WO2006078006A1 - インドール類およびそれを含む医薬組成物 - Google Patents
インドール類およびそれを含む医薬組成物 Download PDFInfo
- Publication number
- WO2006078006A1 WO2006078006A1 PCT/JP2006/300925 JP2006300925W WO2006078006A1 WO 2006078006 A1 WO2006078006 A1 WO 2006078006A1 JP 2006300925 W JP2006300925 W JP 2006300925W WO 2006078006 A1 WO2006078006 A1 WO 2006078006A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- substituted
- alkyl group
- unsubstituted alkyl
- unsubstituted
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a novel indole and a pharmaceutically acceptable salt thereof, which are useful as pharmaceuticals and have excellent oral absorbability.
- the j8 1 adrenergic receptor is mainly present in the heart, and stimulation via the receptor causes an increase in heart rate and an increase in cardiac contractility.
- j8 2 -Adrenergic receptors are mainly present in vascular, bronchial and uterine smooth muscles, and stimulation through these receptors leads to vascular and bronchodilation and suppression of uterine contraction, respectively.
- 8 3-adrenergic receptors are mainly present in adipocytes, gallbladder, and intestinal tract, and are also present in brain, liver, stomach, prostate, etc. It has been reported that stimulating effects induce fat degradation, suppress intestinal motility, promote glucose uptake, and antidepressant.
- j8 3 -adrenergic receptors mainly exist in human bladder, and it has been reported that human bladder smooth muscle is relaxed by ⁇ 3 -adrenergic receptor stimulants.
- Many ⁇ 1 -adrenergic receptor stimulants and j8 2 -adrenergic receptor stimulants have been developed so far, and they are used in medicine as cardiotonic agents, bronchodilators, urgency and premature birth prevention agents.
- ⁇ 3-adrenergic receptor stimulants are effective for obesity, hyperglycemia, hypersensitivity colitis caused by intestinal motility, frequent urination or urinary incontinence, depression, gallstones or increased biliary motility It has been found useful as a preventive or therapeutic agent for diseases caused by it.
- ⁇ 3 adrenoceptor stimulating effect ⁇ 1 and ⁇ or j8 2 -adrenergic receptor stimulating effect, resulting in ⁇ 1 and ⁇ or ⁇ 2 -adrenergic receptor stimulating effect.
- ⁇ 3 adrenoceptor stimulating effect ⁇ 1 and ⁇ or j8 2 -adrenergic receptor stimulating effect, resulting in ⁇ 1 and ⁇ or ⁇ 2 -adrenergic receptor stimulating effect.
- Patent Document 2 reduces the side effects such as increased heart rate and hand tremor due to low ⁇ 1 and ⁇ or ⁇ 2 -adrenergic receptor stimulating action. ing.
- Patent Document 1 Japanese Patent Laid-Open No. 11 255743
- Patent Document 2 Pamphlet of International Publication No. 03Z106418
- the problem to be solved by the present invention is a novel ⁇ 3 adrenergic receptor stimulant having an excellent ⁇ 3 adrenergic receptor stimulating action, more preferably ⁇ 1 and ⁇ or ⁇ 2 adrenergic receptor stimulation. Compared to the action, it has a strong j8 3 -adrenergic receptor stimulating action, and therefore, ⁇ 1 and ⁇ or j8 2 --adrenergic receptor stimulating action causes side effects such as increased heartbeat and finger tremor. Attenuated and highly selective
- the compounds disclosed in WO 03Z106418 have a low ⁇ 1 and ⁇ or ⁇ 2 -adrenoceptor stimulating action, and thus have side effects such as heartbeat enhancement and hand tremor.
- the present inventors found that the compound was excellent as a pharmaceutical product after being attenuated, but there was room for improvement in oral absorbability.
- the present inventors have found that the indole derivative represented by the formula (I) and pharmaceutically acceptable salts thereof have excellent oral absorbability. As a result, the present invention has been accomplished.
- the present invention relates to the following.
- R 1 is not present, or is present in one or more, the same or different, and is a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkoxy group, a halogen atom, a hydroxyl group, or Represents an amino group.
- R 2 represents a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 3 , R 4 , R 5 , and R 6 each independently represent a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 7 is absent, is one or more, is the same or different, and represents a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkoxy group, a halogen atom, a hydroxyl group, or an amino group.
- R 8 represents a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 9 is a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted aralkyl. Represents a substituted group or a substituted or unsubstituted aryl group. Or a pharmaceutically acceptable salt thereof.
- R 2 represents a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 3 , R 4 , R 5 , and R 6 each independently represent a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 8 represents a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 9 is a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted aralkyl. Represents a substituted group or a substituted or unsubstituted aryl group. Or a pharmaceutically acceptable salt thereof.
- R 8 represents a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 9 is a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted aralkyl. Represents a substituted group or a substituted or unsubstituted aryl group. Or a pharmaceutically acceptable salt thereof.
- R 9 is an alkyl group, an alkyl group, an alkyl group, an aralkyl group, an aryl group, Or a substituted alkyl group (the substituent is a halogen atom, a hydroxyl group, an alkanol group, an alkoxy group, an alkoxy carbo group, a cyan group, an alkyl thio group, an aryl thio group, an alkyl sulfo group, an aryl sulfo group, an alkanoic group; A compound selected from the group consisting of a ruoxy group, an alkoxycarbonyloxy group, a cycloalkyloxycarboxoxy group and a carboxy group, which are present in the same or different form. Acceptable salt.
- a pharmaceutical composition comprising the compound according to any one of [1] to [8] or a pharmaceutically acceptable salt thereof as an active ingredient.
- Obesity, hyperglycemia comprising administering to a patient in need of treatment an effective amount of a compound according to any one of [1] to [8] or a pharmaceutically acceptable salt thereof, Treatment methods for hyperinsulinemia, diabetes, hypertriglyceremia, frequent urination, urinary incontinence, overactive bladder syndrome, depression, diseases caused by intestinal hyperactivity, gallstones, or diseases caused by increased biliary motility
- [12] The compound according to any one of [1] to [8] or a pharmaceutically acceptable salt thereof, obesity, hyperglycemia, hyperinsulinemia, diabetes, hypertriglyceridemia, frequent Urine, urinary incontinence, excessive Use for the manufacture of a therapeutic agent for active bladder syndrome, depression, diseases caused by intestinal hyperactivity, gallstones, or diseases caused by biliary hyperactivity.
- R 9 represents the same meaning as described above), and oral absorption is improved.
- the compound represented by the formula (I) or a pharmaceutically acceptable salt thereof is collectively referred to as “the compound of the present invention” as necessary.
- the compound of the present invention is a ⁇ 3 -adrenergic receptor stimulant excellent in oral absorption, such as obesity, hyperglycemia, hyperinsulinemia, diabetes, hypertriglyceridemia, frequent urination, urine Incontinence, overactive bladder syndrome, depression, irritable colitis caused by intestinal hypermotility, gastritis caused by gallstones, or increased bile duct movement, stomach 'duodenal ulcer, enteritis, irritable bowel syndrome, gallbladder' biliary tract It is useful for the treatment of diseases or diseases such as urolithiasis. Brief Description of Drawings
- FIG. 1 shows rat oral absorption evaluation test data of the compound of the present invention.
- ⁇ indicates compound 1
- the X represents compound 2.
- the country represents compound 3.
- ⁇ represents compound 4. ⁇ represents the compound 5
- halogen atom examples include fluorine, chlorine, bromine, iodine and the like.
