BIOFLAVONOL-GLYKOSID-PERESTER und ihre AUFARBEITUNG zu PHARMAKOLOGISCH WIRKSAMEN
KONZENTRATEN und ULTRAMIKROEMULSIONEN
EINLEITUNG
In der Patentanmeldung CH-2088/95 werden Ultramikroemulsionen aus spontan dispergierbaren Konzentraten mit antitumoral, antiviral und viruzid wirksamen Estern von ausgewählten Bioflavonoid-Verbindungen beschrieben. Dank der aufgezeigten Solubilisierung der Wirksubstanzen in der Form von thermostabilen Öl-in-Wasser Emulsionen mit sehr kleinen Mizellen im untersten nm-Bereich lässt sich eine ausgezeichnete Bioverfügbarkeit erzielen. Im Laufe einer intensiven Bearbeitung dieser Verbindungsklasse hat sich erwiesen, dass auch ihren Glykosid-Perestern durchaus vergleichbare pharma- kologische Eigenschaften zukommen, wenn sie angemessen formuliert worden sind. Sie lassen sich dann sowohl in kurativer, wie auch in vorbeugender Hinsicht nutzen. Weil sie nicht-toxisch und dennoch hoch aktiv sind, öffnet sich für diese noch wenig erforschte Naturstoffgruppe dank der Anwendung des Fettschwanzprinzips für die Esterbildung und der Überführung dieser Glykosidester in erfindungsgemässe spontan dispergierbare Konzentrate - wie sie auch bereits in der Patentschrift CH 683'426 und in der CH-Patent- anmeldung 2239-95 im Grundsatz gekennzeichnet wurden - ein breites therapeutisches Fenster. Gegenstand der vorliegenden Erfindung sind demzufolge die nachstehend umschriebenen Flavonolglykosid-Perester selber, ihre Einarbeitung in pharmazeutisch anwendbare Darreichungsformen, sowie ihre Verwendung als Mittel mit Wirksamkeit gegen Tumore, Ekzeme, Psoriasis, virale und parasitäre Erkrankungen, wie auch gegen Stoffwechselstörungen.
BESCHREIBUNG der Erfindung 1.0 Bestimmung des U fangs
Unter Flavonol-Glykosid-Perestern werden für diese Erfindung folgende Ver¬ bindungen verstanden:
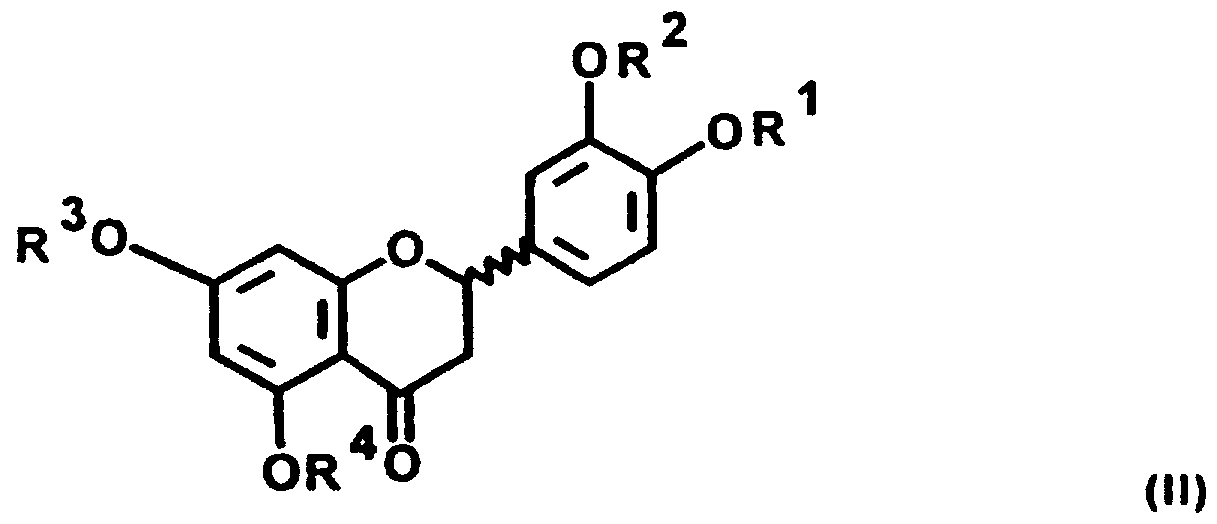
1. QUERCETIN (R1 - R5 = H); Merck-Index 11.8047
2. SPIRAEOSID (R1, R3 - R5 = H; R2 = Glucose) 4'-ß-D-Glucopyranosidoquercetin
3. ISOQUERCETIN (R2 - R5 = H; R1 = Glucose) 3-ß-D-Glucopyranosidoquercetin
4. QUERCITRIN (R2 - R5 = H; R1 = Rhamnose) 3-α-L-Rhamnopyranosidoquercetin; Merck-Index 11.8044
5. RUTIN (R2 - R5 = H; R1 = Rutinose)
3-(α-L-Rhamnopyranosyl (1-. 6)-ß-glucopyranosyl]-quercetin 3-α-L-Rutinosylquercetin; Merck-Index 11.8276
6. ERIODICTYOL (R1 - R4 = H); Merck-Index 11.3616
7. ERIODICTIN (R1, R2, R4 = H; R3 = Rhamnose) 7-α-L-Rhamnopyranosido-eriodictyol
8. HESPERIDIN (R2, R4 - H; (R1 = CH3; R3 = Rutinose) 7-ß-Rutinosyl-4'-methyl-eriodictyol; Merck-Index 11.4591 ,
wobei für alle R=H eine gesättigte oder eine ungesättigte, geradkettige Carbonsäure vom Typus CG-22-Alkyl, C6-22-Alkenyl oder Cc -22-Alkapolyen steht.
