KR20170052631A - 2'-o-푸코실락토오스의 제조 방법 - Google Patents
2'-o-푸코실락토오스의 제조 방법 Download PDFInfo
- Publication number
- KR20170052631A KR20170052631A KR1020177009260A KR20177009260A KR20170052631A KR 20170052631 A KR20170052631 A KR 20170052631A KR 1020177009260 A KR1020177009260 A KR 1020177009260A KR 20177009260 A KR20177009260 A KR 20177009260A KR 20170052631 A KR20170052631 A KR 20170052631A
- Authority
- KR
- South Korea
- Prior art keywords
- formula
- alkyl
- compound
- cycloalkyl
- fucosyllactose
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 89
- LIVQFHIJABPBCM-VHWZWBDXSA-N (3S,4R,5S,6S)-2-[(3R,4S,5S,6R)-2,4-dihydroxy-6-(hydroxymethyl)-5-[(2S,3R,4S,5R,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-3-yl]oxy-6-methyloxane-3,4,5-triol Chemical compound C1([C@@H](O)[C@H](O)[C@H](O)[C@@H](O1)C)O[C@H]1C(O)O[C@@H]([C@H]([C@@H]1O)O[C@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@H](O1)CO)CO LIVQFHIJABPBCM-VHWZWBDXSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 253
- HWHQUWQCBPAQQH-BWRPKUOHSA-N 2-fucosyllactose Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@H]([C@H](O)CO)[C@H](O)[C@@H](O)C=O HWHQUWQCBPAQQH-BWRPKUOHSA-N 0.000 claims abstract description 84
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 81
- 230000008569 process Effects 0.000 claims abstract description 46
- IDIOJRGTRFRIJL-UHFFFAOYSA-N iodosilane Chemical compound I[SiH3] IDIOJRGTRFRIJL-UHFFFAOYSA-N 0.000 claims abstract description 27
- 125000001424 substituent group Chemical group 0.000 claims abstract description 22
- 239000000203 mixture Substances 0.000 claims description 84
- 238000002360 preparation method Methods 0.000 claims description 77
- -1 iodine, iodide salts Chemical class 0.000 claims description 46
- 238000006243 chemical reaction Methods 0.000 claims description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 40
- 239000002585 base Substances 0.000 claims description 38
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 claims description 34
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 claims description 34
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 claims description 34
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 34
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 claims description 32
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 31
- 229910052740 iodine Inorganic materials 0.000 claims description 31
- 239000011630 iodine Substances 0.000 claims description 31
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 31
- 239000001257 hydrogen Substances 0.000 claims description 26
- 229910052739 hydrogen Inorganic materials 0.000 claims description 26
- 239000003153 chemical reaction reagent Substances 0.000 claims description 25
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 23
- 125000004432 carbon atom Chemical group C* 0.000 claims description 22
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical group C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 claims description 22
- 125000006239 protecting group Chemical group 0.000 claims description 20
- 239000011541 reaction mixture Substances 0.000 claims description 20
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 18
- 235000013305 food Nutrition 0.000 claims description 18
- SNOOUWRIMMFWNE-UHFFFAOYSA-M sodium;6-[(3,4,5-trimethoxybenzoyl)amino]hexanoate Chemical compound [Na+].COC1=CC(C(=O)NCCCCCC([O-])=O)=CC(OC)=C1OC SNOOUWRIMMFWNE-UHFFFAOYSA-M 0.000 claims description 18
- 239000012535 impurity Substances 0.000 claims description 16
- 150000001412 amines Chemical class 0.000 claims description 15
- 239000002253 acid Substances 0.000 claims description 12
- 239000003513 alkali Substances 0.000 claims description 12
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 238000005828 desilylation reaction Methods 0.000 claims description 10
- 238000000746 purification Methods 0.000 claims description 10
- 150000004649 carbonic acid derivatives Chemical class 0.000 claims description 9
- 229910052783 alkali metal Inorganic materials 0.000 claims description 8
- PZPGRFITIJYNEJ-UHFFFAOYSA-N disilane Chemical compound [SiH3][SiH3] PZPGRFITIJYNEJ-UHFFFAOYSA-N 0.000 claims description 8
- 150000002431 hydrogen Chemical class 0.000 claims description 8
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 7
- 235000013373 food additive Nutrition 0.000 claims description 7
- 239000002778 food additive Substances 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 7
- XMBWDFGMSWQBCA-UHFFFAOYSA-M iodide Chemical compound [I-] XMBWDFGMSWQBCA-UHFFFAOYSA-M 0.000 claims description 7
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims description 5
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical compound Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 claims description 5
- 229910052723 transition metal Inorganic materials 0.000 claims description 5
- 150000003624 transition metals Chemical class 0.000 claims description 5
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 4
- 150000001340 alkali metals Chemical class 0.000 claims description 4
- 238000011065 in-situ storage Methods 0.000 claims description 4
- 150000003512 tertiary amines Chemical class 0.000 claims description 4
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 claims description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 claims description 3
- 230000008878 coupling Effects 0.000 claims description 3
- 238000010168 coupling process Methods 0.000 claims description 3
- 238000005859 coupling reaction Methods 0.000 claims description 3
- 230000002378 acidificating effect Effects 0.000 claims description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 150000007514 bases Chemical class 0.000 claims 3
- 150000003244 quercetin derivatives Chemical class 0.000 abstract 1
- 235000013350 formula milk Nutrition 0.000 description 192
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 141
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- 239000000243 solution Substances 0.000 description 49
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 45
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 42
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 39
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 36
- 239000000047 product Substances 0.000 description 32
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 27
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- 238000004128 high performance liquid chromatography Methods 0.000 description 24
- 239000012043 crude product Substances 0.000 description 22
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 19
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 239000000741 silica gel Substances 0.000 description 16
- 229910002027 silica gel Inorganic materials 0.000 description 16
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- NEXSMEBSBIABKL-UHFFFAOYSA-N hexamethyldisilane Chemical compound C[Si](C)(C)[Si](C)(C)C NEXSMEBSBIABKL-UHFFFAOYSA-N 0.000 description 13
- 235000009518 sodium iodide Nutrition 0.000 description 13
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 12
- 229910001516 alkali metal iodide Inorganic materials 0.000 description 12
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 12
- 238000010992 reflux Methods 0.000 description 12
- 239000011734 sodium Substances 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 239000007795 chemical reaction product Substances 0.000 description 11
- 239000008101 lactose Substances 0.000 description 11
- 239000003960 organic solvent Substances 0.000 description 11
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 150000001720 carbohydrates Chemical class 0.000 description 9
- 235000014633 carbohydrates Nutrition 0.000 description 9
- 238000001914 filtration Methods 0.000 description 9
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 9
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N 1,1-dimethoxyethane Chemical compound COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 8
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 150000007529 inorganic bases Chemical class 0.000 description 8
- 150000004694 iodide salts Chemical class 0.000 description 8
- 239000002808 molecular sieve Substances 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 8
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 239000000010 aprotic solvent Substances 0.000 description 7
- 229960001701 chloroform Drugs 0.000 description 7
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 7
- 235000019345 sodium thiosulphate Nutrition 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 6
- 238000003776 cleavage reaction Methods 0.000 description 6
- 125000002446 fucosyl group Chemical group C1([C@@H](O)[C@H](O)[C@H](O)[C@@H](O1)C)* 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 230000007017 scission Effects 0.000 description 6
- 238000006884 silylation reaction Methods 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 150000001336 alkenes Chemical class 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 125000004185 ester group Chemical group 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 5
- 235000015097 nutrients Nutrition 0.000 description 5
- 235000016709 nutrition Nutrition 0.000 description 5
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 4
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 150000001350 alkyl halides Chemical class 0.000 description 4
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cis-cyclohexene Natural products C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 238000005984 hydrogenation reaction Methods 0.000 description 4
- 238000007327 hydrogenolysis reaction Methods 0.000 description 4
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Chemical compound [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- 238000006386 neutralization reaction Methods 0.000 description 4
- 235000011181 potassium carbonates Nutrition 0.000 description 4
- 150000003222 pyridines Chemical class 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 238000005809 transesterification reaction Methods 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 102100021935 C-C motif chemokine 26 Human genes 0.000 description 3
- 101100495769 Caenorhabditis elegans che-1 gene Proteins 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- 101000897493 Homo sapiens C-C motif chemokine 26 Proteins 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 3
- 239000011260 aqueous acid Substances 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 150000004292 cyclic ethers Chemical class 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 108010042430 galactose receptor Proteins 0.000 description 3
- 125000001183 hydrocarbyl group Chemical group 0.000 description 3
- 239000012442 inert solvent Substances 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000035764 nutrition Effects 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 3
- 235000015497 potassium bicarbonate Nutrition 0.000 description 3
- 239000011736 potassium bicarbonate Substances 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 238000007127 saponification reaction Methods 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229940001593 sodium carbonate Drugs 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- UQFSVBXCNGCBBW-UHFFFAOYSA-M tetraethylammonium iodide Chemical compound [I-].CC[N+](CC)(CC)CC UQFSVBXCNGCBBW-UHFFFAOYSA-M 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- GKXDJYKZFZVASJ-UHFFFAOYSA-M tetrapropylazanium;iodide Chemical compound [I-].CCC[N+](CCC)(CCC)CCC GKXDJYKZFZVASJ-UHFFFAOYSA-M 0.000 description 3
- UPQQXPKAYZYUKO-UHFFFAOYSA-N 2,2,2-trichloroacetamide Chemical compound OC(=N)C(Cl)(Cl)Cl UPQQXPKAYZYUKO-UHFFFAOYSA-N 0.000 description 2
- MHNNAWXXUZQSNM-UHFFFAOYSA-N 2-methylbut-1-ene Chemical compound CCC(C)=C MHNNAWXXUZQSNM-UHFFFAOYSA-N 0.000 description 2
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 2
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical group CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 2
- 125000005916 2-methylpentyl group Chemical group 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- SVYKKECYCPFKGB-UHFFFAOYSA-N N,N-dimethylcyclohexylamine Chemical compound CN(C)C1CCCCC1 SVYKKECYCPFKGB-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 239000003463 adsorbent Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 2
- 150000008041 alkali metal carbonates Chemical class 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 238000004517 catalytic hydrocracking Methods 0.000 description 2
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 150000001925 cycloalkenes Chemical class 0.000 description 2
- 235000013365 dairy product Nutrition 0.000 description 2
- 150000001983 dialkylethers Chemical class 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000000378 dietary effect Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 150000008267 fucoses Chemical class 0.000 description 2
- 229940052308 general anesthetics halogenated hydrocarbons Drugs 0.000 description 2
- 230000013595 glycosylation Effects 0.000 description 2
- 238000006206 glycosylation reaction Methods 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 229910001385 heavy metal Inorganic materials 0.000 description 2
- 235000020256 human milk Nutrition 0.000 description 2
- 210000004251 human milk Anatomy 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- 125000002346 iodo group Chemical group I* 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 229910052808 lithium carbonate Inorganic materials 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 235000013336 milk Nutrition 0.000 description 2
- 239000008267 milk Substances 0.000 description 2
- 210000004080 milk Anatomy 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- VMOWKUTXPNPTEN-UHFFFAOYSA-N n,n-dimethylpropan-2-amine Chemical compound CC(C)N(C)C VMOWKUTXPNPTEN-UHFFFAOYSA-N 0.000 description 2
- ISRXMEYARGEVIU-UHFFFAOYSA-N n-methyl-n-propan-2-ylpropan-2-amine Chemical compound CC(C)N(C)C(C)C ISRXMEYARGEVIU-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- YWAKXRMUMFPDSH-UHFFFAOYSA-N pentene Chemical compound CCCC=C YWAKXRMUMFPDSH-UHFFFAOYSA-N 0.000 description 2
- QMMOXUPEWRXHJS-UHFFFAOYSA-N pentene-2 Natural products CCC=CC QMMOXUPEWRXHJS-UHFFFAOYSA-N 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 238000002390 rotary evaporation Methods 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 2
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 2
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 2
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical class C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- FIARMZDBEGVMLV-UHFFFAOYSA-N 1,1,2,2,2-pentafluoroethanolate Chemical group [O-]C(F)(F)C(F)(F)F FIARMZDBEGVMLV-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- JLURWBIBHMXAHE-UHFFFAOYSA-N 1-cyclohexyl-n-(cyclohexylmethyl)methanamine Chemical compound C1CCCCC1CNCC1CCCCC1 JLURWBIBHMXAHE-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000004778 2,2-difluoroethyl group Chemical group [H]C([H])(*)C([H])(F)F 0.000 description 1
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 description 1
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 1
- UWKQJZCTQGMHKD-UHFFFAOYSA-N 2,6-di-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=CC(C(C)(C)C)=N1 UWKQJZCTQGMHKD-UHFFFAOYSA-N 0.000 description 1
- 125000004337 3-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical class CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000551546 Minerva Species 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000003302 alkenyloxy group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 230000002925 chemical effect Effects 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000004775 chlorodifluoromethyl group Chemical group FC(F)(Cl)* 0.000 description 1
- 125000004773 chlorofluoromethyl group Chemical group [H]C(F)(Cl)* 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000010549 co-Evaporation Methods 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000000498 cooling water Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- 125000004774 dichlorofluoromethyl group Chemical group FC(Cl)(Cl)* 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- 235000013325 dietary fiber Nutrition 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethyl cyclohexane Natural products CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 230000033581 fucosylation Effects 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 239000011874 heated mixture Substances 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 235000021125 infant nutrition Nutrition 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 150000002496 iodine Chemical class 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 150000002597 lactoses Chemical class 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 238000003541 multi-stage reaction Methods 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- AUONHKJOIZSQGR-UHFFFAOYSA-N oxophosphane Chemical compound P=O AUONHKJOIZSQGR-UHFFFAOYSA-N 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000010517 secondary reaction Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000011182 sodium carbonates Nutrition 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 238000012421 spiking Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 150000004043 trisaccharides Chemical class 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/26—Acyclic or carbocyclic radicals, substituted by hetero rings
-
- A—HUMAN NECESSITIES
- A23—FOODS OR FOODSTUFFS; TREATMENT THEREOF, NOT COVERED BY OTHER CLASSES
- A23L—FOODS, FOODSTUFFS, OR NON-ALCOHOLIC BEVERAGES, NOT COVERED BY SUBCLASSES A21D OR A23B-A23J; THEIR PREPARATION OR TREATMENT, e.g. COOKING, MODIFICATION OF NUTRITIVE QUALITIES, PHYSICAL TREATMENT; PRESERVATION OF FOODS OR FOODSTUFFS, IN GENERAL
- A23L33/00—Modifying nutritive qualities of foods; Dietetic products; Preparation or treatment thereof
- A23L33/10—Modifying nutritive qualities of foods; Dietetic products; Preparation or treatment thereof using additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H3/00—Compounds containing only hydrogen atoms and saccharide radicals having only carbon, hydrogen, and oxygen atoms
- C07H3/06—Oligosaccharides, i.e. having three to five saccharide radicals attached to each other by glycosidic linkages
-
- A—HUMAN NECESSITIES
- A23—FOODS OR FOODSTUFFS; TREATMENT THEREOF, NOT COVERED BY OTHER CLASSES
- A23V—INDEXING SCHEME RELATING TO FOODS, FOODSTUFFS OR NON-ALCOHOLIC BEVERAGES AND LACTIC OR PROPIONIC ACID BACTERIA USED IN FOODSTUFFS OR FOOD PREPARATION
- A23V2002/00—Food compositions, function of food ingredients or processes for food or foodstuffs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Mycology (AREA)
- Nutrition Science (AREA)
- Food Science & Technology (AREA)
- Polymers & Plastics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Coloring Foods And Improving Nutritive Qualities (AREA)
- General Preparation And Processing Of Foods (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Abstract
본 발명은 2'-O-푸코실락토오스의 제조 방법, 상기 방법으로 수득가능한 2'-O-푸코실락토오스 및 이의 용도에 관한 것이다. 방법은 염기 하에서 퍼실릴화, 보호된 하기 식 (I) 의 푸코오스 유도체와 하나 이상의 트리(C1-C6-알킬)실릴 요오다이드를 반응시킨 다음, 이에 따라 수득된 생성물과 일반식 (II) 의 화합물을 반응시키는 것을 포함한다.
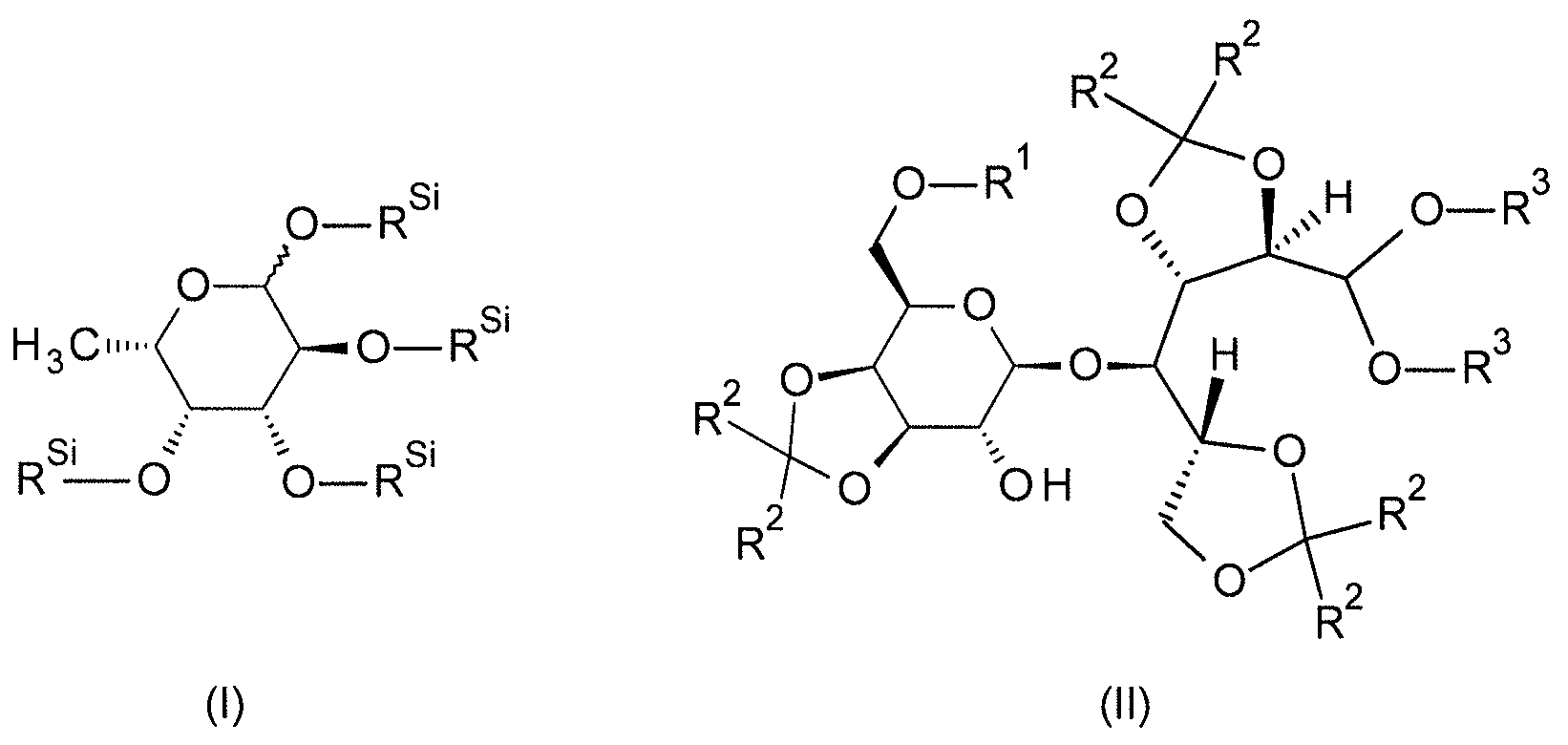
식 (I) 및 (II) 에서, 변수는 각각 하기와 같이 정의된다:
RSi 는 동일 또는 상이하고, 식 SiRaRbRc 의 잔기이고;
R1 은 C(=O)-R11 잔기 또는 SiR12R13R14 잔기이고,
R2 는 동일 또는 상이하고, C1-C8-알킬이거나, 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이하고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성함.
식 (I) 및 (II) 에서, 변수는 각각 하기와 같이 정의된다:
RSi 는 동일 또는 상이하고, 식 SiRaRbRc 의 잔기이고;
R1 은 C(=O)-R11 잔기 또는 SiR12R13R14 잔기이고,
R2 는 동일 또는 상이하고, C1-C8-알킬이거나, 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이하고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성함.
Description
본 발명은 2'-O-푸코실락토오스의 제조 방법, 상기 방법으로 수득가능한 2'-O-푸코실락토오스 및 이의 용도에 관한 것이다.
2'-O-푸코실락토오스 (CAS-No.: 41263-94-9: α-L-푸코피라노실)-(1→2)-O-β-D-갈락토피라노실-(1→4)-D-글루코피라노오스) 는 모유에서 비교적 다량 발견되는 올리고당이다. 모유에 존재하는 2'-O-푸코실락토오스가 모유 수유한 신생아에서 감염의 위험을 인과적으로 감소시킨다는 것이 다양하게 보고되었다 (예를 들어, [Weichert et al, Nutrition Research, 33 (2013), Volume 10, 831-838]; [Jantscher-Krenn et al, Minerva Pediatr. 2012, 64 (1) 83-99]; [Morrow et al, J. Pediatr. 145 (2004) 297-303] 참조). 따라서, 2'-O-푸코실락토오스는 식품 보조제 성분으로서, 특히 유아 영양을 위한 인유화 우유 제품용 첨가제로서 특히 주목받고 있다.
전형적인 화학 또는 생화학적 수단에 의한 2'-O-푸코실락토오스의 제조가 문헌에 다양하게 기재되었다 (예를 들어, [Carbohydrate Res. 88(1) (1981) 51], [Carbohydrate. Res. 154 (1986) 93-101], [Carbohydrate. Res. 212 (1991) C1-C3], [J. Org. Chem. (1997) 62, 992], [Heterocycles 84(1) (2012) 637], US 5,438,124, WO 2010/115934, WO 2010/115935, WO 2010/070616, WO 2012/113404 및 WO 2013/48294 참조). 화학적 제조는 통상적으로 활성화된 푸코실 공여체, 예컨대 메틸 1-티오-2,3,4-트리-O-벤질-β-L-푸코피라노시드, 메틸 3,4-O-이소프로필리덴-2-O-(4-메톡시벤질)-1-티오-L-푸코피라노시드, 펜테닐 3,4-O-이소프로필리덴-2-O-(4-메톡시벤질)-β-L-푸코피라노시드, 페닐 1-티오-2,3,4-트리-O-벤질-β-L-푸코피라노시드, 2,3,4-트리-O-벤질-β-L-푸코피라노실 브로마이드, 또는 2,3,4-트리-O-벤질-β-L-푸코피라노실 트리클로르아세트이미데이트 (푸코오스 공여체에 관하여, 상기 언급된 문헌 및 [Tetrahedron Lett. 31 (1990) 4325] 참조) 를 사용하는, 적합하게 보호된 수용체, 즉 아노머성 OH 기의 위치에 티오알킬 기, 알케닐옥시 기, 트리클로로아세트이미데이트 또는 브롬 원자를 갖는, 2-위치에서 부분 보호, 미보호된 락토오스 유도체, 예를 들어 4-O-(6-O-아세틸-3,4-이소프로필리덴-β-D-갈락토피라노실)-2,3;5,6-비스-O-이소프로필리덴-D-글루코오스 디메틸아세탈의 푸코실화를 기반으로 한다. 단점은 푸코오스 공여체의 복잡한, 일반적으로 다단계식 제조이다. 또 다른 단점은 푸코실화 시약의 벤질 보호기가, 생성물에서 제거하기 어렵고, 식품류에 허용되지 않는 불순물을 야기하는 중금속-함유 촉매를 사용하는 수소첨가분해에 의해 제거되어야 한다는 점이다.
예를 들어, [R. K. Jain et al., Carbohydrate Research, 212 (1991), pp. C1-C3] 은, 푸코실화 시약으로서 메틸 3,4-O-이소프로필리덴-2-O-(4-메톡시벤질)-1-티오-β-L-푸코피라노시드 또는 펜틸 3,4-O-이소프로필리덴-2-O-(4-메톡시벤질)-β-L-푸코피라노시드를 사용하는 4-O-(6-O-아세틸-3,4-이소프로필리덴-β-D-갈락토피라노실)-2,3;5,6-비스-O-이소프로필리덴-D-글루코오스 디메틸아세탈의 푸코실화를 통한 2'-O-푸코실락토오스의 제조 경로를 기재한다. 그러나, 이러한 푸코실화 시약은 제조하기 복잡하다. 유사한 합성이 [J. Org. Chem. 1997, 62, 992] 에 기재되어 있다.
WO 2010/115934 및 WO 2010/115934 는 2-O-벤질화 푸코실 공여체를 사용하는 2-푸코실락토오스의 제조를 기재한다. 푸코실 공여체는 제조하기 복잡하고, 여전히 수소첨가분해에 의해 제거되어야 하는 벤질 기를 갖는다. 유사한 방법이 WO 2010/070616 에 공지되어 있다.
WO 2012/113404 는, 그 중에서도, 글리코실화에서 푸코실 공여체로서 사용될 수 있는 O-보호된 푸코실 포스파이트를 기재한다. 여기에서 또한, 2,3,4-O-보호된 푸코오스 유도체는 먼저 다단계식 반응으로 제조되어야 하는데, 이후 인(III) 트리클로라이드 및 페놀과 반응시켜 해당 푸코실 포스파이트를 수득한다.
요약하면, 현재 공지된 2'-O-푸코실락토오스의 제조 방법은 복잡하고, 따라서 경제적이지 않다는 점이 언급될 수 있다. 나아가, 사용되는 시약은 환경독성학적 관점에서 문제가 있다. 또한, 수득된 2'-O-푸코실락토오스는 완전히 제거될 수 없는 불순물, 예컨대 벤질 보호기의 수소첨가분해 제거로부터의 전이 물질 및 방향족, 및 또한 바람직하지 않은 3당류, 예컨대 2'-O-푸코실락토오스의 β-이성질체, 즉 β-L-푸코피라노실-(1→2)-O-β-D-갈락토피라노실-(1→4)-D-글루코피라노오스를 함유한다. 이러한 불순물은 특히 2'-O-푸코실락토오스가 인체 영양물, 특히 유아 영양물에 사용되는 경우 문제가 된다.
발명의 요약
본 발명의 목적은 선행 기술의 문제점을 갖지 않는 2'-O-푸코실락토오스의 제조 방법을 제공하는 것이다. 방법은 특히 용이하게 제조될 수 있는 개시 물질, 특히 용이하게 이용가능한 푸코실 공여체의 사용을 가능하게 한다. 방법은 추가로 푸코실화에서 양호한 입체선택성 및 양호한 수율을 보장해야 한다. 또한, 방법은 전이 금속 촉매를 통한 수소첨가분해에 의한 임의의 보호기의 제거를 방지하기에 적합해야 한다.
하기 식 (I) 의 퍼실릴화, 보호된 푸코오스 유도체, 특히 하기 식 (I-α) 의 α-아노머:

