WO2011102506A1 - 経口用徐放性固形製剤 - Google Patents
経口用徐放性固形製剤 Download PDFInfo
- Publication number
- WO2011102506A1 WO2011102506A1 PCT/JP2011/053644 JP2011053644W WO2011102506A1 WO 2011102506 A1 WO2011102506 A1 WO 2011102506A1 JP 2011053644 W JP2011053644 W JP 2011053644W WO 2011102506 A1 WO2011102506 A1 WO 2011102506A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- preparation
- drug
- sustained
- preparation according
- weight
- Prior art date
Links
Images










Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2031—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
Definitions
- the present invention relates to a sustained-release solid preparation that reliably exhibits the main pharmacological effect by oral administration once or twice a day.
- Sustained-release preparations for adjusting the blood concentration of drugs are useful in terms of separation of main pharmacological effects and side effects, improvement in medication compliance such as reduction in the number of administrations due to improved drug efficacy, and medical economics. high. For this reason, several techniques relating to sustained-release preparations have been reported. On the other hand, since the chemical properties of compounds that exhibit the main pharmacological action are diverse, several sustained-release techniques for adapting to the various chemical properties of these compounds have been reported, but still insufficient (For example, refer to Patent Documents 1 and 2).
- An acidic drug refers to an acidic compound in which a free form (a free form that does not form a salt with an acidic group of the drug, such as an addition salt of an alkali or an amine) shows acidity.
- a free form a free form that does not form a salt with an acidic group of the drug, such as an addition salt of an alkali or an amine
- the solubility in the upper part of the gastrointestinal tract such as the stomach is low, and in the case of a salt of an acidic compound (such as an alkali or amine addition salt), the problem is that it becomes a free acid with low solubility in an acidic solution. It is as.
- a basic drug refers to a basic compound in which a free form (a free form such as an acid addition salt that does not form a salt with the basic group of the drug) shows basicity, and dissolves well in a strongly acidic aqueous solution.
- a neutral aqueous solution such as a neutral buffer decreases the solubility. That is, when a basic drug is administered orally, it shows good solubility in an acidic stomach, but the solubility in the lower part of the digestive tract, such as the large intestine, which is neutral and has little water, is greatly reduced and the absorption rate is reduced. Concerned.
- sustained-release preparations for oral administration of basic drugs
- the preparations collapse due to coexistence with food in the acidic environment of the upper gastrointestinal tract, which exhibits high water solubility, and mechanical stress due to gastrointestinal motility, etc.
- the overdose of the drug that occurs at the time is a problem.
- the formulation strength is increased by increasing the sustained release base to avoid overdose of the drug
- To maintain the absorption remains as a problem of sustained-release preparations.
- we are satisfied with the sustained release of basic drugs that can avoid excessive drug release in acidic environments such as the upper gastrointestinal tract and can simultaneously achieve sustained elution in the lower gastrointestinal tract, which is a neutral environment. There is nothing you can do.
- the inventors of the present invention contain a pharmacologically active drug, carboxyvinyl polymer, povidone, and a swelling agent, and overrelease the drug in an acidic environment (
- the present invention was completed by finding a sustained-release solid preparation that avoids dose dumping and has improved dissolution in neutrality. That is, the present invention provides the following (1) to (36).
- a sustained-release solid preparation for oral administration comprising the above (A) to (C).
- the sustained-release solid preparation according to (6), wherein the sugar alcohol is mannitol, xylitol or erythritol.
- the sustained-release solid preparation according to (6), wherein the sugar alcohol is xylitol.
- the sustained-release solid preparation according to any one of (5) to (8), wherein the hydration rate of the solid preparation in an acidic aqueous solution is 60% or more.
- the pharmacologically active drug is the following ( ⁇ ) -1- (carbazol-4-yloxy) -3-[[2- (o-methoxyphenoxy) ethyl] amino] -2-propanol ; N 1- (5-chloropyridin-2-yl) -N 2 -((1S, 2R, 4S) -4-[(dimethylamino) carbonyl] -2- ⁇ [(5-methyl-4,5,6 , 7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ cyclohexyl) ethanediamide; and N 1- (5-chloropyridin-2-yl) -N 2 -[(1S, 2R , 4S) -2- ⁇ [(5-Methyl-4,5,6,7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl]
- the content of (A) a pharmacologically active drug in the solid preparation is in the range of 0.1% by weight to 60% by weight, according to any one of (1) to (20) Sustained release solid preparation.
- (5) The preparation according to (24), wherein the component (D) in the preparation is carmellose sodium.
- (26) The preparation according to (24) or (25), wherein the content of component (A) in the preparation is 5 to 35% by weight.
- the component (A) is ( ⁇ ) -1- (carbazol-4-yloxy) -3-[[2- (o-methoxyphenoxy) ethyl] amino] -2-propanol, N 1- (5-chloropyridin-2-yl) -N 2 -((1S, 2R, 4S) -4-[(dimethylamino) carbonyl] -2- ⁇ [(5-methyl-4,5,6 , 7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ cyclohexyl) ethanediamide, and N 1- (5-chloropyridin-2-yl) -N 2 -[(1S, 2R, 4S) -2- ⁇ [(5-methyl-4,5,6,7-tetrahydrothiazolo [5,4 -C] pyridin-2-yl) carbonyl] amino ⁇ -4-([1,3,4]
- a sustained-release pharmaceutical composition for oral administration containing a pharmacologically active drug can be provided. Therefore, for example, a sustained oral sustained-release solid preparation containing an activated blood coagulation factor X (FXa) inhibitor compound (1) as a medicinal ingredient is provided.
- the sustained-release pharmaceutical composition of the present invention has a good tablet strength that prevents excessive release while having a good hydration rate and swelling rate in an acidic solution, and a good dissolution property in a neutral solution. Therefore, the effect of maintaining the continuous elution of the contained pharmacologically active drug from the duodenum and small intestine to the lower digestive tract can be obtained.
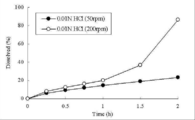
- the tablet of Comparative Example 3 shown in Table 1 was tested at an elution test (paddle method, 0.01 N hydrochloric acid, 900 mL) in an acidic solution at a paddle rotation speed of 200 rpm and 50 rpm, and 200 rpm and 50 rpm until 2 hours later. It is a figure which shows the time-dependent transition of the drug elution rate.
- elution test 0.01 N hydrochloric acid, 900 mL
- FIG. 1 shows a time-dependent transition of a drug elution rate.
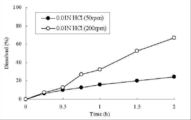
- FIG. About the tablet of the formulation 2a described in Table 4, it tested at the paddle rotation speed 200rpm and 50rpm in the dissolution test (paddle method, 0.01N hydrochloric acid, 900mL) in an acidic solution, and 200rpm and 50rpm of 2 hours later
- the term “acidic solution” means an acidic dissolution test solution used for evaluating dissolution at the upper digestive tract such as the stomach.
- the dissolution test first solution described in the Japanese Pharmacopoeia, US USP 0.1 N hydrochloric acid, 0.01 N hydrochloric acid, Simulated Gastric Fluid without Enzyme and the like described in the pharmacopoeia can be mentioned, but the acidic dissolution test solution is not limited to these.
- the “neutral solution” in the present specification means a neutral dissolution test solution used for evaluating the dissolution property of a drug in the small intestine, the large intestine, etc.
- the dissolution test second solution described in the Japanese Pharmacopoeia Examples include phosphate buffer pH 6.8, USP Phosphate Buffer (pH 6.8) described in US Pharmacopoeia, Simulated Interstitial Fluid without Enzyme, Phosphate Buffer Solution pH 6.8 described in European Pharmacopoeia, etc.
- the “soluble solution” is not limited to a pH 6.8 dissolution test solution.
- the above dissolution test solution is prepared by the method described in the pharmacopoeia of each country.
- the pH of these dissolution test solutions is preferably within ⁇ 0.05 of the pH specified for each dissolution test solution when the dissolution test solution is a buffer solution.
- the paddle method can include a method of performing a dissolution test for 2 hours at 50 rpm and 200 rpm.
- the pharmacologically active drug in the preparation is a basic drug
- the preparation is caused by mechanical stress due to coexistence with food in the acidic environment of the upper gastrointestinal tract showing high water solubility, gastrointestinal motility, etc.
- dose dumping of the drug that occurs due to the collapse of the drug.
- the average dissolution rate of a pharmacologically active drug in an acidic dissolution test solution is to maintain the formulation strength and keep the dissolution rate within a certain range even at 200 rotations per minute of the paddle method and / or 50 rotations per minute of the paddle method. Is preferred.
- the average dissolution rate of the pharmacologically active drug in the dissolution test solution after 2 hours is preferably 50% or less at any of 200 rotations per minute and / or 50 rotations per minute by the paddle method; More preferably 30% or less.
- the difference in the average dissolution rate of pharmacologically active drugs in the dissolution test solution (200 rpm per paddle method—50 rpm per paddle method) when the dissolution test method was performed for 2 hours was 15% or less.
- the ratio of the average dissolution rate of the pharmacologically active drug in the dissolution test solution after 2 hours is preferably 2.0 or less; 1.5 or less Is more preferable; 1.3 or less is particularly preferable.
- the paddle method using a neutral eluate for evaluating the dissolution property in the neutral region of the sustained release solid preparation of the present invention is, for example, 37 ⁇ 0 in phosphate buffer (pH 6.8; 900 mL).
- a method of performing a dissolution test at 50 ° C. per minute at 5 ° C. can be mentioned.
- the average dissolution rate of a pharmacologically active drug in the dissolution test solution is preferably one that shows a continuous dissolution rate even 12 hours after the start of the dissolution test. Further, those that show a continuous dissolution rate even after 12 hours from the start of the dissolution test and show an average dissolution rate of 20% or more are preferred; those that show a dissolution rate of 30% or more are more preferred.
- USP Apparatus 3 which is a dissolution test method under conditions close to the environment in the human digestive tract, may be used.
- the drug concentration in the solution can be quantified using a UV method or the like, and the average dissolution rate and dissolution time of a pharmacologically active drug in the dissolution test solution can be calculated.
- the “average dissolution rate” may be determined by measuring the dissolution rates of at least 2, preferably 6, and more preferably 12 for one kind of solid preparation, and calculating the average value thereof.
- the dissolution of the pharmacologically active drug in the sustained-release solid preparation of the present invention can be confirmed using an in vivo animal test.
- the in vivo animal test include in vivo absorbability evaluation using a dog.
- orally administered preparations are said to remain in the large intestine for a long time after passing through the stomach and small intestine. Therefore, for sustained-release preparations with a long elution time, it is very important that drug release is sustained in the large intestine that stays for a long time.
- canine colon absorbability evaluation in which the preparation is directly administered into the canine large intestine can be mentioned.
- the absorbability in the large intestine of the dog is confirmed by measuring the blood concentration after administration, and evaluated by the relative bioavailability (BA) of each tablet as a ratio to the oral administration of a pharmacologically active drug. be able to.
- BA relative bioavailability
- a neutral compound of a pharmacologically active drug means a compound that does not have a group that ionizes and dissociates even in an acidic state or a basic state in the molecule, and examples of the acidic compound include carboxy group, phenol It means a drug having an acidic group typified by a functional hydroxyl group, phosphoric acid group, sulfonic acid, tetrazolyl group and the like.
- the basic drug means a drug having a basic nitrogen atom represented by amino group, piperidinyl group, piperazinyl group and the like in the molecule.
- a basic drug is particularly suitable.
- Basic drugs have a physicochemical property in which the solubility in the neutral state (7.5> pH> 5) in the small and large intestines is lower than the solubility in the acidic state (pH ⁇ 2) such as the stomach.
- the “basic drug” in the specification of the present application refers to a drug whose solubility in the neutral state is lower than the solubility in the acidic state, and the ratio of the decrease in solubility in the neutral state is as follows: The following ranges can be mentioned.
- (Neutral state solubility) / (Acid state solubility) preferably ranges from 0.00001 to 0.6;
- (Neutral state solubility) / (Acid state solubility) is more preferably in the range of 0.001 to 0.5;
- the range of (solubility in neutral state) / (solubility in acidic state) is more preferably in the range of 0.01 to 0.1, but is not limited to these ranges.
- the solubility is more preferably in the range of 10 to 500 ⁇ g / ml.
- the absolute value of the solubility is preferably a drug whose minimum solubility in a neutral state (in the range of 7.5> pH> 5) is reduced to 3 mg / ml or less; more preferably a drug which decreases to 1 mg / ml or less; More preferred is a drug that lowers to 0.5 mg / ml or less.
- pharmacologically active drugs include anticoagulants.
- an activated blood coagulation factor X (FXa) inhibitor having a basic or weakly basic substituent is preferable.
- FXa inhibitor include the following (a) to (l): Can be mentioned.
- R 1 represents an N, N-dimethylcarbamoyl group or a [1,3,4] oxadiazol-2-yl group.
- the compound represented by the formula [hereinafter sometimes abbreviated as compound (1)] is more preferred.
- Compound (1) may be a free form (free base), a pharmacologically acceptable salt thereof, or a hydrate thereof.
- Examples of the salt of the compound represented by the formula (1) include hydrochloride, sulfate, hydrobromide, hydroiodide, phosphate, nitrate, benzoate, methanesulfonate, 2-hydroxyethane. Sulfonate, p-toluenesulfonate, acetate, propanoate, oxalate, malonate, succinate, glutarate, adipate, tartrate, maleate, fumarate, apple Acid salt, mandelate salt and the like.
- maleic acid, hydrochloride, methanesulfonate, and p-toluenesulfonate are preferable, and maleic acid and p-toluenesulfonate are particularly preferable.
- N 1- 5-chloropyridin-2-yl) -N 2 -[(1S, 2R, 4S) -2- ⁇ [(5-methyl -4,5,6,7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ -4-([1,3,4] oxadiazol-2-yl) cyclohexyl] Ethanediamide monomaleate;
- N 1- (5-chloropyridin-2-yl) -N 2 -((1S, 2R, 4S) -4-[(dimethylamino) carbonyl] -2- ⁇ [(5-methyl-4,5,6 , 7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ cyclohexyl) ethanediamide;
- N 1- (5-chloropyridin-2-yl) -N 2 -[(1S,
- the free base (free form) of compound (1) means, for example, the above-mentioned salt (acid addition salt) and / or acid addition salt “acid” and water forming hydrate. This means a compound excluding “water” in a Japanese product.
- the free base (free form) of compound (1a) and compound (1b) means the following formula (1a-1) and formula (1b-1)
- These compounds (1) can be produced by the method described in the above documents (WO2003-000657 pamphlet; WO2003-000680 pamphlet; WO2003-016302 pamphlet; WO2004-058715 pamphlet) or a method analogous thereto.
- the content of “(A) pharmacologically active drug” in “solid preparation” in the present specification is preferably 0.1 to 60% by weight as “% (A) pharmacologically active drug” by weight; 1 to 50% by weight is more preferable; 5 to 35% by weight is more preferable, but it is not limited to this range.
- “A to B wt%” indicating the content of components in the present specification means “a range of A wt% to B wt%” unless otherwise specified. .
- (B) carboxyvinyl polymer in the present specification, commercially available carbopol (Noveon / CBC) or the like may be used, and various grades having different viscosities are available.
- a preferred carboxyvinyl polymer is Carbopol 974PNF.