1.1 Grundlagen
Ausgewählte Derivate von QUERCETIN laut Formel (l),i mit den Glycosiden 2, 3, 4, 5, sowie seines biogenetischen Vorläufers ERIODICTYOL (II), 6 mit den Glycosiden 7 und 8 wurden in die Untersuchung einbezogen, weil diese Verbindungen erstens in Dicotyledonen weitverbreitet vorkommen (vgl. die Tabellen 8.15 und 11.1 in "The Flavonoids", Ed. J.B. Harborne, T.J. Mabry, H. Mabry, Acade ic Press, N.Y. 1975) und teilweise auch leicht zugänglich sind, und zweitens, weil mehrere davon zudem medizinisch eingehend untersucht worden sind (vgl. V. Cody, E. Middleton, J.B. Harborne: "Plant Flavonoids in Biology and Medicine", A.R. Liss, N.Y., 1986). So sind die Verbindungen 4, 7 und 8 Bestandteil des früher intensiv untersuchten "Vitamins P" (Permeabilitätsvitamin); vgl. H. Vogel: "Chemie und Technik der Vitamine", Enke, Stuttgart, 1940, sowie H. Wagner in "Recent Advances in Phytochemistry", Vol. 12, S. 589; Ed. T. Swain, J.B. Harborne, C.F. VanSumere; Plenum Press, N.Y., 1979. Vgl. auch Jirina Spilkova und Josef Hubik: Biologische Wirkungen von Flavonoiden. Pharmazie in unserer Zeit, 1/1988, 1-9; 4/1992, 174-182.
1.2 Bedeutung der angemessenen Solubilisierung der Wirkprinzipien
Wie im CASE CH-2088/95 für Bioflavonoid-Ester dargelegt, führt die Befolgung des Fettschwanzprinzips bei der Bildung der Derivate zu tiefgreifenden physikochemischen Veränderungen und vorab zu einer wesentlich verstärkten Lipophilie, verbunden mit einer (im Vergleich zu den Grundkörpern) beachtlichen Senkung der Schmelzpunkte; die dergestalt erleichterte Emulgierung setzt sich in eine sehr gute Stabilität der daraus hergestellten, wässerigen Ultramikroemulsionen um. Massgeblich verbessert werden demzufolge auch die Abgabe der Trägermizellen und die gezielte Freisetzung der darin enthaltenen Wirkstoffe, als Bioverfügbarkeit verstanden. Auf diese Weise wird für die erfindungsgemässen Wirkprinzipien volle Bioreaktivität erreicht.
1.3 Herstellung der Perester
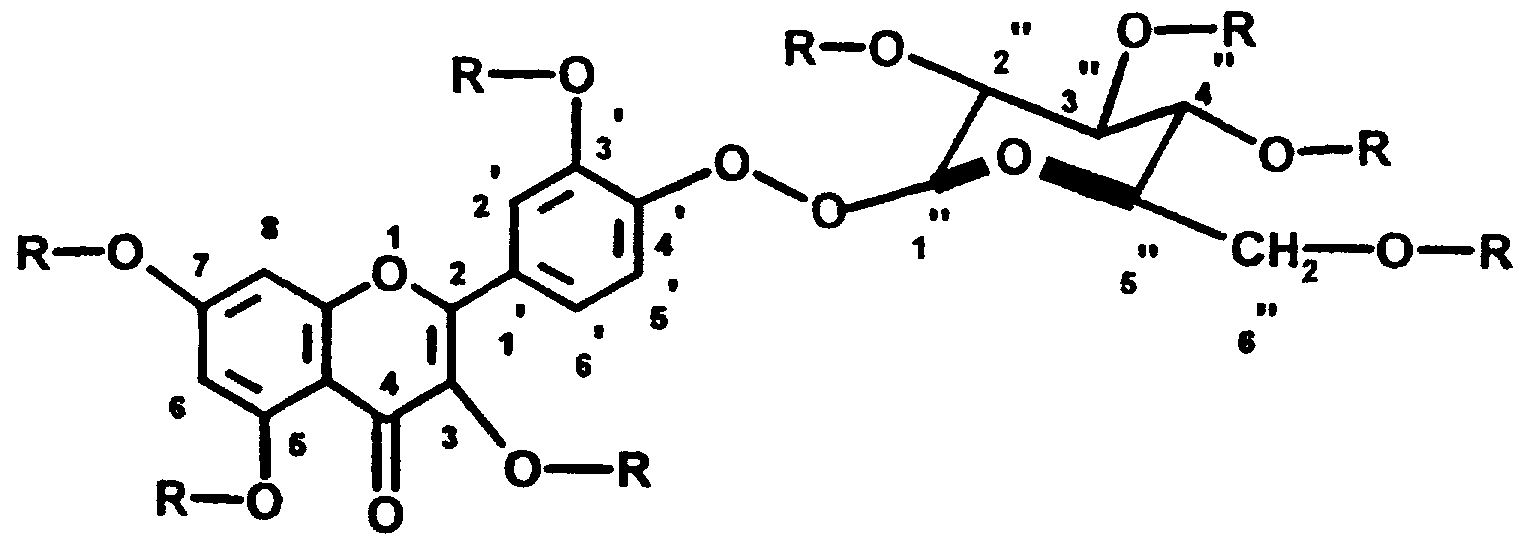
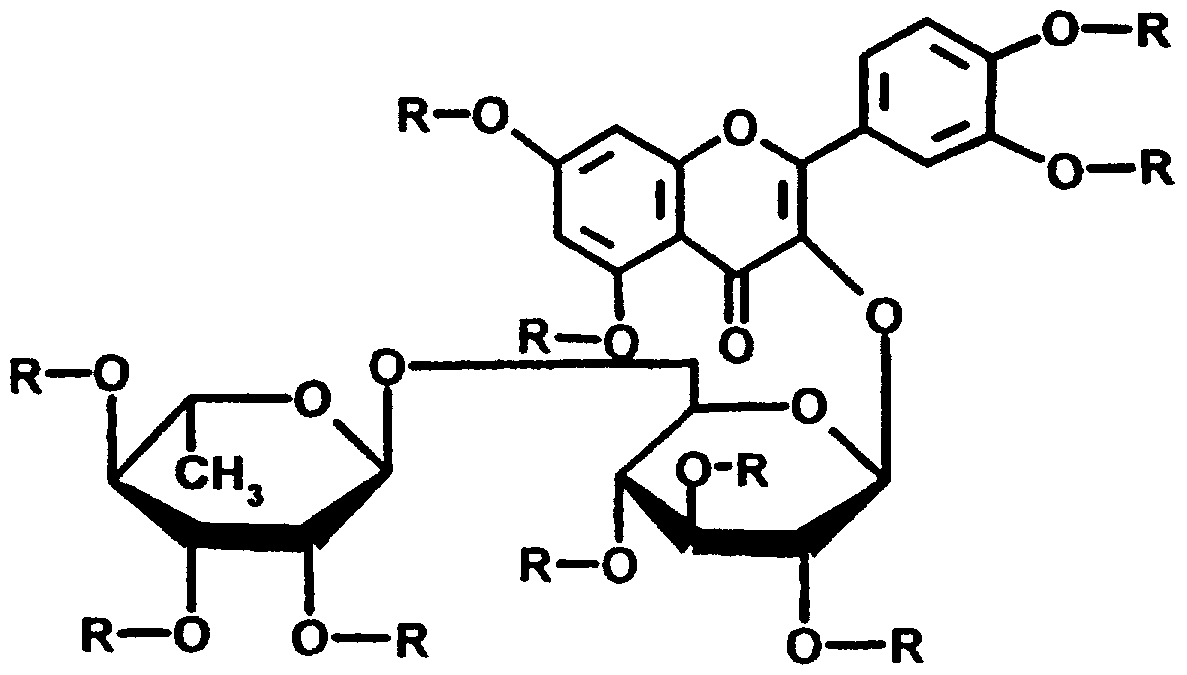
Das neue Verfahren wird auf natürlich vorkommende und in Pflanzen weitverbreitete Flavonolglykoside angewandt. Die Glykosidperester-Verbindun- gen der Formeln (III) und (IV) :
(III)
(IV) lassen sich nach folgenden Verfahren herstellen: 1.31 Fettsäureester von Spiraeosid. laut Formel (I), 2.
