WO2010137349A1 - 神経栄養因子の活性が関与する疾患の治療または予防剤 - Google Patents
神経栄養因子の活性が関与する疾患の治療または予防剤 Download PDFInfo
- Publication number
- WO2010137349A1 WO2010137349A1 PCT/JP2010/003622 JP2010003622W WO2010137349A1 WO 2010137349 A1 WO2010137349 A1 WO 2010137349A1 JP 2010003622 W JP2010003622 W JP 2010003622W WO 2010137349 A1 WO2010137349 A1 WO 2010137349A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- mmol
- added
- optionally substituted
- mixture
- Prior art date
Links
- 0 Cc(cc1)cc2c1[n](*)c(*)n2 Chemical compound Cc(cc1)cc2c1[n](*)c(*)n2 0.000 description 3
- SKUIYRBGLUUSEU-UHFFFAOYSA-N CC(C)(C)OC([n](c(-c(cc1)cc2c1[n](C1CCOCC1)c(C)n2)c1)c2c1cccc2)=O Chemical compound CC(C)(C)OC([n](c(-c(cc1)cc2c1[n](C1CCOCC1)c(C)n2)c1)c2c1cccc2)=O SKUIYRBGLUUSEU-UHFFFAOYSA-N 0.000 description 1
- OVDGUTHABMXVMI-UHFFFAOYSA-N CCCNc(ccc(C(O)=O)c1)c1[N+]([O-])=O Chemical compound CCCNc(ccc(C(O)=O)c1)c1[N+]([O-])=O OVDGUTHABMXVMI-UHFFFAOYSA-N 0.000 description 1
- SZWUXFVTEPUSBH-UHFFFAOYSA-N CN(C)CCNc(c(N)c1)ccc1-c1nc(cccc2)c2[o]1 Chemical compound CN(C)CCNc(c(N)c1)ccc1-c1nc(cccc2)c2[o]1 SZWUXFVTEPUSBH-UHFFFAOYSA-N 0.000 description 1
- RYVPNBSPYULPBX-UHFFFAOYSA-N CN(C)CCNc(c([N+]([O-])=O)c1)ccc1-c1nc(cccc2)c2[o]1 Chemical compound CN(C)CCNc(c([N+]([O-])=O)c1)ccc1-c1nc(cccc2)c2[o]1 RYVPNBSPYULPBX-UHFFFAOYSA-N 0.000 description 1
- MBVSKEYWVHMBDY-UHFFFAOYSA-N COCc1nc(cc(C(F)(F)F)cc2)c2[n]1C1CCOCC1 Chemical compound COCc1nc(cc(C(F)(F)F)cc2)c2[n]1C1CCOCC1 MBVSKEYWVHMBDY-UHFFFAOYSA-N 0.000 description 1
- KPSCYXILHIZIFG-UHFFFAOYSA-N Cc1nc(cc(cc2)-c3nc(cccc4)c4[o]3)c2[n]1CCN(C)C Chemical compound Cc1nc(cc(cc2)-c3nc(cccc4)c4[o]3)c2[n]1CCN(C)C KPSCYXILHIZIFG-UHFFFAOYSA-N 0.000 description 1
- WRRVLSNYSLUPQO-UHFFFAOYSA-N Cc1nc(cc(cc2)Br)c2[n]1C1CCOCC1 Chemical compound Cc1nc(cc(cc2)Br)c2[n]1C1CCOCC1 WRRVLSNYSLUPQO-UHFFFAOYSA-N 0.000 description 1
- AIVUOLKBXGPRCT-UHFFFAOYSA-N Nc(cc(C(F)(F)F)cc1)c1NC1CCOCC1 Chemical compound Nc(cc(C(F)(F)F)cc1)c1NC1CCOCC1 AIVUOLKBXGPRCT-UHFFFAOYSA-N 0.000 description 1
- CXRZFBGEWWPCCK-UHFFFAOYSA-N Nc(cc(cc1)C(O)=O)c1NC1CCOCC1 Chemical compound Nc(cc(cc1)C(O)=O)c1NC1CCOCC1 CXRZFBGEWWPCCK-UHFFFAOYSA-N 0.000 description 1
- HLDFCCHSOZWKAA-UHFFFAOYSA-N [O-][N+](c(cc(C(F)(F)F)cc1)c1F)=O Chemical compound [O-][N+](c(cc(C(F)(F)F)cc1)c1F)=O HLDFCCHSOZWKAA-UHFFFAOYSA-N 0.000 description 1
- ISWGWGDZYATJSK-UHFFFAOYSA-N [O-][N+](c(cc(C(F)(F)F)cc1)c1NC1CCOCC1)=O Chemical compound [O-][N+](c(cc(C(F)(F)F)cc1)c1NC1CCOCC1)=O ISWGWGDZYATJSK-UHFFFAOYSA-N 0.000 description 1
- BOJWTAQWPVBIPG-UHFFFAOYSA-N [O-][N+](c(cc(cc1)C(O)=O)c1F)=O Chemical compound [O-][N+](c(cc(cc1)C(O)=O)c1F)=O BOJWTAQWPVBIPG-UHFFFAOYSA-N 0.000 description 1
- PKPOUWCQDJNQSC-UHFFFAOYSA-N [O-][N+](c1cc(C(O)=O)ccc1NC1CCOCC1)=O Chemical compound [O-][N+](c1cc(C(O)=O)ccc1NC1CCOCC1)=O PKPOUWCQDJNQSC-UHFFFAOYSA-N 0.000 description 1
Images
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/428—Thiazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- Neurotrophic factors such as BDNF and NGF are proteins that play an important role in central and peripheral nervous system cell differentiation, functional maintenance, synapse formation, regeneration and repair upon injury, and neurodegenerative diseases, diabetic Effective for the treatment of neuropathy, amyotrophic lateral sclerosis, multiple sclerosis, cerebral ischemic disease, Alzheimer's disease, Parkinson's disease, Huntington's chorea, depression, peripheral neuropathy caused by cancer chemotherapy, etc. (Non-Patent Documents 2, 3, 4, and 5).
- a neurotrophic factor is a high molecular weight protein having a molecular weight of 10,000 or more, and it is known that when such a high molecular weight protein is used as a therapeutic agent, there are restrictions on the administration method and safety problems (non-patented). From the references 6 and 7), there is an urgent need to provide a low molecular weight compound that enhances the action of a neurotrophic factor with few side effects.
- the expression of certain transcriptional regulatory factors is controlled by neurotrophic factors (see Non-Patent Document 1), and one of the transcriptional regulatory factors whose expression is controlled by neurotrophic factors. NXF is known (see Patent Document 1).
- Patent Document 2 discloses a general formula. The compound represented by these is disclosed.
- Patent Document 4 discloses a general formula. The compound represented by these is disclosed.
- Patent Document 5 discloses a general formula. The compound represented by these is disclosed.
- Patent Document 6 discloses a general formula. The compound represented by these is disclosed.
- R 3 represents a hydrogen atom or a substituent
- R 4 represents a hydrogen atom or a substituent
- Ring A is Represents an optionally substituted benzene ring
- ring B is It represents an optionally substituted benzene ring.
- a pharmaceutically acceptable salt thereof, or a solvate thereof in this specification, sometimes referred to as compound (I). In the present specification, it may be referred to as a compound of the present invention.
- R 1 is (1) C 3-6 alkyl group, (2) (a) a halogen atom, (b) R a -O-, (c) R a —O—CO—, (d) R a —O—CO—NR a —, (e) R a —O—CO—C 2 H 4 —CO—NH—, (f) R a -S-, (g) R a —SO 2 —, (h) R a —CO—O—, (i) R a —CO—NR a —, (j) R a -NR a- , (k) R a -NR a -CS-NR a- , (l) a 5- to 6-membered cyclic group, (m) a carboxy group, (n) a hydroxy group, (o) an amino group, (p) heterocycle-carbonyl group, (q
- composition which is an agent for treating or preventing a disease involving the activity of neurotrophic factor, or an accelerator for a physical therapy effect.
- the treatment or prevention agent for a disease involving the activity of neurotrophic factor, or the accelerator for physiotherapy effect is the treatment or prevention agent for cerebral ischemic disease or diabetic neuropathy according to the above [7].
- Ring A is (a) a halogen atom, (b) a hydroxyl group, (c) a carboxy group, (d) a cyano group, (e) a sulfamoyl group, (f) a monoalkylamide group, (g) a dialkylamide group, (h) an alkyl group optionally substituted by a halogen atom, a benzene ring optionally substituted with one or more substituents selected from the group consisting of (i) a nitro group, and (j) an aryloxy group, Ring B is (a) a halogen atom, (b) a hydroxyl group, (c) a carboxy group, (d) a cyano group, (e) a
- Formula for the manufacture of a therapeutic or prophylactic agent for a disease involving the activity of neurotrophic factor, or an accelerator for a physiotherapy effect [Where: Z represents N or CR 2 ; X is Represents N—R 3 , O, or S; Y is Represents CR 4 or N; R 1 is A hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group; R 2 represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group; R 3 represents a hydrogen atom or a substituent, R 4 represents a hydrogen atom or a substituent, Ring A is Represents an optionally substituted benzene ring, Ring B is It represents an optionally substituted benzene ring. ] Or a pharmaceutically acceptable salt or solvate thereof.
- R 1 is Represents an optionally substituted non-cyclic hydrocarbon group or an optionally substituted cyclic group
- X is N—R 3 (R 3 represents a hydrogen atom or a substituent), O, or S
- Y is C—R 4 (R 4 represents a hydrogen atom or a substituent), or N
- Z is N, or C—R 2 (R 2 represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group)
- Ring A and Ring B are Independently, it represents an optionally substituted benzene ring.
- R 1 is the formula (Wherein R a1 represents a carboxy group, a cyano group, a 1H-tetrazolyl group, a 1-triphenylmethyl-tetrazolyl group, or an alkoxycarbonyl group, and R a2 represents a hydrogen atom, a fluorine atom, a chlorine atom, or bromine. It is not a group represented by. In addition, however, R 1 is is not. ] Or a pharmaceutically acceptable salt or solvate thereof.
- R 1 is a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) a 6 to 10 membered cyclic group which may be substituted with one or more substituents selected from a halogen atom, a C 1-4 alkoxy group, and a C 1-4 alkoxy-carbonyl group;
- X is N—R 3 (R 3 is a hydrogen atom, or R a —O—CO— (R a is a C 1-6 alkyl group)), O, or S;
- Y is CH or N;
- Z is, N, or C-R 2 (Wherein R 2 is a) a hydrogen atom, b) (1) a halogen atom, (2) R b —O—, (3) R b —CO—, (4) R b —CO—O— and (5) R b —NR b — (In the formula, each R b is the same
- R 1 is Represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group
- X is N—R 3 (R 3 represents a hydrogen atom or a substituent), O, or S
- Y is C—R 4 (R 4 represents a hydrogen atom or a substituent), or N
- Z is N, or C-R 2 (.
- R 2 is hydrogen atom, optionally substituted acyclic hydrocarbon group, or a substituted represents an even cyclic group) represents; and ring A and ring B Independently, it represents an optionally substituted benzene ring.
- a pharmaceutically acceptable salt thereof, or a solvate thereof (hereinafter, referred to as compound (I ′).
- Compound (I ′) will be described in accordance with the description of compound (I) above. It is understood in the same manner.
- a therapeutic or prophylactic agent for a disease involving the activity of a neurotrophic factor, or an accelerator for a physical therapy effect [4 '] The agent according to [3 ′] above, which is a therapeutic or preventive agent for cerebral ischemic disease or diabetic neuropathy.
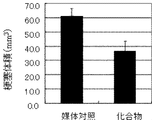
- the graph which shows infarct volume (Test Example 2).
- aryl (group) examples include C 6-14 aryl (group).
- C 6-14 aryl (group) includes phenyl, naphthyl, and anthryl.
- heteroaryl (group) examples include a 5- to 6-membered heteroaryl (group).
- the “5- to 6-membered heteroaryl (group)” includes, for example, 1 to 3 (preferably 1 to 2) heterocycles selected from an oxygen atom, a sulfur atom, and a nitrogen atom as ring-constituting atoms.
- 5- to 6-membered heteroaryl (group) having an atom examples include 1 to 3 (preferably 1 to 2) heterocycles selected from an oxygen atom, a sulfur atom, and a nitrogen atom as ring-constituting atoms.
- furyl eg, 2-furyl, 3-furyl
- thienyl eg, 2-thienyl, 3-thienyl
- pyridyl eg, 2-pyridyl, 3-pyridyl, 4-pyridyl
- pyrimidinyl eg, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, 6-pyrimidinyl
- pyridazinyl eg, 3-pyridazinyl, 4-pyridazinyl
- pyrazinyl eg, 2-pyrazinyl
- pyrrolyl eg, 1-pyrrolyl, 2-pyrrolyl) , 3-pyrrolyl
- imidazolyl eg, 1-imidazoly
- Examples of the “cycloalkenyl (group)” include C 3-10 cycloalkenyl (group).
- Examples of “C 3-10 cycloalkenyl (group)” include cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclononenyl, and cyclodecenyl.
- cycloalkadienyl (group) examples include C 4-10 cycloalkadienyl group.
- Examples of the “C 4-10 cycloalkadienyl group” include cyclobutadienyl, cyclopentadienyl, cyclohexadienyl, cycloheptadienyl, cyclooctadienyl, cyclononadienyl, and cyclodecadienyl. Is mentioned.
- non-aromatic heterocyclic group examples include 5- to 6-membered non-aromatic heterocyclic groups.
- the “5- to 6-membered non-aromatic heterocyclic group” includes, for example, 1 to 3 (preferably 1) selected from an oxygen atom, an optionally oxidized sulfur atom, and a nitrogen atom as ring-constituting atoms.
- alkyl (group) examples include C 1-6 alkyl (group).
- C 1-6 alkyl (group) examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, pentyl, and hexyl.
- alkenyl (group) examples include C 2-6 alkenyl (group).
- Examples of “C 2-6 alkenyl (group)” include vinyl, 2-propenyl, 3-methyl-2-butenyl, and 1,3-butadienyl.
- Examples of “alkynyl (group)” include C 2-6 alkynyl (group).
- Examples of “C 2-6 alkynyl (group)” include ethynyl, 2-propynyl, and 2-penten-4-ynyl.
- Alkylene (chain)” includes, for example, C 1-3 alkylene (chain).
- C 1-3 alkylene (chain) includes, for example, methylene, ethylene, and trimethylene.
- halogen (atom) examples include fluorine, chlorine, bromine, and iodine.
- alkoxy-carbonyl (group) examples include C 1-6 alkoxy-carbonyl (group).
- C 1-6 alkoxy-carbonyl (group) include methoxycarbonyl, ethoxycarbonyl, n-propyloxycarbonyl, isopropyloxycarbonyl, n-butyloxycarbonyl, sec-butyloxycarbonyl, tert-butyloxycarbonyl , N-pentyloxycarbonyl, n-hexyloxycarbonyl group and the like.
- C 1-6 alkoxy-carbonyl (group) is R a —O—CO— (R a is C 1-6 alkyl (group)).
- R 1 is A hydrogen atom, an acyclic hydrocarbon group which may be substituted, or a cyclic group which may be substituted is represented.
- Examples of the “acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 1 include an alkyl group, an alkenyl group, and an alkynyl group.
- Examples of the “alkyl group” include those exemplified above.
- Examples of the “alkenyl group” include those exemplified above.
- Examples of the “alkynyl group” include those exemplified above. Of these, an alkyl group is preferable, and a C 1-6 alkyl group is more preferable.
- the “acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 1 may be substituted with one or more (preferably 1 to 3) substituents. .
- Examples of the “halogen atom” include those exemplified above.
- Examples of the “C 1-6 alkyl group” include those exemplified above.
- Examples of the “optionally substituted cyclic group” include those similar to the “optionally substituted cyclic group” represented by R 1 exemplified below.
- non-aromatic cyclic hydrocarbon group examples include a cycloalkyl group, a cycloalkenyl group, and a cycloalkadienyl group.
- examples of the “cycloalkyl group”, “cycloalkenyl group”, and “cycloalkadienyl group” include those exemplified above.
- Examples of the “non-aromatic heterocyclic group” include those exemplified above.
- cyclic group examples include C 3-10 cycloalkyl, C 3-10 cycloalkenyl, C 4-10 alicyclic hydrocarbon group having 3 to 10 carbon atoms, such as cycloalkadienyl or a 5-6 membered non-aromatic, A group heterocyclic group is preferred.
- the “cyclic group” of the “optionally substituted cyclic group” represented by R 1 may be substituted with one or more (preferably 1 to 3) substituents.
- substituents for example, (1) a halogen atom, (2) Nitro group, (3) a cyano group, (4) a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms, (5) a C 1-6 alkenyl group optionally substituted with one or more (preferably 1 to 3) halogen atoms, (6) a C 1-6 alkynyl group optionally substituted by one or more (preferably 1 to 3) halogen atoms, (7) R a -CO-L-, (8) R a —CO—OL— (9) R a —CO—NR a —L—, (10) R a —OL— (11) R a -O-CO-L-, (12) R a —O—CO—NR a —L—, (13) R
- halogen atom examples include those exemplified above.
- substituent for the non-aromatic cyclic hydrocarbon group particularly an alicyclic hydrocarbon group having 3 to 10 carbon atoms
- non-aromatic heterocyclic group particularly a 5- to 6-membered non-aromatic heterocyclic group
- (a) an oxo group, (b) a C 1-4 alkoxy-carbonyl group, (c) a hydroxy group is preferred, (a) an oxo group, (b) A C 1-4 alkoxy-carbonyl group is more preferred.
- R c is the same or different and represents a hydrogen atom or a C 1-6 alkyl group).
- R 1 is preferably, for example, a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) (1) a halogen atom, (2) a C 1-6 alkoxy group which may be substituted with one or more (preferably 1 to 3) halogen atoms, and (3) optionally substituted with one or more substituents selected from C 1-6 alkoxy-carbonyl groups optionally substituted with one or more (preferably 1 to 3) halogen atoms
- a 3- to 10-membered cyclic group More preferably, for example, a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) optionally substituted with one or more (preferably 1 to 3) substituents selected from a halogen atom, a C 1-4 alkoxy group, and a C 1-4 alkoxy-carbonyl group Member
- X represents N—R 3 (R 3 represents a hydrogen atom or a substituent), O, or S.
- X is more preferably, for example, N—R 3 (R 3 is a hydrogen atom or R a —O—CO— (R a is a C 1-6 alkyl group)), O, Or S.
- X is more preferably NH, O, or S.
- Y represents C—R 4 (R 4 represents a hydrogen atom or a substituent), or N.
- R 4 examples include: (1) a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms, (2) a C 1-6 alkenyl group optionally substituted by one or more (preferably 1 to 3) halogen atoms, (3) a C 1-6 alkynyl group optionally substituted by one or more (preferably 1 to 3) halogen atoms, (4) R a -CO-L-, (5) R a -CO-O -L-, (6) R a —CO—NR a —L—, (7) R a -OL-, (8) R a -O-CO-L-, (9) R a —O—CO—NR a —L—, (10) R a -SL-, (11) R a -SO-L-, (12) R a —SO 2 —L—, (13) R a -NR a -L-, (14) R a —S—
- Y is more preferably, for example, CH or N. Y is more preferably N.
- Z represents N or C—R 2 .
- R 2 represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group. In the compounds of the present invention, preferably Z is C—R 2 .
- Examples of the “acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 2 include an alkyl group, an alkenyl group, and an alkynyl group.
- Examples of the “alkyl group” include those exemplified above.
- Examples of the “alkenyl group” include those exemplified above.
- Examples of the “alkynyl group” include those exemplified above.
- acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 2 may be substituted with one or more (preferably 1 to 3) substituents. .
- each R b is the same or different and represents a hydrogen atom or a C 1-6 alkyl group which may be
- each R b is the same or different and represents a hydrogen atom or a C 1-6 alkyl group which may be substituted with one or more halogens, and n represents an integer of 0 to 2). Is more preferable.
- R 2 is preferably, for example, (1) hydrogen atom, (2) (a) a halogen atom, (b) R b —O—, (c) R b —O—CO—, (d) R b —O—CO—NR b —, (e) R b -S-, (f) R b —SO 2 —, (g) R b —CO—O—, (h) R b —CO—NR b —, (i) R b —NR b —, (j) R b —NR b —R b —S (O) n — (k) phenyl group, (l) a 5- to 6-membered saturated heterocyclic group, (m) a hydroxy group, and (n) an amino group (wherein each R b is the same or different and represents
- a C 1-6 alkyl group which may be substituted with one or more substituents selected from the group consisting of: (3) (a) a halogen atom, (b) R c —O—, (c) R c —O—CO—, and (d) R c —CO—NR c — (Wherein R c are the same or different and each represents a hydrogen atom or a C 1-6 alkyl group.)
- a non-aromatic cyclic hydrocarbon group having 5 to 6 carbon atoms or a 5- to 6-membered non-aromatic heterocyclic group which may be substituted with one or more substituents selected from the group consisting of
- R 2 is a) a hydrogen atom, b) (1) a halogen atom, (2) R b —O—, (3) R b —CO—, (4) R b —CO—O— and (5) R b —NR b —
- each R b is the same or different and represents a hydrogen atom or a C 1-6 alkyl group which may be substituted with one phenyl group.
- a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) substituents selected from: c) 5- to 6-membered cyclic group. ) It is.
- Ring A represents an optionally substituted benzene ring.
- the “benzene ring” of the “optionally substituted benzene ring” represented by ring A may be substituted with one or more (preferably 1 to 3) substituents.
- substituents the thing similar to the substituent illustrated about "the cyclic group which may be substituted" represented by R ⁇ 1 > is mentioned.
- Ring A is preferably, for example, unsubstituted (ie Benzene ring).
- Ring B represents an optionally substituted benzene ring.
- the “benzene ring” of the “optionally substituted benzene ring” represented by ring B may be substituted with one or more (preferably 1 to 3) substituents.
- substituents the thing similar to the substituent illustrated about "the cyclic group which may be substituted" represented by R ⁇ 1 > is mentioned.
- ring B is preferably, for example, (1) a halogen atom, (2) Nitro group, (3) a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms (4) R a -O-CO-L-, (5) R a —SO 2 —NR a —L—, and (6) R a -NR a -L- Wherein each R a is the same or different and is a hydrogen atom or a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms; Is a bond.) Or a benzene ring optionally substituted with one or more (preferably 1 to 3) substituents selected from
- Ring B is more preferably, for example, (1) a halogen atom (preferably chlorine), (2) Nitro group, (3) a C 1-6 alkyl group (preferably methyl), and (4) A benzene ring which may be substituted with one or more (preferably 1 to 3) substituents selected from amino groups.
- a halogen atom preferably chlorine
- Nitro group preferably nitrogen
- C 1-6 alkyl group preferably methyl
- a benzene ring which may be substituted with one or more (preferably 1 to 3) substituents selected from amino groups.
- Compound (I) is more preferably one in which two or more selected from the above-mentioned preferred partial structures, more preferred partial structures, and further preferred partial structures are used in combination.
- the compound (I) is preferably, for example, the following compound A.
- Compound A is a novel compound.
- R 1 is 5- to 6-membered non-aromatic heterocyclic group.
- Z is CR- 2 , Y is N, R 1 is A 5- to 6-membered non-aromatic heterocyclic group, and R 2 is A C 1-6 alkyl group;
- R 1 is a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) a 6-10 membered cyclic group optionally substituted with one or more substituents selected from a halogen atom, one or more C 1-4 alkoxy groups, and a C 1-4 alkoxy-carbonyl group.
- Compound (I) may be a pharmaceutically acceptable salt.
- salts include metal salts, ammonium salts, salts with organic bases, salts with inorganic acids, salts with organic acids, salts with basic or acidic amino acids, and the like.
- metal salt include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt, magnesium salt and barium salt; aluminum salt and the like.
- Preferable examples of the salt with organic base include, for example, trimethylamine, triethylamine, pyridine, picoline, 2,6-lutidine, ethanolamine, diethanolamine, triethanolamine, cyclohexylamine, dicyclohexylamine, N, N′-dibenzylethylenediamine. And the like.
- Preferable examples of the salt with inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfone. And salts with acid, p-toluenesulfonic acid and the like.
- salts with basic amino acids include salts with arginine, lysine, ornithine and the like
- salts with acidic amino acids include salts with aspartic acid and glutamic acid, for example. It is done. Of these, pharmaceutically acceptable salts are preferred.
- the compound (I) has a tautomer, an isomer having a stable structure is preferable.
- the present invention is not limited to this, and does not exclude other isomers. It is within the scope of the present invention.
- compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, or a rotational isomer, any one of the isomers and mixtures are within the scope of the present invention. Included in the compounds of the present invention.
- compound (I) has an optical isomer, the optical isomer resolved from the racemate is also within the scope of the present invention.
- Compound (I) may be crystalline or amorphous and both are within the scope of the present invention.
- Compound (I) labeled or substituted with an isotope is also within the scope of the present invention.
- Compound (I) may be a solvate (such as a hydrate) or a non-solvate.
- each symbol of the compound in the reaction formula represents the same meaning as described above.
- the compound in a formula also includes the case where it forms the salt, as such a salt, the thing similar to the salt of compound (I) is mentioned, for example.
- the compound obtained in each step can be used in the next reaction as a reaction solution or as a crude product, but can be isolated from the reaction mixture according to a conventional method. It can be easily purified by separation means such as extraction, concentration, neutralization, filtration, distillation, recrystallization, distillation, chromatography and the like. Or when the compound in a formula is marketed, a commercial item can also be used as it is. Unless otherwise specified, room temperature herein is about 10 ° C. to about 35 ° C.
- Compound (I) can be produced, for example, by the method shown in the following scheme or a method analogous thereto.
- a halogen atom As the leaving group represented by Xa, for example, A halogen atom; An optionally substituted alkylsulfonyloxy group (eg, a methanesulfonyloxy group, an ethanesulfonyloxy group, and an alkylsulfonyloxy group optionally substituted with a halogen atom such as trifluoromethanesulfonyloxy); and Good arylsulfonyloxy groups (eg, benzenesulfonyloxy, p-toluenesulfonyloxy group, and 2-nitrobenzenesulfonyloxy group) and the like, and a halogen atom is particularly preferable.
- An optionally substituted alkylsulfonyloxy group eg, a methanesulfonyloxy group, an ethanesulfonyloxy group, and an alkylsulfonyl
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from instantaneous to about 24 hours.
- the amount of compound (S-1 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-1).
- the base used include carbonates such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate, cesium carbonate and potassium phosphate; hydroxides such as sodium hydroxide and potassium hydroxide; morpholine, piperidine and pyridine.
- organic amines such as trimethylamine, triethylamine, diisopropylethylamine, 4-dimethylaminopyridine, and the like.
- the amount thereof to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-1).
- the solvent include water; alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and Aprotic solvents such as N-methylpyrrolidone; halogenated hydrocarbons such as chloroform, or mixed solvents thereof are used, and the organic amines and compounds (S-1 ′) described above are used as a solvent. May be.
- the solvent examples include alcohol solvents such as methanol, ethanol and 2-propanol; ether solvents such as tetrahydrofuran; ester solvents such as ethyl acetate; aprotic polar solvents such as N, N-dimethylformamide or the like And the like.
- the reaction can be carried out by reacting at a temperature within a range from about 0 ° C. to the boiling point of the solvent used for 10 minutes to 48 hours.
- the metal reducing agent include tin (II) chloride, reduced iron, titanium trichloride (III), and the like. Is 1 to 5 moles.
- the solvent examples include water; dilute hydrochloric acid; acetic acid; alcohol solvents such as methanol, ethanol and 2-propanol; ether solvents such as tetrahydrofuran and 1,2-dimethoxyethane; ester solvents such as ethyl acetate; acetone, acetonitrile and Examples thereof include aprotic polar solvents such as N, N-dimethylformamide, or mixed solvents thereof.
- an acid chloride compound When an acid chloride compound is used, for example, it can be carried out according to the method described in Zhurnal Obshchei Khimii 1962, 32 (5), 1581-86 (Engl. Transl. Ver .: pp 1565-1569). Specifically, for example, General formula (R 1 , R 3 ′ and ring A are as described above.) A compound (S-3) represented by: Formula R 2 —COCl (R 2 is as described above.) By reacting with an acid chloride compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) In this reaction, the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 24 hours.
- the amount of the acid chloride compound to be used is generally 1 to 5 mol, preferably 1 to 2 mol, per 1 mol of compound (S-3).
- the solvent include ether solvents such as tetrahydrofuran and 1,2-dimethoxyethane, ester solvents such as ethyl acetate, aprotic polar solvents such as acetone, acetonitrile and N, N-dimethylformamide, or a mixture thereof. A solvent etc. are mentioned.
- a carboxylic acid compound when used, for example, it can be carried out according to the method described in Zhurnal Obshchei Khimii 1962, 32 (5), 1581-86 (Engl. Transl. Ver .: pp 1565-1569). Specifically, for example, General formula (R 1 , R 3 ′ and ring A are as described above.) A compound (S-3) represented by: Formula R 2 —COOH (R 2 is as described above.) By reacting with a carboxylic acid compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) (S-4) can be produced.
- the solvent examples include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; or a mixed solvent thereof.
- ether solvents such as tetrahydrofuran and dioxane
- aromatic hydrocarbon solvents such as benzene, toluene and xylene
- a mixed solvent thereof a mixed solvent thereof.
- the carboxylic acid described above is used as the solvent. May be.
- an aldehyde compound when used, it can carry out according to the method as described in Synthesis 2003, No11, 1683-1692, for example.
- General formula (R 1 , R 3 ′ and ring A are as described above.
- S-4 By reacting with an aldehyde compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) (S-4) can be produced.
- the reaction temperature is usually from room temperature to reflux temperature
- the reaction time is usually from instantaneous to about 24 hours.
- the amount of the aldehyde compound to be used is generally 1 to 5 mol, preferably 1 to 2 mol, per 1 mol of compound (S-3).
- the oxidizing agent include m-chloroperbenzoic acid, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), oxone, benzofuroxane, potassium permanganate, hydrogen peroxide, Examples thereof include acetic acid and tert-butyl hydroperoxide, and the amount is usually 0.1 to 5 mol, preferably 0.5 to 2 mol, relative to 1 mol of compound (S-3).
- the solvent examples include water; alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and An aprotic solvent such as N-methylpyrrolidone; a halogenated hydrocarbon such as chloroform, or a mixed solvent thereof is used.
- the amount of the triacetal compound to be used is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound (S-3).
- a catalytic amount of acid may be added to the reaction system.
- the acid used include inorganic acids such as hydrochloric acid, hydrobromic acid, nitric acid, and sulfuric acid, and organic acids such as acetic acid, trifluoroacetic acid, methanesulfonic acid, and p-toluenesulfonic acid.
- the solvent examples include alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and N- An aprotic solvent such as methylpyrrolidone; a halogenated hydrocarbon such as chloroform, or a mixed solvent thereof is used.
- the triacetal compound described above may be used as a solvent.
- an imidate compound when used, for example, it can be carried out according to the method described in JP-A-4-308580.
- the general formula (R 1 , R 3 ′ and ring A are as described above.) (S-3) and a general formula R 2 —C ( ⁇ NH) OR 2 ′ (R 2 and R 2 ′ are as described above.)
- S-4 By reacting with the imidate compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) (S-4) can be produced.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from instantaneous to about 24 hours.
- the amount of the imidate compound to be used is generally 1 to 5 mol, preferably 1 to 2 mol, per 1 mol of compound (S-3).
- the solvent examples include alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and N- An aprotic solvent such as methylpyrrolidone; a halogenated hydrocarbon such as chloroform, or a mixed solvent thereof is used.
- lower fatty acid anhydrides such as acetic anhydride and propionic anhydride
- organic sulfonic acids such as methanesulfonic acid and paratoluenesulfonic acid
- phosphorus oxychloride phosphorus trichloride, diphosphorus pentoxide, sulfuric acid, polyphosphoric acid
- solvent that may use an inorganic acid such as boric acid include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene, and xylene; or a mixed solvent thereof.
- the dehydration condensation agent described above may be used as a solvent.
- the first step is a reaction using a condensing agent or an acid halogenating agent.
- a condensing agent When a condensing agent is used, the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 24 hours.
- the amount of compound (S-5 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-5).
- the condensing agent include Bop (1H-1,2,3-benzotriazol-1-yloxy (tri (dimethylamino)) phosphonium hexafluorophosphate), WSC (1-ethyl-3- (3-dimethylaminopropyl).
- Carbodiimide / hydrochloride Carbodiimide / hydrochloride), DCC (N, N-dicyclohexylcarbodiimide), CDI (carbonyldiimidazole), diethyl phosphoryl cyanide, and the like.
- the amount of the condensing agent to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol with respect to compound (S-5). If necessary, 1 equivalent to an excessive amount of an organic base such as triethylamine may be added to the compound (S-5).
- the solvent examples include halogenated hydrocarbons such as dichloromethane and chloroform, sulfoxides such as dimethyl sulfoxide, esters such as ethyl acetate, ethers such as tetrahydrofuran and 1,4-dioxane, and the like.
- halogenated hydrocarbons such as dichloromethane and chloroform
- sulfoxides such as dimethyl sulfoxide
- esters such as ethyl acetate
- ethers such as tetrahydrofuran and 1,4-dioxane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide.
- the reaction temperature range is usually from room temperature to reflux temperature
- the reaction time range is usually from instantaneous to about 24 hours.
- the amount of compound (S-5 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-5
- the acid halogenating agent examples include thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride and the like.
- the amount of the acid halogenating agent to be used is generally 1 to 10 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-5).
- a catalytic amount of N, N-dimethylformamide may be added to the reaction system.
- the solvent examples include halogenated hydrocarbons such as dichloromethane and chloroform; sulfoxides such as dimethyl sulfoxide; esters such as ethyl acetate; ethers such as tetrahydrofuran and 1,4-dioxane; Examples include amides such as N, N-dimethylformamide and N, N-dimethylacetamide; aromatic hydrocarbons such as benzene and toluene.
- halogenated hydrocarbons such as dichloromethane and chloroform
- sulfoxides such as dimethyl sulfoxide
- esters such as ethyl acetate
- ethers such as tetrahydrofuran and 1,4-dioxane
- aromatic hydrocarbons such as benzene and toluene.
- the second step is a reaction using a dehydration condensing agent.
- the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 48 hours.
- dehydrating condensing agents lower fatty acid anhydrides such as acetic anhydride and propionic anhydride; organic sulfonic acids such as methanesulfonic acid and paratoluenesulfonic acid; phosphorus oxychloride, phosphorus trichloride, diphosphorus pentoxide, sulfuric acid, polyphosphoric acid,
- Examples of the solvent that may use an inorganic acid such as boric acid include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene, and xylene; or a mixed solvent thereof.
- the dehydration condensation agent described above may be used as a solvent.
- nitrosating agent examples include alkali metal nitrites such as sodium nitrite, organic nitrite compounds such as methyl nitrite or isoamyl nitrite, and the amount used is 1 mol of compound (S-3). Usually 1 to 5 mol, preferably 1 to 2 mol.
- the solvent a combination of water and an acid is used.
- the acid include inorganic acids such as hydrochloric acid and sulfuric acid, and organic acids such as acetic acid.
- Cyclization 4 can be performed by methods well known to those skilled in the art (for example, the method described in Synthetic Communications 1998, 28 (22), 4123-4135). Specifically, for example, the general formula (Ya represents a halogen atom, R 1 , R 2 and ring A are as described above. ) (S-8) represented by the general formula (X and ring B are as described above.) Is reacted with a compound (S-5 ′) represented by General formula (R 1 , R 2 , X, ring A and ring B are as described above.) (S-6) can be produced. In this reaction, the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from instantaneous to about 24 hours.
- the amount of compound (S-5 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-8).
- dehydrating condensing agents lower fatty acid anhydrides such as acetic anhydride and propionic anhydride; organic sulfonic acids such as methanesulfonic acid and paratoluenesulfonic acid; phosphorus oxychloride, phosphorus trichloride, diphosphorus pentoxide, sulfuric acid, polyphosphoric acid,
- Examples of the solvent that may use an inorganic acid such as boric acid include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene, and xylene; or a mixed solvent thereof.
- the dehydration condensation agent described above may be used as a solvent.
- Coupling is a method well known to those skilled in the art (for example, translated by Kiyoshi Tomioka, “Organic synthesis strategy learned from personal reaction”, Kagaku Dojin Co., Ltd., August 15, 2006, p258-259: Kumada cross-coupling reaction, p310- 311: Negishi cross coupling reaction, p440-441: Stilley-Kelly coupling reaction, p.448-449: Suzuki-Miyaura coupling reaction).
- the general formula (R 1 , R 2 , Xa and ring A are as described above.
- organotin for example, SnBu 4 (tetrabutyltin)
- X and ring B are as described above.
- General formula (R 1 , R 2 , X, ring A and ring B are as described above.) (S-11) can be produced.
- the reaction is carried out in the presence of a transition metal catalyst, and optionally in the presence of a ligand, base, additive, etc., at a temperature in the range from about 20 ° C. to the boiling point of the solvent used for 10 minutes to 48 hours. This can be done.
- the amount of compound (S-10) to be used is generally 1 to 20 mol, preferably 1 to 5 mol, per 1 mol of compound (S-9).
- transition metal catalyst examples include palladium (II) acetate, palladium (II) chloride, tetrakis (triphenylphosphine) palladium (0), bis (triphenylphosphine) palladium chloride (II), tris (dibenzylideneacetone) dipalladium. (0), or [1,1′-bis (diphenylphosphino) ferrocene] dichloropalladium (II).
- the amount of the transition metal catalyst to be used is generally 0.0001-1 mol, preferably 0.001-1 mol, per 1 mol of compound (S-10).
- Examples of the ligand include triphenylphosphine, tri-o-tolylphosphine, tri-tert-butylphosphine, tri-2-furylphosphine, tri-cyclohexylphosphine, triphenylarsine, 1,1′-bis (diphenylphosphine). Fino) ferrocene (dppf) and the like.
- the amount of the ligand to be used is generally 0.0001-4 mol, preferably 0.001-4 mol, per 1 mol of compound (S-9).
- Examples of the base include organic bases such as triethylamine and diisopropylethylamine And inorganic bases such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate, cesium carbonate, and potassium phosphate.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 4 mol, per 1 mol of compound (S-9).
- Examples of the additive include lithium chloride, cesium fluoride, copper iodide (I) And inorganic salts such as copper (I) bromide.
- Examples of the solvent include water, acetonitrile, chloroform, and dichloromethane.
- the solvent examples include water; alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; benzene, toluene, xylene, and the like.
- Aromatic hydrocarbon solvents include acetonitrile; dimethylformamide; aprotic solvents such as dimethyl sulfoxide or N-methylpyrrolidone; halogenated hydrocarbons such as chloroform, or mixed solvents thereof.
- R 2a represents an alkylcarbonyloxymethyl group or a benzyloxymethyl group
- R 1 , R 2 , X, Y, Ring A and Ring B are as described above.
- the metal catalyst examples include palladium-carbon, palladium hydroxide-carbon, rhodium-carbon, Raney nickel, platinum oxide and the like, and the amount used is based on the compound (S-12). Usually, it is 0.01% to 100% by weight, preferably 0.1 to 10% by weight.
- the solvent include alcohol solvents such as methanol, ethanol and 2-propanol; ether solvents such as tetrahydrofuran; ester solvents such as ethyl acetate; aprotic polar solvents such as N, N-dimethylformamide or the like And the like.
- the second step is a reaction using an acid halogenating agent.
- the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 48 hours.
- the amount of compound (S-12 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of carboxylic acid compound.
- the acid halogenating agent include thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride and the like.
- the amount of the acid halogenating agent to be used is generally 1 to 10 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-12 ′).
- a catalytic amount of N, N-dimethylformamide may be added to the reaction system.
- the solvent include halogenated hydrocarbons such as dichloromethane and chloroform; sulfoxides such as dimethyl sulfoxide; esters such as ethyl acetate; ethers such as tetrahydrofuran and 1,4-dioxane; Examples include amides such as N, N-dimethylformamide and N, N-dimethylacetamide; aromatic hydrocarbons such as benzene and toluene.
- the manufacturing method of compound (I) is a solution, concentration, solvent extraction, fractional distillation, crystallization, recrystallization, and column chromatography. From the reaction mixture, it can be separated and purified by a known technique such as.
- compound (I) when compound (I) is obtained as a free form, it can be converted to the target salt as described above by a method known per se or a method analogous thereto, and conversely, compound (I) When obtained as a salt, it can be converted into a free form or other desired salt by a method known per se or a method analogous thereto.
- the purified compound (I) is converted into inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, acetic acid, citric acid, succinic acid, tartaric acid, fumaric acid, maleic acid, methanesulfonic acid, By reacting with an organic acid such as p-toluenesulfonic acid, a pharmaceutically acceptable acid addition salt can be easily obtained.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, acetic acid, citric acid, succinic acid, tartaric acid, fumaric acid, maleic acid, methanesulfonic acid
- the purified compound (I) can be converted into, for example, lithium hydroxide, lithium methylate, sodium hydride, sodium carbonate, sodium hydroxide, sodium methylate, potassium hydroxide, potassium carbonate, potassium hydroxide, potassium-t-
- an inorganic / organic metal salt such as butyrate
- the compounds of the present invention can be provided in any form suitable for the intended administration. Suitable forms include pharmaceutically (ie physiologically) acceptable salts and predrug and prodrug forms of the compounds of the invention. These predrug and prodrug forms are also within the scope of the present invention.
- the compound of the present invention has an action of enhancing the activity of neurotrophic factor, and in particular, has an action of inducing NXF gene expression that plays an important role in nerve protection. Therefore, the compound of the present invention can be used as a therapeutic or prophylactic agent for diseases involving neurotrophic factor activity. Moreover, the compound of this invention can be used as a promoter of the physical therapy effect for the recovery
- disease is used in the meaning including a disorder and its symptoms.
- treatment is used in the meaning including relief of symptoms.
- diseases involving neurotrophic factor activity include the following cranial neurodegenerative diseases, spinal degenerative diseases, retinal degenerative diseases, peripheral neurodegenerative diseases, and other diseases.
