WO2010137349A1 - Agent de traitement ou de prévention de maladies associées à l'activité d'agents neurotrophiques - Google Patents
Agent de traitement ou de prévention de maladies associées à l'activité d'agents neurotrophiques Download PDFInfo
- Publication number
- WO2010137349A1 WO2010137349A1 PCT/JP2010/003622 JP2010003622W WO2010137349A1 WO 2010137349 A1 WO2010137349 A1 WO 2010137349A1 JP 2010003622 W JP2010003622 W JP 2010003622W WO 2010137349 A1 WO2010137349 A1 WO 2010137349A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- mmol
- added
- optionally substituted
- mixture
- Prior art date
Links
- 0 Cc(cc1)cc2c1[n](*)c(*)n2 Chemical compound Cc(cc1)cc2c1[n](*)c(*)n2 0.000 description 3
- SKUIYRBGLUUSEU-UHFFFAOYSA-N CC(C)(C)OC([n](c(-c(cc1)cc2c1[n](C1CCOCC1)c(C)n2)c1)c2c1cccc2)=O Chemical compound CC(C)(C)OC([n](c(-c(cc1)cc2c1[n](C1CCOCC1)c(C)n2)c1)c2c1cccc2)=O SKUIYRBGLUUSEU-UHFFFAOYSA-N 0.000 description 1
- OVDGUTHABMXVMI-UHFFFAOYSA-N CCCNc(ccc(C(O)=O)c1)c1[N+]([O-])=O Chemical compound CCCNc(ccc(C(O)=O)c1)c1[N+]([O-])=O OVDGUTHABMXVMI-UHFFFAOYSA-N 0.000 description 1
- SZWUXFVTEPUSBH-UHFFFAOYSA-N CN(C)CCNc(c(N)c1)ccc1-c1nc(cccc2)c2[o]1 Chemical compound CN(C)CCNc(c(N)c1)ccc1-c1nc(cccc2)c2[o]1 SZWUXFVTEPUSBH-UHFFFAOYSA-N 0.000 description 1
- RYVPNBSPYULPBX-UHFFFAOYSA-N CN(C)CCNc(c([N+]([O-])=O)c1)ccc1-c1nc(cccc2)c2[o]1 Chemical compound CN(C)CCNc(c([N+]([O-])=O)c1)ccc1-c1nc(cccc2)c2[o]1 RYVPNBSPYULPBX-UHFFFAOYSA-N 0.000 description 1
- MBVSKEYWVHMBDY-UHFFFAOYSA-N COCc1nc(cc(C(F)(F)F)cc2)c2[n]1C1CCOCC1 Chemical compound COCc1nc(cc(C(F)(F)F)cc2)c2[n]1C1CCOCC1 MBVSKEYWVHMBDY-UHFFFAOYSA-N 0.000 description 1
- KPSCYXILHIZIFG-UHFFFAOYSA-N Cc1nc(cc(cc2)-c3nc(cccc4)c4[o]3)c2[n]1CCN(C)C Chemical compound Cc1nc(cc(cc2)-c3nc(cccc4)c4[o]3)c2[n]1CCN(C)C KPSCYXILHIZIFG-UHFFFAOYSA-N 0.000 description 1
- WRRVLSNYSLUPQO-UHFFFAOYSA-N Cc1nc(cc(cc2)Br)c2[n]1C1CCOCC1 Chemical compound Cc1nc(cc(cc2)Br)c2[n]1C1CCOCC1 WRRVLSNYSLUPQO-UHFFFAOYSA-N 0.000 description 1
- AIVUOLKBXGPRCT-UHFFFAOYSA-N Nc(cc(C(F)(F)F)cc1)c1NC1CCOCC1 Chemical compound Nc(cc(C(F)(F)F)cc1)c1NC1CCOCC1 AIVUOLKBXGPRCT-UHFFFAOYSA-N 0.000 description 1
- CXRZFBGEWWPCCK-UHFFFAOYSA-N Nc(cc(cc1)C(O)=O)c1NC1CCOCC1 Chemical compound Nc(cc(cc1)C(O)=O)c1NC1CCOCC1 CXRZFBGEWWPCCK-UHFFFAOYSA-N 0.000 description 1
- HLDFCCHSOZWKAA-UHFFFAOYSA-N [O-][N+](c(cc(C(F)(F)F)cc1)c1F)=O Chemical compound [O-][N+](c(cc(C(F)(F)F)cc1)c1F)=O HLDFCCHSOZWKAA-UHFFFAOYSA-N 0.000 description 1
- ISWGWGDZYATJSK-UHFFFAOYSA-N [O-][N+](c(cc(C(F)(F)F)cc1)c1NC1CCOCC1)=O Chemical compound [O-][N+](c(cc(C(F)(F)F)cc1)c1NC1CCOCC1)=O ISWGWGDZYATJSK-UHFFFAOYSA-N 0.000 description 1
- BOJWTAQWPVBIPG-UHFFFAOYSA-N [O-][N+](c(cc(cc1)C(O)=O)c1F)=O Chemical compound [O-][N+](c(cc(cc1)C(O)=O)c1F)=O BOJWTAQWPVBIPG-UHFFFAOYSA-N 0.000 description 1
- PKPOUWCQDJNQSC-UHFFFAOYSA-N [O-][N+](c1cc(C(O)=O)ccc1NC1CCOCC1)=O Chemical compound [O-][N+](c1cc(C(O)=O)ccc1NC1CCOCC1)=O PKPOUWCQDJNQSC-UHFFFAOYSA-N 0.000 description 1
Images
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/428—Thiazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- Neurotrophic factors such as BDNF and NGF are proteins that play an important role in central and peripheral nervous system cell differentiation, functional maintenance, synapse formation, regeneration and repair upon injury, and neurodegenerative diseases, diabetic Effective for the treatment of neuropathy, amyotrophic lateral sclerosis, multiple sclerosis, cerebral ischemic disease, Alzheimer's disease, Parkinson's disease, Huntington's chorea, depression, peripheral neuropathy caused by cancer chemotherapy, etc. (Non-Patent Documents 2, 3, 4, and 5).
- a neurotrophic factor is a high molecular weight protein having a molecular weight of 10,000 or more, and it is known that when such a high molecular weight protein is used as a therapeutic agent, there are restrictions on the administration method and safety problems (non-patented). From the references 6 and 7), there is an urgent need to provide a low molecular weight compound that enhances the action of a neurotrophic factor with few side effects.
- the expression of certain transcriptional regulatory factors is controlled by neurotrophic factors (see Non-Patent Document 1), and one of the transcriptional regulatory factors whose expression is controlled by neurotrophic factors. NXF is known (see Patent Document 1).
- Patent Document 2 discloses a general formula. The compound represented by these is disclosed.
- Patent Document 4 discloses a general formula. The compound represented by these is disclosed.
- Patent Document 5 discloses a general formula. The compound represented by these is disclosed.
- Patent Document 6 discloses a general formula. The compound represented by these is disclosed.
- R 3 represents a hydrogen atom or a substituent
- R 4 represents a hydrogen atom or a substituent
- Ring A is Represents an optionally substituted benzene ring
- ring B is It represents an optionally substituted benzene ring.
- a pharmaceutically acceptable salt thereof, or a solvate thereof in this specification, sometimes referred to as compound (I). In the present specification, it may be referred to as a compound of the present invention.
- R 1 is (1) C 3-6 alkyl group, (2) (a) a halogen atom, (b) R a -O-, (c) R a —O—CO—, (d) R a —O—CO—NR a —, (e) R a —O—CO—C 2 H 4 —CO—NH—, (f) R a -S-, (g) R a —SO 2 —, (h) R a —CO—O—, (i) R a —CO—NR a —, (j) R a -NR a- , (k) R a -NR a -CS-NR a- , (l) a 5- to 6-membered cyclic group, (m) a carboxy group, (n) a hydroxy group, (o) an amino group, (p) heterocycle-carbonyl group, (q
- composition which is an agent for treating or preventing a disease involving the activity of neurotrophic factor, or an accelerator for a physical therapy effect.
- the treatment or prevention agent for a disease involving the activity of neurotrophic factor, or the accelerator for physiotherapy effect is the treatment or prevention agent for cerebral ischemic disease or diabetic neuropathy according to the above [7].
- Ring A is (a) a halogen atom, (b) a hydroxyl group, (c) a carboxy group, (d) a cyano group, (e) a sulfamoyl group, (f) a monoalkylamide group, (g) a dialkylamide group, (h) an alkyl group optionally substituted by a halogen atom, a benzene ring optionally substituted with one or more substituents selected from the group consisting of (i) a nitro group, and (j) an aryloxy group, Ring B is (a) a halogen atom, (b) a hydroxyl group, (c) a carboxy group, (d) a cyano group, (e) a
- Formula for the manufacture of a therapeutic or prophylactic agent for a disease involving the activity of neurotrophic factor, or an accelerator for a physiotherapy effect [Where: Z represents N or CR 2 ; X is Represents N—R 3 , O, or S; Y is Represents CR 4 or N; R 1 is A hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group; R 2 represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group; R 3 represents a hydrogen atom or a substituent, R 4 represents a hydrogen atom or a substituent, Ring A is Represents an optionally substituted benzene ring, Ring B is It represents an optionally substituted benzene ring. ] Or a pharmaceutically acceptable salt or solvate thereof.
- R 1 is Represents an optionally substituted non-cyclic hydrocarbon group or an optionally substituted cyclic group
- X is N—R 3 (R 3 represents a hydrogen atom or a substituent), O, or S
- Y is C—R 4 (R 4 represents a hydrogen atom or a substituent), or N
- Z is N, or C—R 2 (R 2 represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group)
- Ring A and Ring B are Independently, it represents an optionally substituted benzene ring.
- R 1 is the formula (Wherein R a1 represents a carboxy group, a cyano group, a 1H-tetrazolyl group, a 1-triphenylmethyl-tetrazolyl group, or an alkoxycarbonyl group, and R a2 represents a hydrogen atom, a fluorine atom, a chlorine atom, or bromine. It is not a group represented by. In addition, however, R 1 is is not. ] Or a pharmaceutically acceptable salt or solvate thereof.
- R 1 is a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) a 6 to 10 membered cyclic group which may be substituted with one or more substituents selected from a halogen atom, a C 1-4 alkoxy group, and a C 1-4 alkoxy-carbonyl group;
- X is N—R 3 (R 3 is a hydrogen atom, or R a —O—CO— (R a is a C 1-6 alkyl group)), O, or S;
- Y is CH or N;
- Z is, N, or C-R 2 (Wherein R 2 is a) a hydrogen atom, b) (1) a halogen atom, (2) R b —O—, (3) R b —CO—, (4) R b —CO—O— and (5) R b —NR b — (In the formula, each R b is the same
- R 1 is Represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group
- X is N—R 3 (R 3 represents a hydrogen atom or a substituent), O, or S
- Y is C—R 4 (R 4 represents a hydrogen atom or a substituent), or N
- Z is N, or C-R 2 (.
- R 2 is hydrogen atom, optionally substituted acyclic hydrocarbon group, or a substituted represents an even cyclic group) represents; and ring A and ring B Independently, it represents an optionally substituted benzene ring.
- a pharmaceutically acceptable salt thereof, or a solvate thereof (hereinafter, referred to as compound (I ′).
- Compound (I ′) will be described in accordance with the description of compound (I) above. It is understood in the same manner.
- a therapeutic or prophylactic agent for a disease involving the activity of a neurotrophic factor, or an accelerator for a physical therapy effect [4 '] The agent according to [3 ′] above, which is a therapeutic or preventive agent for cerebral ischemic disease or diabetic neuropathy.
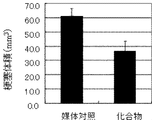
- the graph which shows infarct volume (Test Example 2).
- aryl (group) examples include C 6-14 aryl (group).
- C 6-14 aryl (group) includes phenyl, naphthyl, and anthryl.
- heteroaryl (group) examples include a 5- to 6-membered heteroaryl (group).
- the “5- to 6-membered heteroaryl (group)” includes, for example, 1 to 3 (preferably 1 to 2) heterocycles selected from an oxygen atom, a sulfur atom, and a nitrogen atom as ring-constituting atoms.
- 5- to 6-membered heteroaryl (group) having an atom examples include 1 to 3 (preferably 1 to 2) heterocycles selected from an oxygen atom, a sulfur atom, and a nitrogen atom as ring-constituting atoms.
- furyl eg, 2-furyl, 3-furyl
- thienyl eg, 2-thienyl, 3-thienyl
- pyridyl eg, 2-pyridyl, 3-pyridyl, 4-pyridyl
- pyrimidinyl eg, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, 6-pyrimidinyl
- pyridazinyl eg, 3-pyridazinyl, 4-pyridazinyl
- pyrazinyl eg, 2-pyrazinyl
- pyrrolyl eg, 1-pyrrolyl, 2-pyrrolyl) , 3-pyrrolyl
- imidazolyl eg, 1-imidazoly
- Examples of the “cycloalkenyl (group)” include C 3-10 cycloalkenyl (group).
- Examples of “C 3-10 cycloalkenyl (group)” include cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclononenyl, and cyclodecenyl.
- cycloalkadienyl (group) examples include C 4-10 cycloalkadienyl group.
- Examples of the “C 4-10 cycloalkadienyl group” include cyclobutadienyl, cyclopentadienyl, cyclohexadienyl, cycloheptadienyl, cyclooctadienyl, cyclononadienyl, and cyclodecadienyl. Is mentioned.
- non-aromatic heterocyclic group examples include 5- to 6-membered non-aromatic heterocyclic groups.
- the “5- to 6-membered non-aromatic heterocyclic group” includes, for example, 1 to 3 (preferably 1) selected from an oxygen atom, an optionally oxidized sulfur atom, and a nitrogen atom as ring-constituting atoms.
- alkyl (group) examples include C 1-6 alkyl (group).
- C 1-6 alkyl (group) examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, pentyl, and hexyl.
- alkenyl (group) examples include C 2-6 alkenyl (group).
- Examples of “C 2-6 alkenyl (group)” include vinyl, 2-propenyl, 3-methyl-2-butenyl, and 1,3-butadienyl.
- Examples of “alkynyl (group)” include C 2-6 alkynyl (group).
- Examples of “C 2-6 alkynyl (group)” include ethynyl, 2-propynyl, and 2-penten-4-ynyl.
- Alkylene (chain)” includes, for example, C 1-3 alkylene (chain).
- C 1-3 alkylene (chain) includes, for example, methylene, ethylene, and trimethylene.
- halogen (atom) examples include fluorine, chlorine, bromine, and iodine.
- alkoxy-carbonyl (group) examples include C 1-6 alkoxy-carbonyl (group).
- C 1-6 alkoxy-carbonyl (group) include methoxycarbonyl, ethoxycarbonyl, n-propyloxycarbonyl, isopropyloxycarbonyl, n-butyloxycarbonyl, sec-butyloxycarbonyl, tert-butyloxycarbonyl , N-pentyloxycarbonyl, n-hexyloxycarbonyl group and the like.
- C 1-6 alkoxy-carbonyl (group) is R a —O—CO— (R a is C 1-6 alkyl (group)).
- R 1 is A hydrogen atom, an acyclic hydrocarbon group which may be substituted, or a cyclic group which may be substituted is represented.
- Examples of the “acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 1 include an alkyl group, an alkenyl group, and an alkynyl group.
- Examples of the “alkyl group” include those exemplified above.
- Examples of the “alkenyl group” include those exemplified above.
- Examples of the “alkynyl group” include those exemplified above. Of these, an alkyl group is preferable, and a C 1-6 alkyl group is more preferable.
- the “acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 1 may be substituted with one or more (preferably 1 to 3) substituents. .
- Examples of the “halogen atom” include those exemplified above.
- Examples of the “C 1-6 alkyl group” include those exemplified above.
- Examples of the “optionally substituted cyclic group” include those similar to the “optionally substituted cyclic group” represented by R 1 exemplified below.
- non-aromatic cyclic hydrocarbon group examples include a cycloalkyl group, a cycloalkenyl group, and a cycloalkadienyl group.
- examples of the “cycloalkyl group”, “cycloalkenyl group”, and “cycloalkadienyl group” include those exemplified above.
- Examples of the “non-aromatic heterocyclic group” include those exemplified above.
- cyclic group examples include C 3-10 cycloalkyl, C 3-10 cycloalkenyl, C 4-10 alicyclic hydrocarbon group having 3 to 10 carbon atoms, such as cycloalkadienyl or a 5-6 membered non-aromatic, A group heterocyclic group is preferred.
- the “cyclic group” of the “optionally substituted cyclic group” represented by R 1 may be substituted with one or more (preferably 1 to 3) substituents.
- substituents for example, (1) a halogen atom, (2) Nitro group, (3) a cyano group, (4) a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms, (5) a C 1-6 alkenyl group optionally substituted with one or more (preferably 1 to 3) halogen atoms, (6) a C 1-6 alkynyl group optionally substituted by one or more (preferably 1 to 3) halogen atoms, (7) R a -CO-L-, (8) R a —CO—OL— (9) R a —CO—NR a —L—, (10) R a —OL— (11) R a -O-CO-L-, (12) R a —O—CO—NR a —L—, (13) R
- halogen atom examples include those exemplified above.
- substituent for the non-aromatic cyclic hydrocarbon group particularly an alicyclic hydrocarbon group having 3 to 10 carbon atoms
- non-aromatic heterocyclic group particularly a 5- to 6-membered non-aromatic heterocyclic group
- (a) an oxo group, (b) a C 1-4 alkoxy-carbonyl group, (c) a hydroxy group is preferred, (a) an oxo group, (b) A C 1-4 alkoxy-carbonyl group is more preferred.
- R c is the same or different and represents a hydrogen atom or a C 1-6 alkyl group).
- R 1 is preferably, for example, a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) (1) a halogen atom, (2) a C 1-6 alkoxy group which may be substituted with one or more (preferably 1 to 3) halogen atoms, and (3) optionally substituted with one or more substituents selected from C 1-6 alkoxy-carbonyl groups optionally substituted with one or more (preferably 1 to 3) halogen atoms
- a 3- to 10-membered cyclic group More preferably, for example, a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) optionally substituted with one or more (preferably 1 to 3) substituents selected from a halogen atom, a C 1-4 alkoxy group, and a C 1-4 alkoxy-carbonyl group Member
- X represents N—R 3 (R 3 represents a hydrogen atom or a substituent), O, or S.
- X is more preferably, for example, N—R 3 (R 3 is a hydrogen atom or R a —O—CO— (R a is a C 1-6 alkyl group)), O, Or S.
- X is more preferably NH, O, or S.
- Y represents C—R 4 (R 4 represents a hydrogen atom or a substituent), or N.