Als Spiraeosid wird ein Flavonolglykosid bezeichnet, das zu ca. 1,2% in den getrockneten Blüten von Spiraea Ulmaria L., sive Filipendula Ulmaria (L.) Maxim, vorkommt. Seine Entdeckung erfolgte durch E. Steinegger und P. Caspa- ris [Pharm. Acta Helv. 1945. 20. 154, 174]; spätere Untersuchungen brachten ein umfangreiches Vorkommen im Pflanzenreich zutage, vor allem in solchen Pflanzen, die seit alters für eine volksmedizinische Verwendung bekannt sind, so z.B. zu 1% in Zwiebelschalen lAllium Cepa L.) [K. Hermann, Natur- wiss. 1956. 43. 158; Arch. Pharm. 1958. 291. 238]; zu 3% in getrockneten Blüten von Hamamelis iaponica S. 8ι Z. [L. Hörhammer und R. Griesinger, Na- turwiss. 1959. 46. 427], in den Samen der Rosskastanie (Aesculus Hippocas- tanum L. [J. Wagner, Naturwiss. 1960. 47. 158], in den Blättern verschiedener Hamamelidaceen [K. Egger und H. Reznik, Planta, 1961. 57. 239] und in den Blüten des Stechginsters (Ulex europaeus L. [J.B. Harborne, Phytochem. 1962. i. 203].
Aus neueren Arbeiten sei das Vorkommen erwähnt in Blüten von gärtnerisch verwendeten Fuchsia-Hvbriden [Y. Yasaki, Botanical Magazine (Tokyo) 1976.
89. 45], sowie in zahlreichen Hybriden von Gartenrosen [K. Nayeshiro und CH. Eugster, Helv.Chim.Acta 1989. 72. 985]. In diesen zuletzt angeführten Fällen trägt Spiraeosid entscheidend zur Stabilisierung der farbgebenden An- thocyaninkomplexe bei.
Trotz des verbreiteten Vorkommens in volksmedizinisch verwendeten Pflanzen ist jedoch reines Spiraeosid wissenschaftlich noch kaum auf seine eigentliche Rolle in den populären Anwendungen und schon gar nicht als pharmazeutisch verwendbare Wirksubstanz untersucht worden. Die Struktur von Spiraeosid als 4'-ß-D-Glucopyranosylquercetin ist von E. Steinegger et al. (loc.c.) und von L. Hörhammer und R. Hansel, Arch. Pharm. 19S4. 287. 36 mit klassischen Methoden festgelegt worden. Eine analytische Nachprüfung mittels modernen Geräten, im Zusammenhang mit der Reinheitskontrolle des als Ausgangsmaterial für die erfindungsgemässe Esterbildung frisch isolierten Spiraeosids, ergab die Bestätigung der Struktur gemäss Formet (V) :
(V) Genügende Mengen von (V) lassen sich aus den erwähnten Quellen durch relativ einfache Anreicherungsschritte gewinnen. Als Ausgangsmaterial wurde die kommerziell erhältliche Droge 'Flos Spiraeae Ulmariae' eingesetzt. Durch milde Extraktion, Verteilungsverfahren, Säulenchromatographie und Kristallisation wurde die Verbindung (V) isoliert und anschliessend unter Einsatz von Fettsäurechloriden zu (VI) verestert. Bemerkenswert ist die gegenüber den Quercetinestern erhöhte Stabilität der Esterfunktion an C(5).
