WO2011037207A1 - ストラメノパイルの形質転換方法 - Google Patents
ストラメノパイルの形質転換方法 Download PDFInfo
- Publication number
- WO2011037207A1 WO2011037207A1 PCT/JP2010/066599 JP2010066599W WO2011037207A1 WO 2011037207 A1 WO2011037207 A1 WO 2011037207A1 JP 2010066599 W JP2010066599 W JP 2010066599W WO 2011037207 A1 WO2011037207 A1 WO 2011037207A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- desaturase
- fatty acid
- stramenopile
- schizochytrium
- Prior art date
Links
Images











































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/64—Fats; Fatty oils; Ester-type waxes; Higher fatty acids, i.e. having at least seven carbon atoms in an unbroken chain bound to a carboxyl group; Oxidised oils or fats
- C12P7/6409—Fatty acids
-
- C—CHEMISTRY; METALLURGY
- C11—ANIMAL OR VEGETABLE OILS, FATS, FATTY SUBSTANCES OR WAXES; FATTY ACIDS THEREFROM; DETERGENTS; CANDLES
- C11C—FATTY ACIDS FROM FATS, OILS OR WAXES; CANDLES; FATS, OILS OR FATTY ACIDS BY CHEMICAL MODIFICATION OF FATS, OILS, OR FATTY ACIDS OBTAINED THEREFROM
- C11C3/00—Fats, oils, or fatty acids by chemical modification of fats, oils, or fatty acids obtained therefrom
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8201—Methods for introducing genetic material into plant cells, e.g. DNA, RNA, stable or transient incorporation, tissue culture methods adapted for transformation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8241—Phenotypically and genetically modified plants via recombinant DNA technology
- C12N15/8242—Phenotypically and genetically modified plants via recombinant DNA technology with non-agronomic quality (output) traits, e.g. for industrial processing; Value added, non-agronomic traits
- C12N15/8243—Phenotypically and genetically modified plants via recombinant DNA technology with non-agronomic quality (output) traits, e.g. for industrial processing; Value added, non-agronomic traits involving biosynthetic or metabolic pathways, i.e. metabolic engineering, e.g. nicotine, caffeine
- C12N15/8247—Phenotypically and genetically modified plants via recombinant DNA technology with non-agronomic quality (output) traits, e.g. for industrial processing; Value added, non-agronomic traits involving biosynthetic or metabolic pathways, i.e. metabolic engineering, e.g. nicotine, caffeine involving modified lipid metabolism, e.g. seed oil composition
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0071—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0071—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14)
- C12N9/0083—Miscellaneous (1.14.99)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/64—Fats; Fatty oils; Ester-type waxes; Higher fatty acids, i.e. having at least seven carbon atoms in an unbroken chain bound to a carboxyl group; Oxidised oils or fats
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/64—Fats; Fatty oils; Ester-type waxes; Higher fatty acids, i.e. having at least seven carbon atoms in an unbroken chain bound to a carboxyl group; Oxidised oils or fats
- C12P7/6409—Fatty acids
- C12P7/6427—Polyunsaturated fatty acids [PUFA], i.e. having two or more double bonds in their backbone
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Nutrition Science (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
- Fats And Perfumes (AREA)
Abstract
Description
本発明は、以下の(1)~(22)に記載の技術的事項を要旨とするものである。
(1)ストラメノパイルに外来遺伝子を導入することを特徴とする、ストラメノパイルの形質転換方法。
(2)ストラメノパイルがラビリンチュラ類である、(1)に記載のストラメノパイルの形質転換方法。
(3)ラビリンチュラ類がラビリンチュラ属(Labyrinthula)、アルトルニア属(Altornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、アウランティオキトリウム属(Aurantiochytrium)、ヤブレツボカビ属(Thraustochytrium)、ウルケニア属(Ulkenia)、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属する微生物である、(2)に記載のストラメノパイルの形質転換方法。
(4)微生物がSchizochytrium sp. M-8株(FERM P-19755)、Thraustochytrium aureum ATCC 34304、Thraustochytrium sp. ATCC 26185、Schizochytrium sp. AL1Ac、Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、またはParietichytrium sarkarianum SEK364(FERM ABP-11298)である、(1)ないし(3)のいずれかに記載のストラメノパイルの形質転換方法。
(5)外来遺伝子が抗生物質耐性能、発色タンパク質、および/または脂肪酸不飽和化酵素に関連する遺伝子である、(1)ないし(4)のいずれかに記載のストラメノパイルの形質転換方法。
(6)脂肪酸不飽和化酵素に関連する遺伝子がΔ5デサチュラーゼ遺伝子、Δ12デサチュラーゼ遺伝子、および/またはω3デサチュラーゼ遺伝子である、(1)ないし(5)のいずれかに記載のストラメノパイルの形質転換方法。
(7)エレクトロポレーションまたは遺伝子銃法によって外来遺伝子を導入することを特徴とする、(1)ないし(6)のいずれかに記載のストラメノパイルの形質転換方法。
(8)脂肪酸不飽和化酵素遺伝子を導入し、当該脂肪酸不飽和化酵素を発現することを特徴とする、ストラメノパイルの脂肪酸組成の改変方法。
(9)脂肪酸不飽和化酵素がデサチュラーゼである、(8)に記載の方法。
(10)脂肪酸不飽和化酵素がΔ5デサチュラーゼ、Δ12デサチュラーゼ、またはω3デサチュラーゼである、(8)または(9)に記載の方法。
(11)ストラメノパイルがラビリンチュラ類である、(8)ないし(10)のいずれかに記載の方法。
(12)ラビリンチュラ類がラビリンチュラ属(Labyrinthula)、アルトルニア属(Altornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、アウランティオキトリウム属(Aurantiochytrium)、ヤブレツボカビ属(Thraustochytrium)、ウルケニア属(Ulkenia) 、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属する微生物である、(11)に記載の方法。
(13)微生物がSchizochytrium sp. M-8株(FERM P-19755)、Thraustochytrium aureum ATCC 34304、Thraustochytrium sp. ATCC 26185、Schizochytrium sp. AL1Ac、Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、またはParietichytrium sarkarianum SEK364(FERM ABP-11298)である、(12)に記載の方法。
(14)(8)ないし(13)のいずれかに記載の方法により、ストラメノパイルに高度に脂肪酸を蓄積させる方法。
(15)脂肪酸が不飽和脂肪酸である、(14)に記載の方法。
(16)不飽和脂肪酸が炭素数18~22の不飽和脂肪酸である、(15)に記載の方法。
(17)(14)ないし(16)のいずれかに記載の方法により得られた脂肪酸。
(18)脂肪酸組成の改変を目的として形質転換されたストラメノパイル。
(19)脂肪酸を高度に蓄積させることを目的として形質転換されたストラメノパイル。
(20)ラビリンチュラ類である、(18)または(19)に記載のストラメノパイル。
(21)ラビリンチュラ類がラビリンチュラ属(Labyrinthula)、アルトルニア属(Altornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、アウランティオキトリウム属(Aurantiochytrium)、ヤブレツボカビ属(Thraustochytrium)、ウルケニア属(Ulkenia) 、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属する微生物である、(20)に記載のストラメノパイル。
(22)微生物がSchizochytrium sp. M-8株(FERM P-19755)、Thraustochytrium aureum ATCC 34304、Thraustochytrium sp. ATCC 26185、Schizochytrium sp. AL1Ac、Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、またはParietichytrium sarkarianum SEK364(FERM ABP-11298)である、(21)に記載のストラメノパイル。
[微生物]
本発明の脂肪酸改変方法で用いる微生物としては、脂肪酸不飽和化酵素遺伝子を導入することにより脂肪酸合成を行うであろうと思われるストラメノパイルであれば特に限定されないが、特に好ましくはラビリンチュラ類を対象とする。ラビリンチュラ類としては、ラビリンチュラ属(Labyrinthula)、アルトルニア属(Althornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、ヤブレツボカビ属(Thraustochytrium)、またはウルケニア属(Ulkenia)、アウランティオキトリウム属(Aurantiochytrium)、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属するものが挙げられる。
本発明の脂肪酸不飽和化酵素(デサチュラーゼ、desaturase)は脂肪酸不飽和化酵素としての機能を有するものであれば特に限定されない。脂肪酸不飽和化酵素遺伝子としては動物由来、植物由来等その由来は特に制限されないが、Δ4脂肪酸不飽和化酵素遺伝子、Δ5脂肪酸不飽和化酵素遺伝子、Δ6脂肪酸不飽和化酵素遺伝子、Δ12脂肪酸不飽和化酵素遺伝子等の脂肪酸不飽和化酵素遺伝子を好適に例示することができ、これらは単独でまたは多重に用いることができる。かかるΔ4脂肪酸不飽和化酵素遺伝子、Δ5脂肪酸不飽和化酵素遺伝子、Δ6脂肪酸不飽和化酵素遺伝子、Δ12脂肪酸不飽和化酵素遺伝子は、脂肪酸の末端カルボキシル基の炭素(デルタエンド)から数えて、それぞれ4番目、5番目、6番目、12番目の炭素において不飽和結合を形成するものであり、これらの脂肪酸不飽和化酵素遺伝子としては、例えば微細藻類由来のΔ4脂肪酸不飽和化酵素遺伝子(Tonon, T., Harvey, D., Larson, T. R., and Graham, I. A. Identification of a very long chain polyunsaturated fatty acidΔ4-desaturase from the microalga Pavlova lutheri.非特許文献5)を具体的に挙げることができる。さらに具体的には、Δ5デサチュラーゼとしては、T. aureum由来Δ5デサチュラーゼ、Thraustochytrium sp.ATCC 26185, Dictyostelium discoideum, Rattus norvegicus, Mus musculus, Homo sapiens, Caenorhabditis elegans, Leishmania majorに由来するΔ5デサチュラーゼ等が例示される。また、Δ12デサチュラーゼとしては、ピンギオ藻Pinguiochrysis pyriformis由来Δ12デサチュラーゼ、真菌および原虫由来Δ12デサチュラーゼが例示される。
ストラメノパイル内で発現する脂肪酸不飽和化酵素により生成される不飽和脂肪酸は、例えば、炭素数18~22の不飽和脂肪酸である。用いる脂肪酸不飽和化酵素の種類や脂肪酸基質の種類によって異なるが、ドコサヘキサエン酸(DHA)やエイコサペンタエン酸(EPA)を好適に例示することができ、これらの他にα-リノレン酸(ALA)、オクタデカテトラエン酸(OTA、18:4n-3)、エイコサテトラエン酸(ETA、20:4n-3)、n-3ドコサペンタエン酸(DPA、22:5n-3)、テトラコサペンタエン酸(TPA、24:5n-3)、テトラコサヘキサエン酸(THA、24:6n-3)、リノール酸(LA)、γ-リノレン酸(GLA)、エイコサトリエン酸(20:3n-6)、アラキドン酸(AA)、n-6ドコサペンタエン酸(DPA, 22:5n-6)等を挙げることができる。
本発明において脂肪酸不飽和化酵素遺伝子源として利用できる生物は、段落[0021]に記載のように、特に属、種あるいは株などを限定するものではなく、高度不飽和脂肪酸生産能を有する生物であればいずれのものでも用いることができる。これらの生物は、例えば微生物であれば公的微生物寄託機関で容易に入手することができる。このような微生物としては、モリテラ属(Moritella)に属する細菌モリテラ・マリナ(Moritella marina)MP-1株(ATCC15381)等が例示され、デサチュラーゼおよびエロンガーゼ遺伝子源の例として本株を用いる場合の方法について以下に説明する。しかしながら、ここで示す方法はデサチュラーゼ/エロンガーゼ経路を有する全ての生物種から、その構成遺伝子であるデサチュラーゼおよびエロンガーゼ遺伝子の単離を行う際に適応が可能である。
