TW202128991A - 抗vegf蛋白組成物及其製備方法 - Google Patents
抗vegf蛋白組成物及其製備方法 Download PDFInfo
- Publication number
- TW202128991A TW202128991A TW109128144A TW109128144A TW202128991A TW 202128991 A TW202128991 A TW 202128991A TW 109128144 A TW109128144 A TW 109128144A TW 109128144 A TW109128144 A TW 109128144A TW 202128991 A TW202128991 A TW 202128991A
- Authority
- TW
- Taiwan
- Prior art keywords
- seq
- aflibercept
- protein
- vegf
- cells
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 205
- 238000000034 method Methods 0.000 title claims abstract description 189
- 108090000623 proteins and genes Proteins 0.000 title abstract description 552
- 102000004169 proteins and genes Human genes 0.000 title abstract description 549
- 108010081667 aflibercept Proteins 0.000 claims description 195
- 229960002833 aflibercept Drugs 0.000 claims description 183
- 238000004519 manufacturing process Methods 0.000 claims description 78
- 238000003306 harvesting Methods 0.000 claims description 61
- 239000000872 buffer Substances 0.000 claims description 43
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 39
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 39
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 35
- 239000001301 oxygen Substances 0.000 claims description 35
- 229910052760 oxygen Inorganic materials 0.000 claims description 35
- 108010038807 Oligopeptides Proteins 0.000 claims description 24
- 102000015636 Oligopeptides Human genes 0.000 claims description 24
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 23
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 21
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims description 20
- 238000005571 anion exchange chromatography Methods 0.000 claims description 19
- 235000002639 sodium chloride Nutrition 0.000 claims description 18
- 238000004090 dissolution Methods 0.000 claims description 17
- 230000029087 digestion Effects 0.000 claims description 15
- 102000004190 Enzymes Human genes 0.000 claims description 12
- 108090000790 Enzymes Proteins 0.000 claims description 12
- 238000005406 washing Methods 0.000 claims description 12
- 125000000539 amino acid group Chemical group 0.000 claims description 11
- 239000012530 fluid Substances 0.000 claims description 11
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 10
- 239000011780 sodium chloride Substances 0.000 claims description 9
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 9
- 108090000631 Trypsin Proteins 0.000 claims description 8
- 102000004142 Trypsin Human genes 0.000 claims description 8
- 239000012588 trypsin Substances 0.000 claims description 8
- 125000000151 cysteine group Chemical class N[C@@H](CS)C(=O)* 0.000 claims description 7
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 claims description 6
- 229940025084 amphetamine Drugs 0.000 claims description 5
- 238000004949 mass spectrometry Methods 0.000 claims description 5
- 239000012562 protein A resin Substances 0.000 claims description 5
- 230000006862 enzymatic digestion Effects 0.000 claims description 3
- 235000018102 proteins Nutrition 0.000 description 535
- 210000004027 cell Anatomy 0.000 description 323
- 102000009524 Vascular Endothelial Growth Factor A Human genes 0.000 description 140
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 140
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 138
- 150000001413 amino acids Chemical class 0.000 description 110
- 239000000523 sample Substances 0.000 description 100
- 239000000306 component Substances 0.000 description 81
- 108090000765 processed proteins & peptides Proteins 0.000 description 80
- 230000001186 cumulative effect Effects 0.000 description 76
- 238000005349 anion exchange Methods 0.000 description 74
- 102000004196 processed proteins & peptides Human genes 0.000 description 65
- 238000007254 oxidation reaction Methods 0.000 description 60
- 238000001042 affinity chromatography Methods 0.000 description 59
- 230000003647 oxidation Effects 0.000 description 58
- 229920001184 polypeptide Polymers 0.000 description 56
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 55
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 55
- 238000004587 chromatography analysis Methods 0.000 description 53
- 238000002360 preparation method Methods 0.000 description 53
- 229960002885 histidine Drugs 0.000 description 50
- 230000002378 acidificating effect Effects 0.000 description 43
- 239000000463 material Substances 0.000 description 42
- 229940024606 amino acid Drugs 0.000 description 41
- 235000001014 amino acid Nutrition 0.000 description 41
- 230000027455 binding Effects 0.000 description 40
- 230000036252 glycation Effects 0.000 description 40
- 239000011347 resin Substances 0.000 description 40
- 229920005989 resin Polymers 0.000 description 40
- 239000012634 fragment Substances 0.000 description 36
- 102000035195 Peptidases Human genes 0.000 description 33
- 108091005804 Peptidases Proteins 0.000 description 33
- 239000004365 Protease Substances 0.000 description 33
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 33
- 239000012472 biological sample Substances 0.000 description 33
- 239000002609 medium Substances 0.000 description 30
- 241000699666 Mus <mouse, genus> Species 0.000 description 28
- 230000008569 process Effects 0.000 description 28
- 239000003795 chemical substances by application Substances 0.000 description 27
- 238000000926 separation method Methods 0.000 description 27
- 239000000243 solution Substances 0.000 description 27
- 108091008605 VEGF receptors Proteins 0.000 description 26
- 238000005341 cation exchange Methods 0.000 description 26
- 241000894007 species Species 0.000 description 25
- 239000000126 substance Substances 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 24
- 235000018417 cysteine Nutrition 0.000 description 23
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 22
- 229960002433 cysteine Drugs 0.000 description 22
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 22
- 239000000047 product Substances 0.000 description 22
- 235000019419 proteases Nutrition 0.000 description 22
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 22
- 206010012688 Diabetic retinal oedema Diseases 0.000 description 21
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 21
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 21
- 201000011190 diabetic macular edema Diseases 0.000 description 21
- 230000003993 interaction Effects 0.000 description 21
- 239000007788 liquid Substances 0.000 description 21
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 20
- 239000000427 antigen Substances 0.000 description 20
- 108091007433 antigens Proteins 0.000 description 20
- 102000036639 antigens Human genes 0.000 description 20
- 238000009472 formulation Methods 0.000 description 20
- 239000012535 impurity Substances 0.000 description 20
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical group NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 19
- 238000005516 engineering process Methods 0.000 description 19
- 235000014304 histidine Nutrition 0.000 description 19
- 150000002500 ions Chemical class 0.000 description 19
- 150000007523 nucleic acids Chemical class 0.000 description 19
- 208000004644 retinal vein occlusion Diseases 0.000 description 19
- 239000011534 wash buffer Substances 0.000 description 19
- 206010012689 Diabetic retinopathy Diseases 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 18
- 238000004255 ion exchange chromatography Methods 0.000 description 18
- 230000004048 modification Effects 0.000 description 18
- 238000012986 modification Methods 0.000 description 18
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 17
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 17
- 229960001230 asparagine Drugs 0.000 description 17
- 235000009582 asparagine Nutrition 0.000 description 17
- 238000005342 ion exchange Methods 0.000 description 17
- 108020004707 nucleic acids Proteins 0.000 description 17
- 102000039446 nucleic acids Human genes 0.000 description 17
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 16
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 16
- 239000006167 equilibration buffer Substances 0.000 description 16
- 238000011068 loading method Methods 0.000 description 16
- 229920000642 polymer Polymers 0.000 description 16
- 230000002829 reductive effect Effects 0.000 description 16
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 15
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 15
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 15
- 239000003963 antioxidant agent Substances 0.000 description 15
- 235000006708 antioxidants Nutrition 0.000 description 15
- 238000012258 culturing Methods 0.000 description 15
- 229930182817 methionine Natural products 0.000 description 15
- 235000006109 methionine Nutrition 0.000 description 15
- -1 trihydroxytryptophan Amino acid Chemical class 0.000 description 15
- 102000037865 fusion proteins Human genes 0.000 description 14
- 108020001507 fusion proteins Proteins 0.000 description 14
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 14
- 239000011159 matrix material Substances 0.000 description 14
- 238000004458 analytical method Methods 0.000 description 13
- 239000012149 elution buffer Substances 0.000 description 13
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 13
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 12
- 241000700605 Viruses Species 0.000 description 12
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 12
- 230000002779 inactivation Effects 0.000 description 12
- 238000001155 isoelectric focusing Methods 0.000 description 12
- YGPSJZOEDVAXAB-UHFFFAOYSA-N kynurenine Chemical compound OC(=O)C(N)CC(=O)C1=CC=CC=C1N YGPSJZOEDVAXAB-UHFFFAOYSA-N 0.000 description 12
- 239000012528 membrane Substances 0.000 description 12
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 12
- 235000000346 sugar Nutrition 0.000 description 12
- 239000004475 Arginine Substances 0.000 description 11
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 11
- 239000007983 Tris buffer Substances 0.000 description 11
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 11
- 229960003121 arginine Drugs 0.000 description 11
- 235000009697 arginine Nutrition 0.000 description 11
- 229960005261 aspartic acid Drugs 0.000 description 11
- 235000003704 aspartic acid Nutrition 0.000 description 11
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 229940088598 enzyme Drugs 0.000 description 11
- 210000002919 epithelial cell Anatomy 0.000 description 11
- 238000012434 mixed-mode chromatography Methods 0.000 description 11
- 230000035772 mutation Effects 0.000 description 11
- 239000000546 pharmaceutical excipient Substances 0.000 description 11
- 238000011084 recovery Methods 0.000 description 11
- 238000011144 upstream manufacturing Methods 0.000 description 11
- 208000005590 Choroidal Neovascularization Diseases 0.000 description 10
- 206010060823 Choroidal neovascularisation Diseases 0.000 description 10
- 239000004471 Glycine Substances 0.000 description 10
- 206010028980 Neoplasm Diseases 0.000 description 10
- 208000022873 Ocular disease Diseases 0.000 description 10
- 230000002491 angiogenic effect Effects 0.000 description 10
- 239000011324 bead Substances 0.000 description 10
- 239000000969 carrier Substances 0.000 description 10
- 238000004113 cell culture Methods 0.000 description 10
- 201000010099 disease Diseases 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 229960002449 glycine Drugs 0.000 description 10
- 125000005647 linker group Chemical group 0.000 description 10
- 239000012088 reference solution Substances 0.000 description 10
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 10
- 238000000108 ultra-filtration Methods 0.000 description 10
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 9
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 9
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 239000012501 chromatography medium Substances 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 238000011143 downstream manufacturing Methods 0.000 description 9
- 239000002054 inoculum Substances 0.000 description 9
- 210000003292 kidney cell Anatomy 0.000 description 9
- 239000003446 ligand Substances 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- 150000003839 salts Chemical class 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- FKJQZUQEYSGYFZ-JTQLQIEISA-N N,N-dihydroxy-L-tryptophan Chemical compound C1=CC=C2C(C[C@H](N(O)O)C(O)=O)=CNC2=C1 FKJQZUQEYSGYFZ-JTQLQIEISA-N 0.000 description 8
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 8
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 230000003078 antioxidant effect Effects 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 230000003834 intracellular effect Effects 0.000 description 8
- 230000007246 mechanism Effects 0.000 description 8
- 230000001590 oxidative effect Effects 0.000 description 8
- 230000004481 post-translational protein modification Effects 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 238000003860 storage Methods 0.000 description 8
- 239000000758 substrate Substances 0.000 description 8
- 229960004441 tyrosine Drugs 0.000 description 8
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 7
- 208000010412 Glaucoma Diseases 0.000 description 7
- 108060003951 Immunoglobulin Proteins 0.000 description 7
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 7
- 239000004472 Lysine Substances 0.000 description 7
- 208000001344 Macular Edema Diseases 0.000 description 7
- 206010025415 Macular oedema Diseases 0.000 description 7
- PNBGTYVVHKDDFM-JTQLQIEISA-N N-hydroxy-L-tryptophan Chemical compound C1=CC=C2C(C[C@H](NO)C(O)=O)=CNC2=C1 PNBGTYVVHKDDFM-JTQLQIEISA-N 0.000 description 7
- 201000011510 cancer Diseases 0.000 description 7
- 239000006143 cell culture medium Substances 0.000 description 7
- 229920002678 cellulose Polymers 0.000 description 7
- 230000003247 decreasing effect Effects 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 239000001963 growth medium Substances 0.000 description 7
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 7
- 230000002209 hydrophobic effect Effects 0.000 description 7
- 102000018358 immunoglobulin Human genes 0.000 description 7
- 201000010230 macular retinal edema Diseases 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 239000007790 solid phase Substances 0.000 description 7
- 229920001059 synthetic polymer Polymers 0.000 description 7
- 229920000936 Agarose Polymers 0.000 description 6
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- 108020004414 DNA Proteins 0.000 description 6
- 244000068988 Glycine max Species 0.000 description 6
- 235000010469 Glycine max Nutrition 0.000 description 6
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 6
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 6
- FMYVESSAZXQWAO-JTQLQIEISA-N OC([C@](N)(C(=O)O)O)(C1=CNC2=CC=CC=C12)O Chemical compound OC([C@](N)(C(=O)O)O)(C1=CNC2=CC=CC=C12)O FMYVESSAZXQWAO-JTQLQIEISA-N 0.000 description 6
- 208000002158 Proliferative Vitreoretinopathy Diseases 0.000 description 6
- 208000007135 Retinal Neovascularization Diseases 0.000 description 6
- 206010038934 Retinopathy proliferative Diseases 0.000 description 6
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 6
- MZVQCMJNVPIDEA-UHFFFAOYSA-N [CH2]CN(CC)CC Chemical group [CH2]CN(CC)CC MZVQCMJNVPIDEA-UHFFFAOYSA-N 0.000 description 6
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 6
- 210000004899 c-terminal region Anatomy 0.000 description 6
- 125000002091 cationic group Chemical group 0.000 description 6
- 201000005667 central retinal vein occlusion Diseases 0.000 description 6
- 238000005119 centrifugation Methods 0.000 description 6
- 230000003196 chaotropic effect Effects 0.000 description 6
- 238000003776 cleavage reaction Methods 0.000 description 6
- 239000003599 detergent Substances 0.000 description 6
- RCFKEIREOSXLET-UHFFFAOYSA-N disulfamide Chemical compound CC1=CC(Cl)=C(S(N)(=O)=O)C=C1S(N)(=O)=O RCFKEIREOSXLET-UHFFFAOYSA-N 0.000 description 6
- 230000002255 enzymatic effect Effects 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 230000004927 fusion Effects 0.000 description 6
- 229930182830 galactose Natural products 0.000 description 6
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 229960000789 guanidine hydrochloride Drugs 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 238000011065 in-situ storage Methods 0.000 description 6
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 6
- 230000004060 metabolic process Effects 0.000 description 6
- 229920005615 natural polymer Polymers 0.000 description 6
- 208000021971 neovascular inflammatory vitreoretinopathy Diseases 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 6
- 229960005190 phenylalanine Drugs 0.000 description 6
- 230000006785 proliferative vitreoretinopathy Effects 0.000 description 6
- 230000007017 scission Effects 0.000 description 6
- 238000010561 standard procedure Methods 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 238000004885 tandem mass spectrometry Methods 0.000 description 6
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 6
- 230000014616 translation Effects 0.000 description 6
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 6
- 230000002792 vascular Effects 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 238000003989 weak cation exchange chromatography Methods 0.000 description 6
- 230000005526 G1 to G0 transition Effects 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 206010064930 age-related macular degeneration Diseases 0.000 description 5
- 125000000129 anionic group Chemical group 0.000 description 5
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 5
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 5
- 238000005277 cation exchange chromatography Methods 0.000 description 5
- 239000001913 cellulose Substances 0.000 description 5
- 238000012512 characterization method Methods 0.000 description 5
- 239000002738 chelating agent Substances 0.000 description 5
- 239000000356 contaminant Substances 0.000 description 5
- 238000005520 cutting process Methods 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 238000011026 diafiltration Methods 0.000 description 5
- 238000010828 elution Methods 0.000 description 5
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 5
- 229960002743 glutamine Drugs 0.000 description 5
- 235000004554 glutamine Nutrition 0.000 description 5
- 230000003301 hydrolyzing effect Effects 0.000 description 5
- 208000002780 macular degeneration Diseases 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 230000002207 retinal effect Effects 0.000 description 5
- 239000002002 slurry Substances 0.000 description 5
- 150000008163 sugars Chemical class 0.000 description 5
- 230000001502 supplementing effect Effects 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 4
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 4
- XQIRHEWBKIRPOB-UHFFFAOYSA-N 2-amino-3-(1-hydroxyindol-3-yl)propanoic acid Chemical compound C1=CC=C2C(CC(N)C(O)=O)=CN(O)C2=C1 XQIRHEWBKIRPOB-UHFFFAOYSA-N 0.000 description 4
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 4
- 201000004569 Blindness Diseases 0.000 description 4
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- 108090000288 Glycoproteins Proteins 0.000 description 4
- 102000003886 Glycoproteins Human genes 0.000 description 4
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 description 4
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 description 4
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 4
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 4
- 206010025421 Macule Diseases 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- AIJULSRZWUXGPQ-UHFFFAOYSA-N Methylglyoxal Chemical compound CC(=O)C=O AIJULSRZWUXGPQ-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- DLRVVLDZNNYCBX-UHFFFAOYSA-N Polydextrose Polymers OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(O)O1 DLRVVLDZNNYCBX-UHFFFAOYSA-N 0.000 description 4
- 108010009736 Protein Hydrolysates Proteins 0.000 description 4
- 208000017442 Retinal disease Diseases 0.000 description 4
- 229920002684 Sepharose Polymers 0.000 description 4
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 4
- 230000033115 angiogenesis Effects 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 239000003124 biologic agent Substances 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- 210000001736 capillary Anatomy 0.000 description 4
- 238000002144 chemical decomposition reaction Methods 0.000 description 4
- 238000007385 chemical modification Methods 0.000 description 4
- 239000003638 chemical reducing agent Substances 0.000 description 4
- 239000012539 chromatography resin Substances 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 4
- 229960003957 dexamethasone Drugs 0.000 description 4
- 238000006471 dimerization reaction Methods 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 238000011067 equilibration Methods 0.000 description 4
- 208000030533 eye disease Diseases 0.000 description 4
- 239000000710 homodimer Substances 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- 238000002386 leaching Methods 0.000 description 4
- 229960003136 leucine Drugs 0.000 description 4
- 235000005772 leucine Nutrition 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000012160 loading buffer Substances 0.000 description 4
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- 229960004452 methionine Drugs 0.000 description 4
- 238000012433 multimodal chromatography Methods 0.000 description 4
- 201000003142 neovascular glaucoma Diseases 0.000 description 4
- 238000006386 neutralization reaction Methods 0.000 description 4
- 229910052759 nickel Inorganic materials 0.000 description 4
- 235000015097 nutrients Nutrition 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 239000011148 porous material Substances 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 230000013777 protein digestion Effects 0.000 description 4
- 230000002797 proteolythic effect Effects 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 4
- 239000001488 sodium phosphate Substances 0.000 description 4
- 229910000162 sodium phosphate Inorganic materials 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 230000009469 supplementation Effects 0.000 description 4
- 239000012085 test solution Substances 0.000 description 4
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 4
- 229960004799 tryptophan Drugs 0.000 description 4
- 239000013598 vector Substances 0.000 description 4
- 238000012795 verification Methods 0.000 description 4
- 238000011100 viral filtration Methods 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- 229910052725 zinc Inorganic materials 0.000 description 4
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 3
- CHRJZRDFSQHIFI-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;styrene Chemical compound C=CC1=CC=CC=C1.C=CC1=CC=CC=C1C=C CHRJZRDFSQHIFI-UHFFFAOYSA-N 0.000 description 3
- LDCYZAJDBXYCGN-VIFPVBQESA-N 5-hydroxy-L-tryptophan Chemical compound C1=C(O)C=C2C(C[C@H](N)C(O)=O)=CNC2=C1 LDCYZAJDBXYCGN-VIFPVBQESA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 101100075829 Caenorhabditis elegans mab-3 gene Proteins 0.000 description 3
- 101100476210 Caenorhabditis elegans rnt-1 gene Proteins 0.000 description 3
- 206010055665 Corneal neovascularisation Diseases 0.000 description 3
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- 206010016654 Fibrosis Diseases 0.000 description 3
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 3
- 108010051815 Glutamyl endopeptidase Proteins 0.000 description 3
- 108010024636 Glutathione Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 description 3
- 206010065630 Iris neovascularisation Diseases 0.000 description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 3
- 239000012515 MabSelect SuRe Substances 0.000 description 3
- 206010029113 Neovascularisation Diseases 0.000 description 3
- 241001111421 Pannus Species 0.000 description 3
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 3
- 102100035194 Placenta growth factor Human genes 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 201000002154 Pterygium Diseases 0.000 description 3
- 206010038923 Retinopathy Diseases 0.000 description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 3
- 101710120037 Toxin CcdB Proteins 0.000 description 3
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 3
- 229930003268 Vitamin C Natural products 0.000 description 3
- 238000007792 addition Methods 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 201000007917 background diabetic retinopathy Diseases 0.000 description 3
- 238000010923 batch production Methods 0.000 description 3
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 3
- 230000036983 biotransformation Effects 0.000 description 3
- 238000004422 calculation algorithm Methods 0.000 description 3
- 150000001720 carbohydrates Chemical class 0.000 description 3
- 235000014633 carbohydrates Nutrition 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 201000000159 corneal neovascularization Diseases 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 210000003527 eukaryotic cell Anatomy 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000013604 expression vector Substances 0.000 description 3
- 210000002950 fibroblast Anatomy 0.000 description 3
- 230000004761 fibrosis Effects 0.000 description 3
- 238000013467 fragmentation Methods 0.000 description 3
- 238000006062 fragmentation reaction Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 229960003180 glutathione Drugs 0.000 description 3
- 235000003969 glutathione Nutrition 0.000 description 3
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- VVIUBCNYACGLLV-UHFFFAOYSA-N hypotaurine Chemical compound [NH3+]CCS([O-])=O VVIUBCNYACGLLV-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- AGBQKNBQESQNJD-UHFFFAOYSA-M lipoate Chemical compound [O-]C(=O)CCCCC1CCSS1 AGBQKNBQESQNJD-UHFFFAOYSA-M 0.000 description 3
- 235000019136 lipoic acid Nutrition 0.000 description 3
- 238000004811 liquid chromatography Methods 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 150000002739 metals Chemical class 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 210000002706 plastid Anatomy 0.000 description 3
- 229920005862 polyol Polymers 0.000 description 3
- 150000003077 polyols Chemical class 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 238000011027 product recovery Methods 0.000 description 3
- 201000007914 proliferative diabetic retinopathy Diseases 0.000 description 3
- 230000001172 regenerating effect Effects 0.000 description 3
- 239000013017 sartobind Substances 0.000 description 3
- 229960001153 serine Drugs 0.000 description 3
- 230000009450 sialylation Effects 0.000 description 3
- 238000001542 size-exclusion chromatography Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 230000035882 stress Effects 0.000 description 3
- 239000013589 supplement Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 229960003080 taurine Drugs 0.000 description 3
- 229960002663 thioctic acid Drugs 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 3
- 229940045145 uridine Drugs 0.000 description 3
- 210000003556 vascular endothelial cell Anatomy 0.000 description 3
- 230000004393 visual impairment Effects 0.000 description 3
- 238000011179 visual inspection Methods 0.000 description 3
- 235000019154 vitamin C Nutrition 0.000 description 3
- 239000011718 vitamin C Substances 0.000 description 3
- 238000012784 weak cation exchange Methods 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- KXHSJJRXZAKKRP-BYPYZUCNSA-N (2s)-2-amino-3-(2-oxo-1,3-dihydroimidazol-4-yl)propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CNC(=O)N1 KXHSJJRXZAKKRP-BYPYZUCNSA-N 0.000 description 2
- BMVXCPBXGZKUPN-UHFFFAOYSA-N 1-hexanamine Chemical compound CCCCCCN BMVXCPBXGZKUPN-UHFFFAOYSA-N 0.000 description 2
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- 229940058020 2-amino-2-methyl-1-propanol Drugs 0.000 description 2
- UXFQFBNBSPQBJW-UHFFFAOYSA-N 2-amino-2-methylpropane-1,3-diol Chemical compound OCC(N)(C)CO UXFQFBNBSPQBJW-UHFFFAOYSA-N 0.000 description 2
- IVLXQGJVBGMLRR-UHFFFAOYSA-N 2-aminoacetic acid;hydron;chloride Chemical compound Cl.NCC(O)=O IVLXQGJVBGMLRR-UHFFFAOYSA-N 0.000 description 2
- 239000001763 2-hydroxyethyl(trimethyl)azanium Substances 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 244000247812 Amorphophallus rivieri Species 0.000 description 2
- 235000001206 Amorphophallus rivieri Nutrition 0.000 description 2
- 206010058298 Argininosuccinate synthetase deficiency Diseases 0.000 description 2
- 102000014914 Carrier Proteins Human genes 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- 229920001661 Chitosan Polymers 0.000 description 2
- 235000019743 Choline chloride Nutrition 0.000 description 2
- 108090000317 Chymotrypsin Proteins 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 201000011297 Citrullinemia Diseases 0.000 description 2
- 101100007328 Cocos nucifera COS-1 gene Proteins 0.000 description 2
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 2
- 108010005843 Cysteine Proteases Proteins 0.000 description 2
- 102000005927 Cysteine Proteases Human genes 0.000 description 2
- LEVWYRKDKASIDU-QWWZWVQMSA-N D-cystine Chemical compound OC(=O)[C@H](N)CSSC[C@@H](N)C(O)=O LEVWYRKDKASIDU-QWWZWVQMSA-N 0.000 description 2
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 2
- 108091006020 Fc-tagged proteins Proteins 0.000 description 2
- 229920002148 Gellan gum Polymers 0.000 description 2
- 102000005720 Glutathione transferase Human genes 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000851030 Homo sapiens Vascular endothelial growth factor receptor 3 Proteins 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 101150088608 Kdr gene Proteins 0.000 description 2
- 229920002752 Konjac Polymers 0.000 description 2
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 101710175625 Maltose/maltodextrin-binding periplasmic protein Proteins 0.000 description 2
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 108090000526 Papain Proteins 0.000 description 2
- 108090000284 Pepsin A Proteins 0.000 description 2
- 102000057297 Pepsin A Human genes 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920001100 Polydextrose Polymers 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 108010059712 Pronase Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 229920005654 Sephadex Polymers 0.000 description 2
- 239000012507 Sephadex™ Substances 0.000 description 2
- 241000193996 Streptococcus pyogenes Species 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108090001109 Thermolysin Proteins 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 244000000188 Vaccinium ovalifolium Species 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 108010073925 Vascular Endothelial Growth Factor B Proteins 0.000 description 2
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 2
- 102000016549 Vascular Endothelial Growth Factor Receptor-2 Human genes 0.000 description 2
- 102100038217 Vascular endothelial growth factor B Human genes 0.000 description 2
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 2
- 229930003427 Vitamin E Natural products 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 210000002534 adenoid Anatomy 0.000 description 2
- 229960003767 alanine Drugs 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 150000001412 amines Chemical group 0.000 description 2
- CBTVGIZVANVGBH-UHFFFAOYSA-N aminomethyl propanol Chemical compound CC(C)(N)CO CBTVGIZVANVGBH-UHFFFAOYSA-N 0.000 description 2
- 239000012491 analyte Substances 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical group OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 2
- 108091008324 binding proteins Proteins 0.000 description 2
- 230000008827 biological function Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 210000000424 bronchial epithelial cell Anatomy 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000013019 capto adhere Substances 0.000 description 2
- 235000010418 carrageenan Nutrition 0.000 description 2
- 239000000679 carrageenan Substances 0.000 description 2
- 229920001525 carrageenan Polymers 0.000 description 2
- 229940113118 carrageenan Drugs 0.000 description 2
- 239000012930 cell culture fluid Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 102000021178 chitin binding proteins Human genes 0.000 description 2
- 108091011157 chitin binding proteins Proteins 0.000 description 2
- VXIVSQZSERGHQP-UHFFFAOYSA-N chloroacetamide Chemical compound NC(=O)CCl VXIVSQZSERGHQP-UHFFFAOYSA-N 0.000 description 2
- SGMZJAMFUVOLNK-UHFFFAOYSA-M choline chloride Chemical compound [Cl-].C[N+](C)(C)CCO SGMZJAMFUVOLNK-UHFFFAOYSA-M 0.000 description 2
- 229960003178 choline chloride Drugs 0.000 description 2
- 238000011210 chromatographic step Methods 0.000 description 2
- 229960002376 chymotrypsin Drugs 0.000 description 2
- 238000005352 clarification Methods 0.000 description 2
- 210000001728 clone cell Anatomy 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000004040 coloring Methods 0.000 description 2
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 2
- 229960003067 cystine Drugs 0.000 description 2
- 229960000958 deferoxamine Drugs 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 230000000593 degrading effect Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 239000003398 denaturant Substances 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 238000002845 discoloration Methods 0.000 description 2
- OQALFHMKVSJFRR-UHFFFAOYSA-N dityrosine Chemical compound OC(=O)C(N)CC1=CC=C(O)C(C=2C(=CC=C(CC(N)C(O)=O)C=2)O)=C1 OQALFHMKVSJFRR-UHFFFAOYSA-N 0.000 description 2
- 229940126534 drug product Drugs 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 210000003238 esophagus Anatomy 0.000 description 2
- 230000004438 eyesight Effects 0.000 description 2
- 229940051306 eylea Drugs 0.000 description 2
- 230000033581 fucosylation Effects 0.000 description 2
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 2
- 238000001879 gelation Methods 0.000 description 2
- 235000010492 gellan gum Nutrition 0.000 description 2
- 239000000216 gellan gum Substances 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 210000004907 gland Anatomy 0.000 description 2
- 239000012561 harvest cell culture fluid Substances 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 239000000413 hydrolysate Substances 0.000 description 2
- 125000001165 hydrophobic group Chemical group 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 125000003010 ionic group Chemical group 0.000 description 2
- 235000014705 isoleucine Nutrition 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 210000002510 keratinocyte Anatomy 0.000 description 2
- 239000000252 konjac Substances 0.000 description 2
- 235000010485 konjac Nutrition 0.000 description 2
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000002751 lymph Anatomy 0.000 description 2
- 210000003563 lymphoid tissue Anatomy 0.000 description 2
- 239000012516 mab select resin Substances 0.000 description 2
- 229910052748 manganese Inorganic materials 0.000 description 2
- 239000011572 manganese Substances 0.000 description 2
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 2
- 239000012092 media component Substances 0.000 description 2
- 239000012533 medium component Substances 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 210000000944 nerve tissue Anatomy 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000005457 optimization Methods 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000002741 palatine tonsil Anatomy 0.000 description 2
- 229940055729 papain Drugs 0.000 description 2
- 235000019834 papain Nutrition 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 229940111202 pepsin Drugs 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 239000001259 polydextrose Substances 0.000 description 2
- 235000013856 polydextrose Nutrition 0.000 description 2
- 229940035035 polydextrose Drugs 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 229960002429 proline Drugs 0.000 description 2
- 235000019833 protease Nutrition 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 229940076279 serotonin Drugs 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 239000012798 spherical particle Substances 0.000 description 2
- 210000000278 spinal cord Anatomy 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 239000012086 standard solution Substances 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 150000003440 styrenes Chemical class 0.000 description 2
- 108010059339 submandibular proteinase A Proteins 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 229960002898 threonine Drugs 0.000 description 2
- WRCITXQNXAIKLR-UHFFFAOYSA-N tiadenol Chemical compound OCCSCCCCCCCCCCSCCO WRCITXQNXAIKLR-UHFFFAOYSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 229960004295 valine Drugs 0.000 description 2
- 230000008728 vascular permeability Effects 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 229920001567 vinyl ester resin Polymers 0.000 description 2
- 230000000007 visual effect Effects 0.000 description 2
- 235000019165 vitamin E Nutrition 0.000 description 2
- 229940046009 vitamin E Drugs 0.000 description 2
- 239000011709 vitamin E Substances 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 2
- VIYKYVYAKVNDPS-HKGPVOKGSA-N (2s)-2-azanyl-3-[3,4-bis(oxidanyl)phenyl]propanoic acid Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1.OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 VIYKYVYAKVNDPS-HKGPVOKGSA-N 0.000 description 1
- YLHMQLRPSYHUFC-JTQLQIEISA-N (2s)-3-(1h-indol-3-yl)-2-nitrosopropanoic acid Chemical compound C1=CC=C2C(C[C@@H](C(=O)O)N=O)=CNC2=C1 YLHMQLRPSYHUFC-JTQLQIEISA-N 0.000 description 1
- 229920002818 (Hydroxyethyl)methacrylate Polymers 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- WOUANPHGFPAJCA-UHFFFAOYSA-N 2-[benzyl(methyl)amino]ethanol Chemical compound OCCN(C)CC1=CC=CC=C1 WOUANPHGFPAJCA-UHFFFAOYSA-N 0.000 description 1
- FOWNDZJYGGTHRO-DKWTVANSSA-N 2-aminoacetic acid;(2s)-2-aminobutanedioic acid Chemical compound NCC(O)=O.OC(=O)[C@@H](N)CC(O)=O FOWNDZJYGGTHRO-DKWTVANSSA-N 0.000 description 1
- SRSAMLMFVUJPSR-UHFFFAOYSA-N 2-carboxyethylphosphanium;chloride Chemical compound Cl.OC(=O)CCP SRSAMLMFVUJPSR-UHFFFAOYSA-N 0.000 description 1
- KIUMMUBSPKGMOY-UHFFFAOYSA-N 3,3'-Dithiobis(6-nitrobenzoic acid) Chemical compound C1=C([N+]([O-])=O)C(C(=O)O)=CC(SSC=2C=C(C(=CC=2)[N+]([O-])=O)C(O)=O)=C1 KIUMMUBSPKGMOY-UHFFFAOYSA-N 0.000 description 1
- PBVAJRFEEOIAGW-UHFFFAOYSA-N 3-[bis(2-carboxyethyl)phosphanyl]propanoic acid;hydrochloride Chemical compound Cl.OC(=O)CCP(CCC(O)=O)CCC(O)=O PBVAJRFEEOIAGW-UHFFFAOYSA-N 0.000 description 1
- ZGCHLOWZNKRZSN-NTSWFWBYSA-N 3-deoxyglucosone Chemical compound OC[C@@H](O)[C@@H](O)CC(=O)C=O ZGCHLOWZNKRZSN-NTSWFWBYSA-N 0.000 description 1
- UHPMJDGOAZMIID-UHFFFAOYSA-N 3-deoxyglucosone Natural products OCC1OC(O)C(=O)CC1O UHPMJDGOAZMIID-UHFFFAOYSA-N 0.000 description 1
- KFDVPJUYSDEJTH-UHFFFAOYSA-N 4-ethenylpyridine Chemical compound C=CC1=CC=NC=C1 KFDVPJUYSDEJTH-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 208000003120 Angiofibroma Diseases 0.000 description 1
- 108010009906 Angiopoietins Proteins 0.000 description 1
- 102000009840 Angiopoietins Human genes 0.000 description 1
- KLKHFFMNGWULBN-VKHMYHEASA-N Asn-Gly Chemical compound NC(=O)C[C@H](N)C(=O)NCC(O)=O KLKHFFMNGWULBN-VKHMYHEASA-N 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 241000228251 Aspergillus phoenicis Species 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 239000007989 BIS-Tris Propane buffer Substances 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 1
- 229910000722 Didymium Inorganic materials 0.000 description 1
- 241000224487 Didymium Species 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102000005593 Endopeptidases Human genes 0.000 description 1
- 108010059378 Endopeptidases Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 101001065501 Escherichia phage MS2 Lysis protein Proteins 0.000 description 1
- 102000018389 Exopeptidases Human genes 0.000 description 1
- 108010091443 Exopeptidases Proteins 0.000 description 1
- 101150009958 FLT4 gene Proteins 0.000 description 1
- 108010008177 Fd immunoglobulins Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 1
- 102100031706 Fibroblast growth factor 1 Human genes 0.000 description 1
- 101150048336 Flt1 gene Proteins 0.000 description 1
- 239000012554 Fractogel® EMD TMAE Hicap (M) Substances 0.000 description 1
- 244000182067 Fraxinus ornus Species 0.000 description 1
- 235000002917 Fraxinus ornus Nutrition 0.000 description 1
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101100372760 Homo sapiens FLT1 gene Proteins 0.000 description 1
- 108010020056 Hydrogenase Proteins 0.000 description 1
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 1
- 108010093096 Immobilized Enzymes Proteins 0.000 description 1
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 1
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 1
- 102000016844 Immunoglobulin-like domains Human genes 0.000 description 1
- 108050006430 Immunoglobulin-like domains Proteins 0.000 description 1
- 108700036276 KH902 fusion Proteins 0.000 description 1
- 125000000570 L-alpha-aspartyl group Chemical group [H]OC(=O)C([H])([H])[C@]([H])(N([H])[H])C(*)=O 0.000 description 1
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 1
- QEFRNWWLZKMPFJ-YGVKFDHGSA-N L-methionine (R)-S-oxide group Chemical group N[C@@H](CCS(=O)C)C(=O)O QEFRNWWLZKMPFJ-YGVKFDHGSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010063312 Metalloproteins Proteins 0.000 description 1
- 102000010750 Metalloproteins Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 description 1
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 1
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 description 1
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 1
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 1
- HDFGOPSGAURCEO-UHFFFAOYSA-N N-ethylmaleimide Chemical compound CCN1C(=O)C=CC1=O HDFGOPSGAURCEO-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 108020004485 Nonsense Codon Proteins 0.000 description 1
- 102000011931 Nucleoproteins Human genes 0.000 description 1
- 108010061100 Nucleoproteins Proteins 0.000 description 1
- 101710116435 Outer membrane protein Proteins 0.000 description 1
- 239000012606 POROS 50 HQ resin Substances 0.000 description 1
- 244000131316 Panax pseudoginseng Species 0.000 description 1
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 description 1
- 235000003140 Panax quinquefolius Nutrition 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000000447 Peptide-N4-(N-acetyl-beta-glucosaminyl) Asparagine Amidase Human genes 0.000 description 1
- 108010055817 Peptide-N4-(N-acetyl-beta-glucosaminyl) Asparagine Amidase Proteins 0.000 description 1
- 108010089430 Phosphoproteins Proteins 0.000 description 1
- 102000007982 Phosphoproteins Human genes 0.000 description 1
- 241000235061 Pichia sp. Species 0.000 description 1
- 108010026552 Proteome Proteins 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- GBFLZEXEOZUWRN-VKHMYHEASA-N S-carboxymethyl-L-cysteine Chemical compound OC(=O)[C@@H](N)CSCC(O)=O GBFLZEXEOZUWRN-VKHMYHEASA-N 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- JZRWCGZRTZMZEH-UHFFFAOYSA-N Thiamine Natural products CC1=C(CCO)SC=[N+]1CC1=CN=C(C)N=C1N JZRWCGZRTZMZEH-UHFFFAOYSA-N 0.000 description 1
- 102000035100 Threonine proteases Human genes 0.000 description 1
- 108091005501 Threonine proteases Proteins 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 238000005411 Van der Waals force Methods 0.000 description 1