Isolierung von Spiraeosid (V)
500 g 'Flos Spiraeae' (Dixa St.Gallen, gerebelt) werden im Perkolator mit 4 L Methanol-Wasser 4:1 während 2 Tagen stehen gelassen. Das erhaltene, gelblichbraune Perkolat wird im Vakuum auf ca. 250 ml eingeengt und darauf 4 x mit je 250 ml n-Butanol im Scheidetrichter extrahiert. Die vereinigten Butanol- lösungen werden hierauf bei 5 Torr eingedampft und der lige Rückstand in 250 ml Essigsäure-Wasser-Methanol-Essigsäureethylester 6:34:20:10 gelöst und an einer Säule von 0,8 kg Polyamidpulver (Macherey-Nagel, SC6) oder
wahlweise mit Wasser-Pyridin 97,5:2,5 an Sephadex G-10 (Pharmacia) chroma- tographiert. Die erhaltenen, spiraeosidhaltigen Fraktionen werden zusammen genommen, i.V. eingedampft und der Rückstand aus wenig heisser 15%-iger Essigsäure umkristallisiert.
Ganz reines Spiraeosid kann durch wiederholte Chromatographie an Cellu- losepulver (Avicel®, Merck, Art. 2331 ) mit 13%-iger Essigsäure gewonnen werden. Man erhält hellgelbe Nadeln, Smp. (Vak., unkorr.) 211 ,5 - 212,5 °C.
Herstellung und Charakterisierung von Octalauroylspiraeosid:
(VI)
In einem kleinen Dreihalskolben, versehen mit N2-Einleitrohr, Magnetrührer, Tropftrichter und Kühler werden 214 mg Spiraeosid in 2 ml wasserfreiem Pyri- din durch leichtes Erwärmen gelöst, dann die Lösung mit 5 ml 1,2-Dichlor- ethan verdünnt und mit 10 mg 4-Dimethylaminopyridin versetzt. Nun wird unter N2 auf 0aC gekühlt, dann langsam und unter gutem Rühren tropfenweise mit einer Lösung von 0,87 ml Lauroylchlorid in 5 ml 1,2-Dichlorethan versetzt. Hierauf lässt man auf RT kommen und lässt 12h stehen. Die klare, fast farblose Lösung wird mit Et20 verdünnt und die entstandene Suspension im Scheidetrichter 5 x mit stark verdünnter H2SO4 und zuletzt mit Sole ausgeschüttelt. Eine Behandlung mit verdünnter Hydrogencarbonatlö- sung kann, sofern der Ansatz überschüssige Laurinsäure enthält, zu starker Emulsionsbildung führen. Um dies auf einfache Art und Weise zu vermeiden, empfiehlt sich langsames Filtrieren der getrockneten Lösung durch eine kurze Säule aus Calciumcarbonat, Lösungsmittel: Et20 oder ein Gemisch von Dichlormethan/Essigsäureethylester. Nach dem Trocknen über a2Sθ und der üblichen Aufarbeitung erhält man einen blassgelben, blasigen Schaum, Ausbeute 0,94 g. Er lässt sich aus CH2CI2/CH3CN oder aus 2-Propanol kristallisieren. Kristalle mit schwachem Gelbstich, Smp. 82,5 - 85°C.
fαl -11.70 (c= 1,034; CHCh).
UVι (CH2CI2): 232 (26'900), 254 (20'400), 311 (26'100) iRl (CH2CI2): 3048w, 2932ss, 2853s, 1762s, 1651m, 1622m, 1510m. 1H-NMR: (CDCIj, 300 MHz): 0,8-2,7 (Protonensignale der Esterkomponente); 3,8 bis 5,8 (Signale der Protonen am Zuckerteil); 6,76 (d, J=2,4, H-C(5)). 7,02 {d, J=8,8, H-C(5')}; 7,22 {d, J=2,4, H-C(8)}; 7,44 {d, J=2,2, H-C(2')}; 7,59 (dd, J=8,8 und 2,2, H-C(5')); die ursprünglichen OH-Protonen bei 8,59/9,50/10,79 und 12,41 ppm sind verschwunden.
"C-NMR: (CDCI_. 75 MHz): u.a. 9 Carbonylsignale bei 173,1/172,6/171,9 (dreifache lntensität)/171 , 1/170,7/170,4/169,9 ppm. Verbrennungsanalyse:
Cn7H1t«02o (1922,74) Ber. C 73,08 H 10,28 %
Gef. C 72,89 H 10,91 %
Auf vergleichbare Weise lassen sich auch die Perester mit den Capronsäure-, Undecylensäure-, Palmitinsäure- und Stearinsäurechloriden herstellen.
