WO2012011314A1 - ベンゾフラノン化合物およびそれを含む医薬組成物 - Google Patents
ベンゾフラノン化合物およびそれを含む医薬組成物 Download PDFInfo
- Publication number
- WO2012011314A1 WO2012011314A1 PCT/JP2011/061266 JP2011061266W WO2012011314A1 WO 2012011314 A1 WO2012011314 A1 WO 2012011314A1 JP 2011061266 W JP2011061266 W JP 2011061266W WO 2012011314 A1 WO2012011314 A1 WO 2012011314A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- benzofuran
- cancer
- acetamide
- chloro
- methoxy
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/343—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide condensed with a carbocyclic ring, e.g. coumaran, bufuralol, befunolol, clobenfurol, amiodarone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention relates to a novel benzofuranone compound, a pharmaceutical composition containing the compound as an active ingredient, and a method for producing the novel benzofuran compound.
- Patent Document 1 discloses a benzofuran compound useful as a cancer preventive or cancer therapeutic agent having a leukotriene inhibitory action, particularly a BLT2 antagonistic inhibitory action.
- Patent Documents 2 and 3 disclose benzofuran and benzothiophene derivatives for treating hyperproliferative disorders. On the other hand, various pharmacological actions have been found for compounds having a benzofuran skeleton.
- Patent Document 4 discloses a benzofuran derivative useful for treating allergies and inflammatory diseases.
- Patent Document 5 discloses a benzofuran derivative useful for the prevention and treatment of bone metabolic diseases.
- Patent Document 6 discloses a benzofuran derivative useful for the treatment of inflammatory diseases and autoimmune diseases.
- Patent Document 7 discloses a benzofuran derivative having an antiallergic or anti-inflammatory action. However, until now, no effective benzofuran compound having a broad spectrum of anticancer activity has been found.
- An object of the present invention is to provide a novel compound having a broad spectrum anticancer activity.
- R 1 is an alkoxyalkyl group having 2 to 6 carbon atoms
- the compound of formula (I) is 2-chloro-N- [4- (7-methoxy-4-methoxymethylsulfamoyl-benzofuran-2-yl) -thiazol-2-yl] -acetamide, 2 -Chloro-N- ⁇ 4- [7-methoxy-4- (2-methoxyethylsulfamoyl) -benzofuran-2-yl] -thiazol-2-yl ⁇ -acetamide, 2-chloro-N- ⁇ 4- [7-Methoxy-4- (3-methoxypropylsulfamoyl) -benzofuran-2-yl] -thiazol-2-yl ⁇ -acetamide, 2-chloro-N- ⁇ 4- [7-methoxy-4- ( 4-methoxybutylsulfam
- R 1 is an alkoxyalkyl group having 2 to 6 carbon atoms
- a pharmaceutically acceptable salt thereof comprising: (1) a step of reacting 2-acetyl-7-methoxybenzofuran with chlorosulfonic acid to produce 2-acetyl-7-methoxybenzofuran-4-sulfonyl chloride; (2) The resulting sulfonyl chloride compound is reacted with an alkoxyalkylamine in the presence of a base to give a compound of formula (II):
- the alkoxyalkylamine used in step (2) is methoxymethylamine, 2-methoxyethylamine, 3-methoxypropylamine, 4-methoxybutylamine, 5-methoxypentylamine, ethoxymethylamine, 2-ethoxyethylamine, 3-ethoxypropyl
- the process according to [4] selected from the group consisting of amine, 4-ethoxybutylamine, propoxymethylamine, 2-propoxyethylamine, 3-propoxypropylamine, butoxymethylamine, 2-butoxyethylamine and pentoxymethylamine; [6]
- a pharmaceutical composition for treating cancer comprising a compound represented by formula (I) or
- an anticancer agent having a broad spectrum anticancer activity can be provided.
- the method which can manufacture the anticancer agent industrially can be provided.
- FIG. 3 is a graph showing the growth inhibition of cancer cells (MIA PaCa-2) by the compound of the present invention (— ⁇ —) and the known anticancer agent Gemzar (— ⁇ —).
- 2 is a graph showing the growth inhibition of cancer cells (MCF-7) by the compound of the present invention (— ⁇ —) and the known anticancer agent Gemzar (— ⁇ —).
- the present invention provides the formula (I):
- R 1 is an optionally substituted alkoxyalkyl group having 1 to 6 carbon atoms
- a pharmaceutically acceptable salt thereof
- the compound represented by formula (I) includes 2-chloro-N- [4- (7-methoxy-4-methoxymethylsulfamoyl-benzofuran-2-yl) -thiazol-2-yl] -acetamide, 2-chloro-N- ⁇ 4- [7-methoxy-4- (2-methoxyethylsulfamoyl) -benzofuran-2-yl] -thiazol-2-yl ⁇ -acetamide, 2-chloro-N- ⁇ 4- [7-methoxy-4- (3-methoxypropylsulfamoyl) -benzofuran-2-yl] -thiazol-2-yl ⁇ -acetamide, 2-chloro-N- ⁇ 4- [7-methoxy-4- (4-methoxybutylsulfamoyl) -benzofuran-2-yl] -thiazol-2-yl ⁇ -acetamide
- this invention provides the manufacturing method of the compound represented by Formula (I) in a 2nd aspect.
- the benzofuran compound represented by the formula (I) is (1) reacting 2-acetyl-7-methoxybenzofuran with chlorosulfonic acid to produce 2-acetyl-7-methoxybenzofuran-4-sulfonyl chloride; (2) The resulting sulfonyl chloride compound is reacted with an alkoxyalkylamine in the presence of a base at 0 ° C. to 50 ° C., preferably 10 to 40 ° C., more preferably 20 to 30 ° C. to obtain a compound of formula (II):
- the obtained sulfonic acid amide compound is monohalogenated by reacting a halogenating reagent at 5 to 50 ° C., preferably 15 to 40 ° C., more preferably 20 to 30 ° C .;
- the resulting halide is reacted with thiourea in the presence of a base under heating conditions to give the formula (III):
- alkoxyalkylamine used in step (2) of the above production method examples include methoxymethylamine, 2-methoxyethylamine, 3-methoxypropylamine, 4-methoxybutylamine, 5-methoxypentylamine, ethoxymethylamine, Ethoxyethylamine, 3-ethoxypropylamine, 4-ethoxybutylamine, propoxymethylamine, 2-propoxyethylamine, 3-propoxypropylamine, butoxymethylamine, 2-butoxyethylamine and pentoxymethylamine can be used.
- chlorine, bromine, iodo, etc. can be used for the halogen of the halogenating reagent used at the process (3) of said manufacturing method.
- solvents such as acetonitrile, ethanol, methanol, chloroform, hexane, ethyl acetate, can be used for reaction, liquid-liquid distribution, recrystallization, etc., for example.
- the base include organic bases such as pyridine, diethylamine, and triethylamine, alkali metal alkoxides such as sodium methoxide and sodium ethoxide, and inorganic bases such as sodium hydroxide and potassium hydroxide. it can.
- the present invention also provides, in the third aspect, a pharmaceutical composition for treating cancer, comprising as an active ingredient a compound represented by formula (I) or a pharmaceutically acceptable salt thereof.
