みの理論収量の51.3%であった。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,55,900であり,重量平均分子量は88,200であった。
実施例1に記載した方法でフィルムを作成し,ガラス転移点の測定,引張試験を行った。ガラス転移点は138℃,引張強さは43MPa,弾性率は1.54GPaであり,破断伸びは22%であった。
[合成例3で合成したexo-NB-PentaSXDAとオキシジアニリンから合成されるポリイミド樹脂]
実施例3に比べて反応系の原料濃度(テトラカルボン酸二無水物とジアミンの合計濃度)を高め,より高分子量のポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた200mLの4つ口フラスコに,exo-NB-PentaSXDAを13.3685g(19.51mmol),4,4'-オキシジアニリンを3.9069g(19.51mmol),触媒である2-ヒドロキシピリジンを0.5183g及び1,2-ジクロロベンゼンを90.71g仕込み,加熱攪拌を開始した。反応系のexo-NB-PentaSXDAと4,4'-オキシジアニリンの合計濃度は16.0%であった。
100℃で30分間加熱攪拌して反応液が均一透明液になったことを確認したのち,30分掛けて1,2-ジクロロベンゼンの沸点である179℃に昇温した。この温度で4時間反応を続けた。この間,ディーンスターク分留管上部に水が混じって白濁した1,2-ジクロロベンゼンが溜まったので,これを抜き出し,同量の1,2-ジクロロベンゼンをフラスコに仕込んだ。
4時間経過後,加熱を止めて温度を下げフラスコ内温度が65℃になったときに,アセトン25gを加え,この溶液を300gのメタノールに注ぎ込んだ。ビーカーの底にガム状の塊が沈殿した。この塊を66gのアセトンに溶解させ,このアセトン溶液にメタノールを徐々に滴下した。33gのメタノールを滴下したとき,2層に分離した。上層を除去し,下層に40gのメタノールを加えてガム状の塊を析出させた。この析出物を50gのアセトンに溶解させ,メタノール23gを滴下して2層に分離した。上層を除去し,下層に40gのメタノールを加えてガム状の塊を析出させた。90℃の乾燥器に4時間置いて溶媒を除去した。得られた樹脂の質量は10.98gであり,仕込みの理論収量の66.3%であった。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,89,800であり,重量平均分子量は141,000であった。
実施例1に記載した方法でフィルムを作成し,ガラス転移点の測定,引張試験を行った。ガラス転移点は88℃,引張強さは29MPa,弾性率は1.23GPaであり,破断伸びは127%であった。
[合成例5で得たendo-NB-TriSXDAメソ体とオキシジアニリンから合成されるポリイミド樹脂]
合成例5で得たendo-NB-TriSXDAメソ体を用いてポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,endo-NB-TriSXDAメソ体を6.0239g(11.222mmol),4,4'-オキシジアニリンを2.2471g(11.222mmol),触媒である2-ヒドロキシピリジンを0.2459g及び1,2-ジクロロベンゼンを57.91g仕込み,実施例1とまったく同じ操作で反応させ,樹脂を沈殿させた。
得られた樹脂の質量は5.34gであり,仕込みの理論収量の67.9%であった。GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,11,900であり,重量平均分子量は25,600であった。
しかし,得られた樹脂は,脆いものであり分子量も小さいことから,アセトンに再溶解させ,これにメタノールを加えて樹脂を再沈殿させた。得られた樹脂の質量は2.85gであり,仕込みの理論収量の36.2%であった。GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,19,800であり,重量平均分子量は31,900であった。
図6に1H-NMRスペクトルを示す。繰り返し単位構造を反映したスペクトルであることが確認できる。特に,トリシロキサンの中央のケイ素に結合している2つのメチル基が明瞭に2本に分裂(0.029ppmと0.040ppm)することからメソ体構造が保持されていることを確認した。
このポリイミド樹脂の溶解性を実施例1と同じ方法で調べた。その結果,テトラヒドロフラン,アセトン及びN-メチル-2-ピロリジノンに溶解したが,トルエンでは溶液が白濁し静置後に2層に分離した。
実施例1と同様にこのポリイミド樹脂をN,N-ジメチルアセトアミドに溶解させてフィルムを作成した。しかし,フィルムは脆いため引張試験はもとより,熱機械的分析装置による引張り条件でのガラス転移点の測定はできなかった。そこで,このポリイミド樹脂をメノウ乳鉢で微粉末にし,示差走査熱量計でガラス転移点を測定した。結果を表1に併せて示す。
