WO1995010266A1 - Nitrogen monoxide synthesis inhibitor - Google Patents
Nitrogen monoxide synthesis inhibitor Download PDFInfo
- Publication number
- WO1995010266A1 WO1995010266A1 PCT/JP1994/001695 JP9401695W WO9510266A1 WO 1995010266 A1 WO1995010266 A1 WO 1995010266A1 JP 9401695 W JP9401695 W JP 9401695W WO 9510266 A1 WO9510266 A1 WO 9510266A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- general formula
- hydrogen atom
- nitric oxide
- Prior art date
Links
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 title claims abstract description 69
- 239000003112 inhibitor Substances 0.000 title claims abstract description 19
- 230000015572 biosynthetic process Effects 0.000 title claims abstract description 18
- 238000003786 synthesis reaction Methods 0.000 title claims abstract description 16
- 150000001875 compounds Chemical class 0.000 claims abstract description 146
- 239000004480 active ingredient Substances 0.000 claims abstract description 30
- 150000003839 salts Chemical class 0.000 claims abstract description 16
- 201000010099 disease Diseases 0.000 claims abstract description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 4
- 125000001792 phenanthrenyl group Chemical class C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 claims abstract 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 25
- 125000000217 alkyl group Chemical group 0.000 claims description 24
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 16
- 238000000034 method Methods 0.000 claims description 13
- 125000003545 alkoxy group Chemical group 0.000 claims description 10
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 8
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 7
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 6
- 125000004423 acyloxy group Chemical group 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 claims description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 2
- 125000000687 hydroquinonyl group Chemical group C1(O)=C(C=C(O)C=C1)* 0.000 claims description 2
- 239000003966 growth inhibitor Substances 0.000 claims 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims 1
- 239000012622 synthetic inhibitor Substances 0.000 claims 1
- 206010040070 Septic Shock Diseases 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 84
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 73
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 58
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 56
- 239000000203 mixture Substances 0.000 description 49
- 238000006243 chemical reaction Methods 0.000 description 46
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 42
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 40
- 239000000243 solution Substances 0.000 description 35
- -1 butylyl Chemical group 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 28
- 238000010898 silica gel chromatography Methods 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 24
- 229910052739 hydrogen Inorganic materials 0.000 description 23
- 239000012043 crude product Substances 0.000 description 21
- 239000012044 organic layer Substances 0.000 description 21
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 20
- 235000019341 magnesium sulphate Nutrition 0.000 description 20
- 238000005160 1H NMR spectroscopy Methods 0.000 description 19
- 239000012442 inert solvent Substances 0.000 description 19
- 238000002844 melting Methods 0.000 description 17
- 230000008018 melting Effects 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 16
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 16
- 239000003480 eluent Substances 0.000 description 15
- 238000004519 manufacturing process Methods 0.000 description 14
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 13
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000007864 aqueous solution Substances 0.000 description 12
- 238000001816 cooling Methods 0.000 description 12
- 230000000704 physical effect Effects 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 10
- 230000002829 reductive effect Effects 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 9
- 210000004204 blood vessel Anatomy 0.000 description 8
- 239000012267 brine Substances 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 7
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 239000002158 endotoxin Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 6
- 102000008299 Nitric Oxide Synthase Human genes 0.000 description 6
- 108010021487 Nitric Oxide Synthase Proteins 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 238000000605 extraction Methods 0.000 description 6
- 239000000499 gel Substances 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 229910052744 lithium Inorganic materials 0.000 description 6
- 229910052751 metal Inorganic materials 0.000 description 6
- 239000002184 metal Substances 0.000 description 6
- 239000000825 pharmaceutical preparation Substances 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 238000006722 reduction reaction Methods 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 235000019359 magnesium stearate Nutrition 0.000 description 5
- 235000015097 nutrients Nutrition 0.000 description 5
- 150000002987 phenanthrenes Chemical class 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 229920002261 Corn starch Polymers 0.000 description 4
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 4
- 239000008120 corn starch Substances 0.000 description 4
- 229940099112 cornstarch Drugs 0.000 description 4
- 239000000287 crude extract Substances 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 239000002024 ethyl acetate extract Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 235000006408 oxalic acid Nutrition 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 238000006884 silylation reaction Methods 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 102000015696 Interleukins Human genes 0.000 description 3
- 108010063738 Interleukins Proteins 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 3
- 102000011779 Nitric Oxide Synthase Type II Human genes 0.000 description 3
- 108010076864 Nitric Oxide Synthase Type II Proteins 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 150000007514 bases Chemical class 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 239000012380 dealkylating agent Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 210000003038 endothelium Anatomy 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 229920006008 lipopolysaccharide Polymers 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 101150054634 melk gene Proteins 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000007800 oxidant agent Substances 0.000 description 3
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- VFYFMNCKPJDAPV-UHFFFAOYSA-N 2,2'-(5-oxo-1,3-dioxolan-4,4-diyl)diessigs Chemical compound C1N(C2)CN3CN1CN2C3.OC(=O)CC1(CC(O)=O)OCOC1=O VFYFMNCKPJDAPV-UHFFFAOYSA-N 0.000 description 2
- ANOUKFYBOAKOIR-UHFFFAOYSA-N 3,4-dimethoxyphenylethylamine Chemical compound COC1=CC=C(CCN)C=C1OC ANOUKFYBOAKOIR-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 208000032843 Hemorrhage Diseases 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 229930064664 L-arginine Natural products 0.000 description 2
- 235000014852 L-arginine Nutrition 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000005042 acyloxymethyl group Chemical group 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 208000034158 bleeding Diseases 0.000 description 2
- 230000000740 bleeding effect Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- 239000000066 endothelium dependent relaxing factor Substances 0.000 description 2
- 210000003989 endothelium vascular Anatomy 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 238000004896 high resolution mass spectrometry Methods 0.000 description 2
- 229940047122 interleukins Drugs 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 210000002464 muscle smooth vascular Anatomy 0.000 description 2
- WHQSYGRFZMUQGQ-UHFFFAOYSA-N n,n-dimethylformamide;hydrate Chemical compound O.CN(C)C=O WHQSYGRFZMUQGQ-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- UCUUFSAXZMGPGH-UHFFFAOYSA-N penta-1,4-dien-3-one Chemical class C=CC(=O)C=C UCUUFSAXZMGPGH-UHFFFAOYSA-N 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 2
- 229960001802 phenylephrine Drugs 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 102000003390 tumor necrosis factor Human genes 0.000 description 2
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000002966 varnish Substances 0.000 description 2
- 230000002883 vasorelaxation effect Effects 0.000 description 2
- GEAOJWKXHMGANA-UHFFFAOYSA-N $l^{1}-stannane Chemical compound [SnH] GEAOJWKXHMGANA-UHFFFAOYSA-N 0.000 description 1
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 1
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 1
