Nouveaux dérivés d'indolizine 1,2,3,6,7,8 substituée, inhibiteurs des FGFs, leur procédé de préparation et les compositions pharmaceutiques les contenant.
La présente invention a pour objet de nouveaux dérivés d'indolizine 1,2,3,6,7,8 substituée, qui sont des inhibiteurs des FGFs (Fibroblast Growth Factor), leur procédé de préparation et les compositions pharmaceutiques les contenant. Les FGFs sont une famille de polypeptides synthétisés par un grand nombre de cellules lors du développement embryonnaire et par des cellules des tissus adultes dans diverses conditions pathologiques.
On connaît certains dérivés de naphtyridine diamines et des urées correspondantes qui sont des inhibiteurs sélectifs de FGF-1 (Batley B. et al., Life Sciences, (1998), vol
62_n°2, ppl 43-150 ; Thompson A. et al., J. Med. Chenu, (2000), vol 43, pp4200- 4211).
Certains dérivés d'indolizine sont décrits dans les demandes de brevets et brevets US 4 378 362, FR 2 341 578, GB 2 064 536, EP 0 097 636,
EP 302 792, EP 0 382 628, et EP 0 235 111. Ces composés sont utiles dans le traitement de l'angine de poitrine et de l'arythmie. Des propriétés inhibitrices de la translocation calcique sont décrites pour certains de ces composés. La demande de brevet EP 0 022 762 décrit également certains dérivés d'indolizine qui possèdent une activité inhibitrice de la xanthine oxydase et de l'adénosine désaminase ainsi qu'une activité uricosurique. Ces composés peuvent être utilisés dans le traitement des désordres physiologiques consécutifs à un excès d'acide urique, des perturbations du système immunitaire et aux agents parasitaires.
Il a été maintenant trouvé que certains composés, dérivés d'indolizine, sont des puissants antagonistes de la liaison des FGFs à ses récepteurs. Ainsi, la présente invention a pour objet des nouveaux dérivés d'indolizine de formule I,
dans laquelle : R sur les positions 6, 7 ou 8 de l'indolizine représente un atome d'hydrogène, un atome d'halogène, un radical méthyle, un radical hydroxy, un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, un radical carboxy, un radical alcoxycarbonyle de 2 à 6 atomes de carbone ou un radical de formule : -NR
5R
6 -NH-SO
2-Alk -NH-CO-Alk -NH-CO
2-Alk • -O-Alk-COOR
7 -O-Alk-NR
5R
6 -O-(CH
2)
n-Ph -CO-NR
5R
6 où • Alk représente un radical alkyle ou un radical alkylène linéaire ou ramifié de 1 à 5 atomes de carbone, • n représente un nombre entier de 0 à 5, • R
5 et R
ô identiques ou différents représentent chacun un atome d'hydrogène, un radical alkyle linéaire ou ramifié de 1 à 5 atomes de carbone ou un radical benzyle, • R
7 représente un atome d'hydrogène ou un radical alkyle de 1 à 5 atomes de carbone, • Ph représente un radical phényle éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone, Ri représente • un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone. • un radical carboxy, • un radical alcoxycarbonyle de 2 à 6 atomes de carbone, • un radical phényle éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone, • un radical hétéroaryle à 5 chaînons comportant un hétéroatome choisi parmi un atome de soufre, ou un atome d'oxygène, ou un atome d'azote, comportant
éventuellement un second atome d'azote et étant éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alkyles linéaires ou ramifiés de 1 à 5 atomes de carbone, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone, • ou un radical hétéroaryle à 6 chaînons comportant 1 ou 2 atomes d'azote et pouvant être éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alkyles linéaires ou ramifiés de 1 à 5 atomes de carbone, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone,
• R2 représente un radical alkyle de 1 à 5 atomes de carbone, un radical cycloalkyle de 3 à 6 atomes de carbone ou un radical phényle éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone,
• R et » identiques ou différents représentent chacun un radical hydroxy, un radical alcoxy de 1 à 5 atomes de carbone, un radical amino, un radical carboxy, un radical alcoxycarbonyle de 2 à 6 atomes de carbone, un radical nitro, ou un radical de formule : • - NR5R6 • -NH-CO-Alk • -NH-CO-CF3 • -CO- NR5R6 • -CO- NHOH où • Alk, R5 et Rό ont la signification donnée précédemment pour R, à condition toutefois que lorsque R représente un atome d'hydrogène, Ri ne représente pas un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, un radical carboxy, ou un radical alcoxycarbonyle de 2 à 6 atomes de carbone, sauf dans le cas ou R ou i représente un radical -CO-NR5R6 ou un radical -CO-NHOH, éventuellement sous la forme de l'un de leurs sels pharmaceutiquement acceptables.
On préfère un composé de formule I dans laquelle :
• R sur les positions 6, 7 ou 8 de l'indolizine représente un atome d'hydrogène, un atome d'halogène, un radical hydroxy, un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, un radical carboxy, un radical alcoxycarbonyle de 2 à 6 atomes de carbone ou un radical de formule :
-NR5R6 -NH-SO2-Alk -NH-CO-Alk -NH-CO2-Alk -O-Alk-COOR7 -O-Alk-NR5R6 -CO-NR5R6 où • Alk représente un radical alkyle ou un radical alkylène linéaire ou ramifié de 1 à 5 atomes de carbone, • R5 et R<3 identiques ou différents représentent chacun un atome d'hydrogène, un radical alkyle linéaire ou ramifié de 1 à 5 atomes de carbone ou un radical benzyle, • R représente un atome d'hydrogène ou un radical alkyle de 1 à 5 atomes de carbone, présente • un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, • un radical carboxy, • un radical alcoxycarbonyle de 2 à 6 atomes de carbone, • un radical phényle éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone, • un radical hétéroaryle à 5 chaînons comportant un hétéroatome choisi parmi un atome de soufre, ou un atome d'oxygène, ou un atome d'azote, comportant éventuellement un second atome d'azote et étant éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone, • ou un radical hétéroaryle à 6 chaînons comportant 1 ou 2 atomes d'azote et pouvant être éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone,
• R2 représente un radical alkyle de 1 à 5 atomes de carbone, un radical cycloalkyle de 3 à 6 atomes ou un radical phényle éventuellement substitué par un ou plusieurs ato es d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone,
• R3 et R4 identiques ou différents représentent chacun un radical hydroxy, un radical alcoxy de 1 à 5 atomes de carbone, un radical amino, un radical carboxy, un radical alcoxycarbonyle de 2 à 6 atomes de carbone, un radical nitro, un radical de formule • -NR5R6 • -NH-CO-Alk • -CO- NRsRό • -CO- NHOH où • Alk, R5 et R<5 ont la signification donnée précédemment pour R, à condition toutefois que lorsque R représente un atome d'hydrogène, Ri ne représente pas un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, un radical carboxy, ou un radical alcoxycarbonyle de 2 à 6 atomes de carbone, sauf dans le cas ou R ou R4 représente un radical -CO-NR5R6 ou un radical -CO-NHOH, éventuellement sous la forme de l'un de leurs sels pharmaceutiquement acceptables. On préfère en particulier un composé de formule I dans laquelle :
• R sur les positions 6, 7 ou 8 de l'indolizine représente un atome d'hydrogène, un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, un radical alcoxycarbonyle de 2 à 6 atomes de carbone ou un radical de formule : • -NR5R6 • -CO-NR5R6 où • R5 et R6 identiques ou différents représentent chacun un atome d'hydrogène, un radical alkyle linéaire ou ramifié de 1 à 5 atomes de carbone ou un radical benzyle,
• Ri représente • un radical alcoxy linéaire

ramifié de 1 à 5 atomes de carbone, • un radical carboxy, • un radical phényle éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone,
• un radical hétéroaryle à 5 chaînons comportant un hétéroatome choisi parmi un atome de soufre, ou un atome d'oxygène, ou un atome d'azote, éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone, • ou un radical hétéroaryle à 6 chaînons comportant 1 ou 2 atomes d'azote et pouvant être éventuellement substitué par un ou plusieurs atomes d'halogène, par un ou plusieurs radicaux alcoxy de 1 à 5 atomes de carbone, par un ou plusieurs radicaux carboxy ou par un ou plusieurs radicaux alcoxycarbonyle de 2 à 6 atomes de carbone,
• R2 représente un radical alkyle de 1 à 5 atomes de carbone,
• R3 et R4 identiques ou différents représentent chacun un radical alcoxy de 1 à 5 atomes de carbone, un radical amino, un radical carboxy, un radical hydroxy, ou un radical de formule CO- NR5R6 où R5 et RÔ ont la signification donnée précédemment pour R à condition toutefois que lorsque R représente un atome d'hydrogène, Ri ne représente pas un radical alcoxy linéaire ou ramifié de 1 à 5 atomes de carbone, ou un radical carboxy, sauf dans le cas ou R3 ou 4 représente un radical -CO-NR5R6> éventuellement sous la forme de l'un de leurs sels pharmaceutiquement acceptables.
Parmi les composés de l'invention les composés particulièrement préférés sont les • suivants : - Acide [3-(4-amino-3-méthoxybenzoyl)-6-méthoxy-2-méthylindolizin-l-yl] carboxylique (4-amino-3-méthoxyphényl)[l-(4-méthoxyphényl)-2-méthylindolizin-3-yl] méthanone 3-(4-amino-3-méthoxybenzoyl)-(l-méthoxy-N,2-diméthylindolizin-6-yl) carboxamide acide [3-(4-amino-3-méthoxybenzoyl)-2-méthyl-7-(méthylamino) indolizin-1 - yl] carboxylique acide [3-(4-amino-3-méthoxybenzoyl)-7-(diméthylamino)-2-méthylindolizin-l - yl] carboxylique - acide 2-amino-5-({l-méthoxy-2-méthyl-6-[(méthylamino)carbonyl ]indolizin-3- yl }carbonyl) benzoïque acide 2-amino-5-[(l,6-diméthoxy-2-méthylindolizin-3-yl)carbonyl] benzoïque
2-amino-5-[(l-méthoxy-2-méthylindolizin-3-yl)carbonyl] benzamide [3-(4-amino-3-méthoxybenzoyl)-l-(4-méthoxyphényl)-2-méthylindolizin-6-yl] carboxamide - acide 2-amino-5-{[2-mét yl-l-(2-thiényl)indolizin-3-yl] carbonyl}benzoique (4-amino-3-méthoxyphényl)[2-méthyl-l-(2-thiényl) indolizin-3-yl]méthanone éventuellement sous la forme de l'un de ses sels pharmaceutiquement acceptables.