- Examples of cranial neurodegenerative diseases include: Neurodegenerative diseases (eg, Alzheimer's disease, Parkinson's disease, Huntington's disease, Down syndrome, etc.), cerebral ischemic diseases (stroke, cerebral infarction, transient ischemic attack, subarachnoid hemorrhage, ischemic encephalopathy, cerebral infarction (Such as lacunar infarction, atherothrombotic cerebral infarction, cardiogenic cerebral infarction, hemorrhagic cerebral infarction, other infarction)), traumatic brain injury, leukoencephalopathy, and multiple sclerosis.
- Neurodegenerative diseases eg, Alzheimer's disease, Parkinson's disease, Huntington's disease, Down syndrome, etc.
- cerebral ischemic diseases stroke, cerebral infarction, transient ischemic attack, subarachnoid hemorrhage, ischemic encephalopathy, cerebral infarction (Such as lacunar infarction, atherothrombotic cerebral infarction, cardiogenic cerebral infarction, hemor
- spinal degenerative diseases include: Examples include amyotrophic lateral sclerosis (ALS), spinal cord injury, spinal cord injury due to various causes, spinal progressive muscular atrophy, and spinal cerebral degeneration (SCD).
- ALS amyotrophic lateral sclerosis
- SCD spinal cerebral degeneration
- retinal degenerative diseases include: Examples include age-related macular degeneration (AMD), diabetic retinopathy, retinitis pigmentosa, hypertensive retinopathy, and glaucoma.
- AMD age-related macular degeneration
- diabetic retinopathy examples include diabetic retinopathy, retinitis pigmentosa, hypertensive retinopathy, and glaucoma.
- peripheral neurodegenerative diseases include Diabetic neuropathy, peripheral neuropathy, traumatic peripheral neuropathy, addiction, peripheral neuropathy caused by other toxic substances, peripheral neuropathy caused by cancer chemotherapy, Guillain-Barre syndrome, peripheral neuropathy caused by deficiency of vitamins, Amyloid peripheral neuropathy, ischemic peripheral neuropathy, peripheral neuropathy associated with malignant tumor, uremic peripheral neuropathy, peripheral neuropathy due to physical cause, Charcot-Marie-Tooth disease, alcoholic peripheral neuropathy, autonomic neuropathy (Subconscious hypoglycemia, gastric paresis, neurogenic diarrhea and constipation, erectile dysfunction, orthostatic hypotension, arrhythmia, heart failure, painless myocardial infarction, sweating abnormalities, neurogenic bladder, etc.), bladder dysfunction (e.g., Uninhibited bladder, reflex bladder, autonomous bladder, sensory paralytic bladder, motor paralytic bladder, etc.).
- bladder dysfunction e.g., Uninhibited bladder, reflex bladder, autonomous bladder, sensory paralytic bladder, motor paralytic bladder, etc.
- Other diseases include, for example, Associated with depression, schizophrenia, epilepsy, autism, periodontal disease, diabetes, diabetic cardiomyopathy, diabetic foot lesions, inflammatory bowel disease (eg, ulcerative colitis, Crohn's disease), dementia Problematic behavior (eg, acupuncture, aggressive behavior), anxiety, pain, hearing impairment, bone disease (eg, osteoporosis), joint disease (eg, Charcot joint, osteoarthritis, rheumatism, etc.), Hirsch Examples include Sobrung's disease.
- the compound of this invention is applied suitably with respect to a cerebral ischemic disease or diabetic neuropathy.
- the compound of the present invention can be used as a composition (eg, pharmaceutical composition) formulated as it is or mixed with a pharmaceutically acceptable carrier or the like for diseases involving neurotrophic factor activity.
- a pharmaceutically acceptable carrier or the like for diseases involving neurotrophic factor activity.
- it can be safely administered orally or parenterally to mammals such as humans.
- Parenterally includes intravenous, intramuscular, subcutaneous, intraorgan, intranasal, intradermal, instillation, intracerebral, rectal, intravaginal, intraperitoneal, and the like.
- the composition of the present invention contains, for example, one or more compounds of the present invention and a pharmaceutically acceptable carrier, excipient, and / or additive.
- a pharmaceutically acceptable carrier, excipient, and / or additive eg, pharmaceutical additive, food additive
- Agent, cosmetic additive e.g., cosmetic additive
- the pharmaceutically acceptable carrier, excipient, and / or additive used in the composition of the present invention can be appropriately selected depending on the specific use of the composition.
- the form of the said composition can also be made into forms, such as various solid and liquid, according to a specific use, for example.
- compositions of the present invention or the compound of the present invention when used as a pharmaceutical, specific forms such as powders, fine granules, granules, tablets, syrups, capsules, suspending agents And oral agents such as emulsions, extracts and pills, injections, transdermal solutions (external preparations for skin) such as ointments, suppositories, and parenterals for topical use. .
- Oral preparations include, for example, gelatin, sodium alginate, starch, corn starch, sucrose, lactose, glucose, mannitol, carboxymethylcellulose, dextrin, polyvinylpyrrolidone, crystalline cellulose, soy lecithin, sucrose, fatty acid ester, talc, magnesium stearate, Carriers and excipients such as polyethylene glycol, magnesium silicate, and silicic anhydride, binders, disintegrants, surfactants, lubricants, fluidity promoters, diluents, preservatives, colorants, fragrances, stabilization It can be produced according to a usual method using a pharmaceutical additive such as an agent, a humectant, a preservative, and an antioxidant.
- a pharmaceutical additive such as an agent, a humectant, a preservative, and an antioxidant.
- the dose varies depending on the age, sex, weight, disease level, type of compound of the present invention, dosage form, etc. of the mammal to be administered.
- the amount may be about 1 mg to about 2 g, preferably about 5 mg to about 1 g as the active ingredient amount.
- the above-mentioned daily dose can be administered once or divided into several times.
- injections are water-soluble solvents such as physiological saline and sterile water Ringer's solution, non-water-soluble solvents such as vegetable oil and fatty acid esters, isotonic agents such as glucose and sodium chloride, solubilizing agents, stable It can be produced according to a usual method using a pharmaceutical additive such as an agent, a preservative, a suspending agent, and an emulsifier.
- a pharmaceutical additive such as an agent, a preservative, a suspending agent, and an emulsifier.
- Liquid preparations for external use, percutaneous absorption agents such as gel ointments, suppositories for rectal administration and the like can also be produced according to conventional methods.
- injection subcutaneous, intravenous, etc.
- transdermal administration or rectal administration may be used.
- the topical agent can be produced, for example, by incorporating the compound of the present invention into a pellet of a sustained release polymer such as an ethylene vinyl acetate polymer. This pellet may be surgically implanted into the tissue to be treated.
- the dose varies depending on the age, sex, body weight, degree of disease, the type of the composition of the present invention or the compound of the present invention, the mode of administration, etc. And about 0.1 mg to about 500 mg may be administered as the active ingredient amount.
- the above-mentioned daily dose can be administered once or divided into several times.
- Synthesis example 1 Synthesis Synthesis Example of 5- (Benzothiazol-2-yl) -2-methyl-1-phenylbenzimidazole 1-1 Synthesis of 3-nitro-4-phenylaminobenzoic acid
- 4-fluoro-3-nitrobenzoic acid (2.51 g, 13.56 mmol)
- aniline (2.52 g, 27.12 mmol)
- ethanol 9.5 mL
- the mixture was poured into dilute hydrochloric acid (1M, 50 mL), diluted with distilled water (100 mL), and stirred at room temperature for 30 minutes.
- the precipitated crystals were collected by filtration, washed with dilute hydrochloric acid (1M, about 3 mL ⁇ 3 times), distilled water (about 5 mL ⁇ 2 times), and diethyl ether (about 3 mL). The obtained crystals were heated and dried under reduced pressure to give the title compound (3.39 g, quantitative) as a red-orange solid.
- Synthesis Example 1-2 Synthesis of 3-amino-4-phenylaminobenzoic acid
- 3-nitro-4-phenylaminobenzoic acid see Synthesis Example 1-1) (1.67 g, 6.70 mmol), palladium / carbon (Pd: 10%, 0.170 g), and tetrahydrofuran- (34 mL), and vacuum replacement / hydrogen gas replacement was repeated three times to replace with hydrogen, followed by stirring at room temperature for 2.5 hours. After purging with nitrogen, distilled water (20 mL) was added, and the mixture was stirred for 10 minutes.
- Synthesis Example 1-3 Synthesis of 2-methyl-1-phenylbenzimidazole-5-carboxylic acid
- 3-amino-4-phenylaminobenzoic acid see Synthesis Example 1-2
- Acetyl chloride (1.00 g, 12.70 mmol) / toluene solution (about 2.5 mL) was added dropwise thereto in about 15 minutes, and the mixture was stirred for 2.5 hours under the same conditions.
- the mixture was allowed to cool to room temperature and extracted with dilute aqueous sodium hydroxide solution (10%, 200 mL).
- Synthesis example 2 Synthesis of 5- (benzimidazol-2-yl) -2-methyl-1-phenylbenzimidazole To a two-necked flask (20 mL) with a reflux condenser, 2-methyl-1-phenylbenzimidazole-5-carboxylic acid (see Synthesis Example 1-3) (0.15 g, 0.59 mmol) and polyphosphoric acid (about 2 g) was added and heated to 120 ° C. 1,2-Diaminobenzene (0.09 g, 0.83 mmol) was added thereto, heated to 160 ° C., and stirred for 20 hours under the same conditions.
- Synthesis example 3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-phenylbenzimidazole To a two-necked flask (20 mL) with a reflux condenser, 2-methyl-1-phenylbenzimidazole-5-carboxylic acid (see Synthesis Example 1-3) (0.15 g, 0.59 mmol), 2-aminophenol (0.09) g, 0.82 mmol) and polyphosphoric acid (about 2 g) were added, heated to 160 ° C., and stirred for 20 hours under the same conditions. After cooling to room temperature, ice was added, and the solution was adjusted to about pH 9 with concentrated aqueous ammonia (28%).
- Synthesis example 4 Synthesis of 2-amino-1- (tetrahydropyran-4-yl) amino-4-trifluoromethylbenzene Synthesis Example 4-1 Synthesis of 1- (tetrahydropyran-4-yl) amino-2-nitro-4-trifluoromethylbenzene In a three-necked flask (200 mL), 1-fluoro-2-nitro-4-trifluoromethylbenzene (1.59 g, 7.61 mmol), 4-aminotetrahydropyran- (1.00 g, 9.89 mmol), and pyridine (16 mL) and stirred at 98 ° C. for 23 hours.
- Synthesis Example 4-2 Synthesis of 2-amino-1- (tetrahydropyran-4-yl) amino-4-trifluoromethylbenzene
- 1- (tetrahydropyran-4-yl) amino-2-nitro-4-trifluoromethylbenzene see Synthesis Example 4-1) (1.00 g, 3.45 mmol)
- tetrahydrofuran- (17 mL) were added and shaken to make a homogeneous solution.
- Palladium / carbon (Pd: 10%, 0.09 g) was added thereto and shaken again. Subsequently, reduced pressure / replacement with hydrogen gas was repeated three times to form a hydrogen atmosphere.
- Synthesis example 5 Synthesis Example of 5- (Benzoxazol-2-yl) -1,2-dimethylbenzimidazole5-1 Synthesis of (2-methylaminoanily-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (300 mg, 1.2 mmol), potassium carbonate (50 mg, 3.5 mmol), 40% aqueous monomethylamine (270 mg, 3.5 mmol) ) Containing ethanol (5 mL) solution, and heated and stirred for 2 hours under reflux conditions. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered.
- Synthesis Example 5-2 Synthesis of 5- (benzoxazol-2-yl) -1,2-dimethylbenzimidazole (2-Methylaminoanily-5-yl) benzoxazole (see Synthesis Example 5-2) (75 mg, 0.31 mmol) was dissolved in DMF (2 mL), 90% acetaldehyde aqueous solution (46 mg, 0.94 mmol), and then oxone ( 192 mg, 0.31 mmol) was added, and the mixture was stirred at room temperature for 2.5 hours. The reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- Example 1 Synthesis Example of 5- (Benzothiazol-2-yl) -1-phenyl-2- (phenylmethoxy) methylbenzimidazole
- Example 1-1 Synthesis of 1-phenyl-2- (phenylmethoxy) methylbenzimidazole-5-carboxylic acid To a four-necked flask (300 mL) with a reflux condenser, add 3-amino-4-phenylaminobenzoic acid (see Synthesis Example 1-1) (3.09 g, 13.54 mmol), and dehydrated toluene (45 mL). ) And refluxed.
- Benzyloxyacetyl chloride (5 g, 27.08 mmol) / toluene solution (about 3 mL) was added dropwise thereto in about 10 minutes. The mixture was stirred for 15 hours under the same conditions. The mixture was allowed to cool to room temperature and extracted with dilute aqueous sodium hydroxide solution (10%, 100 mL). After washing with toluene (80 mL) and separating the dark orange aqueous layer, the solution was cooled (5-10 ° C.), and the liquid was adjusted to pH 4 with concentrated hydrochloric acid (12M).
- Example 1-2 Synthesis of 5- (benzothiazol-2-yl) -1-phenyl-2- (phenylmethoxy) methylbenzimidazole
- 1-phenyl-2- (phenylmethoxy) methylbenzimidazole-5-carboxylic acid see Example 1-1) (4.00 g, 11.16 mmol)
- Tetrahydrofuran- 63 mL
- oxalyl chloride (1.65 g, 13.0 mmol) was added, and the mixture was warmed to 50 ° C. and stirred for 3 hr.
- Example 2 Synthesis of 5- (benzothiazol-2-yl) -2-hydroxymethyl-1-phenylbenzimidazole To an eggplant flask (50 mL) with a reflux condenser, add 5- (benzothiazol-2-yl) -1-phenyl-2- (phenylmethoxy) methylbenzimidazole (see Example 1-2) (0.40 g, 0.89 mmol). And dilute hydrochloric acid (6M, 5 mL) were added and refluxed for 2 hours. When the obtained pale yellow transparent solution was ice-cooled, it became a milky white suspension.
- the reaction mixture was allowed to cool to room temperature, and the solvent was distilled off under reduced pressure to prepare the corresponding chloride as a greenish milky white solid.
- dehydrated tetrahydrofuran- (3 mL), and a dimethylamine / tetrahydrofuran-solution (2.0M, 1.1M) was added. mL) was added dropwise.
- the mixture was heated to 40 ° C. and stirred for 2 hours.
- Example 4 Synthesis of 5- (benzothiazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 4-1 Synthesis of 4-((tetrahydropyran-4-yl) amino) -3-nitrobenzoic acid
- 4-Fluoro-3-nitrobenzoic acid (126.4 g, 0.68 mol) was dissolved in ethanol (880 mL).
- Triethylamine (82.6 g, 0.82 mol) and 4-aminotetrahydropyran- (82.7 g, 0.81 mol) were added dropwise in this order, and after the addition, the reaction solution was heated to reflux.
- Example 4-2 Synthesis of 3-amino-4-((tetrahydropyran-4-yl) amino) benzoic acid 4-((Tetrahydropyran-4-yl) amino) -3-nitrobenzoic acid (see Example 4-1) (202 g, 0.76 mol) dissolved in a mixed solution of THF (2.2 L) and methanol (1.5 L) Then, Pd / C (5%, wet, 20 g) was added and a hydrogenation reaction was performed at 5 atm. Since the absorption of hydrogen ceased in 3 hours, the inside of the system was purged with argon, the catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure to obtain the title compound (180 g, quantitative) as a dark gray solid.
- Example 4-3 Synthesis of 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid 3-Amino-4-((tetrahydropyran-4-yl) amino) benzoic acid (see Example 4-2) (90 g, 0.38 mol) was dissolved in dioxane (900 mL), cooled to 10 ° C., and then chlorinated. A solution of acetyl (35.0 g, 0.45 mol) in dioxane (900 mL) was added dropwise over 35 minutes.
- Example 4-4 Synthesis of 5- (benzothiazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole To a four-necked flask (50 mL) with a reflux condenser, add 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.28 g, 1.08 mmol). , 2-aminobenzenethiol (0.14 g, 1.08 mmol) and polyphosphoric acid (about 11 g) were added, heated to 150 ° C. and stirred for 17 hours.
- Chloroform (1 L) was added for liquid separation, and the chloroform layer was washed with water (500 mL) and dehydrated with Na 2 SO 4 .
- Example 6 Synthesis of 5- (benzothiazol-2-yl) -2-methoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 6-1 Synthesis of 2-methoxymethyl-1- (tetrahydropyran-4-yl) -5-trifluoromethylbenzimidazole
- 2-amino-1- (tetrahydropyran-4-yl) amino-4-trifluoromethylbenzene see Synthesis Example 4
- dehydrated 1,4-dioxane 8.5 mL).
- Methoxyacetyl chloride (0.57 g, 5.25 mmol) / 1,4-dioxane solution (about 1.5 mL) was added dropwise thereto in about 10 minutes. The mixture was stirred for 2 hours under the same conditions. The mixture was allowed to cool to room temperature, distilled water was added, and the solvent was distilled off under reduced pressure. Extraction with t-butyl methyl ether, washing with brine and drying over anhydrous sodium sulfate followed by evaporation of the solvent under reduced pressure gave the title compound (0.58 g, yield 77.4%) as a white powder.
- Example 6-2 Synthesis of 5- (benzothiazol-2-yl) -2-methoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- 2-methoxymethyl-1- (tetrahydropyran-4-yl) -5-trifluoromethylbenzimidazole see Example 6-1 (0.102 g, 0.32 mmol)
- 2-aminobenzenethiol 0.041 g, 0.33 mmol
- polyphosphoric acid about 2 g
- Example 7 Synthesis of 2-acetoxymethyl-5- (benzothiazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 7-1 Synthesis of 2-acetoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid
- 3-amino-4-((tetrahydropyran-4-yl) amino) benzoic acid 14.53 g, 45.65 mmol
- dehydrated 1,4-dioxane 226 mL
- Acetoxyacetyl chloride (12.57 g, 92.07 mmol) / 1,4-dioxane solution (100 mL) was added dropwise thereto in about 15 minutes. The mixture was stirred for 13 hours under the same conditions. The mixture was allowed to cool to room temperature, and the precipitated crystals were collected by filtration, washed with distilled water, and dried by heating under reduced pressure to give the title compound (13.46 g, yield 92.6%) as a white powder.
- Example 8 Synthesis of 5- (benzothiazol-2-yl) -2-hydroxymethyl-1- (tetrahydropyran-4-yl) benzimidazole A 4-necked flask (1 L) was charged with 2-acetoxymethyl-5- (benzothiazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 7-2) (5.00 g, 12.27 mmol) and methanol (253 mL) were added, and an aqueous lithium hydroxide solution (1.0 M, 61 mL) was added at room temperature.
- a hydrogen chloride / diethyl ether solution (2.0 M, 2 mL) was gradually added thereto, followed by stirring for 30 minutes under the same conditions.
- the generated crystals were collected by filtration, washed with diethyl ether (about 2 mL ⁇ 3 times), and dried under reduced pressure to give the title compound (0.186 g, yield 96.9%) as a pale yellow powder.
- Example 11 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole An eggplant flask (100 mL) was charged with 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.64 g, 2.46 mmol), 2-aminophenol ( 0.32 g, 2.95 mmol) and polyphosphoric acid (about 18 g) were added, heated to 160 ° C. and stirred for 17 hours. After allowing to cool, ice was added and the solution was adjusted to about pH 9 with concentrated aqueous ammonia (28%).
- Example 12 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-methanesulfonate 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (51.2 g, 0.19 mol), 2-aminophenol (24.0 g, 0.21 mol), dehydration DMF (500 mL) and WSC (45.0 g, 0.23 mol) were stirred for 3 hours under an argon stream. Water (2 L) was added, and the resulting solid was collected by filtration, washed with water, and dried at 50 ° C. under reduced pressure.
- Example 13 Synthesis of 5- (benzoxazol-2-yl) -2-hydroxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 13-1 Synthesis of 2-acetoxymethyl-5- (2-hydroxyanilinocarbonyl) -1- (tetrahydropyran-4-yl) benzimidazole
- 2-acetoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid see Example 7-1) (1.50 g, 4.71 mmol).
- Example 13-2 Synthesis of 5- (benzoxazol-2-yl) -2-hydroxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- An eggplant flask 50 mL was charged with 2- (acetoxymethyl) -5- (2-hydroxyanilinocarbonyl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 13-1) (0.50 g, 1.22 mmol), toluene (6.3 mL), and p-toluenesulfonic acid hydrate (1.60 g, 8.41 mmol) were added and refluxed for 1.5 hours. After standing to cool, the liquid was neutralized with a saturated aqueous sodium hydrogen carbonate solution.
- Example 15 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxyphenyl) -2-methylbenzimidazole
- Example 15-1 Synthesis of 4-fluoro-N- (2-hydroxyphenyl) -3-nitrobenzanilide
- 2-aminophenol (1.18 g, 10.8 mmol)
- 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride Salt (2.17 g, 11.3 mmol)
- triethylamine (1.15 g, 11.3 mmol
- reaction mixture was diluted with ethyl acetate and washed with water, 1N hydrochloric acid and saturated aqueous sodium hydrogen carbonate solution.
- the obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to give the title compound (2.02 g, yield 68%) as orange crystals.
- Example 15-2 Synthesis of 2- (4-fluoro-3-nitrophenyl) benzoxazole
- 4-fluoro-N- (2-hydroxyphenyl) -3-nitrobenzanilide see Example 15-1
- p-toluenesulfonic acid ⁇ 1 Hydrate 2.00 g, 10.9 mmol
- the reaction solution was cooled to room temperature, the solvent was distilled off, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for 10 minutes.
- Example 15-3 Synthesis of 2- (4- (4-methoxyphenylamino) -3-nitrophenyl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- sodium bicarbonate 295 mg, 2.32 mmol
- p. -Anisidine 357 mg, 2.90 mmol
- the mixture was heated with stirring under reflux conditions for 4 hours.
- the mixture was cooled to room temperature, and the precipitated crystals were filtered, washed with water and ethanol, and dried to give the title compound (359 mg, yield 86%) as orange crystals.
- Example 15-4 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxyphenyl) -2-methylbenzimidazole 2- (4- (4-methoxyphenylamino) -3-nitrophenyl) benzoxazole (see Example 15-3) (150 mg, 0.415 mmol) in 10% palladium on carbon (50 mg) in tetrahydrofuran- (5 mL) solution
- the inside of the flask was replaced with hydrogen and stirred at room temperature for 3 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Acetyl chloride (70.8 mg, 0.902 mmol) was added to a toluene (5 mL) solution of the obtained oily substance, and the mixture was heated with stirring under reflux conditions for 2.5 hours. After completion of the reaction, the mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated, and the obtained crystals were purified by silica gel column chromatography to give the title compound (47 mg, yield 32%) as dark brown crystals.
- Example 16 Synthesis of 5- (benzoxazol-2-yl) -1-cyclohexyl-2-methylbenzimidazole
- Example 16-1 Synthesis of 2- (4-cyclohexylamino-3-nitrophenyl) benzoxazole
- sodium bicarbonate 295 mg, 2.32 mmol
- Amine 288 mg, 2.32 mmol
- Example 16-2 Synthesis of 5- (benzoxazol-2-yl) -1-cyclohexyl-2-methylbenzimidazole 2- (4-Cyclohexylamino-3-nitrophenyl) benzoxazole (see Example 16-1) (185 mg, 0.548 mmol) was added to a tetrahydrofuran- (5 mL) solution containing 10% palladium on carbon (50 mg). Was replaced with hydrogen and stirred at room temperature for 5 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Example 17 Synthesis Example of 5- (Benzoxazol-2-yl) -1-benzyl-2-methylbenzimidazole
- Example 17-1 Synthesis of 2- (4-benzylamino-3-nitrophenyl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- benzyl Amine 311 mg, 2.90 mmol
- Example 17-2 Synthesis of 2- (2-benzylaminoaniline-5-yl) benzoxazole 2- (4-Benzylamino-3-nitrophenyl) benzoxazole (see Example 17-1) (340 mg, 0.985 mmol) in 10% aqueous acetic acid (5 mL), ethanol (8 mL), iron powder (165 mg, 2.95) mmol) was added, and the mixture was stirred with heating under reflux conditions for 4 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution and chloroform were added, and the mixture was filtered through celite and extracted with chloroform.
- Example 17-3 Synthesis of 5- (benzoxazol-2-yl) -1-benzyl-2-methylbenzimidazole
- 2- (2-benzylaminoaniline-5-yl) benzoxazole (see Example 17-2) (80.0 mg, 0.254 mmol) in dimethylformamide (2 m) was added acetaldehyde aqueous solution (about 90%, 47.7 ⁇ l, 0.761 mmol) and oxone (102 mg, 0.165 mmol) were added and stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was extracted with ethyl acetate.
- Example 18 Synthesis of 5- (benzothiophen-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 18-1 Synthesis of 5-bromo-2- (tetrahydropyran-4-yl) nitrobenzene
- 5-bromo-2-fluoronitrobenzene 3.0 g, 13.6 mmol
- triethylamine (1.66 g, 16.3 mmol
- 4-aminotetrahydropyran- (1.52 g, 15.0 mmol
- ethanol 60 mL
- Example 18-2 Synthesis of 5-bromo-2- (tetrahydropyran-4-yl) aminoaniline A three-necked flask is charged with 5-bromo-2- (tetrahydropyran-4-yl) nitrobenzene (see Example 18-1) (3.54 g, 11.8 mmol) and 65 mL of 10% aqueous acetic acid, and then electrolytic iron. (6.56 g, 118 mmol) was added and the mixture was refluxed and stirred for 15 minutes. After standing to cool to room temperature, insolubles were filtered off through celite, and the same layer was washed with 65 mL of 10% aqueous acetic acid.
- Example 18-3 Synthesis of 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-hydrochloride
- 5-bromo-2- (tetrahydropyran-4-yl) aminoaniline see Example 18-2
- dehydrated toluene (20 mL)
- Acetyl chloride (1.57 g, 20.0 mmol) / toluene solution (about 2.5 mL) was added dropwise thereto in about 15 minutes, and the mixture was stirred for 2 hours under the same conditions.
- Example 18-4 Synthesis of 5- (benzothiophen-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-hydrochloride (see Example 18-3) (0.38 g, 1.15 mmol), 2-benzothiopheneboronic acid (0.25 g, 1.40) mmol), ethanol (5 mL), toluene (5 mL), 2M aqueous sodium carbonate solution (2.1 mL) were charged and degassed.
- Tetrakis (tophenylphosphine) palladium (0.08 g, 0.07 mmol) was added and heated to reflux for 3 hours. After cooling, ethanol and water were added, and the mixture was filtered through Celite, and the filtered product was washed with ethanol and water. The filtrate was concentrated, and the precipitated crystals were collected by filtration, washed with water and hexane, and dried to give the title compound (315 mg, yield 78.9%) as pale brown crystals.
- Example 19 Synthesis of 5- (benzofuran-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 18-3) (0.41 g, 1.24 mmol), 2-benzofuran-boronic acid (0.25 g, 1.40 mmol), ethanol (5 mL), toluene (5 mL), 2M aqueous sodium carbonate solution (2.1 mL) were charged and degassed. Tetrakis (tophenylphosphine) palladium (0.08 g, 0.07 mmol) was added and heated to reflux for 3 hours.
- Example 20 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 20-1 Synthesis of 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) nitrobenzene
- 5- (Benzoxazol-2-yl) -2-fluoronitrobenzene (0.55 g, 2.13 mmol)
- triethylamine (0.26 g, 2.57 mmol
- aminotetrahydropyran- (0.24 g, 2. 34 mmol) was added to ethanol (10 mL), and the reaction was heated and stirred for 2 hours under reflux conditions.
- Example 20-2 Synthesis of 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) nitrobenzene (see Example 20-1) (1.00 g, 0.76 mol) was dissolved in a mixed solution of 50 mL of THF and 50 mL of methanol, and Pd Hydrogenation reaction was performed by adding / C (5%, wet, 0.5 g). After stirring at room temperature overnight, the catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure to give the title compound (812 mg, yield 89.1%) as a gray solid.
- Example 20-3 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.20 g, 0.646 mmol), triethyl orthoformate (5 mL) in catalytic amount of p -Toluenesulfonic acid monohydrate was added and heated at 100 ° C for 1.5 hours. The reaction mixture was cooled, extracted with ethyl acetate and water.
- Example 21 Synthesis of 5- (benzoxazol-2-yl) -2- (2-pyridyl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 2-pyridinecarboxaldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (1335 mg, yield 70.2%) as white crystals.
- Example 22 Synthesis of 5- (benzoxazol-2-yl) -2-isopropyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, isopropylaldehyde (0.04 g, 0.561 mmol) and oxone (0.19 g, 0.310 mmol) were added, and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (70 mg, yield 43.9%) as white crystals.
- Example 23 Synthesis of 5- (benzoxazol-2-yl) -2-cyclohexyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, cyclohexyl aldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (116 mg, yield 59.6%) as white crystals.
- Example 24 Synthesis of 5- (benzoxazol-2-yl) -2- (3-pyridyl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 3-pyridinecarboxaldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (95.0 mg, yield 49.5%) as white crystals.
- Example 25 Synthesis of 5- (benzoxazol-2-yl) -2-phenyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, phenylaldehyde (0.06 g, 0.561 mmol) and oxone (0.19 g, 0.310 mmol) were added, and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (115 mg, yield 59.9%) as white crystals.
- Example 26 Synthesis of 5- (benzoxazol-2-yl) -2- (4-pyridyl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DM (F 3 mL) and water (0.1 mL) After dissolution, 4-pyridinecarboxaldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (117 mg, yield 60.9%) as white crystals.
- Example 27 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) -2-trifluoromethylbenzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.20 g, 0.646 mmol) in trifluoroacetic acid (7 mL) for 4 hours and a half Heated to reflux. The reaction mixture was cooled, water was added, and the mixture was extracted with ethyl acetate.
- Example 28 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) -benzotriazole
- a solution of 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.35 g, 1.13 mmol) in concentrated hydrochloric acid (2 mL) was brought to 0 ° C.
- an aqueous solution 0.5 mL
- 0.09 g (1.24 mmol) of sodium nitrite was added dropwise.
- Example 29 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-tertbutylbenzimidazole
- Example 29-1 Synthesis of 2- (2-terbutylaminoaniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- Butylamine 212 mg, 2.90 mmol
- the mixture was heated with stirring under reflux conditions for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the mixture was extracted with chloroform.
- the obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- the obtained crystals were added to a tetrahydrofuran- (5 mL) solution containing 10% palladium on carbon (50 mg), the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 8 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the titled compound (50.1 mg, yield 19%) as red crystals.
- Example 29-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-terbutylbenzimidazole
- 2- (2-terbutylaminoaniline-5-yl) benzoxazole see Example 29-1 (45.0 mg, 0.160 mmol) in dimethylformamide (2 mL) was added to an aqueous solution of acetaldehyde (about 90%, 235 ⁇ l, 0.480).
- acetaldehyde about 90%, 235 ⁇ l, 0.480
- oxone (63.9 mg, 0.104 mmol) were added, and the mixture was stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, filtered and washed with water.
- Example 30 Synthesis of 5- (benzoxazol-2-yl) -1- (2-methoxyphenyl) -2-methylbenzimidazole
- Example 30-1 Synthesis of 2- (2- (2-methoxyphenyl) aminoaniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- o -Anisidine 357 mg, 2.90 mmol
- Example 30-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2-methoxyphenyl) -2-methylbenzimidazole
- 2- (2- (2-methoxyphenyl) aminoaniline-5-yl) benzoxazole see Example 30-1 (48.0 mg, 0.135 mmol) in dimethylformamide (2 mL) was added an aqueous acetaldehyde solution (about 90% , 20.5 ⁇ l, 0.405 mmol) and oxone (53.9 mg, 0.0878 mmol) were added, and the mixture was stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, filtered and washed with water.
- Example 31 Synthesis of 5- (benzoxazol-2-yl) -1- (3-methoxyphenyl) -2-methylbenzimidazole
- Example 31-1 Synthesis of 2- (2- (3-methoxyphenyl) aminoaniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- m -Anisidine 357 mg, 2.90 mmol
- Example 31-2 Synthesis of 5- (benzoxazol-2-yl) -1- (3-methoxyphenyl) -2-methylbenzimidazole
- 2- (2- (3-methoxyphenyl) aminoaniline-5-yl) benzoxazole see Example 31-1
- an aqueous acetaldehyde solution about 90% 31.2 ⁇ l, 0.405 mmol
- oxone (66.3 mg, 0.108 mmol) were added and stirred at room temperature for 3 hours.
- an aqueous potassium carbonate solution was added, filtered and washed with water.
- Example 33 Synthesis of 5- (benzoxazol-2-yl) -1-((1-ethoxycarbonyl) piperidin-4-yl) -2-methylbenzimidazole
- Example 33-1 Synthesis of 2- (2- (4- (1-ethoxycarbonyl) piperidinoamino) anilin-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- 4- Ethyl amino-1-piperidinecarboxylate 499 mg, 2.90 mmol
- Example 34 Synthesis of 5- (benzoxazol-2-yl) -1- (4-fluorophenyl) -2-methylbenzimidazole
- Example 34-1 Synthesis of 2- (2- (4-fluorophenyl) aminoanilin-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- 4- Fluoroaniline 322 mg, 2.90 mmol
- Example 34-2 Synthesis of 5- (benzoxazol-2-yl) -1- (4-fluorophenyl) -2-methylbenzimidazole
- 2- (2- (4-fluorophenyl) aminoanilin-5-yl) benzoxazole (see Example 34-1) (38.0 mg, 0.119 mmol) in dimethylformamide (2 mL) was added an aqueous acetaldehyde solution (about 90% , 22.6 ⁇ l, 0.360 mmol) and oxone (73.2 mg, 0.119 mmol) were added and stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was filtered and washed with water.
- Example 35 Synthesis of 5- (benzoxazol-2-yl) -1- (piperidin-4-yl) -2-methylbenzimidazole
- Example 35-1 Synthesis of 2- (2- (4- (1-tertbutoxycarbonyl) piperidinoamino) anilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- sodium bicarbonate 329 mg, 3.88 mmol
- 4- Amino-1-tert-butoxycarbonylpiperidine 970 mg, 4.84 mmol
- Example 35-2 Synthesis of 5- (benzoxazol-2-yl) -1- (piperidin-4-yl) -2-methylbenzimidazole
- 2- (2- (4- (1-tertbutoxycarbonyl) piperidinoamino) anilin-5-yl) benzoxazole (see Example 35-1) (350 mg, 0.857 mmol) in dimethylformamide (3 mL) was added acetaldehyde aqueous solution. (About 90%, 161 ⁇ l, 2.57 mmol) and oxone (527 mg, 0.887 mmol) were added and stirred at room temperature for 1 hour.
- Example 36 Synthesis of 1-adamantyl-5- (benzoxazol-2-yl) -2-methylbenzimidazole
- Example 36-1 Synthesis of 2- (2- (1-adamantylamino) aniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- 1- Adamantamine amine 185 mg, 1.22 mmol was added, and the mixture was heated with stirring under reflux conditions for 4 hours.
- the reaction mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- the obtained crystals were added to a tetrahydrofuran- (5 mL) solution containing 10% palladium on carbon (100 mg), the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 6 hours.
- the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the title compound (67.0 mg, yield 16%) as brown crystals.
- Example 36-2 Synthesis of 1-adamantyl-5- (benzoxazol-2-yl) -2-methylbenzimidazole 2- (2- (1-adamantylamino) anilin-5-yl) benzoxazole (see Example 36-1) (65.0 mg, 0.181 mmol) in dimethylformamide (2 mL) was added to an aqueous acetaldehyde solution (about 90%, 34.0 ⁇ l, 0.542 mmol) and oxone (111 mg, 0.181 mmol) were added, and the mixture was stirred at room temperature for 2 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was extracted with ethyl acetate.
- Example 37 Synthesis of 5- (Nt-butoxycarbonylindol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 18-3) (0.40 g, 1.21 mmol), 2- (Nt-butoxycarbonylindole-) boronic acid ( 0.35 g, 0.242 mmol), ethanol (5 mL), toluene (5 mL), 2M aqueous sodium carbonate solution (1.8 mL) were charged and degassed.
- Tetrakis (tophenylphosphine) palladium (0.07 g, 0.07 mmol) was added, and the mixture was heated to reflux for 3 hours. After cooling, ethanol and water were added, and the mixture was filtered through Celite, and the filtered product was washed with ethanol and water. The filtrate was concentrated, and the precipitated crystals were collected by filtration, washed with water and hexane, and dried to give the title compound (215 mg, yield 41.3%) as white crystals.
- Example 38 Synthesis of 5- (Indol-2-yl) -1- (tetrahydropyran-4-yl) -2-methylbenzimidazole 5- (Nt-butoxycarbonylindol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 38-1) (200 mg, 0.463 mmol) was added to a 1N aqueous hydrochloric acid solution. (10 mL) and stirred at room temperature for 3 hours. After completion of the reaction, the mixture was allowed to stand for 3 days, and the precipitated crystals were collected by filtration, added to saturated aqueous sodium hydrogen carbonate (5 mL), and stirred for 30 minutes.
- Example 39 Synthesis of 5- (5-methylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-4-methylphenol (0.13 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred at room temperature for 3 hours. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 40 Synthesis of 5- (5-chlorobenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-4-chlorophenol (0.15 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred overnight at room temperature. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 41 Synthesis of 5- (6-chlorobenzooxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-5-chlorophenol (0.15 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred overnight at room temperature. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 42 Synthesis of 5- (6-methylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-5-methylphenol (0.13 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred overnight at room temperature. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 43 Synthesis of 5- (benzoxazol-2-yl) -2-ethyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, propylaldehyde (0.03 g, 0.561 mmol) and oxone (0.19 g, 0.310 mmol) were added, and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (44.7 mg, yield 26.6%) as pale brown crystals.
- Example 44 Synthesis of 5- (benzoxazol-2-yl) -2- (imidazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.10 g, 0.32 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 2-imidazole-carbaldehyde (0.03 g, 0.31 mmol) was added followed by oxone (0.13 g, 0.21 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (15 mg, yield 12.1%) as pale brown crystals.
- Example 45 Synthesis of 5- (benzoxazol-2-yl) -2- (thiophen-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.13 g, 0.42 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 2-thiophenecarbaldehyde (0.05 g, 0.45 mmol) was added followed by oxone (0.17 g, 0.28 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- the mixture was extracted with ethyl acetate, washed with water, dried over magnesium sulfate, concentrated and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (60 mg, yield 36%) as pale brown crystals.
- Example 46 Synthesis of 2-methyl-5- (4-methylbenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 46-1 Synthesis of 2-fluoro-5- (4-methylbenzoxazol-2-yl) -nitrobenzene
- Thionyl chloride (0.78 g, 6.56 mmol) was added to a suspension of 3-fluoro-2-nitrobenzoic acid (1.00 g, 5.40 mmol) and 1 drop of DMF in toluene (10 mL), and the mixture was stirred for 3 hours under reflux conditions.
- Example 46-2 Synthesis of 2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole
- 2-fluoro-5- (4-methylbenzoxazol-2-yl) -nitrobenzene see Example 46-1 (0.50 g, 1.8 mmol) in ethanol (10 mL) was added triethylamine (0.22 g, 2.17).
- mmol and aminotetrahydropyran- (0.20 g, 1.9 mmol) were added, and the mixture was heated to reflux for 3 hours.
- Example 46-3 Synthesis of 2-methyl-5- (4-methylbenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 2- (2- (Tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole (see Example 46-2) (0.56 g, 1.7 mmol), dimethylformamide (5 mL) containing water (0.18 mL) Acetaldehyde aqueous solution (about 90%, 90 mg, 1.8 mmol) and oxone (0.69 mg, 1.1 mmol) were added to the solution, and the mixture was stirred at room temperature for 3 hours.
- Example 47 Synthesis of 2-methyl-5- (6-nitrobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 47-1 Synthesis of 5- (2-hydroxy-4-nitroanilinocarbonyl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole
- toluene (10 mL) containing 1 drop of DMF Thionyl chloride (0.41 g, 3.4 mmol) was added, and the mixture was stirred for 7 hours under reflux conditions.
- Example 47-2 Synthesis of 2-methyl-5- (6-nitrobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (2-hydroxy-4-nitroanilinocarbonyl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 47-1) (195 mg, 0.492 mmol) in toluene (20 mL) Then, p-toluenesulfonic acid hydrate (280 mg, 1.47 mmol) was added, and the mixture was refluxed and stirred for 4 hours. After the solvent was concentrated under reduced pressure, multistory water (10 mL) was added and stirred at room temperature for 1 hour.
- Example 48 Synthesis of 2-methyl-5- (6-aminobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-5- (6-nitrobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 47-2) (135 mg, 0.357 mmol) was converted to palladium / carbon (Pd In addition to methanol (20 mL) containing 10%, 0.06 g), hydrogen substitution was repeated by repeating reduced pressure / hydrogen gas replacement three times, followed by stirring at room temperature for 2.5 hours.
- Example 49 Synthesis of 5- (benzoxazol-2-yl) -1- (4-hydroxycyclohexyl) -2-methylbenzimidazole
- Example 49-1 Synthesis of (2- (4-hydroxycyclohexylamino) nitrobenzene-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (0.60 g, 2.3 mmol), triethylamine (0.70 g, 7.0 mmol), 4-aminocyclohexanol hydrochloride (0.53 g, 2.5 mmol) in acetonitrile (20 mL), and the mixture was heated and stirred for 2 hours under reflux conditions.
- Example 49-2 Synthesis of 5- (benzoxazol-2-yl) -1- (4-hydroxycyclohexyl) -2-methylbenzimidazole 2- (2- (2-methoxyphenyl) aminoaniline-5-yl) benzoxazole (see Example 49-1) (0.20 g, 0.62 mmol), methyl imidate hydrochloride (0.07 g, 0.68 mmol) and methanol was stirred with heating under reflux conditions for 5 hours. After completion of the reaction, the mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 50 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole
- Example 50-1 Synthesis of 2- (2-n-propylaminoaniline-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- potassium carbonate 176 mg, 1.28 mmol
- Amine 82.4 mg, 1.39 mmol
- Example 50-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole
- 2- (2-n-propylaminoanilin-5-yl) benzoxazole see Example 50-1) (148 mg, 0.554 mmol) in dimethylformamide (3 ml) was added a solution of acetaldehyde (ca. 90%, 104 ⁇ l, 1.66 mmol) and oxone (341 mg, 0.554 mmol) were added, and the mixture was stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was filtered and washed with water.
- Example 51 Synthesis of 5- (benzoxazol-2-yl) -1- (2-methoxyethyl) -2-methylbenzimidazole
- Example 51-1 Synthesis of 2- (2- (2-methoxyethyl) aminoanilin-5-yl) benzoxazole
- potassium carbonate 176 mg, 1.28 mmol
- Ethylamine 105 mg, 1.39 mmol was added and heated to reflux for 3.5 hours.
- Example 52 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-phenylethyl) benzimidazole
- Example 52-1 Synthesis of 2- (2- (2-phenylethyl) aminoaniline-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (300 mg, 1.16 mmol) in ethanol (5 ml) was added potassium carbonate (176 mg, 1.28 mmol), 2-phenyl.
- Ethylamine 169 mg, 1.39 mmol was added and heated to reflux for 4 hours.
