WO2011121848A1 - ノルボルナン骨格を有するポリアミドイミド及びその製造方法 - Google Patents
ノルボルナン骨格を有するポリアミドイミド及びその製造方法 Download PDFInfo
- Publication number
- WO2011121848A1 WO2011121848A1 PCT/JP2010/071675 JP2010071675W WO2011121848A1 WO 2011121848 A1 WO2011121848 A1 WO 2011121848A1 JP 2010071675 W JP2010071675 W JP 2010071675W WO 2011121848 A1 WO2011121848 A1 WO 2011121848A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- polyamideimide
- compound
- norbornane
- represented
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/30—Low-molecular-weight compounds
- C08G18/34—Carboxylic acids; Esters thereof with monohydroxyl compounds
- C08G18/343—Polycarboxylic acids having at least three carboxylic acid groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/30—Low-molecular-weight compounds
- C08G18/34—Carboxylic acids; Esters thereof with monohydroxyl compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/75—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic
- C08G18/751—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/75—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic
- C08G18/751—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring
- C08G18/752—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring containing at least one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group
- C08G18/753—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring containing at least one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group containing one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group having a primary carbon atom next to the isocyanate or isothiocyanate group
- C08G18/755—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring containing at least one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group containing one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group having a primary carbon atom next to the isocyanate or isothiocyanate group and at least one isocyanate or isothiocyanate group linked to a secondary carbon atom of the cycloaliphatic ring, e.g. isophorone diisocyanate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/76—Polyisocyanates or polyisothiocyanates cyclic aromatic
- C08G18/7657—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings
- C08G18/7664—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings containing alkylene polyphenyl groups
- C08G18/7671—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings containing alkylene polyphenyl groups containing only one alkylene bisphenyl group
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1003—Preparatory processes
- C08G73/1035—Preparatory processes from tetracarboxylic acids or derivatives and diisocyanates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/1075—Partially aromatic polyimides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/14—Polyamide-imides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
- C08L79/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08L79/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
Definitions
- the present invention relates to a polyamideimide having a norbornane skeleton, which is useful as a polymer having high heat resistance and transparency.
- the present invention also relates to a manufacturing method thereof.
- epoxy resin has been widely used as a resin for optical members used in optoelectronic devices and the like because of its mounting process on an electronic substrate and the like, heat resistance under high temperature operation, mechanical properties, and versatility.
- high-intensity laser light, blue light, and near-ultraviolet light has expanded in the field of optoelectronic devices, and a resin that is superior in transparency, heat resistance, and light resistance than ever has been demanded.
- epoxy resin has high transparency in visible light, but sufficient transparency cannot be obtained in the ultraviolet to near ultraviolet region.
- a cured product composed of an alicyclic epoxy resin and an acid anhydride has a relatively high transparency in the near-ultraviolet region, but has a problem that it is easily colored by heat or light.
- improvement in heat resistance and UV resistance is required, and various epoxy resins have been studied (for example, see Patent Documents 1 to 4).
- heat-resistant resins such as polyamide and polyamide-imide are excellent in heat resistance, insulation, light resistance and mechanical properties, and are soluble in various solvents and excellent in workability. It is widely used as a surface protection film of an element, an interlayer insulating film, and the like.
- polyamides having an alicyclic structure are excellent in transparency in the ultraviolet region, and thus have been studied as materials for optoelectronic devices and various displays (for example, see Patent Document 5).
- the above polyamide having an alicyclic structure can be produced by using a dicarboxylic acid having an alicyclic structure as an acid halide and then reacting with a diamine. Further, as a method for producing a polyimide having an alicyclic structure, a method using dicarboxylic acid as an acid halide is also known (see, for example, Patent Document 6).
- the method for producing a polyamide as described in Patent Document 5 is produced by polymerizing a diamine and a carboxylic acid or a derivative thereof. The reaction between the diamine and the dicarboxylic acid is carried out at a high temperature of 240 ° C. to 350 ° C. I need.
- a harmful halogen-based gas is generated during the reaction, so that a processing apparatus or the like is essential. Therefore, there is a problem in manufacturing cost, and an industrially simple manufacturing method is required.
- the present invention has been made to solve the above problems. Specifically, the present invention relates to a polyamideimide having a norbornane skeleton, which is excellent in heat resistance and transparency, and a method for producing the same.
- the present invention is as follows.
- the present invention relates to a polyamideimide having a norbornane skeleton represented by the following general formula (I).
- X is a divalent organic group selected from a divalent aliphatic group having 4 to 16 carbon atoms, a divalent alicyclic group having 4 to 16 carbon atoms, and a divalent aromatic group. is there.
- the present invention relates to a norbornane skeleton represented by the following general formula (I), characterized by reacting a norbornane tricarboxylic acid anhydride represented by the following general formula (II) with a diisocyanate compound in a polar solvent.
- the present invention relates to a method for producing a polyamideimide having
- X is a divalent organic group selected from a divalent aliphatic group having 4 to 16 carbon atoms, a divalent alicyclic group having 4 to 16 carbon atoms, and a divalent aromatic group. is there.
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the diisocyanate compound is a diisocyanate compound represented by the following general formula (III).
- OCN-X-NCO (III) (In the formula, X is a divalent organic group selected from a divalent aliphatic group having 4 to 16 carbon atoms, a divalent alicyclic group having 4 to 16 carbon atoms, and a divalent aromatic group. is there.)
- the present invention provides a process for producing a polyamideimide having a norbornane skeleton, wherein the norbornanetricarboxylic acid anhydride represented by the general formula (II) is obtained by a method comprising the following steps (1) to (3): About.
- R is hydrogen or an alkyl group having 1 to 8 carbon atoms.
- Formic acid ester (HCOOR 2 ) The reaction is carried out in the presence of a catalyst system containing a ruthenium compound, a cobalt compound, and a halide salt to obtain a norbornanetricarboxylic acid derivative represented by the following general formula (V).
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the ruthenium compound is a ruthenium complex having both a carbonyl ligand and a halogen ligand in the molecule.
- the present invention also relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the halide salt is a quaternary ammonium salt.
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the catalyst system further contains a basic compound.
