KR20130110161A - β-세크레타아제(BACE) 저해제로 유용한 4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 유도체 - Google Patents
β-세크레타아제(BACE) 저해제로 유용한 4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 유도체 Download PDFInfo
- Publication number
- KR20130110161A KR20130110161A KR1020137007365A KR20137007365A KR20130110161A KR 20130110161 A KR20130110161 A KR 20130110161A KR 1020137007365 A KR1020137007365 A KR 1020137007365A KR 20137007365 A KR20137007365 A KR 20137007365A KR 20130110161 A KR20130110161 A KR 20130110161A
- Authority
- KR
- South Korea
- Prior art keywords
- mmol
- compound
- alkyl
- mixture
- vacuo
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
본 발명은 베타-부위 아밀로이드 절단 효소, BACE, BACE1, Asp2 또는 메맙신2로도 알려져 있는 β-세크레타아제 저해제로서의 신규 4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 유도체에 관한 것이다. 본 발명은 또한 상기 화합물을 포함하는 약제학적 조성물, 상기 화합물 및 조성물의 제조방법, 및 알츠하이머병 (AD), 경증 인지 장애, 노쇠, 치매, 루이소체 치매, 다운증후군, 뇌졸중 관련 치매, 파킨슨병 관련 치매 또는 베타-아밀로이드 관련 치매 등과 같은 β-세크레타아제가 연루되는 질환을 예방 및 치료하기 위한 상기 화합물 및 조성물의 용도에 관한 것이다.
Description
본 발명은 베타-부위 아밀로이드 절단 효소, BACE, BACE1, Asp2 또는 메맙신2로도 알려져 있는 β-세크레타아제 저해제로서의 신규 4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 유도체에 관한 것이다. 본 발명은 또한 상기 화합물을 포함하는 약제학적 조성물, 상기 화합물 및 조성물의 제조방법, 및 알츠하이머병 (AD), 경증 인지 장애, 노쇠, 치매, 루이소체 치매, 다운증후군, 뇌졸중 관련 치매, 파킨슨병 관련 치매 또는 베타-아밀로이드 관련 치매 등과 같은 β-세크레타아제가 연루되는 질환을 예방 및 치료하기 위한 상기 화합물 및 조성물의 용도에 관한 것이다.
알츠하이머병 (AD)은 노화와 관련된 신경변성 질환이다. AD 환자는 인지 결핍 및 기억 상실뿐 아니라 불안증과 같은 행동 장애를 앓는다. AD로 고통받는 환자 중 90% 이상은 산발적 형태의 질병을 가지며, 10% 미만의 경우는 가족성 또는 유전성이다. 미국에서 65세의 사람 10명 중 약 1명이 AD를 가지나, 85세에는 매 2명의 사람 중 1명이 AD에 걸린다. 처음 진단으로부터의 평균 여명은 7-10년이며, AD 환자는 매우 고가인 노인 원호 생활 시설에서나 가족 구성원에 의한 광범위한 관리를 필요로 한다. AD는 인구 중에 노인 수의 증가와 함께, 의학적 관심이 증대하고 있다. AD에 대한 현재 이용가능한 치료법은 단지 질환의 징후를 치료하는 것이며, 인지 특성을 향상시키기 위한 아세틸콜린에스테라아제 저해제뿐 아니라 이 병과 관련된 행동 장애를 제어하기 위한 항불안제 및 항정신병제를 포함한다.
AD 환자의 뇌에서의 현저한 병리학적 특징은 tau 단백질의 과인산화에 의해 생성되는 신경원섬유 매듭 (neurofibillary tangle) 및 β-아밀로이드1-42 (Aβ1-42)펩티드의 응집에 의해 형성되는 아밀로이드 플라크이다. Aβ1-42 는 올리고머를 형성한 다음, 원섬유를 형성하고, 궁극적으로는 아밀로이드 플라크를 형성한다. 올리고머 및 원섬유는 특히 신경독성인 것으로 여겨지며, AD와 관련된 대부분의 신경 손상을 유발할 수 있다. Aβ1-42의 형성을 예방하는 제제는 AD의 치료용 질환 조절제(disease-modifying agent)일 가능성을 가진다. Aβ1-42는 770개의 아미노산으로 구성되는 아밀로이드 전구체 단백질 (APP)로부터 생성된다. Aβ1-42의 N-말단이 β-세크레타아제 (BACE)에 의해 절단된 다음, γ-세크레타아제가 C-말단을 절단한다. γ-세크레타아제는 또한 Aβ1-42 외에, 주된 절단 산물인 Aβ1-40 뿐 아니라 Aβ1-38 및 Aβ1-43을 유리시킨다. 이들 Aβ는 또한 응집하여 올리고머 및 피브릴을 형성할 수도 있다. 따라서, BACE의 저해제는 Aβ1-42 뿐 아니라 Aβ1-40, Aβ1-38 및 Aβ1-43의 형성을 예방하는 것으로 예상되며, AD의 치료에서의 유력한 치료제일 것이다.
본 발명은 하기 화학식 (I)의 화합물 또는 그의 호변이성체 또는 입체이성체, 및 이들의 부가염 또는 용매화물에 관한 것이다:

상기 식에서,
R1, R2는 독립적으로 수소, 플루오로, 시아노, C1-3알킬, 모노- 및 폴리할로-C1-3알킬, 및 C3-6사이클로알킬로 구성된 군으로부터 선택되거나; 또는
R1 및 R2는 이들이 결합된 탄소원자와 함께, C3-6사이클로알칸디일 환을 형성할 수 있고;
R3는 수소, C1-3알킬, C3-6사이클로알킬, 모노- 및 폴리할로-C1-3알킬, 호모아릴 및 헤테로아릴로 구성된 군으로부터 선택되며;
X1, X2, X3, X4는 독립적으로 C(R4) 또는 N이나, 단 N을 나타내는 것은 두개를 넘지 않고; 각 R4는 수소, 할로, C1-3알킬, 모노- 및 폴리할로-C1-3알킬, 시아노, C1-3알킬옥시, 모노- 및 폴리할로-C1-3알킬옥시로 구성된 군으로부터 선택되며;
L은 결합 또는 -N(R5)CO-이고, 여기서 R5는 수소 또는 C1-3알킬이고;
R6은 수소 또는 트리플루오로메틸이며;
Ar은 호모아릴 또는 헤테로아릴이고;
여기에서, 호모아릴은 페닐, 또는 할로, 시아노, C1-3알킬, C1-3알킬옥시, 모노- 및 폴리할로-C1-3알킬 및 모노- 및 폴리할로-C1-3알킬옥시로 구성된 군으로부터 선택되는 1, 2 또는 3개의 치환체로 치환된 페닐이고;
헤테로아릴은 각각 할로, 시아노, C1-3알킬, C1-3알킬옥시, 모노- 및 폴리할로-C1-3알킬 및 모노- 및 폴리할로-C1-3알킬옥시로 구성된 군으로부터 선택되는 1, 2 또는 3개의 치환체로 임의로 치환된 피리딜, 피리미딜, 피라질, 피리다질, 푸라닐, 티에닐, 피롤릴, 피라졸릴, 이미다졸릴, 트리아졸릴, 티아졸릴, 티아디아졸릴, 옥사졸릴, 및 옥사디아졸릴로 구성된 군으로부터 선택된다.
본 발명의 실례로는 약제학적으로 허용가능한 담체 및 상술된 화합물 중 임의의 것을 포함하는 약제학적 조성물이 있다. 본 발명의 실례로는 상술한 화합물 중 임의의 것과 약제학적으로 허용가능한 담체를 혼합함으로써 제조되는 약제학적 조성물이 있다. 본 발명의 실례로는 상술한 화합물 중 임의의 것과 약제학적으로 허용가능한 담체를 혼합하는 것을 포함하는 약제학적 조성물의 제조 방법이 있다.
본 발명의 예시에는 상술한 화합물 또는 약제학적 조성물 중 임의의 것의 치료적 유효량을 치료를 필요로 하는 대상에 투여하는 것을 포함하는, β-세크레타아제 효소에 의해 매개되는 질환의 치료방법이 있다.
본 발명의 추가의 예시에는 상술한 화합물 또는 약제학적 조성물 중 임의의 것의 치료적 유효량을 β-세크레타아제 효소의 저해를 필요로 하는 대상에 투여하는 것을 포함하는, β-세크레타아제 효소의 저해방법이 있다.
본 발명의 일례는 상술한 화합물 또는 약제학적 조성물 중 임의의 것의 치료적 유효량을 치료를 필요로 하는 대상에 투여하는 것을 포함하는, 알츠하이머병, 경증 인지 장애, 노쇠, 치매, 레비소체치매, 다운증후군, 뇌졸중 관련 치매, 파킨슨병 관련 치매, 베타-아밀로이드 관련 치매로 구성된 군으로부터 선택되는 질환, 바람직하게는 알츠하이머병의 치료방법이다.
본 발명의 또 다른 예는 치료를 필요로 하는 대상에서 (a) 알츠하이머병, (b) 경증 인지 장애, (c) 노쇠, (d) 치매, (e) 레비소체치매, (f) 다운증후군, (g) 뇌졸중 관련 치매, (h) 파킨슨병 관련 치매 및 (i) 베타-아밀로이드 관련 치매의 치료에 사용하기 위한 상술한 화합물 중 임의의 것이다.
본 발명은 상기 정의된 바와 같은 화학식 (I)의 화합물 및 그의 약제학적으로 허용가능한 염 및 용매화물에 관한 것이다. 화학식 (I)의 화합물은 β-세크레타아제 효소 (β-부위 절단 효소, BACE, BACE1, Asp2 또는 메맙신 2로도 공지되어 있음)의 저해제이며, 알츠하이머병, 경증 인지 장애, 노쇠, 치매, 뇌졸중 관련 치매, 레비소체치매, 다운증후군, 파킨슨병 관련 치매, 베타-아밀로이드 관련 치매, 바람직하게는 알츠하이머병, 경증 인지 장애 또는 치매, 더욱 바람직하게는 알츠하이머병의 치료에 유용하다.
본 발명의 일 구체예에 있어서,
R1 및 R2는 독립적으로 수소 및 C1-3알킬로부터 선택되고;
X1, X2, X3, X4는 독립적으로 C(R4)이고, 여기서 각 R4는 수소 및 할로로부터 선택되고;
L은 결합 또는 -N(R5)CO-이고, 여기서 R5는 수소이고;
Ar은 호모아릴 또는 헤테로아릴이고;
호모아릴은 페닐 또는 할로, 시아노, C1-3알킬, 및 C1-3알킬옥시로 구성된 군으로부터 선택되는 1 또는 2개의 치환체로 치환된 페닐이고;
헤테로아릴은 각각 할로, 시아노, C1-3알킬, 및 C1-3알킬옥시로 구성된 군으로부터 선택되는 1 또는 2개의 치환체로 임의로 치환된 피리딜, 피리미딜, 및 피라질 로 구성된 군으로부터 선택되거나; 또는
그의 부가염 또는 용매화물이다.
본 발명의 또다른 구체예에 있어서,
R1 및 R2는 수소이고;
X1, X2, X3, X4는 CH이고;
L은 결합 또는 -N(R5)CO-이고, 여기서 R5는 수소이고;
Ar은 호모아릴 또는 헤테로아릴이고;
호모아릴은 클로로로 치환된 페닐이고;
헤테로아릴은 각각 클로로, 플루오로, 시아노, 메틸, 및 메톡시로 구성된 군으로부터 선택되는 1 또는 2개의 치환체로 임의로 치환된 피리딜 및 피리미딜로 구성된 군으로부터 선택되이거나; 또는
그의 부가염 또는 용매화물이다.
다른 구체예에 있어서, R3으로 치환된 탄소 원자는 R-배열을 가진다.
본 발명의 또다른 구체예에 있어서,
R1 및 R2는 수소이고;
X1은 CH 또는 CF이고, X2, X3, X4는 CH이고;
L은 -N(R5)CO-이고, 여기서 R5는 수소이고;
Ar은 1 또는 2개의 할로 원자로 치환된 피리디닐, 또는 메톡시로 치환된 피라지닐이거나; 또는
그의 부가염 또는 용매화물이다.
정의
"할로"는 플루오로, 클로로 및 브로모를 의미한다;
"C1-3 알킬"은 1, 2 또는 3개의 탄소 원자를 가지는 선형 또는 분지형 포화 알킬 그룹, 예를 들면 메틸, 에틸, 1-프로필 및 2-프로필을 의미한다;
"C1-3알킬옥시"는 C1-3알킬이 상기 정의된 바와 같은 에테르 래디칼을 의미한다;
"모노- 및 폴리할로C1-3알킬"은 1, 2, 3개 또는 경우에 따라 그 이상의 상기 정의된 할로 원자로 치환된 상기 정의된 바와 같은 C1-3알킬을 의미한다;
"모노- 및 폴리할로C1-3알킬옥시"는 모노- 및 폴리할로C1-3알킬이 상기 정의된 바와 같은 에테르 래디칼을 의미한다;
"C3-6사이클로알킬"은 사이클로프로필, 사이클로부틸, 사이클로펜틸 및 사이클로헥실을 의미한다;
"C3-6사이클로알칸디일"은 사이클로프로판디일, 사이클로부탄디일, 사이클로펜탄디일 및 사이클로헥산디일과 같은 2가 래디칼을 의미한다.
본 원에 사용된 용어 "대상"은, 치료, 관찰 또는 실험의 대상이거나 또는 대상이 되는 동물, 바람직하게는 포유동물, 가장 바람직하게는 인간을 의미한다.
본 원에 사용된 용어 "치료적 유효량"은, 치료되는 질환 또는 장애의 증상을 완화시키는 것을 포함하여, 연구원, 수의사, 의사 또는 다른 임상의에 의해 조직계, 동물, 또는 인간에서 생물학 또는 의학적 반응을 이끌어내는 것으로 생각되는 활성 화합물 또는 약제학적 제제의 양을 의미한다.
본 원에 사용된, 용어 "조성물"은 특정 성분들을 특정 양으로 포함하는 생성물뿐만 아니라, 특정 양의 특정 성분들의 조합으로부터 직접 또는 간접적으로 생성되는 임의의 생성물을 포함하고자 한다.
화학식 (I)의 화합물 및 그의 부가염, 수화물 및 용매화물의 일부는 하나 이상의 키랄 중심을 함유할 수 있으며 입체이성체로 존재할 수 있음이 이해될 것이다.
상기 또는 이후에서 사용된 용어 "입체이성체"는 화학식 (I)의 화합물 및 그의 부가염이 가질 수 있는 모든 가능한 입체이성체를 의미한다. 달리 언급되지 않거나 지시가 없으면, 화합물의 화학적 표기는 모든 가능한 입체화학적 이성체의 혼합물을 포함하며, 이들 혼합물은 다른 이성체를 실질적으로 함유하지 않으며, 즉, 다른 이성체를 10% 미만, 바람직하게는 5% 미만, 특히 2% 미만 및 가장 바람직하게는 1% 미만으로 함유하는 기본 분자 구조의 모든 부분입체이성체 및 거울상이성체뿐 아니라 화학식 (I)에 따른 각 개별 이성체 및 그의 염, 용매화물을 함유한다.
본 발명에 따른 화합물이 적어도 하나의 키랄 중심을 가지는 경우, 이들은 거울상이성체로 존재할 수 있다. 화합물이 2 이상의 키랄 중심을 갖는 경우, 이들은 추가로 부분입체이성체로 존재할 수 있다. 모든 이러한 이성체 및 그들의 혼합물이 본 발명의 범주 내에 포함되는 것으로 이해되어야만 한다. 바람직하게는, 화합물이 거울상이성체로 존재하는 경우에, 거울상이성체는 약 80% 이상의 거울상이성체적 과량, 더욱 바람직하게는 약 90% 이상의 거울상이성체적 과량, 보다 바람직하게는 약 95% 이상의 거울상이성체적 과량, 더더욱 바람직하게는 약 98% 이상의 거울상이성체적 과량, 가장 바람직하게는 약 99% 이상의 거울상이성체적 과량으로 존재한다. 유사하게, 화합물이 부분입체이성체로 존재하는 경우에, 부분입체이성체는 약 80% 이상의 부분입체이성체적 과량, 더욱 바람직하게는 약 90% 이상의 부분입체이성체적 과량, 보다 바람직하게는 약 95% 이상의 부분입체이성체적 과량, 더더욱 바람직하게는 약 98% 이상의 부분입체이성체적 과량, 가장 바람직하게는 약 99% 이상의 부분입체이성체적 과량으로 존재한다.
또한, 본 발명의 화합물에 대한 결정질 형태 중 일부는 다형체로 존재할 수 있으며, 그러한 형태는 본 발명에 포함되는 것으로 의도된다. 게다가, 본 발명의 화합물 중 일부는 물과의 용매화물 (즉, 수화물), 또는 일반적인 유기 용매와 용매화물을 형성할 수 있으며, 이러한 용매화물은 또한 본 발명의 범주 내에 포함되는 것으로 의도된다.
의약으로 사용하기 위하여, 본 발명의 화합물의 염은 비독성 "약제학적으로 허용가능한 염"을 말한다. 그러나, 다른 염이 본 발명에 따른 화합물 또는 그들의 약제학적으로 허용가능한 염의 제조에 유용할 수 있다. 화합물의 적합한 약제학적으로 허용가능한 염에는 예를 들어, 화합물의 용액을 염산, 황산, 푸마르산, 말레산, 석신산, 아세트산, 벤조산, 시트르산, 타르타르산, 탄산 또는 인산과 같은 약제학적으로 허용가능한 산의 용액과 혼합함으로써 형성될 수 있는 산 부가염이 포함된다. 또한, 본 발명의 화합물이 산성 부분(moiety)을 지니는 경우, 그들의 적합한 약제학적으로 허용가능한 염에는 알칼리 금속 염, 예컨대 나트륨 또는 칼륨 염; 알칼리 토금속 염, 예컨대 칼슘 또는 마그네슘 염; 및 적합한 유기 리간드와 함께 형성된 염, 이를 테면 4차 암모늄 염이 포함될 수 있다.
약제학적으로 허용가능한 염의 제조에 사용될 수 있는 대표적인 산에는 다음이 포함되나, 이에 한정되는 것은 아니다: 아세트산, 2,2-디클로로아세트산, 아실화된 아미노산, 아디프산, 알긴산, 아스코르브산, L-아스파르트산, 벤젠설폰산, 벤조산, 4-아세트아미도벤조산, (+)-캄포르산, 캄포르설폰산, 카프르산, 카프로산, 카프릴산, 신남산, 시트르산, 사이클람산, 에탄-1,2-디설폰산, 에탄설폰산, 2-하이드록시에탄설폰산, 포름산, 푸마르산, 갈락타르산, 겐티신산, 글루코헵톤산, D-글루콘산, D-글루코론산, L-글루탐산, 베타-옥소-글루타르산, 글리콜산, 히푸루산, 브롬화수소산, 염산, (+)-L-락트산, (±)-DL-락트산, 락토비온산, 말레산, (-)-L-말산, 말론산, (±)-DL-만델산, 메탄설폰산, 나프탈렌-2-설폰산, 나프탈렌-1,5-디설폰산, 1-하이드록시-2-나프토산, 니코틴산, 질산, 올레산, 오로트산, 옥살산, 팔미트산, 파모인산, 인산, L-파이로글루탐산, 살리실산, 4-아미노살리실산, 세바신산, 스테아르산, 석신산, 황산, 타닌산, (+)-L-타르타르산, 티오시안산, p-톨루엔설폰산, 트리플루오로메틸설폰산, 및 운데실렌산. 약제학적으로 허용가능한 염의 제조에 사용될 수 있는 대표적인 염기에는 다음이 포함되나, 이에 한정되는 것은 아니다: 암모니아, L-아르기닌, 베네타민, 벤자틴, 수산화칼슘, 콜린, 디메틸에탄올아민, 디에탄올아민, 디에틸아민, 2-(디에틸아미노)-에탄올, 에탄올아민, 에틸렌디아민, N-메틸글루카민, 하이드라바민, 1H-이미다졸, L-라이신, 수산화마그네슘, 4-(2-하이드록시에틸)모르폴린, 피페라진, 수산화칼륨, 1-(2-하이드록시에틸)-피롤리딘, 2차 아민, 수산화나트륨, 트리에탄올아민, 트로메타민 및 수산화아연.
본 발명의 화합물의 화학명은 CAS(Chemical Abstracts Service) 지침에 따른 명명 규칙에 따라 생성된 것이다.
화학식 (I)의 화합물은 또한 그의 호변이성체로 존재할 수 있다. 이러한 형태는 상기 구조식에 명확히 표시되어 있지 않더라도 본 발명의 범위내에 포함되도록 의도된다.
A. 최종 화합물의 제조
실험 방법 1
화학식 (I)에 따른 최종 화합물은 화학식 (II)의 중간체 화합물을 적절한 암모니아 공급원, 예를 들어, 염화암모늄 또는 수성 암모니아 등과, 반응식 (1)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 적합한 반응-불활성 용매, 예를 들어, 물 또는 메탄올 등중에서, 가열 조건하에, 예를 들면 반응 혼합물을 60 ℃에서, 예컨대 6 시간동안 가열함으로써 수행된다. 반응식 (1)에서, 모든 변수는 화학식 (I)에 정의된 바와 같다.
반응식 1

실험 방법 2
L이 -N(R5)CO-인 화학식 (I-a)에 따른 최종 화합물은 화학식 (III-a)의 중간체 화합물을 화학식 (IV)의 화합물과 반응식 (2)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 적합한 반응-불활성 용매, 예를 들어, N,N-디메틸포름아미드 등중에 적합한 염기, 예를 들어, K3PO4 등, 구리 촉매, 예를 들어, CuI 및 디아민, 예를 들어 (1R,2R)-(-)-l,2-디아미노사이클로헥산 등의 존재하에 가열 조건하에서, 예를 들어, 반응 혼합물을 180 ℃에서, 예컨대 135 분동안 마이크로파 조사하에 가열하여 수행된다. 반응식 (2)에서, 모든 변수는 화학식 (I)에 정의된 바와 같고, W는 할로이다.
반응식 2

실험 방법 3
또한, 화학식 (I-a)에 따른 최종 화합물은 화학식 (III-b)의 중간체 화합물을 화학식 (V)의 화합물과 반응식 (3)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 적합한 반응-불활성 용매, 예를 들어, 디클로로메탄 등중에, 적합한 염기, 예를 들어, 트리에틸아민 등의 존재하, 축합제, 예를 들어 0-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트 [HATU, CAS 148893-10-1]등의 존재하에, 가열 조건하에서, 예를 들어, 반응 혼합물을 25 ℃에서, 예컨대 2 시간동안 가열하여 수행된다. 반응식 (3)에서, 모든 변수는 화학식 (I)에 정의된 바와 같다.
반응식 3
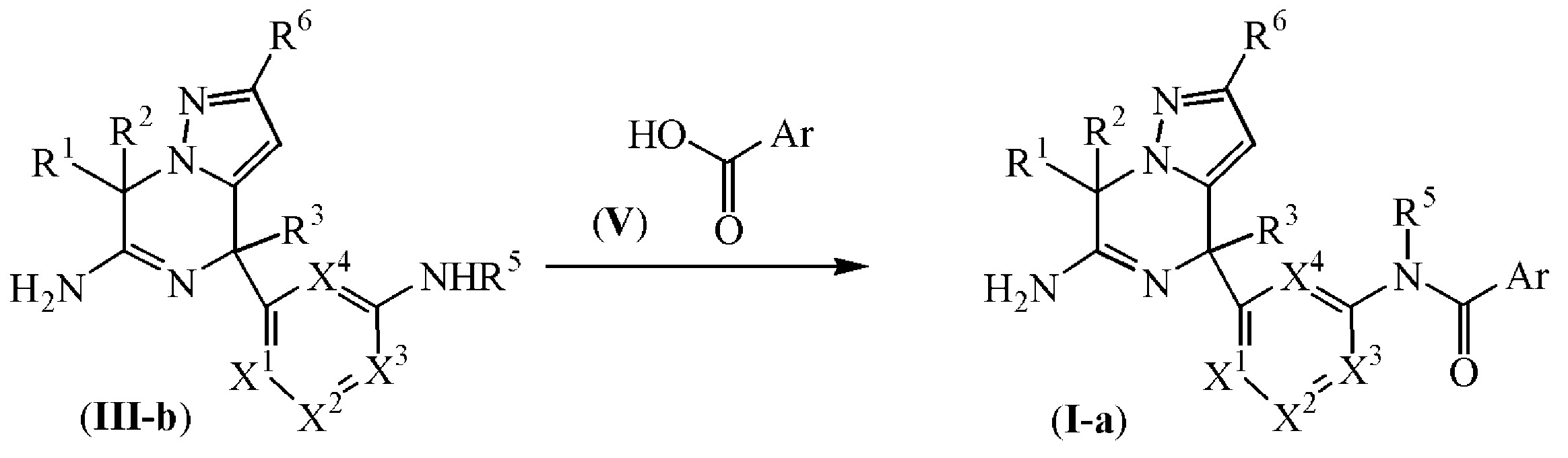
실험 방법 4
또한, 화학식 (I-a)에 따른 최종 화합물은 화학식 (III-b)의 중간체 화합물을 화학식 (VI)의 화합물과 반응식 (4)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 적합한 반응-불활성 용매, 예를 들어, 디클로로메탄 등중, 적합한 염기, 예를 들어, 피리딘 등의 존재하에, 가열 조건하에서, 예를 들어, 반응 혼합물을 25 ℃에서, 예컨대 2 시간동안 가열하여 수행된다. 반응식 (4)에서, 모든 변수는 화학식 (I)에 정의된 바와 같고, Y는 할로이다.
반응식 4