- 108010073923 Vascular Endothelial Growth Factor C Proteins 0.000 description 1
- 108010073919 Vascular Endothelial Growth Factor D Proteins 0.000 description 1
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 1
- 102100038232 Vascular endothelial growth factor C Human genes 0.000 description 1
- 102100038234 Vascular endothelial growth factor D Human genes 0.000 description 1
- 208000000208 Wet Macular Degeneration Diseases 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 239000012572 advanced medium Substances 0.000 description 1
- 238000000787 affinity precipitation Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 239000002870 angiogenesis inducing agent Substances 0.000 description 1
- 239000003011 anion exchange membrane Substances 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- FIVPIPIDMRVLAY-UHFFFAOYSA-N aspergillin Natural products C1C2=CC=CC(O)C2N2C1(SS1)C(=O)N(C)C1(CO)C2=O FIVPIPIDMRVLAY-UHFFFAOYSA-N 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- GIXWDMTZECRIJT-UHFFFAOYSA-N aurintricarboxylic acid Chemical compound C1=CC(=O)C(C(=O)O)=CC1=C(C=1C=C(C(O)=CC=1)C(O)=O)C1=CC=C(O)C(C(O)=O)=C1 GIXWDMTZECRIJT-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- VEZXCJBBBCKRPI-UHFFFAOYSA-N beta-propiolactone Chemical compound O=C1CCO1 VEZXCJBBBCKRPI-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 1
- HHKZCCWKTZRCCL-UHFFFAOYSA-N bis-tris propane Chemical compound OCC(CO)(CO)NCCCNC(CO)(CO)CO HHKZCCWKTZRCCL-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000015624 blood vessel development Effects 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 239000013622 capto Q Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 230000006652 catabolic pathway Effects 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000012832 cell culture technique Methods 0.000 description 1
- 230000007910 cell fusion Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000012461 cellulose resin Substances 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 238000010382 chemical cross-linking Methods 0.000 description 1
- 108700010039 chimeric receptor Proteins 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 230000035071 co-translational protein modification Effects 0.000 description 1
- GVPFVAHMJGGAJG-UHFFFAOYSA-L cobalt dichloride Chemical compound [Cl-].[Cl-].[Co+2] GVPFVAHMJGGAJG-UHFFFAOYSA-L 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 238000000205 computational method Methods 0.000 description 1
- 229950005748 conbercept Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- BPLKXBNWXRMHRE-UHFFFAOYSA-N copper;1,10-phenanthroline Chemical compound [Cu].C1=CN=C2C3=NC=CC=C3C=CC2=C1 BPLKXBNWXRMHRE-UHFFFAOYSA-N 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 1
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical compound NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000005137 deposition process Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- OZRNSSUDZOLUSN-LBPRGKRZSA-N dihydrofolic acid Chemical compound N=1C=2C(=O)NC(N)=NC=2NCC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OZRNSSUDZOLUSN-LBPRGKRZSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000002086 displacement chromatography Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 150000002019 disulfides Chemical class 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 229940066758 endopeptidases Drugs 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000009483 enzymatic pathway Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 238000001976 enzyme digestion Methods 0.000 description 1
- UYMKPFRHYYNDTL-UHFFFAOYSA-N ethenamine Chemical compound NC=C UYMKPFRHYYNDTL-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229960002089 ferrous chloride Drugs 0.000 description 1
- 108091005899 fibrous proteins Proteins 0.000 description 1
- 102000034240 fibrous proteins Human genes 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- FIVPIPIDMRVLAY-RBJBARPLSA-N gliotoxin Chemical compound C1C2=CC=C[C@H](O)[C@H]2N2[C@]1(SS1)C(=O)N(C)[C@@]1(CO)C2=O FIVPIPIDMRVLAY-RBJBARPLSA-N 0.000 description 1
- 229940103893 gliotoxin Drugs 0.000 description 1
- 229930190252 gliotoxin Natural products 0.000 description 1
- 102000034238 globular proteins Human genes 0.000 description 1
- 108091005896 globular proteins Proteins 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 229960004198 guanidine Drugs 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 201000011066 hemangioma Diseases 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 150000002411 histidines Chemical class 0.000 description 1
- 238000000265 homogenisation Methods 0.000 description 1
- 102000055590 human KDR Human genes 0.000 description 1
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 239000012616 hydrophobic interaction chromatography medium Substances 0.000 description 1
- 230000002706 hydrostatic effect Effects 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000000937 inactivator Effects 0.000 description 1
- JGIDSJGZGFYYNX-YUAHOQAQSA-N indian yellow Chemical compound O1[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1OC1=CC=C(OC=2C(=C(O)C=CC=2)C2=O)C2=C1 JGIDSJGZGFYYNX-YUAHOQAQSA-N 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000008863 intramolecular interaction Effects 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000003014 ion exchange membrane Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- NMCUIPGRVMDVDB-UHFFFAOYSA-L iron dichloride Chemical compound Cl[Fe]Cl NMCUIPGRVMDVDB-UHFFFAOYSA-L 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 210000003125 jurkat cell Anatomy 0.000 description 1
- BQINXKOTJQCISL-GRCPKETISA-N keto-neuraminic acid Chemical compound OC(=O)C(=O)C[C@H](O)[C@@H](N)[C@@H](O)[C@H](O)[C@H](O)CO BQINXKOTJQCISL-GRCPKETISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000012633 leachable Substances 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- XIXADJRWDQXREU-UHFFFAOYSA-M lithium acetate Chemical compound [Li+].CC([O-])=O XIXADJRWDQXREU-UHFFFAOYSA-M 0.000 description 1
- MHCFAGZWMAWTNR-UHFFFAOYSA-M lithium perchlorate Chemical compound [Li+].[O-]Cl(=O)(=O)=O MHCFAGZWMAWTNR-UHFFFAOYSA-M 0.000 description 1
- 229910001486 lithium perchlorate Inorganic materials 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229940076783 lucentis Drugs 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 239000013028 medium composition Substances 0.000 description 1
- 238000005374 membrane filtration Methods 0.000 description 1
- 229960003151 mercaptamine Drugs 0.000 description 1
- 150000002734 metacrylic acid derivatives Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- LEBYISUPSSNHTJ-UHFFFAOYSA-N methoxy-methyl-oxo-sulfanylidene-$l^{6}-sulfane Chemical compound COS(C)(=O)=S LEBYISUPSSNHTJ-UHFFFAOYSA-N 0.000 description 1
- CRVGTESFCCXCTH-UHFFFAOYSA-N methyl diethanolamine Chemical compound OCCN(C)CCO CRVGTESFCCXCTH-UHFFFAOYSA-N 0.000 description 1
- 238000001471 micro-filtration Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000003032 molecular docking Methods 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 1
- 229950006780 n-acetylglucosamine Drugs 0.000 description 1
- CERZMXAJYMMUDR-UHFFFAOYSA-N neuraminic acid Natural products NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO CERZMXAJYMMUDR-UHFFFAOYSA-N 0.000 description 1
- 235000013615 non-nutritive sweetener Nutrition 0.000 description 1
- 229910052755 nonmetal Inorganic materials 0.000 description 1
- 230000037434 nonsense mutation Effects 0.000 description 1
- 201000008106 ocular cancer Diseases 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000009928 pasteurization Methods 0.000 description 1
- 239000000813 peptide hormone Substances 0.000 description 1
- 210000001322 periplasm Anatomy 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000000419 plant extract Substances 0.000 description 1
- 210000000557 podocyte Anatomy 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229920002704 polyhistidine Polymers 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 229920001289 polyvinyl ether Polymers 0.000 description 1
- RFAKLMBNSZNUNX-UHFFFAOYSA-N potassium;isothiocyanate Chemical compound [K+].[N-]=C=S RFAKLMBNSZNUNX-UHFFFAOYSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000001023 pro-angiogenic effect Effects 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 229960000380 propiolactone Drugs 0.000 description 1
- 238000012514 protein characterization Methods 0.000 description 1
- 238000000734 protein sequencing Methods 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000007430 reference method Methods 0.000 description 1
- 238000002407 reforming Methods 0.000 description 1
- 239000012492 regenerant Substances 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 230000004233 retinal vasculature Effects 0.000 description 1
- 210000001210 retinal vessel Anatomy 0.000 description 1
- 238000001223 reverse osmosis Methods 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 239000012723 sample buffer Substances 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 238000002305 strong-anion-exchange chromatography Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- RVEZZJVBDQCTEF-UHFFFAOYSA-N sulfenic acid Chemical compound SO RVEZZJVBDQCTEF-UHFFFAOYSA-N 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000004114 suspension culture Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 235000019157 thiamine Nutrition 0.000 description 1
- KYMBYSLLVAOCFI-UHFFFAOYSA-N thiamine Chemical compound CC1=C(CCO)SCN1CC1=CN=C(C)N=C1N KYMBYSLLVAOCFI-UHFFFAOYSA-N 0.000 description 1
- 229960003495 thiamine Drugs 0.000 description 1
- 239000011721 thiamine Substances 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- PIEPQKCYPFFYMG-UHFFFAOYSA-N tris acetate Chemical compound CC(O)=O.OCC(N)(CO)CO PIEPQKCYPFFYMG-UHFFFAOYSA-N 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 239000002525 vasculotropin inhibitor Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000003501 vero cell Anatomy 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000004304 visual acuity Effects 0.000 description 1
- 210000004127 vitreous body Anatomy 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 229960002760 ziv-aflibercept Drugs 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Images






















































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/71—Receptors; Cell surface antigens; Cell surface determinants for growth factors; for growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/177—Receptors; Cell surface antigens; Cell surface determinants
- A61K38/179—Receptors; Cell surface antigens; Cell surface determinants for growth factors; for growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/18—Growth factors; Growth regulators
- A61K38/1858—Platelet-derived growth factor [PDGF]
- A61K38/1866—Vascular endothelial growth factor [VEGF]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/107—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides
- C07K1/113—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides without change of the primary structure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/18—Ion-exchange chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/22—Affinity chromatography or related techniques based upon selective absorption processes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/36—Extraction; Separation; Purification by a combination of two or more processes of different types
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/22—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against growth factors ; against growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/283—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against Fc-receptors, e.g. CD16, CD32, CD64
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/66—General methods for inserting a gene into a vector to form a recombinant vector using cleavage and ligation; Use of non-functional linkers or adaptors, e.g. linkers containing the sequence for a restriction endonuclease
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0018—Culture media for cell or tissue culture
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0018—Culture media for cell or tissue culture
- C12N5/0031—Serum-free culture media
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/06—Preparation of peptides or proteins produced by the hydrolysis of a peptide bond, e.g. hydrolysate products
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21004—Trypsin (3.4.21.4)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/80—Fraction collectors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/33—Fusion polypeptide fusions for targeting to specific cell types, e.g. tissue specific targeting, targeting of a bacterial subspecies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2529/00—Culture process characterised by the use of electromagnetic stimulation
- C12N2529/10—Stimulation by light
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/10—Plasmid DNA
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N2030/022—Column chromatography characterised by the kind of separation mechanism
- G01N2030/027—Liquid chromatography
Abstract
本文係關於包含抗VEGF蛋白之組成物及用於產生此等組成物之方法。
Description
序列表
本申請案含有序列表,該序列表已用ASCII格式以電子方式提交,且藉此以引用之方式全部併入。2020年8月13日創建之該ASCII複本名為070816-02451_SL.txt且大小為134,385位元組。領域
本發明一般係關於抗VEGF組成物及其製備方法。相關申請案之交叉引用
本申請案主張2020年8月13日提出申請之美國臨時專利申請案第63/065,012號之優先權及權益,該案之內容以引用之方式全部併入本文。
基於蛋白質之生物醫藥組成物已成為用於研究、治療眼科疾病、癌症、自體免疫疾病、感染以及其他疾病及病症之重要產品。生物醫藥代表醫藥行業增長最快之產品領域之一。
已識別出一類對血管內皮細胞具有選擇性的細胞衍生之二聚促分裂原,並將其命名為血管內皮細胞生長因子(VEGF)。
持續的血管生成(angiogenesis)可引起或加劇某些疾病,諸如牛皮癬、類風濕性關節炎、血管瘤、血管纖維瘤、糖尿病性視網膜病及新生血管性青光眼。VEGF活性之抑制劑可用於治療此等疾病及其他VEGF誘發之病理性血管生成及血管通透性疾患,諸如腫瘤血管形成(vascularization)。血管生成素及血管內皮生長因子(VEGF)家族之成員為唯一被認為對血管內皮細胞有很大特異性之生長因子。
幾種眼部病症與病理性血管生成有關。例如,年齡相關之黃斑部變性(AMD)之發展與稱為脈絡膜新生血管形成(CNV)之過程有關。CNV之滲漏會引起黃斑部水腫及黃斑部下方積液,從而導致視力喪失。糖尿病性黃斑部水腫(DME)為另一種具有血管生成組分之眼部病症。DME為糖尿病患者中度視力喪失的最普遍原因,並且為糖尿病性視網膜病之常見併發症,糖尿病性視網膜病為一種影響視網膜血管之疾病。當液體滲漏至黃斑部中心時,會發生具有臨床意義之DME,黃斑部為視網膜之光敏部分,負責清晰、直接視力。黃斑部中之液體會導致嚴重視力喪失或失明。
各種VEGF抑制劑,諸如VEGF trap Eylea (阿柏西普(aflibercept)),已經批准用於治療此等眼部病症。
本發明係關於抗VEGF蛋白,其包括作為融合蛋白的VEGF陷阱蛋白阿柏西普(VEGF trap protein aflibercept)。本發明亦係關於新的抗VEGF蛋白,亦即阿柏西普MiniTrap或VEGF MiniTrap (除非另有說明,否則統稱為MiniTrap)。本文揭露製備此等抗VEGF蛋白之方法,包括提供有效及高效手段來產生感興趣之蛋白質的產生模式。在一態樣中,本發明係關於化學成分確定之培養基(CDM)用於產生抗VEGF蛋白之用途。在一具體態樣中,感興趣之CDM係當使用時產生蛋白質樣品之彼等,其中該樣品具有黃棕色且可包含經氧化種類。在本申請案中,亦進一步揭露阿柏西普及VEGF MiniTrap之蛋白質變異體以及附隨產生方法。阿柏西普之產生
本文描述使用細胞培養基產生阿柏西普。在一實施例中,細胞培養基為化學成分確定之培養基(chemically defined medium,「CDM」)。經常使用CDM,因為它為一種不含蛋白質的化學成分確定之配方,不使用動物來源之組分,並且可確定培養基之組成。在另一實施例中,細胞培養基為大豆水解物培養基。
在一實施例中,產生重組蛋白之方法包含:(a)提供經基因工程改造以表現感興趣之重組蛋白的宿主細胞;(b)在合適的條件下在CDM中培養宿主細胞,其中該細胞表現感興趣之重組蛋白;及(c)收穫由該細胞產生之感興趣之重組蛋白的製劑。在一態樣中,感興趣之重組蛋白為抗VEGF蛋白。在一具體態樣中,抗VEGF蛋白係選自由阿柏西普及重組MiniTrap (其實例在美國專利第7,279,159號中揭露)、阿柏西普scFv及其他抗VEGF蛋白組成之群組。在一較佳態樣中,感興趣之重組蛋白為阿柏西普。
在本實施例之一態樣中,阿柏西普在合適的宿主細胞中表現。此等宿主細胞之非限制性實例包括但不限於CHO、CHO K1、EESYR®、NICE®、NS0、Sp2/0、胚胎腎細胞及BHK。
合適的CDM包括經杜爾貝寇改質之伊戈爾(Dulbecco's Modified Eagle's,DME)培養基、漢氏營養混合物、Excell培養基及IS CHO-CD培養基。熟習此項技術者已知之其他CDM亦涵蓋在本發明之範疇內。在一具體態樣中,合適的CDM為CDM1B (Regeneron)或Excell高級培養基(SAFC)。
在一實施例中,使來自包含阿柏西普之CDM培養物的經澄清之收穫物樣品經歷捕獲層析程序。在一態樣中,捕獲步驟為使用例如蛋白A之親和層析程序。在又一態樣中,親和程序之析出液呈現出某種顏色,例如,析出液可呈現出黃棕色。如在下文更詳細地描述,顏色可使用以下手段來評估:(i)歐洲顏色標準「BY」,其中進行定性目視檢查;或(ii)比色檢定,CIE L*, a*, b* (或CIELAB),其比BY系統更具量化性。然而,無論哪種情況,皆應針對蛋白質濃度將多個樣品之間的顏色評估進行標準化,以確保有意義之評估。例如,參看以下實例9,蛋白A析出液具有約2.52之「b*」值,相當於大約BY5之BY值(於蛋白A析出液中以5 g/L之蛋白質濃度量測)。若要將蛋白A析出液之顏色與另一樣品進行比較,則應針對相同蛋白質濃度進行比較。CIELAB顏色空間之b*值用於表示樣品之著色,並覆蓋藍色(-)至黃色(+)。樣品之b*值比另一樣品越高,表明樣品中之黃棕色著色比另一樣品越強。
在一實施例中,使用CDM由經基因工程改造以表現阿柏西普之宿主細胞產生阿柏西普。在一態樣中,亦產生阿柏西普之其他種類或變異體。此等變異體包括包含一或多個經氧化胺基酸殘基之阿柏西普同功型,統稱為側氧變異體(oxo-variant)。使用包含阿柏西普及其側氧變異體之CDM產生的經澄清之收穫物樣品可經歷捕獲層析程序。在一態樣中,捕獲步驟為使用例如蛋白A管柱之親和層析程序。當使用例如液相層析-質譜法(LC-MS)分析自親和析出液中提取之樣品(其可顯示或不顯示黃棕色)時,可偵測到阿柏西普之一或多種經氧化變異體。顯示經修飾之阿柏西普之某些胺基酸殘基經氧化,包括但不限於組胺酸及/或色胺酸殘基。在一態樣中,變異體可包括一或多個甲硫胺酸殘基以及其他殘基之氧化,參見下文。
在另一態樣中,變異體可包括一或多個色胺酸殘基之氧化以形成N-甲醯犬尿胺酸。在又一態樣中,變異體可包括一或多個色胺酸殘基之氧化以形成單羥基色胺酸。在一具體態樣中,蛋白質變異體可包括一或多個色胺酸殘基之氧化以形成二羥基色胺酸。在一具體態樣中,蛋白質變異體可包括一或多個色胺酸殘基之氧化以形成三羥基色胺酸。
在另一態樣中,變異體可包括一或多個選自由以下組成之群組的修飾:例如,一或多個天冬醯胺之去醯胺;一或多個天冬胺酸轉化為異天冬胺酸及/或Asn;一或多個甲硫胺酸之氧化;一或多個色胺酸氧化為N-甲醯犬尿胺酸;一或多個色胺酸氧化為單羥基色胺酸;一或多個色胺酸氧化為二羥基色胺酸;一或多個色胺酸氧化為三羥基色胺酸;一或多個精胺酸之Arg 3-去氧葡醛酮糖化(deoxyglucosonation);C端甘胺酸之移除;以及一或多個非醣化糖位點(non-glycosylated glycosite)之存在。
在另一實施例中,本發明係關於用於產生阿柏西普之方法。在一態樣中,包含阿柏西普及其變異體的經澄清之收穫物樣品經歷捕獲步驟,諸如蛋白A親和層析。在親和步驟之後,親和析出液可經歷離子交換層析。離子交換層析可為陽離子交換層析或陰離子交換層析。混合模式或多模式層析以及其他層析程序亦涵蓋在本實施例之範疇內,下面將進一步討論。在一具體態樣中,離子交換層析為陰離子交換層析(AEX)。採用AEX之合適條件包括但不限於pH值為約8.3至約8.6之Tris鹽酸鹽。在使用例如pH值為約8.3至約8.6之Tris鹽酸鹽進行平衡後,將樣品加載至AEX管柱上。加載管柱後,可使用例如平衡緩衝液將管柱洗滌一次或多次。在一具體態樣中,所用條件可促進阿柏西普及其經氧化變異體之差異層析性能,使得可在流通級分中收集包含阿柏西普而無顯著量之側氧變異體的級分,同時顯著部分之側氧變異體保留在AEX管柱之固相上,並且可在剝離管柱後獲得– 參見以下實例2,圖 11
。參看圖 11
及實例2,可在不同產生步驟之間觀察到側氧變異體之變化。例如,可藉由「色胺酸氧化水準(%)」部分(特別是「W138(+16)」管柱)中之數據來說明此變化。可觀察到,在AEX層析(AEX分離2)後,側氧變異體(具體而言,側氧色胺酸)自加載樣品中之約0.131%變為流通樣品中之約0.070%,表明使用AEX減少阿柏西普之側氧變異體。
可使用離子交換來減弱或最小化顏色。在本實施例之一態樣中,使經澄清之收穫物樣品經歷捕獲層析,例如使用蛋白A親和層析。親和管柱經溶析,並具有第一顏色,該顏色具有為其賦予之特定BY及/或b*值。此蛋白A析出液接著經歷離子交換層析,諸如陰離子交換層析(AEX)。洗滌離子交換管柱,並收集流通液,該流通液具有第二顏色,該顏色具有為其賦予之特定BY及/或b*值。在一具體態樣中,第一顏色之顏色值(「BY」或「b*」)與第二顏色之顏色值不同。在又一態樣中,如由個別BY及/或b*值所反映,與AEX流通液之第二顏色相比,蛋白A析出液之第一顏色具有更黃棕色之顏色。通常,與蛋白A析出液之第一顏色相比,AEX之後的第二顏色之黃棕色減弱。例如,使用陰離子交換將蛋白A析出液樣品中觀察到之黃棕色自約3.06之b*值(第一顏色)減弱至AEX之後的約0.96 (第二顏色)– 參見以下實例2,表 2-3
。
在實施例之一態樣中,用於AEX管柱之平衡緩衝液及洗滌緩衝液之pH值均可為約8.30至約8.60。在另一態樣中,用於AEX管柱之平衡緩衝液及洗滌緩衝液之電導率均可為約1.50至約3.00 mS/cm。
在實施例之一態樣中,平衡緩衝液及洗滌緩衝液可為約50 mM Tris鹽酸鹽。在一態樣中,剝離緩衝液包含2M氯化鈉或1N氫氧化鈉或兩者(參見表 2-2
)。
本實施例可包括不以特定順序添加一或多個步驟,諸如疏水相互作用層析(HIC)、親和層析、多模式層析、病毒滅活(例如,使用低pH值)、病毒過濾及/或超濾/滲濾以及其他熟知之層析步驟。
在一實施例中,抗VEGF蛋白在一或多個天冬醯胺處如下經醣化:G0-GlcNAc醣化;G1-GlcNAc醣化;G1S-GlcNAc醣化;G0醣化;G1醣化;G1S醣化;G2醣化;G2S醣化;G2S2醣化;G0F醣化;G2F2S醣化;G2F2S2醣化;G1F醣化;G1FS醣化;G2F醣化;G2FS醣化;G2FS2醣化;G3FS醣化;G3FS3醣化;G0-2GlcNAc醣化;Man4醣化;Man4_A1G1醣化;Man4_A1G1S1醣化;Man5醣化;Man5_A1G1醣化;Man5_A1G1S1醣化;Man6醣化;Man6_G0+磷酸醣化;Man6+磷酸醣化;及/或Man7醣化。在一態樣中,抗VEGF蛋白可為阿柏西普、抗VEGF抗體或VEGF MiniTrap。
在一態樣中,抗VEGF蛋白之組成物之醣化譜如下:約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約6%至約15%甘露糖-5及約60%至約79%半乳醣化聚醣(參見實例6)。在一態樣中,抗VEGF蛋白在約32.4%天冬醯胺123殘基及/或約27.1%天冬醯胺196殘基處具有Man5醣化。
在一實施例中,該方法可進一步包含使用醫藥學上可接受之賦形劑調配原料藥。在實施例之一態樣中,醫藥學上可接受之賦形劑可選自以下:水、緩衝劑、糖、鹽、界面活性劑、胺基酸、多元醇、螯合劑、乳化劑及防腐劑。熟習此項技術者熟知之其他賦形劑在此實施例之範圍內。
在實施例之一態樣中,調配物可適於向人類個體投與。具體而言,可藉由玻璃體內注射實現投與。在一態樣中,調配物可具有約40至約200 mg/mL感興趣之蛋白質。
調配物可用作治療或預防血管生成性眼部病症之方法,該等病症可包括:年齡相關之黃斑部變性(例如,濕性或乾性)、黃斑部水腫、視網膜靜脈阻塞後之黃斑部水腫、視網膜靜脈阻塞(RVO)、視網膜中央靜脈阻塞(CRVO)、視網膜分支靜脈阻塞(BRVO)、糖尿病性黃斑部水腫(DME)、脈絡膜新生血管形成(CNV)、虹膜新生血管形成、新生血管性青光眼、青光眼手術後纖維化、增生性玻璃體視網膜病(PVR)、視盤新生血管形成、角膜新生血管形成、視網膜新生血管形成、玻璃體新生血管形成、角膜翳、翼狀胬肉、血管性視網膜病、在患有糖尿病性黃斑部水腫之個體中的糖尿病性視網膜病;或糖尿病性視網膜病(例如非增生性糖尿病性視網膜病(例如,以約47或53之糖尿病性視網膜病嚴重程度量表(DRSS)分級為特徵),或增生性糖尿病性視網膜病;例如,在未患有DME之個體中)。VEGF MiniTrap 之產生
本文描述阿柏西普之修飾形式之產生,其中Fc部分經移除或不存在,且係稱為阿柏西普MiniTrap或VEGF MiniTrap。此MiniTrap可在包括化學成分確定之培養基(CDM)或大豆水解物培養基的細胞培養基中產生。
在一實施例中,使用CDM產生MiniTrap。在MiniTrap產生之一態樣中,使用合適的宿主且在合適的條件下產生全長阿柏西普,並進一步加工從而藉由酶促移除Fc部分產生MiniTrap。替代地,可使用合適的宿主細胞在合適的條件下產生編碼MiniTrap之基因(例如,編碼缺少其Fc部分之阿柏西普的核苷酸序列)。
在一實施例中,用於製造MiniTrap之方法包括產生全長阿柏西普融合蛋白,繼之切割Fc區。在一態樣中,方法涉及產生重組蛋白,亦即全長阿柏西普融合蛋白(參見美國專利第7,279,159號,其全部教示以引用方式併入本文),包含:(a)提供經基因工程改造以表現全長阿柏西普之宿主細胞;(b)在合適的條件下在CDM中培養宿主細胞,其中細胞表現全長阿柏西普;(c)收穫由細胞產生之全長阿柏西普的製劑;及(d)對全長阿柏西普進行酶切割,該酶切割對移除融合蛋白之Fc部分具有特異性。在另一態樣中,在熟習此項技術者熟知之合適條件下,由合適的宿主細胞表現編碼阿柏西普減去其Fc部分之核苷酸序列(參見
美國專利第7,279,159號)。
在本實施例之一態樣中,阿柏西普在合適的宿主細胞中表現。此等宿主細胞之非限制性實例包括但不限於CHO、CHO K1、EESYR®、NICE®、NS0、Sp2/0、胚胎腎細胞及BHK。
合適的CDM包括經杜爾貝寇改質之伊戈爾(DME)培養基、漢氏營養混合物、EX-CELL培養基(SAFC)及IS CHO-CD培養基(Irvine)。熟習此項技術者已知之其他CDM亦涵蓋在本發明之範疇內。在一具體態樣中,合適的CDM為CDM1B (Regeneron)或Excell培養基(SAFC)。
在一態樣中,在產生MiniTrap期間,包含感興趣之蛋白質(亦即,阿柏西普融合蛋白及/或MiniTrap)及其變異體(包括側氧變異體)的樣品可顯示某些顏色特性-黃棕色。例如,來自親和層析步驟之析出液樣品可顯示使用BY及/或b*系統量測之某種黃棕色(參見以下實例2及9)。「樣品」之示例性來源可包括親和層析,諸如蛋白A析出液;樣品可自離子交換層析之流通級分獲得;其亦可自離子交換管柱之剝離獲得-在產生過程中存在熟習此項技術者熟知的可由此分析樣品之其他來源。如上所提及且在下文中進一步描述,顏色可使用以下手段來評估:(i)歐洲顏色標準「BY」,其中進行定性目視檢查;或(ii)比色檢定,CIELAB,其比BY系統更具量化性。然而,無論哪種情況,皆應例如使用蛋白質濃度將多個樣品之間的顏色評估進行標準化,以確保樣品之間有意義的評估。
在本實施例之一態樣中,可例如使用採用蛋白酶或其酶活性變異體之蛋白水解消化對全長阿柏西普融合蛋白進行酶處理(「切割活性」)以產生VEGF MiniTrap。在此實施例之一態樣中,蛋白酶可為釀膿鏈球菌(Streptococcus pyogenes)之免疫球蛋白降解酶(IdeS)。在另一態樣中,蛋白酶可為凝血酶胰蛋白酶、胞內蛋白酶Arg-C、胞內蛋白酶Asp-N、胞內蛋白酶Glu-C、外膜蛋白酶T (OmpT)、IdeS、胰凝乳蛋白酶、胃蛋白酶、嗜熱菌蛋白酶、木瓜蛋白酶、鏈黴蛋白酶或來自齋藤麴菌(Aspergillus Saitoi)之蛋白酶。在一態樣中,蛋白酶可為半胱胺酸蛋白酶。在實施例之一具體態樣中,蛋白酶可為IdeS。在另一態樣中,蛋白酶可為IdeS之變異體。IdeS變異體之非限制性實例在下文中描述,並且包括具有由以下組成之群組中列出之胺基酸序列的多肽:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。在一態樣中,蛋白酶可固定在瓊脂糖或另一種合適的基質上。
在一態樣中,使用CDM產生感興趣之蛋白質(連同其變異體)。在一具體態樣中,感興趣之蛋白質包括阿柏西普或MiniTrap。變異體包含一或多個經氧化之胺基酸殘基,統稱為側氧變異體。經氧化殘基之實例包括但不限於一或多個組胺酸及/或色胺酸殘基,亦已使用LC-MS偵測到其他經氧化殘基並且在下文中描述,諸如經氧化之甲硫胺酸。後續層析諸如AEX可用於將給定樣品中之此等側氧變異體與感興趣之蛋白質分離,並且在本文中描述。
在一態樣中,變異體可包括一或多個色胺酸殘基之氧化以形成N-甲醯犬尿胺酸。在又一態樣中,變異體可包括一或多個色胺酸殘基之氧化以形成單羥基色胺酸。在一具體態樣中,蛋白質變異體可包括一或多個色胺酸殘基之氧化以形成二羥基色胺酸。在一具體態樣中,蛋白質變異體可包括一或多個色胺酸殘基之氧化以形成三羥基色胺酸。
在另一態樣中,側氧變異體可包括一或多種選自由以下組成之群組的修飾:一或多個天冬醯胺殘基之去醯胺;一或多個天冬胺酸轉化為異天冬胺酸及/或Asn;一或多個甲硫胺酸殘基之氧化;一或多個色胺酸殘基之氧化以形成N-甲醯犬尿胺酸;一或多個色胺酸殘基之氧化以形成單羥基色胺酸;一或多個色胺酸殘基之氧化以形成二羥基色胺酸;一或多個色胺酸殘基之氧化以形成三羥基色胺酸;一或多個精胺酸殘基之Arg 3-去氧葡醛酮糖化;C端甘胺酸之移除;以及一或多個非醣化糖位點之存在。
在一實施例中,製造MiniTrap蛋白之方法包含(a)在第一層析平台上捕獲全長阿柏西普融合蛋白,及(b)切割阿柏西普,從而形成MiniTrap蛋白,亦即
缺少其Fc域之阿柏西普。在一態樣中,第一層析載體包含親和層析介質、離子交換層析介質或疏水相互作用層析介質。在一具體態樣中,第一層析平台包含親和層析平台,諸如蛋白A。在又一態樣中,將捕獲步驟(a)之蛋白質自第一層析平台中溶析出,之後為切割步驟(b)。在仍又一態樣中,切割步驟(b)之後為第二捕獲步驟。在一具體態樣中,此第二捕獲步驟可藉由親和層析諸如蛋白A親和層析來促進。此第二捕獲步驟之流通液(包含MiniTrap)具有第一顏色,例如黃棕色,並且經量測具有特定的BY及/或b*值–參見例如
以下實例9。另外,此第二捕獲流通液之LC-MS分析可證明存在側氧變異體,其中MiniTrap之一或多個殘基經氧化(參見以下實例9)。
在又一態樣中,第二捕獲流通液可經歷離子交換層析,諸如AEX。可使用合適的緩衝液洗滌此AEX管柱,並且可收集基本上包含MiniTrap之AEX流通級分。此AEX流通級分可具有第二顏色,該顏色具有黃棕色著色,具有特定的BY及/或b*值。在又一態樣中,如藉由BY及/或b*系統所量測,第一顏色(來自第二捕獲步驟之流通液)與第二顏色(離子交換程序之流通液)具有不同顏色。在一態樣中,與第一顏色相比,在AEX之後,使用BY及/或b*值測得之第二顏色顯示出較淡的黃棕色。
在另一實施例中,步驟(b)之切割活性可使用層析管柱進行,其中將切割活性,例如酶活性附著或固定在管柱基質上。步驟(b)中使用之管柱可包含一或多種已經提到且在下文更充分描述之蛋白酶。
在一實施例中,離子交換層析程序可包含陰離子交換(AEX)層析介質。在另一態樣中,離子交換層析介質包含陽離子交換(CEX)層析介質。採用AEX之合適條件包括但不限於pH值為約8.3至約8.6之Tris鹽酸鹽。在使用例如pH值為約8.3至約8.6之Tris鹽酸鹽進行平衡後,將樣品加載至AEX管柱上。加載管柱後,可使用例如平衡緩衝液將管柱洗滌一次或多次。在一具體態樣中,所用條件可促進使用AEX的MiniTrap及其側氧變異體之差異層析性能,從而使MiniTrap實質上位於流通級分中,而側氧變異體則實質上保留在AEX管柱上並可藉由剝離管柱而收集(參見以下實例9)。
在一實例中,分析來自不同產生階段之樣品的顏色及側氧變異體之存在。參看實例9,親和流通池(來自第二蛋白A親和步驟之流通液)具有約1.58之第一b*值(參見表 9-3
)。使此第二親和流通液經歷AEX。AEX流通液具有約0.50之第二b*值,表明使用AEX後黃棕色顯著減弱。AEX管柱之剝離產生剝離樣品並且觀察到約6.10之第三b*值,表明與加載液或流通液相比,此剝離樣品具有更黃棕色之顏色。
再次參看實例9,亦進行側氧變異體分析。所分析之樣品為親和流通池(第二蛋白A親和析出液)、AEX流通液及AEX剝離液。參看表 9-5
及表 9-6
,可在不同產生步驟之間觀察到側氧變異體之變化。例如,可藉由「色胺酸氧化水準(%)」部分(特別是「W58(+16)」管柱)中之數據來說明此變化。可觀察到,在AEX層析之後,側氧變異體(特別是側氧色胺酸)自加載樣品中之約0.055%變為流通樣品中之約0.038%,表明在AEX之後側氧變異性減少。分析AEX剝離液,並且發現側氧色胺酸種類之百分比為約0.089%。當將此剝離值與加載液(以及流通液)進行比較時,似乎此側氧變異體之很大一部分保留在AEX管柱上。
本實施例可包括不以特定順序添加一或多個步驟,諸如疏水相互作用層析、親和層析、多模式層析、病毒滅活(例如,使用低pH值)、病毒過濾及/或超濾/滲濾。
本發明之一實施例係關於一種再生包含樹脂之層析管柱的方法。在實施例之一態樣中,樹脂具有固定之水解劑。在實施例之又另一態樣中,樹脂包含固定之蛋白酶。在實施例之仍另一態樣中,樹脂為FabRICATOR®樹脂或該樹脂之突變體。在實施例之一態樣中,再生包含樹脂之管柱的方法提高樹脂之反應效率。
在實施例之一態樣中,再生包含樹脂之管柱的方法包括將管柱樹脂與乙酸一起孵育。在一態樣中,所用乙酸之濃度為約0.1 M至約2 M。在一態樣中,乙酸濃度為約0.5 M。在一態樣中,將樹脂孵育至少約10分鐘。在另一態樣中,將樹脂孵育至少約30分鐘。在此實施例之仍另一態樣中,將樹脂孵育至少約50分鐘。在此實施例之仍另一態樣中,將樹脂孵育至少約100分鐘。在此實施例之仍另一態樣中,將樹脂孵育至少約200分鐘。在此實施例之仍另一態樣中,將樹脂孵育至少約300分鐘。
視情況,將管柱樹脂進一步與胍鹽酸鹽(Gu-HCl)一起孵育。在一態樣中,使用不含乙酸之Gu-HCl再生管柱樹脂。所用Gu-HCl之濃度為約1 N至約10 N。在另一態樣中,Gu-HCl之濃度為約6 N。在又一態樣中,管柱樹脂可與再生劑(乙酸、Gu-HCl)一起孵育至少約10分鐘。在仍另一態樣中,將樹脂孵育至少約30分鐘。在仍另一態樣中,將樹脂孵育至少約50分鐘。在仍另一態樣中,將該樹脂孵育至少約100分鐘。
在一實施例中,將包含樹脂之管柱儲存在乙醇中。在一態樣中,將管柱儲存在乙醇中,其中乙醇百分比為約5% v/v至約20% v/v。在一具體態樣中,使用20% v/v乙醇儲存管柱。
在一實施例中,該方法可進一步包含使用醫藥學上可接受之賦形劑調配VEGF MiniTrap。在一態樣中,醫藥學上可接受之賦形劑可選自以下:水、緩衝劑、糖、鹽、界面活性劑、胺基酸、多元醇、螯合劑、乳化劑及防腐劑。熟習此項技術者熟知之其他賦形劑在此實施例之範圍內。
本發明之調配物適於向人類個體投與。在本實施例之一態樣中,可藉由玻璃體內注射實現投與。在一態樣中,調配物可具有約40至約200 mg/mL感興趣之蛋白質。在一具體態樣中,感興趣之蛋白質為阿柏西普或阿柏西普MiniTrap。
調配物可用於治療或預防血管生成性眼部病症之方法中,該等病症可包括:年齡相關之黃斑部變性(例如,濕性或乾性)、黃斑部水腫、視網膜靜脈阻塞後之黃斑部水腫、視網膜靜脈阻塞(RVO)、視網膜中央靜脈阻塞(CRVO)、視網膜分支靜脈阻塞(BRVO)、糖尿病性黃斑部水腫(DME)、脈絡膜新生血管形成(CNV)、虹膜新生血管形成、新生血管性青光眼、青光眼手術後纖維化、增生性玻璃體視網膜病(PVR)、視盤新生血管形成、角膜新生血管形成、視網膜新生血管形成、玻璃體新生血管形成、角膜翳、翼狀胬肉、血管性視網膜病、在患有糖尿病性黃斑部水腫之個體中的糖尿病性視網膜病;或糖尿病性視網膜病(例如非增生性糖尿病性視網膜病(例如,以約47或53之糖尿病性視網膜病嚴重程度量表(DRSS)分級為特徵),或增生性糖尿病性視網膜病;例如,在不患有DME之個體中)。IdeS 之變異體
本文描述IdeS (FabRICATOR) (SEQ ID NO.: 1)或作為IdeS變異體(SEQ ID NO.: 2至16)之其他多肽在產生VEGF MiniTrap中的用途。IdeS (SEQ ID NO.: 1)在位置87、130、182及/或274處包括天冬醯胺殘基(在以下SEQ ID NO.: 1中展示為粗體且斜體的「 N*
」)。此等位置處之天冬醯胺可突變為除天冬醯胺以外之胺基酸,以形成IdeS變異體(且突變胺基酸展示為斜體且加下劃線之胺基酸):
SEQ ID NO.: 1
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI N*
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 2
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI N*
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 3
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI N*
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 4
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 5
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTFN*GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINFN*GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFIN*GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDSDGNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 6
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTFDGKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINFRGEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFIN*GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDSN*GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 7
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 8
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI N*
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 9
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N
*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 10
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI N*
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 11
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 12
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS N*
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 13
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI N*
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 14
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF N*
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 15
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF N*
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
SEQ ID NO.: 16
MRKRCYSTSAAVLAAVTLFVLSVDRGVIADSFSANQEIRYSEVTPYHVTSVWTKGVTPPANFTQGEDVFHAPYVANQGWYDITKTF D
GKDDLLCGAATAGNMLHWWFDQNKDQIKRYLEEHPEKQKINF R
GEQMFDVKEAIDTKNHQLDSKLFEYFKEKAFPYLSTKHLGVFPDHVIDMFI L
GYRLSLTNHGPTPVKEGSKDPRGGIFDAVFTRGDQSKLLTSRHDFKEKNLKEISDLIKKELTEGKALGLSHTYANVRINHVINLWGADFDS D
GNLKAIYVTDSDSNASIGMKKYFVGVNSAGKVAISAKEIKEDNIGAQVLGLFTLSTGQDSWNQTN
在一實施例中,多肽具有經分離之胺基酸序列,其在經分離之胺基酸序列之全長上與由以下組成之群組中列出的具有至少70%序列一致性:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。在一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有至少約80%序列一致性。在另一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有至少約90%序列一致性。在另一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有約100%序列一致性。在一態樣中,多肽能夠將靶蛋白切割成片段。在一具體態樣中,靶蛋白為IgG。在另一態樣中,靶蛋白為融合蛋白。在又另一態樣中,片段可包含Fab片段及/或Fc片段。
SEQ ID 1, NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。
本文亦包括經分離之核酸分子,其編碼具有經分離之胺基酸序列的多肽,該胺基酸序列在經分離之胺基酸序列之全長上與由以下組成之群組中列出的具有至少70%序列一致性:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。在一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有至少約80%序列一致性。在另一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有至少約90%序列一致性。在另一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有約100%序列一致性。在一態樣中,多肽能夠將靶蛋白切割成片段。在一具體態樣中,靶蛋白為IgG。在另一具體態樣中,靶蛋白為融合蛋白。在仍另一具體態樣中,片段可包含Fab片段及/或Fc片段。
本文亦包括一種載體,該載體包含編碼具有經分離之胺基酸序列的多肽之核酸,該胺基酸序列在經分離之胺基酸序列之全長上與由以下組成之群組中列出的具有至少70%序列一致性:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。在一態樣中,核酸分子與表現控制序列可操作地連接,該表現控制序列能夠指導其在宿主細胞中之表現。在一態樣中,載體可為質體。在一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有至少約80%序列一致性。在另一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有至少約90%序列一致性。在另一態樣中,經分離之胺基酸序列在經分離之胺基酸序列之全長上具有約100%序列一致性。在一態樣中,多肽能夠將靶蛋白切割成片段。在一具體態樣中,靶蛋白為IgG。在另一態樣中,靶蛋白為融合蛋白。在又另一態樣中,片段可包含Fab片段及/或Fc片段。
在一實施例中,經分離之胺基酸可包含由SEQ ID NO.: 1定義之親本胺基酸序列,其在位置87、130、182及/或274處的天冬醯胺殘基突變為除天冬醯胺以外之胺基酸。在一態樣中,與親本胺基酸序列相比,該突變可使在鹼性pH值下之化學穩定性提高。在另一態樣中,與親本胺基酸序列相比,該突變可使在鹼性pH值下之化學穩定性提高50%。在一態樣中,胺基酸可選自天冬胺酸、白胺酸及精胺酸。在一具體態樣中,位置87處之天冬醯胺殘基突變為天冬胺酸殘基。在另一態樣中,位置130處之天冬醯胺殘基突變為精胺酸殘基。在仍另一態樣中,位置182處之天冬醯胺殘基突變為白胺酸殘基。在仍另一態樣中,位置274處之天冬醯胺殘基突變為天冬胺酸殘基。在仍另一態樣中,位置87及130處之天冬醯胺殘基經突變。在仍另一態樣中,位置87及182處之天冬醯胺殘基經突變。在仍另一態樣中,位置87及274處之天冬醯胺殘基經突變。在仍另一態樣中,位置130及182處之天冬醯胺殘基經突變。在仍另一態樣中,位置130及274處之天冬醯胺殘基經突變。在仍另一態樣中,位置182及274處之天冬醯胺殘基經突變。在仍另一態樣中,位置87、130及182處之天冬醯胺殘基經突變。在仍另一態樣中,位置87、182及274處之天冬醯胺殘基經突變。在仍另一態樣中,位置130、182及274處之天冬醯胺殘基經突變。在仍另一態樣中,位置87、130、182及274處之天冬醯胺殘基經突變。
在一相關實施例中,本文包括經分離之核酸分子,其編碼具有經分離之胺基酸序列的多肽,該胺基酸序列包含由SEQ ID NO.: 1定義之親本胺基酸序列,其在位置87、130、182及/或274處的天冬醯胺殘基突變為除天冬醯胺以外之胺基酸-參見上文。與親本胺基酸序列相比,該突變可使在鹼性pH值下之化學穩定性提高。
在又一相關實施例中,本文包括一種載體,其包含編碼具有經分離之胺基酸序列之多肽的核酸分子,該胺基酸序列包含由SEQ ID NO.: 1定義之親本胺基酸序列,其在位置87、130、182及/或274處的天冬醯胺殘基突變為除天冬醯胺以外之胺基酸-參見上文。與親本胺基酸序列相比,該突變可使在鹼性pH值下之化學穩定性提高。在一態樣中,核酸分子與表現控制序列可操作地連接,該表現控制序列能夠指導其在宿主細胞中之表現。在一態樣中,載體可為質體。基於親和力之產生
本文亦提供使用親和層析在抗VEGF蛋白產生期間減少宿主細胞蛋白以及其他不期望有的蛋白及核酸之方法。
在一實施例中,產生重組蛋白之方法包含:(a)提供經基因工程改造以表現感興趣之重組蛋白的宿主細胞;(b)在宿主細胞表現感興趣之重組蛋白的合適條件下培養宿主細胞;以及(c)收穫由細胞產生的感興趣之重組蛋白的製劑。在一態樣中,感興趣之重組蛋白為抗VEGF蛋白。在一具體態樣中,抗VEGF蛋白係選自由以下組成之群組:阿柏西普、MiniTrap、重組MiniTrap (其實例在美國專利第7,279,159號中揭露)、scFv及其他抗VEGF蛋白。
在本實施例之一態樣中,感興趣之重組蛋白在合適的宿主細胞中表現。合適的宿主細胞之非限制性實例包括但不限於CHO、CHO K1、EESYR®、NICE®、NS0、Sp2/0、胚胎腎細胞及BHK。
在本實施例之一態樣中,感興趣之重組蛋白在CDM中培養。合適的CDM包括經杜爾貝寇改質之伊戈爾(DME)培養基、漢氏營養混合物、Excell培養基、IS CHO-CD培養基及CDM1B。熟習此項技術者已知之其他CDM亦涵蓋在本發明之範疇內。
除了感興趣之重組蛋白外,產生製劑亦可包含至少一種污染物,包括一或多種宿主細胞蛋白。該至少一種污染物可源自細胞受質、細胞培養物或下游製程。
在一實施例中,本發明係關於使用親和層析自生物樣品產生抗VEGF蛋白之方法。在一具體態樣中,本文所揭露之方法可用於將抗VEGF蛋白與一或多種宿主細胞蛋白及在抗VEGF蛋白之培養產生過程中形成之核酸(例如DNA)至少部分地分離。
在一態樣中,該方法可包含在合適的條件下對包含抗VEGF蛋白及伴隨污染物之生物樣品進行親和層析。在一具體態樣中,親和層析可包含能夠選擇性或特異性地結合至抗VEGF蛋白(「捕獲」)之材料。該層析材料之非限制性實例包括:蛋白A、蛋白G、包含例如能夠結合至抗VEGF蛋白之蛋白質的層析材料以及包含Fc結合蛋白之層析材料。在一特定態樣中,能夠與抗VEGF蛋白結合或相互作用的蛋白質可為抗體、融合蛋白或其片段。能夠選擇性或特異性地結合至抗VEGF蛋白的該材料之非限制性實例描述於實例7中。
在本實施例之一態樣中,該方法可包含使包含抗VEGF蛋白及一或多種宿主細胞蛋白/污染物之生物樣品在合適的條件下進行親和層析,其中親和層析固定相包含能夠選擇性或特異性地結合至抗VEGF蛋白之蛋白質。在一具體態樣中,蛋白質可為抗體、融合蛋白、scFv或抗體片段。在一特定態樣中,蛋白質可為VEGF165
、VEGF121
、來自其他物種諸如兔之VEGF形式。例如,如表 7-1
及表 7-10
中所例示,使用VEGF165
作為能夠選擇性或特異性地與抗VEGF蛋白結合或相互作用之蛋白質可成功地產生MT5 (抗VEGF蛋白)、阿柏西普及抗VEGF scFv片段。在另一特定態樣中,蛋白質可為一或多種具有如SEQ ID NO.: 73至80中所示之胺基酸序列的蛋白質。表 7-1
亦揭露使用具有如SEQ ID NO.: 73至80中所示之胺基酸序列的蛋白質作為能夠選擇性或特異性地結合至抗VEGF蛋白(MT5)之蛋白質成功產生MT5。
在本實施例之一態樣中,該方法可包含使包含抗VEGF蛋白及一或多種宿主細胞蛋白/污染物之生物樣品在合適的條件下進行親和層析,其中親和層析固定相包含能夠選擇性或特異性地與抗VEGF蛋白結合或相互作用之蛋白質,其中抗VEGF蛋白可選自阿柏西普、VEGF MiniTrap或抗VEGF抗體。在一具體態樣中,VEGF MiniTrap可獲自VEGF受體組分,進一步,其可藉由在宿主細胞中重組表現VEGF MiniTrap而形成。執行該方法可減少樣品中一或多種宿主細胞蛋白之量。例如,圖 35A
及圖 35B
展示在使用包含以下作為能夠選擇性或特異性地結合至MT5之不同蛋白質之五種不同親和層析管柱時,包含MT5 (抗VEGF蛋白)之樣品中的總宿主細胞蛋白之顯著減少:(i) VEGF165
(SEQ ID NO.: 72);(ii) mAb1 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 73為重鏈,而SEQ ID NO.: 74為輕鏈);(iii) mAb2 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 75為重鏈,而SEQ ID NO.: 76為輕鏈);(iv) mAb3 (小鼠抗VEGFR1 mAb小鼠IgG1,其中SEQ ID NO.: 77為重鏈,而SEQ ID NO.: 78為輕鏈)以及(v) mAb4 (小鼠抗VEGFR1 mAb小鼠IgG1,其中SEQ ID NO.: 79為重鏈,而SEQ ID NO.: 80為輕鏈)。如圖 35A
及圖 35B
所見,來自每個基於親和力之產生過程的析出液將宿主細胞蛋白自7000 ppm以上分別降至約25 ppm及約55 ppm。
使用親和層析之合適條件可包括但不限於使用平衡緩衝液來平衡親和層析管柱。在使用例如pH值為約8.3至約8.6之Tris鹽酸鹽進行平衡後,將生物樣品加載至親和層析管柱上。加載管柱後,可使用例如平衡緩衝液(諸如杜爾貝寇磷酸鹽緩衝鹽水(DPBS))將管柱洗滌一次或多次。在溶析管柱之前,可使用其他洗滌,包括採用不同緩衝液之洗滌。管柱溶析可受到緩衝液類型以及pH值及電導率之影響,可採用熟習此項技術者熟知之其他溶析條件。使用一或多種類型之溶析緩衝液(例如,pH值為約2.0至約3.0之甘胺酸)進行溶析後,可藉由添加中和緩衝液(例如,pH 7.5之1 M Tris)來中和溶析級分。
在實施例之一態樣中,洗滌緩衝液及平衡緩衝液之pH值均可為約7.0至約8.6。在實施例之一態樣中,洗滌緩衝液可為DPBS。在一態樣中,溶析緩衝液可包含100 mM甘胺酸緩衝液,pH值為約2.5。在另一態樣中,溶析緩衝液可為pH值為約2.0至約3.0之緩衝液。在一態樣中,中和緩衝液可包含1 M tris,pH值為約7.5。
在本實施例之一態樣中,該方法可進一步包含用洗滌緩衝液洗滌管柱。在本實施例之一態樣中,該方法可進一步包含用溶析緩衝液溶析管柱以獲得溶析級分。在一具體態樣中,與生物樣品中的宿主細胞蛋白之量相比,溶析級分中的宿主細胞蛋白之量顯著減少,例如,減少了約70%、約80%、90%、約95%、約98%或約99%。
本實施例可包括不以特定順序添加一或多個步驟,諸如疏水相互作用層析、基於親和力之層析、多模式層析、病毒滅活(例如,使用低pH值)、病毒過濾及/或超濾/滲濾。
在一態樣中,抗VEGF蛋白之組成物之醣化譜如下:約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約6%至約15%甘露糖-5及約60%至約79%半乳醣化聚醣。
在此實施例之一態樣中,抗VEGF蛋白在約32.4%天冬醯胺123殘基及/或約27.1%天冬醯胺196殘基上具有Man5醣化。在一特定實施例中,抗VEGF蛋白可為阿柏西普、抗VEGF抗體或VEGF MiniTrap。
在一實施例中,該方法可進一步包含使用醫藥學上可接受之賦形劑調配原料藥。在一態樣中,醫藥學上可接受之賦形劑可選自以下:水、緩衝劑、糖、鹽、界面活性劑、胺基酸、多元醇、螯合劑、乳化劑及防腐劑。熟習此項技術者熟知之其他賦形劑在此實施例之範圍內。
在實施例之一態樣中,調配物可適於向人類個體投與。在本實施例之一態樣中,可藉由玻璃體內注射實現投與。在一態樣中,調配物可具有約40至約200 mg/mL感興趣之蛋白質。在一具體態樣中,感興趣之蛋白質可為阿柏西普、抗VEGF抗體或VEGF MiniTrap。
調配物可用於治療或預防血管生成性眼部病症之方法中,該等病症可包括:年齡相關之黃斑部變性(例如,濕性或乾性)、黃斑部水腫、視網膜靜脈阻塞後之黃斑部水腫、視網膜靜脈阻塞(RVO)、視網膜中央靜脈阻塞(CRVO)、視網膜分支靜脈阻塞(BRVO)、糖尿病性黃斑部水腫(DME)、脈絡膜新生血管形成(CNV)、虹膜新生血管形成、新生血管性青光眼、青光眼手術後纖維化、增生性玻璃體視網膜病(PVR)、視盤新生血管形成、角膜新生血管形成、視網膜新生血管形成、玻璃體新生血管形成、角膜翳、翼狀胬肉、血管性視網膜病、在患有糖尿病性黃斑部水腫之個體中的糖尿病性視網膜病;或糖尿病性視網膜病(例如非增生性糖尿病性視網膜病(例如,以約47或53之糖尿病性視網膜病嚴重程度量表(DRSS)分級為特徵)或增生性糖尿病性視網膜病;例如,在不患有DME之個體中)。側氧種類之合成
本發明之一實施例係關於一或多種使用光來合成經氧化之蛋白質種類的方法。在本實施例之一態樣中,感興趣之蛋白質為抗VEGF蛋白。在一具體態樣中,抗VEGF蛋白為阿柏西普。在另一態樣中,抗VEGF蛋白為包括重組VEGF MiniTrap之VEGF MiniTrap。在本實施例之又另一態樣中,抗VEGF蛋白為單鏈可變片段(scFv)。
在本實施例之一態樣中,樣品包含感興趣之蛋白質,例如阿柏西普融合蛋白,具有最少或沒有側氧變異體。對樣品進行光應力處理以合成阿柏西普之經氧化種類。在一具體態樣中,藉由使用冷白光對樣品進行光應力處理。在另一具體態樣中,藉由使用紫外光對樣品進行光應力處理。
在實施例之一特定態樣中,將包含阿柏西普或另一種抗VEGF蛋白之樣品暴露於冷白光約30小時至約300小時,從而造成經修飾之寡胜肽增加約1.5至約50倍。此等肽經酶消化及分析,包含選自由以下組成之群組的一或多者:
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17)、EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18)、QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19)、TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20)、TNYLTH*R (SEQ ID NO.: 21)、SDTGRPFVEMYSEIPEIIH*MTEGR (SEQ ID NO.: 22)、VH*EKDK (SEQ ID NO.: 23)、SDTGRPFVEM*YSEIPEIIHMTEGR (SEQ ID NO.: 64)、SDTGRPFVEMYSEIPEIIHM*TEGR (SEQ ID NO.: 65)、TQSGSEM*K (SEQ ID NO.: 66)、SDQGLYTC*AASSGLM*TK (SEQ ID NO.: 67)、IIW*DSR/RIIW*DSR/IIW*DSRK (SEQ ID NO.: 28)、TELNVGIDFNW*EYPSSK (SEQ ID NO.: 29)、GFIISNATY*K (SEQ ID NO.: 69)、KF*PLDTLIPDGK (SEQ ID NO.: 70)、F*LSTLTIDGVTR (SEQ ID NO.: 32),其中H*為經氧化為2-側氧基-組胺酸之組胺酸,其中C*為經羧甲基化之半胱胺酸,其中M*為經氧化之甲硫胺酸,其中W*為經氧化之色胺酸,其中Y*為經氧化之酪胺酸,且其中F*為經氧化之苯丙胺酸。可藉由之前提及之蛋白酶例如胰蛋白酶來進行消化。可使用質譜法來分析寡胜肽。
在實施例之一特定態樣中,將包含阿柏西普或其他抗VEGF蛋白之樣品暴露於紫外光約4小時至約40小時,從而造成經修飾之寡胜肽產物增加約1.5至約25倍(在進行消化時獲得),其中樣品包含一或多種選自由以下組成之群組的經修飾之寡胜肽:
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17)、EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18)、QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19)、TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20)、TNYLTH*R (SEQ ID NO.: 21)、SDTGRPFVEMYSEIPEIIH*MTEGR (SEQ ID NO.: 22)、VH*EKDK (SEQ ID NO.: 23)、SDTGRPFVEM*YSEIPEIIHMTEGR (SEQ ID NO.: 64)、SDTGRPFVEMYSEIPEIIHM*TEGR (SEQ ID NO.: 65)、TQSGSEM*K (SEQ ID NO.: 66)、SDQGLYTC*AASSGLM*TK (SEQ ID NO.: 67)、IIW*DSR/RIIW*DSR/IIW*DSRK (SEQ ID NO.: 28)、TELNVGIDFNW*EYPSSK (SEQ ID NO.: 29)、GFIISNATY*K (SEQ ID NO.: 69)、KF*PLDTLIPDGK (SEQ ID NO.: 70)、F*LSTLTIDGVTR (SEQ ID NO.: 32),其中H*為經氧化為2-側氧基-組胺酸之組胺酸,其中C*為經羧甲基化之半胱胺酸,其中M*為經氧化之甲硫胺酸,其中W*為經氧化之色胺酸,其中Y*為經氧化之酪胺酸,且其中F*為經氧化之苯丙胺酸。可藉由之前提及之蛋白酶例如胰蛋白酶來進行消化。可使用質譜法來分析寡胜肽。最小化黃棕色之方法
本文提供在CDM中產生之阿柏西普、MiniTrap或其類似物的產生期間減少黃棕色著色之方法。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,接著收穫包含感興趣之重組蛋白的製劑。在一態樣中,感興趣之重組蛋白為抗VEGF蛋白。在一具體態樣中,抗VEGF蛋白係選自由以下組成之群組:阿柏西普、MiniTrap、重組MiniTrap (其實例在美國專利第7,279,159號中揭露,該專利以引用之方式全部併入本文)、scFv及其他抗VEGF蛋白。在一態樣中,該方法可產生感興趣之重組蛋白的製劑,其中該製劑之顏色使用歐洲BY方法或CIELAB方法(b*)來表徵。另外,可使用例如LC-MS分析側氧變異體之存在。
在本實施例之一態樣中,減弱條件包括提高或降低一或多種培養基組分(例如,胺基酸、金屬或抗氧化物,包括鹽及前驅體)之累積濃度,其對應於顏色以及阿柏西普及VEGF MiniTrap之蛋白質變異體的減少。胺基酸之非限制性實例包括丙胺酸、精胺酸、天冬醯胺、天冬胺酸、半胱胺酸、麩醯胺、麩胺酸、甘胺酸、組胺酸、異白胺酸、白胺酸、離胺酸、甲硫胺酸、苯丙胺酸、脯胺酸、絲胺酸、蘇胺酸、色胺酸、酪胺酸及纈胺酸。在一具體態樣中,減少半胱胺酸可有效減弱製劑之黃棕色。半胱胺酸濃度亦可影響側氧變異體。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普,以及收穫由細胞產生的感興趣之蛋白質的製劑,其中合適的條件部分係藉由將CDM中半胱胺酸之累積濃度降低至小於或等於約10 mM而獲得。合適的培養基之實例包括但不限於CDM1B、Excell或其類似物。如本文所用,術語「累積量」係指在細胞培養過程中添加至生物反應器中以形成CDM的特定組分之總量,包括在培養開始時添加的組分之量(在第0天的CDM)及隨後添加的組分之量。當計算組分之累積量時,亦包括在生物反應器產生之前(亦即,在第0天的CDM之前)添加至種菌培養物或接種物中的組分之量。累積量不受培養過程中組分隨時間損失(例如,經由代謝或化學降解)之影響。因此,具有相同累積量之組分的兩種培養物可能仍具有不同絕對水準,例如,若在不同時間將組分添加至兩種培養物中(例如,若在一種培養物中,所有組分均在開始時添加,而在另一種培養物中,組分係隨時間之推移而添加)。累積量亦不受培養過程中組分隨時間之原位合成(例如,經由代謝或化學轉化)的影響。因此,具有相同累積量之給定組分的兩種培養物可能仍具有不同絕對水準,例如,若組分係經由生物轉化過程在兩種培養物其中一種的原位合成。累積量可以例如組分之克數或莫耳數之單位表示。
如本文所用,術語「累積濃度」係指組分之累積量除以生產批次開始時生物反應器中液體之體積,包括培養物中使用的任何接種物對起始體積之貢獻。例如,若生物反應器在生產批次開始時包含2升細胞培養基,並且在第0、1、2及3天添加1克之組分X,則第3天之後的累積濃度為2 g/L (亦即4克除以2升)。若在第4天向生物反應器中添加另外一升不含組分X之液體,則累積濃度將保持為2 g/L。若在第5天,從生物反應器中損失一些液體(例如,經由蒸發),則累積濃度將保持為2 g/L。累積濃度可以例如克/升或莫耳/升之單位表示。
在此實施例之一態樣中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白;收穫由細胞產生的蛋白質之製劑,其中藉由將累積半胱胺酸濃度與累積總胺基酸濃度之比率自約1:10至1:29降至約1:50至約1:30來獲得合適的條件。
在一實施例中,該方法包含(i)在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普,以及(ii)收穫由細胞產生的感興趣之重組蛋白之製劑,其中藉由將CDM中之鐵累積濃度降至小於約55.0 µM來獲得合適的條件。在此實施例之一態樣中,與藉由其中CDM中之鐵累積濃度大於約55.0 μM的方法獲得之製劑相比,藉由此方法獲得之製劑展示較淺之黃棕色。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普。該方法進一步包含收穫由細胞產生的感興趣之重組蛋白之製劑,其中藉由將CDM中之銅累積濃度降至小於或等於約0.8 µM來獲得合適的條件。在此實施例之一態樣中,與藉由其中CDM中之銅累積濃度大於約0.8 µM的方法獲得之製劑相比,藉由此方法獲得之製劑展示較淺之黃棕色。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普;以及收穫由細胞產生的感興趣之重組蛋白之製劑,其中藉由將CDM中之鎳累積濃度降至小於或等於約0.40 µM來獲得合適的條件。在此實施例之一態樣中,與藉由其中CDM中之鎳累積濃度大於約0.40 µM的方法獲得之製劑相比,藉由此方法獲得之製劑展示較淺之黃棕色。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普。該方法進一步包含收穫由細胞產生的感興趣之重組蛋白之製劑,其中藉由將CDM中之鋅累積濃度降至小於或等於約56 µM來獲得合適的條件。在此實施例之一態樣中,與藉由其中CDM中之鋅累積濃度大於約56 µM的方法獲得之製劑相比,藉由此方法獲得之製劑展示較淺之黃棕色。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普。該方法進一步包含收穫由細胞產生的感興趣之重組蛋白之製劑,其中藉由在CDM中針對單一抗氧化劑存在累積濃度約0.001 mM至約10 mM之抗氧化劑及若在該CDM中添加多種抗氧化劑則不大於約30 mM之累積濃度來獲得合適的條件。在此實施例之一態樣中,與藉由其中在CDM中不存在累積濃度小於約0.01 mM或大於約100 mM之抗氧化劑的方法獲得之製劑相比,藉由此方法獲得之製劑展示較淺之黃棕色。抗氧化劑之非限制性實例可為牛磺酸、次牛磺酸、甘胺酸、硫辛酸、麩胱甘肽、氯化膽鹼、氫皮質酮、維生素C、維生素E、螯合劑、過氧化氫酶、S-羧甲基-L-半胱胺酸及其組合。螯合劑之非限制性實例包括金精三羧酸(ATA)、去鐵胺(DFO)、EDTA及檸檬酸鹽。
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普。該方法進一步包含收穫由細胞產生的感興趣之重組蛋白之製劑,其中合適的條件包括具有以下之CDM:在該CDM中小於約55 μM之鐵累積濃度;在該CDM中小於或等於約0.8 μM之銅累積濃度;在該CDM中小於或等於約0.40 μM之鎳累積濃度;在該CDM中小於或等於約56 μM之鋅累積濃度;在該CDM中小於10 mM之半胱胺酸累積濃度;及/或在該CDM中對於單一抗氧化劑而言濃度為約0.001 mM至約10 mM之抗氧化劑及若在該CDM中添加多種抗氧化劑,則不大於約30 mM之累積濃度。
在本實施例之一態樣中,使用合適的條件獲得之製劑造成阿柏西普及VEGF MiniTrap之蛋白質變異體減少至所需量的阿柏西普及VEGF MiniTrap之蛋白質變異體(稱為阿柏西普及VEGF MiniTrap之蛋白質變異體的「目標值」)。在此實施例之又一態樣中,使用合適的條件獲得之製劑造成當包括阿柏西普及VEGF MiniTrap之變異體的蛋白質製劑經標準化為5 g/L或10 g/L之濃度時,製劑之顏色減小至所需b*值或BY值(分別稱為「目標b*值」、「目標BY值」)。在本實施例之又一態樣中,目標b*值(或目標BY值)及/或變異體之目標值可在力價增加或不明顯減小之製劑中獲得。
當結合以下描述及附圖考慮時,將更好地領會及理解本發明之此等及其他態樣。以下描述雖然指示各種實施例及其許多特定細節,但係藉助於說明而非限制方式給出。可在本發明之範疇內進行許多替代、修改、增加或重新佈置。
血管生成是由先前存在之脈管系統生長新血管,係高度協調之過程,其對於正常的胚胎及產後血管發育至關重要。異常或病理性血管生成係癌症及若干種視網膜疾病之標誌,在此等疾病中,促血管生成因子(諸如血管內皮生長因子(VEGF))之上調導致內皮增生增加、脈管系統形態改變及血管通透性增加。在患有各種眼病之患者的玻璃體液及視網膜脈管系統中發現水準升高之VEGF。阻斷VEGF活性亦已成為治療DME、濕性AMD、CNV、視網膜靜脈阻塞及異常血管生成係潛在病因之其他眼病之首選療法。
如本文所用,阿柏西普為一種該抗VEGF蛋白,其包含全人類胺基酸序列,該序列包含人類VEGFR1之第二Ig域及人類VEGFR2之第三Ig域,表現為與人類IgG1之(Fc)的內聯融合物(inline fusion)。阿柏西普結合所有形式之VEGF-A (VEGF),但另外結合PlGF及VEGF-B。幾種其他同型二聚VEGF MiniTrap已作為阿柏西普之酶切產物產生或直接由宿主細胞株重組表現。VEGF MiniTrap之一個該例子示於圖 1
中。在此圖中,描繪末端離胺酸(k),一些培養過程移除此末端離胺酸,而另一些則沒有。圖 1
示出保留末端離胺酸之過程。通常,阿柏西普涵蓋末端離胺酸之存在及不存在。
如本文所證明,本發明部分揭露使用CDM產生抗VEGF蛋白(實例1)。包含使用某些CDM產生的阿柏西普之溶液之分析顯示出一定顏色特性,諸如強烈的黃棕色。溶液顏色之強度視所用CDM而定。在將溶液標準化至5 g/L之濃度後,並非所有檢查的CDM均產生具有明顯黃棕色之樣品。
在可注射之治療藥物溶液中的顏色(諸如黃棕色)可為不期望之特徵。其可為用於確定藥品是否滿足特定治療劑之預定純度及品質水準的重要參數。在生物製劑之製造過程中觀察到的顏色(諸如黃棕色)可能係由於該生物製劑之化學修飾、調配物賦形劑之降解產物或經由生物製劑及調配物賦形劑之反應而形成的降解產物引起。然而,該資訊對於理解顏色變化之原因可能有價值。其亦可幫助設計短期以及長期儲存條件,以防止促成此一顏色變化之修飾。
發明者觀察到,在抗VEGF蛋白溶液產生期間使用AEX使黃棕色著色最小化。另外,發明者發現可藉由修飾用於產生重組蛋白,諸如阿柏西普或經修飾之阿柏西普像MiniTrap的細胞培養物來減少黃棕色著色。
本發明包括抗VEGF蛋白及使用CDM產生該等抗VEGF蛋白。另外,本發明係基於用於蛋白質產生的上游及下游製程技術之識別及最佳化。
如本文所證明,以下列出之一些實例描述抗VEGF蛋白之產生(實例1)、抗VEGF蛋白之經氧化種類之產生(實例4)、藉由最佳化培養基(實例5)及藉由最佳化產生方法(實例2)來減少抗VEGF蛋白之經氧化種類的方法。
許多近期之專利申請案及授權專利旨在描述各種阿柏西普種類及其產生方法,但沒有一者描述或提出本文所述之抗VEGF組成物及其產生方法。參見,例如,Coherus Biosciences Inc.之美國申請案第16/566,847號、Sam Chun Dang Pharm. Co., Ltd.之美國專利第10,646,546號、Formycon AG之美國專利第10,576,128號、Amgen Inc.之國際申請案第PCT/US2020/015659號以及Momenta Pharmaceuticals, Inc.之美國專利第8,956,830號;第9,217,168號;第9,487,810號;第9,663,810號;第9,926,583號;及第10,144,944號。I. 所選術語之解釋
除非另有描述,否則本文使用之所有技術及科學術語具有與本發明所屬領域之普通技術人員通常所理解之相同含義。與熟練技術人員已知的本文所述之彼等類似或等同之方法及材料可用於實施本文所述之具體實施例。所提及之所有出版物以引用方式全部併入本文。
術語「一」應理解為指代「至少一」,且術語「約」及「近似」應理解為允許標準偏差,如普通熟習此項技術者所理解,並且在提供範圍之情況下,包括端點。
如本文所用,術語「血管生成性眼部病症」係指由血管之生長或增生或由血管滲漏引起或與之相關的任何眼部疾病。
如本文所用,術語「化學成分確定之培養基(chemically defined medium)」或「化學成分確定之培養基(chemically defined media)」(均縮寫為「CDM」)係指其中所有成分之身份及濃度確定的合成生長培養基。化學成分確定之培養基不含細菌、酵母、動物或植物提取物、動物血清或血漿,儘管可添加單個植物或動物衍生之組分(例如蛋白質、多肽等)。化學成分確定之培養基可包含支持生長所需之無機鹽,諸如磷酸鹽、硫酸鹽及其類似物。碳源經確定,且通常為糖,諸如葡萄糖、乳糖、半乳糖及其類似物,或其他化合物,諸如甘油、乳酸鹽、乙酸鹽及其類似物。儘管某些化學成分確定之培養基亦使用磷酸鹽作為緩衝液,但可使用其他緩衝液,諸如碳酸氫鈉、HEPES、檸檬酸鹽、三乙醇胺及其類似物。市售的化學成分確定之培養基之例子包括但不限於各種經杜爾貝寇改質之伊戈爾(DME)培養基(Sigma-Aldrich Co; SAFC Biosciences, Inc.)、漢氏營養混合物(Sigma-Aldrich Co; SAFC Biosciences, Inc.)、各種EX-CELLs培養基(Sigma-Aldrich Co; SAFC Biosciences, Inc.)、各種IS CHO-CD培養基(FUJIFILM Irvine Scientific)、其組合及其類似物。製備化學成分確定之培養基的方法係此項技術中已知的,例如在美國專利第6,171,825號及第6,936,441號、WO 2007/077217及美國專利申請公開案第2008/0009040號及第2007/0212770號,其全部教示皆以引用之方式併入本文。
如本文所用,術語「累積量」係指在細胞培養過程中添加至生物反應器中以形成CDM的特定組分之總量,包括在培養開始時添加的組分之量(在第0天的CDM)及隨後添加的組分之量。當計算組分之累積量時,亦包括在生物反應器產生之前(亦即,在第0天的CDM之前)添加至種菌培養物或接種物中的組分之量。累積量不受培養過程中組分隨時間損失(例如,經由代謝或化學降解)之影響。因此,具有相同累積量之組分的兩種培養物可能仍具有不同絕對水準,例如,若在不同時間將組分添加至兩種培養物中(例如,若在一種培養物中,所有組分均在開始時添加,而在另一種培養物中,組分係隨時間之推移而添加)。累積量亦不受培養過程中組分隨時間之原位合成(例如,經由代謝或化學轉化)的影響。因此,具有相同累積量之給定組分的兩種培養物可能仍具有不同絕對水準,例如,若組分係經由生物轉化過程在兩種培養物其中一種中的原位合成。累積量可以例如組分之克數或莫耳數之單位表示。
如本文所用,術語「累積濃度」係指組分之累積量除以生產批次開始時生物反應器中液體之體積,包括培養物中使用的任何接種物對起始體積之貢獻。例如,若生物反應器在生產批次開始時包含2升細胞培養基,並且在第0、1、2及3天添加1克之組分X,則第3天之後的累積濃度為2 g/L (亦即4克除以2升)。若在第4天將另外1升不含組分X之液體添加至生物反應器中,則累積濃度將保持為2 g/L。若在第5天自生物反應器中損失一些液體(例如經由蒸發),則累積濃度將保持為2 g/L。累積濃度可以例如克/升或莫耳/升之單位表示。
如本文所用,術語「調配物」係指與一或多種醫藥學上可接受之媒劑一起調配的感興趣之蛋白質。在一態樣中,感興趣之蛋白質為阿柏西普及/或MiniTrap。在一些示例性實施例中,調配物中感興趣之蛋白質之量可在約0.01 mg/mL至約600 mg/mL之範圍內。在一些特定實施例中,調配物中感興趣之蛋白質之量可為約0.01 mg/mL、約0.02 mg/mL、約0.03 mg/mL、約0.04 mg/mL、約0.05 mg/mL、約0.06 mg/mL、約0.07 mg/mL、約0.08 mg/mL、約0.09 mg/mL、約0.1 mg/mL、約0.2 mg/mL、約0.3 mg/mL、約0.4 mg/mL、約0.5 mg/mL、約0.6 mg/mL、約0.7 mg/mL、約0.8 mg/mL、約0.9 mg/mL、約1 mg/mL、約2 mg/mL、約3 mg/mL、約4 mg/mL、約5 mg/mL、約6 mg/mL、約7 mg/mL、約8 mg/mL、約9 mg/mL、約10 mg/mL、約15 mg/mL、約20 mg/mL、約25 mg/mL、約30 mg/mL、約35 mg/mL、約40 mg/mL、約45 mg/mL、約50 mg/mL、約55 mg/mL、約60 mg/mL、約65 mg/mL、約70 mg/mL、約75 mg/mL、約80 mg/mL、約85 mg/mL、約90 mg/mL、約100 mg/mL、約110 mg/mL、約120 mg/mL、約130 mg/mL、約140 mg/mL、約150 mg/mL、約160 mg/mL、約170 mg/mL、約180 mg/mL、約190 mg/mL、約200 mg/mL、約225 mg/mL、約250 mg/mL、約275 mg/mL、約300 mg/mL、約325 mg/mL、約350 mg/mL、約375 mg/mL、約400 mg/mL、約425 mg/mL、約450 mg/mL、約475 mg/mL、約500 mg/mL、約525 mg/mL、約550 mg/mL、約575 mg/mL或約600 mg/mL。在一些示例性實施例中,組成物之pH值可大於約5.0。在一示例性實施例中,pH值可大於約5.0、大於約5.5、大於約6、大於約6.5、大於約7、大於約7.5、大於約8或大於約8.5。
如本文所用,術語「資料庫」係指生物資訊學工具,其提供針對資料庫中所有可能序列搜尋未解釋之MS-MS譜的可能性。此等工具之非限制性實例為Mascot (http://www.matrixscience.com)、Spectrum Mill (http://www.chem.agilent.com)、PLGS (http://www.waters.com)、PEAKS (http://www.bioinformaticssolutions.com)、Proteinpilot (http://download.appliedbiosystems.com//proteinpilot)、Phenyx (http://www.phenyx-ms.com)、Sorcerer (http://www.sagenresearch.com)、OMSSA (http://www.pubchem.ncbi.nlm.nih.gov/omssa/)、X!Tandem (http://www.thegpm.org/TANDEM/)、Protein Prospector (http://www. http://prospector.ucsf.edu/ prospector/ mshome.htm)、Byonic (https://www.proteinmetrics.com/products/byonic)或Sequest (http://fields.scripps.edu/sequest)。
如本文所用,術語「超濾」或「UF」可包括類似於逆滲透之膜過濾過程,其使用靜水壓力迫使水通過半透膜。有關超濾之詳情描述於:Leos J. Zeman & Andrew L. Zydney, Microfiltration and ultrafiltration: principles and applications (1996)中,其全部教示併入本文。孔徑小於0.1 μm之過濾器可用於超濾。藉由使用具有如此小孔徑之過濾器,可經由使樣品緩衝液滲透穿過過濾器而將蛋白質保留在過濾器後面來減少樣品之體積。
如本文所用,「滲濾」或「DF」可包括使用超濾器移除及交換鹽、糖及非水溶劑;將遊離種類與結合種類分離;移除低分子量材料及/或引起離子及/或pH環境之快速變化的方法。藉由以近似等於超濾速率之速率向經超濾之溶液中添加溶劑,可最有效地移除微溶質。由此自溶液中以恆定體積洗滌微種。在本發明之某些示例性實施例中,可使用滲濾步驟來交換用於本發明之各種緩衝液,例如,在層析或其他產生步驟之前,以及自蛋白質製劑中移除雜質。如本文所用,術語「下游製程技術」係指在上游製程技術之後用於產生蛋白質之一或多種技術。下游製程技術包括例如使用以下方法產生蛋白質產物:例如,親和層析,包括蛋白A親和層析以及使用固相之親和層析,該固相具有明確限定之分子像VEGF,其可與其同源物像VEGF受體(VEGF R)相互作用;離子交換層析,諸如陰離子或陽離子交換層析;疏水相互作用層析或置換層析。
片語「重組宿主細胞」(或簡稱為「宿主細胞」)包括已引入編碼感興趣之蛋白質的重組表現載體的細胞。應理解,此一術語意欲不僅指代具體主題細胞,而且指代此一細胞之子代。因為由於突變或環境影響,某些修飾可能在後代中發生,所以該子代實際上可與親本細胞不同,但仍包括於如本文所用之術語「宿主細胞」之範疇內。在一實施例中,宿主細胞包括選自任何生命王國之原核及真核細胞。在一態樣中,真核細胞包括原生生物、真菌、植物及動物細胞。在又一態樣中,宿主細胞包括真核細胞,諸如植物及/或動物細胞。細胞可為哺乳動物細胞、魚類細胞、昆蟲細胞、兩棲類細胞或禽類細胞。在一具體態樣中,宿主細胞為哺乳動物細胞。適於在培養物中生長的廣泛多種哺乳動物細胞株可獲自例如美國菌種保存中心(Manassas, Va.)及其他寄存處以及商業供應商。可用於本發明之製程中的細胞包括但不限於MK2.7細胞、PER-C6細胞、中國倉鼠卵巢細胞(CHO)諸如CHO-K1 (ATCC CCL-61)、DG44 (Chasin等人,1986,Som. Cell Molec. Genet.,
12:555-556;Kolkekar 等人, 1997,Biochemistry,
36: 10901-10909;及WO 01/92337 A2)、二氫葉酸還原酶陰性CHO細胞(CHO/-DHFR,Urlaub及Chasin, 1980,Proc. Natl. Acad. Sci. USA,
77:4216)及dp12.CHO細胞(美國專利第5,721,121號);猴腎細胞(CV1,ATCC CCL-70);由SV40轉化之猴腎CV1細胞(COS細胞,COS-7,ATCC CRL-1651);HEK 293細胞及Sp2/0細胞、5L8融合瘤細胞、Daudi細胞、EL4細胞、HeLa細胞、HL-60細胞、K562細胞、Jurkat細胞、THP-1細胞、Sp2/0細胞、原代上皮細胞(例如角質細胞、宮頸上皮細胞、支氣管上皮細胞、氣管上皮細胞、腎上皮細胞及視網膜上皮細胞)以及已建立之細胞株及其品系(例如人類胚胎腎細胞(例如293細胞或經次選殖以便在懸浮培養中生長之293細胞,Graham等人,1977,J. Gen. Virol.
