WO2009100936A2 - Neue aromatische fluorglykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung - Google Patents
Neue aromatische fluorglykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung Download PDFInfo
- Publication number
- WO2009100936A2 WO2009100936A2 PCT/EP2009/001042 EP2009001042W WO2009100936A2 WO 2009100936 A2 WO2009100936 A2 WO 2009100936A2 EP 2009001042 W EP2009001042 W EP 2009001042W WO 2009100936 A2 WO2009100936 A2 WO 2009100936A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- combination
- alkyl
- administered
- Prior art date
Links
- JQSHBVHOMNKWFT-DTORHVGOSA-N C([C@H](CNC1)c2c3)[C@@H]1c2cc1c3nccn1 Chemical compound C([C@H](CNC1)c2c3)[C@@H]1c2cc1c3nccn1 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 1
- YODXTMJXCOLQBV-UHFFFAOYSA-N C/C=[O]\c1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2F)cc2)c2Cl)cc1 Chemical compound C/C=[O]\c1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2F)cc2)c2Cl)cc1 YODXTMJXCOLQBV-UHFFFAOYSA-N 0.000 description 1
- UFVCMTVHRPQAFE-UHFFFAOYSA-N CC(C)Oc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2F)cc2)c2Cl)cc1 Chemical compound CC(C)Oc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2F)cc2)c2Cl)cc1 UFVCMTVHRPQAFE-UHFFFAOYSA-N 0.000 description 1
- JEQQIGMUSXKCMX-UHFFFAOYSA-N CC(CC1Cc(cc2)ccc2OC)(CC=C1C(F)(F)F)C(C(C1O)O)OC(CO)C1(F)F Chemical compound CC(CC1Cc(cc2)ccc2OC)(CC=C1C(F)(F)F)C(C(C1O)O)OC(CO)C1(F)F JEQQIGMUSXKCMX-UHFFFAOYSA-N 0.000 description 1
- YZQLWPMZQVHJED-UHFFFAOYSA-N CCC(CC)CC1(CCCCC1)C(Nc(cccc1)c1SC(C(C)C)=O)=O Chemical compound CCC(CC)CC1(CCCCC1)C(Nc(cccc1)c1SC(C(C)C)=O)=O YZQLWPMZQVHJED-UHFFFAOYSA-N 0.000 description 1
- UNAZAADNBYXMIV-UHFFFAOYSA-N CCNC(CC1)(CCN1c1ncnc2c1nc(-c1ccccc1Cl)[n]2-c(cc1)ccc1Cl)C(N)=O Chemical compound CCNC(CC1)(CCN1c1ncnc2c1nc(-c1ccccc1Cl)[n]2-c(cc1)ccc1Cl)C(N)=O UNAZAADNBYXMIV-UHFFFAOYSA-N 0.000 description 1
- FDJBQOLWDVWIRS-UHFFFAOYSA-N CCOC(C(C)(C)NP(c1ccc(-c2c(CC(C)C)[s]c(N)n2)[o]1)(NC(C)(C)C(OCC)=O)=O)=O Chemical compound CCOC(C(C)(C)NP(c1ccc(-c2c(CC(C)C)[s]c(N)n2)[o]1)(NC(C)(C)C(OCC)=O)=O)=O FDJBQOLWDVWIRS-UHFFFAOYSA-N 0.000 description 1
- JBAKDERJNXLNTL-UHFFFAOYSA-N CCOC(OCC(C(C(C1O)O)(F)F)OC1c(cc1)cc(Cc(cc2)ccc2OC)c1F)=O Chemical compound CCOC(OCC(C(C(C1O)O)(F)F)OC1c(cc1)cc(Cc(cc2)ccc2OC)c1F)=O JBAKDERJNXLNTL-UHFFFAOYSA-N 0.000 description 1
- WROIOYZGAZVAIB-UHFFFAOYSA-N CCOC(OCC(C(C(C1O)O)(F)F)OC1c(cc1)cc(Cc(cc2)ccc2OC)c1OC)=O Chemical compound CCOC(OCC(C(C(C1O)O)(F)F)OC1c(cc1)cc(Cc(cc2)ccc2OC)c1OC)=O WROIOYZGAZVAIB-UHFFFAOYSA-N 0.000 description 1
- FLMGQEQIUBMTMO-UHFFFAOYSA-N CCOC(OCC(C(C(C1O)O)(F)F)OC1c(cc1)cc(Cc(cc2)ccc2OCC)c1Cl)=O Chemical compound CCOC(OCC(C(C(C1O)O)(F)F)OC1c(cc1)cc(Cc(cc2)ccc2OCC)c1Cl)=O FLMGQEQIUBMTMO-UHFFFAOYSA-N 0.000 description 1
- WOZAWWJNQBSRJP-UHFFFAOYSA-N CCOC(OCC(C(C(C1O)O)F)OC1c(cc1)cc(Cc(cc2)ccc2OC)c1F)=O Chemical compound CCOC(OCC(C(C(C1O)O)F)OC1c(cc1)cc(Cc(cc2)ccc2OC)c1F)=O WOZAWWJNQBSRJP-UHFFFAOYSA-N 0.000 description 1
- BLCPSCDMCIPROD-UHFFFAOYSA-N CCOC(OCC(C(C(C1O)O)F)OC1c(cc1)cc(Cc(cc2)ccc2OCC)c1Cl)=O Chemical compound CCOC(OCC(C(C(C1O)O)F)OC1c(cc1)cc(Cc(cc2)ccc2OCC)c1Cl)=O BLCPSCDMCIPROD-UHFFFAOYSA-N 0.000 description 1
- MMFKJQVYHMEMKH-UHFFFAOYSA-N CCOC(OCC(C(OC1CO)(F)F)OC1c1cc(Cc(cc2)ccc2OC)c(C(F)(F)F)cc1)=O Chemical compound CCOC(OCC(C(OC1CO)(F)F)OC1c1cc(Cc(cc2)ccc2OC)c(C(F)(F)F)cc1)=O MMFKJQVYHMEMKH-UHFFFAOYSA-N 0.000 description 1
- OBOGDEXPUZFXRW-UHFFFAOYSA-N CCOc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2(F)F)cc2)c2Cl)cc1 Chemical compound CCOc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2(F)F)cc2)c2Cl)cc1 OBOGDEXPUZFXRW-UHFFFAOYSA-N 0.000 description 1
- HSBUQOGDEDIUAE-UHFFFAOYSA-N CCc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2F)cc2)c2Cl)[s]1 Chemical compound CCc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2F)cc2)c2Cl)[s]1 HSBUQOGDEDIUAE-UHFFFAOYSA-N 0.000 description 1
- QPNWHIMAZBIEMR-UHFFFAOYSA-N CCc1ccc(Cc2cc(C(C(C3O)O)OC(CO)C3O)ccc2Cl)cc1 Chemical compound CCc1ccc(Cc2cc(C(C(C3O)O)OC(CO)C3O)ccc2Cl)cc1 QPNWHIMAZBIEMR-UHFFFAOYSA-N 0.000 description 1
- SCQOFOZWDIIPBO-UHFFFAOYSA-N CCc1ccc(Cc2cc(C(C(C3O)O)OC4C3OC(c3ccccc3)OC4)ccc2Cl)cc1 Chemical compound CCc1ccc(Cc2cc(C(C(C3O)O)OC4C3OC(c3ccccc3)OC4)ccc2Cl)cc1 SCQOFOZWDIIPBO-UHFFFAOYSA-N 0.000 description 1
- YGARPRMHNRAXQN-UHFFFAOYSA-N COc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2(F)F)cc2)c2OC)cc1 Chemical compound COc1ccc(Cc(cc(C(C(C2O)O)OC(CO)C2(F)F)cc2)c2OC)cc1 YGARPRMHNRAXQN-UHFFFAOYSA-N 0.000 description 1
- DFYXNYKWIDKXJO-UHFFFAOYSA-N COc1ccc(Cc2cc(Br)ccc2OC(F)(F)F)cc1 Chemical compound COc1ccc(Cc2cc(Br)ccc2OC(F)(F)F)cc1 DFYXNYKWIDKXJO-UHFFFAOYSA-N 0.000 description 1
- JHBZPLPKKSGANJ-UHFFFAOYSA-N COc1ncc(Cc(cc(cc2)Br)c2Cl)cc1 Chemical compound COc1ncc(Cc(cc(cc2)Br)c2Cl)cc1 JHBZPLPKKSGANJ-UHFFFAOYSA-N 0.000 description 1
- NSMXQKNUPPXBRG-SECBINFHSA-N C[C@H](CCCCN(C(c1c(N2C)nc[n]1C)=O)C2=O)O Chemical compound C[C@H](CCCCN(C(c1c(N2C)nc[n]1C)=O)C2=O)O NSMXQKNUPPXBRG-SECBINFHSA-N 0.000 description 1
- VZOVKVCGZDERPK-TXEJJXNPSA-N N#C[C@@H](CC[C@H]1C#N)N1C(CNC1(CO)CCCC1)=O Chemical compound N#C[C@@H](CC[C@H]1C#N)N1C(CNC1(CO)CCCC1)=O VZOVKVCGZDERPK-TXEJJXNPSA-N 0.000 description 1
- TVMGXKGTVIUPCH-UXUBKHOLSA-N OC([C@H](C1OCc2ccccc2)OCc2ccccc2)OC(COCc2ccccc2)C1F Chemical compound OC([C@H](C1OCc2ccccc2)OCc2ccccc2)OC(COCc2ccccc2)C1F TVMGXKGTVIUPCH-UXUBKHOLSA-N 0.000 description 1
- RTIMCJWREDZAPM-UHFFFAOYSA-N OC(c(cc1)ccc1OC(F)(F)F)c1cc(Br)ccc1Cl Chemical compound OC(c(cc1)ccc1OC(F)(F)F)c1cc(Br)ccc1Cl RTIMCJWREDZAPM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
Definitions
- Novel aromatic fluoroglycoside derivatives Novel aromatic fluoroglycoside derivatives, pharmaceutical compositions containing them and their use
- the invention relates to substituted aromatic fluoroglycoside derivatives, their physiologically acceptable salts and physiologically functional derivatives.
- Glucopyranosyloxy-pyrazoles from Kissei, Bristol-Myers Squibb and Ajinomoto (WO 02068440, WO 02068439, WO 0236602, WO 01016147, WO 02053573, WO 03020737, WO 03090783, WO 04014932, WO 04019958 and WO 04018491) - Bristol O-glycoside benzamides -Myers Squibb (WO 0174835 and WO
- aromatic fluoroglycoside derivatives selectively increase the effect on SGLT 2. These compounds are therefore particularly suitable for the prevention and treatment of diabetes type 1 and type 2.
- the invention therefore relates to compounds of the formula I,
- R1 and R2 are F or R1 H and R2 F;
- R3 hydrogen, F, Cl, Br, CF 3, OCF 3, CN, methyl, ethyl, methoxy, ethoxy, cyclopropyl, CH 2 -cyclopropyl;
- R4, R5, R6, R7 independently of one another are hydrogen, F, Cl 1 Br, J, OH, CF 3 , NO 2 , COOH, COO (C 1 -C 6) -alkyl, CO (C 1 -C 4 ) -alkyl, CONH 2 , CONH (C 1 -
- C 6 ) alkyl CON [(C 1 -C 6 ) alkyl] 2, (C r C 6 ) alkyl, (C 2 -C 6 ) alkenyl, (C 2 -C 6 ) alkynyl, O - (C r C6) alkyl, HO- (C r C6) -alkylene, (Ci-C 6) -alkylene-O- (Ci-C 6) - alkyl, where in the alkyl, alkenyl, alkynyl or O-alkyl radicals, several, or all hydrogen (s) may be replaced by fluorine; SO 2 -NH 2 , SO 2 NH (C 1 -Ce) alkyl, SO 2 N [(C r C 6 ) alkyl] 2 , S (C 1 -Ce) alkyl,
- radicals or substituents can occur several times in the compounds of the formula I, they may all independently of one another have the meanings indicated and be identical or different.
- Ra is -COO- (C 1 -C 6 ) -alkyl
- R1 and R2 are F.
- R3 is hydrogen, F 1 is Cl, Br, CF 3 , OCF 3 , methyl, methoxy, cyclopropyl, CH 2 -cyclopropyl; means.
- R 3 is F 1 Cl 1 Br 1 CF 3 , OCF 3 , methyl, methoxy;
- R 4 an R 4, R 5, R 6 or R 7 F 1 Cl 1 CF 3 , OH 1 COOH 1 (C 1 -C 6 ) -alkyl, (C 2 -C 6 ) -
- R 4, R 5, R 6 or R 7 is F, Cl, CF 3 , OH, (C 1 -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl, O- (C 1 -)
- R4 is Cl, CF 3, OCF 3, ethyl, methoxy, ethoxy;
- R5, R6, R7 are hydrogen.
- Another preferred embodiment are compounds of the formula I in which Cyc1 means.
- alkyl radicals in the substituents R3, R4, R5, R6 and R7 can be both straight-chain and branched.
- Halogen is understood as meaning F, Cl, Br, I, preferably F and Cl.
- the invention relates to compounds of formula I 1 in the form of their tautomers, racemates, racemic mixtures and pure enantiomers and to their
- Diastereomers and mixtures thereof encompasses all these isomeric and optionally tautomeric forms of the compounds of the formula I. These isomeric forms, although not expressly (in part) described expressis verbis, can be obtained by known methods.
- Suitable pharmaceutically acceptable salts are particularly suitable for medical applications because of their higher water solubility compared to the starting or basic compounds. These salts must have a pharmaceutically acceptable anion or cation.
- Suitable pharmaceutically acceptable acid addition salts of the compounds according to the invention are salts of inorganic acids, such as hydrochloric acid, hydrobromic acid, phosphoric acid, metaphosphoric acid, nitric acid and sulfuric acid, and also organic acids, such as, for example, acetic acid, benzenesulfonic, benzoic, citric, ethanesulfonic, fumaric acid.
- Suitable pharmaceutically acceptable basic salts are ammonium _ Salts, Alkalimetallsa ze we atr environmentally un a iumsa ze un ra & ei c V w ⁇ e magnesium and calcium salts) and salts of trometamol (2-amino-2-hydroxymethyl-1, 3-propanediol), diethanolamine, lysine or ethylenediamine.