- R 4 examples include: (1) a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms, (2) a C 1-6 alkenyl group optionally substituted by one or more (preferably 1 to 3) halogen atoms, (3) a C 1-6 alkynyl group optionally substituted by one or more (preferably 1 to 3) halogen atoms, (4) R a -CO-L-, (5) R a -CO-O -L-, (6) R a —CO—NR a —L—, (7) R a -OL-, (8) R a -O-CO-L-, (9) R a —O—CO—NR a —L—, (10) R a -SL-, (11) R a -SO-L-, (12) R a —SO 2 —L—, (13) R a -NR a -L-, (14) R a —S—
- Y is more preferably, for example, CH or N. Y is more preferably N.
- Z represents N or C—R 2 .
- R 2 represents a hydrogen atom, an optionally substituted acyclic hydrocarbon group, or an optionally substituted cyclic group. In the compounds of the present invention, preferably Z is C—R 2 .
- Examples of the “acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 2 include an alkyl group, an alkenyl group, and an alkynyl group.
- Examples of the “alkyl group” include those exemplified above.
- Examples of the “alkenyl group” include those exemplified above.
- Examples of the “alkynyl group” include those exemplified above.
- acyclic hydrocarbon group” of the “optionally substituted acyclic hydrocarbon group” represented by R 2 may be substituted with one or more (preferably 1 to 3) substituents. .
- each R b is the same or different and represents a hydrogen atom or a C 1-6 alkyl group which may be
- each R b is the same or different and represents a hydrogen atom or a C 1-6 alkyl group which may be substituted with one or more halogens, and n represents an integer of 0 to 2). Is more preferable.
- R 2 is preferably, for example, (1) hydrogen atom, (2) (a) a halogen atom, (b) R b —O—, (c) R b —O—CO—, (d) R b —O—CO—NR b —, (e) R b -S-, (f) R b —SO 2 —, (g) R b —CO—O—, (h) R b —CO—NR b —, (i) R b —NR b —, (j) R b —NR b —R b —S (O) n — (k) phenyl group, (l) a 5- to 6-membered saturated heterocyclic group, (m) a hydroxy group, and (n) an amino group (wherein each R b is the same or different and represents
- a C 1-6 alkyl group which may be substituted with one or more substituents selected from the group consisting of: (3) (a) a halogen atom, (b) R c —O—, (c) R c —O—CO—, and (d) R c —CO—NR c — (Wherein R c are the same or different and each represents a hydrogen atom or a C 1-6 alkyl group.)
- a non-aromatic cyclic hydrocarbon group having 5 to 6 carbon atoms or a 5- to 6-membered non-aromatic heterocyclic group which may be substituted with one or more substituents selected from the group consisting of
- R 2 is a) a hydrogen atom, b) (1) a halogen atom, (2) R b —O—, (3) R b —CO—, (4) R b —CO—O— and (5) R b —NR b —
- each R b is the same or different and represents a hydrogen atom or a C 1-6 alkyl group which may be substituted with one phenyl group.
- a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) substituents selected from: c) 5- to 6-membered cyclic group. ) It is.
- Ring A represents an optionally substituted benzene ring.
- the “benzene ring” of the “optionally substituted benzene ring” represented by ring A may be substituted with one or more (preferably 1 to 3) substituents.
- substituents the thing similar to the substituent illustrated about "the cyclic group which may be substituted" represented by R ⁇ 1 > is mentioned.
- Ring A is preferably, for example, unsubstituted (ie Benzene ring).
- Ring B represents an optionally substituted benzene ring.
- the “benzene ring” of the “optionally substituted benzene ring” represented by ring B may be substituted with one or more (preferably 1 to 3) substituents.
- substituents the thing similar to the substituent illustrated about "the cyclic group which may be substituted" represented by R ⁇ 1 > is mentioned.
- ring B is preferably, for example, (1) a halogen atom, (2) Nitro group, (3) a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms (4) R a -O-CO-L-, (5) R a —SO 2 —NR a —L—, and (6) R a -NR a -L- Wherein each R a is the same or different and is a hydrogen atom or a C 1-6 alkyl group which may be substituted with one or more (preferably 1 to 3) halogen atoms; Is a bond.) Or a benzene ring optionally substituted with one or more (preferably 1 to 3) substituents selected from
- Ring B is more preferably, for example, (1) a halogen atom (preferably chlorine), (2) Nitro group, (3) a C 1-6 alkyl group (preferably methyl), and (4) A benzene ring which may be substituted with one or more (preferably 1 to 3) substituents selected from amino groups.
- a halogen atom preferably chlorine
- Nitro group preferably nitrogen
- C 1-6 alkyl group preferably methyl
- a benzene ring which may be substituted with one or more (preferably 1 to 3) substituents selected from amino groups.
- Compound (I) is more preferably one in which two or more selected from the above-mentioned preferred partial structures, more preferred partial structures, and further preferred partial structures are used in combination.
- the compound (I) is preferably, for example, the following compound A.
- Compound A is a novel compound.
- R 1 is 5- to 6-membered non-aromatic heterocyclic group.
- Z is CR- 2 , Y is N, R 1 is A 5- to 6-membered non-aromatic heterocyclic group, and R 2 is A C 1-6 alkyl group;
- R 1 is a) an acyclic hydrocarbon group having 1 to 6 carbon atoms which may be substituted with a 6-membered cyclic group, or b) a 6-10 membered cyclic group optionally substituted with one or more substituents selected from a halogen atom, one or more C 1-4 alkoxy groups, and a C 1-4 alkoxy-carbonyl group.
- Compound (I) may be a pharmaceutically acceptable salt.
- salts include metal salts, ammonium salts, salts with organic bases, salts with inorganic acids, salts with organic acids, salts with basic or acidic amino acids, and the like.
- metal salt include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt, magnesium salt and barium salt; aluminum salt and the like.
- Preferable examples of the salt with organic base include, for example, trimethylamine, triethylamine, pyridine, picoline, 2,6-lutidine, ethanolamine, diethanolamine, triethanolamine, cyclohexylamine, dicyclohexylamine, N, N′-dibenzylethylenediamine. And the like.
- Preferable examples of the salt with inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfone. And salts with acid, p-toluenesulfonic acid and the like.
- salts with basic amino acids include salts with arginine, lysine, ornithine and the like
- salts with acidic amino acids include salts with aspartic acid and glutamic acid, for example. It is done. Of these, pharmaceutically acceptable salts are preferred.
- the compound (I) has a tautomer, an isomer having a stable structure is preferable.
- the present invention is not limited to this, and does not exclude other isomers. It is within the scope of the present invention.
- compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, or a rotational isomer, any one of the isomers and mixtures are within the scope of the present invention. Included in the compounds of the present invention.
- compound (I) has an optical isomer, the optical isomer resolved from the racemate is also within the scope of the present invention.
- Compound (I) may be crystalline or amorphous and both are within the scope of the present invention.
- Compound (I) labeled or substituted with an isotope is also within the scope of the present invention.
- Compound (I) may be a solvate (such as a hydrate) or a non-solvate.
- each symbol of the compound in the reaction formula represents the same meaning as described above.
- the compound in a formula also includes the case where it forms the salt, as such a salt, the thing similar to the salt of compound (I) is mentioned, for example.
- the compound obtained in each step can be used in the next reaction as a reaction solution or as a crude product, but can be isolated from the reaction mixture according to a conventional method. It can be easily purified by separation means such as extraction, concentration, neutralization, filtration, distillation, recrystallization, distillation, chromatography and the like. Or when the compound in a formula is marketed, a commercial item can also be used as it is. Unless otherwise specified, room temperature herein is about 10 ° C. to about 35 ° C.
- Compound (I) can be produced, for example, by the method shown in the following scheme or a method analogous thereto.
- a halogen atom As the leaving group represented by Xa, for example, A halogen atom; An optionally substituted alkylsulfonyloxy group (eg, a methanesulfonyloxy group, an ethanesulfonyloxy group, and an alkylsulfonyloxy group optionally substituted with a halogen atom such as trifluoromethanesulfonyloxy); and Good arylsulfonyloxy groups (eg, benzenesulfonyloxy, p-toluenesulfonyloxy group, and 2-nitrobenzenesulfonyloxy group) and the like, and a halogen atom is particularly preferable.
- An optionally substituted alkylsulfonyloxy group eg, a methanesulfonyloxy group, an ethanesulfonyloxy group, and an alkylsulfonyl
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from instantaneous to about 24 hours.
- the amount of compound (S-1 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-1).
- the base used include carbonates such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate, cesium carbonate and potassium phosphate; hydroxides such as sodium hydroxide and potassium hydroxide; morpholine, piperidine and pyridine.
- organic amines such as trimethylamine, triethylamine, diisopropylethylamine, 4-dimethylaminopyridine, and the like.
- the amount thereof to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-1).
- the solvent include water; alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and Aprotic solvents such as N-methylpyrrolidone; halogenated hydrocarbons such as chloroform, or mixed solvents thereof are used, and the organic amines and compounds (S-1 ′) described above are used as a solvent. May be.
- the solvent examples include alcohol solvents such as methanol, ethanol and 2-propanol; ether solvents such as tetrahydrofuran; ester solvents such as ethyl acetate; aprotic polar solvents such as N, N-dimethylformamide or the like And the like.
- the reaction can be carried out by reacting at a temperature within a range from about 0 ° C. to the boiling point of the solvent used for 10 minutes to 48 hours.
- the metal reducing agent include tin (II) chloride, reduced iron, titanium trichloride (III), and the like. Is 1 to 5 moles.
- the solvent examples include water; dilute hydrochloric acid; acetic acid; alcohol solvents such as methanol, ethanol and 2-propanol; ether solvents such as tetrahydrofuran and 1,2-dimethoxyethane; ester solvents such as ethyl acetate; acetone, acetonitrile and Examples thereof include aprotic polar solvents such as N, N-dimethylformamide, or mixed solvents thereof.
- an acid chloride compound When an acid chloride compound is used, for example, it can be carried out according to the method described in Zhurnal Obshchei Khimii 1962, 32 (5), 1581-86 (Engl. Transl. Ver .: pp 1565-1569). Specifically, for example, General formula (R 1 , R 3 ′ and ring A are as described above.) A compound (S-3) represented by: Formula R 2 —COCl (R 2 is as described above.) By reacting with an acid chloride compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) In this reaction, the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 24 hours.
- the amount of the acid chloride compound to be used is generally 1 to 5 mol, preferably 1 to 2 mol, per 1 mol of compound (S-3).
- the solvent include ether solvents such as tetrahydrofuran and 1,2-dimethoxyethane, ester solvents such as ethyl acetate, aprotic polar solvents such as acetone, acetonitrile and N, N-dimethylformamide, or a mixture thereof. A solvent etc. are mentioned.
- a carboxylic acid compound when used, for example, it can be carried out according to the method described in Zhurnal Obshchei Khimii 1962, 32 (5), 1581-86 (Engl. Transl. Ver .: pp 1565-1569). Specifically, for example, General formula (R 1 , R 3 ′ and ring A are as described above.) A compound (S-3) represented by: Formula R 2 —COOH (R 2 is as described above.) By reacting with a carboxylic acid compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) (S-4) can be produced.
- the solvent examples include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; or a mixed solvent thereof.
- ether solvents such as tetrahydrofuran and dioxane
- aromatic hydrocarbon solvents such as benzene, toluene and xylene
- a mixed solvent thereof a mixed solvent thereof.
- the carboxylic acid described above is used as the solvent. May be.
- an aldehyde compound when used, it can carry out according to the method as described in Synthesis 2003, No11, 1683-1692, for example.
- General formula (R 1 , R 3 ′ and ring A are as described above.
- S-4 By reacting with an aldehyde compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) (S-4) can be produced.
- the reaction temperature is usually from room temperature to reflux temperature
- the reaction time is usually from instantaneous to about 24 hours.
- the amount of the aldehyde compound to be used is generally 1 to 5 mol, preferably 1 to 2 mol, per 1 mol of compound (S-3).
- the oxidizing agent include m-chloroperbenzoic acid, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), oxone, benzofuroxane, potassium permanganate, hydrogen peroxide, Examples thereof include acetic acid and tert-butyl hydroperoxide, and the amount is usually 0.1 to 5 mol, preferably 0.5 to 2 mol, relative to 1 mol of compound (S-3).
- the solvent examples include water; alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and An aprotic solvent such as N-methylpyrrolidone; a halogenated hydrocarbon such as chloroform, or a mixed solvent thereof is used.
- the amount of the triacetal compound to be used is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound (S-3).
- a catalytic amount of acid may be added to the reaction system.
- the acid used include inorganic acids such as hydrochloric acid, hydrobromic acid, nitric acid, and sulfuric acid, and organic acids such as acetic acid, trifluoroacetic acid, methanesulfonic acid, and p-toluenesulfonic acid.
- the solvent examples include alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and N- An aprotic solvent such as methylpyrrolidone; a halogenated hydrocarbon such as chloroform, or a mixed solvent thereof is used.
- the triacetal compound described above may be used as a solvent.
- an imidate compound when used, for example, it can be carried out according to the method described in JP-A-4-308580.
- the general formula (R 1 , R 3 ′ and ring A are as described above.) (S-3) and a general formula R 2 —C ( ⁇ NH) OR 2 ′ (R 2 and R 2 ′ are as described above.)
- S-4 By reacting with the imidate compound represented by General formula (R 1 , R 2 , R 3 ′ and ring A are as described above.) (S-4) can be produced.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from instantaneous to about 24 hours.
- the amount of the imidate compound to be used is generally 1 to 5 mol, preferably 1 to 2 mol, per 1 mol of compound (S-3).
- the solvent examples include alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene and xylene; acetonitrile, dimethylformamide, dimethyl sulfoxide and N- An aprotic solvent such as methylpyrrolidone; a halogenated hydrocarbon such as chloroform, or a mixed solvent thereof is used.
- lower fatty acid anhydrides such as acetic anhydride and propionic anhydride
- organic sulfonic acids such as methanesulfonic acid and paratoluenesulfonic acid
- phosphorus oxychloride phosphorus trichloride, diphosphorus pentoxide, sulfuric acid, polyphosphoric acid
- solvent that may use an inorganic acid such as boric acid include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene, and xylene; or a mixed solvent thereof.
- the dehydration condensation agent described above may be used as a solvent.
- the first step is a reaction using a condensing agent or an acid halogenating agent.
- a condensing agent When a condensing agent is used, the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 24 hours.
- the amount of compound (S-5 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-5).
- the condensing agent include Bop (1H-1,2,3-benzotriazol-1-yloxy (tri (dimethylamino)) phosphonium hexafluorophosphate), WSC (1-ethyl-3- (3-dimethylaminopropyl).
- Carbodiimide / hydrochloride Carbodiimide / hydrochloride), DCC (N, N-dicyclohexylcarbodiimide), CDI (carbonyldiimidazole), diethyl phosphoryl cyanide, and the like.
- the amount of the condensing agent to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol with respect to compound (S-5). If necessary, 1 equivalent to an excessive amount of an organic base such as triethylamine may be added to the compound (S-5).
- the solvent examples include halogenated hydrocarbons such as dichloromethane and chloroform, sulfoxides such as dimethyl sulfoxide, esters such as ethyl acetate, ethers such as tetrahydrofuran and 1,4-dioxane, and the like.
- halogenated hydrocarbons such as dichloromethane and chloroform
- sulfoxides such as dimethyl sulfoxide
- esters such as ethyl acetate
- ethers such as tetrahydrofuran and 1,4-dioxane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide.
- the reaction temperature range is usually from room temperature to reflux temperature
- the reaction time range is usually from instantaneous to about 24 hours.
- the amount of compound (S-5 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-5
- the acid halogenating agent examples include thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride and the like.
- the amount of the acid halogenating agent to be used is generally 1 to 10 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-5).
- a catalytic amount of N, N-dimethylformamide may be added to the reaction system.
- the solvent examples include halogenated hydrocarbons such as dichloromethane and chloroform; sulfoxides such as dimethyl sulfoxide; esters such as ethyl acetate; ethers such as tetrahydrofuran and 1,4-dioxane; Examples include amides such as N, N-dimethylformamide and N, N-dimethylacetamide; aromatic hydrocarbons such as benzene and toluene.
- halogenated hydrocarbons such as dichloromethane and chloroform
- sulfoxides such as dimethyl sulfoxide
- esters such as ethyl acetate
- ethers such as tetrahydrofuran and 1,4-dioxane
- aromatic hydrocarbons such as benzene and toluene.
- the second step is a reaction using a dehydration condensing agent.
- the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 48 hours.
- dehydrating condensing agents lower fatty acid anhydrides such as acetic anhydride and propionic anhydride; organic sulfonic acids such as methanesulfonic acid and paratoluenesulfonic acid; phosphorus oxychloride, phosphorus trichloride, diphosphorus pentoxide, sulfuric acid, polyphosphoric acid,
- Examples of the solvent that may use an inorganic acid such as boric acid include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene, and xylene; or a mixed solvent thereof.
- the dehydration condensation agent described above may be used as a solvent.
- nitrosating agent examples include alkali metal nitrites such as sodium nitrite, organic nitrite compounds such as methyl nitrite or isoamyl nitrite, and the amount used is 1 mol of compound (S-3). Usually 1 to 5 mol, preferably 1 to 2 mol.
- the solvent a combination of water and an acid is used.
- the acid include inorganic acids such as hydrochloric acid and sulfuric acid, and organic acids such as acetic acid.
- Cyclization 4 can be performed by methods well known to those skilled in the art (for example, the method described in Synthetic Communications 1998, 28 (22), 4123-4135). Specifically, for example, the general formula (Ya represents a halogen atom, R 1 , R 2 and ring A are as described above. ) (S-8) represented by the general formula (X and ring B are as described above.) Is reacted with a compound (S-5 ′) represented by General formula (R 1 , R 2 , X, ring A and ring B are as described above.) (S-6) can be produced. In this reaction, the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from instantaneous to about 24 hours.
- the amount of compound (S-5 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-8).
- dehydrating condensing agents lower fatty acid anhydrides such as acetic anhydride and propionic anhydride; organic sulfonic acids such as methanesulfonic acid and paratoluenesulfonic acid; phosphorus oxychloride, phosphorus trichloride, diphosphorus pentoxide, sulfuric acid, polyphosphoric acid,
- Examples of the solvent that may use an inorganic acid such as boric acid include ether solvents such as tetrahydrofuran and dioxane; aromatic hydrocarbon solvents such as benzene, toluene, and xylene; or a mixed solvent thereof.
- the dehydration condensation agent described above may be used as a solvent.