, 36:59);幼倉鼠腎細胞(BHK,ATCC CCL-10);小鼠足細胞(TM4,Mather, 1980, Biol.Reprod., 23:243-251);人類宮頸癌細胞(HELA,ATCC CCL-2);犬腎細胞(MDCK,ATCC CCL-34);人類肺細胞(W138,ATCC CCL-75);人類肝癌細胞(HEP-G2,HB 8065);小鼠乳腺腫瘤細胞(MMT 060562,ATCC CCL-51);水牛鼠幹細胞(BRL 3A,ATCC CRL-1442);TRI細胞(Mather, 1982,Annals NY Acad.Sci.,
383:44-68);MCR 5細胞;FS4細胞;PER-C6視網膜細胞、MDBK (NBL-1)細胞、911細胞、CRFK細胞、MDCK細胞、BeWo細胞、Chang細胞、Detroit 562細胞、HeLa 229細胞、HeLa S3細胞、Hep-2細胞、KB細胞、LS 180細胞、LS 174T細胞、NCI-H-548細胞、RPMI 2650細胞、SW-13細胞、T24細胞、WI-28 VA13, 2RA細胞、WISH細胞、BS-C-I細胞、LLC-MK2細胞、Clone M-3細胞、1-10細胞、RAG細胞、TCMK-1細胞、Y-1細胞、LLC-PK1
細胞、PK(15)細胞、GH1
細胞、GH3
細胞、L2細胞、LLC-RC 256細胞、MH1
C1
細胞、XC細胞、MDOK細胞、VSW細胞及TH-I, B1細胞或其衍生物);來自任何組織或器官(包括但不限於心臟、肝臟、腎臟、結腸、腸、食道、胃、神經組織(腦、脊髓)、肺、血管組織(動脈、靜脈、毛細血管)、淋巴組織(淋巴腺、腺樣體、扁桃體、骨髓及血液)、脾臟)之成纖維細胞、以及成纖維細胞及成纖維細胞樣細胞株(例如,TRG-2細胞、IMR-33細胞、Don細胞、GHK-21細胞、瓜胺酸血症細胞、Dempsey細胞、Detroit 551細胞、Detroit 510細胞、Detroit 525細胞、Detroit 529細胞、Detroit 532細胞、Detroit 539細胞、Detroit 548細胞、Detroit 573細胞、HEL 299細胞、IMR-90細胞、MRC-5細胞、WI-38細胞、WI-26細胞、MiCl1細胞、CV-1細胞、COS-1細胞、COS-3細胞、COS-7細胞、非洲綠猴腎細胞(VERO-76,ATCC CRL-1587;VERO,ATCC CCL-81);DBS-FrhL-2細胞、BALB/3T3細胞、F9細胞、SV-T2細胞、M-MSV-BALB/3T3細胞、K-BALB細胞、BLO-11細胞、NOR-10細胞、C3
H/IOTI/2細胞、HSDM1
C3
細胞、KLN205細胞、McCoy細胞、小鼠L細胞、品系2071 (小鼠L)細胞、L-M品系(小鼠L)細胞、L-MTK (小鼠L)細胞、NCTC純系2472及2555、SCC-PSA1細胞、Swiss/3T3細胞、印度黃麂細胞(Indian muntac cell)、SIRC細胞、CII
細胞及Jensen細胞、或其衍生物)或熟習此項技術者已知之任何其他細胞類型。
如本文所用,術語「宿主細胞蛋白」(host-cell proteins,HCP)包括源自宿主細胞之蛋白,並且可與所需的感興趣之蛋白無關。宿主細胞蛋白可為過程相關之雜質,其可來源於製造過程,並且可包括三個主要類別:細胞受質來源、細胞培養來源及下游來源。細胞受質來源之雜質包括但不限於來源於宿主生物之蛋白質及核酸(宿主細胞基因體、載體或總DNA)。細胞培養來源之雜質包括但不限於誘導劑、抗生素、血清及其他培養基組分。下游來源之雜質包括但不限於酶、化學及生化處理試劑(例如,溴化氰、胍、氧化劑及還原劑)、無機鹽(例如,重金屬、砷、非金屬離子)、溶劑、載劑、配位體(例如單株抗體)及其他可浸出物。
在一些示例性實施例中,宿主細胞蛋白可具有在約4.5至約9.0範圍內之pI。在一示例性實施例中,pI可為約4.5、約5.0、約5.5、約5.6、約5.7、約5.8、約5.9、約6.0、約6.1、約6.2、約6.3、約6.4、約6.5、約6.6、約6.7、約6.8、約6.9、約7.0、約7.1、約7.2、約7.3、約7.4、約7.5、約7.6、約7.7、約7.8、約7.9、約8.0、約8.1、約8.2、約8.3、約8.4、約8.5、約8.6、約8.7、約8.8、約8.9或約9.0。
如本文所用,術語「水解劑」係指可進行蛋白質消化的大量不同試劑中之任何一種或組合。可進行酶消化之水解劑之非限制性實例包括來自佐氏麴菌
之蛋白酶、彈性蛋白酶、枯草桿菌蛋白酶、蛋白酶XIII、胃蛋白酶、胰蛋白酶、Tryp-N、胰凝乳蛋白酶、曲黴菌素I、LysN蛋白酶(Lys-N)、LysC胞內蛋白酶(Lys-C)、胞內蛋白酶Asp-N (Asp-N)、胞內蛋白酶Arg-C (Arg-C)、胞內蛋白酶Glu-C (Glu-C)或外膜蛋白T (OmpT)、釀膿鏈球菌之免疫球蛋白降解酶(IdeS)、嗜熱菌蛋白酶、木瓜蛋白酶、鏈黴蛋白酶、V8蛋白酶或其生物活性片段或同系物或其組合。可進行非酶消化之水解劑之非限制性實例包括使用高溫、微波、超音波、高壓、紅外、溶劑(非限制性實例為乙醇及乙腈)、固定化酶消化(IMER)、磁粒固定化酶及晶片上固定化酶。有關討論可用的蛋白質消化技術之最新評論,參見Switzar 等人, 「Protein Digestion: An Overview of the Available Techniques and Recent Developments」 (Linda Switzar, Martin Giera & Wilfried M. A. Niessen,Protein Digestion: An Overview of the Available Techniques and Recent Developments
, 12 JOURNAL OF PROTEOME RESEARCH 1067-1077 (2013),其全部教示皆併入本文)。一種水解劑或多種水解劑之組合可以序列特異性方式切割蛋白質或多肽中之肽鍵,從而產生較短肽之可預測集合。可適當選擇水解劑與蛋白質之比率及消化所需時間以獲得蛋白質之最佳消化。當酶與受質之比率過高時,相應的高消化速率將不能為肽藉由質譜儀進行分析提供足夠的時間,並且會損害序列覆蓋率。另一方面,低E/S比率將需要長時間消化,且因此需要較長的數據採集時間。酶與受質之比率可在約1:0.5至約 1:200範圍內。如本文所用,術語「消化」係指蛋白質之一或多個肽鍵之水解。有若干種使用適當的水解劑對生物樣品中之蛋白質進行消化之方法,例如,酶消化或非酶消化。用於消化樣品中之蛋白質的一種廣泛接受之方法涉及使用蛋白酶。有許多蛋白酶可用,且每種蛋白酶在特異性、效率及最佳消化條件方面都有其自身特徵。蛋白酶係指內肽酶及外肽酶,根據蛋白酶在肽內的非末端或末端胺基酸上之切割能力進行分類。替代地,蛋白酶亦指代六種不同的類別-天冬胺酸蛋白酶、麩胺酸蛋白酶及金屬蛋白酶、半胱胺酸蛋白酶、絲胺酸蛋白酶及蘇胺酸蛋白酶,根據催化機制進行分類。術語「蛋白酶」與「肽酶」可互換使用,係指水解肽鍵之酶。
術語「與...相關聯」表示可將本發明之組分,亦即抗VEGF組成物與另一種劑諸如抗ANG2一起調配成單一組成物用於同時遞送,或單獨調配成兩種或更多種組成物(例如,包含每種組分之套組)。彼此相關聯投與之組分可在與投與其他組分不同之時間向個體投與;例如,可在給定時間段以一定時間間隔非同時(例如,單獨地或依次地)進行每次投與。彼此相關聯投與之單獨組分亦可在相同投與時段中基本上同時(例如,精確地同時或以非臨床上重要之時間段間隔開)投與。此外,彼此相關聯投與之單獨組分可藉由相同或不同途徑向個體投與,例如,阿柏西普之組成物與另一種劑諸如抗ANG2一起,其中阿柏西普之組成物包含約15%或更少之其變異體。
如本文所用,術語「液相層析」係指以下過程:當組分流過(或流入)固定液相或固相時,由於組分之差異分佈,可將由液體攜帶之生物/化學混合物分離為各組分。液相層析之非限制性實例包括逆相液相層析、離子交換層析、尺寸排阻層析、親和層析、混合模式層析、疏水層析或混合模式層析。
如本文所用,「親和層析」可包括分離,該等分離包括基於兩種物質對層析材料之親和力來分離兩種物質之任何方法。其可包含使物質經歷包含合適的親和層析介質之管柱。該層析介質之非限制性實例包括但不限於蛋白A樹脂、蛋白G樹脂、包含抗原(針對該抗原產生結合分子(例如抗體))之親和載體、能夠結合至感興趣之蛋白質的蛋白質及包含Fc結合蛋白之親和載體。在一態樣中,在樣品加載之前,可用合適的緩衝液平衡親和管柱。合適的緩衝液之實例可為Tris/NaCl緩衝液,pH值約為7.0至8.0。熟練技術人員可開發合適的緩衝液而不會造成不當的負擔。此平衡之後,可將樣品加載至管柱上。加載管柱後,可使用例如平衡緩衝液將管柱洗滌一次或多次。在溶析管柱之前,可使用其他洗滌,包括採用不同緩衝液之洗滌。接著可使用適當的溶析緩衝液溶析親和管柱。合適的溶析緩衝液之一實例可為乙酸/NaCl緩衝液,pH值為約2.0至3.5。熟練技術人員亦可開發適當的溶析緩衝液而不會造成不當的負擔。析出液可使用熟習此項技術者熟知之技術(包括UV)進行監測,例如,可使用280 nm處之吸光度,特別是如果感興趣之樣品包含芳環(例如,具有像色胺酸一樣之芳族胺基酸的蛋白質)。
如本文所用,「離子交換層析」可指代包括任何方法之分離,藉由該方法可基於感興趣之分子及/或層析材料上(整體或局部位於感興趣之分子及/或層析材料之特定區域上)的個別離子電荷之差異來分離兩種物質,且因此可使用陽離子交換材料或陰離子交換材料。離子交換層析根據感興趣之分子之局部電荷與層析材料之局部電荷之間的差異來分離分子。填充式離子交換層析管柱或離子交換膜裝置可在結合-溶析模式、流通模式或混合模式下操作。用平衡緩衝液或另一種緩衝液洗滌管柱或膜裝置之後,可藉由提高溶析緩衝液的離子強度(亦即電導率)以與溶質競爭離子交換基質之帶電位點來實現產物回收。改變pH值且由此改變溶質之電荷可為實現溶質溶析之另一種方式。電導率或pH值之變化可為逐漸的(梯度溶析)或逐步的(分步溶析)。可將陰離子或陽離子取代基附著於基質上,以形成用於層析之陰離子或陽離子載體。陰離子交換取代基之非限制性實例包括二乙胺基乙基(DEAE)、四級胺乙基(QAE)及四級胺(Q)基團。陽離子取代基包括羧甲基(CM)、磺乙基(SE)、磺丙基(SP)、磷酸鹽(P)及磺酸鹽(S)。纖維素離子交換介質或載體可包括可由Whatman Ltd. Maidstone, Kent, U.K獲得之DE23™、DE32™、DE52™、CM-23™、CM-32™及CM-52™。SEPHADEX®基及交聯之離子交換劑亦為已知的。例如,DEAE-、QAE-、CM-及SP-SEPHADEX®以及DEAE-、Q-、CM-及S-SEPHAROSE®以及SEPHAROSE® Fast Flow及Capto™ S均可由GE Healthcare獲得。此外,DEAE及CM衍生化之乙二醇-甲基丙烯酸酯共聚物,諸如TOYOPEARL™ DEAE-650S或M及TOYOPEARL™ CM-650S或M可由Toso Haas Co., Philadelphia, Pa.獲得,或Nuvia S及UNOSphere™ S可由BioRad, Hercules, Calif.獲得,Eshmuno® S可由EMD Millipore, MA獲得。
如本文所用,術語「疏水相互作用層析樹脂」可包括固相,其可經苯基、辛基、丁基或其類似物共價修飾。其可利用疏水性將分子彼此分離。在此類型之層析中,疏水基團(諸如苯基、辛基、己基或丁基)可形成管柱之固定相。諸如蛋白質、肽及其類似物之分子穿過HIC (疏水相互作用層析)管柱,該管柱在其表面上具有一或多個疏水區域或具有疏水袋,並且能夠與構成HIC固定相之疏水基團相互作用。HIC樹脂或載體之實例包括Phenyl sepharose FF、Capto Phenyl (GE Healthcare, Uppsala, Sweden)、Phenyl 650-M (Tosoh Bioscience, Tokyo, Japan)及Sartobind Phenyl (Sartorius corporation, New York, USA)。
如本文所用,術語「混合模式層析」或「多模式層析」(均稱為「MMC」)包括以下層析法:其中溶質經由一種以上相互作用模式或機制與固定相相互作用。MMC可用作傳統逆相(RP)、離子交換(IEX)及正相層析(NP)之替代或補充工具。與RP、NP及IEX層析(其中分別以疏水相互作用、親水相互作用及離子相互作用作為主要相互作用模式)不同,混合模式層析可採用此等相互作用模式中之兩種或更多種之組合。混合模式層析介質可提供無法藉由單模式層析重現之獨特選擇性。與基於親和力之方法相比,混合模式層析亦可節省潛在成本、延長管柱壽命及提高操作靈活性。在一些示例性實施例中,混合模式層析介質可包含直接或經由間隔體與有機或無機載體(有時表示為基礎基質)偶聯之混合模式配位體。載體可呈以下形式:顆粒,諸如基本上呈球形之顆粒、整料、過濾器、膜、表面、毛細管等。在一些示例性實施例中,載體可由天然聚合物諸如交聯碳水化合物材料諸如瓊脂糖、agPV、纖維素、聚葡糖、殼聚醣、蒟蒻、角叉菜膠、結冷膠、藻酸鹽等來製備。為了獲得高吸附能力,載體可為多孔的,且接著將配位體偶聯至外表面以及孔表面。此等天然聚合物載體可根據標準方法製備,諸如反相懸浮凝膠化(S Hjerten: Biochim Biophys Acta 79(2), 393-398 (1964),其全部教示併入本文)。替代地,可由以下物質來製備載體:合成聚合物,諸如交聯合成聚合物,例如苯乙烯或苯乙烯衍生物、二乙烯基苯、丙烯醯胺、丙烯酸酯、甲基丙烯酸酯、乙烯酯、乙烯醯胺及其類似物。此等合成聚合物可根據標準方法產生,例如「藉由懸浮聚合開發的苯乙烯基聚合物載體」(R Arshady: Chimica e L′Industria 70(9), 70-75 (1988),其全部教示併入本文)。多孔的天然或合成聚合物載體亦可由諸如GE Healthcare, Uppsala, Sweden之商業來源獲得。
如本文所用,術語「質譜儀」包括能夠識別特定分子種類並量測其準確質量之裝置。該術語旨在包括可表徵多肽或肽之任何分子偵測器。質譜儀可包括三個主要部分:離子源、質量分析儀及偵測器。離子源之作用係產生氣相離子。可將分析物之原子、分子或簇轉移至氣相中並同時(如以電灑遊離化)或經由單獨過程遊離化。離子源之選擇視應用而定。在一些示例性實施例中,質譜儀可為串聯質譜儀。如本文所用,術語「串聯質譜」包括其中藉由使用質量選擇及質量分離之多個階段獲得關於樣品分子之結構資訊的技術。先決條件係將樣品分子轉化為氣相並遊離化,以便在第一質量選擇步驟之後以可預測及可控制之方式形成片段。多階段MS/MS或MSn
可藉由首先選擇及分離前驅體離子(MS2
),使其片段化,分離初級碎片離子(MS3
),使其片段化,分離次級碎片(MS4
)等方式進行,只要可獲得有意義之資訊,或碎片離子信號為可偵測的。串聯MS已成功地與廣泛多種分析儀組合一起使用。可根據許多不同因素(諸如靈敏度、選擇性及速度,以及尺寸、成本及可用性)來確定為某種應用組合哪種分析儀。串聯MS方法之兩個主要類別為空間串聯及時間串聯,但亦有一些混合方案,其中時間串聯分析儀在空間中或與空間串聯分析儀耦合。空間串聯質譜儀包括離子源、前驅體離子活化裝置及至少兩個非捕獲質量分析儀。特定的m/z分離功能可經設計,以便在儀器之一個部分中選擇離子,在中間區域將其解離,接著將產物離子傳輸至另一分析儀以進行m/z分離及數據採集。在時間串聯質譜儀中,可在同一物理裝置中捕獲、分離、片段化及m/z分離離子源中產生之離子。質譜儀所識別之肽可用作完整蛋白及其轉譯後修飾之替代代表。其可藉由將實驗與理論MS/MS數據相關聯來用於蛋白質表徵,理論MS/MS數據係由蛋白質序列資料庫中之可能的肽產生。該表徵包括但不限於對蛋白質片段之胺基酸進行定序;確定蛋白質定序;確定蛋白質重新(de novo
)定序;定位轉譯後修飾或識別轉譯後修飾;或可比性分析;或其組合。
如本文所用,「Mini-Trap」或「MiniTrap」或「MiniTrap結合分子」能夠結合至VEGF分子。此等MiniTrap可包括(i)嵌合多肽以及(ii)包含兩個或更多個例如經由一或多個二硫橋非共價結合之多肽的多聚(例如,二聚)分子。MiniTrap可經由化學修飾、酶促活性產生或重組製備。
如本文所用,「VEGF MiniTrap」或「VEGF MiniTrap結合分子」可為與VEGF結合且具有一或多組VEGF受體Ig樣域(或其變異體)(例如,VEGFR1 Ig域2及/或VEGFR2 Ig域3及/或4)之分子或分子複合物以及經修飾或不存在之多聚組分(MC),例如,其中MC為經修飾之免疫球蛋白Fc。該修飾可為VEGF trap (例如,阿柏西普或康柏西普(conbercept))之蛋白水解消化或具有經縮短之MC序列的所得多肽鏈之直接表現的結果。(參見圖 1
中描繪之分子結構)圖 1
為VEGF MiniTrap分子之描繪,其為用釀膿鏈球菌IdeS蛋白水解阿柏西普之產物。描繪同型二聚分子,其具有經由兩個平行雙硫鍵連接之Ig鉸鏈域片段。指示VEGFR1域、VEGFR2域及鉸鏈域片段(MC)。阿柏西普中發生IdeS切割之點用「//」指示。亦指示自阿柏西普切下之Fc片段。未經二聚化之單個此等嵌合多肽若具有VEGF結合活性則亦可為VEGF MiniTrap。術語「VEGF MiniTrap」包括單個多肽,其包含第一組一或多個VEGF受體Ig域(或其變異體),該Ig域缺乏MC但以連接子(例如,肽連接子)與一或多個其他組之一或多個VEGF受體Ig域(或其變異體)融合。在本發明之VEGF MiniTrap中的VEGF結合域可彼此相同或不同(參見 WO2005/00895,其全部教示皆併入本文)。
例如,在本發明之一實施例中,未經修飾之免疫球蛋白Fc域包含以下胺基酸序列或其胺基酸1至226:
DKTHTCPX1
CPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKX2
TPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO.: 33;其中X1
為L或P且X2
為A或T)。
VEGF之抑制包括例如VEGF與VEGF受體結合之拮抗作用,例如藉由與VEGF受體競爭VEGF (例如,VEGF110
、VEGF121
及/或VEGF165
)結合。該抑制可造成VEGF介導之VEGFR活化的抑制,例如,表現嵌合VEGF受體(例如,其同型二聚物)之細胞株(例如,HEK293)中螢光素酶表現之抑制,該細胞株在細胞表面上具有與IL18Rα及/或IL18Rβ胞內域融合之VEGFR胞外域且亦具有NFkB-螢光素酶-IRES-eGFP報告基因,例如本文所列舉之細胞株 HEK293/D9/Flt-IL18Rα/Flt-IL18Rβ。
本發明之VEGF MiniTrap之VEGF受體Ig域組分可包括:
(i) VEGFR1 (Flt1)之一或多個免疫球蛋白樣(Ig)域2 (R1D2),
(ii) VEGFR2 (Flk1或KDR)之一或多個Ig域3 (Flk1D3)(R2D3),
(iii) VEGFR2 (Flk1或KDR)之一或多個Ig域4 (Flk1D4)(R2D4),及/或
(iv) VEGFR3 (Flt4)之一或多個Ig域3 (Flt1D3或R3D3)。
VEGF受體之免疫球蛋白樣域在本文中可稱為VEGFR「Ig」域。本文所提及之VEGFR Ig域,例如R1D2 (在本文中可稱為VEGFR1(d2))、R2D3 (在本文中可稱為VEGFR2(d3))、R2D4 (在本文中可稱為VEGFR2(d4))及R3D3 (在本文中可稱為VEGFR3(d3))旨在不僅涵蓋完整的野生型Ig域,而且亦涵蓋實質上保留野生型結構域之功能特徵的其變異體,例如,當併入VEGF MiniTrap中時,保留形成功能性VEGF結合域之能力。熟習此項技術者顯而易知,可獲得上述Ig域之許多變異體,其將保留與野生型結構域實質上相同之功能特徵。
本發明提供包含以下域結構之VEGF MiniTrap多肽:
l ((R1D2)-(R2D3))a
-連接子-((R1D2)-(R2D3))b
;
l ((R1D2)-(R2D3)-(R2D4))c
-連接子-((R1D2)-(R2D3)-(R2D4))d
;
l ((R1D2)-(R2D3))e
-(MC)g
;
l ((R1D2)-(R2D3)-(R2D4))f
-(MC)g
;
其中,
- R1D2為VEGF受體1 (VEGFR1) Ig域2 (D2);
- R2D3為VEGFR2 Ig域3;
- R2D4為VEGFR2 Ig域4;
- MC為多聚組分(例如,IgG鉸鏈域或其片段,例如來自IgG1);
- 連接子為包含約5、6、7、8、9、10、11、12、13、14、15或16個胺基酸之肽,例如(GGGS)g
;
並且,
獨立地,
a = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15;
b = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15;
c = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15;
d = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15;
e = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15;
f = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15;且
g = 1、2、3、4、5、6、7、8、9、10、11、12、13、14或15。
在本發明之一實施例中,R1D2包含胺基酸序列:SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIID (SEQ ID NO.: 34)。在一態樣中,R1D2缺少N端SDT。
在本發明之一實施例中,R1D2包含胺基酸序列:PFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQT (SEQ ID NO.: 35)。
在本發明之一實施例中,R2D3包含胺基酸序列:VVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK (SEQ ID NO.: 36)。
在本發明之一實施例中,R2D4包含胺基酸序列:PFVAFGSGMESLVEATVGERVRIPAKYLGYPPPEIKWYKNGIPLESNHTIKAGHVLTIMEVSERDTGNYTVILTNPISKEKQSHVVSLVVYVPPGPG (SEQ ID NO.: 37)。
在本發明之一實施例中,R2D4包含胺基酸序列:FVAFGSGMESLVEATVGERVRIPAKYLGYPPPEIKWYKNGIPLESNHTIKAGHVLTIMEVSERDTGNYTVILTNPIKSEKQSHVVSLVVYVP (SEQ ID NO.: 38)。
在本發明之一實施例中,用於VEGF MiniTrap中之多聚組分(MC)為肽,例如,經修飾之Fc免疫球蛋白(例如,來自IgG1),其能夠與另一多聚組分結合。在一態樣中,MC為包括免疫球蛋白鉸鏈區的經修飾之Fc免疫球蛋白。例如,在本發明之一實施例中,MC為包含一或多個(例如1、2、3、4、5或6個)半胱胺酸之肽,此等半胱胺酸能夠與另一個MC中之半胱胺酸形成一或多個半胱胺酸橋,例如DKTHTCPPC (SEQ ID NO.: 39)、DKTHTCPPCPPC (SEQ ID NO.: 40)、DKTHTCPPCPPCPPC (SEQ ID NO.: 41)、DKTHTC(PPC)h
(其中h為1、2、3、4或5)、 DKTHTCPPCPAPELLG (SEQ ID NO.: 60)、DKTHTCPLCPAPELLG (SEQ ID NO.: 43)、DKTHTC (SEQ ID NO.: 44)或DKTHTCPLCPAP (SEQ ID NO.: 45)。
本發明亦提供一種包含以下域結構之VEGF MiniTrap多肽:
(i) (R1D2)a
-(R2D3)b
-(MC)c
;或
(ii) (R1D2)a
-(R2D3)b
-(R2D4)c
-(MC)d
;
其可例如藉由每個多肽之MC之間的結合與第二該等多肽同型二聚,
其中
(i) 該等R1D2域配位;
(ii) 該等R2D3域配位;及/或
(iii) 該等R2D4域配位,
以形成二聚VEGF結合域。
在本發明之一實施例中,VEGF MiniTrap多肽包含以下胺基酸序列:
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPN36
ITVTLKKFPLDTLIPDGKRIIWDSRKGFIISN68
ATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLN123
CTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKN196
STFVRVHEKDKTHTCPPCPAPELLG
(SEQ ID NO.: 46;MC加下劃線);GRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHENLSVAFGSGMESLVEATVGERVRIPAKYLGYPPPEIKWYKNGIPLESNHTIKAGHVLTIMEVSERDTGNYTVILTNPISKEKQSHVVSLVVYVPPGPGDKTHTCPLCPAPELLG
(SEQ ID NO.: 47;MC加下劃線);SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPN36
ITVTLKKFPLDTLIPDGKRIIWDSRKGFIISN68
ATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLN123
CTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKN196
STFVRVHEKDKTHTCPPC
(SEQ ID NO.: 48;MC加下劃線);SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPN36
ITVTLKKFPLDTLIPDGKRIIWDSRKGFIISN68
ATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLN123
CTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKN196
STFVRVHEKDKTHTCPPCPPC
(SEQ ID NO.: 49;MC加下劃線);SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPN36
ITVTLKKFPLDTLIPDGKRIIWDSRKGFIISN68
ATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLN123
CTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKN196
STFVRVHEKDKTHTCPPCPPCPPC
(SEQ ID NO.: 50;MC加下劃線);或SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKDKTHTC-(PPC)x
(MC加下劃線;其中x為1、2、3、4或5)。如所討論,該等多肽可經多聚化(例如二聚化(例如,同型二聚化)),其中多肽之間的結合係經由多聚化組分介導。
在本發明之一實施例中,本發明之單體VEGF MiniTrap之VEGFR1 Ig樣域2在N36及/或N68處具有N連接之醣化作用;及/或在C30與C79之間具有鏈內二硫橋;及/或本發明之單體VEGF MiniTrap之VEGFR2 Ig樣域3在N123及/或N196處具有N連接之醣化作用;及/或在C124與C185之間具有鏈內二硫橋。
在本發明之一實施例中,VEGF MiniTrap包含以下結構:
l (R1D2)1
-(R2D3)1
-(G4
S)3
-(R1D2)1
-(R2D3)1
;
l (R1D2)1
-(R2D3)1
-(G4
S)6
-(R1D2)1
-(R2D3)1
;
l (R1D2)1
-(R2D3)1
-(G4
S)9
-(R1D2)1
-(R2D3)1
;或
l (R1D2)1
-(R2D3)1
-(G4
S)12
-(R1D2)1
-(R2D3)1
。
G4
S為-Gly-Gly-Gly-Gly-Ser-
在本發明之一實施例中,VEGF MiniTrap包含以下胺基酸序列:
(i)
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKGGGGSGGGGSGGGGSGGGGSGGGGSGGGGS
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK (SEQ ID NO.: 51;連接子加下劃線);
(iii)
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKGGGGSGGGGSGGGGS
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK (SEQ ID NO.: 52;連接子加下劃線);
(iv)
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGS
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK (SEQ ID NO.: 53;連接子加下劃線);
(v)
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGS
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK (SEQ ID NO.: 54;連接子加下劃線);
或
(vi)
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK-(GGGGS)x -SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEK
(其中x為1、2、3、4、5、6、7、8、9、10、11、12、13、14或15)。如本文所討論,此等多肽可包含二級結構,其中同樣的VEGFR Ig結構域締合以形成鏈內VEGF結合域(例如,圖 2
)。在本發明之一實施例中,兩個或更多個此等多肽多聚化(例如,二聚化(例如,同型二聚化)),其中每條鏈之VEGFR Ig域與另一條鏈之同樣Ig域締合以形成鏈間VEGF結合域。
在本發明之某一實施例中,本發明之VEGF MiniTrap缺乏對VEGF MiniTrap多肽之胺基酸殘基的任何顯著修飾(例如,例如在N端及/或C端處之直接化學修飾,諸如PEG化或碘乙醯胺化)。
在本發明之一實施例中,多肽包含二級結構,其中在單個嵌合多肽(例如,(R1D2)a
-(R2D3)b
-連接子-(R1D2)c
-(R2D3)d
;或(R1D2)a
-(R2D3)b
-(R2D4)c
-連接子-(R1D2)d
-(R2D3)e
-(R2D4)f
)中或在單獨的嵌合多肽(例如,同型二聚物)中之同樣的VEGFR Ig域配位以形成VEGF結合域。例如,其中
(i) 該等R1D2域配位;
(ii) 該等R2D3域配位;及/或
(iii) 該等R2D4域配位,
以形成VEGF結合域。圖 2
為描繪該域配位之單鏈VEGF MiniTrap的說明。指示VEGFR1、VEGFR2及連接子域。所示連接子為(G4
S)6
。本發明包括具有(G4
S)3
;(G4
S)9
或(G4
S)12
連接子之單鏈VEGF MiniTrap。
另外,本發明亦提供一種複合物,其包含與VEGF多肽或其片段或其融合物複合的本文所述之VEGF MiniTrap。在本發明之一實施例中,將VEGF (例如,VEGF165
)同型二聚化及/或將VEGF MiniTrap在2:2複合物(2個VEGF:2個MiniTrap)中同型二聚化及/或將VEGF MiniTrap在1:1複合物中同型二聚化。複合物可包括與同型二聚化之VEGF MiniTrap多肽結合的同型二聚化之VEGF分子。在本發明之一實施例中,複合物係在活體外(例如,固定在固體基質上)或在個體體內。本發明亦包括與VEGF MiniTrap複合的VEGF二聚物(例如,VEGF165
)之複合物的組成物。
如本文所用,術語「蛋白質」或「感興趣之蛋白質」可包括具有共價連接之醯胺鍵的任何胺基酸聚合物。感興趣之蛋白質之實例包括但不限於阿柏西普及MiniTrap。蛋白質包含一或多條胺基酸聚合物鏈,在此項技術中通常稱為「多肽」。「多肽」係指由胺基酸殘基、相關天然存在之結構變異體及其經由肽鍵連接的合成的非天然存在之類似物、相關天然存在之結構變異體及其合成的非天然存在之類似物構成的聚合物。「合成肽或多肽」係指非天然存在之肽或多肽。合成肽或多肽可例如使用自動多肽合成器來合成。各種固相肽合成方法係熟習此項技術者已知的。蛋白質可包含一個或多個多肽以形成單一功能之生物分子。另一示例性態樣,蛋白質可包括抗體片段、奈米抗體、重組抗體嵌合體、細胞介素、趨化介素、肽激素及其類似物。感興趣之蛋白質可包括以下任一者:生物治療性蛋白、用於研究或療法中之重組蛋白、trap蛋白及其他嵌合受體Fc融合蛋白、嵌合蛋白、抗體、單株抗體、多株抗體、人類抗體及雙特異性抗體。在一具體態樣中,感興趣之蛋白質為抗VEGF融合蛋白(例如,阿柏西普或MiniTrap)。可使用以下基於重組細胞之生產系統來產生蛋白質,諸如昆蟲桿狀病毒系統、酵母系統(例如畢赤酵母屬(Pichia sp.))、哺乳動物系統(例如CHO細胞及CHO衍生物,像CHO-K1細胞)。有關討論生物治療性蛋白及其產生之最新評論,參見Ghaderi等人, 「Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation,」 (Darius Ghaderi等人, Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation, 28 BIOTECHNOLOGY AND GENETIC ENGINEERING REVIEWS 147-176 (2012),其全部教示皆併入本文)。在一些示例性實施例中,蛋白質包含修飾、加合物及其他共價連接之部分-此等修飾、加合物及部分包括例如抗生物素蛋白、鏈黴親和素、生物素、聚醣(例如,N-乙醯半乳糖胺、半乳糖、神經胺糖酸、N-乙醯基葡糖胺、岩藻醣、甘露糖及其他單醣)、PEG、聚組胺酸、FLAGtag、麥芽糖結合蛋白(MBP)、幾丁質結合蛋白(CBP)、麩胱甘肽-S-轉移酶(GST) myc表位、螢光標記及其他染料、及其類似物。蛋白質可根據組成及溶解度來分類,且因此可包括簡單蛋白質,諸如球狀蛋白質及纖維狀蛋白質;複合蛋白,諸如核蛋白、醣蛋白、黏蛋白、色蛋白、磷蛋白、金屬蛋白及脂蛋白;以及衍生蛋白,諸如初級衍生蛋白及次級衍生蛋白。
在一些示例性實施例中,感興趣之蛋白質可為重組蛋白、抗體、雙特異性抗體、多特異性抗體、抗體片段、單株抗體、融合蛋白、scFv及其組合。
如本文所用,術語「重組蛋白」係指由於重組表現載體上攜帶之基因之轉錄及翻譯而產生的蛋白,該重組表現載體已引入合適的宿主細胞中。在某些示例性實施例中,重組蛋白可為融合蛋白。在一具體態樣中,重組蛋白為抗VEGF融合蛋白(例如,阿柏西普或MiniTrap)。在某些示例性實施例中,重組蛋白可為抗體,例如,嵌合的、人類化的或完全人類抗體。在某些示例性實施例中,重組蛋白可為選自由以下組成之群組的同種型抗體:IgG、IgM、IgA1、IgA2、IgD或IgE。在某些示例性實施例中,抗體分子為全長抗體(例如,IgG1),或替代地,抗體可為片段(例如,Fc片段或Fab片段)。
如本文所用,術語「抗體」包括包含藉由雙硫鍵互連之四條多肽鏈(亦即兩條重(H)鏈及兩條輕(L)鏈)的免疫球蛋白分子,以及其多聚物(例如,IgM)。每條重鏈包含重鏈可變區(本文中縮寫為HCVR或VH)及重鏈恆定區。重鏈恆定區包含三個域,即CH1、CH2及CH3。每條輕鏈包含輕鏈可變區(本文中縮寫為LCVR或VL)及輕鏈恆定區。輕鏈恆定區包含一個域(CL1)。VH區及VL區可進一步再分成稱為互補決定區(CDR)之高變區,散佈有稱為構架區(FR)之較保守區。各VH及VL由三個CDR及四個FR組成,該等區依以下順序自胺基端至羧基端排列:FR1、CDR1、FR2、CDR2、FR3、CDR3及FR4。在本發明之不同實施例中,抗big-ET-1抗體(或其抗原結合部分)之FR可與人類生殖系序列相同,或者可為天然或人工修飾的。可基於兩個或更多個CDR之並排分析來定義胺基酸共有序列。如本文所用,術語「抗體」亦包括全抗體分子之抗原結合片段。如本文所用,術語抗體之「抗原結合部分」、抗體之「抗原結合片段」及其類似者包括特異性結合抗原以形成複合物之任何天然存在的、以酶促方式獲得的、合成或基因工程改造之多肽或醣蛋白。抗體之抗原結合片段可使用以下任何合適的標準技術例如由全抗體分子衍生而來:諸如蛋白水解消化或重組基因工程技術,其涉及編碼抗體可變域及視情況之恆定域的DNA之操縱及表現。該DNA為已知的及/或可易於由例如商業來源、DNA文庫(包括例如噬菌體-抗體文庫)獲得,或者可經合成。可以化學方式或藉由使用分子生物學技術對DNA進行定序及操縱,例如以將一或多個可變域及/或恆定域排列成合適的構型;或引入密碼子;產生半胱胺酸殘基;修飾、添加或刪除胺基酸等。
如本文所用,「抗體片段」包括完整抗體之一部分,諸如例如抗體之抗原結合或可變區。抗體片段之實例包括但不限於Fab片段、Fab'片段、F(ab’)2片段、scFv片段、Fv片段、dsFv雙抗體、dAb片段、Fd'片段、Fd片段及經分離之互補決定區(CDR)區域,以及由抗體片段形成之三抗體、四抗體、線性抗體、單鏈抗體分子及多特異性抗體。Fv片段為免疫球蛋白重鏈與輕鏈可變區之組合,且ScFv蛋白為重組單鏈多肽分子,其中免疫球蛋白輕鏈與重鏈可變區經由肽連接子連接。在一些示例性實施例中,抗體片段包含親本抗體之足夠的胺基酸序列,其為與親本抗體結合相同抗原之片段;在一些示例性實施例中,片段以與親本抗體相當之親和力結合至抗原及/或與親本抗體競爭結合至抗原。抗體片段可藉由任何方式產生。例如,抗體片段可藉由完整抗體之片段化以酶促或化學方式產生及/或其可由編碼部分抗體序列之基因以重組方式產生。替代或另外,抗體片段可全部或部分合成地產生。抗體片段可視情況包含單鏈抗體片段。替代或另外,抗體片段可包含例如藉由雙硫鍵聯連接在一起之多條鏈。抗體片段可視情況包含多分子複合物。功能性抗體片段通常包含至少約50個胺基酸,且更通常包含至少約200個胺基酸。
術語「雙特異性抗體」包括能夠選擇性地結合兩個或更多個表位之抗體。雙特異性抗體通常包含兩條不同重鏈,每條重鏈特異性結合不同表位-在兩種不同分子(例如,抗原)上或在相同分子(例如,相同抗原)上。若雙特異性抗體能夠選擇性地結合兩個不同表位(第一表位及第二表位),則第一重鏈對第一表位之親和力通常比第一重鏈對第二表位之親和力低至少一個至兩個或三個或四個數量級,且反之亦然。由雙特異性抗體識別之表位可在相同或不同靶標上(例如,在相同或不同蛋白質上)。雙特異性抗體可例如藉由組合識別相同抗原之不同表位的重鏈來製備。例如,可將編碼識別相同抗原之不同表位的重鏈可變序列的核酸序列與編碼不同重鏈恆定區之核酸序列融合,並且可在表現免疫球蛋白輕鏈之細胞中表現該等序列。
典型的雙特異性抗體具有兩條重鏈,每條重鏈均具有三個重鏈CDR,繼之以CH1域、鉸鏈、CH2域及CH3域;及免疫球蛋白輕鏈,其不賦予抗原結合特異性但可與每條重鏈締合,或可與每條重鏈締合且可結合由重鏈抗原結合區結合之一或多個表位,或者可與每條重鏈締合並能夠將一條或兩條重鏈結合至一個或兩個表位。BsAb可分為兩種主要類別,一類為帶有Fc區之抗體(IgG樣),而另一類為缺少Fc區之抗體,後者通常小於包含Fc之IgG及IgG樣雙特異性分子。IgG樣bsAb可具有不同形式,諸如但不限於三功能抗體(triomab)、杵臼IgG (kih IgG)、crossMab、orth-Fab IgG、雙可變域Ig (DVD-Ig)、二合一或雙重作用的Fab (DAF)、IgG單鏈Fv (IgG-scFv)或κλ體。非IgG樣不同形式包括串聯scFv、雙抗體形式、單鏈雙抗體、串聯雙抗體(TandAbs)、雙親和重靶向分子(DART)、DART-Fc、奈米抗體或藉由對接鎖定(DNL)方法產生之抗體(Gaowei Fan, Zujian Wang & Mingju Hao, Bispecific antibodies and their applications, 8 JOURNAL OF HEMATOLOGY & ONCOLOGY 130;Dafne Müller & Roland E. Kontermann, Bispecific Antibodies, HANDBOOK OF THERAPEUTIC ANTIBODIES 265–310 (2014),其全部教示皆併入本文)。產生bsAb之方法不限於基於兩種不同融合瘤細胞株之體細胞融合的四源融合瘤技術、涉及化學交聯劑之化學偶聯以及利用重組DNA技術之遺傳方法。bsAb之實例包括在以下專利申請案(其以引用之方式併入本文)中揭露之彼等:2010年6月25日提出申請之美國第12/823838號;2012年6月5日提出申請之美國第13/488628號;2013年9月19日提出申請之美國第14/031075號;2015年7月24日提出申請之美國第14/808171號;2017年9月22日提出申請之美國第15/713574號;2017年9月22日提出申請之美國第15/713569號;2016年12月21日提出申請之美國第15/386453號;2016年12月21日提出申請之美國第15/386443號;2016年7月29日提出申請之美國第15/22343號;及2017年11月15日提出申請之美國第15814095號。在製造雙特異性抗體之幾個步驟中,可存在少量同型二聚物雜質。當使用完整質量分析進行偵測時,此等同型二聚物雜質之偵測可具有挑戰性,因為同型二聚物雜質之豐度低並且當使用常規液相層析法進行偵測時,此等雜質與主要種類共溶析。
如本文所用,「多特異性抗體」係指對至少兩種不同抗原具有結合特異性之抗體。儘管此等分子通常僅結合兩種抗原(亦即雙特異性抗體,bsAb),但具有額外特異性之抗體(諸如三特異性抗體及KIH三特異性抗體)亦可由本文揭露之系統及方法提出。
如本文所用,術語「單株抗體」不限於經由融合瘤技術產生之抗體。單株抗體可藉由此項技術可獲得或已知之任何手段衍生自單一純系,包括任何真核、原核或噬菌體純系。可使用此項技術已知之廣泛多種技術來製備可用於本文之單株抗體,包括使用融合瘤、重組及噬菌體展示技術或其組合。
在一些示例性實施例中,感興趣之蛋白質可具有在約4.5至約9.0範圍內之pI。在一示例性特定實施例中,pI可為約4.5、約5.0、約5.5、約5.6、約5.7、約5.8、約5.9、約6.0、約6.1、 約6.2、約6.3、約6.4、約6.5、約6.6、約6.7、約6.8、約6.9、約7.0、約7.1、約7.2、約7.3、約7.4、約7.5、約7.6、約7.7、約7.8、約7.9、約8.0、約8.1、約8.2、約8.3、約8.4、約8.5、約8.6、約8.7、約8.8、約8.9或約9.0。在一些示例性實施例中,組成物中感興趣之蛋白質之類型可不止一種。
在一些示例性實施例中,感興趣之蛋白質可由哺乳動物細胞產生。哺乳動物細胞可具有人類來源或非人類來源,可包括原代上皮細胞(例如,角質細胞、宮頸上皮細胞、支氣管上皮細胞、氣管上皮細胞、腎上皮細胞及視網膜上皮細胞);已建立之細胞株及其品系(例如,293胚胎腎細胞、BHK細胞、HeLa宮頸上皮細胞及PER-C6視網膜細胞、MDBK (NBL-1)細胞、911細胞、CRFK細胞、MDCK細胞、CHO細胞、BeWo細胞、Chang細胞、Detroit 562細胞、HeLa 229細胞、HeLa S3細胞、Hep-2細胞、KB細胞、LSI80細胞、LS174T細胞、NCI-H-548細胞、RPMI2650細胞、SW-13細胞、T24細胞、WI-28 VA13, 2RA細胞、WISH細胞、BS-C-I細胞、LLC-MK2細胞、Clone M-3細胞、1-10細胞、RAG細胞、TCMK-1細胞、Y-l細胞、LLC-PKi細胞、PK(15)細胞、GHi細胞、GH3細胞、L2細胞、LLC-RC 256細胞、MHiCi細胞、XC細胞、MDOK細胞、VSW細胞及TH-I, B1細胞、BSC-1細胞、RAf細胞、RK-細胞、PK-15細胞或其衍生物)、來自任何組織或器官(包括但不限於心臟、肝臟、腎臟、結腸、腸、食道、胃、神經組織(腦、脊髓)、肺、血管組織(動脈、靜脈、毛細血管)、淋巴組織(淋巴腺、腺樣體、扁桃體、骨髓及血液)、脾臟)之成纖維細胞、以及成纖維細胞及成纖維細胞樣細胞株(例如,CHO細胞、TRG-2細胞、IMR-33細胞、Don細胞、GHK-21細胞、瓜胺酸血症細胞、Dempsey細胞、Detroit 551細胞、Detroit 510細胞、Detroit 525細胞、Detroit 529細胞、Detroit 532細胞、Detroit 539細胞、Detroit 548細胞、Detroit 573細胞、HEL 299細胞、IMR-90細胞、MRC-5細胞、WI-38細胞、WI-26細胞、Midi細胞、CHO細胞、CV-1細胞、COS-1細胞、COS-3細胞、COS-7細胞、Vero細胞、DBS-FrhL-2細胞、BALB/3T3細胞、F9細胞、SV-T2細胞、M-MSV-BALB/3T3細胞、K-BALB細胞、BLO-11細胞、NOR-10細胞、C3H/IOTI/2細胞、HSDMiC3細胞、KLN205細胞、McCoy細胞、小鼠L細胞、品系2071 (小鼠L)細胞、L-M品系(小鼠L)細胞、L-MTK' (小鼠L)細胞、NCTC純系2472及2555、SCC-PSA1細胞、Swiss/3T3細胞、印度黃麂細胞、SIRC細胞、Cn細胞及Jensen細胞、Sp2/0、NS0、NS1細胞或其衍生物)。
如本文所用,術語「蛋白質烷化劑」係指用於烷化蛋白質中某些遊離胺基酸殘基之試劑。蛋白質烷化劑之非限制性實例為碘乙醯胺(IOA)、氯乙醯胺(CAA)、丙烯醯胺(AA)、N-乙基馬來亞醯胺(NEM)、甲烷硫代磺酸甲酯(MMTS)及4-乙烯基吡啶或其組合。
如本文所用,「蛋白質變性」可指代其中分子之三維形狀自其天然狀態改變之過程。蛋白質變性可使用蛋白質變性劑進行。蛋白質變性劑之非限制性實例包括熱、高或低pH值、還原劑像DTT (參見下文)或暴露於離液劑。幾種離液劑可用作蛋白質變性劑。離液溶質藉由干擾由非共價力(諸如氫鍵、範德華力及疏水作用)介導之分子內相互作用增加系統之熵。離液劑之非限制性實例包括丁醇、乙醇、氯化胍、過氯酸鋰、乙酸鋰、氯化鎂、苯酚、丙醇、十二烷基硫酸鈉、硫脲、N-月桂醯肌胺酸、尿素及其鹽。
如本文所用,術語「蛋白質還原劑」係指用於還原蛋白質中之二硫橋的試劑。用於還原蛋白質之蛋白質還原劑之非限制性實例為二硫蘇糖醇(DTT)、β-巰基乙醇、愛耳門氏試劑(Ellman’s reagent)、鹽酸羥胺、氰基硼氫化鈉、參(2-羧乙基)膦鹽酸鹽(TCEP-HCl)或其組合。
如本文所用,術語多肽(例如,VEGFR Ig域之多肽)之「變異體」係指包含與感興趣之蛋白質的參考或天然胺基酸序列至少約70至99.9% (例如
70%、71%、72%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.5%、99.9%)一致或相似之胺基酸序列的多肽。可藉由例如BLAST算法來執行序列比較,其中選擇算法之參數以在個別參考序列之整個長度上給出個別序列之間的最大匹配(例如,預期閾值:10;字長:3;查詢範圍內之最大匹配:0;BLOSUM 62矩陣;缺口成本:存在11,延伸1;條件組成評分矩陣調整)。多肽(例如,VEGFR Ig域之多肽)之變異體亦可指代除一或多個(例如1、2、3、4、5、6、7、8、9或10個)突變諸如錯義突變(例如保守取代)、無義突變、缺失或插入之外包含參考胺基酸序列之多肽。以下參考文獻涉及經常用於序列分析之BLAST算法:BLAST ALGORITHMS: Altschul等人 (2005) FEBS J. 272(20): 5101-5109;Altschul, S. F.,等人, (1990) J. Mol. Biol. 215:403-410;Gish, W.,等人, (1993) Nature Genet. 3:266-272;Madden, T. L.,等人, (1996) Meth. Enzymol. 266:131-141;Altschul, S. F.,等人, (1997) Nucleic Acids Res. 25:3389-3402;Zhang, J.,等人, (1997) Genome Res. 7:649-656;Wootton, J. C.,等人, (1993) Comput. Chem. 17:149-163;Hancock, J. M.等人, (1994) Comput. Appl. Biosci. 10:67-70;ALIGNMENT SCORING SYSTEMS: Dayhoff, M. O.,等人, 「A model of evolutionary change in proteins.」,Atlas of Protein Sequence and Structure, (1978) 第5卷, 增刊3. M. O. Dayhoff (編), 第345-352頁, Natl. Biomed. Res. Found., Washington, D.C.;Schwartz, R. M.,等人, 「Matrices for detecting distant relationships.」,Atlas of Protein Sequence and Structure, (1978) 第5卷, 增刊3.'' M. O. Dayhoff (編), 第353-358頁, Natl. Biomed. Res. Found., Washington, D.C.;Altschul, S. F., (1991) J. Mol. Biol. 219:555-565;States, D. J.,等人, (1991) Methods 3:66-70;Henikoff, S.,等人, (1992) Proc. Natl. Acad. Sci. USA 89:10915-10919;Altschul, S. F.,等人, (1993) J. Mol. Evol. 36:290-300;ALIGNMENT STATISTICS: Karlin, S.,等人, (1990) Proc. Natl. Acad. Sci. USA 87:2264-2268;Karlin, S.,等人, (1993) Proc. Natl. Acad. Sci. USA 90:5873-5877;Dembo, A.,等人, (1994) Ann. Prob. 22:2022-2039;以及Altschul, S. F. 「Evaluating the statistical significance of multiple distinct local alignments.」,Theoretical and Computational Methods in Genome Research (S. Suhai編), (1997) 第1-14頁, Plenum, N.Y.;其全部教示皆併入本文。
一些變異體可為多肽在其核醣體合成期間(共轉譯修飾)或之後(轉譯後修飾「PTM」)經歷之共價修飾。通常藉由特定的酶或酶途徑引入PTM。許多出現在蛋白質骨架內的特定特徵性蛋白質序列(例如,標籤序列)之位點處。已記錄數百個PTM,且此等修飾始終會影響蛋白質結構或功能之某些態樣(Walsh, G. 「Proteins」(2014) 第二版, 由Wiley and Sons, Ltd.出版, ISBN: 9780470669853,其全部教示皆併入本文)。在某些示例性實施例中,蛋白質組成物可包含一種以上類型的感興趣之蛋白質之蛋白質變異體。
在阿柏西普(及具有阿柏西普結構特徵之蛋白質,例如,阿柏西普之一或多個重鏈或輕鏈區)情況下之蛋白質變異體可包含但不限於可由一或多個在以下各處出現之胺基酸殘基之氧化產生的氧化變異體:例如組胺酸、半胱胺酸、甲硫胺酸、色胺酸、苯丙胺酸及/或酪胺酸殘基;可由天冬醯胺殘基處之去醯胺產生的去醯胺變異體及/或去氧葡醛酮糖化精胺酸殘基產生之變異體。
關於阿柏西普(及具有阿柏西普之結構特徵的蛋白質,例如,阿柏西普之一或多個重鏈或輕鏈區),氧化變異體可包含在His86、His110、His145、His209、His95、His19及/或His203 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的組胺酸殘基之氧化;在Trp58及/或Trp138 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的色胺酸殘基之氧化;在Tyr64 (或具有阿柏西普之某些結構特徵的蛋白質上之等效位置)處的酪胺酸殘基之氧化;在Phe44及/或Phe166 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的苯丙胺酸殘基之氧化;及/或在Met10、Met20、Met163及/或Met192 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的甲硫胺酸殘基之氧化。
關於阿柏西普(及具有阿柏西普之結構特徵的蛋白質,例如阿非柏西之一或多個重鏈或輕鏈區),去醯胺變異體可包含在Asn84及/或Asn99 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的天冬醯胺殘基之去醯胺。
關於阿柏西普(及具有阿柏西普之結構特徵的蛋白質,例如,阿柏西普之一或多個重鏈或輕鏈區),去氧葡醛酮糖化變異體可包含在Arg5 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的精胺酸殘基之3-去氧葡醛酮糖化。
蛋白質變異體可包括酸性種類及鹼性種類。酸性種類係通常比CEX之主峰更早溶析或比AEX之主峰更晚溶析的變異體,而鹼性種類係比CEX之主峰更晚溶析或比AEX之主峰更早溶析的變異體。
如本文所用,術語「酸性種類」、「AS」、「酸性區域」及「AR」係指特徵為總酸性電荷的蛋白質變異體。例如,在重組蛋白製劑中,可藉由以下各種方法偵測此等酸性種類,諸如離子交換,例如WCX-10 HPLC (弱陽離子交換層析)或IEF (等電聚焦)。抗體之酸性種類可包括變異體、結構變異體及/或片段化變異體。示例性變異體可包括但不限於去醯胺變異體、岩藻醣化變異體、氧化變異體、甲基乙二醛(MGO)變異體、醣化變異體及檸檬酸變異體。示例性結構變異體包括但不限於醣化變異體及丙酮化變異體。示例性片段化變異體包括由於肽鏈之解離、酶及/或化學修飾而由靶分子產生的任何經修飾之蛋白質種類,包括但不限於Fc及Fab片段、缺少Fab之片段、缺少重鏈可變域之片段、C端截短變異體、在輕鏈中切除N端Asp之變異體以及具有輕鏈之N端截短的變異體。其他酸性種類變異體包括包含以下各物之變異體:未配對之二硫化物、宿主細胞蛋白及宿主核酸、層析材料及培養基組分。通常,酸性種類在CEX期間比主峰更早溶析或在AEX分析期間比主峰更晚溶析(參見圖 16 及圖 17
)。
在某些實施例中,蛋白質組成物可包含一種以上類型之酸性種類變異體。例如,但不作為限制,可基於出現的峰之層析滯留時間對總酸性種類進行分類。可將全部酸性種類歸類之另一例子可基於變異體之類型-變異體、結構變異體或片段化變異體。
術語「酸性種類」或「AS」不涉及過程相關之雜質。如本文所用,術語「過程相關之雜質」係指存在於包含蛋白質之組成物中但並非衍生自蛋白質本身之雜質。過程相關之雜質包括但不限於宿主細胞蛋白(HCP)、宿主細胞核酸、層析材料及培養基組分。
在一示例性實施例中,與感興趣之蛋白質相比,抗VEGF組成物中酸性種類之量可為最多約 20%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。抗VEGF組成物之實例論述於以下節III中。在一態樣中,抗VEGF組成物可包含選自由以下組成之群組的抗VEGF蛋白:阿柏西普、重組MiniTrap (其實例在美國專利第7,279,159號中揭露)、scFv及其他抗VEGF蛋白。在一較佳態樣中,感興趣之重組蛋白為阿柏西普。
在負責酸性或鹼性種類之化學降解途徑中,蛋白質及肽中最常見的兩種共價修飾為去胺及氧化。甲硫胺酸、半胱胺酸、組胺酸、色胺酸及酪胺酸為最易氧化之胺基酸:Met及Cys係因其硫原子,而His、Trp及Tyr係因其芳環。
如本文所用,術語「氧化種類」、「OS」或「氧化變異體」係指藉由氧化形成的蛋白質變異體。亦可藉由各種方法,諸如離子交換,例如WCX-10 HPLC (弱陽離子交換層析)或IEF (等電聚焦)來偵測此等氧化種類。氧化變異體可由在組胺酸、半胱胺酸、甲硫胺酸、色胺酸、苯丙胺酸及/或酪胺酸殘基處發生之氧化產生。具體而言,關於阿柏西普(及具有阿柏西普之結構特徵的蛋白質,例如阿柏西普之一或多個重鏈或輕鏈區),氧化變異體可包含在His86、His110、His145、His209、His95、His19及/或His203 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的組胺酸殘基之氧化;在Trp58及/或Trp138 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的色胺酸殘基之氧化;在Tyr64 (或具有阿柏西普之某些結構特徵的蛋白質上之等效位置)處的酪胺酸殘基之氧化;在Phe44及/或Phe166 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的苯丙胺酸殘基之氧化;及/或在Met10、Met 20、Met163及/或Met192 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處的甲硫胺酸殘基之氧化。
在一示例性實施例中,與感興趣之蛋白質相比,抗VEGF組成物中氧化種類之量可為最多約15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。抗VEGF組成物之實例論述於以下節III中。在一態樣中,抗VEGF組成物可包含選自由以下組成之群組的抗VEGF蛋白:阿柏西普、重組MiniTrap (其實例在美國專利第7,279,159號中揭露)、scFv及其他抗VEGF蛋白。在一較佳態樣中,感興趣之重組蛋白為阿柏西普或MiniTrap。
半胱胺酸殘基可自發氧化形成分子內或分子間雙硫鍵或單分子副產物(諸如次磺酸)。
組胺酸殘基經由與其咪唑環之反應亦對氧化高度敏感,隨後會生成另外的羥基種類(Li, S, C Schoneich及RT. Borchardt. 1995. Chemical Instability of Protein Pharmaceuticals: Mechanisms of Oxidation and Strategies for Stabilization. Biotechnol. Bioeng. 48:490-500,其全部教示皆併入本文)。所提出的組胺酸氧化機制在圖 2
及圖 3
中突出顯示。詳細機制研究可獲自Anal. Chem. 2014, 86, 4940-4948及J. Pharm. Biomed. Anal. 21 (2000) 1093-1097,其全部教示皆併入本文。
甲硫胺酸之氧化可導致甲硫胺酸亞碸之形成(Li, S, C Schoneich及RT. Borchardt.1995. Chemical Instability of Protein Pharmaceuticals: Mechanisms of Oxidation and Strategies for Stabilization. Biotechnol. Bioeng. 48:490-500)。甲硫胺酸殘基之各種可能的氧化機制已在文獻中討論(Brot, N., Weissbach, H. 1982. The biochemistry of methionine sulfoxide residues in proteins. Trends Biochem. Sci. 7: 137-139,其全部教示皆併入本文)。
色胺酸之氧化可產生復雜的產物混合物。主要產物可為N-甲醯犬尿胺酸及犬尿胺酸以及單氧化、二氧化及/或三氧化產物(圖 4
)。帶有氧化之Trp修飾的肽通常顯示4、16、32及48 Da之質量增加,對應於形成犬尿胺酸(KYN)、羥基色胺酸(Wox1
)及N-甲醯犬尿胺酸/二羥基色胺酸(NFK/Wox2
,亦稱為「雙重氧化之Trp」)、三羥基色胺酸(Wox3
,亦稱為「三重氧化之Trp」)及其組合,諸如羥基犬尿胺酸(KYNox1
,+20 Da)。氧化為羥基色胺酸(Wox1
)(Mass spectrometric identification of oxidative modifications of tryptophan residues in proteins: chemical artifact or post-translational modification? J. Am. Soc. Mass Spectrom. 2010年7月; 21(7): 1114-1117,其全部教示皆併入本文)。已發現色胺酸氧化而非甲硫胺酸及組胺酸氧化會在蛋白質產物中產生顏色變化(Characterization of the Degradation Products of a Color-Changed Monoclonal Antibody: Tryptophan-Derived Chromophores. dx.doi.org/10.1021/ac404218t | Anal. Chem. 2014, 86, 6850−6857)。與色胺酸相似,酪胺酸之氧化主要生成3,4-二羥基苯丙胺酸(DOPA)及二酪胺酸(Li, S, C Schoneich及RT. Borchardt.1995. Chemical Instability of Protein Pharmaceuticals: Mechanisms of Oxidation and Strategies for Stabilization. Biotechnol. Bioeng.48:490-500)。
如本文所用,術語「鹼性種類」、「鹼性區域」及「BR」係指蛋白質,例如抗體或其抗原結合部分之變異體,其特徵為相對於蛋白質中存在的初級電荷變異體種類的總鹼性電荷。例如,在重組蛋白製劑中,可藉由各種方法,諸如離子交換,例如WCX-10 HPLC (弱陽離子交換層析)或IEF (等電聚焦)偵測此等鹼性種類。示例性變異體可包括但不限於離胺酸變異體、天冬胺酸之異構化、天冬醯胺處之琥珀醯亞胺形成、甲硫胺酸氧化、醯胺化、不完全雙硫鍵形成、自絲胺酸至精胺酸之突變、醣化、片段化及聚集。通常,鹼性種類之溶析在CEX期間晚於主峰或在AEX分析期間早於主峰。(Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. MAbs. 2012年9月1日; 4(5): 578–585. doi: 10.4161/mabs.21328,其全部教示皆併入本文。)
在某些實施例中,蛋白質組成物可包含一種以上類型之鹼性種類變異體。例如但不限於,可基於出現的峰之層析滯留時間對總鹼性種類進行劃分。可劃分總鹼性種類之另一例子可基於變異體之類型-變異體、結構變異體或片段化變異體。
如針對酸性種類所討論,術語「鹼性種類」不包括過程相關之雜質,並且鹼性種類可為產物製備之結果(在本文中稱為「製備來源之鹼性種類」)或儲存之結果(在本文中稱為「儲存來源之鹼性種類」)。
在一示例性實施例中,與感興趣之蛋白質相比,抗VEGF組成物中鹼性種類之量可為最多約15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。抗VEGF組成物之實例論述於以下節III中。在一態樣中,抗VEGF組成物可包含選自由以下組成之群組的抗VEGF蛋白:阿柏西普、重組MiniTrap (其實例在美國專利第7,279,159號中揭露)、scFv及其他抗VEGF蛋白。在一較佳態樣中,感興趣之重組蛋白為阿柏西普。
如本文所用,「樣品基質」或「生物樣品」可獲自生物製程之任何步驟,諸如細胞培養液(CCF)、收穫的細胞培養液(HCCF)、下游過程中之任何步驟、原料藥(DS)或包含最終調配產物之藥品(DP)。在一些其他特定示例性實施例中,生物樣品可選自澄清、層析產生、病毒滅活或過濾之下游過程的任何步驟。在一些特定示例性實施例中,藥品可選自診所、運輸、儲存或處理中之製造藥品。
如本文所用,術語「個體」係指哺乳動物(例如,大鼠、小鼠、貓、狗、奶牛、綿羊、馬、山羊、兔),較佳為需要預防及/或治療癌症或血管生成性眼部病症之人類。個體可能患有癌症或血管生成性眼部病症,或易患癌症或血管生成性眼部病症。
就蛋白質調配物而言,如本文所用,術語「穩定的」係指在本文所定義之示例性條件下儲存後,該調配物內的感興趣之蛋白質能夠保持可接受程度之化學結構或生物功能。即使其中所含的感興趣之蛋白質在儲存限定量之時間後未保持100%之其化學結構或生物功能,調配物亦可為穩定的。在某些情境下,儲存限定量之時間後保持約90%、約95%、約96%、約97%、約98%或約99%之蛋白質結構或功能可視為「穩定的」。
術語「治療(treat)」或「治療(treatment)」係指逆轉、穩定或消除不希望有的疾病或病症(例如,血管生成性眼部病症或癌症)之治療措施,例如,藉由使該種疾病或病症之一或多種症狀或症候消退、穩定或消除任何臨床可量測之程度(例如,關於血管生成性眼部病症);藉由使糖尿病性視網膜病嚴重程度評分(DRSS)降低或維持;藉由改善或維持視力(例如,最佳矯正視力,例如,藉由ETDRS字母之增加來量度);增加或維持視野及/或減小或維持中央視網膜厚度;以及就癌症而言,停止或逆轉個體中的癌細胞之生長、存活及/或轉移。通常,治療措施係向患有疾病或病症之個體投與一或多個劑量之治療有效量之VEGF MiniTrap。
在蛋白質製備之情形下,如本文所用,術語「上游製程技術」係指涉及在感興趣之蛋白質的細胞培養期間或之後由細胞產生蛋白質及收集蛋白質的活動。如本文所用,術語「細胞培養」係指用於產生及維持能夠產生感興趣之重組蛋白的宿主細胞群之方法,以及用於最佳化感興趣之蛋白質的產生及收集之方法及技術。例如,一旦將表現載體併入合適的宿主細胞中,則可將宿主細胞維持在適於相關核苷酸編碼序列之表現以及所需重組蛋白之收集及產生的條件下。
當使用本發明之細胞培養技術時,感興趣之蛋白質可在胞內、在周質空間中產生,或直接分泌至培養基中。在胞內產生感興趣之蛋白質的實施例中,可藉由包括但不限於離心或超濾之多種方式移除微粒碎片-宿主細胞或溶解之細胞(例如,由均質化產生之碎片)。當感興趣之蛋白質分泌至培養基中時,可首先使用市售之蛋白質濃縮過濾器(例如,使用Amicon™或Millipore Pellicon™超濾單元)濃縮來自此等表現系統之上清液。在一態樣中,可藉由離心,繼之深度過濾,隨後親和捕獲層析來收穫感興趣之蛋白質。