- alkyl group examples include linear or branched alkyl groups having 1 to 6 carbon atoms, and specific examples include methyl, ethyl, propyl, isopropyl, butyl, 2 butyl, 2 methylpropyl, 1 , 1-dimethylethyl, pentyl, 3-pentyl, 3-methylbutyl, hexyl, 3-hexyl, 4-methylpentyl and the like.
- L and alkyl groups include linear or branched alkyl groups having 1 to 4 carbon atoms.
- alkoxy group examples include groups in which one oxygen atom is bonded to the bond of the alkyl group.
- alkenyl group examples include straight-chain or branched carbon atoms of 6 or less, such as butyl, allyl, probe, 2-probe, butel, pentenyl, hexenyl and the like. Examples include alkenyl groups.
- alkyl group examples include straight-chain or branched alkynyl groups having 6 or less carbon atoms such as ethur, propargyl, 1-buturyl, 2-pentyl, 2-pentyl, 3-pentyl, and 2-hexyl. Etc.
- Examples of the cycloalkyl group include 3- to 8-membered cycloalkyl groups such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
- Examples of the alkanoyl group include carbon such as formyl, acetyl, and propanol. Examples include alkanoyl groups having 1 to 6 atoms.
- the aryl moiety of the aralkyl group is, for example, an aryl group having 10 or less carbon atoms such as phenyl, 1 or 2-naphthyl, and the alkyl moiety is, for example, the number of carbon atoms such as methyl, ethyl, propyl, or butyl. Examples include 6 or less alkyl groups. Representative Examples of typical aralkyl groups include a benzyl group, 1 or 2-phenethyl group, and the like.
- the substituted alkyl group, the substituted alkoxy group, the substituted alkenyl group, the substituted alkynyl group, or the substituted cycloalkyl group may be one or the same or different, and a plurality of substituents may be, for example, a halogen atom , Cyano group, phenoxy group, benzyloxy group, hydroxyl group, alkoxy group, alkanoyloxy group, amino group, monoalkylamino group, dialkylamino group, strong rubamoyl group, alkylaminocarbol group, dialkylaminocarbol group Carboxy group, alkoxy carbo group, alkyl thio group, alkyl sulfiel group, alkyl sulfonyl group, alkanoylamino group, alkyl sulfonyl amino group, phthalimide group, alkanoyl group, allylthio group, allyl sulfonyl
- aryl group examples include aryl groups having 10 or less carbon atoms such as phenyl, 1 or 2-naphthyl, and the like.
- heteroaryl group examples include a 5- to 10-membered monocyclic or bicyclic heteroaryl group containing 1 to 4 heteroatoms selected from the group consisting of a nitrogen atom, an oxygen atom, and a sulfur atom. Can be mentioned. Specific examples include pyridyl, furyl, chenyl, quinolyl, benzimidazolyl, benzthiazolyl, or indolyl.
- the substituents in the substituted aryl group and the substituted aralkyl group may be one or more, the same or different, for example, a halogen atom, an alkyl group which may be substituted with a halogen atom, or a halogen atom. Examples thereof include an alkoxy group and a -tro group. Preferably, the substituent includes an alkoxy group such as methoxy.
- alkylthio group examples include groups in which one sulfur atom is bonded to the bonding site of the alkyl group.
- the alkylsulfier group is represented by the formula: S (O) A group in which one represented group is bonded.
- the alkylsulfol group is represented by the formula: so- at the binding site of the alkyl group.
- Examples of the mono- or dialkylamino group include groups in which one or both hydrogens of an amino group are independently substituted with a substituted or unsubstituted alkyl group.
- alkylsulfo-lumino group is represented by the formula NHSO-
- the sulfur atom side of the group represented can be bonded, and a C1-C8 alkyl group or the like can be substituted on the nitrogen atom.
- arylo group examples include a group in which one sulfur atom is bonded to the bonding site of the aryl group.
- the arylsulfonyl group is represented by the formula: SO— at the above-mentioned aryl group binding site.
- the above-mentioned alkyl group is bonded to the above-mentioned formula: oc (
- the substituent of the substituted alkyl group for R 9 is preferably, for example, an alkanoyloxy group, an alkoxycarboxoxy group, or a cycloalkyloxycarboxyl group. And the like.
- the compound represented by the formula (I) can be obtained by esterifying or transesterifying the compound represented by the formula (III) or a salt thereof by a conventional method.
- the compound represented by the formula (III) is dissolved in alcohol (R 9 OH), and if necessary, a cosolvent is used, and in the presence of an acid, 0 to 100 ° C, preferably 20 to 50 By treating at a temperature of ° C, the compound represented by formula (I) is obtained.
- Acids particularly suitable for this reaction are inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, propion. It is possible to use organic acids such as acid, succinic acid, succinic acid, tartaric acid, fumaric acid, butyric acid, oxalic acid, malonic acid, maleic acid, lactic acid, malic acid, carbonic acid, glutamic acid, and aspartic acid. Examples include inorganic acids such as hydrochloric acid, hydrobromic acid, and sulfuric acid.
- auxiliary solvent examples include ether solvents such as jetyl ether and tetrahydrofuran, halogenated hydrocarbon solvents such as dichloromethane and chloroform, and aprotic polar solvents such as dimethylformamide and dimethyl sulfoxide.
- ether solvents such as jetyl ether and tetrahydrofuran
- halogenated hydrocarbon solvents such as dichloromethane and chloroform
- aprotic polar solvents such as dimethylformamide and dimethyl sulfoxide.
- Such salts include acid addition salts with inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfo Acid addition salts with organic acids such as acid, propionic acid, citrate, succinic acid, tartaric acid, fumaric acid, butyric acid, oxalic acid, malonic acid, maleic acid, lactic acid, malic acid, carbonic acid, glutamic acid, aspartic acid, sodium Examples thereof include inorganic base salts such as salts, potassium salts and calcium salts, and salts with organic bases such as triethylamine, piperidine, morpholine, pyridine and lysine.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid,
- the compounds of the present invention also include solvates with pharmaceutically acceptable solvents such as water and ethanol.
- the compound of the present invention obtained by the above production method can be isolated and purified by conventional separation means such as fractional recrystallization, purification using chromatography, solvent extraction, reprecipitation and the like. .
- the product obtained even in the process of! / ⁇ is in the form of an acid addition salt or free base.
- These products can be converted into the desired acid addition salt or free base form by conventional methods.
- the compound of the present invention or the raw material compound obtained by each of the above production methods is a racemate or a diastereomer mixture, it is separated into each stereoisomer according to a conventional method, for example, the method described in European Patent Application Publication No. 455006. can do.
- the compounds of the present invention can be administered orally or parenterally when they are used as medicaments. That is, it can be administered orally in dosage forms such as powders, granules, tablets, force capsules, syrups, suspensions, etc., which are usually used, or For example, solutions, emulsions and suspensions can be administered parenterally by injection. It can also be administered rectally in the form of a suppository.
- the appropriate dosage form can be produced, for example, by combining the compound of the present invention with an acceptable ordinary carrier, excipient, binder, stabilizer and diluent.
- an acceptable buffer, a solubilizing agent, and an isotonic agent can be added.
- the dose and frequency of administration vary depending on, for example, the target disease, symptoms, age, weight, and dosage form, but usually 0.1 to 2000 mg, preferably 1 to 200 mg per day for adults, once or several times (for example, 2 to (4 times) can be divided and administered.
- Table 2 shows the intravenous administration data (dose is lmgZkg) used in calculating the bioavailability.
- the compound of the present invention has excellent oral absorbability and is useful as a ⁇ 3 -adrenergic receptor stimulant.