- Coupling is a method well known to those skilled in the art (for example, translated by Kiyoshi Tomioka, “Organic synthesis strategy learned from personal reaction”, Kagaku Dojin Co., Ltd., August 15, 2006, p258-259: Kumada cross-coupling reaction, p310- 311: Negishi cross coupling reaction, p440-441: Stilley-Kelly coupling reaction, p.448-449: Suzuki-Miyaura coupling reaction).
- the general formula (R 1 , R 2 , Xa and ring A are as described above.
- organotin for example, SnBu 4 (tetrabutyltin)
- X and ring B are as described above.
- General formula (R 1 , R 2 , X, ring A and ring B are as described above.) (S-11) can be produced.
- the reaction is carried out in the presence of a transition metal catalyst, and optionally in the presence of a ligand, base, additive, etc., at a temperature in the range from about 20 ° C. to the boiling point of the solvent used for 10 minutes to 48 hours. This can be done.
- the amount of compound (S-10) to be used is generally 1 to 20 mol, preferably 1 to 5 mol, per 1 mol of compound (S-9).
- transition metal catalyst examples include palladium (II) acetate, palladium (II) chloride, tetrakis (triphenylphosphine) palladium (0), bis (triphenylphosphine) palladium chloride (II), tris (dibenzylideneacetone) dipalladium. (0), or [1,1′-bis (diphenylphosphino) ferrocene] dichloropalladium (II).
- the amount of the transition metal catalyst to be used is generally 0.0001-1 mol, preferably 0.001-1 mol, per 1 mol of compound (S-10).
- Examples of the ligand include triphenylphosphine, tri-o-tolylphosphine, tri-tert-butylphosphine, tri-2-furylphosphine, tri-cyclohexylphosphine, triphenylarsine, 1,1′-bis (diphenylphosphine). Fino) ferrocene (dppf) and the like.
- the amount of the ligand to be used is generally 0.0001-4 mol, preferably 0.001-4 mol, per 1 mol of compound (S-9).
- Examples of the base include organic bases such as triethylamine and diisopropylethylamine And inorganic bases such as sodium carbonate, sodium hydrogen carbonate, potassium carbonate, cesium carbonate, and potassium phosphate.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 4 mol, per 1 mol of compound (S-9).
- Examples of the additive include lithium chloride, cesium fluoride, copper iodide (I) And inorganic salts such as copper (I) bromide.
- Examples of the solvent include water, acetonitrile, chloroform, and dichloromethane.
- the solvent examples include water; alcohol solvents such as methanol and ethanol; ether solvents such as tetrahydrofuran and dioxane; benzene, toluene, xylene, and the like.
- Aromatic hydrocarbon solvents include acetonitrile; dimethylformamide; aprotic solvents such as dimethyl sulfoxide or N-methylpyrrolidone; halogenated hydrocarbons such as chloroform, or mixed solvents thereof.
- R 2a represents an alkylcarbonyloxymethyl group or a benzyloxymethyl group
- R 1 , R 2 , X, Y, Ring A and Ring B are as described above.
- the metal catalyst examples include palladium-carbon, palladium hydroxide-carbon, rhodium-carbon, Raney nickel, platinum oxide and the like, and the amount used is based on the compound (S-12). Usually, it is 0.01% to 100% by weight, preferably 0.1 to 10% by weight.
- the solvent include alcohol solvents such as methanol, ethanol and 2-propanol; ether solvents such as tetrahydrofuran; ester solvents such as ethyl acetate; aprotic polar solvents such as N, N-dimethylformamide or the like And the like.
- the second step is a reaction using an acid halogenating agent.
- the reaction temperature range is usually from room temperature to reflux temperature, and the reaction time range is usually from instantaneous to about 48 hours.
- the amount of compound (S-12 ′) to be used is generally 1 to 2 mol, preferably 1 to 1.5 mol, per 1 mol of carboxylic acid compound.
- the acid halogenating agent include thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride and the like.
- the amount of the acid halogenating agent to be used is generally 1 to 10 mol, preferably 1 to 1.5 mol, per 1 mol of compound (S-12 ′).
- a catalytic amount of N, N-dimethylformamide may be added to the reaction system.
- the solvent include halogenated hydrocarbons such as dichloromethane and chloroform; sulfoxides such as dimethyl sulfoxide; esters such as ethyl acetate; ethers such as tetrahydrofuran and 1,4-dioxane; Examples include amides such as N, N-dimethylformamide and N, N-dimethylacetamide; aromatic hydrocarbons such as benzene and toluene.
- the manufacturing method of compound (I) is a solution, concentration, solvent extraction, fractional distillation, crystallization, recrystallization, and column chromatography. From the reaction mixture, it can be separated and purified by a known technique such as.
- compound (I) when compound (I) is obtained as a free form, it can be converted to the target salt as described above by a method known per se or a method analogous thereto, and conversely, compound (I) When obtained as a salt, it can be converted into a free form or other desired salt by a method known per se or a method analogous thereto.
- the purified compound (I) is converted into inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, acetic acid, citric acid, succinic acid, tartaric acid, fumaric acid, maleic acid, methanesulfonic acid, By reacting with an organic acid such as p-toluenesulfonic acid, a pharmaceutically acceptable acid addition salt can be easily obtained.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, acetic acid, citric acid, succinic acid, tartaric acid, fumaric acid, maleic acid, methanesulfonic acid
- the purified compound (I) can be converted into, for example, lithium hydroxide, lithium methylate, sodium hydride, sodium carbonate, sodium hydroxide, sodium methylate, potassium hydroxide, potassium carbonate, potassium hydroxide, potassium-t-
- an inorganic / organic metal salt such as butyrate
- the compounds of the present invention can be provided in any form suitable for the intended administration. Suitable forms include pharmaceutically (ie physiologically) acceptable salts and predrug and prodrug forms of the compounds of the invention. These predrug and prodrug forms are also within the scope of the present invention.
- the compound of the present invention has an action of enhancing the activity of neurotrophic factor, and in particular, has an action of inducing NXF gene expression that plays an important role in nerve protection. Therefore, the compound of the present invention can be used as a therapeutic or prophylactic agent for diseases involving neurotrophic factor activity. Moreover, the compound of this invention can be used as a promoter of the physical therapy effect for the recovery
- disease is used in the meaning including a disorder and its symptoms.
- treatment is used in the meaning including relief of symptoms.
- diseases involving neurotrophic factor activity include the following cranial neurodegenerative diseases, spinal degenerative diseases, retinal degenerative diseases, peripheral neurodegenerative diseases, and other diseases.
- Examples of cranial neurodegenerative diseases include: Neurodegenerative diseases (eg, Alzheimer's disease, Parkinson's disease, Huntington's disease, Down syndrome, etc.), cerebral ischemic diseases (stroke, cerebral infarction, transient ischemic attack, subarachnoid hemorrhage, ischemic encephalopathy, cerebral infarction (Such as lacunar infarction, atherothrombotic cerebral infarction, cardiogenic cerebral infarction, hemorrhagic cerebral infarction, other infarction)), traumatic brain injury, leukoencephalopathy, and multiple sclerosis.
- Neurodegenerative diseases eg, Alzheimer's disease, Parkinson's disease, Huntington's disease, Down syndrome, etc.
- cerebral ischemic diseases stroke, cerebral infarction, transient ischemic attack, subarachnoid hemorrhage, ischemic encephalopathy, cerebral infarction (Such as lacunar infarction, atherothrombotic cerebral infarction, cardiogenic cerebral infarction, hemor
- spinal degenerative diseases include: Examples include amyotrophic lateral sclerosis (ALS), spinal cord injury, spinal cord injury due to various causes, spinal progressive muscular atrophy, and spinal cerebral degeneration (SCD).
- ALS amyotrophic lateral sclerosis
- SCD spinal cerebral degeneration
- retinal degenerative diseases include: Examples include age-related macular degeneration (AMD), diabetic retinopathy, retinitis pigmentosa, hypertensive retinopathy, and glaucoma.
- AMD age-related macular degeneration
- diabetic retinopathy examples include diabetic retinopathy, retinitis pigmentosa, hypertensive retinopathy, and glaucoma.
- peripheral neurodegenerative diseases include Diabetic neuropathy, peripheral neuropathy, traumatic peripheral neuropathy, addiction, peripheral neuropathy caused by other toxic substances, peripheral neuropathy caused by cancer chemotherapy, Guillain-Barre syndrome, peripheral neuropathy caused by deficiency of vitamins, Amyloid peripheral neuropathy, ischemic peripheral neuropathy, peripheral neuropathy associated with malignant tumor, uremic peripheral neuropathy, peripheral neuropathy due to physical cause, Charcot-Marie-Tooth disease, alcoholic peripheral neuropathy, autonomic neuropathy (Subconscious hypoglycemia, gastric paresis, neurogenic diarrhea and constipation, erectile dysfunction, orthostatic hypotension, arrhythmia, heart failure, painless myocardial infarction, sweating abnormalities, neurogenic bladder, etc.), bladder dysfunction (e.g., Uninhibited bladder, reflex bladder, autonomous bladder, sensory paralytic bladder, motor paralytic bladder, etc.).
- bladder dysfunction e.g., Uninhibited bladder, reflex bladder, autonomous bladder, sensory paralytic bladder, motor paralytic bladder, etc.
- Other diseases include, for example, Associated with depression, schizophrenia, epilepsy, autism, periodontal disease, diabetes, diabetic cardiomyopathy, diabetic foot lesions, inflammatory bowel disease (eg, ulcerative colitis, Crohn's disease), dementia Problematic behavior (eg, acupuncture, aggressive behavior), anxiety, pain, hearing impairment, bone disease (eg, osteoporosis), joint disease (eg, Charcot joint, osteoarthritis, rheumatism, etc.), Hirsch Examples include Sobrung's disease.
- the compound of this invention is applied suitably with respect to a cerebral ischemic disease or diabetic neuropathy.
- the compound of the present invention can be used as a composition (eg, pharmaceutical composition) formulated as it is or mixed with a pharmaceutically acceptable carrier or the like for diseases involving neurotrophic factor activity.
- a pharmaceutically acceptable carrier or the like for diseases involving neurotrophic factor activity.
- it can be safely administered orally or parenterally to mammals such as humans.
- Parenterally includes intravenous, intramuscular, subcutaneous, intraorgan, intranasal, intradermal, instillation, intracerebral, rectal, intravaginal, intraperitoneal, and the like.
- the composition of the present invention contains, for example, one or more compounds of the present invention and a pharmaceutically acceptable carrier, excipient, and / or additive.
- a pharmaceutically acceptable carrier, excipient, and / or additive eg, pharmaceutical additive, food additive
- Agent, cosmetic additive e.g., cosmetic additive
- the pharmaceutically acceptable carrier, excipient, and / or additive used in the composition of the present invention can be appropriately selected depending on the specific use of the composition.
- the form of the said composition can also be made into forms, such as various solid and liquid, according to a specific use, for example.
- compositions of the present invention or the compound of the present invention when used as a pharmaceutical, specific forms such as powders, fine granules, granules, tablets, syrups, capsules, suspending agents And oral agents such as emulsions, extracts and pills, injections, transdermal solutions (external preparations for skin) such as ointments, suppositories, and parenterals for topical use. .
- Oral preparations include, for example, gelatin, sodium alginate, starch, corn starch, sucrose, lactose, glucose, mannitol, carboxymethylcellulose, dextrin, polyvinylpyrrolidone, crystalline cellulose, soy lecithin, sucrose, fatty acid ester, talc, magnesium stearate, Carriers and excipients such as polyethylene glycol, magnesium silicate, and silicic anhydride, binders, disintegrants, surfactants, lubricants, fluidity promoters, diluents, preservatives, colorants, fragrances, stabilization It can be produced according to a usual method using a pharmaceutical additive such as an agent, a humectant, a preservative, and an antioxidant.
- a pharmaceutical additive such as an agent, a humectant, a preservative, and an antioxidant.
- the dose varies depending on the age, sex, weight, disease level, type of compound of the present invention, dosage form, etc. of the mammal to be administered.
- the amount may be about 1 mg to about 2 g, preferably about 5 mg to about 1 g as the active ingredient amount.
- the above-mentioned daily dose can be administered once or divided into several times.
- injections are water-soluble solvents such as physiological saline and sterile water Ringer's solution, non-water-soluble solvents such as vegetable oil and fatty acid esters, isotonic agents such as glucose and sodium chloride, solubilizing agents, stable It can be produced according to a usual method using a pharmaceutical additive such as an agent, a preservative, a suspending agent, and an emulsifier.
- a pharmaceutical additive such as an agent, a preservative, a suspending agent, and an emulsifier.
- Liquid preparations for external use, percutaneous absorption agents such as gel ointments, suppositories for rectal administration and the like can also be produced according to conventional methods.
- injection subcutaneous, intravenous, etc.
- transdermal administration or rectal administration may be used.
- the topical agent can be produced, for example, by incorporating the compound of the present invention into a pellet of a sustained release polymer such as an ethylene vinyl acetate polymer. This pellet may be surgically implanted into the tissue to be treated.
- the dose varies depending on the age, sex, body weight, degree of disease, the type of the composition of the present invention or the compound of the present invention, the mode of administration, etc. And about 0.1 mg to about 500 mg may be administered as the active ingredient amount.
- the above-mentioned daily dose can be administered once or divided into several times.
- Synthesis example 1 Synthesis Synthesis Example of 5- (Benzothiazol-2-yl) -2-methyl-1-phenylbenzimidazole 1-1 Synthesis of 3-nitro-4-phenylaminobenzoic acid
- 4-fluoro-3-nitrobenzoic acid (2.51 g, 13.56 mmol)
- aniline (2.52 g, 27.12 mmol)
- ethanol 9.5 mL
- the mixture was poured into dilute hydrochloric acid (1M, 50 mL), diluted with distilled water (100 mL), and stirred at room temperature for 30 minutes.
- the precipitated crystals were collected by filtration, washed with dilute hydrochloric acid (1M, about 3 mL ⁇ 3 times), distilled water (about 5 mL ⁇ 2 times), and diethyl ether (about 3 mL). The obtained crystals were heated and dried under reduced pressure to give the title compound (3.39 g, quantitative) as a red-orange solid.
- Synthesis Example 1-2 Synthesis of 3-amino-4-phenylaminobenzoic acid
- 3-nitro-4-phenylaminobenzoic acid see Synthesis Example 1-1) (1.67 g, 6.70 mmol), palladium / carbon (Pd: 10%, 0.170 g), and tetrahydrofuran- (34 mL), and vacuum replacement / hydrogen gas replacement was repeated three times to replace with hydrogen, followed by stirring at room temperature for 2.5 hours. After purging with nitrogen, distilled water (20 mL) was added, and the mixture was stirred for 10 minutes.
- Synthesis Example 1-3 Synthesis of 2-methyl-1-phenylbenzimidazole-5-carboxylic acid
- 3-amino-4-phenylaminobenzoic acid see Synthesis Example 1-2
- Acetyl chloride (1.00 g, 12.70 mmol) / toluene solution (about 2.5 mL) was added dropwise thereto in about 15 minutes, and the mixture was stirred for 2.5 hours under the same conditions.
- the mixture was allowed to cool to room temperature and extracted with dilute aqueous sodium hydroxide solution (10%, 200 mL).
- Synthesis example 2 Synthesis of 5- (benzimidazol-2-yl) -2-methyl-1-phenylbenzimidazole To a two-necked flask (20 mL) with a reflux condenser, 2-methyl-1-phenylbenzimidazole-5-carboxylic acid (see Synthesis Example 1-3) (0.15 g, 0.59 mmol) and polyphosphoric acid (about 2 g) was added and heated to 120 ° C. 1,2-Diaminobenzene (0.09 g, 0.83 mmol) was added thereto, heated to 160 ° C., and stirred for 20 hours under the same conditions.
- Synthesis example 3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-phenylbenzimidazole To a two-necked flask (20 mL) with a reflux condenser, 2-methyl-1-phenylbenzimidazole-5-carboxylic acid (see Synthesis Example 1-3) (0.15 g, 0.59 mmol), 2-aminophenol (0.09) g, 0.82 mmol) and polyphosphoric acid (about 2 g) were added, heated to 160 ° C., and stirred for 20 hours under the same conditions. After cooling to room temperature, ice was added, and the solution was adjusted to about pH 9 with concentrated aqueous ammonia (28%).
- Synthesis example 4 Synthesis of 2-amino-1- (tetrahydropyran-4-yl) amino-4-trifluoromethylbenzene Synthesis Example 4-1 Synthesis of 1- (tetrahydropyran-4-yl) amino-2-nitro-4-trifluoromethylbenzene In a three-necked flask (200 mL), 1-fluoro-2-nitro-4-trifluoromethylbenzene (1.59 g, 7.61 mmol), 4-aminotetrahydropyran- (1.00 g, 9.89 mmol), and pyridine (16 mL) and stirred at 98 ° C. for 23 hours.
- Synthesis Example 4-2 Synthesis of 2-amino-1- (tetrahydropyran-4-yl) amino-4-trifluoromethylbenzene
- 1- (tetrahydropyran-4-yl) amino-2-nitro-4-trifluoromethylbenzene see Synthesis Example 4-1) (1.00 g, 3.45 mmol)
- tetrahydrofuran- (17 mL) were added and shaken to make a homogeneous solution.
- Palladium / carbon (Pd: 10%, 0.09 g) was added thereto and shaken again. Subsequently, reduced pressure / replacement with hydrogen gas was repeated three times to form a hydrogen atmosphere.
- Synthesis example 5 Synthesis Example of 5- (Benzoxazol-2-yl) -1,2-dimethylbenzimidazole5-1 Synthesis of (2-methylaminoanily-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (300 mg, 1.2 mmol), potassium carbonate (50 mg, 3.5 mmol), 40% aqueous monomethylamine (270 mg, 3.5 mmol) ) Containing ethanol (5 mL) solution, and heated and stirred for 2 hours under reflux conditions. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered.
- Synthesis Example 5-2 Synthesis of 5- (benzoxazol-2-yl) -1,2-dimethylbenzimidazole (2-Methylaminoanily-5-yl) benzoxazole (see Synthesis Example 5-2) (75 mg, 0.31 mmol) was dissolved in DMF (2 mL), 90% acetaldehyde aqueous solution (46 mg, 0.94 mmol), and then oxone ( 192 mg, 0.31 mmol) was added, and the mixture was stirred at room temperature for 2.5 hours. The reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- Example 1 Synthesis Example of 5- (Benzothiazol-2-yl) -1-phenyl-2- (phenylmethoxy) methylbenzimidazole
- Example 1-1 Synthesis of 1-phenyl-2- (phenylmethoxy) methylbenzimidazole-5-carboxylic acid To a four-necked flask (300 mL) with a reflux condenser, add 3-amino-4-phenylaminobenzoic acid (see Synthesis Example 1-1) (3.09 g, 13.54 mmol), and dehydrated toluene (45 mL). ) And refluxed.
