WO2009096503A1 - ベンジルフェニルグルコピラノシド誘導体 - Google Patents
ベンジルフェニルグルコピラノシド誘導体 Download PDFInfo
- Publication number
- WO2009096503A1 WO2009096503A1 PCT/JP2009/051532 JP2009051532W WO2009096503A1 WO 2009096503 A1 WO2009096503 A1 WO 2009096503A1 JP 2009051532 W JP2009051532 W JP 2009051532W WO 2009096503 A1 WO2009096503 A1 WO 2009096503A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- deoxy
- phenyl
- fluoro
- hydroxymethyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/203—Monocyclic carbocyclic rings other than cyclohexane rings; Bicyclic carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present invention relates to a compound having a human SGLT1 and / or SGLT2 activity inhibitory action.
- Diabetes is a group of metabolic diseases characterized mainly by chronic hyperglycemia due to insufficient insulin action.
- pharmacotherapy is given along with diet therapy and exercise therapy.
- Diabetes treatment drugs include biguanides and thiazolidinediones that improve insulin resistance, sulfonylureas and glinides that promote insulin secretion from pancreatic ⁇ cells.
- Systemic drugs, ⁇ -glucosidase inhibitors that inhibit sugar absorption, and the like are used.
- Non-Patent Document 1 In recent years, research and development of drugs with a new mechanism that increases glucose excretion in urine and lowers blood glucose levels by inhibiting glucose reabsorption in the kidney has been promoted (for example, Non-Patent Document 1) reference).
- This drug inhibits the reabsorption of sugar from raw urine by inhibiting sodium-dependent glucose transporter 2 (hereinafter referred to as SGLT2) present in the proximal tubule of the kidney It has been shown that blood glucose levels are reduced by suppressing and increasing the excretion of sugar outside the body (see, for example, Non-Patent Document 2).
- SGLT2 sodium-dependent glucose transporter 2
- compounds that inhibit human SGLT2 are expected to normalize blood glucose levels by increasing urinary glucose excretion and are associated with various types of diabetes associated with type 1 and type 2 diabetes and hyperglycemia It becomes an effective drug for diseases.
- anti-obesity effect is expected by reducing sugar accumulation in the body by increasing sugar excretion.
- SGLT1 another subtype of SGLT (Sodium-dependent glucose cotransporter 1: hereinafter referred to as SGLT1), is expressed mainly in the small intestine and is a transporter that absorbs sugar (glucose, galactose) from food.
- SGLT1 sodium-dependent glucose cotransporter 1
- SGLT1 sodium-dependent glucose cotransporter 1
- drugs that suppress human SGLT1 and / or SGLT2 activity are potent type 1 and type 2 diabetes mellitus drugs, anti-obesity drugs, high anti-obesity drugs that have both urinary glucose excretion-increasing activity and glucose absorption inhibition activity from the small intestine. It is expected to be an effective drug for various related diseases accompanying blood glucose.
- O-aryl glucoside compounds have human SGLT2 inhibitory activity (see, for example, Patent Documents 1 to 5).
- Patent Documents 1 to 4 describes a compound having a substituent in the sugar moiety, and further describes or suggests that such a compound has a human SGLT1 inhibitory action. Absent.
- Patent Document 5 discloses a compound having a substituent in the sugar moiety and also describes that it has an SGLT1 inhibitory action. However, it is a patent document filed before the priority date of the present application and published after the priority date. is there.
- the present invention provides a compound having a novel structure and excellent human SGLT1 and / or SGLT2 inhibitory activity with low side effects or a hydrate thereof, type 1 diabetes, type 2 diabetes containing them as active ingredients Gestational diabetes, hyperglycemia due to other factors, impaired glucose tolerance (IGT), diabetes related diseases (eg obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver) Metabolic syndrome, edema, heart failure, angina, myocardial infarction, arteriosclerosis, hyperuricemia, gout, etc. or diabetic complications (eg retinopathy, nephropathy, neuropathy, cataract, foot gangrene, infection)
- ITT impaired glucose tolerance
- diabetes related diseases eg obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver
- Metabolic syndrome edema, heart failure, angina, myocardial infarction, arteriosclerosis, hyperuricemia, gout, etc.
- R 1 is a hydrogen atom or a hydroxyl group
- R 2 is a fluorine atom or a chlorine atom
- R 3 is a methyl group optionally substituted with a halogen atom, an ethyl group optionally substituted with a halogen atom, a cyclopropyl group, or a methoxy group optionally substituted with a halogen atom
- R 4 is a hydrogen atom or a methyl group, n is 1 or 2, m is 0 or 1;
- R 1 is a hydrogen atom
- R 4 is a hydrogen atom, and when R 1 is a hydroxyl group, R 4 is a methyl group.
- R 1 is a hydrogen atom or a hydroxyl group
- R 2 is a fluorine atom or a chlorine atom
- R 3 is a methyl group optionally substituted with a halogen atom, an ethyl group optionally substituted with a halogen atom, a cyclopropyl group, or a methoxy group optionally substituted with a halogen atom
- R 4 is a hydrogen atom or a methyl group, n is 1 or 2, m is 0 or 1;
- R 1 is a hydrogen atom
- R 4 is a hydrogen atom
- R 4 is a methyl group
- R 4 is a methyl group
- R 1 is a hydroxyl group
- R 4 is a methyl group
- R 3 is not a methyl group, an ethyl group, a cyclopropyl group or a methoxy group
- R 3 is not a methyl group, an ethyl group,
- a method for inhibiting human SGLT1 and / or human SGLT2 activity comprising administering to a mammal a therapeutically effective amount of the compound or hydrate thereof according to any one of (1) to (7) above
- (17) A method for treating or preventing a disease comprising administering to a mammal a therapeutically effective amount of the compound or hydrate thereof according to any one of (1) to (7) above
- Diabetes-related diseases are obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver, metabolic syndrome, edema, heart failure, angina, myocardial infarction, arteriosclerosis, high uric acid
- the diabetic complication is retinopathy, nephropathy, neuropathy, cataract, foot gangren
- a compound or hydrate thereof having excellent human SGLT1 and / or SGLT2 inhibitory activity with low side effects.
- these compounds or hydrates thereof are used as active ingredients, and type 1 diabetes, type 2 diabetes, gestational diabetes, hyperglycemia due to other factors, glucose intolerance, diabetes related diseases (for example, Obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver, metabolic syndrome, edema, heart failure, angina, myocardial infarction, arteriosclerosis, hyperuricemia, gout, etc.) or Pharmaceutical compositions for treating and / or preventing diabetic complications (eg, retinopathy, nephropathy, neuropathy, cataract, foot gangrene, infection, ketosis, etc.) can be provided.
- diabetic complications eg, retinopathy, nephropathy, neuropathy, cataract, foot gangrene, infection, ketosis, etc.
- halogen atom refers to a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- Table 1 shows preferred combinations of R 1 , R 2 , R 3 , R 4 , n and m in the general formula (I) or (II).
- hydrate refers to a pharmaceutically acceptable hydrate of the compound of the present invention.
- the compound of the present invention When the compound of the present invention is left in the air or recrystallized, it may absorb moisture and attach adsorbed water or become a hydrate. Such hydrates are also included in the “hydrate” in the present specification.
- the compound of the present invention has an asymmetric carbon atom in the molecule, these isomers in which optical isomers exist, and mixtures of these isomers are all represented by a single formula, that is, the general formula (I) or It is represented by (II). Accordingly, the present invention includes all optical isomers and mixtures of optical isomers in an arbitrary ratio.
- the compound (I) or (II) of the present invention can be produced, for example, according to Method A described later.
- the target compound of each reaction is collected from the reaction mixture according to a conventional method.
- the reaction mixture is appropriately neutralized, and if insolubles are present, after removing by filtration, water and an immiscible organic solvent such as ethyl acetate are added, and the organic layer containing the target compound is separated, After washing with water or the like and drying with anhydrous sodium sulfate or the like, the solvent is distilled off. If necessary, the obtained compound can be separated and purified by a conventional method such as silica gel column chromatography.
- the compound serving as a reaction substrate has a group that inhibits a desired reaction such as an amino group, a hydroxy group, or a carboxyl group
- a protecting group may be introduced into the group and the introduced protecting group may be removed.
- the protecting group is not particularly limited as long as it is a commonly used protecting group.For example, T. H. Greene, P. G. Wuts, Protective Groups in Organic Synthesis. Third Edition, ion1999, John Wiley & Sons, The protecting group described in Inc. etc. is mentioned.
- Such protecting group introduction reaction and removal reaction can be carried out according to a conventional method such as the method described in the above-mentioned literature.
- R 1 , R 2 , R 3 , R 4 , n and m are as defined above, and R 11 , R 12 and R 13 are the same or different and are a hydrogen atom or a protecting group.
- Step A1 is a step for producing compound (2), and is performed by reacting compound (1) with trichloroacetonitrile in the presence of a base in an inert solvent.
- Examples of the inert solvent used in the above reaction include halogenated hydrocarbons and ethers, preferably halogenated hydrocarbons, more preferably methylene chloride.
- Examples of the base used in the above reaction include organic amines, and preferably 1,8-diazabicyclo [5.4.0] -7-undecene.
- the reaction temperature varies depending on the raw material compound, base, inert solvent and the like, but is usually -20 ° C to reflux temperature, preferably 0 ° C to room temperature.
- the reaction time varies depending on the raw material compound, base, inert solvent, reaction temperature, etc., but is usually 15 minutes to 48 hours, preferably 30 minutes to 5 hours.
- Step A2 is a step for producing compound (I). After reacting compound (2) with compound (3) in the presence of a Lewis acid in an inert solvent, R 11 , R 12 and and removing the protecting group represented by R 13.
- Examples of the inert solvent used when the compound (2) is reacted with the compound (3) include halogenated hydrocarbons, aromatic hydrocarbons, ethers, nitriles, and preferably halogens Hydrocarbon, more preferably methylene chloride.
- Lewis acid used in the above reaction examples include boron trifluoride-diethyl ether complex and trimethylsilyl trifluoromethanesulfonate, and boron trifluoride-diethyl ether complex is preferable.
- the reaction temperature varies depending on the raw material compound, Lewis acid, inert solvent and the like, but is usually -30 ° C to reflux temperature, preferably 0 ° C to room temperature.
- the reaction time varies depending on the raw material compound, Lewis acid, inert solvent, reaction temperature, etc., but is usually 5 minutes to 24 hours, preferably 10 minutes to 12 hours.
- Step A3 is a step for producing compound (4), and is performed by reacting compound (1) with hydrobromic acetic acid in an inert solvent.
- Examples of the inert solvent used in the above reaction include halogenated hydrocarbons, and methylene chloride is preferable.
- the reaction temperature varies depending on the raw material compound, the inert solvent and the like, but is usually 0 ° C. to reflux temperature, preferably room temperature.
- the reaction time varies depending on the raw material compound, inert solvent, reaction temperature, etc., but is usually 5 to 50 hours, preferably 15 to 35 hours.
- Step A4 is a step of producing compound (I). After reacting compound (4) with compound (3) in the presence of silver carbonate in an inert solvent, R 11 , R 12 and and removing the protecting group represented by R 13.
- the removal of the protecting group may be performed in the same manner as in step A2.
- Examples of the inert solvent used in the above reaction include halogenated hydrocarbons, aromatic hydrocarbons, ethers, nitriles, etc., preferably halogenated hydrocarbons, more preferably methylene chloride. .
- the reaction temperature varies depending on the raw material compound, the inert solvent and the like, but is usually 0 ° C. to reflux temperature, preferably room temperature.
- the reaction time varies depending on the raw material compound, inert solvent, reaction temperature and the like, but is usually 5 to 150 hours, preferably 10 to 50 hours.
- Compound (1) which is a raw material compound of Method A can be produced by, for example, the following Method B, and Compound (3) can be produced by, for example, the following Method C or Method D.
- R 1 , R 4 , R 11 , R 12 and R 13 are as defined above, and R 14 is a hydrogen atom or a protecting group.
- Step B1 is a step for producing compound (1) and is performed by removing the protecting group represented by R 14 .
- the removal of the protecting group may be performed in the same manner as in step A2.
- the reaction is performed by reacting hydrazine acetate in an inert solvent.
- Examples of the inert solvent used in the above reaction include amides, and preferably dimethylformamide.
- the reaction temperature varies depending on the raw material compound, inert solvent and the like, but is usually 0 to 50 ° C., preferably room temperature.
- the reaction time varies depending on the raw material compound, inert solvent, reaction temperature, etc., but is usually 30 minutes to 35 hours, preferably 1 to 24 hours.
- Step B2 is a step for producing compound (6), and is performed by acetylating R 14 in compound (5) in the presence of an acid catalyst in an inert solvent.
- Examples of the inert solvent used in the above reaction include carboxylic acids and the like, and preferably acetic acid.
- the acid catalyst used in the above reaction is preferably an inorganic acid, more preferably sulfuric acid.
- the reaction temperature varies depending on the raw material compound, acid catalyst, inert solvent and the like, but is usually 0 to 50 ° C., preferably room temperature.
- the reaction time varies depending on the raw material compound, acid catalyst, inert solvent, reaction temperature and the like, but is usually 3 to 48 hours, preferably 6 to 24 hours.
- Step B3 is a step of producing compound (1), and is performed by reacting compound (6) with hydrazine acetate in an inert solvent. This step can be performed in the same manner as step B1.
- Compound (14) in the following scheme is a compound in which n is 1 in compound (3).
- Step C1 is a step for producing compound (9).
- compound (7) is reacted with compound (8) in the presence of a base, and a catalytic amount of trimethylsilylcyanide is added to react. Is done.
- inert solvent used in the above reaction examples include halogenated hydrocarbons, hydrocarbons, aromatic hydrocarbons, ethers, nitriles, etc., preferably nitriles, more preferably acetonitrile. is there.
- Examples of the base used in the above reaction include organic amines, and preferably triethylamine.
- the reaction temperature varies depending on the raw material compound, base, inert solvent and the like, but is usually 0 ° C. to reflux temperature, preferably room temperature to 60 ° C.
- the reaction time varies depending on the raw material compound, base, inert solvent, reaction temperature, etc., but is usually 10 minutes to 12 hours, preferably 2 to 4 hours.
- Step C2 is a step of producing compound (10), and is performed by reacting compound (9) with a halogenating agent in an inert solvent.
- Examples of the inert solvent used in the halogenation reaction that is the first stage of the above reaction include halogenated carbons, and preferably methylene chloride.
- R 2 is fluorine, for example, dimethylaminosulfur trifluoride, 2,2-difluoro-1,3-dimethylimidazolidine, hydrogen fluoride-pyridine, etc.
- R 2 is chlorine, for example, oxalyl dichloride, thionyl chloride, phosphorus oxychloride and the like are preferable, and oxalyl dichloride is preferable.
- the reaction temperature varies depending on the raw material compound, halogenating agent, inert solvent and the like, but is usually -30 to 100 ° C, preferably 0 ° C to room temperature.
- reaction time varies depending on the raw material compound, halogenating agent, inert solvent, reaction temperature, etc., but when R 2 is fluorine, it is usually 30 minutes to 12 hours, preferably 3 hours, and R 2 is chlorine. In this case, it is usually 30 minutes to 24 hours, preferably 1 to 3 hours.
- Step C3 is a step of producing compound (11), and is performed by reacting compound (10) with an oxidizing agent in a basic solvent.
- Examples of the basic solvent used in the above reaction include organic amines, and preferably N-methylmorpholine.
- oxidizing agent used in the above reaction examples include heavy metal salts such as permanganic acid and chromic acid, halogens such as bromine and iodine, and 2,3-dichloro-5,6-dicyano-p-benzoquinone. , Preferably halogen, more preferably iodine.
- the reaction temperature varies depending on the raw material compound, basic solvent, type of oxidizing agent, etc., but is usually 0 to 100 ° C., preferably room temperature.
- the reaction time varies depending on the raw material compound, basic solvent, oxidizing agent, reaction temperature, etc., but is usually 15 minutes to 12 hours, preferably 30 minutes to 2 hours.
- Step C4 is a step for producing compound (12), and is performed by reacting compound (11) with a reducing agent in an inert solvent.
- Examples of the inert solvent used in the above reaction include ethers and alcohols, preferably ethers, more preferably tetrahydrofuran.
- Examples of the reducing agent used in the above reaction include alkali metal borohydrides such as sodium borohydride and lithium borohydride, aluminum hydride compounds such as lithium aluminum hydride and lithium triethoxide aluminum, hydrogen And a hydride reagent such as sodium telluride, preferably an aluminum hydride compound, more preferably lithium aluminum hydride.
- the reaction temperature varies depending on the raw material compound, the reducing agent, the inert solvent and the like, but is usually -30 ° C to reflux temperature, preferably 0 ° C to room temperature.
- the reaction time varies depending on the raw material compound, reducing agent, inert solvent, reaction temperature, etc., but is usually 10 minutes to 10 hours, preferably 30 minutes to 1 hour.
- Step C5 is a step for producing compound (13), and is performed by reacting vinyl acetate with compound (12) in the presence of a bis (dibutyltin chloride) oxide catalyst in an inert solvent.
- Examples of the inert solvent used in the above reaction include ethers, and tetrahydrofuran is preferable.
- the reaction temperature varies depending on the raw material compound, the inert solvent and the like, but is usually 0 to 50 ° C., preferably room temperature to 35 ° C.
- the reaction time varies depending on the raw material compound, inert solvent, reaction temperature and the like, but is usually 1 to 100 hours, preferably 24 to 48 hours.
- Step C6 is a step for producing compound (14), and is performed by reacting compound (13) with a reducing agent in the presence of an acid or a Lewis acid in an inert solvent.
- inert solvent used in the above reaction examples include halogenated hydrocarbons, hydrocarbons, aromatic hydrocarbons, ethers, nitriles, etc., preferably nitriles, more preferably acetonitrile. is there.
- Examples of the acid or Lewis acid used in the above reaction include trifluoroacetic acid, boron trifluoride-diethyl ether complex, aluminum chloride, and the like, and preferably boron trifluoride-diethyl ether complex.
- Examples of the reducing agent used in the above reaction include trialkylsilane, alkali metal borohydride, aluminum hydride compound, etc., preferably trialkylsilane, and more preferably triethylsilane.
- the reaction temperature varies depending on the raw material compound, acid or Lewis acid, reducing agent, inert solvent, etc., but is usually ⁇ 20 to 50 ° C., preferably 0 ° C. to room temperature.
- the reaction time varies depending on the raw material compound, acid or Lewis acid, reducing agent, inert solvent, reaction temperature, etc., but is usually 30 minutes to 10 hours, preferably 1 to 2 hours.
- Compound (22) in the following scheme is a compound in which n is 2 in compound (3).
- Step D1 is a step of producing compound (15), and is performed by reacting compound (14) with a benzylating reagent in the presence of a base in an inert solvent.
- Examples of the inert solvent used in the above reaction include amides, and preferably dimethylformamide.
- Examples of the base used in the above reaction include inorganic bases, and potassium carbonate is preferable.
- benzylating reagent used in the above reaction examples include benzyl halide, benzyl arylsulfonate, benzyl alkylsulfonate, and the like, preferably benzyl halide, more preferably benzyl bromide.
- the reaction temperature varies depending on the raw material compound, base, benzylating reagent, inert solvent, etc., but is usually 0 to 100 ° C., preferably room temperature.
- the reaction time varies depending on the raw material compound, base, benzylating reagent, inert solvent, reaction temperature, etc., but is usually 30 minutes to 48 hours, preferably 2 to 4 hours.
- Step D2 is a step of producing compound (16), and is performed by reacting compound (15) with a base in an inert solvent.
- Examples of the inert solvent used in the above reaction include water and alcohols, preferably alcohols, more preferably methanol.
- Examples of the base used in the above reaction include inorganic bases, and potassium carbonate is preferable.
- the reaction temperature varies depending on the raw material compound, base, inert solvent and the like, but is usually 0 ° C. to reflux temperature, preferably room temperature.
- the reaction time varies depending on the raw material compound, base, inert solvent, reaction temperature and the like, but is usually 10 minutes to 24 hours, preferably 1 to 18 hours.
- Step D3 is a step of producing compound (17), and is performed by reacting compound (16) with an oxidizing agent in an inert solvent.
- Examples of the inert solvent used in the above reaction include hydrocarbons, halogenated hydrocarbons, aromatic hydrocarbons, ethers, esters, etc., preferably halogenated hydrocarbons, more preferably Chloroform.
- the reaction temperature varies depending on the raw material compound, oxidizing agent, inert solvent and the like, but is usually 0 to 200 ° C., preferably the reflux temperature.
- the reaction time varies depending on the raw material compound, oxidizing agent, inert solvent, reaction temperature, etc., but is usually 30 minutes to 48 hours, preferably 2 to 4 hours.
- Step D4 is a step of producing compound (18), and is performed by reacting a phosphonium salt with a base in an inert solvent and further reacting with compound (17).
- Examples of the inert solvent used in the above reaction include ethers, and tetrahydrofuran is preferable.
- Examples of the phosphonium salt used in the above reaction include halogenated (methoxymethyl) triphenylphosphonium, and preferably (methoxymethyl) triphenylphosphonium chloride.
- Examples of the base used in the above reaction include alkali metal bis (trimethylsilyl) amides and alkali metal dialkylamides, preferably alkali metal bis (trimethylsilyl) amide, more preferably lithium bis (trimethylsilyl) amide. It is.
- the reaction temperature varies depending on the raw material compound, phosphonium salt, base, inert solvent and the like, but is usually -78 ° C to reflux temperature, preferably 0 ° C to room temperature.
- the reaction time varies depending on the raw material compound, (methoxymethyl) phosphonium salt, base, inert solvent, reaction temperature, etc., but is usually 30 minutes to 24 hours, preferably 1 to 2 hours.
- Step D5 is a step of producing compound (19), and is performed by hydrolyzing compound (18) in the presence of an acid catalyst in an inert solvent.
- Examples of the inert solvent used in the above reaction include ethers, and preferably 1,4-dioxane.
- Examples of the acid catalyst used in the above reaction include inorganic acids, aryl sulfonic acids, etc., preferably inorganic acids, more preferably hydrochloric acid.
- the reaction temperature varies depending on the raw material compound, acid catalyst, inert solvent and the like, but is usually 0 ° C. to reflux temperature, preferably room temperature.
- the reaction time varies depending on the raw material compound, acid catalyst, inert solvent, reaction temperature, etc., but is usually 5 minutes to 10 hours, preferably 10 minutes to 1 hour.
- Step D6 is a step of producing compound (20), and is performed by reacting compound (19) with a reducing agent in an inert solvent.
- Examples of the inert solvent used in the above reaction include ethers and alcohols, preferably alcohols, more preferably methanol.
- Examples of the reducing agent used in the above reaction include alkali metal borohydrides such as sodium borohydride and lithium borohydride, aluminum hydride compounds such as lithium aluminum hydride and lithium triethoxide aluminum, hydrogen And a hydride reagent such as sodium telluride, preferably an alkali metal borohydride, more preferably sodium borohydride.
- the reaction temperature varies depending on the raw material compound, the reducing agent, the inert solvent and the like, but is usually -30 ° C to reflux temperature, preferably 0 ° C to room temperature.
- the reaction time varies depending on the raw material compound, reducing agent, inert solvent, reaction temperature, etc., but is usually 10 minutes to 10 hours, preferably 30 minutes to 1 hour.
- Step D7 is a step for producing compound (21).
- R 2 is fluorine
- it is carried out by catalytic reduction of compound (20) in the presence of hydrogen and a metal catalyst in an inert solvent.
- R 2 is chlorine
- the reaction is carried out by reacting compound (20) with a Lewis acid in an inert solvent.
- R 2 is fluorine
- examples thereof include ethers and alcohols, preferably alcohols, more preferably methanol, and R 2 is chlorine.
- ethers and alcohols preferably alcohols, more preferably methanol
- R 2 is chlorine.
- nitriles and the like can be mentioned, and acetonitrile is preferable.
- metal catalysts such as platinum, palladium, rhodium and nickel, preferably a palladium catalyst, and more preferably a palladium carbon catalyst.
- Examples of the Lewis acid used in the above reaction when R 2 is chlorine include trialkylsilyl halide, and trimethylsilyl iodide is preferable.
- reaction temperature varies depending on the raw material compound, metal catalyst, inert solvent, etc., but is usually 0 to 100 ° C., preferably room temperature, and when R 2 is chlorine, the raw material Although it varies depending on the compound, Lewis acid, inert solvent and the like, it is usually 0 to 100 ° C., preferably 40 ° C.
- reaction time when R 2 is fluorine, the starting compound, a metal catalyst in an inert solvent, varying reaction temperature and the like, usually, 10 minutes to 24 hours, preferably 30 minutes to 2 hours
- Step D8 is a step for producing compound (22), and is performed by reacting compound (21) with vinyl acetate in the presence of a bis (dibutyltin chloride) oxide catalyst in an inert solvent. The method is carried out in the same manner as in the method of protecting the primary hydroxyl group of compound (12) in step C5 with an acetyl group.
- the compounds of the present invention can be produced by using the above-mentioned methods, and examples described later from known compounds or methods well known in the art (for example, Chem. Ber, 71, 1938, 1843-1849, Carbohydr. Res. 1995). , 273,249-254, Bull.Chem.Soc.Jpn., 1982,55,938-942, Bull.Chem.Soc.Jpn., 1976,49,788-790, Org.Lett., 2003,5,3419-3421, Org. Biomol.Chem, 2003, 1,767-771, J. Chem. Soc., 1956, 2124-2126, WO 02/064606 pamphlet, Liebigs Ann. Chem, GE, 1992, 7, 747-758, etc.) Can be manufactured easily.
- the compound of the present invention or a hydrate thereof exhibits excellent human SGLT1 and / or SGLT2 inhibitory activity with low side effects, type 1 diabetes, type 2 diabetes, gestational diabetes, other factors, hyperglycemia, glucose intolerance, Diabetes-related diseases (eg, obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver, metabolic syndrome, edema, heart failure, angina, myocardial infarction, arteriosclerosis, hyperuricemia , Gout, etc.) or diabetic complications (eg, retinopathy, nephropathy, neuropathy, cataract, foot gangrene, infection, ketosis, etc.) are useful as an active ingredient of a pharmaceutical composition.
- Diabetes-related diseases eg, obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver, metabolic syndrome, edema, heart failure, angina, myocardial infarction, arteriosclerosis, hyperuricemia , Gout, etc
- Such pharmaceutical compositions can be administered to mammals (eg, humans, horses, cows, pigs, preferably humans).
- mammals eg, humans, horses, cows, pigs, preferably humans.
- the administration form may be any of oral administration such as tablets, capsules, granules, powders or syrups, or parenteral administration such as injections or suppositories.
- excipients eg, lactose, corn starch, crystalline cellulose, D-mannitol, anhydrous calcium hydrogen phosphate, sucrose, etc.
- disintegrants eg, corn starch, carmellose calcium, low substituted hydroxypropyl
- binders eg, hydroxypropylcellulose, hydroxypropylmethylcellulose, popidone, corn starch, methylcellulose, etc.
- lubricants eg, magnesium stearate, calcium stearate, talc) , Stearic acid, etc.
- fluidizing agents eg, light anhydrous silicic acid, talc, hydrous silicon dioxide, etc.
- coating agents eg, hydroxypropyl methylcellulose, hydride
- coating agents eg, hydroxypropyl methylcellulose, hydride
- the amount of the compound of the present invention or hydrate thereof used varies depending on symptoms, age, etc., but in the case of oral administration, 1 to 2000 mg, preferably 10 to 400 mg per day, and in the case of intravenous administration, It is desirable to administer 0.1 to 500 mg per day, preferably 1 to 300 mg per day, one to several times per day depending on the symptoms.
- Example 1a 4- (2-Fluoroethyl) benzoyl chloride 4- (2-fluoroethyl) benzoic acid (Bioorg. Med. Chem., 2005, 13, 77-78.) (1.8 g, 11 mmol) was salified The mixture was dissolved in methylene, added with oxalyl chloride (1.1 mL, 13 mmol) and N, N-dimethylformamide (0.1 mL, 1.3 mmol) under ice cooling, and stirred for 3 hours and a half while raising the temperature to room temperature. After completion of the reaction, the solvent was removed under reduced pressure to obtain a crude product (2.1 g) of the title compound as a colorless oil.
- Example 1b Ethyl 4- [4- (2-fluoroethyl) benzoyl] -3-hydroxy-5-oxocyclohex-3-enecarboxylic acid 3-hydroxy-5-oxocyclohex-3-enecarboxylic acid
- Ethyl (EP1571148A1) 2.0 g, 11 mmol
- the crude product obtained in Example 1a 2.1 g, 11 mmol
- triethylamine 4.6 mL, 33 mmol
- Trimethylsilyl cyanide (0.21 mL, 1.6 mmol) was added to this suspension, and the mixture was stirred at 60 ° C. for 2 hours.
- the reaction mixture was cooled to room temperature, diluted with ethyl acetate (50 mL), and washed successively with 2M hydrochloric acid (20 mL) and saturated brine (20 mL, twice).
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (4.3 g) of the title compound.
- Example 1c Ethyl 3-chloro-4- [4- (2-fluoroethyl) benzoyl] -5-oxocyclohex-3-enecarboxylate
- 2-methyl-2-butene 2.8 mL, 26 mmol
- oxalyl dichloride 0.83 mL, 9.8 mmol
- N, N-dimethylformamide 0.13 mmol
- reaction mixture was cooled to room temperature, diluted with methylene chloride (20 mL), and washed with saturated brine (20 mL, twice).
- the organic layer was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the residue was concentrated under reduced pressure twice under azeotropy with toluene (10 mL) to obtain a crude product (2.2 g) of the title compound.
- Example 1d Ethyl 3-chloro-4- [4- (2-fluoroethyl) benzoyl] -5-hydroxybenzoate
- the crude product obtained in Example 1c (2.2 g, 6.5 mmol) was converted to N-methyl. It melt
- the dark brown suspension was filtered through celite, diluted with ethyl acetate (50 mL), and washed successively with 2M hydrochloric acid (50 mL, 2 times), 30% aqueous sodium thiosulfate (20 mL), and saturated brine (20 mL). .
- the organic layer was dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to obtain a crude product (2.2 g) of the title compound.
- Example 1e 3-Chloro-2- ⁇ 1- [4- (2-fluoroethyl) phenyl] -1-hydroxymethyl ⁇ -5- (hydroxymethyl) phenol
- the crude product obtained in Example 1d ( 2.2 g, 6.5 mmol) was dissolved in tetrahydrofuran (65 mL), the gas phase was replaced with nitrogen, and lithium aluminum hydride (1.0 g, 26 mmol) was added in small portions under ice cooling. The mixture was stirred for 1 hour while warming to room temperature, and water (1 mL) was added dropwise under ice cooling to stop the reaction.
- Example 1f Acetic acid 3-chloro-4- ⁇ 1- [4- (2-fluoroethyl) phenyl] -1-hydroxymethyl ⁇ -5-hydroxybenzyl
- Compound obtained in Example 1e 1.0 g, 3.2 mmol
- porcine pancreatic lipase 1.0 g was added.
- the suspension was stirred at 37 ° C. for 8 hours, filtered through celite and washed with ethyl acetate (2 ⁇ 5 mL).
- the obtained filtrate was concentrated under reduced pressure to obtain a crude product of the title compound (1.1 g).
- Example 1g Acetic acid 3-chloro-2- [4- (2-fluoroethyl) benzyl] -5-hydroxybenzyl
- the crude product (0.90 g, 2.5 mmol) obtained in Example 1f was converted to acetonitrile (10 mL).
- triethylsilane (1.2 mL, 7.6 mmol) and boron trifluoride-diethyl ether complex (0.47 mL, 3.8 mmol) were added under ice cooling, and the mixture was stirred at 0 ° C. for 1.5 hours.
- the reaction mixture was diluted with ethyl acetate (20 mL), and washed successively with saturated aqueous sodium hydrogen carbonate solution (10 mL, twice) and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 5: 1, V / V) to give the title compound. (0.76 g, including about 20% of raw material).
- Example 1h 5-Acetoxymethyl-3-chloro-2- [4- (2-fluoroethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231)) (1.1 g, 1.8 mmol) was dissolved in methylene chloride (10 mL).
- Example 1i 3-chloro-2- [4- (2-fluoroethyl) benzyl] -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 1h
- the obtained crude product (1.8 mmol) was dissolved in a solution consisting of tetrahydrofuran (6 mL) and methanol (6 mL), 2M aqueous sodium hydroxide solution (6 mL, 12 mmol) was added, and the mixture was stirred at room temperature for 30 min.
- Example 2a (2-Chloro-4,6-dihydroxyphenyl) (4-ethylphenyl) methanone 1-Chloro-3,5-dimethoxybenzene (8.11 g, 47.0 mmol) was dissolved in toluene (40 mL), Aluminum chloride (6.26 g, 46.9 mmol) and 4-ethylbenzoyl chloride (6.89 mL, 47.0 mmol) were added, and the mixture was stirred at room temperature for 15 minutes, and then stirred at 85 ° C. for 3 hours. After cooling to room temperature, the reaction mixture was added to 1N aqueous hydrochloric acid (300 mL) under ice cooling.
- 1N aqueous hydrochloric acid 300 mL
- Example 2b 5-chloro-2,2-dimethyl-4- (4-ethylphenyl) -4H-benzo [1,3] dioxin-7-ol
- the crude product obtained in Example 2a (1.59 g , 5.75 mmol) was dissolved in methanol (15 mL), sodium borohydride (326 mg, 8.62 mmol) was added under ice cooling, and the mixture was stirred at room temperature for 30 min. After cooling to 0 ° C., saturated aqueous ammonium chloride solution (1 mL) was added, and the solvent was removed under reduced pressure.
- Example 2c Methyl 5-chloro-2,2-dimethyl-4- (4-ethylphenyl) -4H-benzo [1,3] dioxin-7-carboxylate
- methylene chloride 10 mL
- pyridine 0.235 mL, 2.91 mmol
- trifluoromethanesulfonic anhydride 0.90 mL, 2.32 mmol
- methylene chloride 20 mL
- washed with water (20 mL, 3 times The organic layer was dried over anhydrous sodium sulfate, and then the solvent was removed under reduced pressure. Toluene azeotropy was performed to obtain a crude product of triflate (790 mg).
- Example 2d 3-chloro-2- [1- (4-ethylphenyl) -1-methoxymethyl] -5- (hydroxymethyl) phenol Lithium aluminum hydride (103 mg, 2.71 mmol) in tetrahydrofuran (3 mL) After dissolution, under ice cooling, a tetrahydrofuran solution (4 mL) in which the compound obtained in Example 2c (651 mg, 1.80 mmol) was dissolved was added.
- the reaction mixture was stirred at room temperature for 30 minutes, cooled to 0 ° C, water (0.1 mL), 5 mol / L aqueous sodium hydroxide solution (0.1 mL) and water (0.3 mL) were sequentially added, and the mixture was stirred at room temperature for 1 hour. And left at room temperature for 14 hours. After filtration through celite, the solvent was removed under reduced pressure to obtain an oily crude alcohol product (470 mg).
- Example 2e Acetic acid 3-chloro-4- [1- (4-ethylphenyl) -1-methoxymethyl] -5-hydroxybenzyl Compound (401 mg, 1.31 mmol) obtained in Example 2d was added to tetrahydrofuran (4 mL). ), Vinyl acetate (4 mL) and bis (dibutyltin chloride) oxide (144 mg, 0.26 mmol) were added, and the mixture was stirred at room temperature for 72 hours. The solvent was removed under reduced pressure, and finally purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 3: 1, V / V) to obtain the title compound (380 mg) as an oil.
- Example 2f Acetic acid 3-chloro-4- (4-ethylbenzyl) -5-hydroxybenzyl
- the compound (380 mg, 1.09 mmol) obtained in Example 2e was dissolved in acetonitrile (8 mL) and cooled to 0 ° C. Thereafter, triethylsilane (0.520 mL, 3.26 mmol) and boron trifluoride-diethyl ether complex (0.210 mL, 1.67 mmol) were added, and the mixture was stirred at room temperature for 1 hour.
- reaction mixture was diluted with ethyl acetate (40 mL) and washed successively with aqueous sodium hydrogen carbonate solution (40 mL) and saturated brine (40 mL).
- aqueous sodium hydrogen carbonate solution 40 mL
- saturated brine 40 mL
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was removed under reduced pressure.
- the residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 2: 1, V / V) to obtain the title compound (293 mg) as a white solid.
- Example 2g 5-Acetoxymethyl-3-chloro-2- (4-ethylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco -Heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (722 mg, 1.18 mmol) was dissolved in methylene chloride (8 mL) and 0 After cooling to ° C., trichloroacetonitrile (0.597 mL, 5.91 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (0.018 mL, 0.12 mmol) were added, and the mixture was stirred for 1 hour under ice cooling.
- reaction mixture was diluted with ethyl acetate (20 mL) and washed successively with saturated aqueous ammonium chloride (20 mL) and saturated brine (20 mL).
- the organic layer was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. Toluene (2 mL) was added and azeotroped under reduced pressure to give the corresponding imidate as a yellow amorphous.
- Example 2f The compound (290 mg, 0.91 mmol) and imidate (890 mg, 1.18 mmol) obtained in Example 2f were dissolved in methylene chloride (8 mL), and molecular sieve 4A (manufactured by Nacalai Tesque, hereinafter referred to as MS4A) was added.
- MS4A molecular sieve 4A
- Boron trifluoride-diethyl ether complex (0.148 mL, 1.18 mmol) was added dropwise under ice cooling, and the mixture was stirred under ice cooling for 15 minutes and then at room temperature for 15 minutes.
- Example 2h 3-chloro-2- (4-ethylbenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside
- methanol / methylene chloride 16 mL / 4 mL
- potassium carbonate (1.63 g, 11.8 mmol) was added, and the mixture was stirred overnight at room temperature.
- an appropriate amount of acetic acid was added for neutralization, and methanol was removed under reduced pressure.
- Example 3a Ethyl 4- [4- (difluoromethoxy) benzoyl] -3-hydroxy-5-oxocyclohex-3-enecarboxylate ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate ( 2.0 g, 11 mmol) and 4- (difluoromethoxy) benzoyl chloride (J. Org. Chem. USSR, 1981, 1470-1475.) (2.3 g, 11 mmol) (2.3 g, 11 mmol) in the same manner as in Example 1b. Crude product (3.3 g) was obtained.
- Example 3b Ethyl 4- [4- (difluoromethoxy) benzoyl] -3-fluoro-5-oxocyclohex-3-enecarboxylate
- methylene chloride 10 mL
- diethylaminosulfur trifluoride 1.7 mL, 13 mmol
- the reaction mixture was diluted with ethyl acetate (30 mL), and washed successively with saturated brine (10 mL), saturated aqueous sodium hydrogen carbonate solution (20 mL, twice) and saturated brine (10 mL).
- Example 3c Ethyl 4- [4- (difluoromethoxy) benzoyl] -3-fluoro-5-hydroxybenzoate
- the compound obtained in Example 3b (1.2 g, 3.4 mmol) was converted to N-methylmorpholine (15 mL).
- anhydrous sodium sulfate (1.2 g) and iodine (0.97 g, 3.8 mmol) were added at room temperature, followed by stirring at room temperature for 1 hour.
- the dark brown suspension was filtered through celite, diluted with ethyl acetate (50 mL), and washed successively with 2M hydrochloric acid (50 mL, 2 times), 30% aqueous sodium thiosulfate (20 mL), and saturated brine (20 mL). .
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (1.2 g) of the title compound.
- Example 3d Ethyl 4- ⁇ 1- [4- (difluoromethoxy) phenyl] -1-hydroxymethyl ⁇ -3-fluoro-5-hydroxybenzoate
- the crude product obtained in Example 3c (1.5 g, 4.1 mmol) was dissolved in ethanol (20 mL), and sodium borohydride (0.31 g, 8.2 mmol) was added under ice cooling. The mixture was stirred for 2 hours while warming to room temperature, and saturated aqueous ammonium chloride (1 mL) was added dropwise under ice cooling to stop the reaction.
- Example 3e Ethyl 4- [4- (difluoromethoxy) benzyl] -3-fluoro-5-hydroxybenzoate
- the compound obtained in Example 3d (0.82 g, 2.3 mmol) was dissolved in ethanol (8 mL). The solution was stirred for 10 minutes while blowing nitrogen into the solution.
- 2M hydrochloric acid (0.57 mL, 1.1 mmol) and 10% palladium carbon catalyst (containing water, 0.5 g) were added under a nitrogen stream, and the gas phase was replaced with hydrogen, followed by stirring at room temperature for 6 hours. The mixture was filtered through celite, and the solvent was removed under reduced pressure to obtain a crude product of the title compound (0.76 g) as a colorless solid.
- Example 3f 2- [4- (Difluoromethoxy) benzyl] -3-fluoro-5- (hydroxymethyl) phenol
- the crude product obtained in Example 3e (0.76 g, 2.2 mmol) was added to tetrahydrofuran (10 mL). After the gas phase was replaced with nitrogen, lithium aluminum hydride (0.25 g, 6.6 mmol) was added in small portions under ice cooling. The mixture was stirred for 1 hour while warming to room temperature, and water (1 mL) was added dropwise under ice cooling to stop the reaction. The mixture was diluted with ethyl acetate (30 mL), and washed successively with 2M hydrochloric acid (10 mL, twice) and saturated brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (0.65 g) of the title compound as a colorless solid.
- Example 3g Acetic acid 4- [4- (difluoromethoxy) benzyl] -3-fluoro-5-hydroxybenzyl
- the crude product obtained in Example 3f (0.65 g, 2.2 mmol) was added to vinyl acetate (5 mL) and Dissolved in a solution consisting of diisopropyl ether (5 mL), porcine pancreatic lipase (1.0 g) was added.
- the suspension was stirred at 37 ° C. for 8 hours and the suspension was filtered through celite and washed with ethyl acetate (5 mL, 2 ⁇ ).
- Example 3h 5-Acetoxymethyl-2- [4- (difluoromethoxy) benzyl] -3-fluorophenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ - D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.37 g, 0.60 mmol) in methylene chloride (4 mL) Dissolve and prepare an imidate in the same manner as Example 1h using trichloroacetonitrile (0.18 mL, 1.8 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (2.7 ⁇ L, 0.018 mmol).
- Example 3g Similar to Example 1h using the obtained imidate (0.45 g, 0.60 mmol), the compound obtained in Example 3g (0.20 g, 0.60 mmol) and boron trifluoride-diethyl ether complex (37 ⁇ L, 0.30 mmol). By this method, a mixture containing the title compound was obtained.
- the mixture (0.60 mmol) was dissolved in a solution consisting of tetrahydrofuran (2 mL) and methanol (2 mL), 2M aqueous sodium hydroxide solution (2 mL, 4 mmol) was added, and the mixture was stirred at room temperature for 30 min.
- Example 4a Ethyl 3-hydroxy-4- (4-methylbenzoyl) -5-oxocyclohex-3-enecarboxylate
- Ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate (3.0 g, 16.3 mmol) was dissolved in acetonitrile (50 mL), triethylamine (6.8 mL, 48.8 mmol) and p-toluoyl chloride (2.26 mL, 17.1 mmol) were added, and the mixture was stirred at room temperature for 10 minutes. Further, trimethylsilylcyanide (260 ⁇ L, 1.95 mmol) was added and stirred at 60 ° C. for 2 hours.
- Example 4b Ethyl 3-chloro-4- (4-methylbenzoyl) -5-oxocyclohex-3-enecarboxylate
- methylene chloride 10 mL
- Oxalyl dichloride 300 ⁇ L, 3.5 mmol
- 2-methyl-2-butene 1.25 mmol
- N, N-dimethylformamide 100 ⁇ L, 1.29 mmol
- Example 4c Ethyl 3-chloro-5-hydroxy-4- (4-methylbenzoyl) benzoate To the crude product (1.2 g) obtained in Example 4b, N-methylmorpholine (10 mL) and iodine ( 1.68 g, 6.6 mmol) was added, and the mixture was stirred at room temperature for 2 hours. After filtration through celite, the mixture was diluted with toluene (10 mL), and washed successively with aqueous sodium sulfite solution (10 mL) and saturated brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified using silica gel flash column chromatography (hexane: ethyl acetate, 5: 1-4: 1-3: 1, V / V) to give the title compound (0.77 g) as a yellow solid.
- Example 4d Ethyl 3-chloro-5-hydroxy-4- [1-hydroxy-1- (4-methylphenyl) methyl] benzoate
- the compound obtained in Example 4c (0.77 g, 2.4 mmol) was dissolved in methanol. (6 mL) and tetrahydrofuran (1 mL) were dissolved in the solution. Under ice cooling, sodium borohydride (137 mg, 3.6 mmol) was added, and the mixture was stirred at 0 ° C. for 1 hour. Under ice cooling, a saturated aqueous ammonium chloride solution (5 mL) was added to the reaction solution, and the solvent was distilled off under reduced pressure.
- Example 4e Ethyl 5-chloro-2,2-dimethyl-4- (4-methylphenyl) -4H-benzo [1,3] dioxin-7-carboxylate
- acetone 6 mL
- boron trifluoride-diethyl ether complex (220 ⁇ L, 1.8 mmol) was added at ⁇ 10 ° C., and the mixture was stirred for 3 hours while slowly warming to 0 ° C.
- saturated aqueous sodium hydrogen carbonate solution (5 mL) was added to the reaction mixture, diluted with ethyl acetate (20 mL), and washed with saturated brine (10 mL).
- Example 4f 5-chloro-2,2-dimethyl-4- (4-methylphenyl) -4H-benzo [1,3] dioxin-7-ylmethanol
- the compound obtained in Example 4e (0.65 g, 1.8 mmol) was dissolved in tetrahydrofuran (10 mL), and lithium aluminum hydride (101 mg, 2.7 mmol) was added under ice cooling, followed by stirring at room temperature for 2 hours.
- Distilled water (5 mL) was added to the reaction mixture under ice cooling, diluted with ethyl acetate (10 mL), and washed successively with 2M hydrochloric acid (10 mL), saturated aqueous sodium hydrogen carbonate solution (10 mL), and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (0.55 g) of the title compound as a colorless solid.
- Example 4g Acetic acid 5-chloro-2,2-dimethyl-4- (4-methylphenyl) -4H-benzo [1,3] dioxin-7-ylmethyl
- methylene chloride (7 mL) acetic anhydride (200 ⁇ L, 2.1 mmol)
- pyridine 350 ⁇ L, 3.5 mmol
- 4-dimethylaminopyridine 65 mg, 0.5 mmol
- the reaction mixture was diluted with ethyl acetate (15 mL) and washed successively with 2M hydrochloric acid (10 mL), saturated aqueous sodium hydrogen carbonate solution (10 mL) and saturated brine (10 mL).
- 2M hydrochloric acid 10 mL
- saturated aqueous sodium hydrogen carbonate solution 10 mL
- saturated brine 10 mL
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (0.65 g) of the title compound as a colorless oil.
- Example 4h 3-chloro-5-hydroxy-4- (4-methylbenzyl) benzyl acetate
- Acetic acid (0.65 g, 1.8 mmol) obtained in Example 4 g was dissolved in acetonitrile (7 mL) and iced. Under cooling, triethylsilane (850 ⁇ L, 5.3 mmol) and boron trifluoride-diethyl ether complex (340 ⁇ L, 2.7 mmol) were added, and the mixture was stirred at 0 ° C. for 1 hour.
- Example 4i 5-Acetoxymethyl-3-chloro- (4-methylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero-D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231)) (240 mg, 0.39 mmol) in methylene chloride (5 mL ), Trichloroacetonitrile (120 ⁇ L, 1.20 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (6 ⁇ L, 0.04 mmol) were added under ice cooling, and the mixture was stirred at 0 ° C.
- the obtained imidate was dissolved in methylene chloride (5 mL), and the compound obtained in Example 4h (100 mg, 0.33 mmol) and boron trifluoride-diethyl ether complex (50 ⁇ L, 0.39 mmol) were added under ice cooling. And stirred at 0 ° C. for 2 hours. Triethylamine (100 ⁇ L, 0.72 mmol) was added to the reaction mixture, and the mixture was diluted with ethyl acetate (10 mL) and washed with saturated brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (350 mg) of the title compound.
- Example 4j 3-Chloro-5-hydroxymethyl-2- (4-methylbenzyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside
- a solution consisting of tetrahydrofuran (1 mL) and methanol (4 mL) sodium carbonate (46 mg, 0.33 mmol) was added, and the mixture was stirred at room temperature for 2 hours.
- Example 5a Ethyl 4- (3-fluoro-4-methylbenzoyl) -3-hydroxy-5-oxocyclohex-3-enecarboxylate
- Ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate (0.35 g, 1.9 mmol) was dissolved in acetonitrile (4 mL), triethylamine (0.79 mL, 5.7 mmol), 3-fluoro-4-methylbenzoyl chloride (J. Med. Chem., 1997, 40, 2064-2084.
- Example 1b (0.33 g, 1.9 mmol) and trimethylsilylcyanide (0.030 mL, 0.23 mmol) were used in the same manner as in Example 1b to obtain a crude product.
- the crude product was purified using silica gel flash column chromatography (methylene chloride: methanol, 10: 1, V / V) to obtain the title compound (0.61 g).
- Example 5b Ethyl 4- (3-fluoro-4-methylbenzoyl) -3-fluoro-5-oxocyclohex-3-enecarboxylate
- the compound obtained in Example 5a (0.61 g, 1.9 mmol) was obtained.
- the title compound (0.31 g) was obtained by dissolving in methylene chloride (8 mL) and using diethylaminosulfur trifluoride (0.75 mL, 5.7 mmol) in the same manner as Example 3b.
- Example 5c Ethyl 4- (3-fluoro-4-methylbenzoyl) -3-fluoro-5-hydroxybenzoate Dissolve the compound obtained in Example 5b (0.31 g, 0.96 mmol) in acetonitrile (3 mL). Then, ice cooling, triethylamine (0.40 mL, 2.9 mmol) and trimethylsilyl iodide (0.34 mL, 2.4 mmol) were added, and the mixture was stirred at room temperature for 2 hours. After completion of the reaction, the reaction solution was diluted with toluene (10 mL) under ice cooling, and washed twice with a phosphate buffer solution (pH 7, 5 mL).
- the reaction mixture was cooled to room temperature, diluted with ethyl acetate (20 mL), and washed successively with 1M hydrochloric acid (10 mL), saturated aqueous sodium hydrogen carbonate solution (5 mL), and saturated brine (5 mL).
- 1M hydrochloric acid 10 mL
- saturated aqueous sodium hydrogen carbonate solution 5 mL
- saturated brine 5 mL
- the organic layer was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the obtained residue was purified by silica gel flash column chromatography (hexane: ethyl acetate, 3: 1, V / V) to obtain the title compound (0.17 g).
- Example 5d 3-Fluoro-2- [1- (3-fluoro-4-methylphenyl) -1-hydroxymethyl] -5- (hydroxymethyl) phenol
- the compound obtained in Example 5c (0.17 g, 0.53 mmol) was dissolved in tetrahydrofuran (5 mL), and the title compound (0.13 g) was obtained as a crude product in the same manner as in Example 1e using lithium aluminum hydride (60 mg, 1.6 mmol).
- Example 5e Acetic acid 3-fluoro-4- [1- (3-fluoro-4-methylphenyl) -1-hydroxymethyl] -5-hydroxybenzyl
- Crude product obtained in Example 5d (0.13 g, 0.46 mmol) was dissolved in a solution consisting of vinyl acetate (3 mL) and diisopropyl ether (3 mL), porcine pancreatic lipase (0.13 g) was added, and the mixture was stirred at room temperature for 2 days. After completion of the reaction, the mixture was filtered through celite, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel flash column chromatography (hexane: ethyl acetate, 4: 1, V / V) to obtain the title compound (0.12 g).
- Example 5f Acetic acid 3-fluoro-4- (3-fluoro-4-methylbenzyl) -5-hydroxybenzyl
- the compound (0.12 g, 0.37 mmol) obtained in Example 5e was dissolved in acetonitrile (3 mL).
- Triethylsilane (0.18 mL, 1.1 mmol) and boron trifluoride-diethyl ether complex (70 ⁇ L, 0.56 mmol) were added at ⁇ 40 ° C., and the mixture was stirred for 1 hour while warming to room temperature.
- Example 5g 5-acetoxymethyl-3-fluoro-2- (3-fluoro-4-methylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.37 g, 0.60 mmol) in methylene chloride (4 mL) Prepared in the same manner as in Example 1h using trichloroacetonitrile (0.18 mL, 1.8 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (2.7 ⁇ L, 0.018 mmol).
- Example 5f Using this imidate (0.45 g, 0.60 mmol), the compound obtained in Example 5f (85 mg, 0.28 mmol) and boron trifluoride-diethyl ether complex (37 ⁇ L, 0.30 mmol), the same method as in Example 1h To give a mixture containing the title compound.
- Example 5h 3-Fluoro-2- (3-fluoro-4-methylbenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 5g
- the resulting mixture (0.28 mmol) was dissolved in a solution consisting of tetrahydrofuran (2 mL) and methanol (1 mL), 2M aqueous sodium hydroxide solution (2 mL, 4 mmol) was added, and the title compound (84 mg) was prepared in the same manner as in Example 3i. Obtained as a colorless solid.
- Example 6a 3-Fluoro-4-methylbenzoyl chloride 3-fluoro-4-methylbenzoic acid (880 mg, 5.71 mmol), oxalyl dichloride (0.50 mL, 5.71 mmol), catalytic amount of N, N-dimethylformamide
- a crude product of the title compound (950 mg) was obtained as a colorless oil in the same manner as in Example 1a using tetrahydrofuran and tetrahydrofuran (10 mL).
- Example 6b Ethyl 4- (3-fluoro-4-methylbenzoyl) -3-hydroxy-5-oxocyclohex-3-enecarboxylate Ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate (950 mg, 5.16 mmol), using the crude product obtained in Example 6a (950 mg, 11 mmol), acetonitrile (10 mL), triethylamine (2.27 mL, 1.63 mmol) and trimethylsilylcyanide (0.087 mL, 0.65 mmol). In the same manner as in Example 1b, a crude product (1.65 g) of the title compound was obtained.
- Example 6c Ethyl 3-chloro-4- (3-fluoro-4-methylbenzoyl) -5-oxocyclohex-3-enecarboxylate Crude product obtained in Example 6b (1.65 g, 5.15 mmol ), Methylene chloride (20 mL), 2-methyl-2-butene (2.19 mL, 20.6 mmol), oxalyl dichloride (0.46 mL, 5.36 mmol) and a catalytic amount of N, N-dimethylformamide as in Example 1c. By the method, a crude product (1.75 g) of the title compound was obtained.
- Example 6d Ethyl 3-chloro-4- (3-fluoro-4-methylbenzoyl) -5-hydroxybenzoate
- Crude product obtained in Example 6c (1.75 g, 5.15 mmol), N-methylmorpholine (20 mL), anhydrous sodium sulfate (14.6 g) and iodine (1.43 g, 5.63 mmol) were used in the same manner as in Example 1d to obtain a crude product of the title compound.
- purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 3: 1, V / V) to obtain the amorphous title compound (1.28 g).
- Example 6e Ethyl 5-chloro-2,2-dimethyl-4- (3-fluoro-4-methylphenyl) -4H-benzo [1,3] dioxin-7-benzoate Obtained in Example 6d
- a crude diol product (1.28 g) was obtained in the same manner as in Example 2b using the compound (1.28 g, 3.80 mmol), methanol (12 mL) and sodium borohydride (290 mg, 7.67 mmol). Boron trifluoride-diethyl ether complex (0.475 mL, 3.78 mmol) cooled to ⁇ 10 ° C.
- Example 6f 5-Chloro-2,2-dimethyl-4- (3-fluoro-4-methylphenyl) -4H-benzo [1,3] dioxin-7-ylmethanol
- Compound obtained in Example 6e (960 mg, 2.53 mmol), lithium aluminum hydride (96 mg, 2.53 mmol) and tetrahydrofuran (5 mL) were used in the same manner as in Example 2d to obtain a crude product (860 mg) of the title compound as an amorphous product.
- Example 6g Acetic acid 5-chloro-2,2-dimethyl-4- (3-fluoro-4-methylphenyl) -4H-benzo [1,3] dioxin-7-ylmethyl Crude obtained in Example 6f The product (860 mg) was dissolved in pyridine (8.6 mL), acetic anhydride (2.2 mL) was added under ice cooling, and the mixture was stirred at room temperature for 1 hr.
- Example 6h Acetic acid 3-chloro-4- (3-fluoro-4-methylbenzyl) -5-hydroxybenzyl
- acetonitrile 15 mL
- triethylsilane 1.14 mL, 7.09 mmol
- boron fluoride-diethyl ether complex 0.450 mL, 3.58 mmol
- Example 6i 5-Acetoxymethyl-3-chloro-2- (3-fluoro-4-methylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (250 mg, 0.41 mmol), methylene chloride (4 mL), The imidate was prepared in the same manner as in Example 2g using trichloroacetonitrile (0.210 mL, 2.08 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (6 ⁇ L, 0.04 mmol).
- Example 6h Using the obtained imidate (309 mg), the compound obtained in Example 6h (100 mg, 0.41 mmol), methylene chloride (4 mL), MS4A and boron trifluoride-diethyl ether complex (0.051 mL, 0.41 mmol) In the same manner as in Example 2g, a crude product (280 mg) of the title compound was obtained.
- Example 6j 3-chloro-2- (3-fluoro-4-methylbenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 6i Using the crude product (280 mg), methanol / methylene chloride (8 mL / 2 mL) and potassium carbonate (566 mg, 4.10 mmol), the title compound (76 mg) was obtained as a colorless solid in the same manner as in Example 2h. However, solidification was performed from hexane / ethyl acetate.
- Example 7a 3,5-difluoro-4- [1- (3-fluoro-4-methoxyphenyl) -1-hydroxymethyl] benzonitrile Dissolve diisopropylamine (1.20 mL, 8.49 mmol) in tetrahydrofuran (7 mL) Then, n-butyllithium (2.60 mL, 7.19 mmol, 2.77 M n-hexane solution) was added dropwise under ice cooling.
- the reaction mixture was diluted with ethyl acetate (60 mL), and washed successively with water (30 mL) and saturated aqueous sodium hydrogen carbonate solution (30 mL).
- the organic layer was dried over anhydrous magnesium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 3: 1 to 2: 1, V / V). To give the oily title compound (1.59 g).
- Example 7b 3,5-difluoro-4- (3-fluoro-4-methoxybenzyl) benzonitrile
- acetonitrile 16 mL
- boron trifluoride-diethyl ether complex 1.0 mL, 7.9 mmol
- the reaction mixture was diluted with ethyl acetate (60 mL), the organic layer was dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the resulting residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 9: 1 to 2: 1, V / V) to obtain the title compound (1.05 g) as a colorless solid.
- Example 7c 3-Benzyloxy-5-fluoro-4- (3-fluoro-4-methoxybenzyl) benzonitrile
- the compound obtained in Example 7b (1.05 g, 3.79 mmol) and benzyl alcohol (0.53 g, 4.9 mmol) was dissolved in N, N-dimethylformamide (11 mL), and sodium hydride (63%, 0.22 g, 5.7 mmol) was added under ice cooling. After stirring at the same temperature for 2 hours, 2M hydrochloric acid was added dropwise to the reaction solution to stop the reaction.
- reaction solution was diluted with ethyl acetate (100 mL), washed with water (50 mL, twice), and the organic layer was dried over anhydrous magnesium sulfate. After evaporating the solvent under reduced pressure, the resulting residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 4: 1 to 2: 1, V / V) to give a mixture containing the title compound (1.05 g) was obtained.
- Example 7d 3-Benzyloxy-5-fluoro-4- (3-fluoro-4-methoxybenzyl) benzoic acid
- the mixture obtained in Example 7c (1.04 g, 2.85 mmol) was added to ethanol (14.5 mL).
- the precipitated solid was collected by filtration, washed with water, and then dried under reduced pressure to obtain a mixture containing the title compound.
- Example 7e 3-Fluoro-4- (3-fluoro-4-methoxybenzyl) -5-hydroxybenzoic acid
- the mixture (2.85 mmol) obtained in Example 7d was taken from tetrahydrofuran (10 mL) and methanol (10 mL). The solution was dissolved in the solution, and nitrogen substitution was performed while stirring. A 10% palladium carbon catalyst (containing water, 0.20 g) was added to the reaction solution, and the gas phase was replaced with hydrogen, followed by stirring at room temperature for 40 minutes. The mixture was filtered through a membrane filter to remove the catalyst, and then the solvent was distilled off under reduced pressure to obtain a mixture (0.90 g) containing the title compound as a colorless solid.
- Example 7f 3-Fluoro-2- (3-fluoro-4-methoxybenzyl) -5- (hydroxymethyl) phenol
- Lithium aluminum hydride (0.27 g, 7.1 mmol) was suspended in tetrahydrofuran (2 mL) and iced. Under cooling, a tetrahydrofuran solution (28 mL) in which the mixture (0.90 g, 2.85 mmol) obtained in Example 7e was dissolved was added dropwise. After stirring at 50 ° C. for 2 hours, water and 2M hydrochloric acid were added dropwise to the reaction solution under ice cooling to stop the reaction.