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 1
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 1
- IQMTVJBLQXREOP-UHFFFAOYSA-N 2h-phenanthren-1-one Chemical compound C1=CC2=CC=CC=C2C2=C1C(=O)CC=C2 IQMTVJBLQXREOP-UHFFFAOYSA-N 0.000 description 1
- KBARGPSSEIXDQU-UHFFFAOYSA-N 3,4-dihydro-2h-phenanthren-1-one Chemical compound C1=CC2=CC=CC=C2C2=C1C(=O)CCC2 KBARGPSSEIXDQU-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- VUTBELPREDJDDH-UHFFFAOYSA-N 4-amino-5-hydroxymethyl-2-methylpyrimidine Chemical compound CC1=NC=C(CO)C(N)=N1 VUTBELPREDJDDH-UHFFFAOYSA-N 0.000 description 1
- BXNXCVNLLKPXQP-UHFFFAOYSA-N 6-methoxy-2-propan-2-ylnaphthalen-1-ol Chemical compound OC1=C(C(C)C)C=CC2=CC(OC)=CC=C21 BXNXCVNLLKPXQP-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 235000003840 Amygdalus nana Nutrition 0.000 description 1
- 244000296825 Amygdalus nana Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 238000006027 Birch reduction reaction Methods 0.000 description 1
- OWNRRUFOJXFKCU-UHFFFAOYSA-N Bromadiolone Chemical compound C=1C=C(C=2C=CC(Br)=CC=2)C=CC=1C(O)CC(C=1C(OC2=CC=CC=C2C=1O)=O)C1=CC=CC=C1 OWNRRUFOJXFKCU-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 101100524275 Caenorhabditis elegans rege-1 gene Proteins 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- 102000018832 Cytochromes Human genes 0.000 description 1
- 108010052832 Cytochromes Proteins 0.000 description 1
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000032456 Hemorrhagic Shock Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- 229910010082 LiAlH Inorganic materials 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 101100400378 Mus musculus Marveld2 gene Proteins 0.000 description 1
- 101100518501 Mus musculus Spp1 gene Proteins 0.000 description 1
- VQFGRMSVSGVBLR-UHFFFAOYSA-N O=C1C2=C3C=CC(=CC3=CC=C2C(C=C1)=O)C(=O)O Chemical compound O=C1C2=C3C=CC(=CC3=CC=C2C(C=C1)=O)C(=O)O VQFGRMSVSGVBLR-UHFFFAOYSA-N 0.000 description 1
- 241000269799 Perca fluviatilis Species 0.000 description 1
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 235000011432 Prunus Nutrition 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- 206010049771 Shock haemorrhagic Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000705989 Tetrax Species 0.000 description 1
- 241000545405 Tripterygium Species 0.000 description 1
- HGLYTTWJVOQBNH-UHFFFAOYSA-M [F-].F[N+](F)(F)F Chemical compound [F-].F[N+](F)(F)F HGLYTTWJVOQBNH-UHFFFAOYSA-M 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000001800 adrenalinergic effect Effects 0.000 description 1
- 238000005377 adsorption chromatography Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000003935 attention Effects 0.000 description 1
- PRORZGWHZXZQMV-UHFFFAOYSA-N azane;nitric acid Chemical compound N.O[N+]([O-])=O PRORZGWHZXZQMV-UHFFFAOYSA-N 0.000 description 1
- YEESUBCSWGVPCE-UHFFFAOYSA-N azanylidyneoxidanium iron(2+) pentacyanide Chemical compound [Fe++].[C-]#N.[C-]#N.[C-]#N.[C-]#N.[C-]#N.N#[O+] YEESUBCSWGVPCE-UHFFFAOYSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 238000007664 blowing Methods 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- JHRWWRDRBPCWTF-OLQVQODUSA-N captafol Chemical compound C1C=CC[C@H]2C(=O)N(SC(Cl)(Cl)C(Cl)Cl)C(=O)[C@H]21 JHRWWRDRBPCWTF-OLQVQODUSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- KRVSOGSZCMJSLX-UHFFFAOYSA-L chromic acid Substances O[Cr](O)(=O)=O KRVSOGSZCMJSLX-UHFFFAOYSA-L 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000009500 colour coating Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000006900 dealkylation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- AWJWCTOOIBYHON-UHFFFAOYSA-N furo[3,4-b]pyrazine-5,7-dione Chemical compound C1=CN=C2C(=O)OC(=O)C2=N1 AWJWCTOOIBYHON-UHFFFAOYSA-N 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000005935 hexyloxycarbonyl group Chemical group 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 208000023589 ischemic disease Diseases 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 244000145841 kine Species 0.000 description 1
- 229940124280 l-arginine Drugs 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- NBTOZLQBSIZIKS-UHFFFAOYSA-N methoxide Chemical compound [O-]C NBTOZLQBSIZIKS-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- VXAPDXVBDZRZKP-UHFFFAOYSA-N nitric acid phosphoric acid Chemical compound O[N+]([O-])=O.OP(O)(O)=O VXAPDXVBDZRZKP-UHFFFAOYSA-N 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229960002460 nitroprusside Drugs 0.000 description 1
- 230000001129 nonadrenergic effect Effects 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000001148 pentyloxycarbonyl group Chemical group 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- QTWYUOSZMIWHJV-UHFFFAOYSA-N phenanthrene-2-carboxylic acid Chemical compound C1=CC=C2C3=CC=C(C(=O)O)C=C3C=CC2=C1 QTWYUOSZMIWHJV-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229940057847 polyethylene glycol 600 Drugs 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 235000014774 prunus Nutrition 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000002040 relaxant effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 238000000935 solvent evaporation Methods 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/075—Ethers or acetals
- A61K31/085—Ethers or acetals having an ether linkage to aromatic ring nuclear carbon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/095—Sulfur, selenium, or tellurium compounds, e.g. thiols
- A61K31/10—Sulfides; Sulfoxides; Sulfones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/64—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton
- C07C323/65—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton containing sulfur atoms of sulfone or sulfoxide groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/215—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring having unsaturation outside the six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
- C07C45/513—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups the singly bound functional group being an etherified hydroxyl group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/52—Compounds having —CHO groups bound to carbon atoms of six—membered aromatic rings
- C07C47/575—Compounds having —CHO groups bound to carbon atoms of six—membered aromatic rings containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/703—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups
- C07C49/747—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/753—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups
- C07C49/755—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups a keto group being part of a condensed ring system with two or three rings, at least one ring being a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C50/00—Quinones
- C07C50/26—Quinones containing groups having oxygen atoms singly bound to carbon atoms
- C07C50/34—Quinones containing groups having oxygen atoms singly bound to carbon atoms the quinoid structure being part of a condensed ring system having three rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C50/00—Quinones
- C07C50/38—Quinones containing —CHO or non—quinoid keto groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C62/00—Compounds having carboxyl groups bound to carbon atoms of rings other than six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C62/30—Unsaturated compounds
- C07C62/32—Unsaturated compounds containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C62/00—Compounds having carboxyl groups bound to carbon atoms of rings other than six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C62/30—Unsaturated compounds
- C07C62/34—Unsaturated compounds containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C62/00—Compounds having carboxyl groups bound to carbon atoms of rings other than six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C62/30—Unsaturated compounds
- C07C62/38—Unsaturated compounds containing keto groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/50—Organo-phosphines
- C07F9/53—Organo-phosphine oxides; Organo-phosphine thioxides
- C07F9/532—Cycloaliphatic phosphine oxides or thioxides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/22—Ortho- or ortho- and peri-condensed systems containing three rings containing only six-membered rings
- C07C2603/26—Phenanthrenes; Hydrogenated phenanthrenes
Definitions
- Nitric oxide synthesis inhibitor Nitric oxide synthesis inhibitor
- the present invention relates to a nitric oxide synthesis inhibitor useful as a medicament.
- Nitric oxide is the main body of the vascular endothelium-derived relaxing factor (EDRF), and is not adrenergic, non-adrenergic, such as brain, platelet, macrophage, and neutrophil. It has been revealed that tissues other than vascular endothelial cells, such as the active nervous system, function as mediators or modulators.