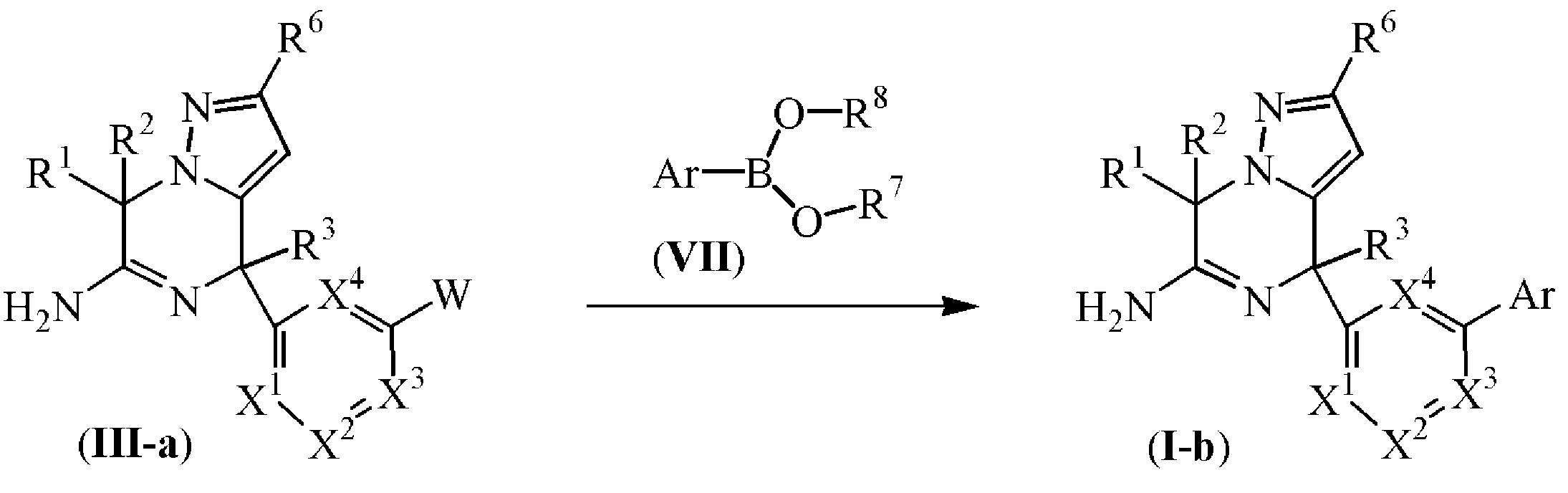
실험 방법 5
L이 결합인 화학식 (I-b)에 따른 최종 화합물은 화학식 (III-a)의 중간체 화합물을 화학식 (VII)의 화합물과 반응식 (5)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 적합한 반응-불활성 용매, 예를 들어, 1,4-디옥산/에탄올과 같은 불활성 용매의 혼합물 등중에, 적합한 염기, 예를 들어, 수성 K3PO4, Pd-착체 촉매, 예를 들어, 테트라키스(트리페닐포스핀)팔라듐(0) [CAS 14221-01-3 등의 존재하에, 가열 조건하에서, 예를 들어, 반응 혼합물을 80 ℃에서, 예컨대 20 시간동안 가열하거나, 또는 예를 들어, 반응 혼합물을 150 ℃에서, 예컨대 10 분 내지 30 분동안 마이크로파 조사하에 가열하여 수행된다. 반응식 (5)에서, 모든 변수는 화학식 (I)에 정의된 바와 같고, W는 할로이다. R7 및 R8은 수소 또는 알킬일 수 있거나, 또는 함께, 예를 들어 식 -CH2CH2-, -CH2CH2CH2-, 또는 -C(CH3)2C(CH3)2-의 2가 래디칼을 형성할 수 있다.
반응식 5
상기 제조에 있어 다수의 중간체 및 출발물질은 업계에 공지된 상기 또는 유사 화합물의 제조방법에 따라 제조될 수 있는 공지 화합물이고, 일부 중간체는 신규하다. 이같은 다수의 제조방법에 대해서는 이후 상세히 설명될 것이다.
B. 중간체 화합물의 제조
실험 방법 6
화학식 (II)에 따른 중간체는 화학식 (VIII)의 중간체 화합물을 티오아미드를 합성하기 위한 적합한 황 공여 시약, 예를 들어, 오황화인 또는 2,4-비스-(4-메톡시페닐)-1,3-디티아-2,4-디포스페탄 2,4-디설파이드 [라벳손 시약, CAS 19172-47-5]와 반응식 (6)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 반응 불활성 용매, 예를 들어, 테트라하이드로푸란 또는 톨루엔 등중에, 적합한 염기, 예를 들어, 피리딘 등의 존재하에, 가열 조건하에서, 예를 들어, 반응 혼합물을 90 ℃에서 예컨대 18 시간동안 가열하여 수행된다. 반응식 (6)에서, 모든 변수는 화학식 (I)에 정의된 바와 같다.
반응식 6
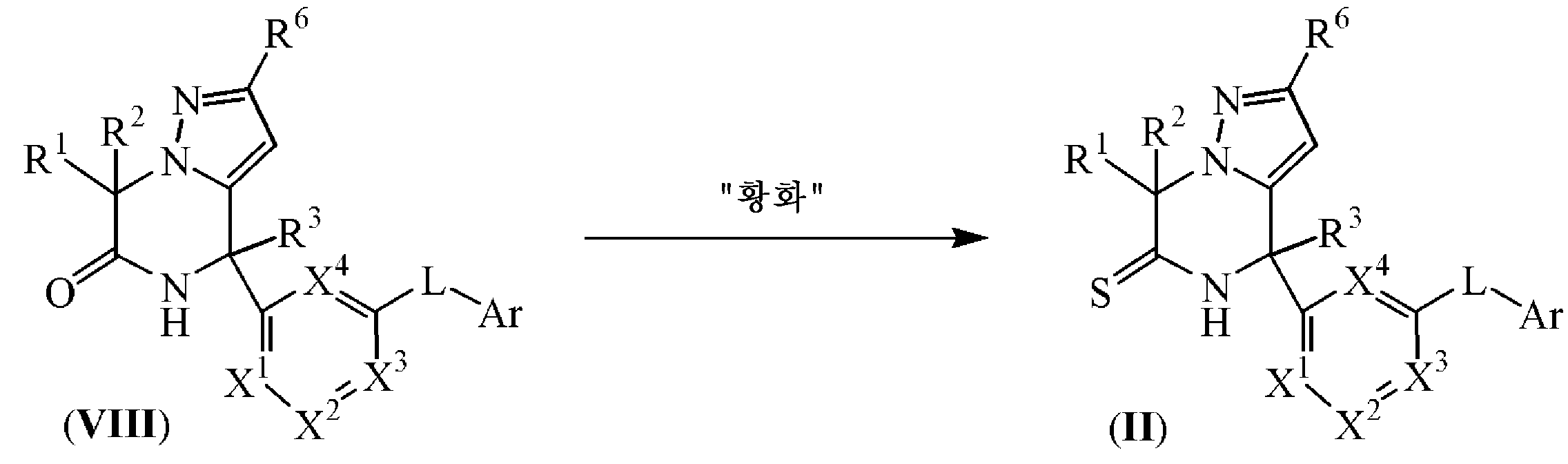
실험 방법 7
L이 결합인 화학식 (VIII)에 따른 중간체는 화학식 (IX-a)의 중간체 화합물을 화학식 (VII)의 화합물과 반응식 (7)에 따라 반응시켜 제조될 수 있으며, 상기 반응은 불활성 용매의 적합한 혼합물, 예를 들어, 1,4-디옥산/물중에, 적합한 염기, 예를 들어, 수성 Na2C03, Pd-착체 촉매, 예를 들어, 테트라키스(트리페닐포스핀)팔라듐(0) [CAS 14221-01-3]의 존재하에 가열 조건하에서, 예를 들어, 반응 혼합물을 80 ℃에서 예컨대 20 시간동안 가열하거나, 또는 예를 들어, 반응 혼합물을 150 ℃에서, 예컨대 15 분동안 마이크로파 조사하에 가열하여 수행된다. 반응식 (7)에서, 모든 변수는 화학식 (I)에 정의된 바와 같고, W는 할로이다. R7 및 R8은 수소 또는 알킬일 수 있거나, 또는 함께, 예를 들어 식 -CH2CH2-, -CH2CH2CH2-, 또는 -C(CH3)2C(CH3)2-의 2가 래디칼을 형성할 수 있다.
반응식 7

실험 방법 8
화학식 (III-b)에 따른 중간체는 상응하는 화학식 (III-a)의 중간체 화합물을 반응식 (8)에 따라 업계에 공지된 부흐발트-하르트빅 타입(Buchwald-Hartwig type) 커플링 반응시켜 제조될 수 있다, 상기 커플링은 적합한 반응-불활성 용매, 예를 들어, 에탄올 또는 1,2-디메톡시에탄/물/에탄올과 같은 불활성 용매의 혼합물 등중에, 적합한 염기, 예를 들어, 수성 K3PO4 또는 CS2CO3 등, Pd-착체 촉매, 예를 들어, [1,1'-비스(디페닐포스피노)페로센]디클로로팔라듐(II) [CAS 72287-26-4] 또는 트랜스-비스(디사이클로헥실아민)팔라듐 디아세테이트 [DAPCy, CAS 628339-96-8] 등의 존재하에, 가열 조건하에서, 예를 들어, 반응 혼합물을 80 ℃에서, 예컨대 20 시간동안 가열하거나, 또는 예를 들어, 반응 혼합물을 130 ℃에서, 예컨대 10 분동안 마이크로파 조사하에 가열하여 수행된다. 반응식 (8)에서, 모든 변수는 화학식 (I)에 정의된 바와 같고, W는 할로이다. R5는 수소 또는 C1-3알킬이다.
반응식 8

실험 방법 9
또한, R5가 수소인 화학식 (III-b)의 중간체는 상응하는 화학식 (III-c)의 중간체로부터 업계에 공지된 니트로를 아미노로 환원하는 방법 (반응 단계 9)에 따라 제조될 수 있다. 상기 환원은 업계에 공지된 촉매적 수소화 방법에 따라 편리하게 수행될 수 있다. 예를 들면, 상기 환원은 반응물을 수소 분위기하 및 적절한 촉매, 예를 들어, 활성탄상 팔라듐, 활성탄상 백금, 라니-니켈 등의 촉매의 존재하에 교반함으로써 수행될 수 있다. 적합한 용매는, 예를 들어, 물, 알칸올, 예를 들면 메탄올, 에탄올 등, 에스테르, 예를 들면 에틸 아세테이트 등이다. 상기 환원 반응의 속도를 향상시키기 위하여, 반응 혼합물의 온도 및/또는 압력을 올리는 것이 유리할 수 있다. 반응물 및 반응 생성물에서 특정 작용기의 원치 않는 추가 수소화는 반응 혼합물에 촉매독, 예를 들어, 티오펜 등을 첨가함으로써 방지할 수 있다. 반응식 (9)에서, 모든 변수는 화학식 (I)에 정의된 바와 같다.
반응식 9

실험 방법 10
화학식 (III-a) 및 (III-c)의 중간체 화합물은 일반적으로 하기 반응식 (10), (11), (12), 및 (13)에 나타내어진 반응 단계에 따라 제조될 수 있다.
반응식 (10)에서 화학식 (III-a) 및 (III-c)의 아미딘 중간체는 상응하는 화학식 (XI-a) 및 (XI-c)의 티오아미드 중간체로부터 업계에 공지된 티오아미드의 아미딘으로의 전환 방법 (반응 단계 A)에 따라 제조될 수 있다. 상기 전환은 상응하는 화학식 (XI-a) 및 (XI-c)의 중간체 화합물을 적합한 반응-불활성 용매, 예를 들어, 물 또는 메탄올 등중에, 가열 조건하에서 암모니아 공급원, 예를 들어, 염화암모늄 또는 수성 암모니아로 처리하여, 예를 들면, 반응 혼합물을 60 ℃에서, 예컨대 6 시간동안 가열함으로써 편리하게 수행될 수 있다.
반응식 (10)에서 화학식 (XI-a) 및 (XI-c)의 티오아미드 유도체는 화학식 (IX-a) 및 (IX-c)의 아미드 유도체로부터 업계에 공지된 황화 방법 (반응 단계 B)에 따라 제조될 수 있다. 상기 전환은 화학식 (XI-a) 및 (XI-c)의 중간체 화합물을 반응 불활성 용매, 예를 들어, 테트라하이드로푸란 또는 1,4-디옥산 등중에서, 가열 조건하에 황화제, 예를 들어, 오황화인 또는 2,4-비스-(4-메톡시페닐)-1,3-디티아-2,4-디포스페탄 2,4-디설파이드 [라벳손 시약, CAS 19172-47-5]로 처리하여, 예를 들어, 반응 혼합물을 50 ℃에서, 예컨대 50 분동안 가열함으로써 편리하게 수행될 수 있다.
반응식 (10)에서 (IX-a) 및 (IX-b)의 아미드 유도체는 상응하는 화학식 (XII-a) 및 (XII-b)의 중간체 화합물로부터 업계에 공지된 폐환 방법 (반응 단계 C)에 따라 제조될 수 있다. 상기 폐환은 편리하게는 화학식 (XII-a) 및 (XII-b)의 중간체 화합물을 적합한 반응 불활성 용매, 예를 들어, 테트라하이드로푸란 등중에 -80 내지 100 ℃, 바람직하게는 -15 내지 25℃에서 적합한 염기, 예를 들어, 수소화나트륨으로 100 시간, 바람직하게는 1 내지 24 시간동안 처리하여 수행될 수 있다.
반응식 10

A: 티오아미드의 아미딘으로의 전환
B: 아미드의 티오아미드로의 전환 (황화)
C: 폐환
D: N-아실화
상기 반응식 (10)에서 화학식 (XII-a) 및 (XII-c)의 중간체 화합물은 상응하는 화학식 (XIII-a) 및 (XIII-c)의 중간체 화합물로부터 업계에 공지된 N-아실화 방법에 따라 제조될 수 있다 (반응 단계 D). 상기 N-아실화는 편리하게는 화학식 (XIII-a) 및 (XIII-c)의 중간체 화합물을 염기, 예컨대 중탄산나트륨, 또는 염기의 혼합물, 예컨대 중탄산나트륨/N,N-디이소프로필에틸아민의 존재하에 적합한 반응 불활성 용매, 예를 들어 에탄올 또는 불활성 용매의 혼합물, 예를 들어, 에탄올/디클로로메탄중에서 -80 ℃ 내지 100 ℃, 바람직하게는 -15 ℃ 내지 25 ℃에서 30 분 내지 100 시간, 바람직하게는 1 시간 내지 24 시간동안 화학식 (XIV)의 중간체 화합물로 처리하여 수행될 수 있다.
반응식 (11)에서 화학식 (XIII-a) 및 (XIII-c)에 따른 중간체는 상응하는 화학식 (XV-a) 및 (XV-c)의 중간체 화합물로부터 업계에 공지된 N-탈보호 방법에 따라 제조될 수 있으며 (반응 단계 E), 여기에서 Z1은 피라졸 시스템의 적합한 보호기, 예를 들어, 디메틸설파모일 그룹이고, Z2는 아민의 적합한 보호기, 예를 들어, tert-부탄설피닐 그룹이다. 상기 N-탈보호는 편리하게는 상응하는 화학식 (XV-a) 및 (XV-c)의 중간체 화합물을 적합한 불활성 용매, 예를 들어, 1,4-디옥산중에 적당한 고온, 예를 들어, 25 ℃에서, 예를 들어 1 시간동안 적합한 산성 시약, 예를 들어, 염산으로 처리하여 수행될 수 있다
반응식 (11)에서 화학식 (XV-a) 및 (XV-c)에 따른 중간체는 화학식 (XVII-a) 및 (XVII-c)의 중간체 화합물을 업계에 공지된 이민의 알킬아민으로의 전환 방법에 따라 반응시킴으로써 제조될 수 있다 (반응 단계 F). 상기 전환은 편리하게는 상응하는 화학식 (XVII-a) 및 (XVII-c)의 중간체 화합물을 적합한 반응-불활성 용매, 예를 들어, 테트라하이드로푸란중에 저온, 예를 들어, 0 ℃에서, 예를 들어 2 시간동안 Y가 할로인 화학식 (XVI)의 중간체 화합물로 처리함으로써 수행될 수 있다.
반응식 11
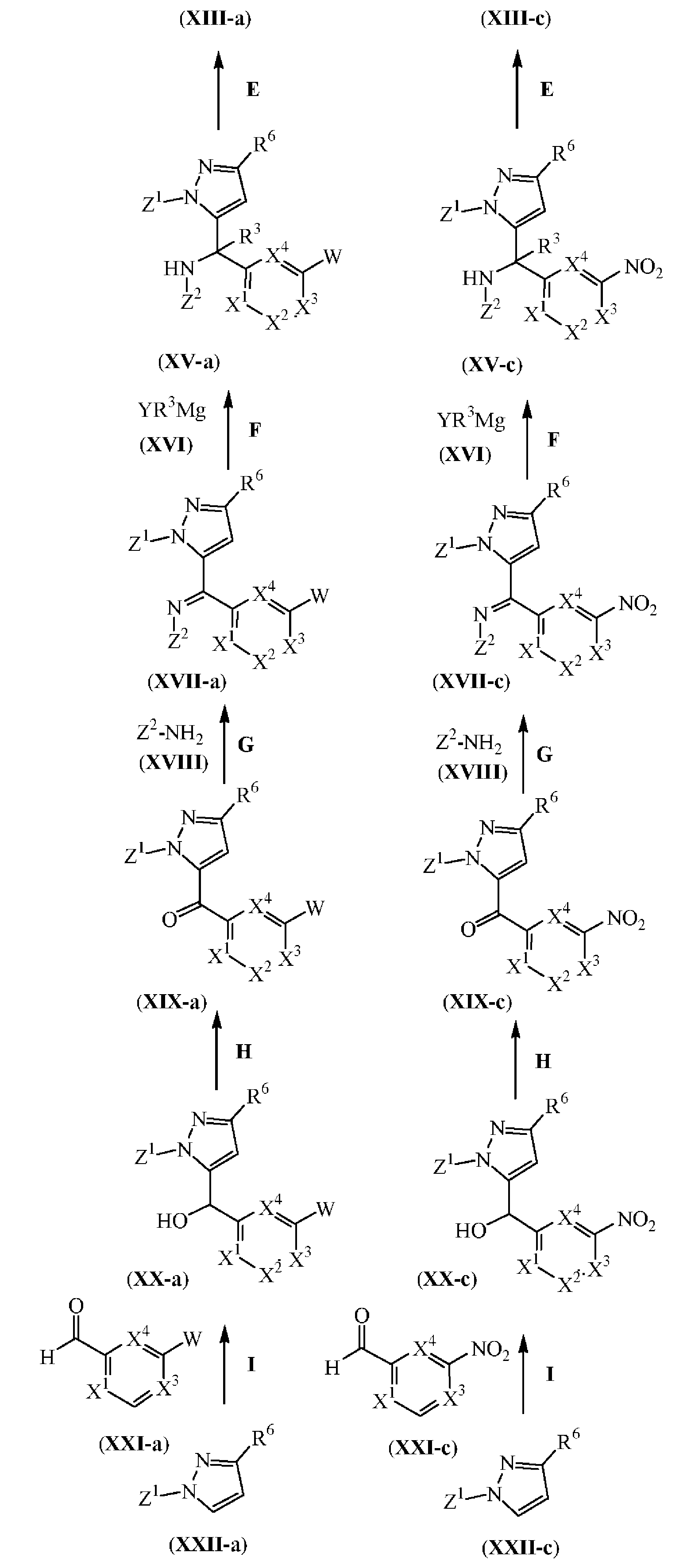
E: N-탈보호
F: 이민의 알킬아민으로의 전환
G: 케톤의 이민으로의 전환
H: 알콜의 카보닐로의 산화
I: 오르토-리튬화-알킬화
상기 반응식 (11)에서 화학식 (XVII-a) 및 (XVII-c)에 따른 중간체는 화학식 (XIX-a) 및 (XIX-c)의 중간체 화합물을 업계에 공지된 케톤으로부터 이민으로 전환시키는 방법에 따라 반응시켜 제조될 수 있다 (반응 단계 G). 상기 전환은 편리하게는 상응하는 화학식 (XIX-a) 및 (XIX-c)의 중간체 화합물을 적합한 루이스산 촉매, 예컨대 티타늄(IV)이소프로폭사이드의 존재하에 적합한 반응-불활성 용매, 예를 들어, 톨루엔중에서, 가열 조건하에 Z2가 알킬설피닐 그룹 예를 들어, tert-부탄설피닐 그룹인 화학식 (XVIII)의 중간체 화합물로 처리하여, 예를 들어, 반응 혼합물을 110 ℃에서, 예를 들어 24 시간동안 가열함으로써 수행될 수 있다
상기 반응식 (11)에서 화학식 (XIX-a) 및 (XIX-c)에 따른 중간체는 화학식 (XX-a) 및 (XX-c)의 중간체 화합물을 업계에 공지된 알콜을 카보닐로 산화시키는 방법에 따라 반응시켜 제조될 수 있다 (반응 단계 H). 상기 산화는 편리하게는 상응하는 화학식 (XX-a) 및 (XX-c)의 중간체 화합물을 적합한 반응-불활성 용매, 예를 들어, 디클로로 메탄중에 산화제, 예를 들어, 데스-마틴 퍼이오디난(Dess-Martin periodinane) [CAS: 87413-09-0]으로 저온 예를 들어, 0 ℃에서, 예를 들어 10 분동안 처리한 뒤, 적당한 고온, 예를 들어, 25 ℃에서, 예를 들어 1 시간동안 처리하여 수행될 수 있다.
상기 반응식 (11)에서 화학식 (XX-a) 및 (XX-c)에 따른 중간체는 화학식 (XXII-a) 및 (XXII-c)의 중간체 화합물을 업계에 공지된 오르토-리튬화-알킬화 방법에 따라 반응시켜 제조될 수 있다 (반응 단계 I). 상기 전환은 편리하게는 상응하는 화학식 (XXII-a) 및 (XXII-c)의 중간체 화합물을 적합한 반응-불활성 용매, 예를 들어, 테트라하이드로푸란중에 저온 예를 들어, -78 ℃에서, 적합한 유기리튬 시약, 예를 들어, n-부틸 리튬으로 예를 들어 45 분동안 처리한 후, 저온 예를 들어, -78 ℃에서 화학식 (XXI-a) 및 (XXI-c)의 중간체 화합물로 예를 들어 45 분동안 처리하여 수행될 수 있다.
Z1이 피라졸 시스템의 적합한 보호기, 예를 들어, 디메틸설파모일 그룹인 화학식 (XXII-a) 및 (XXII-c)의 중간체 화합물은 일반적으로 문헌에 기술되어 있는 업계에 공지된 N-보호 타입 방법에 따라 제조될 수 있다.
반응식 12