如本文所用,「VEGF拮抗劑」為與VEGF結合或相互作用之任何蛋白質或肽。通常,這種與VEGF之結合或相互作用會抑制VEGF與其受體(VEGFR1及VEGFR2)之結合,及/或抑制VEGF之生物信號傳導及活性。VEGF拮抗劑包括干擾VEGF與天然VEGF受體之間的相互作用之分子,例如,與VEGF或VEGF受體結合並阻止或以其他方式阻礙VEGF與VEGF受體之間的相互作用之分子。特定的示例性VEGF拮抗劑包括抗VEGF抗體(例如
蘭尼單抗 [LUCENTIS®])、抗VEGF受體抗體(例如,抗VEGFR1抗體、抗VEGFR2抗體及其類似物)、以及基於VEGF受體之嵌合分子或VEGF抑制融合蛋白(在本文中亦稱為「VEGF-Trap」或「VEGF MiniTrap」),諸如阿柏西普、ziv-阿柏西普及具有SEQ ID NO.: 60之胺基酸的蛋白質。VEGF-Trap之其他實例為ALT-L9、M710、FYB203及CHS-2020。VEGF-Trap之額外實例可見於美國專利第7,070,959號;第7,306,799號;第7,374,757號;第7,374,758號;第7,531,173號;第7,608,261號;第5,952,199號;第6,100,071號;第6,383,486號;第6,897,294號及第7,771,721號中,此等專利具體地以引用之方式全部併入本文。
基於VEGF受體之嵌合分子包括嵌合多肽,其包含VEGF受體諸如VEGFR1 (亦稱為Flt1)及/或VEGFR2 (亦稱為Flk1或KDR)之兩個或更多個免疫球蛋白(Ig)樣域,並且亦可包含多聚域(例如,促進兩種或更多種嵌合多肽之多聚化[例如,二聚化]的Fc域)。示例性的基於VEGF受體之嵌合分子係稱為VEGFR1R2-FcΔC1(a)之分子(亦稱為阿柏西普;以商品名EYLEA®出售)。在某些示例性實施例中,阿柏西普包含如下所列之胺基酸序列:
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFMWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK。(SEQ ID NO.: 55)。
如本文所用,「病毒過濾」可包括使用合適的過濾器進行過濾,該等過濾器包括但不限於來自Asahi Kasei Pharma之Planova 20N™、50 N或BioEx;來自EMD Millipore之Viresolve™過濾器;來自Sartorius之ViroSart CPV;或來自Pall Corporation之Ultipor DV20或DV50™過濾器。對於熟習此項技術之普通技術人員顯而易見的是,選擇合適的過濾器以獲得所需過濾性能。II. 顏色測定
如本文所用,可藉由各種方法來量測在重組蛋白,特別是抗VEGF蛋白之產生期間觀察到的顏色。非限制性實例包括使用碘色號、黑曾(hazen)色號、加德納(gardner)色號、洛維邦德(lovibond)色號、賽氏(Saybolt)色號、礦物油色號、歐洲藥典色號、美國藥典色號、CIE L*, a*, b* (或CIELAB)、凱爾特(Klett)色號、Hess-Ives色號、黃度指數、ADMI色號以及ASBC及EBC啤酒色號。有關此等標度之詳情可見於Lange之申請報告第3.9e號中,其全部教示皆併入本文。
基於歐洲藥典(Ph Eur)(歐洲顏色標準,參見European Pharmacopoeia. 第2.2.2章 Degree of coloration of liquids. 第8版EP,其全部教示皆併入本文)之視覺色彩匹配可包括製備如下中所述之顏色參考溶液:Ph. Eur. (EP 2.2.2. Degree of Coloration of Liquids 2)-製備紅色(氯化鈷(II))、黃色(氯化亞鐵(III))及藍色(硫酸亞銅(II))之三種母液以及1%鹽酸,黃色(Y)、綠黃色(GY)、棕黃色(BY)、棕色(B)及紅色(R)色相之五種顏色參考溶液。依次使用這五種參考溶液,總共製備三十七種顏色參考溶液(Y1-Y7、GY1-GY7、BY1-BY7、B1-B9及R1-R7)。每個參考溶液皆在CIE-Lab色彩空間中由例如明度、色相及色度明確定義。在七個黃棕色標準(BY標準)中,BY1為最暗標準,而BY7為最不暗標準。給定樣品與BY顏色標準之匹配通常係在漫射日光下完成。歐洲黃棕色顏色標準之組成在下表1中描述。表 1. 歐洲棕黃色標準之組成
棕黃色標準溶液(BY):10.8 g/L FeCl3
.6H2
O、6.0 g/L CoCl2
.6H2
O及2.5 g/L CuSO4
.5H2
O
| 參考溶液 | 體積 (mL) | |
| 標準溶液 BY | 鹽酸 (10g/1 HCl) | |
| BY1 | 100.0 | 0.0 |
| BY2 | 75.0 | 25.0 |
| BY3 | 50.0 | 50.0 |
| BY4 | 25.0 | 75.0 |
| BY5 | 12.5 | 87.5 |
| BY6 | 5.0 | 95.0 |
| BY7 | 2.5 | 97.5 |
液體顏色之測試係藉由將測試溶液與標準顏色溶液進行比較來進行。根據測試溶液顏色之色相及強度來選擇標準顏色溶液之組成。通常,比較係在無色、透明、中性玻璃之平底試管中進行,該試管之內徑及所有其他態樣皆應儘可能匹配(例如,直徑約為12、15、16或25 mm之試管)。例如,比較可在2或10 mL之測試溶液與標準顏色溶液之間進行。液體之深度例如可為約15、25、40或50 mm。分配給測試溶液之顏色不應比標準顏色更強烈。顏色比較通常係在針對白色背景之漫射光(例如,日光)下進行。可沿試管之垂直軸或水平軸比較顏色。
與EP顏色量測對比,USP Monograph 1061 Color - Instrumental Measurement引用CIE L*, a*, b* (或CIELAB)顏色量測之使用以便精確及客觀地量化顏色。在美國藥典中定義總共二十種顏色參考溶液(依次由字母A至T識別)。所量測樣品之顏色自動與顏色參考溶液相關。這意味著展示最接近樣品之顏色參考溶液(亦即,與樣品顏色具有最小色差ΔE*之參考溶液)。ΔL*、Δa*及Δb*值給出樣品之L*、a*及b*值與所示USP溶液之L*、a*及b*值之間的定量差異。CIE L*a*b*坐標系,L*代表顏色之明度,標度為0-100,其中0為最暗且100為最亮;a*代表顏色之紅度或綠度(正值a*代表紅色,而負值a*代表綠色),且b*代表樣品之黃度或藍度,其中正值b*代表黃色,而負值b*代表藍色。與標準或評估中之初始樣品的顏色差異可藉由個別顏色組分之變化ΔL*、Δa*及Δb*來表示。複合變化或顏色差異可使用下式來計算作為空間中之簡單歐氏距離(Euclidian distance): 。可例如使用Hunter Labs UltrascanPro (Hunter Associates Laboratory, Reston, Virginia)或BYK Gardner LCS IV (BYK-Gardner, Columbia, Maryland)生成CIE L*, a*, b*顏色坐標。對於Hunter Labs UltraScan Pro,可執行Didymium Filter Test以用於波長校準。儀器在使用前可在具有0.780吋端口插件及DIW之TTRAN中標準化;因此,使用光阱及黑卡建立光度標度之頂部(L = 100)及底部(L = 0)。參見Pack等人, Modernization of Physical Appearance and Solution Color Tests Using Quantitative Tristimulus Colorimetry: Advantages, Harmonization, and Validation Strategies, J. Pharmaceutical Sci. 104: 3299-3313 (2015),其全部教示皆併入本文。BY標準之顏色亦可在CIE L*, a*, b*顏色空間(「CIELAB」或「CIELab」顏色空間)下表示。參見表 2
。表 2. CIE L*, a*, b* 顏色空間中歐洲棕黃色顏色標準之表徵
。可例如使用Hunter Labs UltrascanPro (Hunter Associates Laboratory, Reston, Virginia)或BYK Gardner LCS IV (BYK-Gardner, Columbia, Maryland)生成CIE L*, a*, b*顏色坐標。對於Hunter Labs UltraScan Pro,可執行Didymium Filter Test以用於波長校準。儀器在使用前可在具有0.780吋端口插件及DIW之TTRAN中標準化;因此,使用光阱及黑卡建立光度標度之頂部(L = 100)及底部(L = 0)。參見Pack等人, Modernization of Physical Appearance and Solution Color Tests Using Quantitative Tristimulus Colorimetry: Advantages, Harmonization, and Validation Strategies, J. Pharmaceutical Sci. 104: 3299-3313 (2015),其全部教示皆併入本文。BY標準之顏色亦可在CIE L*, a*, b*顏色空間(「CIELAB」或「CIELab」顏色空間)下表示。參見表 2
。表 2. CIE L*, a*, b* 顏色空間中歐洲棕黃色顏色標準之表徵
^
由Pack等人報告~
本文中以實驗方式量測- L*
及b*
值,針對每個BY顏色標準
| 標準 | L*^ | a*^ | b*^ | L*~ | a*~ | b*~ |
| BY1 | 93.95 | -2.76 | 28.55 | 92.84 | -3.16 | 31.15 |
| BY2 | 94.76 | -2.96 | 22.69 | 94.25 | -3.77 | 26.28 |
| BY3 | 96.47 | -2.84 | 16.41 | 95.92 | -3.44 | 18.52 |
| BY4 | 97.17 | -1.94 | 9.07 | 97.67 | -2.63 | 10.70 |
| BY5 | 98.91 | -1.19 | 4.73 | 98.75 | -1.61 | 5.77 |
| BY6 | 99.47 | -0.59 | 2.09 | 99.47 | -0.71 | 2.38 |
| BY7 | 99.37 | -0.31 | 1.13 | 99.71 | -0.37 | 1.17 |
為了對顏色檢定進行高通量篩選,與BY顏色標準相比,分光光度檢定法(CIELAB)為更適合及更具量化性之量測。如實例部分所述,進一步最佳化替代檢定。
對於評估顏色之任何樣品,必須針對樣品中之蛋白質濃度標準化測試樣品之蛋白質濃度,例如5 g/L、10 g/L及其類似濃度用於比較。III. 抗 VEGF 組成物
調節VEGF信號傳導途徑的VEGF蛋白質家族中有至少有五個成員:VEGF-A、VEGF-B、VEGF-C、VEGF-D及胎盤生長因子(PlGF)。抗VEGF組成物可包含VEGF拮抗劑,其與VEGF蛋白質家族之一或多個成員特異性相互作用,並且抑制其一或多種生物活性,例如其促有絲分裂、血管生成及/或血管通透性活性。
在一實施例中,產生抗VEGF蛋白之方法包含:(a)提供經基因工程改造以表現抗VEGF蛋白之宿主細胞;(b)在適於細胞表現抗VEGF蛋白之條件下在CDM中培養宿主細胞;及(c)收穫由細胞產生的抗VEGF蛋白之製劑。在一態樣中,抗VEGF蛋白係選自由以下組成之群組:阿柏西普、重組MiniTrap (其實例在美國專利第7,279,159號中揭露)、scFv及其他抗VEGF蛋白。在一較佳態樣中,感興趣之重組蛋白為阿柏西普。
發明者發現,在某些CDM中製造抗VEGF蛋白(例如,阿柏西普)產生表現出獨特顏色之生物樣品。在不同製造步驟中且甚至在包含抗VEGF蛋白之最終調配物中皆觀察到不同顏色特性。如實例9中所觀察,為了產生VEGF MiniTrap,在CDM中培養細胞產生具有強烈的黃棕色之抗VEGF蛋白(例如
,阿柏西普)。收穫後的親和捕獲步驟亦產生顯示出某種顏色-黃棕色之析出液。使用AEX之其他產生步驟亦顯示出黃棕色,但強度降低。
如下文中更詳細地描述,顏色可使用以下手段來評估:(i)歐洲顏色標準BY,其中進行定性目視檢查;或(ii)比色檢定,CIELAB,其比BY系統更具量化性。然而,無論哪種情況,皆應針對蛋白質濃度將多個樣品之間的顏色評估進行標準化,以確保有意義之評估/比較。例如,參看實例9,特別是表 9-2
,蛋白A析出液具有約2.52之b*值,大致相當於BY5之BY值(在蛋白A析出液中以5 g/L之蛋白質濃度量測時)。若要將蛋白A析出液之顏色與另一樣品進行比較,則應使用相同的蛋白質濃度進行該比較。因此,將蛋白A析出液與具有約0.74之b*值(在蛋白A析出液中以5 g/L之蛋白質濃度量測時)的AEX池相比較,產生方法顯示在AEX層析之後,樣品之黃棕色自蛋白A析出液至AEX池有實質性降低。
本發明之組成物之特徵可為如本文所討論之黃棕色,例如,不比歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有b*值17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L之抗VEGF蛋白或約10 g/L之抗VEGF蛋白,且其中組成物係自經澄清之收穫物或經澄清之收穫物的蛋白A析出液中作為樣品獲得。
在一實施例中,使用CDM產生的本發明之組成物產生具有明顯黃棕色之生物樣品,其中該樣品可藉由公認的標準顏色表徵來表徵:
(i) 黃棕色不超過歐洲顏色標準BY2;
(ii) 黃棕色不超過歐洲顏色標準BY3;
(iii) 黃棕色不超過歐洲顏色標準BY4;
(iv) 黃棕色不超過歐洲顏色標準BY5;
(v) 介於歐洲顏色標準BY2與BY3之間;
(vi) 介於歐洲顏色標準BY3與BY4之間;
(vii) 介於歐洲顏色標準BY4與BY5之間,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白且其中組成物係自經澄清之收穫物的蛋白A析出液中作為樣品獲得。
在另一實施例中,使用CDM產生的本發明之組成物產生具有明顯黃棕色之生物樣品,其中該組成物藉由CIELAB標度中公認的標準顏色表徵來表徵:
(i) 黃棕色不超過約22至23之b*值;
(ii) 黃棕色不超過約16至17之b*值;
(iii) 黃棕色不超過9至10之b*值;
(iv) 黃棕色不超過4至5之b*值;
(v) 黃棕色不超過2至3之b*值;
(vi) 介於17至23之間的b*值;
(vii) 介於10至17之間的b*值;
(viii) 介於5至10之間的b*值;
(ix) 介於3至5之間的b*值;或
(x) 介於1至3之間的b*值,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白且其中組成物係自經澄清之收穫物的蛋白A析出液中作為樣品獲得。
在一實施例中,使用CDM產生的本發明之組成物可包含抗VEGF蛋白之其他種類或變異體。此等變異體包括抗VEGF蛋白同功型,其包含一或多個氧化之胺基酸殘基,該等同功型總稱為側氧變異體。此等包含抗VEGF蛋白及其側氧變異體之組成物的酶消化物可包含以下一或多者:
EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18),其包含約0.004至0.013%之2-側氧基-組胺酸,
QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),其包含約0.006至0.028%之2-側氧基-組胺酸,
TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20),其包含約0.049至0.085%之2-側氧基-組胺酸,
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17),其包含約0.057至0.092%之2-側氧基-組胺酸,
TNYLTH*R (SEQ ID NO.: 21),其包含約0.008至0.022%之2-側氧基-組胺酸,及/或
IIWDSR (SEQ ID NO.: 56) ,其包含約0.185至0.298%之二氧化色胺酸;
或
EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18),其包含約0.008%之2-側氧基-組胺酸,
QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),其包含約0.02%之2-側氧基-組胺酸,
TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20) ,其包含約0.06%之2-側氧基-組胺酸,
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17),其包含約0.07%之2-側氧基-組胺酸,
TNYLTH*R (SEQ ID NO.: 21),其包含約0.01%之2-側氧基-組胺酸,及/或
IIWDSR (SEQ ID NO.: 56),其包含約0.23%之二-側氧基-色胺酸,其中H*為可經氧化成2-側氧基-組胺酸之組胺酸,且其中C*為可經羧甲基化之半胱胺酸。在一具體實施例中,抗VEGF蛋白為阿柏西普。在另一實施例中,抗VEGF蛋白為VEGF MiniTrap。
在本發明之一示例性實施例中,本發明之組成物可包含抗VEGF蛋白,其中抗VEGF蛋白中不多於約1%、不多於約0.1%或約0.1至1%、0.2至1%、0.3至1%、0.4至1%、0.5至1%、0.6至1%、0.7至1%、0.8至1%或0.9至1%之組胺酸殘基為2-側氧基-組胺酸。在此等組成物中,可存在抗VEGF蛋白變異體之異質群體,每個變異體具有不同量之2-側氧基-組胺酸殘基及未氧化之組胺酸殘基。因此,組成物中的2-側氧基-組胺酸抗VEGF蛋白之百分比係指抗VEGF分子中的位點特異性2-側氧基-組胺酸除以抗VEGF蛋白分子中的總位點特異性組胺酸(氧化加未氧化)乘以100。定量組成物中的2-側氧基-組胺酸水準之一種方法係用蛋白酶(例如,Lys-C及/或胰蛋白酶)消化多肽及藉由例如質譜(ms)分析所得肽中的2-側氧基-組胺酸之量。
在消化抗VEGF蛋白之前,半胱胺酸巰基藉由與碘乙醯胺(IAM)之反應經阻斷,產生由以下化學結構表示之殘基: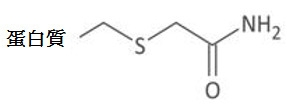 。該修飾保護遊離硫醇免於重整二硫橋,並防止雙硫鍵加擾。本發明包括包含抗VEGF蛋白及其變異體之組成物(例如,水性組成物),當用IAM修飾並用蛋白酶(例如,Lys-C及胰蛋白酶)消化並藉由質譜進行分析時,其包含以下肽:
EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18),其包含約0.004至0.013%之2-側氧基-組胺酸,
QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),其包含約0.006至0.028%之2-側氧基-組胺酸,
TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20),其包含約0.049至0.085%之2-側氧基-組胺酸,
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17),其包含約0.057至0.092%之2-側氧基-組胺酸,
TNYLTH*R (SEQ ID NO.: 21),其包含約0.008至0.022%之2-側氧基-組胺酸,及/或
IIWDSR (SEQ ID NO.: 56),其包含約0.185至0.298%之二氧化色胺酸;
或
EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18),其包含約0.008%之2-側氧基-組胺酸,
QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),其包含約0.02%之2-側氧基-組胺酸,
TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20),其包含約0.06%之2-側氧基-組胺酸,
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17),其包含約0.07%之2-側氧基-組胺酸,
TNYLTH*R (SEQ ID NO.: 21),其包含約0.01%之2-側氧基-組胺酸,及/或
IIWDSR (SEQ ID NO.: 56),其包含約0.23%之二-側氧基-色胺酸,
其中H*為2-側氧基-組胺酸,且其中C*為羧甲基化之半胱胺酸。在本發明之一實施例中,將肽用PNGase F去醣化。
。該修飾保護遊離硫醇免於重整二硫橋,並防止雙硫鍵加擾。本發明包括包含抗VEGF蛋白及其變異體之組成物(例如,水性組成物),當用IAM修飾並用蛋白酶(例如,Lys-C及胰蛋白酶)消化並藉由質譜進行分析時,其包含以下肽:
EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18),其包含約0.004至0.013%之2-側氧基-組胺酸,
QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),其包含約0.006至0.028%之2-側氧基-組胺酸,
TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20),其包含約0.049至0.085%之2-側氧基-組胺酸,
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17),其包含約0.057至0.092%之2-側氧基-組胺酸,
TNYLTH*R (SEQ ID NO.: 21),其包含約0.008至0.022%之2-側氧基-組胺酸,及/或
IIWDSR (SEQ ID NO.: 56),其包含約0.185至0.298%之二氧化色胺酸;
或
EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18),其包含約0.008%之2-側氧基-組胺酸,
QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),其包含約0.02%之2-側氧基-組胺酸,
TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20),其包含約0.06%之2-側氧基-組胺酸,
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17),其包含約0.07%之2-側氧基-組胺酸,
TNYLTH*R (SEQ ID NO.: 21),其包含約0.01%之2-側氧基-組胺酸,及/或
IIWDSR (SEQ ID NO.: 56),其包含約0.23%之二-側氧基-色胺酸,
其中H*為2-側氧基-組胺酸,且其中C*為羧甲基化之半胱胺酸。在本發明之一實施例中,將肽用PNGase F去醣化。
本發明包括包含抗VEGF蛋白之組成物,其中抗VEGF蛋白中所有組胺酸之約0.1%至10%經修飾為2-側氧基-組胺酸。此外,組成物之顏色不比例如歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,替代地,具有如使用CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白。組成物係自經澄清之收穫物或經澄清之收穫物的蛋白A析出液中作為樣品獲得。當使收穫物材料經歷捕獲層析程序時,可自經澄清之收穫物中獲得該等組成物。在一態樣中,捕獲步驟為使用例如蛋白A親和管柱之親和層析程序。當使用液相層析-質譜法(LC-MS)分析親和樣品時,可偵測到一或多種變異體。
本發明包括包含抗VEGF蛋白之組成物,其中抗VEGF蛋白中所有色胺酸之約0.1%至10%經修飾為犬尿胺酸。此外,組成物之顏色不比歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如使用CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L之抗VEGF蛋白或約10 g/L之抗VEGF蛋白。組成物係自經澄清之收穫物或經澄清之收穫物的蛋白A析出液中作為樣品獲得。當經歷捕獲層析程序時,此等組成物可自經澄清之收穫物中獲得。捕獲步驟為使用例如蛋白A親和管柱之親和層析程序。當使用液相層析-質譜法(LC-MS)分析親和樣品時,可偵測到一或多種此等變異體。
本發明包括包含抗VEGF蛋白之組成物,其中抗VEGF蛋白中所有色胺酸之約0.1%至10%經修飾為單羥基色胺酸。此外,組成物之顏色不比歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如藉由CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L之抗VEGF蛋白或約10 g/L之抗VEGF蛋白。組成物係自經澄清之收穫物或經澄清之收穫物的蛋白A析出液中作為樣品獲得。當經歷捕獲層析程序時,此等組成物可自經澄清之收穫物中獲得。捕獲步驟為使用例如蛋白A親和管柱之親和層析程序。當使用液相層析-質譜法(LC-MS)分析由親和步驟中提取之樣品時,可偵測到一或多種此等變異體。
本發明包括包含抗VEGF蛋白之組成物,其中抗VEGF蛋白中所有色胺酸之約0.1%至10%經修飾為二羥基色胺酸。此外,顏色不比歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如使用CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L之抗VEGF蛋白或約10 g/L之抗VEGF蛋白。組成物係自經澄清之收穫物或經澄清之收穫物的蛋白A析出液中作為樣品獲得。該等組成物可由使用包含抗VEGF蛋白及其側氧變異體之CDM製成的經澄清之收穫物中獲得,該經澄清之收穫物經歷捕獲層析程序。捕獲步驟為使用例如蛋白A親和管柱之親和層析程序。當使用液相層析-質譜法(LC-MS)分析由親和步驟中提取之樣品時,可偵測到一或多種此等變異體。
本發明包括包含抗VEGF蛋白之組成物,其中抗VEGF蛋白中所有色胺酸之約0.1%至10%經修飾為三羥基色胺酸。此外,組成物之顏色不比歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如藉由CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L之抗VEGF蛋白或約10 g/L之抗VEGF蛋白。組成物係自經澄清之收穫物或經澄清之收穫物的蛋白A析出液中作為樣品獲得。此等組成物可使用捕獲層析獲得。捕獲步驟為使用例如蛋白A親和管柱之親和層析程序。當使用液相層析-質譜法(LC-MS)分析由親和中提取之樣品時,可偵測到一或多種此等變異體。
在一實施例中,本發明之組成物可包含抗VEGF蛋白,其中抗VEGF蛋白可包含如下一或多個殘基之修飾:一或多個天冬醯胺經去醯胺;一或多個天冬胺酸轉化為異天冬胺酸及/或Asn;一或多個甲硫胺酸經氧化;一或多個色胺酸轉化為N-甲醯犬尿胺酸;一或多個色胺酸為單羥基色胺酸;一或多個色胺酸為二羥基色胺酸;一或多個色胺酸為三羥基色胺酸;一或多個精胺酸轉化為Arg 3-去氧葡醛酮糖;C端甘胺酸不存在;及/或存在一或多個非醣化糖位點。
此等組成物可自使用包含抗VEGF蛋白及其變異體之CDM製成的經澄清之收穫物中獲得,該經澄清之收穫物經歷例如捕獲層析程序。捕獲步驟為使用例如蛋白A管柱之親和層析程序。當使用例如液相層析-質譜法(LC-MS)分析由親和步驟中提取之樣品時,可偵測到一或多種此等變異體。
在示例性實施例中,本發明之組成物可包含具有阿柏西普之結構特徵的抗VEGF蛋白,其可在以下一或多者處經氧化:His86、His110、His145、His209、His95、His19及/或His203 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置);Trp58及/或Trp138 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置);Tyr64 (或具有阿柏西普之某些結構特徵的蛋白質上之等效位置);Phe44及/或Phe166 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置);及/或Met10、Met 20、Met163及/或Met192 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)。此等組成物可自使用包含阿柏西普及其側氧變異體之CDM製成的經澄清之收穫物中獲得,該經澄清之收穫物經歷捕獲層析程序。捕獲步驟可為使用例如蛋白A管柱之親和層析程序。當使用例如液相層析-質譜法(LC-MS)分析由親和步驟中提取之樣品時,可偵測到一或多種此等變異體。
在一實施例中,本發明之組成物可包含具有胺基酸序列SEQ ID NO.: 46之VEGF MiniTrap,其可在以下各處經氧化:His86、His110、His145、His209、His95、His19及/或His203;Trp58及/或Trp138;Tyr64;Phe44及/或Phe166;及/或Met10、Met 20、Met163及/或Met192。此等組成物可自使用包含VEGF MiniTrap及其側氧變異體之CDM製成的經澄清之收穫物中獲得,該經澄清之收穫物經歷捕獲層析程序。捕獲步驟為使用例如蛋白A管柱之親和層析程序-當使用液相層析-質譜法(LC-MS)進行分析時,可偵測到一或多種此等變異體。
在一些示例性實施例中,本發明之組成物可包含抗VEGF蛋白及其變異體(包括側氧變異體),其中組成物中的蛋白質變異體之量為最多約20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。此等組成物可自使用包含抗VEGF蛋白及其變異體之CDM製成的經澄清之收穫物中獲得,該經澄清之收穫物經歷捕獲層析程序。捕獲步驟為使用例如蛋白A管柱之親和層析程序-當使用液相層析-質譜法(LC-MS)進行分析時,可偵測到一或多種此等變異體。在一態樣中,此一組成物之顏色不比例如歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如藉由CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白。
在其他示例性實施例中,本發明之組成物可包含抗VEGF蛋白及其變異體,其中組成物中的蛋白質變異體之量為約0%至約20%,例如,約0%至約20%、約0.05%至約20%、約0.1%至約20%、約0.2%至約20%、約0.3%至約20%、約0.4%至約20%、約0.5%至約20%、約0.6%至約20%、約0.7%至約20%、約0.8%至約20%、約0.9%至約20%、約1%至約20%、約1.5%至約20%、約2%至約20%、約3%至約20%、約4%至約20%、約5%至約20%、約6%至約20%、約7%至約20%、約8%至約20%、約9%至約20%、約10%至約20%、約0%至約10% 、約0.05%至約10%、約0.1%至約10%、約0.2%至約10%、約0.3%至約10%、約0.4%至約10%、約0.5%至約10%、約0.6%至約10%、約0.7%至約10%、約0.8%至約10%、約0.9%至約10%、約1%至約10%、約1.5%至約10%、約2%至約10%、約3%至約10%、約4%至約10%、約5%至約10%、約6%至約10%、約7%至約10%、約8%至約10%、約9%至約10%、約0%至約7.5%、約0.05%至約7.5%、約0.1%至約7.5%、約0.2%至約7.5%、約0.3%至約7.5%、約0.4%至約7.5%、約0.5%至約7.5%、約0.6%至約7.5%、約0.7%至約7.5%、約0.8%至約7.5%、約0.9%至約7.5%、約1%至約7.5%、約1.5%至約7.5%、約2%至約7.5%、約3%至約7.5%、約4%至約7.5%、約5%至約7.5%、約6%至約7.5%、約7%至約7.5%、約0%至約5%、約0.05%至約5%、約0.1%至約5%、約0.2%至約5%、約0.3%至約5%、約0.4%至約5%、約0.5%至約5%、約0.6%至約5%、約0.7%至約5%、約0.8%至約5%、約0.9%至約5%、約1%至約5%、約1.5%至約5%、約2%至約5%、約3%至約5%、約4%至約5%,以及在上述一或多者內之範圍。此等組成物可藉由對收穫物樣品進行捕獲層析而獲得。捕獲步驟為使用例如蛋白A管柱之親和層析程序。當使用液相層析-質譜法(LC-MS)分析樣品時,可偵測到一或多種此等變異體。在一態樣中,此一組成物之顏色不比例如歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如藉由CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白。
在一實施例中,本發明之組成物可包含抗VEGF蛋白,包括其酸性種類,其中組成物中的酸性種類之量可為約20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。如前述所討論,此等酸性種類可藉由各種方法,諸如離子交換,例如,WCX (WCX-10 HPLC,弱陽離子交換層析)或IEF (等電聚焦)來偵測。通常,酸性種類之溶析在CEX期間早於主峰或在AEX分析期間晚於主峰(參見 圖 16
及圖 17
)。包含酸性種類之組成物可使用離子交換層析由生物材料(諸如收穫物或親和產生之材料)獲得。
在一態樣中,此組成物之顏色不比例如歐洲棕黃色顏色標準BY2-BY3、BY3-BY4、BY4-BY5或BY5-BY6暗/強烈,及/或具有如藉由CIE L*, a*, b*所表徵之以下b*值:約17至23、10至17、5至10、3至5或1至3,其中組成物包含約5 g/L或約10 g/L。作為實例,參看圖 16
及圖 17
,級分F1及F2代表包含大部分酸性種類之酸性級分。圖 17
中的MT1之峰1及2包含酸性種類,並且級分F1及F2包含大部分酸性級分。與其他級分相比,包含此等酸性種類之級分(F1及F2)亦顯示出黃棕色(圖 18B
及圖 18C
)。
在另一實施例中,本發明之組成物包含抗VEGF蛋白,包括其酸性種類,其中組成物中的酸性種類之量可為約0%至約20%,例如,約0%至約20%、約0.05%至約20%、約0.1%至約20%、約0.2%至約20%、約0.3%至約20%、約0.4%至約20%、約0.5%至約20%、約0.6%至約20%、約0.7%至約20%、約0.8%至約20%、約0.9%至約20%、約1%至約20%、約1.5%至約20%、約2%至約20%、約3%至約20%、約4%至約20%、約5%至約20%、約6%至約20%、約7%至約20%、約8%至約20%、約9%至約20%、約10%至約20%、約0%至約10%、約0.05%至約10%、約0.1%至約10%、約0.2%至約10%、約0.3%至約10%、約0.4%至約10%、約0.5%至約10%、約0.6%至約10%、約0.7%至約10%、約0.8%至約10%、約0.9%至約10%、約1%至約10%、約1.5%至約10%、約2%至約10%、約3%至約10%、約4%至約10%、約5%至約10%、約6%至約10%、約7%至約10%、約8%至約10%、約9%至約10%、約0%至約7.5%、約0.05%至約7.5%、約0.1%至約7.5%、約0.2%至約7.5%、約0.3%至約7.5%、約0.4%至約7.5%、約0.5%至約7.5%、約0.6%至約7.5%、約0.7%至約7.5%、約0.8%至約7.5%、約0.9%至約7.5%、約1%至約7.5%、約1.5%至約7.5%、約2%至約7.5%、約3%至約7.5%、約4%至約7.5%、約5%至約7.5%、約6%至約7.5%、約7%至約7.5%、約0%至約5%、約0.05%至約5%、約0.1%至約5%、約0.2%至約5%、約0.3%至約5%、約0.4%至約5%、約0.5%至約5%、約0.6%至約5%、約0.7%至約5%、約0.8%至約5%、約0.9%至約5%、約1%至約5%、約1.5%至約5%、約2%至約5%、約3%至約5%、約4%至約5%,以及在上述一或多者內之範圍。如上所述,可藉由各種方法,諸如離子交換,例如WCX (WCX-10 HPLC,弱陽離子交換層析)或IEF (等電聚焦)來偵測此等酸性種類。典型地,酸性種類之溶析在CEX期間早於主峰或在AEX分析期間晚於主峰(參見 圖 16
及圖 17
)。
使用陽離子交換管柱,將感興趣之主峰之前溶析的所有峰累加為酸性區域,而感興趣之蛋白後溶析的所有峰累加為鹼性區域。在示例性實施例中,基於峰之一定滯留時間及所用離子交換管柱,可將酸性種類溶析為兩個或更多個酸性區域,並且可經編號為AR1、AR2、AR3等等。
在一實施例中,組成物可包含抗VEGF蛋白,包括酸性種類,其中AR1為20%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。在一態樣中,組成物可包含抗VEGF蛋白,包括其酸性種類,其中AR1為約0.0%至約10%、約0.0%至約5%、約0.0%至約4%、約0.0%至約3%、約0.0%至約2%、約3%至約5%、約5%至約8%、或約8%至約10%、或約10%至約15%,以及在上述一或多者內之範圍。如上所述,可藉由各種方法,諸如離子交換,例如WCX (WCX-10 HPLC,弱陽離子交換層析)或IEF (等電聚焦)來偵測此等酸性區域。通常,酸性種類之溶析在CEX期間早於主峰或在AEX分析期間晚於主峰(參見圖 16
及圖 17
)。
在另一實施例中,組成物可包含抗VEGF蛋白,包括酸性種類,其中AR2為20%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。在一態樣中,組成物可包含抗VEGF蛋白,包括酸性種類,其中AR2為約0.0%至約10%、約0.0%至約5%、約0.0%至約4%、約0.0%至約3%、約0.0%至約2%、約3%至約5%、約5%至約8%、或約8%至約10%、或約10%至約15%,以及在上述一或多者內之範圍。
在一實施例中,組成物可包含抗VEGF蛋白,包括鹼性種類,其中組成物中的鹼性種類之量可為最多約20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。在一態樣中,組成物可包含抗VEGF蛋白及其鹼性種類,其中與抗VEGF蛋白相比,組成物中的鹼性種類之量可為0%至約20%,例如
,約0%至約20%、約0.05%至約20%、約0.1%至約20%、約0.2%至約20%、約0.3%至約20%、約0.4%至約20%、約0.5%至約20%、約0.6%至約20%、約0.7%至約20%、約0.8%至約20%、約0.9%至約20%、約1%至約20%、約1.5%至約20%、約2%至約20%、約3%至約20%、約4%至約20%、約5%至約20%、約6%至約20%、約7%至約20%、約8%至約20%、約9%至約20%、約10%至約20%、約0%至約10%、約0.05%至約10%、約0.1%至約10%、約0.2%至約10%、約0.3%至約10%、約0.4%至約10%、約0.5%至約10%、約0.6%至約10%、約0.7%至約10%、約0.8%至約10%、約0.9%至約10%、約1%至約10%、約1.5%至約10%、約2%至約10%、約3%至約10%、約4%至約10%、約5%至約10%、約6%至約10%、約7%至約10%、約8%至約10%、約9%至約10%、約0%至約7.5%、約0.05%至約7.5%、約0.1%至約7.5%、約0.2%至約7.5%、約0.3%至約7.5%、約0.4%至約7.5%、約0.5%至約7.5%、約0.6%至約7.5%、約0.7%至約7.5%、約0.8%至約7.5%、約0.9%至約7.5%、約1%至約7.5%、約1.5%至約7.5%、約2%至約7.5%、約3%至約7.5%、約4%至約7.5%、約5%至約7.5%、約6%至約7.5%、約7%至約7.5%、約0%至約5%、約0.05%至約5%、約0.1%至約5%、約0.2%至約5%、約0.3%至約5%、約0.4%至約5%、約0.5%至約5%、約0.6%至約5%、約0.7%至約5%、約0.8%至約5%、約0.9%至約5%、約1%至約5%、約1.5%至約5%、約2%至約5%、約3%至約5%、約4%至約5%,以及在上述一或多者內之範圍。
可將鹼性種類溶析為兩個或更多個鹼性區域,並且可根據峰之一定滯留時間及所用離子交換將其編號為BR1、BR2、BR3等等。
在一實施例中,組成物可包含抗VEGF蛋白,包括其鹼性種類,其中BR1為15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。在一態樣中,組成物可包含抗VEGF蛋白及其鹼性種類,其中BR1為約0.0%至約10%、約0.0%至約5%、約0.0%至約4%、約0.0%至約3%、約0.0%至約2%、約3%至約5%、約5%至約8%、或約8%至約10%、或約10%至約15%,以及在上述一或多者內之範圍。
在另一實施例中,組成物可包含抗VEGF蛋白及其鹼性種類,其中BR2為15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。在一態樣中,組成物可包含抗VEGF蛋白及抗VEGF蛋白之其鹼性種類,其中BR2為約0.0%至約10%、約0.0%至約5%、約0.0%至約4%、約0.0%至約3%、約0.0%至約2%、約3%至約5%、約5%至約8%、或約8%至約10%、或約10%至約15%,以及在上述一或多者內之範圍。
在另一實施例中,組成物可包含抗VEGF蛋白及其鹼性種類,其中BR3為15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4.5%、4%、3.5%、3%、2.5%、2%、1.9%、1.8%、1.7%、1.6%、1.5%、1.4%、1.3%、1.2%、1.1%、1%、0.9%、0.8%、0.7%、0.6%、0.5%、0.4%、0.3%、0.2%、0.1%或0.0%,以及在上述一或多者內之範圍。在一態樣中,組成物可包含抗VEGF蛋白及抗VEGF蛋白之其鹼性種類,其中BR3為約0.0%至約10%、約0.0%至約5%、約0.0%至約4%、約0.0%至約3%、約0.0%至約2%、約3%至約5%、約5%至約8%、或約8%至約10%、或約10%至約15%,以及在上述一或多者內之範圍。阿柏西普之光誘導氧化
除了發現使用CDM產生的抗VEGF蛋白組成物之不同顏色特徵或變異體之外,發明者亦發現可在實驗室中藉由暴露於光來人工產生此等組成物。
可藉由使抗VEGF蛋白暴露於冷白光或紫外光來產生抗VEGF組成物的經修飾(包括經氧化)之變異體。在一態樣中,與樣品相比,抗VEGF組成物可包含一或多種經修飾之寡胜肽約1.5至約50倍之增加,其中該等寡胜肽係選自由以下組成之群組:
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17)、EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18)、QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19)、TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20)、TNYLTH*R (SEQ ID NO.: 21)、SDTGRPFVEMYSEIPEIIH*MTEGR (SEQ ID NO.: 22)、VH*EKDK (SEQ ID NO.: 23)、SDTGRPFVEM*YSEIPEIIHMTEGR (SEQ ID NO.: 64)、SDTGRPFVEMYSEIPEIIHM*TEGR (SEQ ID NO.: 65)、TQSGSEM*K (SEQ ID NO.: 66)、SDQGLYTC*AASSGLM*TK (SEQ ID NO.: 67)、IIW*DSR/RIIW*DSR/IIW*DSRK (SEQ ID NO.: 28)、TELNVGIDFNW*EYPSSK (SEQ ID NO.: 29)、GFIISNATY*K (SEQ ID NO.: 69)、KF*PLDTLIPDGK (SEQ ID NO.: 70)、F*LSTLTIDGVTR (SEQ ID NO.: 32),其中H*為經氧化為2-側氧基-組胺酸之組胺酸,其中C*為經羧甲基化之半胱胺酸,其中M*為經氧化之甲硫胺酸,其中W*為經氧化之色胺酸,其中Y*為經氧化之酪胺酸,且其中F*為經氧化之苯丙胺酸。在又一態樣中,藉由使抗VEGF組成物暴露於冷白光一段時間,例如約30小時,抗VEGF組成物可包含一或多種經修飾之寡胜肽約1.5至約10倍之增加。在另一態樣中,藉由使樣品暴露於冷白光約75小時,抗VEGF組成物可包含一或多種經修飾之寡胜肽約1.5至約10倍之增加。在仍另一態樣中,藉由使樣品暴露於冷白光約100小時,抗VEGF組成物可包含一或多種前述寡胜肽約1.5至約20倍之增加。在仍另一態樣中,藉由使樣品暴露於冷白光約150小時,抗VEGF組成物可包含一或多種前述寡胜肽約1.5至約20倍之增加。在仍另一態樣中,藉由使樣品暴露於冷白光約300小時,抗VEGF組成物可包含一或多種寡胜肽約1.5至約50倍之增加- 參見以下實例4。
藉由使抗VEGF組成物之樣品暴露於紫外光約4小時,抗VEGF組成物可包含一或多種如上所述之寡胜肽約1.5至約3倍之增加。在另一態樣中,藉由使樣品暴露於紫外光約10小時,抗VEGF組成物可包含一或多種寡胜肽約1.5至約10倍之增加。在仍另一態樣中,藉由使樣品暴露於紫外光約16小時,抗VEGF組成物可包含一或多種寡胜肽約1.5至約10倍之增加。在仍另一態樣中,藉由使樣品暴露於紫外光約20小時,抗VEGF組成物可包含一或多種寡胜肽約1.5至約25倍之增加。在仍另一態樣中,藉由使樣品基質暴露於紫外光約40小時,抗VEGF組成物可包含一或多種寡胜肽約1.5至約25倍之增加。參見實例4。醣多樣性 (glycodiversity) – 使用 CDM 產生之抗 VEGF 蛋白
本發明之組成物包含抗VEGF蛋白,其中在CDM中產生之抗VEGF蛋白具有多種醣多樣性。抗VEGF蛋白之不同醣化譜在本發明之範疇內。
在本發明之一些示例性實施例中,組成物可包含在一或多個天冬醯胺處如下經醣化之抗VEGF蛋白:G0-GlcNAc醣化;G1-GlcNAc醣化;G1S-GlcNAc醣化;G0醣化;G1醣化;G1S醣化;G2醣化;G2S醣化;G2S2醣化;G0F醣化;G2F2S醣化;G2F2S2醣化;G1F醣化;G1FS醣化;G2F醣化;G2FS醣化;G2FS2醣化;G3FS醣化;G3FS3醣化;G0-2GlcNAc醣化;Man4醣化;Man4_A1G1醣化;Man4_A1G1S1醣化;Man5醣化;Man5_A1G1醣化;Man5_A1G1S1醣化;Man6醣化;Man6_G0+磷酸醣化;Man6+磷酸醣化;及/或Man7醣化。在一態樣中,感興趣之蛋白質可為阿柏西普、抗VEGF抗體或VEGF MiniTrap。
在一實施例中,組成物可具有如下醣化譜:約40%至約50%總岩藻醣化聚醣、約30%至約50%總唾液酸化聚醣、約6%至約15%甘露糖-5、及約60%至約79%半乳醣化聚醣。(實例6)。
在一實施例中,組成物可包含抗VEGF蛋白,其中感興趣之蛋白質在約32.4%天冬醯胺123殘基及/或約27.1%天冬醯胺196殘基處具有Man5醣化。在一態樣中,感興趣之蛋白質可為阿柏西普、抗VEGF抗體或VEGF MiniTrap。
在另一實施例中,組成物可具有約40%、約41%、約42%、約43%、約44%、約45%、約46%、約47%、約48%、約49%或約50%總岩藻醣化聚醣。
在仍另一實施例中,組成物可具有約30%、約31%、約32%、約33%、約34%、約35%、約36%、約37%、約38%、約39%、約40%、約41%、約42%、約43%、約44%、約45%、約46%、約47%、約48%、約49%或約50%總唾液酸化聚醣。
在一實施例中,組成物可具有約6 %、約7%、約8%、約8%、約10%、約11%、約12%、約13%、約14%或約15%甘露糖-5。
在另一實施例中,組成物可具有約60%、約61%、約62%、約63%、約64%、約65%、約66%、約67%、約68%、約69%、約70%、約71%、約72%、約73%、約74%、約75%、約76%、約77%、約78%或約79%總半乳醣化聚醣。
在一實施例中,抗VEGF蛋白可具有水準降低了約1%、1.2%、1.5%、2%、2.2%、2.5%、3%、3.2%、3.5%、4%、4.2%、4.5%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%之岩藻醣化聚醣。與使用大豆水解物產生的抗VEGF蛋白中之岩藻醣化聚醣水準相比,在一或多個前述值內之範圍:例如,1-10%、1-15%、1-20%、1-25%、1-30%、1-35%、1-40%、1-41%、1-42%、1-43%、1-44%、1-45%、1-46%、1-47%、1-48%、1-49%、1-50%、2-10%、2-15%、2-20%、2-25%、2-30%、2-35%、2-40%、2-41%、2-42%、2-43%、2-44%、2-45%、2-46%、2-47%、2-48%、2-49%、2-50%、3-10%、3-15%、3-20%、3-25%、3-30%、3-35%、3-40%、3-41%、3-42%、3-43%、3-44%、3-45%、3-46%、3-47%、3-48%、3-49%、3-50%、4-10%、4-15%、4-20%、4-25%、4-30%、4-35%、4-40%、4-41%、4-42%、4-43%、4-44%、4-45%、4-46%、4-47%、4-48%、4-49%、4-50%或1-99%。
在一實施例中,抗VEGF蛋白可具有水準降低了約1%、1.2%、1.5%、2%、2.2%、2.5%、3%、3.2%、3.5%、4%、4.2%、4.5%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%之唾液酸化聚醣。與使用大豆水解物產生的抗VEGF蛋白中之唾液酸化聚醣水準相比,在一或多個前述值內之範圍:例如,1-10%、1-15%、1-20%、1-25%、1-30%、1-35%、1-40%、1-41%、1-42%、1-43%、1-44%、1-45%、1-46%、1-47%、1-48%、1-49%、1-50%、2-10%、2-15%、2-20%、2-25%、2-30%、2-35%、2-40%、2-41%、2-42%、2-43%、2-44%、2-45%、2-46%、2-47%、2-48%、2-49%、2-50%、3-10%、3-15%、3-20%、3-25%、3-30%、3-35%、3-40%、3-41%、3-42%、3-43%、3-44%、3-45%、3-46%、3-47%、3-48%、3-49%、3-50%、4-10%、4-15%、4-20%、4-25%、4-30%、4-35%、4-40%、4-41%、4-42%、4-43%、4-44%、4-45%、4-46%、4-47%、4-48%、4-49%、4-50%或1-99%。
在另一實施例中,抗VEGF蛋白可具有水準降低了約1%、1.2%、1.5%、2%、2.2%、2.5%、3%、3.2%、3.5%、4%、4.2%、4.5%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%之半乳醣化聚醣。與使用大豆水解物產生的抗VEGF蛋白中之半乳醣化聚醣水準相比,在一或多個前述值內之範圍:例如,1-10%、1-15%、1-20%、1-25%、1-30%、1-35%、1-40%、1-41%、1-42%、1-43%、1-44%、1-45%、1-46%、1-47%、1-48%、1-49%、1-50%、2-10%、2-15%、2-20%、2-25%、2-30%、2-35%、2-40%、2-41%、2-42%、2-43%、2-44%、2-45%、2-46%、2-47%、2-48%、2-49%、2-50%、3-10%、3-15%、3-20%、3-25%、3-30%、3-35%、3-40%、3-41%、3-42%、3-43%、3-44%、3-45%、3-46%、3-47%、3-48%、3-49%、3-50%、4-10%、4-15%、4-20%、4-25%、4-30%、4-35%、4-40%、4-41%、4-42%、4-43%、4-44%、4-45%、4-46%、4-47%、4-48%、4-49%、4-50%或1-99%。
在一實施例中,抗VEGF蛋白可具有水準提高了約1%、1.2%、1.5%、2%、2.2%、2.5%、3%、3.2%、3.5%、4%、4.2%、4.5%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%之甘露醣化聚醣。與使用大豆水解物產生的抗VEGF蛋白中之甘露醣化聚醣水準相比,在一或多個前述值內之範圍:例如,1-10%、1-15%、1-20%、1-25%、1-30%、1-35%、1-40%、1-41%、1-42%、1-43%、1-44%、1-45%、1-46%、1-47%、1-48%、1-49%、1-50%、2-10%、2-15%、2-20%、2-25%、2-30%、2-35%、2-40%、2-41%、2-42%、2-43%、2-44%、2-45%、2-46%、2-47%、2-48%、2-49%、2-50%、3-10%、3-15%、3-20%、3-25%、3-30%、3-35%、3-40%、3-41%、3-42%、3-43%、3-44%、3-45%、3-46%、3-47%、3-48%、3-49%、3-50%、4-10%、4-15%、4-20%、4-25%、4-30%、4-35%、4-40%、4-41%、4-42%、4-43%、4-44%、4-45%、4-46%、4-47%、4-48%、4-49%、4-50%或1-99%。
此部分中所述之組成物可分別由以下節IV及V中所述之若干個上游及下游參數產生。IV. 使用上游製程技術製備組成物
對於生物製劑而言,需要施行強健而靈活之上游製程。有效的上游製程可實現感興趣之蛋白質的所需產生及按比例放大。發明者發現包含抗VEGF蛋白的本發明之組成物可藉由調節上游蛋白產生期間的條件,諸如CDM之培養基組分的變化來產生。上游製程中之每個步驟皆會影響所製造的蛋白質之品質、純度及數量。
本文提供使用CDM產生的阿柏西普及/或MiniTrap之某些變異體之存在的證據。此等變異體包括包含一或多個經氧化之胺基酸殘基的同功型。經氧化殘基之實例包括但不限於一或多個組胺酸、色胺酸、甲硫胺酸、苯丙胺酸或酪胺酸殘基。藉由使用經修飾之CDM產生的組成物可產生抗VEGF蛋白之製劑,該製劑具有阿柏西普及/或MiniTrap之蛋白質變異體的所需目標值。如上所述,亦可存在與使用CDM產生之級分相關的黃棕色。(如上所提及,並非發明者測試的所有CDM皆表現出明顯變色。)
本發明包括在細胞表現感興趣之重組蛋白的合適條件下在經修飾之CDM中培養宿主細胞,接著收穫由細胞產生的感興趣之重組蛋白之製劑。此一經修飾之CDM可用於產生如上文節III中所述之組成物。(注意,CDM為表現阿柏西普時存在黃棕色之培養基。)
在一實施例中,該方法包含在合適的條件下在CDM中培養宿主細胞,其中宿主細胞表現感興趣之重組蛋白,諸如阿柏西普。該方法進一步包含收穫由細胞產生的感興趣之重組蛋白之製劑,其中合適的條件包括具有以下之CDM:在該CDM中小於約55 μM之鐵累積濃度、在該CDM中小於或等於約0.8 μM之銅累積濃度、在該CDM中小於或等於約0.40 μM之鎳累積濃度、在該CDM中小於或等於約56 μM之鋅累積濃度、在該CDM中小於約10 mM之半胱胺酸累積濃度;及/或在該CDM中對於單一抗氧化劑而言濃度為約0.001 mM至約10 mM之抗氧化劑,及若在該CDM中添加多種抗氧化劑,則不大於約30 mM之累積濃度。
在本實施例之一態樣中,使用合適的條件獲得之製劑造成阿柏西普及VEGF MiniTrap之蛋白質變異體減少至所需量的阿柏西普及VEGF MiniTrap之蛋白質變異體(稱為阿柏西普及VEGF MiniTrap之蛋白質變異體的「目標值」)。在此實施例之又一態樣中,當將包括阿柏西普及VEGF MiniTrap之變異體的蛋白質製劑標準化為5 g/L、10 g/L或甚至更高之濃度時,使用合適的條件獲得之製劑導致製劑之顏色減至所需BY值(稱為「目標BY值」)。
在本實施例之又一態樣中,變異體之目標BY值及/或目標值可在力價增加或不顯著減小之製劑中獲得(參見實例5)。
在一些實施例中,藉由使用經修飾之CDM產生的組成物可產生具有所需目標BY值的抗VEGF蛋白之製劑,其中該製劑之顏色表徵如下:
(i) 黃棕色不超過歐洲顏色標準BY2;
(ii) 黃棕色不超過歐洲顏色標準BY3;
(iii) 黃棕色不超過歐洲顏色標準BY4;
(iv) 黃棕色不超過歐洲顏色標準BY5;
(v) 介於歐洲顏色標準BY2與BY3之間;
(vi) 介於歐洲顏色標準BY3與BY4之間;
(vii) 介於歐洲顏色標準BY4與BY5之間,其中 組成物包含約5 g/L或約10 g/L之抗VEGF蛋白,且其中組成物之樣品可自經澄清之收穫物的蛋白A析出液中作為樣品獲得。如在以下實例9,表 9-3
中所見,包含5 g/L阿柏西普之蛋白A析出液呈黃棕色,經量測為具有1.77之b*值。此一樣品在AEX下游產生時具有0.50之b*值,證明瞭AEX降低樣品之黃棕色著色的效用(表 9-3
)。
藉由使用經修飾之CDM產生的組成物可產生抗VEGF蛋白之製劑,其中製劑之顏色藉由CIELAB標度中公認的標準顏色表徵來表徵:
(i) 黃棕色不超過約22-23之b*值;
(ii) 黃棕色不超過約16-17之b*值;
(iii) 黃棕色不超過9-10之b*值;
(iv) 黃棕色不超過4-5之b*值;
(v) 黃棕色不超過2-3之b*值;
(vi) 介於17至23之間的b*值;
(vii) 介於10至17之間的b*值;
(viii) 介於5至10之間的b*值;
(ix) 介於3至5之間的b*值;或
(x) 介於1至3之間的b*值,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白,且其中組成物自經澄清之收穫物的蛋白A析出液中作為樣品獲得。參見實例9,表 9-3
。
對於添加至細胞培養物中以形成經修飾之CDM的組分,術語「累積量」係指在細胞培養過程中添加至生物反應器中以形成CDM的特定組分之總量,包括在培養開始時添加的組分之量(在第0天的CDM)及隨後添加的組分之量。當計算組分之累積量時,亦包括在生物反應器產生之前(亦即,在第0天的CDM之前)添加至種菌培養物或接種物中的組分之量。累積量不受培養過程中組分隨時間損失(例如,經由代謝或化學降解)之影響。因此,具有相同累積量之組分的兩種培養物可能仍具有不同絕對水準,例如,若在不同時間將組分添加至兩種培養物中(例如,若在一種培養物中,所有組分均在開始時添加,而在另一種培養物中,組分係隨時間之推移而添加)。累積量亦不受培養過程中組分隨時間之原位合成(例如,經由代謝或化學轉化)的影響。因此,具有相同累積量之給定組分的兩種培養物可能仍具有不同絕對水準,例如,若組分係經由生物轉化過程在兩種培養物之一種中原位合成。累積量可以例如組分之克數或莫耳數之單位表示。術語「累積濃度」係指組分之累積量除以生產批次開始時生物反應器中液體之體積,包括培養中使用的任何接種物對起始體積之貢獻。例如,若生物反應器在生產批次開始時包含2升細胞培養基,並且在第0、1、2及3天添加1克之組分X,則第3天之後的累積濃度為2 g/L (亦即4克除以2升)。若在第4天將另外1升不含組分X之液體添加至生物反應器中,則累積濃度將保持為2 g/L。若在第5天自生物反應器中損失一些液體(例如經由蒸發),則累積濃度將保持為2 g/L。累積濃度可以例如克/升或莫耳/升之單位表示。A. 胺基酸:
在一些實施例中,可藉由降低或提高CDM中胺基酸之累積濃度來獲得經修飾之CDM。此等胺基酸之非限制性實例包括丙胺酸、精胺酸、天冬醯胺、天冬胺酸、半胱胺酸、麩醯胺、麩胺酸、甘胺酸、組胺酸、異白胺酸、白胺酸、離胺酸、甲硫胺酸、苯丙胺酸、脯胺酸、絲胺酸、蘇胺酸、色胺酸、酪胺酸及纈胺酸(或其鹽)。與起始CDM相比,經修飾之CDM中的此等胺基酸之累積量之增加或減少可為約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%,以及在上述一或多者內之範圍。替代地,與未經修飾之CDM相比,經修飾之CDM中的一或多種胺基酸之累積量之增加或減少可為約5%至約20%、約10%至約30%、約30%至約40%、約30%至約50%、約40%至約60%、約60%至約70%、約70%至約80%、約80%至約90%、或約90%至約100%,以及在上述一或多者內之範圍(參見圖 25 至 27
及實例5)。
在一些實施例中,可藉由降低CDM中半胱胺酸之累積濃度來獲得經修飾之CDM。與未經修飾之CDM相比,CDM中形成經修飾之CDM的半胱胺酸之量之減少可為約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%,以及在上述一或多者內之範圍。替代地,與CDM相比,經修飾之CDM中的半胱胺酸之累積量之減少可為約5%至約20%、約10%至約30%、約30%至約40%、約30%至約50%、約40%至約60%、約60%至約70%、約70%至約80%、約80%至約90%、或約90%至約100%,以及在上述一或多者內之範圍。在一態樣中,經修飾之CDM中的累積半胱胺酸之量小於約1 mM、約2 mM、約3 mM、約4 mM、約5 mM、約6 mM、約7 mM、約8 mM、約9 mM或約10 mM (參見 圖 25 至 27
及實例5)。
在一些實施例中,可藉由用胱胺酸替換CDM中的至少一定百分比之累積半胱胺酸來獲得經修飾之CDM。與未經修飾之CDM相比,替換可為約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%,以及在上述一或多者內之範圍。替代地,與未經修飾之CDM相比,替換可為約5%至約20%、約10%至約30%、約30%至約40%、約30%至約50%、約40%至約60%、約60%至約70%、約70%至約80%、約80%至約90%、或約90%至約100%,以及在上述一或多者內之範圍(參見圖 25 至 27
及實例5)。
在一些實施例中,可藉由用硫酸半胱胺酸替換CDM中的至少一定百分比之累積半胱胺酸來獲得經修飾之CDM。與未經修飾之CDM相比,替換可為約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%,以及在上述一或多者內之範圍。替代地,與未經修飾之CDM相比,替換可為約5%至約20%、約10%至約30%、約30%至約40%、約30%至約50%、約40%至約60%、約60%至約70%、約70%至約80%、約80%至約90%、或約90%至約100%,以及在上述一或多者內之範圍。B. 金屬:
在一些實施例中,可藉由降低或提高CDM中的金屬之累積濃度來獲得經修飾之CDM。金屬之非限制性實例包括鐵、銅、錳、鉬、鋅、鎳、鈣、鉀及鈉。與未經修飾之CDM相比,經修飾之CDM中的一或多種金屬之量之增加或減少可為約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%,以及在上述一或多者內之範圍。替代地,與未經修飾之CDM相比,經修飾之CDM中的一或多種金屬之累積量之增加或減少可為約5%至約20%、約10%至約30%、約30%至約40%、約30%至約50%、約40%至約60%、約60%至約70%、約70%至約80%、約80%至約90%、或約90%至約100%,以及在上述一或多者內之範圍(參見圖 25 至 27
及實例5)。C. 抗氧化劑:
在一些實施例中,經修飾之CDM包含一或多種抗氧化劑。抗氧化劑之非限制性實例可包括牛磺酸、次牛磺酸、甘胺酸、硫辛酸、麩胱甘肽、氯化膽鹼、氫皮質酮、維生素C、維生素E及其組合(參見圖 28A 至 E
及實例5)。
在一些實施例中,經修飾之CDM包含約0.01 mM至約20 mM牛磺酸,亦即0.01 mM至約1 mM、約0.01 mM至約5 mM、約0.01 mM至約10 mM、0.1 mM至約1 mM、約0.1 mM至約5 mM、約0.1 mM至約10 mM、約1 mM至約5 mM、約1 mM至約10 mM,以及在上述一或多者內之範圍。
在一些實施例中,經修飾之CDM包含約0.01 mM至約20 mM次牛磺酸,亦即0.01 mM至約1 mM、約0.01 mM至約5 mM、約0.01 mM至約10 mM、0.1 mM至約1 mM、約0.1 mM至約5 mM、約0.1 mM至約10 mM、約1 mM至約5 mM、約1 mM至約10 mM,以及在上述一或多者內之範圍。
在一些實施例中,經修飾之CDM包含約0.01 mM至約20 mM甘胺酸,亦即0.01 mM至約1 mM、約0.01 mM至約5 mM、約0.01 mM至約10 mM、0.1 mM至約1 mM、約0.1 mM至約5 mM、約0.1 mM至約10 mM、約1 mM至約5 mM、約1 mM至約10 mM,以及在上述一或多者內之範圍。
在一些實施例中,經修飾之CDM包含約0.01 μM至約5 μM硫辛酸,亦即,約0.01 µM至約0.1 µM、約0.1 µM至約1 µM、約1 µM 至約2.5 µM、約1 µM至約3 µM、約1 µM至約5 µM,以及在上述一或多者內之範圍。
在一些實施例中,經修飾之CDM包含約0.01 M至約5 mM麩胱甘肽,亦即0.01 mM至約1 mM、0.1 mM至約1 mM、約0.1 mM至約5 mM、約1 mM至約5 mM,以及在上述一或多者內之範圍。
在一些實施例中,經修飾之CDM包含約0.01 μM至約5 μM氫皮質酮,亦即,約0.01 µM至約0.1 µM、約0.1 µM至約1 µM、約1 µM 至約2.5 µM、約1 µM至約3 µM、約1 µM至約5 µM,以及在上述一或多者內之範圍。
在一些實施例中,經修飾之CDM包含約1 μM至約50 μM維生素C,亦即,約1 µM至約5 µM、約5 µM至約20 µM、約10 µM至約30 µM、約5 µM至約30 µM、約20 µM至約50 µM、約25 µM至約50 µM,以及在上述一或多者內之範圍。D. 為調節醣化而對培養基之改變:
本文亦包括藉由改變CDM中的某些組分之累積濃度來調節抗VEGF蛋白之醣化的方法。可基於添加至CDM中的組分之累積量來改變總岩藻醣化%、總半乳醣化%、總唾液酸化%及甘露糖-5。
在示例性實施例中,調節抗VEGF蛋白之醣化的方法可包含向CDM中補充尿苷。抗VEGF蛋白可具有約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約2%至約15%甘露糖-5、及約60%至約79%半乳醣化聚醣。(參見以下實例6)。
在一些實施例中,調節抗VEGF蛋白之醣化的方法可包含向CDM中補充錳。在一態樣中,CDM在補充前不含錳。抗VEGF蛋白可具有約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約2%至約15%甘露糖-5、及約60%至約79%半乳醣化聚醣。(參見以下實例6)。
在一些實施例中,調節抗VEGF蛋白之醣化的方法可包含向CDM中補充半乳糖。在一態樣中,CDM在補充前不含半乳糖。抗VEGF蛋白可具有約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約2%至約15%甘露糖-5、及約60%至約79%半乳醣化聚醣。(實例6)。