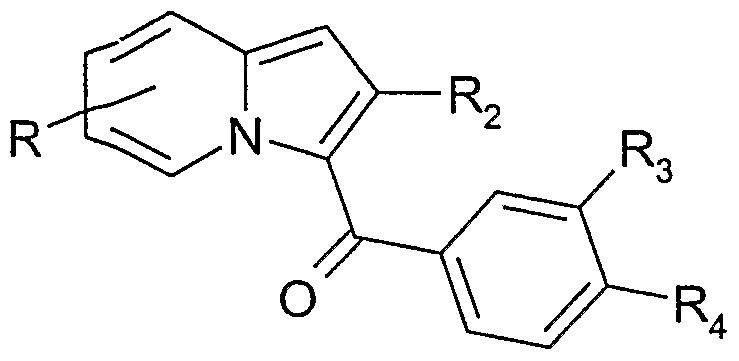
La présente invention concerne également un procédé de préparation des composés de formule I caractérisé en ce que A) on condense un dérivé d'indolizine de formule II,
dans laquelle R, Ri et R
2 ont la signification donnée pour la formule I mais lorsque Ri représente un radical alcoxycarbonyle, R ne représente pas en position 7 un radical -NR
5R\
6, un radical -NH-CO-Alk, un radical -NH-CO
2-Alk ou un radical -NH-SO
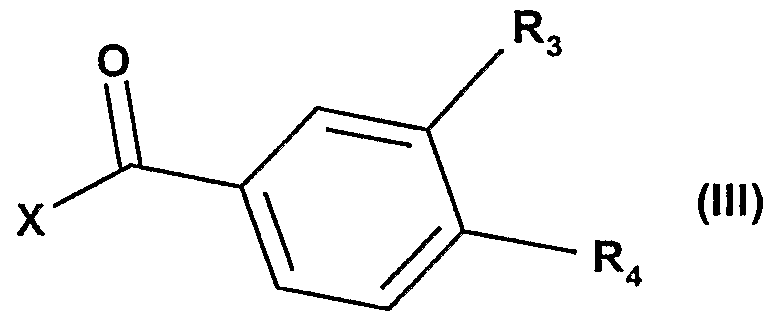
2-Alk, avec un dérivé de formule III :
dans laquelle X représente un atome d'halogène et R
3 ou i représentent indifféremment un radical alcoxy de 1 à 5 atomes de carbone, un radical nitro, ou un radical alcoxycarbonyle de 2 à 6 atomes de carbone, ou un radical trifluoroacétamido pour obtenir les composés de formule la, Ib ou le :
R3 ou R4 = -N02 R3 ou R4 = -C02Alkyl
R
3 ou R
4 = -NH-COCF
j ite, a) on soumet les composés de formule la à une réduction pour obtenir les composés de foπmile Id :
dans laquelle R ou R4 représentent un radical amino, lesquels ensuite composés de formule Id, • sont soumis lorsque R ne représente pas un radical hydroxy à l'action d'un halogénure d'alkyle pour obtenir les composés de formule If pour lesquels
Rt ou R3 représentent un radical -NR5Rό (dans lequel R représente un atome d'hydrogène et R6 représente un radical alkyle de 1 à 5 atomes de carbone) ou sont soumis lorsque R ne représente pas un radical hydroxy à une acylation pour obtenir les composés de formule If pour lesquels R4 ou R représentent un radical -NH-CO-Alk, ou b) on soumet les composés de formule Ib dans laquelle R et / ou R3 et / ou Ri représentent un radical alcoxycarbonyle à une saponification pour obtenir les composés de formule le dans laquelle R et / ou R3 et / ou t représente un radical carboxy, ou c) on soumet lorsque R représente un radical benzyloxy les composés de formule la à une hydrogénation catalytique sous pression, pour obtenir les composés de formule Ig :
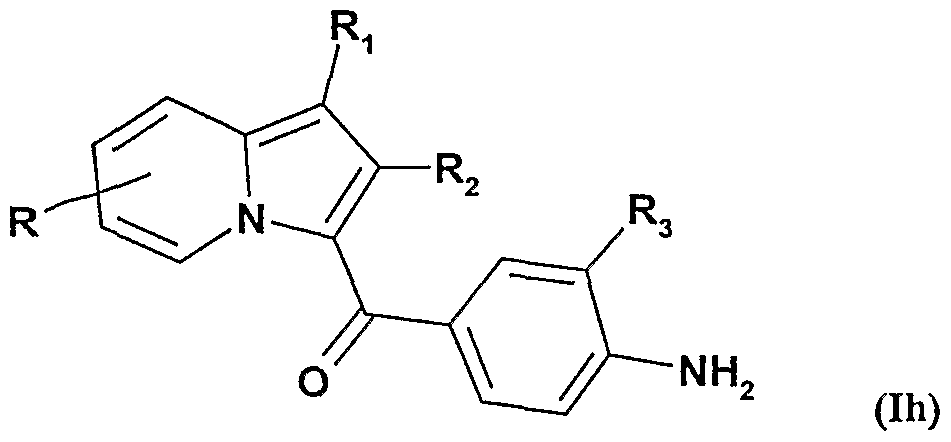
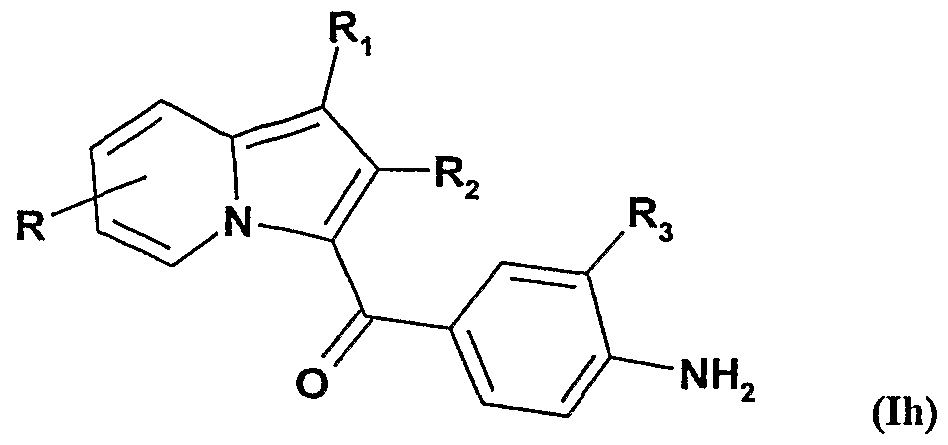
et ensuite on soumet ces composés de formule Ig à une O-alkylation sélective pour obtenir les composés de formule Ih :
ou R représente un radical alcoxy ou un radical de formule -O-Alk-COOR
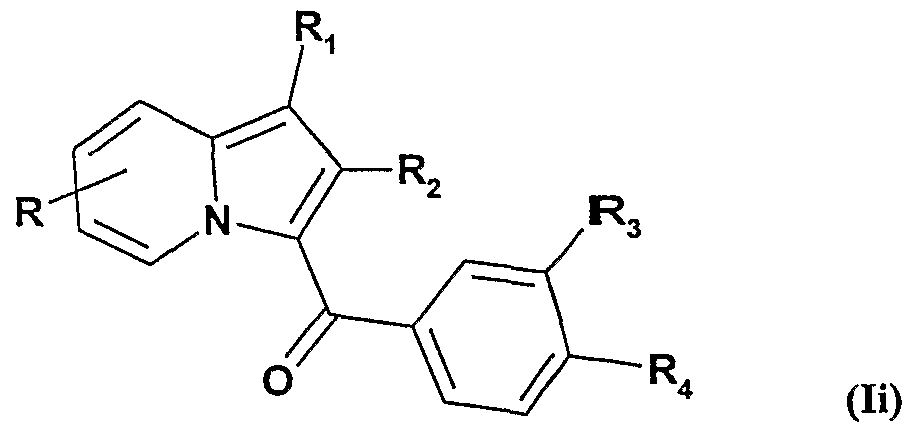
7 qui peut être éventuellement saponifié pour obtenir un radical de formule -O-Alk-COOH. ou d) on soumet lorsque R représente un radical hydroxy les composés de formule la à une O-alkylation, pour obtenir les composés de formule Ii :
dans laquelle R
3 ou i ont les significations données pour la et R représente un radical alcoxy, un radical de formule -0-Alk-NR
5R
6, ou un radical de formule - O-Alk-COOR
7, lequel par la suite peut être éventuellement saponifié pour obtenir un radical de formule -O-Alk-COOH, ou e) on soumet lorsque Ri représente un radical alcoxycarbonyle les composés de formule I à une saponification pour obtenir les composés de formule Ij :
dans laquelle R
3 ou Ri ont les significations données ci-dessus, ou f) on soumet lorsque Ri représente un hydrogène les composés de formule I à une bromation pour obtenir les composés de formule Ik :
lesquels composés sont ensuite soumis lorsque R ne représente pas un halogène tel que brome ou iode à un couplage avec des dérivés phénylboroniques ou hétéroarylboroniques selon les conditions de la réaction de SUZUKI décrite dans Synth. Commun.; (1981), vol 11, p513 pour obtenir les composés de formule II : Ph ou hétéroaryl
dans laquelle R ne représente pas un atome d'halogène tel que brome ou iode et R
2 et R
3 ou Ri ont les significations données ci-dessus et Ri représente un radical phényle substitué ou un hétéroaryle à 5 ou 6 chainons éventuellement substitué, ou g) on soumet lorsque R ne représente pas un radical hydroxy ou amino ou carboxy et R
3 ou
4. représente une fonction carboxy les composés de formule le à un couplage, après activation de la fonction carboxy, avec une amine de formule HNR
5R
6 oα de l'hydroxylamine pour obtenir les composés de formule Ip :
dans laquelle R3 ou -4 représente un radical -CO-NRsRό ou -CO-NHOH,
OU
B) lorsque Ri représente un groupement électro-attracteur et que R représente un radical 7-NH-CO2-Alk on fait réagir les pyridines de formule IV :
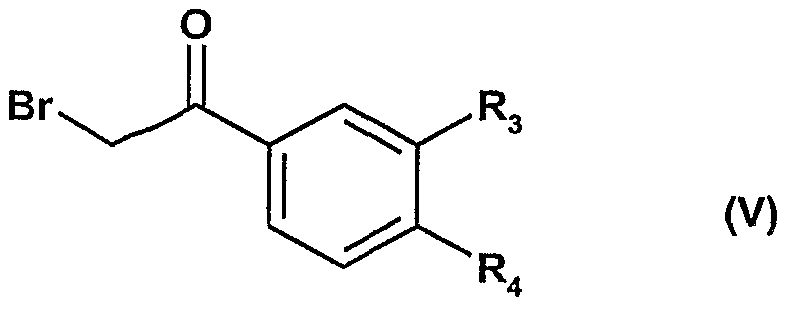
avec une bro oacétophénone de formule V
pour obtenir les composés de formule VI :
lesquels ensuite sont soumis à une cycloaddition 1,3-dipolaire avec l'acrylate de benzyle en présence d'un oxydant pour obtenir les composés de formule la dans laquelle Ri représente un radical benzyloxycarbonyle et R représente un radical de formule -NH-COO-Alk en position 7,
ou
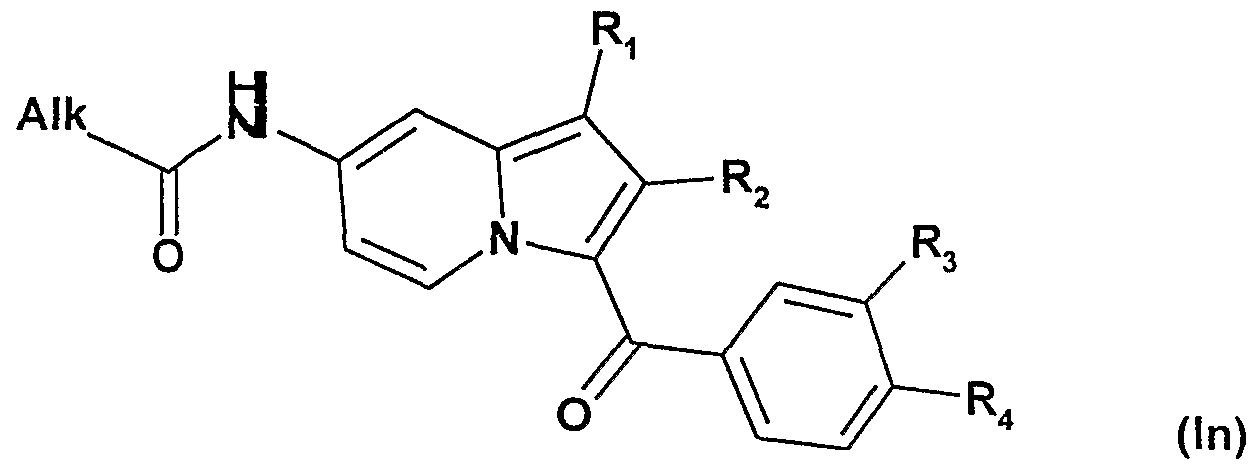
C) on soumet lorsque R représente un radical -NH-CO2tButyle les composés de formule la
soit à une alkylation suivie d'une déprotection et d'une éventuelle deuxiè e alkylation pour obtenir les composés de formule Im :
soit à une déprotection suivie d'une acylation pour obtenir les composés de formule In :
soit à une déprotection suivie d'une sulfonylation pour obtenir les composés de formule Io :
Les figures 1, 2 et 3 donnent les schémas de synthèse des produits la à II et Ip.
FIGURE 1 SCHEMA GENERAL DE SYNTHESE
COMPOSES (If)
R
3 ou R
4 = - R
jRs ou -NHCOAlk = -CONR
jR
g ou -CONHOH
FIGURE 2
COMPOSES Ig COMPOSES Ii R = -OH R
3 ou R
4= NO
z R= -O-Alk, -0-Alk-COOR
7, -0-Alk-NR
5R
6 R'-X O-ALKYLATION SELECTIVE
COMPOSES Ih R= -O-Alk ou 0-Alk-COOR7
FIGURE 3
COMPOSES Ik R., = Br ryl-B(OH)
2 e SUZUKI
COMPOSES II R
1 = Ph ou Heteroaryl
Les composés selon l'invention, quand R
3 ou * représentent un radical nitro sont préparés avec des méthodes connues de benzoylation (Eur. J. Med. Chem. Chim. Ther,
(1983), 18(4"), pp339-346) à partir d'un dérivé d'indolizine de formule II, et un dérivé de chlorure de nitrobenzoyle, de formule III. On obtient ainsi les composés de formule la.
A partir des composés de la formule la par réduction de la fonction nitro on obtient les composés de la formule Id ou Ig dans laquelle R ou R représente un radical amino. En soumettant les composés de formule Id à l'action d'un halogénure d'alkyle on obtient les composés de la formule If pour lesquels R3 ou R4 représente un radical - NR5R6 (dans lequel R5 représente un atome d'hydrogène et R6 a les significations données ci-dessus). Par acylation des composés de formule Id on obtient les composés de formule If pour lesquels R3 ou R4 représente un radical -NH-CO-Alk.
En faisant réagir un dérivé d'indolizine de formule II avec un dérivé de chlorure d'alcoxycarbonylbenzoyle de formule III, on obtient les composés de formule Ib dans laquelle R3 ou R4 représente un radical alcoxycarbonyle. En soumettant ces derniers composés à une saponification on obtient les composés de formule le dans laquelle R3 ou représente un radical carboxy. Si R contient éventuellement une fonction ester, celle ci peut être aussi saponifiée pour obtenir l'acide correspondant. Par couplage des composés de formule le avec des aminés de formule HNR5RÔ OU l'hydroxylamine en présence d'un agent de couplage, on obtient les composés de formule Ip dans laquelle R3 ou R représente un radical -CO-NR5R6 ou -CO-NHOH.
En faisant réagir un dérivé d'indolizine de formule II avec un dérivé de chlorure de trifluoroacétamidobenzoyle de formule III, on obtient les composés de formule le dans laquelle R3 ou R représente un radical trifiuoroacétamide. En soumettant ces derniers composés à une hydrolyse basique on obtient les composés de formule Id dans laquelle R3 et/ou R représente un radical carboxy et/ou amino. Comme représenté dans la figure 2, à partir des composés de formule la dans laquelle R représente un radical benzyloxy et R3 ou R représente un radical nitro, on obtient, en soumettant ces composés à une hydrogénation catalytique sous pression,
les composés de formule Ig, où R représente un radical hydroxy et R3 ou R4 représente un radical amino.
En soumettant les composés de formule Ig à une O-alkylation sélective, on obtient 5 les composés de formule Ih dans laquelle R représente un radical alcoxy linéaire de 1 à 5 atomes de carbone, ou un radical -O-Alk-C00R7 . Ce dernier radical peut être par la suite éventuellement saponifié pour obtenir un radical de formule -O-Alk-COOH.
Comme représenté dans la figure 2, à partir des composés de formule la dans0 laquelle R représente un radical hydroxy et R3 ou R représentent un radical nitro, on obtient, en soumettant ces composés à une O-alkylation, les composés de formule Ii, ou R représente un radical alcoxy de 1 à 5 atomes de carbone, un radical de formule - O-Alk-COOR (qui peut être ensuite éventuellement saponifié pour obtenir un radical de formule -O-Alk-COOH), ou un radical de formule -O-Alk-NR5R6 et R3 ou R 5 représente un radical nitro.
Pour obtenir les composés de formule Ij dans laquelle Ri est un radical carboxy on soumet les composés de formule I dans laquelle Ri est un radical alcoxycarbonyl à une saponification.0 Comme représenté dans la figure 3, à partir des composés de formule I dans laquelle Ri représente un atome d'hydrogène et R, R ou R4 ont les significations données dans la formule générale, on obtient par bromation les composés de formule Ik, ou Ri représente un atome de brorne et R, R3 ou R ont les significations données5 dans la formule générale. Ces derniers composés De soumis (lorsque R ne représente pas un atome de brome ou d'iode) à un couplage de type Suzuki avec des dérivés phénylboroniques ou hétéroarylboroniques conduisent aux dérivés de formule II, ou Ri est un radical phényle ou un hétéroaryle à 5 ou 6 chaînons diversement substitué et R3 ou R4 ont les significations données dans la formule générale et R ne représente pas un0 atome de brome ou d'iode
Les composés de formule II utilisés, lorsque Ri représente un atome d'hydrogène, R2 représente un radical méthyle et R représente un radical -CONH2 ou CO CH3 sont décrits dans J. Chem. Soc. C ; (1969), 901.-> Les composés de formule II, lorsque Ri représente un radical méthoxy, R2 représente un radical méthyle et R représente un radical -CONHCH3 en
position 6 ou -CON(CH3)2 en position 6 sont préparés selon les schémas de synthèse suivants en utilisant la réaction de Tschitschibabin (Synthesis, (1975), p209) pour préparer les indolizines :
Les composés de formule II, lorsque Ri représente un atome d'hydrogène, R2 représente un radical méthyle et R représente soit un radical -CONHCH3 en position 6 soit un radical -NHCOOtBu en position 6 sont préparés selon le schéma de synthèse suivant en utilisant la réaction de Tschitschibabin (Synthesis, (1975), p209) comme précédemment.