MP-1株からデサチュラーゼおよび/もしくはエロンガーゼ遺伝子を単離するためには、それぞれの目的酵素遺伝子のアミノ酸配列において、保存された領域を推定することが必要である。例えば、デサチュラーゼにおいては一つのシトクロムb5ドメインと3つのヒスチジンボックス、エロンガーゼにおいては2つのヒスチジンボックスが生物種を通して保存されていることが知られている。より具体的には、様々な生物種由来の既知のデサチュラーゼもしくはエロンガーゼ遺伝子のアミノ酸配列を、clustal wプログラム (Thompson, J.D., et al. 非特許文献6)を使用した多重アライメントによって比較することで、目的酵素の保存領域を推定することが出来る。さらには、既知の他生物由来デサチュラーゼおよび/もしくはエロンガーゼにおいて、同じ基質特異性を有するデサチュラーゼもしくはエロンガーゼ遺伝子のアミノ酸配列を多重アライメントによって比較することで、同じ基質特異性を有するデサチュラーゼおよび/もしくはエロンガーゼに特異的な保存領域を推定することも可能である。次に、推定した保存領域を元に様々な縮重オリゴヌクレオチドプライマーを作製し、MP-1株由来cDNAライブラリーを鋳型にしたPCR、RACE法などを行うことで、MP-1株由来目的遺伝子の部分配列を増幅する。引き続き、得た増幅産物をプラスミドベクターにクローニング後、常法により塩基配列を決定し、既知の酵素遺伝子と比較することにより、MP-1株より目的酵素遺伝子の一部が単離できたことを確認する。目的酵素遺伝子全長取得には、得た部分配列をプローブとしたハイブリダイゼーション法によるスクリーニング、もしくは目的遺伝子の部分配列から作製したオリゴヌクレオチドプライマーを用いたRACE法を行う。
[その他遺伝子源]
GFP(Green Fluorescent Protein)は 非特許文献7あるいは非特許文献8、EGFP(enhanced GFP)は 特許文献6、ネオマイシン耐性遺伝子は非特許文献9が、それぞれ参考になる。
脂肪酸不飽和化酵素遺伝子導入方法としては、従来の微生物への遺伝子導入により形質転換方法が適用でき、組換え発現ベクターを細胞に導入して形質転換する方法を挙げることができる。本発明におけるストラメノパイルへのデサチュラーゼ遺伝子の導入の詳細については実施例に具体的に記載した。また、形質転換の対象となるストラメノパイルも特に限定されるものではなく、前述するように、ラビリンチュラ類を好適な例として挙げることができる。
なお、本発明での不飽和脂肪酸とは、不飽和脂肪酸を含む物質をも意味し、その含有量、純度、形状、組成、等は特に限定されるものではない。つまり、本発明では、脂肪酸組成が改変された細胞またはその培地自体を、不飽和脂肪酸とみなしてもよい。また、この細胞または培地から不飽和脂肪酸を精製する工程をさらに含んでいてもよい。不飽和脂肪酸を精製する方法としては、不飽和脂肪酸を始めとする脂質(複合脂質を含む)の精製方法として公知である方法を適用することができる。
ストラメノパイルに不飽和脂肪酸を蓄積させるには、形質転換した本発明のストラメパイルを培養することにより達成される。例えば、通常用いられる固体培地あるいは液体培地等で培養する。この時、用いられる培地としては、例えば炭素源としてグルコース、フルクトース、サッカロース、デンプン、グリセリン等、また窒素源として酵母エキス、コーンスティープリカー、ポリペプトン、グルタミン酸ナトリウム、尿素、酢酸アンモニウム、硫酸アンモニウム、硝酸アンモニウム、塩化アンモニウム、硝酸ナトリウム等、また無機塩としてリン酸カリウム等、その他必要な成分を適宜組み合わせた培地であり、ラビリンチュラ類を培養するために通常用いられるものであれば特に限定されないが、特に好ましくは酵母エキス・グルコース寒天培地(GY培地)が用いられる。培地は調製後、pHを3.0~8.0の範囲内に調整した後、オートクレーブ等により殺菌して用いる。培養は、10~40℃、好ましくは15~35℃にて、1~14日間、通気撹拌培養、振とう培養、あるいは静置培養で行えば良い。
また、ある種のラビリンチュラにおける、異種ラビリンチュラ由来Δ5デサチュラーゼの発現試験によると、EPAの含有率が1.4倍程度上昇した。ETAまたはDGLAを含有する培地で培養すると、ETAとDGLAはそれぞれEPAとAAに変換され、不飽和脂肪酸が増加した。ラビリンチュラ内での変換効率は、ラビリンチュラ内での前駆物質の変換効率は酵母の場合よりも高く、ETAで75%、DGLAで63%であった。これらの結果はCG-MSの試験データから得られた。
(1)本発明で使用する菌株
Thraustochytrium aureum ATCC 34304、およびThraustochytrium sp. ATCC 26185はATCCより分譲を受けた。Aurantiochytrium limacinum mh0186、およびSchizochytrium sp. AL1Acは宮崎大学農学部より分譲を受けた。
また、Schizochytrium aggregatum ATCC 28209、およびUlkenia sp. ATCC 28207はATCCより分譲を受けた。Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、およびParietichytrium sarkarianum SEK364は甲南大学理工学部より分譲を受けた。
i.寒天平板培地組成
PDA寒天平板培地
0.78% (w/v) ポテトデキストロース寒天培地 (日水製薬社製)、1.75% (w/v) シーライフ (マリンテック社製) 、1.21% (w/v) 寒天末 (ナカライテスク社製) を混合し、121℃で20分間オートクレーブ滅菌を行った。十分に冷却後に、バクテリアのコンタミネーションを防ぐために終濃度が100 μg/mlとなるようにアンピシリン酸ナトリウム (ナカライテスク社製) を添加し、シャーレに分注して平らな所に静置して固化させた。
GY液体培地
3.18% (w/v) グルコース (ナカライテスク社製)、1.06% (w/v) 乾燥酵母エキス (ナカライテスク社製)、1.75% (w/v) シーライフ (マリンテック社製)を混合し、を混合し、121℃で20分間オートクレーブ滅菌後に、100 μg/mlのアンピシリン酸ナトリウム (ナカライテスク社製) を添加した。
0.48% (w/v) ポテトデキストロース (Difco社製)、1.75% (w/v) シーライフ (マリンテック社製) を混合し、121℃で20分間オートクレーブ滅菌後に、100 μg/mlのアンピシリン酸ナトリウム (ナカライテスク社製) を添加した。
0.2% (w/v)グルコース (ナカライテスク社製)、0.02% (w/v)乾燥酵母エキス (ナカライテスク社製)、0.05%グルタミン酸ナトリウム(ナカライテスク社製)、1.75% (w/v)シーライフ (マリンテック社製)を混合し、121℃で20分間オートクレーブ滅菌後に、100 μg/mlのアンピシリン酸ナトリウム (ナカライテスク社製)を添加した。
i.寒天平板培養
ラビリンチュラ菌体を白金耳、もしくはスプレッダーを用いて接種し、25℃で静置培養することでコロニーを出現させた。その継代培養に関しては、コロニーを白金耳で釣菌して滅菌生理食塩水に懸濁後、その懸濁液を白金耳、もしくはスプレッダーを用いて塗布することで行った。尚、必要に応じて平板上の菌体を液体培地に接種することで、液体培養への転換を行った。
ラビリンチュラ菌体を接種し、三角フラスコ、もしくは試験管を用いて、25℃、150 rpmの回転数で攪拌しながら懸濁培養を行った。その継代培養に関しては、増殖が確認された対数増殖期~定常期までの培養液を、新しいGYもしくはPD液体培地に対して1/200~1/10容添加することで行った。尚、必要に応じて細胞培養液をPDA寒天平板培地に塗布することで、寒天平板培養への転換を行った。
継代培養に加えて、グリセロールストック作製による凍結保存を行った。すなわち、GY液体培地を用いた対数増殖期~定常期の細胞懸濁液に対して、終濃度が15% (v/v) のグリセロール (ナカライテスク社製) を添加し、-80℃のディープフリーザー中にて保存した。
(1)液体培養による感受性を示す抗生物質のスクリーニング
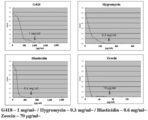
ラビリンチュラ類4株の前培養液を、様々な抗生物質を含有したGY液体培地添加し、150 rpm、25℃で5日間培養後に600nmにおける濁度(OD600)を測定した。使用した抗生物質と濃度、およびその結果を図1に示す。
感受性を示した抗生物質に関して、液体培養におけるMICの決定を行った。ラビリンチュラ類4株の前培養液を、様々な濃度の各種抗生物質を含有するGY液体培地に添加し、150 rpm、25℃で5日間培養後に600nmにおける濁度(OD600)を測定した。T. aureumの結果を図2に、Thraustochytrium sp. ATCC 26185の結果を図3に、A. limacinum mh0186の結果を図4に、Schizochytrium sp. AL1Acの結果を図5にそれぞれ示す。
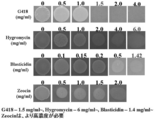
ラビリンチュラ類4株の前培養液5 μlを様々な濃度の各種抗生物質を含有するPDA寒天平板培地に滴下し、25℃で7日間培養後にコロニー形成を観察した。T. aureumの結果を図6に、Thraustochytrium sp. ATCC 26185の結果を図7に、A. limacinum mh0186の結果を図8に、Schizochytrium sp. AL1Acの結果を図9にそれぞれ示す。

(1)T. aureum由来EF-1α遺伝子と遺伝子発現領域の単離
i.T. aureum由来EF-1α遺伝子cDNA配列の単離
GY液体培地で培養したT. aureumの対数増殖期~定常期の菌体を4℃、3,500 x g、10 min遠心することによって集菌した。得た菌体を滅菌生理食塩水に懸濁後に再遠心操作を行うことで菌体を洗浄し、液体窒素によって急速凍結後に乳鉢中で粉末状になるまで擂り潰した。次に、細胞破砕液中からセパゾールRNA I Super (ナカライテスク社製) を使用して全RNAを抽出した。引き続き、OligotexTM-dT30<Super> mRNA Purification Kit (タカラバイオ社製) を使用し、全 RNAからmRNAの精製を行った。
次に、SMARTTM RACE cDNA Amplification Kit (clontech社製) を使用し、5’および3’末端に合成アダプターを付加したcDNAライブラリーを作製した。また既知のEF-1αの保存配列を元にして、DNA合成機 (Applied Biosystems社製) を使用して1つの正方向縮重オリゴヌクレオチドプライマーEF-F1(配列表配列番号1)を作製した。これらを用いて3’ RACEを行ったところ、特異的な増幅産物が確認された。そこで1%アガロースゲルによる電気泳動で分離した該DNA断片を清浄なカッター等で切り出し、非特許文献10に記載の方法に従ってアガロースゲルからDNAを抽出した。次に、pGEMR-T easy Vector System I (Promega社製) を使用して該DNA断片のTAクローニングを行い、Sangerらの方法 (非特許文献11) によってそれらの塩基配列を決定した。具体的には、BigDyeR Terminator v3.1Cyele Sequencing Kitおよび3130ジェネティックアナライザ (Applied Biosystems社製) を使用したダイターミネーター法による塩基配列決定を行った。その結果、得た980 bpの3’ RACE産物(配列表配列番号2)は他生物由来EF-1α遺伝子と高い相同性を示すことから、T. aureum由来EF-1α遺伝子の部分配列であることが強く示唆された。
以上の結果から、今回得たT. aureum由来推定EF-1α遺伝子のcDNA配列は1,396 bp(配列表配列番号7)であり、うちORF領域に関しては、341アミノ酸残基(配列表配列番号8)をコードする1,023 bp(配列表配列番号9)であることが示された。
GY培地で培養したT. aureum菌体を遠心によって集菌した。得た菌体を滅菌生理食塩水に懸濁後に再遠心操作を行うことで菌体を洗浄し、液体窒素で急速凍結後に乳鉢中で粉末状になるまで擂り潰した。その後、非特許文献12に記載の方法に従ってゲノムDNAを抽出し、A260/280を測定することで、抽出したゲノムDNAの純度と濃度の測定を行った。
次に、LA PCR in vitro Cloning Kitを用いて、PCRゲノムウォーキング法によるEF-1α遺伝子ORF上流配列 (プロモーター)、もしくはORF下流配列 (ターミネーター) の単離を試みた。尚、ORF上流配列の増幅に用いた逆方向オリゴヌクレオチドプライマーはr3(配列表配列番号10)、ORF下流配列の増幅に用いた正方向オリゴヌクレオチドプライマーはEF-t-F1(配列表配列番号11)およびEF-t-F2(配列表配列番号12)である。得られた特異的増幅産物の塩基配列を解析したところ、615 bpのT. aureum由来EF-1α遺伝子ORF上流配列(配列表配列番号13)、および1,414 bpのORF下流配列(配列表配列番号14)の単離に成功したことが分かった。以降、前者をEF-1αプロモーター、後者をEF-1αターミネーターと表記する。
i.T. aureum由来ユビキチン遺伝子cDNA配列の単離
SMARTTM RACE cDNA Amplification Kit (clontech社製) を用いて作製したcDNAライブラリーを鋳型として、既知のユビキチン遺伝子の保存配列を元に作製した正方向縮重オリゴヌクレオチドプライマー2F(配列表配列番号15)を使用した3’ RACEを行った。得た増幅産物の塩基配列を解析したところ、278bp(配列表配列番号16)のT. aureum由来推定ユビキチン遺伝子の部分配列であることが分かった。5’ RACEに関しては、種々のオリゴヌクレオチドプライマーの使用、およびPCR条件を試みたにも関わらず、特異的増幅産物を得ることが出来なかった。このことから、高GC含量に由来した目的mRNAの高次構造が、cDNAライブラリー作製時の逆転写反応を阻害している可能性が疑われた。
そこで、熱安定性の高い逆転写酵素を用いる5’ RACE System for Rapid Amplification of cDNA Ends, Version 2.0 (Invitrogen社製) を使用した5’ RACEを行った。尚、逆転写反応に用いた逆方向オリゴヌクレオチドプライマー、およびcDNA合成後のPCR反応に用いた逆方向ヌクレオチドプライマーは、それぞれ1R(配列表配列番号17)、および2R(配列表配列番号18)である。得た増幅産物の塩基配列を解析したところ、260bp(配列表配列番号19)のT. aureum由来推定ユビキチン遺伝子の部分配列であり、3’ RACE産物との重複部分は完全に一致した。以上の結果から、T. aureum由来推定ユビキチン遺伝子cDNA配列の単離に成功したことが分かった。
LA PCR in vitro Cloning Kitを用いて、PCRゲノムウォーキング法によるユビキチン遺伝子ORF上流配列 (プロモーター)、もしくはORF下流配列 (ターミネーター) の単離を試みた。尚、ORF上流配列の増幅に用いた逆方向オリゴヌクレオチドプライマーはREVERS-U PR-1(配列表配列番号22)およびREVERS-U PR-2(配列表配列番号23)、ORF下流配列の増幅に用いた正方向オリゴヌクレオチドプライマーはubqterminalf1(配列表配列番号24)およびter2F(配列表配列番号25)である。得た特異的な増幅産物の塩基配列を解析したところ、801 bpのT. aureum由来ユビキチン遺伝子ORF上流配列(配列表配列番号26)、および584 bpのORF下流配列(配列表配列番号27)の単離に成功したことが分かった。以降、前者をユビキチンプロモーター、後者をユビキチンターミネーターと表記する。
(1)ネオマイシン耐性遺伝子 (Neor) の人工合成
メディビック社に依頼し、codon usage database (www.kazusa.or.jp/codon/) に記載のT. aureumのコドン使用頻度に準じた人工合成Neorを合成した。その塩基配列を配列表配列番号28に示し、コードするアミノ酸配列を配列表配列番号29に示す。
i.EF-1αプロモーター・ターミネーターを用いたNeor発現カセットの構築
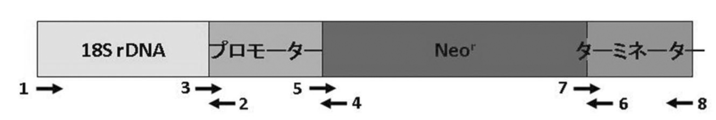
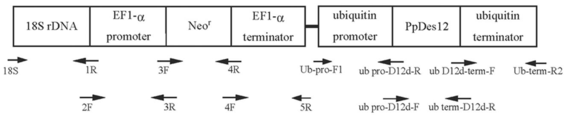
「日本農芸化学会誌 第77巻第2号 (2003年2月) p150-153」記載の方法に従って、fusion PCRによってEF-1αプロモーター/人工合成Neor/ EF-1αターミネーターから成る薬剤耐性遺伝子 (Neor) 発現カセットの上流に、T. aureum由来18S rDNAを連結したDNA断片を、配列表配列番号30~38記載のオリゴヌクレオチドプライマーを用いて作製した。尚、PCR反応条件は変性温度を98℃10秒とし、アニーリングおよび伸長反応はプライマーのTmおよび増幅産物の長さによって適宜調節して行った。