- a pharmaceutical composition for treating cancer comprising as an active ingredient a compound represented by formula (I) or a pharmaceutically acceptable salt thereof.
- the pharmaceutical composition of the present invention can be used, for example, as a therapeutic or prophylactic agent for cancer.
- the pharmaceutical composition of the present invention can be administered orally or parenterally as an anticancer agent. Therefore, so-called prodrugs that are metabolized in vivo to the compound of the present invention or a compound substantially the same as the compound of the present invention are also encompassed by the compound of the present invention.
- the pharmaceutical composition of the present invention is prepared using Compound (I) as an active ingredient or a pharmaceutically acceptable salt thereof and a pharmaceutical carrier, and the pharmaceutical carrier is selected according to the form of the pharmaceutical formulation.
- the pharmaceutical carrier is selected according to the form of the pharmaceutical formulation. Examples thereof include starch, lactose, hydroxypropylcellulose, hydroxypropylmethylcellulose, polyvinylpyrrolidone, aluminum stearate and magnesium stearate.
- the pharmaceutical composition of the present invention When the pharmaceutical composition of the present invention is administered orally, it can be administered in dosage forms such as tablets, capsules, syrups and suspensions.
- dosage forms such as tablets, capsules, syrups and suspensions.
- parenterally for example, solutions, emulsions and suspensions are used as injections, as transdermal agents, as sprays, or tablets as suppositories. Can be administered.
- the pharmaceutical composition of the present invention can also be prepared as a sustained-release preparation.
- a dosage form can be prepared by blending ordinary carriers, excipients, binders, stabilizers and the like with active ingredients by a known dispensing method.
- a buffer, a solubilizing agent, an isotonic agent and the like can be added.
- Cancers to be treated by the pharmaceutical composition include mammals, preferably human pancreatic cancer, breast cancer, brain tumor, glioma, oral cancer, pharyngeal cancer, laryngeal cancer, lung cancer, esophageal cancer. Stomach cancer, kidney cancer, endometrial cancer, cervical cancer, ovarian cancer, retinoblastoma, prostate cancer, testicular tumor, liver cancer, skin cancer, colon cancer and rectal cancer etc. Can be mentioned.
- the dosage and frequency of administration vary depending on various factors such as the target disease, patient symptoms, age, weight, sex, etc., and the dosage form, dosage form, etc.
- the dose of compound (I) as the active ingredient is usually about 1 to 3000 mg, preferably about 1 to 1000 mg or about 500 to 1500 mg, more preferably about 1 to 1000 mg per day for an adult. It can be administered once or divided into several doses to give a range of 10 to 500 mg.
- the dose of compound (I) is usually about 1 to 3000 mg, preferably about 1 to 1000 mg or about 500 to 1500 mg per day, more preferably about 10 to about 1 to adults.
- the 500 mg range can be administered in one or several divided doses.
- the pharmaceutical composition of the present invention can be administered continuously for several weeks to several months as described above, or again after continuous administration for several weeks to several months with a certain drug holiday. Administration can be in a variety of ways, including resuming administration.
- each cultured cell is seeded on a 96-well microplate at a density of 1 ⁇ 10 4 cells (5.0 ⁇ 10 4 cells / mL, 200 ⁇ L) per well, and 24% at 37 ° C. under 5% CO 2. After incubation for a period of time, replace with medium containing compound (180 ⁇ L, maximum compound dose is 10 ⁇ M, final concentration of dimethyl sulfoxide (DMSO, Sigma) adjusted to 0.25%), and further cultured under the same conditions for 72 hours did.
- medium containing compound 180 ⁇ L, maximum compound dose is 10 ⁇ M, final concentration of dimethyl sulfoxide (DMSO, Sigma) adjusted to 0.25%
- D-MEM Dulbecco's modified Eagle's medium
- E-MEM Eagle's minimum essential medium
- MCF-7 MCF-7 culture. Co., Ltd.
- Arama Blue (20 ⁇ L) Iwaki Glass Co., Ltd.
- Excitation wavelength of 530nm and fluorescence wavelength of 590nm using a fluorescence plate reader Spectra Max M5 (Nippon Molecular Device Co., Ltd.)
- the fluorescence value was measured at.
- the fluorescence value immediately before the compound was allowed to act was immediately changed by adding Alama Blue (20 ⁇ L) to the medium, followed by culturing for 3 hours, and measuring the fluorescence value with a fluorescence plate reader.
- Tz the state of cultured cells immediately before the compound was allowed to act
- GI 50 the compound concentration that suppresses the growth of cultured cells to 50% compared to the control
- TGI the same cells as Tz
- LC 50 compound concentration that reduces the number of cultured cells to 50% of Tz
- growth rate growth rate of cultured cells from the time when the compound is acted to measurement of fluorescence value
- the compound MU-1497 of the present invention shows cytotoxicity or growth inhibitory action at a concentration of 2.5 ⁇ M or more in MIA PaCa-2, and the growth rate when 10 ⁇ M (maximum dose) is applied is -95.39%.
- gemcitabine hydrochloride which is a known anticancer agent, showed cytotoxicity or growth inhibitory action at a concentration of 0.0075 ⁇ M or higher in MIA PaCa-2, and the growth rate when applying 10 ⁇ M was 26.47%.
- the compound MU-1497 of the present invention exhibits cytotoxicity or growth inhibitory action at a concentration of 5.0 ⁇ M or higher in MCF-7, and the growth rate when 10 ⁇ M (maximum dose) is applied.
- cytotoxicity or growth inhibitory action was a concentration of 5.0 ⁇ M or higher in MCF-7, and the growth rate when 10 ⁇ M (maximum dose) is applied.
- gemcitabine hydrochloride exhibited cytotoxicity or growth inhibitory action at a concentration of 0.005 ⁇ M or more in MCF-7, and the growth rate when 10 ⁇ M was applied was 59.52%.
- the compound MU-1497 of the present invention showed cytotoxicity or growth inhibitory action on both MIA PaCa-2 and MCF-7 cultured cells (GI 50 value, 4.22 ⁇ M each). And 3.82 ⁇ M).
- 5-fluorouracil a known anticancer agent, does not show any effect on MIA PaCa-2 and MCF-7, and gemcitabine hydrochloride inhibits cytotoxicity or proliferation against MIA PaCa-2 It showed an effect but did not show any effect on MCF-7.
- MU-1497 was found to have an effect of inhibiting or proliferating both MIA PaCa-2 and MCF-7 cells.
- cytotoxicity or growth inhibitory action was observed at 0.0075 ⁇ M in MIA PaCa-2 and 0.005 ⁇ M in MCF-7, but no enhancement was observed at a concentration of 0.05 ⁇ M or more. Therefore, the cytotoxicity or growth inhibitory action of the compound MU-1497 of the present invention was observed in a higher concentration range than gemcitabine hydrochloride, which is a known anticancer agent, but the magnitude of the maximum response was any. It was shown to be superior to gemcitabine hydrochloride. Therefore, according to the present invention, a novel anticancer agent having an anticancer effect exceeding that of known anticancer agents could be obtained.