[식 중,
RSi
는 식 SiRaRbRc 의 잔기 (이때,
Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임] 를,
트리(C1-C6-알킬)실릴 요오다이드와 반응시키고, 이후 하나 이상의 염기 존재 하에서 이에 따라 수득된 푸코오스 공여체 (해당 요오다이드) 를 적합한 락토오스 수용체, 즉 하기에서 보다 상세히 정의되는 일반식 (II) 의 화합물과 반응시키는 것이, 이후 수소첨가 단계를 요구하지 않으면서 2'-O-푸코실락토오스를 수득하기 위해 그 자체로 공지된 방식으로 탈보호될 수 있는 해당, 일반식 (III) 의 보호된 2'-O-푸코실락토오스 유도체를 양호한 수율 및 높은 선택성으로 수득한다는 것을 발견하였다.
따라서, 본 발명은 먼저 하기 단계를 포함하는 2'-O-푸코실락토오스의 제조 방법에 관한 것이다:
a)
하기 일반식 (I) 의 보호된 푸코오스, 특히 하기 식 (I-α) 의 α-아노머:

[식 중, RSi 는 식 SiRaRbRc 의 잔기 (이때,
Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임] 를,
일반식 (II) 의 화합물과 반응시키는 단계

[식 중,
R1
은 C(=O)-R11 잔기 또는 SiR12R13R14 잔기 (이때,
R11
은 수소, C1-C8-알킬, C1-C8-할로알킬, C3-C8-시클로알킬, C3-C8-시클로알킬-C1-C4-알킬이거나 페닐 (이때, 상기 페닐은 미치환되거나 임의로 할로겐, CN, NO2, C1-C4-알킬, C1-C4-알콕시, C1-C4-할로알킬 및 C1-C4-알콕시로부터 선택되는 1 내지 5 개의 치환기를 가짐) 이고,
R12, R13 및 R14 은 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 이고;
R2
는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3
는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성함];
b)
2'-O-푸코실락토오스를 수득하기 위해, 단계 a) 에서 수득된 일반식 (III) 의 커플링 생성물을 탈보호하는 단계

[식 중, RSi, R1, R2 및 R3 는 상기 정의된 바와 같음];
여기서, 단계 a) 는
a.1)
트리(C1-C6-알킬)실릴 요오다이드로의 일반식 (I) 의 보호된 푸코오스의 처리 및
a.2)
하나 이상의 염기 존재 하, 식 (II) 의 화합물과 단계 a.1) 에서 수득된 생성물의 반응
을 포함함.
본 발명은 추가로 일반식 (IIIa), (IIIb), (IIIc) 및 (IV) 의 보호 및 부분 보호된 2'-O-푸코실락토오스 유도체에 관한 것이다:
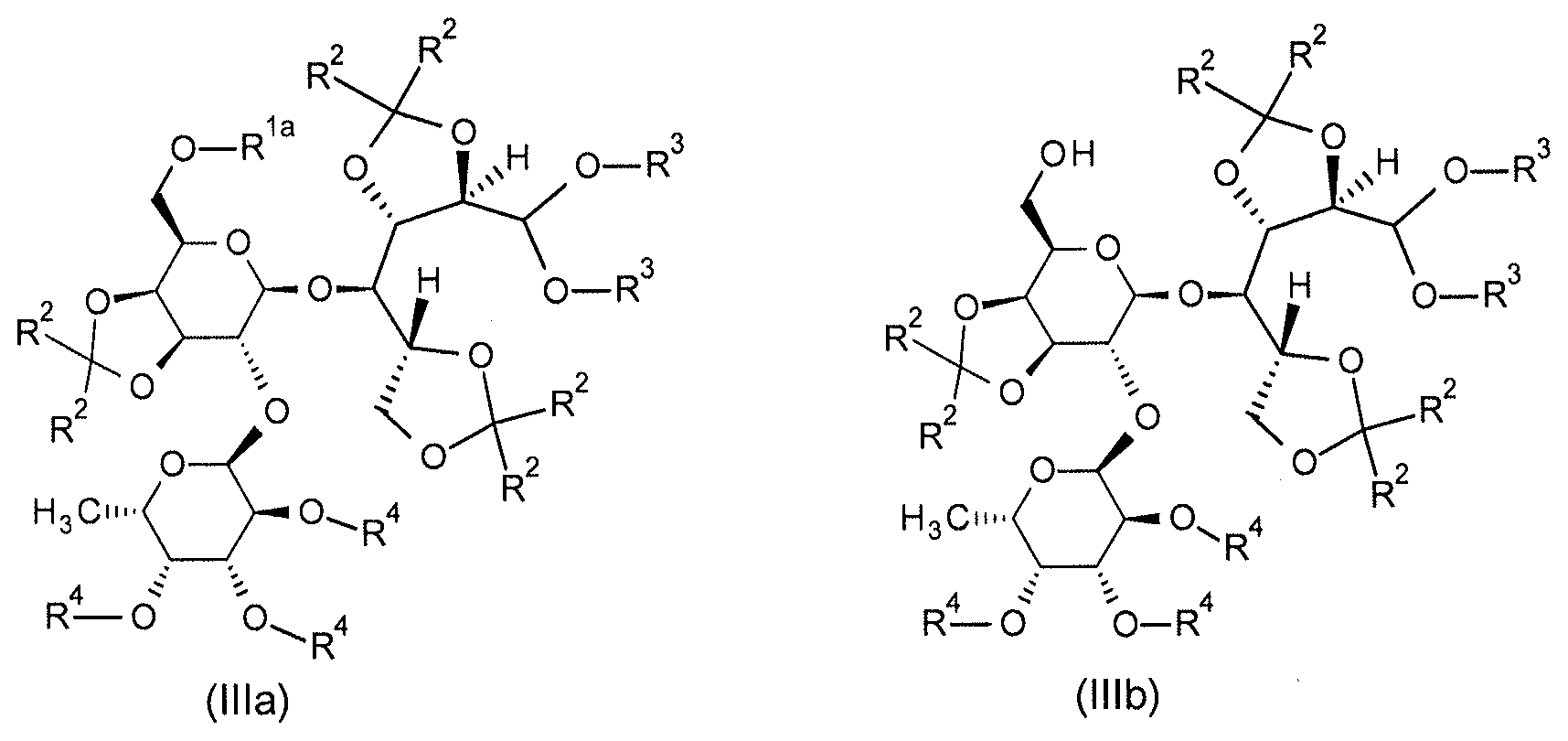

[식 중,
R1a
(식 (IIIa) 에서) 는 SiR12R13R14 잔기 (이때,
R12, R13 및 R14 은 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 이고;
R2
(식 (IIIa), (IIIb) 및 (IIIc) 에서) 는 동일 또는 상이하고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3
(식 (IIIa), (IIIb) 및 (IIIc) 에서) 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R4
(식 (IIIa), (IIIb) 및 (IIIc) 에서) 는 동일 또는 상이하고, 수소이거나 SiRaRbRc 잔기 (이때,
Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 이고;
R11
(식 (IIIc) 및 (IV) 에서) 은 수소, C1-C8-알킬, C1-C8-할로알킬, C3-C8-시클로알킬, C3-C8-시클로알킬-C1-C4-알킬이거나 페닐 (이때, 상기 페닐은 미치환되거나 임의로 할로겐, CN, NO2, C1-C4-알킬, C1-C4-알콕시, C1-C4-할로알킬 및 C1-C4-할로알콕시로부터 선택되는 1 내지 5 개의 치환기를 가짐) 임].
본 발명은 추가로 일반식 (II) 의 부분 보호된 락토오스 유도체 (식 중, R1 은 SiR12R13R14 잔기 (이때, R12, R13 및 R14 은 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임) 에 관한 것이다. 이는 하기 식 (IIa) 로 기재된다:

[식 중, R1a 는 SiR12R13R14 잔기 (이때, R12, R13 및 R14 은 상기 정의된 바와 같음) 이고, R2 및 R3 는 식 (II), (III), (IIIa), (IIIb) 및 (IIIc) 에 대해 정의된 바와 같음].
본 발명의 방법은 일련의 장점과 연관된다. 식 (I) 의 화합물은 푸코오스로부터 한 단계로 제조될 수 있기 때문에, 푸코실 공여체의 복잡한 제조가 회피될 수 있다. 방법은 글리코실화에 관하여 양호한 입체선택성 및 양호한 수율로 식 (III) 의 1차 커플링 생성물을 제공한다. 식 (III) 의 화합물의 보호기의 제거는 전이 금속 촉매를 통한 수소첨가분해에 대한 요구 없이 온화한 가수분해 조건 하에서 가능하다. 수득된 식 (III), 특히 식 (IIIa), (IIIb) 및 (IIIc) 의 중간체는 안정하고, 특히 저장 중 안정하고, 정제될 수 있다. 또한, 방법은 비교적 대규모로 용이하게 수행될 수 있다. 추가의 이점은 공지된 2'-O-푸코실락토오스에 비해, 본 발명에 따른 방법으로 수득가능한 2'-O-푸코실락토오스가, 예를 들어 수소첨가로부터 수득된 중금속 화합물 및 중금속, 및 또한 보호기의 수소첨가에 의해 형성된 알킬 방향족 화합물 (이들 불순물은 제거될 수 없음) 을 포함하지 않거나, 오로지 훨씬 낮은 분율로 포함한다는 점이다. 또한, 본 발명의 방법으로 바람직하지 않은 β-이성질체가 형성되지 않거나, 선행 기술 방법에서 형성된 β-이성질체의 양보다 훨씬 낮은, 매우 낮은 양으로만 오로지 형성된다. 실제로, 식 (I) 의 화합물과 식 (II) 의 화합물의 반응으로, 식 (III) 의 화합물의 바람직하지 않은 β-이성질체는 β-이성질체 (III-β) 대 α-이성질체 (III-α) 의 몰비가 1 : 25 를 초과하지 않고, 특히 1 : 35 내지 1 : 40 범위가 되도록 하는 낮은 양으로 형성된다. 따라서, 본 발명의 방법은 임의로 추가 정제 후 1 중량% 미만, 특히 0.5 중량% 미만의 바람직하지 않은 β-이성질체를 함유하는 목적하는 2'-O-푸코실락토오스를 함유하는 2'-O-푸코실락토오스의 제조를 가능하게 한다.
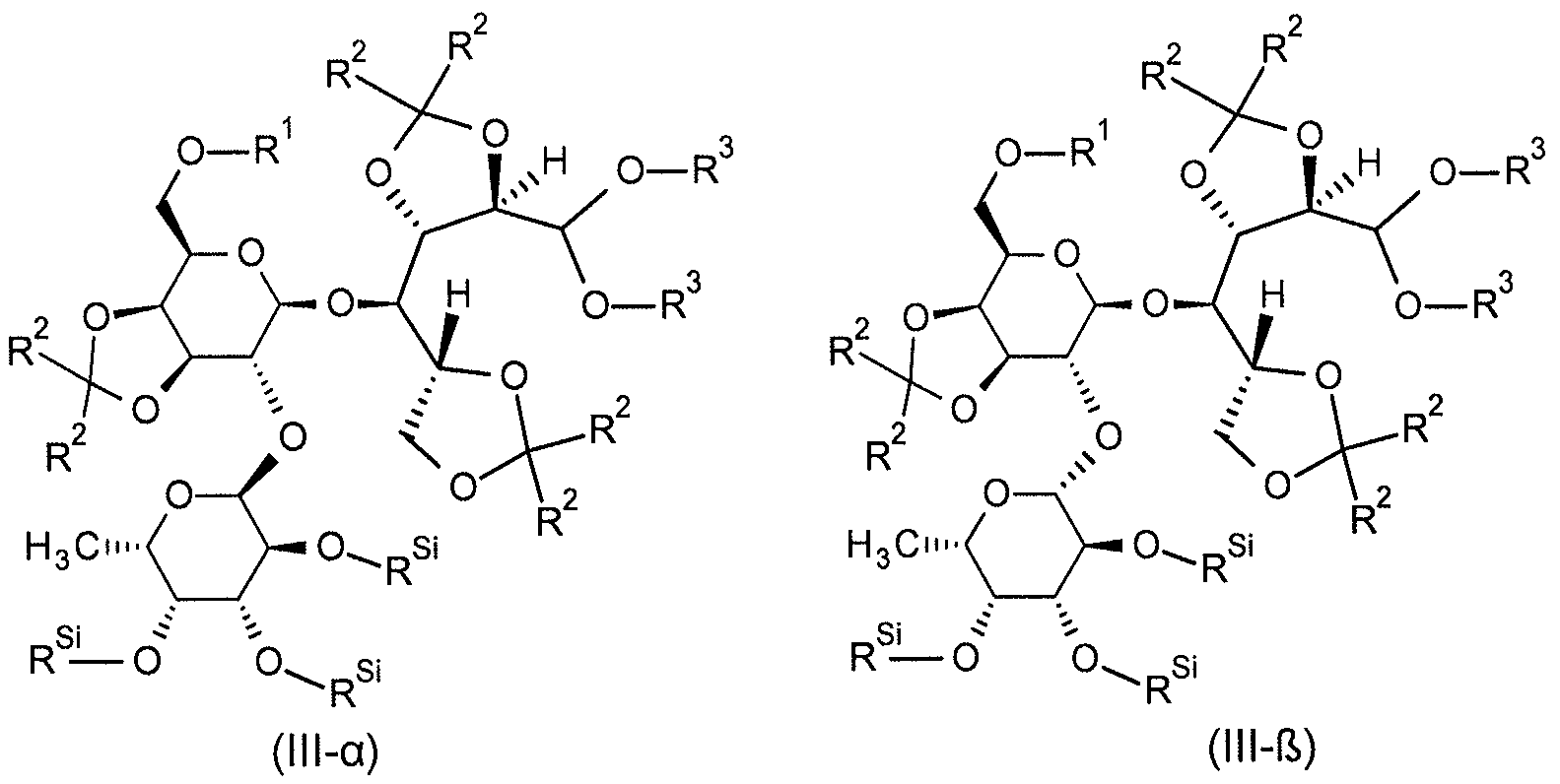
방법 및 방법으로 수득된 식 (IIa) 의 반응물 및 식 (IIIa), (IIIb), (IIIc) 및 (IV) 의 중간체 생성물이 이에 따라 2'-O-푸코실락토오스의 제조에 특히 적합하다. 따라서, 본 발명은 또한 2'-O-푸코실락토오스를 제조하기 위한 일반식 (IIa) 의 화합물의 용도 및 또한 2'-O-푸코실락토오스를 제조하기 위한 일반식 (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물의 용도에 관한 것이다.
본 발명에 따른 방법으로 수득된 2'-O-푸코실락토오스의 품질은 식품류 제조에 특히 적합하다. 따라서, 본 발명은 또한 하기에 관한 것이다:
-
본원에 기재된 방법으로 수득가능한 2'-O-푸코실락토오스;
-
일반식 (IIa), (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물 중 하나 이상을 사용하여 제조된 2'-O-푸코실락토오스;
-
본원에 기재된 방법으로 수득가능한 2'-O-푸코실락토오스의 식품류에서 또는 식품 첨가제로서의 용도;
-
일반식 (IIa), (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물 중 하나 이상을 사용하여 제조된 2'-O-푸코실락토오스의 식품류에서 또는 식품 첨가제로서의 용도;
-
일반식 (IIa), (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물 중 하나 이상으로부터의 2'-O-푸코실락토오스 제조 및 이에 따라 수득가능한 2'-O-푸코실락토오스의 식품류에서의 제형화를 포함하는 식품류 제조 방법;
-
본원에 기재된 방법으로 수득가능한 2'-O-푸코실락토오스 및 식품류에 적합한 하나 이상의 담체를 포함하는 식품류 또는 식품 첨가제.
본 발명의 맥락에서, 사용된 용어는 일반적으로 하기와 같이 정의된다:
접두사 Cx-Cy 는 특정 경우에서 가능한 탄소 원자의 수를 의미한다.
용어 "할로겐" 은 각각의 경우 플루오린, 브롬, 클로린 또는 요오드, 특히 플루오린, 클로린 또는 브롬을 의미한다.
용어 "C1-C4-알킬" 은 1 내지 4 개의 탄소 원자를 포함하는 선형 또는 분지형 알킬 잔기, 예컨대 메틸, 에틸, 프로필, 1-메틸에틸 (이소프로필), 부틸, 1-메틸프로필 (sec-부틸), 2-메틸프로필 (이소부틸) 또는 1,1-디메틸에틸 (tert-부틸) 을 의미한다.
용어 "C1-C8-알킬" 은 1 내지 8 개의 탄소 원자를 포함하는 선형 또는 분지형 알킬 잔기를 의미한다. C1-C4-알킬에 대해 언급된 잔기 이외의 예는 n-펜틸, n-헥실, n-헵틸, n-옥틸, 2-펜틸, 2-헥실, 2-헵틸, 2-옥틸, 3-펜틸, 3-헥실, 3-헵틸, 3-옥틸, 2,2-디메틸프로필, 2-메틸부틸, 3-메틸부틸, 2-에틸부틸, 3-에틸부틸, 2-메틸펜틸, 3-메틸펜틸, 4-메틸펜틸, 2-에틸펜틸, 3-에틸펜틸, 4-에틸펜틸, 2-에틸헥실 및 이의 위치 이성질체이다.
용어 "C1-C8-할로알킬" 은 1 내지 8 개의 탄소 원자, 특히 1 내지 4 개의 탄소 원자 (C1-C4-할로알킬) 를 포함하는 선형 또는 분지형 알킬 잔기 (이의 하나 이상 또는 모든 수소 원자는 할로겐 원자, 특히 플루오린 또는 클로린 원자로 대체됨) 를 의미한다. 이러한 목적에 대한 예는 클로로메틸, 디클로로메틸, 트리클로로메틸, 플루오로메틸, 디플루오로메틸, 트리플루오로메틸, 클로로플루오로메틸, 디클로로플루오로메틸, 클로로디플루오로메틸, 2,2-디플루오로에틸, 2,2,2-트리플루오로에틸, 1,1,2,2-테트라플루오로에틸, 펜타플루오로에틸, 2,2-디플루오로프로필, 3,3-디플루오로프로필, 3,3,3-트리플루오로프로필, 2,2,3,3,3-펜타플루오로프로필, 헵타플루오로프로필 등이다.
용어 "C1-C4-알콕시" 는 산소 원자를 통해 결합되어 있는 1 내지 4 개의 탄소 원자를 포함하는 직쇄 또는 분지형 포화 알킬 기를 의미한다. C1-C4-알콕시의 예는 메톡시, 에톡시, n-프로폭시, 1-메틸에톡시 (이소프로폭시), n-부톡시, 1-메틸프로폭시 (sec-부톡시), 2-메틸프로폭시 (이소부톡시) 및 1,1-디메틸에톡시 (tert-부톡시) 이다.
용어 "C1-C4-할로알콕시" 는 산소 원자를 통해 결합되어 있는 1 내지 4 개의 탄소 원자를 포함하는 직쇄 또는 분지형 포화 할로알킬 기를 의미한다. 이러한 경우의 예는 플루오로메톡시, 디플루오로메톡시, 트리플루오로메톡시, 1-플루오로에톡시, 2-플루오로에톡시, 2,2-디플루오로에톡시, 2,2,2-트리플루오로에톡시, 1,1,2,2-테트라플루오로에톡시, 펜타플루오로에톡시, 3,3,3-트리플루오로프로프-1-옥시, 1,1,1-트리플루오로프로프-2-옥시, 1-플루오로부톡시, 2-플루오로부톡시, 3-플루오로부톡시, 4-플루오로부톡시 등이다.
용어 "C3-C8-시클로알킬" 은 3 내지 8 개의 탄소 원자를 포함하는 시클릭, 포화 히드로카르빌 잔기를 의미한다. 예는 시클로프로필, 시클로부틸, 시클로펜틸, 시클로헥실, 시클로헵틸 및 시클로옥틸이다.
용어 "C3-C8-시클로알킬-C1-C4-알킬" 은 1 내지 4 개의 탄소 원자를 포함하는 선형 또는 분지형 알킬 잔기 (상기 정의된 바와 같이 이의 하나의 수소 원자는 C3-C8-시클로알킬로 대체됨) 를 의미한다.
용어 "선형 C1-C4-알칸디일" 은 1 내지 4 개의 탄소 원자를 갖는 선형, 2가 히드로카르빌 잔기, 예컨대 메틸렌, 에탄-1,2-디일, 프로판-1,3-디일, 및 부탄-1,4-디일을 의미한다.
용어 "선형 C3-C6-알칸디일" 은 3 내지 6 개의 탄소 원자를 갖는 선형, 2가 히드로카르빌 잔기, 예컨대 프로판-1,3-디일, 부탄-1,4-디일, 펜탄-1,5-디일 및 헥산-1,6-디일을 의미한다.
용어 "식품류" 또는 "식품" 은 포유류, 특히 인간을 위한 영양물로 의도되고, 적합한 조성물 및 제형을 의미한다. 본 발명의 맥락에서, 이들은 자연-발생 제품, 예를 들어 유제품, 또한 직접 사용되거나, 임의로 레디-투-유즈 (ready-to-use) 제형으로 전환된 다음 액체를 첨가함으로써 사용될 수 있는, 예를 들어 식이 또는 의약 영양물용 인공 제조 제형 기재의 조성물을 모두 포함한다.
용어 "식품 첨가제" 는 화학적, 물리적 또는 또한 생리학적 효과를 달성하기 위해 식품류와 혼합되는 물질을 의미한다.
본 발명에 따른 방법 및 식 (II), (IIa), (III), (IIIa), (IIIb), (IIIb'), (IIIc) 및 (IIIc') 의 화합물에 대하여, 한 식 내의 변수 R2 는 바람직하게는 각각의 경우 동일한 정의를 갖는다. R2 는 특히 C1-C4-알킬, 특히 메틸이거나 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 1,5-펜탄디일이고, 따라서 이들이 부착되어 있는 탄소 원자와 시클로헥산-1,1-디일 잔기를 형성한다. 모든 R2 잔기는 특히 메틸이다.
본 발명에 따른 방법 및 식 (II), (IIa), (III), (IIIa), (IIIb), (IIIb'), (IIIc) 및 (IIIc') 의 화합물에 대하여, 한 식 내의 변수 R3 는 바람직하게는 각각의 경우 동일한 정의를 갖는다. R3 는 특히 C1-C4-알킬, 특히 메틸이다.
본 발명에 따른 방법 및 식 (IIIa), (IIIb) 및 (IIIc) 의 화합물에 대하여, 한 식 내의 변수 R4 는 바람직하게는 각각의 경우 동일한 정의를 갖는다. R4 는 특히 수소이거나 트리(C1-C4-알킬)실릴, 특히 수소이거나 트리메틸실릴, 즉 SiRaRbRc 잔기에서 잔기 Ra, Rb 및 Rc 가 동일 또는 상이하고, 특히 C1-C4-알킬, 특히 메틸이다. 식 (IIIa) 의 화합물에서, R4 는 특히 트리(C1-C4-알킬)실릴, 특히 트리메틸실릴이다. 식 (IIIb) 의 화합물에서, R4 는 특히 수소이다.
본 발명에 따른 방법 및 식 (I) 및 (II) 의 화합물에 대하여, 한 식 내의 변수 RSi 는 바람직하게는 각각의 경우 동일한 정의를 갖는다. RSi 는 특히 수소 이거나 트리(C1-C4-알킬)실릴, 특히 수소이거나 트리메틸실릴, 즉 SiRaRbRc 잔기에서 잔기 Ra, Rb 및 Rc 가 동일 또는 상이하고, 특히 C1-C4-알킬, 특히 메틸이다.
제 1 구현예는 식 (II) 및 (III) 의 화합물에서, R1 잔기가 SiR12R13R14 잔기, 특히 트리(C1-C4-알킬)실릴, 특히 트리메틸실릴, 즉 SiR12R13R14 잔기에서 잔기 R12, R13 및 R14 이 동일 또는 상이하고, 특히 C1-C4-알킬, 특히 메틸인 방법에 관한 것이다. 따라서, 식 (IIa) 및 (IIIa) 에서, R1a 는 트리(C1-C4-알킬)실릴, 특히 트리메틸실릴이다.
제 2 바람직한 구현예는 식 (II) 및 (III) 의 화합물에서, R1 잔기가 C(=O)-R11 잔기 (이때, R11 은 상기 정의된 바와 같고, 특히 수소, C1-C4-알킬, C1-C4-할로알킬 또는 페닐, 특히 메틸, tert-부틸 또는 페닐임) 인 방법에 관한 것이다. 따라서, 식 (IIIc), (IIIc') 및 (IV) 에서, R11 잔기는 바람직하게는 수소, C1-C4-알킬, C1-C4-할로알킬 또는 페닐, 특히 메틸, tert-부틸 또는 페닐이다. 본 발명의 특정 구현예에서, R11 은 메틸이 아니다. 구현예의 특수한 기에서, R11 은 메틸이다. 구현예의 추가 특수한 기에서, R11 은 tert-부틸이다.
식 (I) 의 특히 바람직한 화합물의 예는 모든 RSi 잔기가 트리메틸실릴인 식 (I) 의 화합물이다.
본 발명의 방법에서, 식 (I) 의 화합물은 통상적으로 이의 α-아노머 (I-α) 형태로 이용된다. 또한, 식 (I) 의 화합물에 대해 이의 β-아노머 (I-β), α-아노머 (I-α) 및 β-아노머 (I-β) 의 혼합물이 가능하다. 식 (I) 의 화합물은 통상적으로 이의 α-아노머 (I-α) 로 본질적으로 이루어진, 즉 α-아노머 대 β-아노머의 비가 적어도 8 : 1 또는 적어도 9 : 1 인 형태로 이용된다. 그러나, 비는 (III) 의 목적하는 이성질체의 형성에 유의한 영향을 미치지 않는다.