- This nitric oxide is produced by nitric oxide synthase (NO synthase) using L-arginine (Arg) as a substrate, and the nitric oxide synthase has at least two types of nitric oxide. It has been reported that an isoform exists. One is a constitutive type that exists in the vascular endothelium and brain
- nitric oxide synthases are available in a variety of cytochromes, such as interleukins (ILs), indium ferroyl-y (IF-r), and tumor necrosis factor (TNF). And endotoxin or it It is known that it is induced by various site proteins derived from them.
- ILs interleukins
- IF-r indium ferroyl-y
- TNF tumor necrosis factor
- endotoxin or it It is known that it is induced by various site proteins derived from them.
- nitric oxide when the above-mentioned nitric oxide is excessively produced and released in a living body, in addition to its vasorelaxant action, various cells or tissues may be formed due to the chemical reactivity of nitric oxide itself. Have been reported to be disabled.
- the above-mentioned inducible nitric oxide synthase which is known to be induced by endotoxin and various cytokins II, has an endotoxin short-chain due to nitric oxide produced thereby. It is known that it is deeply involved in pathogenesis such as bleeding and bleeding, and attention is paid to its involvement in such pathology rather than its physiological role.
- the present inventors have aimed at providing a novel nitric oxide synthesis inhibitor replacing the above-mentioned known nitric oxide synthesis inhibitor or a nitric oxide synthesis inhibitor as the drug.
- the present inventors have conducted a series of studies on a series of phenanthrene derivatives, which were previously developed as substances having interleukin-1 (IL-1) inhibitory activity. It has been found that a salt thereof (see Japanese Patent Application Laid-Open No. 4-212135) has a nitric oxide synthesis inhibitory activity which meets the above-mentioned object, and thus completed the present invention.
- IL-1 interleukin-1
- the present invention comprises as an active ingredient at least one phenanthrene derivative selected from the group consisting of compounds represented by the following formulas (1) to (12) and salts thereof. And a nitric oxide synthesis inhibitor.
- R 1 is a hydrogen atom or a lower alkyl group
- R 2 is a hydrogen atom or a lower alkanol group
- R 3 represents a hydrogen atom or a methyl group
- R 4 represents a lower alkanoyloxy group.
- R 2 is the same as above.
- R 3 and R Q each represent a lower alkoxy group.
- R 1 ⁇ is a lower alkyl group
- R is a formyl group
- R 3A represents a hydroxy group, or R and 3A represent an oxo group, and R 3A may be bonded to R 5A described below to form a double bond.
- R 4A is a lower alkyl group
- R " A is a hydrogen atom, a hydroxy lower alkyl group or R 3A or R 6A described below to form a double bond
- 6 A is a hydrogen atom or 5 ⁇
- R 7 is a hydrogen atom or a lower alkyl group, respectively. You. However, when R 7 is an isopropyl group, R 5A is not a hydroxymethyl group. ]
- R 1A , R and R 7 are as defined above.
- R 2a is a formyl group, tri-lower alkylsilyloxy group, trifluoro
- R 5a is a hydrogen atom, bonded to human Dorokishi lower alkyl or R 3 a or below the R 6 a and each other physician to form a double bond, R 6 a are joined together with a hydrogen atom or R 5a two A heavy bond is formed, R 8 represents a hydrogen atom or a lower alkoxy group, and R 9 represents a lower alkyl group.
- R 7 is an isopropyl group
- R 5a Is assumed not inhibit Dorokishime methyl group
- R 7 and R 8 are simultaneously hydrogen atoms
- R 5 a shall not form a double bond linked together with R G a.
- R lu represents a lower alkyl group.
- R 2B represents a hydroxymethyl group or a carboxyl group.
- R 3 B is a lower alkoxycarbonyl group or a non Doroki caulking butyl group, shows a R 4 B and R 5 d are each a lower alkoxy group.
- each group in the above general formulas (1) to (12) representing the active ingredient compound of the present invention is as follows. That is, examples of the lower alkyl group include linear groups having 1 to 6 carbon atoms such as methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, pentyl, and hexyl. Can be a branched alkyl group.
- the lower alkanol group there can be mentioned, for example, a straight-chain having 1 to 6 carbon atoms such as formyl, acetyl, propionyl, butylyl, isopyryl, pentanoyl and hexanol. Examples thereof include a linear or branched alkanol group.
- the lower alkanoyloxy group for example, a straight or linear group having 2 to 6 carbon atoms such as acetyloxy, propionyloxy, petityloxy, isobutylyloxy, pentanoyloxy, hexanoyloxy group, etc.
- An example is a branched-chain aryloxy group.
- lower alkoxy group examples include a straight-chain or branched group having 1 to 6 carbon atoms such as methoxy, ethoxy, propoxy, isopropoquine, butoxy, tert-butoxy, pentyloxy, and hexyloxy.
- hydroxy lower alkyl group examples include, for example, hydroxymethyl, 1-hydroxyl, 1-hydroxypropyl, 1-hydroxybutyl, 1-hydroxypentyl, 1-hydroxyhexyl, and the like. You.
- Tri-lower alkylsilyloxy groups include, for example, trimethylsilyloxy, triethylsilyloxy, triprovylsilyloxy, tributylsilanoloxy, tritert-butylsilyloxy, and tripentylcy. Examples thereof include riloxy and trihexylsilyloxy groups.
- lower alkoxycarbonyl group examples include methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, tert-butoxycarbonyl, pentyloxycarbonyl, and hexyloxycarbonyl. Groups and the like.
- R 1 , R 3 , R 4 and R. Is the same as R 1A , R 3A , R 4A and R 6A in the general formula (8).
- R 9 represents a lower alkyl group.
- R 2 ′ is the same as R 2A in the general formula (8) or represents a hydrogen atom, a carboxyl group, a lower alkoxycarbonyl group, a hydroxyl group or a lower alkanoloxy group.
- R 5 is the same as R 5A in formula (8) or represents a formyl group, a carboxyl group or a lower alkanoyloxymethyl group.
- the R 7 a is lower Aruki group
- the R 8 a is a lower alkoxy group
- R 12 is also hydrogen atom denotes a hydroxyl group. ]
- the lower alkanoyloxy group and the lower alkanoyloxymethyl group are referred to as the lower alkanol group.
- straight-chain or branched-chain alkanes having 1 to 6 carbon atoms such as formyl, acetyl, propionyl, butyryl, isoptyryl, pentanoyl, and hexanol.
- An example of a noyl group can be given.
- the oxidation reaction of the compound (2a) shown in the above reaction formula 11 is carried out in an inert solvent such as acetonitril, dichloromethan, N, N-dimethylformamide (DMF). , Cellular ammonia nitrate
- Ru can be carried out using an oxidizing agent such as anhydrous chromic acid.
- the amount of the oxidizing agent to be used may be usually about 1 to 3 times the amount of the compound (2a), and the reaction is generally carried out at a temperature of about 0 to 50 ° C for about 10 minutes. It is completed in about 3 hours, and thus the target compound (1a) is obtained.
- R 1 , R 4 and R 9 are the same as above.
- R 5b is hidden Roxy lower alkyl group.
- the dealkylation reaction of the compound (2b) shown in the above reaction scheme 12 is carried out, for example, in an inert solvent such as tetrahydrofuran (THF), acetonitrile, and dichloromethane. It is carried out by treating the compound (2b) with a dealkylating agent such as boron tribromide, anhydrous ammonium chloride, or hydrobromic acid.
- a dealkylating agent such as boron tribromide, anhydrous ammonium chloride, or hydrobromic acid.