- the present invention relates to the pharmaceutical field. Specifically, the present invention relates to the field of novel anticancer agents, and relates to an anticancer agent having a potent anticancer activity and a broad spectrum anticancer activity as compared with known anticancer agents.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
特許文献1にはロイコトリエン阻害作用、特にBLT2拮抗阻害作用を有するがん予防またはがん治療剤として有用なベンゾフラン化合物が開示されている。
特許文献2および3には過剰増殖性障害を処置するためのベンゾフランおよびベンゾチオフェン誘導体が開示されている。
一方、ベンゾフラン骨格を有する化合物には様々な薬理作用が見出されている。
特許文献4にはアレルギーや炎症性疾患を治療するのに有用なベンゾフラン誘導体が開示されている。
特許文献5には骨代謝疾患の予防および治療に有用なベンゾフラン誘導体が開示されている。
特許文献6には炎症性疾患、自己免疫性疾患の治療に有用なベンゾフラン誘導体が開示されている。
特許文献7には抗アレルギーや抗炎症作用を有するベンゾフラン誘導体が開示されている。
しかしながら、現在に至るまで、広いスペクトルの抗がん作用を示す有効なベンゾフラン化合物は見出されていなかった。
すなわち、本発明は、
[1]
式(I):

[式中、R1は炭素数2ないし6個のアルコキシアルキル基である]
で表される化合物、またはその医薬上許容される塩;
[2]
式(I)で表される化合物が、2-クロロ-N-[4-(7-メトキシ-4-メトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-メトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(3-メトキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(4-メトキシブチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(5-メトキシペンチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-[4-(7-メトキシ-4-エトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-エトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(3-エトキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(4-エトキシブチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-[4-(7-メトキシ-4-プロポキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-プロポキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(3-プロポキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-[4-(7-メトキシ-4-ブトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-ブトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、および2-クロロ-N-[4-(7-メトキシ-4-ペントキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミドよりなる群から選択される前記[1]記載の化合物またはその医薬上許容される塩;
[3]
式(I)で表される化合物が2-クロロ-N-{4-[7-メトキシ-4-(2-メトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミドである前記[2]記載の化合物またはその医薬上許容される塩;
[4]
式(I):
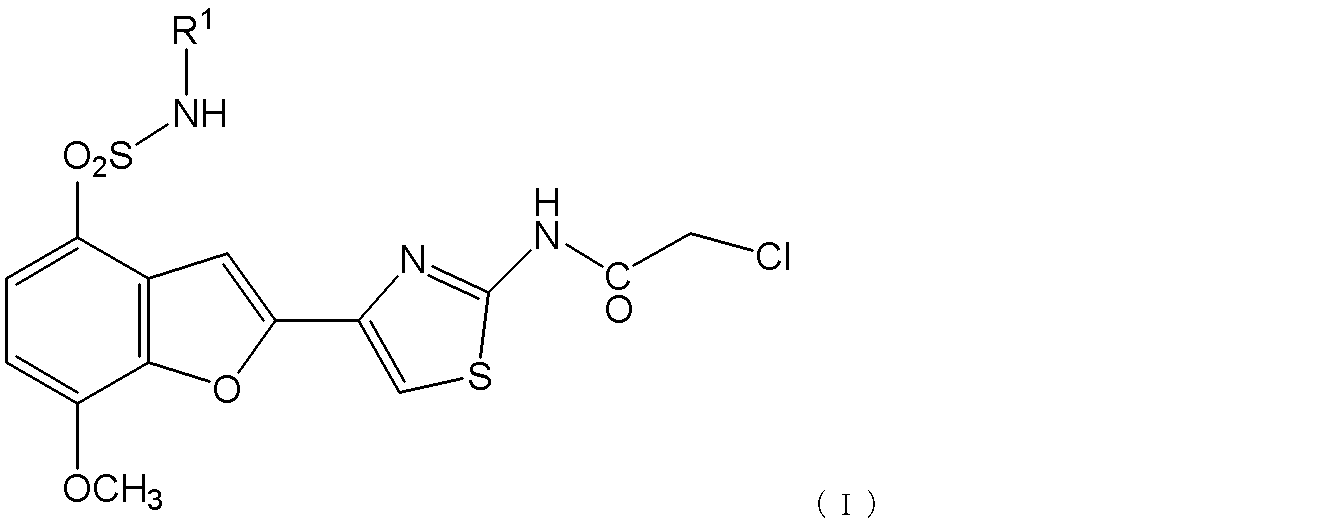
で表される化合物またはその医薬上許容される塩の製法であって、
(1)2-アセチル-7-メトキシベンゾフランにクロロスルホン酸を反応させて2-アセチル-7-メトキシベンゾフラン-4-スルホニルクロリドを生成する工程;
(2)得られたスルホニルクロリド化合物に、塩基存在下で、アルコキシアルキルアミンを反応させて、式(II):

[式中、R1は上記定義に同じである]
で表されるスルホン酸アミド化合物を生成する工程;
(3)得られたスルホン酸アミド化合物にハロゲンを反応させてアセチル基をハロゲン化する工程;
(4)得られたスルホン酸アミドハロゲン化物に、塩基存在下で、チオ尿素を加熱条件下で反応させて式(III):