식 (II) 의 특히 바람직한 화합물의 예는 모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, R1 이 트리메틸실릴인 식 (II) 의 화합물이다. 이러한 화합물은 또한 식 (IIa) 의 화합물의 예이다.
식 (II) 의 추가로 특히 바람직한 화합물의 예는 또한 모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, R1 이 아세틸인 식 (II) 의 화합물이다.
식 (II) 의 추가로 특히 바람직한 화합물의 또 다른 예는 또한 모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, R1 이 벤조일인 식 (II) 의 화합물이다.
식 (II) 의 추가로 특히 바람직한 화합물의 또 다른 예는 또한 모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, R1 이 피발로일, 즉 C(=O)-C(CH3)3 인 식 (II) 의 화합물이다.
식 (III) 의 특히 바람직한 화합물의 예는,
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 RSi 잔기가 트리메틸실릴이고, R1 이 트리메틸실릴인 식 (III) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 RSi 잔기가 트리메틸실릴이고, R1 이 아세틸인 식 (III) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 RSi 잔기가 트리메틸실릴이고, R1 이 피발로일인 식 (III) 의 화합물이다.
식 (IIIa) 의 특히 바람직한 화합물의 예는,
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 트리메틸실릴이고, R1a 가 트리메틸실릴인 식 (IIIa) 의 화합물; 및
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 수소이고, R1a 가 트리메틸실릴인 식 (IIIa) 의 화합물이다.
식 (IIIb) 의 특히 바람직한 화합물의 예는,
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 트리메틸실릴인 식 (IIIb) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 수소인 식 (IIIb) 의 화합물이다.
식 (IIIc) 의 특히 바람직한 화합물의 예는,
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 트리메틸실릴이고, R11 이 수소인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 수소이고, R11 이 수소인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 트리메틸실릴이고, R11 이 메틸인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 수소이고, R11 이 메틸인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 트리메틸실릴이고, R11 이 페닐인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 수소이고, R11 이 페닐인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 트리메틸실릴이고, R11 이 tert-부틸인 식 (IIIc) 의 화합물;
-
모든 R2 잔기가 메틸이고, 모든 R3 잔기가 메틸이고, 모든 R4 잔기가 수소이고, R11 이 tert-부틸인 식 (IIIc) 의 화합물이다.
식 (IV) 의 특히 바람직한 화합물의 예는,
-
R11 이 메틸인 식 (IV) 의 화합물;
-
R11 이 에틸인 식 (IV) 의 화합물;
-
R11 이 페닐인 식 (IV) 의 화합물;
-
R11 이 tert-부틸인 식 (IV) 의 화합물이다.
본 발명에 따른 방법의 단계 a) 는 하나 이상의 트리(C1-C6-알킬)실릴 요오다이드로의 일반식 (I) 의 보호된 푸코오스의 처리를 포함한다. 이러한 경우, 식 (I) 의 화합물은 선택적으로 일반식 (I') 의 해당 요오다이드로 전환된다:

이러한 반응은 또한 하기 단계 a.1) 으로 지칭된다. 수득된 반응 생성물은 이후 식 (II) 의 화합물과 반응하고, 이때 반응은 임의로 반응 (단계 a.2) 에서 형성된 요오드화수소를 제거 (scavenge) 하기 위해 하나 이상의 염기 존재 하에서 수행된다.
사용된 트리(C1-C6-알킬)실릴 요오다이드는 바람직하게는 트리메틸실릴 요오다이드이다.
트리(C1-C6-알킬)실릴 요오다이드는 바람직하게는 식 (I) 의 화합물의 몰 당 0.8 mol 내지 1.4 mol 또는 0.8 mol 내지 1.2 mol 의 양, 특히 0.9 내지 1.1 mol 의 양, 특히 0.9 내지 1 mol 의 양으로 사용된다.
트리(C1-C6-알킬)실릴 요오다이드, 특히 트리메틸실릴 요오다이드가 이와 같이 사용될 수 있다. 트리(C1-C6-알킬)실릴 요오다이드, 특히 트리메틸실릴 요오다이드는 또한 제자리에서 제조될 수 있다.
예를 들어, 트리(C1-C6-알킬)실릴 요오다이드의 제자리 제조는 요오다이드 염, 특히 알칼리 금속 요오다이드, 예컨대 리튬 요오다이드, 칼륨 요오다이드 또는 나트륨 요오다이드로의 해당 트리(C1-C6-알킬)실릴 클로라이드의 처리에 의해 달성될 수 있다. 이에 적합한 방법이, 예를 들어 [Synthesis 1983, p. 459], [Synthesis 1979, p. 740], [Synthesis 1981, p. 67], [Chem. Ber. 1962, 95, p. 174] 및 [Bioorganic and Med. Chem. Lett. 10, 2000, p. 2311] (유사하게 적용될 수 있음) 에 공지되어 있다. 이러한 목적을 위해, 요오다이드 염은 바람직하게는 트리(C1-C6-알킬)실릴 클로라이드를 기준으로 적어도 등몰량으로 사용되고, 특히 트리(C1-C6-알킬)실릴 클로라이드를 기준으로 과량으로 사용된다. 이러한 경우, 바람직한 절차는 트리(C1-C6-알킬)실릴 요오다이드, 특히 트리메틸실릴 요오다이드가 요오다이드 염, 특히 알칼리 금속 요오다이드, 예컨대 리튬 요오다이드, 칼륨 요오다이드 또는 나트륨 요오다이드로의 해당 트리(C1-C6-알킬)실릴 클로라이드의 처리에 의해 초기 제조되고, 반응 생성물이 일반식 (I) 의 화합물에 첨가되는 것이다. 제조는 바람직하게는 적합한 용매, 특히 비양성자성 용매, 예컨대 아세토니트릴 또는 프로피오니트릴에서 수행된다.
트리(C1-C6-알킬)실릴 요오다이드의 제자리 제조는 해당 헥사(C1-C6-알킬)디실란, 특히 헥사메틸디실란 (HMDS) 을 요오드와 반응시킴으로써 달성될 수 있다. 이에 적합한 방법이, 예를 들어 [Synthesis Commun. 1974, p. 740]; [Chem. Commun. 2003, p. 1266]; [Carb. Lett. 1998, 3, p. 179] (유사하게 적용될 수 있음) 에 공지되어 있다.
이러한 목적을 위해, 헥사-(C1-C6-알킬)디실란, 특히 HMDS 를 바람직하게는 제 1 단계에서 요오드 원소와 반응시킨 다음, 식 (I) 의 화합물을 이에 따라 수득된 반응 혼합물에 첨가한다. 헥사-(C1-C6-알킬)디실란, 특히 HMDS 의 요오드와의 반응은 벌크 또는 불활성 유기 용매에서 수행될 수 있다. 적합한 불활성 용매는 특히 할로겐화 탄화수소, 예컨대 트리클로로메탄 및 디클로로메탄을 포함한다. 헥사-(C1-C6-알킬)디실란, 특히 HMDS 의 요오드 원소와의 반응은 0 내지 110℃, 특히 0 내지 60℃ 범위의 온도에서 흔히 수행된다. 대안적으로, 헥사-(C1-C6-알킬)디실란, 특히 HMDS 는 요오드 및 식 (I) 의 화합물과 반응할 수 있다. 이러한 대안은 바람직하게는 불활성 용매에서 수행된다. 적합한 불활성 용매는 특히 할로겐화 탄화수소, 예컨대 트리클로로메탄 및 디클로로메탄을 포함한다. 바람직하게는, 헥사-(C1-C6-알킬)디실란 및 요오드는 0.5 : 1 내지 1 : 0.5 범위의 몰 비, 특히 약 1 : 1 의 몰 비로 반응한다. 바람직하게는, 헥사-(C1-C6-알킬)디실란 및 식 (I) 의 화합물은 0.5 : 1 내지 1 : 1 범위의 몰 비, 특히 0.5 : 1 내지 0.8 : 1 범위의 몰 비로 사용된다. 바람직하게는, 요오드 및 식 (I) 의 화합물은 0.5 : 1 내지 1 : 1 범위의 몰 비, 특히 0.5 : 1 내지 0.8 : 1 범위의 몰 비로 이용된다.
식 (I) 의 화합물은 일반적으로 불활성 유기 용매 또는 희석액에서 트리(C1-C6-알킬)실릴 요오다이드와 반응한다. 바람직한 것은 비양성자성 용매, 특히 낮은 함량의 물, 알코올 또는 산과 같은 양성자성 불순물을 갖는 것이다. 용매 중 양성자성 불순물 함량은 바람직하게는 1000 ppm 미만이다. 바람직하게는, 본 발명에 따른 방법에서 사용되기 전, 비양성자성 용매는 양성자성 불순물, 특히 물의 함량을 감소시키기 위해 적합한 흡착제, 예를 들어 포어 크기가 3 내지 4 옹스트롬인 분자체로 처리함으로써 처리된다. 바람직한 유기 용매는, 알켄 및 시클로알켄, 예컨대 이소부텐, 아밀렌 (1-펜텐, 2-펜텐, 2-메틸부트-1-엔, 3-메틸부트-1-엔 및 이의 혼합물), 시클로펜텐 및 시클로헥센, 할로알칸, 예컨대 디클로로메탄, 트리클로로메탄, 디클로로에탄, 방향족 탄화수소, 예컨대 톨루엔 및 자일렌, 및 또한 알킬 니트릴, 예컨대 아세토니트릴, 및 또한 상기 언급된 용매의 혼합물이다. 용매는 바람직하게는 모든 성분이 용해 형태로 존재하는 것이 선택된다. 식 (I) 의 화합물의 총 농도는 전체 시약 및 용매의 총 중량을 기준으로, 바람직하게는 5 내지 70 중량%, 특히 10 내지 50 중량% 범위이다. 예를 들어, 본 발명의 방법은 알켄이 아닌, 화합물 (I) 을 기준으로 5 내지 100 mol-% 의 안정화 첨가제로서의 알켄 또는 시클로알켄을 임의로 함유하는 비양성자성 용매에서 수행될 수 있다. 또한, 용매로서 하나 이상의 알켄에서 반응을 수행할 수 있다. 또한, 반응을 수행한 후 HI 또는 I2 를 제거하기 위해 안정화제로서 알켄을 첨가할 수 있다.
식 (I) 의 화합물은 바람직하게는 -20 내지 110℃ 범위, 특히 0 내지 80℃ 범위, 특히 20 내지 65℃ 범위의 온도에서 트리(C1-C6-알킬)실릴 요오다이드와 반응한다. 반응은 주변 압력, 감압 또는 승압에서 수행될 수 있다. 반응은 통상적으로 900 내지 1100 mbar 범위의 압력에서 실시된다.
식 (I) 의 화합물과 트리(C1-C6-알킬)실릴 요오다이드의 반응으로부터 수득되는 반응 생성물은 바람직하게는 단리되지 않지만, 추가 단리 또는 정제 없이, 특히 염기 존재 하에서 식 (II) 의 화합물과 반응하여 식 (III) 의 화합물을 수득한다. 식 (I) 의 화합물과 트리(C1-C6-알킬)실릴 요오다이드의 반응으로부터 수득되는 반응 생성물은 또한 예를 들어 바람직하게는 감압 하에서 반응 혼합물로부터 휘발성 성분을 제거함으로써, 및/또는 적합한 저-비등점 물질 (low-boiler), 예를 들어 헥산, 시클로헥산 또는 헵탄과 같은 알칸, 또는 톨루엔과 같은 방향족 화합물과 공-증발함으로써 정제 또는 단리될 수 있다.
임의로, 단계 a.2) 에서의 식 (II) 의 화합물과의 반응 전 단계 a.1) 에서 수득된 반응 생성물에 무기 염기가 첨가될 수 있다. 흔히, 무기 염기는 알칼리 카르보네이트, 알칼리 바이카르보네이트, 알칼리 토금속 카르보네이트 및 알칼리 토금속 바이카르보네이트로부터, 특히 알칼리 카르보네이트, 예컨대 리튬, 나트륨 및 칼륨 카르보네이트, 및 알칼리 바이카르보네이트, 예컨대 나트륨- 또는 칼륨 바이카르보네이트로부터 선택된다. 목적하는 경우, 이러한 무기 염기는 바람직하게는 식 (I) 의 화합물의 몰 당 0.01 내지 0.5 당량의 양, 즉 카르보네이트의 경우 식 (I) 의 화합물의 몰 당 0.005 mol 내지 0.25 mol 의 양, 바이카르보네이트의 경우 식 (I) 의 화합물의 몰 당 0.01 mol 내지 0.5 mol 의 양으로 첨가된다.
단계 a.1, 즉 트리(C1-C6-알킬)실릴 요오다이드로의 화합물 (I) 의 처리로부터 수득된 반응 생성물은 본 발명에 따라 단계 a.2 에서 식 (II) 의 화합물과 반응한다.
단계 a.2 에서의 반응은 하나 이상의 염기 존재 하에 수행된다. 2차 반응을 회피하기 위해, 염기는 바람직하게는 식 (I) 의 화합물을 기준으로 적어도 등몰량으로 사용된다. 특히, 염기는 식 (I) 의 화합물의 몰 당 1 내지 3 mol 의 양, 특히 식 (I) 의 화합물의 몰 당 1 내지 1.5 mol 의 양으로 사용된다.
바람직한 염기는 1차 아민 염기, 특히 3차 아민, 특히 피리딘 염기 및 또한 3차 지방족 또는 시클로지방족 아민이다. 적합한 피리딘 염기는, 예를 들어 피리딘, 퀴놀린 및 C1-C6-알킬-치환 피리딘, 특히 모노-, 디- 및 트리(C1-C6-알킬)피리딘, 예컨대 2,6-디(C1-C6-알킬)피리딘, 예를 들어 2,6-디메틸피리딘 또는 2,6-비스(tert-부틸)피리딘, 및 콜리딘이다. 적합한 3차 지방족 또는 시클로지방족 아민은 트리(C1-C6-알킬)아민, 예컨대 트리메틸아민, 트리에틸아민, 디이소프로필메틸아민, 트리-n-부틸아민 또는 이소프로필디메틸아민, C3-C8-시클로알킬-디(C1-C6-알킬)아민, 예컨대 시클로헥실디메틸아민, N-(C1-C6-알킬)피페리딘, 예컨대 N-메틸피페리딘, 및 디(C3-C8-시클로알킬)-C1-C6-알킬아민, 예컨대 비스시클로헥실메틸아민이다. 특히 바람직한 것은 트리(C1-C6-알킬)아민, 특히 트리메틸아민 및 트리에틸아민이다. 적합한 염기는 또한 알칼리 카르보네이트, 알칼리 바이카르보네이트, 알칼리 토금속 카르보네이트 및 알칼리 토금속 바이카르보네이트로 이루어진 군으로부터, 특히 알칼리 카르보네이트, 예컨대 리튬, 나트륨 및 칼륨 카르보네이트, 및 알칼리 바이카르보네이트, 예컨대 나트륨- 또는 칼륨 바이카르보네이트로부터 선택되는 무기 염기이다.
바람직하게는, 단계 a.2 에서 사용되는 염기는 하나 이상의 아민 염기, 특히 하나 이상의 3차 아민를 포함한다. 특히, 염기는 하나 이상의 아민 염기, 특히 하나 이상의 3차 아민, 및 알칼리 카르보네이트, 알칼리 바이카르보네이트, 알칼리 토금속 카르보네이트 및 알칼리 토금속 바이카르보네이트로 이루어진 군으로부터, 특히 알칼리 카르보네이트 및 알칼리 바이카르보네이트로부터 선택되는 하나 이상의 무기 염기의 조합을 포함한다. 아민 염기 및 무기 염기의 조합이 사용되는 경우, 아민 염기는 바람직하게는 식 (I) 의 화합물의 몰 당 1 내지 2 mol 의 양, 특히 식 (I) 의 화합물의 몰 당 1 내지 1.5 mol 의 양으로 이용된다. 이러한 조합에서, 무기 염기는 바람직하게는 식 (I) 의 화합물의 몰 당 0.01 내지 0.5 당량, 즉 카르보네이트의 경우 식 (I) 의 화합물의 몰 당 0.005 mol 내지 0.25 mol 의 양, 바이카르보네이트의 경우 식 (I) 의 화합물의 몰 당 0.01 mol 내지 0.5 mol 의 양으로 이용된다.
식 (II) 의 화합물은 일반적으로 식 (I) 의 화합물 대 식 (II) 의 화합물의 몰 비가 1 : 3 내지 3 : 1 범위, 특히 1 : 2 내지 2 : 1 범위, 특히 바람직하게는 1 : 1.5 내지 1.5 : 1 범위, 특히 1 : 1.1 내지 1.1 : 1 범위인 양으로 사용된다.
바람직하게는, 단계 a.2) 는 요오드 원소, 요오다이드 염, 트리아릴포스핀 산화물 및 이의 혼합물로부터 선택되는 하나 이상의 추가 시약 존재 하에 수행된다. 적합한 요오드 염은 알칼리금속 요오다이드 및 또한 테트라알킬암모늄 요오다이드, 특히 테트라-C1-C6-알킬암모늄 요오다이드, 예컨대 테트라에틸암모늄 요오다이드 및 테트라프로필암모늄 요오다이드, 특히 테트라부틸암모늄 요오다이드를 포함한다. 바람직한 것은 알칼리금속 요오다이드, 특히 NaI 및 KI 이다. 적합한 트리아릴포스핀산화물은 트리페닐포스핀산화물이다. 특히, 단계 a.2) 는 요오드 원소 및 요오다이드 염으로 이루어진 군으로부터, 특히 요오드 원소 및 알칼리금속 요오다이드 및 이의 혼합물로 이루어진 군으로부터 선택되는 하나 이상의 추가 시약의 존재 하에서 수행된다. 특히, 단계 a.2) 는 요오드 원소 및 요오다이드 염의 혼합물의 존재 하, 특히 요오드 원소 및 알칼리금속 요오다이드 염의 혼합물의 존재 하, 매우 특히 요오드 원소 및 KI 의 혼합물의 존재 하 또는 요오드 원소 및 NaI 의 혼합물의 존재 하에서 수행된다.
본 발명의 제 1 바람직한 구현예 A 에서, 단계 a.2 에서의 반응은 요오드 존재 하에 수행된다. 이러한 구현예에서, 트리(C1-C6-알킬)실릴 요오다이드는 바람직하게는 식 (I) 의 화합물의 몰 당 0.9 내지 1.1 mol 의 양, 특히 0.9 내지 1 mol 의 양으로 사용되고, 요오드는 바람직하게는 식 (I) 의 화합물의 몰 당 0.005 내지 0.5 mol, 특히 0.005 내지 0.1 mol 의 양으로 사용된다.
본 발명의 추가의 바람직한 구현예 B 에서, 단계 a.2 에서의 반응은 요오다이드 염 존재 하에 수행된다. 이러한 구현예에서, 트리(C1-C6-알킬)실릴 요오다이드는 바람직하게는 식 (I) 의 화합물의 몰 당 0.9 내지 1.1 mol 의 양, 특히 0.9 내지 1 mol 의 양으로 사용되고, 요오드 염은 바람직하게는 식 (I) 의 화합물의 몰 당 0.005 내지 0.5 mol, 특히 0.005 내지 0.1 mol 의 양으로 사용된다. 알칼리 금속 요오다이드 이외에 적합한 요오다이드 염은 1차 테트라알킬암모늄 요오다이드, 특히 테트라-C1-C6-알킬암모늄 요오다이드, 예컨대 테트라에틸암모늄 요오다이드, 테트라프로필암모늄 요오다이드, 특히 테트라부틸암모늄 요오다이드이다. 특히 바람직한 것은 알칼리금속 요오다이드, 특히 NaI 및 KI 이다.
본 발명의 추가의 바람직한 구현예 C 에서, 단계 a.2 에서의 반응은 요오드 원소 및 요오다이드 염의 혼합물의 존재 하에 수행된다. 이러한 구현예에서, 트리(C1-C6-알킬)실릴 요오다이드는 바람직하게는 식 (I) 의 화합물의 몰 당 0.9 내지 1.1 mol 의 양, 특히 0.9 내지 1 mol 의 양으로 사용되고, 요오드 원소는 바람직하게는 식 (I) 의 화합물의 몰 당 0.005 내지 0.5 mol, 특히 0.005 내지 0.1 mol 의 양으로 사용되고, 요오드 염은 바람직하게는 식 (I) 의 화합물의 몰 당 0.005 내지 0.5 mol, 특히 0.005 내지 0.1 mol 의 양으로 사용된다. 알칼리 금속 요오다이드 이외에 적합한 요오다이드 염은 1차 테트라알킬암모늄 요오다이드, 특히 테트라-C1-C6-알킬암모늄 요오다이드, 예컨대 테트라에틸암모늄 요오다이드, 테트라프로필암모늄 요오다이드, 특히 테트라부틸암모늄 요오다이드이다. 특히 바람직한 것은 알칼리금속 요오다이드, 특히 NaI 및 KI 이다.
본 발명의 추가의 바람직한 구현예 D 에서, 단계 a.2 에서의 반응은 트리아릴포스핀 산화물의 존재 하에 수행된다. 이러한 구현예에서, 트리(C1-C6-알킬)실릴 요오다이드는 바람직하게는 식 (I) 의 화합물의 몰 당 0.9 내지 1.1 mol 의 양, 특히 0.9 내지 1 mol 의 양으로 사용되고, 트리아릴포스핀 산화물은 바람직하게는 식 (I) 의 화합물의 몰 당 0.005 내지 0.5 mol, 특히 0.005 내지 0.1 mol 의 양으로 사용된다. 적합한 트리아릴포스핀 산화물은 특히 트리페닐포스핀 산화물이다.
본 발명의 동등하게 바람직한 구현예에서, 상기 언급된 추가 시약은 단계 a.2) 중 첨가되지 않는다.
매우 바람직한 구현예에서, 하기 과정의 방법이 실행된다. 먼저, 헥사-(C1-C6-알킬)디실란, 특히 HMDS 를 요오드와 반응시킨 다음 수득된 반응 혼합물을 식 (I) 의 화합물과 반응시킨다. 통상적으로, 이러한 반응은 상기 언급된 조건, 특히 상기 언급된 바람직한 조건 하에서 수행된다. 이후, 특히 알칼리 카르보네이트, 알칼리금속 바이카르보네이트 및 이의 혼합물로부터 선택되는 무기 염기를 이에 따라 수득된 반응 혼합물에 첨가하고, 이에 따라 수득된 혼합물을 이후 아민 염기 존재 하에서 식 (II) 의 화합물과 반응시킨다. 반응 조건에 관하여, 상기 언급된 염기 및 시약의 양은 유사하게 적용된다. 이러한 구현예에 있어서, 단계 a.2) 는 바람직하게는 요오드 원소 및 요오드 염으로 이루어진 군으로부터, 특히 요오드 원소 및 알칼리금속 요오다이드 및 이의 혼합물로부터 선택되는 시약의 존재 하에서 수행된다. 이러한 구현예에 있어서, 단계 a.2) 는 특히 요오드 원소 및 요오드 염의 혼합물의 존재 하, 특히 요오드 원소 및 알칼리금속 요오다이드의 혼합물의 존재 하, 특히 요오드 원소 및 KI 의 혼합물의 존재 하 또는 요오드 원소 및 NaI 의 혼합물의 존재 하에서 수행된다. 이러한 시약의 상대적인 양에 관하여, 구현예 A, B 및 D 에 관한 서술이 유사하게 적용된다.
단계 a.2), 즉 식 (I) 의 화합물의 트리(C1-C6-알킬)실릴 요오다이드로의 처리로부터 수득되는 반응 생성물의 식 (II) 의 화합물과의 반응은 일반적으로 상기 언급된 불활성 유기 용매 또는 희석액 중 하나에서 수행된다. 또한, 여기서 바람직한 것은 상기 언급된 비양성자성 용매, 특히 낮은 함량의 물, 알코올 또는 산과 같은 양성자성 불순물을 갖는 것이다. 용매 중 양성자성 불순물 함량은 바람직하게는 1000 ppm 미만이다. 바람직하게는, 본 발명에 따른 방법에서 사용되기 전, 비양성자성 용매는 양성자성 불순물, 특히 물의 함량을 감소시키기 위해 적합한 흡착제, 예를 들어 포어 크기가 3 내지 4 옹스트롬인 분자체로 처리함으로써 처리된다. 바람직한 유기 용매는 할로알칸, 예컨대 디클로로메탄, 트리클로로메탄, 디클로로에탄, 방향족 탄화수소, 예컨대 톨루엔 및 자일렌, 지방족 카르복실산의 디메틸아미드, 예컨대 디메틸포름아미드 (DMF) 및 디메틸아세트아미드, 및 또한 알킬 니트릴, 예컨대 아세토니트릴, 및 또한 상기 언급된 용매의 혼합물이다. 용매는 바람직하게는 모든 성분이 용해 형태로 존재하는 것이 선택된다. 식 (I) 및 (II) 의 화합물의 총 농도는 전체 시약 및 용매의 총 중량을 기준으로 바람직하게는 5 내지 75 중량%, 특히 10 내지 65 중량%, 또는 15 내지 60 중량% 범위이다.
단계 a.2) 에서의 반응은 바람직하게는 -20 내지 110℃ 범위, 특히 0 내지 80℃ 범위의 온도에서 수행된다. 반응은 주변 압력, 감압 또는 승압에서 수행될 수 있다. 반응은 통상적으로 900 내지 1100 mbar 범위의 압력에서 실시된다.
단계 a) 에서의 반응으로 수득된 식 (III) 의 화합물은 통상 후처리 방법으로 단리될 수 있고, 임의로 결정화 및/또는 크로마토그래피로 정제될 수 있다. 대안적으로, 단계 a) 에서의 반응으로 수득된 식 (III) 의 화합물을 직접 적어도 부분적으로 또는 완전히 탈보호시켜, 식 (IIIa) 의 화합물 (식 중, R4 = H), 식 (IIIc) 의 화합물 (식 중, R4 = H) 또는 식 (IIIb) 또는 (IV) 의 화합물을 수득할 수 있다.
식 (III) 의 화합물의 탈보호는 공지된 탈보호 반응과 유사하게 달성되고, 바람직하게는 가수분해 방법으로 수행된다. 이러한 보호기의 절단을 위한 조건은 예를 들어 [P.G.M Wuts et al., "Greene's Protecting Groups in Organic Synthesis, 4th Edition, Wiley 2006] 및 이에 인용된 문헌, 또는 2'-O-푸코실락토오스의 제조에 대해 처음 인용된 참조문헌으로부터 당업자에게 친숙하다.
본 발명의 제 1 구현예 b.1) 에 있어서, 식 (III) 의 화합물은 산의 존재 하에서 물로 처리된다. 이러한 방식으로, 식 (III) 의 화합물로부터의 전체 보호기의 완전한 절단이 일반적으로 달성되고, 2'-O-푸코실락토오스가 수득된다.
적합한 산은 미네랄 산, 예컨대 염산, 황산, 인산, 미네랄 산의 산성 염, 예컨대 알칼리 금속 히드로겐 포스페이트 및 디히드로겐 포스페이트 또는 알칼리 금속 히드로겐 술페이트, 예를 들어 나트륨 디히드로겐 포스페이트 또는 칼륨 히드로겐 포스페이트, 또한 유기 카르복실산, 예컨대 아세트산, 프로피온산, 디클로로아세트산, 트리클로로아세트산 또는 트리플루오로아세트산, 및 유기 술폰산, 예컨대 메탄술폰산이다. 산은 통상적으로 수성 희산, 예를 들어 5 내지 70 중량% 농도 용액으로서 사용된다. 흔히, 수성 희산은 적합한 유기 용매와 조합되어 사용된다. 이의 예는 물과 혼화성인 유기 용매, 예컨대 C1-C4-알칸올, 예를 들어 메탄올, 에탄올, 이소프로판올, 1-부탄올 또는 tert-부탄올, 시클릭 에테르, 예컨대 테트라히드로푸란 또는 디옥산, 및 또한 물과 단지 제한된 혼화성을 갖는 유기 용매, 예를 들어 할로알칸, 예컨대 디클로로메탄, 트리클로로메탄, 디클로로에탄, 방향족 탄화수소, 예컨대 톨루엔 및 자일렌, 및 또한 디알킬 에테르, 예컨대 디에틸 에테르, 디이소프로필 에테르 또는 메틸 tert-부틸 에테르이다. 요구되는 반응 조건은 당업자에게, 예를 들어 [P. G. M. Wuts et al., loc. cit] 및 이에 인용된 문헌, 또는 2'-O-푸코실락토오스의 제조에 대해 처음 인용된 참조문헌으로부터 공지되어 있다. 보호기의 제거 후, 산은 통상적으로 중화되고, 이후 물을 제거함으로써 생성물은 단리된다. 중화는 이러한 목적에 통상적으로 사용되는, 알칼리금속 수산화물, 알칼리금속 카르보네이트 및 알칼리금속 바이카르보네이트를 포함하는 염기를 사용함으로써 달성될 수 있다. 또한, 중화는 염기성 또는 매우 염기성인 이온-교환 수지를 사용함으로써 달성될 수 있는데, 이는 생성물의 용액에 염을 형성하지 않으면서 중화를 가능하게 할 것이기 때문이다.
구현예 b.1) 에서, 보호기의 절단은 또한 수성 매질 중 산성 이온-교환 수지를 이용하여 달성될 수 있다. 이로 인해, 개별 중화 단계가 회피될 수 있다.
본 발명의 추가 구현예 b.2) 에 있어서, 식 (III) 의 화합물 (식 중, R1 은 SiR12R13R14 잔기임) 은 먼저 탈실릴화 시약으로 처리되고, 이때 식 (IIIb') 의 화합물이 수득된다:

식 (III) 의 화합물 (식 중, R1 은 SiR12R13R14 잔기임) 은 식 (IIIa) 의 화합물 (식 중, R1a 는 SiR12R13R14 잔기이고, R4 는 SiRaRbRc 잔기임) 에 해당한다. 식 (IIIb') 의 화합물은 식 (IIIb) 의 화합물 (식 중, R4 는 수소임) 에 해당한다.
탈실릴화는 두 SiR12R13R14 기 및 SiRaRbRc 기가 동시에 절단되도록 한 단계로 수행될 수 있다. 이는 또한 SiR12R13R14 및 SiRaRbRc 기가 상이한 반응성을 갖는 경우 연속하여 수행될 수 있다.
탈실릴화를 위한 적합한 시약은, 예를 들어 바람직하게는 상기 언급된 C1-C4 알코올, 특히 메탄올 (물 첨가 포함 또는 미포함) 중 하나의 용액 중 상기 언급된 C1-C4 알코올, 특히 메탄올 (물 첨가 포함 또는 미포함), 및 또한 알칼리 금속 또는 알칼리 토금속 카르보네이트 및 히드로겐 카르보네이트, 예컨대 리튬 카르보네이트, 나트륨 카르보네이트, 칼륨 카르보네이트, 나트륨 히드로겐 카르보네이트 및 칼륨 히드로겐 카르보네이트이다. 적합한 탈실릴화 시약은 또한 바람직하게는 극성, 비양성자성 유기 용매, 예를 들어 시클릭 에테르, 예컨대 테트라히드로푸란 또는 디옥산, 또는 지방족 카르복실산의 디-C1-C4-알킬아미드, 예컨대 디메틸포름아미드 또는 디메틸아세트아미드, 또는 알킬 니트릴, 예컨대 아세토니트릴 또는 상기 언급된 극성, 비양성자성 유기 용매의 혼합물 중에서 사용되는 테트라알킬암모늄 플루오라이드이다. 요구되는 반응 조건은 당업자에게, 예를 들어 [P. G. M. Wuts et al., loc. cit.] 및 이에 인용된 문헌으로부터 공지되어 있다.
이어서, 잔류 보호기는 산의 존재 하 식 (IIIb') 의 화합물을 물로 처리함으로써 제거된다. 이는 구현예 b1) 에 기재된 방식으로 실시될 수 있다.
본 발명의 추가 구현예 b.3) 에 있어서, 식 (III) 의 화합물 (식 중, R1 은 C(O)R11 잔기임) 은 먼저 탈실릴화 시약으로 처리되고, 이때 식 (IIIc') 의 화합물이 수득된다:

식 (IIIc') 의 화합물은 식 (IIIc) 의 화합물 (식 중, R4 는 수소임) 에 해당한다. 이어서, C(O)-R11 기 및 잔류 보호기가 동시에 또는 연속하여 제거된다.
식 (III) 의 화합물 (식 중, R1 은 C(O)R11 잔기임) 의 탈실릴화는, 탈실릴화 시약으로 화합물 (III) 을 처리함으로써 구현예 b2) 와 유사하게 달성된다. 탈실릴화에 요구되는 반응 조건은 당업자에게, 예를 들어 [P. G. M. Wuts et al., loc. cit.] 및 이에 인용된 문헌으로부터 공지되어 있다.
에스테르 기 C(=O)-R11 의 후속 절단은 염기성 비누화 또는 염기-촉매화 또는 효소-촉매화 트랜스에스테르화에 의해 그 자체로 공지된 방식으로 달성된다. 이러한 목적을 위한 방법은 예를 들어 [P. G. M. Wuts et al. loc. cit.] 또는 [Kociensky et al. Protective groups, 3rd Edition, Chapter 4.6, Thieme 2005] 으로부터 공지되어 있다. 이후, 잔류 C(R2)2 및 OR3 보호기는 예를 들어 수성 산으로의 처리에 의해, 구현예 b1) 과 관련하여 이미 기재된 바와 같이 그 자체로 공지된 방식으로 제거된다.
추가 구현예 b.4) 에 따라, 대안적으로 절차는 또한 C(R2)2 및 OR3 보호기가 예를 들어 수성 산으로의 처리에 의해, 구현예 b1) 과 관련하여 이미 기재된 바와 같이 식 (IIIc') 의 화합물로부터 초기에 제거되고, 이때 일반식 (IV) 의 화합물이 상기 기재된 바와 같이 수득되는 것일 수 있다. 이후, 에스테르 기 C(=O)-R11 은 예를 들어 염기성 비누화 또는 염기성 트랜스에스테르화 또는 효소-촉매화 트랜스에스테르화에 의해 그 자체로 공지된 방식으로 식 (IV) 의 화합물로부터 절단될 수 있다.
대안적으로, 절차는 또한 C(R2)2 및 OR3 보호기가 예를 들어 수성 산으로의 처리에 의해, 구현예 b1) 과 관련하여 이미 기재된 바와 같이 식 (IIIc') 의 화합물로부터 초기에 제거된 다음, 에스테르 기 C(=O)-R11 이 염기성 비누화 또는 효소-촉매화 트랜스에스테르화에 의해 그 자체로 공지된 방식으로 절단되는 것일 수 있다.
특히 바람직한 구현예 b.5) 에 있어서, 식 (III) 의 화합물은 C1-C4-알칸올 및 알칼리금속 염기로 먼저 처리되어 식 (IIIb') 의 화합물을 수득한 다음 산성 조건 하에서 잔류 보호기가 제거된다. 이러한 구현예에서, R11 은 바람직하게는 C1-C4-알킬, 예컨대 메틸, 에틸 또는 tert-부틸이다. 이로 인해, 탈실릴화 및 에스테르 기 C(=O)-R11 의 제거는 서로 연관될 수 있고, 단일 단계로 절단될 수 있다. 여기서, 적합한 시약은 결과적으로 용매로서 C1-C4-알칸올, 예컨대 메탄올 중 상기 언급된 알칼리 금속 수산화물 및 카르보네이트이다. 이러한 목적에 있어서, 메탄올과 나트륨 카르보네이트 또는 칼륨 카르보네이트의 조합이 특히 유용하다. 이러한 목적에 요구되는 반응 조건은 당업자에게 친숙하고, 통상의 실험에 의해 결정될 수 있다. 바람직하게는, 동시 탈실릴화 및 에스테르 기 C(=O)-R11 의 제거는 20 내지 50℃ 범위의 온도에서, C1-C4-알칸올, 예컨대 메탄올 중 알칼리금속 염기로의 (III) 의 처리에 의해 달성될 수 있다. 알칼리금속 염기, 특히 알칼리금속 카르보네이트의 양은 화합물 (III) 을 기준으로 바람직하게는 3 내지 10 당량, 특히 4 내지 7 당량, 즉 알칼리금속 카르보네이트의 경우 화합물 (III) 의 몰 당 1.5 내지 5 mol, 특히 2 내지 3.5 mol 이다. 보호기 C(R2)2 및 OR3 의 절단은 하기 b.1) 에 기재된 방법과 유사하게 달성될 수 있다.
보호기의 제거 후 수득된 2'-O-푸코실락토오스는 임의로 석탄, 실리카 또는 폴리비닐 피롤리돈과 같은 보조 첨가제와 크로마토그래피 또는 결정화와 같은 종래의 정제 방법을 사용하여 정제될 수 있다. 2'-O-푸코실락토오스의 결정화에 대한 통상의 조건은 [Chem. Ber. 1956, 11, 2513] 에서 확인될 수 있다. 반응 조건 및 정제 방법에 따라, 수득된 2'-O-푸코실락토오스는 예를 들어 생성물의 중량을 기준으로 1% 내지 20% 의 양의 락토오스를 함유할 수 있다. 락토오스를 뺀 2'-O-푸코실락토오스의 화학적 순도는 통상적으로 90% 이상, 특히 적어도 95% 이상이다. 그러나, 소정의 락토오스는 식품에서 2'-O-푸코실락토오스의 사용시 문제 되지 않기 때문에 이는 문제 되는 불순물은 아니다.
특히, 본 발명의 방법은 심지어 후처리 전에 바람직하지 않은 β-이성질체 β-2'-O-푸코실락토오스 (=β-L-푸코피라노실)-(1→2)-O-β-D-갈락토피라노실-(1→4)-D-글루코피라노오스) 의 양이 낮아 반응 생성물의 정제가 2'-O-푸코실락토오스의 총량을 기준으로 1 중량% 미만, 특히 0.5 중량% 미만의 β-2'-O-푸코실락토오스를 함유하는 2'-O-푸코실락토오스를 수득하게 하는 방식의 2'-O-푸코실락토오스 제조를 가능하게 한다. 이는 현재까지 불가능하였다. 선행 기술의 방법에 반해, 본 발명의 방법은 벤질 보호기의 수소첨가분해 절단을 위한 전이 금속 촉매를 요구하지 않고, 따라서 본 발명의 방법으로 수득가능한 2'-O-푸코실락토오스에서의 전이 금속 농도는 흔히 1 ppm 미만, 특히 검출 수준 미만이다.
본 발명에 따른 방법의 단계 a) 에서 사용되는 식 (I) 의 화합물은, 예를 들어 [Synlett. 1996, 499; Org. Lett. 13(3) (2001) 2081-2084]; 및 [Tetrahedron Asym.,16 (1) (2005) 149-158] 로부터 공지되어 있다.
식 (II) 의 화합물 (식 중, R1 은 C(=O)-R11 잔기임) 은, 예를 들어 처음 인용된 참조문헌, 또는 [Tetrahedron Letters, 1981, 22 (50), 5007-5010], WO 2010/115934, WO 2010/115935 및 [Carbohydrate Research, 1981, 88, 51-60] 로부터 공지되어 있거나, 이에 기재된 방법과 유사하게 제조될 수 있다.
식 (II) 의 화합물 (식 중, R1 은 SiR12R13R14 잔기임) 은 식 (IIa) 의 화합물에 해당한다. 상기 화합물은 신규하고, 단 라디칼 R1 은 라디칼 SiR12R13R14 (이때, R12 및 R13 은 메틸이고, R14 은 tert-부틸임) 이 아니고, R2 및 R3 는 메틸이다 ([H. Kogelberg et al. Carbohydrate Research 201 (1990), 161-173] 참조). 따라서, 식 (IIa) 의 화합물은 마찬가지로 본 발명의 주제의 일부이고, 단 R2 및 R3 가 메틸인 경우, 라디칼 R1 은 tert-부틸디메틸실릴이 아니다.
식 (IIa) 의 화합물은 CH2-OH 기의 선택적 실릴화에 의해 식 (IIb) 의 화합물로부터 간단한 방식으로 제조될 수 있다.

식 (IIb) 의 R2 및 R3 는 상기 정의된 바와 같고, 특히 하기 정의된 바와 같다:
R2 는 특히 C1-C4-알킬, 특히 메틸이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기는 함께 1,5-펜탄디일이고, 따라서 이들이 부착되어 있는 탄소 원자와 시클로헥산-1,1-디일 잔기를 형성한다. 모든 R2 잔기는 특히 메틸이다.
R3 는 특히 C1-C4-알킬, 특히 메틸이다.
선택적 실릴화를 위해, 식 (IIb) 의 화합물은 통상적으로 적합한 실릴화 시약, 예를 들어 식 SiXR12R13R14 (이때, R12, R13 및 R14 은 상기 정의된 바와 같고, 특히 메틸이고, X 는 할로겐, 특히 클로린임) 의 화합물과 반응한다. 실릴화 시약과의 반응은 바람직하게는 염기 존재 하에서 수행된다.
선택적 실릴화를 위해, 식 (IIb) 의 화합물의 몰 당 0.9 내지 2 mol, 특히 1 내지 1.5 mol, 특히 약 1.1 mol 의 실릴화 시약이 통상적으로 사용된다.
반응을 선택적으로 진행하기 위해, (IIb) 의 반응은 바람직하게는 -40 내지 +40℃ 범위, 특히 -20 내지 +20℃ 범위, 특히 바람직하게는 -5 내지 +5℃ 범위, 예를 들어 약 0℃ 의 온도에서 수행된다.
적합한 염기는 1차 아민 염기, 특히 2차 및 3차 아민, 특히 피리딘 염기 및 3차 지방족 또는 시클로지방족 아민이다. 적합한 피리딘 염기는, 예를 들어 피리딘, 퀴놀린 및 C1-C6-알킬-치환 피리딘, 특히 모노-, 디- 및 트리(C1-C6-알킬)피리딘, 예컨대 2,6-디(C1-C6-알킬)피리딘 및 콜리딘이다. 적합한 3차 지방족 또는 시클로지방족 아민은 트리(C1-C6-알킬)아민, 예컨대 트리에틸아민, 디이소프로필메틸아민, 트리-n-부틸아민 또는 이소프로필디메틸아민, C3-C8-시클로알킬-디(C1-C6-알킬)아민, 예컨대 시클로헥실디메틸아민, N-(C1-C6-알킬)피페리딘, 예컨대 N-메틸피페리딘 및 디(C3-C8-시클로알킬)-C1-C6-알킬아민, 예컨대 비스시클로헥실메틸아민이다.
염기는 통상적으로 식 (IIb) 의 화합물의 몰 당 0.9 내지 2 mol 의 양, 특히 1 내지 1.5 mol 의 양으로 사용된다.
식 (IIb) 의 화합물은 일반적으로 불활성 유기 용매 또는 희석액에서 실릴화 시약과 반응한다. 바람직한 것은 비양성자성 용매, 특히 낮은 함량의 물, 알코올 또는 산과 같은 양성자성 불순물을 갖는 것이다. 바람직한 유기 용매는 할로알칸, 예컨대 디클로로메탄, 트리클로로메탄, 디클로로에탄, 방향족 탄화수소, 예컨대 톨루엔 및 자일렌, 디알킬 에테르, 예컨대 디에틸 에테르, 디이소프로필 에테르, 메틸 tert-부틸 에테르, 시클릭 에테르, 예컨대 테트라히드로푸란 또는 디옥산, 지방족 카르복실산의 디알킬아미드, 예컨대 디메틸포름아미드 또는 디메틸아세트아미드 및 또한 알킬 니트릴, 예컨대 아세토니트릴, 및 또한 상기 언급된 용매의 혼합물이다. 용매는 바람직하게는 모든 성분이 용해 형태로 존재하는 것이 선택된다. 식 (I) 및 (II) 의 화합물의 총 농도는 전체 시약의 총 중량을 기준으로 바람직하게는 5 내지 50 중량%, 특히 10 내지 40 중량% 범위이다.
식 (IIa) 의 화합물은 여과, 추출 또는 일부 경우 증류로 후처리될 수 있다.
식 (IIb) 의 화합물은, 예를 들어 [Carbohydrate Research, 212 (1991), pp. C1-C3]; [Tetrahedron Lett., 31 (1990) 4325]; [Carbohydrate Research, 75 (1979) C11]; [Carbohydrate Research, 88 (1981) 51]; [Chem. 5 (1999) 1512]; WO 2010/070616, WO 2012/113404, WO 2010/115934 및 WO 2010/115935 로부터 공지되어 있거나, 이에 기재된 방법으로 제조될 수 있다.
이미 언급된 바와 같이, 공지된 2'-O-푸코실락토오스에 비해, 본 발명에 따른 방법으로 수득가능한 2'-O-푸코실락토오스는 제거될 수 없는 불순물을 포함하지 않거나, 오로지 훨씬 낮은 분획을 포함한다는 것을 특징으로 한다. 특히, 본 발명에 따른 방법으로 수득가능한 2'-O-푸코실락토오스는 유의한 양의 불순물, 특히 식품류에서 사용하기에 문제가 될 수 있는 수소첨가로부터 수득되는 불순물을 포함하지 않는다.
따라서, 이러한 2'-O-푸코실락토오스는 그 자체로 식품류로서 적합하고, 또한 식품류용 첨가제로서 적합하다. 2'-O-푸코실락토오스가 사용될 수 있는 식품류의 예는, 예를 들어 처음 인용된 선행 기술로부터 당업자에게 친숙하다. 본원에서, 이는 자연 발생 제품, 예를 들어 유제품, 및 또한 식이 또는 의약 영양물용 인공 제조 제형 기재의 조성물 형태를 취할 수 있다. 후자는 레디-투-유즈 제형일 수 있고, 직접 사용될 수 있거나, 농축 제형, 예를 들어 액체 또는 반-고체 농축물, 또는 액체 특히 물을 첨가함으로써 사용되기 전 레디-투-유즈 제형으로 전환되거나, 종래의 식품류로 혼입되는 과립, 플레이크 또는 분말과 같은 고체 제품 형태를 취할 수 있다.
농축물 및 또한 레디-투-유즈 제형은 고체, 액체 또는 반-고체 제형일 수 있다.
특히, 본 발명에 따른 2'-O-푸코실락토오스가 사용되는 식품류는 특히 신생아 분유 및 특히 유아 분유에서의 아동 영양을 위한 식품류 조성물이다.
일반적으로, 본 발명에 따른 2'-O-푸코실락토오스가 사용되는 식품류는 고체, 반-고체 또는 액체 식품류 조성물, 특히 반-고체 또는 특히 액체 식품류 조성물이다.
식품류 조성물, 즉 레디-투-유즈 식품류 조성물 및 농축물은 본 발명에 따라 수득가능한 2'-O-푸코실락토오스를 식품류 제형 내에 혼입시킴으로써 그 자체로 공지된 방식으로 제조될 수 있다. 이러한 식품류 제형은 2'-O-푸코실락토오스 이외에 기타 영양분을 포함할 수 있고, 일반적으로 식품류에 적합한 하나 이상의 담체 (이는 고체, 액체 또는 반-고체일 수 있음) 를 포함한다. 담체는 식품류 또는 영양적 가치가 있는 물질일 수 있거나, 그 자체로 영양적 가치가 없는 물질, 예를 들어 식이 섬유 또는 물일 수 있다.
하기 실시예는 본 발명을 예시하기 위한 것이다.
하기 약어를 사용하였다:
d:
이중항
s:
단일항
t:
삼중항
m:
다중항
mc.:
중심 다중항
CHE:
시클로헥산
DCM:
디클로로메탄, 바람직하게는 아밀렌으로 안정화되거나, 임의의 안정화제 없이 안정화됨
DMF:
디메틸포름아미드
of th.: 이론의
EE:
에틸 아세테이트
EtOH:
에탄올
eq:
등몰량
b.p.
비등점
MeOH:
메탄올
NEt3:
트리에틸아민
RT:
주변 온도, 약 22℃
Rt.:
체류 시간
TMS:
트리메틸실릴
TMSCl:트리메틸실릴 클로라이드
TMSI:
트리메틸실릴 요오다이드
2'-Fl:2'-O-푸코실락토오스
달리 언급되지 않는 경우, 2'-O-푸코실락토오스 (2'-FL) 는 알파 아노머를 지칭한다.
달리 명시되지 않는 경우, Agilent Series 1200 및 Luna-NH2 컬럼 (3 ㎛; 250 x 4.6 mm, 100 Å) 를 사용하여 HPLC 분석을 수행하였다. 컬럼을 35℃ 에서 유지시키고, 135 bar 에서 작동시켰다.
아세토니트릴/물 75/25 v/v 를 용리액으로서 사용하였고; RID 검출기로 검출하였다. 유속은 1 mL/min, 실행 시간은 10 min 이었다. 샘플 용적은 5 μL 였다.
샘플 제조를 위해, 10 mg 의 샘플을 각각의 경우 75/25 용적비의 1 mL 의 아세토니트릴/물에 용해시켰다.
또한, 하기 고압 액체 크로마토그래피 (HPLC) 방법 (하기에 제시됨) 을 사용하였다.
HPLC 방법 2
Agilent Series 1200 및 Luna-NH2 컬럼 (3 ㎛; 250 x 4.6 mm, 100Å) 를 사용하여 HPLC 분석을 수행하였다. 컬럼을 35℃ 에서 유지시키고, 204 bar 에서 작동시켰다.
아세토니트릴/물 82.5/17.5 v/v 를 용리액으로서 사용하였고; RID 검출기로 검출하였다. 유속은 1 mL/min, 실행 시간은 10 내지 40 min 이었다. 샘플 용적은 5 μL 였다.
샘플 제조를 위해, 100 mg 의 샘플을 각각의 경우 50/50 용적비의 10 mL 의 아세토니트릴/물에 용해시켰다.
HPLC 방법 3
Agilent Series 1200 및 Waters Spherisorb-NH2 컬럼 (3 ㎛; 250 x 4.6 mm, 80Å) 를 사용하여 HPLC 분석을 수행하였다. 컬럼을 35℃ 에서 유지시키고, 112 bar 에서 작동시켰다.
아세토니트릴/물 82.5/17.5 v/v 를 용리액으로서 사용하였고; RID 검출기로 검출하였다. 유속은 1 mL/min, 실행 시간은 10 내지 40 min 이었다. 샘플 용적은 5 μL 였다.
샘플 제조를 위해, 100 mg 의 샘플을 각각의 경우 50/50 용적비의 10 mL 의 아세토니트릴/물에 용해시켰다.
개별 화합물의 체류 시간은 시간에 따라 가변적이고, 가변성의 이유에는 컬럼 분해 및 샘플 조성이 포함된다. 측정 전, 논의되는 개시 물질의 참조 샘플, 논의되는 생성물, 및 논의되는 부산물을 항상 측정하여 실제 체류 시간을 측정하였다.
α/β-비 측정
HPLC 방법 3 을 사용하여, 2'-O-푸코실락토오스의 β-이성질체는 체류 시간 Rt 가 10.2 min 인 반면, 표적 (목적하는) α-이성질체는 체류 시간 Rt 가 11.6 min 이다. 이러한 값은 컬럼 분해 및 샘플 조성으로 인해 시간에 따라 가변적이다. 임의의 경우, β-이성질체는 α-이성질체 이전에 용리된다. 임의의 경우, 두 이성질체의 참조 샘플을 항상 측정하여 실제 체류 시간을 측정하였다.
실시예:
제조예 1: 테트라키스트리메틸실릴푸코오스의 제조:
2.5 g (15.1 mmol) 의 푸코오스를 75 mL 의 DCM (0.2M 용액) 에 충전하였다. 이에 6.78 g (4.4 eq.) 의 트리에틸아민을 첨가하고, 혼합물을 0℃ 로 냉각시켰다. 이후, 7.35 g (4.4 eq.) 의 클로로트리메틸실란을 0℃ 에서 서서히 적가하고, 반응 혼합물을 1 h 동안 0℃ 에서 교반하였다. 혼합물을 RT 로 가온시키고, RT 에서 추가 16 h 후, 75 mL 의 펜탄을 첨가하고, 혼합물을 간단히 교반한 다음, 100 mL 의 냉각수에 첨가하였다. 상 분리 후, 수성 상을 펜탄으로 3 회 재추출하고, 조합된 유기 상을 연속하여 물로 2 회, NaCl 용액으로 3 회 세정하였다. 유기 상을 Na2SO4 로 건조시키고, 여과하고, 감압 하에서 건조될 때까지 농축시켰다. 수율: 6.6 g (97% of th.)
제조예 2: 테트라키스트리메틸실릴푸코오스의 제조:
제조예 1 과 유사한 방식으로, 34.9 g 의 표제 화합물을 386 mL 의 DMF 중 13 g 의 푸코오스와 38.2 g 의 TMSCl 및 35.3 g 의 NEt3 로부터 상기 기재된 방법으로 수득한다.
제조예 3: 4-O-(3,4-이소프로필리덴-β-D-갈락토피라노실)-2,3;5,6-비스-O-이소프로필리덴-D-글루코오스 디메틸 아세탈 (화합물 II-1: 식 (II) 의 화합물 (식 중, R1 = H, R2 = CH3 및 R3 = CH3)) 의 제조
205.4 g (0.6 mol) 의 락토오스를 409 mL 의 1,4-디옥산에 충전하였다. 이에 28.44 g (0.12 mol = 0.2 eq.) 의 DL-캄포술폰산 및 376.4 mL (3 mol = 5 eq.) 의 디메톡시프로판을 첨가하였다. 혼합물을 환류 하 4 h 동안 가열하였다. 10.04 mL 의 트리에틸아민을 이후 첨가하였다. 냉각 후, 혼합물을 감압 (2 mbar) 및 50℃ 하에서 농축시키는 중 각각의 경우 300 mL 의 톨루엔을 2 회 첨가하고, 공동-증류시켰다. 남아 있는 잔류물을 1000 mL 의 메탄올/물 9:1 v/v 에 용해시키고, 60℃ 에서 1 h 동안 교반하였다. 감압 하에서 메탄올을 제거한 후, 600 mL 의 DCM 을 첨가하고, 수득한 용액을 5% NaHCO3 수용액으로 2 회 세정하였다. 감압 하에서 용매를 제거한 후, 잔류물을 50 mL 의 에틸 아세테이트에 용해시키고, -10℃ 에서 50 mL 의 시클로헥산 및 160 mL 의 디이소프로필 에테르를 첨가하여 결정화하였다.
2 x 50 mL 의 저온 디이소프로필 에테르로의 결정 여과 및 세정은 순도 92% 의 118.9 g 의 표제 화합물을 제공한다.

제조예 4: 4-O-(6-O-아세틸-3,4-이소프로필리덴-β-D-갈락토피라노실)-2,3;5,6-비스-O-이소프로필리덴-D-글루코오스 디메틸 아세탈 (화합물 II-2: 식 (II) 의 화합물 (식 중, R1 = 아세틸, R2 = CH3 및 R3 = CH3)) 의 제조
58.8 g (92% 농도 = 0.106 mol) 의 제조예 3 으로부터의 화합물 II-1 을 183 mL 의 DCM 에 용해시켰다. 용액을 25.12 mL (0.181 mol) 의 NEt3 로 처리하고, -5℃ 로 냉각시켰다. 이에 61 mL 의 DCM 중 용해된 60.9 g (0.16 mol) 의 아세틸 클로라이드 용액을 70 min 의 시간에 걸쳐 적가하고, 수득한 혼합물을 0℃ 에서 20 h 동안 교반하였다. 후처리를 위해, 혼합물을 100 mL 의 냉각수로 처리하고, 상을 분리하고, 수성 상을 각각의 경우 50 mL 의 DCM 으로 2 회 추출하였다.
조합된 유기 상을 연속하여 50 mL 의 1N 수성 염산, 50 mL 의 5% NaHCO3 수용액으로 세정하고, Na2SO4 를 통해 건조시키고, 감압 (250 mbar) 하 40℃ 에서 농축시켰다.
표제 화합물 II-2 를 순도 73% 의 65.1 g 의 양으로 수득하였다. 생성물을 추가로 직접 반응시키거나, 시클로헥산으로부터의 2차 성분의 결정화 또는 크로마토그래피에 의해 90% 순도로 정제하였다.