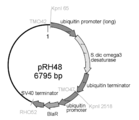
その結果、4,454 bpのT. aureum 18S rDNA/EF-1αプロモーター/人工合成Neor/EF-1αターミネーターの連結に成功した(配列表配列番号39、図10)。
EF-1αプロモーター・ターミネーターを用いたNeor発現カセットの場合と同様の手法により、配列表配列番号40~47記載のオリゴヌクレオチドプライマーを用いて、T. aureum 18S rDNA/ユビキチンプロモーター/人工合成Neor/ユビキチンターミネーターを連結した(図11)。構築したNeor発現カセットをpUC18ベクターのNdeI/KpnIサイトを利用して組み込み、ラビリンチュラ用の選択マーカーとしてユビキチンプロモーター・ターミネーターを用いたNeor発現カセット、および相同組換え部位としてT. aureum由来18S rDNA配列を有するラビリンチュラ-大腸菌シャトルベクターを構築した。以降、これをpUBNeomycinrと表記する (図12)。
(1)遺伝子導入実験に用いたDNA
以降に示す4種のDNAを用いた遺伝子導入実験を行った。
(1) 環状ベクターpUBNeomycinr
(2) 同 pUBNeomycinr
(3) ユビキチンプロモーター・ターミネーターによる直鎖状Neor発現カセット (ub-Neor)
(4) EF-1αプロモーター・ターミネーターによる直鎖状Neor発現カセット(EF-Neor)
(3) に関しては、pUBNeomycinrを鋳型とし、LA taq Hot Start Version (タカラバイオ社製) を使用して1対のオリゴヌクレオチドプライマーUbpro-fug-18s-F(配列表配列番号42)/KpnterR(配列表配列番号47)を用いたPCRを行い、得られた増幅産物をゲル精製したもの、(4) に関してはpEFNeomycinrを鋳型とし、LA taq Hot Start Version (タカラバイオ社製) を使用して1対のオリゴヌクレオチドプライマー2F(配列表配列番号32)/terminator 5R(配列表配列番号33)を用いたPCRを行い、得られた増幅産物をゲル精製したものである。
i.エレクトロポレーション法
GY液体培地を用いて、ラビリンチュラ類を25℃、150 rpmで対数増殖期中期~後期まで培養後に、3,500 x g, 4℃, 10 min遠心し上清を除いた。得られた菌体を滅菌1.75% シーライフ (マリンテック社製) に懸濁後に、再遠心操作を行うことで菌体を洗浄した。次に、5 x 106個の細胞を50 mMスクロース、もしくは使用機器に付属した遺伝子導入試薬に懸濁し、様々な条件の電気パルスを印加した。電気パルスの印加後はただちに1 mlのGY液体培地を加え、25℃で12時間培養を行った。その後、培養液を2 mg/ml (T. aureum, Thraustochytrium sp. ATCC 26185, Schozochytrium sp. AL1Ac) もしくは0.5 mg/ml (A. limacinum mh0186) のG418を含有するPDA寒天平板培地に塗布して25℃で静置培養を行い、G418耐性能が付与された形質転換体のコロニー形成を観察した。
GY液体培地を用いて、ラビリンチュラ類を25℃、150 rpmで対数増殖期中期~後期まで培養後に、3,500 x g, 4℃, 10 min遠心し上清を除いた。次に、得られた菌体を元の培養液の100倍濃縮となるようにGY液体培地に再懸濁し、うち20 μl分の細胞液を、G418を含有する/もしくは含有しない直径5 cmのPDA寒天平板培地上に、直径3 cm程度に薄く均一に塗布して乾燥させた。次に、PDS-1000 /Heシステム (バイオ・ラッド ラボラトリーズ社製) を使用した遺伝子銃法によるDNAの打ち込み操作を行った。打ち込み条件においては、以下に示すように打ち込み圧を変化させた条件を検討した。
・target distance - 6 cm (固定)
・vacuum - 26 inches Hg (固定)
・micro carrier size - 0.6 μm (固定)
・Rupture disk (打ち込み圧) - 450, 900, 1100, 1350, および1,550
G418を含有するPDA寒天平板培地上で打ち込み操作を行った場合は、導入後12時間程度静置培養後、PDA寒天平板に100 μlのPDA液体培地を滴下して菌体を拡げ直して静置培養を続けた。一方、G418を含有しないPDA寒天平板培地上で打ち込み操作を行った場合は、導入後12時間程度静置培養後に菌体を回収し、2 mg/mlもしくは0.5 mg/mlのG418を含有するPDA寒天平板培地に再び塗布して25℃で静置培養を行い、G418耐性能が付与された形質転換体のコロニー形成を観察した。
i.A. limacinum形質転換体
まず、エレクトロポレーション法において以下に示す条件での遺伝子導入を試みた。
・導入DNA・・・pUBNeomycinr およびub-Neor
・遺伝子導入法・・・エレクトロポレーション法
・細胞を懸濁するバッファー・・・50 mMスクロース
・遺伝子導入機器・・・Gene Pulser (バイオ・ラッド ラボラトリーズ社製) 。1 mmギャップキュベットを使用
・パルス条件・・・50 μF/50 Ω/ 0.75 kV、印加回数1回。
この結果、直鎖状DNAであるub-Neorを用いた場合において、~2.4 x 100 cfu/μg DNAの効率でG418耐性能を示すコロニーの出現が見られた。一方、環状DNAであるpUBNeomycinrを用いた場合は、導入操作を複数回行ったのにも関わらず、コロニーの形成は全く見られないことが分かった。
次に、遺伝子導入機器による導入効率の比較検討を行った。Microporator MP-100 (エア・ブラウン社製) もしくはNucleofectorTM (amaxa社製) を使用し、添付の条件検索キットを用いて導入試験を行った。その結果、Microporator MP-100では1個もコロニーが形成されなかった。一方、Nucleofectorを添付の細胞懸濁バッファーNucleofectorR solution Lと共に用いた場合において、~9.5x 100 cfu/μg DNAの効率で再現性よく形質転換体を得られることが分かった。
そこで次に、NucleofectorR solution Lを用いたパルス条件の検討を、Gene Pulser (バイオ・ラッド ラボラトリーズ社製) を使用して行った。その結果、キャパシスタンス50 μF/電気抵抗50 Ω/電場強度0.75 kV、印加回数2回の場合において、~1.2 x 101 cfu/μg DNAの効率で再現よく形質転換体が得られることが分かった。以上の結果をまとめ、以下表2に示す。

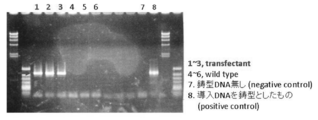
得られた形質転換体は0.5 mg/ml G418含有GY液体培地で培養し、野生株はG418を含有しないGY液体培地で培養した。両株の培養液を、G418を含む (0, 0.2, 0.5, 1, 2, 4mg/ml) PDA寒天平板培地上に10 μlずつスポットし、25℃で2日間培養を行い、寒天平板培地上での生育を観察した。その結果、野生株は0.2 mg/mlのG418で増殖が阻害されるのに対し、形質転換体は4 mg/mlのG418存在下においても増殖することが分かった (図13A)。また、G418を含有しないGY液体培地で形質転換体を5回継代培養し、G418を含む (0, 2, 4, 8, 16, 32 mg/ml) PDA寒天平板培地を用いて同様の実験を行った場合においてもG418耐性能に変化が見られず、G418濃度が32 mg/mlの場合においても増殖が見られる (図13B) ことが分かった。このことから、G418耐性能を指標とした場合、付与された形質は安定していることが確認された。
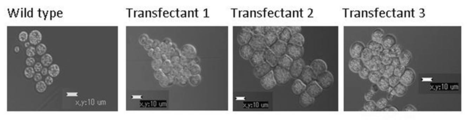
顕微鏡観察(図14A) 、およびnile redで細胞内の油球を染色した後の共焦点レーザー顕微鏡観察(図14B)よって、野生株と今回得た形質転換体との間には、形態的な変化が見られないことが確認された。また、18S rDNA解析によって、形質転換体がA. limacinumであることも確認された。
得られた形質転換体は0.5 mg/ml G418含有GY液体培地で培養し、野生株はG418を含有しないGY液体培地で培養した。ISOPLANT (ナカライテスク社製) を使用して、両株の菌体からゲノムDNAの抽出を行った。引き続いて、得られたゲノムDNAを鋳型として、LA taq Hot Start Version (タカラバイオ社製) を使用したPCRによるNeorの増幅をおこなった。用いたオリゴヌクレオチドプライマーはubproG418fus2F(配列表配列番号45)/G418ubtersus3R(配列表配列番号46)である (PCR cycles : 94℃ 2 min/98℃ 10sec, 68℃ 1 min, 72℃, 30 cycles/4℃)。
この結果、形質転換体においては、野生株に見られないNeorの特異的な増幅が観察された(図15)。このことから、導入したub-Neorは、染色体DNA上に組み込まれていることが示唆された。
定法通り抽出したA. limacinum形質転換体と野生株の2 μgのゲノムDNAを種々の制限酵素で37℃、16時間消化した後に、「DIG説明書集 8th、Roche」に従いDIGラベルNeorをプローブとしたサザンブロッティングを行った。
この結果、図16Aで示すように、Neorのバンドが検出された。このことから、導入したユビキチンプロモーター・ターミネーターによるub-Neorは、染色体DNAに組み込まれていることが示された。また、同一酵素 (PstI) で消化した5つの形質転換体のバンドの分子量が異なることから、導入DNA断片は、染色体DNA上にランダムに組み込まれていることが示された(図16B)。
セパゾールRNA I super (ナカライテスク社製) を用いて、A. limacinum形質転換体と野生株の菌体から全RNAの抽出を行った。続いて、RNeasy plus mini kit (QIAGEN社製) を使用し全RNAのクリーンアップを行った後に、Cloned DNase I (タカラバイオ社製) を使用して、添付のマニュアルに準じて37℃で1時間反応させることで、コンタミしたゲノムDNAの分解を行った。次に、PrimeScript Reverse Transcriptase (タカラバイオ社製) を使用した逆転写反応を行い、逆転写反応によるcDNAの合成を行った。引き続いて、得られたcDNAを鋳型として、LA taq Hot Start Version (タカラバイオ社製) を使用したPCRによるNeorの増幅をおこなった。用いたオリゴヌクレオチドプライマーはubproG418fus2F(配列表配列番号45)/G418ubtersus3R(配列表配列番号46)である (PCR cycles : 94℃ 2 min/98℃ 10sec, 68℃ 1 min, 72℃, 30 cycles/4℃)。
その結果、形質転換体においてNeorの増幅産物が確認された (図17、レーン1~5)。全RNAを鋳型としたPCRを行った場合(図17、レーン8~13)には増幅産物が見られないことから、レーン1-5で観察された産物はゲノムDNAのコンタミではなく、Neor mRNAの逆転写産物 (Neor cDNA) 由来であることが示された。このことから、染色体DNA上に組み込まれたub-Neorは、mRNAへの転写を受けていることが明らかとなった。
導入DNAとして、pUBNeomycinr、もしくはub-Neorの2種のDNAを使用した。様々な条件を検討した結果、エレクトロポレーション法では全く形質転換体が得られないことが判明した。そこで遺伝子銃法を検討したところ、G418耐性能が付与された形質転換体の取得に成功した。その遺伝子導入効率は1,100 psiの打ち込み圧の場合が最も高く、ub-Neorを導入した場合、~1.9 x 102 cfu/μg DNAであることが分かった。またpUBNeomycinrの導入効率は、~1.4 x 101 cfu/μg DNAであり、18S rDNA配列を相同組み換え部位とした環状DNAを導入するよりも、直鎖状DNAを導入したランダムインテグレーションの方が、導入効率が約14倍程度高いことが分かった。また形質転換体は、G418を含有しないGY液体培地で5回継代培養したあとにおいても、そのG418耐性能を維持していることが分かった。
nile redで細胞内の油球を染色した後の共焦点レーザー顕微鏡観察(図18)よって、野生株と今回得た形質転換体との間には、形態的な変化が見られないことが確認された。また18S rDNA解析によって、形質転換体がT. aureumであることが確認された。
A.limacinum形質転換体の場合と同様に、ゲノムDNAを鋳型としたPCR(図19A)、およびサザンブロッティングによるNeorの検出(図19B)によって、ub-Neorが染色体DNAにランダムに組み込まれていることが確認された。
A.limacinum形質転換体の場合と同様に、染色体DNA上に組み込まれたub-Neorは、mRNAへの転写を受けていることが明らかとなった(図20)。
導入DNAとして、EF-1αプロモーター・ターミネーターによる直鎖状Neor発現カセット (EF-Neor) を使用した。条件検討の結果、エレクトロポレーション法において、ごく低い遺伝子導入効率 (10-1 cfu/μg DNA以下) で形質転換体を得た。また形質転換体は、G418を含有しないGY液体培地で5回継代培養したあとにおいても、G418耐性能を維持していることが分かった。
A.limacinum形質転換体の場合と同様に、ゲノムDNAを鋳型としたPCR (図21A、B)、およびサザンブロッティングによるNeorの検出(図21C)によってEF-Neorが染色体DNAにランダムに組み込まれていることが確認された。しかし解析した3つの形質転換体のうちの1つ (Transfectant 2) に関しては、ターミネーター領域の一部欠損が示唆された (図21B、レーン7)。
ターミネーター領域の一部欠損が示唆されたTransfectant 2を含めて(図22、レーン14)、染色体DNA上に組み込まれたEF-Neorは、mRNAへの転写を受けていることが明らかとなった(図22A、B)。
導入DNAとして、ub-Neorを使用した。様々な条件検討にも関わらず、エレクトロポレーション法では形質転換体を得ることが出来なかった。そこで遺伝子銃法の条件検討を行ったところ、1,100 psiの打ち込み圧において、ごく低い遺伝子導入効率 (10-1 cfu/μg DNA以下) で形質転換体を得た。また形質転換体は、G418を含有しないGY液体培地で5回継代培養したあとにおいても、G418耐性能を維持していることが分かった。
A.limacinum形質転換体の場合と同様に、ゲノムDNAを鋳型としたPCR(図23)によって、導入DNA断片が染色体DNAに組み込まれていることが強く示唆された。

(1)オワンクラゲ緑色蛍光タンパク質(Green Fluorescent Protein;GFP)の発現
i.GFP遺伝子のmh0186ゲノムDNAへの組み込み
Thraustochytrium aureum ATCC 34304(American type culture collectionより分譲)由来ユビキチン遺伝子由来プロモーター領域とターミネーター領域、およびEnhanced GFP遺伝子(Clontech社製)をPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたPCRにより増幅した(PCRサイクル: 94℃ 2 min/94℃ 1 min,62℃ 30 sec,72℃ 1 min,30 cycles/4℃)。次に、プロモーター領域とGFP遺伝子をPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたFusion PCRによって連結した(PCRサイクル: 94℃ 2 min/94℃ 1 min,62℃ 30 sec,72℃ 2 min,30 cycles/4℃)。その後、これを鋳型としてPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたFusion PCRを行い、プロモーター領域・GFP遺伝子・ターミネーター領域を連結した(PCRサイクル: 94℃ 2 min/94℃ 1 min,62℃ 30 sec,72℃ 3 min,30 cycles/4℃)。連結したDNA断片は、pGEM-T Easyベクター(Promega社製)に組み込んだ。このプラスミドを鋳型として、プライマーUb-pro-F1(配列表配列番号48)とUb-term-R2(配列表配列番号49)、PrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたPCRを行うことによって、作製したGFP遺伝子カセットの両端にKpnI siteを付加した。その後、人工合成ネオマイシン耐性遺伝子カセット(ユビキチン遺伝子由来プロモーター領域とターミネーター領域)を含むpUC18ベクターのKpnI site(ターミネーター領域の直後)に組み込むことによって、GFP遺伝子/ネオマイシン耐性遺伝子発現カセットを作製した。GFP遺伝子/ネオマイシン耐性遺伝子発現カセットを含むベクターはpNeoGFPと名づけた。