[式中、R1は上記定義に同じである]
で表される2-アミノチアゾリルベンゾフラン化合物を生成する工程;ついで
(5)得られた2-アミノチアゾリルベンゾフラン化合物に塩化クロロアセチルを反応させて式(I)で表される化合物を生成する工程
を含む製法;
[5]
工程(2)で用いるアルコキシアルキルアミンが、メトキシメチルアミン、2-メトキシエチルアミン、3-メトキシプロピルアミン、4-メトキシブチルアミン、5-メトキシペンチルアミン、エトキシメチルアミン、2-エトキシエチルアミン、3-エトキシプロピルアミン、4-エトキシブチルアミン、プロポキシメチルアミン、2-プロポキシエチルアミン、3-プロポキシプロピルアミン、ブトキシメチルアミン、2-ブトキシエチルアミンおよびペントキシメチルアミンよりなる群から選択される前記[4]記載の製法;
[6]
式(I)で表される化合物またはその医薬上許容される塩を有効成分として含む、がんを治療するための医薬組成物;
[7]
がんが膵臓がん、乳がん、脳腫瘍、神経膠腫、口腔がん、咽頭がん、喉頭がん、肺がん、食道がん、胃がん、腎臓がん、子宮体がん、子宮頸がん、卵巣がん、網膜芽細胞腫、前立腺がん、精巣腫瘍、肝臓がん、皮膚がん、結腸がんおよび直腸がんよりなる群から選択される前記[6]記載の医薬組成物を提供する。
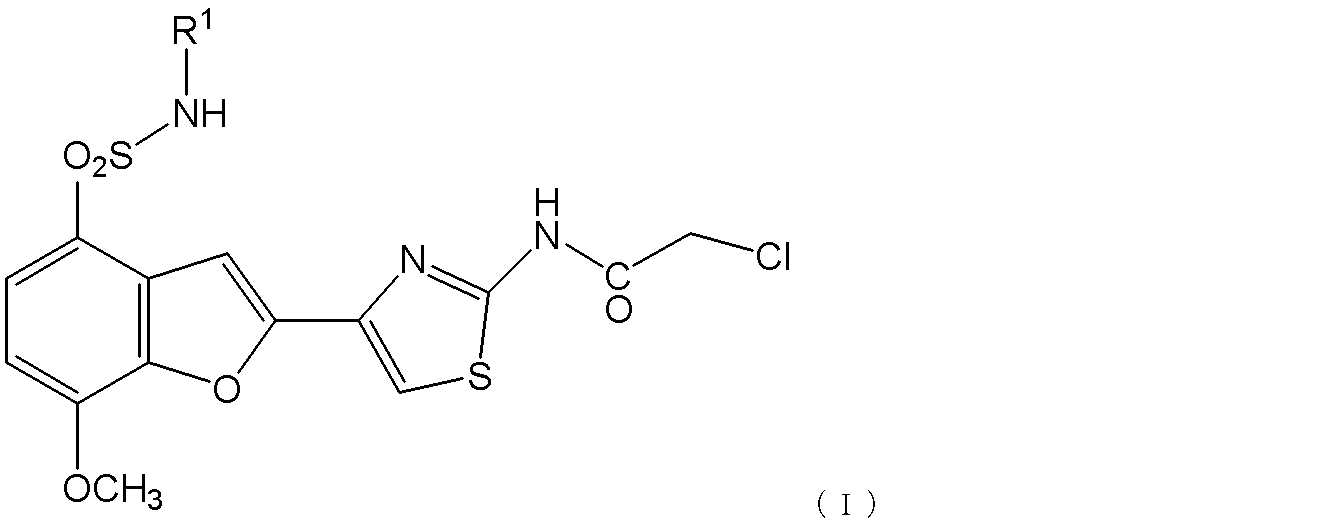
[式中、R1は所望により置換されていてもよい炭素数1ないし6のアルコキシアルキル基である]
で表されるベンゾフラン化合物およびその医薬上許容される塩を提供する。
2-クロロ-N-[4-(7-メトキシ-4-メトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(2-メトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(3-メトキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(4-メトキシブチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(5-メトキシペンチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-[4-(7-メトキシ-4-エトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(2-エトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(3-エトキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(4-エトキシブチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-[4-(7-メトキシ-4-プロポキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(2-プロポキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(3-プロポキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、
2-クロロ-N-[4-(7-メトキシ-4-ブトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、
2-クロロ-N-{4-[7-メトキシ-4-(2-ブトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、および
2-クロロ-N-[4-(7-メトキシ-4-ペントキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、
ならびにこれらの医薬上許容される塩が含まれる。
式(I)で表されるベンゾフラン化合物は、
(1)2-アセチル-7-メトキシベンゾフランにクロロスルホン酸を反応させて2-アセチル-7-メトキシベンゾフラン-4-スルホニルクロリドを生成し;
(2)得られたスルホニルクロリド化合物に、塩基存在下、0℃~50℃、好ましくは10~40℃、より好ましくは20~30℃で、アルコキシアルキルアミンを反応させて、式(II):
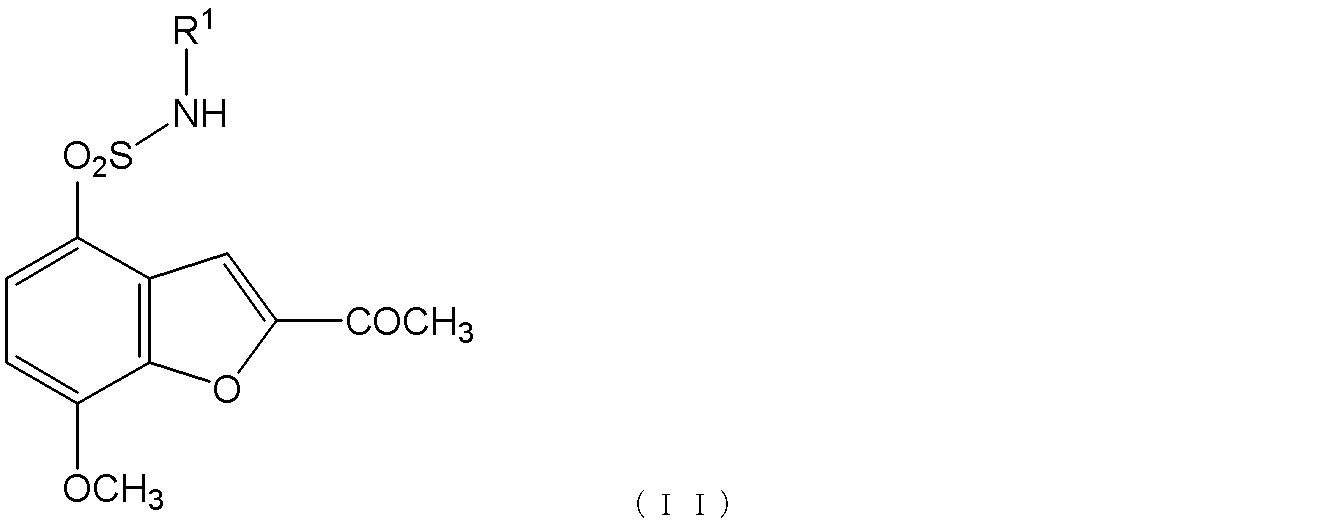
[式中、R1は上記定義に同じである]
で表されるスルホン酸アミド化合物を生成し;
(3)得られたスルホン酸アミド化合物に、5~50℃、好ましくは15~40℃、より好ましくは20~30℃で、ハロゲン化試薬を反応させてアセチル基をモノハロゲン化し;
(4)得られたハロゲン化物に、塩基存在下、チオ尿素を加熱条件下で反応させて式(III):
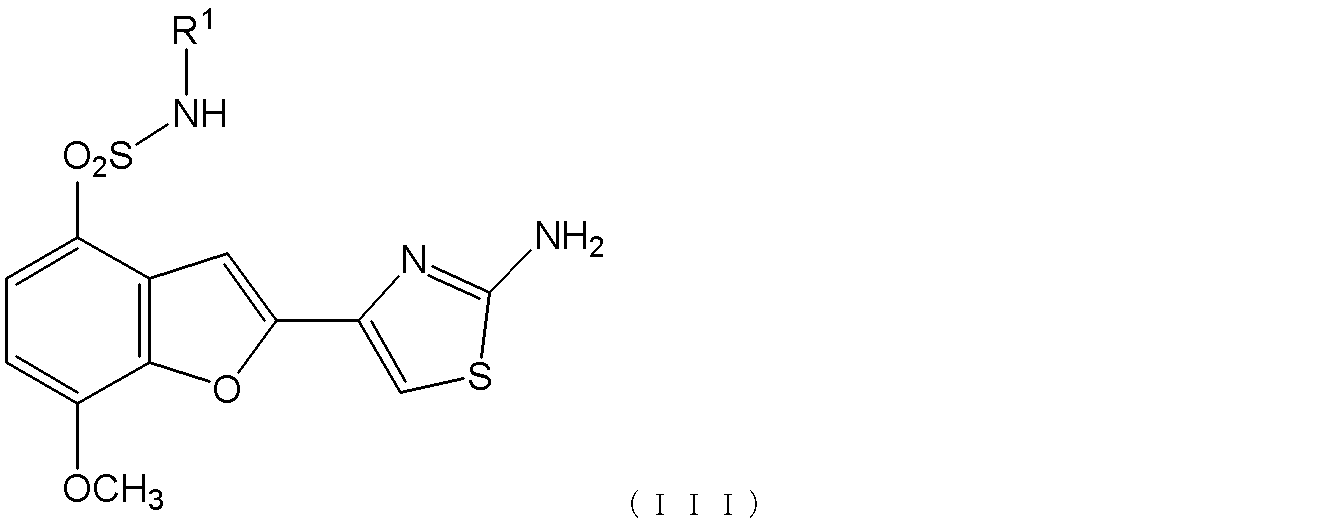
で表される2-アミノチアゾリルベンゾフラン化合物を生成し、ついで
(5)得られた2-アミノチアゾリルベンゾフラン化合物に、0~55℃、好ましくは5~40℃、より好ましくは15~30℃で、塩化クロロアセチルを反応させることによって製造することができる。
また、上記の製法においては、例えば、アセトニトリル、エタノール、メタノール、クロロホルム、ヘキサン、酢酸エチルなどの溶媒を反応、液液分配、再結晶などに用いることができる。