Abstract
経口吸収性の良いβ3-アドレナリン受容体刺激作用を有する化合物として、下記式(I)で表される化合物またはその薬学的に許容される塩を提供する。
(式中、R1は、存在しないか、1つまたは複数、同一もしくは異なって存在し、ハロゲン原子等を表す。R2は水素原子等を表す。R3、R4、R5、およびR6はそれぞれ独立して、水素原子等を表す。R7は、存在しないか、1つまたは複数、同一もしくは異なって存在し、ハロゲン原子等を表す。R8は水素原子、または置換もしくは無置換のアルキル基を表す。R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のアリール基等を表す。)
Description
明 細 書
インドール類およびそれを含む医薬組成物
技術分野
[0001] 本発明は、医薬品として有用であり、経口吸収性に優れた新規なインドール類およ びその薬学的に許容される塩に関するものである。
背景技術
[0002] 交感神経の β アドレナリン受容体には β 1、 β 2および β 3として分類される 3種 類のサブタイプが存在し、それらは特定の生体内組織に分布し、それぞれが特有の 機能を有することが知られて 、る。
例えば、 j8 1 アドレナリン受容体は主に心臓に存在し、当該受容体を介する刺激 は心拍数の増加、心収縮力の増強を引き起こす。 j8 2—アドレナリン受容体は主に 血管、気管支および子宮の平滑筋に存在し、当該受容体を介する刺激はそれぞれ 血管および気管支の拡張および子宮収縮の抑制をもたらす。また、 |8 3—アドレナリ ン受容体は主に脂肪細胞、胆嚢および腸管に存在し、その他に脳、肝臓、胃、前立 腺等にも存在することが知られており、当該受容体を介する刺激により脂肪の分解亢 進作用、腸管運動の抑制作用、グルコースの取り込み促進作用、抗うつ作用等が引 さ起こされることが報告されて ヽる。
また、最近、ヒト膀胱にも主として j8 3—アドレナリン受容体が存在し、 β 3—ァドレ ナリン受容体刺激薬によりヒトの膀胱平滑筋が弛緩することが報告されている。 これまでに多くの β 1 アドレナリン受容体刺激薬および j8 2—アドレナリン受容体 刺激薬が開発されており、強心剤、気管支拡張剤および切迫流,早産防止剤等とし て医療に供されている。
一方、 β 3—アドレナリン受容体刺激薬は、肥満症、高血糖症、腸管運動亢進に起 因する過敏性大腸炎等の疾患、頻尿または尿失禁、うつ病、胆石または胆道運動亢 進に起因する疾患等の予防または治療薬としての有用性が見出されている。現在、 優れた ι8 3—アドレナリン受容体刺激薬の開発に向けて研究開発が盛んに行われ、 β 3—アドレナリン受容体刺激作用を有する化合物が知られているが(例えば、特許
文献 1参照)、 β 3 アドレナリン受容体刺激薬として上巿されるには至っていない。 それ故、優れた β 3—アドレナリン受容体刺激作用を有する新規な β 3—アドレナリ ン受容体刺激薬の開発が大いに望まれている。
より好ましくは、 β 1および Ζまたは j8 2—アドレナリン受容体刺激作用に比し、強力 な β 3 アドレナリン受容体刺激作用を有することにより、 β 1および Ζまたは β 2- アドレナリン受容体刺激作用に起因する、例えば、心悸亢進、手指の振戦等の副作 用が減弱されたより選択性の高い新規な β 3—アドレナリン受容体刺激薬の開発が 望まれている。
また、特許文献 2で開示されている化合物は、 β 1および Ζまたは β 2—アドレナリ ン受容体刺激作用が低いことにより前記心悸亢進、手指の振戦等の副作用が減弱さ れることが記載されている。
[0003] 特許文献 1 :特開平 11 255743号公報
特許文献 2:国際公開第 03Z106418号パンフレット
発明の開示
発明が解決しょうとする課題
[0004] 本発明が解決しょうとする課題は、優れた β 3 アドレナリン受容体刺激作用を有 する新規な β 3 アドレナリン受容体刺激薬、より好ましくは、 β 1および Ζまたは β 2 アドレナリン受容体刺激作用に比し、強力な j8 3—アドレナリン受容体刺激作用を 有することにより、 β 1および Ζまたは j8 2—アドレナリン受容体刺激作用に起因する 、例えば、心悸亢進、手指の振戦等の副作用が減弱された選択性の高い |8 3—アド レナリン受容体刺激薬について、経口吸収性の良い化合物を提供することにある。 課題を解決するための手段
[0005] 従来、国際公開第 03Z106418号パンフレットで開示されている化合物は、 β 1お よび Ζまたは β 2—アドレナリン受容体刺激作用が低いことにより前記心悸亢進、手 指の振戦等の副作用が減弱された、医薬として優れたィ匕合物であつたが、経口吸収 性に改善の余地があることを本発明者らは見出した。
本発明者らは、上記の課題を解決すべく鋭意研究したところ、前記式 (I)で表され るインドール誘導体およびその薬学的に許容される塩が優れた経口吸収性を有する
ことを見出し、本発明を成すに至った。
即ち、本発明は以下のものに関する。
[0006] [1] 式(I) :
[0007] [化 1]
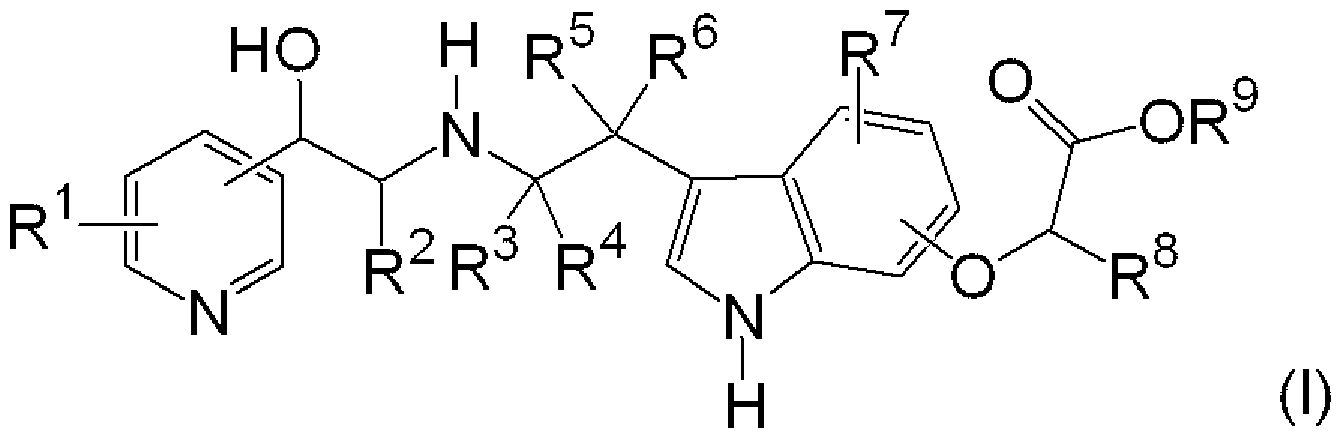
(式中、 R1は、存在しないか、 1つまたは複数、同一もしくは異なって存在し、置換もし くは無置換のアルキル基、置換もしくは無置換のアルコキシ基、ハロゲン原子、水酸 基、またはアミノ基を表す。
R2は水素原子、または置換もしくは無置換のアルキル基を表す。
R3、 R4、 R5、および R6はそれぞれ独立して、水素原子、または置換もしくは無置換 のアルキル基を表す。
R7は、存在しないか、 1つまたは複数、同一もしくは異なって存在し置換もしくは無 置換のアルキル基、置換もしくは無置換のアルコキシ基、ハロゲン原子、水酸基、ま たはアミノ基を表す。
R8は水素原子、または置換もしくは無置換のアルキル基を表す。
R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のァルケ-ル基、置 換もしくは無置換のアルキ-ル基、置換もしくは無置換のシクロアルキル基、置換もし くは無置換のァラルキル基、または置換もしくは無置換のァリール基を表す。)で表さ れる化合物またはその薬学的に許容される塩。
[2] 式 (Π) :
[0008] [化 2]

(式中、 R2は水素原子、または置換もしくは無置換のアルキル基を表す。
R3、 R4、 R5、および R6はそれぞれ独立して、水素原子、または置換もしくは無置換 のアルキル基を表す。
R8は水素原子、または置換もしくは無置換のアルキル基を表す。
R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のァルケ-ル基、置 換もしくは無置換のアルキ-ル基、置換もしくは無置換のシクロアルキル基、置換もし くは無置換のァラルキル基、または置換もしくは無置換のァリール基を表す。)で表さ れる化合物またはその薬学的に許容される塩。
[3] R2、 R3、 R4、 R5、および R6が水素原子である、 [2]記載の化合物またはその薬 学的に許容される塩。
[4] 式 (Ila) :
[化 3]