Les composés de formule II, lorsque Rj représente un radical -CO2CH3, R2 représente un radical méthyle et R soit un radical benzyloxy en position
6 soit un radical benzyloxy en position 8, sont préparés également en utilisant la réaction de Tschitschibabin selon les schémas de synthèse suivants :
Les composés de formule la dans lesquels R représente un radical de formule -NH-COO-Alk en position 7 et Ri est un groupement électro-attracteur tel que benzyloxycarbonyl sont préparés selon des méthodes connues de cycloaddition [J. Heterocyclic Chem., (2001), 38, 853-857].
R, = Cθ
2Bn
La quaternisation des pyridines de formule IN par une bromoacétophénone de formule V convenablement substituée conduit au pyridinium de formule VI. La cycloaddition 1,3-dipolaire de ce dernier est réalisée en présence d'un oxydant comme le dioxyde de manganèse dans un solvant polaire tel que le diméthylformamide.
Les composés de la formule I sont des antagonistes puissants de FGF-1 et 2, Leurs capacités d'inhiber à la fois la formation de nouveaux vaisseaux à partir de cellules endothéliales différenciées et de bloquer la différentiation de cellules de moelle osseuse humaine adulte CD34+ CD133+ en cellules endothéliales a été démontrée in vitro. De plus, leur capacité à inhiber l'angiogénèse pathologique a été démontrée in vivo. Par ailleurs, il a été démontré que les composés de formule I sont des antagonistes puissants du récepteur FGF-1.
De manière générale, les FGFs sont impliqués de façon importante par l'intermédiaire de sécrétions autocrines, paracrines ou juxtacrines dans les phénomènes de dérégulation de la stimulation de la croissance des cellules cancéreuses. De plus, les FGFs affectent l'angiogénèse tumorale qui joue un rôle prépondérant à la fois sur la croissance de la tumeur mais aussi sur les phénomènes de métastasisation.
L'angiogénèse est un processus de génération de nouveaux vaisseaux capillaires à partir de vaisseaux préexistants ou par mobilisation et différentiation de cellules de la moelle osseuse. Ainsi, à la fois une prolifération incontrôlée des cellules endothéliales et une mobilisation d'angioblastes à partir de la moelle osseuse sont observées dans les processus de néo-vascularisation des tumeurs. Il a été montré in vitro et in vivo que plusieurs facteurs de croissance stimulent la prolifération endothéliale, et notamment le FGF-1 ou a-FGF et le FGF-2 ou b-FGF. Ces deux facteurs induisent la prolifération, la migration et la production de protéases par les cellules endothéliales en culture et la néo-vascularisation in vivo. Les récepteurs a-FGF et b-FGF interagissent avec les cellules endothéliales par l'intermédiaire de deux classes de récepteurs, les récepteurs de haute affinité à activité tyrosine kinase (FGFs) et les récepteurs de basse affinité de type héparane sulfate protéoglycane (HSPGs) situés à la surface des cellules et dans les matrices extracellulaires. Alors que le rôle paracrine de ces deux facteurs sur les cellules endothéliales est largement décrit, les a-FGF et b-
FGF pourraient également intervenir sur ces cellules à travers un processus autocrine. Ainsi, les a-FGF et b-FGF et leurs récepteurs représentent des cibles très pertinentes
pour les thérapies visant à inhiber les processus d'angiogenese (Keshet E, Ben-Sasson SA.,. J. Clin. Invest, (1999), vol 501, ppl04-1497 ; Presta M, Rusnati M, Dell'Era P, Tanghetti E, Urbinati C, Giuliani R et al, New York: Plénum Publishers, (2000), pp7- 34, Billottet C, Janji B, Thiery J.P., Jouanneau J, Oncogene, (2002) vol 21, pp8128- 8139).
Par ailleurs, des études systématiques visant à déterminer l'expression due aux a-FGF et b-FGF et de leurs récepteurs (FGFs) sur différents types de cellules tumorales mettent en évidence qu'une réponse cellulaire à ces deux facteurs est fonctionnelle dans une grande majorité de lignées tumorales humaines étudiées. Ces résultats supportent l'hypothèse qu'un antagoniste des a-FGF et b-FGF pourrait également inhiber la prolifération des cellules tumorales (Chandler LA, Sosnowski BA, Greenlees L, Aukerman SL, Baird A, Pierce GF., Int.J. Cancer, (1999), vol 58, pp81-451).
Les a-FGF et b-FGF jouent un rôle important dans la croissance et le maintien des cellules de la prostate. Il a été montré à la fois dans des modèles animaux et chez l'homme qu'une altération de la réponse cellulaire à ces facteurs joue un rôle primordial dans la progression du cancer de la prostate. En effet dans ces pathologies on enregistre à la fois une augmentation de la production des a-FGF et b-FGF par les fibroblastes et les cellules endothéliales présentes au niveau de la tumeur et une augmentation de l'expression des récepteurs FGFs sur les cellules tumorales. Ainsi une stimulation paracrine des cellules cancéreuses de la prostate s'opère, et ce processus serait un composant majeur de cette pathologie. Un composé possédant une activité antagoniste des récepteurs FGFs tels que les composés de la présente invention peut représenter une thérapie de choix dans ces pathologies (Giri D, Ropiquet F., Clin. Cancer Res., (1999), vol 71. pp5-1063 ; Doll JA, Reiher FK, Crawford SE, Pins MR, Campbell SC, Bouck NP., Prostate, (2001), vol 305, pp 49-293). Plusieurs travaux montrent la présence de a-FGF et b-FGF et de leurs récepteurs
FGFRs à la fois dans les lignées tumorales humaines du sein (notamment MCF7) et dans des biopsies de tumeurs. Ces facteurs seraient responsables dans cette pathologie de l'apparition de phénotype très agressif et induisant une forte métastasisation. Ainsi un composé possédant une activité antagoniste des récepteurs FGFRs, comme les composés de la formule I, peut représenter une thérapie de choix dans ces pathologies
(Vercoutter-Edouart A-S, Czeszak X, Crépin M, Lemoine J, Boilly B, Le Bourhis X et al., Exp.Cell Res., (2001), vol 262, pp59-68).
Les mélanomes cancéreux sont des tumeurs qui induisent avec une fréquence importante des métastases et qui sont très résistantes aux différents traitements de chimiothérapie. Les processus d'angiogenese joue un rôle prépondérant dans la progression d'un mélanome cancéreux. De plus, il a été montré que la probabilité d'apparition de métastases augmente très fortement avec l'augmentation de la vascularisation de la tumeur primaire. Les cellules de mélanomes produisent et sécrètent différents facteurs angiogéniques dont le a-FGF et le b-FGF. Par ailleurs, il a été montré qu'une inhibition de l'effet cellulaire de ces deux facteurs par le RÉCEPTEUR FGF-1 soluble bloque in vitro la prolifération et la survie des cellules tumorales de mélanome et bloque in vivo la progression tumorale. Ainsi .un composé possédant une activité antagoniste des récepteurs FGFs comme les composés de la présente invention peut représenter une thérapie de choix dans ces pathologies (Rofstad EK, Halsor EF., Cancer Res., (2000) ; Yayon A, Ma Y-S, Safran M, Klagsbrun M, Halaban R., Oncogene, (1997), vol 14, pp 2999-3009).
Les cellules de gliome produisent in vitro et in vivo du a-FGF et du b-FGF et possèdent à leur surface différents récepteurs FGFs. Cela suggère donc que ces deux facteurs par un effet autocrine et paracrine jouent un rôle pivotai dans la progression de ce type de tumeur. De plus, comme la plupart des tumeurs solides, la progression des gliomes et leur capacité à induire des métastases est très dépendante des processus angiogéniques dans la tumeur primaire. Il a également été montré que des antisens du récepteur FGF-1 bloquent la prolifération d'astrocytomes humains. De plus, des dérivés des naphthalenesulfonates sont décrites pour inhiber les effets cellulaires des a-FGF et b-FGF in vitro et l'angiogénèse induite par ces facteurs de croissance in vivo.
Une injection intracérébrale de ces composés induit une augmentation très significative de l'apoptose et une diminution importante de l'angiogénèse se traduisant par une régression considérable de gliornes chez le rat. Ainsi un composé possédant une activité antagoniste des a-FGF et / ou b-FGF et / ou des récepteurs FGFs, comme les composés de la présente invention, peut représenter une thérapie de choix dans ces pathologies (Yamada SM, Yamaguchi F, Brown R, Berger MS, Morrison RS, Glia , (1999), vol 76, pp28-66 ; Auguste P, Gtïrsel DB, Lemière S, Reimers D, Cuevas P, Carceller F et al., Cancer Res., (2001), vol 26, pp 61-1717). Plus récemment le rôle potentiel d'agents pro angiogéniques dans les leucémies et lymphomes a été documenté. En effet de manière générale il a été rapporté que des clones cellulaires dans ces pathologies peuvent être soit détruits naturellement par le
système immunitaire soit basculer dans un phénotype angiogénique qui favorise leur survie puis leur prolifération. Ce changement de phénotype est induit par une sur expression de facteurs angiogéniques notamment par les macrophages et / ou une mobilisation de ces facteurs à partir de la matrice extracellulaire (Thomas DA, Giles FJ, Cortes J, Albitar M, Kantarjian HM., Acta Haematol, (2001), vol 207, ppl06-190) .
Parmi les facteurs angiogéniques, le b-FGF a été détecté dans de nombreuses lignées cellulaires tumorales lymphoblastiques et hématopoiétiques. Les récepteurs FGFs sont également présents sur la majorité de ces lignées suggérant un possible effet cellulaire autocrine des a-FGF et b-FGF induisant la prolifération de ces cellules. Par ailleurs il a été rapporté que l'angiogénèse de la moelle osseuse par des effets paracrines était corrélée à la progression de certaines de ces pathologies. De manière plus particulière il a été montré dans les cellules CLL (chronic lymphocytic leukemia) que le b-FGF induit une augmentation de l'expression de protéine anti apoptotique (Bcl2) conduisant à une augmentation de la survie de ces cellules et participe donc de manière importante à leur cancérisation. De plus, les taux de b-FGF mesurés dans ces cellules sont très bien corrélés avec le stade d'avancement clinique de la maladie et la résistance à la chimiothérapie appliquée dans cette pathologie (fludarabine). Ainsi, un composé possédant une activité antagoniste des récepteurs FGFs, comme les composés de la présente invention, peut représenter une thérapie de choix soit seul soit en association avec la fludarabine ou d'autres produits actifs dans cette pathologie (Thomas DA, Giles FJ, Cortes J, Albitar M, Kantarjian HM., Acta Haematol, (2001), vol 207, ppl06-190 ; Gabrilove JL, Oncologist, (2001) , vol 6, pp4-7). II existe une corrélation entre le processus d'angiogenese de la moelle osseuse et les
"extramedullar disease" dans les CML (chronic myelomonocytic leukemia) . Différentes études démontrent que l'inhibition de l'angiogénèse, en particulier par un composé possédant une activité antagoniste des récepteurs FGFs, pourrait représenter une thérapeutique de choix dans cette pathologie.
La prolifération et la migration de cellules musculaires lisses vasculaires contribuent à l'hypertrophie intimale des artères et joue ainsi un rôle prépondérant dans l'athérosclérose et dans la resténose après angioplastie et endoarterectomie. Des études in vivo montrent, après lésion de la carotide par "balloon injury", une production locale de a-FGF et de b-FGF. Dans ce même modèle un anticorps neutralisant anti FGF2 inhibe la prolifération des cellules musculaires lisses vasculaires et diminue ainsi l'hypertrophie intimale.
Une protéine chimérique FGF2 liée à une molécule telle que la saporine inhibe la prolifération des cellules musculaires lisses vasculaires in vitro et l'hypertrophie intimale in vivo (Epstein CE, Siegall CB, Biro S, Fu YM, FitzGerald D., Circulation, (1991), vol 87, pp84-778 ; Waltenberger J., Circulation, (1997), pp96-4083). Ainsi, les antagonistes des récepteurs FGFs, tels que les composés de la présente invention représentent une thérapie de choix, soit seul, soit en association avec des composés antagonistes d'autres facteurs de croissance impliqués dans ces pathologies comme le PDGF, dans le traitement des pathologies liées à la prolifération des cellules musculaires lisses vasculaires telles que l'athérosclérose, la resténose post-angioplastie ou suite à la pose de prothèses endovasculaires (stents) ou lors de pontages aorto- coronariens.
L'hypertrophie cardiaque intervient en réponse à un stress de la paroi ventriculaire induit par une surcharge en terme de pression ou de volume. Cette surcharge peut être la conséquence de nombreux états physio pathologiques comme l'hypertension, l'AC
(aortic coarctation), l'infarctus du myocarde, et différents troubles vasculaires. Les conséquences de cette pathologie sont des changements morphologiques, moléculaires et fonctionnels comme l'hypertrophie des myocytes cardiaques, l'accumulation de protéines matricielles et la ré-expression de gènes fœtaux. Le b-FGF est impliqué dans cette pathologie. En effet l'addition de b-FGF à des cultures de cardiomyocytes de rat nouveau-né modifie le profil des gènes correspondants aux protéines contractiles conduisant à un profil de gènes de type fœtaux. De manière complémentaire des myocytes de rat adulte montrent une réponse hypertrophique sous l'effet du b-FGF, cette réponse étant bloquée par des anticorps neutralisants anti b-FGF. Des expériences réalisées in vivo sur des souris transgéniques "knock-out" pour le b-FGF, montrent que le b-FGF est le facteur stimulant majeur de l'hypertrophie des myocyte cardiaque dans cette pathologie (Schultz JeJ, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M et al., J. Clin. Invest., (1999), vol 19. pplO4-709). Ainsi un composé, comme les composés de la présente invention, possédant une activité antagoniste des récepteurs FGFs représente une thérapie de choix dans le traitement de l'insuffisance cardiaque et toute autre pathologie associée à une dégénérescence du tissu cardiaque. Ce traitement pourrait être réalisé seul ou en association avec les traitements courants (beta-bloquants, diurétiques, antagonistes d'angiotensine, antiarrythmiques, anti-calciques, anti-thrombotiques etc ..)
Les troubles vasculaires dus au diabète se caractérisent par une altération de la réactivité vasculaire et du flux sanguin, une hyperperméabilité, une réponse
proliférative exacerbée et une augmentation des dépôts de protéines matricielles. De manière plus précise le a-FGF et le b-FGF sont présents dans les membranes pré rétiniennes de patients ayant des rétinopathies diabétiques, dans les membranes des capillaires sous jacents et dans l'humeur vitrée de malades souffrants de rétinopathies prolifératives. Un récepteur du FGF soluble capable de lier à la fois le a-FGF et le b-FGF est développé dans les troubles vasculaires liés au diabète (Tilton RG, Dixon RAF, Brock TA., Exp. Opin. Invest. Drugs, (1997), vol 84, pp6-1671). Ainsi un composé comme les composés de formule I possédant une activité antagoniste des récepteurs FGFs représente une thérapie de choix soit seul soit en association avec des composés antagonistes d'autres facteurs de croissance impliqués dans ces pathologies comme le VEGF.