辛うじて,紫外可視分光光度計による透過率の測定は可能であった。スペクトルを図2に併せて示した。図2から明らかなように、実施例1のポリイミド樹脂は比較例1のポリイミド樹脂に比べ、約290~350nmの領域において大きな透過率を示し良好な透明性を有する。
[合成例6で得たendo-NB-TriSXDAラセミ体とオキシジアニリンから合成されるポリイミド]
合成例6で得たendo-NB-TriSXDAラセミ体を用いてポリイミド樹脂を下記のように合成した。
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,endo-NB-TriSXDAラセミ体を10.1676g(18.9414mmol),4,4'-オキシジアニリンを3.7924g(18.939mmol),触媒である2-ヒドロキシピリジンを0.4176g及び1,2-ジクロロベンゼンを97.71g仕込み,実施例1とまったく同じ操作で反応させ,樹脂を沈殿させた。さらに,比較例1と同様に樹脂を再沈殿させて,標準ポリスチレン換算の数平均分子量が15,700であり,重量平均分子量が27,500の樹脂を得た。得られた樹脂の質量は6.65gであり,仕込みの理論収量の50.0%であった。
図6に1H-NMRスペクトルを示す。繰り返し単位構造を反映したスペクトルであることが確認できる。トリシロキサンの中央のケイ素に結合している2つのメチル基は等価であり,1本のピーク(0.035ppm)として観測されることからラセミ体構造が保持されていることを確認した。
実施例1と同様にこのポリイミド樹脂をN,N-ジメチルアセトアミドに溶解させてフィルムを作成した。しかし,フィルムは脆いため引張試験はもとより,熱機械的分析装置による引張り条件でのガラス転移点の測定はできなかった。そこで,このポリイミド樹脂をメノウ乳鉢で微粉末にし,示差走査熱量計でガラス転移点を測定した。結果を表1に併せて示した。
[合成例7で得たexo-NB-DiSXDAとオキシジアニリンから合成されるポリイミド樹脂]
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,合成例7で得たexo-NB-DiSXDAを4.3735g(9.453mmol),4,4'-オキシジアニリンを1.8929g(9.453mmol),触媒である2-ヒドロキシピリジンを0.1882g及び1,2-ジクロロベンゼンを43.88g仕込み,加熱攪拌を開始した。反応系のexo-NB-DiSXDAと4,4'-オキシジアニリンの合計濃度は12.5%であった。
実施例1と同様にポリイミド樹脂の合成反応を行ない,実施例6と同様の樹脂精製を行なった。得られた樹脂の質量は2.69gであり,仕込みの理論収量の43.0%であった。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,25,700であり,重量平均分子量は35,700であった。
このポリイミド樹脂の溶解性を実施例1と同じ方法で調べた。その結果,テトラヒドロフラン,アセトン及びN-メチル-2-ピロリジノンに溶解したが,トルエンでは溶液が白濁し静置後に2層に分離した。
実施例1に記載した方法でフィルムを作成し,ガラス転移点の測定,引張試験を行った。ガラス転移点は178℃,引張強さは41MPa,弾性率は1.86GPaであり,破断伸びは2.6%であった。
[合成例8で得たendo-NB-TetraSXDAとオキシジアニリンから合成されるポリイミド樹脂]
攪拌羽根,温度計,ディーンスターク分留管及び窒素導入管を取り付けた100mLの4つ口フラスコに,合成例8で得たendo-NB-TetraSXDAを5.9255g(9.699mmol),4,4'-オキシジアニリンを1.9419g(9.698mmol),触媒である2-ヒドロキシピリジンを0.237g及び1,2-ジクロロベンゼンを55.08g仕込み,加熱攪拌を開始した。反応系のendo-NB-TetraSXDAと4,4'-オキシジアニリンの合計濃度は12.5%であった。
実施例1と同様にポリイミド樹脂の合成反応を行ない,実施例1と同様の樹脂精製を行なった。しかし,得られた樹脂の質量はわずかに0.69gであり,仕込みの理論収量の8.8%であった。
GPCによる分析の結果,標準ポリスチレン換算の数平均分子量は,17,600であり,重量平均分子量は28,200であった。
このポリイミド樹脂の溶解性を実施例1と同じ方法で調べた。その結果,テトラヒドロフラン,アセトン,トルエン及びN-メチル-2-ピロリジノンに溶解した。
実施例1に記載した方法でフィルムを作成しようとしたが,ガラス板上で無数の亀裂が生じ,フィルムは得られなかった。粉末状の試料を使って示差走査熱量計でガラス転移点を測定したところ,104℃の値を得た。

Claims (3)
- 下記一般式(1)で表される繰返し単位構造を有するポリイミド樹脂。

- 下記一般式(2)で表される繰返し単位構造を有するポリアミド酸樹脂。

- 請求項1のポリイミドの製造方法であって,下記一般式(3)で表されるテトラカルボン酸二無水物とジアミン又はジイソシアナートを反応させる工程を含むポリイミドの製造方法。

Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/583,727 US8592546B2 (en) | 2010-08-09 | 2011-04-13 | Silicon-containing alicyclic polyimide resin, polyamic acid resin, and manufacturing method for same |
| JP2012528601A JP5099278B2 (ja) | 2010-08-09 | 2011-04-13 | 新規な含ケイ素脂環式ポリイミド樹脂及びポリアミド酸樹脂並びにそれらの製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010179059 | 2010-08-09 | ||
| JP2010-179059 | 2010-08-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012020584A1 true WO2012020584A1 (ja) | 2012-02-16 |
Family
ID=45567555
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/059209 WO2012020584A1 (ja) | 2010-08-09 | 2011-04-13 | 新規な含ケイ素脂環式ポリイミド樹脂及びポリアミド酸樹脂並びにそれらの製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8592546B2 (ja) |
| JP (1) | JP5099278B2 (ja) |
| TW (1) | TW201213399A (ja) |
| WO (1) | WO2012020584A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015168686A (ja) * | 2014-03-07 | 2015-09-28 | 達興材料股▲ふん▼有限公司 | シロキサン含有三無水物、ポリマー、液晶配向剤、液晶配向膜、および液晶表示装置 |
| CN104946123A (zh) * | 2014-03-24 | 2015-09-30 | 达兴材料股份有限公司 | 用于热阻隔层的组合物 |
| CN110229585A (zh) * | 2019-06-18 | 2019-09-13 | 刘涛 | 一种热处理可快速自修复的涂料及其制备方法 |
| WO2019172460A3 (ja) * | 2018-07-09 | 2019-11-28 | Jxtgエネルギー株式会社 | テトラカルボン酸二無水物、カルボニル化合物、ポリイミド前駆体樹脂、及び、ポリイミド |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2708569B1 (de) * | 2012-09-12 | 2018-05-23 | Ems-Patent Ag | Transparente polyamidimide |
| CN118063772B (zh) * | 2024-04-22 | 2024-07-19 | 潍坊弘润石化科技有限公司 | 一种手性聚酰亚胺及其制备方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0598237A (ja) * | 1991-10-14 | 1993-04-20 | Sumitomo Bakelite Co Ltd | 熱圧着可能なフイルム状接着剤 |
| JP2000143802A (ja) * | 1998-11-17 | 2000-05-26 | Hitachi Chem Co Ltd | ポリアミドイミド系樹脂およびこれを用いた光学用素子 |
| JP2006267800A (ja) * | 2005-03-25 | 2006-10-05 | Kimoto & Co Ltd | 感光性ポリイミド樹脂組成物 |
| JP2006349852A (ja) * | 2005-06-14 | 2006-12-28 | Jsr Corp | 液晶配向剤および液晶表示素子 |
| JP2007302635A (ja) * | 2006-05-15 | 2007-11-22 | Chisso Corp | シルセスキオキサン骨格を有する酸無水物および重合体 |
| WO2010095329A1 (ja) * | 2009-02-18 | 2010-08-26 | 日立化成工業株式会社 | 新規な液状テトラカルボン酸二無水物及びその製造方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4404350A (en) | 1982-07-07 | 1983-09-13 | General Electric Company | Silicone-imide copolymers and method for making |
| JP2004099638A (ja) * | 2002-09-04 | 2004-04-02 | Shin Etsu Chem Co Ltd | イミドシリコーン樹脂およびその製造方法 |