在一些實施例中,調節抗VEGF蛋白之醣化的方法可包含向CDM中補充地塞米松。在一態樣中,CDM在補充前不含地塞米松。抗VEGF蛋白可具有約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約2%至約15%甘露糖-5、及約60%至約79%半乳醣化聚醣。(參見以下實例6)。
在一些實施例中,調節抗VEGF蛋白之醣化的方法可包含向CDM中補充尿苷、錳、半乳糖及地塞米松中之一或多種。在一態樣中,CDM在補充之前不含尿苷、錳、半乳糖及地塞米松中之一或多種。抗VEGF蛋白可具有約40%至約50%總岩藻醣化聚醣、約30%至約55%總唾液酸化聚醣、約2%至約15%甘露糖-5、及約60%至約79%半乳醣化聚醣。(實例6)。V. 使用下游製程技術製備組成物
包含本發明之抗VEGF蛋白的組成物可藉由調節下游蛋白質產生過程中之條件來產生。發明者發現最佳化下游程序可導致抗VEGF蛋白之某些變異體最少化以及變色。下游製程之最佳化可產生具有減少的側氧變異體以及最佳化之顏色特徵的組成物。
下游製程技術可單獨使用或與前述節IV中所述之上游製程技術組合使用。A. 陰離子交換層析:
在一些實施例中,本發明之組成物可涉及一種製程,其包含:在CDM中在宿主細胞中表現抗VEGF蛋白,其中抗VEGF蛋白自宿主細胞分泌至培養基中,及獲得經澄清之收穫物。收穫物經歷以下步驟:(a)將由收穫物獲得之生物樣品加載至陰離子交換層析(AEX)管柱上;(b)用合適的洗滌緩衝液洗滌AEX管柱,(c)收集流通級分,視情況(d)用合適的剝離緩衝液洗滌管柱,及(e)收集剝離級分。
流通級分可包含抗VEGF蛋白之側氧變異體,當與陰離子交換層析管柱之剝離級分中的側氧變異體相比時,其為抗VEGF蛋白樣品之約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%。例如,參看表 9-5
及表 9-6
,流通級分包含抗VEGF蛋白之經氧化變異體,其中與剝離級分中之經氧化變異體相比,若干組胺酸及色胺酸殘基為約1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95% (以及在上述一或多者內之範圍)經氧化。
用於AEX管柱的平衡緩衝液及洗滌緩衝液之pH值均可為約8.20至約8.60。在另一態樣中,用於AEX管柱之平衡緩衝液及洗滌緩衝液之電導率均可為約1.50至約3.0 mS/cm。在一態樣中,平衡緩衝液及洗滌緩衝液可為約50 mM Tris鹽酸鹽。在一態樣中,剝離緩衝液包含2M氯化鈉或1N氫氧化鈉或兩者(參見表 2-2
)。實例2進一步說明最佳化平衡緩衝液及洗滌緩衝液之濃度及電導率。
蛋白質變異體可包括如下一或多個殘基之修飾:一或多個天冬醯胺經去醯胺;一或多個天冬胺酸轉化為異天冬胺酸及/或Asn;一或多個甲硫胺酸經氧化;一或多個色胺酸轉化為N-甲醯犬尿胺酸;一或多個色胺酸為單羥基色胺酸;一或多個色胺酸為二羥基色胺酸;一或多個色胺酸為三羥基色胺酸;一或多個精胺酸轉化為Arg 3-去氧葡醛酮糖;C端甘胺酸不存在;及/或存在一或多個非醣化糖位點。
感興趣之蛋白質可為阿柏西普、抗VEGF抗體或VEGF MiniTrap。可藉由以下方式中之一或多種形成蛋白質變異體:(i)自選自His86、His110、His145、His209、His95、His19及/或His203 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)之組胺酸殘基的組胺酸之氧化;(ii)選自Trp58及/或Trp138 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)處之色胺酸殘基的色胺酸殘基之氧化;(iii) Tyr64 (或具有阿柏西普之某些結構特徵的蛋白質上之等效位置)處的酪胺酸殘基之氧化;(iv)選自Phe44及/或Phe166 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)的苯丙胺酸殘基之氧化;及/或(v)選自Met10、Met 20、Met163及/或Met192 (或具有阿柏西普之某些結構特徵的蛋白質上之等效殘基位置)的甲硫胺酸殘基之氧化。
流通級分可包含以下一或多者:
(a) 一定百分比之已氧化成2-側氧基-組胺酸的組胺酸殘基,其中其顏色表徵如下:
(i) 黃棕色不超過歐洲顏色標準BY2;
(ii) 黃棕色不超過歐洲顏色標準BY3;
(iii) 黃棕色不超過歐洲顏色標準BY4;
(iv) 黃棕色不超過歐洲顏色標準BY5;
(v) 介於歐洲顏色標準BY2與BY3之間;
(vi) 介於歐洲顏色標準BY3與BY4之間;
(vii) 介於歐洲顏色標準BY4與BY5之間,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白,且其中組成物自流通級分中作為樣品獲得。
(b) 一定百分比之已氧化成2-側氧基-組胺酸的組胺酸殘基。此外,其顏色之特徵在於具有近似於BY2、BY3、BY4、BY5、BY6、BY7之黃棕色;或不比BY2暗/強烈、不比BY3暗、不比BY4暗、不比BY5暗、不比BY6暗、不比BY7暗;或介於BY2與BY3之間、介於BY2與BY4之間、介於BY3與BY4之間或介於BY3與BY5之間。
(c) 一定百分比之已氧化成2-側氧基-組胺酸的組胺酸殘基,其中其顏色以如下CIE L*, a*, b*顏色空間中之顏色表徵:
(i) 黃棕色不超過約22-23之b*值;
(ii) 黃棕色不超過約16-17之b*值;
(iii) 黃棕色不超過9-10之b*值;
(iv) 黃棕色不超過4-5之b*值;
(v) 黃棕色不超過2-3之b*值;
(vi) 介於17至23之間的b*值;
(vii) 介於10至17之間的b*值;
(viii) 介於5至10之間的b*值;
(ix) 介於3至5之間的b*值;或
(x) 介於1至3之間的b*值,其中組成物包含約5 g/L或約10 g/L之抗VEGF蛋白,且其中組成物自流通級分中作為樣品獲得。
(d) 組成物中不多於約1%、不多於約0.1%或約0.1-1%、0.2-1%、0.3-1%、0.4-1%、0.5-1%、0.6-1%、0.7-1%、0.8-1%或0.9-1%之組胺酸殘基經氧化成2-側氧基-組胺酸。百分比計算描述於部分II中。B. 親和層析:
在一些實施例中,本發明之組成物可使用包含以下之方法產生:在宿主細胞中表現抗VEGF蛋白,其中抗VEGF蛋白由宿主細胞分泌至培養基中並獲得經澄清之收穫物。收穫物經歷以下步驟,包含:(a)將自經澄清之收穫物獲得的生物樣品加載至親和層析管柱上,其中親和層析包含能夠選擇性或特異性地結合至抗VEGF蛋白之蛋白質;(b)用合適的溶析緩衝液洗滌親和層析管柱,及(c)收集溶析之級分。例如,如表 7-1
及表 7-7
至7-10
所例示,使用VEGF165
作為能夠選擇性或特異性地結合至抗VEGF蛋白之蛋白質並按照上述方法收集溶析之級分,成功產生MT5 (抗VEGF蛋白)、阿柏西普及抗VEGF scFv片段。表 7-1
亦揭露使用以下各物作為能夠選擇性或特異性地結合至MT5之不同蛋白質成功地產生MT5:(i) mAb1 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 73為重鏈,而SEQ ID NO.: 74為輕鏈);(ii) mAb2 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 75為重鏈,而SEQ ID NO.: 76為輕鏈);(iii) mAb3 (小鼠抗VEGF-R1 mAb小鼠IgG1,其中SEQ ID NO.: 77為重鏈,而SEQ ID NO.: 78為輕鏈);以及(iv) mAb4 (小鼠抗VEGFR1 mAb小鼠IgG1,其中SEQ ID NO.: 79為重鏈,而SEQ ID NO.: 80為輕鏈)。
關於上述步驟(a),有待加載至親和管柱上的生物樣品可來自其中經澄清之收穫物可在親和之前經歷生產的樣品,包括但不限於離子交換層析(陰離子或陽離子)。在使用親和步驟之前,亦可採用熟習此項技術者熟知之其他層析程序。重要之處在於包含抗VEGF蛋白之生物樣品可經歷親和層析。
在一些實施例中,本發明之組成物可使用包含以下之方法產生:在宿主細胞中表現VEGF MiniTrap蛋白,其中VEGF MiniTrap由宿主細胞分泌至培養基中,且其中可進一步處理培養基以形成經澄清之收穫物。可藉由已知的層析程序進一步處理此收穫物,從而產生包含VEGF MiniTrap之生物樣品。此生物樣品可藉由採用以下步驟進一步處理,包含:(a)將生物樣品加載至親和層析管柱上,其中親和層析包含能夠與VEGF MiniTrap蛋白選擇性或特異性地結合或相互作用之蛋白;(b)用合適的溶析緩衝液洗滌親和層析管柱,及(c)收集溶析之級分。再次參看表 7-1
,此表中揭露使用以下各物作為能夠選擇性或特異性地結合至MT5或與MT5相互作用之不同蛋白質成功地產生MT5 (VEGF MiniTrap) :(i) VEGF165
;(ii) mAb1 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 73為重鏈,而SEQ ID NO.: 74為輕鏈);(iii) mAb2 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 75為重鏈,而SEQ ID NO.: 76為輕鏈);(iv) mAb3 (小鼠抗VEGF-R1 mAb小鼠IgG1,其中SEQ ID NO.: 77為重鏈,而SEQ ID NO.: 78為輕鏈);以及(v) mAb4 (小鼠抗VEGFR1 mAb小鼠IgG1,其中SEQ ID NO.: 79為重鏈,而SEQ ID NO.: 80為輕鏈)。
在一實施例中,親和層析亦可用於分離其他MiniTrap蛋白。切割阿柏西普之後,可使用對經切割之阿柏西普有特異性之結合物對包含經切割之阿柏西普的樣品進行親和層析。在一態樣中,結合物可為抗體或其部分。
可使用例如用IdeS蛋白酶(FabRICATOR)或其變異體對阿柏西普進行蛋白水解消化來促進阿柏西普之切割,以產生VEGF MiniTrap。用IdeS蛋白酶或其變異體切割阿柏西普可產生包含Fc片段及VEGF MiniTrap的產物之混合物。VEGF MiniTrap可藉由使用本文所述之一或多種生產策略進一步處理。
在一些示例性實施例中,能夠與抗VEGF蛋白諸如阿柏西普或MiniTrap選擇性或特異性結合(「結合物」)或與之相互作用的蛋白質可來源於人類或小鼠。
親和產生過程可進一步包含在加載生物樣品之前使用平衡緩衝液平衡親和管柱。示例性平衡緩衝液可為20 mM磷酸鈉pH 6-8 (尤其是7.2)、10 mM磷酸鈉、500 mM NaCl pH 6-8 (尤其是7.2)、50 mM Tris pH 7-8、DPBS pH 7.4。
可使用合適的緩衝液諸如DPBS來加載生物樣品。
此親和產生過程可進一步包含用一或多種洗滌緩衝液洗滌親和管柱。可洗滌管柱一次或多次。此外,洗滌液亦可作為洗滌級分收集。洗滌緩衝液之pH值可皆為約7.0至約8.60。在一態樣中,洗滌緩衝液可為DPBS。在另一態樣中,洗滌緩衝液可為20 mM磷酸鈉pH 6-8 (尤其是7.2)、10 mM磷酸鈉、500 mM NaCl pH 6-8 (尤其是7.2)、50 mM Tris pH 7-8或DPBS pH 7.4。
此親和過程可進一步包含用一或多種合適的溶析緩衝液洗滌親和管柱及收集溶析之級分。可洗滌管柱一次或多次。此一合適的溶析緩衝液之非限制性實例包括:乙酸銨(pH值為約2.0至約3.0)、乙酸(pH值為約2.0至約3.2)、甘胺酸-HCl (pH值為約2.0至約3.0)、檸檬酸鈉(pH值為約2.0至約3.0)、檸檬酸(pH值為約2.0至約3.0)、異硫氰酸鉀(pH值為約2.0至約3.0)或其組合。
在一些態樣中,可使用中和緩衝液中和溶析級分。此一中和緩衝液之一例子為Tris至Tris-HCl (pH值為約7.0至約9.0)。C. IdeS 突變體:
用於切割Fc融合蛋白(諸如阿柏西普)之IdeS蛋白酶在鹼性pH條件下會迅速喪失酶活性,這可限制其在VEGF MiniTrap製造期間之使用。因此,已開發在鹼性pH下,例如在強鹼諸如NaOH存在下更穩定之變異體。此等鹼性條件可為0.05N NaOH 1小時或0.1N NaOH 0.5小時。
在一些實施例中,IdeS突變體可具有在其全長上與由以下組成之群組中列出之胺基酸序列具有至少約70%序列一致性的胺基酸序列:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。在一些態樣中,胺基酸序列在其全長上與上文剛剛提及之胺基酸序列具有約75%、80%、85%、90%、95%或約100%序列一致性。
在一些實施例中,IdeS突變體可具有編碼多肽的經分離之核酸分子,該多肽之胺基酸序列在其全長上與由以下組成之群組中列出之胺基酸序列具有至少70%序列一致性:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16。在一些態樣中,胺基酸序列在其全長上與上文剛剛提及之胺基酸序列具有約75%、80%、85%、90%、95%或約100%序列一致性。
在一些實施例中,該多肽具有在其全長上與由以下組成之群組中列出之胺基酸序列具有至少70%序列一致性的胺基酸序列:SEQ ID NO.: 2、SEQ ID NO.: 3、SEQ ID NO.: 4、SEQ ID NO.: 5、SEQ ID NO.: 6、SEQ ID NO.: 7、SEQ ID NO.: 8、SEQ ID NO.: 9、SEQ ID NO.: 10、SEQ ID NO.: 11、SEQ ID NO.: 12、SEQ ID NO.: 13、SEQ ID NO.: 14、SEQ ID NO.: 15及SEQ ID NO.: 16,並由具有合適載體之宿主細胞表現,該載體包含編碼所識別之肽的核酸。在一態樣中,核酸分子與表現控制序列可操作地連接,該表現控制序列能夠指導其在宿主細胞中之表現。在一態樣中,載體可為質體。在一些態樣中,胺基酸序列在其全長上與上文剛剛提及之胺基酸序列具有約75%、80%、85%、90%、95%或約100%序列一致性。在一些態樣中,經分離之核酸分子可用於編碼多肽。
在一些實施例中,IdeS突變體可具有包含由SEQ ID NO.: 1 (IdeS)定義之親本胺基酸序列的胺基酸序列,其中在位置87、130、182及/或274處之天冬醯胺殘基突變為除天冬醯胺以外之胺基酸。在一態樣中,與親本胺基酸序列相比,該突變可使在鹼性pH值下之化學穩定性提高。在另一態樣中,與親本胺基酸序列相比,該突變可使在鹼性pH值下之化學穩定性提高50%。在一態樣中,胺基酸可選自天冬胺酸、白胺酸及精胺酸。在一具體態樣中,位置87處之天冬醯胺殘基突變為天冬胺酸殘基。在另一具體態樣中,位置130處之天冬醯胺殘基突變為精胺酸殘基。在仍另一具體態樣中,位置182處之天冬醯胺殘基突變為白胺酸殘基。在仍另一具體態樣中,位置274處之天冬醯胺殘基突變為天冬胺酸殘基。在仍另一具體態樣中,位置87及130處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置87及182處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置87及274處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置130及182處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置130及274處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置182及274處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置87、130及182處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置87、182及274處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置130、182及274處之天冬醯胺殘基經突變。在仍另一具體態樣中,位置87、130、182及274處之天冬醯胺殘基經突變。在一些態樣中,胺基酸序列在其全長上與上述胺基酸序列具有約 75%、80%、85%、90%、95%或約100%序列一致性。在一些態樣中,經分離之核酸分子可用於編碼多肽。
熟悉標準分子生物學技術的熟習此項技術之普通技術人員可在不過度負擔之情況下製備及使用本發明之IdeS突變體。標準技術可用於重組DNA、寡核苷酸合成、組織培養及轉化(例如,電穿孔、脂質轉染)。參見,例如Sambrook等人,Molecular Cloning: A Laboratory Manual, 如前述
,其出於任何目的以引用之方式併入本文。可根據製造商說明書或如本文所述來執行酶反應及產生技術。VI. 一般蛋白質產生
設想多種不同產生技術,包括但不限於親和層析、離子交換層析、混合模式層析、尺寸排阻層析及疏水相互作用層析(單獨或以組合形式)在本發明之範疇內。此等層析步驟視具體的分離形式而定,基於電荷、疏水性程度或尺寸或其組合來分離生物樣品之蛋白質混合物。對於前述提及之每項技術,皆可使用若干種不同的層析樹脂,從而允許根據所涉及之特定蛋白質精確調整生產流程。每種分離方法皆會造成蛋白質以不同速率穿過管柱,從而實現物理分離,隨著蛋白質進一步穿過管柱或選擇性地黏附於分離介質上,物理分離會增加。接著,將蛋白質(i)使用適當的溶析緩衝液進行差異溶析及/或(ii)由所用管柱獲得之流通級分中收集,視情況,用適當的平衡緩衝液洗滌管柱。在一些情況下,當雜質優先黏附於管柱上且感興趣之蛋白質較少黏附時,感興趣之蛋白質便會與雜質(HCP、蛋白質變異體 等)分離,亦即,感興趣之蛋白質不會吸附於特定管柱之固相上,且因此流過管柱。在一些情況下,當雜質無法吸附於管柱上並因此流過管柱時,它們便會與感興趣之蛋白質分離。
在使用上述上游產生方法及/或藉由此項技術中習知的替代產生方法產生重組蛋白後,產生過程可自分離步驟開始。一旦獲得包含感興趣之蛋白質(例如融合蛋白)的經澄清之溶液或混合物,便將感興趣之蛋白質與過程相關之雜質(諸如由細胞(如HCP)產生之其他蛋白質,以及產物相關之物質,諸如酸性或鹼性變異體)分離。可採用一或多種不同產生技術之組合,包括親和層析、離子交換(例如CEX、AEX)層析、混合模式(MM)層析及/或疏水相互作用層析。此等產生步驟基於例如電荷、疏水性程度及/或表觀尺寸分離生物樣品內之組分混合物。對於本文提及之每項層析技術,許多層析樹脂係市售的,從而允許根據所涉及之特定蛋白質精確調整生產流程。每種分離方法皆允許蛋白質以不同速率穿過管柱,從而實現物理分離,隨著蛋白質進一步穿過管柱或選擇性地黏附於分離樹脂(或介質)上,物理分離會增加。接著可差異地收集蛋白質。在一些情況下,當其他組分特異性地吸附於管柱樹脂上,而感興趣之蛋白質不吸附時,感興趣之蛋白質便與生物樣品之組分分離。A. 初步回收及病毒滅活
在某些實施例中,本文揭露之產生方法之初始步驟涉及自生物樣品中澄清及初步回收感興趣之蛋白質。初步回收將包括一或多個離心步驟,以便自宿主細胞及伴隨的細胞碎片中分離出感興趣之蛋白質。樣品之離心可例如但不限於在7,000×g至約12,750×g下進行。在大規模生產之情境下,可以一定流率在線進行該離心,該流率經設定成例如在所得上清液中達成150 NTU之濁度水準。接著可收集該上清液以進行進一步處理,或經由一或多個深度過濾器進行在線過濾,以進一步澄清樣品。
在某些實施例中,初步回收可包括使用一或多個深度過濾步驟來澄清樣品,從而有助於處理感興趣之蛋白質。在其他實施例中,初步回收可包括在離心後使用一或多個深度過濾步驟。可在本發明之上下文中使用的深度過濾器之非限制性實例包括Millistak+ X0HC、F0HC、D0HC、A1HC、B1HC深度過濾器(EMD Millipore);3M™型號30/60ZA、60/90 ZA、VR05、VR07、delipid深度過濾器(3M Corp.)。0.2 μm過濾器,諸如賽多利斯(Sartorius)之0.45/0.2 μm Sartopore™雙層或Millipore之Express SHR或SHC濾筒通常在深度過濾器之後。亦可使用熟習此項技術者熟知之其他過濾器。
在某些實施例中,初步回收過程亦可為減少或滅活可存在於生物樣品中之病毒的關鍵。多種病毒減少/滅活方法中之任何一或多者可在產生初步回收階段使用,包括:熱滅活(巴氏滅菌)、pH滅活、緩衝劑/清潔劑處理、UV及γ射線輻照以及添加某些化學滅活劑諸如β-丙內酯,或例如啡啉銅,如美國專利第4,534,972號中所述,其全部教示皆以引用之方式併入本文。在本發明之某些示例性實施例中,樣品在初步回收階段期間暴露於清潔劑病毒滅活。在其他實施例中,樣品可在初步回收階段期間暴露於低pH滅活。
在採用病毒減少/滅活之彼等實施例中,可對生物樣品進行調整(若需要)以用於其他產生步驟。例如,在低pH病毒滅活之後,在繼續產生過程之前,通常將樣品之pH值調節至更中性之pH值,例如,約4.5至約8.5。另外,可用注射用水(WFI)稀釋混合物以獲得所需電導率。B. 親和層析
在某些示例性實施例中,使生物樣品經歷親和層析對於產生感興趣之蛋白質可為有利的。層析材料能夠選擇性地或特異性地與感興趣之蛋白質結合或相互作用。該層析材料之非限制性實例包括:蛋白A及蛋白G。包含例如蛋白質或其部分之層析材料亦能夠與感興趣之蛋白質結合或相互作用。在一態樣中,感興趣之蛋白質為抗VEGF蛋白,諸如阿柏西普、MiniTrap或與其相關之蛋白質。
親和層析可涉及使生物樣品經歷包含合適的蛋白A樹脂之管柱。當在本文中使用時,術語「蛋白A」涵蓋自其天然來源回收之蛋白A,合成產生之蛋白A (例如,藉由肽合成或藉由重組技術)及其保留結合具有CH
2/CH
3區之蛋白質之能力的變異體。在某些態樣中,蛋白A樹脂藉由與分子之Fc部分(若其具有該區域)特異性相互作用而可用於多種抗體同種型的基於親和力之產生及分離。
蛋白A樹脂有若干商業來源。一種合適的樹脂為來自GE Healthcare之MabSelect™。合適的樹脂包括但不限於:來自GE Healthcare之MabSelect SuRe™、MabSelect SuRe LX、MabSelect、MabSelect SuRe pcc、MabSelect Xtra、rProtein A Sepharose;來自EMD Millipore之ProSep HC、ProSep Ultra及ProSep Ultra Plus;來自Life Technologies之MapCapture。裝填有MabSelect™之合適的管柱之非限制性實例為直徑約1.0 cm x 長度約21.6 cm之管柱(17 mL床體積)。合適的管柱可包含樹脂,諸如MabSelect™SuRe或類似樹脂。蛋白A亦可由Repligen、Pharmacia及Fermatech商購獲得。
樣品加載之前,可使用合適的緩衝液平衡親和管柱。加載管柱後,可使用合適的洗滌緩衝液將管柱洗滌一次或多次。接著可使用適當的溶析緩衝液(例如,甘胺酸-HCl、乙酸或檸檬酸)溶析管柱。可使用熟習此項技術者熟知之技術諸如UV偵測器來監測析出液。可收集析出之感興趣之級分,接著準備以便進一步處理。
在一態樣中,可例如藉由清潔劑或低pH對析出液進行病毒滅活。可選擇合適的清潔劑濃度或pH值(及時間)以獲得所需病毒滅活結果。病毒滅活後,通常對析出液進行pH值及/或電導率調整以便後續產生步驟。
析出液可經由深度過濾器進行過濾以便在額外的層析精製(chromatographic polishing)步驟之前自感興趣之蛋白質中移除濁度及/或各種雜質。合適的深度過濾器之實例包括但不限於Millistak+ XOHC、FOHC、DOHC、AIHC、X0SP及BIHC Pod過濾器(EMD Millipore)或Zeta Plus 30ZA/60ZA、60ZA/90ZA、delipid、VR07及VR05過濾器(3M)。Emphaze AEX Hybrid Purifier多機理過濾器亦可用於澄清析出液。為了由深度過濾步驟實現所需雜質移除及產物回收,可需將析出液池調節至特定的pH值及電導率。C. 陰離子交換層析
在某些實施例中,藉由使生物樣品經歷至少一個陰離子交換分離步驟來產生感興趣之蛋白質。在一種情境下,陰離子交換步驟可在親和層析程序(例如,蛋白A親和)之後進行。在其他情境下,陰離子交換步驟可在親和層析步驟之前進行。在其他方案中,陰離子交換可在親和層析步驟之前及之後進行。在一態樣中,感興趣之蛋白質為阿柏西普或MiniTrap。
相對於陽離子交換材料而言,陰離子交換材料之使用部分基於感興趣之蛋白質的局部電荷。陰離子交換層析可與以下其他層析程序組合使用:諸如親和層析、尺寸排阻層析、疏水相互作用層析以及熟練技術人員已知之其他層析模式。
在進行分離時,可藉由使用多種技術中之任一者,例如使用批式生產技術或層析技術,使初始蛋白質組成物(生物樣品)與陰離子交換材料接觸。
在批式生產之情境下,在所需起始緩衝液中製備或平衡陰離子交換材料。製備後,獲得陰離子交換材料之漿料。使生物樣品與漿料接觸,以使蛋白質吸附於陰離子交換材料上。藉由使漿料沉降並移除上清液,自漿料中分離出包含不與AEX材料結合之酸性種類的溶液。漿料可經歷一或多個洗滌步驟及/或溶析步驟。
在層析分離之情境下,層析管柱用於容納層析載體材料(樹脂或固相)。將包含感興趣之蛋白質的樣品加載至特定層析管柱上。接著管柱可經歷一或多個使用合適的洗滌緩衝液之洗滌步驟。未吸附於樹脂上之樣品組分可能會流過管柱。可使用適當的溶析緩衝液對已吸附於樹脂上的組分進行差異溶析。
通常在AEX層析中使用類似於加載條件之條件或替代地藉由以逐步或線性梯度方式降低洗滌液之pH值及/或提高其離子強度/電導率來進行洗滌步驟。在一態樣中,在加載緩衝液及洗滌緩衝液中使用的鹽水溶液之pH處於或接近感興趣之蛋白質的等電點(pI)。通常,pH比感興趣之蛋白質的pI高或低約0至2個單位,但其範圍可為高或低0至0.5個單位。其亦可位於感興趣之蛋白質的pI處。
陰離子劑可選自由乙酸鹽、氯化物、甲酸鹽及其組合組成之群組。陽離子劑可選自由Tris、精胺酸、鈉及其組合組成之群組。在一具體實例中,緩衝溶液為Tris/甲酸鹽緩衝液。緩沖液可選自由以下組成之群組:吡啶、哌嗪、L-組胺酸,Bis-tris、Bis-Tris丙烷、咪唑、N-乙基嗎啉、TEA (三乙醇胺)、Tris、嗎啉、N-甲基二乙醇胺、AMPD (2-胺基-2-甲基-1,3-丙二醇)、二乙醇胺、乙醇胺、AMP (2-胺基-2-甲基-1-丙醇)、哌嗪、1,3-二胺基丙烷及哌啶。
填充式陰離子交換層析管柱、陰離子交換膜裝置、陰離子交換整體式裝置或深度過濾器介質可以結合-溶析模式、流通模式或混合模式操作,其中蛋白質呈現與層析材料之結合且仍可使用與加載緩衝液相同或實質上相似之緩衝液自該材料上洗去。
在結合-溶析模式下,首先在某些蛋白質會吸附於基於樹脂之基質上之條件下,使用具有適當離子強度及pH值之緩衝液對管柱或膜裝置進行調節。例如,在進料加載期間,由於靜電吸引,感興趣之蛋白質可吸附於樹脂上。在用平衡緩衝液或具有不同pH值及/或電導率之另一種緩衝液洗滌管柱或膜裝置後,藉由提高溶析緩衝液之離子強度(亦即電導率)以便與溶質競爭陰離子交換基質之帶電位點來實現產物回收。改變pH值及由此更改溶質之電荷為達成溶質溶析之另一種方式。電導率或pH值之變化可為逐漸的(梯度溶析)或逐步的(分步溶析)。
在流通模式下,管柱或膜裝置係在選定之pH值及電導率下操作,以使感興趣之蛋白質不會與樹脂或膜結合,而酸性種類將保留在管柱上或與感興趣之蛋白質相比將具有獨特的溶析譜。在此策略之情境下,酸性種類將在合適的條件下與層析材料相互作用或結合,而感興趣之蛋白質及感興趣之蛋白質之某些聚集體及/或片段將流過管柱。
陰離子交換樹脂之非限制性實例包括二乙胺基乙基(DEAE)、四級胺乙基(QAE)及四級胺(Q)基團。另外的非限制性實例包括:Poros 50PI及Poros 50HQ,其為剛性聚合物珠粒,其中主鏈由交聯聚[苯乙烯-二乙烯苯]組成;Capto Q Impres及Capto DEAE,其為高流動性瓊脂糖珠粒;Toyopearl QAE-550、Toyopearl DEAE-650及Toyopearl GigaCap Q-650,其為聚合物基珠粒;Fractogel®
EMD TMAE Hicap,其為具有觸手離子交換劑之合成聚合樹脂;Sartobind STIC®
PA nano,其為具有一級胺配位體之耐鹽層析膜;Sartobind Q nano,其為強陰離子交換層析膜;CUNO BioCap,其為由無機助濾劑、精製纖維素及離子交換樹脂構成之zeta-plus深度過濾器介質;以及XOHC,其為由無機助濾劑、纖維素及混合纖維素酯構成之深度過濾器介質。
在某些實施例中,可將樣品之蛋白質加載量調整至以下管柱之總蛋白質加載量:介於約50 g/L與約500 g/L之間、或介於約75 g/L與約350 g/L之間、或介於約200 g/L與約300 g/L之間。在其他實施例中,將加載蛋白質混合物之蛋白質濃度調整至加載於管柱上的材料之以下蛋白質濃度:介於約0.5 g/L與約50 g/L之間、介於約1 g/L與約20 g/L之間、或介於約3 g/L與約10 g/L之間。在其他實施例中,將加載蛋白質混合物之蛋白質濃度調整至加載於管柱上的材料之約37 g/L蛋白質濃度。
可添加添加劑,諸如聚乙二醇(PEG)、清潔劑、胺基酸、糖、離液劑,以增強分離性能,從而實現更好的分離、回收及/或產物品質。
在某些實施例中,包括與阿柏西普及/或VEGF MiniTrap有關之彼等實施例,本發明之方法可用於選擇性地移除、顯著減少或基本上移除至少10%之蛋白質變異體,從而產生具有減少的蛋白質變異體之蛋白質組成物。
蛋白質變異體可包括如下一或多個殘基之修飾:一或多個天冬醯胺經去醯胺;一或多個天冬胺酸轉化為天冬胺酸-甘胺酸及/或Asn-Gly;一或多個甲硫胺酸經氧化;一或多個色胺酸轉化為N-甲醯犬尿胺酸;一或多個色胺酸為單羥基色胺酸;一或多個色胺酸為二羥基色胺酸;一或多個色胺酸為三羥基色胺酸;一或多個精胺酸轉化為Arg 3-去氧葡醛酮糖;C端甘胺酸不存在;及/或存在一或多個非醣化糖位點。亦觀察到使用AEX可減少該親和析出液中的抗VEGF變異體之氧化及酸性種類。與親和析出液相比,在使用AEX後,流通級分可展示出抗VEGF變異體之氧化及/或酸性種類至少約20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%或5%之減少。
阿柏西普及/或VEGF MiniTrap之蛋白質變異體可包括以下一或多者:(i)來自選自His86、His110、His145、His209、His95、His19及/或His203之組胺酸殘基的經氧化組胺酸;(ii)選自在Trp58及/或Trp138處之色胺酸殘基的經氧化色胺酸殘基;(iii)在Tyr64處之經氧化酪胺酸殘基;(iv)選自Phe44及/或Phe166之經氧化苯丙胺酸殘基;及/或(v)選自Met10、Met 20、Met163及/或Met192之經氧化甲硫胺酸殘基。D. 陽離子交換層析
本發明之組成物可藉由使包含感興趣之蛋白質的生物樣品經歷至少一個陽離子交換(CEX)步驟來產生。在某些示例性實施例中,CEX步驟將為AEX步驟之補充,並且在AEX步驟之前或之後進行。在一態樣中,感興趣之蛋白質為阿柏西普、MiniTrap或與其相關之分子。
陽離子交換材料相對於陰離子交換材料(諸如上文討論之彼等陰離子交換材料)之使用部分基於給定溶液中感興趣之蛋白質的局部電荷及所需分離條件。在使用陰離子交換步驟之前採用陽離子交換步驟,或在使用陽離子交換步驟之前採用陰離子交換步驟皆在本發明之範疇內。此外,僅採用陽離子交換步驟與其他層析程序之組合在本發明之範疇內。
在進行陽離子交換時,可藉由使用以下多種技術中之任一種將包含感興趣之蛋白質的樣品與陽離子交換材料接觸,例如,使用批式生產技術或層析技術,如上文針對AEX所述。
鹽水溶液既可用作加載緩衝液亦可用作洗滌緩衝液,其pH值低於感興趣之蛋白質的等電點(pI)。在一態樣中,pH值比蛋白質之pI低約0至5個單位。在另一態樣中,其在比蛋白質之pI低1-2個單位之範圍內。在仍另一態樣中,其在比蛋白質之pI低1至1.5個單位之範圍內。
在某些實施例中,增加或減少鹽水溶液中陰離子劑之濃度以達到以下pH值:介於約3.5與約10.5之間、或介於約4與約10之間、或介於約4.5與約9.5之間、或介於約5與約9之間、或介於約5.5與約8.5之間、或介於約6與約8之間、或介於約6.5與約7.5之間。在一態樣中,增加或減少鹽水溶液中陰離子劑之濃度以達到5、或5.5、或6、或6.5、或6.8、或7.5之pH值。適用於CEX方法中的緩衝液系統包括但不限於Tris甲酸鹽、Tris乙酸鹽、硫酸銨、氯化鈉及硫酸鈉。
在某些實施例中,藉由提高或降低陽離子劑之濃度來調節鹽水溶液之電導率及pH值。在一態樣中,陽離子劑維持在約20 mM至約500 mM、約50 mM至約350 mM、約100 mM至約300 mM或約100 mM至約200 mM之濃度範圍內。陽離子劑之非限制性實例係選自由以下組成之群組:鈉、Tris、三乙胺、銨、精胺酸及其組合。
填充式陽離子交換層析管柱或陽離子交換膜裝置可以結合-溶析模式、流通模式或混合模式操作,其中產物呈現與層析材料之結合或相互作用,但仍可使用與加載緩衝液相同或實質上相似之緩衝液自該材料上洗去(上面概述此等模式之詳情)。
陽離子取代基包括羧甲基(CM)、磺乙基(SE)、磺丙基(SP)、磷酸鹽(P)及磺酸鹽(S)。另外的陽離子材料包括但不限於:Capto SP ImpRes,其為高流動性瓊脂糖珠粒;CM Hyper D等級 F,其為陶瓷珠粒,用功能化水凝膠包被及滲透,250-400個離子基團μeq/ mL;Eshmuno S,其為具有50-100 μeq/mL離子容量之親水性聚乙烯基醚基礎基質;Nuvia C Prime,其為由大孔高度交聯之親水性聚合物基質構成的疏水性陽離子交換介質,55-75 με/mL;Nuvia S,其具有UNOsphere基礎基質,90 -150 με/mL離子基團;Poros HS,其為剛性聚合物珠粒,其中主鏈由交聯聚[苯乙烯-二乙烯苯]組成;Poros XS,其為剛性聚合物珠粒,其中主鏈由交聯聚[苯乙烯-二乙烯苯]組成;Toyo Pearl Giga Cap CM 650M,其為聚合物基珠粒,具有0.225 meq/mL離子容量;Toyo Pearl Giga Cap S 650M,其為聚合物基珠粒;Toyo Pearl MX TRP,其為聚合物基珠粒。注意到本文所述之CEX層析可與MM樹脂一起使用。
將包含感興趣之蛋白質的樣品之蛋白質加載量調整至以下管柱之總蛋白質加載量:介於約5 g/L與約150 g/L之間、或介於約10 g/L與約100 g/L之間、介於約20 g/L與約80 g/L之間、介於約30 g/L與約50 g/L之間、或介於約40 g/L與約50 g/L之間。在某些實施例中,將加載蛋白質混合物之蛋白質濃度調整至有待加載於管柱上的以下材料之蛋白質濃度:介於約0.5 g/L與約50 g/L之間、或介於約1 g/L與約20 g/L之間。
可添加添加劑,諸如聚乙二醇、清潔劑、胺基酸、糖、離液劑,以增強分離性能,從而實現更好的分離、回收及/或產物品質。
在某些實施例中,包括與阿柏西普或抗VEGF抗體或VEGF MiniTrap有關之彼等實施例,本發明之方法可用於選擇性地移除、顯著減少或基本上移除樣品中所有的側氧變異體,其中感興趣之蛋白質基本上將在CEX程序之流通液中,而側氧變異體實質上將由管柱介質捕獲。E. 混合模式層析
混合模式(「MM」)層析亦可用於製備本發明之組成物。MM層析,在本文中亦稱為「多模式層析」,為一種層析策略,其利用包含配位體之載體,該配位體能夠提供與樣品中的感興趣之分析物或蛋白質之至少兩種不同的相互作用。此等位點中之一者在配位體與感興趣之蛋白質之間提供有吸引力的電荷-電荷相互作用類型,而另一位點提供電子受體-予體相互作用及/或疏水及/或親水相互作用。電子予體-受體相互作用包括諸如氫鍵、π-π、陽離子-π、電荷轉移、偶極-偶極、感應偶極等相互作用。
用於混合模式分離之管柱樹脂可為Capto Adhere。Capto Adhere為具有多模式功能之強陰離子交換劑。其基礎基質為具有配位體(N-芐基-N-甲基乙醇胺)之高度交聯的瓊脂糖,該配位體呈現出不同的相互作用功能,諸如離子相互作用、氫鍵及疏水相互作用。在某些態樣中,用於混合模式分離之樹脂係選自PPA-HyperCel及HEA-HyperCel。PPA-HyperCel及HEA-HyperCel之基礎基質為高孔隙度之交聯纖維素。它們的配位體分別為苯丙胺及己胺。苯丙胺及己胺為蛋白質分離提供不同選擇性及疏水性選擇。額外混合模式層析載體包括但不限於Nuvia C Prime、Toyo Pearl MX Trp 650M及Eshmuno®
HCX。在某些態樣中,混合模式層析樹脂係由直接或經由間隔體與有機或無機載體(有時稱為基礎基質)偶聯之配位體構成。載體可呈以下形式:顆粒,諸如基本上呈球形之顆粒、整料、過濾器、膜、表面、毛細管及其類似物。在某些態樣中,載體係由天然聚合物諸如交聯碳水化合物材料諸如瓊脂糖、瓊脂、纖維素、聚葡糖、殼聚醣、蒟蒻、角叉菜膠、結冷膠、藻酸鹽及其類似物製備。為了獲得高吸附能力,載體可為多孔的,接著將配位體偶聯至外表面以及孔表面。此等天然聚合物載體可根據標準方法製備,諸如反相懸浮凝膠化(S Hjerten: Biochim Biophys Acta 79(2), 393-398 (1964),其全部教示皆以引用之方式併入本文)。替代地,載體可由合成聚合物諸如交聯合成聚合物,例如苯乙烯或苯乙烯衍生物、二乙烯苯、丙烯醯胺、丙烯酸酯、甲基丙烯酸酯、乙烯基酯、乙烯醯胺及其類似物製備。此等合成聚合物可根據標準方法產生,參見「Styrene based polymer supports developed by suspension polymerization」 (R Arshady: Chimica e L’Industria 70(9), 70-75 (1988),其全部教示皆以引用之方式併入本文)。多孔天然或合成聚合物載體亦可由商業來源獲得,諸如GE Healthcare, Uppsala, Sweden。
可將包含感興趣之蛋白質的生物樣品混合物之蛋白質加載量調整至以下管柱之總蛋白質加載量:介於約25 g/L與約750 g/L之間、或介於約75 g/L與約500 g/L之間、或介於約100 g/L與約300 g/L之間。在某些示例性實施例中,將加載蛋白質混合物之蛋白質濃度調整至加載於管柱上的材料之以下蛋白質濃度:介於約1 g/L與約50 g/L之間、或介於約9 g/L與約25 g/L之間。
可添加添加劑,諸如聚乙二醇、清潔劑、胺基酸、糖、離液劑,以增強分離性能,從而實現更好的分離、回收及/或產物品質。
在某些實施例中,包括與阿柏西普及/或MiniTrap有關之彼等實施例,本發明之方法可用於選擇性地移除、顯著減少或基本上移除所有PTM,包括側氧變異體。
用於產生本發明之組成物的方法亦可以連續層析模式實施。在此模式下,使用至少兩個管柱(稱為「第一」管柱及「第二」管柱)。在某些實施例中,可進行此連續層析模式,以使得溶析級分及/或剝離級分(包含PTM,例如側氧變異體)可隨後或同時加載於第二管柱上(有或無稀釋)。
在一實施例中,用於連續模式之介質選擇可為具有側懸疏水及陰離子交換官能基之許多層析樹脂、整料介質、膜吸附介質或深度過濾介質中之一種。F. 疏水相互作用層析
本發明之組成物亦可使用疏水相互作用層析(HIC)來製備。
在進行分離時,例如使用批式生產技術或使用管柱或膜層析使生物樣品與HIC材料接觸。在HIC處理之前,可能需要調整鹽緩衝液之濃度以達成所需蛋白質與樹脂或膜之結合/相互作用。
離子交換層析依賴於感興趣之蛋白質之局部電荷來進行選擇性分離,而疏水相互作用層析則利用蛋白質之疏水特性來實現選擇性分離。蛋白質上或蛋白質內的疏水基團與層析樹脂或膜之疏水基團相互作用。通常,在合適的條件下,蛋白質(或蛋白質之一部分)之疏水性越強,其與管柱或膜之相互作用就越強。因此,在合適的條件下,HIC可用於促進樣品中的過程相關之雜質(例如,HCP)以及產物相關之物質(例如,聚集體及碎片)與感興趣之蛋白質之分離。
像離子交換層析一樣,HIC管柱或HIC膜裝置亦可在溶析模式、流通模式或混合模式下操作,其中產物呈現出與層析材料之結合或相互作用,而仍可使用與加載緩衝液相同或實質上相似之緩衝液自該材料上洗去。(上面結合AEX處理概述了此等模式之詳情。)由於疏水相互作用在高離子強度下最強,所以此種分離形式宜在鹽溶析步驟(諸如通常與離子交換層析結合使用之彼等)之後進行。替代地,可在採用HIC步驟之前將鹽添加至樣品中。高鹽濃度有利於蛋白質吸附於HIC管柱上,但實際濃度可將視感興趣之蛋白質之性質、鹽類型及所選的特定HIC配位體而定在很大範圍內變化。視它們係促進疏水相互作用(鹽析效應)抑或破壞水之結構(離液效應)並導致疏水相互作用減弱而定,各種離子可以所謂的疏溶(soluphobic)系列排列。根據遞增之鹽析效應將陽離子排序為Ba2+
;Ca2+
;Mg2+
;Li+
;Cs+
;Na+
;K+
;Rb+
;NH4+
,而陰離子可根據遞增之離液效應經排序為PO4 3-
;SO4 2-
;CH3
CO3 -
;CI-
;Br-
;NO3 -
;ClO4 -
;I-
;SCN-
。
一般而言,Na+
、K+
或NH4+
硫酸鹽使用HIC有效促進配位體-蛋白質相互作用。可如以下關係所給出來調配影響相互作用強度之鹽:(NH4
)2
SO4
> Na2
SO4
> NaCl > NH4
C1 > NaBr > NaSCN。通常,介於約0.75 M與約2 M之間的硫酸銨或介於約1 M與約4 M之間的NaCl之鹽濃度係有用的。
HIC介質通常包含疏水性配位體(例如
烷基或芳基)與之偶聯的基礎基質(例如,交聯瓊脂糖或合成共聚物材料)。合適的HIC介質包含經苯基官能化之瓊脂糖樹脂或膜(例如,來自GE Healthcare之Phenyl Sepharose™或來自Sartorius之Phenyl Membrane)。許多HIC樹脂係市售的。實例包括但不限於:Capto Phenyl、具有低或高取代之Phenyl Sepharose™ 6 Fast Flow 、Phenyl Sepharose™ High Performance、Octyl Sepharose™ High Performance (GE Healthcare);Fractogel™ EMD Propyl或Fractogel™ EMD Phenyl (E. Merck, Germany);Macro-Prep™甲基或Macro-Prep™第三丁基管柱(Bio-Rad, California);WP HI- Propyl (C3)™ (J. T. Baker, New Jersey);以及Toyopearl™醚、苯基或丁基(TosoHaas, PA);ToyoScreen PPG;ToyoScreen Phenyl;ToyoScreen Butyl;ToyoScreen Hexyl;GE HiScreen及Butyl FF HiScreen Octyl FF。
將包含感興趣之蛋白質的樣品之蛋白質加載量調整至以下管柱之總蛋白質加載量:介於約50 g/L與約1000 g/L之間、介於約5 g/L與約150 g/L之間、介於約10 g/L與約100 g/L之間、介於約20 g/L與約80 g/L之間、介於約30 g/L與約50 g/L之間、或介於約40 g/L與約50 g/L之間。在某些實施例中,將加載蛋白質混合物之蛋白質濃度調整至有待加載於管柱上的以下材料之蛋白質濃度:介於約0.5 g/L與約50 g/L之間、或介於約1 g/L與約20 g/L之間。
由於為任何特定生產過程選擇之pH值必須與蛋白質之穩定性及活性相容,因此特定的pH值條件對每種應用可為特定的。然而,由於在pH 5.0-8.5時,特定的pH值對HIC分離之最終選擇性及解析度影響極小,因此此等條件可為有利的。pH值之增加削弱疏水相互作用,並且在pH值高於8.5或低於5.0時,蛋白質之保留發生更劇烈之變化。此外,離子強度之變化、有機溶劑之存在、溫度及pH值(特別是當沒有淨表面電荷時,在等電點pI下)會影響蛋白質之結構及溶解性,並由此影響與其他疏水性表面(諸如在HIC介質中之彼等)之相互作用,且因此,在某些實施例中,本發明結合生產策略,其中對上述一或多者進行調整,以實現過程相關之雜質及/或產物相關之物質的所需減少。
在某些實施例中,可使用光譜法,諸如UV、NIR、FTIR、螢光、拉曼,從而以隨線(on-line)、近線(at-line)或在線(in-line)模式監測感興趣之蛋白質及雜質,接著可將其用於控制自HIC吸附劑流出物中收集的彙集材料中之聚集體水準。在某些實施例中,可在層析步驟之流出管線上或在收集容器中使用隨線、近線或在線監測方法,以實現所需產物之品質/回收。在某些實施例中,UV信號可用作替代品以實現適當的產物品質/回收,其中UV信號可經適當處理,包括但不限於諸如積分、微分、移動平均值之類的處理技術,以使得可解決正常過程可變性並且可實現目標產物品質。在某些實施例中,此等量測可與在線稀釋方法組合,以使得加載/洗滌液之離子濃度/電導率可藉由反饋來控制,且因此有助於產物品質控制。G. 尺寸排阻層析
尺寸排阻層析或凝膠過濾依賴於隨分子尺寸而變的組分之分離。分離視與流體中之時間相比,物質在多孔固定相中所花費之時間而定。分子將駐留在孔中之概率視分子及孔之尺寸而定。另外,物質滲透至孔中之能力係由大分子之擴散遷移率決定,大分子越小,擴散遷移率越高。極巨大的大分子可能根本不會滲透至固定相之孔中;而且,對於非常小的大分子,滲透之可能性接近均一。較大分子尺寸之組分更快地移動通過固定相,而較小分子尺寸之組分通過固定相之孔的路徑則較長,因此在固定相中之滯留時間更長。
層析材料可包含尺寸排阻材料,其中尺寸排阻材料為樹脂或膜。用於尺寸排阻之基質較佳為惰性凝膠介質,其可為交聯多醣,例如呈球形珠粒形式之交聯瓊脂糖及/或聚葡糖之複合物。交聯度決定溶脹之凝膠珠粒中存在的孔之尺寸。大於某一尺寸之分子不會進入凝膠珠粒,且因此最快移動通過層析床。較小之分子,諸如清潔劑、蛋白質、DNA及其類似物,會根據其尺寸及形狀而不同程度地進入凝膠珠粒,它們通過床時會受到阻礙。因此通常以分子尺寸遞減之順序溶析分子。
適用於病毒之尺寸排阻層析的多孔層析樹脂可由具有不同物理特徵之右旋糖、瓊脂糖、聚丙烯醯胺或二氧化矽製成。亦可使用聚合物組合。最常用的為可自Amersham Biosciences以商品名「 SEPHADEX」獲得之彼等。來自不同構造材料之其他尺寸排阻載體亦為合適的,例如Toyopearl 55F (聚甲基丙烯酸酯,來自Tosoh Bioscience, Montgomery Pa.)及Bio-Gel P-30 Fine (BioRad Laboratories, Hercules, CA)。
可將包含感興趣之蛋白質的樣品之蛋白質加載量調整至以下管柱之總蛋白質加載量:介於約50 g/L至約1000 g/L之間、介於約5 g/L與約150 g/L之間、介於約10 g/L與約100 g/L之間、介於約20 g/L與約80 g/L之間、介於約30 g/L與約50 g/L之間、或介於約40 g/L與約50 g/L之間。在某些實施例中,將加載蛋白質混合物之蛋白質濃度調整至有待加載於管柱上的以下材料之蛋白質濃度:介於約0.5 g/L與約50 g/L之間、或介於約1 g/L與約20 g/L之間。H. 病毒過濾
病毒過濾為產生過程中專用的病毒減少步驟。此步驟通常在層析精製後進行。可藉由使用合適的過濾器來減少病毒,此等過濾器包括但不限於來自Asahi Kasei Pharma之Planova 20N™、50 N或BioEx;來自EMD Millipore之Viresolve™過濾器;來自Sartorius之ViroSart CPV;或來自Pall Corporation之Ultipor DV20或DV50™過濾器。熟習此項技術之普通技術人員顯而易見,選擇合適的過濾器以獲得所需過濾性能。I. 超濾 / 滲濾
本發明之某些實施例採用超濾及滲濾以進一步濃縮及調配感興趣之蛋白質。在以下文獻中詳細描述超濾:Principles and Applications, L. Zeman及A. Zydney (Marcel Dekker, Inc., New York, N.Y., 1996);及Ultrafiltration Handbook, Munir Cheryan (Technomic Publishing, 1986;ISBN號87762- 456-9);其全部教示皆以引用之方式併入本文。一種過濾方法為如題為「Pharmaceutical Process Filtration Catalogue」第177-202頁(Bedford, Mass., 1995/96)之Millipore目錄中所述之切向流過濾,該文獻之全部教示皆以引用之方式併入本文。通常認為超濾係指使用孔徑小於0.1 μm之過濾器進行的過濾。使用具有如此小之孔徑的過濾器,可藉由使樣品緩衝液滲透通過過濾器膜孔來減少樣品之體積,而蛋白質則保留在膜表面之上。
熟習此項技術之普通技術人員可選擇一種合適的膜過濾器裝置用於UF/DF操作。適用於本發明之膜盒(membrane cassette)的實例包括但不限於來自EMD Millipore的具有10 kD、30 kD或50 kD膜之Pellicon 2或Pellicon 3盒;來自GE Healthcare的Kvick 10 kD、30 kD或50 kD膜盒;以及來自Pall Corporation的Centramate或Centrasette 10 kD、30 kD或50 kD盒。J. 示例性生產策略
可藉由依次採用pH降低、離心及過濾自生產生物反應器收穫物中移除細胞及細胞碎片(包括HCP)來進行初步回收。本發明涉及使來自初步回收的包含感興趣之蛋白質之生物樣品經歷一或多個生產步驟,包括(但無特別順序) AEX、CEX、SEC、HIC及/或MM。本發明之某些態樣包括進一步處理步驟。額外的處理程序之實例包括乙醇沉澱、等電聚焦、逆相HPLC、矽膠層析、肝素Sepharose™層析、其他陰離子交換層析及/或其他陽離子交換層析、層析聚焦、SDS-PAGE、硫酸銨沉澱、羥磷灰石層析、凝膠電泳、透析及親和層析(例如,使用蛋白A或G、抗體、特異性受質、配位體或抗原作為捕獲試劑)。在某些態樣中,可獨立地改變管柱溫度(以及其他參數)以提高分離效率及/或任何特定產生步驟之產率。
在某些實施例中,可將未結合之流通及洗滌級分進一步分級分離,並且可彙集提供目標產物純度之級分之組合。
管柱加載及洗滌步驟可藉由以下操作來控制:在管柱流出物或收集池或兩者中在線、近線或離線量測產物相關之雜質/物質水準,以達成特定的目標產物品質及/或產率。在某些實施例中,可藉由用緩衝液或其他溶液在線或分批或連續稀釋來動態控制加載濃度,以實現提高分離效率及/或產率所需之分配。
此等生產程序之實例描繪於圖 5 至 8
中。
圖 5
代表用於產生阿柏西普之一個示例性實施例。參看圖 5
,該方法包含:(a)在CDM中培養之宿主細胞中表現阿柏西普;(b)使用可包括親和捕獲樹脂之第一層析載體捕獲阿柏西普;及(c)使阿柏西普之至少一部分與可包括陰離子交換層析之第二層析載體接觸。步驟(c)可進一步包含洗滌AEX管柱及收集包含阿柏西普之樣品之流通級分。視情況,步驟(c)可包含剝離第二層析載體及收集剝離之級分。該等步驟可藉由常規方法聯合上文提及之方法來進行。應理解,熟習此項技術者可能選擇採用CEX而非AEX或除AEX之外亦使用CEX。亦可不以特定順序採用額外的層析步驟,包括但不限於HIC及SEC。
除了圖 5
中之示例性實施例之外,其他另外的示例性實施例可包括(d)使步驟(c)之該阿柏西普之至少一部分與第三層析載體接觸。在一態樣中,方案可包括(e)使步驟(d)之至少一部分阿柏西普與第四層析載體接觸。在此實施例之一態樣中,方案可視情況包含使步驟(c)之包含阿柏西普的樣品經歷小於5.5之pH值。在一態樣中,本方法包含在步驟(a)之前的澄清步驟。
圖 6
代表用於產生VEGF MiniTrap之一個示例性實施例。此方法包含:(a)在CDM中培養之宿主細胞中表現阿柏西普;(b)使用可包括親和層析樹脂之第一層析載體捕獲阿柏西普;(c)切割阿柏西普,從而移除Fc域並形成包含VEGF MiniTrap之樣品;(d)使步驟(c)之樣品與可為親和層析之第二層析載體接觸,以及(e)使步驟(d)之流通液與可包括陰離子交換層析之第三層析載體接觸。步驟(d)包含流通級分之收集,其中由於缺少Fc域,MiniTrap應駐留,而阿柏西普或具有Fc域之任何其他蛋白質應基本上與步驟(d)之親和管柱相互作用。視情況,步驟(d)可包含剝離第三層析載體及收集剝離之級分。此等步驟可藉由常規方法結合以上概述之方法來進行。不以特定順序,可包括額外的層析步驟,包括但不限於諸如HIC及SEC。
圖 7
代表用於產生阿柏西普之一示例性實施例。此方法包含:(a)在CDM中培養之宿主細胞中表現阿柏西普;(b)使用可包括陽離子交換層析之第一層析載體捕獲阿柏西普;及(c)使步驟(b)之流通液與可包括陰離子交換層析之第二層析載體接觸。視情況,步驟(c)可包含剝離第二層析載體及收集剝離之級分。此等步驟可藉由常規方法結合以上提及之方案來進行。不以特定順序,可採用其他層析程序,包括但不限於HIC及SEC。
圖 8
代表用於產生VEGF MiniTrap之一示例性實施例。此方法包含:(a)在CDM中培養之宿主細胞中表現阿柏西普;(b)使用可包括離子交換層析之第一層析載體捕獲阿柏西普;(c)對(b)之包含阿柏西普的流通級分進行親和層析;溶析,其中溶析液包含阿柏西普;(d)使(c)之阿柏西普經歷切割活性,從而切割Fc域,由此形成VEGF MiniTrap。在一態樣中,步驟(b)之離子交換包含AEX。替代地,步驟(b)可包含CEX。不以特定順序,可包括額外的層析步驟,諸如步驟(d)之後的其他離子交換層析步驟、HIC及/或SEC之添加。VII. 包含組成物之醫藥調配物
本發明亦揭露包含抗VEGF組成物(如上所述)之調配物。抗VEGF蛋白之合適的調配物包括但不限於以下專利中所述之調配物:美國專利第7,608,261號、美國專利第7,807,164號、美國專利第8,092,803號、美國專利第8,481,046號、美國專利第8,802,107號、美國專利第9,340,594號、美國專利第9,914,763號、美國專利第9,580,489號、美國專利第10,400,025號、美國專利第8,110,546號、美國專利第8,404,638號、美國專利第8,710,004號、美國專利第8,921,316號、美國專利第9,416,167號、美國專利第9,511,140號、美國專利第9,636,400號及美國專利第10,406,226號,其皆以引用之方式全部併入本文。
上游製程技術(在上文節IV中描述)及下游製程技術(在上文節V中描述)可單獨使用或彼此組合使用以實現調配物產生。
本發明揭露包含抗VEGF組成物及一或多種成分/賦形劑之調配物以及其使用方法及製備此等組成物之方法。在本發明之一實施例中,本發明之醫藥調配物具有約5.5、5.6、5.7、5.8、5.9、6.0、6.1或6.2之pH值。
為了製備抗VEGF組成物之醫藥調配物,將抗VEGF組成物與醫藥學上可接受之載劑或賦形劑混合。參見,例如Remington’s Pharmaceutical Sciences and U.S. Pharmacopeia: National Formulary, Mack Publishing Company, Easton, Pa. (1984);Hardman等人(2001) Goodman and Gilman’s The Pharmacological Basis of Therapeutics, McGraw-Hill, New York, N.Y.;Gennaro (2000) Remington: The Science and Practice of Pharmacy, Lippincott, Williams, and Wilkins, New York, N.Y.;Avis等人(編著) (1993) Pharmaceutical Dosage Forms: Parenteral Medications, Marcel Dekker, NY;Lieberman等人(編著) (1990) Pharmaceutical Dosage Forms: Tablets, Marcel Dekker, NY;Lieberman等人(編著) (1990) Pharmaceutical Dosage Forms: Disperse Systems, Marcel Dekker, N.Y.;Weiner及Kotkoskie (2000) Excipient Toxicity and Safety, Marcel Dekker, Inc., New York, N.Y.;其全部教示皆以引用之方式併入本文。在本發明之一實施例中,醫藥調配物為無菌的。
本發明之醫藥調配物包括抗VEGF組成物及醫藥學上可接受之載劑,包括例如水、緩衝劑、防腐劑及/或清潔劑。
本發明提供一種醫藥調配物,其包含本文所列之抗VEGF組成物中之任一者及醫藥學上可接受之載劑,例如,其中多肽之濃度為約40 mg/mL、約60 mg/mL、約80 mg/mL、90 mg/mL、約100 mg/mL、約110 mg/mL、約120 mg/mL、約130 mg/mL、約140 mg/mL、約150 mg/mL、約200 mg/mL或約250 mg/mL。
本發明之範疇包括經乾燥、例如經冷凍乾燥之組成物,其包含抗VEGF蛋白及實質上(約85%至約99%或更高)無水之醫藥學上可接受之載劑。
在一實施例中,根據Physicians’ Desk Reference 2003 (Thomson Healthcare; 第57版(2002年11月1日),其全部教示皆以引用之方式併入本文),將與本文揭露之抗VEGF組成物聯合向個體投與之另一種治療劑向個體投與。
本發明提供一種容器(例如,具有蓋之塑料或玻璃小瓶或層析管柱、中空針或注射器筒),其包含本文所述之抗VEGF組成物中之任一者或包含醫藥學上可接受之載劑的醫藥調配物。本發明亦提供一種包含本文所述之抗VEGF組成物或調配物之注射裝置,例如注射器、預填充注射器或自動注射器。在一態樣中,將容器染色(例如棕色)以遮擋自然光或其他光。
本發明包括包含抗VEGF組成物以及一或多種其他治療劑之組合。抗VEGF組成物及其他治療劑可在單一組成物中或在單獨組成物中。例如,治療劑為Ang-2抑制劑(例如奈斯單抗(nesvacumab))、Tie-2受體活化劑、抗PDGF抗體或其抗原結合片段、抗PDGF受體或PDGF受體β抗體或其抗原結合片段及/或另外的VEGF拮抗劑,諸如阿伯西普(abribercept)、康柏西普(conbercept)、貝伐單抗(bevacizumab)、雷尼單抗(ranibizumab);抗VEGF適體,諸如哌加他尼(pegaptanib)(例如,哌加他尼鈉);單鏈(例如,VL-VH)抗VEGF抗體,諸如溴珠單抗(brolucizumab);抗VEGF DARPin,諸如Abicipar Pegol DARPin;雙特異性抗VEGF抗體,例如其亦與ANG2結合,諸如RG7716;或可溶形式的包含胞外域1-3之人類血管內皮生長因子受體3 (VEGFR-3),表示為Fc融合蛋白。VIII. 治療方法
本發明提供用於在個體中治療或預防癌症(例如,其生長及/或轉移至少部分地由VEGF介導,例如VEGF介導之血管生成)或血管生成性眼部病症的方法,其包含投與治療有效量的本文(前述節III)所揭露之組成物。
上游製程技術(前述節IV)及下游製程技術(前述節V及VI)可單獨使用或相互組合使用,以產生如節III中所述之組成物及/或如節VII中所述之調配物,其可用於治療或預防多種病症,包括眼科及腫瘤疾病。
本發明亦提供一種向個體(例如人類)投與本文(節III及VII)所述之組成物的方法,其包含將在不多於約100 µL,例如約50 µL、約70 µL或約100 μL中具有約0.5 mg、2 mg、4 mg、6 mg、8 mg、10 mg、12 mg、14 mg、16 mg、18 mg或20 mg感興趣之蛋白質(例如阿柏西普或MiniTrap)及視情況選用之另一治療劑的組成物藉由例如眼內注射(諸如藉由玻璃體內注射)引入個體體內。
本發明提供一種在有此需要之個體中治療癌症(其生長及/或轉移至少部分地由VEGF介導,例如VEGF介導之血管生成)或血管生成性眼部病症的方法,該方法包含向個體投與治療有效量的本文(以上節III及節VII)所述之組成物,例如,在不多於約100 μl中的2 mg、4 mg、6 mg、8 mg或10 mg感興趣之蛋白質及視情況選用之另一治療劑。在本發明之一實施例中,藉由玻璃體內注射進行投與。可使用本文之方法治療或預防之血管生成性眼部病症之非限制性實例包括:
l 年齡相關之黃斑部變性(例如,濕性或乾性),
l 黃斑部水腫,
l 視網膜靜脈阻塞後之黃斑部水腫,
l 視網膜靜脈阻塞(RVO),
l 視網膜中央靜脈阻塞(CRVO),
l 視網膜分支靜脈阻塞(BRVO),
l 糖尿病性黃斑部水腫(DME),
l 脈絡膜新生血管形成(CNV),
l 虹膜新生血管形成,
l 新生血管性青光眼,
l 青光眼手術後纖維化,
l 增生性玻璃體視網膜病(PVR),
l 視盤新生血管形成,
l 角膜新生血管形成,
l 視網膜新生血管形成,
l 玻璃體新生血管形成,
l 角膜翳,
l 翼狀胬肉,
l 血管性視網膜病,
l 在患有糖尿病性黃斑部水腫之個體中的糖尿病性視網膜病;以及
l 糖尿病性視網膜病(例如非增生性糖尿病性視網膜病(例如,以約47或53之糖尿病性視網膜病嚴重程度量表(DRSS)分級為特徵),或增生性糖尿病性視網膜病;例如,在不患有DME之個體中)。
此等組成物或調配物(節III及節VII)之投與方式可由熟練從業者改變及確定。投與途徑包括腸胃外、非腸胃外、經口、直腸、經黏膜、腸、腸胃外、肌內、皮下、皮內、髓內、鞘內、直接心室內、靜脈內、腹膜內、鼻內、眼內、吸入、吹入、表面、皮膚、眼內、玻璃體內、經皮或動脈內。
在本發明之一實施例中,本發明之醫藥調配物(包括本發明之組成物或調配物)之玻璃體內注射包括以下步驟:用包含調配物之注射器及針(例如30號注射針)刺穿眼睛,及將調配物(例如,小於或等於約100微升;約40、50、55、56、57、57.1、 58、60或70微升)以足量體積注入眼玻璃體中以便遞送本文所述之治療有效量,例如約2、4、6、8.0、8.1、8.2、8.3、8.4、8.5、8.6、8.7、8.8或8.9、10或20 mg的感興趣之蛋白質。視情況,該方法包括以下步驟:向所注射之眼睛投與局部麻醉劑(例如,丙美卡因(proparacaine)、利多卡因(lidocaine)或四卡因(tetracaine))、抗生素(例如,氟喹諾酮(fluoroquinolone))、防腐劑(例如,聚維酮-碘)及/或瞳孔擴張劑。在一態樣中,在注射之前,在有待注射之眼睛周圍建立無菌區。玻璃體內注射後,監測個體之眼內壓升高、發炎及/或血壓。
用於血管生成性眼部病症的感興趣之蛋白質之有效或治療有效量係指足以引起癌症或血管生成性眼部病症之消退、穩定或消除的感興趣之蛋白質之量,例如,藉由使癌症或血管生成性眼部病症之一或多種症狀或症候以任何臨床上可量測之程度消退、穩定或消除(例如,關於血管生成性眼部病症);藉由使糖尿病性視網膜病嚴重程度評分(DRSS)降低或維持;藉由改善或維持視力(例如,最佳矯正視力,如藉由ETDRS字母之增加來量度);增加或維持視野及/或減小或維持中央視網膜厚度;以及就癌症而言,停止或逆轉個體中的癌細胞之生長、存活及/或轉移。
在本發明之一實施例中,用於治療或預防血管生成性眼部病症的感興趣之蛋白質(諸如阿柏西普)的有效或治療有效量為約0.5 mg、2 mg、3 mg、4 mg、5 mg、6 mg、7 mg、7.25 mg、7.7 mg、7.9 mg、8.0 mg、8.1 mg、8.2 mg、8.3 mg、8.4 mg、8.5 mg、8.6 mg、8.7 mg、8.8 mg、8.9 mg、9 mg、10 mg、11 mg、12 mg、13 mg、14 mg、15 mg、16 mg、17 mg、18 mg、19 mg或20 mg,例如,在不多於約100 µL中。該量可根據有待投與之個體之年齡及體格、靶疾病、疾患、投與途徑及其類似因素而變化。在某些示例性實施例中,可在初始劑量之後投與第二或多個後續劑量的感興趣之蛋白質,該第二或多個後續劑量之量可與初始劑量之劑量大致相同或更少或更多,其中後續劑量間隔至少1天至3天、至少一週、至少2週、至少3週、至少4週、至少5週、至少6週、至少7週、至少8週、至少9週、至少10週、至少12週或至少14週。
應注意,劑量值可隨著欲減輕的疾患之類型及嚴重性而變化。應進一步理解對於任何特定個體,具體劑量方案應根據個體需求及投與或監督投與該等組成物之人的專業判斷隨時間加以調節,且本文所列舉之劑量範圍僅為示例性的且不意欲限制所主張之組成物之範疇或實踐。IX. 分析蛋白質變異體之方法
使用本文所述之技術產生的層析樣品中之蛋白質變異體之水準可如以下實例中所述來分析。在某些實施例中,使用帶有經碳氟化合物塗覆之毛細管筒(100 μπι x 5 cm)的iCE3分析儀(ProteinSimple)來採用cIEF方法。兩性電解質溶液係由以下各物在純淨水中之混合物組成:0.35%甲基纖維素(MC)、4% Pharmalyte 3-10載劑兩性電解質、4% Pharmalyte 5-8載劑兩性電解質、10 mM L-精胺酸HCl、24%甲醯胺以及pI標記物5.12及9.77。陽極電解液為80 mM磷酸,而陰極電解液為100 mM氫氧化鈉,均在0.10%甲基纖維素中。將樣品在純淨水中稀釋至10 mg/mL。將樣品與兩性電解質溶液混合,接著藉由引入1500 V之電位持續一分鐘,繼之引入3000 V之電位持續7分鐘來聚焦。藉由使280 nm紫外光穿過毛細管並進入電荷耦合裝置數位相機之鏡頭中,獲得聚焦變異體之影像。隨後分析此影像以確定各種電荷變異體之分佈。熟習此項技術者可改變精確參數,同時仍達成期望結果。
在整個說明書中引用各種出版物,包括專利、專利申請案、公開的專利申請案、登錄號、技術論文及學術論文。此等引用之參考文獻中之每一篇皆以引用之方式全部併入。
本文將藉由參考以下實例更充分地理解。然而,它們不應被視為限製本發明之範疇。實例
在實例中討論之MiniTraps (MT) 1至6如下:
MT1:藉由切割使用CDM1產生之阿柏西普獲得的VEGF MiniTrap。
MT2:藉由切割使用CDM2產生之阿柏西普獲得的VEGF MiniTrap。
MT3:藉由切割使用CDM3產生之阿柏西普獲得的VEGF MiniTrap。
MT4:藉由切割使用大豆水解物產生之阿柏西普獲得的VEGF MiniTrap。
MT5:重組VEGF MiniTrap (二聚物)。
MT6:重組VEGF MiniTrap (scFv)。
MT1、MT5及MT6之表徵描述於以下實例8中。樣品顏色評估
發現量測b*值(CIELAB)之分光光度檢定法適合進行顏色評估。
在可見光譜(380至780 nm)上定量1 mL蛋白質樣品之吸光度,並且使用一組矩陣運算將吸光度曲線轉換為CIELAB顏色空間。該儀器每小時可處理大約6個樣品。該檢定之高通量格式使用CLARIOstar讀板儀(BMG Labtech)。使用需要0.3 mL樣品之96孔板,最多可分析96個樣品。
為了將BY標準轉換為b*值,使用高通量檢定格式對BY參考標準(BY1至BY7)進行定量。
溶液係按照以上討論之BY標準來製備。每個標準之b*值如圖 9
中所示。此方法提供更快的檢定、更少的樣品需求及更短的運行時間,如下表 3
中所示。對於使用此方法評估之所有樣品,將測試樣品之蛋白質濃度標準化為5 g/L或10 g/L。表 3.