제조예 5: 4-O-(6-O-트리메틸실릴-3,4-이소프로필리덴-β-D-갈락토피라노실)-2,3;5,6-비스-O-이소프로필리덴-D-글루코오스 디메틸 아세탈 (화합물 II-3: 식 (II) 의 화합물 (식 중, R1 = TMS, R2 = CH3 및 R3 = CH3)) 의 제조
23 mL 의 DCM 중 5.98 g 의 제조예 3 으로부터의 화합물 II-1 (85% 농도, 1 eq.) 및 1.55 mL (1.5 eq.) 의 NEt3 용액을, 0℃ 에서 6 mL 의 DCM 중 1.2 g (1.1 eq.) 의 TMSCl 용액으로 처리하고, 수득한 혼합물을 0℃ 에서 6 h 동안 교반하였다. 후처리를 위해, 26 mL 의 헵탄 및 50 mL 의 냉각수를 혼합물에 첨가하고, 유기 상을 분리하였다. 포화 NaCl 수용액으로 유기 상을 건조시키고, 용매를 감압 하에서 제거한 후, 5.7 g 의 화합물 II-3 을 추가 반응에 대해 충분히 순수한 미정제 생성물로서 남아있었다. 실리카 겔 상에서의 크로마토그래피 또는 증류에 의한 정제가 달성될 수 있다 (0.1 mbar 에서 b.p. 130-140℃). 에틸 아세테이트/시클로헥산 7/3 v/v 로의 500 mL 의 실리카 겔 상에서의 크로마토그래피는 1.97 g 의 화합물을 제공하였다:
제조예 6: 4-O-(6-O-피발로일-3,4-이소프로필리덴-β-D-갈락토피라노실)-2,3;5,6-비스-O-이소프로필리덴-D-글루코오스-디메틸 아세탈 (화합물 II-4: 식 (II) 의 화합물 (식 중, R1 = C(=O)C(CH3)3, R2 = CH3, 및 R3 = CH3)) 의 제조
150 g (280 mmol) 의 제조예 3 으로부터의 화합물 II-1 (R2 = R3 = CH3) 을 190 mL 의 DCM 에 충전하였다. 이에 57.19 g (565 mmol) 의 트리에틸아민을 첨가하였다. 35 mL 의 DCM 중 49.56 g (411 mmol) 의 피발로일 클로라이드의 용액을 2 h 의 시간에 걸쳐 첨가하여, 온도가 27℃ 초과로 상승하지 못하게 하였다. 이후, 반응 혼합물을 21 h 동안 가열 환류하였다 (내부 온도: 49℃). 냉각 후, 현탁액을 410 mL 의 냉각수에 붓고, 수득한 혼합물을 10 min 동안 교반하였다.
유기 상을 분리하고, 수성 상을 95 mL 의 DCM 으로 1 회 추출하였다. 조합된 유기 상을 연속하여 95 mL 의 물, 95 mL 의 포화 NaCl 용액으로 세정하고, 45 g 의 Na2SO4 를 통해 건조시키고, 고체를 여과하였다. 여과물을 회전 증발 (40℃, 5 mbar) 을 통해 증발시켜, 79.2 중량% 의 표제 화합물을 포함하는 188.65 g 의 생성물을 수득하였다 (89.2 % of th.).

실시예 1: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3 및 R4 = H) 의 제조
3 mL 의 DCM 중 0.8 g 의 테트라키스트리메틸실릴푸코오스 (식 (I) 의 화합물 (식 중, RSi = 트리메틸실릴), 1 eq.) 의 용액에, RT 에서 DCM 중 5.2 mL 의 5 중량% 농도의 트리메틸실릴 요오다이드 용액을 적가하고, 혼합물을 RT 에서 20 min 동안 교반하였다.
이에 따라 수득한 용액을 3 mL 의 DCM 중 0.973 g (1 eq.) 의 제조예 4 로부터의 푸코오스 수용체 II-2 및 0.523 g 의 2,6-디-tert-부틸피리딘의 용액에 0℃ 에서 적가하고, 혼합물을 RT 에서 16 h 동안 교반하였다. 18 mL 의 메탄올을 이에 따라 수득한 반응 혼합물에 첨가하고, 혼합물을 30 min 동안 교반하였다. 이후, 메탄올계 혼합물을 25 mL 의 포화 NaHCO3 수용액에 첨가하고, 상을 분리하였다. 수성 상을 20 mL 의 DCM 으로 한 번 더 추출하고, 조합된 유기 상을 20 mL 의 포화 Na2SO3 수용액으로 세정하였다.
유기 상을 실리카 겔 상에서 크로마토그래피 (200 mL 의 실리카 겔, DCM/MeOH 92/8) 하였고, 이때 0.57 g 의 푸코오스 수용체 II-2 를 회수하고, 0.13 g 의 보호된 식 IIIc 의 표제 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3 및 R4 = H) 을 수득하였다.

실시예 2: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3 및 R4 = Si(CH3)3) 의 제조
실시예 1 과 유사한 방식으로, 1.0 g 의 테트라키스트리메틸실릴푸코오스 및 1.22 g 의 제조예 4 로부터의 화합물 II-2 를 용매로서 아세토니트릴에서 반응시켰다. 메탄올이 첨가되지 않았다는 점에서 후처리는 실시예 1 과 상이했다. 반응 생성물의 실리카 겔 상에서의 크로마토그래피 후, 0.28 g 의 표제 화합물 및 0.67 g 의 사용된 푸코오스 수용체 II-2 를 수득하였다.

실시예 3: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3 및 R4 = Si(CH3)3) 의 제조
실시예 1 과 유사한 방식으로, 2.5 g (5.5 mmol) 의 테트라키스트리메틸실릴푸코오스를 먼저 1.1 g 의 트리메틸실릴 요오다이드와 반응시켰다. 반응 생성물을 0.75 g 의 NEt3, 2.5 g 의 분자체 및 1.4 g 의 요오드로 처리하고, 이어서 8.4 g (0.015 mol) 의 제조예 4 로부터의 화합물 II-2 와 40℃ 에서 48 h 동안 반응시켰다. 실리카 겔 상에서 EE/CHE 1 : 1 v/v 로 여과한 후, 2.1 g 의 표제 화합물을 수득하였다 (이론의 40% 수율).
실시예 4: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3 및 R4 = Si(CH3)3) 의 제조
실시예 3 과 유사한 방식으로, 3.26 g (7.2 mmol) 의 테트라키스트리메틸실릴푸코오스를 먼저 1.44 g 의 트리메틸실릴 요오다이드와 반응시켰다. 20 min. 후, 휘발성 성분을 감압 하에서 제거하였다. 잔류물 (요오다이드) 을 각각의 경우 10 mL 의 n-헵탄과 공증발시켰다. 이후, 잔류물을 10 mL 의 DCM 에 용해시키고, 0.98 g 의 NEt3, 3.3 g 의 분자체 및 2.66 g 의 테트라부틸암모늄 요오다이드의 존재 하, DCM 중 20.79 g (14.4 mmol) 의 제조예 4 로부터의 화합물 II-2 의 38% 농도 용액과 반응시켰다. 실리카 겔 상에서 EE/CHE 1 : 1 v/v 로 여과한 후, 3.8 g 의 표제 화합물을 수득하였다 (이론의 57% 수율).
실시예 5: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3 및 R4 = Si(CH3)3) 의 제조
실시예 3 과 유사한 방식으로, 3.26 g (7.2 mmol) 의 테트라키스트리메틸실릴푸코오스를 먼저 1.44 g 의 트리메틸실릴 요오다이드와 반응시켰다. 20 min. 후, 휘발성 성분을 감압 하에서 제거하였다. 수득한 잔류물을 각각의 경우 10 mL n-헵탄과 2 회 공증발시켰다. 이후, 잔류물을 10 mL 의 DCM 에 용해시키고, 0.94 g 의 NEt3, 3 g 의 분자체 및 1.3 g 의 테트라부틸암모늄 요오다이드로 처리하고, 이어서 5.50 g 의 제조예 4 로부터의 화합물 II-2 와 40℃ 에서 64 h 동안 반응시켰다. 실리카 겔 상에서 EE/CHE 1 : 1 v/v 로 여과한 후, 2.1 g 의 표제 화합물을 수득하였다 (이론의 64% 수율).
실시예 6: 식 IIIb 의 화합물 (식 중, R2 = CH3 및 R3 = CH3 및 R4 = H) 의 제조
실시예 3 과 유사한 방식으로, 1.5 g (3.3 mmol) 의 테트라키스트리메틸실릴푸코오스를 먼저 DCM 중 3.27 g 의 29 중량% 농도의 트리메틸실릴 요오다이드 용액과 20 min 동안 교반하였다. 이후, 반응 용액을 12 mL 의 DCM 중 0.45 g 의 NEt3, 0.12 g 의 테트라부틸암모늄 요오다이드 및 1.8 g (3.1 mmol) 의 제조예 4 로부터의 화합물 II-2 에 첨가하고, 혼합물을 40℃ 에서 24 h 동안 반응시켰다. 이후, 10 mL 의 메탄올을 반응 혼합물에 첨가하고, 이를 30 분 동안 RT 에서 교반한 다음, 50 mg 의 K2CO3 로 처리하고, 추가 3 h 동안 교반하였다. 수득한 혼합물을 20 mL 의 DCM 으로 희석한 후, 유기 상을 분리하고, 포화 NaHCO3 수용액으로 세정하고, 이어서 건조될 때까지 증발시켰다. 실리카 겔 상에서 CHE/EE 100/0 → CHE/EE/MeOH 0/80/20 의 구배로 잔류물을 여과하여, 163 mg 의 표제 화합물을 수득하였다.

실시예 7: 2'-O-푸코실락토오스의 제조
3 mL 의 MeOH 중 0.3 g 의 실시예 5 로부터의 화합물 IIIc 를 0.3 mL 의 물및 2.3 eq. 의 K2CO3 와 RT 에서 16 h 동안 교반하였다. HPLC 크로마토그램은 98% 의 화합물 IIIb (식 중, R2 = R3 = CH3 및 R4 = H) 를 보였고, 이는 이의 체류 시간 Rt = 3.46 min. 및 실시예 6 으로부터의 샘플과의 스파이킹 (spiking) 에 의해 판별되었다. 이후, 이에 9 g 의 60 중량% 아세트산 수용액을 첨가하고, HPLC 에 따라 완전한 전환이 달성될 때까지 혼합물을 60℃ 에서 23 h 동안 교반하였다.
반응 혼합물의 증발 및 실리카 겔 상에서 클로로포름/메탄올 8/2 로의 잔류물의 여과 후, 54 mg 의 2'-O-푸코실락토오스를 수득하고, 이는 HPLC 중 이의 체류 시간 (Rt = 8.16 min.) 및 시판 2'-O-푸코실락토오스와의 비교에 의해 판별되었다.
아노머성 α-이성질체의 혼합물로서의 13C
이성질체 I 
이성질체 II 
실시예 8: 2'-O-푸코실락토오스의 제조
0.3 g 의 실시예 5 로부터의 화합물 IIIc 를 7 mL 의 0.5% 수성 HCl 에서 24 h 동안 교반하였다.
HPLC 에 따라, 화합물 IIIc 를 정량적으로 2'-O-푸코실락토오스로 전환하고, 이는 실시예 7 과 동일한 이의 체류 시간에 의해 판별되었다.
실시예 9: 식 IIIa 의 화합물 (식 중, R1a = CH3, R2 = CH3, R3 = CH3, 및 R4 = Si(CH3)3) 의 제조
a)
3.4 g 의 테트라키스트리메틸실릴푸코오스 (식 (I) 의 화합물 (식 중, RSi = Si(CH3)3), 92% 농도, 6.9 mmol) 를 10 mL 의 DCM 중에 용해시킨 다음, 1.38 g (6.9 mmol) 의 TMSI 를 적가하고, 반응 혼합물을 20 min 동안 교반하였다. 이후, 휘발성 성분을 40℃ 에서 진공 중 제거하고, 잔류물을 각각의 경우 10 mL 의 톨루엔으로 2 회 공동증류시켰다. 미정제 생성물을 10 mL 의 DMF 에 용해시켰다.
b)
제 2 플라스크에서, 7.8 g 의 DMF 중 진공에서 사전 가열된 3.4 g 의 분자체 (4 옹스트롬), 0.11 g (0.7 mmol) 의 KI, 0.18 g (0.7 mmol) 의 I2, 0.92 g (9.1 mmol) 의 NEt3 및 5.7 g (10.4 mmol, 1.3 eq.) 의 제조예 4 로부터의 화합물 II-2 의 현탁액을 50℃ 에서 가열하였다. 이후, 단계 a) 로부터의 DMF 중 트리스(트리메틸실릴)푸코실 요오다이드의 용액을 적가하고, 혼합물을 24 h 동안 50℃ 에서 교반하였다.
혼합물의 불용성 성분을 여과하고, 휘발성 성분을 진공에서 제거하여, 13.8 g 의 생성물 함량이 32.3% 인 미정제 생성물을 수득하였다 (정량적 HPLC 분석에 따름). 미정제 수율: 이론의 77.3%.
미정제 생성물을 1% NEt3 를 갖는 시클로헥산/에틸 아세테이트 2:1 → 1% NEt3 를 갖는 시클로헥산/에틸 아세테이트 1:1 의 구배로 실리카 겔 (1000 mL) 상에서 크로마토그래피로 추가 정제하여, 순도가 95% 인 5.1 g 의 표제 화합물을 수득하였다 (HPLC 방법에 따름; 10.3 min).
실시예 10: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3, 및 R4 = 트리메틸실릴) 의 제조
a)
1.08 g (7.2 mmol) 의 헥사메틸디실란 (98% 농도) 및 1.86 g (7.2 mmol) 의 요오드 (98% 농도) 를 65℃ 에서 가열하고, 발열 반응이 약화될 때까지 혼합물을 상기 온도에서 남아있었다. 반응 혼합물을 약 110℃ 에서 환류될 때까지 서서히 가열하고, 90 min 동안 교반하였다. 반응 혼합물을 RT 로 냉각시켰다. 이후, 2 mL 의 DCM 중 3.58 g (7.2 mmol) 의 테트라키스트리메틸실릴푸코오스 (92% 농도) 를 첨가하고, 반응 혼합물을 20 min 동안 교반하였다. 휘발성 성분을 40℃ 에서 진공 중 제거하고, 잔류물을 각각의 경우 10 mL 의 톨루엔으로 3 회 공동증류시켰다. 잔류물을 10 mL 의 DCM 에 용해시켰다.
b)
제 2 플라스크에서, 3.5 g 의 DCM 중 진공에서 사전 가열된 3.4 g 분자체 (4 옹스트롬), 0.053 g (0.36 mmol) 의 NaI, 0.091 g (0.36 mmol) 의 I2, 0.95 g (9.4 mmol) 의 트리에틸아민 및 3.97 g (7.2 mmol, 1 eq.) 의 제조예 4 로부터의 식 II-2 의 화합물의 현탁액을 50℃ 에서 가열하고, 단계 a) 로부터의 요오드-함유 푸코오스 구성 요소의 용액을 적가하고, 혼합물을 24 h 동안 50℃ 에서 교반하였다.
불용성 성분을 여과하고, 휘발성 성분을 진공에서 제거하여, 59.6 중량% 의 표제 화합물을 포함하는 8.1 g 의 미정제 생성물을 수득하였다 (수율: 78%).
후처리를 위해, 미정제 생성물을 10 mL 의 DCM 에 다시 용해시키고, 10 mL 의 10% 나트륨 티오술페이트 용액으로 세정하였다. 진공에서 증발시킨 후, 71.6 중량% 의 표제 화합물을 포함하는 6.6 g 의 미정제 화합물을 수득하였다.
실시예 9 와 유사한 방식으로, 미정제 표제 화합물을 실리카 겔 상에서 크로마토그래피로 정제하여, 순도가 99% 인 4.8 g 의 표제 화합물을 수득하였다 (수율: 75.8%, HPLC 방법 2; 10.3 min).
실시예 11: 식 IIIc 의 화합물 (식 중, R11 = CH3, R2 = CH3, R3 = CH3, 및 R4 = 트리메틸실릴) 의 제조
a)
2.02 g (13.5 mmol) 의 헥사메틸디실란 (98% 농도) 및 3.47 g (13.5 mmol) 의 요오드 (98% 농도) 를 4.2 g 의 DCM 중에서 180 min 동안 RT 에서 교반한 다음, 4.2 g 의 DCM 중 11 g (22.5 mmol) 테트라키스트리메틸실릴푸코오스 (92.8% 농도) 를 첨가하였다. RT 에서 180 min 교반 후, 휘발성 성분을 진공 중 40℃ 에서 제거하고, 잔류물을 각각의 경우 10 mL 의 톨루엔으로 3 회 공동증류시켰다. 이후, 미정제 생성물을 3.8 mL 의 DCM 에 용해시켰다.
b)
제 2 플라스크에서, 10 g 의 DCM 중 0.169 g (1.1 mmol) 의 NaI, 2.96 g (29.3 mmol) 의 NEt3 및 12.42 g (22.5 mmol, 1 eq.) 의 제조예 4 로부터의 화합물 II-2 의 현탁액을 제조하고, 단계 a) 로부터의 요오드-함유 푸코오스 구성 요소의 용액을 적가하였다. 50-64℃ 의 내부 온도로 24 h 동안 가열한 후, 혼합물을 냉각시키고, 불용성 성분을 여과하였다. 이후, 여과물을 13.4 g 의 DCM 으로 희석하고, 10% 나트륨 티오술페이트 용액 (14 g) 으로 세정하였다.
농축 후, 72.5 중량% 의 표제 화합물을 포함하는 21.7 g 의 미정제 생성물을 수득하였다 (HPLC 방법 2 에 따름: 10.3 min).
실시예 12: 식 IIIc 의 화합물 (식 중, R11 = C(CH3)3, R2 = CH3, R3 = CH3, 및 R4 = 트리메틸실릴) 의 제조
3 mL 의 DCM 중 3.33 g (13.1 mmol) 의 요오드 및 1.94 g (13.1 mmol) 의 헥사메틸디실란을 3 h 동안 RT 에서 교반하였다. 이에 3 mL 의 DCM 중 11 g (21.6 mmol) 의 89% 농도 테트라키스트리메틸실릴푸코오스 () 를 첨가하고, 추가 20 min 동안 RT 에서 교반을 지속하였다. 휘발성 성분을 진공 중 40℃ 및 30 mbar 에서 제거하고, 잔류물을 각각의 경우 10 mL 의 톨루엔으로 3 회 공증발시켰다. 잔류물을 1 mL 의 DCM 에서 용해시켰다.
7.5 g 의 DCM 중 2.84 g (28 mmol) 의 트리에틸아민, 0.16 g (1.1 mmol) 의 NaI, 12.81 g (21.6 mmol) 의 제조예 6 으로부터의 화합물 II-4 의 환류 하 가열된 혼합물에 상기 용액을 적가하였다. 이후, 반응 혼합물을 환류 하 20 h 동안 교반하면서 가열하였다. 후처리를 위해, 10 mL 의 10 % 나트륨 티오술페이트 용액을 첨가하고, 혼합물을 5 min 동안 격렬하게 교반하고, 상을 분리하였다. 유기 상을 5 mL 의 물로 세정하고, Na2SO4 를 통해 건조시키고, 농축시켰다.
특징화 및 정제를 위해, 17.0 g 의 미정제 생성물을 시클로헥산/에틸 아세테이트 5:1 에 용해시키고, 1% 트리에틸아민으로 처리하고, 실리카 겔 컬럼 (컬럼 치수: 직경 d = 9 cm, 높이 h = 37 cm, 용적 V ~ 2.3 L) 의 상부에 이동시켰다. 컬럼을 약한 압력 하에서 용리하였다. 생성물 분획을 45℃ 및 5 mbar 에서 회전 증발 이후 1 h 동안 오일 펌프 진공을 통해 조합하고, 농축시켜 11.42 g 의 표제 화합물을 수득하였다.

실시예 13: 식 IIIc 의 화합물 (식 중, R11 = C(CH3)3, R2 = CH3, R3 = CH3, 및 R4 = 트리메틸실릴) 의 제조
a)
7.08 g (14 mmol) 의 89.4 중량% 농도 테트라키스트리메틸실릴푸코오스를 4 mL 의 DCM 에 용해시키고, 2.88 g (13.5 mmol) 의 TMSI (97%) 로 처리하였다. 20 min 동안 교반한 후, 휘발성 성분을 진공 중 제거하고, 이후 잔류물을 각각의 경우 10 mL 의 톨루엔으로 3 회 공동증류시켰다. 미정제 생성물을 2 mL 의 DCM 에 용해시켰다.
b)
제 2 플라스크에서, 4 g 의 DCM 중 8.28 g (14 mmol) 의 제조예 6 으로부터의 화합물 II-4 를 1.86 g 의 트리에틸아민 (푸코오스 구성 요소를 기준으로 1.3 eq.), 105 mg 의 NaI (푸코오스 구성 요소를 기준으로 0.05 eq.) 및 0.177 g 의 요오드 (푸코오스 구성 요소를 기준으로 0.05 eq.) 로 처리하고, 이어서 단계 a) 로부터의 트리메틸실릴푸코오스 요오다이드 용액을 50 내지 65℃ 에서 적가하였다. 환류 하 추가 24 h 동안 교반 및 가열한 후, 혼합물을 냉각시키고, 불용성 성분을 여과하였다.
유기 상을 연속하여 10 mL 의 10% 나트륨 티오술페이트 용액 및 10 mL 의 물로 세정한 다음 Na2SO4 를 통해 건조시켰다.
여과 및 농축 후, 69.5 중량% 의 표제 화합물을 포함하는 13.5 g 의 미정제 생성물을 수득하였다 (71% 수율, HPLC 방법 2 에 따름: 15.04 min).
실시예 14: 식 IIIc 의 화합물 (식 중, R11 = C(CH3)3, R2 = CH3, R3 = CH3, R4 = 트리메틸실릴) 의 제조
1.28 g (8.7 mmol) 의 헥사메틸디실란 (98%) 및 2.2 g (8.7 mmol) 의 요오드 (98%) 를 2 mL 의 DCM 중에서 180 min 동안 RT 에서 교반하였다. 이에 1 mL 의 DCM 중 8 g (14.3 mmol) 의 테트라키스트리메틸실릴푸코오스 (81% 농도) 를 첨가하였다. 180 min 동안 RT 에서 교반한 후, 0.573 g (5 mmol) KHCO3 를 첨가하고, 혼합물을 60 min 동안 RT 에서 교반하였다. 1.88 g (18.6 mmol) 의 트리에틸아민을 이후 첨가하고, 혼합물을 환류 하 가열하였다. 15.92 g 의 제조예 6 으로부터의 화합물 II-4 (14.3 mmol) 의 53 중량% 디클로로메탄 용액을 첨가하고, 혼합물을 교반 및 환류 하 24 h 동안 가열하였다.
냉각 후, 불용성 성분을 여과하고, 이어서 여과물을 10 g 의 10% 나트륨 티오술페이트 용액 및 물로 세정하였다. Na2SO4 를 통해 건조시키고, 건조될 때 까지 농축시킨 후, 61.5 중량% 의 표제 화합물을 포함하는 16 g 의 미정제 생성물을 수득하였다 (HPLC 방법 2 에 따름) (미정제 수율: 71.5%).
실시예 15: 2'-O-푸코실락토오스의 제조를 위한 원-포트 (one-pot) 반응
2 mL 의 DCM 중 1.28 g 의 헥사메틸디실란 (8.7 mmol) 및 2.2 g 의 요오드 (8.7 mmol) 를 3 h 동안 RT 에서 교반하였다. 이후, 7.25 g (14.3 mmol) 의 테트라키스트리메틸실릴푸코오스 (89.4% 농도) 를 첨가하고, 혼합물을 20 min 동안 교반하였다. 휘발성 성분을 진공 중 제거하고, 잔류물을 각각의 경우 10 mL 의 톨루엔으로 3 회 증발시켰다. 잔류물을 1.4 mL 의 톨루엔에 용해시키고, 5.4 g 의 DCM 중 1.88 g (18.6 mmol) 의 트리에틸아민, 0.107 g (0.7 mmol) 의 NaI 및 8.47 g 의 제조예 6 으로부터의 화합물 II-4 의 용액에 적가하고, 용액을 환류 하 가열하였다. 혼합물을 교반하고, 환류 하 24 h 동안 가열하고, 여과하고, 농축시켰다.
잔류물을 약 10 mL 의 DCM 에 용해시키고, 10 mL 의 10% 나트륨 카르보네이트 용액 및 10 mL 의 10% 나트륨 티오술페이트 용액으로 세정하였다. 조합된 수성 상을 6 mL 의 DCM 으로 추출하였다. 조합된 유기 상을 Na2SO4 를 통해 건조시키고, 농축시켜 13.9 g 의 미정제 생성물 IIIc (식 중, R11 = C(CH3)3, R2 = CH3, R3 = CH3 및 R4 = Si(CH3)3) 를 수득하였다.
44.8 mL 의 메탄올 중 13.8 g 의 수득한 미정제 생성물 IIIc 를 2.82 g (20 mmol) 의 K2CO3 로 처리하고, 16 h 동안 교반하였다. 메탄올을 온도가 38℃ 가 될 때까지 300 mbar 에서 증류시켰다. 이후, 28 mL 의 DCM 을 첨가하고, 혼합물을 13.5 mL 의 물로 세정하였다. 수성 상을 6.6 mL 의 DCM 으로 재추출하고, 조합된 유기 상을 Na2SO4 를 통해 건조시켰다. 용매를 제거한 후, HPLC 분석을 통해 미정제 생성물에 대한 완전한 전환을 확인한 다음, 28 mL 의 DCM 에 용해시켰다. 이에 28 mL 의 물을 첨가하고, DCM 을 증류시켰다. 이후, 6.6 mL 의 물 및 34.8 mL 의 1 N HCl 을 첨가하고, 혼합물을 24 h 동안 RT 에서 교반하였다.
이어서, 혼합물을 53 mL 의 이온 교환기 IMAC HP 661 로 채워진 컬럼을 통해 용리하여 중화시킨 다음, 26 mL 의 물로 다시 세정하고, 조합된 수성 상을 6 mL 의 DCM 으로 다시 세정하였다. 진공 중 증발시킨 후, 67.2 중량% 의 2'-O-푸코실락토오스를 갖는 6.2 g 의 미정제 생성물을 수득하였다 (HPLC 방법 2). 표적 (1-2-α) 생성물 대 (1-2-β) 생성물의 비는 29:1 (면적 백분율) 이었다.
실시예 16: 식 IIIc 의 화합물 (식 중, R11 = C(CH3)3, R2 = CH3, R3 = CH3, 및 R4 = Si(CH3)3) 의 제조
30 mL 의 DCM 중 17.97 g (0.120 mol) 의 헥사메틸디실란 및 30.84 g 의 요오드 (0.120 mol) 를 3 h 동안 RT 에서 교반하였다. 102.3 g 의 테트라키스트리메틸실릴푸코오스 (89 중량%, 0.205 mol) 를 첨가하고, 혼합물을 20 min 동안 RT 에서 교반하였다. 휘발성 성분을 진공 중 제거하고, 잔류물을 각각의 경우 75 mL 의 톨루엔으로 3 회 공동증류시켰다.
이에 따라 수득한 미정제 트리스(트리메틸실릴)푸코실 요오다이드를 75 g 의 DCM 중 118.83 g 의 제조예 6 으로부터의 화합물 II-4, 26.38 g (0.2607 mol) 의 트리에틸아민 및 1.5 g 의 NaI (0.01 mol) 의 용액에 적가하고, 용액을 환류 하 가열한 다음, 30 mL 의 DCM 으로 헹궜다. 환류 하 24 h 동안 가열한 후, 반응 혼합물을 RT 로 냉각시키고, 150 mL 의 DCM 및 200 mL 의 5% 나트륨 티오술페이트 용액을 첨가하고, 혼합물을 10 min 동안 격렬하게 교반하였다. 유기 상을 분리하고, 50 mL 의 물로 세정하고, Na2SO4 를 통해 건조시키고, 여과하고, 농축시켰다. HPLC 분석에 따라, 76.7% 의 표제 생성물을 수득하였다.
미정제 생성물을 각각 100 g 의 두 분율로 분배하고, 두 분획을 시클로헥산/에틸 아세테이트 5:1 및 1% 의 트리에틸아민을 사용하는 실리카 겔 상에서의 컬럼 크로마토그래피 (컬럼: 직경 d = 12.5 cm, 높이 h = 40 cm, 용적 V~4.9 L) 로 정제하여, 94.6% (제 1 컬럼) 의 순도를 갖는 70.9 g 의 생성물 분획 및 87.5% (제 2 컬럼) 의 순도를 갖는 71.9 g 의 생성물 분획을 수득하였다. 두 생성물 분획을 조합하였다.
실시예 17: 식 IIIb 의 화합물 (식 중, R2 = CH3, R3 = CH3 및 R4 = Si(CH3)3) 의 제조
155.38 g 의 화합물 IIIc (식 중, R11 = C(CH3)3, R2 = CH3, R3 = CH3, R4 = Si(CH3)3 (함량 85.7 중량%, 실시예 16 에 따라 제조됨)) 를 569 mL 의 MeOH 에서 38.5 g K2CO3 와 22 h 동안 RT 에서 교반하였다. 휘발성 성분을 진공 중 제거하고, 잔류물을 350 mL 의 DCM 에 용해시키고, 각각의 경우 150 mL 의 물로 3 회 세정하였다. 조합된 수성 상을 DCM 으로 1 회 재추출하고, 조합된 유기 상을 Na2SO4 를 통해 건조시켰다. 진공 중 여과 및 농축시킨 후, 90.4% 의 함량을 갖는 118.2 g 의 미정제 생성물 IIIb 를 수득하였다.
추가 반응의 용이한 처리를 위해, 미정제 생성물을 100 mL 의 DCM 에 용해시켰다.
실시예 18: 2'-O-푸코실락토오스의 제조
125 g 의 실시예 17 로부터의 미정제 생성물 용액 (DCM 중 59.1 g 의 화합물 IIIb) 을 175 mL 의 물로 처리하고, DCM 을 50℃ 및 400 mbar 에서 회전 증발기를 이용하여 증류시켰다. 수성 혼합물을 RT 로 냉각시키고, 추가로 191 mL 의 물 및 122 mL 의 2N HCl (HCl 의 총 농도는 0.5 mol/L 에 상응함) 을 첨가하고, 밤새 RT 에서 교반하였다. 이후, 용액 (535 g) 을 약 염기성 이온 교환기 Lewatit MP62 를 통해 여과시킨 다음, 각각의 경우 160 mL 의 물로 3 회 다시 세정하였다. 모액 및 세척액을 조합하고, 30℃ 및 5 mbar 에서 회전 증발기 및 1 h 동안 오일 펌프 진공을 이용하여 증발시켜, 90.7% 의 순도를 갖는 38.9 g 의 생성물을 수득하고; 생성물 함량은 78.3 중량% (HPLC 방법 3 에 따름) 이었다. 표적 (1-2-α) 생성물 대 (1-2-β) 생성물의 비는 49:1 (중량 백분율) 이었다.
실시예 19: 2'-O-푸코실락토오스의 제조
실시예 18 과 유사한 방식으로, 실시예 17 에서 수득된 DCM 중 25 g 의 화합물 IIIb 의 용액을 수성 염산으로 처리한 다음, 80 mL 의 Amberlyst A21 을 통해 여과시켜, 락토오스와 상이한 성분을 기준으로 88.7% 의 순도를 갖는 8.05 g 의 2'-O-푸코실락토오스를 수득하였다. 2'-O-푸코실락토오스의 함량은 72 중량% 였다.
실시예 20: 2'-O-푸코실락토오스의 제조
실시예 18 과 유사한 방식으로, 실시예 17 에서 수득된 DCM 중 25 g 의 화합물 IIIb 의 용액을 수성 염산으로 처리한 다음, IMAC HP 661 을 통해 여과시켜, 락토오스와 상이한 성분을 기준으로 91.1% 의 순도를 갖는 7.7 g 의 미정제 생성물을 수득하였다. 2'-O-푸코실락토오스의 함량은 78 중량% 였다.
5.0 g 의 2'-O-푸코실락토오스를 가열 하 15 mL 의 MeOH 에 용해시키고, 불용성 성분을 가열 하 여과하였다. 표제 생성물을 45 mL 의 저온 EtOH 를 첨가함으로써 여과물로부터 침전시키고, 침전물을 여과하고, EtOH 로 2 회 세정하였다. 표제 생성물을 순도 92% 로 수득하였고, 순도는 락토오스를 포함하지 않고 측정된다. 수율은 78.8% 였다.
HPLC: 0.05 중량% 의 푸코오스; 8.9 중량% 의 락토오스; 0.2 중량% 의 2'-Fl β-아노머; 69.0 중량% 의 2'-Fl; 1.75 면적-% 의 3당류
정제 1: 메탄올을 사용하는 2'-O-푸코실락토오스의 정제:
35 mL 의 MeOH 중 5.0 g 의 실시예 18 로부터의 2'-O-푸코실락토오스의 미정제 생성물을 환류 하 가열하고, 현탁액을 총 용적 10 mL 로 농축시켰다. 이후, 혼합물을 -5℃ 로 냉각시키고, 30 min 동안 교반하였다. 수득한 고체를 흡인시키고, 각각의 경우 2 mL 의 저온 메탄올로 2 회 및 각각의 경우 2 mL 의 저온 아세톤으로 2 회 세정하였다. 수율이 83.7 중량% 인 3.41 g 의 표제 생성물을 수득하였다.
표제 생성물은 11.5 중량% 의 락토오스를 함유하였고, HPLC 방법 3 에 따라 및 락토오스 함량을 고려하여 순도는 97.4 면적-% 였다.
정제 2: H2O/아세톤을 사용하는 2'-O-푸코실락토오스의 정제:
5.0 g 의 실시예 18 로부터의 2'-O-푸코실락토오스를 75℃ 에서 3 mL 의 물에 용해시켰다. 이후, 용액을 냉각시키고, 1 mL 의 아세톤 및 5 mL 의 EtOH 를첨가하고, 혼합물을 16 h 동안 교반하였다. 혼합물을 여과하고, 수득한 고체를 각각의 경우 3 mL 의 EtOH/H2O (96.5:3.5) 로 3 회 세정하였다. 2.14 g 의 표제 생성물 (수율: 50.0%) 을 수득하였다. 락토오스의 함량은 HPLC 분석에 따라 12% 였다.
실시예 21 (비교예): 비교 화합물, 2'-FL (2'-O-α-L-푸코피라노실락토오스) 의 β-이성질체의 제조:
2,3,4-트리-O-아세틸-β-L-푸코피라노실트리클로로아세트이미데이트를 [Liebigs Ann Chemie, 1991, 121] 에 서술된 바와 같이 제조하였다.
단계 1: O-(2,3,4-트리-O-아세틸-β-L-푸코피라노실)-(1-2)-O-(6-O-아세틸-3,4-이소프로필리덴-β-D-갈락토피라노실)-(1-4)-2,3:5,6-디-O-이소프로필리덴-D-글루코오스-디메틸아세탈
1.5 g 의 분자체 4Å 로 채워진 플라스크를 진공 중 가열하고, 아르곤으로 퍼징하였다. 이에 2 g 의 CH2Cl2 중 2.77 g (5 mmol) 의 제조예 4 로부터의 화합물 II-2 의 용액 및 이후 -5 - 0℃ 에서 1.46 g (3.3 mmol) 의 3,4-트리-O-아세틸-β-L-푸코피라노실트리클로로아세트이미데이트를 첨가하고, 혼합물을 1 h 동안 0℃ 에서 교반하였다. 이후, 1 mL 의 CH2Cl2 중 48 mg 의 BF3·OEt2 를 0 내지 5℃ 에서 적가하고, 혼합물을 6 h 동안 0℃ 에서, 이후 밤새 0 내지 20℃ 에서 교반하였다. 반응 혼합물을 여과하고, 10 mL 의 CH2Cl2 로 세정하였다. 조합된 유기 상을 10 mL 의 포화 NaHCO3 용액으로 세정하고, Na2SO4 를 통해 건조시키고, 여과하고, 농축시켰다. 실리카 겔 (500 mL) 상에서 시클로헥산/에틸 아세테이트 2:1 내지 1:1 로의 크로마토그래피에 의해 정제된 3.85 g 의 미정제 생성물을 수득하여, 1 g 의 표제 화합물을 수득하였다.

단계 2: O-β-L-푸코피라노실-(1-2)-O-(3,4-이소프로필리덴-β-D-갈락토피라노실)-(1-4)-2,3:5,6-디-O-이소프로필리덴-D-글루코오스-디메틸아세탈
1.87 g 의 단계 1 로부터의 화합물을 10 mL 의 메탄올 및 406 mg 의 나트륨 메틸레이트로 16 h 동안 RT 에서 교반 하 처리하였다. 이후, 불용성 성분을 여과하고, 혼합물을 진공 중 농축시키고, 100 mL 의 실리카 겔 상에서 디클로로메탄/MeOH 95:5 로의 크로마토그래피에 의해 정제하여, 1.1 g 의 표제 화합물을 수득하였다.

단계 3: β-2'-O-푸코실락토오스
0.97 g 의 단계 2 로부터의 생성물을 21.6 mL 의 수성 염산 (0.5 M) 으로 처리하고, 20 h 동안 RT 에서 교반하였다. 이후, 반응 혼합물을 7.8 mL 의 이온 교환기 IMAC HP661 로 중화시키고, 여과하고, 10 mL 의 물로 세정하였다. 진공 중 휘발성 성분을 제거한 후, 잔류물을 고 진공 하 건조시키고, 환원 말단기에서의 아노머 혼합물로서 0.66 g 의 2'-O-푸코실락토오스의 β-이성질체를 수득하였다.
아노머 I 
아노머 II 
Claims (32)
- 하기 단계를 포함하는, 2'-O-푸코실락토오스의 제조 방법:
a) 일반식 (I) 의 보호된 푸코오스를,

[식 중, RSi 는 동일 또는 상이할 수 있고, 식 SiRaRbRc 의 잔기 (이때,
Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임];
일반식 (II) 의 화합물과 반응시키는 단계

[식 중,
R1 은 C(=O)-R11 잔기 또는 SiR12R13R14 잔기 (이때,
R11 은 수소, C1-C8-알킬, C1-C8-할로알킬, C3-C8-시클로알킬, C3-C8-시클로알킬-C1-C4-알킬이거나 페닐 (이때, 상기 페닐은 미치환되거나 임의로 할로겐, CN, NO2, C1-C4-알킬, C1-C4-알콕시, C1-C4-할로알킬 및 C1-C4-할로알콕시로부터 선택되는 1 내지 5 개의 치환기를 가짐) 이고,
R12, R13 및 R14 은 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 이고;
R2 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성함];
b) 2'-O-푸코실락토오스를 수득하기 위해, 단계 a) 에서 수득된 일반식 (III) 의 커플링 생성물을 탈보호하는 단계

[식 중, RSi, R1, R2 및 R3 는 상기 정의된 바와 같음];
여기서, 단계 a) 는
a.1) 트리(C1-C6-알킬)실릴 요오다이드로의 일반식 (I) 의 보호된 푸코오스의 처리
a.2) 하나 이상의 염기 존재 하, 식 (II) 의 화합물과 단계 a.1) 에서 수득된 생성물의 반응
을 포함함. - 제 1 항에 있어서, 트리(C1-C6-알킬)실릴 요오다이드가 트리메틸실릴 요오다이드인 방법.
- 제 1 항 또는 제 2 항에 있어서, 트리(C1-C6-알킬)실릴 요오다이드가 식 (I) 의 화합물의 몰 당 0.8 내지 1.4 mol 의 양으로 사용되는 방법.
- 제 1 항 내지 제 3 항 중 어느 한 항에 있어서, 트리(C1-C6-알킬)실릴 요오다이드가 요오다이드 염으로 해당 트리(C1-C6-알킬)실릴 클로라이드를 처리함으로써 제자리에서 생성되는 방법.
- 제 1 항 내지 제 3 항 중 어느 한 항에 있어서, 트리(C1-C6-알킬)실릴 요오다이드가 해당 헥사(C1-C6-알킬)디실란과 요오드를 반응시킴으로써 제자리에서 생성되는 방법.
- 제 5 항에 있어서, 먼저 단계 a.1) 에서 헥사(C1-C6-알킬)디실란이 요오드와 반응한 다음, 수득된 반응 혼합물이 식 (I) 의 화합물과 반응하는 방법.
- 제 1 항 내지 제 6 항 중 어느 한 항에 있어서, 단계 a) 에서:
a.1) 식 (I) 의 화합물이 먼저 트리(C1-C6-알킬)실릴 요오다이드로 처리되고;
a.2) 염기 존재 하, 수득된 생성물이 추가 정제 없이 식 (II) 의 화합물과 반응하는 방법. - 제 1 항 내지 제 7 항 중 어느 한 항에 있어서, 염기가 식 (I) 의 화합물을 기준으로 적어도 등몰량으로 사용되는 방법.
- 제 1 항 내지 제 8 항 중 어느 한 항에 있어서, 염기가 아민 염기, 특히 3차 아민으로부터 선택되는 하나 이상의 염기성 화합물을 포함하는 방법.
- 제 9 항에 있어서, 염기가 부가적으로 알칼리 카르보네이트, 알칼리 히드로겐 카르보네이트 및 이의 혼합물로부터 선택되는 염기성 화합물을 포함하는 방법.
- 제 10 항에 있어서,
a.1) 먼저, 헥사(C1-C6-알킬)디실란이 요오드와 반응한 다음, 수득된 반응 혼합물이 식 (I) 의 화합물과 반응하고;
a.2) 단계 a.1) 의 반응 혼합물이 알칼리 카르보네이트, 알칼리 히드로겐 카르보네이트 및 이의 혼합물로부터 선택되는 염기성 화합물로 처리되고, 이후 수득된 혼합물이 아민 염기 존재 하에서 식 (II) 의 화합물과 반응하는 방법. - 제 1 항 내지 제 11 항 중 어느 한 항에 있어서, 단계 a.2) 가 요오드, 요오다이드 염 및 트리아릴포스핀 산화물 및 이의 혼합물로부터 선택되는 하나 이상의 시약 존재 하에서 수행되는 방법.
- 제 1 항 내지 제 12 항 중 어느 한 항에 있어서, 식 (I) 의 화합물 및 식 (II) 의 화합물이 1 : 3 내지 3 : 1 범위의 (I) : (II) 몰 비로 반응하는 방법.
- 제 1 항 내지 제 13 항 중 어느 한 항에 있어서, 단계 b) 에서:
b.1) 산 존재 하에서 식 (III) 의 화합물이 물로 처리되거나;
또는
b.2) 식 (III) 의 화합물 (식 중, R1 은 SiR12R13R14 잔기임) 이 먼저 탈실릴화 시약으로 처리되고, 이때 식 (IIIb') 의 화합물이 수득되고:
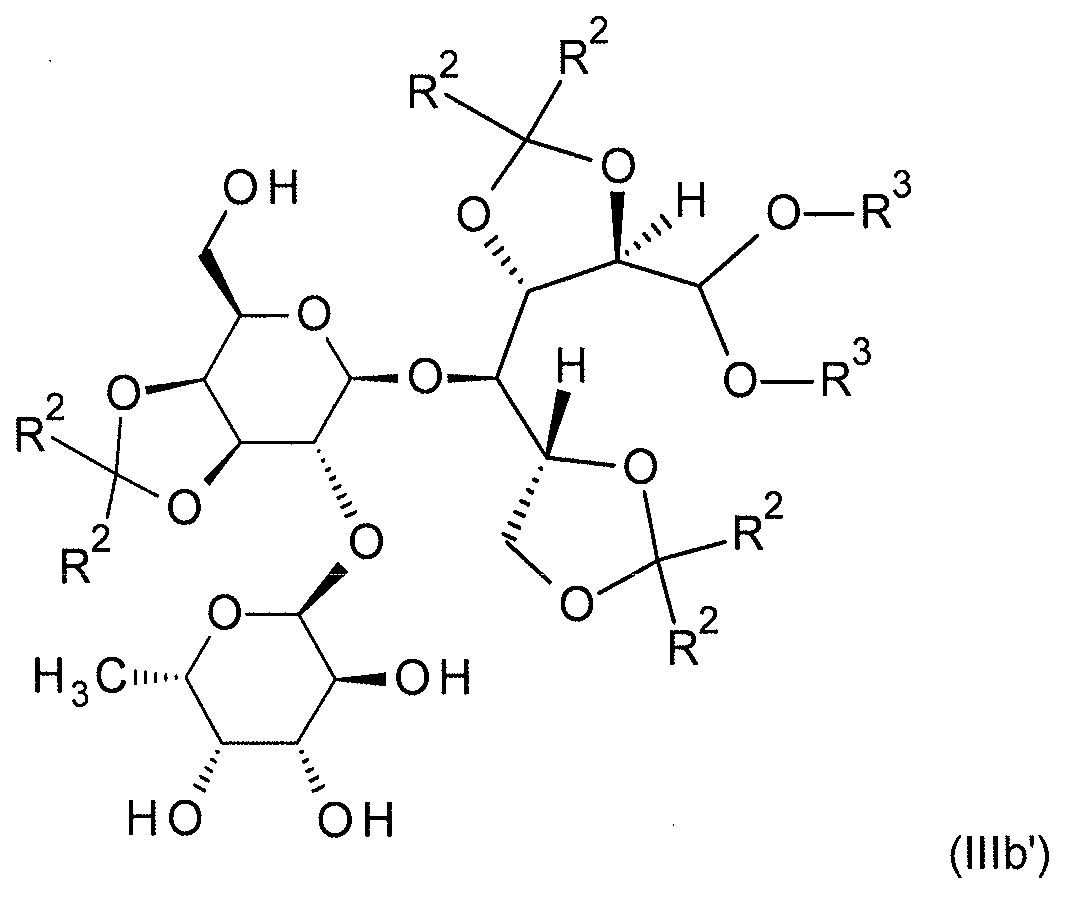
이후, 잔류 보호기가 산 존재 하에서 식 (IIIb') 의 화합물을 물로 처리함으로써 제거되거나;
또는
b.3) 식 (III) 의 화합물 (식 중, R1 은 C(O)R11 잔기임) 이 먼저 탈실릴화 시약으로 처리되고, 이때 식 (IIIc') 의 화합물이 수득되고:

이후, C(O)-R11 기 및 잔류 보호기가 연속적으로 제거되거나;
또는
b.4) 보호기 C(R2)2 및 OR3 가 먼저 식 (III) 의 화합물 (식 중, R1 은 C(O)R11 잔기임) 로부터 제거되고, 이때 식 (IV) 의 화합물이 수득되고:

이후, C(O)-R11 기가 제거되거나,
또는
b.5) 식 (III) 의 화합물 (식 중, R1 은 라디칼 C(O)-R11 임) 이 먼저 C1-C4-알칸올 및 알칼리금속 염기로 처리되어 식 (IIIb') 의 화합물을 수득하고, 이후 잔류 보호기가 산성 반응 조건 하에서 식 (IIIb') 의 화합물을 처리함으로써 제거되는 방법. - 제 1 항 내지 제 14 항 중 어느 한 항에 있어서, 식 (I) 및 (III) 의 RSi 잔기가 트리메틸실릴인 방법.
- 제 1 항 내지 제 15 항 중 어느 한 항에 있어서, 식 (II) 및 (III) 의 R1 잔기가 트리메틸실릴인 방법.
- 제 1 항 내지 제 15 항 중 어느 한 항에 있어서, 식 (II) 및 (III) 의 R1 잔기가 아세틸, 피발로일 또는 벤조일인 방법.
- 제 1 항 내지 제 17 항 중 어느 한 항에 있어서, 식 (II) 및 (III) 의 R2 잔기가 메틸인 방법.
- 제 1 항 내지 제 18 항 중 어느 한 항에 있어서, 식 (II) 및 (III) 의 R3 잔기가 메틸인 방법.
- 일반식 (IIIa) 의 화합물:

[식 중,
R1a 는 SiR12R13R14 잔기 (이때,
R12, R13 및 R14 은 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 이고;
R2 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R4 는 동일 또는 상이하고, 수소이거나 SiRaRbRc 잔기 (이때,
Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임]. - 일반식 (IIIb) 의 화합물:

[식 중,
R2 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R4 는 동일 또는 상이하고, 수소이거나 SiRaRbRc 잔기 (이때,
Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임]. - 일반식 (IIIc) 의 화합물:
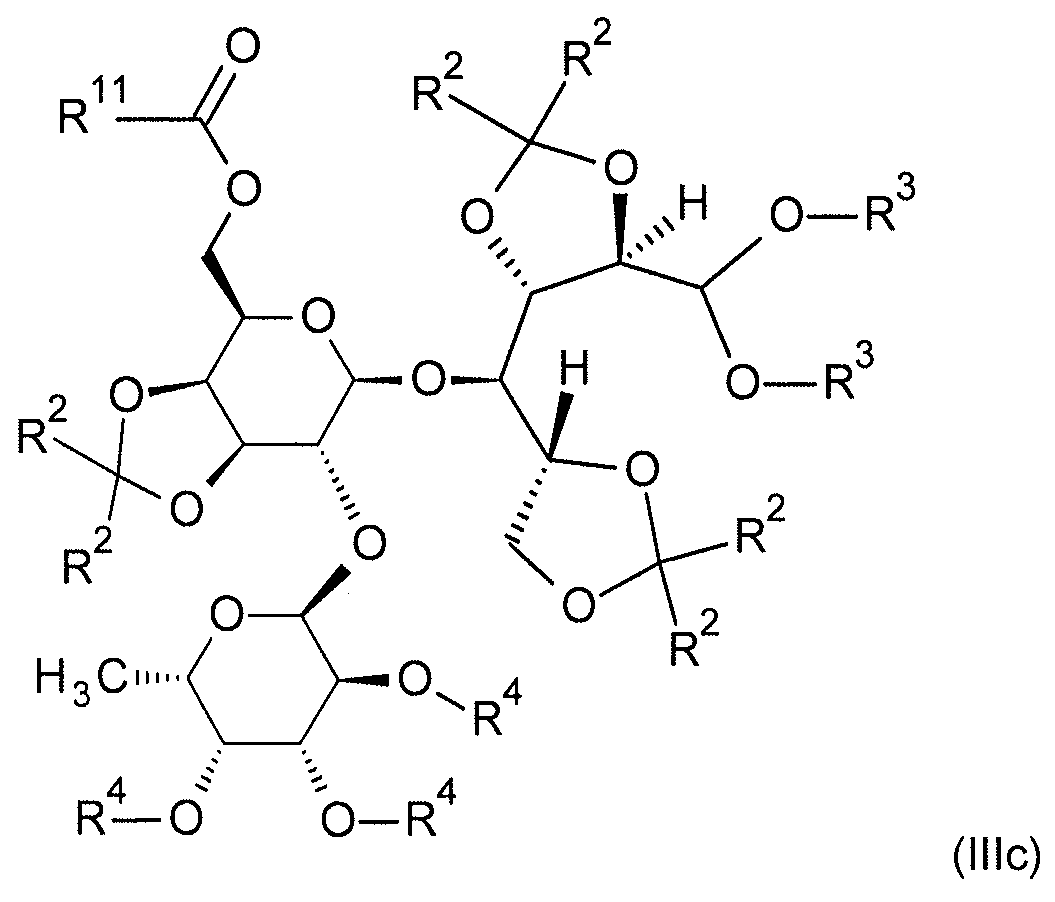
[식 중,
R11 은 수소, C1-C8-알킬, C1-C8-할로알킬, C3-C8-시클로알킬, C3-C8-시클로알킬-C1-C4-알킬이거나 페닐 (이때, 상기 페닐은 미치환되거나 임의로 할로겐, CN, NO2, C1-C4-알킬, C1-C4-알콕시, C1-C4-할로알킬 및 C1-C4-할로알콕시로부터 선택되는 1 내지 5 개의 치환기를 가짐) 이고;
R2 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R4 는 동일 또는 상이하고, 수소이거나 SiRaRbRc 잔기 (이때, Ra, Rb 및 Rc 는 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 임]. - R1a 가 tert.부틸디메틸실릴이고, R2 및 R3 가 모두 메틸인 식 (IIa) 의 화합물을 제외한 일반식 (IIa) 의 화합물:

[식 중,
R1a 는 SiR12R13R14 잔기 (이때, R12, R13 및 R14 은 동일 또는 상이하고, C1-C8-알킬, C3-C8-시클로알킬, 페닐 및 C3-C8-시클로알킬-C1-C4-알킬로부터 선택됨) 이고;
R2 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 동일한 탄소 원자에 부착되어 있는 2 개의 R2 잔기가 함께 선형 C3-C6-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성하고;
R3 는 동일 또는 상이할 수 있고, C1-C8-알킬이거나, 함께 선형 C1-C4-알칸디일 (이는 미치환되거나 치환기로서 1 내지 6 개의 메틸기를 가짐) 을 형성함]. - 일반식 (IV) 의 화합물:
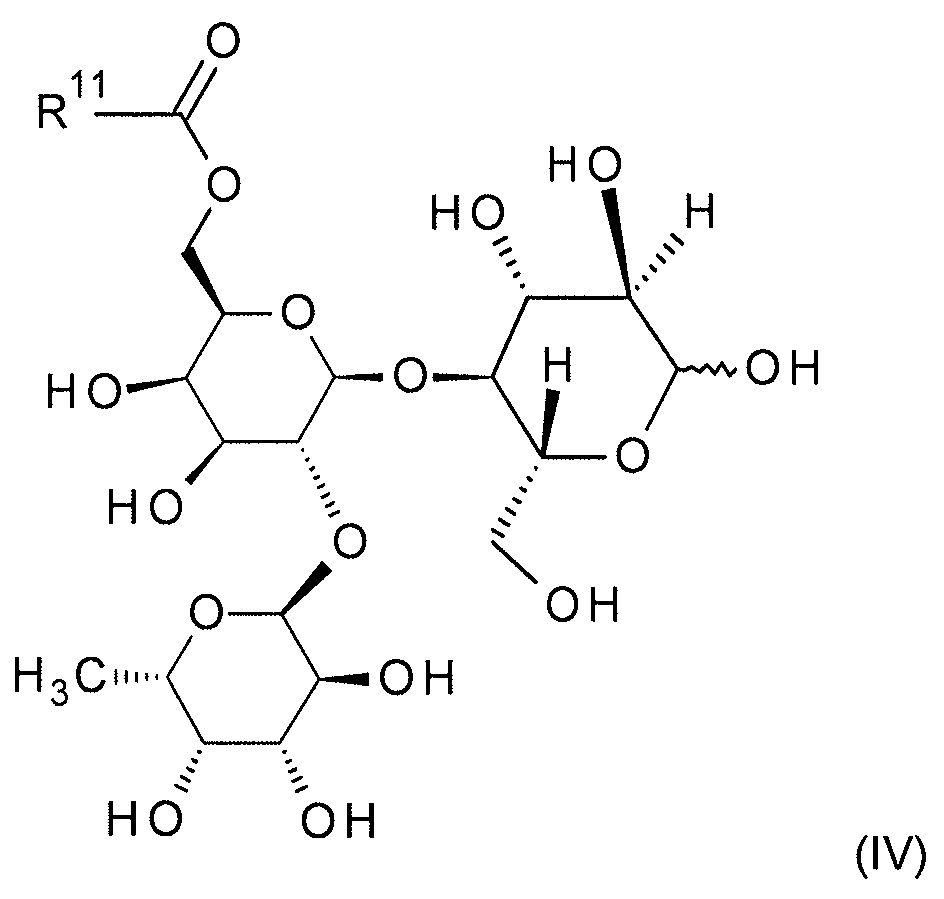
[식 중,
R11 은 수소, C2-C8-알킬, C1-C8-할로알킬, C3-C8-시클로알킬, C3-C8-시클로알킬-C1-C4-알킬이거나 페닐 (이때, 상기 페닐은 미치환되거나 임의로 할로겐, CN, NO2, C1-C4-알킬, C1-C4-알콕시, C1-C4-할로알킬 및 C1-C4-할로알콕시로부터 선택되는 1 내지 5 개의 치환기를 가짐) 임]. - 제 1 항 내지 제 19 항 중 어느 한 항에 따른 방법으로 수득가능한 2'-O-푸코실락토오스.
- 제 25 항에 있어서, 1 중량% 미만의 β 아노머 β-2'-O-푸코실락토오스를 함유하는 2'-O-푸코실락토오스.
- 제 25 항에 있어서, 전이 금속 불순물의 양이 검출가능한 수준 미만인 2'-O-푸코실락토오스.
- 2'-O-푸코실락토오스를 제조하기 위한, 제 20 항 내지 제 24 항에 따른 일반식 (IIa), (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물의 용도.
- 식품류에서 또는 식품 첨가제로서의, 제 1 항 내지 제 19 항 중 어느 한 항에 따른 방법으로 수득가능한 2'-O-푸코실락토오스의 용도.
- 일반식 (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물 중 하나 이상으로부터의 2'-O-푸코실락토오스의 제조를 포함하는, 식품류 및 식품 첨가제를 제조하기 위한 제 20 항 내지 제 24 항에 따른 일반식 (IIa), (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물의 용도.
- 제 20 항 내지 제 24 항에 정의된 일반식 (IIa), (IIIa), (IIIb), (IIIc) 또는 (IV) 의 화합물 중 하나 이상으로부터의 2'-O-푸코실락토오스의 제조 및 식품류에서의 2'-O-푸코실락토오스의 제형화를 포함하는 식품류 제조 방법.
- 제 1 항 내지 제 19 항 중 어느 한 항에 따른 방법으로 수득가능한 2'-O-푸코실락토오스, 및 식품류에 적합한 하나 이상의 담체를 포함하는 식품류 또는 식품 첨가제.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14184606 | 2014-09-12 | ||
| EP14184606.3 | 2014-09-12 | ||
| PCT/EP2015/070844 WO2016038192A1 (en) | 2014-09-12 | 2015-09-11 | Method for preparing 2'-o-fucosyllactose |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20170052631A true KR20170052631A (ko) | 2017-05-12 |
Family
ID=51687775
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020177009260A KR20170052631A (ko) | 2014-09-12 | 2015-09-11 | 2'-o-푸코실락토오스의 제조 방법 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US11098075B2 (ko) |
| EP (1) | EP3191498B1 (ko) |
| JP (1) | JP6637492B2 (ko) |
| KR (1) | KR20170052631A (ko) |
| CN (1) | CN106795193B (ko) |
| AU (1) | AU2015314165B2 (ko) |
| BR (1) | BR112017004667A2 (ko) |
| CA (1) | CA2960056A1 (ko) |
| CR (1) | CR20170134A (ko) |
| DK (1) | DK3191498T3 (ko) |
| HU (1) | HUE051855T2 (ko) |
| IL (1) | IL250682A0 (ko) |
| MX (1) | MX2017003238A (ko) |
| WO (1) | WO2016038192A1 (ko) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102584658B1 (ko) | 2015-03-26 | 2023-10-04 | 바스프 에스이 | L-푸코오스의 생체촉매적 생산 |
| MX2017014148A (es) | 2015-05-04 | 2018-03-15 | Basf Se | Proceso para la preparacion de melonal. |
| CR20180414A (es) * | 2016-02-03 | 2018-11-29 | Basf Se | Proceso para producir 2´-o-fucosil lactosa |
| WO2017140909A1 (de) | 2016-02-19 | 2017-08-24 | Basf Se | Enzymatische zyklisierung von homofarnesylsäure |
| ES2844182T3 (es) | 2016-06-07 | 2021-07-21 | Basf Se | Procedimiento para la preparación de alcoholes con insaturación en posición 2,3 |
| BR112018075784B1 (pt) | 2016-06-29 | 2022-06-28 | Basf Se | Processo para preparação de aldeídos alfa, beta insaturados |
| CN109788781A (zh) * | 2016-08-31 | 2019-05-21 | 奥利格科学生物技术有限公司 | 人乳低聚糖在犊牛育肥中的用途 |
| WO2020079146A1 (en) | 2018-10-18 | 2020-04-23 | Basf Se | Crystalline form ii of 2'-o-fucosyllactose, process for its preparation, nutritional, cosmetic or pharmaceutical formulation containing the same |
| WO2024038195A1 (en) * | 2022-08-19 | 2024-02-22 | Dsm Ip Assets B.V. | Synthesis of hmo propionate |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5438124A (en) | 1991-07-16 | 1995-08-01 | Health Research, Inc. | Glycosylating reagent for the synthesis of linear and other α-L-fucosyl oligosaccharides |
| IT1392456B1 (it) | 2008-12-18 | 2012-03-09 | Inalco Spa | Processo per la sintesi di di- o oligosaccaridi l-fucosilati e loro nuovi intermedi 2,3,4-tribenzil-fucosil-derivati. |
| WO2010115934A1 (en) | 2009-04-07 | 2010-10-14 | Glycom A/S | Synthesis of 2'-o-fucosyllactose |
| US20140057868A1 (en) | 2011-02-21 | 2014-02-27 | Glycom A/S | Catalytic hydrogenolysis of a composition of a mixture of oligosaccharide precursors and uses thereof |
| US9567361B2 (en) * | 2011-05-13 | 2017-02-14 | Glycosyn LLC | Use of purified 2′-fucosyllactose, 3-fucosyllactose and lactodifucotetraose as prebiotics |
| GB201111263D0 (en) | 2011-07-01 | 2011-08-17 | Nat Univ Ireland | Multivalent oligosaccharides |
| WO2013048294A1 (en) | 2011-09-30 | 2013-04-04 | Volvo Technology Corporation | Exhaust gas after treatment system comprising multiple catalytic objects |
-
2015
- 2015-09-11 WO PCT/EP2015/070844 patent/WO2016038192A1/en active Application Filing
- 2015-09-11 KR KR1020177009260A patent/KR20170052631A/ko unknown
- 2015-09-11 JP JP2017513743A patent/JP6637492B2/ja active Active
- 2015-09-11 EP EP15763307.4A patent/EP3191498B1/en active Active
- 2015-09-11 BR BR112017004667A patent/BR112017004667A2/pt not_active IP Right Cessation
- 2015-09-11 US US15/510,268 patent/US11098075B2/en active Active
- 2015-09-11 AU AU2015314165A patent/AU2015314165B2/en not_active Ceased
- 2015-09-11 MX MX2017003238A patent/MX2017003238A/es unknown
- 2015-09-11 DK DK15763307.4T patent/DK3191498T3/da active
- 2015-09-11 CA CA2960056A patent/CA2960056A1/en not_active Abandoned
- 2015-09-11 CR CR20170134A patent/CR20170134A/es unknown
- 2015-09-11 CN CN201580048101.9A patent/CN106795193B/zh active Active
- 2015-09-11 HU HUE15763307A patent/HUE051855T2/hu unknown
-
2017
- 2017-02-20 IL IL250682A patent/IL250682A0/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| DK3191498T3 (da) | 2020-12-21 |
| AU2015314165A1 (en) | 2017-03-16 |
| BR112017004667A2 (pt) | 2018-01-30 |
| US20180230176A1 (en) | 2018-08-16 |
| IL250682A0 (en) | 2017-04-30 |
| CN106795193B (zh) | 2021-02-26 |
| WO2016038192A1 (en) | 2016-03-17 |
| MX2017003238A (es) | 2017-06-29 |
| HUE051855T2 (hu) | 2021-03-29 |
| EP3191498B1 (en) | 2020-09-30 |
| EP3191498A1 (en) | 2017-07-19 |
| CR20170134A (es) | 2017-06-13 |
| CN106795193A (zh) | 2017-05-31 |
| JP6637492B2 (ja) | 2020-01-29 |
| CA2960056A1 (en) | 2016-03-17 |
| US11098075B2 (en) | 2021-08-24 |
| JP2017534575A (ja) | 2017-11-24 |
| AU2015314165B2 (en) | 2019-06-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20170052631A (ko) | 2'-o-푸코실락토오스의 제조 방법 | |
| AU596644B2 (en) | C(29)-carbonyloxymilbemycin derivatives for controlling parasitic pests of animals and plants | |
| US8450293B2 (en) | Synthesis and characterization of C8 analogs of c-di-GMP | |
| EP2417144B1 (en) | Synthesis of 2'-o-fucosyllactose | |
| CN103703012A (zh) | 乳糖-n-四糖的制造 | |
| US11365211B2 (en) | Stereoselective synthesis and process for the manufacturing of 2′-deoxynucleosides | |
| WO2007099392A2 (en) | Convergent synthesis of carbohydrate building blocks | |
| KR20180104751A (ko) | 2'-o-푸코실락토오스의 제조 방법 | |
| JP2019509285A (ja) | 2’−o−フコシルラクトースの製造方法 | |
| JP4773336B2 (ja) | Lpsアンタゴニストb1287およびその立体異性体の調製のための試薬および方法 | |
| US5342929A (en) | Process for the preparation of glycosides | |
| JP4762973B2 (ja) | 1,2−トランスグリコシド化合物の製造方法 | |
| JPH04352794A (ja) | 抗寄生虫剤として有用なジサッカライドとアグリコンの間に挿入されたスペーサーを含むアベルメクチン誘導体 | |
| Pavashe et al. | Methyl 3, 4, 6-tri-O-acetyl-2-deoxy-2-[(bis-methoxycarbonyl) methyl]-β-d-glucopyranoside | |
| JP2002511478A (ja) | ラミナリビオースの合成方法 | |
| Pavashe et al. | acetyl-2-deoxy-2-[(bis-methoxycarbonyl) methyl]-β-D-glucopyranoside | |
| CN116199657A (zh) | 灰黄霉素4位醚化衍生物及其应用 | |
| CN102099366A (zh) | 结晶型糖衍生物 | |
| US20080177049A1 (en) | Process for Producing 1,2-Trans-Glycoside Compound | |
| CS259423B1 (cs) | Způsob přípravy bisulfitových derivátů 3-fenoxybenzaldehydu | |
| JPS6152152B2 (ko) |