L'arthrite rhumatoïde (RA) est une maladie chronique avec une étiologie inconnue. Alors qu'elle affecte de nombreux organes, la forme la plus sévère de RA est une inflammation synoviale des articulations progressive aboutissant à la destruction.
L'angiogénèse semble affecter de manière importante la progression de cette pathologie. Ainsi le a-FGF et le b-FGF ont été détectés dans le tissu synovial et dans le fluide articulaire de patients atteints de RA, indiquant que ce facteur de croissance intervient dans l'initiation et / ou la progression de cette pathologie. Dans des modèles de AI A (adjuvant-induced model of artliritis) chez le rat, il a été montré que la surexpression de b-FGF augmente la sévérité de la maladie alors qu'un anticorps neutralisant anti b-FGF bloque la progression de la RA (Yamashita A, Yonemitsu Y, Okano S, Nakagawa K, Nakashima Y, Irisa T et al., J.Immunol., (2002), vol 57, pp 168-450 ; Manabe N, Oda H, Nakamura K, Kuga Y, Uchida S, Kawaguchi H, Rheumatol, (1999), vol.20, pp38-714). Ainsi les composés selon l'invention représentent une thérapie de choix dans cette pathologie.
Les IBD ( inflammatory bowel disease) comprennent deux formes de maladies inflammatoires chroniques de l'intestin : les UC ( ulcerative colitis) et la maladie de Crohn's (CD). Les IBD sont caractérisées par une dysfonction immunitaire se traduisant par une production inappropriée de cytokines inflammatoires induisant l'établissement d'un système micro-vasculaire local. Cette angiogénèse d'origine inflammatoire a pour conséquence une ischémie intestinale induite par vasoconstriction. Des taux circulants et locaux de b-FGF importants ont été mesurés chez des patients atteints de ces pathologies (Kanazawa S, Tsunoda T, Onuma E,
Majima T, Kagiyama M, Kkuchi K., American Journal of Gastroenterology, (2001), vol 28, pp 96-822 ; Thorn M, Raab Y, Larsson A, Gerdin B, Hallgren R.,
Scandinavian Journal of Gastroenterology, (2000), vol 12, pp35-408). Les composés de l'invention présentant une activité anti angiogénique importante dans un modèle d'angiogenese inflammatoire représentent une thérapie de choix dans ces pathologies. Les récepteurs FGF-1 2 et 3 sont impliqués dans les processus de chronogénèse et osteogénèse. Des mutations conduisant à l'expression de FGFRs toujours activés ont été reliées à un grand nombre de maladies génétiques humaines se traduisant par des malformations du squelette comme les syndromes de Pfeiffer, Crouzon, Apert, Jackson- Weiss et Bear-Stevenson cutis gyrata. Certaines de ces mutations affectant plus particulièrement le récepteur FGF-3 conduisent notamment à des achondroplasies
(ACH), des hypochondroplasies (HCH) et des TD (Thanatophoric dysplasia); ACH étant la forme la plus courante de nanisme. D'un point de voie biochimique l'activation soutenue de ces récepteurs s'effectue par une dimérisation du récepteur en absence de ligand (Chen L., Adar R. , Yang X. Monsonego E.O., LI C, Hauschka P.N, Yagon A. and Deng C.X., (1999), The Journ. Of Clin. Invest., vol 104 n° 11, pp 1517-1525).
Ainsi les composés de l'invention présentant une activité antagoniste de la liaison du b-FGF au récepteur FGF et inhibant ainsi la dimérisation du récepteur représentent une thérapie de choix dans ces pathologies. Par ailleurs, on connaît que le tissu adipeux est un des rares tissus qui chez l'adulte peut se développer ou régresser. Ce tissu est très vascularisé et un réseau très dense de micro vaisseaux entoure chaque adipocyte. Ces observations ont conduit à tester l'effet d'agent anti angiogéniques sur le développement du tissu adipeux chez l'adulte. Ainsi il apparaît que dans des modèles pharmacologiques chez la souris ob/ob, l'inhibition de l'angiogénèse se traduit par une perte significative de poids des souris (Rupnick MA et al, (2002), PNAS, Vol 99 n°16, pp 10730-10735). Ainsi un composé antagoniste des récepteurs FGFs possédant une activité anti angiogénique puissante peuvent représenter une thérapie de choix dans les pathologies liées à l'obésité. Grâce à leur faible toxicité et leurs propriétés pharmacologiques et biologiques, les composés de la présente invention trouvent leur application dans le traitement de tout carcinome ayant un degré de vascularisation important (poumon, sein, prostate, œsophage) ou induisant des métastases (colon, estomac, mélanome) ou étant sensibles au a-FGF ou au b-FGF de manière autocrine ou enfin dans des pathologies de type lymphomes et leucémies. Ces composés représentent une thérapie de choix soit seul soit en association avec une chimiothérapie adaptée. Les composés selon l'invention trouvent également leur application dans le traitement de maladies cardiovasculaires
comme l'athérosclérose, la resténose post angioplastie dans le traitement des maladies liés aux complications apparaissant suite à la pose de prothèses endovasculaires et/ou de pontages aorto-coronariens ou d'autres greffes vasculaires et l'hypertrophie cardiaque ou de complications vasculaires du diabète comme les rétinopathies diabétiques. Les composés selon l'invention trouvent également leur application dans le traitement de maladies inflammatoires chroniques comme l'arthrite rhumatoïde ou les IBD. Enfin les composés selon l'invention peuvent être utilisés dans le traitement des achondroplasies (ACH), des hypochondroplasies (HCH) et des TD (Thanatophoric dysplasia), comme également dans le traitement de l'obésité.
Les produits selon l'invention trouvent également leur application dans le traitement de la dégénérescence maculaire. Un caractère majeur de la perte de la vision chez l'adulte est la néo-vascularisation et les hémorragies consécutives qui causent des désordres fonctionnels importants au niveau de l'oeil et qui se traduisent par une cécité précoce. Récemment, l'étude des mécanismes impliqués dans les phénomènes de néo- vascularisation oculaire a permis de mettre en évidence l'implication de facteur pro- angiogéniques dans ces pathologies. En mettant en œuvre un modèle de néoangiogenèse choroïdienne induite par laser, il a été possible de confirmer que les produits selon l'invention permettent également de moduler la néo- vascularisation de la choroïde.
Par ailleurs, les produits de l'invention peuvent être utilisés dans le traitement ou la prévention des tl rombopénies dues notamment à une chimiothérapie anti-cancéreuse. Il a été en effet démontré que les produits de l'invention peuvent améliorer les taux des plaquettes circulantes lors d'une chimiothérapie.
Selon un autre de ses aspects, la présente invention a pour objet une composition pharmaceutique contenant, en tant que principe actif, un composé de formule I selon l'invention ou un de ses sels pharmaceutiquement acceptables, éventuellement en association avec un ou plusieurs excipients inertes et appropriés.
Lesdits excipients sont choisis selon la forme pharmaceutique et le mode d'administration souhaitée : orale, sublinguale, sous-cutanée, intramusculaire, intraveineuse, transdemiique, transmuqueux, locale ou rectale.
Les compositions pharmaceutiques selon la présente invention sont administrées de préférence par voie orale.
Dans les compositions pharmaceutiques de la présente invention pour l'administration orale, les principes actifs peuvent être administrés sous forme unitaire d'administration, en mélange avec des supports pharmaceutiques classiques. Les formes unitaires d'administration appropriées comprennent par exemple les comprimés éventuellement sécables, les gélules, les poudres, les granules et les solutions ou suspensions orales.
Lorsqu'on prépare une composition solide sous forme de comprimés, on mélange l'ingrédient actif principal avec un véhicule pharmaceutique tel que la gélatine, l'amidon, le lactose, le stéarate de magnésium, le talc, la gomme arabique ou analogues. On peut enrober les comprimés de saccharose ou d'autres matières appropriées ou encore on peut les traiter de telle sorte qu'ils aient une activité prolongée ou retardée et qu'ils libèrent d'une façon continue une quantité prédéterminée de principe actif.
On obtient une préparation en gélules en mélangeant l'ingrédient actif avec un diluant et en versant le mélange obtenu dans des gélules molles ou dures. Une préparation sous forme de sirop ou d'élixir peut contenir l'ingrédient actif conjointement avec un édulcorant, acalorique de préférence, du méthylparaben et du propylparaben comme antiseptiques, ainsi qu'un agent donnant du goût et un colorant approprié. Les poudres ou les granules dispersibles dans l'eau peuvent contenir l'ingrédient actif en mélange avec des agents de dispersion ou des agents mouillants ou des agents de mise en suspension, comme la polyvinylpyrrolidone, de même qu'avec des édulcorants ou des correcteurs du goût. Le principe actif peut être formulé également sous forme de microcapsules, éventuellement avec un ou plusieurs supports ou additifs.
Dans les compositions pharmaceutiques selon la présente invention, le principe actif peut être aussi sous forme de complexe d'inclusion dans des cyclodextrines, leurs éthers ou leurs esters.
La quantité de principe actif à administrer dépend, comme toujours, du degré de progression de la maladie ainsi que de l'âge et du poids du patient.
Les compositions selon l'invention, pour une administration orale, contiennent donc des doses recommandées de 0,01 à 700 mg.
Les exemples suivants, donnés à titre non limitatif, illustrent la présente invention.
PREPARATIONS
Préparations I et II
Synthèse de 6-(méthoxyméthyl)-N-méthylnicotmamide et de 6-(méthoxyméthyl)- N^V-diméthylnicotinamide A 1,56g (42,19 m.moles) d'hydrure de sodium - à 65% en dispersion dans l'huile - dans 23ml de diméthylformamide, on ajoute goutte à goutte à 0°C une solution de
3,21g (21,10 m.moles) de 6-(hydroxyméthyl)nicotinamide [décrit dans Bull. Chem.
Soc. Jpn.; (1988), 61(8), 2837-2846] dans 90ml de diméthylformamide et on agite à 0°C pendant 0,5 heure. Toujours à la même température on ajoute ensuite goutte à goutte 2,63ml (42,19 m.moles) d'iodure de méthyle dans 7ml de diméthylformamide.
L'introduction terminée, on laisse revenir à température ambiante et on agite pendant 2 heures. Le milieu réactiormel est versé sur de l'eau et de l'acétate d'éthyle. La phase organique est décantée, lavée avec une solxition aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. On recueille un mélange contenant les deux produits désirés que l'on sépare par chromatographie sur gel de silice en éluant avec un mélange de dichlorométhane et de méthanol (95-5 puis 9-1). Les deux fractions recueillies sont ensuite évaporées.
A ) Première fraction : Préparation I 6-(méthoxyméthyl)-N^V-diméthylnicotinamide
On obtient 1,27g d'une huile jaune Rendement : 31%
Spectrométrie de Masse (Mode ES+) MH+ = 195,3
B) Deuxième fraction : Préparation II 6-(méthoxyméthyl)-N-méthylnicotinamide
On obtient 1,06g d'une huile jaune
Rendement : 28%
Spectrométrie de Masse (Mode ES+) MH+ = 181 ,2
Préparation III
Synthèse de [l-méthoxy-NN,2-triméthylindolizin-6-yl]carboxamide A une solution de 547mg (6,29 m.moles) de bromure de lithium dans 10ml d'acétonitrile on ajoute 501 μl (6,29 m.moles) de chloroacétone et agite 15minutes à température ambiante puis on ajoute 0,94g (4, 84 m.moles) de 6-(méthoxyméthyl)- TVN-diméthylnicotinamide, obtenu à la préparation I, dissout dans 10ml d'acétonitrile et chauffé à reflux 24 heures. Le milieu réactionnel est évaporé à sec. Le résidu est repris par 20ml d'eau et lavé à l'éther éthylique. Après décantation, on récupère la phase aqueuse, on ajoute 1,34g (9,71 m.moles) de carbonate de potassium et on chauffe à 80°C pendant 2 heures. On ajoute de l'acétate d'éthyle, on décante, on lave la phase organique avec une solution aqueuse saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. Le résidu est purifié par chromatographie sur gel de silice en éluant avec un mélange de dichlorométhane et de méthanol (95-5). Après évaporation on recueille 960mg d'un solide jaune. Rendement : 67%
Point de fusion : 112,5°C
Préparation IV Synthèse de [l-méthoxy-N,2-diméthyIindoliziιι-6-yl]carboxamide Ce composé est obtenu selon le même mode opératoire que le composé de la préparation III en utilisant la réaction de Tschitschibabin, en partant de 710mg de
6-(méthoxyméthyl)-N-méthylnicotinamide obtenu dans la préparation II et de chloroacétone. On obtient 1,03g d'un solide jaune. Rendement : 84%
Point de fusion : 127°C
Préparation V
Synthèse de [8-(benzyIoxy)-2-méthylindolizin-l-yl]carboxylate de méthyle Ce composé est obtenu selon le même mode opératoire que le composé de la préparation III en utilisant la réaction de Tschitschibabin, en partant de 25,82g de 2-[3-(benzyloxy)pyridin-2-yl]acétate de méthyle [selon J Med. Chem.; (1996), 39(19), 3636-3658] et de chloroacétone. On obtient 19,1 lg d'un solide jaune. Rendement : 65% Spectrométrie de Masse (Mode ES+) MH+- = 296
Préparation VI
Synthèse de [6-(benzyloxy)-2-méthyIindoIizin-l-yI]carboxylate de méthyle Ce composé est obtenu selon le même mode opératoire que le composé de la préparation III en utilisant la réaction de Tschitschibabin, en partant de 19g de
2-[5-(benzyloxy)pyridin-2-yl]acétate de méthyle [selon Bull. Pol. Acad. Sci. Chem.;
(1990), 38(1-12), 17-27] et de chloroacétone. On obtient 15,01g d'un solide orange.
Rendement : 69%
Point de fusion : 143°C
Préparation VII
Synthèse de [N,2-diméthylindolizin-6-yl] carboxamide Ce composé est obtenu selon le même mode opératoire que le composé de la préparation III en utilisant la réaction de Tschitschibabin, en partant de 5.4g de
6-méthyl-N-méthylnicotinamide [décrit dans J. Org. Chem. ; (1959), 24, 1189-1191] et de chloroacétone en utilisant la dibutylamine comme base. On obtient 2.04g d'un solide jaune.
Rendement : 32% Spectrométrie de Masse (Mode ES+) MH+- ≈ 189.4
Préparation VIII
Synthèse de [2-méthyIindoIizin-6-yl]carbamate de tert-butyle Ce composé est obtenu selon le même mode opératoire que le composé de la préparation III en utilisant la réaction de Tschitschibabin, en partant de 5.79g de
(6-mélhylpyridin-3-yl)carbamate de tert.butyle [décrit dans J.Med. Chem. ; (2000), 43,
5017-5029] et de chloroacétone. On obtient 1.96g d'un résidu pâteux.