- Benzyloxyacetyl chloride (5 g, 27.08 mmol) / toluene solution (about 3 mL) was added dropwise thereto in about 10 minutes. The mixture was stirred for 15 hours under the same conditions. The mixture was allowed to cool to room temperature and extracted with dilute aqueous sodium hydroxide solution (10%, 100 mL). After washing with toluene (80 mL) and separating the dark orange aqueous layer, the solution was cooled (5-10 ° C.), and the liquid was adjusted to pH 4 with concentrated hydrochloric acid (12M).
- Example 1-2 Synthesis of 5- (benzothiazol-2-yl) -1-phenyl-2- (phenylmethoxy) methylbenzimidazole
- 1-phenyl-2- (phenylmethoxy) methylbenzimidazole-5-carboxylic acid see Example 1-1) (4.00 g, 11.16 mmol)
- Tetrahydrofuran- 63 mL
- oxalyl chloride (1.65 g, 13.0 mmol) was added, and the mixture was warmed to 50 ° C. and stirred for 3 hr.
- Example 2 Synthesis of 5- (benzothiazol-2-yl) -2-hydroxymethyl-1-phenylbenzimidazole To an eggplant flask (50 mL) with a reflux condenser, add 5- (benzothiazol-2-yl) -1-phenyl-2- (phenylmethoxy) methylbenzimidazole (see Example 1-2) (0.40 g, 0.89 mmol). And dilute hydrochloric acid (6M, 5 mL) were added and refluxed for 2 hours. When the obtained pale yellow transparent solution was ice-cooled, it became a milky white suspension.
- the reaction mixture was allowed to cool to room temperature, and the solvent was distilled off under reduced pressure to prepare the corresponding chloride as a greenish milky white solid.
- dehydrated tetrahydrofuran- (3 mL), and a dimethylamine / tetrahydrofuran-solution (2.0M, 1.1M) was added. mL) was added dropwise.
- the mixture was heated to 40 ° C. and stirred for 2 hours.
- Example 4 Synthesis of 5- (benzothiazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 4-1 Synthesis of 4-((tetrahydropyran-4-yl) amino) -3-nitrobenzoic acid
- 4-Fluoro-3-nitrobenzoic acid (126.4 g, 0.68 mol) was dissolved in ethanol (880 mL).
- Triethylamine (82.6 g, 0.82 mol) and 4-aminotetrahydropyran- (82.7 g, 0.81 mol) were added dropwise in this order, and after the addition, the reaction solution was heated to reflux.
- Example 4-2 Synthesis of 3-amino-4-((tetrahydropyran-4-yl) amino) benzoic acid 4-((Tetrahydropyran-4-yl) amino) -3-nitrobenzoic acid (see Example 4-1) (202 g, 0.76 mol) dissolved in a mixed solution of THF (2.2 L) and methanol (1.5 L) Then, Pd / C (5%, wet, 20 g) was added and a hydrogenation reaction was performed at 5 atm. Since the absorption of hydrogen ceased in 3 hours, the inside of the system was purged with argon, the catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure to obtain the title compound (180 g, quantitative) as a dark gray solid.
- Example 4-3 Synthesis of 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid 3-Amino-4-((tetrahydropyran-4-yl) amino) benzoic acid (see Example 4-2) (90 g, 0.38 mol) was dissolved in dioxane (900 mL), cooled to 10 ° C., and then chlorinated. A solution of acetyl (35.0 g, 0.45 mol) in dioxane (900 mL) was added dropwise over 35 minutes.
- Example 4-4 Synthesis of 5- (benzothiazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole To a four-necked flask (50 mL) with a reflux condenser, add 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.28 g, 1.08 mmol). , 2-aminobenzenethiol (0.14 g, 1.08 mmol) and polyphosphoric acid (about 11 g) were added, heated to 150 ° C. and stirred for 17 hours.
- Chloroform (1 L) was added for liquid separation, and the chloroform layer was washed with water (500 mL) and dehydrated with Na 2 SO 4 .
- Example 6 Synthesis of 5- (benzothiazol-2-yl) -2-methoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 6-1 Synthesis of 2-methoxymethyl-1- (tetrahydropyran-4-yl) -5-trifluoromethylbenzimidazole
- 2-amino-1- (tetrahydropyran-4-yl) amino-4-trifluoromethylbenzene see Synthesis Example 4
- dehydrated 1,4-dioxane 8.5 mL).
- Methoxyacetyl chloride (0.57 g, 5.25 mmol) / 1,4-dioxane solution (about 1.5 mL) was added dropwise thereto in about 10 minutes. The mixture was stirred for 2 hours under the same conditions. The mixture was allowed to cool to room temperature, distilled water was added, and the solvent was distilled off under reduced pressure. Extraction with t-butyl methyl ether, washing with brine and drying over anhydrous sodium sulfate followed by evaporation of the solvent under reduced pressure gave the title compound (0.58 g, yield 77.4%) as a white powder.
- Example 6-2 Synthesis of 5- (benzothiazol-2-yl) -2-methoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- 2-methoxymethyl-1- (tetrahydropyran-4-yl) -5-trifluoromethylbenzimidazole see Example 6-1 (0.102 g, 0.32 mmol)
- 2-aminobenzenethiol 0.041 g, 0.33 mmol
- polyphosphoric acid about 2 g
- Example 7 Synthesis of 2-acetoxymethyl-5- (benzothiazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 7-1 Synthesis of 2-acetoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid
- 3-amino-4-((tetrahydropyran-4-yl) amino) benzoic acid 14.53 g, 45.65 mmol
- dehydrated 1,4-dioxane 226 mL
- Acetoxyacetyl chloride (12.57 g, 92.07 mmol) / 1,4-dioxane solution (100 mL) was added dropwise thereto in about 15 minutes. The mixture was stirred for 13 hours under the same conditions. The mixture was allowed to cool to room temperature, and the precipitated crystals were collected by filtration, washed with distilled water, and dried by heating under reduced pressure to give the title compound (13.46 g, yield 92.6%) as a white powder.
- Example 8 Synthesis of 5- (benzothiazol-2-yl) -2-hydroxymethyl-1- (tetrahydropyran-4-yl) benzimidazole A 4-necked flask (1 L) was charged with 2-acetoxymethyl-5- (benzothiazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 7-2) (5.00 g, 12.27 mmol) and methanol (253 mL) were added, and an aqueous lithium hydroxide solution (1.0 M, 61 mL) was added at room temperature.
- a hydrogen chloride / diethyl ether solution (2.0 M, 2 mL) was gradually added thereto, followed by stirring for 30 minutes under the same conditions.
- the generated crystals were collected by filtration, washed with diethyl ether (about 2 mL ⁇ 3 times), and dried under reduced pressure to give the title compound (0.186 g, yield 96.9%) as a pale yellow powder.
- Example 11 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole An eggplant flask (100 mL) was charged with 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.64 g, 2.46 mmol), 2-aminophenol ( 0.32 g, 2.95 mmol) and polyphosphoric acid (about 18 g) were added, heated to 160 ° C. and stirred for 17 hours. After allowing to cool, ice was added and the solution was adjusted to about pH 9 with concentrated aqueous ammonia (28%).
- Example 12 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-methanesulfonate 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (51.2 g, 0.19 mol), 2-aminophenol (24.0 g, 0.21 mol), dehydration DMF (500 mL) and WSC (45.0 g, 0.23 mol) were stirred for 3 hours under an argon stream. Water (2 L) was added, and the resulting solid was collected by filtration, washed with water, and dried at 50 ° C. under reduced pressure.
- Example 13 Synthesis of 5- (benzoxazol-2-yl) -2-hydroxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 13-1 Synthesis of 2-acetoxymethyl-5- (2-hydroxyanilinocarbonyl) -1- (tetrahydropyran-4-yl) benzimidazole
- 2-acetoxymethyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid see Example 7-1) (1.50 g, 4.71 mmol).
- Example 13-2 Synthesis of 5- (benzoxazol-2-yl) -2-hydroxymethyl-1- (tetrahydropyran-4-yl) benzimidazole
- An eggplant flask 50 mL was charged with 2- (acetoxymethyl) -5- (2-hydroxyanilinocarbonyl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 13-1) (0.50 g, 1.22 mmol), toluene (6.3 mL), and p-toluenesulfonic acid hydrate (1.60 g, 8.41 mmol) were added and refluxed for 1.5 hours. After standing to cool, the liquid was neutralized with a saturated aqueous sodium hydrogen carbonate solution.
- Example 15 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxyphenyl) -2-methylbenzimidazole
- Example 15-1 Synthesis of 4-fluoro-N- (2-hydroxyphenyl) -3-nitrobenzanilide
- 2-aminophenol (1.18 g, 10.8 mmol)
- 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride Salt (2.17 g, 11.3 mmol)
- triethylamine (1.15 g, 11.3 mmol
- reaction mixture was diluted with ethyl acetate and washed with water, 1N hydrochloric acid and saturated aqueous sodium hydrogen carbonate solution.
- the obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to give the title compound (2.02 g, yield 68%) as orange crystals.
- Example 15-2 Synthesis of 2- (4-fluoro-3-nitrophenyl) benzoxazole
- 4-fluoro-N- (2-hydroxyphenyl) -3-nitrobenzanilide see Example 15-1
- p-toluenesulfonic acid ⁇ 1 Hydrate 2.00 g, 10.9 mmol
- the reaction solution was cooled to room temperature, the solvent was distilled off, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for 10 minutes.
- Example 15-3 Synthesis of 2- (4- (4-methoxyphenylamino) -3-nitrophenyl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- sodium bicarbonate 295 mg, 2.32 mmol
- p. -Anisidine 357 mg, 2.90 mmol
- the mixture was heated with stirring under reflux conditions for 4 hours.
- the mixture was cooled to room temperature, and the precipitated crystals were filtered, washed with water and ethanol, and dried to give the title compound (359 mg, yield 86%) as orange crystals.
- Example 15-4 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxyphenyl) -2-methylbenzimidazole 2- (4- (4-methoxyphenylamino) -3-nitrophenyl) benzoxazole (see Example 15-3) (150 mg, 0.415 mmol) in 10% palladium on carbon (50 mg) in tetrahydrofuran- (5 mL) solution
- the inside of the flask was replaced with hydrogen and stirred at room temperature for 3 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Acetyl chloride (70.8 mg, 0.902 mmol) was added to a toluene (5 mL) solution of the obtained oily substance, and the mixture was heated with stirring under reflux conditions for 2.5 hours. After completion of the reaction, the mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated, and the obtained crystals were purified by silica gel column chromatography to give the title compound (47 mg, yield 32%) as dark brown crystals.
- Example 16 Synthesis of 5- (benzoxazol-2-yl) -1-cyclohexyl-2-methylbenzimidazole
- Example 16-1 Synthesis of 2- (4-cyclohexylamino-3-nitrophenyl) benzoxazole
- sodium bicarbonate 295 mg, 2.32 mmol
- Amine 288 mg, 2.32 mmol
- Example 16-2 Synthesis of 5- (benzoxazol-2-yl) -1-cyclohexyl-2-methylbenzimidazole 2- (4-Cyclohexylamino-3-nitrophenyl) benzoxazole (see Example 16-1) (185 mg, 0.548 mmol) was added to a tetrahydrofuran- (5 mL) solution containing 10% palladium on carbon (50 mg). Was replaced with hydrogen and stirred at room temperature for 5 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Example 17 Synthesis Example of 5- (Benzoxazol-2-yl) -1-benzyl-2-methylbenzimidazole
- Example 17-1 Synthesis of 2- (4-benzylamino-3-nitrophenyl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- benzyl Amine 311 mg, 2.90 mmol
- Example 17-2 Synthesis of 2- (2-benzylaminoaniline-5-yl) benzoxazole 2- (4-Benzylamino-3-nitrophenyl) benzoxazole (see Example 17-1) (340 mg, 0.985 mmol) in 10% aqueous acetic acid (5 mL), ethanol (8 mL), iron powder (165 mg, 2.95) mmol) was added, and the mixture was stirred with heating under reflux conditions for 4 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution and chloroform were added, and the mixture was filtered through celite and extracted with chloroform.
- Example 17-3 Synthesis of 5- (benzoxazol-2-yl) -1-benzyl-2-methylbenzimidazole
- 2- (2-benzylaminoaniline-5-yl) benzoxazole (see Example 17-2) (80.0 mg, 0.254 mmol) in dimethylformamide (2 m) was added acetaldehyde aqueous solution (about 90%, 47.7 ⁇ l, 0.761 mmol) and oxone (102 mg, 0.165 mmol) were added and stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was extracted with ethyl acetate.
- Example 18 Synthesis of 5- (benzothiophen-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 18-1 Synthesis of 5-bromo-2- (tetrahydropyran-4-yl) nitrobenzene
- 5-bromo-2-fluoronitrobenzene 3.0 g, 13.6 mmol
- triethylamine (1.66 g, 16.3 mmol
- 4-aminotetrahydropyran- (1.52 g, 15.0 mmol
- ethanol 60 mL
- Example 18-2 Synthesis of 5-bromo-2- (tetrahydropyran-4-yl) aminoaniline A three-necked flask is charged with 5-bromo-2- (tetrahydropyran-4-yl) nitrobenzene (see Example 18-1) (3.54 g, 11.8 mmol) and 65 mL of 10% aqueous acetic acid, and then electrolytic iron. (6.56 g, 118 mmol) was added and the mixture was refluxed and stirred for 15 minutes. After standing to cool to room temperature, insolubles were filtered off through celite, and the same layer was washed with 65 mL of 10% aqueous acetic acid.
- Example 18-3 Synthesis of 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-hydrochloride
- 5-bromo-2- (tetrahydropyran-4-yl) aminoaniline see Example 18-2
- dehydrated toluene (20 mL)
- Acetyl chloride (1.57 g, 20.0 mmol) / toluene solution (about 2.5 mL) was added dropwise thereto in about 15 minutes, and the mixture was stirred for 2 hours under the same conditions.
- Example 18-4 Synthesis of 5- (benzothiophen-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-hydrochloride (see Example 18-3) (0.38 g, 1.15 mmol), 2-benzothiopheneboronic acid (0.25 g, 1.40) mmol), ethanol (5 mL), toluene (5 mL), 2M aqueous sodium carbonate solution (2.1 mL) were charged and degassed.
- Tetrakis (tophenylphosphine) palladium (0.08 g, 0.07 mmol) was added and heated to reflux for 3 hours. After cooling, ethanol and water were added, and the mixture was filtered through Celite, and the filtered product was washed with ethanol and water. The filtrate was concentrated, and the precipitated crystals were collected by filtration, washed with water and hexane, and dried to give the title compound (315 mg, yield 78.9%) as pale brown crystals.
- Example 19 Synthesis of 5- (benzofuran-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 18-3) (0.41 g, 1.24 mmol), 2-benzofuran-boronic acid (0.25 g, 1.40 mmol), ethanol (5 mL), toluene (5 mL), 2M aqueous sodium carbonate solution (2.1 mL) were charged and degassed. Tetrakis (tophenylphosphine) palladium (0.08 g, 0.07 mmol) was added and heated to reflux for 3 hours.
- Example 20 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 20-1 Synthesis of 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) nitrobenzene
- 5- (Benzoxazol-2-yl) -2-fluoronitrobenzene (0.55 g, 2.13 mmol)
- triethylamine (0.26 g, 2.57 mmol
- aminotetrahydropyran- (0.24 g, 2. 34 mmol) was added to ethanol (10 mL), and the reaction was heated and stirred for 2 hours under reflux conditions.
- Example 20-2 Synthesis of 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) nitrobenzene (see Example 20-1) (1.00 g, 0.76 mol) was dissolved in a mixed solution of 50 mL of THF and 50 mL of methanol, and Pd Hydrogenation reaction was performed by adding / C (5%, wet, 0.5 g). After stirring at room temperature overnight, the catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure to give the title compound (812 mg, yield 89.1%) as a gray solid.
- Example 20-3 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.20 g, 0.646 mmol), triethyl orthoformate (5 mL) in catalytic amount of p -Toluenesulfonic acid monohydrate was added and heated at 100 ° C for 1.5 hours. The reaction mixture was cooled, extracted with ethyl acetate and water.
- Example 21 Synthesis of 5- (benzoxazol-2-yl) -2- (2-pyridyl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 2-pyridinecarboxaldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (1335 mg, yield 70.2%) as white crystals.
- Example 22 Synthesis of 5- (benzoxazol-2-yl) -2-isopropyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, isopropylaldehyde (0.04 g, 0.561 mmol) and oxone (0.19 g, 0.310 mmol) were added, and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (70 mg, yield 43.9%) as white crystals.
- Example 23 Synthesis of 5- (benzoxazol-2-yl) -2-cyclohexyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, cyclohexyl aldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (116 mg, yield 59.6%) as white crystals.
- Example 24 Synthesis of 5- (benzoxazol-2-yl) -2- (3-pyridyl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 3-pyridinecarboxaldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (95.0 mg, yield 49.5%) as white crystals.
- Example 25 Synthesis of 5- (benzoxazol-2-yl) -2-phenyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, phenylaldehyde (0.06 g, 0.561 mmol) and oxone (0.19 g, 0.310 mmol) were added, and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (115 mg, yield 59.9%) as white crystals.
- Example 26 Synthesis of 5- (benzoxazol-2-yl) -2- (4-pyridyl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DM (F 3 mL) and water (0.1 mL) After dissolution, 4-pyridinecarboxaldehyde (0.06 g, 0.561 mmol) was added followed by oxone (0.19 g, 0.310 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.09 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (117 mg, yield 60.9%) as white crystals.
- Example 27 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) -2-trifluoromethylbenzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.20 g, 0.646 mmol) in trifluoroacetic acid (7 mL) for 4 hours and a half Heated to reflux. The reaction mixture was cooled, water was added, and the mixture was extracted with ethyl acetate.
- Example 28 Synthesis of 5- (benzoxazol-2-yl) -1- (tetrahydropyran-4-yl) -benzotriazole
- a solution of 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.35 g, 1.13 mmol) in concentrated hydrochloric acid (2 mL) was brought to 0 ° C.
- an aqueous solution 0.5 mL
- 0.09 g (1.24 mmol) of sodium nitrite was added dropwise.
- Example 29 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-tertbutylbenzimidazole
- Example 29-1 Synthesis of 2- (2-terbutylaminoaniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- Butylamine 212 mg, 2.90 mmol
- the mixture was heated with stirring under reflux conditions for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the mixture was extracted with chloroform.