- Example 53 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylbenzimidazole
- Example 53-1 Synthesis of 2- (2-cyc-propylaminoaniline-5-yl) benzoxazole
- potassium carbonate 176 mg, 1.28 mmol
- cyclopropylamine 99.3 mg, 1.74 mmol
- Example 53-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylbenzimidazole
- 2- (2-cyc-propylaminoanilin-5-yl) benzoxazole (see Example 53-1) (140 mg, 0.425 mmol) in methanol (5 ml) was added 1,1,1-trimethoxyethane (75 mg , 0.624 mmol) was added and the mixture was heated to reflux for 6 hours. After completion of the reaction, the reaction mixture was concentrated and the resulting residue was purified by silica gel column chromatography to obtain 63.5 mg (42%) of imidazole.
- Example 54 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylmethylbenzimidazole
- Example 54-1 Synthesis of 2- (2-cyc-propylmethylaminoanilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (300 mg, 1.16 mmol) in ethanol (5 ml) was added potassium carbonate (176 mg, 1.28 mmol), cyclopropylmethyl.
- Amine 176 mg, 1.28 mmol was added and heated to reflux for 7 hours.
- Example 54-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylmethylbenzimidazole
- 2- (2-cyc-propylmethylaminoanilin-5-yl) benzoxazole (see Example 54-1) (140 mg, 0.425 mmol) in methanol (5 ml) was added 1,1,1-trimethoxyethane ( 75 mg, 0.624 mmol) was added, and the mixture was heated to reflux for 10 hours. After completion of the reaction, the reaction mixture was concentrated, and the resulting residue was purified by silica gel column chromatography to obtain the title compound (81.9 mg, yield 54%) as pink crystals.
- Example 55 Synthesis of 5- (benzoxazol-2-yl) -1- (2- (tert-butoxycarbonylamino) ethyl) -2-methylbenzimidazole
- Example 55-1 Synthesis of 2- (tert-butoxycarbonylamino) ethylaminoanilin-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (600 mg, 2.32 mmol) was converted to triethylamine (469 mg, 4.65 mmol) and 2- (tert-butoxycarbonylamino) ethylamine (447 mg, 2.79 mmol) in acetonitrile (10 ml) was added and heated to reflux for 5 hours.
- Example 55-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2- (tert-butoxycarbonylamino) ethyl) -2-methylbenzimidazole
- 2- (tert-butoxycarbonylamino) ethylaminoanilin-5-yl) benzoxazole 200 mg, 0.567 mmol
- methyl acetimidate hydrochloride 102 mg, 0.851 mmol
- Example 56 Synthesis of 1- (2-aminoethyl) -5- (benzoxazol-2-yl) -2-methylbenzimidazole hydrochloride 5- (Benzoxazol-2-yl) -1- (2- (tert-butoxycarbonylamino) ethyl) -2-methylbenzimidazole (see Example 55-2) (100 mg, 0.255 mmol) was added to 4N hydrochloric acid-dioxane. It was added to the solution (10 ml) and heated to reflux for 5 hours. After completion of the reaction, the title compound (95 mg, quantitative) was obtained as white crystals by concentration to dryness.
- Example 59 Synthesis of 5- (5-ter-butylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (250 mg, 0.842 mmol), 2-amino-4-tert-butylphenol (139 mg, 0.842 mmol) ), DMF (2 mL), chloroform (5 mL) and WSC (178 mg, 0.926 mmol) were added and stirred for 22 hours. Water was added, and the resulting solid was collected by filtration, washed with water, and dried.
- Example 60 Synthesis of 5- (5-nitrobenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (264 mg, 0.888 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 62 Synthesis of 5- (benzoxazol-2-yl) -2-trans-cinnam-1- (tetrahydropyran-4-yl) benzimidazole
- 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline see Example 20-2
- trans-cinnamaldehyde 111 mg, 0.840 mmol
- oxone 258 mg, 0.420 mmol
- Example 66 Synthesis of 5- (5-methoxybenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (250 mg, 0.842 mmol), 2-amino-3-methoxyphenol (139 mg, 0.842 mmol) , DMF (5 mL) and WSC (178 mg, 0.926 mmol) were added and stirred for 22 hours. Water was added and extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- Methanesulfonic acid (283 mg, 2.95 mmol) was added to the obtained solid dioxane (5 mL) solution, and the mixture was heated to reflux for 18 hours. After cooling the reaction solution, water and saturated aqueous sodium hydrogen carbonate solution were added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The obtained residue was purified by silica gel chromatography to give the title compound (10 mg, yield 3.3%) as brown crystals.
- Example 70 Synthesis of 5- (5-ethylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 72 Synthesis of 5- (5-cyc-hexylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 74 Synthesis Example 5-4-1 of 5- (Benzoxazol-2-yl) -1-n-butyl-2-methylbenzimidazole Synthesis of 2- (4-n-butylcyamino-3-nitrophenyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (300 mg, 1.16 mmol) in ethanol (5 ml) suspension in potassium carbonate (321 mg, 2.32 mmol), butylamine (170 mg , 2.32 mmol) was added and heated to reflux for 4 hours.
- Example 74-2 2- (2-n-Butylaminoaniline-5-yl) benzoxazole
- 2- (4-n-butylcyamino-3-nitrophenyl) benzoxazole (see Example 74-1) (353 mg, 1.13 mmol) in tetrahydrofuran (5 ml) was added 10% palladium on carbon (50 mg), and Was replaced with hydrogen and stirred at room temperature for 15 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to obtain the title compound (305 mg, yield: 96%).
- Example 74-3 5- (Benzoxazol-2-yl) -1-n-butyl-2-methylbenzimidazole
- 2- (2-n-butylcyaminoaniline-5-yl) benzoxazole (see Example 74-2) (300 mg, 1.07 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (175 mg, 1.60 mmol).
- methyl acetimidate hydrochloride (175 mg, 1.60 mmol).
- a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- Example 75 Synthesis of 5- (5-cyanobenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 76 Synthesis of 5- (5-trifluoromethoxybenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 79 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1- (2-picolyl) benzimidazole
- Example 79-1 Synthesis of 2- (2- (2-picolyl) aminoanilin-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (500 mg, 1.9 mmol) was added to a suspension of 2-picolylamine (520 mg, 4.8 mmol) in acetonitrile (5 ml). And heated to reflux for 2.5 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- Example 79-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-picolyl) benzimidazole
- 2- (2- (2-picolyl) aminoanilin-5-yl) benzoxazole (see Example 79-1) (250 mg, 0.79 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (100 mg, 0.87 mmol) was added and heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 80 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-iso-propylbenzimidazole 80-1 Synthesis of 2- (4-iso-propylamino-3-nitrophenyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (200 mg, 0.774 mmol) in ethanol (5 ml) suspension in potassium carbonate (214 mg, 1.55 mmol), isopropylamine ( 137 mg, 2.32 mmol) was added and the mixture was heated to reflux for 4 hours.
- Example 80-2 Synthesis of 2- (2-iso-propylaminoaniline-5-yl) benzoxazole
- 2- (4-iso-propylamino-3-nitrophenyl) benzoxazole see Example 80-1) (224 mg, 0.753 mmol) in tetrahydrofuran (5 ml) was added 10% palladium on carbon (40 mg), and the flask was The inside was replaced with hydrogen and stirred at room temperature for 15 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the title compound (184 mg, yield 91%).
- Example 80-3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-iso-propylbenzimidazole To a solution of 2- (2-iso-propylaminoaniline-5-yl) benzoxazole (see Example 80-2) (180 mg, 0.673 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (111 mg, 1.01 mmol).
- Example 81 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-neo-pentylbenzimidazole
- Example 81-1 Synthesis of 2- (4-neo-pentylamino-3-nitrophenyl) benzoxazole
- 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (200 mg, 0.774 mmol) in ethanol (5 ml) suspension in potassium carbonate (214 mg, 1.55 mmol), neopentylamine (135 mg, 1.55 mmol) was added and heated to reflux for 4 hours.
- Example 81-2 Synthesis of 2- (2-neo-pentylaminoaniline-5-yl) benzoxazole
- 2- (4-neo-pentylamino-3-nitrophenyl) benzoxazole see Example 81-1) (242 mg, 0.744 mmol) in tetrahydrofuran (5 ml) was added 10% palladium on carbon (40 mg), and the flask was The inside was replaced with hydrogen and stirred at room temperature for 15 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the title compound (196 mg, yield 89%).
- Example 81-3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-neo-pentylbenzimidazole To a solution of 2- (2-neo-pentylaminoanilin-5-yl) benzoxazole (see Example 81-2) (192 mg, 0.650 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (107 mg, 0.975 mmol). And heated to reflux for 2 hours.
- Example 82 Synthesis of Example 8-2-1 of 5- (5-aminobenzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole Synthesis of 3-nitro-4-n-propylaminobenzoic acid To a suspension of 4-trifluoro-3-nitrobenzoic acid (2.0 g, 10.8 mmol) in ethanol (20 ml) was added potassium carbonate (2.34 mg, 16.2 mmol) and propylamine (1.27 g, 21.6 mmol). Heated to reflux for hours.
- Example 82-2 3-Amino-4-n-propylaminobenzoic acid To a solution of 3-nitro-4-n-propylaminobenzoic acid (see Example 82-1) (1.94 g, 10.8 mmol) in tetrahydrofuran (20 ml) was added 10% palladium carbon (200 mg), and the flask was filled with hydrogen. Replace and stir at room temperature for 6 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated to obtain the title compound (1.68 g, quantitative).
- Example 82-3 2-Methyl-1-n-propylbenzimidazole-5-carboxylic acid To a solution of 3-amino-4-n-propylaminobenzoic acid (see Example 82-2) (1.68 g, 8.65 mmol) in methanol (17 ml) was added methyl acetimidate hydrochloride (1.14 g, 10.4 mmol), The mixture was heated to reflux for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, diethyl ether was added, and the mixture was stirred at room temperature for 10 minutes. The obtained crystals were filtered, washed with diethyl ether and dried to give the title compound (2.20 g, quantitative).
- Example 82-4 Synthesis of 5- (5-aminobenzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole 2-Methyl-1-n-propylbenzimidazole-5-carboxylic acid (see Example 82-3) (1.0 g, 3.93 mmol) and thionyl chloride (8 ml) were added, and the mixture was stirred under reflux conditions for 3 hours. After completion of the reaction, 2-aminophenol (456 mg, 2.96 mmol), triethylamine (899 mg, 8.88 mmol) and tetrahydrofuran (10 mL) were added to the residue obtained by concentration under reduced pressure, and the mixture was stirred at room temperature for 14 hours.
- the reaction solution was cooled, 1N aqueous sodium hydroxide solution and chloroform were added, the mixture was filtered through celite, and the obtained filtrate was extracted. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The obtained residue was purified by silica gel column chromatography. The obtained oil was dissolved in tetrahydrofuran (3 ml), and 1M hydrogen chloride diethyl ether solution (1 ml) was added. The precipitated crystals were filtered, washed with tetrahydrofuran and dried to give the title compound (168 mg, yield 25%) as pale green crystals.
- Example 83 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2- (tetrahydropyran-4-yl) ethyl) benzimidazole
- Example 83-1 Synthesis of 2- (2- (tetrahydropyran-4-yl) ethyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (800 mg, 3.1 mmol) was replaced with 2- (tetrahydropyran-4-yl) ethylamine (520 mg, 3.9 mmol) and triethylamine (500 mg , 3.9 mmol) was added to a solution of acetonitrile (5 ml), and the mixture was heated to reflux for 3 hours.
- Example 83-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2- (tetrahydropyran-4-yl) ethyl) benzimidazole
- 2- (2- (tetrahydropyran-4-yl) ethyl) benzoxazole (see Example 83-1) (300 mg, 0.89 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (220 mg, 1.96 mmol). And heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 84 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-((tetrahydropyran-4-yl) methyl) benzimidazole
- Example 84-1 Synthesis of 2-((tetrahydropyran-4-yl) methyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (900 mg, 3.5 mmol) was added to (tetrahydropyran-4-yl) methylamine (520 mg, 3.9 mmol) and triethylamine (500 mg , 4.3 mmol) was added to an acetonitrile (5 ml) solution, and the mixture was heated to reflux for 3 hours.
- Example 84-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-((tetrahydropyran-4-yl) methyl) benzimidazole
- 2-((tetrahydropyran-4-yl) methyl) benzoxazole see Example 84-1 (300 mg, 0.93 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (120 mg, 1.11 mmol). The mixture was heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 85 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole
- Example 85-1 Synthesis of 2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (500 mg, 3.5 mmol) (tetrahydropyran-4-yl) methylamine (360 mg, 2.4 mmol) and triethylamine (590 mg, 7.3 mmol) was added to an acetonitrile (5 ml) solution, and the mixture was heated to reflux for 3 hours.
- Example 85-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole
- 2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole (see Example 85-1) (300 mg, 0.93 mmol) in methanol (5 ml) was added methyl acetimidate.
- Hydrochloride 120 mg, 1.11 mmol was added and heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 87 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (1,2,3,4-tetrahydronaphthalen-1-yl) benzimidazole
- Example 87-1 Synthesis of 2- (2- (1,2,3,4-tetrahydronaphthalen-1-yl) aminoanilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole (0.50 g, 1.9 mmol) in acetonitrile (10 ml) was added 1-amino-1,2,3, 4-tetrahydronaphthalene (0.31 g, 2.1 mmol) and triethylamine (0.29 g, 3.2 mmol) were added, and the mixture was heated to reflux for 6 hours.
- Example 87-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (1,2,3,4-tetrahydronaphthalen-1-yl) benzimidazole 2- (2- (1,2,3,4-tetrahydronaphthalen-1-yl) aminoanilin-5-yl) benzoxazole (see Example 87-1) (0.30 g, 0.8 mmol) in methanol (5 ml ) To the solution was added methyl acetimidate hydrochloride (0.10 g, 0.9 mmol), and the mixture was heated to reflux for 3 hours. The reaction solution was cooled, water was added, and the mixture was extracted with chloroform.
- Example 88-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2-hydroxyethyl) -2-methylbenzimidazole
- 2- (2- (2-hydroxyethyl) aminoanilin-5-yl) benzoxazole (0.10 g, 0.4 mmol) in methanol (2 ml) was added methyl acetimidate hydrochloride ( 0.08 g, 0.7 mmol) was added and the mixture was heated to reflux for 3 hours.
- the reaction solution was cooled, water was added, and the precipitated crystals were filtered, washed with water, and dried to give the title compound (0.05 g, yield 49%) as white crystals.
- Example 89-2 Synthesis of 5- (benzoxazol-2-yl) -1-diphenylmethyl-2-methylbenzimidazole
- 2- (2-diphenylmethylaminoanilin-5-yl) benzoxazole (0.10 g, 0.3 mmol) in methanol (2 ml) was added methyl acetimidate hydrochloride (0.06 g, 0.5 mmol).
- methyl acetimidate hydrochloride (0.06 g, 0.5 mmol).
- the reaction solution was cooled, water was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- Example 91 Synthesis of 5- (benzoxazol-2-yl) -2-tert-butoxycarbonylmethyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 91-1 Synthesis of 2- (2-tert-butoxycarbonylmethylamino) aniline-5-yl) benzoxazole
- acetonitrile (10 ml) was added
- glycine tert-butyl ester hydrochloride (0.60 g 3.6 mmol
- triethylamine (0.90 g, 9.0 mmol
- Example 91-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-tert-butoxycarbonylmethylbenzimidazole
- 2- (2- (tert-butoxycarbonylmethylaminoanilin-5-yl) benzoxazole (see Example 91-1) (120 mg, 0.353 mmol) in dimethylformamide (2 ml) was added acetaldehyde (ca. 90% , 66 ⁇ l, 1.06 mmol) and oxone (217 mg, 0.353 mmol) were added, and the mixture was stirred at room temperature for 2 hours After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was filtered and washed with water.
- Example 92 Synthesis of 5- (benzoxazol-2-yl) -1-carboxymethyl-2-methylbenzimidazole To a solution of 5- (benzoxazol-2-yl) -2-methyl-1-tert-butoxycarbonylmethylbenzimidazole (see Example 91-2) (100 mg, 0.275 mmol) in chloroform (3 ml) was added an aqueous sodium hydroxide solution. (1M, 0.55 ml, 0.55 mmol) was added. Methanol was added to the two-layer solution until uniform and stirred at room temperature for 2 hours. After completion of the reaction, the solution was concentrated, an aqueous acetic acid solution was added, and the mixture was stirred at room temperature.
- Example 93-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2- (thiomorpholin-1,1-dioxido-4-yl) ethyl) benzimidazole 2- (N- (2- (thiomorpholin-1,1-dioxido-4-yl) ethyl) -2-nitroanilin-4-yl) benzoxazole (see Example 93-1) (290 mg, 0.696 mmol ) In tetrahydrofuran (5 ml) was added 10% palladium on carbon (50 mg), the flask was purged with hydrogen, and the mixture was stirred at room temperature for 17 hours.
- Example 94-2 Synthesis of 2- (2- (tetrahydrofuran-2-yl) amino-3-trifluoromethylanilin-5-yl) benzoxazole
- 2- (2-fluoro-3-trifluoromethylnitrobenzene-5-yl) benzoxazole see Example 94-1 (0.40 g, 1.2 mmol) in acetonitrile (8 ml) was added 4-aminotetrahydro Pyran (0.30 g, 2.9 mmol) was added and heated to reflux for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- Example 94-3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) -4-trifluoromethylbenzimidazole
- 2- (2- (tetrahydrofuran-2-yl) amino-3-trifluoromethylanilin-5-yl) benzoxazole (0.20 g, 0.5 mmol) in methanol (4 ml)
- Methyl acetimidate hydrochloride (0.06 g, 0.6 mmol
- Example 95 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydrofuran-2-yl) methylbenzimidazole
- Example 95-1 Synthesis of 2- (2- (tetrahydrofuran-2-yl) methylaminoaniline-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole (0.40 mg, 1.5 mmol) in acetonitrile (8 ml) was added 2-aminomethyltetrahydrofuran (0.36 g, 3.6 mmol) was added and heated to reflux for 7 hours.
- Example 95-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydrofuran-2-yl) methylbenzimidazole
- 2- (tetrahydropyran-2-yl) methylaminoanilin-5-yl) benzoxazole (0.15 g, 0.4 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride ( 0.05 mg, 0.4 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 96 Synthesis of 5- (benzoxazol-2-yl) -2methyl-1- (3- (morpholin-4-yl) propyl) benzimidazole
- Example 96-1 Synthesis of 2- (N- (2- (morpholin-4-yl) -n-propyl) -2-nitroanilin-4-yl) benzoxazole
- potassium carbonate 500 mg, 1.94 mmol
- N- (3-Amino-n-propyl) morpholine (336 mg, 2.33 mmol) was added and heated to reflux for 4 hours.
- Example 96-2 Synthesis of 2- (2- (3- (morpholin-4-yl) -n-propyl) aminoanilin-4-yl) benzoxazole 2- (N- (2- (morpholin-4-yl) -n-propyl) -2-nitroanilin-4-yl) benzoxazole (see Example 96-1) (665 mg, 1.74 mmol) in tetrahydrofuran ( 10 ml), 10% palladium on carbon (70 mg) was added, the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 17 hours. After completion of the reaction, the mixture was filtered through celite, and the filtrate was concentrated to obtain the title compound (605 mg, 99%).
- Example 96-3 Synthesis of 5- (benzoxazol-2-yl) -2methyl-1- (3- (morpholin-4-yl) -n-propyl) benzimidazole 2- (2- (3- (morpholin-4-yl) -n-propyl) aminoanilin-4-yl) benzoxazole (see Example 96-2) (300 mg, 0.851 mmol) in methanol (5 ml) Was added with methyl acetimidate hydrochloride (103 mg, 0.936 mmol) and heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate.
- Example 100 Synthesis of 2- (4-acetylaminophenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 100-1 Synthesis of 5-chloro-2- (2-fluoronitrobenzene-5-yl) benzoxazole
- 4-fluoro-3-nitrobenzoic acid (25.0 g, 135 mmol)
- 2-amino-4-chlorophenol (21.3 g, 149 mmol)
- CHCl3 500 mL
- WSC 28.5 g, 149 mmol
- Example 100-2 Synthesis of 5-chloro-2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole
- 5-chloro-2- (2-fluoronitrobenzene-5-yl) benzoxazole see Example 100-1 (5.00 g, 17.1 mmol) in acetonitrile (20 ml) was added 4-aminotetrahydropyran ( 3.81 g, 38.0 mmol) was added, and the mixture was heated to reflux for 3 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- Example 100-3 Synthesis of 2- (4-acetylaminophenyl) -5- (5-chlorobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- 5-chloro-2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole see Example 100-2
- 4-Formylacetanilide 142 mg, 0.872 mmol
- oxone 313 mg, 0.509 mmol
- Example 102 Synthesis of 2- (4-carboxylphenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (5-Chlorobenzoxazol-2-yl) -2- (4-methoxycarbonylphenyl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 101) (220 mg, 0.451 mmol) in chloroform To the (3 ml) solution was added aqueous sodium hydroxide (1M, 1 ml, 1 mmol). Methanol was added to the two-layer solution until uniform and stirred at room temperature for 3 hours.
- aqueous sodium hydroxide (1M, 1 ml, 1 mmol
- Example 105 Synthesis of 2- (3-carboxylphenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (5-Chlorobenzoxazol-2-yl) -2- (3-methoxycarbonylphenyl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 103) (280 mg, 0.574 mmol) in chloroform To the (3 ml) solution was added 1M aqueous sodium hydroxide (1 ml, 1 mmol). Methanol was added to the two-layer solution until uniform and stirred at room temperature for 3 hours.
- Example 106 Synthesis of 2- (3-acetylaminophenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- 5-chloro-2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole see Example 100-2
- 3-acetamide benzoic acid 130 mg, 0.727 mmol
- WSC 167 mg, 0.872 mmol
- Example 107 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-((4-morpholinyl) carbonylmethyl) benzimidazole
- Example 107-1 Synthesis of 2- (2-((4-morpholinyl) carbonylmethyl) aminoanilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole (1.20 g, 4.6 mmol) in acetonitrile (24 ml) was added triethylamine (1.17 g, 11.6 mmol), amino Acetylmorpholine (0.73 g, 5.1 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 107-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (4-morpholinyl) benzimidazole
- 2- (4-morpholinyl) aminoanilin-5-yl) benzoxazole see Example 107-1 (0.08 g, 0.2 mmol) in methanol (2 ml) was added methyl acetimidate hydrochloride (0.03 g, 3 mmol).
- methyl acetimidate hydrochloride (0.03 g, 3 mmol).
- the reaction solution was cooled, water was added, and the precipitated crystals were filtered, washed with water, and dried to give the title compound (0.10 g, quantitative) as white crystals.
- Example 109 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-trifluoroacetylaminoethyl) benzimidazole 1- (2-aminoethyl) -5- (benzoxazol-2-yl) -2-methylbenzimidazole hydrochloride (see Example 56) (0.20 g, 0.6 mmol), trifluoroacetic anhydride (0.17 g 0.8 mmol), triethylamine (0.22 g, 2.2 mmol), and CHCl3 (2 mL) were added, and the mixture was stirred at room temperature for 1 hour. After cooling, chloroform and water were added for extraction.
- Example 110 Synthesis of 3- (2- (5- (benzoxazol-2-yl) -2-methylbenzimidazol-1-yl) ethyl) -1-methylthiourea 1- (2-aminoethyl) -5- (benzoxazol-2-yl) -2-methylbenzimidazole hydrochloride (see Example 56) (0.20 g, 0.6 mmol), methyl isothiocyanate (0.17 g, 0.6 mmol), triethylamine (0.13 g, 13 mmol), and THF (2 mL) were added, and the mixture was stirred at room temperature for 1 hour. After cooling, chloroform and water were added for extraction.
- the obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- p-toluenesulfonic acid monohydrate (0.18 g, 1.8 mmol) was added, and the mixture was heated to reflux for 40 hours.
- the reaction solution was concentrated, water was added, and the precipitated crystals were filtered, washed with water, and dried to give the title compound (1.40 g, yield 26%) as yellowish crystals.
- Example 112-2 Synthesis of 2- (4-chloro-2- (tetrahydropyran-4-yl) aminonitrobenzene-5-yl) benzoxazole
- 2- (4-chloro-2-fluoronitrobenzene-5-yl) benzoxazole see Example 112-1
- 4-aminotetrahydropyran 0.30 g, 2.9 mmol
- Example 112-3 Synthesis of 2- (4-chloro-2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole
- 2- (4-Chloro-2- (tetrahydropyran-4-yl) aminonitrobenzene-5-yl) benzoxazole (0.10 g, 0.8 mmol) to iron powder (0.33 g, 5.8 mmol) and acetic acid (50 mL) were added, and the mixture was heated to reflux for 2 hours.
- Example 112-4 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) -6-chlorobenzimidazole 2- (4-Chloro-2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole (see Example 112-3) (0.04 g, 0.2 mmol) in methanol (1.2 ml) , Methyl acetimidate hydrochloride (0.06 g, 0.6 mmol) was added and heated to reflux for 3 hours.
- Example 113 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxycarbonylphenylmethyl) -2-methylbenzimidazole
- Example 113-1 Synthesis of 2- (2- (4-methoxycarbonylphenylmethyl) aminonitrobenzene-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added triethylamine (245 mg, 2.42 mmol), 4-carbohydrate.
- Methoxybenzylamine hydrochloride (215 mg, 1.07 mmol) was added and heated to reflux for 2 hours.
- Example 113-2 Synthesis of 2- (2- (4-methoxycarbonylphenylmethyl) aminoaniline-5-yl) benzoxazole 2- (2- (4-Methoxycarbonylphenylmethyl) aminonitrobenzene-5-yl) benzoxazole (see Example 113-2) (367 mg, 0.910 mmol) to iron powder (102 mg, 1.82 mmol), 10% acetic acid An aqueous solution (5 mL) and methanol (7 mL) were added, and the mixture was heated to reflux for 16 hours. The reaction solution was cooled, saturated aqueous sodium hydrogen carbonate solution and chloroform were added, the mixture was filtered through celite, and the resulting filtrate was extracted.
- Example 113-3 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxycarbonylphenylmethyl) -2-methylbenzimidazole 2- (2- (4-Methoxycarbonylphenylmethyl) aminoaniline-5-yl) benzoxazol (see Example 113-3)
- Amine 59.0 mg, 0.159 mmol
- methanol 3 ml
- Hydrochloride (19 mg, 0.174 mmol) was added and heated to reflux for 1.5 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for several minutes.
- Example 114 Synthesis of 5- (benzoxazol-2-yl) -1- (4-carboxylphenylmethyl) -2-methylbenzimidazole
- 5- (benzoxazol-2-yl) -1- (4-methoxycarbonylphenylmethyl) -2-methylbenzimidazole (see Example 113-4) (55 mg, 0.138 mmol) in chloroform (3 ml)
- Aqueous sodium hydroxide (1M, 0.6 ml, 0.6 mmol) was added.
- Methanol was added to the two-layer solution until uniform and stirred at room temperature for 20 hours. After completion of the reaction, the solution was concentrated, an aqueous acetic acid solution was added, and the mixture was stirred at room temperature.
- Example 115 Synthesis Example of 5- (1-Benzoxazol-2-yl) -1- (1-butanol-2-yl) -2-methylbenzimidazole 115-1 Synthesis of 2- (2- (1-n-butanol-2-yl) aminonitrobenzene-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added potassium carbonate (268 mg, 1.94 mmol), 2- Amino-1-n-butanol (95.4 mg, 1.07 mmol) was added and heated to reflux for 4 hours.
- Example 115-2 Synthesis of 5- (1-benzoxazol-2-yl) -1- (1-n-butanol-2-yl) -2-methylbenzimidazole Synthesis of 2- (2- (1-n-butanol-2-yl) aminonitrobenzene-5-yl) benzoxazole (see Example 115-1) (295 mg, 0.901 mmol) in ethanol, ethyl acetate (1: The solution was added 10% palladium on carbon (50 mg) to the solution, and the flask was replaced with hydrogen, followed by stirring at room temperature for 19 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Ethyl acetimidate hydrochloride (133 mg, 1.08 mmol) was added to a solution of the obtained residue in ethanol (5 ml), and the mixture was heated to reflux for 2 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for several minutes. The obtained crystals were filtered, washed with water, and dried to give the title compound (232 mg, 72%) as white crystals.
- Example 116 Synthesis Example of 5- (1-Benzoxazol-2-yl) -1- (2-n-propanol-1-yl) -2-methylbenzimidazole 116-1 Synthesis of 2- (2- (2-n-propanol-1-yl) aminonitrobenzene-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added potassium carbonate (268 mg, 1.94 mmol), 1- Amino-2-n-propanol (80.4 mg, 1.07 mmol) was added and heated to reflux for 4 hours.
- Example 116-2 Synthesis of 5- (1-benzoxazol-2-yl) -1- (2-n-propanol-1-yl) -2-methylbenzimidazole 2- (2- (2-n-propanol-1-yl) aminonitrobenzene-5-yl) benzoxazole (Example 116-1) (280 mg, 0.894 mmol) in ethanol, ethyl acetate (1: 1, 6 ml) ) 10% palladium carbon (50 mg) was added to the solution, the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 19 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Ethyl acetimidate hydrochloride (133 mg, 1.08 mmol) was added to a solution of the obtained residue in ethanol (5 ml), and the mixture was heated to reflux for 2 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for several minutes. The obtained crystals were filtered, washed with water, and dried to give the title compound (254 mg, 92%) as white crystals.
- Example 117-2 Synthesis of 5- (benzoxazol-2-yl) -1- (3-methoxycarbonylphenylmethyl) -2-methylbenzimidazole 2- (2- (3-methoxycarbonylphenylmethyl) aminonitrobenzene-5-yl) benzoxazole (Example 117-1) (220 mg, 0.545 mmol) to iron powder (91.4 mg, 1.64 mmol), acetic acid (5 mL ) And heated to reflux for 12 hours. After cooling the reaction solution, the reaction solution was concentrated. The obtained residue was purified by silica gel column chromatography to give the titled compound (139 mg, 64%) as white crystals.
- Example 118 Synthesis Example of 5- (1-Benzoxazol-2-yl) -1- (1-acetoxy-2-phenylethane-2-yl) -2-methylbenzimidazole 118-1 Synthesis of 2- (2- (2-phenyl-1-ethanol-2-yl) aminonitrobenzene-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added potassium carbonate (268 mg, 1.94 mmol), 2- Phenylglycinol (146 mg, 1.07 mmol) was added and heated to reflux for 2 hours.
- Example 118-2 Synthesis of 5- (1-benzoxazol-2-yl) -1- (1-acetoxy-2-phenylethan-2-yl) -2-methylbenzimidazole 2- (2- (2-Phenyl-1-ethanol-2-yl) aminonitrobenzene-5-yl) benzoxazole (Example 118-1) (340 mg, 0.545 mmol) to iron powder (152 mg, 2.72 mmol) Acetic acid (5 mL) was added, and the mixture was heated to reflux for 12 hours. After cooling the reaction solution, the reaction solution was concentrated. The obtained residue was purified by silica gel column chromatography to give the titled compound (214 mg, 57%) as a colorless amorphous.
- Example 119 Synthesis of 5- (benzoxazol-2-yl) -1- (3-carboxylphenylmethyl) -2-methylbenzimidazole
- 5- (benzoxazol-2-yl) -1- (3-methoxycarbonylphenylmethyl) -2-methylbenzimidazole see Example 117-2
- Aqueous sodium hydroxide (1M, 1 ml, 1 mmol) was added.
- Methanol was added to the two-layer solution until uniform and stirred at room temperature for 20 hours. After completion of the reaction, the solution was concentrated, an aqueous acetic acid solution was added, and the mixture was stirred at room temperature.
- Example 120 Synthesis of 5- (1-benzoxazol-2-yl) -1- (2-phenylethanol-2-yl) -2-methylbenzimidazole 5- (1-Benzoxazol-2-yl) -1- (1-acetoxy-2-phenylethan-2-yl) -2-methylbenzimidazole (see Example 118-2) (200 mg, 0.486 mmol ) In methanol (5 ml) was added aqueous sodium hydroxide solution (1M, 1.5 ml, 1.5 mmol) and stirred at room temperature for 20 hours. After completion of the reaction, the solution was concentrated, water was added, and the mixture was stirred at room temperature.
- Example 121 Synthesis Example 5-1-1 of 5- (benzoxazol-2-yl) -2-methyl-1- (2-tert-butoxycarbonyl-n-propyl) benzimidazole Synthesis of 2- (2-tert-butoxycarbonyl-n-propylamino) aniline-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (0.50 g, 1.9 mmol) in acetonitrile (10 ml) was added 2-tert-butoxycarbonyl-1-propyl.
- Example 121-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-tert-butoxycarbonyl-n-propyl) benzimidazole
- 2- (2-tert-butoxycarbonyl-n-propylamino) anilin-5-yl) benzoxazole (0.62 g, 1.7 mmol) in THF (20 ml) was added methyl acetimidate.
- Hydrochloride (0.37 g, 3.4 mmol) was added and heated to reflux for 3 hours.
- Example 129 Synthesis Example of 5- (Benzoxazol-2-yl) -1- (2- (piperidin-1-yl) ethyl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 129-1 Synthesis of N- (2- (piperidin-1-yl) ethyl) -4- (benzoxazol-2-yl) -2-nitroaniline
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (200 mg, 0.775 mmol) in acetonitrile (5 ml) was added potassium carbonate (214 mg, 1.55 mmol), 1- (2-Aminoethyl) piperidine (119 mg, 0.93 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 130 Synthesis Example of 5- (Benzoxazol-2-yl) -1- (2-dimethylaminoethyl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 130-1 Synthesis of N- (2-dimethylaminoethyl) -4- (benzoxazol-2-yl) -2-nitroaniline
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (200 mg, 0.775 mmol) in acetonitrile (5 ml) was added potassium carbonate (214 mg, 1.55 mmol), N, N-dimethylethylenediamine (82 mg, 0.93 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 131 Synthesis Example of 5- (Benzoxazol-2-yl) -1- (2- (methylthio) ethyl) -2-methylbenzimidazole
- Example 131-1 Synthesis of N- (2- (methylthio) ethyl) -4- (benzoxazol-2-yl) -2-nitroaniline
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- potassium carbonate 321 mg, 1.55 mmol
- 2- (Methylthio) ethylamine 127 mg, 1.39 mmol
- Example 131-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2- (methylthio) ethyl) -2-methylbenzimidazole Iron powder (188 mg, 3.37 mmol) and acetic acid (5 mL) were added to N- (2- (methylthio) ethyl) -4- (benzoxazol-2-yl) -2-nitroaniline (370 mg, 1.12 mmol), and 10 Heated to reflux for hours. After cooling the reaction solution, the reaction solution was concentrated. The obtained residue was purified by silica gel column chromatography to obtain 305 mg (84%) of the title compound as pink crystals.
- Example 135 Synthesis Example of 5- (5-Chlorobenzoxazol-2-yl) -1-n-propylbenzimidazole
- Example 135-1 Synthesis of 5- (5-chlorobenzooxazol-2-yl) -2-n-propylaminoaniline
- To a suspension of 5-chloro-2- (2-fluoro-nitrobenzene-5-yl) benzoxazole (see Example 100-1) (1.5 g, 5.13 mmol) in acetonitrile (10 ml) was added triethylamine (779 mg, 7.70). mmol) and propylamine (364 mg, 6.16 mmol) were added, and the mixture was heated to reflux for 4 hours.
- Example 135-2 To a solution of 5- (5-chlorobenzooxazol-2-yl) -2-n-propylaminoaniline (see Example 135-1) (300 mg, 0.994 mmol) in triethyl orthoformate (5 ml) was added p-toluenesulfonic acid. (10 mg) was added and stirred at 100 ° C. for 2 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated, and the resulting crude crystals were washed with diethyl ether to obtain 146 mg (47%) of the title compound as pale yellow crystals.
- Example 137 Synthesis of 5- (5-chlorobenzooxazol-2-yl) -2-methyl-1-n-propylbenzimidazole
- a solution of 5- (5-chlorobenzooxazol-2-yl) -2-n-propylaminoaniline (see Example 143-1) (200 mg, 0.663 mmol) in ethanol (5 ml) was added ethyl acetimidate hydrochloride (123 mg , 0.995 mmol) was added and heated to reflux for 5 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 138 Synthesis Example of 5- (5-Chlorobenzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole
- Example 138-1 Synthesis of 5-chloro-2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole
- To a suspension of 5-chloro-2- (2-fluoronitrobenzene-5-yl) benzoxazole (see Example 100-1) (815 mg, 2.79 mmol) in acetonitrile (10 ml) was added triethylamine (847 mg, 8.37 mmol).
- 3,3,3-Trifluoropropylamine hydrochloride 500 mg, 3.34 mmol was added and heated to reflux for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried. To a solution of the obtained crystals in ethanol and tetrahydrofuran (1: 2, 15 ml), 10% palladium on carbon (100 mg) was added, the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 18 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated to obtain 993 mg (100%) of the title compound as orange crystals.
- Example 138-2 Synthesis of 5- (5-chlorobenzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole 5-Chloro-2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole (see Example 138-1) (200 mg, 0.562 mmol) in ethanol (5 ml) Was added ethyl acetimidate hydrochloride (104 mg, 0.843 mmol), and the mixture was heated to reflux for 5 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 139 Synthesis of 5- (5-chlorobenzoxazol-2-yl) -1- (3,3,3-trifluoropropyl) benzimidazole 5-chloro-2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole (see Example 138-1) (200 mg, 0.562 mmol) triethyl orthoformate (5 ml P-Toluenesulfonic acid (10 mg) was added to the solution and stirred at 100 ° C. for 4 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 140 Synthesis of 5- (benzimidazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole To a suspension of 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (500 mg, 1.86 mmol), toluene (10 mL) was added thionyl chloride (3 mL). 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid chloride hydrochloride (533 mg, 1.69) obtained by stirring under reflux conditions for 1.5 hours and concentrating under reduced pressure.
- Test example 1 (Preparation of compound evaluation cells)
- forward primer (5'-tccagtatttgagaaaaggagccaggagtctccat-3 ')
- reverse primer (5'-ggaggcttcctctttgcttcccggtcttttcg-3') as PCR primers
- the region from about 5 kbp upstream of the NXF gene promoter partial translation region to the vicinity of the transcription start site was isolated by PCR.
- the resulting NXF promoter partial fragment is introduced into the SmaI site of the pGL3 Basic vector (Promega) so that the luciferase gene is located downstream of the promoter, and a reporter plasmid in which the luciferase enzyme is expressed by the action of the NXF promoter was made.
- PC12 cells purchased from ATCC were prepared in RPMI medium (GIBCO-BRL), 5% FCS (GIBCO-BRL), 15% horse serum (GIBCO-BRL), and 1 mM sodium pyruvate (GIBCO- BRL was added and cultured at 37 ° C., 5% in the presence of CO 2 .
- the PC12 of the reporter plasmid 12 ⁇ g and 1 ⁇ g against 106 cells pRC / RSV vector (Invitrogen Corp.) were transfected sheet transfected using Lipofectamine2000 reagent (Invitrogen Corporation), 40 mg / L after 2 days transfection
- the medium was replaced with a medium containing Geneticin sulfate (GIBCO) and the culture was continued.
- a culture solution containing only a final concentration of 0.1% DMSO was used as a negative control group
- a culture group containing a saturated concentration of NGF was used as a positive control group
- 200 ng / ml NGF corresponding to a compound concentration of 0 was added.
- an experimental group containing 0.1% DMSO was placed, and the compound solution (dissolved in DMSO to a final DMSO concentration of 0.1%) was added to 200 ng / ml NGF, based on the luciferase activity.
- the relative luciferase activity of the experimental group to which (prepared as described above) was added was calculated, and this was defined as the activity of enhancing the activity of 200 ng / ml NGF by the compound.
- the test results of NGF activity enhancing activity are listed in the table below. +: Relatively weak potentiating activity (enhancing activity only at 10 ⁇ M) ++: shows enhancing activity ++: shows particularly strong enhancing activity (increases the activity of NGF 200 ng / ml more than 2 times at 1 ⁇ M)
- Test example 2 Male 8-week-old C57BL / 6J mice are normally bred for 12 days, then the left common neck and external common artery are ligated in advance, and the middle cerebral artery is occluded for 90 minutes and then reopened. A model was created. In the same model, the compound was administered into the tail vein at 30 mg / kg 3 hours after occlusion and 48 hours after 48 hours. After 3 days of occlusion / reopening, the head was decapitated, the skull was cut from the cerebellum side, and the brain was removed. The removed brain was placed in a Buan solution (pH 3.5 to 4.0) and fixed. From the fixed brain, coronal sections were cut out, and after cutting, dehydration and penetration, and embedded in paraffin.
- Buan solution pH 3.5 to 4.0
- Infarct volume was calculated as follows. A digital image was captured using a camera (MCD-350, Olympus Corporation) attached to an optical microscope (objective lens ⁇ 1). The captured digital image was pasted on a single mount (new layer) in Adobe Photoshop 2.0.
- Area MR, mm 2 ) was measured, and non-infarct area (ML, mm 2 ) [infarct side (left) area (mm 2 ) ⁇ infarct area (mm 2 )] on the infarct side (left) was calculated.
- the infarct volume (SV, mm 3 ), the infarct side (left) non-infarct volume (MLV, mm 3 ), and the non-infarct side (right) volume (MRV, mm 3 ) were calculated by the following equations.
- test result (infarct volume) of the compound of Example 12 is shown in FIG. Thus, a decrease in infarct volume was confirmed in the compound administration group.
- Test example 3 The prophylactic effect of the compounds on the latency delay of motor nerve transmission velocity (MNCV) in the sciatic nerve in streptozotocin-induced diabetic rats was examined.
- the group composition was a non-diabetic control group, a diabetic control group, and a compound administration group.
- the non-diabetic control group and the diabetic control group had 5 ml / kg of water for injection, and the compound administration group had a compound solution prepared at 10 mg / ml with water for injection at 5 ml / kg.
- Oral administration once a day for 4 weeks.
- MNCV measurement was performed 3 times for each group. The measurement time is the first day before administration of the streptozotocin solution or pH 4.5 0.75 mmol / L citrate buffer solution, and the second time is 4 weeks after administration of the streptozotocin solution or pH 4.5 0.75 mmol / L citrate buffer solution. Thereafter, the third measurement was performed on the day after the last day of administration of the water or compound solution for 4 weeks.
- the test results (motor nerve transmission rate) of the compound of Example 12 are shown in FIG. 2 (in the figure, STZ represents streptozotocin). Thus, recovery of motor nerve transmission rate was recognized in the compound administration group compared to the solvent control administration group.
- Test example 4 In the same manner as in Test Example 1, the enhancing activity of Example NGF activity of the compounds shown in Table 2 was evaluated. The results are shown in Table 2. +: Relatively weak potentiating activity (enhancing activity only at 10 ⁇ M) ++: shows enhancing activity ++: shows particularly strong enhancing activity (increases the activity of NGF 200 ng / ml more than 2 times at 1 ⁇ M)
- the compound of the present invention is effective for treatment or prevention of diseases involving the activity of neurotrophic factor.
- SEQ ID NO: 1 is a forward primer.
- SEQ ID NO: 2 is a reverse primer.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Epidemiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Hospice & Palliative Care (AREA)
- Endocrinology (AREA)
- Psychology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Obesity (AREA)
- Immunology (AREA)
- Gynecology & Obstetrics (AREA)
- Reproductive Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
一方で、神経栄養因子によってある種の転写調節因子の発現が制御されていることが知られており(非特許文献1参照)、神経栄養因子によって発現が制御されている転写調節因子の1つとしてNXFが知られている(特許文献1参照)。
また、NXFが欠損したマウスでは、神経変性疾患の原因の一つとして考えられているグルタミン酸毒性を惹起した際に神経変性疾患の症状が重篤にあらわれることから、NXFが神経保護作用に重要な役割を持つことが知られている(特許文献2参照)。
ところで、特許文献3には、一般式