- the content of the carboxyvinyl polymer in the “solid preparation” is preferably 1 to 50% by weight; more preferably 5 to 30% by weight; and even more preferably 10 to 30% by weight.
- (C) povidone (polyvinylpyrrolidone) is a nonionic water-soluble polymer, which means a linear polymer of 1-vinyl-2-pyrrolidone, and 1-vinyl-2- It does not contain crospovidone which is a cross-linked polymer of pyrrolidone.
- Povidone can be obtained from various grades of K value (viscosity characteristic value correlating with molecular weight) from BASF Japan and the like, and preferred examples include commercially available Kollidon 30 (BASF Japan).
- the content of povidone in the “solid preparation” is preferably 1 to 70% by weight; more preferably 6 to 70% by weight; and even more preferably 10 to 70% by weight.
- (1) and (2) are preferable as the contents of “(B) carboxyvinyl polymer” and “(C) povidone” in the “solid preparation” of the present specification, (1) Povidone content is 10 to 70% by weight when the carboxyvinyl polymer content is 15 to 25% by weight; and (2) Povidone when the carboxyvinyl polymer content is 10 to less than 15% by weight.
- the content of is 20 to 70% by weight.
- water-soluble excipient in the present specification, fructose, purified sucrose, sucrose, purified sucrose spherical granules, lactose, anhydrous lactose, sucrose / starch spheroid granules, semi-digested starch, glucose, glucose hydrate, powder
- sugars such as sugar, pullulan, ⁇ -cyclodextrin, mannitol, xylitol, and erythritol.
- sugar alcohols such as mannitol, xylitol, erythritol are more preferred; xylitol is particularly preferred.
- the content of the water-soluble excipient in the “solid preparation” is preferably 0 to 80% by weight; more preferably 0 to 60% by weight.
- xanthan gum sodium carboxymethyl starch, guar gum, alginic acid, sodium alginate, chitosan, agar, starch, dextrin, gum arabic, gelatin, carmellose sodium and the like; carmellose sodium, xanthan gum, And carboxymethyl starch sodium is preferred; carmellose sodium is preferred.
- carmellose sodium for example, commercially available products such as Sunrose (trade name: Nippon Paper Chemicals) can be used, and those having various viscosities and etherification degrees can be obtained.
- the content of the swelling agent in the “solid preparation” is preferably 1 to 40% by weight; more preferably 5 to 15% by weight; and even more preferably 7 to 15% by weight.
- the effect of the present invention is affected.
- a disintegrating agent, a binder, a fluidizing agent, a lubricant, a coloring agent, a brightening agent and the like may be blended.
- Disintegrants include adipic acid, alginic acid, pregelatinized starch, sodium carboxymethyl starch, hydrous silicon dioxide, calcium citrate, light anhydrous silicic acid, synthetic aluminum silicate, wheat starch, rice starch, calcium stearate, corn starch, tragacanth Powder, potato starch, hydroxypropyl starch, partially pregelatinized starch, monosodium fumarate, anhydrous citric acid, calcium dihydrogen phosphate, croscarmellose sodium, crospovidone, carmellose, carmellose calcium and the like.
- binders include candy powder, gum arabic, gum arabic powder, sodium alginate, propylene glycol alginate, hydrolyzed gelatin powder, hydrolyzed starch, light anhydrous silicic acid, fructose, carboxymethyl ethyl cellulose, hydrous silicon dioxide, agar powder, Light anhydrous silicic acid, light anhydrous silicic acid-containing hydroxypropyl cellulose, crystalline cellulose, synthetic aluminum silicate, copolydon, wheat flour, wheat starch, rice flour, rice starch, vinyl acetate resin, cellulose acetate phthalate, dioctyl sodium sulfosuccinate, Dihydroxyaluminum aminoacetate, potassium sodium tartrate, normal water, sucrose fatty acid ester, purified gelatin, purified sucrose, gelatin, D-sorbitol, dextrin, starch, corn Pung, tragacanth, tragacanth powder, lactose, concentrated glycerin, sucrose,
- Fluidizing agents include hydrous silicon dioxide, light anhydrous silicic acid, crystalline cellulose, synthetic aluminum silicate, titanium oxide, stearic acid, calcium stearate, magnesium stearate, tricalcium phosphate, talc, corn starch, magnesium metasilicate aluminate Etc.
- Lubricants include cocoa butter, carnauba wax, hydrous silicon dioxide, dry aluminum hydroxide gel, glycerin fatty acid ester, magnesium silicate, light anhydrous silicic acid, crystalline cellulose, hydrogenated oil, synthetic aluminum silicate, white beeswax, magnesium oxide , Sodium potassium tartrate, sucrose fatty acid ester, stearic acid, calcium stearate, magnesium stearate, stearyl alcohol, polyoxyl 40 stearate, hardened soybean oil, gelatin, talc, magnesium carbonate, precipitated calcium carbonate, corn starch, potato starch , Fumaric acid, sodium stearyl fumarate, polyethylene glycol 600, polyethylene glycol 4000, polyethylene glycol 6000, beeswax, Magnesium Takei aluminometasilicate, sodium laurate, and magnesium sulfate.
- Coloring agents include yellow iron sesquioxide, iron sesquioxide, titanium oxide, orange essence, brown iron oxide, ⁇ -carotene, black iron oxide, edible blue No. 1, edible blue No. 2, edible red No. 2, edible red 3 No., Edible Red No. 102, Edible Yellow No. 4, Edible Yellow No. 5 and the like.
- Examples thereof include medium gold foil, white shellac, paraffin, povidone (polyvinylpyrrolidone), polyethylene glycol 1500, polyethylene glycol 4000, polyethylene glycol 6000, beeswax, glyceryl monostearate, and rosin.
- the sustained-release solid preparation of the present invention is not particularly limited as long as it is a solid preparation that can be administered orally, but granules and tablets are preferred.
- a matrix tablet is preferable.
- the sustained-release solid preparation of the present invention comprises (A) a pharmacologically active drug, (B) carboxyvinyl polymer, and (C) povidone, and the above-mentioned water-soluble excipient and swelling agent, If necessary, disintegrating agents, binders, fluidizers, lubricants, colorants, brighteners, etc. are blended, and manufactured by, for example, the method for producing solid preparations described in the General Formulation of the Japanese Pharmacopoeia be able to.
- an additive may be added to the fluidized bed.
- a granulator such as a granulator
- granulation such as wet granulation can be performed to obtain granulated granules.
- the granulated granules can be made into tablets for tableting by adding additives such as lubricants and mixing, and can be tableted by tableting.
- the resulting tablets can be coated with a coating agent as necessary. It can also be used as a film-coated tablet.
- the dosage form of the sustained-release solid preparation of the present invention is a tablet
- (A) a pharmacologically active drug (B) a carboxyvinyl polymer, and (C) povidone, a water-soluble excipient, a swelling agent, And (B) carboxyvinyl polymer; (C) a combination of povidone, a water-soluble excipient; and a swelling agent, and a pharmaceutical product.
- the shape of the tablet is not particularly limited, but a lens shape, a disk shape, a circle shape, an ellipse shape, and a polygonal shape such as a triangle or a rhombus are preferable.
- the content ratio of the pharmacologically active drug in the pharmaceutical composition of the present invention is preferably in the range of 0.1% by weight to 60% by weight in terms of free form of the pharmacologically active drug; % By weight is more preferred; 5 to 35% by weight is even more preferred.
- the dosage form of the pharmaceutical composition of the present invention is a tablet
- the content of the pharmacologically active drug contained in each tablet is in the range of 0.5 to 500 mg in terms of free form of the pharmacologically active drug. 1-100 mg is preferred; 5-75 mg is more preferred; 15-60 mg is even more preferred.
- each tablet is not hydrated after standing for 2 hours at 37 ⁇ 0.5 ° C. in 0.01 N hydrochloric acid. Obtain the volume of the part and use the following formula 1
- the hydration rate was calculated by the formula represented by:
- the hydration rate is preferably 60% or more; more preferably 90% or more.
- the swelling rate is preferably 150% or more; more preferably 180% or more; and particularly preferably 200% or more.
- the sustained-release solid preparation of the present invention contains (A) a pharmacologically active drug, (B) carboxyvinyl polymer, (C) povidone, and (D) carmellose sodium, xanthan gum or carboxymethyl starch sodium. .
- the “pharmacologically active drug” that is the component (A) used in the preparation of the present invention is not particularly limited, and the drug may be a prodrug that is converted into a pharmacologically active drug in the body. Good.
- Examples of the component (A) of the present invention include those described above as preferred, but are not limited thereto.
- the content of the component (A) in the preparation of the present invention is preferably 0.1 to 60% by weight, more preferably 1 to 50% by weight, still more preferably 5 to 35% by weight, but is not limited to this range. It is not something.
- carbopol 974PNF carbopol 974PNF
- the content of the component (B) in the preparation of the present invention is preferably 1 to 50% by weight, more preferably 5 to 30% by weight, and even more preferably 10 to 30% by weight.
- Podone which is the component (C) used in the preparation of the present invention, the above-mentioned thing is meant, and commercially available Kollidon 30 (BASF Japan) is preferable.
- the content of the component (C) in the preparation of the present invention is preferably 10 to 70% by weight.
- a content rate of (B) component and (C) component in the formulation of this invention following (1) and (2) are preferable.
- the content of component (C) in the case of less than 20 is 70 to 70% by weight.
- the component (D) used in the preparation of the present invention is carmellose sodium, xanthan gum or carboxymethyl starch sodium, preferably carmellose sodium.
- the content of component (D) in the preparation of the present invention is preferably 5 to 15% by weight; more preferably 7 to 15% by weight.
- the preparation of the present invention preferably contains a sugar alcohol in addition to (A) to (D).
- Sugar alcohols include those described above, with mannitol, xylitol or erythritol being preferred; xylitol being more preferred.
- the content of sugar alcohol in the preparation of the present invention is preferably 10 to 40% by weight.
- additives such as the above-mentioned binders, fluidizing agents, lubricants, coloring agents, brightening agents and the like may be blended within the range not affecting the effects of the present invention. .
- the dosage form of the preparation of the present invention is not particularly limited as long as it can be administered orally, but tablets are preferred.
- a matrix tablet is preferable.
- the preparation of the present invention can be obtained by tableting after mixing (A) to (D), or by tableting after mixing (A) to (D) and granulating. Mixing, granulation, and tableting may be performed using methods well known in the art.
- tableting is performed after mixing (A) to (D) and sugar alcohol, or granulation is performed by mixing (A) to (D) and sugar alcohol. And then obtained by tableting.
- the shape of the tablet is not particularly limited, but a lens shape, a disk shape, a circular shape, an elliptical shape, and a polygonal shape such as a triangular shape and a rhombus shape are preferable.
- the preparation of the present invention may be subjected to film coating using a coating agent as necessary. Mixing, granulation, tableting, and coating may be performed using methods well known in the art.
- additives When other additives are blended in the preparation of the present invention, they may be blended in any of the mixing step, granulating step, tableting step or coating step.
- the preparation of the present invention thus obtained has a good tablet strength that prevents excessive release while having a good hydration rate and swelling rate in an acidic solution, as described in the above-mentioned effects of the invention. From the duodenum and small intestine to the lower gastrointestinal tract, the effect of maintaining the continuous dissolution of the contained pharmacologically active drug can be obtained.
- the hydration rate and swelling rate of the preparation of the present invention can be calculated by the above formulas 1 and 2.
- the hydration rate is more preferably 90% or more, and the swelling rate Is preferably 150% or more; more preferably 180% or more; and particularly preferably 200% or more.
- the dissolution test in an acidic solution or a neutral solution was performed as follows. (Dissolution test in acidic solution) The dissolution test was carried out at 37 ⁇ 0.5 ° C. in 0.01 N hydrochloric acid (900 mL) at 50 and 200 rpm for the paddle method, and the average drug dissolution rate in the eluate was calculated over time.
- the ratio of the rates was calculated.
- the hydration rate and swelling rate of the tablets were measured as follows. (Evaluation of tablet hydration rate) As a method for evaluating the hydration rate, each tablet was allowed to stand at 37 ⁇ 0.5 ° C. in 0.01 N hydrochloric acid for 2 hours and then calculated using the above formula 1.
- Example 1 Manufacture of tablets
- Table 1 Each component shown in Table 1 was mixed using an agate mortar, and 200 mg of this mixed powder was tableted [tablet diameter: 8.0 mm (flat tablet)] with a single tableting machine (N-30E, Okada Seiko). And used as a sample.
- Example 2 About each prescription shown in Table 4, each component was mixed using an agate mortar, and 200 mg of this mixed powder was tableted using a single tableting machine (N-30E, Okada Seiko) to give a sample [ Tablet diameter: 8.0 mm ⁇ (6.5R)].
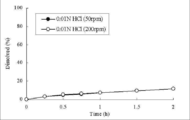
- the obtained sample was subjected to an elution test in an acidic solution, and the results are shown in FIGS.
- Example 3 The ingredients shown in Table 6 were mixed using an agate mortar, and 200 mg of this mixed powder was tableted with a single tableting machine (N-30E, Okada Seiko) to give a sample [tablet diameter: 8.0 mm ⁇ ( 6.5R)].
- FIG. 4, FIG. 8 and Table 7 show the dissolution test results in an acidic solution for Formulation 1 with a formulation ratio of carboxyvinyl polymer of 25% and Formulation 3 with 15%.
- Example 4 For the formulations shown in Table 8, each component was mixed using an agate mortar, and tableted with a single tableting machine (N-30E) to produce tablets (tablet diameter: 10.0 mm ⁇ (8R)).
- Table 9 and FIGS. 9 to 12 show the results of dissolution test in acidic solution and neutral solution, and the measurement results of hydration rate and swelling rate of each formulation.
- Example 5 About the prescription 4 and the prescription 5 shown in Table 8, the large intestine absorbability in the fasting dog was evaluated.
- the formulation shown in Table 8 was reduced to about 2/3, and evaluation was performed using a formulation (formulation 4 ′ and formulation 5 ′) having a compound (1a-1) content of 20 mg and a total amount of 240 mg.
- one tablet soaked in 0.01 N hydrochloric acid for 2 hours is set in an endoscope for animals (Olympus), and about 30 cm from the anus while observing the inside of the canine large intestine with an endoscope. It was administered to the site. Before administration, the amount of drug partly dissolved in 0.01N hydrochloric acid was measured from the tablet, and the plasma drug concentration was converted so that the dose was 20 mg / head as compound (1a-1). did. The transition of plasma drug concentration at this time is shown in FIG.
- Example 6 Compound (1b), carboxyvinyl polymer, carmellose sodium, povidone, polyethylene glycol 6000, xylitol, and sodium stearyl fumarate were placed in a mortar so as to have a weight ratio shown in Table 10, and mixed for 3 minutes to obtain a mixed powder. A predetermined amount of one tablet shown in Table 10 was weighed out from this mixed powder, and tableted using a single tableting machine (NE-30, Okada Seiko) () shape: 10 mm ⁇ , flat type).
- a single tableting machine NE-30, Okada Seiko
- Table 11 shows the dissolution test results in acidic aqueous solutions for Formulation 8, Comparative Example 4 and Comparative Example 5.
- Example 7 Since the above-mentioned formulation 5 is not easily affected by the paddle rotation number in an acidic solution, exhibits a continuous dissolution property in a neutral solution, and exhibits a good swelling rate and hydration rate, Table 13 The prescription preparation shown in FIG. That is, the drug (1a), carboxyvinyl polymer, xylitol, povidone (part), carmellose sodium, and other additives were added to the fluidized bed granulator at the ratio shown in Table 13 and mixed, and then the povidone solution (part) Wet granulation was carried out by spraying the remainder excluding) as a binder solution.
- the obtained granulated product was dried to obtain granulated granules, and then sodium stearyl fumarate was added thereto and mixed using a V-type mixer to obtain granules for tableting.