作製したGFP遺伝子/ネオマイシン耐性遺伝子発現カセットを、プライマーUb18Spro-F2(配列表配列番号50)とpUC18-R(配列表配列番号51)、PrimeSTAR GC polymerase kit(タカラバイオ社製)を用いて増幅し、精製した。精製したDNA断片5μgをmh0186株に導入した。導入は、200 mlのGY液体培地で3日間培養した細胞を使用して、最終細胞懸濁液として0.3 Mソルビトール(和光純薬工業社製)、またはNucleofector Solution L(lonza社製)に懸濁し、0.75 kV、50Ω、50 μFの条件でGENE PULSER(登録商標) II(バイオ・ラッド ラボラトリーズ社製)を用いたエレクトロポレーション法による遺伝子導入操作を行った。また、同様に精製したDNA断片0.625μgを、200 mlのGY液体培地で5日間培養したT. aureumに、Standard Pressure Kit(バイオ・ラッド ラボラトリーズ社製)とPDS-1000/Heシステム(バイオ・ラッド ラボラトリーズ社製)を用いた遺伝子銃法によって導入した。導入条件としては、0.6 Micronの金粒子、Target distance 6 cm、Vacuum 26 mmHg、Rupture disk 1,100 PSIで行い、PDA寒天平板培地(2 mg/ml G418含有)に塗布した菌体に打ち込んだ。
mh0186株に関しては3日間100 mlのGY液体培地(0.5 mg/ml G418含有)で培養した菌体、T. aureumに関しては7日間100 mlからのGY液体培地(2 mg/ml G418含有)で培養した菌体からゲノムDNAを抽出し、Ultrospec 3000(Amersham Pharmacia Biotech社製)を用いてA260/280を測定することで、抽出したゲノムDNAの純度と濃度の測定を行った。抽出したゲノムDNAを鋳型として、プライマー3F(配列表配列番号52)、4R(配列表配列番号53)、Ub-GFP-F(配列表配列番号54)、UB-GFP-R(配列表配列番号55)、 およびLA Taq HS polymerase Kit(タカラバイオ社製)を用いたPCRを行った(PCRサイクル: 98℃ 2 min/98℃ 20 sec,60℃ 30 sec,72℃ 1 min,30 cycles/4℃)。

mh0186株に関しては3日間100 mlのGY液体培地(0.5 mg/ml G418含有)で本培養した菌体、T. aureumに関しては7日間100 mlからのGY液体培地(2 mg/ml G418含有)で培養した菌体から、セパゾールRNAISuper(ナカライテスク社製)を用いてTotal RNAを抽出した。次に、RNeasy Mini Kit(QIAGEN社製)を用いてTotal RNAのクリーンアップを行った。さらに、Cloned DNaseI(タカラバイオ社製)を用いたDNase処理を行うことによって、純度の高いTotal RNAを精製し、Ultrospec 3000(Amersham Pharmacia Biotech社製)を用いてA260/280を測定することで、精製したTotal RNAの純度と濃度の測定を行った。その後、精製したTotal RNAから、PrimeScriptTM Reverse Transcriptase(タカラバイオ社製)を用いてcDNAを作製した。作製したcDNAを鋳型として、プライマー3F(配列表配列番号52)、4R(配列表配列番号53)、Ub-GFP-F(配列表配列番号54)、UB-GFP-R(配列表配列番号55)、LA Taq HS polymerase kit(タカラバイオ社製)を用いてPCRを行った(PCRサイクル: 98℃ 2 min/98℃ 20 sec,60℃ 30 sec,72℃ 1 min,30 cycles/4℃)。
この結果、組み込まれたGFP遺伝子およびネオマイシン耐性遺伝子は、mRNAへと転写されていることが示された(図26)。
mh0186株に関しては3日間3 mlのGY液体培地(0.5 mg/ml G418含有)で培養した菌体、T. aureumに関しては7日間100 mlからのGY液体培地(2 mg/ml G418含有)で培養した菌体1 mlを室温、3,500 x g、10 min遠心して集菌した。集菌した菌体を500 μlの滅菌SEA LIFEで2回洗浄し、共焦点レーザー顕微鏡(ECLIPSE TE2000-U;ニコン社製)下(40×60倍、油浸レンズ、励起光Arレーザー 488 nm)で観察し、EZ-C1ソフトウェア(ニコン社製)を用いて画像を取得した。
共焦点レーザー顕微鏡観察の結果、得られたGFP遺伝子/ネオマイシン耐性遺伝子発現カセット形質転換体では、wild-typeにはみられないGFPの蛍光が観察された(図27)。
i.Δ12デサチュラーゼのクローニング
Pinguiochrysis pyriformis MBIC 10872(Marine Biotechnology Institute Culture collectionより分譲)をESM培地(NIESコレクションの培地リストに記載の方法で作製)で培養した後、体数増殖期後期の菌体を4℃、6,000 x g、15 min遠心することによって集菌した。集菌した菌体を液体窒素により凍結し、フェノール/SDS/LiCl法(1)によってTotal RNAを抽出した。得られたTotal RNAから、mRNA Purification Kit(GE healthcare Bio-sciences社製)を用いてpoly (A)+RNAを精製した。精製したpoly (A)+RNAから、Ready-To-Go T-Primed First-Strand Kit(GE healthcare Bio-sciences社製)を用いて一本鎖cDNAを作製した。作製したcDNAを鋳型として、既知のΔ12デサチュラーゼの保存配列を元に作製したプライマーF1(配列表配列番号56)、R1(配列表配列番号57)、およびAdvantageTM 2 PCR Kit(Clontech社製)を用いたPCRを行った(PCRサイクル: 95℃ 30 sec,50℃ 30 sec,68℃ 2 min,40 cycles/4℃)。増幅したPCR産物は、pGEM-T easy vector(Promega社製)に組み込んだ後に、エレクトロポレーション法によりコンピタントセル DH5α(東洋紡績社製)に導入した。抽出した形質転換体プラスミドを鋳型とし、Dye Terminator Cycle Sequencing Kit(BECKMAN COULTER社製)を用いたシーケンスにより塩基配列を解析した。P. pyriformisのcDNAライブラリーを、Lambda cDNA Library Construction Kits(Stratagene社製)を用いて構築した。ECL Direct Nucleic Acid Labeling and Detection System(GE healthcare Bio-sciences社製)を用いたプラークハイブリダイゼーションにより、ポジティブクローンのスクリーニングを行った。プローブとのインキュベーション条件は、8 ng/mlの濃度で添加した標識プローブと42℃で3時間、洗浄条件は、primary wash(ウレア無し)は55℃で10分間を2回、secondary wash(ウレア無し)は室温で5分間を2回、とした。プローブには、取得した部分配列を含むプラスミドを鋳型とし、プライマーSP1/F(配列表配列番号58)、SP1/R(配列表配列番号59)、およびAdvantageTM 2 PCR Kit(Clontech社製)を用いたPCRにより増幅した314 bpのcDNA断片を用いた(PCRサイクル: 94℃ 3 min/94℃ 30 sec,56℃ 30 sec,68℃ 1 min,35 cycles/4℃)。数回のスクリーニング後、得られたポジティブクローンはExAssistヘルパーファージ(Stratagene社製)を用いてλファージからpBluescript(Stratagene社製)に移し変えた。
この結果、515 bpの推定Δ12デサチュラーゼ遺伝子部分配列の増幅に成功した。取得したDNA断片をプローブとしたプラークハイブリダイゼーションによる、目的遺伝子全長を含むポジティブクローンのスクリーニングを行った結果、5.5×106個のクローンから7つの陽性クローンの単離に成功した。これらの配列を解析した結果、取得した遺伝子は、437アミノ酸をコードする1,314 bpのORFを含む遺伝子であることが示唆された。
P. pyriformis、真菌および原虫由来Δ12デサチュラーゼのアミノ酸配列を対象として、ClustalW 1.81とESPript 2.2(http://espript.ibcp.fr/ESPript/cgi-bin /ESPript.cgi)による多重アライメント解析を行った。
この結果、取得した遺伝子のアミノ酸配列は、他生物由来のΔ12デサチュラーゼ遺伝子のアミノ酸配列と高い同一性を示すことが明らかとなった(図28)。また、取得した遺伝子の推定アミノ酸配列中には、デサチュラーゼ全般において保存されている、3つのヒスチジンボックスが保存されていた(図28)。
P. pyriformis由来Δ12デサチュラーゼを含む、Δ12デサチュラーゼおよびΔ12/Δ15デサチュラーゼの分子系統樹を、MOLPHY version 2.3 computer program package(非特許文献13)を用いた最尤法によって作成した。まず、全てのアミノ酸配列を用いて、ClustalW 1.81による多重アライメントを行った。次に、不確定部分を除去した後に、近隣結合法(2)による系統樹を初期系統樹とした最尤系統樹の探索を行った。
この結果、取得した推定Δ12デサチュラーゼ、他生物由来のΔ12デサチュラーゼおよびΔ12/Δ15デサチュラーゼは、fungal & nematode Δ12デサチュラーゼ群、plant Δ12デサチュラーゼ群、cyanobacterial and chloroplast-localized plant Δ12デサチュラーゼ群の3つの系統群に分類された。そして、取得した推定Δ12デサチュラーゼは、fungal & nematode Δ12デサチュラーゼ群に分類され、Saprolegnia diclina由来Δ12デサチュラーゼと最も近縁であることが明らかとなった(図29)。
P. pyriformis由来Δ12デサチュラーゼ遺伝子全長を含むプラスミドを鋳型として、プライマーPry-F(配列表配列番号60)、Pyr-R(配列表配列番号61)、およびPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたPCRを行い、両端にHindIII制限酵素サイトとXbaI制限酵素サイトを付加した。増幅断片をpGEM-T-Easyベクター(Promega社製)に組み込んで配列解析を行い、増幅ミスが無かったプラスミドからHindIII/XbaI処理によってΔ12デサチュラーゼ遺伝子を切り出し、同様に制限酵素処理した酵母用ベクターpYES2/CT(Invitrogen社製)に組み込み、Δ12デサチュラーゼ遺伝子発現ベクターpYpΔ12Desを構築した。構築したΔ12デサチュラーゼ遺伝子発現ベクターpYpΔ12DesおよびpYES2/CTを「Current Protocols in Molecular Biology, Unit 13 (Ausubel et al., 1994)」および「Guide to Yeast Genetics and Molecular Biology (Gutherie and Fink, 1991)」記載の方法に従い、酢酸リチウム法によって出芽酵母Saccharomyces cerevisiaeに導入し形質転換体の選別を行った。次に、得た形質転換体 (pYpΔ12Des導入株およびmock導入株) をQiuらの方法 (Qiu, X., et al. J. Biol. Chem. (2001) 276, 31561-6) に準じて培養し、菌体由来脂肪酸の抽出とメチルエステル化を行った。GC解析にはガスクロマトグラフGC-2014(島津製作所社製)を使用し、カラム:HR-SS-10(30 m x 0.25 mm;信和化工社製)、カラム温度:150℃ →(5℃/min)→ 220℃(10 min)、キャリアガス:He(1.3 mL/min)の条件で行った。また、GC-MS解析には、GC-17A 及びGCMS-QP-5000(島津製作所社製)を使用し、DB-1キャピラリーカラム(0.25 mmi.d. x 30 m, film thickness 0.25 μm;アジレント社製)を用い、カラム温度160℃ →(4℃/min)→260℃、インジェクターポート温度250℃の条件で行った。また、さらに解析が困難であったピークに関しては、ピコリニルエステル化した脂肪酸を前記と同じ装置及びカラムを用い、240℃→(2.5℃/min)→260℃(15min→(2.5℃/min )→280℃の温度条件で解析を行った。
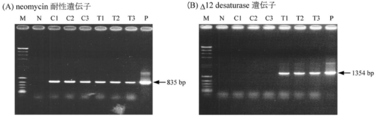
まず、ユビキチン遺伝子由来プロモーター領域とターミネーター領域、およびΔ12デサチュラーゼ遺伝子をPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたPCRにより増幅した(PCRサイクル: 94℃ 2 min/94℃ 1 min,62℃ 30 sec,72℃ 1.5 min,30 cycles/4℃)。次に、プロモーター領域とΔ12デサチュラーゼ遺伝子をPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたFusion PCRによって連結した(PCRサイクル: 94℃ 2 min/94℃ 1 min,62℃ 30 sec,72℃ 2.5 min,30 cycles/4℃)。その後、これを鋳型としてPrimeSTAR GC polymerase kit(タカラバイオ社製)を用いたFusion PCRを行い、プロモーター領域・GFP遺伝子・ターミネーター領域を連結した(PCRサイクル: 94℃ 2 min/94℃ 1 min,62℃ 30 sec,72℃ 3 min,30 cycles/4℃)。連結したDNA断片は、pGEM-T Easyベクター(Promega社製)に組み込んだ。このプラスミドを鋳型とし、PrimeSTAR MAX DNA polymerase(タカラバイオ社製)を用いて、Δ12デサチュラーゼ遺伝子の配列中にあるKpnIsiteに一塩基変異を導入した。続いて、KpnI処理によって連結断片を切り出し、人工合成ネオマイシン耐性遺伝子カセット(両端にEF1-α遺伝子由来プロモーター領域およびターミネーター領域を連結)を含むpUC18ベクター(タカラバイオ社製)のKpnIsiteに組み込んだ。Δ12デサチュラーゼ遺伝子/ネオマイシン耐性遺伝子発現カセットを含むベクターは、pNeoDes12と名づけた。作製に要したPCRプライマーの配列は、表6に示した。作製したΔ12デサチュラーゼ遺伝子/ネオマイシン耐性遺伝子発現カセットを、プライマー2F(配列表配列番号62)とpUC18-R(配列表配列番号51)、PrimeSTAR GC polymerase kitを用いて増幅し、精製した。精製したDNA断片5 μgを、(1)-1.同様に、mh0186株に導入した。最終細胞懸濁液としては、Nucleofector Solution L(lonza社製)を用いた。3日間、100 mlのGY液体培地(0.5 or 2 mg/ml G418含有)で本培養した菌体からゲノムDNAを抽出し、Ultrospec 3000(Amersham Pharmacia Biotech社製)を用いてA260/280を測定することで、抽出したゲノムDNAの純度と濃度の測定を行った。抽出したゲノムDNAを鋳型として、プライマー3F(配列表配列番号52)、4R(配列表配列番号53)、ub pro-D12d-F(配列表配列番号63)、ub term-Δ12d-R(配列表配列番号64)、およびLA Taq HS polymerase Kit(タカラバイオ社製)を用いたPCRを行った(PCRサイクル: 98℃ 2 min/98℃ 20 sec,60℃ 30 sec,72℃ 1.5 min,30 cycles/4℃)。