- the obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- the obtained crystals were added to a tetrahydrofuran- (5 mL) solution containing 10% palladium on carbon (50 mg), the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 8 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the titled compound (50.1 mg, yield 19%) as red crystals.
- Example 29-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-terbutylbenzimidazole
- 2- (2-terbutylaminoaniline-5-yl) benzoxazole see Example 29-1 (45.0 mg, 0.160 mmol) in dimethylformamide (2 mL) was added to an aqueous solution of acetaldehyde (about 90%, 235 ⁇ l, 0.480).
- acetaldehyde about 90%, 235 ⁇ l, 0.480
- oxone (63.9 mg, 0.104 mmol) were added, and the mixture was stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, filtered and washed with water.
- Example 30 Synthesis of 5- (benzoxazol-2-yl) -1- (2-methoxyphenyl) -2-methylbenzimidazole
- Example 30-1 Synthesis of 2- (2- (2-methoxyphenyl) aminoaniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- o -Anisidine 357 mg, 2.90 mmol
- Example 30-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2-methoxyphenyl) -2-methylbenzimidazole
- 2- (2- (2-methoxyphenyl) aminoaniline-5-yl) benzoxazole see Example 30-1 (48.0 mg, 0.135 mmol) in dimethylformamide (2 mL) was added an aqueous acetaldehyde solution (about 90% , 20.5 ⁇ l, 0.405 mmol) and oxone (53.9 mg, 0.0878 mmol) were added, and the mixture was stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, filtered and washed with water.
- Example 31 Synthesis of 5- (benzoxazol-2-yl) -1- (3-methoxyphenyl) -2-methylbenzimidazole
- Example 31-1 Synthesis of 2- (2- (3-methoxyphenyl) aminoaniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- m -Anisidine 357 mg, 2.90 mmol
- Example 31-2 Synthesis of 5- (benzoxazol-2-yl) -1- (3-methoxyphenyl) -2-methylbenzimidazole
- 2- (2- (3-methoxyphenyl) aminoaniline-5-yl) benzoxazole see Example 31-1
- an aqueous acetaldehyde solution about 90% 31.2 ⁇ l, 0.405 mmol
- oxone (66.3 mg, 0.108 mmol) were added and stirred at room temperature for 3 hours.
- an aqueous potassium carbonate solution was added, filtered and washed with water.
- Example 33 Synthesis of 5- (benzoxazol-2-yl) -1-((1-ethoxycarbonyl) piperidin-4-yl) -2-methylbenzimidazole
- Example 33-1 Synthesis of 2- (2- (4- (1-ethoxycarbonyl) piperidinoamino) anilin-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- 4- Ethyl amino-1-piperidinecarboxylate 499 mg, 2.90 mmol
- Example 34 Synthesis of 5- (benzoxazol-2-yl) -1- (4-fluorophenyl) -2-methylbenzimidazole
- Example 34-1 Synthesis of 2- (2- (4-fluorophenyl) aminoanilin-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- 4- Fluoroaniline 322 mg, 2.90 mmol
- Example 34-2 Synthesis of 5- (benzoxazol-2-yl) -1- (4-fluorophenyl) -2-methylbenzimidazole
- 2- (2- (4-fluorophenyl) aminoanilin-5-yl) benzoxazole (see Example 34-1) (38.0 mg, 0.119 mmol) in dimethylformamide (2 mL) was added an aqueous acetaldehyde solution (about 90% , 22.6 ⁇ l, 0.360 mmol) and oxone (73.2 mg, 0.119 mmol) were added and stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was filtered and washed with water.
- Example 35 Synthesis of 5- (benzoxazol-2-yl) -1- (piperidin-4-yl) -2-methylbenzimidazole
- Example 35-1 Synthesis of 2- (2- (4- (1-tertbutoxycarbonyl) piperidinoamino) anilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- sodium bicarbonate 329 mg, 3.88 mmol
- 4- Amino-1-tert-butoxycarbonylpiperidine 970 mg, 4.84 mmol
- Example 35-2 Synthesis of 5- (benzoxazol-2-yl) -1- (piperidin-4-yl) -2-methylbenzimidazole
- 2- (2- (4- (1-tertbutoxycarbonyl) piperidinoamino) anilin-5-yl) benzoxazole (see Example 35-1) (350 mg, 0.857 mmol) in dimethylformamide (3 mL) was added acetaldehyde aqueous solution. (About 90%, 161 ⁇ l, 2.57 mmol) and oxone (527 mg, 0.887 mmol) were added and stirred at room temperature for 1 hour.
- Example 36 Synthesis of 1-adamantyl-5- (benzoxazol-2-yl) -2-methylbenzimidazole
- Example 36-1 Synthesis of 2- (2- (1-adamantylamino) aniline-5-yl) benzoxazole
- sodium bicarbonate 195 mg, 2.32 mmol
- 1- Adamantamine amine 185 mg, 1.22 mmol was added, and the mixture was heated with stirring under reflux conditions for 4 hours.
- the reaction mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- the obtained crystals were added to a tetrahydrofuran- (5 mL) solution containing 10% palladium on carbon (100 mg), the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 6 hours.
- the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the title compound (67.0 mg, yield 16%) as brown crystals.
- Example 36-2 Synthesis of 1-adamantyl-5- (benzoxazol-2-yl) -2-methylbenzimidazole 2- (2- (1-adamantylamino) anilin-5-yl) benzoxazole (see Example 36-1) (65.0 mg, 0.181 mmol) in dimethylformamide (2 mL) was added to an aqueous acetaldehyde solution (about 90%, 34.0 ⁇ l, 0.542 mmol) and oxone (111 mg, 0.181 mmol) were added, and the mixture was stirred at room temperature for 2 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was extracted with ethyl acetate.
- Example 37 Synthesis of 5- (Nt-butoxycarbonylindol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 5-bromo-2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 18-3) (0.40 g, 1.21 mmol), 2- (Nt-butoxycarbonylindole-) boronic acid ( 0.35 g, 0.242 mmol), ethanol (5 mL), toluene (5 mL), 2M aqueous sodium carbonate solution (1.8 mL) were charged and degassed.
- Tetrakis (tophenylphosphine) palladium (0.07 g, 0.07 mmol) was added, and the mixture was heated to reflux for 3 hours. After cooling, ethanol and water were added, and the mixture was filtered through Celite, and the filtered product was washed with ethanol and water. The filtrate was concentrated, and the precipitated crystals were collected by filtration, washed with water and hexane, and dried to give the title compound (215 mg, yield 41.3%) as white crystals.
- Example 38 Synthesis of 5- (Indol-2-yl) -1- (tetrahydropyran-4-yl) -2-methylbenzimidazole 5- (Nt-butoxycarbonylindol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 38-1) (200 mg, 0.463 mmol) was added to a 1N aqueous hydrochloric acid solution. (10 mL) and stirred at room temperature for 3 hours. After completion of the reaction, the mixture was allowed to stand for 3 days, and the precipitated crystals were collected by filtration, added to saturated aqueous sodium hydrogen carbonate (5 mL), and stirred for 30 minutes.
- Example 39 Synthesis of 5- (5-methylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-4-methylphenol (0.13 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred at room temperature for 3 hours. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 40 Synthesis of 5- (5-chlorobenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-4-chlorophenol (0.15 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred overnight at room temperature. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 41 Synthesis of 5- (6-chlorobenzooxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-5-chlorophenol (0.15 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred overnight at room temperature. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 42 Synthesis of 5- (6-methylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (0.25 g, 0.96 mmol), 2-amino-5-methylphenol (0.13 g, 1.05) mmol), dehydrated DMF (10 mL) and WSC (0.22 g, 1.14 mmol) were stirred overnight at room temperature. After completion of the reaction, water (50 mL) was added, the precipitated crystals were filtered, and the filter residue was extracted with water / chloroform.
- Example 43 Synthesis of 5- (benzoxazol-2-yl) -2-ethyl-1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.15 g, 0.484 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, propylaldehyde (0.03 g, 0.561 mmol) and oxone (0.19 g, 0.310 mmol) were added, and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (44.7 mg, yield 26.6%) as pale brown crystals.
- Example 44 Synthesis of 5- (benzoxazol-2-yl) -2- (imidazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.10 g, 0.32 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 2-imidazole-carbaldehyde (0.03 g, 0.31 mmol) was added followed by oxone (0.13 g, 0.21 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- the mixture was extracted with chloroform, washed with water, dried over magnesium sulfate, concentrated, and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (15 mg, yield 12.1%) as pale brown crystals.
- Example 45 Synthesis of 5- (benzoxazol-2-yl) -2- (thiophen-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (Benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline (see Example 20-2) (0.13 g, 0.42 mmol) in DMF (3 mL) and water (0.1 mL) After dissolution, 2-thiophenecarbaldehyde (0.05 g, 0.45 mmol) was added followed by oxone (0.17 g, 0.28 mmol), and the mixture was stirred at room temperature for 2.5 hours.
- the reaction solution was added to an aqueous potassium carbonate solution (0.10 g / 15 mL).
- the mixture was extracted with ethyl acetate, washed with water, dried over magnesium sulfate, concentrated and purified by silica gel column chromatography.
- the obtained crystals were washed with hexane and a small amount of ethyl acetate and dried to give the title compound (60 mg, yield 36%) as pale brown crystals.
- Example 46 Synthesis of 2-methyl-5- (4-methylbenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 46-1 Synthesis of 2-fluoro-5- (4-methylbenzoxazol-2-yl) -nitrobenzene
- Thionyl chloride (0.78 g, 6.56 mmol) was added to a suspension of 3-fluoro-2-nitrobenzoic acid (1.00 g, 5.40 mmol) and 1 drop of DMF in toluene (10 mL), and the mixture was stirred for 3 hours under reflux conditions.
- Example 46-2 Synthesis of 2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole
- 2-fluoro-5- (4-methylbenzoxazol-2-yl) -nitrobenzene see Example 46-1 (0.50 g, 1.8 mmol) in ethanol (10 mL) was added triethylamine (0.22 g, 2.17).
- mmol and aminotetrahydropyran- (0.20 g, 1.9 mmol) were added, and the mixture was heated to reflux for 3 hours.
- Example 46-3 Synthesis of 2-methyl-5- (4-methylbenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 2- (2- (Tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole (see Example 46-2) (0.56 g, 1.7 mmol), dimethylformamide (5 mL) containing water (0.18 mL) Acetaldehyde aqueous solution (about 90%, 90 mg, 1.8 mmol) and oxone (0.69 mg, 1.1 mmol) were added to the solution, and the mixture was stirred at room temperature for 3 hours.
- Example 47 Synthesis of 2-methyl-5- (6-nitrobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 47-1 Synthesis of 5- (2-hydroxy-4-nitroanilinocarbonyl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole
- toluene (10 mL) containing 1 drop of DMF Thionyl chloride (0.41 g, 3.4 mmol) was added, and the mixture was stirred for 7 hours under reflux conditions.
- Example 47-2 Synthesis of 2-methyl-5- (6-nitrobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (2-hydroxy-4-nitroanilinocarbonyl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole (see Example 47-1) (195 mg, 0.492 mmol) in toluene (20 mL) Then, p-toluenesulfonic acid hydrate (280 mg, 1.47 mmol) was added, and the mixture was refluxed and stirred for 4 hours. After the solvent was concentrated under reduced pressure, multistory water (10 mL) was added and stirred at room temperature for 1 hour.
- Example 48 Synthesis of 2-methyl-5- (6-aminobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-5- (6-nitrobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 47-2) (135 mg, 0.357 mmol) was converted to palladium / carbon (Pd In addition to methanol (20 mL) containing 10%, 0.06 g), hydrogen substitution was repeated by repeating reduced pressure / hydrogen gas replacement three times, followed by stirring at room temperature for 2.5 hours.
- Example 49 Synthesis of 5- (benzoxazol-2-yl) -1- (4-hydroxycyclohexyl) -2-methylbenzimidazole
- Example 49-1 Synthesis of (2- (4-hydroxycyclohexylamino) nitrobenzene-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (0.60 g, 2.3 mmol), triethylamine (0.70 g, 7.0 mmol), 4-aminocyclohexanol hydrochloride (0.53 g, 2.5 mmol) in acetonitrile (20 mL), and the mixture was heated and stirred for 2 hours under reflux conditions.
- Example 49-2 Synthesis of 5- (benzoxazol-2-yl) -1- (4-hydroxycyclohexyl) -2-methylbenzimidazole 2- (2- (2-methoxyphenyl) aminoaniline-5-yl) benzoxazole (see Example 49-1) (0.20 g, 0.62 mmol), methyl imidate hydrochloride (0.07 g, 0.68 mmol) and methanol was stirred with heating under reflux conditions for 5 hours. After completion of the reaction, the mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 50 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole
- Example 50-1 Synthesis of 2- (2-n-propylaminoaniline-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- potassium carbonate 176 mg, 1.28 mmol
- Amine 82.4 mg, 1.39 mmol
- Example 50-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole
- 2- (2-n-propylaminoanilin-5-yl) benzoxazole see Example 50-1) (148 mg, 0.554 mmol) in dimethylformamide (3 ml) was added a solution of acetaldehyde (ca. 90%, 104 ⁇ l, 1.66 mmol) and oxone (341 mg, 0.554 mmol) were added, and the mixture was stirred at room temperature for 3 hours. After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was filtered and washed with water.
- Example 51 Synthesis of 5- (benzoxazol-2-yl) -1- (2-methoxyethyl) -2-methylbenzimidazole
- Example 51-1 Synthesis of 2- (2- (2-methoxyethyl) aminoanilin-5-yl) benzoxazole
- potassium carbonate 176 mg, 1.28 mmol
- Ethylamine 105 mg, 1.39 mmol was added and heated to reflux for 3.5 hours.
- Example 52 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-phenylethyl) benzimidazole
- Example 52-1 Synthesis of 2- (2- (2-phenylethyl) aminoaniline-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (300 mg, 1.16 mmol) in ethanol (5 ml) was added potassium carbonate (176 mg, 1.28 mmol), 2-phenyl.
- Ethylamine 169 mg, 1.39 mmol was added and heated to reflux for 4 hours.
- Example 53 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylbenzimidazole
- Example 53-1 Synthesis of 2- (2-cyc-propylaminoaniline-5-yl) benzoxazole
- potassium carbonate 176 mg, 1.28 mmol
- cyclopropylamine 99.3 mg, 1.74 mmol
- Example 53-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylbenzimidazole
- 2- (2-cyc-propylaminoanilin-5-yl) benzoxazole (see Example 53-1) (140 mg, 0.425 mmol) in methanol (5 ml) was added 1,1,1-trimethoxyethane (75 mg , 0.624 mmol) was added and the mixture was heated to reflux for 6 hours. After completion of the reaction, the reaction mixture was concentrated and the resulting residue was purified by silica gel column chromatography to obtain 63.5 mg (42%) of imidazole.
- Example 54 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylmethylbenzimidazole
- Example 54-1 Synthesis of 2- (2-cyc-propylmethylaminoanilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (300 mg, 1.16 mmol) in ethanol (5 ml) was added potassium carbonate (176 mg, 1.28 mmol), cyclopropylmethyl.
- Amine 176 mg, 1.28 mmol was added and heated to reflux for 7 hours.
- Example 54-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-cyc-propylmethylbenzimidazole
- 2- (2-cyc-propylmethylaminoanilin-5-yl) benzoxazole (see Example 54-1) (140 mg, 0.425 mmol) in methanol (5 ml) was added 1,1,1-trimethoxyethane ( 75 mg, 0.624 mmol) was added, and the mixture was heated to reflux for 10 hours. After completion of the reaction, the reaction mixture was concentrated, and the resulting residue was purified by silica gel column chromatography to obtain the title compound (81.9 mg, yield 54%) as pink crystals.
- Example 55 Synthesis of 5- (benzoxazol-2-yl) -1- (2- (tert-butoxycarbonylamino) ethyl) -2-methylbenzimidazole
- Example 55-1 Synthesis of 2- (tert-butoxycarbonylamino) ethylaminoanilin-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (600 mg, 2.32 mmol) was converted to triethylamine (469 mg, 4.65 mmol) and 2- (tert-butoxycarbonylamino) ethylamine (447 mg, 2.79 mmol) in acetonitrile (10 ml) was added and heated to reflux for 5 hours.
- Example 55-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2- (tert-butoxycarbonylamino) ethyl) -2-methylbenzimidazole
- 2- (tert-butoxycarbonylamino) ethylaminoanilin-5-yl) benzoxazole 200 mg, 0.567 mmol
- methyl acetimidate hydrochloride 102 mg, 0.851 mmol
- Example 56 Synthesis of 1- (2-aminoethyl) -5- (benzoxazol-2-yl) -2-methylbenzimidazole hydrochloride 5- (Benzoxazol-2-yl) -1- (2- (tert-butoxycarbonylamino) ethyl) -2-methylbenzimidazole (see Example 55-2) (100 mg, 0.255 mmol) was added to 4N hydrochloric acid-dioxane. It was added to the solution (10 ml) and heated to reflux for 5 hours. After completion of the reaction, the title compound (95 mg, quantitative) was obtained as white crystals by concentration to dryness.
- Example 59 Synthesis of 5- (5-ter-butylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (250 mg, 0.842 mmol), 2-amino-4-tert-butylphenol (139 mg, 0.842 mmol) ), DMF (2 mL), chloroform (5 mL) and WSC (178 mg, 0.926 mmol) were added and stirred for 22 hours. Water was added, and the resulting solid was collected by filtration, washed with water, and dried.
- Example 60 Synthesis of 5- (5-nitrobenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (264 mg, 0.888 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 62 Synthesis of 5- (benzoxazol-2-yl) -2-trans-cinnam-1- (tetrahydropyran-4-yl) benzimidazole
- 5- (benzoxazol-2-yl) -2- (tetrahydropyran-4-yl) aminoaniline see Example 20-2
- trans-cinnamaldehyde 111 mg, 0.840 mmol
- oxone 258 mg, 0.420 mmol
- Example 66 Synthesis of 5- (5-methoxybenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (250 mg, 0.842 mmol), 2-amino-3-methoxyphenol (139 mg, 0.842 mmol) , DMF (5 mL) and WSC (178 mg, 0.926 mmol) were added and stirred for 22 hours. Water was added and extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- Methanesulfonic acid (283 mg, 2.95 mmol) was added to the obtained solid dioxane (5 mL) solution, and the mixture was heated to reflux for 18 hours. After cooling the reaction solution, water and saturated aqueous sodium hydrogen carbonate solution were added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The obtained residue was purified by silica gel chromatography to give the title compound (10 mg, yield 3.3%) as brown crystals.