1.32 Fettsäureester von Vitamin P (Quercetin-3-rutinosid) Herstellung von Rutindecalaurat
Käufliches Rutintrihydrat (Fluka 84082) ge äss Formel (VI), wobei R » H ist
100°C/0.01 Torr getrocknet. 0.92 g wasserfreies Rutin wurden dann in 5 ml abs. Pyridin und 5 ml abs. Dimethylformamid durch Erwärmen gelöst und hiernach mit 10 ml 1 ,2-Dichlorethan verdünnt. Nach Kühlen unter N2 und Rühren erfolgte die tröpfchenweise Zugabe von 3,59 ml Lauroylchlorid in 10 ml 1 ,2-Dichlorethan. Anschliessend lässt man den Ansatz langsam auf RT kommen, worauf man ihn unter stetem Rühren während 20h stehen lässt. Dann gibt man genügend Ether zu, damit sich beim Ausschütteln mit stark verdünnter H2S04 und Wasser eine gut abgetrennte obere Phase bildet. Nach Trocknen über Na2S04 und üblicher Aufarbeitung, sowie Trocknen des Rückstandes im Hochvakuum erhält man ein honigartiges, hellbräunliches Öl. Ausbeute ca. 90%. Analytische Daten:
UV: (Hexan, qualitativ): λmax 202 nm, 250, sh 293, sh 306 fαlrv = -28,5° (c = 0.82 in Chloroform) ißi (Dichlorethan): keine Banden im OH-Bereich; 3045w, 2625ss, 2855s, 1750s, 1646m, 1629m, 1504w.
2.0 Herstellung von spontan disoeroierbaren Konzentraten und von wässerigen Ultramikroemulsionen
Die beschriebenen Verbindungen der Formeln (I) bis (VI) sind als Folge ihrer ausgeprägten Lipophiiie wasserunlöslich. Damit sie durch die Membranbar¬ riere von Tumor- oder Wirtzellen und durch die Proteinhülle (das Capsid) von Viren diffundieren und im Zellplasma, bzw. im Virus-Innenkörper wirksam werden können, müssen sie vorerst in geeigneter Weise im wässerigen Medium solubllisiert werden. Dies geschieht erfindungsgemäss über zwei Stufen: die Herstellung von spontan disper gierbaren Konzentraten, welche die Wirksubstanz als integrierenden Systembestandteil enthalten, und die Zugabe von destilliertem Wasser, 5%-Glucoselösung oder physiologischer Kochsalzlösung (Ringerlösung) im Verhältnis 1 :20 bis 1 :1 '000'000.
Im Wege der Bildung von thermodynamisch stabilen Oel-in-Wasser Ultramikroemulsionen mithilfe von besonders ausgewählten Cotensiden oder Lösungsvermittlern einerseits und geeigneten Tensiden anderseits gelingt es, einen optimalen Solubilisierungsgrad der bezeichneten Ester zu erzielen. Alle experimentellen Beobachtungen an dergestalt ausgebildeten Ultramikroemulsionen lassen sich einheitlich durch die Annahme deuten, dass die ausgewählten Tenside und Cotenside, als ausgewogenes System genommen, in der wässerigen Phase organisierte Aggregate, sog. Mizellen bilden. Diese besitzen mehr oder weniger kugelförmige Gestalt, mit einem hydrodynamischen Radius von 2.2 bis 3.0 nm. Vgl. "Mode-selective dynamic light scattering; theory versus experimental realization", Thomas Gisler, et al., Applied Optics/Vol. 34, No. 18/ 20 June 1995, pp. 3546-3553.
Die Tenside und Hydrotrope (Cotenside) lassen zwischen der äusseren, wässerigen Phase und der inneren, öligen Phase der Mikroemulsion [enthaltend die Ester der Formeln (I) bis (VI), gelöst im biotensiden Lösungsvermittler] eine Grenzschicht entstehen, wodurch die Mischung dieser beiden Phasen unterbleibt. In der öligen, inneren Phase liegen die Moleküle der ausgewählten Ester in monomerer oder in oligomer agglomerierter Form vor. Die Mizellen der Flavonolglykosidester-haltigen inneren Phase der erfin- dungsgemässen Ultramikroemulsionen sind demnach an ihrer Oberfläche, d.h. an ihrer Grenzschicht mit einem Tensidmantel geschützt, was sie instand setzt, leicht und selektiv durch die Zellmembran ins Innere der tumoralen oder Virus-infizierten und somit "defekten" oder "abnormen" Zellen zu diffundieren. Diese Diffusion erfolgt ausschliesslich aufgrund thermischer Molekularbewegungen.
Die Richtung, die ein konkreter Diffusionsvorgang einschlägt, wird vom Konzentrationsunterschied bestimmt, welcher an der (fraktalen) Plasmamembran zwischen ausserhalb und innerhalb der Tumor- oder Wirtzelle besteht. Die Diffusion verläuft solange entlang dem Konzentrationsgefälle, bis es abgebaut ist. Zwischen der extrazellulären Zone und dem Inneren der einzelnen Wirtzelle oder des Virus wird die Konzentration an Wirksubstanz, bzw. des Wirkstoffsystems ("multiple drug system") ausgeglichen, wobei auch verzögerte Abgabeeffekte auftreten können. Derartige Diffusionsvorgänge verlaufen unabhängig von jeglicher Energiezufuhr. Sie haben keinen Bezug auf die zelluläre Stoffwechselenergie.
Wie aus der nachstehenden Tabelle ersichtlich ist, hat eine globuläre "Mizelle" mit einem hydrodynamischen Radius von einem Centi meter ein Volumen von 4,189 cm3 und eine Phasenoberfläche von 12,564 cm2.
Demgegenüber weisen 1018 Mizellen mit einem hydrodynamischen Radius von nur 1 x 10-6 cm (10 nm), welche zusammen das gleiche Volumen von 4,187 cm3 ausmachen, schon eine Gesamt-Phasenoberfläche von 1 *256,4 π.2 auf.
MIZELLEN: VERHÄLTNIS VOLUMEN ZU GESAMTOBERFLÄCHE
Kugelvolumen = — π i"3 3
Kugeloberfläche = 4 π f2
Fazit: Durch die enorm grosse Phasenoberfläche, welche die Mizellen mit einem hydrodynamischen Radius von 1.8 bis 5 nm in Ultramikroemulsionen ausbilden, wird zusätzlich zu deren gesteigertem Diffusionsvermögen die rheologlsche Verteilung ("spreadlng") und somit die Bioverfügbarkelt und Bioaktivität der Wirkstoffe, welche in der Inneren Phase der Mizellen In mono- merer oder in ollgomer agglomerierter Form vorliegen, ebenfalls stark verbessert. Das kann eine beträchtliche Ermässigung der kritischen Dosierung erlauben und damit unerwünschte Nebenwirkungen ganz vermeiden oder wenigstens verringern helfen.