また、特許文献4には、一般式

また、特許文献5には、一般式

また、特許文献6には、一般式

式

ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表す。)を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、及び
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物(本明細書中、化合物(I)と称する場合がある。また、本明細書中、本発明の化合物と称する場合がある。)が神経栄養因子の活性を増強し、かつ神経細胞保護作用することを見出し、本発明に至った。
[1]
式

R1は、
(1)C3-6アルキル基、
(2)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O-、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(式中、各Raは、同一または相異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されたC1-6アルキル基、、又は、
(3)
(a)オキソ基、及び
(b)C1-4アルコキシ-カルボニル基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、又は
(4)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換された芳香族炭化水素環基を表し、
Xは、
NH、O、又はSを表し、
Yは、
CH、又はNを表し、
Zは、
N、又はC-R2を表し、
R2は、
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-S-、
(f)Rb-SO2-、
(g)Rb-CO-O-、
(h)Rb-CO-NRb-、
(i)Rb-NRb-、
(j)Rb-NRb-Rb-S(O)n-
(k)フェニル基、
(l)5~6員飽和複素環基、
(m)ヒドロキシ基、及び
(n)アミノ基、(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基でそれぞれ置換されていてもよいC1-6アルキル基、C2-6アルケニル基、又はC2-6アルキニル基、又は、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基
を表し、
環Aは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)ハロゲン原子で置換されていてもよいアルキル基、
(i)ニトロ基、及び
(j)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環を表し、
環Bは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)アミド基、
(i)エステル基、
(j)ハロゲン原子で置換されていてもよいアルキル基、
(k)ニトロ基、及び
(l)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、またはその溶媒和物。
[2]
Zは、
C-R2であり、及び
Yは、
Nである
前記[1]に記載の化合物。
[3]
R1は、
5~6員非芳香族複素環基である
前記[1]に記載の化合物。
[4]
R2は、
C1-6アルキル基である
前記[1]に記載の化合物。
[5]
Zは、
C-R2であり、
Yは、
Nであり、
R1は、
5~6員非芳香族複素環基であり、及び
R2は、
C1-6アルキル基である
前記[1]に記載の化合物。
[6]
前記[1]に記載の化合物を含有する医薬組成物。
[7]
神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、または理学療法効果の促進剤である前記[6]に記載の医薬組成物。
[8]
神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、または理学療法効果の促進剤が、脳虚血性疾患、または糖尿病性神経障害の治療、または予防剤である前記[7]に記載の医薬組成物。
[9]
式

ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物を有効成分として含有する、神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、又は理学療法効果の促進剤。
[10]
環Aは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)ハロゲン原子で置換されていてもよいアルキル基、
(i)ニトロ基、及び
(j)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環であり、
環Bは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)アミド基、
(i)エステル基、
(j)ハロゲン原子で置換されていてもよいアルキル基、
(k)ニトロ基、及び
(l)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環であり、
ZはN、又はC-R2であり、
Xは、
NH、O、又はSであり、
Yは、
CH、又はNであり、
R1は、
(1)水素原子、
(2)C1-6アルキル基、
(3)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O-、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(式中、各Raは、同一または相異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されたC1-6アルキル基、
(4)
(a)オキソ基、
(b)C1-4アルコキシ-カルボニル基、及び
(c)ヒドロキシ基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、又は
(5)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換されていてもよい芳香族炭化水素環基を表し、
R2は、
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-S-、
(f)Rb-SO2-、
(g)Rb-CO-O-、
(h)Rb-CO-NRb-、
(i)Rb-NRb-、
(j)Rb-NRb-Rb-S(O)n-
(k)フェニル基、
(l)5~6員飽和複素環基、
(m)ヒドロキシ基、及び
(n)アミノ基
(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基でそれぞれ置換されていてもよいC1-6アルキル基、C2-6アルケニル基、又はC2-6アルキニル基、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基、又は
(4)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
フェニル基であるか、あるいは、
R1およびR2は、これらが隣接するNおよびCと一緒になって、5~8員含窒素非芳香族複素環を形成してもよく、
X1は、
O、又はNHであり、および
R2Zは、
C1-6アルキル基である
前記[9]に記載の剤。
[11]
脳虚血性疾患、または糖尿病性神経障害の治療、または予防剤である、前記[9]に記載の剤。
[12]
式

ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物の有効量を哺乳動物に投与することを特徴とする、神経栄養因子の活性が関与する疾患の治療、若しくは予防方法、又は理学療法効果の促進方法。
[13]
神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、又は理学療法効果の促進剤を製造するための、式

ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物の使用。
[1’]
式

R1は、
置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し;
Xは、
N-R3(R3は、水素原子、又は置換基を表す。)、O、又はSを表し;
Yは、
C-R4(R4は、水素原子、又は置換基を表す。)、又はNを表し;
Zは、
N、又はC-R2(R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表す。)を表し;及び
環A及び環Bは独立して、置換されていてもよいベンゼン環を表す。
但し、
R1は、式
(式中、Ra1は、カルボキシ基、シアノ基、1H-テトラゾリル基、1-トリフェニルメチル-テトラゾリル基、又はアルコキシカルボニル基を表し、Ra2は、水素原子、フッ素原子、塩素原子、又は臭素原子を表す。)で表される基ではない。
更に、但し、
R1は、

で示される化合物、その薬学的に許容される塩、又はその溶媒和物。
(但し、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-(3-フェニルプロピル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-(2-フラニルメチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-シクロペンチル-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-(2-フラニルメチル)-1-(1-メチルエチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-[(4-メトキシフェニル)メチル]-1-(1-メチルエチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-シクロペンチル-1-(2-フラニルメチル)-2'-フェニル-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-シクロペンチル-1-(2-フラニルメチル)-2'-[4-(メチルチオ)フェニル]-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 2'-シクロヘキシル-1'-シクロペンチル-1-(2-フラニルメチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-シクロペンチル-1-(2-フラニルメチル)-2'-(3-チエニル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-シクロペンチル-2'-[4-(メチルチオ)フェニル]-1-[3-(4-モルホリニル)プロピル]-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 2'-(4-クロロフェニル)-1'-シクロペンチル-1-[3-(4-モルホリニル)プロピル]-, メチル エステル、
2,5'-ビ-1H-ベンゾイミダゾール, 6-フェニル-1,1'-ビス(フェニルメチル)-、
2,5':2',5''-Ter-1H-ベンゾイミダゾール, 6-フェニル-1,1',1''-トリス(フェニルメチル)-、
2,5'-ビ-1H-ベンゾイミダゾール, 1,1',2'-トリス(フェニルメチル)-、
2,5'-ビ-1H-ベンゾイミダゾール, 2'-(3-クロロフェニル)-1'-[(2,5-ジフルオロフェニル)メチル]-1-(フェニルメチル)-、
ベンゾチアゾール, 2-(2-メチル-1-フェニル-1H-ベンゾイミダゾール-5-イル)-, 4-メチルベンゼンスルホナート (1:1)、
ベンゾチアゾール, 2-(2-メチル-1-フェニル-1H-ベンゾイミダゾール-5-イル)-、
ベンゾオキサゾール, 2-(2-メチル-1-フェニル-1H-ベンゾイミダゾール-5-イル)-、
2,5'-ビ-1H-ベンゾイミダゾール, 2'-メチル-1'-フェニル-、
2,5'-ビ-1H-ベンゾイミダゾール, 1-エチル-2'-メチル-1'-フェニル-、
ベンゾオキサゾール, 2-(2-メチル-1-フェニル-1H-ベンゾイミダゾール-5-イル)-, 4-メチルベンゼンスルホナート (1:1)、
[1,1'-ビフェニル]-2-カルボン酸, 4'-[(1,7'-ジメチル-2'-プロピル[2,5'-ビ-1H-ベンゾイミダゾール]-1'-イル)メチル]-, 1,1-ジメチルエチル エステル、
[1,1'-ビフェニル]-2-カルボン酸, 4'-[(1,7'-ジメチル-2'-プロピル[2,5'-ビ-1H-ベンゾイミダゾール]-1'-イル)メチル]-、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-シクロペンチル-1'-(1-メチルエチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-(2-メトキシエチル)-1'-(2-メチルプロピル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-(1-メチルエチル)-1-(2-メチルプロピル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1'-ブチル-1-(2-メチルプロピル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-(2-メトキシエチル)-1'-(1-メチルエチル)-, メチル エステル、
フェノール, 4-[1,1'-ジメチル-5-(4-メチル-1-ピペラジニル)[2,5'-ビ-1H-ベンゾイミダゾール]-2'-イル]-、
[2,5'-ビ-1H-ベンゾイミダゾール]-4,4',7,7'-テトラオール, 2'-(3,4-ジヒドロキシフェニル)-1,1'-ジメチル-5-(4-メチル-1-ピペラジニル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-4,4',7,7'-テトラオール, 2'-(4-ヒドロキシフェニル)-1,1'-ジメチル-5-(4-メチル-1-ピペラジニル)-、
2,5'-ビ-1H-ベンゾイミダゾール, 4',7'-ジメトキシ-2'-(メトキシメチル)-1'-メチル-6-(4-メチル-1-ピペラジニル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-2'-メタノール, 4',7'-ジメトキシ-1'-メチル-6-(4-メチル-1-ピペラジニル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-4',7'-ジオール, 2'-(ヒドロキシメチル)-1'-メチル-6-(4-メチル-1-ピペラジニル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-2'-メタノール, 4',7'-ジメトキシ-1'-メチル-6-(4-メチル-1-ピペラジニル)-, 2'-アセタート、
2,5'-ビ-1H-ベンゾイミダゾール, 2'-(クロロメチル)-4',7'-ジメトキシ-1'-メチル-6-(4-メチル-1-ピペラジニル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-2'-メタノール, 7'-ヒドロキシ-4'-メトキシ-1'-メチル-6-(4-メチル-1-ピペラジニル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-4',7'-ジオール, 2'-(クロロメチル)-1'-メチル-6-(4-メチル-1-ピペラジニル)-、
2,5'-ビ-1H-ベンゾイミダゾール, 2'-メチル-1'-(1-メチルエチル)-1-(フェニルメチル)-、
2,5'-ビ-1H-ベンゾイミダゾール, 2'-(3-クロロフェニル)-1-(フェニルメチル)-1'-[2-[(フェニルメチル)チオ]エチル]-、
[2,5'-ビ-1H-ベンゾイミダゾール]-6-カルボキシミドアミド, 2'-(4-フルオロフェニル)-1'-メチル-N-(1-メチルエチル)-、
1H-ベンゾイミダゾール-6-カルボキシミドアミド, 2-[4-[1'-ブチル-6-[イミノ[(1-メチルエチル)アミノ]メチル][2,5'-ビ-1H-ベンゾイミダゾール]-2'-イル]フェニル]-N-(1-メチルエチル)-、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-2'-(2-フルオロフェニル)-1'-(1-メチルエチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-2'-シクロヘキシル-1'-(1-メチルエチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-(1-メチルエチル)-2'-(3-チエニル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-(1-メチルエチル)-2'-(4-ニトロフェニル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-(1-メチルエチル)-2'-フェニル-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 2'-(2-ブロモフェニル)-1-ブチル-1'-(1-メチルエチル)-, メチル エステル、
[2,5'-ビ-1H-ベンゾイミダゾール]-5-カルボン酸, 1-ブチル-1'-(1-メチルエチル)-, メチル エステル、
ベンゾオキサゾール, 2-(1,2-ジメチル-1H-ベンゾイミダゾール-5-イル)-、
ベンゾオキサゾール, 2-(1-エチル-2-メチル-1H-ベンゾイミダゾール-5-イル)-、
ベンゾチアゾール, 2-(1-エチル-2-メチル-1H-ベンゾイミダゾール-5-イル)-、および
ベンゾチアゾール, 2-(1,2-ジメチル-1H-ベンゾイミダゾール-5-イル)-
を除く)。
[2’]
R1は
a)6員の環状基で置換されていてもよい炭素数1~6の非環状炭化水素基、または
b)ハロゲン原子、C1-4アルコキシ基、及びC1-4アルコキシ-カルボニル基から選択される1個以上の置換基で置換されていてもよい6~10員の環状基
であり、
Xが、N-R3(R3は、水素原子、又はRa-O-CO-(RaはC1-6アルキル基である。))、O、又はSであり、
Yが、CH、又はNであり、
Zが、N、又はC-R2
(式中、R2は、
a)水素原子、
b)
(1)ハロゲン原子、
(2)Rb-O-、
(3)Rb-CO-、
(4)Rb-CO-O-、および
(5)Rb-NRb-
(式中、各Rbは、同一または異なって、水素原子、1個のフェニル基で置換されていてもよいC1-6アルキル基を表す。)
から選択される1個以上(好ましくは1~3個)の置換基で
置換されていてもよいC1-6アルキル基、又は
c)5~6員の環状基
であり、
環Aがベンゼン環であり、および
環Bがハロゲン原子、ニトロ基、C1-6アルキル基、およびアミノ基から選択される1個以上の置換基で置換されていてもよいベンゼン環である前記[1’]に記載の化合物。
[3’]
式

R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し;
Xは、
N-R3(R3は、水素原子、又は置換基を表す。)、O、又はSを表し;
Yは、
C-R4(R4は、水素原子、又は置換基を表す。)、又はNを表し;
Zは、
N、又はC-R2(R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表す。)を表し;及び
環A及び環Bは独立して、置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物(以下、化合物(I’)と称する場合がある。化合物(I’)については、上記化合物(I)の説明によって、同様に理解される。)を有効成分として含有する、神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、又は理学療法効果の促進剤。
[4’]
脳虚血性疾患、または糖尿病性神経障害の治療、または予防剤である、前記[3’]に記載の剤。
「環構成原子として、酸素原子、硫黄原子、および窒素原子から選択される1~3個(好ましくは1~2個)のへテロ原子を有する5~6員のヘテロアリール(基)」としては、例えば、フリル(例、2-フリル、3-フリル)、チエニル(例、2-チエニル、3-チエニル)、ピリジル(例、2-ピリジル、3-ピリジル、4-ピリジル)、ピリミジニル(例、2-ピリミジニル、4-ピリミジニル、5-ピリミジニル、6-ピリミジニル)、ピリダジニル(例、3-ピリダジニル、4-ピリダジニル)、ピラジニル(例、2-ピラジニル)、ピロリル(例、1-ピロリル、2-ピロリル、3-ピロリル)、イミダゾリル(例、1-イミダゾリル、2-イミダゾリル、4-イミダゾリル、5-イミダゾリル)、ピラゾリル(例、1-ピラゾリル、3-ピラゾリル、4-ピラゾリル)、チアゾリル(例、2-チアゾリル、4-チアゾリル、5-チアゾリル)、イソチアゾリル、オキサゾリル(例、2-オキサゾリル、4-オキサゾリル、5-オキサゾリル)、イソオキサゾリル、オキサジアゾリル(例、1,2,4-オキサジアゾール-5-イル、1,3,4-オキサジアゾール-2-イル)、チアジアゾリル(例、1,3,4-チアジアゾール-2-イル)、トリアゾリル(例、1,2,4-トリアゾール-1-イル、1,2,4-トリアゾール-3-イル、1,2,3-トリアゾール-1-イル、1,2,3-トリアゾール-2-イル、1,2,3-トリアゾール-4-イル)、テトラゾリル、およびトリアジニル等が挙げられる。
「C3-10シクロアルキル(基)」としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル、およびアダマンチルが挙げられる。
「C3-10シクロアルケニル(基)」としては、例えば、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル、シクロノネニル、およびシクロデセニルが挙げられる。
「C4-10シクロアルカジエニル基」としては、例えば、シクロブタジエニル、シクロペンタジエニル、シクロヘキサジエニル、シクロヘプタジエニル、シクロオクタジエニル、シクロノナジエニル、およびシクロデカジエニルが挙げられる。
「環構成原子として、酸素原子、酸化されていてもよい硫黄原子、および窒素原子から選択される1~3個(好ましくは1~2個)のへテロ原子を有する5~6員の非芳香族複素環基」としては、例えば、
ピロリジニル(例、1-ピロリジニル、2-ピロリジニル)、
ピペリジニル(例、ピペリジノ、2-ピペリジニル、3-ピペリジニル、4-ピペリジニル)、
モルホリニル(例、モルホリノ)、
チオモルホリニル(例、チオモルホリノ)、
ピペラジニル(例、1-ピペラジニル、2-ピペラジニル、3-ピペラジニル)、
ヘキサメチレンイミニル(例、ヘキサメチレンイミン-1-イル)、
オキサゾリジニル(例、オキサゾリジン-2-イル)、
チアゾリジニル(例、チアゾリジン-2-イル)、
イミダゾリジニル(例、イミダゾリジン-2-イル、イミダゾリジン-3-イル)、
オキサゾリニル(例、オキサゾリン-2-イル)、
チアゾリニル(例、チアゾリン-2-イル)、
イミダゾリニル(例、イミダゾリン-2-イル、イミダゾリン-3-イル)、
ジオキソリル(例、1,3-ジオキソール-4-イル)、ジオキソラニル(例、1,3-ジオキソラン-4-イル)、
ジヒドロオキサジアゾリル(例、4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)、
ピラニル(例、4-ピラニル)、
テトラヒドロピラニル(例、2-テトラヒドロピラニル、3-テトラヒドロピラニル、4-テトラヒドロピラニル)、
チオピラニル(例、4-チオピラニル)、
テトラヒドロチオピラニル(例、2-テトラヒドロチオピラニル、3-テトラヒドロチオピラニル、4-テトラヒドロチオピラニル)、
1-オキシドテトラヒドロチオピラニル(例、1-オキシドテトラヒドロチオピラン-4-イル)、
1,1-ジオキシドテトラヒドロチオピラニル(例、1,1-ジオキシドテトラヒドロチオピラン-4-イル)、
テトラヒドロフリル(例、テトラヒドロフラン-3-イル、テトラヒドロフラン-2-イル)、
ピラゾリジニル(例、ピラゾリジン-1-イル、ピラゾリジン-3-イル)、
ピラゾリニル(例、ピラゾリン-1-イル)、
テトラヒドロピリミジニル(例、テトラヒドロピリミジン-1-イル)、
ジヒドロトリアゾリル(例、2,3-ジヒドロ-1H-1,2,3-トリアゾール-1-イル)、および
テトラヒドロトリアゾリル(例、2,3,4,5-テトラヒドロ-1H-1,2,3-トリアゾール-1-イル)、
ジヒドロオキサゼピニル等が挙げられる。
「アルキニル(基)」としては、例えば、C2-6アルキニル(基)が挙げられる。「C2-6アルキニル(基)」としては、例えば、エチニル、2-プロピニル、および2-ペンテン-4-イニルが挙げられる。
「アルキレン(鎖)」としては、例えば、C1-3アルキレン(鎖)が挙げられる。C1-3アルキレン(鎖)としては、例えば、メチレン、エチレン、およびトリメチレンが挙げられる。
「C1-6アルコキシ(基)」としては、例えば、メトキシ、エトキシ、n-プロピルオキシ、イソプロピルオキシ、n-ブチルオキシ、sec-ブチルオキシ、tert-ブチルオキシ、n-ペンチルオキシ、およびn-ヘキシルオキシ基等が挙げられる。
C1-6アルコキシ(基)は、すなわち、Ra-O-(Raは、C1-6アルキル(基))である。
「C1-6アルコキシ-カルボニル(基)」としては、例えば、メトキシカルボニル、エトキシカルボニル、n-プロピルオキシカルボニル、イソプロピルオキシカルボニル、n-ブチルオキシカルボニル、sec-ブチルオキシカルボニル、tert-ブチルオキシカルボニル、n-ペンチルオキシカルボニル、およびn-ヘキシルオキシカルボニル基等が挙げられる。
C1-6アルコキシ-カルボニル(基)は、すなわち、Ra-O-CO-(Raは、C1-6アルキル(基))である。
水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表す。
当該「アルキル基」としては、前記で例示したものが挙げられる。
当該「アルケニル基」としては、前記で例示したものが挙げられる。
当該「アルキニル基」としては、前記で例示したものが挙げられる。
なかでも、アルキル基が好ましく、C1-6アルキル基がより好ましい。
R1で表される「置換されていてもよい非環状炭化水素基」の「非環状炭化水素基」は、1個以上(好ましくは1~3個)の置換基で置換されていてもよい。
(1)ハロゲン原子、
(2)ニトロ基、
(3)シアノ基、
(4)Ra-CO-、
(5)Ra-CO-O-、
(6)Ra-CO-NRa-、
(7)Ra-O-、
(8)Ra-O-CO-、
(9)Ra-O-CO-NRa-、
(10)Ra-S-、
(11)Ra-SO-、
(12)Ra-SO2-、
(13)Ra-NRa-、
(14)Ra-S-NRa-、
(15)Ra-SO-NRa-、
(16)Ra-SO2-NRa-、
(17)Ra-NRa-、
(18)Ra-NRa-CO-、
(19)Ra-NRa-CO-NRa-、
(20)Ra-NRa-S-、
(21)Ra-NRa-SO-、
(22)Ra-NRa-SO2-、
(23)Ra-NRa-S-NRa-、
(24)Ra-NRa-SO-NRa-、
(25)Ra-NRa-SO2-NRa-、
(26)置換されていてもよい環状基、
(27)Ra-O-CO-C2H4-CO-NH-、および
(28)Ra-NRa-CS-NRa-L-
(これらの式中、各Raは、同一または異なって、水素原子、ハロゲン原子およびフェニル基から選択される1個以上(好ましくは、1~3個)の置換基で置換されていてもよいC1-6アルキル基を表す。)が挙げられる。
当該「ハロゲン原子」としては、前記で例示したものが挙げられる。
当該「C1-6アルキル基」としては、前記で例示したものが挙げられる。
当該「置換されていてもよい環状基」としては、後記で例示する、R1で表される「置換されていてもよい環状基」と同様のものが挙げられる。
前記置換基としては、なかでも、
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O―、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(これらの式中、各Raは、同一または異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
が好ましく、
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-CO-O―、
(g)Ra-CO-NRa-、
(h)Ra-NRa-、
(i)Ra-NRa-CS-NRa-、
(j)5~6員の環状基
(式中、各Raは、同一または異なって、1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表す。)
がより好ましい。
また、R1で表される「置換されていてもよい非環状炭化水素基」の「非環状炭化水素基」は、無置換であることもまた、好ましい。
当該「アリール基」としては、前記で例示したものが挙げられる。
当該「ヘテロアリール基」としては、前記で例示したものが挙げられる。
当該「非芳香族環状炭化水素基」としては、例えば、シクロアルキル基、シクロアルケニル基、およびシクロアルカジエニル基が挙げられる。
当該「シクロアルキル基」、「シクロアルケニル基」、および「シクロアルカジエニル基」としては、それぞれ前記で例示したものが挙げられる。
当該「非芳香族複素環基」としては、前記で例示したものが挙げられる。
前記環状基としては、C3-10シクロアルキル、C3-10シクロアルケニル、C4-10シクロアルカジエニル等の炭素数3~10の脂環式炭化水素基、または5~6員非芳香族複素環基が好ましい。
当該置換基としては、例えば、
(1)ハロゲン原子、
(2)ニトロ基、
(3)シアノ基、
(4)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基、
(5)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルケニル基、
(6)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキニル基、
(7)Ra-CO-L-、
(8)Ra-CO-O-L-、
(9)Ra-CO-NRa-L-、
(10)Ra-O-L-、
(11)Ra-O-CO-L-、
(12)Ra-O-CO-NRa-L-、
(13)Ra-S-L-、
(14)Ra-SO-L-、
(15)Ra-SO2-L-、
(16)Ra-NRa-L-、
(17)Ra-S-NRa-L-、
(18)Ra-SO-NRa-L-、
(19)Ra-SO2-NRa-L-、
(20)Ra-NRa-L-、
(21)Ra-NRa-CO-L-、
(22)Ra-NRa-CO-NRa-L-、
(23)Ra-NRa-S-L-、
(24)Ra-NRa-SO-L-、
(25)Ra-NRa-SO2-L-、
(26)Ra-NRa-S-NRa-L-、
(27)Ra-NRa-SO-NRa-L-、
(28)Ra-NRa-SO2-NRa-L-、
(29)Ra-O-CO-C2H4-CO-NH-、および
(30)Ra-NRa-CS-NRa-L-
(これらの式中、各Raは、同一または異なって、水素原子、または1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基を表し、Lは、結合手またはアルキレン鎖を表す。)が挙げられる。
前記非芳香族環状炭化水素基(特に、炭素数3~10の脂環式炭化水素基)および前記非芳香族複素環基(特に、5~6員非芳香族複素環基)の置換基としては、なかでも、
(a)オキソ基、
(b)C1-4アルコキシ-カルボニル基、
(c)ヒドロキシ基
が好ましく、
(a)オキソ基、
(b)C1-4アルコキシ-カルボニル基
がより好ましい。
また、前記アリール基(特に、フェニル基)の置換基としては、なかでも、
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、
(d)Rc-CO-NRc-
(これらの式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)が好ましい。
好ましくは、例えば、
(1)水素原子、
(2)C1-6アルキル基、
(3)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O―、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(式中、各Raは、同一または異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよいC1-6アルキル基、
(4)
(a)オキソ基、
(b)C1-4アルコキシ-カルボニル基、及び
(c)ヒドロキシ基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、又は
(5)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換されていてもよい芳香族炭化水素環基であり、
より好ましくは、例えば、
(1)C3-6アルキル基、
(2)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-CO-O-、
(g)Ra-CO-NRa-、
(h)Ra-NRa-、
(i)Ra-NRa-CS-NRa-、及び
(j)炭素数5~6の炭素環基
(式中、各Raは、同一または異なって、1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選ばれる1個以上の置換基で置換されたC1-6アルキル基、又は、
(3)
(a)オキソ基、及び
(b)C1-4アルコキシ-カルボニル基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、
(4)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換された芳香族炭化水素環基であり、
更に好ましくは、例えば、5~6員非芳香族複素環基である。
a)6員の環状基で置換されていてもよい炭素数1~6の非環状炭化水素基、または
b)
(1)ハロゲン原子、
(2)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルコキシ基、及び
(3)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基
から選択される1個以上の置換基で置換されていてもよい3~10員の環状基であり、
より好ましくは、例えば、
a)6員の環状基で置換されていてもよい炭素数1~6の非環状炭化水素基、または
b)ハロゲン原子、C1-4アルコキシ基、及びC1-4アルコキシ-カルボニル基から選択される1個以上(好ましくは、1~3個)の置換基で置換されていてもよい6~10員の環状基であり、更に好ましくは、例えば、
a)6員の環状基で置換されていてもよいC1-4アルキル基、または
b)1個以上(好ましくは、1~3個)のC1-4アルコキシ基で置換されていてもよい6員の環状基である。
(1)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基、
(2)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルケニル基、
(3)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキニル基、
(4)Ra-CO-L-、
(5)Ra-CO-O-L-、
(6)Ra-CO-NRa-L-、
(7)Ra-O-L-、
(8)Ra-O-CO-L-、
(9)Ra-O-CO-NRa-L-、
(10)Ra-S-L-、
(11)Ra-SO-L-、
(12)Ra-SO2-L-、
(13)Ra-NRa-L-、
(14)Ra-S-NRa-L-、
(15)Ra-SO-NRa-L-、
(16)Ra-SO2-NRa-L-、
(17)Ra-NRa-L-、
(18)Ra-NRa-CO-L-、
(19)Ra-NRa-CO-NRa-L-、
(20)Ra-NRa-S-L-、
(21)Ra-NRa-SO-L-、
(22)Ra-NRa-SO2-L-、
(23)Ra-NRa-S-NRa-L-、
(24)Ra-NRa-SO-NRa-L-、および
(25)Ra-NRa-SO2-NRa-L-
(式中、各Raは、同一または異なって、水素原子、または1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基を表し、Lは、結合手またはアルキレン鎖を表す。)が挙げられる。
当該「ハロゲン原子」、「C1-6アルキル基」、「C1-6アルケニル基」、「C1-6アルキニル基」、および「アルキレン鎖」としては、それぞれ前記で例示したものが挙げられる。
Xは、更に好ましくは、NH、O、又はSである。
(1)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基、
(2)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルケニル基、
(3)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキニル基、
(4)Ra-CO-L-、
(5)Ra-CO-O-L-、
(6)Ra-CO-NRa-L-、
(7)Ra-O-L-、
(8)Ra-O-CO-L-、
(9)Ra-O-CO-NRa-L-、
(10)Ra-S-L-、
(11)Ra-SO-L-、
(12)Ra-SO2-L-、
(13)Ra-NRa-L-、
(14)Ra-S-NRa-L-、
(15)Ra-SO-NRa-L-、
(16)Ra-SO2-NRa-L-、
(17)Ra-NRa-L-、
(18)Ra-NRa-CO-L-、
(19)Ra-NRa-CO-NRa-L-、
(20)Ra-NRa-S-L-、
(21)Ra-NRa-SO-L-、
(22)Ra-NRa-SO2-L-、
(23)Ra-NRa-S-NRa-L-、
(24)Ra-NRa-SO-NRa-L-、および
(25)Ra-NRa-SO2-NRa-L-
(式中、各Raは、同一または異なって、水素原子、または1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基を表し、Lは、結合手またはアルキレン鎖を表す。)が挙げられる。
当該「ハロゲン原子」、「C1-6アルキル基」、「C1-6アルケニル基」、「C1-6アルキニル基」、および「アルキレン鎖」としては、それぞれ前記で例示したものが挙げられる。
Yは、更に好ましくは、Nである。
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表す。
本発明の化合物において、好ましくは、Zは、C-R2である。
当該「アルキル基」としては、前記で例示したものが挙げられる。
当該「アルケニル基」としては、前記で例示したものが挙げられる。
当該「アルキニル基」としては、前記で例示したものが挙げられる。
(1)ハロゲン原子、
(2)ニトロ基、
(3)シアノ基、
(4)Rb-CO-、
(5)Rb-CO-O-、
(6)Rb-CO-NRb-、
(7)Rb-O-、
(8)Rb-O-CO-、
(9)Rb-O-CO-NRb-、
(10)Rb-S-、
(11)Rb-SO-、
(12)Rb-SO2-、
(13)Rb-NRb-、
(14)Rb-S-NRb-、
(15)Rb-SO-NRb-、
(16)Rb-SO2-NRb-、
(17)Rb-NRb-、
(18)Rb-NRb-CO-、
(19)Rb-NRb-CO-NRb-、
(20)Rb-NRb-S-、
(21)Rb-NRb-SO-、
(22)Rb-NRb-SO2-、
(23)Rb-NRb-S-NRb-、
(24)Rb-NRb-SO-NRb-、および
(25)Rb-NRb-SO2-NRb-
(これらの式中、各Rbは、同一または異なって、水素原子、ハロゲン原子およびフェニル基から選択される1個以上(好ましくは、1~3個)の置換基で置換されていてもよいC1-6アルキル基を表す。)が挙げられる。
当該「ハロゲン原子」としては、前記で例示したものが挙げられる。
当該「C1-6アルキル基」としては、前記で例示したものが挙げられる。
前記置換基としては、なかでも、例えば
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-CO-O-、
(f)Rb-CO-NRb-、
(g)Rb-NRb-、
(h)Rb-NRb-Rb-S(O)n-
(i)フェニル、
(j)5~6員飽和複素環基、及び
(k)ヒドロキシ、
(l)アミノ、
(これらの式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
が好ましく、
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、及び
(e)Rb-CO-NRb-、
(f)Rb-NRb-Rb-S(O)n-
(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
がより好ましい。
R2は、好ましくは、例えば
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-S-、
(f)Rb-SO2-、
(g)Rb-CO-O-、
(h)Rb-CO-NRb-、
(i)Rb-NRb-、
(j)Rb-NRb-Rb-S(O)n-
(k)フェニル基、
(l)5~6員飽和複素環基、
(m)ヒドロキシ基、及び
(n)アミノ基(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基でそれぞれ置換されていてもよいC1-6アルキル基、C2-6アルケニル基、又はC2-6アルキニル基、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基、又は
(4)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
フェニル基であり、
より好ましくは、
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、及び
(e)Rb-CO-NRb-、
(f)Rb-NRb-Rb-S(O)n-
(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよいC1-6アルキル基、又は、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基
である。
N、又は
C-R2
(R2は、
a)水素原子、
b)
(1)ハロゲン原子、
(2)Rb-O-、
(3)Rb-O-CO-、
(4)Rb-O-CO-NRb-、
(5)Rb-CO-、
(6)Rb-CO-O-、および
(7)Rb-NRb-
(これらの式中、各Rbは、同一または異なって、水素原子、ハロゲン原子およびフェニル基から選択される1個以上(好ましくは、1~3個)の置換基で置換されていてもよいC1-6アルキル基を表す。)
から選択される1個以上(好ましくは1~3個)の置換基で
置換されていてもよいC1-6アルキル基、又は
c)
(1)ハロゲン原子、
(2)Ra-O-(式中、RaはC1-4アルキル基を表す。)、
(3)Ra-S-L-(式中、Raは、水素原子、またはC1-3アルキル基を表し、Lは、結合手またはC1-2アルキレン鎖である。)、および
から選択される1個以上(好ましくは、1個)の置換基で置換されていてもよい
5~6員の環状基である。)
であり、より好ましくは、例えば、N、又はC-R2
(R2は、
a)水素原子、
b)
(1)ハロゲン原子、
(2)Rb-O-、
(3)Rb-CO-、
(4)Rb-CO-O-、および
(5)Rb-NRb-
(これらの式中、各Rbは、同一または異なって、水素原子、1個のフェニル基で置換されていてもよいC1-6アルキル基を表す。)
から選択される1個以上(好ましくは1~3個)の置換基で
置換されていてもよいC1-6アルキル基、又は
c)5~6員の環状基である。)
である。
当該置換基としては、R1で表される「置換されていてもよい環状基」について例示した置換基と同様のものが挙げられる。
なかでも、例えば
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)ハロゲン原子で置換されていてもよい、
(i)ニトロ基、
(j)アリールオキシ基
が好ましい。

当該置換基としては、R1で表される「置換されていてもよい環状基」について例示した置換基と同様のものが挙げられる。
なかでも、例えば
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)アミド基、
(i)アルコキシカルボニル基、
(j)ハロゲン原子で置換されていてもよいアルキル基、
(k)ニトロ基、
(l)アリールオキシ基
が好ましい。
環Bは、好ましくは、無置換のベンゼン環である。
(1)ハロゲン原子、
(2)ニトロ基、
(3)1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基
(4)Ra-O-CO-L-、
(5)Ra-SO2-NRa-L-、および
(6)Ra-NRa-L-
(式中、各Raは、同一または異なって、水素原子、または1個以上(好ましくは、1~3個)のハロゲン原子で置換されていてもよいC1-6アルキル基であり、Lは、結合手である。)
から選択された1個以上(好ましくは、1~3個)の置換基で置換されていてもよいベンゼン環である。
(1)ハロゲン原子(好ましくは、塩素)、
(2)ニトロ基、
(3)C1-6アルキル基(好ましくは、メチル)、および
(4)アミノ基
から選択された1個以上(好ましくは、1~3個)の置換基で置換されていてもよいベンゼン環である。

で示される化合物、その薬学的に許容される塩、又はその溶媒和物が除外される。
なお、化合物Aは新規化合物である。
(化合物A)
式

R1は、
(1)C3-6アルキル基、
(2)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O-、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(式中、各Raは、同一または相異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されたC1-6アルキル基、
(3)
(a)オキソ基、及び
(b)C1-4アルコキシ-カルボニル基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、又は
(4)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換された芳香族炭化水素環基を表し、
Xは、
NH、O、又はSを表し、
Yは、
CH、又はNを表し、
ZはN、又はC-R2を表し、
R2は、
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-S-、
(f)Rb-SO2-、
(g)Rb-CO-O-、
(h)Rb-CO-NRb-、
(i)Rb-NRb-、
(j)Rb-NRb-Rb-S(O)n-
(k)フェニル基、
(l)5~6員飽和複素環基、
(m)ヒドロキシ基、及び
(n)アミノ基、
(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基でそれぞれ置換されていてもよいC1-6アルキル基、C2-6アルケニル基、又はC2-6アルキニル基、又は、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基
を表し、
環Aは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)ハロゲン原子で置換されていてもよいアルキル基、
(i)ニトロ基、及び
(j)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環を表し、
環Bは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)アミド基、
(i)アルコキシカルボニル基、
(j)ハロゲン原子で置換されていてもよいアルキル基、
(k)ニトロ基、及び
(l)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、またはその溶媒和物。
Zは、
C-R2であり、及び
Yは、
Nである。
R1は、
5~6員非芳香族複素環基である。
R2は、
C1-6アルキル基である。
Zは、
C-R2であり、
Yは、
Nであり、
R1は、
5~6員非芳香族複素環基であり、及び
R2は、
C1-6アルキル基である。
R1は
a)6員の環状基で置換されていてもよい炭素数1~6の非環状炭化水素基、または
b)ハロゲン原子、1個以上のC1-4アルコキシ基、及びC1-4アルコキシ-カルボニル基から選択される1個以上の置換基で置換されていてもよい6~10員の環状基
であり、
Xが、N-R3(R3は、水素原子、又はRa-O-CO-(RaはC1-6アルキル基である。))、O、又はSであり、
Yが、CH、又はNであり、
Zが、N、又はC-R2
(式中、R2は、
a)水素原子、
b)
(1)ハロゲン原子、
(2)Rb-O-、
(3)Rb-CO-、
(4)Rb-CO-O-、および
(5)Rb-NRb-
(式中、各Rbは、同一または異なって、水素原子、1個のフェニル基で置換されていてもよいC1-6アルキル基を表す。)
から選択される1個以上(好ましくは1~3個)の置換基で
置換されていてもよいC1-6アルキル基、又は
c)5~6員の環状基
であり、
環Aが(無置換の)ベンゼン環であり、および
環Bがハロゲン原子、C1-6アルキル基、ニトロ基、およびアミノ基から選択される1個以上(好ましくは1~3個)の置換基で置換されていてもよいベンゼン環である
化合物である。
化合物(I)は、互変異性体を有する場合、安定構造をとる異性体が好ましいが、本発明は、これに限定されるものではなく、他の異性体を排除するものではなく、どちらも本発明の範囲内である。
また、化合物(I)が、光学異性体、立体異性体、位置異性体、回転異性体などの異性体を有する場合には、いずれか一方の異性体も混合物も本発明の範囲内である。本発明の化合物に包含される。さらに、化合物(I)に光学異性体が存在する場合には、ラセミ体から分割された光学異性体も本発明の範囲内である。
化合物(I)は、結晶であっても非結晶であってもよく、いずれも本発明の範囲内である。
同位元素(例、2H,3H,14C,35S,125Iなど)などで標識または置換された化合物(I)も、本発明の範囲内である。
化合物(I)は、溶媒和物(例えば、水和物など)であっても、無溶媒和物であってもよい。
特に記載の無い限り、反応式中の化合物の各記号は、上記と同様の意味を表す。なお、式中の化合物は、塩を形成している場合も含み、このような塩としては、例えば化合物(I)の塩と同様のものが挙げられる。また、各工程で得られた化合物は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、それ自体が公知の手段、例えば、抽出、濃縮、中和、濾過、蒸留、再結晶、蒸留、クロマトグラフィーなどの分離手段により容易に精製することができる。あるいは、式中の化合物が市販されている場合には市販品をそのまま用いることもできる。
特に記載の無い限り、本明細書中、室温は、約10℃~約35℃である。



カルボキシル基、
ハロゲン原子、
トリハロメチル基、または一般式

Xaは脱離基を表し、
X、Y、環Aおよび環Bは前記の通りである。)
で表される化合物(S-1)と、
一般式
H2N-R1
(R1は前記の通りである。)
で表される化合物(S-1’)およびその塩と
を反応させることにより、
一般式

で表される化合物(S-2)を製造することができる。
ハロゲン原子;
置換されていてもよいアルキルスルホニルオキシ基(例、メタンスルホニルオキシ基、エタンスルホニルオキシ基、およびトリフルオロメタンスルホニルオキシ等のハロゲン原子で置換されていてもよいアルキルスルホニルオキシ基);および 置換されてもよいアリールスルホニルオキシ基(例、ベンゼンスルホニルオキシ、p-トルエンスルホニルオキシ基、および2-ニトロベンゼンスルホニルオキシ基)等が挙げられ、特にハロゲン原子が好ましい。
化合物(S-1’)の使用量は、化合物(S-1)1モルに対し、通常1~2モル、好ましくは1~1.5モルである。
用いられる塩基としては、例えば、例えば、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、リン酸カリウム等の炭酸塩類;水酸化ナトリウム、水酸化カリウム等の水酸化物類;モルホリン、ピペリジン、ピリジン、トリメチルアミン、トリエチルアミン、ジイソプロピルエチルアミン、4-ジメチルアミノピリジンのような有機アミン等が挙げられる。その使用量は、化合物(S-1)1モルに対して、通常1~2モル、好ましくは1~1.5モルである。
溶媒としては、例えば、水;メタノール、およびエタノール等のアルコール系溶媒;テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシドおよびN-メチルピロリドン等の非プロトン性溶媒;クロロホルム等のハロゲン化炭化水素類、またはこれらの混合溶媒等が用いられ、先に記載の有機アミン類や化合物(S-1’)を溶媒と使用してもよい。


で表される化合物(S-2)を、接触還元法または金属還元試薬を用いることで、一般式

で表される化合物(S-3)を製造することができる。
金属触媒としては、例えばパラジウム-炭素、水酸化パラジウム-炭素、ロジウム-炭素、ラネーニッケル、酸化白金等が挙げられ、その使用量は化合物(S-2)に対してる。通常0.01%~100%重量、好ましくは0.1~50%重量である。
溶媒としては、例えば、メタノール、エタノール、2-プロパノールのようなアルコール系溶媒;テトラヒドロフラン等のエーテル系溶媒;酢酸エチル等のエステル系溶媒;N,N-ジメチルホルムアミド等の非プロトン性極性溶媒またはこれらの混合溶媒等が挙げられる。
金属還元試剤を用いる場合、約0℃~用いた溶媒の沸点までの範囲内の温度で、10分間~48時間反応させることにより行うことができる。
金属還元試剤としては、例えば塩化スズ(II)、還元鉄または三塩化チタン(III)等が挙げら、その使用量は化合物(S-2)1モルに対して、通常1~20モル、好ましくは1~5モルである。
溶媒としては、例えば水;希塩酸;酢酸;メタノール、エタノールおよび2-プロパノール等のアルコール系溶媒、テトラヒドロフラン、1,2-ジメトキシエタン等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、アセトン、アセトニトリルおよびN,N-ジメチルホルムアミド等の非プロトン性極性溶媒、またはこれらの混合溶媒等が挙げられる。