J: 웨인렙(Weinreb) 아미드의 케톤으로의 전환
K: 웨인렙 아미드 형성
또한, 상기 반응식 (12)에서 R6이 수소인 화학식 (XIX-a) 및 (XIX-c)에 따른 중간체는 화학식 (XXIV-a) 및 (XXIV-c)의 중간체 화합물을 업계에 공지된 웨인렙 아미드의 케톤으로의 전환 방법에 따라 반응시켜 제조될 수 있다 (반응 단계 J). 상기 전환은 편리하게는 상응하는 화학식 (XXIV-a) 및 (XXIV-c)의 중간체 화합물을 Y가 할로인 화학식 (XXIII-a) 및 (XXIII-c)의 중간체 화합물로 적합한 반응-불활성 용매, 예를 들어, 테트라하이드로푸란중에 저온, 예를 들어, -78 ℃에서, 예를 들어 1 시간동안 처리한 후, 적당한 고온 예를 들어, 25 ℃에서, 예를 들어 5 시간동안 처리하여 수행될 수 있다.
상기 반응식 (12)에서 화학식 (XXIV-a) 및 (XXIV-c)에 따른 중간체는 화학식 (XXV-a) 및 (XXV-c)의 중간체 화합물을 업계에 공지된 웨인렙 아미드 형성 방법에 따라 반응시켜 제조될 수 있다 (반응 단계 K). 상기 전환은 편리하게는 상응하는 화학식 (XXV-a) 및 (XXV-c)의 중간체 화합물을 N,O-디메틸하이드록실아민으로 적합한 염기, 예를 들어, 이소프로필염화마그네슘의 존재하에 적합한 반응-불활성 용매, 예를 들어, 디클로로메탄중에서 저온 예를 들어, -78 ℃에서, 예를 들어 1 시간동안 처리한 후, 적당한 고온 예를 들어, 25 ℃에서, 예를 들어 24 시간동안 처리하여 수행될 수 있다.
Z1이 피라졸 시스템의 적합한 보호기, 예를 들어, 디메틸설파모일 그룹인 화학식 (XXV-a) 및 (XXV-c)의 중간체 화합물은 상업적으로 입수가능하다.
또한, 반응식 (13)에서 R6이 수소인 화학식 (XIII-a) 및 (XIII-c)에 따른 중간체는 Z3이 아민의 보호기, 예를 들어, tert-부톡시카보닐 그룹인 상응하는 화학식 (XXVI-a) 및 (XXVI-c)의 중간체 화합물 (반응 단계 E)로부터 업계에 공지된 N-탈보호 방법, 예컨대 반응식 (11)에 기술된 방법 (반응 단계 E)에 따라 제조될 수 있다.
반응식 (13)에서 화학식 (XXVI-a) 및 (XXVI-c)에 따른 중간체는 화학식 (XXVII-a) 및 (XXVII-c)의 중간체 화합물을 업계에 공지된 피라졸 환 형성 방법에 따라 반응시켜 제조될 수 있다 (반응 단계 L). 상기 피라졸 환 형성은 편리하게는 상응하는 화학식 (XXVII-a) 및 (XXVII-c)의 중간체 화합물을 적합한 불활성 용매 예를 들어, 에탄올중에 히드라진의 존재하에서 적당한 고온 예를 들어, 25 ℃에서, 예를 들어 1 시간동안 처리하여 수행될 수 있다.
반응식 (13)에서 화학식 (XXVII-a) 및 (XXVII-c)에 따른 중간체는 화학식 (XXVIII-a) 및 (XXVIII-c)의 중간체 화합물을 업계에 공지된 알콜의 카보닐로의 산화 방법, 예컨대 반응식 (11)에 기술된 방법 (반응 단계 H)에 따라 반응시켜 제조될 수 있다.
반응식 13
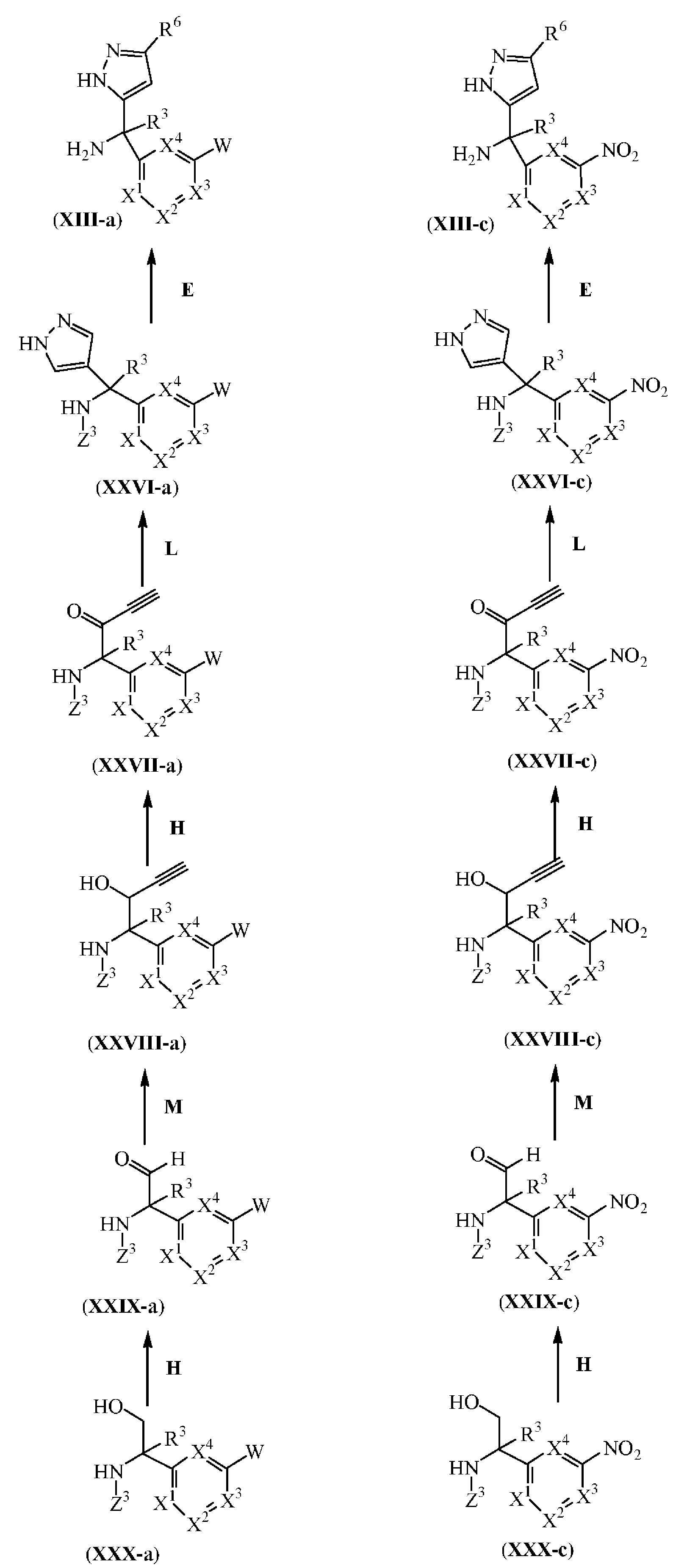
E: N-탈보호
L: 피라졸 환 형성
H: 알콜의 카보닐로의 산화
M: 알데히드의 하이드록시알키닐로의 전환
반응식 (13)에서 화학식 (XXVIII-a) 및 (XXVIII-c) 에 따른 중간체는 화학식 (XXIX-a) 및 (XXIX-c)의 중간체 화합물을 업계에 공지된 알데히드를 하이드록시알키닐로 전환시키는 방법에 따라 제조될 수 있다 (반응 단계 M). 상기 전환은 편리하게는 상응하는 화학식 (XXIX-a) 및 (XXIX-c)의 중간체 화합물을 적합한 반응-불활성 용매, 예를 들어, 테트라하이드로푸란중에 저온 예를 들어, 0 ℃에서 적합한 마그네슘 시약, 예를 들어, 에티닐브롬화마그네슘으로 예를 들어 10 분동안 처리한 후, 적당한 고온 예를 들어, 25 ℃에서 예를 들어 30 분동안 처리하여 수행될 수 있다.
반응식 (13)에서 화학식 (XXIX-a) 및 (XXIX-c)에 따른 중간체는 화학식 (XXX-a) 및 (XXX-c)의 중간체 화합물을 업계에 공지된 알콜을 카보닐로 산화시키는 방법, 예컨대 반응식 (11)에 기술된 방법 (반응 단계 H)에 따라 반응시켜 제조될 수 있다.
Z3이 아민의 보호기, 예를 들어, tert-부톡시카보닐 그룹인 화학식 (XXX-a) 및 (XXX-c)의 중간체 화합물은 일반적으로 문헌에 기술되어 있는 업계에 공지된 스트렉커(Strecker) 타입 방법에 따라 제조될 수 있다.
약제학적 조성물
본 발명은 또한 알츠하이머병 (AD), 경증 인지 장애, 노쇠, 치매, 루이소체 치매, 다운증후군, 뇌졸중 관련 치매, 파킨슨병 관련 치매 및 베타-아밀로이드 관련 치매 등과 같은 β-세크레타아제 저해가 유익한 질환의 예방 또는 치료용 조성물을 제공한다. 이 조성물은 치료적 유효량의 화학식 (I)의 화합물 및 약제학적으로 허용가능한 담체 또는 희석제를 포함한다.
활성 성분은 단독으로 투여될 수 있으나, 약제학적 조성물로서 존재하는 것이 바람직하다. 따라서, 본 발명은 또한 약제학적으로 허용가능한 담체 또는 희석제와 함께, 활성 성분으로서, 본 발명에 따른 화합물을 포함하는 약제학적 조성물을 제공한다. 담체 또는 희석제는 조성물의 다른 성분과 상용성을 나타내고 수용자에게 유해하지 않다는 점에서 "허용가능"하여야 한다.
본 발명의 약제학적 조성물은 제약 업계에 잘 알려져 있는 임의의 방법으로 제조될 수 있다. 활성 성분으로서 치료적 유효량의 염기 또는 부가염 형태의 특정 화합물이, 투여에 필요한 제제의 형태에 따라 각종 다양한 형태를 취할 수 있는 약제학적으로 허용가능한 담체와 친밀히 혼합된다. 이들 약제학적 조성물은 바람직하게는 전신 투여, 특히 경구 투여, 경피 투여, 비경구 투여, 또는 국소 투여(예를 들면 흡입, 비강내 스프레이, 점안제 또는 크림, 겔, 샴푸 등을 통해)에 적절한 단위 제형인 것이 바람직하다. 예를 들면, 조성물을 경구 제형으로 제조함에 있어서, 예를 들면, 현탁제, 시럽, 엘릭시르 및 용액 등의 경구 액체 제제의 경우에는 물, 글리콜, 오일, 알콜 등; 또는 분제, 환약, 캡슐 및 정제의 경우에는 전분, 당, 카올린, 희석제, 윤활제, 결합제, 붕해제 등의 고체 담체와 같은 통상의 약제학적 매질 중 임의의 것을 사용할 수 있다. 투여의 용이성으로 인해, 정제 및 캡슐이 가장 유리한 경구 복용 단위형을 나타내는데, 이 경우에는 고체 약제학적 담체가 명백히 사용된다. 비경구 조성물의 경우에, 담체는 예를 들면 용해성을 돕기 위해 다른 성분, 보통은 적어도 대부분을 멸균수로 포함할 것이다. 예를 들면, 담체가 식염수, 글루코스 용액, 또는 식염수와 글루코스 용액의 혼합물을 포함하도록 주사 용액이 제조될 수 있다. 주사용 현탁제도 제조될 수 있으며, 이 경우에는 적당한 액체 담체, 현탁화제 등이 사용될 수 있다. 경피 투여에 적합한 조성물에 있어서, 담체는 임의로 소정 특성을 지니는 적절한 첨가제와 소량의 비율로 혼합된 침투촉진제 및/또는 적당한 습윤제를 포함하고, 이때 첨가제는 피부에 심각한 유해 효과를 끼치지 않는 것이다. 상기 첨가제는 피부에의 투여를 용이하게 하고/하거나 원하는 조성물을 제조하는데 도움이 될 수 있다. 이들 조성물은 다양한 방법, 예를 들면 경피 패치, 스팟 온제(spot-on) 또는 연고로서 투여될 수 있다.
투여의 용이성 및 용량의 균일성을 위해 상술한 약제학적 조성물을 단위 복용 형태로 제형화하는 것이 특히 유리하다. 본 원에서 사용되는 단위 복용 형태는 단위 용량으로서 적합한 물리적으로 분리된 단위이며, 각각의 단위는 필요한 약제학적 담체와 함께 원하는 치료 효과를 산출하도록 계산된 소정량의 활성 성분을 함유한다. 이러한 단위 복용 형태의 예로는 정제 (분할정 또는 코팅정을 포함), 캡슐, 환약, 분말 패킷, 웨이퍼, 주사 용액 또는 주사용 현탁제, 티스푼형 (teaspoonful), 테이블스푼형 (tablespoonful) 등, 및 이들의 분리된 다중회분 (segregated multiples)이 있다.
정확한 투여 용량 및 빈도는 당업계에 주지된 바와 같이, 사용되는 화학식 (I)의 특정 화합물, 치료될 특정 증상, 치료될 증상의 중증도, 특정 환자의 연령, 체중, 성별, 질병의 범위 및 전신적인 신체 상태 및 개개인이 취할 수 있는 기타 약제에 따라 달라진다. 또한, 상기 일일 유효량이 치료되는 대상의 반응 및/또는 본 발명의 화합물을 처방하는 의사의 평가에 따라 가감될 수 있음이 명백하다.
투여 방식에 따라, 약제학적 조성물은 0.05 내지 99 중량%, 바람직하게는 0.1 내지 70 중량%, 더욱 바람직하게는 0.1 내지 50 중량%의 활성 성분 및 1 내지 99.95 중량%, 바람직하게는 30 내지 99.9 중량%, 더욱 바람직하게는 50 내지 99.9 중량%의 약제학적으로 허용가능한 담체를 포함할 것이며, 이때 모든 비율은 조성물의 총 중량에 대한 것이다.
본 발명의 화합물은 경구, 경피 또는 비경구 투여와 같은 전신 투여; 또는 흡입, 비강내 스프레이, 점안제 또는 크림, 겔, 샴푸 등을 통한 국소 투여용으로 사용될 수 있다. 화합물은 바람직하게는 경구 투여된다. 정확한 투여 용량 및 빈도는 당업계에 주지된 바와 같이, 사용되는 화학식 (I)의 특정 화합물, 치료될 특정 증상, 치료될 증상의 중증도, 특정 환자의 연령, 체중, 성별, 질병의 범위 및 전신적인 신체 상태 및 개개인이 취할 수 있는 기타 약제에 따라 달라진다. 또한, 상기 일일 유효량이 치료되는 대상의 반응 및/또는 본 발명의 화합물을 처방하는 의사의 평가에 따라 가감될 수 있음이 명백하다.
단일 복용형을 산출하기 위해 담체 물질과 배합될 수 있는 화학식 (I)의 화합물의 양은 치료하고자 하는 질환, 포유동물종 및 특정 투여 방식에 따라 달라질 것이다. 그러나, 일반 지침으로서, 본 발명의 화합물에 적합한 단위 용량은, 예를 들어 바람직하게는 활성 화합물을 0.1 mg 내지 약 1000 mg으로 함유할 수 있다. 바람직한 단위 용량은 1 mg 내지 약 500 mg 범위이다. 더욱 바람직한 단위 용량은 1 mg 내지 약 300 mg이다. 더욱 더 바람직한 단위 용량은 1 mg 내지 약 100 mg이다. 이 단위 용량은 하루에 수 회, 예를 들면 1일 2, 3, 4, 5 또는 6 회, 바람직하게는 1일 1 또는 2회 투여될 수 있으며, 70 kg 성인의 총 투약량은 0.001 내지 약 15 mg/대상 체중 kg/투여이다. 바람직한 투약량은 0.01 내지 약 1.5 mg/대상 체중 kg/투여이고, 이러한 치료법은 수 주 또는 수 개월, 및 일부의 경우는 수 년에 걸칠 수 있다. 그러나, 임의의 특정 환자에 특정적인 용량 수준은, 당업자들에게 명백한 바와 같이, 사용되는 특정 화합물의 활성; 치료 대상의 연령, 체중, 전체적인 건강, 성별 및 식이; 투여 시기 및 경로; 배출률; 이전에 투여된 다른 약물; 및 치료를 받고 있는 특정 질환의 중증도에 따라 변화될 것임이 이해될 것이다.
당업자들에게 명백한 바와 같이, 일부의 경우에는 투약량이 상기 범위를 벗어나는 것이 필요할 수 있다. 또한, 임상의나 치료 담당의는 개별 환자 반응에 답해 치료를 언제 어떻게 개시, 중단, 조정 또는 종료할 것인지를 알 것이다.
다음의 실시예로 본 발명을 구체적으로 설명하고자 하나, 본 발명의 범위가 이로 제한되지 않는다.
실험 부분
이후, 용어 "m.p."는 융점을 의미하고, "aq."는 수성을 의미하고, "r.m."은 반응 혼합물을 의미하고, "r.t."는 실온을 의미하고, 'DIPEA'는 디이소프로필에틸아민을 의미하고, 'DIPE'는 디이소프로필에테르를 의미하고, Et20는 디에틸 에테르를 의미하고, 'THF'는 테트라하이드로푸란을 의미하고, 'DMF'는 디메틸포름아미드를 의미하고, 'DCM'은 디클로로메탄을 의미하고, 'AcOEt'는 에틸아세테이트를 의미하고, 'AcOH'는 아세트산을 의미하고, 'MeOH'는 메탄올을 의미하고, 'EtOH'는 에탄올을 의미하고, 'rac'는 라세믹을 의미하고, 'sat.'는 포화를 의미하고, 'SFC'는 초임계 유체 크로마토그래피를 의미하고, 'SFC-MS'는 초임계 유체 크로마토그래피/질량 분광법을 의미하고, 'LCMS'는 액체 크로마토그래피/질량 분광법을 의미하고, 'HPLC'는 고성능 액체 크로마토그래피를 의미하고, 'DMTMM'은 4-(4,6-디메톡시-1,3,5-트리아진-2-일)-4-메틸모르폴리늄 클로라이드를 의미하고, 'HATU'는 0-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트를 의미한다.
A. 중간체의 제조
실시예 A1
중간체 1
: rac-2-아미노-2-(3-브로모페닐)프로판니트릴의 제조
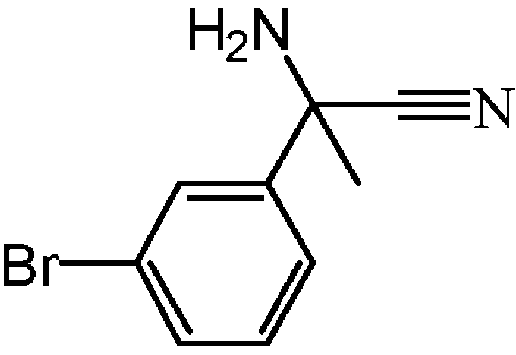
트리메틸실릴시아나이드 (20 g, 200 mmol)를 H3/MeOH (400 mL) 중 3-브로모아세토페논 (20 g, 100 mmol) 및 NH4Cl (11 g, 200 mmol)의 교반 용액에 첨가하였다. 혼합물을 실온에서 4 일동안 교반하였다. 이어, 용매를 진공중에 증발시키고, 잔사를 AcOEt (100 mL)에 취하였다. 고체를 여과하고, 여액을 진공중에 증발시켜 rac-2-아미노-2-(3-브로모페닐)프로피오니트릴 (20 g, 86% 수율)을 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A2
중간체 2
: rac-메틸 2-아미노-2-(3-브로모페닐)프로파노에이트의 제조

rac-2-아미노-2-(3-브로모페닐)프로피오니트릴 (20 g, 88.9 mmol)을 HCl/MeOH (500 mL)에 용해시키고, 혼합물을 4 일동안 환류시켰다. 실온으로 냉각 후, AcOEt (100 mL) 및 물 (100 mL)을 첨가하고, 혼합물을 AcOEt로 추출하였다 (2 x 100 mL). 수성층을 모아 암모니아 수용액으로 pH 8이 될 때까지 염기화하고, AcOEt로 추출하였다 (5 x 100 mL). 유기층을 모아 건조시키고 (Na2S04), 여과한 다음, 용매를 진공중에 증발시켜 rac-2-아미노-2-(3-브로모페닐)프로피온산 메틸 에스테르 (10.6 g, 46% 수율)를 오일로 수득하였다.
실시예 A3
중간체 3
: rac-2-아미노-2-(3-브로모페닐)프로판-1-올의 제조
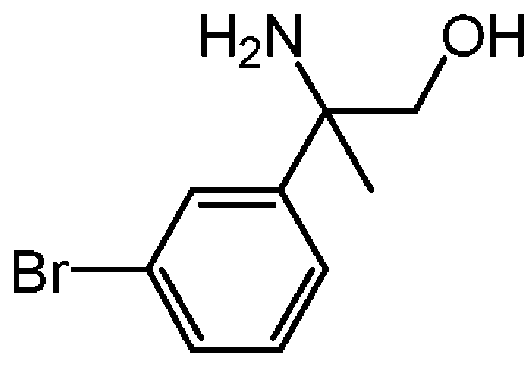
리튬 알루미늄 하이드라이드 (THF중 1 M; 22 mL, 22 mmol)를 -15 ℃에서 THF (200 mL) 중 rac-2-아미노-2-(3-브로모페닐)프로피온산 메틸 에스테르 (7.5 g, 29.1 mmol)의 교반 용액에 적가하였다. 혼합물을 1 시간동안 0 ℃로 서서히 가온하였다. 이어, THF (150 mL)를 추가하고, sat. Na2S04를 수소가 더이상 형성되지 않을 때까지 적가하였다. 이어, 무수 Na2S04를 첨가하고, 실온에서 밤새 교반하였다. 혼합물을 규조토를 통해 여과하고, THF로 헹군 후, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하여 rac-2-아미노-2-(3-브로모페닐)프로판-1-올 (5.70 g, 85% 수율)을 오일로 수득하였다.
실시예 A4
중간체 4
: (R)-2-아미노-2-(3-브로모페닐)프로판-1-올의 제조

rac-2-아미노-2-(3-브로모페닐)프로판-1-올 (15.4 g)의 샘플을 (Chiralpak® Daicel AD x 250 mm) 상에서 분취용 SFC에 의해 상응하는 거울상이성체로 분리하여 (이동상 (C02, MeOH + 0.2% iPrNH2)) (R)-2-아미노-2-(3-브로모페닐)프로판-1-올 (7.21 g, 40% 수율)을 수득하였다.
αD: -14.9 0 (589 nm, c 0.2946 w/v %, MeOH, 20 ℃).
실시예 A5
중간체 5
: rac-tert-부틸 N-[1-(3-브로모페닐)-2-하이드록시-1-메틸에틸]카바메이트의 제조
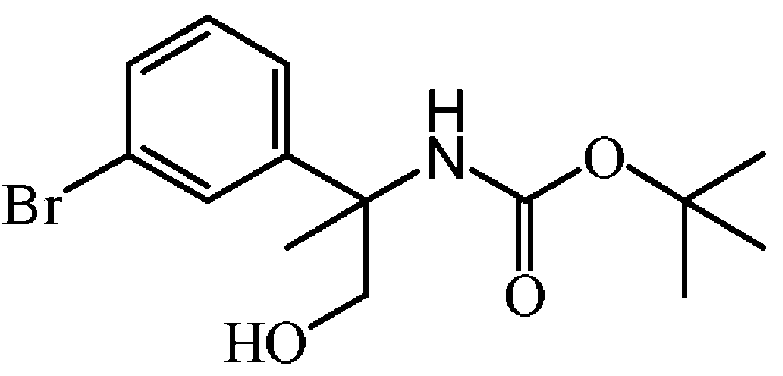
디-tert-부틸디카보네이트 (4.84 g, 22.16 mmol)를 0 ℃에서 sat. NaHC03 (15 mL) 및 THF (15 mL)의 혼합물중 rac-2-아미노-2-(3-브로모페닐)프로판-1-올 (1.7 g, 7.39 mmol)의 교반 용액에 조금씩 첨가하였다. 혼합물을 0 ℃에서 10 분동안 교반한 다음, 실온에서 15 시간동안 교반하였다. 혼합물을 빙수조에서 냉각하고, 교반하면서 KHSO4로 pH 1-2까지 산성화하였다. 유기층을 분리하고, 수성층을 AcOEt로 추출하였다. 유기층을 모아 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카; AcOEt/DCM 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하여 rac-[1-(3-브로모페닐)-2-하이드록시-1-메틸에틸]카밤산 tert-부틸 에스테르 (2.36 g, 93% 수율)를 무색 오일로 수득하였다.
실시예 A6
중간체 6
: rac-tert-부틸 N-[1-(3-브로모페닐)-1-메틸-2-옥소에틸]카바메이트의 제조

데스-마틴 퍼이오디난 (3.55 g, 8.36 mmol)을 0 ℃에서 무수 DCM (45 mL) 중 rac-[1-(3-브로모페닐)-2-하이드록시-1-메틸에틸]-카밤산 tert-부틸 에스테르 (2.3 g, 6.97 mmol)의 용액에 5 분에 걸쳐 조금씩 첨가하였다. 혼합물을 0 ℃에서 10 분동안 교반한 다음, 실온에서 1 시간동안 교반하였다. 반응 혼합물을 NaHC03 (포화 수용액)에 이어 NaHS03 (포화 수용액)로 퀀칭하였다. 이어, Et20를 첨가하고, 혼합물을 실온에서 30 분동안 교반하였다. 유기층을 분리하고, 수성층을 Et20로 추출하였다. 유기층을 모아 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; DCM). 목적 분획을 수집하고, 진공중에 농축하여 rac-[1-(3-브로모페닐)-1-메틸-2-옥소에틸]카밤산 tert-부틸 에스테르 (2 g, 88% 수율)를 무색 오일로 수득하였다.
실시예 A7
중간체 7
: rac-tert-부틸 N-[1-(3-브로모페닐)-2-하이드록시-1-메틸부트-3-이닐]카바메이트의 제조
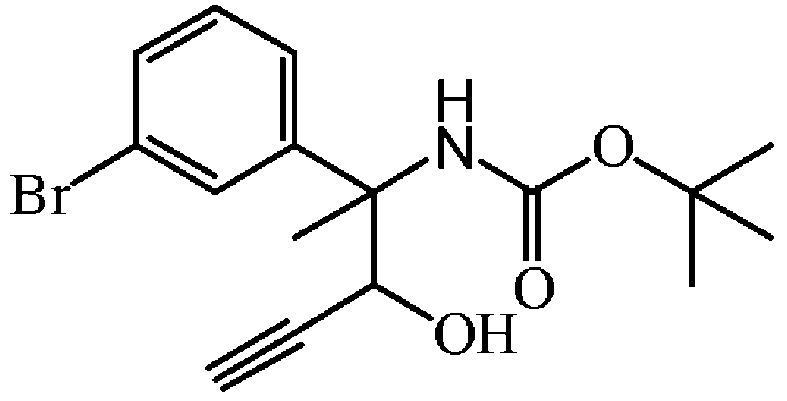
THF 중 에티닐브롬화마그네슘 0.5 M (23.89 mL, 11.94 mmol)을 0 ℃에서 질소하에 THF (60 mL) 중 rac-[1-(3-브로모페닐)-1-메틸-2-옥소에틸]-카밤산 tert-부틸 에스테르 (1.96 g, 5.97 mmol)의 용액에 적가하였다. 혼합물을 0 ℃에서 15 분동안 교반한 다음, 실온에서 30 분동안 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켜 rac-[1-(3-브로모페닐)-2-하이드록시-1-메틸부트-3-이닐]-카밤산 tert-부틸 에스테르 (2.11 g, 99% 수율)를 오일로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A8
중간체 8
: rac-tert-부틸 N-[1-(3-브로모페닐)-1-메틸-2-옥소부트-3-이닐]카바메이트의 제조