また、塩基としては、例えば、ピリジン、ジエチルアミン、トリエチルアミンなどの有機塩基類、ナトリウムメトキシド、ナトリウムエトキシドなどのアルカリ金属アルコキシド類、水酸化ナトリウム、水酸化カリウムなどの無機塩基類などを用いることができる。
また、本発明の医薬組成物は、上記の用量を、数週間ないし数ヶ月間連続して投与することができ、または、数週間ないし数ヶ月連続投与した後に一定の休薬期間を挟んで再度投与を再開するなど、様々な様式で投与することができる。
0℃に氷冷した約2mLのクロロスルホン酸に、攪拌しつつ、0.15gの2-アセチル-7-メトキシベンゾフラン(0.79mmol)を徐々に加えた。40分後、反応を薄層クロマトグラフィー(E.Merck シリカゲル板(0.5mm, 60F-254)、展開液CHCl3:AcOEt= 3:2)で確認したところ原料の消失が確認されたため反応を終了した。得られた生成物を氷にあけてクロロホルムで抽出し、MgSO4上で乾燥後、濾過し、溶媒を留去させて2-アセチル-7-メトキシベンゾフラン-4-スルホニルクロリドを白色の固体として得た。
計算値 C,45.86;H,3.14
実測値 C, 45.74 ; H, 3.21
質量分析
計算値:288.70
実測値:287.99.
Rf=0.32 (CHCl3:AcOEt=5:2)
m.p. 153.6-155.1℃
元素分析C14H17NO6Sとして
計算値:C,51.37;H,5.23;N,4.28
実測値:C,51.28;H,5.28;N,4.23.
1H-NMR (500 MHz, CDCl3)δ:2.65 (3H, s, -COCH 3), 3.11 (2H, q, J=5.5 Hz, -SO2NHCH 2CH2OCH3), 3.23 (3H, s, -SO2NHCH2CH2OCH 3), 3.37 (2H, t, J=5.1 Hz, -SO2NHCH2CH 2OCH3), 4.09 (3H, s, -OCH 3), 4.89 (1H, t, J=6.0 Hz, -SO2NHCH2CH2OCH3), 6.98 (1H, d, J=8.3 Hz, Bf-6H), 7.81 (1H, d, J=8.2 Hz, Bf-5H), 7.87 (1H, s, Bf-3H).
EIMS(70eV)m/z(rel.int.,%):327 (M+, 19.35), 282 (15.68),253 (79.83), 189 (34.55), 86 (100.00).
Rf=0.45 (CHCl3:AcOEt=5:2)
m.p. 154.7-157.6℃
EIMS(70eV)m/z(rel.int.,%):407 (M+2, 29.27), 405 (M+, 27.86), 362 (35.24), 333 (100.00).
Rf=0.89 (CHCl3:MeOH=5:2)
m.p. 205.7-207.3℃
元素分析C15H17N3O5S2として
計算値:C,46.98;H,4.47;N,10.96
実測値:C,47.00;H,4.39;N,10.77.
1H-NMR (400 MHz, DMSO-D6)δ:2.88 (2H, t, J=5.9 Hz, -SO2NHCH 2 CH2OCH3), 3.10 (3H, s, -SO2NHCH2CH2OCH 3 ), 3.25 (2H, t, J=5.9 Hz, -SO2NHCH2CH 2 OCH3), 4.03 (3H, s, -OCH3), 7.08 (1H, d, J=8.4 Hz, Bf-6H), 7.15 (1H, s, Bf-3H or thiazol-H), 7.21 (2H, s, -NH2), 7.32 (1H, s, Bf-3H or thiazol-H), 7.63 (1H, d, J=8.4 Hz, Bf-5H), 7.70 (1H, br-s, -SO2NH-).
EIMS(70eV)m/z(rel.int.,%):383 (M+, 85.57), 309 (80.39), 245 (100.00).
Rf=0.83 (CHCl3:AcOEt=7:3)
m.p. 182.0-184.5℃
元素分析C17H18ClN3O6S2として
計算値:C,44.39;H,3.94;N,9.14
実測値:C,44.38;H,3.99;N,8.96.
1H-NMR (400 MHz, CDCl3)δ:3.11 (2H, q, J=5.5Hz, -SO2NHCH 2CH2OCH3), 3.21 (3H, S, -SO2NHCH2CH2OCH 3), 3.34 (2H, t, J=5.1Hz, -SO2NHCH2CH 2OCH3), 4.09 (3H, s, -OCH3),4.31 (2H, s, -NHCOCH 2Cl), 4.93 (1H, t, J=6.0Hz, -SO2NHCH2CH2OCH3), 6.76 (1H, d, J=8.4Hz, Bf-6H), 7.49 (1H, s, Bf-3H or thiazol-H), 7.55 (1H, s, Bf-3H or thiazol-H), 7.75 (1H, d, J=8.4Hz, Bf-5H), 9.77 (1H, s, -NHCOCH2Cl).
EIMS(70eV)m/z(rel.int.,%): 461 (M+2, 33.19), 459 (M+, 74.83), 321 (100.00).