- Example 70 Synthesis of 5- (5-ethylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 72 Synthesis of 5- (5-cyc-hexylbenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 74 Synthesis Example 5-4-1 of 5- (Benzoxazol-2-yl) -1-n-butyl-2-methylbenzimidazole Synthesis of 2- (4-n-butylcyamino-3-nitrophenyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (300 mg, 1.16 mmol) in ethanol (5 ml) suspension in potassium carbonate (321 mg, 2.32 mmol), butylamine (170 mg , 2.32 mmol) was added and heated to reflux for 4 hours.
- Example 74-2 2- (2-n-Butylaminoaniline-5-yl) benzoxazole
- 2- (4-n-butylcyamino-3-nitrophenyl) benzoxazole (see Example 74-1) (353 mg, 1.13 mmol) in tetrahydrofuran (5 ml) was added 10% palladium on carbon (50 mg), and Was replaced with hydrogen and stirred at room temperature for 15 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to obtain the title compound (305 mg, yield: 96%).
- Example 74-3 5- (Benzoxazol-2-yl) -1-n-butyl-2-methylbenzimidazole
- 2- (2-n-butylcyaminoaniline-5-yl) benzoxazole (see Example 74-2) (300 mg, 1.07 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (175 mg, 1.60 mmol).
- methyl acetimidate hydrochloride (175 mg, 1.60 mmol).
- a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- Example 75 Synthesis of 5- (5-cyanobenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 76 Synthesis of 5- (5-trifluoromethoxybenzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole 2-Methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (182 mg, 0.612 mmol) and thionyl chloride (2 ml) were added and stirred for 3 hours under reflux conditions. did.
- Example 79 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1- (2-picolyl) benzimidazole
- Example 79-1 Synthesis of 2- (2- (2-picolyl) aminoanilin-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (500 mg, 1.9 mmol) was added to a suspension of 2-picolylamine (520 mg, 4.8 mmol) in acetonitrile (5 ml). And heated to reflux for 2.5 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- Example 79-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-picolyl) benzimidazole
- 2- (2- (2-picolyl) aminoanilin-5-yl) benzoxazole (see Example 79-1) (250 mg, 0.79 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (100 mg, 0.87 mmol) was added and heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 80 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-iso-propylbenzimidazole 80-1 Synthesis of 2- (4-iso-propylamino-3-nitrophenyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (200 mg, 0.774 mmol) in ethanol (5 ml) suspension in potassium carbonate (214 mg, 1.55 mmol), isopropylamine ( 137 mg, 2.32 mmol) was added and the mixture was heated to reflux for 4 hours.
- Example 80-2 Synthesis of 2- (2-iso-propylaminoaniline-5-yl) benzoxazole
- 2- (4-iso-propylamino-3-nitrophenyl) benzoxazole see Example 80-1) (224 mg, 0.753 mmol) in tetrahydrofuran (5 ml) was added 10% palladium on carbon (40 mg), and the flask was The inside was replaced with hydrogen and stirred at room temperature for 15 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the title compound (184 mg, yield 91%).
- Example 80-3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-iso-propylbenzimidazole To a solution of 2- (2-iso-propylaminoaniline-5-yl) benzoxazole (see Example 80-2) (180 mg, 0.673 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (111 mg, 1.01 mmol).
- Example 81 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-neo-pentylbenzimidazole
- Example 81-1 Synthesis of 2- (4-neo-pentylamino-3-nitrophenyl) benzoxazole
- 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (200 mg, 0.774 mmol) in ethanol (5 ml) suspension in potassium carbonate (214 mg, 1.55 mmol), neopentylamine (135 mg, 1.55 mmol) was added and heated to reflux for 4 hours.
- Example 81-2 Synthesis of 2- (2-neo-pentylaminoaniline-5-yl) benzoxazole
- 2- (4-neo-pentylamino-3-nitrophenyl) benzoxazole see Example 81-1) (242 mg, 0.744 mmol) in tetrahydrofuran (5 ml) was added 10% palladium on carbon (40 mg), and the flask was The inside was replaced with hydrogen and stirred at room temperature for 15 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography to give the title compound (196 mg, yield 89%).
- Example 81-3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-neo-pentylbenzimidazole To a solution of 2- (2-neo-pentylaminoanilin-5-yl) benzoxazole (see Example 81-2) (192 mg, 0.650 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (107 mg, 0.975 mmol). And heated to reflux for 2 hours.
- Example 82 Synthesis of Example 8-2-1 of 5- (5-aminobenzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole Synthesis of 3-nitro-4-n-propylaminobenzoic acid To a suspension of 4-trifluoro-3-nitrobenzoic acid (2.0 g, 10.8 mmol) in ethanol (20 ml) was added potassium carbonate (2.34 mg, 16.2 mmol) and propylamine (1.27 g, 21.6 mmol). Heated to reflux for hours.
- Example 82-2 3-Amino-4-n-propylaminobenzoic acid To a solution of 3-nitro-4-n-propylaminobenzoic acid (see Example 82-1) (1.94 g, 10.8 mmol) in tetrahydrofuran (20 ml) was added 10% palladium carbon (200 mg), and the flask was filled with hydrogen. Replace and stir at room temperature for 6 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated to obtain the title compound (1.68 g, quantitative).
- Example 82-3 2-Methyl-1-n-propylbenzimidazole-5-carboxylic acid To a solution of 3-amino-4-n-propylaminobenzoic acid (see Example 82-2) (1.68 g, 8.65 mmol) in methanol (17 ml) was added methyl acetimidate hydrochloride (1.14 g, 10.4 mmol), The mixture was heated to reflux for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, diethyl ether was added, and the mixture was stirred at room temperature for 10 minutes. The obtained crystals were filtered, washed with diethyl ether and dried to give the title compound (2.20 g, quantitative).
- Example 82-4 Synthesis of 5- (5-aminobenzoxazol-2-yl) -2-methyl-1-n-propylbenzimidazole 2-Methyl-1-n-propylbenzimidazole-5-carboxylic acid (see Example 82-3) (1.0 g, 3.93 mmol) and thionyl chloride (8 ml) were added, and the mixture was stirred under reflux conditions for 3 hours. After completion of the reaction, 2-aminophenol (456 mg, 2.96 mmol), triethylamine (899 mg, 8.88 mmol) and tetrahydrofuran (10 mL) were added to the residue obtained by concentration under reduced pressure, and the mixture was stirred at room temperature for 14 hours.
- the reaction solution was cooled, 1N aqueous sodium hydroxide solution and chloroform were added, the mixture was filtered through celite, and the obtained filtrate was extracted. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The obtained residue was purified by silica gel column chromatography. The obtained oil was dissolved in tetrahydrofuran (3 ml), and 1M hydrogen chloride diethyl ether solution (1 ml) was added. The precipitated crystals were filtered, washed with tetrahydrofuran and dried to give the title compound (168 mg, yield 25%) as pale green crystals.
- Example 83 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2- (tetrahydropyran-4-yl) ethyl) benzimidazole
- Example 83-1 Synthesis of 2- (2- (tetrahydropyran-4-yl) ethyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (800 mg, 3.1 mmol) was replaced with 2- (tetrahydropyran-4-yl) ethylamine (520 mg, 3.9 mmol) and triethylamine (500 mg , 3.9 mmol) was added to a solution of acetonitrile (5 ml), and the mixture was heated to reflux for 3 hours.
- Example 83-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2- (tetrahydropyran-4-yl) ethyl) benzimidazole
- 2- (2- (tetrahydropyran-4-yl) ethyl) benzoxazole (see Example 83-1) (300 mg, 0.89 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (220 mg, 1.96 mmol). And heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 84 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1-((tetrahydropyran-4-yl) methyl) benzimidazole
- Example 84-1 Synthesis of 2-((tetrahydropyran-4-yl) methyl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (900 mg, 3.5 mmol) was added to (tetrahydropyran-4-yl) methylamine (520 mg, 3.9 mmol) and triethylamine (500 mg , 4.3 mmol) was added to an acetonitrile (5 ml) solution, and the mixture was heated to reflux for 3 hours.
- Example 84-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-((tetrahydropyran-4-yl) methyl) benzimidazole
- 2-((tetrahydropyran-4-yl) methyl) benzoxazole see Example 84-1 (300 mg, 0.93 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride (120 mg, 1.11 mmol). The mixture was heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 85 Synthesis Example of 5- (Benzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole
- Example 85-1 Synthesis of 2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole 2- (4-Fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (500 mg, 3.5 mmol) (tetrahydropyran-4-yl) methylamine (360 mg, 2.4 mmol) and triethylamine (590 mg, 7.3 mmol) was added to an acetonitrile (5 ml) solution, and the mixture was heated to reflux for 3 hours.
- Example 85-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole
- 2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole (see Example 85-1) (300 mg, 0.93 mmol) in methanol (5 ml) was added methyl acetimidate.
- Hydrochloride 120 mg, 1.11 mmol was added and heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 87 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (1,2,3,4-tetrahydronaphthalen-1-yl) benzimidazole
- Example 87-1 Synthesis of 2- (2- (1,2,3,4-tetrahydronaphthalen-1-yl) aminoanilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole (0.50 g, 1.9 mmol) in acetonitrile (10 ml) was added 1-amino-1,2,3, 4-tetrahydronaphthalene (0.31 g, 2.1 mmol) and triethylamine (0.29 g, 3.2 mmol) were added, and the mixture was heated to reflux for 6 hours.
- Example 87-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (1,2,3,4-tetrahydronaphthalen-1-yl) benzimidazole 2- (2- (1,2,3,4-tetrahydronaphthalen-1-yl) aminoanilin-5-yl) benzoxazole (see Example 87-1) (0.30 g, 0.8 mmol) in methanol (5 ml ) To the solution was added methyl acetimidate hydrochloride (0.10 g, 0.9 mmol), and the mixture was heated to reflux for 3 hours. The reaction solution was cooled, water was added, and the mixture was extracted with chloroform.
- Example 88-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2-hydroxyethyl) -2-methylbenzimidazole
- 2- (2- (2-hydroxyethyl) aminoanilin-5-yl) benzoxazole (0.10 g, 0.4 mmol) in methanol (2 ml) was added methyl acetimidate hydrochloride ( 0.08 g, 0.7 mmol) was added and the mixture was heated to reflux for 3 hours.
- the reaction solution was cooled, water was added, and the precipitated crystals were filtered, washed with water, and dried to give the title compound (0.05 g, yield 49%) as white crystals.
- Example 89-2 Synthesis of 5- (benzoxazol-2-yl) -1-diphenylmethyl-2-methylbenzimidazole
- 2- (2-diphenylmethylaminoanilin-5-yl) benzoxazole (0.10 g, 0.3 mmol) in methanol (2 ml) was added methyl acetimidate hydrochloride (0.06 g, 0.5 mmol).
- methyl acetimidate hydrochloride (0.06 g, 0.5 mmol).
- the reaction solution was cooled, water was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- Example 91 Synthesis of 5- (benzoxazol-2-yl) -2-tert-butoxycarbonylmethyl-1- (tetrahydropyran-4-yl) benzimidazole
- Example 91-1 Synthesis of 2- (2-tert-butoxycarbonylmethylamino) aniline-5-yl) benzoxazole
- acetonitrile (10 ml) was added
- glycine tert-butyl ester hydrochloride (0.60 g 3.6 mmol
- triethylamine (0.90 g, 9.0 mmol
- Example 91-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-tert-butoxycarbonylmethylbenzimidazole
- 2- (2- (tert-butoxycarbonylmethylaminoanilin-5-yl) benzoxazole (see Example 91-1) (120 mg, 0.353 mmol) in dimethylformamide (2 ml) was added acetaldehyde (ca. 90% , 66 ⁇ l, 1.06 mmol) and oxone (217 mg, 0.353 mmol) were added, and the mixture was stirred at room temperature for 2 hours After completion of the reaction, an aqueous potassium carbonate solution was added, and the mixture was filtered and washed with water.
- Example 92 Synthesis of 5- (benzoxazol-2-yl) -1-carboxymethyl-2-methylbenzimidazole To a solution of 5- (benzoxazol-2-yl) -2-methyl-1-tert-butoxycarbonylmethylbenzimidazole (see Example 91-2) (100 mg, 0.275 mmol) in chloroform (3 ml) was added an aqueous sodium hydroxide solution. (1M, 0.55 ml, 0.55 mmol) was added. Methanol was added to the two-layer solution until uniform and stirred at room temperature for 2 hours. After completion of the reaction, the solution was concentrated, an aqueous acetic acid solution was added, and the mixture was stirred at room temperature.
- Example 93-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2- (thiomorpholin-1,1-dioxido-4-yl) ethyl) benzimidazole 2- (N- (2- (thiomorpholin-1,1-dioxido-4-yl) ethyl) -2-nitroanilin-4-yl) benzoxazole (see Example 93-1) (290 mg, 0.696 mmol ) In tetrahydrofuran (5 ml) was added 10% palladium on carbon (50 mg), the flask was purged with hydrogen, and the mixture was stirred at room temperature for 17 hours.
- Example 94-2 Synthesis of 2- (2- (tetrahydrofuran-2-yl) amino-3-trifluoromethylanilin-5-yl) benzoxazole
- 2- (2-fluoro-3-trifluoromethylnitrobenzene-5-yl) benzoxazole see Example 94-1 (0.40 g, 1.2 mmol) in acetonitrile (8 ml) was added 4-aminotetrahydro Pyran (0.30 g, 2.9 mmol) was added and heated to reflux for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- Example 94-3 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) -4-trifluoromethylbenzimidazole
- 2- (2- (tetrahydrofuran-2-yl) amino-3-trifluoromethylanilin-5-yl) benzoxazole (0.20 g, 0.5 mmol) in methanol (4 ml)
- Methyl acetimidate hydrochloride (0.06 g, 0.6 mmol
- Example 95 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydrofuran-2-yl) methylbenzimidazole
- Example 95-1 Synthesis of 2- (2- (tetrahydrofuran-2-yl) methylaminoaniline-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole (0.40 mg, 1.5 mmol) in acetonitrile (8 ml) was added 2-aminomethyltetrahydrofuran (0.36 g, 3.6 mmol) was added and heated to reflux for 7 hours.
- Example 95-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydrofuran-2-yl) methylbenzimidazole
- 2- (tetrahydropyran-2-yl) methylaminoanilin-5-yl) benzoxazole (0.15 g, 0.4 mmol) in methanol (5 ml) was added methyl acetimidate hydrochloride ( 0.05 mg, 0.4 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 96 Synthesis of 5- (benzoxazol-2-yl) -2methyl-1- (3- (morpholin-4-yl) propyl) benzimidazole
- Example 96-1 Synthesis of 2- (N- (2- (morpholin-4-yl) -n-propyl) -2-nitroanilin-4-yl) benzoxazole
- potassium carbonate 500 mg, 1.94 mmol
- N- (3-Amino-n-propyl) morpholine (336 mg, 2.33 mmol) was added and heated to reflux for 4 hours.
- Example 96-2 Synthesis of 2- (2- (3- (morpholin-4-yl) -n-propyl) aminoanilin-4-yl) benzoxazole 2- (N- (2- (morpholin-4-yl) -n-propyl) -2-nitroanilin-4-yl) benzoxazole (see Example 96-1) (665 mg, 1.74 mmol) in tetrahydrofuran ( 10 ml), 10% palladium on carbon (70 mg) was added, the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 17 hours. After completion of the reaction, the mixture was filtered through celite, and the filtrate was concentrated to obtain the title compound (605 mg, 99%).
- Example 96-3 Synthesis of 5- (benzoxazol-2-yl) -2methyl-1- (3- (morpholin-4-yl) -n-propyl) benzimidazole 2- (2- (3- (morpholin-4-yl) -n-propyl) aminoanilin-4-yl) benzoxazole (see Example 96-2) (300 mg, 0.851 mmol) in methanol (5 ml) Was added with methyl acetimidate hydrochloride (103 mg, 0.936 mmol) and heated to reflux for 3 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with ethyl acetate.
- Example 100 Synthesis of 2- (4-acetylaminophenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 100-1 Synthesis of 5-chloro-2- (2-fluoronitrobenzene-5-yl) benzoxazole
- 4-fluoro-3-nitrobenzoic acid (25.0 g, 135 mmol)
- 2-amino-4-chlorophenol (21.3 g, 149 mmol)
- CHCl3 500 mL
- WSC 28.5 g, 149 mmol
- Example 100-2 Synthesis of 5-chloro-2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole
- 5-chloro-2- (2-fluoronitrobenzene-5-yl) benzoxazole see Example 100-1 (5.00 g, 17.1 mmol) in acetonitrile (20 ml) was added 4-aminotetrahydropyran ( 3.81 g, 38.0 mmol) was added, and the mixture was heated to reflux for 3 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried.
- Example 100-3 Synthesis of 2- (4-acetylaminophenyl) -5- (5-chlorobenzoxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- 5-chloro-2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole see Example 100-2
- 4-Formylacetanilide 142 mg, 0.872 mmol
- oxone 313 mg, 0.509 mmol
- Example 102 Synthesis of 2- (4-carboxylphenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (5-Chlorobenzoxazol-2-yl) -2- (4-methoxycarbonylphenyl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 101) (220 mg, 0.451 mmol) in chloroform To the (3 ml) solution was added aqueous sodium hydroxide (1M, 1 ml, 1 mmol). Methanol was added to the two-layer solution until uniform and stirred at room temperature for 3 hours.
- aqueous sodium hydroxide (1M, 1 ml, 1 mmol
- Example 105 Synthesis of 2- (3-carboxylphenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole 5- (5-Chlorobenzoxazol-2-yl) -2- (3-methoxycarbonylphenyl) -1- (tetrahydropyran-4-yl) benzimidazole (see Example 103) (280 mg, 0.574 mmol) in chloroform To the (3 ml) solution was added 1M aqueous sodium hydroxide (1 ml, 1 mmol). Methanol was added to the two-layer solution until uniform and stirred at room temperature for 3 hours.
- Example 106 Synthesis of 2- (3-acetylaminophenyl) -5- (5-chlorobenzooxazol-2-yl) -1- (tetrahydropyran-4-yl) benzimidazole
- 5-chloro-2- (2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole see Example 100-2
- 3-acetamide benzoic acid 130 mg, 0.727 mmol
- WSC 167 mg, 0.872 mmol
- Example 107 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1-((4-morpholinyl) carbonylmethyl) benzimidazole
- Example 107-1 Synthesis of 2- (2-((4-morpholinyl) carbonylmethyl) aminoanilin-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole (1.20 g, 4.6 mmol) in acetonitrile (24 ml) was added triethylamine (1.17 g, 11.6 mmol), amino Acetylmorpholine (0.73 g, 5.1 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 107-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (4-morpholinyl) benzimidazole
- 2- (4-morpholinyl) aminoanilin-5-yl) benzoxazole see Example 107-1 (0.08 g, 0.2 mmol) in methanol (2 ml) was added methyl acetimidate hydrochloride (0.03 g, 3 mmol).