데스-마틴 퍼이오디난 (3.04 g, 7.16 mmol)을 0 ℃에서 무수 DCM (20 mL) 중 rac-[1-(3-브로모페닐)-2-하이드록시-1-메틸부트-3-이닐]-카밤산 tert-부틸 에스테르 (2.12 g, 5.97 mmol)의 용액에 5 분에 걸쳐 조금씩 첨가하였다. 혼합물을 0 ℃에서 10 분동안 교반한 다음, 실온에서 1 시간동안 교반하였다. 반응 혼합물을 NaHC03 (포화 수용액)에 이어 NaHS03 (포화 수용액)로 퀀칭하였다. 이어, Et20를 첨가하고, 혼합물을 실온에서 30 분동안 교반하였다. 유기층을 분리하고, 수성층을 Et20로 추출하였다. 유기층을 모아 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; DCM). 목적 분획을 수집하고, 진공중에 농축하여 rac-[1-(3-브로모페닐)-1-메틸-2-옥소부트-3-이닐]-카밤산 tert-부틸 에스테르 (1.89 g, 90% 수율)를 오일로 수득하였다.
실시예 A9
중간체 9
: rac-tert-부틸 N-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]카바메이트의 제조

히드라진 하이드레이트(2.48 mL, 51.10 mmol)를 EtOH (30 mL) 중 rac-[1-(3-브로모페닐)-1-메틸-2-옥소부트-3-이닐]-카밤산 tert-부틸 에스테르 (1.8 g, 5.11 mmol)의 용액에 첨가하고, 혼합물을 실온에서 1 시간동안 교반하였다. 용매를 진공중에 제거하고, 잔사를 DCM에 용해시킨 뒤, 물로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 50/50). 목적 분획을 수집하고, 진공중에 농축하여 rac-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]-카밤산 tert-부틸 에스테르 (1.62 g, 87% 수율)를 백색 고체로 수득하였다.
실시예 A10
중간체 10:
rac-1-(3-브로모페닐)-1-(1H-피라졸-3-일)에탄아민의 제조

디옥산중 염산 4 M (7.88 mL, 31.54 mmol)을 실온에서 rac-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]-카밤산 tert-부틸 에스테르 (1.65 g, 4.51 mmol)에 첨가하였다. 혼합물을 실온에서 1 시간동안 교반하였다. 용매를 진공중에 증발시켰다. 잔사를 DCM에 현탁시키고, NaHC03 (포화 수용액)로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켜 rac-1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸아민 (1.2 g, 100% 수율)을 백색 고체로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A11
중간체 11
: rac-N-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]-2-클로로아세트아미드의 제조

DIPEA (1.18 mL, 6.77 mmol)를 DCM (20 mL) 중 rac-1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸아민 (1.2 g, 4.51 mmol)의 용액에 첨가하고, 혼합물을 빙조에서 냉각하였다. 이어, 클로로아세틸 클로라이드 (0.40 mL, 4.96 mmol)를 첨가하고, 혼합물을 0 ℃에서 3 시간동안 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하였다. 잔사를 EtOH (10 mL) 및 NaHC03 (포화 수용액) (1 mL)에 용해시키고, 혼합물을 실온에서 30 분동안 교반하였다. 혼합물을 물로 희석하고, 생성물을 DCM으로 추출하였다. 유기층을 모아 진공중에 농축하여 rac-N-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]-2-클로로아세트아미드 (1.22 g, 79% 수율)를 무색 오일로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A12
중간체 12:
rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

THF (40 mL) 중 rac-N-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]-2-클로로아세트아미드 (1.22 g, 3.56 mmol)의 용액을 0 ℃에서 질소하에 THF (40 mL) 중 수소화나트륨 (0.28 g, 7.12 mmol)의 현탁액에 적가하였다. 혼합물을 0 ℃에서 1 시간동안 교반하였다. 혼합물을 물로 희석하고, 생성물을 DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 50/50 - 100/0). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.7 g, 64% 수율)을 백색 고체로 수득하였다.
실시예 A13
중간체 13
: rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조

오황화인 (1.02 g, 4.57 mmol)을 피리딘 (10 mL) 중 rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.7 g, 2.29 mmol)의 용액에 첨가하고, 혼합물을 95 ℃에서 18 시간동안 가열하였다. 이어, 용매를 진공중에 증발시키고, 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 100/0). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온 (0.45 g, 61% 수율)을 백색 고체로 수득하였다.
실시예 A14
중간체 14
: rac-4-(3-브로모페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

NH4Cl (0.15 g, 2.79 mmol)을 EtOH (50 mL) 중 rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온 (0.45 g, 1.40 mmol)의 교반 용액에 첨가하고, 혼합물을 80 ℃에서 28 시간동안 가열하였다. 용매를 진공중에 제거하고, 잔사를 DCM에 용해시킨 다음, 물로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/AcOEt 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-(3-브로모페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 (0.42 g, 99% 수율)을 황색 고체로 수득하였다.
실시예 A15
중간체 15
: rac-4-[3-(벤즈히드릴리덴아미노)페닐]-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

톨루엔 (10 mL)을 밀봉 튜브중의 rac-4-(3-브로모페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 (0.39 g, 1.28 mmol), 트리스(디벤질리덴아세톤)디팔라듐(0) (0.12 g, 0.13 mmol), rac-2,2'-비스(디페닐포스피노)-1,1'-비나프틸 (0.24 g, 0.38 mmol) 및 소듐 tert-부톡사이드 (0.22 g, 2.3 mmol)의 혼합물에 질소하에 실온에서 첨가하였다. 혼합물을 질소로 수분간 플러싱하고, 벤조페논 이민 (0.43 mL, 2.56 mmol)을 첨가한 후, 혼합물을 100 ℃에서 2 시간동안 교반하였다. 냉각후, 혼합물을 물로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카; 메탄올중 암모니아 7M 용액/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-[3-(벤즈히드릴리덴아미노)페닐]-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 (0.37 g, 70% 수율)을 황색 포움으로 수득하였다.
실시예 A16
중간체 16:
rac-4-(3-아미노페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조
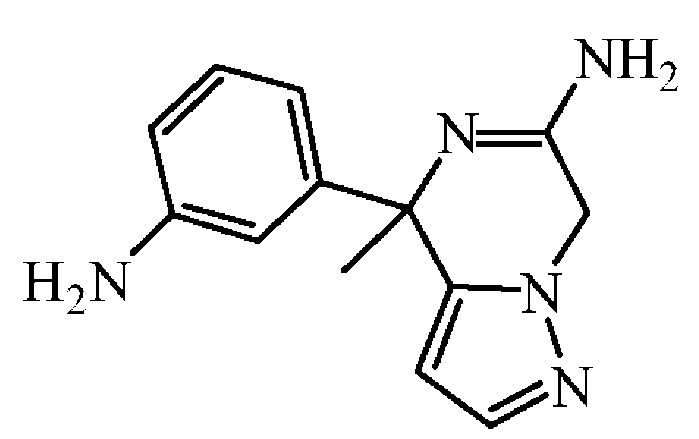
H20 중 염산 37% (0.14 mL)를 이소프로판올 (10 mL) 중 rac-4-[3-(벤즈히드릴리덴아미노)페닐]-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 (0.37 g, 0.9 mmol)의 용액에 첨가하였다. 혼합물을 실온에서 3 시간동안 교반하였다. Et20를 첨가하고, 혼합물을 15 분동안 교반하였다. 침전된 고체를 여과하여 Et20로 세척하고, 진공중에 건조시켰다. 잔사를 DCM에 현탁시키고, NaHC03 (포화 수용액)로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켜 (0.21 g, 97% 수율) 백색 고체를 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A17
중간체 17
: 1H-피라졸-3-카복실산의 제조

수 (150 mL) 중 과망간산칼륨 (16.17 g, 102.31 mmol) 용액을 수 (100 mL) 중 3-메틸피라졸 (4.2 g, 51.15 mmol)의 용액에 첨가하고, 혼합물을 밤새 환류시켰다. 실온으로 냉각 후, 불용 물질을 여과로 제거하였다. 여액을 30 mL가 되도록 농축하고, 2 N HCl을 고체가 침전될 때까지 첨가하였다. 고체를 여과하고, 냉수로 세척한 후, 진공중에 건조시켜 1H-피라졸-3-카복실산 (3.1 g, 54% 수율)을 백색 고체로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A18
중간체 18
: 메틸 1H-피라졸-3-카복실레이트의 제조

황산 (5.8 mL)을 0 ℃에서 MeOH (65 mL) 중 1H-피라졸-3-카복실산 (1 g, 8.92 mmol)의 교반 용액에 적가하였다. 적가를 마친 후, 혼합물을 실온으로 가온하고, 18 시간동안 교반하였다. 혼합물을 진공중에 농축하고, 잔사를 물에 용해시킨 후, NaHC03 (포화 수용액)로 염기화하였다. 혼합물을 AcOEt로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켜 1H-피라졸-3-카복실산 메틸 에스테르 (0.7 g, 62% 수율)를 백색 고체로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A19
중간체 19
: 메틸 1-(디메틸설파모일)-1H-피라졸-3-카복실레이트의 제조

수소화나트륨 (1.57 g, 41.03 mmol)을 0 ℃에서 THF (20 mL) 중 1H-피라졸-3-카복실산 메틸 에스테르 (3.45 g, 27.36 mmol)의 용액에 첨가하였다. 혼합물을 0 ℃에서 30 분동안 교반하였다. 이어, 디메틸 설파모일 클로라이드 (4.41 mL, 41.03 mmol)를 첨가하고, 혼합물을 실온으로 가온한 뒤, 18 시간동안 교반하였다. 혼합물을 물로 희석하고, 생성물을 AcOEt로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 1-디메틸설파모일-1H-피라졸-3-카복실산 메틸 에스테르 (4.8 g, 75% 수율)를 무색 오일로 수득하였다.
실시예 A20
중간체 20
: 1-(디메틸설파모일)-N-메톡시-N-메틸-1H-피라졸-3-카복실레이트의 제조

1-디메틸설파모일-1H-피라졸-3-카복실산 메틸 에스테르 (4 g, 17.15 mmol) 및 N,O-디메틸하이드록실아민 하이드로클로라이드 (2.18 g, 22.29 mmol)를 DCM (20 mL)에서 슬러리화하였다. 혼합물을 질소로 플러싱하고, -78 ℃로 냉각하였다. 이어, 이소프로필염화마그네슘 (THF중 2M) (24.01 mL, 48.02 mmol) 용액을 적가하였다. 적가를 마친 후, 혼합물을 실온으로 가온하고, 밤새 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 퀀칭하고, 생성물을 AcOEt로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 100/0). 목적 분획을 수집하고, 진공중에 농축하여 1-디메틸설파모일-1H-피라졸-3-카복실산 메톡시메틸아미드 (3.2 g, 71% 수율)를 담황색 오일로 수득하였다.
실시예 A21
중간체 21
: 3-(3-클로로페닐)카보닐-N,N-디메틸-1H-피라졸-1-설폰아미드의 제조

3-클로로페닐브롬화마그네슘 (THF중 0.5 M) (15.89 mL, 7.95 mmol) 용액을 THF (20 mL) 중 1-디메틸설파모일-1H-피라졸-3-카복실산 메톡시메틸아미드 (1.60 g, 6.11 mmol) 의 용액에 -78 ℃에서 질소하에 첨가하였다. 혼합물을 -78 ℃에서 1 시간동안 교반한 후, 실온에서 5 시간 더 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 퀀칭하고, 생성물을 AcOEt로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 3-(3-클로로벤조일)-1H-피라졸-1-설폰산 디메틸아미드 (1.68 g, 88% 수율) 담황색 고체로 수득하였다.
실시예 A22
중간체 22
: 3-{[(tert-부틸설피닐)이미노](3-클로로페닐)메틸}-N,N-디메틸-1H-피라졸-1-설폰아미드의 제조
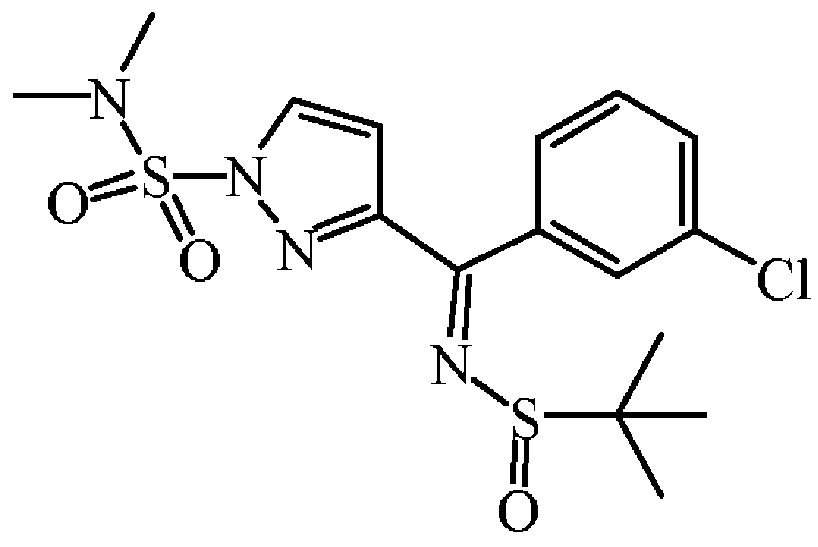
티타늄(IV)이소프로폭사이드(3.22 mL, 10.71 mmol)를 톨루엔 (32 mL) 중 3-(3-클로로벤조일)-l H-피라졸-1-설폰산 디메틸아미드 (1.68 g, 5.35 mmol) 및 2-메틸-2-프로판설핀아미드 (0.71 g, 5.89 mmol)의 혼합물에 질소하에 첨가하였다. 혼합물을 110 ℃에서 24 시간동안 교반하였다. 혼합물을 냉각하고, 빠르게 교반하면서 염수에 부었다. 혼합물을 규조토를 통해 여과하고, 필터 케이크를 AcOEt로 세척하였다. 여액을 분별 깔때기로 옮겨 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 3-[(3-클로로페닐)-(2-메틸프로판-2-설피닐이미노)메틸]피라졸-1-설폰산 디메틸아미드 (2.17 g, 97% 수율)를 황색 오일로 수득하였다.
실시예 A23
중간체 23
: 3-[1-(tert-부틸설피닐아미노)-1-(3-클로로페닐)에틸]-N,N-디메틸-1H-피라졸-1-설폰아미드의 제조

메틸브롬화마그네슘 (15.08 mL, 21.11 mmol)을 0 ℃에서 질소하에 THF (25 mL) 중 3-[(3-클로로페닐)-(2-메틸프로판-2-설피닐이미노)메틸]피라졸-1-설폰산 디메틸아미드 (2.2 g, 5.28 mmol)의 용액에 첨가하였다. 혼합물을 0 ℃에서 2 시간동안 교반하고, NH4Cl (포화 수용액)로 퀀칭한 후, 생성물을 DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 100/0). 목적 분획을 수집하고, 진공중에 농축하여 3-[1-(3-클로로페닐)-1-(2-메틸프로판-2-설피닐아미노)에틸]피라졸-1-설폰산 디메틸아미드 (2.28 g, 99% 수율)를 방치하면 고체로 되는 무색 오일로 얻었다.
실시예 A24
중간체 24
: rac-1-(3-클로로페닐)-1-(1H-피라졸-3-일)에탄아민의 제조

디옥산중 염산 4 M (19.79 mL, 79.15 mmol)을 MeOH (5 mL) 중 3-[1-(3-클로로페닐)-1-(2-메틸프로판-2-설피닐아미노)에틸]피라졸-1-설폰산 디메틸아미드 (2.29 g, 5.28 mmol)의 용액에 첨가하고, 혼합물을 밀봉 튜브중에 80 ℃에서 18 시간동안 교반하였다. 용매를 진공중에 증발시켰다. 잔사를 NaHC03 (포화 수용액)에 부은 후, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgSO4), 여과한 다음, 용매를 진공중에 증발시켜 rac-1-(3-클로로페닐)-1-(1H-피라졸-3-일)에틸아민 (1 g, 86% 수율)을 담황색 고체로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A25
중간체 25
: rac-2-클로로-N-[1-(3-클로로페닐)-1-(1H-피라졸-3-일)에틸]아세트아미드의 제조

중간체 25를 실시예 A11에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 24 (1 g, 4.51 mmol)로부터 출발하여 중간체 25 (0.73 g, 54% 수율)를 백색 고체로 수득하였다.
실시예 A26
중간체 26
: rac-4-(3-클로로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

중간체 26을 실시예 A12에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 25 (0.73 g, 2.43 mmol)로부터 출발하여 중간체 26 (0.45 g, 71% 수율)을 백색 고체로 수득하였다.
실시예 A27
중간체 27
: rac-4-메틸-4-(3-피리미딘-5-일페닐)-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조
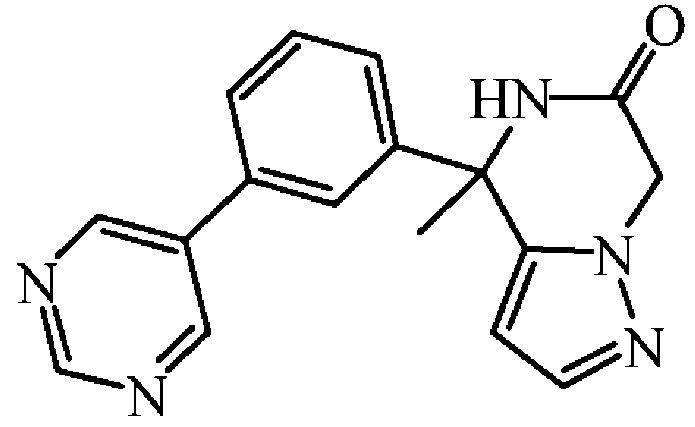
팔라듐(II) 아세테이트 (0.017 g, 0.075 mmol)를 실온에서 질소하에 톨루엔 (5 mL) 및 EtOH (0.5 mL) 중 rac-4-(3-클로로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.13 g, 0.50 mmol), 피리미딘-5-보론산 (0.19 g, 1.49 mmol), 2-디사이클로헥실포스피노-2',6'-디메톡시비페닐 (0.061 g, 0.149 mmol) 및 인산칼륨 (0.21 g, 0.99 mmol)의 교반 현탁액에 첨가하였다. 혼합물을 150 ℃에서 30 분동안 마이크로파 조사하에 교반하였다 이어, 혼합물을 규조토를 통해 여과하고, AcOEt로 세척하였다. 여액을 진공중에 증발시켰다. 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-메틸-4-(3-피리미딘-5-일-페닐)-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.09 g, 59% 수율)을 백색 고체로 수득하였다.
실시예 A28
중간체 28
: rac-4-메틸-4-(3-피리미딘-5-일페닐)-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조

라벳손 시약 (0.14 g, 0.35 mmol)을 실온에서 피리딘 (2 mL) 중 rac-4-메틸-4-(3-피리미딘-5-일-페닐)-4, 5-디하이드로피라졸로[1,5-a]피라진-6-티온 (0.09 g, 0.30 mmol)의 교반 용액에 첨가하였다. 혼합물을 95 ℃에서 18 시간동안 가열하였다. 용매를 진공중에 증발시키고, 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-메틸-4-(3-피리미딘-5-일-페닐)-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온 (0.02 g, 21% 수율)을 백색 고체로 수득하였다.
실시예 A29
중간체 29
: rac-4-[3-(5-메톡시피리딘-3-일)페닐]-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

팔라듐(II) 아세테이트 (0.022 g, 0.097 mmol)를 실온에서 질소하에 톨루엔 (2 mL) 및 EtOH (0.2 mL) 중 rac-4-(3-클로로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.17 g, 0.65 mmol), 5-메톡시피리딘-3-보론산 (0.15 g, 0.97 mmol), 2-디사이클로헥실-포스피노-2',6'-디메톡시비페닐 (0.080 g, 0.195 mmol) 및 인산칼륨 (0.28 g, 1.30 mmol)의 교반 현탁액에 첨가하였다. 혼합물을 150 ℃에서 30 분동안 마이크로파 조사하에 교반하였다. 이어, 혼합물을 규조토를 통해 여과하고, AcOEt로 세척하였다. 여액을 진공중에 증발시켰다. 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-[3-(5-메톡시-피리딘-3-일)페닐]-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.11 g, 51% 수율)을 백색 고체로 수득하였다.
실시예 A30
중간체 30
: rac-4 3-(5-메톡시피리딘-3-일)페닐]-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조

피리딘 (3 mL)을 rac-4-[3-(5-메톡시-피리딘-3-일)페닐]-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (0.11 g, 0.31 mmol) 및 오황화인 (0.07 g, 0.31 mmol)의 혼합물에 첨가하고, 혼합물을 80 ℃에서 5 시간동안 가열하였다. 이어, 오황화인 (0.07 g, 0.31 mmol)을 추가하고, 혼합물을 100 ℃에서 18 시간동안 가열하였다. 이어, 용매를 진공중에 증발시키고, 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; MeOH/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-[3-(5-메톡시-피리딘-3-일)페닐]-4-메틸-4, 5-디하이드로피라졸로[1,5-a]피라진-6-티온 (0.1 g, 93% 수율)을 백색 고체로 수득하였다.
실시예 A31
중간체 31
: rac-4-(5-브로모-2,4-디플루오로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조

오황화인 (2.53 g, 11.40 mmol)을 피리딘 (30 mL) 중의 중간체 rac-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온에 대해 전술한 것과 동일한 방법으로 제조된 rac-4-(5-브로모-2,4-디플루오로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온 (3 g, 8.77 mmol)의 용액에 첨가하고, 혼합물을 95 ℃에서 18 시간동안 가열하였다. 이어, 용매를 진공중에 증발시키고, 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 40/60). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-(5-브로모-2,4-디플루오로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온 (2.4 g, 76% 수율)을 백색 고체로 수득하였다.
실시예 A32
중간체 32
: rac-4-(5-브로모-2,4-디플루오로페닐)-4-메틸 디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

NH4Cl (0.72 g, 13.4 mmol)을 암모니아 2 M/EtOH (67 mL) 중 rac-4-(5-브로모-2,4-디플루오로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온 (2.4 g, 6.7 mmol)의 교반 현탁액에 첨가하고, 혼합물을 85 ℃에서 18 시간동안 가열하였다. 용매를 진공중에 제거하고, 잔사를 DCM에 현탁시킨 뒤, 물로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/AcOEt 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하여 rac-4-(5-브로모-2,4-디플루오로페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-일아민 (1.8 g, 78% 수율)을 황색 고체로 수득하였다.
실시예 A33
중간체 33
: (R)-tert-부틸 N-[1-(3-브로모페닐)-2-하이드록시-1-메틸에틸]카바메이트의 제조

중간체 33을 실시예 A5에 기술된 방법과 동일한 방식으로 합성하였다. (R)-2-아미노-2-(3-브로모페닐)프로판-1-올 (4.7 g, 20.43 mmol)로부터 출발하여 중간체 33 (6.4 g, 95% 수율)을 방치하면 고체로 되는 무색 오일로 수득하였다.
실시예 A34
중간체 34
: (R)-tert-부틸 N-[1-(3-브로모페닐)-1-메틸-2-옥소에틸]카바메이트의 제조

중간체 34를 실시예 A6에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 33 (6.4 g, 19.38 mmol)으로부터 출발하여 중간체 34 (5.7 g, 90% 수율)를 방치하면 고체로 되는 무색 오일로 수득하였다.
실시예 A35
중간체 35
: (1R,2R) 및 (1R,2S)-tert-부틸 N-[1-(3-브로모페닐)-2-하이드록시-1-메틸부트-3-이닐]카바메이트의 부분입체이성체 혼합물의 제조

중간체 35를 실시예 A7에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 34 (5.7 g, 17.38 mmol)로부터 출발하여 중간체 35 (5.4 g, 88% 수율)를 오일의 부분입체이성체 혼합물로서 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A36
중간체 36
: (R)-tert-부틸 N-[1-(3-브로모페닐)-1-메틸-2-옥소부트-3-이닐]카바메이트의 제조