作製したΔ12デサチュラーゼ遺伝子/ネオマイシン耐性遺伝子発現カセットを、プライマー2F(配列表配列番号62)とpUC18-R(配列表配列番号51)、PrimeSTAR GC polymerase kitを用いて増幅し、精製した。精製したDNA断片0.625 μgを、200 mlのGY液体培地で5日間培養した菌体に、Standard Pressure Kit(バイオ・ラッド ラボラトリーズ社製)とPDS-1000/Heシステム(バイオ・ラッド ラボラトリーズ社製)を用いた遺伝子銃法によって導入した。導入条件としては、0.6 Micronの金粒子、Target distance 6 cm、Vacuum 26 mmHg、Rupture disk 1,100 PSIで行い、PDA寒天平板培地(2 mg/ml G418含有)に塗布した菌体に打ち込んだ。7日間、100 mlのGY液体培地(2 mg/ml G418含有)で培養した菌体からゲノムDNAを抽出し、Ultrospec 3000(Amersham Pharmacia Biotech社製)を用いてA260/280を測定することで、抽出したゲノムDNAの純度と濃度の測定を行った。抽出したゲノムDNAを鋳型として、プライマー3F(配列表配列番号52)、4R(配列表配列番号53)、ub pro-D12d-F(配列表配列番号63)、ub term-D12d-R(配列表配列番号64)、およびLA Taq HS polymerase Kit(タカラバイオ社製)を用いたPCRを行った(PCRサイクル: 98℃ 2 min/98℃ 20 sec,60℃ 30 sec,72℃ 1.5 min,30 cycles/4℃)。
3日間、100 mlのGY液体培地(0.5 mg/ml G418含有)で本培養した菌体から、セパゾールRNAISuper(ナカライテスク社製)を用いてTotal RNAを抽出した。実施例1(1)-2.と同様に、純度の高いTotal RNAを精製し、cDNAを作製した。作製したcDNAを鋳型として、プライマープライマー3F(配列表配列番号52)、4R(配列表配列番号53)、ub pro-D12d-F(配列表配列番号63)、ub term-D12d-R(配列表配列番号64)、およびLA Taq HS polymerase kit(タカラバイオ社製)を用いてPCRを行った(PCRサイクル: 98℃ 2 min/98℃ 20 sec,60℃ 30 sec,72℃ 1.5 min,30 cycles/4℃)。
この結果、cDNAを鋳型としたPCRの結果から、組み込まれたΔ12デサチュラーゼ遺伝子およびネオマイシン耐性遺伝子は、mRNAへと転写されていることが示された(図35)。
7日間、100 mlのGY液体培地(2 mg/ml G418含有)で本培養した菌体から、セパゾールRNAISuper(ナカライテスク社製)を用いてTotal RNAを抽出した。実施例1(1)-2.と同様に、純度の高いTotal RNAを精製し、cDNAを作製した。作製したcDNAを鋳型として、プライマープライマー3F(配列表配列番号52)、4R(配列表配列番号53)、ub pro-D12d-F(配列表配列番号63)、ub term-D12d-R(配列表配列番号64)、およびLA Taq HS polymerase kit(タカラバイオ社製)を用いてPCRを行った(PCRサイクル: 98℃ 2 min/98℃ 20 sec,60℃ 30 sec,72℃ 1.5 min,30 cycles/4℃)。
この結果、cDNAを鋳型としたPCRの結果から、組み込まれたΔ12デサチュラーゼ遺伝子およびネオマイシン耐性遺伝子は、mRNAへと転写されていることが示された(図34)。
i.Δ5デサチュラーゼのクローニング
Thraustochytrium aureum ATCC 34304に近種のThraustochytrium sp. ATCC 26185が持つΔ5デサチュラーゼの配列中に存在する保存領域において、プライマー3F(配列表配列番号65)および1R(配列表配列番号66)を作製した。次にAdvantage 2 PCR Kit (Clontech社製)を用いて、T. aureum由来のRACE cDNAライブラリを鋳型としnested-PCR(PCRサイクル:94℃ 30 sec, 50℃ 30 sec, 72℃ 2 min, 30 cycle)を行った結果、挟み込みのプライマーセット(1R NES:配列表配列番号67)により目的サイズの増幅断片が得られた。
この挟み込みのプライマーにより得た予想サイズのDNA断片(550bp)を解析したところ、T. aureumのもつΔ5デサチュラーゼであったため、この増幅断片から100%マッチのプライマー(RACEd5F:配列表配列番号68、およびRACEd5FNES:配列表配列番号69)を作製し、Advantage 2 PCR Kit を用いてRACE PCR(PCRサイクル:94℃ 30 sec, 50℃ 30 sec, 72℃ 2 min, 30 cycle)を行ったところ、700bpのΔ5デサチュラーゼの3'末端が得られた。
次にこの既知配列からReverseプライマーGSP1(配列表配列番号70)を作製し5' RACE PCRを行ったが、得られた5' RACE産物は予想サイズより短いものであった(PCRサイクル:94℃ 30 sec/72℃ 3 min, 5cycle、94℃ 30 sec/70℃ 30 sec/72℃ 3 min, 5cycle、94℃ 30 sec/68℃ 30 sec/72℃ 3 min, 20cycle)。そのためcDNAを鋳型としたPCRをやめ、ゲノムカセットライブラリー (TaKaRa LA PCR in vitro Cloning Kit)を鋳型としたPCR(プライマーGSP2(配列表配列番号71))を前述同様に行った。その結果、BglIIカセットライブラリーを鋳型としたPCRを行ったところプライマー作製部位よりおよそ2.5kbp上流のゲノム配列を取得することが出来た。
ゲノムウォーキング法を用いて得た上流配列はΔ5デサチュラーゼの開始コドンを含んでおり、解析したゲノム途中にイントロンの存在は認められなかった。また3'-RACEおよび5'-RACEより得た配列によりΔ5デサチュラーゼの全長配列情報を取得できた。その結果得た全長配列は1320 bpのORFを有する439アミノ酸からなり、Δ5デサチュラーゼに高度に保存される一つのcytochrome b5 ドメイン (HPGGSI)と、三つのヒスチジンボックス (HECGH, HSKHH and QIEHH)を有していた。この情報をもとに、ORF末端で以下のプライマーを作成しcDNAを鋳型としたPCR(プライマーd5fulllengthF(配列表配列番号72)およびd5fulllengthR(配列表配列番号73)、PCRサイクル:94°C for 30 s, 60°C for 30 s, and 72°C for 2 min, 30cycle)を行うことで、完全長のT. aureum由来Δ5デサチュラーゼを単離した。
T. aureum由来Δ5デサチュラーゼおよびThraustochytrium sp.ATCC 26185, Dictyostelium discoideum, Rattus norvegicus, Mus musculus, Homo sapiens, Caenorhabditis elegans, Leishmania major由来Δ5デサチュラーゼのアミノ酸配列を用いて、ClustalX-1.83.1による多重アラインメントを行った (図35)。
この結果、T. aureum由来Δ5デサチュラーゼは他生物由来のΔ5デサチュラーゼ遺伝子とアミノ酸レベルで有意な相同性を示した (D. discoideum: 34%, R. norvegicus: 28%, M. musculus: 28%, H. sapiens: 26%)。特に、同属のThraustochytrium sp.とは、57%と高い同一性を示した。
T. aureum由来Δ5デサチュラーゼを含む全てのデサチュラーゼ遺伝子の分子系統樹を、molphyを用いた最尤法によって作成した。まず、全ての配列をFasta形式にし、clustalWを用いた多重アラインメントを行った。次に、不確定部分を除去した後に、近隣結合法による系統樹を初期系統樹とした最尤系統樹の探索を行った。
この結果、取得した遺伝子は原生動物由来のデサチュラーゼ群と近縁であり、Thraustochytrium sp. ATCC 26185およびL. major由来Δ5デサチュラーゼと、同一の系統群に分類されることが明らかになった (図36)。
取得遺伝子が確かにΔ5デサチュラーゼであるかを検証するために、出芽酵母S. cerevisiaeを宿主とした過剰発現実験を行った。まず最初に、取得した遺伝子を酵母用ベクターであるpYES2/CT (Invitrogen社製)のEcoRI/XhoIサイトに組み込み、発現ベクターpYΔ5desを構築した。次に、構築した発現ベクターpY5ΔdesをS. cerevisiaeに導入し、酢酸リチウム法によって得た形質転換体の脂肪酸を抽出後にメチル化しGC解析を行った。GC解析にはガスクロマトグラフGC-2014(島津製作所社製)を使用し、カラム:HR-SS-10(30 m x 0.25 mm;信和化工社製)、カラム温度:150℃ →(5℃/min)→ 220℃(10 min)、キャリアガス:He(1.3 mL/min)の条件で行った。また、GC-MS解析には、GC-17A 及びGCMS-QP-5000(島津製作所社製)を使用し、DB-1キャピラリーカラム(0.25 mmi.d. x 30 m, film thickness 0.25 μm;アジレント社製)を用い、カラム温度160℃ →(4℃/min)→260℃、インジェクターポート温度250℃の条件で行った。また、さらに解析が困難であったピークに関しては、ピコリニルエステル化した脂肪酸を前記と同じ装置及びカラムを用い、240℃→(2.5℃/min)→260℃(15min→(2.5℃/min )→280℃の温度条件で解析を行った。
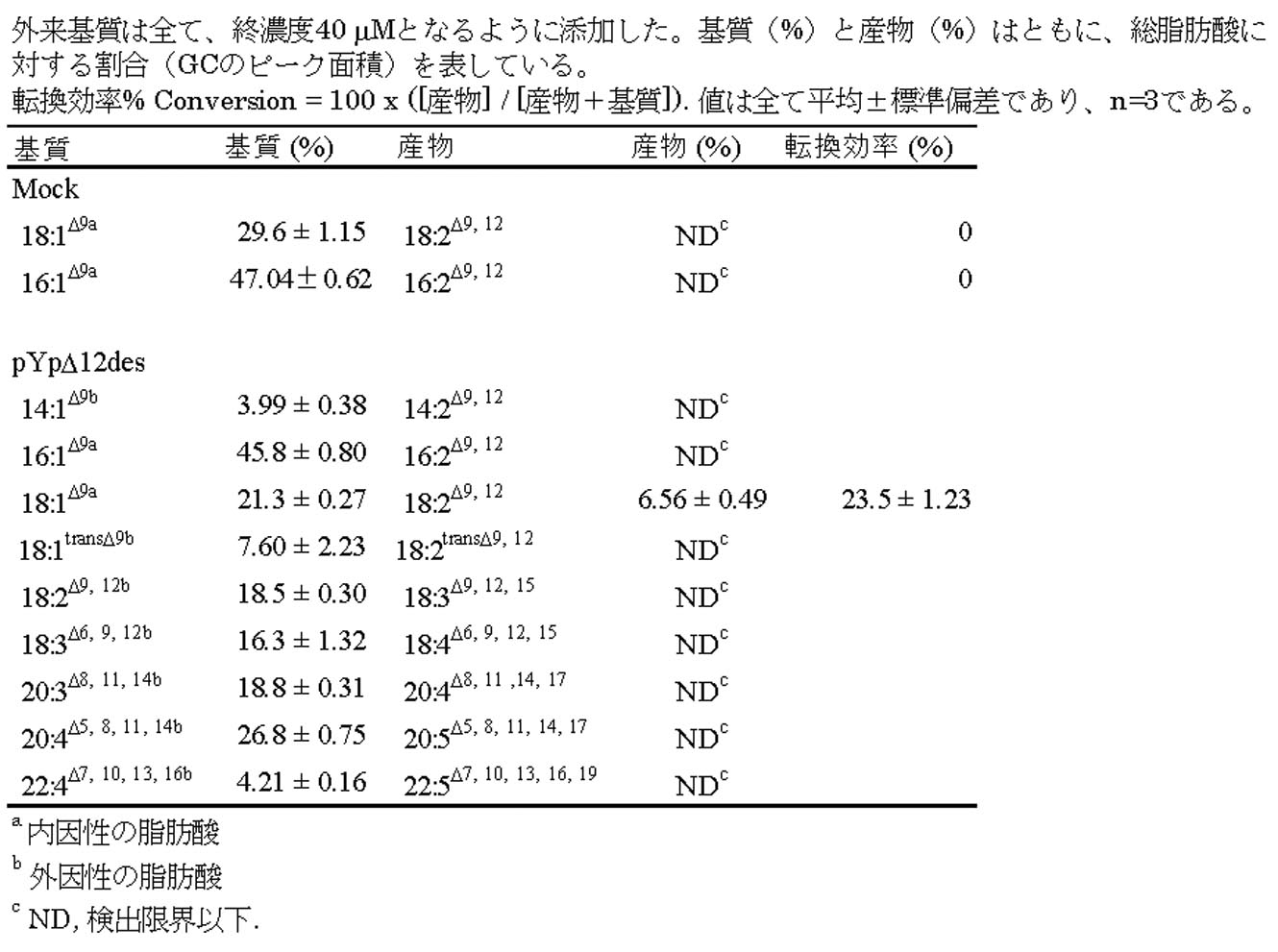
この結果、Δ5デサチュラーゼの前駆物質として知られるエイコサテトラエン酸(Eicosatetraenoic acid (ETA), C20:4 Δ8,11,14,17) およびジホモ-γ-リノレン酸(dihomo-γ-linoleic acid (DGLA), C20:3 Δ8,11,14)が、それぞれEPAとアラキドン酸(Arachidonic acid (AA); C20:4 n-6) に変換された。変換効率はそれぞれ32%および27%であった。また、他の基質に対する特異性は見られなかった。変換により生じたEPAとAAに関してはGC-MSによる構造の確認も行い、確かに構造も一致していた(図37a~c、表7)

発現ベクターの構築のためのT. aureum ATCC 34304 由来ユビキチン遺伝子プロモーター/ターミネーターの単離を行った。それにはまず、以下の様なRACE法を用いたユビキチン遺伝子の単離を行った。始めに縮重プライマー2F(配列表配列番号74)を用い、cDNAを鋳型としたPCRによってユビキチン遺伝子の3'側の断片を増幅した。
次に、5'-RACE産物を得る為に、5'RACE System for Rapid Amplification of cDNA Ends,version 2.0 (invitrogen社製)を用い、逆転写用プライマー1R(配列表配列番号17)と5'-RACE用プライマー(配列表配列番号75)を作成しキットの操作を行った。
得られたユビキチン遺伝子ORFの配列をもとに、プライマーREVERS-U PR-1(配列表配列番号22)およびREVERS-U PR-2(配列表配列番号23)を作成し、ゲノムウォーキング法によりPCR(PCRサイクル:98℃ 30 sec/60℃ 30 sec/72℃ 2 min,30cycle)を行った。この結果、SalIカセットライブラリーを鋳型としたPCRにより812 bpのプロモーター領域を単離した。
次に、同様の方法でターミネーターの単離を行い、その結果612 bpのDNA断片を得た。
尚、プライマーとして1st PCRではubqterminalf1(配列表配列番号24)を、2nd PCRではter2F(配列表配列番号25)を用い、PCRサイクルは94℃ 30 sec/60℃ 30 sec/72℃ 3 min, 30 cycleで行った。得られた増幅断片をフュージョンPCRにより連結しpUC18に組み込み図38aに示す様な環状ベクターを作成後、PCRによって図38bに示す導入遺伝子断片を調整した。
続いて得た形質転換体がゲノムDNAに組み込まれている事を確認するために、Δ5デサチュラーゼ増幅プライマーとしてd5fulllengthF(配列表配列番号72)およびd5fulllengthR(配列表配列番号73)、ネオマイシン耐性遺伝子増幅プライマーとしてFU2FA(配列表配列番号76)およびFU2RA(配列表配列番号77)を用い、98℃ 10 sec/98℃ 10 sec/60℃ 30 sec/72℃ 1.5 min,30cycleのPCRプログラムでゲノムDNAを鋳型としたPCRを行った。
この結果、導入した遺伝子の増幅が確認され、確かにゲノム中に取り込まれている事が確認された (図39)。
Aurantiochytrium sp. mh0186形質転換体を培養後、RNA抽出 (セパゾールRNA I super; ナカライテスク社製)を、キットの操作方法を参照して行った。まず各クローンから得たtotalRNAをPrimeScript Reverse Transcriptase (タカラバイオ社製) を用いて逆転写しcDNAの合成を行った。得たcDNAを鋳型として次に示す様な条件でPCRを行った。(PCRサイクル:98℃ 10 sec/55℃ 30 sec/72℃ 1.5 min,30cycle)
この結果、各目的遺伝子の増幅が確認できた (図40)。このことから、得た形質転換体は導入遺伝子がmRNAへと転写され発現している事が確認された。
(1)ピンギオ藻由来Δ12デサチュラーゼの発現による脂肪酸組成の改変
実施例6(2)v.で得られた形質転換体クローンを10 mlのGY液体培地(0.5 mg/ml G418含有)で2日間培養した後、オレイン酸を終濃度が50μMとなるように添加して、さらに1日培養した。培養後は、実施例6(2)iv.同様にGCおよびGC-MSを用いて脂肪酸組成を解析した。GC解析にはガスクロマトグラフGC-2014(島津製作所社製)を使用し、カラム:HR-SS-10(30 m x 0.25 mm;信和化工社製)、カラム温度:150℃ →(5℃/min)→ 220℃(10 min)、キャリアガス:He(1.3 mL/min)の条件で行った。また、GC-MS解析には、GC-17A 及びGCMS-QP-5000(島津製作所社製)を使用し、DB-1キャピラリーカラム(0.25 mmi.d. x 30 m, film thickness 0.25 μm;アジレント社製)を用い、カラム温度160℃ →(4℃/min)→260℃、インジェクターポート温度250℃の条件で行った。また、さらに解析が困難であったピークに関しては、ピコリニルエステル化した脂肪酸を前記と同じ装置及びカラムを用い、240℃→(2.5℃/min)→260℃(15min→(2.5℃/min )→280℃の温度条件で解析を行った。コントロールとしては、ネオマイシン耐性遺伝子カセットのみを導入した形質転換体を用いた。
得られた形質転換体の脂肪酸組成のGC解析を行った結果、Δ12デサチュラーゼ遺伝子/ネオマイシン耐性遺伝子発現カセット導入株において、リノール酸のリテンションタイムに相当する位置に、コントロール株にはみられない新規ピークが確認された(図41)。この新規ピークのGC-MS解析を行った結果、その分子量およびフラグメントパターンは、標品のリノール酸メチルエステルのものと一致することが明らかとなった(図42)。オレイン酸からリノール酸への転換効率は30.1 ± 6.64 %であり、他の脂肪酸組成への影響は確認されなかった(図43)。
実施例6(3)v.で得られた形質転換体クローンを3日間培養後、脂肪酸メチルエステルの抽出を行いGCを用いてmhneorおよびmhΔ5neorを解析した。また外因性のΔ5デサチュラーゼが基質とする脂肪酸であるETAまたはDGLAを0.1 mMだけ培地に添加し、ラビリンチュラに取り込ませ、酵母における過剰発現実験と同様にして脂肪酸を抽出後、GC解析を行った。GC解析にはガスクロマトグラフGC-2014(島津製作所社製)を使用し、カラム:HR-SS-10(30 m x 0.25 mm;信和化工社製)、カラム温度:150℃ →(5℃/min)→ 220℃(10 min)、キャリアガス:He(1.3 mL/min)の条件で行った。
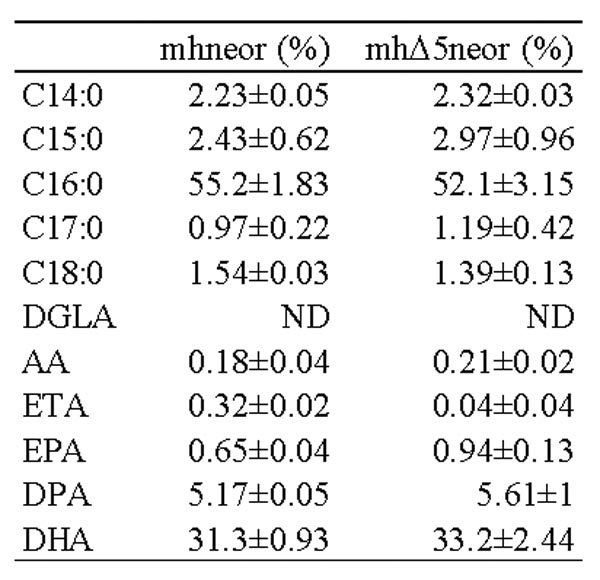
Aurantiochytrium sp. mh0186株に内在性のETAが、導入したΔ5デサチュラーゼの働きにより変換され、EPAの含有率がmhneorと比較して1.4倍程度高くなっている事が分かった (図44、表8)。またΔ5デサチュラーゼが基質とするETAまたはDGLAを0.1 mMだけ培地に添加し、ラビリンチュラに取り込ませたところ、酵母におけるΔ5デサチュラーゼ発現実験で観察された様に前駆物質がラビリンチュラ内でEPAとAAに変換されその含有率が増大した (表9、10)。またラビリンチュラ内での前駆物質の変換効率は前述の酵母の場合よりも高く、それぞれETAが75.2 %、DGLAが62.9 %であった。また、本実験は3回以上の再現性確認実験を行っており、いずれの場合も結果は一致していた。


Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、及びParietichytrium sarkarianum SEK364に対し、遺伝子導入実験を行った。
前記ラビリンチュラ類6株の前培養液5μlを様々な濃度のG418を含有するPDA寒天平板培地に滴下し、28℃で7日間培養後にコロニー形成を観察した。これらの結果、抗生物質感受性の結果と真核生物形質転換系で用いられる選択マーカー遺伝子に鑑みて、ラビリンチュラ類の形質転換系に使用出来る選択マーカー遺伝子は、G418が有効であると示された。
T. aureum由来ユビキチン遺伝子及びその遺伝子発現調節領域の単離は、実施例3と同様に行った。
薬剤耐性遺伝子発現カセットの作製は、実施例4と同様に行った。
ユビキチンプロモーター・ターミネーターによる直鎖状Neor発現カセット (ub-Neor)を用いた遺伝子導入実験を行った。このカセットは、実施例4-ii.で得られたpUBNeomycin rを鋳型とし、LA taq Hot Start Version (タカラバイオ社製)を使用して1対のオリゴヌクレオチドプライマーNeoF(配列表配列番号78)/NeoR(配列表配列番号79)を用いたPCRを行い、得られた増幅産物をゲル精製したものである。
この結果、直鎖状DNAであるub-Neorを用いた場合において、~1.6 x 100 cfu/μg DNAの効率でG418耐性能を示すコロニーの出現が見られた。また、得られた形質転換体は、G418を含有しないGY液体培地で5回継代培養したあとにおいても、そのG418耐性能を維持していることが分かり、G418耐性能を指標とした場合、付与された形質は安定していることが確認された。
得られた形質転換体は1.0および2.0mg/ml G418含有GY液体培地で培養し、野生株はG418を含有しないGY液体培地で培養した。ISOPLANT (ナカライテスク社製)を使用して、両株の菌体からゲノムDNAの抽出を行った。引き続いて、得られたゲノムDNAを鋳型として、KOD FX(東洋紡ライフサイエンス社製) を使用したPCRによるNeorの増幅をおこなった。用いたオリゴヌクレオチドプライマーはNeoF(配列表配列番号78)/NeoR(配列表配列番号79)である (PCR cycles : 94℃ 2 min/98℃ 10sec, 68℃ 30 sec,72℃ 2 min, 30 cycles/4℃)。
この結果、形質転換体においては、野生株に見られないNeorの特異的な増幅が観察された(図45)。このことから、導入したub-Neorは、染色体DNA上に組み込まれていることが示唆された。
[実施例9-1] SV40 terminator配列のサブクローニング
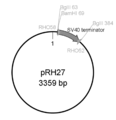
pcDNA 3.1 Myc-His vectorを鋳型に、PrimeSTAR polymerase (タカラバイオ社製) によりSV40 terminator配列を増幅した。用いたPCR primer は下記の通りで、RHO58はSV40 terminator配列上に設定し、BglIIおよびBamHIリンカー配列を含む。RHO52はSV40 terminator配列上に設定し、BglII配列を含む。 [RHO58: 34mer: 5’- CAG ATC TGG ATC CGC GAA ATG ACC GAC CAA GCG A-3’ (配列番号:80)、 RHO52: 24mer: 5’- ACG CAA TTA ATG TGA GAT CTA GCT -3’ (配列番号:81) ]。下記の条件で増幅後、pGEM-T easy vector (プロメガ社製) にクローニングした。[PCR cycles: 98℃ 2 min/ 98℃ 30 sec, 60℃ 30 sec, 72℃ 1 min, 30 cycles/ 72℃ 1min]。大腸菌にて増幅後、Dye Terminator Cycle Sequencing Kit を用いて配列を確認した。これをpRH27と名付けた。
サブクローニングしたSV40 terminator配列 (342 bp、配列番号:82) を含むプラスミド (pRH27) を図46に示す。
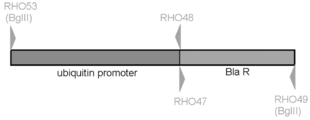
Thraustochytrium aureum ATCC 34304よりゲノムDNAを鋳型に、PrimeSTAR GC polymeraseによりubiquitin promoter 配列 (618 bp、配列番号83) を増幅した。用いたPCR primer は下記の通りで、RHO53はubiquitin promoter 配列上に設定、BglIIリンカー配列を含む。RHO48はubiquitin promoter 配列とブラストサイジン耐性遺伝子配列を含む。 [RHO53: 36mer: 5’- CCC AGA TCT GCC GCA GCG CCT GGT GCA CCC GCC GGG -3’ (配列番号:84)、RHO48: 58mer: 5’- CTT CTT GAG ACA AAG GCT TGG CCA TGT TGG CTA GTG TTG CTT AGG TCG CTT GCT GCT G -3’ (配列番号:85) ]。[PCR cycles: 98℃ 2min/ 98℃ 10 sec, 68℃ 1 min, 30 cycles/ 68℃ 1min]。
pTracer-CMV/Bsd/lacZ を鋳型に、PrimeSTAR GC polymeraseによりブラストサイジン耐性遺伝子(432 bp、配列番号:86) を増幅した。用いたPCR primer は下記の通りで、RHO47はubiquitin promoter 配列とブラストサイジン耐性遺伝子配列を含む。RHO49はブラストサイジン耐性遺伝子配列を含み、BglIIリンカー配列を有する。[RHO47: 54mer:5’- AGC GAC CTA AGC AAC ACT AGC CAA CAT GGC CAA GCC TTT GTC TCA AGA AGA ATC -3’ (配列番号:87)、RHO49: 38mer: 5’- CCC AGA TCT TAG CCC TCC CAC ACA TAA CCA GAG GGC AG -3’ (配列番号:88)]。[PCR cycles: 98℃ 2min/ 98℃ 10 sec, 68℃ 1 min, 30 cycles/ 68℃ 1min]。
上記のようにしてFusionしたThraustochytrium aureum ATCC 34304 由来ubiquitin promoter-pTracer-CMV/Bsd/lacZ由来ブラストサイジン耐性遺伝子 (1000 bp、配列番号:89) を、BglIIにて消化し、これを実施例9-1記載pRH27 (図46) のBamHIサイトに繋いだ。出来たプラスミドを大腸菌にて増幅後、Dye Terminator Cycle Sequencing Kit を用いて配列を確認した。これをpRH38と名付けた。
作製したブラストサイジン耐性遺伝子カセット (pRH38) を図48に示す。
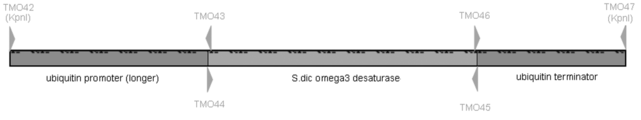
Thraustochytrium aureum ATCC 34304よりゲノムDNAを鋳型に、LA taq GC II polymeraseによりubiquitin promoter 配列 (longer) (812 bp、配列番号:90) を増幅した。用いたPCR primer は下記の通りで、TMO42はubiquitin promoter 配列上、RHO53 (実施例9-2、配列番号:84) よりも上流に設定、KpnIリンカー配列を含む。TMO43はubiquitin promoter 配列とSaprolegnia diclina由来ω3 デサチュラーゼ遺伝子配列を含む。 [TMO42: 29mer: 5’- TCG GTA CCC GTT AGA ACG CGT AAT ACG AC -3’ (配列番号:91)、TMO43: 45mer: 5’- TTC GTC TTA TCC TCA GTC ATG TTG GCT AGT GTT GCT TAG GTC GCT -3’ (配列番号:92) ]。[PCR cycles: 96℃ 2min/ 98℃ 20 sec, 60℃ 30 sec, 72℃ 1 min, 30 cycles/ 72℃ 1min]。
次に、Saprolegnia diclinaを、1LあたりD-Glucose 31.8g、Yesat Extract 10.6g、を含有する培地(脱イオン水で調整)で培養した。対数増殖期後期の細胞を4℃、3,500×g、5 min 遠心してペレットにし、液体窒素にて凍結破砕した。細胞破砕液をフェノール抽出後、エタノール沈殿させ、沈殿を TE 溶液に溶解した。TE 溶液に溶解した核酸を37℃、30 min RNase 処理して RNA を分解し、再度フェノール抽出後、エタノール沈殿させ、沈殿を TE 溶液に溶解した。A260/280 を測定し、DNA 純度、および濃度を算出した。このようにして得られたSaprolegnia diclinaのゲノムDNAを鋳型に、LA taq GC II polymeraseによりSaprolegnia diclina由来ω3 デサチュラーゼ遺伝子配列 (1116 bp、配列番号:93) を増幅した。用いたPCR primer は下記の通りで、TMO44はubiquitin promoter 配列とSaprolegnia diclina由来ω3 デサチュラーゼ遺伝子配列を含む。TMO45はSaprolegnia diclina由来ω3 デサチュラーゼ遺伝子配列およびubiquitin terminatorを含む。 [TMO44: 43mer: 5’- CCT AAG CAA CAC TAG CCA ACA TGA CTG AGG ATA AGA CGA AGG T -3’ (配列番号:94)、TMO45: 40mer: 5’- ATA CTA CAG ATA GCT TAG TTT TAG TCC GAC TTG GCC TTG G -3’ (配列番号:95) ]。[PCR cycles: 96℃ 2min/ 98℃ 20 sec, 60℃ 30 sec, 72℃ 1 min 30 sec, 30 cycles/ 72℃ 1min 30 sec]。
配列番号90、93、96を鋳型にTMO42 (配列番号:91) およびTMO47 (配列番号:98) を用いてFusion PCRを行った。酵素はLA taq GC II polymeraseを使用し、下記の条件で増幅した。 [PCR cycles: 96℃ 2min/ 98℃ 20 sec, 55℃ 30 sec , 68℃ 3 min, 30 cycles/ 68℃ 3min。ただし55℃から68℃へは1℃/ 10sec とした] (図49、2463 bp、配列番号:99)。
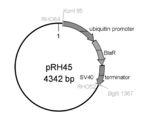
図49でFusionしたThraustochytrium aureum ATCC 34304 由来ubiquitin promoter-Saprolegnia diclina由来ω3 デサチュラーゼ遺伝子-Thraustochytrium aureum ATCC 34304 由来ubiquitin terminator (配列番号:99) を、KpnIにて消化し、これをpRH45 (図50) のKpnIサイトに繋いだ。出来たプラスミドを大腸菌にて増幅後、Dye Terminator Cycle Sequencing Kit を用いて配列を確認した。これをpRH48と名付けた。
作製したSaprolegnia diclina由来ω3 デサチュラーゼ遺伝子発現プラスミド (pRH48) を図51に示す。
実施例9-3で作製したターゲティングベクターを鋳型に、TMO42 (実施例9-3、配列番号:91) およびRHO52 (実施例9-1、配列番号:81) をprimerとして用いて、Prime STAR Max polymeraseによりDNAを増幅した。 [PCR cycles: 94℃ 30 sec, 72℃ 1 min, 5 cycles/ 94℃ 30 sec, 70℃ 30 sec, 72℃ 1 min, 5 cycles/ 94℃ 30 sec, 68℃ 30 sec, 72℃ 1 min, 25 cycles / 72℃ 2 min]。1.0% アガロースゲルより増幅産物を回収し、エタノール沈殿させ、沈殿を0.1×TEに溶解した。A260/280を測定しDNA濃度を算出した。PCRによって得られる導入断片は 3777 bpで、ubiquitin promoter-ω3デサチュラーゼ遺伝子-ubiquitin terminator-ubiquitin promoter-ブラストサイジン耐性遺伝子配列-SV40 terminator 配列という並びになる (配列番号:101)。
Thraustochytrium aureum をGY培地中で4日間培養し、対数増殖期にある細胞を遺伝子導入に使用した。OD600 = 1~1.5相当分の細胞に対し、0.625?gのDNA断片を遺伝子銃法(マイクロキャリア:0.6 micronの金粒子、target distance:6 cm、chamber vacuum:26 mmHg、Rupture disk:1,100 PSI)により導入した。遺伝子導入した細胞は4~6時間のリカバリータイムの後、0.2 mg/ml ブラストサイジン含有PDA寒天平板培地に塗布した。
1撃ちこみあたり、20から30個の薬剤耐性株を得た。
実施例9-4で得られたω3デサチュラーゼ遺伝子発現株からゲノムDNAを抽出後、A260/280を測定しDNA濃度を算出した。これを鋳型にし、LA taq Hot start version を用いてゲノム構造確認のPCRを行った。用いたprimerの位置、増幅に用いる組み合わせ、増幅産物の予想サイズを図52に示す。TMO42 (実施例9-3、配列番号:91) はubiquitin promoter上、RHO49 (実施例9-2、配列番号:88)はブラストサイジン耐性遺伝子上に設定した。[PCR cycles: 98℃ 2min/ 98℃ 10 sec, 68℃ 4 min, 30 cycles/ 68℃ 7min]。
予想されるサイズに増幅するバンドが確認できた(図53)。導入した発現断片がゲノムに安定に導入された株が単離できた。
Thraustochytrium aureumおよび実施例9-5で得られたω3 デサチュラーゼ発現株を培養、凍結乾燥後、脂肪酸をメチルエステル化し、GCを用いて解析した。GC解析にはガスクロマトグラフGC-2014(島津製作所社製)を使用し、カラム:HR-SS-10(30 m x 0.25 mm;信和化工社製)、カラム温度:150℃ →(5℃/min)→ 220℃(10 min)、キャリアガス:He(1.3mL/min)の条件で行った。
ω3 デサチュラーゼ発現株ではn-6系列の脂肪酸が減少し、n-3系列の脂肪酸に増加傾向が見られた (図54)。また、 図55に野生型株を100%とした時の割合を示す。
これらの結果、アラキドン酸が約1/10、DPAが約1/7に減少し、またEPAが約1.8倍、DHAが約1.2倍に増加した。
Claims (22)
- ストラメノパイルに外来遺伝子を導入することを特徴とする、ストラメノパイルの形質転換方法。
- ストラメノパイルがラビリンチュラ類である、請求項1に記載のストラメノパイルの形質転換方法。
- ラビリンチュラ類がラビリンチュラ属(Labyrinthula)、アルトルニア属(Altornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、アウランティオキトリウム属(Aurantiochytrium)、ヤブレツボカビ属(Thraustochytrium)、ウルケニア属(Ulkenia)、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属する微生物である、請求項2に記載のストラメノパイルの形質転換方法。
- 微生物がSchizochytrium sp. M-8株(FERM P-19755)、Thraustochytrium aureum ATCC 34304、Thraustochytrium sp. ATCC 26185、Schizochytrium sp. AL1Ac、Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、またはParietichytrium sarkarianum SEK364(FERM ABP-11298)である、請求項1ないし3のいずれかに記載のストラメノパイルの形質転換方法。
- 外来遺伝子が抗生物質耐性能、発色タンパク質、および/または脂肪酸不飽和化酵素に関連する遺伝子である、請求項1ないし4のいずれかに記載のストラメノパイルの形質転換方法。
- 脂肪酸不飽和化酵素に関連する遺伝子がΔ5デサチュラーゼ遺伝子、Δ12デサチュラーゼ遺伝子、および/またはω3デサチュラーゼ遺伝子である、請求項1ないし5のいずれかに記載のストラメノパイルの形質転換方法。
- エレクトロポレーションまたは遺伝子銃法によって外来遺伝子を導入することを特徴とする、請求項1ないし6のいずれかに記載のストラメノパイルの形質転換方法。
- 脂肪酸不飽和化酵素遺伝子を導入し、当該脂肪酸不飽和化酵素を発現することを特徴とする、ストラメノパイルの脂肪酸組成の改変方法。
- 脂肪酸不飽和化酵素がデサチュラーゼである、請求項8に記載の方法。
- 脂肪酸不飽和化酵素がΔ5デサチュラーゼ、Δ12デサチュラーゼ、またはω3デサチュラーゼである、請求項8または9に記載の方法。
- ストラメノパイルがラビリンチュラ類である、請求項8ないし10のいずれかに記載の方法。
- ラビリンチュラ類がラビリンチュラ属(Labyrinthula)、アルトルニア属(Altornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、アウランティオキトリウム属(Aurantiochytrium)、ヤブレツボカビ属(Thraustochytrium)、ウルケニア属(Ulkenia) 、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属する微生物である、請求項11に記載の方法。
- 微生物がSchizochytrium sp. M-8株(FERM P-19755)、Thraustochytrium aureum ATCC 34304、Thraustochytrium sp. ATCC 26185、Schizochytrium sp. AL1Ac、Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、またはParietichytrium sarkarianum SEK364(FERM ABP-11298)である、請求項12に記載の方法。
- 請求項8ないし13のいずれかに記載の方法により、ストラメノパイルに高度に脂肪酸を蓄積させる方法。
- 脂肪酸が不飽和脂肪酸である、請求項14に記載の方法。
- 不飽和脂肪酸が炭素数18~22の不飽和脂肪酸である、請求項15に記載の方法。
- 請求項14ないし16のいずれかに記載の方法により得られた脂肪酸。
- 脂肪酸組成の改変を目的として形質転換されたストラメノパイル。
- 脂肪酸を高度に蓄積させることを目的として形質転換されたストラメノパイル。
- ラビリンチュラ類である、請求項18または19に記載のストラメノパイル。
- ラビリンチュラ類がラビリンチュラ属(Labyrinthula)、アルトルニア属(Altornia)、アプラノキトリウム属(Aplanochytrium)、イァポノキトリウム属(Japonochytrium)、ラビリンチュロイデス属(Labyrinthuloides)、シゾキトリウム属(Schizochytrium)、アウランティオキトリウム属(Aurantiochytrium)、ヤブレツボカビ属(Thraustochytrium)、ウルケニア属(Ulkenia)、オブロンギキトリウム属(Oblongichytrium)、ボトリオキトリウム属(Botryochytrium)、パリエティキトリウム属(Parietichytrium)、またはシクヨイドキトリウム属(Sicyoidochytrium)に属する微生物である、請求項20に記載のストラメノパイル。
- 微生物がSchizochytrium sp. M-8株(FERM P-19755)、Thraustochytrium aureum ATCC 34304、Thraustochytrium sp. ATCC 26185、Schizochytrium sp. AL1Ac、Schizochytrium aggregatum ATCC 28209、Ulkenia sp. ATCC 28207、Schizochytrium sp. SEK210(NBRC 102615)、Schizochytrium sp. SEK345(NBRC 102616)、Botryochytrium radiatum SEK353(NBRC 104107)、またはParietichytrium sarkarianum SEK364(FERM ABP-11298)である、請求項21に記載のストラメノパイル。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2807754A CA2807754C (en) | 2009-09-24 | 2010-09-24 | Method for transforming stramenopiles |
| KR1020177027743A KR20170116226A (ko) | 2009-09-24 | 2010-09-24 | 스트라메노파일의 형질전환 방법 |
| KR1020127010459A KR20130026411A (en) | 2009-09-24 | 2010-09-24 | Method for transforming stramenopile |
| EP10818879.8A EP2481799A4 (en) | 2009-09-24 | 2010-09-24 | METHOD FOR THE TRANSFORMATION OF STRAMENOPILES |
| CN201080042308.2A CN102725405B (zh) | 2009-09-24 | 2010-09-24 | 原生藻菌的转化方法 |
| US13/497,894 US9150891B2 (en) | 2009-09-24 | 2010-09-24 | Method for transforming stramenopiles |
| JP2011533057A JP5894794B2 (ja) | 2009-09-24 | 2010-09-24 | ストラメノパイルの形質転換方法 |
| US14/832,595 US20150353944A1 (en) | 2009-09-24 | 2015-08-21 | Method for transforming stramenopile |
| US15/886,599 US10815505B2 (en) | 2009-09-24 | 2018-02-01 | Microbial oil extracted from stramenopiles |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009219820 | 2009-09-24 | ||
| JP2009-219820 | 2009-09-24 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/497,894 A-371-Of-International US9150891B2 (en) | 2009-09-24 | 2010-09-24 | Method for transforming stramenopiles |
| US14/832,595 Division US20150353944A1 (en) | 2009-09-24 | 2015-08-21 | Method for transforming stramenopile |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011037207A1 true WO2011037207A1 (ja) | 2011-03-31 |
Family
ID=43795949
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/066599 WO2011037207A1 (ja) | 2009-09-24 | 2010-09-24 | ストラメノパイルの形質転換方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (3) | US9150891B2 (ja) |
| EP (2) | EP2481799A4 (ja) |
| JP (1) | JP5894794B2 (ja) |
| KR (2) | KR20130026411A (ja) |
| CN (2) | CN102725405B (ja) |
| CA (2) | CA3121944A1 (ja) |
| WO (1) | WO2011037207A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012043826A1 (ja) * | 2010-10-01 | 2012-04-05 | 国立大学法人九州大学 | ストラメノパイルの形質転換方法 |
| WO2012120375A1 (en) * | 2011-03-07 | 2012-09-13 | Ocean Nutrition Canada Limited | Engineering thraustochytrid microorganisms |
| WO2017006918A1 (ja) * | 2015-07-03 | 2017-01-12 | 国立大学法人九州大学 | 微生物油産生ラビリンチュラ類、微生物油、ならびにそれらの作成方法およびそれらの使用 |
| JPWO2016111368A1 (ja) * | 2015-01-09 | 2017-11-24 | 国立大学法人 宮崎大学 | ヤブレツボカビ類を用いたリグニン分解活性を有するタンパク質の製造方法 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102725405B (zh) | 2009-09-24 | 2015-08-19 | 国立大学法人九州大学 | 原生藻菌的转化方法 |
| JP6135144B2 (ja) * | 2013-01-22 | 2017-05-31 | 富士電機株式会社 | 油脂製造方法 |
| US9932599B2 (en) | 2015-03-02 | 2018-04-03 | Synthetic Genomics, Inc. | Regulatory elements from labyrinthulomycetes microorganisms |
| CN106916796B (zh) * | 2015-12-28 | 2021-06-29 | 丰益(上海)生物技术研发中心有限公司 | 裂殖壶菌delta-12脂肪酸脱饱和酶相关序列及其应用 |
| EP3795586A3 (en) | 2016-03-16 | 2021-06-23 | Conagen Inc. | Production of proteins in labyrinthulomycetes |
| US10633454B2 (en) | 2016-11-01 | 2020-04-28 | Conagen Inc. | Expression of modified glycoproteins and glycopeptides |
| CN109628349B (zh) * | 2019-01-04 | 2022-05-17 | 自然资源部第一海洋研究所 | 一株具有羽毛降解活性的南极菌及其应用 |
| CN113215176B (zh) * | 2021-06-10 | 2023-08-08 | 广东省农业科学院动物卫生研究所 | 核酸分子、pcr引物对及用于检测禽毛滴虫ef1a基因的试剂盒 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005503125A (ja) * | 2001-04-16 | 2005-02-03 | マーテック・バイオサイエンスィズ・コーポレーション | トラウストチトリアレス微生物の形質転換についての産物および方法 |
| JP2005102680A (ja) | 2002-12-27 | 2005-04-21 | Fuji Photo Film Co Ltd | ドコサヘキサエン酸生産能を有する微生物及びその利用 |
| JP2005287380A (ja) | 2004-03-31 | 2005-10-20 | Masahiro Hayashi | 動物プランクトン用飼料及びそれを用いた動物プランクトンの培養方法 |
| JP2006304686A (ja) | 2005-04-28 | 2006-11-09 | Fuji Photo Film Co Ltd | ラビリンチュラ類への遺伝子導入法 |
| JP2006304685A (ja) | 2005-04-28 | 2006-11-09 | Fuji Photo Film Co Ltd | ラビリンチュラ類を形質転換可能なベクター |
| JP2007143479A (ja) | 2005-11-28 | 2007-06-14 | Kyushu Univ | スキゾキトリウム属の微生物を用いた長鎖高度不飽和脂肪酸含有リン脂質の製造方法 |
| JP2007527716A (ja) * | 2004-02-27 | 2007-10-04 | ビーエーエスエフ プラント サイエンス ゲーエムベーハー | トランスジェニック植物における多価不飽和脂肪酸の製造方法 |
| WO2008006202A1 (en) * | 2006-07-11 | 2008-01-17 | Bioriginal Food & Science Corporation | Fatty acid desaturases and uses thereof |
| WO2008144473A2 (en) * | 2007-05-16 | 2008-11-27 | Martek Biosciences Corporation | Chimeric pufa polyketide synthase systems and uses thereof |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1254238T3 (da) * | 2000-02-09 | 2009-11-30 | Basf Se | Nyt elongasegen og fremgangsmåde til fremstilling af flerumættede fedtsyrer |
| CA2421267C (en) * | 2000-09-28 | 2009-01-06 | Bioriginal Food & Science Corporation | Fad4, fad5, fad5-2, and fad6, novel fatty acid desaturase family members and uses thereof |
| EP1654344B1 (de) | 2003-08-01 | 2018-10-17 | BASF Plant Science GmbH | Verfahren zur herstellung mehrfach ungesättigter fettsäuren in transgenen |
| CA2559360A1 (en) * | 2004-02-27 | 2005-09-09 | Basf Plant Science Gmbh | Method for producing c18, c20 and c22 polyunsaturated fatty acids in transgenic plants |
| US7678560B2 (en) | 2006-05-17 | 2010-03-16 | E.I. Du Pont De Nemours And Company | Δ 5 desaturase and its use in making polyunsaturated fatty acids |
| WO2009016202A2 (de) * | 2007-07-31 | 2009-02-05 | Basf Plant Science Gmbh | Desaturasen und verfahren zur herstellung mehrfach ungesättigter fettsäuren in transgenen organismen |
| CN102725405B (zh) | 2009-09-24 | 2015-08-19 | 国立大学法人九州大学 | 原生藻菌的转化方法 |
-
2010
- 2010-09-24 CN CN201080042308.2A patent/CN102725405B/zh not_active Expired - Fee Related
- 2010-09-24 KR KR1020127010459A patent/KR20130026411A/ko active Search and Examination
- 2010-09-24 CA CA3121944A patent/CA3121944A1/en not_active Abandoned
- 2010-09-24 US US13/497,894 patent/US9150891B2/en not_active Expired - Fee Related
- 2010-09-24 CN CN201510430733.3A patent/CN105368726A/zh active Pending
- 2010-09-24 EP EP10818879.8A patent/EP2481799A4/en not_active Withdrawn
- 2010-09-24 JP JP2011533057A patent/JP5894794B2/ja active Active
- 2010-09-24 KR KR1020177027743A patent/KR20170116226A/ko active Search and Examination
- 2010-09-24 EP EP15178199.4A patent/EP2966169A1/en not_active Withdrawn
- 2010-09-24 CA CA2807754A patent/CA2807754C/en active Active
- 2010-09-24 WO PCT/JP2010/066599 patent/WO2011037207A1/ja active Application Filing
-
2015
- 2015-08-21 US US14/832,595 patent/US20150353944A1/en not_active Abandoned
-
2018
- 2018-02-01 US US15/886,599 patent/US10815505B2/en active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005503125A (ja) * | 2001-04-16 | 2005-02-03 | マーテック・バイオサイエンスィズ・コーポレーション | トラウストチトリアレス微生物の形質転換についての産物および方法 |
| JP2005102680A (ja) | 2002-12-27 | 2005-04-21 | Fuji Photo Film Co Ltd | ドコサヘキサエン酸生産能を有する微生物及びその利用 |
| JP2007527716A (ja) * | 2004-02-27 | 2007-10-04 | ビーエーエスエフ プラント サイエンス ゲーエムベーハー | トランスジェニック植物における多価不飽和脂肪酸の製造方法 |
| JP2005287380A (ja) | 2004-03-31 | 2005-10-20 | Masahiro Hayashi | 動物プランクトン用飼料及びそれを用いた動物プランクトンの培養方法 |
| JP2006304686A (ja) | 2005-04-28 | 2006-11-09 | Fuji Photo Film Co Ltd | ラビリンチュラ類への遺伝子導入法 |
| JP2006304685A (ja) | 2005-04-28 | 2006-11-09 | Fuji Photo Film Co Ltd | ラビリンチュラ類を形質転換可能なベクター |
| JP2007143479A (ja) | 2005-11-28 | 2007-06-14 | Kyushu Univ | スキゾキトリウム属の微生物を用いた長鎖高度不飽和脂肪酸含有リン脂質の製造方法 |
| WO2008006202A1 (en) * | 2006-07-11 | 2008-01-17 | Bioriginal Food & Science Corporation | Fatty acid desaturases and uses thereof |
| WO2008144473A2 (en) * | 2007-05-16 | 2008-11-27 | Martek Biosciences Corporation | Chimeric pufa polyketide synthase systems and uses thereof |
Non-Patent Citations (22)
| Title |
|---|
| "Abstracts of the Annual Meeting of the Society for Biotechnology, Japan", vol. 2005, 2005, article TSUNEHIRO AKI ET AL.: "Development of transformation system of oleaginous microorganisum, Labyrinthulida", pages: 199, XP008160071 * |
| "Abstracts of the Annual Meeting of the Society for Biotechnology, Japan", vol. 60, 2008, article HIROAKI IWASAKA ET AL.: "Modification of lipid composition by genetic engineering in oleaginous microorganism, Labyrinthulida", pages: 136, XP008160072 * |
| ADACHI, J. ET AL., COMPUT. SCI. MONOGR., 1996, pages 28 |
| CHALFIE M. ET AL., SCIENCE, vol. 263, 1994, pages 802 - 805 |
| FEBS LETT., vol. 553, 2003, pages 440 - 444 |
| HORROCKS, LA; YEO YK, PHARMACOL RES, vol. 40, 1999, pages 211 - 225 |
| LECTURE SUMMARY FOR THE 60TH CONFERENCE OF THE SOCIETY FOR BIOTECHNOLOGY, 2008, pages 136 |
| LIPPMEIER JC ET AL., LIPIDS, vol. 44, no. 7, 3 June 2009 (2009-06-03), pages 621 - 30 |
| NAOKI NAGANO ET AL.: "Labyrinthulea no Kodo Fuhowa Shibosan Seigosei Keiro no Hanbetsuho no Kaihatsu", NIPPON NOGEI KAGAKUKAI TAIKAI KOEN YOSHISHU, vol. 2009, March 2009 (2009-03-01), pages 35, XP008160075 * |
| NIPPON NOGEIKAGAKU KAISHI, vol. 77, no. 2, February 2003 (2003-02-01), pages 150 - 153 |
| NUCLEIC ACIDS RES., vol. 22, 1994, pages 4673 - 4680 |
| POULOS, A, LIPIDS, vol. 30, 1995, pages 1 - 14 |
| PRASHER, D. C. ET AL., GENE, vol. 111, no. 2, 1992, pages 229 - 233 |
| QIU, X. ET AL., J. BIOL. CHEM., vol. 276, 2001, pages 31561 - 6 |
| SANGER, F. ET AL., PROC. NATL. ACAD. SCI, vol. 74, 1977, pages 5463 |
| See also references of EP2481799A4 * |
| SHUJUNSHA: "Bio-Experiment Illustrated 2", FUNDAMENTALS OF GENE ANALYSIS, pages 63 - 68 |
| SHUJUNSHA: "Bio-Experiment Illustrated 2", FUNDAMENTALS OF GENE ANALYSIS, pages LL7 - 128 |
| SOUTHERN, P. J.; BERG, P., J. MOLEC. APPL. GEN., vol. 1, 1982, pages 327 - 339 |
| TAKUMI KOBAYASHI ET AL.: "A5desaturase Idenshi no Hatsugen ni yoru Labyrinthulea no Shibosan Sosei no Kaihen", DAI 82 KAI THE JAPANESE BIOCHEMICAL SOCIETY TAIKAI PROGRAM KOEN YOSHISHU, 25 September 2009 (2009-09-25), pages 2P-200, XP008160073 * |
| TAKUMI KOBAYASHI ET AL.: "Labyrinthulea no Kodo Fuhowa Shibosan Gosei ni Kansuru Idenshi no Shutoku to Kino Kaiseki", NIPPON NOGEI KAGAKUKAI TAIKAI KOEN YOSHISHU, vol. 2008, 2008, pages 22, XP008160074 * |
| YOKOYAMA R.; HONDA D., MYCOSCIENCE, vol. 48, 2007, pages 199 - 211 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012043826A1 (ja) * | 2010-10-01 | 2012-04-05 | 国立大学法人九州大学 | ストラメノパイルの形質転換方法 |
| US9062315B2 (en) | 2010-10-01 | 2015-06-23 | Kyushu University, National University Corporation | Method for transformation of stramenopile |
| US11203763B2 (en) | 2010-10-01 | 2021-12-21 | Kyushu University, National University Corporation | Microbial oil containing fatty acids obtained from stramenopile and method of producing the same |
| WO2012120375A1 (en) * | 2011-03-07 | 2012-09-13 | Ocean Nutrition Canada Limited | Engineering thraustochytrid microorganisms |
| US9133463B2 (en) | 2011-03-07 | 2015-09-15 | Dsm Nutritional Products Ag | Engineering microorganisms |
| US9758787B2 (en) | 2011-03-07 | 2017-09-12 | Dsm Nutritional Products Ag | Engineering microorganisms |
| JPWO2016111368A1 (ja) * | 2015-01-09 | 2017-11-24 | 国立大学法人 宮崎大学 | ヤブレツボカビ類を用いたリグニン分解活性を有するタンパク質の製造方法 |
| WO2017006918A1 (ja) * | 2015-07-03 | 2017-01-12 | 国立大学法人九州大学 | 微生物油産生ラビリンチュラ類、微生物油、ならびにそれらの作成方法およびそれらの使用 |
| JPWO2017006918A1 (ja) * | 2015-07-03 | 2018-05-24 | 国立大学法人九州大学 | 微生物油産生ラビリンチュラ類、微生物油、ならびにそれらの作成方法およびそれらの使用 |
| US20180320208A1 (en) * | 2015-07-03 | 2018-11-08 | Kyushu University, National University Corporation | Labyrinthulid microorganism capable of producing microbial oil, microbial oil, methods for producing said microorganism and for producing said microbial oil, and uses of said microorganism and said microbial oil |
| US11898190B2 (en) | 2015-07-03 | 2024-02-13 | Kyushu University, National University Corporation | Labyrinthulid microorganism capable of producing microbial oil, microbial oil, methods for producing said microorganism and for producing said microbial oil, and uses of said microorganism and said microbial oil |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102725405A (zh) | 2012-10-10 |
| JPWO2011037207A1 (ja) | 2013-02-21 |
| KR20170116226A (ko) | 2017-10-18 |
| CN102725405B (zh) | 2015-08-19 |
| EP2481799A9 (en) | 2013-02-13 |
| US20150353944A1 (en) | 2015-12-10 |
| EP2481799A4 (en) | 2013-08-07 |
| US10815505B2 (en) | 2020-10-27 |
| CA3121944A1 (en) | 2011-03-31 |
| EP2966169A1 (en) | 2016-01-13 |
| JP5894794B2 (ja) | 2016-03-30 |
| US9150891B2 (en) | 2015-10-06 |
| KR20130026411A (en) | 2013-03-13 |
| CA2807754C (en) | 2021-07-27 |
| CN105368726A (zh) | 2016-03-02 |
| CA2807754A1 (en) | 2011-03-31 |
| US20120322116A1 (en) | 2012-12-20 |
| US20180291407A1 (en) | 2018-10-11 |
| EP2481799A1 (en) | 2012-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5894794B2 (ja) | ストラメノパイルの形質転換方法 | |
| US11203763B2 (en) | Microbial oil containing fatty acids obtained from stramenopile and method of producing the same | |
| DK1620543T3 (en) | PREPARATION OF POLYUM Saturated FAT ACIDS IN OIL Yeast | |
| EP0978563A1 (en) | Delta 9-desaturase gene | |
| US11898190B2 (en) | Labyrinthulid microorganism capable of producing microbial oil, microbial oil, methods for producing said microorganism and for producing said microbial oil, and uses of said microorganism and said microbial oil | |
| JP5278891B2 (ja) | 高度不飽和脂肪酸の生産蓄積性を有する形質転換微生物 | |
| WO2010101220A1 (ja) | 新規酵素及びそれをコードするdna | |
| Tan | Cloning and functional analysis of the genes from entomopathogenic fungi involved in the biosynthesis of eicosatetraenoic acid (ETA) | |
| 松田高宜 | Study on the biosynthetic pathway of polyunsaturated fatty acids in unicellular marine eukaryotes aiming for industrial applications | |
| Okuda | Biochemical analysis and molecular breeding of oleaginous microorganisms for ω3 polyunsaturated fatty acid production |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080042308.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10818879 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2011533057 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20127010459 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010818879 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13497894 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2807754 Country of ref document: CA |