- the amount of the dealkylating agent to be used may be generally about 1 to 3 times the molar amount of the compound (2b), and the reaction is generally carried out at a temperature of about 178 ° C to 50 ° C. It takes about 30 minutes to 5 hours.
- the desired compound (lb) can be obtained by oxidizing the compound (3b) obtained above.
- the oxidation reaction is carried out, for example, in an inert solvent such as ethanol or dichloromethane, as an oxidizing agent such as potassium dinitrosulfonate, potassium nitrorodisulfonate or chromium.
- the acid or the like is usually used in an amount of about 1 to 3 times the molar amount of the compound (3b), and if necessary, an additive such as hydrogen phosphate dihydrogen or the like is added to obtain a mixture of about 0 to 5 times. It takes about 5 to 20 hours at 0 ° C.
- the compounds (2a) and (2b) used as starting materials in the above reaction schemes 11 and 12 are both novel compounds, and some of them are chloro. It can be obtained by extraction and isolation from Prunus elegans (Tripterygium wi 1 fordii Hookfi 1 var, rege 1 ii Maki no), or by subsequent appropriate chemical treatment.
- Compounds (2c) to (2f), which belong to compound (2a) and compound (2b), respectively, must be produced according to the methods shown in the following reaction schemes. [Reaction process formula 1]
- R 1 , R 4 , R 7a , R 8a , R 9 and R 1 Q are the same as above.
- Ph represents a phenyl group.
- R 11 represents a lower alkyl group, X represents a halogen atom, and the broken line represents the presence of one double bond.
- the halogenation reaction of the compound (4b) is performed by reacting a halogenating agent such as N-bromosuccinic acid imide (NBS) or bromine with the compound (4b) in an inert solvent such as DMF or chloroform. It is carried out in a temperature condition of about 0 to 50 ° C, using about 1 to 1.5 times the molar amount, and about 1 to 20 hours.
- NBS N-bromosuccinic acid imide
- the resulting compound (5b) is converted to compound (6b) by alkylating it.
- the alkylation reaction is carried out without solvent or in a suitable inert solvent such as getyl ether, acetate, or the like, if necessary, in the presence of a deoxidizing agent such as an aqueous solution of a hydroxylic power or potassium carbonate.
- the reaction is carried out using an alkylating agent such as dimethyl sulfate, diazomethane, or methyl iodide, and the reaction is carried out at a temperature of about 0 to 30 ° C for about 30 minutes to 2 hours. Complete.
- an alkylating agent such as dimethyl sulfate, diazomethane, or methyl iodide
- the conversion reaction of the compound (6b) into the compound (7b) is carried out in an inert solvent such as DMF, methanol, or the like, in the presence of copper (I) iodide, and sodium. It is carried out by using a metal lower alkoxide such as um methoxide and sodium ethoxide. As the reaction conditions, a temperature of about 50 to 100 ° C. for about 30 minutes to 5 hours is employed.
- the reduction reaction of the compound (7b) is carried out in a solvent such as methanol, ethanol, liquid ammonia or the like, with an alkali metal, preferably a metal sodium, for about 3 hours. It is carried out by treating at a temperature of 0 to 80 ° C for 10 minutes to 1 hour.
- a solvent such as methanol, ethanol, liquid ammonia or the like
- an alkali metal preferably a metal sodium
- the resulting compound (8b) is hydrolyzed with an acid such as oxalic acid or hydrochloric acid in an inert solvent such as methanol or ethanol to give the compound (9b). Is converted to The reaction is completed in 5 to 24 hours at a temperature of about 50 to 80 ° C O
- the compound (9b) is treated with an halogen such as pyridyl or pyridin in an inert solvent such as benzene, dioxane or toluene in the presence of an amine such as pyrrolidine or piperidine.
- an alkylated compound By treatment with an alkylated compound, it is converted to an alkylated compound (10b).
- the compound (9b) is first reacted with the above-mentioned amine in the above-mentioned inert solvent at a temperature of 20 to 50 ° C for 3 to 6 hours, and then the above-mentioned alkyl halide is added. The reaction is carried out at a temperature of about 50 to 80 ° C for 10 to 50 hours.
- the compound (10b) can be converted to the target compound (2c) by performing a cyclization reaction with a vinyl ketone derivative (lib).
- the cyclization reaction is carried out in an inert solvent such as methanol, ethanol, or THF, in the presence of a base such as an aqueous solution of a hydroxylic solvent or an aqueous solution of sodium hydroxide. This is carried out using a vinyl ketone derivative (lib) in an amount of 1 to 1.3 times the amount of 10).
- the reaction is carried out at a temperature of about 130 to 30 ° C. for 5 to 15 hours (compound (2c) is reacted with compound (12b)).
- the reaction is carried out in an inert solvent such as THF, in an inert solvent such as THF, alkyllithium such as n-butyllithium and diisopropylamine, and
- the reaction is carried out in the presence of an amine such as tilamine using the compound (12b) in an amount of 1 to 1.5 times the amount of the compound (2c) by a molar amount of about ⁇ 78 to ⁇ 60.
- an amine such as tilamine
- the compound (2d) obtained above can be reduced with a hydride compound such as potassium hydride or sodium hydride in an inert solvent such as DMF, THF, and getyl ether. Is converted to the compound (2e).
- the reduction reaction is carried out at a temperature of about ⁇ 30 to 10 ° C. for 30 minutes to 2 hours.
- the compound (2f) can be obtained by hydrolyzing the compound (2e).
- the hydrolysis reaction is carried out in an inert solvent such as methanol or ethanol using an aqueous solution of oxalic acid or an acid such as hydrochloric acid.
- the solvent and the acid must all be degassed, and the reaction must be performed in an inert gas atmosphere such as argon or nitrogen.
- the reaction is carried out at a temperature of 50 ° C to the boiling point of the solvent. Completed in ⁇ 6 hours.
- R 1 , R 4 and R 7a are the same as above.
- R 8b indicates whether or lower alkanoyloxy noisy Ruo alkoxy group is the same as R 8 a.
- R 9 is the same as R 9 or represents a hydrogen atom or a lower alkanol group.
- R 2b represents a tri-lower alkylsiloxy group.
- the compound (2g) can be obtained.
- the reduction reaction is carried out with 1 mole of hydrogen in an inert solvent.
- the reaction is carried out by catalytic reduction using an alkali metal such as metallic sodium or metallic lithium in liquid ammonia (birch reduction).
- the state of the metal enolate obtained by removing the ammonia without isolating the perch reduction product of the compound (2c) is obtained.
- the reaction can be carried out by performing a silylation reaction.
- the silylation reaction is carried out in an inert solvent such as THF or getyl ale, in the presence of a deoxidizing agent such as triethylamine, pyridin or the like, in the presence of a dealkylating agent such as trialkylsilyl chloride. It takes about 10 minutes to 1 hour at a temperature of about -10 to 10 ° C using an agent.
- the obtained compound (2h) was obtained in an inert solvent such as THF and ether in the presence of alkyllithium such as methyllithium and butyllithium. After treating for 30 minutes to 1 hour at a temperature of C to 30 ° C, it is brought to a temperature of about 178 to 10 ° C with N-phenyl trifluoromethanesulfonimide (PhNTi 2 ). The compound is converted to the compound (2i) by treating for 5 to 15 hours.
- an inert solvent such as THF and ether
- alkyllithium such as methyllithium and butyllithium.