중간체 36을 실시예 A8에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 35 (5.4 g, 15.24 mmol)로부터 출발하여 중간체 36 (5.3 g, 99% 수율)을 담황색 오일로 수득하였다.
실시예 A37
중간체 37
: (R)-tert-부틸 N-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]카바메이트의 제조
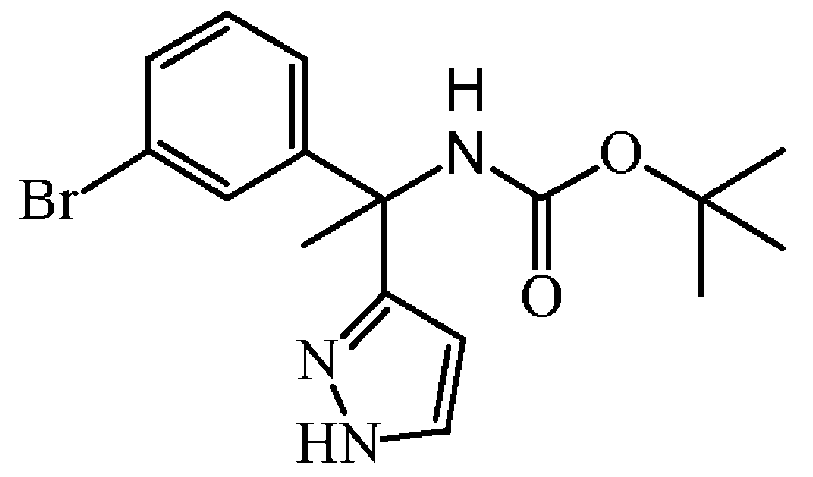
중간체 37을 실시예 A9에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 36 (5.3 g, 15.05 mmol)으로부터 출발하여 중간체 37 (5 g, 91% 수율)을 포움으로 수득하였다.
실시예 A38
중간체 38
: (R)-1-(3-브로모페닐)-1-(1H-피라졸-3-일)에탄아민의 제조

중간체 38을 실시예 A10에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 37 (5 g, 13.65 mmol)로부터 출발하여 중간체 38 (3.5 g, 96% 수율)을 백색 고체로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A39
중간체 39:
(R)-N-[1-(3-브로모페닐)-1-(1H-피라졸-3-일)에틸]-2-클로로아세트아미드의 제조

중간체 39를 실시예 A11에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 38 (3.5 g, 13.15 mmol)로부터 출발하여 중간체 39 (3.5 g, 78% 수율)를 무색 오일로 수득하였다.
실시예 A40
중간체 40
: (R)-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조
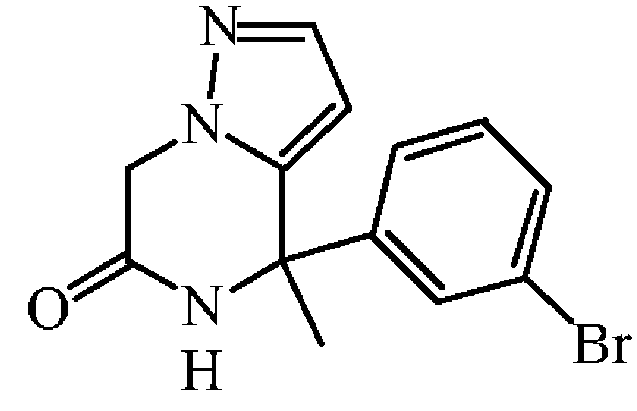
중간체 40을 실시예 A12에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 39 (3.5 g, 10.22 mmol)로부터 출발하여 중간체 40 (2.15 g, 69% 수율)을 백색 고체로 수득하였다.
실시예 A41
중간체 41
: (R)-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조

중간체 41을 실시예 A13에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 40 (2.1 g, 6.86 mmol)으로부터 출발하여 중간체 41 (1.8 g, 81% 수율)을 포움으로 수득하였다.
실시예 A42
중간체 42
: (R)-4-(3-브로모페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

32% 암모니아 수용액 (11.9 mL, 201.1 mmol)을 밀봉 튜브중에 암모니아 7 N 용액/메탄올 (11.97 mL, 83.79 mmol) 중 (R)-4-(3-브로모페닐)-4-메틸-4,5-디하이드로피라졸로[1,5-a]피라진-6-티온 (1.8 g, 5.59 mmol)의 교반 혼합물에 첨가하고, 혼합물을 60 ℃에서 90 분동안 교반하였다. 실온으로 냉각 후, 혼합물을 물로 희석하고, Na2CO3 (포화 수용액) 및 DCM으로 추출하였다. 유기층을 분리하고, 건조시키고 (Na2S04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 2/98 - 3/97 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 (R)-4-(3-브로모페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 (1.4 g, 82% 수율)을 황색 고체로 수득하였다.
실시예 A43
중간체 43
: N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조

1,4-디아자비사이클로[2.2.2]옥탄 (5.44 g, 48.5 mmol) 및 디메틸설파모일 클로라이드 (4.76 mL, 44.46 mmol)를 0 ℃에서 아세토니트릴 (50 mL) 중 3-(트리플루오로메틸)피라졸 (5.5 g, 40.42 mmol)의 용액에 첨가하였다. 혼합물을 실온으로 가온하고, 18 시간동안 교반하였다. 혼합물을 진공중에 농축하고, 잔사를 물로 희석하였다. 생성물을 AcOEt로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; DCM). 목적 분획을 수집하고, 진공중에 농축하여 중간체 43 (9.4 g, 95% 수율)을 무색 오일로 수득하였다.
실시예 A44
중간체 44:
5-[(3-브로모페닐)(하이드록실)메틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조
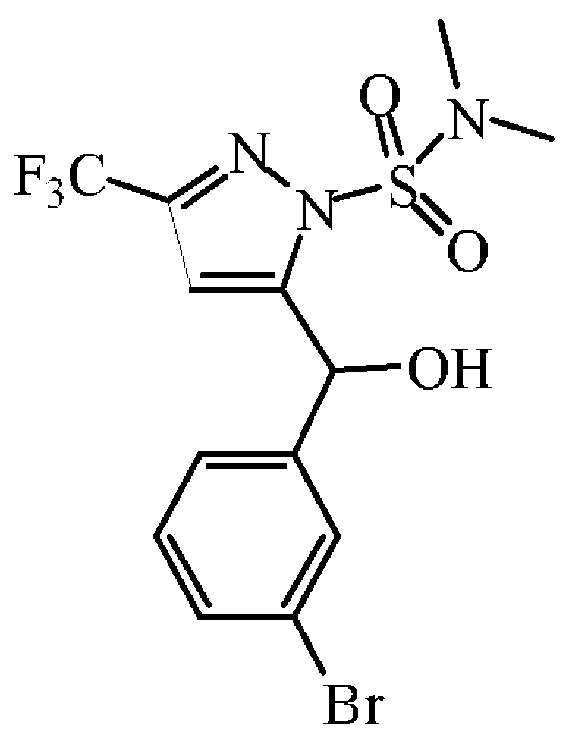
부틸 리튬 용액(헥산중 2.5 M) (15.2 mL, 37.9 mmol)을 -78 ℃에서 질소하에 THF (125 mL) 중 중간체 43 (8.4 g, 34.54 mmol)의 용액에 첨가하였다. 혼합물을 -78 ℃에서 45 분동안 교반한 후, 2-브로모벤즈알데히드 (6 mL, 51.8 mmol)를 적가하였다. 반응 혼합물을 -78 ℃에서 30 분동안 교반한 후, 실온으로 가온하고, 1 시간동안 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 퀀칭하고, 생성물을 AcOEt로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; DCM/헵탄 0/100 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 중간체 44 (13.2 g, 89% 수율)를 무색 오일로 수득하였다.
실시예 A45
중간체 45
: 5-[(3-브로모페닐)카보닐]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조
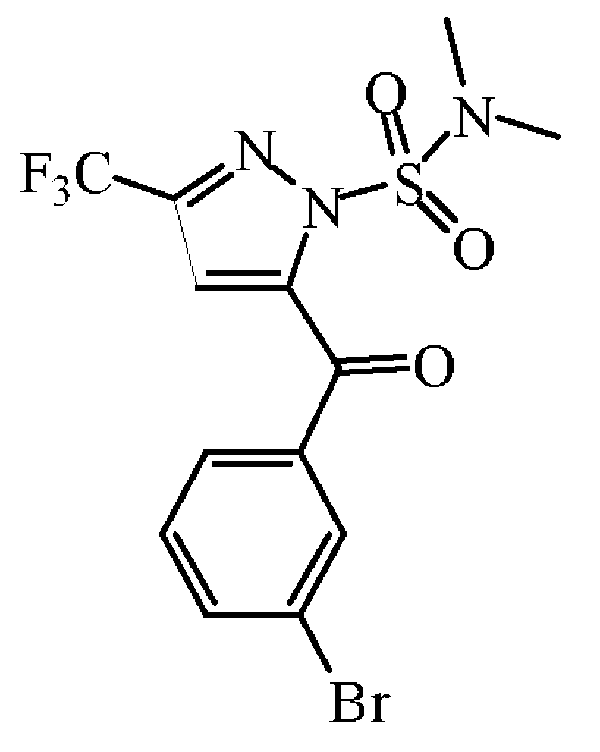
이산화망간 (15.4 g, 169.3 mmol)을 1,4-디옥산 (150 mL) 중 중간체 44 (14.5 g, 33.86 mmol)의 용액에 첨가하였다. 혼합물을 120 ℃에서 3 시간동안 교반하였다. 반응 혼합물을 40 ℃로 냉각하고, 규조토를 통해 여과하였다. 용매를 진공중에 증발시켜 중간체 45 (25.6 g, 97% 수율)를 백색 고체로 얻고, 다음 단계에 그대로 사용하였다.
실시예 A46
중간체 46
: 5-[(3-브로모페닐)[(tert-부틸설피닐)이미노1메틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조
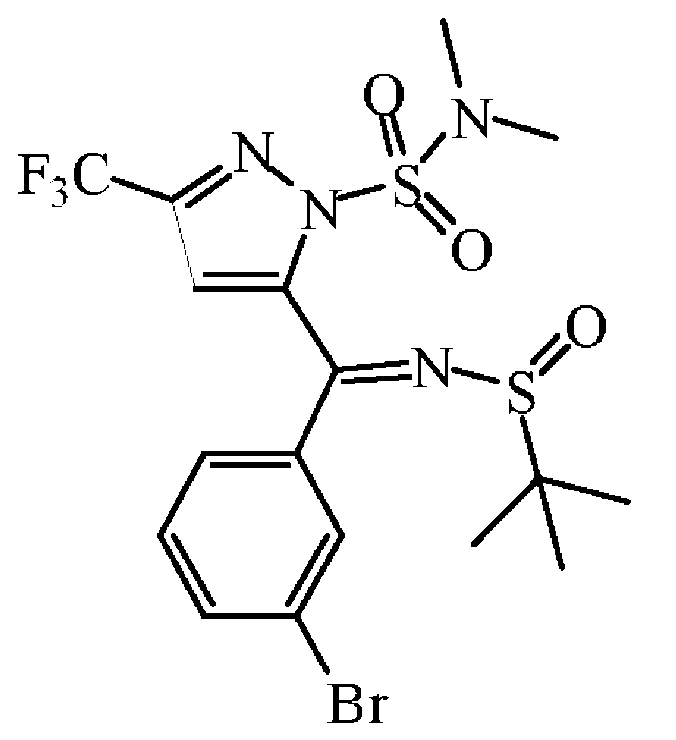
티타늄(IV)이소프로폭사이드(11.35 mL, 46.9 mmol)를 톨루엔 (140 mL) 중 중간체 45 (10 g, 23.46 mmol) 및 2-메틸-2-프로판설핀아미드 (3.128 g, 25.81 mmol)의 혼합물에 질소하에 첨가하였다. 혼합물을 110 ℃에서 8 시간동안 교반하였다. 혼합물을 냉각하고, 빠르게 교반하면서 염수에 부었다. 혼합물을 규조토를 통해 여과하고, 필터 케이크를 AcOEt로 세척하였다. 여액을 분별 깔때기로 옮겨 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 중간체 46 (4 g, 32% 수율)을 황색 오일로 수득하였다.
실시예 A47
중간체 47
: rac-5-[1-(3-브로모페닐)-1-[(tert-부틸설핀일)아미노]에틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조

메틸브롬화마그네슘 (디에틸 에테르중 3 M, 6.3 mL, 18.89 mmol)을 -78 ℃에서 질소하에 THF (56 mL) 중 중간체 46 (4 g, 7.56 mmol)의 용액에 첨가하였다. 혼합물을 -78 ℃에서 30 분동안 교반한 후, 실온으로 가온하고, 18 시간동안 교반하였다. 반응 혼합물을 NH4Cl (포화 수용액)로 퀀칭하고, 생성물을 DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 100/0). 목적 분획을 수집하고, 진공중에 농축하여 중간체 47 (3.6 g, 87% 수율)을 방치하면 고체로 되는 무색 오일로 수득하였다.
실시예 A48
중간체 48
: rac-5-[1-아미노-1-(3-브로모페닐)에틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조

디옥산중 염산 4 M (24.7 mL, 99 mmol)을 MeOH (5 mL) 중 중간체 47 (3.6 g, 6.6 mmol)의 용액에 첨가하고, 혼합물을 밀봉 튜브중에 80 ℃에서 18 시간동안 교반하였다. 용매를 진공중에 증발시켰다. 잔사를 NaHC03 (포화 수용액)에 붓고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올/DCM 0/100 - 2/98). 목적 분획을 수집하고, 진공중에 농축하여 중간체 48 (1.5 g, 54% 수율)을 담황색 고체로 수득하였다.
실시예 A49
중간체 49
: rac-N-{1-(3-브로모페닐)-1-[3-(트리플루오로메틸)-1H-피라졸-5-일]에틸}-2-클로로아세트아미드의 제조

DIPEA (1.9 mL, 11.2 mmol)를 DCM (20 mL) 중 중간체 48 (1.5 g, 4.49 mmol)의 용액에 첨가하고, 혼합물을 빙조에서 냉각하였다. 이어, 클로로아세틸 클로라이드 (0.429 mL, 5.38 mmol)를 첨가하고, 혼합물을 0 ℃에서 3 시간동안 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 30/70). 목적 분획을 수집하고, 진공중에 농축하여 중간체 49 (1.1 g, 60% 수율)를 담황색 고체로 수득하였다.
실시예 A50
중간체 50
: rac-4-(3-브로모페닐)-4-메틸-2-(트리플루오로메틸)-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

THF (40 mL) 중 중간체 49 (1.1 g, 2.68 mmol)의 용액을 0 ℃에서 질소하에 THF (40 mL) 중 수소화나트륨 (0.214 g, 5.36 mmol)의 현탁액에 적가하였다. 혼합물을 0 ℃에서 1 시간동안 교반하였다. 혼합물을 물로 희석하고, 생성물을 DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하여 중간체 50 (0.92 g, 92% 수율)을 백색 고체로 수득하였다.
실시예 A51
중간체 51
: rac-4-(3-브로모페닐)-4-메틸-2-(트리플루오로메틸)-4, 5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조
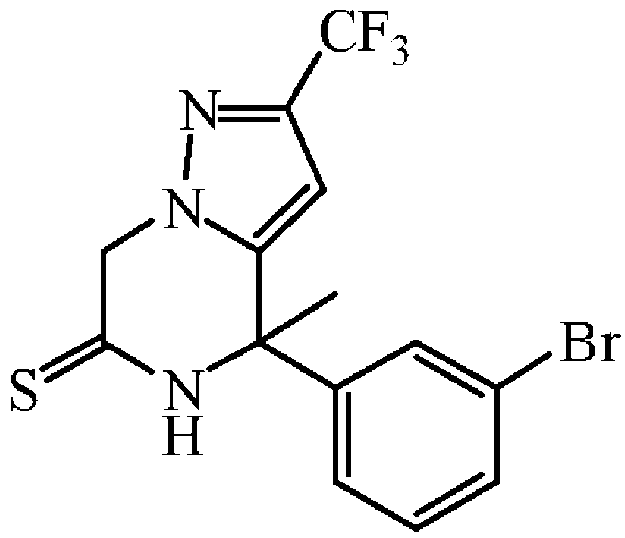
오황화인 (0.82 g, 3.69 mmol)을 피리딘 (10 mL) 중 중간체 50 (0.92 g, 2.46 mmol)의 용액에 첨가하고, 혼합물을 90 ℃에서 18 시간동안 가열하였다. 반응 혼합물을 진공중에 농축하고, 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; AcOEt/DCM 0/100 - 100/0). 목적 분획을 수집하고, 진공중에 농축하여 중간체 51 (0.27 g, 28% 수율)을 담황색 고체로 수득하였다.
실시예 A52
중간체 52
: rac-4-(3-브로모페닐)-4-메틸-2-(트리플루오로메틸)-4,5-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

NH4Cl (0.148 g, 2.77 mmol)을 암모니아 2 M/EtOH (6 mL) 중 중간체 51 (0.27 g, 0.69 mmol)의 교반 현탁액에 첨가하고, 혼합물을 80 ℃에서 18 시간동안 가열하였다. 용매를 진공중에 제거하고, 잔사를 DCM에 현탁시킨 후, 물로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 2/98). 목적 분획을 수집하고, 진공중에 농축하여 중간체 52 (0.195 g, 75% 수율)를 베이지색 고체로 수득하였다.
실시예 A53
중간체 53
: rac-5-[(5-브로모-2-플루오로페닐)(하이드록시)메틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조

중간체 53을 실시예 A44에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 43 (17 g, 69.9 mmol)으로부터 출발하여 중간체 53 (28 g, 76% 수율)을 무색 오일로 수득하였다.
실시예 A54
중간체 54
: rac-5-[(5-브로모-2-플루오로페닐)카보닐]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조
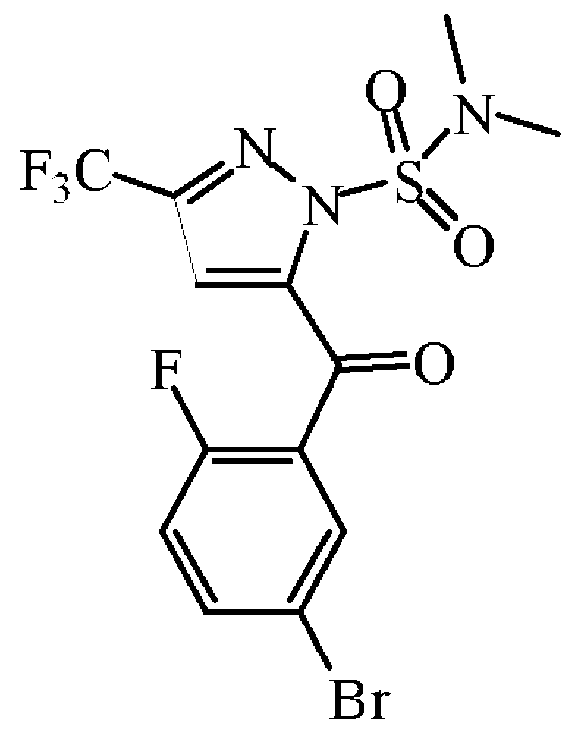
중간체 54를 실시예 A45에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 53 (28 g, 53.3 mmol)으로부터 출발하여 중간체 54 (25.6 g, 97% 수율)를 백색 고체로 얻고 다음 단계에 그대로 사용하였다.
실시예 A55
중간체 55
: rac-5-[(5-브로모-2-플루오로페닐)[(tert-부틸설피닐)이미노]메틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조

중간체 55를 실시예 A46에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 54 (25.6 g, 57.6 mmol)로부터 출발하여 중간체 55 (21 g, 67% 수율)를 담황색 고체로 수득하였다.
실시예 A56
중간체 56
: rac-5-[1-(5-브로모-2-플루오로페닐)-1-[(tert-부틸설핀일)아미노]에틸]-N,N-디메틸-3-(트리플루오로메틸)-1H-피라졸-1-설폰아미드의 제조

중간체 56을 실시예 A47에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 55 (21 g, 38.36 mmol)로부터 출발하여 중간체 56 (19 g, 88% 수율)을 방치하면 고체로 되는 무색 오일로 수득하였다.
실시예 A57
중간체 57
: rac-1-(5-브로모-2-플루오로페닐)-1-[3-(트리플루오로메틸)-1H-피라졸-5-일]에탄아민의 제조
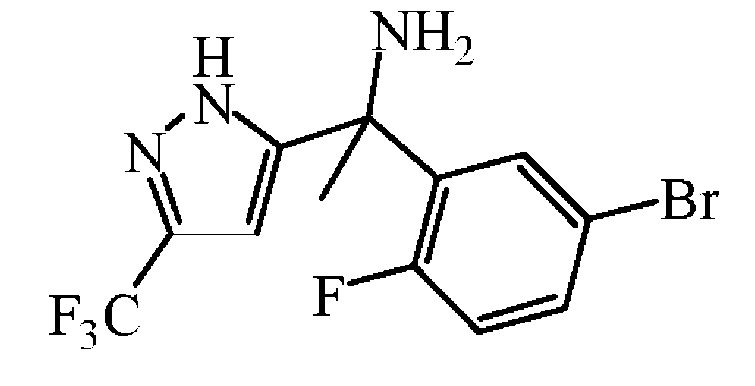
메탄올중 염산 1.25 M (159 mL, 199 mmol)을 중간체 56 (18.7 g, 33.2 mmol)에 첨가하고, 혼합물을 밀봉 튜브중 60 ℃에서 3 시간동안 교반하였다. 용매를 진공중에 증발시켰다. 잔사를 NaHC03 (포화 수용액)에 붓고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켜 중간체 57 (11.5 g, 98% 수율)을 담황색 고체로 얻고 다음 단계에 추가 정제없이 그대로 사용하였다.
실시예 A58
중간체 58
: rac-N-{1-(5-브로모-2-플루오로페닐)-1-[3-(트리플루오로메틸)-1H-피라졸-5-일]에틸}-2-클로로아세트아미드의 제조
중간체 58을 실시예 A49에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 57 (11.5 g, 32.66 mmol)로부터 출발하여 중간체 58 (6.6 g, 47% 수율)을 담황색 고체로 수득하였다.
실시예 A59
중간체 59
: rac-4-(5-브로모-2-플루오로페닐)-4-메틸-2-(트리플루오로메틸)-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

중간체 59를 실시예 A50에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 58 (6.6 g, 15.4 mmol)로부터 출발하여 중간체 59 (6 g, 99% 수율)를 백색 고체로 수득하였다.
실시예 A60
중간체 60
: rac-4-(5-브로모-2-플루오로페닐)-4-메틸-2-(트리플루오로메틸)-4, 5-디하이드로피라졸로[1,5-a]피라진-6-티온의 제조
오황화인 (2.27 g, 10.2 mmol)을 디옥산 (80 mL) 중 중간체 59 (4 g, 10.2 mmol)의 용액에 첨가하고, 혼합물을 80 ℃에서 18 시간동안 가열하였다. 반응 혼합물을 규조토를 통해 여과하였다. 이어, 여액을 진공중에 증발시키고, 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; DCM). 목적 분획을 수집하고, 진공중에 농축하여 중간체 60 (2.3 g, 55% 수율)을 담황색 고체로 수득하였다.
실시예 A61
중간체 61
: rac-4-(5-브로모-2-플루오로페닐)-4-메틸-2-(트리플루오로메틸)-4,5-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

중간체 61을 실시예 A52에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 60 (2 g, 4.9 mmol)으로부터 출발하여 중간체 61 (1.5 g, 78% 수율)을 백색 고체로 수득하였다.
실시예 A62
중간체 62:
rac-4-[5-(벤즈히드릴리덴아미노)-2-플루오로페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

중간체 62를 실시예 A15에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 61 (0.67 g, 1.59 mmol)로부터 출발하여 중간체 62 (0.53 g, 67% 수율)를 담황색 고체로 수득하였다.
실시예 A63
중간체 63
: rac-4-(5-아미노-2-플루오로페닐)-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

중간체 63을 실시예 A16에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 62 (0.525 g, 1.07 mmol)로부터 출발하여 중간체 63 (0.225 g, 64% 수율)을 담황색 고체로 수득하였다.
실시예 A64
중간체 64
: (5-브로모-2-플루오로페닐)(옥소)아세트산의 제조

5'-브로모-2'-플루오로아세토페논 [(CAS 198477-89-3), 70 g, 322 mmol) 및 이산화셀레늄 (71.6 g, 645 mmol)을 피리딘 (520 mL)에 용해시켰다. 반응 혼합물을 100 ℃에서 2 시간동안 교반하였다. 용매를 증발시키고, HCl 1N 수용액을 첨가하였다. 수성층을 EtOAc로 추출하였다. 유기층을 모아 건조시킨 후 (Mg2S04), 여과한 다음, 진공중에 농축하여 중간체 64 (62 g, 78% 수율)를 얻고, 다음 반응에 그대로 사용하였다.
실시예 A65
중간체 65
: tert-부틸 (5-브로모-2-플루오로페닐)(옥소)아세테이트의 제조