-
2011
- 2011-04-13 JP JP2012528601A patent/JP5099278B2/ja active Active
- 2011-04-13 WO PCT/JP2011/059209 patent/WO2012020584A1/ja active Application Filing
- 2011-04-13 US US13/583,727 patent/US8592546B2/en active Active
- 2011-04-26 TW TW100114436A patent/TW201213399A/zh unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0598237A (ja) * | 1991-10-14 | 1993-04-20 | Sumitomo Bakelite Co Ltd | 熱圧着可能なフイルム状接着剤 |
| JP2000143802A (ja) * | 1998-11-17 | 2000-05-26 | Hitachi Chem Co Ltd | ポリアミドイミド系樹脂およびこれを用いた光学用素子 |
| JP2006267800A (ja) * | 2005-03-25 | 2006-10-05 | Kimoto & Co Ltd | 感光性ポリイミド樹脂組成物 |
| JP2006349852A (ja) * | 2005-06-14 | 2006-12-28 | Jsr Corp | 液晶配向剤および液晶表示素子 |
| JP2007302635A (ja) * | 2006-05-15 | 2007-11-22 | Chisso Corp | シルセスキオキサン骨格を有する酸無水物および重合体 |
| WO2010095329A1 (ja) * | 2009-02-18 | 2010-08-26 | 日立化成工業株式会社 | 新規な液状テトラカルボン酸二無水物及びその製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| SWINT, S.A. ET AL.: "Synthesis of poly (imidosiloxanes) via disiloxane equilibration reactions.", MACROMOLECULES, vol. 23, no. 21, 1990, pages 4514 - 4518, XP000174802, DOI: doi:10.1021/ma00223a003 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015168686A (ja) * | 2014-03-07 | 2015-09-28 | 達興材料股▲ふん▼有限公司 | シロキサン含有三無水物、ポリマー、液晶配向剤、液晶配向膜、および液晶表示装置 |
| CN104946123A (zh) * | 2014-03-24 | 2015-09-30 | 达兴材料股份有限公司 | 用于热阻隔层的组合物 |
| JP2015183187A (ja) * | 2014-03-24 | 2015-10-22 | 達興材料股▲ふん▼有限公司 | 耐熱膜形成用樹脂組成物、並びに、それにより形成された耐熱膜及び該耐熱膜を有する電池 |
| WO2019172460A3 (ja) * | 2018-07-09 | 2019-11-28 | Jxtgエネルギー株式会社 | テトラカルボン酸二無水物、カルボニル化合物、ポリイミド前駆体樹脂、及び、ポリイミド |
| JPWO2019172460A1 (ja) * | 2018-07-09 | 2021-08-12 | Eneos株式会社 | テトラカルボン酸二無水物、カルボニル化合物、ポリイミド前駆体樹脂、及び、ポリイミド |
| CN110229585A (zh) * | 2019-06-18 | 2019-09-13 | 刘涛 | 一种热处理可快速自修复的涂料及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20130158225A1 (en) | 2013-06-20 |
| TW201213399A (en) | 2012-04-01 |
| JPWO2012020584A1 (ja) | 2013-10-28 |
| US8592546B2 (en) | 2013-11-26 |
| JP5099278B2 (ja) | 2012-12-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5099278B2 (ja) | 新規な含ケイ素脂環式ポリイミド樹脂及びポリアミド酸樹脂並びにそれらの製造方法 | |
| WO2013121917A1 (ja) | ジアミン、ポリイミド、ならびに、ポリイミドフィルムおよびその利用 | |
| Seino et al. | Synthesis of fully aliphatic polyimides | |