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the basic compound is a tertiary amine compound.
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the catalyst system further contains a phenol compound.
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton, wherein the catalyst system further contains an organic halogen compound.
- the polyamideimide having a norbornane skeleton of the present invention is excellent in heat resistance and transparency, optical materials represented by electronic parts, optical fibers, optical lenses, etc. used in semiconductors and liquid crystals, display-related materials, and medical use Can be used as material. Moreover, the polyamideimide having a norbornane skeleton can be produced under industrially advantageous conditions by the production method of the present invention.
- Polyamideimide having norbornane skeleton of the present invention relates to a polyamideimide having a norbornane skeleton represented by the following general formula (I).
- X is a divalent organic group selected from a divalent aliphatic group having 4 to 16 carbon atoms, a divalent alicyclic group having 4 to 16 carbon atoms, and a divalent aromatic group. is there.
- X in the above formula is the same as X in the diisocyanate compound represented by the general formula (III) described later. Details will be described in ⁇ 2> below.
- n 2 to 800.
- the polyamideimide having a norbornane skeleton of the present invention preferably has a number average molecular weight of 2,000 to 250,000, more preferably 3,000 to 220,000.
- the number average molecular weight is less than 2,000, heat resistance and the like tend to decrease, and when it exceeds 250,000, solubility in a solvent tends to decrease.
- the polyamide having a norbornane skeleton In order to make the number average molecular weight of the polyamide having a norbornane skeleton within the above range, it may be produced by the production method of the present application.
- the number average molecular weight is measured using gel permeation chromatography (hereinafter abbreviated as “GPC”) under the following conditions, and is calculated using a standard polystyrene calibration curve.
- GPC gel permeation chromatography
- a norbornane tricarboxylic acid anhydride represented by the following general formula (II) is reacted with a diisocyanate compound in a polar solvent.
- the present invention relates to a method for producing a polyamideimide having a norbornane skeleton represented by the above general formula (I).
- diisocyanate compound examples of the diisocyanate compound to be reacted with the norbornane tricarboxylic acid anhydride represented by the general formula (II) in the present invention include an aliphatic and / or alicyclic diisocyanate compound represented by the following general formula (III), and an aromatic Group diisocyanate compounds.
- OCN-X-NCO (III)
- X is a divalent organic group selected from a divalent aliphatic group having 4 to 16 carbon atoms, a divalent alicyclic group having 4 to 16 carbon atoms, and a divalent aromatic group. is there.
- the aliphatic isocyanate compound include those in which X in the general formula (III) is a divalent aliphatic group having 4 to 16 carbon atoms. Specific examples include hexamethylene diisocyanate, 2,2,4, and the like. -Trimethylhexamethylene diisocyanate, lysine diisocyanate, etc. can be used, and these can be used alone or in admixture of two or more.
- alicyclic isocyanate compound examples include those in which X in the general formula (III) is a divalent alicyclic group having 4 to 16 carbon atoms. Specific examples thereof include isophorone diisocyanate, 4,4′- Dicyclohexylmethane diisocyanate, cyclohexylene diisocyanate and the like can be used, and these can be used alone or in admixture of two or more.
- aromatic isocyanate examples include those in which X in the general formula (III) is a divalent aromatic group. Specific examples include 4,4′-diphenylmethane diisocyanate, tolylene diisocyanate, xylylene diisocyanate, 4,4′-diphenyl ether diisocyanate, 4,4 ′-[2,2-bis (4-phenoxyphenyl) propane] diisocyanate, biphenyl-4,4′-diisocyanate, biphenyl-3,3′-diisocyanate, biphenyl-3 , 4'-diisocyanate, 3,3'-dimethylbiphenyl-4,4'-diisocyanate, 2,2'-dimethylbiphenyl-4,4'-diisocyanate, 3,3'-diethylbiphenyl-4,4'-diisocyanate 2,2'-diethylbiphenyl-4,4'-di Socyanate,
- aliphatic isocyanate compounds aliphatic isocyanate compounds
- alicyclic isocyanate compounds aromatic isocyanate compounds
- the amount of the norbornane tricarboxylic acid anhydride represented by the general formula (II) and the diisocyanate compound represented by the general formula (III) in the present invention is the total number of moles of carboxyl groups of the norbornane tricarboxylic acid anhydride. Is preferably 0.7 to 2.0, more preferably 0.8 to 1.7, still more preferably 0.9 to 1.5, and still more preferably 0.8 to 1.7. It is particularly preferably 95 to 1.3.
- the norbornanetricarboxylic acid anhydride represented by the general formula (II) is preferably obtained by a method including the following steps (1) to (3).
- R is hydrogen or an alkyl group having 1 to 8 carbon atoms.
- Formic acid ester (HCOOR 2 ) The reaction is carried out in the presence of a catalyst system containing a ruthenium compound, a cobalt compound, and a halide salt to obtain a norbornanetricarboxylic acid derivative represented by the following general formula (V).
- the formic acid ester (HCOOR 2 ) to be reacted with the norbornene dicarboxylic acid derivative represented by the general formula (IV) is particularly limited.
- methyl formate, ethyl formate, propyl formate, isopropyl formate, butyl formate, isobutyl formate, amyl formate, isoamyl formate, allyl formate, vinyl formate, benzyl formate and the like can be used.
- linear alkyl formate such as methyl formate and ethyl formate is preferred, and methyl formate is more preferred.
- ester moiety of the formic acid ester (R 2) corresponds to R 2 in the formula (V).
- R in the above formula (IV) is the same as R in the above formula (V).
- the catalyst system for the above reaction includes a ruthenium compound, a cobalt compound, and a halide salt.
- the “catalyst system” includes not only the catalyst itself, but also additives, sensitizers, and the like that assist the action of the catalyst.
- the ruthenium compound is not particularly limited as long as it contains ruthenium.
- suitable ruthenium compounds include [Ru (CO) 3 Cl 2 ] 2 , [Ru (CO) 2 Cl 2 ] n , [Ru (CO) 3 Cl 3 ] ⁇ , [Ru 3 (CO) 11 Cl ] -, [Ru 4 (CO ) 13 Cl] - , such as, ruthenium compounds having both a carbonyl ligand and halogen ligands in the molecule. of these, from the viewpoint of the reaction rate increase, [ Ru (CO) 3 Cl 2 ] 2 , [Ru (CO) 2 Cl 2 ] n and the like are more preferable.
- the ruthenium compounds include RuCl 3 , Ru 3 (CO) 12 , RuCl 2 (C 8 H 12 ), Ru (CO) 3 (C 8 H 8 ), Ru (CO) 3 (C 8 H 12 ), and Ru.
- (C 8 H 10 ) (C 8 H 12 ) or the like is used as a precursor compound, and the ruthenium compound is converted into the precursor before or during the reaction to obtain the norbornanetricarboxylic acid derivative represented by the general formula (V). It may be prepared from the body compound and introduced into the reaction system.
- the amount of the ruthenium compound used is preferably 1/10000 to 1 equivalent, more preferably 1/1000 to 1/50 equivalent, relative to the norbornene dicarboxylic acid derivative represented by the general formula (IV) as a raw material. . Considering the production cost, it is preferable that the amount of the ruthenium compound used is smaller, but if it is less than 1/10000 equivalent, the reaction tends to become extremely slow.
- the cobalt compound is not particularly limited as long as it contains cobalt.
- suitable cobalt compounds include cobalt compounds having a carbonyl ligand such as Co 2 (CO) 8 , Co (CO) 4 , Co 4 (CO) 12 ; cobalt acetate, cobalt propionate, cobalt benzoate, Examples include cobalt compounds having a carboxylic acid compound such as cobalt oxide as a ligand; cobalt phosphate and the like. Of these, Co 2 (CO) 8 , cobalt acetate, cobalt citrate and the like are more preferable from the viewpoint of improving the reaction rate.