イン・ビトロ(in vitro)におけるヒト膵がん細胞(MIA PaCa-2)および乳がん細胞(MCF-7)に対する、新規に合成した化合物(MU-1497)の細胞増殖抑制作用を検討した。陽性対照薬として5-フルオロウラシル(5-FU、Sigma社)およびゲムシタビン塩酸塩(ジェムザール、GEM、日本イーライリリー(株))を供試した。実験は文部科学省の制がん剤スクリーニング委員会が行っている方法に準じて各実験区とも9回繰り返し実験を行い、その平均値を求めた。具体的には、各培養細胞を96ウェルマイクロプレートに1ウェル当たり1×104細胞(5.0×104細胞/mL、200μL)の密度で播種し、5%CO2下、37℃にて24時間培養した後、化合物を含む培地(180μL、化合物の最高用量は10μM、ジメチルスルフォキシド(DMSO、Sigma社)の終濃度は0.25%に調整)に交換し、さらに同条件下で72時間培養した。なお、MIA PaCa-2の培養にはダルベッコ改変イーグル培地(D-MEM、和光純薬工業(株))を、MCF-7の培養にはイーグル最小必須培地(E-MEM、和光純薬工業(株))を用いた。
培養後、培地にアラマブルー(20μL)(岩城硝子(株))を加えて攪拌した後に3時間培養し、蛍光プレートリーダー(Spectra Max M5(日本モレキュラーデバイス(株))により励起波長530nm、蛍光波長590nmにて蛍光値を測定した。
また、化合物を作用させる直前における蛍光値は、培地を交換した後、直ちに培地にアラマブルー(20μL)を加えて攪拌した後に3時間培養し、蛍光プレートリーダーにより蛍光値を測定した。
なお、各化合物の溶解度の問題から最高濃度を10μMとし、そのためLC50値が算出できない化合物については>10μMとした。
また、図2に示されるように、本発明の化合物MU-1497は、MCF-7において5.0μM以上の濃度で細胞傷害または増殖抑制作用を示し、10μM(最高用量)を適用した場合の増殖率は-99.71%であった。一方、ゲムシタビン塩酸塩は、MCF-7において0.005μM以上の濃度で細胞傷害または増殖抑制作用を示し、10μMを適用した場合の増殖率は59.52%であった。
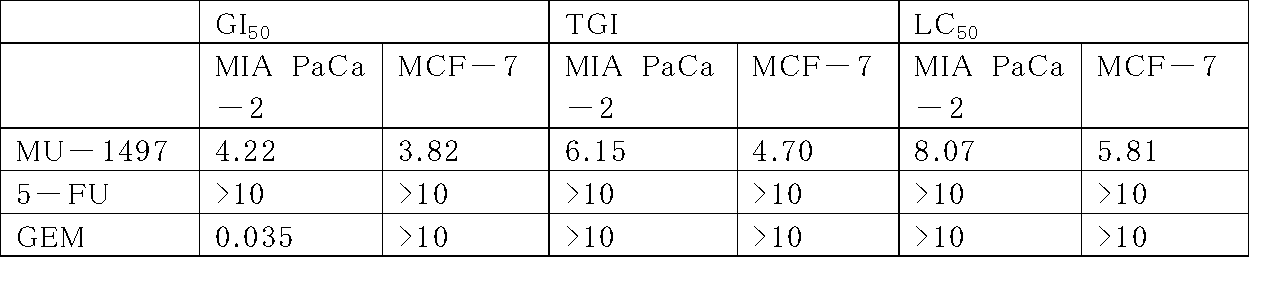
したがって、本発明の化合物MU-1497の細胞傷害または増殖抑制作用は、公知の抗がん剤であるゲムシタビン塩酸塩と比較して高濃度域で認められたが、最大反応の大きさはいずれもゲムシタビン塩酸塩を上回ることが示された。よって、本発明により、公知の抗がん剤を上回る制がん作用を有する新規な抗がん剤を得ることができた。
Claims (7)
- 式(I):

[式中、R1は炭素数2ないし6個のアルコキシアルキル基である]
で表される化合物、またはその医薬上許容される塩。 - 式(I)で表される化合物が、2-クロロ-N-[4-(7-メトキシ-4-メトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-メトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(3-メトキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(4-メトキシブチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(5-メトキシペンチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-[4-(7-メトキシ-4-エトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-エトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(3-エトキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(4-エトキシブチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-[4-(7-メトキシ-4-プロポキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-プロポキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(3-プロポキシプロピルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、2-クロロ-N-[4-(7-メトキシ-4-ブトキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミド、2-クロロ-N-{4-[7-メトキシ-4-(2-ブトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミド、および2-クロロ-N-[4-(7-メトキシ-4-ペントキシメチルスルファモイル-ベンゾフラン-2-イル)-チアゾール-2-イル]-アセトアミドよりなる群から選択される請求項1記載の化合物またはその医薬上許容される塩。
- 式(I)で表される化合物が2-クロロ-N-{4-[7-メトキシ-4-(2-メトキシエチルスルファモイル)-ベンゾフラン-2-イル]-チアゾール-2-イル}-アセトアミドである請求項2記載の化合物またはその医薬上許容される塩。
- 式(I):

で表される化合物またはその医薬上許容される塩の製法であって、
(1)2-アセチル-7-メトキシベンゾフランにクロロスルホン酸を反応させて2-アセチル-7-メトキシベンゾフラン-4-スルホニルクロリドを生成する工程;
(2)得られたスルホニルクロリド化合物に、塩基存在下で、アルコキシアルキルアミンを反応させて、式(II):
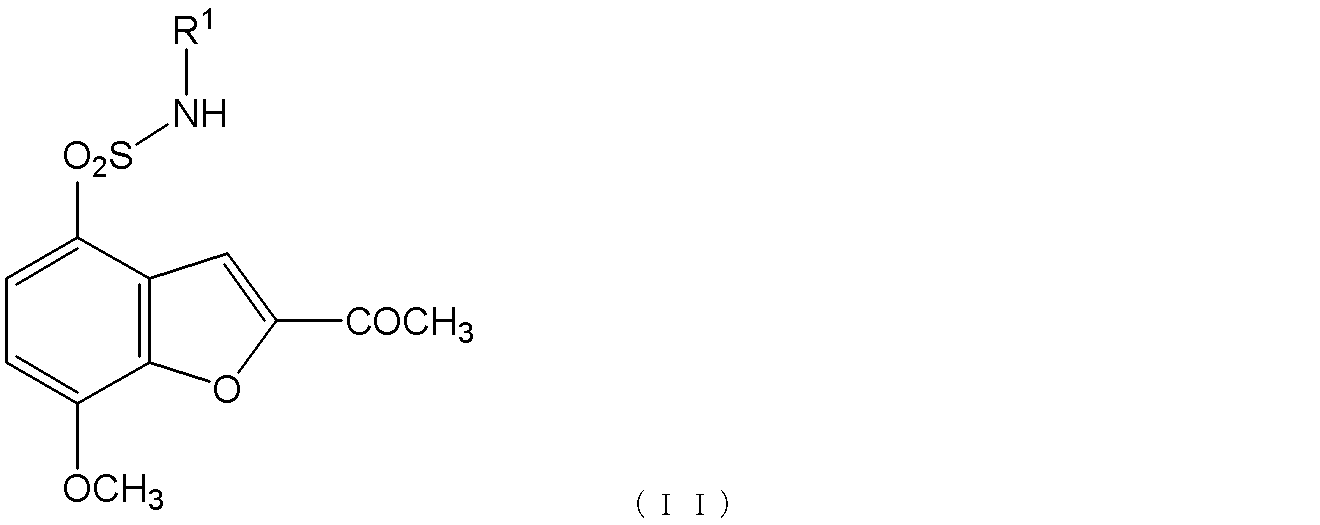
[式中、R1は上記定義に同じである]
で表されるスルホン酸アミド化合物を生成する工程;
(3)得られたスルホン酸アミド化合物にハロゲンを反応させてアセチル基をハロゲン化する工程;
(4)得られたスルホン酸アミドハロゲン化物に、塩基存在下で、チオ尿素を加熱条件下で反応させて式(III):
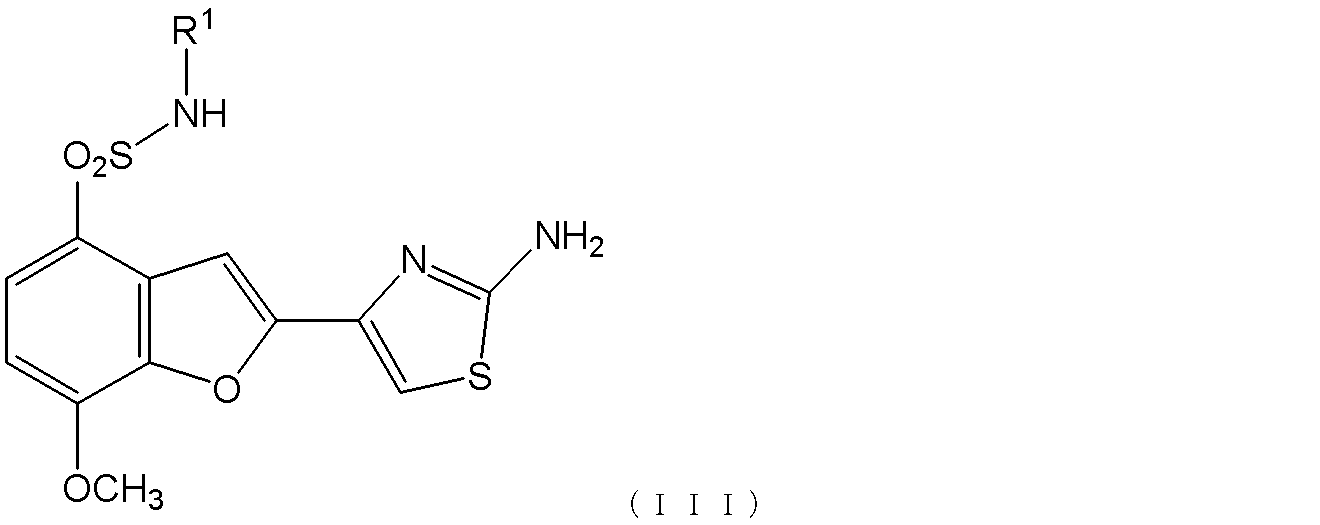
[式中、R1は上記定義に同じである]
で表される2-アミノチアゾリルベンゾフラン化合物を生成する工程;ついで
(5)得られた2-アミノチアゾリルベンゾフラン化合物に塩化クロロアセチルを反応させて式(I)で表される化合物を生成する工程
を含む製法。 - 工程(2)で用いるアルコキシアルキルアミンが、メトキシメチルアミン、2-メトキシエチルアミン、3-メトキシプロピルアミン、4-メトキシブチルアミン、5-メトキシペンチルアミン、エトキシメチルアミン、2-エトキシエチルアミン、3-エトキシプロピルアミン、4-エトキシブチルアミン、プロポキシメチルアミン、2-プロポキシエチルアミン、3-プロポキシプロピルアミン、ブトキシメチルアミン、2-ブトキシエチルアミンおよびペントキシメチルアミンよりなる群から選択される請求項4記載の製法。
- 式(I)で表される化合物またはその医薬上許容される塩を有効成分として含む、がんを治療するための医薬組成物。
- がんが膵臓がん、乳がん、脳腫瘍、神経膠腫、口腔がん、咽頭がん、喉頭がん、肺がん、食道がん、胃がん、腎臓がん、子宮体がん、子宮頸がん、卵巣がん、網膜芽細胞腫、前立腺がん、精巣腫瘍、肝臓がん、皮膚がん、結腸がんおよび直腸がんよりなる群から選択される請求項6記載の医薬組成物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11809488.7A EP2597093B1 (en) | 2010-07-22 | 2011-05-17 | Benzofuranone compound and pharmaceutical composition containing same |
| CN201180035250.3A CN103003270B (zh) | 2010-07-22 | 2011-05-17 | 苯并呋喃酮化合物以及含有该化合物的药物组合物 |