| KR20070099676A (ko) | 실록산 변성 다분기 폴리이미드 | |
| Wu et al. | Novel soluble and optically active polyimides containing axially asymmetric 9, 9′-spirobifluorene units: synthesis, thermal, optical and chiral properties | |
| KR20170125973A (ko) | 신규 테트라카르복시산 이무수물, 및 산이무수물로부터 얻어지는 폴리이미드 및 폴리이미드 공중합체 | |
| CN111187414B (zh) | 高性能透明聚酰亚胺薄膜及其制备方法 | |
| KR102060190B1 (ko) | 폴리이미드계 랜덤 공중합체 및 이를 포함하는 폴리이미드계 필름 | |
| KR100717377B1 (ko) | 지방족 폴리이미드-실록산 및 그 제조방법 | |
| JP3026201B2 (ja) | アルコキシ置換基を有する新規の可溶性ポリイミド樹脂及びその製造方法 | |
| JP2932052B2 (ja) | 新規なポリイミド及びその製造方法 | |
| JP6905227B2 (ja) | ジアミンおよびポリイミド、並びにそれらの利用 | |
| JP2016196630A (ja) | 新規なポリイミド共重合体 | |
| Wu et al. | Synthesis and characterization of organosoluble polysiloxaneimides derived from siloxane-containing aliphatic dianhydride and various aromatic diamines | |
| Li et al. | Synthesis and characterization of fluorinated poly (amide imide) s derived from 1, 4‐bis (2′‐trifluoromethyl‐4′‐trimellitimidophenoxy) benzene and aromatic diamines | |
| JPWO2019172460A1 (ja) | テトラカルボン酸二無水物、カルボニル化合物、ポリイミド前駆体樹脂、及び、ポリイミド | |
| Liaw et al. | Synthesis and characterization of new highly organosoluble poly (ether imide) s based on 3, 3′, 5, 5′‐tetramethyl‐2, 2‐bis [4‐(4‐dicarboxyphenoxy) phenyl] propane dianhydride | |
| US7282553B2 (en) | Flexible isopropylidene and tetramethyl-containing fluoropolyamide and fluoropolymide and preparation method thereof | |
| KR101827801B1 (ko) | 아이소헥사이드 다이안하이드라이드 단량체, 이로부터 제조된 폴리이미드 및 이들의 제조방법 | |
| RU2751883C1 (ru) | Полиимиды и сополиимиды как диэлектрические материалы | |
| EP3539999B1 (en) | Oligomer, composition including oligomer, article prepared from the composition, method for preparing article, and display device including the article | |
| WO2005061585A1 (ja) | ポリアミック酸およびポリイミド | |
| KR100700749B1 (ko) | 이미드 올리고머, 그 제조방법 및 상기 이미드 올리고머의 가교반응을 통해서 제조된 폴리이미드 박막 | |
| TW202305035A (zh) | 脂環族二酸酐單體及其製造方法與應用 | |
| KR101092600B1 (ko) | 고리형 이미드 화합물 및 이를 이용한 나노기공을 갖는 고분자 겔의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11816250 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012528601 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13583727 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11816250 Country of ref document: EP Kind code of ref document: A1 |