- the compound (2i) was synthesized according to the method of Still et al. (Sti 11 et a 1.) [J. Am. Chem. Soc., 108 (3), 452 (1986)]. And tetrakis (triphenylphosphine) The presence of palladium [Pd (Ph 3 P) 4 ], by the this reaction with ⁇ high dry de such carbon monoxide and hydrogen tin preparative-butyl, can be converted to the compound (2 f). As the reaction temperature and time, about 30 to 60 hours and about 15 to 60 hours are employed, respectively.
- R 1 , R 4 , R 7a , R 8a , R 9 and R i Q are as defined above.
- the oxidation reaction of the compound (2e) shown in the above reaction scheme 15 is carried out according to the method of Schmidt et al. [Angev. Chem., U, 116 (1959)]. That is, the compound (2e) is added to the complex of palladium chloride (11) and copper chloride (11) in the presence of oxygen (air) in a water-containing inert solvent such as DMF-water, and the compound (2e) is added to about 50
- the compound (2j) can be obtained by acting for about 10 to 24 hours at a temperature of about 100 ° C. [Reaction process formula 1 6]
- the compound (13) is reduced and then silylated to obtain the compound (14).
- the reduction reaction and the silylation reaction can be carried out in the same manner as the reduction reaction and the silylation reaction of the compound (2c) in Reaction Scheme 14.
- the compound (15) is treated with an inert solvent such as THF or ether in the presence of a desilylating agent such as tetrafluoroammonium fluoride (TBAF) to formaldehyde gas or the like.
- a desilylating agent such as tetrafluoroammonium fluoride (TBAF) to formaldehyde gas or the like.
- TBAF tetrafluoroammonium fluoride
- the funinanthrene derivative obtained by the method shown in each of the above reaction schemes can be isolated and purified by conventional separation means.
- the means include adsorption chromatography, recrystallization, solvent evaporation, precipitation, and solvent extraction.
- Each of the compounds represented by (10) to (12) can be produced by the method described in the above-mentioned JP-A-6-192555.
- the active ingredient of the nitric oxide synthesis inhibitor of the present invention not only the above-mentioned finnanthrene derivative but also an acidic group, that is, a phenolic hydroxyl group and / or a carboquinol group among the derivatives.
- a salt formed by a general salt forming reaction between the compound and a basic compound is also included.
- the basic compound used in the production of the above salt include sodium hydroxide, potassium hydroxide, calcium hydroxide, sodium carbonate, and carbonated lithium.
- organic amines such as methylamine, ethylamine, isopamine pyramine, morpholine, pyrazine, pyridine, 3,4—dimethoxyphenethylamine, and the like. Can also be used as the salt-forming basic compound.
- the active ingredient compound of the present invention may be a stereoisomer or an optical isomer of each of the above-mentioned finnanthrene derivatives and salts thereof.
- the phenanthrene derivative and a salt thereof used as an active ingredient in the present invention can exert an action of inhibiting the biosynthesis of nitric oxide.
- the drug is used to treat various conditions caused by nitric oxide, more specifically, endotoxin shock, hemorrhagic shock, various ischemic diseases, and various conditions associated with hypotension. It can be applied effectively to prevention.
- the inhibitor of the present invention has an effect of inhibiting the induction of inducible nitric oxide synthase as described above, and thus can specifically inhibit nitric oxide produced by the enzyme. It has more favorable characteristics in that it can suppress such abnormal or excessively produced and released nitric oxide.
- particularly preferred active ingredient compounds have quinone and hydroquinone structures. To choose from.
- One specific example is a compound represented by the above general formula (1).
- Specific examples of other preferred effective component compounds include the above-mentioned general formula (4) (otherwise, R 3 is a hydrogen atom).
- R 3 is a hydrogen atom.
- a compound represented by the above general formula (11) can be exemplified.
- the inhibitor of the present invention is prepared in the form of a general pharmaceutical composition using a conventional pharmaceutical carrier, and can be administered to humans and other animals.
- Pharmaceutical formulation carrier, formulation form
- Dosage unit form can be the same as those of ordinary pharmaceutical preparations. That is, the above pharmaceutical preparations include tablets, pills, powders, solutions, suspensions, milks, granules, capsules, suppositories, injections (solutions) containing an effective amount of the active ingredient compound of the present invention. , Suspending agents, etc.).
- the preparations in the above various forms are all prepared according to a conventional method, and the carriers used in this case may be various commonly used ones.
- tablets are prepared by using the compound of the present invention as an active ingredient and excipients such as gelatin, starch, lactose, magnesium stearate, talc, and gum arabic. Is mixed with and shaped.
- Capsules are prepared by mixing the active ingredient with an inert filler or diluent and filling into hard gelatin capsules, soft capsules, and the like.
- Parenteral preparations such as injections are produced by dissolving or suspending the compound of the present invention as an active ingredient in a sterilized liquid carrier.
- Preferred liquid carriers used in this case are water and physiological saline.
- Injectable preparations and the like to be prepared may further contain ordinary solubilizing agents, buffers, soothing agents and the like.
- a coloring agent, a preservative, a fragrance, a flavoring agent, a sweetening agent and the like and other pharmaceuticals can be contained as necessary.
- the amount of the funinancelen derivative and its salt to be contained as an active ingredient in the preparation of the present invention is not particularly limited and can be appropriately selected from a wide range, but is usually 1 to 70 in the total pharmaceutical preparation composition. % By weight, preferably about 1 to 30% by weight.
- the administration method of the pharmaceutical preparation prepared above is not particularly limited. For example, tablets, pills, powders, granules, capsules and the like are administered orally, and injections (solutions, suspensions, etc.) are used alone. It is administered intravenously, or as a mixture with normal rehydration fluids such as glucose and amino acids, or as needed alone intramuscularly, intradermally, subcutaneously or intraperitoneally.
- the dosage of the above pharmaceutical preparation depends on the usage, age, sex and other conditions of the patient.
- the amount of the compound of the present invention, which is usually an active ingredient, is preferably about 0.1 to 100 mg / kg of body weight per day, and is appropriately selected depending on the degree of the disease. It can be administered 1 to 4 times a day.
- the dosage unit form preferably contains about 1 to 60 mg of the active ingredient.
- finolemco consisting of castor oil and methanol. Coating with a coating agent to produce film-coated tablets.
- the above active ingredient compound, citrate, lactose, dicalcium phosphate, pull nick F-68 and sodium raurylsulfate are mixed, and the above mixture is N 0.60
- the mixture is sieved with a screen and wet-granulated with an alcoholic solution containing polyvinylpyrrolidone, Carbox 1500 and Carbox 600. Add alcohol as needed, pulverize powder into paste mass, add corn starch, continue mixing until uniform particles are formed. Pass through a No. 10 screen, place in a tray, and dry in an oven at 100 ° C for 12 to 14 hours.
- the dried particles are sieved with an N 0.16 screen, dried sodium sulfate and magnesium stearate are added and mixed, and compression-molded into the desired shape using a tableting machine. To create the core.
- the core is treated with a varnish, talc is sprayed to prevent moisture absorption, an undercoat layer is coated around the core, and a sufficient number of varnish coatings are performed for internal use. Layer formation and smooth coating are repeated, color coating is performed until a desired color is obtained, and dried to obtain coated tablets.
- THF (13.5 ml) in which 0.65 ml of diisopropylamine was dissolved was cooled to 178 ° C, and a THF solution (1.6 M) of n-butyl lipium was added. 1 ml was added and stirred for 25 minutes. This can be done at 178 ° C at 13.5 ml of THF in which 1.15 g of phenylphosphonoxide was dissolved was added, and the mixture was stirred at 178 ° C for 25 minutes. To this was added a solution of compound (750 mg) obtained in Example 1 in THF (6 m 1), and the mixture was stirred at ⁇ 78 ° C. for 2.5 hours.
- Example 3 2.1 g of the compound obtained in Example 3 was dissolved in 45 ml of degassed methanol, and an aqueous solution (7 ml) of degassed oxalic acid (690 mg) was added. For 4.5 hours. After cooling, water was added, extracted with ethyl acetate, and the organic layer was washed with saturated saline, dried over magnesium sulfate, and concentrated. The obtained crude product was purified by silica gel force chromatography (eluent: getyl ether: n-hexane: black-mouthed form: 20: 300: 3).
- tetrabutylammonium fluoride in THF solution (1.0 M) was introduced at 178 ° C while formaldehyde gas was passed through the reaction vessel together with nitrogen gas.
- R5a and R 6A ⁇ R 7 : — CH (CH 3 ) 2 ,
- R 8 -0CH 3
- R 9 one CH 3
- R 1A -CH 3
- R 2a Ph 2
- R 3a —OH
- R4A —CH 3
- R5a and R6a Two keys—mystery! ⁇
- R 7 -CH (CH 3 ) 2, R 8 : -OCH 3 ⁇ R 9 : — CH 3
- R 1A :-CH 3 , R 2a and R 3a : CH-OCH 3 , R 4A : -CH:
- R 7 — CH (CH 3 ) 2, R 8 :-OCH 3 ⁇ R 9 : One CH 3
- R 1A ⁇ -CH 3
- R 2a —CH0
- R 6a ⁇ -H ⁇
- R 7 One CH (CH 3 ) 2
- R 8 -OCH 3 ⁇
- R 1A yS-CH 3
- R 2a —CHO
- R3a and R5a Two! ! ⁇ Join Ito) ⁇
- R 4A — CH 3 ,
- R 6a / 3 - H
- R 7 -CH (CH 3) 2
- R 8 - OCH 3
- R 1A; 9 CH 3 , R 2a and R3a: 0, R 4A : a-CH 3 ,
- R 5a - H, R 6a a- H, R 7 -CH (CH 3) 2, R 8 -OCH 3 R9 -CH 3
- R 1A -CH 3
- R 2a — CHO
- R3a -OH.
- R4A; -CH 3
- R5a and R6a two fi ⁇ ⁇ - ⁇ : ⁇ —CH (CH 3 ) 2 ,
- R 5a a-CH 2 OH
- R 6a a_H
- R 7 H
- R8 H
- R 1A ⁇ -CUs .R 2a : -OS i (CH 3 ) 3 ,
- R 3A and R 5A Combined ⁇ R 4A : — CH 3 , R6a: ⁇ - H, R 7 : —CH (CH 3 ) 2, R 8 : — OCH 3 , R9: —CH 3
- RiA ⁇ - ⁇ 3
- R 2a -OS0 2 CF 3
- R 3A and R 5A ⁇ R 4A : ⁇ CH 3 ,: aU,
- R7 one CH (CH 3) 2
- R 8 - ⁇ _CH 3
- R9 -CH 3
- R 1A ⁇ CH 3
- R 2 — CHO
- R 4A — CH 3
- R3 and R5 two (-m ⁇ R 6: over H ⁇
- R 7 One CH (CH 3 ) 2
- R 1A —CH 3
- R 4A —CH 3
- R 5 ⁇ one H
- R6 a-H
- R 7 - CH (CH 3) 2
- R A -CH 3
- R 2 one CHO
- R3 one OH
- R 4A - CH 3
- Example 16 One hundred and eight kilograms of a closle stalk is cut into pieces, and extracted with methanol 201 at room temperature for 7 days. The extract is concentrated under reduced pressure to obtain a crude extract. This crude extract was suspended in water 201, and extracted three times with 201 parts each of ethyl acetate. The ethyl acetate layers were combined and concentrated under reduced pressure to obtain 130 g of an ethyl acetate extract.
- fraction (10) was distilled off under reduced pressure, and 6.37 g of 6.5 g of the obtained residue was separated with Sephadex LH-2. 0 (500 ml, Pharmaceutical Co., Ltd.), chromatographic chromatography once, fractionated and eluted with methanol 21 (10-1) to (10-7) were obtained.
- Example 19 38 mg of the compound obtained in Example 19 was dissolved in 0.5 ml of dried THF, and the solution was dried.
- One of the stainless wires was fixed to a support device, and the other was connected to an isometric transducer (Isometr tric ransducer, Nihon Kohden, TB-651T).
- the sample was pre-loaded with a static tension of 1.0 g and maintained at this tension until the end of the experiment.
- a crease solution was used, and a mixed gas consisting of 95% 09 and 5% C 00 was aerated.
- the pH of the crep solution is about 7.4 when the mixed gas is vented, and the composition is as follows.
- the nutrient solution should always contain 100 ng Zm1 or recombinant lipopolysaccharide (LPS).
- Interleukin lyg (rIL-1 ⁇ ) 0.3 ng / m1 was allowed to coexist.
- the specimen was pre-contracted with (phenylphrine) (1 / zM), and the drug was applied cumulatively when the tension became constant.
- Vascular relaxation was determined as a percentage of the contraction caused by Phenylephrine 1 / M.
- each of the compounds 1 to 6 of the active ingredient of the present invention shown in Table 10 below was applied (coexisting in a nutrient solution). Observation results obtained on the blood vessels 8 to 10 hours after the removal (Table 11 to Table 16 (—Nitric oxide synthase) for each test compound were used to determine the relaxation rate (%) and the relative percentage of 100% contraction due to phenylephrine 1 / M. When LPS is used as an induction promoter). In addition, Table 17 shows the same results (using the active ingredient compound 1 of the present invention) when rIL_l; 5 was used as the nitric oxide synthase-inducing promoter.
- Table 18 shows the results of the determination of the effect of the active ingredient compound of the present invention on the relaxation reaction by nitroprusside (II itr0prUsside) in the same manner as in the above test.
- the active ingredient compound of the present invention does not have any effect on the relaxing action of the two-port plunge, which acts directly on smooth muscle to relax blood vessels.
- the nitric oxide synthesis inhibitor of the present invention is useful for preventing and treating various diseases caused by nitric oxide such as endotoxin.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description









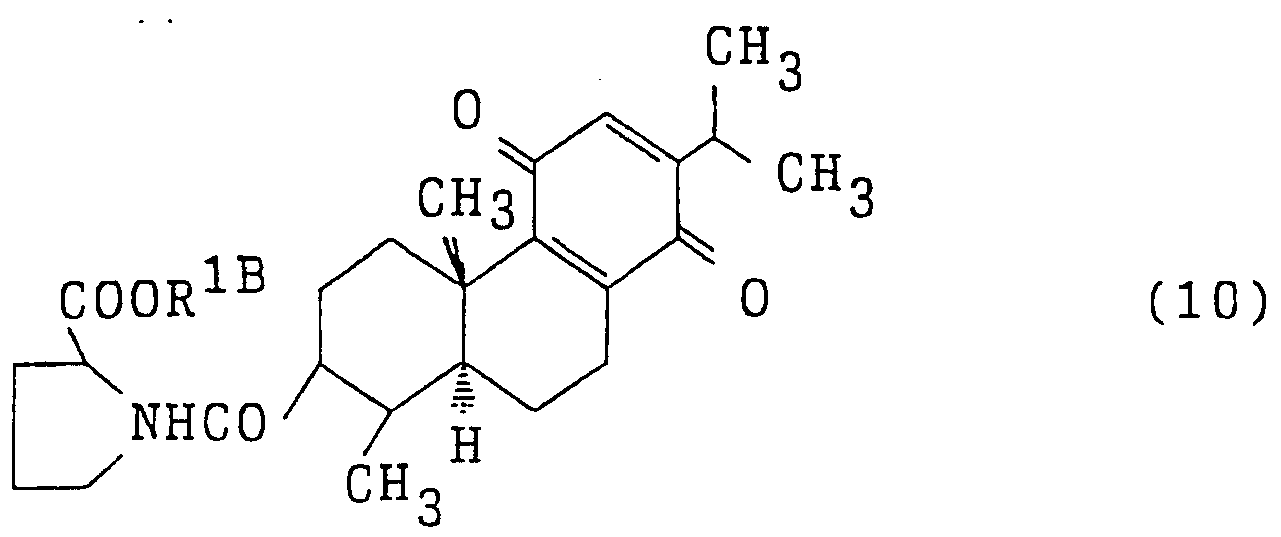


















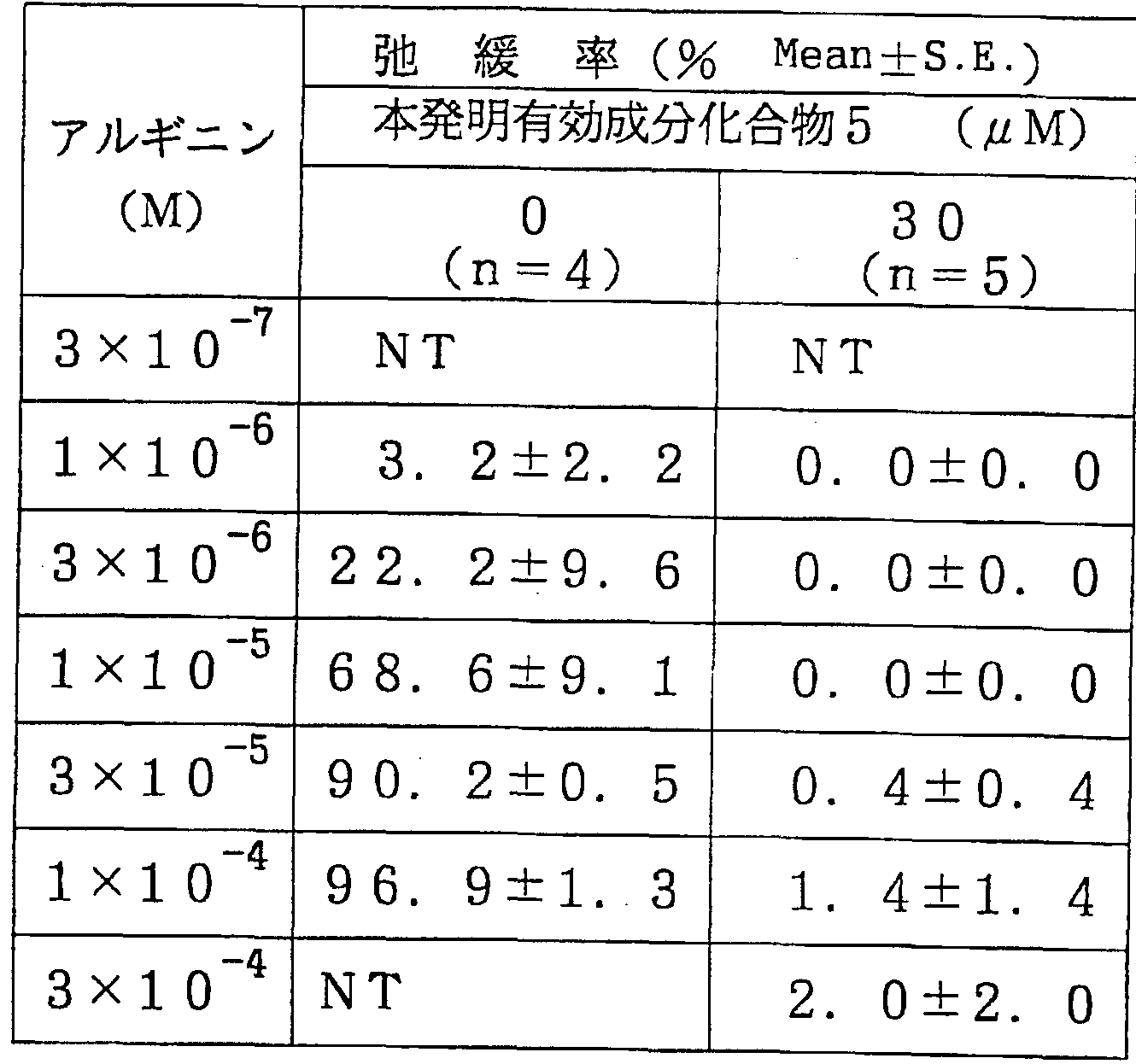



Claims












Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1019950702413A KR950703941A (ko) | 1993-10-13 | 1994-10-11 | 일산화질소 합성 억제제(nitrogen monoxide synthesis inhibitor) |
| EP94929028A EP0676196A1 (en) | 1993-10-13 | 1994-10-11 | Nitrogen monoxide synthesis inhibitor |
| US08/495,645 US5654343A (en) | 1993-10-13 | 1994-10-11 | Method of treating a nitric oxide-associated disease with phenanthrene derivatives |
| AU78224/94A AU684553B2 (en) | 1993-10-13 | 1994-10-11 | Nitrogen monoxide synthesis inhibitor |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP25594493 | 1993-10-13 | ||
| JP5/255944 | 1993-10-13 | ||
| JP6/94559 | 1994-05-06 | ||
| JP9455994 | 1994-05-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995010266A1 true WO1995010266A1 (en) | 1995-04-20 |
Family
ID=26435841
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1994/001695 WO1995010266A1 (en) | 1993-10-13 | 1994-10-11 | Nitrogen monoxide synthesis inhibitor |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US5654343A (ja) |
| EP (1) | EP0676196A1 (ja) |
| KR (1) | KR950703941A (ja) |
| AU (1) | AU684553B2 (ja) |
| TW (1) | TW312623B (ja) |
| WO (1) | WO1995010266A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5807886A (en) * | 1994-05-07 | 1998-09-15 | Astra Aktiebolag | Bicyclic amidine dervatives as inhibitors of nitric oxide synthetase |
| US5849782A (en) * | 1995-01-13 | 1998-12-15 | The General Hospital Corporation | Methods of inhibiting neurodegenerative diseases |
| US6380223B1 (en) | 1999-04-30 | 2002-04-30 | Pfizer Inc. | Glucocorticoid receptor modulators |
| US6852719B2 (en) | 2000-10-30 | 2005-02-08 | Pfizer Inc. | Glucocorticoid receptor modulators |
| US7713989B2 (en) | 2000-04-27 | 2010-05-11 | Dow Robert L | Glucocorticoid receptor modulators |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU688811B2 (en) | 1993-10-21 | 1998-03-19 | G.D. Searle & Co. | Amidino derivatives useful as nitric oxide synthase inhibitors |
| CA2216882A1 (en) * | 1995-04-20 | 1996-10-24 | G.D. Searle & Co. | Cyclic amidino agents useful as nitric oxide synthase inhibitors |
| US5945408A (en) * | 1996-03-06 | 1999-08-31 | G.D. Searle & Co. | Hydroxyanidino derivatives useful as nitric oxide synthase inhibitors |
| US5981556A (en) * | 1997-07-22 | 1999-11-09 | G.D. Searle & Co. | 1,3-diazolino and 1,3-diazolidino heterocycles as useful nitric oxide synthase inhibitors |
| JP2002517502A (ja) | 1998-06-10 | 2002-06-18 | ジー・ディー・サール・アンド・カンパニー | 複素二環状及び三環状一酸化窒素シンターゼ阻害剤 |
| US6344473B1 (en) | 2000-08-07 | 2002-02-05 | G.D. Searle & Co. | Imidazoles useful as nitric oxide synthase inhibitors |
| US20090137687A1 (en) * | 2003-02-28 | 2009-05-28 | Oxigene, Inc. | Compositions and Methods With Enhanced Therapeutic Activity |
| CN102924411B (zh) * | 2012-11-14 | 2016-08-17 | 兰州大学 | 雷公藤内酯醇中间体的合成方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH061746A (ja) * | 1992-04-20 | 1994-01-11 | Otsuka Pharmaceut Factory Inc | フェナンスレン誘導体及びジオキソフェナンスレン誘導体の製造法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991013855A1 (en) * | 1990-03-06 | 1991-09-19 | Otsuka Pharmaceutical Co., Ltd. | Phenanthrene derivative |
| JPH07110830B2 (ja) * | 1990-03-06 | 1995-11-29 | 大塚製薬株式会社 | フェナンスレン誘導体 |
| JPH06263688A (ja) * | 1991-09-04 | 1994-09-20 | Otsuka Pharmaceut Factory Inc | フェナンスレン誘導体 |
| JPH06192155A (ja) * | 1992-12-24 | 1994-07-12 | Otsuka Pharmaceut Co Ltd | フェナンスレン誘導体 |
| CA2111138A1 (en) * | 1993-01-15 | 1994-07-16 | Thierry Godel | Octahydrophenanthrene derivatives |
-
1994
- 1994-10-11 WO PCT/JP1994/001695 patent/WO1995010266A1/ja not_active Application Discontinuation
- 1994-10-11 EP EP94929028A patent/EP0676196A1/en not_active Withdrawn
- 1994-10-11 US US08/495,645 patent/US5654343A/en not_active Expired - Fee Related
- 1994-10-11 KR KR1019950702413A patent/KR950703941A/ko not_active Application Discontinuation
- 1994-10-11 AU AU78224/94A patent/AU684553B2/en not_active Ceased