- methyl acetimidate hydrochloride (0.03 g, 3 mmol).
- the reaction solution was cooled, water was added, and the precipitated crystals were filtered, washed with water, and dried to give the title compound (0.10 g, quantitative) as white crystals.
- Example 109 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-trifluoroacetylaminoethyl) benzimidazole 1- (2-aminoethyl) -5- (benzoxazol-2-yl) -2-methylbenzimidazole hydrochloride (see Example 56) (0.20 g, 0.6 mmol), trifluoroacetic anhydride (0.17 g 0.8 mmol), triethylamine (0.22 g, 2.2 mmol), and CHCl3 (2 mL) were added, and the mixture was stirred at room temperature for 1 hour. After cooling, chloroform and water were added for extraction.
- Example 110 Synthesis of 3- (2- (5- (benzoxazol-2-yl) -2-methylbenzimidazol-1-yl) ethyl) -1-methylthiourea 1- (2-aminoethyl) -5- (benzoxazol-2-yl) -2-methylbenzimidazole hydrochloride (see Example 56) (0.20 g, 0.6 mmol), methyl isothiocyanate (0.17 g, 0.6 mmol), triethylamine (0.13 g, 13 mmol), and THF (2 mL) were added, and the mixture was stirred at room temperature for 1 hour. After cooling, chloroform and water were added for extraction.
- the obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated.
- p-toluenesulfonic acid monohydrate (0.18 g, 1.8 mmol) was added, and the mixture was heated to reflux for 40 hours.
- the reaction solution was concentrated, water was added, and the precipitated crystals were filtered, washed with water, and dried to give the title compound (1.40 g, yield 26%) as yellowish crystals.
- Example 112-2 Synthesis of 2- (4-chloro-2- (tetrahydropyran-4-yl) aminonitrobenzene-5-yl) benzoxazole
- 2- (4-chloro-2-fluoronitrobenzene-5-yl) benzoxazole see Example 112-1
- 4-aminotetrahydropyran 0.30 g, 2.9 mmol
- Example 112-3 Synthesis of 2- (4-chloro-2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole
- 2- (4-Chloro-2- (tetrahydropyran-4-yl) aminonitrobenzene-5-yl) benzoxazole (0.10 g, 0.8 mmol) to iron powder (0.33 g, 5.8 mmol) and acetic acid (50 mL) were added, and the mixture was heated to reflux for 2 hours.
- Example 112-4 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) -6-chlorobenzimidazole 2- (4-Chloro-2- (tetrahydropyran-4-yl) aminoanilin-5-yl) benzoxazole (see Example 112-3) (0.04 g, 0.2 mmol) in methanol (1.2 ml) , Methyl acetimidate hydrochloride (0.06 g, 0.6 mmol) was added and heated to reflux for 3 hours.
- Example 113 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxycarbonylphenylmethyl) -2-methylbenzimidazole
- Example 113-1 Synthesis of 2- (2- (4-methoxycarbonylphenylmethyl) aminonitrobenzene-5-yl) benzoxazole
- 2- (4-fluoro-3-nitrophenyl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added triethylamine (245 mg, 2.42 mmol), 4-carbohydrate.
- Methoxybenzylamine hydrochloride (215 mg, 1.07 mmol) was added and heated to reflux for 2 hours.
- Example 113-2 Synthesis of 2- (2- (4-methoxycarbonylphenylmethyl) aminoaniline-5-yl) benzoxazole 2- (2- (4-Methoxycarbonylphenylmethyl) aminonitrobenzene-5-yl) benzoxazole (see Example 113-2) (367 mg, 0.910 mmol) to iron powder (102 mg, 1.82 mmol), 10% acetic acid An aqueous solution (5 mL) and methanol (7 mL) were added, and the mixture was heated to reflux for 16 hours. The reaction solution was cooled, saturated aqueous sodium hydrogen carbonate solution and chloroform were added, the mixture was filtered through celite, and the resulting filtrate was extracted.
- Example 113-3 Synthesis of 5- (benzoxazol-2-yl) -1- (4-methoxycarbonylphenylmethyl) -2-methylbenzimidazole 2- (2- (4-Methoxycarbonylphenylmethyl) aminoaniline-5-yl) benzoxazol (see Example 113-3)
- Amine 59.0 mg, 0.159 mmol
- methanol 3 ml
- Hydrochloride (19 mg, 0.174 mmol) was added and heated to reflux for 1.5 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for several minutes.
- Example 114 Synthesis of 5- (benzoxazol-2-yl) -1- (4-carboxylphenylmethyl) -2-methylbenzimidazole
- 5- (benzoxazol-2-yl) -1- (4-methoxycarbonylphenylmethyl) -2-methylbenzimidazole (see Example 113-4) (55 mg, 0.138 mmol) in chloroform (3 ml)
- Aqueous sodium hydroxide (1M, 0.6 ml, 0.6 mmol) was added.
- Methanol was added to the two-layer solution until uniform and stirred at room temperature for 20 hours. After completion of the reaction, the solution was concentrated, an aqueous acetic acid solution was added, and the mixture was stirred at room temperature.
- Example 115 Synthesis Example of 5- (1-Benzoxazol-2-yl) -1- (1-butanol-2-yl) -2-methylbenzimidazole 115-1 Synthesis of 2- (2- (1-n-butanol-2-yl) aminonitrobenzene-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added potassium carbonate (268 mg, 1.94 mmol), 2- Amino-1-n-butanol (95.4 mg, 1.07 mmol) was added and heated to reflux for 4 hours.
- Example 115-2 Synthesis of 5- (1-benzoxazol-2-yl) -1- (1-n-butanol-2-yl) -2-methylbenzimidazole Synthesis of 2- (2- (1-n-butanol-2-yl) aminonitrobenzene-5-yl) benzoxazole (see Example 115-1) (295 mg, 0.901 mmol) in ethanol, ethyl acetate (1: The solution was added 10% palladium on carbon (50 mg) to the solution, and the flask was replaced with hydrogen, followed by stirring at room temperature for 19 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Ethyl acetimidate hydrochloride (133 mg, 1.08 mmol) was added to a solution of the obtained residue in ethanol (5 ml), and the mixture was heated to reflux for 2 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for several minutes. The obtained crystals were filtered, washed with water, and dried to give the title compound (232 mg, 72%) as white crystals.
- Example 116 Synthesis Example of 5- (1-Benzoxazol-2-yl) -1- (2-n-propanol-1-yl) -2-methylbenzimidazole 116-1 Synthesis of 2- (2- (2-n-propanol-1-yl) aminonitrobenzene-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added potassium carbonate (268 mg, 1.94 mmol), 1- Amino-2-n-propanol (80.4 mg, 1.07 mmol) was added and heated to reflux for 4 hours.
- Example 116-2 Synthesis of 5- (1-benzoxazol-2-yl) -1- (2-n-propanol-1-yl) -2-methylbenzimidazole 2- (2- (2-n-propanol-1-yl) aminonitrobenzene-5-yl) benzoxazole (Example 116-1) (280 mg, 0.894 mmol) in ethanol, ethyl acetate (1: 1, 6 ml) ) 10% palladium carbon (50 mg) was added to the solution, the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 19 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated.
- Ethyl acetimidate hydrochloride (133 mg, 1.08 mmol) was added to a solution of the obtained residue in ethanol (5 ml), and the mixture was heated to reflux for 2 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was stirred at room temperature for several minutes. The obtained crystals were filtered, washed with water, and dried to give the title compound (254 mg, 92%) as white crystals.
- Example 117-2 Synthesis of 5- (benzoxazol-2-yl) -1- (3-methoxycarbonylphenylmethyl) -2-methylbenzimidazole 2- (2- (3-methoxycarbonylphenylmethyl) aminonitrobenzene-5-yl) benzoxazole (Example 117-1) (220 mg, 0.545 mmol) to iron powder (91.4 mg, 1.64 mmol), acetic acid (5 mL ) And heated to reflux for 12 hours. After cooling the reaction solution, the reaction solution was concentrated. The obtained residue was purified by silica gel column chromatography to give the titled compound (139 mg, 64%) as white crystals.
- Example 118 Synthesis Example of 5- (1-Benzoxazol-2-yl) -1- (1-acetoxy-2-phenylethane-2-yl) -2-methylbenzimidazole 118-1 Synthesis of 2- (2- (2-phenyl-1-ethanol-2-yl) aminonitrobenzene-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (250 mg, 0.968 mmol) in acetonitrile (5 ml) was added potassium carbonate (268 mg, 1.94 mmol), 2- Phenylglycinol (146 mg, 1.07 mmol) was added and heated to reflux for 2 hours.
- Example 118-2 Synthesis of 5- (1-benzoxazol-2-yl) -1- (1-acetoxy-2-phenylethan-2-yl) -2-methylbenzimidazole 2- (2- (2-Phenyl-1-ethanol-2-yl) aminonitrobenzene-5-yl) benzoxazole (Example 118-1) (340 mg, 0.545 mmol) to iron powder (152 mg, 2.72 mmol) Acetic acid (5 mL) was added, and the mixture was heated to reflux for 12 hours. After cooling the reaction solution, the reaction solution was concentrated. The obtained residue was purified by silica gel column chromatography to give the titled compound (214 mg, 57%) as a colorless amorphous.
- Example 119 Synthesis of 5- (benzoxazol-2-yl) -1- (3-carboxylphenylmethyl) -2-methylbenzimidazole
- 5- (benzoxazol-2-yl) -1- (3-methoxycarbonylphenylmethyl) -2-methylbenzimidazole see Example 117-2
- Aqueous sodium hydroxide (1M, 1 ml, 1 mmol) was added.
- Methanol was added to the two-layer solution until uniform and stirred at room temperature for 20 hours. After completion of the reaction, the solution was concentrated, an aqueous acetic acid solution was added, and the mixture was stirred at room temperature.
- Example 120 Synthesis of 5- (1-benzoxazol-2-yl) -1- (2-phenylethanol-2-yl) -2-methylbenzimidazole 5- (1-Benzoxazol-2-yl) -1- (1-acetoxy-2-phenylethan-2-yl) -2-methylbenzimidazole (see Example 118-2) (200 mg, 0.486 mmol ) In methanol (5 ml) was added aqueous sodium hydroxide solution (1M, 1.5 ml, 1.5 mmol) and stirred at room temperature for 20 hours. After completion of the reaction, the solution was concentrated, water was added, and the mixture was stirred at room temperature.
- Example 121 Synthesis Example 5-1-1 of 5- (benzoxazol-2-yl) -2-methyl-1- (2-tert-butoxycarbonyl-n-propyl) benzimidazole Synthesis of 2- (2-tert-butoxycarbonyl-n-propylamino) aniline-5-yl) benzoxazole To a suspension of 2- (4-fluoro-3-nitrophenyl) benzoxazole (see Example 15-2) (0.50 g, 1.9 mmol) in acetonitrile (10 ml) was added 2-tert-butoxycarbonyl-1-propyl.
- Example 121-2 Synthesis of 5- (benzoxazol-2-yl) -2-methyl-1- (2-tert-butoxycarbonyl-n-propyl) benzimidazole
- 2- (2-tert-butoxycarbonyl-n-propylamino) anilin-5-yl) benzoxazole (0.62 g, 1.7 mmol) in THF (20 ml) was added methyl acetimidate.
- Hydrochloride (0.37 g, 3.4 mmol) was added and heated to reflux for 3 hours.
- Example 129 Synthesis Example of 5- (Benzoxazol-2-yl) -1- (2- (piperidin-1-yl) ethyl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 129-1 Synthesis of N- (2- (piperidin-1-yl) ethyl) -4- (benzoxazol-2-yl) -2-nitroaniline
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (200 mg, 0.775 mmol) in acetonitrile (5 ml) was added potassium carbonate (214 mg, 1.55 mmol), 1- (2-Aminoethyl) piperidine (119 mg, 0.93 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 130 Synthesis Example of 5- (Benzoxazol-2-yl) -1- (2-dimethylaminoethyl) -1- (tetrahydropyran-4-yl) benzimidazole
- Example 130-1 Synthesis of N- (2-dimethylaminoethyl) -4- (benzoxazol-2-yl) -2-nitroaniline
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2) (200 mg, 0.775 mmol) in acetonitrile (5 ml) was added potassium carbonate (214 mg, 1.55 mmol), N, N-dimethylethylenediamine (82 mg, 0.93 mmol) was added, and the mixture was heated to reflux for 3 hours.
- Example 131 Synthesis Example of 5- (Benzoxazol-2-yl) -1- (2- (methylthio) ethyl) -2-methylbenzimidazole
- Example 131-1 Synthesis of N- (2- (methylthio) ethyl) -4- (benzoxazol-2-yl) -2-nitroaniline
- 2- (4-fluoro-3-nitrophenyl) benzoxazole see Example 15-2
- potassium carbonate 321 mg, 1.55 mmol
- 2- (Methylthio) ethylamine 127 mg, 1.39 mmol
- Example 131-2 Synthesis of 5- (benzoxazol-2-yl) -1- (2- (methylthio) ethyl) -2-methylbenzimidazole Iron powder (188 mg, 3.37 mmol) and acetic acid (5 mL) were added to N- (2- (methylthio) ethyl) -4- (benzoxazol-2-yl) -2-nitroaniline (370 mg, 1.12 mmol), and 10 Heated to reflux for hours. After cooling the reaction solution, the reaction solution was concentrated. The obtained residue was purified by silica gel column chromatography to obtain 305 mg (84%) of the title compound as pink crystals.
- Example 135 Synthesis Example of 5- (5-Chlorobenzoxazol-2-yl) -1-n-propylbenzimidazole
- Example 135-1 Synthesis of 5- (5-chlorobenzooxazol-2-yl) -2-n-propylaminoaniline
- To a suspension of 5-chloro-2- (2-fluoro-nitrobenzene-5-yl) benzoxazole (see Example 100-1) (1.5 g, 5.13 mmol) in acetonitrile (10 ml) was added triethylamine (779 mg, 7.70). mmol) and propylamine (364 mg, 6.16 mmol) were added, and the mixture was heated to reflux for 4 hours.
- Example 135-2 To a solution of 5- (5-chlorobenzooxazol-2-yl) -2-n-propylaminoaniline (see Example 135-1) (300 mg, 0.994 mmol) in triethyl orthoformate (5 ml) was added p-toluenesulfonic acid. (10 mg) was added and stirred at 100 ° C. for 2 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform. The obtained organic layer was dried over anhydrous sodium sulfate, filtered and concentrated, and the resulting crude crystals were washed with diethyl ether to obtain 146 mg (47%) of the title compound as pale yellow crystals.
- Example 137 Synthesis of 5- (5-chlorobenzooxazol-2-yl) -2-methyl-1-n-propylbenzimidazole
- a solution of 5- (5-chlorobenzooxazol-2-yl) -2-n-propylaminoaniline (see Example 143-1) (200 mg, 0.663 mmol) in ethanol (5 ml) was added ethyl acetimidate hydrochloride (123 mg , 0.995 mmol) was added and heated to reflux for 5 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 138 Synthesis Example of 5- (5-Chlorobenzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole
- Example 138-1 Synthesis of 5-chloro-2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole
- To a suspension of 5-chloro-2- (2-fluoronitrobenzene-5-yl) benzoxazole (see Example 100-1) (815 mg, 2.79 mmol) in acetonitrile (10 ml) was added triethylamine (847 mg, 8.37 mmol).
- 3,3,3-Trifluoropropylamine hydrochloride 500 mg, 3.34 mmol was added and heated to reflux for 4 hours. After completion of the reaction, the mixture was cooled to room temperature, water was added, and the precipitated crystals were filtered, washed with water, and dried. To a solution of the obtained crystals in ethanol and tetrahydrofuran (1: 2, 15 ml), 10% palladium on carbon (100 mg) was added, the inside of the flask was replaced with hydrogen, and the mixture was stirred at room temperature for 18 hours. After completion of the reaction, the mixture was filtered through Celite, and the filtrate was concentrated to obtain 993 mg (100%) of the title compound as orange crystals.
- Example 138-2 Synthesis of 5- (5-chlorobenzoxazol-2-yl) -2-methyl-1- (3,3,3-trifluoropropyl) benzimidazole 5-Chloro-2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole (see Example 138-1) (200 mg, 0.562 mmol) in ethanol (5 ml) Was added ethyl acetimidate hydrochloride (104 mg, 0.843 mmol), and the mixture was heated to reflux for 5 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 139 Synthesis of 5- (5-chlorobenzoxazol-2-yl) -1- (3,3,3-trifluoropropyl) benzimidazole 5-chloro-2- (2- (3,3,3-trifluoropropyl) aminoanilin-5-yl) benzoxazole (see Example 138-1) (200 mg, 0.562 mmol) triethyl orthoformate (5 ml P-Toluenesulfonic acid (10 mg) was added to the solution and stirred at 100 ° C. for 4 hours. After completion of the reaction, a saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform.
- Example 140 Synthesis of 5- (benzimidazol-2-yl) -2-methyl-1- (tetrahydropyran-4-yl) benzimidazole To a suspension of 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid (see Example 4-3) (500 mg, 1.86 mmol), toluene (10 mL) was added thionyl chloride (3 mL). 2-methyl-1- (tetrahydropyran-4-yl) benzimidazole-5-carboxylic acid chloride hydrochloride (533 mg, 1.69) obtained by stirring under reflux conditions for 1.5 hours and concentrating under reduced pressure.
- Test example 1 (Preparation of compound evaluation cells)
- forward primer (5'-tccagtatttgagaaaaggagccaggagtctccat-3 ')
- reverse primer (5'-ggaggcttcctctttgcttcccggtcttttcg-3') as PCR primers
- the region from about 5 kbp upstream of the NXF gene promoter partial translation region to the vicinity of the transcription start site was isolated by PCR.
- the resulting NXF promoter partial fragment is introduced into the SmaI site of the pGL3 Basic vector (Promega) so that the luciferase gene is located downstream of the promoter, and a reporter plasmid in which the luciferase enzyme is expressed by the action of the NXF promoter was made.
- PC12 cells purchased from ATCC were prepared in RPMI medium (GIBCO-BRL), 5% FCS (GIBCO-BRL), 15% horse serum (GIBCO-BRL), and 1 mM sodium pyruvate (GIBCO- BRL was added and cultured at 37 ° C., 5% in the presence of CO 2 .
- the PC12 of the reporter plasmid 12 ⁇ g and 1 ⁇ g against 106 cells pRC / RSV vector (Invitrogen Corp.) were transfected sheet transfected using Lipofectamine2000 reagent (Invitrogen Corporation), 40 mg / L after 2 days transfection
- the medium was replaced with a medium containing Geneticin sulfate (GIBCO) and the culture was continued.