Rendement : 31 %
Spectrométrie de Masse (Mode ES+) MH+ = 247.1
Préparation IX Synthèse de [l-méthoxy-2-méthylindolizin-6-yI]carboxylate de méthyle Etape A 6-(méthoxyméthyI)nicotinate de méthyle A une solution de méthylate de sodium obtenue par ajout de lg (0.044 atome/g) de sodium dans 40 ml de méthanol, on ajoute à température ambiante 5.1g (0.022 mole) de 6-(bromométhyl)nicotinate de méthyle [décrit dans J Med. Chem. ; (2002, 45(23),
5005-5022] dissout dans 60ml de méthanol. Le milieu réactionnel est ensuite porté à reflux 2 heures. Le milieu réactionnel est concentré sous vide et le résidu est repris par une solution aqueuse d'hydrogénosulfate de potassium et de dichloromethane. La phase organique est décantée, séchée sur sulfate de sodium et évaporée à sec. On obtient 3.4g d'un solide jaune.
Rendement : 85%» Point de fusion : 33°C Etape B A 1.5g (0.0174 mole) de bromure de lithium dans 10ml d'acétone on ajoute 1.46ml (0.0174 mole) de chloroacétone et on agite à température ambiante 15 minutes puis ajoute 1.5g (0.00828 mole) de 6-(méthoxyméthyl)nicotinate de méthyle et porte au reflux une nuit. On ajoute au milieu réactionnel 1.5g (O.0174 mole) de bromure de lithium et 1.46ml (0.0174 mole) de chloroacétone et maintient le reflux 5 heures. On évapore à sec et le résidu obtenu est repris avec un mélange dichlorométhane- méthanol (9-1). L'insoluble obtenu est filtré, puis on ajoute au filtrat 4.62ml
(0.033mole) de triéthylamine et agite à température ambiante une heure. Le milieu réactionnel est concentré. Le résidu obtenu est repris au toluène et le produit est purifié par filtration sur lit de silice en éluant au toluène. Après évaporation on obtient 450mg d'un solide jaune. Rendement : 25%
Point de fusion : 51°C
EXEMPLES
Exemples 1 et 2 [8-(benzyloxy)-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin-l- yl]carboxylate de méthyle (exemple 1) et [8-hydroxy-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin-l-yl]carboxylate de méthyle (exemple 2) A 19,1g (0,0647 mole) de [8-(benzyloxy)-2-méthylindolizin-l-yl]carboxylate de méthyle dissout dans 100ml de 1 ,2-dichloroéthane, on ajoute 18,1g (0,0841 mole) de chlorure de 3-méthoxy -4-nitrobenzoyle et on agite à température ambiante pendant 60 heures. Le milieu réactionnel est évaporé à sec. On recueille un mélange de deux produits que l'on sépare par chromatographie sur gel de silice en éluant avec un mélange de toluène-acétate d'éthyle (9-1). Cette séparation est suivie par chromatographie sur couche mince (CCM) en utilisant comme éluant un mélange de dichloromethane et de méthanol (9-1). On évapore ensuite chacune des deux fractions :
A) PREMIERE FRACTION
[8-(benzyIoxy)-3-(3-méthoxy-4-nitrobenzoyl)-2-méthyIindolizin-l-yl]carboxylate de méthyle
On obtient 8,71g d'un solide orange. Rendement : 28% Point de fusion : 125°C
B) DEUXIEME FRACTION [8-hydroxy-3-(3-méthoxy-4-nitrobenzoyl)-2-mιéthyImdolizin-l-yl]carboxylate de méthyle On obtient 7,68g d'un solide orange.
Rendement : 31% Point de fusion : 150°C
Exemples 3 à 17 En procédant selon la préparation décrite ci-dessus, on synthétise les composés de formule I, décrits dans le Tableau I ci-dessous, par benzoylation de la position 3 des indolizines, diversement substituées en position 1, 2, 6, 7, et 8, avec les chlorures de benzoyle substitués adéquats.
TABLEAU I
* benzoylation en présence de triéthylamine (1 équivalent) Bn = benzyle Me = méthyle BOC = tert-butyloxycarbonyle
Exemple 18 [l-méthoxy-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin-6-yI]carboxamide
Etape A Acide [l-méthoxy-3-(3-méthoxy-4-nitrobenzoyI)-2-méthylindolizin-6-yl] carboxylique A 440mg (0.0011 mole) de [l-méthoxy-3-(3-méthoxy-4-nitrobenzoyl)-2- méthylindolizin-6-yl]carboxylate de méthyle (obtenu à l'exemple 10) dans 10ml de méthanol on ajoute 1.21ml (0.00121 mole) de soude IN et agiter à température ambiante une nuit. Le milieu réactionnel est concentré sous vide et le résidu est repris avec de l'eau et de l'acétate d'éthyle. La phase aqueuse est décantée puis acidifiée avec
1.21ml d'acide chlorhydrique IN. Le précipité formé est filtré, lavé à l'eau et séché. On obtient 280mg d'un solide orange.
Rendement : 66%
Point de fusion : 287°C
Etape B A 260mg (0.66 m.mole) d'acide [l-méthoxy-3-(3-méthoxy-4-nitrobenzoyl)-2- méthylindolizin-6-yl] carboxylique dans 5ml de iV,N-diméthylformamide on ajoutel04μl (0.74 m.mole) de triéthylamine puis 330mg (0.74 m.mole) de benzotriazol-l-yloxy-tris(diméthylamino)phosphonium liexafluorophosphate (BOP) et agite une heure à température ambiante. On ajoute au ilieu réactionnel 0.9ml (2.71 m.moles) d'une solution d'ammoniac 3Ν dans le tetrahydrofurane et on laisse agiter à température ambiante une nuit. On ajoute de l'eau et une solution aqueuse saturée de bicarbonate de sodium. On extrait plusieurs fois à l'acétate d'éthyle. La phase organique est décantée, séchée sur sulfate de sodium et concentrée sous vide. Le résidu solide obtenu est repris à l'éther isopropylique, filtré, lavé à l'éther isopropylique puis séché. On obtient 210mg d'un solide rouge.
Rendement : 81%»
Point de fusion : 244°C
Exemple 19
[8-méthoxy-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylin<lolizin-l-yl]carboxylate de méthyle A un mélange de 705mg (1,84 m.mole) de [8-hydroxy-3-(3-méthoxy-4- nitrobenzoyl)-2-méthylindolizin-l-yl]carboxylate de méthyle obtenu à l'exemple 2, dans 10ml d'acétone avec 330mg (2,39 m.mole) de carbonate de potassium on ajoute 0,19ml (2,02 m.mole) de diméthylsulfate et on chauffe à 60°C pendant 6 heures. Le milieu est évaporé. L'huile obtenue est reprise à l'acétate d'éthyle et à l'eau. La phase organique est décantée, lavée à l'eau puis avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Le solide obtenu est repris à l'éther isopropylique, filtré, lavé à l'éther isopropylique puis séché. On recueille 649mg d'un solide jaune. Rendement : 88%o Point de fusion : 73°C
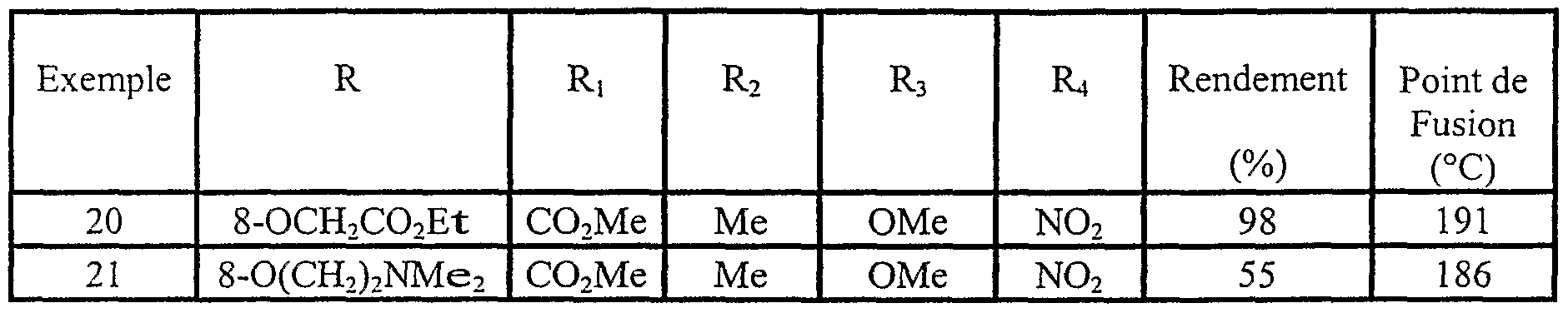
Exemples 20 et 21 En procédant selon la préparation décrite ci-dessus, on synthétise les composés de formule Ii, décrits dans le Tableau II ci-dessous, par O-alkylation de l'hydroxy sur la position 8 des indolizines diversement substituées avec les halogenures d'alkyles adéquats.
TABLEAU II
Exemple 22 (3-méthoxy-4-nitrophényI)[l-(4-méthoxyphényI)-2-méthylindolizin-3-yl] methanone ETAPE A (3-méthoxy-4-nitrophényl)( 2-méthylindolizin-3-yI) methanone Ce composé est obtenu en procédant selon la préparation décrite dans l'exemple 1 , par benzoylation de la 2-méthylindolizine, décrite dans Pharmazie ; (1980), vol 35(4), pp 203-204, avec le chlorure de 3-méthoxy -4-nitrobenzoyle. On obtient 6,52g d'un solide orange. Rendement : 92% Point de fusion : 161°C
ETAPE B (3-méthoxy-4-nitrophényl)(l-bromo-2-méthylindolizin-3-yI) methanone A 5,47g (17,63 m.moles) de (3-méthoxy-4-nitrophényl)( 2-méthylindolizin-3-yl) methanone, obtenu à l'étape A ci-dessus, dans 56ml de dioxanne, on ajoute goutte à goutte une solution de 993 μl (19,39 m.moles) de brome dans 35ml de dioxanne en maintenant le milieu réactionnel à température ambiante. L'introduction terminée on agile 1 heure de plus à la même température. Le milieu réactionnel est versé sur une solution aqueuse saturée de bicarbonate de sodium et extrait à l'acétate d'éthyle. La phase organique est décantée, lavée avec une
solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Le résidu est repris au dichloromethane et le produit est purifié par filtration sur un lit de gel de silice. Après évaporation on recueille 6,75g d'un solide jaune.
Rendement : 98% Point de fusion : 166°C
ETAPE C (3-méthoxy-4-nitrophényl)[l-(4-méthoxyph.ényl)-2-méthylindoIizin-3-yl] methanone A 500mg (1,28 m.mole) de (3-méthoxy-4-nitrophényl)(l-bromo-2-méthylindolizin- 3-yl) methanone, obtenu à l'étape B précédente, dans 4,3ml de diméthoxyéthane, sous atmosphère d'argon, en présence de 3ml d'une solution aqueuse IN de carbonate de sodium, on ajoute 59,4mg (0,05 m.mole) de tetrakis(triphénylphosphine)palladium(0) puis 238mg (1,57 m.mole) d'acide 4-méthoxypb.énylboronique et on chauffe à reflux pendant 2 heures. Le milieu réactionnel est versé sur de l'eau et extrait à l'acétate d'éthyle. La phase organique est décantée, lavée avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Le produit est purifié par chromatographie sur gel de silice en éluant avec un mélange de toluène et d'acétate d'éthyle (92-8). Après évaporation on recueille 500mg d'un solide orange. Rendement : 93,5% Point de fusion : 169°C
Exemples 23 à 29 En procédant selon la préparation décrite ci-dessus (Exemple 22 Etape B), on synthétise les composés de formules Ik , décrits dans le Tableau III ci-dessous, par bromation des composés de formule I (avec Ri = H) avec du brome.
TABLEAU III
** la bromation est effectuée en présence d'acétate de sodium
Exemples 30 à 46
En procédant selon la préparation décrite dans l'Exemple 22 Etape C, on synthétise les composés de formules II , décrits dans le Tableau IV ci-dessous, par couplage de type Suzuki des composés bromes de formule générale Ik avec des dérivés phenylboroniques ou hétéroarylboroniques en faisant varier les conditions expérimentales (catalyseurs, ligands, bases) selon les composés à obtenir.
TABLEAU IV
Ph = phényle Bu = butyle * on utilise les dérivés pinacol boronates au lieu des acides boroniques correspondants ** perte du groupement protecteur trifluoroacétyl au cours de la synthèse en milieu basique
Exemple 47 [7-{N-(ter/-butoxycarbonyl)amino}-3-(3-nιéthoxy-4-nitrobenzoyl)-2-méthyI- indolizin-l-yI]carboxylate de benzyle ETAPE A Bromure de 4-[N-(ter -butoxycarbonyl)amino]-l-[2-(3-méthoxy-4-nitrophényl)-2- oxoéthyl]pyridinium
On ajoute 2,90g (10,6 m.moles) de 2-bromo-l-(3-méthoxy-4-nitrophényl)-l- éthanone [décrit dans Bull. Soc. Chim. Fr., (1962), pp2255-2261] par portion à 2,07g (10,6 m.moles) de 4-[N-(tert-butoxycarbonyl)amino]pyridine [décrit dans Tetrahedron; (2001), vol 57(43), pp9033-9044] en suspension dans 10ml d'acétonitrile et 20ml d'acétone. Le milieu réactionnel devient homogène en agitant à température ambiante puis une précipitation apparaît. On agite 1 heure à température ambiante. Le précipité formé est filtré, lavé à l'acétone et séché. On obtient 4,60g d'une poudre blanche.
Rendement : 93% Point de fusion : 224°C
ETAPE B A une solution de 5,0g (10,67 m.moles) de bromure de 4-[N-(tert-butoxycarbonyl) amino]-l-[2-(3-méthoxy-4-nitrophényl)-2-oxoéthyl]pyridinium dans 40ml diméthyl- formamide, on ajoute successivement 9,4g (53,38 m.moles) de crotonate de benzyle,
1,8ml (12,8 m.moles) de triéthylamine et 3,7g (42,68 m.moles) d'oxyde de manganèse.
Le mélange réactionnel est ensuite chauffé à 90 °C pendant 4 heures puis filtré sur lit de silice en éluant avec de l'acétate d'éthyle. Le filtrat est versé sur de l'eau et extrait à l'acétate d'éthyle. Après décantation, la phase organique est lavée avec une solution saturée de chlorure de sodium, séchée sur sulfate de sodium puis concentrée sous pression réduite. Le produit est purifié par chromatographie sur gel de silice en éluant avec un mélange toluène/acétate d'éthyle (95/5 à 70/30). On obtient 2,6g d'une poudre orange.