| KR1020137003240A KR101734868B1 (ko) | 2010-07-22 | 2011-05-17 | 벤조푸라논 화합물 및 그것을 포함하는 의약 조성물 |
| US13/810,796 US8563743B2 (en) | 2010-07-22 | 2011-05-17 | Benzofuranone compound and pharmaceutical composition containing same |
| JP2012525338A JP5648058B2 (ja) | 2010-07-22 | 2011-05-17 | ベンゾフラノン化合物およびそれを含む医薬組成物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010165081 | 2010-07-22 | ||
| JP2010-165081 | 2010-07-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012011314A1 true WO2012011314A1 (ja) | 2012-01-26 |
Family
ID=45496743
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/061266 WO2012011314A1 (ja) | 2010-07-22 | 2011-05-17 | ベンゾフラノン化合物およびそれを含む医薬組成物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8563743B2 (ja) |
| EP (1) | EP2597093B1 (ja) |
| JP (1) | JP5648058B2 (ja) |
| KR (1) | KR101734868B1 (ja) |
| CN (1) | CN103003270B (ja) |
| WO (1) | WO2012011314A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019064976A (ja) * | 2017-10-03 | 2019-04-25 | 国立大学法人 熊本大学 | 抗がん剤 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020139044A1 (ko) * | 2018-12-27 | 2020-07-02 | 홀로스메딕 주식회사 | 신규한 화합물 및 이를 포함하는 항암 활성 증진용 약학 조성물 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62155269A (ja) * | 1984-12-29 | 1987-07-10 | Kaken Pharmaceut Co Ltd | ベンゾフラン誘導体 |
| JP2000501411A (ja) | 1995-12-05 | 2000-02-08 | ダーウィン・ディスカバリー・リミテッド | ベンゾフランカルボキサミドおよびスルホンアミド |
| WO2004078751A1 (ja) | 2003-03-07 | 2004-09-16 | Kowa Co., Ltd. | ベンゾフラン誘導体 |
| JP2005008631A (ja) | 2003-05-29 | 2005-01-13 | New Industry Research Organization | ベンゾフラン化合物、およびそれを含有してなる医薬組成物 |
| JP2006507215A (ja) | 2002-02-22 | 2006-03-02 | バイエル・フアーマシユーチカルズ・コーポレーシヨン | 過剰増殖性障害の処置に有用なベンゾフランおよびベンゾチオフェン誘導体 |
| WO2006054793A1 (ja) | 2004-11-19 | 2006-05-26 | The New Industry Research Organization | ベンゾフラン化合物、およびそれを含有してなる医薬組成物 |
| JP2006225303A (ja) | 2005-02-16 | 2006-08-31 | New Industry Research Organization | ベンゾフラン誘導体を含有してなる、骨代謝疾患を予防または治療するための組成物 |
| JP2007501796A (ja) | 2003-08-07 | 2007-02-01 | バイエル・フアーマシユーチカルズ・コーポレーシヨン | 過剰増殖性障害の処置に有用なベンゾフラン誘導体 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20070049655A (ko) | 2004-08-13 | 2007-05-11 | 제넨테크, 인크. | Atp-이용 효소 억제 활성을 나타내는2-아미도-티아졸-기재 화합물과 조성물 및 이들의 용도 |
| SE0402735D0 (sv) * | 2004-11-09 | 2004-11-09 | Astrazeneca Ab | Novel compounds |
-
2011
- 2011-05-17 CN CN201180035250.3A patent/CN103003270B/zh active Active
- 2011-05-17 WO PCT/JP2011/061266 patent/WO2012011314A1/ja active Application Filing
- 2011-05-17 EP EP11809488.7A patent/EP2597093B1/en active Active
- 2011-05-17 US US13/810,796 patent/US8563743B2/en active Active
- 2011-05-17 JP JP2012525338A patent/JP5648058B2/ja active Active
- 2011-05-17 KR KR1020137003240A patent/KR101734868B1/ko active IP Right Grant
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62155269A (ja) * | 1984-12-29 | 1987-07-10 | Kaken Pharmaceut Co Ltd | ベンゾフラン誘導体 |
| JP2000501411A (ja) | 1995-12-05 | 2000-02-08 | ダーウィン・ディスカバリー・リミテッド | ベンゾフランカルボキサミドおよびスルホンアミド |
| JP2006507215A (ja) | 2002-02-22 | 2006-03-02 | バイエル・フアーマシユーチカルズ・コーポレーシヨン | 過剰増殖性障害の処置に有用なベンゾフランおよびベンゾチオフェン誘導体 |
| WO2004078751A1 (ja) | 2003-03-07 | 2004-09-16 | Kowa Co., Ltd. | ベンゾフラン誘導体 |
| JP2005008631A (ja) | 2003-05-29 | 2005-01-13 | New Industry Research Organization | ベンゾフラン化合物、およびそれを含有してなる医薬組成物 |
| JP2007501796A (ja) | 2003-08-07 | 2007-02-01 | バイエル・フアーマシユーチカルズ・コーポレーシヨン | 過剰増殖性障害の処置に有用なベンゾフラン誘導体 |
| WO2006054793A1 (ja) | 2004-11-19 | 2006-05-26 | The New Industry Research Organization | ベンゾフラン化合物、およびそれを含有してなる医薬組成物 |