實例 1 :使用化學成分確定之培養基產生蛋白質 1.1 細胞來源及收穫
| 原始 | 高通量 | |
| 量 / 樣品 | 1 mL | 0.3 mL |
| 量測格式 | 比色管(單個) | 96孔板(整體) |
| 運行時間 | 6個樣品/小時 | 96個樣品/5分鐘 |
| 資料輸入 | 手動 | 自動 |
| 資料存儲 | Excel | LIMS |
在本研究中使用產生阿柏西普之細胞株。使用化學成分確定之培養基(CDM)培養及收穫產生阿柏西普之細胞株。1.2 阿柏西普之蛋白水解切割
將具有經固定之IdeS酶(獲自Genovis (Cambridge, MA)之FabRICATOR®)的管柱用於產生MT1。將獲自細胞培養收穫物之阿柏西普(在1.0 mL切割緩衝液中之20 mg)添加至管柱中,並在18℃下孵育30 min。30 min後,用切割緩衝液(1.0 mL)洗滌管柱。將消化混合物與洗滌溶液合併。將混合物加載於分析型蛋白A親和管柱(Applied Biosystems™,POROS™ 20 µM蛋白A筒2.1x30 mm,0.1 mL (目錄號2-1001-00)上並自其溶析。根據用於POROS™ 20 µM蛋白A筒2.1x30 mm,0.1 mL (目錄號2-1001-00)之Applied Biosystems’™方案進行處理。管柱高度為20±1.0 cm,滯留時間為15分鐘,且使用40 mM Tris、54 mM乙酸鹽pH 7.0±0.1進行平衡/洗滌。實例 2. 陰離子交換層析 (AEX) 用於使顏色最小化 (A) 使用 AEX 減少顏色形成
進行AEX層析以移除在使用CDM1表現的阿柏西普之產生期間獲得的著色。2.1 設計
如表 2-1
中所詳示,使用表 2-2
中所述之AEX方案對本研究進行五次AEX分離。將15.7 mL Q Sepharose Fast Flow管柱(19.5 cm床高,1.0 cm I.D.)及14.1 mL POROS 50 HQ管柱(18.0 cm床高,1.0 cm I.D.)整合至AKTA Avant台式液相層析控制器中。
使用2 M Tris鹼或2 M乙酸將AEX加載液之pH值調節至目標±0.05個pH單位。使用5 M氯化鈉或去離子水將AEX加載液之電導率調節至目標±0.1 mS/cm。分析所有彙集樣品之高分子量(HMW)、顏色及產率。表 2-1. 用於 AEX 顏色減少之研究設計的概述
表 2-2. 用於顏色減少之 AEX 方案
2.2 結果
| AEX 分離 | 評估條件 | 樹脂 |
| 1 | pH 8.30 – 8.50,1.90 – 2.10 mS/cm | POROS 50 HQ |
| 2 | pH 7.90 – 8.10,2.40 – 2.60 mS/cm | Q Sepharose FF |
| 3 | pH 7.90 – 8.10,2.40 – 2.60 mS/cm | POROS 50 HQ |
| 4 | pH 7.70 – 7.90,3.90 – 4.10 mS/cm | Q Sepharose FF |
| 5 | pH 7.70 – 7.90,3.90 – 4.10 mS/cm | POROS 50 HQ |
| 步驟 | 說明 | 流動相 | 管柱體積 (CV) | 線速度 (cm/h) |
| 1 | 預平衡 | 2 M氯化鈉(NaCl) | 2 | 200 |
| 2 | 平衡 | 50 mM Tris,可變mM NaCl 可變pH值及電導率 | 2 | 200 |
| 3 | 加載 | AEX加載液 可變pH值及電導率 | 40 g/L-樹脂 | 200 |
| 4 | FT/洗滌 | 50 mM Tris,可變mM NaCl 可變pH值及電導率 | 2 | 200 |
| 5 | 剝離1 | 2 M氯化鈉(NaCl) | 2 | 200 |
| 6 | 剝離2 | 1 N氫氧化鈉(NaOH) | 2 | 200 |
將AEX分離用於產生會呈現顏色之顯著減少。(表2-3)。例如,如表 2-3
中所見,在AEX分離1 (pH 8.30-8.50,1.90-2.10 mS/cm)之流通液(FT)及洗滌液中觀察到的顏色具有1.05之b*值,而用於AEX之加載液(「AEX加載液」)之顏色具有3.06之b*值。b*值之增加反映樣品之黃棕色著色的強度。
進行五次AEX分離以評估樹脂(Q Sepharose FF或POROS 50 HQ)以及pH及電導率設定點(pH 8.40及2.00 mS/cm、pH 8.00及2.50 mS/cm、或pH 7.80及4.00 mS/cm)對顏色減少之影響。對於POROS 50 HQ,產率(64.4%、81.9%及91.4%)及彙集HMW水準(1.02%、1.29%及1.83%)隨著設定點變化為較低pH值及較高電導率而提高。顏色(b*值)亦隨著設定點變化為較低pH值及較高電導率而增加(1.05、1.33及1.55)。對於POROS 50 HQ之AEX分離,較高之pH值及較低之電導率提供最大程度之顏色減少。
對於Q Sepharose Fast Flow,產率(49.5%及77.7%)及彙集HMW水準(0.59%及1.25%)亦隨著設定點變化為較低pH值及較高電導率而提高。顏色(b*值)亦隨著設定點變化為較低pH值及較高電導率而增加(0.96及1.35)。
AEX之使用減少黃棕色著色-參見表 2-3
。另外,對於在兩種樹脂上評估之兩個設定點,確定Q Sepharose Fast Flow減少之顏色比POROS 50 HQ多。在pH 8.00及2.50 mS/cm設定點下,POROS 50 HQ池具有1.33之b*值,而Q Sepharose Fast Flow池具有0.96之b*值。類似地,在pH 7.80及4.00 mS/cm設定點下,POROS 50 HQ池具有1.55之b*值,而Q Sepharose Fast Flow池具有1.35之b*值(表 2-3
)。表 2-3. AEX 顏色減少研究之實驗結果的概述
AEX,陰離子交換層析;HMW,高分子量種類;N/A,不適用
將級分調節至10 g/L之蛋白質濃度以用於顏色量測。2.3 結論
| AEX 分離 | 級分 | 產率 (%) | HMW (%) | 顏色 (L*) | 顏色 (a*) | 顏色 (b*) |
| 1 | FT/洗滌 | 64.4 | 1.02 | 98.89 | 0.01 | 1.05 |
| 2 | FT/洗滌 | 49.5 | 0.59 | 98.30 | -0.03 | 0.96 |
| 3 | FT/洗滌 | 81.9 | 1.29 | 99.07 | -0.07 | 1.33 |
| 4 | FT/洗滌 | 77.7 | 1.25 | 99.42 | -0.04 | 1.35 |
| 5 | FT/洗滌 | 91.4 | 1.83 | 99.19 | -0.09 | 1.55 |
| - | 過濾池(AEX加載液) | - | 3.66 – 3.98 | 98.73 | -0.21 | 3.06 |
發現AEX之使用減少黃棕色著色,參見 表 2-3
。參看表 2-3
,AEX加載液具有3.06之b*值,但在經歷AEX層析(AEX分離1-5)時,b*值減小表明黃棕色著色之減少。再者,隨著b*值之減小,著色亦隨之減少;隨著b*值之增大,其反映了給定樣品中黃棕色之增加。
使用兩種AEX樹脂(POROS 50 HQ及Q Sepharose Fast Flow)以及三個設定點(pH 8.40及2.00 mS/cm、pH 8.00及2.50 mS/cm、以及pH 7.80及4.00 mS/cm)評估顏色減少。在這兩種樹脂下,對於較高pH值及較低電導率之設定點,顏色減少較多。此外,在對兩種樹脂評估之兩個設定點(pH 8.00及2.50 mS/cm以及pH 7.80及4.00 mS/cm)下,Q Sepharose Fast Flow比POROS 50 HQ提供更多的顏色減少。然而,與用於AEX之加載溶液(「AEX加載液」)相比,所有五種AEX分離方法均導致顯著的顏色減少,表明在用CDM表現之阿柏西普的產生過程中AEX產生之重要性。AEX加載液(濃度為10 g/L)之初始b*值可在約0.5至約30之範圍內、更具體在約1.0至約25.0之範圍內、及甚至更具體在約2.0至約20.0之範圍內。使用AEX之後,流通液(濃度為10 g/L)之b*值可在0.5至約10.0之範圍內、更具體在約0.5至約7.0之範圍內、及甚至更具體在約0.5至約5.0之範圍內。2.4 肽作圖 (Peptide Mapping)
樣品製備
。進行自上述實驗(表 2-3
)之AEX加載液及流通液獲得的還原及烷化阿柏西普樣品之胰蛋白酶作圖(tryptic mapping),以識別及定量轉譯後修飾(post-translational modification,PTM)。將每個樣品(加載液及流通液)之等分試樣用8.0 M尿素、0.1 M Tris-HCl pH 7.5變性,用DTT還原,接著用碘乙醯胺烷化。首先將經變性、還原及烷化之樣品在37℃下用重組Lys-C (rLys-C)以1:100 (w/w)之酶與受質比率消化30分鐘,用0.1 M Tris-HCl pH 7.5稀釋,以使最終尿素濃度為1.8 M,隨後在37℃下用胰蛋白酶以1:20 (w/w)之酶與受質比率消化2小時,隨後在37℃下用PNGase F以1:5 (w/w)之酶與受質比率去醣化1小時。藉由使用甲酸(FA)使pH值低於2.0來中止消化。
LC-MS 分析
。將由每個樣品得到的rLys-C/胰蛋白酶肽之20 µg等分試樣藉由以下來分離及分析:使用Waters ACQUITY UPLC CSH C18管柱(130 Å,1.7 µm,2.1×150 mm)進行逆相超高效液相層析(UPLC),繼之隨線PDA偵測(在280 nm、320 nm及350 nm之波長下)及質譜分析。流動相A為在水中之0.1% FA,而流動相B為在乙腈中之0.1% FA。樣品注入後,梯度以在0.1% B下保持5分鐘開始,繼之在75分鐘內線性增加至35% B,以實現最佳肽分離。使用Thermo Scientific Q Exactive Hybrid Quadrupole-Orbitrap質譜儀進行MS及MS/MS實驗,該質譜儀採用高能碰撞解離(HCD)進行用於MS/MS實驗之肽片段化。肽身份分配係基於在全MS譜中的給定肽之經實驗確定之精確質量以及在相應HCD MS/MS譜中之b及y碎片離子。生成來自加載液及流通液之肽之提取離子層析圖(參見圖 10
)。如圖 10
中之提取離子層析圖中所見,展示在來自AEX分離1-5的「AEX加載液」及「AEX FT/洗滌液」中識別之肽片段(如表 2-3
中所識別)。來自肽作圖分析的在圖 10
中識別之此等肽中之一些的相對豐度示於圖 11
中。
參看圖 11
,此圖識別所分析之各種肽片段及其相對氧化水準。具體而言,第三管柱識別經分離及分析之肽片段之胺基酸殘基(「肽序列」)。每個肽序列皆具有加下劃線之胺基酸殘基。加下劃線之胺基酸殘基識別肽序列中經氧化之胺基酸。經氧化之胺基酸對應於組胺酸(H)氧化或色胺酸(W)氧化。此圖中亦描繪每個肽序列右側之列,展示經氧化種類之豐度。列中之陰影表示使用在相應的行標題中識別之不同AEX分離,在特定樣品中之經氧化殘基之相對量的差異。例如,參看圖 11
中之第二肽(EIGLLTCEATVNGHLYK),當以水平方式讀取時,在特定樣品(「阿柏西普AEX加載液」)中經氧化之此肽之相對總群體大致0.013%經氧化。當在同一列上進行時,陰影發生偏移,表明經氧化種類之相對豐度發生改變。例如,使用此相同的肽序列,當遵循不同AEX分離方案時,用於AEX分離之經氧化種類之相對豐度為0.006%至0.010%。因此,可理解,AEX層析降低經氧化種類之豐度。(B) 使用 AEX 來減少 MiniTrap 產生中的顏色形成
進行AEX層析以移除在MT1之產生期間獲得之著色,該著色係在進行使用CDM1表現之全長阿柏西普的切割後獲得。2.5 設計
如表 2-4
中所述,本研究進行4次AEX分離。自MT1之過濾樣品(「MT1過濾池」)獲得AEX加載液。將15.7 mL Capto Q管柱(床高20.0 cm,1.0 cm I.D.)、14.1 mL POROS 50 HQ管柱(床高18.0 cm,1.0 cm I.D.)及16.5 mL Q Sepharose FF管柱(床高21.0 cm,1.0 cm I.D.)整合至用於本實驗之AKTA Avant台式液相層析控制器中。
使用2 M tris鹼或2 M乙酸將AEX加載液之pH值調節至目標±0.05個pH單位。使用5 M氯化鈉或去離子水將AEX加載液之電導率調節至目標±0.1 mS/cm。分析所有彙集樣品之HMW、顏色及產率。表 2-4. AEX 顏色減少研究之研究設計的概述
表 2-5. 用於顏色減少研究之流通 AEX 方案
AEX,陰離子交換層析;CV,管柱體積表 2-6. 用於顏色減少研究之結合及溶析 AEX 方案
AEX,陰離子交換層析;CV,管柱體積2.6 結果
| AEX 分離 | 樹脂 | AEX 方案 |
| 1 | Capto Q | 表2-6 |
| 2 | POROS 50 HQ | 表2-6 |
| 3 | Q Sepharose FF | 表2-6 |
| 4 | POROS 50 HQ | 表2-5 |
| 步驟 | 說明 | 流動相 | 管柱體積 (CV) | 線速度 (cm/h) |
| 1 | 預平衡 | 2 M氯化鈉(NaCl) | 2 | 200 |
| 2 | 平衡 | 50 mM Tris,40 mM NaCl pH 7.90 – 8.10,6.50 – 7.50 mS/cm | 2 | 200 |
| 3 | 加載 | AEX加載液 pH 7.90 – 8.10,6.50 – 7.50 mS/cm | 30 g/L-樹脂 | 200 |
| 4 | 洗滌 | 50 mM Tris,40 mM NaCl pH 7.90 – 8.10,6.50 – 7.50 mS/cm | 2 | 200 |
| 5 | 剝離1 | 2 M氯化鈉(NaCl) | 2 | 200 |
| 6 | 剝離2 | 1 N氫氧化鈉(NaOH) | 2 | 200 |
| 步驟 | 說明 | 流動相 | 管柱體積(CV) | 線速度 (cm/h) |
| 1 | 預平衡 | 2 M氯化鈉(NaCl) | 2 | 200 |
| 2 | 平衡 | 50 mM Tris pH 8.30 – 8.50,1.90 – 2.10 mS/cm | 2 | 200 |
| 3 | 加載 | AEX加載液 pH 8.30 – 8.50,1.90 – 2.10 mS/cm | 30 g/L-樹脂 | 200 |
| 4 | 洗滌 | 50 mM Tris pH 8.30 – 8.50,1.90 – 2.10 mS/cm | 2 | 200 |
| 5 | 溶析 | 50 mM Tris,70 mM NaCl pH 8.30 – 8.50,8.50 – 9.50 mS/cm | 2 | 200 |
| 6 | 剝離1 | 2 M氯化鈉(NaCl) | 2 | 200 |
| 7 | 剝離2 | 1 N氫氧化鈉(NaOH) | 2 | 200 |
如對於AEX分離1-4之流通液及洗滌液之著色(表 2-7
)所見,所有四次AEX分離均導致顏色減少。雖然前三次AEX分離係在結合及溶析模式下評估(表 2-6
),但觀察到大部分產物存在於加載及洗滌區(62%– 94%)中。
前三次分離評估用於Capto Q、POROS 50 HQ及Q Sepharose FF樹脂之pH 8.4及2.0 mS/cm設定點。所有三次分離均具有良好的產率(> 80%)。POROS 50 HQ AEX池展示在AEX池中之最低黃色(b*值為2.09),繼之為Q Sepharose FF AEX池(b*值為2.22)及Capto Q AEX池(b*值為2.55)。表 2-7. AEX 顏色減少研究之實驗結果的概述
AEX,陰離子交換層析;HMW,高分子量種類。
將級分調節至5 g/L之蛋白質濃度以用於顏色量測。2.7 結論
| AEX 分離 | 級分 | 產率 (%) | HMW (%) | 顏色 (L*) | 顏色 (a*) | 顏色 (b*) |
| 1 | FT/洗滌 | 90.7 | 0.49 | 99.11 | -0.27 | 2.55 |
| 2 | FT/洗滌 | 93.8 | 0.33 | 99.20 | -0.28 | 2.09 |
| 3 | FT/洗滌 | 86.7 | 0.23 | 98.88 | -0.23 | 2.22 |
| 4 | FT/洗滌 | 99.5 | 1.13 | 98.90 | -0.39 | 3.40 |
| - | MT1過濾池(AEX加載液) | - | 0.65 | 98.18 | -0.37 | 4.17 |
如對於阿柏西普所見(參見以上部分2.3),發現使用AEX可減少MiniTrap產生之黃棕色著色(表 2-7
)。參看表 2-7
,AEX加載液具有4.17之b*值,但在經歷AEX層析(AEX分離1-4)時,b*值減小,表明黃棕色著色之減少。再者,隨著b*值之減小,著色亦隨之減少。AEX加載液(濃度為5 g/L)之初始b*值可在約0.5至約25之範圍內、更具體在約1.0至約20.0之範圍內、及甚至更具體在約1.5至約15.0之範圍內。使用AEX之後,流通液(濃度為5 g/L)之b*值可在0.5至約10.0之範圍內、更具體在約0.5至約7.0之範圍內、及甚至更具體在約0.5至約5.0之範圍內。實例 3. 氧化肽研究
3.1
肽作圖
樣品製備
。進行還原及烷化MiniTrap (MT1)及MT4 (類似於MT1之MiniTrap,使用採用大豆水解物細胞培養產生的不同全長阿柏西普)樣品之胰蛋白酶作圖,以識別及定量轉譯後修飾。將樣品之等分試樣使用在0.1 M Tris-HCl, pH 7.5中之8.0 M尿素變性,用DTT還原,接著用碘乙醯胺烷化。將經變性、還原及烷化之原料藥首先在37℃下用重組Lys-C (rLys-C)以1:100 (w/w)之酶與受質比率消化30分鐘,用0.1 M Tris-HCl pH 7.5稀釋,以使最終尿素濃度為1.8 M,隨後在37℃下用胰蛋白酶以1:20 (w/w)之酶與受質比率消化2小時,接著在37℃下用PNGase F以1:5 (w/w)之酶受質比率去醣化1小時。藉由使用甲酸(FA)使pH值低於2.0來中止消化。
LC-MS 分析
。將由每個樣品得到的rLys-C/胰蛋白酶肽之20 µg等分試樣藉由以下來分離及分析:使用Waters ACQUITY UPLC CSH C18管柱(130 Å,1.7 µm,2.1×150 mm)進行逆相超高效液相層析(UPLC),繼之隨線PDA偵測(在280 nm、320 nm及350 nm之波長下)及質譜分析。流動相A為在水中之0.1% FA,而流動相B為在乙腈中之0.1% FA。樣品注入後,梯度以在0.1% B下保持5分鐘開始,繼之在75分鐘內線性增加至35% B,以實現最佳肽分離。在Thermo Scientific Q Exactive Hybrid Quadrupole-Orbitrap質譜儀上進行MS及MS/MS實驗,該質譜儀採用高能碰撞解離(HCD)進行用於MS/MS實驗之肽片段化。肽身份分配係基於在全MS譜中的給定肽之經實驗確定之精確質量以及在相應HCD MS/MS譜中之b及y碎片離子。生成氧化肽及相應的天然肽之提取離子層析圖,並對峰面積積分,以計算MT1樣品中一或多個經氧化胺基酸殘基之位點特異性百分比。
肽片段與增加的350 nm吸光度有關
在比較MT1與MT4之胰蛋白酶肽圖後,觀察到MT1上之PTM (圖 12A
展示自20.0至75分鐘溶析之肽的吸光度)。突顯具有變化之UV峰的肽。層析圖之展開視圖示於圖 12B
中,該圖展示自16至30分鐘溶析之肽的吸光度。在MT1與MT4之間的UV吸光度具有強對比之肽為TNYLTH*R、IIW(+4)DSR及IIIW(+132)DSR (*或下劃線表示殘基之氧化)。此外,層析圖之展開視圖示於圖 12C
中,該圖展示自30至75分鐘溶析之肽的吸光度。在MT1與MT4之間的UV吸光度具有強對比之肽為DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17)、TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20)、EIGLLTCEATVNGH*LYK (SEQ ID NO.: 18)及QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19) (*表示殘基之氧化)。肽作圖揭露在VEGF MiniTrap之間的豐度顯著不同的肽之身份。由肽作圖分析識別的肽之相對豐度示於表 3-1
中。MT1 (在CDM中產生)中之2-側氧基-組胺酸之量高於MT4 (在大豆水解物中產生),表明用於表現阿柏西普之培養基可對具有氧化組胺酸或氧化色胺酸之肽之相對豐度具有顯著影響。例如,對於肽QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19),該肽在MT1 (在CDM中產生)中之相對豐度百分比為0.015%,與肽在MT4 (在大豆水解物中產生;其比MT1小約15倍)中之相對豐度百分比形成對比。表 3-1.
| 肽 | 肽修飾之序列 | MT1 | MT4 | 倍數變化 MT1/MT4 |
| EIGLLTCEATVNGHLYK (SEQ ID NO.: 57) | EIGLLTC[+57]EATVNGH[+14]LYK (SEQ ID NO.: 18) | 0.011% | 0.004% | 2.75 |
| QTNTIIDVVLSPSHGIELSVGEK (SEQ ID NO.: 58) | QTNTIIDVVLSPSH[+14]GIELSVGEK (SEQ ID NO.: 19) | 0.015% | 0.001% | 15.00 |
| TELNVGIDFNWEYPSSKHQHK (SEQ ID NO.: 59) | TELNVGIDFNWEYPSSKH[+14]QHK (SEQ ID NO.: 20) | 0.204% | 0.026% | 7.85 |
| DKTHTCPPCPAPELLG (SEQ ID NO.: 60) | DKTH[+14]TC[+57]PPC[+57]PAPELLG (SEQ ID NO.: 17) | 0.115% | 0.018% | 6.39 |
| TNYLTHR (SEQ ID NO.: 61) | TNYLTH[+14]R (SEQ ID NO.: 21) | 0.130% | 0.020% | 6.50 |
顏色及 2- 側氧基 - 組胺酸之定量
。亦展示藉由蛋白酶消化所產生之寡胜肽中的2-側氧基-組胺酸之百分比,如藉由質譜法所量測(表 3-2
)。(將值針對未經修飾之肽進行標準化。) 表3-2 (I)
展示針對以下所觀察到的氧化組胺酸/色胺酸之百分比:MT1批次1之AEX流通液、MT1批次2之AEX流通液及MT1批次3之AEX流通液。表 3-2 (II)
展示針對以下所觀察到的氧化組胺酸/色胺酸之百分比:酸性級分1、酸性級分2及針對MT1批次3進行CEX分離所獲得之主要級分。自此表中可清楚地看出,酸性變異體包含經氧化種類。自表 3-2(I)
中可明顯看出,構成肽/蛋白質之2-側氧基-組胺酸及色胺酸二氧化之%與AEX剝離液相比在AEX流通液中係降低的。顯然,剝離AEX管柱可富集此等經修飾之肽之百分比。例如,經修飾之肽 「EIGLLTC[+57]EATVNGH[+14]LYK (SEQ ID NO.: 18)」之%在AEX流通液(MT1批次1)中為0.013%,而在「AEX剝離液」中為0.080%。由此亦證實AEX管柱捕獲經修飾之肽,從而降低AEX流通液中經修飾之肽之百分比。表 3-2 (I). 2- 側氧基 - 組胺酸 / 色胺酸之百分比
表 3-2 (II). 2- 側氧基 - 組胺酸 / 色胺酸之百分比
| 經修飾之肽 | AEX 剝離液 | AEX 流通液 | ||
| 強烈的黃色 | MT1 批次 1 BY1 , 110 mg/mL | MT1 批次 2 ≤ BY3 , 110 mg/mL | MT1 批次 3 ≤ BY3 , 110 mg/mL | |
| EIGLLTC[+57]EATVNGH[+14]LYK (SEQ ID NO.: 18) | 0.080% | 0.013% | 0.008% | 0.006% |
| QTNTIIDVVLSPSH[+14]GIELSVGEK (SEQ ID NO.: 19) | 0.054% | 0.028% | 0.023% | 0.019% |
| TELNVGIDFNWEYPSSKH[+14]QHK (SEQ ID NO.: 20 ) | 0.235% | 0.085% | 0.049% | 0.049% |
| DKTH[+14]TC[+57]PPC[+57]PAPELLG (SEQ ID NO.: 17) | 0.544% | 0.092% | 0.077% | 0.057% |
| TNYLTH[+14]R (SEQ ID NO.: 21) | 0.089% | 0.022% | 0.011% | 0.010% |
| IIW[+32]DSR (SEQ ID NO.: 28) | 0.738% | 0.252% | 0.198% | 0.298% |
| 經修飾之肽 | CEX 流通液 | ||
| 來自 MT1 批次 3 之酸性級分 1 黃色 | 來自 MT1 批次 3 之酸性級分 2 黃色 | 來自 MT1 批次 3 之主要級分 無顏色 | |
| EIGLLTC[+57]EATVNGH[+14]LYK (SEQ ID NO.: 18) | 0.009% | 0.008% | 0.004% |
| QTNTIIDVVLSPSH[+14]GIELSVGEK (SEQ ID NO.: 19) | 0.013% | 0.015% | 0.006% |
| TELNVGIDFNWEYPSSKH[+14]QHK (SEQ ID NO.: 20 ) | 0.131% | 0.151% | 0.049% |
| DKTH[+14]TC[+57]PPC[+57]PAPELLG (SEQ ID NO.: 17) | 0.117% | 0.132% | 0.068% |
| TNYLTH[+14]R (SEQ ID NO.: 21) | 0.014% | 0.008% | 0.008% |
| IIW[+32]DSR (SEQ ID NO.: 28) | 0.458% | 0.269% | 0.185% |
在表 3-2(II)
中,[+57]表示藉由碘乙醯胺使半胱胺酸烷化將在半胱胺酸上添加羧甲基胺部分,其淨質量與未經修飾之半胱胺酸相比增加了約+57: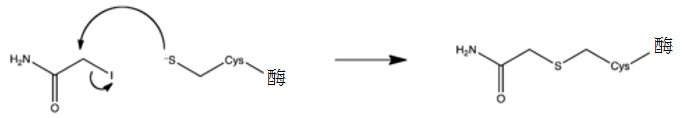
在表 3-2(II)
中,[+14]表示由His轉化為2-側氧基-His,一個氧原子添加至碳2上,但失去兩個氫原子(一個來自碳2,另一個來自氮3),其淨質量與未經修飾之組胺酸相比增加了約+14。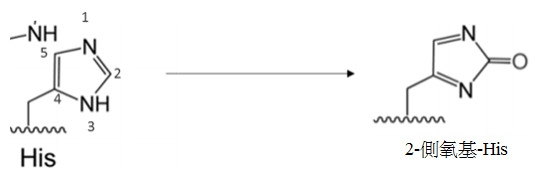
在表 3-2(II)
中,[+32]表示色胺酸二氧化導致形成N-甲醯犬尿胺酸,其淨質量與未經修飾之色胺酸相比增加了約+32 (圖 4
)。
進行第二組實驗以評估藉由AEX層析處理的經蛋白酶消化之FabRICATOR切割之阿柏西普(MT4)的寡胜肽中2-側氧基-組胺酸(及色胺酸二氧化)之百分比(以下圖 13
及表 3-3
)。在經蛋白酶消化之FabRICATOR切割之阿柏西普(MT4)的寡胜肽之AEX剝離液中2-側氧基-組胺酸及色胺酸二氧化之百分比顯著高於AEX流通液中2-側氧基-組胺酸及色胺酸二氧化之百分比(參看下表 3-3
中之「MT1」)。表 3-3. 2- 側氧基 - 組胺酸之百分比
a
使用全長阿柏西普之不同肽計算的值,因為C端肽與MiniTrap不同。
| 經修飾之肽 | 全長阿柏西普 | MT1 | 來自 MT1 之 AEX 剝離液 | AEX 剝離液 /MT1 之變化倍數 |
| IIW[+32]DSR (SEQ ID NO.: 28) | 0.22% | 0.34% | 0.81% | 2.4 |
| EIGLLTC[+57]EATVNGH[+14]LYK (SEQ ID NO.: 18) | 0.00% | 0.02% | 0.08% | 4.0 |
| QTNTIIDVVLSPSH[+14]GIELSVGEK (SEQ ID NO.: 19) | 0.01% | 0.04% | 0.07% | 1.8 |
| TELNVGIDFNWEYPSSKH[+14]QHK (SEQ ID NO.: 20) | 0.01% | 0.19% | 0.42% | 2.2 |
| DKTH[+14]TC[+57]PPC[+57]PAPELLG (SEQ ID NO.: 17) | 0.01%a | 0.11% | 0.63% | 5.7 |
| TNYLTH[+14]R (SEQ ID NO.: 21) | 0.00% | 0.03% | 0.10% | 3.3 |
在MT1產生期間,AEX剝離液中2-側氧基-組胺酸及色胺酸二氧化之百分比顯著高於AEX流通液中2-側氧基-組胺酸及色胺酸二氧化之百分比(參看上表 3-3
中之「MT1」)。與表 3-2
相比,表 3-3
展示相似的結果:剝離AEX管柱所產生之樣品中的2-側氧基-組胺酸及色胺酸二氧化百分比顯著高於AEX流通液中2-側氧基-組胺酸及色胺酸二氧化百分比,表明在分離期間2-側氧基-組胺酸及色胺酸二氧化種類與AEX管柱結合,並在剝離AEX管柱後移除。這在提取離子層析圖中更明顯,如圖 14
中所見。
強陽離子交換層析(CEX)
為了識別包含抗VEGF蛋白之樣品中存在的酸性種類及其他變異體,進行一系列實驗。
使用MonoS (10/100) GL管柱(GE Life Sciences, Marlborough, MA)進行強陽離子交換層析。對於樣品分離,所用流動相為20 mM 2-(N-嗎啉代)乙磺酸(MES) pH 5.7 (流動相A)及40 mM磷酸鈉、100 mM氯化鈉pH 9.0 (流動相B)。將非線性pH梯度用於溶析MT1之電荷變異體,並在280 nm處偵測。在相對滯留時間早於主峰溶析之峰在本文中稱為酸性種類。
在任何富集之前,來自MT1批次2 (≤BY3)之樣品皆使用圖 15
中所示之方法進行CEX。為了降低MT1變異體之複雜性,對樣品進行去唾液酸化處理。繼之進行製備型SEC處理(Superdex 200製備級XK26/100),使用1X DPBS pH 7.2±0.2作為流動相。將得自包含去唾液酸化MiniTrap (dsMT1)之製備型SEC管柱之級分合併,並進一步進行強陽離子交換(SCX)層析,以便使用雙重鹽-pH梯度富集MT1之電荷變異體。程序產生總共7個級分(F1-F7;MC表示方法對照,圖 16
及圖 17
)。
進行CEX時,酸性種類之溶析時間早於主峰,而鹼性種類之溶析時間晚於主峰。如圖 17
中所觀察,峰3-5為主峰。在溶析MT1之主要種類(峰3-5)之前,溶析出峰1及2,且因此包含酸性種類。在溶析MT1之主要種類(峰3-5)之後,溶析出峰6,且因此包含鹼性種類。表 3-4
展示在MC中的峰之相對豐度(如圖 16
中所識別)。例如,表 3-4
之第二列(標記為MC)展示MC中酸性種類之總相對量為約19.8% (亦即峰1 + 峰2)。表 3-4
亦展示每個單獨級分之峰之相對豐度。儘管在不同級分中有重疊種類(如圖 16
及圖 17
所反映),但級分F1及F2之大部分為酸性種類(亦即峰1及峰2)。例如,級分F1包含63.7%峰1及19.2%峰2 (總共82.9%之酸性種類)。級分F2包含9.6%峰1及75.9%峰2 (總共85.5%之酸性種類)。級分F3-F5之大部分為MT1之主要種類(峰3-5)。最後,級分F6-F7之大部分為鹼性種類(峰6),但確實包括一些部分之主要種類(例如峰4及5)。
亦觀察到,級分F1及F2 (包含酸性種類)與級分F3-F5 (包含主要種類或「MT1」)相比具有強烈的黃棕色著色。在濃度≥13 mg/mL下檢查所有級分之顏色。自此實例中明顯看出,樣品中酸性種類之存在係以黃棕色著色之出現來追蹤,該著色之移除(或最小化)可藉由自MT1中移除(或最少化)酸性種類來完成。表 3-4. 基於分析 CEX 的峰之相對豐度
ND:未偵測到
| 樣品 | 峰面積 (%) | |||||
| 峰 1 | 峰 2 | 峰 3 | 峰 4 | 峰 5 | 峰 6 | |
| MC | 5.9 | 13.9 | 15.0 | 25.4 | 20.9 | 19.0 |
| MT1 F1 | 63.7 | 19.2 | 17.1 | ND | ND | ND |
| MT1 F2 | 9.6 | 75.9 | 10.6 | 2.2 | 1.6 | ND |
| MT1 F3 | ND | 5.0 | 57.2 | 37.8 | ND | ND |
| MT1 F4 | ND | ND | 16.3 | 56.3 | 27.4 | ND |
| MT1 F5 | ND | ND | ND | 33.1 | 50.4 | 16.5 |
| MT1 F6 | ND | ND | ND | 16.0 | 27.7 | 56.3 |
| MT1 F7 | 2.8 | 7.7 | 8.0 | 16.1 | 16.5 | 48.9 |
MT1批次2及級分F1至F7之3D層析圖示於圖 18A 至 H
中。MT1批次2未呈現任何顯著的光譜特徵(圖 18A
)。級分1及2 (包含酸性種類)呈現介於320至360 nm之間的光譜特徵(參見圖 18B
中之圓圈)。與級分2相比,此特徵在級分1中更為突出(圖 18B
及圖 18C
),而在級分3及級分4至7 (主要種類,MT1)中則不存在(圖 18D
及圖 18H
),此等級分未呈現黃棕色著色。
因此,如上所觀察,CEX實現酸性種類/酸性級分(級分1及2)之識別,它們與主要種類/級分(級分3至6)相比展示強烈的黃棕色著色。此結果亦以級分F1至F2之3D層析圖中存在而級分F3-F7中不存在的獨特光譜特徵之形式觀察到。
成像毛細管等電聚焦(icIEF)電泳圖
藉由icIEF進一步評估級分F1至7及MC (來自CEX後之MT1-批次2)中之變異體分佈(圖 19
)。
級分F1至7及MC (來自CEX後之MT1-批次2)之變異體分佈係使用具有經碳氟化合物塗覆之毛細管筒(100 μm x 5 cm)的iCE3分析儀(ProteinSimple)藉由icIEF來進一步評估。兩性電解質溶液係由以下各物於純淨水中之混合物組成:0.35%甲基纖維素(MC)、0.75% Pharmalyte 3-10載劑兩性電解質、4.2% Pharmalyte 8-10.5載劑兩性電解質以及0.2% pI標記物7.40及0.15% pI標記物9.77。陽極電解液為80 mM磷酸,而陰極電解液為100 mM氫氧化鈉,均在0.10%甲基纖維素中。將樣品在純淨水中稀釋,並以1:200 (每毫克MT1之唾液酸酶A之單位數)之酶與受質比率將唾液酸酶A添加至每個經稀釋之樣品中,繼之在環境溫度下孵育大約16小時。將經唾液酸酶A處理之樣品與兩性電解質溶液混合,接著藉由引入1500 V之電位持續1分鐘,隨後引入3000 V之電位持續7分鐘來聚焦。經聚焦之MT1變異體之影像係藉由使280 nm紫外光通過毛細管並進入電荷耦合裝置數位相機之鏡頭中而獲得。接著分析此影像以確定各種電荷變異體之分佈。(圖 19
)。參看圖 19
,級分F1及F2 (或酸性級分)展示不存在MT1之峰,該峰在MC及級分F3至F7 (主要種類,MT1)中清楚地觀察到。因此,認為icIEF電泳圖能夠偵測及確定所考慮之蛋白質(在此情況下為MT1)的不同電荷變異體之分佈。因此,很明顯,進行CEX分析時,酸性級分展示(a) 2-側氧基-組胺酸或二側氧基-色胺酸之相對豐度百分比之增加(表 3-2 (II)
);(b)黃棕色著色之增加(數據未顯示);及(c)如級分1及2之3D層析圖中所見的光譜特徵之存在(圖 18B
及圖 18C
)。實例 4. 光誘導研究
在此實例中,藉由使蛋白質樣品暴露於變化量之冷白(CW)螢光或紫外線A (UVA)光來進行VEGF MiniTrap (MT)(例如MT1)之光誘導。測定光暴露之樣品之顏色及經氧化胺基酸含量。如上所述,在暴露後進行LCMS分析。MT暴露於冷白光或UVA光將造成經氧化胺基酸殘基(例如組胺酸)之增加(表 4-1 、表 4-2
及表 4-3
)。表 4-1. 光誘導研究設計
ICH係指ICH協調三方指南(ICH Harmonised Tripartite Guideline):穩定性測試:新原料藥及產品Q1B之光穩定性測試(Photostability Testing of New Drug Substances and Products Q1B),其中規定在不小於120萬lux*小時冷白螢光及近紫外線能量不小於200 W*hr/m2
下進行光穩定性研究。
| 累積暴露 | 0.2 (xICH) | 0.5 (x ICH) | 0.8 (xICH) | 1.0 (xICH)H | 2.0 (xICH) |
| CW 螢光暴露 (lux*hr) | 0.24百萬lux*hr | 0.6百萬lux*hr | 0.96百萬lux*hr | 1.2百萬lux*hr | 2.4百萬lux*hr |
| CW 螢光燈之孵育時間 (8 klux) | 30小時 | 75小時 | 100小時 | 150小時 | 300小時 |
| UVA 暴露 (W*hr/m2 ) | 40 | 100 | 160 | 200 | 400 |
| 在 UVA 下之孵育時間 (10 W/m2 ) | 4小時 | 10小時 | 16小時 | 20小時 | 40小時 |
表 4-2
描述暴露於冷白光及紫外光的MT樣品之著色的增加。例如,樣品(t=0)之b值為9.58。此樣品在暴露於2.4百萬lux*hr下之冷白光時,b值增至22.14。b值之此增加表明使MT暴露於2.4百萬lux*hr下之冷白光與樣品(t=0)相比,增加樣品之黃棕色著色。類似地,在MT樣品(t=0)暴露於400 W*h/m2下之紫外光時,b值自9.58增至10.72。b值之此增加表明使MT樣品暴露於400 W*h/m2下之紫外光與樣品(t=0)相比,增加樣品之黃棕色著色。表 4-2. 暴露於冷白光及紫外光之樣品的著色
樣品顏色係使用CIELAB顏色空間(L*, a*及b*變量)並相對於EP BY顏色標準表示;L* = 白色至黑色(L*為明度);a* = 洋紅色至淺綠色;b* = 黃色至藍色,b值越高,黃色越強。表 4-3 (I). 來自經紫外光應力處理之 MiniTrap 的肽中之 2- 側氧基 -His 水準
表 4-3 (II). 來自經冷白光應力處理之 MiniTrap 的肽中之 2- 側氧基 -His 水準
| 光暴露xICH (lux*hr) | L* | a* | b* | BY 值 |
| 冷白光 | ||||
| T=0 | 97.37 | -1.12 | 9.58 | 4.0 |
| 0.2x (0.24百萬lux*hr) | 96.46 | -0.72 | 11.75 | 3.7 |
| 0.5x (0.6百萬lux*hr) | 95.47 | -0.4 | 11.3 | 3.7 |
| 0.8x (0.96百萬lux*hr) | 95.33 | -0.38 | 11.96 | 3.6 |
| 1.0x (1.2百萬lux*hr) | 94.42 | -0.2 | 13.72 | 3.3 |
| 2.0x (2.4百萬lux*hr) | 92.70 | 0.41 | 22.14 | 2.0 |
| UVA | ||||
| 0.2x (40 W*h/m2 ) | 97.26 | -0.92 | 12.66 | 3.5 |
| 0.5x (100 W*h/m2 ) | 100.39 | -1.01 | 11.83 | 3.7 |
| 0.8x (160 W*h/m2 ) | 79.69 | -0.18 | 10.1 | 3.6 |
| 1.0x (200 W*h/m2 ) | 97.48 | -0.95 | 11.36 | 3.7 |
| 2.0x (400 W*h/m2 ) | 97.76 | -0.98 | 10.72 | 3.8 |
| 肽 | 位點 | t0 | UV_4h | UV_10h | UV_16h | UV_20h | UV_40h |
| DKTHTCPPCPAPELLG (SEQ ID NO.: 17) | H209 | 0.056% | 0.067% | 0.081% | 0.088% | 0.077% | 0.091% |
| EIGLLTCEATVNGHLYK (SEQ ID NO.: 18) | H86 | 0.010% | 0.020% | 0.034% | 0.037% | 0.033% | 0.035% |
| QTNTIIDVVLSPSHGIELSVGEK (SEQ ID NO.: 19) | H110 | 0.024% | 0.031% | 0.028% | 0.028% | 0.027% | 0.027% |
| TELNVGIDFNWEYPSSKHQHK (SEQ ID NO.: 20) | H145 | 0.096% | 0.147% | 0.163% | 0.173% | 0.147% | 0.125% |
| TNYLTHR (SEQ ID NO.: 21) | H95 | 0.014% | 0.032% | 0.044% | 0.056% | 0.058% | 0.078% |
| SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 22) | H19 | 0.007% | 0.013% | 0.021% | 0.025% | 0.024% | 0.034% |
| VHEKDK (SEQ ID NO.: 23) | H203 | 0.040% | 0.105% | 0.238% | 0.255% | 0.269% | 0.324% |
| 肽 | 位點 | t0 | CW_30h | CW_75h | CW_100h | CW_150h | CW_300h |
| DKTHTCPPCPAPELLG (SEQ ID NO.: 17) | H209 | 0.056% | 0.152% | 0.220% | 0.243% | 0.258% | 0.399% |
| EIGLLTCEATVNGHLYK (SEQ ID NO.: 18) | H86 | 0.010% | 0.063% | 0.110% | 0.132% | 0.170% | 0.308% |
| QTNTIIDVVLSPSHGIELSVGEK (SEQ ID NO.: 19) | H110 | 0.024% | 0.085% | 0.120% | 0.128% | 0.148% | 0.180% |
| TELNVGIDFNWEYPSSKHQHK (SEQ ID NO.: 20) | H145 | 0.096% | 0.423% | 0.585% | 0.634% | 0.697% | 0.748% |
| TNYLTHR (SEQ ID NO.: 21) | H95 | 0.014% | 0.103% | 0.175% | 0.198% | 0.267% | 0.437% |
| SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 22) | H19 | 0.007% | 0.025% | 0.043% | 0.049% | 0.058% | 0.115% |
| VHEKDK (SEQ ID NO.: 23) | H203 | 0.040% | 0.426% | 0.542% | 0.622% | 0.702% | 1.309% |
阿柏西普MT於冷白光或UVA光之暴露係以經氧化組胺酸(2-側氧基-his)之出現來追蹤(表 4-3
)。參看表 4-3
,在MT樣品(t=0)中具有側氧基-組胺酸之肽「SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 22)」為0.007%, 而在紫外光下暴露40小時,其豐度增至0.324% (表 4-3(I)
),而在冷白光下暴露300小時,其豐度增至1.309% (表 4-3(II)
)。
觀察到2-側氧基-組胺酸之兩種種類,亦即13.98 Da種類(如圖 2
所示)及15.99 Da種類(如圖 3
所示),其中13.98 Da種類在經光應力處理之MiniTrap樣品中佔優勢。已知15.99 Da種類為銅金屬催化過程之產物。Schöneich, J. Pharm. Biomed Anal. 21:1093-1097 (2000)。此外,13.98 Da種類為光驅動過程之產物。Liu等人, Anal. Chem. 86(10: 4940-4948 (2014))。
類似於暴露於冷白光及UVA光之樣品中的經氧化組胺酸之豐度增加,使MT暴露於冷白光或UVA光亦誘導其他PTM之形成(表 4-4
及表 4-5
)。表 4-4 (I). 在來自經紫外光應力處理之 MiniTrap 的肽中之其他 PTM
表 4-4 (II). 來自經冷白光應力處理之 MiniTrap 的肽中之其他 PTM
表 4-5 (I). 來自經紫外光應力處理之 MiniTrap 的肽中之色胺酸 / 酪胺酸 / 苯丙胺酸之氧化水準
表 4-5 (II). 來自經冷白光應力處理之 MiniTrap 的肽中之色胺酸 / 酪胺酸 / 苯丙胺酸之氧化水準
| 肽 | 位點 | t0 | UV_4h | UV_10h | UV_16h | UV_20h | UV_40h |
| 去醯胺 | |||||||
| EIGLLTCEATVNGHLYK (SEQ ID NO.: 62) | N84 | 20.8% | 21.7% | 21.5% | 21.5% | 22.7% | 22.4% |
| QTNTIIDVVLSPSHGIELSVGEK (SEQ ID NO.: 63) | N99 | 5.3% | 5.4% | 5.5% | 5.4% | 5.5% | 5.6% |
| 氧化 | |||||||
| SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 64) | M10 | 4.5% | 8.2% | 11.1% | 13.3% | 13.8% | 19.3% |
| SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 65) | M20 | 1.1% | 2.0% | 2.8% | 3.4% | 3.4% | 4.6% |
| TQSGSEMK (SEQ ID NO.: 66) | M163 | 2.0% | 2.7% | 4.1% | 4.6% | 7.9% | 8.7% |
| SDQGLYTCAASSGLMTK (SEQ ID NO.: 67) | M192 | 5.4% | 8.1% | 10.9% | 12.1% | 12.5% | 18.3% |
| 3-去氧葡醛酮糖 | |||||||
| SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 68) | R5 | 9.9% | 10.0% | 9.9% | 9.7% | 9.8% | 9.3% |
| 肽 | 位點 | t0 | CW_30h | CW_75h | CW_100h | CW_150h | CW_300h |
| 去醯胺 | |||||||
| EIGLLTC EATVN GHLYK (SEQ ID NO.: 62) | N84 | 20.8% | 22.0% | 22.9% | 20.3% | 21.8% | 21.3% |
| QTN TIIDVVLSPSHGIELSVGEK (SEQ ID NO.: 63) | N99 | 5.3% | 5.6% | 5.2% | 5.6% | 5.5% | 5.8% |
| 氧化 | |||||||
| SDTGRPFVEM YSEIPEIIHMTEGR (SEQ ID NO.: 64) | M10 | 4.5% | 11.7% | 17.3% | 19.9% | 25.1% | 39.7% |
| SDTGRPFVEMYSEIPEIIHM TEGR (SEQ ID NO.: 65) | M20 | 1.1% | 3.1% | 4.3% | 5.1% | 6.1% | 8.2% |
| TQSGSEM K (SEQ ID NO.: 66) | M163 | 2.0% | 3.3% | 15.7% | 11.7% | 26.4% | 20.5% |
| SDQGLYTC AASSGLM TK (SEQ ID NO.: 67) | M192 | 5.4% | 10.7% | 15.3% | 18.7% | 22.8% | 37.6% |
| 3-去氧葡醛酮糖 | |||||||
| SDTGR PFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 68) | R5 | 9.9% | 9.9% | 9.6% | 9.3% | 9.3% | 9.0% |
| 肽 | 修飾 | 位點 | t0 | UV_4h | UV_10h | UV_16h | UV_20h | UV_40h |
| SDQGLYTCAASSGLM TK (SEQ ID NO.: 67) | +4 | W58 | 0.016% | 0.049% | 0.089% | 0.119% | 0.132% | 0.221% |
| +16 | 0.047% | 0.109% | 0.177% | 0.225% | 0.242% | 0.514% | ||
| +32 | 0.200% | 0.487% | 0.415% | 0.481% | 0.423% | 0.498% | ||
| +48 | 0.000% | 0.000% | 0.000% | 0.001% | 0.001% | 0.001% | ||
| TELNVGIDFNW EYPSSK (SEQ ID NO.: 29) | +4 | W138 | 0.435% | 0.462% | 0.550% | 0.557% | 0.502% | 0.512% |
| +16 | 0.083% | 0.100% | 0.161% | 0.206% | 0.239% | 0.448% | ||
| +32 | 0.009% | 0.018% | 0.027% | 0.039% | 0.044% | 0.115% | ||
| +48 | 0.284% | 0.278% | 0.270% | 0.302% | 0.343% | 0.275% | ||
| GFIISNATY K (SEQ ID NO.: 69) | +16 | Y64 | 0.032% | 0.041% | 0.046% | 0.053% | 0.054% | 0.073% |
| KF PLDTLIPDGK (SEQ ID NO.: 70) | +16 | F44 | 0.068% | 0.077% | 0.087% | 0.084% | 0.070% | 0.096% |
| F LSTLTIDGVTR (SEQ ID NO.: 71) | +16 | F166 | 0.066% | 0.075% | 0.085% | 0.089% | 0.089% | 0.124% |
| 肽 | 修飾 | 位點 | t0 | CW_30h | CW_75h | CW_100h | CW_150h | CW_300h |
| SDQGLYTCAASSGLM TK (SEQ ID NO.: 67) | +4 | W58 | 0.016% | 0.063% | 0.124% | 0.161% | 0.228% | 0.526% |
| +16 | 0.047% | 0.129% | 0.227% | 0.283% | 0.377% | 0.795% | ||
| +32 | 0.200% | 1.601% | 2.706% | 3.139% | 3.925% | 6.974% | ||
| +48 | 0.000% | 0.001% | 0.002% | 0.002% | 0.003% | 0.005% | ||
| TELNVGIDFNW EYPSSK (SEQ ID NO.: 29) | +4 | W138 | 0.435% | 0.555% | 0.481% | 0.490% | 0.429% | 0.522% |
| +16 | 0.083% | 0.109% | 0.364% | 0.251% | 0.399% | 0.753% | ||
| +32 | 0.009% | 0.019% | 0.027% | 0.033% | 0.048% | 0.135% | ||
| +48 | 0.284% | 0.284% | 0.330% | 0.308% | 0.347% | 0.316% | ||
| GFIISNATY K (SEQ ID NO.: 69) | +16 | Y64 | 0.032% | 0.043% | 0.057% | 0.063% | 0.078% | 0.127% |
| KF PLDTLIPDGK (SEQ ID NO.: 70) | +16 | F44 | 0.068% | 0.087% | 0.072% | 0.088% | 0.079% | 0.144% |
| F LSTLTIDGVTR (SEQ ID NO.: 71) | +16 | F166 | 0.066% | 0.091% | 0.088% | 0.101% | 0.112% | 0.168% |
因此,MT於冷白光或UVA光之暴露係以經氧化殘基(諸如組胺酸/色胺酸(側氧基-Trp))之出現來追蹤。觀察到側氧基-trp之四種種類:+4 Da、+16 Da、+32 Da及+48 Da。+4 Da種類係由犬尿胺酸之形成來解釋(圖 4
),而16 Da、+32 Da及+48 Da為色胺酸殘基之單氧化、二氧化及三氧化。在320 nm處監測的MT樣品之胰蛋白酶消化物之肽作圖如圖 20
中所示。可在圖 20
中比較構成肽的經氧化殘基之相對存在。例如,對於肽IIW(+4)DSRK而言,可看出在MT樣品t=0、及暴露於UVA 40小時之MT樣品及暴露於CWL 300小時之MT1樣品中,其存在具有顯著差異。
亦評估MT在暴露於冷白光或UVA光下,HMW/低分子量(LMW)種類之存在(表 4-6
)。表 4-6. 在延長的 UVA 及 CWL 應力處理之後產生 HMW/LMW 種類
| 樣品: MT1 , 80 mg/mL , pH 5.8 | ||||||
| 光暴露之樣品 | 深色對照樣品 | 光暴露之樣品 | 深色對照樣品 | 光暴露之樣品 | 深色對照樣品 | |
| 累積 UVA 暴露 (x ICH) | ||||||
| HMW% | 天然% | LMW% | ||||
| t=0 | 2.1 | NA | 96.7 | NA | 1.2 | NA |
| 0.2x ICH (40 W*h/m2) | 2.1 | 2.1 | 96.7 | 96.7 | 1.2 | 1.3 |
| 0.5x ICH (100 W*h/m2) | 11.6 | 2.2 | 86.5 | 96.6 | 1.9 | 1.2 |
| 0.8x ICH (160 W*h/m2) | 14.5 | 2.2 | 83.4 | 96.6 | 2.1 | 1.2 |
| 1.0x ICH (200 W*h/m2) | 15.8 | 2.2 | 81.9 | 96.6 | 2.3 | 1.3 |
| 2.0x ICH (400 W*h/m2) | 22.7 | 2.3 | 74.5 | 96.7 | 2.8 | 1.0 |
| 累積 CWL 暴露 (x ICH) | ||||||
| HMW% | 天然% | LMW% | ||||
| 0.2x ICH (0.24百萬lux* h) | 12.1 | 2.2 | 86.6 | 96.6 | 1.4 | 1.2 |
| 0.5x ICH (0.6百萬lux*hr) | 20.4 | 2.3 | 77.9 | 96.4 | 1.6 | 1.3 |
| 0.8x ICH (0.96百萬lux*hr) | 23.2 | 2.4 | 75.1 | 96.2 | 1.7 | 1.4 |
| 1.0x ICH (1.2百萬lux*hr) | 30.1 | 2.6 | 68.1 | 96.2 | 1.9 | 1.3 |
| 2.0x ICH (2.4百萬lux*hr) | 45.0 | 2.9 | 52.6 | 95.8 | 2.4 | 1.4 |
為了追蹤關於每個樣品之HMW/LMW種類之著色,如上所示對所有應力處理之樣品(CWL及UVA)執行具有全譜PDA偵測之分析型尺寸排阻層析(SEC-PDA)。SEC-PDA對經CWL應力處理之MT的分析揭露,除了LMW種類外的所有尺寸變異體在~350 nm處之吸光度的顯著增加(圖 21
),而對經UVA應力處理之MT的SEC-PDA則未顯示出在~350 nm處之吸光度增加(圖 22
)。與CWL應力處理之樣品不同,經UVA應力處理之樣品不產生任何顯著可量化之黃棕色。
在研究經UVA及CWL應力處理之樣品在320 nm與280 nm處的吸光度比之後,獲得相似的結果。藉由原始強度或總峰面積分析之A320/A280比率在經CWL應力處理之樣品中以黃色強度之增加來追蹤(圖 23
),而A320/A280比率在經UVA應力處理之樣品中不以黃色強度之增加來追蹤(圖 24
)。這證實了先前之觀察結果,即經UVA應力處理之MT1樣品在CWL應力處理後觀察不到相同的黃棕色。實例 5. 減少著色之上游方法 5.1 化學成分確定之培養基的孵育研究
考察摻入包含阿柏西普之新鮮的化學成分確定之培養基(CDM)中的各種成分對於著色之影響。
將一或多個具有10 mL工作體積(新鮮CDM1)之50 mL加透氣蓋(vent-capped)之振盪管孵育7天,分別在第0天及第7天取樣。將阿柏西普樣品(在緩衝水溶液pH 6.2中之阿柏西普重組蛋白,該緩衝水溶液包含5 mM磷酸鈉、5 mM檸檬酸鈉及100 mM氯化鈉)以6 g/L之濃度摻入振盪管中。
添加組分以達到累積濃度:
l 半胱胺酸:16.6 mM
l 鐵:0.23 mM
l 銅:0.0071 mM
l 鋅:0.54 mM
所添加之每種成分對b*值(CIE L*, a*, b*顏色空間)之按比例效應在圖 25A
中列出,並且實際b*值針對預測b*值之繪圖在圖 25B
中列出。半胱胺酸之添加導致最大的黃棕色增加。鐵及鋅亦產生顏色。葉酸及B族維生素(包括硫胺素、菸鹼醯胺、D-泛酸、D-生物素及吡哆醇)增加黃棕色。核黃素及維生素B12在統計學上沒有影響顏色。5.2 減少半胱胺酸及金屬對 b* 值之影響
由產阿柏西普之細胞株的種菌培養物接種生物反應器(例如2 L)。使經接種之培養物在35.5℃之溫度、pH 7.1±0.25及22 ccm之空氣噴射設定點下生長。根據需要向生物反應器中補充葡萄糖、消泡劑及基礎進料。在CDM1中評估表現阿柏西普時降低半胱胺酸及金屬之濃度對顏色之影響。
第0天之培養基 = CDM1,包括1.48 mM之半胱胺酸
l 營養進料:
o 第2天 = 化學成分確定之進料(CDF) + 1.3-2.1 mM半胱胺酸
o 第4天 = CDF + 1.6-1.7 mM半胱胺酸
o 第6天 = CDF + 1.6-1.7 mM半胱胺酸
o 第8天 = CDF + 1.6-1.7 mM半胱胺酸
生物反應器之條件如下:
l 以每升培養物約6-7毫莫耳、每升培養物8-9毫莫耳或每升培養物10-11毫莫耳之累積濃度添加半胱胺酸。
l 下面列出CDM1 (0.5x、1x或1.5x CDM1水準)中1x水準之金屬(其中濃度為接種物添加之前的濃度):
o Fe = 每升培養物68-83微莫耳
o Zn = 每升培養物6-7微莫耳
o Cu = 每升培養物0.1-0.2微莫耳
o Ni = 每升培養物0.5-1微莫耳
累積半胱胺酸水準降至6-7毫莫耳/L會減少黃棕色,其中對力價無明顯影響。培養基中之金屬濃度降至0.5x會減少顏色,其中力價有顯著提高。對力價、VCC (活細胞濃度)、生存力、氨或重量滲透濃度之影響最小(參見圖 26A-E
)。金屬含量及半胱胺酸對b*值及力價之預測的按比例效應在圖 27
中列出。5.3 抗氧化劑對 b* 值之影響的評價
評估摻入包含阿柏西普之廢CDM中的抗氧化劑、牛磺酸、次牛磺酸、硫辛酸、麩胱甘肽、甘胺酸及維生素C對顏色之影響。將一或多個具有10 mL工作體積(CDM1)之50 mL加透氣蓋之振盪管孵育7天,在第0天及第7天取樣。
向廢CDM1中添加組分之條件如下:
l 阿柏西普樣品(在緩衝水溶液pH 6.2中之阿柏西普重組蛋白,其包含5 mM磷酸鈉、5 mM檸檬酸鈉及100 mM氯化鈉)以6 g/L之濃度摻入振盪管中
l 以下列濃度添加至廢CDM1中的抗氧化劑:
o 牛磺酸 = 10 mM之培養物
o 次牛磺酸 = 10 mM之培養物
o ○ 甘胺酸 = 10 mM之培養物
o 硫辛酸 = 0.0024 mM之培養物
o ○ 還原麩胱甘肽 = 2 mM之培養物
o 氫皮質酮 = 0.0014 mM之培養物
o 維生素C(抗壞血酸) = 0.028 mM之培養物
多重抗氧化劑減少廢培養基中之顏色形成:次牛磺酸、牛磺酸及甘胺酸之組合;硫辛酸;以及維生素C。麩胱甘肽增加b*值。表 5-1. 抗氧化劑對 MiniTrap 之顏色形成之影響的概述
*
顯著減小b*值之抗氧化劑:次牛磺酸/牛磺酸/甘胺酸、 硫辛酸、維生素C。
| 條件 | b* 值 |
| 第0天之廢培養基 | 0.37 |
| 第7天之廢培養基對照 | 1.47 |
| 第7天之廢培養基 + 抗氧化劑 * | 1.02 |
各種抗氧化劑對b*值(CIE L*, a*, b*顏色空間)之預測效應的概述在圖 28 (A 至 C)
中列出。
評估摻入包含阿柏西普之廢CDM中的抗氧化劑之進一步添加對顏色之影響。將一或多個具有10 mL工作體積(CDM1)之50 mL加透氣蓋之振盪管孵育7天,在第0天及第7天取樣。
向廢CDM1中添加組分之條件如下:
l 阿柏西普樣品(在緩衝水溶液pH 6.2中之阿柏西普重組蛋白,該緩衝水溶液包含5 mM磷酸鈉、5 mM檸檬酸鈉及100 mM氯化鈉)以6 g/L之濃度摻入振盪管中
l 運行兩個DOE實驗:
l (i) 抗氧化劑以下列濃度添加至廢CDM1中:
o 牛磺酸 = 10 mM之培養物
o 次牛磺酸= 10 mM之培養物
o 甘胺酸 = 10 mM之培養物
o 硫辛酸= 0.0024 mM之培養物
o 維生素C (抗壞血酸) = 0.028 mM之培養物
l (ii) 添加抗氧化劑以達到以下累積濃度:
o ATA = 2.5 µM – 5 µM
o 甲磺酸去鐵胺(DFO) = 5 µM – 10 µM
o 過氧化氫酶= 101.5 mg/L
o S-羧甲基-L-半胱胺酸 = 10 mM
發現次牛磺酸減少廢培養基中之顏色形成(圖 28D
)。DFO亦顯著減少廢培養基中之顏色形成(圖 28D
)。其他抗氧化劑對顏色形成沒有統計學上之影響。表 5-2. 抗氧化劑對 MiniTrap 之顏色形成之影響的概述
振盪瓶抗氧化劑研究:
| 條件 | b* 值 |
| 第0天之廢培養基 | 0.44 |
| 第7天之廢培養基對照 | 1.73 |
| 第7天之廢培養基 +次牛磺酸 | 1.32 |
| 第7天之廢培養基 + DFO | 0.92 |
對牛磺酸、次牛磺酸、甘胺酸、硫辛酸及維生素C關於減少細胞培養物中顏色形成之能力進行單獨及組合評估(表 5-3
)。
由產阿柏西普之細胞株的種菌培養物接種250 mL振盪瓶。使經接種之細胞在35.5℃下在5% CO2
控制下之孵育箱中生長。根據需要向振盪瓶中補充葡萄糖及基礎進料。使用上述製程,其中金屬在CDM1中以0.5x濃度存在,且半胱胺酸以6至7 mM之累積濃度添加。表 5-3.