(式中、 R8は水素原子、または置換もしくは無置換のアルキル基を表す。
R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のァルケ-ル基、置 換もしくは無置換のアルキ-ル基、置換もしくは無置換のシクロアルキル基、置換もし くは無置換のァラルキル基、または置換もしくは無置換のァリール基を表す。)で表さ れる化合物またはその薬学的に許容される塩。
[5] R9が、アルキル基、ァルケ-ル基、アルキ-ル基、ァラルキル基、ァリール基、
または置換アルキル基 (該置換基は、ハロゲン原子、水酸基、アルカノィル基、アル コキシ基、アルコキシカルボ-ル基、シァノ基、アルキルチオ基、ァリールチオ基、ァ ルキルスルホ -ル基、ァリールスルホ-ル基、アルカノィルォキシ基、アルコキシカル ボニルォキシ基、シクロアルキルォキシカルボ-ルォキシ基およびカルボキシ基から 選ばれ、 1または複数、同一または異なって存在する。)である、 [4]記載の化合物ま たはその薬学的に許容される塩。
[6] (2S)—2— ( (3— (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチ ル)ァミノ)ェチル) 1H インドールー 7 ィル)ォキシ)プロパン酸ェチルである、 [ 1]記載の化合物またはその薬学的に許容される塩。
[7] (2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチ ル)ァミノ)ェチル) 1H—インドールー 7 ィル)ォキシ)ー3 メチルブタン酸ェチ ルである、 [1]記載の化合物またはその薬学的に許容される塩。
[8] (2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチ ル)ァミノ)ェチル) 1H—インドールー 7 ィル)ォキシ)ペンタン酸ェチルである、 [ 1]記載の化合物またはその薬学的に許容される塩。
[9] [ 1]〜 [8]の 、ずれかに記載の化合物またはその薬学的に許容される塩を有 効成分として含有する医薬組成物。
[10] [ 1 ]〜 [8]の 、ずれかに記載の化合物またはその薬学的に許容される塩を有 効成分として含有する、肥満症、高血糖症、高インスリン血症、糖尿病、高中性脂肪 血症、頻尿、尿失禁、過活動膀胱症候群、うつ病、腸管運動亢進に起因する疾患、 胆石、または胆管運動亢進に起因する疾患の治療剤。
[11] 治療が必要な患者に、 [1]〜[8]のいずれかに記載の化合物またはその薬学 的に許容される塩の有効量を投与することからなる、肥満症、高血糖症、高インスリン 血症、糖尿病、高中性脂肪血症、頻尿、尿失禁、過活動膀胱症候群、うつ病、腸管 運動亢進に起因する疾患、胆石、または胆管運動亢進に起因する疾患の治療方法
[12] [ 1 ]〜 [8]の 、ずれかに記載の化合物またはその薬学的に許容される塩の、 肥満症、高血糖症、高インスリン血症、糖尿病、高中性脂肪血症、頻尿、尿失禁、過
活動膀胱症候群、うつ病、腸管運動亢進に起因する疾患、胆石、または胆管運動亢 進に起因する疾患の治療剤の製造のための使用。
[0010] 式(III) :
[0011] [化 4]

(式中、
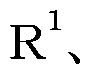 R2、 R3、 R4、 R5、 R6、 R7および R8は上記と同じ意味を表す)で表される力 ルボン酸誘導体は経口吸収性が十分ではな力つたが、式 (III)で表される化合物の カルボキシ基をエステルイ匕することにより得られる式 (I):
R2、 R3、 R4、 R5、 R6、 R7および R8は上記と同じ意味を表す)で表される力 ルボン酸誘導体は経口吸収性が十分ではな力つたが、式 (III)で表される化合物の カルボキシ基をエステルイ匕することにより得られる式 (I):
[0012]

[0013] 以下、式 (I)で表される化合物またはその薬学的に許容される塩を、必要に応じ「本 発明化合物」と総称する。
発明の効果
[0014] 本発明化合物は、経口吸収性に優れた β 3—アドレナリン受容体刺激薬として、例 えば肥満症、高血糖症、高インスリン血症、糖尿病、高中性脂肪血症、頻尿、尿失禁 、過活動膀胱症候群、うつ病、腸管運動亢進に起因する過敏性大腸炎等の疾患、 胆石、または胆管運動亢進に起因する胃炎、胃'十二指腸潰瘍、腸炎、過敏性大腸 症候群、胆のう '胆道疾患、または尿路結石症等の疾患の治療等に有用である。 図面の簡単な説明
[0015] [図 1]本発明化合物のラット経口吸収性評価試験データを示す。 ♦は化合物 1を表
す。 Xは化合物 2を表す。 國は化合物 3を表す。 ▲は化合物 4を表す。 參はィ匕 合物 5を表す。
発明を実施するための最良の形態
[0016] 本発明における各種の用語を詳細に説明すると次の通りである。なお、特に指示の ない限り、各々の基の説明は他の基の一部である場合も含む。
[0017] ハロゲン原子としては例えばフッ素、塩素、臭素、ヨウ素等が挙げられる。
アルキル基としては、例えば直鎖または分枝した炭素原子数 1〜6個のアルキル基 等が挙げられ、具体的には例えばメチル、ェチル、プロピル、イソプロピル、ブチル、 2 ブチル、 2 メチルプロピル、 1, 1ージメチルェチル、ペンチル、 3 ペンチル、 3 ーメチルブチル、へキシル、 3—へキシル、 4ーメチルペンチル等が挙げられる。好ま L 、アルキル基としては、例えば直鎖または分枝した炭素原子数 1〜4個のアルキル 基等が挙げられる。
アルコキシ基としては、上記アルキル基の結合手に酸素原子が一つ結合した基が 挙げられる。
[0018] ァルケ-ル基としては、例えばビュル、ァリル、プロべ-ル、 2—プロべ-ル、ブテ- ル、ペンテニル、へキセニル等の直鎖または分枝した炭素原子数 6以下のァルケ- ル基等が挙げられる。
アルキ-ル基としては、例えばェチュル、プロパルギル、 1ーブチュル、 2—プチ- ル、 2 ペンチ-ル、 3 ペンチ-ル、 2 へキシュル等の直鎖または分枝した炭素 原子数 6以下のアルキニル基等が挙げられる。
シクロアルキル基としては、例えばシクロプロピル、シクロブチル、シクロペンチル、 シクロへキシル、シクロへプチルなどの 3〜8員環のシクロアルキル基等が挙げられる アルカノィル基としては、例えばホルミル、ァセチルまたはプロパノィル等の炭素原 子数 1〜6のアルカノィル基等が挙げられる。
ァラルキル基のァリール部分としては、例えばフエ-ル、 1 または 2—ナフチル等 の炭素原子数 10以下のァリール基等が、アルキル部分としては、例えばメチル、ェ チル、プロピル、ブチル等の炭素原子数 6以下のアルキル基等が挙げられる。代表
的なァラルキル基としては、例えばべンジル基、 1 または 2—フエネチル基等が挙 げられる。
[0019] 置換アルキル基、置換アルコキシ基、置換アルケニル基、置換アルキニル基、また は置換シクロアルキル基の置換基は一個または同一もしくは異なって複数個あっても よぐ置換基としては、例えばハロゲン原子、シァノ基、フエノキシ基、ベンジルォキシ 基、水酸基、アルコキシ基、アルカノィルォキシ基、アミノ基、モノアルキルアミノ基、 ジアルキルアミノ基、力ルバモイル基、アルキルアミノカルボ-ル基、ジアルキルアミノ カルボ-ル基、カルボキシ基、アルコキシカルボ-ル基、アルキルチオ基、アルキル スルフィエル基、アルキルスルホ-ル基、アルカノィルァミノ基、アルキルスルホ-ル アミノ基、フタルイミド基、アルカノィル基、ァリールチオ基、ァリールスルホ-ル基、ァ ルコキシカルボ-ルォキシ基、シクロアルキルォキシカルボ-ルォキシ基、ァリール 基 (ハロゲン原子、アルキル基、またはアルコキシ基等によって、 1または複数、同一 または異なって置換されて!ヽてもよ ヽ)またはへテロアリール基 (ノヽロゲン原子、アル キル基、またはアルコキシ基等によって、 1または複数、同一または異なって置換され て!、てもよ 、)等が挙げられる。
[0020] ァリール基としては、例えばフエニル、 1 または 2—ナフチル等の炭素原子数 10 以下のァリール基等が挙げられる。
ヘテロァリール基としては、例えば窒素原子、酸素原子、硫黄原子からなる群から 選ばれる 1〜4個のへテロ原子を含有する、 5〜10員の単環または二環のへテロァリ ール基等が挙げられる。具体的には例えば、ピリジル、フリル、チェニル、キノリル、 ベンズイミダゾリル、ベンズチアゾリル、またはインドリル等が挙げられる。
[0021] 置換ァリール基および置換ァラルキル基における置換基としては、 1または複数、 同一または異なって、例えばノヽロゲン原子、ハロゲン原子で置換されてもよいアルキ ル基、ハロゲン原子で置換されてもよいアルコキシ基、または-トロ基等が挙げられる 。好まし 、置換基としてはメトキシ等のアルコキシ基が挙げられる。
[0022] アルキルチオ基としては、上記アルキル基の結合部位に硫黄原子が 1つ結合した 基が挙げられる。
アルキルスルフィエル基としては、上記アルキル基の結合部位に式: S (O) で
表される基が 1つ結合した基が挙げられる。
アルキルスルホ-ル基としては、上記アルキル基の結合部位に式: so—で表さ
2 れる基が 1つ結合した基が挙げられる。
アルコキシカルボ-ル基としては、上記アルキル基の結合部位に式: oc(=o) 一で表される基の酸素原子側が結合した基が挙げられる。
アルカノィル基としては、上記アルキル基の結合部位に式: c(=o)—で表され る基が 1つ結合した基が挙げられる。
アルカノィルァミノ基としては、上記アルキル基の結合部位に式: NHC ( = 0)— で表される基の炭素原子側が結合し、さらに窒素原子上に C1〜C8アルキル基等が 置換して 、てもよ 、基が挙げられる。
アルキルアミノカルボ-ル基としては、上記アルキル基の結合部位に式: c( = o ) NH で表される基の窒素原子側が結合し、さらに窒素原子上に C1〜C8アルキル 基等が置換して 、てもよ 、基が挙げられる。
モノもしくはジアルキルアミノ基としては、ァミノ基の水素が一つまたは両方が独立 に、置換もしくは無置換のアルキル基で置換された基が挙げられる。
アルキルスルホ -ルァミノ基は、上記アルキル基の結合部位に式: NHSO—で
2 表される基の硫黄原子側が結合し、さらに窒素原子上に C1〜C8アルキル基等が置 換して 、てもよ 、基が挙げられる。
ァリールチオ基としては、上記ァリール基の結合部位に硫黄原子が 1つ結合した基 が挙げられる。
ァリールスルホ-ル基としては、上記ァリール基の結合部位に式: SO—で表さ
2 れる基が 1つ結合した基が挙げられる。
アルカノィルォキシ基としては、上記アルキル基の結合部位に式: oc(=o)— で表される基の炭素原子側が結合した基が挙げられる。
アルコキシカルボ-ルォキシ基としては、上記アルキル基の結合部位に式: oc(
= O) 0—で表される基が結合した基が挙げられる。
R9における置換アルキル基の置換基としては、好ましくは、例えば、アルカノィルォ キシ基、アルコキシカルボ-ルォキシ基またはシクロアルキルォキシカルボ-ルォキ
シ基等が挙げられる。
[0024] 次に本発明の合成方法および合成中間体について詳細に説明する。
(A)式 (I)で表される化合物の合成方法
[0025] [化 6]

(式中、 R\ R2、 R3、 R4、 R5、 R6、 R R8および R9は上記と同じ意味を表す) 式 (III)で表される化合物は、文献に開示されている方法で合成できる。(例えば、 WO03Z106418等参照。;)
式 (I)で表される化合物は、式 (III)で表される化合物またはその塩を、常法によりェ ステル化、またはエステル交換することにより得ることができる。
例えば、式 (III)で表される化合物をアルコール (R9OH)に溶解し、必要に応じて補 助溶媒を使用し、酸の存在下で、 0〜100°C、好ましくは 20〜50°Cの温度で処理す ることで式 (I)で表される化合物が得られる。
この反応にとくに適する酸は、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン 酸等の無機酸、ギ酸、酢酸、メタンスルホン酸、ベンゼンスルホン酸、 p—トルエンス ルホン酸、プロピオン酸、クェン酸、コハク酸、酒石酸、フマル酸、酪酸、シユウ酸、マ ロン酸、マレイン酸、乳酸、リンゴ酸、炭酸、グルタミン酸、ァスパラギン酸等の有機酸 などを用いることが可能であり、好ましくは塩酸、臭化水素酸、硫酸などの無機酸が 挙げられる。
補助溶媒としては、例えばジェチルエーテル、テトラヒドロフランなどのエーテル系 溶媒、ジクロロメタン、クロ口ホルムなどのハロゲン化炭化水素系溶媒、ジメチルホル ムアミド、ジメチルスルホキシドなどの非プロトン性極性溶媒等が挙げられる。
[0026] 本発明化合物は、常法に従いその薬学的に許容される塩とすることができる。この ような塩としては塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等の無機酸と の酸付加塩、ギ酸、酢酸、メタンスルホン酸、ベンゼンスルホン酸、 p—トルエンスルホ ン酸、プロピオン酸、クェン酸、コハク酸、酒石酸、フマル酸、酪酸、シユウ酸、マロン 酸、マレイン酸、乳酸、リンゴ酸、炭酸、グルタミン酸、ァスパラギン酸等の有機酸との 酸付加塩、ナトリウム塩、カリウム塩、カルシウム塩等の無機塩基塩、トリェチルァミン 、ピぺリジン、モルホリン、ピリジン、リジン等の有機塩基との塩を挙げることができる。
[0027] 本発明化合物には水やエタノール等の薬学的に許容される溶媒との溶媒和物も含 まれる。
[0028] 前記製造方法により得られる本発明化合物は、慣用の分離手段である分別再結晶 法、クロマトグラフィーを用いた精製方法、溶媒抽出法、再沈殿等により単離精製す ることがでさる。
また!/ヽずれの製法にお!ヽても得られる生成物は、反応条件により酸付加塩または 遊離塩基の形をとる。これらの生成物は常法により所望の酸付加塩または遊離塩基 の形に変換することができる。
前記各製法によって得られる本発明の化合物または原料化合物がラセミ体または ジァステレオマー混合物である場合には、常法、例えば欧州特許出願公開第 4550 06号明細書に記載の方法に従って各立体異性体に分離することができる。
なお、以上説明した反応において、特定の保護基を例示した場合に限らず、各出 発化合物がカルボキシ基や水酸基、ァミノ基のような、反応に活性な基を有する場合 には、これらの基を予め適当な保護基で保護しておき、本反応を実施した後に保護 基を除去することにより、 目的化合物を製造することができる。保護、脱保護の方法と しては各々の保護基に応じ、文献(例えば、 Green, T. W.および Wuts, P. G. M. , Protective Groups m urganic Syntnesis, John Wiley & Sons, Inc. (1999)等)記載の方法により行うことができる。
[0029] 本発明化合物は、これらを医薬として用いるにあたり経口的または非経口的に投与 することができる。すなわち通常用いられる投与形態、例えば粉末、顆粒、錠剤、力 プセル剤、シロップ剤、懸濁液等の剤型で経口的に投与することができ、あるいは、
例えば、その溶液、乳剤、懸濁液の剤型にしたものを注射の型で非経口投与するこ とができる。坐剤の型で直腸投与することもできる。前記の適当な投与剤型は、例え ば、許容される通常の担体、賦型剤、結合剤、安定剤、希釈剤に本発明化合物を配 合すること〖こより製造することができる。注射剤型で用いる場合には、例えば、許容さ れる緩衝剤、溶解補助剤、等張剤を添加することもできる。投与量および投与回数は 、例えば、対象疾患、症状、年齢、体重、投与形態によって異なるが、通常は成人に 対し 1日あたり 0.1〜2000mg好ましくは l〜200mgを 1回または数回(例えば 2〜4回) に分けて投与することができる。
[0030] 以下に、参考例、実施例および試験例により、本発明をさらに詳細に説明するが、 本発明はこれに限定されるものではない。
実施例 1
[0031] 実施例 1
(2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)アミ ノ)ェチル) 1H インドールー 7 ィル)ォキシ)プロパン酸ェチル (化合物 2)
(2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)ァ ミノ)ェチル) 1H—インドールー 7 ィル)ォキシ)プロパン酸(ィ匕合物 1) (3. 273g , 8. 86mmol)のエタノール(65mL)溶液に、 4規定塩酸ジォキサン溶液(26mL)を 加えて室温で 2時間攪拌した。反応液の溶媒を留去した後、残渣に飽和重曹水とク ロロホルムを加えて分配抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウ ムで乾燥したのち、セライトを用いて濾過した。濾液の溶媒を留去して、アモルファス 状の表題化合物(3. 47g,収率 99%)を得た。
'H-NMRCCDCl ) δ :1.26(3H,t,J=7.1Hz),1.69(3H,d,J=6.8Hz),2.66(lH,dd,J=12.3,9.4H
3
z),2.91-3.06(5H,m),4.22(2H,q,J=7.1Hz),4.67(lH,dd,J=9.3,3.5Hz), 4.88(lH,q,J=6.8H z),6.58(lH,d,J=7.7Hz),6.98(lH,dd,J=7.9,7.7Hz),7.03(lH,d,J=2.1Hz),7.24-7.26(2H, m),7.68(lH,ddd,J=7.8,2.0,1.6Hz),8.51(lH,dd,J=4.8,1.6Hz),8.56(lH,d,J=2.0Hz),8.58 (lH'brs).
[0032] 実施例 2
(2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)アミ
ノ)ェチル)—1H—インドール— 7—ィル)ォキシ)—3—メチルブタン酸ェチル (ィ匕 合物 3)
(2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)ァ ミノ)ェチル) 1H—インドールー 7 ィル)ォキシ) 3 メチルブタン酸(ィ匕合物 5) (lOOmg, 0. 252mmol)のエタノール(5mL)溶液に、 4規定塩酸ジォキサン溶液( 2mL)を加えて 40°Cで 3. 5時間攪拌した。反応液の溶媒を留去した後、残渣に飽和 重曹水とクロ口ホルムを加えて分配抽出した。有機層を無水硫酸ナトリウムで乾燥、ろ 過したのち、溶媒を留去して、残渣をシリカゲルカラムクロマトグラフィー(エタノール Zクロ口ホルム = 1Z5〜5Z1)にて精製してアモルファス状の表題化合物(101 mg,収率 94%)を得た。
'H-NMRCCDCl ) δ :1.13(3H,d,J=6.9Hz),1.13(3H,d,J=6.8Hz),1.23(3H,t,J=7.1Hz),2.3
3
3-2.37(lH,m),2.66(lH,dd,J=12.3,9.4Hz),2.88-3.03(5H,m),4.19(2H,q,J=7.1Hz),4.52( lH,d,J=5.5Hz),4.68(lH,dd,J=9.3,3.5Hz),6.57(lH,d,J=7.7Hz),6.95-6.99(2H,m),7.22- 7.25(2H,m),7.67(lH,ddd,J=7.8,2.1,1.8Hz),8.48(lH,dd,J=4.8,1.8Hz),8.55(lH,d,J=2.1 Hz),8.69(lH,brs).
実施例 3
(2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)アミ ノ)ェチル) 1H インドールー 7 ィル)ォキシ)ペンタン酸ェチル (化合物 4) (2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)アミ ノ)ェチル) 1H—インドールー 7 ィル)ォキシ)ペンタン酸(ィ匕合物 6) (205mg, 0 . 516mmol)のエタノール(5mL)溶液に、 1規定塩酸ジェチルエーテル溶液(lmL )をカ卩えて 50°Cで 10時間攪拌した。反応液を飽和重曹水に注ぎ、クロ口ホルムをカロ えて分配抽出した。有機層を無水硫酸マグネシウムで乾燥、ろ過したのち、溶媒を留 去して、残渣をシリカゲルカラムクロマトグラフィー(エタノール Zクロ口ホルム = 1 Z5〜: LZ3)にて精製して表題ィ匕合物(166mg,収率 75%)を得た。
1H-NMR(CDC1 ) δ :1.01(3H, t, J= 7.4 Hz), 1.24 (3H, t, J= 7.1 Hz), 1.54—1.68 (2H,
3
m), 1.93-2.04 (2H, m), 2.66 (1H, dd, J= 12.2, 9.4 Hz), 2.92-3.07 (5H, m), 4.20 (2h, q, J= 7.1 Hz), 4.67 (1H, dd, J= 9.4, 3.6 Hz), 4.75 (1H, dd, J= 8.0, 4.8 Hz), 6.56 (1
H, d, J= 7.7 Hz), 6.98 (1H, dd, J= 8.0, 7.7 Hz), 7.03 (lh, d, J= 2.1 Hz), 7.23-7.26 ( 2H, m), 7.69 (1H, ddd, J= 7.9, 2.1, 1.6 Hz), 8.51 (1H, dd, J= 4.8, 1.6 Hz), 8.53 (1H , br s), 8.57 (1H, d, J= 2.1 Hz).
[0034] 試験例 1
化合物 1〜5を使用し、 Crj:CD(SD)系雄性ラット(日本チヤ一ルス'リバ一) 7週齢に 非絶食下、それぞれ 10mg/kgで経口投与した。投与後 15、 30分、 1、 2、 4、 6、 24時間 後にエーテル麻酔下採血し、室温で約 30分放置後、遠心分離 (3,000 r.p.m., 10分) して血清を得た。得られた血清は、分析まで- 20°Cで保存した。血清 50 μ Lに内部標 準 (I.S.)溶液 100 Lを加えて攪拌後、遠心分離(10,000 r.p.m., 2分)した。上清約 10 0 μ Lに水 100 μ Lをカ卩えて攪拌したものをセントリカット(倉敷紡績)で遠心濾過(10,00 0 r.p.m., 1分)し、濾液 10 /z Lを LC-MS/MSで分析した。その結果、化合物 2を経口 投与したときの生物学的利用率 (BA)は 29.4%、化合物 3を経口投与したときの BAは 1
I.8%、化合物 4を経口投与したときの BAは 11.0%となり、カルボン酸体である化合物 1 を経口投与した場合 (生物学的利用率: 1.2%)および化合物 5を経口投与した場合 ( 生物学的利用率: 0.1%)と比べて、優れた経口吸収性を持つことが確認された。表 1 および図 1に血清中薬物濃度推移データを示した。なお、試験化合物 2を投与した いずれのラットの血液からも当該試験化合物は一切認められずィヒ合物 1のみが検出 され、試験化合物 3を投与したいずれのラットの血液からも当該試験化合物は一切認 められず化合物 5のみが検出され、試験化合物 4を投与したいずれのラットの血液か らも当該試験化合物は一切認められずィ匕合物 6のみが検出された。
BAは静脈内投与のデータ力 算出した (下記表 2参照)。
[0035] [表 1]
ラット経ロヨ 与後の血淸中薬物濃度 〔ii g /mL)
経口投与後 化合物 1 化合物 2 化合物 3 化合物 4 化合物 5 時間 (分)
1 5 0. 0 1 0. 6 8 5 0. 98 0. 3 8 3 0. 0 0 7
30 0. 0 1 6 0. 5 6 5 0. 9 5 2 0. 1 8 7 0. 0 0 6
6 0 0. 0 1 7 0. 2 5 3 0. 40 3 0. 04 1 0. 0 0 5
1 2 0 0. 0 1 5 0. 1 0 5 0. 1 5 5 0. 0 2 0. 0 0 6
24 0 0. 0 1 5 0. 1 2 4 0. 1 3 8 0. 0 5 0. 0 0 1
3 6 0 0. 0 1 3 0. 08 9 0. 1 0 8 0. 0 3 5 0. 0 0 2
1 44 0 N. D . 0. 0 2 0. 0 2 1 0. 0 1 4 N. D.
N. D. :検出限界以下
[0036] 生物学的利用率算出の際に用いた静脈内投与データ (投与量は lmgZkg)を表 2 に示す。
[0037] [表 2]

N. D. :検出限界以下
産業上の利用可能性
[0038] 本発明の化合物は経口吸収性に優れており、 β 3—アドレナリン受容体刺激薬とし て有用である。
Claims
請求の範囲
[1] 式 (I)
[化 1]

(式中、 R1は、存在しないか、 1つまたは複数、同一もしくは異なって存在し、置換もし くは無置換のアルキル基、置換もしくは無置換のアルコキシ基、ハロゲン原子、水酸 基、またはアミノ基を表す。
R2は水素原子、または置換もしくは無置換のアルキル基を表す。
R3、 R4、 R5、および R6はそれぞれ独立して、水素原子、または置換もしくは無置換 のアルキル基を表す。
R7は、存在しないか、 1つまたは複数、同一もしくは異なって存在し置換もしくは無 置換のアルキル基、置換もしくは無置換のアルコキシ基、ハロゲン原子、水酸基、ま たはアミノ基を表す。
R8は水素原子、または置換もしくは無置換のアルキル基を表す。
R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のァルケ-ル基、置 換もしくは無置換のアルキ-ル基、置換もしくは無置換のシクロアルキル基、置換もし くは無置換のァラルキル基、または置換もしくは無置換のァリール基を表す。)で表さ れる化合物またはその薬学的に許容される塩。
[2] 式 (Π) :
[化 2]
HO H R5 R6
R2 R3 R4
(式中、 R2は水素原子、または置換もしくは無置換のアルキル基を表す。
R3、 R4、 R5、および R6はそれぞれ独立して、水素原子、または置換もしくは無置換 のアルキル基を表す。
R8は水素原子、または置換もしくは無置換のアルキル基を表す。
R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のァルケ-ル基、置 換もしくは無置換のアルキ-ル基、置換もしくは無置換のシクロアルキル基、置換もし くは無置換のァラルキル基、または置換もしくは無置換のァリール基を表す。)で表さ れる化合物またはその薬学的に許容される塩。
[3] R2、 R3、 R4、 R5、および R6が水素原子である、請求項 2記載の化合物またはその薬 学的に許容される塩。
[4] 式 (Ila) :
[化 3]

(式中、 R8は水素原子、または置換もしくは無置換のアルキル基を表す。
R9は、置換もしくは無置換のアルキル基、置換もしくは無置換のァルケ-ル基、置 換もしくは無置換のアルキ-ル基、置換もしくは無置換のシクロアルキル基、置換もし くは無置換のァラルキル基、または置換もしくは無置換のァリール基を表す。)で表さ れる化合物またはその薬学的に許容される塩。
R9が、アルキル基、ァルケ-ル基、アルキニル基、ァラルキル基、ァリール基、または 置換アルキル基 (該置換基は、ハロゲン原子、水酸基、アルカノィル基、アルコキシ 基、アルコキシカルボ-ル基、シァノ基、アルキルチオ基、ァリールチオ基、アルキル スルホ-ル基、ァリールスルホ-ル基、アルカノィルォキシ基、アルコキシカルボ-ル ォキシ基、シクロアルキルォキシカルボニルォキシ基およびカルボキシ基から選ばれ 、 1または複数、同一または異なって存在する。)である、請求項 4記載の化合物また
はその薬学的に許容される塩。
[6] (2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ一 2 ピリジン一 3—ィルェチル)アミ ノ)ェチル) 1H—インドールー 7 ィル)ォキシ)プロパン酸ェチルである、請求項 1 記載の化合物またはその薬学的に許容される塩。
[7] (2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ— 2 ピリジン— 3—ィルェチル)アミ ノ)ェチル) 1H インドールー 7 ィル)ォキシ) 3 メチルブタン酸ェチルである
、請求項 1記載の化合物またはその薬学的に許容される塩。
[8] (2S) - 2- ( (3- (2- ( ( (2R)—2 ヒドロキシ— 2 ピリジン— 3—ィルェチル)アミ ノ)ェチル) 1H—インドールー 7 ィル)ォキシ)ペンタン酸ェチルである、請求項 1 記載の化合物またはその薬学的に許容される塩。
[9] 請求項 1〜8のいずれか一項に記載の化合物またはその薬学的に許容される塩を有 効成分として含有する医薬組成物。
[10] 請求項 1〜8のいずれか一項に記載の化合物またはその薬学的に許容される塩を有 効成分として含有する、肥満症、高血糖症、高インスリン血症、糖尿病、高中性脂肪 血症、頻尿、尿失禁、過活動膀胱症候群、うつ病、腸管運動亢進に起因する疾患、 胆石、または胆管運動亢進に起因する疾患の治療剤。
[11] 治療が必要な患者に、請求項 1〜8のいずれか一項に記載の化合物またはその薬 学的に許容される塩の有効量を投与することからなる、肥満症、高血糖症、高インス リン血症、糖尿病、高中性脂肪血症、頻尿、尿失禁、過活動膀胱症候群、うつ病、腸 管運動亢進に起因する疾患、胆石、または胆管運動亢進に起因する疾患の治療方 法。
[12] 請求項 1〜8のいずれか一項に記載の化合物またはその薬学的に許容される塩の、 肥満症、高血糖症、高インスリン血症、糖尿病、高中性脂肪血症、頻尿、尿失禁、過 活動膀胱症候群、うつ病、腸管運動亢進に起因する疾患、胆石、または胆管運動亢 進に起因する疾患の治療剤の製造のための使用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005014983A JP2008100916A (ja) | 2005-01-24 | 2005-01-24 | インドール類およびそれを含む医薬組成物 |
| JP2005-014983 | 2005-01-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006078006A1 true WO2006078006A1 (ja) | 2006-07-27 |
Family
ID=36692378
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/300925 WO2006078006A1 (ja) | 2005-01-24 | 2006-01-23 | インドール類およびそれを含む医薬組成物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2008100916A (ja) |
| WO (1) | WO2006078006A1 (ja) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015108039A1 (ja) * | 2014-01-14 | 2015-07-23 | アステラス製薬株式会社 | インドール化合物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994029290A1 (en) * | 1993-06-14 | 1994-12-22 | Pfizer Inc. | Secondary amines as antidiabetic and antiobesity agents |