- Salts with a non-pharmaceutically acceptable anion are also within the scope of the invention as useful intermediates for the preparation or purification of pharmaceutically acceptable salts and / or for use in non-therapeutic, for example, in vitro applications.
- physiologically functional derivative refers to any physiologically acceptable derivative of a compound of formula I 1, for example, an ester capable of (directly or indirectly), when administered to a mammal, such as a human Formula I or to form an active metabolite thereof.
- the physiologically functional derivatives also include prodrugs of the compounds according to the invention, as described, for example, in H. Okada et al., Chem. Pharm. Bull. 1994, 42, 57-61. Such prodrugs can be metabolized in vivo to a compound of the invention. These prodrugs may or may not be effective.
- the compounds of the invention may also be in various polymorphic forms, e.g. as amorphous and crystalline polymorphic forms. All polymorphic forms of the compounds of the invention are within the scope of the invention and are a further aspect of the invention.
- This invention further relates to the use of compounds of formula I and their pharmaceutical compositions for inhibiting SGLT 2 (sodium dependent glucose transporter 2).
- SGLT2 is responsible for the reconstitution of D-glucose from the glomerular filtrate of the kidney (Wright, Wright et al., Am J. Physiol 2001, 263: F459-F465.).
- inhibitors of SGLT2 are useful for the treatment, control and prophylaxis of metabolic diseases, particularly diabetes mellitus.
- the compounds of the invention are also characterized by a particularly high selectivity for SGLT2 over the SGLT1 receptor. This selectivity is increased again in the di-fluorine compounds.
- Compounds according to the invention which are esterified on the glucose unit act as prodrugs. They show in in vitro test procedures bad IC50 values for SGLT2. Nevertheless, they are selective SGLT2 inhibitors, as evidenced by the glucose excretion data of the in vivo tests.
- the compounds of the formula I are distinguished by favorable effects on the glucose metabolism, in particular they lower the blood sugar level and are suitable for the treatment of type 1 and type 2 diabetes.
- the compounds can therefore be used alone or in combination with other blood sugar-lowering agents (antidiabetics).
- the compounds of the formula I are also suitable for the prevention and treatment of diabetic late damage, such as nephropathy, retinopathy, neuropathy and syndrome X, obesity, myocardial infarction, peripheral arterial occlusive diseases, thrombosis, arteriosclerosis, inflammation, immune diseases, autoimmune diseases, such as AIDS, asthma, osteoporosis, cancer, psoriasis, Alzheimer's disease, schizophrenia and infectious diseases, preferred are the treatment of type 1 and type 2 diabetes as well as for the prevention and treatment of diabetic late damage, syndrome X and obesity.
- diabetic late damage such as nephropathy, retinopathy, neuropathy and syndrome X, obesity, myocardial infarction, peripheral arterial occlusive diseases, thrombosis, arteriosclerosis, inflammation, immune diseases, autoimmune diseases, such as AIDS, asthma, osteoporosis, cancer, psoriasis, Alzheimer's disease, schizophrenia and infectious diseases
- diabetic late damage such as nephropathy, reti
- the amount of a compound of Formula I required to achieve the desired biological effect is dependent upon a number of factors, e.g. the chosen specific compound, the intended
- the daily dose is in the range of 0.3 mg to 100 mg (typically 3 mg and 50 mg) per day per kilogram of body weight, e.g. 3-10 mg / kg / day.
- Orally administrable dosage unit formulations such as tablets or capsules, may contain, for example, from 1.0 to 1000 mg, more typically from 10 to 600 mg.
- the compounds according to formula I can themselves be used as compound, but they are preferably present with a compatible carrier in the form of a pharmaceutical composition.
- the carrier must of course be compatible in the sense that it is compatible with the other ingredients of the composition and is not harmful to the patient.
- the carrier may be a solid or a liquid, or both, and is preferably formulated with the compound as a single dose, for example, as a tablet, which may contain from 0.05% to 95% by weight of the active ingredient.
- Other pharmaceutically active substances may likewise be present, including further compounds of the formula I.
- the pharmaceutical compositions according to the invention can be prepared by one of the known pharmaceutical methods, which essentially consist in mixing the ingredients with pharmacologically acceptable carriers and / or excipients ,
- compositions according to the invention are those which are suitable for oral, rectal, peroral (eg sublingual) and administration, although the most suitable mode of administration in each individual case depends on the nature and severity of the condition to be treated and on the nature of the particular compound of formula I used is. Also coated formulations and sugar-coated
- Retard formulations are within the scope of the invention. Preference is given to acid and enteric formulations. Suitable enteric coatings include cellulose acetate phthalate, polyvinyl acetate phthalate, Hydroxypropylmethylcellulosephthalat and anionic polymers of methacrylic acid and methyl methacrylate.
- Suitable pharmaceutical preparations for oral administration may be in separate units, such as capsules, cachets,
- Lozenges or tablets each containing a certain amount of the compound of formula I; as a powder or granules; as a solution or suspension in an aqueous or non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion.
- these compositions may be prepared by any suitable pharmaceutical method comprising a step of contacting the active ingredient and the carrier (which may consist of one or more additional ingredients).
- the compositions are prepared by uniformly and homogeneously mixing the active ingredient with a liquid and / or finely divided solid carrier, after which the product is molded, if necessary.
- a tablet can be made by compressing or molding a powder or granules of the compound, optionally with one or more additional ingredients.
- Pressed tablets may be prepared by tableting the compound in free-flowing form, such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, and / or one or more surface active / dispersing agents in a suitable machine.
- Molded tablets may be prepared by shaping the powdered compound moistened with an inert liquid diluent in a suitable machine.
- compositions suitable for peroral (sublingual) administration include lozenges containing a compound of Formula I with a flavor, usually sucrose and gum arabic or tragacanth, and lozenges containing the compound in an inert base such as gelatin and glycerol or sucrose and gum arabic.
- Suitable pharmaceutical compositions for rectal administration are preferably as single dose suppositories. These can be prepared by reacting a compound according to formula I with one or more conventional solid carriers, such as cocoa butter, mixes and brings the resulting mixture in the form.
- the compounds of the invention may be administered alone or in combination with one or more other pharmacologically active substances which, for example, have beneficial effects on metabolic disorders or diseases frequently associated therewith. They can be combined with the compounds of the formula I according to the invention in particular for the synergistic effect improvement.
- the administration of the active ingredient combination can be carried out either by separate administration of the active ingredients to the patient or in the form of combination preparations in which several active ingredients are present in a pharmaceutical preparation. If the administration of the active ingredients by separate administration of the active ingredients, so this can be done simultaneously or sequentially.
- Antidiabetics include insulin and insulin derivatives, such as Lantus ® (see www.lantus.com) or HMR 1964 or Levemir® (insulin detemir), Humalog (R) (insulin lispro), Humulin (R), VIAject TM, SuliXen (R) or those as described in WO2005005477 (Novo Nordisk), fast-acting insulins (see US 6,221, 633), inhalable insulins such.
- Lantus ® see www.lantus.com
- HMR 1964 Levemir® (insulin detemir), Humalog (R) (insulin lispro), Humulin (R), VIAject TM, SuliXen (R) or those as described in WO2005005477 (Novo Nordisk), fast-acting insulins (see US 6,221, 633), inhalable insulins such.
- Levemir® insulin detemir
- Humalog (R) insulin lispro
- Humulin R
- IN-105 Nobex
- Oral-lyn TM Geneex Biotechnology
- Technosphere (R) insulin MannKind
- Cobalamin TM oral insulin or insulins as described in WO2007128815, WO2007128817, WO2008034881, WO2008049711 or insulins
- GLP-1 derivatives and GLP-1 agonists such as exenatides or special preparations thereof, as described, for example, in WO2008061355, liraglutide, Taspoglutide (R-1583), albiglutide, lixisenatide or those described in WO 98/08871, WO2005027978, WO2006037811 WO2006037810 of Novo Nordisk A / S, in WO 01/04156 of Zealand or in WO 00/34331 of Beaufour-Ipsen, Pramlintide acetate (Symlin; Amylin Pharmaceuticals), AVE-0010, B
- Antidiabetic agents also include agonists of the glucose-dependent insulinotropic polypeptide (GIP) receptor as described e.g. in WO2006121860 are described.
- GIP glucose-dependent insulinotropic polypeptide
- Antidiabetics also include the glucose-dependent insulinotropic polypeptide (GIP) as well as analogous compounds as described e.g. in WO2008021560 are described.
- GIP glucose-dependent insulinotropic polypeptide
- Antidiabetics also include analogs and derivatives of fibroblast growth factor 21 (FGF-21, fibroblast growth factor 21).
- FGF-21 fibroblast growth factor 21
- the orally active hypoglycemic agents preferably comprise
- GAT glutamine-fructose 6-phosphate amidotransferase
- Potassium channel opener e.g. Pinacidil, cromakalim, diazoxide or those like them
- DPP-IV dipeptidyl peptidase-IV
- Insulin sensitizers inhibitors of liver enzymes involved in the stimulation of gluconeogenesis and / or
- Modulators of sodium-dependent glucose transporters 1 or 2 (SGLT1, SGLT2), inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 (11 ⁇ -HSD1),
- PTP-1 B protein tyrosine phosphatase 1 B
- Nicotinic receptor agonists
- Inhibitors of acetyl-CoA carboxylase ACC1 and / or ACC2
- Inhibitors of G - eta Inhibitors of G - eta.
- lipid metabolism-altering compounds such as antihyperlipidemic agents and antilipidemic agents.
- HMGCoA reductase inhibitors HMGCoA reductase inhibitors, farnesoid X receptor (FXR) modulators,
- SST5 receptor Antagonists of the somatostatin 5 receptor
- the compound of the formula I is administered in combination with insulin.
- the compound of formula I is administered in combination with an agent that acts on the ATP-dependent potassium channel of beta cells, e.g. Sulfonylureas, e.g. Tolbutamide, glibenclamide, glipizide, gliclazide or glimepiride.
- an agent that acts on the ATP-dependent potassium channel of beta cells e.g. Sulfonylureas, e.g. Tolbutamide, glibenclamide, glipizide, gliclazide or glimepiride.
- the compound of formula I is administered in combination with a tablet containing both glimepride which is rapidly released and contains metformin which is released over a prolonged period of time (e.g., as described in US2007264331, WO2008050987, WO2008062273).
- the compound of formula I is used in combination with a biguanide, e.g. Metformin, administered.
- a biguanide e.g. Metformin
- the compound of formula I is administered in combination with a meglitinide such as repaglinide, nateglinide or mitiglinide.
- a meglitinide such as repaglinide, nateglinide or mitiglinide.
- the compound of formula I is administered with a combination of mitiglinides with a glitazone, eg, pioglitazone hydrochloride.
- the compound of formula I is administered with a combination of mitiglinides with an alpha-glucosidase inhibitor.
- the compound of the formula I is administered in combination with antidiabetic compounds, as described in WO2007095462, WO2007101060, WO2007105650.
- the compound of the formula I is administered in combination with antihypoglycemic compounds, as described in WO2007137008, WO2008020607.
- the compound of formula I is used in combination with a thiazolidinedione, e.g. Troglitazone, ciglitazone, pioglitazone, rosiglitazone or those described in WO 97/41097 by Dr. med. Reddy's Research Foundation disclosed compounds, particularly 5 - [[4 - [(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy] phenyl] methyl] -2,4-thiazolidinedione.
- the compound of formula I is used in combination with a PPAR gamma agonist, such as rosiglitazone, pioglitazone, JTT-501, GI 262570, R-483, CS-011 (rivoglitazone), DRL-17564, DRF-2593 (Balaglitazone), INT-131, T-2384 or those as described in WO2005086904, WO2007060992, WO2007100027, WO2007103252, WO2007122970, WO2007138485, WO2008006319, WO2008006969, WO2008010238, WO2008017398, WO2008028188, WO2008066356, WO2008084303, WO2008089461-WO2008089464, WO2008093639, WO2008096769 , WO2008096820, WO2008096829, US2008194617, WO2008099944, WO2008108602, WO2008
- the compound of formula I is administered in combination with Tandemact TM, a solid combination of pioglitazone with glimepride.
- the compound of the formula I in combination with a solid combination of pioglitazone hydrochloride with an angiotensin II agonist, e.g. TAK-536 administered.
- the compound of the formula I is administered in combination with a PPAR alpha agonist or mixed PPAR alpha / PPAR delta agonists, such as e.g. GW9578, GW-590735, K-111, LY-674, KRP-101, DRF-10945, LY-518674, CP-900691, BMS-687453, BMS-711939 or those as described in WO2001040207, WO2002096894, WO2005097076, WO2007056771, WO2007087448, WO2007089667, WO2007089557, WO2007102515, WO2007103252, JP2007246474, WO2007118963, WO2007118964, WO2007126043, WO2008006043, WO2008006044, WO2008012470, WO2008035359, WO2008087365, WO2008087366, WO2008087367, WO2008117982.
- the compound of formula I is used in combination with a mixed PPAR alpha / gamma agonist, e.g.
- the compound of the formula I is administered in combination with a PPAR delta agonist such as GW-501516 or as described in WO2006059744, WO2006084176, WO2006029699, WO200
- the compound of formula I is used in combination with a pan-SPPARM (selective PPAR modulator alpha, gamma, delta), e.g. GFT-505 or those as described in WO2008035359 administered.
- a pan-SPPARM selective PPAR modulator alpha, gamma, delta
- the compound of formula I is administered in combination with metaglidases or with MBX-2044 or other partial PPAR gamma agonist / antagonist.
- the compound of formula I is administered in combination with an ⁇ -glucosidase inhibitor, e.g. Miglitol or acarbose or those as described e.g. in WO2007114532, WO2007140230, US2007287674, US2008103201, WO2008065796, WO2008082017.
- an ⁇ -glucosidase inhibitor e.g. Miglitol or acarbose or those as described e.g. in WO2007114532, WO2007140230, US2007287674, US2008103201, WO2008065796, WO2008082017.
- the compound of formula I is used in combination with a glycogen phosphorylase inhibitor, e.g. PSN-357 or FR-258900 or those as described in WO2003084922, WO2004007455, WO2005073229-31, WO2005067932, WO2008062739, WO2008099000, WO2008113760.
- a glycogen phosphorylase inhibitor e.g. PSN-357 or FR-258900 or those as described in WO2003084922, WO2004007455, WO2005073229-31, WO2005067932, WO2008062739, WO2008099000, WO2008113760.
- the compound of the formula I is used in combination with glucagon receptor antagonists, such as eg A-770077 or NNC-25-2504 or as in WO2004100875, WO2005065680, WO2006086488, WO2007047177, WO2007106181, WO2007111864, WO2007120270, WO2007120284,
- glucagon receptor antagonists such as eg A-770077 or NNC-25-2504 or as in WO2004100875, WO2005065680, WO2006086488, WO2007047177, WO2007106181, WO2007111864, WO2007120270, WO2007120284,
- the compound of formula I is used in combination with an antisense compound, e.g. ISIS-325568, which inhibits the production of the glucagon receptor.
- an antisense compound e.g. ISIS-325568
- the compound of the formula I in combination with activators of glucokinase such as. LY-2121260 (WO2004063179), PSN-105, PSN-110, GKA-50, or those as described e.g. B.
- the compound of the formula I in combination with an inhibitor of gluconeogenesis as z.
- an inhibitor of gluconeogenesis as described in FR-225654, WO2008053446.
- the compound of formula I is used in combination with inhibitors of fructose-1, 6-bisphosphatase (FBPase) such as MB-07729, CS-917 (MB-06322) or MB-07803 or those as described in WO2006023), WO2006104030, WO2007014619, WO2007137962, WO2008019309, WO2008037628.
- FBPase 6-bisphosphatase
- the compound of the formula I in combination with modulators of the glucose transporter-4 (GLUT4), such as. KST-48 (D.O. Lee et al .: Arzneim.-Research, Drug Res. 54 (12), 835 (2004)).
- the compound of formula I is used in combination with inhibitors of glutamine-fructose 6-phosphate amidotransferase (GFAT), as described e.g. As described in WO2004101528 administered.
- GFAT glutamine-fructose 6-phosphate amidotransferase
- the compound of formula I in combination with inhibitors of dipeptidyl peptidase-IV in combination with inhibitors of dipeptidyl peptidase-IV (DPP-IV), such as. Vildagliptin (LAF-237), sitagliptin (MK-0431), sitagliptin phosphate, saxagliptin ((BMS-477118), GSK-IV
- DPP-IV dipeptidyl peptidase-IV
- the compound of formula I is administered in combination with Janumet TM, a solid combination of sitagliptin phosphate with metformin hydrochloride.
- the compound of formula I is administered in combination with Eucreas (R) , a solid combination of vildagliptin with metformin hydrochloride.
- the compound of formula I is administered in combination with a solid combination of alogliptin benzoate with pioglitazone.
- the compound of formula I is administered in combination with a solid combination of a salt of sitagliptin with metformin hydrochloride.
- the compound of formula I is administered in combination with a combination of a DPP-IV inhibitor with omega-3 or omega-3 fatty acid esters, e.g. in WO2007128801, administered.
- the compound of formula I is administered in combination with a solid combination of a salt of sitagliptin with metformin hydrochloride.
- the compound of formula I in combination with an insulin secretion enhancing substance such as. KCP-265 (WO2003097064) or those as described in WO2007026761, WO2008045484, US2008194617.
- the compound of the formula I in combination with agonists of the glucose-dependent insulinotropic receptor (GDIR) such. B. APD-668 administered.
- the compound of formula I is used in combination with an ATP citrate lyase inhibitor, e.g. SB-204990 administered.
- an ATP citrate lyase inhibitor e.g. SB-204990 administered.
- the compound of formula I is used in combination with modulators of the sodium-dependent glucose transporter 1 or 2 (SGLT1, SGLT2), e.g. KGA-2727, T-1095, SGL-0010, AVE 2268, SAR 7226, SGL-5083, SGL-5085, SGL-5094, ISIS-388626, sergliflozin or dapagliflozin, or as described e.g. B.
- modulators of the sodium-dependent glucose transporter 1 or 2 SGLT1, SGLT2
- SGLT1 sodium-dependent glucose transporter 1 or 2
- the compound of the formula I in combination with inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 (11 ß-HSD1), such.
- 11 ß-HSD1 11-beta-hydroxysteroid dehydrogenase-1
- WO2004041264 WO2004037251, WO2004056744, WO2004058730, WO2004065351, WO2004089367, WO2004089380, WO2004089470-71, WO2004089896, WO2005016877, WO2005063247, WO2005097759, WO2006010546, WO2006012227, WO2006012173, WO2006017542, WO2006034804, WO2006040329, WO2006051662, WO2006048750, WO2006049952, WO2006048331, WO2006050908, WO2006024627, WO2006040329, WO2006066109, WO2006074244, WO2006078006, WO2006106423, WO2006132436, WO2006134481, WO2006134467, WO2006135795, WO2006136502, WO2006138508, WO2006138695, WO2006133926,
- the compound of the formula I is used in combination with inhibitors of protein tyrosine phosphatase-1B (PTP-1B), as described, for.
- PTP-1B protein tyrosine phosphatase-1B
- WO200119830-31 WO200117516, WO2004506446, WO2005012295, WO2005116003, WO2005116003, WO2006007959, DE 10 2004 060542.4, WO2007009911, WO2007028145, WO2007067612-615, WO20 ⁇ / U ⁇ 1 fi ⁇ , WO2007115058, US2008004325, WO2008033455, WO2008033931, WO2008033932, WO2008033934, WO2008089581.
- PTP-1B protein tyrosine phosphatase-1B
- GPR109A HM74A receptor agonists; NAR agonists (nicotinic acid receptor agonists)
- Nicotinic acid or "extended release niacin” in association with MK-0524A (laropiprant) or MK-0524 or such compounds as described in WO2004041274, WO2006045565, WO2006045564, WO2006069242, WO2006085108, WO2006085112, WO2006085113, WO2006124490, WO2006113150, WO2007017261, WO2007017262, WO2007017265 , WO2007015744, WO2007027532, WO2007092364, WO2007120575, WO2007134986, WO2007150025, WO2007150026, WO2008016968, WO2008051403, WO2008086949, WO2008091338, WO2008097535, WO20080994
- the compound of formula I is administered in combination with a solid combination of niacin with simvastatin.
- the compound of formula I is administered in combination with nicotinic acid or extended release niacin in association with MK-0524A (laropiprant).
- the compound of the formula I is administered in combination with nicotinic acid or extended release niacin in conjunction with MK-0524A (laropiprant) and with simvastatin.
- the compound of formula I is administered in combination with nicotinic acid or another nicotinic acid receptor agonist and a prostaglandin DP receptor antagonist, such as those described in WO2008039882.
- a prostaglandin DP receptor antagonist such as those described in WO2008039882.
- the compound of formula I is used in combination with modulators of GPR40, as described, e.g. in WO2007013689, WO2007033002, WO2007106469, US2007265332, WO2007123225, WO2007131619, WO2007131620, WO2007131621, US2007265332, WO2007131622, WO2007136572, WO2008001931, WO2008030520, WO2008030618, WO2008054674, WO2008054675, WO2008066097, US2008176912.
- the compound of formula I is used in combination with modulators of GPR119 (G protein-coupled glucose-dependent insulinotropic receptor), such as e.g. PSN-119-1, PSN-821, PSN-119-2, MBX-2982 or such.
- GPR119 G protein-coupled glucose-dependent insulinotropic receptor
- the compound of formula I is used in combination with modulators of the GPR120, e.g. in EP1688138, WO2008066131,
- the compound of the formula I in combination with inhibitors of hormone-sensitive lipase (HSL) and / or phospholipases, such.
- HSL hormone-sensitive lipase
- WO2007042178 WO2007119837, WO2008122352, WO2008122357.
- the compound of the formula I in combination with inhibitors of endothelial lipase, such as.
- the compound of formula I is used in combination with a phospholipase A2 inhibitor, e.g. Darapladib or A-002 or those as described in WO2008048866, WO20080488867 administered.
- a phospholipase A2 inhibitor e.g. Darapladib or A-002 or those as described in WO2008048866, WO20080488867 administered.
- the compound of the formula I is administered in combination with myricitrin, a lipase inhibitor (WO2007119827).
- the compound of the formula I in combination with an inhibitor of glycogen synthase kinase-3 beta (GSK-3 beta), such as. B. in US2005222220, WO2005085230, WO2005111018, WO2003078403, WO2004022544, WO2003106410, WO2005058908, US2005038023, WO2005009997, US2005026984, WO2005000836, WO2004106343, EP1460075, WO2004014910, WO2003076442, WO2005087727, WO2004046117, WO2007073117, WO2007083978, WO2007120102, WO2007122634, WO2007125109, WO2007125110, US2007281949 , WO2008002244, WO2008002245, WO2008016123, WO2008023239, WO2008044700, WO2008056266, WO2008057940, WO2008077138, EP193919
- the compound of formula I is used in combination with an inhibitor of phosphoenolpyruvate carboxykinase (PEPCK), e.g. such as described in WO2004074288 administered.
- PPCK phosphoenolpyruvate carboxykinase
- the compound of formula I is administered in combination with an inhibitor of phosphoinositide kinase-3 (PI3K), such as those described in WO2008027584, WO2008070150, WO2008125833, WO2008125835, WO2008125839.
- PI3K phosphoinositide kinase-3
- the compound of the formula I is used in combination with a serum / glucocorticoid regulated kinase (SGK) inhibitor, such as, e.g. As described in WO2006072354, WO2007093264, WO2008009335, WO2008086854.
- SGK serum / glucocorticoid regulated kinase
- the compound of formula I in combination with a modulator of the glucocorticoid receptor, such.
- a modulator of the glucocorticoid receptor such as WO2008057855, WO2008057856, WO2008057857, WO2008057859, WO2008057862, WO2008059867, WO2008059866, WO2008059865, WO2008070507, WO2008124665, WO2008124745.
- the compound of formula I in combination with a modulator of the mineralocorticoid receptor (MR), such as.
- MR mineralocorticoid receptor
- drospirenones or those as described in WO2008104306, WO2008119918 administered.
- the compound of formula I in combination with an inhibitor of protein kinase C beta (PKC beta), such as. Ruboxistaurin, or those as described in WO2008096260, WO2008125945 administered.
- PLC beta protein kinase C beta
- the compound of formula I in combination with an inhibitor of protein kinase D such as. B. Doxazosin (WO2008088006) administered.
- the compound of the formula I in combination with an activator of AMP-activated protein kinase (AMPK), as described, for.
- AMPK AMP-activated protein kinase
- the compound of the formula I in combination with an inhibitor of ceramide kinase, as z.
- an inhibitor of ceramide kinase as described in WO2007112914, WO2007149865.
- the compound of the formula I is administered in combination with an inhibitor of the MAPK-interacting kinase 1 or 2 (MNK1 or 2), as described, for example, in WO2007104053, WO2007115822, WO2008008547, WO2008075741.
- the compound of the formula I is used in combination with inhibitors of "I-kappaB kinase" (IKK inhibitors), as described, for example, in WO2001000610, WO2001030774, WO2004022057, WO2004022553, WO2005097129, WO2005113544, US2007244140, WO2008099072, WO2008099073, WO2008099073, WO2008099074, WO2008099075 described, administered.
- IKK inhibitors inhibitors of "I-kappaB kinase”
- the compound of formula I in combination with inhibitors of NF-kappaB (NFKB) activation as described, for. As salsalates administered.
- the compound of the formula I in combination with inhibitors of ASK-1 (apoptosis signal-regulating kinase 1), as z. As described in WO2008016131 administered.
- the compounds of formula I are used in combination with an HMGCoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin, pitavastatin, L-659699, BMS-644950 or those described in US2007249583 , WO2008083551.
- an HMGCoA reductase inhibitor such as simvastatin, fluvastatin, pravastatin, lovastatin, atorvastatin, cerivastatin, rosuvastatin, pitavastatin, L-659699, BMS-644950 or those described in US2007249583 , WO2008083551.
- the compound of formula I in combination with a Famesoid X receptor (FXR) modulator e.g. WAY-362450 or those as in WO2003099821, WO2005056554, WO2007052843, WO2007070796, WO2007092751, JP2007230909, WO2007095174, WO2007140174, WO2007140183, WO2008000643, WO2008002573,
- FXR Famesoid X receptor
- the compound of the formula I is used in combination with a ligand of the liver X receptor (LXR), as described, for example, in WO2007092965, WO2008041003, WO2008049047, WO2008065754, WO2008073825, US2008242677.
- LXR liver X receptor
- the compound of formula I is used in combination with a fibrate, e.g. Fenofibrate, clofibrate, bezafibrate, or those as described in WO2008093655.
- a fibrate e.g. Fenofibrate, clofibrate, bezafibrate, or those as described in WO2008093655.
- the compound of formula I is used in combination with fibrates, e.g. the choline salt of fenofibrate (SLV-348).
- fibrates e.g. the choline salt of fenofibrate (SLV-348).
- the compound of formula I is used in combination with fibrates, e.g. the choline salt of fenofibrate and a HMGCoA reductase inhibitor, e.g. Rosuvastatin, administered.
- fibrates e.g. the choline salt of fenofibrate and a HMGCoA reductase inhibitor, e.g. Rosuvastatin, administered.
- the compound of the formula I is administered in combination with bezafibrate and diflunisal.
- the compound of formula I is administered in combination with a fixed combination of fenofibrate or a salt thereof with simvastatin, rosuvastatin, fluvastatin, lovastatin, cerivastatin, pravastatin, pitavastatin or atorvastatin.
- the compound of formula I is administered in combination with Synordia (R), a fixed combination of fenofibrate with metformin.
- the compound of formula I is used in combination with a cholesterol absorption inhibitor such as ezetimibe, tiqueside, pamaqueside, FM-VP4 (sitostanol / campesterol ascorbyl phosphate; Forbes Medi-Tech, WO2005042692, WO2005005453), MD-0727 (Microbia Inc., WO2005021497, WO2005021495) or with compounds as described in WO2002066464, WO2005000353 (Kotobuki Pharmaceutical Co.