- the amount of the cobalt compound used is 1/100 to 10 equivalents, preferably 1/10 to 5 equivalents, relative to the ruthenium compound. Whether the ratio of the cobalt compound to the ruthenium compound is lower than 1/100 or higher than 10, the norbornanetricarboxylic acid derivative represented by the general formula (V) (hereinafter also referred to as “ester compound”) The amount produced tends to decrease significantly.
- the halide salt is not particularly limited as long as it is a compound composed of a halogen ion such as chloride ion, bromide ion and iodide ion and a cation.
- the cation may be either an inorganic ion or an organic ion.
- the halide salt may contain one or more halogen ions in the molecule.
- the inorganic ions constituting the halide salt may be one metal ion selected from alkali metals and alkaline earth metals. Specific examples include ions of lithium, sodium, potassium, rubidium, cesium, calcium, strontium and the like.
- the organic ion may be a monovalent or higher valent organic group derived from an organic compound.
- examples include ions such as ammonium, phosphonium, pyrrolidinium, pyridium, imidazolium, and iminium, and the hydrogen atom of these ions may be substituted with a hydrocarbon group such as an alkyl group and an aryl group.
- suitable organic ions include tetramethylammonium, tetraethylammonium, tetrapropylammonium, tetrabutylammonium, tetrapentylammonium, tetrahexylammonium, tetraheptylammonium, tetraoctylammonium, and trioctyl.
- ions of quaternary ammonium salts such as butylmethylpyrrolidinium chloride, bis (triphenylphosphine) iminium iodide, riooctylmethylammonium chloride are more preferable.
- the halide salt used in the present invention does not need to be a solid salt, and an ionic liquid containing halide ions that becomes liquid near room temperature or in a temperature range of 100 ° C. or less may be used.
- ionic liquids include 1-ethyl 3-methylimidazolium, 1-propyl-3-methylimidazolium, 1-butyl-3-methylimidazolium, 1-pentyl-3- Methylimidazolium, 1-hexyl-3-methylimidazolium, 1-heptyl-3-methylimidazolium, 1-octyl-3-methylimidazolium, 1-decyl-3-methylimidazolium, 1-dodecyl-3- Methylimidazolium, 1-tetradecyl-3-methylimidazolium, 1-hexadecyl-3-methylimidazolium, 1-octadecyl-3-methylimidazolium,
- the above-described halide salts may be used alone or in combination.
- halide salts are chloride salts, bromide salts, and iodide salts, and the cation is an organic ion.
- specific examples of the halide salt suitable in the present invention include butylmethylpyrrolidinium chloride, bis (triphenylphosphine) iminium iodide, trioctylmethylammonium chloride and the like.
- the added amount of the halide salt is, for example, 1 to 1000 equivalents, preferably 2 to 50 equivalents, relative to the ruthenium compound.
- the addition amount is, for example, 1 to 1000 equivalents, preferably 2 to 50 equivalents, relative to the ruthenium compound.
- a catalyst system containing the ruthenium compound, cobalt compound and halide salt is used. If necessary, by adding any one or more of a basic compound, a phenol compound, and an organic halogen compound, the effect of promoting the reaction by the catalyst system can be further enhanced.
- the basic compound used in the present invention may be an inorganic compound or an organic compound.
- Specific examples of the basic inorganic compound include alkali metal and alkaline earth metal carbonates, hydrogen carbonates, hydroxide salts, alkoxides, and the like.
- Specific examples of the basic organic compound include primary amine compounds, secondary amine compounds, tertiary amine compounds, pyridine compounds, imidazole compounds, quinoline compounds, and the like.
- tertiary amine compounds are preferred from the viewpoint of the reaction promoting effect.
- suitable tertiary amine compounds include trialkylamine, N-alkylpyrrolidine, quinuclidine, and triethylenediamine.
- the amount of the basic compound added is not particularly limited, but is, for example, 1 to 1000 equivalents, preferably 2 to 200 equivalents, relative to the ruthenium compound.
- the addition amount 1 equivalent or more By making the addition amount 1 equivalent or more, the expression of the promoting effect tends to become more prominent.
- the addition amount exceeds 1000 equivalents even if the addition amount is further increased, there is a tendency that a further improvement effect of reaction promotion cannot be obtained.
- the phenol compound used in the present invention is not particularly limited. Specific examples of usable phenol compounds include phenol, cresol, alkylphenol, methoxyphenol, phenoxyphenol, chlorophenol, trifluoromethylphenol, hydroquinone and catechol.
- the amount of the phenol compound added is not particularly limited, but is, for example, 1 to 1000 equivalents, preferably 2 to 200 equivalents, relative to the ruthenium compound.
- the addition amount 1 equivalent or more By making the addition amount 1 equivalent or more, the expression of the promoting effect tends to become more prominent.
- the addition amount exceeds 1000 equivalents even if the addition amount is further increased, there is a tendency that a further improvement effect of reaction promotion cannot be obtained.
- the organic halogen compound used in the present invention is not particularly limited, and specific examples of usable organic halogen compounds include methyl halide, dihalogen methane, dihalogen ethane, trihalogen methane, tetrahalogen carbon, halogenated benzene and the like. It is done.
- the amount of the organic halogen compound added is not particularly limited, but is, for example, 1 to 1000 equivalents, preferably 2 to 200 equivalents, relative to the ruthenium compound.
- the addition amount 1 equivalent or more By making the addition amount 1 equivalent or more, the expression of the promoting effect tends to become more prominent.
- the addition amount exceeds 1000 equivalents even if the addition amount is further increased, there is a tendency that a further improvement effect of reaction promotion cannot be obtained.
- the reaction can proceed without using any solvent.
- a solvent may be used.
- the usable solvent is not particularly limited as long as it can dissolve a compound used as a raw material, a norbornene monocarboxylic acid derivative dicyclopentadiene, formic acid ester (HCOOR 2 ), and the like.
- solvents that can be suitably used include n-pentane, n-hexane, n-heptane, cyclohexane, benzene, toluene, o-xylene, p-xylene, m-xylene, ethylbenzene, cumene, tetrahydrofuran, and N-methylpyrrolidone.
- Dimethylformamide, dimethylacetamide, dimethylimidazolidinone ethylene glycol dimethyl ether, diethylene glycol dimethyl ether, triethylene glycol dimethyl ether, tetralin and the like.
- the reaction of the norbornene dicarboxylic acid derivative represented by the general formula (IV) and formic acid ester (HCOOR 2 ) in the present invention is preferably carried out in a temperature range of 80 ° C. to 200 ° C.
- the above reaction is more preferably carried out in the temperature range of 100 ° C to 160 ° C.
- the reaction rate is increased and the reaction is facilitated efficiently.
- by controlling the reaction temperature to 200 ° C. or lower decomposition of formic acid ester (HCOOR 2 ) used as a raw material can be suppressed.
- the formic acid ester (HCOOR 2 ) is decomposed, the addition of an ester group to the norbornene dicarboxylic acid derivative represented by the general formula (IV) cannot be achieved, so that a too high reaction temperature is not desirable.
- reaction temperature exceeds the boiling point of either the norbornene dicarboxylic acid derivative or formic acid ester (HCOOR 2 ) represented by the general formula (IV) used as a raw material, it is necessary to carry out the reaction in a pressure resistant vessel.
- the completion of the reaction can be confirmed using a well-known analytical technique such as gas chromatography or NMR.
- the norbornanetricarboxylic acid derivative represented by the general formula (V) obtained as described above can be isolated by distillation or the like as necessary, and used as a raw material for the hydrolysis step of the following step (2).
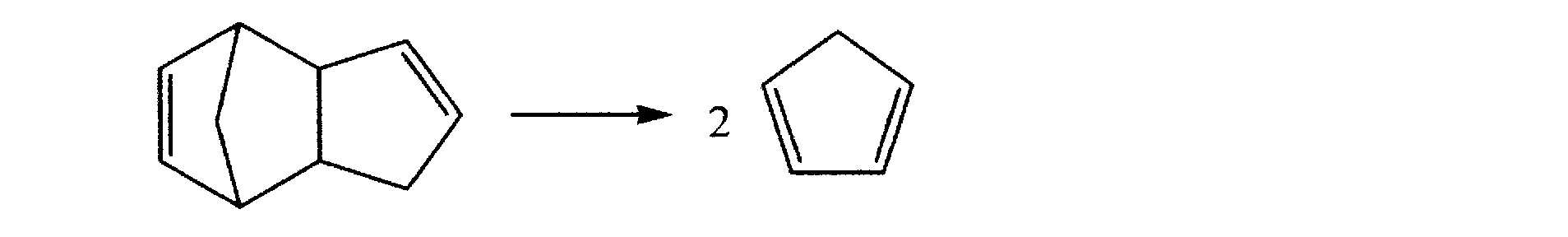