| JPH11255743A (ja) * | 1998-03-06 | 1999-09-21 | Dainippon Pharmaceut Co Ltd | 光学活性のインドール誘導体の製造方法及びその製造中間体 |
| WO2003044017A1 (en) * | 2001-11-20 | 2003-05-30 | Eli Lilly And Company | Beta 3 adrenergic agonists |
| WO2003106418A1 (ja) * | 2002-06-01 | 2003-12-24 | 住友製薬株式会社 | インドール、インダゾール、およびベンズアゾール類 |
| JP2005194266A (ja) * | 2003-12-11 | 2005-07-21 | Sumitomo Pharmaceut Co Ltd | インドール類の合成方法および合成中間体 |
-
2005
- 2005-01-24 JP JP2005014983A patent/JP2008100916A/ja active Pending
-
2006
- 2006-01-23 WO PCT/JP2006/300925 patent/WO2006078006A1/ja not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994029290A1 (en) * | 1993-06-14 | 1994-12-22 | Pfizer Inc. | Secondary amines as antidiabetic and antiobesity agents |
| JPH11255743A (ja) * | 1998-03-06 | 1999-09-21 | Dainippon Pharmaceut Co Ltd | 光学活性のインドール誘導体の製造方法及びその製造中間体 |
| WO2003044017A1 (en) * | 2001-11-20 | 2003-05-30 | Eli Lilly And Company | Beta 3 adrenergic agonists |
| WO2003106418A1 (ja) * | 2002-06-01 | 2003-12-24 | 住友製薬株式会社 | インドール、インダゾール、およびベンズアゾール類 |
| JP2005194266A (ja) * | 2003-12-11 | 2005-07-21 | Sumitomo Pharmaceut Co Ltd | インドール類の合成方法および合成中間体 |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015108039A1 (ja) * | 2014-01-14 | 2015-07-23 | アステラス製薬株式会社 | インドール化合物 |
| CN105916840A (zh) * | 2014-01-14 | 2016-08-31 | 安斯泰来制药株式会社 | 吲哚化合物 |
| JPWO2015108039A1 (ja) * | 2014-01-14 | 2017-03-23 | アステラス製薬株式会社 | インドール化合物 |
| US9637451B2 (en) | 2014-01-14 | 2017-05-02 | Astellas Pharma Inc. | Indole compound |
| CN105916840B (zh) * | 2014-01-14 | 2019-01-08 | 安斯泰来制药株式会社 | 吲哚化合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2008100916A (ja) | 2008-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006078006A1 (ja) | インドール類およびそれを含む医薬組成物 | |
| US6599929B2 (en) | 1H-indole derivatives as a highly selective cyclooxygenase-2 inhibitor | |
| AU2009331179B2 (en) | Novel bicyclic heterocyclic compound | |
| EP2393809B1 (fr) | Derives d'azaspiranyl-alkylcarbamates d'heterocycles a 5 chainons, leur preparation et leur application en therapeutique | |
| JP3643107B2 (ja) | (s)−4−アミノ−5−クロロ−2−メトキシ−n−[1−[1−(2−テトラヒドロフリルカルボニル)−4−ピペリジニルメチル]−4−ピペリジニル]ベンズアミド、その製造方法、それを含有する医薬組成物及び該化合物の中間体 | |
| US20040266755A1 (en) | Prodrugs of 1-(1-hydroxy-5-isoquinolinesulfonyl) homopiperazine | |
| EA007008B1 (ru) | Триамидзамещённые индолы, бензофураны и бензотиофены в качестве ингибиторов микросомального белка, переносящего триглицериды, (мтр) и/или ингибиторов секреции аполипопротеина в (аро в) | |
| WO2005075426A1 (en) | Novel dipeptidyl peptidase iv inhibitors; processes for their preparation and compositions thereof | |
| WO2008069242A1 (ja) | 新規2環性複素環化合物 | |
| US20220073516A1 (en) | Crystalline spirocyclic compound, a dosage form containing, a method for using in treatment of disease, and a method for recrystallizing | |
| EP2429998B1 (fr) | Dérivés de cyclopenta[c]pyrrolylalkylcarbamates d'hétérocycles à 5 chaînons, leur préparation et leur application en thérapeutique | |
| CA2731789A1 (fr) | Derives de carbamates d'alkylthiazoles, leur preparation et leur application en therapeutique | |
| JP2006111553A (ja) | スルホニルオキシインドール誘導体及びそれを含有する医薬組成物 | |
| JPH08165276A (ja) | 2−アルキルアミノ−1−フェニルエタノール誘導体 | |
| WO2006121104A1 (ja) | ピペリジン環を有するインドール誘導体の結晶およびその製法 | |
| JP3162523B2 (ja) | ピペリジルメチル−置換クロマン誘導体 | |
| JPH07267954A (ja) | 新規の3−フェニルスルホニル−3,7−ジアザビシクロ[3,3,1ノナン−化合物、その製法及び抗不整脈剤 | |
| JPH10152460A (ja) | 2−アミノ−1−(4−ヒドロキシ−2−メチルフェニル)プロパノール誘導体 | |
| JP2005082508A (ja) | 2−アルコキシ−6−アミノ−5−ハロゲノ−n−(1−置換−4−ピペリジニル)ピリジン−3−カルボキサミド誘導体およびそれを含有する医薬組成物 | |
| WO2010041568A1 (ja) | インダゾール誘導体 | |
| US6696574B2 (en) | Processes and intermediates for preparing glycogen phosphorylase inhibitors | |
| WO1999031097A1 (fr) | Derives d'imidazole en tant qu'antagonistes des recepteurs muscariniques m3 | |
| TW200418850A (en) | Acyl derivatives of 5-(2-(4-(1,2 benzisothiazole-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2H-indol-2-one having neuroleptic activity | |
| CN116082259B (zh) | 氨基甲酸酯基或氨甲酰基取代的5-ht2b拮抗剂 | |
| JP2002501064A (ja) | 複素芳香環縮合シクロペンテノピリジン誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06712141 Country of ref document: EP Kind code of ref document: A1 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 6712141 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: JP |