티오닐 클로라이드 (72 g, 607 mmol)를 0 ℃에서 톨루엔 (500 mL) 중 중간체 64 (50 g, 202 mmol) 의 교반 용액에 적가하였다. 혼합물을 60 ℃에서 1.5 시간동안 교반하였다. 용매를 진공중에 증발시켰다. DCM을 첨가한 후, 혼합물을 진공중에서 재농축하였다. 조 물질을 DCM (100 mL)에 용해시켰다. tert-부탄올 (30 g, 404 mmol), 피리딘 (16 mL, 202 mmol) 및 무수 DCM (100 mL)을 주의하여 첨가하였다. 혼합물을 rt에서 1.5 시간동안 교반하였다. 용매를 진공중에 증발시켰다. 조 물질을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; DCM). 목적 분획을 수집하고, 진공중에 농축하여 중간체 65 (45.5 g, 74% 수율)를 수득하였다.
실시예 A66
중간체 66
: (S)-1-메틸에틸 (5-브로모-2-플루오로페닐)[(tert-부틸설피닐)이미노]아세테이트의 제조

티타늄(IV) 이소프로폭사이드(85 mL, 283 mmol)를 n-헵탄 (1000 mL) 중 중간체 65 (43 g, 142 mmol) 및 (S)-2-메틸-2-프로판설핀아미드 (25.8 g, 212 mmol)의 교반 혼합물에 첨가하였다. 혼합물을 80 ℃에서 18 시간동안 교반하였다. 혼합물을 진공중에 부분적으로 농축한 후, EtOAc로 희석하였다. 혼합물을 실온으로 냉각하고, 물을 첨가하였다. 생성된 혼합물을 규조토 패드를 통해 여과하고, EtOAc 및 물로 헹구었다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 진공중에 농축하였다. 잔사를 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 용리제: n-헵탄/EtOAc 90/10 - 80/20). 목적 분획을 수집하고, 진공중에 농축하여 중간체 66 (44 g 79% 수율)을 수득하였다.
실시예 A67
중간체 67
: 이소프로필 (2R)-2-(5-브로모-2-플루오로페닐)-2-[[(S)-tert-부틸설피닐]아미노]-2-사이클로프로필아세테이트의 제조
사이클로프로필브롬화마그네슘 (0.5 M, 174 mL, 87 mmol)을 -78 ℃에서 DCM (388 mL) 중 중간체 66의 교반 용액에 적가하였다. 혼합물을 이 온도에서 30 분동안 교반한 후, NH4Cl 포화 수용액에 이어 물을 가하여 반응을 중단시켰다. 혼합물을 DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켜 중간체 67 (26.4, 98% 수율)을 누르스름한 오일로 얻고, 다음 단계에 그대로 사용하였다.
실시예 A68
중간체 68
: 이소프로필 (2R)-2-아미노-2-(5-브로모-2-플루오로페닐)-2-사이클로프로필아세테이트의 제조

4M HCL 용액/디옥산 (27 mL) 중 중간체 67 (23.9 g, 55 mmol)의 용액을 r.t.에서 15 분동안 교반하였다. 용매를 진공중에 농축하였다. 조 물질을 EtOAc에 용해시키고, sat. NaHC03을 첨가하였다. 혼합물을 1 시간동안 교반하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 진공중에 농축하여 중간체 68 (16.6, 91% 수율)을 누르스름한 오일로 얻고 다음 단계에 그대로 사용하였다.
실시예 A69
중간체 69
: (2R)-2-아미노-2-(5-브로모-2-플루오로페닐)-2-사이클로프로필에탄올의 제조

리튬 알루미늄 하이드라이드 (THF중 1M, 38 mL, 38 mmol)를 -15 ℃에서 THF (346 mL) 중 중간체 68 (16.6 g, 50.2 mmol)의 교반 용액에 적가하였다. 혼합물을 0 ℃로 서서히 가온하면서 1 시간동안 교반하였다. 고체 Na2S04 10 수화물을 가스 발생이 더이상 관찰되지 않을 때까지 혼합물에 첨가하였다. 혼합물을 30 분동안 실온에서 교반하였다. 혼합물을 규조토 패드를 통해 여과하고, THF로 헹구었다. 유기층을 모아 진공중에 증발로 건조시키고, 생성된 조 물질을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; MeOH중 7N NH3/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하여 중간체 69를 황색 오일로 수득하였다 (13.8 g, 정량적 수율).
실시예 A70
중간체 70
: tert-부틸 [(1R)-1-(5-브로모-2-플루오로페닐)-1-사이클로프로필-2-하이드록시에틸]카바메이트의 제조
중간체 70을 실시예 A5에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 69 (4.35 g, 6.03 mmol)로부터 출발하여 중간체 70 (3.29 g)을 방치하면 고체로 되는 황색 오일로 수득하였다.
실시예 A71
중간체 71
: tert-부틸 [(1R)-1-(5-브로모-2-플루오로페닐)-1-사이클로프로필-2-옥소에틸]카바메이트의 제조

중간체 71을 실시예 A6에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 70 (4.52 g, 12.08 mmol)으로부터 출발하여 중간체 71 (4 g, 89% 수율)을 담황색 오일로 수득하였다.
실시예 A72
중간체 72
: tert-부틸 [(1R)-1-(5-브로모-2-플루오로페닐)-1-사이클로프로필-2-하이드록시부트-3-인-1-일]카바메이트의 제조
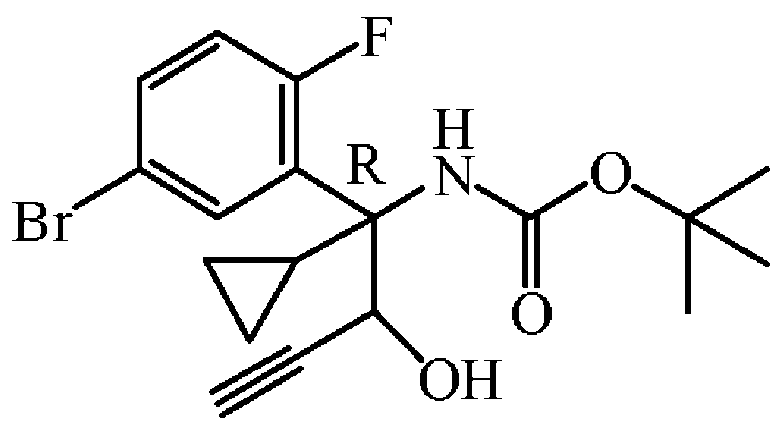
중간체 72를 실시예 A7에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 71 (4 g, 10.75 mmol)로부터 출발하여 중간체 72 (3.9 g, 91% 수율)를 부분입체이성체 혼합물로서 오일로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A73
중간체 73
: tert-부틸 [(1R)-1-(5-브로모-2-플루오로페닐)-1-사이클로프로필-2-옥소부트-3-인-1-일]카바메이트의 제조
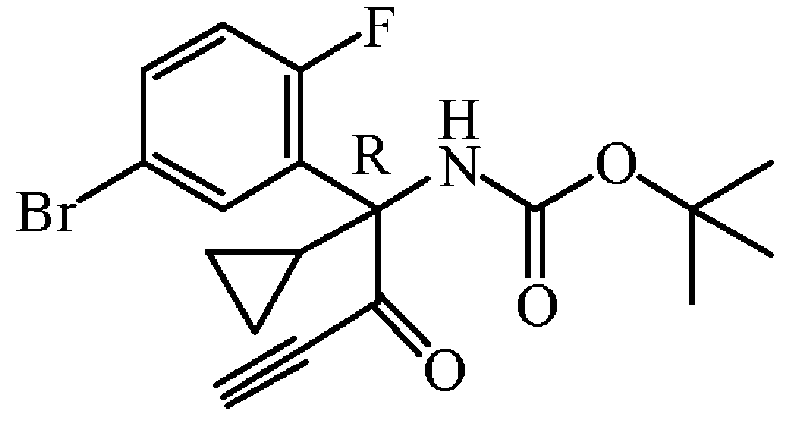
중간체 73을 실시예 A8에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 72 (3.9 g, 9.8 mmol)로부터 출발하여 중간체 73 (3.4 g, 88% 수율)을 황색 오일로 수득하였다.
실시예 A74
중간체 74
: tert-부틸 N-[(R-(5-브로모-2-플루오로페닐)사이클로프로필-(1H-피라졸-3-일)메틸]카바메이트의 제조

중간체 74를 실시예 A9에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 73 (3.4 g, 8.58 mmol)으로부터 출발하여 중간체 74 (3.45 g, 98% 수율)를 포움으로 수득하였다.
실시예 A75
중간체 75
: (R)-(5-브로모-2-플루오로페닐)사이클로프로필-(1H-피라졸-3-일)메탄아민의 제조

중간체 75를 실시예 A10에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 74 (3.45 g, 8.41 mmol)로부터 출발하여 중간체 75 (2.85 g)를 황색 포움으로 얻고, 다음 단계에 추가 정제없이 사용하였다.
실시예 A76
중간체 76
: N-[(R)-(5-브로모-2-플루오로페닐)사이클로프로필-(1H-피라졸-3-일)메틸]-2-클로로아세트아미드의 제조

중간체 76을 실시예 A11에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 75 (2.8 g, 9.03 mmol)로부터 출발하여 중간체 76 (1.03 g, 30% 수율)을 고체로 수득하였다.
실시예 A77
중간체 77
: (R)-4-(5-브로모-2-플루오로페닐)-4-사이클로프로필-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

중간체 77을 실시예 A12에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 76 (0.765 g, 1.98 mmol)으로부터 출발하여 중간체 77 (0.52 g, 75% 수율)을 백색 고체로 수득하였다.
실시예 A78
중간체 78
: (R)-4-(5-브로모-2-플루오로페닐)-4-사이클로프로필-4,5-디하이드로피라졸로[1,5-a]피라진-6-온의 제조

중간체 78을 실시예 A13에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 77 (0.62 g, 1.77 mmol)로부터 출발하여 중간체 78 (0.46 g, 70% 수율)을 담적색 고체로 수득하였다.
실시예 A79
중간체 79
: (R)-4-(5-브로모-2-플루오로페닐)-4-사이클로프로필-4,5-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조
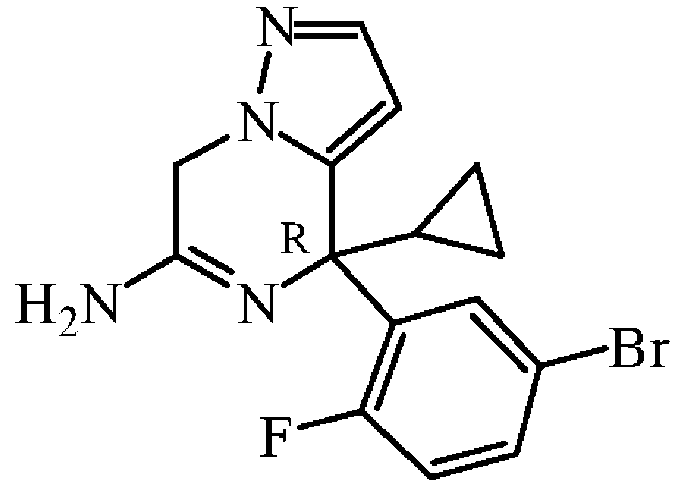
32% 암모니아 수용액 (3 mL, 50.7 mmol)을 밀봉 튜브중에 암모니아 7 N 용액/메탄올 (7 mL, 49 mmol) 중 중간체 78 (0.46 g, 1.26 mmol)의 교반 혼합물에 첨가하였다. 혼합물을 70 ℃에서 8 시간동안 교반하였다. 실온으로 냉각 후, 혼합물을 물로 희석하고, Na2CO3 (포화 수용액) 및 DCM으로 추출하였다. 유기층을 분리하고, 건조시키고 (Na2S04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 2/98 - 3/97 - 10/90). 목적 분획을 수집하고, 진공중에 농축하여 중간체 79 (0.34 g, 78% 수율)를 황색 포움으로 수득하였다.
실시예 A80
중간체 80
: (R)-4-[5-(벤즈히드릴리덴아미노)-2-플루오로페닐]-4-사이클로프로필-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조
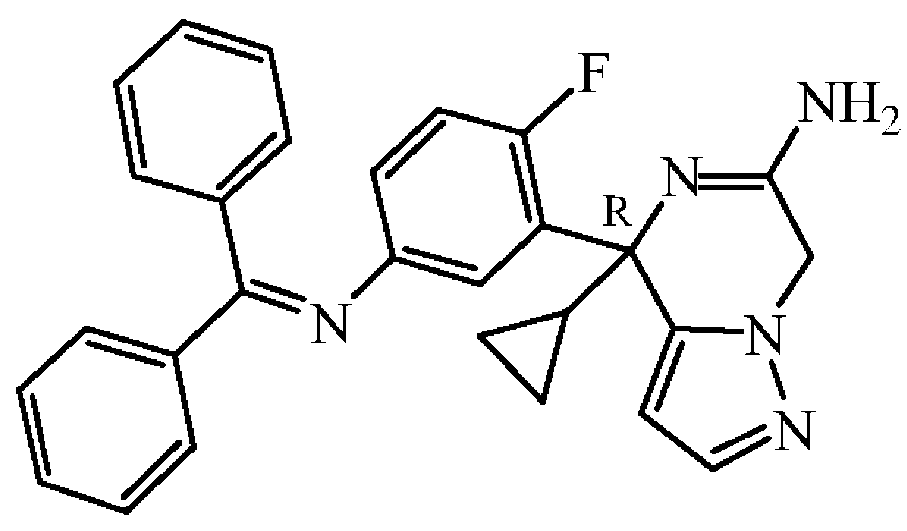
중간체 80을 실시예 A15에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 79 (0.34 g, 0.974 mmol)로부터 출발하여 중간체 80 (0.38 g, 61% 수율)을 황색 고체로 수득하였다.
실시예 A81
중간체 81
: (R)-4-(5-아미노-2-플루오로페닐)-4-사이클로프로필-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

중간체 81을 실시예 A16에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 80 (0.38 g, 0.845 mmol)으로부터 출발하여 중간체 81 (0.16 g, 66% 수율)을 담황색 고체로 수득하였다.
최종 화합물의 제조
실시예 B1
화합물 1
: rac-4-메틸-4-(3-피리미딘-5-일페닐)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조
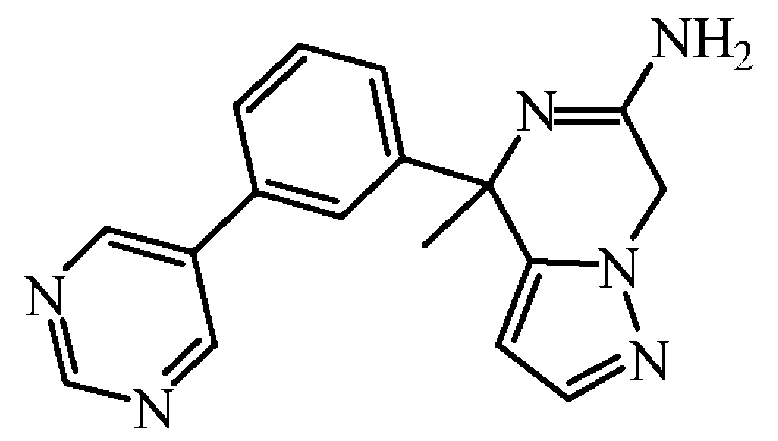
NH4Cl (0.007 g, 0.121 mmol)을 EtOH (3 mL) 중 중간체 28 (0.026 g, 0.081 mmol)의 교반 용액에 첨가하고, 혼합물을 75 ℃에서 18 시간동안 가열하였다. 용매를 진공중에 제거하고, 잔사를 DCM에 용해시킨 후, 물로 세척하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/AcOEt 0/100 - 20/80). 목적 분획을 수집하고, 진공중에 농축하여 화합물 1 (0.02 g, 81% 수율)을 담황색 고체로 수득하였다.
실시예 B2
화합물 2
: rac-4-[3-(5-메톡시피리딘-3-일)페닐]-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

화합물 2를 실시예 B1에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 30 (0.1 g, 0.285 mmol)으로부터 출발하여 화합물 2 (0.06 g, 63%) 수율)를 백색 고체로 수득하였다.
실시예 B3
화합물 3
: rac-N-[3-(6-아미노-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-4-일)페닐]-5-클로로피리딘-2-카복사미드의 제조

N,N-디메틸아닐린 (0.24 mL, 1.92 mmol)을 DCM (15 mL) 중 5-클로로-2-피리딘카복실산 (0.15 g, 0.96 mmol) 및 HATU (0.40 g, 1.04 mmol)의 현탁액에 첨가하였다. 혼합물을 실온에서 10 분동안 교반하였다. 이어, 중간체 16 (0.21 g, 0.87 mmol)을 첨가하고, 혼합물을 실온에서 18 시간동안 교반하였다. 혼합물을 NH4Cl (포화 수용액)로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 5/95). 목적 분획을 수집하고, 진공중에 농축하여 화합물 3 (0.070 g, 21% 수율)을 백색 고체로 수득하였다.
실시예 B4
화합물 4
: rac-4-(2,4-디플루오로-5-피리미딘-5-일-페닐)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

테트라키스(트리페닐포스핀)팔라듐(0) (0.025 g, 0.02 mmol)을 1,4-디옥산 (4 mL) 및 에탄올 (0.4 mL) 중 중간체 32 (0.15 g, 0.44 mmol), 피리미딘-5-보론산 (0.16 g, 1.32 mmol) 및 탄산칼륨 (0.18 g, 1.32 mmol)의 교반 현탁액에 실온에서 질소하에 첨가하였다. 혼합물을 150 ℃에서 30 분동안 마이크로파 조사하에 교반하였다. 이어, 혼합물을 물로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하여 화합물 4 (0.081 g, 54% 수율)를 백색 고체로 수득하였다.
실시예 B5
화합물 7
: (R)-4-(3'-메톡시비페닐-3-일)-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조

테트라키스(트리페닐포스핀)팔라듐(0) (0.028 g, 0.025 mmol)을 1,4-디옥산 (4 mL) 및 에탄올 (0.4 mL) 중 중간체 42 (0.15 g, 0.49 mmol), 5-메톡시피리딘-3-보론산 (0.23 g, 1.48 mmol) 및 탄산칼륨 (0.20 g, 1.48 mmol)의 교반 현탁액에 실온에서 질소하에 첨가하였다. 혼합물을 150 ℃에서 30 분동안 마이크로파 조사하에 교반하였다. 이어, 혼합물을 물로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하여 화합물 7 (0.12 g, 73% 수율)을 백색 고체로 수득하였다.
실시예 B6
화합물 10
: (S*)-N-[3-(6-아미노-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-4-일)-4-플루오로페닐]-5-클로로피리딘-2-카복사미드 및
화합물 11
: (R*)-N-[3-(6-아미노-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-4-일)-4-플루오로페닐]-5-클로로피리딘-2-카복사미드의 제조

rac-5-클로로-피리딘-2-카복실산[3-(6-아미노-4-메틸-4,7-디하이드로피라졸로[1,5-a]피라진-4-일)-4-플루오로페닐]아미드 (0.182 g)의 샘플을 35 ℃에서 3.0 ml/min의 유속으로 Chiralpak® AD Daicel 칼럼 (10 ㎛, 4.6 × 250 mm) 상에서 분취용 SFC에 의해 상응하는 거울상이성체로 분리하여 (이동상: C02, 50% 에탄올, 50% EtOH (0.3% iPrNH2 함유)) 화합물 11 (0.07 g; 38% 수율) 및 화합물 10 (0.06 g, 33% 수율)을 수득하였다.
실시예 B7
화합물 21
: rac-4-[3-(5-메톡시피리딘-3-일)페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민 및
화합물 22
: (R*)-4-[3-(5-메톡시피리딘-3-일)페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민 및
화합물 23
: (S*)-4-[3-(5-메톡시피리딘-3-일)페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조
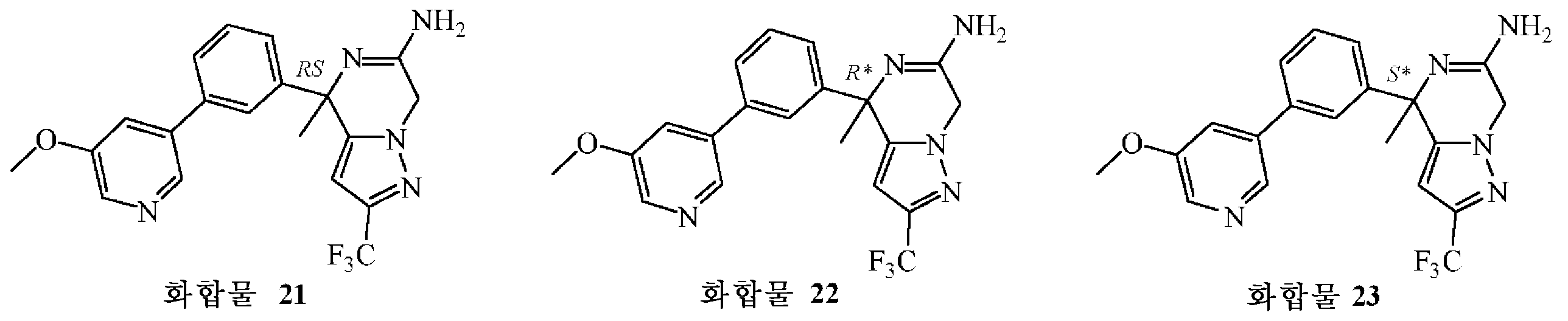
테트라키스(트리페닐포스핀)팔라듐(0) (0.031 g, 0.027 mmol)을 1,4-디옥산 (6 mL) 및 에탄올 (0.6 mL) 중 중간체 52 (0.2 g, 0.54 mmol), 5-메톡시피리딘-3-보론산 (0.163 g, 1.07 mmol) 및 탄산칼륨 (0.222 g, 1.61 mmol)의 교반 현탁액에 실온에서 질소하에 첨가하였다. 혼합물을 80 ℃에서 24 시간동안 교반하였다. 이어, 혼합물을 물로 희석하고, DCM으로 추출하였다. 유기층을 분리하고, 건조시킨 후 (MgS04), 여과한 다음, 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7M 용액/DCM 0/100 - 3/97). 목적 분획을 수집하고, 진공중에 농축하였다. 잔사를 디에틸 에테르에서 연마하고, 초음파처리한 후, 여과한 다음, 50 ℃에서 진공중에 건조시켜 화합물 21 (0.13 g, 60% 수율)을 백색 고체로 수득하였다. 이어, 이 라세믹 화합물을 Chiralpak® AD-H 칼럼 (20 x 250 mm) 상에서 분취용 SFC에 의해 정제하여 (이동상 (C02, iPrOH + 0.3% iPrNH2)) 화합물 22 (0.047 g, 22% 수율) 및 화합물 23 (0.051 g, 24% 수율)을 순수한 거울상이성체로 수득하였다 (둘 다 고체 화합물).
실시예 B8
화합물 24
: rac-4-[5-(5-클로로피리딘-3-일)-2-플루오로페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민 및
화합물 25
: (R*)-4-[5-(5-클로로피리딘-3-일)-2-플루오로페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민 및
화합물 26
: (S*)-4-[5-(5-클로로피리딘-3-일)-2-플루오로페닐]-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-6-아민의 제조
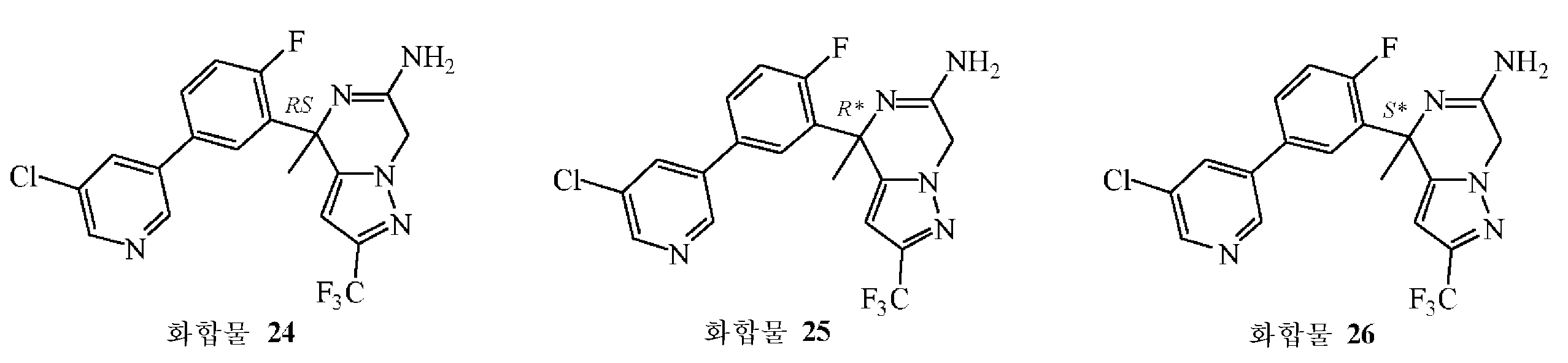
화합물 24를 실시예 B7에 기술된 방법과 동일한 방식으로 합성하였다. 중간체 61 (0.3 g, 0.77 mmol)로부터 출발하여 화합물 24 (0.21 g, 64% 수율)를 백색 고체로 수득하였다. 이어, 이 라세믹 화합물을 Chiralpak® AD-H 칼럼 (20 x 250 mm) 상에서 분취용 SFC에 의해 정제하여 (이동상 (C02, iPrOH + 0.3% iPrNH2)) 화합물 25 (0.089 g, 27% 수율) 및 화합물 26 (0.092 g, 28% 수율)을 순수한 거울상이성체로 수득하였다 (둘 다 고체 화합물).
실시예 B9
화합물 27
: rac-N-{3-[6-아미노-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-4-일]-4-플루오로페닐}-3,5-디클로로피리딘-2-카복사미드 및
화합물 28
: (R*)-N-{3-[6-아미노-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-4-일]-4-플루오로페닐}-3,5-디클로로피리딘-2-카복사미드 및
화합물 29
: (S*)-N-{3-[6-아미노-4-메틸-2-(트리플루오로메틸)-4,7-디하이드로피라졸로[1,5-a]피라진-4-일]-4-플루오로페닐}-3,5-디클로로피리딘-2-카복사미드의 제조
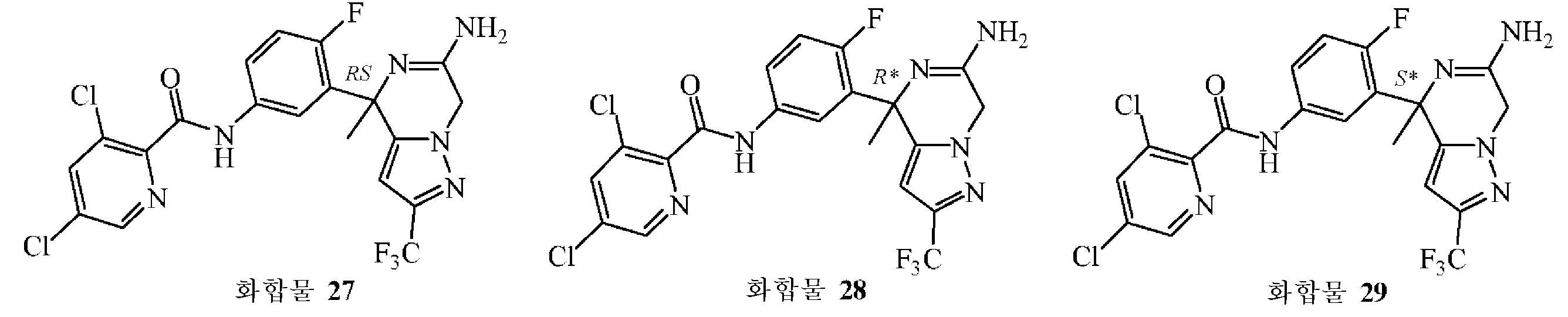
3,5-디클로로-2-피리딘카복실산 (0.54 g, 0.81 mmol)을 MeOH (4 mL)에 용해시키고, DMTMM (0.223 g, 0.81 mmol)을 첨가하였다. 혼합물을 5 분동안 교반한 후, MeOH (4 mL) 중 중간체 63 (0.22 g, 0.67 mmol)의 용액을 0 ℃에서 첨가하고, 혼합물을 4 시간 교반하였다. 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7 M 용액/DCM 0/100 - 5/95). 목적 분획을 수집하고, 진공중에 농축하였다. 잔사를 DIPE로부터 결정화하여 화합물 27 (0.124 g, 37% 수율)을 백색 고체로 수득하였다. 이어, 이 라세믹 화합물을 Chiralpak® AD-H 칼럼 (20 x 250 mm) 상에서 분취용 SFC에 의해 정제하여 (이동상 (C02, EtOH + 0.3% iPrNH2)) 화합물 28 (0.038 g, 11%) 수율) 및 화합물 29 (0.036 g, 11% 수율)을 순수한 거울상이성체로 수득하였다 (둘 다 고체 화합물).
실시예 B1O
화합물 33
: (R)-N-{3-[6-아미노-4-사이클로프로필-4,7-디하이드로피라졸로[1,5-a]피라진-4-일]-4-플루오로페닐}-5-클로로-3-플루오로피리딘-2-카복사미드의 제조
5-클로로-3-플루오로피리딘-2-카복실산 (0.052 g, 0.26 mmol)을 MeOH (1.5 mL)에 용해시키고, DMTMM (0.086 g, 0.31 mmol)을 첨가하였다. 혼합물을 5 분동안 교반한 후, MeOH (1.5 mL) 중 중간체 81 (0.074 g, 0.26 mmol)의 용액을 0 ℃에서 첨가하고, 혼합물을 24 시간 교반하였다. 용매를 진공중에 증발시켰다. 조생성물을 플래쉬 칼럼 크로마토그래피로 정제하였다 (실리카겔; 메탄올중 암모니아 7 M 용액/DCM 0/100 - 5/95). 목적 분획을 수집하고, 진공중에 농축하였다. 잔사를 진공하에 건조하여 화합물 33 (0.084 g, 73% 수율)을 백색 고체로 수득하였다.



분석 파트
LCMS
본 발명의 화합물의 (LC)MS-특정을 위해 다음의 방법들이 이용되었다.
일반적인 방법 A
탈기장치가 있는 펌프 (쿼터너리 또는 바이너리), 오토샘플러, 칼럼 오븐, 다이오드-어레이 검출기 (DAD) 및 각 방법에 명시되어 있는 칼럼을 포함하는 HP 1100 (Agilent Technologies) 시스템을 사용하여 HPLC 측정을 수행하였다. MS 검출기에는 전기스프레이 이온화 공급원이 설치되어 있다. 네뷸라이저 가스로서 질소를 사용하였다. 공급원 온도는 140 ℃ 또는 100 ℃로 유지하였다. 데이터는 MassLynx-Openlynx 소프트웨어 (Waters)로 수집하였다.
일반적인 방법 B
샘플 오가나이저, 탈기장치가 있는 바이너리 펌프, 4 칼럼 오븐, 다이오드 어레이 검출기 (DAD) 및 각 방법에 명시되어 있는 칼럼을 포함하는 Acquity UPLC (Waters) 시스템을 사용하여 UPLC (초성능 액체 크로마토그래피) 측정을 수행하였다. MS 검출기에는 ESCI 이중 이온화 공급원 (대기압 화학 이온화와 결합된 전기스프레이)이 설치되어 있다. 네뷸라이저 가스로서 질소를 사용하였다. 공급원 온도는 140 ℃로 유지하였다. 데이터는 MassLynx-Openlynx 소프트웨어 (Waters)로 수집하였다.
일반적인 방법 C
탈기장치가 있는 바이너리 펌프, 오토샘플러, 다이오드 어레이 검출기 (DAD) 및 하기 각 방법에 명시되어 있는 칼럼을 포함하는 Acquity UPLC (초고성능 액체 크로마토그래피) (Waters) 시스템을 사용하여 LC 측정을 수행하고, 칼럼은 40 ℃의 온도로 유지하였다. 칼럼으로부터의 유동을 MS 분광계로 분배하였다. MS 검출기에는 전자스프레이 이온화 공급원이 설치되어 있다. 모세 바늘 전압은 3 kV이며 공급원 온도는 Quattro (Waters사 제품인 트리플 쿼드러폴 질량 분광계) 상에서 130 ℃로 유지하였다. 네뷸라이저 가스로서 질소를 사용하였다. 데이터는 MassLynx-Openlynx 소프트웨어 (Waters)로 수집하였다.
방법 1:
일반적인 방법 A 외에: 역상 HPLC를 Agilent사 제품인 Eclipse Plus-C18 칼럼 (3.5 μm, 2.1 × 30 mm) 상에서 1.0 ㎖/분의 유속으로 60 ℃에서 수행하였다. 다음과 같은 구배 조건을 이용하였다: 95% A (0.5 g/l 암모늄 아세테이트 용액 + 5% 아세토니트릴), 5% B (아세토니트릴) - 100% B, 5.0 분, 5.15 분까지 유지 및 5.30 분에서 7.0 분까지 초기 조건으로 평형화. 주입 용적 2 ㎕. 0.3초의 체류시간을 사용하여 0.5초에 100 내지 750회 스캔하여 고분해능 질량 스펙트럼 (비행시간, TOF 검출기)을 얻었다. 모세 바늘 전압은 포지티브 이온화 모드의 경우 2.5kV이고 네가티브 이온화 모드의 경우 2.9kV이었다. 콘 전압은 포지티브 및 네가티브 이온화 모드 모두 20V이었다. 류신-엔케팔린을 록 질량 보정을 위한 표준 물질로 사용하였다.
방법 2: 방법 1과 동일한 HPLC 구배
0.1초의 체류시간을 사용하여 0.5초에 100 내지 750회 스캔하여 고분해능 질량 스펙트럼 (비행시간, TOF 검출기)을 포지티브 이온화 모드로만 얻었다. 모세 바늘 전압은 포지티브 이온화 모드의 경우 2.5kV이고, 콘 전압은 20V이었다. 류신-엔케팔린을 록 질량 보정을 위한 표준 물질로 사용하였다.
방법 3:
일반적인 방법 A 외에: 역상 HPLC를 Agilent사 제품인 Eclipse Plus-C18 칼럼 (3.5 μm, 2.1 × 30 mm) 상에서 1.0 ㎖/분의 유속으로 60 ℃에서 MS 검출기로의 분리없이 수행하였다. 다음과 같은 구배 조건을 이용하였다: 95% A (0.5 g/l 암모늄 아세테이트 용액 + 5% 아세토니트릴), 5% B (아세토니트릴/메탄올, 1/1의 혼합물) - 0.2 분 유지, 3.0 분에 100% B, 3.15 분까지 유지 및 3.30 분에서 5.0 분까지 초기 조건으로 평형화. 주입 용적 2 ㎕. 0.08초의 채널간 지연을 이용하여 0.1초에 100 내지 1000회 스캔하여 저분해능 질량 스펙트럼 (단일 쿼드러폴, SQD 검출기)을 얻었다. 모세 바늘 전압은 3kV이었다. 콘 전압은 포지티브 이온화 모드의 경우 20V 및 50V이고 네가티브 이온화 모드의 경우 30V이었다.
방법 4:
일반적인 방법 B 외에: 역상 UPLC를 Waters사 제품인 RRHD Eclipse Plus-C18 (1.8 μm, 2.1 × 50 mm) 상에서 1.0 ㎖/분의 유속으로 50 ℃에서 MS 검출기로의 분리없이 수행하였다. 다음과 같은 구배 조건을 이용하였다: 95% A (0.5 g/l 암모늄 아세테이트 용액 + 5% 아세토니트릴), 5% B (아세토니트릴) - 40% A, 60% B, 3.8 분 - 5% A, 95% B, 4.6 분, 5.0 분까지 유지. 주입 용적 2 ㎕. 0.08초의 채널간 지연을 이용하여 0.1초에 100 내지 1000회 스캔하여 저분해능 질량 스펙트럼 (단일 쿼드러폴, SQD 검출기)을 얻었다. 모세 바늘 전압은 3kV이었다. 콘 전압은 포지티브 이온화 모드의 경우 25V이고 네가티브 이온화 모드의 경우 30V이었다.
방법 5 : 방법 4와 동일한 구배; 사용된 칼럼: Agilent사 제품인 RRHD Eclipse Plus-C18 (1.8 μm, 2.1 × 50 mm).
방법 6:
일반적인 방법 C 외에: 역상 UPLC를 Waters Acquity BEH (가교된 에틸실록산/실리카 하이브리드) 페닐-헥실 칼럼 (1.7 μm, 2.1 × 100 mm) 상에서 0.343 ml/분의 유속으로 수행하였다. 2종의 이동상 (이동상 A: 95% 7 mM 암모늄 아세테이트/5% 아세토니트릴; 이동상 B: 100% 아세토니트릴)을 84.2% A 및 15.8% B (0.49 분 유지) - 10.5% A 및 89.5% B, 2.18 분, 1.94 분 유지 및 0.73 분내에 다시 초기 조건으로, 0.73 분 유지의 구배조건으로 사용하였다. 주입 용적 2 ml가 사용되었다. 콘 전압은 포지티브 및 네가티브 이온화 모드 모두 20V이었다. 0.1초의 채널간 지연을 이용하여 0.2초에 100 내지 1000회 스캔하여 질량 스펙트럼을 얻었다.
방법 7:
일반적인 방법 A 외에: 역상 UPLC를 Agilent사 제품인 Eclipse Plus-C18 (3.5 μm, 2.1 × 30 mm) 상에서 1.0 ㎖/분의 유속으로 60 ℃에서 MS 검출기로의 분리없이 수행하였다. 다음과 같은 구배 조건을 이용하였다: 95% A (0.5 g/l 암모늄 아세테이트 용액 + 5% 아세토니트릴), 5% B (아세토니트릴/메탄올, 1/1의 혼합물) - 0.5 분에 100% B, 5.15 분까지 유지, 5.30 분에서 7.0 분까지 초기 조건으로 평형화. 주입 용적 2 ㎕. 0.08초의 채널간 지연을 이용하여 0.1초에 100 내지 1000회 스캔하여 저분해능 질량 스펙트럼 (단일 쿼드러폴, SQD 검출기)을 얻었다. 모세 바늘 전압은 3kV이었다. 콘 전압은 포지티브 이온화 모드의 경우 20V이고 네가티브 이온화 모드의 경우 30V이었다.
융점
수치는 피크값 또는 융점 범위이고, 이러한 분석 방법에서 보통 일어나는 실험 불확실성으로 얻어진다.
Mettler FP 81HT/FP90 장치 (표 3에서 FP90으로 표시)
다수의 화합물에 있어, 융점은 오픈 모세관에서 Mettler FP81HT/FP90 장치로 측정하였다. 융점은 1, 3, 5 또는 10 ℃/분의 온도 구배로 측정하였다. 최대 온도는 300 ℃이었다. 융점은 디지털 디스플레이에서 읽었다.
표 2: 분석 데이터 - Rt는 체류 시간(분)을 의미하고, [M+H]+는 화합물의 양자화 질량을 의미하며, 방법은 (LC)MS를 위해 사용된 방법을 가리킨다.


(n.d.는 결정되지 않았음을 의미한다)
선광도:
선광도는 나트륨 램프를 사용하는 Perkin Elmer 341 편광계에서 측정하였다.
표 3: 분석 데이터 - 거울상이성체적으로 순수한 화합물에 대한 선광도 값

SFCMS - 방법:
SFC-MS 방법을 위한 일반적인 방법
SFC 측정은, 이산화탄소(CO2) 및 모디파이어(modifier) 전달용 FCM-1200 듀얼 펌프 유체 제어 모듈, CTC Analytics 자동 액체 샘플러, 실온에서 80 ℃ 까지 칼럼을 가열하기 위한 TCM-20000 열 제어 모듈을 구비한 Berger Instruments사 (Newark, DE, USA) 제품인 분석 SFC 시스템을 사용하여 수행하였다. 고압 유동 셀 (최대 400 bar)이 장착된 Agilent 1100 UV 광다이오드 어레이 검출기가 사용되었다. 칼럼으로부터의 유동을 MS 분광계로 분배하였다. MS 검출기에는 대기압 이온화 공급원이 설치되었다. Waters ZQ 질량 분광계의 이온화 파라미터는 다음과 같다: 코로나: 9μa, 공급원 온도: 140 ℃, 콘: 30 V, 프로브 온도 450 ℃, 추출기 3 V, 탈용매화 가스 400 L/hr, 콘 가스 70 L/hr. 네뷸라이저 가스로서 질소를 사용하였다. 데이타 수집은 Waters-Micromass MassLynx-Openlynx 데이타 시스템으로 행하였다.
방법 1:
일반적인 방법 외에: SFC에서의 키랄 분리를 CHIRALPAK AD DAICEL 칼럼 (10 ㎛, 4.6 × 250 mm) 상에서 35 ℃에서 3.0 ml/분의 유속으로 수행하였다. 이동상으로 C02, 50% 에탄올, 50% EtOH (0.3% iPrNH2 함유)을 7 분 유지하여 사용하였다.
방법 2:
일반적인 방법 A 외에: SFC에서의 키랄 분리를 CHIRALPAK AD DAICEL 칼럼 (10 ㎛, 4.6 × 250 mm) 상에서 35 ℃에서 3.0 ml/분의 유속으로 수행하였다. 이동상으로 C02, 20% 메탄올 (0.3% iPrNH2 함유)을 7 분 유지하여 사용하였다.
방법 3:
일반적인 방법 A 외에: SFC에서의 키랄 분리를 CHIRALPAK AD DAICEL 칼럼 (10 ㎛, 4.6 × 250 mm) 상에서 35 ℃에서 3.0 ml/분의 유속으로 수행하였다. 이동상으로 C02, 25% 이소프로판올 (0.3% iPrNH2 함유)을 7 분 유지하여 사용하였다.
방법 4:
일반적인 방법 A 외에: SFC에서의 키랄 분리를 CHIRALPAK AD DAICEL 칼럼 (10 ㎛, 4.6 × 250 mm) 상에서 35 ℃에서 3.0 ml/분의 유속으로 수행하였다. 이동상으로 C02, 30% 에탄올 (0.3% iPrNH2 함유)을 7 분 유지하여 사용하였다.
표 4: 분석 SFC 데이터 - Rt는 체류 시간(분)을 의미하고, [M+H]+는 화합물의 양자화 질량을 의미하며, 방법은 거울상이성체적으로 순수한 화합물의 SFC/MS 분석에 이용된 방법을 가리킨다.

약물학적 실시예
본 발명으로 제공되는 화합물은 베타-부위 APP-절단 효소 1 (BACE1)의 저해제이다. 아스파라긴산 프로테아제인 BACE1 저해는 알츠하이머 병 (AD)의 치료와 관련이 있을 것으로 판단된다. 베타-아밀로이드 전구체 단백질 (APP)로부터 베타-아밀로이드 펩티드 (Aβ)의 생산 및 축적은 AD의 발병 및 진행에 중요한 역할을 담당할 것으로 여겨진다. Aβ는 아밀로이드 전구체 단백질 (APP)로부터 Aβ 도메인의 N- 및 C-말단에서 각각 β-세크레타아제 및 γ-세크레타아제에 의해 연속 절단됨으로써 생산된다.
화학식 (I)의 화합물은 효소 활성의 저해능으로 인해 BACE1에서 실질적으로 그의 효과를 발휘할 것으로 기대된다. 상기 화합물, 특히 화학식 (I)에 따른 화합물의 동정에 적합한 후술하는 생화학적 형광 공명 에너지 전이 (FRET) 기반 검정 및 SKNBE2 세포에서의 세포 αLisa 검정을 이용하여 시험한 저해제 거동을 표 1에 나타내었다.
생화학적 FRET 기반 검정
이 검정은 형광 공명 에너지 전이 검정 (FRET)에 기반한 검정이다. 이 검정에 사용되는 기질은 아밀로이드 전구체 단백질 (APP) β-세크레타아제 절단 부위의 'Swedish' Lys-Met/Asn-Leu 돌연변이를 가지는 APP 유래 13 아미노산 펩티드이다. 상기 기질은 또한 다음과 같은 두개의 형광단을 가진다: (7-메톡시쿠마린-4-일) 아세트산 (Mca)은 여기 파장이 320 nm이고 발광 파장이 405 nm인 형광 공여체이고, 2,4-디니트로페닐 (Dnp)은 독점적 소광제 수용체이다. 이들 두 그룹 간의 거리는 광 여기시 공여 형광 에너지가 공명 에너지 전이를 통해 수용체에 의해 상당히 소광되도록 선택된다. BACE1에 의해 절단 시, 형광단 Mca는 소거 그룹 Dnp으로부터 분리되고, 공여체의 완전 형광 수율을 회복한다. 형광 증가는 단백질가수분해와 선형적인 관계에 있다 (Koike H et al. J Biochem. 1999, 126, 235-42).
요약하면, 384-웰 포맷에서 최종 농도 1 μg/ml의 재조합 BACE1 단백질을 실온에서 인큐베이션 완충제 (40 mM 시트레이트 완충제 pH 5.0, 0.04% PEG, 4% DMSO) 중에 10 ㎛ 기질과 120 분동안 화합물의 부재 또는 존재하에서 인큐베이션한다. 이어, 단백질가수분해량을 T=0 및 T=120에서 형광 측정 (320 nm에서 여기 및 405 nm에서 발광)으로 직접 측정한다. 결과를 T120와 TO 사이의 차이로서 RFU (상대 형광 단위)로 나타내었다.
최소 제곱합의 방법으로 화합물 농도에 대해 대조군최소%(%Controlmin)를 플롯팅하여 최고의 적합 곡선을 조정하였다. 이로부터 IC50 값 (활성을 50% 저해하는 저해 농도)을 구할 수 있다.
LC = 대조군 저치의 중간
= 저 대조군: 효소 부재하의 반응
HC = 대조군 고치의 중간
= 고 대조군: 효소 존재하의 반응
효과% = 100-[(샘플-LC)/(HC-LC)×100]
대조군% = (샘플/HC)×100
대조군최소% = (샘플-LC)/(HC-LC)×100
하기 예시된 화합물들에 대해서 실질적으로 상술된 바와 같이 시험하고 하기 활성을 얻었다:
표 5


SKNBE2 세포에서 세포 αLisa 검정
두 αLisa 검정에서 인간 신경모세포종 SKNBE2의 배지에 생산되고 분비된 Aβtotal 및 Aβ42의 수준을 정량하였다. 이 검정은 야생형 아밀로이드 전구체 단백질 (hAPP695)을 발현하는 인간 신경모세포종 SKBE2를 기반으로 한다. 화합물을 희석하여 이 세포에 첨가한 후, 18 시간동안 인큐베이션하고, Aβ42 및 Aβtotal을 측정하였다. Aβtotal 및 Aβ42는 샌드위치 αLisa로 측정하였다. αLisa는 스트렙타비딘 코팅 비드에 부착된 비오티닐화 항체 AbN/25 및 각각 Aβtotal 및 Aβ42를 검출하기 위한 항체 Ab4G8 또는 cAb42/26 접합 수용체 비드를 사용하는 샌드위치 검정이다. Aβtotal 또는 Aβ42의 존재하에, 비드는 근접한다. 공여 비드의 여기는 수용체 비드에서 에너지 전이 케스케이드를 촉발하여 광 방출을 일어나게 하는 단일 산소 분자의 방출을 유도한다. 광 방출은 1 시간 인큐베이션 후에 측정한다 (650 nm에서 여기 및 615 nm에서 방출).
최소 제곱합의 방법으로 화합물 농도에 대해 대조군최소%(%Controlmin)를 플롯팅하여 최고의 적합 곡선을 조정하였다. 이로부터 IC50 값 (활성을 50% 저해하는 저해 농도)을 구할 수 있다.
LC = 대조군 저치의 중간
= 저 대조군: αLisa에서 비오티닐화 Ab없이 화합물 부재하에 예비인큐베이션된 세포
HC = 대조군 고치의 중간
= 고 대조군: 화합물 부재하에 예비인큐베이션된 세포
효과% = 100-[(샘플-LC)/(HC-LC)×100]
대조군% = (샘플/HC)×100
대조군최소% = (샘플-LC)/(HC-LC)×100
하기 예시된 화합물들에 대해서 실질적으로 상술된 바와 같이 시험하고 하기 활성을 얻었다:
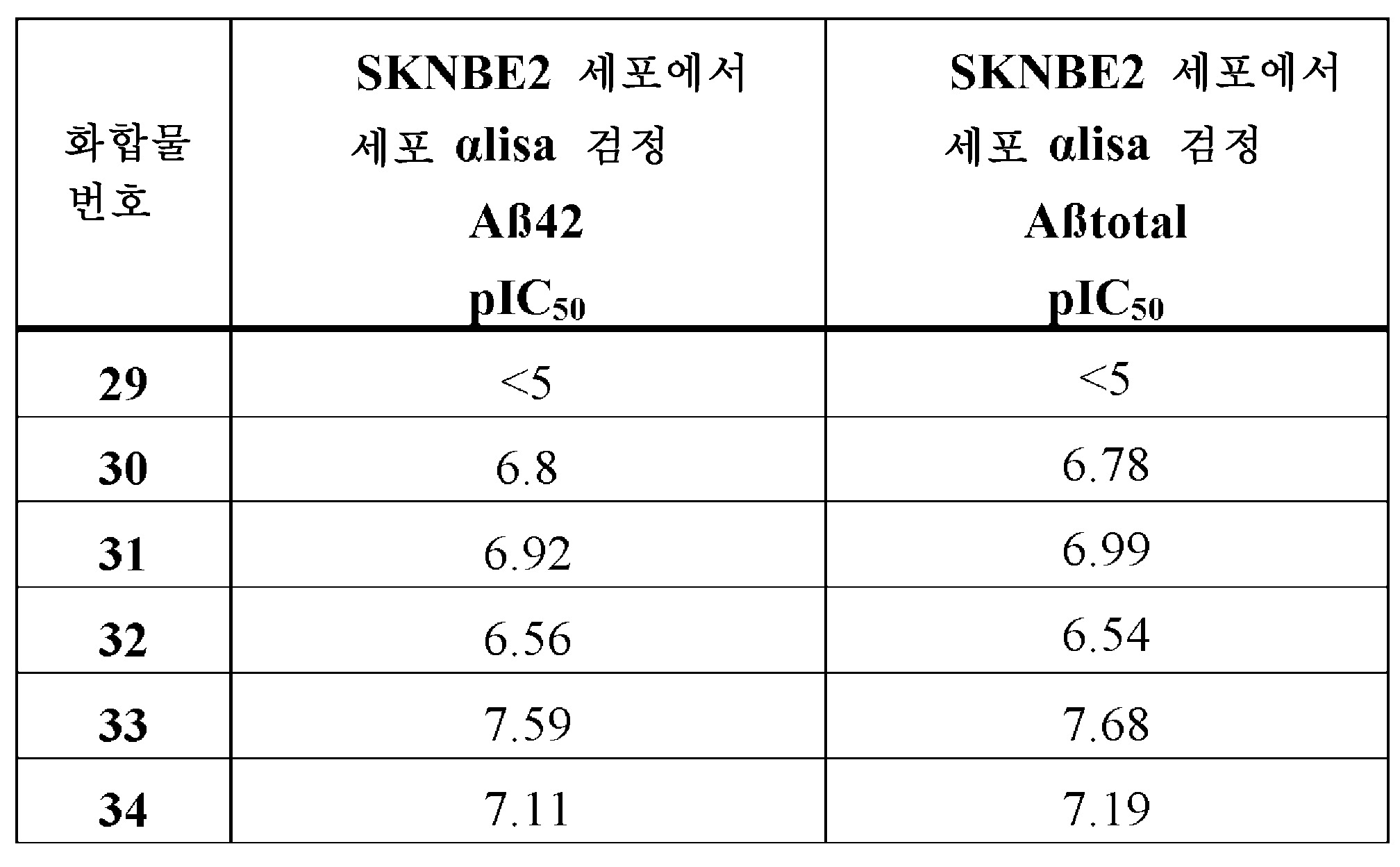
표 6