- This was tableted with a rotary tableting machine ( ⁇ type: 10 mm ⁇ ) to obtain an uncoated tablet.
- a film-coated tablet was obtained by spraying an uncoated tablet with an aqueous dispersion of a coating base composed of hypromellose 2910, talc, titanium oxide and polyethylene glycol in a pan coating machine.
- the obtained preparation was subjected to dissolution test in acidic solution, hydration rate, and swelling rate measurement, and the results are shown in Tables 14 and 15.
- Formulations 11a to 11c were prepared by granulation and film coating, and it was confirmed that the evaluation results of the average dissolution rate in an acidic solution were almost the same as those of tablets.
- the preparations of Formulas 11a to 11c like the tablets of Examples 4 and 5, showed a decrease in Cmax and an increase in blood concentration after 24 hours compared with the administration of an aqueous solution of the same drug amount in the human administration test, and sustained release. It was confirmed that the preparation showed a good profile.
- the present invention can be used for the production of a sustained-release solid preparation for oral administration containing a pharmacologically active drug such as compound (1).
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
薬物の過量放出を回避し、消化管下部における薬剤の溶出性を改善することにより、1日1回又は2回の経口投与で確実に主薬理効果を発揮する経口投与用の徐放性固形製剤を提供する。 (A)薬理上活性な薬物、(B)カルボキシビニルポリマー、(C)ポビドン、及び、(D)カルメロースナトリウム、キサンタンガム又はカルボキシメチルスターチナトリウム、を含有する徐放性固形製剤。
Description
本発明は、1日1回又は2回の経口投与で確実に主薬理効果を発揮する、徐放性固形製剤に関する。
薬物の血中濃度を調節するための徐放製剤は、主薬理作用と副作用の分離、薬効の持続性向上による投与回数の削減などの服用コンプライアンス向上、さらに医療経済性などの点から有用性が高い。このことから、徐放製剤に関する技術がいくつかの報告されている。一方、主薬理作用を発現する化合物の化学的性質が多様であることから、それらの化合物の多様な化学的性質に適応するための徐放技術がいくつか報告されているが、未だ十分なものではない(例えば、特許文献1、2参照)。
薬物それ自体の性質としては、大きく中性、酸性、塩基性に大別することができ、それらの中でも水に対する溶解性(溶解度)は化合物ごとに大きく異なり、水溶性の低い化合物では、溶出特性を改善するための製剤設計における問題点が多い。酸性薬物とは、遊離体(アルカリ又はアミンの付加塩など、薬物の酸性基との塩を形成していないフリー体)が酸性を示す酸性化合物を指し、酸性の薬物の問題点としては、酸性溶液、例えば胃などの消化管上部における溶解性が低い問題点があり、酸性化合物の塩(アルカリ又はアミン付加塩など)では、酸性溶液中では、溶解性の低い遊離酸になる点が問題点としてある。また、塩基性薬物とは、遊離体(酸付加塩など、薬物の塩基性基との塩を形成していないフリー体)が塩基性を示す塩基性化合物を指し、強酸性水溶液では良好な溶解性を示すが、中性の緩衝液などの中性水溶液では溶解性が低下することが知られている。すなわち、塩基性薬物を経口投与した場合、酸性である胃では良好な溶解性を示すものの、中性でかつ水分の少ない大腸などの消化管下部での溶解性が大きく低下し吸収率の低下が懸念される。
例えば、塩基性薬物の経口投与用の徐放製剤の設計においては、高い水溶性を示す消化管上部の酸性環境における食物との共存、消化管運動などによる機械的ストレスにより、製剤が崩壊するために生じる薬物の過量放出(dose dumping)が問題となる。さらに、薬物の過量放出を回避するため徐放基剤を増量するなどして製剤強度を高めても、中性領域で水溶性が低下する塩基性薬物を含む製剤の消化管下部での溶出性を改善し、吸収を維持させることが徐放性製剤の問題点として残る。現在までに、塩基性薬物の徐放剤で、消化管上部などの酸性環境での薬物の過量放出を回避し、かつ中性環境である消化管下部での持続溶出を同時に達成できる技術として満足できるものはない。
本発明の課題は、消化管上部、特に胃の酸性環境と食物との共存下での消化管運動による機械的ストレスによる薬物の過量放出(dose dumping)を回避し、かつ中性領域である消化管下部における薬物の溶出性を改善することにより、1日1回又は2回の経口投与で確実に主薬理効果を発揮する薬物を主薬効成分として含有する経口投与用の徐放性製剤を提供することにある。
本発明者らは、経口投与用の徐放製剤化の検討を重ねた結果、薬理上活性な薬物、カルボキシビニルポリマー、ポビドン、及び膨潤剤を含有し、酸性環境下での薬物の過量放出(dose dumping)を回避し、かつ中性における溶出性を改善した徐放性固形製剤を見出し、本発明を完成した。
すなわち、本発明は、以下の(1)~(36)を提供するものである。
(1):
(A)薬理上活性な薬物;
(B)カルボキシビニルポリマー;及び
(C)ポビドン;
上記(A)~(C)を含有することを特徴とする経口投与用の徐放性固形製剤。
(2):0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転及び毎分200回転で2時間溶出試験を行ったときの、溶出試験液中における薬理上活性な薬物の平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転)が10%以下であるか、又は、溶出試験液中における薬理上活性な薬物の平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転)が2.0以下である(1)に記載の徐放性固形製剤。
(3):溶出試験液中における薬理上活性な薬物の平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転)が、5%以下である(2)に記載の徐放性固形製剤。
(4):溶出試験液中における薬理上活性な薬物の平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転)が、1.5以下である(2)又は(3)に記載の徐放性固形製剤。
(5):さらに、水溶性賦形剤を含有する(1)~(4)のいずれか1に記載の徐放性固形製剤。
(6):水溶性賦形剤が、糖アルコール類である(5)に記載の徐放性固形製剤。
(7):糖アルコール類が、マンニトール、キシリトール又はエリスリトールである(6)に記載の徐放性固形製剤。
(8):糖アルコール類が、キシリトールである(6)に記載の徐放性固形製剤。
(9):酸性水溶液中での固形製剤の水和率が、60%以上になることを特徴とする(5)~(8)のいずれか1に記載の徐放性固形製剤。
(10):酸性水溶液中での固形製剤の水和率が、90%以上であることを特徴とする(5)~(8)のいずれか1に記載の徐放性固形製剤。
(11):さらに、膨潤剤を含有する(1)~(10)のいずれか1に記載の徐放性固形製剤。
(12):膨潤剤が、カルメロースナトリウムである(11)に記載の徐放性固形製剤。
(13):酸性水溶液中での固形製剤の膨潤率が、150%以上である(11)又は(12)に記載の徐放性固形製剤。
(14):酸性水溶液中での固形製剤の膨潤率が、180%以上である(11)~(13)のいずれか1に記載の徐放性固形製剤。
(15):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲である。
(16):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲である。
(17):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、3mg/ml以下である。
(18):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、0.5mg/ml以下である。
(19):(A)薬理上活性な薬物が、塩基性薬物である(1)~(18)のいずれか1に記載の徐放性固形製剤。
(20):(A)薬理上活性な薬物が、下記の
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;及び
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド;
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である(1)~(14)のいずれか1に記載の徐放性固形製剤。
(21):固形製剤中における、(A)薬理上活性な薬物の含有率が、0.1重量%以上~60重量%以下の範囲である(1)~(20)のいずれか1に記載の徐放性固形製剤。
(22):固形製剤が、錠剤である(1)~(21)のいずれか1に記載の徐放性固形製剤。
(23):錠剤が、マトリックス錠剤である(22)に記載の徐放性固形製剤。
すなわち、本発明は、以下の(1)~(36)を提供するものである。
(1):
(A)薬理上活性な薬物;
(B)カルボキシビニルポリマー;及び
(C)ポビドン;
上記(A)~(C)を含有することを特徴とする経口投与用の徐放性固形製剤。
(2):0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転及び毎分200回転で2時間溶出試験を行ったときの、溶出試験液中における薬理上活性な薬物の平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転)が10%以下であるか、又は、溶出試験液中における薬理上活性な薬物の平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転)が2.0以下である(1)に記載の徐放性固形製剤。
(3):溶出試験液中における薬理上活性な薬物の平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転)が、5%以下である(2)に記載の徐放性固形製剤。
(4):溶出試験液中における薬理上活性な薬物の平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転)が、1.5以下である(2)又は(3)に記載の徐放性固形製剤。
(5):さらに、水溶性賦形剤を含有する(1)~(4)のいずれか1に記載の徐放性固形製剤。
(6):水溶性賦形剤が、糖アルコール類である(5)に記載の徐放性固形製剤。
(7):糖アルコール類が、マンニトール、キシリトール又はエリスリトールである(6)に記載の徐放性固形製剤。
(8):糖アルコール類が、キシリトールである(6)に記載の徐放性固形製剤。
(9):酸性水溶液中での固形製剤の水和率が、60%以上になることを特徴とする(5)~(8)のいずれか1に記載の徐放性固形製剤。
(10):酸性水溶液中での固形製剤の水和率が、90%以上であることを特徴とする(5)~(8)のいずれか1に記載の徐放性固形製剤。
(11):さらに、膨潤剤を含有する(1)~(10)のいずれか1に記載の徐放性固形製剤。
(12):膨潤剤が、カルメロースナトリウムである(11)に記載の徐放性固形製剤。
(13):酸性水溶液中での固形製剤の膨潤率が、150%以上である(11)又は(12)に記載の徐放性固形製剤。
(14):酸性水溶液中での固形製剤の膨潤率が、180%以上である(11)~(13)のいずれか1に記載の徐放性固形製剤。
(15):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲である。
(16):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲である。
(17):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、3mg/ml以下である。
(18):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(14)のいずれか1に記載の徐放性固形製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、0.5mg/ml以下である。
(19):(A)薬理上活性な薬物が、塩基性薬物である(1)~(18)のいずれか1に記載の徐放性固形製剤。
(20):(A)薬理上活性な薬物が、下記の
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;及び
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド;
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である(1)~(14)のいずれか1に記載の徐放性固形製剤。
(21):固形製剤中における、(A)薬理上活性な薬物の含有率が、0.1重量%以上~60重量%以下の範囲である(1)~(20)のいずれか1に記載の徐放性固形製剤。
(22):固形製剤が、錠剤である(1)~(21)のいずれか1に記載の徐放性固形製剤。
(23):錠剤が、マトリックス錠剤である(22)に記載の徐放性固形製剤。
(24):(A)薬理上活性な薬物、(B)カルボキシビニルポリマー、(C)ポビドン、及び、(D)カルメロースナトリウム、キサンタンガム又はカルボキシメチルスターチナトリウム、を含有する徐放性固形製剤。
(25):製剤中における(D)成分がカルメロースナトリウムである、(24)に記載の製剤。
(26)製剤中における(A)成分の含有率が5~35重量%である、(24)又は(25)に記載の製剤。
(27):製剤中における(B)成分の含有率が5~30重量%である、(24)~(26)のいずれか1に記載の製剤。
(28):製剤中における(C)成分の含有率が10~70重量%である、(24)~(27)のいずれか1に記載の製剤。
(29):製剤中における(D)成分の含有率が5~15重量%である、(24)~(28)のいずれか1に記載の製剤。
(30):さらに、糖アルコールを含有する、(24)~(29)のいずれか1に記載の製剤。
(31):製剤中における糖アルコールの含有率が10~40重量%である、(30)に記載の製剤。
(32):糖アルコールがマンニトール、キシリトール又はエリスリトールである、(30)又は(31)に記載の製剤。
(33):糖アルコールがキシリトールである、(30)又は(31)に記載の製剤。
(34):(A)成分が塩基性薬物である、(24)~(33)のいずれか1に記載の製剤。
(35):(A)成分が、
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール、
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、及び、
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である、(24)~(34)のいずれか1に記載の製剤。
(36):剤型が錠剤である、(24)~(35)のいずれか1に記載の製剤。
(25):製剤中における(D)成分がカルメロースナトリウムである、(24)に記載の製剤。
(26)製剤中における(A)成分の含有率が5~35重量%である、(24)又は(25)に記載の製剤。
(27):製剤中における(B)成分の含有率が5~30重量%である、(24)~(26)のいずれか1に記載の製剤。
(28):製剤中における(C)成分の含有率が10~70重量%である、(24)~(27)のいずれか1に記載の製剤。
(29):製剤中における(D)成分の含有率が5~15重量%である、(24)~(28)のいずれか1に記載の製剤。
(30):さらに、糖アルコールを含有する、(24)~(29)のいずれか1に記載の製剤。
(31):製剤中における糖アルコールの含有率が10~40重量%である、(30)に記載の製剤。
(32):糖アルコールがマンニトール、キシリトール又はエリスリトールである、(30)又は(31)に記載の製剤。
(33):糖アルコールがキシリトールである、(30)又は(31)に記載の製剤。
(34):(A)成分が塩基性薬物である、(24)~(33)のいずれか1に記載の製剤。
(35):(A)成分が、
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール、
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、及び、
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である、(24)~(34)のいずれか1に記載の製剤。
(36):剤型が錠剤である、(24)~(35)のいずれか1に記載の製剤。
本発明によれば、薬理上活性な薬物を含有する、経口投与用の徐放性医薬組成物の提供が可能になる。従って、例えば、活性化血液凝固第X因子(FXa)阻害剤の化合物(1)を薬効成分として含有する持続性の経口徐放性固形製剤が提供される。本発明の徐放性医薬組成物は、酸性溶液中における良好な水和率、膨潤率を有しつつ過剰放出を防ぐ良好な錠剤強度を持ち、かつ中性溶液中で良好な溶出性を持つことから、十二指腸、小腸から下部消化管に至るまで、含有する薬理上活性な薬物の持続的な溶出を維持する効果が得られる。
以下に本発明を詳細に説明する。
本願明細書における「酸性溶液」としては、胃などの消化管上部での溶出性を評価するために用いる酸性の溶出試験液を意味し、例えば、日本薬局方記載の溶出試験第1液、米国薬局方に記載のUSP 0.1規定塩酸、0.01規定塩酸、Simulated Gastric Fluid without Enzyme等を挙げることができるが、酸性の溶出試験液は、これらに限られるものではない。
本願明細書における「中性溶液」としては、小腸、大腸などにおける薬物の溶出性を評価するために用いる中性の溶出試験液を意味し、例えば、日本薬局方記載の溶出試験第2液やリン酸塩緩衝液pH6.8、米国薬局方記載のUSP Phosphate Buffer(pH6.8)、Simulated Interstinal Fluid without Enzyme、欧州薬局方記載のPhosphate Buffer Solution pH6.8等を挙げることができるが、「中性溶液」はなんらpH6.8溶出試験液に限られるものではない。
上記の溶出試験液は、各国の薬局方等に記載された方法で調製される。これらの溶出試験液のpHは、溶出試験液が緩衝液の場合、各溶出試験液に規定されたpHの±0.05以内となることが好ましい。
本願発明の徐放性固形製剤の、消化管上部における溶出性評価のための酸性溶出液を用いたパドル法としては、例えば0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転及び200回転で2時間溶出試験を行う方法を挙げることができる。上記のように、製剤中の薬理上活性な薬物が塩基性薬物の場合には、高い水溶性を示す消化管上部の酸性環境における食物との共存、消化管運動などによる機械的ストレスにより、製剤が崩壊するために生じる薬物の過量放出(dose dumping)の問題がある。したがって、酸性溶出試験液中における薬理上活性な薬物の平均溶出率は、パドル法毎分200回転及び/又はパドル法毎分50回転においても、製剤強度を保ち、一定の範囲に溶出率を抑えるのが好ましい。2時間後の溶出試験液中における薬理上活性な薬物の平均溶出率が、パドル法で毎分200回転及び/又は毎分50回転のいずれにおいても、50%以下が好ましく;40%以下のものがより好ましく;30%以下のものがさらに好ましい。また、2時間溶出試験方法を行ったときの、溶出試験液中における薬理上活性な薬物の平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転)が、15%以下が好ましく;10%以下がより好ましく;5%以下がさらに好ましい。また、2時間後の溶出試験液中における薬理上活性な薬物の平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転)が、2.0以下が好ましく;1.5以下がより好ましく;1.3以下が特に好ましい。
本願発明の徐放性固形製剤の、中性領域における溶出性評価のための中性溶出液を用いたパドル法としては、例えばリン酸塩緩衝液(pH6.8;900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行う方法を挙げることができる。当該溶出試験液中における薬理上活性な薬物の平均溶出率としては、溶出試験開始後12時間でも持続的な溶出率を示しているものが好ましい。また、溶出試験開始後12時間でも持続的な溶出率を示し、かつ20%以上の平均溶出率を示すものが好ましく;30%以上の溶出率を示すものがより好ましい。
溶出試験法としては、ヒト消化管内環境に近い条件での溶出試験法であるUSP Apparatus3(Bio-Dis法)を用いてもよい。
溶液中の薬物濃度は、UV法などを用いて定量する事が可能であり、溶出試験液中における薬理上活性な薬物の平均溶出率、溶出時間を算出することができる。
なお、上記「平均溶出率」とは、1種の固形製剤について、少なくとも2個、好ましくは6個、さらに好ましくは12個の溶出率を測定し、それらの平均値を求めればよい。
また、本願発明の徐放性固形製剤における、上記薬理上活性な薬物の溶出性は、in vivo動物試験を用いで確認することができる。in vivo動物試験としては、例えばイヌを用いたin vivo吸収性評価を挙げることができる。一般的に、経口投与された製剤は、胃及び小腸を通過後、大腸に長時間滞留するとされている。そのため、溶出時間の長い徐放性製剤にとって、長時間滞留する大腸において薬物放出が持続することは、非常に重要である。薬理上活性な薬物を含有する製剤の大腸内の吸収性を確認する方法として、イヌ大腸内に製剤を直接投与するイヌ大腸吸収性評価を挙げることができる。すなわち、投与後の血中濃度測定から、イヌ大腸内での吸収性を確認し、薬理上活性な薬物の水溶液経口投与時に対する比として、各錠剤の相対バイオアベイラビリティ(BA)等で、評価することができる。
本願明細書における「薬理上活性な薬物」としては、製剤の処方中で主薬理効果を発揮する薬物であって、比較的水溶性の低い薬物が好ましい。薬理上活性な薬物の、中性化合物としては、分子中に酸性状態又は塩基性の状態においてもイオン化して解離する基を持たない化合物を意味し、また、酸性化合物としては、カルボキシ基、フェノール性水酸基、リン酸基、スルホン酸、テトラゾリル基などに代表される酸性基を有する薬物を意味する。さらに、塩基性薬物としては、分子内にアミノ基、ピペリジニル基、ピペラジニル基などに代表される塩基性窒素原子を有する薬物を意味する。本発明においては、特に塩基性薬物が好適である。塩基性薬物は、小腸、大腸での中性状態(7.5>pH>5)での溶解度が、胃などの酸性状態(pH≦2)での溶解度と比較して低下する物理化学的性質を有する。
上記のように、本願明細書における「塩基性薬物」とは、上記の酸性状態における溶解度に比べて中性状態における溶解度が低下する薬物を指し、中性状態における溶解度の低下の割合としては、下記の範囲を挙げることができる。
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲のものが好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲のものがより好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.01~0.1の範囲のものがさらに好ましいが、これら範囲になんら限定されるものではない。
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲のものが好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲のものがより好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.01~0.1の範囲のものがさらに好ましいが、これら範囲になんら限定されるものではない。
本願明細書における「塩基性薬物」の酸性領域(日本薬局方溶出試験第1液:pH=1.2,20±5℃)での溶解度としては、1~500mg/mlの範囲であり;かつ中性領域(日本薬局方溶出試験第2液:pH=6.8,20±5℃)での溶解度が、0.01~3000μg/mlの範囲が好ましく;
酸性領域(日本薬局方溶出試験第1液:pH=1.2,20±5℃)での溶解度が、1~500mg/mlの範囲であり;かつ中性領域(日本薬局方溶出試験第2液:pH=6.8,20±5℃)での溶解度が、10~500μg/mlの範囲であるものがより好ましい。また、溶解度の絶対値としては、中性状態(7.5>pH>5の範囲)における最低溶解度が3mg/ml以下に低下する薬物が好ましく;1mg/ml以下に低下する薬物がより好ましく;0.5mg/ml以下に低下する薬物がさらに好ましい。
「薬理上活性な薬物」の具体的な例としては、抗凝固剤を挙げることができる。
抗凝固剤としては、塩基性若しくは弱塩基性置換基を有する活性化血液凝固第X因子(FXa)阻害剤が好ましく、FXa阻害剤の具体例としては、下記の(a)~(l)を挙げることができる。
(a)Darexaban Maleate(tanexaban),(N-[2-ヒドロキシ-6-メトキシベンズアミド]フェニル]-4-(4-メチル-1,4-ジアゼパン-1-イル)ベンズアミド)[薬食審査発1111第1号(平成22年11月11日);Pre-publication copy,Proposed INN: List 101;Research and development pipeline.Yamanouchi Pharmaceutical Co Ltd.Company World Wide Web site,11 Feb 2004,参照];
(b)rivaroxaban,(5-クロロ-N-({(5S)-2-オキソ-3-[4-(3-オキソ-4-モルホリニル)フェニル]-1,3-オキサゾリジン-5-イル}メチル)-2-チオフェンカルボキサミド)[WFO Drug Information,Vol.18,No.3,2004,page 260;Susanne R,et al,J.Med.Chem.,2005,48,5900-5908;D.Kubitza et al,Multiple dose escalation study investigating the pharmacodynamics,safety,and pharmacokinetic of Bay59-7939,an oral,direct Factor Xa inhibitor,in healthy male subjects.Blood,2003,102;Abstract 3004.参照];
(c)apixaban,(1-(4-メトキシフェニル)-7-オキソ-6-[4-(2-オキソピペリジン-1-イル)フェニル]-4,5,6,7-テトラヒドロ-1H-ピラゾロ[3,4-c]ピリジン-3-カルボキサミド)[WFO Drug Information,Vol.20, No.1,2006,page 38;Pinto DJP, Orwat MJ, Lam PYS,et al,Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247),a highly potent, selective, efficacious,and orally bioavailable inhibitor of blood coagulation factor Xa,J.Med.Chem.,50(22),5339-56,2007,参照];
(d)Betrixaban,(N-(5-クロロピリジン-2-イル)-2-[4-(N,N-ジメチルカルバムイミドイル)ベンズアミド]-5-メトキシベンズアミド)[WFO Drug Information,Vol.22,No.3,2008,page 226-227;Zhang P, Huang W,Zhu BY,et al.,Discovery of betrixaban (PRT054021),N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide,a highly potent,selective, and orally efficacious factor Xa inhibitor,Bioorg Med.Chem.Lett.19(8),2179-85,2009,参照];
(e)AX-1826[S.Takehana et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P;T.Kayahara
et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P];
(f)HMR-2906[XVIIth Congress of the International Society for Thrombosis
and Haemostasis,Washington D.C.,USA,14-21 Aug 1999;Generating greater value from our products and pipeline.Aventis SA Company Presentation,05 Feb 2004];
(g)Otamixaban((2R,3R)-2-(3-カルバムイミドイルベンジル)-3-[[4-(1-オキシドピリジン-4-イル)ベンゾイル]アミモ]ブタン酸メチル)[WFO Drug Information,Vol.16,No.3,2002,page 257,参照];
(h)BIBT-986(プロドラッグ:BIBT-1011)[American Chemical Society-226th National Meeting,New York City,NY,USA,2003];
(i)DPC-602[J.R.Pruitt et al.J.Med.Chem.2003,46,5298-5313];
(j)LY517717(N-[(1R)-2-[4-(1-メチル-4-ピペリジニル)-1-ピペラジニル]-2-オキソ-1-フェニルエチル]-1H-インドール-6-カルボキサミド)[S.Young,Medicinal Chemistry-12th RSC-SCI Symposium,7-10 September 2003,Cambridge,UK;M.Wiley et al.228th ACS National Meeting,Philadelphia,August 22-26,2004,MEDI-252&254,参照];
(k)N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開2004/058715号パンフレット,参照];及び
(l)N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開03/000657号パンフレット;国際公開03/000680号パンフレット;国際公開03/016302号パンフレット,参照]。
(a)Darexaban Maleate(tanexaban),(N-[2-ヒドロキシ-6-メトキシベンズアミド]フェニル]-4-(4-メチル-1,4-ジアゼパン-1-イル)ベンズアミド)[薬食審査発1111第1号(平成22年11月11日);Pre-publication copy,Proposed INN: List 101;Research and development pipeline.Yamanouchi Pharmaceutical Co Ltd.Company World Wide Web site,11 Feb 2004,参照];
(b)rivaroxaban,(5-クロロ-N-({(5S)-2-オキソ-3-[4-(3-オキソ-4-モルホリニル)フェニル]-1,3-オキサゾリジン-5-イル}メチル)-2-チオフェンカルボキサミド)[WFO Drug Information,Vol.18,No.3,2004,page 260;Susanne R,et al,J.Med.Chem.,2005,48,5900-5908;D.Kubitza et al,Multiple dose escalation study investigating the pharmacodynamics,safety,and pharmacokinetic of Bay59-7939,an oral,direct Factor Xa inhibitor,in healthy male subjects.Blood,2003,102;Abstract 3004.参照];
(c)apixaban,(1-(4-メトキシフェニル)-7-オキソ-6-[4-(2-オキソピペリジン-1-イル)フェニル]-4,5,6,7-テトラヒドロ-1H-ピラゾロ[3,4-c]ピリジン-3-カルボキサミド)[WFO Drug Information,Vol.20, No.1,2006,page 38;Pinto DJP, Orwat MJ, Lam PYS,et al,Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247),a highly potent, selective, efficacious,and orally bioavailable inhibitor of blood coagulation factor Xa,J.Med.Chem.,50(22),5339-56,2007,参照];
(d)Betrixaban,(N-(5-クロロピリジン-2-イル)-2-[4-(N,N-ジメチルカルバムイミドイル)ベンズアミド]-5-メトキシベンズアミド)[WFO Drug Information,Vol.22,No.3,2008,page 226-227;Zhang P, Huang W,Zhu BY,et al.,Discovery of betrixaban (PRT054021),N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide,a highly potent,selective, and orally efficacious factor Xa inhibitor,Bioorg Med.Chem.Lett.19(8),2179-85,2009,参照];
(e)AX-1826[S.Takehana et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P;T.Kayahara
et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P];
(f)HMR-2906[XVIIth Congress of the International Society for Thrombosis
and Haemostasis,Washington D.C.,USA,14-21 Aug 1999;Generating greater value from our products and pipeline.Aventis SA Company Presentation,05 Feb 2004];
(g)Otamixaban((2R,3R)-2-(3-カルバムイミドイルベンジル)-3-[[4-(1-オキシドピリジン-4-イル)ベンゾイル]アミモ]ブタン酸メチル)[WFO Drug Information,Vol.16,No.3,2002,page 257,参照];
(h)BIBT-986(プロドラッグ:BIBT-1011)[American Chemical Society-226th National Meeting,New York City,NY,USA,2003];
(i)DPC-602[J.R.Pruitt et al.J.Med.Chem.2003,46,5298-5313];
(j)LY517717(N-[(1R)-2-[4-(1-メチル-4-ピペリジニル)-1-ピペラジニル]-2-オキソ-1-フェニルエチル]-1H-インドール-6-カルボキサミド)[S.Young,Medicinal Chemistry-12th RSC-SCI Symposium,7-10 September 2003,Cambridge,UK;M.Wiley et al.228th ACS National Meeting,Philadelphia,August 22-26,2004,MEDI-252&254,参照];
(k)N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開2004/058715号パンフレット,参照];及び
(l)N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開03/000657号パンフレット;国際公開03/000680号パンフレット;国際公開03/016302号パンフレット,参照]。
上記の活性化血液凝固第X因子(FXa)阻害剤としては、下記の式(1)