具体的には、例えば、
一般式

で表される化合物(S-3)と、
一般式
R2-COCl
(R2は前記の通りである。)
で表される酸クロライド化合物と
を反応させることで、
一般式

で表される化合物(S-4)を製造することができる
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
酸クロライド化合物の使用量は、化合物(S-3)1モルに対し、通常1~5モル、好ましくは1~2モルである。
溶媒としては、例えば、テトラヒドロフラン、1,2-ジメトキシエタン等のエーテル系溶媒、酢酸エチル等のエステル系溶媒、アセトン、アセトニトリルおよびN,N-ジメチルホルムアミド等の非プロトン性極性溶媒、またはこれらの混合溶媒等が挙げられる。
具体的には、例えば、
一般式

で表される化合物(S-3)と、
一般式
R2-COOH
(R2は前記の通りである。)
で表されるカルボン酸化合物と
を反応させることで、
一般式

で表される化合物(S-4)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
カルボン酸化合物の使用量は、化合物(S-3)1モルに対し、通常1~5モル、好ましくは1~2モルである。
脱水縮合剤として、無水酢酸、無水プロピオン酸等の低級脂肪酸無水物;メタンスルホン酸、パラトルエンスルホン酸等の有機スルホン酸;オキシ塩化リン、三塩化リン、五酸化二りん、硫酸、ポリリン酸、ホウ酸等の無機酸を用いてもよい。
溶媒としては、例えば、テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;またはこれらの混合溶媒等が用いられ、先に記載のカルボン酸を溶媒と使用してもよい。
具体的には、例えば、
一般式

で表される化合物(S-3)と、
一般式
R2-CHO
(R2は前記のとおりであり、R2’はアルキル鎖を表す。)
で表されるアルデヒド化合物と
を反応させることで、
一般式

で表される化合物(S-4)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
アルデヒド化合物の使用量は、化合物(S-3)1モルに対し、通常1~5モル、好ましくは1~2モルである。
酸化剤としては、例えばm-クロロ過安息香酸、2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン(DDQ)、オキソン、ベンゾフロキサン、過マンガン酸カリウム、過酸化水素、過酢酸、tert-ブチルハイドロパーオキサイド等があげられ、化合物(S-3)1モルに対し、通常0.1~5モル、好ましくは0.5~2モルである。
溶媒としては、例えば、水;メタノール、およびエタノール等のアルコール系溶媒;テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシドおよびN-メチルピロリドン等の非プロトン性溶媒;クロロホルム等のハロゲン化炭化水素類、またはこれらの混合溶媒等が用いられる。
具体的には、例えば、
一般式

で表される化合物(S-3)と、
一般式
R2-C(OR2’)3
(R2は前記のとおりであり、R2’はアルキル鎖を表す。)
で表されるトリアセタール化合物と
を反応させることで、一般式

で表される化合物(S-4)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
トリアセタール化合物の使用量は、化合物(S-3)1モルに対し、通常1~10モル、好ましくは1~5モルである。
反応系中に触媒量の酸を加えてもよい。用いられる酸の種類としては、塩酸、臭化水素酸、硝酸酸、硫酸等の無機酸類や、酢酸、トリフルオロ酢酸、メタンスルホン酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
溶媒としては、例えば、メタノール、およびエタノール等のアルコール系溶媒;テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシドおよびN-メチルピロリドン等の非プロトン性溶媒;クロロホルム等のハロゲン化炭化水素類、またはこれらの混合溶媒等が用いられる。先に記載のトリアセタール化合物を溶媒と使用してもよい。

で表される化合物(S-3)と一般式
R2-C(=NH)OR2’
(R2およびR2’は前記の通りである。)
で表されるイミデート化合物と反応させることで、
一般式

で表される化合物(S-4)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
イミデート化合物の使用量は、化合物(S-3)1モルに対し、通常1~5モル、好ましくは1~2モルである。
溶媒としては、例えば、メタノール、およびエタノール等のアルコール系溶媒;テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシドおよびN-メチルピロリドン等の非プロトン性溶媒;クロロホルム等のハロゲン化炭化水素類、またはこれらの混合溶媒等が用いられる。

直接環化は、例えば、Zhurnal Obshchei Khimii 1962, 32(5), 1581-86 (Engl. Transl. Ver.: pp 1565-1569に記載に記載の方法に準じ、上記の、カルボン酸を用いた場合の環化1と同様な方法で製造することができる。
具体的には、例えば、
一般式

で表される酸化合物(S-5)と
一般式

で表される化合物(S-5’)と
を反応させることで、
一般式

で表される化合物(S-6)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
化合物(S-5’)の使用量は、化合物(S-5)モルに対し、通常1~5モル、好ましくは1~2モルである。
脱水縮合剤として、無水酢酸、無水プロピオン酸等の低級脂肪酸無水物;メタンスルホン酸、パラトルエンスルホン酸等の有機スルホン酸;オキシ塩化リン、三塩化リン、五酸化二りん、硫酸、ポリリン酸、ホウ酸等の無機酸を用いてもよい
溶媒としては、例えば、テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;またはこれらの混合溶媒等が用いられ、先に記載の脱水縮合剤を溶媒と使用してもよい。
Bioorganic & Medicinal Chemistry 2004, 12(1), 17-21に記載の方法に準じて行うことができる。
具体的には、例えば、一般式

で表される化合物(S-5)と
一般式

で表される化合物(S-5’)とから、
一般式

で表される化合物(S-6’)を合成し、これを変換することで、
一般式

で表される化合物(S-6)を製造することができる。
縮合剤を用いる場合、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
化合物(S-5’)の使用量は、化合物(S-5)1モルに対し、通常1~2モル、好ましくは1~1.5モルである。
縮合剤としては、例えば、Bop(1H-1,2,3-ベンゾトリアゾール-1-イルオキシ(トリ(ジメチルアミノ))ホスホニウム ヘキサフルオロホスフェート)、WSC(1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド・塩酸塩)、DCC(N,N-ジシクロヘキシルカルボジイミド)、CDI(カルボニルジイミダゾール)、ジエチルホスホリルシアニド等が挙げられる。縮合剤の使用量は、化合物(S-5)に対して1モルに対し、通常1~2モル、好ましくは1~1.5モルである。
また必要に応じて化合物(S-5)に対して1当量から過剰量の有機塩基、例えばトリエチルアミン等を加えてもよい。
溶媒として、例えば、ジクロロメタン、クロロホルム等のハロゲン化炭化水素類、例えば、ジメチルスルホキシド等のスルホキシド類、例えば、酢酸エチル等のエステル類、例えば、テトラヒドロフラン、1,4-ジオキサン等のエーテル類、例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類等が挙げられる。
酸ハロゲン化剤を用いる場合、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
化合物(S-5’)の使用量は、化合物(S-5)1モルに対し、通常1~2モル、好ましくは1~1.5モルである。
酸ハロゲン化剤としては、例えば、チオニルクロリド、オキザリルクロリド、三塩化リン、五塩化リン等が挙げられる。酸ハロゲン化剤の使用量は、化合物(S-5)に対して1モルに対し、通常1~10モル、好ましくは1~1.5モルである。
反応系中に触媒量のN,N-ジメチルホルムアミドを加えてもよい。
溶媒として、例えば、ジクロロメタン、クロロホルム等のハロゲン化炭化水素類;例えば、ジメチルスルホキシド等のスルホキシド類;例えば、酢酸エチル等のエステル類、例えば、テトラヒドロフラン、1,4-ジオキサン等のエーテル類;例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類;べンゼン、トルエン等の芳香族炭化水素類等が挙げられる。
反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約48時間である。
脱水縮合剤として、無水酢酸、無水プロピオン酸等の低級脂肪酸無水物;メタンスルホン酸、パラトルエンスルホン酸等の有機スルホン酸;オキシ塩化リン、三塩化リン、五酸化二りん、硫酸、ポリリン酸、ホウ酸等の無機酸を用いてもよい
溶媒としては、例えば、テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;またはこれらの混合溶媒等が用いられ、先に記載の脱水縮合剤を溶媒と使用してもよい。

具体的には、例えば、一般式

で表される化合物(S-3)をニトロソ化剤と
を反応させることで、
一般式

で表される化合物(S-7)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
ニトロソ化剤としては、亜硝酸ナトリウムといったアルカリ金属亜硝酸塩類、亜硝酸メチルエステルあるいは亜硝酸イソアミルといった有機亜硝酸化合物類が上げられ、その使用量は、化合物(S-3)1モルに対し、通常1~5モル、好ましくは1~2モルである。
溶媒としては、水と酸の組み合わせが用いられる。酸としては、塩酸や硫酸といった無機酸類や酢酸といった有機酸類が挙げられる。

具体的には、例えば、一般式

R1、R2および環Aは前記の通りである。)
で表される化合物(S-8)と
一般式

で表される化合物(S-5’)と
を反応させることで、
一般式

で表される化合物(S-6)を製造することができる。
本反応において、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
化合物(S-5’)の使用量は、化合物(S-8)1モルに対し、通常1~2モル、好ましくは1~1.5モルである。
脱水縮合剤として、無水酢酸、無水プロピオン酸等の低級脂肪酸無水物;メタンスルホン酸、パラトルエンスルホン酸等の有機スルホン酸;オキシ塩化リン、三塩化リン、五酸化二りん、硫酸、ポリリン酸、ホウ酸等の無機酸を用いてもよい
溶媒としては、例えば、テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;またはこれらの混合溶媒等が用いられ、先に記載の脱水縮合剤を溶媒と使用してもよい。

具体的には、例えば、一般式

で表される化合物(S-9)と
一般式
X、および環Bは前記の通りである。)
で表される化合物(S-10)と
を反応させることで、
一般式

で示される化合物(S-11)を製造することができる。
反応は、遷移金属触媒の存在下、また、場合により配位子、塩基、添加剤等の存在下、約20℃~用いた溶媒の沸点までの範囲内の温度で、10分間~48時間反応させることにより行うことができる。
化合物(S-10)の使用量は、化合物(S-9)1モルに対し、通常1~20モル、好ましくは1~5モルである。
遷移金属触媒としては、例えば酢酸パラジウム(II)、塩化パラジウム(II)、テトラキス(トリフェニルホスフィン)パラジウム(0)、ビス(トリフェニルホスフィン)パラジウムクロリド(II)、トリス(ジベンジリデンアセトン)ジパラジウム(0)、または[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)などが挙げられる。遷移金属触媒の使用量は、化合物(S-10)1モルに対し、通常0.0001~1モル、好ましくは0.001~1モルである。
配位子としては、例えばトリフェニルホスフィン、トリ-o-トリルホスフィン、トリ-tert-ブチルホスフィン、トリ-2-フリルホスフィン、トリ-シクロヘキシルホスフィン、トリフェニルアルシン、1,1'-ビス(ジフェニルホスフィノ)フェロセン(dppf)等が挙げられる。配位子の使用量は、化合物(S-9)1モルに対し、通常0.0001~4モル、好ましくは0.001~4モルである
塩基としては、例えばトリエチルアミン、ジイソプロピルエチルアミン等の有機塩基、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシウム、リン酸カリウム等の無機塩基等が挙げられる。塩基の使用量は、化合物(S-9)1モルに対し、通常1~10モル、好ましくは1~4モルである
添加剤としては、例えば塩化リチウム、フッ化セシウム、ヨウ化銅(I)、臭化銅(I)等の無機塩が挙げられる。
溶媒としては、例えば水、アセトニトリルや、クロロホルムまたはジクロロメタン等の溶媒としては、例えば、水;メタノール、およびエタノール等のアルコール系溶媒;テトラヒドロフラン、およびジオキサン等のエーテル系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;アセトニトリル;ジメチルホルムアミド;ジメチルスルホキシドまたはN-メチルピロリドン等の非プロトン性溶媒;クロロホルム等のハロゲン化炭化水素類、またはこれらの混合溶媒等が挙げられる。

一般式

R1、R2、X、Y、環Aおよび環Bは前記の通りである。)
で表される化合物(S-12)から、
一般式

で表される化合物(S-12’)を得て、さらにこれを変換することで、
一般式

R1、X、Y、環Aおよび環Bは前記の通りである。)
で表される化合物(S-12’’)を製造することができる。
R2aがアルキルカルボニルオキシメチル基の場合、反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約24時間である。
用いられるアルカリの種類は、水酸化リチウム、水酸化ナトリウム等の水酸化物が挙げられ、その使用量は、化合物(S-12)1モルに対し、通常1~10モル、好ましくは1~3モルである。
溶媒として、例えば、水と、メタノール、エタノールといったアルコール溶媒類;例えば、ジメチルスルホキシド等のスルホキシド類;例えば、テトラヒドロフラン、1,4-ジオキサン等のエーテル類、例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類等の混合溶媒等が用いられる。
R2aがベンジルオキシメチル基の場合、1~5気圧の水素雰囲気下、または、場合により水素の代わりにギ酸アンモニウムを用い、金属触媒の存在下、約0℃~用いた溶媒の沸点までの範囲内の温度で、10分間~48時間反応させることにより行うことができる。
金属触媒としては、例えばパラジウム-炭素、水酸化パラジウム-炭素、ロジウム-炭素、ラネーニッケル、酸化白金等が挙げられ、その使用量は化合物(S-12)に対してる。通常0.01%~100%重量、好ましくは0.1~10%重量である。
溶媒としては、例えば、メタノール、エタノール、2-プロパノールのようなアルコール系溶媒;テトラヒドロフラン等のエーテル系溶媒;酢酸エチル等のエステル系溶媒;N,N-ジメチルホルムアミド等の非プロトン性極性溶媒またはこれらの混合溶媒等が挙げられる。
反応温度の範囲は、通常室温から還流温度であり、反応時間の範囲は、通常瞬時~約48時間である。
化合物(S-12’)の使用量は、カルボン酸化合物1モルに対し、通常1~2モル、好ましくは1~1.5モルである。
酸ハロゲン化剤としては、例えば、チオニルクロリド、オキザリルクロリド、三塩化リン、五塩化リン等が挙げられる。酸ハロゲン化剤の使用量は、化合物(S-12’)に対して1モルに対し、通常1~10モル、好ましくは1~1.5モルである。
反応系中に触媒量のN,N-ジメチルホルムアミドを加えてもよい。
溶媒として、例えば、ジクロロメタン、クロロホルム等のハロゲン化炭化水素類;例えば、ジメチルスルホキシド等のスルホキシド類;例えば、酢酸エチル等のエステル類、例えば、テトラヒドロフラン、1,4-ジオキサン等のエーテル類;例えば、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類;べンゼン、トルエン等の芳香族炭化水素類等が挙げられる。
また、化合物(I)が遊離体として得られた場合には、自体公知の方法あるいはそれに準ずる方法によって、目的とする、上記のような塩に変換することができ、逆に化合物(I)の塩として得られた場合には、自体公知の方法あるいはそれに準ずる方法により、遊離体または目的とする他の塩に変換することができる。
例えば、精製された化合物(I)を、例えば、塩酸、臭化水素酸、リン酸、硫酸等の無機酸類や、酢酸、クエン酸、コハク酸、酒石酸、フマル酸、マレイン酸、メタンスルホン酸、p-トルエンスルホン酸等の有機酸類と反応させることにより、薬剤として許容し得る酸付加塩を容易に得ることが出来る。
また、精製された化合物(I)を、例えば水酸化リチウム、リチウムメチラート、水素化ナトリウム、炭酸ナトリウム、水酸化ナトリウム、ナトリウムメチラート、水酸化カリウム、炭酸カリウム、水酸化カリウム、カリウム-t-ブチラートのような無機・有機金属塩と
を反応させることで、薬剤として許容し得る金属塩を容易に得ることが出来る。
従って、本発明の化合物は、神経栄養因子の活性が関与する疾患の治療または予防剤として使用できる。
また、本発明の化合物は、身体機能の回復のための理学療法効果の促進剤として使用できる。
本明細書中、疾患は障害およびその症状を含む意味で用いられる。
また、本明細書中、治療は症状の緩和を含む意味で用いられる。
神経変性疾患(例、アルツハイマー病、パーキンソン病、ハンチントン舞踏病、ダウン症候群など)、脳虚血性疾患(脳卒中、脳梗塞、一過性脳虚血発作、クモ膜下出血、虚血性脳症、脳梗塞(ラクナ梗塞、アテローム血栓性脳梗塞、心原性脳梗塞症、出血性脳梗塞、その他の梗塞)など)、外傷性脳損傷、白質脳症、および多発性硬化症が挙げられる。
筋萎縮性側索硬化症(ALS)、脊髄損傷、種々の原因による脊髄障害、脊髄性進行性筋萎縮症、および脊髄性脳変性症(SCD)が挙げられる。
加齢黄斑変性症(AMD)、糖尿病性網膜症、網膜色素変性症、高血圧性網膜症、および緑内症が挙げられる。
糖尿病性神経障害、末梢神経損傷、外傷性末梢神経障害、中毒、他の毒性物質による末梢神経障害、がん化学治療法による末梢神経障害、Guillain-Barre 症候群、ビタミン等の欠乏による末梢神経障害、アミロイド末梢神経障害、虚血性末梢神経障害、悪性腫瘍に伴う末梢神経障害、尿毒症性末梢神経障害、物理的原因による末梢神経障害、Charcot-Marie-Tooth病、アルコール性末梢神経障害、自律神経異常(無自覚性低血糖、胃不全麻痺、神経因性下痢および便秘、勃起不全、起立性低血圧、不整脈、心不全、無痛性心筋梗塞、発汗異常、神経因性膀胱など)、膀胱機能障害(例、無抑制膀胱、反射性膀胱、自律性膀胱、知覚麻痺性膀胱、運動麻痺性膀胱など)が挙げられる。
うつ病、統合失調症、てんかん、自閉症、歯周病、糖尿病、糖尿病性心筋症、糖尿病性足病変、炎症性腸疾患(例、潰瘍性大腸炎、クローン病など)、認知症に伴う問題行動(例、徘徊、攻撃的行為など)、不安症、疼痛、聴覚障害、骨疾患(例、骨粗しょう症など)、関節疾患(例、シャルコー関節、変形性関節症、リウマチなど)、ヒルシュスブルング病が挙げられる。
本発明の組成物は、その形態に応じて、公知の方法により、本発明の化合物と、薬学的に許容される担体、賦形剤、および/または添加剤(例、医薬添加剤、食品添加剤、化粧品添加剤)等とを混合することにより、製造することができる。
本発明の組成物に用いられる、薬学的に許容される担体、賦形剤、および/または添加剤等は、前記組成物の具体的用途に応じて適宜選択することができる。また、当該組成物の形態も、具体的用途に応じて、例えば、種々の固体、液体等の形態とすることができる。
例えば、本発明の医薬組成物または本発明の化合物を医薬品として用いる場合には、具体的な形態として、例えば、散剤、細粒剤、顆粒剤、錠剤、シロップ剤、カプセル剤、懸濁化剤、エマルジョン剤、エキス剤および丸剤等の経口剤、注射剤、外用液剤、軟膏剤等の経皮吸収剤(皮膚外用剤)、坐剤および局所等への非経口剤等をあげることができる。
経口剤は、例えば、ゼラチン、アルギン酸ナトリウム、澱粉、コーンスターチ、白糖、乳糖、ぶどう糖、マンニット、カルボキシメチルセルロース、デキストリン、ポリビニルピロリドン、結晶セルロース、大豆レシチン、ショ糖、脂肪酸エステル、タルク、ステアリン酸マグネシウム、ポリエチレングリコール、ケイ酸マグネシウム、無水ケイ酸等の担体や賦形剤、結合剤、崩壊剤、界面活性剤、滑沢剤、流動性促進剤、希釈剤、保存剤、着色剤、香料、安定化剤、保湿剤、防腐剤、酸化防止剤等の医薬品添加剤を用いて、通常の方法に従って製造することができる。
投与量は、投与される哺乳動物の年令、性別、体重、疾患の程度、本発明の化合物の種類、投与形態等によって異なるが、通常は経口の場合にはヒト成人で1日あたり有効成分量として約1mg~約2g、好ましくは有効成分量として約5mg~約1gを投与すればよい。また、前記の1日の投与量を1回または数回に分けて投与することができる。
投与量は、投与される哺乳動物の年令、性別、体重、疾患の程度、本発明の組成物または本発明の化合物の種類、投与形態等によって異なるが、通常は注射の場合にはヒト成人で有効成分量として約0.1mg~約500mgを投与すればよい。また、前記の1日の投与量を1回または数回に分けて投与することができる。
5-(ベンゾチアゾール-2-イル)-2-メチル-1-フェニルベンゾイミダゾールの合成
合成例1-1
3-ニトロ-4-フェニルアミノ安息香酸の合成

1H-NMR (DMSO-d6)δ(ppm): 7.13 (d, 1 H, J= 9.0 Hz), 7.30 (t, 1 H, J= 7.3 Hz), 7.37 (d, 1 H, J= 7.5 Hz), 7.48 (t, 2 H, J= 7.4 Hz), 7.93 (dd, 1 H, J= 2.0, 9.0 Hz), 8.64 (d, 1 H, J= 2.1 Hz), 9.80 (s, 1 H), 13.00 (brs, 1 H).
3-アミノ-4-フェニルアミノ安息香酸の合成

1H-NMR (DMSO-d6)δ(ppm): :4.98 (brs, 2 H), 6.83 (dt, 1 H, J= 7.4, 1.0 Hz), 6.97 (dd, 2 H, J= 7.6, 1.0 Hz), 7.06 (d, 1 H, J= 8.3 Hz), 7.15 (dd, 1 H, J= 8.2, 1.9 Hz ), 7.23 (t, 2 H, J= 7.4 Hz), 7.33(d, 1 H, J= 1.9Hz), 7.35 (s, 1 H), 12.25 (brs, 1 H).
2-メチル-1-フェニルベンゾイミダゾール-5-カルボン酸の合成

1H-NMR (DMSO-d6)δ(ppm): :2.47 (s, 3 H), 7.18 (d, 1 H, J= 8.4 Hz), 7.58 (d, 2 H, J= 7.0 Hz), 7.59 (dt, 1 H, J= 9.1, 1.5 Hz), 7.67 (t, 2 H, J= 7.7 Hz), 7.83 (dd, 1 H, J= 8.5, 1.4 Hz), 8.21 (d, 1 H, J= 1.0 Hz), 12.75 (brs, 1 H).
5-(ベンゾチアゾール-2-イル)-2-メチル-1-フェニルベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): :2.55 (s, 3 H), 7.21 (d, 1 H, J= 8.4 Hz), 7.37 (dt, 1 H, J= 7.1, 1.1 Hz), 7.40-7.43 (m, 2 H), 7.48 (dt, 1 H, J= 8.3, 1.2 Hz), 7.53-7.58 (m, 1 H), 7.58-7.67 (m, 2 H), 7.90 (d, 1 H, J= 7.9 Hz), 8.06 (dd, 1 H, J= 8.5, 1.6 Hz), 8.07 (d, 1 H, J= 8.0 Hz), 8.41 (d, 1 H, J= 1.3 Hz).
5-(ベンゾイミダゾール-2-イル)-2-メチル-1-フェニルベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 2.50 (s, 3 H), 7.15-7.23 (m, 2 H), 7.27 (d, 1 H, J= 8.5 Hz), 7.58-7.65 (m, 4 H), 7.65-7.73 (m, 3 H), 8.09 (dd, 1 H, J= 8.5, 1.5 Hz), 8.44 (d, 1 H, J= 0.9 Hz), 12.87 (brs, 1 H).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-フェニルベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 2.56 (s, 3 H), 7.24 (d, 1 H, J= 8.4 Hz), 7.32-7.38 (m, 2 H), 7.39-7.44 (m, 2 H), 7.57-7.65 (m, 4 H), 7.75-7.79 (m, 1 H), 8.17 (dd, 1 H, J= 8.5, 1.6 Hz), 8.63 (d, 1 H, J= 1.1 Hz).
2-アミノ-1-(テトラヒドロピラン-4-イル)アミノ-4-トリフルオロメチルベンゼンの合成
合成例4-1
1-(テトラヒドロピラン-4-イル)アミノ-2-ニトロ-4-トリフルオロメチルベンゼンの合成

1H-NMR (CDCl3)δ(ppm): 1.66-1.76 (m, 2 H), 2.06-2.10 (m, 2 H), 3.56-3.62 (m, 2 H), 3.77-3.80 (m, 1 H), 4.02-4.07 (m, 2 H), 6.97 (d, 1 H, J= 9.1 Hz), 7.61 (dd, 1 H, J= 9.1, 2.3 Hz), 8.33 (brd, 1 H, J= 6.3 Hz), 8.49 (d, 1 H, J= 1.2 Hz).
2-アミノ-1-(テトラヒドロピラン-4-イル)アミノ-4-トリフルオロメチルベンゼンの合成

1H-NMR (CDCl3)δ(ppm): 1.53-1.57 (m, 2 H), 2.04-2.07 (m, 2 H), 3.35 (brs, 2 H), 3.51-3.58 (m, 3 H), 3.65 (brs, 1 H), 4.00-4.05 (m, 2 H), 6.65 (d, 1 H, J= 8.3 Hz), 6.95 (d, 1 H, J= 1.9 Hz), 7.07 (dd, 1 H, J= 8.3, 1.1 Hz).
5-(ベンゾオキサゾール-2-イル)- 1,2-ジメチルベンゾイミダゾールの合成
合成例5-1
(2-メチルアミノアニリ-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.95 (3H, s), 6.71 (1H, d, J = 8.2 Hz), 7.24-7.34 (3H, m), 7.50-7.53 (1H, m), 7.63 (1H, d, J = 1.8 Hz), 7.69-7.72 (1H, m), 7.80 (1H, dd, J = 8.2, 1.8 Hz).
5-(ベンゾオキサゾール-2-イル)- 1,2-ジメチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.66 (3H, s), 3.78 (3H, s), 7.33-7.35 (2H, m), 7.41 (1H, d, J = 8.6 Hz), 7.59-7.62 (1H, m), 7.74-7.80 (1H, m), 8.22 (1H, dd, J = 8.6, 1.4 Hz), 8.56 (1H, d, J = 1.4 Hz).

1H-NMR (DMSO-D6) δ: 2.36 (3H, s), 2.83 (3H, s), 7.42-7.51 (2H, m), 7.83-7.86 (2H, m), 7.98 (1H, d, J = 8.6 Hz), 8.33 (1H, d, J = 8.6 Hz), 8.52 (1H, s).
5-(ベンゾオキサゾール-2-イル)- 1-エチル-2-メチルベンゾイミダゾールの合成
2-(4-エチルアミノ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.43 (3H, t, J = 7.3 Hz), 3.45 (2H, ddd, J = 14.3, 7.2, 5.2 Hz), 6.99 (1H, d, J = 9.1 Hz), 7.32-7.36 (2H, m), 7.55-7.58 (1H, m), 7.71-7.74 (1H, m), 8.27-8.31 (2H, m), 9.05 (1H, d, J = 2.1 Hz).
2-(2-エチルアミノアニリン-5-イル)ベンゾオキサゾール

1H-NMR (CDCl3) δ: 1.35 (3H, t, J = 7.2 Hz), 3.26 (2H, q, J = 7.1 Hz), 6.71 (1H, d, J = 8.4 Hz), 7.24-7.33 (2H, m), 7.50-7.79 (4H, m).
1H-NMR (CDCl3) δ: 1.45 (3H, t, J = 7.3 Hz), 2.65 (3H, s), 4.20 (2H, q, J = 7.3 Hz), 7.26-7.43 (3H, m), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 8.21 (1H, dd, J = 8.6, 1.5 Hz), 8.56 (1H, s).
5-(ベンゾチアゾール-2-イル)-1-フェニル-2-(フェニルメトキシ)メチルベンゾイミダゾールの合成
実施例1-1
1-フェニル-2-(フェニルメトキシ)メチルベンゾイミダゾール-5-カルボン酸の合成

1H-NMR (DMSO-d6)δ(ppm): 4.46 (s, 2 H), 4.71 (s, 2 H), 7.14 (dd, 2 H, J= 7.9, 1.5 Hz), 7.25-7.32 (m, 4 H), 7.58-7.68 (m, 5 H), 7.91 (dd, 1 H, J= 8.6, 1.5 Hz), 8.33 (d, 1 H, J= 1.5 Hz), 12.90 (brs, 1 H).
実施例1-2
5-(ベンゾチアゾール-2-イル)-1-フェニル-2-(フェニルメトキシ)メチルベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 4.48 (s, 2 H), 4.72 (s, 2 H), 7.16 (dd, 2 H, J= 7.9, 1.9 Hz), 7.27-7.33 (m, 3 H), 7.38 (d, 1 H, J= 8.5 Hz), 7.46 (dt, 1 H, J= 8.1, 1.1 Hz), 7.55 (dt, 1 H, 8.3, 1.1 Hz), 7.60-7.70 (m, 5 H), 8.06 (d, 1 H, J= 7.8 Hz), 8.08 (dd, 1 H, J= 8.5, 1.7 Hz), 8.14 (d, 1 H, J= 7.5 Hz), 8.44 (d, 1 H, J= 1.3 Hz).
5-(ベンゾチアゾール-2-イル)-2-ヒドロキシメチルー1-フェニルベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 4.73-4.77 (m, 2 H), 7.49 (d, 1 H, J= 8.6 Hz), 7.49 (t, 1 H, J= 7.3 Hz), 7.58 (t, 1 H, J= 7.2 Hz), 7.64-7.77 (m, 5 H), 8.10 (d, 1 H, J= 8.2 Hz), 8.15 (dd, 1 H, J= 8.6, 1.6 Hz), 8.17 (d, 1 H, J= 8.1 Hz), 8.45-8.50 (m, 1 H).
5-(ベンゾチアゾール-2-イル)-2-(N,N-ジメチルアミノ)メチル-1-フェニルベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 2.31 (s, 6 H), 3.59 (s, 2 H), 7.28 (d, 1 H, J= 8.6 Hz), 7.37 (dt, 1 H, J= 7.6, 1.1 Hz), 7.49 (dt, 1 H, J= 7.7, 1.1 Hz), 7.50-7.66 (m, 5 H), 7.90 (d, 1 H, J= 7.9 Hz), 8.06 (d, 1 H, J= 8.1 Hz), 8.11 (d, 1 H, J= 8.5 Hz), 8.48 (s, 1 H).
5-(ベンゾチアゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例4-1
4-((テトラヒドロピラン-4-イル)アミノ)-3-ニトロ安息香酸の合成

1H-NMR (DMSO-D6) δ(ppm): 1.55-1.70(2H, m), 1.95 (2H, d, J = 10.9 Hz), 3.44-3.53 (2H, m), 3.84-4.01 (3H, m), 7.27 (1H, d, J = 9.2 Hz), 7.96 (1H, dd, J = 9.2, 2.0 Hz), 8.21 (1H, d, J = 7.7 Hz), 8.62 (1H, d, J = 2.0 Hz).
実施例4-2
3-アミノ-4-((テトラヒドロピラン-4-イル)アミノ)安息香酸の合成

1H-NMR (DMSO-D6) δ(ppm): 1.36-1.51 (2H, m), 1.87-1.95 (2H, m), 3.38-3.48 (2H, m), 3.85-3.92 (2H, m), 4.92 (1H, d, J = 6.1 Hz), 6.52 (1H, d, J = 8.9 Hz), 7.16-7.20 (2H, m).
実施例4-3
2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール-5-カルボン酸の合成

1H-NMR (DMSO-D6) δ(ppm): 1.95-2.02 (2H, m), 2.49-2.52 (2H, m), 2.96 (3H, s), 3.53-3.64 (2H, m), 4.06 (2H, dd, J = 11.6, 3.9 Hz), 4.82-4.91 (1H, m), 8.04 (1H, dd, J = 8.8, 1.3 Hz), 8.15 (1H, d, J = 8.8 Hz), 8.27 (1H, d, J = 1.3 Hz).
実施例4-4
5-(ベンゾチアゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 1.87-1.91 (m, 2 H), 2.55-2.65 (m, 2 H), 2.70 (s, 3 H), 3.57-3.64 (m, 2 H), 4.21-4.25 (m, 2 H), 4.40-4.50 (m, 1 H), 7.37 (dt, 1 H, J= 7.2, 1.1 Hz), 7.48 (dt, 1 H, J= 8.3, 1.1 Hz), 7.63 (d, 1 H, J= 8.6 Hz), 7.91 (d, 1 H, J= 8.0 Hz), 8.07 (d, 1 H, J= 8.6 Hz), 8.08 (d, 1 H, J= 8.6 Hz), 8.35 (d, 1 H, J= 1.8 Hz).
5-(ベンゾチアゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール・メタンスルホン酸塩の合成

1H-NMR (DMSO-D6) δ(ppm): 1.99-2.07(2H, m), 2.36 (3H, s), 2.39-2.52 (2H, m), 2.95 (3H, s), 3.61 (2H, t, J = 11.4 Hz), 4.09 (2H, dd, J = 11.4 3.8 Hz), 4.85-4.96 (1H, m), 7.48-7.63 (2H, m), 8.12 (1H, d, J = 7.4 Hz), 8.18-8.24 (3H, m), 8.46 (1H, d, J = 1.2 Hz).
5-(ベンゾチアゾール-2-イル)-2-メトキシメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例6-1
2-メトキシメチル-1-(テトラヒドロピラン-4-イル)-5-トリフルオロメチルベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 1.89-1.92 (m, 2 H), 2.37-2.50 (m, 2 H), 3.40 (s, 3 H), 3.50-3.58 (m, 2 H), 4.04-4.08 (m, 2 H), 4.75-4.85 (m, 1 H), 4.95 (s, 2 H), 7.68 (dd, 1 H, J= 8.7, 1.5 Hz), 8.07 (s, 1 H), 8.12 (d, 1 H, J= 8.7 Hz).
実施例6-2
5-(ベンゾチアゾール-2-イル)-2-メトキシメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 1.90-1.94 (m, 2 H), 2.58-2.70 (m, 2 H), 3.41 (s, 3 H), 3.58-3.65 (m, 2 H), 4.20-4.24 (m, 2 H), 4.69-4.78 (m, 1 H), 4.81 (s, 2 H), 7.38 (dt, 1 H, J= 8.1, 1.1 Hz), 7.49 (dt, 1 H, J= 7.5, 1.2 Hz), 7.72 (d, 1 H, J= 8.8 Hz), 7.90 (d, 1 H, J= 8.0 Hz), 8.08 (d, 1 H, J= 7.7 Hz), 8.14 (dd, 1 H, J= 8.7, 1.7 Hz), 8.42 (d, 1 H, J= 1.4 Hz).
2-アセトキシメチル-5-(ベンゾチアゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例7-1
2-アセトキシメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール-5-カルボン酸の合成

1H-NMR (DMSO-d6)δ(ppm): 1.89-1.92 (m, 2 H), 2.13 (s, 3 H), 2.38-2.46 (m, 2 H), 3.53-3.59 (m, 2 H), 4.03-4.07 (m, 2 H), 4.70-4.82 (m, 1 H), 5.52 (s, 2 H), 7.94-7.96 (m, 2 H), 8.26 (s, 1 H).
実施例7-2
2-アセトキシメチル-5-(ベンゾチアゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 1.90-1.94 (m, 2 H), 2.15 (s, 3 H), 2.59-2.70 (m, 2 H), 3.56-3.62 (m, 2 H), 4.21-4.30 (m, 2 H), 4.52-4.62 (m, 1 H), 5.45 (s, 2 H), 7.38 (dt, 1 H, J= 7.6, 1.1 Hz), 7.49 (dt, 1 H, J= 7.7, 1.2 Hz), 7.72 (d, 1 H, J= 8.7 Hz), 7.90 (d, 1 H, J= 8.0 Hz), 8.07 (d, 1 H, J= 8.1 Hz), 8.16 (dd, 1 H, J= 8.6, 1.7 Hz), 8.45 (d, 1 H, J= 1.4 Hz).
5-(ベンゾチアゾール-2-イル)- 2-ヒドロキシメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 2.00-2.03 (m, 2 H), 2.38-2.45 (m, 2 H), 3.55-3.61 (m, 2 H), 4.06-4.10 (m, 2 H), 4.82-4.86 (m, 1 H), 5.12 (s, 2 H), 7.50 (dt, 1 H, J= 8.1, 1.2 Hz), 7.59 (dt, 1 H, J= 8.3, 1.3 Hz), 8.12 (d, 1 H, J= 7.8 Hz), 8.15-8.23 (m, 3 H), 8.42 (s, 1 H).
5-(ベンゾチアゾール-2-イル)-2-(N,N-ジメチルアミノメチル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 1.86-1.90 (m, 2 H), 2.29 (s, 6 H), 2.55-2.66 (m, 2 H), 3.53-3.61 (m, 2 H), 3.76 (s, 2 H), 4.18-4.23 (m, 2 H), 4.84-4.93 (m, 1 H), 7.35 (t, 1 H, J= 7.3 Hz), 7.48 (t, 1 H, J= 7.4 Hz), 7.69 (d, 1 H, J= 8.6 Hz), 7.89 (d, 1 H, J= 7.8 Hz), 8.06 (d, 1 H, J= 7.9 Hz), 8.11 (dd, 1 H, J= 8.6, 1.4 Hz), 8.38 (d, 1 H, J= 1.5 Hz).
5-(ベンゾチアゾール-2-イル)-2-(N,N-ジメチルアミノメチル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール 塩酸塩 の合成

1H-NMR (DMSO-d6)δ(ppm): 1.95-2.02 (m, 2 H), 2.35-2.55 (m, 2 H), 3.01 (s, 6 H), 3.56-3.65 (m, 2 H), 4.02-4.11 (m, 2 H), 4.71-4.85 (m, 1 H), 4.93 (s, 2 H), 7.46 (dt, 1 H, J= 7.6, 1.2 Hz), 7.56 (dt, 1 H, J= 7.7, 1.3 Hz), 8.01 (d, 1 H, J= 8.7 Hz), 8.06 (d, 1 H, J= 8.5 Hz), 8.08 (dd, 1 H, J= 8.5, 1.7 Hz), 8.15 (d, 1 H, J= 8.0 Hz), 8.37 (d, 1 H, J= 1.6 Hz), 10.95 (brs, 1 H).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3)δ(ppm): 1.88-1.92 (m, 2 H), 2.58-2.68 (m, 2 H), 2.70 (s, 3 H), 3.57-3.64 (m, 2 H), 4.21-4.25 (m, 2 H), 4.43-4.49 (m, 1 H), 7.29(d, 1H, J= 9.2 Hz), 7.33-7.35 (m, 2 H), 7.59-7.62 (m, 1 H), 7.76-7.78 (m, 1 H), 8.18 (dd, 1 H, J= 8.6, 1.6 Hz), 8.57 (d, 1 H, J= 1.4 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール-・メタンスルホン酸塩の合成

1H-NMR (DMSO-D6) δ(ppm): 1.99-2.07(2H, m), 2.36 (3H, s), 2.39-2.51 (2H, m), 2.95 (3H, s), 3.61 (2H, t, J = 11.4 Hz), 4.09 (2H, dd, J = 11.4, 3.8 Hz), 4.87-4.96 (1H, m), 7.42-7.52 (2H, m), 7.82-7.89 (2H, m), 8.31-8.32 (2H, m), 8.54 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-ヒドロキシメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例13-1
2-アセトキシメチル-5-(2-ヒドロキシアニリノカルボニル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 1.90-1.93 (m, 2 H), 2.14 (s, 3 H), 2.41-2.53 (m, 2 H), 3.54-3.60 (m, 2 H), 4.05-4.09 (m, 2 H), 4.76-4.82 (m, 1 H), 5.54 (s, 2 H), 6.85 (dt, 1 H, J= 7.7, 1.5 Hz), 6.94 (dd, 1 H, J= 8.1, 1.4 Hz), 7.05 (dt, 1 H, J= 8.1, 1.4 Hz), 7.69 (dd, 1 H, J= 7.9, 1.5 Hz), 7.98-8.00 (m, 2 H), 8.36 (s, 1 H), 9.68(s, 1 H), 10.00(brs, 1H).
実施例13-2
5-(ベンゾオキサゾール-2-イル)-2-ヒドロキシメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-d6)δ(ppm): 1.99-2.02 (m, 2 H), 2.40-2.50 (m, 2 H), 3.54-3.60 (m, 2 H), 4.06-4.10 (m, 2 H), 4.82-4.89 (m, 1 H), 5.09 (s, 2 H), 7.41-7.49 (m, 2 H), 7.82-7.88 (m, 2 H), 8.23 (d, 1 H, J= 8.8 Hz), 8.29 (dd, 1 H, J= 8.8, 1.5 Hz), 8.48 (d, 1 H, J= 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(N,N-ジメチルアミノメチル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (acetone-d6)δ(ppm): 2.07 (s, 6 H), 2.10-2.16 (m, 2 H), 2.54-2.63 (m, 2 H), 3.76-3.80 (m, 2 H), 3.79 (s, 2 H), 4.06-4.10 (m, 2 H), 5.02-5.08 (m, 1 H), 7.38-7.46 (m, 2 H), 7.70-7.75 (m, 1 H), 7.75-7.80 (m, 1 H), 8.06 (d, 1 H, J= 8.7 Hz), 8.27 (dd, 1 H, J= 8.7, 1.6 Hz), 8.56 (d, 1 H, 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-(4-メトキシフェニル)-2-メチルベンゾイミダゾールの合成
実施例15-1
4-フルオロ-N-(2-ヒドロキシフェニル)-3-ニトロベンズアニリドの合成

1H-NMR (DMSO-D6) δ(ppm): 6.84 (1H, td, J = 7.6, 1.4 Hz), 6.93 (1H, dd, J = 8.1, 1.5 Hz), 7.08 (1H, ddd, J = 8.5, 6.9, 1.2 Hz), 7.54 (1H, dd, J = 7.8, 1.6 Hz), 7.76 (1H, dd, J = 11.2, 8.7 Hz), 8.39 (1H, ddd, J = 8.7, 4.2, 2.3 Hz), 8.75 (1H, dd, J = 7.3, 2.3 Hz), 9.69 (1H, br s), 9.94 (1H, br s).
実施例15-2
2-(4-フルオロ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 7.44-7.53 (2H, m), 7.82-7.89 (3H, m), 8.55-8.61 (1H, m), 8.80-8.84 (1H, m).
実施例15-3
2-(4-(4-メトキシフェニルアミノ)-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 3.81 (3H, s), 7.05-7.11 (3H, m), 7.30-7.44 (4H, m), 7.73-7.81 (2H, m), 8.17 (1H, dd, J = 9.1, 2.1 Hz), 8.85 (1H, d, J = 2.1 Hz), 9.83 (1H, br s).
実施例15-4
5-(ベンゾオキサゾール-2-イル)- 1-(4-メトキシフェニル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.53 (3H, s), 3.90 (3H, s), 7.05-7.37 (7H, m), 7.59-7.64 (1H, m), 7.74-7.80 (1H, m), 8.16 (1H, dd, J = 8.4, 1.5 Hz), 8.62 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-シクロヘキシル-2-メチルベンゾイミダゾールの合成
実施例16-1
2-(4-シクロヘキシルアミノ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.36-2.11 (10H, m), 3.56-3.66 (1H, m), 7.01 (1H, d, J = 9.2 Hz), 7.32-7.36 (2H, m), 7.55-7.59 (1H, m), 7.71-7.75 (1H, m), 8.26 (1H, ddd, J = 9.2, 3.1, 0.5 Hz), 8.46 (1H, d, J = 7.3 Hz), 9.06 (1H, d, J = 2.0 Hz).
実施例16-2
5-(ベンゾオキサゾール-2-イル)- 1-シクロヘキシル-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.26-2.30 (10H, m), 2.68 (3H, s), 4.17-4.26 (1H, m), 7.30-7.37 (2H, m), 7.58-7.64 (2H, m), 7.75-7.79 (1H, m), 8.15 (1H, dd, J = 8.6, 1.6 Hz), 8.54 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-ベンジル-2-メチルベンゾイミダゾールの合成
実施例17-1
2-(4-ベンジルアミノ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 4.64 (2H, d, J = 5.6 Hz), 6.99 (1H, d, J = 8.9 Hz), 7.31-7.44 (7H, m), 7.55-7.58 (1H, m), 7.71-7.75 (1H, m), 8.24-8.28 (1H, m), 8.72 (1H, br s), 9.09 (1H, d, J = 2.1 Hz).
実施例17-2 2-(2-ベンジルアミノアニリンー5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 4.42 (2H, s), 6.74 (2H, d, J = 8.1 Hz), 7.24-7.76 (10H, m).
実施例17-3
5-(ベンゾオキサゾール-2-イル)- 1-ベンジル-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.62 (3H, s), 5.38 (2H, s), 7.06-7.10 (2H, m), 7.30-7.37 (6H, m), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 8.18 (1H, dd, J = 8.5, 1.6 Hz), 8.60 (1H, d, J = 1.6 Hz).
5-(ベンゾチオフェンー2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例18-1
5-ブロモ-2-(テトラヒドロピラン-4-イル)ニトロベンゼンの合成

1H-NMR (CDCl3) δ(ppm): 1.60-1.74 (2H, m), 2.04-2.08 (2H, m), 3.52-3.78 (3H, m), 4.03 (2H, td, J = 8.0, 4.1 Hz), 6.79 (1H, d, J = 9.2 Hz), 7.49 (1H, ddd, J = 9.2, 2.4, 0.6 Hz), 8.06-8.08 (1H, m), 8.33 (1H, d, J = 2.4 Hz).
実施例18-2
5-ブロモ-2-(テトラヒドロピラン-4-イル)アミノアニリンの合成

1H-NMR (CDCl3) δ(ppm): 1.43-1.58 (2H, m), 1.98-2.05 (2H, m), 3.36-3.56 (3H, m), 4.00 (2H, dt, J = 11.8, 3.6 Hz), 6.53 (1H, d, J = 8.1 Hz), 6.85-6.90 (2H, m).
実施例18-3
5-ブロモ2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール-塩酸塩の合成

1H-NMR (DMSO-D6) δ(ppm): 1.94-1.97 (2H, m), 2.30-2.49(2H, m), 2.87(3H,s), 3.57 (2H, t, J = 11.5 Hz), 4.05 (2H, dd, J = 11.5, 4.0 Hz), 4.79-4.88(1H, m), 7.64 (1H, dd, J = 8.9, 1.8 Hz), 8.00-8.03 (2H, m).
実施例18-4
5-(ベンゾチオフェンー2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.88 (2H, dd, J = 12.7, 2.1 Hz), 2.56-2.62 (2H, m), 2.68 (3H, s), 3.60 (2H, dd, J = 12.1, 10.3 Hz), 4.20-4.25 (2H, m), 4.39-4.45 (1H, m), 7.29-7.39 (3H, m), 7.52-7.59 (2H, m), 7.78-7.82 (2H, m), 8.04 (1H, s).
5-(ベンゾフラン-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.86-1.90 (2H, m), 2.57-2.63 (2H,m), 2.67 (3H, s), 3.60 (2H, td, J = 11.8, 1.8 Hz), 4.22 (2H, dd, J = 11.8, 4.6 Hz), 4.41-4.45 (1H, m), 7.01 (1H, d, J = 0.8 Hz), 7.22-7.27 (2H, m), 7.57-7.59 (3H, m), 7.75 (1H, dd, J = 8.6, 1.4 Hz), 8.18 (1H, d, J = 1.4 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例20-1
5-(ベンゾオキサゾール-2-イル)-2-(テトラヒドロピラン-4-イル)ニトロベンゼンの合成

1H-NMR (CDCl3) δ(ppm): 1.67-1.81 (2H, m), 2.07-2.17 (2H, m), 3.7-3.67(2H, m), 3.76-3.77 (1H, m), 4.03-4.10 (2H, m),7.01 (1H, t, J = 8.6 Hz), 7.35 (2H, tt, J = 6.5, 2.5 Hz), 7.54-7.59 (1H, m), 7.71-7.77 (1H, m), 8.30 (1H, dd, J = 9.1, 2.1 Hz), 8.43 (1H, d, J = 7.4 Hz), 9.08 (1H, d, J = 2.1 Hz)
また本中間体は以下のように4-((テトラヒドロピラン-4-イル)アミノ)-3-ニトロ安息香酸(実施例4-1参照)から得ることもできる。

実施例20-2
5-(ベンゾオキサゾール-2-イル)-2-(テトラヒドロピラン-4-イル)アミノアニリンの合成

1H-NMR (CDCl3) δ(ppm): 1.56-1.61 (2H, m), 1.90-2.27 (2H, m), 3.25-3.83 (5H, m), 4.06-4.11 (2H, m), 6.73 (1H, d, J = 8.4 Hz), 7.24-7.31 (2H, m), 7.47-7.55 (1H, m), 7.59-7.77 (3H, m).
実施例20-3
5-(ベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.21-2.30 (4H, m), 3.65 (2H, dt, J = 16.0, 8.0 Hz), 4.16-4.24 (2H, m), 4.45-4.57 (1H, m), 7.32-7.39 (2H, m), 7.59-7.62 (2H, m), 7.77-7.80 (1H, m), 8.11 (1H, s), 8.28 (1H, dd, J = 8.6, 1.5 Hz), 8.71 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-(2-ピリジル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.03-2.08 (2H, m), 2.65-2.77 (2H, m), 3.53-3.62 (2H, m), 4.18-4.24 (2H, m), 5.85-5.92 (1H, m), 7.31-7.44 (3H, m), 7.59-7.65 (1H, m), 7.76-7.82 (1H, m), 7.88-7.93 (2H, m), 8.24-8.35 (2H, m), 8.71-8.75 (2H, m).
5-(ベンゾオキサゾール-2-イル)-2-イソプロピル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.50 (6H, d, J = 6.8 Hz), 1.88 (2H, d, J = 11.0 Hz), 2.65-2.75 (2H, m), 3.23-3.33 (1H, m), 3.61 (2H, t, J = 11.7 Hz), 4.24 (2H, dd, J = 11.7, 4.1 Hz), 4.50-4.54 (1H, m), 7.33-7.34 (2H, m), 7.61-7.75 (3H, m), 8.18 (1H, d, J = 8.6 Hz), 8.63 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-シクロヘキシル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.35-1.55 (3H, m), 1.86-2.05 (9H, m), 2.61-2.77 (2H, m), 2.84-2.93 (1H, m), 3.63 (2H, t, J = 11.2 Hz), 4.24 (2H, dd, J = 11.8, 4.2 Hz), 4.45-4.54 (1H, m), 7.30-7.33 (2H, m), 7.56-7.78 (3H, m), 8.18 (1H, dd, J = 8.6, 1.6 Hz), 8.62 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(3-ピリジル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.93 (2H, dd, J = 12.5, 2.6 Hz), 2.68-2.83 (2H, m), 3.43-3.52 (2H, m), 4.19 (2H, dd, J = 11.5, 4.3 Hz), 4.51-4.63 (1H, m), 7.33-7.40 (2H, m), 7.52-7.57 (1H, m), 7.60-7.64 (1H, m), 7.77-7.86 (2H, m), 8.07 (1H, dt, J = 7.9, 1.8 Hz), 8.29 (1H, dd, J = 8.7, 1.8 Hz), 8.71 (1H, d, J = 1.2 Hz), 8.83 (1H, dd, J = 4.9, 1.8 Hz), 8.91 (1H, d, J = 1.2 Hz).
5-(ベンゾオキサゾール-2-イル)-2-フェニル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.91 (2H, dd, J = 13.2, 3.1 Hz), 2.66-2.81 (2H, m), 3.43-3.48 (2H, m), 4.15-4.19 (2H, m), 4.61-4.66 (1H, m), 7.32-7.39 (2H, m), 7.56-7.68 (6H, m), 7.78-7.82 (2H, m), 8.26 (1H, dd, J = 8.7, 1.6 Hz), 8.69-8.69 (1H, m).
5-(ベンゾオキサゾール-2-イル)-2-(4-ピリジル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.93 (2H, dd, J = 12.8, 2.7 Hz), 2.67-2.83 (2H, m), 3.45-3.53(2H, m), 4.20 (2H, dd, J = 11.8, 4.5 Hz), 4.54-4.66 (1H, m), 7.33-7.40 (2H, m), 7.59-7.65 (3H, m), 7.78-7.86 (2H, m), 8.30 (1H, dd, J = 8.6, 1.6 Hz), 8.71-8.72 (1H, m), 8.87 (2H, dd, J = 4.3, 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)-2-トリフルオロメチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.99 (2H, dd, J = 12.4, 2.9 Hz), 2.60-2.76 (2H, m), 3.61 (2H, td, J = 12.0, 1.8 Hz), 4.24 (2H, dd, J = 11.9, 4.6 Hz), 4.67-4.79 (1H, m), 7.34-7.41 (2H, m), 7.59-7.66 (1H, m), 7.76-7.87 (2H, m), 8.37 (1H, dd, J = 8.7, 1.6 Hz), 8.76-8.77 (1H, m).
5-(ベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)-ベンゾトリアゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.17-2.22 (2H, m), 2.53-2.59 (2H, m), 3.69 (2H, td, J = 11.9, 2.1 Hz), 4.22-4.27 (2H, m), 4.96-5.01 (1H, m), 7.36-7.43 (2H, m), 7.60-7.67 (1H, m), 7.74 (1H, dd, J = 8.8, 0.7 Hz), 7.79-7.82 (1H, m), 8.45 (1H, dd, J = 8.9, 1.5 Hz), 8.96 (1H, dd, J = 1.4, 0.7 Hz).
5-(ベンゾオキサゾール-2-イル)- 2-メチル-1-tertブチルベンゾイミダゾールの合成
実施例29-1
2-(2-terブチルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.45 (9H, m), 6.96 (1H, d, J = 8.4 Hz), 7.19-7.33 (2H, m), 7.47-7.54 (1H, m), 7.64 (1H, d, J = 2.0 Hz), 7.67-7.73 (2H, m).
実施例29-2
5-(ベンゾオキサゾール-2-イル)- 2-メチル-1-terブチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.86 (9H, s), 2.85 (3H, s), 7.32-7.37 (2H, m), 7.57-7.62 (1H, m), 7.75-7.79 (2H, m), 8.12 (1H, dd, J = 8.8, 1.7 Hz), 8.51 (1H, d, J = 1.7 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-(2-メトキシフェニル)-2-メチルベンゾイミダゾールの合成
実施例30-1
2-(2-(2-メトキシフェニル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 3.92 (3H, s), 6.87-7.00 (4H, m), 7.30-7.35 (3H, m), 7.54-7.57 (1H, m), 7.65-7.75 (3H, m).
実施例30-2
5-(ベンゾオキサゾール-2-イル)- 1-(2-メトキシフェニル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.46 (3H, s), 3.78 (3H, s), 7.12-7.16 (3H, m), 7.32-7.37 (3H, m), 7.51-7.62 (2H, m), 7.74-7.79 (1H, m), 8.15 (1H, dd, J = 8.5, 1.6 Hz), 8.61-8.62 (1H, m).
5-(ベンゾオキサゾール-2-イル)- 1-(3-メトキシフェニル)-2-メチルベンゾイミダゾールの合成
実施例31-1
2-(2-(3-メトキシフェニル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 3.79 (3H, s), 6.50-6.58 (3H, m), 7.16-7.35 (4H, m), 7.54-7.57 (1H, m), 7.65-7.76 (3H, m).
実施例31-2
5-(ベンゾオキサゾール-2-イル)- 1-(3-メトキシフェニル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.57 (3H, s), 3.88 (3H, s), 6.92 (1H, t, J = 2.1 Hz), 6.96-7.00 (1H, m), 7.09 (1H, dd, J = 8.4, 2.5 Hz), 7.25-7.38 (3H, m), 7.52 (1H, t, J = 8.1 Hz), 7.59-7.63 (1H, m), 7.75-7.79 (1H, m), 8.17 (1H, dd, J = 8.6, 1.5 Hz), 8.62 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-ベンジルアミノメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.83-1.91 (2H, m), 2.54-2.66 (2H, m), 3.48-3.58 (2H, m), 3.98 (2H, s), 4.11-4.21 (4H, m), 4.60-4.74 (1H, m), 7.31-7.43 (7H, m), 7.56-7.61 (1H, m), 7.69-7.78 (2H, m), 8.20 (1H, dd, J = 8.7, 1.6 Hz), 8.59 (1H, d, J = 1.2 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-((1-エトキシカルボニル)ピペリジン-4-イル)-2-メチルベンゾイミダゾールの合成
実施例33-1
2-(2-(4-(1-エトキシカルボニル)ピペリジノアミノ)アニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.28 (3H, t, J = 7.1 Hz), 1.38-1.53 (2H, m), 2.08-2.14 (2H, m), 3.00-3.10 (2H, m), 3.52-3.60 (1H, m), 4.08-4.20 (4H, m), 6.72 (1H, d, J = 8.4 Hz), 7.27-7.32 (2H, m), 7.51-7.54 (1H, m), 7.65 (1H, d, J = 1.8 Hz), 7.67-7.71 (1H, m), 7.76 (1H, dd, J = 8.4, 2.0 Hz).
実施33-2
5-(ベンゾオキサゾール-2-イル)- 1-((1-エトキシカルボニル)ピペリジン-4-イル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.34 (3H, t, J = 7.1 Hz), 1.96 (2H, d, J = 10.9 Hz), 2.36-2.52 (2H, m), 2.70 (3H, s), 2.88-3.00 (2H, m), 4.23 (2H, q, J = 7.1 Hz), 4.33-4.54 (3H, m), 7.33-7.36 (2H, m), 7.54-7.62 (2H, m), 7.75-7.79 (1H, m), 8.16 (1H, d, J = 8.6 Hz), 8.55 (1H, s).
5-(ベンゾオキサゾール-2-イル)- 1-(4-フルオロフェニル)-2-メチルベンゾイミダゾールの合成
実施例34-1
2-(2-(4-フルオロフェニル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 6.92-7.05 (4H, m), 7.15 (1H, d, J = 8.4 Hz), 7.29-7.36 (2H, m), 7.53-7.57 (1H, m), 7.65-7.75 (3H, m).
5-(ベンゾオキサゾール-2-イル)- 1-(4-フルオロフェニル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.54 (3H, s), 7.20 (1H, d, J = 8.6 Hz), 7.28-7.43 (6H, m), 7.59-7.63 (1H, m), 7.75-7.79 (1H, m), 8.18 (1H, dd, J = 8.5, 1.6 Hz), 8.62 (1H, d, J = 1.0 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-(ピペリジン-4-イル)-2-メチルベンゾイミダゾールの合成
実施例35-1
2-(2-(4-(1-tertブトキシカルボニル)ピペリジノアミノ)アニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.37-1.45 (2H, m), 1.48 (9H, s), 2.05-2.12 (2H, m), 2.99 (2H, t, J = 11.5 Hz), 3.50-3.58 (1H, m), 4.05-4.16 (2H, m), 7.27-7.31 (3H, m), 7.50-7.54 (1H, m), 7.65 (1H, d, J = 2.0 Hz), 7.68-7.71 (1H, m), 7.75 (1H, dd, J = 8.3, 1.9 Hz).
実施例35-2
5-(ベンゾオキサゾール-2-イル)- 1-(ピペリジン-4-イル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.93-2.03 (2H, m), 2.48-2.64 (2H, m), 2.70 (3H, s), 2.85-2.96 (2H, m), 3.41 (2H, d, J = 12.0 Hz), 4.29-4.38 (1H, m), 7.33-7.36 (2H, m), 7.59-7.63 (1H, m), 7.70-7.79 (2H, m), 8.15-8.20 (1H, m), 8.56 (1H, d, J = 1.5 Hz).
1-アダマンチル-5-(ベンゾオキサゾール-2-イル)-2-メチルベンゾイミダゾールの合成
実施例36-1
2-(2-(1-アダマンチルアミノ)アニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.72 (6H, br s), 2.01 (6H, br s), 2.16 (3H, br s), 7.03 (1H, d, J = 8.2 Hz), 7.26-7.31 (2H, m), 7.49-7.53 (1H, m), 7.62-7.71 (3H, m).
実施例36-2
1-アダマンチル-5-(ベンゾオキサゾール-2-イル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.86 (6H, br s), 2.34 (3H, br s), 2.55 (6H, br s), 2.89 (3H, s), 7.32-7.35 (2H, m), 7.58-7.62 (1H, m), 7.75-7.79 (1H, m), 7.83 (1H, d, J = 8.9 Hz), 8.09 (1H, dd, J = 8.9, 1.7 Hz), 8.51 (1H, d, J = 1.7 Hz).
5-(N-t-ブトキシカルボニルインドール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.31 (9H, s), 1.87 (2H, dd, J = 12.5, 2.6 Hz), 2.59-2.68 (2H, m), 2.59 (3H, s), 3.60 (2H, td, J = 12.1, 1.9 Hz), 4.22 (2H, dd, J = 11.7, 4.5 Hz), 4.41-4.46 (1H, m), 6.57 (1H, d, J = 0.7 Hz), 7.22-7.35 (3H, m), 7.55 (2H, d, J = 8.4 Hz), 7.76 (1H, d, J = 1.5 Hz), 8.19 (1H, d, J = 8.4 Hz).
5-(インドールー2-イル)- 1-(テトラヒドロピラン-4-イル)-2-メチルベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 2.00 (2H, d, J = 9.6 Hz), 2.40-2.45 (2H, m), 2.93 (3H, s), 3.60 (2H, t, J = 11.3 Hz), 4.08 (2H, dd, J = 11.3, 3.6 Hz), 4.84-4.88 (1H, m), 7.04-7.12 (3H, m), 7.44 (1H, d, J = 7.9 Hz), 7.56 (1H, d, J = 7.9 Hz), 8.01-8.13 (2H, m), 8.24 (1H, s), 11.88 (1H, s).
5-(5-メチルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.88 (2H, d, J = 9.4 Hz), 2.30-2.45 (5H, m), 2.67 (3H, s), 3.58 (2H, t, J = 11.2 Hz), 4.06 (2H, dd, J = 11.2, 4.1 Hz), 4.61-4.70 (1H, m), 7.21 (1H, dd, J = 8.6 1.3 Hz), 7.58 (1H, s), 7.65 (1H, d, J = 8.6 Hz), 7.88 (1H, d, J = 8.6 Hz), 8.03 (1H, d, J = 1.3 Hz), 8.30 (1H, d, J = 1.3 Hz).
5-(5-クロロベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.88 (2H, d, J = 10.6 Hz), 2.36-2.41 (2H, m), 2.67 (3H, s), 3.58 (2H, t, J = 11.0 Hz), 4.02-4.09 (2H, m), 4.64-4.69 (1H, m), 7.43-7.46 (1H, m), 7.83 (1H, d, J = 8.7 Hz), 7.89-7.91 (2H, m), 8.05 (1H, dd, J = 8.7, 1.6 Hz), 8.32 (1H, d, J = 1.2 Hz).
5-(6-クロロベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.88 (2H, d, J = 9.4 Hz), 2.38-2.43 (2H, m), 2.67 (3H, s), 3.58 (2H, t, J = 11.0 Hz), 4.02-4.08 (2H, m), 4.65-4.68 (1H, m), 7.45 (1H, dd, J = 8.6, 2.0 Hz), 7.80 (1H, d, J = 8.6 Hz), 7.90 (1H, d, J = 8.6 Hz), 7.99 (1H, d, J = 2.0 Hz), 8.03 (1H, dd, J = 8.6, 1.6 Hz), 8.31 (1H, d, J = 1.6 Hz).
5-(6-メチルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.85-1.90 (2H, m), 2.34-2.52 (8H, m), 3.58 (2H, t, J = 11.0 Hz), 4.00-4.08 (2H, m), 4.62-4.72 (1H, m), 7.22 (1H, d, J = 8.1 Hz), 7.60 (1H, s), 7.65 (1H, d, J = 8.1 Hz), 7.87 (1H, d, J = 8.8 Hz), 8.03 (1H, dd, J = 8.8, 1.6 Hz), 8.29 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-エチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.37 (3H, t, J = 7.4 Hz), 1.86 (2H, d, J = 9.1 Hz), 2.39-2.49(2H, m), 3.03 (2H, q, J = 7.4 Hz), 3.59 (2H, t, J = 11.1 Hz), 4.02-4.08 (2H, m), 4.63-4.72 (1H, m), 7.39-7.44 (2H, m), 7.76-7.81 (2H, m), 7.90 (1H, d, J = 8.6 Hz), 8.06 (1H, dd, J = 8.6, 1.5 Hz), 8.36 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(イミダゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 2.13 (2H, dd, J = 12.4, 3.4 Hz), 2.66-2.82 (2H, m), 3.78 (2H, t, J = 11.5Hz), 4.26 (2H, dd, J = 11.5, 4.0 Hz), 6.66-6.79 (1H, m), 7.22-7.40(4H, m), 7.55-7.59(1H, m), 7.72-7.76(1H, m), 7.91 (1H, d, J = 8.7 Hz), 8.23 (1H, dd, J = 8.7, 1.4 Hz), 8.53 (1H, d, J = 1.4 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(チオフェン-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.96 (2H, dd, J = 12.9, 2.8 Hz), 2.71-2.80 (2H, m), 3.55 (2H, td, J = 12.0, 1.8 Hz), 4.22 (2H, dd, J = 11.6, 4.7 Hz), 4.93-4.98 (1H, m),7.23-7.26 (1H, m), 7.34-7.37 (2H, m), 7.46 (1H, dd, J = 3.6, 1.2 Hz), 7.59-7.65 (2H, m), 7.77-7.81 (2H, m), 8.25 (1H, dd, J = 8.7, 1.6 Hz), 8.68 (1H, d, J = 1.6 Hz).
2-メチル-5-(4-メチルベンゾオキサゾール-2-イル)- 1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例46-1
2-フルオロ-5-(4-メチルベンゾオキサゾール-2-イル)- ニトロベンゼンの合成

1H-NMR (CDCl3) δ(ppm): 2.67 (3H, s), 7.26-7.33 (1H, m), 7.27-7.32 (1H, m), 7.41-7.49 (2H, m), 8.52-8.55 (1H, m), 8.95 (1H, dd, J = 7.1, 2.1 Hz).
実施例46-2
2-(2-(テトラヒドロピラン-4-イル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.71-1.77 (2H, m), 2.12 (2H, d, J = 12.9 Hz), 2.66 (3H, s), 3.57-3.66 (2H, m), 3.80-3.90 (1H, m), 4.05-4.08 (2H, m), 7.01 (1H, d, J = 9.2 Hz), 7.14 (1H, d, J = 7.9 Hz), 7.20-7.23 (1H, m), 7.39 (1H, d, J = 7.9 Hz), 8.31 (1H, dd, J = 9.2, 2.0 Hz), 8.42 (1H, d, J = 7.9 Hz), 9.08 (1H, d, J = 2.0 Hz).
実施例46-3
2-メチル-5-(4-メチルベンゾオキサゾール-2-イル)- 1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.85-1.91 (2H, m), 2.39 (2H, td, J = 12.4, 4.4 Hz), 2.60 (3H, s), 2.67 (3H, s), 3.58 (2.2H, t, J = 11.3 Hz), 4.06 (2H, dd, J = 11.3, 4.0 Hz), 4.62-4.71 (1H, m), 7.20 (1H, d, J = 7.7 Hz), 7.29 (1H, t, J = 7.7 Hz), 7.58 (1H, d, J = 7.7 Hz), 7.88 (1H, d, J = 8.6 Hz), 8.06 (1H, dd, J = 8.6, 1.5Hz), 8.31 (1H, d, J = 1.5 Hz).
2-メチル-5-(6-ニトロベンゾオキサゾール-2-イル)- 1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例47-1
5-(2-ヒドロキシ-4-ニトロアニリノカルボニル)- 2-メチル- 1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.99 (2H, d, J = 9.5 Hz), 2.49-2.52 (2H, m), 2.92 (3H, s), 3.60 (2H, t, J = 11.2 Hz), 4.08 (2H, dd, J = 11.2, 3.8 Hz), 4.85-4.89 (1H, m), 7.78-7.83 (2H, m), 8.08 (1H, dd, J = 8.8, 1.5 Hz), 8.16-8.20 (2H, m), 8.38 (1H, d, J = 1.5 Hz), 9.95 (1H, s), 11.22 (1H, s).
実施例47-2
2-メチル-5-(6-ニトロベンゾオキサゾール-2-イル)- 1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ(ppm): 1.87 (2H, d, J = 12.7 Hz), 2.34-2.51 (2H, m), 2.67 (3H, s), 3.57 (2H, t, J = 11.0 Hz), 4.05 (2H, d, J = 7.6 Hz), 4.60-4.75 (1H, m), 7.92-8.00 (2H, m), 8.09 (1H, d, J = 8.6 Hz), 8.31 (1H, dd, J = 9.0, 2.1 Hz), 8.37 (1H, s), 8.73 (1H, d, J = 2.1 Hz).
2-メチル-5-(6-アミノベンゾオキサゾール-2-イル)- 1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.87 (2H, dd, J = 12.6, 2.6 Hz), 2.58-2.69 (2H, m), 2.63 (3H, s), 3.59 (2H, t, J = 11.5 Hz), 4.21 (2H, dd, J = 11.5, 4.5 Hz), 4.41-4.46 (1H, m), 6.68 (1H, dd, J = 8.4, 2.0 Hz), 6.89 (1H, d, J = 2.0 Hz), 7.50 (1H, d, J = 8.5 Hz), 7.62 (1H, d, J = 8.5 Hz), 8.09 (1H, d, J = 8.5 Hz), 8.47 (1H, s).
5-(ベンゾオキサゾール-2-イル)- 1-(4-ヒドロキシシクロヘキシル)- 2-メチルベンゾイミダゾールの合成
実施例49-1
(2-(4-ヒドロキシシクロヘキシルアミノ)ニトロベンゼン-5-イル)ベンゾオキサゾールの合成

実施例49-2
5-(ベンゾオキサゾール-2-イル)- 1-(4-ヒドロキシシクロヘキシル)- 2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ(ppm): 1.58-2.38 (9H, m), 2.68 (3H, s), 3.86-3.94 (1H, m), 4.26 (1H, tt, J = 12.2, 4.1 Hz), 7.31-7.38 (2H, m), 7.56 (1H, d, J = 8.6 Hz), 7.59-7.64 (1H, m), 7.73-7.80 (1H, m), 8.15 (1H, dd, J = 8.6, 1.6 Hz), 8.55 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-n-プロピルベンゾイミダゾールの合成
実施例50-1
2-(2-n-プロピルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.06 (3H, t, J = 7.3 Hz), 1.67-1.77 (2H, m), 3.18 (2H, t, J = 7.2 Hz), 6.71 (1H, d, J = 8.2 Hz), 7.26-7.33 (2H, m), 7.50-7.79 (4H, m).
実施例50-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-n-プロピルベンゾイミダゾールの合成
1H-NMR (CDCl3) δ: 1.01 (3H, t, J = 7.4 Hz), 1.82-1.95 (2H, m), 2.65 (3H, s), 4.12 (2H, t, J = 7.3 Hz), 7.30-7.43 (3H, m), 7.58-7.62 (1H, m), 7.74-7.78 (1H, m), 8.20 (1H, dd, J = 8.6, 1.2 Hz), 8.55 (1H, d, J = 1.2 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-メトキシエチル)-2-メチルベンゾイミダゾールの合成
実施例51-1
2-(2-(2-メトキシエチル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 3.35-3.42 (5H, m), 3.67-3.71 (2H, m), 6.72 (1H, d, J = 8.2 Hz), 7.26-7.31 (2H, m), 7.50-7.77 (4H, m).
実施例51-2
5-(ベンゾオキサゾール-2-イル)-1-(2-メトキシエチル)-2-メチルベンゾイミダゾールの合成
1H-NMR (CDCl3) δ: 2.67 (3H, s), 3.29 (3H, s), 3.72 (2H, t, J = 5.4 Hz), 4.33 (2H, t, J = 5.4 Hz), 7.32-7.44 (3H, m), 7.58-7.63 (1H, m), 7.75-7.78 (1H, m), 8.20 (1H, dd, J = 8.5, 1.5 Hz), 8.56 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-フェニルエチル)ベンゾイミダゾールの合成
実施例52-1
2-(2-(2-フェニルエチル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 3.01 (2H, t, J = 7.0 Hz), 3.50 (2H, t, J = 7.0 Hz), 6.75 (1H, d, J = 8.4 Hz), 7.25-7.79 (11H, m).
実施例52-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-フェニルエチル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 2.22 (3H, s), 3.10 (2H, t, J = 6.8 Hz), 4.35 (2H, t, J = 6.8 Hz), 6.94-6.98 (2H, m), 7.22-7.37 (6H, m), 7.59-7.62 (1H, m), 7.76-7.79 (1H, m), 8.19 (1H, dd, J = 8.5, 1.2 Hz), 8.55 (1H, d, J = 1.2 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-cyc-プロピルベンゾイミダゾールの合成
実施例53-1
2-(2-cyc-プロピルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 0.56-0.62 (2H, m), 0.79-0.86 (2H, m), 2.50-2.55 (1H, m), 7.12 (1H, d, J = 8.4 Hz), 7.26-7.33 (2H, m), 7.51-7.81 (4H, m).
実施例53-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-cyc-プロピルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.07-1.13 (2H, m), 1.23-1.33 (2H, m), 2.71 (3H, s), 3.22-3.30 (1H, m), 7.32-7.35 (2H, m), 7.58-7.62 (2H, m), 7.75-7.78 (1H, m), 8.19 (1H, dd, J = 8.5, 1.2 Hz), 8.52 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-cyc-プロピルメチルベンゾイミダゾールの合成
実施例54-1
2-(2-cyc-プロピルメチルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 0.27-0.32 (2H, m), 0.58-0.64 (2H, m), 1.13-1.23 (1H, m), 3.05 (2H, d, J = 7.1 Hz), 6.68 (1H, d, J = 8.4 Hz), 7.27-7.32 (2H, m), 7.50-7.77 (4H, m).
実施例54-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-cyc-プロピルメチルベンゾイミダゾールの合成
1H-NMR (CDCl3) δ: 0.38-0.44 (2H, m), 0.61-0.68 (2H, m), 1.20-1.28 (1H, m), 2.67 (3H, s), 4.05 (2H, d, J = 6.6 Hz), 7.30-7.45 (3H, m), 7.57-7.62 (1H, m), 7.75-7.78 (1H, m), 8.20 (1H, dd, J = 8.5, 1.2 Hz), 8.56 (1H, d, J = 1.2 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-(tert-ブトキシカルボニルアミノ)エチル)-2-メチルベンゾイミダゾールの合成
実施例55-1
2-(tert-ブトキシカルボニルアミノ)エチルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.46 (9H, s), 3.34-3.49 (5H, m), 4.48 (1H, br s), 4.87 (1H, br s), 6.67 (1H, d, J = 8.4 Hz), 7.26-7.33 (2H, m), 7.50-7.54 (1H, m), 7.61 (1H, d, J = 1.8 Hz), 7.68-7.76 (2H, m).
実施例55-2
5-(ベンゾオキサゾール-2-イル)-1-(2-(tert-ブトキシカルボニルアミノ)エチル)-2-メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.47 (9H, s), 2.64 (3H, s), 3.53-3.55 (2H, m), 4.33 (2H, t, J = 5.4 Hz), 5.12 (1H, s), 7.33-7.36 (2H, m), 7.41 (1H, d, J = 8.4 Hz), 7.56-7.59 (1H, m), 7.72-7.77 (1H, m), 8.14 (1H, d, J = 8.4 Hz), 8.45 (1H, s).
1-(2-アミノエチル)-5-(ベンゾオキサゾール-2-イル)- 2-メチルベンゾイミダゾール塩酸塩の合成

1H-NMR (D2O) δ: 2.72 (3H, s), 3.40 (2H, t, J = 6.8 Hz), 4.54 (2H, t, J = 6.8 Hz), 7.21-7.32 (2H, m), 7.43-7.46 (2H, m), 7.68 (1H, d, J = 8.9 Hz), 8.06-8.11 (2H, m).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(3-フェニルプロピル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 2.11-2.22 (2H, m), 2.55 (3H, s), 2.72 (2H, t, J = 7.5 Hz), 4.12 (2H, t, J = 7.5 Hz), 7.16-7.35 (8H, m), 7.58-7.61 (1H, m), 7.75-7.78 (1H, m), 8.17 (1H, d, J = 8.2 Hz), 8.54 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-(tert-ブトキシカルバミドメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.49 (9H, s), 1.83-1.88 (2H, m), 2.53-2.68 (2H, m), 3.57-3.67 (2H, m), 4.16-4.22 (2H, m), 4.70-4.83 (3H, m), 5.59 (1H, br s), 7.33-7.39 (2H, m), 7.59-7.62 (1H, m), 7.71-7.79 (2H, m), 8.21 (1H, dd, J = 8.7, 1.5 Hz), 8.60 (1H, d, J = 1.5 Hz).
5-(5-ter-ブチルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

として得た。
1H-NMR (CDCl3) δ: 1.40 (9H, s), 1.85-1.92 (2H, m), 2.54-2.69 (5H, m), 3.60 (2H, t, J = 11.6 Hz), 4.22 (2H, dd, J = 11.6, 4.1 Hz), 4.40-4.50 (1H, m), 7.39 (1H, dd, J = 8.6, 1.5 Hz), 7.51 (1H, d, J = 8.6 Hz), 7.66 (1H, d, J = 8.6 Hz), 7.80 (1H, s), 8.16 (1H, d, J = 8.6 Hz), 8.54 (1H, s).
5-(5-ニトロベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.91 (2H, d, J = 13.0 Hz), 2.59-2.72 (5H, m), 3.61 (3H, t, J = 12.0 Hz), 4.21-4.28 (3H, m), 4.42-4.53 (1H, m), 7.70 (2H, d, J = 8.7 Hz), 8.18 (1H, d, J = 8.6 Hz), 8.30 (1H, d, J = 8.7 Hz), 8.56 (1H, s), 8.63 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-ベンジル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.39 (2H, dd, J = 12.5, 2.6 Hz), 2.37-2.53 (2H, m), 3.24-3.34 (2H, m), 4.05 (2H, dd, J = 11.5, 4.5 Hz), 4.32-4.42 (3H, m), 7.22-7.38 (7H, m), 7.56-7.68 (2H, m), 7.76-7.80 (1H, m), 8.19 (1H, dd, J = 8.6, 1.3Hz), 8.65 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-trans-シンナム-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.94-2.01 (2H, m), 2.61-2.77 (2H, m), 3.61-3.71 (2H, m), 4.26 (2H, dd, J = 11.6, 4.4 Hz), 4.64-4.75 (1H, m), 7.18 (1H, d, J = 15.7 Hz), 7.32-7.48 (5H, m), 7.59-7.72 (4H, m), 7.77-7.80 (1H, m), 8.03 (1H, d, J = 15.7 Hz), 8.22 (1H, dd, J = 8.6, 1.3 Hz), 8.64 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(フラン-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 2.00 (2H, dd, J = 12.8, 2.6 Hz), 2.63-2.79 (2H, m), 3.56-3.65 (2H, m), 4.23 (2H, dd, J = 11.6, 4.4 Hz), 5.09-5.21 (1H, m), 6.66 (1H, dd, J = 3.5, 1.8 Hz), 7.19 (1H, dd, J = 3.5, 0.7 Hz), 7.32-7.39 (2H, m), 7.59-7.65 (1H, m), 7.69 (1H, dd, J = 1.8, 0.7 Hz), 7.76-7.81 (2H, m), 8.24 (1H, dd, J = 8.7, 1.6 Hz), 8.66 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(フラン-3-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.87-1.94 (2H, m), 2.65-2.81 (2H, m), 3.50-3.59 (2H, m), 4.22 (2H, dd, J = 11.5, 4.5 Hz), 4.70-4.79 (1H, m), 6.79 (1H, dd, J = 1.9, 0.7 Hz), 7.34-7.37 (2H, m), 7.60-7.65 (2H, m), 7.77-7.80 (2H, m), 7.93 (1H, dd, J = 1.5, 0.7 Hz), 8.24 (1H, dd, J = 8.7, 1.6 Hz), 8.66 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(3-メトキシフェニル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.92-1.98 (2H, m), 2.50-2.57 (2H, m), 3.35-3.45 (5H, m), 3.86 (3H, s), 3.99-4.04 (2H, m), 4.56-4.67 (1H, m), 7.06-7.57 (6H, m), 7.79-7.84 (2H, m), 8.06 (1H, d, J = 8.7 Hz), 8.18 (1H, dd, J = 8.7, 1.5 Hz), 8.49 (1H, d, J = 1.5 Hz).
5-(5-メトキシベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.86-1.93 (2H, m), 2.55-2.70 (5H, m), 3.56-3.65 (2H, m), 3.88 (3H, s), 4.23 (2H, dd, J = 11.8, 4.5 Hz), 4.41-4.50 (1H, m), 6.93 (1H, dd, J = 8.7, 2.6 Hz), 7.26-7.28 (1H, m), 7.48 (1H, d, J = 8.7 Hz), 7.65 (1H, d, J = 8.7 Hz), 8.15 (1H, dd, J = 8.6, 1.5 Hz), 8.53 (1H, d, J = 1.5 Hz).
2-アミノメチル-5-(ベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾール塩酸塩の合成

1H-NMR (D2O) δ: 1.77-1.84 (2H, m), 2.22-2.38 (2H, m), 3.50-3.62 (2H, m), 4.04 (2H, dd, J = 11.5, 3.8 Hz), 4.35-4.46 (3H, m), 7.02-7.13 (2H, m), 7.26-7.31 (2H, m), 7.51 (1H, d, J = 8.9 Hz), 7.62 (1H, dd, J = 8.9, 1.3 Hz), 7.91 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(2-フェニルエチル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.56 (2H, dd, J = 12.5, 2.6 Hz), 2.45-2.61 (2H, m), 3.27 (4H, s), 3.40-3.50 (2H, m), 4.14 (2H, dd, J = 11.6, 4.5 Hz), 4.23-4.35 (1H, m), 7.20-7.38 (7H, m), 7.56-7.67 (2H, m), 7.75-7.81 (1H, m), 8.18 (1H, dd, J = 8.6, 1.3 Hz), 8.63 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(2-フェニルエチニル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.98-2.04 (2H, m), 2.68-2.84 (2H, m), 3.60-3.70 (2H, m), 4.26 (2H, dd, J = 11.7, 4.5 Hz), 4.85-4.94 (1H, m), 7.33-7.49 (5H, m), 7.59-7.80 (5H, m), 8.29 (1H, dd, J = 8.7, 1.5 Hz), 8.65 (1H, d, J = 1.5 Hz).
5-(5-エチルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.31 (3H, t, J = 7.6 Hz), 1.86-1.93 (2H, m), 2.55-2.70 (5H, m), 2.79 (2H, q, J = 7.6 Hz), 3.56-3.65 (2H, m), 4.23 (2H, dd, J = 11.9, 4.6 Hz), 4.41-4.50 (1H, m), 7.17 (1H, dd, J = 8.3, 1.6 Hz), 7.49 (1H, d, J = 8.3 Hz), 7.59 (1H, br s), 7.66 (1H, d, J = 8.6 Hz), 8.17 (1H, dd, J = 8.6, 1.5 Hz), 8.55 (1H, d, J = 1.5 Hz).
5-(5-トリフルオロメチルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.88-1.93 (2H, m), 2.55-2.72 (5H, m), 3.56-3.65 (2H, m), 4.24 (2H, dd, J = 11.7, 4.6 Hz), 4.41-4.53 (1H, m), 7.61 (1H, dd, J = 8.6, 1.6 Hz), 7.69 (2H, dd, J = 8.6, 2.6 Hz), 8.04 (1H, br s), 8.18 (1H, dd, J = 8.6, 1.5 Hz), 8.57 (1H, d, J = 1.5 Hz).
5-(5-cyc-ヘキシルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.18-1.92 (13H, m), 2.56-2.70 (5H, m), 3.56-3.65 (2H, m), 4.23 (2H, dd, J = 11.5, 4.5 Hz), 4.41-4.50 (1H, m), 7.19 (1H, dd, J = 8.6, 1.6 Hz), 7.49 (1H, d, J = 8.6Hz), 7.60-7.67 (2H, m), 8.16 (1H, dd, J = 8.6, 1.6 Hz), 8.54 (1H, d, J = 1.6 Hz).
5-(5-メトキシカルボニルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.88-1.94 (2H, m), 2.55-2.72 (5H, m), 3.56-3.65 (2H, m), 3.97 (3H, s), 4.24 (2H, dd, J = 11.9, 4.5 Hz), 4.42-4.52 (1H, m), 7.63 (1H, d, J = 8.6 Hz), 7.68 (1H, d, J = 8.6 Hz), 8.10 (1H, dd, J = 8.6, 1.6 Hz), 8.18 (1H, dd, J = 8.6, 1.6 Hz), 8.46 (1H, d, J = 1.6 Hz), 8.56 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-1-n-ブチル-2-メチルベンゾイミダゾールの合成
実施例74-1
2-(4-n-ブチルシアミノ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.02 (3H, t, J = 7.3 Hz), 1.45-1.59 (2H, m), 1.72-1.83 (2H, m), 3.40 (2H, td, J = 7.0, 5.3 Hz), 7.00 (1H, d, J = 9.1 Hz), 7.32-7.36 (2H, m), 7.55-7.59 (1H, m), 7.71-7.75 (1H, m), 8.29 (1H, dd, J = 9.1, 2.0 Hz), 8.38 (1H, br s), 9.06 (1H, d, J = 2.0 Hz).
実施例74-2
2-(2-n-ブチルアミノアニリン-5-イル)ベンゾオキサゾール

1H-NMR (CDCl3) δ: 0.99 (3H, t, J = 7.3 Hz), 1.42-1.56 (2H, m), 1.64-1.75 (2H, m), 3.21 (2H, t, J = 7.1 Hz), 6.71 (1H, d, J = 8.4 Hz), 7.23-7.33 (2H, m), 7.50-7.79 (4H, m).
実施例74-3
5-(ベンゾオキサゾール-2-イル)-1-n-ブチル-2-メチルベンゾイミダゾール

1H-NMR (CDCl3) δ: 0.99 (3H, t, J = 7.2 Hz), 1.35-1.50 (2H, m), 1.76-1.88 (2H, m), 2.65 (3H, s), 4.14 (2H, t, J = 7.3 Hz), 7.27-7.42 (3H, m), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 8.20 (1H, d, J = 8.2 Hz), 8.56 (1H, s).
5-(5-シアノベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.89-1.95 (2H, m), 2.54-2.72 (5H, m), 3.56-3.67 (2H, m), 4.24 (2H, dd, J = 11.9, 4.6 Hz), 4.42-4.54 (1H, m), 7.61-7.71 (3H, m), 8.05-8.06 (1H, m), 8.17 (1H, dd, J = 8.7, 1.5 Hz), 8.56 (1H, d, J = 1.5 Hz).
5-(5-トリフルオロメトキシベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.87-1.93 (2H, m), 2.54-2.72 (5H, m), 3.56-3.66 (2H, m), 4.24 (2H, dd, J = 11.8, 4.4 Hz), 4.40-4.54 (1H, m), 7.20-7.24 (1H, m), 7.57-7.70 (3H, m), 8.16 (1H, dd, J = 8.6, 1.3 Hz), 8.56 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)-2-トリクロロメチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 2.04-2.11 (2H, m), 2.63-2.79 (2H, m), 3.57-3.66 (2H, m), 4.25 (2H, dd, J = 11.7, 4.6 Hz), 5.29-5.38 (1H, m), 7.34-7.40 (2H, m), 7.59-7.64 (1H, m), 7.75-7.85 (2H, m), 8.34 (1H, dd, J = 8.7, 1.3 Hz), 8.77 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-(シクロヘキシサノン-4-イル)- 2-メチルベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.73-2.77 (9H, m), 5.03 (1H, br), 7.39-7.42 (2H, m), 7.77-7.81 (2H, m), 8.00-8.15 (2H, m), 8.36 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-ピコリル)ベンゾイミダゾールの合成
実施例79-1
2-(2-(2-ピコリル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (DMSO-D6) δ: 4.50 (2H, d, J = 5.8 Hz), 4.97 (2H, s), 6.10 (1H, t, J = 5.8 Hz), 6.43 (1H, d, J = 8.4 Hz), 7.24-7.37 (5H, m), 7.44 (1H, d, J = 2.0 Hz), 7.61-7.65 (2H, m), 7.75 (1H, td, J = 7.7, 2.0 Hz), 8.55 (1H, d, J = 4.1 Hz).
実施例79-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-ピコリル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.62 (3H, s), 5.64 (2H, s), 7.33-7.38 (4H, m), 7.74-7.79 (4H, m), 8.04 (1H, dd, J = 8.4, 1.3 Hz), 8.35 (1H, d, J = 1.3 Hz), 8.50 (1H, d, J = 4.9 Hz).
5-(ベンゾオキサゾール-2-イル) -2-メチル-1-iso-プロピルベンゾイミダゾールの合成
実施例80-1
2-(4-iso-プロピルアミノ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.38 (3H, s), 1.41 (3H, s), 3.88-4.01 (1H, m), 7.01 (1H, d, J = 9.2 Hz), 7.31-7.39 (2H, m), 7.55-7.59 (1H, m), 7.71-7.75 (1H, m), 8.26-8.36 (2H, m), 9.06 (1H, d, J = 2.1 Hz).
実施例80-2
2-(2-iso-プロピルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.28 (3H, s), 1.31 (3H, s), 3.68-3.78 (1H, m), 6.71 (1H, d, J = 8.4 Hz), 7.26-7.33 (2H, m), 7.50-7.78 (4H, m).
実施例80-3
5-(ベンゾオキサゾール-2-イル) -2-メチル-1-iso-プロピルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.67 (3H, s), 1.70 (3H, s), 2.67 (3H, s), 4.66-4.76 (1H, m), 7.30-7.37 (2H, m), 7.58-7.62 (2H, m), 7.74-7.79 (1H, m), 8.16 (1H, dd, J = 8.6, 1.5 Hz), 8.55 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル) -2-メチル-1-neo-ペンチルベンゾイミダゾールの合成
実施例81-1
2-(4-neo-ペンチルアミノ-3-ニトロフェニル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.11 (9H, s), 3.19 (2H, d, J = 5.1 Hz), 7.02 (1H, d, J = 9.1 Hz), 7.33-7.37 (2H, m), 7.55-7.59 (1H, m), 7.71-7.75 (1H, m), 8.28 (1H, dd, J = 9.1, 2.0 Hz), 8.60 (1H, br s), 9.07 (1H, d, J = 2.0 Hz).
実施例81-2
2-(2-neo-ペンチルアミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.06 (9H, s), 2.98 (2H, s), 6.73 (1H, d, J = 8.4 Hz), 7.23-7.33 (2H, m), 7.49-7.79 (4H, m).
実施例81-3
5-(ベンゾオキサゾール-2-イル) -2-メチル-1-neo-ペンチルベンゾイミダゾールの合成
1H-NMR (CDCl3) δ: 1.08 (9H, s), 2.67 (3H, s), 3.97 (2H, s), 7.32-7.37 (2H, m), 7.45 (1H, d, J = 8.6 Hz), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 8.18 (1H, dd, J = 8.6, 1.5 Hz), 8.55 (1H, d, J = 1.5 Hz).
5-(5-アミノベンゾオキサゾール-2-イル) -2-メチル-1-n-プロピルベンゾイミダゾールの合成
実施例82-1
3-ニトロ-4-n-プロピルアミノ安息香酸の合成

1H-NMR (CDCl3) δ: 1.08 (3H, t, J = 7.4 Hz), 1.74-1.84 (2H, m), 3.32-3.39 (2H, m), 6.90 (1H, d, J = 9.1 Hz), 8.09 (1H, dd, J = 9.1, 2.0 Hz), 8.45 (1H, br s), 8.97 (1H, d, J = 2.0 Hz).
実施例82-2
3-アミノ-4-n-プロピルアミノ安息香酸

1H-NMR (DMSO-D6) δ: 0.96 (3H, t, J = 7.3 Hz), 1.54-1.65 (2H, m), 3.02-3.10 (2H, m), 5.10 (1H, br s), 6.40 (1H, d, J = 8.2 Hz), 7.13-7.20 (2H, m).
実施例82-3
2-メチル-1-n-プロピルベンゾイミダゾール-5-カルボン酸

1H-NMR (DMSO-D6) δ: 0.94 (3H, t, J = 7.3 Hz), 1.76-1.89 (2H, m), 2.86 (3H, s), 4.39 (2H, t, J = 7.3 Hz), 8.03-8.11 (2H, m), 8.27 (1H, s).
実施例82-4
5-(5-アミノベンゾオキサゾール-2-イル) -2-メチル-1-n-プロピルベンゾイミダゾールの合成

1H-NMR (D2O) δ: 0.85 (3H, t, J = 7.4 Hz), 1.73-1.86 (2H, m), 2.72 (3H, s), 4.23 (2H, t, J = 7.3 Hz), 7.28 (1H, dd, J = 8.6, 2.1 Hz), 7.56 (1H, br s), 7.64 (1H, d, J = 8.6 Hz), 7.80 (1H, d, J = 8.6 Hz), 8.13 (1H, d, J = 8.6 Hz), 8.27 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-(テトラヒドロピラン-4-イル)エチル)ベンゾイミダゾールの合成
実施例83-1
2-(2-(テトラヒドロピラン-4-イル)エチル)ベンゾオキサゾールの合成

1H-NMR (DMSO-D6) δ: 1.07-1.66 (7H, m), 2.95-3.34 (4H, m), 3.85 (2H, dd, J = 11.0, 3.5 Hz), 4.91 (2H, br), 5.18 (1H, t, J = 5.2 Hz), 6.56 (1H, d, J = 8.1 Hz), 7.26-7.43 (5H, m), 7.62-7.67 (2H, m).
実施例83-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-(テトラヒドロピラン-4-イル)エチル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.21-1.34 (2H, m), 1.60-1.66 (5H, m), 2.60 (3H, s), 3.24-3.40 (2H, m), 3.85 (2H, dd, J = 11.4, 3.3 Hz), 4.27 (2H, t, J = 7.4 Hz), 7.38-7.42 (2H, m), 7.71-7.82 (3H, m), 8.08 (1H, dd, J = 8.5, 1.5 Hz), 8.33 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-((テトラヒドロピラン-4-イル)メチル)ベンゾイミダゾールの合成
実施例84-1
2-((テトラヒドロピラン-4-イル)メチル)ベンゾオキサゾールの合成
1H-NMR (DMSO-D6) δ: 1.22-1.275 (2H, m), 1.73 (2H, d, J = 12.7 Hz), 1.82-1.95 (1H, m), 3.05 (2H, t, J = 6.0 Hz), 3.27-3.32 (2H, m), 3.88 (2H, dd, J = 11.4, 2.8 Hz), 4.92 (2H, s), 5.27 (1H, t, J = 5.4 Hz), 6.57 (1H, d, J = 8.9 Hz), 7.26-7.35 (2H, m), 7.39-7.43 (2H, m), 7.60-7.69 (2H, m).
実施例84-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-((テトラヒドロピラン-4-イル)メチル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.37-1.41 (4H, m), 2.06-2.12 (1H, m), 2.61 (3H, s), 3.18-3.28 (2H, m), 3.83 (2H, d, J = 11.0 Hz), 4.16 (2H, d, J = 7.4 Hz), 7.39-7.41 (2H, m), 7.77-7.82 (3H, m), 8.06-8.09 (1H, m), 8.32 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(3,3,3-トリフルオロプロピル)ベンゾイミダゾールの合成
実施例85-1
2-(2-(3,3,3-トリフルオロプロピル)アミノアニリン-5-イル)ベンゾオキサゾールの合成

1H-NMR (CDCl3) δ: 1.59 (1H, br s), 2.47-2.55 (2H, m), 3.34 (1H, br s), 3.54 (2H, t, J = 6.4 Hz), 4.09 (1H, br s), 6.71 (1H, d, J = 8.2 Hz), 7.22-7.35 (2H, m), 7.52-7.56 (1H, m), 7.66-7.72 (2H, m), 7.78 (1H, dd, J = 8.2, 2.0 Hz).
実施例85-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(3,3,3-トリフルオロプロピル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.63 (3H, s), 2.83-3.00 (2H, m), 4.55 (2H, t, J = 6.9 Hz), 7.36-7.40 (2H, m), 7.75-7.82 (3H, m), 8.10 (1H, dd, J = 8.5, 1.3 Hz), 8.34 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(酢酸エチルエステル-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.30 (3H, t, J = 7.1 Hz), 1.92-2.00 (2H, m), 2.58-2.73 (2H, m), 3.53-3.63 (2H, m), 4.12 (2H, s), 4.19-4.27 (4H, m), 4.41-4.54 (1H, m), 7.33-7.37 (2H, m), 7.58-7.66 (1H, m), 7.73-7.80 (2H, m), 8.22 (1H, dd, J = 8.6, 1.4 Hz), 8.61 (1H, d, J = 1.4 Hz).
実施例87-1
2-(2-(1,2,3,4-テトラヒドロナフタレン-1-イル)アミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.80-2.05 (4H, m), 2.82-2.88 (2H, m), 3.28 (2H, br s), 4.20 (1H, d, J = 6.9 Hz), 4.71-4.79 (1H, br m), 6.88 (1H, d, J = 8.4 Hz), 7.15-7.39 (6H, m), 7.50-7.53 (1H, m), 7.67-7.70 (2H, m), 7.80 (1H, dd, J = 8.4, 2.0 Hz).
実施例87-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(1,2,3,4-テトラヒドロナフタレン-1イル)ベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 1.90-2.34 (4H, m), 2.64 (3H, s), 2.93-3.16 (2H, m), 5.71 (1H, t, J = 8.6 Hz), 6.72 (1H, d, J = 7.7 Hz), 6.82-6.90 (1H, br m), 7.01-7.07 (1H, m), 7.24-7.34 (4H, m), 7.56-7.60 (1H, m), 7.72-7.78 (1H, m), 7.96 (1H, d, J = 8.5 Hz), 8.57 (1H, t, J = 2.3 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-ヒドロキシエチル)-2-メチルベンゾイミダゾ-ルの合成
実施例88-1
2-(2-(2-ヒドロキシエチル)アミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.87 (1H, s), 3.41 (4H, br), 3.94 (2H, d, J = 4.5 Hz), 4.26 (1H, s), 6.74 (1H, d, J = 8.2 Hz), 7.26-7.34 (2H, m), 7.51-7.55 (1H, m), 7.64 (1H, d, J = 2.0 Hz), 7.68-7.72 (1H, m), 7.76 (1H, dd, J = 8.2, 2.0 Hz).
実施例88-2
5-(ベンゾオキサゾール-2-イル)-1-(2-ヒドロキシエチル)-2-メチルベンゾイミダゾ-ルの合成

1H-NMR (DMSO-D6) δ: 2.61 (3H, s), 3.75 (2H, q, J = 5.3 Hz), 4.31 (2H, t, J = 5.3 Hz), 4.99 (1H, t, J = 5.3 Hz), 7.38-7.41 (2H, m), 7.73-7.79 (3H, m), 8.07 (1H, dd, J = 8.4, 1.5 Hz), 8.32 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-1-ジフェニルメチル-2-メチルベンゾイミダゾ-ルの合成
実施例89-1
2-(2-ジフェニルメチルアミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 3.41 (2H, br s), 4.49 (1H, br s), 5.63 (1H, br s), 6.52 (1H, d, J = 8.4 Hz), 7.27-7.35 (12H, m), 7.47-7.50 (1H, m), 7.60 (1H, dd, J = 8.2, 2.0 Hz), 7.65-7.69 (2H, m).
実施例89-2
5-(ベンゾオキサゾール-2-イル)-1-ジフェニルメチル-2-メチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 2.59 (3H, s), 6.66 (1H, d, J = 8.7 Hz), 6.91 (1H, s), 7.14-7.18 (4H, m), 7.29-7.39 (8H, m), 7.54-7.60 (1H, m), 7.72-7.78 (1H, m), 7.91 (1H, dd, J = 8.7, 1.6 Hz), 8.56 (1H, d, J = 1.2 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(テトラヒドロフラン-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.86-2.45 (5H, m), 2.52-2.76 (2H, m), 2.90-3.02 (1H, m), 3.55-3.67 (2H, m), 3.86-4.00 (2H, m), 4.21 (2H, dd, J = 11.6, 4.5 Hz), 4.83-4.96 (1H, m), 5.23 (1H, dd, J = 7.1, 6.1 Hz), 7.30-7.38 (2H, m), 7.58-7.62 (1H, m), 7.72-7.80 (2H, m), 8.21 (1H, dd, J = 8.7, 1.5 Hz), 8.65 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-tert-ブトキシカルボニルメチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例91-1
2-(2-tert-ブトキシカルボニルメチルアミノ)アニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.52 (9H, s), 3.72-3.80 (3H, m), 3.90 (2H, s), 6.59 (1H, d, J = 8.2 Hz), 7.29-7.31 (2H, m), 7.50-7.52 (1H, m), 7.64 (1H, d, J = 1.8 Hz), 7.68-7.77 (2H, m).
実施例91-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-tert-ブトキシカルボニルメチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.45 (9H, s), 2.63 (3H, s), 4.76 (2H, s), 7.32-7.37 (3H, m), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 8.23 (1H, dd, J = 8.5, 1.5 Hz), 8.57 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-1-カルボキシメチル-2-メチルベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.51 (3H, s), 5.04 (2H, s), 7.38-7.42 (2H, m), 7.67-7.70 (1H, m), 7.77-7.81 (2H, m), 8.04-8.08 (1H, m), 8.32-8.33 (1H, m).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-(チオモルホリン-1,1-ジオキシド-4-イル)エチル)ベンゾイミダゾールの合成
実施例93-1
2-(N-(2-(チオモルホリン-1,1-ジオキシド-4-イル)エチル)-2-ニトロアニリン-4-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 2.96 (2H, t, J = 5.9 Hz), 3.15 (8H, s), 3.48 (2H, q, J = 5.5 Hz), 6.96 (1H, d, J = 8.9 Hz), 7.34-7.39 (2H, m), 7.56-7.60 (1H, m), 7.72-7.75 (1H, m), 8.33 (1H, dd, J = 8.9, 2.0 Hz), 8.82 (1H, br s), 9.07 (1H, d, J = 2.0 Hz).
実施例93-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-(チオモルホリン-1,1-ジオキシド-4-イル)エチル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.67 (3H, s), 3.26-3.31 (2H, m), 3.42-3.48 (2H, m), 3.77 (2H, t, J = 7.3 Hz), 3.90-3.99 (4H, m), 4.84 (2H, t, J = 7.3 Hz), 7.39-7.42 (2H, m), 7.78-7.84 (3H, m), 8.11 (1H, dd, J = 8.4, 1.4 Hz), 8.34 (1H, d, J = 1.4 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)-4-トリフルオロメチルベンゾイミダゾ-ルの合成
実施例94-1
2-(2-フルオロ-3-トリフルオロメチルニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 7.44-7.55 (2H, m), 7.85-7.92 (2H, m), 8.74 (1H, dd, J = 6.0, 1.8 Hz), 9.04 (1H, dd, J = 6.6, 1.8 Hz).
実施例94-2
2-(2-(テトラヒドロフラン-2-イル)アミノ-3-トリフルオロメチルアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.56-1.59 (2H, m), 1.83 (2H, d, J = 13.0 Hz), 3.40 (2H, t, J = 11.4 Hz), 3.61 (2H, s), 3.98-4.02 (4H, m), 7.33-7.38 (2H, m), 7.54-7.59 (1H, m), 7.75 (2H, t, J = 4.3 Hz), 7.89 (1H, s).
実施例94-3
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)-4-トリフルオロメチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 1.96 (2H, dd, J = 12.1, 2.1 Hz), 2.40-2.49 (2H, m), 2.88 (3H, s), 3.59 (2H, td, J = 11.8, 1.8 Hz), 4.21 (2H, dd, J = 11.8, 4.5 Hz), 4.97-5.02 (1H, m), 7.33-7.41 (2H, m), 7.63 (1H, dd, J = 6.1, 3.1 Hz), 7.80 (1H, dd, J = 5.9, 3.1 Hz), 8.57 (1H, d, J = 1.5 Hz), 8.71 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロフラン-2-イル)メチルベンゾイミダゾ-ルの合成
実施例95-1
2-(2-(テトラヒドロフラン-2-イル)メチルアミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.64-1.77 (1H, m), 1.92-2.02 (2H, m), 2.04-2.17 (1H, m), 3.17 (1H, dd, J = 12.3, 7.8 Hz), 3.34 (1H, dd, J = 12.3, 3.7 Hz), 3.78-3.96 (2H, m), 4.17-4.27 (1H, m), 6.71 (1H, d, J = 8.2 Hz), 7.27-7.34 (2H, m), 7.50-7.54 (1H, m), 7.62 (1H, d, J = 2.0 Hz), 7.68-7.71 (1H, m), 7.75 (1H, dd, J = 8.4, 2.0 Hz).
実施例95-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロフラン-2-イル)メチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 1.56-2.14 (4H, m), 2.69 (3H, s), 3.77-3.83 (2H, m), 4.18-4.29 (3H, m), 7.30-7.37 (2H, m), 7.46 (1H, d, J = 8.6 Hz), 7.58-7.62 (1H, m), 7.75-7.79 (1H, m), 8.20 (1H, dd, J = 8.6, 1.5 Hz), 8.55 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2メチル-1-(3-(モルホリン-4-イル)プロピル)ベンゾイミダゾールの合成
実施例96-1
2-(N-(2-(モルホリン-4-イル)-n-プロピル)-2-ニトロアニリン-4-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.89-1.99 (2H, m), 2.47-2.55 (6H, m), 3.47-3.54 (2H, m), 3.79 (4H, t, J = 4.7 Hz), 7.05 (1H, d, J = 9.1 Hz), 7.31-7.39 (2H, m), 7.55-7.60 (1H, m), 7.71-7.75 (1H, m), 8.29 (1H, dd, J = 9.1, 2.1 Hz), 8.74 (1H, br s), 9.07 (1H, d, J = 2.1 Hz).
実施例96-2
2-(2-(3-(モルホリン-4-イル)-n-プロピル)アミノアニリン-4-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.85-1.95 (2H, m), 2.49-2.57 (6H, m), 3.31 (2H, t, J = 6.2 Hz), 3.38 (1H, br s), 3.77 (4H, t, J = 4.7 Hz), 6.68 (1H, d, J = 8.4 Hz), 7.26-7.33 (2H, m), 7.50-7.77 (4H, m).
実施例96-3
5-(ベンゾオキサゾール-2-イル)-2メチル-1-(3-(モルホリン-4-イル)-n-プロピル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.97-2.06 (2H, m), 2.31 (2H, t, J = 6.5 Hz), 2.40 (4H, t, J = 4.5 Hz), 2.68 (3H, s), 3.73 (4H, t, J = 4.7 Hz), 4.27 (2H, t, J = 6.7 Hz), 7.32-7.36 (2H, m), 7.50 (1H, d, J = 8.6 Hz), 7.59-7.62 (1H, m), 7.75-7.78 (1H, m), 8.20 (1H, dd, J = 8.6, 1.5 Hz), 8.55 (1H, d, J = 1.5 Hz).
5-(5-カルボキシルベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.85-1.91 (2H, m), 2.35-2.44 (2H, m), 2.68 (3H, s), 3.58 (2H, t, J = 11.0 Hz), 4.06 (2H, dd, J = 11.0, 3.8 Hz), 4.61-4.73 (1H, m), 7.87-7.92 (2H, m), 8.01-8.09 (2H, m), 8.29 (1H, d, J = 1.4 Hz), 8.35 (1H, d, J = 1.4 Hz).
5-(5-アセチルアミノベンゾオキサゾール-2-イル)-2-メチル-1-n-プロピルベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 0.91 (3H, t, J = 7.3 Hz), 1.70-1.85 (2H, m), 2.08 (3H, s), 2.60 (3H, s), 4.22 (2H, t, J = 7.3 Hz), 7.48 (1H, dd, J = 8.7, 2.1 Hz), 7.68-7.76 (2H, m), 8.00-8.10 (2H, m), 8.30 (1H, d, J = 1.3 Hz), 10.10 (1H, s).
2-メチル-5-(5-n-プロピルアミノカルボニルベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.02 (3H, t, J = 7.3 Hz), 1.61-1.73 (2H, m), 1.90 (2H, dd, J = 12.9, 2.9 Hz), 2.55-2.69 (2H, m), 2.71 (3H, s), 3.47 (2H, dd, J = 13.6, 6.3 Hz), 3.56-3.66 (2H, m), 4.24 (2H, dd, J = 11.8, 4.5 Hz), 4.42-4.51 (1H, m), 6.21 (1H, t, J = 4.9 Hz), 7.62-7.70 (2H, m), 7.84 (1H, dd, J = 8.6, 1.6 Hz), 8.11 (1H, d, J = 1.6 Hz), 8.17 (1H, dd, J = 8.6, 1.6 Hz), 8.56 (1H, d, J = 1.6 Hz).
2-(4-アセチルアミノフェニル)-5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
実施例100-1
5-クロロ-2-(2-フルオロニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 7.54 (1H, d, J = 8.4 Hz), 7.84-7.89 (2H, m), 7.99 (1H, s), 8.54-8.59 (1H, m), 8.80 (1H, d, J = 8.4 Hz).
実施例100-2
5-クロロ-2-(2-(テトラヒドロピラン-4-イル)アミノアニリン-5-イル) ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 1.40-1.54 (2H, m), 1.94 (2H, d, J = 12.4 Hz), 3.46 (2H, t, J = 11.4 Hz), 3.60 (1H, br s), 3.90 (2H, d, J = 11.4 Hz), 4.97 (2H, s), 5.12 (1H, d, J = 7.6 Hz), 6.67 (1H, d, J = 8.3 Hz), 7.33 (1H, dd, J = 8.3, 1.9 Hz), 7.38-7.41 (2H, m), 7.68-7.74 (2H, m).
実施例100-3
2-(4-アセチルアミノフェニル)-5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.86-1.93 (2H, m), 2.25 (3H, s), 2.66-2.80 (2H, m), 3.47 (2H, t, J = 11.4 Hz), 4.11-4.20 (2H, m), 4.60-4.69 (1H, m), 7.32 (1H, dd, J = 8.7, 2.1 Hz), 7.52-7.83 (7H, m), 8.22 (1H, dd, J = 8.7, 1.5 Hz), 8.65 (1H, d, J = 1.5 Hz).
5-(5-クロロベンゾオキサゾール-2-イル)-2-(4-メトキシカルボニルフェニル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.88-1.96 (2H, m), 2.65-2.81 (2H, m), 3.40-3.50 (2H, m), 4.00 (3H, s), 4.13-4.22 (2H, m), 4.55-4.64 (1H, m), 7.33 (1H, dd, J = 8.6, 2.1 Hz), 7.54 (1H, d, J = 9.1 Hz), 7.74-7.77 (3H, m), 7.84 (1H, d, J = 8.7 Hz), 8.23-8.27 (3H, m), 8.68 (1H, d, J = 1.3 Hz).
2-(4-カルボキシルフェニル)-5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.93-2.02 (2H, m), 2.51-2.59 (2H, m), 3.35-3.47 (2H, m), 3.98-4.05 (2H, m), 4.55-4.67 (1H, m), 7.47 (1H, dd, J = 8.7, 2.1 Hz), 7.84-7.93 (4H, m), 8.08-8.20 (4H, m), 8.50 (1H, s).
5-(5-クロロベンゾオキサゾール-2-イル)-2-(3-メトキシカルボニルフェニル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成
5-クロロ-2-(2-(テトラヒドロピラン-4-イル)アミノアニリン-5-イル) ベンゾオキサゾ-ル(実施例100-2参照)(250mg、0.727mmol)のジメチルホルムアミド(3ml)溶液に、3-ホルミル安息香酸メチル(143mg、0.872mmol)、オキソン(313mg、0.509mmol)を加え、室温で2時間攪拌した。反応終了後、炭酸カリウム水溶液を加え、ろ過、水洗浄後、得られた結晶をシリカゲルカラムクロマトグラフィーで精製することで表題化合物(220mg、62%)を黄色結晶として得た。
1H-NMR (CDCl3) δ: 1.91-1.97 (2H, m), 2.66-2.81 (2H, m), 3.41-3.51 (2H, m), 3.97 (3H, s), 4.13-4.21 (2H, m), 4.55-4.64 (1H, m), 7.32 (1H, dd, J = 8.6, 2.1 Hz), 7.54 (1H, d, J = 8.6 Hz), 7.67 (1H, t, J = 7.7 Hz), 7.75-7.91 (3H, m), 8.22-8.27 (2H, m), 8.34 (1H, t, J = 1.5 Hz), 8.67 (1H, d, J = 1.5 Hz).
2-アセチルアミノメチル-5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.83-1.91 (2H, m), 2.13 (3H, s), 2.53-2.69 (2H, m), 3.57-3.66 (2H, m), 4.21 (2H, dd, J = 11.7, 4.3 Hz), 4.60-4.72 (1H, m), 4.77 (2H, d, J = 4.9 Hz), 6.82 (1H, br s), 7.32 (1H, dd, J = 8.6, 2.1 Hz), 7.52 (1H, d, J = 8.6 Hz), 7.72-7.75 (2H, m), 8.19 (1H, dd, J = 8.6, 1.5 Hz), 8.57 (1H, d, J = 1.5 Hz).
2-(3-カルボキシルフェニル)-5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.93-2.01 (2H, m), 2.51-2.59 (2H, m), 3.33-3.45 (2H, m), 3.99-4.06 (2H, m), 4.55-4.67 (1H, m), 7.45-7.49 (1H, m), 7.73-8.00 (4H, m), 8.07-8.19 (3H, m), 8.28 (1H, br s), 8.50 (1H, br s).
2-(3-アセチルアミノフェニル)-5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.91-1.98 (2H, m), 2.20 (3H, s), 2.63-2.79 (2H, m), 3.47-3.56 (2H, m), 4.14-4.20 (2H, m), 4.71-4.84 (1H, m), 7.32 (1H, dd, J = 8.7, 2.1 Hz), 7.41-7.60 (4H, m), 7.75 (1H, d, J = 2.0 Hz), 7.84 (1H, d, J = 8.7 Hz), 8.00 (1H, br s), 8.22 (1H, dd, J = 8.7, 1.6 Hz), 8.65 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-((4-モルホリニル)カルボニルメチル)ベンゾイミダゾ-ルの合成
実施例107-1
2-(2-((4-モルホリニル)カルボニルメチル)アミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 3.48-3.73 (10H, m), 3.96 (2H, d, J = 4.1 Hz), 5.09 (1H, t, J = 4.1 Hz), 6.58 (1H, dd, J = 8.3, 2.1 Hz), 7.26-7.35 (2H, m), 7.49-7.54 (1H, m), 7.62 (1H, t, J = 1.8 Hz), 7.66-7.83 (2H, m).
実施例107-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(4-モルホリニル)ベンゾイミダゾ-ルの合成

1H-NMR (DMSO-D6) δ: 2.47 (3H, s), 3.39-3.84 (8H, m), 5.34 (2H, s), 7.39-7.41 (2H, m), 7.67 (1H, d, J = 8.6 Hz), 7.78-7.80 (2H, m), 8.07 (1H, d, J = 8.6 Hz), 8.34 (1H, s).
4-オキソ-4-(2-(5-(ベンゾオキサゾール-2-イル)-2-メチルベンゾイミダゾ-ル-1-イル)エチル)アミノ酪酸の合成

1H-NMR (DMSO-D6) δ: 2.22 (2H, t, J = 6.8 Hz), 2.36 (2H, t, J = 6.8 Hz), 2.57 (3H, s), 3.41-3.45 (2H, m), 4.29 (2H, t, J = 5.9 Hz), 7.39-7.43 (2H, m), 7.68 (1H, d, J = 8.6 Hz), 7.78-7.80 (2H, m), 8.07-8.11 (2H, m), 8.33 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-トリフルオロアセチルアミノエチル)ベンゾイミダゾ-ルの合成

1H-NMR (DMSO-D6) δ: 2.58 (3H, s), 3.58-3.63 (2H, m), 4.42 (2H, t, J = 5.6 Hz), 7.41 (2H, dd, J = 5.4, 3.8 Hz), 7.69 (1H, d, J = 8.4 Hz), 7.78-7.80 (2H, m), 8.07-8.09 (1H, m), 8.33 (1H, s), 9.66 (1H, br).
3-(2-(5-(ベンゾオキサゾール-2-イル)-2-メチルベンゾイミダゾ-ル-1-イル)エチル)-1-メチルチオ尿素の合成

1H-NMR (DMSO-D6) δ: 2.50-2.62 (6H, m), 2.76 (1H, br s), 3.79-3.81 (2H, m), 4.43 (2H, t, J = 5.5 Hz), 7.39-7.42 (2H, m), 7.54 (1H, br s), 7.74-7.79 (3H, m), 8.07 (1H, dd, J = 8.5, 1.4 Hz), 8.33 (1H, d, J = 1.4 Hz).
5-(ベンゾオキサゾール-2-イル) -1-(テトラヒドロピラン-4-イル)-2-(テトラヒドロピラン-4-イル)メチルベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.41-1.89 (6H, m), 2.20-2.32 (1H, m), 2.60-2.76 (2H, m), 2.90 (2H, d, J = 7.3 Hz), 3.45 (2H, td, J = 11.7, 1.9 Hz), 3.61 (2H, t, J = 11.3 Hz), 4.00 (2H, dd, J = 11.1, 3.5 Hz), 4.25 (2H, dd, J = 11.8, 4.2 Hz), 4.41-4.54 (1H, m), 7.33-7.36 (2H, m), 7.57-7.80 (3H, m), 8.19 (1H, dd, J = 8.7, 1.6 Hz), 8.59 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)-6-クロロベンゾイミダゾ-ルの合成
実施例112-1
2-(4-クロロ-2-フルオロニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 7.44-7.56 (2H, m), 7.85-7.92 (2H, m), 8.21 (1H, d, J = 11.0 Hz), 8.87 (1H, d, J = 7.9 Hz).
実施例112-2
2-(4-クロロ-2-(テトラヒドロピラン-4-イル)アミノニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.66-1.81 (2H, m), 2.10-2.17 (2H, m), 3.57-3.66 (2H, m), 3.76-3.81 (1H, m), 4.07 (2H, dt, J = 12.1, 3.8 Hz), 7.05 (1H, s), 7.35-7.42 (2H, m), 7.56-7.67 (1H, m), 7.78-7.84 (1H, m), 8.28 (1H, d, J = 7.3 Hz), 9.09 (1H, t, J = 2.9 Hz).
実施例112-3
2-(4-クロロ-2-(テトラヒドロピラン-4-イル)アミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.50-1.64 (2H, m), 2.04-2.11(2H, m), 3.31(2H, br), 3.61 (2H, t, J = 11.4 Hz), 3.95-4.08(3H, m), 6.72(1H, s), 7.26-7.36 (2H, m), 7.53-7.59 (2H, m), 7.74-7.81(1H, m).
実施例112-4
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)-6-クロロベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 1.92-1.97 (2H, m), 2.50-2.66(2H, m), 2.78 (3H, s), 3.61 (2H, t, J = 11.5 Hz), 4.25 (2H, dd, J = 11.5, 4.7 Hz), 4.42-4.45 (1H, m), 7.36-7.43 (2H, m), 7.62-7.65 (1H, m), 7.79 (1H, s), 7.84-7.88 (1H, m), 8.47 (1H, s).
5-(ベンゾオキサゾール-2-イル)- 1-(4-メトキシカルボニルフェニルメチル)- 2-メチルベンゾイミダゾ-ルの合成
実施例113-1
2-(2-(4-メトキシカルボニルフェニルメチル)アミノニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 3.84 (3H, s), 4.84 (2H, d, J = 6.3 Hz), 7.07 (1H, d, J = 9.2 Hz), 7.35-7.42 (2H, m), 7.54 (2H, d, J = 8.2 Hz), 7.72-7.78 (2H, m), 7.95 (2H, d, J = 8.2 Hz), 8.15 (1H, dd, J = 9.1, 2.1 Hz), 8.85 (1H, d, J = 2.1 Hz), 9.20 (1H, t, J = 6.3 Hz).
実施例113-2
2-(2-(4-メトキシカルボニルフェニルメチル)アミノアニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 3.92 (3H, s), 4.50 (2H, s), 6.65 (1H, d, J = 8.2 Hz), 7.26-7.32 (2H, m), 7.45-7.53 (3H, m), 7.67-7.73 (3H, m), 8.04 (2H, d, J = 8.2 Hz).
実施例113-3
5-(ベンゾオキサゾール-2-イル)- 1-(4-メトキシカルボニルフェニルメチル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (DMSO-D6) δ: 2.56 (3H, s), 3.83 (3H, s), 5.67 (2H, s), 7.28 (2H, d, J = 8.2 Hz), 7.38-7.42 5(2H, m), 7.70 (1H, d, J = 8.5 Hz), 7.77-7.81 (2H, m), 7.94 (2H, d, J = 8.2 Hz), 8.07 (1H, dd, J = 8.5, 1.6 Hz), 8.38 (1H, s).
5-(ベンゾオキサゾール-2-イル)- 1-(4-カルボキシルフェニルメチル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (DMSO-D6) δ: 2.57 (3H, s), 5.66 (2H, s), 7.24 (2H, d, J = 8.2 Hz), 7.38-7.42 (2H, m), 7.70 (1H, d, J = 8.5 Hz), 7.77-7.81 (2H, m), 7.91 (2H, d, J = 8.2 Hz), 8.07 (1H, dd, J = 8.5, 1.6 Hz), 8.38 (1H, s).
5-(1-ベンゾオキサゾール-2-イル)- 1-(1-ブタノール-2-イル)- 2-メチルベンゾイミダゾ-ルの合成
実施例115-1
2-(2-(1-n-ブタノール-2-イル)アミノニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.06 (3H, t, J = 7.4 Hz), 1.65-1.90 (2H, m), 2.00-2.04 (1H, m), 3.75-3.90 (3H, m), 7.10 (1H, d, J = 9.1 Hz), 7.33-7.37 (2H, m), 7.54-7.59 (1H, m), 7.70-7.76 (1H, m), 8.25 (1H, dd, J = 9.1, 2.1 Hz), 8.47 (1H, d, J = 7.1 Hz), 9.04 (1H, d, J = 2.1 Hz).
実施例115-2
5-(1-ベンゾオキサゾール-2-イル)- 1-(1-n-ブタノール-2-イル)- 2-メチルベンゾイミダゾ-ルの合成

(実施例115-1参照)(295mg、0.901mmol)のエタノール、酢酸エチル(1:1, 6ml)溶液に、10%パラジウム炭素(50mg)を加え、フラスコ内を水素で置換し、室温で19時間攪拌した。反応終了後、セライト濾過し、濾液を濃縮した。得られた残渣のエタノール(5ml)溶液に、アセトイミド酸エチル塩酸塩(133mg、1.08mmol)を加え、2時間加熱還流した。反応終了後、飽和炭酸水素ナトリウム水溶液を加え、室温で数分攪拌した。得られた結晶を濾過、水で洗浄後、乾燥することで表題化合物(232mg、72%)を白色結晶として得た。
1H-NMR (CDCl3) δ: 0.84 (3H, t, J = 7.4 Hz), 1.93-2.20 (2H, m), 2.61 (3H, s), 3.96-4.02 (1H, m), 4.32-4.49 (2H, m), 7.29-7.44 (4H, m), 7.65-7.68 (1H, m), 7.73 (1H, dd, J = 8.7, 1.6 Hz), 7.87 (1H, d, J = 1.5 Hz).
5-(1-ベンゾオキサゾール-2-イル)- 1-(2-n-プロパノール-1-イル)- 2-メチルベンゾイミダゾ-ルの合成
実施例116-1
2-(2-(2-n-プロパノール-1-イル)アミノニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 1.18 (3H, d, J = 6.1 Hz), 3.26-3.34 (1H, m), 3.48-3.56 (1H, m), 3.88-3.99 (1H, m), 5.13 (1H, d, J = 4.9 Hz), 7.33 (1H, d, J = 9.2 Hz), 7.38-7.42 (2H, m), 7.74-7.79 (2H, m), 8.23 (1H, dd, J = 9.1, 1.8 Hz), 8.68 (1H, t, J = 5.4 Hz), 8.82 (1H, d, J = 2.1 Hz).
実施例116-2
5-(1-ベンゾオキサゾール-2-イル)- 1-(2-n-プロパノール-1-イル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 1.48 (3H, d, J = 6.3 Hz), 2.64 (3H, s), 3.90-4.15 (2H, m), 4.45-4.52 (1H, m), 5.63 (1H, br s), 7.29-7.41 (4H, m), 7.66-7.79 (3H, m).
5-(ベンゾオキサゾール-2-イル)- 1-(3-メトキシカルボニルフェニルメチル)- 2-メチルベンゾイミダゾ-ルの合成
実施例117-1
2-(2-(3-メトキシカルボニルフェニルメチル)アミノニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (DMSO-D6) δ: 3.85 (3H, s), 4.82 (2H, d, J = 6.3 Hz), 7.14 (1H, d, J = 9.1 Hz), 7.37-7.40 (2H, m), 7.52 (1H, t, J = 7.8 Hz), 7.68-7.77 (3H, m), 7.87 (1H, d, J = 7.8 Hz), 8.03 (1H, s), 8.15 (1H, dd, J = 9.1, 2.1 Hz), 8.84 (1H, d, J = 2.1 Hz), 9.21 (1H, t, J = 6.5 Hz).
実施例117-2
5-(ベンゾオキサゾール-2-イル)- 1-(3-メトキシカルボニルフェニルメチル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 2.63 (3H, s), 3.91 (3H, s), 5.42 (2H, s), 7.18 (1H, d, J = 7.8 Hz), 7.32-7.44 (4H, m), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 7.89 (1H, s), 7.99 (1H, d, J = 7.8 Hz), 8.19 (1H, dd, J = 8.6, 1.5 Hz), 8.62 (1H, d, J = 1.5 Hz).
5-(1-ベンゾオキサゾール-2-イル)- 1-(1-アセトキシ-2-フェニルエタン-2-イル)- 2-メチルベンゾイミダゾ-ルの合成
実施例118-1
2-(2-(2-フェニル-1-エタノール-2-イル)アミノニトロベンゼン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.93 (1H, br s), 3.95-4.13 (2H, m), 4.82 (1H, dd, J = 10.4, 5.9 Hz), 6.77 (1H, d, J = 9.1 Hz), 7.29-7.44 (7H, m), 7.51-7.56 (1H, m), 7.68-7.72 (1H, m), 8.12 (1H, dd, J = 9.1, 2.1 Hz), 9.06-9.12 (2H, m).
実施例118-2
5-(1-ベンゾオキサゾール-2-イル)- 1-(1-アセトキシ-2-フェニルエタン-2-イル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 1.96 (3H, s), 2.66 (3H, s), 4.91 (1H, dd, J = 11.5, 9.4 Hz), 5.08 (1H, dd, J = 11.5, 5.0 Hz), 5.95 (1H, dd, J = 9.4, 5.0 Hz), 7.11 (1H, d, J = 8.6 Hz), 7.23-7.26 (2H, m), 7.30-7.43 (5H, m), 7.57-7.61 (1H, m), 7.74-7.77 (1H, m), 8.04 (1H, dd, J = 8.6, 1.5 Hz), 8.58 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)- 1-(3-カルボキシルフェニルメチル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (DMSO-D6) δ: 2.57 (3H, s), 5.65 (2H, s), 7.34-7.50 (4H, m), 7.72-7.87 (5H, m), 8.07 (1H, dd, J = 8.4, 1.4 Hz), 8.38 (1H, d, J = 1.4 Hz),.
5-(1-ベンゾオキサゾール-2-イル)- 1-(2-フェニルエタノール-2-イル)- 2-メチルベンゾイミダゾ-ルの合成

1H-NMR (CDCl3) δ: 2.67 (3H, s), 4.67 (1H, dd, J = 12.0, 4.0 Hz), 4.80 (1H, dd, J = 12.0, 9.4 Hz), 5.76 (1H, dd, J = 9.4, 4.0 Hz), 6.95 (1H, d, J = 8.6 Hz), 7.20-7.24 (2H, m), 7.28-7.39 (6H, m), 7.55 (1H, dd, J = 8.6, 1.4 Hz), 7.60-7.65 (1H, m), 7.79 (1H, d, J = 1.4 Hz).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-tert-ブトキシカルボニル-n-プロピル)ベンゾイミダゾールの合成
実施例121-1
2-(2-tert-ブトキシカルボニル-n-プロピルアミノ)アニリン-5-イル)ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 1.25 (3H, d, J = 7.1 Hz), 1.46 (9H, s), 2.68-2.82 (1H, m), 3.24-3.49 (4H, m), 4.31 (1H, br), 6.71 (1H, d, J = 8.4 Hz), 7.24-7.31 (2H, m), 7.48-7.55 (1H, m), 7.62 (1H, d, J = 2.0 Hz), 7.64-7.77 (2H, m).
実施例121-2
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-tert-ブトキシカルボニル-n-プロピル)ベンゾイミダゾールの合成