- a culture solution containing only a final concentration of 0.1% DMSO was used as a negative control group
- a culture group containing a saturated concentration of NGF was used as a positive control group
- 200 ng / ml NGF corresponding to a compound concentration of 0 was added.
- an experimental group containing 0.1% DMSO was placed, and the compound solution (dissolved in DMSO to a final DMSO concentration of 0.1%) was added to 200 ng / ml NGF, based on the luciferase activity.
- the relative luciferase activity of the experimental group to which (prepared as described above) was added was calculated, and this was defined as the activity of enhancing the activity of 200 ng / ml NGF by the compound.
- the test results of NGF activity enhancing activity are listed in the table below. +: Relatively weak potentiating activity (enhancing activity only at 10 ⁇ M) ++: shows enhancing activity ++: shows particularly strong enhancing activity (increases the activity of NGF 200 ng / ml more than 2 times at 1 ⁇ M)
- Test example 2 Male 8-week-old C57BL / 6J mice are normally bred for 12 days, then the left common neck and external common artery are ligated in advance, and the middle cerebral artery is occluded for 90 minutes and then reopened. A model was created. In the same model, the compound was administered into the tail vein at 30 mg / kg 3 hours after occlusion and 48 hours after 48 hours. After 3 days of occlusion / reopening, the head was decapitated, the skull was cut from the cerebellum side, and the brain was removed. The removed brain was placed in a Buan solution (pH 3.5 to 4.0) and fixed. From the fixed brain, coronal sections were cut out, and after cutting, dehydration and penetration, and embedded in paraffin.
- Buan solution pH 3.5 to 4.0
- Infarct volume was calculated as follows. A digital image was captured using a camera (MCD-350, Olympus Corporation) attached to an optical microscope (objective lens ⁇ 1). The captured digital image was pasted on a single mount (new layer) in Adobe Photoshop 2.0.
- Area MR, mm 2 ) was measured, and non-infarct area (ML, mm 2 ) [infarct side (left) area (mm 2 ) ⁇ infarct area (mm 2 )] on the infarct side (left) was calculated.
- the infarct volume (SV, mm 3 ), the infarct side (left) non-infarct volume (MLV, mm 3 ), and the non-infarct side (right) volume (MRV, mm 3 ) were calculated by the following equations.
- test result (infarct volume) of the compound of Example 12 is shown in FIG. Thus, a decrease in infarct volume was confirmed in the compound administration group.
- Test example 3 The prophylactic effect of the compounds on the latency delay of motor nerve transmission velocity (MNCV) in the sciatic nerve in streptozotocin-induced diabetic rats was examined.
- the group composition was a non-diabetic control group, a diabetic control group, and a compound administration group.
- the non-diabetic control group and the diabetic control group had 5 ml / kg of water for injection, and the compound administration group had a compound solution prepared at 10 mg / ml with water for injection at 5 ml / kg.
- Oral administration once a day for 4 weeks.
- MNCV measurement was performed 3 times for each group. The measurement time is the first day before administration of the streptozotocin solution or pH 4.5 0.75 mmol / L citrate buffer solution, and the second time is 4 weeks after administration of the streptozotocin solution or pH 4.5 0.75 mmol / L citrate buffer solution. Thereafter, the third measurement was performed on the day after the last day of administration of the water or compound solution for 4 weeks.
- the test results (motor nerve transmission rate) of the compound of Example 12 are shown in FIG. 2 (in the figure, STZ represents streptozotocin). Thus, recovery of motor nerve transmission rate was recognized in the compound administration group compared to the solvent control administration group.
- Test example 4 In the same manner as in Test Example 1, the enhancing activity of Example NGF activity of the compounds shown in Table 2 was evaluated. The results are shown in Table 2. +: Relatively weak potentiating activity (enhancing activity only at 10 ⁇ M) ++: shows enhancing activity ++: shows particularly strong enhancing activity (increases the activity of NGF 200 ng / ml more than 2 times at 1 ⁇ M)
- the compound of the present invention is effective for treatment or prevention of diseases involving the activity of neurotrophic factor.
- SEQ ID NO: 1 is a forward primer.
- SEQ ID NO: 2 is a reverse primer.
Abstract
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI1011961A BRPI1011961A2 (pt) | 2009-05-29 | 2010-05-28 | agente para o tratamento ou prevenção de doenças associadas com a atividade de fatores neurotróficos. |
| AU2010253336A AU2010253336A1 (en) | 2009-05-29 | 2010-05-28 | Agent for treatment or prevention of diseases associated with activity of neurotrophic factors |
| US13/322,888 US8829035B2 (en) | 2009-05-29 | 2010-05-28 | Agent for treatment or prevention of diseases associated with activity of neurotrophic factors |
| EP10780309.0A EP2436683B1 (fr) | 2009-05-29 | 2010-05-28 | Agent de traitement ou de prévention de maladies associées à l'activité d'agents neurotrophiques |
| ES10780309.0T ES2484169T3 (es) | 2009-05-29 | 2010-05-28 | Agente para el tratamiento o la prevención de enfermedades asociadas a la actividad de factores neurotróficos |
| CN2010800218228A CN102428080A (zh) | 2009-05-29 | 2010-05-28 | 神经营养因子活性参与的疾病的治疗或预防剂 |
| MX2011012537A MX2011012537A (es) | 2009-05-29 | 2010-05-28 | Agente para el tratamiento o prevencion de enfermedades asociadas con actividad de factores neurotroficos. |
| CA2762620A CA2762620A1 (fr) | 2009-05-29 | 2010-05-28 | Agent de traitement ou de prevention de maladies associees a l'activite d'agents neurotrophiques |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-131333 | 2009-05-29 | ||
| JP2009131333 | 2009-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010137349A1 true WO2010137349A1 (fr) | 2010-12-02 |
Family
ID=43222475
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/003622 WO2010137349A1 (fr) | 2009-05-29 | 2010-05-28 | Agent de traitement ou de prévention de maladies associées à l'activité d'agents neurotrophiques |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US8829035B2 (fr) |
| EP (1) | EP2436683B1 (fr) |
| JP (1) | JP2011006409A (fr) |
| KR (1) | KR20120093066A (fr) |
| CN (1) | CN102428080A (fr) |
| AU (1) | AU2010253336A1 (fr) |
| BR (1) | BRPI1011961A2 (fr) |
| CA (1) | CA2762620A1 (fr) |
| ES (1) | ES2484169T3 (fr) |
| MX (1) | MX2011012537A (fr) |
| WO (1) | WO2010137349A1 (fr) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017055147A1 (fr) | 2015-09-28 | 2017-04-06 | Syngenta Participations Ag | Dérivés hétérocycliques à activité pesticide comportant des substituants contenant du soufre |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2951153B1 (fr) | 2013-02-01 | 2019-04-03 | Regenacy Pharmaceuticals, LLC | Inhibiteurs hdac3 sélectifs |
| KR102460388B1 (ko) * | 2013-09-05 | 2022-10-27 | 미쓰비시 타나베 파마 코퍼레이션 | 신규한 결정성 아릴알킬아민 화합물 및 그의 제조 방법 |
| JP5829741B2 (ja) * | 2013-09-05 | 2015-12-09 | 田辺三菱製薬株式会社 | 新規な結晶性アリールアルキルアミン化合物およびその製造方法 |
| EP3177604A1 (fr) * | 2014-08-05 | 2017-06-14 | Bristol-Myers Squibb Company | Inhibiteurs de kinase hétérocycliques |
| CN108299413A (zh) * | 2018-01-22 | 2018-07-20 | 复旦大学 | 一种无金属催化制备2-(胺基苯基)苯并噁唑及其衍生物的方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0468470A1 (fr) | 1990-07-23 | 1992-01-29 | Dr. Karl Thomae GmbH | Benzimidazoles, médicaments les contenant et procédé pour leur préparation |
| JPH04308580A (ja) | 1991-04-05 | 1992-10-30 | Sumitomo Chem Co Ltd | ベンズイミダゾール誘導体、その製造法およびそれを有効成分とする農園芸用殺菌剤 |
| JPH0881464A (ja) * | 1994-09-12 | 1996-03-26 | Lilly Ind Ltd | インドール誘導体及び該誘導体を含む医薬用組成物 |
| DE19920936A1 (de) | 1999-05-07 | 2000-11-09 | Basf Ag | Heterozyklisch substituierte Benzimidazole, deren Herstellung und Anwendung |
| WO2001000213A1 (fr) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Composes inhibiteurs de la src kinase |
| WO2003053344A2 (fr) | 2001-12-10 | 2003-07-03 | Bristol-Myers Squibb Company | 2-methyl-benzimidazole substitues en tant qu'agents antiviraux du virus respiratoire syncytial |
| WO2005082901A1 (fr) | 2004-02-25 | 2005-09-09 | Smithkline Beecham Corporation | Nouveaux composes chimiques |
| WO2007115408A1 (fr) * | 2006-04-10 | 2007-10-18 | Painceptor Pharma Corporation | Compositions et procédés destinés à moduler des canaux ioniques commandés |
| JP2007282502A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | 細胞変性制御能力の検定方法 |
| JP2007282501A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | アポトーシス制御能力の検定方法 |
| WO2008073451A2 (fr) * | 2006-12-11 | 2008-06-19 | Sirtris Pharmaceuticals, Inc. | Composés modulateurs de la sirtuine |
| WO2008153701A1 (fr) | 2007-05-24 | 2008-12-18 | Schering Corporation | Composés d'inhibition de l'activité de ksp kinésine |
| US20090156613A1 (en) * | 2007-12-18 | 2009-06-18 | Kindrachuk David E | Bicyclic heteroaryl-substituted imidazoles as modulators of the histamine H4 receptor |
| JP2010047517A (ja) * | 2008-08-21 | 2010-03-04 | Sumitomo Chemical Co Ltd | 神経栄養因子の活性が関与する疾患の治療または予防剤、およびその製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1063028B (de) | 1957-09-10 | 1959-08-06 | Wolfen Filmfab Veb | Verfahren zur Sensibilisierung von Halogensilberemulsionen |
| DE4023369A1 (de) | 1990-07-23 | 1992-01-30 | Thomae Gmbh Dr K | Benzimidazole, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung |
| GB9418326D0 (en) | 1994-09-12 | 1994-11-02 | Lilly Industries Ltd | Pharmaceutical compounds |
| AU4979700A (en) | 1999-05-03 | 2000-11-17 | Battelle Memorial Institute | Compositions for aerosolization and inhalation |
| CN1623992A (zh) * | 2003-12-03 | 2005-06-08 | 北京师范大学 | 新型非肽类血管紧张素ⅱ受体拮抗剂类药物、它们的制备及应用 |
-
2010
- 2010-05-28 ES ES10780309.0T patent/ES2484169T3/es active Active
- 2010-05-28 KR KR1020117031347A patent/KR20120093066A/ko not_active Application Discontinuation
- 2010-05-28 JP JP2010123614A patent/JP2011006409A/ja not_active Ceased
- 2010-05-28 CA CA2762620A patent/CA2762620A1/fr not_active Abandoned
- 2010-05-28 AU AU2010253336A patent/AU2010253336A1/en not_active Abandoned
- 2010-05-28 MX MX2011012537A patent/MX2011012537A/es not_active Application Discontinuation
- 2010-05-28 EP EP10780309.0A patent/EP2436683B1/fr active Active
- 2010-05-28 CN CN2010800218228A patent/CN102428080A/zh active Pending
- 2010-05-28 WO PCT/JP2010/003622 patent/WO2010137349A1/fr active Application Filing
- 2010-05-28 BR BRPI1011961A patent/BRPI1011961A2/pt not_active IP Right Cessation
- 2010-05-28 US US13/322,888 patent/US8829035B2/en not_active Expired - Fee Related
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0468470A1 (fr) | 1990-07-23 | 1992-01-29 | Dr. Karl Thomae GmbH | Benzimidazoles, médicaments les contenant et procédé pour leur préparation |
| JPH04308580A (ja) | 1991-04-05 | 1992-10-30 | Sumitomo Chem Co Ltd | ベンズイミダゾール誘導体、その製造法およびそれを有効成分とする農園芸用殺菌剤 |
| JPH0881464A (ja) * | 1994-09-12 | 1996-03-26 | Lilly Ind Ltd | インドール誘導体及び該誘導体を含む医薬用組成物 |
| DE19920936A1 (de) | 1999-05-07 | 2000-11-09 | Basf Ag | Heterozyklisch substituierte Benzimidazole, deren Herstellung und Anwendung |
| JP2002544199A (ja) * | 1999-05-07 | 2002-12-24 | ビーエーエスエフ アクチェンゲゼルシャフト | ヘテロ環置換されたベンズイミダゾール、その製造方法およびその使用 |
| WO2001000213A1 (fr) | 1999-06-30 | 2001-01-04 | Merck & Co., Inc. | Composes inhibiteurs de la src kinase |
| WO2003053344A2 (fr) | 2001-12-10 | 2003-07-03 | Bristol-Myers Squibb Company | 2-methyl-benzimidazole substitues en tant qu'agents antiviraux du virus respiratoire syncytial |
| WO2005082901A1 (fr) | 2004-02-25 | 2005-09-09 | Smithkline Beecham Corporation | Nouveaux composes chimiques |
| JP2007282502A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | 細胞変性制御能力の検定方法 |
| JP2007282501A (ja) | 2006-03-24 | 2007-11-01 | Sumitomo Chemical Co Ltd | アポトーシス制御能力の検定方法 |
| WO2007115408A1 (fr) * | 2006-04-10 | 2007-10-18 | Painceptor Pharma Corporation | Compositions et procédés destinés à moduler des canaux ioniques commandés |
| WO2008073451A2 (fr) * | 2006-12-11 | 2008-06-19 | Sirtris Pharmaceuticals, Inc. | Composés modulateurs de la sirtuine |
| WO2008153701A1 (fr) | 2007-05-24 | 2008-12-18 | Schering Corporation | Composés d'inhibition de l'activité de ksp kinésine |
| US20090156613A1 (en) * | 2007-12-18 | 2009-06-18 | Kindrachuk David E | Bicyclic heteroaryl-substituted imidazoles as modulators of the histamine H4 receptor |
| JP2010047517A (ja) * | 2008-08-21 | 2010-03-04 | Sumitomo Chemical Co Ltd | 神経栄養因子の活性が関与する疾患の治療または予防剤、およびその製造方法 |
Non-Patent Citations (16)
| Title |
|---|
| "From the hyperacute phase to prevention", vol. 1, 2006, article "Studies on Intervention Periods for Stroke", pages: 649 - 654 |
| ANNUAL REVIEW OF NEUROSCIENCE, vol. 24, 2001, pages 1217 - 1281 |
| BIOORGANIC & MEDICINAL CHEMISTRY, vol. 200412, no. 1, pages 17 - 21 |
| BRAIN RESEARCH REVIEWS, vol. 30, 1999, pages 176 - 188 |
| CURRENT MEDICINE JAPAN, vol. 54, no. 7, 1999, pages 88 - 94 |
| ENGL. TRANSL. VER., pages 1565 - 1569 |
| JOURNAL OF MEDICINAL CHEMISTRY, vol. 31, no. 9, 1988, pages 1778 - 85 |
| KIYOSHI TOMIOKA: "Strategic Applications of Named Reactions in Organic Synthesis", 15 August 2006, KAGAKU DOJIN, pages: 258 - 259,310- |
| NATURE, vol. 322, 1986, pages 552 - 555 |
| PROGRESS IN BRAIN RESEARCH, vol. 146, 2004, pages 387 - 401 |
| See also references of EP2436683A4 * |
| SYNTHESIS, 2003, pages 1683 - 1692 |
| SYNTHESIS, 2008, pages 387 - 394 |
| SYNTHETIC COMMUNICATIONS, vol. 28, no. 22, 1998, pages 4123 - 4135 |
| THE JOURNAL OF THE AMERICAN SOCIETY FOR EXPERIMENTAL NEUROTHERAPEUTICS, vol. 2, 2005, pages 120 - 128 |
| ZHURNAL OBSHCHEI KHIMII, vol. 32, no. 5, 1962, pages 1581 - 86 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017055147A1 (fr) | 2015-09-28 | 2017-04-06 | Syngenta Participations Ag | Dérivés hétérocycliques à activité pesticide comportant des substituants contenant du soufre |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102428080A (zh) | 2012-04-25 |
| ES2484169T3 (es) | 2014-08-11 |
| JP2011006409A (ja) | 2011-01-13 |
| AU2010253336A1 (en) | 2011-12-08 |
| EP2436683B1 (fr) | 2014-05-14 |
| US8829035B2 (en) | 2014-09-09 |
| US20120115856A1 (en) | 2012-05-10 |
| EP2436683A4 (fr) | 2012-05-16 |
| KR20120093066A (ko) | 2012-08-22 |
| MX2011012537A (es) | 2011-12-14 |
| BRPI1011961A2 (pt) | 2016-04-26 |
| EP2436683A1 (fr) | 2012-04-04 |
| CA2762620A1 (fr) | 2010-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11814376B2 (en) | Hepatitis b core protein modulators | |
| DK2380881T3 (en) | NEW BICYCLIC HETEROCYCLIC COMPOUND | |
| AU2015229174B2 (en) | Hepatitis B core protein allosteric modulators | |
| JP5221453B2 (ja) | イミダゾール誘導体 | |
| TWI434842B (zh) | Azole compounds | |
| AU2010294214B2 (en) | Heterocylcic derivatives as inhibitors of glutaminyl cyclase | |
| WO2010137349A1 (fr) | Agent de traitement ou de prévention de maladies associées à l'activité d'agents neurotrophiques | |
| WO2010137350A1 (fr) | Agent de traitement ou de prévention de maladies associées à l'activité de facteurs neurotrophiques | |
| EP2250165A1 (fr) | Dérivés de triazoles et d'oxadiazoles | |
| JP5667934B2 (ja) | 新規2環性複素環化合物からなる医薬 | |
| TW202031644A (zh) | 苯并咪唑衍生物 | |
| JP2019508390A (ja) | mGluR7調節薬としてのインダン誘導体 | |
| EP3541812B1 (fr) | Composés agonistes de mglur7 pour le traitement de maladies, troubles ou états régulés par mglur7 | |
| OA19643A (en) | Hepatitis B core protein modulators. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080021822.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10780309 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4708/KOLNP/2011 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010253336 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2762620 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/012537 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13322888 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2010253336 Country of ref document: AU Date of ref document: 20100528 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010780309 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20117031347 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2011153381 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1011961 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1011961 Country of ref document: BR Kind code of ref document: A2 Effective date: 20111128 |