Rendement : 44% Point de fusion : 95°C
Exemple 48
[7-{N-(éthoxycarbonyl)amino}-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin-l- yl]carboxyIate de benzyle
ETAPE A
Bromure de 4-[N-(éthoxycarbonyl)amino]-l-[2-(3-méthoxy-4-nitrophényI)-2-oxo- éthyl]pyridinium Ce composé est préparé selon le même procédé que celui décrit dans l'exemple 47 Etape A par quaternisation de 1,21g (7,33 m.moles) de 4-pyridinylcarbamate d'éthyle
[décrit dans J Chem. Soc; (1962), 2379-2381 ] avec la 2-bromo-l-(3-méthoxy-4-
nitrophényl)l-éthanone dans le dichloromethane. On obtient 3,24g d'un précipité blanc.
Rendement : 100%
Spectrométrie de masse (Mode ES+) MH+ = 360,3
ETAPE B Ce composé est préparé selon le même procédé que celui décrit dans l'exemple 47
Etape B à partir de 1,8g (4,1 m.moles) de bromure de 4-[N-(éthoxycarbonyl)amino]-l-
[2-(3-méthoxy-4-nitrophényl)-2-oxoéthyl]pyridinium avec le crotonate de benzyle en présence d'oxyde de manganèse comme oxydant. On obtient 573mg d'une poudre jaune.
Rendement : 26%
Point de fusion : 182°C
Exemple 49
[7-amino-3-(3-méthoxy-4-nitrobenzoyI)-2-méthylindolizin-l-yl]carboxylate de benzyle A 8,34g (14,9 m.moles) de [7-N-[(te^t-butoxycarbonyl)amino]-3-(3-méthoxy-4- nitrobenzoyl)-2-méthylindolizin-l-yl]carboxylate de benzyle en solution dans 40ml dichloromethane, on ajoute 26ml d'acide trifluoroacétique. On agite la solution pendant 2 heures à température ambiante, on verse le milieu réactionnel sur une solution aqueuse saturée de bicarbonate de sodium. On filtre le solide rouge brique obtenu et on le lave abondamment à l'eau puis on le sèche. Le produit est adsorbé sur silice et purifié par chromatographie sur gel de silice en éluant avec dichlorométhane- méthanol (97-3). On obtient 6,3g d'un solide rouge. Rendement : 92% Point de fusion : 260-274°C (décomposition)
Exemple 50
(4-amino-3-méthoxyphényl)[6-amino-l-(4-méthoxyphényl)-2-méthylindolizin-3- l] methanone Ce composé est préparé selon le même procédé que celui décrit dans l'exemple 49 par déboculation du [3-(4-amino-3-méthoxybenzoyl)-l-(4-méthoxyphényl)-2- méthylindolizin-6-yl]carbamate de tert-butyle avec de l'acide trifluoro acétique On obtient 317mg d'un solide jaune.
Rendement : quantitatif Point de fusion : 182°C
Exemple 51
[7-(acétylamino)-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin-l-yl] carboxylate de benzyle Une suspension de 1,5 g (3,26 m.moles) de [7-amino-3-(3-méthoxy-4- nitrobenzoyl)-2-méthylindolizin-l-yl] carboxylate de benzyle dans 10ml d'anhydride acétique est chauffée à 100 °C pendant 15 minutes. Le mélange devient homogène. Le mélange réactionnel est refroidi puis le précipité formé est filtré. Le solide obtenu est lavé avec du dichloromethane et de l'éther isopropylique puis adsorbé sur silice et purifié par chromatographie sur gel de silice en éluant avec un mélange de toluène et d'acétate d'éthyle (100/0 à 50/50). On obtient 994mg d'un solide orange. Rendement : 61 % Point de fusion : 227 °C
Exemple 52
[3-(3-méthoxy-4-nitrobenzoyl)-2-méthyl-7-[(méthylsulfonyl)amino)]indolizin-l- yl] carboxylate de benzyle A 1,22g (2,66 m.moles) de [7-amino-3-(3 -méthoxy-4-nitrobenzoyl)-2- méthylindolizin-1-yl] carboxylate de benzyle dans 15ml de pyridine, on ajoute 410μl
(5,32 m.moles) de chlorure de mésyle. Le mélange réactionnel est chauffé à 70°C pendant 2,5 heures, puis refroidi à température ambiante et concentré sous pression réduite. Le résidu obtenu est dissout dans du dichloromethane et lavé avec une solution molaire d'acide chlorhydrique. Après décantation, la phase organique est lavée avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Le produit est adsorbé sur silice et purifié par chromatographie sur gel de silice en éluant avec un mélange de dichloromethane et de méthanol (9/1). On obtient 348mg d'une poudre marron.
Rendement : 24 % Point de fusion : 221 °C
Exemple 53
[7-[(N-tert-butoxycarbonyI)(N-méthyl)amino]-3-(3-méthoxy-4-nitrobenzoyI)-2- méthyIindolizin-l-yl]carboxylate de benzyle A 1,33g (2,34 m.moles) de [7-[(N-ter£-butoxycarbonyl)amino]-3-(3-méthoxy-4- nitrobenzoyl)-2-méthylindolizin-l-yl] carboxylate de benzyle dans 10ml de tetrahydrofurane, on ajoute par portion 154-mg (3,54 m.moles) d'hydrure de sodium
(60% en dispersion dans l'huile) à température ambiante. Après 15 minutes, on ajoute à la solution 220μl (3,54 m.moles) d'iodure de méthyle. Le mélange réactionnel est agité à 40°C pendant 2 heures, puis refroidi et versé dans une solution molaire d'acide chlorhydrique puis extrait à l'acétate d'éthyle. La phase organique est lavée avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Le produit est adsorbé sur silice et purifié par chromatographie sur gel de silice en éluant avec dichlorométhane/méthanol (9/1). On obtient 505mg d'un solide orange. Rendement : 64 % Point de fusion : 117 °C
Exemple 54
[3-(3-méthoxy-4-nitrobenzoyl)-2-méthyl-7-(méthylamino)indolizin-l- yI]carboxylate de benzyle Ce composé est obtenu selon le même procédé que le composé de l'exemple 49 par déprotection de la fonction amino de 849 mg (1,48 m.mole) de [7-[(N-tert- butoxycarbonyl)(N-méthyl)amino]-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin- 1-yl] carboxylate de benzyle avec de l'acide trifluoroacétique. On obtient 549mg d'une poudre orange.
Rendement : 78% Point de fusion : 228°C
Exemple 55
[7-(diméthylamino)-3-(3-méthoxy-4-nitrobenzoyl)-2-méthylindolizin-l-yl] carboxylate de benzyle A 525mg (1,11 m.mole) de [3-(3-méthoxy-4-nitrobenzoyl)-2-méthyl-7- méthylaminoindolizin-l-yl]carboxylate de benzyle en solution dans 10ml de diméthylformamide, on ajoute par portions 80mg (1,67 m.mole) d'hydrure de sodium (60% en dispersion dans l'huile) à température ambiante. Après 1O minutes, on ajoute 138μl (2,22 m.moles) d'iodure de méthyle au mélange réactionnel. On agite à 40°C pendant 2 heures, on verse dans une solution molaire d'acide chlorhydrique puis on extrait à l'acétate d'éthyle. La phase organique est lavée avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Le solide obtenu est repris dans du dichloromethane et filtré. On obtient 439mg d'un solide orange clair. Rendement : 81 %
Point de fusion : 167 °C
Exemple 56 (4-amino-3-méthoxyphényl)[l-(4-méthoxyphényl)-2-méthylindolizin-3-yl] methanone A 460mg (1,1 m.mole) de (3-méthoxy-4-nitrophényl)[l-(4-méthoxyphényl)-2- méthylindolizin-3-yl] methanone, composé de l'exemple 22, dans 9ml d'éthanol, on ajoute 92mg de Pd/C à 10% puis 1,12ml (10,05 m.moles) de cyclohexène et on chauffe à reflux 4 heures. Le milieu réactionnel est refroidi, filtré sur talc et lave le catalyseur au dichloromethane. Le filtrat est concentré sous pression réduite. Le produit est purifié par chromatographie sur gel de silice en éluant avec un mélange de toluène et d'acétate d'éthyle (9-1 puis 8-2). On obtient 400mg d'une poudre jaune. Le produit est salifié par dissolution de la poudre précédemment obtenue dans le dioxanne puis ajout de 1,18ml (1,2 équivalent) d'acide chlorhydrique IN dans l'éther éthylique. Après addition d'éther éthylique, le précipité obtenu est filtré, lavé à l'éther éthylique puis séché. On recueille 400mg d'une poudre jaune sous forme de chlorhydrate. Rendement : 94%
Point de fusion : 222,5°C
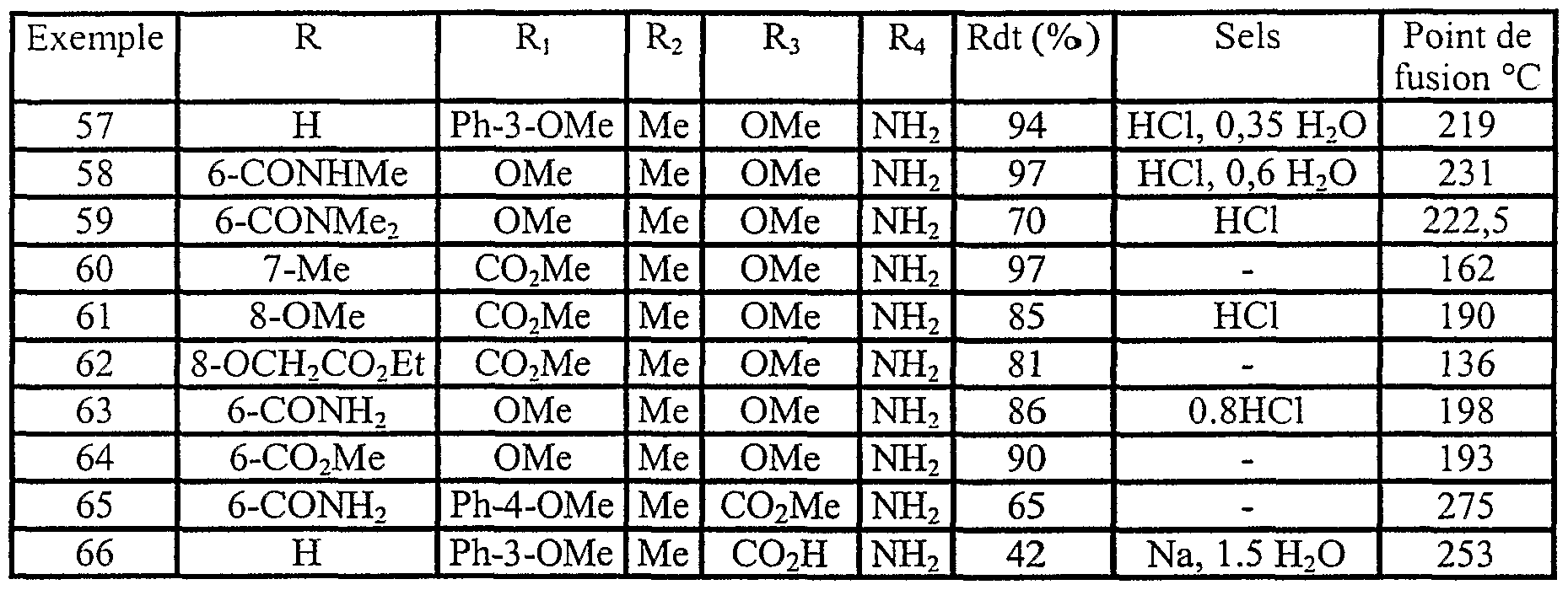
Exemples 57 à 66 En procédant selon la préparation décrite ci-dessus, on synthétise les composés décrits dans le Tableau V ci-dessous, par réduction de la fonction nitro des composés de formule la avec le cyclohexène en présence de Pd/C à 10% comme catalyseur.
TABLEAU V
Exemple 67 [3-(4-amino-3-méthoxybenzoyl)-6-(benzyloxy)-2-méth rlindolizin-l-yl] carboxylate de méthyle A 441mg (0,93 m.mole) de [6-(benzyloxy)-3-(3 -méthoxy-4-nitrobenzoyl)-2- méthylindolizin-1-yl] carboxylate de méthyle en solution dans un mélange de 4ml d'eau et 8ml d'éthanol, on ajoute 0,4ml d'acide acétique et 200mg de fer. On chauffe à 70°C pendant 3 heures puis on laisse revenir à température ambiante avant de verser le milieu réactionnel sur de l'eau et d'extraire au dichloromethane. La phase organique est lavée avec une solution saturée de chlorure de sodium, séchée sur sulfate de sodium puis concentrée sous pression réduite. Les 410mg de solide jaune sont salifiés sous forme de chlorhydrate. On obtient un solide jaune (chlorhydrate) Rendement : 80% Point de fusion : 204°C
Exemples 68 et 69 En procédant selon la préparation décrite ci-dessus, on synthétise les composés décrits dans le Tableau VI ci-dessous, par réduction de la fonction nitro des composés de formule la avec le fer et l'acide acétique dans l'éthanol
TABLEAU VI
Exemple 70 [3-(4-amino-3-méthoxybenzoyl) -l-(4-méthoxyphényl)-2-méthylindolizin-6-yl] carboxamide A 880mg (1.92 m.mole) de [3-(3-méthoxy-4-nitrobenzoyl)-l-(4-méthoxyphényl)-2- méthylindolizin-6-yl]carboxamide dans 15ml de méthanol et 13ml de dichloromethane, on ajoute d'abord 184mg de Pd/C à 10% en suspension dans 4ml de méthanol puis 470μl (9.60 m.moles) d'hydrate d'hydrazine et on agite à température ambiante pendant une nuit. Le milieu réactionnel est filtré sur talc et on lave le catalyseur au méthanol. Le filtrat est concentré sous pression réduite. Le résidu est purifié par chromatographie flash sur colonne de silice en éluant avec un mélange dichloromethane - méthanol (98-2) puis (95-5). On concentre sous pression réduite les fractions pures et on recueille 610mg d'une poudre jaune. Le produit est salifié par ajout d'acide chlorhydrique IN dans l'éther éthylique. Après addition d'éther éthylique, le précipité obtenu est filtré, lavé à l'éther éthylique puis séché. On recueille un solide jaune sous forme de chlorhydrate hydraté (1.35 H2O). Rendement : 74% Point de fusion : 213°C
Exemple 71 à 77 En procédant selon la préparation décrite à l'exemple 70, on synthétise les composés décrits dans le Tableau VII ci-dessous, par réduction de la fonction nitro des composés de formule la avec l'hydrate d'hydrazine en présence de Pd/C à 10%> comme catalyseur.
TABLEAU VII
Exemple 78 [3-(4-amino-3-méthoxybenzoyl)-6-hydroxy-2-méthylindolizin-l-yI]carboxylate de méthyle A 1,0g (2,17 m.moles) de [6-(benzyloxy)-3-(3-méthoxy-4-nitrobenzoyl)-2- méthylindolizin-1-yl] carboxylate de méthyle en suspension dans 30ml de diméthylformamide et 30ml de tetrahydrofurane, on ajoute lOOr g de Pd/C (10%). On agite le mélange réactionnel sous 10 bars d'hydrogène pendant 48 heures. On réalise une ultrafiltration (filtre Millipore™ 5μM) et on lave le catalyseur avec du diméthylformamide. On concentre le filtrat sous pression réduite pour obtenir 780rng d'un solide jaune. Rendement : 91% Point de fusion : 184°C
Exemple 79 Acide [3-(4-amino-3-méthoxybenzoyl)-6-hydroxy-2-méthylimdolizin-l-yl] carboxylique
Ce composé est obtenu selon le même procédé que le composé de l'exemple 78 ci- dessus par débenzylation de la fonction hydroxy et réduction de la fonction nitro, par hydrogénation catalytique sous pression du composé de l'exemple 97 On obtient un solide jaune que l'on salifie sous forme de sel de sodium. On obtient un solide jaune (sel de Na, l,5H2O) Rendement : 70% Point de fusion : 214°C
Exemple 80
Acide [3-(4-amino-3-méthoxybenzoyl)- 8 {2-(diméthylamino) éthoxy}-2- méthy!indolizin-l-yl]carboxylique Ce composé est obtenu selon le même procédé que le composé de l'exemple 78 par réduction de la fonction nitro, par hydrogénation catalytique sous pression du composé de l'exemple 98. On obtient un solide jaune salifié sous forme de sel de lithium hydraté (2,3H2O). Rendement : 76%> Point de fusion : 178°C
Exemple 81
[3-(4-amino-3-méthoxybenzoyl)-6-méthoxy-2-méthylindolizin-l-yl]carboxylate de méthyle A 460mg (1,29 m.mole) de [3-(4-amino-3-méthoxybenzoyl)-6-hydroxy-2- méthylindolizin-l-yl]carboxylate de méthyle en suspension dans 20ml d'acétone, on ajoute 503mg (1,54 m.mole) de carbonate de césium et 123μl (1,29 m.mole) de diméthylsulfate. Le mélange réactionnel est agité à température ambiante pendant 30 minutes puis versé sur de l'eau et extrait avec de l'acétate d'éthyle. La phase organique est lavée avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium puis concentrée sous pression réduite. Le produit est purifié par chromatographie sur gel de silice en éluant avec un mélange de dichloromethane et de méthanol (100/0 à 98/2). On obtient 470mg d'un solide jaune. Rendement : 98%
Point de fusion : 95°C
Exemple 82
[3-(4-amino-3-méthoxybenzoyl)-6-(2-éthoxy-2-oxoéthoxy)-2-méthylindolizin-l- yl]carboxylate de méthyle Ce composé est obtenu en procédant selon la préparation décrite dans l'exemple ci- dessus, par alkylation de [3-(4-amino-3-méthoxybenzoyl)-6-hydroxy-2- méthylindolizin-1-yl] carboxylate de méthyle avec le bromoacétate d'éthyle. On obtient une poudre jaune. Rendement : 84% Point de fusion : 192°C
Exemple 83 acide [7-(acétylamino)-3-(4-amino-3-méthoxybenzoyl)-2-méthylindolizîn-l-yl] carboxylique A 855mg (1,76 m.mole) de [7-(acétylamino)-3-(3-méthoxy-4-nitrobenzoyl)-2- méthylindolizin-1-yl] carboxylate de benzyle en suspension dans 9ml de diméthylformamide, on ajoute 450 mg de Pd/C (10%). On agite le mélange réactionnel sous 10 bars d'hydrogène pendant 5 heures. On filtre sur un lit de silice en éluant avec un mélange de dichloromethane et de méthanol (9-1) puis on concentre le filtrat pour obtenir un solide jaune qui est repris dans l'éthanol, puis filtré et séché. On obtient 561 mg d'une poudre jaune que l'on met en suspension dans 6 ml de méthanol puis on ajoute 1,40 ml de soude IN (1 équivalent). La solution est concentrée sous pression réduite et le solide obtenu est lavé à l'acétone. Après séchage, on obtient 596mg d'un solide vert (sel de Na, 3,7 H2O). Rendement : 84%
Point de fusion : 288-291°C (décomposition)
Exemple 84 à 88 En procédant selon la procédure décrite ci-dessus, on synthétise les composés décrits dans le tableau VIII ci-dessous par hydrogénation sous pression de l'ester benzylique de Rj et réduction du nitro de R3 ou Ri
TABLEAU VIII
Exemple 89
Acide 2-{[3-(4-amino-3-méthoxybenzoyl)-l-(méthoxycarbonyl)-2-méthyl- indolizin-6-yI] oxy} acétique A 600mg (1,36 m.mole) de [3-(4-amino-3-méthoxybenzoyl)-6-(2-éthoxy-2- oxoéthoxy)-2-méthylindolizin-l-yl]carboxylate de méthyle en solution dans 20ml de dioxanne on ajoute 272mg (6,81 m.moles) de soude en pastilles et on chauffe à reflux pendant 1 heure. Le milieu réactionnel est concentré sous pression réduite. Le résidu est repris à l'eau et la solution obtenue est acidifiée à pH 3-4 avec une solution aqueuse à 10%> d'hydrogénosulfate de potassium puis extrait à l'acétate d'éthyle. La phase organique est décantée, lavée avec une solution aqueuse saturée en chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. Les 398mg de solide jaune obtenu sont ensuite salifiés, sous forme de sel de sodium.
On obtient un solide jaune ( sel de Na, l,5H2O) Rendement : 68% Point de fusion : 182°C
Exemple 90 Acide [3-(4-amino-3-méthoxybenzoyl)-l-méthoxy-2-méthylindolizin-6-yl] carboxylique A 225mg (0.61 m.mole) de [3-(4-amino-3-méthoxybenzoyl)-l-méthoxy-2- méthylindolizin-6-yl]carboxylate de méthyle dans un mélange de 20ml de méthanol- dioxanne (1-1), on ajoute 2.01ml (2.01 m.moles) de soude I N et chauffe à 50°C pendant 6 heures. Le milieu réactionnel est concentré sous vide et le résidu est repris avec de l'eau et de l'acétate d'éthyle. La phase aqueuse est décantée, lavée à l'acétate d'éthyle puis acidifiée avec 2ml d'acide chlorhydrique IN.
Le précipité formé est filtré, lavé à l'eau puis séché. Les 189mg de solide orange obtenu sont ensuite salifiés, sous forme de sel de sodium.
On obtient un solide orange ( sel de Na) Rendement : 87% Point de fusion : 290°C
Exemple 91 Acide [3-(4-amino-3-méthoxybenzoyl)-6-(benzyloxy)-2-méthyIindolizin-l-yl] carboxylique A 800mg (1,79 m.mole) de [3-(4-amino-3-méthoxybenzoyl)-6-(benzyloxy)-2- méthylindolizin-l-yl]carboxylate de méthyle en solution dans 30ml de dioxanne et 10ml de méthanol on ajoute 358mg (8,95 m.moles) de soude en pastilles et on chauffe à reflux pendant 16 heures. Le milieu réactionnel est concentré sous pression réduite. Le résidu est repris à l'eau et la solution obtenue est acidifiée à pH 5 avec une solution aqueuse à 10% d'hydrogénosulfate de potassium. Le précipité formé est filtré, lavé à l'eau puis séché. Les 760mg de solide jaune obtenu sont ensuite salifiés, sous forme de sel de sodium.
On obtient un solide jaune (sel de Na) Rendement : 98% Point de fusion : 258°C
Exemples 92 et 93 En procédant selon la procédure décrite ci-dessus, on synthétise les composés de formule Ij décrits dans le tableau IX ci-dessous par saponification de la fonction ester contenue dans le substituant Ri des composés de formule Ib avec de la soude.
TABLEAU IX
Exemple 94 Acide [3-(4-amino-3-méthoxybenzoyl)-6-(carbométhoxy)-2-méthylindolizin-l- yl] carboxylique A 800mg (1,81 m.mole) de [3-(4-amino-3-méthoxybenzoyl)-6-(2-éthoxy-2- oxoéthoxy)-2-méthylindolizin-l-yl] carboxylate de méthyle en solution dans 40ml de dioxanne et 20ml de méthanol on ajoute 1,81g (45,40 m.moles) de soude en pastilles et on chauffe à reflux pendant 72 heures. Le milieu réactionnel est refroidi puis acidifié à pH 5 avec une solution aqueuse à 10% d'hydrogénosulfate de potassium. Le précipité formé est filtré, lavé à l'eau puis séché. Les 390mg de solide jaune obtenu sont ensuite salifiés, sous forme de sel de sodium. On obtient un solide jaune ( disel de Na, 4H2O) Rendement : 54% Point de fusion : 273°C
Exemple 95 Acide [3-(4-amino-3-méthoxybenzoyl)-8-(carbométhoxy)-2-méthylindolizin-l- yl] carboxylique Ce composé est obtenu en procédant selon la préparation décrite dans l'exemple 94 ci-dessus, par saponification des 2 fonctions ester du composé [3-(4-amino-3- méthoxybenzoyl)-8-(2-éthoxy-2-oxoéthoxy)-2-méthylindolizin- 1 -yl]carb oxylate de méthyle avec de la soude. On obtient après salification une poudre jaune (disel de Na, 3H2O) Rendement : 45% Point de fusion : 268°C
Exemple 96
Acide [3-(4-amino-3-méthoxybenzoyI)-6-(méthoxy)-2-méthylindolizin-l-yl] carboxylique A 420mg (1,14 m.mole) de [3-(4-amino-3-méthoxybenzoyl)-6-(méthoxy)-2- méthylindolizin-1-yl] carboxylate de méthyle dissous dans 12ml de dioxanne, on ajoute 48mg (11,4 m.moles) d'hydroxyde de lithium. Le mélange réactionnel est chauffé à 70°C pendant 13 heures puis refroidi à température ambiante, versé dans de l'eau et extrait à l'acétate d'éthyle. La solution aqueuse est acidifiée jusqu'à pH 5 par ajout d'une solution aqueuse d'hydrogénosulfate de sodium à 10%>. On extrait avec de l'acétate d'éthyle. On lave la phase organique avec une solution saturée de chlorure de sodium, on la sèche sur sulfate de sodium puis on la concentre sous pression réduite. Les 232mg de solide jaune sont ensuite salifiés sous forme de sel de sodium, l,5H2O Rendement : 80% Point de fusion : 242°C
Exemples 97 et 98 En procédant selon la procédure décrite ci-dessus, on synthétise les composés de formule Ij décrits dans le tableau X ci-dessous par saponification de la fonction ester contenue dans le substituant Ri des composés de formule la avec de la soude.
TABLEAU X
Exemple 99 acide 2-amino-5-({l-méthoxy-2-méthyl-6-[(méthylarnino)carbonyl]indolizin-3- yl}carbonyl)benzoique A 710mg (1,44 m.moles) de 5-({l-méthoxy-2-méthyl-6-[(méthylamino) carbonyl]indolizin-3-yl}carbonyl)-2-[2,2,2-trifluoroacétyl)amino] benzoate de méthyle en solution dans 12ml de dioxanne, on ajoute 3,03ml d'une solution de soude IN. On chauffe le milieu réactionnel à 60°C pendant 16 heures, puis laisse revenir à température ambiante et concentre sous pression réduite. Le résidu est repris à l'eau, et
on lave à l'éther éthylique. Après décantation, on acidifie la phase aqueuse avec une solution molaire d'acide chlorhydrique et on extrait à l'acétate d'éthyle. La phase organique est décantée, lavée à l'eau, séchée sur sulfate de sodium et concentrée sous pression réduite Les 400mg de solide jaune sont ensuite salifié sous forme de sel de sodium dihydraté (2 H2O). Rendement : 73% Point de fusion : 311°C
Exemples 100 et 101 En procédant selon la procédure décrite ci-dessus, on synthétise les composés de formule le décrits dans le tableau XI ci-dessous par hydrolyse basique de la fonction ester et déprotection de la fonction amino contenue dans les substituant R3 et/ouR4 des composés de formule le avec de la soude.
TABLEAU XI
Exemple 102 Acide 2-amino-5-{[2-méthyl-l-(2-thiényl)indolizin-3-yl]carbonyl}benzoïque A 0.29g (0.74 m.mole) de 2-amino-5-{[2-méthyl-l-(2-thiényl)indolizin-3- yl]carbonyl}benzoate de méthyle dans un mélange de 16ml de dioxane-méthanol (1-1) on ajoute 2.34ml (2.34 m.moles) de soude IN et chauffe à 70°C pendant 6 heures. Le milieu réactionnel est évaporé à sec. Le résidu est repris à l'acide chlorhydrique IN et extrait à l'acétate d'éthyle. La phase organique est lavée à l'eau, séchée sur sul fate de sodium et évaporée sous pression réduite. On purifie le produit par chromatographie flash sur gel de silice en éluant avec un mélange de dichlorométhane-méthanol (98-2). Les 280mg de solide jaune obtenu sont ensuite salifiés sous forme de sel de sodium, 1.25 H2O Rendement : quantitatif Point de fusion : 294°C
Exemples 103 à 109 En procédant selon la procédure décrite ci-dessus, on synthétise les composés de formule le décrits dans le tableau XII ci-dessous par saponification de la fonction ester contenue dans les substituant R ou des composés de formule Ib et éventuellement de la fonction ester du substituant R avec de la soude IN.
TABLEAU XII
Exemple 110 2-amino-5-[(l-méthoxy-2-méthyIindolizin-3-yI)carbonyl]benzamide A 500mg (14,4 m.moles) 2-amino-5-[(l-méthoxy-2-méthylindolizin-3- yl)carbonyl] benzoate de sodium dans 8ml de diméthylformamide, on ajoute 702,4mg (15,9 m.moles) de benzotriazol-l-yloxy-tris(diméthylamino)phosphonium hexafluorophosphate (BOP) et agite une heure à température ambiante puis on fait barboter dans le milieu réactionnel un courant d'ammoniac pendant 5 minutes et on laisse agiter à température ambiante une nuit. On ajoute de l'eau et on extrait à l'acétate d'éthyle. La phase organique est lavée avec une solution aqueuse saturée de chlorure de sodium, séchée sur sulfate de sodium et concentrée sous pression réduite. On purifie le produit par chromatographie flash sur gel de silice en éluant avec un mélange de dichloromethane et de méthanol (99-1 puis 98-2). Après évaporation on cristallise le produit dans l'éther éthylique. On obtient 3 lOmg d'une poudre jaune. Rendement : 67% Point de fusion : 156°C
Exemple 111 2-amino-N-hydroxy-5-[(l-méthoxy-2-méthylindolizin-3-yl)carboπyl]benzamide Ce composé est obtenu en procédant selon la préparation décrite dans l'exemple 1 10 ci-dessus, par couplage de l'hydroxylamine sur la fonction acide du composé 2- amino-5-[(l-méthoxy-2-méthylindolizin-3-yl)carbonyl] benzoate de sodium après activation de celle-ci au BOP
On obtient une poudre jaune.