Die "Packungsdichte" eines spontan dispergierbaren, stabilen MARIGENOL®- Konzentrates nimmt in exponentieiler Funktion mit der kleiner werdenden Teilchengrösse der Mizellen zu. Entscheidend sind die richtige Ausbildung der inneren Phase (d.h. des mizellaren Kerns), ihr ausgewogenes Verhältnis zum Gesamtkonzentrat und die Auswahl der je dazu passenden Tenside.
2.1 Die erfindungsgemäss spontan disperαierbaren Konzentrate enthalten: 0,5 bis 5 Gewichts-% einzelner Ester der Formeln (I) oder (II), bzw. eine Kombination solcher Ester, sowie
1 bis 25 Gewichts-% eines als Hydrotrop, bzw. Co-Emulgator dienenden, pharmaverträglichen Lösungsmittels oder Lösungsmittelgemisches, 0 bis 5 Gewichts-% eines Good-Puffers oder 3-[(3-Cholamido-propyl)-dime- thylammonio]-propansulfonat (CHAPS) und/oder DMSO (Dimethylsulfoxyd), 5 bis 90 Gewichts-% eines pharmaverträglichen Tensides oder Tensidgemisches, und wahlweise
0 bis 10 Gewichts-% eines Vitamins oder Provitamins,
0 bis 10 Gewichts-% eines Stabilisators, Radikalfängers oder Penetrations- verbesserers.
Die erfindungsgemäss anwendbaren, wässerigen Ultramikroemulsionen enthalten:
0,1 bis 5 Gewichts-% eines spontan dispergierbaren Konzentrates wie oben beschrieben,
85 bis 99,9 Gewichts-% dest. Wasser, physiologische Kochsalzlösung (Ringerlösung) oder 5% Glucoselösung, 0 bis 10 Gewichts-% pharmazeutische Hilfsmittel.
Diese wässerigen Ultramikroemulsionen zeichnen sich aus durch eine ermäs- sigte Oberflächenspannung von 28-32 mNm- , einen geringen inneren Reibungswiderstand (mit einer dynamischen Viskosität η bei 20*C um 1.0 cP = 10-3 Pa.s), eine sehr kleine Teilchengrösse und eine gute thermodynamische Stabilität (kein Koaleszieren, bzw. Agglomerieren der Mizellen, bzw. keine Autokondensation der Tenside; d.h. es tritt kein "seif assembly" infolge einer chemischen Veränderung der Tenside ein). Die Wirksubstanzen finden sich demnach monomer oder oligomer gelöst in der inneren Phase der Mizellen vor, im mizellaren Kern ("micellar core") verpackt. Wegen des ausgewogenen Verhältnisses bleibt auch trotz starker Verdünnung der die einzelnen Mizellen umhüllende Tensidmantel unversehrt; demnach unterbleiben sog. "instability or Marangoni effects". Dank diesen Eigenschaften lässt sich mit derartigen Ultramikroemulsionen eine hohe kapillare Diffusion erreichen. Sie besitzen ein ausgeprägtes Permeationsvermögen an der Zellmembran von defekten oder abnormen Zellen und eine sehr gute Spreitung (rheologische Verteilung, "spreading") im Zellinneren. Dies führt zu einer ausgezeichneten Biover-
- 1 2 --
fügbarkeit und als Folge davon auch zu einer ausgeprägten Bioreaktivität der darin enthaltenen Wirkstoffe, gekoppelt mit geringer oder keiner Toxizität.
Die erfindungsgemäss einzusetzenden Tenside oder Tensidgemische können anionaktiv, kationaktiv, amphoter oder nicht-ionogen sein. Bevorzugt sind sie amphoter oder nicht-ionogen und haben ein HLB-Verhältnis (d.h. eine "hydro- philic-lipophilic balance") zwischen 2 und 18; bei Gemischen liegt es vorzugsweise zwischen 2 bis 6 einerseits und 10 bis 15 anderseits. In hohem Masse bevorzugt zur Herstellung von erfindungsgemässen, spontan dispergierbaren Konzentraten sind einerseits Phosphorsäureestertenside, wie z.B.: das praktisch wasserfreie Tristyrylphenolpolyoxyethylen-18-phosphor- säure
(Soprophor® FL, RHÖNE-POULENC); Diphasol® 3873 (CIBA-GEIGY), bzw. das identische Sermul® EA 188 (SERVO), ein Mischemulgator, bestehend aus je 50 % der beiden Verbindungen mit den Formeln:
Diphasol® 3873 (CIBA-GEIGY), ein Alkylphenol-polyglycolether-phos- phat-Tensid
Tensid 508 (CIBA-GEIGY)
(Tensid 508, CIBA-GEIGY);
Tinovetin® JU (CIBA-GEIGY), ein Hydroxybiphenyl-10-ethoxy-phosphorsäure- ester
Butyl-mono-4-ethoxy-phosphorsäureester (Zerostat® AT, CIBA-GEIGY), bzw.
O
CH,-{ CH2 Js-CM-CHj-O (-CH2— CH2-O )6- IPI OCH,
C_H5
OCH,
(Zerostat ® AN, CIBA-GEIGY) und anderseits Betainverbindungen, d.h. amphotere, salz- und wasserfreie Imidazolderivate, wie z.B.:
worin Mel+1 für Wasserstoff, Alkali- und Erdalkaliatome und R
x für eine Cι.
32- Alkyl- oder eine C
2>32-Alkenylgruppe stehen.
Verwendung finden auch sog. "multi-functional glucose derivatives", wie z.B. Glucate® SS (Methyl-Glucose-Sesquistβarat, in der CTFA-Classification) und Glucamate® SSE-20 (PEG-20 Methyl-Glucose-Sesquistearat, in der CTFA- Classification) von Amerchol, Edison, N.J., U.S.A.
Gute Ergebnisse sind fallweise auch zu erzielen unter Mitverwendung von nicht-ionogenen Tensiden der "TWEEN®"-Reihe (Atlas Chem. Ind. Inc., bzw. ICI Speciality Chemicals), sog. Polyoxyethylen-Sorbitan-Monoestern oder "Polysorbate" 20-85 in der CTFA Classification.
Als Hydrotrop, bzw. als Co-Emulgator dienende, pharmaverträgliche Lösungsmittel lassen sich einsetzen, z.B.:
Ester eines aliphatischen Alkohols (C3.18) mit einer aliphatischen Carbonsäure (C10.22), wie etwa Isopropyllaurat, Hexyllaurat, Decyllaurat, Iso-
propyimyristat, Isopropylpalmitat und Laurylmyristat; Kohlenwasserstoffe mit einer geraden Kohlenstoffkette (C12-32). welche mit 6-16 Methylgruppen substituiert ist und 1 -6 Doppelbindungen aufweisen kann, wofür Terpene wie Polymethylbutane und Polymethylbutene als Beispiele dienen mögen. Mono-Ester aus Ethylenglykol oder Propylenglykol mit einer aliphatischen Carbonsäure (Cβ.22), wie etwa Propylenglykolmonolaurat und Propylen- glykolmonomyristat.
Ester aus einem aliphatischen Alkohol (C12_22) mit Milchsäure, wie z.B.
Myristyl- oder vorzugsweise Lauryl-Lactat; Mono-, Di- oder Triester des Gly- cerins mit einer aliphatischen Carbonsäure (C6.22), wie z.B. Glyceryl-Caprylat, oder Miglyol® 812 Neutralöl (Oleum neutrale).
Ester aus einem Poly(2-7)ethylenglykolglyzerinether mit mindestens einer freien Hydroxylgruppe mit einer aliphatischen Carbonsäure (Cβ.22), wie z.B. aliphatische Alkohole (C12.22), somit u.a. Dodecanol, Tetradecanol, Oleylal- kohol, 2-Hexyldecanol und 2-Octyldecanol.
Ester mit mindestens einer freien Hydroxylgruppe, aus Poly-(2-10)glykol mit einer aliphatischen Carbonsäure (Cβ.22), Monoether aus einem Polyethyleπ- glykol mit einem aliphatischen Alkohol (C12.18), wie z.B. Polyoxyethylen (C10)- octylether.
Heterocyclische Verbindungen, wie z.B. 1 -Methyl-2-pyrrolidon. Biotenside Terpinylester der allgemeinen Formel (VII) : R6 COO R 7
(VII) worin R6 eine C2_31-Alkyl, eine C,.31-Alkenyl- oder eine C,,31-Alkapolyen- gruppe und R7 Citronellyl-, Farnesyl-, Geranyl-, Isophytyl- oder Phytyl- bedeuten.
Alle technischen Tenside wurden vor dem Eintrag in die spontan dispergierbaren Konzentrate mittels Filtration, bzw. Chromatographie über neutralem Aluminiumoxyd mit einem inerten Lösungsmittel wie z.B. Tetrahydro- furan, Ethanol oder Dichlormethan gereinigt.
2.2 ZUSAMMENSETZUNGSBEISPIELE von erfindungsgemässen, spontan dispergierbaren KONZENTRATEN
a) 0,1 bis 5 Gewichts-% einer Verbindung gemäss Formel (I) oder (II),
1 bis 25 Gewichts-% Isopropylmyristat, Isopropylpalmitat, oder Neutralöl, wie z.B. Miglyol® 812 (Dynamit Nobel oder Hüls)
0 bis 5 Gewichts-% eines Good-Puffers oder 3-[(3-Choiamido-propyl)-dime- thylammonio]-propansulfonat (CHAPS) und/oder DMSO (Dimethylsulfoxyd), 5 bis 45 Gewichts-% eines Phosphorsäureester-Tensides, wie z.B. Diphasol® 3873 (CIBA-GEIGY), Tensid 508 (CIBA-GEIGY), Zerostat® AN oder AT (CIBA- GEIGY), Tinovetin® JU (CIBA-GEIGY), Soprophor® FL (RHONE-POULENC), und/oder Tween®-20 (Polyoxyethylen-(20)-sorbitan-monolaurat, ICI), 5 bis 90 Gewichts-% Invadin JFC 800% (CIBA-GEIGY),
0 bis 10 Gewichts-% eines Vitamins oder Provitamins.
b) 0,1 bis 5 Gewichts-% einer Verbindung laut Formel (I) oder (II),
1 bis 25 Gewichts-% eines oder mehrerer biotensider Terpinylester der allgemeinen Formel (VII):
R6 COO— R (V||)
worin R6 eine C2.31-Alkyl-, eine C3.31-Alkenyl- oder eine C, 31-Alkapolyen- gruppe ist und R für Citronellyl-, Farnesyl-, Geranyl-, Isophytyl- oder Phytyl- steht,
30 bis 45 Gewichts-% Invadin® JFC 800% und/oder Tween®-20,
30 bis 45 Gewichts-% Soprophor® FL oder Diphasol® 3873.
c) 1 Gewichts-% einer Verbindung gemäss den Formeln (I) oder (II), 4 bis 9 Gewichts-% 2-Pentanol oder Glycerin wasserfrei oder DMSO
15 bis 20 Gewichts-% Citronellyl-10-undecenoat (Cn .i-Citronellylester) 30 Gewichts-% Invadin® JFC 800% und/oder Tween®-20, 45 Gewichts-% Soprophor® FL oder Diphasol® 3873.
N.B.: INVADIN® JFC 800% (CIBA-GEIGY) ist ein wasserfreies tert. Octyl- phenylpolyoxyethylenether-Tensid mit 9 bis 10 Oxyethylengruppen.
2.3 BEISPIEL für die pharmazeutische Herstellung eines Systempräparates mit erfindungsgemässen Konzentraten in der Form von "multiple units". a) Granulierung
Metolose® 90 SH-4000 (Shin-Etsu Chemical) 90.0 g
Avicel® PH-101 80.3 g
Erfindungsgemässes MARIGENOL®-KONZENTRAT 139.4 g
Aerosil® 200 80.3 g
∑ 390.0 g
Granulieren/formen im Schnellmixer oder im Rotationsbett unter Zusatz von 110 g Ethanol, brechen, sieben 18 bis 42 mesh, trocknen 24 h bei 40 °C.
b) MSR- und RETARD-Ausrüstung im Rotationsbett mit AQOAT® AS-HG (Shin-Etsu Chemical) und Talk
c) Zusammensetzung fertiges Granulat/bzw. Micropellets Kernmaterial 44 % Erfindungsgemässes KONZENTRAT 25 % MSR-Beschichtung 31 %
Σ 100 %
N.B.: MSR = Magensaft-Resistenz. Die Pellets/Granulate gemäss a) können auch ohne Befilmung unmittelbar in Kapseln abgefüllt werden, welche aus AQOAT® (HPMC-AS-M oder HPMC-AS-N) hergestellt sind, mit Aceton/Ethanol 1 :1 verschlossen werden und so die Funktionen der MSR und der verzögerten Abgabe (Retard-Formulierung) angemessen steuern.
3.0 BIOLOGISCHE PRÜFUNGEN
Die antitumorale Wirkung von spontan dispergierbaren Konzentraten mit Wirkstoffen gemäss den a) bis c) wird anhand folgender Prüfungsergebnisse bestätigt:
3.1 In vltro-Tests auf Zytotoxizltät mit geeigneten Tumorzell-Linien
Es wurde ein biologisches Assay-System entwickelt, das mit Mikrotiter- platten und Verdünnungsreihen arbeitet. Angesetzt werden je 1θ4/ml Tumorzellen in Kulturmedium RPMI 1640 mit 10% fötalem Kalbserum inaktiviert (GIBCO); sie werden so undicht ausgesät, dass sie während des Assays wachsen können, in sog. nichtkonfluenten Monolayers. Die Probenzugabe erfolgt nach 6 bis 24 Stunden, mit 100 μl pro Reihe, die man im 1. Loch mit 100 μl Medium versetzt. Davon wird die Hälfte entnommen und in das folgende Loch eingebracht, wieder mit 100 μl Medium versetzt, usf. Es entsteht eine geometrische Verdünnungsreihe n%.
Die Proben werden im Plaque Assay während 3 bis 5 Tagen bei 37*C mit 3%% CO2 inkubiert. Anschliessend färben/fixieren mit 0,1% Kristallviolett (Fluka, Buchs) in einer Lösung von 70% Methanol, 1% Formaldehyd, 29% Wasser. Die Auswertung wird am Mikroskop vorgenommen, Vergrösserung 300-fach. Man bestimmt die grösste cytotoxische Verdünnung. Die quantitative Auswertung lässt sich auch mittels Scanning und Absorptionsmessung am Spektrophoto- meter vornehmen.
3.2 Prüfung auf Zelltoxizltät
3.21 Zelltoxizität der MARIGENOL®-KONZENTRATE geprüft an Py6-Zellen (Polyoma Virus transformierten 3T3 Maus-Fibrobiasten)
Py6 Zytotoxizltäts-Test 11. - 15.12.1995
1%-Konzentrate 24 h 48 h 72 h Exposition Exposition Exposition Konzentrat Konzentrat Konzentrat
W.S. .S. W.S.
OCTALAUROYL- SPIRAEOSID 32*000 64*000 128*000 (mit CHAPS) 3.2 Mio. 6.4 Mio. 12.8 Mio.
OCTALAUROYL- SPIRAEOSID 16*000 64*000 128*000 (mit Tween-20) 1.6 Mio. 6.4 Mio. 12.8 Mio.
Grösste zytotoxische Verdünnung: auf Konzentrat, bzw. W.S. -Gehalt berechnet.
Zu den eternalisierten Py6-Zellen vgl.: "Biochemistry", Coordinating Editor Geoffrey L. Zubay, Addison-Wesiey Publishing Company, 1983. p. 1079. Vgl. auch "Molecular Cell Biology", second Edition, by J. Darnell, H. Lodish, D. Baltimore; Scientific American Books, Chapter 5: Viruses, Structure and Functions, pp. 177-188. New York, 1990 (W.H. Freeman & Co.)
3.22 Zelltoxizität der MARIGENOL®-KONZENTRATE (Fortsetzung)
Py6 Zytotoxizitäts-Test 28.3. - 2.4.1996
Grösste zytotoxische Verdünnung: auf Konzentrat, bzw. W.S.-Gehalt berechnet; Konzentrations-Angabe in μMol. N.B.: Zusammensetzung der 1%-Konzentrate: 1 Gewichts-% Wirksubstanz
12 Gewichts-% Cn:i-CITRONELLYL-ESTER
87 Gewichts-% Invadin JFC 800%/Soprophor FL 1 :1