| 抗氧化劑 | 水準 1 0x | 水準 1 0.5x | 水準 1 1x |
| 牛磺酸 | 0 | 3.75 mM | 7.5 mM |
| 次牛磺酸 | 0 | 3.75 mM | 7.5 mM |
| 甘胺酸 | 2.0 mM | 5.75 mM | 9.5 mM |
| 硫辛酸 | 1.0 µm | 1.9 µm | 2.8 µm |
| 維生素C | 0 | 11.0 µm | 21.0 µm |
圖 28E
展示表 5-3
之抗氧化劑對b*值(CIE L*, a*, b*顏色空間)及最終力價之預測效應。牛磺酸、次牛磺酸、甘胺酸顯著降低b*值,而不會對力價產生負面影響。實例 6. 使用 CDM 產生阿柏西普之醣化及生存力研究
由產阿柏西普之細胞株的種菌培養物接種生物反應器(例如2 L)。使經接種之培養物在35.5℃之溫度、pH 7.1±0.25及22 ccm之空氣噴射設定點下生長。根據需要向生物反應器中補充葡萄糖、消泡劑及基礎進料。阿柏西普蛋白之產生係使用CDM1 (專屬)進行。表現阿柏西普融合蛋白之宿主細胞株之產生係使用CDM 1 (專屬)、CDM 2 (商購獲得)及CDM 3 (商購獲得)進行。使用CDM 1、2及3進行一組實驗,其中沒有額外的培養基組分。使用CDM 1至3 (將錳(四水合氯化錳,Sigma,3.2 mg/L)、半乳糖(Sigma,8 g/L)及尿苷(Sigma,6 g/L)添加至進料中以修飾半乳醣化譜)進行另一組實驗。最後,使用CDM 1至3 (將錳(四水合氯化錳,Sigma,3.2 mg/L)、半乳糖(Sigma,8 g/L)及尿苷(Sigma,6 g/L)添加至進料中以修飾半乳醣化譜,並將地塞米松(Sigma,12 mg/L)添加至進料中以修飾組成物之唾液酸化譜)進行一組實驗。藉由離心,繼之0.45 µm過濾,即可製備出使用每種CDM之經澄清之收穫物。
在N-聚醣分析之前,用蛋白A處理樣品。
力價量測
在整個此等實例中,除非另有說明,否則每天使用Agilent (Santa Clara, CA) 1200系列HPLC或等效物量測阿柏西普力價,其中在低pH值及逐步溶析梯度下操作,在280 nm處偵測。相對於參考標準校準曲線指定濃度。
活細胞密度(VCD)及細胞生存力值
在整個此等實例中,除非另有說明,否則藉由Nova BioProfile Flex自動細胞計數器(Nova Biomedical,Waltham,MA)經由台盼藍排除法量測活細胞密度(VCD)及細胞生存力值。葡萄糖、乳酸鹽、離線pH值、溶解氧(DO)、pCO2量測值及重量滲透濃度係用Nova BioProfile Flex (Nova Biomedical, Waltham, MA)量測。
N-聚醣寡醣譜分析
根據Waters GlycoWorks方案,使用GlycoWorks快速去醣化及GlycoWorks RapiFluor-MS Label套組(分別為Waters零件號186008939及186008091),製備大約15 μg來自CDM 1至3收穫物之經蛋白A處理之樣品以用於N-聚醣分析。藉由在50.5℃下用PNGase-F處理樣品5分鐘,接著在25℃下冷卻5分鐘,自阿柏西普蛋白中移除N-聚醣。經由在室溫下反應5分鐘,用RapiFluor-MS螢光染料標記釋放的聚醣。藉由向反應混合物中添加乙腈使蛋白質沉澱,並藉由在2,204 xg下離心10分鐘沉澱至孔底。收集包含經標記之聚醣的上清液,並在UPLC上使用具有管柱後螢光偵測之親水相互作用液相層析(Waters BEH Amide管柱)進行分析。在結合至管柱後,分離經標記之聚醣,並使用由乙腈及50 mM甲酸銨水溶液(pH 4.4)構成之二元流動相梯度來溶析。使用螢光偵測器偵測經標記之聚醣,該螢光偵測器之激發波長為265 nm,發射波長為425 nm。使用所得層析圖中N-聚醣峰之相對面積百分比,將N-聚醣分佈報告為符合以下條件之N-聚醣之總百分比:(1)含有核心岩藻醣殘基(總岩藻醣化,表 6-1
),(2)含有至少一個唾液酸殘基(總唾液酸化,表 6-2
),(3)識別為甘露糖-5 (甘露糖-5,表 6-3
),(4)含有至少一個半乳糖殘基(總半乳醣化,表 6-4
)及(5)具有已知身份(總識別峰,表 6-5
)。
結果
對於CDM 1至3 (有或無額外組分),活細胞計數(VCC)、生存力及收穫物力價之結果示於圖 29-31
中。
在這九種培養物中,包含尿苷、錳及半乳糖之CDM1培養物在12天時展示最高力價(5.5 g/L)。無額外組分之CDM1在12天時與其他七種培養物相比亦展示高力價(約4.25 g/L)(圖 29
)。
直至處理第6天,在各種條件下之細胞生存力結果均相似。在處理第7天後,有或無額外培養基組分之CDM2及CDM3培養物均展示大於約90%生存力(圖 30
)。
具有尿苷、錳及半乳糖之CDM1培養物在第6天左右展示最高VCC (圖 31
)。
培養物及補充劑對總體N-聚醣分佈有重要影響(表 6-1
至6-5
)。使用由大豆水解物製成之經蛋白A處理之阿柏西普(評估兩個樣品)比較聚醣水準。表 6-5
中列出總識別峰。表 6-1. 總岩藻醣化 (%)
U為尿苷,M為錳,G為半乳糖,Dex為地塞米松表 6-2. 總唾液酸化 (%)
U為尿苷,M為錳,G為半乳糖,Dex為地塞米松表 6-3. 甘露糖 -5 (%)
U為尿苷,M為錳,G為半乳糖,Dex為地塞米松表 6-4. 總半乳醣化 (%)
U為尿苷,M為錳,G為半乳糖,Dex為地塞米松表 6-5. 總識別峰 (%)
U為尿苷,M為錳,G為半乳糖,Dex為地塞米松
| 條件 | 第 6 天 | 第 10 天 | 第 12 天 |
| CDM1 | 48.75 | - | 46.26 |
| CDM1 +UMG | 49.21 | - | 44.38 |
| CDM1 + UMG + Dex | 48.88 | - | 46.23 |
| CDM2 | - | 45.68 | 45.14 |
| CDM2 +UMG | - | 46.36 | 45.27 |
| CDM2 + UMG + Dex | - | 46.92 | - |
| CDM3 | 49.24 | - | 45.59 |
| CDM3 +UMG | 48.71 | - | 42.61 |
| CDM3 + UMG + Dex | 49.36 | - | 44.56 |
| 大豆水解物1 | 51.37 | ||
| 大豆水解物2 | 52.43 |
| 條件 | 第 6 天 | 第 10 天 | 第 12 天 |
| CDM1 | 44.06 | - | 39.14 |
| CDM1 +UMG | 43.72 | - | 35.8 |
| CDM1 + UMG + Dex | 43.2 | - | 36.72 |
| CDM2 | - | 37.62 | 36.67 |
| CDM2 +UMG | - | 37.57 | 36.29 |
| CDM2 + UMG + Dex | - | 38.06 | - |
| CDM3 | 44 | - | 31.21 |
| CDM3 +UMG | 42.48 | - | 30.84 |
| CDM3 + UMG + Dex | 43.82 | - | 32.74 |
| 大豆水解物1 | 58.24 | ||
| 大豆水解物2 | 59.23 |
| 條件 | 第 6 天 | 第 10 天 | 第 12 天 |
| CDM1 | 6.76 | - | 10.1 |
| CDM1 +UMG | 6.9 | - | 13.17 |
| CDM1 + UMG + Dex | 6.23 | - | 8.86 |
| CDM2 | - | 9.71 | 11.96 |
| CDM2 +UMG | - | 9.44 | 10.93 |
| CDM2 + UMG + Dex | - | 8.21 | - |
| CDM3 | 2.31 | - | 12.63 |
| CDM3 +UMG | 2.71 | - | 13.38 |
| CDM3 + UMG + Dex | 2.05 | - | 11.98 |
| 大豆水解物1 | 5.19 | ||
| 大豆水解物2 | 5.24 |
| 條件 | 第6天 | 第10天 | 第12天 |
| CDM1 | 68.44 | - | 62.9 |
| CDM1 +UMG | 69.25 | - | 59.02 |
| CDM1 + UMG + Dex | 69.05 | - | 63.26 |
| CDM2 | - | 65.33 | 63.68 |
| CDM2 +UMG | - | 68.13 | 66 |
| CDM2 + UMG + Dex | - | 69.35 | - |
| CDM3 | 74.57 | - | 62.28 |
| CDM3 +UMG | 74.82 | - | 62.2 |
| CDM3 + UMG + Dex | 76.48 | - | 65.18 |
| 大豆水解物1 | 79.64 | ||
| 大豆水解物2 | 80.55 |
| 條件 | 第 6 天 | 第 10 天 | 第 12 天 |
| CDM1 | 87.28 | - | 84.67 |
| CDM1 +UMG | 88.43 | - | 83.82 |
| CDM1 + UMG + Dex | 87.36 | - | 83.44 |
| CDM2 | - | 86.23 | 86.67 |
| CDM2 +UMG | - | 87.81 | 86.87 |
| CDM2 + UMG + Dex | - | 87.53 | - |
| CDM3 | 86.38 | - | 86.31 |
| CDM3 +UMG | 87.07 | - | 86.13 |
| CDM3 + UMG + Dex | 87.18 | - | 87.43 |
| 大豆水解物1 | 93.93 | ||
| 大豆水解物2 | 94.74 |
在各種CDM之培養物中,在第12天觀察到的總岩藻醣化、總唾液酸化、總半乳醣化及甘露糖-5分別為42.61%至46.26%、30.84%至39.14%、59.02至66%及8.86%至13.38%。此等醣化值不同於使用大豆水解物獲得之醣化值。
最後,對經澄清之收穫物進行顏色量測,此等收穫物係由在補充有尿苷、錳及半乳糖之CDM1、CDM2及CDM3中表現阿柏西普之細胞獲得。生物反應器研究步驟之操作參數將為熟習此項技術之普通技術人員所知。實例 7. 抗 VEGF 蛋白之親和產生
7.1 VEGF MiniTrap之表現
將重組VEGF MiniTrap (例如,MT5,SEQ ID NO.: 46)之編碼區與信號序列可操作地連接,選殖至哺乳動物表現載體中,並轉染至中國倉鼠卵巢(CHO-K1)細胞中;在用400 µg/mL潮黴素選擇12天之後分離經穩定轉染之池。將在化學成分確定的不含蛋白質之培養基中生長的穩定CHO細胞池用於產生蛋白質以進行測試。重組多肽自細胞分泌至生長培養基中。
VEGF MiniTrap之組成域序列
l 人類Flt1 (登錄號NP_001153392.1)
l 人類Flk1 (登錄號NP_002244.1)
l 人類Fc (IGHG1,登錄號P01857-1)
自此製程獲得具有(MT5)之重組VEGF MiniTrap,並進一步處理。
7.2 親和層析管柱之製備
評價能夠與VEGF MiniTrap (MT5)結合之五種不同蛋白質。所用蛋白質包括VEGF165
(SEQ ID NO.: 72)、mAb1 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 73為重鏈,而SEQ ID NO.: 74為輕鏈);mAb2 (小鼠抗VEGFR1 mAb人類IgG1,其中SEQ ID NO.: 75為重鏈,而SEQ ID NO.: 76為輕鏈);mAb3 (小鼠抗VEGF-R1 mAb小鼠IgG1,其中SEQ ID NO.: 77為重鏈,而SEQ ID NO.: 78為輕鏈)及mAb4 (小鼠抗VEGFR1 mAb小鼠IgG1,其中SEQ ID NO.: 79為重鏈,而SEQ ID NO.: 80為輕鏈)。
藉由用6個管柱體積(CV)之1 mM冰冷的鹽酸以不超過1 mL/min之流率洗滌管柱來活化管柱。將10 mg每種蛋白質加載至三個HiTrap NHS活化之HP親和管柱(1 mL,GE Healthcare, 目錄號17-0716-01)上,並封閉管柱以允許偶合在室溫下進行30分鐘。用18個管柱體積之0.5 M乙酸鈉、0.5 M NaCl pH 4.0洗滌管柱,並用18個管柱體積之0.5 M Tris-HCl、0.5 M NaCl pH 8.3封閉開放位點(按以下順序進行洗滌:6個管柱體積之0.5 M Tris-HCl、0.5 M NaCl pH 8.3;6個管柱體積之0.5 M乙酸鈉(乙酸鈉:JT Baker, 目錄號3470-01)、0.5 M NaCl pH 4.0;6個管柱體積之0.5 M Tris pH 8.3;在室溫下孵育管柱30分鐘;6個管柱體積之0.5 M乙酸鈉緩衝液、0.5 M NaCl pH 4.0;6個管柱體積之0.5 M Tris-HCl、0.5 NaCl pH 8.3;以及6個管柱體積之0.5 M乙酸鈉緩衝液、0.5 M NaCl pH 4.0)。將管柱儲存在DPBS pH 7.5中。所評估之五個管柱分別指定為管柱1 (包含VEGF165
)、管柱2 (包含mAb1)、管柱3 (包含mAb2)、管柱4 (包含mAb3)及管柱5 (包含mAb4)。
7.3 使用親和層析產生MiniTrap
樣品製備
。對MiniTrap執行兩種不同產生製程。在一種情況下,使用每個親和管柱產生包含MiniTrap樣品之材料,其中將母體材料(MiniTrap)在1X DPBS緩衝液中稀釋至20 mg/mL並將其施加於管柱上,並在室溫下孵育30分鐘。使用親和管柱,自~7000 ppm之HCP中分離MiniTrap。
替代地,使用在上清液中包含0.4 mg/mL蛋白質之收穫的培養物上清液,並且單獨加載至不同親和管柱(1至5)上。不再進行稀釋。接著將親和管柱用9 CV之1x DPBS緩衝液洗滌,繼之用IgG溶析緩衝液pH 2.8 (Thermo,目錄號21009)溶析。
隨後,將如上所述獲得的MiniTrap材料經由0.45 µm過濾器過濾或離心,之後加載至如以上部分7.2中所述製備之管柱上。將25 mL包含約0.4 mg/mL蛋白質之加載溶液加載至每個管柱上並且孵育20分鐘。用9 CV之DPBS (Invitrogen,目錄號14190-144)洗滌每個管柱,之後溶析以進行平衡。洗滌級分中的MT5之量示於表 7-1
中。洗滌後使用6 CV之pH 2.8 (商用溶析緩衝液(Thermo,目錄號21009))及100 mM甘胺酸緩衝液pH 2.5來溶析,並藉由添加1 M Tris pH 7.5 (Invitrogen目錄號15567-027)迅速中和各級分。溶析級分中的MiniTrap之量亦示於表 7-1
中。
自所有五個親和管柱成功產生MiniTrap (MT5)。與mAb1及mAb2管柱相比,使用VEGF165
之管柱的產率更高。包含人類化抗VEGFR1 mAb之mAb3及mAb4亦展示MT5之成功產生,其產率與mAb1及mAb2相似。在表 7-1
中,基於100%偶聯效率及親和捕獲蛋白與MT5之1:1莫耳比來計算預期產率。表 7-1.
7.4 管柱穩定性研究
| 親和管柱 | 管柱 1 VEGF165 | 管柱 2 mAb1 | 管柱 3 mAb2 | 管柱 4 mAb3 | 管柱 5 mAb4 |
| 偶聯量 (mg) | 10 | 10 | 10 | 10 | 10 |
| 加載液(mg) | 21.2 | 21.2 | 21.2 | 20.1 | 20.1 |
| 預期(mg) | ~12 | ~3.2 | ~3.2 | ~3.2 | ~3.2 |
| 洗滌液(mg) | 14.9 | 13.2 | 12.6 | 15.2 | 14.7 |
| 析出液(mg) | 4.8 | 1.6 | 1.8 | 1.5 | 1.6 |
按照部分7.3中論述之方法,使用管柱1及2進行多次運行(針對管柱1之表 7-2
及針對管柱2之表 7-3
)。表 7-2
表 7-3
| 生產產率 | |||||||
| 運行編號 | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 加載液 (mg) | 21.2 | 19.7 | 19.7 | 19.7 | 19.6 | 19.6 | 19.6 |
| 洗滌液 (mg) | 14.9 | 13.5 | 15.0 | 15.0 | 14.6 | 14.6 | 14.3 |
| 析出液 (mg) | 4.8 | 5.4 | 5.2 | 4.8 | 5.2 | 4.6 | 4.8 |
| 生產產率 | |||||||
| 運行編號 | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 加載液 (mg) | 21.2 | 19.7 | 19.7 | 19.7 | 19.6 | 19.6 | 19.6 |
| 洗滌液 (mg) | 13.2 | 16.0 | 16.4 | 16.4 | 17.1 | 17.2 | 17.2 |
| 析出液 (mg) | 1.6 | 1.8 | 1.8 | 1.8 | 2.0 | 2.0 | 2.0 |
將管柱在4℃下儲存約5週。自每次產生中溶析出相似量之MT5,表明管柱具有良好穩定性。
7.5 所產生之VEGF MiniTrap之穩定性研究
對來自三個管柱(管柱1、管柱2及管柱3)之溶析級分執行SDS-PAGE分析。在非還原及還原SDS-PAGE樣品緩衝液中製備樣品,並使用1X MES (目錄號NP0322,Invitrogen, Carlsbad, CA)在4至12%梯度之NuPage bis-Tris凝膠上運行。
向孔中加載:(1)分子量標準品,(2)加載溶液,(3)來自管柱1之管柱洗滌液,(4)來自管柱2之溶析級分,(5)來自管柱1之溶析級分,(6)來自管柱3之溶析級分,(7)在pH 2.8下儲存1分鐘之MT5,(8)在pH 2.8下儲存30分鐘之MT5,以及(9)分子量標準品(圖 33
及圖 34
)。分析表明,自所有三個親和管柱(管柱1至3)之溶析級分中獲得的級分顯示出相似的尺寸分佈,並且親和管柱之使用不會使MiniTrap不穩定。
7.6 宿主細胞蛋白水準之計算
使用CHO HCP ELISA套組,3G (F550) (Cygus Technologies)獲得宿主細胞蛋白濃度之標準曲線(圖 32
及表 7-4
)。使用如圖 32
所示之標準曲線及表 7-4
中列出之曲線公式計算加載溶液及溶析級分中的HCP之量。表 7-4.
| 曲線公式 | 低漸近線 | 斜率 | EC50 (ng/mL) | 高漸近線 | R2 |
| Y = (A-D)/(1+(X/C)^B) + D | 0.2 | 1.9 | 32.9 | 2.3 | 1 |
使用標準曲線計算總HCP,並且具有宿主細胞蛋白之總量的圖表在圖 35A
中示出。圖 35B
亦展示與來自管柱1、2、4及5之洗滌液及溶析級分相比,加載液中的宿主細胞蛋白之總量。使用管柱進行多次運行並且圖 35B
中之(#)表示評估級分的運行。
使用能夠結合至MiniTrap之蛋白質進行的親和捕獲展示HCP自約7000 ppm有效降至約25-50 ppm。如針對產率所觀察到的,具有VEGF165
之管柱展示來自HCP之MiniTrap純度高於mAb1及mAb2管柱所示的。
7.7 在親和產生之前及之後的VEGF MiniTrap之SEC譜圖
將來自三個管柱(管柱1至3)之溶析級分的SEC譜圖與加載溶液中MiniTrap之SEC譜圖相比較。如圖 36
及表 7-5
中所見,在親和產生之前或之後的MT5之SEC譜圖高度相似。表 7-5.
7.8 VEGF MiniTrap管柱前及管柱後樣品與mAb1、mAb2及VEGF165
結合的動力學
| 如圖 36 中之峰編號 | 滯留時間 | 峰面積 % | 滯留時間 | 峰面積 % | 滯留時間 | 峰面積 % | 滯留時間 | 峰面積 % |
| 加載溶液 | 管柱 1 析出液 | 管柱 2 析出液 | 管柱 3 析出液 | |||||
| 1 | 6.8 | 1.8 | 6.8 | 1.2 | 6.9 | 1.1 | 7.0 | 1.2 |
| 2 | 7.8 | 94.6 | 7.9 | 97.2 | 7.9 | 97.3 | 7.9 | 98.3 |
| 3 | 9.4 | 3.6 | 10.2 | 1.7 | 11.4 | 1.6 | 11.3 | 0.5 |
使用Biacore T200儀器進行動力學研究。
使用Biacore T200儀器,使用即時表面電漿子共振生物感測器測定VEGF165與來自管柱1及2之析出液及加載溶液中之MiniTrap結合的平衡解離常數(KD
值)。所有結合研究均在25℃下在10 mM HEPES、150 mM NaCl、3 mM EDTA及0.05% v/v界面活性劑Tween-20 pH 7.4 (HBS-ET)運行緩衝液中進行。首先藉由與mAb1偶合之胺衍生化Biacore感測器表面,以捕獲MT5。結合研究之草圖表示在圖 37
中示出。
簡而言之,將來自管柱之析出液及加載溶液稀釋至HBS-EP (Biacore)緩衝液中,並以~70 RU之捕獲水準跨固定的蛋白質基質注入。接著以50 µL/min之流率注入VEGF165
。將等價濃度之分析物同時注入未經處理之參考表面上,以充當空白感測圖,用於減去總體折射率背景。在各週期之間,經由以25 µL/min兩次5-min注入10 mM甘胺酸來使感測器晶片表面再生。接著使用BIA評估4.0.1軟體評估所得的實驗結合感測圖,以確定動力學速率參數。將每個樣品之資料集擬合至1:1朗繆爾模型(Langmuir model)。對於此等研究,在Global Fit Analysis方案下分析結合及解離數據,同時選擇局部擬合最大分析物結合能力(RU)或Rmax屬性。在此情況下,軟體計算單個解離常數(kd)、締合常數(ka)及親和常數(Kd)。平衡解離常數為KD
=kd/ka。藉由使用在0.03-2 nM範圍內之不同VEGF165
濃度來確定動力學結合速率、動力學解離速率及總親和力(表 7-6
)。根據動力學速率常數將解離半衰期(t½)計算為:t1/2
= ln(2)/60*Kd。表 7-6
展示在25℃下親和層析生產之前及之後獲得的MT5與VEGF165
之結合動力學參數。
將由親和層析產生的MT5之親和力(KD)、結合速率(ka,M-1s-1)及解離速率(kd)與加載溶液比較,以評估親和層析步驟之效果,顯示來自不同樣品之MT5的動力學沒有變化。VEGF MiniTrap構築體之SPR感測圖示於圖 38
中。表 7-6.
7.9 複數生產週期
| VEGF MiniTrap 樣品 | ka (106 M-1 s-1 ) | kd (10-5 s-1 ) | KD (10-12 M) | t½ (min) | Chi2 (RU2 ) | Rmax (RU) |
| 加載溶液 | 9.44 | 1.74 | 1.84 | 664 | 0.10 | 20 |
| 管柱2析出液 | 8.83 | 1.49 | 1.69 | 775 | 0.17 | 28 |
| 管柱1析出液 | 12.18 | 1.80 | 1.48 | 641 | 0.18 | 19 |
如7.3所示,使用管柱1 (hVEGF165
)及管柱2 (mAb1)進行如藉由步驟7.1所得的收穫物之層析產生。將管柱用於複數層析週期。管柱中之產率沒有由於進行額外的運行而顯著變化,表明管柱保留結合能力(表 7-7
)。表 7-7.
| 生產產率 | ||||||||
| 親和管柱 | 管柱 1 | 管柱 2 | ||||||
| 運行編號 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 |
| 加載液 (mg) | 21.2 | 19.7 | 19.7 | 19.7 | 21.2 | 19.7 | 19.7 | 19.7 |
| 洗滌液 (mg) | 14.9 | 13.5 | 15.0 | 15.0 | 13.2 | 16.0 | 16.4 | 16.4 |
| 析出液 (mg) | 4.8 | 5.4 | 5.2 | 4.8 | 1.6 | 1.8 | 1.8 | 1.8 |
使用7.4中所述之方法獲得管柱1及2之加載溶液、洗滌級分及溶析級分中的HCP計算(圖 39
)。所計算之總HCP展示,重複使用管柱不會降低管柱與MiniTrap結合之能力。
7.10 最佳化親和層析管柱
使用管柱1 (VEGF165
)及管柱2 (mAb1)進行如部分7.1中獲得的收穫物材料之層析產生。為了進行最佳化研究,將14 mg或45 mg而非10 mg之VEGF165或抗VEGF R1 mAb加載至兩個HiTrap NHS活化之HP親和管柱(1 mL,GE Healthcare)上,並封閉管柱以允許偶合在室溫下進行30分鐘。如上文7.2及7.3中所討論,進行管柱製備及包括MiniTrap的收穫物之產生。洗滌及溶析級分中的MT5之量示於表 7-8
中。用14 mg或45 mg (VEGF165
或抗VEGF R1 mAb (mAb1))偶聯量代替10 mg的親和管柱之比較展示兩個管柱之MiniTrap的產率均提高。因此,可藉由最佳化蛋白質與管柱之比率或藉由更改pH值、孵育時間、孵育溫度等來提高偶聯效率,來提高使用所概述方法之管柱產率。表 7-8.
7.11 CEX與親和層析之結合使用
| 親和管柱 | (hVEGF165 ) | ( 小鼠抗 VEGF R1 mAb) | ||
| MW (kDa) | ~ 40 ( 二聚物 ) | 145 | ||
| 偶聯量 (mg) | 10 | 14 | 10 | 45 |
| 加載液 (mg) | 21.2 | 45.5 | 21.2 | 45.5 |
| 洗滌液 (mg) | 14.9 | 36.8 | 13.2 | 29.2 |
| 析出液 (mg) | ~5.0 | 7.6 | ~1.8 | 5.5 |
如上文部分7.3中所討論,使用管柱1產生來自MT5表現之細胞培養物樣品。使所獲得之析出液經歷陽離子交換層析(CEX)管柱(HiTrap Capto S,1 mL)。管柱之操作條件示於表 7-9
中。表 7-9.
| 步驟 | 親和力 | 陽離子交換 (CEX) |
| 管柱 | 親和管柱,1 mL | HiTrap Capto S,1 mL |
| 加載 | MT5 CM2926 | 20 mM乙酸鹽,pH 5.0 (加載/洗滌液1) |
| 洗滌 | 1X DPBS pH 7.2 | 10 mM磷酸鹽,pH 7.0 |
| 溶析 | PierceTM IgG溶析緩衝液 | 50 mM Tris,62.5 mM (NH4 )2 SO4 ,pH 8.5 |
| 再生/剝離 | 10 mM甘胺酸pH 2.5 | 50 mM Tris,1 M (NH4 )2 SO4 ,pH 8.5 |
在原始/起始細胞培養物樣品、親和層析管柱1析出液及CEX析出液中之總HCP分別為約230,000 ng/mL、約9,000 ng/mL及約850 ng/mL。如上所提及,使用Cygnus CHO HCP ELISA套組3G定量測定HCP量。
7.12 使用親和層析產生其他抗VEGF蛋白
評價管柱1以研究其產生其他抗VEGF蛋白之能力。將阿柏西普及具有VEGF結合潛力之scFv片段用於此項研究。產生製程如部分7.3中所討論進行。表 7-10
證明管柱1成功結合並溶析其他抗VEGF蛋白。表 7-10.
實例 8. VEGF MiniTrap 構築體的基於質譜之表徵 材料
。VEGF MiniTrap (MT1)如實例1中所述由阿柏西普產生。VEGF MiniTrap 5 (MT5)如實例7中所述產生。VEGF MiniTrap (MT6)藉由以下方法產生:將重組VEGF MiniTrap (MT5)之編碼區與信號序列可操作地連接並選殖至哺乳動物表現載體中,轉染至中國倉鼠卵巢(CHO-K1)細胞中,並在用400 µg/mL潮黴素選擇12天後分離經穩定轉染之池。將在CDM中生長的穩定CHO細胞池用於產生蛋白質以進行分析。8.1 醣蛋白之去醣化
| 親和管柱 1 | scFv | 阿柏西普 |
| 加載液 (mg) | 10 | 20 |
| 洗滌液 (mg) | 4.5 | 10.6 |
| 析出液 (mg) | 3.6 | 10.2 |
將來自MT1、MT2及MT3之經澄清收穫物的樣品在1% (w/v) RG界面活性劑(RapiGest SF, Waters, Milford, MA)及50 mM HEPES (pH 7.9)之28.8 μL溶液中稀釋或復原至0.52 mg/mL之濃度。將此等溶液經2分鐘加熱至大約95℃,允許冷卻至50℃,並與1.2 μL之PNGase F溶液(GlycoWorks Rapid PNGase F, Waters, Milford, MA)混合。藉由將樣品在50℃下孵育5分鐘來完成去醣化。8.2 HILIC- 螢光 -ESI-MS (MS/MS) 分析
經由HILIC分離組合螢光及質譜偵測對MT1進行分析。僅使用HILIC分析MT2及MT3。使用配備有光電二極體陣列及螢光(FLR)偵測器並與Waters Synapt G2-S質譜儀(MS條件)連接之Waters 2D Acquity UPLC進行層析。將親水相互作用層析(HILIC)分離模式與150 × 2.1 mm, 1.7 μm之Waters UPLC Glycan BEH Amide管柱一起使用。管柱溫度設定為60℃,而自動進樣器溫度設定為5℃。注入體積為50 μL。光電二極體陣列之掃描範圍為190–700 nm。FLR設定為:對於RapiFluor標記之聚醣而言,激發265 nm,發射425 nm;對於醣肽中存在之酪胺酸而言,激發274 nm,發射303 nm。初始流率為0.4 mL/min,其中流動相A包含100 mM甲酸銨(pH 4.4),而流動相B為乙腈。8.3 MS 條件
液相層析/質譜(LC/MS)實驗係使用Waters Synapt G2-S質譜儀進行。掃描範圍針對正離子模式及負離子模式分析為質荷比100-2400。掃描時間為1 s,並且恆定輸注(2 µL/min) glu-血纖維肽B作為校準物(「鎖定質量(lock mass)」)。毛細管電壓設定為2.5 kV,其中源溫度為120℃,去溶劑化溫度為500℃。氮氣噴霧器氣體流量設定為700 l/h。8.4 天然 SEC-MS
將ACQUITY UPLC I類系統(Waters, Milford, MA)與Q Exactive HF混合四極桿Orbitrap質譜儀(Thermo Scientific, Bremen, Germany)耦合,以用於所有隨線SEC-MS分析。將ACQUITY UPLC Protein BEH SEC管柱(200Å, 1.7 µm, 4.6 × 300 mm)設定在30℃下,並用於蛋白質分離。流動相為pH 6.8之100 mM乙酸銨。每次分離為30分鐘,其中流率為0.3 mL/min,且注入量設定為40 μg。以下MS參數用於隨線SEC-nano-ESI-MS資料獲取。每次獲取為25分鐘,在樣品注入後立即開始。將去醣化樣品在3 kV噴霧電壓、200℃毛細管溫度及70 S鏡頭RF位準下以正模式遊離化。源內(In-source) CID設定為75 eV。以15 K解析力獲取全MS掃描,質量範圍介於m/z
2000-8000之間。將100 ms之最大注入時間、3e6之自動增益控制目標值及10次微掃描用於全MS掃描。8.5 肽作圖
用於肽作圖之樣品製備
。藉由添加500 mmol/L二硫蘇糖醇(DTT)至5 mmol/L之最終濃度,繼之在4℃下孵育60分鐘來達成還原。藉由添加500 mmol/L碘乙醯胺(IAM)至10 mmol/L之最終濃度並在黑暗中於4℃下孵育60分鐘來進行烷化。根據製造商說明書,使用Zeba™ Spin 7 K MWCO尺寸排阻去鹽管柱(P/N 89882) (Thermo Scientific, Waltham, MA)將變性緩衝液更換為消化緩衝液 (1 mol/L尿素,在0.1 mol/L Tris pH 7.8中)。將重組豬胰蛋白酶(購自Sigma,目錄號03708985001)以1:18 (酶:樣品)質量比(基於VEGF MiniTrap蛋白濃度,如在緩衝液交換後藉由UV-Vis分光光度法所量測)添加,將VEGF MiniTrap蛋白之濃度調節至0.5 μg/μL並允許在室溫下孵育4小時期間進行消化。消化完成時,以1:1體積比添加在LC-MS級水中之0.1%甲酸。將消化物在-80℃下儲存直至分析。
胰蛋白酶消化物之 LC-MS/MS 分析
。經由自動進樣器將一或多份2.5 μg (10 μL)之肽消化物加載至封閉在設定為40℃之恆溫管柱烘箱中的C18管柱上。樣品在排隊注入時保持在7℃。層析方法係以98%流動相A (於水中0.1%體積分數之甲酸)及2%流動相B (於乙腈中0.1%體積分數之甲酸)開始,其中流率設定在恆定為0.200 mL/min。洗滌10分鐘後,將肽在110分鐘之梯度上溶析,其中流動相B含量以每分鐘0.39%之速率升高,以達到包含45%流動相B之最終組成。在下一次樣品注入之前,將管柱用97%流動相B洗滌15分鐘,接著在98%流動相A中平衡25分鐘。在運行之前1.5分鐘及最後5分鐘將析出液轉移至廢液。藉由在214 nm下之UV吸收,繼之在LTQ Orbitrap Elite或Discovery XL上之質譜法,分析自層析管柱溶析之肽。收集PS 8670及RM 8671樣品之複製肽作圖資料,以便各自包括三個串聯MS (MS/MS)分析及一個僅有MS之分析。MS/MS分析係以資料依賴性模式進行以用於肽識別,其中一個實驗週期由以下組成:300m/z
至2000m/z
之一次全MS掃描,繼之五個順序MS/MS事件,它們係在啟動該週期之MS掃描中以500之最小閾值計數偵測到的第一至第五個最強離子上進行。順序質譜法(MSn
) AGC目標設定為1E4,其中微掃描 = 3。在質心模式下以正常掃描速率使用離子阱來分析MS/MS片段。使用高解析度FTMS分析儀(R
= 30,000)以輪廓圖模式收集全MS掃描,其中全掃描AGC目標為1E6且微掃描= 1。選擇離子以便使用2 Da之隔離寬度進行MS/MS,接著使用35之標準化CID能量、0.25之活化Q值及10毫秒之活化時間,藉由與氦氣之碰撞誘導解離(CID)進行片段化。默認電荷狀態設定在z
= 2。若前驅體離子在30 s內兩次觸發事件,則將資料依賴性質量放置在排除清單中持續45 s;排除質量寬度設定在±1 Da。對未經分配之電荷狀態啟用電荷狀態拒絕。拒絕質量清單包括在以下各處之常見污染物:122.08m/z
、185.94m/z
、355.00m/z
、371.00m/z
、391.00m/z
、413.30m/z
、803.10m/z
、1222.10m/z
、1322.10m/z
、1422.10m/z
、1522.10m/z
、1622.10m/z
、1722.10m/z
、1822.10m/z
及1922.10m/z
。進行僅有MS之分析以生成TIC非還原肽圖及還原圖。8.6 結果
VEGF MiniTrap 構築體之結構
。VEGF MiniTrap MT1、MT5及MT6之結構描繪於圖 40
、圖 41
、圖 43
及圖 44
中。
使用SEC-MS進行的初始質量分析證實去醣化之後在完整蛋白質層面上所有三個分子之身份(圖 42
)。天然SEC-MS分析之總離子層析圖(TIC)證明在約12-13分鐘時偵測到完整的VEGF MiniTrap分子。TIC之低分子量(LMW)區域之擴展顯示在所有三個蛋白質樣品中均存在LMW雜質。
VEGF MiniTrap之反摺積質譜進一步證實它們的身份,並提供資料來闡明主要PTM存在於包含MT1及MT5 (圖 43
)(其為二聚物)及MT6 (圖 44
)(其為單鏈蛋白)之樣品中。
MT1 樣品之分析
。提取由包含MT1之樣品的SEC-MS分析之TIC中識別的LMW種類,以檢查三種不同的LMW雜質- LMW1、LMW2及LMW3 (圖 45A
及圖 45B
)。LMW1種類包含阿柏西普之經截短之種類。LMW2種類包含樣品中存在之Fc雜質,此等雜質係藉由阿柏西普之切割產生,進行該切割以產生MiniTrap。LMW3種類包含可能由MT1 (二聚物)分子上切割之單體。
MT1樣品未顯示FabRICATOR酶之存在,該酶已用於切割阿柏西普以形成MiniTrap蛋白。該酶若存在,則在約11.5及12.5分鐘時偵測到。在MT1樣品之SEC-MS分析期間未偵測到該峰(圖 46
)。
MT5 樣品之分析
。提取由包含MT5之樣品的SEC-MS分析之TIC中識別的LMW種類,以檢查兩種不同的LMW雜質- LMW1及LMW2之存在(圖 47
)。
MT6 樣品之分析
。提取由包含MT1之樣品的SEC-MS分析之TIC中識別的LMW種類,以檢查三種不同的LMW雜質– LMW1、LMW2及LMW3之存在(圖 48
)。LMW2種類包含MT6之片段,其中切割產生帶有G4S連接子之VEGF MiniTrap之片段。LMW5種類包含MT6之片段,其中切割發生在G4S連接子之前或之後。
MT6樣品中之聚醣係在HILIC層析法中使用由Waters and the National Institute for Bioprocessing Research and Training (Dublin, Ireland)率先提出之葡萄糖單位值藉由其質量及溶析順序來識別(圖 49A
及圖 49B
)。
遊離硫醇之定量
。VEGF MiniTrap構築體之半胱胺酸殘基可參與分子內及分子間雙硫鍵之形成,或它們可以遊離硫醇形式存在。肽及蛋白質中硫鍵之存在已顯示會對蛋白質施加構象剛性。硫醇可藉由多種試劑及分離技術來偵測。針對三種VEGF MiniTrap構築體的極低水準之遊離硫醇的分析示於表 8-1
中。表 8-1.
| 位置 | 肽 ( 遊離半胱胺酸之位點 ) | MT1 | MT5 | MT6 |
| VEGF R1 | ELVIPCR (SEQ ID NO.: 81) | <0.1% | <0.1% | <0.1% |
| VEGF R2 | LVLNCTAR (SEQ ID NO.: 82) | 0.3% | 0.3% | 0.3% |
| Fc 鉸鏈 | THTCPPCPAPELLG (SEQ ID NO.: 83) | 0.0% | 0.0% | N/A |
三硫化物定量
。與VEGF MiniTrap構築體之Cys殘基中的遊離硫醇相似,三硫鍵可影響蛋白質之結構。在具有極低遊離硫醇水準之條件下三種VEGF MiniTrap構築體之分析示於表 8-2
中。表 8-2.
| 位置 | 肽 | MT1 | MT5 | MT6 |
| VEGF R1 | ELVIPCR – EIGLLTCEATVNGHLYK (SEQ ID NO.: 84) | 0.1% | <0.1% | 0.1% |
| VEGF R2 | LVLNCTAR - SDQGLYTCAASSGLMTK(K) (SEQ ID NO.: 85) | <0.1% | <0.1% | <0.1% |
| Fc 鉸鏈 | THTCPPCPAPELLG - THTCPPCPAPELL(G) (SEQ ID NO.: 86) | 1.5% | 3.7% | N/A |
鉸鏈區中之鏈內雙硫鍵
。鉸鏈區中的錯配雙硫鍵可對VEGF MiniTrap構築體之結構、功能及穩定性產生影響。對三種VEGF MiniTrap構築體關於在VEGF MiniTrap構築體[THTC*PPC*PAPELLG,C*展示可形成鏈內雙硫鍵之處]之鉸鏈區中的極低或不存在之鏈內雙硫鍵進行的分析示於表 8-3
中。表 8-3.
| 肽 | MT1 | MT5 | MT6 |
| 二硫化物 | <0.1% | <0.1% | N/A |
| 三硫化物 | <0.1% | <0.1% | N/A |
交叉及平行雙硫鍵聯異構物之定量
。對於MT1及MT5 (它們為由鉸鏈區中之平行雙硫鍵連接的二聚物),存在異構物之可能性,其中鉸鏈區中之雙硫鍵可交叉(圖 50
)。
雙硫鍵類型(平行對比交叉)之定量顯示,與作為FabRICATOR消化之分子的MT1相比,MT2重組表現之蛋白在Fc鉸鏈區中具有稍微較高水準之交叉二硫橋(表 8-4
)。表 8-4.
| 二硫化物 | MT1 | MT5 | MT6 |
| 交叉 | 0.2% | 3.9% | N/A |
| 平行 | 99.8% | 96.1% | N/A |
轉譯後修飾
(PTM)
。
表
8-5.
| PTM | 位點 | 經修飾之肽 | MT1 | MT5 | MT6 | |
| 去醯胺 | Asn84 (Asn319) | EIGLLTCEATV N GHLYK (SEQ ID NO.: 87) | 琥珀醯亞胺 | 3.1% | 3.2% | 3.2% |
| Asp/iso Asp | 21.9% | 18.9% | 20.9% | |||
| Asn99 (Asn334) | QT N TIIDVVLSPSHGIELSVGEK (SEQ ID NO.: 88) | 琥珀醯亞胺 | 4.6% | 4.6% | 4.0% | |
| Asp/iso Asp | 0.7% | 0.5% | 0.6% | |||
| 氧化 | Met10 | SDTGRPFVE M YSEIPEIIHMTEGR (SEQ ID NO.: 64) | 1.8% | 2.1% | 2.1% | |
| Met20 | SDTGRPFVEMYSEIPEIIH M TEGR (SEQ ID NO.: 65) | 2.9% | 3.0% | 2.7% | ||
| Met245 | GGGGSGGGGSGGGGSGGGGSGGGGSGGGGSSDTGRPFVE M YSEIPEIIHMTEGR (SEQ ID NO.: 91) | - | - | 1.4% | ||
| Met255 | GGGGSGGGGSGGGGSGGGGSGGGGSGGGGSSDTGRPFVEMYSEIPEIIH M TEGR (SEQ ID NO.: 92) | - | - | 2.7% | ||
| Met163 (Met398) | TQSGSE M K (SEQ ID NO.: 66) | 4.3% | 4.3% | 3.8% | ||
| Met192 (Met427) | SDQGLYTCAASSGL M TK (SEQ ID NO.: 94) | 5.0% | 5.0% | 4.2% | ||
| C 端甘胺酸損失 | Gly211 | THTCPPCPAPELL G (SEQ ID NO.: 95) | 0.1% | 2.0% | - |
對所有三種VEGF MiniTrap構築體中之PTM的評估展示水準相當之PTM (表 8-5
)。在Asn84處觀察到的形成琥珀醯亞胺之去醯胺在約3.1至3.2%之範圍內,並且形成天冬胺酸/異天冬胺酸之去醯胺為18.9- 21.9%。對於所有三種VEGF MiniTrap構築體,觀察到幾個甲硫胺酸殘基(例如,Met10、Met 20m、Met163及Met192)之氧化在約0.7-6.8%之範圍內。與MT1及MT5相比包含連接子之MT6展示在該連接子上的甲硫胺酸殘基(例如Met245及Met255)之額外氧化。MT1及MT5中約0.1%及2.0%之C端甘胺酸(Gly211)發生甘胺酸損失。未觀察到缺少C端甘胺酸之MT6。
高級醣化終產物修飾與離胺酸及精胺酸醣化有關。VEGF MiniTrap構築體之醣化會改變其結構及功能,從而導致抗VEGF活性受損。表 8-6.
| 位點 | PTM | MT1 | MT5 | MT6 |
| Arg5 | 3-去氧葡醛酮糖 | 8.0% | 8.1% | 9.2% |
| 醣化 | 0.1% | 0.1% | 0.1% | |
| 羧甲基化 | 1.5% | 1.4% | 1.4% | |
| Arg153 | 3-去氧葡醛酮糖 | <0.1% | <0.1% | <0.1% |
| Arg96 | 3-去氧葡醛酮糖 | <0.1% | <0.1% | <0.1% |
| Lys62 | 醣化 | 1.1% | 1.1% | 1.3% |
| 羧甲基化 | <0.1% | <0.1% | <0.1% | |
| Lys68 | 醣化 | 0.4% | 0.3% | 0.5% |
| Lys149 | 醣化 | 0.6% | 0.5% | 0.6% |
| 羧甲基化 | <0.1% | <0.1% | <0.1% | |
| Lys185 | 醣化 | <0.1% | <0.1% | <0.1% |
對所有三種VEGF MiniTrap構築體中之修飾的評估展示相當之水準(表 8-6
)。
經修飾之位點
。如按照部分8.4之完整質量分析所闡明,VEGF MiniTrap構築體上的經修飾之位點使用如部分8.5中所說明之還原肽作圖得到證實及定量(表 8-7
)。對於肽序列TNYLTHR之位點T90N91,**表示在截短後天冬醯胺轉化為天冬胺酸,而對於肽序列 QTNTIIDVVLSPSHGIELSVGEK之位點N99T100,*表示胰蛋白酶之高水準的無特異性切割。在評估MT1及MT5期間,發現這兩個截短位點形成LMW種類雜質。僅在具有獨特連接子之MT3上發現M245Y246處之截短,並且該截短係在MT3製備期間出現LMW2種類雜質之原因。表 8-7.
| 位點 | 肽序列 | MT1 | MT5 | MT6 |
| N99T100 | QTNTIIDVVLSPSHGIELSVGEK* (SEQ ID NO.: 96) | 12.6% | 13.2% | 13.6% |
| T90N91 | TNYLTHR** (SEQ ID NO.: 97) | 0.5% | 0.1% | 0.3% |
| M245Y246 | GGGGSGGGGSGGGGSGGGGSGGGGSGGGGSSDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 98) | - | - | 1.8% |
| M10Y11 | SDTGRPFVEMYSEIPEIIHMTEGR (SEQ ID NO.: 99) | 0.2% | 1.5% | 1.7% |
糖位點佔用率之量化
。N-醣化為常見的PTM。表徵位點特異性N-醣化(包括N-聚醣宏觀異質性(醣化位點佔用)及微觀異質性(位點特異性聚醣結構))對於理解醣蛋白之生物合成及功能很重要。醣化程度可視蛋白質之表現方式而變化。對於所有三種VEGF MiniTrap,在N36處之醣化水準係相似的 (表 8-8
及圖 51
)。類似地,對於所有三種VEGF MiniTrap,在N68處之醣化水準亦為相似的(表 8-8
及圖 52
)。對於所有三種VEGF MiniTrap,在N123處之醣化水準亦為相似的(表 8-8
及圖 53
),但在MT1製劑中發現甘露糖-5升高。對於VEGF MiniTrap構築體,與MT1相比,MT5及MT6在Asn196處之醣化程度較低(表 8-8
及圖 54
)。另外,與MT4及MT6製劑相比,MT1製劑中之甘露糖-5亦為升高的。表 8-8.
| 位點 | 肽 | MT1 | MT5 | MT6 |
| N36 | (R)VTSPNITVTLK (SEQ ID NO.: 100) | 98.3% | 98.1% | 99.4% |
| N68 | (K)GFIISNATYK (SEQ ID NO.: 101) | 51.9% | 55.4% | 64.9% |
| N123 | (K)LVLNCTAR (SEQ ID NO.: 102) | 99.9% | 99.4% | 98.4% |
| N196 | (K)NSTFVR (SEQ ID NO.: 103) | 98.6% | 44.5% | 55.1% |
N- 聚醣之分析
。N36處之醣化示於表 8-9
。G2F、G2FS、G2FS2為在所有三種VEGF MiniTrap中所見的主要N-聚醣。對於表 8-10
所示的N68處之醣化,G2F及G2FS為在所有三種VEGF MiniTrap中所見的主要N-聚醣。對於表 8-11
的N123處之醣化,G2F及G2S為在所有三種VEGF MiniTrap中所見的主要N-聚醣,並且與MT5及MT6相比,在MT1中偵測到高水準之甘露糖-5。對於表 8-12
中所示的N196處之醣化,G2、G2S、G2S2為在所有三種VEGF MiniTrap中所見的主要N-聚醣,並且與MT5及MT6相比,在MT1中偵測到高水準之甘露糖-5。表 8-9.
表 8-10.
表 8-11.
表 8-12.
| N36 處之聚醣 | MT1 | MT5 | MT6 |
| G0F-GlcNAc | 2.0% | 1.8% | 1.8% |
| G1F | 3.2% | 1.0% | 1.4% |
| G1F-GlcNAc | 4.8% | 4.6% | 4.9% |
| G1FS-GlcNAc | 3.1% | 3.8% | 3.1% |
| G2F | 17.4% | 15.1% | 19.8% |
| G2F2S | 1.7% | 2.0% | 2.2% |
| G2FS | 34.2% | 31.5% | 31.9% |
| G2FS2 | 20.4% | 25.8% | 19.0% |
| G3FS | 2.3% | 4.0% | 5.5% |
| G3FS2 | 2.6% | 4.7% | 5.0% |
| G3FS3 | 1.1% | 2.4% | 1.9% |
| G1_Man5+Phos | 1.2% | 0.3% | 0.2% |
| Man6+Phos | 5.7% | 2.5% | 2.8% |
| N68 處之聚醣 | MT1 | MT5 | MT6 |
| G0F-GlcNAc | 1.2% | 1.1% | 1.1% |
| G1F | 5.1% | 1.4% | 1.7% |
| G1F-GlcNAc | 3.9% | 3.9% | 4.0% |
| G1FS | 1.2% | 0.4% | 0.4% |
| G1FS1-GlcNAc | 1.2% | 1.6% | 1.4% |
| G2F | 27.4% | 23.6% | 28.6% |
| G2F2S | 2.2% | 3.0% | 3.4% |
| G2FS | 52.4% | 55.2% | 50.2% |
| G2FS2 | 3.9% | 6.9% | 5.8% |
| G3FS | 0.5% | 1.2% | 1.6% |
| G3FS2 | 0.4% | 1.1% | 1.2% |
| N123 處之聚醣 | MT1 | MT5 | MT6 |
| G0-GlcNAc | 3.5% | 3.7% | 3.5% |
| G1-GlcNAc | 6.2% | 6.8% | 6.4% |
| G1S-GlcNAc | 4.1% | 3.5% | 2.8% |
| G2 | 10.6% | 16.7% | 17.1% |
| G2F | 1.5% | 7.2% | 7.0% |
| G2FS | 2.1% | 13.6% | 14.2% |
| G2S | 12.7% | 26.1% | 25.5% |
| G2S2 | 1.3% | 5.0% | 6.6% |
| G1_Man4 | 3.8% | 1.3% | 1.4% |
| G1S_Man4 | 3.9% | 2.1% | 1.8% |
| G1_Man5 | 4.0% | 1.2% | 1.1% |
| G1S_Man5 | 3.2% | 1.4% | 1.4% |
| Man4 | 2.6% | 1.9% | 1.8% |
| Man5 | 35.5% | 4.3% | 3.1% |
| Man6 | 1.1% | 0.1% | 0.1% |
| Man7 | 2.8% | 0.1% | 0.1% |
| N196 處之聚醣 | MT1 | MT5 | MT6 |
| G0-GlcNAc | 1.9% | 1.8% | 1.9% |
| G1 | 4.1% | 3.6% | 4.2% |
| G1-GlcNAc | 1.9% | 2.5% | 2.4% |
| G1S-GlcNAc | 2.9% | 2.6% | 1.8% |
| G2 | 20.7% | 28.2% | 32.1% |
| G2F | 2.0% | 5.1% | 6.0% |
| G2FS | 2.0% | 6.1% | 6.2% |
| G2FS2 | 0.5% | 1.6% | 1.3% |
| G2S | 17.7% | 31.2% | 29.9% |
| G2S2 | 4.4% | 9.7% | 6.7% |
| G3S | 0.1% | 0.7% | 1.0% |
| G1S_Man4 | 1.0% | 0.3% | 0.3% |
| G1_Man5 | 2.3% | 0.5% | 0.5% |
| Man3 | 3.1% | 0.7% | 0.6% |
| Man4 | 2.7% | 0.8% | 0.6% |
| Man5 | 30.4% | 3.6% | 3.4% |
MT3 之連接子處的 O- 聚醣
。評估MT3之GS連接子以研究MT3上之O-聚醣。O-木醣化見於位於MT3 (GGGGS
GGGGS
GGGGS
GGGGS
GGGGS
GGGGS
SDTGRPFVEMYSEIPEIIHMTEGR,加下劃線之絲胺酸殘基經醣化)之GS連接子上的絲胺酸殘基上。O-聚醣之組成示於表 8-13
中。表 8-13.
| 組成 | 質量 | 註解 | 數量 | 水準 |
| 木醣化 | +132.0 |  | 三 | <0.1% |
| 二 | 1.5% | |||
| 單 | 15% | |||
| 木糖 + 半乳糖 | +294.1 |  | 單 | 0.9% |
| 木糖 + 半乳糖 + 唾液酸 | +585.2 |  | 單 | 0.7% |
HILIC-FLR-MS 分析
。如部分8.2中所述,對所有VEGF MiniTrap蛋白進行HILIC-FLR-MS分析。分析表明,MT5及MT6之N-連接聚醣相似,但與針對MT1所獲得之N-連接聚醣不同(圖 55
展示全標度及疊加式層析圖,圖 56
展示全標度及重疊式層析圖,且圖 57
展示全標度、疊加式及標準化層析圖)。
最後,表 8-14
及圖 58A 至 C
中分別列出所有三種VEGF MiniTrap蛋白之醣化百分比以及詳細的聚醣識別及定量。如在所有聚醣分析中所觀察到的,MT5與MT6之醣化譜及甘露糖水準相似,但與MT1不同。表 8-14.
實例 9. 使用上游培養基之生產及顏色定量以及進料製程最佳化 (A) 未最佳化之 CDM ( 對照生物反應器 )
| MT1 | MT2 | MT3 | |
| 岩藻醣化 % | 42.9% | 57.8% | 57.2% |
| 半乳醣化 % | 71.6% | 92.9% | 93.7% |
| 唾液酸化 % | 33.1% | 47.6% | 44.8% |
| 高甘露糖 % | 17.6% | 2.6% | 2.3% |
| 平分 % (% Bisecting) | 1.9% | 0.4% | 0.4% |
使用實例5中所述的MiniTrap之製造。
研究步驟之操作參數係熟習此項技術之普通技術人員已知的。
第0天之培養基 = CDM1,並且包括以下營養素、抗氧化劑及金屬:
l 以8-9 mM之累積濃度添加半胱胺酸
l 下面以1x濃度(此處之濃度係在接種物添加之前)列出起始培養基中之金屬:
o Fe = 每升培養物68-83微莫耳
o Zn = 每升培養物6-7微莫耳
o Cu = 每升培養物0.1-0.2微莫耳
o Ni = 每升培養物0.5-1微莫耳
在收穫MT1時,遵循如圖 59
中所示之生產程序。層析之操作參數係熟習此項技術之普通技術人員已知的。用於親和捕獲(圖 59
之步驟3)、親和流通(圖 59
之步驟5)、AEX (圖 59
之步驟8)及HIC (圖 59
之步驟9)之操作參數概述於表 9-1
中。在親和捕獲及過濾步驟之後,使用實例1.2中概述之程序進行阿伯西普之蛋白水解切割。表 9-1.
| 步驟 | 親和捕獲 | 親和流通 | AEX | HIC |
| 樹脂 | MabSelect SuRe | MabSelect SuRe | POROS 50 HQ | Capto Phenyl HS |
| 加載 | 30 g/L樹脂 | 30 g/L樹脂pH 6.80 – 7.20 | 40 g/L樹脂 pH 8.30 – 8.50,1.90 – 2.10 mS/cm | 100 g/L樹脂pH 4.40 – 4.60 7.50 – 10.50 mS/cm |
| 平衡 | 20 mM磷酸鈉pH 7.10 – 7.30,2.60 – 3.20 mS/cm | 26 mM Tris,16 mM磷酸鈉,18 mM乙酸鹽pH 6.90 – 7.10,2.00 – 4.00 mS/cm | 50 mM Tris pH 8.30 – 8.50,1.90 – 2.10 mS/cm | 40 mM Tris,30 mM檸檬酸鈉,74 mM乙酸鹽pH 4.40 – 4.60,7.50 – 10.50 mS/cm |
| 洗滌1 | 10 mM磷酸鈉,500 mM NaCl pH 7.10 – 7.30,40 – 50 mS/cm | 26 mM Tris,16 mM磷酸鈉,18 mM乙酸鹽pH 6.90 – 7.10,2.00 – 4.00 mS/cm | 50 mM Tris pH 8.30 – 8.50,1.90 – 2.10 mS/cm | 40 mM Tris,30 mM檸檬酸鈉,74 mM乙酸鹽pH 4.40 – 4.60,7.50 – 10.50 mS/cm |
| 洗滌2 | 20 mM磷酸鈉pH 7.10 – 7.30,2.60 – 3.20 mS/cm | N/A | N/A | N/A |
| 溶析 | 40 mM乙酸pH 2.80 – 3.20,0.28 – 0.36 mS/cm | 40 mM乙酸pH 2.80 – 3.20,0.28 – 0.36 mS/cm | N/A | N/A |
| 再生/剝離1 | 500 mM乙酸,pH 2.25 – 2.65,0.90 – 1.25 mS/cm | 500 mM乙酸,pH 2.25 – 2.65,0.90 – 1.25 mS/cm | 2 M氯化鈉(NaCl) | 專屬緩衝液 |
| 再生/剝離2 | N/A | N/A | 1 N氫氧化鈉(NaOH) | N/A |
表 9-2
展示在進行各種層析步驟時獲得的池之顏色定量。使用來自蛋白質濃度為5 g/L之池的樣品進行顏色定量。
親和捕獲池係指在進行親和捕獲步驟(圖 59
之步驟3)時收集之析出液。酶池係指在進行酶切割步驟(圖 59
之步驟4)時收集之流通液。親和流通池係指在進行親和流通步驟(圖 59
之步驟5)時收集之流通液,而親和流通析出液係指在進行親和流通步驟(圖 59
之步驟5)時收集之析出液。AEX池及AEX剝離液係指在進行陰離子交換層析步驟(圖 59
之步驟8)時獲得之流通及剝離級分。HIC池係指在進行疏水相互作用層析步驟(圖 59
之步驟9)時收集之流通液。
如表 9-2
中所見的每個步驟均展示著色減少(如由池之b*值減小所觀察到)。例如,在進行親和流通層析時,流通級分具有2.16之b*值(與由親和捕獲步驟收集之流通液的2.52之b*值相比減小的)。AEX分離後之流通液及洗滌液進一步減少著色,如藉由b*值自2.16降至0.74所觀察到。如所預期的,剝離AEX管柱產生帶有黃棕色之樣品,該顏色比來自AEX分離後之流通液及洗滌液之著色顯著更強,如由b*值可見(8.10對比0.74)。最後,HIC步驟進一步提供著色減少(b*值可針對5 g/L蛋白質濃度進行標準化,該b*值來自在28.5 g/L蛋白質濃度下之HIC池所獲得之b*值)。表 9-2. 各個生產步驟處之樣品的顏色定量
| 樣品 | 濃度 (g/L) | L* | a* | b* |
| 親和捕獲池 | 5.0 ± 0.1 | 98.75 | -0.12 | 2.52 |
| 酶切割池 | 5.0 ± 0.1 | 99.03 | -0.07 | 1.61 |
| 親和流通池 | 5.0 ± 0.1 | 98.95 | -0.08 | 2.16 |
| 親和流通析出液 | 5.0 ± 0.1 | 98.92 | -0.01 | 0.83 |
| AEX池 | 5.0 ± 0.1 | 99.72 | -0.03 | 0.74 |
| AEX 2 M NaCl剝離液 | 5.0 ± 0.1 | 96.25 | -0.42 | 8.10 |
| HIC池 | 28.5 | 98.78 | -0.28 | 3.11 |
(B)
最佳化之
CDM (
低半胱胺酸、低金屬及增加之抗氧化劑生物反應器
)
使用以下方案評估降低之半胱胺酸濃度、降低之金屬濃度及增加之抗氧化劑對著色之影響:
第0天之培養基= CDM1
o 以5-6 mM之累積濃度添加半胱胺酸
l 將抗氧化劑添加至CDM1中,以達到以下累積濃度(其中濃度為接種物添加之前的濃度):
o 牛磺酸 = 10 mM之培養物
o 甘胺酸 = 10 mM之培養物
o 硫辛酸 = 0.0024 mM之培養物
o 維生素C (抗壞血酸) = 0.028 mM之培養物
l 下面以1x水準列出起始培養基中之金屬。
o Fe = 每升培養物68-83微莫耳
o Zn = 每升培養物6-7微莫耳
o Cu = 每升培養物0.1-0.2微莫耳
o Ni = 每升培養物0.5-1微莫耳。
o 所有金屬之減少,包括使用0.25x上文針對培養基所示之濃度。
在收穫MT1樣品時,遵循如圖 59
所示之生產程序。層析之操作參數係熟習此項技術之普通技術人員已知的。表 9-1
中概述用於親和捕獲、親和流通及HIC之操作參數。在親和捕獲及過濾步驟之後,使用實例1.2中概述之程序進行阿伯西普之蛋白水解切割。
表 9-3
展示在進行各種層析步驟時獲得的池之顏色定量。使用來自蛋白質濃度為5 g/L之池的樣品進行顏色定量。如表 9-3
中所示之步驟提供與表 9-2
中之步驟所見類似的生產。表 9-3. MiniTrap 之各個生產步驟處之樣品的顏色定量
| 樣品 | 濃度 (g/L) | L* | a* | b* |
| 親和捕獲池 | 5.0 ± 0.1 | 99.18 | -0.09 | 1.77 |
| 酶切割池 | 5.0 ± 0.1 | 99.44 | -0.06 | 1.17 |
| 親和流通池 | 5.0 ± 0.1 | 99.32 | -0.10 | 1.58 |
| 親和流通析出液 | 5.0 ± 0.1 | 99.74 | -0.05 | 0.60 |
| AEX池 | 5.0 ± 0.1 | 99.63 | -0.07 | 0.50 |
| AEX 2 M NaCl剝離液 | 5.0 ± 0.1 | 97.63 | -0.49 | 6.10 |
| HIC池 | 27.6 | 99.07 | -0.29 | 2.32 |
比較表 9-2
與表 9-3
,很明顯,與「對照生物反應器條件」 (b*值為2.52)相比,「低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件」在親和捕獲池中具有較低的顏色(b*值為1.77)。
預計濃度為160 g/L之MT樣品(使用表 9-2
及表 9-3
中列出之步驟形成MT)對於「低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件」具有13.45之b*值且對於「對照生物反應器條件」具有17.45之b*值。經由最佳化上游培養基及進料,達成23%顏色減少。類似地,預計濃度為110 g/L之MT樣品(使用表 9-2
及表 9-3
中列出之步驟形成MT)對於「低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件」具有9.25之b*值且對於「對照生物反應器條件」具有12之b*值。
為了理解每個生產單元操作如何有助於減少顏色,將每個生產製程中間物之b*值計算為親和捕獲池之顏色百分比(表 9-4
)。表 9-4.
| 樣品 | 濃度(g/L) | b* | Δb* | b* ( 作為親和捕獲池之%) | |
| 對照生物反應器 | 親和捕獲池 | 5.0 ± 0.1 | 2.52 | N/A | 100.0 |
| 酶切割池 | 5.0 ± 0.1 | 1.61 | -0.91 | 63.8 | |
| 親和流通池 | 5.0 ± 0.1 | 2.16 | 0.55 | 85.7 | |
| AEX池 | 5.0 ± 0.1 | 0.74 | -1.42 | 29.4 | |
| HIC池 | 5.0 ± 0.1 | 0.55 | -0.19 | 21.8 | |
| 低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器 | 親和捕獲池 | 5.0 ± 0.1 | 1.77 | N/A | 100.0 |
| 酶切割池 | 5.0 ± 0.1 | 1.17 | -0.60 | 66.1 | |
| 親和流通池 | 5.0 ± 0.1 | 1.58 | 0.41 | 89.2 | |
| AEX池 | 5.0 ± 0.1 | 0.50 | -1.08 | 28.2 | |
| HIC池 | 5.0 ± 0.1 | 0.42 | -0.08 | 23.7 |
AEX單元操作提供最多的顏色減少(b*值變化自1.08至1.42),而HIC單元操作提供一些額外的顏色減少(b*值變化自0.08至0.19)。所評估之單元操作總體上移除親和捕獲池中存在的76.3%至78.2%之顏色。
亦關於以下指標對「對照生物反應器條件」以及「低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件」之各種生產製程中間物之顏色進行研究:藉由蛋白酶消化產生的寡胜肽中之2-側氧基-組胺酸之百分比及側氧基-色胺酸之百分比,如藉由質譜法所量測,如表 9-5
及表 9-6
中分別所示。如實例3中所討論進行肽作圖。
參看表 9-5
,在比較不同生產步驟中池中之組胺酸氧化水準時,很明顯,隨著生產過程之進展,池中形成的MT之組胺酸氧化水準百分比之相對豐度降低。例如,對於「對照生物反應器條件」下之H209,組胺酸氧化水準之百分比對於酶切割池為0.062,且對於AEX流通液,其降至0.029,並且對於HIC池,進一步降至0.020。類似地,對於「低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件」下之H209,組胺酸氧化水準之百分比對於酶切割池為0.039,且對於AEX流通液,其降至0.023,並且對於HIC池,進一步降至0.016。因此,生產策略導致MT中組胺酸氧化水準之百分比的降低。隨著著色之減少,樣品中一些經氧化殘基之存在亦減少。與組胺酸氧化相似,在「對照生物反應器條件」以及「低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件」下的不同生產步驟中,亦對池之色胺酸氧化水準進行追蹤(表 9-6
)。表 9-5
表 9-6.