1H-NMR (CDCl3) δ: 1.23 (3H, d, J = 7.1 Hz), 1.30 (9H, s), 2.67 (3H, s), 2.91-3.08 (1H, m), 4.09 (1H, dd, J = 14.7, 7.1 Hz), 4.46 (1H, dd, J = 14.7, 8.3 Hz), 7.27 (1H, s), 7.32-7.37 (1H, m), 7.45 (1H, d, J = 8.4 Hz), 7.57-7.62 (1H, m), 7.75-7.79 (1H, m), 8.20 (1H, dd, J = 8.4, 1.2 Hz), 8.55 (1H, s).
5-(ベンゾオキサゾール-2-イル)-2-メチル-1-(2-カルボニル-n-プロピル)ベンゾイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.15 (3H, d, J = 6.9 Hz), 2.61 (3H, s), 2.97-3.06 (1H, m), 4.28 (1H, dd, J = 14.8, 6.8 Hz), 4.49 (1H, dd, J = 14.8, 8.6 Hz), 7.39-7.42 (2H, m), 7.77-7.80 (3H, m), 8.08 (1H, dd, J = 8.6, 1.3 Hz), 8.33 (1H, d, J = 1.3 Hz).
5-(5-クロロベンゾオキサゾール-2-イル)-2-(4-n-プロピルアミノカルボニルフェニル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.01 (3H, t, J = 7.4 Hz), 1.61-1.74 (2H, m), 1.89-1.96 (2H, m), 2.65-2.81 (2H, m), 3.42-3.51 (4H, m), 4.18 (2H, dd, J = 11.7, 4.3 Hz), 4.57-4.66 (1H, m), 6.37 (1H, br s), 7.33 (1H, dd, J = 8.7, 2.1 Hz), 7.54 (1H, d, J = 8.6 Hz), 7.66 (1H, t, J = 7.7 Hz), 7.74-7.78 (2H, m), 7.84 (1H, d, J = 8.9 Hz), 7.99 (1H, dt, J = 7.7, 1.5 Hz), 8.10 (1H, s), 8.25 (1H, dd, J = 8.7, 1.6 Hz), 8.67 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(4-メトキシフェニル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.87-1.93 (2H, m), 2.70-2.79 (2H, m), 3.43-3.51 (2H, m), 3.92(3H, s), 4.18 (2H, dd, J = 11.7, 4.6 Hz), 4.60-4.69 (1H, m), 7.07-7.11 (2H, m), 7.34-7.39 (2H, m), 7.58-7.64 (3H, m), 7.77-7.82 (2H, m), 8.24 (1H, dd, J = 8.7, 1.6 Hz), 8.67 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(2-メトキシフェニル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 2.02 (2H, br s), 2.63 (2H, br s), 3.36-3.46 (2H, m), 3.82 (3H, s), 4.13-4.30 (3H, m), 7.07 (1H, d, J = 8.4 Hz), 7.15 (1H, t, J = 7.5 Hz), 7.32-7.39 (2H, m), 7.52-7.64 (3H, m), 7.77-7.82 (2H, m), 8.25 (1H, dd, J = 8.6, 1.6 Hz), 8.68 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(2-クロロフェニル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.69-1.73 (1H, m), 2.15-2.20 (1H, m), 2.51-2.75 (2H, m), 3.32-3.49 (2H, m), 4.07-4.21 (3H, m), 7.33-7.65 (7H, m), 7.78-7.84 (2H, m), 8.29 (1H, dd, J = 8.6, 1.6 Hz), 8.71 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(3-クロロフェニル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.91 (2H, dd, J = 12.9, 3.3 Hz), 2.66-2.81 (2H, m), 3.43-3.53 (2H, m), 4.19 (2H, dd, J = 11.8, 4.4 Hz), 4.53-4.65 (1H, m), 7.33-7.39 (2H, m), 7.49-7.86 (7H, m), 8.28 (1H, dd, J = 8.7, 1.6 Hz), 8.70 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(4-クロロフェニル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.90 (2H, dd, J = 13.0, 3.3 Hz), 2.65-2.81 (2H, m), 3.42-3.52 (2H, m), 4.18 (2H, dd, J = 11.7, 4.6 Hz), 4.51-4.64 (1H, m), 7.33-7.41 (2H, m), 7.55-7.64 (5H, m), 7.77-7.84 (2H, m), 8.27 (1H, dd, J = 8.7, 1.6 Hz), 8.69 (1H, d, J = 1.6 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-(ピペリジン-1-イル)エチル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成
実施例129-1
N-(2-(ピペリジン-1-イル)エチル)-4-(ベンゾオキサゾール-2-イル)-2-ニトロアニリンの合成

1H-NMR (CDCl3) δ: 1.45-1.69 (6H, m), 2.48 (4H, t, J = 4.9 Hz), 2.70 (2H, t, J = 6.2 Hz), 3.44 (2H, dd, J = 10.8, 6.2 Hz), 6.97 (1H, d, J = 9.1 Hz), 7.28-7.37 (2H, m), 7.54-7.58 (1H, m), 7.71-7.76 (1H, m), 8.29 (1H, dd, J = 9.0, 2.1 Hz), 8.92 (1H, br s), 9.07 (1H, d, J = 2.1 Hz).
実施例129-2
5-(ベンゾオキサゾール-2-イル)-1-(2-(ピペリジン-1-イル)エチル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.44-1.62 (6H, m), 2.43-2.47 (4H, m), 2.67 (2H, t, J = 6.8 Hz), 2.68 (3H, s), 4.24 (2H, t, J = 6.8 Hz), 7.32-7.36 (2H, m), 7.42 (1H, d, J = 8.6 Hz), 7.57-7.80 (2H, m), 8.20 (1H, dd, J = 8.6, 1.5 Hz), 8.55 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-ジメチルアミノエチル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成
実施例130-1
N-(2-ジメチルアミノエチル)-4-(ベンゾオキサゾール-2-イル)-2-ニトロアニリンの合成

1G3058451NON_E2_FT.als
1H-NMR (CDCl3) δ: 2.34 (6H, s), 2.68 (2H, t, J = 6.1 Hz), 3.44 (2H, dd, J = 10.9, 6.1 Hz), 6.98 (1H, d, J = 9.1 Hz), 7.32-7.38 (2H, m), 7.55-7.59 (1H, m), 7.71-7.75 (1H, m), 8.30 (1H, dd, J = 9.1, 2.0 Hz), 8.72 (1H, br s), 9.07 (1H, d, J = 2.0 Hz).
実施例130-2
5-(ベンゾオキサゾール-2-イル)-1-(2-ジメチルアミノエチル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 2.33 (6H, s), 2.67 (2H, t, J = 7.1 Hz), 2.68 (3H, s), 4.23 (2H, t, J = 7.1 Hz), 7.32-7.36 (2H, m), 7.42 (1H, d, J = 8.4 Hz), 7.59-7.62 (1H, m), 7.75-7.79 (1H, m), 8.21 (1H, dd, J = 8.4, 1.5 Hz), 8.56 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-(メチルチオ)エチル)-2-メチルベンズイミダゾールの合成
実施例131-1
N-(2-(メチルチオ)エチル)-4-(ベンゾオキサゾール-2-イル)-2-ニトロアニリンの合成

1H-NMR (CDCl3) δ: 2.20 (3H, s), 2.89 (2H, t, J = 6.7 Hz), 3.61-3.68 (2H, m), 7.01 (1H, d, J = 9.1 Hz), 7.31-7.38 (2H, m), 7.56-7.59 (1H, m), 7.72-7.75 (1H, m), 8.32 (1H, dd, J = 9.1, 2.0 Hz), 8.61 (1H, br s), 9.08 (1H, d, J = 2.0 Hz).
実施例131-2
5-(ベンゾオキサゾール-2-イル)-1-(2-(メチルチオ)エチル)-2-メチルベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 2.08 (3H, s), 2.72 (3H, s), 2.93 (2H, t, J = 7.1 Hz), 4.38 (2H, t, J = 7.1 Hz), 7.32-7.36 (2H, m), 7.43 (1H, d, J = 8.6 Hz), 7.59-7.63 (1H, m), 7.74-7.79 (1H, m), 8.23 (1H, dd, J = 8.6, 1.5 Hz), 8.57 (1H, d, J = 1.5 Hz).
5-(ベンゾオキサゾール-2-イル)-1-(2-(メチルスルホニル)チオエチル)-2-メチルベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 2.74 (3H, s), 2.80 (3H, s), 3.52 (2H, t, J = 6.9 Hz), 4.73 (2H, t, J = 6.9 Hz), 7.34-7.39 (2H, m), 7.47 (1H, d, J = 8.4 Hz), 7.59-7.63 (1H, m), 7.76-7.79 (1H, m), 8.26 (1H, dd, J = 8.5, 1.4 Hz), 8.58 (1H, d, J = 1.3 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(2-(メチルチオ)エチル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.91 (2H, dd, J = 13.0, 3.3 Hz), 2.20 (3H, s), 2.59-2.75 (2H, m), 3.07-3.13 (2H, m), 3.21-3.29 (2H, m), 3.61 (2H, td, J = 12.0, 1.8 Hz), 4.24 (2H, dd, J = 11.7, 4.5 Hz), 4.46-4.55 (1H, m), 7.31-7.38 (2H, m), 7.57-7.63 (1H, m), 7.70 (1H, dd, J = 8.7, 0.5 Hz), 7.74-7.81 (1H, m), 8.20 (1H, dd, J = 8.7, 1.6 Hz), 8.60 (1H, dd, J = 1.6, 0.5 Hz).
5-(ベンゾオキサゾール-2-イル)-2-(2-(メチルスルホニル)エチル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (CDCl3) δ: 1.86-1.94 (2H, m), 2.56-2.72 (2H, m), 3.05 (3H, s), 3.48 (2H, t, J = 7.5 Hz), 3.61 (2H, t, J = 11.5 Hz), 3.86 (2H, t, J = 7.5 Hz), 4.19-4.26 (2H, m), 4.47-4.56 (1H, m), 7.33-7.38 (2H, m), 7.58-7.79 (3H, m), 8.21 (1H, dd, J = 8.7, 1.6 Hz), 8.57 (1H, s).
5-(5-クロロベンゾオキサゾール-2-イル)-1-n-プロピルベンズイミダゾールの合成
実施例135-1
5-(5-クロロベンゾオキサゾール-2-イル)-2-n-プロピルアミノアニリンの合成

1H-NMR (CDCl3) δ: 1.05 (3H, t, J = 7.5 Hz), 1.67-1.80 (2H, m), 3.18 (2H, t, J = 7.1 Hz), 6.70 (1H, d, J = 8.5 Hz), 7.23 (1H, dd, J = 8.5, 1.9Hz), 7.41 (1H, d, J = 8.8 Hz), 7.59-7.65 (2H, m), 7.73 (1H, dd, J = 8.5, 1.9 Hz).
実施例135-2

1H-NMR (DMSO-D6) δ: 0.87 (3H, t, J = 7.3 Hz), 1.79-1.92 (2H, m), 4.29 (2H, t, J = 7.1 Hz), 7.45 (1H, dd, J = 8.8, 2.2 Hz), 7.82-7.91 (3H, m), 8.13 (1H, dd, J = 8.4, 1.5 Hz), 8.45-8.46 (2H, m).
5-(5-クロロベンゾオキサゾール-2-イル)-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.02-2.22 (4H, m), 3.58 (2H, td, J = 11.5, 2.2 Hz), 4.04 (2H, dd, J = 11.5, 3.1 Hz), 4.71-4.83 (1H, m), 7.45 (1H, dd, J = 8.7, 2.1 Hz), 7.83 (1H, d, J = 8.6 Hz), 7.90 (1H, d, J = 2.1 Hz), 7.97 (1H, d, J = 8.6 Hz), 8.14 (1H, dd, J = 8.6, 1.6 Hz), 8.47 (1H, d, J = 1.5 Hz), 8.60 (1H, s).
5-(5-クロロベンゾオキサゾール-2-イル)-2-メチル-1-n-プロピルベンズイミダゾールの合成

1H-NMR (DMSO-D6) δ: 0.90 (3H, t, J = 7.4 Hz), 1.71-1.81 (2H, m), 2.60 (3H, s), 4.22 (2H, t, J = 7.3 Hz), 7.44 (1H, dd, J = 8.6, 2.1 Hz), 7.76 (1H, d, J = 8.6 Hz), 7.82 (1H, d, J = 8.7 Hz), 7.89 (1H, d, J = 2.1 Hz), 8.06 (1H, dd, J = 8.5, 1.6 Hz), 8.32 (1H, d, J = 1.2 Hz).
5-(5-クロロベンゾオキサゾール-2-イル)-2-メチル-1-(3,3,3-トリフルオロプロピル)ベンズイミダゾールの合成
実施例138-1
5-クロロ-2-(2-(3,3,3-トリフルオロプロピル)アミノアニリン-5-イル) ベンゾオキサゾ-ルの合成

1H-NMR (CDCl3) δ: 2.45-2.57 (2H, m), 3.54 (2H, t, J = 7.0 Hz), 6.70 (1H, d, J = 8.4 Hz), 7.25 (1H, dd, J = 8.7, 1.9 Hz), 7.43 (1H, dd, J = 8.6, 0.5 Hz), 7.62-7.66 (2H, m), 7.76 (1H, dd, J = 8.4, 2.0 Hz).
実施例138-2
5-(5-クロロベンゾオキサゾール-2-イル)-2-メチル-1-(3,3,3-トリフルオロプロピル)ベンズイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.63 (3H, s), 2.82-3.00 (2H, m), 4.55 (2H, t, J = 6.9 Hz), 7.45 (1H, dd, J = 8.7, 2.1 Hz), 7.77 (1H, d, J = 8.4 Hz), 7.83 (1H, d, J = 8.7 Hz), 7.90 (1H, d, J = 2.1 Hz), 8.09 (1H, dd, J = 8.5, 1.6 Hz), 8.33 (1H, d, J = 1.6 Hz).
5-(5-クロロベンゾオキサゾール-2-イル) -1-(3,3,3-トリフルオロプロピル)ベンズイミダゾールの合成

1H-NMR (DMSO-D6) δ: 2.91-3.09 (2H, m), 4.63 (2H, t, J = 6.8 Hz), 7.46 (1H, dd, J = 8.6, 1.3 Hz), 7.84 (1H, d, J = 8.7 Hz), 7.91-7.94 (2H, m), 8.16 (1H, d, J = 8.4 Hz), 8.47-8.50 (2H, m).
5-(ベンズイミダゾール-2-イル)-2-メチル-1-(テトラヒドロピラン-4-イル)ベンズイミダゾールの合成

1H-NMR (DMSO-D6) δ: 1.86 (2H, dd, J = 12.3, 2.7 Hz), 2.36-2.45 (2H, m), 2.65 (3H, s), 3.53-3.62 (2H, m), 4.03-4.08 (2H, m), 4.58-4.69 (1H, m), 7.15-7.21 (2H, m), 7.49-7.67 (2H, m), 7.81 (1H, d, J = 8.4 Hz), 8.06 (1H, dd, J = 8.6, 1.6 Hz), 8.33 (1H, d, J = 1.5 Hz), 12.82 (1H, br s).
(化合物評価用細胞の作製)
NXF遺伝子のゲノム配列(Accession No.AB054577およびNC_000085)の配列情報に従い、PCRプライマーとしてフォワードプライマー: (5’-tccagtatttgagaaaaggagccaggagtctccat-3’)、リバースプライマー: (5’-ggaggcttcctcttccttgcttcccggtcttttcg-3’) を作製し、NXF遺伝子プロモーター部分翻訳領域の上流約5kbpから転写開始点近傍までをPCR法で単離した。この際、マウスゲノムDNA(タカラ社製)1μgを鋳型にし、PCR反応の変性条件として95℃で1分、アニーリング&伸張条件として68℃で8分の2ステップ反応を35回繰り返すことで、PCR産物を得た。得られたNXFのプロモーター部分断片を同プロモーターの下流にルシフェラーゼ遺伝子が配置されるようにpGL3 Basicベクター(Promega社製)のSmaIサイトに導入し、NXFのプロモーターの作用でルシフェラーゼ酵素が発現するレポータープラスミドを作製した。
PC12細胞(ATCCより購入)は、RPMI培地(GIBCO-BRL社製)に5%FCS(GIBCO-BRL社製)と15%ウマ血清(GIBCO-BRL社製)、及び1mMピルビン酸ナトリウム(GIBCO-BRL社製)を添加したものを用いて、37℃、5%、CO2存在下で培養した。このPC12細胞106個に対して上記のレポータープラスミド12μgおよび1μgのpRC/RSVベクター(Invitrogen社製)をLipofectamine2000試薬(Invitrogen社製)を用いてトランスシクションし、トランスフェクション2日後に40mg/LのGeneticin sulfate(GIBCO社製)を含んだ培養液に交換して培養を継続した。数日ごとに40mg/LのGeneticin sulfate(GIBCO社製)を含んだ培養液で培地交換を続けながら約1ヶ月間培養を継続して、結果として得られた細胞コロニーを複数をピックアップした。このうち、40ng/mlのNGF(和光純薬社製)存在下にルシフェラーゼ活性が誘導される一つのコロニーを選択し、以下の化合物活性評価用細胞とした。
96穴プレートに8割コンフルエントになった同細胞を準備し、Promega社製Steady Glo luciferaseアッセイキットを用いて、ルシフェラーゼレポーターアッセイを実施した。化合物を含む培養液による処理は終夜(約16時間)の条件で実施し、化合物を含む培養液を全部除いた後に残った細胞に対してPBSで半分に希釈したSteady Glo基質液を添加し、30分放置後、そのそれぞれの発光量をルミノメーター(パーキンエルマー社製Envision)で測定した。
評価対象化合物の活性は、200ng/mlのNGFの活性の増強活性として測定した。対照実験区としては、最終濃度0.1%DMSOのみを含む培養液を陰性対照区、飽和濃度のNGFを含む培養区を陽性対照区とし、また化合物濃度0に相当する200ng/mlのNGFに加えて0.1%DMSOを含む実験区を置き、そのルシフェラーゼ活性を基準としたときの、200ng/mlのNGFに加えて化合物溶液(DMSOに溶解させ、DMSOの最終濃度が0.1%になるように調製)を添加した実験区の相対的ルシフェラーゼ活性を算出し、それを化合物による200ng/mlのNGFの活性の増強活性とした。
NGFの活性の増強活性の試験結果を以下の表に記載する。
+ :比較的弱い増強活性を示す(10μMでのみ増強活性を示す)
++ :増強活性を示す
+++:特に強い増強活性を示す(1μMでNGF200ng/mlの活性を2倍以上増強する)

雄性8週齢C57BL/6J系マウスを12日間通常飼育した後に、あらかじめ左総頚、外総動脈を結紮し、中大脳動脈を90分間閉塞した後に再開通することにより中大脳動脈閉塞・再開通モデルを作成した。同モデルに化合物を閉塞3時間後、24時間後48時間後に30mg/kgで尾静脈内投与した。閉塞・再開通3日後に断頭し、頭骨を小脳側より切断して脳を摘出した。摘出した脳をブアン液(pH3.5~4.0)に入れ固定した。固定した脳より、冠状断の切り出しを行い、切り出し後、脱水、透徹後、パラフィン包埋した。薄切は、Bregma 1.95mm、Bregma 1.0mm、Bregma -1.40mmおよびBregma -3.8mm付近の4部位とした。厚さ約6μmの切片を作製し、HE染色を行った。梗塞体積の算出は以下のように実施した。
光学顕微鏡 (対物レンズ×1)に取り付けたカメラ(MCD-350、オリンパス株式会社) を用いてデジタル画像を取り込んだ。取り込んだデジタル画像をAdobe Photoshop 2.0に1枚の台紙(新しいレイヤー)に貼り付けた。台紙に貼り付けた画像を基にして画像解析装置(Win ROOF V5.6 三谷商事株式会社)で梗塞面積(S、mm2)、梗塞側(左)面積(mm2)および非梗塞側(右)面積MR、mm2)を測定し、梗塞側(左)の非梗塞面積(ML、mm2)[梗塞側(左)面積(mm2)-梗塞面積(mm2)]を算出した。
以下の式で梗塞体積(SV、mm3)、梗塞側(左)の非梗塞体積(MLV、mm3)および非梗塞側(右)体積(MRV、mm3)を算出した。
SV=1/3Σ(S1+S2+S1 1/2×S2 1/2)
MLV=1/3Σ(ML1+ML2+ML1 1/2×ML2 1/2)
MRV=1/3Σ(MR1+MR2+MR1 1/2×MR2 1/2)
SV=2/3Σ(Sn+Sn+1+Sn 1/2×Sn+1 1/2)
MLV=2/3Σ(MLn+MLn+1+MLn 1/2×MLn+1 1/2)
MRV=2/3Σ(MRn+MRn+1+MRn 1/2×MRn+1 1/2)(n=1、2、3、4)
ストレプトゾトシン(Streptozotocin)誘発糖尿病ラットにおける坐骨神経での運動神経伝達速度(MNCV)の潜時の遅延に対する化合物の予防効果を検討した。
群構成は非糖尿病対照群、糖尿病対照群および化合物投与群とした。雄性8週齢Slc:Wistar系ラットを7または8日間通常飼育した後に、非糖尿病対照群ではpH4.5の0.75mmol/Lクエン酸緩衝溶液を2ml/kgで、糖尿病対照群および化合物投与群では0.75mmol/Lクエン酸緩衝溶液でストレプトゾトシン(化学名:N-(Methylnitrosocarbamoyl)-α-D-glucosamine、製造元:SIGMA)を20mg/mlに調製した溶液を2ml/kgでそれぞれ1回尾静脈内投与した。その後4週飼育した後に、非糖尿病対照群および糖尿病対照群には注射用水を5ml/kgで、化合物投与群には注射用水で10mg/mlに調製した化合物溶液を5ml/kgで各群とも、1日1回経口投与で4週間投与した。MNCVの測定は各群ともに3回実施した。測定時期は、1回目をストレプトゾトシン溶液またはpH4.5の0.75mmol/Lクエン酸緩衝溶液の投与前日、2回目をストレプトゾトシン溶液またはpH4.5の0.75mmol/Lクエン酸緩衝溶液投与の4週間後、3回目を4週間の注射用水または化合物溶液投与の最終日の翌日に測定した。
実施例12の化合物の試験結果(運動神経伝達速度)を図2(図中、STZはストレプトゾトシンを表す。)に示す。この様に化合物投与群において溶媒対照投与群と比較して、運動神経伝達速度の回復が認められた。
試験例1と同様にして、表2に示す化合物の実施例NGF活性の増強活性を評価した。結果を表2に示す。
+ :比較的弱い増強活性を示す(10μMでのみ増強活性を示す)
++ :増強活性を示す
+++:特に強い増強活性を示す(1μMでNGF200ng/mlの活性を2倍以上増強する)

配列番号2は、リバースプライマーである。
Claims (13)
- 式
[式中、
R1は、
(1)C3-6アルキル基、
(2)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O-、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(式中、各Raは、同一または相異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されたC1-6アルキル基、
(3)
(a)オキソ基、及び
(b)C1-4アルコキシ-カルボニル基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、又は
(4)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換された芳香族炭化水素環基を表し、
Xは、
NH、O、又はSを表し、
Yは、
CH、又はNを表し、
Zは、
N、又はC-R2を表し、
R2は、
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-S-、
(f)Rb-SO2-、
(g)Rb-CO-O-、
(h)Rb-CO-NRb-、
(i)Rb-NRb-、
(j)Rb-NRb-Rb-S(O)n-
(k)フェニル基、
(l)5~6員飽和複素環基、
(m)ヒドロキシ基、及び
(n)アミノ基、
(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基でそれぞれ置換されていてもよいC1-6アルキル基、C2-6アルケニル基、又はC2-6アルキニル基、又は、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基
を表し、
環Aは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)ハロゲン原子で置換されていてもよいアルキル基、
(i)ニトロ基、及び
(j)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環を表し、
環Bは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)アミド基、
(i)エステル基、
(j)ハロゲン原子で置換されていてもよいアルキル基、
(k)ニトロ基、及び
(l)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、またはその溶媒和物。 - Zは、
C-R2であり、及び
Yは、
Nである
請求項1に記載の化合物。 - R1は、
5~6員非芳香族複素環基である
請求項1に記載の化合物。 - R2は、
C1-6アルキル基である
請求項1に記載の化合物。 - Zは、
C-R2であり、
Yは、
Nであり、
R1は、
5~6員非芳香族複素環基であり、及び
R2は、
C1-6アルキル基である
請求項1に記載の化合物。 - 請求項1に記載の化合物を含有する医薬組成物。
- 神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、または理学療法効果の促進剤である請求項6に記載の医薬組成物。
- 神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、または理学療法効果の促進剤が、脳虚血性疾患、または糖尿病性神経障害の治療、または予防剤である請求項7に記載の医薬組成物。
- 式
[式中、
ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物を有効成分として含有する、神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、又は理学療法効果の促進剤。 - 環Aは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)ハロゲン原子で置換されていてもよいアルキル基、
(i)ニトロ基、及び
(j)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環であり、
環Bは、
(a)ハロゲン原子、
(b)水酸基、
(c)カルボキシ基、
(d)シアノ基、
(e)スルファモイル基、
(f)モノアルキルアミド基、
(g)ジアルキルアミド基、
(h)アミド基、
(i)エステル基、
(j)ハロゲン原子で置換されていてもよいアルキル基、
(k)ニトロ基、及び
(l)アリールオキシ基
からなる群から選ばれる1個以上の置換基で置換されていてもよいベンゼン環であり、
ZはN、又はC-R2であり、
Xは、
NH、O、又はSであり、
Yは、
CH、又はNであり、
R1は、
(1)水素原子、
(2)C1-6アルキル基、
(3)
(a)ハロゲン原子、
(b)Ra-O-、
(c)Ra-O-CO-、
(d)Ra-O-CO-NRa-、
(e)Ra-O-CO-C2H4-CO-NH-、
(f)Ra-S-、
(g)Ra-SO2-、
(h)Ra-CO-O-、
(i)Ra-CO-NRa-、
(j)Ra-NRa-、
(k)Ra-NRa-CS-NRa-、
(l)5~6員の環状基、
(m)カルボキシ基、
(n)ヒドロキシ基、
(o)アミノ基、
(p)複素環-カルボニル基、
(q)HO-CO-C2H4-CO-NH-
(式中、各Raは、同一または相異なって、ハロゲンで置換されていてもよいC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されたC1-6アルキル基、
(4)
(a)オキソ基、
(b)C1-4アルコキシ-カルボニル基、及び
(c)ヒドロキシ基
からなる群から選ばれる1個以上の置換基で、それぞれ置換されていてもよい、炭素数3~10の非芳香族環状炭化水素基または5~6員非芳香族複素環基、又は
(5)ハロゲン原子及びC1-4アルコキシ基からなる群から選ばれる1以上の基で置換されていてもよい芳香族炭化水素環基を表し、
R2は、
(1)水素原子、
(2)
(a)ハロゲン原子、
(b)Rb-O-、
(c)Rb-O-CO-、
(d)Rb-O-CO-NRb-、
(e)Rb-S-、
(f)Rb-SO2-、
(g)Rb-CO-O-、
(h)Rb-CO-NRb-、
(i)Rb-NRb-、
(j)Rb-NRb-Rb-S(O)n-
(k)フェニル基、
(l)5~6員飽和複素環基、
(m)ヒドロキシ基、及び
(n)アミノ基
(式中、各Rbは、同一または異なって、水素原子又は1個以上のハロゲンで置換されていてもよいC1-6アルキル基を表し、nは0から2の整数を表す。)
からなる群から選択される1個以上の置換基でそれぞれ置換されていてもよいC1-6アルキル基、C2-6アルケニル基、又はC2-6アルキニル基、
(3)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
炭素数5~6の非芳香族環状炭化水素基又は5~6員非芳香族複素環基、又は
(4)
(a)ハロゲン原子、
(b)Rc-O-、
(c)Rc-O-CO-、及び
(d)Rc-CO-NRc-
(式中、Rcは、同一または異なって、水素原子又はC1-6アルキル基を表す。)
からなる群から選択される1個以上の置換基で置換されていてもよい
フェニル基であるか、あるいは、
R1およびR2は、これらが隣接するNおよびCと一緒になって、5~8員含窒素非芳香族複素環を形成してもよく、
X1は、
O、又はNHであり、および
R2Zは、
C1-6アルキル基である
請求項9に記載の剤。 - 脳虚血性疾患、または糖尿病性神経障害の治療、または予防剤である、請求項9に記載の剤。
- 式
[式中、
ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物、又はその有効量を哺乳動物に投与することを特徴とする、神経栄養因子の活性が関与する疾患の治療、若しくは予防方法、又は理学療法効果の促進方法。 - 神経栄養因子の活性が関与する疾患の治療、若しくは予防剤、又は理学療法効果の促進剤を製造するための、式
[式中、
ZはN、又はC-R2を表し、
Xは、
N-R3、O、又はSを表し、
Yは、
C-R4、又はNを表し、
R1は、
水素原子、置換されていてもよい非環状炭化水素基、または置換されていてもよい環状基を表し、
R2は、水素原子、置換されていてもよい非環状炭化水素基、又は置換されていてもよい環状基を表し、
R3は、水素原子、又は置換基を表し、
R4は、水素原子、又は置換基を表し、
環Aは、
置換されていてもよいベンゼン環を表し、
環Bは、
置換されていてもよいベンゼン環を表す。]
で示される化合物、その薬学的に許容される塩、又はその溶媒和物の使用。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES10780309.0T ES2484169T3 (es) | 2009-05-29 | 2010-05-28 | Agente para el tratamiento o la prevención de enfermedades asociadas a la actividad de factores neurotróficos |
| BRPI1011961A BRPI1011961A2 (pt) | 2009-05-29 | 2010-05-28 | agente para o tratamento ou prevenção de doenças associadas com a atividade de fatores neurotróficos. |
| AU2010253336A AU2010253336A1 (en) | 2009-05-29 | 2010-05-28 | Agent for treatment or prevention of diseases associated with activity of neurotrophic factors |
| EP10780309.0A EP2436683B1 (en) | 2009-05-29 | 2010-05-28 | Agent for treatment or prevention of diseases associated with activity of neurotrophic factors |
| CN2010800218228A CN102428080A (zh) | 2009-05-29 | 2010-05-28 | 神经营养因子活性参与的疾病的治疗或预防剂 |
| CA2762620A CA2762620A1 (en) | 2009-05-29 | 2010-05-28 | Agent for treatment or prevention of diseases associated with activity of neurotrophic factors |
| MX2011012537A MX2011012537A (es) | 2009-05-29 | 2010-05-28 | Agente para el tratamiento o prevencion de enfermedades asociadas con actividad de factores neurotroficos. |
| US13/322,888 US8829035B2 (en) | 2009-05-29 | 2010-05-28 | Agent for treatment or prevention of diseases associated with activity of neurotrophic factors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009131333 | 2009-05-29 | ||
| JP2009-131333 | 2009-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010137349A1 true WO2010137349A1 (ja) | 2010-12-02 |
Family
ID=43222475
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/003622 WO2010137349A1 (ja) | 2009-05-29 | 2010-05-28 | 神経栄養因子の活性が関与する疾患の治療または予防剤 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US8829035B2 (ja) |
| EP (1) | EP2436683B1 (ja) |
| JP (1) | JP2011006409A (ja) |
| KR (1) | KR20120093066A (ja) |
| CN (1) | CN102428080A (ja) |
| AU (1) | AU2010253336A1 (ja) |
| BR (1) | BRPI1011961A2 (ja) |
| CA (1) | CA2762620A1 (ja) |
| ES (1) | ES2484169T3 (ja) |
| MX (1) | MX2011012537A (ja) |
| WO (1) | WO2010137349A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017055147A1 (en) | 2015-09-28 | 2017-04-06 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9096549B2 (en) | 2013-02-01 | 2015-08-04 | Acetylon Pharmaceuticals, Inc. | Selective HDAC3 inhibitors |
| KR20200093705A (ko) * | 2013-09-05 | 2020-08-05 | 미쓰비시 타나베 파마 코퍼레이션 | 신규한 결정성 아릴알킬아민 화합물 및 그의 제조 방법 |
| JP5829741B2 (ja) * | 2013-09-05 | 2015-12-09 | 田辺三菱製薬株式会社 | 新規な結晶性アリールアルキルアミン化合物およびその製造方法 |
| EP3177604A1 (en) * | 2014-08-05 | 2017-06-14 | Bristol-Myers Squibb Company | Heterocyclic kinase inhibitors |
| CN108299413A (zh) * | 2018-01-22 | 2018-07-20 | 复旦大学 | 一种无金属催化制备2-(胺基苯基)苯并噁唑及其衍生物的方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0468470A1 (de) | 1990-07-23 | 1992-01-29 | Dr. Karl Thomae GmbH | Benzimidazole, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung |
| JPH04308580A (ja) | 1991-04-05 | 1992-10-30 | Sumitomo Chem Co Ltd | ベンズイミダゾール誘導体、その製造法およびそれを有効成分とする農園芸用殺菌剤 |
| JPH0881464A (ja) * | 1994-09-12 | 1996-03-26 | Lilly Ind Ltd | インドール誘導体及び該誘導体を含む医薬用組成物 |
| DE19920936A1 (de) | 1999-05-07 | 2000-11-09 | Basf Ag | Heterozyklisch substituierte Benzimidazole, deren Herstellung und Anwendung |
| WO2001000213A1 (en) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| WO2003053344A2 (en) | 2001-12-10 | 2003-07-03 | Bristol-Myers Squibb Company | Substituted 2-methyl-benzimidazole respiratory syncytial virus antiviral agents |
| WO2005082901A1 (en) | 2004-02-25 | 2005-09-09 | Smithkline Beecham Corporation | Novel chemical compounds |
| WO2007115408A1 (en) * | 2006-04-10 | 2007-10-18 | Painceptor Pharma Corporation | Compositions and methods for modulating gated ion channels |
| JP2007282502A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | 細胞変性制御能力の検定方法 |
| JP2007282501A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | アポトーシス制御能力の検定方法 |
| WO2008073451A2 (en) * | 2006-12-11 | 2008-06-19 | Sirtris Pharmaceuticals, Inc. | Benzoimidazole derivatives as sirtuin (sir) modulating compounds |
| WO2008153701A1 (en) | 2007-05-24 | 2008-12-18 | Schering Corporation | Compounds for inhibiting ksp kinesin activity |
| US20090156613A1 (en) * | 2007-12-18 | 2009-06-18 | Kindrachuk David E | Bicyclic heteroaryl-substituted imidazoles as modulators of the histamine H4 receptor |
| JP2010047517A (ja) * | 2008-08-21 | 2010-03-04 | Sumitomo Chemical Co Ltd | 神経栄養因子の活性が関与する疾患の治療または予防剤、およびその製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1063028B (de) * | 1957-09-10 | 1959-08-06 | Wolfen Filmfab Veb | Verfahren zur Sensibilisierung von Halogensilberemulsionen |
| DE4023369A1 (de) | 1990-07-23 | 1992-01-30 | Thomae Gmbh Dr K | Benzimidazole, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| GB9418326D0 (en) | 1994-09-12 | 1994-11-02 | Lilly Industries Ltd | Pharmaceutical compounds |
| BR0010262A (pt) | 1999-05-03 | 2002-01-15 | Battelle Memorial Institute | Composição para formar um aerossol, processo de preparação e de aerossolização de uma composição, e, dispositivo gerador de aerossol |
| CN1623992A (zh) * | 2003-12-03 | 2005-06-08 | 北京师范大学 | 新型非肽类血管紧张素ⅱ受体拮抗剂类药物、它们的制备及应用 |
-
2010
- 2010-05-28 WO PCT/JP2010/003622 patent/WO2010137349A1/ja active Application Filing
- 2010-05-28 CN CN2010800218228A patent/CN102428080A/zh active Pending
- 2010-05-28 JP JP2010123614A patent/JP2011006409A/ja not_active Ceased
- 2010-05-28 AU AU2010253336A patent/AU2010253336A1/en not_active Abandoned
- 2010-05-28 EP EP10780309.0A patent/EP2436683B1/en active Active
- 2010-05-28 MX MX2011012537A patent/MX2011012537A/es not_active Application Discontinuation
- 2010-05-28 CA CA2762620A patent/CA2762620A1/en not_active Abandoned
- 2010-05-28 BR BRPI1011961A patent/BRPI1011961A2/pt not_active IP Right Cessation
- 2010-05-28 ES ES10780309.0T patent/ES2484169T3/es active Active
- 2010-05-28 KR KR1020117031347A patent/KR20120093066A/ko not_active Application Discontinuation
- 2010-05-28 US US13/322,888 patent/US8829035B2/en not_active Expired - Fee Related
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0468470A1 (de) | 1990-07-23 | 1992-01-29 | Dr. Karl Thomae GmbH | Benzimidazole, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung |
| JPH04308580A (ja) | 1991-04-05 | 1992-10-30 | Sumitomo Chem Co Ltd | ベンズイミダゾール誘導体、その製造法およびそれを有効成分とする農園芸用殺菌剤 |
| JPH0881464A (ja) * | 1994-09-12 | 1996-03-26 | Lilly Ind Ltd | インドール誘導体及び該誘導体を含む医薬用組成物 |
| DE19920936A1 (de) | 1999-05-07 | 2000-11-09 | Basf Ag | Heterozyklisch substituierte Benzimidazole, deren Herstellung und Anwendung |
| JP2002544199A (ja) * | 1999-05-07 | 2002-12-24 | ビーエーエスエフ アクチェンゲゼルシャフト | ヘテロ環置換されたベンズイミダゾール、その製造方法およびその使用 |
| WO2001000213A1 (en) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| WO2003053344A2 (en) | 2001-12-10 | 2003-07-03 | Bristol-Myers Squibb Company | Substituted 2-methyl-benzimidazole respiratory syncytial virus antiviral agents |
| WO2005082901A1 (en) | 2004-02-25 | 2005-09-09 | Smithkline Beecham Corporation | Novel chemical compounds |
| JP2007282502A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | 細胞変性制御能力の検定方法 |
| JP2007282501A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | アポトーシス制御能力の検定方法 |
| WO2007115408A1 (en) * | 2006-04-10 | 2007-10-18 | Painceptor Pharma Corporation | Compositions and methods for modulating gated ion channels |
| WO2008073451A2 (en) * | 2006-12-11 | 2008-06-19 | Sirtris Pharmaceuticals, Inc. | Benzoimidazole derivatives as sirtuin (sir) modulating compounds |
| WO2008153701A1 (en) | 2007-05-24 | 2008-12-18 | Schering Corporation | Compounds for inhibiting ksp kinesin activity |
| US20090156613A1 (en) * | 2007-12-18 | 2009-06-18 | Kindrachuk David E | Bicyclic heteroaryl-substituted imidazoles as modulators of the histamine H4 receptor |
| JP2010047517A (ja) * | 2008-08-21 | 2010-03-04 | Sumitomo Chemical Co Ltd | 神経栄養因子の活性が関与する疾患の治療または予防剤、およびその製造方法 |
Non-Patent Citations (16)
| Title |
|---|
| "From the hyperacute phase to prevention", vol. 1, 2006, article "Studies on Intervention Periods for Stroke", pages: 649 - 654 |
| ANNUAL REVIEW OF NEUROSCIENCE, vol. 24, 2001, pages 1217 - 1281 |
| BIOORGANIC & MEDICINAL CHEMISTRY, vol. 200412, no. 1, pages 17 - 21 |
| BRAIN RESEARCH REVIEWS, vol. 30, 1999, pages 176 - 188 |
| CURRENT MEDICINE JAPAN, vol. 54, no. 7, 1999, pages 88 - 94 |
| ENGL. TRANSL. VER., pages 1565 - 1569 |
| JOURNAL OF MEDICINAL CHEMISTRY, vol. 31, no. 9, 1988, pages 1778 - 85 |
| KIYOSHI TOMIOKA: "Strategic Applications of Named Reactions in Organic Synthesis", 15 August 2006, KAGAKU DOJIN, pages: 258 - 259,310- |
| NATURE, vol. 322, 1986, pages 552 - 555 |
| PROGRESS IN BRAIN RESEARCH, vol. 146, 2004, pages 387 - 401 |
| See also references of EP2436683A4 * |
| SYNTHESIS, 2003, pages 1683 - 1692 |
| SYNTHESIS, 2008, pages 387 - 394 |
| SYNTHETIC COMMUNICATIONS, vol. 28, no. 22, 1998, pages 4123 - 4135 |
| THE JOURNAL OF THE AMERICAN SOCIETY FOR EXPERIMENTAL NEUROTHERAPEUTICS, vol. 2, 2005, pages 120 - 128 |
| ZHURNAL OBSHCHEI KHIMII, vol. 32, no. 5, 1962, pages 1581 - 86 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017055147A1 (en) | 2015-09-28 | 2017-04-06 | Syngenta Participations Ag | Pesticidally active heterocyclic derivatives with sulphur containing substituents |
Also Published As
| Publication number | Publication date |
|---|---|
| US20120115856A1 (en) | 2012-05-10 |
| CA2762620A1 (en) | 2010-12-02 |
| US8829035B2 (en) | 2014-09-09 |
| JP2011006409A (ja) | 2011-01-13 |
| AU2010253336A1 (en) | 2011-12-08 |
| BRPI1011961A2 (pt) | 2016-04-26 |
| ES2484169T3 (es) | 2014-08-11 |
| MX2011012537A (es) | 2011-12-14 |
| EP2436683A1 (en) | 2012-04-04 |
| CN102428080A (zh) | 2012-04-25 |
| EP2436683A4 (en) | 2012-05-16 |
| EP2436683B1 (en) | 2014-05-14 |
| KR20120093066A (ko) | 2012-08-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11814376B2 (en) | Hepatitis b core protein modulators | |
| DK2380881T3 (en) | NEW BICYCLIC HETEROCYCLIC COMPOUND | |
| AU2015229174B2 (en) | Hepatitis B core protein allosteric modulators | |
| JP5221453B2 (ja) | イミダゾール誘導体 | |
| TWI434842B (zh) | Azole compounds | |
| AU2010294214B2 (en) | Heterocylcic derivatives as inhibitors of glutaminyl cyclase | |
| WO2010137349A1 (ja) | 神経栄養因子の活性が関与する疾患の治療または予防剤 | |
| WO2010137350A1 (ja) | 神経栄養因子の活性が関与する疾患の治療または予防剤 | |
| EP2250165A1 (en) | Triazole oxadiazoles derivatives | |
| JP5667934B2 (ja) | 新規2環性複素環化合物からなる医薬 | |
| EP3541812B1 (en) | Mglur7 agonist compounds for treating mglur7- regulated diseases, disorders, or conditions | |
| TW202031644A (zh) | 苯并咪唑衍生物 | |
| JP2019508390A (ja) | mGluR7調節薬としてのインダン誘導体 | |
| OA19643A (en) | Hepatitis B core protein modulators. | |
| BR112018005178B1 (pt) | Compostos moduladores de proteína base de hepatite b, composições farmacêuticas compreendendo tais compostos e seus usos |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080021822.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10780309 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4708/KOLNP/2011 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010253336 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2762620 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/012537 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13322888 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2010253336 Country of ref document: AU Date of ref document: 20100528 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010780309 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20117031347 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2011153381 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1011961 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1011961 Country of ref document: BR Kind code of ref document: A2 Effective date: 20111128 |