- 1994-10-12 TW TW083109450A patent/TW312623B/zh active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH061746A (ja) * | 1992-04-20 | 1994-01-11 | Otsuka Pharmaceut Factory Inc | フェナンスレン誘導体及びジオキソフェナンスレン誘導体の製造法 |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, Vol. 118 (1993), Abstract No. 4551. * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5807886A (en) * | 1994-05-07 | 1998-09-15 | Astra Aktiebolag | Bicyclic amidine dervatives as inhibitors of nitric oxide synthetase |
| US6117898A (en) * | 1994-05-07 | 2000-09-12 | Astra Aktiebolag | Bicyclic amidine derivatives as inhibitors of nitric oxide synthetase |
| US5849782A (en) * | 1995-01-13 | 1998-12-15 | The General Hospital Corporation | Methods of inhibiting neurodegenerative diseases |
| US6133306A (en) * | 1995-01-13 | 2000-10-17 | The General Hospital Corporation | Methods of inhibiting neurodegerative diseases |
| US6380223B1 (en) | 1999-04-30 | 2002-04-30 | Pfizer Inc. | Glucocorticoid receptor modulators |
| US6699893B2 (en) | 1999-04-30 | 2004-03-02 | Pfizer Inc | Glucocorticoid receptor modulators |
| US7166593B2 (en) | 1999-04-30 | 2007-01-23 | Pfizer, Inc. | Glucocorticoid receptor modulators |
| BG66055B1 (bg) * | 1999-04-30 | 2010-12-30 | Pfizer Products Inc. | Използване на модулатори на глюкокортикоидния рецептор |
| US7713989B2 (en) | 2000-04-27 | 2010-05-11 | Dow Robert L | Glucocorticoid receptor modulators |
| US6852719B2 (en) | 2000-10-30 | 2005-02-08 | Pfizer Inc. | Glucocorticoid receptor modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| KR950703941A (ko) | 1995-11-17 |
| TW312623B (ja) | 1997-08-11 |
| EP0676196A1 (en) | 1995-10-11 |
| AU684553B2 (en) | 1997-12-18 |
| AU7822494A (en) | 1995-05-04 |
| US5654343A (en) | 1997-08-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0045118B1 (en) | Intermediates for the synthesis of bicyclo (2,2,1) heptanes and bicyclo (2,2,1) hept-2z-enes | |
| EP0011928B1 (en) | 3,5-dihydroxypentanoic ester derivatives having antihyperlipaemic activity, their manufacture and pharmaceutical compositions containing them | |
| FI87792B (fi) | Tigogenin-cellubiosid-okta-acetat | |
| DeShong et al. | A general approach to the stereoselective synthesis of spiroketals. A total synthesis of the pheromones of the olive fruit fly and related compounds | |
| WO1995010266A1 (en) | Nitrogen monoxide synthesis inhibitor | |
| EP0747348A1 (en) | Aminoalkylcyclopropane derivative | |
| GB1599863A (en) | Pharmaceutical compositions for use in the inhibition of the biosynthesis of mevalonic acid | |
| CH621803A5 (ja) | ||
| GB1574246A (en) | Prostaglandin derivatives and process for preparing the same | |
| US5116960A (en) | Amphotericin b derivatives | |
| US3524867A (en) | Process for producing cyclopenta (b)pyrans | |
| US5047574A (en) | Certain optically active mono esters of dicarboxylic acids | |
| PL163128B1 (pl) | Sposób wytwarzania adduktu aldehydu PL | |
| EP0100900B1 (en) | Novel (3.2.0.) bicycloheptanone oxime ethers with valuable therapeutic properties | |
| JPS6317077B2 (ja) | ||
| JP2743121B2 (ja) | 一酸化窒素合成阻害剤 | |
| US4060691A (en) | 7-{3-Hydroxy-2-[4-hydroxy-4-(lower alkyl)-trans-1-octen-1-yl]-5-oxocyclopent-1-yl}heptanoic acids and esters | |
| JPS5910359B2 (ja) | シンキナチツソガンユウタカンカゴウブツ ノ チヨウセイホウホウ | |
| HU197870B (en) | Process for producing 8-/lower alkyl/bicyclo/4.2.0/oktan derivatives, salts and esters and pharmaceutical compositions containing them as active components | |
| US5183909A (en) | Mono ester of dicarboxylic acid | |
| Ando et al. | Synthetic Studies of Sesquiterpenes with a Cis-Fused Decalin System, 5. A Synthetic Approach to the Study of Structure-Activity Relationships of the Termiticidal Norsesquiterpenoids, Chamaecynone and Related Compounds | |
| JPH02167284A (ja) | プロスタグランジン前駆体およびその製法 | |
| JP2640239B2 (ja) | プロトスタ−13(17)エン−3,16−ジオン化合物 | |
| US5175341A (en) | Mono ester of dicarboxylic acid and process thereof | |
| JPS6248639A (ja) | 薬剤化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08495645 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994929028 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWP | Wipo information: published in national office |
Ref document number: 1994929028 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994929028 Country of ref document: EP |