| JP2006225303A (ja) | 2005-02-16 | 2006-08-31 | New Industry Research Organization | ベンゾフラン誘導体を含有してなる、骨代謝疾患を予防または治療するための組成物 |
Non-Patent Citations (3)
| Title |
|---|
| KURAMOTO M. ET AL.: "Preparation of leukotriene B4 inhibitory active 2- and 3-(2-aminothiazol- 4-yl)benzo[b]furan derivatives and their growth inhibitory activity on human pancreatic cancer cells", ORGANIC & BIOMOLECULAR CHEMISTRY, vol. 6, no. 15, 2008, pages 2772 - 2781, XP055073459 * |
| See also references of EP2597093A4 * |
| YANG X.F. ET AL.: "Synthesis of 2-aryl-5-alkyl- 7-methoxylbenzo[b]furan derivatives", CHINESE CHEMICAL LETTERS, vol. 18, no. 4, 2007, pages 380 - 382, XP022004590 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019064976A (ja) * | 2017-10-03 | 2019-04-25 | 国立大学法人 熊本大学 | 抗がん剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20130042565A (ko) | 2013-04-26 |
| EP2597093A4 (en) | 2013-12-04 |
| EP2597093A1 (en) | 2013-05-29 |
| JP5648058B2 (ja) | 2015-01-07 |
| CN103003270A (zh) | 2013-03-27 |
| CN103003270B (zh) | 2014-11-12 |
| US20130123313A1 (en) | 2013-05-16 |
| JPWO2012011314A1 (ja) | 2013-09-09 |
| KR101734868B1 (ko) | 2017-05-12 |
| US8563743B2 (en) | 2013-10-22 |
| EP2597093B1 (en) | 2014-11-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Fallah-Tafti et al. | Thiazolyl N-benzyl-substituted acetamide derivatives: synthesis, Src kinase inhibitory and anticancer activities | |
| RU2528408C2 (ru) | Сп0соб получения соединений дигидроинденамида, фармацевтические композии, содержащие данные соединение и их применение в качестве ингибитора протеинкиназы | |
| CN114516867B (zh) | 一类含氧五元杂环化合物、合成方法、药物组合物及用途 | |
| Shi et al. | Anthranilic acid-based diamides derivatives incorporating aryl-isoxazoline pharmacophore as potential anticancer agents: Design, synthesis and biological evaluation | |
| JP4890723B2 (ja) | TNFαインヒビターとして有用なクマリン誘導体 | |
| BR112016023382B1 (pt) | Composto, medicamento, e, uso de um composto | |
| CZ2002534A3 (cs) | Inhibitory protein-kináz, farmaceutické prostředky, které je obsahují, a jejich pouľití | |
| GB2449293A (en) | Compounds having Hsp90 inhibitory activity | |
| MX2010011258A (es) | Inhibidores de molecula pequeña del dominio de homologia de pleckstrin y metodos para usar los mismos. | |
| CA2802130C (en) | Cyanoquinoline derivatives | |
| JP6559699B2 (ja) | 置換含窒素複素環誘導体、それを含む医薬組成物及びその抗腫瘍性の適用 | |
| TW201728579A (zh) | 作為檢測點激酶1 (chk1)抑制劑之3,5-二取代吡唑及其製備及應用 | |
| AU2012302723A1 (en) | Pyrazole compound and use thereof for medical purposes | |
| JP2013530130A (ja) | ヘテロアリール(アルキル)ジチオカルバメート化合物、その調製方法及び使用 | |
| CN105705493A (zh) | 喹唑啉衍生物、其制备方法、药物组合物和应用 | |
| JP2008542242A (ja) | 新規のテトラヒドロピリドチオフェン | |
| CA2901155A1 (en) | Camkii inhibitors and uses thereof | |
| BR112019013273A2 (pt) | agente antitumoral e inibidor de bromodomínio | |
| JP5469604B2 (ja) | 新規テトラヒドロ融合ピリジン | |
| JP5648058B2 (ja) | ベンゾフラノン化合物およびそれを含む医薬組成物 | |
| AU2020410900A1 (en) | Compound used as RET kinase inhibitor and application thereof | |
| CN102432622B (zh) | 4-胺基噁二唑表鬼臼毒素衍生物及其制备方法和用途 | |
| CN103980252A (zh) | 制备含1,2,4-三氮唑的吡唑类席夫碱治疗肿瘤的药物 | |
| JP4370291B2 (ja) | 4,8−ジヒドロベンゾジチオフェン−4,8−ジオンの誘導体およびこれを含む抗癌剤組成物 | |
| KR101630243B1 (ko) | 신규한 화합물, 이의 약학적으로 허용가능한 염 또는 이의 광학 이성질체, 이의 제조방법 및 이를 유효성분으로 함유하는 바이러스성 질환의 예방 또는 치료용 약학적 조성물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11809488 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012525338 Country of ref document: JP Ref document number: 2011809488 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13810796 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137003240 Country of ref document: KR Kind code of ref document: A |