- a cholesterol absorption inhibitor such as ezetimibe, tiqueside, pamaqueside, FM-VP4 (sitostanol / campesterol ascorbyl phosphate; Forbes Medi-Tech, WO2005042692, WO2005005453), MD-0727 (Microbia Inc., WO2005021497, WO2005021495) or with compounds as described in WO2002066464, WO2005000353 (Kotobuki Pharmaceutical Co.
- WO2005044256 or WO2005062824 Merck & Co.
- WO2005061451 and WO2005061452 stra eneco un enom xo er ⁇ U, WI UU (Lipi ⁇ eon Biotechnology AG) or as in WO2002050060, WO2002050068, WO2004000803, WO2004000804, WO2004000805, WO2004087655, WO2004097655, WO2005047248, WO2006086562, WO2006102674, WO2006116499, WO2006121861, WO2006122186, WO2006122216, WO2006127893, WO2006137794, WO2006137796, WO2006137782, WO2006137793, WO2006137797, WO2006137795, WO2006137792, WO2006138163, WO2007059871, US2007232688, WO2007126358, WO2008033431, WO2008033465
- the compound of formula I is administered in combination with an NPC1L1 antagonist, e.g. those as described in WO2008033464, WO2008033465, administered.
- an NPC1L1 antagonist e.g. those as described in WO2008033464, WO2008033465, administered.
- the compound of formula I is administered in combination with Vytorin TM, a fixed combination of ezetimibe with simvastatin.
- the compound of formula I is administered in combination with a fixed combination of ezetimibe with atorvastatin.
- the compound of formula I is administered in combination with a fixed combination of ezetimibe with fenofibrate.
- the further active ingredient is a diphenylazetidinone derivative, e.g. in US 6,992,067 or US 7,205,290.
- the further active ingredient is a
- Diphenylazetidinonderivat as described for example in US 6,992,067 or US 7,205,290 combined with a statin such as simvastatin, fluvastatin, pravastatin, lovastatin, cerivastatin, atorvastatin, pitavastatin or rosuvastatin.
- a statin such as simvastatin, fluvastatin, pravastatin, lovastatin, cerivastatin, atorvastatin, pitavastatin or rosuvastatin.
- the compound of hormel I is administered in combination with a solid combination of Lapaquistat, a squalene synthase inhibitor, with atorvastatin.
- a CETP inhibitor e.g. Torcetrapib, JTT-705 or anacetrapib (Dalcetrapib) or those described in WO2006002342, WO2006010422, WO2006012093, WO2006073973, WO2006072362, WO2007088996, WO2007088999, US2007185058, US2007185113, US2007185154, US2007185182, WO2006097169, WO2007041494, WO2007090752, WO2007107243, WO2007120621, US2007265252, US2007265304 , WO2007128568, WO2007132906, WO2008006257, WO2008009435, WO2008018529, WO2008058961, WO2008058967, WO2008059513, WO2008070496, WO2008115442, WO2008111604.
- a CETP inhibitor e.g. Torcetrapib, JTT-705 or anacetrapi
- the compound of formula I is used in combination with bile acid resorption inhibitors (inhibitors of the intestinal bile acid transporter (IBAT)) (see, for example, US 6,245,744, US 6,221,897 or WO00 / 61568), e.g. HMR 1741 or those described in DE 10 2005 033099.1 and DE 10 2005 033100.9, DE 10 2006 053635, DE 10 2006 053637, WO2007009655- 56, WO2008058628, WO2008058629, WO2008058630, WO2008058631.
- IBAT intestinal bile acid transporter
- the compound of formula I is used in combination with agonists of GPBAR1 (G protein-coupled bile acid receptoM; TGR5) as described e.g. in US20060199795, WO2007110237, WO2007127505, WO2008009407, WO2008067219, WO2008067222, FR2908310, WO2008091540, WO2008097976.
- GPBAR1 G protein-coupled bile acid receptoM; TGR5
- the compound of the formula I is administered in combination with inhibitors of the TRPM5 channel (TRP cation channel M ⁇ ), as described, for example, in WO2008097504.
- the compound of formula I is administered in combination with a polymeric bile acid adsorber such as cholestyramine, colesevelam hydrochloride.
- the compound of the formula I is administered in combination with colesevelam hydrochloride and metformin or a sulfonylurea or insulin.
- the compound of formula I is administered in combination with a phytosterol-containing chewing gum (Reductol TM).
- the compound of formula I is used in combination with an inhibitor of the microsomal triglyceride transfer protein (MTP inhibitor), e.g. Implitapide, BMS-201038, R-103757, AS-1552133, SLx-4090, AEGR-733 or those as described in WO2005085226, WO2005121091, WO2006010423, WO2006113910, WO2007143164, WO2008049806, WO2008049808, WO2008090198, WO2008100423.
- MTP inhibitor microsomal triglyceride transfer protein
- the compound of formula I is used in combination with a combination of a cholesterol absorption inhibitor, e.g. Ezetimibe, and an inhibitor of the triglyceride transfer protein (MTP inhibitor), such as. Implitapide as described in WO2008030382 or WO2008079398 described.
- a cholesterol absorption inhibitor e.g. Ezetimibe
- MTP inhibitor an inhibitor of the triglyceride transfer protein
- the compound of formula I is administered in combination with an antihypertriglyceridemic agent, e.g. such as those described in WO2008032980 administered.
- an antihypertriglyceridemic agent e.g. such as those described in WO2008032980 administered.
- the compound of the formula I is administered in combination with an antagonist of the somatostatin 5 receptor (SST5 receptor), such as those described in WO2006094682.
- the compound of the formula I is administered in combination with an ACAT inhibitor, such as, for example, Avasimibe, SMP-797 or KY-382 or those as described in WO2008087029, WO2008087030, WO2008095189.
- the compound of the formula I in combination with an inhibitor of hepatic-cardiin palmitoyltransferase-1 (L-CPT1), as described e.g. in WO2007063012, WO2007096251 (ST-3473), WO2008015081, US2008103182, WO2008074692.
- L-CPT1 hepatic-cardiin palmitoyltransferase-1
- the compound of formula I is used in combination with a modulator of serine palmitoyltransferase (SPT), as described e.g. in WO2008031032, WO2008046071, WO2008083280, WO2008084300.
- SPT serine palmitoyltransferase
- the compound of formula I is used in combination with a squalene synthetase inhibitor, e.g. BMS-188494, TAK-475 (Lapaquistat acetate) or as described in WO2005077907, JP2007022943, WO2008003424.
- a squalene synthetase inhibitor e.g. BMS-188494, TAK-475 (Lapaquistat acetate) or as described in WO2005077907, JP2007022943, WO2008003424.
- the compound of formula I is administered in combination with ISIS-301012 (mipomersen), an antisense oligonucleotide capable of regulating the apolipoprotein B gene.
- Combination with a stimulator of the ApoA-1 gene, as described e.g. in WO2008092231 is administered.
- the compound of the formula I is administered in combination with an LDL receptor inducer (see US Pat. No. 6,342,512), for example HMR1171, HMR1586 or those as described in WO2005097738, WO2008020607. , _, _ ,. ", _.
- an LDL receptor inducer see US Pat. No. 6,342,512
- the compound of the hormel I in combination with an HDL-cholesterol increasing agent such as e.g. those as described in WO2008040651, WO2008099278 administered.
- the compound of formula I is used in combination with an ABCA1 expression enhancer, e.g. in WO2006072393, WO2008062830, administered.
- the compound of the formula I is administered in combination with a lipoprotein-lipase modulator, e.g. Ibrolipim (NO-1886).
- a lipoprotein-lipase modulator e.g. Ibrolipim (NO-1886).
- the compound of formula I in combination with a lipoprotein (a) antagonist such as e.g. Gemcabene (CI-1027).
- the compound of formula I is administered in combination with a lipase inhibitor, e.g. Orlistat or cetilistat (ATL-962).
- a lipase inhibitor e.g. Orlistat or cetilistat (ATL-962).
- the compound of the formula I is administered in combination with an adenosine A1 receptor agonist (adenosine A1 R), as described, for example, in US Pat. in EP1258247, EP1375508, WO2008028590, WO2008077050.
- an adenosine A1 receptor agonist as described, for example, in US Pat. in EP1258247, EP1375508, WO2008028590, WO2008077050.
- the compound of formula I is used in combination with adenosine A2B receptor agonist (adenosine A2B R), e.g. ATL-801 administered.
- adenosine A2B receptor agonist e.g. ATL-801 administered.
- the compound of the formula I is administered in combination with a modulator of the adenosine A2A and / or adenosine A3 receptors, as described, for example, in WO2007111954, WO2007121918, WO2007121921, WO2007121923, WO2008070661.
- adenosine A2B receptor antagonist adenosine A2B R
- the compound of the formula I is used in combination with
- Inhibitors of acetyl-CoA carboxylase such as those as described in W0199946262, WO200372197, WO2003072197, WO2005044814, WO2005108370, JP2006131559, WO2007011809, WO2007011811, WO2007013691, WO2007095601 -603, WO2007119833, WO2008065508, WO2008069500, WO2008070609, WO2008072850, WO2008079610, WO2008088688, WO2008088689, WO2008088692, US2008171761, WO2008090944, JP2008179621 , US2008200461, WO2008102749, WO2008103382, WO2008121592.
- the compound of the formula I is used in combination with modulators of the microsomal acyl-CoA: glycerol-3-phosphate acyltransferase 3 (GPAT3, described in WO2007100789) or with modulators of the microsomal acyl-CoA: glycerol-3-phosphate Acyltransferase 4 (GPAT4, described in WO2007100833) administered.
- modulators of the microsomal acyl-CoA glycerol-3-phosphate acyltransferase 3
- modulators of the microsomal acyl-CoA glycerol-3-phosphate Acyltransferase 4
- the compound of the formula I is administered in combination with modulators of xanthine oxidoreductase (XOR).
- the compound of the formula I is administered in combination with inhibitors of soluble epoxide hydrolase (sEH), as described, for example, in WO2008051873, WO2008051875, WO2008073623, WO2008094869, WO2008112022.
- the compound of the formula I is used in combination with CART modulators (see “cocaine-amphetamine-regulated transcript-influenced transient-energy metabolism, anxiety and gastric emptying in mice" Asakawa, A. et al .: Hormone and Metabolism Research (2001 ), 33 (9), 554-558);
- NPY antagonists e.g. Naphthalene-1-sulfonic acid ⁇ 4 - [(4-amino-quinazolin-2-ylamino) -methyl] -cyclohexylmethyl ⁇ -amide hydrochloride (CGP 71683A) or Velneperite;
- NPY-5 receptor antagonists such as L-152804 or the compound "NPY-5-BY” from Banyu or as described, for example, in WO2006001318, WO2007103295, WO2007125952, WO2008026563, WO2008026564, WO2008052769, WO2008092887, WO2008092888, WO2008092891;
- NPY-4 receptor antagonists as they are e.g. As described in WO2007038942;
- NPY-2 receptor antagonists such as. As described in WO2007038943;
- Peptide YY 3-36 PYY3-36 or analogous compounds such.
- CJC-1682 PYY3-36 conjugated to human serum albumin via Cys34
- CJC-1643 derivative of PYY3-36 conjugated to serum albumin in vivo
- CB1R Cannabinoid Receptor 1 antagonists such as Rimonabant, Surinabant (SR147778), SLV-319 (Ibipinabant), AVE-1625, Taranabant (MK-0364) or salts thereof, Otenabant (CP-945,598), Rosonabant, V-24343 or such compounds as in z.
- WO2008081009, WO2008084057, EP1944295, US2008090809, US2008090810, WO2008092816, WO2008094473, WO2008094476, WO2008099076, WO2008099139, WO2008101995, US2008207704, WO2008107179, WO2008109027, WO2008112674, WO2008115705, WO2008118414, WO2008119999, WO200812000, WO2008121257, WO2008127585 are described;
- Cannabinoid Receptor 1 / Cannabinoid Receptor 2 (CB1 / CB2) modulating compounds e.g. delta-9-tetrahydrocannabivarin or those as described e.g. in WO2007001939, WO2007044215, WO2007047737, WO2007095513, WO2007096764, WO2007112399, WO2007112402, WO2008122618 are described;
- FAAH fatty acid amide hydrolase
- Inhibitors of fatty acid synthase e.g. in WO2008057585, WO2008059214, WO2008075064, WO2008075070, WO2008075077 are described;
- LCE Long chain fatty acid elongase
- Vanilloid-1 receptor modulators (modulators of TRPV1), e.g. in WO2007091948, WO2007129188, WO2007133637, WO2008007780, WO2008010061, WO2008007211, WO2008010061, WO2008015335, WO2008018827, WO2008024433, WO2008024438, WO2008032204, WO2008050199, WO2008059339, WO2008059370, WO2008066664, WO2008075150, WO2008090382, WO2008090434, WO2008093024, WO2008107543, WO2008107544, are described in WO2008110863;
- Modulators, antagonists or inverse agonists of opioid receptors such as e.g. GSK-982 or such as e.g. WO2007047397, WO2008021849, WO2008021851, WO2008032156, WO2008059335;
- MC4 receptor agonists (melanocortin-4 receptor agonists, MC4R agonists such as 1-amino-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [2- (3a-benzyl-2-methyl-3-oxo) 2,3,3a, 4,6,7-hexahydro-pyrazolo [4,3-c] pyridin-5-yl) -1- (4-chlorophenyl) -2-oxoethyl] -amide; (WO 01/91752)) or LB53280, LB53279, LB53278 or THIQ, MB243, RY764, CHIR-785, PT-141, MK-0493 or those as described in WO2005060985, WO2005009950, WO2004087159, WO2004078717, WO2004078716, WO2004024720, US20050124652, WO2005051391, WO2004112793, WOUS200
- Orexin receptor 1 antagonists (OX1 R antagonists), Orexin receptor 2 antagonists (OX2R antagonists), or mixed OX1 R / OX2R antagonists (eg, 1 - (2-methyl-benzoxazol-6-yl) -3- [1, 5] naphthyridin-4-ylurea hydrochloride (SB-334867-A) or those which are described, for example, in WO200196302, WO200185693, WO2004085403, WO2005075458, WO2006067224, WO2007085718, WO2007088276, WO2007116374, WO2007122591, WO2007126934, WO2007126935, WO2008008517, WO2008008518 , WO2008008551, WO2008020405, WO2008026149, WO2008038251, US2008132490, WO2008065626, WO2008078291, WO2008087611, WO20080813
- Histamine H1 / histamine H3 modulators such as. B. Betahistine or its dihydrochloride;
- Histamine H4 modulators as described e.g. in WO2007117399 are described;
- CRF antagonists eg [2-methyl-9- (2,4,6-trimethyl-phenyl) -9 H -1,3,9-triaza-fluoren-4-yl] -dipropyl-amine (WO 00/66585) or those CRF1 antagonists as described in WO2007105113, WO2007133756, WO2008036541, WO2008036579, WO2008083070);
- CRF BP antagonists e.g., urocortin
- Urocortin agonists Modulators of the beta-3 adrenoceptor such as 1- (4-chloro-3-methanesulfonylmethyl-phenyl) -2- [2- (2 ) 3-dimethyl-1H-indol-6-yloxy) -ethylamino] -ethanol hydrochloride (WO 01/83451) or Solabegron (GW-427353) or N-5984 (KRP-204) or those as described in JP2006111553, WO2002038543, WO2002038544, WO2007048840-843, WO2008015558, EP1947103;
- Modulators of the beta-3 adrenoceptor such as 1- (4-chloro-3-methanesulfonylmethyl-phenyl) -2- [2- (2 ) 3-dimethyl-1H-indol-6-yloxy) -ethylamino] -ethanol hydrochloride (WO 01/83451) or Sol
- MSH melanocyte-stimulating hormone
- MCH (melanin-concentrating hormone) receptor antagonists such as NBI-845, A-761, A-665798, A-798, ATC-0175, T-226296, T-71 (AMG-071, AMG-076 ), GW-856464, NGD-4715, ATC-0453, ATC-0759, GW-803430 or such compounds as described in WO2005085200, WO2005019240, WO2004011438, WO2004012648, WO2003015769, WO2004072025, WO2005070898, WO2005070925, WO2004039780, WO2004092181, WO2003033476, WO2002006245,
- CCK-A (CCK-1) agonists / modulators such as ⁇ 2- [4- (4-chloro-2,5-dimethoxyphenyl) -5- (2-cyclohexyl-ethyl) -thiazol-2-ylcarbamoyl] -5,7-dimethyl-indol-1-yl ⁇ -acetic acid trifluoroacetic acid salt (WO 99/15525) or SR-146131 (WO 0244150) or SSR-125180) or those as described in WO2005116034, WO2007120655, WO2007120688, WO2007120718, WO2008091631 are described; Serotonin reuptake inhibitors (eg dexfenfluramines) or those as described in WO2007148341, WO2008034142, WO2008081477, WO2008120761;
- modulators such as ⁇ 2- [4- (4-chloro-2,5-dimethoxyphenyl) -5- (2
- mixed serotonin / dopamine reuptake inhibitors e.g., bupropion
- mixed reuptake inhibitors such as e.g. DOV 21,947;
- mixed sertonine and noradrenergic compounds e.g., WO 00/71549
- 5-HT receptor agonists e.g. 1- (3-ethyl-benzofuran-7-yl) -piperazine oxalic acid salt (WO 01/09111);
- mixed dopamine / norepinephrine / acetylcholine reuptake inhibitors e.g., tesofensins
- those as described e.g. in WO2006085118 e.g., WO2006085118;
- Norepinephrine reuptake inhibitors as described e.g. in US2008076724;
- 5-HT2A receptor antagonists as described e.g. in WO2007138343 are described;
- 5-HT2C receptor agonists such as Lorcaserin hydrochloride (APD-356) or BVT-933 or those as described in WO200077010, WO200077001-02, WO2005019180, WO2003064423, WO200242304, WO2005035533, WO2005082859, WO2006004937, US2006025601, WO2006028961, WO2006077025, WO2006103511 , WO2007028132, WO2007084622, US2007249709, WO2007132841, WO2007140213, WO2008007661, WO2008007664, WO2008009125, WO2008010073, WO2008108445); 5-HT6 receptor modulators such as E-6837, BVT-74316 or PRX-07034 or those as described, for example, in WO2005058858, WO2007054257, WO2007107373, WO2007108569, WO2007108742-744, WO
- estrogen receptor gamma e.g. in WO2007131005, WO2008052709;
- estrogen receptor alpha (ERR ⁇ / ERR1 agonists), as described e.g. in WO2008109727 are described;
- Muscarinic 3 receptor (M3R) antagonists as described e.g. in WO2007110782, WO2008041184 are described;
- Bombesin receptor agonists (BRS-3 agonists), as described e.g. in WO2008051404, WO2008051405, WO2008051406, WO2008073311 are described;
- Growth hormone e.g., human growth hormone or AOD-9604
- human growth hormone e.g., human growth hormone or AOD-9604
- Growth Hormone Secretagogue Receptor Antagonists such as A-778193 or those as described in WO2005030734, WO2007127457, WO2008008286; Growth Hormone Secretagogue Receptor Modulators (ghrelin modulators) such as JMV-2959, JMV-3002, JMV-2810, JMV-2951 or those as described in WO2006012577 (eg YIL-781 or YIL-870), WO2007079239, WO2008092681 ;
- TRH agonists see, e.g., EP 0462 884
- Leptin agonists see, eg, Lee, Daniel W, Leinung, Matthew C, Rozhavskaya Arena, Marina, Grasso, Patricia, Leptin agonists as a Potential Approach to the Treatment of Obesity, Drugs of the Future (2001), 26 (9), 873-881);
- DA agonists bromocriptine, doprexin
- Lipase / amylase inhibitors e.g., WO 00/40569, WO2008107184
- Inhibitors of diacylglycerol O-acyltransferases such.
- Inhibitors of stearoyl-CoA delta- ⁇ desaturase as described e.g. in WO2007009236, WO2007044085, WO2007046867, WO2007046868, WO20070501124, WO2007056846, WO2007071023, WO2007130075, WO2007134457, WO2007136746, WO2007143597, WO2007143823, WO2007143824, WO2008003753, WO2008017161, WO2008024390, WO2008029266, WO2008036715, WO2008043087, WO2008044767, WO2008046226, WO2008056687, WO2008062276, WO2008064474, WO2008074824 , WO2008074832, WO2008074833, WO2008074834, WO2008074835, WO2008089580, WO2008096746, WO2008104524, WO2008
- hypoglycemic / hypertriglyceridemic indoline compounds as described in WO2008039087;
- Activators of adiponectin secretion e.g. in WO2006082978, WO2008105533;
- Promoters of adiponectin production e.g. in WO2007125946, WO2008038712 described; modified adiponectins such as e.g. described in WO2008121009;
- Oleoyl-estrone or agonists or partial agonists of the thyroid hormone receptor agonists (eg, thyroid hormone receptor agonists).
- TR-beta thyroid hormone receptor beta
- the compound of the formula I is administered in combination with a combination of Ezetimibe Eprotiromes.
- the compound of formula I is used in combination with an inhibitor of Site-1 protease (S1 P), e.g. PF-429242 administered.
- S1 P Site-1 protease
- the compound of formula I is used in combination with a modulator of the "Trace Amine Associated Receptor-1" (TAAR1), as described e.g. in US2008146523, WO2008092785.
- TAAR1 Race Amine Associated Receptor-1
- the compound of formula I is used in combination with an inhibitor of growth factor receptor Bound protein-2 (GRB2), e.g. in WO2008067270, administered.
- GRB2 growth factor receptor Bound protein-2
- the compound of the formula I is administered in combination with an RNAi (siRNA) therapeutic which is directed against PCSK9 (proprotein convertase subtilisin / kexin type 9).
- RNAi siRNA
- PCSK9 proprotein convertase subtilisin / kexin type 9
- the compound of formula I is administered in combination with Omacor® or Lovaza TM (omega-3 fatty acid esters, high-concentration ethyl esters of eicosapentaenoic acid and docosahexaenoic acid).
- the compound of the formula I is administered in combination with lycopene.
- the compound of formula I is used in combination with an antioxidant, e.g. OPC-14117, AGI-1067 (succinobucol), probucol, tocopherol, ascorbic acid, beta-carotene or selenium.
- an antioxidant e.g. OPC-14117, AGI-1067 (succinobucol), probucol, tocopherol, ascorbic acid, beta-carotene or selenium.
- the compound of the formula I in combination with a vitamin, such as. As vitamin B6 or vitamin B12 administered.
- the compound of formula I is used in combination with more than one of the aforementioned compounds, e.g. in combination with a sulfonylurea and metformin, a sulfonylurea and acarbose, repaglinide and metformin (PrandiMet (TM)), insulin and a sulfonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc.
- a sulfonylurea and metformin e.g. in combination with a sulfonylurea and metformin, a sulfonylurea and acarbose, repaglinide and metformin (PrandiMet (TM)), insulin and a sulfonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc.
- TM repaglinide and metformin
- the compound of formula I is used in combination with an inhibitor of carbonic anhydrase type 2, such as carbonic anhydrase type 2, e.g. such as described in WO2007065948 administered.
- an inhibitor of carbonic anhydrase type 2 such as carbonic anhydrase type 2, e.g. such as described in WO2007065948 administered.
- the compound of formula I is administered in combination with topiramate or a derivative thereof as described in WO2008027557.
- the compound of formula I is administered in combination with a solid combination of topiramate with phentermine (Qnexa TM).
- the compound of the formula I is administered in combination with an antisense compound, eg ISIS-377131, which inhibits the production of the glucocorticoid receptor.
- the compound of formula I is administered in combination with an aldosterone synthase inhibitor and an antagonist of the glucocorticoid receptor, a cortisol synthesis inhibitor and / or an antagonist of the corticotropin releasing factor, such as corticotropin releasing factor, e.g. described in EP1886695, WO2008119744.
- the compound of formula I in combination with an agonist of the RUP3 receptor, such.
- an agonist of the RUP3 receptor such as described in WO2007035355, WO2008005576.
- the compound of the formula I in combination with an activator of the gene coding for the Ataxia Telangiectasia Mutated (ATM) protein kinase such as. As chloroquine administered.
- ATM Ataxia Telangiectasia Mutated
- the compound of the formula I in combination with a tau protein kinase 1 inhibitor (TPK1 inhibitor), such as. As described in WO2007119463 administered.
- TPK1 inhibitor tau protein kinase 1 inhibitor
- the compound of the formula I is administered in combination with a "c-Jun N-terminal kinase” inhibitor (JNK inhibitor), as described, for example, in WO2007125405, WO2008028860, WO2008118626.
- JNK inhibitor c-Jun N-terminal kinase inhibitor
- the compound of formula I in combination with an endothelin A receptor antagonist, such as. B. avosentan (SPP-301).
- an endothelin A receptor antagonist such as. B. avosentan (SPP-301).
- the compound of formula I is used in combination with modulators of the glucocorticoid receptor (GR), such as KB-3305 or such compounds as e.g. As described in WO2005090336, WO2006071609, WO2006135826, WO2007105766, WO2008120661.
- the other active ingredient is varenicline tartrate, a partial agonist of the alpha 4-beta 2 nicotinic acetylcholine receptor.
- the other active ingredient is trodusquemine.
- the further active ingredient is a modulator of the enzyme SIRT1 and / or SIRT3 (an NAD + -dependent protein deacetylase); this active substance may be, for example, resveratrol in suitable formulations, or such compounds as are mentioned in WO2007019416 (eg SRT-1720), WO2008073451.
- the further active ingredient is DM-71 (N-acetyl-L-cysteine with bethanechol).
- the compound of formula I is used in combination with anti-hypercholesterolemic compounds, such as those described e.g. in WO2007107587, WO2007111994, WO2008106600, WO2008113796.
- the compound of the formula I is used in combination with inhibitors of the SREBP (sterol regulatory element-binding protein), as described, for example, in US Pat. in WO2008097835.
- SREBP sterol regulatory element-binding protein
- the compound of formula I is used in combination with a cyclic peptide agonist of the VPAC2 receptor, as described e.g. in WO2007101146, WO2007133828.
- the compound of formula I is administered in combination with an agonist of the endothelin receptor, as described e.g. in WO2007112069 are administered.
- the compound of the formula I is administered in combination with AKP-020 (bis (ethylmaltolato) oxovanadium-IV).
- the compound of the formula I is administered in ovoination with tissue-selective androgen receptor modulators (SARM), as described, for example, in WO2007099200, WO2007137874.
- SARM tissue-selective androgen receptor modulators
- the compound of formula I is used in combination with an AGE (advanced glycation endproduct) inhibitor, e.g. in JP2008024673.
- an AGE advanced glycation endproduct
- the further active ingredient is leptin; see, e.g. "Perspectives in the therapeutic use of leptin", Salvador, Javier; Gomez-Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001), 2 (10), 1615-1622.
- the further active ingredient is metreleptin (recombinant methionyl-leptin) combined with pramlintide.
- the further active ingredient is the tetrapeptide ISF-402.
- the other active ingredient is dexamphetamine or amphetamine.
- the other active ingredient is fenfluramine or dexfenfluramine.
- the other active ingredient is sibutramine or such derivatives as described in WO2008034142.
- the other active ingredient is mazindol or phentermine.
- the further active ingredient is geniposidic acid (geniposidic acid, WO2007100104) or derivatives thereof (JP2008106008).
- the further active ingredient is a nasally administered calcium channel blocker such as diltiazem or those described in US 7,138,107.
- the further active ingredient is an inhibitor of sodium-calcium ion exchange such as e.g. those as described in WO2008028958, WO2008085711.
- the further active ingredient is a blocker of calcium channels, e.g. of the CaV3.2 or CaV2.2 as described in WO2008033431, WO2008033447, WO2008033356, WO2008033460, WO2008033464, WO2008033465, WO2008033468, WO2008073461.
- the further active ingredient is a modulator of a calcium channel, e.g. those as described in WO2008073934, WO2008073936.
- the further active ingredient is a blocker of the "T-type calcium channel" as described, for example, in WO2008033431, WO2008110008.
- the further active ingredient is an inhibitor of KCNQ potassium channel-2 or -3, e.g. those as described in US2008027049, US2008027090.
- the further active ingredient is an inhibitor of the potassium Kv1.3 ion channel, e.g. those as described in WO2008040057, WO2008040058, WO2008046065.
- the further active ingredient is a modulator of the MCP-1 receptor (monocyte chemoattractant protein-1 (MCP-1)) such as those described in WO2008014360, WO2008014381.
- MCP-1 receptor monocyte chemoattractant protein-1 (MCP-1)
- MCP-1 receptor monocyte chemoattractant protein-1 (MCP-1)
- MCP-1 receptor monocyte chemoattractant protein-1 (MCP-1)
- MCP-1 receptor monocyte chemoattractant protein-1 (MCP-1)
- MCP-1 receptor monocyte chemoattractant protein-1
- SSTR5 somatostatin receptor 5
- the further active ingredient is a modulator of
- Somatostatin receptor 2 (SSTR2), e.g. those as described in WO2008051272.
- the further active ingredient is an erythropoietin-mimetic peptide which acts as an erythropoietin (EPO) receptor agonist.
- EPO erythropoietin
- the further active ingredient is an anorectic / hypoglycemic compound, e.g. those as described in WO2008035305, WO2008035306, WO2008035686.
- the further active ingredient is an inducer of lipoic acid synthetase, e.g. those as described in WO2008036966, WO2008036967.
- the further active ingredient is a stimulator of endothelial nitric oxide synthase (eNOS), such as e.g. those as described in WO2008058641, WO2008074413.
- eNOS endothelial nitric oxide synthase
- the further active ingredient is a modulator of carbohydrate and / or lipid metabolism, e.g. those as described in WO2008059023, WO2008059024, WO2008059025, WO2008059026.
- the further active ingredient is an angiotensin II receptor antagonist such as, for example, those described in WO2008062905, WO2008067378, WO2008062905.
- the further active ingredient is an agonist of the sphingosine-1-phosphate receptor (S1P), such as those described in WO2008064315, WO2008074820.
- S1P sphingosine-1-phosphate receptor
- the further active ingredient is an agent which retards gastric emptying such as e.g. 4-hydroxyisoleucine (WO2008044770).
- the further active ingredient is a muscle relaxant substance as described e.g. in WO2008090200 is described.
- the further active ingredient is an inhibitor of monoamine oxidase B (MAO-B), e.g. those as described in WO2008092091.
- MAO-B monoamine oxidase B
- the further active ingredient is an inhibitor of the binding of cholesterol and / or triglycerides to the SCP-2 protein (sterol carrier protein-2), e.g. those as described in US2008194658.
- the other active ingredient is lisofylline, which prevents autoimmune damage to insulin-producing cells.
- the compound of formula I in combination with bulking agents preferably insoluble bulking agents
- bulking agents preferably insoluble bulking agents
- Caromax is a carob-containing product of the company Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark availability, 65926 Frankfurt / Main)) administered.
- the combination with Caromax can be carried out in one preparation, or by separate administration of compounds of formula I and Caromax ® .
- Caromax ® can also be administered in the form of food, such as in baked goods or muesli bars.
- Red List 2007, chapter 39 all coronary and gastrointestinal agents mentioned in the Red List 2007, chapters 55 and 60; all migraine, neuropathy and Parkinson's drugs listed in the Red List
- the cDNA for human SGLT2 was prepared by standard molecular biological methods as described in Sambrook et al. (Molecular Cloning, A Laboratory Manual, Second Edition), into the pcDNA4 / TO vector (Invitrogen). Subsequent sequencing of the insert revealed complete identity with bases 21 to 2039 of Wells et al. and stored in the GenBank sequence database base sequence for human SGLT2 (GenBank Accesion number: M95549). Bases 21 through 2039 correspond to the complete coding region of human SGLT2.
- the expression vector for human SGLT2 was introduced into CHO-TREx cells (Invitrogen) by FuGeneo lipofection (Roche). For the selection of
- Single cell clones were supplemented to cell culture medium (Nutrient Mixture F-12 (urine), (Invitrogen) supplemented with 10% fetal calf serum (FBS Gold, PAA), 10 ⁇ g / ml blasticidin S (CN Biosciences), 100 units / ml penicillin, 100 units / ml Streptomycin) 600 ⁇ g / ml Zeocin (Invitrogen).
- F-12 fetal calf serum
- FBS Gold fetal calf serum
- FBS Gold fetal calf serum
- CHO-TRex-hSGLT2 The one cell clone with the highest uptake activity for methyl- ⁇ -D-glucopyranoside, hereinafter referred to as CHO-TRex-hSGLT2, was selected for the further experiments and further cultured in the presence of 600 ⁇ g / ml zeocin.
- CHO-TRex-hSGLT2 cells were seeded in cell culture medium at a concentration of 50,000 cells per well in Cytostar-T Scintillating 96-WeII plates (Amersham Biosciences) and cultured for 24 h.
- the expression of the recombinant human SGLT2 was induced by addition of 1 ⁇ g / ml tetracycline for a further 24 h.
- the cells were washed with PBS and then starved for one hour in starvation medium (PBS supplemented with 10% fetal calf serum) at 37 ° C.
- Transport Assay Buffers 14 mM sodium chloride, 2 mM potassium chloride, 1 mM magnesium chloride, 1 mM calcium chloride, 10 mM HEPES / Tris, pH 7.5
- the test substances were diluted starting from a 10 mM stock solution in dimethyl sulphoxide correspondingly in transport assay buffer (40 ⁇ l / well).
- the assay was then started by adding 10 ⁇ l / well of a mixture of radioactively labeled methyl ⁇ -D- [U- 14 C] glucopyranoside (Amersham) and unlabelled methyl ⁇ -D-glucopyranoside (Acros).
- the final concentration of methyl ⁇ -D-glucopyranoside in the assay was 50 ⁇ M.
- the reaction was stopped by adding 50 ⁇ l / well of 10 mM methyl ⁇ -D-glucopyranoside in transport assay buffer (4 ° C) and the radioactivity absorbed into the cells in a MicroBeta Scintillation Microplate Reader (Wallac) determined.
- the half-maximal inhibitory effect of the test substances (IC50 value) was determined as follows:
- Buffer 14 oMM choline chloride, 2 mM potassium chloride, 1 mM magnesium chloride, 1 mM calcium chloride, 10 mM HEPESH®, pH 7.5).
- Test substance can be determined, which reduces the uptake of methyl ⁇ -D-glucopyranoside by 50% (IC50 value).
- the expression vector for human SGLT1 was introduced into CHO-TRex cells (Invitrogen) by FuGene® lipofection (Roche).
- the cell culture medium Nutrient Mixture F-12 (urine), (Invitrogen) supplemented with 10% fetal calf serum (BD Biosciences), 10 ⁇ g / ml blasticidin S (CN Biosciences), 100 units / ml penicillin, 100 units / ml streptomycin) 600 ⁇ g / ml Zeocin (Invitrogen).
- the functionality of the individual cell clones resulting from the selection was tested by their uptake activity for radiolabeled methyl- ⁇ -D-glucopyranoside.
- CHO-TRex-hSGLT1 The cell clone with the highest uptake activity for methyl- ⁇ -D-glucopyranoside, hereinafter referred to as CHO-TRex-hSGLT1, was selected for further experiments and further cultured in the presence of 600 ⁇ g / ml zeocin.
- CHO-TRex-hSGLT1 cells were seeded in cell culture medium at a concentration of 50,000 cells per well in Cytostar-T scintillating 96-well plates (Amersham Biosciences) and cultured for 24 h.
- the expression of recombinant human SGLT1 was monitored by addition of 1 ⁇ g / ml tetracycline for a further 24 h induced.
- the cells were washed with PBS and then starved for one hour in starvation medium (PBS supplemented with 10% fetal calf serum) at 37 ° C.
- transport assay buffer 14 mM sodium chloride, 2 mM potassium chloride, 1 mM magnesium chloride, 1 mM calcium chloride, 10 mM HEPES / Tris, pH 7.5
- the cells were incubated for 15 min at room temperature either in the absence or presence of test substances of different concentration.
- the test substances were diluted starting from a 10 mM stock solution in dimethyl sulphoxide correspondingly in transport assay buffer (40 ⁇ l / l_och).
- the assay was then started by adding 10 ⁇ l of a mixture of radiolabeled methyl ⁇ -D- [U- 14 C] glucopyranoside (Amersham) and unlabeled methyl ⁇ -D-glucopyranoside (Acros). The final concentration of methyl ⁇ -D-glucopyranoside in the assay was 50 ⁇ M. After an incubation period of 30 min at room temperature, the reaction was stopped by adding 50 ⁇ l / well of 10 mM methyl ⁇ -D-glucopyranoside in transport assay buffer (4 ° C.) and the cells were taken up in the cells
- test compounds for administration Each compound was dissolved in water containing 5% Solutol and 0.5% Tylose. From this solution, 5 ml / kg were orally administered to rats and 20 ml / kg to mice.
- the compounds were orally administered in doses of 3, 10 and 30 mg / kg.
- Urine volume (U VO ⁇ ) and urine glucose concentration were measured to be the
- the ID 50 (mg / kg) values were calculated from the corresponding regression line based on 50% inhibition of maximal renal glucose filtration (RGF, renal glucose filtration) of the untreated healthy animals.
- Creatinine in serum and urine was analyzed with Crea plus
- the invention further relates to processes for the preparation of
- Reaction solution is then added to a mixture of 100 ml of water and 150 ml
- peracyl compound 8 520 mg (0.96 mmol) of peracyl compound 8 are taken up in 3 ml of methylene chloride and 20 ml of methanol, and 1.5 ml of 1 M NaOMe / MeOH are added. After a
- Residue is purified by chromatography on silica gel (methylene chloride
- Example 2 Starting from 2.25 g (5.5 mmol) of C-glycoside 9 (BMS Patent US 2003/0114390 A1), 320 mg of the difluoro-C-glycoside 15 (Example 2) are obtained via the same reaction sequence as shown for the preparation of Example 1 colorless solid.
- reaction solution is then poured onto a mixture of 100 ml of 10% aqueous ammonium chloride solution and 100 ml of ethyl acetate.
- the organic phase is washed once more with aqueous NaCl solution, filtered through a little silica gel and concentrated. This gives 11.2 g of crude product which is dissolved in 150 ml of acetonitrile and 20 ml of triethylsilane and then cooled under argon to -40 0 C.
- 10 ml boron trifluoride etherate is stirred for 30 minutes at -40 0 C and then the reaction solution is added to a mixture of 100 ml of water and 150 ml of ethyl acetate.
- reaction solution is then poured onto a mixture of 20 ml of 10% aqueous ammonium chloride solution and 20 ml of ethyl acetate.
- the organic phase is washed once more with aqueous NaCl solution, filtered through a little silica gel and concentrated.
- the alcohol 53 is oxidized analogously to the literature procedure (Helvetica Chimica Acta-Vol. 89 (2006) page 648, compound 17) to lactone 53 (96% yield).
- bromide 48 3.3 g (11.2 mmol) of bromide 48 is dissolved in 60 ml of dry tetrahydrofuran (THF) and cooled to -78 ° C with an acetone / dry ice mixture under an argon atmosphere. After addition of 5 ml of a 2.6 molar n-butyllithium solution in toluene
- the C-glycosides 64 and 65 are analogous to the procedure for the synthesis of
- Example 8 and 9 starting from 4-bromoanisole and 5-bromo-2-methoxybenzaldehyde, prepared over 9 stages in similar yields.
- the C-glycosides 66 and 67 are prepared analogously to the procedure for the synthesis of Example 10 and 11, starting from 4-bromoanisole and 5-bromo-2-methoxybenzaldehyde, over 10 stages with similar yields.
- MS for compound 66 C 24 H 28 F 2 O 8 (482.48), MS (ESI +) 500.20 (M + NH 4 +).
- MS for Compound 67 C 2 H 24 F 2 O 6 (410.42), MS (ESI + ) 428.22 (M + NH 4 + ).
- the C-glycosides 68 and 69 are prepared analogously to the procedure for the synthesis of Example 8 and 9, starting from 4-bromo-1-chloro- (4-ethoxy-benzyl) -benzene and lactone 53, with similar yields.
- MS for compound 68 C 24 H 28 CIFO 7 (482.94), MS (ESI +) 482.16 (M - H 2 O + NH 4 +).
- MS for compound 69 C 21 H 24 CIFO 5 (410.87), MS (ESI +) 428.42 (M + NH 4 +).
- the C-glycosides 73 and 74 are prepared analogously to the procedure for the synthesis of Example 10 and 11, starting from 4-bromo-1-chloro- (4-methoxy-benzyl) -benzene and lactone 53, with similar yields.
- MS for Compound 73 C 23 H 25 CIF 2 O 7 (486.90), MS (ESI + ) 469.15 (M - H 2 O + H + ).
- MS for Compound 74 C 20 H 21 CIF 2 O 5 (414.18), MS (ESI + ) 432.18 (M + NH 4 + ).
- the bromide 76 is prepared analogously to the procedure for the synthesis of bromide 47, starting from 4-bromo-1-chloro-2-iodobenzene and p-trifluoromethoxy-benzaldehyde, in similar yields.
- the benzyl alcohol 76 does not directly deoxygenate. Therefore, he is successfully deoxygenated with a newly developed method of activation by trichloroacetimidate.
- the C-glycosides 79 and 80 are prepared analogously to the procedure for the synthesis of Example 8 and 9, starting from 4-bromo-1-chloro- (4-trifluoro-methoxy-benzyl) -benzene 78 and lactone 53, with similar yields , MS for Compound 79: C 23 H 23 CIF 4 O 7 (522.88), MS (ESI + ) 540.42 (M + NH 4 + ). MS for Compound 80: C 20 H 19 CIF 4 O 5 (450.82), MS (ESI + ) 468.06 (M + NH 4 + ).
- the C-glycosides 81 and 82 are prepared analogously to the procedure for the synthesis of Example 10 and 11, starting from 4-bromo-1-chloro- (4-trifluoro-methoxy-benzyl) -benzene 78 and lactone 53, with similar yields , MS for Compound 81: C 23 H 22 CIF 5 O 7 (540.87), MS (ESI + ) 523.07 (M -H 2 O + H + ). MS for Compound 82: C 20 Hi 8 CIF 5 O 5 (468.81), MS (ESI + ) 486.05 (M + NH 4 + ).
- the C-glycosides 84 and 85 are prepared analogously to the procedure for the synthesis of Example 8 and 9, starting from bromide 83 and lactone 53, with similar yields.
- the C-glycoside 86 is prepared analogously to the procedure for the synthesis of Example 1, starting from 2- (4-benzyloxy-benzyl) -4-bromo-1-chloro-benzene, with similar yields.
- the bromide 91 is prepared analogously to the procedure for the synthesis of bromide 78, starting from 4-bromo-1-chloro-2-iodobenzene and 5-methoxy-pyridine-2-carbaldehyde, with similar yields.
- the C-glycosides 94 and 95 are analogous to the procedure for the synthesis of
- iodide 96 6.0 g (16.4 mmol) of iodide 96 are dissolved in 50 ml of dry tetrahydrofuran (THF) and cooled to -78 ° C. with an acetone / dry ice mixture under an argon atmosphere. After addition of 8.8 ml of a 2.6 molar n-butyllithium solution in toluene (22.9 mmol), the reaction solution is stirred for 20 minutes at -78 C C. To the reaction solution is then added dropwise a solution of 3.2 g (22.9 mmol) of anisaldehyde in 20 ml of THF and stirred at -78 ° C for one hour.
- THF dry tetrahydrofuran
- reaction solution to stir for 30 minutes at -40 0C.
- the reaction solution is then poured onto a mixture of 100 ml of saturated sodium chloride solution and 100 ml of ethyl acetate.
- the organic phase is washed once more with aqueous NaCl solution, filtered through a little silica gel and concentrated. The residue is through
- the C-glycosides 99 and 100 are analogous to the procedure for the synthesis of
- the C-glycosides 101 and 102 are prepared analogously to the procedure for the synthesis of Example 10 and 11, starting from bromide 98 and lactone 53, with similar yields.
- MS for compound 101 C 24 H 25 F 5 O 8 (536.45), MS (ESI +) 537.07 (M + H +).
- MS for compound 102 C 21 H 21 F 5 O 6 (464.39), MS (ESI +) 482.07 (M + NH 4 +).
- the bromide 103 is prepared analogously to the procedure for the synthesis of bromide 78, starting from 4-bromo-1-chloro-2-iodobenzene and p-chlorobenzaldehyde, with similar yields.
- Example 43 and 44 (compound 104 and 105)
- the C-glycosides 104 and 105 are prepared analogously to the procedure for the synthesis of Example 8 and 9, starting from bromide 103 and lactone 53, with similar yields.
- MS for compound 104 C 22 H 23 Cl 2 FO 6 (473.33), MS (ESI +) 495.12 (M + NH 4 +).
- MS for Compound 105 C 19 H 1 QiCl 2 FO 4 (401.27), MS (ESO 446.07 (M + HCOO).
- the C-glycosides 106 and 107 are prepared analogously to the procedure for the synthesis of Example 10 and 11, starting from bromide 103 and lactone 53, with similar yields.
- MS for compound 106 C 22 H 22 Cl 2 F 2 O 6 (491.32), MS (ESI +) 513.10 (M + Na +).
- MS for compound 107 C 19 H 18 Cl 2 F 2 O 4 (419.26), MS (ESI +) 437.22 (M + NH 4 +).
- the C-glycosides 111 and 112 are analogous to the procedure for the synthesis of
- the C-glycosides 113 and 114 are analogous to the protocol for the synthesis of
- the C-glycosides 117 and 118 are analogous to the procedure for the synthesis of
- the C-glycosides 119 and 120 are prepared analogously to the procedure for the synthesis of Example 10 and 11, starting from bromide 116 and lactone 53, with similar yields.
- MS for compound 119 C 25 H 27 F 5 O 6 (518.48), MS (ESI + ) 536.16 (M + NH 4 + ).
- MS for compound 120 C 21 H 2I F 5 O 5 (446.42), MS (ESI +) 464.08 (M + NH 4 +).
- the C-glycosides 121 and 122 are analogous to the procedure for the synthesis of
- Lactone 53 prepared with similar yields.
- the C-glycosides 123 and 124 are analogous to the procedure for the synthesis of
- the C-glycosides 126 and 127 are analogous to the procedure for the synthesis of
- the C-glycosides 128 and 129 are analogous to the protocol for the synthesis of
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Endocrinology (AREA)
- Biochemistry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyrane Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description









































































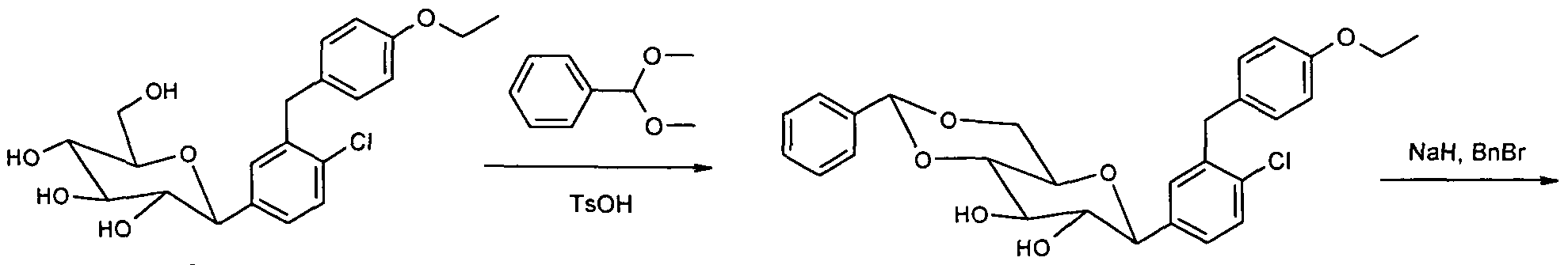


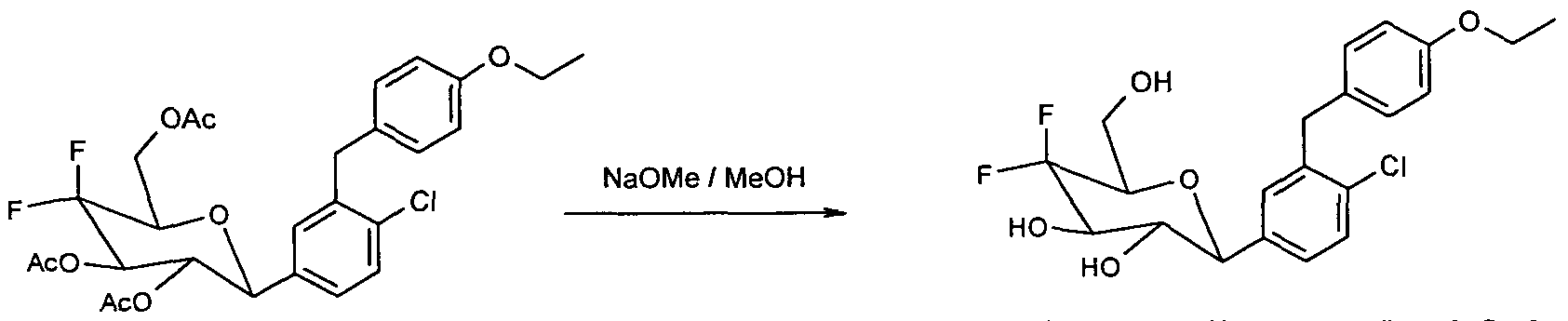























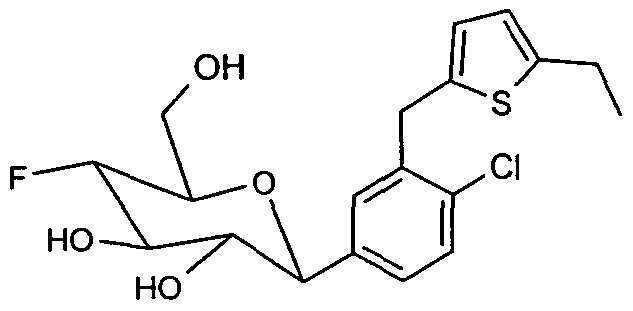




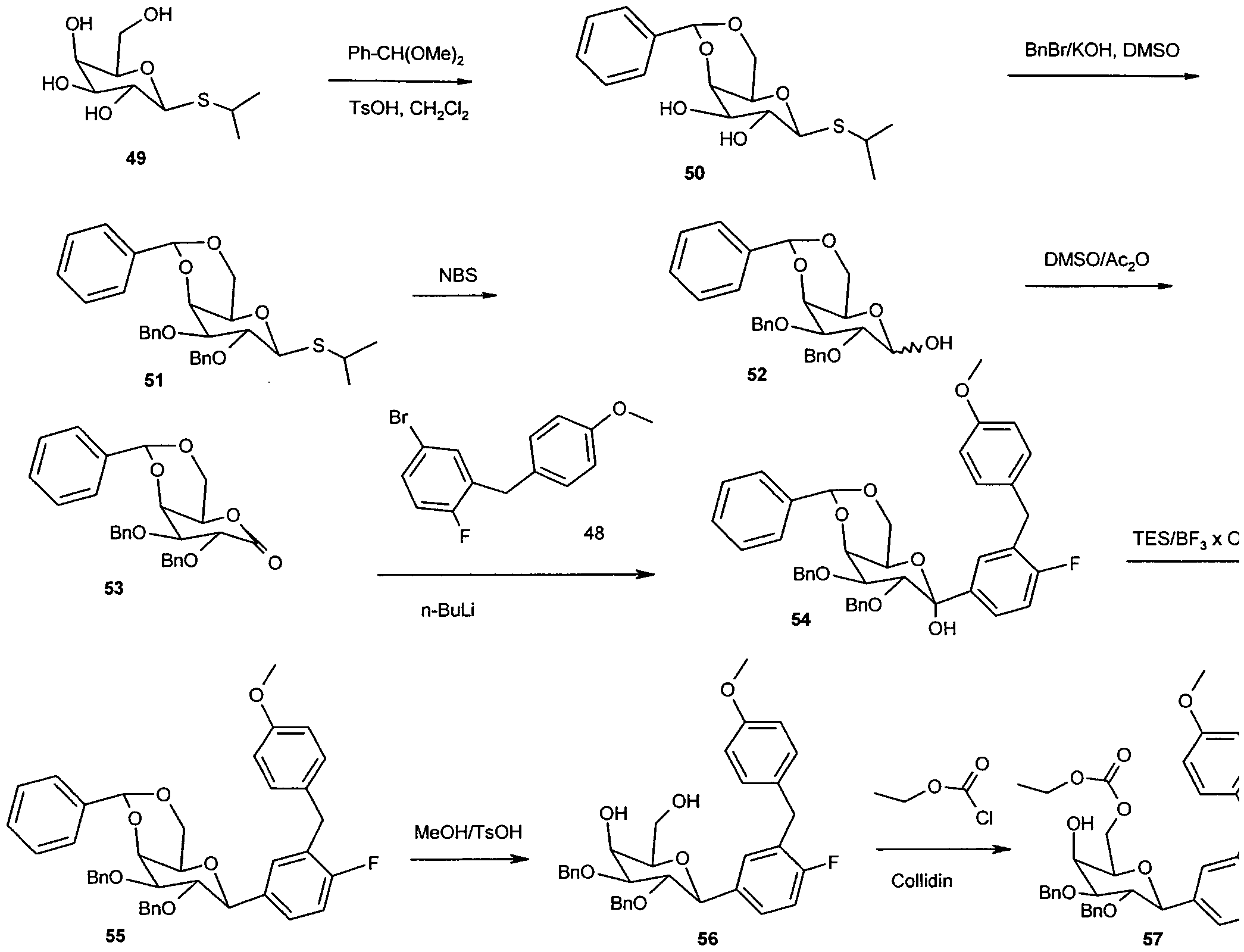



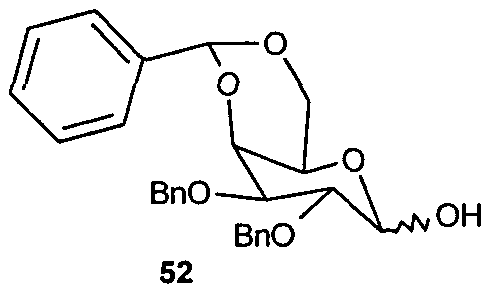
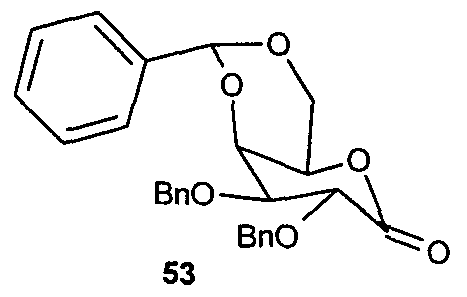


































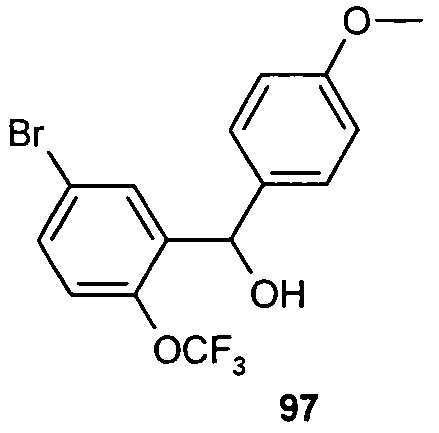















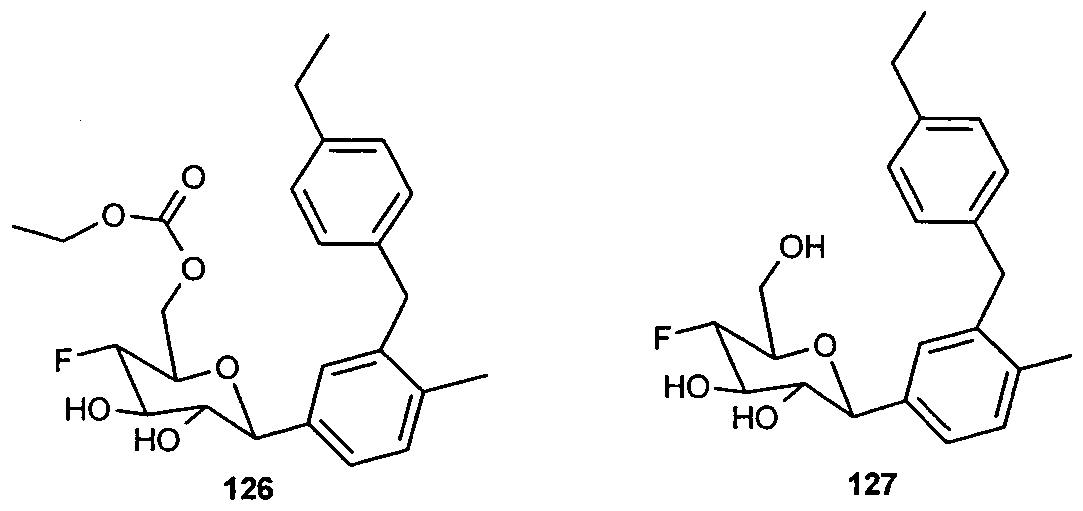




Claims
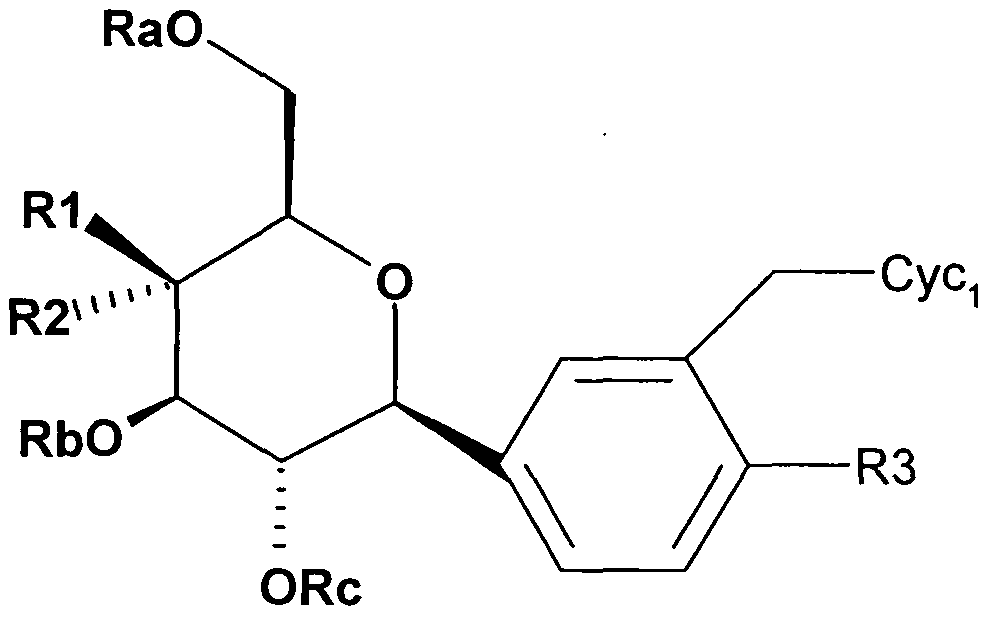



Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09711333A EP2268653A2 (de) | 2008-02-13 | 2009-02-13 | Neue aromatische fluorglykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| JP2010546266A JP2011511820A (ja) | 2008-02-13 | 2009-02-13 | 新規な芳香族フルオログリコシド誘導体、該化合物を含む薬剤及びその使用 |
| BRPI0907572-0A BRPI0907572A2 (pt) | 2008-02-13 | 2009-02-13 | Derivados de fluorglicosídeos aromáticos, fármacos compreendendo os referidos compostos e uso dos mesmos |
| CN2009801125891A CN101998962A (zh) | 2008-02-13 | 2009-02-13 | 新的芳香氟代糖苷衍生物,包含所述化合物的药物及其用途 |
| MX2010008051A MX2010008051A (es) | 2008-02-13 | 2009-02-13 | Nuevos derivados de fluoroglicosidos aromaticos, productos farmaceuticos que comprenden dichos compuestos y uso de los mismos. |
| CA2714110A CA2714110A1 (en) | 2008-02-13 | 2009-02-13 | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof |
| AU2009214278A AU2009214278A1 (en) | 2008-02-13 | 2009-02-13 | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds, and the use thereof |
| IL207201A IL207201A0 (en) | 2008-02-13 | 2010-07-25 | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof |
| US12/851,944 US20110059910A1 (en) | 2008-02-13 | 2010-08-06 | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08290152.1 | 2008-02-13 | ||
| EP08290152 | 2008-02-13 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/851,944 Continuation US20110059910A1 (en) | 2008-02-13 | 2010-08-06 | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009100936A2 true WO2009100936A2 (de) | 2009-08-20 |
| WO2009100936A3 WO2009100936A3 (de) | 2009-10-22 |
Family
ID=39617750
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/001042 WO2009100936A2 (de) | 2008-02-13 | 2009-02-13 | Neue aromatische fluorglykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US20110059910A1 (de) |
| EP (1) | EP2268653A2 (de) |
| JP (1) | JP2011511820A (de) |
| KR (1) | KR20100121615A (de) |
| CN (1) | CN101998962A (de) |
| AR (1) | AR070701A1 (de) |
| AU (1) | AU2009214278A1 (de) |
| BR (1) | BRPI0907572A2 (de) |
| CA (1) | CA2714110A1 (de) |
| CL (1) | CL2009000309A1 (de) |
| IL (1) | IL207201A0 (de) |
| MX (1) | MX2010008051A (de) |
| TW (1) | TW201000494A (de) |
| UY (1) | UY31651A1 (de) |
| WO (1) | WO2009100936A2 (de) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2016041470A1 (en) * | 2014-09-15 | 2016-03-24 | National Institute Of Biological Sciences, Beijing | Sglt-2 inhibitors |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011153712A1 (en) * | 2010-06-12 | 2011-12-15 | Theracos, Inc. | Crystalline form of benzylbenzene sglt2 inhibitor |
| US8614195B2 (en) * | 2011-04-14 | 2013-12-24 | Novartis Ag | Glycoside derivatives and uses thereof |
| CN104513283B (zh) | 2013-09-27 | 2018-01-16 | 广东东阳光药业有限公司 | 吡喃葡萄糖基衍生物及其在医药上的应用 |
| WO2018029264A1 (en) | 2016-08-10 | 2018-02-15 | Amneal Pharmaceuticals Company Gmbh | Process for preparation of dapagliflozin and intermediates thereof |
| US10696662B2 (en) | 2017-08-21 | 2020-06-30 | Janssen Pharmaceutica Nv | 5-fluoro-C-(aryl or heterocyclyl)-glycoside derivatives useful as dual SGLT1 / SGLT2 modulators |
| US20210238170A1 (en) | 2018-05-09 | 2021-08-05 | Janssen Pharmaceutica Nv | 5,5-Difluoro- and 5-Fluoro-5-Methyl-C-Glycoside Derivatives Useful As Dual SGLT1 / SGLT2 Modulators |
| CN109206331A (zh) * | 2018-09-17 | 2019-01-15 | 康化(上海)新药研发有限公司 | 一种2-甲基丝氨酸的制备方法 |
| CN113429379A (zh) * | 2021-06-28 | 2021-09-24 | 江苏法安德医药科技有限公司 | 一种lh-1801中间体及其制备方法和应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004052902A1 (de) * | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Neue aromatische fluorglycosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| EP1609785A1 (de) * | 2003-03-14 | 2005-12-28 | Astellas Pharma Inc. | C-glycosidderivate und deren salze |
| WO2006037537A2 (de) * | 2004-10-01 | 2006-04-13 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituierte phenyle, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| WO2008013321A1 (en) * | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI1651658T2 (sl) * | 2003-08-01 | 2020-12-31 | Mitsubishi Tanabe Pharma Corporation | Nove spojine, ki imajo inhibitorno aktivnost proti transporterju, ki je odvisen od natrija |
-
2009
- 2009-02-11 TW TW098104261A patent/TW201000494A/zh unknown
- 2009-02-11 UY UY031651A patent/UY31651A1/es not_active Application Discontinuation
- 2009-02-11 CL CL2009000309A patent/CL2009000309A1/es unknown
- 2009-02-11 AR ARP090100471A patent/AR070701A1/es unknown
- 2009-02-13 AU AU2009214278A patent/AU2009214278A1/en not_active Abandoned
- 2009-02-13 CN CN2009801125891A patent/CN101998962A/zh active Pending
- 2009-02-13 MX MX2010008051A patent/MX2010008051A/es unknown
- 2009-02-13 EP EP09711333A patent/EP2268653A2/de not_active Withdrawn
- 2009-02-13 BR BRPI0907572-0A patent/BRPI0907572A2/pt not_active IP Right Cessation
- 2009-02-13 WO PCT/EP2009/001042 patent/WO2009100936A2/de active Application Filing
- 2009-02-13 CA CA2714110A patent/CA2714110A1/en not_active Abandoned
- 2009-02-13 JP JP2010546266A patent/JP2011511820A/ja not_active Abandoned
- 2009-02-13 KR KR1020107017950A patent/KR20100121615A/ko not_active Application Discontinuation
-
2010
- 2010-07-25 IL IL207201A patent/IL207201A0/en unknown
- 2010-08-06 US US12/851,944 patent/US20110059910A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004052902A1 (de) * | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Neue aromatische fluorglycosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| EP1609785A1 (de) * | 2003-03-14 | 2005-12-28 | Astellas Pharma Inc. | C-glycosidderivate und deren salze |
| WO2006037537A2 (de) * | 2004-10-01 | 2006-04-13 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituierte phenyle, diese verbindungen enthaltende arzneimittel, deren verwendung und verfahren zu ihrer herstellung |
| WO2008013321A1 (en) * | 2006-07-28 | 2008-01-31 | Mitsubishi Tanabe Pharma Corporation | Novel sglt inhibitors |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2016041470A1 (en) * | 2014-09-15 | 2016-03-24 | National Institute Of Biological Sciences, Beijing | Sglt-2 inhibitors |
| CN107108539A (zh) * | 2014-09-15 | 2017-08-29 | 北京生命科学研究所 | 钠‑葡萄糖协同转运蛋白2(sglt‑2)抑制剂 |
Also Published As
| Publication number | Publication date |
|---|---|
| UY31651A1 (es) | 2009-09-30 |
| IL207201A0 (en) | 2010-12-30 |
| CL2009000309A1 (es) | 2009-06-26 |
| WO2009100936A3 (de) | 2009-10-22 |
| US20110059910A1 (en) | 2011-03-10 |
| JP2011511820A (ja) | 2011-04-14 |
| AU2009214278A1 (en) | 2009-08-20 |
| MX2010008051A (es) | 2010-08-10 |
| CN101998962A (zh) | 2011-03-30 |
| KR20100121615A (ko) | 2010-11-18 |
| AR070701A1 (es) | 2010-04-28 |
| BRPI0907572A2 (pt) | 2015-07-21 |
| EP2268653A2 (de) | 2011-01-05 |
| TW201000494A (en) | 2010-01-01 |
| CA2714110A1 (en) | 2009-08-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE102006053637B4 (de) | Neue mit Fluor substituierte 1,4-Benzothiepin-1,1-Dioxidderivate, diese Verbindungen enthaltende Arzneimittel und deren Verwendung | |
| DE102006053635B4 (de) | Neue mit Benzylresten substituierte 1,4-Benzothiepin-1,1-Dioxidderivate, diese Verbindungen enthaltende Arzneimittel und deren Verwendung | |
| DE102006053636B4 (de) | Neue mit Cyclohexylresten substituierte 1,4-Benzothiepin-1,1-Dioxidderivate und deren Verwendung | |
| EP2203416B1 (de) | (carboxylalkylen-phenyl)-phenyl-oxalamide, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2203415B1 (de) | (cyclopropyl-phenyl)-phenyl-oxalamide, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2083832B1 (de) | Neue 1,4-benzothiepin-1,1-dioxidderivate mit verbesserten eigenschaften, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| US20110059910A1 (en) | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof | |
| EP2590930B1 (de) | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| DE102006021878A1 (de) | Phenylamino-benzoxazol substituierte Carbonsäuren, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| US20090264402A1 (en) | Novel diphenylazetidinone substituted by piperazine-1-sulfonic acid and having improved pharmacological properties | |
| EP2220040B1 (de) | Neue kristalline diphenylazetidinonhydrate, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2252592A1 (de) | Arylchalcogeno-arylalkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung | |
| WO2009097996A1 (de) | Verwendung von substituierten phenylimidazolidinen zur herstellung von arzneimitteln zur behandlung des metabolischen syndroms | |
| EP2252591B1 (de) | Aryl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung | |
| WO2011107494A1 (de) | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2683704B1 (de) | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung | |
| WO2010000401A1 (de) | Verwendung von substituierten pyridinoncarbonsäurederivaten zur herstellung von medikamenten zur behandlung der dislipidämie | |
| DE102010015123A1 (de) | Benzylamidische Diphenylazetidinone, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980112589.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09711333 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009711333 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/008051 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 207201 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009214278 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2714110 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4922/CHENP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20107017950 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010546266 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PI 2010003309 Country of ref document: MY |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2009214278 Country of ref document: AU Date of ref document: 20090213 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0907572 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100812 |