n.t. 검사되지 않음을 의미한다.
생체내 효과 입증
본 발명의 Aβ 펩티드 저하제는 인간과 같은 포유동물에서 AD를 치료하기 위해 사용될 수 있거나, 마우스, 래트 또는 기니피그가 예시되나 이들에만 한정되지 않는 동물 모델에서 효과를 입증하는데 사용될 수 있다. 포유동물은 AD로 진단되지 않았거나, AD에 대한 유전적 소질을 가지지 않았으나, AD에 걸린 인간에서 나타나는 것과 유사한 방식으로 Aβ를 과생산하여 마침내는 이를 침착시키도록 유전자이식된 것일 수 있다.
Aβ 펩티드 저하제는 임의의 표준 방법을 이용하여 임의의 표준 형태로 투여될 수 있다. 예를 들어, Aβ 펩티드 저하제는 경구 또는 주사로 취해지는 액체, 정제 또는 캡슐제의 형태일 수 있으나, 이들로 제한되는 것은 아니다. Aβ 펩티드 저하제는 혈액, 혈장, 혈청, 뇌척수액 (CSF) 또는 뇌에서 Aβ 펩티드 수준을 현저하게 감소시키기에 충분한 임의적 용량으로 투여될 수 있다.
Aβ 펩티드 저하제의 급성 투여가 생체내에서 Aβ 펩티드 수준을 감소시킬 수 있는지를 결정하기 위하여, 비-유전자이식 설치류, 예를 들면, 마우스 또는 래트를 사용하였다. Aβ 펩티드 저하제로 처리된 동물을 비처리 또는 비히클로 처리된 동물과 비교 조사하고, 가용성 Aβ42 및 전체 Aβ의 뇌 수준을 표준 기술, 예를 들어, ELISA를 사용하여 정량할 수 있다. 처리 기간을 수시간 에서 수일로 변화시키고, 일단 효과 개시 기간이 확정되면, Aβ42 저하 결과에 기초해 조정한다.
생체내 Aβ42 저하를 측정하기 위한 전형적인 프로토콜이 제시되지만, 검출가능한 Aβ 수준을 최적화하기 위해 사용될 수 있는 수 많은 변형방법 중 하나일 뿐이다. 예를 들어, Aβ 펩티드 저하 화합물을 20% 하이드록시프로필 β 사이클로덱스트린에서 제제화한다. Aβ 펩티드 저하제를 밤새 굶긴 동물에 단일 경구 투여(p.o.) 또는 단일 피하 투여(s.c.) 경로로 투여한다. 특정 시간, 보통 2 내지 4 시간(표 19에 제시된 바와 같이) 후, 동물을 희생시키고 Aβ42 수준을 분석한다.
단두하고 방혈시켜 혈액을 EDTA-처리된 수집관에 모은다. 혈액을 4 ℃에서 10 분간 1900 g으로 원심분리하고, 혈장을 회수하여 나중 분석을 위하여 급냉동시킨다. 두개골과 후뇌로부터 뇌를 제거한다. 소뇌를 제거하여 좌 및 우측 반구로 분리한다. 좌측 반구는 시험 화합물 수준의 정량적 분석을 위하여 -18 ℃에서 보관한다. 우측 반구는 인산염-완충 염수 (PBS) 완충액으로 세정하여 드라이 아이스상에서 급냉동시켜 생화학적 분석을 위하여 균질화시킬 때까지 -80 ℃에서 보관한다.
비-유전자이식 동물 유래 마우스의 뇌를 예를 들어, 뇌 0.158 g에 대해 조직 1 그램 당 8 배 용적의 0.4% DEA (디에틸아민)/50 mM NaCl 함유 프로테아제 저해제 (Roche-11873580001 또는 0469315900)에 재현탁시키고, 0.4% DEA 1.264 ㎖를 가한다. 모든 샘플을 20 초동안 6 m/s로 용해 매트릭스 D(MPBio #6913-100)를 사용하여 FastPrep-24 시스템(MP Biomedicals)에서 균질화한다. 균질물을 221,300 x g으로 50 분간 원심분리시킨다. 이어서, 생성된 고속 상등물을 새로운 에펜도르프 시험관으로 옮긴다. 상등액의 9부를 1부 0.5M Tris-HCl(pH6.8)로 중성화시키고 Aβtotal 및 Aβ42를 정량하는데 사용한다.
뇌 균질물의 가용성 분획에서 Aβtotal 및 Aβ42의 양을 정량하기 위하여, 효소-결합-면역흡착측정법을 사용한다. 간략하면, 표준물질 (합성 Aβ1-40 및 Aβ1-42의 희석액, Bachem)을 최종 농도 범위 10000 내지 0.3 pg/㎖로, Ultraculture 중의 1.5 ㎖ 에펜도르프 시험관에 준비한다. 샘플 및 표준물질을 Aβ42 검출을 위해 HPRO-표지 N-말단 항체 및 Aβtotal 검출을 위해 비오티닐화 중-도메인 항체 4G8과 공배양한다. 이어, 접합체/샘플 또는 접합체/표준물 혼합물 50 ㎕를 항체-코팅 플레이트 (캡쳐 항체는 Aβ42 검출을 위해 Aβ42의 C-종결 말단, 항체 JRF/cAβ42/26 및 Aβtotal 검출을 위해 Aβ의 N-종결 말단, 항체 JRF/rAβ42/2을 선택적으로 인식한다)에 가한다. 상기 플레이트를 4 ℃에서 밤새 배양시켜 항체-아밀로이드 복합체를 형성시킨다. 상기 배양 및 후속 세척 단계 후, 제조업자의 지시에 따라 Quanta Blu 형광발생성 퍼옥시다제 기질 (Pierce Corp., Rockford, Il)을 가해 Aβ42 정량화를 위한 ELISA를 마친다. 10 내지 15 분후 판독한다 (여기 320/방출 420).
Aβtotal의 검출을 위해, 스트렙타비딘-퍼옥시다제-접합체를 첨가하고, 60 분후 추가 세척 단계 후, 제조업자의 지시에 따라 Quanta Blu 형광발생성 퍼옥시다제 기질 (Pierce Corp., Rockford, Il)을 가한다. 10 내지 15 분후 판독한다 (여기 320/방출 420).
상기 모델에서는, 비처리 동물과 비교하여 Aβ42를 적어도 20% 저하시키는 것이 유리하다.
하기 예시된 화합물을 실질적으로 상술된 바와 같이 시험하고 하기 활성을 얻었다:
표 20

s.c.는 피하 투여를 의미히고; p.o.는 경구 투여를 의미한다.
Claims (8)
- 화학식 (I)의 화합물 또는 그의 호변이성체 또는 입체이성체, 또는 이들의 부가염 또는 용매화물:

상기 식에서,
R1 및 R2는 독립적으로 수소, 플루오로, 시아노, C1-3알킬, 모노- 및 폴리할로-C1-3알킬 및 C3-6사이클로알킬로 구성된 군으로부터 선택되거나; 또는
R1 및 R2는 이들이 결합된 탄소원자와 함께, C3-6사이클로알칸디일 환을 형성할 수 있고;
R3는 수소, C1-3알킬, C3-6사이클로알킬, 모노- 및 폴리할로-C1-3알킬, 호모아릴 및 헤테로아릴로 구성된 군으로부터 선택되며;
X1, X2, X3, X4는 독립적으로 C(R4) 또는 N이나, 단 N을 나타내는 것은 두개를 넘지 않고; 각 R4는 수소, 할로, C1-3알킬, 모노- 및 폴리할로-C1-3알킬, 시아노, C1-3알킬옥시, 모노- 및 폴리할로-C1-3알킬옥시로 구성된 군으로부터 선택되며;
L은 결합 또는 -N(R5)CO-이고, 여기서 R5는 수소 또는 C1-3알킬이고;
R6은 수소 또는 트리플루오로메틸이며;
Ar은 호모아릴 또는 헤테로아릴이고;
여기에서, 호모아릴은 페닐, 또는 할로, 시아노, C1-3알킬, C1-3알킬옥시, 모노 및 폴리할로-C1-3알킬; 및 모노- 및 폴리할로-C1-3알킬옥시로 구성된 군으로부터 선택되는 1, 2 또는 3개의 치환체로 치환된 페닐이고;
헤테로아릴은 각각 할로, 시아노, C1-3알킬, C1-3알킬옥시, 모노- 및 폴리할로-C1-3알킬 및 모노- 및 폴리할로-C1-3알킬옥시로 구성된 군으로부터 선택되는 1, 2 또는 3개의 치환체로 임의로 치환된 피리딜, 피리미딜, 피라질, 피리다질, 푸라닐, 티에닐, 피롤릴, 피라졸릴, 이미다졸릴, 트리아졸릴, 티아졸릴, 티아디아졸릴, 옥사졸릴, 및 옥사디아졸릴로 구성된 군으로부터 선택된다. - 제 1 항에 있어서,
R1 및 R2는 독립적으로 수소 및 C1-3알킬로부터 선택되고;
X1, X2, X3, X4는 독립적으로 C(R4)이고, 여기서 각 R4는 수소 및 할로로부터 선택되며;
L은 결합 또는 -N(R5)CO-이고, 여기서 R5는 수소이고;
Ar은 호모아릴 또는 헤테로아릴이며;
호모아릴은 페닐, 또는 할로, 시아노, C1-3알킬, 및 C1-3알킬옥시로 구성된 군으로부터 선택되는 1 또는 2개의 치환체로 치환된 페닐이고;
헤테로아릴은 각각 할로, 시아노, C1-3알킬, 및 C1-3알킬옥시로 구성된 군으로부터 선택되는 1 또는 2개의 치환체로 임의로 치환된 피리딜, 피리미딜 및 피라질로 구성된 군으로부터 선택되는
화합물; 또는 그의 부가염 또는 용매화물. - 제 1 항에 있어서,
R1 및 R2는 수소이고;
X1, X2, X3, X4는 CH이며;
L은 결합 또는 -N(R5)CO-이고, 여기서 R5는 수소이고;
Ar은 호모아릴 또는 헤테로아릴이며;
호모아릴은 클로로로 치환된 페닐이고;
헤테로아릴은 각각 클로로, 플루오로, 시아노, 메틸, 및 메톡시로 구성된 군으로부터 선택되는 1 또는 2개의 치환체로 임의로 치환된 피리딜 및 피리미딜로 구성된 군으로부터 선택되는
화합물; 또는 그의 부가염 또는 용매화물. - 제 1 항에 있어서, R3으로 치환된 탄소 원자가 R-배열을 갖는 화합물.
- 제 1 항 내지 4 항중 어느 한항에 정의된 화합물의 치료적 유효량 및 약제학적으로 허용가능한 담체를 포함하는 약제학적 조성물.
- 약제학적으로 허용가능한 담체를 제 1 항 내지 4 항중 어느 한항에 정의된 화합물의 치료적 유효량과 충분히 혼합하는 것을 포함하는 제 5 항에 정의된 약제학적 조성물의 제조방법.
- 제 1 항 내지 4 항중 어느 한항에 있어서, 알츠하이머병 (AD), 경증 인지 장애, 노쇠, 치매, 레비소체치매, 다운증후군, 뇌졸중 관련 치매, 파킨슨병 관련 치매 또는 베타-아밀로이드 관련 치매를 치료, 방지 또는 예방하는데 사용하기 위한 화합물.
- 치료적 유효량의 제 1 항 내지 4 항중 어느 한항에 정의된 화합물 또는 제 5 항에 정의된 약제학적 조성물을 치료를 필요로 하는 대상에 투여하는 것을 포함하는, 알츠하이머병, 경증 인지 장애, 노쇠, 치매, 레비소체치매, 다운증후군, 뇌졸중 관련 치매, 파킨슨병 관련 치매 및 베타-아밀로이드 관련 치매로부터 선택되는 질환의 치료방법.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10178315.7 | 2010-09-22 | ||
| EP10178315 | 2010-09-22 | ||
| EP11157858.9 | 2011-03-11 | ||
| EP11157858 | 2011-03-11 | ||
| PCT/EP2011/066343 WO2012038438A1 (en) | 2010-09-22 | 2011-09-20 | 4,7-DIHYDRO-PYRAZOLO[1,5-a]PYRAZIN-6-YLAMINE DERIVATIVES USEFUL AS INHIBITORS OF BETA-SECRETASE (BACE) |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20130110161A true KR20130110161A (ko) | 2013-10-08 |
| KR101962216B1 KR101962216B1 (ko) | 2019-03-26 |
Family
ID=45873470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020137007365A KR101962216B1 (ko) | 2010-09-22 | 2011-09-20 | β-세크레타아제(BACE) 저해제로 유용한 4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 유도체 |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US8609660B2 (ko) |
| EP (1) | EP2619207B1 (ko) |
| JP (1) | JP5797756B2 (ko) |
| KR (1) | KR101962216B1 (ko) |
| CN (1) | CN103097389B (ko) |
| AU (1) | AU2011306941B2 (ko) |
| BR (1) | BR112013006353A2 (ko) |
| CA (1) | CA2807904C (ko) |
| CL (1) | CL2013000782A1 (ko) |
| CO (1) | CO6660450A2 (ko) |
| EA (1) | EA021723B1 (ko) |
| ES (1) | ES2529641T3 (ko) |
| HK (1) | HK1182391A1 (ko) |
| IL (1) | IL225349A (ko) |
| IN (1) | IN2013MN00782A (ko) |
| MX (1) | MX2013003246A (ko) |
| MY (1) | MY160138A (ko) |
| NZ (1) | NZ606848A (ko) |
| SG (1) | SG188338A1 (ko) |
| WO (1) | WO2012038438A1 (ko) |
| ZA (1) | ZA201302128B (ko) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2572263T3 (es) | 2005-10-25 | 2016-05-31 | Shionogi & Co | Derivados de dihidrooxazina y tetrahidropirimidina como inhibidores de BACE 1 |
| EP2147914B1 (en) | 2007-04-24 | 2014-06-04 | Shionogi&Co., Ltd. | Aminodihydrothiazine derivatives substituted with cyclic groups |
| EP2151435A4 (en) | 2007-04-24 | 2011-09-14 | Shionogi & Co | PHARMACEUTICAL COMPOSITION FOR THE TREATMENT OF ALZHEIMER'S DISEASE |
| KR101324426B1 (ko) | 2008-06-13 | 2013-10-31 | 시오노기세야쿠 가부시키가이샤 | β 세크레타제 저해 작용을 갖는 황 함유 복소환 유도체 |
| EP2360155A4 (en) | 2008-10-22 | 2012-06-20 | Shionogi & Co | 2-AMINOPYRIDIN-4-ON AND 2-AMINOPYRIDINE DERIVATIVE WITH BACE1-HEMDERING EFFECT |
| CN102834384A (zh) | 2009-12-11 | 2012-12-19 | 盐野义制药株式会社 | *嗪衍生物 |
| BR112012031094A2 (pt) | 2010-06-09 | 2016-10-25 | Janssen Pharmaceutica Nv | derivados de 5,6-di-hidro-2h-[1,4]oxazin-3-il-maina úteis como inibidores de beta-secretase (bace) |
| US9018219B2 (en) | 2010-10-29 | 2015-04-28 | Shionogi & Co., Ltd. | Fused aminodihydropyrimidine derivative |
| WO2012057248A1 (ja) | 2010-10-29 | 2012-05-03 | 塩野義製薬株式会社 | ナフチリジン誘導体 |
| JP5834091B2 (ja) | 2010-12-22 | 2015-12-16 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプJanssen Pharmaceutica Naamloze Vennootschap | ベータ−セクレターゼ(BACE)の阻害剤として有用な5,6−ジヒドロ−イミダゾ[1,2−a]ピラジン−8−イルアミン誘導体 |
| CN103415521B (zh) | 2011-03-09 | 2016-01-06 | 詹森药业有限公司 | 用作β分泌酶(BACE)抑制剂的3,4-二氢-吡咯并[1,2-a]吡嗪-1-基胺衍生物 |
| EP2703399A4 (en) | 2011-04-26 | 2014-10-15 | Shionogi & Co | OXAZINE DERIVATIVE AND BACE-1 HEMMER THEREOF |
| CA2840196C (en) | 2011-06-27 | 2022-09-20 | Eisai R&D Management Co., Ltd. | Microrna biomarkers indicative of alzheimer's disease |
| WO2014065434A1 (en) | 2012-10-24 | 2014-05-01 | Shionogi & Co., Ltd. | Dihydrooxazine or oxazepine derivatives having bace1 inhibitory activity |
| KR102243135B1 (ko) | 2013-06-12 | 2021-04-22 | 얀센 파마슈티카 엔.브이. | 베타-세크레타제(bace) 저해제로서의 4-아미노-6-페닐-5,6-디하이드로이미다조[1,5-a]피라진-3(2h)-온 유도체 |
| EA028775B1 (ru) | 2013-06-12 | 2017-12-29 | Янссен Фармацевтика Нв | Производные 4-амино-6-фенил-6,7-дигидро[1,2,3]триазоло[1,5-a]пиразина в качестве ингибиторов бета-секретазы (bace) |
| CN105283457B (zh) | 2013-06-12 | 2018-09-18 | 詹森药业有限公司 | 作为β-分泌酶(BACE)抑制剂的4-氨基-6-苯基-5,6-二氢咪唑并[1,5-A]吡嗪衍生物 |
| ES2768823T3 (es) | 2014-12-18 | 2020-06-23 | Janssen Pharmaceutica Nv | Derivados de 2,3,4,5-tetrahidropiridin-6-amina y 3,4-dihidro-2H-pirrol-5-amina útiles como inhibidores de beta-secretasa |
| WO2018165520A1 (en) | 2017-03-10 | 2018-09-13 | Vps-3, Inc. | Metalloenzyme inhibitor compounds |
| WO2019025250A1 (en) | 2017-08-04 | 2019-02-07 | Basf Se | SUBSTITUTED TRIFLUOROMETHYLOXADIAZOLES FOR COMBATING PHYTOPATHOGENIC FUNGI |
| CN108440320A (zh) * | 2018-05-09 | 2018-08-24 | 上海凌凯医药科技有限公司 | 一种高手性选择性合成α-双取代α-氨基酸的方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007058583A2 (en) * | 2005-11-15 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-heterocycles useful in the treatment of abeta-related pathologies |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003301226A1 (en) * | 2002-12-20 | 2004-07-22 | Pharmacia Corp | Acyclic pyrazole compounds for the inhibition of mitogen activated protein kinase-activated protein kinase-2 |
| BRPI0612545A2 (pt) * | 2005-06-14 | 2010-11-23 | Schering Corp | compostos inibidores de protease, composições farmacêuticas e uso dos mesmos |
| WO2011002409A1 (en) * | 2009-07-02 | 2011-01-06 | Astrazeneca Ab | 5h-pyrrolo[3,4-£>]pyrazin-7-amine derivatives inhibitors of beta-secretase |
-
2011
- 2011-09-20 KR KR1020137007365A patent/KR101962216B1/ko active IP Right Grant
- 2011-09-20 JP JP2013528719A patent/JP5797756B2/ja not_active Expired - Fee Related
- 2011-09-20 WO PCT/EP2011/066343 patent/WO2012038438A1/en active Application Filing
- 2011-09-20 IN IN782MUN2013 patent/IN2013MN00782A/en unknown
- 2011-09-20 NZ NZ606848A patent/NZ606848A/en not_active IP Right Cessation
- 2011-09-20 EP EP11757886.4A patent/EP2619207B1/en active Active
- 2011-09-20 BR BR112013006353A patent/BR112013006353A2/pt not_active IP Right Cessation
- 2011-09-20 AU AU2011306941A patent/AU2011306941B2/en not_active Ceased
- 2011-09-20 MY MYPI2013000589A patent/MY160138A/en unknown
- 2011-09-20 SG SG2013015136A patent/SG188338A1/en unknown
- 2011-09-20 EA EA201390419A patent/EA021723B1/ru not_active IP Right Cessation
- 2011-09-20 MX MX2013003246A patent/MX2013003246A/es active IP Right Grant
- 2011-09-20 US US13/825,139 patent/US8609660B2/en not_active Expired - Fee Related
- 2011-09-20 CA CA2807904A patent/CA2807904C/en not_active Expired - Fee Related
- 2011-09-20 ES ES11757886.4T patent/ES2529641T3/es active Active
- 2011-09-20 CN CN201180045440.3A patent/CN103097389B/zh not_active Expired - Fee Related
-
2013
- 2013-03-20 CO CO13055809A patent/CO6660450A2/es unknown
- 2013-03-20 ZA ZA2013/02128A patent/ZA201302128B/en unknown
- 2013-03-20 IL IL225349A patent/IL225349A/en active IP Right Grant
- 2013-03-21 CL CL2013000782A patent/CL2013000782A1/es unknown
- 2013-08-21 HK HK13109786.4A patent/HK1182391A1/zh not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007058583A2 (en) * | 2005-11-15 | 2007-05-24 | Astrazeneca Ab | Novel 2-amino-heterocycles useful in the treatment of abeta-related pathologies |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2807904A1 (en) | 2012-03-29 |
| CA2807904C (en) | 2018-09-11 |
| BR112013006353A2 (pt) | 2024-01-16 |
| ES2529641T3 (es) | 2015-02-24 |
| MY160138A (en) | 2017-02-28 |
| SG188338A1 (en) | 2013-04-30 |
| CO6660450A2 (es) | 2013-04-30 |
| CL2013000782A1 (es) | 2013-08-09 |
| NZ606848A (en) | 2015-01-30 |
| IL225349A0 (en) | 2013-06-27 |
| AU2011306941A1 (en) | 2013-02-28 |
| WO2012038438A1 (en) | 2012-03-29 |
| EA201390419A1 (ru) | 2013-07-30 |
| EP2619207A1 (en) | 2013-07-31 |
| AU2011306941B2 (en) | 2014-06-12 |
| CN103097389B (zh) | 2015-10-14 |
| JP2013540740A (ja) | 2013-11-07 |
| MX2013003246A (es) | 2013-05-22 |
| HK1182391A1 (zh) | 2013-11-29 |
| IL225349A (en) | 2016-07-31 |
| US8609660B2 (en) | 2013-12-17 |
| KR101962216B1 (ko) | 2019-03-26 |
| EP2619207B1 (en) | 2014-11-12 |
| US20130190318A1 (en) | 2013-07-25 |
| IN2013MN00782A (ko) | 2015-06-12 |
| EA021723B1 (ru) | 2015-08-31 |
| CN103097389A (zh) | 2013-05-08 |
| ZA201302128B (en) | 2014-08-27 |
| JP5797756B2 (ja) | 2015-10-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101962216B1 (ko) | β-세크레타아제(BACE) 저해제로 유용한 4,7-디하이드로피라졸로[1,5-a]피라진-6-일아민 유도체 | |
| KR102012675B1 (ko) | 베타-세크레타제(BACE)의 억제제로서 유용한 3,4-디하이드로-피롤로[1,2-a]피라진-1-일아민 유도체 | |
| KR101866987B1 (ko) | 베타-세크레타아제(BACE) 저해제로 유용한 5,6-디하이드로-이미다조[1,2-a]피라진-8-일-아민 유도체 | |
| JP5853033B2 (ja) | β−セクレターゼ(BACE)の阻害剤として有用な6,7−ジヒドロ−ピラゾロ[1,5−a]ピラジン−4−イルアミン誘導体 | |
| NZ614551B2 (en) | 6,7-dihydro-pyrazolo[1,5-a]pyrazin-4-ylamine derivatives useful as inhibitors of beta-secretase (bace) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant |