作为二肽肽激酶- I V抑制剂的
N -取代硫吗啉衍生物及其医药用途 技术领域
本发明涉及作为二肽肽激酶 IV (Dipeptidyl peptidase IV (DPP-IV))抑制剂的 N-取代硫吗啉化合物, 其所有可能的异构体、 其药学上可接受的盐、 其溶剂化物或水合物或其前体药物; 以及 式 I化合物的制备方法、 含有式 I化合物的药物组合物及 述化 合物在医疗领域的用途, 特别是用于制备治疗和预防糖尿病 (尤 其是 Π型糖尿病) 、 高血糖症、 X综合征、 高胰岛素血症、 肥胖 症、 动脉粥样硬化以及各种免疫调节性疾病的药物的用途。 背景技术
二肽肽激酶 IV(DPP-IV) 是一种在多种哺乳动物的组织中广 泛表达多功能 Π型跨膜糖基^蛋白, 它是一种 T 细胞活化抗原 CD26 ,也是一种腺苷脱氨酶 (ADA) 结合蛋白。 人类 DPP- IV ( hDPP-IV) 的单链由 766 个氨基酸组成,分为 5 个结构区域: 胞 质区 (1 - 6) 、 跨膜区(7 - 28) 、 高度糖基化区 (29 - 323) 、 牟胱氨 酸富集区(324 - 551) 和催化区(552 - 766),不同物种间这些 S域的 长度稍有差异。 可溶性 DPP-IV是一种大约为 210 ~ 290 kDa;的同 型二聚体,也能聚合成高达 900 kDa 的复合物。 DPP-IV通过氨基 端的一个由高度糖基化区和半胱氨酸富集区形成的疏水性螵旋与 膜结合,羧基端的丝氨酸蛋白酶区域与 al δ水解酶同源。二聚体形 式是其产生活性的前提 (异型二聚体则是一种成纤维细胞活性蛋 白 FAP ) 。
普遍认为 DPP-IV在神经肽代谢、 T 细胞活化、 癌细胞与内 皮的附着以及 HIV进入淋巴细胞中起着重要的作用。 DPP-IV能
特异性的将二肽从其中倒数笫二个氨基酸主要是脯氨酸、 丙氨酸 或羟脯氨酸的肽的 N-末端裂解下来。 它的作用底物包括在 T2DM 免疫应答信号传导过程中起重要作用的两种肠降血糖素: 高血 糖素样肽 1 的片段(GLP-l7 -36) 和抑胃肽(GIP ) 。 GLP?1 和 GIP 分别是由胃黏膜 L 细胞及 K 细胞为了应答摄入的碳水化合 物及脂肪而分泌的肠降血糖素,在餐后血糖浓度的稳定上 着关 键性作用。用餐之后,胃黏膜应激分泌 GLP-1 和 GIP,二者作用于 胰腺以增强葡萄糖诱导的胰岛素分泌,调节血糖浓度。 而体内的 DPP-IV则可将其水解后,产生相应的氨基端切断形式的 GIP3.42和 GLP-19 . 36,失去其胰岛素诱导活性。 由此可见,抑制 DPP-ΙΫ就可 以增强 GIP 和 GLP-1 的活性,葡萄糖耐受氷平将相应得以改善。
Marguet 等和 Omarello 等的 DPP-IV缺陷小鼠实验 果表 明, DPP-IV缺小鼠完全能够存活并具有正常的表型,同时与野生型 小鼠相比 ,DPP- IV缺陷小鼠还有较高的葡萄糖耐受能力及 高的 血中胰岛素、 GLP-1 漆度。
综上所述, 抑制血浆 DPP-IV活性具有降血糖作用, 其作用机 理至少有以下三个: 其一保护胰岛素的活性, 生理情况下循环血 中完整 GLP-1的半衰期不足 lmin, DPP-IV降解 GLP-1后的无活 性代谢物能与 GLP-1 受体结合拮抗活性 GLP-1,使单独注射 GLP-1 的效果短暂, 而 DPP- IV抑制剂能完全保护内源性甚至外 源 -性的 GLP-1不被 DPP-IV所灭活。 已知 GLP-1具有多种生理活 性, 包括促进胰岛素基因的表达, 促进 细胞的生长, 抑剁胰升 血糖素的分泌, 增加饱腹感, 减少摄食, 抑制胃拍空, 使血糖水 平趋于正常化, 而且 DPP-IV抑制剂除能提高 GLP-1的水平, 也 减少 GLP-1代谢物的拮抗作用。此外小肠上部 k细胞分泌的 GIP 通过作用于 G蛋白偶联受体, 引起腺苷酸环化酶活性提高, 激活 酶 A2.升高细胞内钙离子水平, 促进胰岛素舞放。 GIP也鸽促进
前胰岛素基因的转录与翻译, 上调质膜的葡萄糖转运和增加 β细 胞己糖激酶活性, 因此 GIP对 Π型糖尿病(T2DM)有治疗作用 。 但与 GLP-1相似,内源性的 GIP也迅速被 DPP-IV灭活。 Dkcon 等对猪静脉注射 GIP后,放射免疫法显示完整的 GIP免疫活性只 有 14.5%。 当使用 DPP-IV抑制剂后 GIP免疫活性提高到 49%, 显示 DPP-IV虽然不是体内唯一降解 GIP的酶, 但在其灭 中起 重要作用。可见 DPP-IV抑制剂能保护活性的肠促胰岛素而发挥其 作用。
其二刺激胰岛 细胞再生, Pospisilik 等对链脲佐菌章诱导 DM雄性鼠给予 P32/98 (—种 DPP-IV抑制剂) , 2次 /d, 7;周后 用免疫组化分析发现胰岛数目比对照组增加 35%, 总 细胞数增 加 120%, 胰岛 细胞分数增加产品品种 12%,且血浆胰岛素水平 接近正常。 DPP-IV抑制剂刺激胰岛素再生, 提高 细胞存活的作 用,可能是提高 GLP-1与胰岛内巢蛋白阳性的胰岛衍生前体细胞 (NIP)表面的 GLP-1受体转合而促进 NIP细胞分化为胰岛细胞。
其三改善糖耐量及胰岛素敏感性,研究表明 DPP- IV抑制剂不 仅对 DM有治疗作用,而且对延緩 DM的发生和发展有预防作用。
Sudre等以 FE999011 (—种长效 DPP-IV抑制剂)治疗肥胖小鼠, 证实 FE999011呈剂量依赖性地减慢葡萄糖的释放, 以 10m /kg, 2次 /d应用 FE999011能提高糖耐量。 以上述同等剂量长期治疗, 能使肥胖小鼠高血糖的发生推迟 21d, 同时可改善多饮多食等症 状, 减少高甘油三酯血症的发生, 防止血中游离脂肪酸升高, 且 治疗后基础血浆 GLP-1水平提高, 胰腺的 GLP-1受体基因表达 明显上调。因此研究者认为 DPP-IV抑制剂能延緩由于糖耐量异常 发展为 Π型糖尿病。 另有研究提示应用 DPP-IV抑制剂后, 可改善 糖耐量异常, 提高胰岛素敏感性, 并使 细胞对葡萄糖的 应得 到改善。
所以,抑制 DPP-IV可以提供 II型糖尿病性和其他 DPPrl 调 节疾病的治疗方法。 发明内容
本发明的目的是提供一类新颖结构类型的二肽肽 酶 IV ( DPP-W )抑制剂, 道过抑制二肽肽激酶 IV活性, 保护肠 岛素 不被降解, 改善糖耐量异常及增加胰岛素敏感性, 达到降僞血糖 的目的, 从而可以有效的治疗糖尿病和其他 DPP-I 调节的疾病。
本发明提供如权利要求中所定义的通式 I的新型化合 , 或 其所有可能的异构体, 其药学上可接受的盐、 其溶剂化物 7J合 物, 或其前体药物,
其中:
R1选自氢、 氰基、 卤素或者三氟甲基;
A为氨基酸, 所述氨基酸侧链至少含有一个官能团;
B为以共价键与 A侧链官能团相连接的化合物,选自由 0 - 5 个氨基酸組成的多肽。
在本发明的一个实施方式中, 通式 I中的 A为 a -氨基酸。 在本发明的另一个实 方式中, 通式 I中的 A为天然 α -氨 基酸。
在本发明的另一个实施方式中, 通式 I中的 A为非天然氨基
在本发明的另一个实施方式中, 通式 I中的 A选自亮氨酸、 缬氨酸、 甘氨酸、 丙氨酸、 缬氨酸、 异亮氨酸、 苯丙氨酸、 脯氨 酸、 色氨酸、 丝氨酸、 酪氨酸、 半胱氨酸, 优选缬氨酸。
本发明最优选的化合物为:
化合物 1: (R)-3-氰基 -4-(2-氨基 -3-甲基-丁酰基) 硫吗啉盐酸 盐;或
化合物 2: (R)-3-氰基 -4-(2-氨基 -4-甲基-戊酰基) 硫吗啉盐酸 盐;
或其所有可能的异构体、 可药用盐、 溶剂合物或水合物, 或其前 体药物。 本发明的另一方面涉及通式 I化合物或其药用盐或水合物的 制备方法。
本发明通式 I 化合物可通过下面的反应路线制备, 使经过 BOC保护的 A与 00 - 3-酰胺硫吗啉按下式进行反应:
其中, A为氨基酸, 所迷氨基酸侧链至少含有一个官能闳, 优 选缬氨酸和亮氨酸。
具体来说,将氨基酸进行 B0C保护后以等摩尔和 (R) -3-酰胺 硫吗啉溶于干燥的 THF中, 加入 1. 5倍量的 EDCI和 1. 5倍量的 H0BT, 再滴加 2倍量二异丙基乙胺, 室温搅拌 12h ~ 16h。 反应完 毕后减压浓缩, 加水稀释, 用乙酸乙酯萃取, 有机相用无水硫酸
钠干燥 4h后, 减压除去溶剂, 残留物溶于干燥的 THF中, 加入 2 倍量的三氟醋酐, 搅拌 lh后, 用饱和的 NaHC03 水洗, 直 没有 气泡出现, 乙酸乙酯萃取, 无水硫酸钠千燥 4h, 硅胶柱层析分离 (乙酸乙酯: 环己烷 =1: 4), 得无色油状物, 溶于 3N的盐 乙酸 乙酯中, 搅拌 5h, 得到本发明化合物。 如有需要, 将式 (ί )化 合物按照本领域常规的方法转变为其药学上可接受的盐或其溶剂 化物或水合物或前体药物。
本发明另一方面涉及式 III化合物的制备方法,式 III化合物 是合成式 I化合物的关键中间体, 本发明提供一种新型的简便易 行、 适合大规模合成的立体专一性成环反应, 可以得到立^专一 的式 III化合物, 式 III化合物再通过氨化转变成式 II化^物, 式 II化合物进而可与 Α反应得到式 I化合物。
丄、
式 III 式 II 本发明通式 III化合物可通过下面的反应路线制备:
式 IV 式 III
具体来说, 在水中、 在缚酸剂碱作用下, 使式 IV化合物进行 立体选择性关环得到式 III化合物。 所述的缚酸剂碱包括但不限 于氢氧化钠、 氢氧化钾、 三乙胺、 碳酸氢钠、 碳酸钠等, 优选碳
酸氢钠。
具体的式 III 化合物 (R)-3-羧酸甲酯硫吗啉盐酸盐的制备可 参见实施例 1。 本发明还涉及含有式(I )化合物或其所有可能异构体、 其药 学上可接受的盐、 其溶剂化物或水合物或前体药物以及一种或多 种药学上可接受的载体或赋形剂的药物组合物。
"药物组合物" 是指一种或多种活性成分, 和一种或多种构 成载体的惰性成分, 以及直接或间接地由这些成分中的任意两种 或多种的结合、 络合或聚集, 或由一种或多种成分的分解, 或由 一种或多种成分的其它类型反应或相互作用产生的任何产物。 因 此, 本发明的药物组合物包括将本发明化合物与药学上可接受的 载体混合制成的任何組合物。
本发明的药物组合物中还可以包含一种或多种其它化合物作 为活性成分,例如一种或多种其它 DPP-IV抑制剂, 所述的其它活 性成分选自胰岛素敏化剂、 PPAR.激动剂、 双胍类或 PTP-1B抑制 剂等。
本发明的另一方面涉及本发明式( I )化合物或其所有可能异 构体、 其药学上可接受的盐、 其溶剂化物或水合物或其前侔药物 用于制备与二肽肽激酶 IV相关疾病的药物的用途,所述与二肽肽 激酶 IV相关的疾病包括但不限于糖尿病 (尤其是 II型糖尿病 Ϊ、 高 血糖症、 X综合征、 高胰岛素血症、 肥胖症、 动脉粥样硬化以及 各种免疫调节性疾病。
本发明的另一方面涉及与二肽肽激酶 Π 相关疾病的治疗方 法,所述方法包括给予有此需要的患者治疗有效量的本发明式( I ) 化合物或其所有可能异构体、 其药学上可接受的盐、 其溶 ¾化物 或水合物或其前体药物,所述与二肽肽激酶 IV相关的疾病包括但
不限于糖尿病 (尤其是 II型糖尿病) 、 高血糖症、 X综合征、 高胰 岛素血症、 肥胖症、 动脉粥样硬化以及备种免疫调节性疾病。 具体实施方式
下面的具体实施例是本发明的优选实施方案, 用于说明本发 明, 但不对本发明构成任何限制。
化合物的熔点由 RY-1熔点仪测定, 温度计未较正。 质谱由 Micromass ZabSpec 高分辨率质谱仪测定; 1H-NM 由 JNM-ECA-400超导 NMR仪测定, 工作频率 1H-NMR 400MHz。 实施例 1
步骤 1 2-羟乙基半胱氨酸的制备
取 L-半胱氨酸 109.5g(0.9mol), 置于 2000ml的烧瓶中,;溶于 1000 ml水中。 再加入 lmol/1的 NaOH溶液 24ml, 置冰浴申, 取 环氧乙烷 96ml(1.8mol) , 水浴下緩慢滴加半胱氨酸溶液中,:搅拌 一小时, 撤去冰浴恢复至室温再反应一小时。 将混合物用无水乙 醚 1000ml 分四次提取, 留水层。 蒸干水层得黄色晶体, 用水: 乙醇 =85ml:350ml重结晶, 滤出后用 95%的乙醇 2000ml冼涤, 烘干得白色晶体 121.8g,收率为 74.3%。
D
20) δ: 2.80 (t, 2H, /= 6.036Hz) , 3.08 (dd, 1H, J,=7.48Hz, 2=14.80Hz) ,3 18(dd,lH,/!=4.27Hz, J
2= 14.81Hz ) , 3.77-3.81 (m, 2H) 3.96 (dd, 1H,
步骤 2 2-氯乙基半胱氨酸的制备
取出 40g(0.24mol)2-羟乙基半胱氨酸,置于 1000ml的烧軏中, 加入浓盐酸 550ml,加热回流 7小时,回流结束,室温静置析出白色 晶体。滤出烘干得白色晶体 41.6g,收率为 QS o H-NMR OOMHz, D
20) δ:, 3.01- 3.04(m, 2H) , 3.12(dd,lH,
, 3.26 (dd,lH,
, 3.78- 3.84 (m,2H) 4.27-4.30 (m,lH) 步骤 3 2-氯乙基半胱酸甲酯盐酸盐的制备
取 2-氯乙基半胱氨酸 39g(0.21mol),溶于 500ml无水甲醇中, 冰浴冷却。 在冰浴的条件下緩慢滴 80ml(l.lmol)氯化亚砜, 加毕, 升至室温, 在室温下反 24小时。 减压蒸去溶剂, 用无水甲醇 (200 mi x 2)溶解后再蒸干, 以除去多余的氯化亚砜。 所得固体用: 50ml 无水甲醇和 160ml无水乙醚重结晶, 得白色晶体 37.5g, 收率为 76.1%。 。 H-NMR (400MHz, D
20) δ: 2.99-3.049 (m, 2H) 3.20(dd, 1H,
15.034Hz), , 3.78-3.82(m, 2H), 3. 90(s, 3H) 4.45 (dd,lH, /!=4.50Hz,
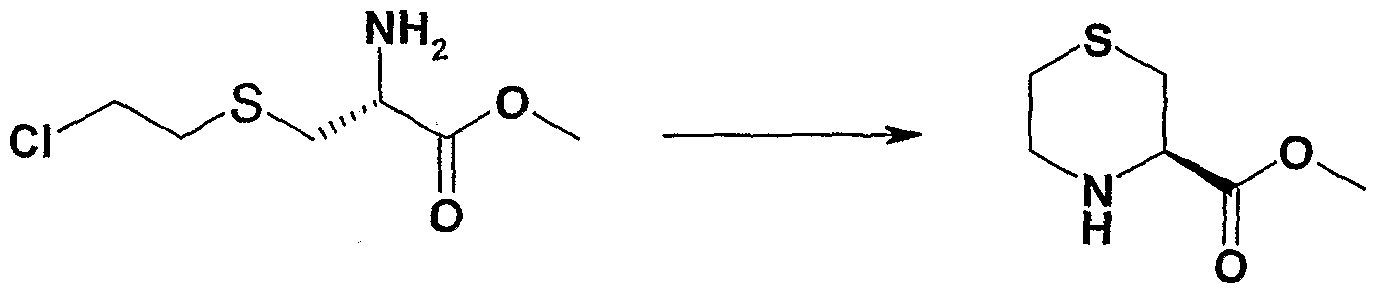
2=7.48Hz) 步骤 4 (R)-3-羧酸甲酯硫吗啉盐酸盐的制备
取 20g(0.085mol)2-氯乙基半胱氨酸甲酯盐酸盐, 溶于 200 ml 水中, 冰浴下滴加 7.2g(0.085mol)碳酸氢钠于 120 ml水的爝液, 冰浴下再恒温反应 1 小时后停止反应, 用乙酸乙酯 (100ml ^ 3)萃 取三次, 合并酯层, 用无水硫酸钠千燥 4小时, 减压蒸除¾剂, 残留物溶于 400ml无水甲醇中, 常温下搅拌反应 5天, 减 θέ蒸去 溶剂, 残留物用无水甲醇 /乙酸乙酯重结晶,得白色晶体 8.9g,;收率 为 49.2%。 iH-NMR OOM HZ, DMSO-d6) δ: 2.86-2.88(m,lH) , 2.96-2.99(m,lH) 3.06-3.22(m, 3H) 3.48-3.50 (m,lH),, 3.78 (s, 3H|) 4.42
(dd, 1H, !=3.52Hz, J2= 8.56Hz) 10.1(brs, 2H) 步骤 5 (R)-3-酰胺硫吗啉的制备
取( R )-3-羧酸甲醋硫吗啉盐酸盐 8.9g( 0.042mol )溶于 ^OOml 的无水甲醇中, 通入氨气反应 48h后, 蒸干溶剂, 残留物用无水 乙醇溶解, 滤出不溶物, 蒸干溶剂, 残留物用无水甲醇 /无水乙醚 重结晶, 得白色晶体 5.44g, 收率为 89.2%。 iH-NMR OOM Mz, CDC13) S:】H-NMR (400HMZ, CDCI3) δρρπι: 2.45 ~ 2.47(d, 1H J=8.4Hz) 2.50一 2.55(m, 2H) 2.56 ~ 2.58(m, 1H ) 3.21 ~ 3.28(m, 2H) 5.33(s,lH) 7.08(s,lH) 7.26(s,lH) 实施例 2
化合物 l: (R)-3-氰基 -4-(2-氨基 -3-甲基-丁酰基) 硫吗啉盐酸 盐的制备
取 Boc-VaL 0.217g(0.001mol)和实施例 1制备 (R)-3-瞵胺硫 吗啉 0.147g(0.001mol)溶于 20ml 干燥的 THF 中 , 加入 0.29g(0.0015mol)EDCI和 0.18g HOBT(0.0015mol),再滴加 · 异丙 基乙胺 0.35ml(0.002mol), 室温搅拌 12h。 反应完毕后减压浓缩, 加 20ml的水稀幹, 用乙酸乙酯 50ml x 3萃取,有机相用无水硫酸 钠干燥 4h后, 減压除去溶剂, 残留物溶于 20ml干燥的 THF中, 加入 2.8ml(0.002mol)三氟乙酸酐,搅拌 lh后,用饱和的 NaSC03 水溶液洗涤, 直到没有气泡出现,用乙酸乙酯 (50ml X 3)萃取,无水 硫酸钠干燥 4h, 硅胶柱层析分离(AcOEt: Cyclohexane =1:4), 得 无色油状物, 溶于 30ml 3N的盐酸乙酸乙酯中, 搅拌 5h,出现白
色沉淀, 滤出后用乙酸乙酯洗涤滤饼, 得到目标物为白色固体
0.072g, 收率为 27.2%。 ^-NMR (400HMZ, DMSO) 6ppm:
0.90 ~ 0.95(m, 6H) 2.01 ~ 2.06 (m, 1H) 2.50(s, 1H) 2.85 -
3.01(m, 3H) 3.39 - 3.44(m, 1H) 4.33 ~ 4.44(m, 2H) 6.15(s, 1H) 8.43(brs, 3H) FAB-MS m/e:228 [M+l] 实施例 3
化合物 2: (R)-3-氰基 -4-(2-氨基 -4-甲基-戊酰基) 硫吗啉盐酸 盐的制备
取 Boc-Leu 0.231g(0.001mol)和实施例 1制备的 (R)-3-酰胺 硫吗啉 0.147g(0.001mol)溶 20ml 干燥的 THF 中 , 加入 0.29g(0.0015mol)EDCI和 0.18g HOBT(0.0015mol),再滴加二异丙 基乙胺 0.35ml(0.002mol), 室温搅拌 12h。 反应完毕后减压浓缩, 加 20ml的水稀释, 用乙酸乙酯 (50ml < 3)萃取,有机相用无水硫酸 钠干燥 4h后, 减压除去溶剂, 残留物溶于 20ml干燥的 THF中, 加入 2.8ml(0.002mol)三氟醋酐,搅拌 lh后,用饱和的 NaHC03水 溶液洗涤, 直到没有气泡出现, 乙酸乙酯 (50ml X 3)萃取,无水硫酸 钠干燥 4h, 硅胶柱层析分离(乙酸乙酯: 环己垸 =1:4), 得无色油 状物, 溶于 30ml 3N的盐酸乙酸乙酯中, 搅拌 5h,出现白色沉淀, 滤出后用乙酸乙酯洗涤滤饼, 得到目标物为白色固体 0.096g, 收 率为 34.8%。 iH-NMR (400HMZ, D20) δρριιι: 0.77 ~ 0.81(m, 6H) 1.41 ~ 1.61 (m, 1H) 2.48 - 2.52(d, 1H J=7.6Hz) 2.68 ~ 2.28(m, 2H) 2,90 - 2.95(m, 1H) 3.51 ~ 3.57(t, 2H J=12.4Hz)
3.88 - 3.93(d, 1H J=14.4Hz) 3.35 ~ 3.70 (m, 1H) 5.97(s; 1H) EI-MS m/e:241【M+】。 实施例 4 测定化合物 1和 2的 DI -IV肽酶活性的抑制率和
ICso
步骤 1 DPP-IV制备
人结肠癌细 包系细胞 ( Caco-2 ) 培养: Caco-2 细胞培养于 DMEM (高糖, 含 10%胎牛血清, 1%NAA )培养基中, 按照 1: 1或 1: 2的比例传代细胞, 使细胞生长接近融合, 继续培养 2-3 周, 每 2-3天换液一次, 观察细胞出现刷状缘突起表明细胞已分 化, 收集细胞。
制备: 用预冷的 PBS洗细胞 2-3次, 每培养瓶细胞加入 0.5-lml 冰冷的 10mMTris-Hcl (含 0.15MNaCl,0.04t.i.u 抑肽酶 aprotinin,O.5%非离子去垢剂 P40, PH8), 使细胞溶解, 收乘至离 心管中, 4°C, 35000g离心 30min,取上清液, 测定蛋白含量, 低 温冰箱保存。 步骤 2: DPP-IV活性测定及化合物筛选
实臉分为: 酶对照组, 底物对照组, 酶反应对照组, 牝合物 组, 在 96孔板中进行, 反应体系 150μ1。 在 96孔板中化会物组 加入稀释好的受试化合物, 30 1/孑 ; 在酶对照组中加入 20μ1 DPP-IV酶液, 化合物組中也加入 20μ1 DPP-IV酶液; 加 N分析 緩冲液( 25Mm Tris-Hcl ΡΗ7·4,含 140mM NaCl,10Mm KCl,i%牛 血清白蛋白) , 酶对照组加 130 μ 1, 底物对照组加 125μ1, 酶反 应对照组加 105μ1, 化合物组加 75μ1; 加入 25μ1底物( )开 始反应,反应时间 lOmin在室温下进行。 酶对照組不加底物; 反应 lOmin后加入 19μ1/孔 25%水醋酸终止反应; 酶标仪 405nM:波长
测定吸光度, 计算的抑制率见下表。
化合物編号 浓度 ( n ) 抑制率(% ) ICso
10 7.65±0.68
30 13.15±1.92
化合物 1 100 40.27±3.09 188.7±26.4
300 73.02±1.74
1000 101.01±1.36
100 24.07±2,41
化合物 1
300 47.16±0.64