(式中、R1は、N,N-ジメチルカルバモイル基又は[1,3,4]オキサジアゾール-2-イル基を示す。)
で表される化合物[以下、化合物(1)と略する場合がある]が、より好ましい。化合物(1)はフリー体(遊離塩基)、又はその薬理学上許容される塩、それらの水和物であってもよい。
で表される化合物[以下、化合物(1)と略する場合がある]が、より好ましい。化合物(1)はフリー体(遊離塩基)、又はその薬理学上許容される塩、それらの水和物であってもよい。
式(1)で表される化合物の塩としては、塩酸塩、硫酸塩、臭化水素酸塩、ヨウ化水素酸塩、燐酸塩、硝酸塩、安息香酸塩、メタンスルホン酸塩、2-ヒドロキシエタンスルホン酸塩、p-トルエンスルホン酸塩、酢酸塩、プロパン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、グルタル酸塩、アジピン酸塩、酒石酸塩、マレイン酸塩、フマル酸塩、リンゴ酸塩、マンデル酸塩等が挙げられる。
式(1)で表される化合物の塩としては、マレイン酸、塩酸塩、メタンスルホン酸塩、p-トルエンスルホン酸塩が好ましく、マレイン酸及びp-トルエンスルホン酸塩が特に好ましい。
式(1)で表される化合物として好ましいものとして、以下の
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド モノマレイン酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド 塩酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩;及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物;
を挙げることができる。
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド モノマレイン酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド 塩酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩;及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物;
を挙げることができる。
上記の好ましい化合物の中で、下記の式(1a)[以下、化合物(1a)と略する場合がある]及び式(1b)[以下、化合物(1b)と略する場合がある]