枚舉之實例
| 級分 | 顏色(b*) | 組胺酸氧化水準(%) | ||||||
| H19 (+14) | H86 (+14) | H95 (+14) | H110 (+14) | H145 (+14) | H209 (+14) | |||
| 對照生物反應器條件 | 酶切割池 | 1.61 | 0.023 | 0.018 | 0.011 | 0.014 | 0.007 | 0.062 |
| 親和流通池(AEX加載液) | 2.16 | 0.030 | 0.027 | 0.018 | 0.015 | 0.011 | 0.067 | |
| 親和流通析出液 | 0.83 | 0.030 | 0.022 | 0.000 | 0.018 | 0.004 | 0.046 | |
| AEX流通液 | 0.74 | 0.026 | 0.025 | 0.013 | 0.016 | 0.010 | 0.029 | |
| AEX 2 M NaCl剝離液 | 8.10 | 0.024 | 0.063 | 0.033 | 0.019 | 0.012 | 0.063 | |
| HIC池 | 0.55 | 0.018 | 0.009 | 0.002 | 0.021 | 0.005 | 0.020 | |
| 低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件 | 酶切割池 | 1.17 | 0.019 | 0.017 | 0.009 | 0.014 | 0.008 | 0.039 |
| 親和流通池(AEX加載液) | 1.58 | 0.026 | 0.025 | 0.013 | 0.014 | 0.010 | 0.043 | |
| 親和流通析出液 | 0.60 | 0.031 | 0.017 | 0.007 | 0.020 | 0.003 | 0.016 | |
| AEX流通液 | 0.50 | 0.020 | 0.022 | 0.009 | 0.014 | 0.010 | 0.023 | |
| AEX 2 M NaCl剝離液 | 6.10 | 0.020 | 0.055 | 0.025 | 0.016 | 0.011 | 0.042 | |
| HIC池 | 0.42 | 0.013 | 0.009 | 0.002 | 0.017 | 0.003 | 0.016 |
| 級分 | 顏色(b*) | 色胺酸氧化水準(%) | |||||||
| W58 (+4) | W58 (+16) | W58 (+32) | W58 (+48) | W138 (+4) | W138 (+16) | W138 (+32) | |||
| 對照生物反應器條件 | 酶切割池 | 1.61 | 0.006 | 0.032 | 0.289 | 0.000 | 0.020 | 1.093 | 0.106 |
| 親和流通池(AEX加載液) | 2.16 | 0.016 | 0.055 | 0.327 | 0.000 | 0.017 | 0.771 | 0.111 | |
| 親和流通析出液 | 0.83 | 0.009 | 0.031 | 0.453 | 0.000 | 0.025 | 1.039 | 0.132 | |
| AEX流通液 | 0.74 | 0.014 | 0.038 | 0.283 | 0.000 | 0.023 | 0.720 | 0.120 | |
| AEX 2 M NaCl剝離液 | 8.10 | 0.043 | 0.089 | 0.462 | 0.000 | 0.031 | 0.620 | 0.175 | |
| HIC池 | 0.55 | 0.037 | 0.126 | 0.413 | 0.000 | 0.020 | 0.656 | 0.274 | |
| 低半胱胺酸、低金屬及增加的抗氧化劑之生物反應器條件 | 酶切割池 | 1.17 | 0.009 | 0.027 | 0.239 | 0.001 | 0.027 | 1.026 | 0.136 |
| 親和流通池(AEX加載液) | 1.58 | 0.013 | 0.045 | 0.284 | 0.000 | 0.021 | 0.628 | 0.107 | |
| 親和流通析出液 | 0.60 | 0.003 | 0.026 | 0.421 | 0.021 | 0.025 | 1.032 | 0.132 | |
| AEX流通液 | 0.50 | 0.011 | 0.031 | 0.235 | 0.000 | 0.022 | 0.676 | 0.102 | |
| AEX 2 M NaCl剝離液 | 6.10 | 0.034 | 0.073 | 0.478 | 0.000 | 0.032 | 0.635 | 0.169 | |
| HIC池 | 0.42 | 0.029 | 0.122 | 0.355 | 0.000 | 0.022 | 0.800 | 0.236 |
下文提供之示例性枚舉實例不旨在限制,而應理解,基於如熟習此項技術者所瞭解的本文中之描述,使用CDM及修改條件以減少或增加蛋白質變異體及黃棕色來產生抗VEGF組成物(包括阿柏西普及VEGF MiniTrap)之另外的變化方案可按照本文中之描述實施。
1. 一種包含側氧-MiniTrap之組成物,其中MiniTrap之一或多個胺基酸殘基經氧化。
2. 如請求項1之組成物,其中該一或多個胺基酸殘基為組胺酸及/或色胺酸。
3. 如請求項1之組成物,其中該側氧-MiniTrap經酶消化,產生一或多種寡胜肽,且其中該一或多種寡胜肽選自由以下組成之群組:SEQ ID NO. 17、SEQ ID NO. 18、SEQ ID NO. 19、SEQ ID NO. 20、SEQ ID NO. 21、SEQ ID NO. 22、SEQ ID NO. 23、SEQ ID NO. 24、SEQ ID NO. 25、SEQ ID NO. 26、SEQ ID NO. 27、SEQ ID NO. 28、SEQ ID NO. 29、SEQ ID NO. 30、SEQ ID NO. 31、SEQ ID NO. 32及其組合。
4. 如請求項3之組成物,其中該酶消化係使用胰蛋白酶進行。
5. 一種產生MiniTrap之經氧化種類的方法,該方法包含使具有MiniTrap之樣品經歷自約0.24百萬lux*hr至約2.4百萬lux*hr之冷白光。
6. 一種產生MiniTrap之經氧化種類的方法,該方法包含:
a. 使包含MiniTrap之樣品經歷自約0.24百萬lux*hr至約2.4百萬lux*hr之冷白光;以及
b. 進行(a)之消化,形成寡胜肽,其中該等寡胜肽包含以下寡胜肽中之一或多者:
DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17)、EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18)、QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19)、TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20)、TNYLTH*R (SEQ ID NO.: 21)、SDTGRPFVEMYSEIPEIIH*MTEGR (SEQ ID NO.: 22)、VH*EKDK (SEQ ID NO.: 23)、SDTGRPFVEM*YSEIPEIIHMTEGR (SEQ ID NO.: 64)、SDTGRPFVEMYSEIPEIIHM*TEGR (SEQ ID NO.: 65)、TQSGSEM*K (SEQ ID NO.: 66)、SDQGLYTC*AASSGLM*TK (SEQ ID NO.: 67)、IIW*DSR/RIIW*DSR/IIW*DSRK (SEQ ID NO.: 28)、TELNVGIDFNW*EYPSSK (SEQ ID NO.: 29)、GFIISNATY*K (SEQ ID NO.: 69)、KF*PLDTLIPDGK (SEQ ID NO.: 70)、F*LSTLTIDGVTR (SEQ ID NO.: 32),其中H*為經氧化為2-側氧基-組胺酸之組胺酸,其中C*為經羧甲基化之半胱胺酸,其中M*為經氧化之甲硫胺酸,其中W*為經氧化之色胺酸,其中Y*為經氧化之酪胺酸,且其中F*為經氧化之苯丙胺酸。
7. 如請求項6之方法,其中MiniTrap之經氧化種類之量在暴露於冷白光之後與未經處理之MiniTrap相比增加了約1.5至約50倍。
8. 如請求項6之方法,其中該消化係使用胰蛋白酶進行。
9. 如請求項6之方法,其中該等寡胜肽係使用質譜法來分析。
10. 如請求項6之方法,其中該冷白光具有約8 klux之強度。
11. 如請求項5或6之方法,其中該MiniTrap經歷約0.24百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之量與未經處理之MiniTrap相比增加了約1.5至約10倍。
12. 如請求項5或6之方法,其中該MiniTrap經歷約0.96百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之量與未經處理之MiniTrap相比增加了約1.5至約20倍。
13. 如請求項5或6之方法,其中該MiniTrap經歷約1.2百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之量與未經處理之MiniTrap相比增加了約1.5至約20倍。
14. 如請求項5或6之方法,其中該MiniTrap經歷約2.4百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之量與未經處理之MiniTrap相比增加了約1.5至約50倍。
15. 一種產生MiniTrap之經氧化種類的方法,該方法包含:
a. 在化學成分確定之培養基(CDM)中培養經基因工程改造以表現阿柏西普之細胞;
b. 使來自該經澄清之收穫物的阿柏西普與蛋白A樹脂結合;
c. 溶析步驟(b)之該阿柏西普並且使該阿柏西普經歷酶切割以移除其Fc域,從而形成具有第一量之MiniTrap之經氧化種類的MiniTrap;
d. 使來自步驟(c)之MiniTrap經歷第二捕獲層析,其中該第二捕獲層析步驟經歷一次或多次洗滌,且其中第一流通級分包含MiniTrap;
e. 使步驟(d)之該第一流通液經歷陰離子交換層析(AEX);以及
f. 洗滌來自步驟(e)之該AEX管柱並且收集第二流通液;以及
g. 使用剝離緩衝液剝離步驟(f)之AEX管柱,其中在該AEX管柱之該剝離之後在該流通液中的該MiniTrap包含第二量之經氧化之MiniTrap。
16. 如請求項15之方法,其中該剝離緩衝液選自包含以下之群組:2 M氯化鈉(NaCl)、1 N氫氧化鈉(NaOH)或其組合。
17. 如請求項15之方法,使來自步驟(c)之該MiniTrap進一步經歷自約0.24百萬lux*hr至約2.4百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之該量增加了約1.5至約50倍。
18. 如請求項15之方法,使來自步驟(c)之該經析出之MiniTrap進一步經歷約0.96百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之該第二量增加了約1.5至約20倍。
19. 如請求項15之方法,使來自步驟(c)之該經析出之MiniTrap進一步經歷約1.2百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之該第二量增加了約1.5至約20倍。
20. 如請求項15之方法,使來自步驟(c)之該經析出之MiniTrap進一步經歷約2.4百萬lux*hr之冷白光且其中MiniTrap之經氧化種類之該第二量增加了約1.5至約50倍。
無
圖1描繪使用一示例性實施例產生之VEGF MiniTrap,其包括VEGFR1 (SEQ ID NO.: 34)、VEGFR2 (SEQ ID NO.: 36)、鉸鏈域片段(SEQ ID NO.: 60)及自阿柏西普切下之Fc片段(SEQ ID NO.: 55)。
圖2描繪所提出的組胺酸氧化為2-側氧基-組胺酸(14 Da)之機制。
圖3描繪所提出的組胺酸氧化為2-側氧基-組胺酸(16 Da)之機制。
圖4描繪所提出的色胺酸氧化為N-甲醯犬尿胺酸及犬尿胺酸之機制。
圖5描繪用於產生阿柏西普之示例性實施例。
圖6描繪用於產生VEGF MiniTrap之示例性實施例。
圖7描繪用於產生阿柏西普之示例性實施例。
圖8描繪用於產生VEGF MiniTrap之示例性實施例。
圖9描繪作為示例性實施例計算得出的BY標準對比所計算之b*值之圖表。
圖10描繪為評估來自蛋白酶消化之AEX加載液及流通液的寡胜肽(包括還原及烷化阿柏西普之片段(SEQ ID NO.: 55))中的2-側氧基-組胺酸及色胺酸之氧化百分比(其中下劃線表示殘基之氧化)而進行的實驗之結果。
圖11描繪由使用來自蛋白酶消化之AEX加載液及流通液的寡胜肽(包括阿柏西普之片段(SEQ ID NO.: 55))進行的肽作圖分析(其中下劃線表示肽序列中殘基之氧化)所識別的肽之相對豐度。
圖12A
描繪在350 nm處MT4及MT1之吸光度對時間(分鐘)之層析圖的全視圖。
圖12B
描繪在350 nm處MT4及MT1 (包括SEQ ID NO.: 21及28)之吸光度對時間(16至30分鐘)之層析圖的展開視圖。
圖12C
描繪在350 nm處MT4及MT1 (包括SEQ ID NO.: 17、18、19、20)之吸光度對時間(30至75分鐘)之層析圖的展開視圖。
圖13描繪為評估來自已藉由AEX層析處理的蛋白酶消化之MT1的寡胜肽及來自已由AEX層析剝離的蛋白酶消化之MT1的寡胜肽(包括SEQ ID NO.: 17、18、19、20、21、28)中的2-側氧基-組胺酸(及色胺酸二氧化)百分比而進行的實驗之結果。
圖14描繪為比較MT1之不同產生批次中存在的酸性種類及在進行強陽離子交換(CEX)層析時獲得的酸性級分而進行的實驗之結果,包括SEQ ID NO.: 17、18、19、20、21、28。
圖15描繪使用強陽離子交換層析富集細胞培養物收穫物樣品中存在的酸性種類及其他變異體之示例性方法。
圖16描繪根據一示例性實施例來自進行強陽離子交換層析之級分。
圖17描繪使用雙重鹽-pH梯度針對經歷CEX之MT1產生(在任何生產程序之前,≤BY3)及針對去唾液酸化MiniTrap (dsMT1)之富集變異體根據一示例性實施例進行的強陽離子交換層析圖。
圖18A
描繪根據示例性實施例藉由強陽離子交換層析進行的針對未分級分離之親本對照之3D層析圖。
圖18B
描繪根據示例性實施例藉由強陽離子交換層析進行的針對MT1級分1 (代表實驗之一些拖尾特徵)之3D層析圖。
圖18C
描繪根據示例性實施例藉由強陽離子交換層析進行的針對MT1級分2特徵之3D層析圖。
圖18D
描繪根據示例性實施例藉由強陽離子交換層析進行的針對MT1級分3特徵之3D層析圖。
圖18E
描繪根據示例性實施例藉由強陽離子交換層析進行的針對MT1級分4特徵之3D層析圖。
圖18F
描繪根據一示例性實施例藉由強陽離子交換層析進行的針對MT1級分5特徵之3D層析圖。
圖18G
描繪根據一示例性實施例藉由強陽離子交換層析進行的針對MT1級分6特徵之3D層析圖。
圖18H
描繪根據一示例性實施例藉由強陽離子交換層析進行的針對MT1級分7特徵之3D層析圖。
圖19描繪根據用於MT1產生之一示例性實施例進行的成像毛細管等電聚焦(icIEF)電泳圖。
圖20描繪MT1冷白光或UVA光之暴露與經氧化之胺基酸殘基的出現(包括SEQ ID NO.: 17、18、19、20、21、28、29及83)相關聯之研究的結果。
圖21描繪根據一示例性實施例具有~350 nm之吸光度(參見,例如圓圈突出顯示~350 nm)的經CWL應力處理之MT1的3D SEC-PDA (與光電二極體陣列偵測耦合之尺寸排阻層析)層析圖,其中A
展示T=0時之層析圖,B
展示在0.5x ICH處之層析圖。C
展示在2.0x ICH處之層析圖,且D
展示由CWL在不同時間間隔下進行應力處理的小瓶(標準化為80 mg/mL)中之MT1影像。
圖22描繪根據示例性實施例具有~350 nm之吸光度(參見,例如圓圈突出顯示~350 nm)的經UVA應力處理之MT1的3D SEC-PDA層析圖,其中A
展示T=0時之層析圖,B
展示在0.5x ICH處之層析圖。C
展示在2.0x ICH處之層析圖,且D
展示由UVA在不同時間間隔下進行應力處理的小瓶(標準化為80 mg/mL)中之MT1影像。
圖23A
描繪自使用CWL進行應力處理之樣品的SEC-PDA層析圖中定量得到的A320/280吸光度比(上圖)且圖23B
描繪使用CWL進行應力處理之樣品中的尺寸變異體之A320/280吸光度比之圖表(下圖),其中根據示例性實施例對樣品進行應力處理。
圖24A
描繪自使用UVA進行應力處理之樣品的SEC-PDA層析圖中定量得到的A320/280吸光度比(上圖)且圖24B
描繪使用UVA進行應力處理之樣品中的尺寸變異體之A320/280吸光度比之圖表(下圖),其中根據一示例性實施例對樣品進行應力處理。
圖25A
描繪將各種組分與阿柏西普一起孵育對顏色產生(CIE L*, a*, b*預測之b值)之影響的按比例估算值;且圖25B
描繪實際b值相對於預測b值之圖。
圖26描繪包含低半胱胺酸及低金屬之CDM對阿柏西普力價(A
)、活細胞濃度(B
)、生存力(C
)、氨(D
)及重量滲透濃度(E
)之影響。
圖27為展示根據一示例性實施例隨著金屬及半胱胺酸濃度之增加/減少,收穫物顏色之預測曲線的圖表(視為第13天b*值)。
圖28 (A-B
)描繪將各種組分與阿柏西普在廢CDM中一起孵育對顏色產生(CIE L*, a*, b*預測之b值)之影響(A
);及對b值的按比例預測之影響的圖(B
)。
圖28C
描繪在振盪瓶培養中將各種組分與阿柏西普在CDM中一起孵育對顏色產生(CIE L*, a*, b*預測之b值)之按比例估算之影響。
圖28D
描繪將次牛磺酸及甲磺酸去鐵胺鹽(DFO)與阿柏西普在廢CDM中一起孵育對顏色產生(CIE L*, a*, b*預測之「b」值)之影響。
圖28E
描繪來自振盪瓶培養的各種組分與阿柏西普個別孵育對顏色產生(CIE L*, a*, b*預測之「b」值)之影響。
圖29為展示在CDM中添加尿苷、錳、半乳糖及地塞米鬆對所產生的阿柏西普之產生力價的影響之圖表。
圖30為展示在CDM中添加尿苷、錳、半乳糖及地塞米鬆對表現阿柏西普之細胞的生存力之影響的圖表,其中產生了阿柏西普。
圖31為展示在CDM中添加尿苷、錳、半乳糖及地塞米鬆對表現阿柏西普之細胞的活細胞計數之影響的圖表,其中產生了阿柏西普。
圖32為展示使用來自Cygnus 3G (F550)之標準宿主細胞蛋白溶液所製備的吸光度對宿主細胞蛋白濃度(ng/mL)之標準曲線的圖表。
圖33為使用非還原SDS-PAGE樣品緩衝液進行的SDS-PAGE分析之影像。
圖34為使用還原SDS-PAGE樣品緩衝液進行的SDS-PAGE分析之影像。
圖35A
為在加載溶液、分別自包含VEGF165
、mAb1及mAb2之親和層析管柱1至3溶析之級分中偵測到的總宿主細胞蛋白之圖表。
圖35B
為在加載溶液、分別自包含VEGF165
、mAb1、mAb3及mAb4之親和層析管柱1、2、4及5溶析之級分中偵測到的總宿主細胞蛋白之圖表。
圖36描繪在進行親和層析產生之前A
及之後B
VEGF MiniTrap之SEC譜。
圖37描繪VEGF MiniTrap至VEGF165
之動力學研究的草圖表示,其中所研究之VEGF MiniTrap構築體來自根據一些示例性實施例進行親和層析產生之前及之後。
圖38描繪VEGF MiniTrap至VEGF165
之動力學研究的SPR感測圖,其中所研究之VEGF MiniTrap構築體來自根據一些示例性實施例進行親和層析產生之前及之後。
圖39為在加載溶液、自重複用於包含VEGF165
、mAb1及mAb2之管柱的親和層析管柱溶析之級分中偵測到的總宿主細胞蛋白之圖表。
圖40描繪根據一示例性實施例的VEGF MiniTrap MT1 (SEQ ID NO.: 46)之結構。
圖41描繪根據一示例性實施例的VEGF MiniTrap MT6 (SEQ ID NO.: 51)之結構。
圖42描繪MT1、MT5及MT6之天然SEC-MS分析的相對吸光度對時間(分鐘)之總離子層析圖(TIC),以及來自TIC之低分子量區域的放大視圖。
圖43描繪MT1及MT5主峰之反摺積質譜,以藉由闡明一些PTM來證實MiniTrap二聚物身份。
圖44描繪MT6主峰之反摺積質譜,以藉由闡明一些PTM來證實單鏈MiniTrap身份。
圖45A
描繪在MT1中之低分子量雜質的相對吸光度對時間(分鐘)之圖表。
圖45B
描繪在MT1中之低分子量雜質的質譜。
圖46描繪MT1之相對吸光度對時間(分鐘)之圖,其顯示不存在用於將阿柏西普切割成MT1之FabRICATOR酶。
圖47描繪MT5中之低分子量雜質的相對吸光度對時間(分鐘)之圖。
圖48描繪MT6中之低分子量雜質的相對吸光度對時間(分鐘)之圖。
圖49A
描繪在對VEGF MiniTrap MT6執行HILIC-UV/MS時獲得的吸光度對時間(分鐘)之圖表,其中該圖表展示在21分鐘時主峰之溶析及在21.5分鐘左右時O-聚醣之溶析。
圖49B
描繪對VEGF MiniTrap MT6執行HILIC-UV/MS時獲得的質譜,其展示在47985.8 Da處之主峰。
圖49C
描繪在進行HILIC-UV/MS時獲得的VEGF MiniTrap MT6之O-聚醣的質譜。
圖50為VEGF MiniTrap二聚物之影像,其中VEGF MiniTrap之鉸鏈區(SEQ ID NO.: 83)中的二硫橋可以平行或交叉。
圖51描繪在MT1、MT5及MT6中在Asn36 (包括SEQ ID NO.: 100)處觀察到的聚醣分佈之相對豐度。
圖52描繪在MT1、MT5及MT6中在Asn68 (包括SEQ ID NO.: 101)處觀察到的聚醣分佈之相對豐度。
圖53描繪在MT1、MT5及MT6中在Asn123 (包括SEQ ID NO.: 102)處觀察到的聚醣分佈之相對豐度。
圖54描繪在MT1、MT5及MT6中在Asn196 (包括SEQ ID NO.: 103)處觀察到的聚醣分佈之相對豐度。
圖55描繪藉由親水相互作用層析(HILIC)與螢光偵測及質譜分析(全標度及疊加式)耦合得到的釋放的N-連接之聚醣分析。
圖56描繪MT1、MT5及MT6之HILIC-FLR層析圖。
圖57描繪藉由HILIC與螢光偵測及質譜分析(全標度、疊加式及標準化)耦合得到的釋放的N-連接之聚醣分析。
圖58A
為來自VEGF MiniTrap樣品MT1、MT5及MT6之詳細聚醣識別及定量的表。
圖58B
為來自VEGF MiniTrap樣品MT1、MT5及MT6之詳細聚醣識別及定量的表。
圖58C
為來自VEGF MiniTrap樣品MT1、MT5及MT6之詳細聚醣識別及定量的表。
圖59描繪根據一示例性實施例製造MiniTrap之示例性生產程序。































































































Claims (20)
- 一種包含側氧阿柏西普(oxo-aflibercept)之組成物,其中阿柏西普之一或多個胺基酸殘基經氧化。
- 如請求項1之組成物,其中該一或多個胺基酸殘基為組胺酸及/或色胺酸。
- 如請求項1之組成物,其中該側氧阿柏西普經酶消化,產生一或多種寡胜肽,且其中該一或多種寡胜肽選自由以下組成之群組:SEQ ID NO. 17、SEQ ID NO. 18、SEQ ID NO. 19、SEQ ID NO. 20、SEQ ID NO. 21、SEQ ID NO. 22、SEQ ID NO. 23、SEQ ID NO. 24、SEQ ID NO. 25、SEQ ID NO. 26、SEQ ID NO. 27、SEQ ID NO. 28、SEQ ID NO. 29、SEQ ID NO. 30、SEQ ID NO. 31、SEQ ID NO. 32及其組合。
- 如請求項3之組成物,其中該酶消化係使用胰蛋白酶進行。
- 一種產生阿柏西普之經氧化種類的方法,該方法包含使具有阿柏西普之樣品經歷自約0.24百萬lux*hr至約2.4百萬lux*hr之冷白光。
- 一種產生阿柏西普之經氧化種類的方法,該方法包含: a. 使包含阿柏西普之樣品經歷自約0.24百萬lux*hr至約2.4百萬lux*hr之冷白光;以及 b. 進行(a)之消化,形成寡胜肽,其中該等寡胜肽包含以下寡胜肽中之一或多者: DKTH*TC*PPC*PAPELLG (SEQ ID NO.: 17)、EIGLLTC*EATVNGH*LYK (SEQ ID NO.: 18)、QTNTIIDVVLSPSH*GIELSVGEK (SEQ ID NO.: 19)、TELNVGIDFNWEYPSSKH*QHK (SEQ ID NO.: 20)、TNYLTH*R (SEQ ID NO.: 21)、SDTGRPFVEMYSEIPEIIH*MTEGR (SEQ ID NO.: 22)、VH*EKDK (SEQ ID NO.: 23)、SDTGRPFVEM*YSEIPEIIHMTEGR (SEQ ID NO.: 64)、SDTGRPFVEMYSEIPEIIHM*TEGR (SEQ ID NO.: 65)、TQSGSEM*K (SEQ ID NO.: 66)、SDQGLYTC*AASSGLM*TK (SEQ ID NO.: 67)、IIW*DSR/RIIW*DSR/IIW*DSRK (SEQ ID NO.: 28)、TELNVGIDFNW*EYPSSK (SEQ ID NO.: 29)、GFIISNATY*K (SEQ ID NO.: 69)、KF*PLDTLIPDGK (SEQ ID NO.: 70)、F*LSTLTIDGVTR (SEQ ID NO.: 32),其中H*為經氧化為2-側氧基-組胺酸之組胺酸,其中C*為經羧甲基化之半胱胺酸,其中M*為經氧化之甲硫胺酸,其中W*為經氧化之色胺酸,其中Y*為經氧化之酪胺酸,且其中F*為經氧化之苯丙胺酸。
- 如請求項6之方法,其中阿柏西普之經氧化種類之量在暴露於冷白光之後與未經處理之阿柏西普相比增加了約1.5至約50倍。
- 如請求項6之方法,其中該消化係使用胰蛋白酶進行。
- 如請求項6之方法,其中該等寡胜肽係使用質譜法來分析。
- 如請求項6之方法,其中該冷白光具有約8 klux之強度。
- 如請求項5或6之方法,其中該阿柏西普經歷約0.24百萬lux*hr之冷白光且其中阿柏西普之經氧化種類之量與未經處理之阿柏西普相比增加了約1.5至約10倍。
- 如請求項5或6之方法,其中該阿柏西普經歷約0.96百萬lux*hr之冷白光且其中阿柏西普之經氧化種類之量與未經處理之阿柏西普相比增加了約1.5至約20倍。
- 如請求項5或6之方法,其中該阿柏西普經歷約1.2百萬lux*hr之冷白光且其中阿柏西普之經氧化種類之量與未經處理之阿柏西普相比增加了約1.5至約20倍。
- 如請求項5或6之方法,其中該阿柏西普經歷約2.4百萬lux*hr之冷白光,其中阿柏西普之經氧化種類之量與未經處理之阿柏西普相比增加了約1.5至約50倍。
- 一種產生阿柏西普之經氧化種類的方法,該方法包含: a. 在化學成分確定之培養基(CDM)中培養經基因工程改造以表現阿柏西普之細胞; b. 使來自該經澄清之收穫物的阿柏西普與蛋白A樹脂結合; c. 溶析步驟(b)之該阿柏西普,其中該經析出之阿柏西普具有第一量之經氧化之寡胜肽; d. 使步驟(c)之該經析出之阿柏西普經歷陰離子交換層析(AEX); e. 洗滌步驟(d)之該AEX管柱;以及 f. 使用剝離緩衝液(stripping buffer)來剝離(stripping)步驟(e)之AEX管柱,其中在該AEX管柱之該剝離後之該流通液中的該阿柏西普包含第二量之經氧化之阿柏西普。
- 如請求項15之方法,其中該剝離緩衝液選自包含以下之群組:2 M氯化鈉(NaCl)、1 N氫氧化鈉(NaOH)或其組合。
- 如請求項15之方法,使來自步驟(c)之該經析出之阿柏西普進一步經歷自約0.24百萬lux*hr至約2.4百萬lux*hr之冷白光且其中當將濃度標準化時,阿柏西普之經氧化種類之該第二量與阿柏西普之經氧化種類之該第一量相比增加了約1.5至約50倍。
- 如請求項15之方法,使來自步驟(c)之該經析出之阿柏西普進一步經歷約0.96百萬lux*hr之冷白光且其中當將濃度標準化時,阿柏西普之經氧化種類之該第二量與阿柏西普之經氧化種類之該第一量相比增加了約1.5至約20倍。
- 如請求項15之方法,使來自步驟(c)之該經析出之阿柏西普進一步經歷約1.2百萬lux*hr之冷白光且其中當將濃度標準化時,阿柏西普之經氧化種類之該第二量與阿柏西普之經氧化種類之該第一量相比增加了約1.5至約20倍。
- 如請求項15之方法,使來自步驟(c)之該經析出之阿柏西普進一步經歷約2.4百萬lux*hr之冷白光且其中當將濃度標準化時,阿柏西普之經氧化種類之該第二量與阿柏西普之經氧化種類之該第一量相比增加了約1.5至約50倍。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962944635P | 2019-12-06 | 2019-12-06 | |
| US62/944,635 | 2019-12-06 | ||
| US202063065012P | 2020-08-13 | 2020-08-13 | |
| US63/065,012 | 2020-08-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| TW202128991A true TW202128991A (zh) | 2021-08-01 |
Family
ID=76210828
Family Applications (6)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109128142A TW202134256A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組成物及其製備方法 |
| TW109128143A TW202122417A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
| TW109128140A TW202128744A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
| TW109128144A TW202128991A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組成物及其製備方法 |
| TW109128139A TW202128743A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
| TW109128145A TW202128745A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109128142A TW202134256A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組成物及其製備方法 |
| TW109128143A TW202122417A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
| TW109128140A TW202128744A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109128139A TW202128743A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
| TW109128145A TW202128745A (zh) | 2019-12-06 | 2020-08-18 | 抗vegf蛋白組合物及其製備方法 |
Country Status (18)
| Country | Link |
|---|---|
| US (31) | US11104715B2 (zh) |
| EP (6) | EP3906303A1 (zh) |
| JP (11) | JP2022548818A (zh) |
| KR (12) | KR20240007293A (zh) |
| CN (6) | CN114401994A (zh) |
| AU (12) | AU2020398547C1 (zh) |
| BR (6) | BR112021025769A2 (zh) |
| CA (4) | CA3172625A1 (zh) |
| CO (6) | CO2021018234A2 (zh) |
| IL (16) | IL288360B2 (zh) |
| MX (9) | MX2021014862A (zh) |
| MY (12) | MY193353A (zh) |
| NZ (6) | NZ781136A (zh) |
| PE (6) | PE20221789A1 (zh) |
| SG (6) | SG11202110972UA (zh) |
| TW (6) | TW202134256A (zh) |
| WO (7) | WO2021112927A1 (zh) |
| ZA (3) | ZA202107647B (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112021025769A2 (pt) * | 2019-12-06 | 2022-04-12 | Regeneron Pharma | Composições de proteína anti-vegf e métodos para a produção das mesmas |
| WO2022234412A1 (en) | 2021-05-03 | 2022-11-10 | Lupin Limited | A process for purification of fc-fusion proteins |
| WO2023039457A1 (en) * | 2021-09-08 | 2023-03-16 | Regeneron Pharmaceuticals, Inc. | A high-throughput and mass-spectrometry-based method for quantitating antibodies and other fc-containing proteins |
| WO2023137143A1 (en) * | 2022-01-14 | 2023-07-20 | Shattuck Labs, Inc. | Methods of contaminant removal from protein isolates |
| WO2023230486A2 (en) * | 2022-05-23 | 2023-11-30 | Inhalon Biopharma, Inc. | Compositions and methods for inhalable therapeutics |
Family Cites Families (131)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1522343A (en) | 1923-05-02 | 1925-01-06 | Thom Clarence | Magnetic separator |
| US4534972A (en) | 1983-03-29 | 1985-08-13 | Miles Laboratories, Inc. | Protein compositions substantially free from infectious agents |
| DE3871206D1 (de) | 1987-03-24 | 1992-06-25 | Grace W R & Co | Basisnaehrmedium fuer eine zellkultur. |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| GB9022545D0 (en) | 1990-10-17 | 1990-11-28 | Wellcome Found | Culture medium |
| US20030108545A1 (en) | 1994-02-10 | 2003-06-12 | Patricia Rockwell | Combination methods of inhibiting tumor growth with a vascular endothelial growth factor receptor antagonist |
| AU3374795A (en) * | 1994-08-29 | 1996-03-22 | Prizm Pharmaceuticals, Inc. | Conjugates of vascular endothelial growth factor with targeted agents |
| US5705364A (en) | 1995-06-06 | 1998-01-06 | Genentech, Inc. | Mammalian cell culture process |
| US5721121A (en) | 1995-06-06 | 1998-02-24 | Genentech, Inc. | Mammalian cell culture process for producing a tumor necrosis factor receptor immunoglobulin chimeric protein |
| US6100071A (en) | 1996-05-07 | 2000-08-08 | Genentech, Inc. | Receptors as novel inhibitors of vascular endothelial growth factor activity and processes for their production |
| EP1482031B1 (en) | 1996-08-30 | 2015-10-28 | Life Technologies Corporation | Serum-free mammalian cell culture medium, and uses thereof |
| US5804420A (en) | 1997-04-18 | 1998-09-08 | Bayer Corporation | Preparation of recombinant Factor VIII in a protein free medium |
| US6475725B1 (en) | 1997-06-20 | 2002-11-05 | Baxter Aktiengesellschaft | Recombinant cell clones having increased stability and methods of making and using the same |
| SE519827C2 (sv) | 1998-03-30 | 2003-04-15 | Viranative Ab | Näringsmedium innehållande metionin samt användning av detta |
| US6833349B2 (en) | 1999-06-08 | 2004-12-21 | Regeneron Pharmaceuticals, Inc. | Methods of treating inflammatory skin diseases |
| US7070959B1 (en) | 1999-06-08 | 2006-07-04 | Regeneron Pharmaceuticals, Inc. | Modified chimeric polypeptides with improved pharmacokinetic properties |
| US7396664B2 (en) | 1999-06-08 | 2008-07-08 | Regeneron Pharmaceuticals, Inc. | VEGF-binding fusion proteins and nucleic acids encoding the same |
| US7303746B2 (en) | 1999-06-08 | 2007-12-04 | Regeneron Pharmaceuticals, Inc. | Methods of treating eye disorders with modified chimeric polypeptides |
| US7306799B2 (en) | 1999-06-08 | 2007-12-11 | Regeneron Pharmaceuticals, Inc. | Use of VEGF inhibitors for treatment of eye disorders |
| IL146890A0 (en) * | 1999-06-08 | 2002-08-14 | Regeneron Pharma | Modified chimeric polypeptides with improved pharmacokinetic properties |
| US7087411B2 (en) * | 1999-06-08 | 2006-08-08 | Regeneron Pharmaceuticals, Inc. | Fusion protein capable of binding VEGF |
| AU2669501A (en) * | 1999-11-30 | 2001-06-12 | Smithkline Beecham Plc | New use |
| WO2001092337A2 (en) | 2000-05-26 | 2001-12-06 | Bristol-Myers Squibb Company | Soluble ctla4 mutant molecules and uses thereof |
| WO2002066603A2 (en) | 2001-02-15 | 2002-08-29 | Centocor, Inc. | Chemically defined medium for cultured mammalian cells |
| IL158328A0 (en) | 2001-04-13 | 2004-05-12 | Wyeth Corp | Surface proteins of streptococcus pyogenes |
| AU2002316230A1 (en) | 2001-06-13 | 2002-12-23 | Genentech, Inc. | Methods of culturing animal cells and polypeptide production in animal cells |
| GB0130228D0 (en) | 2001-12-18 | 2002-02-06 | Hansa Medica Ab | Protein |
| US8777906B1 (en) | 2002-01-24 | 2014-07-15 | Robin Scott Gray | Syringe with inspection window |
| CA2508925C (en) | 2002-12-23 | 2012-08-28 | Bristol-Myers Squibb Company | Product quality enhancement in mammalian cell culture processes for protein production |
| US7399612B2 (en) | 2003-06-30 | 2008-07-15 | Regeneron Pharmaceuticals, Inc. | VEGF-binding fusion proteins and nucleic acids encoding the same |
| CN100537751C (zh) | 2004-05-12 | 2009-09-09 | 华东理工大学 | 一种适合中国仓鼠卵巢细胞培养的无血清培养基 |
| US7335491B2 (en) | 2004-08-27 | 2008-02-26 | Wyeth Research Ireland Limited | Production of anti-abeta |
| WO2006050050A2 (en) | 2004-10-29 | 2006-05-11 | Centocor, Inc. | Chemically defined media compositions |
| US20060094104A1 (en) | 2004-10-29 | 2006-05-04 | Leopold Grillberger | Animal protein-free media for cultivation of cells |
| WO2006088650A2 (en) | 2005-02-02 | 2006-08-24 | Regeneron Pharmaceuticals, Inc. | Method of treating eye injury with local administration of a vegf inhibitor |
| LT2586459T (lt) | 2005-03-25 | 2017-09-25 | Regeneron Pharmaceuticals, Inc. | Vegf antagonisto kompozicijos |
| GB0511769D0 (en) * | 2005-06-09 | 2005-07-20 | Hansa Medical Ab | Treatment |
| EP1901773B1 (en) | 2005-06-09 | 2012-03-07 | Hansa Medical AB | Use of the ides proteinase (from s. pyogenes) for treating autoimmune diseases and graft rejection |
| CN1778903A (zh) | 2005-09-29 | 2006-05-31 | 华东理工大学 | 一种动物细胞高密度连续灌注培养方法 |
| RU2486236C2 (ru) | 2006-01-04 | 2013-06-27 | Бакстер Интернэшнл Инк. | Способ экспрессии белка |
| CN100502945C (zh) | 2006-03-31 | 2009-06-24 | 成都康弘生物科技有限公司 | Vegf受体融合蛋白在治疗眼睛疾病中的应用 |
| LT2944306T (lt) | 2006-06-16 | 2021-02-25 | Regeneron Pharmaceuticals, Inc. | Vfgf antagonisto kompozicijos, tinkamos įvedimui intravitrealiniu būdu |
| DK2041270T3 (da) | 2006-07-13 | 2014-01-27 | Wyeth Llc | Fremstilling af glycoproteiner |
| WO2008025747A1 (en) * | 2006-08-28 | 2008-03-06 | Ares Trading S.A. | Process for the purification of fc-fusion proteins |
| ES2620261T3 (es) | 2006-09-10 | 2017-06-28 | Glycotope Gmbh | Uso de células humanas de origen leucémico mieloide para expresión de anticuerpos |
| US10259860B2 (en) | 2007-02-27 | 2019-04-16 | Aprogen Inc. | Fusion proteins binding to VEGF and angiopoietin |
| CA2682738A1 (en) | 2007-04-16 | 2008-10-23 | Momenta Pharmaceuticals, Inc. | Defined glycoprotein products and related methods |
| US20100221823A1 (en) | 2007-06-11 | 2010-09-02 | Amgen Inc. | Method for culturing mammalian cells to improve recombinant protein production |
| CN101784670A (zh) | 2007-08-31 | 2010-07-21 | 弗·哈夫曼-拉罗切有限公司 | 糖基化谱分析 |
| EP2225273B1 (en) * | 2007-12-21 | 2012-05-23 | Roche Glycart AG | Stability testing of antibodies |
| KR101415099B1 (ko) | 2008-07-31 | 2014-07-08 | 고쿠리츠 다이가쿠 호진 교토 다이가쿠 | 불포화 폴리에스테르 수지와 마이크로피브릴화 식물 섬유를 함유하는 성형 재료 |
| CN102224239A (zh) | 2008-09-26 | 2011-10-19 | 先灵公司 | 高效价抗体生产 |
| WO2010138502A2 (en) | 2009-05-26 | 2010-12-02 | Momenta Pharmaceuticals, Inc. | Production of glycoproteins |
| AU2010265933B2 (en) | 2009-06-26 | 2015-05-14 | Regeneron Pharmaceuticals, Inc. | Readily isolated bispecific antibodies with native immunoglobulin format |
| KR101756354B1 (ko) | 2009-07-31 | 2017-07-26 | 백스터 인터내셔널 인코포레이티드 | Adamts 단백질 발현을 위한 세포 배양 배지 |
| RS60577B1 (sr) * | 2009-10-20 | 2020-08-31 | Abbvie Inc | Izolacija i prečišćavanje anti-il-13 antitela upotrebom protein a afinitetne hromatografije |
| EP2325296A1 (en) | 2009-11-20 | 2011-05-25 | LEK Pharmaceuticals d.d. | Production of glycoproteins with low N-glycolylneuraminic acid (Neu5Gc) content |
| US20130045492A1 (en) | 2010-02-08 | 2013-02-21 | Regeneron Pharmaceuticals, Inc. | Methods For Making Fully Human Bispecific Antibodies Using A Common Light Chain |
| KR20200077622A (ko) | 2011-01-13 | 2020-06-30 | 리제너론 파아마슈티컬스, 인크. | 혈관신생 눈 장애를 치료하기 위한 vegf 길항제의 용도 |
| CN102653729B (zh) | 2011-03-04 | 2014-07-16 | 上海赛金生物医药有限公司 | 一种用于中国仓鼠卵巢细胞的培养基 |
| EP2697361A1 (en) | 2011-04-11 | 2014-02-19 | Cellcura Asa | Protein-free culture media products for manufacturing viral-based vaccines |
| US9670519B2 (en) | 2011-04-29 | 2017-06-06 | Biocon Limited | Methods for reducing accumulation of lactate during culturing and method for producing polypeptide |
| KR101591671B1 (ko) | 2011-04-29 | 2016-02-04 | 바이오콘 리서치 리미티드 | 항체의 이질성을 감소시키는 방법 및 그 항체의 제조방법 |
| SG185920A1 (en) | 2011-05-27 | 2012-12-28 | Abbott Biotherapeutics Corp | Dac hyp compositions and methods |
| WO2013028330A2 (en) | 2011-08-19 | 2013-02-28 | Emd Millipore Corporation | Methods of reducing level of one of more impurities in a sample during protein purification |
| WO2013054250A1 (en) * | 2011-10-10 | 2013-04-18 | Dr Reddy's Laboratories Limited | Purification method |
| CN108771655A (zh) | 2011-10-28 | 2018-11-09 | 诚信生物公司 | 含有氨基酸的蛋白质制剂 |
| US20130281355A1 (en) | 2012-04-24 | 2013-10-24 | Genentech, Inc. | Cell culture compositions and methods for polypeptide production |
| CN102757496B (zh) * | 2012-06-07 | 2014-06-18 | 山东泉港药业有限公司 | 一种抗vegf抗体片段的纯化制备方法 |
| AU2012101677B4 (en) * | 2012-07-03 | 2012-12-20 | Novartis Ag | Device |
| EP3354750A1 (en) * | 2012-07-18 | 2018-08-01 | Siemens Healthcare Diagnostics Inc. | Kit of normalizing biological samples |
| SG11201500480TA (en) * | 2012-08-02 | 2015-02-27 | Sanofi Sa | Article of manufacture comprising aflibercept or ziv-aflibercept |
| KR20150043523A (ko) | 2012-09-02 | 2015-04-22 | 애브비 인코포레이티드 | 단백질 불균일성의 제어 방법 |
| JOP20200236A1 (ar) | 2012-09-21 | 2017-06-16 | Regeneron Pharma | الأجسام المضادة لمضاد cd3 وجزيئات ربط الأنتيجين ثنائية التحديد التي تربط cd3 وcd20 واستخداماتها |
| EP2722673B1 (en) * | 2012-10-17 | 2017-07-12 | Hexal AG | Improved method of mapping glycans of glycoproteins |
| US8956830B2 (en) | 2013-03-14 | 2015-02-17 | Momenta Pharmaceuticals, Inc. | Methods of cell culture |
| US9217168B2 (en) | 2013-03-14 | 2015-12-22 | Momenta Pharmaceuticals, Inc. | Methods of cell culture |
| US20140271622A1 (en) | 2013-03-14 | 2014-09-18 | Momenta Pharmaceuticals, Inc. | Methods of cell culture |
| CN103146648A (zh) | 2013-03-14 | 2013-06-12 | 北京京蒙高科干细胞技术有限公司 | 一种无动物源的淋巴细胞无血清培养基 |
| AR095196A1 (es) | 2013-03-15 | 2015-09-30 | Regeneron Pharma | Medio de cultivo celular libre de suero |
| WO2014145098A1 (en) | 2013-03-15 | 2014-09-18 | Genentech, Inc. | Cell culture compositions with antioxidants and methods for polypeptide production |
| PT3611180T (pt) * | 2013-03-15 | 2022-03-15 | Biomolecular Holdings Llc | Imunoglobulina híbrida que contém uma ligação não peptídica |
| CN103773732B (zh) | 2013-06-08 | 2016-05-11 | 李锋 | 一种化学成分确定的培养基、其应用及大规模培养哺乳动物细胞的生产工艺 |
| US10154971B2 (en) | 2013-06-17 | 2018-12-18 | Adamas Pharma, Llc | Methods of administering amantadine |
| US9085618B2 (en) | 2013-10-18 | 2015-07-21 | Abbvie, Inc. | Low acidic species compositions and methods for producing and using the same |
| WO2015058369A1 (en) | 2013-10-23 | 2015-04-30 | Sanofi (China) Investment Co., Ltd. | Use of aflibercept and docetaxel for the treatment of nasopharyngeal carcinoma |
| EP3102589A1 (en) | 2014-02-04 | 2016-12-14 | Biogen MA Inc. | Use of cation-exchange chromatography in the flow-through mode to enrich post-translational modifications |
| CN104073464A (zh) | 2014-07-08 | 2014-10-01 | 西藏天虹科技股份有限责任公司 | 一种cho细胞无血清培养基及其制备方法 |
| AR101262A1 (es) | 2014-07-26 | 2016-12-07 | Regeneron Pharma | Plataforma de purificación para anticuerpos biespecíficos |
| GB201502305D0 (en) * | 2015-02-12 | 2015-04-01 | Hansa Medical Ab | Protein |
| GB201502306D0 (en) | 2015-02-12 | 2015-04-01 | Hansa Medical Ab | Protein |
| CN107428817B (zh) * | 2015-03-12 | 2022-07-12 | 免疫医疗有限责任公司 | 纯化白蛋白融合蛋白的方法 |
| KR101867134B1 (ko) | 2015-03-23 | 2018-06-12 | 한양대학교 산학협력단 | 포유류 세포를 이용하여 목적 물질을 고효율로 생산하기 위한 세포 배양 배지, 이를 이용한 세포 배양 방법 및 목적 물질의 생산 방법 |
| CA3208857A1 (en) | 2015-04-01 | 2016-10-06 | Boehringer Ingelheim International Gmbh | Cell culture medium |
| CN105002242A (zh) | 2015-07-23 | 2015-10-28 | 苏州康聚生物科技有限公司 | 用于cho细胞中高效表达重组人促甲状腺激素的无血清培养基及其应用 |
| TWI797060B (zh) | 2015-08-04 | 2023-04-01 | 美商再生元醫藥公司 | 補充牛磺酸之細胞培養基及用法 |
| US20180230540A1 (en) | 2015-08-12 | 2018-08-16 | Novartis Ag | Methods of treating ophthalmic disorders |
| CN108291220B (zh) * | 2015-10-22 | 2022-10-28 | 普罗泰纳瓦株式会社 | 免疫球蛋白结合多肽 |
| EP3377100A1 (en) * | 2015-11-18 | 2018-09-26 | Formycon AG | Pre-filled pharmaceutical package comprising a liquid formulation of a vegf-antagonist |
| US20180326126A1 (en) | 2015-11-18 | 2018-11-15 | Formycon Ag | Pre-filled plastic syringe containing a vegf antagonist |
| CN105368808B (zh) * | 2015-11-30 | 2019-04-30 | 苏州康聚生物科技有限公司 | 一种IdeS蛋白酶、其制备方法及应用 |
| ES2954153T3 (es) | 2015-12-22 | 2023-11-20 | Regeneron Pharma | Combinación de anticuerpos anti-PD-1 y anticuerpos biespecíficos anti-CD20/anti-CD3 para tratar el cáncer |
| WO2017112762A1 (en) | 2015-12-22 | 2017-06-29 | Regeneron Pharmaceuticals, Inc. | Bispecific anti-cd20/anti-cd3 antibodies to treat acute lymphoblastic leukemia |
| EP3196646B1 (en) * | 2016-01-19 | 2019-12-18 | Hexal AG | Methods of mapping protein variants |
| EP3407868A1 (en) | 2016-01-26 | 2018-12-05 | Formycon AG | Liquid formulation of a vegf antagonist |
| WO2017168296A1 (en) * | 2016-03-29 | 2017-10-05 | Navya Biologicals Pvt. Ltd | A process for purification of fc-fusion proteins |
| KR101685532B1 (ko) * | 2016-04-26 | 2016-12-13 | 한국프라임제약주식회사 | 혈관내피성장인자 수용체 융합단백질 |
| CN106279412A (zh) * | 2016-09-12 | 2017-01-04 | 广东东阳光药业有限公司 | 一种抗vegf类单克隆抗体的纯化方法 |
| KR102520731B1 (ko) | 2016-09-23 | 2023-04-14 | 리제너론 파아마슈티컬스, 인크. | 항-steap2 항체, 항체-약물 컨쥬게이트, 및 steap2 및 cd3에 결합하는 이중특이적 항원-결합 분자, 및 이의 용도 |
| WO2018067331A1 (en) | 2016-09-23 | 2018-04-12 | Regeneron Pharmaceuticals, Inc. | Bi specific anti-muc16-cd3 antibodies and nti-muc16 drug conjugates |
| TWI782930B (zh) | 2016-11-16 | 2022-11-11 | 美商再生元醫藥公司 | 抗met抗體,結合met之雙特異性抗原結合分子及其使用方法 |
| WO2018094316A1 (en) | 2016-11-21 | 2018-05-24 | Just Biotherapeutics, Inc. | Aflibercept formulations and uses thereof |
| PE20191436A1 (es) * | 2016-12-23 | 2019-10-14 | Serum Institute Of India Pvt Ltd | Metodos mejorados para estimular la productividad de anticuerpos en el cultivo de celulas de mamiferos y reducir la agregacion durante los procesos de formulacion post-tratamiento (downstream) y formulaciones de anticuerpos estables obtenidas a partir de los mismos |
| WO2018147960A1 (en) | 2017-02-08 | 2018-08-16 | Imaginab, Inc. | Extension sequences for diabodies |
| US10941178B2 (en) * | 2017-03-17 | 2021-03-09 | Gilead Sciences, Inc. | Method of purifying an antibody |
| KR101861163B1 (ko) | 2017-04-26 | 2018-05-25 | 삼천당제약주식회사 | 안과용 약학 조성물 |
| US20210228738A1 (en) | 2017-07-17 | 2021-07-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Compositions and methods for increasing or enhancing transduction of gene therapy vectors and for removing or reducing immunoglobulins |
| DK3668985T3 (da) * | 2017-08-17 | 2021-09-20 | Just Evotec Biologics Inc | Fremgangsmåde til rensning af glycosyleret protein fra værtscellegalectiner og andre urenheder |
| MX2020003945A (es) * | 2017-10-18 | 2020-11-09 | Regenxbio Inc | Tratamiento de enfermedades oculares y cancer de colon metastasico con vegf-trap humano modificado post-traduccionalmente . |
| WO2019099965A1 (en) * | 2017-11-20 | 2019-05-23 | Just Biotherapeutics, Inc. | Aflibercept formulations containing a lysine salt as tonicifying agent and uses thereof |
| CN109929027B (zh) * | 2017-12-15 | 2020-10-09 | 山东博安生物技术有限公司 | 采用线性洗脱步骤的重组融合蛋白纯化方法 |
| CN109929038B (zh) * | 2017-12-15 | 2020-10-09 | 山东博安生物技术有限公司 | Vegf捕获剂融合蛋白的纯化方法 |
| WO2019173767A1 (en) * | 2018-03-08 | 2019-09-12 | Coherus Biosciences Inc. | Stable aqueous formulations of aflibercept |
| US11426446B2 (en) | 2018-03-08 | 2022-08-30 | Coherus Biosciences, Inc. | Stable aqueous formulations of aflibercept |
| AU2019234739A1 (en) | 2018-03-13 | 2020-09-03 | Amgen Inc. | Methods for the preparation of trypsin-resistant polypeptides for mass spectrometric analysis |
| JP7235770B2 (ja) | 2018-05-10 | 2023-03-08 | リジェネロン・ファーマシューティカルズ・インコーポレイテッド | 高濃度vegf受容体融合タンパク質を含む製剤 |
| WO2020102740A2 (en) * | 2018-11-16 | 2020-05-22 | Spark Therapeutics, Inc. | Compositions and methods for increasing or enhancing transduction of gene therapy vectors and for removing or reducing immunoglobulins |
| US20220098279A1 (en) | 2019-01-30 | 2022-03-31 | Amgen Inc. | Aflibercept attributes and methods of characterizing and modifying thereof |
| US20220204920A1 (en) | 2019-05-16 | 2022-06-30 | Formycon Ag | Method for reducing methionine oxidation in recombinant proteins |
| KR102286892B1 (ko) * | 2019-07-08 | 2021-08-06 | 삼천당제약주식회사 | 안과용 단백질 제제의 정제방법 |
| BR112021025769A2 (pt) * | 2019-12-06 | 2022-04-12 | Regeneron Pharma | Composições de proteína anti-vegf e métodos para a produção das mesmas |
-
2020
- 2020-08-18 BR BR112021025769A patent/BR112021025769A2/pt active Search and Examination
- 2020-08-18 IL IL288360A patent/IL288360B2/en unknown
- 2020-08-18 JP JP2022506131A patent/JP2022548818A/ja active Pending
- 2020-08-18 IL IL301888A patent/IL301888A/en unknown
- 2020-08-18 SG SG11202110972UA patent/SG11202110972UA/en unknown
- 2020-08-18 CN CN202080058870.8A patent/CN114401994A/zh active Pending
- 2020-08-18 KR KR1020237044969A patent/KR20240007293A/ko active Application Filing
- 2020-08-18 WO PCT/US2020/046842 patent/WO2021112927A1/en active Application Filing
- 2020-08-18 PE PE2022001023A patent/PE20221789A1/es unknown
- 2020-08-18 SG SG11202110964QA patent/SG11202110964QA/en unknown
- 2020-08-18 BR BR112021025359A patent/BR112021025359A2/pt unknown
- 2020-08-18 SG SG11202110968VA patent/SG11202110968VA/en unknown
- 2020-08-18 KR KR1020237010909A patent/KR20230051296A/ko active Search and Examination
- 2020-08-18 CN CN202080055377.0A patent/CN114206914A/zh active Pending
- 2020-08-18 MX MX2021014862A patent/MX2021014862A/es unknown
- 2020-08-18 KR KR1020227002039A patent/KR102519234B1/ko active IP Right Grant
- 2020-08-18 IL IL288352A patent/IL288352B2/en unknown
- 2020-08-18 IL IL288357A patent/IL288357B2/en unknown
- 2020-08-18 JP JP2022506125A patent/JP2022548197A/ja active Pending
- 2020-08-18 BR BR112022001016A patent/BR112022001016A2/pt unknown
- 2020-08-18 EP EP20765402.1A patent/EP3906303A1/en active Pending
- 2020-08-18 CA CA3172625A patent/CA3172625A1/en active Pending
- 2020-08-18 EP EP20764873.4A patent/EP3906249A1/en active Pending
- 2020-08-18 PE PE2022001024A patent/PE20221261A1/es unknown
- 2020-08-18 US US16/996,030 patent/US11104715B2/en active Active
- 2020-08-18 JP JP2022506067A patent/JP2022550930A/ja active Pending
- 2020-08-18 AU AU2020398547A patent/AU2020398547C1/en active Active
- 2020-08-18 KR KR1020237044978A patent/KR20240005222A/ko active Application Filing
- 2020-08-18 EP EP20775971.3A patent/EP3931204A1/en active Pending
- 2020-08-18 PE PE2022001022A patent/PE20221788A1/es unknown
- 2020-08-18 AU AU2020398830A patent/AU2020398830C1/en active Active
- 2020-08-18 EP EP20765400.5A patent/EP3906250A1/en active Pending
- 2020-08-18 NZ NZ781136A patent/NZ781136A/en unknown
- 2020-08-18 NZ NZ781142A patent/NZ781142A/en unknown
- 2020-08-18 US US16/996,127 patent/US11180540B2/en active Active
- 2020-08-18 IL IL288367A patent/IL288367B2/en unknown
- 2020-08-18 IL IL302921A patent/IL302921A/en unknown
- 2020-08-18 WO PCT/US2020/046809 patent/WO2021112923A1/en active Application Filing
- 2020-08-18 JP JP2022506127A patent/JP2022547652A/ja active Pending
- 2020-08-18 AU AU2020396490A patent/AU2020396490C1/en active Active
- 2020-08-18 MX MX2022006758A patent/MX2022006758A/es unknown
- 2020-08-18 SG SG11202110953UA patent/SG11202110953UA/en unknown
- 2020-08-18 IL IL296864A patent/IL296864B2/en unknown
- 2020-08-18 JP JP2022506072A patent/JP2022547651A/ja active Pending
- 2020-08-18 MY MYPI2022002113A patent/MY193353A/en unknown
- 2020-08-18 WO PCT/US2020/046823 patent/WO2021112924A1/en active Application Filing
- 2020-08-18 TW TW109128142A patent/TW202134256A/zh unknown
- 2020-08-18 MY MYPI2022002104A patent/MY193351A/en unknown
- 2020-08-18 KR KR1020237044758A patent/KR20240005206A/ko active Application Filing
- 2020-08-18 IL IL296893A patent/IL296893B1/en unknown
- 2020-08-18 KR KR1020227002041A patent/KR102519236B1/ko active IP Right Grant
- 2020-08-18 NZ NZ781145A patent/NZ781145A/en unknown
- 2020-08-18 KR KR1020237010912A patent/KR20230051297A/ko active Application Filing
- 2020-08-18 MY MYPI2021006226A patent/MY190686A/en unknown
- 2020-08-18 CN CN202080055633.6A patent/CN114206925A/zh active Pending
- 2020-08-18 TW TW109128143A patent/TW202122417A/zh unknown
- 2020-08-18 IL IL296512A patent/IL296512B2/en unknown
- 2020-08-18 AU AU2020398125A patent/AU2020398125C1/en active Active
- 2020-08-18 AU AU2020397865A patent/AU2020397865C1/en active Active
- 2020-08-18 CN CN202080055352.0A patent/CN114206907A/zh active Pending
- 2020-08-18 US US16/996,042 patent/US11098112B2/en active Active
- 2020-08-18 IL IL288355A patent/IL288355B2/en unknown
- 2020-08-18 US US16/996,293 patent/US11186625B2/en active Active
- 2020-08-18 MX MX2021014863A patent/MX2021014863A/es unknown
- 2020-08-18 PE PE2022001020A patent/PE20221770A1/es unknown
- 2020-08-18 MY MYPI2021006235A patent/MY190627A/en unknown
- 2020-08-18 WO PCT/US2020/046846 patent/WO2021112928A1/en active Application Filing
- 2020-08-18 AU AU2020397803A patent/AU2020397803C1/en active Active
- 2020-08-18 KR KR1020227002040A patent/KR20220038062A/ko not_active IP Right Cessation
- 2020-08-18 MY MYPI2021006201A patent/MY190625A/en unknown
- 2020-08-18 TW TW109128140A patent/TW202128744A/zh unknown
- 2020-08-18 MY MYPI2021006189A patent/MY190624A/en unknown
- 2020-08-18 KR KR1020227002043A patent/KR20220038349A/ko not_active IP Right Cessation
- 2020-08-18 IL IL309491A patent/IL309491A/en unknown
- 2020-08-18 BR BR112021025432A patent/BR112021025432A2/pt unknown
- 2020-08-18 BR BR112021025158A patent/BR112021025158A2/pt not_active Application Discontinuation
- 2020-08-18 CA CA3172631A patent/CA3172631C/en active Active
- 2020-08-18 IL IL309560A patent/IL309560A/en unknown
- 2020-08-18 WO PCT/US2020/046831 patent/WO2021112925A1/en active Application Filing
- 2020-08-18 MY MYPI2022002103A patent/MY193354A/en unknown
- 2020-08-18 SG SG11202110955VA patent/SG11202110955VA/en unknown
- 2020-08-18 TW TW109128144A patent/TW202128991A/zh unknown
- 2020-08-18 US US16/996,103 patent/US11098311B2/en active Active
- 2020-08-18 IL IL302538A patent/IL302538A/en unknown
- 2020-08-18 SG SG11202110958QA patent/SG11202110958QA/en unknown
- 2020-08-18 WO PCT/US2020/046862 patent/WO2021112929A1/en active Application Filing
- 2020-08-18 NZ NZ781143A patent/NZ781143A/en unknown
- 2020-08-18 MY MYPI2022002100A patent/MY193355A/en unknown
- 2020-08-18 EP EP20765397.3A patent/EP3906302A1/en active Pending
- 2020-08-18 KR KR1020227002038A patent/KR102619866B1/ko active IP Right Grant
- 2020-08-18 WO PCT/US2020/046834 patent/WO2021112926A1/en active Application Filing
- 2020-08-18 IL IL288351A patent/IL288351B1/en unknown
- 2020-08-18 PE PE2022001026A patent/PE20221791A1/es unknown
- 2020-08-18 MY MYPI2022002191A patent/MY193349A/en unknown
- 2020-08-18 NZ NZ781138A patent/NZ781138A/en unknown
- 2020-08-18 CN CN202080055314.5A patent/CN114423783A/zh active Pending
- 2020-08-18 MY MYPI2022002109A patent/MY193350A/en unknown
- 2020-08-18 US US16/996,007 patent/US11053280B2/en active Active
- 2020-08-18 CA CA3159586A patent/CA3159586A1/en active Pending
- 2020-08-18 PE PE2022001025A patent/PE20221790A1/es unknown
- 2020-08-18 MX MX2022006760A patent/MX2022006760A/es unknown
- 2020-08-18 TW TW109128139A patent/TW202128743A/zh unknown
- 2020-08-18 MX MX2022006759A patent/MX2022006759A/es unknown
- 2020-08-18 NZ NZ781137A patent/NZ781137A/en unknown
- 2020-08-18 MY MYPI2021006216A patent/MY190626A/en unknown
- 2020-08-18 MX MX2022006788A patent/MX2022006788A/es unknown
- 2020-08-18 MY MYPI2021006223A patent/MY190623A/en unknown
- 2020-08-18 CN CN202080055393.XA patent/CN114206924A/zh active Pending
- 2020-08-18 EP EP20765403.9A patent/EP3931303A1/en active Pending
- 2020-08-18 KR KR1020227002042A patent/KR20220035393A/ko active Application Filing
- 2020-08-18 IL IL296863A patent/IL296863B2/en unknown
- 2020-08-18 CA CA3129193A patent/CA3129193C/en active Active
- 2020-08-18 JP JP2022506130A patent/JP2022552052A/ja active Pending
- 2020-08-18 TW TW109128145A patent/TW202128745A/zh unknown
- 2020-08-18 BR BR112021025438A patent/BR112021025438A2/pt unknown
- 2020-08-18 KR KR1020237036668A patent/KR20230152810A/ko active Application Filing
-
2021
- 2021-03-18 US US17/205,745 patent/US11174283B2/en active Active
- 2021-03-24 US US17/211,333 patent/US11286290B2/en active Active
- 2021-07-16 US US17/378,362 patent/US11440950B2/en active Active
- 2021-07-22 US US17/383,322 patent/US11535663B2/en active Active
- 2021-08-26 US US17/458,519 patent/US20220098280A1/en active Pending
- 2021-08-30 US US17/460,578 patent/US11306135B2/en active Active
- 2021-09-29 US US17/489,495 patent/US11299532B2/en active Active
- 2021-10-01 US US17/492,440 patent/US11407813B2/en active Active
- 2021-10-11 ZA ZA2021/07647A patent/ZA202107647B/en unknown
- 2021-10-11 ZA ZA2021/07644A patent/ZA202107644B/en unknown
- 2021-10-11 ZA ZA2021/07646A patent/ZA202107646B/en unknown
- 2021-10-13 US US17/500,735 patent/US11459373B2/en active Active
- 2021-12-02 MX MX2023005421A patent/MX2023005421A/es unknown
- 2021-12-02 MX MX2022011973A patent/MX2022011973A/es unknown
- 2021-12-02 MX MX2022012042A patent/MX2022012042A/es unknown
- 2021-12-21 US US17/557,904 patent/US11472861B2/en active Active
- 2021-12-30 CO CONC2021/0018234A patent/CO2021018234A2/es unknown
- 2021-12-30 CO CONC2021/0018192A patent/CO2021018192A2/es unknown
- 2021-12-30 CO CONC2021/0018181A patent/CO2021018181A2/es unknown
- 2021-12-30 CO CONC2021/0018214A patent/CO2021018214A2/es unknown
- 2021-12-30 CO CONC2021/0018203A patent/CO2021018203A2/es unknown
-
2022
- 2022-01-13 US US17/575,425 patent/US11485770B2/en active Active
- 2022-01-13 US US17/575,585 patent/US11649273B2/en active Active
- 2022-01-25 CO CONC2022/0000660A patent/CO2022000660A2/es unknown
- 2022-02-08 US US17/667,298 patent/US11505593B2/en active Active
- 2022-02-08 US US17/667,330 patent/US11459374B2/en active Active
- 2022-04-01 US US17/711,723 patent/US11505594B2/en active Active
- 2022-06-15 US US17/841,349 patent/US11548932B2/en active Active
- 2022-06-16 US US17/842,286 patent/US11542317B1/en active Active
- 2022-07-06 US US17/858,629 patent/US20230041349A1/en active Pending
- 2022-07-06 US US17/858,724 patent/US11753459B2/en active Active
- 2022-11-25 US US17/994,223 patent/US11732025B2/en active Active
-
2023
- 2023-03-01 US US18/116,166 patent/US20230235019A1/en active Pending
- 2023-03-07 US US18/118,558 patent/US20230272044A1/en active Pending
- 2023-03-14 AU AU2023201589A patent/AU2023201589A1/en active Pending
- 2023-03-14 AU AU2023201586A patent/AU2023201586A1/en active Pending
- 2023-03-14 AU AU2023201588A patent/AU2023201588A1/en active Pending
- 2023-03-14 AU AU2023201585A patent/AU2023201585A1/en active Pending
- 2023-03-14 AU AU2023201590A patent/AU2023201590A1/en active Pending
- 2023-03-21 AU AU2023201748A patent/AU2023201748A1/en active Pending
- 2023-06-23 US US18/340,707 patent/US20240067701A1/en active Pending
- 2023-06-23 US US18/213,779 patent/US11958894B2/en active Active
- 2023-06-26 US US18/214,282 patent/US20230357357A1/en active Pending
- 2023-07-11 IL IL304390A patent/IL304390A/en unknown
- 2023-09-08 JP JP2023145749A patent/JP2023171771A/ja active Pending
- 2023-09-22 JP JP2023156642A patent/JP2023179531A/ja active Pending
- 2023-10-12 JP JP2023176817A patent/JP2024009949A/ja active Pending
- 2023-10-12 JP JP2023176837A patent/JP2024009950A/ja active Pending
- 2023-11-20 JP JP2023196342A patent/JP2024026116A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11440950B2 (en) | Anti-VEGF protein compositions and methods for producing the same |