- the reaction mixture was diluted with ethyl acetate (60 mL), washed with saturated brine (20 mL, twice), and the organic layer was dried over anhydrous magnesium sulfate.
- the residue obtained by evaporating the solvent under reduced pressure was purified using silica gel flash chromatography (hexane: ethyl acetate, 1: 1, V / V) to obtain the title compound (0.61 g) as a colorless solid. .
- Example 7g 3-fluoro-4- (3-fluoro-4-methoxybenzyl) -5-hydroxybenzyl acetate
- the compound (0.60 g, 2.14 mmol) obtained in Example 7f was dissolved in tetrahydrofuran (3 mL).
- Vinyl acetate (3 mL) and bis (dibutyltin chloride) oxide (0.35 g, 0.63 mmol) were added, and the mixture was stirred at room temperature for 3 days.
- Example 7h 5-Acetoxymethyl-3-fluoro-2- (3-fluoro-4-methoxybenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.68 g, 1.1 mmol), methylene chloride (14 mL) , Trichloroacetonitrile (0.34 mL, 3.4 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (15 ⁇ L, 0.10 mmol) were used to prepare an imidate in the same manner as Example 1h.
- Example 1h A crude product of the title compound was obtained in the same manner as above.
- Example 7i 3-Fluoro-2- (3-fluoro-4-methoxybenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 7h
- the crude product was dissolved in a solution consisting of tetrahydrofuran (2 mL) and methanol (6 mL), and a methanol solution (3 mL) in which sodium methoxide (28% methanol solution, 0.36 g, 1.9 mmol) was dissolved was added at room temperature. I left it all night.
- Example 8a Ethyl 4- (3-fluoro-4-methoxybenzoyl) -3-hydroxy-5-oxocyclohex-3-enecarboxylate
- Ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate 800 mg, 4.34 mmol
- 3-fluoro-4-methoxybenzoyl chloride 819 mg, 4.34 mmol
- acetonitrile 14 mL
- triethylamine (1.82 mL, 13.1 mmol
- trimethylsilylcyanide 0.070 mL, 0.52 mmol
- Example 8b Ethyl 3-chloro-4- (3-fluoro-4-methoxybenzoyl) -5-oxocyclohex-3-enecarboxylate
- Methylene chloride (18 mL
- 2-methyl-2-butene (1.74 mL, 16.4 mmol)
- oxalyl dichloride 0.348 mL, 4.06 mmol
- a crude product (1.37 g) of the title compound was obtained.
- Example 8c Ethyl 3-chloro-4- (3-fluoro-4-methoxybenzoyl) -5-hydroxybenzoate
- Crude product obtained in Example 8b (1.37 g, 3.86 mmol), N-methylmorpholine (15.6 mL), anhydrous sodium sulfate (11.0 g) and iodine (1.18 g, 4.65 mmol) were used to give a crude product of the title compound in the same manner as in Example 1d.
- purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 3: 1, V / V) to obtain the amorphous title compound (672 mg).
- Example 8d Ethyl 3-chloro-4- (3-fluoro-4-methoxybenzyl) -5-hydroxybenzoate Compound (670 mg, 1.90 mmol) obtained in Example 8c, methanol (8 mL) and hydrogenation 3-chloro-4- [1- (3-fluoro-4-methoxyphenyl) -1-hydroxymethyl] -5-hydroxybenzoate was prepared in the same manner as in Example 2b using sodium boron (144 mg, 3.81 mmol). Ethyl acid (670 mg) was obtained as a crude product and used in the next reaction without purification.
- Example 8e 3-chloro-4- (3-fluoro-4-methoxybenzyl) -5-hydroxybenzyl acetate
- Acetic acid mixture (525 mg), lithium aluminum hydride (118 mg, 3.11 mmol) and Tetrahydrofuran (8 mL) was used to obtain a mixture (333 mg) as amorphous by the same method as in Example 2d.
- this mixture (322 mg) was dissolved in acetonitrile (4 mL), triethylsilane (0.430 mL, 2.70 mmol) was added, and after cooling to 0 ° C., boron trifluoride-diethyl ether complex (0.170 mL, 1.35 mmol) was added. And stirred at room temperature for 3 hours. Ethyl acetate (40 mL) was added to the reaction mixture, and the mixture was washed with saturated aqueous sodium hydrogen carbonate solution (20 mL) and saturated brine (20 mL).
- Example 8f 5-Acetoxymethyl-3-chloro-2- (3-fluoro-4-methoxybenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (328 mg, 0.54 mmol), methylene chloride (5 mL), The imidate was prepared in the same manner as Example 2g using trichloroacetonitrile (0.272 mL, 2.69 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (7 ⁇ L, 0.05 mmol).
- Example 8e Using the resulting imidate (406 mg), the compound obtained in Example 8e (140 mg, 0.41 mmol), methylene chloride (5 mL), MS4A and boron trifluoride-diethyl ether complex (0.068 mL, 0.54 mmol) In the same manner as in Example 2g, a crude product (540 mg) of the title compound was obtained.
- Example 8g 3-chloro-2- (3-fluoro-4-methoxybenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 8f
- the title compound 73 mg was obtained as a colorless solid in the same manner as in Example 2h.
- solidification was performed from hexane / ethyl acetate / methanol.
- Example 9a 3,5-difluoro-4- [1- (2-fluoro-4-methoxyphenyl) -1-hydroxymethyl] benzonitrile diisopropylamine (1.20 mL, 8.49 mmol), n-butyllithium (2.60 mL, 7.19mmol, 2.77M n-hexane solution), 3,5-difluorobenzonitrile (1.00g, 7.19mmol), 2-fluoro-4-methoxybenzaldehyde (1.11g, 7.20mmol) and tetrahydrofuran (17mL)
- the title compound (1.66 g) was obtained as a pale yellow solid in the same manner as in Example 7a.
- the purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 9: 1 to 4: 1 to 2: 1, V / V).
- Example 9b 3,5-difluoro-4- (2-fluoro-4-methoxybenzyl) benzonitrile
- the compound obtained in Example 9a (1.66 g, 5.66 mmol), triethylsilane (2.7 mL, 17 mmol),
- the title compound (1.29 g) was obtained as a colorless solid in the same manner as in Example 7b using boron trifluoride-diethyl ether complex (1.1 mL, 8.7 mmol) and acetonitrile (17 mL).
- the purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 9: 1, V / V).
- Example 9c 3-Benzyloxy-5-fluoro-4- (2-fluoro-4-methoxybenzyl) benzonitrile
- the compound obtained in Example 9b (1.28 g, 4.62 mmol), sodium hydride (63% 0.26 g, 6.8 mmol), benzyl alcohol (0.66 g, 6.1 mmol) and N, N-dimethylformamide (13 mL) were used to prepare a crude product of the title compound (0.95 g) in the same manner as in Example 7c. Obtained as a yellow solid.
- Example 9d 3-Benzyloxy-5-fluoro-4- (2-fluoro-4-methoxybenzyl) benzoic acid
- Crude product obtained in Example 9c (0.95 g, 2.6 mmol)
- 5M sodium hydroxide A crude product (0.97 g) of the title compound was obtained as a pale yellow solid in the same manner as in Example 7d using an aqueous solution (2.6 mL, 13 mmol) and ethanol (13 mL).
- Example 9e 3-Fluoro-4- (2-fluoro-4-methoxybenzyl) -5-hydroxybenzoic acid
- Crude product obtained in Example 9d (0.97 g, 2.5 mmol), 10% palladium on carbon catalyst (Water content, 0.20 g), tetrahydrofuran (10 mL), methanol (10 mL) and hydrogen were used in the same manner as in Example 7e to obtain a crude product (0.75 g) of the title compound as a pale yellow solid.
- Example 9f 3-Fluoro-2- (2-fluoro-4-methoxybenzyl) -5- (hydroxymethyl) phenol
- Crude product obtained in Example 9e (465 mg, 1.58 mmol), lithium aluminum hydride (0.12 g, 3.2 mmol) and tetrahydrofuran (12 mL) were used, and the title compound (0.37 g) was obtained as a colorless solid in the same manner as in Example 7f.
- the purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 1: 1 to 2: 3, V / V).
- Example 9g Acetic acid 3-fluoro-4- (2-fluoro-4-methoxybenzyl) -5-hydroxybenzyl
- bis (dibutyltin chloride) oxide (0.22 g, 0.40 mmol)
- vinyl acetate (2 mL)
- tetrahydrofuran (2 mL)
- the purification was performed using silica gel flash chromatography (hexane: ethyl acetate, 2: 1 to 3: 2, V / V).
- Example 9h 5-Acetoxymethyl-3-fluoro-2- (2-fluoro-4-methoxybenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.74 g, 1.2 mmol), methylene chloride (7.5 mL ), Trichloroacetonitrile (0.36 mL, 3.6 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (20 ⁇ L, 0.13 mmol) were prepared in the same manner as in Example 1h.
- Example 1h A crude product of the title compound was obtained in the same manner as above.
- Example 10a Ethyl 3-chloro-4- [4- (difluoromethoxy) benzoyl] -5-oxocyclohex-3-enecarboxylate
- the compound obtained in Example 3b (1.7 g, 5.0 mmol) was salified. Dissolved in methylene (20 mL) and performed with 2-methyl-2-butene (2.2 mL, 20 mmol), oxalyl chloride (0.45 mL, 5.2 mmol) and N, N-dimethylformamide (0.1 mL, 0.13 mmol) A crude product of the title compound (1.8 g) was obtained in the same manner as in Example 1c.
- Example 10b Ethyl 3-chloro-4- [4- (difluoromethoxy) benzoyl] -5-hydroxybenzoate
- the crude product obtained in Example 10a (1.8 g, 6.5 mmol) was converted to N-methylmorpholine ( 20 mL), and a crude product (1.9 g) of the title compound was obtained in the same manner as in Example 1d using anhydrous sodium sulfate (1.8 g) and iodine (1.5 g, 6.0 mmol).
- Example 10c 3-Chloro-2- ⁇ 1- [4- (difluoromethoxy) phenyl] -1-hydroxymethyl ⁇ -5- (hydroxymethyl) phenol
- the crude product obtained in Example 10b (1.8 g 4.9 mmol) was dissolved in tetrahydrofuran (55 mL), and the title compound (1.3 g) was obtained as a yellow solid in the same manner as in Example 1e using lithium aluminum hydride (0.57 g, 15 mmol).
- Example 10d Acetic acid 3-chloro-4- ⁇ 1- [4- (difluoromethoxy) phenyl] -1-hydroxymethyl ⁇ -5-hydroxybenzyl
- Example 10c 1.3 g, 4.0 mmol
- the crude product obtained by the same method as in Example 1f is purified by silica gel flash chromatography using porcine pancreatic lipase (0.6 g). Purification using hexane: ethyl acetate, 4: 1, V / V) gave the title compound (0.99 g).
- Example 10e Acetic acid 3-chloro-4- [4- (difluoromethoxy) benzyl] -5-hydroxybenzyl
- the compound (0.99 g, 2.56 mmol) obtained in Example 10d was dissolved in acetonitrile (10 mL).
- acetonitrile 10 mL
- boron trifluoride-diethyl ether complex 0.49 mL, 3.9 mmol
- Example 10f 5-Acetoxymethyl-3-chloro-2- [4- (difluoromethoxy) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ - D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.61 g, 1.0 mmol) in methylene chloride (4 mL) Dissolve and prepare the imidate by the same method as Example 1h using trichloroacetonitrile (0.3 mL, 3.0 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (4.3 ⁇ L, 0.03 mmol).
- Example 10g 3-Chloro-2- [4- (difluoromethoxy) benzyl] -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 10f
- the mixture (0.40 mmol) was dissolved in a solution consisting of tetrahydrofuran (3 mL) and methanol (3 mL), and the title compound (2) was added in a manner similar to Example 3i using 2M aqueous sodium hydroxide solution (3.0 mL, 6.0 mmol). 32 mg) was obtained as a colorless solid.
- Example 11a Ethyl 3-fluoro-4- [4- (2-fluoroethyl) benzoyl] -5-oxocyclohex-3-enecarboxylate
- the crude product obtained in Example 1b (2.0 g, 6.2 mmol) was dissolved in methylene chloride (18 mL), and the title compound (1.6 g) was obtained in the same manner as in Example 3b using diethylaminosulfur trifluoride (2.5 mL, 19 mmol).
- Example 11b Ethyl 3-fluoro-4- [4- (2-fluoroethyl) benzoyl] -5-hydroxybenzoate
- the compound obtained in Example 11a (1.6 g, 4.9 mmol) was converted to N-methylmorpholine ( 16 mL) and the crude product (1.6 g) of the title compound was obtained in the same manner as in Example 3c using anhydrous sodium sulfate (1.6 g) and iodine (1.4 g, 5.4 mmol).
- Example 11c Ethyl 3-fluoro-4- ⁇ 1- [4- (2-fluoroethyl) phenyl] -1-hydroxymethyl ⁇ -5-hydroxybenzoate
- the crude product obtained in Example 11b (1.6 g, 4.9 mmol) was dissolved in ethanol (20 mL), and the title compound (0.83 g) was obtained as a colorless solid in the same manner as in Example 3d using sodium borohydride (0.37 g, 9.8 mmol).
- Example 11d Ethyl 3-fluoro-4- [4- (2-fluoroethyl) benzyl] -5-hydroxybenzoate
- the compound (0.83 g, 2.5 mmol) obtained in Example 11c was added to ethanol (15 mL). Dissolved, and the crude product of the title compound was obtained in the same manner as in Example 3e using 10% hydrogen chloride methanol (0.5 mL, 2.5 mmol) and 10% palladium carbon catalyst (containing water, 0.5 g).
- the resulting crude product was purified using silica gel flash chromatography (hexane: ethyl acetate, 5: 1, V / V) to obtain the title compound (0.37 g) as a colorless solid.
- Example 11e 3-Fluoro-2- [4- (2-fluoroethyl) benzyl] -5- (hydroxymethyl) phenol
- the compound (0.37 g, 1.2 mmol) obtained in Example 11d was added to tetrahydrofuran (15 mL).
- the title compound (0.33 g) was obtained as a crude product in the same manner as in Example 3f using lithium aluminum hydride (0.13 g, 3.5 mmol).
- Example 11f Acetic acid 3-fluoro-4- [4- (2-fluoroethyl) benzyl] -5-hydroxybenzyl
- the crude product obtained in Example 11e (0.33 g, 1.2 mmol) was converted into vinyl acetate (5 mL).
- diisopropyl ether (5 mL)
- the porcine pancreatic lipase 1.0 g
- the title compound (0.35 g) was obtained as a pale brown solid in the same manner as in Example 3 g.
- Example 11g 5-Acetoxymethyl-3-fluoro-2- [4- (2-fluoroethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.61 g, 1.0 mmol) in methylene chloride (4 mL
- the imidate was prepared in the same manner as in Example 1h using trichloroacetonitrile (0.30 mL, 3.0 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (4 ⁇ L, 0.03 mmol).
- Example 11f Using this imidate (0.75 g, 1.0 mmol), the compound obtained in Example 11f (0.25 g, 0.78 mmol) and boron trifluoride-diethyl ether complex (39 ⁇ L, 0.39 mmol), the same method as in Example 1h Gave a mixture containing the title compound.
- Example 11h 3-Fluoro-2- [4- (2-fluoroethyl) benzyl] -5-hydroxymethyl-phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 11g
- the title compound was dissolved in a solution consisting of tetrahydrofuran (3 mL) and methanol (3 mL) in the same manner as in Example 3i using 2M aqueous sodium hydroxide solution (3.0 mL, 6.0 mmol). (0.21 g) was obtained as a colorless solid.
- Example 12 3-Fluoro-5-hydroxymethyl-2- (4-trifluoromethoxybenzyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside (No. 3 in Table 1)
- Example 12a Ethyl 3-hydroxy-5-oxo-4- [4- (trifluoromethoxy) benzoyl] cyclohex-3-enecarboxylate ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate ( 1.3 g, 7.3 mmol) and 4- (trifluoromethoxy) benzoyl chloride (J. Med. Chem., 45 (14), 2002, 3112-3129.) (1.6 g, 7.3 mmol) In the same manner, a crude product (2.4 g) of the title compound was obtained.
- Example 12b 3-Fluoro-5-oxo-4- [4- (trifluoromethoxy) benzoyl] cyclohex-3-enecarboxylic acid ethyl ester
- Crude product obtained in Example 12a (1.8 g, 5.4 mmol ) was dissolved in methylene chloride (15 mL), and the title compound (1.5 g) was obtained as a yellow oil by the same method as Example 3b using diethylaminosulfur trifluoride (2.1 mL, 16 mmol).
- Example 12c Ethyl 3-fluoro-5-hydroxy-4- [4- (trifluoromethoxy) benzoyl] benzoate
- the compound obtained in Example 12b (1.5 g, 4.5 mmol) was converted to N-methylmorpholine (18 mL).
- Example 1d using anhydrous sodium sulfate (1.5 g) and iodine (1.3 g, 5.0 mmol), a crude product (1.5 g) of the title compound was obtained.
- Example 12d 3-Fluoro-5-hydroxy-4- ⁇ 1-hydroxy-1- [4- (trifluoromethoxy) phenyl] methyl ⁇ ethyl benzoate
- the crude product obtained in Example 12c (1.5 g 4.5 mmol) was dissolved in ethanol (15 mL), and the title compound (1.0 g) was obtained in the same manner as in Example 3d using sodium borohydride (0.34 g, 9.0 mmol).
- Example 12e 3-Fluoro-5-hydroxymethyl-2- ⁇ 1-hydroxy-1- [4- (trifluoromethoxy) phenyl] methyl ⁇ phenol
- the compound obtained in Example 12d (1.0 g, 3.0 mmol ) was dissolved in tetrahydrofuran (20 mL), and the title compound (1.0 g) was obtained in the same manner as in Example 1e (but without purification) using lithium aluminum hydride (0.46 g, 12 mmol). Obtained as a crude product.
- Example 12f 3-fluoro-5-hydroxy-4- ⁇ 1-hydroxy-1- [4- (trifluoromethoxy) phenyl] methyl ⁇ benzyl acetate
- Acetic acid 1.0 g, 3.0 mmol
- vinyl acetate 5 mL
- diisopropyl ether 5 mL
- the crude product obtained in the same manner as in Example 1f was purified by silica gel flash using porcine pancreatic lipase (1.0 g). Purification using chromatography (hexane: ethyl acetate, 4: 1, V / V) gave the title compound (0.65 g).
- Example 12g 3-fluoro-5-hydroxy-4- [4- (trifluoromethoxy) benzyl] benzyl acetate
- the compound (0.65 g, 1.7 mmol) obtained in Example 12f was dissolved in acetonitrile (10 mL).
- Triethylsilane (0.83 mL, 5.2 mmol) and boron trifluoride-diethyl ether complex (0.33 mL, 2.6 mmol) were used in the same manner as in Example 1g to obtain the title compound (0.18 g).
- Example 12h 5-Acetoxymethyl-3-fluoro-2- [4- (trifluoromethoxy) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.61 g, 1.0 mmol) in methylene chloride (4 mL)
- the imidate was prepared in the same manner as Example 1h using trichloroacetonitrile (0.3 mL, 3.0 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (4.3 ⁇ L, 0.03 mmol).
- Example 12i 3-Fluoro-5-hydroxymethyl-2- [4- (trifluoromethoxy) benzyl] phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside Mixture obtained in Example 12h (0.50 mmol) was dissolved in a solution consisting of tetrahydrofuran (3 mL) and methanol (3 mL), and the title compound (0.12 g) was prepared in the same manner as in Example 3i using 2M aqueous sodium hydroxide solution (3.0 mL, 6.0 mmol). ) was obtained as a colorless solid.
- Example 13a 5-Acetoxymethyl-3-fluoro-2- (4-methylbenzyl) phenyl 4-deoxy-2,3,6-tri-O-benzoyl- ⁇ -D-glucopyranoside 4-deoxy-2, 3,6-tri-4-O-benzoyl-D-glucopyranoside (Liebig Ann.
- Example 4i using 5-hydroxybenzyl (WO2008 / 016132 (PCT / JP2007 / 65231)) (100 mg, 0.35 mmol), boron trifluoride-diethyl ether complex (44 ⁇ L, 0.35 mmol) and methylene chloride (5 mL) In the same manner as above, a crude product (285 mg) of the title compound was obtained.
- 5-hydroxybenzyl WO2008 / 016132 (PCT / JP2007 / 65231)
- boron trifluoride-diethyl ether complex 44 ⁇ L, 0.35 mmol
- methylene chloride 5 mL
- Example 13b 3-Fluoro-5-hydroxymethyl-2- (4-methylbenzyl) phenyl 4-deoxy- ⁇ -D-glucopyranoside
- Crude product obtained in Example 13a (285 mg, 0.35 mmol), tetrahydrofuran (1 mL), methanol (4 mL) and calcium carbonate (48 mg, 0.35 mmol) were used in the same manner as in Example 4j to obtain the title compound (90 mg) as a colorless solid.
- Example 4h The compound obtained in Example 4h (100 mg, 0.33 mmol), boron trifluoride-diethyl A crude product (291 mg) of the title compound was obtained in the same manner as in Example 4i using an ether complex (41 ⁇ L, 0.33 mmol) and methylene chloride (5 mL).
- Example 14b 3-Chloro-5-hydroxymethyl-2- (4-methylbenzyl) phenyl 4-deoxy- ⁇ -D-glucopyranoside
- Crude product obtained in Example 14a (291 mg, 0.33 mmol), tetrahydrofuran (1 mL), methanol (4 mL) and potassium carbonate (46 mg, 0.33 mmol) were used in the same manner as in Example 4j to obtain the title compound (106 mg) as a colorless solid.
- Example 15a 5-Acetoxymethyl-2- (4-ethylbenzyl) -3-fluorophenyl 4-deoxy-2,3,6-tri-O-benzoyl- ⁇ -D-glucopyranoside 4-deoxy-2, 3,6-tri-4-O-benzoyl-D-glucopyranoside (189 mg, 0.40 mmol), trichloroacetonitrile (120 ⁇ L, 1.20 mmol), 1,8-diazabicyclo [5.4.0] -7-undecene (6 ⁇ L, 0.04 mmol) ) And methylene chloride (5 mL), and an imidate was prepared in the same manner as in Example 4i.
- Example 15b 2- (4-Ethylbenzyl) -3-fluoro-5- (hydroxymethyl) phenyl 4-deoxy- ⁇ -D-glucopyranoside
- Example 15a 3-(2-Ethylbenzyl) -3-fluoro-5- (hydroxymethyl) phenyl 4-deoxy- ⁇ -D-glucopyranoside
- Tetrahydrofuran (1 mL
- methanol 4 mL
- calcium carbonate 46 mg, 0.33 mmol
- Example 2f Using the resulting imidate (248 mg), the compound obtained in Example 2f (100 mg, 0.31 mmol), methylene chloride (4 mL), MS4A and boron trifluoride-diethyl ether complex (0.050 mL, 0.40 mmol) In the same manner as in Example 2g, a crude product (270 mg) of the title compound was obtained.
- Example 16b 3-chloro-2- (4-ethylbenzyl) -5- (hydroxymethyl) phenyl 4-deoxy- ⁇ -D-glucopyranoside
- Crude product obtained in Example 16a 270 mg
- the title compound (126 mg) was obtained as a colorless solid in the same manner as in Example 2h using methylene chloride (8 mL / 2 mL) and potassium carbonate (550 mg, 3.98 mmol). However, solidification was performed from hexane / ethyl acetate.
- Example 17b 2- (4-Cyclopropylbenzyl) -3-fluoro-5- (hydroxymethyl) phenyl 4-deoxy- ⁇ -D-glucopyranoside
- the crude product obtained in Example 17a (301 mg, 0.32 mmol) ), Tetrahydrofuran (1 mL), methanol (4 mL) and potassium carbonate (44 mg, 0.32 mmol) were obtained in the same manner as in Example 4j to obtain the title compound (107 mg) as a colorless solid.
- Example 18a 4- (2,2,2-trifluoroethyl) benzoyl chloride 4- (2,2,2-trifluoroethyl) benzoic acid (Liebigs Ann. Chem., 1983, 9, 1510-1523. ) (786 mg, 3.85 mmol), oxalyl dichloride (0.385 mL, 4.42 mmol), catalytic amounts of N, N-dimethylformamide and tetrahydrofuran (6 mL) in the same manner as in Example 1a to give a crude product of the title compound Product (857 mg) was obtained.
- Example 18b Ethyl 3-hydroxy-5-oxo-4- [4- (2,2,2-trifluoroethyl) benzoyl] cyclohex-3-enecarboxylate 3-hydroxy-5-oxocyclohexa-3 -Ethylencarboxylate (709 mg, 3.85 mmol), crude product obtained in Example 18a (857 mg, 3.85 mmol), acetonitrile (10 mL), triethylamine (1.61 mL, 11.5 mmol) and trimethylsilylcyanide (0.062 mL, 0.46 mmol) was used in the same manner as in Example 1b to obtain a crude product of the title compound (1.42 g).
- Example 18c Ethyl 3-fluoro-5-oxo-4- [4- (2,2,2-trifluoroethyl) benzoyl] cyclohex-3-enecarboxylate
- the crude product obtained in Example 18b ( 1.42 g, 3.61 mmol), methylene chloride (15 mL) and diethylaminosulfur trifluoride (1.51 mL, 11.5 mmol) were used to give the title compound (953 mg) as a white solid in the same manner as in Example 3b.
- Example 18c Using the compound obtained in Example 18c (953 mg, 2.56 mmol), N-methylmorpholine (12 mL), anhydrous sodium sulfate (7.27 g) and iodine (779 mg, 3.07 mmol), the title was prepared in the same manner as in Example 1d. A crude product of the compound was obtained. The resulting crude product was purified using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 3: 1, V / V) to give the amorphous title compound (614 mg).
- Example 18e 3-Fluoro-5-hydroxymethyl-2- [1-hydroxy-1- ⁇ 4- (2,2,2-trifluoroethyl) phenyl ⁇ methyl] phenol
- Compound obtained in Example 18d (610 mg, 1.65 mmol) was dissolved in tetrahydrofuran (15 mL), cooled to 0 ° C., lithium aluminum hydride (187 mg, 4.93 mmol) was added, and the mixture was stirred at room temperature for 30 min.
- Example 18f 3-Fluoro-5-hydroxymethyl-2- [1-methoxy-1- ⁇ 4- (2,2,2-trifluoroethyl) phenyl ⁇ methyl] phenol
- the product (540 mg, 1.64 mmol) was dissolved in methanol (10 mL), paratoluenesulfonic acid monohydrate (157 mg, 0.83 mmol) was added, and the mixture was stirred at 50 ° C. for 2 hr.
- the reaction mixture was cooled to room temperature, triethylamine was added, and the mixture was concentrated under reduced pressure.
- the residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 9: 1 to 1: 4, V / V) to give the title compound (508 mg) as an oil.
- Example 18g Acetic acid 3-fluoro-5-hydroxy-4- ⁇ 1-methoxy-1- [4- (2,2,2-trifluoroethyl) phenyl] methyl ⁇ benzyl
- Compound obtained in Example 18f (508 mg, 1.48 mmol), tetrahydrofuran (5 mL), vinyl acetate (5 mL) and bis (dibutyltin chloride) oxide (325 mg, 0.59 mmol) were used in the same manner as in Example 2e to give the oily title compound (525 mg). Obtained.
- Example 18h 3-Fluoro-5-hydroxy-4- [4- (2,2,2-trifluoroethyl) benzyl] benzyl acetate
- Acetic acid 525 mg, 1.36 mmol
- acetonitrile 10 mL
- triethylsilane 0.650 mL, 4.08 mmol
- boron trifluoride-diethyl ether complex 0.256 mL, 2.04 mmol
- Example 18i 5-Acetoxymethyl-3-fluoro-2- [4- (2,2,2-trifluoroethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl -D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (267 mg, 0.44 mmol), The imidate was prepared in the same manner as in Example 2g using methylene chloride (5 mL), trichloroacetonitrile (0.221 mL, 2.19 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (7 ⁇ L, 0.05 mmol).
- Example 18j 3-Fluoro-5-hydroxymethyl-2- [4- (2,2,2-trifluoroethyl) benzyl] phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside
- methanol / methylene chloride 8 mL / 2 mL
- potassium carbonate 604 mg, 4.34 mmol
- the title compound was obtained as a white solid. Obtained. However, solidification was performed from hexane / ethyl acetate / methanol.
- Example 19a Methyl 4-acetyl-3-fluorobenzoate 4-acetyl-3-fluorophenyl sulfonate (Org. Lett., 2002, 4 (26), 4717-4718.) (8.69 g, 30.4 mmol) in N, N-dimethylformaldehyde (120 mL), methanol (24.6 mL, 606 mmol), palladium acetate (682 mg, 3.03 mmol), 1,3-bis (diphenylphosphino) propane (1.25 g, 3.03 mmol) ) And triethylamine (84.0 mL, 606 mol) were added, and the mixture was stirred at room temperature for 16 hours under a carbon monoxide atmosphere.
- Example 19b Methyl 3-fluoro-4- (1-hydroxyethyl) benzoate
- the compound obtained in Example 19a (3.67 g, 18.7 mmol) was dissolved in methanol (40 mL) and cooled to 0 ° C.
- Sodium borohydride (850 mg, 22.5 mmol) was added and stirred at 0 ° C. for 1 hour.
- a saturated aqueous ammonium chloride solution (2 mL) was added to the reaction solution, and the solvent was removed under reduced pressure.
- Example 19c 4-Ethyl-3-fluorobenzoic acid
- the compound obtained in Example 19b (3.67 g, 18.7 mmol) was dissolved in methanol (35 mL), 2 mol / L hydrochloric acid (1.09 mL) and 10% wet Palladium / carbon (600 mg) was added, and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere.
- the reaction solution was filtered through celite to remove palladium. Subsequently, a 5 mol / L aqueous sodium hydroxide solution (8 mL) and an appropriate amount of tetrahydrofuran were added to the reaction solution, and the mixture was stirred at 50 ° C. for 1 hour.
- Example 19d 4-ethyl-3-fluorobenzoyl chloride The compound obtained in Example 19c (730 mg, 4.34 mmol), oxalyl dichloride (0.43 mL, 4.95 mmol), catalytic amounts of N, N-dimethylformamide and A crude product of the title compound (810 mg) was obtained as a colorless oil by a method similar to Example 1a using tetrahydrofuran (6 mL).
- Example 19e Ethyl 4- (4-ethyl-3-fluorobenzoyl) -3-hydroxy-5-oxocyclohex-3-enecarboxylate
- Ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate 800 mg, 4.34 mmol
- acetonitrile 9 mL
- triethylamine 1.82 mL, 13.1 mmol
- trimethylsilylcyanide 0.070 mL, 0.52 mmol
- Example 19f Ethyl 3-chloro-4- (4-ethyl-3-fluorobenzoyl) -5-oxocyclohex-3-enecarboxylate
- Crude product obtained in Example 19e (1.45 g, 4.34 mmol ), Methylene chloride (18 mL), 2-methyl-2-butene (1.84 mL, 17.3 mmol), oxalyl dichloride (0.39 mL, 4.55 mmol) and a catalytic amount of N, N-dimethylformamide and
- a crude product (1.52 g) of the title compound was obtained.
- Example 19g Ethyl 3-chloro-4- (4-ethyl-3-fluorobenzoyl) -5-hydroxybenzoate
- Crude product obtained in Example 19f (1.52 g, 4.34 mmol), N-methylmorpholine (18 mL), anhydrous sodium sulfate (12.3 g) and iodine (1.32 g, 5.20 mmol) were used to give a crude product of the title compound in the same manner as in Example 1d.
- the resulting crude product was purified using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 3: 1, V / V) to give the amorphous title compound (1.04 g).
- Example 19h Ethyl 5-chloro-4- (4-ethyl-3-fluorophenyl) -2,2-dimethyl-4H-benzo [1,3] dioxin-7-benzoate obtained in Example 19g
- a crude diol product (1.04 g) was obtained in the same manner as in Example 2b, using the compound (1.04 g, 2.96 mmol), methanol (12 mL) and sodium borohydride (220 mg, 5.82 mmol).
- Example 19i 5-chloro-4- (4-ethyl-3-fluorophenyl) -2,2-dimethyl-4H-benzo [1,3] dioxin-7-ylmethanol
- Compound obtained in Example 19h (730 mg, 1.86 mmol), lithium aluminum hydride (71 mg, 1.87 mmol) and tetrahydrofuran (8 mL) were used in the same manner as in Example 2d to obtain a crude product of the title compound (664 mg) as an oil.
- Example 19j Acetic acid 5-chloro-4- (4-ethyl-3-fluorophenyl) -2,2-dimethyl-4H-benzo [1,3] dioxin-7-ylmethyl Crude obtained in Example 19i Using the product (664 mg), pyridine (6.6 mL) and acetic anhydride (1.7 mL), the title compound (720 mg) was obtained in the same manner as in Example 6g.
- Example 19k Acetic acid 3-chloro-4- (4-ethyl-3-fluorobenzyl) -5-hydroxybenzyl Compound (720 mg, 1.83 mmol) obtained in Example 19j, acetonitrile (12 mL), triethylsilane ( 0.880 mL, 5.47 mmol) and boron trifluoride-diethyl ether complex (0.350 mL, 2.79 mmol) were used in the same manner as in Example 2f to obtain the title compound (491 mg) as a colorless solid.
- Example 19l 5-acetoxymethyl-3-chloro-2- (4-ethyl-3-fluorobenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231)) 318 mg, 0.52 mmol), methylene chloride (5 mL), trichloroacetonitrile (0.263 mL, 2.60 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (7 ⁇ L, 0.05 mmol) as in Example 2g
- the imidate was prepared by the method.
- Example 19k Performed using the obtained imidate (393 mg), the compound obtained in Example 19k (135 mg, 0.40 mmol), methylene chloride (5 mL), MS4A and boron trifluoride-diethyl ether complex (0.065 mL, 0.52 mmol). In the same manner as in Example 2g, a crude product (380 mg) of the title compound was obtained.
- Example 19m 3-Chloro-2- (4-ethyl-3-fluorobenzyl) -5-hydroxymethylphenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside Crude obtained in Example 19l
- the title compound (67 mg) was obtained as a white solid in the same manner as in Example 2h using the product (380 mg), methanol / methylene chloride (8 mL / 2 mL) and potassium carbonate (720 mg, 5.21 mmol). However, solidification was performed from hexane / ethyl acetate / methanol.
- the reaction mixture was diluted with ethyl acetate (50 mL), and washed successively with 10% brine (10 mL, 3 times) and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (1.2 g) of the title compound.
- Example 20b 3-Benzyloxy-5-fluoro-4- (4-methoxybenzyl) benzaldehyde
- the crude product obtained in Example 20a (1.2 g, 3.3 mmol) was dissolved in chloroform (12 mL).
- Manganese (4.3 mL, 50 mmol) was added, and the mixture was stirred with heating under reflux for 2.5 hours.
- the suspension was filtered through celite, and the filtrate was concentrated under reduced pressure to obtain a crude product (1.2 g) of the title compound.
- Example 20c 1-benzyloxy-3-fluoro-2- (4-methoxybenzyl) -5- (2-methoxyvinyl) benzene (methoxymethyl) triphenylphosphonium chloride (3.4 g, 10 mmol) was added to tetrahydrofuran (8 mL). And stirred under ice cooling. Lithium bis (trimethylsilyl) amide (1M tetrahydrofuran solution, 10 mL, 10 mmol) was added dropwise to this suspension, and the mixture was stirred for 30 minutes while warming to room temperature.
- a tetrahydrofuran solution (8 mL) in which the crude product (1.2 g, 3.3 mmol) obtained in Example 20b was dissolved was added dropwise to the reaction mixture, and the mixture was stirred at room temperature for 3 hours.
- a saturated aqueous ammonium chloride solution (10 mL) was added to the reaction mixture, followed by extraction with ethyl acetate (20 mL, twice), and the organic layer was washed with saturated brine (10 mL, twice).
- Example 20d 3-Benzyloxy-5-fluoro-4- (4-methoxybenzyl) phenylacetaldehyde
- the compound obtained in Example 20c (0.92 g, 2.4 mmol) was dissolved in 1,4-dioxane (10 mL). Under ice-cooling, water (0.43 mL, 24 mmol) and 4M hydrogen chloride 1,4-dioxane solution (6.0 mL, 24 mmol) were added, and the mixture was stirred for 1 hour while warming to room temperature.
- the solution was diluted with ethyl acetate (50 mL), and washed successively with 10% brine (10 mL, twice), saturated aqueous sodium hydrogen carbonate solution (10 mL) and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (0.87 g) of the title compound.
- Example 20e 2- [3-Benzyloxy-5-fluoro-4- (4-methoxybenzyl) phenyl] ethanol
- the crude product obtained in Example 20d (0.87 g, 2.4 mmol) was added to tetrahydrofuran (4 mL).
- tetrahydrofuran (4 mL)
- sodium borohydride (0.14 g, 3.6 mmol) was added under ice cooling.
- the reaction solution was stirred for 40 minutes while warming to room temperature, and saturated aqueous ammonium chloride solution (1 mL) was added dropwise under ice cooling to stop the reaction.
- Example 20f 3-Fluoro-5- (2-hydroxyethyl) -2- (4-methoxybenzyl) phenol
- the compound obtained in Example 20e (0.57 g, 1.6 mmol) was mixed with tetrahydrofuran (3 mL) and methanol ( 3 mL), and stirred for 10 minutes while blowing nitrogen into the solution.
- 10% palladium carbon catalyst (containing water, 0.12 g) was added under a nitrogen stream, and the gas phase was replaced with hydrogen, followed by stirring at room temperature for 1 hour.
- the mixture was filtered through celite, and the solvent was removed under reduced pressure to obtain a crude product of the title compound (0.44 g).
- Example 20g 2- [3-Fluoro-5-hydroxy-4- (4-methoxybenzyl) phenyl] ethyl acetate
- the crude product obtained in Example 20f (0.44 g, 1.6 mmol) was dissolved in collidine (5.0 mL). ), Acetyl chloride (0.11 mL, 1.6 mmol) was added at ⁇ 20 ° C., and the mixture was stirred for 2 hours while raising the temperature to 0 ° C.
- Water (1 mL) was added to the reaction mixture, diluted with ethyl acetate (30 mL), and washed successively with 30% aqueous citric acid solution (10 mL, twice) and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product of the title compound (0.51 g, including raw materials).
- Example 20h 5- (2-acetoxyethyl) -3-fluoro-2- (4-methoxybenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (0.43 g, 0.71 mmol) in methylene chloride (4 mL)
- the imidate was prepared in the same manner as in Example 1h using trichloroacetonitrile (0.20 mL, 2.1 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (3 ⁇ L, 0.02 mmol).
- Example 11f Similar to Example 1h using this imidate (0.75 g, 1.0 mmol), the compound obtained in Example 11f (0.51 g, 1.6 mmol) and boron trifluoride-diethyl ether complex (0.05 mL, 0.4 mmol). By the method, a mixture containing the title compound was obtained.
- Example 20i 3-Fluoro-5- (2-hydroxyethyl) -2- (4-methoxybenzyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside Mixture obtained in Example 20h (1.6 mmol) was dissolved in a solution consisting of tetrahydrofuran (3 mL) and methanol (3 mL), and the title compound (64 mg) was prepared in the same manner as in Example 3i using 2M aqueous sodium hydroxide solution (3.0 mL, 6.0 mmol). Was obtained as a colorless solid.
- Example 21a Acetic acid 3-benzyloxy-5-chloro-4- (4-methylbenzyl) benzyl Compound (500 mg, 3.3 mmol) obtained in Example 4h was dissolved in N, N-dimethylformamide (10 mL). Benzyl bromide (430 ⁇ L, 3.6 mmol) and potassium carbonate (680 mg, 4.9 mmol) were added, and the mixture was stirred at room temperature for 4 hours. The reaction mixture was diluted with ethyl acetate (30 mL) and washed with distilled water (10 mL) and saturated brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (1.13 g) of the title compound as a yellow oily substance.
- Example 21b 3-Benzyloxy-5-chloro-4- (4-methylbenzyl) benzyl alcohol
- the crude product obtained in Example 21a (1.13 g, 2.9 mmol) was dissolved in methanol (12 mL).
- 1M aqueous potassium hydroxide solution (3.3 mL, 3.3 mmol) was added, and the mixture was stirred at room temperature for 18 hours.
- the solvent was distilled off under reduced pressure, the residue was diluted with ethyl acetate (10 mL), and washed successively with distilled water (10 mL) and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, and then the solvent was distilled off under reduced pressure to obtain a crude product (0.91 g) of the title compound as a yellow oily substance.
- Example 21c 3-Benzyloxy-5-chloro-4- (4-methylbenzyl) benzaldehyde
- the crude product (0.91 g, 2.6 mmol) obtained in Example 21b was dissolved in chloroform (10 mL) Manganese (2.85 g, 32.8 mmol) was added, and the mixture was stirred at 60 ° C. for 4 hours. After filtration through celite, the solvent was distilled off under reduced pressure to obtain a crude product of the title compound (0.68 g) as a brown solid.
- Example 21d 1-benzyloxy-3-chloro-5- (2-methoxyvinyl) -2- (4-methylbenzyl) benzene (methoxymethyl) triphenylphosphonium chloride (2.0 g, 5.4 mmol) in toluene ( 5 mL) was added, and the solvent was distilled off under reduced pressure. To the residue were added tetrahydrofuran (4 mL) and lithium bis (trimethylsilyl) amide (1M tetrahydrofuran solution, 5.9 mL, 5.9 mmol) under ice cooling, and the mixture was stirred at room temperature for 1 hour.
- Example 21c a tetrahydrofuran solution (6 mL) in which the crude product obtained in Example 21c (0.68 g, 1.9 mmol) was dissolved was added dropwise to the reaction solution, followed by stirring at room temperature for 1 hour.
- the reaction mixture was diluted with ethyl acetate (10 mL) and washed successively with distilled water (10 mL) and saturated brine (5 mL).
- the organic layer was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was purified using silica gel flash column chromatography (hexane: ethyl acetate, 20: 1 to 5: 1, V / V) to obtain the title compound (0.95 g, quant.) As a yellow solid.
- Example 21e 3-Benzyloxy-5-chloro-4- (4-methylbenzyl) phenylacetaldehyde
- the compound obtained in Example 21d (0.95 g, 1.9 mmol) was dissolved in 1,4-dioxane (5 mL).
- Distilled water (350 ⁇ L) and 4N hydrochloric acid 1,4-dioxane solution (5 mL) were added, and the mixture was stirred at room temperature for 10 minutes.
- the reaction mixture was diluted with ethyl acetate (10 mL) and washed successively with distilled water (5 mL) and saturated brine (10 mL).
- the organic layer was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- Example 21f 2- [3-Benzyloxy-5-chloro-4- (4-methylbenzyl) phenyl] ethanol
- methanol 5 mL
- sodium borohydride 60 mg, 1.6 mmol
- the reaction mixture was stirred at 0 ° C. for 30 min.
- saturated aqueous ammonium chloride solution 5 mL
- saturated brine 10 mL
- Example 21g 3-Chloro-5- (2-hydroxyethyl) -2- (4-methylbenzyl) phenol
- the crude product (0.41 g, 1.1 mmol) obtained in Example 21f was added to acetonitrile (5 mL).
- trimethylsilyl iodide (320 ⁇ L, 2.2 mmol) was added, and the mixture was stirred at 40 ° C. for 2 hours.
- the reaction mixture was diluted with ethyl acetate (15 mL) and washed successively with 2M hydrochloric acid (10 mL), saturated aqueous sodium hydrogen carbonate solution (10 mL) and saturated brine (10 mL).
- Example 21h 2- ⁇ [3-Chloro-5-hydroxy-4- (4-methylbenzyl)] phenyl ⁇ ethyl acetate
- the compound (122 mg, 0.4 mmol) obtained in Example 21 g was added to tetrahydrofuran (2 mL). Dissolved, vinyl acetate (2 mL, 21.6 mmol) and bis (dibutyltin chloride) oxide (24 mg, 0.04 mmol) were added and stirred at room temperature for 24 hours.
- Example 21i 5- (2-acetoxyethyl) -3-chloro-2- (4-methylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero-D-gluco-heptopyranoside (255 mg, 0.42 mmol), trichloroacetonitrile (125 ⁇ L, 1.25 mmol), 1 , 8-diazabicyclo [5.4.0] -7-undecene (6 ⁇ L, 0.04 mmol) and methylene chloride (5 mL) were used to prepare an imidate in the same manner as in Example 4i, and the compound obtained in Example 21h (111 mg, 0.35 mmol), boron trifluoride-diethyl ether complex (44 ⁇ L, 0.35 mmol
- Example 21j 3-Chloro-5- (2-hydroxyethyl) -2- (4-methylbenzyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside
- Compound obtained in Example 21i (391 mg, 0.35 mmol), tetrahydrofuran (1 mL), methanol (4 mL) and calcium carbonate (48 mg, 0.35 mmol) were used in the same manner as in Example 4j to obtain the title compound (104 mg) as a colorless solid.
- Example 22a 3-Chloro-4- (4-methoxybenzyl) -5- (methoxymethoxy) benzyl alcohol Acetic acid 3-chloro-5-hydroxy-4- (4-methoxybenzyl) benzyl (WO2008 / 016132 (PCT / JP2007 / 65231)) (500 mg, 1.6 mmol) dissolved in N, N-dimethylformamide (5 mL), and ice-cooled, chloromethyl methyl ether (180 ⁇ L, 2.4 mmol) and calcium carbonate (1.08 g, 7.8 mmol) And stirred at room temperature for 4 hours. Methanol (5 mL) was added to the reaction solution, and the mixture was stirred at room temperature for 1 hour.
- Example 22b 3-Chloro-4- (4-methoxybenzyl) -5- (methoxymethoxy) benzaldehyde
- the crude product obtained in Example 22a (511 mg, 1.6 mmol), chloroform (10 mL) and manganese dioxide ( 1.36 g (15.6 mmol) was used in the same manner as in Example 21c to obtain a crude product of the title compound (480 mg) as a pale yellow oily substance.
- Example 22c 1-chloro-2- (4-methoxybenzyl) -3-methoxymethoxy-5- (2-methoxyvinyl) benzene
- Crude product obtained in Example 22b (480 mg, 1.5 mmol), chloride (Methylmethoxy) triphenylphosphonium (1.60 g, 4.8 mmol), lithium bis (trimethylsilyl) amide (1M tetrahydrofuran solution, 4.7 mL, 4.7 mmol) and tetrahydrofuran (8 mL) were used in the same manner as in Example 21d.
- the compound (810 mg, quant.) was obtained as a yellow oil.
- the purification was performed using silica gel flash column chromatography (hexane: ethyl acetate, 20: 1 to 15: 1 to 10: 1, V / V).
- Example 22d 3-chloro-5-hydroxy-4- (4-methoxybenzyl) phenylacetaldehyde
- the compound obtained in Example 22c (810 mg, 1.5 mmol), 1,4-dioxane solution (4 mL) in 4N hydrochloric acid,
- the title compound (210 mg) was obtained as a yellow oil in the same manner as in Example 21e using distilled water (280 ⁇ L) and 1,4-dioxane (4 mL).
- the purification was performed using silica gel flash column chromatography (hexane: ethyl acetate, 8: 1 to 6: 1 to 4: 1, V / V).
- Example 22e 3-Chloro-5- (2-hydroxyethyl) -2- (4-methoxybenzyl) phenol
- the compound obtained in Example 22d (210 mg, 0.72 mmol), sodium borohydride (33 mg, 0.87) mmol) and methanol (2.5 mL) were used to give the title compound (157 mg) as a colorless oil in the same manner as in Example 21f.
- Example 22f 2- ⁇ [3-Chloro-5-hydroxy-4- (4-methoxybenzyl)] phenyl ⁇ ethyl acetate
- the compound obtained in Example 22e (157 mg, 0.54 mmol), vinyl acetate (2 mL, 21.61 mmol), bis (dibutyltin chloride) oxide (38 mg, 0.07 mmol) and tetrahydrofuran (2 mL) were used in the same manner as in Example 21h to obtain the title compound (218 mg) as a colorless solid.
- the purification was performed using silica gel flash column chromatography (hexane: ethyl acetate, 4: 1 to 3: 1, V / V).
- Example 22g 5-Acetoxymethyl-3-chloro-2- (4-methoxybenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco -Heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero-D-gluco-heptopyranoside (220 mg, 0.36 mmol), trichloroacetonitrile (110 ⁇ L, 1.10 mmol), 1,8-diazabicyclo [5.4.0] -7-Undecene (6 ⁇ L, 0.04 mmol) and methylene chloride (5 mL) were used to prepare an imidate in the same manner as in Example 4i, and the compound (100 mg, 0.30) obtained in Example 22f was prepared.
- Example 22h 3-Chloro-5- (2-hydroxyethyl) -2- (4-methoxybenzyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside Crude obtained in Example 22g Using the product (320 mg, 0.30 mmol), tetrahydrofuran (1 mL), methanol (4 mL) and calcium carbonate (41 mg, 0.30 mmol), the title compound (81 mg) was obtained as a colorless solid in the same manner as in Example 4j. .
- Example 23a Ethyl 3-chloro-5-oxo-4- [4- (2,2,2-trifluoroethyl) benzoyl] cyclohex-3-enecarboxylate
- the crude product obtained in Example 18b ( 1.34 g, 3.61 mmol), methylene chloride (15 mL), 2-methyl-2-butene (1.54 mL, 14.5 mmol), oxalyl dichloride (0.32 mL, 3.73 mmol) and a catalytic amount of N, N-dimethylformamide
- a crude product (1.40 g) of the title compound was obtained.
- Example 23b Ethyl 3-chloro-5-hydroxy-4- [4- (2,2,2-trifluoroethyl) benzoyl] benzoate
- Crude product obtained in Example 23a (1.40 g, 3.60 mmol ), N-methylmorpholine (16 mL), anhydrous sodium sulfate (10.3 g) and iodine (1.10 g, 4.33 mmol) were used to give a crude product of the title compound in the same manner as in Example 1d.
- the resulting crude product was purified using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 3: 1, V / V) to give the amorphous title compound (1.02 g).
- Example 23c 3-Chloro-5-hydroxymethyl-2- [1-hydroxy-1- ⁇ 4- (2,2,2-trifluoroethyl) phenyl ⁇ methyl] phenol Compound obtained in Example 23b (1.02 g, 2.64 mmol), tetrahydrofuran (30 mL) and lithium aluminum hydride (300 mg, 7.91 mmol) were used in the same manner as in Example 18e to obtain a crude product (866 mg) of the title compound.
- Example 23d 3-chloro-5-hydroxymethyl-2- [1-methoxy-1- ⁇ 4- (2,2,2-trifluoroethyl) phenyl ⁇ methyl] phenol Crude obtained in Example 23c Using the product (866 mg, 2.50 mmol), methanol (16 mL) and paratoluenesulfonic acid monohydrate (238 mg, 1.25 mmol), the title compound (654 mg) as an oil was obtained in the same manner as in Example 18f. .
- Example 23e Acetic acid 3-chloro-4- [1-methoxy-1- ⁇ 4- (2,2,2-trifluoroethyl) phenyl ⁇ methyl] -5-hydroxybenzyl
- Compound obtained in Example 23d (654 mg, 1.81 mmol), tetrahydrofuran (6 mL), vinyl acetate (6 mL) and bis (dibutyltin chloride) oxide (302 mg, 0.55 mmol) were used in the same manner as in Example 2e to give the oily title compound (703 mg). Obtained.
- Example 23f Acetic acid 3-chloro-4- [4- (2,2,2-trifluoroethyl) benzyl] -5-hydroxybenzyl
- acetonitrile 15 mL
- triethylsilane 0.34 mL, 5.24 mmol
- boron trifluoride-diethyl ether complex 0.329 mL, 2.62 mmol
- Example 23g 5-Acetoxymethyl-3-chloro-2- [4- (2,2,2-trifluoroethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl -D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (213 mg, 0.35 mmol), The imidate was prepared in the same manner as in Example 2g using methylene chloride (4 mL), trichloroacetonitrile (0.176 mL, 1.74 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (5 ⁇ L, 0.03 mmol).
- Example 23f the compound obtained in Example 23f (105 mg, 0.23 mmol), methylene chloride (4 mL), MS4A and boron trifluoride-diethyl ether complex (0.044 mL, 0.34 mmol)
- a crude product 280 mg of the title compound was obtained.
- Example 23h 3-Chloro-5-hydroxymethyl-2- [4- (2,2,2-trifluoroethyl) benzyl] phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside
- the title compound (84 mg) was obtained as a white solid in the same manner as in Example 2h using 23 g of the crude product (280 mg), methanol / methylene chloride (8 mL / 2 mL) and potassium carbonate (480 mg, 3.47 mmol). Obtained. However, solidification was performed from hexane / ethyl acetate.
- Example 24a 2-Fluoro-4-methylbenzoyl chloride 2-fluoro-4-methylbenzoic acid (500 mg, 3.24 mmol), oxalyl dichloride (0.31 mL, 3.57 mmol) and a catalytic amount of N, N-dimethylformamide , Tetrahydrofuran (10 mL) was used to give a crude product of the title compound (560 mg) as a colorless oil in the same manner as in Example 1a.
- Example 24b (2-chloro-6-hydroxy-4-methoxyphenyl) (2-fluoro-4-methylphenyl) methanone 1-chloro-3,5-dimethoxybenzene (560 mg, 3.24 mmol), toluene (3 mL ), Aluminum chloride (480 mg, 3.60 mmol) and the crude product obtained in Example 24a (560 mg, 3.24 mmol) using the same method as in Example 2a to obtain the crude product of the title compound (630 mg). Obtained as a solid.
- Example 24c (2-Chloro-4,6-dihydroxyphenyl) (2-fluoro-4-methylphenyl) methanone
- the crude product obtained in Example 24b (510 mg, 1.73 mmol) was converted to 1,2-dichloroethane. (5 mL), aluminum chloride (460 mg, 3.45 mmol) was added, and the mixture was stirred at 80 ° C. for 1.5 hr.
- the reaction mixture was cooled to 0 ° C., added with an appropriate amount of ice, stirred for 5 minutes, extracted with ethyl acetate (40 mL), and washed with 2 mol / L hydrochloric acid (10 mL) and saturated sodium hydrogen carbonate solution (20 mL).
- Example 24d 5-chloro-4- (2-fluoro-4-methylphenyl) -2,2-dimethyl-4H-benzo [1,3] dioxin-7-ol
- the product (366 mg, 1.30 mmol), methanol (5 mL) and sodium borohydride (123 mg, 3.25 mmol) were used in the same manner as in Example 2b to obtain a crude triol product (360 mg).
- Example 24e Methyl 5-chloro-4- (2-fluoro-4-methylphenyl) -2,2-dimethyl-4H-benzo [1,3] dioxin-7-carboxylate obtained in Example 24d Using the compound (310 mg, 0.96 mmol), methylene chloride (6 mL), pyridine (0.120 mL, 1.49 mmol) and trifluoromethanesulfonic anhydride (0.190 mL, 1.13 mmol), the triflate form was prepared in the same manner as in Example 2c. Crude product (437 mg) was obtained.
- Example 24g 3-Chloro-2- ⁇ [1- (2-fluoro-4-methylphenyl) -1-methoxy] methyl ⁇ -5- (hydroxymethyl) phenol
- Crude product obtained in Example 24f (241 mg, 0.72 mmol) was dissolved in methanol (5 mL), paratoluenesulfonic acid monohydrate (68 mg, 0.36 mmol) was added, and the mixture was stirred at 50 ° C. for 2 hr. Triethylamine was added to the reaction solution, and the solvent was removed under reduced pressure.
- purification using silica gel flash chromatography (hexane: ethyl acetate, 9: 1 to 3: 2, V / V) gave the title compound (210 mg) as an oil.
- Example 24h 3-Chloro-4- ⁇ [1- (2-fluoro-4-methylphenyl) -1-methoxy] methyl ⁇ -5-hydroxybenzyl acetate
- bis (dibutyltin chloride) oxide (112 mg, 0.20 mmol) were used in the same manner as in Example 2e to obtain the amorphous title compound (238 mg). It was.
- Example 24i Acetic acid 3-chloro-4- (2-fluoro-4-methylbenzyl) -5-hydroxybenzyl
- acetonitrile 5 mL
- triethylsilane 0.322 mL, 2.01 mmol
- boron trifluoride-diethyl ether complex 0.127 mL, 1.01 mmol
- Example 24j 5-Acetoxymethyl-3-chloro-2- (2-fluoro-4-methylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (236 mg, 0.39 mmol), methylene chloride (4 mL), The imidate was prepared in the same manner as Example 2g using trichloroacetonitrile (0.195 mL, 1.93 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (6 ⁇ L, 0.04 mmol).
- Example 24i Using the resulting imidate (292 mg), the compound obtained in Example 24i (96 mg, 0.30 mmol), methylene chloride (4 mL), MS4A and boron trifluoride-diethyl ether complex (0.049 mL, 0.39 mmol) In the same manner as in Example 2g, a crude product (435 mg) of the title compound was obtained.
- Example 24k 3-Chloro-2- (2-fluoro-4-methylbenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 24j Using the same crude product (435 mg), methanol / methylene chloride (8 mL / 2 mL) and potassium carbonate (534 mg, 3.89 mmol) as in Example 2h, the title compound (86 mg) was obtained as a white solid. However, solidification was performed from hexane / ethyl acetate / methanol.
- Example 25a (2-chloro-6-hydroxy-4-methoxyphenyl) (4-ethyl-2-fluorophenyl) methanone 1-chloro-3,5-dimethoxybenzene (855 mg, 4.95 mmol), toluene (5 mL ), Aluminum chloride (727 mg, 5.45 mmol) and 4-ethyl-2-fluorobenzoyl chloride (US2006 / 20146A1) (918 mg, 4.92 mmol) in the same manner as in Example 2a, the crude product (1.02 g) was obtained as a solid.
- Example 25b (2-Chloro-4,6-dihydroxyphenyl) (4-ethyl-2-fluorophenyl) methanone Crude product obtained in Example 25a (1.02 g, 3.30 mmol), 1,2- A crude product of the title compound (824 mg) was obtained as a solid in the same manner as in Example 24c using dichloroethane (10 mL) and aluminum chloride (880 mg, 6.60 mmol).
- Example 25c 5-Chloro-2,2-dimethyl-4- (4-ethyl-2-fluorophenyl) -4H-benzo [1,3] dioxin-7-ol
- Product 815 mg, 2.77 mmol
- sodium borohydride 262 mg, 6.93 mmol
- Example 25d 5-Chloro-2,2-dimethyl-4- (4-ethyl-2-fluorophenyl) -4H-benzo [1,3] dioxin-7-carboxylate methyl obtained in Example 25c Using the compound (515 mg, 1.53 mmol), methylene chloride (10 mL), pyridine (0.185 mL, 2.29 mmol) and trifluoromethanesulfonic anhydride (0.308 mL, 1.83 mmol) in the same manner as in Example 2c, Crude product (717 mg) was obtained.
- Example 25d Using the compound obtained in Example 25d (465 mg, 1.23 mmol), lithium aluminum hydride (70 mg, 1.84 mmol) and tetrahydrofuran (7 mL) in the same manner as in Example 2d, the crude product of the title compound (432 mg) was obtained as amorphous.
- Example 25f 3-Chloro-2- [1- (4-ethyl-2-fluorophenyl) -1-methoxymethyl] -5- (hydroxymethyl) phenol
- the crude product obtained in Example 25e (430 mg 1.23 mmol), methanol (10 mL), and paratoluenesulfonic acid monohydrate (116 mg, 0.61 mmol) were used to give the title compound (377 mg) as an oil in the same manner as in Example 24g.
- Example 25g Acetic acid 3-chloro-4- [1- (4-ethyl-2-fluorophenyl) -1-methoxymethyl] -5-hydroxybenzyl
- Compound obtained in Example 25f (377 mg, 1.16 mmol)
- Tetrahydrofuran (4 mL)
- vinyl acetate (4 mL)
- bis (dibutyltin chloride) oxide (192 mg, 0.35 mmol) were used in the same manner as in Example 2e to obtain the oily title compound (414 mg).
- Example 25h Acetic acid 3-chloro-4- (4-ethyl-2-fluorobenzyl) -5-hydroxybenzyl
- Compound (414 mg, 1.13 mmol) obtained in Example 25 g, acetonitrile (8 mL), triethylsilane ( 0.539 mL, 3.38 mmol) and boron trifluoride-diethyl ether complex (0.213 mL, 1.70 mmol) were used in the same manner as in Example 2f to obtain the title compound (271 mg) as a white solid.
- Example 25i 5-Acetoxymethyl-3-chloro-2- (4-ethyl-2-fluorobenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (236 mg, 0.39 mmol), methylene chloride (4 mL), The imidate was prepared in the same manner as Example 2g using trichloroacetonitrile (0.195 mL, 1.93 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (6 ⁇ L, 0.04 mmol).
- Example 25h Using the resulting imidate (292 mg), the compound obtained in Example 25h (100 mg, 0.30 mmol), methylene chloride (4 mL), MS4A and boron trifluoride-diethyl ether complex (0.049 mL, 0.39 mmol) In the same manner as in Example 2g, a crude product (409 mg) of the title compound was obtained.
- Example 25j 3-Chloro-2- (4-ethyl-2-fluorobenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 25i Using the same crude product (409 mg), methanol / methylene chloride (8 mL / 2 mL) and potassium carbonate (534 mg, 3.89 mmol), the title compound (52 mg) was obtained as a white solid in the same manner as in Example 2h. However, solidification was performed from hexane / ethyl acetate / methanol.
- Example 26a Ethyl 3-hydroxy-5-oxo-4- [4- (trifluoromethyl) benzoyl] cyclohex-3-enecarboxylate ethyl 3-hydroxy-5-oxocyclohex-3-enecarboxylate ( 1.0 g, 5.4 mmol), 4-trifluoromethylbenzoic acid chloride (1.16 g, 5.6 mmol), triethylamine (2.3 mL, 16.5 mmol), trimethylsilylcyanide (90 ⁇ L, 0.67 mmol) and acetonitrile (15 mL) In the same manner as in Example 4a, a crude product of the title compound (2.25 g) was obtained as a pale yellow solid.
- Example 26b Ethyl 3-fluoro-5-oxo-4- [4- (trifluoromethyl) benzoyl] cyclohex-3-enecarboxylate
- Crude product obtained in Example 26a (2.25 g, 5.4 mmol) was dissolved in methylene chloride (20 mL), diethylaminosulfur trifluoride (2.2 mL, 16.7 mmol) was added under ice cooling, and the mixture was stirred at room temperature for 2 hr. Distilled water (5 mL) was added dropwise to the reaction solution, diluted with methylene chloride (10 mL), and washed successively with distilled water (10 mL, twice) and saturated brine (10 mL).
- Example 26c Ethyl 3-fluoro-5-hydroxy-4- [4- (trifluoromethyl) benzoyl] benzoate Compound obtained in Example 26b (1.31 g, 3.7 mmol), triethylamine (1.5 mL, 10.8 mmol), acetonitrile (13 mL) and trimethylsilane iodide (1.3 mL, 9.1 mmol) were used in the same manner as in Example 5c to obtain a silyl enol ether as an oily crude product.
- Example 5c the obtained oily crude product was sequentially treated with toluene (13 mL), silica gel SK-85 (5.2 g), potassium carbonate (506 mg, 3.7 mmol) and ethanol (13 mL). Purification using silica gel flash column chromatography (hexane: ethyl acetate, 5: 1-4: 1, V / V) gave the title compound (0.82 g) as a pale yellow solid.
- Example 26d 3-Fluoro-5-hydroxymethyl-2- ⁇ 1-hydroxy-1- [4- (trifluoromethyl) phenyl] methyl ⁇ phenol
- the compound obtained in Example 26c (0.82 g, 2.3 mmol ) was dissolved in tetrahydrofuran (12 mL), lithium aluminum hydride (0.26 g, 6.5 mmol) was added under ice cooling, and the mixture was stirred at room temperature for 15 minutes.
- Example 26e 3-Fluoro-5-hydroxymethyl-2- ⁇ 1-methoxy-1- [4- (trifluoromethyl) phenyl] methyl ⁇ phenol
- the crude product obtained in Example 26d (0.66 g, 2.1 mmol) was dissolved in methanol (10 mL), paratoluenesulfonic acid monohydrate (0.2 g, 1.1 mmol) was added, and the mixture was stirred at 50 ° C. for 4 hr.
- Triethylamine (290 ⁇ L, 2.1 mmol) was added to the reaction solution under ice cooling.
- Example 26f Acetic acid 3-fluoro-5-hydroxy-4- ⁇ 1-methoxy-1- [4- (trifluoromethyl) phenyl] methyl ⁇ benzyl
- Crude product obtained in Example 26e (0.74 g, 2.1 mol) was dissolved in diisopropyl ether (4 mL), vinyl acetate (4 mL, 43.2 mmol) and porcine pancreatic lipase (0.37 g) were added, and the mixture was stirred at 35 ° C. for 24 hours. After filtration, the solvent was distilled off under reduced pressure.
- Example 26h 3-Fluoro-5-hydroxy-4- [4- (trifluoromethyl) benzyl] benzyl acetate
- the compound (0.43 g, 1.2 mmol) obtained in Example 26g was dissolved in acetonitrile (5 mL). Under ice cooling, triethylsilane (550 ⁇ L, 3.5 mmol) and boron trifluoride-diethyl ether complex (440 ⁇ L, 3.5 mmol) were added, and the mixture was stirred at 50 ° C. for 4 hours.
- Example 26i 5-Acetoxymethyl-3-fluoro- [4- (trifluoromethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D -Gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero-D-gluco-heptopyranoside (275 mg, 0.45 mmol), trichloroacetonitrile (135 ⁇ L, 1.35 mmol), 1,8 -Imidabicyclo [5.4.0] -7-undecene (7 ⁇ L, 0.05 mmol) and methylene chloride (5 mL) were used to prepare an imidate in the same manner as in Example 4i.
- Example 26h The compound (0.14 g) obtained in Example 26h 0.41 mmol), boron trifluoride-diethyl ether complex (51 ⁇ L, 0.41 mmol) and methylene chloride (5 mL) were obtained in the same manner as in Example 4i to obtain a crude product (339 mg) of the title compound.
- Example 26j 3-Fluoro-5-hydroxymethyl-2- [4- (trifluoromethyl) benzyl] phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside Crude obtained in Example 26i Using the product (339 mg, 0.36 mmol), tetrahydrofuran (1 mL), methanol (4 mL) and potassium carbonate (56 mg, 0.41 mmol), the title compound (100 mg) was obtained as a colorless solid in the same manner as in Example 4j. .
- Example 27a Ethyl 3-chloro-5-oxo-4- [4- (trifluoromethyl) benzoyl] cyclohex-3-enecarboxylate Crude product obtained in Example 26a (2.25 g, 5.4 mmol)
- Example 4b oxalyl dichloride (500 ⁇ L, 3.8 mmol), 2-methyl-2-butene (2.3 mL, 21.7 mmol), N, N-dimethylformamide (100 ⁇ L, 1.29 mmol) and methylene chloride (20 mL).
- a crude product (2.5 g) of the title compound was obtained as a brown oily substance.
- Example 27b Ethyl 3-chloro-5-hydroxy-4- [4- (trifluoromethyl) benzoyl] benzoate
- Crude product obtained in Example 27a (2.5 g), N-methylmorpholine (10 mL)
- the title compound (1.66 g) was obtained as a brown solid in the same manner as in Example 4c, using iodine and iodine (1.65 g, 6.5 mmol).
- purification was performed using silica gel flash column chromatography (hexane: ethyl acetate, 5: 1-4: 1-3: 1, V / V).
- Example 27c 3-Chloro-5-hydroxymethyl-2- ⁇ 1-hydroxy-1- [4- (trifluoromethyl) phenyl] methyl ⁇ phenol
- the compound obtained in Example 27b (1.66 g, 4.5 mmol)
- Lithium aluminum hydride (0.51 g, 13.4 mmol)
- tetrahydrofuran (17 mL) were obtained in the same manner as in Example 26d to obtain a crude product of the title compound (1.44 g) as a colorless solid.
- Example 27d 3-chloro-5-hydroxymethyl-2- ⁇ 1-methoxy-1- [4- (trifluoromethyl) phenyl] methyl ⁇ phenol
- the crude product obtained in Example 27c (1.44 g, 4.3 mmol), paratoluenesulfonic acid monohydrate (0.41 g, 2.2 mmol) and methanol (15 mL) were used in the same manner as in Example 26e to obtain a crude product of the title compound.
- the product was purified by silica gel flash column chromatography (hexane: ethyl acetate, 4: 1-3: 1-2: 1, V / V) to give the title compound (0.31 g) as a colorless oil.
- Example 27e 3-chloro-5-hydroxy-4- ⁇ 1-methoxy-1- [4- (trifluoromethyl) phenyl] methyl ⁇ benzyl acetate
- Acetic acid (0.31 g, 0.89 mmol) obtained in Example 27d
- Vinyl acetate (3 mL, 32.4 mmol)
- porcine pancreatic lipase (0.16 g)
- diisopropyl ether 3 mL
- the purification was performed using silica gel flash column chromatography (hexane: ethyl acetate, 6: 1-5: 1-4: 1, V / V).
- Example 27f Acetic acid 3-chloro-5-hydroxy-2- [4- (trifluoromethyl) benzyl] benzyl Compound obtained in Example 27e (0.33 g, 0.85 mmol), triethylsilane (400 ⁇ L, 2.5 mmol) ), Boron trifluoride-diethyl ether complex (320 ⁇ L, 2.5 mmol) and acetonitrile (3 mL) by a method similar to that in Example 26h, 3-chloro-5-hydroxymethyl-2- [4- (tri Fluoromethyl) benzyl] phenol (100 mg) was obtained.
- Example 27g 5-Acetoxymethyl-3-chloro-2- [4- (trifluoromethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero-D-gluco-heptopyranoside (194 mg, 0.32 mmol), trichloroacetonitrile (95 ⁇ L, 0.95 mmol), 1 , 8-diazabicyclo [5.4.0] -7-undecene (5 ⁇ L, 0.03 mmol) and methylene chloride (5 mL) were used to prepare an imidate in the same manner as in Example 4i, and the compound obtained in Example 27f ( 95 mg, 0.26 mmol), boron trifluoride-diethyl ether complex (33 ⁇ L, 0.26 m
- Example 27h 3-Chloro-5-hydroxymethyl-2- [4- (trifluoromethyl) benzyl] phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside Crude obtained in Example 27g Using the product (313 mg, 0.26 mmol), tetrahydrofuran (1 mL), methanol (4 mL) and potassium carbonate (36 mg, 0.26 mmol), the title compound (84 mg) was obtained as a colorless solid in the same manner as in Example 4j. .
- Example 28a Methyl 4- (2,2-difluoroacetyl) benzoate
- Methyl 4-acetylbenzoate (1.0 g, 5.6 mmol) was dissolved in a solution consisting of benzene (28 mL) and hexane (56 mL), and morpholine ( Using a hexane solution (15 mL) in which 2.3 mL, 26 mmol) and titanium trichloride (0.42 mL, 3.9 mmol) are dissolved, the literature is known (J. Org. Chem., 2005, 70 (14), 5763.) The method yielded the corresponding crude enamine product as a yellow oil.
- Example 28b Methyl 4- (2,2-difluoro-1-hydroxyethyl) benzoate
- the crude product obtained in Example 28a (1.2 g, 5.6 mmol) was obtained from methanol (5 mL) and tetrahydrofuran (5 mL).
- the resulting solution was dissolved in sodium borohydride (0.32 g, 8.4 mmol) under ice cooling.
- the reaction mixture was stirred for 2 hours while warming to room temperature, and saturated aqueous ammonium chloride solution (1 mL) was added dropwise under ice cooling to stop the reaction.
- Example 28c Methyl 4- (2,2-difluoroethyl) benzoate
- the compound synthesized in Example 28b (0.69 g, 3.2 mmol) was dissolved in tetrahydrofuran (10 mL), and thiocarbonyldiimidazole (0.86 g) was dissolved at room temperature. 4.8 mmol), and the mixture was stirred for 1 hour while heating under reflux.
- the mixture was cooled, diluted with ethyl acetate (50 mL), and washed successively with 1M hydrochloric acid (5 mL), saturated sodium bicarbonate (10 mL), and saturated brine (10 mL).
- Example 28d 4- (2,2-difluoroethyl) benzoic acid
- the compound obtained in Example 28c (0.53 g, 2.6 mmol) was dissolved in 1,4-dioxane (3.4 mL), and 2M water at room temperature.
- An aqueous sodium oxide solution (3.4 mL, 6.8 mmol) was added, and the mixture was stirred at 35 ° C. for 1 hour.
- the reaction mixture was poured into 1M hydrochloric acid (20 mL), extracted with ethyl acetate (50 mL), and the organic layer was washed with saturated brine (10 mL). After drying over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure to obtain a crude product of the title compound (0.48 g) as a colorless solid.
- Example 28e 4- (2,2-difluoroethyl) benzoyl chloride
- the crude product (0.48 g, 2.6 mmol) obtained in Example 28d was dissolved in methylene chloride, and oxalyl chloride (1.1 mL) was cooled with ice. 13 mmol) and N, N-dimethylformamide (0.1 mL, 1.3 mmol) were added, and the mixture was stirred for 3 and a half hours while warming to room temperature. After completion of the reaction, the solvent was removed under reduced pressure to obtain a crude product (0.53 g) of the title compound as a colorless oil.
- Example 28f Ethyl 4- [4- (2,2-difluoroethyl) benzoyl] -3-hydroxy-5-oxocyclohex-3-enecarboxylate 3-hydroxy-5-oxocyclohex-3-ene Using the ethyl carboxylate (EP1571148A1) (0.48 g, 2.6 mmol) and the crude product obtained in Example 28e (0.53 g, 2.6 mmol) in the same manner as in Example 1b, the crude product of the title compound (0.92 g) was obtained.
- Example 28g Ethyl 4- [4- (2,2-difluoroethyl) benzoyl] -3-fluoro-5-oxocyclohex-3-enecarboxylate
- the crude product obtained in Example 28f (0.95 g 2.7 mmol) was dissolved in methylene chloride (10 mL), and the title compound (0.47 g) was obtained in the same manner as in Example 3b using diethylaminosulfur trifluoride (1.1 mL, 8.1 mmol).
- Example 28h Ethyl 4- [4- (2,2-difluoroethyl) benzoyl] -3-fluoro-5-hydroxybenzoate
- the compound obtained in Example 28g (0.47 g, 1.3 mmol) was converted to N-methyl. It was dissolved in morpholine (6 mL), and a crude product (0.46 g) of the title compound was obtained in the same manner as in Example 3c using anhydrous sodium sulfate (0.47 g) and iodine (0.40 g, 1.6 mmol).
- Example 28i Ethyl 4- ⁇ 1- [4- (2,2-difluoroethyl) phenyl] -1-hydroxymethyl ⁇ -3-fluoro-5-hydroxybenzoate
- Crude product obtained in Example 28h (0.46 g, 1.3 mmol) was dissolved in a solution of tetrahydrofuran (3 mL) and methanol (2 mL), and the title compound (0.33 g) was prepared in the same manner as in Example 3d using sodium borohydride (98 mg, 2.6 mmol). ) was obtained as a colorless solid.
- Example 28j Ethyl 4- [4- (2,2-difluoroethyl) benzyl] -3-fluoro-5-hydroxybenzoate
- the compound obtained in Example 28i (0.33 g, 0.93 mmol) was added to ethanol (10 mL).
- ethanol 10 mL
- the crude product of the title compound (0.31 g) Got.
- the crude product obtained in Example 28j (0.31 g, 0.93 mmol)
- the product was dissolved in tetrahydrofuran (10 mL), and the crude product (0.31 g) of the title compound was obtained as a colorless solid in the same manner as in Example 3f using lithium aluminum hydride (0.14 g, 3.7 mmol).
- Example 28l Acetic acid 4- [4- (2,2-difluoroethyl) benzyl] -3-fluoro-5-hydroxybenzyl
- the crude product obtained in Example 28k (0.31 g, 0.93 mmol) was converted into vinyl acetate. (3 mL) and diisopropyl ether (3 mL) were dissolved in the solution, and the title compound (0.30 g) was obtained as a pale brown solid by the same method as Example 3 g using porcine pancreatic lipase (1.0 g).
- Example 28m 5-Acetoxymethyl-3-fluoro-2- [4- (2,2-difluoroethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D- Glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231 )) (0.61 g, 1.0 mmol) dissolved in methylene chloride (4 mL) and trichloroacetonitrile (0.30 mL, 3.0 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (4 ⁇ L, 0.03 mmol)
- the imidate was prepared in the same manner as in Example 1h.
- Example 28l Using this imidate (0.75 g, 1.0 mmol), the compound obtained in Example 28l (0.30 g, 0.89 mmol) and boron trifluoride-diethyl ether complex (56 ⁇ L, 0.44 mmol), the same method as in Example 1h Gave a mixture containing the title compound.
- Example 28n 3-Fluoro-2- [4- (2,2-difluoroethyl) benzyl] -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside
- Example 28m The mixture obtained in step (0.89 mmol) was dissolved in a solution consisting of tetrahydrofuran (3 mL) and methanol (3 mL), and 2M aqueous sodium hydroxide solution (3.0 mL, 6.0 mmol) was used in the same manner as in Example 3i. The title compound (0.12 g) was obtained as a colorless solid.
- Example 29a Ethyl 3-chloro-4- [4- (2,2-difluoroethyl) benzoyl] -5-oxocyclohex-3-enecarboxylate
- the crude product obtained in Example 28f (0.92 g , 2.6 mmol) in methylene chloride (10 mL), 2-methyl-2-butene (1.1 mL, 10 mmol), oxalyl dichloride (0.23 mL, 2.7 mmol) and N, N-dimethylformamide (0.05 mL, 0.07).
- the crude product (0.96 g) of the title compound was obtained in the same manner as in Example 1b.
- Example 29b Ethyl 3-chloro-4- [4- (2,2-difluoroethyl) benzoyl] -5-hydroxybenzoate
- the crude product obtained in Example 29a (0.96 g, 2.6 mmol) was converted to N -Dissolved in methylmorpholine (12 mL), and obtained the crude product (0.96 g) of the title compound in the same manner as in Example 3c using anhydrous sodium sulfate (0.96 g) and iodine (0.79 g, 3.1 mmol). .
- Example 29c 3-Chloro-2- ⁇ 1- [4- (2,2-difluoroethyl) phenyl] -1-hydroxymethyl ⁇ -5- (hydroxymethyl) phenol
- the product (0.96 g, 2.6 mmol) was dissolved in tetrahydrofuran (30 mL) and the title compound (0.85 g) was obtained as a crude product in the same manner as in Example 1e using lithium aluminum hydride (0.38 g, 10 mmol). Obtained.
- Example 29d Acetic acid 3-chloro-4- ⁇ 1- [4- (2,2-difluoroethyl) phenyl] -1-hydroxymethyl ⁇ -5-hydroxybenzyl
- Crude product obtained in Example 29c 0.85 g, 2.6 mmol
- the title compound (0.51 g) was purified by the same method as Example 3 g using porcine pancreatic lipase (2.0 g). Obtained as a solid.
- Example 29e 3-chloro-4- [4- (2,2-difluoroethyl) benzyl] -5-hydroxybenzyl acetate
- Acetic acid (10 mL) was prepared from the compound (0.40 g, 1.1 mmol) obtained in Example 29d.
- triethylsilane (0.53 mL, 3.3 mmol) and boron trifluoride-diethyl ether complex (0.28 mL, 2.2 mmol) were used in the same manner as in Example 5f (but no purification was performed). To give a mixture containing the title compound.
- Example 29f 5-Acetoxymethyl-3-chloro-2- [4- (2,2-difluoroethyl) benzyl] phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D- Glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231 )) (0.37 g, 0.60 mmol) dissolved in methylene chloride (4 mL) and trichloroacetonitrile (0.18 mL, 1.8 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (2 ⁇ L, 0.02 mmol)
- the imidate was prepared in the same manner as in Example 1h.
- Example 29e Using this imidate (0.45 g, 0.60 mmol), the compound obtained in Example 29e (0.13 g, 0.36 mmol) and boron trifluoride-diethyl ether complex (46 ⁇ L, 0.37 mmol), the same method as in Example 1h Gave a mixture containing the title compound.
- Example 29g 3-chloro-2- [4- (2,2-difluoroethyl) benzyl] -5-hydroxymethyl-phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D -Glycero- ⁇ -D-gluco-heptopyranoside
- the mixture (0.37 mmol) obtained in Example 29f was dissolved in tetrahydrofuran (3 mL) and methanol (3 mL), and 2M aqueous sodium hydroxide solution (3.0 mL, 6.0 mmol) was used.
- the title compound (0.12 g) was obtained as a colorless solid.
- Example 30a 3,5-difluoro-4- [1- (2-fluoro-4-methylphenyl) -1-hydroxymethyl] benzonitrile diisopropylamine (1.12 mL, 7.92 mmol), n-butyllithium (2.59 mL, 7.17 mmol, 2.77 M n-hexane solution), 3,5-difluorobenzonitrile (1.00 g, 7.19 mmol), 2-fluoro-4-methylbenzaldehyde (1.00 g, 7.24 mmol) and tetrahydrofuran (20 mL)
- the title compound (1.44 g) was obtained as a pale yellow solid in the same manner as in Example 7a.
- Example 30b 3,5-Difluoro-4- (2-fluoro-4-methylbenzyl) benzonitrile
- Sodium borohydride (1.69 g, 44.7 mmol) was dissolved in trifluoroacetic acid (30 mL) and brought to 0 ° C.
- a methylene chloride solution (7 mL) in which the compound obtained in Example 30a (1.24 g, 4.47 mmol) was dissolved was added. Thereafter, the reaction solution was stirred at room temperature for 30 hours. However, during that time, sodium borohydride (2.00 g, 52.9 mmol) and trifluoroacetic acid (20 mL) were added in several portions until the reaction was completed.
- reaction solution was added to a 10% aqueous sodium hydroxide solution (300 mL) cooled to 0 ° C.
- the mixture was extracted with diethyl ether (300 mL), and washed successively with water (100 mL), saturated aqueous ammonium chloride (100 mL), and saturated brine (100 mL).
- the organic layer was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 10: 1, V / V).
- the title compound (756 mg) was obtained as a white solid.
- Example 30c 3-Benzyloxy-5-fluoro-4- (2-fluoro-4-methylbenzyl) benzonitrile
- the compound obtained in Example 30b (756 mg, 2.89 mmol), sodium hydride (63%, 165 mg, 4.33 mmol), benzyl alcohol (387 mg, 3.58 mmol) and N, N-dimethylformamide (10 mL) were used in the same manner as in Example 7c to obtain the title compound (768 mg) as a white solid.
- the purification was performed using silica gel flash chromatography (hexane: ethyl acetate 19: 1 to 10: 1, V / V).
- Example 30d 3-Benzyloxy-5-fluoro-4- (2-fluoro-4-methylbenzyl) benzoic acid
- 5M aqueous sodium hydroxide solution 2.2 mL, 11.0 mmol
- ethanol 10 mL
- Example 30e 3-Fluoro-4- (2-fluoro-4-methylbenzyl) -5-hydroxybenzoic acid
- Crude product obtained in Example 30d (740 mg, 2.01 mmol), 10% palladium on carbon catalyst (
- a crude product (553 mg) of the title compound was obtained in the same manner as in Example 7e using water (150 mg), tetrahydrofuran (7 mL), methanol (7 mL) and hydrogen.
- Example 30f 3-Fluoro-2- (2-fluoro-4-methylbenzyl) -5- (hydroxymethyl) phenol
- Crude product obtained in Example 30e (553 mg, 1.99 mmol), lithium aluminum hydride (188 mg, 4.95 mmol) and tetrahydrofuran (20 mL) were used to give a crude product of the title compound (498 mg) in the same manner as in Example 7f.
- Example 30g 3-fluoro-4- (2-fluoro-4-methylbenzyl) -5-hydroxybenzyl acetate
- Acetic product (498 mg, 1.88 mmol) obtained in Example 30f, bis (dibutyltin chloride)
- the title compound (522 mg) was obtained in the same manner as in Example 7g using oxide (312 mg, 0.56 mmol), vinyl acetate (5 mL) and tetrahydrofuran (5 mL).
- Example 30h 5-Acetoxymethyl-3-fluoro-2- (2-fluoro-4-methylbenzyl) phenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231)) 311 mg, 0.51 mmol), methylene chloride (6 mL), trichloroacetonitrile (0.257 mL, 2.56 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (8 ⁇ L, 0.05 mmol) as in Example 1h
- the imidate was prepared by the method.
- Example 30g Same as Example 1h, using the compound obtained in Example 30g (122 mg, 0.40 mmol), imidate (0.51 mmol), methylene chloride (6 mL) and boron trifluoride-diethyl ether complex (0.064 mL, 0.51 mmol). In this way, a crude product of the title compound was obtained.
- Example 30i 3-Fluoro-2- (2-fluoro-4-methylbenzyl) -5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 30h Using the crude product, methanol / methylene chloride (10 mL / 2.5 mL) and potassium carbonate (700 mg, 5.06 mmol), a crude product of the title compound was obtained in the same manner as in Example 2h. The resulting crude product was purified using silica gel flash chromatography (hexane: ethyl acetate, 1: 1 to methylene chloride: methanol, 10: 1, V / V) to give the title compound (87 mg).
- Example 31a (4-Ethyl-2-fluorophenyl) methanol 4-ethyl-2-fluorobenzoyl chloride (US2006 / 20146 A1) (200 mg, 1.19 mmol), lithium aluminum hydride (90 mg, 2.37 mmol) and tetrahydrofuran (20 mL) was used to give a crude product of the title compound (194 mg) in the same manner as in Example 2d.
- Example 31b 4-Ethyl-2-fluorobenzaldehyde
- the crude product obtained in Example 31a (194 mg) was dissolved in methylene chloride (10 mL), and cooled with ice, pyridinium chlorochromate (540 mg, 2.51 mmol). And stirred at room temperature for 2 hours.
- the reaction mixture was filtered through celite, the solvent was evaporated under reduced pressure, and the obtained residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 9: 1, V / V) to give the oily title compound ( 158 mg) was obtained.
- Example 31c 3,5-difluoro-4- [1- (4-ethyl-2-fluorophenyl) -1-hydroxymethyl] benzonitrile diisopropylamine (0.85 mL, 6.01 mmol), n-butyllithium (1.97 mL, 5.46 mmol, 2.77 M n-hexane solution), 3,5-difluorobenzonitrile (759 mg, 5.46 mmol), the compound obtained in Example 31b (830 mg, 5.45 mmol) and tetrahydrofuran (12 mL) In the same manner as in Example 7a, the oily title compound (1.49 g) was obtained.
- Example 31d 3,5-Difluoro-4- (4-ethyl-2-fluorobenzyl) benzonitrile
- Sodium borohydride (1.94 g, 51.2 mmol) was dissolved in trifluoroacetic acid (35 mL) and brought to 0 ° C. After cooling, a methylene chloride solution (7 mL) in which the compound obtained in Example 31c (1.49 g, 5.12 mmol) was dissolved was added. Then, it stirred at room temperature for 30 hours. However, during that time, sodium borohydride (1.94 g, 51.2 mmol) and trifluoroacetic acid (20 mL) were added in several portions until the reaction was completed.
- reaction solution was added to a 10% aqueous sodium hydroxide solution (330 mL) cooled to 0 ° C.
- the mixture was extracted with diethyl ether (300 mL), and washed successively with water (100 mL), saturated aqueous ammonium chloride (100 mL), and saturated brine (100 mL).
- the organic layer was dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and the resulting residue was purified using silica gel flash chromatography (hexane: ethyl acetate, 19: 1 to 10: 1, V / V).
- the title compound (947 mg) was obtained as a white solid.
- Example 31e 3-Benzyloxy-4- (4-ethyl-2-fluorobenzyl) -5-fluorobenzonitrile
- the compound obtained in Example 31d (940 mg, 3.41 mmol), sodium hydride (63%, 195 mg, 5.12 mmol), benzyl alcohol (0.46 mL, 4.47 mmol) and N, N-dimethylformamide (12 mL) were used in the same manner as in Example 7c to obtain the title compound (1.11 g) as a white solid.
- the purification was performed using silica gel flash chromatography (hexane: ethyl acetate 19: 1 to 10: 1, V / V).
- Example 31f 3-Benzyloxy-4- (4-ethyl-2-fluorobenzyl) -5-fluorobenzoic acid
- the compound obtained in Example 31e (1.11 g, 3.05 mmol), 5M aqueous sodium hydroxide solution
- the crude product (1.01 g) of the title compound was obtained in the same manner as in Example 7d using 3.37 mL, 16.8 mmol) and ethanol (10 mL).
- Example 31g 4- (4-Ethyl-2-fluorobenzyl) -3-fluoro-5-hydroxybenzoic acid Crude product obtained in Example 31f (1.01 g, 2.64 mmol), 10% palladium on carbon catalyst (Water content, 300 mg), tetrahydrofuran (10 mL), methanol (10 mL) and hydrogen were used in the same manner as in Example 7e to obtain a crude product of the title compound (806 mg).
- Example 31h 2- (4-Ethyl-2-fluorobenzyl) -3-fluoro-5- (hydroxymethyl) phenol
- Example 31g 4-Ethyl-2-fluorobenzyl) -3-fluoro-5- (hydroxymethyl) phenol
- Li aluminum hydride 342 mg, 9.0 mmol
- tetrahydrofuran 10 mL
- Example 31i 2- (4-Ethyl-2-fluorobenzyl) -3-fluoro-5-hydroxybenzyl acetate
- the crude product obtained in Example 31h (827 mg, 2.97 mmol), bis (dibutyltin chloride)
- the title compound (871 mg) was obtained in the same manner as in Example 7g using oxide (164 mg, 0.30 mmol), vinyl acetate (10 mL) and tetrahydrofuran (10 mL).
- Example 31j 5-Acetoxymethyl-2- (4-ethyl-2-fluorobenzyl) -3-fluorophenyl 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ -D-gluco-heptopyranoside 7-deoxy-2,3,4,6-tetra-O-benzoyl-D-glycero- ⁇ , ⁇ -D-gluco-heptopyranoside (WO2008 / 016132 (PCT / JP2007 / 65231)) 2.5 g, 4.08 mmol), methylene chloride (20 mL), trichloroacetonitrile (1.2 mL, 12.2 mmol) and 1,8-diazabicyclo [5.4.0] -7-undecene (61 ⁇ L, 0.41 mmol) and Example 1h In the same manner, imidate was prepared.
- Example 1h using the compound obtained in Example 31i (870 mg, 2.7 mmol), imidate (4.1 mmol), methylene chloride (30 mL) and boron trifluoride-diethyl ether complex (0.38 mL, 3.0 mmol). In this way, a crude product of the title compound was obtained.
- Example 31k 2- (4-Ethyl-2-fluorobenzyl) -3-fluoro-5- (hydroxymethyl) phenyl 7-deoxy-D-glycero- ⁇ -D-gluco-heptopyranoside obtained in Example 31j Using the crude product, methanol / methylene chloride (16 mL / 4 mL) and sodium methoxide (162.1 mg, 3.0 mmol), a crude product of the title compound was obtained in the same manner as in Example 2h.
- the target DNA fragment is recovered from a single band corresponding to 2013 bases, cleaved with restriction enzymes HindIII and XhoI, and then placed on the HindIII / XhoI site of the vector pCMV-Script (Stratagene).
- the SGLT1 expression plasmid pCMV-SGLT1 was introduced.
- a HindIII / XhoI fragment was excised from pCMV-SGLT1 and introduced into the BamHI / XhoI site of pENTR1A (Gateway, Invitrogen) to create pENTR-SGLT1.
- SGLT1-expressing retroviral vector pLPCX-SGLT1 was prepared using the retroviral vector pLPCX (Clontech) into which Gateway Vector Conversion System Cassette A (Invitrogen) was introduced as the destination plasmid.
- the retrovirus pLPCX-SGLT1 obtained in 1) was transfected into integrin ⁇ v ⁇ 3-expressing HEK-293 cells, followed by antibiotic G418 (trade name: Geneticin, manufactured by Invitrogen), puromycin ( The cells were treated with Clontech), and stable expression cells HEK-SGLT1 of the target vector having resistance were obtained. Culture and maintenance of stably expressing cells were performed using DMEM medium containing 250 mg / ml G418, 1 mg / ml puromycin, 3 mM KGT-1075, 10% FBS.
- HEK-SGLT1 cells were suspended in DMEM medium containing 250 mg / ml G418, 1 mg / ml puromycin and 10% FBS at a density of 10 6 cells / ml, and a 96-well culture plate coated with type I collagen ( 100 ⁇ l was seeded in each well of Corning).
- the target DNA fragment is recovered from a single band corresponding to 2037 bases, cleaved with restriction enzymes HindIII and XhoI, and then placed on the HindIII / XhoI site of the vector pCMV-Script (Stratagene). Introduced. This was designated as SGLT2 expression plasmid pCMV-SGLT2.
- a HindIII / XhoI fragment was excised from pCMV-SGLT2 and introduced into the BamHI / XhoI site of pENTR1A (Gateway, Invitrogen) to create pENTR-SGLT2.
- SGLT2-expressing retrovirus vector pLPCX-SGLT2 was prepared using the retrovirus vector pLPCX (Clontech) into which Gateway Vector Conversion System Cassette A (Invitrogen) was introduced as a destination plasmid.
- the retrovirus pLPCX-SGLT2 obtained in 1) was transfected into integrin ⁇ v ⁇ 3-expressing HEK-293 cells, followed by antibiotic G418 (trade name: Geneticin, manufactured by Invitrogen), puromycin ( The cells were treated with Clontech), and stable expression cells HEK-SGLT2 of the target vector having resistance were obtained.
- Culture and maintenance of stably expressing cells were performed using DMEM medium containing 250 mg / ml G418, 1 mg / ml puromycin, 3 mM KGT-1075, 10% FBS.
- HEK-SGLT2 cells were suspended in DMEM medium containing 250 mg / ml G418, 1 mg / ml puromycin and 10% FBS at a density of 10 6 cells / ml, and a 96-well culture plate coated with type I collagen ( 100 ⁇ l was seeded in each well of Corning).
- Test Example 3 In vivo test Each test compound is suspended or dissolved in a solvent (0.5% methylcellulose solution) to a dose of 10 mL / kg, and multiple doses (preferably in the range of 0.03 to 10 mg / kg). (Included) is orally administered to C57BL / 6NCrlCrlj mice (7-10 weeks old, male) fasted overnight. In the control group, the solvent is orally administered at 10 mL / kg. 10 minutes after administration, glucose is orally administered at 2 g / 10 mL / kg.
- the blood glucose level was measured over time (before glucose administration, 20, 40, 60, 120 minutes after administration), the area under the curve of the blood glucose level was calculated, and the rate of decrease in the area under the blood glucose level curve for each group from the control group
- the efficacy of each test compound in the living body is evaluated by obtaining a 50% effective dose ED50 by using as an index.
- the compound of the present invention or a hydrate thereof exhibits excellent human SGLT1 and / or SGLT2 inhibitory activity with low side effects, type 1 diabetes, type 2 diabetes, gestational diabetes, other factors, hyperglycemia, glucose intolerance ( impaired glucose tolerance (IGT), diabetes related diseases (eg obesity, hyperlipidemia, hypercholesterolemia, dyslipidemia, hypertension, fatty liver, metabolic syndrome, edema, heart failure, angina, myocardial infarction, arteries Sclerosis, hyperuricemia, gout, etc.) or diabetic complications (eg retinopathy, nephropathy, neuropathy, cataract, foot gangrene, infection, ketosis, etc.) It is useful as a pharmaceutical composition for the prevention or treatment of mammals (eg, humans, horses, cows or pigs, preferably humans).
- mammals eg, humans, horses, cows or pigs, preferably humans.
- SEQ ID NO: 1 PCR sense primer for human SGLT1
- SEQ ID NO: 2 PCR antisense primer for human SGLT1
- SEQ ID NO: 3 PCR sense primer for human SGLT2
- SEQ ID NO: 4 PCR antisense primer for human SGLT2
Abstract
新規な構造を有し、副作用が低く優れたヒトSGLT1および/またはSGLT2阻害活性を有する化合物またはその水和物、それらを有効成分として含有する、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全(impaired glucose tolerance:IGT)、糖尿病関連疾患または糖尿病合併症を治療および/または予防するための医薬組成物を提供すること。
【化1】 で表される化合物またはその水和物
Description
本発明は、ヒトSGLT1および/またはSGLT2活性阻害作用を有する化合物に関する。
糖尿病は、インスリン作用不足による慢性の高血糖状態を主たる特徴とする代謝疾患群である。糖尿病治療には、食事療法、運動療法と共に薬物療法が施され、糖尿病治療薬としては、インスリン抵抗性を改善するビグアナイド剤やチアゾリジンジオン剤、膵臓β細胞からのインスリン分泌を促進するスルホニルウレア剤やグリニド系薬剤、糖吸収を阻害するα-グルコシダーゼ阻害剤などが使用されている。しかし、ビグアナイド剤は乳酸アシドーシス、チアゾリジンジオン剤は体重増加と浮腫、スルホニルウレア剤やグリニド系薬剤は低血糖や長期使用による2次無効、α-グルコシダーゼ阻害剤には下痢といった副作用があることが報告されており、このような問題を解決した新しい作用機序の抗糖尿病薬の開発が望まれている。
近年、腎臓において糖再吸収を阻害することによって、尿中への糖排泄を増加させ、血糖値を低下させる新しい機序の薬剤の研究開発が進められている(例えば、非特許文献1などを参照)。本薬剤は、腎臓の近位尿細管に存在するナトリウム依存性グルコース供輸送体2(Sodium-dependent glucose cotransporter 2:以下、SGLT2という。)を阻害することで、原尿からの糖の再吸収を抑制し、体外への糖排泄を増加させることで血糖値を低下させることが示されている(例えば、非特許文献2などを参照)。こうした背景から、ヒトSGLT2を阻害する化合物は、尿中糖排泄量を増加させることで血糖値を正常化させることが期待され、1型および2型糖尿病、また高血糖に付随して起こる各種関連疾患に有効な薬剤となる。また、糖の排出を増すことで、体内の糖蓄積を減らすことによる抗肥満効果も期待される。
一方、SGLTの別のサブタイプであるSGLT1(Sodium-dependent glucose cotransporter 1:以下、SGLT1という。)は、主に小腸に発現し、食物から糖(グルコース、ガラクトース)を体内に吸収するトランスポーターとして働いている(例えば、非特許文献3などを参照)。SGLT1が先天的に欠損しているヒトでは糖吸収不良が起こることが知られている(例えば、非特許文献4などを参照)。こうした知見から、SGLT1を阻害する薬剤は、小腸からの糖の吸収を阻害・遅延させることで、食後過血糖抑制作用を示すと考えられる。また、体内への糖の流入を抑制することで抗肥満効果も期待出来る。
以上から、ヒトSGLT1および/またはSGLT2活性を抑制する薬剤は、尿中への糖排泄増加作用と小腸からの糖吸収阻害作用を併せ持つ強力な1型および2型糖尿病治療薬、抗肥満薬、高血糖に付随して起こる各種関連疾患に有効な薬剤となることが期待される。
O-アリールグルコシド化合物がヒトSGLT2阻害作用を有することは知られている(例えば、特許文献1~5などを参照)。
国際公開第01/68660号パンフレット
国際公開第02/28872号パンフレット
国際公開第02/44192号パンフレット
国際公開第02/64606号パンフレット
国際公開第08/016132号パンフレット
J. Clin. Invest., Vol.79, pp. 1510-1515(1987)
J. Clin. Invest., Vol.93, pp. 397-404(1994)
Am J Clin Nutr. Vol.59(3 Suppl) pp.690S-698S(1994)
Nature, Vol.350, pp.354-356(1991)
しかしながら、上記特許文献1~4にはいずれも、糖部分に置換基を有する化合物については全く記載されておらず、さらにそのような化合物がヒトSGLT1阻害作用を有することは記載も示唆もされていない。一方、上記特許文献5には糖部分に置換基を有する化合物について開示され、SGLT1阻害作用を有することも記載されているが、本願の優先日前に出願され、優先日後に公開された特許文献である。
従って、本発明は、新規な構造を有し、副作用が低く優れたヒトSGLT1および/またはSGLT2阻害活性を有する化合物またはその水和物、それらを有効成分として含有する、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全(impaired glucose tolerance:IGT)、糖尿病関連疾患(例えば、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症、痛風など)または糖尿病合併症(例えば、網膜症、腎症、神経障害、白内障、足壊疽、感染症、ケトーシスなど)を治療および/または予防するための医薬組成物を提供することを目的とする。
本発明は、
(1)一般式(I):
(1)一般式(I):
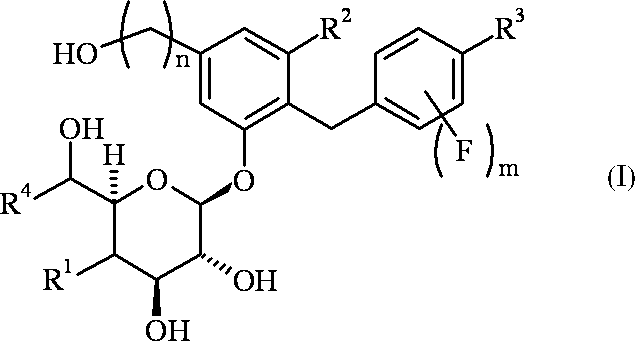
(式中、R1は水素原子または水酸基であり、
R2はフッ素原子または塩素原子であり、
R3はハロゲン原子で置換されていてもよいメチル基、ハロゲン原子で置換されていてもよいエチル基、シクロプロピル基、または、ハロゲン原子で置換されていてもよいメトキシ基であり、
R4は水素原子またはメチル基であり、
nは1または2であり、
mは0または1である、
但し、R1が水素原子である場合、R4は水素原子であり、R1が水酸基である場合、R4はメチル基である)
で表される化合物またはその水和物、
(2)一般式(II):

[式中、R1は水素原子または水酸基であり、
R2はフッ素原子または塩素原子であり、
R3はハロゲン原子で置換されていてもよいメチル基、ハロゲン原子で置換されていてもよいエチル基、シクロプロピル基、または、ハロゲン原子で置換されていてもよいメトキシ基であり、
R4は水素原子またはメチル基であり、
nは1または2であり、
mは0または1である、
但し、R1が水素原子である場合、R4は水素原子であり、R1が水酸基である場合、R4はメチル基である
(但し、(イ)R1が水酸基であり、R2がフッ素原子であり、mが0であり、かつ、nが1である場合、R3はメチル基、エチル基、シクロプロピル基またはメトキシ基ではない、
(ロ)R1が水酸基であり、R2が塩素原子であり、mが0であり、かつ、nが1である場合、R3はメトキシ基ではない、あるいは、
(ハ)R1が水素原子であり、R2がフッ素原子または塩素原子であり、mが0であり、かつ、nが1である場合、R3はメトキシ基ではない)]
で表される化合物またはその水和物、
(3)R1が水酸基である、前記(2)に記載の化合物、
(4)R3がハロゲン原子で置換されていてもよいメチル基またはハロゲン原子で置換されていてもよいメトキシ基である、前記(2)または(3)に記載の化合物、
(5)R4がメチル基である、前記(2)~(4)いずれか1つに記載の化合物、
(6)nが1である、前記(2)~(5)いずれか1つに記載の化合物、
(7)3-クロロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、2-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-2-(2-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-[4-(ジフルオロメトキシ)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-5-ヒドロキシメチル-2-[(4-トリフルオロメトキシ)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド、3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド、2-(4-エチルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド、3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド、2-(4-シクロプロピルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド、3-フルオロ-5-ヒドロキシメチルフェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-(4-エチル-3-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-5-(ヒドロキシメチル)-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-(4-エチル-2-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-5-(ヒドロキシメチル)-2-[(4-トリフルオロメチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-5-(ヒドロキシメチル)-2-[(4-トリフルオロメチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、2-[4-(2,2-ジフルオロエチル)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-クロロ-2-[4-(2,2-ジフルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、3-フルオロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、および、2-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド、からなる群より選択される化合物またはその水和物、
(8)前記(1)~(7)いずれか1つに記載の化合物またはその水和物を有効成分として含有する医薬組成物、
(9)ヒトSGLT1および/またはヒトSGLT2活性を阻害するための、前記(8)に記載の医薬組成物、
(10)1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全、糖尿病関連疾患もしくは糖尿病合併症の治療または予防のための、前記(8)または(9)に記載の医薬組成物、
(11)糖尿病関連疾患が、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症または痛風であり、あるいは、糖尿病合併症が、網膜症、腎症、神経障害、白内障、足壊疽、感染症またはケトーシスである、前記(10)に記載の医薬組成物、
(12)医薬組成物を製造するための、前記(1)~(7)いずれか1つに記載の化合物またはその水和物の使用、
(13)医薬組成物が、ヒトSGLT1および/またはヒトSGLT2活性を阻害するための組成物である、前記(12)に記載の使用、
(14)医薬組成物が、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全、糖尿病関連疾患もしくは糖尿病合併症の治療または予防のための組成物である、前記(12)または(13)に記載の使用、
(15)糖尿病関連疾患が、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症または痛風であり、あるいは、糖尿病合併症が、網膜症、腎症、神経障害、白内障、足壊疽、感染症またはケトーシスである、前記(14)に記載の使用、
(16)前記(1)~(7)いずれか1つに記載の化合物またはその水和物の治療有効量を哺乳動物に投与することを含む、ヒトSGLT1および/またはヒトSGLT2活性を阻害する方法、
(17)前記(1)~(7)いずれか1つに記載の化合物またはその水和物の治療有効量を哺乳動物に投与することを含む、疾病の治療または予防方法、
(18)疾病が、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全、糖尿病関連疾患または糖尿病合併症である、前記(17)に記載の方法、
(19)糖尿病関連疾患が、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症または痛風であり、あるいは、糖尿病合併症が、網膜症、腎症、神経障害、白内障、足壊疽、感染症またはケトーシスである、前記(18)に記載の方法、ならびに、
(20)哺乳動物がヒトである、前記(17)~(19)いずれか1つに記載の方法、
を提供する。
本発明により、副作用が低く優れたヒトSGLT1および/またはSGLT2阻害活性を有する化合物またはその水和物を提供することができる。また、本発明により、これらの化合物またはその水和物を有効成分として、含有する、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全、糖尿病関連疾患(例えば、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症、痛風など)または糖尿病合併症(例えば、網膜症、腎症、神経障害、白内障、足壊疽、感染症、ケトーシスなど)を治療および/または予防するための医薬組成物を提供することができる。
本明細書において、「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子またはヨウ素原子をいう。
一般式(I)または(II)におけるR1、R2、R3、R4、nおよびmの好ましい組み合わせを表1に示す。

本明細書において「水和物」とは、本発明の化合物の薬学的に許容され得る水和物をいう。本発明の化合物は、大気中に放置されたり、再結晶することにより、水分を吸収して、吸着水が付いたり、水和物となる場合がある。かかる水和物も本明細書における「水和物」に包含される。
本発明の化合物は、その分子内に不斉炭素原子を有するので、光学異性体が存在するこれらの異性体、およびこれらの異性体の混合物がすべて単一の式、すなわち一般式(I)または(II)で表されている。従って、本発明は光学異性体および光学異性体の任意の割合の混合物をもすべて包含するものである。
本発明の化合物(I)または(II)は、例えば、後述するA法に従って製造することができる。
なお、A法および後述のB法~D法において、各反応終了後、各反応の目的化合物は常法に従って、反応混合物から採取される。例えば、反応混合物を適宜中和し、また、不溶物が存在する場合には濾過により除去した後、水と酢酸エチルのような混和しない有機溶媒を加え、目的化合物を含む有機層を分離し、水などで洗浄後、無水硫酸ナトリウムなどで乾燥後、溶剤を留去することによって得られる。得られた化合物は、必要ならば、常法、例えばシリカゲルカラムクロマトグラフィーで分離・精製することができる。
さらに、A法および後述のB法~D法において、反応基質となる化合物が、アミノ基、ヒドロキシ基、カルボキシル基などの目的の反応を阻害する基を有する場合、必要に応じて適宜、それらの基への保護基の導入および導入した保護基の除去を行なってもよい。かかる保護基としては、通常用いられる保護基であれば特に限定はなく、例えば、T. H. Greene, P. G. Wuts, Protective Groups in Organic Synthesis. Third Edition, 1999年, John Wiley & Sons, Inc.などに記載された保護基が挙げられる。かかる保護基の導入反応および除去反応は、上記文献に記載された方法のような常法に従って行うことができる。
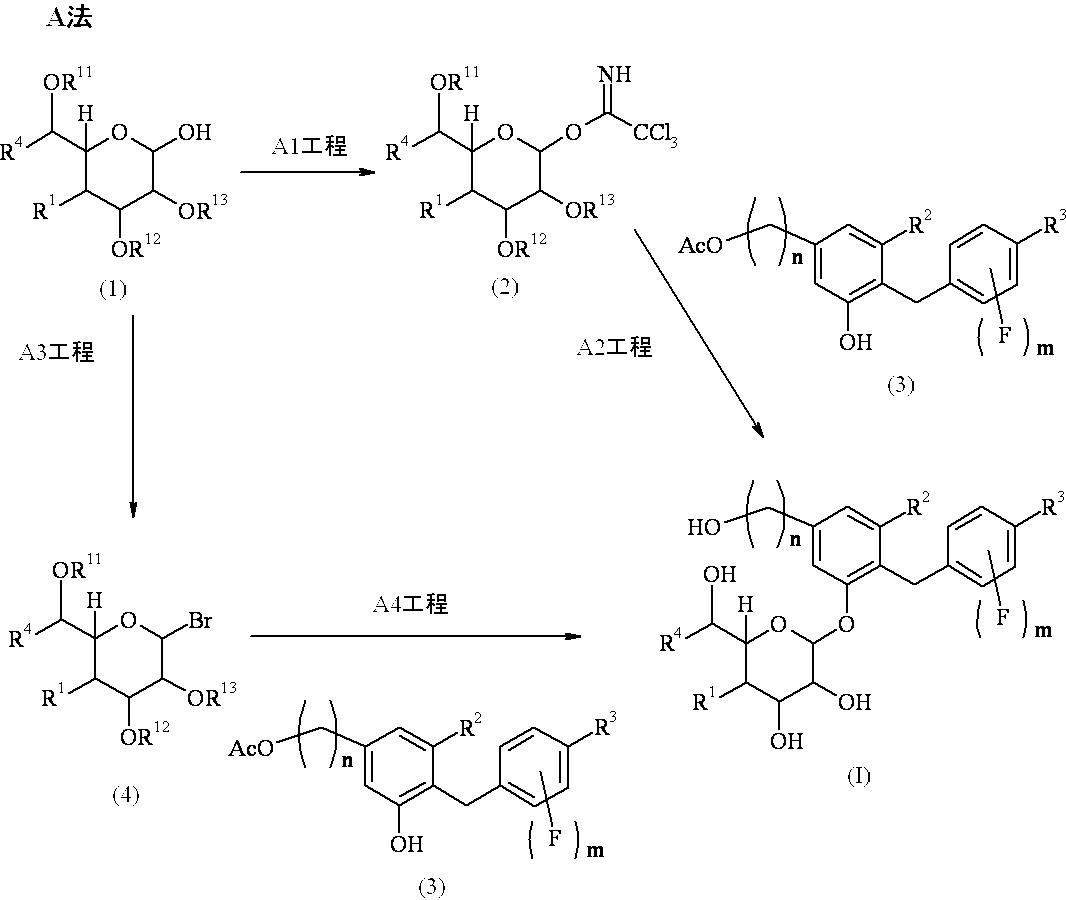
(式中、R1、R2、R3、R4、nおよびmは前記の通りであり、R11、R12およびR13は、同一または異なって、水素原子または保護基である。)
A1工程は、化合物(2)を製造する工程であり、不活性溶媒中、塩基の存在下、化合物(1)をトリクロロアセトニトリルと反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、ハロゲン化炭化水素類、エーテル類などが挙げられ、好ましくはハロゲン化炭化水素、より好ましくはメチレンクロリドである。
上記反応に使用される塩基としては、例えば、有機アミン類などが挙げられ、好ましくは1,8-ジアザビシクロ[5.4.0]-7-ウンデセンである。
反応温度は、原料化合物、塩基、不活性溶媒などにより異なるが、通常、-20℃~還流温度、好ましくは0℃~室温である。
反応時間は、原料化合物、塩基、不活性溶媒、反応温度などにより異なるが、通常、15分間~48時間、好ましくは30分間~5時間である。
A2工程は、化合物(I)を製造する工程であり、不活性溶媒中、ルイス酸の存在下、化合物(2)を化合物(3)と反応させた後、所望により、R11、R12およびR13で表される保護基を除去することにより行われる。
保護基の除去はその種類によって異なるが、上述したように、一般に有機合成化学の技術において周知の方法、例えば、T. H. Greene, P. G. Wuts, Protective Groups in Organic Synthesis. Third Edition, 1999年, John Wiley & Sons, Inc.などに記載された方法のような常法に従って行うことができる。
化合物(2)を化合物(3)と反応させる際に使用される不活性溶媒としては、例えば、ハロゲン化炭化水素類、芳香族炭化水素類、エーテル類、ニトリル類などが挙げられ、好ましくはハロゲン化炭化水素、より好ましくはメチレンクロリドである。
上記反応に使用されるルイス酸としては、例えば、三フッ化ホウ素-ジエチルエーテル錯体、トリフルオロメタンスルホン酸トリメチルシリルなどが挙げられ、好ましくは三フッ化ホウ素-ジエチルエーテル錯体である。
反応温度は、原料化合物、ルイス酸、不活性溶媒などにより異なるが、通常、-30℃~還流温度、好ましくは0℃~室温である。
反応時間は、原料化合物、ルイス酸、不活性溶媒、反応温度などにより異なるが、通常、5分間~24時間、好ましくは10分間~12時間である。
A3工程は、化合物(4)を製造する工程であり、不活性溶媒中、化合物(1)を臭化水素酢酸と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、ハロゲン化炭化水素類などが挙げられ、好ましくはメチレンクロリドである。
反応温度は、原料化合物、不活性溶媒などにより異なるが、通常、0℃~還流温度、好ましくは室温である。
反応時間は、原料化合物、不活性溶媒、反応温度などにより異なるが、通常、5~50時間、好ましくは15~35時間である。
A4工程は、化合物(I)を製造する工程であり、不活性溶媒中、炭酸銀の存在下、化合物(4)を化合物(3)と反応させた後、所望により、R11、R12およびR13で表される保護基を除去することにより行われる。
保護基の除去はA2工程と同様に行えばよい。
上記反応に使用される不活性溶媒としては、例えば、ハロゲン化炭化水素類、芳香族炭化水素類、エーテル類、ニトリル類などが挙げられ、好ましくはハロゲン化炭化水素、より好ましくはメチレンクロリドである。
反応温度は、原料化合物、不活性溶媒などにより異なるが、通常、0℃~還流温度、好ましくは室温である。
反応時間は、原料化合物、不活性溶媒、反応温度などにより異なるが、通常、5~150時間、好ましくは10~50時間である。
A法の原料化合物である化合物(1)は例えば下記B法、化合物(3)は例えば下記C法またはD法によって製造することができる。
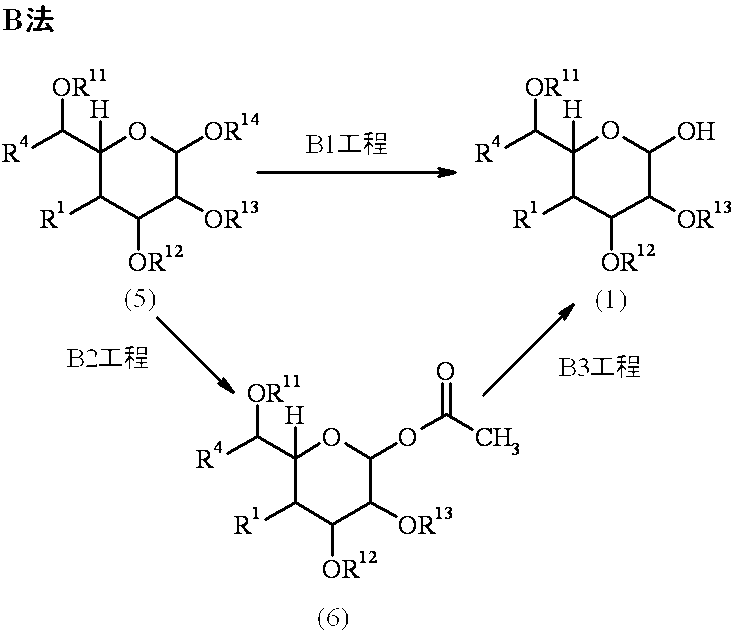
(式中、R1、R4、R11、R12およびR13は前記の通りであり、R14は水素原子または保護基である。)
B1工程は、化合物(1)を製造する工程であり、R14で表される保護基を除去することにより行われる。
保護基の除去はA2工程と同様に行えばよい。
例えば、R14で表される保護基がベンゾイル基である場合、不活性溶媒中、ヒドラジン酢酸塩を作用させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、アミド類などが挙げられ、好ましくはジメチルホルムアミドである。
反応温度は、原料化合物、不活性溶媒などにより異なるが、通常、0~50℃、好ましくは室温である。
反応時間は、原料化合物、不活性溶媒、反応温度などにより異なるが、通常、30分間~35時間、好ましくは1~24時間である。
B2工程は、化合物(6)を製造する工程であり、不活性溶媒中、酸触媒の存在下、化合物(5)におけるR14をアセチル化することにより行われる。
上記反応に使用される不活性溶媒としては、例えば、カルボン酸類などが挙げられ、好ましくは酢酸である。
上記反応に使用される酸触媒は、好ましくは無機酸、より好ましくは硫酸である。
反応温度は、原料化合物、酸触媒、不活性溶媒などにより異なるが、通常、0~50℃、好ましくは室温である。
反応時間は、原料化合物、酸触媒、不活性溶媒、反応温度などにより異なるが、通常、3~48時間、好ましくは6~24時間である。
B3工程は、化合物(1)を製造する工程であり、不活性溶媒中、化合物(6)をヒドラジン酢酸塩と反応させることにより行われる。本工程は、B1工程と同様に行うことができる。
次に、C法について説明する。下記スキーム中の化合物(14)は化合物(3)におけるnが1である化合物である。

(式中、R2、R3およびmは前記の通りである。)
C1工程は、化合物(9)を製造する工程であり、不活性溶媒中、塩基の存在下、化合物(7)を化合物(8)と反応させ、さらに触媒量のトリメチルシリルシアニドを添加し、反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、ハロゲン化炭化水素類、炭化水素類、芳香族炭化水素類、エーテル類、ニトリル類などが挙げられ、好ましくはニトリル類、より好ましくはアセトニトリルである。
上記反応に使用される塩基としては、例えば、有機アミン類などが挙げられ、好ましくはトリエチルアミンである。
反応温度は、原料化合物、塩基、不活性溶媒などにより異なるが、通常、0℃~還流温度、好ましくは室温~60℃である。
反応時間は、原料化合物、塩基、不活性溶媒、反応温度などにより異なるが、通常、10分間~12時間、好ましくは2~4時間である。
C2工程は、化合物(10)を製造する工程であり、不活性溶媒中、化合物(9)をハロゲン化剤と反応させることにより行われる。
上記反応の第一段階であるハロゲン化反応に使用される不活性溶媒としては、例えば、ハロゲン化炭素類などが挙げられ、好ましくは塩化メチレンである。
上記反応に使用されるハロゲン化剤としては、R2がフッ素の場合には、例えば、ジメチルアミノ硫黄三フッ化物、2,2-ジフルオロ-1,3-ジメチルイミダゾリジン、フッ化水素-ピリジンなどが挙げられ、好ましくはジメチルアミノ硫黄三フッ化物であり、R2が塩素の場合には、例えば、二塩化オキサリル、塩化チオニル、オキシ塩化リンなどが挙げられ、好ましくは二塩化オキサリルである。
反応温度は、原料化合物、ハロゲン化剤、不活性溶媒などにより異なるが、通常、-30~100℃、好ましくは0℃~室温である。
反応時間は、原料化合物、ハロゲン化剤、不活性溶媒、反応温度などにより異なるが、R2がフッ素の場合には、通常、30分間~12時間、好ましくは3時間であり、R2が塩素の場合には、通常、30分間~24時間、好ましくは1~3時間である。
C3工程は、化合物(11)を製造する工程であり、塩基性溶媒中、化合物(10)を酸化剤と反応させることにより行われる。
上記反応に使用される塩基性溶媒としては、例えば、有機アミン類などが挙げられ、好ましくはN-メチルモルホリンである。
上記反応に使用される酸化剤としては、例えば、過マンガン酸やクロム酸などの重金属塩、臭素やヨウ素などのハロゲン、2,3-ジクロロ-5,6-ジシアノ-p-ベンゾキノンなどが挙げられ、好ましくはハロゲン、より好ましくはヨウ素である。
反応温度は、原料化合物、塩基性溶媒、酸化剤の種類などにより異なるが、通常、0~100℃、好ましくは室温である。
反応時間は、原料化合物、塩基性溶媒、酸化剤、反応温度などにより異なるが、通常、15分間~12時間、好ましくは30分間~2時間である。
C4工程は、化合物(12)を製造する工程であり、不活性溶媒中、化合物(11)を還元剤と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、エーテル類、アルコール類などが挙げられ、好ましくはエーテル類、より好ましくはテトラヒドロフランである。
上記反応に使用される還元剤としては、例えば、水素化ホウ素ナトリウム、水素化ホウ素リチウムなどの水素化ホウ素アルカリ金属、水素化アルミニウムリチウム、水素化リチウムトリエトキシドアルミニウムなどの水素化アルミニウム化合物、水素化テルルナトリウムなどのヒドリド試薬などが挙げられ、好ましくは水素化アルミニウム化合物、より好ましくは水素化アルミニウムリチウムである。
反応温度は、原料化合物、還元剤、不活性溶媒などにより異なるが、通常、-30℃~還流温度、好ましくは0℃~室温である。
反応時間は、原料化合物、還元剤、不活性溶媒、反応温度などにより異なるが、通常、10分間~10時間、好ましくは30分間~1時間である。
C5工程は、化合物(13)を製造する工程であり、不活性溶媒中、ビス(ジブチル塩化スズ)オキシド触媒の存在下、化合物(12)に酢酸ビニルを反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、エーテル類などが挙げられ、好ましくはテトラヒドロフランである。
反応温度は、原料化合物、不活性溶媒などにより異なるが、通常、0~50℃、好ましくは室温~35℃である。
反応時間は、原料化合物、不活性溶媒、反応温度などにより異なるが、通常、1~100時間、好ましくは24~48時間である。
C6工程は、化合物(14)を製造する工程であり、不活性溶媒中、酸またはルイス酸の存在下、化合物(13)を還元剤と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、ハロゲン化炭化水素類、炭化水素類、芳香族炭化水素類、エーテル類、ニトリル類などが挙げられ、好ましくはニトリル類、より好ましくはアセトニトリルである。
上記反応に使用される酸またはルイス酸としては、例えば、トリフルオロ酢酸、三フッ化ホウ素-ジエチルエーテル錯体、塩化アルミニウムなどが挙げられ、好ましくは三フッ化ホウ素-ジエチルエーテル錯体である。
上記反応に使用される還元剤としては、例えば、トリアルキルシラン、水素化ホウ素アルカリ金属、水素化アルミニウム化合物などが挙げられ、好ましくはトリアルキルシラン、より好ましくはトリエチルシランである。
反応温度は、原料化合物、酸またはルイス酸、還元剤、不活性溶媒などにより異なるが、通常、-20~50℃、好ましくは0℃~室温である。
反応時間は、原料化合物、酸またはルイス酸、還元剤、不活性溶媒、反応温度などにより異なるが、通常、30分間~10時間、好ましくは1~2時間である。
最後に、D法について説明する。下記スキーム中の化合物(22)は化合物(3)におけるnが2である化合物である。

(式中、R2、R3およびmは前記の通りである。)
D1工程は、化合物(15)を製造する工程であり、不活性溶媒中、塩基の存在下、化合物(14)をベンジル化試薬と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、アミド類などが挙げられ、好ましくはジメチルホルムアミドである。
上記反応に使用される塩基としては、例えば、無機塩基類などが挙げられ、好ましくは炭酸カリウムである。
上記反応に使用されるベンジル化試薬としては、例えば、ハロゲン化ベンジル、アリールスルホン酸ベンジル、アルキルスルホン酸ベンジルなどが挙げられ、好ましくはハロゲン化ベンジル、より好ましくは臭化ベンジルである。
反応温度は、原料化合物、塩基、ベンジル化試薬、不活性溶媒などにより異なるが、通常、0~100℃、好ましくは室温である。
反応時間は、原料化合物、塩基、ベンジル化試薬、不活性溶媒、反応温度などにより異なるが、通常、30分間~48時間、好ましくは2~4時間である。
D2工程は、化合物(16)を製造する工程であり、不活性溶媒中、化合物(15)を塩基と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、水、アルコール類などが挙げられ、好ましくはアルコール類、より好ましくはメタノールである。
上記反応に使用される塩基としては、例えば、無機塩基類などが挙げられ、好ましくは炭酸カリウムである。
反応温度は、原料化合物、塩基、不活性溶媒などにより異なるが、通常、0℃~還流温度、好ましくは室温である。
反応時間は、原料化合物、塩基、不活性溶媒、反応温度などにより異なるが、通常、10分間~24時間、好ましくは1~18時間である。
D3工程は、化合物(17)を製造する工程であり、不活性溶媒中、化合物(16)を酸化剤と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、炭化水素類、ハロゲン化炭化水素類、芳香族炭化水素類、エーテル類、エステル類などが挙げられ、好ましくはハロゲン化炭化水素、より好ましくはクロロホルムである。
反応温度は、原料化合物、酸化剤、不活性溶媒などにより異なるが、通常、0~200℃、好ましくは還流温度である。
反応時間は、原料化合物、酸化剤、不活性溶媒、反応温度などにより異なるが、通常、30分間~48時間、好ましくは2~4時間である。
D4工程は、化合物(18)を製造する工程であり、不活性溶媒中、ホスホニウム塩を塩基と反応させた後、さらに化合物(17)と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、エーテル類などが挙げられ、好ましくはテトラヒドロフランである。
上記反応に使用されるホスホニウム塩としては、例えば、ハロゲン化(メトキシメチル)トリフェニルホスホニウムなどが挙げられ、好ましくは塩化(メトキシメチル)トリフェニルホスホニウムである。
上記反応に使用される塩基としては、例えば、アルカリ金属ビス(トリメチルシリル)アミド類、アルカリ金属ジアルキルアミド類などが挙げられ、好ましくはアルカリ金属ビス(トリメチルシリル)アミド、さらに好ましくはリチウム ビス(トリメチルシリル)アミドである。
反応温度は、原料化合物、ホスホニウム塩、塩基、不活性溶媒などにより異なるが、通常、-78℃~還流温度、好ましくは0℃~室温である。
反応時間は、原料化合物、(メトキシメチル)ホスホニウム塩、塩基、不活性溶媒、反応温度などにより異なるが、通常、30分間~24時間、好ましくは1~2時間である。
D5工程は、化合物(19)を製造する工程であり、不活性溶媒中、酸触媒存在下、化合物(18)を加水分解することにより行われる。
上記反応に使用される不活性溶媒としては、例えば、エーテル類などが挙げられ、好ましくは1,4-ジオキサンである。
上記反応に使用される酸触媒としては、例えば、無機酸、アリールスルホン酸などが挙げられ、好ましくは無機酸、より好ましくは塩酸である。
反応温度は、原料化合物、酸触媒、不活性溶媒などにより異なるが、通常、0℃~還流温度、好ましくは室温である。
反応時間は、原料化合物、酸触媒、不活性溶媒、反応温度などにより異なるが、通常、5分間~10時間、好ましくは10分間~1時間である。
D6工程は、化合物(20)を製造する工程であり、不活性溶媒中、化合物(19)を還元剤と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、例えば、エーテル類、アルコール類などが挙げられ、好ましくはアルコール類、より好ましくはメタノールである。
上記反応に使用される還元剤としては、例えば、水素化ホウ素ナトリウム、水素化ホウ素リチウムなどの水素化ホウ素アルカリ金属、水素化アルミニウムリチウム、水素化リチウムトリエトキシドアルミニウムなどの水素化アルミニウム化合物、水素化テルルナトリウムなどのヒドリド試薬などが挙げられ、好ましくは水素化ホウ素アルカリ金属、より好ましくは水素化ホウ素ナトリウムである。
反応温度は、原料化合物、還元剤、不活性溶媒などにより異なるが、通常、-30℃~還流温度、好ましくは0℃~室温である。
反応時間は、原料化合物、還元剤、不活性溶媒、反応温度などにより異なるが、通常、10分間~10時間、好ましくは30分間~1時間である。
D7工程は、化合物(21)を製造する工程であり、R2がフッ素の場合には、不活性溶媒中、水素および金属触媒存在下、化合物(20)を接触還元することにより行われる。一方、R2が塩素の場合には、不活性溶媒中、化合物(20)をルイス酸と反応させることにより行われる。
上記反応に使用される不活性溶媒としては、R2がフッ素の場合には、例えば、エーテル類、アルコール類などが挙げられ、好ましくはアルコール類、より好ましくはメタノールであり、R2が塩素の場合には、例えば、ニトリル類などが挙げられ、好ましくはアセトニトリルである。
R2がフッ素の場合に使用される金属触媒としては、例えば、白金、パラジウム、ロジウム、ニッケルなどの金属触媒などが挙げられ、好ましくはパラジウム触媒、さらに好ましくはパラジウム炭素触媒である。
R2が塩素の場合に上記反応に使用されるルイス酸としては、例えば、ハロゲン化トリアルキルシリルなどが挙げられ、好ましくはヨウ化トリメチルシリルである。
反応温度は、R2がフッ素の場合には、原料化合物、金属触媒、不活性溶媒などにより異なるが、通常、0~100℃、好ましくは室温であり、R2が塩素の場合には、原料化合物、ルイス酸、不活性溶媒などにより異なるが、通常、0~100℃、好ましくは40℃である。
反応時間は、R2がフッ素の場合には、原料化合物、金属触媒、不活性溶媒、反応温度などにより異なるが、通常、10分間~24時間、好ましくは30分間~2時間であり、R2が塩素の場合には、原料化合物、ルイス酸、不活性溶媒、反応温度などにより異なるが、通常、30分間~24時間、好ましくは1~3時間である。
D8工程は、化合物(22)を製造する工程であり、不活性溶媒中、ビス(ジブチル塩化スズ)オキシド触媒の存在下、化合物(21)を酢酸ビニルと反応させることにより行われ、本工程は、C5工程における化合物(12)の一級水酸基をアセチル基で保護する方法と同様に行われる。
本発明の化合物は、上記方法を用いて製造できる他、公知の化合物から後述する実施例または当該分野で周知の方法(例えば、Chem.Ber,71,1938,1843-1849、Carbohydr.Res.1995,273,249-254、Bull.Chem.Soc.Jpn.,1982,55,938-942、Bull.Chem.Soc.Jpn.,1976,49,788-790、Org.Lett.,2003,5,3419-3421、Org.Biomol.Chem,2003,1,767-771、J.Chem.Soc.,1956,2124-2126、国際公開02/064606号パンフレット、Liebigs Ann.Chem,GE,1992,7,747-758などを参照のこと)に従って、容易に製造することができる。
本発明の化合物またはその水和物は、副作用が低く優れたヒトSGLT1および/またはSGLT2阻害活性を示し、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全、糖尿病関連疾患(例えば、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症、痛風など)、または糖尿病合併症(例えば、網膜症、腎症、神経障害、白内障、足壊疽、感染症、ケトーシスなど)の治療または予防のための医薬組成物の有効成分として有用である。
かかる医薬組成物は哺乳動物(例えば、ヒト、ウマ、ウシ、ブタ、好ましくはヒト)に投与することができる。
その投与形態は、例えば、錠剤、カプセル剤、顆粒剤、散剤もしくはシロップ剤などによる経口的投与または注射剤もしくは坐剤などによる非経口的投与のいずれでもよい。
これらの製剤は、賦形剤(例えば、乳糖、トウモロコシデンプン、結晶セルロース、D-マンニトール、無水リン酸水素カルシウム、白糖など)、崩壊剤(例えば、トウモロコシデンプン、カルメロースカルシウム、低置換度ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウムなど)、結合剤(例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポピドン、トウモロコシデンプン、メチルセルロースなど)、滑沢剤(例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク、ステアリン酸など)、流動化剤(例えば、軽質無水ケイ酸、タルク、含水二酸化ケイ素など)、コーティング剤(例えば、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ポリビニルアセタールジエチルアミノアセテート、アミノアルキルメタアクリレートコポリマーE、メタアクリル酸コポリマーL、メタアクリル酸コポリマーLD、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、エチルセルロース、アミノアルキルメタアクリレートコポリマーRSなど)、懸濁化剤(例えば、アルギン酸ナトリウム、カルメロースナトリウム、メチルセルロース、ポリソルベート80など)、乳化剤(例えば、モノステアリン酸グリセリン、ショ糖脂肪酸エステルなど)、安定化剤(例えば、ジブチルヒドロキシトルエン、トコフェロール、亜硫酸塩など)、保存剤(例えば、パラオキシ安息香酸エステル類など)、等張化剤(例えば、塩化ナトリウム、グリセリンなど)、嬌味剤(例えば、白糖、D-ソルビトール、キシリトール、カンゾウ、サッカリンナトリウム、アスパルテーム、ステビアなど)などの添加剤を用いて当該分野で周知の方法で製造される。
本発明の化合物またはその水和物の使用量は、症状、年齢などにより異なるが、経口投与の場合には、1日1~2000mg、好ましくは10~400mgを、静脈内投与の場合には、1日0.1~500mg、好ましくは1~300mgを成人に対して、1日あたり1~数回に分けて、症状に応じて投与することが望ましい。
(実施例1)3-クロロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.12)
(実施例1a)塩化4-(2-フルオロエチル)ベンゾイル
4-(2-フルオロエチル)安息香酸(Bioorg.Med.Chem.,2005,13,77-78.)(1.8g、11mmol)を塩化メチレンに溶解し、氷冷下、塩化オキサリル(1.1mL、13mmol)およびN,N-ジメチルホルムアミド(0.1mL、1.3mmol)を加え、室温に昇温しつつ3時間半撹拌した。反応終了後、減圧下溶媒を除去し、標記化合物の粗生成物(2.1g)を無色油状物質として得た。
4-(2-フルオロエチル)安息香酸(Bioorg.Med.Chem.,2005,13,77-78.)(1.8g、11mmol)を塩化メチレンに溶解し、氷冷下、塩化オキサリル(1.1mL、13mmol)およびN,N-ジメチルホルムアミド(0.1mL、1.3mmol)を加え、室温に昇温しつつ3時間半撹拌した。反応終了後、減圧下溶媒を除去し、標記化合物の粗生成物(2.1g)を無色油状物質として得た。
(実施例1b)4-[4-(2-フルオロエチル)ベンゾイル]-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(EP1571148A1)(2.0g、11mmol)および実施例1aで得られた粗生成物(2.1g、11mmol)をアセトニトリル(20mL)に溶解し、氷冷下、トリエチルアミン(4.6mL、33mmol)を滴下した。10分後に反応の終了を確認し、この懸濁液にトリメチルシリルシアニド(0.21mL、1.6mmol)を加え60℃で2時間撹拌した。反応液を室温に冷却後、酢酸エチル(50mL)で希釈し、2M塩酸(20mL)および飽和食塩水(20mL、2回)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(4.3g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(EP1571148A1)(2.0g、11mmol)および実施例1aで得られた粗生成物(2.1g、11mmol)をアセトニトリル(20mL)に溶解し、氷冷下、トリエチルアミン(4.6mL、33mmol)を滴下した。10分後に反応の終了を確認し、この懸濁液にトリメチルシリルシアニド(0.21mL、1.6mmol)を加え60℃で2時間撹拌した。反応液を室温に冷却後、酢酸エチル(50mL)で希釈し、2M塩酸(20mL)および飽和食塩水(20mL、2回)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(4.3g)を得た。
(実施例1c)3-クロロ-4-[4-(2-フルオロエチル)ベンゾイル]-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例1bで得られた粗生成物(2.1g、6.5mmol)を塩化メチレン(20mL)に溶解し、氷冷下、2-メチル-2-ブテン(2.8mL、26mmol)、二塩化オキサリル(0.83mL、9.8mmol)およびN,N-ジメチルホルムアミド(0.1mL、0.13mmol)を順次加え、室温まで昇温しつつ2時間撹拌した。反応液を室温に冷却後、塩化メチレン(20mL)で希釈し、飽和食塩水(20mL、2回)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、トルエン(10mL)共沸下で2回減圧濃縮を行って、標記化合物の粗生成物(2.2g)を得た。
実施例1bで得られた粗生成物(2.1g、6.5mmol)を塩化メチレン(20mL)に溶解し、氷冷下、2-メチル-2-ブテン(2.8mL、26mmol)、二塩化オキサリル(0.83mL、9.8mmol)およびN,N-ジメチルホルムアミド(0.1mL、0.13mmol)を順次加え、室温まで昇温しつつ2時間撹拌した。反応液を室温に冷却後、塩化メチレン(20mL)で希釈し、飽和食塩水(20mL、2回)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、トルエン(10mL)共沸下で2回減圧濃縮を行って、標記化合物の粗生成物(2.2g)を得た。
(実施例1d)3-クロロ-4-[4-(2-フルオロエチル)ベンゾイル]-5-ヒドロキシ安息香酸エチル
実施例1cで得られた粗生成物(2.2g、6.5mmol)をN-メチルモルホリン(26mL)に溶解し、無水硫酸ナトリウム(1.1g)およびヨウ素(2.0g、7.8mmol)を加えて室温で1時間撹拌した。暗褐色の懸濁液をセライトで濾過し、酢酸エチル(50mL)で希釈後、2M塩酸(50mL、2回)、30%チオ硫酸ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(2.2g)を得た。
実施例1cで得られた粗生成物(2.2g、6.5mmol)をN-メチルモルホリン(26mL)に溶解し、無水硫酸ナトリウム(1.1g)およびヨウ素(2.0g、7.8mmol)を加えて室温で1時間撹拌した。暗褐色の懸濁液をセライトで濾過し、酢酸エチル(50mL)で希釈後、2M塩酸(50mL、2回)、30%チオ硫酸ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(2.2g)を得た。
(実施例1e)3-クロロ-2-{1-[4-(2-フルオロエチル)フェニル]-1-ヒドロキシメチル}-5-(ヒドロキシメチル)フェノール
実施例1dで得られた粗生成物(2.2g、6.5mmol)をテトラヒドロフラン(65mL)に溶解し、気相を窒素置換後、氷冷下、水素化アルミニウムリチウム(1.0g、26mmol)を少量ずつ加えた。室温に昇温しつつ1時間撹拌し、氷冷下、水(1mL)を滴下して反応を停止した。混合物を酢酸エチル(50mL)で希釈後、2M塩酸(20mL、2回)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1、V/V)を用いて精製して、標記化合物(1.0g)を得た。
実施例1dで得られた粗生成物(2.2g、6.5mmol)をテトラヒドロフラン(65mL)に溶解し、気相を窒素置換後、氷冷下、水素化アルミニウムリチウム(1.0g、26mmol)を少量ずつ加えた。室温に昇温しつつ1時間撹拌し、氷冷下、水(1mL)を滴下して反応を停止した。混合物を酢酸エチル(50mL)で希釈後、2M塩酸(20mL、2回)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1、V/V)を用いて精製して、標記化合物(1.0g)を得た。
(実施例1f)酢酸 3-クロロ-4-{1-[4-(2-フルオロエチル)フェニル]-1-ヒドロキシメチル}-5-ヒドロキシベンジル
実施例1eで得られた化合物(1.0g、3.2mmol)を酢酸ビニル(10mL)およびジイソプロピルエーテル(10mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を加えた。この懸濁液を37℃で8時間撹拌し、セライトで濾過し、酢酸エチル(5mL、2回)で洗浄した。得られた濾液を減圧下濃縮して、標記化合物(1.1g)の粗生成物を得た。
実施例1eで得られた化合物(1.0g、3.2mmol)を酢酸ビニル(10mL)およびジイソプロピルエーテル(10mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を加えた。この懸濁液を37℃で8時間撹拌し、セライトで濾過し、酢酸エチル(5mL、2回)で洗浄した。得られた濾液を減圧下濃縮して、標記化合物(1.1g)の粗生成物を得た。
(実施例1g)酢酸 3-クロロ-2-[4-(2-フルオロエチル)ベンジル]-5-ヒドロキシベンジル
実施例1fで得られた粗生成物(0.90g、2.5mmol)をアセトニトリル(10mL)に溶解し、氷冷下、トリエチルシラン(1.2mL、7.6mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.47mL、3.8mmol)を加え、0℃で1時間半撹拌した。反応液を酢酸エチル(20mL)で希釈後、飽和炭酸水素ナトリウム水溶液(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、5:1、V/V)を用いて精製して、標記化合物(0.76g、ただし20%程度の原料を含む)を得た。
実施例1fで得られた粗生成物(0.90g、2.5mmol)をアセトニトリル(10mL)に溶解し、氷冷下、トリエチルシラン(1.2mL、7.6mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.47mL、3.8mmol)を加え、0℃で1時間半撹拌した。反応液を酢酸エチル(20mL)で希釈後、飽和炭酸水素ナトリウム水溶液(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、5:1、V/V)を用いて精製して、標記化合物(0.76g、ただし20%程度の原料を含む)を得た。
(実施例1h)5-アセトキシメチル-3-クロロ-2-[4-(2-フルオロエチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(1.1g、1.8mmol)を塩化メチレン(10mL)に溶解し、氷冷下、トリクロロアセトニトリル(0.50mL、5.41mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7.5μL、0.05mmol)を加え、1時間攪拌した。この溶液を塩化メチレン(20mL)で希釈後、飽和塩化アンモニウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。トルエン(2mL)を加え、減圧下共沸を行って、相当するイミダートの粗生成物を黄色アモルファスとして得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(1.1g、1.8mmol)を塩化メチレン(10mL)に溶解し、氷冷下、トリクロロアセトニトリル(0.50mL、5.41mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7.5μL、0.05mmol)を加え、1時間攪拌した。この溶液を塩化メチレン(20mL)で希釈後、飽和塩化アンモニウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。トルエン(2mL)を加え、減圧下共沸を行って、相当するイミダートの粗生成物を黄色アモルファスとして得た。
このイミダート(1.36g、1.8mmol)および実施例1gで得られた粗生成物(0.66g、1.8mmol)を塩化メチレン(5mL)に溶解し、氷冷下、三フッ化ホウ素-ジエチルエーテル錯体(0.11mL、0.90mmol)を加え、0℃で30分間撹拌した。混合物にトリエチルアミン(0.14mL、1.0mmol)を加えて反応を停止し、酢酸エチル(20mL)で希釈後、飽和塩化アンモニウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物を得た。
(実施例1i)3-クロロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例1hで得られた粗生成物(1.8mmol)をテトラヒドロフラン(6mL)およびメタノール(6mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(6mL、12mmol)を加えて室温で30分間撹拌した。混合物に水(20mL)を加えた後、酢酸エチル(20mL、3回)で抽出し、有機層を飽和食塩水(10mL)で2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、10:1、V/V)を用いて精製した。得られた固体を酢酸エチル:ジイソプロピルエーテル:ヘキサン(1:1:2、V/V)に溶解した後、冷却して沈殿物を得た。この沈殿物を回収し、標記化合物(0.19g)を無色固体として得た。
1H NMR (500 MHz, CDCl3):δ 1.20 (3H, d, J = 6.4 Hz), 2.91 (2H, dt, J = 22.9, 6.6 Hz), 3.32-3.45 (4H, m), 4.04-4.06 (1H, m), 4.12 (1H, d, J = 14.6 Hz), 4.25 (1H, d, J = 14.6 Hz), 4.53 (2H, dt, J = 47.4, 6.6 Hz), 4.55 (2H, s), 4.93 (1H, d, J = 6.8 Hz), 7.07 (2H, d, J = 7.8 Hz), 7.09 (1H, s), 7.12 (1H, s), 7.18 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 471 (M+H)+, 493 (M+Na)+.。
実施例1hで得られた粗生成物(1.8mmol)をテトラヒドロフラン(6mL)およびメタノール(6mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(6mL、12mmol)を加えて室温で30分間撹拌した。混合物に水(20mL)を加えた後、酢酸エチル(20mL、3回)で抽出し、有機層を飽和食塩水(10mL)で2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、10:1、V/V)を用いて精製した。得られた固体を酢酸エチル:ジイソプロピルエーテル:ヘキサン(1:1:2、V/V)に溶解した後、冷却して沈殿物を得た。この沈殿物を回収し、標記化合物(0.19g)を無色固体として得た。
1H NMR (500 MHz, CDCl3):δ 1.20 (3H, d, J = 6.4 Hz), 2.91 (2H, dt, J = 22.9, 6.6 Hz), 3.32-3.45 (4H, m), 4.04-4.06 (1H, m), 4.12 (1H, d, J = 14.6 Hz), 4.25 (1H, d, J = 14.6 Hz), 4.53 (2H, dt, J = 47.4, 6.6 Hz), 4.55 (2H, s), 4.93 (1H, d, J = 6.8 Hz), 7.07 (2H, d, J = 7.8 Hz), 7.09 (1H, s), 7.12 (1H, s), 7.18 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 471 (M+H)+, 493 (M+Na)+.。
(実施例2)3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.13)
(実施例2a)(2-クロロ-4,6-ジヒドロキシフェニル)(4-エチルフェニル)メタノン
1-クロロ-3,5-ジメトキシベンゼン(8.11g、47.0mmol)をトルエン(40mL)に溶解し、塩化アルミニウム(6.26g、46.9mmol)および塩化4-エチルベンゾイル(6.89mL、47.0mmol)を加え、室温で15分攪拌した後に、85℃で3時間攪拌した。室温に冷却後、反応液を氷冷下、1N塩酸水溶液(300mL)に加えた。トルエン(100mL)で抽出後、飽和食塩水(100mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。残渣をヘキサンで洗浄し、濾過して、固体の粗生成物(10.3g)を得た。
1-クロロ-3,5-ジメトキシベンゼン(8.11g、47.0mmol)をトルエン(40mL)に溶解し、塩化アルミニウム(6.26g、46.9mmol)および塩化4-エチルベンゾイル(6.89mL、47.0mmol)を加え、室温で15分攪拌した後に、85℃で3時間攪拌した。室温に冷却後、反応液を氷冷下、1N塩酸水溶液(300mL)に加えた。トルエン(100mL)で抽出後、飽和食塩水(100mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。残渣をヘキサンで洗浄し、濾過して、固体の粗生成物(10.3g)を得た。
得られた粗生成物(10.3g)を塩化メチレン(100mL)に溶解して-78℃に冷却後、1mol/L三臭化ホウ素塩化メチレン溶液(106mL、106mmol)を加え、室温で16時間攪拌した。反応液を氷冷下、氷(300g)に加えた。塩化メチレン(100mL、2回)で抽出後、水(200mL)および飽和炭酸水素ナトリウム溶液(200mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。残渣を塩化メチレン/ヘキサンで洗浄し、濾過して、標記化合物の粗生成物(7.53g)を固体として得た。
(実施例2b)5-クロロ-2,2-ジメチル-4-(4-エチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-オール
実施例2aで得られた粗生成物(1.59g、5.75mmol)をメタノール(15mL)に溶解し、氷冷下、水素化ホウ素ナトリウム(326mg、8.62mmol)を加え、室温で30分攪拌した。0℃に冷却後、飽和塩化アンモニウム水溶液(1mL)を加え、減圧下、溶媒を除去した。残渣を酢酸エチル(20mL)で溶解し、飽和塩化アンモニウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去して、粗生成物(1.17g)を得た。
実施例2aで得られた粗生成物(1.59g、5.75mmol)をメタノール(15mL)に溶解し、氷冷下、水素化ホウ素ナトリウム(326mg、8.62mmol)を加え、室温で30分攪拌した。0℃に冷却後、飽和塩化アンモニウム水溶液(1mL)を加え、減圧下、溶媒を除去した。残渣を酢酸エチル(20mL)で溶解し、飽和塩化アンモニウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去して、粗生成物(1.17g)を得た。
粗生成物(1.17g)をアセトン(25mL)に溶解し、パラトルエンスルホン酸一水和物(0.24g、1.26mmol)を加え、室温で14時間攪拌した。セライトで濾過後、適量のトリエチルアミンを加え、減圧下、溶媒を除去した。残渣を酢酸エチル(20mL)で溶解し、飽和炭酸水素ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~5:1、V/V)を用いて精製して、アモルファス状の標記化合物(620mg)を得た。
(実施例2c)5-クロロ-2,2-ジメチル-4-(4-エチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-カルボン酸メチル
実施例2bで得られた化合物(620mg、1.94mmol)を塩化メチレン(10mL)に溶解し、ピリジン(0.235mL、2.91mmol)を加え、0℃に冷却後、無水トリフルオロメタンスルホン酸(0.390mL、2.32mmol)を加え、20分攪拌した。さらに、塩化メチレン(20mL)を加え、水(20mL、3回)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。トルエン共沸を行って、トリフラート体の粗生成物(790mg)を得た。
実施例2bで得られた化合物(620mg、1.94mmol)を塩化メチレン(10mL)に溶解し、ピリジン(0.235mL、2.91mmol)を加え、0℃に冷却後、無水トリフルオロメタンスルホン酸(0.390mL、2.32mmol)を加え、20分攪拌した。さらに、塩化メチレン(20mL)を加え、水(20mL、3回)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。トルエン共沸を行って、トリフラート体の粗生成物(790mg)を得た。
得られた粗生成物(790mg)をN,N-ジメチルホルムアルデヒド(8mL)に溶解し、メタノール(1.42mL、35.0mmol)、酢酸パラジウム(39mg、0.17mmol)、1,3-ビス(ジフェニルホスフィノ)プロパン(72mg、0.17mmol)およびトリエチルアミン(4.86mL、35.0mmol)を加え、一酸化炭素雰囲気下、55℃で3時間攪拌した。室温に冷却後、反応液に酢酸エチル(60mL)で希釈し、1mol/L塩酸水溶液(30mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~4:1、V/V)を用いて精製して、油状の標記化合物(584mg)を得た。
(実施例2d)3-クロロ-2-[1-(4-エチルフェニル)-1-メトキシメチル]-5-(ヒドロキシメチル)フェノール
水素化リチウムアルミニウム(103mg、2.71mmol)をテトラヒドロフラン(3mL)に溶解し、氷冷下、実施例2cで得られた化合物(651mg、1.80mmol)を溶解させたテトラヒドロフラン溶液(4mL)を加えた。反応液を室温で30分攪拌した後に、0℃に冷却後、水(0.1mL)、5mol/L水酸化ナトリウム水溶液(0.1mL)および水(0.3mL)を順次加え、室温で1時間攪拌し、室温で、14時間静置した。セライトで濾過後、減圧下溶媒を除去して、油状のアルコール体粗生成物(470mg)を得た。
水素化リチウムアルミニウム(103mg、2.71mmol)をテトラヒドロフラン(3mL)に溶解し、氷冷下、実施例2cで得られた化合物(651mg、1.80mmol)を溶解させたテトラヒドロフラン溶液(4mL)を加えた。反応液を室温で30分攪拌した後に、0℃に冷却後、水(0.1mL)、5mol/L水酸化ナトリウム水溶液(0.1mL)および水(0.3mL)を順次加え、室温で1時間攪拌し、室温で、14時間静置した。セライトで濾過後、減圧下溶媒を除去して、油状のアルコール体粗生成物(470mg)を得た。
得られた粗生成物(470mg)をメタノール(10mL)に溶解し、パラトルエンスルホン酸一水和物(130mg、0.68mmol)および水(1mL)を加え50℃で3時間攪拌した。反応液を減圧下濃縮後、残渣を酢酸エチル(20mL)に溶解し、炭酸水素ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~3:2、V/V)を用いて精製して、油状の標記化合物(401mg)を得た。
(実施例2e)酢酸 3-クロロ-4-[1-(4-エチルフェニル)-1-メトキシメチル]-5-ヒドロキシベンジル
実施例2dで得られた化合物(401mg、1.31mmol)をテトラヒドロフラン(4mL)に溶解し、酢酸ビニル(4mL)およびビス(ジブチル塩化スズ)オキシド(144mg、0.26mmol)を加え、室温で、72時間攪拌した。減圧下溶媒を除去し、最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、油状の標記化合物(380mg)を得た。
実施例2dで得られた化合物(401mg、1.31mmol)をテトラヒドロフラン(4mL)に溶解し、酢酸ビニル(4mL)およびビス(ジブチル塩化スズ)オキシド(144mg、0.26mmol)を加え、室温で、72時間攪拌した。減圧下溶媒を除去し、最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、油状の標記化合物(380mg)を得た。
(実施例2f)酢酸 3-クロロ-4-(4-エチルベンジル)-5-ヒドロキシベンジル
実施例2eで得られた化合物(380mg、1.09mmol)をアセトニトリル(8mL)に溶解し、0℃に冷却後、トリエチルシラン(0.520mL、3.26mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.210mL、1.67mmol)を加え、室温で1時間攪拌した。反応液を酢酸エチル(40mL)で希釈し、炭酸水素ナトリウム水溶液(40mL)および飽和食塩水(40mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~2:1、V/V)を用いて精製して、標記化合物(293mg)を白色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, t, J = 7.7 Hz), 2.11 (3H, s), 2.60 (2H, q, J = 7.7 Hz), 4.15 (2H, s), 5.00 (2H, s), 6.72 (1H, d, J = 1.6 Hz), 7.02 (1H, d, J = 1.6 Hz), 7.11 (2H, d, J = 8.0 Hz), 7.18 (2H, d, J = 8.0 Hz);
MS (EI) m/z: 318, 320 (M)+.。
実施例2eで得られた化合物(380mg、1.09mmol)をアセトニトリル(8mL)に溶解し、0℃に冷却後、トリエチルシラン(0.520mL、3.26mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.210mL、1.67mmol)を加え、室温で1時間攪拌した。反応液を酢酸エチル(40mL)で希釈し、炭酸水素ナトリウム水溶液(40mL)および飽和食塩水(40mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~2:1、V/V)を用いて精製して、標記化合物(293mg)を白色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, t, J = 7.7 Hz), 2.11 (3H, s), 2.60 (2H, q, J = 7.7 Hz), 4.15 (2H, s), 5.00 (2H, s), 6.72 (1H, d, J = 1.6 Hz), 7.02 (1H, d, J = 1.6 Hz), 7.11 (2H, d, J = 8.0 Hz), 7.18 (2H, d, J = 8.0 Hz);
MS (EI) m/z: 318, 320 (M)+.。
(実施例2g)5-アセトキシメチル-3-クロロ-2-(4-エチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(722mg、1.18mmol)を塩化メチレン(8mL)に溶解し、0℃に冷却後、トリクロロアセトニトリル(0.597mL、5.91mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(0.018mL、0.12mmol)を加え、氷冷下1時間攪拌した。反応液を酢酸エチル(20mL)で希釈し、飽和塩化アンモニウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。トルエン(2mL)を加え、減圧下共沸を行って、相当するイミダートを黄色アモルファスとして得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(722mg、1.18mmol)を塩化メチレン(8mL)に溶解し、0℃に冷却後、トリクロロアセトニトリル(0.597mL、5.91mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(0.018mL、0.12mmol)を加え、氷冷下1時間攪拌した。反応液を酢酸エチル(20mL)で希釈し、飽和塩化アンモニウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。トルエン(2mL)を加え、減圧下共沸を行って、相当するイミダートを黄色アモルファスとして得た。
実施例2fで得られた化合物(290mg、0.91mmol)およびイミダート(890mg、1.18mmol)を塩化メチレン(8mL)に溶解し、モレキュラーシーブ4A(ナカライテスク社製、以下、MS4Aという。)を加え、三フッ化ホウ素-ジエチルエーテル錯体(0.148mL、1.18mmol)を氷冷下滴下し、氷冷下15分、次いで、室温で15分攪拌した。反応液に飽和炭酸水素ナトリウム(5mL)を加え、酢酸エチル(20mL)で希釈し、飽和炭酸水素ナトリウム水(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(850mg)を得た。
(実施例2h)3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例2gで得られた粗生成物(850mg)をメタノール/塩化メチレン(16mL/4mL)に溶解し、炭酸カリウム(1.63g、11.8mmol)を加え、室温で一晩攪拌した。セライトで濾過を行い過剰の炭酸カリウムを除去した後、適量の酢酸を加え中和し、減圧下メタノールを除去した。残渣を酢酸エチル(20mL)に溶解し、飽和炭酸水素ナトリウム水(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をヘキサン/酢酸エチル/メタノールから固体化し、標記化合物(196mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.17 (3H, t, J = 7.5 Hz), 1.21 (3H, d, J = 6.3 Hz), 2.56 (2H, q, J = 7.5 Hz), 3.36-3.38 (2H, m), 3.44-3.46 (2H, m), 4.05 (1H, dd, J = 6.3, 3.5 Hz), 4.11 (1H, d, J = 14.5 Hz), 4.24 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.93 (1H, d, J = 7.8 Hz), 7.02 (2H, d, J = 8.2 Hz), 7.10 (1H, s), 7.12 (1H, s), 7.15 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 475 (M+Na)+.。
実施例2gで得られた粗生成物(850mg)をメタノール/塩化メチレン(16mL/4mL)に溶解し、炭酸カリウム(1.63g、11.8mmol)を加え、室温で一晩攪拌した。セライトで濾過を行い過剰の炭酸カリウムを除去した後、適量の酢酸を加え中和し、減圧下メタノールを除去した。残渣を酢酸エチル(20mL)に溶解し、飽和炭酸水素ナトリウム水(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をヘキサン/酢酸エチル/メタノールから固体化し、標記化合物(196mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.17 (3H, t, J = 7.5 Hz), 1.21 (3H, d, J = 6.3 Hz), 2.56 (2H, q, J = 7.5 Hz), 3.36-3.38 (2H, m), 3.44-3.46 (2H, m), 4.05 (1H, dd, J = 6.3, 3.5 Hz), 4.11 (1H, d, J = 14.5 Hz), 4.24 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.93 (1H, d, J = 7.8 Hz), 7.02 (2H, d, J = 8.2 Hz), 7.10 (1H, s), 7.12 (1H, s), 7.15 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 475 (M+Na)+.。
(実施例3)2-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.1)
(実施例3a)4-[4-(ジフルオロメトキシ)ベンゾイル]-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(2.0g、11mmol)および塩化4-(ジフルオロメトキシ)ベンゾイル(J.Org.Chem.USSR,1981,1470-1475.)(2.3g、11mmol)を用い、実施例1bと同様の方法により標記化合物の粗生成物(3.3g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(2.0g、11mmol)および塩化4-(ジフルオロメトキシ)ベンゾイル(J.Org.Chem.USSR,1981,1470-1475.)(2.3g、11mmol)を用い、実施例1bと同様の方法により標記化合物の粗生成物(3.3g)を得た。
(実施例3b)4-[4-(ジフルオロメトキシ)ベンゾイル]-3-フルオロ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例3aで得られた粗生成物(1.6g、4.4mmol)を塩化メチレン(10mL)に溶解し、氷冷下、ジエチルアミノ硫黄三フッ化物(1.7mL、13mmol)を加え、室温まで昇温しつつ5時間撹拌した。反応液を酢酸エチル(30mL)で希釈し、飽和食塩水(10mL)、飽和炭酸水素ナトリウム水溶液(20mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(1.5g)を得た。
実施例3aで得られた粗生成物(1.6g、4.4mmol)を塩化メチレン(10mL)に溶解し、氷冷下、ジエチルアミノ硫黄三フッ化物(1.7mL、13mmol)を加え、室温まで昇温しつつ5時間撹拌した。反応液を酢酸エチル(30mL)で希釈し、飽和食塩水(10mL)、飽和炭酸水素ナトリウム水溶液(20mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(1.5g)を得た。
(実施例3c)4-[4-(ジフルオロメトキシ)ベンゾイル]-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例3bで得られた化合物(1.2g、3.4mmol)をN-メチルモルホリン(15mL)に溶解し、室温で無水硫酸ナトリウム(1.2g)およびヨウ素(0.97g、3.8mmol)を加えて室温で1時間撹拌した。暗褐色の懸濁液をセライトで濾過し、酢酸エチル(50mL)で希釈後、2M塩酸(50mL、2回)、30%チオ硫酸ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(1.2g)を得た。
実施例3bで得られた化合物(1.2g、3.4mmol)をN-メチルモルホリン(15mL)に溶解し、室温で無水硫酸ナトリウム(1.2g)およびヨウ素(0.97g、3.8mmol)を加えて室温で1時間撹拌した。暗褐色の懸濁液をセライトで濾過し、酢酸エチル(50mL)で希釈後、2M塩酸(50mL、2回)、30%チオ硫酸ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(1.2g)を得た。
(実施例3d)4-{1-[4-(ジフルオロメトキシ)フェニル]-1-ヒドロキシメチル}-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例3cで得られた粗生成物(1.5g、4.1mmol)をエタノール(20mL)に溶解し、氷冷下、水素化ホウ素ナトリウム(0.31g、8.2mmol)を加えた。室温に昇温しつつ2時間撹拌し、氷冷下、飽和塩化アンモニウム水溶液(1mL)を滴下して反応を停止した。混合物を酢酸エチル(50mL)で希釈後、飽和炭酸水素ナトリウム(20mL、2回)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.82g)を無色固体として得た。
実施例3cで得られた粗生成物(1.5g、4.1mmol)をエタノール(20mL)に溶解し、氷冷下、水素化ホウ素ナトリウム(0.31g、8.2mmol)を加えた。室温に昇温しつつ2時間撹拌し、氷冷下、飽和塩化アンモニウム水溶液(1mL)を滴下して反応を停止した。混合物を酢酸エチル(50mL)で希釈後、飽和炭酸水素ナトリウム(20mL、2回)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.82g)を無色固体として得た。
(実施例3e)4-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例3dで得られた化合物(0.82g、2.3mmol)をエタノール(8mL)に溶解し、溶液中に窒素を吹き込みながら10分間撹拌した。この溶液に2M塩酸(0.57mL、1.1mmol)および10%パラジウム炭素触媒(含水、0.5g)を窒素気流下で加え、気相を水素置換後、室温で6時間撹拌した。混合物をセライトで濾過し、減圧下溶媒を除去して、標記化合物の粗生成物(0.76g)を無色固体として得た。
実施例3dで得られた化合物(0.82g、2.3mmol)をエタノール(8mL)に溶解し、溶液中に窒素を吹き込みながら10分間撹拌した。この溶液に2M塩酸(0.57mL、1.1mmol)および10%パラジウム炭素触媒(含水、0.5g)を窒素気流下で加え、気相を水素置換後、室温で6時間撹拌した。混合物をセライトで濾過し、減圧下溶媒を除去して、標記化合物の粗生成物(0.76g)を無色固体として得た。
(実施例3f)2-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェノール
実施例3eで得られた粗生成物(0.76g、2.2mmol)をテトラヒドロフラン(10mL)に溶解し、気相を窒素置換後、氷冷下、水素化アルミニウムリチウム(0.25g、6.6mmol)を少量ずつ加えた。室温に昇温しつつ1時間撹拌し、氷冷下、水(1mL)を滴下して反応を停止した。混合物を酢酸エチル(30mL)で希釈後、2M塩酸(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.65g)を無色固体として得た。
実施例3eで得られた粗生成物(0.76g、2.2mmol)をテトラヒドロフラン(10mL)に溶解し、気相を窒素置換後、氷冷下、水素化アルミニウムリチウム(0.25g、6.6mmol)を少量ずつ加えた。室温に昇温しつつ1時間撹拌し、氷冷下、水(1mL)を滴下して反応を停止した。混合物を酢酸エチル(30mL)で希釈後、2M塩酸(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.65g)を無色固体として得た。
(実施例3g)酢酸 4-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-ヒドロキシベンジル
実施例3fで得られた粗生成物(0.65g、2.2mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を加えた。この懸濁液を37℃で8時間撹拌し、懸濁液をセライトで濾過し、酢酸エチル(5mL、2回)で洗浄した。得られた濾液を減圧下濃縮し、残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.75g、quant.)を微黄色固体として得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 3.98 (2H, s), 5.00 (2H, s), 6.45 (1H, t, J = 74.2 Hz), 6.60 (1H, s), 6.69 (1H, d, J = 9.3 Hz), 7.01 (2H, d, J = 7.3 Hz), 7.27 (2H, d, J = 7.3 Hz);
MS (FAB) m/z: 340 (M)+.。
実施例3fで得られた粗生成物(0.65g、2.2mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を加えた。この懸濁液を37℃で8時間撹拌し、懸濁液をセライトで濾過し、酢酸エチル(5mL、2回)で洗浄した。得られた濾液を減圧下濃縮し、残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.75g、quant.)を微黄色固体として得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 3.98 (2H, s), 5.00 (2H, s), 6.45 (1H, t, J = 74.2 Hz), 6.60 (1H, s), 6.69 (1H, d, J = 9.3 Hz), 7.01 (2H, d, J = 7.3 Hz), 7.27 (2H, d, J = 7.3 Hz);
MS (FAB) m/z: 340 (M)+.。
(実施例3h)5-アセトキシメチル-2-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロフェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.37g、0.60mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.18mL、1.8mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(2.7μL、0.018mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。得られたイミダート(0.45g、0.60mmol)、実施例3gで得られた化合物(0.20g、0.60mmol)および三フッ化ホウ素-ジエチルエーテル錯体(37μL、0.30mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.37g、0.60mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.18mL、1.8mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(2.7μL、0.018mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。得られたイミダート(0.45g、0.60mmol)、実施例3gで得られた化合物(0.20g、0.60mmol)および三フッ化ホウ素-ジエチルエーテル錯体(37μL、0.30mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例3i)2-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例3hで得られた混合物(0.60mmol)をテトラヒドロフラン(2mL)およびメタノール(2mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(2mL、4mmol)を加えて室温で30分間撹拌した。混合物に水(10mL)を加えた後、酢酸エチル(20mL、3回)で抽出し、有機層を飽和食塩水(10mL)で2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、10:1、V/V)を用いて精製して、標記化合物(0.11g)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.6 Hz), 3.36-3.51 (4H, m), 4.00 (1H, d, J = 15.1 Hz), 4.06-4.07 (1H, m), 4.08 (1H, d, J = 15.1 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.4 Hz), 6.71 (1H, t, J = 74.5 Hz), 6.80 (1H, d, J = 9.8 Hz), 6.98 (2H, d, J = 8.6 Hz), 6.99 (1H, s), 7.32 (2H, d, J = 8.6 Hz);
MS (FAB) m/z: 497 (M+Na)+.。
実施例3hで得られた混合物(0.60mmol)をテトラヒドロフラン(2mL)およびメタノール(2mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(2mL、4mmol)を加えて室温で30分間撹拌した。混合物に水(10mL)を加えた後、酢酸エチル(20mL、3回)で抽出し、有機層を飽和食塩水(10mL)で2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、10:1、V/V)を用いて精製して、標記化合物(0.11g)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.6 Hz), 3.36-3.51 (4H, m), 4.00 (1H, d, J = 15.1 Hz), 4.06-4.07 (1H, m), 4.08 (1H, d, J = 15.1 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.4 Hz), 6.71 (1H, t, J = 74.5 Hz), 6.80 (1H, d, J = 9.8 Hz), 6.98 (2H, d, J = 8.6 Hz), 6.99 (1H, s), 7.32 (2H, d, J = 8.6 Hz);
MS (FAB) m/z: 497 (M+Na)+.。
(実施例4)3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.9)
(実施例4a)3-ヒドロキシ-4-(4-メチルベンゾイル)-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(3.0g、16.3mmol)をアセトニトリル(50mL)に溶解し、トリエチルアミン(6.8mL、48.8mmol)および塩化p-トルオイル(2.26mL、17.1mmol)を加え、室温で10分間撹拌した。さらに、トリメチルシリルシアニド(260μL、1.95mmol)を加え、60℃で2時間撹拌した。減圧下溶媒を留去した後、残渣を酢酸エチル(30mL)に溶解し、1M塩酸(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(3.47g)を褐色油状物質として得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(3.0g、16.3mmol)をアセトニトリル(50mL)に溶解し、トリエチルアミン(6.8mL、48.8mmol)および塩化p-トルオイル(2.26mL、17.1mmol)を加え、室温で10分間撹拌した。さらに、トリメチルシリルシアニド(260μL、1.95mmol)を加え、60℃で2時間撹拌した。減圧下溶媒を留去した後、残渣を酢酸エチル(30mL)に溶解し、1M塩酸(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(3.47g)を褐色油状物質として得た。
(実施例4b)3-クロロ-4-(4-メチルベンゾイル)-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例4aで得られた化合物(1.0g、3.3mmol)を塩化メチレン(10mL)に溶解し、氷冷下、二塩化オキサリル(300μL、3.5mmol)、2-メチル-2-ブテン(1.4mL、13.2mmol)およびN,N-ジメチルホルムアミド(100μL、1.29mmol)を加え、室温で2時間攪拌した。反応液に蒸留水(5mL)を加え、塩化メチレン(10mL)で希釈し、蒸留水(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣にトルエン(10mL)を加えて減圧下溶媒留去を2回行い、標記化合物の粗生成物(1.2g)を褐色油状物質として得た。
実施例4aで得られた化合物(1.0g、3.3mmol)を塩化メチレン(10mL)に溶解し、氷冷下、二塩化オキサリル(300μL、3.5mmol)、2-メチル-2-ブテン(1.4mL、13.2mmol)およびN,N-ジメチルホルムアミド(100μL、1.29mmol)を加え、室温で2時間攪拌した。反応液に蒸留水(5mL)を加え、塩化メチレン(10mL)で希釈し、蒸留水(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣にトルエン(10mL)を加えて減圧下溶媒留去を2回行い、標記化合物の粗生成物(1.2g)を褐色油状物質として得た。
(実施例4c)3-クロロ-5-ヒドロキシ-4-(4-メチルベンゾイル)安息香酸エチル
実施例4bで得られた粗生成物(1.2g)に、N-メチルモルホリン(10mL)およびヨウ素(1.68g、6.6mmol)を加え、室温で2時間攪拌した。セライトで濾過後、トルエン(10mL)で希釈し、亜硫酸ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1~3:1、V/V)を用いて精製して、標記化合物(0.77g)を黄色固体として得た。
実施例4bで得られた粗生成物(1.2g)に、N-メチルモルホリン(10mL)およびヨウ素(1.68g、6.6mmol)を加え、室温で2時間攪拌した。セライトで濾過後、トルエン(10mL)で希釈し、亜硫酸ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1~3:1、V/V)を用いて精製して、標記化合物(0.77g)を黄色固体として得た。
(実施例4d)3-クロロ-5-ヒドロキシ-4-[1-ヒドロキシ-1-(4-メチルフェニル)メチル]安息香酸エチル
実施例4cで得られた化合物(0.77g、2.4mmol)をメタノール(6mL)およびテトラヒドロフラン(1mL)からなる溶液に溶解し、氷冷下、水素化ホウ素ナトリウム(137mg、3.6mmol)を加え、0℃で1時間攪拌した。氷冷下、反応液に飽和塩化アンモニウム水溶液(5mL)を加え、減圧下溶媒を留去した。得られた残渣を酢酸エチル(10mL)に溶解し、飽和塩化アンモニウム水溶液(5mL)、飽和炭酸水素ナトリウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1、V/V)を用いて精製して、標記化合物(0.57g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.38 (3H, t, J = 7.3 Hz), 2.34 (3H, s), 2.97 (1H, brs), 4.36 (2H, q, J = 7.2 Hz), 6.47 (1H, s), 7.17 (2H, d, J = 8.0 Hz), 7.32 (2H, d, J = 8.0 Hz), 7.51 (1H, s), 7.56 (1H, s), 9.15 (1H, brs).。
実施例4cで得られた化合物(0.77g、2.4mmol)をメタノール(6mL)およびテトラヒドロフラン(1mL)からなる溶液に溶解し、氷冷下、水素化ホウ素ナトリウム(137mg、3.6mmol)を加え、0℃で1時間攪拌した。氷冷下、反応液に飽和塩化アンモニウム水溶液(5mL)を加え、減圧下溶媒を留去した。得られた残渣を酢酸エチル(10mL)に溶解し、飽和塩化アンモニウム水溶液(5mL)、飽和炭酸水素ナトリウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1、V/V)を用いて精製して、標記化合物(0.57g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.38 (3H, t, J = 7.3 Hz), 2.34 (3H, s), 2.97 (1H, brs), 4.36 (2H, q, J = 7.2 Hz), 6.47 (1H, s), 7.17 (2H, d, J = 8.0 Hz), 7.32 (2H, d, J = 8.0 Hz), 7.51 (1H, s), 7.56 (1H, s), 9.15 (1H, brs).。
(実施例4e)5-クロロ-2,2-ジメチル-4-(4-メチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-カルボン酸エチル
実施例4dで得られた化合物(0.57g、1.8mmol)をアセトン(6mL)に溶解し、-10℃で、三フッ化ホウ素-ジエチルエーテル錯体(220μL、1.8mmol)を加え、0℃までゆっくりと昇温しながら3時間撹拌した。氷冷下、反応液に飽和炭酸水素ナトリウム水溶液(5mL)を加え、酢酸エチル(20mL)で希釈し、飽和食塩水(10mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、10:1、V/V)を用いて精製して、標記化合物(0.65g、quant.)を無色油状物質として得た。
実施例4dで得られた化合物(0.57g、1.8mmol)をアセトン(6mL)に溶解し、-10℃で、三フッ化ホウ素-ジエチルエーテル錯体(220μL、1.8mmol)を加え、0℃までゆっくりと昇温しながら3時間撹拌した。氷冷下、反応液に飽和炭酸水素ナトリウム水溶液(5mL)を加え、酢酸エチル(20mL)で希釈し、飽和食塩水(10mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、10:1、V/V)を用いて精製して、標記化合物(0.65g、quant.)を無色油状物質として得た。
(実施例4f)5-クロロ-2,2-ジメチル-4-(4-メチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-イルメタノール
実施例4eで得られた化合物(0.65g、1.8mmol)をテトラヒドロフラン(10mL)に溶解し、氷冷下、水素化アルミニウムリチウム(101mg、2.7mmol)を加え、室温で2時間撹拌した。氷冷下、反応液に蒸留水(5mL)を加え、酢酸エチル(10mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.55g)を無色固体として得た。
実施例4eで得られた化合物(0.65g、1.8mmol)をテトラヒドロフラン(10mL)に溶解し、氷冷下、水素化アルミニウムリチウム(101mg、2.7mmol)を加え、室温で2時間撹拌した。氷冷下、反応液に蒸留水(5mL)を加え、酢酸エチル(10mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.55g)を無色固体として得た。
(実施例4g)酢酸 5-クロロ-2,2-ジメチル-4-(4-メチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-イルメチル
実施例4fで得られた粗生成物(0.55g、1.8mmol)を塩化メチレン(7mL)に溶解し、氷冷下、無水酢酸(200μL、2.1mmol)、ピリジン(350μL、3.5mmol)および4-ジメチルアミノピリジン(65mg、0.5mmol)を加え、室温で20時間撹拌した。反応液を酢酸エチル(15mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.65g)を無色油状物質として得た。
実施例4fで得られた粗生成物(0.55g、1.8mmol)を塩化メチレン(7mL)に溶解し、氷冷下、無水酢酸(200μL、2.1mmol)、ピリジン(350μL、3.5mmol)および4-ジメチルアミノピリジン(65mg、0.5mmol)を加え、室温で20時間撹拌した。反応液を酢酸エチル(15mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.65g)を無色油状物質として得た。
(実施例4h)酢酸 3-クロロ-5-ヒドロキシ-4-(4-メチルベンジル)ベンジル
実施例4gで得られた粗生成物(0.65g、1.8mmol)をアセトニトリル(7mL)に溶解し、氷冷下、トリエチルシラン(850μL、5.3mmol)および三フッ化ホウ素-ジエチルエーテル錯体(340μL、2.7mmol)を加え、0℃で1時間撹拌した。氷冷下、反応液に飽和炭酸水素ナトリウム水溶液(5mL)を加え、酢酸エチル(10mL)で希釈し、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、8:1~6:1~4:1、V/V)を用いて精製して、標記化合物(376mg)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.12 (3H, s), 2.30 (3H, s), 4.14 (2H, s), 4.93 (1H, s), 5.00 (2H, s), 6.72 (1H, s), 7.02 (1H, s), 7.09 (2H, d, J = 7.8 Hz), 7.15 (2H, d, J = 7.8 Hz);
MS (EI) m/z: 304 (M)+.。
実施例4gで得られた粗生成物(0.65g、1.8mmol)をアセトニトリル(7mL)に溶解し、氷冷下、トリエチルシラン(850μL、5.3mmol)および三フッ化ホウ素-ジエチルエーテル錯体(340μL、2.7mmol)を加え、0℃で1時間撹拌した。氷冷下、反応液に飽和炭酸水素ナトリウム水溶液(5mL)を加え、酢酸エチル(10mL)で希釈し、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、8:1~6:1~4:1、V/V)を用いて精製して、標記化合物(376mg)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.12 (3H, s), 2.30 (3H, s), 4.14 (2H, s), 4.93 (1H, s), 5.00 (2H, s), 6.72 (1H, s), 7.02 (1H, s), 7.09 (2H, d, J = 7.8 Hz), 7.15 (2H, d, J = 7.8 Hz);
MS (EI) m/z: 304 (M)+.。
(実施例4i)5-アセトキシメチル-3-クロロ-(4-メチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(240mg、0.39mmol)を塩化メチレン(5mL)に溶解し、氷冷下、トリクロロアセトニトリル(120μL、1.20mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を加えて、0℃で1時間撹拌した。反応液を塩化メチレン(10mL)で希釈し、飽和塩化アンモニウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、相当するイミダートの粗生成物を黄色アモルファスとして得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(240mg、0.39mmol)を塩化メチレン(5mL)に溶解し、氷冷下、トリクロロアセトニトリル(120μL、1.20mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を加えて、0℃で1時間撹拌した。反応液を塩化メチレン(10mL)で希釈し、飽和塩化アンモニウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、相当するイミダートの粗生成物を黄色アモルファスとして得た。
得られたイミダートを塩化メチレン(5mL)に溶解し、氷冷下、実施例4hで得られた化合物(100mg、0.33mmol)および三フッ化ホウ素-ジエチルエーテル錯体(50μL、0.39mmol)を加えて、0℃で2時間撹拌した。反応液にトリエチルアミン(100μL、0.72mmol)を加えた後に、酢酸エチル(10mL)で希釈し、飽和食塩水(10mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(350mg)を得た。
(実施例4j)3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例4iで得られた粗生成物(350mg、0.33mmol)をテトラヒドロフラン(1mL)およびメタノール(4mL)からなる溶液に溶解し、炭酸ナトリウム(46mg、0.33mmol)を加え、室温で2時間攪拌した。減圧下溶媒を留去した後、得られた残渣を酢酸エチル(10mL)に溶解し、飽和塩化アンモニウム水溶液(10mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(塩化メチレン:メタノール、20:1~15:1~10:1、V/V)を用いて精製して、標記化合物(76mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 2.25 (3H, s), 3.37-3.38 (2H, m), 3.43-3.45 (2H, m), 4.05 (1H, dd, J = 6.7 Hz, 3.5 Hz), 4.10 (1H, d, J = 14.4 Hz), 4.23 (1H, d, J = 14.4 Hz), 4.55 (2H, s), 4.92 (1H, d, J = 7.4 Hz), 7.00 (2H, d, J = 7.9 Hz), 7.09-7.12 (4H, m);
MS (FAB) m/z: 438 (M+Na)+.。
実施例4iで得られた粗生成物(350mg、0.33mmol)をテトラヒドロフラン(1mL)およびメタノール(4mL)からなる溶液に溶解し、炭酸ナトリウム(46mg、0.33mmol)を加え、室温で2時間攪拌した。減圧下溶媒を留去した後、得られた残渣を酢酸エチル(10mL)に溶解し、飽和塩化アンモニウム水溶液(10mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(塩化メチレン:メタノール、20:1~15:1~10:1、V/V)を用いて精製して、標記化合物(76mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 2.25 (3H, s), 3.37-3.38 (2H, m), 3.43-3.45 (2H, m), 4.05 (1H, dd, J = 6.7 Hz, 3.5 Hz), 4.10 (1H, d, J = 14.4 Hz), 4.23 (1H, d, J = 14.4 Hz), 4.55 (2H, s), 4.92 (1H, d, J = 7.4 Hz), 7.00 (2H, d, J = 7.9 Hz), 7.09-7.12 (4H, m);
MS (FAB) m/z: 438 (M+Na)+.。
(実施例5)3-フルオロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.7)
(実施例5a)4-(3-フルオロ-4-メチルベンゾイル)-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(0.35g、1.9mmol)をアセトニトリル(4mL)に溶解し、トリエチルアミン(0.79mL、5.7mmol)、塩化3-フルオロ-4-メチルベンゾイル(J.Med.Chem.,1997,40,2064-2084.)(0.33g、1.9mmol)およびトリメチルシリルシアニド(0.030mL、0.23mmol)を用い、実施例1bと同様の方法により、粗生成物を得た。この粗生成物をシリカゲルフラッシュカラムクロマトグラフィー(塩化メチレン:メタノール、10:1、V/V)を用いて精製して、標記化合物(0.61g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(0.35g、1.9mmol)をアセトニトリル(4mL)に溶解し、トリエチルアミン(0.79mL、5.7mmol)、塩化3-フルオロ-4-メチルベンゾイル(J.Med.Chem.,1997,40,2064-2084.)(0.33g、1.9mmol)およびトリメチルシリルシアニド(0.030mL、0.23mmol)を用い、実施例1bと同様の方法により、粗生成物を得た。この粗生成物をシリカゲルフラッシュカラムクロマトグラフィー(塩化メチレン:メタノール、10:1、V/V)を用いて精製して、標記化合物(0.61g)を得た。
(実施例5b)4-(3-フルオロ-4-メチルベンゾイル)-3-フルオロ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例5aで得られた化合物(0.61g、1.9mmol)を塩化メチレン(8mL)に溶解し、ジエチルアミノ硫黄三フッ化物(0.75mL、5.7mmol)を用いて、実施例3bと同様の方法により、標記化合物(0.31g)を得た。
実施例5aで得られた化合物(0.61g、1.9mmol)を塩化メチレン(8mL)に溶解し、ジエチルアミノ硫黄三フッ化物(0.75mL、5.7mmol)を用いて、実施例3bと同様の方法により、標記化合物(0.31g)を得た。
(実施例5c)4-(3-フルオロ-4-メチルベンゾイル)-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例5bで得られた化合物(0.31g、0.96mmol)をアセトニトリル(3mL)に溶解し、氷冷化、トリエチルアミン(0.40mL、2.9mmol)およびヨウ化トリメチルシリル(0.34mL、2.4mmol)を加え、室温で2時間撹拌した。反応終了後、氷冷下、反応液をトルエン(10mL)で希釈し、リン酸緩衝溶液(pH7、5mL)で2回洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去して、得られた残渣をトルエン(10mL)で希釈した。この溶液に、シリカゲル(関東化学社製、シリカゲル60(球状)、40~100μm、1g)を加え、室温で1時間撹拌した。この混合物をセライトで濾過して、減圧下溶媒を留去し、エタノール(10mL)および炭酸カリウム(0.41g、3.0mmol)を加えて、50℃で1時間撹拌した。反応液を室温に冷却後、酢酸エチル(20mL)で希釈し、1M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.17g)を得た。
実施例5bで得られた化合物(0.31g、0.96mmol)をアセトニトリル(3mL)に溶解し、氷冷化、トリエチルアミン(0.40mL、2.9mmol)およびヨウ化トリメチルシリル(0.34mL、2.4mmol)を加え、室温で2時間撹拌した。反応終了後、氷冷下、反応液をトルエン(10mL)で希釈し、リン酸緩衝溶液(pH7、5mL)で2回洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去して、得られた残渣をトルエン(10mL)で希釈した。この溶液に、シリカゲル(関東化学社製、シリカゲル60(球状)、40~100μm、1g)を加え、室温で1時間撹拌した。この混合物をセライトで濾過して、減圧下溶媒を留去し、エタノール(10mL)および炭酸カリウム(0.41g、3.0mmol)を加えて、50℃で1時間撹拌した。反応液を室温に冷却後、酢酸エチル(20mL)で希釈し、1M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.17g)を得た。
(実施例5d)3-フルオロ-2-[1-(3-フルオロ-4-メチルフェニル)-1-ヒドロキシメチル]-5-(ヒドロキシメチル)フェノール
実施例5cで得られた化合物(0.17g、0.53mmol)をテトラヒドロフラン(5mL)に溶解し、水素化アルミニウムリチウム(60mg、1.6mmol)を用いて、実施例1eと同様の方法により、標記化合物(0.13g)を粗生成物として得た。
実施例5cで得られた化合物(0.17g、0.53mmol)をテトラヒドロフラン(5mL)に溶解し、水素化アルミニウムリチウム(60mg、1.6mmol)を用いて、実施例1eと同様の方法により、標記化合物(0.13g)を粗生成物として得た。
(実施例5e)酢酸 3-フルオロ-4-[1-(3-フルオロ-4-メチルフェニル)-1-ヒドロキシメチル]-5-ヒドロキシベンジル
実施例5dで得られた粗生成物(0.13g、0.46mmol)を酢酸ビニル(3mL)およびジイソプロピルエーテル(3mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(0.13g)を加え、室温で2日間撹拌した。反応終了後、混合物をセライトで濾過し、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.12g)を得た。
実施例5dで得られた粗生成物(0.13g、0.46mmol)を酢酸ビニル(3mL)およびジイソプロピルエーテル(3mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(0.13g)を加え、室温で2日間撹拌した。反応終了後、混合物をセライトで濾過し、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.12g)を得た。
(実施例5f)酢酸 3-フルオロ-4-(3-フルオロ-4-メチルベンジル)-5-ヒドロキシベンジル
実施例5eで得られた化合物(0.12g、0.37mmol)をアセトニトリル(3mL)に溶解し、-40℃でトリエチルシラン(0.18mL、1.1mmol)および三フッ化ホウ素-ジエチルエーテル錯体(70μL、0.56mmol)を加え、室温まで昇温しながら1時間撹拌した。反応終了後、氷冷下、反応液にトリエチルアミン(0.15mL、1.1mmol)を加え、酢酸エチル(10mL)で希釈し、飽和塩化アンモニウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去して得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1、V/V)を用いて精製して、油状の標記化合物(85mg)を得た。
1H NMR (500 MHz, CDCl3): δ 2.12 (3H, s), 2.22 (3H, s), 3.96 (2H, s), 5.01 (2H, s), 6.61 (1H, s), 6.68 (1H, d, J = 9.8 Hz), 6.91-6.97 (2H, m), 7.07 (1H, m);
MS (EI) m/z: 306 (M)+.。
実施例5eで得られた化合物(0.12g、0.37mmol)をアセトニトリル(3mL)に溶解し、-40℃でトリエチルシラン(0.18mL、1.1mmol)および三フッ化ホウ素-ジエチルエーテル錯体(70μL、0.56mmol)を加え、室温まで昇温しながら1時間撹拌した。反応終了後、氷冷下、反応液にトリエチルアミン(0.15mL、1.1mmol)を加え、酢酸エチル(10mL)で希釈し、飽和塩化アンモニウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去して得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1、V/V)を用いて精製して、油状の標記化合物(85mg)を得た。
1H NMR (500 MHz, CDCl3): δ 2.12 (3H, s), 2.22 (3H, s), 3.96 (2H, s), 5.01 (2H, s), 6.61 (1H, s), 6.68 (1H, d, J = 9.8 Hz), 6.91-6.97 (2H, m), 7.07 (1H, m);
MS (EI) m/z: 306 (M)+.。
(実施例5g)5-アセトキシメチル-3-フルオロ-2-(3-フルオロ-4-メチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.37g、0.60mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.18mL、1.8mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(2.7μL、0.018mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.45g、0.60mmol)、実施例5fで得られた化合物(85mg、0.28mmol)および三フッ化ホウ素-ジエチルエーテル錯体(37μL、0.30mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.37g、0.60mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.18mL、1.8mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(2.7μL、0.018mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.45g、0.60mmol)、実施例5fで得られた化合物(85mg、0.28mmol)および三フッ化ホウ素-ジエチルエーテル錯体(37μL、0.30mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例5h)3-フルオロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例5gで得られた混合物(0.28mmol)をテトラヒドロフラン(2mL)およびメタノール(1mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(2mL、4mmol)を加えて実施例3iと同様の方法により標記化合物(84mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.7 Hz), 2.17 (3H, s), 3.38-3.47 (4H, m), 3.97 (1H, d, J = 14 Hz), 4.03-4.05 (1H, m), 4.05 (1H, d, J = 14 Hz), 4.56 (2H, s), 4.95 (1H, d, J = 7.4 Hz), 6.80 (1H, d, J = 9.3 Hz), 6.91(1H, d, J = 11 Hz), 6.96-7.06 (3H, m);
MS (FAB) m/z: 441 (M+H)+, 463 (M+Na)+.。
実施例5gで得られた混合物(0.28mmol)をテトラヒドロフラン(2mL)およびメタノール(1mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(2mL、4mmol)を加えて実施例3iと同様の方法により標記化合物(84mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.7 Hz), 2.17 (3H, s), 3.38-3.47 (4H, m), 3.97 (1H, d, J = 14 Hz), 4.03-4.05 (1H, m), 4.05 (1H, d, J = 14 Hz), 4.56 (2H, s), 4.95 (1H, d, J = 7.4 Hz), 6.80 (1H, d, J = 9.3 Hz), 6.91(1H, d, J = 11 Hz), 6.96-7.06 (3H, m);
MS (FAB) m/z: 441 (M+H)+, 463 (M+Na)+.。
(実施例6)3-クロロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.17)
(実施例6a)塩化3-フルオロ-4-メチルベンゾイル
3-フルオロ-4-メチル安息香酸(880mg、5.71mmol)、二塩化オキサリル(0.50mL、5.71mmol)、触媒量のN,N-ジメチルホルムアミドおよびテトラヒドロフラン(10mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(950mg)を無色油状物質として得た。
3-フルオロ-4-メチル安息香酸(880mg、5.71mmol)、二塩化オキサリル(0.50mL、5.71mmol)、触媒量のN,N-ジメチルホルムアミドおよびテトラヒドロフラン(10mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(950mg)を無色油状物質として得た。
(実施例6b)4-(3-フルオロ-4-メチルベンゾイル)-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(950mg、5.16mmol)、実施例6aで得られた粗生成物(950mg、11mmol)、アセトニトリル(10mL)、トリエチルアミン(2.27mL、1.63mmol)およびトリメチルシリルシアニド(0.087mL、0.65mmol)を用いて実施例1bと同様の方法により、標記化合物の粗生成物(1.65g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(950mg、5.16mmol)、実施例6aで得られた粗生成物(950mg、11mmol)、アセトニトリル(10mL)、トリエチルアミン(2.27mL、1.63mmol)およびトリメチルシリルシアニド(0.087mL、0.65mmol)を用いて実施例1bと同様の方法により、標記化合物の粗生成物(1.65g)を得た。
(実施例6c)3-クロロ-4-(3-フルオロ-4-メチルベンゾイル)-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例6bで得られた粗生成物(1.65g、5.15mmol)、塩化メチレン(20mL)、2-メチル-2-ブテン(2.19mL、20.6mmol)、二塩化オキサリル(0.46mL、5.36mmol)および触媒量のN,N-ジメチルホルムアミドを用い実施例1cと同様の方法により、標記化合物の粗生成物(1.75g)を得た。
実施例6bで得られた粗生成物(1.65g、5.15mmol)、塩化メチレン(20mL)、2-メチル-2-ブテン(2.19mL、20.6mmol)、二塩化オキサリル(0.46mL、5.36mmol)および触媒量のN,N-ジメチルホルムアミドを用い実施例1cと同様の方法により、標記化合物の粗生成物(1.75g)を得た。
(実施例6d)3-クロロ-4-(3-フルオロ-4-メチルベンゾイル)-5-ヒドロキシ安息香酸エチル
実施例6cで得られた粗生成物(1.75g、5.15mmol)、N-メチルモルホリン(20mL)、無水硫酸ナトリウム(14.6g)およびヨウ素(1.43g、5.63mmol)を用い、実施例1dと同様の方法により、標記化合物の粗生成物を得た。次いで、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、アモルファス状の標記化合物(1.28g)を得た。
実施例6cで得られた粗生成物(1.75g、5.15mmol)、N-メチルモルホリン(20mL)、無水硫酸ナトリウム(14.6g)およびヨウ素(1.43g、5.63mmol)を用い、実施例1dと同様の方法により、標記化合物の粗生成物を得た。次いで、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、アモルファス状の標記化合物(1.28g)を得た。
(実施例6e)5-クロロ-2,2-ジメチル-4-(3-フルオロ-4-メチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-安息香酸エチル
実施例6dで得られた化合物(1.28g、3.80mmol)、メタノール(12mL)および水素化ホウ素ナトリウム(290mg、7.67mmol)を用い、実施例2bと同様の方法により、ジオール体粗生成物(1.28g)を得た。-10℃に冷却した三フッ化ホウ素-ジエチルエーテル錯体(0.475mL、3.78mmol)をアセトン(10mL)に溶解し、ジオール体粗生成物(1.28g)を溶解させたアセトン溶液(15mL)をゆっくり加えた。その後1時間かけて反応液を10℃まで昇温した。反応液に酢酸エチル(50mL)を加え、飽和炭酸水素ナトリウム水溶液(30mL)および飽和食塩水(30mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~5:1、V/V)を用いて精製して、油状の標記化合物(964mg)を得た。
実施例6dで得られた化合物(1.28g、3.80mmol)、メタノール(12mL)および水素化ホウ素ナトリウム(290mg、7.67mmol)を用い、実施例2bと同様の方法により、ジオール体粗生成物(1.28g)を得た。-10℃に冷却した三フッ化ホウ素-ジエチルエーテル錯体(0.475mL、3.78mmol)をアセトン(10mL)に溶解し、ジオール体粗生成物(1.28g)を溶解させたアセトン溶液(15mL)をゆっくり加えた。その後1時間かけて反応液を10℃まで昇温した。反応液に酢酸エチル(50mL)を加え、飽和炭酸水素ナトリウム水溶液(30mL)および飽和食塩水(30mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~5:1、V/V)を用いて精製して、油状の標記化合物(964mg)を得た。
(実施例6f)5-クロロ-2,2-ジメチル-4-(3-フルオロ-4-メチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-イルメタノール
実施例6eで得られた化合物(960mg、2.53mmol)、水素化リチウムアルミニウム(96mg、2.53mmol)およびテトラヒドロフラン(5mL)を用い、実施例2dと同様の方法により、標記化合物の粗生成物(860mg)をアモルファスとして得た。
実施例6eで得られた化合物(960mg、2.53mmol)、水素化リチウムアルミニウム(96mg、2.53mmol)およびテトラヒドロフラン(5mL)を用い、実施例2dと同様の方法により、標記化合物の粗生成物(860mg)をアモルファスとして得た。
(実施例6g)酢酸 5-クロロ-2,2-ジメチル-4-(3-フルオロ-4-メチルフェニル)-4H-ベンゾ[1,3]ジオキシン-7-イルメチル
実施例6fで得られた粗生成物(860mg)をピリジン(8.6mL)に溶解し、氷冷下、無水酢酸(2.2mL)を加え、室温で1時間攪拌した。反応液にメタノール(2.2mL)を加え、15分間攪拌した後に、酢酸エチル(40mL)を加え、30%クエン酸水溶液(10mL、2回)、飽和炭酸水素ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、油状の標記化合物(900mg)を得た。
実施例6fで得られた粗生成物(860mg)をピリジン(8.6mL)に溶解し、氷冷下、無水酢酸(2.2mL)を加え、室温で1時間攪拌した。反応液にメタノール(2.2mL)を加え、15分間攪拌した後に、酢酸エチル(40mL)を加え、30%クエン酸水溶液(10mL、2回)、飽和炭酸水素ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、油状の標記化合物(900mg)を得た。
(実施例6h)酢酸 3-クロロ-4-(3-フルオロ-4-メチルベンジル)-5-ヒドロキシベンジル
実施例6fで得られた化合物(900mg、2.38mmol)、アセトニトリル(15mL)、トリエチルシラン(1.14mL、7.09mmol)およびフッ化ホウ素-ジエチルエーテル錯体(0.450mL、3.58mmol)を用い、実施例2fと同様の方法により、標記化合物(631mg)を無色固体として得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 2.21 (3H, s), 4.12 (2H, s), 5.00 (2H, s), 5.28 (1H, brs), 6.71 (1H, s), 6.89 (1H, d, J = 10.8 Hz), 6.94 (1H, d, J = 8.0 Hz), 7.01 (1H, s), 7.06 (1H, t, J = 8.0 Hz);
MS (FAB) m/z: 345 (M+Na)+.
実施例6fで得られた化合物(900mg、2.38mmol)、アセトニトリル(15mL)、トリエチルシラン(1.14mL、7.09mmol)およびフッ化ホウ素-ジエチルエーテル錯体(0.450mL、3.58mmol)を用い、実施例2fと同様の方法により、標記化合物(631mg)を無色固体として得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 2.21 (3H, s), 4.12 (2H, s), 5.00 (2H, s), 5.28 (1H, brs), 6.71 (1H, s), 6.89 (1H, d, J = 10.8 Hz), 6.94 (1H, d, J = 8.0 Hz), 7.01 (1H, s), 7.06 (1H, t, J = 8.0 Hz);
MS (FAB) m/z: 345 (M+Na)+.
(実施例6i)5-アセトキシメチル-3-クロロ-2-(3-フルオロ-4-メチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(250mg、0.41mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.210mL、2.08mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(309mg)、実施例6hで得られた化合物(100mg、0.41mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.051mL、0.41mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(280mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(250mg、0.41mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.210mL、2.08mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(309mg)、実施例6hで得られた化合物(100mg、0.41mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.051mL、0.41mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(280mg)を得た。
(実施例6j)3-クロロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例6iで得られた粗生成物(280mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(566mg、4.10mmol)を用い、実施例2hと同様の方法により、標記化合物(76mg)を無色固体として得た。ただし、固体化はヘキサン/酢酸エチルから行った。
1H NMR (500 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 2.18 (3H, s), 3.35-3.41 (2H, m), 3.44-3.46 (2H, m), 4.03-4.08 (1H, m), 4.12 (1H, d, J = 15.0 Hz), 4.24 (1H, d, J = 15.0 Hz), 4.56 (2H, s), 4.94 (1H, d, J = 7.8 Hz), 6.87 (1H, d, J = 11.2 Hz), 6.95 (1H, d, J = 7.9 Hz), 7.04 (1H, t, J = 7.9 Hz), 7.11 (1H, s), 7.14 (1H, s);
MS (FAB) m/z: 479 (M+Na)+.。
実施例6iで得られた粗生成物(280mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(566mg、4.10mmol)を用い、実施例2hと同様の方法により、標記化合物(76mg)を無色固体として得た。ただし、固体化はヘキサン/酢酸エチルから行った。
1H NMR (500 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 2.18 (3H, s), 3.35-3.41 (2H, m), 3.44-3.46 (2H, m), 4.03-4.08 (1H, m), 4.12 (1H, d, J = 15.0 Hz), 4.24 (1H, d, J = 15.0 Hz), 4.56 (2H, s), 4.94 (1H, d, J = 7.8 Hz), 6.87 (1H, d, J = 11.2 Hz), 6.95 (1H, d, J = 7.9 Hz), 7.04 (1H, t, J = 7.9 Hz), 7.11 (1H, s), 7.14 (1H, s);
MS (FAB) m/z: 479 (M+Na)+.。
(実施例7)3-フルオロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.8)
(実施例7a)3,5-ジフルオロ-4-[1-(3-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]ベンゾニトリル
ジイソプロピルアミン(1.20mL、8.49mmol)をテトラヒドロフラン(7mL)に溶解し、氷冷下、n-ブチルリチウム(2.60mL、7.19mmol、2.77M n-ヘキサン溶液)を滴下した。同じ温度で10分間攪拌した後、-78℃に冷却し、3,5-ジフルオロベンゾニトリル(1.00g、7.19mmol)を溶解させたテトラヒドロフラン溶液(5mL)を反応液にゆっくりと滴下した。同じ温度で1時間攪拌した後、3-フルオロ-4-メトキシベンズアルデヒド(1.11g、7.20mmol)を溶解させたテトラヒドロフラン溶液(5mL)を反応液にゆっくりと滴下し、同温度で2時間攪拌した。-20℃まで昇温した後、反応液に0.5M塩酸を加えて反応を停止した。反応液を酢酸エチル(60mL)で希釈し、水(30mL)および飽和炭酸水素ナトリウム水溶液(30mL)で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1~2:1、V/V)を用いて精製して、油状の標記化合物を(1.59g)を得た。
ジイソプロピルアミン(1.20mL、8.49mmol)をテトラヒドロフラン(7mL)に溶解し、氷冷下、n-ブチルリチウム(2.60mL、7.19mmol、2.77M n-ヘキサン溶液)を滴下した。同じ温度で10分間攪拌した後、-78℃に冷却し、3,5-ジフルオロベンゾニトリル(1.00g、7.19mmol)を溶解させたテトラヒドロフラン溶液(5mL)を反応液にゆっくりと滴下した。同じ温度で1時間攪拌した後、3-フルオロ-4-メトキシベンズアルデヒド(1.11g、7.20mmol)を溶解させたテトラヒドロフラン溶液(5mL)を反応液にゆっくりと滴下し、同温度で2時間攪拌した。-20℃まで昇温した後、反応液に0.5M塩酸を加えて反応を停止した。反応液を酢酸エチル(60mL)で希釈し、水(30mL)および飽和炭酸水素ナトリウム水溶液(30mL)で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1~2:1、V/V)を用いて精製して、油状の標記化合物を(1.59g)を得た。
(実施例7b)3,5-ジフルオロ-4-(3-フルオロ-4-メトキシベンジル)ベンゾニトリル
実施例7aで得られた化合物(1.59g、5.42mmol)をアセトニトリル(16mL)に溶解し、氷冷下、トリエチルシラン(2.6mL、16mmol)および三フッ化ホウ素-ジエチルエーテル錯体(1.0mL、7.9mmol)を加えた。室温で2時間半撹拌した後、反応液に飽和炭酸水素ナトリウム水溶液を加えて反応を停止した。反応液を酢酸エチル(60mL)で希釈し、有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~2:1、V/V)を用いて精製して、標記化合物(1.05g)を無色固体として得た。
実施例7aで得られた化合物(1.59g、5.42mmol)をアセトニトリル(16mL)に溶解し、氷冷下、トリエチルシラン(2.6mL、16mmol)および三フッ化ホウ素-ジエチルエーテル錯体(1.0mL、7.9mmol)を加えた。室温で2時間半撹拌した後、反応液に飽和炭酸水素ナトリウム水溶液を加えて反応を停止した。反応液を酢酸エチル(60mL)で希釈し、有機層を無水硫酸マグネシウムで乾燥後、減圧下溶媒を留去した。得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~2:1、V/V)を用いて精製して、標記化合物(1.05g)を無色固体として得た。
(実施例7c)3-ベンジルオキシ-5-フルオロ-4-(3-フルオロ-4-メトキシベンジル)ベンゾニトリル
実施例7bで得られた化合物(1.05g、3.79mmol)およびベンジルアルコール(0.53g、4.9mmol)をN,N-ジメチルホルムアミド(11mL)に溶解し、氷冷下、水素化ナトリウム(63%、0.22g、5.7mmol)を加えた。同温度で2時間撹拌した後、反応液に2M塩酸を滴下して反応を停止した。反応液を酢酸エチル(100mL)で希釈し、水(50mL、2回)で洗浄し、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去した後、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1~2:1、V/V)を用いて精製して、標記化合物を含む混合物(1.05g)を得た。
実施例7bで得られた化合物(1.05g、3.79mmol)およびベンジルアルコール(0.53g、4.9mmol)をN,N-ジメチルホルムアミド(11mL)に溶解し、氷冷下、水素化ナトリウム(63%、0.22g、5.7mmol)を加えた。同温度で2時間撹拌した後、反応液に2M塩酸を滴下して反応を停止した。反応液を酢酸エチル(100mL)で希釈し、水(50mL、2回)で洗浄し、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去した後、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1~2:1、V/V)を用いて精製して、標記化合物を含む混合物(1.05g)を得た。
(実施例7d)3-ベンジルオキシ-5-フルオロ-4-(3-フルオロ-4-メトキシベンジル)安息香酸
実施例7cで得られた混合物(1.04g、2.85mmol)をエタノール(14.5mL)に溶解し、室温で5M水酸化ナトリウム水溶液(2.9mL、15mmol)を加えた。加熱還流条件下で2時間撹拌した後、室温まで冷却し、反応液に2M塩酸(8mL)を加えた(pH=1)。析出した固体を濾取し、水で洗浄した後、減圧下乾燥して、標記化合物を含む混合物を得た。
実施例7cで得られた混合物(1.04g、2.85mmol)をエタノール(14.5mL)に溶解し、室温で5M水酸化ナトリウム水溶液(2.9mL、15mmol)を加えた。加熱還流条件下で2時間撹拌した後、室温まで冷却し、反応液に2M塩酸(8mL)を加えた(pH=1)。析出した固体を濾取し、水で洗浄した後、減圧下乾燥して、標記化合物を含む混合物を得た。
(実施例7e)3-フルオロ-4-(3-フルオロ-4-メトキシベンジル)-5-ヒドロキシ安息香酸
実施例7dで得られた混合物(2.85mmol)をテトラヒドロフラン(10mL)およびメタノール(10mL)からなる溶液に溶解し、攪拌しながら窒素置換を行なった。反応液に10%パラジウム炭素触媒(含水、0.20g)を加え、気相を水素置換した後、室温で40分間撹拌した。混合物をメンブレンフィルターで濾過して触媒を除去した後、減圧下溶媒を留去して標記化合物を含む混合物(0.90g)を無色固体として得た。
実施例7dで得られた混合物(2.85mmol)をテトラヒドロフラン(10mL)およびメタノール(10mL)からなる溶液に溶解し、攪拌しながら窒素置換を行なった。反応液に10%パラジウム炭素触媒(含水、0.20g)を加え、気相を水素置換した後、室温で40分間撹拌した。混合物をメンブレンフィルターで濾過して触媒を除去した後、減圧下溶媒を留去して標記化合物を含む混合物(0.90g)を無色固体として得た。
(実施例7f)3-フルオロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェノール
水素化アルミニウムリチウム(0.27g、7.1mmol)をテトラヒドロフラン(2mL)に懸濁し、氷冷下、実施例7eで得られた混合物(0.90g、2.85mmol)を溶解させたテトラヒドロフラン溶液(28mL)を滴下した。50℃で2時間撹拌した後、氷冷下、反応液に水および2M塩酸を滴下して反応を停止した。反応液を酢酸エチル(60mL)で希釈し、飽和食塩水(20mL、2回)で洗浄した後、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去して得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1、V/V)を用いて精製して、標記化合物(0.61g)を無色固体として得た。
水素化アルミニウムリチウム(0.27g、7.1mmol)をテトラヒドロフラン(2mL)に懸濁し、氷冷下、実施例7eで得られた混合物(0.90g、2.85mmol)を溶解させたテトラヒドロフラン溶液(28mL)を滴下した。50℃で2時間撹拌した後、氷冷下、反応液に水および2M塩酸を滴下して反応を停止した。反応液を酢酸エチル(60mL)で希釈し、飽和食塩水(20mL、2回)で洗浄した後、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去して得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1、V/V)を用いて精製して、標記化合物(0.61g)を無色固体として得た。
(実施例7g)酢酸 3-フルオロ-4-(3-フルオロ-4-メトキシベンジル)-5-ヒドロキシベンジル
実施例7fで得られた化合物(0.60g、2.14mmol)をテトラヒドロフラン(3mL)に溶解し、酢酸ビニル(3mL)およびビス(ジブチル塩化スズ)オキシド(0.35g、0.63mmol)を加え、室温で3日間攪拌した。減圧下溶媒を除去し、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、2:1~1:1、V/V)を用いて精製して、標記化合物を(0.69g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.11 (3H, s), 3.85 (3H, s), 3.93 (2H, s), 5.00 (2H, s), 5.17 (1H, d, J = 1.2 Hz), 6.60 (1H, brs), 6.69 (1H, dd, J = 9.5 Hz, 1.4 Hz), 6.86 (1H, t, J = 8.6 Hz), 6.97-7.03 (2H, m);
MS (FAB) m/z: 322 (M)+.
実施例7fで得られた化合物(0.60g、2.14mmol)をテトラヒドロフラン(3mL)に溶解し、酢酸ビニル(3mL)およびビス(ジブチル塩化スズ)オキシド(0.35g、0.63mmol)を加え、室温で3日間攪拌した。減圧下溶媒を除去し、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、2:1~1:1、V/V)を用いて精製して、標記化合物を(0.69g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.11 (3H, s), 3.85 (3H, s), 3.93 (2H, s), 5.00 (2H, s), 5.17 (1H, d, J = 1.2 Hz), 6.60 (1H, brs), 6.69 (1H, dd, J = 9.5 Hz, 1.4 Hz), 6.86 (1H, t, J = 8.6 Hz), 6.97-7.03 (2H, m);
MS (FAB) m/z: 322 (M)+.
(実施例7h)5-アセトキシメチル-3-フルオロ-2-(3-フルオロ-4-メトキシベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.68g、1.1mmol)、塩化メチレン(14mL)、トリクロロアセトニトリル(0.34mL、3.4mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(15μL、0.10mmol)を用い、実施例1hと同様の方法により、イミダートを調製した。得られたイミダート(1.1mmol)、実施例7gで得られた化合物(300mg、0.931mmol)、塩化メチレン(5mL)および三フッ化ホウ素-ジエチルエーテル錯体(60μL、0.47mmol)を用い、実施例1hと同様の方法により、標記化合物の粗生成物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.68g、1.1mmol)、塩化メチレン(14mL)、トリクロロアセトニトリル(0.34mL、3.4mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(15μL、0.10mmol)を用い、実施例1hと同様の方法により、イミダートを調製した。得られたイミダート(1.1mmol)、実施例7gで得られた化合物(300mg、0.931mmol)、塩化メチレン(5mL)および三フッ化ホウ素-ジエチルエーテル錯体(60μL、0.47mmol)を用い、実施例1hと同様の方法により、標記化合物の粗生成物を得た。
(実施例7i)3-フルオロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例7hで得られた粗生成物をテトラヒドロフラン(2mL)およびメタノール(6mL)からなる溶液に溶解し、ナトリウムメトキシド(28%メタノール溶液、0.36g、1.9mmol)を溶解させたメタノール溶液(3mL)を室温で加えて終夜静置した。氷冷下、反応液に2M塩酸(0.95mL、1.9mmol)および飽和食塩水を加えた後、酢酸エチル(50mL、2回)で抽出し、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去した後、得られた残渣をシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、9:1~5:1、V/V)を用いて精製して、標記化合物(343mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.6 Hz), 3.37-3.40 (2H, m), 3.44-3.51 (2H, m), 3.80 (3H, s), 3.94 (1H, d, J = 14.5 Hz), 3.99-4.08 (2H, m), 4.55 (2H, s), 4.95 (1H, d, J = 7.4 Hz), 6.80 (1H, dd, J = 10.4 Hz, 1.0 Hz), 6.91 (1H, t, J = 8.8 Hz), 6.98-7.03 (3H, m);
MS (FAB) m/z: 479 (M+Na)+.。
実施例7hで得られた粗生成物をテトラヒドロフラン(2mL)およびメタノール(6mL)からなる溶液に溶解し、ナトリウムメトキシド(28%メタノール溶液、0.36g、1.9mmol)を溶解させたメタノール溶液(3mL)を室温で加えて終夜静置した。氷冷下、反応液に2M塩酸(0.95mL、1.9mmol)および飽和食塩水を加えた後、酢酸エチル(50mL、2回)で抽出し、有機層を無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去した後、得られた残渣をシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、9:1~5:1、V/V)を用いて精製して、標記化合物(343mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.6 Hz), 3.37-3.40 (2H, m), 3.44-3.51 (2H, m), 3.80 (3H, s), 3.94 (1H, d, J = 14.5 Hz), 3.99-4.08 (2H, m), 4.55 (2H, s), 4.95 (1H, d, J = 7.4 Hz), 6.80 (1H, dd, J = 10.4 Hz, 1.0 Hz), 6.91 (1H, t, J = 8.8 Hz), 6.98-7.03 (3H, m);
MS (FAB) m/z: 479 (M+Na)+.。
(実施例8)3-クロロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.19)
(実施例8a)4-(3-フルオロ-4-メトキシベンゾイル)-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(800mg、4.34mmol)、塩化3-フルオロ-4-メトキシベンゾイル(819mg、4.34mmol)、アセトニトリル(14mL)、トリエチルアミン(1.82mL、13.1mmol)およびトリメチルシリルシアニド(0.070mL、0.52mmol)を用い、実施例1bと同様の方法により、標記化合物の粗生成物(1.30g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(800mg、4.34mmol)、塩化3-フルオロ-4-メトキシベンゾイル(819mg、4.34mmol)、アセトニトリル(14mL)、トリエチルアミン(1.82mL、13.1mmol)およびトリメチルシリルシアニド(0.070mL、0.52mmol)を用い、実施例1bと同様の方法により、標記化合物の粗生成物(1.30g)を得た。
(実施例8b)3-クロロ-4-(3-フルオロ-4-メトキシベンゾイル)-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例8aで得られた粗生成物(1.30g、3.87mmol)、塩化メチレン(18mL)、2-メチル-2-ブテン(1.74mL、16.4mmol)、二塩化オキサリル(0.348mL、4.06mmol)および触媒量のN,N-ジメチルホルムアミドを用い、実施例1cと同様の方法により、標記化合物の粗生成物(1.37g)を得た。
実施例8aで得られた粗生成物(1.30g、3.87mmol)、塩化メチレン(18mL)、2-メチル-2-ブテン(1.74mL、16.4mmol)、二塩化オキサリル(0.348mL、4.06mmol)および触媒量のN,N-ジメチルホルムアミドを用い、実施例1cと同様の方法により、標記化合物の粗生成物(1.37g)を得た。
(実施例8c)3-クロロ-4-(3-フルオロ-4-メトキシベンゾイル)-5-ヒドロキシ安息香酸エチル
実施例8bで得られた粗生成物(1.37g、3.86mmol)、N-メチルモルホリン(15.6mL)、無水硫酸ナトリウム(11.0g)およびヨウ素(1.18g、4.65mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。次いで、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(672mg)を得た。
実施例8bで得られた粗生成物(1.37g、3.86mmol)、N-メチルモルホリン(15.6mL)、無水硫酸ナトリウム(11.0g)およびヨウ素(1.18g、4.65mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。次いで、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(672mg)を得た。
(実施例8d)3-クロロ-4-(3-フルオロ-4-メトキシベンジル)-5-ヒドロキシ安息香酸エチル
実施例8cで得られた化合物(670mg、1.90mmol)、メタノール(8mL)および水素化ホウ素ナトリウム(144mg、3.81mmol)を用い、実施例2bと同様の方法により、3-クロロ-4-[1-(3-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]-5-ヒドロキシ安息香酸エチル(670mg)を粗生成物として得、精製せずに次の反応に用いた。
実施例8cで得られた化合物(670mg、1.90mmol)、メタノール(8mL)および水素化ホウ素ナトリウム(144mg、3.81mmol)を用い、実施例2bと同様の方法により、3-クロロ-4-[1-(3-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]-5-ヒドロキシ安息香酸エチル(670mg)を粗生成物として得、精製せずに次の反応に用いた。
この粗生成物(670mg、1.89mmol)をアセトニトリル(8mL)に溶解し、トリエチルシラン(0.900mL、5.65mmol)を加え、0℃に冷却後、三フッ化ホウ素-ジエチルエーテル錯体(0.356mL、2.87mmol)をさらに加え、室温で3時間攪拌した。反応液に酢酸エチル(40mL)を加え、飽和炭酸水素ナトリウム水溶液(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去して、標記化合物と3-クロロ-4-[1-(3-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]-5-ヒドロキシ安息香酸エチルとを含む混合物(525mg)を得た。
(実施例8e)酢酸 3-クロロ-4-(3-フルオロ-4-メトキシベンジル)-5-ヒドロキシベンジル
実施例8dで得られた混合物(525mg)、水素化リチウムアルミニウム(118mg、3.11mmol)およびテトラヒドロフラン(8mL)を用い、実施例2dと同様の方法により混合物(333mg)をアモルファスとして得た。得られた混合物(333mg)、テトラヒドロフラン(4mL)、酢酸ビニル(4mL)およびビス(ジブチル塩化スズ)オキシド(186mg、0.34mmol)を用い、実施例2eと同様の方法により、標記化合物および酢酸 3-クロロ-4-[1-(3-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]-5-ヒドロキシベンジルを含む混合物(322mg)を得た。
実施例8dで得られた混合物(525mg)、水素化リチウムアルミニウム(118mg、3.11mmol)およびテトラヒドロフラン(8mL)を用い、実施例2dと同様の方法により混合物(333mg)をアモルファスとして得た。得られた混合物(333mg)、テトラヒドロフラン(4mL)、酢酸ビニル(4mL)およびビス(ジブチル塩化スズ)オキシド(186mg、0.34mmol)を用い、実施例2eと同様の方法により、標記化合物および酢酸 3-クロロ-4-[1-(3-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]-5-ヒドロキシベンジルを含む混合物(322mg)を得た。
さらにこの混合物(322mg)をアセトニトリル(4mL)に溶解し、トリエチルシラン(0.430mL、2.70mmol)を加え、0℃に冷却後、三フッ化ホウ素-ジエチルエーテル錯体(0.170mL、1.35mmol)を加え、室温で3時間攪拌した。反応液に酢酸エチル(40mL)を加え、飽和炭酸水素ナトリウム水溶液(20mL)および飽和食塩水(20mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去し、最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~2:1、V/V)を用いて精製して、標記化合物(252mg)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.12 (3H, s), 3.85 (3H, s), 4.09 (2H, s), 5.00 (2H, s), 5.19 (1H, s), 6.72 (1H, d, J = 1.5 Hz), 6.86 (1H, t, J = 8.6 Hz), 6.98-7.02 (3H, m);
MS (FAB) m/z: 377 (M+K)+.。
1H NMR (400 MHz, CDCl3): δ 2.12 (3H, s), 3.85 (3H, s), 4.09 (2H, s), 5.00 (2H, s), 5.19 (1H, s), 6.72 (1H, d, J = 1.5 Hz), 6.86 (1H, t, J = 8.6 Hz), 6.98-7.02 (3H, m);
MS (FAB) m/z: 377 (M+K)+.。
(実施例8f)5-アセトキシメチル-3-クロロ-2-(3-フルオロ-4-メトキシベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(328mg、0.54mmol)、塩化メチレン(5mL)、トリクロロアセトニトリル(0.272mL、2.69mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(406mg)、実施例8eで得られた化合物(140mg、0.41mmol)、塩化メチレン(5mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.068mL、0.54mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(540mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(328mg、0.54mmol)、塩化メチレン(5mL)、トリクロロアセトニトリル(0.272mL、2.69mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(406mg)、実施例8eで得られた化合物(140mg、0.41mmol)、塩化メチレン(5mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.068mL、0.54mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(540mg)を得た。
(実施例8g)3-クロロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例8fで得られた粗生成物(540mg)、メタノール/塩化メチレン(10mL/5mL)および炭酸カリウム(570mg、4.12mmol)を用い、実施例2hと同様の方法により、標記化合物(73mg)を無色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CDCl3): δ 1.21 (3H, d, J = 6.2 Hz), 3.35-3.42 (2H, m), 3.45-3.47 (2H, m), 3.80 (3H, s), 4.06 (1H, dd, J = 6.5 Hz, 3.7 Hz), 4.09 (1H, d, J = 14.5 Hz), 4.20 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.5 Hz), 6.89-7.02 (3H, m), 7.11 (1H, s), 7.13 (1H, s);
MS (FAB) m/z: 495 (M+Na)+.。
実施例8fで得られた粗生成物(540mg)、メタノール/塩化メチレン(10mL/5mL)および炭酸カリウム(570mg、4.12mmol)を用い、実施例2hと同様の方法により、標記化合物(73mg)を無色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CDCl3): δ 1.21 (3H, d, J = 6.2 Hz), 3.35-3.42 (2H, m), 3.45-3.47 (2H, m), 3.80 (3H, s), 4.06 (1H, dd, J = 6.5 Hz, 3.7 Hz), 4.09 (1H, d, J = 14.5 Hz), 4.20 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.5 Hz), 6.89-7.02 (3H, m), 7.11 (1H, s), 7.13 (1H, s);
MS (FAB) m/z: 495 (M+Na)+.。
(実施例9)3-フルオロ-2-(2-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.8)
(実施例9a)3,5-ジフルオロ-4-[1-(2-フルオロ-4-メトキシフェニル)-1-ヒドロキシメチル]ベンゾニトリル
ジイソプロピルアミン(1.20mL、8.49mmol)、n-ブチルリチウム(2.60mL、7.19mmol、2.77M n-ヘキサン溶液)、3,5-ジフルオロベンゾニトリル(1.00g、7.19mmol)、2-フルオロ-4-メトキシベンズアルデヒド(1.11g、7.20mmol)およびテトラヒドロフラン(17mL)を用い、実施例7aと同様の方法により標記化合物(1.66g)を淡黄色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~4:1~2:1、V/V)を用いて行った。
ジイソプロピルアミン(1.20mL、8.49mmol)、n-ブチルリチウム(2.60mL、7.19mmol、2.77M n-ヘキサン溶液)、3,5-ジフルオロベンゾニトリル(1.00g、7.19mmol)、2-フルオロ-4-メトキシベンズアルデヒド(1.11g、7.20mmol)およびテトラヒドロフラン(17mL)を用い、実施例7aと同様の方法により標記化合物(1.66g)を淡黄色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~4:1~2:1、V/V)を用いて行った。
(実施例9b)3,5-ジフルオロ-4-(2-フルオロ-4-メトキシベンジル)ベンゾニトリル
実施例9aで得られた化合物(1.66g、5.66mmol)、トリエチルシラン(2.7mL、17mmol)、三フッ化ホウ素-ジエチルエーテル錯体(1.1mL、8.7mmol)およびアセトニトリル(17mL)を用い、実施例7bと同様の方法により標記化合物(1.29g)を無色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1、V/V)を用いて行った。
実施例9aで得られた化合物(1.66g、5.66mmol)、トリエチルシラン(2.7mL、17mmol)、三フッ化ホウ素-ジエチルエーテル錯体(1.1mL、8.7mmol)およびアセトニトリル(17mL)を用い、実施例7bと同様の方法により標記化合物(1.29g)を無色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1、V/V)を用いて行った。
(実施例9c)3-ベンジルオキシ-5-フルオロ-4-(2-フルオロ-4-メトキシベンジル)ベンゾニトリル
実施例9bで得られた化合物(1.28g、4.62mmol)、水素化ナトリウム(63%、0.26g、6.8mmol)、ベンジルアルコール(0.66g、6.1mmol)およびN,N-ジメチルホルムアミド(13mL)を用い、実施例7cと同様の方法により標記化合物の粗生成物(0.95g)を淡黄色固体として得た。
実施例9bで得られた化合物(1.28g、4.62mmol)、水素化ナトリウム(63%、0.26g、6.8mmol)、ベンジルアルコール(0.66g、6.1mmol)およびN,N-ジメチルホルムアミド(13mL)を用い、実施例7cと同様の方法により標記化合物の粗生成物(0.95g)を淡黄色固体として得た。
(実施例9d)3-ベンジルオキシ-5-フルオロ-4-(2-フルオロ-4-メトキシベンジル)安息香酸
実施例9cで得られた粗生成物(0.95g、2.6mmol)、5M水酸化ナトリウム水溶液(2.6mL、13mmol)およびエタノール(13mL)を用い、実施例7dと同様の方法により標記化合物の粗生成物(0.97g)を淡黄色固体として得た。
実施例9cで得られた粗生成物(0.95g、2.6mmol)、5M水酸化ナトリウム水溶液(2.6mL、13mmol)およびエタノール(13mL)を用い、実施例7dと同様の方法により標記化合物の粗生成物(0.97g)を淡黄色固体として得た。
(実施例9e)3-フルオロ-4-(2-フルオロ-4-メトキシベンジル)-5-ヒドロキシ安息香酸
実施例9dで得られた粗生成物(0.97g、2.5mmol)、10%パラジウム炭素触媒(含水、0.20g)、テトラヒドロフラン(10mL)、メタノール(10mL)および水素を用い、実施例7eと同様の方法により標記化合物の粗生成物(0.75g)を淡黄色固体として得た。
実施例9dで得られた粗生成物(0.97g、2.5mmol)、10%パラジウム炭素触媒(含水、0.20g)、テトラヒドロフラン(10mL)、メタノール(10mL)および水素を用い、実施例7eと同様の方法により標記化合物の粗生成物(0.75g)を淡黄色固体として得た。
(実施例9f)3-フルオロ-2-(2-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェノール
実施例9eで得られた粗生成物(465mg、1.58mmol)、水素化アルミニウムリチウム(0.12g、3.2mmol)およびテトラヒドロフラン(12mL)を用い、実施例7fと同様の方法により標記化合物(0.37g)を無色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1~2:3、V/V)を用いて行った。
実施例9eで得られた粗生成物(465mg、1.58mmol)、水素化アルミニウムリチウム(0.12g、3.2mmol)およびテトラヒドロフラン(12mL)を用い、実施例7fと同様の方法により標記化合物(0.37g)を無色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1~2:3、V/V)を用いて行った。
(実施例9g)酢酸 3-フルオロ-4-(2-フルオロ-4-メトキシベンジル)-5-ヒドロキシベンジル
実施例9fで得られた化合物(0.37g、1.32mmol)、ビス(ジブチル塩化スズ)オキシド(0.22g、0.40mmol)、酢酸ビニル(2mL)およびテトラヒドロフラン(2mL)を用い、実施例7gと同様の方法により標記化合物(0.39g)を淡黄色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、2:1~3:2、V/V)を用いて行った。
1H NMR (400 MHz, CDCl3):δ 2.11 (3H, s), 3.76 (3H, s), 3.93 (2H, s), 5.01 (2H, s), 5.20-5.21 (1H, m), 6.59-6.64 (3H, m), 6.69 (1H, dd, J = 9.6 Hz, 1.3 Hz), 7.06 (1H, t, J = 8.8 Hz);
MS (FAB) m/z: 332 (M)+.。
実施例9fで得られた化合物(0.37g、1.32mmol)、ビス(ジブチル塩化スズ)オキシド(0.22g、0.40mmol)、酢酸ビニル(2mL)およびテトラヒドロフラン(2mL)を用い、実施例7gと同様の方法により標記化合物(0.39g)を淡黄色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、2:1~3:2、V/V)を用いて行った。
1H NMR (400 MHz, CDCl3):δ 2.11 (3H, s), 3.76 (3H, s), 3.93 (2H, s), 5.01 (2H, s), 5.20-5.21 (1H, m), 6.59-6.64 (3H, m), 6.69 (1H, dd, J = 9.6 Hz, 1.3 Hz), 7.06 (1H, t, J = 8.8 Hz);
MS (FAB) m/z: 332 (M)+.。
(実施例9h)5-アセトキシメチル-3-フルオロ-2-(2-フルオロ-4-メトキシベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.74g、1.2mmol)、塩化メチレン(7.5mL)、トリクロロアセトニトリル(0.36mL、3.6mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(20μL、0.13mmol)を用い、実施例1hと同様の方法により、イミダートを調製した。得られたイミダート(1.2mmol)、実施例9gで得られた化合物(300mg、0.931mmol)、塩化メチレン(7mL)および三フッ化ホウ素-ジエチルエーテル錯体(60μL、0.47mmol)を用い、実施例1hと同様の方法により、標記化合物の粗生成物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.74g、1.2mmol)、塩化メチレン(7.5mL)、トリクロロアセトニトリル(0.36mL、3.6mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(20μL、0.13mmol)を用い、実施例1hと同様の方法により、イミダートを調製した。得られたイミダート(1.2mmol)、実施例9gで得られた化合物(300mg、0.931mmol)、塩化メチレン(7mL)および三フッ化ホウ素-ジエチルエーテル錯体(60μL、0.47mmol)を用い、実施例1hと同様の方法により、標記化合物の粗生成物を得た。
(実施例9i)3-フルオロ-2-(2-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例9hで得られた粗生成物、ナトリウムメトキシド(28%メタノール溶液、0.36g、1.9mmol)、テトラヒドロフラン(2mL)およびメタノール(6mL)を用い、実施例7iと同様の方法により標記化合物(342mg)を無色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(塩化メチレン:メタノール、7:1~5:1、V/V)を用いて行った。
1H NMR (400 MHz, CD3OD): δ 1.19 (3H, d, J = 6.3 Hz), 3.34-3.39 (2H, m), 3.42-3.48 (2H, m), 3.73 (3H, s), 3.97 (1H, d, J = 14.9 Hz), 4.01-4.07 (2H, m), 4.56 (2H, s), 4.93-4.95 (1H, m), 6.56 (1H, dd, J = 8.6 Hz, 2.4 Hz), 6.60 (1H, dd, J = 11.8 Hz, 2.3 Hz), 6.80 (1H, d, J = 9.8 Hz), 6.99-7.03 (2H, m);
MS (FAB) m/z: 479 (M+Na)+.。
1H NMR (400 MHz, CD3OD): δ 1.19 (3H, d, J = 6.3 Hz), 3.34-3.39 (2H, m), 3.42-3.48 (2H, m), 3.73 (3H, s), 3.97 (1H, d, J = 14.9 Hz), 4.01-4.07 (2H, m), 4.56 (2H, s), 4.93-4.95 (1H, m), 6.56 (1H, dd, J = 8.6 Hz, 2.4 Hz), 6.60 (1H, dd, J = 11.8 Hz, 2.3 Hz), 6.80 (1H, d, J = 9.8 Hz), 6.99-7.03 (2H, m);
MS (FAB) m/z: 479 (M+Na)+.。
(実施例10)3-クロロ-2-[4-(ジフルオロメトキシ)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.10)
(実施例10a)3-クロロ-4-[4-(ジフルオロメトキシ)ベンゾイル]-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例3bで得られた化合物(1.7g、5.0mmol)を塩化メチレン(20mL)に溶解し、2-メチル-2-ブテン(2.2mL、20mmol)、塩化オキサリル(0.45mL、5.2mmol)およびN,N-ジメチルホルムアミド(0.1mL、0.13mmol)を用いて、実施例1cと同様の方法により標記化合物の粗生成物(1.8g)を得た。
実施例3bで得られた化合物(1.7g、5.0mmol)を塩化メチレン(20mL)に溶解し、2-メチル-2-ブテン(2.2mL、20mmol)、塩化オキサリル(0.45mL、5.2mmol)およびN,N-ジメチルホルムアミド(0.1mL、0.13mmol)を用いて、実施例1cと同様の方法により標記化合物の粗生成物(1.8g)を得た。
(実施例10b)3-クロロ-4-[4-(ジフルオロメトキシ)ベンゾイル]-5-ヒドロキシ安息香酸エチル
実施例10aで得られた粗生成物(1.8g、6.5mmol)をN-メチルモルホリン(20mL)に溶解し、無水硫酸ナトリウム(1.8g)およびヨウ素(1.5g、6.0mmol)を用いて、実施例1dと同様の方法により標記化合物の粗生成物(1.9g)を得た。
実施例10aで得られた粗生成物(1.8g、6.5mmol)をN-メチルモルホリン(20mL)に溶解し、無水硫酸ナトリウム(1.8g)およびヨウ素(1.5g、6.0mmol)を用いて、実施例1dと同様の方法により標記化合物の粗生成物(1.9g)を得た。
(実施例10c)3-クロロ-2-{1-[4-(ジフルオロメトキシ)フェニル]-1-ヒドロキシメチル}-5-(ヒドロキシメチル)フェノール
実施例10bで得られた粗生成物(1.8g、4.9mmol)をテトラヒドロフラン(55mL)に溶解し、水素化アルミニウムリチウム(0.57g、15mmol)を用いて、実施例1eと同様の方法により、標記化合物(1.3g)を黄色固体として得た。
実施例10bで得られた粗生成物(1.8g、4.9mmol)をテトラヒドロフラン(55mL)に溶解し、水素化アルミニウムリチウム(0.57g、15mmol)を用いて、実施例1eと同様の方法により、標記化合物(1.3g)を黄色固体として得た。
(実施例10d)酢酸 3-クロロ-4-{1-[4-(ジフルオロメトキシ)フェニル]-1-ヒドロキシメチル}-5-ヒドロキシベンジル
実施例10cで得られた化合物(1.3g、4.0mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(0.6g)を用いて、実施例1fと同様の方法により得られた粗生成物をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.99g)を得た。
実施例10cで得られた化合物(1.3g、4.0mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(0.6g)を用いて、実施例1fと同様の方法により得られた粗生成物をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.99g)を得た。
(実施例10e)酢酸 3-クロロ-4-[4-(ジフルオロメトキシ)ベンジル]-5-ヒドロキシベンジル
実施例10dで得られた化合物(0.99g、2.56mmol)をアセトニトリル(10mL)に溶解し、トリエチルシラン(1.3mL、7.9mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.49mL、3.9mmol)を用いて、実施例1gと同様の方法により、標記化合物(0.58g、ただし28%程度の原料を含む)を得た。
実施例10dで得られた化合物(0.99g、2.56mmol)をアセトニトリル(10mL)に溶解し、トリエチルシラン(1.3mL、7.9mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.49mL、3.9mmol)を用いて、実施例1gと同様の方法により、標記化合物(0.58g、ただし28%程度の原料を含む)を得た。
(実施例10f)5-アセトキシメチル-3-クロロ-2-[4-(ジフルオロメトキシ)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.3mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4.3μL、0.03mmol)を用いて、実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.76g、1.0mmol)および実施例10eで得られた化合物(0.20g、実質0.40mmol)を塩化メチレン(5mL)に溶解し、三フッ化ホウ素-ジエチルエーテル錯体(63μL、0.50mmol)を用いて、実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.3mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4.3μL、0.03mmol)を用いて、実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.76g、1.0mmol)および実施例10eで得られた化合物(0.20g、実質0.40mmol)を塩化メチレン(5mL)に溶解し、三フッ化ホウ素-ジエチルエーテル錯体(63μL、0.50mmol)を用いて、実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例10g)3-クロロ-2-[4-(ジフルオロメトキシ)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例10fで得られた混合物(0.40mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて、実施例3iと同様の方法により、標記化合物(32mg)を無色固体として得た。
1H NMR (500 MHz, CD3OD): δ 1.21 (3H, d, J = 6.4 Hz), 3.34-3.46 (4H, m), 4.15(1H, d, J = 14.3 Hz), 4.25 (1H, d, J = 14.3 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.3 Hz), 6.70 (1H, t, J = 74.4 Hz), 6.96 (2H, d, J = 8.6 Hz), 7.10 (1H, s), 7.13 (1H, s), 7.29 (2H, d, J = 8.6 Hz);
MS (FAB) m/z: 491 (M+H)+, 513 (M+Na)+.。
実施例10fで得られた混合物(0.40mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて、実施例3iと同様の方法により、標記化合物(32mg)を無色固体として得た。
1H NMR (500 MHz, CD3OD): δ 1.21 (3H, d, J = 6.4 Hz), 3.34-3.46 (4H, m), 4.15(1H, d, J = 14.3 Hz), 4.25 (1H, d, J = 14.3 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.3 Hz), 6.70 (1H, t, J = 74.4 Hz), 6.96 (2H, d, J = 8.6 Hz), 7.10 (1H, s), 7.13 (1H, s), 7.29 (2H, d, J = 8.6 Hz);
MS (FAB) m/z: 491 (M+H)+, 513 (M+Na)+.。
(実施例11)3-フルオロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.2)
(実施例11a)3-フルオロ-4-[4-(2-フルオロエチル)ベンゾイル]-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例1bで得られた粗生成物(2.0g、6.2mmol)を塩化メチレン(18mL)に溶解し、ジエチルアミノ硫黄三フッ化物(2.5mL、19mmol)を用いて、実施例3bと同様の方法により、標記化合物(1.6g)を得た。
実施例1bで得られた粗生成物(2.0g、6.2mmol)を塩化メチレン(18mL)に溶解し、ジエチルアミノ硫黄三フッ化物(2.5mL、19mmol)を用いて、実施例3bと同様の方法により、標記化合物(1.6g)を得た。
(実施例11b)3-フルオロ-4-[4-(2-フルオロエチル)ベンゾイル]-5-ヒドロキシ安息香酸エチル
実施例11aで得られた化合物(1.6g、4.9mmol)をN-メチルモルホリン(16mL)に溶解し、無水硫酸ナトリウム(1.6g)およびヨウ素(1.4g、5.4mmol)を用いて、実施例3cと同様の方法により標記化合物の粗生成物(1.6g)を得た。
実施例11aで得られた化合物(1.6g、4.9mmol)をN-メチルモルホリン(16mL)に溶解し、無水硫酸ナトリウム(1.6g)およびヨウ素(1.4g、5.4mmol)を用いて、実施例3cと同様の方法により標記化合物の粗生成物(1.6g)を得た。
(実施例11c)3-フルオロ-4-{1-[4-(2-フルオロエチル)フェニル]-1-ヒドロキシメチル}-5-ヒドロキシ安息香酸エチル
実施例11bで得られた粗生成物(1.6g、4.9mmol)をエタノール(20mL)に溶解し、水素化ホウ素ナトリウム(0.37g、9.8mmol)を用いて、実施例3dと同様の方法により標記化合物(0.83g)を無色固体として得た。
実施例11bで得られた粗生成物(1.6g、4.9mmol)をエタノール(20mL)に溶解し、水素化ホウ素ナトリウム(0.37g、9.8mmol)を用いて、実施例3dと同様の方法により標記化合物(0.83g)を無色固体として得た。
(実施例11d)3-フルオロ-4-[4-(2-フルオロエチル)ベンジル]-5-ヒドロキシ安息香酸エチル
実施例11cで得られた化合物(0.83g、2.5mmol)をエタノール(15mL)に溶解し、10%塩化水素メタノール(0.5mL、2.5mmol)および10%パラジウム炭素触媒(含水、0.5g)を用い、実施例3eと同様の方法により、標記化合物の粗生成物を得た。得られた粗生成物をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、5:1、V/V)を用いて精製して、標記化合物(0.37g)を無色固体として得た。
実施例11cで得られた化合物(0.83g、2.5mmol)をエタノール(15mL)に溶解し、10%塩化水素メタノール(0.5mL、2.5mmol)および10%パラジウム炭素触媒(含水、0.5g)を用い、実施例3eと同様の方法により、標記化合物の粗生成物を得た。得られた粗生成物をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、5:1、V/V)を用いて精製して、標記化合物(0.37g)を無色固体として得た。
(実施例11e)3-フルオロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェノール
実施例11dで得られた化合物(0.37g、1.2mmol)をテトラヒドロフラン(15mL)に溶解し、水素化アルミニウムリチウム(0.13g、3.5mmol)を用い、実施例3fと同様の方法により標記化合物(0.33g)を粗生成物として得た。
実施例11dで得られた化合物(0.37g、1.2mmol)をテトラヒドロフラン(15mL)に溶解し、水素化アルミニウムリチウム(0.13g、3.5mmol)を用い、実施例3fと同様の方法により標記化合物(0.33g)を粗生成物として得た。
(実施例11f)酢酸 3-フルオロ-4-[4-(2-フルオロエチル)ベンジル]-5-ヒドロキシベンジル
実施例11eで得られた粗生成物(0.33g、1.2mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を用い、実施例3gと同様の方法により標記化合物(0.35g)を微褐色固体として得た。
1H NMR (400 MHz, CDCl3):δ 2.11 (3H, s), 2.97 (2H, dt, J = 22.8, 6.5 Hz), 3.99 (2H, s), 4.60 (2H, dt, J = 46.9, 6.5 Hz), 5.01 (2H, s), 6.60 (1H, s), 6.69 (1H, d, J = 9.8 Hz), 7.14 (2H, d, J = 8.1 Hz), 7.23 (2H, d, J = 8.1 Hz);
MS (FAB) m/z: 320 (M)+.。
実施例11eで得られた粗生成物(0.33g、1.2mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を用い、実施例3gと同様の方法により標記化合物(0.35g)を微褐色固体として得た。
1H NMR (400 MHz, CDCl3):δ 2.11 (3H, s), 2.97 (2H, dt, J = 22.8, 6.5 Hz), 3.99 (2H, s), 4.60 (2H, dt, J = 46.9, 6.5 Hz), 5.01 (2H, s), 6.60 (1H, s), 6.69 (1H, d, J = 9.8 Hz), 7.14 (2H, d, J = 8.1 Hz), 7.23 (2H, d, J = 8.1 Hz);
MS (FAB) m/z: 320 (M)+.。
(実施例11g)5-アセトキシメチル-3-フルオロ-2-[4-(2-フルオロエチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.30mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4μL、0.03mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.75g、1.0mmol)、実施例11fで得られた化合物(0.25g、0.78mmol)および三フッ化ホウ素-ジエチルエーテル錯体(39μL、0.39mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.30mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4μL、0.03mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.75g、1.0mmol)、実施例11fで得られた化合物(0.25g、0.78mmol)および三フッ化ホウ素-ジエチルエーテル錯体(39μL、0.39mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例11h)3-フルオロ-2-[4-(2-フルオロエチル)ベンジル]-5-ヒドロキシメチル-フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例11gで得られた混合物(0.78mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて、実施例3iと同様の方法により、標記化合物(0.21g)を無色固体として得た。
1H NMR (500 MHz, CD3OD): δ 1.20 (3H, d, J = 6.4 Hz), 2.90 (2H, dt, J = 22.9, 6.6 Hz), 3.34-3.50 (4H, m), 3.97 (1H, d, J = 14.2 Hz), 4.05-4.06 (1H, m), 4.07 (1H, d, J = 7.8 Hz), 4.52 (2H, dt, J = 47.4, 6.6 Hz), 4.54 (2H, s), 4.93 (1H, d, J = 7.4 Hz), 6.78 (1H, d, J = 9.8 Hz), 6.97 (1H, s), 7.08 (2H, d, J = 7.9 Hz), 7.21 (2H, d, J = 7.9 Hz);
MS (FAB) m/z: 493 (M+K)+.。
実施例11gで得られた混合物(0.78mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて、実施例3iと同様の方法により、標記化合物(0.21g)を無色固体として得た。
1H NMR (500 MHz, CD3OD): δ 1.20 (3H, d, J = 6.4 Hz), 2.90 (2H, dt, J = 22.9, 6.6 Hz), 3.34-3.50 (4H, m), 3.97 (1H, d, J = 14.2 Hz), 4.05-4.06 (1H, m), 4.07 (1H, d, J = 7.8 Hz), 4.52 (2H, dt, J = 47.4, 6.6 Hz), 4.54 (2H, s), 4.93 (1H, d, J = 7.4 Hz), 6.78 (1H, d, J = 9.8 Hz), 6.97 (1H, s), 7.08 (2H, d, J = 7.9 Hz), 7.21 (2H, d, J = 7.9 Hz);
MS (FAB) m/z: 493 (M+K)+.。
(実施例12)3-フルオロ-5-ヒドロキシメチル-2-(4-トリフルオロメトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.3)
(実施例12a)3-ヒドロキシ-5-オキソ-4-[4-(トリフルオロメトキシ)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(1.3g、7.3mmol)および塩化4-(トリフルオロメトキシ)ベンゾイル(J.Med.Chem.,45(14),2002,3112-3129.)(1.6g、7.3mmol)を用い、実施例1bと同様の方法により、標記化合物の粗生成物(2.4g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(1.3g、7.3mmol)および塩化4-(トリフルオロメトキシ)ベンゾイル(J.Med.Chem.,45(14),2002,3112-3129.)(1.6g、7.3mmol)を用い、実施例1bと同様の方法により、標記化合物の粗生成物(2.4g)を得た。
(実施例12b)3-フルオロ-5-オキソ-4-[4-(トリフルオロメトキシ)ベンゾイル]シクロヘキサ-3-エンカルボン酸 エチルエステル
実施例12aで得られた粗生成物(1.8g、5.4mmol)を塩化メチレン(15mL)に溶解し、ジエチルアミノ硫黄三フッ化物(2.1mL、16mmol)を用いて実施例3bと同様の方法により標記化合物(1.5g)を黄色油状物質として得た。
実施例12aで得られた粗生成物(1.8g、5.4mmol)を塩化メチレン(15mL)に溶解し、ジエチルアミノ硫黄三フッ化物(2.1mL、16mmol)を用いて実施例3bと同様の方法により標記化合物(1.5g)を黄色油状物質として得た。
(実施例12c)3-フルオロ-5-ヒドロキシ-4-[4-(トリフルオロメトキシ)ベンゾイル]安息香酸エチル
実施例12bで得られた化合物(1.5g、4.5mmol)をN-メチルモルホリン(18mL)に溶解し、無水硫酸ナトリウム(1.5g)およびヨウ素(1.3g、5.0mmol)を用いて、実施例1dと同様の方法により、標記化合物の粗生成物(1.5g)を得た。
実施例12bで得られた化合物(1.5g、4.5mmol)をN-メチルモルホリン(18mL)に溶解し、無水硫酸ナトリウム(1.5g)およびヨウ素(1.3g、5.0mmol)を用いて、実施例1dと同様の方法により、標記化合物の粗生成物(1.5g)を得た。
(実施例12d)3-フルオロ-5-ヒドロキシ-4-{1-ヒドロキシ-1-[4-(トリフルオロメトキシ)フェニル]メチル}安息香酸エチル
実施例12cで得られた粗生成物(1.5g、4.5mmol)をエタノール(15mL)に溶解し、水素化ホウ素ナトリウム(0.34g、9.0mmol)を用いて実施例3dと同様の方法により標記化合物(1.0g)を得た。
実施例12cで得られた粗生成物(1.5g、4.5mmol)をエタノール(15mL)に溶解し、水素化ホウ素ナトリウム(0.34g、9.0mmol)を用いて実施例3dと同様の方法により標記化合物(1.0g)を得た。
(実施例12e)3-フルオロ-5-ヒドロキシメチル-2-{1-ヒドロキシ-1-[4-(トリフルオロメトキシ)フェニル]メチル}フェノール
実施例12dで得られた化合物(1.0g、3.0mmol)をテトラヒドロフラン(20mL)に溶解し、水素化アルミニウムリチウム(0.46g、12mmol)を用いて、実施例1eと同様の方法(ただし、精製は行っていない。)により、標記化合物(1.0g)を粗生成物として得た。
実施例12dで得られた化合物(1.0g、3.0mmol)をテトラヒドロフラン(20mL)に溶解し、水素化アルミニウムリチウム(0.46g、12mmol)を用いて、実施例1eと同様の方法(ただし、精製は行っていない。)により、標記化合物(1.0g)を粗生成物として得た。
(実施例12f)酢酸 3-フルオロ-5-ヒドロキシ-4-{1-ヒドロキシ-1-[4-(トリフルオロメトキシ)フェニル]メチル}ベンジル
実施例12eで得られた粗生成物(1.0g、3.0mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を用いて、実施例1fと同様の方法により得られた粗生成物をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.65g)を得た。
実施例12eで得られた粗生成物(1.0g、3.0mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を用いて、実施例1fと同様の方法により得られた粗生成物をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、4:1、V/V)を用いて精製して、標記化合物(0.65g)を得た。
(実施例12g)酢酸 3-フルオロ-5-ヒドロキシ-4-[4-(トリフルオロメトキシ)ベンジル]ベンジル
実施例12fで得られた化合物(0.65g、1.7mmol)をアセトニトリル(10mL)に溶解し、トリエチルシラン(0.83mL、5.2mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.33mL、2.6mmol)を用いて実施例1gと同様の方法により、標記化合物(0.18g)を得た。
1H NMR (400 MHz, CDCl3): δ 2.11 (3H, s), 4.00 (2H, s), 5.01 (2H, s), 5.19 (1H, brs), 6.60 (1H, s), 6.70 (1H, dd, J = 9.6, 1.4 Hz), 7.11 (2H, d, J = 8.1 Hz), 7.30 (2H, d, J = 8.1 Hz);
MS (FAB) m/z: 358 (M)+.。
実施例12fで得られた化合物(0.65g、1.7mmol)をアセトニトリル(10mL)に溶解し、トリエチルシラン(0.83mL、5.2mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.33mL、2.6mmol)を用いて実施例1gと同様の方法により、標記化合物(0.18g)を得た。
1H NMR (400 MHz, CDCl3): δ 2.11 (3H, s), 4.00 (2H, s), 5.01 (2H, s), 5.19 (1H, brs), 6.60 (1H, s), 6.70 (1H, dd, J = 9.6, 1.4 Hz), 7.11 (2H, d, J = 8.1 Hz), 7.30 (2H, d, J = 8.1 Hz);
MS (FAB) m/z: 358 (M)+.。
(実施例12h)5-アセトキシメチル-3-フルオロ-2-[4-(トリフルオロメトキシ)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.3mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4.3μL、0.03mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.76g、1.0mmol)および実施例12gで得られた化合物(0.18g、0.50mmol)を塩化メチレン(5mL)に溶解し、三フッ化ホウ素-ジエチルエーテル錯体(63μL、0.50mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.3mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4.3μL、0.03mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.76g、1.0mmol)および実施例12gで得られた化合物(0.18g、0.50mmol)を塩化メチレン(5mL)に溶解し、三フッ化ホウ素-ジエチルエーテル錯体(63μL、0.50mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例12i)3-フルオロ-5-ヒドロキシメチル-2-[4-(トリフルオロメトキシ)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例12hで得られた混合物(0.50mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(0.12g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, d, J = 6.7 Hz), 3.36-3.51 (4H, m), 4.04 (1H, d, J = 14.2 Hz), 4.06-4.07 (1H, m), 4.11 (1H, d, J = 14.2 Hz), 4.55 (2H, s), 4.96 (1H, d, J = 7.5 Hz), 6.81 (1H, d, J = 10.2 Hz), 7.00 (1H, s), 7.11 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz);
MS (FAB) m/z: 515 (M+Na)+.。
実施例12hで得られた混合物(0.50mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(0.12g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, d, J = 6.7 Hz), 3.36-3.51 (4H, m), 4.04 (1H, d, J = 14.2 Hz), 4.06-4.07 (1H, m), 4.11 (1H, d, J = 14.2 Hz), 4.55 (2H, s), 4.96 (1H, d, J = 7.5 Hz), 6.81 (1H, d, J = 10.2 Hz), 7.00 (1H, s), 7.11 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz);
MS (FAB) m/z: 515 (M+Na)+.。
(実施例13)3-フルオロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド(表1のNo.25)
(実施例13a)5-アセトキシメチル-3-フルオロ-2-(4-メチルベンジル)フェニル 4-デオキシ-2,3,6-トリ-O-ベンゾイル-β-D-グルコピラノシド
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(Liebig Ann.Chem.,1992,7,747-758.)(200mg、0.42mmol)、トリクロロアセトニトリル(125μL、1.25mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、酢酸 3-フルオロ-4-(4-メチルベンジル)-5-ヒドロキシベンジル(WO2008/016132(PCT/JP2007/65231))(100mg、0.35mmol)、三フッ化ホウ素-ジエチルエーテル錯体(44μL、0.35mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(285mg)を得た。
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(Liebig Ann.Chem.,1992,7,747-758.)(200mg、0.42mmol)、トリクロロアセトニトリル(125μL、1.25mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、酢酸 3-フルオロ-4-(4-メチルベンジル)-5-ヒドロキシベンジル(WO2008/016132(PCT/JP2007/65231))(100mg、0.35mmol)、三フッ化ホウ素-ジエチルエーテル錯体(44μL、0.35mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(285mg)を得た。
(実施例13b)3-フルオロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド
実施例13aで得られた粗生成物(285mg、0.35mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(48mg、0.35mmol)を用い、実施例4jと同様の方法により、標記化合物(90mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.47 (1H, q, J = 11.8 Hz), 1.95-2.00 (1H, m), 2.25 (3H, s), 3.39 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.74 (2H, m), 3.94 (1H, d, J = 13.7 Hz), 4.06 (1H, d, J = 13.7 Hz), 4.55 (2H, s), 4.89 (1H, d, J = 7.4 Hz), 6.78 (1H, d, J = 9.8 Hz), 6.98 (1H, s), 7.00 (2H, d, J = 8.0 Hz), 7.14 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 393 (M+H)+, 415 (M+Na)+.。
実施例13aで得られた粗生成物(285mg、0.35mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(48mg、0.35mmol)を用い、実施例4jと同様の方法により、標記化合物(90mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.47 (1H, q, J = 11.8 Hz), 1.95-2.00 (1H, m), 2.25 (3H, s), 3.39 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.74 (2H, m), 3.94 (1H, d, J = 13.7 Hz), 4.06 (1H, d, J = 13.7 Hz), 4.55 (2H, s), 4.89 (1H, d, J = 7.4 Hz), 6.78 (1H, d, J = 9.8 Hz), 6.98 (1H, s), 7.00 (2H, d, J = 8.0 Hz), 7.14 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 393 (M+H)+, 415 (M+Na)+.。
(実施例14)3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド(表1のNo.26)
(実施例14a)5-アセトキシメチル-3-クロロ-2-(4-メチルベンジル)フェニル 4-デオキシ-2,3,6-トリ-O-ベンゾイル-β-D-グルコピラノシド
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(188mg、0.39mmol)、トリクロロアセトニトリル(120μL、1.20mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、実施例4hで得られた化合物(100mg、0.33mmol)、三フッ化ホウ素-ジエチルエーテル錯体(41μL、0.33mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(291mg)を得た。
(実施例14b)3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド
実施例14aで得られた粗生成物(291mg、0.33mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(46mg、0.33mmol)を用い、実施例4jと同様の方法により、標記化合物(106mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.46 (1H, q, J = 11.8 Hz), 1.95-2.00 (1H, m), 2.25 (3H, s), 3.37 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.72 (2H, m), 4.09 (1H, d, J = 14.5 Hz), 4.24 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 7.00 (2H, d, J = 7.8 Hz), 7.10 (2H, J = 7.8 Hz), 7.13 (2H, s);
MS (FAB) m/z: 431 (M+Na)+.。
実施例14aで得られた粗生成物(291mg、0.33mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(46mg、0.33mmol)を用い、実施例4jと同様の方法により、標記化合物(106mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.46 (1H, q, J = 11.8 Hz), 1.95-2.00 (1H, m), 2.25 (3H, s), 3.37 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.72 (2H, m), 4.09 (1H, d, J = 14.5 Hz), 4.24 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 7.00 (2H, d, J = 7.8 Hz), 7.10 (2H, J = 7.8 Hz), 7.13 (2H, s);
MS (FAB) m/z: 431 (M+Na)+.。
(実施例15)2-(4-エチルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド(表1のNo.24)
(実施例15a)5-アセトキシメチル-2-(4-エチルベンジル)-3-フルオロフェニル 4-デオキシ-2,3,6-トリ-O-ベンゾイル-β-D-グルコピラノシド
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(189mg、0.40mmol)、トリクロロアセトニトリル(120μL、1.20mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、酢酸 4-(4-エチルベンジル)-3-フルオロ-5-ヒドロキシベンジル(WO2008/016132(PCT/JP2007/65231))(100mg、0.33mmol)、三フッ化ホウ素-ジエチルエーテル錯体(42μL、0.33mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(323mg)を得た。
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(189mg、0.40mmol)、トリクロロアセトニトリル(120μL、1.20mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、酢酸 4-(4-エチルベンジル)-3-フルオロ-5-ヒドロキシベンジル(WO2008/016132(PCT/JP2007/65231))(100mg、0.33mmol)、三フッ化ホウ素-ジエチルエーテル錯体(42μL、0.33mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(323mg)を得た。
(実施例15b)2-(4-エチルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド
実施例15aで得られた粗生成物(323mg、0.33mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(46mg、0.33mmol)を用い、実施例4jと同様の方法により、標記化合物(100mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.17 (3H, t, J = 7.6 Hz), 1.47 (1H, q, J = 11.7 Hz), 1.95-2.00 (1H, m), 2.56 (2H, q, J = 7.6 Hz), 3.40 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.67-3.74 (2H, m), 3.95 (1H, d, J = 14.5 Hz), 4.07 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.90 (1H, d, J = 7.5 Hz), 6.78 (1H, d, J = 9.7 Hz), 6.99 (1H, s), 7.03 (2H, d, J = 8.0 Hz), 7.17 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 407 (M+H)+, 429 (M+Na)+.。
実施例15aで得られた粗生成物(323mg、0.33mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(46mg、0.33mmol)を用い、実施例4jと同様の方法により、標記化合物(100mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.17 (3H, t, J = 7.6 Hz), 1.47 (1H, q, J = 11.7 Hz), 1.95-2.00 (1H, m), 2.56 (2H, q, J = 7.6 Hz), 3.40 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.67-3.74 (2H, m), 3.95 (1H, d, J = 14.5 Hz), 4.07 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.90 (1H, d, J = 7.5 Hz), 6.78 (1H, d, J = 9.7 Hz), 6.99 (1H, s), 7.03 (2H, d, J = 8.0 Hz), 7.17 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 407 (M+H)+, 429 (M+Na)+.。
(実施例16)3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド(表1のNo.27)
(実施例16a)5-アセトキシメチル-3-クロロ-2-(4-エチルベンジル)フェニル 4-デオキシ-2,3,6-トリ-O-ベンゾイル-β-D-グルコピラノシド
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(190mg、0.40mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.200mL、1.98mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(248mg)、実施例2fで得られた化合物(100mg、0.31mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.050mL、0.40mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(270mg)を得た。
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(190mg、0.40mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.200mL、1.98mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(248mg)、実施例2fで得られた化合物(100mg、0.31mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.050mL、0.40mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(270mg)を得た。
(実施例16b)3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド
実施例16aで得られた粗生成物(270mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(550mg、3.98mmol)を用い実施例2hと同様の方法により、標記化合物(126mg)を無色固体として得た。ただし、固体化はヘキサン/酢酸エチルから行った。
1H NMR (400 MHz, CD3OD): δ 1.18 (3H, t, J = 7.5 Hz), 1.46 (1H, q, J = 11.8 Hz), 1.95-2.00 (1H, m), 2.56 (2H, q, J = 7.5 Hz), 3.37 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.72 (2H, m), 4.10 (1H, d, J = 14.5 Hz), 4.25 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.89 (1H, d, J = 7.4 Hz), 7.03 (2H, d, J = 8.4 Hz), 7.09 (1H, s), 7.13 (1H, s), 7.15 (2H, d, J = 8.4 Hz);
MS (FAB) m/z: 445 (M+Na)+.。
実施例16aで得られた粗生成物(270mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(550mg、3.98mmol)を用い実施例2hと同様の方法により、標記化合物(126mg)を無色固体として得た。ただし、固体化はヘキサン/酢酸エチルから行った。
1H NMR (400 MHz, CD3OD): δ 1.18 (3H, t, J = 7.5 Hz), 1.46 (1H, q, J = 11.8 Hz), 1.95-2.00 (1H, m), 2.56 (2H, q, J = 7.5 Hz), 3.37 (1H, dd, J = 9.0, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.72 (2H, m), 4.10 (1H, d, J = 14.5 Hz), 4.25 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.89 (1H, d, J = 7.4 Hz), 7.03 (2H, d, J = 8.4 Hz), 7.09 (1H, s), 7.13 (1H, s), 7.15 (2H, d, J = 8.4 Hz);
MS (FAB) m/z: 445 (M+Na)+.。
(実施例17)2-(4-シクロプロピルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド(表1のNo.23)
(実施例17a)5-アセトキシメチル-2-(4-シクロプロピルベンジル)-3-フルオロフェニル 4-デオキシ-2,3,6-トリ-O-ベンゾイル-β-D-グルコピラノシド
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(192mg、0.40mmol)、トリクロロアセトニトリル(120μL、1.20mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、酢酸 4-(4-シクロプロピルベンジル)-3-フルオロ-5-ヒドロキシベンジル(WO2008/016132(PCT/JP2007/65231))(100mg、0.32mmol)、三フッ化ホウ素-ジエチルエーテル錯体(40μL、0.32mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(301mg)を得た。
4-デオキシ-2,3,6-トリ-4-O-ベンゾイル-D-グルコピラノシド(192mg、0.40mmol)、トリクロロアセトニトリル(120μL、1.20mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、酢酸 4-(4-シクロプロピルベンジル)-3-フルオロ-5-ヒドロキシベンジル(WO2008/016132(PCT/JP2007/65231))(100mg、0.32mmol)、三フッ化ホウ素-ジエチルエーテル錯体(40μL、0.32mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(301mg)を得た。
(実施例17b)2-(4-シクロプロピルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド
実施例17aで得られた粗生成物(301mg、0.32mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(44mg、0.32mmol)を用い、実施例4jと同様の方法により、標記化合物(107mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 0.57-0.61 (2H, m), 0.85-0.90 (2H, m), 1.47 (1H, q, J = 11.8 Hz), 1.78-1.85 (1H, m), 1.95-2.00 (1H, m), 3.39 (1H, dd, J = 9.0 Hz, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.74 (2H, m), 3.93 (1H, d, J = 14.5 Hz), 4.05 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.89 (1H, d, J = 7.8 Hz), 6.77 (1H, d, J = 10.2 Hz), 6.91 (2H, d, J = 8.0 Hz), 6.98 (1H, s), 7.14 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 419 (M+H)+, 441 (M+Na)+.。
実施例17aで得られた粗生成物(301mg、0.32mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(44mg、0.32mmol)を用い、実施例4jと同様の方法により、標記化合物(107mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 0.57-0.61 (2H, m), 0.85-0.90 (2H, m), 1.47 (1H, q, J = 11.8 Hz), 1.78-1.85 (1H, m), 1.95-2.00 (1H, m), 3.39 (1H, dd, J = 9.0 Hz, 7.8 Hz), 3.57-3.59 (2H, m), 3.66-3.74 (2H, m), 3.93 (1H, d, J = 14.5 Hz), 4.05 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.89 (1H, d, J = 7.8 Hz), 6.77 (1H, d, J = 10.2 Hz), 6.91 (2H, d, J = 8.0 Hz), 6.98 (1H, s), 7.14 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 419 (M+H)+, 441 (M+Na)+.。
(実施例18)3-フルオロ-5-ヒドロキシメチル-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.4)
(実施例18a)塩化4-(2,2,2-トリフルオロエチル)ベンゾイル
4-(2,2,2-トリフルオロエチル)安息香酸(Liebigs Ann.Chem.,1983,9,1510-1523.)(786mg、3.85mmol)、二塩化オキサリル(0.385mL、4.42mmol)、触媒量のN,N-ジメチルホルムアミドおよびテトラヒドロフラン(6mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(857mg)を得た。
4-(2,2,2-トリフルオロエチル)安息香酸(Liebigs Ann.Chem.,1983,9,1510-1523.)(786mg、3.85mmol)、二塩化オキサリル(0.385mL、4.42mmol)、触媒量のN,N-ジメチルホルムアミドおよびテトラヒドロフラン(6mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(857mg)を得た。
(実施例18b)3-ヒドロキシ-5-オキソ-4-[4-(2,2,2-トリフルオロエチル)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(709mg、3.85mmol)、実施例18aで得られた粗生成物(857mg、3.85mmol)、アセトニトリル(10mL)、トリエチルアミン(1.61mL、11.5mmol)およびトリメチルシリルシアニド(0.062mL、0.46mmol)を用いて、実施例1bと同様の方法により、標記化合物の粗生成物(1.42g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(709mg、3.85mmol)、実施例18aで得られた粗生成物(857mg、3.85mmol)、アセトニトリル(10mL)、トリエチルアミン(1.61mL、11.5mmol)およびトリメチルシリルシアニド(0.062mL、0.46mmol)を用いて、実施例1bと同様の方法により、標記化合物の粗生成物(1.42g)を得た。
(実施例18c)3-フルオロ-5-オキソ-4-[4-(2,2,2-トリフルオロエチル)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
実施例18bで得られた粗生成物(1.42g、3.61mmol)、塩化メチレン(15mL)およびジエチルアミノ硫黄三フッ化物(1.51mL、11.5mmol)を用い、実施例3bと同様の方法により標記化合物(953mg)を白色固体として得た。
実施例18bで得られた粗生成物(1.42g、3.61mmol)、塩化メチレン(15mL)およびジエチルアミノ硫黄三フッ化物(1.51mL、11.5mmol)を用い、実施例3bと同様の方法により標記化合物(953mg)を白色固体として得た。
(実施例18d)3-フルオロ-5-ヒドロキシ-4-[4-(2,2,2-トリフルオロエチル)ベンゾイル]安息香酸エチル
実施例18cで得られた化合物(953mg、2.56mmol)、N-メチルモルホリン(12mL)、無水硫酸ナトリウム(7.27g)およびヨウ素(779mg、3.07mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(614mg)を得た。
(実施例18e)3-フルオロ-5-ヒドロキシメチル-2-[1-ヒドロキシ-1-{4-(2,2,2-トリフルオロエチル)フェニル}メチル]フェノール
実施例18dで得られた化合物(610mg、1.65mmol)をテトラヒドロフラン(15mL)に溶解して0℃に冷却後、水素化リチウムアルミニウム(187mg、4.93mmol)を加え、室温で30分間攪拌した。再び0℃に冷却後、反応液に水(0.19mL)、5mol/L水酸化ナトリウム水溶液(0.19mL)および水(0.57mL)を順次加え、室温で1時間攪拌した。反応液に酢酸エチル(20mL)を加え、2mol/L塩酸(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去して、標記化合物の粗生成物(540mg)を得た。
実施例18dで得られた化合物(610mg、1.65mmol)をテトラヒドロフラン(15mL)に溶解して0℃に冷却後、水素化リチウムアルミニウム(187mg、4.93mmol)を加え、室温で30分間攪拌した。再び0℃に冷却後、反応液に水(0.19mL)、5mol/L水酸化ナトリウム水溶液(0.19mL)および水(0.57mL)を順次加え、室温で1時間攪拌した。反応液に酢酸エチル(20mL)を加え、2mol/L塩酸(20mL)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去して、標記化合物の粗生成物(540mg)を得た。
(実施例18f)3-フルオロ-5-ヒドロキシメチル-2-[1-メトキシ-1-{4-(2,2,2-トリフルオロエチル)フェニル}メチル]フェノール
実施例18eで得られた粗生成物(540mg、1.64mmol)をメタノール(10mL)に溶解し、パラトルエンスルホン酸一水和物(157mg、0.83mmol)を加え、50℃で2時間攪拌した。反応液を室温に冷却後、トリエチルアミンを加え、減圧下濃縮した。残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~1:4、V/V)を用いて精製して、油状の標記化合物(508mg)を得た。
実施例18eで得られた粗生成物(540mg、1.64mmol)をメタノール(10mL)に溶解し、パラトルエンスルホン酸一水和物(157mg、0.83mmol)を加え、50℃で2時間攪拌した。反応液を室温に冷却後、トリエチルアミンを加え、減圧下濃縮した。残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~1:4、V/V)を用いて精製して、油状の標記化合物(508mg)を得た。
(実施例18g)酢酸 3-フルオロ-5-ヒドロキシ-4-{1-メトキシ-1-[4-(2,2,2-トリフルオロエチル)フェニル]メチル}ベンジル
実施例18fで得られた化合物(508mg、1.48mmol)、テトラヒドロフラン(5mL)、酢酸ビニル(5mL)およびビス(ジブチル塩化スズ)オキシド(325mg、0.59mmol)を用い実施例2eと同様の方法により、油状の標記化合物(525mg)を得た。
実施例18fで得られた化合物(508mg、1.48mmol)、テトラヒドロフラン(5mL)、酢酸ビニル(5mL)およびビス(ジブチル塩化スズ)オキシド(325mg、0.59mmol)を用い実施例2eと同様の方法により、油状の標記化合物(525mg)を得た。
(実施例18h)酢酸 3-フルオロ-5-ヒドロキシ-4-[4-(2,2,2-トリフルオロエチル)ベンジル]ベンジル
実施例18gで得られた化合物(525mg、1.36mmol)、アセトニトリル(10mL)、トリエチルシラン(0.650mL、4.08mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.256mL、2.04mmol)を用い、実施例2fと同様の方法により標記化合物(363mg)を白色固体として得た。
1H NMR (400 MHz, CDCl3):δ 2.11 ( 3H, s ), 3.32 (2H, q, J = 10.8 Hz), 4.00 (2H, s), 5.01 (2H, s), 5.06 (1H, brs), 6.61 (1H, s), 6.70 (1H, d, J = 9.4 Hz ), 7.20 (2H, d, J = 7.8 Hz), 7.27 (2H, d, J = 7.8 Hz);
MS (FAB) m/z: 356 (M)+.。
実施例18gで得られた化合物(525mg、1.36mmol)、アセトニトリル(10mL)、トリエチルシラン(0.650mL、4.08mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.256mL、2.04mmol)を用い、実施例2fと同様の方法により標記化合物(363mg)を白色固体として得た。
1H NMR (400 MHz, CDCl3):δ 2.11 ( 3H, s ), 3.32 (2H, q, J = 10.8 Hz), 4.00 (2H, s), 5.01 (2H, s), 5.06 (1H, brs), 6.61 (1H, s), 6.70 (1H, d, J = 9.4 Hz ), 7.20 (2H, d, J = 7.8 Hz), 7.27 (2H, d, J = 7.8 Hz);
MS (FAB) m/z: 356 (M)+.。
(実施例18i)5-アセトキシメチル-3-フルオロ-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(267mg、0.44mmol)、塩化メチレン(5mL)、トリクロロアセトニトリル(0.221mL、2.19mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(330mg)、実施例18hで得られた化合物(120mg、0.34mmol)、塩化メチレン(5mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.055mL、0.44mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(460mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(267mg、0.44mmol)、塩化メチレン(5mL)、トリクロロアセトニトリル(0.221mL、2.19mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(330mg)、実施例18hで得られた化合物(120mg、0.34mmol)、塩化メチレン(5mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.055mL、0.44mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(460mg)を得た。
(実施例18j)3-フルオロ-5-ヒドロキシメチル-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例18iで得られた粗生成物(460mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(604mg、4.34mmol)を用い実施例2hと同様の方法により、標記化合物(58mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, d, J = 6.3 Hz), 3.34-3.49 (6H, m), 4.01 (1H, d, J = 14.5 Hz), 4.06 (1H, dd, J = 6.3 Hz, 3.7 Hz), 4.09 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.5 Hz), 6.80 (1H, d, J = 9.8 Hz), 6.99 (1H, s), 7.17 (2H, d, J = 8.2 Hz), 7.28 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 529 (M+K)+.。
実施例18iで得られた粗生成物(460mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(604mg、4.34mmol)を用い実施例2hと同様の方法により、標記化合物(58mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, d, J = 6.3 Hz), 3.34-3.49 (6H, m), 4.01 (1H, d, J = 14.5 Hz), 4.06 (1H, dd, J = 6.3 Hz, 3.7 Hz), 4.09 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.5 Hz), 6.80 (1H, d, J = 9.8 Hz), 6.99 (1H, s), 7.17 (2H, d, J = 8.2 Hz), 7.28 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 529 (M+K)+.。
(実施例19)3-クロロ-2-(4-エチル-3-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.18)
(実施例19a)4-アセチル-3-フルオロ安息香酸メチル
トリフルオロメタンスルホン酸 4-アセチル-3-フルオロフェニル(Org.Lett.,2002,4(26),4717-4718.)(8.69g、30.4mmol)をN,N-ジメチルホルムアルデヒド(120mL)に溶解させ、メタノール(24.6mL、606mmol)、酢酸パラジウム(682mg、3.03mmol)、1,3-ビス(ジフェニルホスフィノ)プロパン(1.25g、3.03mmol)およびトリエチルアミン(84.0mL、606mol)を加え、一酸化炭素雰囲気下、室温で16時間攪拌した。減圧下溶媒をある程度除去し、反応液に酢酸エチル(300mL)を加え、1mol/L塩酸水溶液(150mL)および飽和食塩水(100mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、標記化合物(3.67g)を白色固体として得た。
トリフルオロメタンスルホン酸 4-アセチル-3-フルオロフェニル(Org.Lett.,2002,4(26),4717-4718.)(8.69g、30.4mmol)をN,N-ジメチルホルムアルデヒド(120mL)に溶解させ、メタノール(24.6mL、606mmol)、酢酸パラジウム(682mg、3.03mmol)、1,3-ビス(ジフェニルホスフィノ)プロパン(1.25g、3.03mmol)およびトリエチルアミン(84.0mL、606mol)を加え、一酸化炭素雰囲気下、室温で16時間攪拌した。減圧下溶媒をある程度除去し、反応液に酢酸エチル(300mL)を加え、1mol/L塩酸水溶液(150mL)および飽和食塩水(100mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製して、標記化合物(3.67g)を白色固体として得た。
(実施例19b)3-フルオロ-4-(1-ヒドロキシエチル)安息香酸メチル
実施例19aで得られた化合物(3.67g、18.7mmol)をメタノール(40mL)に溶解して0℃に冷却後、水素化ホウ素ナトリウム(850mg、22.5mmol)を加え、0℃で1時間攪拌した。反応液に飽和塩化アンモニウム水溶液(2mL)を加え、減圧下、溶媒を除去した。残渣に酢酸エチル(50mL)を加え、飽和塩化アンモニウム水溶液(50mL)、飽和水酸化ナトリウム水溶液(50mL)および飽和食塩水(50mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:2、V/V)を用いて精製して、油状の標記化合物(3.59g)を得た。
実施例19aで得られた化合物(3.67g、18.7mmol)をメタノール(40mL)に溶解して0℃に冷却後、水素化ホウ素ナトリウム(850mg、22.5mmol)を加え、0℃で1時間攪拌した。反応液に飽和塩化アンモニウム水溶液(2mL)を加え、減圧下、溶媒を除去した。残渣に酢酸エチル(50mL)を加え、飽和塩化アンモニウム水溶液(50mL)、飽和水酸化ナトリウム水溶液(50mL)および飽和食塩水(50mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:2、V/V)を用いて精製して、油状の標記化合物(3.59g)を得た。
(実施例19c)4-エチル-3-フルオロ安息香酸
実施例19bで得られた化合物(3.67g、18.7mmol)をメタノール(35mL)に溶解し、2mol/L塩酸(1.09mL)および10%wetパラジウム/炭素(600mg)を加え、水素雰囲気下、室温で3時間攪拌した。反応液に対してセライトで濾過を行うことでパラジウムを除去した。続いて、反応液に5mol/L水酸化ナトリウム水溶液(8mL)と適量のテトラヒドロフランを加え、50℃で1時間攪拌した。反応液を0℃に冷却後、適量の2mol/L塩酸を加えてpHを3未満に調製し、減圧下溶媒を除去した。残渣に酢酸エチル(50mL)を加え、飽和食塩水(50mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。得られた残渣をヘキサンで洗浄し、濾過して得られた固体を減圧下乾燥して、標記化合物(2.98g)を白色固体として得た。
実施例19bで得られた化合物(3.67g、18.7mmol)をメタノール(35mL)に溶解し、2mol/L塩酸(1.09mL)および10%wetパラジウム/炭素(600mg)を加え、水素雰囲気下、室温で3時間攪拌した。反応液に対してセライトで濾過を行うことでパラジウムを除去した。続いて、反応液に5mol/L水酸化ナトリウム水溶液(8mL)と適量のテトラヒドロフランを加え、50℃で1時間攪拌した。反応液を0℃に冷却後、適量の2mol/L塩酸を加えてpHを3未満に調製し、減圧下溶媒を除去した。残渣に酢酸エチル(50mL)を加え、飽和食塩水(50mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。得られた残渣をヘキサンで洗浄し、濾過して得られた固体を減圧下乾燥して、標記化合物(2.98g)を白色固体として得た。
(実施例19d)塩化4-エチル-3-フルオロベンゾイル
実施例19cで得られた化合物(730mg、4.34mmol)、二塩化オキサリル(0.43mL、4.95mmol)、触媒量のN,N-ジメチルホルムアミドおよびテトラヒドロフラン(6mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(810mg)を無色油状物質として得た。
実施例19cで得られた化合物(730mg、4.34mmol)、二塩化オキサリル(0.43mL、4.95mmol)、触媒量のN,N-ジメチルホルムアミドおよびテトラヒドロフラン(6mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(810mg)を無色油状物質として得た。
(実施例19e)4-(4-エチル-3-フルオロベンゾイル)-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(800mg、4.34mmol)、実施例19dで得られた粗生成物(810mg、4.34mmol)、アセトニトリル(9mL)、トリエチルアミン(1.82mL、13.1mmol)およびトリメチルシリルシアニド(0.070mL、0.52mmol)を用いて、実施例1bと同様の方法により、標記化合物の粗生成物(1.45g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(800mg、4.34mmol)、実施例19dで得られた粗生成物(810mg、4.34mmol)、アセトニトリル(9mL)、トリエチルアミン(1.82mL、13.1mmol)およびトリメチルシリルシアニド(0.070mL、0.52mmol)を用いて、実施例1bと同様の方法により、標記化合物の粗生成物(1.45g)を得た。
(実施例19f)3-クロロ-4-(4-エチル-3-フルオロベンゾイル)-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例19eで得られた粗生成物(1.45g、4.34mmol)、塩化メチレン(18mL)、2-メチル-2-ブテン(1.84mL、17.3mmol)、二塩化オキサリル(0.39mL、4.55mmol)および触媒量のN,N-ジメチルホルムアミドを用い、実施例1cと同様の方法により標記化合物の粗生成物(1.52g)を得た。
実施例19eで得られた粗生成物(1.45g、4.34mmol)、塩化メチレン(18mL)、2-メチル-2-ブテン(1.84mL、17.3mmol)、二塩化オキサリル(0.39mL、4.55mmol)および触媒量のN,N-ジメチルホルムアミドを用い、実施例1cと同様の方法により標記化合物の粗生成物(1.52g)を得た。
(実施例19g)3-クロロ-4-(4-エチル-3-フルオロベンゾイル)-5-ヒドロキシ安息香酸エチル
実施例19fで得られた粗生成物(1.52g、4.34mmol)、N-メチルモルホリン(18mL)、無水硫酸ナトリウム(12.3g)およびヨウ素(1.32g、5.20mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(1.04g)を得た。
実施例19fで得られた粗生成物(1.52g、4.34mmol)、N-メチルモルホリン(18mL)、無水硫酸ナトリウム(12.3g)およびヨウ素(1.32g、5.20mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(1.04g)を得た。
(実施例19h)5-クロロ-4-(4-エチル-3-フルオロフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-安息香酸エチル
実施例19gで得られた化合物(1.04g、2.96mmol)、メタノール(12mL)および水素化ホウ素ナトリウム(220mg、5.82mmol)を用い、実施例2bと同様の方法により、ジオール体粗生成物(1.04g)を得た。
実施例19gで得られた化合物(1.04g、2.96mmol)、メタノール(12mL)および水素化ホウ素ナトリウム(220mg、5.82mmol)を用い、実施例2bと同様の方法により、ジオール体粗生成物(1.04g)を得た。
三フッ化ホウ素-ジエチルエーテル錯体(0.370mL、2.95mmol)、ジオール体粗生成物(1.04g、2.95mmol)およびアセトン(16mL)を用い、実施例6eと同様の方法により、油状の標記化合物(730mg)を得た。
(実施例19i)5-クロロ-4-(4-エチル-3-フルオロフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-イルメタノール
実施例19hで得られた化合物(730mg、1.86mmol)、水素化リチウムアルミニウム(71mg、1.87mmol)およびテトラヒドロフラン(8mL)を用い、実施例2dと同様の方法により、油状の標記化合物の粗生成物(664mg)を得た。
実施例19hで得られた化合物(730mg、1.86mmol)、水素化リチウムアルミニウム(71mg、1.87mmol)およびテトラヒドロフラン(8mL)を用い、実施例2dと同様の方法により、油状の標記化合物の粗生成物(664mg)を得た。
(実施例19j)酢酸 5-クロロ-4-(4-エチル-3-フルオロフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-イルメチル
実施例19iで得られた粗生成物(664mg)、ピリジン(6.6mL)および無水酢酸(1.7mL)を用いて、実施例6gと同様の方法により、油状の標記化合物(720mg)を得た。
実施例19iで得られた粗生成物(664mg)、ピリジン(6.6mL)および無水酢酸(1.7mL)を用いて、実施例6gと同様の方法により、油状の標記化合物(720mg)を得た。
(実施例19k)酢酸 3-クロロ-4-(4-エチル-3-フルオロベンジル)-5-ヒドロキシベンジル
実施例19jで得られた化合物(720mg、1.83mmol)、アセトニトリル(12mL)、トリエチルシラン(0.880mL、5.47mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.350mL、2.79mmol)を用いて、実施例2fと同様の方法により、標記化合物(491mg)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.19 (3H, t, J = 7.7 Hz), 2.12 (3H, s), 2.61 (2H, q, J= 7.7 Hz), 4.13 (2H, s), 5.00 (2H, s), 6.72 (1H, d, J = 1.5 Hz), 6.90 (1H, dd, J = 11.2 Hz, 1.8 Hz), 6.98 (1H, dd, J = 7.8 Hz, 1.8 Hz), 7.02 (1H, d, J = 1.5 Hz), 7.09 (1H, t, J = 7.8 Hz);
MS (FAB) m/z: 336 (M)+.。
実施例19jで得られた化合物(720mg、1.83mmol)、アセトニトリル(12mL)、トリエチルシラン(0.880mL、5.47mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.350mL、2.79mmol)を用いて、実施例2fと同様の方法により、標記化合物(491mg)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 1.19 (3H, t, J = 7.7 Hz), 2.12 (3H, s), 2.61 (2H, q, J= 7.7 Hz), 4.13 (2H, s), 5.00 (2H, s), 6.72 (1H, d, J = 1.5 Hz), 6.90 (1H, dd, J = 11.2 Hz, 1.8 Hz), 6.98 (1H, dd, J = 7.8 Hz, 1.8 Hz), 7.02 (1H, d, J = 1.5 Hz), 7.09 (1H, t, J = 7.8 Hz);
MS (FAB) m/z: 336 (M)+.。
(実施例19l)5-アセトキシメチル-3-クロロ-2-(4-エチル-3-フルオロベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(318mg、0.52mmol)、塩化メチレン(5mL)、トリクロロアセトニトリル(0.263mL、2.60mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(393mg)、実施例19kで得られた化合物(135mg、0.40mmol)、塩化メチレン(5mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.065mL、0.52mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(380mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(318mg、0.52mmol)、塩化メチレン(5mL)、トリクロロアセトニトリル(0.263mL、2.60mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(393mg)、実施例19kで得られた化合物(135mg、0.40mmol)、塩化メチレン(5mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.065mL、0.52mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(380mg)を得た。
(実施例19m)3-クロロ-2-(4-エチル-3-フルオロベンジル)-5-ヒドロキシメチルフェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例19lで得られた粗生成物(380mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(720mg、5.21mmol)を用い実施例2hと同様の方法により、標記化合物(67mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CD3OD): δ 1.16 (3H, t, J = 7.6 Hz), 1.20 (3H, d, J = 6.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.35-3.42 (2H, m), 3.44-3.46 (2H, m), 4.06 (1H, dd, J = 6.6 Hz, 3.6 Hz), 4.12 (1H, d, J = 14.5 Hz), 4.24 (1H, d, J = 14.5 Hz), 4.56 (2H, s), 4.95 (1H, d, J = 7.5 Hz), 6.89 (1H, d, J = 1.2 Hz), 6.99 (1H, dd, J = 7.9 Hz, 1.6 Hz), 7.06 (1H, t, J = 7.9 Hz), 7.11 (1H, d, J = 1.6 Hz), 7.14 (1H, d, J = 1.2 Hz);
MS (FAB) m/z: 509 (M+K)+.。
実施例19lで得られた粗生成物(380mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(720mg、5.21mmol)を用い実施例2hと同様の方法により、標記化合物(67mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CD3OD): δ 1.16 (3H, t, J = 7.6 Hz), 1.20 (3H, d, J = 6.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.35-3.42 (2H, m), 3.44-3.46 (2H, m), 4.06 (1H, dd, J = 6.6 Hz, 3.6 Hz), 4.12 (1H, d, J = 14.5 Hz), 4.24 (1H, d, J = 14.5 Hz), 4.56 (2H, s), 4.95 (1H, d, J = 7.5 Hz), 6.89 (1H, d, J = 1.2 Hz), 6.99 (1H, dd, J = 7.9 Hz, 1.6 Hz), 7.06 (1H, t, J = 7.9 Hz), 7.11 (1H, d, J = 1.6 Hz), 7.14 (1H, d, J = 1.2 Hz);
MS (FAB) m/z: 509 (M+K)+.。
(実施例20)3-フルオロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.6)
(実施例20a)3-ベンジルオキシ-5-フルオロ-4-(4-メトキシベンジル)ベンジルアルコール
酢酸 3-フルオロ-5-ヒドロキシ-4-(4-メトキシベンジル)ベンジル(WO2008/016132(PCT/JP2007/65231))(1.0g、3.3mmol)をN,N-ジメチルホルムアミド(10mL)に溶解し、氷冷下、臭化ベンジル(0.58mL、4.9mmol)および炭酸カリウム(2.3g、17mmol)を加えた。懸濁液を室温に昇温しつつ1時間撹拌し、メタノール(1.0mL)を加えてさらに1時間半撹拌した。反応液を酢酸エチル(50mL)で希釈後、10%食塩水(10mL、3回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(1.2g)を得た。
酢酸 3-フルオロ-5-ヒドロキシ-4-(4-メトキシベンジル)ベンジル(WO2008/016132(PCT/JP2007/65231))(1.0g、3.3mmol)をN,N-ジメチルホルムアミド(10mL)に溶解し、氷冷下、臭化ベンジル(0.58mL、4.9mmol)および炭酸カリウム(2.3g、17mmol)を加えた。懸濁液を室温に昇温しつつ1時間撹拌し、メタノール(1.0mL)を加えてさらに1時間半撹拌した。反応液を酢酸エチル(50mL)で希釈後、10%食塩水(10mL、3回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(1.2g)を得た。
(実施例20b)3-ベンジルオキシ-5-フルオロ-4-(4-メトキシベンジル)ベンズアルデヒド
実施例20aで得られた粗生成物(1.2g、3.3mmol)をクロロホルム(12mL)に溶解し、二酸化マンガン(4.3mL、50mmol)を加えて、加熱還流下、2時間半撹拌した。この懸濁液をセライトで濾過した後、濾液を減圧下濃縮して、標記化合物の粗生成物(1.2g)を得た。
実施例20aで得られた粗生成物(1.2g、3.3mmol)をクロロホルム(12mL)に溶解し、二酸化マンガン(4.3mL、50mmol)を加えて、加熱還流下、2時間半撹拌した。この懸濁液をセライトで濾過した後、濾液を減圧下濃縮して、標記化合物の粗生成物(1.2g)を得た。
(実施例20c)1-ベンジルオキシ-3-フルオロ-2-(4-メトキシベンジル)-5-(2-メトキシビニル)ベンゼン
塩化(メトキシメチル)トリフェニルホスホニウム(3.4g、10mmol)をテトラヒドロフラン(8mL)に懸濁し、氷冷下、撹拌を行った。この懸濁液にリチウム ビス(トリメチルシリル)アミド(1Mテトラヒドロフラン溶液、10mL、10mmol)を滴下し、室温まで昇温しつつ30分間撹拌を行った。実施例20bで得られた粗生成物(1.2g、3.3mmol)を溶解させたテトラヒドロフラン溶液(8mL)をこの反応混合物に滴下し、室温で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液(10mL)を加えた後、酢酸エチル(20mL、2回)で抽出し、有機層を飽和食塩水(10mL、2回)で洗浄した。この有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、10:1、V/V)で精製して、標記化合物(0.92g)を得た。
塩化(メトキシメチル)トリフェニルホスホニウム(3.4g、10mmol)をテトラヒドロフラン(8mL)に懸濁し、氷冷下、撹拌を行った。この懸濁液にリチウム ビス(トリメチルシリル)アミド(1Mテトラヒドロフラン溶液、10mL、10mmol)を滴下し、室温まで昇温しつつ30分間撹拌を行った。実施例20bで得られた粗生成物(1.2g、3.3mmol)を溶解させたテトラヒドロフラン溶液(8mL)をこの反応混合物に滴下し、室温で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液(10mL)を加えた後、酢酸エチル(20mL、2回)で抽出し、有機層を飽和食塩水(10mL、2回)で洗浄した。この有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、10:1、V/V)で精製して、標記化合物(0.92g)を得た。
(実施例20d)3-ベンジルオキシ-5-フルオロ-4-(4-メトキシベンジル)フェニルアセトアルデヒド
実施例20cで得られた化合物(0.92g、2.4mmol)を1,4-ジオキサン(10mL)に溶解し、氷冷下、水(0.43mL、24mmol)および4M塩化水素1,4-ジオキサン溶液(6.0mL、24mmol)を加えた後、室温まで昇温しつつ1時間撹拌した。この溶液を酢酸エチル(50mL)で希釈し、10%食塩水(10mL、2回)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.87g)を得た。
実施例20cで得られた化合物(0.92g、2.4mmol)を1,4-ジオキサン(10mL)に溶解し、氷冷下、水(0.43mL、24mmol)および4M塩化水素1,4-ジオキサン溶液(6.0mL、24mmol)を加えた後、室温まで昇温しつつ1時間撹拌した。この溶液を酢酸エチル(50mL)で希釈し、10%食塩水(10mL、2回)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.87g)を得た。
(実施例20e)2-[3-ベンジルオキシ-5-フルオロ-4-(4-メトキシベンジル)フェニル]エタノール
実施例20dで得られた粗生成物(0.87g、2.4mmol)をテトラヒドロフラン(4mL)およびメタノール(4mL)からなる溶液に溶解し、氷冷下、水素化ホウ素ナトリウム(0.14g、3.6mmol)を加えた。反応液を室温に昇温しつつ40分間撹拌し、氷冷下、飽和塩化アンモニウム水溶液(1mL)を滴下して反応を停止した。混合物を酢酸エチル(40mL)で希釈後、飽和炭酸水素ナトリウム(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.57g)を得た。
実施例20dで得られた粗生成物(0.87g、2.4mmol)をテトラヒドロフラン(4mL)およびメタノール(4mL)からなる溶液に溶解し、氷冷下、水素化ホウ素ナトリウム(0.14g、3.6mmol)を加えた。反応液を室温に昇温しつつ40分間撹拌し、氷冷下、飽和塩化アンモニウム水溶液(1mL)を滴下して反応を停止した。混合物を酢酸エチル(40mL)で希釈後、飽和炭酸水素ナトリウム(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.57g)を得た。
(実施例20f)3-フルオロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェノール
実施例20eで得られた化合物(0.57g、1.6mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、溶液中に窒素を吹き込みながら10分間撹拌した。この溶液に10%パラジウム炭素触媒(含水、0.12g)を窒素気流下で加え、気相を水素置換後、室温で1時間撹拌した。混合物をセライトで濾過し、減圧下溶媒を除去して、標記化合物の粗生成物(0.44g)を得た。
実施例20eで得られた化合物(0.57g、1.6mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、溶液中に窒素を吹き込みながら10分間撹拌した。この溶液に10%パラジウム炭素触媒(含水、0.12g)を窒素気流下で加え、気相を水素置換後、室温で1時間撹拌した。混合物をセライトで濾過し、減圧下溶媒を除去して、標記化合物の粗生成物(0.44g)を得た。
(実施例20g)酢酸 2-[3-フルオロ-5-ヒドロキシ-4-(4-メトキシベンジル)フェニル]エチル
実施例20fで得られた粗生成物(0.44g、1.6mmol)をコリジン(5.0mL)に溶解し、-20℃で塩化アセチル(0.11mL、1.6mmol)を加えて0℃まで昇温しつつ2時間撹拌した。反応液に水(1mL)を加え、酢酸エチル(30mL)で希釈後、30%クエン酸水溶液(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.51g、ただし原料を含む)を得た。
実施例20fで得られた粗生成物(0.44g、1.6mmol)をコリジン(5.0mL)に溶解し、-20℃で塩化アセチル(0.11mL、1.6mmol)を加えて0℃まで昇温しつつ2時間撹拌した。反応液に水(1mL)を加え、酢酸エチル(30mL)で希釈後、30%クエン酸水溶液(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.51g、ただし原料を含む)を得た。
(実施例20h)5-(2-アセトキシエチル)-3-フルオロ-2-(4-メトキシベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.43g、0.71mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.20mL、2.1mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(3μL、0.02mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.75g、1.0mmol)、実施例11fで得られた化合物(0.51g、1.6mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.05mL、0.4mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(0.43g、0.71mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.20mL、2.1mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(3μL、0.02mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.75g、1.0mmol)、実施例11fで得られた化合物(0.51g、1.6mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.05mL、0.4mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例20i)3-フルオロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例20hで得られた混合物(1.6mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(64mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.7 Hz), 2.77 (2H, t, J = 6.8 Hz), 3.35-3.51 (4H, m), 3.72 (3H, s), 3.75 (2H, t, J = 6.8 Hz), 3.91 (1H, d, J = 14.7 Hz), 4.00 (1H, d, J = 14.7 Hz), 4.05-4.07 (1H, m), 4.93 (1H, d, J = 7.4 Hz), 6.67 (1H, d, J = 9.4 Hz), 6.75 (2H, d, J = 8.8 Hz), 6.88 (1H, s), 7.19 (2H, d, J = 8.8 Hz);
MS (FAB) m/z: 475 (M+Na)+.。
実施例20hで得られた混合物(1.6mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(64mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.7 Hz), 2.77 (2H, t, J = 6.8 Hz), 3.35-3.51 (4H, m), 3.72 (3H, s), 3.75 (2H, t, J = 6.8 Hz), 3.91 (1H, d, J = 14.7 Hz), 4.00 (1H, d, J = 14.7 Hz), 4.05-4.07 (1H, m), 4.93 (1H, d, J = 7.4 Hz), 6.67 (1H, d, J = 9.4 Hz), 6.75 (2H, d, J = 8.8 Hz), 6.88 (1H, s), 7.19 (2H, d, J = 8.8 Hz);
MS (FAB) m/z: 475 (M+Na)+.。
(実施例21)3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.15)
(実施例21a)酢酸 3-ベンジルオキシ-5-クロロ-4-(4-メチルベンジル)ベンジル
実施例4hで得られた化合物(500mg、3.3mmol)をN,N-ジメチルホルムアミド(10mL)に溶解し、臭化ベンジル(430μL、3.6mmol)および炭酸カリウム(680mg、4.9mmol)を加え、室温で4時間撹拌した。反応液を酢酸エチル(30mL)で希釈し、蒸留水(10mL)および飽和食塩水(10mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(1.13g)を黄色油状物質として得た。
実施例4hで得られた化合物(500mg、3.3mmol)をN,N-ジメチルホルムアミド(10mL)に溶解し、臭化ベンジル(430μL、3.6mmol)および炭酸カリウム(680mg、4.9mmol)を加え、室温で4時間撹拌した。反応液を酢酸エチル(30mL)で希釈し、蒸留水(10mL)および飽和食塩水(10mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(1.13g)を黄色油状物質として得た。
(実施例21b)3-ベンジルオキシ-5-クロロ-4-(4-メチルベンジル)ベンジルアルコール
実施例21aで得られた粗生成物(1.13g、2.9mmol)をメタノール(12mL)に溶解し、1M水酸化カリウム水溶液(3.3mL、3.3mmol)を加え、室温で18時間攪拌した。減圧下溶媒を留去し、残渣を酢酸エチル(10mL)で希釈し、蒸留水(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.91g)を黄色油状物質として得た。
実施例21aで得られた粗生成物(1.13g、2.9mmol)をメタノール(12mL)に溶解し、1M水酸化カリウム水溶液(3.3mL、3.3mmol)を加え、室温で18時間攪拌した。減圧下溶媒を留去し、残渣を酢酸エチル(10mL)で希釈し、蒸留水(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去して、標記化合物の粗生成物(0.91g)を黄色油状物質として得た。
(実施例21c)3-ベンジルオキシ-5-クロロ-4-(4-メチルベンジル)ベンズアルデヒド
実施例21bで得られた粗生成物(0.91g、2.6mmol)をクロロホルム(10mL)に溶解し、二酸化マンガン(2.85g、32.8mmol)を加え、60℃で4時間攪拌した。セライトで濾過後、減圧下溶媒を留去して、標記化合物の粗生成物(0.68g)を褐色固体として得た。
実施例21bで得られた粗生成物(0.91g、2.6mmol)をクロロホルム(10mL)に溶解し、二酸化マンガン(2.85g、32.8mmol)を加え、60℃で4時間攪拌した。セライトで濾過後、減圧下溶媒を留去して、標記化合物の粗生成物(0.68g)を褐色固体として得た。
(実施例21d)1-ベンジルオキシ-3-クロロ-5-(2-メトキシビニル)-2-(4-メチルベンジル)ベンゼン
塩化(メトキシメチル)トリフェニルホスホニウム(2.0g、5.4mmol)にトルエン(5mL)を加え、減圧下溶媒を留去した。残渣に、氷冷下、テトラヒドロフラン(4mL)およびリチウム ビス(トリメチルシリル)アミド(1Mテトラヒドロフラン溶液、5.9mL、5.9mmol)を加え、室温で1時間撹拌した。さらに、実施例21cで得られた粗生成物(0.68g、1.9mmol)を溶解させたテトラヒドロフラン溶液(6mL)を反応液に滴下し、室温で1時間攪拌した。反応液を酢酸エチル(10mL)で希釈し、蒸留水(10mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、20:1~5:1、V/V)を用いて精製して、標記化合物(0.95g、quant.)を黄色固体として得た。
塩化(メトキシメチル)トリフェニルホスホニウム(2.0g、5.4mmol)にトルエン(5mL)を加え、減圧下溶媒を留去した。残渣に、氷冷下、テトラヒドロフラン(4mL)およびリチウム ビス(トリメチルシリル)アミド(1Mテトラヒドロフラン溶液、5.9mL、5.9mmol)を加え、室温で1時間撹拌した。さらに、実施例21cで得られた粗生成物(0.68g、1.9mmol)を溶解させたテトラヒドロフラン溶液(6mL)を反応液に滴下し、室温で1時間攪拌した。反応液を酢酸エチル(10mL)で希釈し、蒸留水(10mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、20:1~5:1、V/V)を用いて精製して、標記化合物(0.95g、quant.)を黄色固体として得た。
(実施例21e)3-ベンジルオキシ-5-クロロ-4-(4-メチルベンジル)フェニルアセトアルデヒド
実施例21dで得られた化合物(0.95g、1.9mmol)を1,4-ジオキサン(5mL)に溶解し、蒸留水(350μL)および4N塩酸 1,4-ジオキサン溶液(5mL)を加え、室温で10分撹拌した。反応液を酢酸エチル(10mL)で希釈し、蒸留水(5mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、20:1~10:1~4:1、V/V)を用いて精製して、標記化合物(0.48g)を黄色油状物質として得た。
実施例21dで得られた化合物(0.95g、1.9mmol)を1,4-ジオキサン(5mL)に溶解し、蒸留水(350μL)および4N塩酸 1,4-ジオキサン溶液(5mL)を加え、室温で10分撹拌した。反応液を酢酸エチル(10mL)で希釈し、蒸留水(5mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、20:1~10:1~4:1、V/V)を用いて精製して、標記化合物(0.48g)を黄色油状物質として得た。
(実施例21f)2-[3-ベンジルオキシ-5-クロロ-4-(4-メチルベンジル)フェニル]エタノール
実施例21eで得られた化合物(0.48g、1.3mmol)をメタノール(5mL)に溶解し、氷冷下、水素化ホウ素ナトリウム(60mg、1.6mmol)を加え、0℃で30分間撹拌した。氷冷下、反応液に飽和塩化アンモニウム水溶液(5mL)を加え、酢酸エチル(10mL)で希釈し、飽和塩化アンモニウム水溶液(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(0.41g)を無色固体として得た。
実施例21eで得られた化合物(0.48g、1.3mmol)をメタノール(5mL)に溶解し、氷冷下、水素化ホウ素ナトリウム(60mg、1.6mmol)を加え、0℃で30分間撹拌した。氷冷下、反応液に飽和塩化アンモニウム水溶液(5mL)を加え、酢酸エチル(10mL)で希釈し、飽和塩化アンモニウム水溶液(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(0.41g)を無色固体として得た。
(実施例21g)3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メチルベンジル)フェノール
実施例21fで得られた粗生成物(0.41g、1.1mmol)をアセトニトリル(5mL)に溶解し、ヨウ化トリメチルシリル(320μL、2.2mmol)を加え、40℃で2時間撹拌した。室温に冷却後、反応液を酢酸エチル(15mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~3:1~2:1、V/V)を用いて精製して、標記化合物(122mg)を無色固体として得た。
実施例21fで得られた粗生成物(0.41g、1.1mmol)をアセトニトリル(5mL)に溶解し、ヨウ化トリメチルシリル(320μL、2.2mmol)を加え、40℃で2時間撹拌した。室温に冷却後、反応液を酢酸エチル(15mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~3:1~2:1、V/V)を用いて精製して、標記化合物(122mg)を無色固体として得た。
(実施例21h)酢酸 2-{[3-クロロ-5-ヒドロキシ-4-(4-メチルベンジル)]フェニル}エチル
実施例21gで得られた化合物(122mg、0.4mmol)をテトラヒドロフラン(2mL)に溶解し、酢酸ビニル(2mL、21.6mmol)およびビス(ジブチル塩化スズ)オキシド(24mg、0.04mmol)を加え、室温で24時間撹拌した。減圧下溶媒を留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1、V/V)を用いて精製して、標記化合物(111mg)を無色固体として得た。
実施例21gで得られた化合物(122mg、0.4mmol)をテトラヒドロフラン(2mL)に溶解し、酢酸ビニル(2mL、21.6mmol)およびビス(ジブチル塩化スズ)オキシド(24mg、0.04mmol)を加え、室温で24時間撹拌した。減圧下溶媒を留去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1、V/V)を用いて精製して、標記化合物(111mg)を無色固体として得た。
(実施例21i)5-(2-アセトキシエチル)-3-クロロ-2-(4-メチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(255mg、0.42mmol)、トリクロロアセトニトリル(125μL、1.25mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用いて、実施例4iと同様の方法によりイミダートを調製し、実施例21hで得られた化合物(111mg、0.35mmol)、三フッ化ホウ素-ジエチルエーテル錯体(44μL、0.35mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(391mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(255mg、0.42mmol)、トリクロロアセトニトリル(125μL、1.25mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用いて、実施例4iと同様の方法によりイミダートを調製し、実施例21hで得られた化合物(111mg、0.35mmol)、三フッ化ホウ素-ジエチルエーテル錯体(44μL、0.35mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(391mg)を得た。
(実施例21j)3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例21iで得られた化合物(391mg、0.35mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(48mg、0.35mmol)を用い、実施例4jと同様の方法により、標記化合物(104mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.2 Hz), 2.25 (3H, s), 2.77 (2H, t, J = 6.9 Hz), 3.35-3.36 (2H, m), 3.43-3.45 (2H, m), 3.76 (2H, t, J = 6.9 Hz), 4.03-4.05 (1H, m), 4.08(1H, d, J = 14.9 Hz), 4.21 (1H, d, J = 14.9 Hz), 4.91 (1H, d, J = 7.4 Hz), 6.99 (2H, s), 7.02 (2H, d, J = 7.8 Hz), 7.12 (2H, d, J = 7.8 Hz);
MS (FAB) m/z: 453 (M+H)+, 475 (M+Na)+.。
実施例21iで得られた化合物(391mg、0.35mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(48mg、0.35mmol)を用い、実施例4jと同様の方法により、標記化合物(104mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.2 Hz), 2.25 (3H, s), 2.77 (2H, t, J = 6.9 Hz), 3.35-3.36 (2H, m), 3.43-3.45 (2H, m), 3.76 (2H, t, J = 6.9 Hz), 4.03-4.05 (1H, m), 4.08(1H, d, J = 14.9 Hz), 4.21 (1H, d, J = 14.9 Hz), 4.91 (1H, d, J = 7.4 Hz), 6.99 (2H, s), 7.02 (2H, d, J = 7.8 Hz), 7.12 (2H, d, J = 7.8 Hz);
MS (FAB) m/z: 453 (M+H)+, 475 (M+Na)+.。
(実施例22)3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.16)
(実施例22a)3-クロロ-4-(4-メトキシベンジル)-5-(メトキシメトキシ)ベンジルアルコール
酢酸 3-クロロ-5-ヒドロキシ-4-(4-メトキシベンジル)ベンジル(WO2008/016132(PCT/JP2007/65231))(500mg、1.6mmol)をN,N-ジメチルホルムアミド(5mL)に溶解し、氷冷下、クロロメチルメチルエーテル(180μL、2.4mmol)および炭酸カルシウム(1.08g、7.8mmol)を加え、室温で4時間撹拌した。反応液にメタノール(5mL)を加え、室温で1時間撹拌した。減圧下溶媒を留去した後、残渣を酢酸エチル(10mL)で希釈し、蒸留水(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(511mg)を無色固体として得た。
酢酸 3-クロロ-5-ヒドロキシ-4-(4-メトキシベンジル)ベンジル(WO2008/016132(PCT/JP2007/65231))(500mg、1.6mmol)をN,N-ジメチルホルムアミド(5mL)に溶解し、氷冷下、クロロメチルメチルエーテル(180μL、2.4mmol)および炭酸カルシウム(1.08g、7.8mmol)を加え、室温で4時間撹拌した。反応液にメタノール(5mL)を加え、室温で1時間撹拌した。減圧下溶媒を留去した後、残渣を酢酸エチル(10mL)で希釈し、蒸留水(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(511mg)を無色固体として得た。
(実施例22b)3-クロロ-4-(4-メトキシベンジル)-5-(メトキシメトキシ)ベンズアルデヒド
実施例22aで得られた粗生成物(511mg、1.6mmol)、クロロホルム(10mL)および二酸化マンガン(1.36g、15.6mmol)を用い、実施例21cと同様の方法により、標記化合物の粗生成物(480mg)を淡黄色油状物質として得た。
実施例22aで得られた粗生成物(511mg、1.6mmol)、クロロホルム(10mL)および二酸化マンガン(1.36g、15.6mmol)を用い、実施例21cと同様の方法により、標記化合物の粗生成物(480mg)を淡黄色油状物質として得た。
(実施例22c)1-クロロ-2-(4-メトキシベンジル)-3-メトキシメトキシ-5-(2-メトキシビニル)ベンゼン
実施例22bで得られた粗生成物(480mg、1.5mmol)、塩化(メチルメトキシ)トリフェニルホスホニウム(1.60g、4.8mmol)、リチウム ビス(トリメチルシリル)アミド(1Mテトラヒドロフラン溶液、4.7mL、4.7mmol)およびテトラヒドロフラン(8mL)を用い、実施例21dと同様の方法により、標記化合物(810mg、quant.)を黄色油状物質として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、20:1~15:1~10:1、V/V)を用いて行った。
実施例22bで得られた粗生成物(480mg、1.5mmol)、塩化(メチルメトキシ)トリフェニルホスホニウム(1.60g、4.8mmol)、リチウム ビス(トリメチルシリル)アミド(1Mテトラヒドロフラン溶液、4.7mL、4.7mmol)およびテトラヒドロフラン(8mL)を用い、実施例21dと同様の方法により、標記化合物(810mg、quant.)を黄色油状物質として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、20:1~15:1~10:1、V/V)を用いて行った。
(実施例22d)3-クロロ-5-ヒドロキシ-4-(4-メトキシベンジル)フェニルアセトアルデヒド
実施例22cで得られた化合物(810mg、1.5mmol)、4N塩酸 1,4-ジオキサン溶液(4mL)、蒸留水(280μL)および1,4-ジオキサン(4mL)を用い、実施例21eと同様の方法により、標記化合物(210mg)を黄色油状物質として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、8:1~6:1~4:1、V/V)を用いて行った。
実施例22cで得られた化合物(810mg、1.5mmol)、4N塩酸 1,4-ジオキサン溶液(4mL)、蒸留水(280μL)および1,4-ジオキサン(4mL)を用い、実施例21eと同様の方法により、標記化合物(210mg)を黄色油状物質として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、8:1~6:1~4:1、V/V)を用いて行った。
(実施例22e)3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェノール
実施例22dで得られた化合物(210mg、0.72mmol)、水素化ホウ素ナトリウム(33mg、0.87mmol)およびメタノール(2.5mL)を用い、実施例21fと同様の方法により、標記化合物(157mg)を無色油状物質として得た。
実施例22dで得られた化合物(210mg、0.72mmol)、水素化ホウ素ナトリウム(33mg、0.87mmol)およびメタノール(2.5mL)を用い、実施例21fと同様の方法により、標記化合物(157mg)を無色油状物質として得た。
(実施例22f)酢酸 2-{[3-クロロ-5-ヒドロキシ-4-(4-メトキシベンジル)]フェニル}エチル
実施例22eで得られた化合物(157mg、0.54mmol)、酢酸ビニル(2mL、21.61mmol)、ビス(ジブチル塩化スズ)オキシド(38mg、0.07mmol)およびテトラヒドロフラン(2mL)を用い、実施例21hと同様の方法により、標記化合物(218mg)を無色固体として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、4:1~3:1、V/V)を用いて行った。
実施例22eで得られた化合物(157mg、0.54mmol)、酢酸ビニル(2mL、21.61mmol)、ビス(ジブチル塩化スズ)オキシド(38mg、0.07mmol)およびテトラヒドロフラン(2mL)を用い、実施例21hと同様の方法により、標記化合物(218mg)を無色固体として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、4:1~3:1、V/V)を用いて行った。
(実施例22g)5-アセトキシメチル-3-クロロ-2-(4-メトキシベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(220mg、0.36mmol)、トリクロロアセトニトリル(110μL、1.10mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用いて、実施例4iと同様の方法によりイミダートを調製し、実施例22fで得られた化合物(100mg、0.30mmol)、三フッ化ホウ素-ジエチルエーテル錯体(38μL、0.40mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(320mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(220mg、0.36mmol)、トリクロロアセトニトリル(110μL、1.10mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)および塩化メチレン(5mL)を用いて、実施例4iと同様の方法によりイミダートを調製し、実施例22fで得られた化合物(100mg、0.30mmol)、三フッ化ホウ素-ジエチルエーテル錯体(38μL、0.40mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(320mg)を得た。
(実施例22h)3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例22gで得られた粗生成物(320mg、0.30mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(41mg、0.30mmol)を用い、実施例4jと同様の方法により、標記化合物(81mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.6 Hz), 2.76 (2H, t, J = 6.6 Hz), 3.35-3.38 (2H, m), 3.44-3.49 (2H, m), 3.72 (3H, s), 3.75 (2H, t, J = 6.6 Hz), 4.03-4.09 (1H, m), 4.06 (1H, d, J = 14.5 Hz), 4.18 (1H, d, J = 14.5 Hz), 4.93 (1H, d, J = 4.3 Hz), 6.75 (2H, d, J = 9.0 Hz), 6.98 (1H, d, J = 1.6 Hz), 7.03 (1H, d, J = 1.6 Hz), 7.18 (2H, d, J = 9.0 Hz);
MS (FAB) m/z: 469 (M+H)+, 491 (M+Na)+.。
実施例22gで得られた粗生成物(320mg、0.30mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カルシウム(41mg、0.30mmol)を用い、実施例4jと同様の方法により、標記化合物(81mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.6 Hz), 2.76 (2H, t, J = 6.6 Hz), 3.35-3.38 (2H, m), 3.44-3.49 (2H, m), 3.72 (3H, s), 3.75 (2H, t, J = 6.6 Hz), 4.03-4.09 (1H, m), 4.06 (1H, d, J = 14.5 Hz), 4.18 (1H, d, J = 14.5 Hz), 4.93 (1H, d, J = 4.3 Hz), 6.75 (2H, d, J = 9.0 Hz), 6.98 (1H, d, J = 1.6 Hz), 7.03 (1H, d, J = 1.6 Hz), 7.18 (2H, d, J = 9.0 Hz);
MS (FAB) m/z: 469 (M+H)+, 491 (M+Na)+.。
(実施例23)3-クロロ-2-[4-(2,2,2-トリフルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.14)
(実施例23a)3-クロロ-5-オキソ-4-[4-(2,2,2-トリフルオロエチル)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
実施例18bで得られた粗生成物(1.34g、3.61mmol)、塩化メチレン(15mL)、2-メチル-2-ブテン(1.54mL、14.5mmol)、二塩化オキサリル(0.32mL、3.73mmol)および触媒量のN,N-ジメチルホルムアミドを用い、実施例1cと同様の方法により標記化合物の粗生成物(1.40g)を得た。
実施例18bで得られた粗生成物(1.34g、3.61mmol)、塩化メチレン(15mL)、2-メチル-2-ブテン(1.54mL、14.5mmol)、二塩化オキサリル(0.32mL、3.73mmol)および触媒量のN,N-ジメチルホルムアミドを用い、実施例1cと同様の方法により標記化合物の粗生成物(1.40g)を得た。
(実施例23b)3-クロロ-5-ヒドロキシ-4-[4-(2,2,2-トリフルオロエチル)ベンゾイル]安息香酸エチル
実施例23aで得られた粗生成物(1.40g、3.60mmol)、N-メチルモルホリン(16mL)、無水硫酸ナトリウム(10.3g)およびヨウ素(1.10g、4.33mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(1.02g)を得た。
実施例23aで得られた粗生成物(1.40g、3.60mmol)、N-メチルモルホリン(16mL)、無水硫酸ナトリウム(10.3g)およびヨウ素(1.10g、4.33mmol)を用い、実施例1dと同様の方法により標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~3:1、V/V)を用いて精製してアモルファス状の標記化合物(1.02g)を得た。
(実施例23c)3-クロロ-5-ヒドロキシメチル-2-[1-ヒドロキシ-1-{4-(2,2,2-トリフルオロエチル)フェニル}メチル]フェノール
実施例23bで得られた化合物(1.02g、2.64mmol)、テトラヒドロフラン(30mL)および水素化リチウムアルミニウム(300mg、7.91mmol)を用い、実施例18eと同様の方法により標記化合物の粗生成物(866mg)を得た。
実施例23bで得られた化合物(1.02g、2.64mmol)、テトラヒドロフラン(30mL)および水素化リチウムアルミニウム(300mg、7.91mmol)を用い、実施例18eと同様の方法により標記化合物の粗生成物(866mg)を得た。
(実施例23d)3-クロロ-5-ヒドロキシメチル-2-[1-メトキシ-1-{4-(2,2,2-トリフルオロエチル)フェニル}メチル]フェノール
実施例23cで得られた粗生成物(866mg、2.50mmol)、メタノール(16mL)およびパラトルエンスルホン酸一水和物(238mg、1.25mmol)を用い、実施例18fと同様の方法により、油状の標記化合物(654mg)を得た。
実施例23cで得られた粗生成物(866mg、2.50mmol)、メタノール(16mL)およびパラトルエンスルホン酸一水和物(238mg、1.25mmol)を用い、実施例18fと同様の方法により、油状の標記化合物(654mg)を得た。
(実施例23e)酢酸 3-クロロ-4-[1-メトキシ-1-{4-(2,2,2-トリフルオロエチル)フェニル}メチル]-5-ヒドロキシベンジル
実施例23dで得られた化合物(654mg、1.81mmol)、テトラヒドロフラン(6mL)、酢酸ビニル(6mL)およびビス(ジブチル塩化スズ)オキシド(302mg、0.55mmol)を用い実施例2eと同様の方法により、油状の標記化合物(703mg)を得た。
実施例23dで得られた化合物(654mg、1.81mmol)、テトラヒドロフラン(6mL)、酢酸ビニル(6mL)およびビス(ジブチル塩化スズ)オキシド(302mg、0.55mmol)を用い実施例2eと同様の方法により、油状の標記化合物(703mg)を得た。
(実施例23f)酢酸 3-クロロ-4-[4-(2,2,2-トリフルオロエチル)ベンジル]-5-ヒドロキシベンジル
実施例23eで得られた化合物(703mg、1.75mmol)、アセトニトリル(15mL)、トリエチルシラン(0.834mL、5.24mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.329mL、2.62mmol)を用い、実施例2fと同様の方法により標記化合物(306mg)を白色固体として得た。
実施例23eで得られた化合物(703mg、1.75mmol)、アセトニトリル(15mL)、トリエチルシラン(0.834mL、5.24mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.329mL、2.62mmol)を用い、実施例2fと同様の方法により標記化合物(306mg)を白色固体として得た。
(実施例23g)5-アセトキシメチル-3-クロロ-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(213mg、0.35mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.176mL、1.74mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(5μL、0.03mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(263mg)、実施例23fで得られた化合物(105mg、0.23mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.044mL、0.34mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(280mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(213mg、0.35mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.176mL、1.74mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(5μL、0.03mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(263mg)、実施例23fで得られた化合物(105mg、0.23mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.044mL、0.34mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(280mg)を得た。
(実施例23h)3-クロロ-5-ヒドロキシメチル-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例23gで得られた粗生成物(280mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(480mg、3.47mmol)を用い実施例2hと同様の方法により、標記化合物(84mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチルから行った。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.4 Hz), 3.34-3.46 (6H, m), 4.06 (1H, dd, J = 6.4 Hz, 3.7 Hz), 4.17 (1H, d, J = 14.5 Hz), 4.28 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.8 Hz), 7.11 (1H, s), 7.13 (1H, s), 7.16 (2H, d, J = 8.3 Hz), 7.26 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 545 (M+K)+.。
実施例23gで得られた粗生成物(280mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(480mg、3.47mmol)を用い実施例2hと同様の方法により、標記化合物(84mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチルから行った。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.4 Hz), 3.34-3.46 (6H, m), 4.06 (1H, dd, J = 6.4 Hz, 3.7 Hz), 4.17 (1H, d, J = 14.5 Hz), 4.28 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.8 Hz), 7.11 (1H, s), 7.13 (1H, s), 7.16 (2H, d, J = 8.3 Hz), 7.26 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 545 (M+K)+.。
(実施例24)3-クロロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.17)
(実施例24a)塩化2-フルオロ-4-メチルベンゾイル
2-フルオロ-4-メチル安息香酸(500mg、3.24mmol)、二塩化オキサリル(0.31mL、3.57mmol)および触媒量のN,N-ジメチルホルムアミド、テトラヒドロフラン(10mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(560mg)を無色油状物質として得た。
2-フルオロ-4-メチル安息香酸(500mg、3.24mmol)、二塩化オキサリル(0.31mL、3.57mmol)および触媒量のN,N-ジメチルホルムアミド、テトラヒドロフラン(10mL)を用いて実施例1aと同様の方法により、標記化合物の粗生成物(560mg)を無色油状物質として得た。
(実施例24b)(2-クロロ-6-ヒドロキシ-4-メトキシフェニル)(2-フルオロ-4-メチルフェニル)メタノン
1-クロロ-3,5-ジメトキシベンゼン(560mg、3.24mmol)、トルエン(3mL)、塩化アルミニウム(480mg、3.60mmol)および実施例24aで得られた粗生成物(560mg、3.24mmol)を用いて実施例2aと同様の方法を用いて標記化合物の粗生成物(630mg)を固体として得た。
1-クロロ-3,5-ジメトキシベンゼン(560mg、3.24mmol)、トルエン(3mL)、塩化アルミニウム(480mg、3.60mmol)および実施例24aで得られた粗生成物(560mg、3.24mmol)を用いて実施例2aと同様の方法を用いて標記化合物の粗生成物(630mg)を固体として得た。
(実施例24c)(2-クロロ-4,6-ジヒドロキシフェニル)(2-フルオロ-4-メチルフェニル)メタノン
実施例24bで得られた粗生成物(510mg、1.73mmol)を1,2-ジクロロエタン(5mL)に溶解し、塩化アルミニウム(460mg、3.45mmol)を加え、80℃で1.5時間攪拌した。反応液を0℃に冷却後、適量の氷を加え5分間攪拌し、酢酸エチル(40mL)で抽出後、2mol/L塩酸(10mL)および飽和炭酸水素ナトリウム溶液(20mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。残渣を塩化メチレン/ヘキサンで洗浄し、濾過後、標記化合物の粗生成物(366mg)を固体として得た
実施例24bで得られた粗生成物(510mg、1.73mmol)を1,2-ジクロロエタン(5mL)に溶解し、塩化アルミニウム(460mg、3.45mmol)を加え、80℃で1.5時間攪拌した。反応液を0℃に冷却後、適量の氷を加え5分間攪拌し、酢酸エチル(40mL)で抽出後、2mol/L塩酸(10mL)および飽和炭酸水素ナトリウム溶液(20mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下、溶媒を除去した。残渣を塩化メチレン/ヘキサンで洗浄し、濾過後、標記化合物の粗生成物(366mg)を固体として得た
(実施例24d)5-クロロ-4-(2-フルオロ-4-メチルフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-オール
実施例24cで得られた粗生成物(366mg、1.30mmol)、メタノール(5mL)および水素化ホウ素ナトリウム(123mg、3.25mmol)を用いて、実施例2bと同様の方法によりトリオール体の粗生成物(360mg)を得た。
実施例24cで得られた粗生成物(366mg、1.30mmol)、メタノール(5mL)および水素化ホウ素ナトリウム(123mg、3.25mmol)を用いて、実施例2bと同様の方法によりトリオール体の粗生成物(360mg)を得た。
粗生成物(360mg)、アセトン(5mL)および三フッ化ホウ素-ジエチルエーテル錯体(0.160mL、1.27mmol)を用い、実施例6eと同様の方法により、標記化合物(310mg)をアモルファスとして得た。
(実施例24e)5-クロロ-4-(2-フルオロ-4-メチルフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-カルボン酸メチル
実施例24dで得られた化合物(310mg、0.96mmol)、塩化メチレン(6mL)、ピリジン(0.120mL、1.49mmol)および無水トリフルオロメタンスルホン酸(0.190mL、1.13mmol)を用いて、実施例2cと同様の方法によりトリフラート体の粗生成物(437mg)を得た。
実施例24dで得られた化合物(310mg、0.96mmol)、塩化メチレン(6mL)、ピリジン(0.120mL、1.49mmol)および無水トリフルオロメタンスルホン酸(0.190mL、1.13mmol)を用いて、実施例2cと同様の方法によりトリフラート体の粗生成物(437mg)を得た。
得られた粗生成物(437mg、0.96mmol)、N,N-ジメチルホルムアルデヒド(4mL)、メタノール(0.780mL、19.2mmol)、酢酸パラジウム(22mg、0.10mmol)、1,3-ビス(ジフェニルホスフィノ)プロパン(40mg、0.10mmol)、トリエチルアミン(2.66mL、19.2mmol)および一酸化炭素を用いて、実施例2cと同様の方法により、アモルファス状の標記化合物(261mg)を得た。
(実施例24f)5-クロロ-4-(2-フルオロ-4-メチルフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-イルメタノール
実施例24eで得られた化合物(261mg、0.72mmol)、水素化リチウムアルミニウム(41mg、1.08mmol)およびテトラヒドロフラン(4mL)を用い、実施例2dと同様の方法により標記化合物の粗生成物(241mg)をアモルファスとして得た。
(実施例24g)3-クロロ-2-{[1-(2-フルオロ-4-メチルフェニル)-1-メトキシ]メチル}-5-(ヒドロキシメチル)フェノール
実施例24fで得られた粗生成物(241mg、0.72mmol)をメタノール(5mL)に溶解し、パラトルエンスルホン酸一水和物(68mg、0.36mmol)を加え50℃で2時間攪拌した。反応液にトリエチルアミンを加え、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~3:2、V/V)を用いて精製して、油状の標記化合物(210mg)を得た。
実施例24fで得られた粗生成物(241mg、0.72mmol)をメタノール(5mL)に溶解し、パラトルエンスルホン酸一水和物(68mg、0.36mmol)を加え50℃で2時間攪拌した。反応液にトリエチルアミンを加え、減圧下、溶媒を除去した。最後にシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1~3:2、V/V)を用いて精製して、油状の標記化合物(210mg)を得た。
(実施例24h)酢酸 3-クロロ-4-{[1-(2-フルオロ-4-メチルフェニル)-1-メトキシ]メチル}-5-ヒドロキシベンジル
実施例24gで得られた化合物(210mg、0.68mmol)、テトラヒドロフラン(2mL)、酢酸ビニル(2mL)およびビス(ジブチル塩化スズ)オキシド(112mg、0.20mmol)を用いて、実施例2eと同様の方法により、アモルファス状の標記化合物(238mg)を得た。
実施例24gで得られた化合物(210mg、0.68mmol)、テトラヒドロフラン(2mL)、酢酸ビニル(2mL)およびビス(ジブチル塩化スズ)オキシド(112mg、0.20mmol)を用いて、実施例2eと同様の方法により、アモルファス状の標記化合物(238mg)を得た。
(実施例24i)酢酸 3-クロロ-4-(2-フルオロ-4-メチルベンジル)-5-ヒドロキシベンジル
実施例24hで得られた化合物(238mg、0.67mmol)、アセトニトリル(5mL)、トリエチルシラン(0.322mL、2.01mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.127mL、1.01mmol)を用いて、実施例2fと同様の方法により、標記化合物(96mg)を白色固体として得た。
実施例24hで得られた化合物(238mg、0.67mmol)、アセトニトリル(5mL)、トリエチルシラン(0.322mL、2.01mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.127mL、1.01mmol)を用いて、実施例2fと同様の方法により、標記化合物(96mg)を白色固体として得た。
(実施例24j)5-アセトキシメチル-3-クロロ-2-(2-フルオロ-4-メチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(236mg、0.39mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.195mL、1.93mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(292mg)、実施例24iで得られた化合物(96mg、0.30mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.049mL、0.39mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(435mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(236mg、0.39mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.195mL、1.93mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(292mg)、実施例24iで得られた化合物(96mg、0.30mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.049mL、0.39mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(435mg)を得た。
(実施例24k)3-クロロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例24jで得られた粗生成物(435mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(534mg、3.89mmol)を用い実施例2hと同様の方法により、標記化合物(86mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.4 Hz), 2.27 (3H, s), 3.34-3.43 (4H, m), 4.05 (1H, dd, J = 6.4 Hz, 3.6 Hz), 4.15 (1H, d, J = 15.4 Hz), 4.23 (1H, d, J = 15.4 Hz), 4.58 (2H, s), 4.93 (1H, d, J = 7.4 Hz), 6.75-6.76 (2H, m), 6.85 (1H, d, J = 11.3 Hz), 7.13 (1H, s), 7.15 (1H, s);
MS (FAB) m/z: 495 (M+K)+.。
実施例24jで得られた粗生成物(435mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(534mg、3.89mmol)を用い実施例2hと同様の方法により、標記化合物(86mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.4 Hz), 2.27 (3H, s), 3.34-3.43 (4H, m), 4.05 (1H, dd, J = 6.4 Hz, 3.6 Hz), 4.15 (1H, d, J = 15.4 Hz), 4.23 (1H, d, J = 15.4 Hz), 4.58 (2H, s), 4.93 (1H, d, J = 7.4 Hz), 6.75-6.76 (2H, m), 6.85 (1H, d, J = 11.3 Hz), 7.13 (1H, s), 7.15 (1H, s);
MS (FAB) m/z: 495 (M+K)+.。
(実施例25)3-クロロ-2-(4-エチル-2-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.18)
(実施例25a)(2-クロロ-6-ヒドロキシ-4-メトキシフェニル)(4-エチル-2-フルオロフェニル)メタノン
1-クロロ-3,5-ジメトキシベンゼン(855mg、4.95mmol)、トルエン(5mL)、塩化アルミニウム(727mg、5.45mmol)および塩化4-エチル-2-フルオロベンゾイル(US2006/20146A1)(918mg、4.92mmol)を用いて実施例2aと同様の方法により標記化合物の粗生成物(1.02g)を固体として得た。
1-クロロ-3,5-ジメトキシベンゼン(855mg、4.95mmol)、トルエン(5mL)、塩化アルミニウム(727mg、5.45mmol)および塩化4-エチル-2-フルオロベンゾイル(US2006/20146A1)(918mg、4.92mmol)を用いて実施例2aと同様の方法により標記化合物の粗生成物(1.02g)を固体として得た。
(実施例25b)(2-クロロ-4,6-ジヒドロキシフェニル)(4-エチル-2-フルオロフェニル)メタノン
実施例25aで得られた粗生成物(1.02g、3.30mmol)、1,2-ジクロロエタン(10mL)および塩化アルミニウム(880mg、6.60mmol)を用いて、実施例24cと同様の方法により、標記化合物の粗生成物(824mg)を固体として得た。
実施例25aで得られた粗生成物(1.02g、3.30mmol)、1,2-ジクロロエタン(10mL)および塩化アルミニウム(880mg、6.60mmol)を用いて、実施例24cと同様の方法により、標記化合物の粗生成物(824mg)を固体として得た。
(実施例25c)5-クロロ-2,2-ジメチル-4-(4-エチル-2-フルオロフェニル)-4H-ベンゾ[1,3]ジオキシン-7-オール
実施例25bで得られた粗生成物(815mg、2.77mmol)、メタノール(10mL)および水素化ホウ素ナトリウム(262mg、6.93mmol)を用いて、実施例2bと同様の方法によりトリオール体の粗生成物(564mg)を得た。
実施例25bで得られた粗生成物(815mg、2.77mmol)、メタノール(10mL)および水素化ホウ素ナトリウム(262mg、6.93mmol)を用いて、実施例2bと同様の方法によりトリオール体の粗生成物(564mg)を得た。
粗生成物(564mg)、アセトン(6mL)および三フッ化ホウ素-ジエチルエーテル錯体(0.300mL、2.39mmol)を用い、実施例6eと同様の方法により、標記化合物(515mg)をアモルファスとして得た。
(実施例25d)5-クロロ-2,2-ジメチル-4-(4-エチル-2-フルオロフェニル)-4H-ベンゾ[1,3]ジオキシン-7-カルボン酸メチル
実施例25cで得られた化合物(515mg、1.53mmol)、塩化メチレン(10mL)、ピリジン(0.185mL、2.29mmol)および無水トリフルオロメタンスルホン酸(0.308mL、1.83mmol)を用いて、実施例2cと同様の方法によりトリフラート体の粗生成物(717mg)を得た。
実施例25cで得られた化合物(515mg、1.53mmol)、塩化メチレン(10mL)、ピリジン(0.185mL、2.29mmol)および無水トリフルオロメタンスルホン酸(0.308mL、1.83mmol)を用いて、実施例2cと同様の方法によりトリフラート体の粗生成物(717mg)を得た。
得られた粗生成物(717mg、1.53mmol)、N,N-ジメチルホルムアルデヒド(6.4mL)、メタノール(1.24mL、30.5mmol)、酢酸パラジウム(34mg、0.15mmol)、1,3-ビス(ジフェニルホスフィノ)プロパン(63mg、0.15mmol)、トリエチルアミン(4.24mL、30.6mmol)および一酸化炭素を用いて、実施例2cと同様の方法により、標記化合物(465mg)を白色固体として得た。
(実施例25e)5-クロロ-4-(4-エチル-2-フルオロフェニル)-2,2-ジメチル-4H-ベンゾ[1,3]ジオキシン-7-イルメタノール
実施例25dで得られた化合物(465mg、1.23mmol)、水素化リチウムアルミニウム(70mg、1.84mmol)およびテトラヒドロフラン(7mL)を用い、実施例2dと同様の方法により標記化合物の粗生成物(432mg)をアモルファスとして得た。
(実施例25f)3-クロロ-2-[1-(4-エチル-2-フルオロフェニル)-1-メトキシメチル]-5-(ヒドロキシメチル)フェノール
実施例25eで得られた粗生成物(430mg、1.23mmol)、メタノール(10mL)およびパラトルエンスルホン酸一水和物(116mg、0.61mmol)を用い、実施例24gと同様の方法により、油状の標記化合物(377mg)を得た。
実施例25eで得られた粗生成物(430mg、1.23mmol)、メタノール(10mL)およびパラトルエンスルホン酸一水和物(116mg、0.61mmol)を用い、実施例24gと同様の方法により、油状の標記化合物(377mg)を得た。
(実施例25g)酢酸 3-クロロ-4-[1-(4-エチル-2-フルオロフェニル)-1-メトキシメチル]-5-ヒドロキシベンジル
実施例25fで得られた化合物(377mg、1.16mmol)、テトラヒドロフラン(4mL)、酢酸ビニル(4mL)およびビス(ジブチル塩化スズ)オキシド(192mg、0.35mmol)を用いて、実施例2eと同様の方法により、油状の標記化合物(414mg)を得た。
実施例25fで得られた化合物(377mg、1.16mmol)、テトラヒドロフラン(4mL)、酢酸ビニル(4mL)およびビス(ジブチル塩化スズ)オキシド(192mg、0.35mmol)を用いて、実施例2eと同様の方法により、油状の標記化合物(414mg)を得た。
(実施例25h)酢酸 3-クロロ-4-(4-エチル-2-フルオロベンジル)-5-ヒドロキシベンジル
実施例25gで得られた化合物(414mg、1.13mmol)、アセトニトリル(8mL)、トリエチルシラン(0.539mL、3.38mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.213mL、1.70mmol)を用いて、実施例2fと同様の方法により、標記化合物(271mg)を白色固体として得た。
実施例25gで得られた化合物(414mg、1.13mmol)、アセトニトリル(8mL)、トリエチルシラン(0.539mL、3.38mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.213mL、1.70mmol)を用いて、実施例2fと同様の方法により、標記化合物(271mg)を白色固体として得た。
(実施例25i)5-アセトキシメチル-3-クロロ-2-(4-エチル-2-フルオロベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(236mg、0.39mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.195mL、1.93mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(292mg)、実施例25hで得られた化合物(100mg、0.30mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.049mL、0.39mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(409mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(236mg、0.39mmol)、塩化メチレン(4mL)、トリクロロアセトニトリル(0.195mL、1.93mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(6μL、0.04mmol)を用い、実施例2gと同様の方法により、イミダートを調製した。得られたイミダート(292mg)、実施例25hで得られた化合物(100mg、0.30mmol)、塩化メチレン(4mL)、MS4Aおよび三フッ化ホウ素-ジエチルエーテル錯体(0.049mL、0.39mmol)を用い、実施例2gと同様の方法により、標記化合物の粗生成物(409mg)を得た。
(実施例25j)3-クロロ-2-(4-エチル-2-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例25iで得られた粗生成物(409mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(534mg、3.89mmol)を用い実施例2hと同様の方法により、標記化合物(52mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400MHz, CD3OD): δ 1.17-1.20 (6H, m), 2.58 (2H, q, J = 7.6 Hz), 3.34-3.43 (4H, m), 4.05 (1H, dd, J = 6.4 Hz, 3.7 Hz), 4.15 (1H, d, J = 15.2 Hz), 4.24 (1H, d, J = 15.2 Hz), 4.57 (2H, s), 4.93 (1H, d, J = 7.4Hz), 6.77-6.79 (2H, m), 6.87 (1H, d, J = 11.3 Hz), 7.13 (1H, s), 7.15 (1H, s);
MS (FAB) m/z: 509 (M+K)+.。
実施例25iで得られた粗生成物(409mg)、メタノール/塩化メチレン(8mL/2mL)および炭酸カリウム(534mg、3.89mmol)を用い実施例2hと同様の方法により、標記化合物(52mg)を白色固体として得た。ただし、固体化はヘキサン/酢酸エチル/メタノールから行った。
1H NMR (400MHz, CD3OD): δ 1.17-1.20 (6H, m), 2.58 (2H, q, J = 7.6 Hz), 3.34-3.43 (4H, m), 4.05 (1H, dd, J = 6.4 Hz, 3.7 Hz), 4.15 (1H, d, J = 15.2 Hz), 4.24 (1H, d, J = 15.2 Hz), 4.57 (2H, s), 4.93 (1H, d, J = 7.4Hz), 6.77-6.79 (2H, m), 6.87 (1H, d, J = 11.3 Hz), 7.13 (1H, s), 7.15 (1H, s);
MS (FAB) m/z: 509 (M+K)+.。
(実施例26)3-フルオロ-2-(4-トリフルオロメチル)ベンジル-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.5)
(実施例26a)3-ヒドロキシ-5-オキソ-4-[4-(トリフルオロメチル)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(1.0g、5.4mmol)、塩化4-トリフルオロメチル安息香酸(1.16g、5.6mmol)、トリエチルアミン(2.3mL、16.5mmol)、トリメチルシリルシアニド(90μL、0.67mmol)およびアセトニトリル(15mL)を用いて、実施例4aと同様の方法により、標記化合物の粗生成物(2.25g)を淡黄色固体として得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(1.0g、5.4mmol)、塩化4-トリフルオロメチル安息香酸(1.16g、5.6mmol)、トリエチルアミン(2.3mL、16.5mmol)、トリメチルシリルシアニド(90μL、0.67mmol)およびアセトニトリル(15mL)を用いて、実施例4aと同様の方法により、標記化合物の粗生成物(2.25g)を淡黄色固体として得た。
(実施例26b)3-フルオロ-5-オキソ-4-[4-(トリフルオロメチル)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
実施例26aで得られた粗生成物(2.25g、5.4mmol)を塩化メチレン(20mL)に溶解し、氷冷下、ジエチルアミノ硫黄三フッ化物(2.2mL、16.7mmol)を加え、室温で2時間攪拌した。反応液に蒸留水(5mL)を滴下し、塩化メチレン(10mL)で希釈し、蒸留水(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1~4:1、V/V)を用いて精製して、標記化合物(1.31g)を黄色固体として得た。
実施例26aで得られた粗生成物(2.25g、5.4mmol)を塩化メチレン(20mL)に溶解し、氷冷下、ジエチルアミノ硫黄三フッ化物(2.2mL、16.7mmol)を加え、室温で2時間攪拌した。反応液に蒸留水(5mL)を滴下し、塩化メチレン(10mL)で希釈し、蒸留水(10mL、2回)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1~4:1、V/V)を用いて精製して、標記化合物(1.31g)を黄色固体として得た。
(実施例26c)3-フルオロ-5-ヒドロキシ-4-[4-(トリフルオロメチル)ベンゾイル]安息香酸エチル
実施例26bで得られた化合物(1.31g、3.7mmol)、トリエチルアミン(1.5mL、10.8mmol)、アセトニトリル(13mL)およびヨウ化トリメチルシラン(1.3mL、9.1mmol)を用い、実施例5cと同様の方法により、油状粗生成物であるシリルエノールエーテル体を得た。さらに実施例5cと同様の方法に従い、得られた油状粗生成物を、トルエン(13mL)、シリカゲルSK-85(5.2g)、炭酸カリウム(506mg、3.7mmol)およびエタノール(13mL)で順次処理し、シリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1、V/V)を用いて精製して標記化合物(0.82g)を淡黄色個体として得た。
実施例26bで得られた化合物(1.31g、3.7mmol)、トリエチルアミン(1.5mL、10.8mmol)、アセトニトリル(13mL)およびヨウ化トリメチルシラン(1.3mL、9.1mmol)を用い、実施例5cと同様の方法により、油状粗生成物であるシリルエノールエーテル体を得た。さらに実施例5cと同様の方法に従い、得られた油状粗生成物を、トルエン(13mL)、シリカゲルSK-85(5.2g)、炭酸カリウム(506mg、3.7mmol)およびエタノール(13mL)で順次処理し、シリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1、V/V)を用いて精製して標記化合物(0.82g)を淡黄色個体として得た。
(実施例26d)3-フルオロ-5-ヒドロキシメチル-2-{1-ヒドロキシ-1-[4-(トリフルオロメチル)フェニル]メチル}フェノール
実施例26cで得られた化合物(0.82g、2.3mmol)をテトラヒドロフラン(12mL)に溶解し、氷冷下、水素化リチウムアルミニウム(0.26g、6.5mmol)を加え、室温で15分間攪拌した。氷冷下、反応液に2M塩酸(5mL)を加え、酢酸エチル(20mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(5mL)で順次洗浄し、標記化合物の粗生成物(0.66g)を褐色油状物質として得た。
実施例26cで得られた化合物(0.82g、2.3mmol)をテトラヒドロフラン(12mL)に溶解し、氷冷下、水素化リチウムアルミニウム(0.26g、6.5mmol)を加え、室温で15分間攪拌した。氷冷下、反応液に2M塩酸(5mL)を加え、酢酸エチル(20mL)で希釈し、2M塩酸(10mL)、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(5mL)で順次洗浄し、標記化合物の粗生成物(0.66g)を褐色油状物質として得た。
(実施例26e)3-フルオロ-5-ヒドロキシメチル-2-{1-メトキシ-1-[4-(トリフルオロメチル)フェニル]メチル}フェノール
実施例26dで得られた粗生成物(0.66g、2.1mmol)をメタノール(10mL)に溶解し、パラトルエンスルホン酸一水和物(0.2g、1.1mmol)を加え、50℃で4時間撹拌した。氷冷下、反応液にトリエチルアミン(290μL、2.1mmol)を加えた。減圧下溶媒を留去した後、残渣を酢酸エチル(20mL)で希釈し、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(0.74g)を黄色油状物質として得た。
実施例26dで得られた粗生成物(0.66g、2.1mmol)をメタノール(10mL)に溶解し、パラトルエンスルホン酸一水和物(0.2g、1.1mmol)を加え、50℃で4時間撹拌した。氷冷下、反応液にトリエチルアミン(290μL、2.1mmol)を加えた。減圧下溶媒を留去した後、残渣を酢酸エチル(20mL)で希釈し、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(0.74g)を黄色油状物質として得た。
(実施例26f)酢酸 3-フルオロ-5-ヒドロキシ-4-{1-メトキシ-1-[4-(トリフルオロメチル)フェニル]メチル}ベンジル
実施例26eで得られた粗生成物(0.74g、2.1mol)をジイソプロピルエーテル(4mL)に溶解し、酢酸ビニル(4mL、43.2mmol)およびブタ膵臓リパーゼ(0.37g)を加え、35℃で24時間撹拌した。濾過後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1~2:1、V/V)を用いて精製して、標記化合物(0.43g)を淡黄色油状物質として得た。
実施例26eで得られた粗生成物(0.74g、2.1mol)をジイソプロピルエーテル(4mL)に溶解し、酢酸ビニル(4mL、43.2mmol)およびブタ膵臓リパーゼ(0.37g)を加え、35℃で24時間撹拌した。濾過後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1~2:1、V/V)を用いて精製して、標記化合物(0.43g)を淡黄色油状物質として得た。
(実施例26h)酢酸 3-フルオロ-5-ヒドロキシ-4-[4-(トリフルオロメチル)ベンジル]ベンジル
実施例26gで得られた化合物(0.43g、1.2mmol)をアセトニトリル(5mL)に溶解し、氷冷下、トリエチルシラン(550μL、3.5mmol)および三フッ化ホウ素-ジエチルエーテル錯体(440μL、3.5mmol)を加え、50℃で4時間撹拌した。氷冷下、反応液に飽和炭酸水素ナトリウム水溶液(5mL)を加え、酢酸エチル(20mL)で希釈し、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1~4:1、V/V)を用いて精製して、標記化合物(0.14g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.12 (3H, s), 4.05 (2H, s), 5.01 (2H, s), 5.13 (1H, brs), 6.61 (1H, s), 6.71 (1H, d, J = 9.8 Hz), 7.39 (2H, d, J = 8.0 Hz), 7.52 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 342 (M)+.
実施例26gで得られた化合物(0.43g、1.2mmol)をアセトニトリル(5mL)に溶解し、氷冷下、トリエチルシラン(550μL、3.5mmol)および三フッ化ホウ素-ジエチルエーテル錯体(440μL、3.5mmol)を加え、50℃で4時間撹拌した。氷冷下、反応液に飽和炭酸水素ナトリウム水溶液(5mL)を加え、酢酸エチル(20mL)で希釈し、飽和炭酸水素ナトリウム水溶液(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1~4:1、V/V)を用いて精製して、標記化合物(0.14g)を無色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.12 (3H, s), 4.05 (2H, s), 5.01 (2H, s), 5.13 (1H, brs), 6.61 (1H, s), 6.71 (1H, d, J = 9.8 Hz), 7.39 (2H, d, J = 8.0 Hz), 7.52 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 342 (M)+.
(実施例26i)5-アセトキシメチル-3-フルオロ-[4-(トリフルオロメチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(275mg、0.45mmol)、トリクロロアセトニトリル(135μL、1.35mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、実施例26hで得られた化合物(0.14g、0.41mmol)、三フッ化ホウ素-ジエチルエーテル錯体(51μL、0.41mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(339mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(275mg、0.45mmol)、トリクロロアセトニトリル(135μL、1.35mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(7μL、0.05mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、実施例26hで得られた化合物(0.14g、0.41mmol)、三フッ化ホウ素-ジエチルエーテル錯体(51μL、0.41mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(339mg)を得た。
(実施例26j)3-フルオロ-5-ヒドロキシメチル-2-[4-(トリフルオロメチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例26iで得られた粗生成物(339mg、0.36mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(56mg、0.41mmol)を用い、実施例4jと同様の方法により、標記化合物(100mg)を無色固体として得た。
1H NMR (500 MHz, CD3OD): δ 1.19 (3H, d, J=6.4 Hz), 3.33-3.40 (2H, m), 3.43-3.49 (2H, m), 4.03-4.07 (1H, m), 4.10 (1H, d, J = 14.6 Hz), 4.16 (1H, d, J = 14.6 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.3 Hz), 6.81 (1H, d, J = 10.3 Hz), 7.00 (1H, s), 7.46 (2H, d, J = 8.3 Hz), 7.49 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 477 (M+H)+, 499 (M+Na)+.。
実施例26iで得られた粗生成物(339mg、0.36mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(56mg、0.41mmol)を用い、実施例4jと同様の方法により、標記化合物(100mg)を無色固体として得た。
1H NMR (500 MHz, CD3OD): δ 1.19 (3H, d, J=6.4 Hz), 3.33-3.40 (2H, m), 3.43-3.49 (2H, m), 4.03-4.07 (1H, m), 4.10 (1H, d, J = 14.6 Hz), 4.16 (1H, d, J = 14.6 Hz), 4.55 (2H, s), 4.95 (1H, d, J = 7.3 Hz), 6.81 (1H, d, J = 10.3 Hz), 7.00 (1H, s), 7.46 (2H, d, J = 8.3 Hz), 7.49 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 477 (M+H)+, 499 (M+Na)+.。
(実施例27)3-クロロ-2-(4-トリフルオロメチル)ベンジル-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.11)
(実施例27a)3-クロロ-5-オキソ-4-[4-(トリフルオロメチル)ベンゾイル]シクロヘキサ-3-エンカルボン酸エチル
実施例26aで得られた粗生成物(2.25g、5.4mmol)、二塩化オキサリル(500μL、3.8mmol)、2-メチル-2-ブテン(2.3mL、21.7mmol)、N,N-ジメチルホルムアミド(100μL、1.29mmol)および塩化メチレン(20mL)を用い、実施例4bと同様の方法により、標記化合物の粗生成物(2.5g)を褐色油状物質として得た。
実施例26aで得られた粗生成物(2.25g、5.4mmol)、二塩化オキサリル(500μL、3.8mmol)、2-メチル-2-ブテン(2.3mL、21.7mmol)、N,N-ジメチルホルムアミド(100μL、1.29mmol)および塩化メチレン(20mL)を用い、実施例4bと同様の方法により、標記化合物の粗生成物(2.5g)を褐色油状物質として得た。
(実施例27b)3-クロロ-5-ヒドロキシ-4-[4-(トリフルオロメチル)ベンゾイル]安息香酸エチル
実施例27aで得られた粗生成物(2.5g)、N-メチルモルホリン(10mL)およびヨウ素(1.65g、6.5mmol)を用い、実施例4cと同様の方法により、標記化合物(1.66g)を褐色固体として得た。ただし、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1~3:1、V/V)を用いて行った。
実施例27aで得られた粗生成物(2.5g)、N-メチルモルホリン(10mL)およびヨウ素(1.65g、6.5mmol)を用い、実施例4cと同様の方法により、標記化合物(1.66g)を褐色固体として得た。ただし、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、5:1~4:1~3:1、V/V)を用いて行った。
(実施例27c)3-クロロ-5-ヒドロキシメチル-2-{1-ヒドロキシ-1-[4-(トリフルオロメチル)フェニル]メチル}フェノール
実施例27bで得られた化合物(1.66g、4.5mmol)、水素化リチウムアルミニウム(0.51g、13.4mmol)およびテトラヒドロフラン(17mL)を用い、実施例26dと同様の方法により、標記化合物の粗生成物(1.44g)を無色固体として得た。
実施例27bで得られた化合物(1.66g、4.5mmol)、水素化リチウムアルミニウム(0.51g、13.4mmol)およびテトラヒドロフラン(17mL)を用い、実施例26dと同様の方法により、標記化合物の粗生成物(1.44g)を無色固体として得た。
(実施例27d)3-クロロ-5-ヒドロキシメチル-2-{1-メトキシ-1-[4-(トリフルオロメチル)フェニル]メチル}フェノール
実施例27cで得られた粗生成物(1.44g、4.3mmol)、パラトルエンスルホン酸一水和物(0.41g、2.2mmol)およびメタノール(15mL)を用いて、実施例26eと同様の方法により、標記化合物の粗生成物を得た。次いで、シリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、4:1~3:1~2:1、V/V)を用いて精製して標記化合物(0.31g)を無色油状物質として得た。
実施例27cで得られた粗生成物(1.44g、4.3mmol)、パラトルエンスルホン酸一水和物(0.41g、2.2mmol)およびメタノール(15mL)を用いて、実施例26eと同様の方法により、標記化合物の粗生成物を得た。次いで、シリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、4:1~3:1~2:1、V/V)を用いて精製して標記化合物(0.31g)を無色油状物質として得た。
(実施例27e)酢酸 3-クロロ-5-ヒドロキシ-4-{1-メトキシ-1-[4-(トリフルオロメチル)フェニル]メチル}ベンジル
実施例27dで得られた化合物(0.31g、0.89mmol)、酢酸ビニル(3mL、32.4mmol)、ブタ膵臓リパーゼ(0.16g)およびジイソプロピルエーテル(3mL)を用いて、実施例26fと同様の方法により、標記化合物(0.33g)を黄色油状物質として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1~4:1、V/V)を用いて行った。
実施例27dで得られた化合物(0.31g、0.89mmol)、酢酸ビニル(3mL、32.4mmol)、ブタ膵臓リパーゼ(0.16g)およびジイソプロピルエーテル(3mL)を用いて、実施例26fと同様の方法により、標記化合物(0.33g)を黄色油状物質として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1~4:1、V/V)を用いて行った。
(実施例27f)酢酸 3-クロロ-5-ヒドロキシ-2-[4-(トリフルオロメチル)ベンジル]ベンジル
実施例27eで得られた化合物(0.33g、0.85mmol)、トリエチルシラン(400μL、2.5mmol)、三フッ化ホウ素-ジエチルエーテル錯体(320μL、2.5mmol)およびアセトニトリル(3mL)を用いて、実施例26hと同様の方法により、3-クロロ-5-ヒドロキシメチル-2-[4-(トリフルオロメチル)ベンジル]フェノール(100mg)を得た。この化合物、酢酸ビニル(2mL、21.6mmol)、ビス(ジブチル塩化スズ)オキシド(16mg、0.03mmol)およびテトラヒドロフラン(2mL)を用いて、実施例21hと同様の方法により、標記化合物(95mg)を無色固体として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1、V/V)を用いて行った。
1H NMR (500 MHz, CDCl3): δ 2.12 (3H, s), 4.21 (2H, s), 5.00 (2H, s), 5.09 (1H, s), 6.72 (1H, s), 7.02 (1H, s), 7.37 (2H, d, J = 8.0 Hz), 7.50 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 358 (M)+.。
実施例27eで得られた化合物(0.33g、0.85mmol)、トリエチルシラン(400μL、2.5mmol)、三フッ化ホウ素-ジエチルエーテル錯体(320μL、2.5mmol)およびアセトニトリル(3mL)を用いて、実施例26hと同様の方法により、3-クロロ-5-ヒドロキシメチル-2-[4-(トリフルオロメチル)ベンジル]フェノール(100mg)を得た。この化合物、酢酸ビニル(2mL、21.6mmol)、ビス(ジブチル塩化スズ)オキシド(16mg、0.03mmol)およびテトラヒドロフラン(2mL)を用いて、実施例21hと同様の方法により、標記化合物(95mg)を無色固体として得た。なお、精製はシリカゲルフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル、6:1~5:1、V/V)を用いて行った。
1H NMR (500 MHz, CDCl3): δ 2.12 (3H, s), 4.21 (2H, s), 5.00 (2H, s), 5.09 (1H, s), 6.72 (1H, s), 7.02 (1H, s), 7.37 (2H, d, J = 8.0 Hz), 7.50 (2H, d, J = 8.0 Hz);
MS (FAB) m/z: 358 (M)+.。
(実施例27g)5-アセトキシメチル-3-クロロ-2-[4-(トリフルオロメチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(194mg、0.32mmol)、トリクロロアセトニトリル(95μL、0.95mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(5μL、0.03mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、実施例27fで得られた化合物(95mg、0.26mmol)、三フッ化ホウ素-ジエチルエーテル錯体(33μL、0.26mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(313mg)を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-D-グルコ-ヘプトピラノシド(194mg、0.32mmol)、トリクロロアセトニトリル(95μL、0.95mmol)、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(5μL、0.03mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法によりイミダートを調製し、実施例27fで得られた化合物(95mg、0.26mmol)、三フッ化ホウ素-ジエチルエーテル錯体(33μL、0.26mmol)および塩化メチレン(5mL)を用い、実施例4iと同様の方法により、標記化合物の粗生成物(313mg)を得た。
(実施例27h)3-クロロ-5-ヒドロキシメチル-2-[4-(トリフルオロメチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例27gで得られた粗生成物(313mg、0.26mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(36mg、0.26mmol)を用い、実施例4jと同様の方法により、標記化合物(84mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.7 Hz), 3.34-3.46 (4H, m), 4.03-4.09 (1H, m), 4.26 (1H, d, J = 14.8 Hz), 4.35 (1H, d, J = 14.8 Hz), 4.56 (2H, s), 4.96 (1H, d, J = 7.5 Hz), 7.13 (1H, s), 7.16 (1H, s), 7.44 (2H, d, J = 8.2 Hz), 7.50 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 493 (M+H)+, 515 (M+Na)+.。
実施例27gで得られた粗生成物(313mg、0.26mmol)、テトラヒドロフラン(1mL)、メタノール(4mL)および炭酸カリウム(36mg、0.26mmol)を用い、実施例4jと同様の方法により、標記化合物(84mg)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.20 (3H, d, J = 6.7 Hz), 3.34-3.46 (4H, m), 4.03-4.09 (1H, m), 4.26 (1H, d, J = 14.8 Hz), 4.35 (1H, d, J = 14.8 Hz), 4.56 (2H, s), 4.96 (1H, d, J = 7.5 Hz), 7.13 (1H, s), 7.16 (1H, s), 7.44 (2H, d, J = 8.2 Hz), 7.50 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 493 (M+H)+, 515 (M+Na)+.。
(実施例28)2-[4-(2,2-ジフルオロエチル)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.20)
(実施例28a)4-(2,2-ジフルオロアセチル)安息香酸メチル
4-アセチル安息香酸メチル(1.0g、5.6mmol)をベンゼン(28mL)およびヘキサン(56mL)からなる溶液に溶解し、モルホリン(2.3mL、26mmol)および三塩化チタン(0.42mL、3.9mmol)を溶解させたヘキサン溶液(15mL)を用いて、文献既知(J.Org.Chem.,2005,70(14),5763.)の方法により、相当するエナミンの粗生成物を黄色油状物質として得た。このエナミンを精製せず、アセトニトリル(120mL)に溶解し、MS4A(10g)および1-クロロメチル-4-フルオロ-1,4-ジアゾニアビシクロ[2.2.2]オクタン ビス(テトラフルオロホウ酸)(Aldrich社から購入、Selectfluor、3.9g、11mmol)を用いて、上記文献に従い、標記化合物の粗生成物(1.2g)を得た。
4-アセチル安息香酸メチル(1.0g、5.6mmol)をベンゼン(28mL)およびヘキサン(56mL)からなる溶液に溶解し、モルホリン(2.3mL、26mmol)および三塩化チタン(0.42mL、3.9mmol)を溶解させたヘキサン溶液(15mL)を用いて、文献既知(J.Org.Chem.,2005,70(14),5763.)の方法により、相当するエナミンの粗生成物を黄色油状物質として得た。このエナミンを精製せず、アセトニトリル(120mL)に溶解し、MS4A(10g)および1-クロロメチル-4-フルオロ-1,4-ジアゾニアビシクロ[2.2.2]オクタン ビス(テトラフルオロホウ酸)(Aldrich社から購入、Selectfluor、3.9g、11mmol)を用いて、上記文献に従い、標記化合物の粗生成物(1.2g)を得た。
(実施例28b)4-(2,2-ジフルオロ-1-ヒドロキシエチル)安息香酸メチル
実施例28aで得られた粗生成物(1.2g、5.6mmol)をメタノール(5mL)およびテトラヒドロフラン(5mL)からなる溶液に溶解し、氷冷下、水素化ホウ素ナトリウム(0.32g、8.4mmol)を加えた。反応液を室温に昇温しつつ2時間撹拌し、氷冷下、飽和塩化アンモニウム水溶液(1mL)を滴下して反応を停止した。混合物を酢酸エチル(50mL)で希釈後、飽和炭酸水素ナトリウム(20mL、2回)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.74g)を無色固体として得た。
実施例28aで得られた粗生成物(1.2g、5.6mmol)をメタノール(5mL)およびテトラヒドロフラン(5mL)からなる溶液に溶解し、氷冷下、水素化ホウ素ナトリウム(0.32g、8.4mmol)を加えた。反応液を室温に昇温しつつ2時間撹拌し、氷冷下、飽和塩化アンモニウム水溶液(1mL)を滴下して反応を停止した。混合物を酢酸エチル(50mL)で希釈後、飽和炭酸水素ナトリウム(20mL、2回)および飽和食塩水(20mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、3:1、V/V)を用いて精製して、標記化合物(0.74g)を無色固体として得た。
(実施例28c)4-(2,2-ジフルオロエチル)安息香酸メチル
実施例28bで合成した化合物(0.69g、3.2mmol)をテトラヒドロフラン(10mL)に溶解し、室温でチオカルボニルジイミダゾール(0.86g、4.8mmol)を加え、加熱還流下、1時間撹拌した。混合物を冷却後、酢酸エチル(50mL)で希釈し、1M塩酸(5mL)、飽和炭酸水素ナトリウム(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、キサンタートの粗生成物を得た。この粗生成物を精製せずトルエン(10mL)に溶解し、脱気後、窒素気流下で水素化トリブチルスズ(1.2mL、4.6mmol)およびアゾビスイソブチロニトリル(0.10g、0.61mmol)を加えた。混合物を90℃で2時間撹拌し、反応液を直接シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、10:1、V/V)を用いて精製して、標記化合物(0.54g)を無色固体として得た。
実施例28bで合成した化合物(0.69g、3.2mmol)をテトラヒドロフラン(10mL)に溶解し、室温でチオカルボニルジイミダゾール(0.86g、4.8mmol)を加え、加熱還流下、1時間撹拌した。混合物を冷却後、酢酸エチル(50mL)で希釈し、1M塩酸(5mL)、飽和炭酸水素ナトリウム(10mL)および飽和食塩水(10mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、キサンタートの粗生成物を得た。この粗生成物を精製せずトルエン(10mL)に溶解し、脱気後、窒素気流下で水素化トリブチルスズ(1.2mL、4.6mmol)およびアゾビスイソブチロニトリル(0.10g、0.61mmol)を加えた。混合物を90℃で2時間撹拌し、反応液を直接シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、10:1、V/V)を用いて精製して、標記化合物(0.54g)を無色固体として得た。
(実施例28d)4-(2,2-ジフルオロエチル)安息香酸
実施例28cで得られた化合物(0.53g、2.6mmol)を1,4-ジオキサン(3.4mL)に溶解し、室温で2M水酸化ナトリウム水溶液(3.4mL、6.8mmol)を加えて35℃で1時間撹拌した。反応液を1M塩酸(20mL)に注ぎ、酢酸エチル(50mL)で抽出後、有機層を飽和食塩水(10mL)で洗浄した。これを無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(0.48g)を無色固体として得た。
実施例28cで得られた化合物(0.53g、2.6mmol)を1,4-ジオキサン(3.4mL)に溶解し、室温で2M水酸化ナトリウム水溶液(3.4mL、6.8mmol)を加えて35℃で1時間撹拌した。反応液を1M塩酸(20mL)に注ぎ、酢酸エチル(50mL)で抽出後、有機層を飽和食塩水(10mL)で洗浄した。これを無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、標記化合物の粗生成物(0.48g)を無色固体として得た。
(実施例28e)塩化4-(2,2-ジフルオロエチル)ベンゾイル
実施例28dで得られた粗生成物(0.48g、2.6mmol)を塩化メチレンに溶解し、氷冷下、塩化オキサリル(1.1mL、13mmol)およびN,N-ジメチルホルムアミド(0.1mL、1.3mmol)を加え、室温に昇温しつつ3時間半撹拌した。反応終了後、減圧下溶媒を除去し、標記化合物の粗生成物(0.53g)を無色油状物質として得た。
実施例28dで得られた粗生成物(0.48g、2.6mmol)を塩化メチレンに溶解し、氷冷下、塩化オキサリル(1.1mL、13mmol)およびN,N-ジメチルホルムアミド(0.1mL、1.3mmol)を加え、室温に昇温しつつ3時間半撹拌した。反応終了後、減圧下溶媒を除去し、標記化合物の粗生成物(0.53g)を無色油状物質として得た。
(実施例28f)4-[4-(2,2-ジフルオロエチル)ベンゾイル]-3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(EP1571148A1)(0.48g、2.6mmol)および実施例28eで得られた粗生成物(0.53g、2.6mmol)を用い、実施例1bと同様の方法で標記化合物の粗生成物(0.92g)を得た。
3-ヒドロキシ-5-オキソシクロヘキサ-3-エンカルボン酸エチル(EP1571148A1)(0.48g、2.6mmol)および実施例28eで得られた粗生成物(0.53g、2.6mmol)を用い、実施例1bと同様の方法で標記化合物の粗生成物(0.92g)を得た。
(実施例28g)4-[4-(2,2-ジフルオロエチル)ベンゾイル]-3-フルオロ-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例28fで得られた粗生成物(0.95g、2.7mmol)を塩化メチレン(10mL)に溶解し、ジエチルアミノ硫黄三フッ化物(1.1mL、8.1mmol)を用いて実施例3bと同様の方法により、標記化合物(0.47g)を得た。
実施例28fで得られた粗生成物(0.95g、2.7mmol)を塩化メチレン(10mL)に溶解し、ジエチルアミノ硫黄三フッ化物(1.1mL、8.1mmol)を用いて実施例3bと同様の方法により、標記化合物(0.47g)を得た。
(実施例28h)4-[4-(2,2-ジフルオロエチル)ベンゾイル]-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例28gで得られた化合物(0.47g、1.3mmol)をN-メチルモルホリン(6mL)に溶解し、無水硫酸ナトリウム(0.47g)およびヨウ素(0.40g、1.6mmol)を用いて実施例3cと同様の方法により標記化合物の粗生成物(0.46g)を得た。
実施例28gで得られた化合物(0.47g、1.3mmol)をN-メチルモルホリン(6mL)に溶解し、無水硫酸ナトリウム(0.47g)およびヨウ素(0.40g、1.6mmol)を用いて実施例3cと同様の方法により標記化合物の粗生成物(0.46g)を得た。
(実施例28i)4-{1-[4-(2,2-ジフルオロエチル)フェニル]-1-ヒドロキシメチル}-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例28hで得られた粗生成物(0.46g、1.3mmol)をテトラヒドロフラン(3mL)およびメタノール(2mL)からなる溶液に溶解し、水素化ホウ素ナトリウム(98mg、2.6mmol)を用いて実施例3dと同様の方法により標記化合物(0.33g)を無色固体として得た。
実施例28hで得られた粗生成物(0.46g、1.3mmol)をテトラヒドロフラン(3mL)およびメタノール(2mL)からなる溶液に溶解し、水素化ホウ素ナトリウム(98mg、2.6mmol)を用いて実施例3dと同様の方法により標記化合物(0.33g)を無色固体として得た。
(実施例28j)4-[4-(2,2-ジフルオロエチル)ベンジル]-3-フルオロ-5-ヒドロキシ安息香酸エチル
実施例28iで得られた化合物(0.33g、0.93mmol)をエタノール(10mL)に溶解し、10%塩化水素メタノール(0.33mL、0.93mmol)および10%パラジウム炭素触媒(含水、0.5g)を用い実施例3eと同様の方法により、標記化合物の粗生成物(0.31g)を得た。
実施例28iで得られた化合物(0.33g、0.93mmol)をエタノール(10mL)に溶解し、10%塩化水素メタノール(0.33mL、0.93mmol)および10%パラジウム炭素触媒(含水、0.5g)を用い実施例3eと同様の方法により、標記化合物の粗生成物(0.31g)を得た。
(実施例28k)2-[4-(2,2-ジフルオロエチル)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェノール
実施例28jで得られた粗生成物(0.31g、0.93mmol)をテトラヒドロフラン(10mL)に溶解し、水素化アルミニウムリチウム(0.14g、3.7mmol)を用い実施例3fと同様の方法により標記化合物の粗生成物(0.31g)を無色固体として得た。
実施例28jで得られた粗生成物(0.31g、0.93mmol)をテトラヒドロフラン(10mL)に溶解し、水素化アルミニウムリチウム(0.14g、3.7mmol)を用い実施例3fと同様の方法により標記化合物の粗生成物(0.31g)を無色固体として得た。
(実施例28l)酢酸 4-[4-(2,2-ジフルオロエチル)ベンジル]-3-フルオロ-5-ヒドロキシベンジル
実施例28kで得られた粗生成物(0.31g、0.93mmol)を酢酸ビニル(3mL)およびジイソプロピルエーテル(3mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を用い実施例3gと同様の方法により標記化合物(0.30g)を微褐色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.11 (3H, s), 3.09 (2H, dt, J = 17.4, 4.3 Hz), 3.99 (2H, s), 5.01 (2H, s), 5.88 (1H, tt, J = 56.7, 4.1 Hz), 6.60 (1H, s), 6.70 (1H, d, J = 9.4 Hz), 7.16 (2H, d, J = 7.8 Hz), 7.26 (2H, d, J = 7.8 Hz).。
実施例28kで得られた粗生成物(0.31g、0.93mmol)を酢酸ビニル(3mL)およびジイソプロピルエーテル(3mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(1.0g)を用い実施例3gと同様の方法により標記化合物(0.30g)を微褐色固体として得た。
1H NMR (400 MHz, CDCl3): δ 2.11 (3H, s), 3.09 (2H, dt, J = 17.4, 4.3 Hz), 3.99 (2H, s), 5.01 (2H, s), 5.88 (1H, tt, J = 56.7, 4.1 Hz), 6.60 (1H, s), 6.70 (1H, d, J = 9.4 Hz), 7.16 (2H, d, J = 7.8 Hz), 7.26 (2H, d, J = 7.8 Hz).。
(実施例28m)5-アセトキシメチル-3-フルオロ-2-[4-(2,2-ジフルオロエチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.30mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4μL、0.03mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.75g、1.0mmol)、実施例28lで得られた化合物(0.30g、0.89mmol)および三フッ化ホウ素-ジエチルエーテル錯体(56μL、0.44mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(0.61g、1.0mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.30mL、3.0mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(4μL、0.03mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.75g、1.0mmol)、実施例28lで得られた化合物(0.30g、0.89mmol)および三フッ化ホウ素-ジエチルエーテル錯体(56μL、0.44mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例28n)3-フルオロ-2-[4-(2,2-ジフルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例28mで得られた混合物(0.89mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(0.12g)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 3.05 (2H, dt, J = 17.5, 4.6 Hz), 3.33-3.50 (4H, m), 3.99 (1H, d, J = 14.5 Hz), 4.03-4.08 (1H, m), 4.08 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.94 (1H, d, J = 7.5 Hz), 5.92 (1H, tt, J = 56.8, 4.6 Hz), 6.79 (1H, d, J = 10.1 Hz), 6.99 (1H, s), 7.12 (2H, d, J = 8.2 Hz), 7.25 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 495 (M+Na)+.。
実施例28mで得られた混合物(0.89mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)からなる溶液に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(0.12g)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 3.05 (2H, dt, J = 17.5, 4.6 Hz), 3.33-3.50 (4H, m), 3.99 (1H, d, J = 14.5 Hz), 4.03-4.08 (1H, m), 4.08 (1H, d, J = 14.5 Hz), 4.55 (2H, s), 4.94 (1H, d, J = 7.5 Hz), 5.92 (1H, tt, J = 56.8, 4.6 Hz), 6.79 (1H, d, J = 10.1 Hz), 6.99 (1H, s), 7.12 (2H, d, J = 8.2 Hz), 7.25 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 495 (M+Na)+.。
(実施例29)3-クロロ-2-[4-(2,2-ジフルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.21)
(実施例29a)3-クロロ-4-[4-(2,2-ジフルオロエチル)ベンゾイル]-5-オキソシクロヘキサ-3-エンカルボン酸エチル
実施例28fで得られた粗生成物(0.92g、2.6mmol)を塩化メチレン(10mL)に溶解し、2-メチル-2-ブテン(1.1mL、10mmol)、二塩化オキサリル(0.23mL、2.7mmol)およびN,N-ジメチルホルムアミド(0.05mL、0.07mmol)を用いて実施例1bと同様の方法により、標記化合物の粗生成物(0.96g)を得た。
実施例28fで得られた粗生成物(0.92g、2.6mmol)を塩化メチレン(10mL)に溶解し、2-メチル-2-ブテン(1.1mL、10mmol)、二塩化オキサリル(0.23mL、2.7mmol)およびN,N-ジメチルホルムアミド(0.05mL、0.07mmol)を用いて実施例1bと同様の方法により、標記化合物の粗生成物(0.96g)を得た。
(実施例29b)3-クロロ-4-[4-(2,2-ジフルオロエチル)ベンゾイル]-5-ヒドロキシ安息香酸エチル
実施例29aで得られた粗生成物(0.96g、2.6mmol)をN-メチルモルホリン(12mL)に溶解し、無水硫酸ナトリウム(0.96g)およびヨウ素(0.79g、3.1mmol)を用いて実施例3cと同様の方法により標記化合物の粗生成物(0.96g)を得た。
実施例29aで得られた粗生成物(0.96g、2.6mmol)をN-メチルモルホリン(12mL)に溶解し、無水硫酸ナトリウム(0.96g)およびヨウ素(0.79g、3.1mmol)を用いて実施例3cと同様の方法により標記化合物の粗生成物(0.96g)を得た。
(実施例29c)3-クロロ-2-{1-[4-(2,2-ジフルオロエチル)フェニル]-1-ヒドロキシメチル}-5-(ヒドロキシメチル)フェノール
実施例29bで得られた粗生成物(0.96g、2.6mmol)をテトラヒドロフラン(30mL)に溶解し、水素化アルミニウムリチウム(0.38g、10mmol)を用いて実施例1eと同様の方法により、標記化合物(0.85g)を粗生成物として得た。
実施例29bで得られた粗生成物(0.96g、2.6mmol)をテトラヒドロフラン(30mL)に溶解し、水素化アルミニウムリチウム(0.38g、10mmol)を用いて実施例1eと同様の方法により、標記化合物(0.85g)を粗生成物として得た。
(実施例29d)酢酸 3-クロロ-4-{1-[4-(2,2-ジフルオロエチル)フェニル]-1-ヒドロキシメチル}-5-ヒドロキシベンジル
実施例29cで得られた粗生成物(0.85g、2.6mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(2.0g)を用い実施例3gと同様の方法により標記化合物(0.51g)を無色固体として得た。
実施例29cで得られた粗生成物(0.85g、2.6mmol)を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)からなる溶液に溶解し、ブタ膵臓リパーゼ(2.0g)を用い実施例3gと同様の方法により標記化合物(0.51g)を無色固体として得た。
(実施例29e)酢酸 3-クロロ-4-[4-(2,2-ジフルオロエチル)ベンジル]-5-ヒドロキシベンジル
実施例29dで得られた化合物(0.40g、1.1mmol)をアセトニトリル(10mL)に溶解し、トリエチルシラン(0.53mL、3.3mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.28mL、2.2mmol)を用いて実施例5fと同様の方法(ただし、精製は行っていない。)により、標記化合物を含む混合物を得た。
実施例29dで得られた化合物(0.40g、1.1mmol)をアセトニトリル(10mL)に溶解し、トリエチルシラン(0.53mL、3.3mmol)および三フッ化ホウ素-ジエチルエーテル錯体(0.28mL、2.2mmol)を用いて実施例5fと同様の方法(ただし、精製は行っていない。)により、標記化合物を含む混合物を得た。
得られた混合物をメタノール(2.5mL)に溶解し、p-トルエンスルホン酸一水和物(72mg、0.38mmol)を加え50℃で1時間半撹拌し、酢酸エチル(20mL)で希釈後、飽和炭酸水素ナトリウム水溶液(5mL)および飽和食塩水(5mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去して得られた残渣をシリカゲルフラッシュカラムクロマトグラフィー(塩化メチレン:メタノール、98:2、V/V)を用いて精製し、標記化合物の脱アセチル体(0.16g)を無色固体として得た。
この脱アセチル体を酢酸ビニル(5mL)およびジイソプロピルエーテル(5mL)に溶解し、ブタ膵臓リパーゼ(2.0g)を用い、実施例3gと同様の方法により標記化合物(0.13g)を無色固体として得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 3.09 (2H, dt, J = 17.4, 4.6 Hz), 4.15 (2H, s), 5.00 (2H, s), 5.05 (1H, s), 5.88 (1H, tt, J = 56.7, 4.6 Hz), 6.72 (1H, d, J = 1.5 Hz), 7.02 (1H, d, J = 1.5 Hz), 7.15 (2H, d, J = 8.3 Hz), 7.23 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 354 (M)+.。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 3.09 (2H, dt, J = 17.4, 4.6 Hz), 4.15 (2H, s), 5.00 (2H, s), 5.05 (1H, s), 5.88 (1H, tt, J = 56.7, 4.6 Hz), 6.72 (1H, d, J = 1.5 Hz), 7.02 (1H, d, J = 1.5 Hz), 7.15 (2H, d, J = 8.3 Hz), 7.23 (2H, d, J = 8.3 Hz);
MS (FAB) m/z: 354 (M)+.。
(実施例29f)5-アセトキシメチル-3-クロロ-2-[4-(2,2-ジフルオロエチル)ベンジル]フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(0.37g、0.60mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.18mL、1.8mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(2μL、0.02mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.45g、0.60mmol)、実施例29eで得られた化合物(0.13g、0.36mmol)および三フッ化ホウ素-ジエチルエーテル錯体(46μL、0.37mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(0.37g、0.60mmol)を塩化メチレン(4mL)に溶解し、トリクロロアセトニトリル(0.18mL、1.8mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(2μL、0.02mmol)を用いて実施例1hと同様の方法により、イミダートを調製した。このイミダート(0.45g、0.60mmol)、実施例29eで得られた化合物(0.13g、0.36mmol)および三フッ化ホウ素-ジエチルエーテル錯体(46μL、0.37mmol)を用いて実施例1hと同様の方法により、標記化合物を含む混合物を得た。
(実施例29g)3-クロロ-2-[4-(2,2-ジフルオロエチル)ベンジル]-5-ヒドロキシメチル-フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例29fで得られた混合物(0.37mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(0.12g)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 3.05 (2H, dt, J = 17.5, 4.6 Hz), 3.34-3.46 (4H, m), 4.03-4.09 (1H, m), 4.14 (1H, d, J = 14.7 Hz), 4.27 (1H, d, J = 14.7 Hz), 4.55 (2H, s), 4.94 (1H, d, J = 7.5 Hz), 5.93 (1H, tt, J = 56.7, 4.7 Hz), 7.10-7.13 (4H, m), 7.22 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 511 (M+Na)+.。
実施例29fで得られた混合物(0.37mmol)をテトラヒドロフラン(3mL)およびメタノール(3mL)に溶解し、2M水酸化ナトリウム水溶液(3.0mL、6.0mmol)を用いて実施例3iと同様の方法により、標記化合物(0.12g)を無色固体として得た。
1H NMR (400 MHz, CD3OD): δ 1.21 (3H, d, J = 6.3 Hz), 3.05 (2H, dt, J = 17.5, 4.6 Hz), 3.34-3.46 (4H, m), 4.03-4.09 (1H, m), 4.14 (1H, d, J = 14.7 Hz), 4.27 (1H, d, J = 14.7 Hz), 4.55 (2H, s), 4.94 (1H, d, J = 7.5 Hz), 5.93 (1H, tt, J = 56.7, 4.7 Hz), 7.10-7.13 (4H, m), 7.22 (2H, d, J = 8.2 Hz);
MS (FAB) m/z: 511 (M+Na)+.。
(実施例30)3-フルオロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.7)
(実施例30a)3,5-ジフルオロ-4-[1-(2-フルオロ-4-メチルフェニル)-1-ヒドロキシメチル]ベンゾニトリル
ジイソプロピルアミン(1.12mL、7.92mmol)、n-ブチルリチウム(2.59mL、7.17mmol、2.77M n-ヘキサン溶液)、3,5-ジフルオロベンゾニトリル(1.00g、7.19mmol)、2-フルオロ-4-メチルベンズアルデヒド(1.00g、7.24mmol)およびテトラヒドロフラン(20mL)を用い、実施例7aと同様の方法で標記化合物(1.44g)を淡黄色固体として得た。
ジイソプロピルアミン(1.12mL、7.92mmol)、n-ブチルリチウム(2.59mL、7.17mmol、2.77M n-ヘキサン溶液)、3,5-ジフルオロベンゾニトリル(1.00g、7.19mmol)、2-フルオロ-4-メチルベンズアルデヒド(1.00g、7.24mmol)およびテトラヒドロフラン(20mL)を用い、実施例7aと同様の方法で標記化合物(1.44g)を淡黄色固体として得た。
(実施例30b)3,5-ジフルオロ-4-(2-フルオロ-4-メチルベンジル)ベンゾニトリル
水素化ホウ素ナトリウム(1.69g、44.7mmol)をトリフルオロ酢酸(30mL)に溶解して0℃に冷却後、実施例30aで得られた化合物(1.24g、4.47mmol)を溶解させた塩化メチレン溶液(7mL)を加えた。その後、反応液を室温で30時間攪拌した。ただし、その間、反応が終了するまで、数回に分けて水素化ホウ素ナトリウム(2.00g、52.9mmol)およびトリフルオロ酢酸(20mL)を追加した。続いて、反応液を、0℃に冷却した10%水酸化ナトリウム水溶液(300mL)に加えた。ジエチルエーテル(300mL)で抽出し、水(100mL)、飽和塩化アンモニウム水溶液(100mL)および飽和食塩水(100mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~10:1、V/V)を用いて精製して、標記化合物(756mg)を白色固体として得た。
水素化ホウ素ナトリウム(1.69g、44.7mmol)をトリフルオロ酢酸(30mL)に溶解して0℃に冷却後、実施例30aで得られた化合物(1.24g、4.47mmol)を溶解させた塩化メチレン溶液(7mL)を加えた。その後、反応液を室温で30時間攪拌した。ただし、その間、反応が終了するまで、数回に分けて水素化ホウ素ナトリウム(2.00g、52.9mmol)およびトリフルオロ酢酸(20mL)を追加した。続いて、反応液を、0℃に冷却した10%水酸化ナトリウム水溶液(300mL)に加えた。ジエチルエーテル(300mL)で抽出し、水(100mL)、飽和塩化アンモニウム水溶液(100mL)および飽和食塩水(100mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~10:1、V/V)を用いて精製して、標記化合物(756mg)を白色固体として得た。
(実施例30c)3-ベンジルオキシ-5-フルオロ-4-(2-フルオロ-4-メチルベンジル)ベンゾニトリル
実施例30bで得られた化合物(756mg、2.89mmol)、水素化ナトリウム(63%、165mg、4.33mmol)、ベンジルアルコール(387mg、3.58mmol)およびN,N-ジメチルホルムアミド(10mL)を用い、実施例7cと同様の方法で、標記化合物(768mg)を白色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル19:1~10:1、V/V)を用いて行った。
実施例30bで得られた化合物(756mg、2.89mmol)、水素化ナトリウム(63%、165mg、4.33mmol)、ベンジルアルコール(387mg、3.58mmol)およびN,N-ジメチルホルムアミド(10mL)を用い、実施例7cと同様の方法で、標記化合物(768mg)を白色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル19:1~10:1、V/V)を用いて行った。
(実施例30d)3-ベンジルオキシ-5-フルオロ-4-(2-フルオロ-4-メチルベンジル)安息香酸
実施例30cで得られた化合物(768mg、2.20mmol)、5M水酸化ナトリウム水溶液(2.2mL、11.0mmol)およびエタノール(10mL)を用い、実施例7dと同様の方法で標記化合物の粗生成物(740mg)を得た。
実施例30cで得られた化合物(768mg、2.20mmol)、5M水酸化ナトリウム水溶液(2.2mL、11.0mmol)およびエタノール(10mL)を用い、実施例7dと同様の方法で標記化合物の粗生成物(740mg)を得た。
(実施例30e)3-フルオロ-4-(2-フルオロ-4-メチルベンジル)-5-ヒドロキシ安息香酸
実施例30dで得られた粗生成物(740mg、2.01mmol)、10%パラジウム炭素触媒(含水、150mg)、テトラヒドロフラン(7mL)、メタノール(7mL)および水素を用い、実施例7eと同様の方法で標記化合物の粗生成物(553mg)を得た。
実施例30dで得られた粗生成物(740mg、2.01mmol)、10%パラジウム炭素触媒(含水、150mg)、テトラヒドロフラン(7mL)、メタノール(7mL)および水素を用い、実施例7eと同様の方法で標記化合物の粗生成物(553mg)を得た。
(実施例30f)3-フルオロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェノール
実施例30eで得られた粗生成物(553mg、1.99mmol)、水素化アルミニウムリチウム(188mg、4.95mmol)およびテトラヒドロフラン(20mL)を用い、実施例7fと同様の方法で標記化合物の粗生成物(498mg)を得た。
実施例30eで得られた粗生成物(553mg、1.99mmol)、水素化アルミニウムリチウム(188mg、4.95mmol)およびテトラヒドロフラン(20mL)を用い、実施例7fと同様の方法で標記化合物の粗生成物(498mg)を得た。
(実施例30g)酢酸 3-フルオロ-4-(2-フルオロ-4-メチルベンジル)-5-ヒドロキシベンジル
実施例30fで得られた粗生成物(498mg、1.88mmol)、ビス(ジブチル塩化スズ)オキシド(312mg、0.56mmol)、酢酸ビニル(5mL)およびテトラヒドロフラン(5mL)を用い、実施例7gと同様の方法で標記化合物(522mg)を得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 2.29 (3H, s), 3.96 (2H, s), 5.00 (2H, s), 5.22 (1H, d, J = 3.9 Hz), 6.63 (1H, s), 6.68 (1H, d, J = 9.2 Hz), 6.83-6.87 (2H, m), 7.03 (1H, t, J = 7.9 Hz);
MS (EI) m/z: 306 (M)+.。
実施例30fで得られた粗生成物(498mg、1.88mmol)、ビス(ジブチル塩化スズ)オキシド(312mg、0.56mmol)、酢酸ビニル(5mL)およびテトラヒドロフラン(5mL)を用い、実施例7gと同様の方法で標記化合物(522mg)を得た。
1H NMR (500 MHz, CDCl3): δ 2.11 (3H, s), 2.29 (3H, s), 3.96 (2H, s), 5.00 (2H, s), 5.22 (1H, d, J = 3.9 Hz), 6.63 (1H, s), 6.68 (1H, d, J = 9.2 Hz), 6.83-6.87 (2H, m), 7.03 (1H, t, J = 7.9 Hz);
MS (EI) m/z: 306 (M)+.。
(実施例30h)5-アセトキシメチル-3-フルオロ-2-(2-フルオロ-4-メチルベンジル)フェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(311mg、0.51mmol)、塩化メチレン(6mL)、トリクロロアセトニトリル(0.257mL、2.56mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(8μL、0.05mmol)を用い、実施例1hと同様の方法で、イミダートを調製した。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(311mg、0.51mmol)、塩化メチレン(6mL)、トリクロロアセトニトリル(0.257mL、2.56mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(8μL、0.05mmol)を用い、実施例1hと同様の方法で、イミダートを調製した。
実施例30gで得られた化合物(122mg、0.40mmol)、イミダート(0.51mmol)、塩化メチレン(6mL)および三フッ化ホウ素-ジエチルエーテル錯体(0.064mL、0.51mmol)を用い、実施例1hと同様の方法で、標記化合物の粗生成物を得た。
(実施例30i)3-フルオロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例30hで得られた粗生成物、メタノール/塩化メチレン(10mL/2.5mL)および炭酸カリウム(700mg、5.06mmol)を用い、実施例2hと同様の方法で、標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1~塩化メチレン:メタノール、10:1、V/V)を用いて精製して標記化合物(87mg)を得た。
1H NMR (400 MHz, CD3OD): δ 1.19 (3H, d, J = 6.6 Hz), 2.27 (3H, s), 3.33-3.38 (2H, m), 3.43-3.45 (2H, m), 3.98-4.05 (3H, m), 4.57 (2H, s), 4.93 (1H, d, J = 7.4 Hz), 6.78-6.84 (3H, m), 6.95 (1H, t, J = 7.8 Hz), 7.00 (1H, s);
MS (FAB) m/z:479 (M+K)+.。
実施例30hで得られた粗生成物、メタノール/塩化メチレン(10mL/2.5mL)および炭酸カリウム(700mg、5.06mmol)を用い、実施例2hと同様の方法で、標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1~塩化メチレン:メタノール、10:1、V/V)を用いて精製して標記化合物(87mg)を得た。
1H NMR (400 MHz, CD3OD): δ 1.19 (3H, d, J = 6.6 Hz), 2.27 (3H, s), 3.33-3.38 (2H, m), 3.43-3.45 (2H, m), 3.98-4.05 (3H, m), 4.57 (2H, s), 4.93 (1H, d, J = 7.4 Hz), 6.78-6.84 (3H, m), 6.95 (1H, t, J = 7.8 Hz), 7.00 (1H, s);
MS (FAB) m/z:479 (M+K)+.。
(実施例31)2-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド(表1のNo.22)
(実施例31a)(4-エチル-2-フルオロフェニル)メタノール
塩化4-エチル-2-フルオロベンゾイル(US2006/20146 A1)(200mg、1.19mmol)、水素化アルミニウムリチウム(90mg、2.37mmol)およびテトラヒドロフラン(20mL)を用い、実施例2dと同様の方法により標記化合物の粗生成物(194mg)を得た。
塩化4-エチル-2-フルオロベンゾイル(US2006/20146 A1)(200mg、1.19mmol)、水素化アルミニウムリチウム(90mg、2.37mmol)およびテトラヒドロフラン(20mL)を用い、実施例2dと同様の方法により標記化合物の粗生成物(194mg)を得た。
(実施例31b)4-エチル-2-フルオロベンズアルデヒド
実施例31aで得られた粗生成物(194mg)を塩化メチレン(10mL)に溶解し、氷冷下、クロロクロム酸ピリジニウム(540mg、2.51mmol)を加え、室温で2時間攪拌した。反応液をセライトで濾過後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1、V/V)を用いて精製し、油状の標記化合物(158mg)を得た。
実施例31aで得られた粗生成物(194mg)を塩化メチレン(10mL)に溶解し、氷冷下、クロロクロム酸ピリジニウム(540mg、2.51mmol)を加え、室温で2時間攪拌した。反応液をセライトで濾過後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、9:1、V/V)を用いて精製し、油状の標記化合物(158mg)を得た。
(実施例31c)3,5-ジフルオロ-4-[1-(4-エチル-2-フルオロフェニル)-1-ヒドロキシメチル]ベンゾニトリル
ジイソプロピルアミン(0.85mL、6.01mmol)、n-ブチルリチウム(1.97mL、5.46mmol、2.77M n-ヘキサン溶液)、3,5-ジフルオロベンゾニトリル(759mg、5.46mmol)、実施例31bで得られた化合物(830mg、5.45mmol)およびテトラヒドロフラン(12mL)を用い、実施例7aと同様の方法で油状の標記化合物(1.49g)を得た。
ジイソプロピルアミン(0.85mL、6.01mmol)、n-ブチルリチウム(1.97mL、5.46mmol、2.77M n-ヘキサン溶液)、3,5-ジフルオロベンゾニトリル(759mg、5.46mmol)、実施例31bで得られた化合物(830mg、5.45mmol)およびテトラヒドロフラン(12mL)を用い、実施例7aと同様の方法で油状の標記化合物(1.49g)を得た。
(実施例31d)3,5-ジフルオロ-4-(4-エチル-2-フルオロベンジル)ベンゾニトリル
水素化ホウ素ナトリウム(1.94g、51.2mmol)をトリフルオロ酢酸(35mL)に溶解し、0℃に冷却後、実施例31cで得られた化合物(1.49g、5.12mmol)を溶解させた塩化メチレン溶液(7mL)を加えた。その後、室温で30時間攪拌した。ただし、その間、反応が終了するまで、数回に分けて水素化ホウ素ナトリウム(1.94g、51.2mmol)およびトリフルオロ酢酸(20mL)を追加した。続いて、反応溶液を、0℃に冷却した10%水酸化ナトリウム水溶液(330mL)に加えた。ジエチルエーテル(300mL)で抽出し、水(100mL)、飽和塩化アンモニウム水溶液(100mL)および飽和食塩水(100mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~10:1、V/V)を用いて精製し、標記化合物(947mg)を白色固体として得た。
水素化ホウ素ナトリウム(1.94g、51.2mmol)をトリフルオロ酢酸(35mL)に溶解し、0℃に冷却後、実施例31cで得られた化合物(1.49g、5.12mmol)を溶解させた塩化メチレン溶液(7mL)を加えた。その後、室温で30時間攪拌した。ただし、その間、反応が終了するまで、数回に分けて水素化ホウ素ナトリウム(1.94g、51.2mmol)およびトリフルオロ酢酸(20mL)を追加した。続いて、反応溶液を、0℃に冷却した10%水酸化ナトリウム水溶液(330mL)に加えた。ジエチルエーテル(300mL)で抽出し、水(100mL)、飽和塩化アンモニウム水溶液(100mL)および飽和食塩水(100mL)で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去し、得られた残渣をシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、19:1~10:1、V/V)を用いて精製し、標記化合物(947mg)を白色固体として得た。
(実施例31e)3-ベンジルオキシ-4-(4-エチル-2-フルオロベンジル)-5-フルオロベンゾニトリル
実施例31dで得られた化合物(940mg、3.41mmol)、水素化ナトリウム(63%、195mg、5.12mmol)、ベンジルアルコール(0.46mL、4.47mmol)およびN,N-ジメチルホルムアミド(12mL)を用い、実施例7cと同様の方法で、標記化合物(1.11g)を白色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル19:1~10:1、V/V)を用いて行った。
実施例31dで得られた化合物(940mg、3.41mmol)、水素化ナトリウム(63%、195mg、5.12mmol)、ベンジルアルコール(0.46mL、4.47mmol)およびN,N-ジメチルホルムアミド(12mL)を用い、実施例7cと同様の方法で、標記化合物(1.11g)を白色固体として得た。なお、精製はシリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル19:1~10:1、V/V)を用いて行った。
(実施例31f)3-ベンジルオキシ-4-(4-エチル-2-フルオロベンジル)-5-フルオロ安息香酸
実施例31eで得られた化合物(1.11g、3.05mmol)、5M水酸化ナトリウム水溶液(3.37mL、16.8mmol)およびエタノール(10mL)を用い、実施例7dと同様の方法で標記化合物の粗生成物(1.01g)を得た。
実施例31eで得られた化合物(1.11g、3.05mmol)、5M水酸化ナトリウム水溶液(3.37mL、16.8mmol)およびエタノール(10mL)を用い、実施例7dと同様の方法で標記化合物の粗生成物(1.01g)を得た。
(実施例31g)4-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-ヒドロキシ安息香酸
実施例31fで得られた粗生成物(1.01g、2.64mmol)、10%パラジウム炭素触媒(含水、300mg)、テトラヒドロフラン(10mL)、メタノール(10mL)および水素を用い、実施例7eと同様の方法で標記化合物の粗生成物(806mg)を得た。
実施例31fで得られた粗生成物(1.01g、2.64mmol)、10%パラジウム炭素触媒(含水、300mg)、テトラヒドロフラン(10mL)、メタノール(10mL)および水素を用い、実施例7eと同様の方法で標記化合物の粗生成物(806mg)を得た。
(実施例31h)2-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェノール
実施例31gで得られた粗生成物(806mg、2.76mmol)、水素化アルミニウムリチウム(342mg、9.0mmol)およびテトラヒドロフラン(10mL)を用い、実施例7fと同様の方法で標記化合物の粗生成物(827mg)を得た。
実施例31gで得られた粗生成物(806mg、2.76mmol)、水素化アルミニウムリチウム(342mg、9.0mmol)およびテトラヒドロフラン(10mL)を用い、実施例7fと同様の方法で標記化合物の粗生成物(827mg)を得た。
(実施例31i)酢酸 2-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-ヒドロキシベンジル
実施例31hで得られた粗生成物(827mg、2.97mmol)、ビス(ジブチル塩化スズ)オキシド(164mg、0.30mmol)、酢酸ビニル(10mL)およびテトラヒドロフラン(10mL)を用い、実施例7gと同様の方法で標記化合物(871mg)を得た。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, t, J = 7.6 Hz), 2.11 (3H, s), 2.60 (2H, q, J = 7.4 Hz), 3.97 (2H, s), 5.01 (2H, s), 5.38 (1H, brs), 6.64 (1H, s), 6.69 (1H, d, J = 9.4 Hz), 6.88-6.90 (2H, m), 7.06 (1H, t, J =8.0 Hz).。
実施例31hで得られた粗生成物(827mg、2.97mmol)、ビス(ジブチル塩化スズ)オキシド(164mg、0.30mmol)、酢酸ビニル(10mL)およびテトラヒドロフラン(10mL)を用い、実施例7gと同様の方法で標記化合物(871mg)を得た。
1H NMR (400 MHz, CDCl3): δ 1.20 (3H, t, J = 7.6 Hz), 2.11 (3H, s), 2.60 (2H, q, J = 7.4 Hz), 3.97 (2H, s), 5.01 (2H, s), 5.38 (1H, brs), 6.64 (1H, s), 6.69 (1H, d, J = 9.4 Hz), 6.88-6.90 (2H, m), 7.06 (1H, t, J =8.0 Hz).。
(実施例31j)5-アセトキシメチル-2-(4-エチル-2-フルオロベンジル)-3-フルオロフェニル 7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-β-D-グルコ-ヘプトピラノシド
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(2.5g、4.08mmol)、塩化メチレン(20mL)、トリクロロアセトニトリル(1.2mL、12.2mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(61μL、0.41mmol)を用い、実施例1hと同様の方法で、イミダートを調製した。
7-デオキシ-2,3,4,6-テトラ-O-ベンゾイル-D-グリセロ-α,β-D-グルコ-ヘプトピラノシド(WO2008/016132(PCT/JP2007/65231))(2.5g、4.08mmol)、塩化メチレン(20mL)、トリクロロアセトニトリル(1.2mL、12.2mmol)および1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(61μL、0.41mmol)を用い、実施例1hと同様の方法で、イミダートを調製した。
実施例31iで得られた化合物(870mg、2.7mmol)、イミダート(4.1mmol)、塩化メチレン(30mL)および三フッ化ホウ素-ジエチルエーテル錯体(0.38mL、3.0mmol)を用い、実施例1hと同様の方法で、標記化合物の粗生成物を得た。
(実施例31k)2-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
実施例31jで得られた粗生成物、メタノール/塩化メチレン(16mL/4mL)およびナトリウムメトキシド(162.1mg、3.0mmol)を用い、実施例2hと同様の方法で、標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1~塩化メチレン:メタノール、10:1、V/V)を用いて精製して標記化合物(829.5mg)を得た。
1H NMR (400 MHz, CD3OD): δ 1.18 (3H, t, J = 7.5 Hz), 1.19 (3H, d, J = 6.6 Hz), 2.58 (2H, q, J = 7.4 Hz), 3.33-3.36 (2H, m), 3.43-3.45 (2H, m), 3.99-4.09 (2H, m), 4.57 (2H, s), 4.93-4.95 (1H, m), 6.79-6.87 (3H, m), 6.97 (1H, d, J = 7.8 Hz), 7.00 (1H, s);
MS (FAB) m/z:477 (M+Na)+.。
実施例31jで得られた粗生成物、メタノール/塩化メチレン(16mL/4mL)およびナトリウムメトキシド(162.1mg、3.0mmol)を用い、実施例2hと同様の方法で、標記化合物の粗生成物を得た。得られた粗生成物を、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル、1:1~塩化メチレン:メタノール、10:1、V/V)を用いて精製して標記化合物(829.5mg)を得た。
1H NMR (400 MHz, CD3OD): δ 1.18 (3H, t, J = 7.5 Hz), 1.19 (3H, d, J = 6.6 Hz), 2.58 (2H, q, J = 7.4 Hz), 3.33-3.36 (2H, m), 3.43-3.45 (2H, m), 3.99-4.09 (2H, m), 4.57 (2H, s), 4.93-4.95 (1H, m), 6.79-6.87 (3H, m), 6.97 (1H, d, J = 7.8 Hz), 7.00 (1H, s);
MS (FAB) m/z:477 (M+Na)+.。
(試験例1)ヒトSGLT1発現細胞を用いたSGLT1阻害活性の測定
1)ヒトSGLT1 cDNAの動物細胞発現用ベクターの作成
ヒトSGLT1のcDNAクローン(Origene社Clone Number:TC119918;GenBank accession number:NM_000343)を鋳型としPCR法を用いて増幅した。PCR法のセンス・オリゴヌクレオチド・プライマーとして、
5’-ttaagcttaccatggacagtagcacctggagccc-3’(Primer 1:配列表の配列番号1)、
アンチセンス・オリゴヌクレオチド・プライマーとして、
5’-ttctcgagtcaggcaaaatatgcatggcaa-3’(Primer 2:配列表の配列番号2)
を用いた。PCR反応産物をアガロース電気泳動後、2013塩基に相当する単一バンドより目的のDNA断片を回収し、制限酵素HindIII、XhoIで切断した後、ベクターpCMV-Script(Stratagene社)のHindIII/XhoIサイトに導入し、SGLT1発現プラスミドpCMV-SGLT1とした。pCMV-SGLT1よりHindIII/XhoIフラグメントを切り出し、pENTR1A(Gateway、Invitrogen社)のBamHI/XhoIサイトへ導入し、pENTR-SGLT1を作成した。Gateway Vector Conversion System Cassette A(Invitrogen社)を導入したレトロ・ウイルス・ベクターpLPCX(Clontech社)をDestination Plasmidとして、SGLT1発現レトロ・ウイルス・ベクターpLPCX-SGLT1を作成した。
1)ヒトSGLT1 cDNAの動物細胞発現用ベクターの作成
ヒトSGLT1のcDNAクローン(Origene社Clone Number:TC119918;GenBank accession number:NM_000343)を鋳型としPCR法を用いて増幅した。PCR法のセンス・オリゴヌクレオチド・プライマーとして、
5’-ttaagcttaccatggacagtagcacctggagccc-3’(Primer 1:配列表の配列番号1)、
アンチセンス・オリゴヌクレオチド・プライマーとして、
5’-ttctcgagtcaggcaaaatatgcatggcaa-3’(Primer 2:配列表の配列番号2)
を用いた。PCR反応産物をアガロース電気泳動後、2013塩基に相当する単一バンドより目的のDNA断片を回収し、制限酵素HindIII、XhoIで切断した後、ベクターpCMV-Script(Stratagene社)のHindIII/XhoIサイトに導入し、SGLT1発現プラスミドpCMV-SGLT1とした。pCMV-SGLT1よりHindIII/XhoIフラグメントを切り出し、pENTR1A(Gateway、Invitrogen社)のBamHI/XhoIサイトへ導入し、pENTR-SGLT1を作成した。Gateway Vector Conversion System Cassette A(Invitrogen社)を導入したレトロ・ウイルス・ベクターpLPCX(Clontech社)をDestination Plasmidとして、SGLT1発現レトロ・ウイルス・ベクターpLPCX-SGLT1を作成した。
2)ヒトSGLT1発現細胞の樹立
1)で得られたレトロ・ウイルスpLPCX-SGLT1をインテグリンαvβ3発現HEK-293細胞にトランスフェクションした後、抗生物質G418(商品名:Geneticin、Invitrogen社製)、puromycin(Clontech社製)で細胞を処理し、耐性を有する目的ベクターの安定発現細胞HEK-SGLT1を取得した。安定発現細胞の培養、維持は、250mg/ml G418、1mg/ml puromycin、3mM KGT-1075、10%FBS含有DMEM培地を用いて行った。
1)で得られたレトロ・ウイルスpLPCX-SGLT1をインテグリンαvβ3発現HEK-293細胞にトランスフェクションした後、抗生物質G418(商品名:Geneticin、Invitrogen社製)、puromycin(Clontech社製)で細胞を処理し、耐性を有する目的ベクターの安定発現細胞HEK-SGLT1を取得した。安定発現細胞の培養、維持は、250mg/ml G418、1mg/ml puromycin、3mM KGT-1075、10%FBS含有DMEM培地を用いて行った。
3)SGLT1阻害活性の測定
HEK-SGLT1細胞を250mg/ml G418、1mg/ml puromycin、10%FBS含有DMEM培地で106cells/mlの密度で懸濁し、タイプIコーラゲンコートした96穴培養プレート(コーニング社製)の各ウェルに100μlずつ播種した。翌日に培地を糖取り込みバッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM NaCl、2mM KCl、1mM CaCl2、1mM MgCl2)に換え、1mM [14C]-α-methyl-D-glucopyranoside(0.1m Ci)および評価化合物と共に30分間37℃培養した後、洗浄バッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM Choline Chloride、2mM KCl、1mM CaCl2、1mM MgCl2)で3回洗浄した。96穴プレートの各ウェルに100μlずつ液体シンチレーションカクテル(商品名:Supermix、パーキンエルマー社製)を加え、10分間攪拌した後、液体シンチレーションカウンターの一種であるマイクロβ(パーキンエルマー社製)により、放射活性を測定した。過剰量のSGLT1阻害化合物存在下での放射活性をバックグランドとして各測定値から差し引いた値を糖取り込み活性とし、試験化合物を用いずに求めたコントロールの糖取り込み活性と一定濃度の試験化合物を用いた時の糖取り込み活性から阻害率(%)を求め、糖取り込み活性を50%阻害する試験化合物の濃度を調べた。その結果を表2に示す。下記結果より、本発明の化合物は優れたSGLT1阻害活性を示した。
HEK-SGLT1細胞を250mg/ml G418、1mg/ml puromycin、10%FBS含有DMEM培地で106cells/mlの密度で懸濁し、タイプIコーラゲンコートした96穴培養プレート(コーニング社製)の各ウェルに100μlずつ播種した。翌日に培地を糖取り込みバッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM NaCl、2mM KCl、1mM CaCl2、1mM MgCl2)に換え、1mM [14C]-α-methyl-D-glucopyranoside(0.1m Ci)および評価化合物と共に30分間37℃培養した後、洗浄バッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM Choline Chloride、2mM KCl、1mM CaCl2、1mM MgCl2)で3回洗浄した。96穴プレートの各ウェルに100μlずつ液体シンチレーションカクテル(商品名:Supermix、パーキンエルマー社製)を加え、10分間攪拌した後、液体シンチレーションカウンターの一種であるマイクロβ(パーキンエルマー社製)により、放射活性を測定した。過剰量のSGLT1阻害化合物存在下での放射活性をバックグランドとして各測定値から差し引いた値を糖取り込み活性とし、試験化合物を用いずに求めたコントロールの糖取り込み活性と一定濃度の試験化合物を用いた時の糖取り込み活性から阻害率(%)を求め、糖取り込み活性を50%阻害する試験化合物の濃度を調べた。その結果を表2に示す。下記結果より、本発明の化合物は優れたSGLT1阻害活性を示した。

(試験例2)ヒトSGLT2発現細胞を用いたSGLT2阻害活性の測定
1)ヒトSGLT2 cDNAの動物細胞発現用ベクターへの作成
ヒトSGLT2のcDNAクローン(Origene社Clone Number:TC303267;GenBank accession number:NM_003041)を鋳型としPCR法を用いて増幅した。PCR法のセンス・オリゴヌクレオチド・プライマーとして、
5’-ttaagcttaccatggaggagcacacagaggcagg-3’(Primer 3:配列表の配列番号3)、
アンチセンス・オリゴヌクレオチド・プライマーとして、
5’-ttctcgagttaggcatagaagccccagagg-3’(Primer 4:配列表の配列番号4)
を用いた。PCR反応産物をアガロース電気泳動後、2037塩基に相当する単一バンドより目的のDNA断片を回収し、制限酵素HindIII、XhoIで切断した後、ベクターpCMV-Script(Stratagene社)のHindIII/XhoIサイトに導入した。SGLT2発現プラスミドpCMV-SGLT2とした。pCMV-SGLT2よりHindIII/XhoIフラグメントを切り出し、pENTR1A(Gateway、Invitrogen社)のBamHI/XhoIサイトへ導入し、pENTR-SGLT2を作成した。Gateway Vector Conversion System Cassette A(Invitrogen社)を導入したレトロ・ウイルス・ベクターpLPCX(Clontech社)をDestination Plasmidとして、SGLT2発現レトロ・ウイルス・ベクターpLPCX-SGLT2を作成した。
1)ヒトSGLT2 cDNAの動物細胞発現用ベクターへの作成
ヒトSGLT2のcDNAクローン(Origene社Clone Number:TC303267;GenBank accession number:NM_003041)を鋳型としPCR法を用いて増幅した。PCR法のセンス・オリゴヌクレオチド・プライマーとして、
5’-ttaagcttaccatggaggagcacacagaggcagg-3’(Primer 3:配列表の配列番号3)、
アンチセンス・オリゴヌクレオチド・プライマーとして、
5’-ttctcgagttaggcatagaagccccagagg-3’(Primer 4:配列表の配列番号4)
を用いた。PCR反応産物をアガロース電気泳動後、2037塩基に相当する単一バンドより目的のDNA断片を回収し、制限酵素HindIII、XhoIで切断した後、ベクターpCMV-Script(Stratagene社)のHindIII/XhoIサイトに導入した。SGLT2発現プラスミドpCMV-SGLT2とした。pCMV-SGLT2よりHindIII/XhoIフラグメントを切り出し、pENTR1A(Gateway、Invitrogen社)のBamHI/XhoIサイトへ導入し、pENTR-SGLT2を作成した。Gateway Vector Conversion System Cassette A(Invitrogen社)を導入したレトロ・ウイルス・ベクターpLPCX(Clontech社)をDestination Plasmidとして、SGLT2発現レトロ・ウイルス・ベクターpLPCX-SGLT2を作成した。
2)ヒトSGLT2発現細胞の樹立
1)で得られたレトロ・ウイルスpLPCX-SGLT2をインテグリンαvβ3発現HEK-293細胞にトランスフェクションした後、抗生物質G418(商品名:Geneticin、Invitrogen社製)、puromycin(Clontech社製)で細胞を処理し、耐性を有する目的ベクターの安定発現細胞HEK-SGLT2を取得した。安定発現細胞の培養、維持は、250mg/ml G418、1mg/ml puromycin、3mM KGT-1075、10%FBS含有DMEM培地を用いて行った。
1)で得られたレトロ・ウイルスpLPCX-SGLT2をインテグリンαvβ3発現HEK-293細胞にトランスフェクションした後、抗生物質G418(商品名:Geneticin、Invitrogen社製)、puromycin(Clontech社製)で細胞を処理し、耐性を有する目的ベクターの安定発現細胞HEK-SGLT2を取得した。安定発現細胞の培養、維持は、250mg/ml G418、1mg/ml puromycin、3mM KGT-1075、10%FBS含有DMEM培地を用いて行った。
3)SGLT2阻害活性の測定
HEK-SGLT2細胞を250mg/ml G418、1mg/ml puromycin、10%FBS含有DMEM培地で106cells/mlの密度で懸濁し、タイプIコーラゲンコートした96穴培養プレート(コーニング社製)の各ウェルに100μlずつ播種した。翌日に培地を糖取り込みバッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM NaCl、2mM KCl、1mM CaCl2、1mM MgCl2)に換え、1mM [14C]-α-methyl-D-glucopyranoside(0.1m Ci)および評価化合物と共に30分間37℃培養した後、洗浄バッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM Choline Chloride、2mM KCl、1mM CaCl2、1mM MgCl2)で3回洗浄した。96穴プレートの各ウェルに100μlずつ液体シンチレーションカクテル(商品名:Supermix、パーキンエルマー社製)を加え、10分間攪拌した後、液体シンチレーションカウンターの一種であるマイクロβ(パーキンエルマー社製)により、放射活性を測定した。過剰量のSGLT2阻害化合物存在下での放射活性をバックグランドとして各測定値から差し引いた値を糖取り込み活性とし、試験化合物を用いずに求めたコントロールの糖取り込み活性と一定濃度の試験化合物を用いた時の糖取り込み活性から阻害率(%)を求め、糖取り込み活性を50%阻害する試験化合物の濃度を調べた。その結果を表3に示す。下記結果より、本発明の化合物は優れたSGLT2阻害活性を示した。
HEK-SGLT2細胞を250mg/ml G418、1mg/ml puromycin、10%FBS含有DMEM培地で106cells/mlの密度で懸濁し、タイプIコーラゲンコートした96穴培養プレート(コーニング社製)の各ウェルに100μlずつ播種した。翌日に培地を糖取り込みバッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM NaCl、2mM KCl、1mM CaCl2、1mM MgCl2)に換え、1mM [14C]-α-methyl-D-glucopyranoside(0.1m Ci)および評価化合物と共に30分間37℃培養した後、洗浄バッファー(10mM HEPES(pH7.5)、5mM Tris-HCl(pH7.5)、140mM Choline Chloride、2mM KCl、1mM CaCl2、1mM MgCl2)で3回洗浄した。96穴プレートの各ウェルに100μlずつ液体シンチレーションカクテル(商品名:Supermix、パーキンエルマー社製)を加え、10分間攪拌した後、液体シンチレーションカウンターの一種であるマイクロβ(パーキンエルマー社製)により、放射活性を測定した。過剰量のSGLT2阻害化合物存在下での放射活性をバックグランドとして各測定値から差し引いた値を糖取り込み活性とし、試験化合物を用いずに求めたコントロールの糖取り込み活性と一定濃度の試験化合物を用いた時の糖取り込み活性から阻害率(%)を求め、糖取り込み活性を50%阻害する試験化合物の濃度を調べた。その結果を表3に示す。下記結果より、本発明の化合物は優れたSGLT2阻害活性を示した。

(試験例3)in vivo試験
各被検化合物を溶媒(0.5%メチルセルロース液)に10mL/kgの投与容量になるように懸濁あるいは溶解し、複数用量(好ましくは0.03~10mg/kgの範囲に含まれる)を一晩絶食したC57BL/6NCrlCrljマウス(7~10週齡、雄性)に経口投与する。対照群には溶媒を10mL/kg経口投与する。投与10分後にグルコースを2g/10mL/kg経口投与する。血糖値を経時的(グルコース投与前、投与20、40、60、120分後)に測定し、血糖値の曲線下面積を算出し、各群の血糖値曲線下面積の対照群からの低下率を指標に50%有効用量ED50を求めることで各被検化合物の生体での有効性を評価する。
各被検化合物を溶媒(0.5%メチルセルロース液)に10mL/kgの投与容量になるように懸濁あるいは溶解し、複数用量(好ましくは0.03~10mg/kgの範囲に含まれる)を一晩絶食したC57BL/6NCrlCrljマウス(7~10週齡、雄性)に経口投与する。対照群には溶媒を10mL/kg経口投与する。投与10分後にグルコースを2g/10mL/kg経口投与する。血糖値を経時的(グルコース投与前、投与20、40、60、120分後)に測定し、血糖値の曲線下面積を算出し、各群の血糖値曲線下面積の対照群からの低下率を指標に50%有効用量ED50を求めることで各被検化合物の生体での有効性を評価する。
(製剤例)錠剤
実施例の化合物5g、乳糖90g、トウモロコシデンプン34g、結晶セルロース20gおよびステアリン酸マグネシウム1gをブレンダーで混合した後、打錠機で打錠することにより、錠剤が得られる。
実施例の化合物5g、乳糖90g、トウモロコシデンプン34g、結晶セルロース20gおよびステアリン酸マグネシウム1gをブレンダーで混合した後、打錠機で打錠することにより、錠剤が得られる。
本発明の化合物またはその水和物は、副作用が低く優れたヒトSGLT1および/またはSGLT2阻害活性を示し、1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全(impaired glucose tolerance:IGT)、糖尿病関連疾患(例えば、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症、痛風など)、または糖尿病合併症(例えば、網膜症、腎症、神経障害、白内障、足壊疽、感染症、ケトーシスなど)の治療薬または予防薬として有用であり、哺乳動物(例えば、ヒト、ウマ、ウシまたはブタ、好ましくはヒト)の予防もしくは治療のための医薬組成物として有用である。
配列番号1:ヒトSGLT1のPCRセンスプライマー
配列番号2:ヒトSGLT1のPCRアンチセンスプライマー
配列番号3:ヒトSGLT2のPCRセンスプライマー
配列番号4:ヒトSGLT2のPCRアンチセンスプライマー
配列番号2:ヒトSGLT1のPCRアンチセンスプライマー
配列番号3:ヒトSGLT2のPCRセンスプライマー
配列番号4:ヒトSGLT2のPCRアンチセンスプライマー
Claims (15)
- 一般式(I):
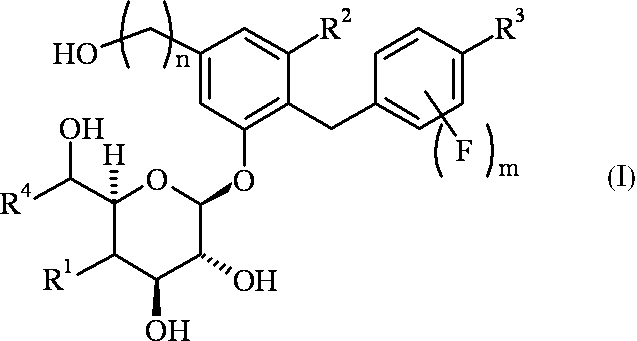
(式中、R1は水素原子または水酸基であり、
R2はフッ素原子または塩素原子であり、
R3はハロゲン原子で置換されていてもよいメチル基、ハロゲン原子で置換されていてもよいエチル基、シクロプロピル基、または、ハロゲン原子で置換されていてもよいメトキシ基であり、
R4は水素原子またはメチル基であり、
nは1または2であり、
mは0または1である、
但し、R1が水素原子である場合、R4は水素原子であり、R1が水酸基である場合、R4はメチル基である)
で表される化合物またはその水和物。 - 一般式(II):
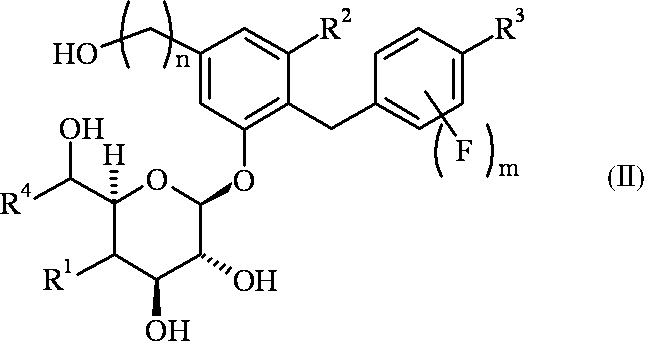
[式中、R1は水素原子または水酸基であり、
R2はフッ素原子または塩素原子であり、
R3はハロゲン原子で置換されていてもよいメチル基、ハロゲン原子で置換されていてもよいエチル基、シクロプロピル基、または、ハロゲン原子で置換されていてもよいメトキシ基であり、
R4は水素原子またはメチル基であり、
nは1または2であり、
mは0または1である、
但し、R1が水素原子である場合、R4は水素原子であり、R1が水酸基である場合、R4はメチル基である
(但し、(イ)R1が水酸基であり、R2がフッ素原子であり、mが0であり、かつ、nが1である場合、R3はメチル基、エチル基、シクロプロピル基またはメトキシ基ではない、
(ロ)R1が水酸基であり、R2が塩素原子であり、mが0であり、かつ、nが1である場合、R3はメトキシ基ではない、あるいは、
(ハ)R1が水素原子であり、R2がフッ素原子または塩素原子であり、mが0であり、かつ、nが1である場合、R3はメトキシ基ではない)]
で表される化合物またはその水和物。 - R1が水酸基である、請求項2に記載の化合物。
- R3がハロゲン原子で置換されていてもよいメチル基またはハロゲン原子で置換されていてもよいメトキシ基である、請求項2または3に記載の化合物。
- R4がメチル基である、請求項2~4いずれか1項に記載の化合物。
- nが1である、請求項2~5いずれか1項に記載の化合物。
- 3-クロロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
2-[4-(ジフルオロメトキシ)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-(3-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-(3-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-2-(2-フルオロ-4-メトキシベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-[4-(ジフルオロメトキシ)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-2-[4-(2-フルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-5-ヒドロキシメチル-2-[(4-トリフルオロメトキシ)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド;
3-クロロ-5-ヒドロキシメチル-2-(4-メチルベンジル)フェニル 4-デオキシ-β-D-グルコピラノシド;
2-(4-エチルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド;
3-クロロ-2-(4-エチルベンジル)-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド;
2-(4-シクロプロピルベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 4-デオキシ-β-D-グルコピラノシド;
3-フルオロ-5-ヒドロキシメチル-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-(4-エチル-3-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メチルベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-5-(2-ヒドロキシエチル)-2-(4-メトキシベンジル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-5-(ヒドロキシメチル)-2-[4-(2,2,2-トリフルオロエチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-(4-エチル-2-フルオロベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-5-(ヒドロキシメチル)-2-[(4-トリフルオロメチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-5-(ヒドロキシメチル)-2-[(4-トリフルオロメチル)ベンジル]フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
2-[4-(2,2-ジフルオロエチル)ベンジル]-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-クロロ-2-[4-(2,2-ジフルオロエチル)ベンジル]-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;
3-フルオロ-2-(2-フルオロ-4-メチルベンジル)-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド;および
2-(4-エチル-2-フルオロベンジル)-3-フルオロ-5-(ヒドロキシメチル)フェニル 7-デオキシ-D-グリセロ-β-D-グルコ-ヘプトピラノシド
からなる群より選択される化合物またはその水和物。 - 請求項1~7いずれか1項に記載の化合物またはその水和物を有効成分として含有する医薬組成物。
- ヒトSGLT1および/またはヒトSGLT2活性を阻害するための、請求項8に記載の医薬組成物。
- 1型糖尿病、2型糖尿病、妊娠糖尿病、その他の要因による高血糖症、耐糖能不全、糖尿病関連疾患もしくは糖尿病合併症の治療または予防のための、請求項8または9に記載の医薬組成物。
- 糖尿病関連疾患が、肥満、高脂血症、高コレステロール血症、脂質代謝異常、高血圧症、脂肪肝、メタボリックシンドローム、浮腫、心不全、狭心症、心筋梗塞、動脈硬化症、高尿酸血症または痛風であり、あるいは、糖尿病合併症が、網膜症、腎症、神経障害、白内障、足壊疽、感染症またはケトーシスである、請求項10に記載の医薬組成物。
- 医薬組成物を製造するための、請求項1~7いずれか1項に記載の化合物またはその水和物の使用。
- 請求項1~7いずれか1項に記載の化合物またはその水和物の治療有効量を哺乳動物に投与することを含む、ヒトSGLT1および/またはヒトSGLT2活性を阻害する方法。
- 請求項1~7いずれか1項に記載の化合物またはその水和物の治療有効量を哺乳動物に投与することを含む、疾病の治療または予防方法。
- 哺乳動物がヒトである、請求項14に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008021879 | 2008-01-31 | ||
| JP2008-021879 | 2008-01-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009096503A1 true WO2009096503A1 (ja) | 2009-08-06 |
Family
ID=40912846
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/051532 WO2009096503A1 (ja) | 2008-01-31 | 2009-01-30 | ベンジルフェニルグルコピラノシド誘導体 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW200934503A (ja) |
| WO (1) | WO2009096503A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001068660A1 (fr) * | 2000-03-17 | 2001-09-20 | Kissei Pharmaceutical Co., Ltd. | Derives glucopyranosyloxybenzylbenzene, preparations medicinales les contenant et intermediaires pour la preparation desdits derives |
| WO2002028872A1 (fr) * | 2000-09-29 | 2002-04-11 | Kissei Pharmaceutical Co., Ltd. | Derives de glucopyranosiloxybenzylbenzene et compositions therapeutiques contenant ces composes |
| WO2002044192A1 (fr) * | 2000-11-30 | 2002-06-06 | Kissei Pharmaceutical Co., Ltd. | Derives de glucopyranosyloxybenzylbenzene, compositions medicinales contenant ces derives et produits intermediaires obtenus lors de l'elaboration de ces compositions |
| WO2002064606A1 (fr) * | 2001-02-14 | 2002-08-22 | Kissei Pharmaceutical Co., Ltd. | Derives glucopyranosyloxybenzylbenzene et leur utilisation medicale |
| JP2004500416A (ja) * | 2000-03-30 | 2004-01-08 | ブリストル−マイヤーズ スクイブ カンパニー | O−アリールグルコシドsglt2抑制剤および方法 |
| WO2005095429A1 (ja) * | 2004-03-31 | 2005-10-13 | Kissei Pharmaceutical Co., Ltd. | フェノール誘導体、それを含有する医薬組成物及びその医薬用途 |
| JP2007515441A (ja) * | 2003-12-22 | 2007-06-14 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グルコピラノシロキシ置換芳香族化合物、前記化合物を含む医薬、それらの使用及びそれらの製造方法 |
| WO2008016132A1 (en) * | 2006-08-04 | 2008-02-07 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
-
2009
- 2009-01-30 WO PCT/JP2009/051532 patent/WO2009096503A1/ja active Application Filing
- 2009-02-02 TW TW098103224A patent/TW200934503A/zh unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001068660A1 (fr) * | 2000-03-17 | 2001-09-20 | Kissei Pharmaceutical Co., Ltd. | Derives glucopyranosyloxybenzylbenzene, preparations medicinales les contenant et intermediaires pour la preparation desdits derives |
| JP2004500416A (ja) * | 2000-03-30 | 2004-01-08 | ブリストル−マイヤーズ スクイブ カンパニー | O−アリールグルコシドsglt2抑制剤および方法 |
| WO2002028872A1 (fr) * | 2000-09-29 | 2002-04-11 | Kissei Pharmaceutical Co., Ltd. | Derives de glucopyranosiloxybenzylbenzene et compositions therapeutiques contenant ces composes |
| WO2002044192A1 (fr) * | 2000-11-30 | 2002-06-06 | Kissei Pharmaceutical Co., Ltd. | Derives de glucopyranosyloxybenzylbenzene, compositions medicinales contenant ces derives et produits intermediaires obtenus lors de l'elaboration de ces compositions |
| WO2002064606A1 (fr) * | 2001-02-14 | 2002-08-22 | Kissei Pharmaceutical Co., Ltd. | Derives glucopyranosyloxybenzylbenzene et leur utilisation medicale |
| JP2007515441A (ja) * | 2003-12-22 | 2007-06-14 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グルコピラノシロキシ置換芳香族化合物、前記化合物を含む医薬、それらの使用及びそれらの製造方法 |
| WO2005095429A1 (ja) * | 2004-03-31 | 2005-10-13 | Kissei Pharmaceutical Co., Ltd. | フェノール誘導体、それを含有する医薬組成物及びその医薬用途 |
| WO2008016132A1 (en) * | 2006-08-04 | 2008-02-07 | Daiichi Sankyo Company, Limited | Benzyl phenyl glucopyranoside derivative |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200934503A (en) | 2009-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009096503A1 (ja) | ベンジルフェニルグルコピラノシド誘導体 | |
| CN101448791B (zh) | Fxr激动剂 | |
| TWI377210B (en) | Fused heterocyclic derivatives, pharmaceutical compositions comprising the same, and pharmaceutical uses thereof | |
| TWI734736B (zh) | Fxr受體激動劑 | |
| TWI534144B (zh) | 甲狀腺荷爾蒙β受體作動藥 | |
| CN101657471B (zh) | 二环化合物及其作为抗糖尿病药的用途 | |
| TWI290556B (en) | Glucopyranosyloxybenzyl benzene derivatives, medicinal compositions containing the same and intermediates in the production thereof | |
| JP5371062B2 (ja) | 代謝障害の処置のための化合物 | |
| CN1040323C (zh) | 噻唑烷衍生物 | |
| KR101751325B1 (ko) | 잔틴 옥시다아제 저해제로서 효과적인 신규 화합물, 그 제조방법 및 그를 함유하는 약제학적 조성물 | |
| WO2006011469A1 (ja) | 新規なシクロヘキサン誘導体、そのプロドラッグおよびその塩、ならびにそれらを含有する糖尿病治療薬 | |
| WO2008016132A1 (en) | Benzyl phenyl glucopyranoside derivative | |
| JP4505753B2 (ja) | ラクタム化合物及びその医薬用途 | |
| TW200426139A (en) | 5-substituted-six-membered heteroaromatic glucokinase activators | |
| WO1996038428A1 (fr) | Derives de n-benzyldioxothiazolidylbenzamide et leur procede de production | |
| TW200410980A (en) | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof | |
| CN105555783A (zh) | 包含杂芳族环-苄基-酰胺-环核心的自分泌运动因子抑制剂 | |
| JP2004520290A (ja) | 新規マンデル酸誘導体とそれらのトロンビン阻害剤としての使用 | |
| EA013003B1 (ru) | Антагонисты рецептора глюкагона, их получение и терапевтическое применение | |
| JP4837557B2 (ja) | 代謝障害の処置のための化合物 | |
| WO2004089966A1 (ja) | 選択的なヘテロアリール 5-チオ-β-D-アルドヘキソピラノシドの製造法 | |
| CN101304983A (zh) | 作为ppar调节剂的化合物和组合物 | |
| WO2015024448A1 (zh) | 苯并哌啶环与苯并吗啉环类化合物、其制法及医药应用 | |
| JP2006117651A (ja) | Sglt2の活性阻害剤 | |
| CN101679248A (zh) | 具有cPLA2抑制活性的吲哚衍生物及其用途以及制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09706389 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09706389 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |