WO2010032731A1 - 結晶態様のベンゾイミダゾール化合物又はその塩 - Google Patents
結晶態様のベンゾイミダゾール化合物又はその塩 Download PDFInfo
- Publication number
- WO2010032731A1 WO2010032731A1 PCT/JP2009/066116 JP2009066116W WO2010032731A1 WO 2010032731 A1 WO2010032731 A1 WO 2010032731A1 JP 2009066116 W JP2009066116 W JP 2009066116W WO 2010032731 A1 WO2010032731 A1 WO 2010032731A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- salt
- compound
- acenaphthen
- piperidin
- dihydro
- Prior art date
Links
Images











Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention provides a crystalline embodiment of (R) -2- ⁇ 3- [1- (acenaphthen-1-yl) piperidin-4-yl] -2,3-dihydro-2-oxo-benzimidazol-1-yl ⁇ It relates to —N-methylacetamide (hereinafter referred to as the present compound) or a salt thereof.
- This compound is a compound represented by the following structural formula and exhibiting ORL-1 (opioid receptor-like 1) receptor agonist activity (Patent Document 1). This compound is produced in Example 18 of Patent Document 1.
- Compounds with ORL-1 receptor agonist activity are useful for the treatment of psychiatric, neurological and physiological disorders, especially anxiety and stress disorders, depression, traumatic disorders, memory loss due to Alzheimer's disease or other dementia, epilepsy and Symptoms of convulsions, acute and / or chronic pain symptoms, remission of drug withdrawal symptoms including withdrawal symptoms that occur during withdrawal of abused drugs, alcohol abuse, control of water balance, Na + excretion, arterial blood pressure disorder, obesity and anorexia Therefore, the present compound is also useful for the prevention and / or treatment of the above-mentioned diseases.
- An object of the present invention is to provide a stable form of the present compound which has no problems such as moisture adsorption and exhibits excellent water solubility.
- the present inventors have found that the present crystalline compound or a salt thereof is in a stable form with almost no change in weight due to moisture adsorption, and the salt of the present compound. was found to have excellent water solubility, and the present invention was completed. That is, the present invention is as follows. [1] (R) -2- ⁇ 3- [1- (Acenaphthen-1-yl) piperidin-4-yl] -2,3-dihydro-2-oxo-benzoimidazol-1-yl ⁇ -in crystalline form N-methylacetamide salt.
- a pharmaceutical composition comprising the salt or crystal according to any one of [1] to [14] as an active ingredient.
- the pharmaceutical composition according to [16] which is used for prevention and / or treatment of a disease associated with the ORL-1 receptor.
- the pharmaceutical composition according to [16] which is used for the prevention and / or treatment of a central nervous system disease involving the ORL-1 receptor.
- a method for preventing and / or treating a disease associated with the ORL-1 receptor comprising administering an effective amount of the salt or crystal according to any one of [1] to [14] to a subject.
- a method for preventing and / or treating a central nervous system disease involving ORL-1 receptor comprising administering an effective amount of the salt or crystal according to any one of [1] to [14] to a subject .
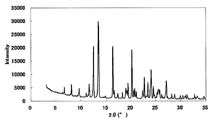
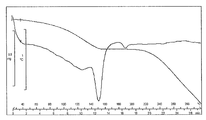
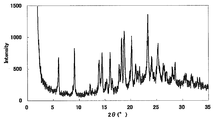
- FIG. 1 is a diagram showing an XRD pattern of a monohydrochloride monohydrate of the present compound of Example 1.
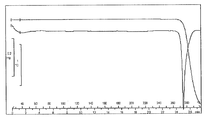
- FIG. 1 is a diagram showing a TG / DTA curve of monohydrochloride monohydrate of the present compound of Example 1.
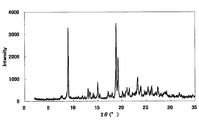
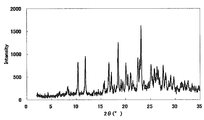
- FIG. 2 is a diagram showing an XRD pattern of a monohydrochloride salt of the present compound of Example 2.
- FIG. 4 is a diagram showing a TG / DTA curve of monohydrochloride of the present compound of Example 2.
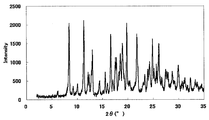
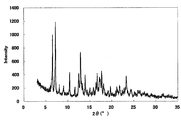
- FIG. 3 is a diagram showing an XRD pattern of a monohydrochloride ⁇ 2-3 trihydrate of the present compound of Example 3.
- FIG. 3 is a diagram showing a TG / DTA curve of monohydrochloride ⁇ 2-3 trihydrate of the present compound of Example 3.
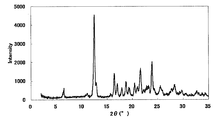
- FIG. 4 is a diagram showing an XRD pattern of a type I crystal of monohydrochloride / 1acetate solvate of the present compound of Example 4.
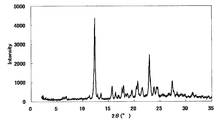
- FIG. 6 is a diagram showing an XRD pattern of a type II crystal of monohydrochloride / 1acetate solvate of the present compound of Example 5.
- FIG. 4 is a diagram showing an XRD pattern of 1 methanesulfonate of the present compound of Example 6.
- FIG. 2 is a diagram showing a TG / DTA curve of 1 methanesulfonate of the present compound of Example 6.
- FIG. FIG. 4 is a diagram showing an XRD pattern of 1 methanesulfonate ⁇ 1/2 to 1 hydrate of the present compound of Example 7.
- FIG. 3 is a graph showing a TG / DTA curve of 1 methanesulfonate ⁇ 1/2 to 1 hydrate of the present compound of Example 7.
- FIG. 4 is a diagram showing an XRD pattern of monomethanesulfonate trihydrate of the present compound of Example 8.
- 4 is a diagram showing an XRD pattern of a monomethanesulfonate dihydrate of the present compound of Example 9.
- FIG. 1 is a diagram showing an XRD pattern of a 1 ⁇ 2 fumarate salt / 3/2 hydrate of the present compound of Example 10.
- FIG. FIG. 3 is a diagram showing a TG / DTA curve of 1/2 fumarate salt / 3/2 hydrate of the present compound of Example 10.
- FIG. 3 is a diagram showing an XRD pattern of a monofumarate salt of the present compound of Example 11 and 1 to 2 hydrate.
- 2 is a diagram showing an XRD pattern of a monohydrobromide salt of the present compound of Example 12.
- FIG. 4 shows an XRD pattern of 1/2 citrate of the present compound of Example 13.
- FIG. 4 shows an XRD pattern of 1 citrate of the present compound of Example 14.
- FIG. 4 is a diagram showing an XRD pattern of 1/2 tartrate salt 1/2 hydrate of the present compound of Example 15.
- FIG. 4 is a diagram showing an XRD pattern of a crystal of the present compound of Example 16.
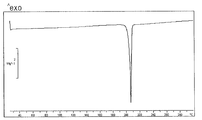
- FIG. 4 shows a DSC curve of the crystal of the present compound of Example 16.
- FIG. 4 is a diagram showing an XRD pattern of crystals of the present compound of Example 17.
- 2 is a diagram showing a DSC curve of a crystal of the present compound of Example 17.
- FIG. 2 is a diagram showing an XRD pattern of crystals of the present compound of Example 18.
- FIG. 2 is a diagram showing a DSC curve of a crystal of the present compound of Example 18.
- FIG. 4 shows an XRD pattern of the monohydrate of the present compound of Example 19.
- 2 is a diagram showing an XRD pattern of crystals of the present compound of Example 20.
- FIG. FIG. 4 is a diagram showing a DSC curve of the crystal of the present compound of Example 20.
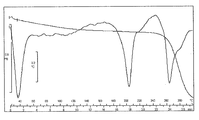
- 2 is a graph showing the results of moisture adsorption measurement of the monohydrochloride monohydrate of the present compound of Example 1.
- FIG. 2 is a graph showing the results of water adsorption measurement of monohydrochloride of the present compound of Example 2.
- FIG. It is a figure which shows the result of the water
- FIG. 1 is a graph showing the results of water adsorption measurement of monohydrochloride of the present compound of Example 2.
- Examples of the salt of the present compound in the crystalline form include hydrochloride, methanesulfonate, fumarate, hydrobromide, tartrate, citrate and the like. These salts can exist in both unsolvated and solvated forms.
- solvates such as water, methanol, ethanol, isopropyl alcohol, acetone, acetonitrile, and ethyl acetate can be mentioned. preferable.
- solvates such as hemi-, mono-, di-, tri-, tetra-, penta- and hexa- can be obtained depending on the number of solvents for the present compound.
- a hydrate it is preferably a hydrate of 3 or less, more preferably a monohydrate or a dihydrate.
- the salt of the present compound in the form of crystals include hydrochloride salt, monohydrate, hydrochloride salt, anhydrous salt, hydrochloride salt, 2- to 3-hydrate salt, hydrochloride salt, monoacetonitrile, type I crystal, II Type crystals, methanesulfonate / anhydride, methanesulfonate / trihydrate, methanesulfonate / dihydrate, methanesulfonate / 1/2 to 1 hydrate, 1/2 fumaric acid Salt, 3/2 hydrate, 1 fumarate, 1-2 hydrate, hydrobromide, 1/2 citrate, 1 citrate, 1/2 DL-tartaric acid, 1/2 hydrate Thing etc.
- hydrochloride / monohydrate hydrochloride / anhydride, methanesulfonate / anhydride
- hydrochloride / monohydrate preferred is hydrochloride / monohydrate.
- the salt of the present compound in crystalline form has a markedly superior effect of being stable and having very high water solubility.
- hydrochloride / monohydrate, hydrochloride / anhydride, and methanesulfonate / anhydride have no problems such as moisture adsorption, no chargeability, good fluidity, and high bioactivity when administered orally. It also has an excellent effect of having availability (biological utilization rate).
- the salt of this compound in crystalline form is mixed with an excess of an organic acid or inorganic acid such as hydrochloric acid, methanesulfonic acid, fumaric acid, hydrobromic acid, tartaric acid, citric acid and the like in an equivalent amount to form a salt. It can be obtained by crystallization.
- an organic acid or inorganic acid such as hydrochloric acid, methanesulfonic acid, fumaric acid, hydrobromic acid, tartaric acid, citric acid and the like in an equivalent amount to form a salt.
- an organic acid or inorganic acid such as hydrochloric acid, methanesulfonic acid, fumaric acid, hydrobromic acid, tartaric acid, citric acid and the like in an equivalent amount to form a salt.
- any solvent can be used as the solvent used to form the salt, it is preferable to select a solvent that can be used as a solvent used for subsequent crystallization.
- solvent examples include water, alcohol (methanol, ethanol, 1-propanol, isopropyl alcohol, butanol, etc.), ketone (acetone, methyl ethyl ketone, etc.), nitrile (acetonitrile, propionitrile, etc.), ester (ethyl formate, acetic acid, etc.).
- alcohol methanol, ethanol, 1-propanol, isopropyl alcohol, butanol, etc.
- ketone acetone, methyl ethyl ketone, etc.
- nitrile acetonitrile, propionitrile, etc.
- ester ethyl formate, acetic acid, etc.
- hydrochloride monohydrate for example, hydrochloride is formed in a mixed solvent of water and an organic solvent such as alcohol (methanol, ethanol, 1-propanol, isopropyl alcohol, etc.), ketone (acetone, methyl ethyl ketone, etc.), etc. And can be produced by crystallization. During crystallization, esters (ethyl formate, ethyl acetate, isopropyl acetate, etc.) may be added, thereby improving the yield.
- alcohol methanol, ethanol, 1-propanol, isopropyl alcohol, etc.
- ketone acetone, methyl ethyl ketone, etc.
- esters ethyl formate, ethyl acetate, isopropyl acetate, etc.
- a hydrochloride is formed in an organic solvent such as alcohol (methanol, ethanol, 1-propanol, isopropyl alcohol, etc.) and a ketone (acetone, methyl ethyl ketone, etc.) that does not contain moisture, It can be manufactured by crystallization.
- alcohol methanol, ethanol, 1-propanol, isopropyl alcohol, etc.
- ketone acetone, methyl ethyl ketone, etc.
- it can be crystallized under anhydrous conditions using azeotropic dehydration, a dehydrating agent, or the like.
- methanesulfonate / anhydride for example, methanesulfonate is used in an organic solvent such as alcohol (methanol, ethanol, 1-propanol, isopropyl alcohol, etc.) and ketone (acetone, methyl ethyl ketone, etc.) that does not contain moisture. It can be produced by crystallization and crystallization. Moreover, after producing
- alcohol methanol, ethanol, 1-propanol, isopropyl alcohol, etc.
- ketone acetone, methyl ethyl ketone, etc.
- Examples of the present crystalline compound include type I crystals, type II crystals, type III crystals, type IV (monohydrate) crystals, and type V crystals. Preferable examples include type I crystals and type II crystals.
- the compound can be crystallized by, for example, heating to dissolve the compound and then cooling to crystallize, or adding a poor solvent to the solution of the compound. Solvents used include water, alcohol (methanol, ethanol, 1-propanol, isopropyl alcohol, butanol, etc.), ketones (acetone, methyl ethyl ketone, etc.), nitriles (acetonitrile, propionitrile, etc.), esters (ethyl formate, ethyl acetate).
- Particularly preferable solvents include water, a mixed solvent of water and alcohol (methanol, ethanol, 1-propanol, isopropyl alcohol, etc.), a mixed solvent of water and ketone (acetone, methyl ethyl ketone, etc.), or an ester thereof (ethyl formate). , Ethyl acetate, isopropyl acetate, etc.).
- the prophylactic and / or therapeutic agent for example, involved in ORL-1 receptor
- ORL-1 receptor the prophylactic and / or therapeutic agent
- the pharmaceutical preparation for the prevention and / or treatment of diseases (especially central nervous system diseases), particularly for sleep disorders, alcoholism, drug addiction, anxiety, stress disorders Can do.
- diseases especially central nervous system diseases
- the pharmaceutical preparation include pharmaceutical preparations described in WO03 / 082333 and WO2008 / 050698, which are manufactured and administered according to the method described in the publication.
- Dosage should take into account age, weight, general health, sex, diet, time of administration, method of administration, excretion rate, combination of drugs, degree of medical condition being treated at the time of the patient, and other factors It is decided.
- the daily dose is, for example, 0.01 to 1000 mg / kg body weight / day orally, and is administered once to several times a day, or about 0.01 to 100 mg / kg parenterally. It is preferable to administer kg body weight / day in 1 to several times a day.
- the preventive drug is a drug administered to a healthy person who has not developed a disease, for example, a drug administered for the purpose of preventing the onset of the disease.
- a therapeutic drug is a drug administered to a person (patient) diagnosed as having developed a disease by a doctor. For example, it is administered for the purpose of alleviating the disease or symptoms, or restoring health. It is a medicine. Moreover, even if the purpose of administration is prevention of worsening of diseases and symptoms, or prevention of seizures, it is a therapeutic agent if administered to a patient.
- This compound was synthesized according to the method described in WO03 / 082333.
- Powder X-ray diffraction (XRD) was performed using Cu K ⁇ 1 as an X-ray tube at room temperature using a powder X-ray diffractometer RINT2200 / Ultima + (Rigaku) or X'Pert Pro MPD (PANallytical).
- a diffraction angle (2 ⁇ ) range of ⁇ 35 ° was measured.
- the measurement conditions using each diffraction device are as follows.
- Tube current 40 mA, tube voltage: 40 kV, scanning speed: 4 ° / min
- Diffraction device X'Pert Pro MPD (PANallytical)
- Tube current 40 mA, tube voltage: 45 kV, scanning speed: 40.1 ° / min.
- Tube current 30 mA, tube voltage: 40 kV, scanning speed: 12.3 ° / min. In general, a variation of about ⁇ 0.2 ° is observed, but a larger error may occur depending on the measurement conditions.
- Thermal analysis was performed in a stream of 40 mL of dry nitrogen gas per minute using a differential thermogravimetric simultaneous measurement device TG / SDTA851e (TG / DTA) or a differential scanning calorimetry device DSC821e (DSC) manufactured by METTLER TOLEDO, It measured on the temperature increase rate conditions of 10 degree-C / min.
- TG / DTA differential thermogravimetric simultaneous measurement device
- DSC821e DSC821e
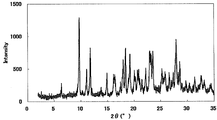
- Example 1 Monohydrochloride monohydrate of this compound Monohydrochloride monohydrate was obtained according to the following (1) to (4).
- a hydrogen chloride solution (7 ml of a solution of 10 ml of 4M hydrochloric acid-ethyl acetate solution diluted with 30 ml of acetonitrile) was added dropwise with stirring at 40 ° C. The mixture was further stirred at 40 ° C. for 10 minutes and allowed to stand for 6 hours when it became a homogeneous solution. Then, 2.6 g of precipitated I-hydrochloride monoacetate crystals of this compound were collected by filtration.
- the obtained homogeneous solution was concentrated under reduced pressure to about half at 55 ° C., and then 5 ml of isopropyl acetate was added dropwise in 30 minutes.
- the obtained suspension was stirred at 55 ° C. for 1.5 hours and at room temperature for 3 hours, and then the precipitated crystals were collected by filtration and dried at 60 ° C. under vacuum to give 1.0 g of the title compound as white crystals. Obtained.
- Example 2 Monohydrochloride / anhydride of this compound To a suspension of 2.64 g of this compound and 29 ml of isopropyl alcohol, 1.04 ml (1.05 equivalents) of 6M hydrochloric acid was added at room temperature with stirring. After heating, insoluble matters were filtered, 32 ml of isopropyl alcohol was added to the filtrate, and stirring was continued at room temperature. After 35 ml of the solvent was distilled off by atmospheric distillation (removal of water by azeotropic distillation), seed crystals were added, 15 ml of the solvent was distilled off, and “25 ml of isopropyl alcohol and 25 ml of solvent were distilled off” were repeated twice.
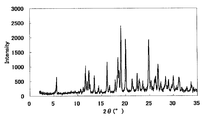
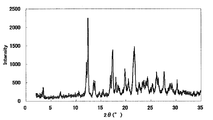
- Example 3 Monohydrochloride ⁇ 2-3 hydrate of this compound 15 mL of water was added to 500 mg of the monohydrochloride ⁇ monohydrate of Example 1, and the mixture was heated to about 75 ° C. and cooled again to around room temperature. Seed crystals were added to this suspension, and the mixture was stirred and washed at room temperature for 1 week under light shielding. The precipitated crystals were collected by filtration to give the title compound as a crystalline white powder. [XRD (diffractometer: RINT2200 / Ultima +)] The XRD pattern is shown in FIG.
- Characteristic peaks are 6.4 °, 9.7 °, 11.8 °, 14.9 °, 18.4 °, and 19.2 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ). It was near.
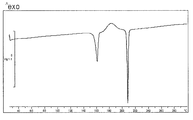
- [TG / DTA] From around 40 ° C., a weight loss due to desorption of crystal water (about 7 to 11%, which varies depending on the environmental humidity at the time of measurement), and an endothermic peak due to it were observed. In this measurement method, the temperature at which the weight loss due to desorption of crystallization water and the endothermic peak resulting from it decrease and the range thereof may vary depending on the crystal particle size, crystal habit, and the like of the title compound. Further, an endothermic peak having a peak top around 215 ° C. was observed, and then a significant weight reduction due to decomposition was observed. The obtained TG / DTA curve is shown in FIG.
- Example 4 Form 1 crystals of monohydrochloride and monoacetonitrile solvate of this compound
- 1.1 mL of 90% acetonitrile solution was added and heated to 65 ° C. While stirring this suspension, an ethanol solution of hydrochloric acid corresponding to 1.2 equivalents was added and dissolved completely, and then slowly cooled to 25 ° C. Seed crystals were added thereto, and the mixture was stirred at room temperature for 1 day, and the precipitated crystals were collected by filtration to obtain 361 mg of the title compound as a crystalline white powder.
- XRD diffractometer: RINT2200 / Ultima +
- the XRD pattern is shown in FIG. Characteristic peaks were around 9.7 °, 18.1 °, 21.9 °, 25.6 °, and 27.8 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 5 Type II crystals of monohydrochloride monoacetate solvate of this compound 2.2 mL of acetonitrile was added to 5 mg monohydrochloride monohydrate of Example 1, and dissolved by heating to 60 ° C. After hot filtration, the filtrate was allowed to stand overnight at 20 ° C. to obtain a crystalline white solid.
- XRD X'Pert Pro MPD [Condition 2]
- the XRD pattern is shown in FIG. The characteristic peaks were around 12.6 °, 13.6 °, 16.5 °, 20.3 ° and 24.1 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 6 1 Methanesulfonate / anhydride of this compound According to the following (1) and (2), 1methanesulfonate / anhydride was obtained.
- 1M-methanesulfonic acid ethanol solution 5.4 ml 1.2 equivalents
- Characteristic peaks are 7.6 °, 11.5 °, 17.6 °, 18.4 °, 19.9 °, and 23.5 ° ( ⁇ 0.2 ° respectively) as diffraction angles (2 ⁇ ). It was near.
- [TG / DTA] Melting and / or decomposition peaks were observed at 271 ° C. (extrapolated onset temperature) and significant weight loss due to decomposition was observed. The obtained TG / DTA curve is shown in FIG.
- Example 7 1 Methanesulfonic acid salt of this compound ⁇ 1/2 to 1 hydrate 0.1 mL of 90% toluene / methanol solution was added to 15 mg of 1methanesulfonic acid salt ⁇ anhydride of this compound. The mixture was capped and stirred for about 1 month at room temperature under light shielding to gradually evaporate the solvent, and the title compound was obtained as a crystalline white powder.
- XRD diffractometer: RINT2200 / Ultima +
- the XRD pattern is shown in FIG. The characteristic peaks were around 10.7 °, 11.4 °, 16.9 °, 21.4 ° and 25.5 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 8 1 Methanesulfonic acid salt / trihydrate of this compound 2 mL of water was added to 200 mg of 1methanesulfonic acid salt / anhydride of this compound. After heating and dissolving at 80 ° C., the mixture was cooled to room temperature, and a small amount of seed crystals was added thereto, followed by stirring at room temperature. This was covered and aged at room temperature for 4 days in the dark, and the precipitate was filtered and placed in a sealed container to give the title compound as a crystalline white solid. [XRD (diffractometer: RINT2200 / Ultima +)] The XRD pattern is shown in FIG. The characteristic peaks were around 8.9 °, 15.0 °, 18.8 ° and 19.2 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 9 Monomethanesulfonate dihydrate of this compound Monomethanesulfonate trihydrate of this compound of Example 8 was dried under reduced pressure at room temperature for about 30 minutes to obtain the title compound as a crystalline white powder. It was. [XRD (diffractometer: RINT2200 / Ultima +)] The XRD pattern is shown in FIG. The characteristic peaks were around 9.4 °, 10.6 °, 16.1 °, 18.1 °, and 19.8 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 10 1 ⁇ 2 fumarate salt of this compound , 3/2 hydrate
- a suspension of 2 g of this compound and 20 ml of aqueous ethanol (18 ml of ethanol + 2 ml of water) 10.8 ml (1. 2 equivalents) was added dropwise with stirring at 70 ° C. After complete dissolution, the mixture was cooled to room temperature and stirred as it was overnight. The precipitated crystals were collected by filtration and dried under vacuum at 40 ° C. for 2 hours to obtain 1.52 g of the title compound as white crystals.
- XRD Powder X-ray diffraction
- Characteristic peaks are around 8.4 °, 11.2 °, 18.0 °, 19.2 °, 21.1 ° and 23.1 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Met. [TG / DTA] From about 50 ° C., a weight loss of 5.5% accompanying the desorption of water of crystallization was gradually observed, and a gentle endothermic peak was observed. In this measurement method, the temperature at which the weight loss due to desorption of crystallization water and the endothermic peak resulting from it decrease and the range thereof may vary depending on the crystal particle size, crystal habit, and the like of the title compound. Further, an endothermic peak having a peak top was observed around 150 ° C., and a significant weight reduction due to decomposition was observed from around 220 ° C. The obtained TG / DTA curve is shown in FIG.
- Example 11 1 fumarate salt of this compound, 1 to 2 hydrate 2 mL of methanol was added to 200 mg of this compound, and 2.4 equivalent of a fumaric acid ethanol solution was added dropwise at 60 ° C. with heating and stirring. A small amount of precipitate was observed when 0.9 mL of methanol was added and completely dissolved while stirring at a temperature of 65 ° C. and then slowly cooled to room temperature. The crystals were aged by stirring at room temperature for about 4 days, and the precipitated crystals were collected by filtration. Drying at 40 ° C. under vacuum for 1 hour gave 116 mg of the title compound as a crystalline white powder.
- Example 12 Monohydrobromide salt of this compound 0.5 mL of 90% ethanol solution was added to 50 mg of this compound and heated to 60 ° C. While stirring this suspension, an ethanol solution of hydrobromic acid equivalent to 1.2 equivalents was added and completely dissolved, and then slowly cooled to room temperature. Seed crystals were added thereto, and the mixture was stirred at room temperature for one day and night. The precipitated crystals were collected by filtration and air-dried for 10 minutes to obtain 35 mg of the title compound as a crystalline white powder.
- XRD diffractometer: RINT2200 / Ultima +
- the XRD pattern is shown in FIG. Characteristic peaks were around 8.3 °, 10.4 °, 11.9 °, 18.5 ° and 23.2 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 13 1/2 citrate of this compound 2 mL of 80 mM citric acid aqueous solution was added to 150 mg of this compound, followed by shaking at 200 ° C. for about 24 hours (200 rpm). This was allowed to stand at room temperature for 5 days, and the solid component was collected by filtration to obtain 139 mg of the title compound as a crystalline white powder.
- XRD diffractometer: RINT2200 / Ultima +
- the XRD pattern is shown in FIG. Characteristic peaks are around 8.4 °, 11.3 °, 13.0 °, 16.6 °, 19.8 °, and 21.8 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ). Met.
- Example 14 Citrate of this compound 1 mL of 1.7 M citric acid aqueous solution was added to 100 mg of 1/2 citrate of this compound, and the mixture was shaken (200 rpm) at 37 ° C. for about 24 hours. The solid component was collected by filtration to give the title compound as a crystalline white powder.
- XRD diffractometer: RINT2200 / Ultima +
- Characteristic peaks were around 8.0 °, 9.1 °, 16.0 °, 18.2 °, and 24.4 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ).
- Example 15 1 / 2DL-tartrate 1/2 hydrate of this compound A suspension of 440 mg of this compound and 20 ml of methanol was heated and stirred at 80 ° C., and 10 m of methanol was added. After complete dissolution, 2.4 ml (2.4 equivalents) of 1M-DL-ethanolic tartrate solution was added dropwise with stirring at 80 ° C. The mixture was cooled to room temperature and stirred as it was for 3 hours. The precipitated crystals were collected by filtration and dried under vacuum at 60 ° C. for 2 hours to obtain 274.3 mg of the title compound as white crystals.
- Example 16 150 g of ethanol was added to 42 g of this compound of type I crystals of this compound, and the mixture was heated and stirred at 70 ° C. After complete dissolution, 600 ml of ethyl acetate was added, cooled to room temperature, and stirred as it was for a whole day and night. The precipitated crystals were collected by filtration and dried at 60 ° C. under reduced pressure for 10 hours to obtain 31 g of the title compound as slightly pink crystals. [XRD (diffractometer: RINT2200 / Ultima +)] The XRD pattern is shown in FIG.
- Example 17 Type II crystals of this compound To 2 g of the compound obtained in Example 1, 400 ml of ethyl acetate was added and heated to reflux. After completely dissolving, it was cooled to room temperature and left as it was for a whole day and night. The precipitated crystals were collected by filtration and dried at 60 ° C. under reduced pressure for 5 hours to obtain 0.3 g of the title compound as white crystals.
- XRD diffractometer: RINT2200 / Ultima +
- the XRD pattern is shown in FIG. Characteristic peaks are 6.3 °, 12.6 °, 13.7 °, 14.4 °, 16.7 °, 20.9 °, and 23.5 ° ( ⁇ each) as diffraction angles (2 ⁇ ). 0.2 °).
- [DSC] A melting point (extrapolated onset temperature) was observed around 204 ° C. The DSC curve is shown in FIG.
- Example 18 Type III crystals of this compound 200 ml of ethanol was added to 25 g of this compound, and the mixture was heated and stirred at 70 ° C. After complete dissolution, the solution was concentrated under reduced pressure until a solid precipitated. It was cooled to room temperature and left as it is for a whole day and night. The precipitated crystals were collected by filtration and dried at 60 ° C. under reduced pressure for 8 hours to obtain 20 g of the title compound as slightly yellow crystals.
- XRD diffractometer: RINT2200 / Ultima +
- the characteristic peaks are 6.2 °, 6.4 °, 6.8 °, 12.3 °, 15.7 °, and 23.0 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ). It was near.
- [DSC] A melting point (extrapolated onset temperature) was observed around 191 ° C. In addition, following the melting peak, an exothermic peak was observed so as to overlap, and then a second endothermic peak was observed around about 204 ° C. (extrapolation start temperature). The DSC curve is shown in FIG.
- Example 19 Form IV (monohydrate) crystals of this compound According to the following (1) to (2), type IV (monohydrate) crystals were obtained.
- a suspension obtained by adding 3 mL of sodium phosphate buffer (pH 6.8) to about 15 mg of the hydrochloride monohydrate of Example 1 was shaken at 37 ° C. for 24 hours. The solid component of the suspension was quickly filtered and stored in a sealed container to give the title compound as white crystals.
- Example 20 V-type crystals of this compound Several mg of the compound of Example 19 was dried (for example, dried at 40 ° C. under reduced pressure for 1 hour) to give the title compound as white crystals.
- XRD diffractometer: RINT2200 / Ultima +
- the XRD pattern is shown in FIG. Characteristic peaks are around 3.4 °, 6.9 °, 12.1 °, 12.4 °, 17.4 ° and 21.7 ° (each ⁇ 0.2 °) as diffraction angles (2 ⁇ ). Met.
- [DSC] A melting point (extrapolated onset temperature) was observed around 157 ° C. Following the melting peak, an exothermic peak was observed near about 172 ° C. (extrapolation start temperature), and then a second endothermic peak was observed near about 207 ° C. (extrapolation start temperature).
- the DSC curve is shown in FIG.
- Test example 1 Moisture adsorption measurement
- Moisture adsorption measurement which is an index of hygroscopicity
- the relative humidity was varied in the range from 0% to 95% relative humidity.
- the change in weight was recorded for each set relative humidity, and converted into a change (%) based on the weight at 0% relative humidity.
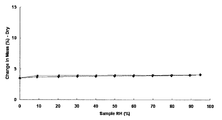
- the monohydrochloride monohydrate of Example 1 is a hydrate with very stable water of crystallization, and the weight loss does not become 0 even when left at a relative humidity of 0% RH for 15 hours. It was.
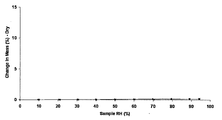
- the 1/2 fumarate salt 3/2 hydrate of Example 10 and the 1 methanesulfonic acid trihydrate of Example 8 were 1.3% and 2.0% by weight absorption, respectively. Showed an increase.
- monohydrochloride ⁇ 2-3 hydrate of Example 3 monomethanesulfonate ⁇ 1 / 2–1 hydrate of Example 7, monofumarate of Example 11, 1–2 hydrate
- the citrate salt of Example 13, the citrate salt of Example 13, the citrate salt of Example 14, and the 1 ⁇ 2 tartrate salt 1 ⁇ 2 hydrate of Example 15 are all increased in weight by absorption of 4% or more. was there.
- Test example 2 Solubility in water
- HPLC high performance liquid chromatography
- Example 16 0.01 mg / ml
- Example 17 0.01 mg / ml
- Example 6 > 2 mg / ml
- Example 1 > 2 mg / ml
- Example 10 0.3 mg / ml
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Addiction (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Anesthesiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

即ち、本発明は以下の通りである。
[1] 結晶態様の(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの塩。
[2] 塩酸塩、メタンスルホン酸塩、フマル酸塩、臭化水素酸塩、酒石酸塩又はクエン酸塩である[1]記載の塩。
[3] 粉末X線回折スペクトルにおいて回折角度(2θ)で5.6°、16.2°、19.0°、20.1°、及び24.9°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの1塩酸塩・1水和物。
[4] 図1の粉末X線回折パターン及び/又は図2の示差熱熱重量同時測定(TG/DTA)曲線を示す[3]記載の1塩酸塩・1水和物。
[5] 粉末X線回折スペクトルにおいて回折角度(2θ)で5.2°、6.8°、9.1°、10.5°及び15.7°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの1塩酸塩。
[6] 図3の粉末X線回折パターン及び/又は図4の示差熱熱重量同時測定(TG/DTA)曲線を示す[5]記載の1塩酸塩。
[7] 粉末X線回折スペクトルにおいて回折角度(2θ)で7.6°、11.5°、17.6°、18.4°、19.9°、及び23.5°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドのメタンスルホン酸塩。
[8] 図9の粉末X線回折パターン及び/又は図10の示差熱熱重量同時測定(TG/DTA)曲線を示す[7]記載のメタンスルホン酸塩。
[10] 図15の粉末X線回折パターン及び/又は図16の示差熱熱重量同時測定(TG/DTA)曲線を示す[9]記載の1/2フマル酸塩・3/2水和物。
[11] 粉末X線回折スペクトルにおいて回折角度(2θ)で6.4°、12.5°、12.8°、16.5°、18.7°、21.6°、及び23.9°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの結晶。
[12] 図22の粉末X線回折パターン及び/又は図23の示差走査熱分析(DSC)曲線Bを示す[11]記載の結晶。
[13] 粉末X線回折スペクトルにおいて回折角度(2θ)で6.3°、12.6°、13.7°、14.4°、16.7°、20.9°、及び23.5°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの結晶。
[14] 図24の粉末X線回折パターン及び/又は図25の示差走査熱分析(DSC)曲線を示す[13]記載の結晶。
[15] [1]~[14]のいずれか記載の塩又は結晶から成る医薬。
[16] [1]~[14]のいずれか記載の塩又は結晶を有効成分として含有する医薬組成物。
[17] ORL-1受容体に関与する疾患の予防及び/又は治療に用いる[16]記載の医薬組成物。
[18] ORL-1受容体に関与する中枢神経系疾患の予防及び/又は治療に用いる[16]記載の医薬組成物。
[19] 睡眠障害、アルコール依存症、薬物依存症、不安又はストレス障害の予防及び/又は治療に用いる[16]記載の医薬組成物。
[20] ORL-1受容体に関与する疾患の予防及び/又は治療薬の製造のための、[1]~[14]のいずれか記載の塩又は結晶の使用。
[21] ORL-1受容体に関与する中枢神経系疾患の予防及び/又は治療薬の製造のための、[1]~[14]のいずれか記載の塩又は結晶の使用。
[22] 睡眠障害、アルコール依存症、薬物依存症、不安又はストレス障害の予防及び/又は治療薬の製造のための、[1]~[14]のいずれか記載の塩又は結晶の使用。
[23] [1]~[14]のいずれか記載の塩又は結晶の有効量を、対象に投与することを含む、ORL-1受容体に関与する疾患の予防及び/又は治療方法。
[24] [1]~[14]のいずれか記載の塩又は結晶の有効量を、対象に投与することを含む、ORL-1受容体に関与する中枢神経系疾患の予防及び/又は治療方法。
[25] [1]~[14]のいずれか記載の塩又は結晶の有効量を、対象に投与することを含む、睡眠障害、アルコール依存症、薬物依存症、不安又はストレス障害の予防及び/又は治療方法。
結晶態様の本化合物の塩としては、例えば、塩酸塩・1水和物、塩酸塩・無水物、塩酸塩・2~3水和物、塩酸塩・1アセトニトリル和物のI型結晶、同II型結晶、メタンスルホン酸塩・無水物、メタンスルホン酸塩・3水和物、メタンスルホン酸塩・2水和物、メタンスルホン酸塩・1/2~1水和物、1/2フマル酸塩・3/2水和物、1フマル酸塩・1~2水和物、臭化水素酸塩、1/2クエン酸塩、1クエン酸塩、1/2DL-酒石酸・1/2水和物等が挙げられる。これらのうち、好ましいものとして塩酸塩・1水和物、塩酸塩・無水物、メタンスルホン酸塩・無水物等が挙げられ、特に好ましくは塩酸塩・1水和物が挙げられる。
結晶態様の本化合物の塩は、安定であり、非常に高い水溶解性を有するという顕著に優れた効果を有する。特に、塩酸塩・1水和物、塩酸塩・無水物、メタンスルホン酸塩・無水物は水分吸着性等の問題がなく、帯電性が無く流動性が良く、また経口投与した場合に高いバイオアベイラビリテイ(生物学的利用率)を有するという優れた効果をも有している。
塩酸塩・1水和物の場合は、例えば、アルコール(メタノール、エタノール、1-プロパノール、イソプロピルアルコール等)、ケトン(アセトン、メチルエチルケトン等)等の有機溶媒と水の混合溶媒中で塩酸塩を生成させて、結晶化することで、製造することができる。結晶化の際、エステル(ギ酸エチル、酢酸エチル、酢酸イソプロピル等)を加えてもよく、それによって収率が向上する。
塩酸塩・無水物の場合は、例えば、水分を含まないアルコール(メタノール、エタノール、1-プロパノール、イソプロピルアルコール等)、ケトン(アセトン、メチルエチルケトン等)等の有機溶媒中で塩酸塩を生成させて、結晶化することで、製造することができる。また、含水条件下で塩酸塩を生成させた後、共沸脱水、脱水剤等を用いて無水条件とし、結晶化することもできる。
メタンスルホン酸塩・無水物の場合は、例えば、水分を含まないアルコール(メタノール、エタノール、1-プロパノール、イソプロピルアルコール等)、ケトン(アセトン、メチルエチルケトン等)等の有機溶媒中でメタンスルホン酸塩を生成させて、結晶化することで、製造することができる。また、含水条件下でメタンスルホン酸塩を生成させた後、共沸脱水、脱水剤等を用いて無水条件とし、結晶化することもできる。
本化合物の結晶化は、例えば加温して本化合物を溶解した後、冷却して結晶化するか、又は本化合物の溶液に貧溶媒を添加することで結晶化することができる。用いられる溶媒としては、水、アルコール(メタノール、エタノール、1-プロパノール、イソプロピルアルコール、ブタノール等)、ケトン(アセトン、メチルエチルケトン等)、ニトリル(アセトニトリル、プロピオニトリル等)、エステル(ギ酸エチル、酢酸エチル、酢酸イソプロピル等)、エーテル(ジエチルエーテル、ジイソプロピルエーテル、t-ブチルメチルエーテル、1,4-ジオキサン、THF等)、アミド(ホルムアミド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素(ジクロロメタン、クロロホルム、1,2-ジクロロエタン等)、炭化水素(n-ヘキサン、シクロヘキサン、ベンゼン、トルエン等)、スルホキシド(ジメチルスルホキシド等)等、又はこれらの混合溶媒等が挙げられる。特に好ましい溶媒としては、水、水とアルコール(メタノール、エタノール、1-プロパノール、イソプロピルアルコール等)との混合溶媒、水とケトン(アセトン、メチルエチルケトン等)との混合溶媒、又はこれらとエステル(ギ酸エチル、酢酸エチル、酢酸イソプロピル等)との混合溶媒等が挙げられる。
粉末X線回折(XRD)は、粉末X線回折装置RINT2200/Ultima+(リガク社)、またはX’Pert Pro MPD(PANalytical社)を用いて、室温でX線管球としてCu Kα1を用いて、2~35°の回析角度(2θ)範囲を測定した。各回折装置を用いた測定条件は以下の通りである。
・回折装置:RINT2200/Ultima+(リガク社)
管電流:40mA、管電圧:40kV、走査速度:4°/分
・回折装置:X’Pert Pro MPD(PANalytical社)
[条件1]管電流:40mA、管電圧:45kV、走査速度:40.1°/分
[条件2]管電流:30mA、管電圧:40kV、走査速度:12.3°/分
なお、2θ値は一般的には±0.2°程度のバラツキが観察されるが、測定条件などによってより大きな誤差を生じる場合がある。
熱分析は、メトラー・トレド社の示差熱熱重量同時測定装置TG/SDTA851e(TG/DTA)、または示差走査熱量測定装置DSC821e(DSC)を用いて、毎分40mLの乾燥窒素ガスの気流中、毎分10℃の昇温速度条件にて測定した。
本化合物の1塩酸塩・1水和物
以下の(1)~(4)に従って、1塩酸塩・1水和物を得た。
(1)本化合物2.5gおよびアセトニトリル80mlの懸濁溶液に、1.23当量の塩化水素溶液(4M塩酸-酢酸エチル溶液10mlをアセトニトリル30mlで希釈した溶液7ml)を40℃攪拌下滴下した。さらに40℃で10分攪拌し均一溶液となったところで6時間静置したのち、析出した本化合物の1塩酸塩・1アセトニトリル和物のI型結晶2.6gをろ取した。ろ取した結晶をメタノール40mlに溶解し、減圧下メタノールを留去したのち、含水アセトン10ml(アセトン9ml+水1ml)を加え均一な溶液とし、2日間静置した。析出した結晶をろ取し、真空下60℃で2時間乾燥することにより表題化合物2.2gを白色結晶として得た。
(2)本化合物37gにアセトン400mlおよび水40mlを加えた懸濁溶液に、6M塩酸15ml(1.07当量)を室温攪拌下滴下した。60℃で攪拌し均一溶液となったのち、室温まで放冷することにより塩酸塩の析出が生じることを確認したところで酢酸エチル100mlを室温攪拌下滴下した。1時間室温で攪拌したのち、さらに酢酸エチル100mlを滴下し、その後4時間室温で攪拌した。析出した結晶をろ取し、真空下70℃で3時間乾燥することにより表題化合物31gを白色結晶として得た。
(3)本化合物138.03g、アセトン1518ml、水152mlの懸濁液に攪拌下室温で6M塩酸54.83ml(1.05当量)を添加した。内温48℃まで加熱後不溶物をろ過し、ろ液を室温で攪拌した。30分後(内温40℃)に種晶(0.2g)を添加し、室温で3時間攪拌後、内温5~10℃で酢酸イソプロピル373ml、さらに1時間後に373mlを滴下した。一夜放置後、ろ過、冷アセトン(180ml×2)で洗浄、60℃で5時間乾燥し、白色の粉末118.83gを得た。この粉末を飽和食塩水存在下のデシケータ内(相対湿度:~75%)に42時間静置し、白色粉末120.04gを得た。
(4)本化合物1.0g、1-プロパノール6.9mlおよび水1.2mlの懸濁溶液に、1.1当量の濃塩酸(0.26g)を室温で添加し攪拌した。得られた均一溶液を55℃で約半量に減圧濃縮した後、酢酸イソプロピル5mlを30分かけて分割滴下した。得られた懸濁液を55℃で1.5時間、室温にて3時間攪拌した後、析出した結晶をろ取し、真空下60℃で乾燥することにより表題化合物1.0gを白色結晶として得た。
XRDパターンを図1に示した。特徴的なピークは、回折角度(2θ)として5.6°、16.2°、19.0°、20.1°、及び24.9°(それぞれ±0.2°)付近であった。
[TG/DTA]
120℃付近より徐々に結晶水の脱離に伴う3.6%の重量減少、及びそれに起因する吸熱ピークが認められた。なお、本測定法において結晶水の脱離による重量減少およびそれに起因する吸熱ピークの生じる温度ならびにその範囲は、表題化合物の結晶の粒子径や晶癖などにより変わる可能性がある。また、220℃付近にピークトップを持つ吸熱ピークが認められ、その後分解による著しい重量減少が観察された。得られたTG/DTA曲線を図2に示した。
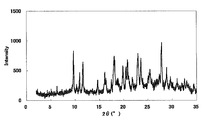
本化合物の1塩酸塩・無水物
本化合物2.64g、イソプロピルアルコール29mlの懸濁液に攪拌下室温で6M塩酸1.04ml(1.05当量)を添加した。加熱後不溶物をろ過し、ろ液にイソプロピルアルコール32mlを添加、室温で攪拌を継続した。常圧蒸留(共沸による水の除去)で溶媒を35ml留去後に種晶を添加し、さらに溶媒を15ml留去、「イソプロピルアルコール25ml添加、溶媒25mlを留去」を2回繰り返した。室温まで冷却後析出結晶をろ過、イソプロピルアルコールで洗浄、60℃で4時間乾燥し、白色の粉末2.624gを得た。この粉末を飽和食塩水存在下のデシケータ内(相対湿度:~75%)に24時間静置し、結晶性の白色粉末2.627gを得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図3に示した。特徴的なピークは、回折角度(2θ)として5.2°、6.8°、9.1°、10.5°及び15.7°(それぞれ±0.2°)付近であった。
[TG/DTA]
融解及び/又は分解ピークが259℃(補外開始温度)に認められ、分解による著しい重量減少が観察された。得られたTG/DTA曲線を図4に示した。
本化合物の1塩酸塩・2~3水和物
実施例1の1塩酸塩・1水和物500mgに15mLの水を加えて約75℃まで加熱し、再び室温付近まで冷却した。この懸濁液に種晶を加え、遮光下室温で1週間攪拌懸洗した。析出した結晶をろ取し、表題化合物を結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図5に示した。特徴的なピークは、回折角度(2θ)として6.4°、9.7°、11.8°、14.9°、18.4°、及び19.2°(それぞれ±0.2°)付近であった。
[TG/DTA]
40℃付近より結晶水の脱離に伴う重量減少(約7~11%、測定時の環境湿度により変化)、及びそれに起因する吸熱ピークが認められた。なお、本測定法において結晶水の脱離による重量減少およびそれに起因する吸熱ピークの生じる温度ならびにその範囲は、表題化合物の結晶の粒子径や晶癖などにより変わる可能性がある。また、215℃付近にピークトップを持つ吸熱ピークが認められ、その後分解による著しい重量減少が観察された。得られたTG/DTA曲線を図6に示した。
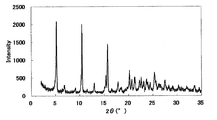
本化合物の1塩酸塩・1アセトニトリル和物のI型結晶
本化合物400mgに1.1mLの90%アセトニトリル溶液を加え、65℃に加熱した。この懸濁液を攪拌しながら1.2当量相当の塩酸エタノール溶液を加え、完全に溶解した後25℃までゆっくり冷却した。これに種晶を加えて室温で1昼夜攪拌し、析出した結晶をろ取して表題化合物361mgを結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図7に示した。特徴的なピークは、回折角度(2θ)として9.7°、18.1°、21.9°、25.6°及び27.8°(それぞれ±0.2°)付近であった。
本化合物の1塩酸塩・1アセトニトリル和物のII型結晶
実施例1の1塩酸塩・1水和物5mgに2.2mLのアセトニトリルを加え、60℃に加熱して溶解させた。これを熱ろ過した後にろ液を20℃で一昼夜静置して、結晶性の白色固体を得た。
[XRD(回折装置:X’Pert Pro MPD[条件2])]
XRDパターンを図8に示した。特徴的なピークは、回折角度(2θ)として12.6°、13.6°、16.5°、20.3°及び24.1°(それぞれ±0.2°)付近であった。
本化合物の1メタンスルホン酸塩・無水物
以下の(1)及び(2)に従って、1メタンスルホン酸塩・無水物を得た。
(1)本化合物2gおよび含水エタノール5ml(エタノール4.5ml+水0.5ml)の懸濁溶液に、1M-メタンスルホン酸エタノール溶液5.4ml(1.2当量)を70℃加熱攪拌下滴下した。完全に溶解した後、室温まで冷却し、そのまま一昼夜攪拌した。析出した結晶をろ取し、真空下50℃で10時間乾燥することにより表題化合物1.54gを白色結晶として得た。
(2)本化合物18gおよびメタノール300mlの懸濁溶液に、メタンスルホン酸4.68g(1.2当量)のメタノール45ml溶液を室温攪拌下滴下した。減圧下メタノールを留去したのち、含水アセトン60ml(アセトン54ml+水6ml)を加え均一な溶液とし、2日間静置した。析出した結晶をろ取し、真空下60℃で4時間乾燥することにより表題化合物11.5gを白色結晶として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図9に示した。特徴的なピークは、回折角度(2θ)として7.6°、11.5°、17.6°、18.4°、19.9°、及び23.5°(それぞれ±0.2°)付近であった。
[TG/DTA ]
融解及び/又は分解ピークが271℃(補外開始温度)に認められ、分解による著しい重量減少が観察された。得られたTG/DTA曲線を図10に示した。
本化合物の1メタンスルホン酸塩・1/2~1水和物
本化合物の1メタンスルホン酸塩・無水物15mgに0.1mLの90%トルエン/メタノール溶液を加えた。これに蓋をして、遮光下室温で約1ヶ月間攪拌すると徐々に溶媒が蒸発し、表題化合物を結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図11に示した。特徴的なピークは、回折角度(2θ)として10.7°、11.4°、16.9°、21.4°及び25.5°(それぞれ±0.2°)付近であった。
[TG/DTA]
30℃付近より徐々に結晶水の脱離に伴う約2.9%の重量減少、及びそれに起因するゆるやかな吸熱ピークが認められた。なお、本測定法において結晶水の脱離による重量減少およびそれに起因する吸熱ピークの生じる温度ならびにその範囲は、表題化合物の結晶の粒子径や晶癖などにより変わる可能性がある。また、192℃付近(補外開始温度)に吸熱ピークが認められ、その後発熱ピーク及び分解による著しい重量減少が観察された。得られたTG/DTA曲線を図12に示した。
本化合物の1メタンスルホン酸塩・3水和物
本化合物の1メタンスルホン酸塩・無水物200mgに2mLの水を加えた。80℃で加熱溶解後に室温まで冷却し、これに種晶を少量加えて室温で攪拌すると白濁した。これに蓋をして、遮光下に室温で4日間熟成させ、沈殿物をろ過して密閉容器に入れ、表題化合物を結晶性の白色固体として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図13に示した。特徴的なピークは、回折角度(2θ)として8.9°、15.0°、18.8°及び19.2°(それぞれ±0.2°)付近であった。
本化合物の1メタンスルホン酸塩・2水和物
実施例8の本化合物の1メタンスルホン酸塩・3水和物を室温で約30分間減圧乾燥し、表題化合物を結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図14に示した。特徴的なピークは、回折角度(2θ)として9.4°、10.6°、16.1°、18.1°及び19.8°(それぞれ±0.2°)付近であった。
本化合物の1/2フマル酸塩・3/2水和物
本化合物2gおよび含水エタノール20ml(エタノール18ml+水2ml)の懸濁溶液に、0.5M-フマル酸エタノール溶液10.8ml(1.2当量)を70℃加熱攪拌下滴下した。完全に溶解した後、室温まで冷却し、そのまま一昼夜攪拌した。析出した結晶をろ取し、真空下40℃で2時間乾燥することにより表題化合物1.52gを白色結晶として得た。
[粉末X線回折(XRD)分析]
XRDパターンを図15に示した。特徴的なピークは、回折角度(2θ)として8.4°、11.2°、18.0°、19.2°、21.1°及び23.1°(それぞれ±0.2°)付近であった。
[TG/DTA]
50℃付近より徐々に結晶水の脱離に伴う5.5%の重量減少、及びそれに起因するゆるやかな吸熱ピークが認められた。なお、本測定法において結晶水の脱離による重量減少およびそれに起因する吸熱ピークの生じる温度ならびにその範囲は、表題化合物の結晶の粒子径や晶癖などにより変わる可能性がある。また、150℃付近にピークトップを持つ吸熱ピークが認められ、220℃付近より分解による著しい重量減少が観察された。得られたTG/DTA曲線を図16に示した。
本化合物の1フマル酸塩・1~2水和物
本化合物200mgに2mLのメタノールを加え、60℃で加熱攪拌下2.4当量相当のフマル酸エタノール溶液を滴下した。温度65℃で攪拌しながらメタノールを0.9mL加えて完全に溶解した後、室温までゆっくり冷却すると少量の析出物が観察された。そのまま室温で約4日間攪拌して結晶を熟成させ、析出した結晶をろ取した。真空下40℃で1時間乾燥することにより表題化合物116mgを結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図17に示した。特徴的なピークは、回折角度(2θ)として6.0°、9.1°、18.8°、及び23.3°(それぞれ±0.2°)付近であった。
本化合物の1臭化水素酸塩
本化合物50mgに0.5mLの90%エタノール溶液を加え、60℃に加熱した。この懸濁液を攪拌しながら1.2当量相当の臭化水素酸エタノール溶液を加え、完全に溶解した後室温までゆっくり冷却した。これに種晶を加えて室温で1昼夜攪拌し、析出した結晶をろ取して10分間風乾して表題化合物35mgを結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図18に示した。特徴的なピークは、回折角度(2θ)として8.3°、10.4°、11.9°、18.5°及び23.2°(それぞれ±0.2°)付近であった。
本化合物の1/2クエン酸塩
本化合物150mgに2mLの80mMクエン酸水溶液を加え、37℃で約24時間振とう(200rpm)した。これを室温で5日間静置し、固体成分をろ取して表題化合物139mgを結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図19に示した。特徴的なピークは、回折角度(2θ)として8.4°、11.3°、13.0°、16.6°、19.8°及び21.8°(それぞれ±0.2°)付近であった。
本化合物の1クエン酸塩
本化合物の1/2クエン酸塩100mgに1mLの1.7Mクエン酸水溶液を加え、37℃で約24時間振とう(200rpm)した。固体成分をろ取して表題化合物を結晶性の白色粉末として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図20に示した。特徴的なピークは、回折角度(2θ)として8.0°、9.1°、16.0°、18.2°及び24.4°(それぞれ±0.2°)付近であった。
本化合物の1/2DL-酒石酸塩・1/2水和物
本化合物440mgおよびメタノール20mlの懸濁溶液を80℃加熱攪拌したところに、メタノール10mを加えた。完全に溶解した後、1M-DL-酒石酸エタノール溶液2.4ml(2.4当量)を80℃加熱攪拌下滴下した。室温まで冷却し、そのまま3時間攪拌した。析出した結晶をろ取し、真空下60℃で2時間乾燥することにより表題化合物274.3mgを白色結晶として得た。
[XRD(回折装置:X’Pert Pro MPD[条件1])]
測定条件を管電流:40mA、管電圧:45kVに変更して測定した。XRDパターンを図21に示した。特徴的なピークは、回折角度(2θ)として6.5°、7.2°、10.4°、12.9°及び13.9°(それぞれ±0.2°)付近であった。
本化合物のI型結晶
本化合物42gにエタノール150mlを加え70℃で加熱攪拌した。完全に溶解した後、酢酸エチル600mlを加え室温まで冷却し、そのまま一昼夜攪拌した。析出した結晶をろ取し、減圧下60℃で10時間乾燥することにより表題化合物31gを微桃色結晶として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図22に示した。特徴的なピークは、回折角度(2θ)として6.4°、12.5°、12.8°、16.5°、18.7°、21.6°、及び23.9°(それぞれ±0.2°)付近であった。
[DSC]
融点(補外開始温度)が約206℃付近に認められた。DSC曲線を図23に示した。
本化合物のII型結晶
実施例1で得られた化合物2gに酢酸エチル400mlを加え加熱還流した。完全に溶解した後、室温まで冷却し、そのまま一昼夜放置した。析出した結晶をろ取し、減圧下60℃で5時間乾燥することにより表題化合物0.3gを白色結晶として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図24に示した。特徴的なピークは、回折角度(2θ)として6.3°、12.6°、13.7°、14.4°、16.7°、20.9°、及び23.5°(それぞれ±0.2°)付近であった。
[DSC]
融点(補外開始温度)が約204℃付近に認められた。DSC曲線を図25に示した。
本化合物のIII型結晶
本化合物25gにエタノール200mlを加え70℃で加熱攪拌した。完全に溶解した後、固体が析出するまで減圧濃縮した。室温まで冷却し、そのまま一昼夜放置した。析出した結晶をろ取し、減圧下60℃で8時間乾燥することにより表題化合物20gを微黄色結晶として得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図26に示した。特徴的なピークは、回折角度(2θ)として6.2°、6.4°、6.8°、12.3°、15.7°、及び23.0°(それぞれ±0.2°)付近であった。
[DSC]
融点(補外開始温度)が約191℃付近に認められた。なお、融解ピークに引き続いて、重なるように発熱ピークが観察され、その後約204℃(補外開始温度)付近に2つ目の吸熱ピークが認められた。DSC曲線を図27に示した。
本化合物のIV型(1水和物)結晶
以下の(1)~(2)に従って、IV型(1水和物)結晶を得た。
(1)実施例1の塩酸塩・1水和物約15mgにリン酸ナトリウム緩衝液(pH6.8)3mLを加えた懸濁溶液を、37℃で24時間振とうした。懸濁液の固体成分を手早くろ過して密閉容器内に保存し、表題化合物の白色結晶を得た。
(2)本化合物5mgに250μLのジメチルホルムアミドを加え、60℃に加熱して攪拌し溶解させた。これを熱ろ過した後にろ液に1mLの水を加えて、20℃で一昼夜静置して、結晶性の白色固体を得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図28に示した。特徴的なピークは、回折角度(2θ)として3.5°、11.5°、13.5°、18.4°及び19.9°(それぞれ±0.2°)付近であった。
本化合物のV型結晶
実施例19の化合物 数mgを乾燥(例えば40℃で減圧乾燥1時間)することにより、表題化合物の白色結晶を得た。
[XRD(回折装置:RINT2200/Ultima+)]
XRDパターンを図29に示した。特徴的なピークは、回折角度(2θ)として3.4°、6.9°、12.1°、12.4°、17.4°及び21.7°(それぞれ±0.2°)付近であった。
[DSC]
融点(補外開始温度)が約157℃付近に認められた。なお、融解ピークに引き続いて約172℃(補外開始温度)付近に発熱ピークが観察され、その後約207℃(補外開始温度)付近に2つ目の吸熱ピークが認められた。DSC曲線を図30に示した。
水分吸着測定
吸湿性の指標となる水分吸着測定を、DVS-1型水分吸着装置(SMS社製)を用い、以下の条件で行った。試料約6~12mgを用いて、相対湿度0%から95%までの範囲について相対湿度を変化させた。設定した相対湿度毎に重量変化を記録し、相対湿度0%における重量をもとに変化量(%)に換算をした。なお、実施例1の1塩酸塩・1水和物は、非常に安定な結晶水を持つ水和物であり、相対湿度0%RHで15時間静置しても重量減少が0にならなかった。この化合物については、カールフィッシャー法により測定した試料中の水分値を元にゼロ点を補正し、変化量(%)に換算をした。
実施例1の1塩酸塩・1水和物、実施例2の1塩酸塩・無水物、及び実施例6の1メタンスルホン酸塩・無水物の水分吸着測定結果は、それぞれ図31、図32、及び図33の通りであり、それぞれ0.2%、0.2%、及び0.4%の重量増加を示した。これらの化合物は、湿度変化による重量変化が少なく、医薬品の原薬として望ましい形態であることが判明した。
他方、実施例10の1/2フマル酸塩・3/2水和物、及び実施例8の1メタンスルホン酸・3水和物は、それぞれ1.3%及び2.0%の吸湿による重量増加を示した。また、実施例3の1塩酸塩・2~3水和物、実施例7の1メタンスルホン酸塩・1/2~1水和物、実施例11の1フマル酸塩・1~2水和物、実施例13の1/2クエン酸塩、実施例14の1クエン酸塩、及び実施例15の1/2酒石酸塩・1/2水和物はいずれも4%以上の吸湿による重量増加があった。
水に対する溶解度
得られた化合物の37℃の水に対する溶解度を以下の条件で測定した。各試料を適当量採取し、それぞれに水を加えて37℃で4時間振とうした。上澄み液をフィルターでろ過し、必要に応じてTFA/アセトニトリル/水=(0.05:30:70)の混液で希釈して試料溶液とした。高速液体クロマトグラフィー(HPLC)を用いて検量線法にて試料溶液の濃度(mg/ml)を測定し、これを37℃の水に対する溶解度とした。
HPLC分析条件
装置:HPLC System Class-VP(島津製作所)
検出器:フォトダイオードアレイ検出器
測定波長範囲:200~370nm
固定波長:220nm
カラム:Inertsil ODS-3V(4.6mmφ×150mm)
カラム温度:40℃
移動相:A液 0.05%トリフルオロ酢酸水溶液
B液 0.05%トリフルオロ酢酸アセトニトリル溶液
A:B=70:30(Isocratic Elution)
流量:1.0mL/分
以下に、各試料の水に対する溶解度を記す。
・実施例16 :0.01mg/ml
・実施例17 :0.01mg/ml
・実施例6 : > 2mg/ml
・実施例1 : > 2mg/ml
・実施例10 :0.3mg/ml
Claims (19)
- 結晶態様の(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの塩。
- 塩酸塩、メタンスルホン酸塩、フマル酸塩、臭化水素酸塩、酒石酸塩又はクエン酸塩である請求項1記載の塩。
- 粉末X線回折スペクトルにおいて回折角度(2θ)で5.6°、16.2°、19.0°、20.1°、及び24.9°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの1塩酸塩・1水和物。
- 粉末X線回折スペクトルにおいて回折角度(2θ)で5.2°、6.8°、9.1°、10.5°及び15.7°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの1塩酸塩。
- 粉末X線回折スペクトルにおいて回折角度(2θ)で7.6°、11.5°、17.6°、18.4°、19.9°、及び23.5°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドのメタンスルホン酸塩。
- 粉末X線回折スペクトルにおいて回折角度(2θ)で8.4°、11.2°、18.0°、19.2°、21.1°及び23.1°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの1/2フマル酸塩・3/2水和物。
- 粉末X線回折スペクトルにおいて回折角度(2θ)で6.4°、12.5°、12.8°、16.5°、18.7°、21.6°、及び23.9°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの結晶。
- 粉末X線回折スペクトルにおいて回折角度(2θ)で6.3°、12.6°、13.7°、14.4°、16.7°、20.9°、及び23.5°(それぞれ±0.2°)付近にピークを示す(R)-2-{3-[1-(アセナフテン-1-イル)ピペリジン-4-イル]-2,3-ジヒドロ-2-オキソ-ベンゾイミダゾール-1-イル}-N-メチルアセトアミドの結晶。
- 請求項1~8のいずれか記載の塩又は結晶から成る医薬。
- 請求項1~8のいずれか記載の塩又は結晶を有効成分として含有する医薬組成物。
- ORL-1受容体に関与する疾患の予防及び/又は治療に用いる請求項10記載の医薬組成物。
- ORL-1受容体に関与する中枢神経系疾患の予防及び/又は治療に用いる請求項10記載の医薬組成物。
- 睡眠障害、アルコール依存症、薬物依存症、不安又はストレス障害の予防及び/又は治療に用いる請求項10記載の医薬組成物。
- ORL-1受容体に関与する疾患の予防及び/又は治療薬の製造のための、請求項1~8のいずれか記載の塩又は結晶の使用。
- ORL-1受容体に関与する中枢神経系疾患の予防及び/又は治療薬の製造のための、請求項1~8のいずれか記載の塩又は結晶の使用。
- 睡眠障害、アルコール依存症、薬物依存症、不安又はストレス障害の予防及び/又は治療薬の製造のための、請求項1~8のいずれか記載の塩又は結晶の使用。
- 請求項1~8のいずれか記載の塩又は結晶の有効量を、対象に投与することを含む、ORL-1受容体に関与する疾患の予防及び/又は治療方法。
- 請求項1~8のいずれか記載の塩又は結晶の有効量を、対象に投与することを含む、ORL-1受容体に関与する中枢神経系疾患の予防及び/又は治療方法。
- 請求項1~8のいずれか記載の塩又は結晶の有効量を、対象に投与することを含む、睡眠障害、アルコール依存症、薬物依存症、不安又はストレス障害の予防及び/又は治療方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09814576A EP2345649A4 (en) | 2008-09-16 | 2009-09-16 | BENZIMIDAZOLE COMPOUND IN CRYSTALLINE FORM AND SALT THEREOF |
| US13/119,242 US8471032B2 (en) | 2008-09-16 | 2009-09-16 | Benzimidazole compound in crystal form and salt thereof |
| CN200980136325XA CN102159565A (zh) | 2008-09-16 | 2009-09-16 | 晶体形式的苯并咪唑化合物及其盐 |
| JP2010529765A JPWO2010032731A1 (ja) | 2008-09-16 | 2009-09-16 | 結晶態様のベンゾイミダゾール化合物又はその塩 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-235846 | 2008-09-16 | ||
| JP2008235846 | 2008-09-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010032731A1 true WO2010032731A1 (ja) | 2010-03-25 |
Family
ID=42039555
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/066116 WO2010032731A1 (ja) | 2008-09-16 | 2009-09-16 | 結晶態様のベンゾイミダゾール化合物又はその塩 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8471032B2 (ja) |
| EP (1) | EP2345649A4 (ja) |
| JP (1) | JPWO2010032731A1 (ja) |
| CN (1) | CN102159565A (ja) |
| TW (1) | TW201016675A (ja) |
| WO (1) | WO2010032731A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0717161A2 (pt) * | 2006-10-16 | 2013-10-15 | Mitsubishi Tanabe Pharma Corp | Agente para profilaxia ou tratamento de abuso e dependência de substância. |
| CN112225732B (zh) * | 2019-07-15 | 2024-01-09 | 四川科瑞德制药股份有限公司 | 一种盐酸哌罗匹隆水合物晶型及其制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003082333A1 (fr) * | 2002-03-29 | 2003-10-09 | Mitsubishi Pharma Corporation | Remede contre les troubles du sommeil |
| JP2006508909A (ja) * | 2002-08-06 | 2006-03-16 | テバ ファーマシューティカル インダストリーズ リミティド | ガチフロキサシンの新規な結晶形 |
| JP2006199700A (ja) * | 2005-01-21 | 2006-08-03 | Pfizer Ltd | 結晶形態 |
| JP2008534436A (ja) * | 2005-04-07 | 2008-08-28 | 帝人ファーマ株式会社 | アミノピロリジン誘導体の結晶及びその製造方法 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2226058C (en) | 1997-01-30 | 2008-01-29 | F. Hoffmann-La Roche Ag | 8-substituted-1,3,8-triaza-spiro[4.5]decan-4-one derivatives |
| ATE276242T1 (de) | 1997-05-30 | 2004-10-15 | Banyu Pharma Co Ltd | 2-oxoimidazol-derivate |
| PT921125E (pt) | 1997-12-05 | 2002-06-28 | Hoffmann La Roche | Derivados de 1,38-triaza-espiro 4,5 decan-4-ona |
| DK1049689T3 (da) | 1998-01-19 | 2002-07-22 | Pfizer | 4-(2-keto-1-benzimidazolinyl)piperidinderivater som ORL1-receptoragonister |
| ES2194402T3 (es) | 1998-06-12 | 2003-11-16 | Hoffmann La Roche | Derivados de di - o triaza-espiro(4,5)decano.. |
| ID29137A (id) | 1998-07-27 | 2001-08-02 | Schering Corp | Ligan-ligan afinitas tinggi untuk reseptor nosiseptin orl-1 |
| US20020009486A1 (en) | 1999-11-30 | 2002-01-24 | 3M Innovative Properties Company | Therapeutic agent delivery incorporating reflective optical film |
| JP2003524634A (ja) | 1999-12-06 | 2003-08-19 | ユーロ−セルティーク,エス.エイ. | ノシセプチン受容体親和性を有するベンズイミダゾール化合物 |
| CA2396079A1 (en) * | 2000-01-07 | 2001-07-19 | Transform Pharmaceuticals, Inc. | High-throughput formation, identification, and analysis of diverse solid-forms |
| US7423153B2 (en) | 2002-05-10 | 2008-09-09 | Teva Pharmaceutical Industries Ltd. | Crystalline forms of gatifloxacin |
| US8067603B2 (en) | 2003-09-25 | 2011-11-29 | Solvay Pharmaceuticals B.V. | Benzimidazolone and quinazolinone derivatives as agonists on human ORL1 receptors |
| BRPI0717161A2 (pt) | 2006-10-16 | 2013-10-15 | Mitsubishi Tanabe Pharma Corp | Agente para profilaxia ou tratamento de abuso e dependência de substância. |
| JPWO2008102859A1 (ja) * | 2007-02-22 | 2010-05-27 | 田辺三菱製薬株式会社 | 不安障害の予防および/または治療薬 |
| JP5313865B2 (ja) | 2007-03-01 | 2013-10-09 | 田辺三菱製薬株式会社 | ベンゾイミダゾール化合物およびその医薬用途 |
-
2009
- 2009-09-15 TW TW098131012A patent/TW201016675A/zh unknown
- 2009-09-16 JP JP2010529765A patent/JPWO2010032731A1/ja active Pending
- 2009-09-16 WO PCT/JP2009/066116 patent/WO2010032731A1/ja active Application Filing
- 2009-09-16 US US13/119,242 patent/US8471032B2/en not_active Expired - Fee Related
- 2009-09-16 EP EP09814576A patent/EP2345649A4/en not_active Withdrawn
- 2009-09-16 CN CN200980136325XA patent/CN102159565A/zh active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003082333A1 (fr) * | 2002-03-29 | 2003-10-09 | Mitsubishi Pharma Corporation | Remede contre les troubles du sommeil |
| JP2006508909A (ja) * | 2002-08-06 | 2006-03-16 | テバ ファーマシューティカル インダストリーズ リミティド | ガチフロキサシンの新規な結晶形 |
| JP2006199700A (ja) * | 2005-01-21 | 2006-08-03 | Pfizer Ltd | 結晶形態 |
| JP2008534436A (ja) * | 2005-04-07 | 2008-08-28 | 帝人ファーマ株式会社 | アミノピロリジン誘導体の結晶及びその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20110178128A1 (en) | 2011-07-21 |
| EP2345649A4 (en) | 2012-07-04 |
| TW201016675A (en) | 2010-05-01 |
| JPWO2010032731A1 (ja) | 2012-02-09 |
| EP2345649A1 (en) | 2011-07-20 |
| US8471032B2 (en) | 2013-06-25 |
| CN102159565A (zh) | 2011-08-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2016253550B2 (en) | A crystalline form of (R)-7-chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2- carboxamide hydrochloride monohydrate | |
| CA2728541C (en) | Crystalline forms of thiazolidinedione compound and its manufacturing method | |
| TWI548630B (zh) | 1-(3-氰基-1-異丙基-吲哚-5-基)吡唑-4-羧酸結晶型及其製造方法 | |
| CN104470911B (zh) | 氨基甲酸酯/脲衍生物 | |
| JP2009517353A (ja) | メシル酸イマチニブのf、g、h、iおよびk結晶形 | |
| JP2022504943A (ja) | アミノピリミジン誘導体又はその塩を含む経口投与用医薬組成物 | |
| TW202308991A (zh) | 三苯化合物之固體形式 | |
| JP6610793B2 (ja) | 環状アミン誘導体の結晶及びその医薬用途 | |
| JP2023548429A (ja) | パーキンソン病、特にl-dopa誘発性ジスキネジアのための医薬品に対する感作の予防または軽減における使用のための[2-(3-フルオロ-5-メタンスルホニルフェノキシ)エチル](プロピル)アミン(メスドペタム) | |
| WO2006121104A1 (ja) | ピペリジン環を有するインドール誘導体の結晶およびその製法 | |
| WO2010032731A1 (ja) | 結晶態様のベンゾイミダゾール化合物又はその塩 | |
| WO2010111951A1 (zh) | 普拉格雷氢溴酸盐的晶体 | |
| JP5888612B2 (ja) | 縮合ピリジン化合物塩の結晶 | |
| WO2008080290A1 (fr) | Antagoniste selectif du recepteur m4 et utilisation medicale associee | |
| CN102666528B (zh) | 晶体cdc7 抑制剂盐 | |
| US9981912B2 (en) | Cocrystal of lorcaserin, preparation methods, pharmaceutical compositions and uses thereof | |
| WO2024009977A1 (ja) | 5H-ピロロ[2,3-d]ピリミジン-6(7H)-オン及びその塩体の結晶 | |
| JP4435090B2 (ja) | スルホンアミド含有インドール化合物の結晶およびその製造方法 | |
| WO2015107545A1 (en) | Water soluble salts of dasatinib hydrate | |
| TW201904972A (zh) | 一種咪唑并異吲哚類衍生物游離鹼的晶型及其製備方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980136325.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09814576 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010529765 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13119242 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009814576 Country of ref document: EP |