- the norbornene dicarboxylic acid derivative represented by the general formula (IV) in the present invention is directly synthesized by a usual method, that is, Diels-Alder reaction of dicyclopentadiene or cyclopentadiene with maleic acid or a diester compound thereof.
- Diels-Alder reaction of dicyclopentadiene or cyclopentadiene with maleic acid or a diester compound thereof.
- Decomposition of dicyclopentadiene to cyclopentadiene is described in, for example, Org. Syn, 1963, Vol. 4, P238, Org. Syn, 1962, Vol. 42, P50, Organic Synthesis Handbook, 1990, P501 and the like can be used. Specifically, use a method of recovering cyclopentadiene flowing out at 42-46 ° C by charging dicyclopentadiene into a flask equipped with a sneader or Vigreux fractionating tube and heating to 150-170 ° C. Can do.
- the Diels-Alder reaction method of cyclopentadiene and maleic acid or its diester compound is not particularly limited, but a method in which cyclopentadiene is dropped after charging maleic acid or its diester compound in a flask is preferred.
- the Diels-Alder reaction method of cyclopentadiene and maleic acid or its diester compound can proceed without using any solvent.
- a solvent may be used.
- the solvent that can be used is not particularly limited as long as the compound used as a raw material can be dissolved.
- Specific examples of solvents that can be suitably used include n-pentane, n-hexane, n-heptane, cyclohexane, benzene, toluene, o-xylene, p-xylene, m-xylene, ethylbenzene, cumene, tetrahydrofuran, and N-methylpyrrolidone.
- Dimethylformamide, dimethylacetamide, dimethylimidazolidinone ethylene glycol dimethyl ether, diethylene glycol dimethyl ether, triethylene glycol dimethyl ether, tetralin and the like.
- the reaction temperature of Diels-Alder reaction between cyclopentadiene and maleic acid or a diester compound thereof is preferably 20 to 50 ° C, more preferably 20 to 40 ° C, and particularly preferably 30 to 40 ° C.
- the reaction temperature is less than 20 ° C, the reaction time tends to be long. Moreover, when it exceeds 50 degreeC, side reactions, such as dimerization of cyclopentadiene, may occur.
- the reaction time can be appropriately selected depending on the scale of the batch and the reaction conditions employed.
- the maleic diester compound to be used is not particularly limited, and for example, dimethyl maleate, diethyl maleate, dipropyl maleate, dibutyl maleate, diamyl maleate, dioctyl maleate and the like can be used. In view of properties and the like, dimethyl maleate is preferable.
- Step 2 hydrolysis step of norbornanetricarboxylic acid derivative represented by the above general formula (V)
- the norbornanetricarboxylic acid derivative represented by the above general formula (V) is hydrolyzed to give the following general formula (VI).
- the norbornane tricarboxylic acid represented by () is not particularly limited, and for example, acid hydrolysis, alkali hydrolysis, etc. described in Japanese Patent No. 2591492, Japanese Patent Application Laid-Open No. 2008-31406, etc. are used. be able to. Alternatively, it can be hydrolyzed by heating at a high temperature of 140 ° C. or higher in the presence of moisture in a heat-resistant container without adding an acid component or an alkali component.
- Dehydration and ring closure of norbornanetricarboxylic acid represented by the above general formula (VI) is represented by the following general formula (II).
- the thermal ring closure method is preferable in consideration of the production cost and the residual ionic impurity concentration in the resulting polyamideimide.
- reaction conditions for obtaining polyamideimide (I) having norbornane skeleton A polar solvent is used for the reaction between the norbornanetricarboxylic acid anhydride represented by the general formula (II) and the diisocyanate compound represented by the general formula (III).
- the polar solvent that can be used is not particularly limited as long as it can dissolve the compound used as a raw material.
- polar solvents that can be suitably used include N-methylpyrrolidone, N, N′-dimethylacetamide, N, N′-dimethylformamide, 1,3-dimethyl-3,4,5,6-tetrahydro-2 ( 1H) -nitrogenous solvents such as pyrimidinone; Ether solvents such as diethylene glycol dimethyl ether, diethylene glycol diethyl ether, triethylene glycol dimethyl ether, triethylene glycol diethyl ether; Sulfur-containing solvents such as dimethyl sulfoxide, diethyl sulfoxide, dimethyl sulfone, sulfolane; ester solvents such as ⁇ -butyrolactone and cellosolve acetate; Ketone solvents such as cyclohexanone and methyl ethyl ketone; and the like can be used.
- Ether solvents such as diethylene glycol dimethyl ether, diethylene glycol diethyl ether, triethylene glyco
- the amount of the polar solvent used is 20 to 500 parts by mass with respect to 100 parts by mass of the total amount of the norbornanetricarboxylic acid anhydride represented by the general formula (II) and the diisocyanate compound represented by the general formula (III).
- the amount is preferably 30 to 300 parts by mass, more preferably 50 to 200 parts by mass.
- the amount used is less than 20 parts by mass, the raw materials are not sufficiently dissolved, and the reaction rate tends to be slow. Even when the amount exceeds 500 parts by mass, the yield of the polyamide per batch is reduced, which is particularly advantageous. There is no.
- the reaction temperature is preferably 80 to 200 ° C, more preferably 90 to 190 ° C, and particularly preferably 100 to 180 ° C.
- the reaction time can be appropriately selected depending on the scale of the batch and the reaction conditions employed.
- the polyamide having a norbornane skeleton in the range of 2,000 to 250,000, it may be produced by the production method of the present application.
- reaction vessel After adding 50 mL, the reaction vessel was purged with nitrogen gas at 0.5 MPa and maintained at 120 ° C. for 8 hours. Thereafter, the reaction apparatus was cooled to room temperature and released, and a part of the remaining organic phase was extracted and analyzed using a gas chromatograph.
- methyl norbornane tricarboxylate produced by the reaction was 90. 3 mmol (yield 90.3% based on methyl norbornane tricarboxylate).
- the resulting methyl norbornanetricarboxylate was isolated by distillation under reduced pressure.
- the white powder was filtered, washed with water and dried to obtain 135 g of norbornane tricarboxylic acid anhydride.
- the obtained norbornanetricarboxylic acid anhydride was analyzed by H 1 -NMR. As a result, the peaks of methylene and methine groups of norbornane (tricyclodecane) were around 1.1 to 3.0 ppm. A peak was confirmed in the vicinity of 12.4 ppm, and the integrated intensity ratio was 9.00 / 0.98 (theoretical value: 9/1).
- the obtained polyamideimide (PAI-1) having a norbornane skeleton was applied onto a Teflon (registered trademark) substrate, heated at 250 ° C. to dry the organic solvent, and a coating film having a thickness of 30 ⁇ m was formed.
- the glass transition temperature (Tg) and thermal decomposition start temperature (5% mass reduction temperature, Td 5 ) of this coating film were measured under the following conditions. The results are shown in Table 1.
- Tg Glass transition temperature
- Measurement mode Extension measurement span: 10 mm Load: 10g Temperature increase rate: 5 ° C / min Atmosphere: Air (2) Thermal decomposition start temperature (5% mass loss temperature, Td 5 ) It was measured with a differential thermal balance (Seiko Electronics Co., Ltd., Model 5200 TG-DTA).
- the polyamideimides having norbornane skeletons obtained in Examples 1 to 4 have good heat resistance and high light transmittance, whereas the polyamideimides obtained in Comparative Examples 1 and 2 are The light transmittance was inferior.
- a polyamideimide having a norbornane skeleton excellent in heat resistance and transparency can be obtained. Therefore, it can be used as an electronic material used for semiconductors and liquid crystals, an optical material typified by an optical fiber, an optical lens, and the like, a display-related material, and a medical material.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polyurethanes Or Polyureas (AREA)
Abstract
Description

本発明は、下記一般式(II)で表されるノルボルナントリカルボン酸無水物と、ジイソシアネート化合物と、を極性溶媒中で反応させることを特徴とする、下記一般式(I)で表されるノルボルナン骨格を有するポリアミドイミドの製造方法に関する。


本発明は、上記ジイソシアネート化合物が、下記一般式(III)で表されるジイソシアネート化合物であることを特徴とする、ノルボルナン骨格を有するポリアミドイミドの製造方法に関する。
(但し、式中Xは、炭素数4~16の2価の脂肪族基、炭素数4~16の2価の脂環族基及び2価の芳香族基から選ばれる2価の有機基である。)
本発明は、上記一般式(II)で表されるノルボルナントリカルボン酸無水物が、下記(1)~(3)工程を含む方法で得られることを特徴とするノルボルナン骨格を有するポリアミドイミドの製造方法に関する。

ギ酸エステル(HCOOR2)とを、
ルテニウム化合物と、コバルト化合物と、ハロゲン化物塩と、を含む触媒系の存在下で反応させて、下記一般式(V)で表されるノルボルナントリカルボン酸誘導体とする。

(2)工程:上記一般式(V)で表されるノルボルナントリカルボン酸誘導体のアルコキシカルボニル基を加水分解して、下記一般式(VI)で表されるノルボルナントリカルボン酸を得る。

本発明は、下記一般式(I)で表されるノルボルナン骨格を有するポリアミドイミドに関する。

なお、上記式中のXは、後述の一般式(III)で表されるジイソシアネート化合物の中のXと同じである。詳細については、下記<2>において説明する。
カラム:昭和電工(株)製、Shodex KD-806M×1本
溶離液:N-メチル-2-ピロリドン 1.0ml/min
検出器:UV(280nm)
<2>本発明のノルボルナン骨格を有するポリアミドイミドの製造方法
本発明の一実施形態は、下記一般式(II)で表されるノルボルナントリカルボン酸無水物と、ジイソシアネート化合物と、を極性溶媒中で反応させることを特徴とする、上記一般式(I)で表されるノルボルナン骨格を有するポリアミドイミドの製造方法に関する。

本発明における一般式(II)で表されるノルボルナントリカルボン酸無水物と反応させるジイソシアネート化合物としては、下記一般式(III)で表される、脂肪族及び/又は脂環族ジイソシアネート化合物、及び、芳香族ジイソシアネート化合物が挙げられる。
(但し、式中Xは、炭素数4~16の2価の脂肪族基、炭素数4~16の2価の脂環族基及び2価の芳香族基から選ばれる2価の有機基である。)
脂肪族イソシアネート化合物としては、上記一般式(III)中のXが炭素数4~16の2価の脂肪族基であるものが挙げられ、具体的に例えば、ヘキサメチレンジイソシアネート、2,2,4-トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート等を使用することができ、これらは、単独又は2種以上を混合して使用することもできる。
上記一般式(II)で表されるノルボルナントリカルボン酸無水物は、下記の(1)~(3)の工程を含む方法で得られたものが好ましい。

ギ酸エステル(HCOOR2)とを、
ルテニウム化合物と、コバルト化合物と、ハロゲン化物塩と、を含む触媒系の存在下で反応させて、下記一般式(V)で表されるノルボルナントリカルボン酸誘導体とする。

(2)工程:その後、上記一般式(V)で表されるノルボルナントリカルボン酸誘導体のアルコキシカルボニル基を加水分解して、下記一般式(VI)で表されるノルボルナントリカルボン酸とする。

上記一般式(IV)で表されるノルボルネンジカルボン酸誘導体と反応させるギ酸エステル(HCOOR2)としては、特に制限は無く、例えば、ギ酸メチル、ギ酸エチル、ギ酸プロピル、ギ酸イソプロピル、ギ酸ブチル、ギ酸イソブチル、ギ酸アミル、ギ酸イソアミル、ギ酸アリル、ギ酸ビニル、ギ酸ベンジル等から適宜選択して使用することができる。コスト及び反応性の観点から、ギ酸メチル、ギ酸エチル等の直鎖状のアルキルギ酸エステルが好ましく、ギ酸メチルがより好適である。
また、上記一般式(V)で表されるノルボルナントリカルボン酸誘導体を加水分解して、下記一般式(VI)で表されるノルボルナントリカルボン酸とする方法には、特に制限は無く、例えば、特許第2591492号、特開2008-31406号公報等に記載されている酸加水分解、アルカリ加水分解等を使用することができる。あるいは、酸成分又はアルカリ成分を加えること無しに、耐熱容器内で水分存在下、140℃以上の高温で加熱することによっても加水分解することができる。
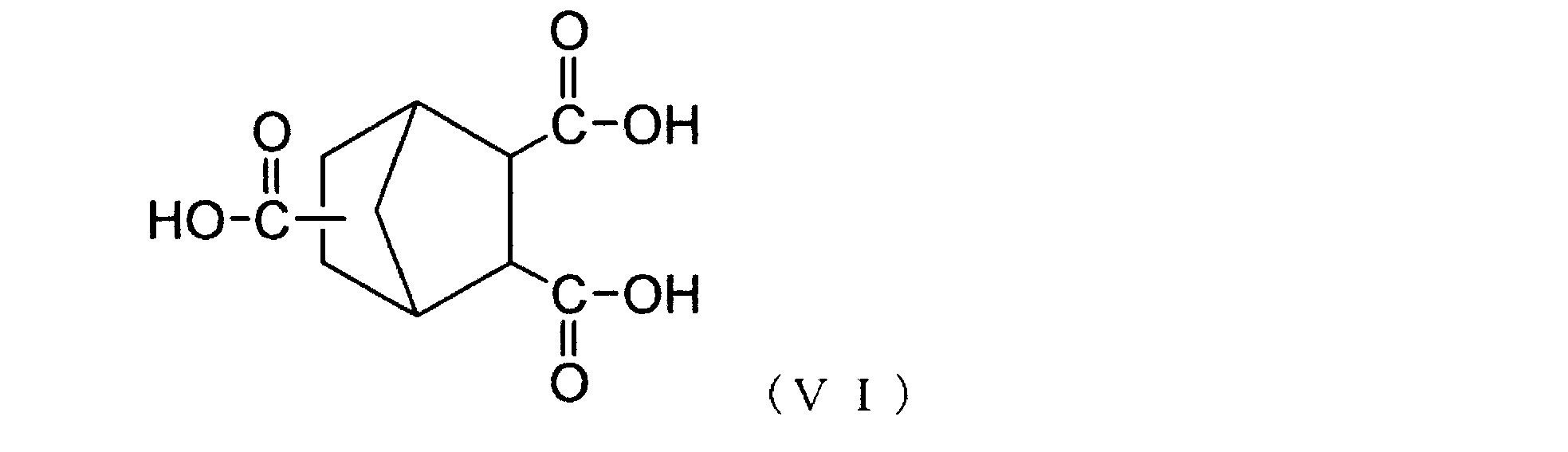
上記一般式(VI)で表されるノルボルナントリカルボン酸を脱水閉環して、下記一般式(II)で表されるノルボルナントリカルボン酸無水物を得る方法には、特に制限は無く、例えば、無水酢酸又は五酸化リン等の脱水剤を用いる化学閉環法、減圧化加熱又は溶媒存在下で加熱還流する熱閉環法等を用いることができる。製造コストや得られるポリアミドイミド中の残存イオン性不純物濃度等を考慮すると、熱閉環法が好ましい。

一般式(II)で表されるノルボルナントリカルボン酸無水物と、一般式(III)で表されるジイソシアネート化合物と、の反応には、極性溶媒を使用する。使用可能な極性溶媒は、原料として使用する化合物を溶解できればよく、特に限定されない。
ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、トリエチレングリコールジメチルエーテル、トリエチレングリコールジエチルエーテル等のエーテル系溶媒;
ジメチルスルホキシド、ジエチルスルホキシド、ジメチルスルホン、スルホラン等の含硫黄系溶媒;
γ-ブチロラクトン、酢酸セロソルブ等のエステル系溶媒;
シクロヘキサノン、メチルエチルケトン等のケトン系溶媒;等を使用することができる。
(合成例1)〔シクロペンタジエンの生成〕

(合成例3)〔ノルボルナントリカルボン酸メチルの合成〕

(合成例4)〔ノルボルナントリカルボン酸の合成〕
冷却管を取り付けた1リットル成す型フラスコに、合成例3で得られたノルボルナントリカルボン酸メチル 30g及びメタノール 200gを投入して均一溶液とした後、10%水酸化ナトリウム溶液 200gを仕込み、100℃のオイルバスに入れ、6時間加熱還流した。その後、反応液量が140gになるまでメタノールを留去し、これに36%塩酸 48mlを加え、pHを1としたところ、白色粉末が沈殿した。この白色粉末をろ過、水洗、乾燥し、ノルボルナンジカルボン酸 22gを得た。得られたノルボルナントリカルボン酸を、H1-NMRで分析した結果、ノルボルナン(トリシクロデカン)のメチレン及びメチン基のピークが1.1~3.0ppm付近に、カルボン酸に起因する水酸基のピークが12.4ppm付近に確認でき、その積分強度比が9.00/3.03(理論値:9/3)であった。
(合成例5)〔ノルボルナントリカルボン酸無水物の合成〕
撹拌機、温度計、窒素導入管及び油水分離器付き冷却管を備えた1リットルフラスコに、合成例4で得られたノルボルナントリカルボン酸 150g及び1,2,3,4-テトラヒドロナフタレン 450gを仕込み、215℃のオイルバスに入れ、3時間加熱還流した。その後、室温まで冷却すると白色粉末が析出したので、この白色粉末をろ過、水洗、乾燥し、ノルボルナントリカルボン酸無水物 135gを得た。得られたノルボルナントリカルボン酸無水物を、H1-NMRで分析した結果、ノルボルナン(トリシクロデカン)のメチレン及びメチン基のピークが1.1~3.0ppm付近に、カルボン酸に起因する水酸基のピークが12.4ppm付近に確認でき、その積分強度比が9.00/0.98(理論値:9/1)であった。
(実施例1)〔ノルボルナン骨格を有するポリアミドイミド(PAI-1)の合成〕
攪拌機、温度計、窒素導入管及び冷却管を備えた500mlフラスコに、合成例5で得られたノルボルナントリカルボン酸無水物 63.00g(0.300モル)、ヘキサメチレンジイソシアネート 51.41g(0.306モル)(トリカルボン酸無水物/ジイソシアネート(モル比)=1.00/1.02)及びN-メチルピロリドン 171.61gを仕込み、120℃まで昇温した後、5時間反応させて、数平均分子量が95,000のノルボルナン骨格を持つポリアミドイミド(PAI-1)を得た。
熱機械分析装置(セイコー電子(株)製、5200型 TMA)で測定した。
測定スパン:10mm
荷重:10g
昇温速度:5℃/min
雰囲気:空気
(2)熱分解開始温度(5%質量減少温度、Td5)
示差熱天秤(セイコー電子(株)製、5200型 TG-DTA)で測定した。
雰囲気:空気
(3)光線透過率
また、得られたノルボルナン骨格を有するポリアミドイミド(PAI-1)の各波長における光線透過率を、日本分光(株)製 V-570型UV/VISスペクトロフォトメーターで測定した。評価結果をまとめて表1に示す。
攪拌機、温度計、窒素導入管及び冷却管を備えた500mlフラスコに、合成例4で得られたノルボルナントリカルボン酸無水物 54.60g(0.260モル)、4,4’-シクロヘキシルメタンジイソシアネート 69.48g(0.265モル)(トリカルボン酸無水物/ジイソシアネート(モル比)=1.00/1.02)及びN-メチルピロリドン 186.12gを仕込み、120℃まで昇温した後、5時間反応させて、数平均分子量が102,000のノルボルナン骨格を持つポリアミドイミド(PAI-2)を得た。
攪拌機、温度計、窒素導入管及び冷却管を備えた500mlフラスコに、合成例4で得られたノルボルナントリカルボン酸無水物 58.80g(0.280モル)、イソホロンジイソシアネート 63.40g(0.286モル)(トリカルボン酸無水物/ジイソシアネート(モル比)=1.00/1.02)及びN-メチルピロリドン 183.30gを仕込み、120℃まで昇温した後、5時間反応させて、数平均分子量が94,000のノルボルナン骨格を持つポリアミドイミド(PAI-3)を得た。
攪拌機、温度計、窒素導入管及び冷却管を備えた500mlフラスコに、合成例4で得られたノルボルナントリカルボン酸無水物 53.46g(0.260モル)、4,4’-ジフェニルメタンジイソシアネート 66.30g(0.265モル)(トリカルボン酸無水物/ジイソシアネート(モル比)=1.00/1.02)及びN-メチルピロリドン 181.35gを仕込み、120℃まで昇温した後、5時間反応させて、数平均分子量が98,000のノルボルナン骨格を有するポリアミドイミド(PAI-4)を得た。
攪拌機、温度計、窒素導入管及び冷却管を備えた500mlフラスコに、トリメリット酸無水物 51.84g(0.270モル)、4,4’-ジフェニルメタンジイソシアネート 68.85g(0.275モル)(トリカルボン酸無水物/ジイソシアネート(モル比)=1.00/1.02)及びN-メチルピロリドン 181.04gを仕込み、120℃まで昇温した後、5時間反応させて、数平均分子量が110,000の芳香族ポリアミドイミド(PAI-5)を得た。
攪拌機、温度計、窒素導入管及び冷却管を備えた500mlフラスコに、トリメリット酸無水物 49.92g(0.260モル)、4,4’-シクロヘキシルメタンジイソシアネート69.48g(0.265モル)(トリカルボン酸無水物/ジイソシアネート(モル比)=1.00/1.02)及びN-メチルピロリドン 179.10gを仕込み、120℃まで昇温した後、5時間反応させて、数平均分子量が76,000のポリアミドイミド(PA-6)を得た。
まとめて表1に示す。

Claims (10)
- 下記一般式(I)で表されるノルボルナン骨格を有するポリアミドイミド。

- 下記一般式(II)で表されるノルボルナントリカルボン酸無水物と、ジイソシアネート化合物と、を極性溶媒中で反応させることを特徴とする、下記一般式(I)で表されるノルボルナン骨格を有するポリアミドイミドの製造方法。


- 前記ジイソシアネート化合物が、下記一般式(III)で表されるジイソシアネート化合物であることを特徴とする請求項2に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
OCN-X-NCO (III)
(但し、式中Xは、炭素数4~16の2価の脂肪族基、炭素数4~16の2価の脂環族基及び2価の芳香族基から選ばれる2価の有機基である。) - 前記一般式(II)で表されるノルボルナントリカルボン酸無水物が、下記(1)~(3)工程を含む方法で得られることを特徴とする請求項2又は3に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
(1)工程:下記一般式(IV)で表されるノルボルネンジカルボン酸誘導体と、

ギ酸エステル(HCOOR2)とを、
ルテニウム化合物と、コバルト化合物と、ハロゲン化物塩と、を含む触媒系の存在下で反応させて、下記一般式(V)で表されるノルボルナントリカルボン酸誘導体とする。

(2)工程:前記一般式(V)で表されるノルボルナントリカルボン酸誘導体のアルコキシカルボニル基を加水分解して、下記一般式(VI)で表されるノルボルナントリカルボン酸を得る。

- 前記ルテニウム化合物が、分子内にカルボニル配位子とハロゲン配位子とを合わせ持つルテニウム錯体である請求項4に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
- 前記ハロゲン化物塩が、四級アンモニウム塩である請求項4又は5に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
- 前記触媒系がさらに塩基性化合物を含む請求項4~6のいずれか一項に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
- 前記塩基性化合物が、三級アミン化合物である請求項7記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
- 前記触媒系がさらにフェノール化合物を含む請求項4~8のいずれか一項に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
- 前記触媒系がさらに有機ハロゲン化合物を含む請求項4~9のいずれか一項に記載のノルボルナン骨格を有するポリアミドイミドの製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201080065292.7A CN102791770B (zh) | 2010-03-29 | 2010-12-03 | 具有降冰片烷骨架的聚酰胺酰亚胺及其制造方法 |
| JP2012508024A JP5532123B2 (ja) | 2010-03-29 | 2010-12-03 | ノルボルナン骨格を有するポリアミドイミド及びその製造方法 |
| KR1020127027547A KR20120140682A (ko) | 2010-03-29 | 2010-12-03 | 노르보르난 골격을 갖는 폴리아미드이미드 및 그의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010074522 | 2010-03-29 | ||
| JP2010-074522 | 2010-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011121848A1 true WO2011121848A1 (ja) | 2011-10-06 |
Family
ID=44711621
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/071675 WO2011121848A1 (ja) | 2010-03-29 | 2010-12-03 | ノルボルナン骨格を有するポリアミドイミド及びその製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP5532123B2 (ja) |
| KR (1) | KR20120140682A (ja) |
| CN (1) | CN102791770B (ja) |
| TW (1) | TW201132678A (ja) |
| WO (1) | WO2011121848A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI746707B (zh) * | 2017-01-31 | 2021-11-21 | 日商日本化藥股份有限公司 | 反應性多元羧酸化合物、使用該化合物的活性能量線硬化型樹脂組成物、該組成物的硬化物及該硬化物的用途 |
| JP7236812B2 (ja) * | 2017-04-27 | 2023-03-10 | 日本化薬株式会社 | 反応性ポリカルボン酸化合物、それを用いた活性エネルギー線硬化型樹脂組成物、その硬化物及びその用途 |
| JP7236813B2 (ja) * | 2017-04-28 | 2023-03-10 | 日本化薬株式会社 | 反応性ポリカルボン酸化合物、それを用いた活性エネルギー線硬化型樹脂組成物、その硬化物及びその用途 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3413317A (en) * | 1966-04-25 | 1968-11-26 | Universal Oil Prod Co | Bicycloheptane tricarboxylic acids, anhydrides and alkyl esters thereof |
| JPS582333A (ja) * | 1981-06-30 | 1983-01-07 | Toutoku Toryo Kk | ポリアミドイミドの製造方法 |
| JP2000143802A (ja) * | 1998-11-17 | 2000-05-26 | Hitachi Chem Co Ltd | ポリアミドイミド系樹脂およびこれを用いた光学用素子 |
| WO2008072495A1 (ja) * | 2006-12-12 | 2008-06-19 | Toyo Boseki Kabushiki Kaisha | ポリアミドイミド樹脂、それから得られる無色透明フレキシブル金属張積層体および配線板 |
| JP2008163090A (ja) * | 2006-12-27 | 2008-07-17 | Mitsubishi Chemicals Corp | テトラカルボン酸二無水物およびその製造方法並びに重合物 |
| JP2008308550A (ja) * | 2007-06-13 | 2008-12-25 | Mitsui Chemicals Inc | 新規ポリアミド酸、ポリイミド並びにその用途 |
| WO2009101885A1 (ja) * | 2008-02-14 | 2009-08-20 | Kyowa Hakko Chemical Co., Ltd. | ポリイミド |
| JP2009292940A (ja) * | 2008-06-05 | 2009-12-17 | Nissan Chem Ind Ltd | ポリアミック酸およびポリイミドフィルム |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0582333A (ja) * | 1991-09-19 | 1993-04-02 | Hitachi Ltd | 核磁気共鳴診断装置 |
-
2010
- 2010-12-03 KR KR1020127027547A patent/KR20120140682A/ko active IP Right Grant
- 2010-12-03 CN CN201080065292.7A patent/CN102791770B/zh not_active Expired - Fee Related
- 2010-12-03 WO PCT/JP2010/071675 patent/WO2011121848A1/ja active Application Filing
- 2010-12-03 JP JP2012508024A patent/JP5532123B2/ja not_active Expired - Fee Related
- 2010-12-14 TW TW099143758A patent/TW201132678A/zh unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3413317A (en) * | 1966-04-25 | 1968-11-26 | Universal Oil Prod Co | Bicycloheptane tricarboxylic acids, anhydrides and alkyl esters thereof |
| JPS582333A (ja) * | 1981-06-30 | 1983-01-07 | Toutoku Toryo Kk | ポリアミドイミドの製造方法 |
| JP2000143802A (ja) * | 1998-11-17 | 2000-05-26 | Hitachi Chem Co Ltd | ポリアミドイミド系樹脂およびこれを用いた光学用素子 |
| WO2008072495A1 (ja) * | 2006-12-12 | 2008-06-19 | Toyo Boseki Kabushiki Kaisha | ポリアミドイミド樹脂、それから得られる無色透明フレキシブル金属張積層体および配線板 |
| JP2008163090A (ja) * | 2006-12-27 | 2008-07-17 | Mitsubishi Chemicals Corp | テトラカルボン酸二無水物およびその製造方法並びに重合物 |
| JP2008308550A (ja) * | 2007-06-13 | 2008-12-25 | Mitsui Chemicals Inc | 新規ポリアミド酸、ポリイミド並びにその用途 |
| WO2009101885A1 (ja) * | 2008-02-14 | 2009-08-20 | Kyowa Hakko Chemical Co., Ltd. | ポリイミド |
| JP2009292940A (ja) * | 2008-06-05 | 2009-12-17 | Nissan Chem Ind Ltd | ポリアミック酸およびポリイミドフィルム |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102791770B (zh) | 2014-05-07 |
| JP5532123B2 (ja) | 2014-06-25 |
| JPWO2011121848A1 (ja) | 2013-07-04 |
| TW201132678A (en) | 2011-10-01 |
| CN102791770A (zh) | 2012-11-21 |
| KR20120140682A (ko) | 2012-12-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7795370B2 (en) | Tetracarboxylic acid compound, polyimide thereof, and production method thereof | |
| WO2012035874A1 (ja) | ノルボルナン骨格を有するポリエステル及びその製造方法 | |
| JP5532123B2 (ja) | ノルボルナン骨格を有するポリアミドイミド及びその製造方法 | |
| US11525037B2 (en) | Tetracarboxylic dianhydride, polyimide precursor resin and solution thereof, and polyimide and solution thereof | |
| JP5586030B2 (ja) | トリシクロデカンモノメタノールモノカルボン酸誘導体の製造方法 | |
| JP5590117B2 (ja) | ノルボルナン骨格を有するポリアミドの製造方法及びそれにより得られるノルボルナン骨格を有するポリアミド | |
| WO2011121850A1 (ja) | ナジイミド骨格を有するポリアミドイミド及びその製造方法 | |
| JP5790386B2 (ja) | ノルボルナン骨格含有ポリアミドの製造方法 | |
| JP5811753B2 (ja) | ノルボルナン骨格含有ポリアミドの製造方法 | |
| JP5556301B2 (ja) | ノルボルナン骨格を有するポリアミド及びその製造方法 | |
| JP5796416B2 (ja) | ノルボルナン骨格を含有するポリアミドイミドの製造方法 | |
| US8653301B2 (en) | Tricyclodecane monomethanol monocarboxylic acid and derivatives thereof | |
| WO2012035875A1 (ja) | 脂環式ポリエステル及びその製造方法 | |
| JP2012240981A (ja) | ビスナジイミドのジヒドロジエステル体 | |
| WO2011121847A1 (ja) | ナジイミド骨格を有するポリアミドイミドの製造方法 | |
| JP2013079350A (ja) | エキソ体比率の高いノルボルナン骨格含有ポリアミド | |
| US20210122724A1 (en) | Tetracarboxylic dianhydride, carbonyl compound, polyimide precursor resin, and polyimide | |
| JP2012240980A (ja) | ビスナジイミドのジヒドロジエステル体 | |
| WO2012141313A1 (ja) | ノルボルナンジカルボン酸エステルの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080065292.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10849019 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012508024 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20127027547 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10849019 Country of ref document: EP Kind code of ref document: A1 |