Rendement : 71% Point de fusion : 205 °C
Exemple 112
Etude de la liaison 125I - b-FGF au Récepteur purifié FGF R α Hic par la méthode de scintillation de proximité Des plaques NBS (NBS plate 96 well solid white CORNING 3600) sont coatées avec 100 μl de gélatine 0.1 % par puit, 2 heures à 37°C. Au terme de l'incubation, on élimine le coating, on rince et on sèche bien les plaques. On distribue 100 μl de tampon de binding (Tampon Bis Tris 40 mM pH 7.0) dans les plaques. Les dilutions des composés de l'invention sont réparties dans les puits à raison de 10 μl / puit. On distribue ensuite 10 μl / puit de b-FGF (AMERSHAM ARM 35050) et 10 μl / puit de FGF R α III c (R&D Systems 658 FR). On ajoute après 10 μl / puit de 125I - bFGF (Dupont NEN NEX 268 - Activité spécifique > 70 μCi) et 50 μl / puit des billes SPA (AMERSHAM RPQN 00019). On agite quelques secondes la plaque et on l'incube 60 minutes à 37°C et à l'abri de la lumière.
Au terme de l'incubation, la plaque est lue dans un compteur de radioactivité MIBROBETA TRILUX (: WALLAC - PERKINELMER)
Les composés de l'invention ont démontré une activité spécifique comprise entre
10"6M et lO"9M.
Exemple 113
Effets des composés de la formule I sur la prolifération d'HUVECs versus 30 ng/ml de b-FGF ou 10 ng/ l de a-FGF
Coater les plaques 24 puits (FALCON PRIMARIA) avec 200 μl d'une solution de fibronectine (50 μg / ml préparée dans du PBS) / puits. Ensemencer à raison de 30000 cellules / ml / puit dans un milieu RPMI 1640 + 10 % SVF + 1% Glutamine + mélange Héparine-ECGF (HE). Incuber à 37 °C , 5 % CO2 le temps que les cellules adhèrent.
Dissoudre les produits et préparer des solutions dans le DMSO / milieu réactionnel ayant une concentration finale de 1 μM final à 10"7M. Après adhésion des cellules pendant 6 heures à 37 °C en présence de 5% C02, le milieu est remplacé par du RPMI 1640 0.1 % SVF + Glutamine +HE.
Pour la dérivatisation, on utilise comme contrôle négatif 0.1 % SVF, comme contrôle positif 0 % SVF et comme témoin 0.1 % SVF + 30 ng/ml de b-FGF ou 10 ng/ml de a-FGF. On incube ensuite 24 heures à 37 °C en présence de
5 % CO2.
Le deuxième jour les cellules sont rincées par 1 ml de PBS et 200 μl de trypsine, puis elles sont récupérées dans l'isoton. On procède à la numération (n> 9μm)
Dans ce test de prolifération des cellules endothéliales induite par le b-FGF ou le a- FGF les composés de l'invention ont démontré une activité spécifique comprise entre 10"5M et lO"9M.
Exemple 114
Modèle d'angiogenese in-vitro Préparer les gels en distribuant dans chaque puits de chamberslide (Biocoat
Cellware rat tail collagen, Type I, 8-well culturesides: Becton Dickinson 354630) 160 μl de matrigel dilué au 1/6 (Growth factor reduced Matrigel: Becton Dickinson
356230) dans du collagen (Rat Tail Collagène, type I: Becton Dickinson 354236).
Laisser gélifier pendant 1 heure à 37°C. Ensemencer les cellules endothéliales veineuses humaines (HUVEC ref:C-015-10C
- cascade Biologics, INC) ou artérielles de porc (PAEC) à 15.103 cellules/puits dans 400 μl de milieu EBM (Clonetics C3121) + 2% FBS + hEGF 10 μg/ml pour les
HUVEC et DMEM + 3% SVF + Glutamine 2 mM + Sodium pyruvate 1 mM + Acides
Aminés Non Essentiels 1% (GIBCO) pour les PAEC.
Stimuler avec le b-FGF (TEBU/Peprotech) 10 ng/ml ou le a-FGF (TEBU/Peprotech) 10 ng/ml en présence ou non des produits de l'intention pendant 24h à 37°C en présence de 5% CO2. Après 24 heures, fixer les cellules et colorer la lame au trichrome de Masson avant observation au microscope objectif X4 et analyse d'image (BIOCOM- logiciel Visiolab 2000).
Pour le test d'angiogenese in vitro induite par le b-FGF ou le a-FGF, les composés 7 1 1 de l'invention ont démontré une activité spécifique comprise entre 10" M et 10" M.
Exemple 115 Modèle d'angiogenese inflammatoire chez la souris L'angiogénèse est requise pour le développement des maladies inflammatoires chroniques comme l'arthrite rhumatoïde, les IBD, mais aussi pour le développement des tumeurs solides. La formation de nouveaux vaisseaux permet non seulement la perfusion des tissus pathologiques mais également le transport de cytokines responsables de l'établissement de la chronicité de la maladie. Le modèle décrit par Colville-Nash P et al (D. JPET., 1995, vol 274 n°3, ppl463- 1472) permet d'étudier des agents pharmacologiques susceptibles de moduler l'apparition de l'angiogénèse.
Les animaux, souris blanches non consanguines de 25g environ sont anesthésiés avec du pentobarbital sodique (60 mg/kg ; Sanofi Nutrition Santé animale) par voie intrapéritonéale. Une poche d'air est créée sur le dos de la souris par injection de 3 ml d'air en sous- cutanée. Après le réveil, les animaux reçoivent un traitement en général par gavage et reçoivent une injection de 0.5 ml d'adjuvant de Freund (Sigma) avec 0.1 % d'huile de croton (Sigma) dans la poche. Sept jours après, les souris sont à nouveau anesthésiées et placées sur une plaque chauffante à 40°C. Un ml de rouge carmin (5% dans 10 % de gélatine - Aldrich Chemicals) est injecté à la veine de la queue. Les animaux sont ensuite mis à 4°C pendant 2-3 heures. Les peaux sont ensuite prélevées et mises à sécher pendant 48 heures dans une étuve à 56°C. Les tissus secs sont pesés et mis dans 1.8 ml de tampon digestif
(dithiothreitol 2 mM, Na2HPO4 20mM, EDTA 1 mM, papaïne 12U/ml) pendant 24 heures. Le colorant est alors dissout dans 0.2 ml de NaOH 5M. Les peaux sont centrifugées à 2000g pendant 10 mn. Les surnageants sont filtrés sur membranes d'acétate de cellulose 0.2 μm. Les filtrats sont lus dans un spectrophotomètre à 492 nm. contre une gamme étalon de rouge carmin.
Deux paramètres sont étudiés : le poids sec du granulome et la quantité de colorant après digestion des tissus.
Les résultats sont exprimés en valeurs moyennes (± sem). Les différences entre les groupes sont testées avec une ANOVA suivie d'un test de Dunnet dont le groupe de référence est le groupe "témoin solvant". Les composés de l'invention sont actifs par voie orale à des doses de 0.1 à lOOmg/kg.
Exemple 116 Modèle d'angiogenese MATRIGEL chez la souris Le modèle décrit par Passaniti and al. (Laboratory Investigation (1992) 67 (4) pp519-524) permet d'étudier des agents pharmacologiques susceptibles de moduler l'apparition de l'angiogénèse spécifiquement induite par le bFGF. A du Matrigel (Beckton Dickinson) maintenu sous forme liquide à 4°C est ajouté du FGF2 (Peprotecch) à raison de 300 ng/ml. Après homogénéisation, le mélange (0.5ml) est injecté par voie sous-cutanée dans le bas du dos de souris noires femelles(C57/B16) de 20g environ préalablement anesthésiées avec du pentobarbital sodique (60 mg/kg ; Sanofi Nutrition Santé animale) par voie intrapéritonéale. Les animaux sont traités par gavage. Après 5 jours les souris sont à nouveau anesthésiées et la peau du bas du dos est prélevée, à ce stade, les différences qualitatives de vascularisation du granulome sont évaluées (scorées) et les granulomes sont photographiés. Un dosage de DNA dans les granulomes est ensuite effectué pour quantifier la cellularité de celui-ci. Pour cela, les granulomes isolés sont digérés par de la collagénase (3mg/ml) toute une nuit à 37°C. Après centrifugation à 850 g durant 10 min, le surnageant est écarté et le culot est remis en solution dans 1.2 ml de tampon PBS contenant 1 mM CaCl2, 1 mM
MgCL et 5 mM de glucose. La quantité de DNA présente est mesurée à l'aide d'un kit (Cyquant-GR®, Molecular probe) suivant les instructions du fournisseur.
Les résultats sont exprimés en valeurs moyennes (± sem). Les différences entre les groupes sont testées avec une ANOVA suivie d'un test de Dunnet dont le groupe de référence est le groupe "témoin solvant".
Pour les études histologiques, les granulomes sont prélevés avec le muscle et la peau, fixés une nuit dans une solution de formaldéhyde à 10% et inclus dans de la paraffine (Embedder Leica®). Les granulomes sont ensuite coupés à l'aide d'un microtome (Leica) et colorés avec le colorant Trichrome de Mason. La néo vascularisation des granulomes est alors évaluée. Les niveaux de vascularisation sont compris entre une valeur 0 et 5.
Les composés de l'invention sont actifs par voie orale à des doses de 0.1 à lOOmg/kg
Exemple 117 Modèle d'angiogenese tumorale chez la souris Ce modèle permet d'étudier des agents pharmacologiques susceptibles de moduler l'apparition de l'angiogénèse spécifiquement induite par le développement tumoral.
Des souris C56/B16 de 20g environ sont anesthésiées avec du pentobarbital sodique (60 mg/kg ; Sanofi Nutrition Santé animale) par voie intrapéritonéale. Les tumeurs sont établies par injection sous cutanée sur le dos de cellules Lewis Lung de souris à raison de 2 105 cellules/souris. Après 5 jours, les souris sont traitées tous les jours par gavage. La taille des tumeurs est mesurée 2 fois par semaine durant 21 jours et le volume tumoral est calculé en utilisant la formule : [π/6(α>ι x ω2 x ω2)], où α>ι représente le plus grand diamètre et ω2 représente le plus petit diamètre.
Les résultats sont exprimés en valeurs moyennes (± sem). Les différences entre les groupes sont testées avec une ANOVA suivie d'un test de Dunnet dont le groupe de référence est le groupe "témoin solvant". Les composés de l'invention sont actifs par voie orale à des doses de 0.1 à lOOmg/kg
Exemple 118 Effet sur la Thrombopénie La thrombopénie reste une pathologie pour laquelle il existe peu de traitements efficaces à part la transfusion de concentrés plaquettaires et la thrombopoïétine
(Kaushansky, K. New EngJMed (1998), 339, pp746-754). La chimiothérapie anti-cancéreuse constitue une des causes majeures de thrombopénie. Un des agents de chimiothérapie, le carboplatine., a été largement utilisé pour induire une thrombopénie chez la souris et pouvoir ainsi caractériser l'effet de composés capables d'améliorer le taux de plaquettes comme par exemple la thrombopoïétine (Hokom MM et al, Blood (1995), 86, ρp4486-4492).
150 mg/kg de carboplatine ont été administrés par voie intrapéritonéale à des souris balbC ayant un poids de 20g. Un prélèvement sanguin est effectué périodiquement par ponction rétro-orbitale et le taux de plaquettes circulantes est déterminé par un automate d'hématologie (MS9™ de Melet-Schloesing Laboratoires, Cergy-Pontoise, France). Dans ces conditions, on observe une thrombopénie réversible avec un nadir situé 9 à 10 jours après l'administration de carboplatine (diminution du taux de plaquettes circulantes de 50-60%).
Les composés selon l'invention ou leur solvant (un blanc - témoin) sont administrés par voie orale pendant 5 jours en commençant le traitement 7 jours avant l'administration de carboplatine. Les expériences sont effectuées sur des lots comprenant 10-12 souris et les résultats sont exprimés en moyenne ± erreur standard. Dans ces conditions, les composés de l'invention augmentent le taux de plaquettes circulantes à des doses de 0.1 à 100 mg/kg.
Exemple 119 Modèle de CNV (choroïdal neo vascularization) induite par laser argon chez la souris : Un caractère majeur de la perte de transparence oculaire est la néo-vascularisation et les hémorragies consécutives qui causent des désordres fonctionnels importants au niveau de l'œil et qui se traduisent par une cécité précoce. Récemment, l'étude des mécanismes impliqués dans les phénomènes de néo-vascularisation oculaire a permis de mettre en évidence l'implication de facteur pro-angiogéniques dans ces pathologies.
Le modèle de néo-angiogenèse choroïdienne induite par laser décrit par Rakic JM ét al, dans Invest Ophthalmol Vis Sci. (2003) Jul;44(7), pp3186-3193) permet d'étudier des agents pharmacologiques susceptibles de moduler la néo-vascularisation de la choroïde. Les souris sont anesthésiées par injection intrapéritonéale d'Avertin™. Les deux pupilles sont dilatées avec une solution à 1%> de tropicamide en application topique, et trois lésions sont réalisées autour du disque optique à l'aide d'un laser argon (532 nm; diamètre "spot size" 50 μm; durée 0.05 sec; 400 mW) . Le disque optique est ensuite recouvert d'une lentille.
14 jours après, les souris sont sacrifiées et les yeux énucléés et fixés dans un tampon contenant 3.5% de Formalin™, enrobés dans du Tissue TeK™ (Miles Laboratories, Naperville, Illinois) et congeler dans l'azote liquide de manière à pouvoir réaliser des coupes à l'aide d'un cryostat.
La quantification de la néo-vascularisation choroïdienne a été réalisée par une étude morphométrique quantitative permettant d'évaluer l'épaisseur du réseaux de néovaisseaux présents au niveau de la choroïde, à l'aide d'un système d'analyse d'image assisté par un ordinateur (Olympus Micro Image version 3.0 for Windows 95/NT, Olympus Optical CO. Europe GmBH).
La néo-vascularisation est estimée par le rapport (B/C) de l'épaisseur de la couche pigmentée de la choroïde au niveau de la lésion (B) à l'épaisseur de cette même couche pigmentée dans une région adjacente à la lésion (C). Les résultats sont exprimés en valeurs moyennes (± sem). Les différences entre les groupes traités et les groupes contrôle sont testées avec une ANOVA suivi d'un test Dunnet dont le groupe de référence est le groupe "témoin solvant".
Les composés de l'invention sont actifs par voie orale à des doses de 0.1 à 100 mg/kg.