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド モノマレイン酸塩(1a);及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物(1b)が特に好ましい。
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物(1b)が特に好ましい。
化合物(1)の遊離塩基(フリー体)とは、化合物(1)と、たとえば上記の塩(酸付加塩)、及び/又は水和物を形成している酸付加塩の「酸」及び水和物の「水」を除いた化合物を意味し、例えば化合物(1a)及び化合物(1b)の遊離塩基(フリー体)とは、下記の式(1a-1)及び式(1b-1)

で表されるN1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1a-1)及びN1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド(1b-1)を意味する。
これらの化合物(1)は、上記文献(WO2003-000657号パンフレット;WO2003-000680号パンフレット;WO2003-016302号パンフレット;WO2004-058715号パンフレット)に記載の方法又はそれに準じる方法によって製造することができる。
また、本発明の(A)薬理上活性な薬物の好ましい例として、下記の式(2)[以下、化合物(2)と略する場合がある]

で表される(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール(2)(CAS Number:72956-09-3)、その薬理上許容される塩、又はそれらの水和物を挙げることができる。
本願明細書における「(A)薬理上活性な薬物」の「固形製剤」中の含有率としては、「(A)薬理上活性な薬物」重量%として、0.1~60重量%が好ましく;1~50重量%がより好ましく;5~35重量%がさらに好ましいが、なんらこの範囲に限定されるものではない。
ここで、本願明細書における成分の含有量を示す「A~B重量%」とは、特別に記載されていない限り、「A重量%以上~B重量%以下の範囲」を意味するものである。
ここで、本願明細書における成分の含有量を示す「A~B重量%」とは、特別に記載されていない限り、「A重量%以上~B重量%以下の範囲」を意味するものである。
本願明細書における「(B)カルボキシビニルポリマー」としては、市販のカーボポール(Noveon/CBC)等を使用すればよく、粘度の異なる種々のグレードのものが入手可能である。好ましいカルボキシビニルポリマーとしては、カーボポール974PNFを挙げることができる。
本願明細書におけるカルボキシビニルポリマーの「固形製剤」中の含有率としては、1~50重量%が好ましく;5~30重量%がより好ましく;10~30重量%がさらに好ましい。
本願明細書における「(C)ポビドン(ポリビニルピロリドン)」は非イオン性の水溶性ポリマーであり、1-ビニル-2-ピロリドンの直鎖重合物を意味するものであり、1-ビニル-2-ピロリドンの架橋重合物であるクロスポビドンを含むものではない。ポビドンは、BASFジャパン等から種々のK値(分子量と相関する粘性特性値)のグレードのものを入手することができ、好ましいものとしては、市販のKollidon30(BASFジャパン)を挙げることができる。
本発明におけるポビドンの「固形製剤」中の含有率としては、1~70重量%が好ましく;6~70重量%がより好ましく;10~70重量%がさらに好ましい。
また、本願明細書の「固形製剤」における「(B)カルボキシビニルポリマー」及び「(C)ポビドン」の含有率としては下記の(1)及び(2)が好ましい、
(1)カルボキシビニルポリマーの含有率が15~25重量%の場合におけるポビドンの含有率は10~70重量%;及び
(2)カルボキシビニルポリマーの含有率が10~15重量%未満の場合におけるポビドンの含有率は、20~70重量%。
(1)カルボキシビニルポリマーの含有率が15~25重量%の場合におけるポビドンの含有率は10~70重量%;及び
(2)カルボキシビニルポリマーの含有率が10~15重量%未満の場合におけるポビドンの含有率は、20~70重量%。
本願明細書における「水溶性賦形剤」としては、果糖、精製白糖、白糖、精製白糖球状顆粒、乳糖、無水乳糖、白糖・デンプン球状顆粒、半消化体デンプン、ブドウ糖、ブドウ糖水和物、粉糖、プルラン、β-シクロデキストリン、マンニトール、キシリトール、エリスリトール等の糖類を好ましいものとして挙げることができる。
糖類としては、マンニトール、キシリトール、エリスリトール等の糖アルコールがより好ましく;キシリトールが特に好ましい。
本願明細書における水溶性賦形剤の「固形製剤」中の含有率としては、0~80重量%が好ましく;0~60重量%がより好ましい。
本願明細書における膨潤剤としては、キサンタンガム、カルボキシメチルスターチナトリウム、グアーガム、アルギン酸、アルギン酸ナトリウム、キトサン、寒天、澱粉、デキストリン、アラビアゴム、ゼラチン、カルメロースナトリウム等が考えられ;カルメロースナトリウム、キサンタンガム、及びカルボキシメチルスターチナトリウムが好ましく;カルメロースナトリウムが得に好ましい。カルメロースナトリウムは、例えばサンローズ(商品名:日本製紙ケミカル)等の市販のものが使用でき、各種の粘度、エーテル化度のものを入手することができる。
本願明細書における膨潤剤の「固形製剤」中の含有率としては、1~40重量%が好ましく;5~15重量%がより好ましく;7~15重量%がさらに好ましい。
本願発明においては、上記の(A)薬理上活性な薬物、(B)カルボキシビニルポリマー、及び(C)ポビドン、及び上記の、水溶性賦形剤、膨潤剤に加え、本発明の効果に影響を与えない範囲内で、崩壊剤、結合剤、流動化剤、滑沢剤、着色剤、光沢化剤等を配合してもよい。
崩壊剤としては、アジピン酸、アルギン酸、アルファー化デンプン、カルボキシメチルスターチナトリウム、含水二酸化ケイ素、クエン酸カルシウム、軽質無水ケイ酸、合成ケイ酸アルミニウム、コムギデンプン、コメデンプン、ステアリン酸カルシウム、トウモロコシデンプン、トラガント末、バレイショデンプン、ヒドロキシプロピルスターチ、部分アルファー化デンプン、フマル酸一ナトリウム、無水クエン酸、リン酸二水素カルシウム、クロスカルメロースナトリウム、クロスポビドン、カルメロース、カルメロースカルシウム等が挙げられる。
結合剤としては、アメ粉、アラビアゴム、アラビアゴム末、アルギン酸ナトリウム、アルギン酸プロピレングリコールエステル、加水分解ゼラチン末、加水分解デンプン加軽質無水ケイ酸、果糖、カルボキシメチルエチルセルロース、含水二酸化ケイ素、カンテン末、軽質無水ケイ酸、軽質無水ケイ酸含有ヒドロキシプロピルセルロース、結晶セルロース、合成ケイ酸アルミニウム、コポリドン、小麦粉、コムギデンプン、米粉、コメデンプン、酢酸ビニル樹脂、酢酸フタル酸セルロース、ジオクチルソジウムスルホサクシネート、ジヒドロキシアルミニウムアミノアセテート、酒石酸ナトリウムカリウム、常水、ショ糖脂肪酸エステル、精製ゼラチン、精製白糖、ゼラチン、D-ソルビトール、デキストリン、デンプン、トウモロコシデンプン、トラガント、トラガント末、乳糖、濃グリセリン、白糖、バレイショデンプン、ヒドロキシエチルセルロース、ヒドロキシエチルメチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルスターチ、ヒドロキシプロピルメチルセルロース2208、ヒドロキシプロピルメチルセルロース2906、ヒドロキシプロピルメチルセルロース2910、ヒドロキシプロピルメチルセルロースフタレート、ビニルピロリドン・酢酸ビニル共重合物、ピペロニルブトキシド、ブドウ糖、部分アルファー化デンプン、フマル酸、フマル酸ステアリン酸・ポリビニルアセタールジエチルアミノアセテート・ヒドロキシプロピルメチルセルロース2910混合物、プルラン、ポリビニルアルコール(完全ケン化物)、ポリビニルアルコール(部分ケン化物)、ポリリン酸ナトリウム、ポリエチレングリコール4000、ポリエチレングリコール6000、ポリエチレングリコール20000、D-マンニトール、メチルセルロース等が挙げられる。
流動化剤としては、含水二酸化ケイ素、軽質無水ケイ酸、結晶セルロース、合成ケイ酸アルミニウム、酸化チタン、ステアリン酸、ステアリン酸カルシウム、ステアリン酸マグネシウム、第三リン酸カルシウム、タルク、トウモロコシデンプン、メタケイ酸アルミン酸マグネシウム等を挙げることができる。
滑沢剤としては、カカオ脂、カルナウバロウ、含水二酸化ケイ素、乾燥水酸化アルミニウムゲル、グリセリン脂肪酸エステル、ケイ酸マグネシウム、軽質無水ケイ酸、結晶セルロース、硬化油、合成ケイ酸アルミニウム、サラシミツロウ、酸化マグネシウム、酒石酸ナトリウムカリウム、ショ糖脂肪酸エステル、ステアリン酸、ステアリン酸カルシウム、ステアリン酸マグネシウム、ステアリルアルコール、ステアリン酸ポリオキシル40、セタノール、ダイズ硬化油、ゼラチン、タルク、炭酸マグネシウム、沈降炭酸カルシウム、トウモロコシデンプン、バレイショデンプン、フマル酸、フマル酸ステアリルナトリウム、ポリエチレングリコール600、ポリエチレングリコール4000、ポリエチレングリコール6000、ミツロウ、メタケイ酸アルミン酸マグネシウム、ラウリル酸ナトリウム、硫酸マグネシウム等が挙げられる。
着色剤としては、黄色三二酸化鉄、三二酸化鉄、酸化チタン、オレンジエッセンス、褐色酸化鉄、β-カロチン、黒酸化鉄、食用青色1号、食用青色2号、食用赤色2号、食用赤色3号、食用赤色102号、食用黄色4号、食用黄色5号等を挙げることができる。
光沢化剤としては、カルナウバロウ、硬化油、酢酸ビニル樹脂、サラシミツロウ、酸化チタン、ステアリン酸、ステアリン酸カルシウム、ステアリン酸ポリオキシル40、ステアリン酸マグネシウム、精製セラック、精製パラフィン・カルナウバロウ混合ワックス、セタノール、タルク、中金箔、白色セラック、パラフィン、ポビドン(ポリビニルピロリドン)、ポリエチレングリコール1500、ポリエチレングリコール4000、ポリエチレングリコール6000、ミツロウ、モノステアリン酸グリセリン、ロジン等が挙げられる。
本願発明の徐放性固形製剤は、経口投与可能な固形製剤であればその剤形は特に制限されないが、顆粒剤、錠剤が好ましい。錠剤としてはマトリックス錠剤が好ましい。
また、本願発明の徐放性固形製剤の製造方法は、固形製剤の周知の製造方法を採用することができる。例えば、本願発明の徐放性固形製剤は、(A)薬理上活性な薬物、(B)カルボキシビニルポリマー、及び(C)ポビドン、及び上記の、水溶性賦形剤、膨潤剤に加え、さらに必要に応じて崩壊剤、結合剤、流動化剤、滑沢剤、着色剤及び光沢化剤等を配合し、例えば、日本薬局方の製剤総則に記載されている固形製剤の製造法により製造することができる。
本発明の徐放性固形製剤の剤形が顆粒剤の場合、(A)~(C)、及び上記の、水溶性賦形剤、膨潤剤に加え、さらに必要に応じて添加剤を流動層造粒機等の造粒装置で混合後、湿式造粒等の造粒を行って造粒顆粒を得ることができる。造粒顆粒は、滑沢剤等の添加剤を加えて混合後、打錠用顆粒とし、これを打錠して錠剤とすることもでき、得られた錠剤は、必要に応じてコーティング剤を用いてフィルムコーティング錠剤とすることも可能である。
また、本発明の徐放性固形製剤の剤形が錠剤の場合、(A)薬理上活性な薬物、(B)カルボキシビニルポリマー、及び(C)ポビドン、さらに水溶性賦形剤、膨潤剤との組合せに加え、医薬品添加物の混合末、好ましくは(A)薬理上活性な薬物;(B)カルボキシビニルポリマー;(C)ポビドン、水溶性賦形剤;及び膨潤剤との組合せ、並びに医薬品添加物の混合末をそのまま圧縮成型することにより製造してもよい。また、錠剤の形状は、特に制限はないが、レンズ型、円盤型、円形、楕円形及び三角形やひし形等の多角形のものが好ましい。さらに、パンコーティング機にて、得られた錠剤にコーティング剤の懸濁液/溶解液を噴霧してコーティングすることも可能である。
(A)薬理上活性な薬物の本発明医薬組成物中の含有比は、薬理上活性な薬物のフリー体換算で、0.1重量%以上~60重量%以下の範囲が好ましく;1~50重量%がより好ましく;5~35重量%がさらに好ましい。特に、本発明医薬組成物の剤形が錠剤の場合、1錠あたりに含有される薬理上活性な薬物の含有量は、薬理上活性な薬物のフリー体換算で、0.5~500mgの範囲であり;1~100mgが好ましく;5~75mgがより好ましく;15~60mgがさらに好ましい。
また、本願発明の徐放性固形製剤の水和率の評価方法としては、各錠剤を、0.01規定塩酸中、37±0.5℃において、2時間静置後の水和していない部分の体積を求め、下記の式1

で表される式で水和率を算出した。水和率としては、60%以上のものが好ましく;90%以上のものがより好ましい。
本発明の医薬組成物の膨潤率の評価方法としては、下記の式2

で表される式により算出することができる。
膨潤率としては、150%以上が好ましく;180%以上がより好ましく;200%以上が特に好ましい。
本願発明の別の態様について以下に説明する。
本願発明の徐放性固形製剤は、(A)薬理上活性な薬物、(B)カルボキシビニルポリマー、(C)ポビドン、及び、(D)カルメロースナトリウム、キサンタンガム又はカルボキシメチルスターチナトリウム、を含有する。
本願発明の製剤に使用される(A)成分である「薬理上活性な薬物」は特に限定されるものではなく、また薬物は体内で薬理上活性な薬物に変換されるプロドラッグであってもよい。本願発明の(A)成分の例示としては上述のものを好ましいものとして挙げることができるが、なんらこれらに限定されるものではない。本願発明の製剤における(A)成分の含有率としては、0.1~60重量%が好ましく、1~50重量%がより好ましく、5~35重量%がさらに好ましいが、なんらこの範囲に限定されるものではない。
本願発明の製剤に使用される(B)成分である「カルボキシビニルポリマー」としては、上述のものを使用でき、市販のカーボポール974PNFが好ましい。本願発明の製剤における(B)成分の含有率としては、1~50重量%が好ましく、5~30重量%がより好ましく、10~30重量%がさらに好ましい。
本願発明の製剤に使用される(C)成分である「ポビドン」としては、上述のものを意味し、市販のKollidon30(BASFジャパン)が好ましい。本願発明の製剤における(C)成分の含有率としては、10~70重量%が好ましい。
また、本願発明の製剤における(B)成分及び(C)成分の含有率としては、下記の(1)及び(2)が好ましい。
(1)(B)成分の含有率が15~30重量%の場合における(C)成分の含有率は10~70重量%;及び
(2)(B)成分の含有率が10~15重量%未満の場合における(C)成分の含有率は、20~70重量%。
(1)(B)成分の含有率が15~30重量%の場合における(C)成分の含有率は10~70重量%;及び
(2)(B)成分の含有率が10~15重量%未満の場合における(C)成分の含有率は、20~70重量%。
本願発明の製剤に使用される(D)成分は、カルメロースナトリウム、キサンタンガムまたはカルボキシメチルスターチナトリウムであり、好ましくはカルメロースナトリウムである。本願発明の製剤における(D)成分の含有率としては、5~15重量%が好ましく;7~15重量%がより好ましい。
本願発明の製剤は、(A)~(D)に加えて糖アルコールを含有することが好ましい。糖アルコールとしては上述のものが挙げられるが、マンニトール、キシリトールまたはエリスリトールが好ましく;キシリトールがより好ましい。本願発明の製剤における糖アルコールの含有率としては、10~40重量%が好ましい。
本願発明の製剤には、本発明の効果に影響を与えない範囲内で、上述の、結合剤、流動化剤、滑沢剤、着色剤、光沢化剤等の添加剤を配合してもよい。
本願発明の製剤は、経口投与可能な製剤であればその剤型は特に制限されないが、錠剤が好ましい。錠剤としてはマトリックス錠剤が好ましい。
本願発明の製剤は、(A)~(D)を混合した後に打錠するか、または、(A)~(D)を混合して造粒した後に打錠して得られる。混合、造粒および打錠は、当該分野で周知の方法を用いて行えばよい。
本願発明の製剤が糖アルコールを含有する場合は、(A)~(D)および糖アルコールを混合した後に打錠するか、または、(A)~(D)および糖アルコールを混合して造粒した後に打錠して得られる。
錠剤の形状としては特に制限はないが、レンズ型、円盤型、円形、楕円形、及び、三角形、ひし形等の多角形のものが好ましい。本願発明の製剤には、必要に応じて、コーティング剤を用いてフィルムコーティングを施してもよい。混合、造粒、打錠およびコーティングは、当該分野で周知の方法を用いて行えばよい。
本願発明の製剤にその他の添加剤を配合する場合、混合工程、造粒工程、打錠工程またはコーティング工程のいずれの工程で配合しても良い。
かくして得られた本願発明の製剤は、上述の発明の効果に記載したように、酸性溶液中における良好な水和率、膨潤率を有しつつ過剰放出を防ぐ良好な錠剤強度を持ち、かつ中性溶液中で良好な溶出性を持つことから、十二指腸、小腸から下部消化管に至るまで、含有する薬理上活性な薬物の持続的な溶出を維持する効果が得られる。
また、本願発明の製剤の水和率及び膨潤率の評価方法としては上記の式1、及び式2で算出することができ、水和率としては、90%以上のものがより好ましく、膨潤率としては、150%以上が好ましく;180%以上がより好ましく;200%以上が特に好ましい。
次に実施例を挙げて本発明を詳細に説明するが、本発明は何らこれら実施例に限定されるものではない。
酸性溶液または中性溶液中での溶出性試験は次のように行った。
(酸性溶液中での溶出試験)
0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転及び200回転で溶出試験を行い、経時的に溶出液中の薬物平均溶出率を算出した。各回転数での平均溶出率、2時間溶出試験を行い、薬物平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転:D2h,200rpm-D2h,50rpm)、平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転:D2h,200rpm/D2h,50rpm)を算出した。
(酸性溶液中での溶出試験)
0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転及び200回転で溶出試験を行い、経時的に溶出液中の薬物平均溶出率を算出した。各回転数での平均溶出率、2時間溶出試験を行い、薬物平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転:D2h,200rpm-D2h,50rpm)、平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転:D2h,200rpm/D2h,50rpm)を算出した。
(中性溶液中での溶出試験)
pH6.8のリン酸緩衝液(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行い経時的に溶出液中の薬物平均溶出率を算出した。
pH6.8のリン酸緩衝液(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行い経時的に溶出液中の薬物平均溶出率を算出した。
錠剤の水和率及び膨潤率の測定は次のように行った。
(錠剤の水和率の評価)
水和率の評価方法としては、各錠剤を、0.01規定塩酸中、37±0.5℃において、2時間静置後、上記の式1を用いて算出した。
(錠剤の水和率の評価)
水和率の評価方法としては、各錠剤を、0.01規定塩酸中、37±0.5℃において、2時間静置後、上記の式1を用いて算出した。
(膨潤率の評価)
錠剤を0.01規定塩酸(1000mL)中で2時間静置後、錠剤を取り出し、静置後の体積から上記の式2により膨潤率を算出した。
錠剤を0.01規定塩酸(1000mL)中で2時間静置後、錠剤を取り出し、静置後の体積から上記の式2により膨潤率を算出した。
(実施例1)
(錠剤の製造)
メノウ乳鉢を用いて表1に記載の各成分を混合し、この混合末200mgを単発打錠機(N-30E、岡田精工)により打錠[錠径:8.0mm・(平錠)]して試料とした。
(錠剤の製造)
メノウ乳鉢を用いて表1に記載の各成分を混合し、この混合末200mgを単発打錠機(N-30E、岡田精工)により打錠[錠径:8.0mm・(平錠)]して試料とした。


<評価結果>
表2に示したように、いずれの処方においても錠剤の水和率は90%以上であり、錠剤内部のほとんどが水和していることが確認された。
表2に示したように、いずれの処方においても錠剤の水和率は90%以上であり、錠剤内部のほとんどが水和していることが確認された。
(表1の処方の酸性溶液中における溶出性評価)
表1に示した各処方について酸性溶液中における溶出性試験を行った。その結果を、表3、図1~図4に示した。
表1に示した各処方について酸性溶液中における溶出性試験を行った。その結果を、表3、図1~図4に示した。

<評価結果>
処方1の錠剤は、比較例1~3の処方の錠剤と比較して、薬物放出を抑制し、パドル回転数の影響を受け難いことを確認したことから、カルボキシビニルポリマー及びポビドンの同時添加が、酸性溶液中における錠剤強度の維持に有効であることがわかった。
処方1の錠剤は、比較例1~3の処方の錠剤と比較して、薬物放出を抑制し、パドル回転数の影響を受け難いことを確認したことから、カルボキシビニルポリマー及びポビドンの同時添加が、酸性溶液中における錠剤強度の維持に有効であることがわかった。
処方1の錠剤について中性溶液中における溶出性試験を行った。その結果を図5に示した。
<評価結果>
図5に示すように処方1の錠剤は、中性溶液中で持続的な溶出性を示した。
図5に示すように処方1の錠剤は、中性溶液中で持続的な溶出性を示した。
(実施例2)
表4に示した各処方について、各成分を、メノウ乳鉢を用いて混合し、この混合末200mgを、単発打錠機(N-30E、岡田精工)を用いて打錠して試料とした[錠径:8.0mmφ(6.5R)]。
表4に示した各処方について、各成分を、メノウ乳鉢を用いて混合し、この混合末200mgを、単発打錠機(N-30E、岡田精工)を用いて打錠して試料とした[錠径:8.0mmφ(6.5R)]。
得られた試料について、酸性溶液中における溶出試験を行い、その結果を図6、図7及び表5に示した。


<試験結果>
図6、図7及び表5に示したように、酸性溶液での溶出に対するパドル回転数の影響について、処方2bでは小さかったが、処方2aでは比較的大きかった。この結果から、ポビドンを10重量%以上添加することにより、良好な錠剤強度が得られることがわかった。
図6、図7及び表5に示したように、酸性溶液での溶出に対するパドル回転数の影響について、処方2bでは小さかったが、処方2aでは比較的大きかった。この結果から、ポビドンを10重量%以上添加することにより、良好な錠剤強度が得られることがわかった。
(実施例3)
表6に示す処方についてメノウ乳鉢を用いて各成分を混合し、この混合末200mgを単発打錠機(N-30E、岡田精工)により打錠して試料とした[錠径:8.0mmφ(6.5R)]。
表6に示す処方についてメノウ乳鉢を用いて各成分を混合し、この混合末200mgを単発打錠機(N-30E、岡田精工)により打錠して試料とした[錠径:8.0mmφ(6.5R)]。
カルボキシビニルポリマーの処方比率が25%の処方1、15%の処方3について、酸性溶液中における溶出試験結果を図4、図8及び表7に示した。


<評価結果>
図4、図8及び表7の溶出挙動に示すように、カルボキシビニルポリマーを25%添加した処方1、及び15%添加する処方3ともに酸性溶液中でのパドル回転数の影響は小さかった。
図4、図8及び表7の溶出挙動に示すように、カルボキシビニルポリマーを25%添加した処方1、及び15%添加する処方3ともに酸性溶液中でのパドル回転数の影響は小さかった。
(実施例4)
表8に示した処方について、メノウ乳鉢を用いて各成分を混合し、単発打錠機(N-30E)で打錠して錠剤(錠径:10.0mmφ(8R))を製造した。
表8に示した処方について、メノウ乳鉢を用いて各成分を混合し、単発打錠機(N-30E)で打錠して錠剤(錠径:10.0mmφ(8R))を製造した。
酸性溶液及び中性溶液中における溶出性の試験結果、各処方の水和率及び膨潤率の測定結果を表9、及び図9~12に示した。


<試験結果>
図9~12に示すように、いずれの処方も中性で持続的な溶出挙動を示した。また、表9に示すように、処方4の場合、速やかに水和したものの錠剤はほとんど膨潤せず、2時間後の膨潤率は109%であった。一方、膨潤剤として、カルメロースナトリウム、キサンタンガム、又はカルボキシメチルスターチナトリウムを添加した処方5及~処方7の場合、錠剤は速やかに水和し、さらにイニシャルと比較して2時間で約1.6倍~約2.5倍に膨潤することが確認された。処方5及~処方7は、酸性溶液中における良好な水和率、膨潤率を有するとともに、酸性溶液中におけるパドル回転数の影響が小さかった、特にカルメロースナトリウムを添加した処方5は、酸性溶液中における水和率、膨潤率、及び錠剤強度の面で優れていた。
図9~12に示すように、いずれの処方も中性で持続的な溶出挙動を示した。また、表9に示すように、処方4の場合、速やかに水和したものの錠剤はほとんど膨潤せず、2時間後の膨潤率は109%であった。一方、膨潤剤として、カルメロースナトリウム、キサンタンガム、又はカルボキシメチルスターチナトリウムを添加した処方5及~処方7の場合、錠剤は速やかに水和し、さらにイニシャルと比較して2時間で約1.6倍~約2.5倍に膨潤することが確認された。処方5及~処方7は、酸性溶液中における良好な水和率、膨潤率を有するとともに、酸性溶液中におけるパドル回転数の影響が小さかった、特にカルメロースナトリウムを添加した処方5は、酸性溶液中における水和率、膨潤率、及び錠剤強度の面で優れていた。
(実施例5)
表8に示す処方4及び処方5について、絶食下イヌにおける大腸吸収性を評価した。なお、ここでは、表8に示す処方をおよそ2/3にし、化合物(1a-1)含量として20mg、総量240mgの製剤(処方4’及び処方5’)を用いて評価した。
表8に示す処方4及び処方5について、絶食下イヌにおける大腸吸収性を評価した。なお、ここでは、表8に示す処方をおよそ2/3にし、化合物(1a-1)含量として20mg、総量240mgの製剤(処方4’及び処方5’)を用いて評価した。
大腸内への製剤投与は、0.01規定塩酸中に2時間浸した錠剤1錠を動物用内視鏡(オリンパス)にセットし、内視鏡でイヌ大腸内を観察しながら肛門から約30cmの部位に投与した。なお、投与前に錠剤から0.01規定塩酸中に薬物の一部が溶出する量を測定し、血漿中薬物濃度は、投与量が化合物(1a-1)として20mg/headとなるように換算した。この時の血漿中薬物濃度推移を図13に示した。
<評価結果>
図13に示すように、膨潤剤としてカルメロースナトリウムを含む処方5’の薬物大腸吸収性は、カルメロースナトリウムを含まない処方4’よりも大きく、水膨潤効果が吸収性向上に寄与することが確認できた。この結果から、消化管下部における持続的な吸収性の向上のために膨潤剤の存在が重要であることがわかった。
図13に示すように、膨潤剤としてカルメロースナトリウムを含む処方5’の薬物大腸吸収性は、カルメロースナトリウムを含まない処方4’よりも大きく、水膨潤効果が吸収性向上に寄与することが確認できた。この結果から、消化管下部における持続的な吸収性の向上のために膨潤剤の存在が重要であることがわかった。
(実施例6)
化合物(1b)、カルボキシビニルポリマー、カルメロースナトリウム、ポビドン、ポリエチレングリコール6000、キシリトール、フマル酸ステアリルナトリウムについて、表10示す重量比率になるように乳鉢にとり、3分間混合し混合末を得た。本混合末について表10に示す1錠の所定量を量り、単発打錠機(NE-30、岡田精工)を用いて打錠した(杵形状:10mmφ、平型)。
化合物(1b)、カルボキシビニルポリマー、カルメロースナトリウム、ポビドン、ポリエチレングリコール6000、キシリトール、フマル酸ステアリルナトリウムについて、表10示す重量比率になるように乳鉢にとり、3分間混合し混合末を得た。本混合末について表10に示す1錠の所定量を量り、単発打錠機(NE-30、岡田精工)を用いて打錠した(杵形状:10mmφ、平型)。
処方8、比較例4及び比較例5について、酸性水溶液における溶出性の試験結果を表11に示した。
処方8、9及び10について、水和率及び膨潤率を測定し、その結果を表12に示した。



<評価結果>
比較例4及び比較例5ではでは、比(パドル回転数毎分50回転での溶出率/パドル回転数毎分200回転での溶出率)は、それぞれ1.9及び3.5であったのに対し、処方8では1.2であり、回転数の影響が小さかった。また、処方9及び処方10では、膨潤率はそれぞれ116%及び163%、水和率はそれぞれ94.5%及び75.9%であったのに対し、処方8では、膨潤率は196%、水和率は100%であり、膨潤剤であるカルメロースナトリウム添加の効果が確認できた。また、水溶性賦形剤としてキシリトールを含有していない処方10の水和率は、キシリトールを含有する処方8、9に比較して低かった。
比較例4及び比較例5ではでは、比(パドル回転数毎分50回転での溶出率/パドル回転数毎分200回転での溶出率)は、それぞれ1.9及び3.5であったのに対し、処方8では1.2であり、回転数の影響が小さかった。また、処方9及び処方10では、膨潤率はそれぞれ116%及び163%、水和率はそれぞれ94.5%及び75.9%であったのに対し、処方8では、膨潤率は196%、水和率は100%であり、膨潤剤であるカルメロースナトリウム添加の効果が確認できた。また、水溶性賦形剤としてキシリトールを含有していない処方10の水和率は、キシリトールを含有する処方8、9に比較して低かった。
(実施例7)
上記の処方5が、酸性溶液中においてパドル回転数の影響を受け難く、かつ中性溶液中における持続的な溶出性を示し、かつ良好な膨潤率及び水和率を示したことから、表13に示す処方製剤を製造した。すなわち、表13示す比率で薬物(1a)、カルボキシビニルポリマー、キシリトール、ポビドン(一部)、カルメロースナトリウム、その他の添加剤を流動層造粒機に投入して混合後、ポビドン溶液(一部を除いた残り)を結合液として噴霧して湿式造粒を行った。得られた造粒物を乾燥して造粒顆粒を得た後、これにフマル酸ステアリルナトリウムを加え、V型混合機を用いて混合し打錠用顆粒とした。これをロータリー打錠機にて打錠し(杵型:10mmφ)、素錠を得た。素錠に、ヒプロメロース2910、タルク、酸化チタン及びポリエチレングリコールからなるコーティング基剤の水分散液を、パンコーティング機にて噴霧することにより、フィルムコーティング錠を得た。
上記の処方5が、酸性溶液中においてパドル回転数の影響を受け難く、かつ中性溶液中における持続的な溶出性を示し、かつ良好な膨潤率及び水和率を示したことから、表13に示す処方製剤を製造した。すなわち、表13示す比率で薬物(1a)、カルボキシビニルポリマー、キシリトール、ポビドン(一部)、カルメロースナトリウム、その他の添加剤を流動層造粒機に投入して混合後、ポビドン溶液(一部を除いた残り)を結合液として噴霧して湿式造粒を行った。得られた造粒物を乾燥して造粒顆粒を得た後、これにフマル酸ステアリルナトリウムを加え、V型混合機を用いて混合し打錠用顆粒とした。これをロータリー打錠機にて打錠し(杵型:10mmφ)、素錠を得た。素錠に、ヒプロメロース2910、タルク、酸化チタン及びポリエチレングリコールからなるコーティング基剤の水分散液を、パンコーティング機にて噴霧することにより、フィルムコーティング錠を得た。
得られた製剤につき、酸性溶液中における溶出性試験、水和率、膨潤率の測定を行い、結果を表14及び15に示した。



<試験結果>
処方11a~11cは、造粒及びフィルムコーティングを行った製剤であるが、酸性溶液中における平均溶出率の評価結果は錠剤とほぼ同等の効果であることを確認できた。また、処方11a~11cの製剤は実施例4,5の錠剤と同様に、ヒト投与試験において同一薬物量の水溶液投与と比較しCmax低減及び24時間後の血中濃度の増大を示し、徐放製剤として良好なプロファイルを示すことが確認された。
処方11a~11cは、造粒及びフィルムコーティングを行った製剤であるが、酸性溶液中における平均溶出率の評価結果は錠剤とほぼ同等の効果であることを確認できた。また、処方11a~11cの製剤は実施例4,5の錠剤と同様に、ヒト投与試験において同一薬物量の水溶液投与と比較しCmax低減及び24時間後の血中濃度の増大を示し、徐放製剤として良好なプロファイルを示すことが確認された。
(製剤例1)
薬物として化合物(2)を用いた表16に示す処方製剤を製造した。
薬物として化合物(2)を用いた表16に示す処方製剤を製造した。

<試験結果>
処方12の製剤のイヌにおけるバイオアベイラビリティー(BA)は、化合物(2)の既存徐放製剤(Coreg CR)と比較して1.54倍の高いパフォーマンスを示し、かつ最高血中濃度到達時間(Tmax)が既存徐放製剤と比較して2倍以上延長したことから、徐放性製剤として好ましいプロファイルを持つことがわかった。
処方12の製剤のイヌにおけるバイオアベイラビリティー(BA)は、化合物(2)の既存徐放製剤(Coreg CR)と比較して1.54倍の高いパフォーマンスを示し、かつ最高血中濃度到達時間(Tmax)が既存徐放製剤と比較して2倍以上延長したことから、徐放性製剤として好ましいプロファイルを持つことがわかった。
本発明は、化合物(1)などの薬理上活性な薬物を含有する、経口投与用の徐放性固形製剤の製造に利用することができる。
Claims (13)
- (A)薬理上活性な薬物、(B)カルボキシビニルポリマー、(C)ポビドン、及び、(D)カルメロースナトリウム、キサンタンガム又はカルボキシメチルスターチナトリウム、を含有する徐放性固形製剤。
- 製剤中における(D)成分がカルメロースナトリウムである、請求項1に記載の製剤。
- 製剤中における(A)成分の含有率が5~35重量%である、請求項1又は2に記載の製剤。
- 製剤中における(B)成分の含有率が5~30重量%である請求項1~3のいずれか1項に記載の製剤。
- 製剤中における(C)成分の含有率が10~70重量%である、請求項1~4のいずれか1項に記載の製剤。
- 製剤中における(D)成分の含有率が5~15重量%である、請求項1~5のいずれか1項に記載の製剤。
- さらに、糖アルコールを含有する、請求項1~6のいずれか1項に記載の製剤。
- 製剤中における糖アルコールの含有率が10~40重量%である、請求項7に記載の製剤。
- 糖アルコールがマンニトール、キシリトール又はエリスリトールである、請求項7又は8に記載の製剤。
- 糖アルコールがキシリトールである、請求項7又は8に記載の製剤。
- (A)成分が塩基性薬物である、請求項1~10のいずれか1項に記載の製剤。
- (A)成分が、
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール、
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、及び、
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である、請求項1~10のいずれか1項に記載の製剤。 - 剤型が錠剤である、請求項1~12のいずれか1項に記載の製剤。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES11744792.0T ES2600894T3 (es) | 2010-02-22 | 2011-02-21 | Preparación sólida de liberación sostenida para uso oral |
| EP11744792.0A EP2540294B1 (en) | 2010-02-22 | 2011-02-21 | Sustained-release solid preparation for oral use |
| JP2012500677A JP5714562B2 (ja) | 2010-02-22 | 2011-02-21 | 経口用徐放性固形製剤 |
| US13/591,981 US20130005763A1 (en) | 2010-02-22 | 2012-08-22 | Sustained-release solid preparation for oral use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010035884 | 2010-02-22 | ||
| JP2010-035884 | 2010-02-22 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/591,981 Continuation US20130005763A1 (en) | 2010-02-22 | 2012-08-22 | Sustained-release solid preparation for oral use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011102506A1 true WO2011102506A1 (ja) | 2011-08-25 |
Family
ID=44483089
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/053644 WO2011102506A1 (ja) | 2010-02-22 | 2011-02-21 | 経口用徐放性固形製剤 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20130005763A1 (ja) |
| EP (1) | EP2540294B1 (ja) |
| JP (1) | JP5714562B2 (ja) |
| ES (1) | ES2600894T3 (ja) |
| TW (1) | TW201200165A (ja) |
| WO (1) | WO2011102506A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013022059A1 (ja) | 2011-08-10 | 2013-02-14 | 第一三共株式会社 | ジアミン誘導体含有医薬組成物 |
| JP2015500812A (ja) * | 2011-12-02 | 2015-01-08 | シンクロニューロン インコーポレイテッド | アカンプロサート製剤、その使用方法、及びそれを含む合剤 |
| KR101535586B1 (ko) * | 2014-08-01 | 2015-07-09 | 에스케이케미칼주식회사 | 무정형 또는 열역학적 준 안정형 리바록사반을 포함하는 약학 제제 |
| US9918975B2 (en) | 2010-03-19 | 2018-03-20 | Daiichi Sankyo Company, Limited | Method for improving dissolution of anticoagulant agent |
| US10166207B2 (en) | 2013-06-05 | 2019-01-01 | Synchroneuron, Inc. | Acamprosate formulations, methods of using the same, and combinations comprising the same |
| US10512621B2 (en) | 2011-12-02 | 2019-12-24 | Synchroneuron, Inc. | Methods of treating posttraumatic stress disorder with acamprosate salts |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5749247B2 (ja) | 2010-02-22 | 2015-07-15 | 第一三共株式会社 | 経口用徐放性固形製剤 |
| BR112015004190A2 (pt) | 2012-09-03 | 2017-07-04 | Daiichi Sankyo Co Ltd | composição farmacêutica de liberação sustentada, e, método para produzir uma composição farmacêutica de liberação sustentada |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001513752A (ja) * | 1996-09-12 | 2001-09-04 | ロシュ ダイアグノスティクス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 迅速崩壊ペレット |
| JP2001270821A (ja) * | 2000-03-23 | 2001-10-02 | Eisai Co Ltd | 服用感が優れた散剤 |
| JP2001522794A (ja) * | 1997-11-12 | 2001-11-20 | ベーリンガー・マンハイム・ファーマシューティカルズ・コーポレイション−スミスクライン・ベックマン・コーポレイション・リミテッド・パートナーシップ・ナンバー1 | カルベジロールの新規経口剤形 |
| WO2003000657A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003000680A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| JP2003507413A (ja) * | 1999-08-26 | 2003-02-25 | ニューロクライン バイオサイエンシーズ, インコーポレイテッド | 徐放性鎮静催眠組成物およびそれに関連する方法 |
| WO2003016302A1 (fr) | 2001-08-09 | 2003-02-27 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| JP2004518676A (ja) | 2000-12-20 | 2004-06-24 | シャイア ラボラトリーズ,インコーポレイテッド | 最小化pH依存性溶解プロフィールを有する徐放性薬学的剤形 |
| WO2004058715A1 (ja) | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| JP2006507216A (ja) | 2002-02-11 | 2006-03-02 | プリバ イストラツイヴアンイエ イ ラツヴオイ ディ.オー.オー | 用量ダンピングの恐れを低減させた新規な薬物送達システムとしての徐放性固形製剤 |
| WO2008041553A1 (fr) * | 2006-09-26 | 2008-04-10 | Astellas Pharma Inc. | Préparation à libération entretenue de tacrolimus |
| JP2008169173A (ja) * | 2007-01-15 | 2008-07-24 | Kissei Pharmaceut Co Ltd | 炭水化物分解酵素阻害剤の胃内滞留型徐放性製剤 |
| WO2008100107A1 (en) * | 2007-02-15 | 2008-08-21 | Amorepacific Corporation | Controlled-release preparation containing cilostazol and process for the preparation thereof |
| JP2009500317A (ja) * | 2005-06-29 | 2009-01-08 | パナセア バイオテック リミテッド | 放出特性改良医薬組成物およびその製造方法 |
| WO2009047802A2 (en) * | 2007-10-10 | 2009-04-16 | Lupin Limited | Novel colon targeted modified release bioadhesive formulation of 5-amino salicylic acid or its salts and metabolites thereof |
| WO2009057138A2 (en) * | 2007-10-29 | 2009-05-07 | Lupin Limited | Controlled release pharmaceutical compositions of tolterodine |
| JP2009532462A (ja) * | 2006-04-05 | 2009-09-10 | カディラ・ヘルスケア・リミテッド | 調節放出クロピドグレル製剤 |
| JP2009535351A (ja) * | 2006-04-26 | 2009-10-01 | スパーナス ファーマシューティカルズ インコーポレイテッド | シグモイド放出プロファイルを有するオキサカルバゼピンの制御放出製剤 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8601204D0 (en) * | 1986-01-18 | 1986-02-19 | Boots Co Plc | Therapeutic agents |
| US5783212A (en) * | 1996-02-02 | 1998-07-21 | Temple University--of the Commonwealth System of Higher Education | Controlled release drug delivery system |
| JP2002212062A (ja) * | 2001-01-24 | 2002-07-31 | Teijin Ltd | 遅延放出制御組成物 |
| IN191028B (ja) * | 2001-05-17 | 2003-09-13 | Sun Pharmaceutical Ind Ltd | |
| US7125563B2 (en) * | 2002-04-12 | 2006-10-24 | Dava Pharmaceuticals, Inc. | Sustained release pharmaceutical preparations and methods for producing the same |
| US20050186276A1 (en) * | 2003-07-17 | 2005-08-25 | Pfizer Inc | Pharmaceutical formulations |
| US9173847B2 (en) * | 2003-10-10 | 2015-11-03 | Veloxis Pharmaceuticals A/S | Tablet comprising a fibrate |
| US20050096365A1 (en) * | 2003-11-03 | 2005-05-05 | David Fikstad | Pharmaceutical compositions with synchronized solubilizer release |
| US8007826B2 (en) * | 2003-12-11 | 2011-08-30 | Acorda Therapeutics, Inc. | Sustained release aminopyridine composition |
| FR2874325B1 (fr) * | 2004-08-19 | 2006-10-20 | Sanofi Synthelabo | Composition pharmaceutique sous forme de comprime a residence gastrique contenant de l'alfuzosine |
| US20080274180A1 (en) * | 2005-08-30 | 2008-11-06 | Nicholas Piramal India Limited | Extended Release Pharmaceutical Composition of Metformin and a Process for Producing It |
| JP2007106759A (ja) * | 2005-09-16 | 2007-04-26 | Dai Ichi Seiyaku Co Ltd | 光学活性なジアミン誘導体およびその製造方法 |
| WO2007090881A2 (en) * | 2006-02-10 | 2007-08-16 | Boehringer Ingelheim International Gmbh | Modified release formulation |
| CA2569776A1 (en) * | 2006-02-17 | 2007-08-17 | Kos Life Sciences, Inc. | Low flush niacin formulation |
| US20080004260A1 (en) * | 2006-06-29 | 2008-01-03 | Transcept Pharmaceuticals, Inc. | Compositions of 5-HT3 antagonists and dopamine D2 antagonists for treatment of dopamine-associated chronic conditions |
| CN100563637C (zh) * | 2006-10-13 | 2009-12-02 | 北京红林制药有限公司 | 一种控释给药的药芯组合物和控释制剂及其制备方法 |
| FI2140867T4 (fi) * | 2007-03-29 | 2023-09-14 | Farmaseuttinen koostumus |
-
2011
- 2011-02-21 TW TW100105563A patent/TW201200165A/zh unknown
- 2011-02-21 JP JP2012500677A patent/JP5714562B2/ja active Active
- 2011-02-21 WO PCT/JP2011/053644 patent/WO2011102506A1/ja active Application Filing
- 2011-02-21 EP EP11744792.0A patent/EP2540294B1/en not_active Not-in-force
- 2011-02-21 ES ES11744792.0T patent/ES2600894T3/es active Active
-
2012
- 2012-08-22 US US13/591,981 patent/US20130005763A1/en not_active Abandoned
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001513752A (ja) * | 1996-09-12 | 2001-09-04 | ロシュ ダイアグノスティクス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 迅速崩壊ペレット |
| JP2001522794A (ja) * | 1997-11-12 | 2001-11-20 | ベーリンガー・マンハイム・ファーマシューティカルズ・コーポレイション−スミスクライン・ベックマン・コーポレイション・リミテッド・パートナーシップ・ナンバー1 | カルベジロールの新規経口剤形 |
| JP2003507413A (ja) * | 1999-08-26 | 2003-02-25 | ニューロクライン バイオサイエンシーズ, インコーポレイテッド | 徐放性鎮静催眠組成物およびそれに関連する方法 |
| JP2001270821A (ja) * | 2000-03-23 | 2001-10-02 | Eisai Co Ltd | 服用感が優れた散剤 |
| JP2004518676A (ja) | 2000-12-20 | 2004-06-24 | シャイア ラボラトリーズ,インコーポレイテッド | 最小化pH依存性溶解プロフィールを有する徐放性薬学的剤形 |
| WO2003000657A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003000680A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003016302A1 (fr) | 2001-08-09 | 2003-02-27 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| JP2006507216A (ja) | 2002-02-11 | 2006-03-02 | プリバ イストラツイヴアンイエ イ ラツヴオイ ディ.オー.オー | 用量ダンピングの恐れを低減させた新規な薬物送達システムとしての徐放性固形製剤 |
| WO2004058715A1 (ja) | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| JP2009500317A (ja) * | 2005-06-29 | 2009-01-08 | パナセア バイオテック リミテッド | 放出特性改良医薬組成物およびその製造方法 |
| JP2009532462A (ja) * | 2006-04-05 | 2009-09-10 | カディラ・ヘルスケア・リミテッド | 調節放出クロピドグレル製剤 |
| JP2009535351A (ja) * | 2006-04-26 | 2009-10-01 | スパーナス ファーマシューティカルズ インコーポレイテッド | シグモイド放出プロファイルを有するオキサカルバゼピンの制御放出製剤 |
| WO2008041553A1 (fr) * | 2006-09-26 | 2008-04-10 | Astellas Pharma Inc. | Préparation à libération entretenue de tacrolimus |
| JP2008169173A (ja) * | 2007-01-15 | 2008-07-24 | Kissei Pharmaceut Co Ltd | 炭水化物分解酵素阻害剤の胃内滞留型徐放性製剤 |
| WO2008100107A1 (en) * | 2007-02-15 | 2008-08-21 | Amorepacific Corporation | Controlled-release preparation containing cilostazol and process for the preparation thereof |
| WO2009047802A2 (en) * | 2007-10-10 | 2009-04-16 | Lupin Limited | Novel colon targeted modified release bioadhesive formulation of 5-amino salicylic acid or its salts and metabolites thereof |
| WO2009057138A2 (en) * | 2007-10-29 | 2009-05-07 | Lupin Limited | Controlled release pharmaceutical compositions of tolterodine |
Non-Patent Citations (17)
| Title |
|---|
| AMERICAN CHEMICAL SOCIETY-226TH NATIONAL MEETING, NEW YORK CITY, NY, USA, 2003 |
| BLOOD, 2003, pages 102 |
| GENERATING GREATER VALUE FROM OUR PRODUCTS AND PIPELINE. AVENTIS SA COMPANY PRESENTATION, 5 February 2004 (2004-02-05) |
| J. R. PRUITT ET AL., J. MED. CHEM., vol. 46, 2003, pages 5298 - 5313 |
| M. WILEY ET AL., 228TH ACS NATIONAL MEETING, PHILADELPHIA, 22 August 2004 (2004-08-22), pages 252,254 |
| PINTO DJP; ORWAT MJ; LAM PYS ET AL.: "Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa", J. MED. CHEM., vol. 50, no. 22, 2007, pages 5339 - 56, XP007915445, DOI: doi:10.1021/jm070245n |
| S. TAKEHANA ET AL., JAPANESE JOURNAL OF PHARMACOLOGY, vol. 82, no. 1, 2000, pages 213P |
| S. YOUNG, MEDICINAL CHEMISTRY-12TH RSC-SCI SYMPOSIUM, 7 September 2003 (2003-09-07) |
| See also references of EP2540294A4 |
| SUSANNE R ET AL., J. MED. CHEM., vol. 48, 2005, pages 5900 - 5908 |
| T. KAYAHARA ET AL., JAPANESE JOURNAL OF PHARMACOLOGY, vol. 82, no. 1, 2000, pages 213P |
| WFO DRUG INFORMATION, vol. 16, no. 3, 2002, pages 257 |
| WFO DRUG INFORMATION, vol. 18, no. 3, 2004, pages 260 |
| WFO DRUG INFORMATION, vol. 20, no. 1, 2006, pages 38 |
| WFO DRUG INFORMATION, vol. 22, no. 3, 2008, pages 226 - 227 |
| XVIITH CONGRESS OF THE INTERNATIONAL SOCIETY FOR THROMBOSIS AND HAEMOSTASIS, WASHINGTON D. C., USA, 14 August 1999 (1999-08-14) |
| ZHANG P; HUANG W; ZHU BY ET AL.: "Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide, a highly potent, selective, and orally efficacious factor Xa inhibitor", BIOORG MED. CHEM. LETT., vol. 19, no. 8, 2009, pages 2179 - 85, XP026079433, DOI: doi:10.1016/j.bmcl.2009.02.111 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9918975B2 (en) | 2010-03-19 | 2018-03-20 | Daiichi Sankyo Company, Limited | Method for improving dissolution of anticoagulant agent |
| WO2013022059A1 (ja) | 2011-08-10 | 2013-02-14 | 第一三共株式会社 | ジアミン誘導体含有医薬組成物 |
| KR20140054029A (ko) | 2011-08-10 | 2014-05-08 | 다이이찌 산쿄 가부시키가이샤 | 디아민 유도체 함유 의약 조성물 |
| US9402907B2 (en) | 2011-08-10 | 2016-08-02 | Daiichi Sankyo Company, Limited | Pharmaceutical composition containing diamine derivative |
| JP2015500812A (ja) * | 2011-12-02 | 2015-01-08 | シンクロニューロン インコーポレイテッド | アカンプロサート製剤、その使用方法、及びそれを含む合剤 |
| US10512621B2 (en) | 2011-12-02 | 2019-12-24 | Synchroneuron, Inc. | Methods of treating posttraumatic stress disorder with acamprosate salts |
| US10166207B2 (en) | 2013-06-05 | 2019-01-01 | Synchroneuron, Inc. | Acamprosate formulations, methods of using the same, and combinations comprising the same |
| KR101535586B1 (ko) * | 2014-08-01 | 2015-07-09 | 에스케이케미칼주식회사 | 무정형 또는 열역학적 준 안정형 리바록사반을 포함하는 약학 제제 |
| WO2016017993A1 (ko) * | 2014-08-01 | 2016-02-04 | 에스케이케미칼 주식회사 | 무정형 또는 열역학적 준 안정형 리바록사반을 포함하는 약학 제제 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5714562B2 (ja) | 2015-05-07 |
| TW201200165A (en) | 2012-01-01 |
| US20130005763A1 (en) | 2013-01-03 |
| EP2540294A1 (en) | 2013-01-02 |
| JPWO2011102506A1 (ja) | 2013-06-17 |
| EP2540294A4 (en) | 2013-12-04 |
| EP2540294B1 (en) | 2016-08-03 |
| ES2600894T3 (es) | 2017-02-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5714562B2 (ja) | 経口用徐放性固形製剤 | |
| KR101424843B1 (ko) | 의약 조성물 | |
| EP1441713B1 (en) | Modified release tamsulosin tablets | |
| US9629808B2 (en) | Sustained-release solid preparation for oral use | |
| FI117373B (fi) | Parasetamolia ja domperidonia sisältävä kalvopäällysteinen tabletti | |
| JPWO2013022059A1 (ja) | ジアミン誘導体含有医薬組成物 | |
| WO2010147169A1 (ja) | 溶出性の改善された医薬組成物 | |
| JP2013532651A (ja) | シロドシンと塩基性コポリマーとの混合物を含む経口投与用医薬品 | |
| JP5870023B2 (ja) | 経口用徐放性固形製剤 | |
| CN109996542A (zh) | 包含二胺衍生物的口腔崩解片 | |
| AU2013365715B2 (en) | A pharmaceutical composition containing candesartan cilexetil and amlodipine | |
| JP2016117738A (ja) | シロドシン−シクロデキストリン包接化合物 | |
| TW201431569A (zh) | 具有高含量菲索芬那定(fexofenadine)之固態單位及其製備方法 | |
| WO2016012898A1 (en) | Oral pharmaceutical composition of lurasidone | |
| JP2022113667A (ja) | エドキサバン含有医薬組成物 | |
| JP6116847B2 (ja) | シクロデキストリンとの混合体を含有する錠剤 | |
| MX2007016238A (es) | Formulacion de liberacion prolongada de principios activos que presentan una solubilidad dependiente de ph. | |
| JP2020520892A (ja) | ダビガトランエテキシラート又は医薬的に許容されるその塩を含む錠剤及びその製造方法 | |
| JP6858873B2 (ja) | セレコキシブを含む錠剤 | |
| TWI810656B (zh) | 二氟甲基鳥氨酸及舒林酸(sulindac),固定劑量組合調配物 | |
| BR112015018952B1 (pt) | Composições farmacêuticas contendo dexcetoprofeno e tramadol, seu método de fabricação e seu uso | |
| JP2018024632A (ja) | 口腔内崩壊性被覆錠剤及び口腔内崩壊性被覆錠剤の被覆層用組成物 | |
| AU2007201830A1 (en) | High drug load tablet |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11744792 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012500677 Country of ref document: JP |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011744792 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011744792 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |