WO2006133611A1 - Dérivés de purine substituée par un groupe n2-quinoléyle ou isoquinoléyle, leurs préparations et utilisations - Google Patents
Dérivés de purine substituée par un groupe n2-quinoléyle ou isoquinoléyle, leurs préparations et utilisations Download PDFInfo
- Publication number
- WO2006133611A1 WO2006133611A1 PCT/CN2006/000113 CN2006000113W WO2006133611A1 WO 2006133611 A1 WO2006133611 A1 WO 2006133611A1 CN 2006000113 W CN2006000113 W CN 2006000113W WO 2006133611 A1 WO2006133611 A1 WO 2006133611A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- cancer
- salt
- hydrate
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
Definitions
- N 2 -quinoline or isoquinoline substituted anthracene derivative preparation method thereof and use thereof
- the present invention relates to medicinal chemistry, and in particular to N 2 -quinoline or isoquinoline substituted anthracene derivatives, and processes for their preparation and use. Background technique
- Cancer is a major threat to human health. Most of human cancers are caused by external environmental factors. The number of people who die of cancer every year in the world is no less than 5 million. Although there are some treatments, such as surgery, radiotherapy, chemotherapy, etc., the patient can be cured, and the cure rate is not good. The prevention and treatment of cancer with chemical drugs is currently one of the most effective methods of subduing tumors.
- the pyrimidine or purine or other heterocyclic substituent is located at the 1 position of the sugar ring, which corresponds to the 2 position of the hydroxyhydrofuran derivative. Recently, there have been reports of such derivatives as effective drugs against tumor or/and antiviral activity.
- N 2 - butylphenyl -2 - deoxy purine derivatives discloses N 2 - butylphenyl -2 - deoxy purine derivatives, the compounds having active eukaryotic cell a DNA polymerase.
- Tetrahedron Letters (1998, Vol. 39, 1827-1830) discloses 2,6,9-trisubstituted anthracene derivatives.
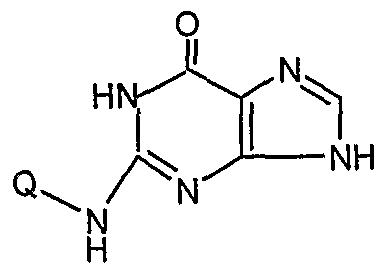
- the above hydrazine compounds are identical or similar in structure to the compounds of the present application, but these quinone compounds do not have a good antitumor effect.
- the technical problem to be solved by the present invention is to study and design an N 2 -substituted anthracene compound with low toxicity, wide anticancer spectrum, high anticancer activity and good stability.
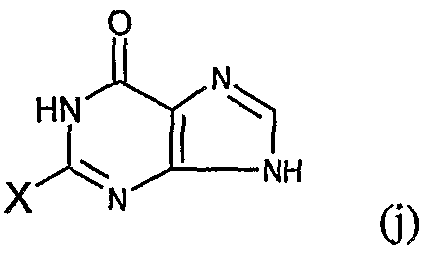
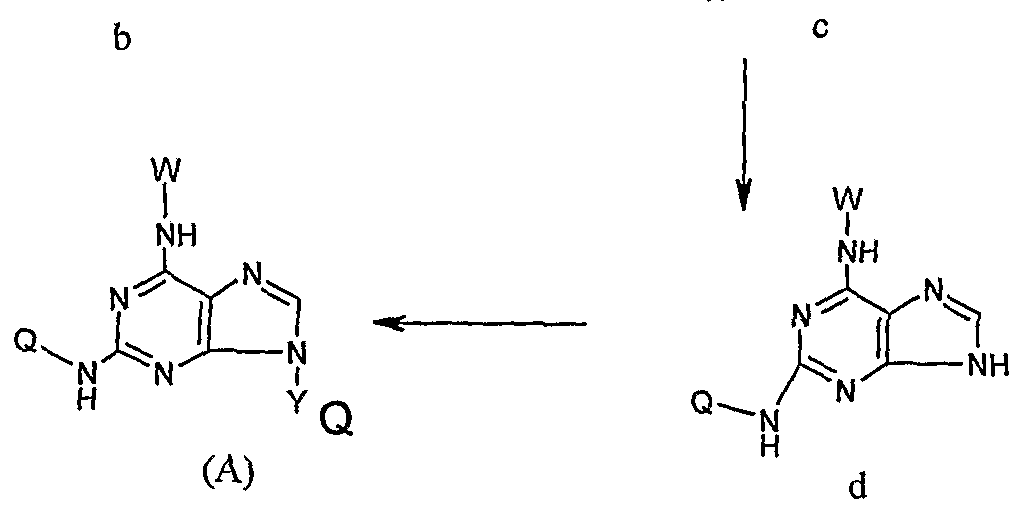
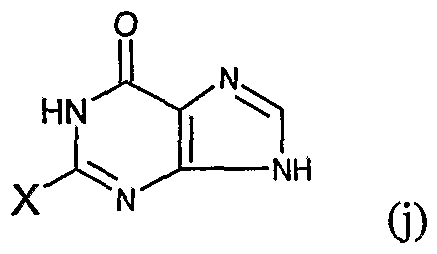
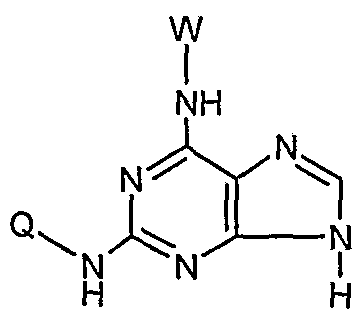
- the present invention provides an N 2 -quinoline or isoxaline substituted indole compound of the formula (A) or a salt thereof or a hydrate thereof:
- W is H or an optionally substituted C r C 6 linear or branched fluorenyl or substituted C 3 -C 6 cycloalkyl group, said substituent being a -C6 linear or branched alkyl group or halogen;
- Y is H or a pharmaceutically acceptable glycosyl group, wherein the glycosyl group is preferably of the formula:
- Z is the following group 0
- B, E, G, R, T, M are a straight or branched alkyl or haloalkyl group of H or CC 6 , a C 3 -C 6 cyclodecyl group, a halogen, CN or NH 2 .
- W is H or the following groups
- the Q of the compound is the following group:
- the substituents B, E, G, R, T, M of the compound are H, F, CH 3 , CF 3 , CN or NH 2 ; especially H.
- the ⁇ is 11.
- the invention specifically provides the following compounds:
- Another object of the present invention is to provide a pharmaceutical composition of the compound of the formula XVDI, which is composed of a compound of any one of formulas I-Formula XWI, or a salt, hydrate thereof and pharmaceutically acceptable excipient thereof,
- the composition is a pharmaceutical composition suitable for enteral, topical and parenteral administration, or administration to a mammal by inhalation spray.
- the composition is a tablet, a capsule, a pill, an oral liquid preparation, a granule, a powder, an injection, an implant or an external preparation.
- a halogenating agent is added, and an organic solvent is reacted at 50 to 150 ° C for 1 to 72 hours, cooled, water is added, and the pH is adjusted to 2 to 5 with an acid, followed by cooling;
- reaction is carried out at ° C for 1-72 hours, and the organic solvent is distilled off;
- the present application also provides another method of preparing the above compound or a salt thereof, the method comprising the steps of:
- the salt of the compound of the formula (A) includes pharmaceutically acceptable salts, for example, acid addition salts obtained from inorganic or organic acids, such as hydrochloride, hydrobromide, hydroiodide, p-toluenesulfonate, Phosphates, sulfates, perchlorates, acetates, trifluoroacetates, propionates, citrates, malonates, succinates, lactates, oxalates, tartrates and Benzoate.
- inorganic or organic acids such as hydrochloride, hydrobromide, hydroiodide, p-toluenesulfonate, Phosphates, sulfates, perchlorates, acetates, trifluoroacetates, propionates, citrates, malonates, succinates, lactates, oxalates, tartrates and Benzoate.
- the salt may also be a salt formed with a base, such a salt including a salt obtained from an inorganic base or an organic base, such as an alkaline earth metal salt such as a magnesium salt or a calcium salt, an organic amine salt such as morpholine, piperidine, dimethylamine or two. Ethylamine salt.
- a base such as a salt including a salt obtained from an inorganic base or an organic base, such as an alkaline earth metal salt such as a magnesium salt or a calcium salt, an organic amine salt such as morpholine, piperidine, dimethylamine or two. Ethylamine salt.
- the present invention further relates to a pharmaceutical composition suitable for administration to the mammal (including human) by enteral (for example, oral or rectal administration), topical and parenteral administration, or by inhalation spray, for example, orally. , injection, implantation, external use, etc.
- enteral for example, oral or rectal administration
- topical and parenteral administration for example, orally.
- inhalation spray for example, orally.
- Oral including tablets (ordinary tablets, tablets, sublingual tablets, oral patches, chewable tablets, dispersible tablets, soluble tablets, effervescent tablets, vaginal tablets or vaginal effervescent tablets, sustained release tablets, controlled release tablets, enteric dissolution) Tablets, oral immediate release tablets, etc.; capsules (hard capsules, soft capsules, sustained release capsules, controlled release capsules, intestinal sols) Pellets, etc.; pills (drop pills, sugar pills, pellets); oral liquid preparations (syrups; oral solutions; oral suspensions; oral emulsions); granules (suspension granules, effervescent granules, enteric granules, Slow release granules, controlled release granules, etc.); powder.
- tablets ordinary tablets, tablets, sublingual tablets, oral patches, chewable tablets, dispersible tablets, soluble tablets, effervescent tablets, vaginal tablets or vaginal effervescent tablets, sustained release tablets, controlled
- the injection includes an injection solution, a sterile powder for injection or a sterile mass (including a process such as solvent crystallization, spray drying or freeze drying), a concentrated solution for injection; an implant; the external preparation includes: a suppository; Aerosol; powder; spray; film; gel; patch;
- compositions of the present invention can be prepared according to any of the method dosage forms known in the art for preparing pharmaceutical compositions.
- the composition is useful for anti-tumor and for the treatment of a related condition, the composition being used alone or in combination with one or more other anti-tumor drugs.
- the amount of active ingredient which may be combined with carrier materials to produce a single dosage form may vary depending upon the host treated and the particular mode of administration.
- the application also provides the use of the compound in the preparation of a medicament.
- the compound is used for preventing or treating abnormal cell growth, and the abnormal cell growth may be expressed as a tumor, and the tumor may be lung cancer, liver cancer, blood cancer, bone cancer, pancreatic cancer, skin cancer, melanoma, uterine cancer, ovary Cancer, rectal cancer, gastric cancer, colon cancer, breast cancer, uterine cancer, fallopian tube cancer, endometrial cancer, cervical cancer, vaginal cancer, vulvar cancer, esophageal cancer, small intestine cancer, endocrine system cancer, soft tissue sarcoma, urethral cancer, Prostate cancer, lymphoma, bladder cancer, kidney or ureteral cancer, spinal tumor, brain stem glioma, pituitary adenoma, especially lung cancer, liver cancer, blood cancer, pancreatic cancer and breast cancer.
- the compound By measuring the compound of the present invention, the compound has a significant growth inhibitory effect on human lung cancer, liver cancer, and blood cancer cells cultured in vitro in an experiment for inhibiting tumor cell growth in vitro, and shows a dose- Effect relationship.
- the drug of the present invention has an LD 5 o of 160 mg/kg in mice upon intraperitoneal injection.
- liver cancer H 22 mice The tumor inhibition test of liver cancer H 22 mice was 80m g /kg, and the tumor inhibition rate was 69%.
- the compound can be combined with chemotherapy, radiotherapy and biotechnological therapy to treat tumors with synergistic effects and reduced drug side effects.
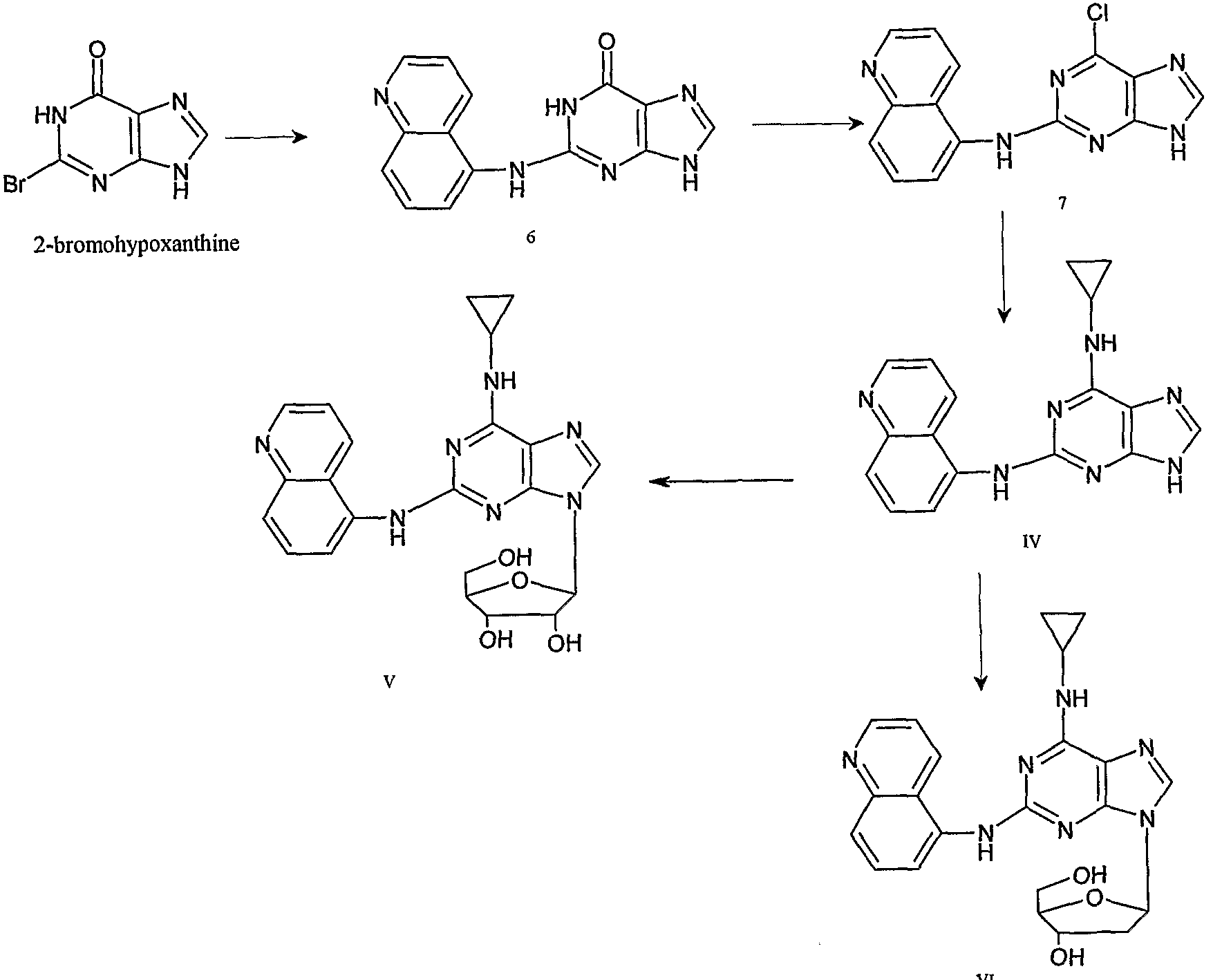
- Example 8 The same compound IV as in Example 5 was prepared and prepared by the following route.
- Preparation process Accurately weigh the prescribed amount of compound I and lactose, and then add a prescribed amount of micro-silica gel to help increase the fluidity of the main drug. After adding other auxiliary materials and fully mixing the powder, the powder is directly compressed, that is, it is obtained.
- Preparation process Accurately weigh the prescribed amount of compound I and Tween-80 mixed and grind, and add 0.3% of the drug solution to dissolve.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicinal Preparation (AREA)
Description

































Claims

















Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008516106A JP2008546653A (ja) | 2005-06-16 | 2006-01-23 | N2−キノリン又はイソキノリン置換のプリン誘導体及びその製造方法並びにその用途 |
| CA002611865A CA2611865A1 (en) | 2005-06-16 | 2006-01-23 | N2-quinolyl or isoquinolyl substituted purine derivatives, the preparation and uses thereof |
| AU2006257583A AU2006257583B2 (en) | 2005-06-16 | 2006-01-23 | N2-quinolyl or isoquinolyl substituted purine derivatives, the preparation and uses thereof |
| EP06705534.3A EP1897882B1 (en) | 2005-06-16 | 2006-01-23 | N2-quinolyl or isoquinolyl substituted purine derivatives, the preparation and uses thereof |
| BRPI0611564-0A BRPI0611564A2 (pt) | 2005-06-16 | 2006-01-23 | derivados da purina por substituição de n2-quinolina ou isoquinolina e respectivos métodos de preparação e usos |
| ES06705534.3T ES2529915T3 (es) | 2005-06-16 | 2006-01-23 | Derivados de purina sustituidos con N2-quinolilo o isoquinolilo, su preparación y sus usos |
| US11/452,955 US7399754B2 (en) | 2005-06-16 | 2006-06-15 | N2-quinoline or isoquinoline substituted purine derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200510026846.3 | 2005-06-16 | ||
| CNB2005100268463A CN100526315C (zh) | 2005-06-16 | 2005-06-16 | N2-喹啉或异喹啉取代的嘌呤衍生物及其制备方法和其用途 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/452,955 Continuation US7399754B2 (en) | 2005-06-16 | 2006-06-15 | N2-quinoline or isoquinoline substituted purine derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006133611A1 true WO2006133611A1 (fr) | 2006-12-21 |
Family
ID=37518697
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2006/000113 WO2006133611A1 (fr) | 2005-06-16 | 2006-01-23 | Dérivés de purine substituée par un groupe n2-quinoléyle ou isoquinoléyle, leurs préparations et utilisations |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US7399754B2 (zh) |
| EP (1) | EP1897882B1 (zh) |
| JP (1) | JP2008546653A (zh) |
| CN (1) | CN100526315C (zh) |
| AU (1) | AU2006257583B2 (zh) |
| BR (1) | BRPI0611564A2 (zh) |
| CA (1) | CA2611865A1 (zh) |
| ES (1) | ES2529915T3 (zh) |
| RU (1) | RU2417230C2 (zh) |
| WO (1) | WO2006133611A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008135232A1 (en) * | 2007-05-02 | 2008-11-13 | Riccardo Cortese | Use and compositions of purine derivatives for the treatment of proliferative disorders |
| JP2010524862A (ja) * | 2007-04-20 | 2010-07-22 | ヂェ ジィァン メディスン カンパニー リミテッド シィンシャン ファーマシューティカル ファクトリー | 2,6−ジ含窒素置換したプリン誘導体及びその製造方法と使用 |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007070872A1 (en) | 2005-12-15 | 2007-06-21 | Rigel Pharmaceuticals, Inc. | Kinase inhibitors and their uses |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| NZ587051A (en) | 2008-01-04 | 2012-12-21 | Intellikine Llc | Isoquinolinone derivatives, compositions and methods of inhibiting phosphatidyl inositol-3 kinase (pi3 kinase) |
| CN101497615B (zh) * | 2008-01-29 | 2010-12-15 | 浙江医药股份有限公司新昌制药厂 | 取代嘌呤,其制备方法及在医学中的应用 |
| US8703778B2 (en) | 2008-09-26 | 2014-04-22 | Intellikine Llc | Heterocyclic kinase inhibitors |
| CA2760791C (en) | 2009-05-07 | 2017-06-20 | Intellikine, Inc. | Heterocyclic compounds and uses thereof |
| WO2011032050A2 (en) * | 2009-09-11 | 2011-03-17 | Trius Therapeutics, Inc. | Gyrase inhibitors |
| ES2593256T3 (es) | 2010-05-21 | 2016-12-07 | Infinity Pharmaceuticals, Inc. | Compuestos químicos, composiciones y métodos para las modulaciones de cinasas |
| WO2012064973A2 (en) | 2010-11-10 | 2012-05-18 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN103648499B (zh) | 2011-01-10 | 2017-02-15 | 无限药品股份有限公司 | 用于制备异喹啉酮的方法及异喹啉酮的固体形式 |
| EP2734520B1 (en) | 2011-07-19 | 2016-09-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CA2842190A1 (en) | 2011-07-19 | 2013-01-24 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013032591A1 (en) | 2011-08-29 | 2013-03-07 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| CN104418858B (zh) * | 2013-08-30 | 2018-12-11 | 浙江医药股份有限公司新昌制药厂 | 2,6-二含氮取代的嘌呤衍生物及其制备方法和其药物组合物与应用 |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CA2925944C (en) | 2013-10-04 | 2023-01-10 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| EP4066834A1 (en) | 2014-03-19 | 2022-10-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders |
| WO2015160975A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| WO2016054491A1 (en) | 2014-10-03 | 2016-04-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN108349985A (zh) | 2015-09-14 | 2018-07-31 | 无限药品股份有限公司 | 异喹啉酮的固体形式、其制备方法、包含其的组合物及其使用方法 |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2017223422A1 (en) | 2016-06-24 | 2017-12-28 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| CN114980900A (zh) * | 2020-01-13 | 2022-08-30 | 上海华禹生物科技有限公司 | N2-喹啉或异喹啉取代的嘌呤衍生物在癌症治疗中的用途 |
| WO2021225926A1 (en) * | 2020-05-07 | 2021-11-11 | Zhanggui Wu | Combination cancer therapy using n2-quinoline or isoquinoline substituted purine derivatives |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0173624A2 (en) | 1984-08-24 | 1986-03-05 | Merck & Co. Inc. | 4-(guanin-9-yl) butanals and antiviral compositions containing them |
| EP0253412A2 (en) | 1986-07-18 | 1988-01-20 | Ceskoslovenska akademie ved | N-Phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases, methods for their preparation and pharmaceutical compositions therefrom with antiviral activity |
| US4853386A (en) | 1985-08-17 | 1989-08-01 | Boehringer Mannheim Gmbh | N6 -disubstituted purine derivatives, and pharmaceutical compositions containing them, useful for treating allergic diseases, bronchospastic and bronchoconstrictory conditions |
| EP0353955A2 (en) | 1988-08-02 | 1990-02-07 | Beecham Group Plc | Novel compounds |
| WO1991013898A1 (en) | 1990-03-13 | 1991-09-19 | The United States Of America, Represented By The Secretary, United States Department Of Commerce | O6-benzylated guanine, guanosine and 2'-deoxyguanosine compounds possessing o6-alkylguanine-dna alkyltransferase depleting activity |
| WO1992001698A1 (en) | 1990-07-19 | 1992-02-06 | Beecham Group Plc | Antiviral phosphono-alken derivatives of purines |
| US5091430A (en) | 1990-03-13 | 1992-02-25 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | O6 -substituted guanine compounds and methods for depleting O6 -alkylguanine-DNA alkyltransferase levels |
| EP0481214A1 (en) | 1990-09-14 | 1992-04-22 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Prodrugs of phosphonates |
| WO2000055161A1 (en) * | 1999-03-17 | 2000-09-21 | Albany Molecular Research, Inc. | 6-substituted biaryl purine derivatives as potent cyclin/cdk inhibitors and antiproliferative agents |
| JP2003055377A (ja) | 2001-08-16 | 2003-02-26 | Mitsui Chemicals Inc | 2−アミノ−6−シクロプロピルアミノ−9h−プリンの製造法 |
| JP2003119197A (ja) | 2001-10-15 | 2003-04-23 | Mitsui Chemicals Inc | 2−アミノ−6−シクロプロピルアミノ−9h−プリンの製造法 |
| WO2005016528A2 (en) | 2003-08-15 | 2005-02-24 | Irm Llc | 6-substituted anilino purines as rtk inhibitors |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0704215A3 (en) * | 1994-06-02 | 1998-04-01 | Takeda Chemical Industries, Ltd. | Inhibitor of vascular permeability enhancer |
| JPH0848631A (ja) * | 1994-06-02 | 1996-02-20 | Takeda Chem Ind Ltd | 血管透過性亢進抑制剤 |
| GB9918035D0 (en) * | 1999-07-30 | 1999-09-29 | Novartis Ag | Organic compounds |
| GB0407723D0 (en) * | 2004-04-05 | 2004-05-12 | Novartis Ag | Organic compounds |
-
2005
- 2005-06-16 CN CNB2005100268463A patent/CN100526315C/zh not_active Expired - Fee Related
-
2006
- 2006-01-23 AU AU2006257583A patent/AU2006257583B2/en not_active Ceased
- 2006-01-23 CA CA002611865A patent/CA2611865A1/en not_active Abandoned
- 2006-01-23 EP EP06705534.3A patent/EP1897882B1/en not_active Not-in-force
- 2006-01-23 ES ES06705534.3T patent/ES2529915T3/es active Active
- 2006-01-23 RU RU2007148681/04A patent/RU2417230C2/ru not_active IP Right Cessation
- 2006-01-23 JP JP2008516106A patent/JP2008546653A/ja active Pending
- 2006-01-23 WO PCT/CN2006/000113 patent/WO2006133611A1/zh active Application Filing
- 2006-01-23 BR BRPI0611564-0A patent/BRPI0611564A2/pt not_active IP Right Cessation
- 2006-06-15 US US11/452,955 patent/US7399754B2/en active Active - Reinstated
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0173624A2 (en) | 1984-08-24 | 1986-03-05 | Merck & Co. Inc. | 4-(guanin-9-yl) butanals and antiviral compositions containing them |
| US4853386A (en) | 1985-08-17 | 1989-08-01 | Boehringer Mannheim Gmbh | N6 -disubstituted purine derivatives, and pharmaceutical compositions containing them, useful for treating allergic diseases, bronchospastic and bronchoconstrictory conditions |
| EP0253412A2 (en) | 1986-07-18 | 1988-01-20 | Ceskoslovenska akademie ved | N-Phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases, methods for their preparation and pharmaceutical compositions therefrom with antiviral activity |
| EP0353955A2 (en) | 1988-08-02 | 1990-02-07 | Beecham Group Plc | Novel compounds |
| WO1991013898A1 (en) | 1990-03-13 | 1991-09-19 | The United States Of America, Represented By The Secretary, United States Department Of Commerce | O6-benzylated guanine, guanosine and 2'-deoxyguanosine compounds possessing o6-alkylguanine-dna alkyltransferase depleting activity |
| US5091430A (en) | 1990-03-13 | 1992-02-25 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | O6 -substituted guanine compounds and methods for depleting O6 -alkylguanine-DNA alkyltransferase levels |
| WO1992001698A1 (en) | 1990-07-19 | 1992-02-06 | Beecham Group Plc | Antiviral phosphono-alken derivatives of purines |
| EP0481214A1 (en) | 1990-09-14 | 1992-04-22 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Prodrugs of phosphonates |
| WO2000055161A1 (en) * | 1999-03-17 | 2000-09-21 | Albany Molecular Research, Inc. | 6-substituted biaryl purine derivatives as potent cyclin/cdk inhibitors and antiproliferative agents |
| JP2003055377A (ja) | 2001-08-16 | 2003-02-26 | Mitsui Chemicals Inc | 2−アミノ−6−シクロプロピルアミノ−9h−プリンの製造法 |
| JP2003119197A (ja) | 2001-10-15 | 2003-04-23 | Mitsui Chemicals Inc | 2−アミノ−6−シクロプロピルアミノ−9h−プリンの製造法 |
| WO2005016528A2 (en) | 2003-08-15 | 2005-02-24 | Irm Llc | 6-substituted anilino purines as rtk inhibitors |
Non-Patent Citations (4)
| Title |
|---|
| J. MED. CHEM, vol. 27, 1984, pages 175 - 181 |
| J. ORG. CHEM, vol. 69, 2004, pages 3212 - 3215 |
| See also references of EP1897882A4 |
| TETRAHEDRON LETTERS, vol. 39, 1998, pages 1827 - 1830 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010524862A (ja) * | 2007-04-20 | 2010-07-22 | ヂェ ジィァン メディスン カンパニー リミテッド シィンシャン ファーマシューティカル ファクトリー | 2,6−ジ含窒素置換したプリン誘導体及びその製造方法と使用 |
| RU2498987C2 (ru) * | 2007-04-20 | 2013-11-20 | Чжэ Цзян Медисин Ко., Лтд, Синьчан Фармасеутикал Фэктори | 2,6-динитросодержащие замещенные производные пурина, способ их получения и использование |
| WO2008135232A1 (en) * | 2007-05-02 | 2008-11-13 | Riccardo Cortese | Use and compositions of purine derivatives for the treatment of proliferative disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1897882A1 (en) | 2008-03-12 |
| US7399754B2 (en) | 2008-07-15 |
| RU2417230C2 (ru) | 2011-04-27 |
| EP1897882B1 (en) | 2014-12-10 |
| EP1897882A4 (en) | 2009-12-23 |
| BRPI0611564A2 (pt) | 2010-09-21 |
| AU2006257583B2 (en) | 2013-01-24 |
| RU2007148681A (ru) | 2009-07-27 |
| JP2008546653A (ja) | 2008-12-25 |
| AU2006257583A1 (en) | 2006-12-21 |
| ES2529915T3 (es) | 2015-02-25 |
| US20060293274A1 (en) | 2006-12-28 |
| CN1880315A (zh) | 2006-12-20 |
| CN100526315C (zh) | 2009-08-12 |
| CA2611865A1 (en) | 2006-12-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006133611A1 (fr) | Dérivés de purine substituée par un groupe n2-quinoléyle ou isoquinoléyle, leurs préparations et utilisations | |
| TWI683813B (zh) | 苯并咪唑衍生物及其醫藥組合物及使用方法 | |
| CN105153122B (zh) | [(吲哚-3-基)嘧啶-2-基]氨基苯基丙-2-烯酰胺衍生物及盐、制备方法、应用 | |
| KR101210361B1 (ko) | 다형의 gabaa 효능제 | |
| CN103087133B (zh) | 嘌呤衍生物作为腺苷a1受体激动剂及其用法 | |
| CN109516999B (zh) | 用作蛋白质激酶调节剂的化合物及其应用 | |
| ES2772498T3 (es) | Preparación de un inhibidor mek y formulación que comprende el mismo | |
| TWI705967B (zh) | 苯并咪唑衍生物及其醫藥組合物及使用方法 | |
| CN111655693A (zh) | 抑制瞬时型感受器电位a1离子通道 | |
| KR101563371B1 (ko) | 2,6 질소 함유 이치환 푸린 유도체 | |
| US10913765B2 (en) | Liver specific delivery-based gemcitabine prodrug nucleoside cyclic phosphate compound, and application thereof | |
| JPS63107995A (ja) | ヌクレオシド類縁体 | |
| CN115109058B (zh) | 一种用于治疗胃癌的药物及其制备方法 | |
| TWI794576B (zh) | 一類含氟取代的苯并噻吩類化合物及其藥物組合物及應用 | |
| CZ288366B6 (en) | 2-Aminoalkyl-5-aminoalkylamino substituted isoquinoindazol-6-(2H)-ones exhibiting antitumor activity, their use, pharmaceutical preparation in which they are comprised and process of their preparation | |
| CN114031561B (zh) | 含4-苯氧基喹唑啉类化合物及其应用 | |
| WO2021047672A1 (en) | Triterpenoid compounds, pharmaceutical compositions thereof, and their use for treating nuclear receptor subfamily 4 group member 1-mediated disease | |
| TW200817426A (en) | 2'-cyanopyrimidine nucleoside compound | |
| WO2020114455A1 (zh) | 含有哌嗪酮的喹唑啉二酮盐类化合物、其制备方法、药物组合物和用途 | |
| CN115772156A (zh) | 用作hpk1激酶抑制剂的化合物及其制备方法和应用 | |
| CN116751240A (zh) | 2’-去氧-2’,2’-二氟胞苷碳酸酯酰肼的制备及其应用 | |
| CN115784987A (zh) | 一种与肺炎相关的化合物及其制备方法和应用以及一种药物组合物 | |
| TW201343663A (zh) | 新穎嘧啶核苷化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 11452955 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 11452955 Country of ref document: US |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2611865 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/016206 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008516106 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006257583 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006705534 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007148681 Country of ref document: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006257583 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006257583 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006705534 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0611564 Country of ref document: BR Kind code of ref document: A2 |